Область, к которой относится изобретение
Изобретение относится к медицине, а именно к веществам, являющимися ингибиторами дипептидилпептидазы-4, из группы амидов бета-аминокислот - N-ацильных производных 3-(3-азабицикло[2.2.1]гептан-2-ил)-1,2,4-оксадиазола и 3-азабицикло[2.2.1]гептен-2-карбонитрила для лечения сахарного диабета.
Уровень техники
Сахарный диабет является одной из серьезнейших медико-социальных проблем в связи с высокой распространенностью, стремительным ростом заболеваемости, высокой частотой инвалидизации и сохраняющейся высокой летальностью из-за развития осложнений этого заболевания. Существуют две формы диабета. Сахарный диабет 1-го типа или инсулин-зависимый, в котором инсулин - гормон, регулирующий утилизацию глюкозы, не образуется или образуется в малых количествах. Инсулин-независимый диабет 2-го типа характеризуется тем, что его уровень в крови такой же или чуть выше, чем у людей, не страдающих диабетом, однако, при этом присутствует устойчивость к стимулирующему действию инсулина на метаболизм глюкозы в инсулин-чувствительных тканях. Причиной же является не снижение количества рецепторов к инсулину, а следующий за связыванием инсулина с рецептором дефект, который и приводит к недостаточности активации инсулином утилизации в тканях глюкозы и неадекватному подавлению инсулином липолиза в жировой ткани.
По данным ВОЗ сахарный диабет 2-го типа (СД-2), в свою очередь выявлен у ~90% всех больных сахарным диабетом. Во всем мире значительно увеличилось количество ранее редких случаев заболевания детей диабетом 2-го типа. По прогнозам ВОЗ в 2030 году диабет станет седьмой по значимости причиной смерти.
В настоящее время на фармацевтическом рынке имеется ряд препаратов для коррекции состояния лиц, страдающих сахарным диабетом 2-го типа.
Бигуаниды, сахароснижающее действие которых обусловлено механизмами действия, не связанными с секрецией инсулина β-клетками. Препараты сульфонилмочевины (СМ) - основной механизм действия препаратов СМ заключается в стимуляции секреции инсулина. Прандиальные регуляторы (глиниды) - короткодействующие препараты, реализующие свои сахароснижающие свойства путем острой стимуляции секреции инсулина после принятия пищи. Ингибиторы α-глюкозидазы - к этой группе препаратов относятся средства, которые конкурируют с пищевыми углеводами за связывающие центры ферментов желудочно-кишечного тракта, участвующих в расщеплении и всасывании углеводов. Ингибиторы дипептидилпептидазы-4 и инкретиномиметики - механизм действия этих препаратов тесно связан с основными биологическими эффектами гормонов желудочно-кишечного тракта и состоит в усилении глюкозозависимого инсулинового ответа и одновременном подавлении глюкозозависимой секреции глюкагона на фоне повышения уровня глюкозы крови. Глитазоны - PPAR-гамма агонисты - препараты этой группы принадлежат к новому классу пероральных сахароснижающих агентов, действующих на уровне рецепторов, активируемых пролиферацией пероксисом (PPARγ).
Одним из востребованных подходов для лечения симптомов сахарного диабета являются ингибиторы дипептидилпептидазы-4.
Механизм действия этих препаратов, как и действие инкретиномиметиков, тесно связан с основными биологическими эффектами гормонов желудочно-кишечного тракта и состоит в усилении глюкозозависимого инсулинового ответа и одновременном подавлении глюкозозависимой секреции глюкагона на фоне повышения уровня глюкозы крови.
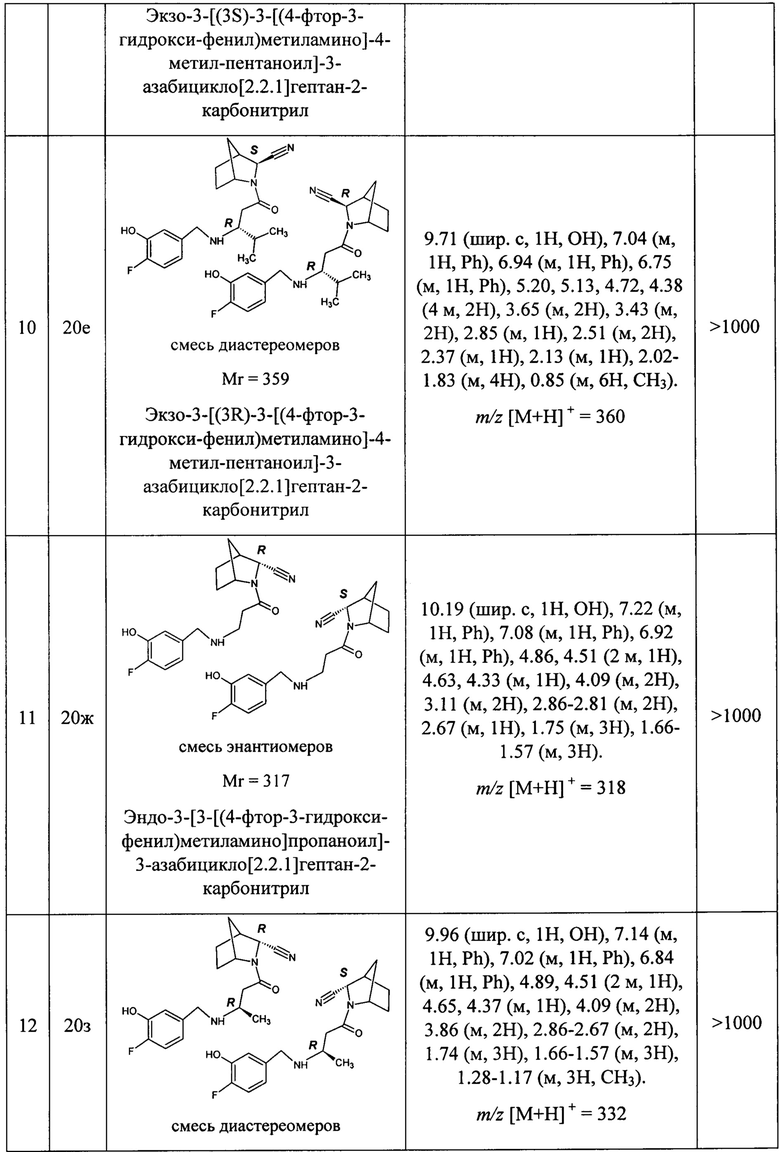
Поскольку прием большинства препаратов сопровождается серьезными побочными эффектами в виде гипогликемии, лактатацидоза, гепатотоксичности, тошноты, диареи, сердечной недостаточности, отечности, остеопороза и др., что существенно ухудшает качество жизни пациентов, особую актуальность приобретает разработка новых, более эффективных, патогенетически обоснованных подходов к терапии этого заболевания.
Одним из звеньев патогенеза СД 2-го типа является нарушение функции инкретинов - гормонов желудочно-кишечного тракта, вырабатываемых в ответ на прием пищи и вызывающих стимуляцию секреции инсулина (инкретин - от англ. INCRETIN - INtestine seCRETion of INsulin). В организме человека известны два основных инкретина: глюкозозависимый инсулинотропный полипептид, также известный как гастроингибиторньй пептид (ГИП) или желудочный ингибиторный пептид (ЖИП), вырабатываемый К-клетками двенадцатиперстной и тощей кишок, а также глюкагон-подобный пептид-1 (ГПП-1), секретируемый энтероэндокринными L-клетками. То есть, оба гормона вырабатываются эндокринными клетками, расположенными в эпителии тонкой кишки, а их высвобождение регулируется аналогичным образом другими гормонами желудочно-кишечного тракта. Увеличение концентрации вещества в просвете желудочно-кишечного тракта (в данном случае, глюкозы) действует как триггер для секреции гормона. Исследования, свидетельствующие, что у больных СД 2-го типа нарушено не только количественное содержание инкретинов, но и механизм их действия, позволили задуматься о создании группы препаратов, влияющих на уровень инкретинов. Наличие подобных средств смогло бы улучшить гликемический контроль путем воздействия на иные патогенетические звенья данного заболевания, в отличие от существующих групп антидиабетических препаратов.
Снижение эффекта инкретинов, находящихся в кровеносном русле, является следствием их быстрого разрушения и выведения из организма. Причина деградации инкретинов и потери их функции кроется в их структуре - наличии остатка аланина (Ala) во 2-м с N-конца положении. Подобные аминокислотные последовательности являются субстратами для сериновой протеазы - дипептидилпептидазы-4 (ДПП-4).
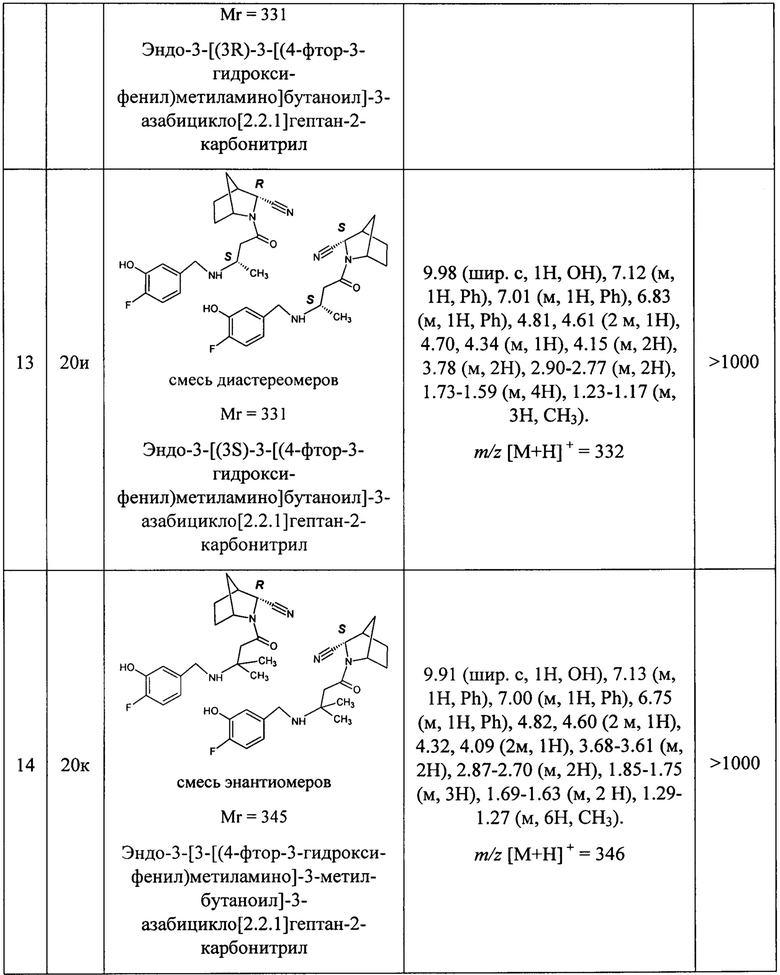
Раскрытие роли ДПП-4 в метаболизме инкретинов явилось ключевым моментом в создании препаратов, увеличивающих время действия эндогенных ГИП и ГПП-1. Такими препаратами являются ингибиторы ДПП-4. Результаты экспериментов на животных моделях продемонстрировали сохранение концентрации инкретинов в крови под влиянием ингибиторов ДПП-4. Измерение концентрации эндогенных инкретинов в плазме крови среди пациентов с СД 2-го типа, у которых прием ингибиторов ДПП-4 снижал уровень гликемии, подтвердило ранее полученные результаты.
Таким образом, разработка новых лекарственных препаратов, являющихся антагонистами ДПП-4, имеет высокую значимость и перспективу получения высокоселективных ингибиторов дипептидилпептидазы-4.
Известны ингибиторы дипептидилпептидазы-4, патенты РФ №2180901, №2251544 «N-замещенные 2-цианопирролидины», заявка US 20130204012.
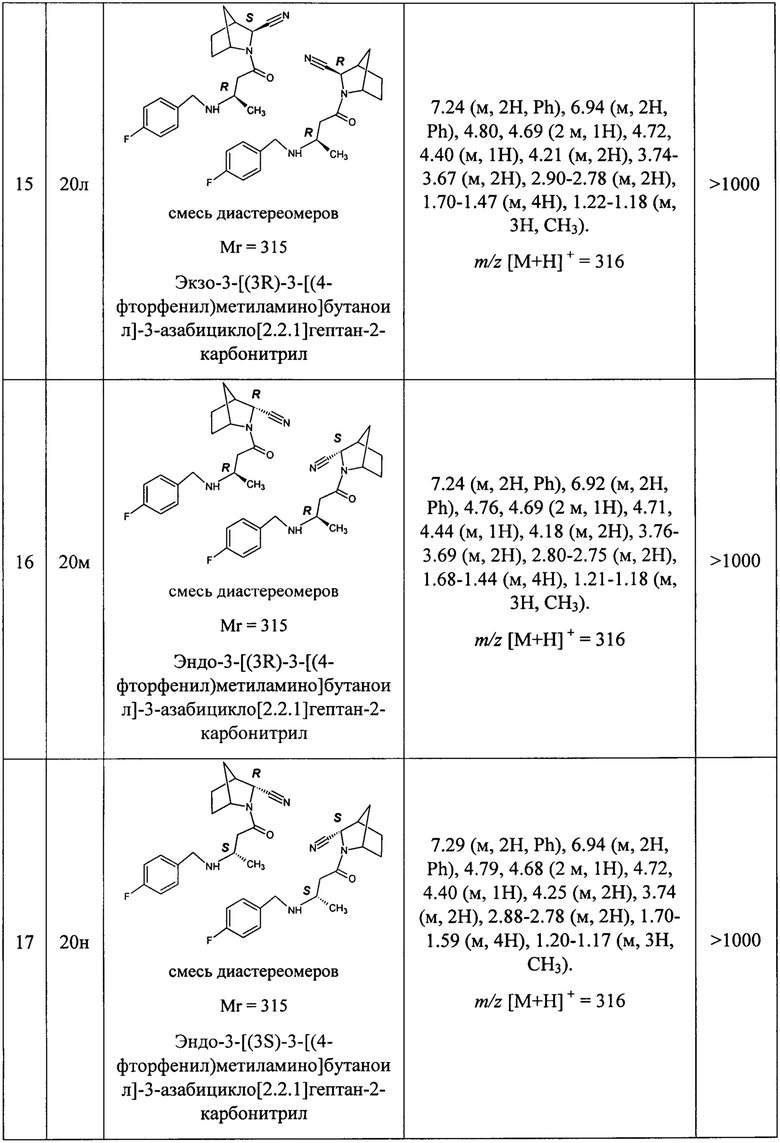
Известен патент РФ №2286986 «Ингибиторы дипептидилпептидазы-4 на основе конденсированных циклопропилпирролидинов и способ их применения».
Известен патент РФ на изобретение №2443687, «Новые ингибиторы дипептидилпептидазы IV, способы их получения и содержащие их фармацевтические композиции».
Известен патент РФ 2483716.
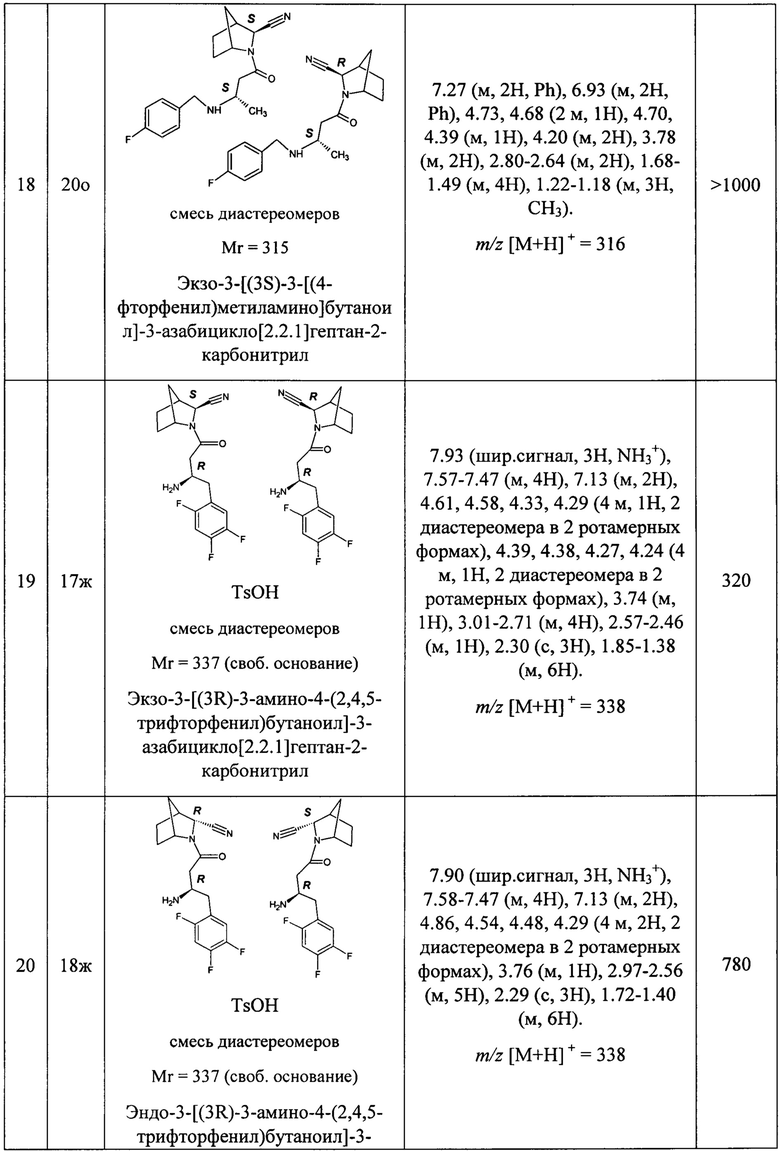
Изобретение относится к медицине и фармацевтической промышленности и касается фармацевтического состава, включающего ингибитор дипептидилпептидазы-4, предпочтительно, вилдаглиптин от 1.5 до 20% и метформин от 80 до 98.5%. При этом активные ингредиенты составляют от 60 до 98% композиции. В качестве связующего вещества используются целлюлоза или ее производные в количестве от 1 до 20%.
Синтезировано новое аминопроизводное пирролидина в качестве ингибитора дипептидилпептидазы-4.
Известно изобретение по заявке US №20130023671 А1, в котором заявлен способ производства саксаглиптина - ингибитора дипептидилпептидазы-4, являющегося производным пирролидина.
Саксаглиптин - селективный обратимый конкурентный ингибитор дипептидилпептидазы-4 (ДПП-4). У пациентов с сахарным диабетом 2 типа прием саксаглиптина приводит к подавлению активности фермента ДПП-4 в течение 24 часов.
После приема внутрь ингибирование ДПП-4 приводит к 2-3 кратному увеличению концентрации глюкагоноподобного пептида-1 (ГПП-1) и глюкозозависимого инсулинотропного пептида (ГИП), уменьшению концентрации глюкагона и усилению глюкозозависимой ответной реакции бета-клеток, что приводит к повышению концентрации инсулина и С-пептида и, соответственно, к снижению гликемии натощак и постпрандиальной гликемии.
В качестве ингибиторов дипептидилпептидазы-4 на фармацевтическом рынке представлены: ситаглиптин, вилдаглиптин, саксаглиптин, линаглиптин, обладающие несколько отличающимися свойствами.
Известен патент, RU 2628573, в котором ингибитор дипептидилпептидазы-4 из группы N-ацильных производных аминоацил-2-цианопирролидина, представляет вещество:
(R/S)-3-амино-1-[(S)-5-фенил-[1,2,4]оксадиазол-3-ил)-пирролидин-1-ил]-3-(4-фторфенил)-пропан-1-он, соответствующее структурной формуле:
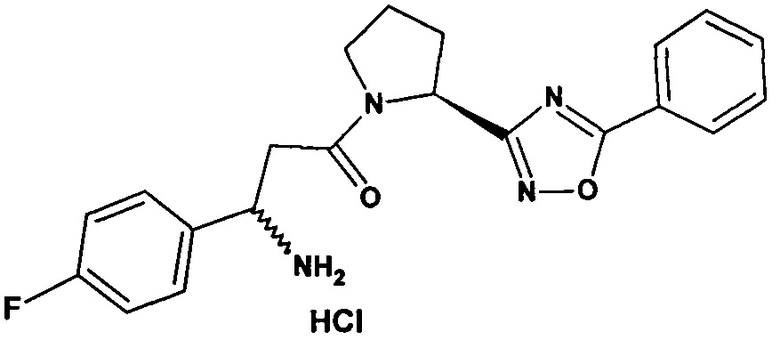
эффективно для лечения сахарного диабета 2 типа, обладает наряду с высокой эффективностью также высокой стабильностью (Прототип)
Несмотря на наличие на рынке коммерческих противодиабетических средств - ингибиторов ДПП-4, есть необходимость в создании новых ингибиторов дипептидилпептидазы-4, сохраняющих свою химическую и пространственную структуру в естественных условиях человеческого организма. При этом соединение должно быть в первую очередь стабильным, даже при не очень высокой его активности, как ингибитора.
Раскрытие изобретения
В настоящее время сохраняется необходимость в создании новых ингибиторов дипептидилпептидазы-4 для расширения линейки известных ингибиторов дипептидилпептидазы-4 с целью удовлетворения потребности в данном препарате.
Задачей заявляемого изобретения является создание ингибиторов дипептидилпептидазы-4, устойчивых к внутримолекулярной циклизации, для лечения больных сахарным диабетом 2-ого типа и расширение линейки известных ингибиторов дипептидилпептидазы-4.
Указанная задача решается за счет того, что
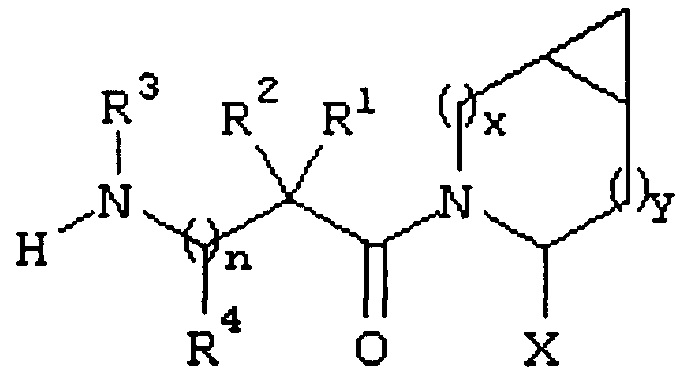
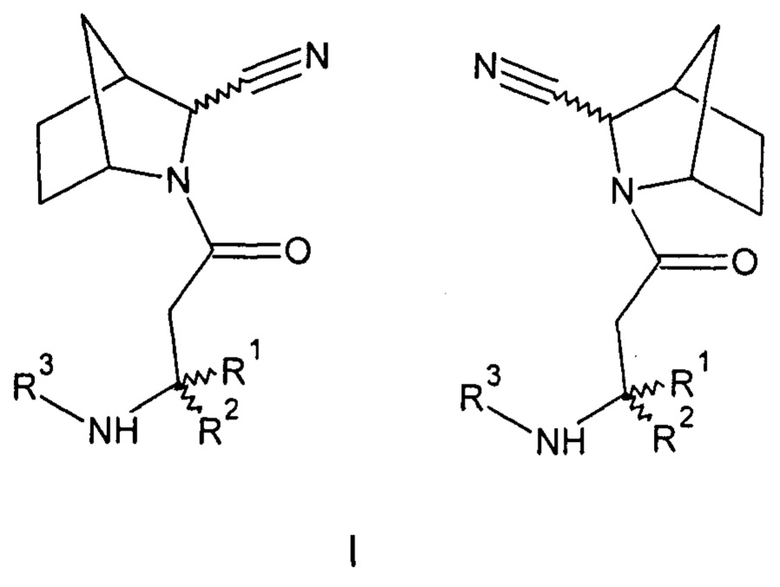
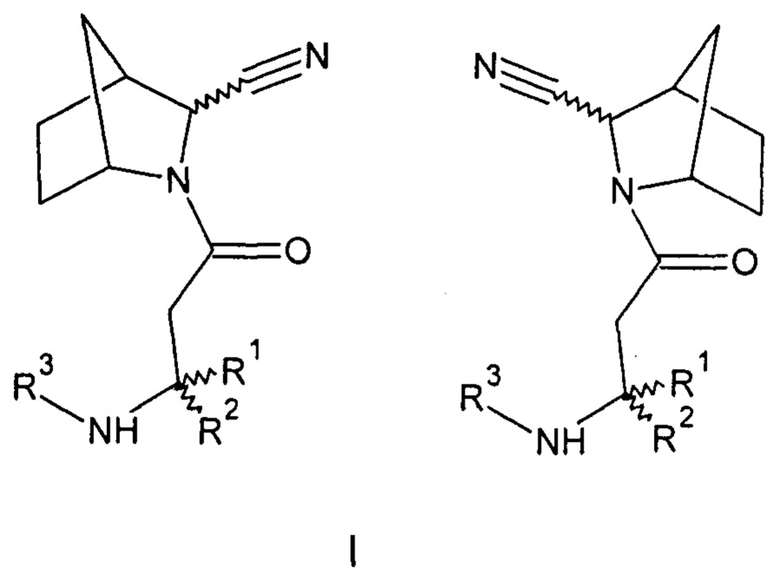
Созданы соединения общей формулы (I) - амиды бета-аминокислот 3-азабицикло[2.2.1]гептен-2-карбонитрила (I) или их фармацевтически приемлемые соли.
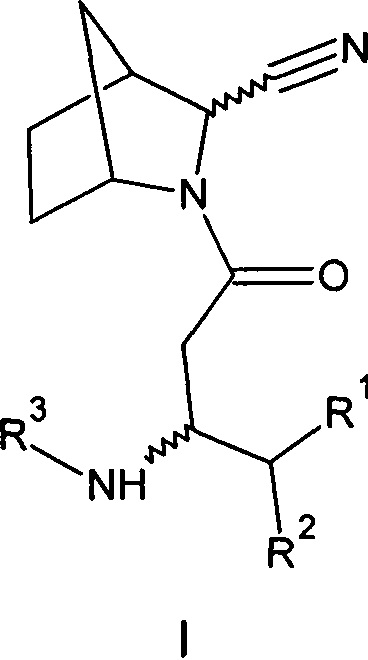
Где:
R1=Н - водород; С1-5 - алкил, в котором каждый углерод независимо может иметь один или два заместителя; арил, в котором каждый углерод ароматического кольца может иметь заместитель вместе или независимо, выбранные из: гидрокси- (ОН-), галоген (F, Cl, Br,);
R2=Н - водород; С1-5 - алкил, в котором каждый углерод независимо может иметь один или два заместителя; адамантил, в котором каждый углерод вместе или независимо может иметь заместитель, выбранный из: гидрокси (ОН-), галоген (F, Cl, Br,); фенил- (С6Н5-), в котором каждый углерод ароматического кольца может иметь заместитель вместе или независимо, выбранный из: гидрокси- (ОН-), галоген (F, Cl, Br,); бензил- (С6Н5-СН2-), в котором каждый углерод ароматического кольца может иметь заместитель вместе или независимо, выбранный из: гидрокси- (ОН-), галоген (F, Cl, Br,);
R3=Н - водород, С1-5 - алкил, в котором каждый углерод независимо может иметь один или два заместителя; бензил- (С6Н5-СН2-), в котором каждый углерод ароматического кольца может иметь заместитель вместе или независимо, выбранный из: гидрокси- (ОН-), галоген (F, Cl, Br,) (вариант I).
При этом синтезированы соединения, где:
R1=H, R2=H, R3=гидроксиадамантил-экзо-3-[3-[(3-гидрокси-1-адамантил)амино]пропаноил]-3-азабицикло[2.2.1]гептен-2-карбонитрил (12а), или
R1=H, R2=H, R3=гидроксиадамантил-экзо-3-[3-[(3-гидрокси-1-адамантил)амино]пропаноил]-3-азабицикло[2.2.1]гептен-2-карбонитрил (12б), или
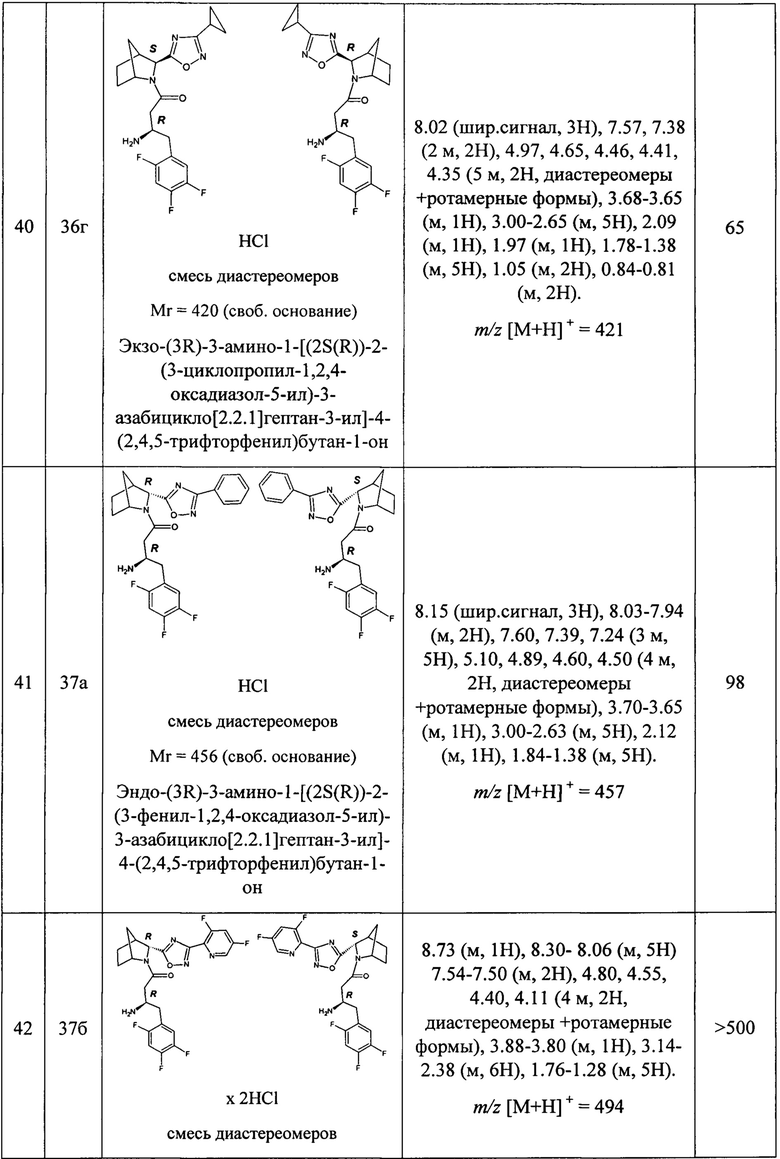
R1=Н, R2=H, R3=гидроксиадамантил-эндо-3-[3-[(3-гидрокси-1-адамантил)амино]пропаноил]-3-азабицикло[2.2.1]гептан-2-карбонитрил (13а), или
R1=H, R2=H, R3=4-фтор-3-гидроксибензил эндо-3-[3-[(3-гидрокси-1-адамантил) амино]пропаноил]-3-азабицикло[2.2.1]гептан-2-карбонитрил (13б), или
R1=метил, R2=H, R3=4-фтор-3-гидроксибензил-экзо-3-[3-[(4-фторо-3-гидрокси-фенил)метиламино]пропаноил]-3-азабицикло[2.2.1]гептан-2-карбонитрил (20а), или
R1=метил, R2=Н, R3=4-фтор-3-гидроксибензил-экзо-3-[(3R)-3-[(4-фтор-3-гидрокси-фенил)метиламино]бутаноил]-3-азабицикло[2.2.1]гептан-2-карбонитрил (20б), или
R1=метил, R2=H, R3=4-фтор-3-гидроксибензил-экзо-3-[(3S)-3-[(4-фтор-3-гидрокси-фенил)метиламино]бутаноил] -3-азабицикло[2.2.1]гептан-2-карбонитрил (20в), или
R1=метил, R2=метил, R3=4-фтор-3-гидроксибензил-экзо-3-[3-[(4-фтор-3-гидрокси-фенил)метиламино]-3-метил-бутаноил]-3-азабицикло[2.2.1]гептан-2-карбонитрил (20г), или
R1=изопропил, R2=Н, R3=4-фтор-3-гидроксибензил-экзо-3-[(3S)-3-[(4-фтор-3-гидрокси-фенил)метиламино]-4-метил-пентаноил]-3-азабицикло[2.2.1]гептан-2-карбонитрил (20д), или
R1=изопропил, R2=Н, R3=4-фтор-3-гидроксибензил-экзо-3-[(3R)-3-[(4-фтор-3-гидрокси-фенил)метиламино]-4-метил-пентаноил]-3-азабицикло[2.2.1]гептан-2-карбонитрил (20е), или
R1=Н, R2=H, R3=4-фтор-3-гидроксибензил-эндо-3-[3-[(4-фтор-3-гидрокси-фенил)метиламино]пропаноил]-3-азабицикло[2.2.1]гептан-2-карбонитрил (20ж), или
R1=метил, R2=Н, R3=4-фтор-3-гидроксибензил-эндо-3-[(3R)-3-[(4-фтор-3-гидрокси-фенил)метиламино]бутаноил]-3-азабицикло[2.2.1]гептан-2-карбонитрил (20з), или
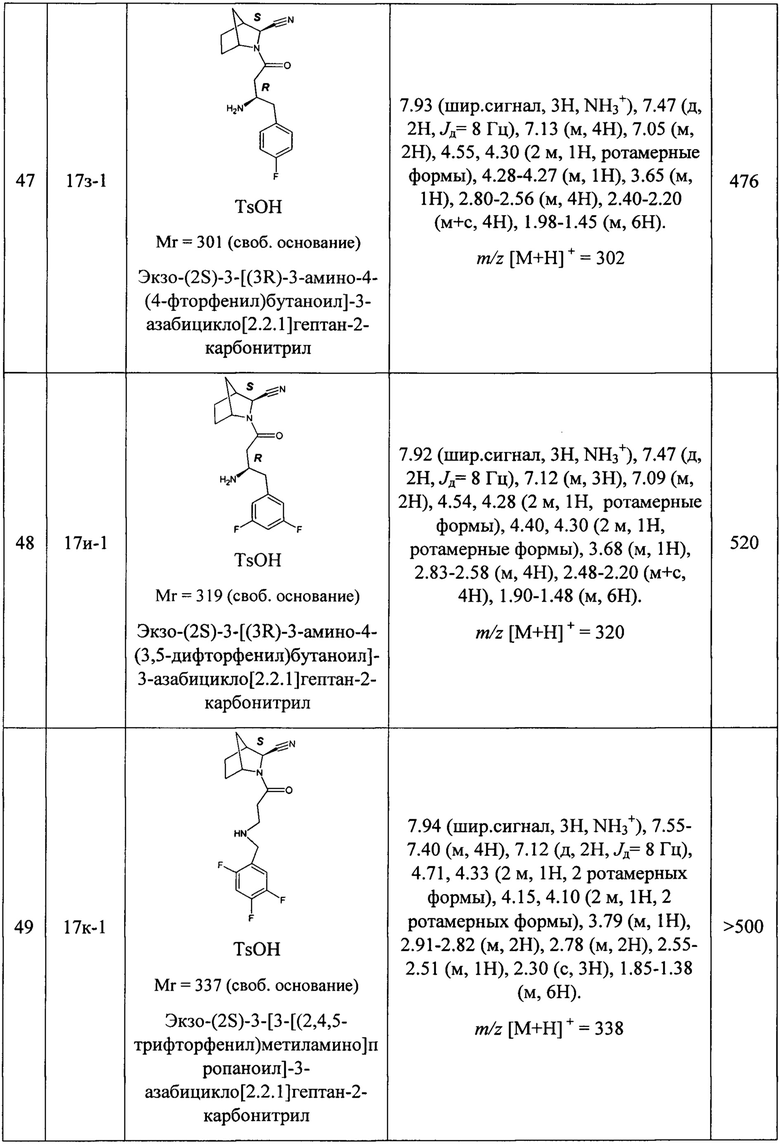
R1=метил, R2=Н, R3=4-фтор-3-гидроксибензил
эндо-3-[(3S)-3-[(4-фтор-3-гидрокси-фенил)метиламино]бутаноил]-3-азабицикло[2.2.1]гептан-2-карбонитрил (20и), или
R1=метил, R2=метил, R3=4-фтор-3-гидроксибензил-эндо-3-[3-[(4-фтор-3-гидрокси-фенил)метиламино]-3-метил-бутаноил]-3-азабицикло[2.2.1]гептан-2-карбонитрил (20к), или
R1=метил, R2=H, R3=4-фторбензил-экзо-3-[(3R)-3-[(4-фторфенил)метиламино]бутаноил]-3-азабицикло[2.2.1]гептан-2-карбонитрил (20л), или
R1=метил, R2=Н, R3=4-фторбензил-эндо-3-[(3R)-3-[(4-фторфенил)метиламино]бутаноил]-3-азабицикло[2.2.1]гептан-2-карбонитрил (20м), или
R1=метил, R2=H, R3=4-фторбензил-эндо-3-[(3S)-3-[(4-фторофенил)метиламино]бутаноил]-3-азабицикло[2.2.1]гептан-2-карбонитрил (20н), или
R1=метил, R2=H, R3=4-фторбензил-экзо-3-[(3S)-3-[(4-фторфенил)метиламино]бутаноил]-3-азабицикло[2.2.1]гептан-2-карбонитрил (20о), или
R1=Н, R2=2,4,5-трифторбензил, R3=H-экзо-3-[(3R)-3-амино-4-(2,4,5-трифторфенил)бутаноил]-3-азабицикло[2.2.1]гептан-2-карбонитрил (17ж), или
R1=Н, R2=2,4,5-трифторбензил, R3=H-эндо-3-[(3R)-3-амино-4-(2,4,5-трифторфенил)бутаноил]-3-азабицикло[2.2.1]гептан-2-карбонитрил (18ж), или
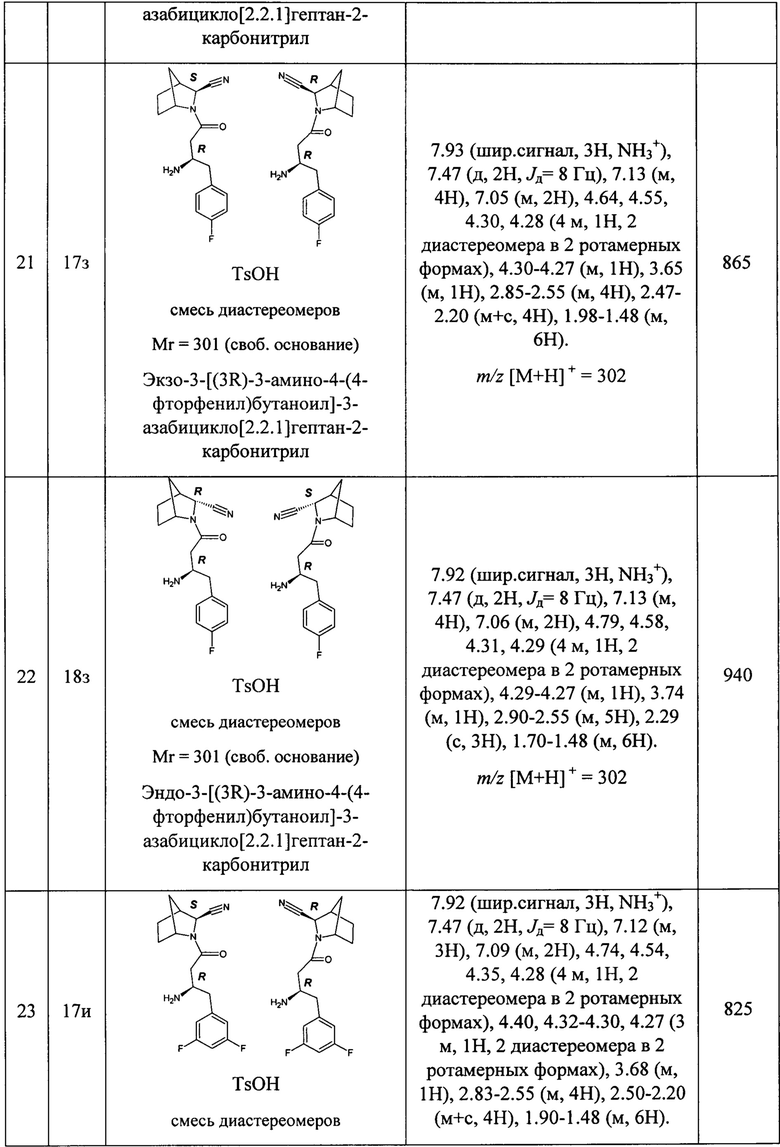
R1=Н, R2=4-фторбензил, R3=Н-экзо-3-[(3R)-3-амино-4-(4-фторфенил)бутаноил]-3-азабицикло[2.2.1]гептан-2-карбонитрил (17з), или
R1=Н, R2=4-фторбензил, R3=Н-эндо-3-[(3R)-3-амино-4-(4-фторфенил)бутаноил]-3-азабицикло[2.2.1]гептан-2-карбонитрил (18з), или
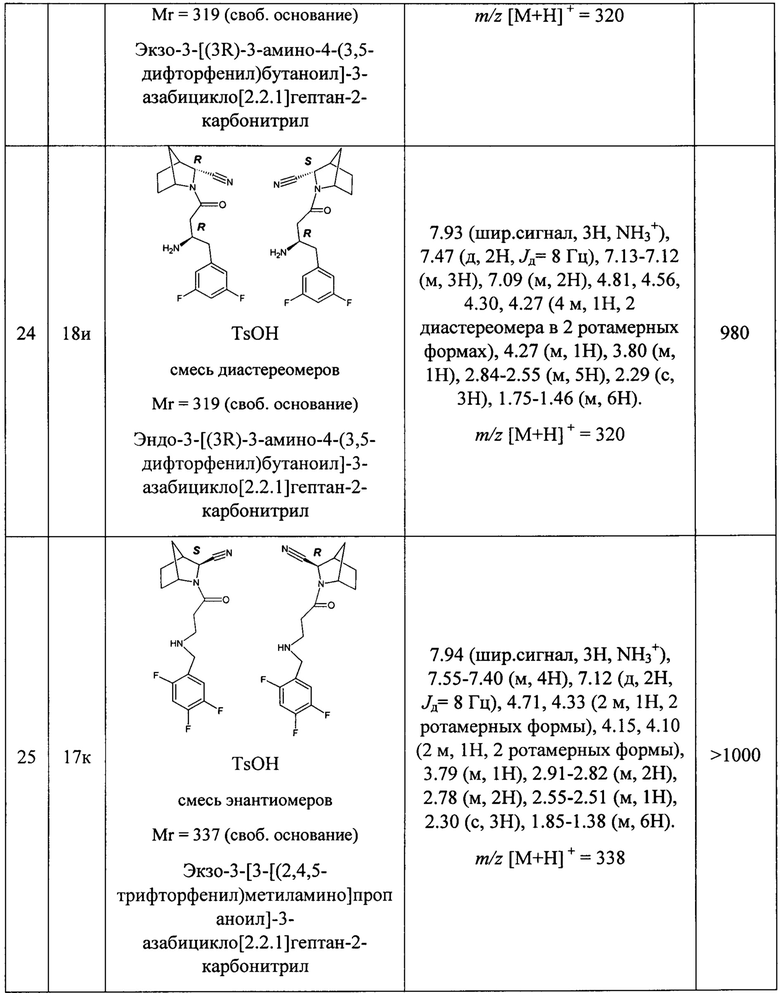
R1=Н, R2=3,5-дифторбензил, R3=Н,
экзо-3-[(3R)-3-амино-4-(3,5-дифторфенил)бутаноил]-3-азабицикло[2.2.1]гептан-2-карбонитрил (17и), или
R1=Н, R2=3,5-дифторбензил, R3=Н-эндо-3-[(3R)-3-амино-4-(3,5-дифторфенил)бутаноил]-3-азабицикло[2.2.1]гептан-2-карбонитрил (18и), или
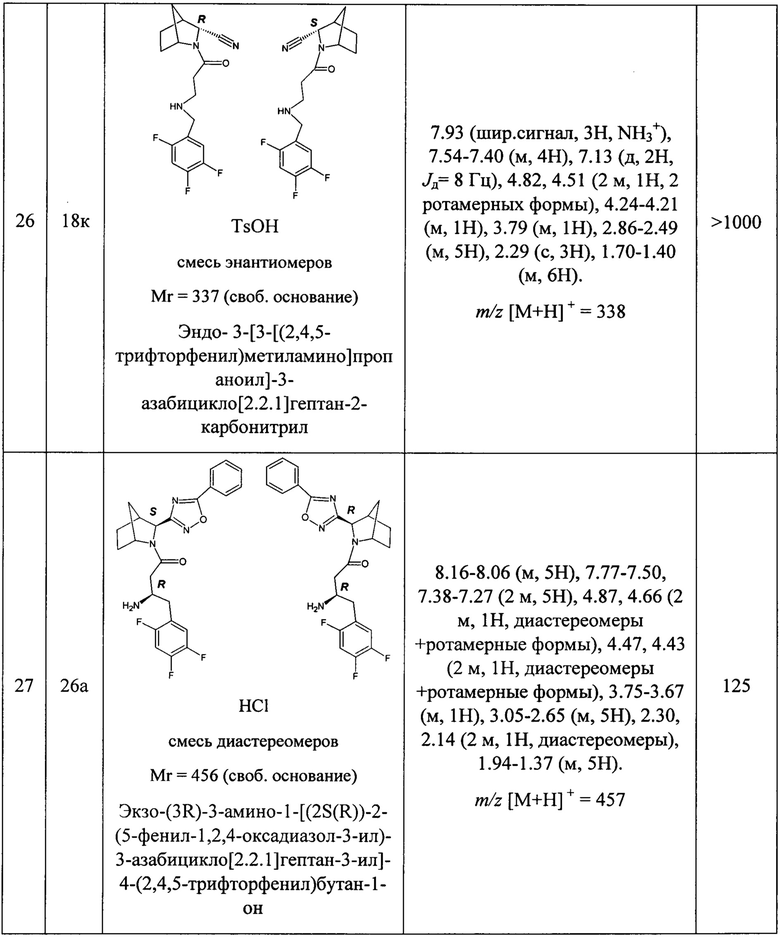
R1=Н, R2=H, R3=2,4,5-трифторбензил-экзо- 3-[3-[(2,4,5-трифторфенил)метиламино]пропаноил]-3-азабицикло[2.2.1]гептан-2-карбонитрил (17к), или
R1=Н, R2=H, R3=2,4,5-трифторбензил-эндо- 3-[3-[(2,4,5-трифторфенил)метиламино]пропаноил]-3-азабицикло[2.2.1]гептан-2-карбонитрил (18к), или
энантиомерно чистые соединения:
R1=Н, R2=2,4,5-трифторбензил, R3=H-экзо-(2S)-3-[(3R)-3-амино-4-(2,4,5-трифторфенил)бутаноил]-3-азабицикло[2.2.1]гептан-2-карбонитрил (17ж-1), или
R1=Н, R2=2,4,5-трифторбензил, R3=H-экзо-(2R)-3-[(3R)-3-амино-4-(2,4,5-трифторфенил)бутаноил]-3-азабицикло[2.2.1]гептан-2-карбонитрил (17ж-2), или
R1=Н, R2=4-фторбензил, R3=Н-экзо-(2S)-3-[(3R)-3-амино-4-(4-фторфенил)бутаноил]-3-азабицикло[2.2.1]гептан-2-карбонитрил (17з-1), или
R1=Н, R2=3,5-дифторбензил, R3=Н-экзо-(2S)-3-[(3R)-3-амино-4-(3,5-дифторфенил)бутаноил]-3-азабицикло [2.2.1]гептан-2-карбонитрил (17и-1), или
R1=Н, R2=H, R3=2,4,5-трифторбензил
экзо-(2S)-3-[3-[(2,4,5-трифторфенил)метиламино]пропаноил]-3-азабицикло[2.2.1]гептан-2-карбонитрил (17к-1).
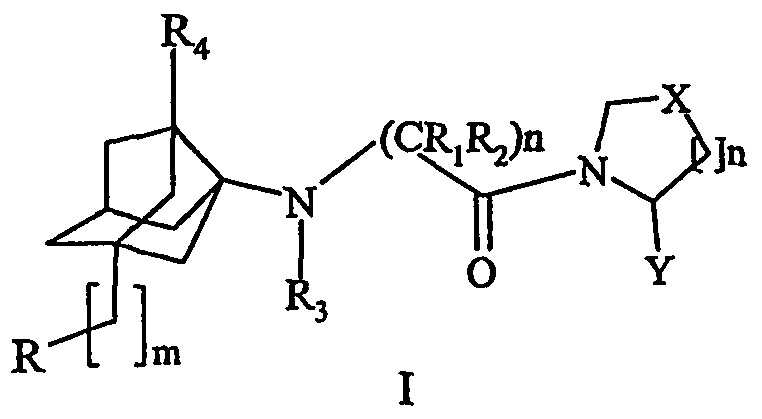
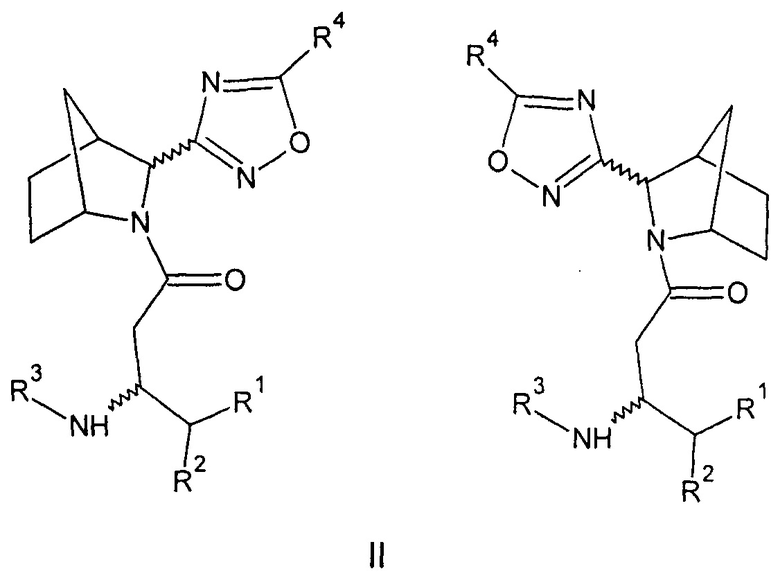
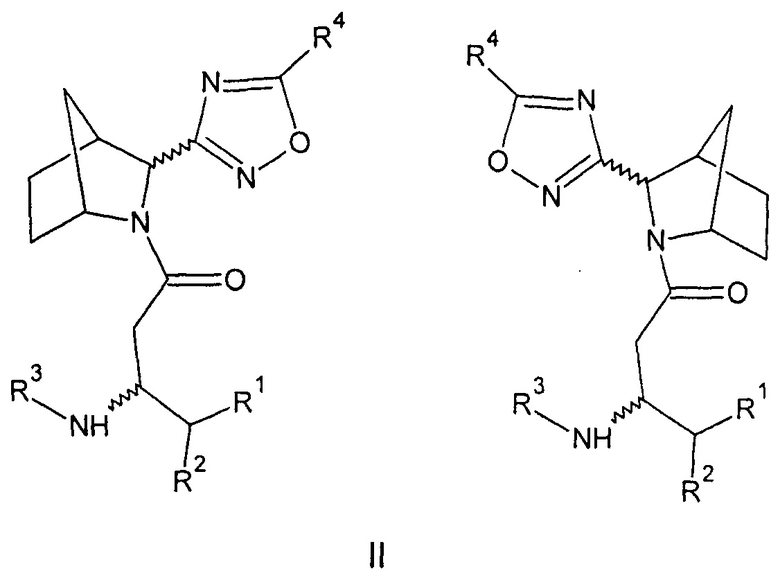
Созданы также соединения общей формулы (II) - амиды бета-аминокислот 3-(3-азабицикло[2.2.1]гептан-2-ил)-1,2,4-оксадиазола, или их фармацевтически приемлемые соли.
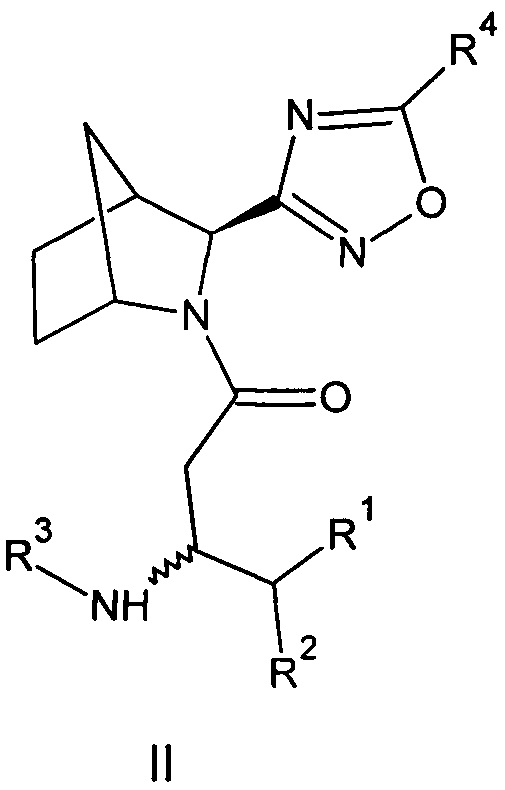
Где:
R1=Н - водород; С1-5 - алкил, в котором каждый углерод независимо может иметь один или два заместителя; арил, в котором каждый углерод ароматического кольца может иметь заместитель вместе или независимо, выбранный из: гидрокси- (ОН-), галоген (F, С1, Br,);
R2=Н - водород; С1-5 - алкил, в котором каждый углерод независимо может иметь один или два заместителя; адамантил, в котором каждый углерод вместе или независимо может иметь заместитель, выбранный из: гидрокси (ОН-), галоген (F, О, Br,); фенил- (C6H5-), в котором каждый углерод ароматического кольца может иметь заместитель вместе или независимо, выбранный из: гидрокси- (ОН-), галоген (F, С1, Br,); бензил- (С6Н5-СН2-), в котором каждый углерод ароматического кольца может иметь заместитель вместе или независимо, выбранный из: гидрокси- (ОН-), галоген (F, Cl, Br,);
R3=Н - водород, С1-5 - алкил, в котором каждый углерод независимо может иметь один или два заместителя; бензил- (С6Н5-СН2-), в котором каждый углерод ароматического кольца может иметь заместитель вместе или независимо, выбранный из: гидрокси- (ОН-), галоген (F, Cl, Br,);
R4=Н - водород, С1-5 - алкил, циклопропил, изопропил в котором каждый углерод независимо может иметь один или два заместителя; бензил- (С6Н5-СН2-), в котором каждый углерод ароматического кольца может иметь заместитель вместе или независимо, выбранный из: гидрокси- (ОН-), галоген (F, Cl, Br,), гетарил (C5H4N-CH2-), в котором каждый углерод ароматического кольца может иметь заместитель вместе или независимо, выбранный из: гидрокси- (ОН-), галоген (F, Cl, Br,) (вариант 2).
При этом синтезированы соединения, где:
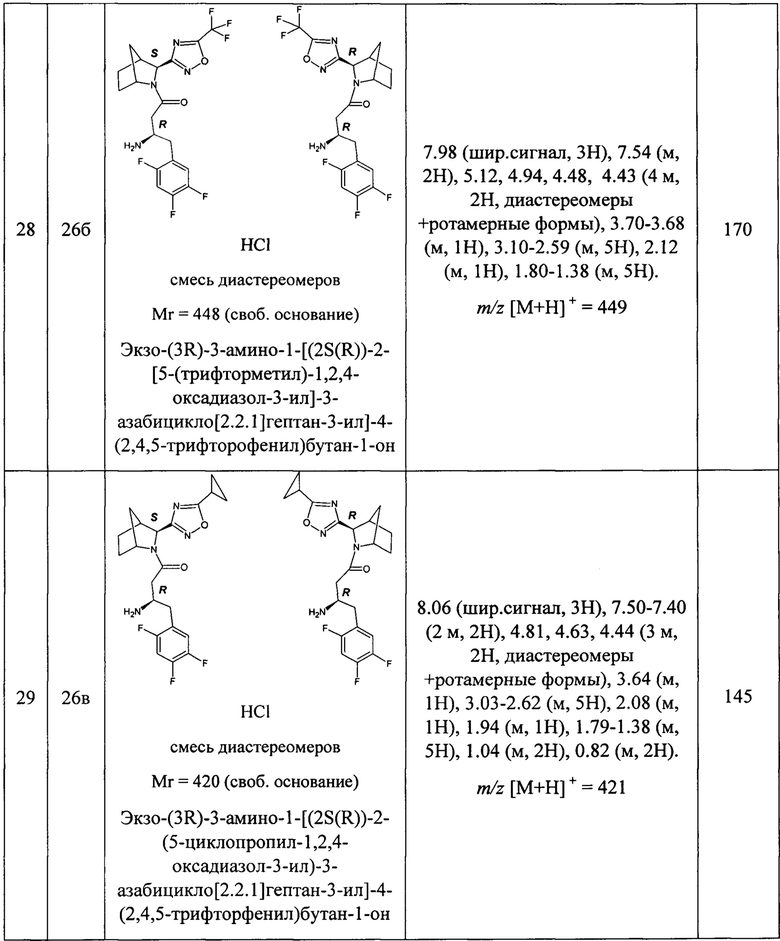
R1=H, R2=2,4,5-трифторбензил, R3=H, R4-фенил-экзо-(3R)-3-амино-1-[(2S(R))-2-(5-фенил-1,2,4-оксадиазол-3-ил)-3-азабицикло[2.2.1]гептан-3-ил]-4-(2,4,5-трифторфенил)бутан-1-он (26a), или
R1=H, R2=2,4,5-трифторбензил, R3=H, R4=трифторметил-экзо-(3R)-3-амино-1-[(2S(R))-2-[5-(трифторметил)-1,2,4-оксадиазол-3-ил]-3-азабицикло[2.2.1]гептан-3-ил]-4-(2,4,5-трифторфенил)бутан-1-он (26б), или
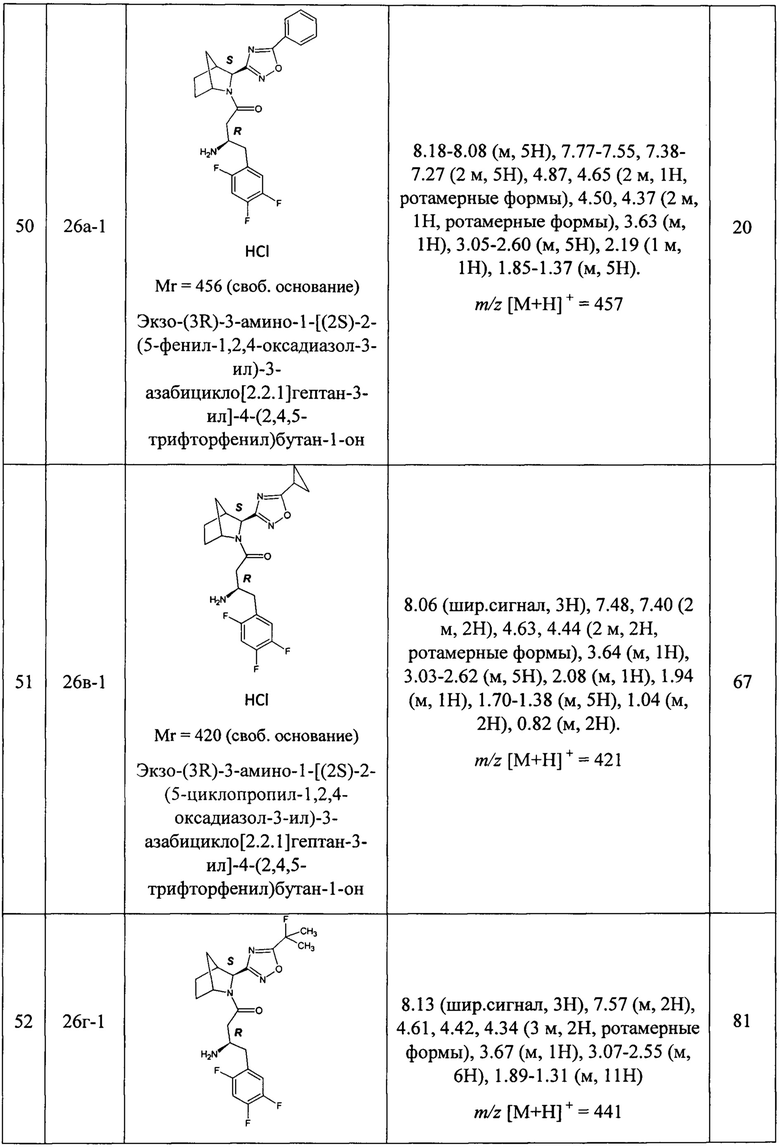
R1=H, R2=2,4,5-трифторбензил, R3=H, R4=циклопропил-экзо-(3R)-3-амино-1-[(2S(R))-2-(5-циклопропил-1,2,4-оксадиазол-3-ил)-3-азабицикло[2.2.1]гептан-3-ил]-4-(2,4,5-трифторфенил)бутан-1-он (26в), или
R1=H, R2=2,4,5-трифторбензил, R3=H, R4=фторпропил-экзо-(3R)-3-амино-1-[(2S(R))-2-[5-(1-фтор-1-метил-этил)-1,2,4-оксадиазол-3-ил]-3-азабицикло[2.2.1]гептан-3-ил]-4-(2,4,5-трифторфенил)бутан-1-он (26г), или
R1=H, R2=2,4,5-трифторбензил, R3=H, R4=изопропил-экзо-(3R)-3-амино-1-[(2S(R))-2-(5-изопропил-1,2,4-оксадиазол-3-ил)-3-азабицикло[2.2.1]гептан-3-ил]-4-(2,4,5-трифторфенил)бутан-1-он (26д), или
R1=H, R2=2,4,5-трифторбензил, R3=H, R4=фенил-эндо-(3R)-3-амино-1-[(2S(R))-2-(5-фенил-1,2,4-оксадиазол-3-ил)-3-азабицикло[2.2.1]гептан-3-ил]-4-(2,4,5-трифторфенил)бутан-1-он (27а), или
R1=H, R2=2,4,5-трифторбензил, R3=H, R4=трифторметил-эндо-(3R)-3-амино-1-[(2S(R))-2-[5-(трифторметил)-1,2,4-оксадиазол-3-ил]-3-азабицикло[2.2.1]гептан-3-ил]-4-(2,4,5-трифторофенил)бутан-1-он (27б), или
R1=H, R2=2,4,5-трифторбензил, R3=H, R4=циклопропил-эндо-(3R)-3-амино-1-[(2S(R))-2-(5-циклопропил-1,2,4-оксадиазол-3-ил)-3-азабицикло[2.2.1]гептан-3-ил]-4-(2,4,5-трифторфенил)бутан-1-он (27в), или
R1=H, R2=2,4,5-трифторбензил, R3=H, R4=2-фторпропил-эндо-(3R)-3-амино-1-[(2S(R))-2-[5-(1-фтор-1-метил-этил)-1,2,4-оксадиазол-3-ил]-3-азабицикло[2.2.1]гептан-3-ил]-4-(2,4,5-трифторфенил)бутан-1-он (27г), или
R1=H, R2=2,4,5-трифторбензил, R3=H, R4=изопропил-эндо-(3R)-3-амино-1-[(2S(R))-2-(5-изопропил-1,2,4-оксадиазол-3-ил)-3-азабицикло[2.2.1]гептан-3-ил]-4-(2,4,5-трифторфенил)бутан-1-он (27д), или
энантиомерно чистые соединения, где:
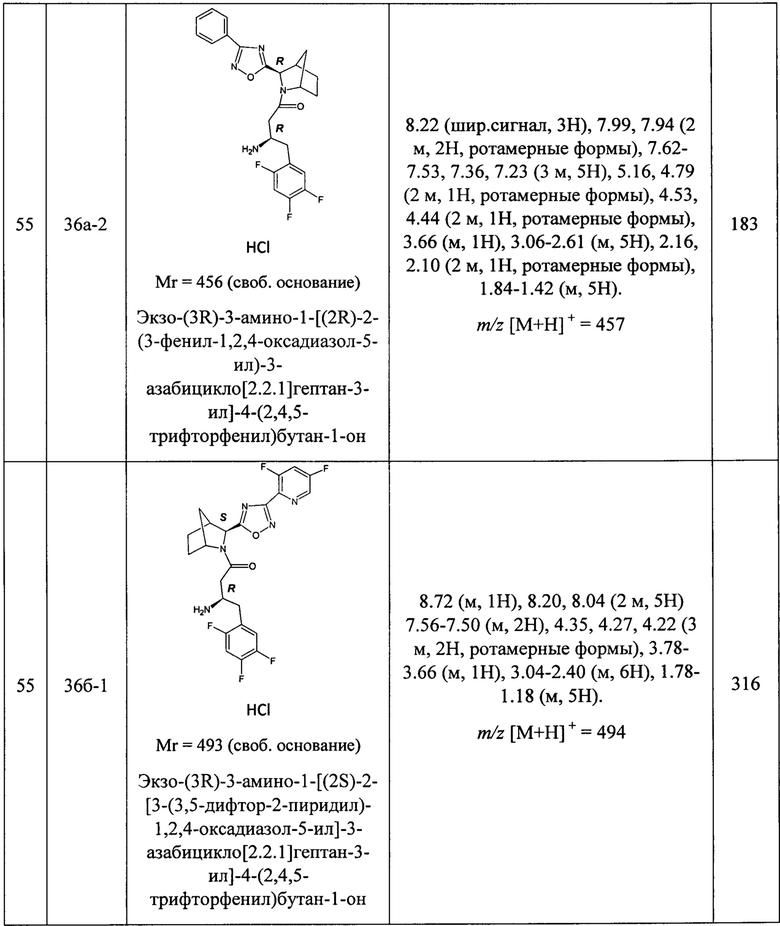
R1=H, R2=2,4,5-трифторбензил, R3=H, R4=фенил-экзо-(3R)-3-амино-1-[(2S)-2-(5-фенил-1,2,4-оксадиазол-3-ил)-3-азабицикло[2.2.1]гептан-3-ил]-4-(2,4,5-трифторфенил)бутан-1-он (26а-1), или
R1=H, R2=2,4,5-трифторбензил, R3=H, R4=циклопропил-экзо-(3R)-3-амино-1-[(2S)-2-(5-циклопропил-1,2,4-оксадиазол-3-ил)-3-азабицикло[2.2.1]гептан-3-ил]-4-(2,4,5-трифторфенил)бутан-1-он (26в-1), или
R1=H, R2=2,4,5-трифторбензил, R3=H, R4=2-фторпропил-экзо-(3R)-3-амино-1-[(2S)-2-[5-(1-фтор-1-метил-этил)-1,2,4-оксадиазол-3-ил]-3-азабицикло[2.2.1]гептан-3-ил]-4-(2,4,5-трифторфенил)бутан-1-он (27г-1), или
R1=H, R2=2,4,5-трифторбензил, R3=H, R4=2-фторпропил-экзо-(3R)-3-амино-1-[(2S)-2-(5-изоопропил-1,2,4-оксадиазол-3-ил)-3-азабицикло[2.2.1]гептан-3-ил]-4-(2,4,5-трифторфенил)бутан-1-он (27г-1), или
R1=H, R2=2,4,5-трифторбензил, R3=H, R4=изопропил-эндо-(3R)-3-амино-1-[(2S)-2-(5-изопропил-1,2,4-оксадиазол-3-ил)-3-азабицикло[2.2.1]гептан-3-ил]-4-(2,4,5-трифторфенил)бутан-1-он (27д-1).
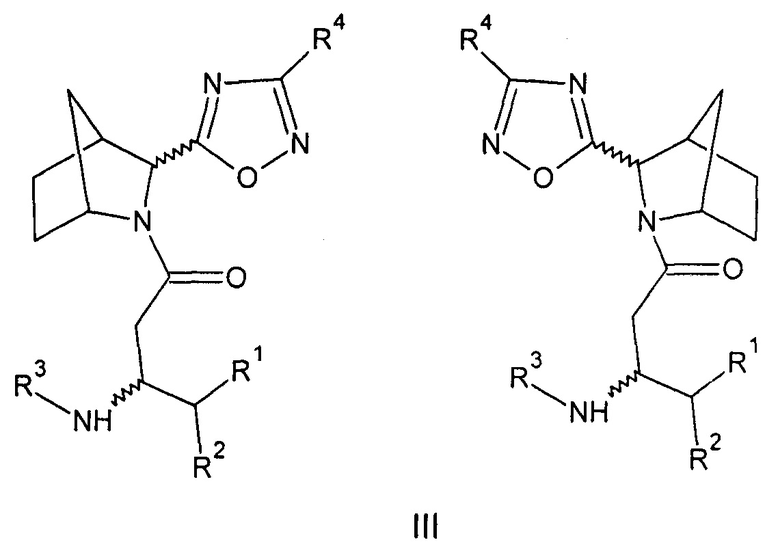
Созданы также соединения общей формулы (III) - амиды бета-аминокислот 5-(3-азабицикло[2.2.1]гептан-2-ил)-1,2,4-оксадиазола или их фармацевтически приемлемые соли.
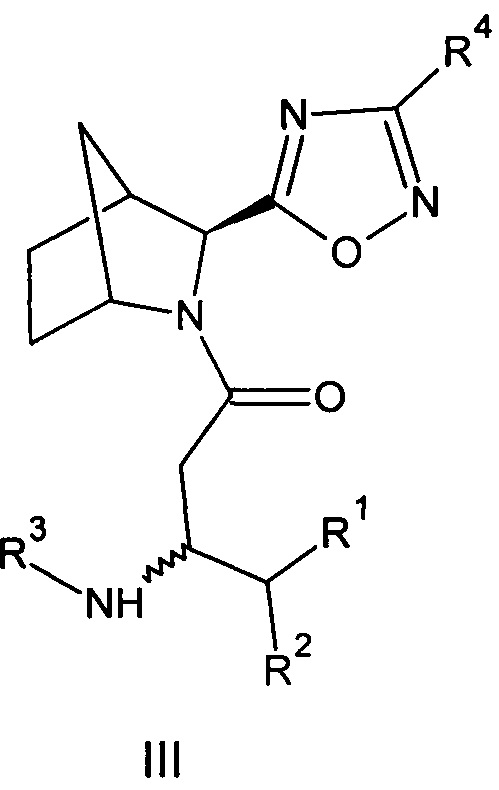
Где:
R1=Н - водород; С1-5 - алкил, в котором каждый углерод независимо может иметь один или два заместителя; арил, в котором каждый углерод ароматического кольца может иметь заместитель вместе или независимо, выбранный из: гидрокси- (ОН-), галоген (F, Cl, Br,);
R2=Н - водород; С1-5 - алкил, в котором каждый углерод независимо может иметь один или два заместителя; адамантил, в котором каждый углерод вместе или независимо может иметь заместитель, выбранный из: гидрокси (ОН-), галоген (F, Cl, Br,); фенил- (С6Н5-), в котором каждый углерод ароматического кольца может иметь заместитель вместе или независимо, выбранный из: гидрокси- (ОН-), галоген (F, Cl, Br,); бензил- (С6Н5-СН2-), в котором каждый углерод ароматического кольца может иметь заместитель вместе или независимо, выбранный из: гидрокси- (ОН-), галоген (F, Cl, Br,);
R3=Н - водород, С1-5 - алкил, в котором каждый углерод независимо может иметь один или два заместителя; бензил- (С6Н5-СН2-), в котором каждый углерод ароматического кольца может иметь заместитель вместе или независимо, выбранный из: гидрокси- (ОН-), галоген (F, Cl, Br,);
R4=Н - водород, С1-5 - алкил, циклопропил, изопропил в котором каждый углерод независимо может иметь один или два заместителя; бензил- (С6Н5-СН2-), в котором каждый углерод ароматического кольца может иметь заместитель вместе или независимо, выбранный из: гидрокси- (ОН-), галоген (F, Cl, Br,), гетарил (C5H4N-CH2-), в котором каждый углерод ароматического кольца может иметь заместитель вместе или независимо, выбранный из: гидрокси- (ОН-), галоген (F, Cl, Br,) (вариант 3).
При этом синтезированы соединения, где:
R1=H, R2=2,4,5-трифторбензил, R3=H, R4=фенил-экзо-(3R)-3-амино-1-[(2S(R))-2-(3-фенил-1,2,4-оксадиазол-5-ил)-3-азабицикло[2.2.1]гептан-3-ил]-4-(2,4,5-трифторфенил)бутан-1-он (36а), или
R1=H, R2=2,4,5-трифторбензил, R3=H, R4=3,5-дифтор-2-пиридил-экзо-(3R)-3-амино-1-[(2S(R))-2-[3-(3,5-дифтор-2-пиридил)-1,2,4-оксадиазол-5-ил]-3-азабицикло[2.2.1]гептан-3-ил]-4-(2,4,5-трифторфенил)бутан-1-он (36б), или
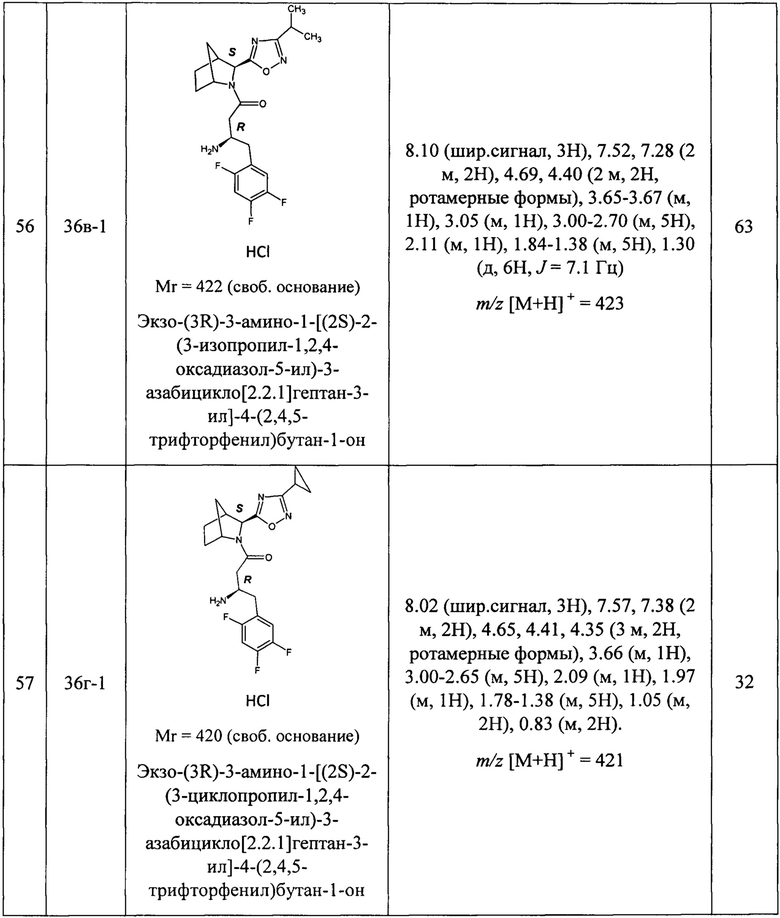
R1=H, R2=2,4,5-трифторбензил, R3=H, R4=изопропил-экзо-(3R)-3-амино-1-[(2S(R))-2-(3-изопропил-1,2,4-оксадиазол-5-ил)-3-азабицикло[2.2.1]гептан-3-ил]-4-(2,4,5-трифторфенил)бутан-1-он (36в), или
R1=H, R2=2,4,5-трифторбензил, R3=H, R4=3,5-циклопропил-экзо-(3R)-3-амино-1-[(2S(R))-2-(3-циклопропил-1,2,4-оксадиазол-5-ил)-3-азабицикло[2.2.1]гептан-3-ил]-4-(2,4,5-трифторофенил)бутан-1-он (36г), или
R1=H, R2=2,4,5-трифторбензил, R3=H, R4=фенил-эндо-(3R)-3-амино-1-[(2S(R))-2-(3-фенил-1,2,4-оксадиазол-5-ил)-3-азабицикло[2.2.1]гептан-3-ил]-4-(2,4,5-трифторфенил)бутан-1-он (37а), или
R1=H, R2=2,4,5-трифторбензил, R3=H, R4=3,5-дифторо-2-пиридил
эндо-(3R)-3-амино-1-[(2S(R))-2-[3-(3,5-дифтор-2-пиридил)-1,2,4-оксадиазол-5-ил]-3-азабицикло[2.2.1]гептан-3-ил]-4-(2,4,5-трифторфенил)бутан-1-он (37б), или
R1=H, R2=2,4,5-трифторбензил, R3=H, R4=изопропил-эндо-(3R)-3-амино-1-[(2S(R))-2-(3-изопропил-1,2,4-оксадиазол-5-ил)-3-азабицикло[2.2.1]гептан-3-ил]-4-(2,4,5-трифторфенил)бутан-1-он (37в), или
R1=H, R2=2,4,5-трифторбензил, R3=H, R4=циклопропил-эндо-(3R)-3-амино-1-[(2S(R))-2-(3-циклопропил-1,2,4-оксадиазол-5-ил)-3-азабицикло[2.2.1]гептан-3-ил]-4-(2,4,5-трифторфенил)бутан-1-он (37г), или
энантиомерно чистые соединения, где:
R1=H, R2=2,4,5-трифторбензил, R3=H, R4=фенил-экзо-(3R)-3-амино-1-[(2S)-2-(3-фенил-1,2,4-оксадиазол-5-ил)-3-азабицикло[2.2.1]гептан-3-ил]-4-(2,4,5-трифторфенил)бутан-1-он (36а-1), или
R1=H, R2=2,4,5-трифторбензил, R3=H, R4=фенил-экзо-(3R)-3-амино-1-[(2R)-2-(3-фенил-1,2,4-оксадиазол-5-ил)-3-азабицикло[2.2.1]гептан-3-ил]-4-(2,4,5-трифторфенил)бутан-1-он (36а-2), или
R1=H, R2=2,4,5-трифторбензил, R3=H, R4=3,5-дифтор-2-пиридил-экзо-(3R)-3-амино-1-[(2S)-2-[3-(3,5-дифторо-2-пиридил)-1,2,4-оксадиазол-5-ил]-3-азабицикло[2.2.1]гептан-3-ил]-4-(2,4,5-трифторфенил)бутан-1-он (36б-1), или
R1=H, R2=2,4,5-трифторбензил, R3=H, R4=изопропил-экзо-(3R)-3-амино-1-[(2S)-2-(3-изопропил-1,2,4-оксадиазол-5-ил)-3-азабицикло[2.2.1]гептан-3-ил]-4-(2,4,5-трифторфенил)бутан-1-он (36в-1), или
R1=H, R2=2,4,5-трифторбензил, R3=H, R4=3,5-циклопропил-экзо-(3R)-3-амино-1-[(2S)-2-(3-циклопропил-1,2,4-оксадиазол-5-ил)-3-азабицикло[2.2.1]гептан-3-ил]-4-(2,4,5-трифторфенил)бутан-1-он (36г-1).
Созданы также соединения, выбранные из фармацевтически приемлемых солей соединений перечисленных выше.
При этом ингибитор дипептидилпептидазы-4 для лечения сахарного диабета 2-го типа или метаболического синдрома выбирают из соединений, общей формулы I, или II, или III, или из их фармацевтически приемлемых солей.
Реализация изобретения
Известно, что у больных СД 2-го типа нарушено не только количественное содержание инкретинов (ГПП-1 и ГИП), но и механизм их действия, поэтому очевидна необходимость создания группы препаратов, влияющих на уровень инкретинов. Наличие подобных средств улучшает гликемический контроль путем воздействия на иные патогенетические звенья данного заболевания, в отличие от других существующих групп антидиабетических препаратов.
Влияние дипептидилпептидазы-4. Снижение эффекта инкретинов, находящихся в кровеносном русле, является следствием их быстрого разрушения и выведения из организма. Причина деградации инкретинов и потери их функции кроется в их структуре - наличии остатка аланина (Ala) во 2-м с N-конца положении. Подобные аминокислотные последовательности являются субстратами для сериновой протеазы - дипептидилпептидазы-4 (ДПП-4).
Инкретины ГПП-1 и ГИП представляют собой пептидные гормоны, зрелые формы которых являются результатом расщепления предшественников в К- и L-клетках тонкого кишечника. ГИП состоит из 42 аминокислот и образуется в результате посттрансляционной модификации про-ГИП прогормон-протеазой PC1/3 в энтероэндокринных K-клетках. Форма ГИП является единственной известной активной формой ГИП у человека, крысы, мыши, свиньи и крупного рогатого скота. ГПП-1 представляет собой результат посттрансляционного расщепления прогормон-протеазой PC1/3 продукта гена проглюкагона в энтероэндокринных L-клетках. ГПП-1 является основной формой биологически активного циркулирующего ГПП-1 в организме человека. В результате протеолитической обработки ГПП-1 и ГИП во 2-м N-концевом положении их последовательностей появляется остаток аланина. Получающиеся структуры являются субстратами для дипептидилпептидазы-4, отщепляющей от полипептидной цепи N-концевые дипептиды, содержащие остатки Pro или Ala во 2-м с N-конца положении.
Еще одним доказательством роли дипептидилпептидазы-4 в патогенезе сахарного диабета 2-го типа является влияние функции генов, регулирующих экспрессию гена ДПП-4. Генами, снижающими экспрессию указанного гена, являются печеночные ядерные транскрипционные факторы 1-α и 1-β члены семейства гомеодомен-содержащих белков. Наличие мутаций, снижающих функцию любого из этих генов и, соответственно, повышающих уровень экспрессии ДПП-4, приводит к развитию сахарного диабета.
Понимание роли ДПП-4 в метаболизме инкретинов дало основание для создания препаратов, увеличивающих время действия эндогенных ГИП и ГПП-1 - ингибиторов ДПП-4. Результаты экспериментов на животных моделях продемонстрировали сохранение концентрации инкретинов в крови под влиянием ингибиторов ДПП-4.
Ингибиторы ДПП-4 имеют и другое преимущество перед применяющимися в настоящее время гипогликемическими препаратами. Так как инкретин-опосредованные эффекты на биосинтез и высвобождение инсулина являются глюкозозависимыми, при применении ингибиторов ДПП-4 риск возникновения гипогликемии значительно ниже, чем при применении инсулина, препаратов сульфонилмочевины или меглитинидов. Кроме того, в отличие от указанных препаратов, ингибиторы ДПП-4 не вызывают прибавку массы тела. Благодаря особенностям их действия, ингибиторы ДПП-4 представляют особый интерес в применении их на ранних стадиях СД 2-го типа, как в виде монотерапии, так и в комбинации с другими препаратами, так как они могут способствовать протекции β-клеток.
Экзопептидазная каталитическая активность дипептидилпептидазы-4, отщепляющей от полипептидной цепи определенные N-концевые дипептиды, содержащие остатки Pro или Ala во 2-м с N-конца положении, определяется строением ее активного центра.
ГПП-1 и ГИП обеспечивают от 60 до 70% общего 1, 3 инсулинового ответа после приема пищи (эффект инкретина) у здоровых людей. В клинических исследованиях пациентов с сахарным диабетом 2 типа эффект инкретина присутствовал, но был значительно снижен по сравнению со здоровыми людьми. У таких пациентов после перорального введения глюкозы инсулиновый ответ был не только отсрочен во времени, но и снижен.
Лекарственные средства, ингибирующие каталитическую активность ДПП-4 (ингибиторы ДПП-4) и взаимодействующие с ее активным центром, должны отвечать следующим критериям:
- субструктуры, образованные остатком Ser630 в каталитическом центре фермента и соседние аминокислоты, как правило, связывают гидрофобные кольцевые группы, такие как три-фторбензил группа ситаглиптина или бензонитрил алоглиптина;
- обязательна стабилизация двух кислотных остатков - Glu205 и Glu206 - в сайте связывания ДПП-4 аминогруппой ингибитора;
- в дополнение к этим обязательным взаимодействиям, потенциальные ингибиторы могут содержать различные функциональные группы с целью усиления связывания с ДПП-4.
Согласно указанным выше критериям, эффективными ингибиторами ДПП-4 могут быть соединения, N-ацилированные альфа-аминокислотами производные цианопирролидина. Однако данная группа веществ в водных растворах при рН 7,4 способна к внутримолекулярной циклизации. Этот феномен приводит в естественных условиях человеческого организма к функциональной инактивации лекарственных средств - N-ацилированных альфа-аминокислотами производных цианопирролидина. Процесс циклизации таких цианопирролидинов отражен ниже.
Большинство описанных препаратов класса цианопирролидинов обладает высокой эффективностью за счет ковалентного связывания циано-фрагмента с гидрокси-группой Ser630 с образованием соответствующего имидата. Однако присутствие в молекуле ингибитора α-аминокислотного фрагмента создает вероятность внутримолекулярной циклизации с получением соответствующего дикетопиперазина и, как следствие, потерю активности.
Ниже представлена внутримолекулярная циклизация из цис-формы N-ацилцианопирролидина.
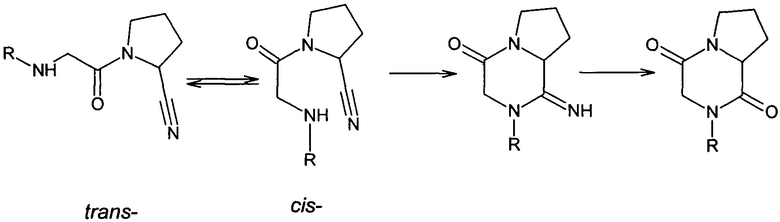
Процесс внутримолекулярной циклизации N-ацилцианопирролидина в физиологических условиях говорит о том, что для сохранения химической структуры потенциального лекарственного средства необходимо предотвратить данное свойство подобных производных цианопирролидина. Например, включать в ацильную часть цианопирролидина пространственно объемные группы, препятствующие быстрой циклизации или совмещать наличие объемных групп с увеличением алифатической цепи, связывающей цианопирролидин с амино-функцией, участвующей в связывании в домене связывания лиганда таким образом, чтобы циклизация оказалась термодинамически затруднена.
Структуры соединений - кандидатов-претендентов отбирались, прежде всего, на стерические соответствия структур, обеспечивающих взаимодействия с молекулой дипептилпептидазы-4, с предположением двух ключевых допущений: (i) при наличии циано-группы ожидается ковалентное связывание с Ser630 и (ii) кислород при двойной связи лиганда должен образовывать водородные связи с атомами белка в ближайшем окружении домена связывания лиганда.
Ключевым решением явилась возможность образования ковалентной связи между углеродом с тройной связью на атом азота и кислородом Ser630 (так, как происходит при связывании вилдаглиптина, а также сходного с вилдаглиптином пространственного размещения группы лиганда N-(C=O)-C с карбонильным кислородом, вовлеченным в водородные связи при связывании с DPP-4. Для всех предложенных лигандов геометрия последовательности связей (C≡N)-C-N-(C=O)-C соответствует такой последовательности в молекуле вилдаглиптина.

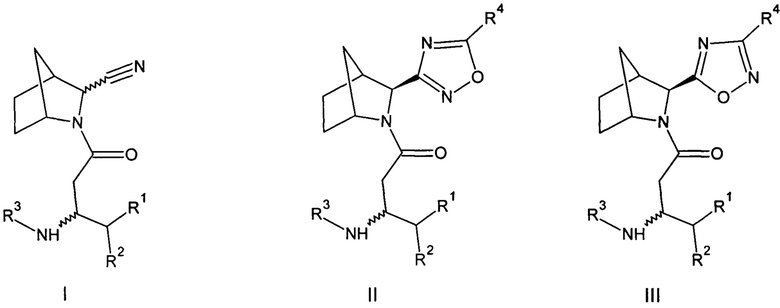
Настоящее изобретение представляет собой разработку ряда ингибиторов дипептидилпептидазы-4 из группы амидов бета-аминокислот - N-ацильных производных 3-азабицикло[2.2.1]гептен-2-карбонитрила (I), 5-(3-азабицикло[2.2.1]гептан-2-ил)-1,2,4-оксадиазола (II) и 3-(3-азабицикло[2.2.1]гептан-2-ил)-1,2,4-оксадиазола (III), объединенных общими формулами:
Пример 1
Синтез ключевых исходных реагентов экзо- и эндо- 2-аза-бицикло[2.2.1]гептан-3-карбонитрилов 7а,б производился по разработанной синтетической схеме, приведенной ниже. Экзо- и эндо-3-азабицикло[2.2.1]гептан-2-карбонитрилы 7а,б были получены в виде рацемических смесей.
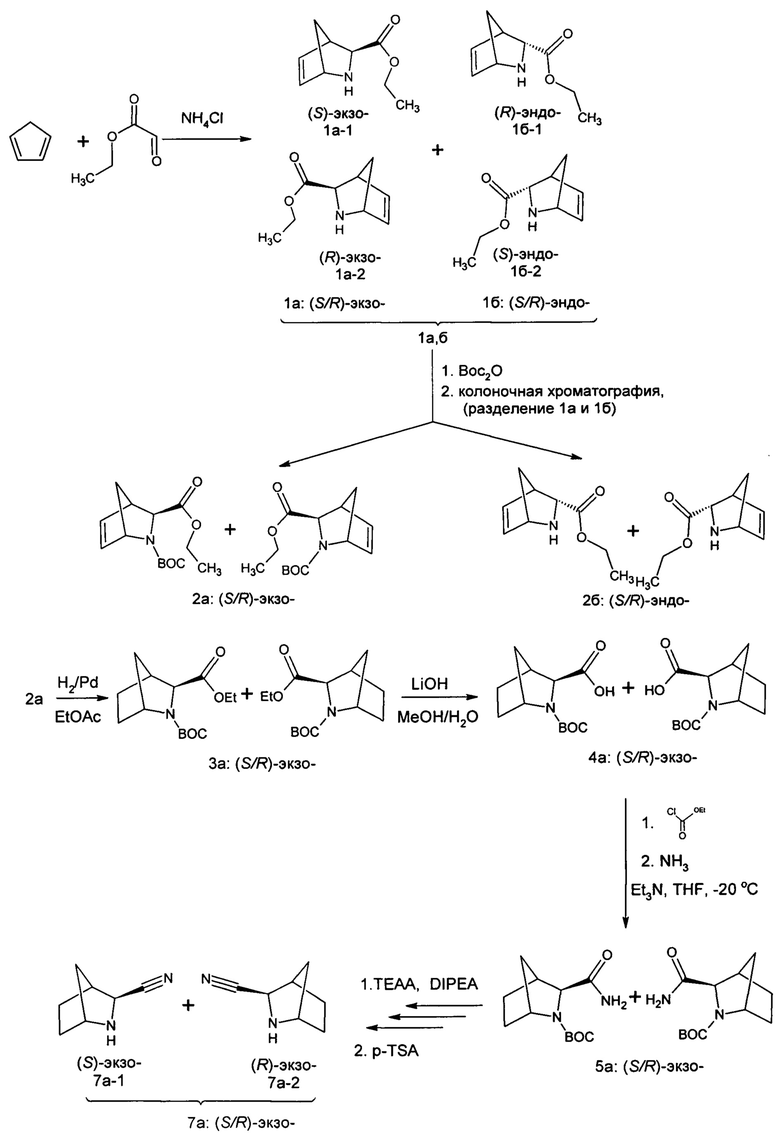

Процедура синтеза 2-аза-бицикло[2.2.1]гептан-3-карбонитрила 7а,б.
Экзо/эндо-этил-2-азабицикло[2.2.1]гепт-5-ен-3-карбоксилат (1а,б).
К смеси насыщенного раствора хлорида аммония (39.3 г) и толуольного раствора этилглиоксилата (50%, 150 г) при температуре 19-20°С порциями добавили свежеперегнанный циклопентадиен 64.7 г. Реакционную массу перемешивали 12 часов при комнатной температуре, затем подвергали экстракции смесью МТБЕ:ПЭ 1:3, подщелачивали до рН 8-9 раствором NaOH (50%), экстрагировали МТБЕ, сушили над безв. Na2SO4. После отгонки растворителя получили 67 г (53%) продукта - смеси 1а,б в виде желтого густого масла.
(R/S)-Экзо-2-третбутил-3-этил-2-азабицикло[2.2.1]гепт-5-ен-2,3-дикарбоксилат (2а) и
(R/S)-эндо-2-третбутил-3-этил-2-азабицикло[2.2.1]гепт-5-ен-2,3-дикарбоксилат (2б).
К раствору этил-2-азабицикло[2.2.1]гепт-5-ен-3-карбоксилата 1а,б (38 г) в ТГФ (200 мл) при охлаждении льдом добавили по каплям раствор БОК ангидрида (55 г) в ТГФ (200 мл). Реакционную смесь оставляли на ночь при перемешивании (комнатная температура), затем упаривали растворяли остаток в смеси ПЭ:EtOAc 1:1 и промывали водой. Органический слой сушили над сульфатом натрия и разделяли экзо- и эндо- изомеры на колонке. Элюент - ПЭ→ПЭ:EtOAc 30%. Получено после разделения 20 г чистого эндо-изомера 2б и 65г смеси экзо- и эндо-, последнюю подвергали дальнейшему разделению и выделяли 17 г экзо- изомера 2а. Общий выход смеси изомеров 77%.
(R/S)-Экзо-2-третбутил-3-этил-2-азабицикло[2.2.1]гептан-2,3-дикарбоксилат (3а)
17 г исходного карбоксилата 2а гидрировали 1.5 ч при 55-60°С и давлении 45 PSI в растворе этанола в присутствии 0.8 г 10% Pd/C. Полученная реакционная смесь была профильтрована через целит, для отделения 10% Pd/C, и растворитель был упарен при пониженном давлении на ротационном растворителе. Получили 3а (14.8 г, 86%) в виде желтого масла.
(R/S)-Эндо-2-третбутил-3-этил-2-азабицикло[2.2.1]гептан-2,3-дикарбоксилат (3б)
Гидрировали 26 г исходного карбоксилата 2б при температуре 55-60°С 1.5 часа в растворе этилацетата и давлении 45-50 PSI, 0.5 г Pd/C. Полученная реакционная смесь была профильтрована через целит, для отделения 10% Pd/C, и растворитель был упарен при пониженном давлении на ротационном растворителе. Получили 3б (23 г, 87%) в виде желтого масла.
(R/S)-Экзо-2-(трет-бутоксикарбонил)-2-азабицикло[2.2.1]гептан-3-карбоновая кислота (4а)
Перемешивали водно-метанольную эмульсию исходного эфира 3а (14.8 г) с литиевой щелочью (моногидрат, 6.58 г). Перемешивали в течение ночи при комнатной температуре, по ТСХ реакция прошла не полностью. Добавили еще 1.5 эквивалента литиевой щелочи и нагревали реакцию в течение 2-х часов при 40-50°С. Затем отгоняли метанол, разбавляли реакционную смесь водой, экстрагировали этилацетатом, затем подкисляли водный слой лимонной кислотой до рН=4, экстрагировали хлористым метиленом. После сушки сульфатом натрия и удаления растворителя получили 11.9 г (89%) целевой кислоты 4а.
(R/S)-Эндо-2-(трет-бутоксикарбонил)-2-азабицикло[2.2.1]гептан-3-карбоновая кислота (4б)
Перемешивали водно-метанольную эмульсию Вос-аминоэфира 3б (15.7 г) с литиевой щелочью (7 г) при кипячении в течение 3 часов. Обрабатывали аналогично экзо-изомеру 4а. Получили 12.6 г эндо-кислоты 4б (89%).
(R/S)-Экзо-трет-бутил-3-карбамоил-2-азабицикло[2.2.1] гептан-2-карбоксилат (5а)
К раствору исходной кислоты 4а (10.3 г) в сухом ТГФ при охлаждении до -20°С в атмосфере аргона добавляли триэтиламин (6.54 мл, 4.75 г). Затем по каплям добавляли этилхлорформиат (5.10 г) в течение 10 мин. Выдерживали реакционную смесь при охлаждении в течение 40 мин и добавляли по каплям при охлаждении водный аммиак (8.36 г). Через 1 ч упарили ТГФ, обработали остаток раствором лимонной кислоты до рН=4, экстрагировали этилацетатом, этилацетатные экстракты промывали раствором соды и сушили сульфатом натрия и концентрировали. Получили 10 г бесцветного кристаллического остатка 5а. Выход количественный.
(R/S)-Эндо-трет-бутил-3-карбамоил-2-азабицикло[2.2.1]гептан-2-карбоксилат (5б)
К раствору исходной эндо-кислоты 4б (12.3 г) в сухом ТГФ при охлаждении до -20°С добавили ТЭА (8 мл) и прикапали этилхлорформиат (6.1 г). После часовой выдержки при охлаждении обрабатывали водным аммиаком (10 г). Реакцию проводили и обрабатывали аналогично экзо-изомеру 5а. Получили 11.8 г бесцветного кристаллического вещества 5б. Выход количественный.
(R/S)-Экзо-трет-бутил-3-циано-2-азабицикло[2.2.1]гептан-2-карбоксилат (6а)
К суспензии исходного амида 5а (10.3 г) в сухом ТГФ, при температуре не выше 4°С добавляли ангидрид трифторуксусной кислоты (14.4 г) в течение 10 минут. По ТСХ присутствовал исходный, добавили еще 9 г трифторуксусного ангидрида. Реакционную смесь выдерживали при охлаждении в течение 3 часов, затем порциями добавили к реакционной смеси при охлаждении гидрокарбонат аммония (45 г). Реакционную смесь наносили на силикагель и разделяли на хроматографической колонке. Элюент - смесь ПЭ:EtOAc 4:1. Получили 8.3 г (87%) целевого нитрила 6а в виде бледно-желтого густого маслообразного вещества.
(R/S)-Эндо-третбутил-3-циано-2-азабицикло[2.2.1]гептан-2-карбоксилат (6б)
Реакция проводилась аналогично экзо-изомеру 6а. Исходный амид 5б (11.8 г), трифторуксусный ангидрид (16.5 г + 10.7 г), гидрокарбонат аммония - 51 г. Получено после очистки на хроматографической колонке 6 г (54%) нитрила 6б в виде светло-желтого масла.
(R/S)-Экзо-2-азабицикло[2.2.1]гептан-3-карбонитрил (7а)
К исходному БОК-нитрилу 6а (7 г) в 30 мл ацетонитрила добавили 12 г (двукратный избыток) п-толуолсульфоновой кислоты (п-ТСК) и оставляли перемешиваться на ночь. Отгоняли ацетонитрил, остаток растирали с диэтиловым эфиром (3-4 обработки с декантацией). Упаривали эфир, остаток растворяли в хлористом метилене и насыщали аммиаком из баллона. Выпавший осадок аммонийной соли п-ТСК отфильтровывали. Фильтрат упаривали и остаток хроматографировали. Элюент - хлористый метилен после экстракции водного аммиака в соотношении 1:10 (100 мл DCM экстрагировали 10 мл водного аммиака). Получили после колонки целевой продукт 7а (4.2 г) в виде желтоватого масла.
(R/S)-Эндо-2-азабицикло[2.2.1]гептан-3-карбонитрил (7б)
Получали аналогично экзо-изомеру 7а из БОК-нитрила 6б (6 г), п-ТСК (10.3 г). Получили после хроматографирования 3.4 г целевого амина (7б) в виде желтоватого масла.
Для синтеза целевых соединений были предварительно синтезированы модифицированные по аминогруппе β-аминокислоты 11а,б.
Примеры:
Синтез 3-(3-гидрокси-адамантан-1-иламино)-пропионовых кислот.
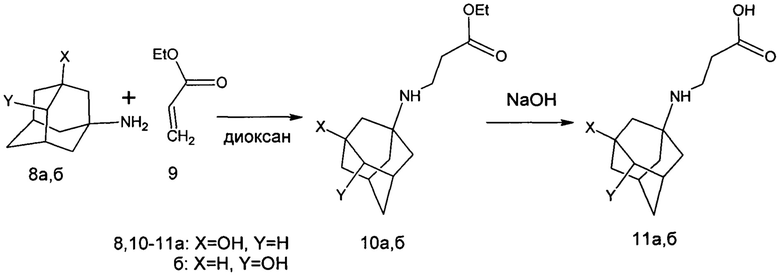
Процедура синтеза 3-(3-гидрокси-адамантан-1-иламино)-пропионовой кислоты 11а.
К 3-амино-адамантан -1-олу 8а в виде свободного основания 16.7г (0.1 моль) было добавлено 57 г (0.5 моль) этилакрилата в 100 мл диоксана. Смесь нагревалась при 50°С в течение 24 часов. По окончании нагревания смесь была упарена досуха, снова был добавлен диоксан в объеме 100 мл, и смесь снова была упарена досуха в вакууме на ротационном испарителе. Образовавшийся остаток был растворен в смеси вода/метанол 1:1 (150 мл). Натрия гидроксид 6 г (0.15 моль) был добавлен к раствору, и смесь перемешивалась при комнатной температуре в течение 12 часов. Затем смесь была сконцентрирована до объема 75 мл и нейтрализована титрованным раствором содержащим 0.15 моль HCl в воде. Выход 3-(3-гидрокси-адамантан-1-иламино)-пропионовой кислоты 11а 16.1 г (67%).
Синтез кислоты 11б (10 г, 55%) осуществляли из 8б аналогично процедуре, приведенной для 11а.
Синтез целевых соединений 12а,б и 13а,б осуществляли из рацемических экзо- и эндо-изомеров 3-азабицикло[2.2.1]гептан-2-карбонитрилов 7а и 7б, согласно схеме, изображенной ниже.
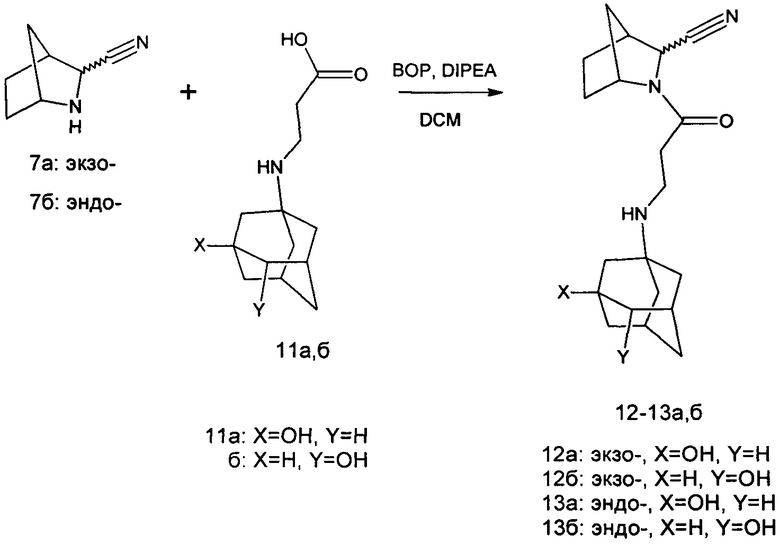
Процедура синтеза целевых продуктов. 12а,б и 13а,б.
К кислоте 11а 0.239 г (1 ммоль) в 10 мл дихлорметана добавляли ВОР (0.534 г, 1.2 ммоль) и рацемический амин 7а (0.122 г, 1 ммоль). Смесь перемешивали при комнатной температуре в течение ночи. Промывали 10% раствором поташа, водой, сушили над безводным сульфатом натрия и упаривали досуха на ротационном испарителе. Полученный остаток очищали колоночной хроматографией в системе этилацетат-гексан, 4:1. Выход 12а-0.123 г (36%).
Синтез 12б (0.085 г, 25%) осуществляли из 11б и 7а аналогично процедуре, приведенной для 12а.
Синтез 13а (0.154 г, 45%%) осуществляли из 11а и 7б аналогично процедуре, приведенной для 12а.
Синтез 13б (0.188 г, 55%) осуществляли из 11б и 7б аналогично процедуре, приведенной для 12а.
Для простоты восприятия здесь и далее, во всех случаях использования рацемических смесей 7а и 7б, на схемах будет представлено только по одному энантиомеру исходного реактива и по одному, соответствующему ему, стереоизомерному продукту на каждой стадии.
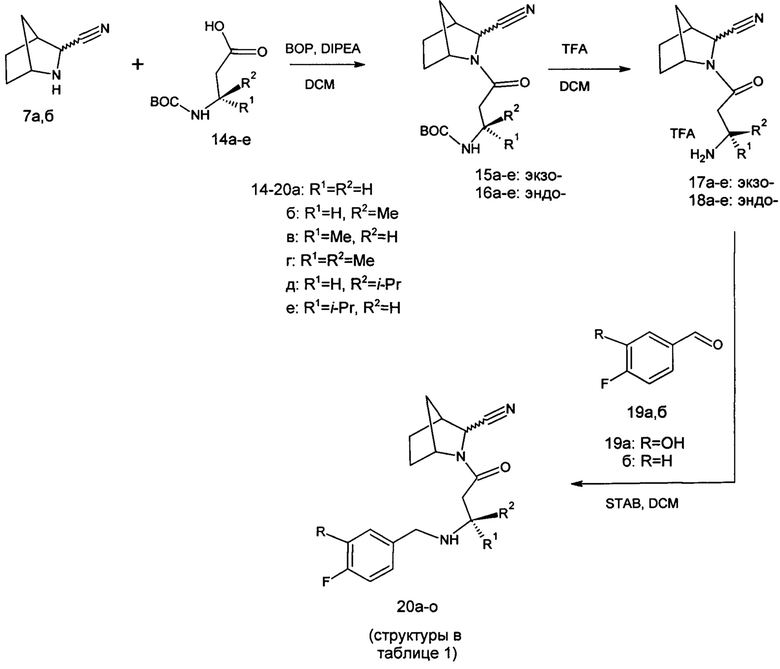
Синтез целевых соединений 20а-о осуществляли из рацемических экзо- и эндо-изомеров 3-азабицикло[2.2.1]гептан-2-карбонитрила 7а,б согласно схеме, изображенной ниже.
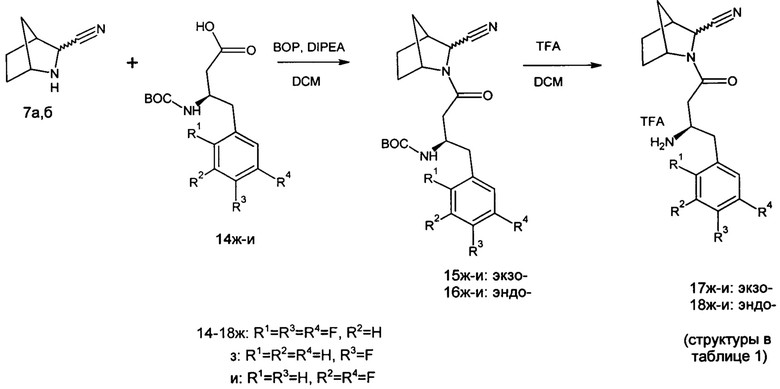
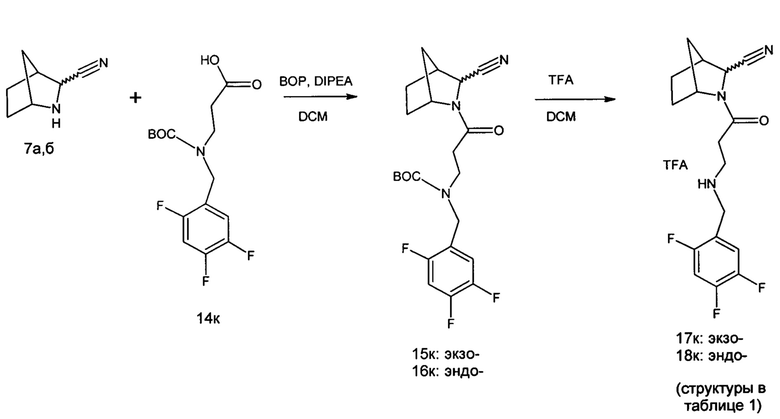
Синтез целевых соединений 17,18ж-к осуществляли из рацемических экзо- и эндо-изомеров 3-азабицикло[2.2.1]гептан-2-карбонитрила 7а,б, согласно схемам, изображенным ниже.
Синтез целевых соединений 26,27а-д и 36,37а-г осуществляли из рацемических смесей экзо- и эндо-изомеров 3-азабицикло[2.2.1]гептан-2-карбонитрилов 7а,б, согласно схемам, изображенным ниже.
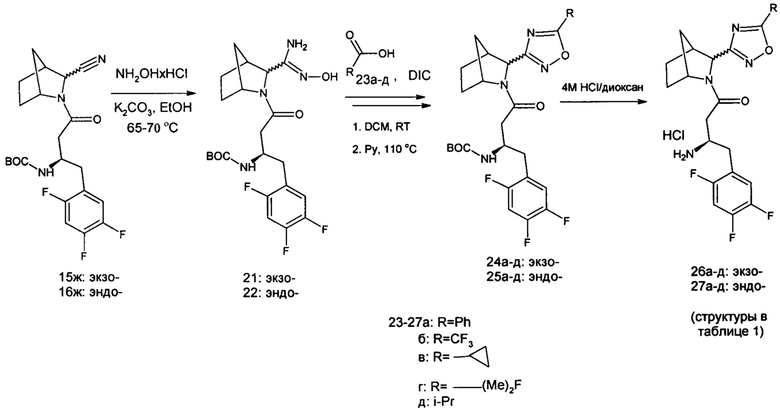
Процедура синтеза целевых продуктов 17,18ж-к и 20а-о.
К кислоте 14а 0.19 г (1 ммоль) в DCM (20 мл) добавляли DIPEA (0.13 г, 1 ммоль), ВОР (0.44 г, 1 ммоль) и рацемический амин 7а (0.12 г, 1 ммоль). Смесь перемешивали при комнатной температуре в течение ночи. Промывали 5% водным раствором лимонной кислоты (3×10 мл) и 10% водным раствором NaHCO3 (3×10 мл). Органический слой сушили над безв. Na2SO4. Растворитель упаривали на роторном испарителе досуха. Остаток растворяли в CHCl3 и проводили очистку методом колоночной хроматографии на силикагеле (элюент 1-5% МеОН/CHCl3). Выход 15а- 0.21 г (65%).
Синтез 15б (0.18 г, 60%) осуществляли из 14б и 7а аналогично процедуре, приведенной для 15а.
Синтез 15в (0.19 г, 62%) осуществляли из 14в и 7а аналогично процедуре, приведенной для 15а.
Синтез 15г (0.22 г, 70%) осуществляли из 14г и 7а аналогично процедуре, приведенной для 15а.
Синтез 15д (0.19 г, 58%) осуществляли из 14д и 7а аналогично процедуре, приведенной для 15а.
Синтез 15е (0.18 г, 55%) осуществляли из 14е и 7а аналогично процедуре, приведенной для 15а.
Синтез 15ж (0.30 г, 70%) осуществляли из 14ж и 7а аналогично процедуре, приведенной для 15а.
Синтез 15з (0.30 г, 75%) осуществляли из 14з и 7а аналогично процедуре, приведенной для 15а.
Синтез 15и (0.28 г, 68%) осуществляли из 14и и 7а аналогично процедуре, приведенной для 15а.
Синтез 15к (0.34 г, 80%) осуществляли из 14и и 7а аналогично процедуре, приведенной для 15а.
Синтез 16а (0.17 г, 57%) осуществляли из 14а и 7б аналогично процедуре, приведенной для 15 а.
Синтез 16б (0.17 г, 48%) осуществляли из 14б и 7б аналогично процедуре, приведенной для 15а.
Синтез 16в (0.18 г, 59%) осуществляли из 14в и 7б аналогично процедуре, приведенной для 15а.
Синтез 16г (0.19 г, 60%) осуществляли из 14г и 7б аналогично процедуре, приведенной для 15а.
Синтез 16д (0.19 г, 57%) осуществляли из 14д и 7б аналогично процедуре, приведенной для 15а.
Синтез 16е (0.18 г, 55%) осуществляли из 14е и 7б аналогично процедуре, приведенной для 15а.
Синтез 16ж (0.26 г, 60%) осуществляли из 14ж и 7б аналогично процедуре, приведенной для 15а.
Синтез 16з (0.25 г, 62%) осуществляли из 14з и 7б аналогично процедуре, приведенной для 15а.
Синтез 16и (0.24 г, 60%) осуществляли из 14и и 7б аналогично процедуре, приведенной для 15а.
Синтез 16к (0.32 г, 75%) осуществляли из 14к и 7б аналогично процедуре, приведенной для 15а.
Метод А.
Соединение 15а (0.21 г, 0.65 ммоль) растворяли в DCM (20 мл) и добавляли TFA (0.5 мл). Смесь перемешивали при комнатной температуре в течение ночи. Растворитель упаривали на роторном испарителе досуха. К остатку добавляли Et2O (20 мл). Выпавший осадок отфильтровывали и сушили в вакууме. Выход 17а в виде TFA-соли - 0.18 г (90%).
Синтез 17б (0.16 г, 85%) осуществляли из 156 аналогично процедуре, приведенной для 17а.
Синтез 17в (0.18 г, 90%) осуществляли из 15в аналогично процедуре, приведенной для 17а.
Синтез 17г (0.18 г, 80%) осуществляли из 15г аналогично процедуре, приведенной для 17а.
Синтез 17д (0.18 г, 90%) осуществляли из 15д аналогично процедуре, приведенной для 17а.
Синтез 17е (0.18 г, 91%) осуществляли из 15е аналогично процедуре, приведенной для 17а.
Синтез 18а (0.16 г, 84%) осуществляли из 16а аналогично процедуре, приведенной для 17а.
Синтез 18б (0.15 г, 81%) осуществляли из 16б аналогично процедуре, приведенной для 17а.
Синтез 18в (0.18 г, 90%) осуществляли из 16в аналогично процедуре, приведенной для 17а.
Синтез 18г (0.18 г, 80%) осуществляли из 16г аналогично процедуре, приведенной для 17а.
Синтез 18д (0.18 г, 90%) осуществляли из 16д аналогично процедуре, приведенной для 17а.
Синтез 18е (0.18 г, 91%) осуществляли из 16е аналогично процедуре, приведенной для 17а.
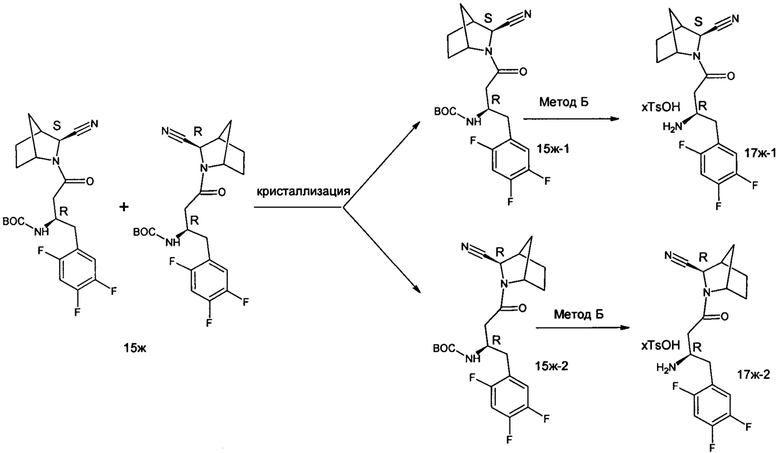
Метод Б.
Соединение 15ж (0.30 г, 0.69 ммоль) растворяли в ацетонитриле (20 мл) и добавляли пара-толуолсульфокислоту (2 экв.). Смесь перемешивали при комнатной температуре в течение ночи. Выпавший осадок отфильтровывали, промывали Et2O и сушили в вакууме. Выход 17ж в виде п-ТСК-соли - 0.30 г (85%).
Синтез 17з (0.23 г, 65%) осуществляли из 15з аналогично процедуре, приведенной для 17а.
Синтез 17и (0.22 г, 67%) осуществляли из 15и аналогично процедуре, приведенной для 17а.
Синтез 17к (0.21 г, 53%) осуществляли из 15к аналогично процедуре, приведенной для 17а.
Синтез 18ж (0.25 г, 82%) осуществляли из 16ж аналогично процедуре, приведенной для 17а.
Синтез 18з (0.22 г, 74%) осуществляли из 16з аналогично процедуре, приведенной для 17а.
Синтез 18и (0.19 г, 67%) осуществляли из 16и аналогично процедуре, приведенной для 17а.
Синтез 18к (0.24 г, 64%) осуществляли из 16к аналогично процедуре, приведенной для 17а.
К раствору 17а 0.18 г (0.58 ммоль) в DCM (20 мл) добавляли альдегид 19а (2 экв.) и STAB (5 экв.). Смесь перемешивали при комнатной температуре в течение ночи, упаривали на роторном испарителе до ~3-4 мл объема и наносили на колонку с силикагелем. Проводили очистку методом колоночной хроматографии (элюент 10-50% МеОН/CHCl3). Выход 20а - 0.06 г (30%).
Синтез 20б (0.056 г, 30%) осуществляли из 17б и 19а аналогично процедуре, приведенной для 20а.
Синтез 20б (0.05 г, 29%) осуществляли из 17б и 19а аналогично процедуре, приведенной для 20а.
Синтез 20в (0.06 г, 31%) осуществляли из 17в и 19а аналогично процедуре, приведенной для 20а.
Синтез 20г (0.05 г, 28%) осуществляли из 17г и 19а аналогично процедуре, приведенной для 20а.
Синтез 20д (0.05 г, 25%) осуществляли из 17д и 19а аналогично процедуре, приведенной для 20а.
Синтез 20е (0.06 г, 31%) осуществляли из 17е и 19а аналогично процедуре, приведенной для 20а.
Синтез 20ж (0.06 г, 29%) осуществляли из 18а и 19а аналогично процедуре, приведенной для 20а.
Синтез 20з (0.05 г, 25%) осуществляли из 18б и 19а аналогично процедуре, приведенной для 20а.
Синтез 20и (0.06 г, 30%) осуществляли из 18в и 19а аналогично процедуре, приведенной для 20а.
Синтез 20к (0.06 г, 31%) осуществляли из 18г и 19а аналогично процедуре, приведенной для 20а.
Синтез 20л (0.06 г, 30%) осуществляли из 17б и 19б аналогично процедуре, приведенной для 20а.
Синтез 20м (0.05 г, 26%) осуществляли из 18б и 19б аналогично процедуре, приведенной для 20а.
Синтез 20н (0.06 г, 31%) осуществляли из 18в и 19б аналогично процедуре, приведенной для 20а.
Синтез 20о (0.06 г, 31%) осуществляли из 17в и 19б аналогично процедуре, приведенной для 20а.
Процедура синтеза целевых продуктов 26-27а-д.
К нитрилу 15ж (0.43 г, 1 ммоль) в EtOH (20 мл) добавляли NH2OH×HCl (0.5 г, 7 ммоль) и K2CO3 (0.7 г, 5 ммоль). Смесь перемешивали при температуре 65-70°С в течение ночи. Охлаждали до комнатной температуры. Добавляли DCM (50 мл). Отфильтровывали осадок. Маточный раствор упаривали досуха на роторном испарителе. Остаток (21) использовали далее без очистки.
К кислоте 23а (0.13 г, 1.1 ммоль) в DCM (20 мл) добавили DIC (0.26 г, 2.5 экв). Смесь перемешивали при комнатной температуре 1 ч. Добавляли соединение 21. Перемешивание продолжали еще 1 ч. Растворитель упаривали на роторном испарителе. К остатку добавляли пиридин (25 мл). Смесь перемешивали при температуре 110°С в течение ночи. Охлаждали до комнатной температуры. Растворитель упаривали досуха на роторном испарителе. К остатку добавляли DCM (30 мл). Промывали 5% водным раствором лимонной кислоты (3×10 мл) и 10% водным раствором NaHCO3 (3×10 мл). Органический слой сушили над безв. Na2SO4. Растворитель упаривали на роторном испарителе. Остаток растворяли в DCM и проводили очистку методом колоночной хроматографии на силикагеле (элюент 0-20% Et2O/DCM). Выход 24а - 0.2 г (37% на 3 стадии).
Синтез 24б (0.11 г, 20%) осуществляли из 15ж и 23б аналогично процедуре, приведенной для 24а.
Синтез 24в (0.21 г, 40%) осуществляли из 15ж и 23в аналогично процедуре, приведенной для 24а.
Синтез 24г (0.15 г, 27%) осуществляли из 15ж и 23г аналогично процедуре, приведенной для 24а.
Синтез 24д (0.16 г, 30%) осуществляли из 15ж и 23д аналогично процедуре, приведенной для 24а.
Синтез 25а (0.19 г, 35%) осуществляли из 16ж и 23а аналогично процедуре, приведенной для 24а.
Синтез 25б (0.10 г, 18%) осуществляли из 16ж и 23б аналогично процедуре, приведенной для 24а.
Синтез 25в (0.19 г, 36%) осуществляли из 16ж и 23в аналогично процедуре, приведенной для 24а.
Синтез 25г (0.13 г, 23%) осуществляли из 16ж и 23г аналогично процедуре, приведенной для 24а.
Синтез 25д (0.14 г, 26%) осуществляли из 15ж и 23д аналогично процедуре, приведенной для 24а.
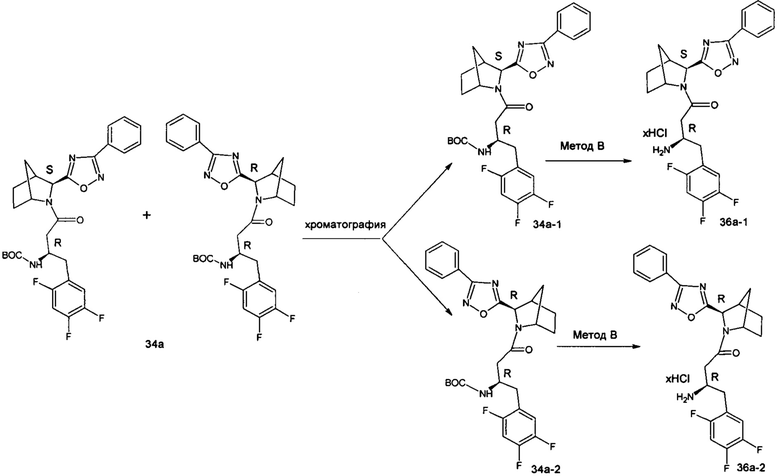
Метод В.
К соединению 24а (0.2 г, 0.36 ммоль) добавляли 4М HCl в диоксане (15 мл). Смесь перемешивали при комнатной температуре в течение ночи. Растворитель упаривали на роторном испарителе досуха. К остатку добавляли Et2O (20 мл). Выпавший осадок отфильтровывали и сушили в вакууме. Выход 26а в виде HCl-соли - 0.16 г (93%).
Синтез 26б (0.09 г, 93%) осуществляли из 24б аналогично процедуре, приведенной для 26а.
Синтез 26в (0.17 г, 92%) осуществляли из 24в аналогично процедуре, приведенной для 26а.
Синтез 26 г (0.12 г, 94%) осуществляли из 24г аналогично процедуре, приведенной для 26а.
Синтез 26д (0.12 г, 90%) осуществляли из 24д аналогично процедуре, приведенной для 26а.
Синтез 27а (0.15 г, 91%) осуществляли из 25а аналогично процедуре, приведенной для 26а.
Синтез 27б (0.08 г, 93%) осуществляли из 25б аналогично процедуре, приведенной для 26а.
Синтез 27в (0.15 г, 90%) осуществляли из 25в аналогично процедуре, приведенной для 26а.
Синтез 27г (0.10 г, 91%) осуществляли из 25г аналогично процедуре, приведенной для 26а.
Синтез 27д (0.11 г, 93%) осуществляли из 25д аналогично процедуре, приведенной для 26а.
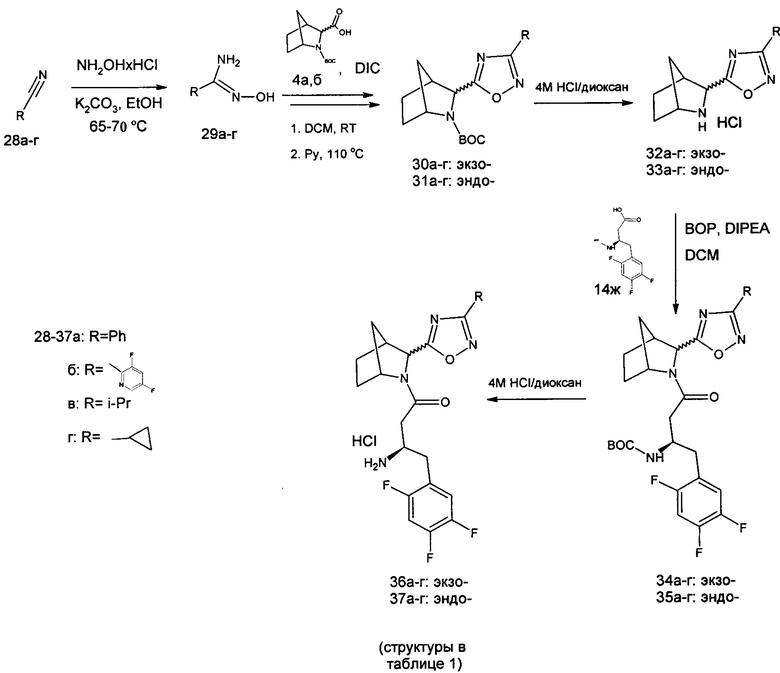
Процедура синтеза целевых продуктов 36-37а-д.
К нитрилу 28а (0.5 г, 5 ммоль) в EtOH (20 мл) добавляли NH2OH×HCl (1.38 г, 20 ммоль) и K2CO3 (2.05 г, 15 ммоль). Смесь перемешивали при температуре 65-70°С в течение ночи. Охлаждали до комнатной температуры. Добавляли DCM (50 мл). Отфильтровывали осадок. Маточный раствор упаривали досуха на роторном испарителе. Остаток (29а) использовали далее без очистки.
К кислоте 4а (0.9 г, 4 ммоль) в DCM (50 мл) добавили DIC (1.17 г, 2.5 экв). Смесь перемешивали при комнатной температуре 1 ч. Добавляли соединение 29а. Перемешивание продолжали еще 1 ч. Растворитель упаривали на роторном испарителе. К остатку добавляли пиридин (40 мл). Смесь перемешивали при температуре 110°С в течение ночи. Охлаждали до комнатной температуры. Растворитель упаривали досуха на роторном испарителе. К остатку добавляли DCM (30 мл). Промывали 5% водным раствором лимонной кислоты (3×10 мл) и 10% водным раствором NaHCO3 (3×10 мл). Органический слой сушили над безв. Na2SO4. Растворитель упаривали на роторном испарителе. Остаток растворяли в CHCl3 и проводили очистку методом колоночной хроматографии на силикагеле (элюент - CHCl3). Выход 30а - 0.7 г (55% на 3 стадии).
Синтез 30б (0.5 г, 35%) осуществляли из 28б и 4а аналогично процедуре, приведенной для 20а, с небольшими модификациями. Активацию кислоты и ацилирование полупродукта 29б проводили в пиридине (40 мл). Смесь перемешивали при комнатной температуре 2 ч. Затем нагревали до температуры 110°С и продолжали перемешивание в течение ночи. Охлаждали до комнатной температуры. Растворитель упаривали досуха на роторном испарителе. К остатку добавляли DCM (5 мл). Остаток растворяли в CHCl3 и проводили очистку методом колоночной хроматографии на силикагеле (элюент - 0-5% EtOH/CHCl3).
Синтез 30в (0.7 г, 61%) осуществляли из 28в и 4а аналогично процедуре, приведенной для 30а.
Синтез 30г (0.74 г, 66%) осуществляли из 28г и 4а аналогично процедуре, приведенной для 30а.
Синтез 31а (0.6 г, 47%) осуществляли из 28а и 4б аналогично процедуре, приведенной для 30а.
Синтез 31б (0.4 г, 28%) осуществляли из 28б и 4б аналогично процедуре, приведенной для 30б.
Синтез 31в (0.6 г, 52%) осуществляли из 28в и 4б аналогично процедуре, приведенной для 30а.
Синтез 31г (0.65 г, 57%) осуществляли из 28г и 4г аналогично процедуре, приведенной для 30а.
Синтез 32а (0.55 г, 96%) осуществляли из 30а аналогично процедуре (Метод В), приведенной для 26а.
Синтез 32б (0.40 г, 86%) осуществляли из 30б аналогично процедуре (Метод В), приведенной для 26а. Продукт получен в виде дигидрохлорида (×2HCl).
Синтез 32в (0.48 г, 86%) осуществляли из 30в аналогично процедуре (Метод В), приведенной для 26а.
Синтез 32г (0.52 г, 89%) осуществляли из 30г аналогично процедуре (Метод В), приведенной для 26а.
Синтез 33а (0.41 г, 84%) осуществляли из 31а аналогично процедуре (Метод В), приведенной для 26а.
Синтез 33б (0.31 г, 83%) осуществляли из 31б аналогично процедуре (Метод В), приведенной для 26а. Продукт получен в виде дигидрохлорида (×2HCl).
Синтез 33в (0.44 г, 92%) осуществляли из 31в аналогично процедуре (Метод В), приведенной для 26а.
Синтез 33г (0.46 г, 89%) осуществляли из 31г аналогично процедуре, приведенной для 26а.
Синтез 34а (0.8 г, 73%) осуществляли из 32а и 14ж аналогично процедуре, приведенной для 15а. Очистку методом колоночной хроматографии проводили в элюенте 0-20% Et2O/DCM.
Синтез 34б (0.40 г, 59%) осуществляли из 32б и 14ж аналогично процедуре, приведенной для 15а, исключив промывку реакционной смеси раствором лимонной кислоты.
Синтез 34в (0.69 г, 67%) осуществляли из 32в и 14ж аналогично процедуре, приведенной для 15а. Очистку методом колоночной хроматографии проводили в элюенте 0-20% Et2O/DCM.
Синтез 34г (0.71 г, 63%) осуществляли из 32г и 14ж аналогично процедуре, приведенной для 15а. Очистку методом колоночной хроматографии проводили в элюенте 0-20% Et2O/DCM.
Синтез 35а (0.51 г, 61%) осуществляли из 33а и 14ж аналогично процедуре, приведенной для 15а. Очистку методом колоночной хроматографии проводили в элюенте 0-20% Et2O/DCM.
Синтез 35б (0.26 г, 50%) осуществляли из 33б и 14ж аналогично процедуре, приведенной для 15а, исключив промывку реакционной смеси раствором лимонной кислоты.
Синтез 35в (0.51 г, 55%) осуществляли из 33в и 14ж аналогично процедуре, приведенной для 15а. Очистку методом колоночной хроматографии проводили в элюенте 0-20% Et2O/DCM.
Синтез 35г (0.57 г, 58%) осуществляли из 33г и 14ж аналогично процедуре, приведенной для 15а. Очистку методом колоночной хроматографии проводили в элюенте 0-20% Et2O/DCM.
В таблице 1 представлены структуры полученных соединений, их названия, аналитические характеристики и показатели ДПП-4 ингибирующей активности (IC50) целевых продуктов.

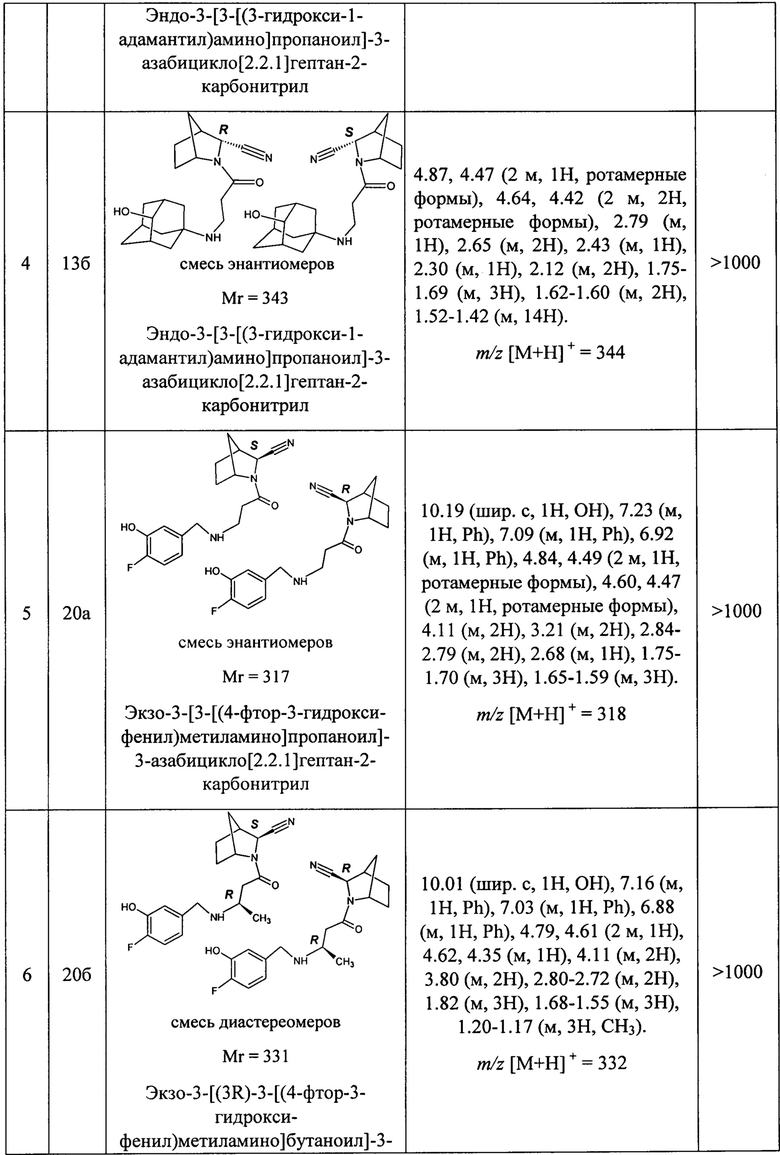
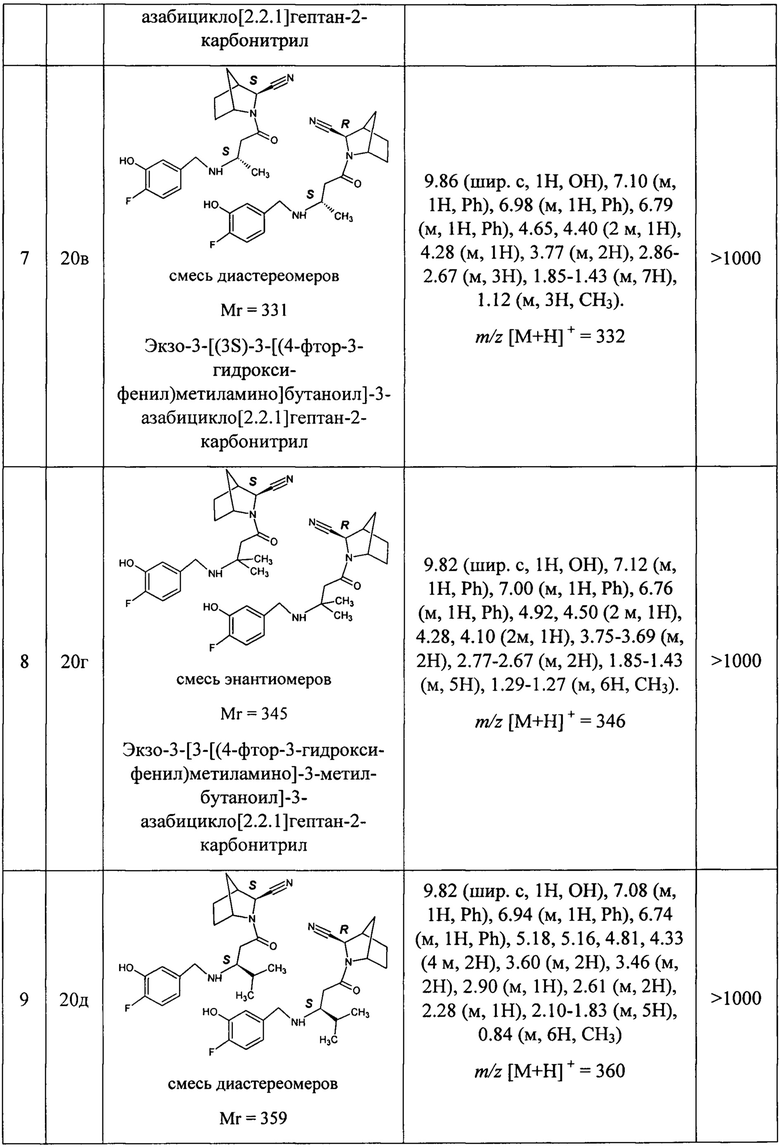
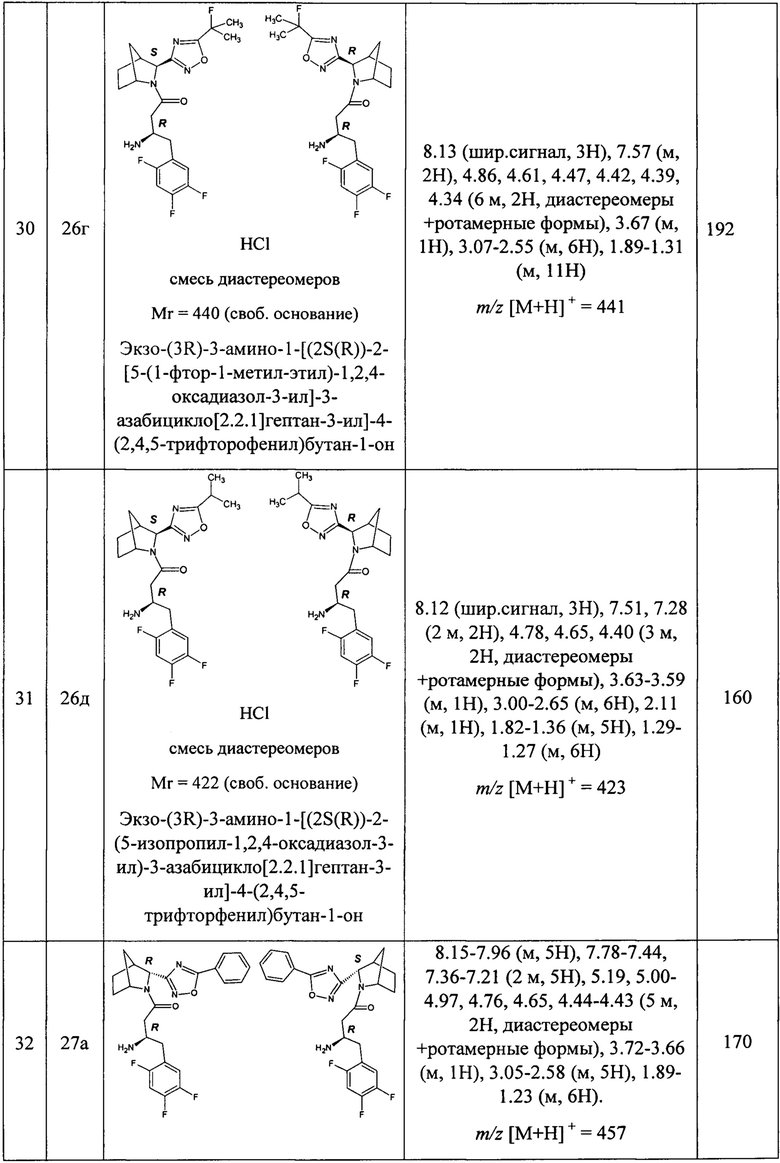
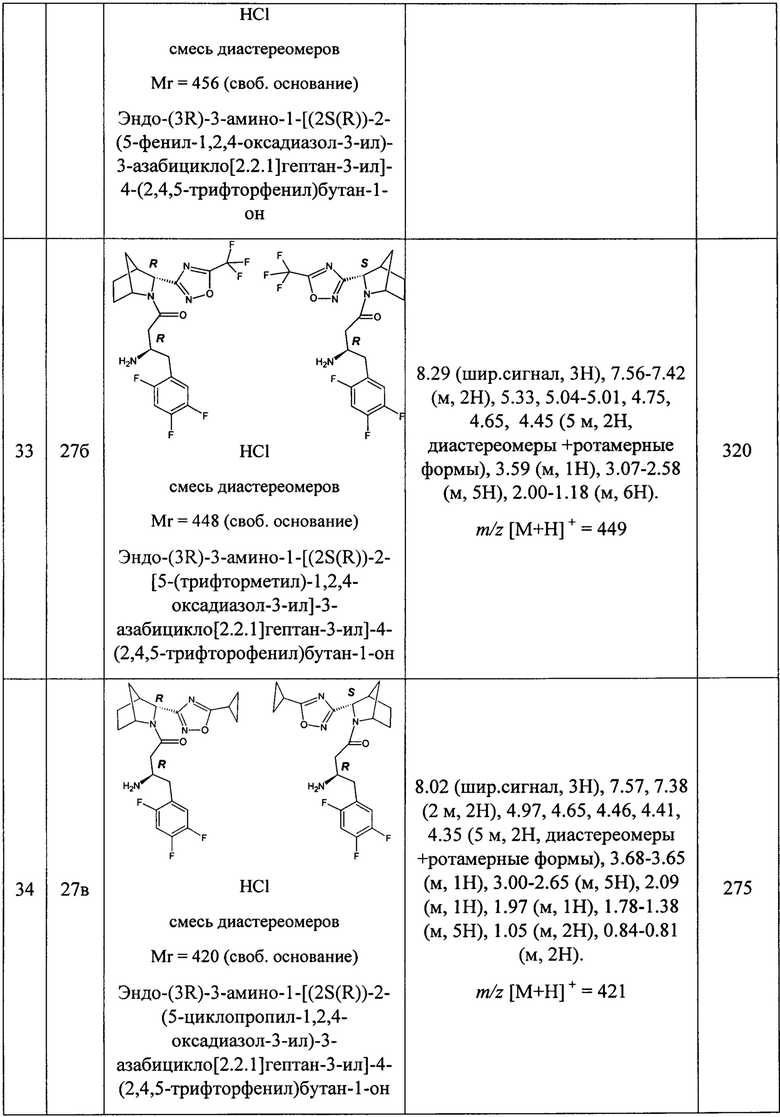
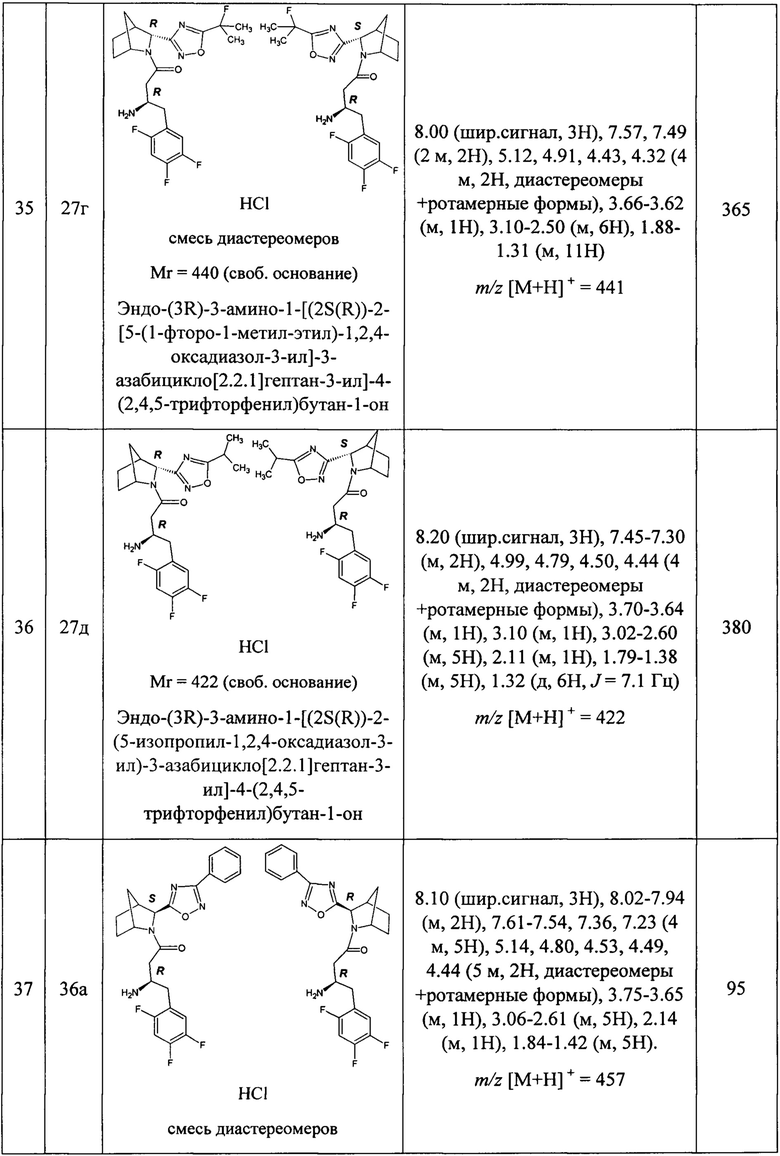
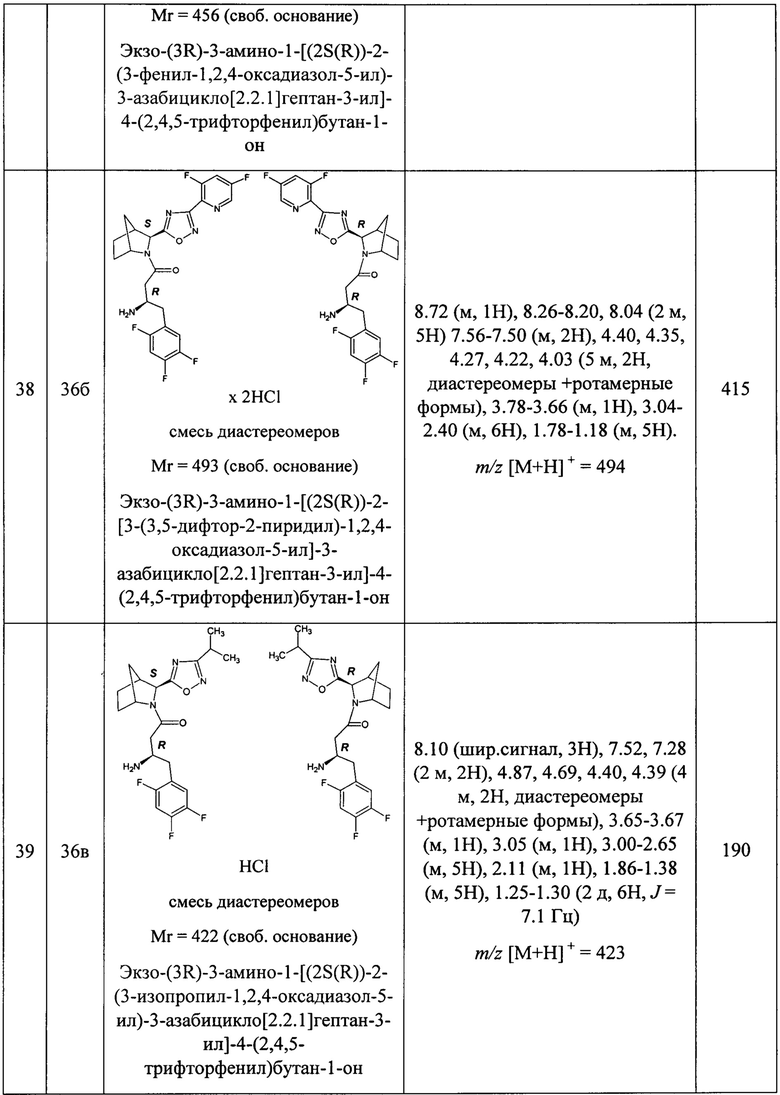

Условия проведения анализа: колонка - Onix С18 50×4.6 мм; элюент 1 - 0.1% TFA в воде; элюент 2 - 0.1% TFA в ацетонитриле, градиент - элюент 1 - 2.9 мин, элюент 2 - 0.2 мин, элюент 1 - промывка, скорость потока - 3.75 мл/мин, детекция - УФ (254 нм) и масс-спектрометрия.
Были также разработаны методы получения индивидуальных диастереомеров целевых соединений.
Метод Г (кристаллизация).
Индивидуальные диастереомеры 15ж-1 и 15ж-2 были получены из изомерной смеси 15ж последовательной 5-кратной кристаллизацией из смеси DCM/Et2O. Удаление защитной группы (метод Б) в 15ж-1 и 15ж-2 приводило к целевым диастереомерно чистым продуктам 17ж-1 и 17ж-2 (структуры в таблице 2).
Метод Д (колоночная хроматография на силикагеле).
Индивидуальные диастереомеры 34а-1 и 34а-2 были получены из изомерной смеси 34а колоночной хроматографией на силикагеле в элюенте 1:1->2:1 Et2O/н-гексан. Удаление защитной группы (метод В) в 34а-1 и 34а-2 приводило к целевым диастереомерно чистым продуктам 36а-1 и 36а-2 (структуры в таблице 2).
Метод Е (стереоспецифичный синтез энантиомерно чистого экзо-3-азабицикло[2.2.1]гептан-2-карбонитрила 7а-1.
Синтез энантиомерно чистого (S)-экзо-3-азабицикло[2.2.1]гептан-2-карбонитрила 7а-1 был осуществлен согласно схеме:
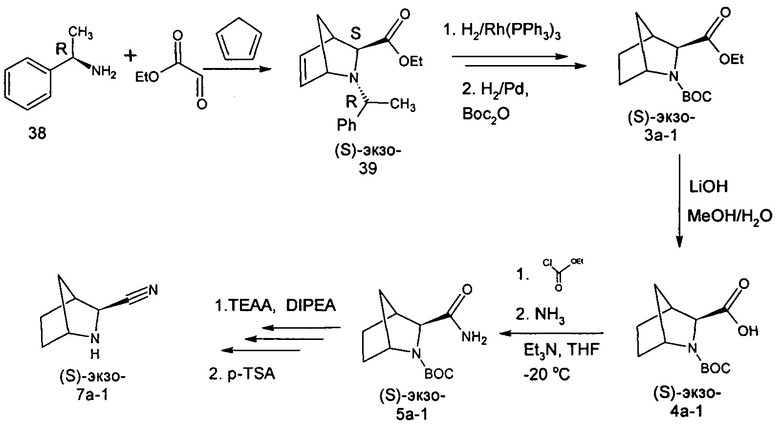
Процедура синтеза (S)-экзо-3-азабицикло[2.2.1]гептан-2-карбонитрила 7а-1.
Этил-(2S,3S)-3-(1-фенилэтил)-3-азабицикло [2.2.1]гепт-5-ен-2-карбоксилат (39)
В литровую колбу поместили 50%-ный раствор этилглиоксилата в толуоле (19.6 мл, 0.096 моль), толуол (300 мл) и бензиламин 38 (10.3 мл, 0.08 моль), перемешивали в течении 2 часов до выделения воды, затем воду удалили при помощи сульфата натрия и растворитель упарили. Полученный остаток растворили в диметилформамиде (100 мл), прибавили при охлаждении в ледяной бане раствор трифторуксусной кислоты (6.12 мл, 0.08 моль) в диметилформамиде. Перемешивали реакционную смесь при комнатной температуре в течение 1 часа. Затем при охлаждении в ледяной бане одной порцией прибавили свежеперегнанный циклопентадиен (10.6 г, 0.16 моль). Выдержали реакционную смесь 24 часа при комнатной температуре.
Через 24 часа реакционную смесь разбавили 400 мл 10%-ного раствора поташа при охлаждении в ледяной бане, экстрагировали этилацетатом 3 раза по 100 мл, органический слой высушили сульфатом натрия и упарили при температуре бани не выше 30°С. Колоночной хроматографией на силикагеле в системе этилацетат : петролейный фир 1:10 выделили 8.1 г (0.0299 моль) чистой фракции экзо-изомера 39. Выход 37.3%.
(2S)-3-трет-бутоксикарбонил-3-азабицикло[2.2.1]гептен-2-карбоновой кислоты этиловый эфир (3а-1).
В 200 мл бензола растворили экзо-изомер 39 (8.1 г, 0.0299 моль) и прибавили трис(трифенилфосфин)родийхлорид (0.405 г, 5% масс). Гидрировали на аппарате Парра при 20psi до прекращения поглощения водорода (около 4 часов). По данным LCMS контролировали полноту протекания гидрирования. После окончания реакции раствор пропустили через слой силикагеля, смывая продукт системой этилацетат : петролейный эфир 1:4. Полученный раствор упарили, добавили 200 мл этанола, 5%-ный Pd на угле (0.8 г) и Бок-ангидрид (6.9 г, 0.0316 моль). Далее гидрировали при 20psi на аппарате Парра. Контроль полноты реакции осуществляли методом LCMS. Полученную реакционную смесь использовали в следующей стадии.
(2S)-3-трет-бутоксикарбонил-3-азабицикло[2.2.1]гептен-2-карбоновая кислота (4а-1)
Этанольный раствор Бок-эфира экзо-изомера 3а-1 с предыдущей стадии после завершения гидрирования отфильтровали от катализатора и прибавили к нему 50 мл (0.075 моль) водного раствора гидроксида натрия. Полученный раствор нагревали 1 час при 60°С. После полной конверсии эфира (контроль тсх) упарили этанол, растворили остаток в воде, подкислили раствор 2М соляной кислотой до рН 3, экстрагировали продукт этилацетатом, промыли органические вытяжки водой, высушили сульфатом натрия и упарили. Продукт перекристаллизовывали из гексана. Получили 5.42 г (0.022 моль) экзо-кислоты 4а-1. Выход составил 90%. Далее использовали этот продукт без дополнительной очистки.
(2S)-Экзо-трет-бутил-3-карбамоил-2-азабицикло[2.2.1]гептан-2-карбоксилат (5а-1)
К раствору исходной кислоты 4а-1 (5.42 г) в сухом ТГФ при охлаждении до -20°С в атмосфере аргона добавляли триэтиламин 3.45 мл (2.5 г) затем по каплям добавляли этилхлорформиат (2.68 г, 0.0247 моль) в течение 10 мин. Выдерживали реакционную смесь при охлаждении в течение 40 мин. Затем пропускали аммиак из баллона 1 ч. Упарили ТГФ, обработали остаток раствором лимонной кислоты до рН 4, экстрагировали этилацетатом, этилацетатные экстракты промывали раствором соды, сушили сульфатом натрия и концентрировали. Получили 5.29 г бесцветного кристаллического остатка 5а-1. Выход количественный.
(2S)-Экзо-третбутил-3-циано-2-азабицикло[2.2.1]гептан-2-карбоксилат (6а-1)
К суспензии исходного амида 5а-1 (5.29 г) в сухом ТГФ, при температуре не выше 4°С добавляли 2 эквивалента триэтиламина (6.13 мл, 4.45 г), ангидрид трифторуксусной кислоты (6.93 г, 0.033 моль) в течение 10 минут, по ТСХ контролировали степень протекания реакции. Реакционную смесь выдерживали при охлаждении в течение 3 часов. Реакционную смесь упаривали, наносили на силикагель и разделяли на хроматографической колонке. Элюент - смесь петролейный эфир : этилацетат 4:1. Получили 4.29 г (87%) целевого нитрила 6а-1 в виде бледно-желтого густого маслообразного вещества.
(2S)-Экзо-2-азабицикло[2.2.1]гептан-3-карбонитрил (7а-1)
К исходному БОК-нитрилу 6а-1 (4.29 г, 0.019 моль) в 30 мл ацетонитрила добавили двукратный избыток n-толилсульфоновой кислоты (ПТСК) (6.54 г, 0.038 моль) и оставили перемешиваться на ночь. Отгоняли ацетонитрил, остаток растирали с диэтиловым эфиром (3-4 обработки с декантацией). Упаривали эфир. Получили 4.58 г кристаллического целевого вещества 7а-1.
трет-бутил-N-[(1R)-3-[(2S)-2-циано-3-азабицикло[2.2.1]гептан-3-ил]-3-оксо-1-[(2,4,5-трифторфенил)метил]пропил]карбамат (15ж-1) (получали из энантиомерно чистого (S)-экзо-изомера 3-азабицикло[2.2.1]гептан-2-карбонитрила 7а-1, аналогично 7а.
К раствору кислоты 14ж (5.19 г, 0.0156 моль) в 60 мл дихлорметана прибавили ВОР (8.27 г, 0.0187 моль) и триэтиламин (4.74 мл, 0.0468 моль, 3 экв.). Через 15 минут при комнатной температуре прибавили тозилат амина 7а-1 (4.57 г, 0.0156 моль). Через 2 часа LCMS показала полную конверсию. Дихлорметан упарили, остаток растворили в этилацетате и промыли 10%-ным раствором поташа, затем водой, высушили сульфатом натрия и упарили. Добавили к остатку небольшое количество петролейного эфира и выкристаллизовавшийся продукт отфильтровали. Получили 5.08 г кристаллов 15ж-1. Выход 75%
(2S)-3-[(3R)-3-амино-4-(2,4,5-трифторфенил)бутаноил]-3-азабицикло[2.2.1]гептен-2-карбонитрил (17ж-1)
Для дебокирования суспендировали бок-производное 15ж-1 (5.08 г) в 100 мл ацетонитрила и прибавили двукратный избыток гидрата толуолсульфокислоты (4.02 г, 0.0234 моль). Перемешивали ночь при комнатной температуре. Отфильтровали осадок и промыли малым количеством ацетонитрила. Получили 5.18 г тозилата продукта 17ж-1. Выход 87%
Синтез перечисленных ниже диастереомерно (или энантиомерно) чистых целевых соединений (структуры в таблице 2) осуществляли аналогично 17ж-1:
17ж-1, 17з-1, 17и-1 - из энантиомерно чистого (S)-экзо-изомера 3-азабицикло[2.2.1]гептан-2-карбонитрила 7а-1.
17к-1 - из энантиомерно чистого (S)-экзо-изомера 3-азабицикло[2.2.1]гептан-2-карбонитрила 7а-1.
17ж-2 - из энантиомерно чистого (R)-экзо-изомера 3-азабицикло[2.2.1]гептан-2-карбонитрила 7а-2.
26а-1, 26в-1, 26г-1 и 26д-1 - из диастереомерно чистого 15ж-1.
36а-1, 36б-1, 36в-1 и 36г-1 - из энантиомерно чистой кислоты 4а-1.
В таблице 2 представлены структуры полученных соединений, названия, аналитические характеристики и показатели ДПП-4 ингибирующей активности (IC50) целевых продуктов - индивидуальных диастереомеров или энантиомеров.


Условия проведения анализа: колонка - Onix С18 50×4.6 мм; элюент 1 - 0.1% TFA в воде; элюент 2-0.1% TFA в ацетонитриле, градиент - элюент 1 - 2.9 мин, элюент 2 - 0.2 мин, элюент 1 - промывка, скорость потока - 3.75 мл/мин, детекция - УФ (254 нм) и масс-спектрометрия.
Структурным элементом молекул всех полученных ингибиторов ДПП-4 является слабоосновная функциональная аминогруппа аминокислотного фрагмента. Специалисту среднего уровня понятно, что все вещества могут быть получены, как в виде бессолевых форм (свободного основания), так и солевых форм с противоионами кислотной природы - такие как:
ацетат, аспартат, бензилсульфонат, бензоат, бикарбонат, тартрат, глутамат, гликолат, сахаринат, гексаноат, гексилрезорцинат, мукат, напсилат, нитрат, октаноат, олеат, памоат, пантотенат, фосфат/дигидрофосфат, камсилат, карбонат, изетионат, гидрохлорид, полилактоуронат, пропионат, цитрат, гидрохлорид, лактат, салицилат, стеарат, малат, деканоат, малеат, мезилат, сульфат, фумарат, малеат, тозилат, метилсульфат, глюконат и др.
Бессолевые формы могут быть получены выделением свободного основания после нейтрализации солевых форм, образующихся в результате удаления БОК-защитной группы.
Солевые формы могут быть получены в результате:
- удаления БОК-защитной группы:
- n-толуолсульфокислотой в органических растворителях,
- соляной кислотой в спиртовых или водных средах,
- раствором хлористого водорода (HCl) в диоксане,
- трифторуксусной кислотой в органических растворителях;
- добавления к свободным основаниям (бессолевым формам) кислот из числа перечисленных выше;
- солевым обменом кислот из числа перечисленных выше.
Исследование ДПП-4-ингибирующей активности синтезированных соединений.
Ингибирующую активность синтезированных образцов в отношении ДПП-4 проводили в сравнении активности в отсутствии и в присутствии в системе анализируемого образца (субстрата). Оценку осуществляли флуоресцентным методом с использованием сертифицированного набора реагентов (Kit-ы) «Dipeptidyl peptidase IV Inhibitor Screening Assay Kit (ab 133081)». Все манипуляции проводили в полном соответствии с инструкцией к набору. Каждое вещество проанализировано в диапазоне концентраций от 10-4 до 10-11 М, в диапазоне флуоресценции 450-465 нм.
Поставленная задача заявляемого изобретения - создание ингибиторов дипептидилпептидазы-4, устойчивых к внутримолекулярной циклизации, для лечения больных сахарным диабетом 2-ого типа и расширение линейки известных ингибиторов дипептидилпептидазы-4 решена за счет создания ряда ингибиторов дипептидилпептидазы-4, устойчивых к внутримолекулярной циклизации за счет как введения в пирролидиновый фрагмент стерического фактора, препятствующего циклизации, так и введением заместителей, при которых циклизация невозможна.
Задача решена путем синтеза и исследования перечисленных в таблицах 1 и 2 соединений. В результате исследований было обнаружено, что все соединения проявляют ингибирующий эффект по отношению к ДПП-4, при этом наиболее активными оказались соединения: экзо-(2S)-3-[(3R)-3-амино-4-(2,4,5-трифторфенил)бутаноил]-3-азабицикло[2.2.1]гептан-2-карбонитрил (17ж-1), экзо-(3R)-3-амино-1-[(2S)-2-(5-фенил-1,2,4-оксадиазол-3-ил)-3-азабицикло[2.2.1]гептан-3-ил]-4-(2,4,5-трифторфенил)бутан-1-он (26а-1), и экзо-(3R)-3-амино-1-[(2S)-2-(3-фенил-1,2,4-оксадиазол-5-ил)-3-азабицикло [2.2.1]гептан-3-ил]-4-(2,4,5-трифторфенил)бутан-1-он (36а-1).
Промышленная применимость доказана и подтверждена примерами синтеза и анализа полученных веществ.
Список сокращений.
СД - сахарный диабет
СМ - сульфонилмочевина
ДПП-4 - дипептидилпептидаза-4
ГИП - гастроингибиторньй пептид
ЖИП - желудочный ингибиторный пептид
ГПП-1 - глюкагон-подобный пептид-1
Ala - аланин
STAB - натрий триацетоксиборгидрид
DCM - дихлорметан (хлористый метилен)
МеОН - метанол
EtOH - этанол
CHCl - хлороформ
п-ТСК - пара-толуолсульфокислота
NH2OH×HCl - гидроксиламина гидрохлорид
К2СО3 - калия карбонат (поташ)
HCl - хлористый водород
МТБЕ - метил-трет-бутиловый эфир
NaOH - натрия гидроксид
ПЭ - петролейный эфир
EtOAc - этилацетат
ТГФ - тетрагидрофуран
ТЭА - триэтиламин
Pd/C - палладий на угле
DIC - диизопропилкарбодиимид
Et2O - диэтиловый эфир
TFA - трифторуксусная кислота
NaHCO3 - натрия гидрокарбонат
DIPEA - диизопропилэтиламин
Na2SO4 - натрия сульфат
ВОР - бензотриазол-1-илокси-трис(диметиламино)фосфоний гексафторфосфат
ТСХ - тонкослойная хроматография
LC/MS - высокоэффективная жидкостная хроматография с масс-детекцией
ЯМР - ядерный магнитный резонанс
DMSO - диметилсульфоксид
Mr - молекулярная масса
IC50 - концентрация ингибитора, при которой соответствующий биологический процесс в условиях in vitro ингибирован на 50%
шир. - широкий
с - синглет
д - дуплет
м - мультиплет
| название | год | авторы | номер документа |
|---|---|---|---|
| СПОСОБ ПРОГНОЗИРОВАНИЯ ТЕРАПЕВТИЧЕСКОГО ЭФФЕКТА ИНГИБИТОРА LSD1 НА ОСНОВЕ ЭКСПРЕССИИ INSM1 | 2018 |
|
RU2789449C2 |
| УСИЛИТЕЛЬ ПРОТИВООПУХОЛЕВОГО ЭФФЕКТА С ПРИМЕНЕНИЕМ НОВОГО СОЕДИНЕНИЯ БИФЕНИЛА | 2018 |
|
RU2765153C2 |
| НОВОЕ СОЕДИНЕНИЕ БИФЕНИЛА ИЛИ ЕГО СОЛЬ | 2018 |
|
RU2765152C2 |
| НОВОЕ СОЕДИНЕНИЕ БИФЕНИЛА ИЛИ ЕГО СОЛЬ | 2016 |
|
RU2726622C2 |
| ПРОИЗВОДНЫЕ ПИРАЗОЛА, СПОСОБ ИХ ПОЛУЧЕНИЯ, СОДЕРЖАЩАЯ ИХ ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ И ПРОМЕЖУТОЧНЫЕ СОЕДИНЕНИЯ СИНТЕЗА | 1993 |
|
RU2119917C1 |
| ГЕТЕРОЦИКЛИЧЕСКИЕ ИНГИБИТОРЫ ЯНУС-КИНАЗЫ 3 | 2006 |
|
RU2434013C2 |
| ИНГИБИТОРЫ ДИПЕПТИДИЛПЕПТИДАЗЫ IV | 2010 |
|
RU2574410C2 |
| Фармацевтическая композиция на основе действующего вещества, ингибитора дипептидилпептидазы-4, для предупреждения развития и лечения сахарного диабета 2 типа | 2020 |
|
RU2727898C1 |
| ПРОИЗВОДНЫЕ 3β-АЛКЕНИЛПЕНАМА И ИХ ФАРМАЦЕВТИЧЕСКИ СОВМЕСТИМЫЕ СОЛИ | 1994 |
|
RU2139290C1 |
| ПРОИЗВОДНЫЕ ГЕКСАГИДРОДИАЗЕПИНОНА, ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ, ИХ СОДЕРЖАЩАЯ, И ПРИМЕНЕНИЕ ДЛЯ ПОЛУЧЕНИЯ ЛЕКАРСТВЕННОГО СРЕДСТВА ДЛЯ ЛЕЧЕНИЯ ИНСУЛИННЕЗАВИСИМОГО ДИАБЕТА | 2003 |
|
RU2301803C2 |
Изобретение относится к области органической химии, а именно к новым соединениям, представляющим собой амиды бета-аминокислот стереоизомеров 3-азабицикло[2.2.1]гептен-2-карбонитрила формулы (I), или их фармацевтически приемлемым солям, где R1 = Н - водород; С1-5 - алкил; R2 = Н - водород; С1-5 - алкил; бензил (C6H5-CH2), замещенный одним, двумя или тремя атомами галогена (F); R3 = Н - водород; адамантил, в котором каждый углерод может иметь заместитель, выбранный из: гидрокси (ОН-); бензил (C6H5-CH2), в котором могут быть заместители: гидрокси- (ОН-) и один галоген (F). Также изобретение относится к амиду бета-аминокислот стереоизомеров 3-(3-азабицикло[2.2.1]гептан-2-ил)-1,2,4-оксадиазола формулы II и амиду бета-аминокислот стереоизомеров 5-(3-азабицикло[2.2.1]гептан-2-ил)-1,2,4-оксадиазола формулы III, ингибитору дипептидилпептидазы-4 на основе указанных соединений. Технический результат: создание ингибиторов дипептидилпептидазы-4 для лечения больных сахарным диабетом 2-го типа. 4 н. и 4 з.п. ф-лы, 99 пр., 2 табл.
 ,
, 
1. Соединения общей формулы (I) - амиды бета-аминокислот стереоизомеров 3-азабицикло[2.2.1]гептен-2-карбонитрила (I), или их фармацевтически приемлемые соли,
где:
R1 = Н - водород; С1-5 - алкил;
R2 = Н - водород; С1-5 - алкил; бензил (C6H5-CH2), замещенный одним, двумя или тремя атомами галогена (F);
R3 = Н - водород; адамантил, в котором каждый углерод может иметь заместитель, выбранный из: гидрокси (ОН-); бензил (C6H5-CH2), в котором могут быть заместители: гидрокси- (ОН-) и один галоген (F).
2. Соединения по п. 1, где:
R1 = H, R2 = H, R3 = гидроксиадамантил-
экзо-3-[3-[(3-гидрокси-1-адамантил)амино]пропаноил]-3-азабицикло[2.2.1]гептен-2-карбонитрил (12а), или
R1 = H, R2 = H, R3 = гидроксиадамантил-
экзо-3-[3-[(10-гидрокси-1-адамантил)амино]пропаноил]-3-азабицикло[2.2.1]гептен-2-карбонитрил (12б), или
R1 = Н, R2 = H, R3 = гидроксиадамантил-
эндо-3-[3-[(3-гидрокси-1-адамантил)амино]пропаноил]-3-азабицикло[2.2.1]гептан-2-карбонитрил (13а), или
R1 = Н, R2 = H, R3 = гидроксиадамантил-
эндо-3-[3-[(10-гидрокси-1-адамантил)амино]пропаноил]-3-азабицикло[2.2.1]гептан-2-карбонитрил (13б), или
R1 = Н, R2 = Н, R3 = 4-фтор-3-гидроксибензил-
экзо-3-[3-[(4-фтор-3-гидроксифенил)метиламино]пропаноил]-3-азабицикло[2.2.1]гептан-2-карбонитрил (20а), или
R1 = метил, R2 = Н, R3 = 4-фтор-3-гидроксибензил-
экзо-3-[(3R)-3-[(4-фтор-3-гидроксифенил)метиламино]бутаноил]-3-азабицикло[2.2.1]гептан-2-карбонитрил (20б), или
R1 = Н, R2 = метил, R3 = 4-фтор-3-гидроксибензил-
экзо-3-[(3S)-3-[(4-фтор-3-гидроксифенил)метиламино]бутаноил]-3-азабицикло[2.2.1]гептан-2-карбонитрил (20в), или
R1 = метил, R2 = метил, R3 = 4-фтор-3-гидроксибензил-
экзо-3-[3-[(4-фтор-3-гидроксифенил)метиламино]-3-метил-бутаноил]-3-азабицикло[2.2.1]гептан-2-карбонитрил (20г), или
R1 = изопропил, R2 = H, R3 = 4-фтор-3-гидроксибензил-
экзо-3-[(3S)-3-[(4-фтор-3-гидрокси-фенил)метиламино]-4-метил-пентаноил]-3-азабицикло[2.2.1]гептан-2-карбонитрил (20д), или
R1 = Н, R2 = изопропил, R3 = 4-фтор-3-гидроксибензил-
экзо-3-[(3R)-3-[(4-фтор-3-гидроксифенил)метиламино]-4-метил-пентаноил]-3-азабицикло[2.2.1]гептан-2-карбонитрил (20е), или
R1 = Н, R2 = H, R3 = 4-фтор-3-гидроксибензил-
эндо-3-[3-[(4-фтор-3-гидроксифенил)метиламино]пропаноил]-3-азабицикло[2.2.1]гептан-2-карбонитрил (20ж), или
R1 = метил, R2 = H, R3 = 4-фтор-3-гидроксибензил-
эндо-3-[(3R)-3-[(4-фтор-3-гидроксифенил)метиламино]бутаноил]-3-азабицикло[2.2.1]гептан-2-карбонитрил (20з), или
R1 = Н, R2 = метил, R3 = 4-фтор-3-гидроксибензил
эндо-3-[(3S)-3-[(4-фтор-3-гидроксифенил)метиламино]бутаноил]-3-азабицикло[2.2.1]гептан-2-карбонитрил (20и), или
R1 = метил, R2 = метил, R3 = 4-фтор-3-гидроксибензил-
эндо-3-[3-[(4-фтор-3-гидроксифенил)метиламино]-3-метил-бутаноил]-3-азабицикло[2.2.1]гептан-2-карбонитрил (20к), или
R1 = метил, R2 = H, R3 = 4-фторбензил-
экзо-3-[(3R)-3-[(4-фторфенил)метиламино]бутаноил]-3-азабицикло[2.2.1]гептан-2-карбонитрил (20л), или
R1 = метил, R2 = H, R3 = 4-фторбензил-
эндо-3-[(3R)-3-[(4-фторфенил)метиламино]бутаноил]-3-азабицикло[2.2.1]гептан-2-карбонитрил (20м), или
R1 = Н, R2 = метил, R3 = 4-фторбензил-
эндо-3-[(3S)-3-[(4-фторфенил)метиламино]бутаноил]-3-азабицикло[2.2.1]гептан-2-карбонитрил (20н), или
R1 = Н, R2 = метил, R3 = 4-фторбензил-
экзо-3-[(3S)-3-[(4-фторфенил)метиламино]бутаноил]-3-азабицикло[2.2.1]гептан-2-карбонитрил (20о), или
R1 = Н, R2 = 2,4,5-трифторбензил, R3 = H-
зкзо-3-[(3R)-3-амино-4-(2,4,5-трифторфенил)бутаноил]-3-азабицикло[2.2.1]гептан-2-карбонитрил (17ж), или
R1 = Н, R2 = 2,4,5-трифторбензил, R3 = H-
эндо-3-[(3R)-3-амино-4-(2,4,5-трифторфенил)бутаноил]-3-азабицикло[2.2.1]гептан-2-карбонитрил (18ж), или
R1 = Н, R2 = 4-фторбензил, R3 = Н-
экзо-3-[(3R)-3-амино-4-(4-фторфенил)бутаноил]-3-азабицикло[2.2.1]гептан-2-карбонитрил (17з), или
R1 = Н, R2 = 4-фторбензил, R3 = Н-
эндо-3-[(3R)-3-амино-4-(4-фторфенил)бутаноил]-3-азабицикло[2.2.1]гептан-2-карбонитрил (18з), или
R1 = Н, R2 = 3,5-дифторбензил, R3 = Н,
экзо-3-[(3R)-3-амино-4-(3,5-дифторфенил)бутаноил]-3-азабицикло[2.2.1]гептан-2-карбонитрил (17и), или
R1 = Н, R2 = 3,5-дифторбензил, R3 = Н-
эндо-3-[(3R)-3-амино-4-(3,5-дифторфенил)бутаноил]-3-азабицикло[2.2.1]гептан-2-карбонитрил (18и), или
R1 = Н, R2 = H, R3 = 2,4,5-трифторбензил-
экзо-3-[3-[(2,4,5-трифторфенил)метиламино]пропаноил]-3-азабицикло[2.2.1]гептан-2-карбонитрил (17к), или
R1 = Н, R2 = H, R3 = 2,4,5-трифторбензил-
эндо-3-[3-[(2,4,5-трифторфенил)метиламино]пропаноил]-3-азабицикло[2.2.1]гептан-2-карбонитрил (18к), или
энантиомерно чистые соединения:
R1 = Н, R2 = 2,4,5-трифторбензил, R3 = H-
экзо-(2S)-3-[(3R)-3-амино-4-(2,4,5-трифторфенил)бутаноил]-3-азабицикло[2.2.1]гептан-2-карбонитрил (17ж-1), или
R1 = Н, R2 = 2,4,5-трифторбензил, R3 = H-
экзо-(2R)-3-[(3R)-3-амино-4-(2,4,5-трифторфенил)бутаноил]-3-азабицикло[2.2.1]гептан-2-карбонитрил (17ж-2), или
R1 = Н, R2 = 4-фторбензил, R3 = Н-
экзо-(2S)-3-[(3R)-3-амино-4-(4-фторфенил)бутаноил]-3-азабицикло[2.2.1]гептан-2-карбонитрил (17з-1), или
R1 = Н, R2 = 3,5-дифторбензил, R3 = Н-
экзо-(2S)-3-[(3R)-3-амино-4-(3,5-дифторфенил)бутаноил]-3-азабицикло[2.2.1]гептан-2-карбонитрил (17и-1), или
R1 = Н, R2 = H, R3 = 2,4,5-трифторбензил
экзо-(2S)-3-[3-[(2,4,5-трифторфенил)метиламино]пропаноил]-3-азабицикло[2.2.1]гептан-2-карбонитрил (17к-1).
3. Соединения общей формулы (II) - амиды бета-аминокислот стереоизомеров 3-(3-азабицикло[2.2.1]гептан-2-ил)-1,2,4-оксадиазола, или их фармацевтически приемлемые соли,
где:
R1 = Н - водород;
R2 = фенил - (C6H5-), замещенный тремя атомами галогена (F);
R3 = Н - водород;
R4 = метил, в котором может быть три заместителя, выбранных из галогена (F); циклопропил, изопропил, в котором каждый углерод независимо может иметь один заместитель галогена (F); фенил (C6H5-).
4. Соединения по п. 3, где:
R1 = H, R2 = 2,4,5-трифторфенил, R3 = H, R4 = фенил-
экзо-(3R)-3-амино-1-[(2S(R))-2-(5-фенил-1,2,4-оксадиазол-3-ил)-3-азабицикло[2.2.1]гептан-3-ил]-4-(2,4,5-трифторфенил)бутан-1-он (26а), или
R1 = H, R2 = 2,4,5-трифторфенил, R3 = H, R4 = трифторметил-
экзо-(3R)-3-амино-1-[(2S(R))-2-[5-(трифторметил)-1,2,4-оксадиазол-3-ил]-3-азабицикло[2.2.1]гептан-3-ил]-4-(2,4,5-трифторфенил)бутан-1-он (26б), или
R1 = H, R2 = 2,4,5-трифторфенил, R3 = H, R4 = циклопропил-
экзо-(3R)-3-амино-1-[(2S(R))-2-(5-циклопропил-1,2,4-оксадиазол-3-ил)-3-азабицикло[2.2.1]гептан-3-ил]-4-(2,4,5-трифторфенил)бутан-1-он (26в), или
R1 = H, R2 = 2,4,5-трифторфенил, R3 = H, R4 = 2-фтор-изопропил-
экзо-(3R)-3-амино-1-[(2S(R))-2-[5-(1-фтор-1-метил-этил)-1,2,4-оксадиазол-3-ил]-3-азабицикло[2.2.1]гептан-3-ил]-4-(2,4,5-трифторфенил)бутан-1-он (26г), или
R1 = H, R2 = 2,4,5-трифторфенил, R3 = H, R4 = изопропил-
экзо-(3R)-3-амино-1-[(2S(R))-2-(5-изопропил-1,2,4-оксадиазол-3-ил)-3-азабицикло[2.2.1]гептан-3-ил]-4-(2,4,5-трифторфенил)бутан-1-он (26д), или
R1 = H, R2 = 2,4,5-трифторфенил, R3 = H, R4 = фенил-
эндо-(3R)-3-амино-1-[(2S(R))-2-(5-фенил-1,2,4-оксадиазол-3-ил)-3-азабицикло[2.2.1]гептан-3-ил]-4-(2,4,5-трифторфенил)бутан-1-он (27а), или
R1 = Н, R2 = 2,4,5-трифторфенил, R3 = Н, R4 = трифторметил-
эндо-(3R)-3-амино-1-[(2S(R))-2-[5-(трифторметил)-1,2,4-оксадиазол-3-ил]-3-азабицикло[2.2.1]гептан-3-ил]-4-(2,4,5-трифторофенил)бутан-1-он (27б), или
R1 = H, R2 = 2,4,5-трифторфенил, R3 = H, R4 = циклопропил-
эндо-(3R)-3-амино-1-[(2S(R))-2-(5-циклопропил-1,2,4-оксадиазол-3-ил)-3-азабицикло[2.2.1]гептан-3-ил]-4-(2,4,5-трифторфенил)бутан-1-он (27в), или
R1 = H, R2 = 2,4,5-трифторфенил, R3 = H, R4 = 2-фтор-изопропил-
эндо-(3R)-3-амино-1-[(2S(R))-2-[5-(1-фтор-1-метил-этил)-1,2,4-оксадиазол-3-ил]-3-азабицикло[2.2.1]гептан-3-ил]-4-(2,4,5-трифторфенил)бутан-1-он (27г), или
R1 = H, R2 = 2,4,5-трифторфенил, R3 = H, R4 = изопропил-
эндо-(3R)-3-амино-1-[(2S(R))-2-(5-изопропил-1,2,4-оксадиазол-3-ил)-3-азабицикло[2.2.1]гептан-3-ил]-4-(2,4,5-трифторфенил)бутан-1-он (27д), или
энантиомерно чистые соединения, где:
R1 = H, R2=2,4,5-трифторфенил. R3 = H, R4 = фенил-
экзо-(3R)-3-амино-1-[(2S)-2-(5-фенил-1,2,4-оксадиазол-3-ил)-3-азабицикло[2.2.1]гептан-3-ил]-4-(2,4,5-трифторфенил)бутан-1-он (26а-1), или
R1 = H, R2 = 2,4,5-трифторфенил, R3 = H, R4 = циклопропил-
экзо-(3R)-3-амино-1-[(2S)-2-(5-циклопропил-1,2,4-оксадиазол-3-ил)-3-азабицикло[2.2.1]гептан-3-ил]-4-(2,4,5-трифторфенил)бутан-1-он (26в-1), или
R1 = H, R2 = 2,4,5-трифторфенил, R3 = H, R4 = 2-фтор-изопропил-
экзо-(3R)-3-амино-1-[(2S)-2-[5-(1-фтор-1-метил-этил)-1,2,4-оксадиазол-3-ил]-3-азабицикло[2.2.1]гептан-3-ил]-4-(2,4,5-трифторфенил)бутан-1-он (26г-1), или
R1 = H, R2 = 2,4,5-трифторфенил, R3 = H, R4 = изопропил-
экзо-(3R)-3-амино-1-[(2S)-2-(5-изоопропил-1,2,4-оксадиазол-3-ил)-3-азабицикло[2.2.1]гептан-3-ил]-4-(2,4,5-трифторфенил)бутан-1-он (26д-1).
5. Соединения общей формулы (III) - амиды бета-аминокислот стереоизомеров 5-(3-азабицикло[2.2.1]гептан-2-ил)-1,2,4-оксадиазола, или их фармацевтически приемлемые соли,
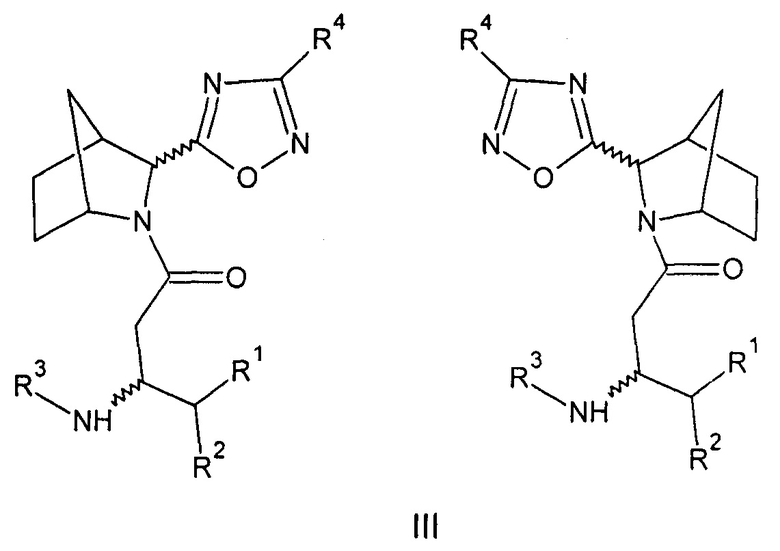
где:
R1 = Н - водород;
R2 = фенил- (C6H5-), замещенный тремя атомами галогена (F);
R3 = Н - водород;
R4 = циклопропил, изопропил, фенил, гетарил (C5H4N-), замещенный двумя атомами галогена (F).
6. Соединения по п. 5, где:
R1 = H, R2 = 2,4,5-трифторфенил, R3 = H, R4 = фенил-
экзо-(3R)-3-амино-1-[(2S(R))-2-(3-фенил-1,2,4-оксадиазол-5-ил)-3-азабицикло[2.2.1]гептан-3-ил]-4-(2,4,5-трифторфенил)бутан-1-он (36а), или
R1 = H, R2 = 2,4,5-трифторфенил, R3 = H, R4 = 3,5-дифтор-2-пиридил-
экзо-(3R)-3-амино-1-[(2S(R))-2-[3-(3,5-дифтор-2-пиридил)-1,2,4-оксадиазол-5-ил]-3-азабицикло[2.2.1]гептан-3-ил]-4-(2,4,5-трифторфенил)бутан-1-он (36б), или
R1 = H, R2 = 2,4,5-трифторфенил, R3 = H, R4 = изопропил-
экзо-(3R)-3-амино-1-[(2S(R))-2-(3-изопропил-1,2,4-оксадиазол-5-ил)-3-азабицикло[2.2.1]гептан-3-ил]-4-(2,4,5-трифторфенил)бутан-1-он (36в), или
R1 = H, R2 = 2,4,5-трифторфенил, R3 = H, R4 = циклопропил-
экзо-(3R)-3-амино-1-[(2S(R))-2-(3-циклопропил-1,2,4-оксадиазол-5-ил)-3-азабицикло[2.2.1]гептан-3-ил]-4-(2,4,5-трифторофенил)бутан-1-он (36г), или
R1 = H, R2 = 2,4,5-трифторфенил, R3 = H, R4 = фенил-
эндо-(3R)-3-амино-1-[(2S(R))-2-(3-фенил-1,2,4-оксадиазол-5-ил)-3-азабицикло[2.2.1]гептан-3-ил]-4-(2,4,5-трифторфенил)бутан-1-он (37а), или
R1 = H, R2 = 2,4,5-трифторфенил, R3 = H, R4 = 3,5-дифтор-2-пиридил
эндо-(3R)-3-амино-1-[(2S(R))-2-[3-(3,5-дифтор-2-пиридил)-1,2,4-оксадиазол-5-ил]-3-азабицикло[2.2.1]гептан-3-ил]-4-(2,4,5-трифторфенил)бутан-1-он (37б), или
R1 = H, R2 = 2,4,5-трифторфенил, R3 = H, R4 = изопропил-
эндо-(3R)-3-амино-1-[(2S(R))-2-(3-изопропил-1,2,4-оксадиазол-5-ил)-3-азабицикло[2.2.1]гептан-3-ил]-4-(2,4,5-трифторфенил)бутан-1-он (37в), или
R1 = H, R2 = 2,4,5-трифторфенил, R3 = H, R4 = циклопропил-
эндо-(3R)-3-амино-1-[(2S(R))-2-(3-циклопропил-1,2,4-оксадиазол-5-ил)-3-азабицикло[2.2.1]гептан-3-ил]-4-(2,4,5-трифторфенил)бутан-1-он (37 г), или
энантиомерно чистые соединения, где:
R1 = H, R2 = 2,4,5-трифторфенил, R3 = H, R4 = фенил-
экзо-(3R)-3-амино-1-[(2S)-2-(3-фенил-1,2,4-оксадиазол-5-ил)-3-азабицикло[2.2.1]гептан-3-ил]-4-(2,4,5-трифторфенил)бутан-1-он (36а-1), или
R1 = Н, R2 = 2,4,5-трифторфенил, R3 = H, R4 = фенил-
экзо-(3R)-3-амино-1-[(2R)-2-(3-фенил-1,2,4-оксадиазол-5-ил)-3-азабицикло[2.2.1]гептан-3-ил]-4-(2,4,5-трифторфенил)бутан-1-он (36а-2), или
R1 = H, R2 = 2,4,5-трифторфенил, R3 = H, R4 = 3,5-дифтор-2-пиридил-
экзо-(3R)-3-амино-1-[(2S)-2-[3-(3,5-дифтор-2-пиридил)-1,2,4-оксадиазол-5-ил]-3-азабицикло[2.2.1]гептан-3-ил]-4-(2,4,5-трифторфенил)бутан-1-он (36б-1), или
R1 = H, R2 = 2,4,5-трифторфенил, R3 = H, R4 = изопропил-
экзо-(3R)-3-амино-1-[(2S)-2-(3-изопропил-1,2,4-оксадиазол-5-ил)-3-азабицикло[2.2.1]гептан-3-ил]-4-(2,4,5-трифторфенил)бутан-1-он (36в-1), или
R1 = H, R2 = 2,4,5-трифторфенил, R3 = H, R4 = 3,5-циклопропил-
экзо-(3R)-3-амино-1-[(2S)-2-(3-циклопропил-1,2,4-оксадиазол-5-ил)-3-азабицикло[2.2.1]гептан-3-ил]-4-(2,4,5-трифторфенил)бутан-1-он (36г-1).
7. Соединение, выбранное по любому из предыдущих пунктов, и его фармацевтически приемлемые солевые формы.
8. Ингибитор дипептидилпептидазы-4 для лечения сахарного диабета 2-го типа или метаболического синдрома по пп. 1-7, выбранный из соединений общей формулы I, или II, или III или из их фармацевтически приемлемых солей.
| НОВЫЙ СОСТАВ | 2006 |
|
RU2483716C2 |
| WO 2007113634 A1, 11.10.2007. | |||

