Область техники
Данное изобретение относится к соединениям замещенного пирролидина и родственным соединениям, имеющим активность антагониста мускариновых рецепторов или антихолинергическую активность. Данное изобретение также относится к фармацевтическим композициям, содержащим указанные соединения; способам использования указанных соединений для лечения клинических состояний, опосредованных мускариновыми рецепторами; и способам и промежуточным соединениям, используемым для получения вышеуказанных соединений.
Предшествующий уровень техники
Легочные нарушения, такие как хроническая обструктивная болезнь легких (COPD) и астма, поражают много миллионов людей во всем мире и указанные расстройства являются основной причиной заболеваемости и смертности.
Антагонисты мускариновых рецепторов, как известно, обеспечивают бронхозащитные эффекты, и поэтому указанные соединения пригодны для лечения нарушений дыхания, таких как COPD и астма. При использовании для лечения таких нарушений антагонисты мускариновых рецепторов, как правило, вводят путем ингаляции. Однако даже при введении ингаляцией значительное количество антагониста мускариновых рецепторов зачастую абсорбируется в системное кровообращение, что приводит к системным побочным эффектам, таким как ксеростомия (сухость во рту), задержка мочи, мидриаз (расширение зрачка) и сердечно-сосудистые побочные эффекты.
Кроме того, многие вводимые ингаляцией антагонисты мускариновых рецепторов имеют относительно короткую продолжительность действия, что требует их введения несколько раз в день. Такая схема многократного ежедневного приема лекарственного средства не только неудобна, но и создает значительный риск неадекватного лечения из-за несоблюдения пациентом необходимой схемы частого приема лекарственного средства.
В соответствии с этим существует потребность в новых антагонистах мускариновых рецепторов. В частности, существует потребность в новых антагонистах мускариновых рецепторов, имеющих высокую эффективность и демонстрирующих ослабление проявления системных побочных эффектов при введении ингаляцией. Кроме того, существует потребность во вдыхаемых антагонистах мускариновых рецепторов, имеющих продолжительный период действия, тем самым делая возможным прием дозы лекарственного средства раз в день или даже один раз в неделю. Ожидается, что такие соединения будут, в частности, эффективными для лечения легочных нарушений, таких как COPD и астма, при этом уменьшая или ликвидируя проявление побочных действий, таких как ксеростомия.
Краткое изложение существа изобретения
Настоящее изобретение предлагает новые соединения замещенного пирролидина и родственные соединения, которые имеют активность антагониста мускариновых рецепторов или антихолинергическую активность. Среди других свойств соединения по данному изобретению, как было установлено, обладают удивительной и неожиданной аффинностью связывания с подтипами hM2 и hM3 мускариновых рецепторов по сравнению с родственными соединениями. Кроме того, соединения по данному изобретению, как было обнаружено, имеют удивительную и неожиданную легочную селективность при введении ингаляцией, тем самым приводя к снижению системных побочных эффектов. Кроме того, соединения по данному изобретению, как было установлено, обладают удивительной и неожиданной продолжительностью действия бронхозащиты при введении ингаляцией.
Соответственно, в одном из составляющих аспектов, данное изобретение предлагает соединение формулы I:
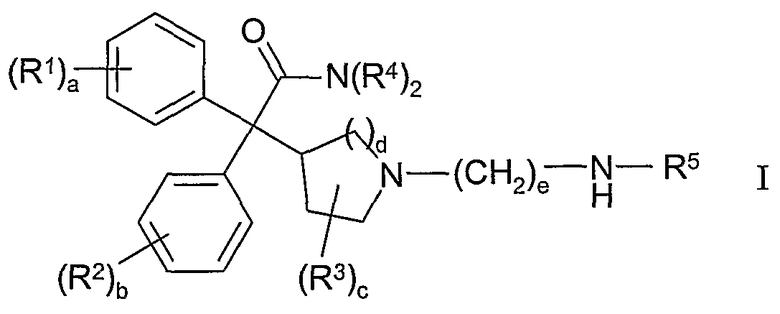
где
каждый R1 и R2 независимо выбран из C1-4алкила, С2-4алкенила, C2-4алкинила, C3-6циклоалкила, циано, галогена, -ORа, -SRа, -S(О)Rа, -S(O)2Ra и -NRbRc; или две соседние группы R1 или две соседние группы R2 соединены вместе с образованием С3-6алкилена, -(C2-4алкилен)-О- или -О-(C1-4алкилен)-О-;
каждый R3 независимо выбран из C1-4алкила и фтора;
каждый R4 независимо выбран из водорода, C1-6алкила, C2-6алкенила, C2-6алкинила, C3-6циклоалкила, C6-10арила, C2-9гетероарила, C3-6гетероциклила (гетероциклической группы), -CH2-R6 и -CH2CH2-R7; или обе группы R4 соединены вместе с атомом азота, с которым они связаны, с образованием С3-6гетероциклила;
R5 выбран из C1-6алкила, C2-6алкенила, С2-6алкинила, C3-6циклоалкила и -CH2-R8, где каждая алкильная, алкенильная и алкинильная группа необязательно замещена -OH или 1-5 фтор-заместителями;
каждый R6 независимо выбран из C3-6циклоалкила, C6-10арила, C2-9гетероарила и C3-6гетероциклила;
каждый R7 независимо выбран из C3-6циклоалкила, C6-10арила, C2-9гетероарила, C3-6гетероциклила, -OH, -О(С1-6алкила), -(C3-6циклоалкила), -О(С6-10арила), -О(С2-9гетероарила), -S(C1-6алкила), -S(О)(C1-6алкила), -S(О)2(C1-6алкила), -S(C3-6циклоалкила), -S(О)(C3-6циклоалкила), -S(О)2(C3-6циклоалкила), -S(C6-10арила), -S(О)(C6-10арила), -S(O)2(C6-10арила), -S(C2-9гетероарила), -S(О)(C2-9гетероарила) и -S(O)2(C2-9гетероарила);
каждый R8 независимо выбран из C3-6циклоалкила, C6-10арила, C2-9гетероарила и C3-6гетероциклила;
каждый Rа независимо выбран из водорода, C1-4алкила, С2-4алкенила, С2-4алкинила и С3-6циклоалкила;
каждый Rb и Rc независимо выбран из водорода, C1-4алкила, С2-4алкенила, С2-4алкинила, и С3-6циклоалкила; или Rb и Rc соединены вместе с атомом азота, с которым они связаны, образуя С3-6гетероциклил;
a означает целое число от 0 до 3;
b означает целое число от 0 до 3;
c означает целое число от 0 до 4;
d равно 1 или 2;
e равно 8 или 9;
где каждая алкильная, алкиленовая, алкенильная, алкинильная и циклоалкильная группа в R1, R2, R3, R4, R7, Rа, Rb и Rc необязательно замещена 1-5 фтор-заместителями; каждая арильная, циклоалкильная, гетероарильная и гетероциклическая группа в R1, R2, R4, R5, R6, R7, R8, Rа, Rb и Rc необязательно замещена 1-3 заместителями, независимо выбранными из C1-4алкила, C2-4алкенила, C2-4алкинила, циано, галогена, -О(С1-4алкила), -S(C1-4алкила), -S(О)(C1-4алкила), -S(О)2(C1-4алкила), -NH2, -NH(C1-4алкила) и -N(C1-4алкил)2, где каждая алкильная, алкиленовая, алкенильная и алкинильная группа необязательно замещена 1-5 фтор-заместителями; и каждая -CH2- группа в -(CH2)е- необязательно замещена 1 или 2 заместителями, независимо выбранными из C1-2алкила, -OH и фтора;
или его фармацевтически приемлемую соль, или его сольват, или его стереоизомер.
В другом из составляющих аспектов данное изобретение предлагает соединение формулы II:
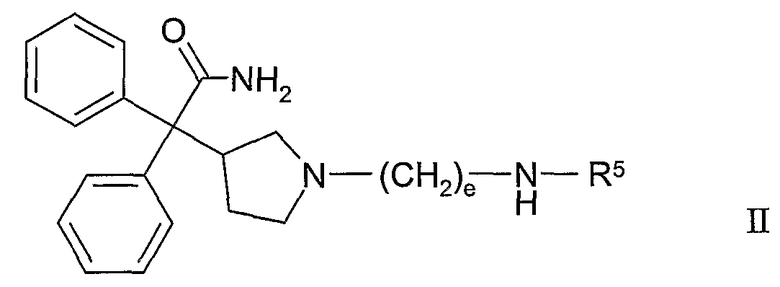
где R5 и e такие, как определено выше; или его фармацевтически приемлемую соль, или его сольват, или его стереоизомер.
В отдельных и различных вариантах данное изобретение касается также соединений формулы II, где стереохимия в 3-положении пирролидинового кольца имеет (R)-конфигурацию; и соединений формулы II, где стереохимия в 3-положении пирролидинового кольца имеет (S)-конфигурацию.
В другом из составляющих аспектов данное изобретение предлагает фармацевтическую композицию, содержащую фармацевтически приемлемый носитель и терапевтически эффективное количество соединения формулы I, или его фармацевтически приемлемой соли, или его сольвата, или его стереоизомера. Указанные фармацевтические композиции могут необязательно содержать другие терапевтические средства. Соответственно, в одном варианте, данное изобретение относится к такой фармацевтической композиции, где композиция дополнительно содержит терапевтически эффективное количество стероидного противовоспалительного средства, такого как кортикостероид; агонист β2-адренергического рецептора; ингибитор фосфодиэстеразы-4; или их комбинацию.
Соединения по данному изобретению представляют собой антагонисты мускариновых рецепторов. Соответственно, в одном из аспектов способа данное изобретение предлагает способ лечения млекопитающего, имеющего клиническое состояние, которое облегчается лечением антагонистом мускариновых рецепторов, и этот способ включает введение млекопитающему терапевтически эффективного количества соединения формулы I, или его фармацевтически приемлемой соли, или его сольвата, или его стереоизомера.
В другом из аспектов способа данное изобретение предлагает способ лечения легочного нарушения, и этот способ включает введение пациенту, нуждающемуся в таком лечении, терапевтически эффективного количества соединения формулы I, или его фармацевтически приемлемой соли, или его сольвата, или его стереоизомера.
В очередном из аспектов способа данное изобретение предлагает способ бронходилатации у пациента, и этот способ включает введение путем ингаляции пациенту вызывающего бронходилатацию количества соединения формулы I, или его фармацевтически приемлемой соли, или его сольвата, или его стереоизомера.
В следующем из аспектов способа данное изобретение предлагает способ лечения хронической обструктивной болезни легких или астмы, и этот способ включает введение пациенту, нуждающемся в таком лечении, терапевтически эффективного количества соединения формулы I, или его фармацевтически приемлемой соли, или его сольвата, или его стереоизомера.
Так как соединения по данному изобретению обладают активностью антагониста мускариновых рецепторов, указанные соединения также полезны в качестве инструментальных средств для изучения биологических систем или образцов, имеющих мускариновый рецептор, или для изучения активности других химических соединений. Соответственно, в очередном из аспектов способа данное изобретение предлагает способ применения соединения формулы I или его фармацевтически приемлемой соли, или его сольвата, или его стереоизомера в качестве инструментального средства для изучения биологической системы или образца, или для обнаружения новых химических соединений, имеющих активность антагониста мускариновых рецепторов.
Данное изобретение относится также к способам и новым промежуточным соединениям, используемым для получения соединений формулы I или их соли, или их сольвата, или их стеризомера. Соответственно в другом из аспектов способа данное изобретение предлагает способ получения соединения формулы I, и этот способ включает:
(a) взаимодействие соединения формулы III с соединением формулы IV в присутствии восстановителя;
(b) взаимодействие соединения формулы V с соединением формулы VI в присутствии восстановителя;
(c) взаимодействие соединения формулы VII с соединением формулы IV;
или
(d) взаимодействие соединения формулы V с соединением VIII;
и затем
(e) удаление любых защитных групп с получением соединения формулы I или его соли; где соединения формул I и III-VIII такие, как определены в данном описании.
В одном варианте вышеупомянутый способ дополнительно включает дополнительную стадию получения фармацевтически приемлемой соли соединения формулы I.
В другом из аспектов способа данное изобретение предлагает способ получения фармацевтически приемлемой соли соединения формулы I, и этот способ включает контактирование соединения формулы IX:
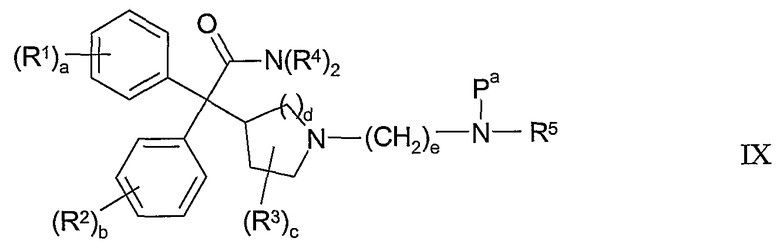
где R1-R5 и a-e такие, как определены выше; и Pа представляет собой неустойчивую к действию кислоты амино-защитную группу; с фармацевтически приемлемой кислотой, получая фармацевтически приемлемую соль соединения формулы I.
В других вариантах данное изобретение относится к другим способам, описанным здесь; и к продукту, полученному любым из способов, описанных здесь.
В другом из составляющих аспектов данное изобретение предлагает соединение формулы X:
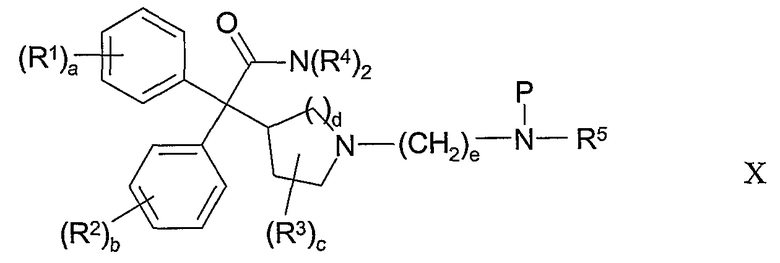
где R1-R5 и a-e такие, как определены здесь; и P представляет собой амино-защитную группу; или его соль, или его сольват, или его стереоизомер; для использования в качестве промежуточного соединения для получения соединений формулы I.
В другом из составляющих аспектов данное изобретение предлагает соединение формулы XI:

где R5 и e такие, как определены выше; P представляет собой амино-защитную группу; и G выбран из -CHO, -CH(ORf)2, -CH2OH и -2-L, где каждый Rf представляет независимо С1-6алкил или обе группы Rf соединены вместе с образованием С2-6алкилена; и L представляет собой удаляемую группу; или его соль, или его стереоизомер; для использования в качестве промежуточного соединения для получения соединений формулы I; при условии, что когда L представляет собой хлор, Р не является этоксикарбонилом (т.е. CH3CH2ОС(О)-).
В дополнительных отдельных и различных аспектах данное изобретение обеспечивает:
соединение формулы I или его фармацевтически приемлемую соль, или его сольват, или его стереоизомер для применения при лечении;
соединение формулы I или его фармацевтически приемлемую соль, или его сольват, или его стереоизомер для применения в качестве лекарственного средства;
соединение формулы I или его фармацевтически приемлемую соль, или его сольват, или его стереоизомер для применения при лечении легочных нарушений, включая хроническую обструктивную болезнь легких и астму;
лекарственное средство, содержащее соединение формулы I или его фармацевтически приемлемую соль, или его сольват, или его стереоизомер;
применение соединения формулы I или его фармацевтически приемлемой соли, или его сольвата, или его стереоизомера для лечения легочных нарушений, включая хроническую обструктивную болезнь легких и астму;
применение соединения формулы I или его фармацевтически приемлемой соли, или его сольвата, или его стереоизомера в качестве лекарственного средства для лечения легочных нарушений, включая хроническую обструктивную болезнь легких и астму;
применение соединения формулы I или его фармацевтически приемлемой соли, или его сольвата, или его стереоизомера для изготовления лекарственного средства; и
применение соединения формулы I или его фармацевтически приемлемой соли, или его сольвата, или его стереоизомера для изготовления лекарственного средства для лечения легочных нарушений, включая хроническую обструктивную болезнь легких и астму.
Подробное описание изобретения
Данное изобретение обеспечивает новые соединения замещенного пирролидина и родственные соединения формулы I или их фармацевтически приемлемые соли, или их сольваты, или их стереоизомеры. Указанные соединения могут содержать один или несколько хиральных центров и, когда такой хиральный центр или центры присутствуют, данное изобретение касается и рацемических смесей; чистых стереоизомеров (т.е. индивидуальных энантиомеров или диастереомеров); и стереоизомер-обогащенных смесей таких изомеров, если не оговорено особо. В тех случаях, когда представлен конкретный стереоизомер, для квалифицированных специалистов в данной области техники должно быть очевидно, что в композициях по данному изобретению могут присутствовать незначительные количества других стереоизомеров, если не оговорено особо, при условии, что присутствие в композиции таких других изомеров не устраняет ее полезности в целом.
Соединения по данному изобретению также содержат несколько основных групп (например, аминогрупп), и поэтому соединения формулы I могут существовать в виде свободного основания или в различных солевых формах. Все такие формы выходят в объем данного изобретения. В объем данного изобретения также включены фармацевтически приемлемые сольваты соединений формулы I или их соли.
Дополнительно, где это применимо, все цис-транс или E/Z изомеры (геометрические изомеры), таутомерные формы и топоизомерные формы соединений формулы I входят в объем данного изобретения, если не оговорено иначе.
Номенклатура, используемая в настоящем описании для названия соединений по данному изобретению, наглядно представлена в нижеприведенных Примерах. Обычно эту номенклатуру получали, используя коммерчески доступное программное обеспечение AutoNom software (MDL, San Leandro, California).
Типичные варианты осуществления изобретения
Подразумевается, что нижеследующие заместители и значения обеспечивают типичные примеры и варианты различных аспектов данного изобретения. Эти типичные значения предназначены для более подробного определения таких аспектов и вариантов осуществления и не подразумевают исключения других вариантов осуществления или ограничения объема данного изобретения. В этом отношении констатация того, что конкретное значение или заместитель является предпочтительным, не подразумевает, ни в каком смысле, исключения других значений или заместителей из данного изобретения, если не оговорено конкретно.
В конкретном варианте, R1 или R2, когда присутствуют, независимо выбраны из C1-4алкила, фтора, хлора и -ORа; где каждая алкильная группа необязательно замещена 1-3 фтор-заместителями. В другом конкретном варианте, каждый R1 и R2 представляет собой С1-2алкил или фтор. Типичные группы R1 и R2 включают, но не ограничиваясь ими, метил, этил, н-пропил, изопропил, дифторметил, трифторметил, 2,2,2-трифторэтил, фтор, хлор, метокси, этокси, дифторметокси и трифторметокси.
В конкретном варианте, каждый R3, когда присутствует, независимо выбран из С1-2алкила и фтора; где каждая алкильная группа необязательно замещена 1-3 фтор-заместителями. Когда присутствуют два заместителя R3, они могут находиться на одном и том же или различных атомах углерода. Типичные группы R3 включают, но не ограничиваясь ими, метил, этил, дифторметил, трифторметил и фтор.
В конкретных вариантах, каждый R4 представляет собой независимо водород или С1-4алкил; или каждый R4 представляет собой независимо водород или С1-2алкил; или каждый R4 представляет собой водород. Типичные группы R4 включают, но не ограничиваясь ими, водород, метил и этил.
Альтернативно, в другом конкретном варианте, обе группы R4 соединены вместе с атомом азота, с которым они связаны, образуя С3-5гетероциклическое кольцо, необязательно содержащее один дополнительный гетероатом, выбранный из азота, кислорода или серы. Типичные гетероциклические кольца включают, но не ограничиваются ими, пирролидин-1-ил, пиперидин-1-ил, пиперазин-1-ил, морфолин-4-ил и тиоморфолин-4-ил.
В конкретных вариантах, R5 представляет собой С1-5алкил; или R5 представляет собой С1-4алкил; или R5 представляет собой С1-3алкил; или R5 представляет собой C1-2алкил; где алкильная группа необязательно замещена -OH или 1-3 фтор-заместителями. Типичные группы R5 в этом варианте включают, но не ограничиваются ими, метил, этил, 2-гидроксиэтил, 2,2,2-трифторэтил, н-пропил, изопропил, 1-гидроксипроп-2-ил, н-бутил и изобутил. В одном варианте, R5 представляет собой метил.
В других конкретных вариантах, R5 представляет собой С3-5циклоалкил; или R5 представляет собой С3-4циклоалкил; где циклоалкильная группа необязательно замещена -OH или 1-3 фтор-заместителями. Типичные группы R5 в этом варианте включают, но не ограничиваются ими, циклопропил, циклобутил и циклопентил.
В другом конкретном варианте, R5 представляет собой -CH2-R8, где R8 такой, как определено здесь. В отдельных аспектах этого варианта, R5 (т.е. -CH2-R8) выбран из:
(a) -CH2-(C3-5циклоалкила); или -CH2-(С3-4циклоалкила); где циклоалкильная группа необязательно замещена -OH или 1-3 фтор-заместителями;
(b) -CH2-(фенила), т.е. бензила, где фенильная группа необязательно замещена 1-3 заместителями, независимо выбранными из С1-4алкила, циано, фтора, хлора, -О(С1-4алкила), -S(C1-4алкила) и -S(О)2(C1-4алкила); где каждая алкильная группа необязательно замещена 1-3 фтор-заместителями.
Типичные группы R5 в этом варианте включают, но не ограничиваясь ими, циклопропилметил, циклобутилметил и циклопентилметил; и бензил, 4-цианобензил, 3-метилбензил, 4-метилбензил, 4-трифторметоксибензил, 3-фторбензил и 4-фторбензил.
В конкретном варианте, каждый R6 представляет собой независимо фенил; где каждая фенильная группа необязательно замещена 1-3 заместителями, независимо выбранными из С1-4алкила, циано, фтора, хлора, -О(C1-4алкила), -S(C1-4алкила) и -S(O)2(C1-4алкила); где каждая алкильная группа необязательно замещена 1-3 фтор-заместителями.
В конкретном варианте, каждый R7 независимо выбран из фенила, -OH и -О(C1-2алкила); где каждая алкильная группа необязательно замещена 1-3 фтор-заместителями; и каждая фенильная группа необязательно замещена 1-3 заместителями, независимо выбранными из C1-4алкила, циано, фтора, хлора, -(C1-4алкила), -S(C1-4алкила) и -S(О)2(C1-4алкила); где каждая алкильная группа необязательно замещена 1-3 фтор-заместителями.
В конкретных вариантах, каждый Rа независимо выбран из водорода и C1-3алкила; или водорода и C1-2алкила; где каждая алкильная группа необязательно замещена 1-3 фтор-заместителями. Типичные группы Rа включают, но не ограничиваются ими, метил, этил, н-пропил, изопропил, дифторметил, трифторметил и 2,2,2-трифторэтил.
В конкретных вариантах, каждый Rb и Rс независимо выбран из водорода и C1-3алкила; или водорода и C1-2алкила; где каждая алкильная группа необязательно замещена 1-3 фтор-заместителями. Типичные группы Rа и Rb включают, но не ограничиваясь ими, метил, этил, н-пропил, изопропил, дифторметил, трифторметил и 2,2,2-трифторэтил.
Альтернативно, в другом конкретном варианте, Rа и Rb соединены вместе с атомом азота, с которым они связаны, образуя С3-5гетероциклическое кольцо, необязательно содержащее один дополнительный гетероатом, выбранный из азота, кислорода или серы. Типичные гетероциклические кольца включают, но не ограничиваясь ими, пирролидин-1-ил, пиперидин-1-ил, пиперазин-1-ил, морфолин-4-ил и тиоморфолин-4-ил.
В конкретных вариантах, a равно 0, 1 или 2; или a равно 0 или 1; или a равно 0.
В конкретных вариантах, b равно 0, 1 или 2; или b равно 0 или 1; или b равно 0.
В конкретных вариантах, c равно 0, 1 или 2; или c равно 0 или 1; или c равно 0.
Когда d равно 1, т.е. когда кольцо, определяемое согласно d, представляет собой пирролидиновое кольцо, тогда в одном варианте, стереоцентр в 3-положении пирролидинового кольца (т.е. углеродный атом, несущий 1-карбамоил-1,1-дифенилметильную группу) имеет (S)-стереохимию. В другом варианте этот стереоцентр имеет (R)-стереохимию.
В одном варианте e равно 8. В другом варианте e равно 9.
Конкретный вариант настоящего изобретения представляет собой соединения формулы I, где обе группы R4 представляют собой водород, a, b и c равны 0; d равно 1; e равно 8 или 9; и R5 представляет собой С1-3алкил; или C1-2алкил; или их фармацевтически приемлемую соль, или их сольват, или их стереоизомер.
Другим конкретным вариантом настоящего изобретения являются соединения формулы I, где обе группы R4 представляют собой водород, a, b и c равны 0; d равно 1; e равно 8 или 9; и R5 представляет собой C3-5циклоалкил; или C3-4циклоалкил; или их фармацевтически приемлемую соль, или их сольват, или их стереоизомер.
Очередным конкретным вариантом настоящего изобретения являются соединения формулы I, где R5 представляет собой метил; и R1, R2, R3, R4, a, b, c, d и e такие, как определены здесь; или их фармацевтически приемлемую соль, или их сольват, или их стереоизомер.
Другие конкретные варианты настоящего изобретения представляют собой соединения формулы IIa:
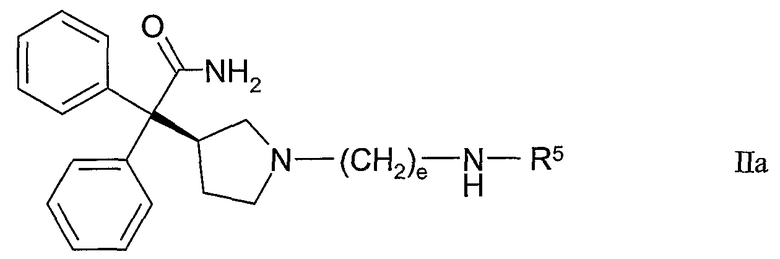
где R5 и e такие, как определены в таблице I, или их фармацевтически приемлемую соль, или их сольват.
Следующими конкретными вариантами воплощения настоящего изобретения являются соединения формулы IIb:
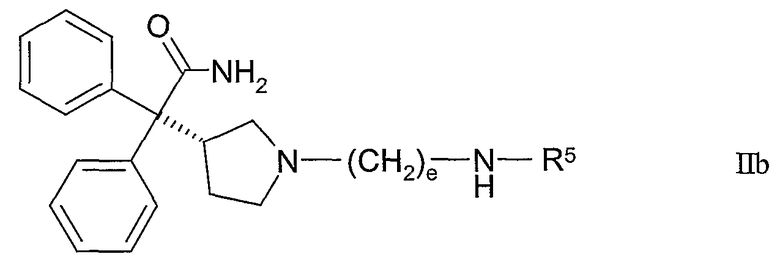
где R5 и e такие, как определены в таблице II, или их фармацевтически приемлемая соль или их сольват.
Следующие конкретные варианты воплощения настоящего изобретения представляют собой соединения формулы XII:
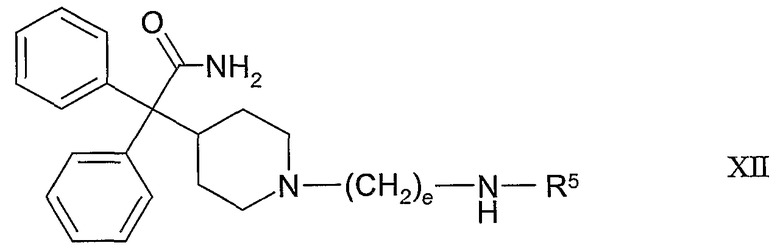
где R5 и e такие, как определены в таблице III, или их фармацевтически приемлемая соль или их сольват.
В соединениях формул X и XI P представляет собой амино-защитную группу. В одном варианте P представляет собой неустойчивую к действию кислоты амино-защитную группу (Pа). В другом варианте P выбран из бензила, 4-метоксибензила, 2,4-диметоксибензила, дифенилметила, трифенилметила, метоксикарбонила, этоксикарбонила, трет-бутоксикарбонила, бензилоксикарбонила, п-метоксибензилоксикарбонила, 9-флуоренилметоксикарбонила, формила, ацетила, триметилсилила и трет-бутилдиметилсилила. В отдельном варианте P представляет собой трет-бутоксикарбонил.
В соединениях формулы XI L представляет собой удаляемую группу. В одном варианте L представляет собой хлор, бром или иод. В другом варианте L представляет собой метансульфонилокси (мезилат) или п-толуолсульфонилокси (тозилат). В отдельном варианте L представляет собой п-толуолсульфонилокси.
В одном варианте Rf представляет собой метил или этил. В другом варианте обе группы Rf соединены вместе, образуя -(CH2)2- или -(CH2)3-.
Конкретными соединениями формулы X, представляющими интерес, являются:
2-[(S)-1-(8-N-бензил-N-метиламинооктил)пирролидин-3-ил]-2,2-дифенилацетамид; и
2-{(S)-1-[8-(N-трет-бутоксикарбонил-N-метиламино)октил]пирролидин-3-ил}-2,2-дифенилацетамид.
Конкретными соединениями формулы XI, представляющими интерес, являются:
8-(N-бензил-N-метиламино)октан-1-ол;
8-(N-трет-бутоксикарбонил-N-метиламино)октан-1-ол; и
8-(N-трет-бутоксикарбонил-N-метиламино)октиловый эфир толуол-4-сульфокислоты.
Определения
При описании соединений, композиций, способов и процессов по данному изобретению нижеуказанные термины имеют нижеследующие значения, если не оговорено противное.
Термин "алкил" означает одновалентную насыщенную углеводородную группу, которая может быть прямой (линейной) или разветвленной. Если не определено иначе, такие алкильные группы обычно содержат от 1 до 10 углеродных атомов. Типичные алкильные группы включают, в качестве примера, метил, этил, н-пропил, изопропил, н-бутил, втор-бутил, изобутил, трет-бутил, н-пентил, н-гексил, н-гептил, н-октил, н-нонил, н-децил и т.п.
Термин "алкилен" означает двухвалентную насыщенную углеводородную группу, которая может быть прямой или разветвленной. Если не оговорено иначе, такие алкиленовые группы обычно содержат от 1 до 10 углеродных атомов. Типичные алкиленовые группы включают, в качестве примера, метилен, этан-1,2-диил ("этилен"), пропан-1,2-диил, пропан-1,3-диил, бутан-1,4-диил, пентан-1,5-диил и т.п.
Термин "алкенил" означает одновалентную ненасыщенную углеводородную группу, которая может быть прямой или разветвленной и которая имеет, по крайней мере, одну, и обычно 1, 2 или 3, углерод-углеродных двойных связей. Если не определено иначе, такие алкенильные группы обычно содержат от 2 до 10 углеродных атомов. Типичные алкенильные группы включают, в качестве примера, этенил, н-пропенил, изопропенил, н-бут-2-енил, н-гекс-3-енил и т.п.
Термин "алкинил" означает одновалентную ненасыщенную углеводородную группу, которая может быть прямой или разветвленной и которая имеет, по крайней мере, одну, и обычно 1, 2 или 3, углерод-углеродных тройных связей. Если не оговорено иначе, такие алкинильные группы обычно содержат от 2 до 10 углеродных атомов. Типичные алкинильные группы включают, в качестве примера, этинил, н-пропинил, н-бут-2-инил, н-гекс-3-инил и т.п.
Термин "арил" означает одновалентный ароматический углеводород, имеющий одно кольцо (т.е. фенил) или конденсированные кольца (т.е. нафталин). Если не оговорено иначе, такие арильные группы обычно содержат от 6 до 10 углеродных кольцевых атомов. Типичные арильные группы включают, в качестве примера, фенил и нафталин-1-ил, нафталин-2-ил и т.п.
Термин "циклоалкил" означает одновалентную насыщенную карбоциклическую углеводородную группу. Если не оговорено иначе, такие циклоалкильные группы обычно содержат от 3 до 10 углеродных атомов. Типичные циклоалкильные группы включают, в качестве примера, циклопропил, циклобутил, циклопентил, циклогексил и т.п.
Термин "галоген" означает фтор, хлор, бром и иод.
Термин "гетероарил" означает одновалентную ароматическую группу, имеющую одно кольцо или два конденсированных кольца и содержащую в кольце, по крайней мере, один гетероатом (обычно от 1 до 3 гетероатомов), выбранный из азота, кислорода или серы. Если не оговорено иначе, такие гетероарильные группы обычно содержат, суммарно, от 5 до 10 атомов в кольце. Типичные гетероарильные группы включают, в качестве примера, одновалентные группы пиррола, имидазола, тиазола, оксазола, фурана, тиофена, триазола, пиразола, изоксазола, изотиазола, пиридина, пиразина, пиридазина, пиримидина, триазина, индола, бензофурана, бензотиофена, бензимидазола, бензтиазола, хинолина, изохинолина, хиназолина, хиноксалина и т.п., где точка присоединения находится на любом доступном атоме углерода или азота кольца.
Термин "гетероциклил" или "гетероциклическая группа" означает одновалентную насыщенную или ненасыщенную (неароматическую) группу, имеющую одно кольцо или несколько конденсированных колец и содержащую в кольце, по крайней мере, один гетероатом (обычно от 1 до 3 гетероатомов), выбранный из азота, кислорода или серы. Если не оговорено иначе, такие гетероциклические группы обычно содержат, суммарно, от 2 до 9 атомов в кольце. Типичные гетероциклические группы включают, в качестве примера, одновалентные группы пирролидина, имидазолидина, пиразолидина, пиперидина, 1,4-диоксана, морфолина, тиоморфолина, пиперазина, 3-пирролина и т.п., где точка присоединения находится на любом доступном атоме углерода или азота кольца.
Термин "фармацевтически приемлемая соль" означает соль, которая приемлема для введения пациенту, как, например, млекопитающему (например, соли, являющиеся допустимо безопасными для данной схемы приема лекарственного средства). Такие соли можно получить из фармацевтически приемлемых неорганических или органических оснований и из фармацевтически приемлемых неорганических или органических кислот. Соли, полученные из фармацевтически приемлемых неорганических оснований, включают соли аммония, кальция, меди, железные (содержащие трехвалентное железо), железистые (содержащие двухвалентное железо), лития, магния, марганцовые (содержащие трехвалентный марганец), марганцовистые (содержащие двухвалентный марганец), калия, натрия, цинка и т.п. Конкретными солями, представляющими интерес, являются соли аммония, кальция, магния, калия и натрия. Соли, полученные из фармацевтически приемлемых органических оснований, включают соли первичного, вторичного и третичного аминов, включая замещенные амины, циклические амины, амины природного происхождения и т.п., такие как соли аргинина, бетаина, кофеина, холина, N,N'-дибензилэтилендиамина, диэтиламина, 2-диэтиламиноэтанола, 2-диметиламиноэтанола, этаноламина, этилендиамина, N-этилморфолина, N-этилпиперидина, глюкамина, глюкозамина, гистидина, гидрабамина, изопропиламина, лизина, метилглюкамина, морфолина, пиперазина, пиперадина, полиаминовых смол, прокаина, пуринов, теобромина, триэтиламина, триметиламина, трипропиламина, трометамина и т.п. Соли, полученные из фармацевтически приемлемых кислот, включают соли уксусной, аскорбиновой, бензолсульфоновой, бензойной, камфорсульфоновой, лимонной, этансульфоновой, edisylic, фумаровой, глюконовой, глюкуроновой, глутаминовой, гиппуровой, бромистоводородной, хлористоводородной, изэтионовой, молочной, лактобионовой, малеиновой, яблочной, миндальной, метансульфоновой, слизевой, нафталинсульфоновой, нафталин-1,5-дисульфоновой, нафталин-2,6-дисульфоновой, никотиновой, азотной, памовой, пантотеновой, фосфорной, янтарной, серной, винной, п-толуолсульфоновой, ксинафовой и т.п. Конкретными солями, представляющими интерес, являются соли лимонной, бромистоводородной, хлористоводородной, изэтиновой, малеиновой, фосфорной, серной и винной кислот.
Термин "его соль" означает соединение, полученное в результате замены водорода кислоты на катион, такой как катион металла или органический катион и т.п. Предпочтительно, соль представляет собой фармацевтически приемлемую соль, хотя это не требуется для солей промежуточных соединений, которые не предназначаются для введения пациенту.
Термин "сольват" означает комплекс или агрегат, образованный одной или несколькими молекулами растворенного вещества, т.е. соединения формулы I или его фармацевтически приемлемой соли, и одной или несколькими молекулами растворителя. Такие сольваты обычно представляют собой кристаллические твердые вещества, имеющие, по существу, постоянное молярное соотношение растворенного вещества и растворителя. Этот термин также включает клатраты, включая клатраты с водой. Типичные растворители включают, в качестве примера, воду, метанол, этанол, изопропанол, уксусную кислоту и т.д. В тех случаях, когда растворителем является вода, образуемый сольват представляет собой гидрат.
Термин "бронхозащита" или "бронхозащитный" означает предотвращение, улучшение, подавление или облегчение симптомов респираторного заболевания или нарушения. Для определения продолжительности бронхозащиты используют модель ацетилхолин-индуцируемого бронхостеноза у морской свинки, если не оговорено иначе.
Термин "терапевтически эффективное количество" означает количество, достаточное для осуществления лечения при введении пациенту, нуждающемуся в таком лечении.
Используемый здесь термин "лечение" означает лечение заболевания или клинического состояния (как, например, COPD или астма) у пациента, такого как млекопитающее (в частности, человека или домашнего животного), которое включает:
(a) предотвращение возникновения заболевания или клинического состояния, т.е. профилактическое лечение пациента;
(b) улучшение (уменьшение интенсивности симптомов) заболевания или клинического состояния, т.е. ликвидация или достижение состояния регрессии заболевания или клинического состояния у пациента;
(c) подавление заболевания или клинического состояния, т.е. замедление или купирование развития заболевания или клинического состояния у пациента; или
(d) облегчение симптомов заболевания или клинического состояния у пациента.
Термин "удаляемая группа" означает функциональную группу или атом, который может быть замещен другой функциональной группой или атомом в реакции замещения, такой как реакция нуклеофильного замещения. В качестве примера, типичные удаляемые группы включают группы хлора, брома и иода; сложноэфирные группы сульфоновых кислот, такие как мезилат, тозилат, брозилат, нозилат и т.п.; и ацилоксигруппы, такие как ацетокси, трифторацетокси и т.п.
Термин "его защищенные производные" означает производное определенного соединения, в котором одна или несколько функциональных групп соединения защищены от протекания нежелательных реакций защитной или блокирующей группой. Функциональные группы, которые могут быть защищены, включают, в качестве примера, группы карбоновых кислот, аминогруппы, гидроксильные группы, тиольные группы, карбонильные группы и т.п. Типичные защитные группы для карбоновых кислот включают сложные эфиры (такой как п-метоксибензиловый сложный эфир), амиды и гидразиды; для аминогрупп, карбаматы (такие как трет-бутоксикарбонил) и амиды; для гидроксильных групп, простые эфиры и сложные эфиры; для тиольных групп, простые и сложные тиоэфиры; для карбонильных групп, ацетали и кетали; и т.п. Такие защитные группы являются общеизвестными для специалистов в данной области техники и они описаны, например, в T.W. Greene and G.M. Wuts, Protecting Groups in Organic Synthesis, Third Edition, Wiley, New York, 1999, и ссылках, цитируемых там.
Термин "амино-защитная группа" означает защитную группу, подходящую для предотвращения протекания нежелательных реакций с участием аминогруппы. Типичные амино-защитные группы включают, но не ограничиваются ими, бензил, трет-бутоксикарбонил (ВОС), тритил (Tr), бензилоксикарбонил (Cbz), п-метоксибензилоксикарбонил (Moz), 9-флуоренилметоксикарбонил (Fmoc), формил, ацетил, триметилсилил (TMS), трет-бутилдиметилсилил (TBS) и т.п. Термин "неустойчивая к кислоте амино-защитная группа" означает амино-защитную группу, которую удаляют обработкой кислотой, включая, например, минеральную кислоту или органическую кислоту, такую как карбоновая кислота или сульфоновая кислота. Типичные неустойчивые к кислоте амино-защитные группы включают, но не ограничиваются ими, карбаматы, такие как трет-бутоксикарбонил (ВОС), п-метоксибензилоксикарбонил (Moz) и т.п.
Термин "карбокси-защитная группа" означает защитную группу, подходящую для предотвращения протекания нежелательных реакций с участием карбоксигруппы. Типичные карбокси-защитные группы включают, но, не ограничиваясь ими, сложные эфиры, такие как метиловый, этиловый, трет-бутиловый, бензиловый (Bn), п-метоксибензиловый (PMB), 9-флуоренилметиловый (Fm), триметилсилиловый (TMS), трет-бутилдиметилсилиловый (TBS), дифенилметиловый (бензилгидрил, DPM) и т.п.
Термин "необязательно замещенный" означает, что конкретная группа или фрагмент, такой как алкильная группа, фенильная группа и т.п., может быть незамещенной или замещенной указанными заместителями.
Общие способы синтеза
Соединения замещенного пирролидина и родственные соединения по данному изобретению можно получить, исходя из легко доступных исходных продуктов, используя нижеследующие общие способы и методики. Несмотря на то, что конкретный вариант осуществления настоящего изобретения может быть проиллюстрирован или описан схемами, представленными ниже, специалистам в данной области техники должно быть очевидно, что все варианты или аспекты данного изобретения можно осуществить, используя способы, описанные здесь, или используя другие способы, реагенты и исходные продукты, известные специалистам в данной области техники. Следует также иметь в виду, что в тех случаях, когда приводятся типичные или предпочтительные условия способа (т.е. температуры реакций, времена протекания реакций, мольные соотношения реагирующих веществ, растворители, давления и т.д.), могут быть использованы и другие условия способа, если не оговорено иначе. Оптимальные условия проведения реакции могут варьироваться в зависимости от конкретных используемых реагирующих веществ или растворителей, однако, такие условия могут быть легко определены специалистом в данной области, используя обычные методики оптимизации процесса.
Кроме того, как очевидно для специалистов в данной области техники, для предотвращения протекания нежелательных реакций с участием некоторых функциональных групп может быть необходимо или желательно использование обычных защитных групп. Выбор подходящей защитной группы для конкретной функциональной группы, а также подходящих условий для осуществления защиты или снятия защиты с указанных функциональных групп общеизвестны в данной области техники. При желании, могут быть использованы защитные группы, отличающиеся от тех, которые проиллюстрированы в способах, описанных здесь. Например, многочисленные защитные группы и их введение и удаление описаны в T.W. Greene and G.M. Wuts, Protecting Group in Organic Synthesis, Third Edition, Wiley, New York, 1999 и цитируемых там ссылках.
Соединения формулы I и их соли могут быть получены способом, включающим:
(а) взаимодействие соединения формулы III
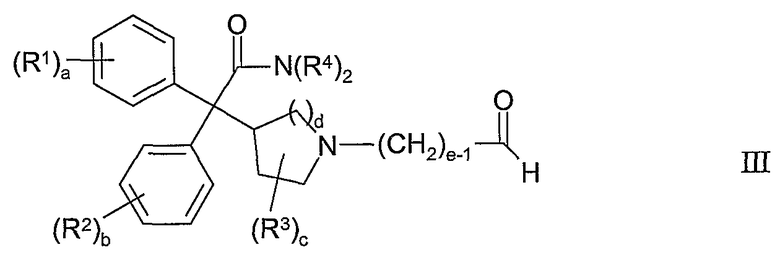
с соединением формулы IV:

где P1 представляет собой амино-защитную группу, в присутствии восстановителя;
(b) взаимодействие соединения формулы V:
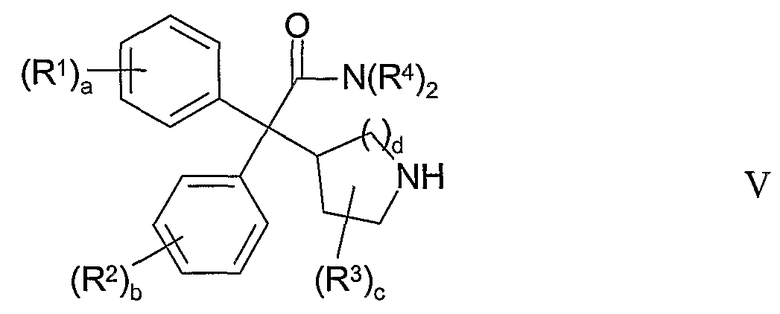
с соединением формулы VI:
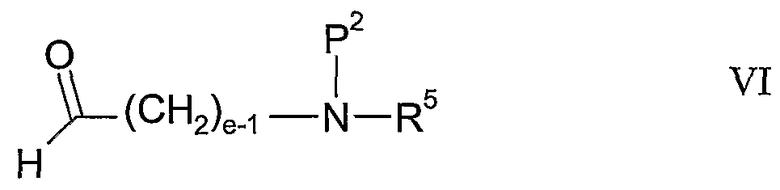
где P2 представляет собой амино-защитную группу, в присутствии восстановителя;
(с) взаимодействие соединения формулы VII:
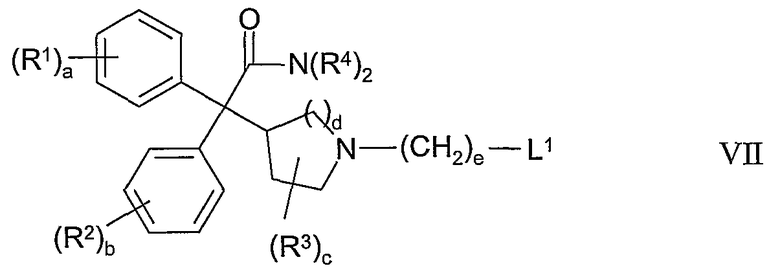
где L1 представляет собой удаляемую группу, с соединением формулы IV; или
(d) взаимодействие соединения формулы V с соединением формулы VIII;
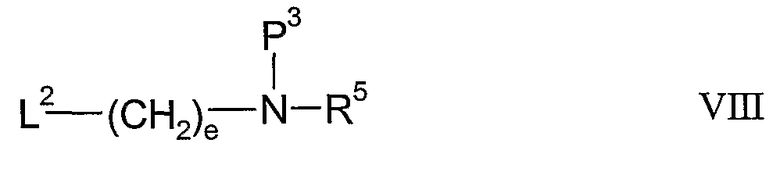
где L2 представляет собой удаляемую группу и Р3 представляет собой амино-защитную группу; и затем
(е) удаление защитной группы Р1, Р2 или Р3 с получением соединения формулы I или его соли; где R1-5 и а-е такие, как определены здесь.
Необязательно, фармацевтически приемлемую соль соединения формулы I можно получить непосредственно на стадии (е) или на отдельной дополнительной стадии из продукта стадии (е).
В способе (а) Р1 может представлять собой любую подходящую амино-защитную группу, такую как бензильная и т.п. Восстановитель может представлять собой любой подходящий восстановитель, включая металлгидридные восстановители, такие как натрийтриацетоксиборгидрид, натрийцианоборгидрид и т.п. После завершения реакции амино-защитная группа Р1 может быть удалена, используя обычные способы и реагенты. Например, бензильную защитную группу можно удалить гидрогенолизом в присутствии катализатора, такого как палладий.
В способе (b) Р2 может представлять собой любую подходящую амино-защитную группу, такую как бензил, трет-бутоксикарбонил, бензилоксикарбонил, 9-флуоренилметоксикарбонил, трет-бутилдиметилсилил и т.п. Восстановителем может быть любой подходящий восстановитель, включая металлгидридные восстановители, такие как натрийтриацетоксиборгидрид, натрийцианоборгидрид и т.п. После завершения реакции амино-защитная группа Р2 может быть удалена, используя обычные способы и реагенты. Например, бензильную защитную группу можно удалить гидрогенолизом в присутствии катализатора, такого как палладий; трет-бутоксикарбонильная группа может быть удалена обработкой кислотой, такой как хлористоводородная кислота, п-толуолсульфокислота и т.п.; трет-бутилдиметилсилильная группа может быть удалена обработкой источником фторидных ионов, таким как триэтиламина тригидрофторид.
В способе (с) L1 может представлять собой любую подходящую удаляемую группу, включая, но не ограниваясь ими, галоген, такой как хлор, бром или иод, или сложноэфирную группу сульфокислот, такую как мезилат, тозилат и т.п.; и Р1, такая как определено здесь.
В способе (d) L2 может представлять собой любую подходящую удаляемую группу, включая, но не ограниваясь ими, галоген, такой как хлор, бром или иод, или сложноэфирную группу сульфокислот, такую как мезилат, тозилат и т.п.; и Р3 может быть любой подходящей амино-защитной группой, такой как бензил, трет-бутоксикарбонил, бензилоксикарбонил, 9-флуоренилметоксикарбонил, трет-бутилдиметилсилил и т.п. Восстановителем может быть любой подходящий восстановитель, включая металлгидридные восстановители, такие как натрийтриацетоксиборгидрид, натрийцианоборгидрид и т.п. После завершения реакции амино-защитная группа Р2 может быть удалена, используя обычные способы и реагенты. Например, бензильную защитную группу можно удалить гидрогенолизом в присутствии катализатора, такого как палладий; трет-бутоксикарбонильная группа может быть удалена обработкой кислотой, такой как хлористоводородная кислота, п-толуолсульфокислота и т.п.; трет-бутилдиметилсилильная группа может быть удалена обработкой источником фторидных ионов, таким как триэтиламина тригидрофторид.
В конкретных вариантах способов (b) и (d) Р2 и Р3 представляют собой трет-бутоксикарбонильную группу, которую удаляют обработкой фармацевтически приемлемой кислотой, получая in situ фармацевтически приемлемую соль соединения формулы I.
В качестве дополнительной иллюстрации, получение типичных соединений формулы I представлено на схеме А (где заместители и переменные, показанные на нижеследующих схемах, имеют предусмотренные здесь определения, если не оговорено особо).
Схема А
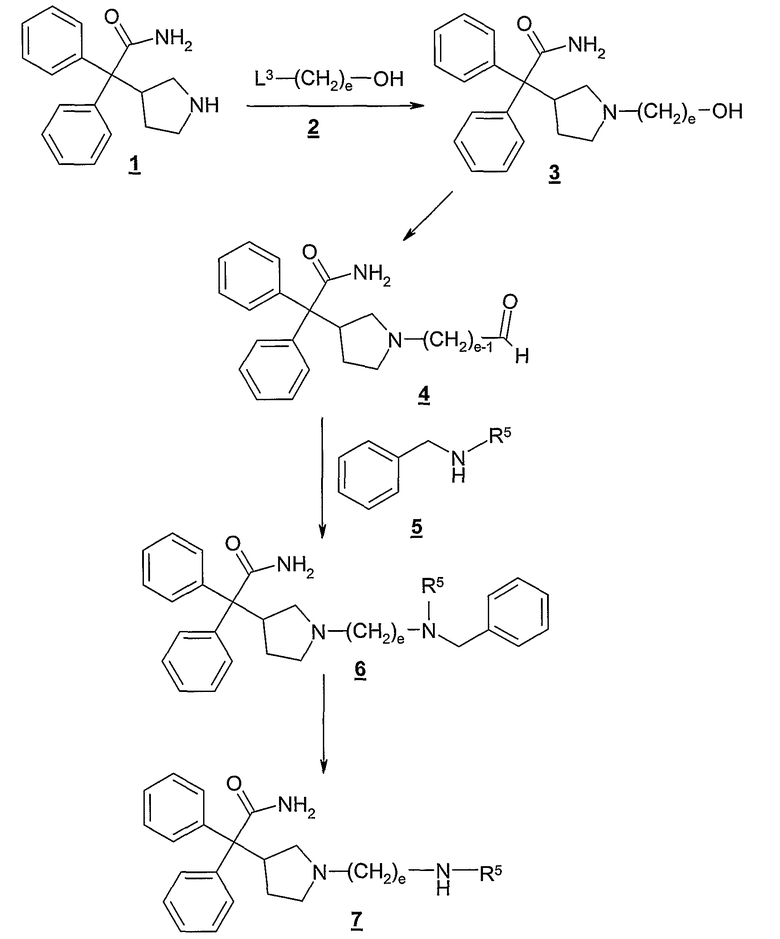
Как показано на Схеме А, соединение формулы 1 сначала подвергают взаимодействию со спиртом 2, где L3 представляет собой подходящую удаляемую группу, такую как хлор, бром, иод, тозил, мезил и т.п., с получением промежуточного соединения 3. Как правило, эту реакцию проводят путем контактирования 1 с, по крайней мере, одним эквивалентом, предпочтительно с от около 1,0 до около 1,1 эквивалентами, спирта 2 в инертном растворителе, таком как ацетонитрил и т.п. Указанную реакцию обычно проводят в присутствии избытка основания; предпочтительно, в присутствии от около 2 до около 4 эквивалентов основания, такого как триалкиламин, предпочтительно триэтиламин. Как правило, указанную реакцию проводят при температуре в диапазоне от около 0°С до около 80°С, предпочтительно от около 40°С до 50°С, в течение от около 1 до 24 часов, или до практически полного завершения реакции. При желании, полученное промежуточное соединение 3 очищают обычными способами, таким как хроматография, перекристаллизация и т.п.
Спирты формулы 2, используемые в этой реакции, являются либо коммерчески доступными, либо их можно получить из коммерчески доступных исходных продуктов и реагентов, используя общеизвестные способы. Типичные спирты формулы 2 включают, в качестве примера, 8-хлор-1-октанол, 9-хлор-1-нонанол, 8-бром-1-октанол, 9-бром-1-нонанол, 8-иод-1-октанол, 9-иод-1-нонанол и т.п.
Гидроксильную группу промежуточного соединения 3 затем окисляют в соответствующий альдегид, получая промежуточное соединение 4. Эту реакцию обычно проводят, контактируя 3 с избыточным количеством подходящего окисляющего агента. Для этой реакции может быть использован любой окислитель, способный окислять гидроксильную группу в альдегид, включая реагенты хрома (VI), такие как дипиридин оксид хрома (VI), хлорхромат пиридиния, дихромат пиридиния и т.п.; активированные диметилсульфоксидные реагенты, такие как оксалилхлорид/ДМСО, комплекс триоксид серы-пиридин/ДМСО/триалкиламин и т.п.
Предпочтительно, указанную реакцию проводят, используя избыток комплекса триоксид серы-пиридин и диметилсульфоксида в присутствии триалкиламина, такого как триэтиламин, диизопропилэтиламин и т.п. Как правило, эту реакцию проводят путем контактирования 3 с от около 2,5 до около 3,5 эквивалентами комплекса триоксид серы-пиридин и избытком, предпочтительно около 10 эквивалентов, диметилсульфоксида в присутствии избытка, предпочтительно около 5 эквивалентов, диизопропилэтиламина в инертном растворителе, таком как дихлорметан. Эту реакцию обычно проводят при температуре в диапазоне от около -30°С до около 0°С, предпочтительно от около -10°С до около -20°С, в течение от около 0,25 до около 2 часов, или до практически полного завершения реакции. Необязательно, полученное альдегидное промежуточное соединение 4 затем очищают, используя обычные способы, такие как хроматография, перекристаллизация и т.п.
Альтернативно, альдегидное промежуточное соединение 4 можно получить сначала взаимодействием 1 с соединением формулы:
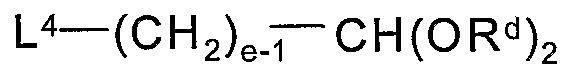
или
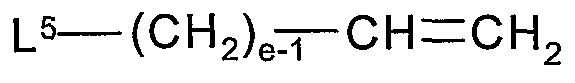
или
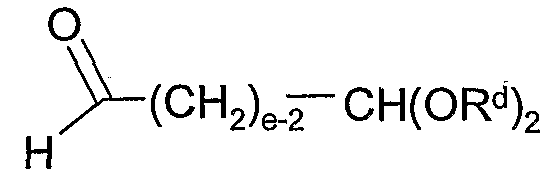
где L4 и L5 представляют собой подходящие удаляемые группы, такие как хлор, бром, иод, тозил, мезил и т.п., е является таким, как определено здесь, и каждый Rd представляет собой, независимо, С1-6алкил или обе группы Rd соединены вместе, образуя С2-6алкилен. Впоследствии гидролиз ацеталя (т.е. использование водной кислоты) или озонолиз олефина (т.е. использование О3, с последующим разложением озонида восстановителем, таким как триметилфосфит, диметилсульфид и т.п.) дает альдегид 4.
Затем альдегидное промежуточное соединение 4 связывают с амином 5, получая соединение формулы 6. Как правило, эту реакцию проводят, контактируя альдегид 4 с избытком, таким как от около 1,0 до около 1,2 эквивалента, соединения 5 в присутствии избытка, предпочтительно от около 1,2 до около 1,5 эквивалента, подходящего восстановителя в инертном разбавителе, таком как дихлорметан. Подходящие восстановители включают, в качестве иллюстрации, натрийтриацетоксиборгидрид, натрийцианоборгидрид и т.п. Предпочтительно, восстановитель представляет собой натрийтриацетоксиборгидрид. Обычно эту реакцию проводят при температуре в диапазоне от около 0°С до около 30°С в течение от около 6 до около 24 часов, или до практически полного завершения реакции. Полученное соединение формулы 6, как правило, очищают, используя обычные способы, такие как хроматография, перекристаллизация и т.п.
Удаление бензильной группы из 6, с использованием обычных реагентов и условий реакции, далее дает 7. Например, гидрогенолиз 6, с использованием катализатора, такого как палладий на углероде и/или гидроксид палладия, легко удаляет бензильную группу с получением 7. Обычно эту реакцию проводят, контактируя 6 с водородом при давлении в диапазоне от около 40 до около 60 фунт/дюйм2 в присутствии катализатора, такого как 10% палладий на углероде. Эту реакцию обычно проводят в инертном растворителе, таком как этанол или изопропанол, при температуре окружающей среды в течение от около 12 до около 120 часов, или до практически полного завершения реакции.
Альтернативно, альдегидное промежуточное соединение 5 может быть подвергнуто взаимодействию с амином формулы R5-NH2, где R5 такой, как определен здесь, с получением непосредственно соединения 7. Альтернативно, при желании, вместо бензильной группы на схеме А могут быть использованы другие амино-защитные группы.
Аминовые соединения, подходящие для использования в описанных здесь реакциях, являются либо коммерчески доступными, либо их можно получить из коммерчески доступных исходных продуктов и реагентов, используя общеизвестные способы. Типичные амины, подходящие для использования, включают, но ими не ограничиваются, N-метил-N-бензиламин, N-этил-N-бензиламин, метиламин, этиламин, н-пропиламин, изопропиламин, 2-гидроксиэтиламин, DL-2-амино-1-пропанол, (R)-(-)-2-амино-1-пропанол, (S)-(+)-2-амино-1-пропанол, 2,2,2-трифторэтиламин, бензиламин, циклопропиламин, циклобутиламин, циклопентиламин и т.п.
Соединения формулы 1, используемые в описанных здесь реакциях, легко получают способами, иллюстрируемыми на схеме В.
Схема В
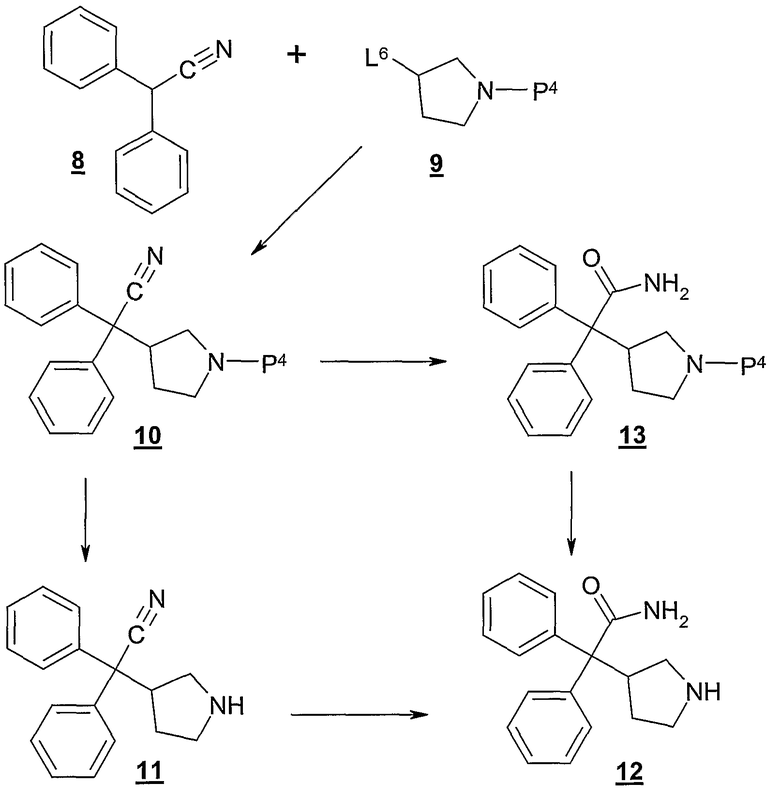
Как проиллюстрировано на Схеме В, дифенилацетонитрил 8 подвергают взаимодействию с промежуточным соединением 9, где L6 представляет собой подходящую удаляемую группу, такую как хлор, бром, иод, тозил, мезил и т.п., и P4 представляет собой амино-защитную группу, такую как бензил, 4-метоксибензил, 4-нитробензил, этоксикарбонил, фенилкарбонил и т.п., с получением промежуточного соединения 10. Обычно эту реакцию проводят сначала получая анион соединения 8 контактированием 8 с избытком, предпочтительно от около 1,4 до около 1,6 эквивалентов, сильного основания, такого как трет-бутоксид калия, в инертном разбавителе, таком как тетрагидрофуран, при температуре в диапазоне от около -10°С до около 10°С в течение от около 0,5 до около 2,0 часов. Полученный анион затем подвергают взаимодействию in situ с от около 0,95 до около 1,05 эквивалентов 9 при температуре в диапазоне от около 20°С до около 50°С в течение от около 10 до около 48 часов, или до практически полного завершения реакции. Соединения формулы 9, где L6 представляет собой сульфонатную сложноэфирную удаляемую группу, легко получают из соответствующего спирта, используя обычные способы и реагенты. Например, (S)-1-бензил-3-пирролидинол легко превращают в (S)-1-бензил-3-(п-толуолсульфонилокси)пирролидин обработкой около 1,1 эквивалентами п-толуолсульфонилхлорида и около 1,2 эквивалентами 1,4-диазабицикло[2.2.2]октана (DABCO). Другие соединения формулы 9 можно получить аналогичными способами, используя коммерчески доступные исходные продукты и реагенты.
Затем из соединения 10 снимают защиту, используя обычные способы и реагенты, получая соединение 11. Например, если Р4 в соединении 10 представляет собой бензильную защитную группу, бензильную группу легко удаляют гидрогенолизом, используя источник водорода, такой как формиат аммония, и катализатор, такой как палладий на углероде. Предпочтительно, эту реакцию проводят, используя гидрохлоридную или гидробромидную соль соединения 10 или в присутствии кислоты, такой как хлористоводородная кислота, бромистоводородная кислота, муравьиная кислота, серная кислота, фосфорная кислота, п-толуолсульфокислота, уксусная кислота, щавелевая кислота и т.п. Указанную реакцию гидрогенолиза можно также проводить, используя водород и катализатор в присутствии кислоты. См., например, патент США 6005119, выданный 21 декабря 1999 N. Mori et al.
Затем нитрильную группу соединения 11 гидролизуют в соответствующий амид (т.е. -С(О)NH2), получая соединение формулы 10. Эту реакцию, как правило, проводят путем контактирования 11 с водной серной кислотой, предпочтительно 80% серной кислотой, при температуре в диапазоне от около 70°С до около 100°С, предпочтительно около 90°С, в течение от около 12 до около 36 часов, или до практически полного завершения реакции. Как показано на схеме В, гидролиз нитрильной группы в амид может быть также осуществлен до удаления защитной группы, с получением 13, из которого затем может быть снята защита с получением соединения 12.
При желании, нитрильная группа соединения 10 или 11 может быть гидролизована до соответствующей карбоновой кислоты (т.е. -ООН), используя, например, водный гидроксид натрия, содержащий от около 6 до около 12% пероксида водорода. Затем полученную карбоновую кислоту можно сочетать с различными аминами (т.е. ReReNH, где Re такой, как определен здесь) с получением замещенных амидов, используя общеизвестные способы и реагенты.
Соединения по данному изобретению можно также получить способом, проиллюстрированном на схеме С.
Схема С
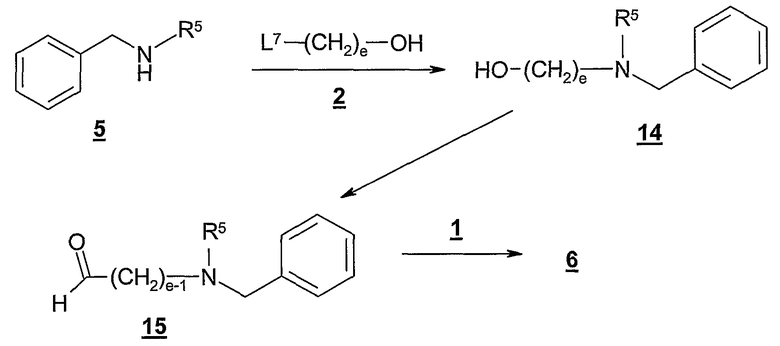
Как показано на Схеме С, спирт 2, где L7 представляет собой подходящую удаляемую группу, такую как хлор, бром, иод, тозил, мезил и т.п., может быть подвергнут взаимодействию с бензиламином 5 с получением промежуточного соединения 14. Типично эту реакцию проводят, контактируя спирт 2 с, по крайней мере, одним эквивалентом, предпочтительно с от около 1,0 до около 1,1 эквивалентами, бензиламина 5 в инертном растворителе, таком как ацетонитрил и т.п. Эту реакцию обычно проводят в присутствии избытка основания; предпочтительно, в присутствии от около 2 до около 4 эквивалентов основания, такого как триалкиламин, предпочтительно триэтиламин. Типично эту реакцию проводят при температуре в диапазоне от около 0°С до около 80°С, предпочтительно от около 40°С до около 60°С, в течение от около 1 до около 24 часов, или до практически полного завершения реакции. При желании полученное промежуточное соединение 14 легко очищают обычными способами, такими как хроматография, перекристаллизация и т.п.
Затем гидроксильную группу промежуточного соединения 14 окисляют в соответствующий альдегид, получая промежуточное соединение 15. Эту реакцию обычно проводят, подвергая контакту 14 с избыточным количеством подходящего окислителя. В этой реакции может быть использован любой окислитель, способный окислить гидроксильную группу в альдегид, включая реагенты хрома (VI), такие как дипиридин-оксид хрома (VI), хлорхромат пиридиния, дихромат пиридиния и т.п.; и активированные диметилсульфоксидные реагенты, такие как оксалилхлорид/ДМСО, комплекс триоксид серы-пиридин/ДМСО/триалкиламин и т.п.
Предпочтительно, эту реакцию проводят, используя избыток комплекса триоксид серы-пиридин и диметилсульфоксида в присутствии триалкиламина, такого как триэтиламин, диизопропилэтиламин и т.п. Типично эту реакцию проводят, контактируя 14 с от около 2,5 до около 3,5 эквивалентами комплекса триоксид серы-пиридин и избытком, предпочтительно около 10 эквивалентов, диметилсульфоксида в присутствии избытка, предпочтительно около 5 эквивалентов, диизопропилэтиламина в инертном растворителе, таком как дихлорметан. Эту реакцию обычно проводят при температуре в диапазоне от около -30°С до около 0°С, предпочтительно при от около -10°С до около -20°С, в течение от около 0,25 до около 6 часов, или до практически полного завершения реакции. Необязательно затем полученное альдегидное промежуточное соединение 15 очищают, используя обычные способы, такие как хроматография, перекристаллизация и т.п.
Затем альдегидное промежуточное соединение 15 сочетают с 1, получая соединение формулы 6. Типично эту реакцию проводят, контактируя альдегид 15 с, по крайней мере, приблизительно одним эквивалентом 1 в присутствии избытка, предпочтительно от около 1,2 до около 1,5 эквивалентов, подходящего восстановителя в инертном растворителе, таком как дихлорметан. Подходящие восстановители включают, в качестве иллюстрации, натрийтриацетоксиборгидрид, натрийцианоборгидрид и т.п. Предпочтительно, восстановитель представляет собой натрийтриацетоксиборгидрид. Обычно эту реакцию проводят при температуре в диапазоне от около 0°С до около 30°С в течение от около 2 до около 24 часов, или до практически полного завершения реакции. Полученное соединение формулы 6, как правило, очищают, используя обычные способы, такие как хроматография, перекристаллизация и т.п. Затем из соединения 6 можно удалить бензильную группу, получая 7, как обсуждено выше.
Кроме того, для специалистов в данной области техники должно быть очевидно, что для получения соединения формулы 7 синтетические стадии, проиллюстрированные на схемах А, В и С, могут быть выполнены в порядке, отличном от представленного на указанных схемах, или используя реагенты, отличающиеся от описанных. Например, вместо окисления гидроксильной группы промежуточного соединения 3 или 14 в альдегид эти гидроксильные группы могут быть превращены в удаляемую группу, такую как хлор, бром, иод, мезилат или тозилат, используя обычные реагенты и реакционные способы. Затем полученную удаляемую группу легко замещают амином 5 или промежуточным соединением 1, получая соединение 6.
В качестве другого примера, типичные соединения формулы I можно получить, как проиллюстрировано на схеме D.
Схема D
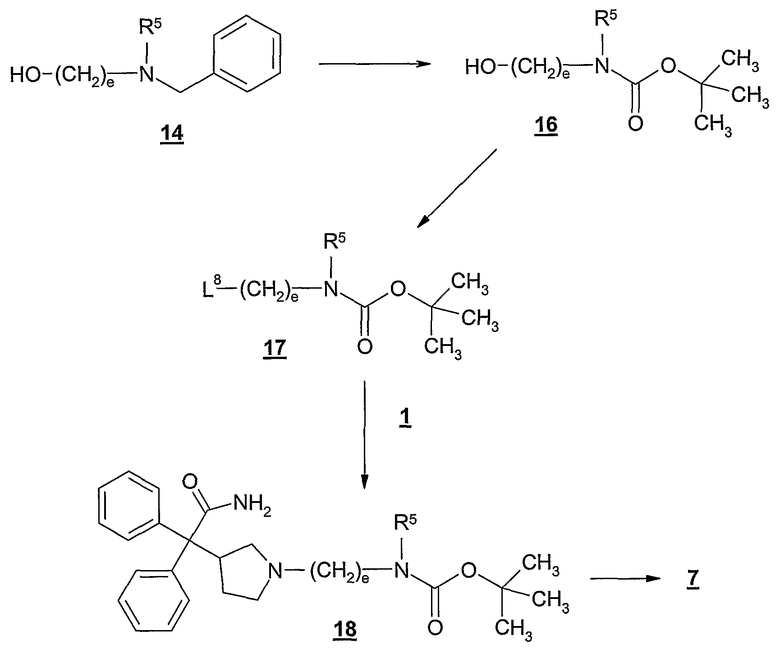
Как показано на Схеме D, чтобы получить соединение 16, бензильную амино-защитную группу соединения 14 можно удалить и заменить трет-бутоксикарбонильной амино-защитной группой, используя обычные способы и реагенты (т.е. гидрогенолиз, чтобы удалить бензильную группу, и ди-трет-бутилдикарбонат, чтобы получить трет-бутоксикарбонильную группу).
Затем гидроксильную группу соединения 16 превращают в удаляемую группу, такую как хлор, бром, иод, мезилат или тозилат, используя обычные реагенты и реакционные способы, получая соединение формулы 17. Например, гидроксильную группу превращают в тозилатную удаляемую группу реакцией с тозилхлоридом (п-толуолсульфонилхлорид) в присутствии подходящего основания, включая третичные амины, такие как 1,4-диазабицикло[2.2.2]октан. Эту реакцию, как правило, проводят в инертном растворителе, таком как метил-трет-бутиловый эфир, при температуре в диапазоне от около 0°С до около 30°С в течение от 0,5 до 6 часов, или до практически полного завершения реакции.
Затем удаляемую группу соединения 17 заменяют соединением формулы 1, получая соединение формулы 18. Эту реакцию, как правило, проводят, контактируя 17 с от около 0,95 до около 1,1 молярными эквивалентами 1 в присутствии третичного амина, такого как диизопропилэтиламин. Реакцию обычно проводят в инертном растворителе, таком как ацетонитрил, при температуре в диапазоне от около 25°С до около 100°С в течение от около 2 до около 12 часов, или до практически полного завершения реакции.
Затем трет-бутоксикарбонильную амино-защитную группу соединения 18 удаляют, используя обычные реагенты и условия реакции, получая соединение формулы 7 или его соль. Например, трет-бутоксикарбонильную амино-защитную группу можно легко удалить обработкой кислотой, такой как хлористоводородная кислота, трифторуксусная кислота, п-толуолсульфокислота и т.п.
В одном варианте, соединение формулы 18 подвергают контактированию с фармацевтически приемлемой кислотой, получая сразу фармацевтически приемлемую соль соединения 7, без выделения свободного основания. Например, соединение 18 может быть введено в контакт с нафталин-1,5-дисульфокислотой с получением соли нафталин-1,5-дисульфокислоты соединения 7. Эту реакцию, как правило, проводят, контактируя 18 с от около 1 до около 3 эквивалентами, как, например, 2 эквивалентами, нафталин-1,5-дисульфокислоты в инертном растворителе, таком как изопропанол. В одном варианте, для получения кристаллической соли нафталин-1,5-дисульфокислоты в качестве растворителя используют изопропанол, содержащий от около 2 до около 10%, по объему, воды.
Дополнительные детали в отношении конкретных условий реакций и других способов получения типичных соединений по данному изобретению или промежуточных соединений, используемых для их синтеза, описаны в примерах, приведенных ниже.
Фармацевтические композиции
Соединения замещенного пирролидина и родственные соединения по данному изобретению обычно вводят пациенту в форме фармацевтической композиции. Такие фармацевтические композиции могут вводиться пациенту любым приемлемым путем введения, включая, но не ограничиваясь им, пероральный, ингаляционный, назальный, местный (включая трансдермальный) и парентеральный способы введения.
Должно быть очевидно, что любая форма соединений по данному изобретению (т.е. свободное основание, фармацевтически приемлемая соль или сольват), которая является подходящей для конкретного способа введения, может быть использована в фармацевтических композициях, обсуждаемых здесь.
Соответственно, в одном из составляющих аспектов, данное изобретение касается фармацевтической композиции, содержащей фармацевтически приемлемый носитель или эксципиент и терапевтически эффективное количество соединения формулы I или II, или его фармацевтически приемлемой соли. Необязательно, при желании, указанные фармацевтические композиции могут содержать другие терапевтические и/или составляющие композицию (вспомогательные) средства.
Фармацевтические композиции по данному изобретению обычно содержат терапевтически эффективное количество соединения по данному изобретению или его фармацевтически приемлемой соли. Обычно, такие фармацевтические композиции могут содержать от около 0,01 до около 95 мас.% активного средства; включая, от около 0,01 до около 30 мас.%; как, например, от около 0,01 до около 10 мас.% активного средства.
В фармацевтических композициях по данному изобретению может быть использован любой обычный носитель или эксципиент. Выбор конкретного носителя или эксципиента, или комбинаций носителей или эксципиентов обычно зависит от способа введения, который используют для лечения конкретного пациента, или типа клинического состояния, или болезненного состояния. В этом отношении получение подходящей фармацевтической композиции для конкретного способа введения находится в пределах квалификации специалистов в области фармации. Кроме того, компоненты для таких композиций коммерчески доступны от, например, Sigma, P.O. Box 14508, St. Louis, MO 63178. В качестве дополнительной иллюстрации, обычные способы получения конкретных фармацевтических композиций изложены в Remington: The Science and Practice of Pharmacy, 20th Edition, Lippincott Williams & White, Baltimore, Maryland (2000); and H.C. Ansel et al., Pharmaceutical Dosage Forms and Drug Delivery Systems, 7th Edition, Lippincott Williams & White, Baltimore, Maryland (1999).
Типичные примеры материалов, которые могут служить в качестве фармацевтически приемлемых носителей, включают, но ими не ограничиваясь, нижеследующие: (1) сахара, такие как лактоза, глюкоза и сахароза; (2) крахмалы, такие как кукурузный крахмал и картофельный крахмал; (3) целлюлозу и ее производные, такие как натрий-карбоксиметилцеллюлоза, этилцеллюлоза и ацетат целлюлозы; (4) порошкообразный трагакант; (5) солод; (6) желатин; (7) тальк; (8) эксципиенты, такие как масло какао и воски для суппозитория; (9) масла, такие как арахисовое масло, хлопковое масло, сафлоровое масло, кунжутное масло, оливковое масло, кукурузное масло и соевое масло; (10) гликоли, такие как пропиленгликоль; (11) полиолы, такие как глицерин, сорбит, маннит и полиэтиленгликоль; (12) сложные эфиры, такие как этилолеат и этиллаурат; (13) агар; (14) средства буферизации, такие как гидроксид магния и гидроксид алюминия; (15) альгиновую кислоту; (16) апирогенную воду; (17) изотонический физиологический раствор; (18) раствор Рингера; (19) этиловый спирт; (20) растворы фосфатного буфера; (21) сжатые газы-вытеснители, такие как хлорфторуглероды и гидрофторуглероды; и (22) другие нетоксические совместимые вещества, используемые в фармацевтических композициях.
Фармацевтические композиции согласно данному изобретению обычно получают тщательным смешением до гомогенного состояния или компаундированием соединения по данному изобретению с фармацевтически приемлемым носителем и одним или несколькими необязательными компонентами. Если это необходимо или по желанию, полученная однородная компаундированная смесь затем может быть подвергнута формованию или загрузке в таблетки, капсулы, пилюли, фильтрующие (поглотительные) коробки, картриджи, дозаторы и т.п., используя обычные способы и оборудование.
В одном варианте, фармацевтические композиции по данному изобретению являются подходящими для введения ингаляцией. Подходящие фармацевтические композиции для ингаляционного введения обычно находятся в форме аэрозоля или порошка. Такие композиции обычно вводят, используя общеизвестные устройства для доставки, такие как распылитель-ингалятор, ингалятор с отмеренной дозой (MDI), ингалятор сухого порошка (DPI) или другое устройство доставки подобного типа.
В конкретном варианте данного изобретения фармацевтическую композицию, содержащую активное средство, вводят ингаляцией, используя распылитель-ингалятор. Такие распылители-ингаляторы обычно создают высокоскоростной поток воздуха, который вынуждает фармацевтическую композицию, содержащую активное средство, распыляться в виде аэрозоля, который поступает в дыхательные пути пациента. Соответственно, при составлении композиции для использования в распылителе-ингаляторе активное средство, как правило, растворяют в подходящем носителе с получением раствора. Альтернативно, активное средство может быть подвергнуто тонкому измельчению и смешению с подходящим носителем с образованием суспензии тонкоизмельченных частиц вдыхаемого размера, где термин "тонкоизмельченный материал" обычно подразумевает наличие около 90% или больше частиц с диаметром меньше чем около 10 мкм. Подходящие распыляющие устройства обеспечены коммерчески, например PARI GmbH (Stamberg, Германия). Другие распыляющие устройства раскрыты, например, в патенте США 6123068 и WO 97/12687.
Типичная фармацевтическая композиция для использования в распылителе-ингаляторе включает изотонический водный солевой раствор, содержащий от около 0,05 мкг/мл до около 10 мг/мл соединения формулы I или его фармацевтически приемлемой соли, или его сольвата, или его стереоизомера. В одном варианте, рН этой композиции находится в диапазоне от около 4 до около 6. В конкретном варианте, указанную композицию необязательно подвергают буферизации до рН около 5, используя цитратный буфер.
В другом конкретном варианте данного изобретения фармацевтическую композицию, содержащую активное средство, вводят ингаляцией, используя ингалятор сухого порошка. Такие ингаляторы сухого порошка обычно содержат активное средство в виде свободно-текучего порошка, который диспергируется в потоке воздуха во время вдоха пациента. Для получения свободно-текучего порошка активное средство обычно объединяют в состав с подходящим эксципиентом, таким как лактоза или крахмал.
Типичная фармацевтическая композиция для использования в ингаляторе сухого порошка содержит лактозу, имеющую размер частиц от около 1 мкм до около 100 мкм, и тонкоизмельченные частицы соединения формулы I или его фармацевтически приемлемой соли, или его сольвата, или его стереоизомера.
Такая композиция сухого порошка может быть приготовлена, например, объединением лактозы с активным средством и затем сухим смешением компонентов. Альтернативно, при желании, активное средство можно формулировать без эксципиента. Затем фармацевтическую композицию обычно загружают в дозатор сухого порошка, или в картриджи, или капсулы для ингаляции для последующего их использования в устройствах доставки сухого порошка.
Примеры устройств для доставки сухого порошка ингаляцией включают Diskhaler (GlaxoSmithKline, Research Triangle Park, NC) (см., например, патент США 5035237); Diskus (GlaxoSmithKline) (см., например, патент США 6378519; Turbuhaler (AstraZeneca, Wilmington, DE) (см., например, патент США 4524769); и Rotahaler (GlaxoSmithKline) (см., например, патент США 4353365). Дополнительные примеры подходящих устройств DPI описаны в патентах США 5415162, 5239993 и 5715810 и ссылках, цитированных там.
В очередном конкретном варианте данного изобретения фармацевтическую композицию, содержащую активное средство, вводят ингаляцией, используя ингалятор с отмеренной дозой. Такие ингаляторы с отмеренной дозой обычно поставляют отмеренное количество активного средства или его фармацевтически приемлемой соли, используя сжатый газ-вытеснитель. Соответственно, фармацевтические композиции, вводимые с использованием ингалятора с отмеренной дозой, обычно содержат раствор или суспензию активного средства в сжиженном газе-вытеснителе. Может быть использован любой подходящий сжиженный газ-вытеснитель, включая хлорфторуглероды, такой как CCl3F, и гидрофторалканы (HFA), такие как 1,1,1,2-тетрафторэтан (HFA 134a) и 1,1,1,2,3,3,3-гептафтор-н-пропан, (HFA 227). Из-за опасений по воздействию хлорфторуглеродов на озоновый слой обычно отдается предпочтение препаратам, содержащим HFA. Дополнительные необязательные компоненты препаратов с HFA включают сорастворители, такие как этанол или пентан, и поверхностно-активные вещества, такие как сорбиттриолеат, олеиновая кислота, лецитин и глицерин. См., например, патент США 5225183, EP 0717987 A2 и WO 92/22286.
Типичная фармацевтическая композиция для использования в ингаляторе с отмеренной дозой содержит от около 0,01% до около 5 мас.% соединения формулы I, или его фармацевтически приемлемой соли, или его сольвата, или его стереоизомера; от около 0% до около 20 мас.% этанола; и от около 0% до около 5 мас.% поверхностно-активного вещества; при этом остаток составляет газ-вытеснитель HFA.
Такие композиции обычно получают, добавляя охлажденный или находящийся под давлением гидрофторалкан в подходящий контейнер, содержащий активное средство, этанол (если присутствует) и поверхностно-активное вещество (если присутствует). Для получения суспензии активное средство подвергают тонкому измельчению и затем объединяют с газом-вытеснителем. Затем состав загружают в аэрозольную фильтрующую (поглотительную) коробку, которая является частью устройства-ингалятора с отмеренной дозой. Примеры устройств-ингаляторов с отмеренной дозой, разработанных специально для использования с газами-вытеснителями типа HFA, представлены в патентах США 6006745 и 6143277. Альтернативно, композицию в виде суспензии можно получить распылительной сушкой слоя поверхностно-активного вещества на поверхности тонкоизмельченных частиц активного средства. См., например, WO 99/53901 и WO 00/61108.
В качестве дополнительных примеров способов получения вдыхаемых частиц и композиций (препаратов) и устройств для дозирования ингаляцией см. патенты США 6268533, 5983956, 5874063 и 6221398, и WO 99/55319 и WO 00/30614.
В другом варианте, фармацевтические композиции по данному изобретению являются подходящими для перорального введения. Подходящие фармацевтические композиции для перорального введения могут быть в форме капсул, таблеток, пилюль, леденцов, саше, драже, порошков, гранул; или в виде раствора или суспензии в водной или неводной жидкости; или в виде жидкой эмульсии масло-в-воде или вода-в-масле; или в виде эликсира или сиропа; и т.п.; при этом каждая форма содержит заранее установленное количество соединения по данному изобретению в качестве активного компонента.
В случае предназначения для перорального введения в твердой дозированной форме (т.е. капсулы, таблетки, пилюли и т.п.), фармацевтические композиции согласно данному изобретению обычно содержат в качестве активного компонента соединение по данному изобретению и один или несколько фармацевтически приемлемых носителей, таких как цитрат натрия и вторичный кислый фосфат кальция. Необязательно или альтернативно, указанные твердые дозированные формы могут также содержать: (1) наполнители или разбавители, такие как крахмалы, лактоза, сахароза, глюкоза, маннит и/или кремниевая кислота; (2) связующие, такие как карбоксиметилцеллюлоза, альгинаты, желатин, поливинилпирролидон, сахароза и/или акация; (3) увлажнители, такие как глицерин; (4) дезинтегрирующие средства, такие как агар-агар, карбонат кальция, картофельный или маниоковый крахмал, альгиновая кислота, некоторые силикаты и/или карбонат натрия; (5) ингибиторы растворобразования, такой как парафин; (6) ускорители абсорбции (всасывания), такие как соединения четвертичного аммония; (7) смачивающие вещества, такие как цетиловый спирт и/или глицеролмоностеарат; (8) абсорбенты, такие как каолин и/или бентонитовая глина; (9) лубриканты, такие как тальк, стеарат кальция, стеарат магния, твердые полиэтиленгликоли, лаурилсульфат натрия, и/или их смеси; (10) красители; и (11) средства буферизации.
В фармацевтических композициях согласно данному изобретению могут также присутствовать средства, способствующие высвобождению, смачивающие средства, средства для покрытия, подслащивающие вещества, вкусовые вещества и ароматизаторы (отдушки), консерванты и антиоксиданты. Примеры фармацевтически приемлемых антиоксидантов включают: (1) растворимые в воде антиоксиданты, такие как аскорбиновая кислота, цистеин гидрохлорид, бисульфат натрия, метабисульфат натрия, сульфит натрия и т.п.; (2) маслорастворимые антиоксиданты, такие как аскорбилпальмитат, бутилированный гидроксианизол (BHA), бутилированный гидрокситолуол (BHT), лецитин, пропилгаллат, альфа-токоферол и т.п.; и (3) металл-хелатирующие средства, такие как лимонная кислота, этилендиаминтетрауксусная кислота (EDDA), сорбит, винная кислота, фосфорная кислота и т.п. Средства для покрытия таблеток, капсул, пилюль и т.п. включают вещества, используемые для энтеросолюбильных покрытий, такие как фталат ацетилцеллюлозы (CAP), поливинилацетатфталат (PAP), фталат гидроксипропилметилцеллюлозы, сополимеры метакриловая кислота-сложный эфир метакриловой кислоты, тримеллитат ацетатцеллюлозы (CAT), карбоксиметилэтилцеллюлозу (CMEC), ацетатсукцинат гидроксипропилметилцеллюлозы (HPMCAS) и т.п.
При желании, фармацевтические композиции согласно данному изобретению могут быть также составлены в препарат, обеспечивающий пролонгированное или контролируемое высвобождение активного компонента, используя, в качестве примера, гидроксипропилметилцеллюлозу в различных пропорциях; или другие полимерные матрицы, липосомы и/или микросферы.
Кроме того, фармацевтические композиции согласно настоящему изобретению могут необязательно содержать контрастные средства и могут быть составлены в препарат таким образом, чтобы высвобождать только активный компонент, или предпочтительно, в определенной части желудочно-кишечного тракта, необязательно, прологированным образом. Примеры таких удерживающих композиций, которые могут быть использованы, включают полимерные материалы и воски. Активный компонент может также находиться в микроинкапсулированной форме, если это целесообразно, с одним или несколькими вышеописанными эксципиентами.
Подходящие жидкие дозированные формы для перорального введения включают, в качестве иллюстрации, фармацевтически приемлемые эмульсии, микроэмульсии, растворы, суспензии, сиропы и эликсиры. Такие жидкие дозированные формы обычно включают активный компонент и инертный разбавитель, такой как, например, вода, или другие растворители, солюбилизирующие средства и эмульгаторы, такие как этиловый спирт, изопропиловый спирт, этилкарбонат, этилацетат, бензиловый спирт, бензилбензоат, пропиленгликоль, 1,3-бутиленгликоль, масла (главным образом, хлопковое, арахисовое, кукурузное, проростков семян, оливковое, касторовое и кунжутное масла), глицерин, фурфуриловый спирт, полиэтиленгликоли и сложные эфиры жирных кислот и сорбита, и их смеси. Суспензии, помимо активного компонента, могут содержать суспендирующие средства, такие как, например, этоксилированные изостеариловые спирты, полиоксиэтиленсорбит и сложные эфиры сорбитана, микрокристаллическая целлюлоза, метагидроксид алюминия, бентонит, агар-агар и трагакант, и их смеси.
В случае предназначения для перорального введения фармацевтические композиции по данному изобретению предпочтительно упаковывают в единичную дозированную форму. Термин "единичная дозированная форма" означает физически дискретную единицу, подходящую для дозирования пациенту, т.е. каждая единица содержит заранее установленное количество активного средства, рассчитанное так, чтобы обеспечить проявление желательного терапевтического действия, либо в виде одной единицы, либо в комбинации с одной или несколькими дополнительными единицами. Например, такими единичными дозированными формами могут быть капсулы, таблетки, пилюли и т.п.
Соединения согласно данному изобретению можно также вводить трансдермально, используя известные системы и эксципиенты для трансдермальной доставки. Например, соединение по данному изобретению может быть смешано с усилителями проникновения, такими как пропиленгликоль, полиэтиленгликольмонолаурат, азациклоалкан-2-оны и т.п., и включены в пластырь или другую подобную систему доставки. При желании, в таких трансдермальных композициях могут быть использованы дополнительные эксципиенты, включая гелеобразующие средства, эмульгаторы и буферы.
Фармацевтические композиции согласно данному изобретению могут дополнительно содержать другие терапевтические средства, которые вводят совместно с соединением формулы I, или его фармацевтически приемлемой солью, или его сольватом, или его стереоизомером. Например, фармацевтические композиции согласно данному изобретению могут дополнительно включать одно или несколько терапевтических средств, выбранных из агонистов β2-адренергических рецепторов, противовоспалительных средств (например, кортикостероиды и нестероидные противовоспалительные средства (NSAID)), других антагонистов мускариновых рецепторов (т.е. антихолинергические средства), антиинфекционых средств (например, антибиотики или противовирусные средства) и антигистаминов. Другие терапевтические средства могут быть использованы в форме фармацевтически приемлемых солей или сольватов. Кроме того, если это целесообразно, другие терапевтические средства могут быть использованы в виде оптически чистых стереоизомеров.
Типичные агонисты β2-адренергических рецепторов, которые могут быть использованы в комбинации с соединениями по данному изобретению, включают, но не ограничиваясь ими, салметерол, салбутамол, формотерол, салмефамол, фенотерол, тербуталин, албутерол, изоэтарин, метапротеренол, битолтерол, пирбутерол, левалбутерол и т.п., или их фармацевтически приемлемые соли. Другие агонисты β2-адренергических рецепторов, которые могут быть использованы в комбинации с соединениями по данному изобретению, включают, но не ограничиваются ими, 3-(4-{[6-({(2R)-2-гидрокси-2-[4-гидрокси-3-(гидроксиметил)фенил]этил}мино)гексил]окси}утил)бензолсульфонамид и 3-(-3-{[7-({(2R)-2-гидрокси-2-[4-гидрокси-3-(гидроксиметил)фенил]этил}мино)гептил]окси}ропил)бензолсульфонамид и родственные соединения, раскрытые в WO 02/066422, опубликованной 29 августа 2002; 3-[3-(4-{[6-([(2R)-2-гидрокси-2-[4-гидрокси-3-(гидроксиметил)фенил]этил}мино)гексил]окси}утил)фенил]имидазолидин-2,4-дион и родственные соединения, раскрытые в WO 02/070490, опубликованной 12 сентября 2002; 3-(4-{[6-({(2R)-2-[3-(формиламино)-4-гидроксифенил]-2-гидроксиэтил}мино)гексил]окси}утил)бензолсульфонамид, 3-(4-{[6-({(2S)-2-[3-(формиламино)-4-гидроксифенил]-2-гидроксиэтил}мино)гексил]окси}утил)бензолсульфонамид, 3-(4-{[6-({(2R/S)-2-[3-(формиламино)-4-гидроксифенил]-2-гидроксиэтил}мино)гексил]окси}утил)бензолсульфонамид, N-(трет-бутил)-3-(4-{[6-({(2R)-2-[3-(формиламино)-4-гидроксифенил]-2-гидроксиэтил}мино)гексил]окси}бутил)бензолсульфонамид, N-(трет-бутил)-3-(4-{[6-({(2S)-2-[3-(формиламино)-4-гидроксифенил]-2-гидроксиэтил}мино)гексил]окси}утил)бензолсульфонамид, N-(трет-бутил)-3-(4-{[6-({(2R/S)-2-[3-(формиламино)-4-гидроксифенил]-2-гидроксиэтил}мино)гексил]окси}утил)бензолсульфонамид и родственные соединения, раскрытые в WO 02/076933, опубликованной 3 октября 2002; 4-{(1R)-2-[(6-{2-[(2,6-дихлорбензил)окси]этокси}ексил)амино]-1-гидроксиэтил}-2-(гидроксиметил)фенол и родственные соединения, раскрытые в WO 03/024439, опубликованной 27 марта 2003; N-{2-[4-((R)-2-гидрокси-2-фенилэтиламино)фенил]этил}-(R)-2-гидрокси-2-(3-формамидо-4-гидроксифенил)этиламин и родственные соединения, раскрытые в патенте США 6576793 B1, выданном 10 июня 2003; N-{2-[4-(3-фенил-4-метоксифенил)аминофенил]этил}-(R)-2-гидрокси-2-(8-гидрокси-2(1Н)-хинолинон-5-ил)этиламин и родственные соединения, раскрытые в патенте США 6653323 B2, выданном 25 ноября 2003; и их фармацевтически приемлемые соли. При использовании агонист β2-адренергического рецептора обычно присутствует в фармацевтической композиции в терапевтически эффективном количестве. Типично, агонист β2-адренергического рецептора может присутствовать в количестве, достаточном для получения дозы от около 0,05 мкг до около 500 мкг.
Типичные кортикостероиды, которые могут быть использованы в комбинации с соединениями по данному изобретению, включают, но не ограничиваясь ими, метилпреднизолон, преднизолон, дексаметазон, флутиказонпропионат, S-фторметиловый эфир 6α,9α-дифтор-17α-[(2-фуранилкарбонил)окси]-11β-гидрокси-16α-метил-3-оксоандроста-1,4-диен-17β-карботиокислоты, S-(2-оксотетрагидрофуран-3S-ил)овый эфир 6α,9α-дифтор-11β-гидрокси-16α-метил-3-оксо-17α-пропионилоксиандроста-1,4-диен-17β-карботиокислоты, сложные эфиры беклометазона (например, 17-пропионатный сложный эфир или 17,21-дипропионатный сложный эфир), будезонид, флунизолид, сложные эфиры мометазона (например, фуроатный сложный эфир), триамцинолон-ацетонид, рофлепонид, циклезонид, бутиксокорт-пропионат, RPR-106541, ST-126 и т.п., или их фармацевтически приемлемые соли. При использовании кортикостероид обычно присутствует в фармацевтической композиции в терапевтически эффективном количестве. Типично, стероидное противовоспалительное средство может присутствовать в количестве, достаточном для получения дозы от около 0,5 мкг до около 500 мкг.
Другие подходящие комбинации включают, например, другие противовоспалительные средства, например, NSAID (такие как кромогликат натрия; недокромил-натрий; ингибиторы фосфодиэстеразы (PDE) (например, теофиллин, ингибиторы PDE4 или смешанные ингибиторы (общие для) PDE3/PDE4); антагонисты лейкотриена (например, монтелейкаст); ингибиторы синтеза лейкотриена; ингибиторы iNOS; ингибиторы протеаз, такие как ингибиторы триптазы и эластазы; антагонисты бета-2 интегрина и агонисты или антагонисты аденозинового рецептора (например, агонисты аденозина 2а); антагонисты цитокинов (например, антагонисты хемокинов, такие как антитело к интерлейкину (αIL антитело), в частности, терапия антителами αIL-4, терапия αIL-13, или их комбинация); или ингибиторы синтеза цитокина.
Например, типичные ингибиторы фосфодиэстеразы-4 (PDE4) или смешанные ингибиторы PDE3/PDE4, которые могут быть использованы в комбинации с соединениями по данному изобретению, включают, но ими не ограничиваясь, цис-4-циано-4-(3-циклопентилокси-4-метоксифенил)циклогексан-1-карбоновую кислоту, 2-карбометокси-4-циано-4-(3-циклопропилметокси-4-дифторметоксифенил)циклогексан-1-он; цис-[4-циано-4-(3-циклопропилметокси-4-дифторметоксифенил)циклогексан-1-ол]; цис-4-циано-4-[3-(циклопентилокси)-4-метоксифенил]циклогексан-1-карбоновую кислоту и т.п., или их фармацевтически приемлемые соли. Другие типичные ингибиторы PDE4 или смешанные ингибиторы PDE4/PDE3 включают AWD-12-281 (elbion); NCS-613 (INSERM); D-4418 (Chiroscience and Schering-Plough); CI-1018 или PD-168787 (Pfizer); соединения бензодиоксола, раскрытые в WO 99/16766 (Kyowa Hakko); K-34 (Kyowa Hakko); V-11294A (Napp); рофлумиласт (Byk-Gulden); соединения фталазинона, раскрытые в WO 99/47505 (Byk-Gulden); пумафентрин (Byk-Gulden, now Altana); арофиллин (Almirall-Prodesfarma); VM554/UM565 (Vernalis); T-440 (Tanabe Seiyaku); и T2585 (Tanabe Seiyaku).
Типичные мускариновые антагонисты (т.е. антихолинергические средства), которые могут быть использованы в комбинации с и в дополнение к соединениям по данному изобретению, включают, но не ограничиваясь ими, атропин, атропинсульфат, атропиноксид, метилатропиннитрат, гоматропина гидробромид, гиосциамина (d, l) гидробромид, скополамина гидробромид, ипратропийбромид, окситропийбромид, тиотропийбромид, метантелин, пропантелинбромид, анизотропинметилбромид, клидинийбромид, копирролат (Robinul), изопропамидиодид, мепензолатбромид, тридигексэтилхлорид (Pathilone), гексоциклийметилсульфат, циклопентолата гидрохлорид, тропикамид, тригексифенидила гидрохлорид, пирензепин, телензепин, AF-DX 116 и метострамин и т.п., или их фармацевтически приемлемую соль; или, для соединений, перечисленных в виде соли, их альтернативную фармацевтически приемлемую соль.
Типичные антигистамины (т.е. антагонисты H1-рецепторов), которые могут быть использованы в комбинации с соединениями по данному изобретению, включают, но не ограничиваясь ими, этаноламины, такие как карбиноксаминмалеат, клемастинфумарат, гидрохлорид и дименгидринат дифенилгидрамина; этилендиамины, такие как пириламинамлеат, трипеленнамина гидрохлорид и трипеленнаминцитрат; алкиламины, такие как хлорфенирамин и акривастин; пиперазины, такие как гидроксизина гидрохлорид, гидроксизинпамоат, циклизина гидрохлорид, циклизинлактат, меклизина гидрохлорид и цетиризина гидрохлорид; пиперидины, такие как астемизол, левокабастин гидрохлорид, лоратадин или его дезкарбоэтокси-аналог, терфенадин- и фексофенадин-гидрохлорид; азеластина гидрохлорид; и т.п., или их фармацевтически приемлемую соль; или, для соединений, перечисленных в виде соли, их альтернативную фармацевтически приемлемую соль.
Подходящие дозы для других терапевтических средств, вводимых в комбинации с соединением по данному изобретению, находятся в диапазоне от около 0,05 мкг/день до около 100 мг/день.
Нижеследующие композиции (препараты) иллюстрируют типичные фармацевтические композиции по данному изобретению:
Пример композиции А
Твердые желатиновые капсулы для перорального введения получают следующим образом:
Типичный способ: Компоненты тщательно смешивают и затем загружают в твердую желатиновую капсулу (460 мг композиции на капсулу).
Пример композиции В
Твердые желатиновые капсулы для перорального введения получают следующим образом:
Типичный способ: Компоненты тщательно смешивают и затем пропускают через сито U.S., размером № 45 меш, и загружают в твердую желатиновую капсулу (200 мг композиции на капсулу).
Пример композиции С
Капсулы для перорального введения получают следующим образом:
Типичный способ: Компоненты тщательно смешивают и затем загружают в желатиновую капсулу (300 мг композиции на капсулу).
Пример композиции D
Таблетки для перорального введения получают следующим образом:
Типичный способ: Соединение по изобретению, крахмал и целлюлозу пропускают через сито U.S., размером № 45 меш, и тщательно смешивают. Раствор поливинилпирролидона смешивают с полученными порошками и эту смесь затем пропускают через сито U.S., размером № 14 меш. Полученные таким образом гранулы сушат при 50-60°С и пропускают через сито U.S., размером № 18 меш. Затем к гранулам добавляют натрий-карбоксиметилкрахмал, стеарат магния и тальк (предварительно пропущенный через сито U.S., размером № 60 меш). После смешения смесь подвергают прессованию на таблетировочной машине, получая таблетку массой 100 мг.
Пример композиции Е
Таблетки для перорального введения получают следующим образом:
Типичный способ: Компоненты тщательно смешивают и затем подвергают прессованию с получением таблеток (665 мг композиции на таблетку).
Пример композиции F
Таблетки с одной насечкой для перорального введения получают следующим образом:
Типичный способ: Компоненты тщательно смешивают и прессуют с получением таблетки с одной насечкой (600 мг композиции на таблетку).
Пример композиции G
Суспензию для перорального введения получают следующим образом:
Типичный способ: Компоненты смешивают, получая суспензию, содержащую 100 мг активного компонента на 10 мл суспензии.
Пример композиции H
Сухой порошок для введения ингаляцией получают следующим образом:
Типичный способ: Соединение по изобретению тонко измельчают и затем смешивают с лактозой. Эту компаундированную смесь затем загружают в желатиновый картридж для ингаляции. Содержимое картриджа вводят, используя порошковый ингалятор.
Пример композиции I
Композицию сухого порошка для использования в устройстве-ингаляторе сухого порошка получают следующим образом:
Типичный способ: Получают фармацевтическую композицию, имеющую объемное отношение компонентов тонкоизмельченного соединения по изобретению к лактозе 1:200. Композицию упаковывают в аппарат для ингаляции сухого порошка, способный доставлять от около 10 до около 100 мкг соединения по изобретению на дозу.
Пример композиции J
Сухой порошок для введения ингаляцией через ингалятор с отмеренной дозой получают следующим образом:
Типичный способ: Суспензию, содержащую 5 мас.% соединения по изобретению и 0,1 мас.% лецитина, получают диспергированием 10 г соединения по изобретению в виде тонкоизмельченных частиц со средним размером меньше чем 10 мкм в растворе, полученном из 0,2 г лецитина, растворенного в 200 мл деминерализованной воды. Суспензию сушат распылением и полученный продукт подвергают тонкому измельчению до частиц, имеющих средний диаметр меньше чем 1,5 мкм. Частицы загружают в картриджи с находящимся под давлением 1,1,1,2-тетрафторэтаном.
Пример композиции K
Фармацевтическую композицию для использования в ингаляторе с отмеренной дозой получают следующим образом:
Типичный способ: Суспензию, содержащую 5% соединения по изобретению, 0,5% лецитина и 0,5% трегалозы, получают диспергированием 5 г активного компонента в виде тонкоизмельченных частиц со средним размером меньше чем 10 мкм в коллоидном растворе, полученном из 0,5 г трегалозы и 0,5 г лецитина, растворенных в 100 мл деминерализованной воды. Суспензию сушат распылением и полученный продукт подвергают тонкому измельчению до частиц, имеющих средний диаметр меньше чем 1,5 мкм. Частицы загружают в фильтрующие [поглотительные] коробки с находящимся под давлением 1,1,1,2-тетрафторэтаном.
Пример композиции L
Фармацевтическую композицию для использования в распылителе-ингаляторе получают следующим образом:
Типичный способ: Водный аэрозольный состав для использования в распылителе получают, растворяя 0,1 мг соединения по данному изобретению в 1 мл 0,9% растворе хлорида натрия, подкисленного лимонной кислотой. Смесь перемешивают и обрабатывают ультразвуком до тех пор, пока активный компонент не растворится. рН раствора доводят до значения около 5, медленно добавляя NaOH.
Пример композиции M
Инъецируемую композицию получают следующим образом:
Типичный способ: Вышеуказанные компоненты смешивают и рН доводят до 4±0,5, используя 0,5н. HCl или 0,5н. NaOH.
Полезность
Соединения замещенного пирролидина и родственные соединения по данному изобретению являются полезными в качестве антагонистов мускариновых рецепторов, и поэтому указанные соединения используют для лечения клинических состояний, опосредованных мускариновыми рецепторами, т.е. клинических состояний, интенсивность симптомов которых уменьшается лечением антагонистом мускариновых рецепторов. Такие клинические состояния включают, в качестве примера, нарушения функции дыхательных путей, такие как хроническая обструктивная болезнь легких, астма, пневмосклероз, аллергический ринит, ринорея; нарушения функции мочеполовой системы, такие как учащенное мочеиспускание (гиперестезия мочевого пузыря) или гиперрефлексия (гиперактивность) детрузора и их симптомы; нарушения желудочно-кишечного тракта, такие как синдром раздраженного кишечника, дивертикулез, ахалазия, гиперкинез желудочно-кишечного тракта и диарея; сердечная аритмия, такая как синусовая брадикардия; болезнь Паркинсона; расстройства познавательных способностей, такие как болезнь Альцгеймера; дисменорея; и т.п.
В одном варианте, соединения по данному изобретению полезны для лечения нарушений функции (тонуса) гладкой мускулатуры у млекопитающих, включая людей и их домашних животных (например, собаки, кошки и т.д.). Такие нарушения функции гладкой мускулатуры включают, в качестве примера, учащенное мочеиспускание, хроническую обструктивную болезнь легких и синдром раздраженного кишечника.
При использовании для лечения нарушений функции гладкой мускулатуры и других состояний, опосредованных мускариновыми рецепторами, соединения по данному изобретению обычно вводят перорально, ректально, парентерально или путем ингаляции в единичной суточной дозе или в виде множественных доз в течение дня. Количество активного средства, вводимого на дозу, или суммарное количество активного средства, вводимого в день, обычно может быть определено лечащим врачом пациента и обычно зависит от таких факторов, как природа и тяжесть состояния пациентов; состояние, подлежащее лечению, возраст и общее состояние здоровья пациента, толерантность пациента к активному средству, путь введения и т.п.
Обычно, подходящие дозы для лечения нарушений функции гладкой мускулатуры и других нарушений, опосредованных мускариновыми рецепторами, могут изменяться от около 0,14 мкг/кг/день до около 7 мг/кг/день активного средства; включая от около 0,15 мкг/кг/день до около 5 мг/кг/день. Для человека со средней массой тела 70 кг это составит от около 10 мкг в день до около 500 мг в день активного средства.
В конкретном варианте, соединения по данному изобретению полезны для лечения нарушений дыхания, таких как COPD или астма, у млекопитающих, включая людей. При использовании для лечения нарушений дыхания, соединения по данному изобретению обычно вводят ингаляцией по схеме приема множественных доз в течение дня, однократной суточной дозы или одной дозы в неделю. Обычно, доза для лечения нарушения дыхания может меняться от около 10 мкг/день до около 200 мкг/день.
При использовании для лечения нарушения дыхания соединения по данному изобретению необязательно вводят в комбинации с другими терапевтическими средствами, такими как агонист β2-адренергического рецептора; кортикостероид, нестероидное противовоспалительное средство, или их комбинации.
В другом варианте, соединения по данному изобретению используют для лечения учащенного мочеиспускания. При использовании для лечения учащенного мочеиспускания соединения по данному изобретению обычно вводят перорально в единичной суточной дозе или в виде множественных доз в течение дня; предпочтительно в единичной суточной дозе. Предпочтительно, доза для лечения учащенного мочеиспускания обычно варьирует от около 1,0 мг/кг до около 500 мг/день.
В очередном варианте соединения по данному изобретению используют для лечения синдрома раздраженного кишечника. При использовании для лечения синдрома раздраженного кишечника соединения по данному изобретению обычно можно вводить перорально или ректально в единичной суточной дозе или в виде множественных доз в течение дня. Предпочтительно, доза для лечения синдрома раздраженного кишечника обычно варьируется от около 1,0 мг/день до около 500 мг/день.
Так как соединения по данному изобретению являются антагонистами мускариновых рецепторов, указанные соединения также полезны в качестве инструментальных средств для исследования или изучения биологических систем или образцов, имеющих мускариновые рецепторы. Такие биологические системы или образцы могут содержать мускариновые рецепторы M1, M2, M3, M4 и/или M5. В таких исследованиях, которые могут выполняться либо in vitro, либо in vivo, может быть использована любая биологическая система или образец, имеющий мускариновые рецепторы. Типичные системы или образцы, подходящие для таких исследований, включают, но не ограничиваясь ими, клетки, клеточные экстракты, плазменные мембраны, тканевые образцы, млекопитающих (таких как мыши, крысы, морские свинки, кролики, собаки, свиньи и т.д.), и т.п.
В этом варианте биологическую систему или образец, содержащий мускариновый рецептор, приводят в контакт с антагонизирующим мускариновый рецептор количеством соединения по данному изобретению. Затем оценивают действие антагониста на мускариновый рецептор, используя обычные методы и оборудование, такие как анализ связывания меченного радиоактивным изотопом лиганда и функциональные анализы. Такие функциональные анализы включают лиганд-опосредованные изменения во внутриклеточном циклическом аденозинмонофосфате (цАМФ, cAMP), лиганд-опосредованные изменения в активности фермента - аденилилциклазы (которая синтезирует cAMP), лиганд-опосредованные изменения во включении гуанозин 5'-О-(-тио)трифосфата ([35S]GTP) в изолированные мембраны посредством катализируемого рецептором обмена [35S]GTP на GDP, лиганд-опосредованные изменения в содержании внутриклеточного кальция в виде свободных ионов (измеренного, например, посредством планшет-ридера (спектрофотометр для прочтения планшетов) с детектированием по флуоресцентной метке или FLIPR® от Molecular Devices, Inc.). Соединение по данному изобретению обычно антагонизирует или уменьшает активацию мускариновых рецепторов в любом из функциональных анализов, перечисленных выше, или анализах подобной природы. Антагонизирующее мускариновый рецептор количество соединения по данному изобретению обычно находится в диапазоне от 0,1 наномолей до около 100 наномолей.
Кроме того, соединения по данному изобретению могут быть использованы в качестве инструментальных средств для обнаружения новых соединений, которые имеют активность антагонистов мускариновых рецепторов. В этом варианте данные по связыванию с мускариновым рецептором (например, определенные согласно анализам in vitro по вытеснению радиоактивного лиганда) для испытуемого соединения или группы испытуемых соединений сравнивают с данными по связыванию с мускариновым рецептором для соединения по данному изобретению с целью идентифицирования тех испытуемых соединений, которые обладают приблизительно равным или превосходящим сродством к связыванию с мускариновым рецептором, если оно вообще имеется. Этот аспект изобретения включает, в отдельных вариантах, как получение данных сравнения (используя соответствующие анализы), так и анализ данных испытаний с целью идентифицирования испытуемых соединений, представляющих интерес.
Среди других свойств, соединения по данному изобретению, как было обнаружено, являются сильнодействующими ингибиторами активности мускариновых рецепторов M3. Соответственно, в конкретных вариантах, данное изобретение относится к соединением формулы I, имеющим константу ингибирования диссоциации для подтипа рецептора M3 меньше, чем или равную 100 нМ; или меньше чем или равную 50 нМ; или меньше чем или равную 10 нМ (определенную согласно испытаниям in vitro по вытеснению лиганда, меченного радиоактивным изотопом).
Дополнительно, соединения по данному изобретению, как было обнаружено, обладают удивительной и неожиданной продолжительностью бронхозащиты при введении путем ингаляции. Соответственно, в другом конкретном варианте, данное изобретение относится к соединениям формулы I, имеющим продолжительность бронхозащиты больше чем около 24 часов, включая от около 24 часов до около 72 часов, при введении ингаляцией. Термин "продолжительность бронхозащиты" означает отрезок времени, на протяжении которого соединение обеспечивает бронхозащитный эффект на модели ацетилхолин-индуцированного бронхостеноза у морской свинки.
Кроме того, соединения по данному изобретению, как было установлено, обладают удивительной и неожиданной селективностью действия в отношении легких, при введении путем ингаляции. Соответственно, в другом конкретном варианте, данное изобретение относится к соединениям формулы I, имеющим кажущийся показатель селективности по отношению к легким больше чем 10 как через 1,5 часа, так и через 24 часа после дозирования путем ингаляции. Термин "кажущийся показатель селективности по отношению к легким" означает либо (а) отношение ID50 для средства, подавляющего слюноотделение (доза, требуемая для подавления пилокарипин-индуцированного слюноотделения на 50%), к бронхозащитной ID50 (доза, требуемая для подавления ацетилхолин-индуцированного бронхостеноза на 50%); или (b) отношение ID50 для анти-депрессора (доза, требуемая для подавления метахолин-индуцированного снижения среднего артериального давления на 50%) к бронхозащитной ID50 (доза, требуемая для подавления ацетилхолин-индуцированного бронхостеноза на 50%).
Вышеуказанные свойства, а также полезность соединений по данному изобретению, могут быть продемонстрированы, используя различные испытания in vitro и in vivo, общеизвестные специалистам в данной области. Например, типичные испытания описаны более подробно в нижеследующих примерах.
ПРИМЕРЫ
Нижеследующие синтетические и биологические примеры представлены с целью иллюстрации данного изобретения и их не следует рассматривать как ограничивающие каким-либо образом объем данного изобретения. В представленных ниже примерах нижеперечисленные аббревиатуры имеют нижеследующие значения, если не оговорено особо. Аббревиатуры, которые не приведены ниже, имеют свое общепринятое значение.
Все температуры, сообщаемые в нижеследующих примерах, представлены в градусах Цельсия (°С), если не оговорено особо. Кроме того, если не отмечено особо, реагенты, исходные продукты и растворители закупали у коммерческих поставщиков (таких фирм, как Aldrich, Fluka, Sigma и т.п.) и использовали без дополнительной очистки.
Пример А
Получение 2,2-дифенил-2-(S)-пирролидин-3-илацетамида
Стадия А - Получение (S)-1-бензил-3-(п-толуолсульфонилокси)пирролидина
К перемешиваемому раствору (S)-1-бензил-3-пирролидинола (44,3 г, 0,25 моль) и 1,4-диазабицикло[2.2.2]октана (33,7 г, 0,3 моль) в 250 мл простого трет-бутилметилового эфира в атмосфере азота при 0°С добавляют порциями на протяжении 20 мин п-толуолсульфонилхлорид (52,4 г, 0,275 моль). Реакционную смесь перемешивают при 0°С в течение 1 ч. Баню со льдом убирают и смесь перемешивают при температуре окружающей среды на протяжении ночи (20±5 ч). Добавляют этилацетат (100 мл), а затем насыщенный водный раствор бикарбоната натрия (250 мл). Полученную смесь перемешивают при температуре окружающей среды в течение 1 ч. Слои разделяют и органический слой промывают насыщенным водным раствором бикарбоната натрия (250 мл); насыщенным водным раствором хлорида аммония (250 мл); насыщенным водным раствором хлорида натрия (250 мл); и затем сушат над сульфатом натрия (80 г). Сульфат натрия отфильтровывают и промывают этилацетатом (20 мл) и растворитель удаляют в вакууме, получая 78,2 г указанного в заголовке промежуточного соединения в виде беловатого твердого вещества (выход 94%; чистота 95% согласно ВЭЖХ).
Стадия В - Получение (S)-1-бензил-3-(1-циано-1,1-дифенилметил)пирролидина
К перемешиваемому раствору дифенилацетонитрила (12,18 г, 61,8 ммоль) в безводном ТГФ (120 мл) при 0°С добавляют трет-бутоксид калия (10,60 г, 94,6 ммоль) на протяжении 5 мин. Реакционную смесь перемешивают при 0°С в течение 1 ч. К реакционной смеси при 0°С добавляют (S)-1-бензил-3-(п-толуолсульфонилокси)пирролидин (20,48 г, 61,3 ммоль) одной порцией. Холодную баню убирают и реакционную смесь перемешивают в течение от 5 до 10 мин, и в этот период времени реакционная смесь превращается в коричневый гомогенный раствор. Затем реакционную смесь нагревают при 40°С на протяжении ночи (20±5 ч). Реакционной смеси (ярко-желтая суспензия) дают возможность охладиться до комнатной температуры, прежде чем добавить воду (150 мл). Затем бульшую часть ТГФ удаляют в вакууме и добавляют изопропилацетат (200 мл). Слои разделяют и органический слой промывают насыщенным водным раствором хлорида аммония (150 мл); насыщенным водным раствором хлорида натрия (150 мл); и затем сушат над сульфатом натрия (50 г). Сульфат натрия отфильтровывают и промывают изопропилацетатом (20 мл) и растворитель удаляют в вакууме, получая 23,88 г указанного в заголовке промежуточного соединения в виде светло-коричневого масла (выход >99%, чистота согласно ВЭЖХ 75%, загрязненное, в основном, избытком дифенилацетонитрила).
Стадия С - Получение (S)-3-(1-циано-1,1-дифенилметил)пирролидина
(S)-1-Бензил-3-(1-циано-1,1-дифенилметил)пирролидин растворяют в изопропилацетате (приблизительно 1 г/10 мл) и раствор смешивают с равным объемом 1н. водной хлористоводородной кислоты. Полученные слои разделяют и водный слой экстрагируют равным объемом изопропилацетата. Органические слои объединяют, сушат над сульфатом натрия и фильтруют. Растворитель удаляют в вакууме, получая гидрохлорид (S)-1-бензил-3-(1-циано-1,1-дифенилметил)пирролидина в виде светло-желтого пенистого твердого вещества. (Примечание: Эту гидрохлоридную соль можно также получить во время обработки стадии В).
К перемешиваемому раствору гидрохлорида (S)-1-бензил-3-(1-циано-1,1-дифенилметил)пирролидина (8,55 г, 21,98 ммоль) в метаноле (44 мл) добавляют палладий на углероде (1,71 г) и формиат аммония (6,93 г, 109,9 ммоль). Реакционную смесь нагревают до 50°С при перемешивании в течение 3 ч. Реакционную смесь охлаждают до температуры окружающей среды и добавляют воду (20 мл). Полученную смесь фильтруют через слой целита, промывая метанолом (20 мл). Фильтрат собирают и бульшую часть метанола удаляют в вакууме. Остаток смешивают с изопропилацетатом (100 мл) и 10% водным карбонатом натрия (50 мл). Полученные слои разделяют и водный слой экстрагируют изопропилацетатом (50 мл). Органические слои объединяют и сушат над сульфатом натрия (20 г). Сульфат натрия отфильтровывают и промывают изопропилацетатом (20 мл). Растворитель удаляют в вакууме, получая 5,75 г указанного в заголовке промежуточного соединения в виде светло-желтого масла (выход 99,7%, чистота согласно ВЭЖХ 71%).
Стадия D - Получение 2,2-дифенил-2-(S)-пирролидин-3-илацетамида
Колбу объемом 200 мл с магнитной мешалкой и входом для азота загружают (S)-3-(1-циано-1,1-дифенилметил)пирролидином (2,51 г) и 80% H2SO4 (19,2 мл; предварительно полученная с 16 мл 96% H2SO4 и 3,2 мл H2O). Затем реакционную смесь нагревают при 90°С в течение 24 ч или до тех пор, пока, согласно ВЭЖХ, исходный продукт не будет израсходован. Реакционной смеси дают возможность охладиться до комнатной температуры и затем выливают на лед (приблизительно, 50 мл по объему). К смеси медленно, при перемешивании на ледяной бане, добавляют 50% водный раствор гидроксида натрия до тех пор, пока не установится рН около 12. Дихлорметан (200 мл) добавляют и смешивают с водным раствором, и в это время осаждается сульфат натрия и его отфильтровывают. Фильтрат собирают и слои разделяют. Водный слой экстрагируют дихлорметаном (100 мл) и органические слои объединяют и сушат над сульфатом натрия (5 г). Сульфат натрия отфильтровывают и промывают дихлорметаном (10 мл). Растворитель удаляют в вакууме, получая неочищенный продукт в виде светло-желтого пенистого твердого вещества (приблизительно 2,2 г, чистота согласно ВЭЖХ 86%).
Неочищенный продукт растворяют в этаноле (18 мл) при перемешивании. К этому раствору добавляют теплый раствор L-винной кислоты (1,8 г) в этаноле (14 мл) и полученную смесь перемешивают на протяжении ночи (15±5 ч). Полученный осадок выделяют фильтрацией, получая беловатое твердое вещество (приблизительно 3,2 г, чистота >95% согласно ВЭЖХ). К этому твердому веществу добавляют метанол (15 мл) и полученную суспензию перемешивают при 70°С на протяжении ночи (15 ч). Суспензии дают возможность охладиться до температуры окружающей среды и после фильтрации получают белое твердое вещество (˜2,6 г, чистота >99% согласно ВЭЖХ). К этому твердому веществу добавляют этилацетат (30 мл) и 1н. водный гидроксид натрия (25 мл). Указанную смесь смешивают до тех пор, пока не образуются два отдельных слоя и затем эти слои разделяют и водный слой экстрагируют этилацетатом (20 мл). Органические слои объединяют и сушат над сульфатом натрия (10 г). Сульфат натрия удаляют фильтрацией и растворитель выпаривают в вакууме, получая 1,55 г указанного в заголовке промежуточного соединения в виде беловатого пенистого твердого вещества (выход 58%; чистота согласно ВЭЖХ >99%).
Пример 1
Синтез 2-[(S)-1-(8-метиламинооктил)пирролидин-3-ил]-2,2-дифенилацетамида
Стадия А - Получение 8-(N-бензил-N-метиламино)октан-1-ола
8-Бром-1-октанол (25 г, 119,6 ммоль) в ацетонитриле (50 мл) добавляют к перемешиваемому раствору N-бензил-N-метиламина (43,49 г, 358,9 ммоль) и карбоната калия (49,52 г, 358,9 ммоль) в ацетонитриле (250 мл) при 35°С. Затем реакционную смесь перемешивают при 35°С в течение 7 ч и затем охлаждают до температуры окружающей среды. Карбонат калия отфильтровывают и фильтрат концентрируют при пониженном давлении. Неочищенный остаток растворяют в MTBE (400 мл) и органическую фазу промывают водой, насыщенным раствором соли и сушат над сульфатом магния. Добавляют N-метил-2-пирролидин и смесь концентрируют при пониженном давлении, удаляя избыток N-бензил-N-метиламина. Добавляют MTBE (400 мл) и органическую фазу промывают водой, насыщенным раствором соли и сушат над сульфатом магния, фильтруют и концентрируют при пониженном давлении, получая указанное в заголовке промежуточное соединение в виде масла (конверсия ˜100%).
Данные анализа: МС m/z 250,3 (МН+).
Стадия В - Получение 8-(N-бензил-N-метиламино)октаналя
Диметилсульфоксид (22,71 мл, 320 ммоль) и затем диизопропилэтиламин (55,74 мл, 320 ммоль) добавляют к перемешиваемому раствору промежуточного соединения со стадии А (20 г, 80 ммоль) в дихлорметане (200 мл) при -10°С. Реакционную смесь перемешивают при -10°С в течение 30 мин, затем порциями добавляют комплекс триоксид серы-пиридин (38 г, 240 ммоль). Реакционную смесь перемешивают в течение еще 1 ч при -10°С и затем добавляют воду (200 мл). Органический слой отделяют и промывают водой (200 мл), насыщенным раствором соли (30 мл), сушат над сульфатом магния и затем концентрируют при пониженном давлении. Толуол (100 мл) добавляют и удаляют при пониженном давлении, получая указанное в заголовке промежуточное соединение в виде масла (конверсия ˜100%).
Стадия С - Получение 2-[(S)-1-(8-N-бензил-N-метиламинооктил)пирролидин-3-ил]-2,2-дифенилацетамида
Раствор промежуточного соединения со стадии В (5,95 г, 24 ммоль) и 2,2-дифенил-2-(S)-пирролидин-3-илацетамида (7,4 г, 26,4 ммоль) в дихлорметане (250 мл) охлаждают при 0°С и перемешивают в течение 10 мин. Добавляют при 0°С, порциями, натрийтриацетоксиборгидрид (8,4 г, 36 ммоль) и реакционную смесь перемешивают при комнатной температуре в течение 4 ч. Добавляют дихлорметан и органическую фазу промывают бикарбонатом натрия (2x), насыщенным раствором соли (1x), сушат над сульфатом магния и концентрируют при пониженном давлении. Неочищенный продукт очищают флеш хроматографией (элюент DCM/MeOH/NH4OH=90/9/1), получая 9 г указанного в заголовке промежуточного соединения в виде масла (выход 75%).
Данные анализа: МС m/z 512,8 (МН+).
Стадия D - Получение 2-[(S)-1-(8-метиламинооктил)пирролидин-3-ил]-2,2-дифенилацетамида
К перемешиваемому раствору промежуточного соединения со стадии С (9 г, 17,6 ммоль) в уксусной кислоте (170 мл) в атмосфере азота добавляют палладий на углероде (10 мас.%, 600 мг) и гидроксид палладия на углероде (20 мас.%, влажный, 600 мг). Реакционную смесь продувают азотом три раза и затем присоединяют к водород-содержащему баллону и выдерживают в течение 3 дней при комнатной температуре. Реакционную смесь фильтруют через целит, промывая уксусной кислотой, и растворитель удаляют при пониженном давлении. Полученный остаток очищают препаративной ВЭЖХ, получая 4,02 г указанного в заголовке соединения в виде его соли бис-трифторуксусной кислоты, которая представляет собой масло (выход 35%).
Данные анализа: МС m/z 422,2 (МН+).
Альтернативно, указанное в заголовке соединение получают следующим образом:
Стадия А - Получение 8-(N-бензил-N-метиламино)октан-1-ола
Из 8-бромоктан-1-ола: В 250-мл колбу загружают N-бензил-N-метиламин (24,3 г, 200 ммоль), карбонат калия (28 г, 200 ммоль), 8-бромоктан-1-ол (14 г, 67 ммоль) и ацетонитрил (150 мл). Эту реакционную смесь перемешивают при 35-40°С в течение 5 ч. Затем твердое вещество отфильтровывают и фильтрат подвергают перегонке в высоком вакууме, удаляя избыток N-бензил-N-метиламина, с получением масла. Остаток растворяют в 150 мл MTBE и промывают 15% раствором хлорида аммония (2x100 мл), насыщенным раствором соли (100 мл), сушат над 20 г сульфата натрия, фильтруют и подвергают перегонке в вакууме, получая 13,2 г указанного в заголовке промежуточного соединения в виде масла (выход 79%).
Из 8-хлороктан-1-ола: Двухлитровую колбу загружают бензилметиламином (270 г, 2,23 моль), карбонатом натрия (157 г, 1,48 моль), иодидом натрия (11,1 г, 0,074 моль), 8-хлороктанолом (122 г, 0,74 моль) и ацетонитрилом (1000 мл) и полученную суспензию перемешивают при 80°С в течение 20-30 ч. Затем реакционную смесь концентрируют до объема около 500 мл и добавляют воду (600 мл) и простой трет-бутилметиловый эфир (1000 мл). Затем MTBE-слой отделяют и промывают водой (500 мл). MTBE-раствор концентрируют дистилляцией в вакууме, получая масло и затем масло дополнительно концентрируют дистилляцией в высоком вакууме, удаляя избыток бензилметиламина. Затем к оставшемуся маслу добавляют N-метил-2-пирролидон (300 мл) и этот раствор концентрируют дистилляцией в высоком вакууме, получая масло. Масло растворяют в MTBE (1000 мл) и полученный раствор промывают водой (2x500 мл), насыщенным раствором соли (500 мл), сушат сульфатом натрия (100 г), фильтруют и концентрируют дистилляцией, получая указанное в заголовке соединение в виде масла (178 г, выход 96%, чистота >95%).
Стадия В - Получение 8-(N-бензил-N-метиламино)октан-1-илового эфира толуолсульфокислоты
В 250-мл колбу загружают промежуточное соединение со стадии А (10 г), DABCO (6,72 г) и MTBE (100 мл). Реакционную смесь охлаждают до <10°С и добавляют при <15°С раствор хлорангидрида толуолсульфокислоты (9,2 г) в 60 мл MTBE. Реакционную смесь перемешивают при комнатной температуре в течение 2 ч и затем добавляют гептан (40 мл) и смесь фильтруют. Фильтрат перегоняют в вакууме, получая 16 г указанного в заголовке промежуточного соединения в виде масла (выход 99%).
Стадия С - Получение 2-[(S)-1-(8-N-бензил-N-метиламинооктил)пирролидин-3-ил]-2,2-дифенилацетамида
1000-мл колбу с вводом для азота загружают промежуточным соединением со стадии В (16 г), 2,2-дифенил-2-(S)-пирролидин-3-илацетамидом (10 г), диизопропилэтиламином (10,3 г) и ацетонитрилом (200 мл). Реакционную смесь перемешивают при 45-50°С в течение 20 ч и затем добавляют уксусный ангидрид (2 г) и смесь перемешивают при комнатной температуре в течение 2 часов. Добавляют простой трет-бутилметиловый эфир (300 мл) и воду (400 мл) и MTBE-слой отделяют и промывают водой (2x150 мл) и затем 1н. HCl (1x150 мл). Водный слой отделяют и промывают MTBE (3x100 мл) и затем подщелачивают 27% раствором гидроксида аммония до рН >12. Затем щелочной водный слой экстрагируют MTBE (2x200 мл) и MTBE-слой промывают водой (200 мл), насыщенным раствором соли (200 мл), сушат над сульфатом натрия (20 г), фильтруют и подвергают перегонке, получая 16,5 г указанного в заголовке промежуточного соединения в виде масла (выход 90%). При желании эту реакцию можно проводить в N-метилпирролидоне в качестве растворителя. Кроме того, вместо диизопропилэтиламина можно использовать карбонат калия или карбонат натрия и в реакционную смесь необязательно может быть добавлен иодид натрия.
Стадия D - Получение 2-[(S)-1-(8-метиламинооктил)пирролидин-3-ил]-2,2-дифенилацетамида
250-мл колбу загружают промежуточным соединением со стадии С (24 г), палладием на углероде (10% палладий на углероде с 50% воды, 5,3 г), изопропанолом (160 мл) и 3М раствором HCl (30 мл). Реакционную смесь дегазируют азотом и затем подвергают гидрированию (45-50 фунт/дюйм2) при комнатной температуре в течение 16 ч. Затем смесь фильтруют через слой целита и фильтрат дистиллируют до объема около 50 мл. Остаток растворяют в 1н. HCl (100 мл) и промывают дихлорметаном (2x100 мл). Водный слой доводят до рН >12, добавляя гидроксид аммония, и затем экстрагируют, используя MTBE (2x150 мл). Затем MTBE-раствор промывают водой (100 мл), насыщенным раствором соли (100 мл), сушат над сульфатом натрия (30 г), фильтруют и дистиллируют, получая масло, которое сушат в высоком вакууме, получая 16,5 г указанного в заголовке соединения (выход 91%).
Альтернативно, указанное в заголовке соединение получают, как следует ниже:
Стадия А - Получение 8,8-диметоксиоктаналя
Колбу, объемом 1 л, загружают циклооктеном (50 г), метанолом (250 мл) и дихлорметаном (250 мл). Озон барботируют в раствор при -70°С в течение 8 ч. Затем добавляют толуолсульфокислоту (3 г) и реакционную смесь перемешивают при -70°С в течение 6 ч. Затем добавляют бикарбонат натрия (20 г) и реакционную смесь перемешивают в течение еще 2 ч при -60°С. Наконец, добавляют диметилсульфит (56 г) при -60°С и реакционную смесь перемешивают при комнатной температуре в течение 16 ч. Образовавшееся твердое вещество отфильтровывают и фильтрат упаривают до масла. Масло растворяют в дихлорметане (300 мл) и промывают 1% раствором бикарбоната натрия (2x150 мл). Затем раствор дихлорметана сушат над сульфатом натрия (50 г), фильтруют и подвергают перегонке, получая 60,3 г указанного в заголовке промежуточного соединения в виде масла (выход 71%).
Стадия В - Получение 2-[(S)-1-(8-оксооктил)пирролидин-3-ил]-2,2-дифенилацетамида
100-мл колбу загружают 2,2-дифенил-2-(S)-пирролидин-3-илацетамидом (2,8 г), 8,8-диметоксиоктаналем (2,1 г) и дихлорметаном (20 мл) и эту смесь перемешивают при комнатной температуре в течение 1 ч. Добавляют натрийтриацетоксиборгидрид (3,18 г) и реакционную смесь перемешивают при комнатной температуре в течение 14 ч. Затем добавляют 5% раствор бикарбоната натрия (350 мл) и эту смесь перемешивают в течение 0,5 часов. Слои разделяют и водный слой экстрагируют дихлорметаном (20 мл). Объединенный дихлорметановый раствор концентрируют до объема около 20 мл, фильтруют через слой силикагеля (10 г) и промывают 10% метанолом в дихлорметане (100 мл). Раствор продукта концентрируют до масла и масло растворяют в 50 мл ацетонитрила и перемешивают с 1% HCl (30 мл) в течение 16 ч. Смесь подщелачивают до приблизительно рН >12, добавляя 28% раствор гидроксида аммония, и затем экстрагируют MTBE (2x100 мл). MTBE-слой промывают насыщенным раствором соли (100 мл), сушат над сульфатом натрия (10 г), фильтруют и концентрируют в вакууме, получая 3,8 г указанного в заголовке промежуточного соединения в виде масла (выход 93%).
Стадия С - Получение 2-[(S)-1-(8-N-бензил-N-метиламинооктил)пирролидин-3-ил]-2,2-дифенилацетамида
100-мл колбу с вводом для азота загружают промежуточным соединением со стадии В (3 г), N-бензил-N-метиламином (2,1 г) и дихлорметаном (20 мл) и эту смесь перемешивают при комнатной температуре в течение 1 ч. Добавляют натрийтриацетоксиборгидрид (3,18 г) и реакционную смесь перемешивают при комнатной температуре в течение 14 ч. Затем реакционную смесь гасят добавлением 50 мл 5% HCl и полученную смесь перемешивают в течение 0,5 часов. Слои разделяют и водный слой промывают дихлорметаном (20 мл). Водный слой доводят до рН >13, добавляя 50% гидроксид калия, и экстрагируют MTBE (2x100 мл). Объединенный MTBE-раствор промывают насыщенным раствором соли (100 мл), сушат сульфатом натрия (10 г), фильтруют и концентрируют, получая 2,8 г указанного в заголовке промежуточного соединения в виде масла (выход 75%). Используя методику, описанную в вышеупомянутой стадии D, указанное промежуточное соединение превращают в указанное в заголовке соединение.
Альтернативно, указанное в заголовке соединение получают в виде соли нафталин-1,5-дисульфокислоты, используя нижеследующий способ:
Стадия А - Получение 8-(N-трет-бутоксикарбонил-N-метиламино)октан-1-ола
1-литровую колбу загружают 8-(бензилметиламино)октан-1-олом (49 г, 0,20 моль), изопропанолом (400 мл), 2н. водной хлористоводородной кислотой (100 мл) и активированным углем (5 г, DARCO) и полученную смесь перемешивают в течение 30 мин. Затем смесь фильтруют, удаляя активированный уголь, и к фильтрату добавляют палладий на углероде (5 г, 10% сухая масса). Полученную смесь дегазируют три раза азотом и затем дважды водородом; и затем смесь подвергают гидрированию в аппарате Парра при давлении водорода 20-30 фунт/дюйм2 в течение 12-24 часов. Затем смесь фильтруют через слой целита (20 г) и концентрируют перегонкой до объема около 100 мл. Добавляют изопропанол (200 мл) и этот раствор снова концентрируют перегонкой в вакууме до объема 100 мл. Эту процедуру повторяют еще два раза, получая раствор, содержащий гидрохлорид 8-етиламинооктан-1-ола.
1-литровую колбу загружают раствором гидрохлорида 8-етиламинооктанола в изопропаноле, полученным выше, и триэтиламином (30,3 г, 0,30 моль) и к этой смеси порциями добавляют ди-трет-бутилдикарбонат (48 г, 0,22 моль). Полученную смесь перемешивают при комнатной температуре в течение 2-5 ч и затем смесь концентрируют до объема около 300 мл. Добавляют воду (200 мл) и этилацетат (400 мл) и эту смесь перемешивают в течение 15 мин. Затем органический слой отделяют и промывают водой (300 мл), насыщенным раствором соли (300 мл), сушат над Na2SO4 (50 г), фильтруют и растворитель удаляют в вакууме, получая указанное в заголовке соединение в виде светло-желтого масла (40 г, выход 77%, чистота ˜95%).
Стадия В - Получение 8-(N-трет-бутоксикарбонил-N-метиламино)октилового эфира толуол-4-сульфокислоты
В 250-мл колбе раствор продукта со стадии А (5,2 г, 20 ммоль) и DABCO (3,13 г, 2,8 ммоль) в MTBE (30 мл) охлаждают до около 10°С и добавляют раствор п-толуолсульфонилхлорида (4,2 г, 22 ммоль) в MTBE (20 мл), поддерживая температуру реакционной смеси при 20°С или ниже. Затем полученный раствор перемешивают при комнатной температуре в течение 2 ч. Добавляют воду (100 мл) и смесь перемешивают в течение 15 минут. Органический слой отделяют, промывают водой (100 мл), насыщенным раствором соли (100 мл) и затем концентрируют дистилляцией, получая указанное в заголовке соединение в виде масла.
Стадия С - Получение 2-{(S)-1-[8-(N-трет-бутоксикарбонил-N-метиламино)октил]пирролидин-3-ил}-2,2-дифенилацетамида
В 500-мл колбу добавляют продукт со стадии В (17,68 г, 43 ммоль), продукт из Получения 1 (12 г, 43 ммоль), диизопропилэтиламин (16,55 г, 128 ммоль) и ацетонитрил (100 мл). Полученную смесь перемешивают при температуре 60-65°С в течение 5-7 часов и затем охлаждают до комнатной температуры. Растворитель выпаривают в вакууме и добавляют изопропилацетат (100 мл) для растворения остатка. Полученный раствор промывают водой (100 мл), насыщенным раствором NaHCO3 (100 мл), насыщенным раствором соли (100 мл), сушат над MgSO4 (5 г) и фильтруют, получая оранжевый раствор.
Слой силикагеля (115 г, 280-400 меш) предварительно обрабатывают 400 мл изопропилацетата, содержащего 1% триэтиламина, а затем 250 мл изопропилацетата (слой силикагеля имеет размеры, диаметр около 6,4 см и высоту около 10,2 см). Вышеупомянутый фильтрат (около 150 мл, по объему) нагружают на предварительно обработанный слой силикагеля и элюируют изопропилацетатом (400 мл) и затем 20% изопропанолом в изопропилацетате (1000 мл). Фракции продукта объединяют и концентрируют, получая указанное в заголовке соединение в виде масла (17,16 г, выход 77%, чистота 97%).
Стадия D - Получение соли нафталин-1,5-дисульфокислоты 2-[(S)-1-(8-метиламинооктил)пирролидин-3-ил]-2,2-дифенилацетамида
В 1000-мл колбу добавляют продукт со стадии С (9,88 г, 19 ммоль), тетрагидрат 1,5-нафталиндисульфокислоты (13,69 г, 38 ммоль) и изопропанол, содержащий 3% воды (497 мл). Эту смесь нагревают до 85°С в течение от 3 до 5 часов, затем медленно охлаждают до комнатной температуры на протяжении 4-часового периода и затем перемешивают при комнатной температуре в течение от 12 до 24 часов. Полученное твердое вещество отфильтровывают и промывают изопропанолом, содержащим 3% воды, по объему, (400 мл) и сушат в вакууме в течение от 10 до 15 часов при комнатной температуре, получая указанное в заголовке соединение в виде кристаллического твердого вещества (12,59 г, выход 95%, чистота ˜99%).
При желании, указанную соль можно дополнительно очистить, следуя нижеследующему способу:
В 1-литровую колбу добавляют соль нафталин-1,5-дисульфокислоты 2-[(S)-1-(8-метиламинооктил)пирролидин-3-ил]-2,2-дифенилацетамида (21,4 г, 30,1 ммоль) и изопропанол, содержащий 3% воды, по объему, (637 мл). Полученную суспензию перемешивают при 80°С в течение 2 часов и затем медленно охлаждают до комнатной температуры и затем перемешивают при комнатной температуре в течение 12 часов. Полученную кристаллическую соль отфильтровывают, промывают изопропанолом (600 мл) и затем сушат в вакууме и в атмосфере азота в течение 16 часов при комнатной температуре, получая указанное в заголовке соединение в виде белого кристаллического твердого вещества (20,4 г, выход 96%).
Пример 2
Синтез 2-[(S)-1-(8-изопропиламинооктил)пирролидин-3-ил]-2,2-дифенилацетамида
Стадия А - Получение 2-[(S)-1-(8-гидроксиоктил)пирролидин-3-ил]-2,2-дифенилацетамида
8-Бром-1-октанол (2,51 г, 12 ммоль) в ацетонитриле (10 мл) добавляют к перемешиваемому раствору 2,2-дифенил-2-(S)-пирролидин-3-илацетамида (2,8 г, 10 ммоль) и триэтиламина (4,27 мл, 30 ммоль) в ацетонитриле (90 мл) при 40°С. Реакционную смесь нагревают при 55°С в течение 16 часов и затем охлаждают до температуры окружающей среды. Затем растворитель удаляют при пониженном давлении. Неочищенный остаток растворяют в этилацетате (100 мл) и органическую фазу промывают насыщенным водным бикарбонатом натрия (50 мл), сушат над сульфатом магния, фильтруют и концентрируют при пониженном давлении. Неочищенный продукт очищают флэш-хроматографией (элюент: DCM/MeOH/NH4OH=90/9/1), получая 1,8 г указанного в заголовке промежуточного соединения в виде масла (выход 44%).
Стадия В - Получение 2-[(S)-1-(8-оксооктил)пирролидин-3-ил]-2,2-дифенилацетамида
Диметилсульфоксид (1,57 мл, 22,1 ммоль), а затем диизопропилэтиламин (3,85 мл, 22,1 ммоль) добавляют к перемешиваемому раствору промежуточного соединения со стадии А (1,8 г, 4,4 ммоль) в дихлорметане (44 мл) при 0°С. Реакционную смесь перемешивают при -10°С в течение 15 мин и затем добавляют комплекс триоксид серы-пиридин (2,1 г, 13,2 ммоль). Реакционную смесь перемешивают в течение еще 2 ч. при -10°С. Добавляют воду (50 мл) и DCM (50 мл) и органический слой отделяют и промывают насыщенным водным бикарбонатом натрия (2x30 мл), насыщенным водным раствором сульфата меди (II) (2x15 мл) и насыщенным раствором соли (30 мл). Затем органический слой сушат над сульфатом магния и концентрируют при пониженном давлении, получая 1,5 г указанного в заголовке промежуточного соединения в виде масла (выход 84%).
Стадия С - Получение 2-[(S)-1-(8-изопропиламинооктил)пирролидин-3-ил]-2,2-дифенилацетамида
Промежуточное соединение со стадии В (40,6 мг, 0,1 ммоль) и изопропиламин (10,2 мкл, 0,12 ммоль) в 1,2-дихлорэтане (1 мл) перемешивают при комнатной температуре в течение 1 ч и затем добавляют натрийтриацетоксиборгидрид (35,1 мг, 1,5 ммоль). Реакционную смесь перемешивают в течение 16 ч и затем растворитель удаляют при пониженном давлении. Остаток очищают ВЭЖХ, получая указанное в заголовке соединение в виде его соли бис-трифторуксусной кислоты.
Данные анализа: МС m/z 450,3 (MH+).
Используя описанные здесь способы и соответствующие исходные продукты, получают соединения, представленные в таблице IV:
‡ NA - Не имеется в наличии.
Кроме того, используя способы, описанные здесь, и соответствующие исходные продукты, можно получить соединения, представленные в таблице V.
ил)-2,2-дифенилацетамид
Сравнительный пример А
Синтез 2-[(S)-1-(8-диметиламинооктил)пирролидин-3-ил]-2,2-дифенилацетамида
50-мл сухую круглодонную колбу загружают 2,2-дифенил-2-(S)-пирролидин-3-илацетамидом (200 мг, 0,714 ммоль) и хлороформом (20 мл) и затем продувают азотом. Добавляют диметиламин (535 мкл, 1,071 ммоль), а затем по каплям добавляют 1,8-дибромоктан (131 мкл, 0,714 ммоль). Реакционную смесь нагревают до 50°С и перемешивают в течение примерно 60 часов. Желтую гомогенную смесь охлаждают до комнатной температуры и экстрагируют 1,0 М водным хлористым водородом, который затем промывают свежим хлороформом. К кислому водному слою добавляют этилацетат и смесь подщелачивают до рН 13 10,0 М водным гидроксидом натрия. Щелочной водный слой затем экстрагируют дополнительным этилацетатом (2x15 мл). Затем объединенные органические слои промывают насыщенным водным хлоридом натрия, сушат над сульфатом натрия, фильтруют и упаривают, получая неочищенный продукт. Неочищенный продукт (223,0 мг) очищают препаративной ВЭЖХ и лиофилизуют, получая указанное в заголовке соединение в виде его бис(трифторацетатной) соли, которая представляет собой белое гигроскопичное твердое вещество.
Данные анализа: МС m/z 436,4 (C28H41N3O+H)+; выч. 463,3.
Сравнительный пример В
Синтез 2-[(S)-1-(9-диметиламинононил)пирролидин-3-ил]-2,2-дифенилацетамида
Используя описанные здесь способы и соответствующие исходные продукты, получают указанное в заголовке соединение в виде его бис(трифторацетатной) соли, которая представляет собой белое гигроскопичное твердое вещество.
Данные анализа: МС m/z 450,4 (C29H43N3O+Н)+; выч. 450,3.
Сравнительный пример С
Синтез 2-{(S)-1-[8-N-(2-гидроксиэтил)-N-метиламинононил]пирролидин-3-ил}-2,2-дифенилацетамида
Используя описанные здесь способы и соответствующие исходные продукты, получают указанное в заголовке соединение в виде его бис(трифторацетатной) соли, которая представляет собой белое гигроскопичное твердое вещество.
Данные анализа: МС m/z 480,2; выч. 480,4.
Сравнительный пример D
Синтез 2-[(S)-1-(8-аминооктил)пирролидин-3-ил]-2,2-дифенилацетамида
Стадия А - Получение 2-[(S)-1-(8-бромоктил)пирролидин-3-ил]-2,2-дифенилацетамида
К раствору 2,2-дифенил-2-(S)-пирролидин-3-илацетамида (1,2 г, 0,004 моль) и диизопропилэтиламина (0,74 мл, 0,004 моль) в смеси 1:1 (об./об.) ацетона и ДМФА (20 мл) добавляют 1,8-дибромоктан (0,99 мл, 0,005 моль). Смесь нагревают до 40°С в течение пяти часов и затем концентрируют досуха и разбавляют дихлорметаном (20 мл). Полученную смесь промывают насыщенным бикарбонатом натрия (2x20 мл), насыщенным раствором соли (1x20 мл), сушат над сульфатом магния, фильтруют и концентрируют. Остаток очищают хроматографией на силикагеле, элюируя смесью 5% метанол/дихлорметан, получая 315 мг указанного в заголовке промежуточного соединения в виде белого твердого вещества (выход 15%).
Данные анализа: МС m/z 472,5 (MH+); выч. 472,2.
Стадия В - Получение 2-[(S)-1-(8-ди-трет-ВОС-аминооктил)пирролидин-3-ил]-2,2-дифенилацетамида
К раствору ди-трет-бутилиминодикарбоксилата (61 мг, 0,28 ммоль) в 5 мл ДМФА при -10°С добавляют гидрид натрия (11 мг, 0,28 ммоль; 60% в минеральном масле). Раствору дают возможность медленно нагреваться до комнатной температуры и после перемешивания в течение 2 часов добавляют промежуточное соединение со стадии А (0,095 мг, 0,20 моль) в диметилформамиде (5 мл). Затем реакционной смеси дают возможность перемешиваться при комнатной температуре на протяжении ночи и затем смесь концентрируют в вакууме, разбавляют 10 мл дихлорметана и эту смесь промывают насыщенным бикарбонатом натрия (2x10 мл), насыщенным раствором соли (1х110 мл), сушат над сульфатом магния, фильтруют и концентрируют, получая 120 мг указанного в заголовке промежуточного соединения в виде белого твердого вещества (выход 99%).
Данные анализа: МС m/z 608,8 (MH+); выч. 608,5.
Стадия С - Получение 2-[(S)-1-(8-аминооктил)пирролидин-3-ил]-2,2-дифенилацетамида
К промежуточному соединению со стадии В (120 мг, 0,16 ммоль) добавляют смесь трифторуксусной кислоты (0,08 мл) в дихлорметане (0,720 мл) и реакционную смесь перемешивают при комнатной температуре в течение четырех часов. Затем реакционную смесь концентрируют досуха в вакууме, разбавляют дихлорметаном (10 мл) и медленно добавляют 1н. гидроксид натрия до тех пор, пока рН не достигнет 14. Органический слой отделяют и промывают насыщенным бикарбонатом натрия (2x10 мл), насыщенным раствором соли (1x110 мл), сушат над сульфатом магния, фильтруют, концентрируют. Остаток очищают препаративной ВЭЖХ, получая 27 мг указанного в заголовке соединения в виде его соли бис-трифторуксусной кислоты, которая представляет собой белое твердое вещество.
Данные анализа: МС m/z 408,6 (MH+); выч. 408,3.
Испытание 1
Анализ связывания меченного радиоактивным изотопом лиганда (радиолиганд)
A. Препараты мембран из клеток, экспрессирующих подтипы мускариновых рецепторов hM1, hM2, hM3 и hM4
Клеточные линии CHO (яичник китайского хомячка), стабильно экспрессирующие клонированные подтипы мускариновых рецепторов человека hM1, hM2, hM3 и hM4, соответственно, культивировали до состояния, близкого к конфлюентности, в среде, состоящей из F-12 HAM, дополненной 10% FBS (фетальная телячья сыворотка) и 250 мкг/мл генетицина. Клетки культивировали в инкубаторе в 5% CO2 при 37°C и извлекали с помощью 2 мм EDTA в dPBS. Клетки собирали 5-минутным центрифугированием при 650 x g, и дебрис (осадок клеток) либо хранили в замороженном состоянии при -80°С, либо сразу получали мембраны. Для получения мембран дебрис ресуспендировали в буфере для лизиса и гомогенезировали с помощью тканевого дезинтегратора Polytron PT-2100 (Kinematica AG; 20 секунд x 2 захода). Неочищенные мембраны центрифугировали при 40000 x g в течение 15 минут при 4°C. Затем осадок мембран ресуспендировали буфером для ресуспендирования и снова гомогенизировали с помощью тканевого дезинтегратора Polytron. Концентрацию белка в суспензии мембран определяли методом, описанным Lowry, О. et al., 1951, Journal of Biochemistry: 193, 265. Все мембраны хранили в замороженном состоянии в аликвотах при -80°C или сразу использовали. Аликвоты полученных мембран рецепторов hM5 приобретали от Perkin Elmer и хранили при -80°C до использования.
B. Метод определения радиолиганд-связывающей способности подтипов мускариновых рецепторов hM1, hM2, hM3, hM4 и hM5
Испытания связывания радиоактивного лиганда осуществляли в 96-луночных титрационных микропланшетах с общим объемом пробы 100 мкл. Мембраны CHO, стабильно экспрессирующие либо мускариновый подтип hM1, hM2, hM3, hM4, либо мускариновый подтип hM5, разбавляли в буфере для анализа до нижеуказанных концентраций конкретного белка-мишени (мкг/лунку): 10 мкг для hM1, 10-15 мкг для hM2, 10-20 мкг для hM3, 10-20 мкг для hM4 и 10-12 мкг для hM5. Мембраны кратковременно (10 секунд) гомогенизировали, используя тканевый дезинтегратор Polytron, до добавления в аналитический планшет. Изучение реакций связывания до насыщения для определения значений KD радиоактивного лиганда выполняли, используя L-[N-метил-3H]скополаминметилхлорид ([3H]-NMS) (TRK666, 84,0 Ки/ммоль, Amersham Pharmacia Biotech, Buckinghamshire, England) при концентрациях в диапазоне от 0,001 нМ до 20 нМ. Анализы, связанные с вытеснением радиолиганда, для определения значений Ki для испытуемых соединений выполняли с [3H]-NMS при концентрации 1 нМ и одиннадцати различных концентрациях испытуемых соединений. Испытуемые соединения сначала растворяли до концентрации 400 мкМ в буфере для разбавления и затем серийно разбавляли 5x буфером для разбавления до конечных концентраций в диапазоне от 10 пМ до 100 мкМ. Порядок и объемы добавления в аналитические планшеты были следующими: 25 мкл меченого лиганда, 25 мкл разбавленного испытуемого соединения и 500 мкл мембран. Аналитические планшеты инкубировали в течение 60 минут при 37°C. Реакции связывания прекращали быстрой фильтрацией через стекловолоконные (GF/B) фильтровальные планшеты (PerkinElmer Inc., Wellesley, MA), предварительно обработанные в 1% BSA. Фильтровальные планшеты промывали три раза буфером для промывки (10 мм HEPES), чтобы удалить несвязанную радиоактивность. Затем планшеты сушили на воздухе и в каждую лунку добавляли 50 мкл сцинтилляционной жидкости Microscint-20 (PerkinElmer Inc., Wellesley, MA). Затем планшеты считывали в сцинтилляционном счетчике PerkinElmer Topcount (PerkinElmer Inc., Wellesley, MA). Данные связывания анализировали методом нелинейной регрессии с помощью пакета программного обеспечения GraphPad Prism (GraphPad Software, Inc., San Diego, CA), используя модель односайтового конкурентного связывания. Значения Кi для испытуемых соединений рассчитывали из наблюдаемых значений IC50 и значения KD радиоактивного лиганда, используя уравнение Cheng-Prusoff (Cheng Y; Prusoff WH. (1973) Biochemical Pharmacology, 22(23):3099-108). Значения Кi преобразовывали в значения pKi, чтобы определить среднее геометрическое и 95% доверительные интервалы. Эти итоговые статистические данные снова преобразовывали в значения Кi для представления результатов испытания.
В этом испытании более низкое значение Кi свидетельствует о том, что испытуемое соединение имеет более высокую аффинность связывания с используемым в испытании рецептором. В этом испытании было установлено, что соединение формулы I имеет значение Кi приблизительно 0,96 нМ для подтипа мускаринового рецептора М3.
Испытуемые соединения, имеющие в этом испытании более низкое значение Кi, имеют более высокую аффинность связывания с мускариновым рецептором. Соединения по данному изобретению, которые были испытаны в данном анализе, имели значение Кi для hM2 в диапазоне от около 200 нМ до меньше чем 1 нМ; обычно в диапазоне от около 100 нМ до меньше чем 1 нМ; и значение Кi для hM3 в диапазоне от около 100 нМ до меньше чем 1 нМ; обычно в диапазоне от около 50 нМ до меньше чем 1 нМ. Например, соединения примеров 1-11, 14, 26, 27 и 39 имели значение Кi для hM3 меньше чем 50 нМ. Таким образом, соединения по данному изобретению, как было установлено в этом испытании, достаточно убедительно связываются с подтипами рецепторов hM2 и hM3 (т.е. обладают высокой аффинностью к ним).
Дополнительно, аффинность связывания для соединений формулы:
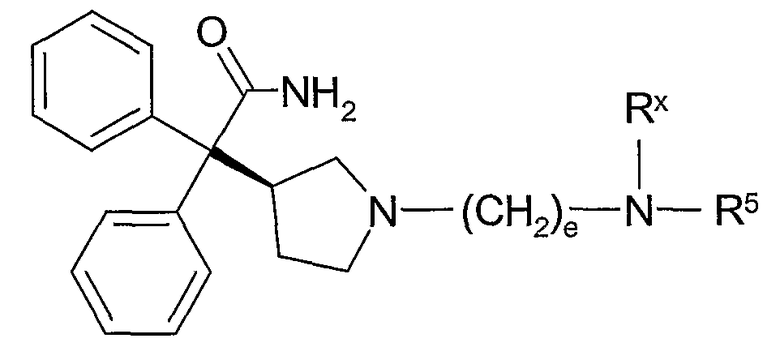
представлена в Таблице VI (где R5, Rх и e такие, как указаны в таблице VI):
Пр. №.
x 39
x 52
Данные в Таблице V наглядно показывают, что замещение концевой аминогруппы дополнительной алкильной группой, такой как метильная, значительно уменьшает аффинность связывания (лиганда) с подтипами рецептора hM2 и hM3. Кроме того, данные в таблице V наглядно показывают, что удаление алкильной группы, такой как метильная, из концевой аминогруппы существенно уменьшает аффинность связывания (лиганда) с подтипами рецептора hM2 и hM3.
Испытание 2
Анализы функциональной эффективности мускариновых рецепторов
А. Блокада агонист-опосредованного ингибирования аккумуляции цАМФ (cAMP)
В этом испытании определяли функциональную эффективность испытуемого соединения, измеряя способность испытуемого соединения блокировать ингибирование оксотреморином форсколин-опосредованной аккумуляции cAMP в клетках CHO-K1, экспрессирующих рецептор hM2.
Анализы cAMP выполняли в формате радиоиммуноанализа, используя the Flashplate Adenylyl Cyclase Activation Assay System 125I-cAMP (NEN SMP004B, PerkinElmer Life Sciences Inc., Boston, MA), в соответствии с инструкциями производителя.
Клетки промывали один раз dPBS и извлекали раствором Трипсин-EDDA (0,05% трипсин/0,53 мМ EDDA), как описано выше в разделе Получения клеточной культуры и мембран. Отдельные (отделенные друг от друга) клетки промывали дважды, центрифугируя при 650 x g в течение пяти минут в 50 мл dPBS. Дебрис затем ресуспендировали в 10 мл dPBS и клетки подсчитывали с помощью счетчика частиц Coulter Zl Dual Particle Counter (Beckman Coulter, Fullerton, CA). Клетки снова центрифугировали при 650 x g в течение пяти минут и вновь ресуспендировали в буфере для стимуляции до концентрации в пробе 1,6x106 - 2,8x106 клеток/мл.
Испытуемое соединение сначала растворяли до концентрации 400 мкМ в буфере для разбавления (dPBS, дополненный 1 мг/мл BSA (0,1%)) и затем серийно разбавляли буфером для разбавления до конечных молярных концентраций в диапазоне от 100 мкМ до 0,1 нМ. Оксотреморин разбавляли подобным образом.
Для измерения ингибирования оксотреморином активности аденилилциклазы (AC), 25 мкл форсколина (25 мкМ конечная концентрация разбавления в dPBS), 25 мкл разбавленного оксотреморина и 50 мкл клеток добавляли в лунки для анализа агониста. Для измерения способности испытуемого соединения блокировать оксотреморин-ингибируемую активность AC, 25 мкл форсколина и оксотреморина (25 мкМ и 5 мкМ конечные концентрации, соответственно, разбавленные в dPBS), 25 мкл разбавленного испытуемого соединения и 50 мкл клеток добавляли в оставшиеся лунки для анализа.
Реакционные смеси инкубировали в течение 10 минут при 37°C и реакции прекращали добавлением 100 мкл ледяного буфера для детектирования. Планшеты герметизировали, инкубировали на протяжении ночи при комнатной температуре и на следующее утро подсчитывали с помощью сцинтилляционного счетчика PerkinElmer TopCount (PerkinElmer Inc., Wellesley, MA). Количество полученного cAMP (пмоль/лунку) рассчитывали, исходя из импульсов, наблюдаемых для образцов и стандартов cAMP, как описано в руководстве производителя для пользователя. Данные анализировали методом нелинейной регрессии с помощью пакета программного обеспечения GraphPad Prism (GraphPad Software, Inc., San Diego, CA), используя нелинейную регрессию, уравнение для односайтового конкурентного связывания. Уравнение Cheng-Prusoff использовали для количественного определения Ki, используя EC50 кривой зависимости "концентрация оксотреморина-ответ" и концентрацию оксотреморина в испытании как KD и [L], соответственно. Значения Ki преобразовывали в значения pKi, чтобы определить среднее геометрическое и 95% доверительные интервалы. Эти итоговые статистические данные снова преобразовывали в значения Кi для представления результатов испытания.
В этом испытании более низкое значение Ki свидетельствует о том, что испытуемое соединение имеет более высокую функциональную активность в отношении используемого в испытании рецептора. Соединение примера 1, как было установлено, имеет значение Ki меньше чем 5 нМ для блокады оксотреморин-ингибирования форсколин-опосредованной аккумуляции cAMP в клетках CHO-K1, экспрессирующих рецептор hM2.
B. Блокада агонист-опосредованного связывания GTP[35S]
Во втором функциональном анализе определяли функциональную эффективность испытуемого соединения, измеряя способность соединения блокировать оксотреморин-стимулируемое связывание [35S]GTP в клетках CHO-K1, экспрессирующих рецептор hM2. Во время использования замороженные мембраны оттаивали и затем разбавляли в буфере для анализа с конечной концентрацией ткани-мишени 5-10 мкг белка на лунку. Мембраны кратковременно гомогенизировали, используя тканевый дезинтегратор Polytron PT-2100 и затем добавляли в аналитические планшеты.
В каждом эксперименте определяли значение EC90 (эффективная концентрация для 90% максимального ответа) для стимуляции связывания [35S]GTP агонистом, оксотреморином.
Для определения способности испытуемого соединения ингибировать оксотреморином стимулируемое связывание [35S]GTP, в каждую лунку 96-луночных планшетов добавляли нижеследующее: 25 мкл буфера для анализа с [35S]GTP (0,4 нМ), 25 мкл оксотреморина (EC90) и GTP (3 мкМ), 25 мкл разбавленного испытуемого соединения и 25 мкл клеточных мембран CHO, экспрессирующих рецептор hM2. Затем аналитические планшеты инкубировали при 37°C в течение 60 минут. Аналитические планшеты фильтровали через предварительно обработанные 1% BSA фильтры GF/B, используя 96-луночный харвестер PerkinElmer. Планшеты промывали ледяным буфером для промывки 3 раза, каждый раз в течение 3 секунд, и затем сушили на воздухе или в вакууме. В каждую лунку добавляли сцинтилляционную жидкость Microscint-20 (50 мкл), и каждый планшет герметизировали и подсчитывали радиоактивность с помощью счетчика (topcounter (PerkinElmer)). Данные анализировали методом нелинейной регрессии с помощью пакета программного обеспечения GraphPad Prism (GraphPad Software, Inc., San Diego, CA), используя нелинейную регрессию, уравнение для односайтового конкурентного связывания. Уравнение Cheng-Prusoff использовали для количественного определения Ki, используя значения IC50 кривой зависимости концентрация-ответ для испытуемого соединения и концентрацию оксотреморина в испытании как KD и [L], концентрация лиганда, соответственно.
В этом испытании более низкое значение Ki указывает на то, что испытуемое соединение имеет более высокую функциональную активность на используемом в испытании рецепторе. Соединение примера 1, как было установлено, имеет значение Ki меньше чем 5 нМ для блокады оксотреморином стимулируемого связывания [35S]GTP в клетках CHO-K1, экспрессирующих рецептор hM2.
C. Блокада агонист-опосредованного высвобождения кальция с помощью метода FLIPR
Подтипы мускариновых рецепторов (рецепторы M1, M3 и M5), которые связываются с белками Gq, активируют путь фосфолипазы C (PLC) после связывания агониста с рецептором. В результате, активированная PLC гидролизует фосфатил-инозитолдифосфат (PIP2) в диацилглицерол (DAG) и фосфатидил-1,4,5-трифосфат (IP3), который, в свою очередь, генерирует высвобождение кальция из внутриклеточных запасов, т.е. эндоплазматического и саркоплазматического ретикулума. Анализ FLIPR (Molecular Devices, Sunnyvale, CA) основан на использовании этого увеличения во внутриклеточном кальции с применением чувствительного к кальцию красителя (Fluo-4AM, Molecular Probes, Eugene, OR), который флуоресцирует, когда связывает свободный кальций. Это явление флуоресценции измеряют в реальном масштабе времени с помощью FLIPR, который детектирует изменение флуоресценции в монослое клеток, клонированных человеческими рецепторами M1 и M3 и рецепторами M5 шимпанзе. Антагонистическую эффективность определяют по способности антагонистов ингибировать агонист-опосредованное увеличение во внутриклеточном кальции.
В случае анализа стимуляции (высвобождения) кальция методом FLIPR, клетки CHO, стабильно экспрессирующие рецепторы hM1, hM3 и cM5, засевали в 96-луночные FLIPR планшеты в ночь перед проведением испытания. Выращенные клетки промывали дважды Cellwash (MTX Labsystems, Inc.) буфером для PLIPR (10 мм HEPES, pH 7,4, 2 мм хлорида кальция, 2,5 мм пробенецида в сбалансированном солевом растворе Хэнка (HBSS) без кальция и магния), удаляя среду для роста и оставляя 50 мкл/лунку буфера для FLIPR. Затем клетки инкубировали с 50 мкл/лунку 4 мкМ FLUO-4AM (готовили 2x раствор) в течение 40 минут при 37°C, 5% диоксида углерода. После периода инкубации с красителем клетки промывали два раза буфером для FLIPR, оставляя конечный объем 50 мкл/лунку.
Для определения антагонистической активности сначала устанавливали доза-зависимую стимуляцию высвобождения внутриклеточного кальция для оксотреморина с тем, чтобы позже можно было определить антагонистическую активность в отношении стимуляции оксотреморином при концентрации EC90. Клетки сначала инкубировали с буфером для разбавления соединения в течение 20 минут, с последующим введением агониста, эту операцию выполняли, используя FLIPR. Значение EC90 для оксотреморина получали согласно способу, подробно описанному в нижеследующем разделе измерения и обработки данных FLIPR, в связи с формулой ECF=((F/100-F)^1/H)·EC50. Концентрацию оксотреморина 3 x ECF получали в стимуляционных планшетах с тем, чтобы концентрацию оксотреморина EC90 добавить в каждую лунку планшетов для анализа подавления антагонистом стимуляции высвобождения кальция.
Параметрами, используемыми для FLIPR, были длительность экспозиции 0,4 секунд, мощность лазера 0,5 ватт, длина волны возбуждения 480 нм и длина волны излучения 550 нм. Базовую линию определяли, измеряя изменение флуоресценции в течение 10 секунд до добавления агониста. После стимуляции агонистом PLIPR непрерывно измерял изменение флуоресценции через каждые 0,5-1 секунд в течение 1,5 минут, чтобы уловить максимальное изменение во флуоресценции.
Изменение флуоресценции выражали как максимальную флуоресценцию минус базовая флуоресценция для каждой лунки. Необработанные данные анализировали в зависимости от логарифма концентрации лекарственного средства методом нелинейной регрессии с помощью пакета программного обеспечения GraphPad Prism (GraphPad Software, Inc., San Diego, CA), используя встроенную модель для сигмоидальной кривой (зависимости) доза-ответ. Значения Ki для антагониста определяли согласно Prism, используя значение EC50 для оксотреморина как KD и EC90 оксотреморина для концентрации лиганда согласно уравнению Cheng-Prusoff (Cheng & Prusoff, 1973).
В этом испытании более низкое значение Ki указывает на то, что испытуемое соединение имеет более высокую функциональную активность в отношении используемого в испытании рецептора. Соединение формулы I, как было обнаружено, имеет значение Ki меньше чем 5 нМ для блокады агонист-опосредованного высвобождения кальция в клетках CHO, стабильно экспрессирующих рецептор hM3.
Испытание 3
Определение продолжительности бронхозащиты на модели ацетилхолин-индуцированного бронхостеноза у морских свинок
Этот анализ in vivo использовали для оценки бронхозащитных воздействий испытуемых соединений, проявляющих антагонистическую активность в отношении мускариновых рецепторов.
Группы из шести морских свинок, самцов (Duncan-Hartley (HsdPoc:DH) Harlan, Madison, WI), весом от 250 до 350 г, были индивидуально отождествлены карточками с обозначением клетки. На протяжении всего исследования животные имели доступ к пище и воде, по желанию.
Испытуемое соединение вводили ингаляцией на протяжении 10 минут в камере экспозиционного дозированного распыления лекарственного препарата (R&S Molds, San Carlos, CA). Камеры для дозированного распыления были расположены таким образом, чтобы аэрозоль одновременно поступал в 6 индивидуальных камер из центрального коллектора. Морских свинок подвергали воздействию аэрозоля испытуемого соединения или эксципиента (WFI). Эти аэрозоли генерировали из водных растворов, используя установку для распыления звездообразной формы LC (Model 22F51, PARI Respiratory Equipment, Inc. Midlothian, VA), приводимую в действие смесью газов (CO2=5%, О2=21% и N2=74%) при давлении 22 фунт/дюйм2. Скорость газового потока через распылитель при указанном рабочем давлении составляла приблизительно 3 л/мин. Генерированные аэрозоли подавали в камеры под давлением выше атмосферного. Во время подачи растворов для аэрозолизации никакого разбавления воздухом не использовали. Во время распыления на протяжении 10 минут распылялось приблизительно 1,8 мл раствора. Эту величину определяли гравитометрически, сравнивая массы заполненного распылителя перед и после распыления.
Бронхозащитные действия испытуемого соединения, вводимого ингаляцией, оценивали, используя плетизмографию всего тела (регистрация изменений объема всего тела) через 1,5; 24, 48 и 72 часа после введения дозы.
За сорок пять минут до начала оценки дыхательной функции легких каждую морскую свинку анестизировали внутримышечной инъекцией кетамина (43,75 мг/кг), ксилазина (3,50 мг/кг) и ацепромазина (1,05 мг/кг). После того как место для оперативного вмешательства было выбрито и очищено 70% спиртом, делали 2-3 см срединный разрез вентральной стороны шеи. Затем яремную вену отделяли и подвергали канюлированию полиэтиленовым катетером, заполненным физиологическим раствором (PE-50, Becton Dickinson, Sparks, MD), делая возможным внутривенные вливания ацетилхолина (Ach) (Sigma, St. Louis, MO) в физиологическом растворе. Затем высвобождали трахею и подвергали канюлированию тефлоновой трубкой 14G (#NE-014, Small Parts, Miami Lakes, FL). При необходимости анестезию поддерживали дополнительными внутримышечными инъекциями вышеупомянутой анестезирующей смеси. Глубину анестезии контролировали и корректировали, если животное реагировало на сдавливание его лапки или если частота дыхания была больше чем 100 дыханий/минуту.
Сразу после завершения катетеризаций животное помещали в плетизмограф (#PLY3114, Buxco Electronics, Inc., Sharon, CT) и вставляли пищеводный зонд для измерения (внутриплеврального) давления (PE-160, Becton Dickinson, Sparks, MD), чтобы измерить давление, движущее легкие (включающее легочную вентиляцию) (давление). Тефлоновую трахеальную трубку присоединяли к отверстию плетизмографа, чтобы позволить морской свинке вдыхать воздух из помещения снаружи камеры. Затем камеру герметизировали. Для поддержания температуры тела использовали нагревающую лампу, и чтобы не допустить возникновения коллапса нижних воздухоносных путей и гипервентиляцию легких у животного, легкие морской свинки 3 раза накачивали 4 мл воздуха, используя калибровочный шприц объемом 10 мл (#5520 Series, Hans Rudolph, Kansas City, MO).
Как только было установлено, что базовые значения для растяжимости легких находятся в диапазоне 0,3-0,9 мл/см вод. ст. и для сопротивления дыхательных путей в диапазоне 0,1-0,199 см вод.ст./мл в секунду, начинали оценку дыхательной функции легких. Компьютерная программа измерения показателей функции легких Buxco включала сбор и деривацию значений легочных показателей.
Начало этой программы инициировал экспериментальный протокол и сбор данных. Изменения в объеме за некоторое время, которые происходят внутри плетизмографа с каждым дыханием, измеряли посредством датчика давления Buxco. Интегрируя этот сигнал по времени, оценивали параметры потока (воздуха) для каждого дыхания. Этот сигнал, вместе с изменениями давления, движущего легкие, которые собирали, используя датчик давления Sensym (#TRD4100), соединяли через предусилитель Buxco (MAX 2270) с интерфейсом сбора данных (# SFT3400 и SFT3813). Все другие легочные показатели получали из этих двух исходных данных.
Базовые значения собирали в течение 5 минут и после этого времени морским свинкам вводили провокационную пробу Ach. Ach (Sigma-Aldrich, St. Louis, MO) (0,1 мг/мл), инфузировали внутривенно в течение 1 минуты из насоса-шприца (sp210iw. World Precision Instruments, Inc., Sarasota, FL) в указанных ниже дозах и в предписанные времена от начала эксперимента: 1,9 мкг/минута через 5 минут, 3,8 мкг/минута через 10 минут, 7,5 мкг/минута через 15 минут, 15,0 мкг/минута через 20 минут, 30 мкг/минута через 25 минут и 60 мкг/минута через 30 минут. Если сопротивление дыхательных путей или растяжимость легких не возвращались к базовым значениям через 3 минуты после каждой дозы Ach, легкие морской свинки 3 раза накачивали 4 мл воздуха, используя калибровочный шприц объемом 10 мл. Регистрируемые легочные параметры включали частоту дыхания (дыханий/минуту), растяжимость легких (мл/см вод.ст.) и сопротивление дыхательных путей (см вод.ст./мл в секунду). После завершения измерений показателей функции легких на 35 минуте по данному протоколу, морскую свинку извлекали из плетизмографа и безболезненно умерщвляли асфиксией диоксидом углерода.
Данные оценивали одним или обоими из нижеследующих способов:
(a) Сопротивление дыхательных путей (RL, см вод.ст./мл в секунду) рассчитывали из отношения "изменение в давлении" к "изменение в потоке (воздуха)".
Ответ RL на ACh (60 мкг/мин, ингаляция(IH)) вычисляли для групп, получавших наполнитель и испытуемое соединение. Рассчитывали средний ответ на ACh для животных, обработанных наполнителем, в каждое время до лечения, и использовали для вычисления % подавления ответа на ACh, во время, соответствующее времени до лечения, для каждой дозы испытуемого соединения. Кривые подавления доза-ответ для 'RL' были описаны логистическим уравнением с четырьмя параметрами, используя программное обеспечение GraphPad Prism, версия 3.00 для Windows (GraphPad Software, San Diego, California), чтобы оценить бронхозащитную ID50 (дозу, необходимую для подавления ответа на бронхоконстриктор ACh (60 мкг/мин) на 50%). Используемое уравнение было следующим:
Y=Min+(Max-Min)/(1+10((LOG ID50-X)·Hillslope)),
где X означает логарифм дозы, Y представляет собой ответ (% подавления индуцируемого ACh увеличения в RL). Y начинается в Min и приближается асимптотически к Max по сигмоидальной кривой.
(b) Рассчитывают величину PD2, которую определяют как количество Ach или гистамина, необходимое для того, чтобы вызвать удвоение базового сопротивления дыхательных путей, используя значения сопротивления дыхательных путей, полученные из "потока (воздуха)" и "давления" в диапазоне введения провокационных доз Ach или гистамина, используя нижеследующее уравнение (которое получено из уравнения, используемого для вычисления значений PC20, описанного в American Thoracic Society. Guidelines for methacholine and exercise challenge testing - 1999. Am J Respir Crit Care Med. 2000; 161:309-329):
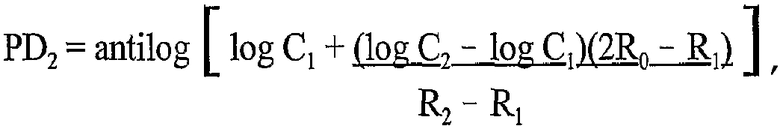
где:
C1 = концентрация Ach или гистамина, предшествующая C2;
C2 = концентрация Ach или гистамина, приводящая к, по крайней мере, 2-кратному увеличению в сопротивлении дыхательных путей (RL);
R0 = базовое значение RL;
R1 = значение RL после C1;
R2 = значение RL после C2.
Эффективную дозу определяют как дозу, которая ограничивала реакцию бронхостеноза на Ach при дозе 50 мкг/мл до удвоения базового сопротивления дыхательных путей (PD2(50)).
Статистический анализ данных выполняли, используя двусторонне ограниченный критерий Стьюдента. P-значение <0,05 считали значимым.
Обычно, в этом испытании испытуемое соединение, имеющее PD2(50) меньше чем около 200 мкг/мл для ACh-индуцируемого бронхостеноза через 1,5 часа после введения дозы, является предпочтительным. Соединение формулы I, как было установлено, имеет PD2(50) меньше чем около 200 мкг/мл для ACh-индуцируемого бронхостеноза через 1,5 часа после введения.
Испытание 4
Ингаляционный тест слюноотделения у морских свинок
Морские свинки (Charles River, Wilmington, MA), весом 200-350 г, были подвергнуты акклиматизации в условиях пребывания внутри собственной колонии в течение, по крайней мере, 3 дней после прибытия. Испытуемое соединение или эксципиент дозировали ингаляцией (IH) на протяжении периода 10 минут в камере дозированного распыления (препарата) круглой формы (R+S Molds, San Carlos, CA). Испытуемые соединения растворяли в стерильной воде и доставляли, используя распылитель, наполненный 5,0 мл раствора для дозирования. Морских свинок удерживали в ингаляционной камере в течение 30 минут. На протяжении этого времени морские свинки были ограничены площадью примерно 100 см2. Это пространство было достаточным для того, чтобы животные могли свободно поворачиваться, самостоятельно перемещаться, и предусматривало возможность чистки, ухода за поверхностью тела. После 20 минут акклиматизации морских свинок подвергали воздействию аэрозоля, генерируемого из распыляющего агрегата звездообразной формы LS (Model 22F51, PARI Respiratory Equipment, Inc. Midlothian, VA), приводимого в действие воздухом помещения при давлении 22 фунт/дюйм2. После завершения распыления состояние морских свинок оценивали через 1,5; 6, 12, 24, 48 или 72 часа после обработки.
За час до испытания морских свинок подвергали анестезии внутримышечной (IM) инъекцией смеси кетамина 43,75 мг/кг, ксилазина 3,5 мг/кг и ацепромазина 1,05 мг/кг при объеме 0,88 мл/кг. Животных размещали брюшной стороной вверх на поверхности с обогреваемым (37°C) покрытием, наклоненной под углом 20 градусов, с запрокинутой назад головой. В рот морской свинки помещали 4-слойную марлевую мягкую прокладку, размером 2x2 дюйма, (тампоны из Nu- марли общего пользования, Johnson and Johnson, Arlington, TX). Через пять минут после этого вводили мускариновый агонист, пилокарпин (3,0 мг/кг, подкожно) и марлевую мягкую прокладку сразу же отбрасывали и заменяли новой предварительно взвешенной марлевой мягкой прокладкой. Слюну собирали в течение 10 минут и по истечении этого времени марлевую прокладку взвешивали и фиксировали разницу в массе, чтобы определить количество накопленной слюны (в мг). Рассчитывали среднее количество слюны, собранной у животных, получавших наполнитель и получавших каждую дозу испытуемого соединения. Считали, что среднее (значение) для группы, получавшей наполнитель, является 100% слюноотделением. Результаты рассчитывали, используя средние результатов (n=3 или больше). Доверительные интервалы (95%) рассчитывали для каждой дозы в каждый момент времени, используя двухсторонний ANOVA (two-way ANOVA). Эта модель представляет собой модифицированную версию способа, описанного в Rechter, "Estimation of anticholinergic drug effects in mice by antagonism against pilocarpine-induced salivation" Ata Pharmacol Toxicol, 1996, 24:243-254.
Рассчитывали среднюю массу слюны у животных, обработанных наполнителем, в каждый момент времени до лечения, и использовали для вычисления % подавления слюноотделения, во время, соответствующее времени до лечения, для каждой дозы. Данные подавления "доза-ответ" были описаны логистическим уравнением с четырьмя параметрами, используя программное обеспечение GraphPad Prism, версия 3.00 для Windows (GraphPad Software, San Diego, California), чтобы оценить ID50 (дозу, необходимую для подавления пилокарпин-вызываемого слюноотделения на 50%) для средства, подавляющего слюноотделение. Используемое уравнение было следующим:
Y=Min+(Max-Min)/(1+10((LOG ID50-X)·Hillslope))
где X означает логарифм дозы, Y представляет собой ответ (% подавления слюноотделения). Y начинается в Min и приближается асимптотически к Max по сигмоидальной кривой.
Отношение ID50 для средства, подавляющего слюноотделения, к бронхозащитной ID50 использовали для вычисления показателя кажущейся селективности испытуемого соединения по отношению к легким. Обычно соединения, имеющие показатель кажущейся селективности испытуемого соединения по отношению к легким больше чем около 5, являются предпочтительными. В этом испытании соединение формулы I имело показатель кажущейся селективности испытуемого соединения по отношению к легким больше чем около 5.
Испытание 5
Метахолин-индуцируемые депрессорные ответы у находящихся в сознании морских свинок
В этих исследованиях использовали здоровых, взрослых морских свинок-самцов линии Sprague-Dawley (Harlan, Indianapolis, IN), весом от 200 до 300 г. Под анестезией изофлураном (под ее действием) животных снабжали обычными катетерами (PE-50 трубка) через сонную артерию и яремную вену. Катетеры были выведены наружу, используя подкожный туннель в подлопаточную область. Все хирургические разрезы зашивали шелковой нитью 4-0 Ethicon и катетеры блокировали гепарином (1000 единиц/мл). По окончании хирургического вмешательства каждому животному вводили физиологический раствор (3 мл, подкожно), а также бупренорфин (0,05 мг/кг, внутримышечно). Животным предоставляли возможность прийти в себя на электрогрелке до того, как возвратить их в помещение содержания.
Приблизительно после 18-20 часов после оперативного вмешательства животных взвешивали и катетер через сонную артерию соединяли с датчиком для регистрации артериального давления. Артериальное давление и частоту сердечных сокращений фиксировали, используя систему сбора данных Biopac MP-100. Животным предоставляли возможность акклиматизироваться и стабилизировать свое состояние в течение 20 минут.
Каждому животному вводили провокационную пробу метилхолина (MCh) (0,3 мг/кг, внутривенно (в.в.)) через линию яремной вены и ответы сердечно-сосудистой системы контролировали на протяжении 10 минут. Затем животных помещали в камеру дозированного распыления (препарата), которую соединяли с распылителем, содержащим раствор испытуемого соединения или наполнителя. Раствор распыляли в течение 10 минут, используя газовую смесь из вдыхаемого воздуха и 5% диоксида углерода, со скоростью потока 3 литра/минуту. Затем животных удаляли из камеры дозированного распыления (препарата) и возвращали в их соответствующие клетки. Через 1,5 и 24 часа после дозирования животным вновь вводили провокационную дозу MCh (0,3 мг/кг, в.в.) и фиксировали гемодинамический ответ. После этого животных подвергали эвтаназии пентобарбиталом натрия (150 мг/кг, в.в.).
MCh вызывает снижение среднего артериального давления (MAP) и замедление сердечных сокращений (брадикардия). Максимальное уменьшение, исходя из базового, в MAP (депрессорные ответы) измеряли для каждого введения провокационной дозы MCh (до и после дозирования IH). Изменения в брадикардических симптомах не использовали для анализа, поскольку эти ответы не были устойчивыми и воспроизводимыми. Воздействие лечения на ответы MCh выражали как % ингибирования (среднее+/-ст.ош.ср.) контрольных депрессорных ответов. Двухсторонний тест ANOVA с соответствующей обработкой данных использовали для тестирования влияния лечения и времени предварительного лечения. Депрессорные ответы на MCh относительно не изменялись через 1,5 и 24 часа после дозирования ингаляцией наполнителя.
Отношение ID50 антидепрессора к бронхозащитной ID50 использовали для вычисления кажущейся селективности испытуемого соединения по отношению к легким. Обычно соединения, имеющие показатель кажущейся селективности испытуемого соединения по отношению к легким больше чем около 5, являются предпочтительными. В этом испытании соединение формулы I имело показатель кажущейся селективности испытуемого соединения по отношению к легким больше чем 5.
Хотя настоящее изобретение было описано выше, ссылаясь на его конкретные аспекты или конкретные варианты воплощения, для специалистов в данной области, очевидно, что в него могут быть внесены различные изменения или использованы эквиваленты, не выходящие за рамки существа и объема изобретения. Кроме того, в степени, разрешенной соответствующим (действующим) статусом патента и регламетированием порядка его выдачи, все публикации, патенты и патентные заявки, цитируемые здесь, входят в настоящее описание в полном объеме посредством ссылки в такой же степени, как если бы каждый документ был индивидуально включен в него посредством ссылки.
| название | год | авторы | номер документа |
|---|---|---|---|
| ЧЕТВЕРТИЧНЫЕ АММОНИЕВЫЕ ДИФЕНИЛМЕТИЛСОЕДИНЕНИЯ, ПРИМЕНИМЫЕ В КАЧЕСТВЕ АНТАГОНИСТОВ МУСКАРИНОВЫХ РЕЦЕПТОРОВ | 2008 |
|
RU2452728C2 |
| ГУАНИДИНСОДЕРЖАЩИЕ СОЕДИНЕНИЯ, ПРИМЕНИМЫЕ В КАЧЕСТВЕ АНТАГОНИСТОВ МУСКАРИНОВЫХ РЕЦЕПТОРОВ | 2008 |
|
RU2480458C2 |
| БИФЕНИЛЬНЫЕ СОЕДИНЕНИЯ, ПОЛЕЗНЫЕ В КАЧЕСТВЕ АНТАГОНИСТОВ МУСКАРИНОВЫХ РЕЦЕПТОРОВ | 2005 |
|
RU2366656C2 |
| ПРОИЗВОДНЫЕ БИФЕНИЛА | 2004 |
|
RU2330841C2 |
| ПРОИЗВОДНЫЕ ПИРРОЛИДИНИЯ В КАЧЕСТВЕ МУСКАРИНОВЫХ РЕЦЕПТОРОВ М3 | 2005 |
|
RU2412183C2 |
| ПРОИЗВОДНЫЕ 4-ПИПЕРИДИНИЛАЛКИЛАМИНА И ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ, ОБЛАДАЮЩАЯ АНТАГОНИСТИЧЕСКИМ ДЕЙСТВИЕМ В ОТНОШЕНИИ МУСКАРИНОВЫХ РЕЦЕПТОРОВ | 2002 |
|
RU2300524C2 |
| ПРОИЗВОДНЫЕ БИЦИКЛО[2,2,1]ГЕПТ-7-ИЛАМИНА И ИХ ПРИМЕНЕНИЯ | 2006 |
|
RU2442771C2 |
| КОМПОЗИЦИИ И СПОСОБЫ ДЛЯ ЛЕЧЕНИЯ КОГНИТИВНЫХ РАССТРОЙСТВ | 2005 |
|
RU2420318C2 |
| ПРОИЗВОДНЫЕ ПИПЕРАЗИНИЛ ПИРИМИДИНА, СПОСОБ ИХ ПОЛУЧЕНИЯ И ИХ ПРИМЕНЕНИЕ | 2013 |
|
RU2608315C2 |
| ЗАМЕЩЕННЫЕ 1-АМИНОАЛКИЛЛАКТАМЫ, ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ НА ИХ ОСНОВЕ И СПОСОБ ИХ ПОЛУЧЕНИЯ | 2001 |
|
RU2243222C2 |
Настоящая группа изобретений может найти применение в медицине и относится к биологически активным соединениям, охарактеризованным формулой (I), их фармацевтически приемлемым солям, сольватам и стереоизомерам, фармацевтическим композициям, содержащим указанные соединения, их способу получения, промежуточным соединениям, используемым для получения соединений формулы (I), и способам лечения болезненных состояний, опосредованных мускариновыми рецепторами с использованием указанных соединений.
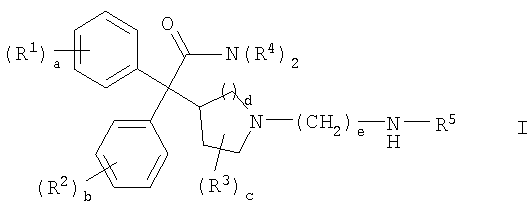
где R4 представляет собой водород; R5 выбран из С1-6алкила, С3-6циклоалкила и -CH2-R8; где алкильная группа необязательно замещена -ОН или 1-5 фтор-заместителями; R8 выбран из С3-5циклоалкила или фенила; a, b и с равны 0; d равно 1; е равно 8 или 9. Технический результат - получение новых биологически активных соединений для лечения ряда заболеваний, опосредованных мускариновыми рецепторами. 10 н.п. и 9 з.п. ф-лы, 15 табл.
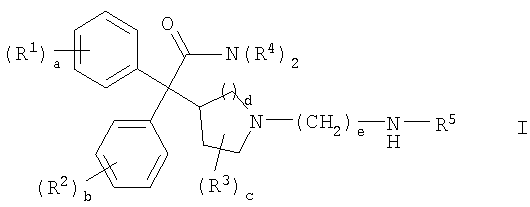
где R4 представляет собой водород;
R5 выбран из C1-6алкила, С3-6циклоалкила и -CH2-R8; где алкильная группа необязательно замещена -ОН или 1-5 фтор-заместителями; R8 выбран из С3-6циклоалкила или фенила;
a, b и с равны 0; d равно 1; е равно 8 или 9;
или его фармацевтически приемлемая соль, или его сольват, или его стереоизомер.
2-[(S)-1-(8-метиламинооктил)пирролидин-3-ил]-2,2-дифенилацетамида;
2-[(S)-1-(8-изопропиламинооктил)пирролидин-3-ил]-2,2-дифенилацетамида;
2-[(S)-1-(8-проп-1-иламинооктил)пирролидин-3-ил]-2,2-дифенилацетамида;
2-[(S)-1-(8-циклопропиламинооктил)пирролидин-3-ил]-2,2-дифенилацетамида;
2-[(S)-1-(8-циклобутиламинооктил)пирролидин-3-ил]-2,2-дифенилацетамида;
2-[(S)-1-(8-циклопентиламинооктил)пирролидин]-3-ил]-2,2-дифенилацетамида;
2-[(S)-1-(8-этиламинооктил)пирролидин-3-ил]-2,2-дифенилацетамида;
2-{(S)-1-[8-(2-гидроксиэтил)аминооктил]пирролидин-3-ил}-2,2-дифенилацетамида;
2-{(S)-1-[8-(R)-(1-гидроксипроп-2-ил)аминооктил]пирролидин-3-ил}-2,2-дифенилацетамида;
2-{(S)-1-[8-(1-гидроксипроп-2-ил)аминооктил]пирролидин-3-ил}-2,2-дифенилацетамида;
2-{(S)-1-[8-(5)-(1-гидроксипроп-2-ил)аминооктил]пирролидин-3-ил}-2,2-дифенилацетамида;
2-{(S)-1-[8-(2,2,2-трифторэтил)аминооктил]пирролидин-3-ил}-2,2-дифенилацетамида;
2-[(S)-1-(8-бензиламинооктил)пирролидин-3-ил]-2,2-дифенилацетамида;
2-[(R)-1-(8-метиламинооктил)пирролидин-3-ил]-2,2-дифенилацетамида;
2-[(R)-1-(8-изопропиламинооктил)пирролидин-3-ил]-2,2-дифенилацетамида;
2-[(R)-1-(8-проп-1-иламинооктил)пирролидин-3-ил]-2,2-дифенилацетамида;
2-[(R)-1-(8-циклопропиламинооктил)пирролидин-3-ил]-2,2-дифенилацетамида;
2-[(R)-1-(8-циклобутиламинооктил)пирролидин-3-ил]-2,2-дифенилацетамида;
2-[(R)-1-(8-циклопентиламинооктил)пирролидин-3-ил]-2,2-дифенилацетамида;
2-[(R)-1-(8-этиламинооктил)пирролидин-3-ил]-2,2-дифенилацетамида;
2-{(R)-1-[8-(2-гидроксиэтил)аминооктил]пирролидин-3-ил}-2,2-дифенилацетамида;
2-{(R)-1-[8-(R)-(1-гидроксипроп-2-ил)аминооктил]пирролидин-3-ил}-2,2-дифенилацетамида;
2-{(R)-1-[8-(1-гидроксипроп-2-ил)аминооктил]пирролидин-3-ил}-2,2-дифенилацетамида;
2-{(R)-1-[8-(S)-(1-гидроксипроп-2-ил)аминооктил]пирролидин-3-ил}-2,2-дифенилацетамида;
2-{(R)-1-[8-(2,2,2-трифторэтил)аминооктил]пирролидин-3-ил}-2,2-дифенилацетамида;
2-[(R)-1-(8-бензиламинооктил)пирролидин-3-ил]-2,2-дифенилацетамида;
2-[(S)-1-(9-метиламинононил)пирролидин-3-ил]-2,2-дифенилацетамида;
2-[(S)-1-(9-изопропиламинононил)пирролидин-3-ил]-2,2-дифенилацетамида;
2-[(S)-1-(9-проп-1-иламинононил)пирролидин-3-ил]-2,2-дифенилацетамида;
2-[(S)-1-(9-циклопропиламинононил)пирролидин-3-ил]-2,2-дифенилацетамида;
2-[(S)-1-(9-циклобутиламинононил)пирролидин-3-ил]-2,2-дифенилацетамида;
2-[(S)-1-(9-этиламинононил)пирролидин-3-ил]-2,2-дифенилацетамида;
2-{(S)-1-[9-(2-гидроксиэтил)аминононил]пирролидин-3-ил}-2,2-дифенилацетамида;
2-{(S)-1-[9-(R)-(1-гидроксипроп-2-ил)аминононил]пирролидин-3-ил}-2,2-дифенилацетамида;
2-{(S)-1-[9-(1-гидроксипроп-2-ил)аминононил]пирролидин-3-ил}-2,2-дифенилацетамида;
2-{(S)-1-[9-(S)-(1-гидроксипроп-2-ил)аминононил]пирролидин-3-ил}-2,2-дифенилацетамида;
2-{(S)-1-[9-(2,2,2-трифторэтил)аминононил]пирролидин-3-ил}-2,2-дифенилацетамида;
2-[(S)-1-(9-бензиламинононил)пирролидин-3-ил]-2,2-дифенилацетамида;
2-[(R)-1-(9-метиламинононил)пирролидин-3-ил]-2,2-дифенилацетамид;
2-[(R)-1-(9-изопропиламинононил)пирролидин-3-ил]-2,2-дифенилацетамида;
2-[(R)-1-(9-проп-1-иламинононил)пирролидин-3-ил]-2,2-дифенилацетамида;
2-[(R)-1-(9-циклопропиламинононил)пирролидин-3-ил]-2,2-дифенилацетамида;
2-[(R)-1-(9-циклобутиламинононил)пирролидин-3-ил]-2,2-дифенилацетамида;
2-[(R)-1-(9-циклопентиламинононил)пирролидин-3-ил]-2,2-дифенилацетамида;
2-[(R)-1-(9-этиламинононил)пирролидин-3-ил]-2,2-дифенилацетамида;
2-{(R)-1-[9-(2-гидроксиэтил)аминононил]пирролидин-3-ил}-2,2-дифенилацетамида;
2-{(R)-1-[9-(R)-(1-гидроксипроп-2-ил)аминононил]пирролидин-3-ил}-2,2-дифенилацетамида;
2-{(R)-1-[9-(1-гидроксипроп-2-ил)аминононил]пирролидин-3-ил}-2,2-дифенилацетамида;
2-{(R)-1-[9-(S)-(1-гидроксипроп-2-ил)аминононил]пирролидин-3-ил}-2,2-дифенилацетамида;
2-{(R)-1-[9-(2,2,2-трифторэтил)аминононил]пирролидин-3-ил}-2,2-дифенилацетамида;
2-[(R)-1-(9-бензиламинононил)пирролидин-3-ил]-2,2-дифенилацетамида;
2-[1-(8-метиламинооктил)пирролидин-3-ил]-2,2-дифенилацетамида;
или их фармацевтически приемлемой соли, или их сольвата.
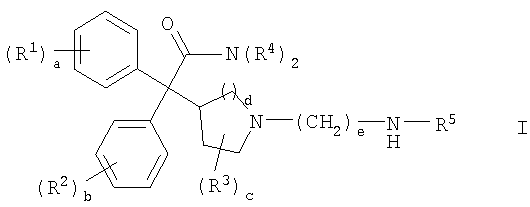
или его соли, или его сольвата, или его стереоизомера;
где способ включает:
а) взаимодействие соединения формулы V
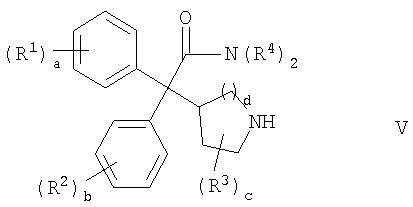
с соединением формулы VIII:
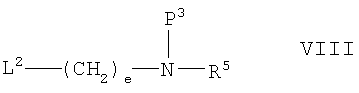
где L2 представляет собой удаляемую группу, предпочтительно хлор, бром, йод, ацетокси, трифторацетокси, тозил, мезил, брозил, нозил;
Р3 представляет собой амино-защитную группу, предпочтительно бензил, трет-бутоксикарбонил, бензилоксикарбонил, 9-флуоренилметоксикарбонил, трет-бутилдиметилсилил, с получением промежуточного соединения;
b) удаление защитной группы Р3 у промежуточного соединения, полученного на стадии (а), предпочтительно гидрогенолизом в присутствии палладия, если Р3 представляет собой бензил; или обработкой фармацевтически приемлемой кислотой, если Р3 представляет собой трет-бутоксикарбонил, бензилоксикарбонил, 9-флуоренилметоксикарбонил; или триэтиламина тригидрофторидом, если Р3 представляет собой трет-бутилдиметилсилил;
c) при необходимости дополнительное взаимодействие продукта стадии (b) с фармацевтически приемлемой кислотой с получением фармацевтически приемлемой соли соединения формулы I;
где R4 представляет собой водород; R5 выбран из C1-6алкила, С3-6циклоалкила и -CH2-R8; где алкильная группа необязательно замещена -ОН или 1-5 фтор-заместителями; R8 выбран из С3-6циклоалкила или фенила;
а, b и с равны 0; d равно 1; е равно 8 или 9.
фармацевтически приемлемой соли соединения формулы I.
или его сольвата, или его стереоизомера;
где способ включает взаимодействие соединения формулы IX:
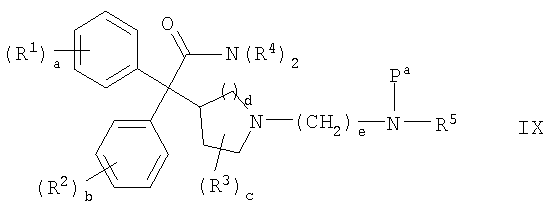
где Рa представляет собой неустойчивую в кислоте амино-защитную группу, предпочтительно трет-бутоксикарбонил, п-метоксибензилоксикарбонил, бензилоксикарбонил, 9-флуоренилметоксикарбонил,
с фармацевтически приемлемой кислотой с получением фармацевтически приемлемой соли соединения формулы I;
где R4 представляет собой водород; R5 выбран из C1-6алкила, С3-6циклоалкила и -СН2-R8; где алкильная группа необязательно замещена -ОН или 1-5 фтор-заместителями; R8 выбран из С3-6циклоалкила или фенила;
a, b и с равны 0; d равно 1; е равно 8 или 9.
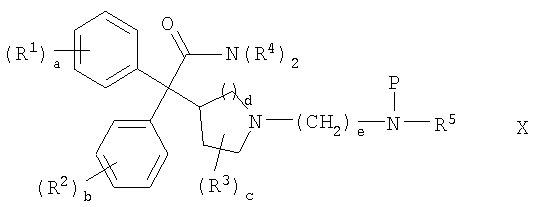
где R4 представляет собой водород; R5 выбран из C1-6алкила, С3-6циклоалкила и -СН2-R8; где алкильная группа необязательно замещена -ОН или 1-5 фтор-заместителями; R8 выбран из С3-5циклоалкила или фенила;
a, b и с равны 0; d равно 1; е равно 8 или 9,
и Р представляет собой бензил, трет-бутоксикарбонил, тритил, бензилоксикарбонил, п-метоксибензилоксикарбонил, 9-флуоренилметоксикарбонил, формил, ацетил, триметилсилил, трет-бутилдиметилсилил;
или его соль, или его сольват, или его стереоизомер.
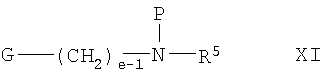
где R5 выбран из C1-6алкила, С3-6циклоалкила и -СН2-R8; где алкильная группа необязательно замещена -ОН или 1-5 фтор-заместителями; R8 выбран из С3-6циклоалкила или фенила; е равно 8 или 9,
Р представляет собой бензил, трет-бутоксикарбонил, тритил, бензилоксикарбонил, п-метоксибензилоксикарбонил, 9-флуоренилметоксикарбонил, формил, ацетил, триметилсилил, трет-бутилдиметилсилил;
G представляет собой -СНО, хлор, бром, йод, ацетокси, трифторацетокси, тозил, мезил, брозил, нозил;
или его соль, или его стереоизомер.
| JP 04090559 (RICOH KK, NEOS KK) 21.05.2004 | |||
| JP 04151168 (RICOH КК, NEOS KK) 25.05.1992 | |||
| СТАЛЬ | 0 |
|
SU388054A1 |
| ГЕТЕРОЦИКЛИЧЕСКИЕ СОЕДИНЕНИЯ, ПРИМЕНИМЫЕ В КАЧЕСТВЕ АЛЛОСТЕРИЧЕСКИХ ЭФФЕКТОРОВ ПРИ МУСКАРИНОВЫХ РЕЦЕПТОРАХ | 1995 |
|
RU2152385C1 |

