Настоящее изобретение относится к производным спиро[2Н-1-бензопиран-2,4’-пиперидина], к фармацевтическим композициям, включающим их, а также к применению указанных производных спиро[2Н-1-бензопиран-2,4’-пиперидина] в терапии.
Простейшая α-аминокислота глицин имеет множество важных функций в центральной нервной системе (ЦНС) млекопитающих. Вместе с γ-аминомасляной кислотой (ГАМК) она является основным пост-синаптическим ингибитором проведения нервного импульса в спинном мозге и в стволе головного мозга, действуя через лиганд, находящийся у входа в ионные каналы. Антагонистом взаимодействия глицина с указанными рецепторами, может быть алкалоид стрихнин. В этой связи указанные рецепторы рассматриваются как рецепторы глицина, “чувствительные к стрихнину”. Глицинергическая нейротрансмиссия важна для осуществления и регуляции зрительной, слуховой и двигательной сигнальных систем. Глицин также является обязательным соагонистом, действующим вместе с глютаматом на рецептор N-метил-D-аспартата (NMDA). Таким образом, глицин функционирует в цепи проведения нервного возбуждения модулируя действие глютамата - основного нейромедиатора в цепи проведения нервного возбуждения в ЦНС. Кроме того, указанная аминокислота играет роль в метаболизме пептидов и белков, включая обмен моноуглеродных соединений.
Регуляция доступности глицина для участия в указанных выше процессах может оказывать влияние на их функционирование и представлять собой средство для лечения множества заболеваний и состояний. Одним из основных процессов, контролирующих концентрацию свободного глицина вблизи “стрихнин-чувствительных” и “стрихнин-нечувствительных” рецепторов глицина, является, кроме метаболизма, функционирование селективных переносчиков с высокой аффинностью к глицину. Указанные белки могут активно ограничивать распространение глицина за пределы непосредственного окружения рецепторов, поддерживая таким образом пространственную и временную точность активации рецептора. Быстрое выделение медиатора в нейрональные или глиальные клетки с помощью переносчика способствует также сохранению глицина для последующего высвобождения.
В результате клонирования переносчиков глицина были выявлены два основных класса: GlyT-1 и GlyT-2. GlyT-1 экспрессируется всем мозгом, при этом наибольший уровень мРНК обнаруживается в каудальных зонах и клетки локализуются преимущественно в глиальной области. Кимом с соавт (Kirn et al., Molecular Pharm. 1994, 45, 608-617) были идентифицированы три изоформы GlyT-1: 1a, 1b и 1с, возникающие в результате дифференциального сплайсинга и использования экзона. Недавно в заявке на европейский патент ЕР 951543 (Allelix Neuroscience, Inc.) были раскрыты клонирование и экспрессия другой человеческой изоформы GlyT-1d.
По данным иммунохимических исследований распределение GlyT-2 четко соответствует локализации ингибитора “стрихнин-чувствительных” рецепторов глицина, в особенности в спинном мозге.
Можно ожидать, что посредством регуляции уровня глицина в синапсах, переносчики GlyT-1 и GlyT-2 будут селективно воздействовать, соответственно, на активность рецепторов NMDA и “стрихнин-чувствительных” рецепторов глицина.
Соединения, которые изменяют функциональную активность переносчиков глицина могут приводить к изменению уровня глицина в тканях, и в этой связи могут использоваться при лечении многих болезненных состояний. Такие болезненные состояния включают те состояния, которые связаны с пониженным или ухудшенным функционированием рецепторов NMDA, а именно:
психоз, депрессия, деменция и другие формы нарушения познавательной способности, такие как расстройство внимания. Рецепторы NMDA участвуют также в патогенезе состояний, возникающих в результате гибели нейрональных клеток и нейродегенерации, таких как, например, кровоизлияние (травма головы), болезнь Альцгеймера, болезнь Паркинсона и болезнь Хантингтона. Повышенное ингибирование глицинергического проведения нервного импульса, возникающее при ингибировании активности GlyT-2 или GlyT-1, может использоваться при лечении мышечной гиперактивности, связанной с мышечной спастичностыо, миоклонией и эпилепсией. Соединения, повышающие уровень глицина в спинном мозге, могут также обладать анальготическими свойствами.
Соединения, ингибирующие транспорт глицина через переносчиков Gly-T1 или Gly-T2, раскрыты в WO 97/45115 (Trophix Pharm. Inc.), в WO 97/45423 (Trophix Pharm. Inc.), в WO 99/34790 (Allelix Neuroscience Inc.) и в WO 00/07978 (Akzo Nodel N.V.) в качестве соединений, полезных при лечении указанных выше неврологических и нейропсихиатрических заболеваний. Исходя из сказанного, имеется потребность в дополнительных соединениях, пригодных для лечения психических и неврологических заболеваний, особенно в таких соединениях, которые обладают селективным фармакологическим профилем.
Было обнаружено, что производные спиро[2Н-1-бензопиран-2,4'-пиперидина], имеющие общую формулу I
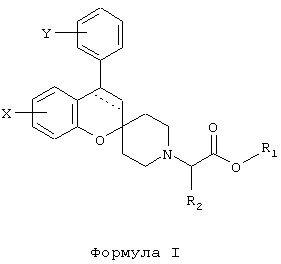
где
пунктирная линия обозначает необязательную связь;
Y обозначает 1-4 заместителя, независимо выбираемые из водорода, галогена, (C1-6)алкила (необязательно замещенного одним или более галогенами), (C1-6)алкилокси (необязательно замещенного одним или более галогенами или (С3-6)циклоалкилом), (С2-6)алкенилокси, (С2-6)алкинилокси, (С3-6)циклоалкилокси, (С6-12)арилокси, (C7-15)арилалкилокси, гетероарилокси, гетероарилалкилокси, SR3, NR3R4, OSO2R5 и NR3SO2R4;
два заместителя Y могут вместе образовывать O-(СН2)n-O или O-(CF2)n-O, где n принимает значение 1 или 2; или Y обозначает конденсированную (C5-6)арильную группу;
Х обозначает 1-3 заместителя, независимо выбираемые из водорода, галогена, гидрокси, (C1-4)алкилокси, SR3, NR3SO2R4 и (C1-4)алкила, необязательно замещенного галогеном;
r1 обозначает водород, (С1-4)алкил или (С6-12)арил;
R2, R3 и R4 обозначают независимо водород или (С1-4)алкил;
r5 обозначает (С1-4)алкил (необязательно замещенный одним или более галогенами) или (С6-12)арил (необязательно замещенный (C1-4)алкилом);
или их фармацевтически приемлемая соль
селективно ингибируют транспорт глицина с помощью переносчика GlyT-1 человека, по сравнению с участием переносчика GlyT-2 человека, и могут применяться при лечении или профилактике шизофрении, депрессии, деменции и других формах нарушения познавательной способности или нейродегенеративных заболеваний, таких как болезнь Альцгеймера, болезнь Паркинсона и болезнь Хантингтона, или мышечной гиперактивности, связанной с мышечной спастичностыо, миоклонией и эпилепсией.
Термин "(C1-6)алкил" в контексте определения формулы I обозначает алкильную группу с разветвленной или прямой цепью, содержащую 1-6 атомов углерода, типа гексила, пентила, неопентила, (2,2-диметилпропила), бутила, изобутила, третичного бутила, пропила, изопропила, этила и метила. Аналогично, термин (C1-4)алкил относится к алкильной группе, содержащей 1-4 атома углерода.
В термине "(C1-6)алкилокси" (C1-6)алкил обозначает разветвленную или неразветвленную алкильную группу, определенную выше. (C1-6)алкилокси группа может быть замещена 1-3 галогенами или (С3-6)циклоалкилом, который означает циклическую алкильную группу, содержащую 3-6 атомов углерода, типа циклопропила, циклобутила, циклопентила или диклогексила. Примеры таких замещенных (C1-6)алкилокси групп включают трифторметилокси и циклопропилметилокси.
Термин “галоген” обозначает F, Cl, Вr или I. В том случае, когда галоген представляет собой заместитель на алкильной группе, предпочтителен F. Предпочтительной галогензамещенной группой является трифторметил.
Термин "(С2-6)алкенил", такой как используемый в термине (С2-6)алкенилокси, означает разветвленную или прямоцепочечную алкенильную группу, содержащую 2-6 атомов углерода, такую как этенил (винил), 2-пропенил (аллил), изопропенил и 2-бутенил.
Термин "(С2-6)алкинил", такой как используемый в термине (С2-6)алкинилокси, означает разветвленную или прямоцепочечную алкинильную группу, содержащую 2-6 атомов углерода, такую как пропаргил.
В термине "(С6-12)арилокси", используемом в определении формулы I, (С6-12)арил означает ароматическую углеводородную группу, содержащую 6-12 атомов углерода, такую как фенил, нафтил, тетрагидронафтил, инденил или бифенил. Указанные ароматические группы могут быть замещены галогеном или (C1-4)алкилом или (C1-4)алкилокси, где (С1-4)алкил имеет указанные ранее значения и может быть замещен галогеном или (C1-4)алкилокси.
Термин "(C7-15)арилалкил", используемый в определении формулы I, означает арилалкильную группу, содержащую от 7 до 15 атомов углерода, причем указанная алкильная группа представляет (C1-6)алкильную группу и указанная арильная группа представляет (С6-12)арил, определенные ранее. Фенил(C1-6)алкильные группы представляют собой предпочтительно арилалкильные группы, такие как бензил.
Термин “гетероарил”, используемый в термине гетероарилокси, означает замещенную или незамещенную ароматическую группу, содержащую 6-12 атомов углерода и включающую по меньшей мере один гетероатом, выбираемый из N, О и S, типа, например, имидазолила, тиенила, бензтиенила, хинолинила и индолила. Гетероарильная группа может нести заместители, приведенные для арильной группы.
Гетероарилалкильные группы представляют собой аналоги (7-15)арилалкильных групп, включающих по меньшей мере один гетероатом, выбранный из N, О и S.
В определении формулы I Y может обозначать конденсированную (C5-6)арильную группу, что означает, что Y представляет собой 5- или 6-членное ароматическое кольцо, конденсированное с бензольным кольцом, к которому Х присоединяется с образованием (C11-12) ароматической кольцевой системы, наподобие нафталинового или инденового кольца.
В дополнение к определению R1, O-R1 группа в формуле I может представлять собой любую другую группу, из которой может быть получена (in vivo) свободная кислота (R1 обозначает водород). В технике известны такие альтернативные предшественники кислоты или пролекарства, такие как сложноэфирные или амидные производные, которые входят в объем настоящего изобретения.
Производные спиро [2Н-1-бензопиран-2,4’-пиперидина] формулы I и их соли могут содержать один или более стереогенных центров и могут существовать в виде стереоизомеров. В объем настоящего изобретения включены указанные стереоизомеры, а также энантиомеры соединений формулы I и их солей, которые по существу свободны от другого энантиомера, то есть содержат его менее, чем на 5%, предпочтительно, менее, чем на 2%, и в особенности, менее, чем на 1%, а также смеси таких стереоизомеров в любых пропорциях, включая рацемические смеси, содержащие по существу равные количества двух энантиомеров.
Предпочтительными являются производные спиро[2Н-1-бензопиран-2,4’-пиперидина] формулы I, в которых пунктирная линия обозначает связь, и более предпочтительны те соединения, в которых, кроме того, R1 и R2, оба, обозначают водород.
Особенно предпочтительные производные спиро[2Н-1-бензопиран-2,4’-пиперидина] согласно настоящему изобретению и их соли соответствуют соединениям формулы I, в которых пунктирная линия обозначает связь, R1 и R2 обозначают водород и Y обозначает заместитель в пара-положении, выбираемый из хлора, брома, (C1-4)алкилокси, (C1-4)алкенилокси, (C1-4)алкинилокси и NR3R4, и 1 или 2 заместителя в мета-положении, выбираемые из галогенов, при этом предпочтительным является фтор. Конкретные примеры предпочтительного замещения для Y включают: 3-фтор-4-метил, 3-фтор-4-хлор, 3-фтор-4-диметиламино и 3,3-дифтор-4-диметиламино. Особенно предпочтительны соединения формулы I, в которых Y обозначает 3-фтор-4-алкилокси, в частности 3-фтор-4-н-пропокси и 3-фтор-4-н-бутокси, и 3,5-дифтор-4-алкилокси.
Производные спиро[2Н-1-бензопиран-2,4’-пиперидина] общей формулы I могут быть получены с помощью последовательности реакций, в которых 2’-гидроксиацетофеноновые производные формулы II, где Х имеет указанные выше значения, используются в качестве исходных материалов, и которые либо легко доступны коммерчески, либо могут быть получены синтезом с помощью методов, известных специалистам в области органического синтеза. Проводят конденсацию 2’-гидроксиацетофеноновых производных II с 1-метил-4-пиперидоном [R обозначает метил; в качестве альтернативы может использоваться 1-бензил-4-пиперидон (R обозначает бензил), бензильная группа зачастую легче удаляется, чем метильная группа (см. схему С)] в метанольном растворе в присутствии пирролидина для получения производных N-метил-спиро[2Н-1-бензопиран-2,4’-пиперидин]-4(3Н)-она, имеющего формулу III, показанную на схеме А:
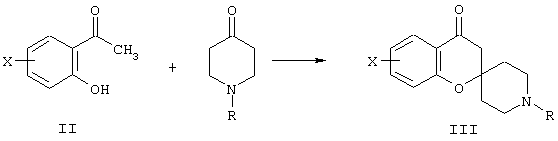
Схема А.
Спиро-кето производные формулы III. затем подвергают обработке, как показано на схеме В, с использованием реагента Гриньяра формулы IV, где Y имеет указанное выше значение, в подходящем растворителе, таком как тетрагидрофуран, с получением после обработки кислотой производных 4-арил-N-метил- или N-бензил-спиро[2Н-1-бензопиран-2,4’-пиперидина] формулы V.

Схема В.
Альтернативно, соединения формулы V могут быть получены при превращении спиро-кето производных формулы III, где R обозначает Н, метил или бензил, в енолтрифлатное производное III’’ с последующим проведением реакции связывания Сузуки с производным фенилбороновой кислоты IV’’ (схема В’’).
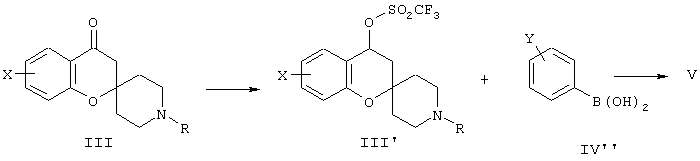
Схема В”.
N-дезалкилирование соединений формулы V с использованием 1-хлорэтилхлорформиата в хлорсодержащем растворителе, таком как 1,3-дихлорпропан или дихлорметан, приводит к получению интермедиата в виде производных 4-арил-спиро[2Н-1-бензопиран-2,4’-пиперидина] формулы VI, которые далее подвергаются алкилированию в реакции с HalCH2R2-COOR1, где R1 может представлять (С1-4)алкил или (С6-12)арил, R2 имеет указанное ранее значение и Hal обозначает галоген, предпочтительно бром, с получением производных 4-арил-спиро[2Н-1-бензопиран-2,4’-пиперидина] формулы I, как показано на схеме С, сложноэфирная группа которого гидролизуется с образованием соединений формулы I, где R1 обозначает водород.
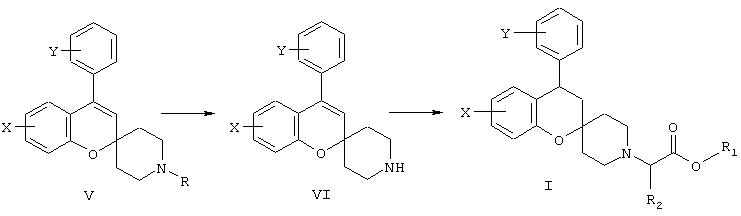
Схема С.
В том случае, когда требуемый реагент Гриньяра формулы IV недоступен коммерчески, его получают из соответствующего бромарена с помощью стандартной процедуры (The Handbook of Grignard Reagents, ed. G.S. Silverman and P.E. Rakita, 1996, Marcel Dekker, New York).
Соединения согласно настоящему изобретению могут использоваться при лечении шизофрении, депрессии, деменции и других форм нарушения познавательной способности, для лечения или профилактики нейродегенеративных расстройств, возникающих после кровоизлияний(инсульта) или травмы головы, для лечения нейродегенеративных заболеваний, таких как болезни Альцгеймера, Паркинсона и Хантингтона, для лечения мышечной гиперактивности, связанной с мышечной спастичностью, миоклонией и эпилепсией, для лечения или профилактики боли, расстройств настроения или расстройств, связанных с нарушением способности к обучению.
Соединения согласно настоящему изобретению могут обладать одним или более стереогенными центрами и могут быть получены в виде чистых стереоизомеров или в виде смеси стереоизомеров. В уровне технике известны методы ассиметричного синтеза, в результате которых получают чистые стереоизомеры и которые включают, например, синтез с хиральной индукцией, энантиоселективный ферментативный гидролиз сложного эфира, кристаллизацию солей, полученных из оптически активных кислот и рацемической смеси, разделение стереоизомеров или энантиомеров с помощью хроматографии на хиральных средах или хроматографии в прямой фазе или в обращенной фазе. Указанные методы описаны, например, в Chirality in Industry (edited by A.N. Collins, G.N. Sheldrake and J. Crosby, 1992; John Wiley).
Фармацевтически приемлемые соли соединений формулы I могут быть получены обработкой свободного основания соединений формулы I минеральной кислотой, такой как хлористоводородная кислота, фосфорная кислота, серная кислота, предпочтительно хлористоводородная кислота, или органической кислотой, такой как, например, аскорбиновая кислота, лимонная кислота, винная кислота, молочная кислота, малеиновая кислота, малоновая кислота, фумаровая кислота, гликолевая кислота, янтарная кислота, пропионовая кислота, уксусная кислота, метансульфоновая кислота и др.
Фармацевтически приемлемые соли соединения формулы I, в которых R1 обозначает водород, могут быть получены обработкой кислоты или цвиттерионной формы указанных соединения органическим основанием или неорганическим основанием, таким как гидроксид натрия, калия или лития.
Другим объектом изобретения являются фармацевтические композиции, включающие производное спиро[2Н-1-бензопиран-2,4’-пиперидина] формулы I или его фармацевтически приемлемые соли в смеси с фармацевтически приемлемыми добавками.
Фармацевтические композиции, применимые согласно настоящему изобретению, включают производное спиро[2Н-1-бензопиран-2,4’-пиперидина] формулы I или его фармацевтически приемлемую соль в смеси с фармацевтически приемлемыми добавками и необязательно с другими терапевтическими средствами. Термин “приемлемый” означает совместимость данного агента с другими ингредиентами композиции и безвредность для реципиента. Композиции могут быть получены с помощью стандартных методик, таких как, например, описаны в стандартном руководстве Gennaro et al., Remington’s Pharmaceutical Science, (18 th ed., Mack Publishing Company, 1990, см. в особенности Part 8: Pharmaceutical Preparations and Their Manufacture).
Композиции включают, например, такие композиции, которые пригодны для перорального, сублингвального, интраназального, подкожного, внутривенного внутримышечного, местного и ректального и др. введения, в виде стандартных дозированных форм.
Для случая перорального введения активный ингредиент может быть представлен в виде дискретных единиц, таких как таблетки, капсулы, порошки, гранулированные средства, растворы и суспензии.
Для случая парентерального введения фармацевтическая композиция согласно настоящему изобретению может быть представлена в виде разовой дозы или в виде контейнеров с многократной дозой, в частности, для инъекции жидкостей в заданных количествах, например, в запаянных флаконах и ампулах, а также может храниться в высушенном замораживанием (лиофилизированном) виде, которое требует лишь добавления стерильного жидкого носителя, например, воды, перед использованием.
Соединения согласно настоящему изобретению могут вводиться людям в дозе 0,001-50 мг на кг веса тела, предпочтительно, в дозе 0,01-20 мг на кг веса тела.
Настоящее изобретение также включает фармацевтическую композицию, указанную выше, в сочетании с упаковочным материалом, подходящим для данной композиции, причем указанная упаковка включает инструкцию по употреблению композиции с целью описанного выше применения.
Изобретение иллюстрируется с помощью приведенных ниже примеров.
Основные примечания.
Все масс-спектрометрические анализы проведены либо с использованием приборов РЕ SCIEX API 150EX, либо РЕ SCIEX API
365. Точки плавления не корректированы и определялись с использованием либо приборов системы Leica Galen III, либо Leica VMHB System Kofler.
Пример 1
Гидрохлорид 1’-карбоксиметил-7-метокси-4-фенилспиро[2Н-1-бензопиран-2,4’-пиперидина]
Стадия А: 7-метокси-Н-метилспиро[2Н-1-бензопиран-2,4’-пиперидин]-4(3Н)-он
К перемешиваемому раствору 2-гидрокси-4-метоксиацетофенона (4,08 г, 24,58 ммоль) в безводном метиловом спирте (60 см3) в атмосфере сухого азота добавляют 1-метил-4-пиперидинон (3 см3, 24,58 ммоль) и затем пирролидин (4 см3, 47,92 ммоль) и раствор нагревают до температуры кипения с обратным холодильником. Через 7 часов добавляют еще порцию 1-метил-4-пиперидинона (0,6 см3, 4,76 ммоль) и смесь нагревают до температуры кипения с обратным холодильником в течение еще 4,5 часов. Затем смеси позволяют охладиться до комнатной температуры, после чего летучие фракции удаляют при пониженном давлении. Образовавшееся масло обрабатывают дихлорметаном (100 см3), промывают водой (5×100 см3) и сушат над безводным сульфатом натрия с получением темного вязкого масла (6,33 г, 24,25 ммоль), которое частично кристаллизуется при стоянии в течение длительного периода времени.
Стадия В: 7-метокси-N-метил-4-фенилспиро[2Н-1-бензопиран-2,4'-пиперидин]
К перемешиваемому раствору 7-метокси-N-метилспиро[2Н-1-бензопиран-2,4’-пиперидин]-4(3Н)-она (6,19 г, 23,72 ммоль) в безводном тетрагидрофуране (80 см3) в атмосфере безводного азота добавляют по каплям раствор фенилбромида магния в тетрагидрофуране (40 см3, 1,0 М, 40 ммоль), поддерживая температуру реакции ниже 30°С. После завершения добавления реакционную смесь перемешивают при комнатной температуре в течение 2,5 часа, к этому времени реакция еще не завершается, но реагент Гриньяра отсутствует. Далее осторожно добавляют еще порцию фенилбромида магния (13,3 см3) и реакционную смесь перемешивают в течение ночи. Добавляют воду (30 см3) и затем насыщенный водный раствор хлорида аммония (30 см3). В вакууме удаляют летучие продукты и затем полученный материал обрабатывают диэтиловым эфиром (100 см3) и водой (100 см3). Отделяют органический слой и водную часть экстрагируют диэтиловым эфиром (2×100 см3). Объединенные экстракты промывают водой (3×100 см3), сушат над сульфатом натрия и эфир удаляют в вакууме. Остаток растирают с небольшим количеством диэтилового эфира и полученные кристаллы отделяют посредством фильтрования в вакууме (4,57 г, 12,15 ммоль), 51%). Твердый материал поглощают этиловым спиртом (75 см3) и обрабатывают хлористоводородной кислотой (75 см3, 2 н), после чего начинают нагревание до температуры кипения с обратным холодильником в течение 1,5 часа. Раствор концентрируют при пониженном давлении до начала кристаллизации. Затем смесь охлаждают и твердый материал отделяют фильтрованием в вакууме. Затем указанный материал обрабатывают смесью воды (300 см3), насыщенного водного раствора бикарбоната калия (50 см3) и диэтилового эфира (650 см3) и встряхивают. Водный слой отделяют и экстрагируют диэтиловым эфиром (2×100 см3) и затем объединенные экстракты промывают водой (3×250 см3), сушат над сульфатом натрия и летучие продукты удаляют в вакууме с получением указанного в заголовке соединения (3,91 г, 12,18 ммоль, 51% из кетона).
Стадия С: 7-метокси-4-фенилспиро[2Н-1-бензопиран-2,4’-пиперидин]
К перемешиваемому раствору 7-метокси-N-метил-4-фенилспиро[2Н-1-бензопиран-2,4’-пиперидина] (3,76 г, 11,71 ммоль) в безводном 1,2-дихлорпропане (150 см3) добавляют карбонат калия (4,04 г, 29,23 ммоль) и полученную суспензию охлаждают на ледяной бане, после чего добавляют по каплям 1-хлорэтилхлорформиат (1,58 см3, 14,64 ммоль). Реакционную смесь нагревают при температуре кипения с обратным холодильником в течение ночи и добавляют еще порцию 1-хлорэтилхлорформиата (0,8 см3, 7,4 ммоль) и смесь нагревают до температуры кипения с обратным холодильником в течение еще 24 часов. При охлаждении реакционную смесь фильтруют через хлопчатобумажный фильтр, который впоследствии промывают дихлорметаном (50 см3) и летучие фракции удаляют в вакууме. Полученный интермедиат обрабатывают метиловым спиртом (200 см3) и смесь нагревают при температуре кипения с обратным холодильником в течение ночи. При охлаждении летучие продукты удаляют в вакууме, после чего образовавшийся твердый материал растворяют в смеси дихлорметана (150 см3) и водного раствора карбоната натрия (5%, 30 см3). Органический слой отделяют, промывают водой (2×50 см3), сушат над сульфатом натрия и растворитель удаляют в вакууме с получением указанного в заголовке соединения в виде камеди (3,93 г).
Стадия D: этил 7-метокси-4-фенилспиро[2Н-1-бензопиран-2,4’-пиперидин]-1’-ацетат
К раствору 7-метокси-4-фенилспиро[2Н-1-бензопиран-2,4’-пиперидина] (3,85 г, 12,54 ммоль) в безводном N,N-диметилформамиде (75 см3) добавляют карбонат калия (4,32 г, 31,30 ммоль) и затем этилбромацетат (1,39 см3, 12,53 ммоль) и смесь нагревают до 100°С в атмосфере азота в течение 2 часов. Полученную смесь выливают в воду (600 см3) и экстрагируют этилацетатом (3×150 см3). Объединенные органические экстракты промывают водой (3×300 см3), сушат над сульфатом натрия и летучие продукты удаляют в вакууме. Указанный неочищенный продукт очищают хроматографией на колонке (силикагель, при элюировании смесью дихлорметан-этилацетат в соотношении от 9:1 до 4:1), что дает чистый этиловый сложный эфир (3,51 г, 71%).
Стадия Е:
Смесь этил 7-метокси-4-фенилспиро[2Н-1-бензопиран-2,4’-пиперидин]-1’-ацетата (3,38 г; 8,60 ммоль), этилового спирта (250 см3) и водного раствора гидроксида лития (2 н, 6,44 см3 12,88 ммоль) нагревают при температуре кипения с обратным холодильником в течение 3,5 часа. При охлаждении смесь обрабатывают соляной кислотой (5 н, 70 см3) и некоторое количество этилового спирта удаляют до начала выпадения осадка. Затем смесь охлаждают до 4°С до тех пор, пока не завершится кристаллизация. Твердый материал удаляют фильтрованием в вакууме с получением указанного в заголовке продукта в виде белого твердого вещества, Тпл. 195-230°С; положительный ион ESI (M+H)+ 366,4.
Приведенные ниже соединения получают аналогичным способом с использованием соответствующего производного 2’-гидроксиацетофенона формулы II (Схема А) и реагента Гриньяра формулы IV (Схема В):
Пример 2: Гидрохлорид 1’-карбоксиметил-4-(4-хлорфенил)-спиро[2Н-1-бензопиран-2,4’-пиперидина]: Тпл. 259-265°С; положительный ион ESI (M+H)+ 370,0.
Пример 3: 4-(4-фторфенил)спиро[2Н-1-бензопиран-2,4’-пиперидин]-1'-ацетат лития:
Литиевую соль получают с помощью процедуры, аналогичной описанной в примере 1 (стадия Е) за исключением того, что гидролиз сложного этилового эфира проводят с использованием 1,02 молярных эквивалентов водного раствора гидроксида лития (2,0 н) и по завершению реакции летучие продукты удаляют в вакууме. Тпл. 285-291°С (разлож.); положительный ион ESI (M+H)+ 354/2.
Пример 4: Гидрохлорид 1’-карбоксиметил-4-(4-метилфенил)-спиро[2Н-1-бензопиран-2,4’-пиперидина]: Тпл. 247-249°С; положительный ион ESI (M+H)+ 352,2.
Пример 5: 6-Фтор-4-фенилспиро[2Н-1-бензопиран-2,4’-пиперидин]-1’-ацетат лития: Тпл. 293-298°С (разлож.); отрицательный ион ESI (M+H)+ 354,2.
Пример 6: Гидрохлорид 1’-карбоксиметил-6-метил-4-фенилспиро[2Н-1-бензопиран-2,4’-пиперидина]: Тпл. 240-244°С; положительный ион ESI (M+H)+ 350,2.
Пример 7: Гидрохлорид 1’-карбоксиметил-7-фтор-4-фенилспиро[2Н-1-бензопиран-2,4’-пиперидина]: Тпл. 237-242°С; положительный ион ESI (M+H)+ 354,2.
Пример 8: Гидрохлорид 1’-карбоксиметил-4-(4-хлор-3-фторфенил)спиро[2Н-1-бензопиран-2,4’-пиперидина]: Тпл. 263-274°С; положительный ион ESI (M+H)+ 388,2.
Пример 9: Гидрохлорид 1’-карбоксиметил-4-(1-нафтил)-спиро[2Н-1-бензопиран-2,4’-пиперидина]: Тпл. 237-252°С; положительный ион ESI (M+H)+ 385,9.
Пример 10: Гидрохлорид 1’-карбоксиметил-4-(2-нафтил)-спиро[2Н-1-бензопиран-2,4’-пиперидина]: Тпл. 253-264°С; положительный ион ESI (M+H)+ 386,2.
Пример 11: Гидрохлорид 1’-карбоксиметил-4-(3-фтор-4-метоксифенил)спиро[2Н-1-бензопиран-2,4’-пиперидина] Тпл. 252-261°С; положительный ион ESI (M+H)+ 384,2.
Пример 12: Гидрохлорид 1’-карбоксиметил-4-(4-трет-бутилфенил)спиро[2Н-1-бензопиран-2,4’-пиперидина]: положительный ион ESI (M+H)+ 392,2.
Пример 13: Гидрохлорид 1’-карбоксиметил-4-(3-фторфенил)спиро[2Н-1-бензопиран-2,4’-пиперидина]: положительный ион ESI (M+H)+ 354,4.
Пример 14: Гидрохлорид 4-(1,3-бензодиоксоло)-1’-карбоксиметилспиро[2Н-1-бензопиран-2,4’-пиперидина]: Тпл. 260-265°С; положительный ион ESI (M+H)+ 380,4.
Пример 15: Гидрохлорид 1’-карбоксиметил-4-(3,4-диметилфенил)спиро[2Н-1-бензопиран-2,4’-пиперидина]: Тпл. 224-234°С; положительный ион ESI (М+Н)+ 361,9.
Пример 16: Гидрохлорид 1’-карбоксиметил-4-(3,4-дихлорфенил)спиро[2Н-1-бензопиран-2,4’-пиперидина]: положительный ион ESI (M+H)+ 404,0.
Пример 17: Гидрохлорид 1’-карбоксиметил-4-(3,4-диметоксифенил)спиро[2Н-1-бензопиран-2,4’-пиперидина]: положительный ион ESI (M+H)+ 396,2.
Пример 18: Гидрохлорид 1’-карбоксиметил-4-(3,4,5-трифторфенил)спиро[2Н-1-бензопиран-2,4’-пиперидина]: Тпл. 242-250°С; положительный ион ESI (М+H)+ 390,1.
Пример 19: Гидрохлорид 1’-карбоксиметил-7-фтор-4-(4-метилфенил)спиро[2Н-1-бензопиран-2,4’-пиперидина]: Тпл. 153-163°С; положительный ион ESI (M+H)+ 368,0.
Пример 20: Гидрохлорид 1’-карбоксиметил-4-(4-метоксифенил)спиро[2Н-1-бензопиран-2,4’-пиперидина]: положительный ион ESI (M+H)+ 399,6.
Пример 21: Гидрохлорид 1’-карбоксиметил-4-(3-хлорфенил)-спиро[2Н-1-бензопиран-2,4’-пиперидина]: положительный ион ESI (M+H)+ 370,2.
Пример 22: Гидрохлорид 1’-карбоксиметил-4-(3-метокси-фенил)спиро[2Н-1-бензопиран-2,4’-пиперидина]: положительный ион ESI (М+Н)+ 366,4.
Пример 23: Гидрохлорид 1’-карбоксиметил-4-(3-метилфенил)спиро[2Н-1-бензопиран-2,4’-пиперидина]: положительный ион ESI (M+H)+ 350,0.
Пример 24: Гидрохлорид 1’-карбоксиметил-4-[4-(N,N-диметиламино)фенил]спиро[2Н-1-бензопиран-2,4’-пиперидина]
Указанное соединение получают с использованием методов, указанных в примере 1, за исключением того, что реакцию гидролиза на заключительной стадии Е проводят с 1,5 эквивалентами водного раствора гидроксида натрия (1 М). Как только реакция завершается, большую часть этилового спирта удаляют и полученные кристаллы собирают фильтрованием. Полученное твердое вещество обрабатывают избытком метанольного раствора хлористого водорода в течение 1 часа при комнатной температуре. Метиловый спирт удаляют в вакууме, а полученный продукт обрабатывают смесью 2-пропанол/метанол (1:1) и добавляют по каплям диэтиловый эфир до осаждения продукта. Указанный продукт отделяют фильтрованием и сушат; Тпл. 238-250°С; положительный ион ESI (M+H)+ 379,4.
Пример 25: Гидрохлорид 1’-карбоксиметил-4-(4-этилфенил)-спиро[2Н-1-бензопиран-2,4’-пиперидина]: положительный ион ESI (М+Н)+ 364,4.
Пример 26: Гидрохлорид 4-(4-бифенил)-1’-карбоксиметил-спиро[2Н-1-бензопиран-2,4’-пиперидина]: положительный ион ESI (М+Н)+ 412,2.
Пример 27: Гидрохлорид 1’-карбоксиметил-4-(4-феноксифенил)спиро[2Н-1-бензопиран-2,4’-пиперидина]: положительный ион ESI (М+H)+ 428,2.
Пример 28: Гидрохлорид 1’-карбоксиметил-4-(3-фтор-4-метилфенил)спиро[2Н-1-бензопиран-2,4’-пиперидина]: положительный ион ESI (М+Н)+ 368,2.
Пример 29: Гидрохлорид 1’-карбоксиметил-7-хлор-4-фенил-спиро[2Н-1-бензопиран-2,4’-пиперидина]: положительный ион ESI (M+H)+ 370,2.
Пример 30: Гидрохлорид 1’-карбоксиметил-6-хлор-4-фенил-спиро[2Н-1-бензопиран-2,4’-пиперидина]: положительный ион ESI (М+Н)+ 370,2.
Пример 31: Гидрохлорид 1’-карбоксиметил-7-хлор-4-(4-этилфенил)спиро[2Н-1-бензопиран-2,4’-пиперидина]: положительный ион ESI (М+Н)+ 398,2.
Пример 32: Гидрохлорид 1’-карбоксиметил-7-хлор-4-(4-пропилфенил)спиро[2Н-1-бензопиран-2,4’-пиперидина]: положительный ион ESI (М+Н)+ 412,2.
Пример 3 3: Гидрохлорид 1’-карбоксиметил-4-(2,2-дифтор-1,3-бензодиоксоло)спиро[2Н-1-бензопиран-2,4’-пиперидина]: Тпл. 248-250°С; положительный ион ESI (M+H)+ 416,2.
Пример 34: Гидрохлорид 1’-карбоксиметил-4-(2,3,5-трифторфенил)спиро[2Н-1-бензопиран-2,4’-пиперидина]: Тпл. 206-210°С; положительный ион ESI (M+H)+ 390,4.
Пример 35: Гидрохлорид 1’-карбоксиметил-7-хлор-4-(3-фтор-4-метилфенил)спиро[2Н-1-бензопиран-2,4’-пиперидина]: положительный ион ESI (M+H)+ 402,3.
Пример 36: Гидрохлорид 1’-карбоксиметил-4-(3,5-дифтор-4-метоксифенил)спиро[2Н-1-бензопиран-2,4'-пиперидина]: положительный ион ESI (M+H)+ 402,2 4-бром-2,6-дифторанизол, исходный материал для приготовления реагента Гриньяра формулы IV (схема В), получают из 4-бром-2,6-дифторфенола. К раствору фенола (49,0 г, 234 ммоль) в безводном ацетоне (980 см3) добавляют метилиодид (29,4 см3, 468 ммоль) и затем карбонат калия (80,85 г, 585 ммоль). Перемешиваемую смесь нагревают при температуре кипения с обратным холодильником в течение 2 часов, после чего позволяют смеси охладиться. Образовавшийся твердый материал отфильтровывают и фильтрат выпаривают в вакууме. Остаток переносят в диэтиловый эфир (1000 см3) и раствор промывают водой (3×300 см3), сушат (Na2SO4) и удаляют эфир в вакууме с получением неочищенного продукта, который используют для получения требуемого реагента Гриньяра без дополнительной очистки (49,4 г, 222 ммоль, 95%).
Пример 37: Гидрохлорид 1’-карбоксиметил-4-(4-диметиламино-3-фторфенил)спиро[2Н-1-бензопиран-2,4’-пиперидина]: Тпл. 231-239°С; положительный ион ESI (M+H)+ 397,4.
Пример 38: Гидрохлорид 1’-карбоксиметил-4-(3,5-дифтор-4-диметиламинофенил)спиро[2Н-1-бензопиран-2,4’-пиперидина]: Тпл. 231-242°С; положительный ион ESI (M+H)+ 415,0.
Пример 39: Гидрохлорид 4-(4-бром-3-фторфенил)-1’-карбоксиметил)спиро[2Н-1-бензопиран-2,4’-пиперидина]: положительный ион ESI (М+Н)+ 432,5.
Пример 40: Гидрохлорид 1’-карбоксиметил-4-(3-бром-4-метоксифенил)спиро[2Н-1-бензопиран-2,4’-пиперидина]: положительный ион ESI (М+H)+ 444,1.
Пример 41: Гидрохлорид 1’-карбоксиметил-4-(3,5-дифторфенил)спиро[2Н-1-бензопиран-2,4’-пиперидина]: Тпл. 230-264°С; положительный ион ESI (M+H)4 372,2.
Пример 42: Гидрохлорид 1’-карбоксиметил-4-(3,5-дифтор-4-этоксифенил)спиро[2Н-1-бензопиран-2,4’-пиперидина]:
Стадия А: Гидробромид 1’-карбоксиметил-4-(3,5-дифтор-4-гидроксифенил)спиро[2Н-1-бензопиран-2,4’-пиперидина]
Смесь метилового эфира (2,35 г) в уксусной кислоте (15 см3) и 47% бромистоводородной кислоты перемешивают при 120°С в течение 40 часов. Полученную суспензию охлаждают в водяной бане со льдом и затем добавляют воду (30 см3). Твердый материал отделяют фильтрованием, промывают уксусной кислотой и затем водой, после чего сушат в вакууме.
Стадия В: Этил (3,5-дифтор-4-гидроксифенил)спиро[2Н-1-бензопиран-2,4’-пиперидин]-1’-ацетат
Гидробромид 1’-карбоксиметил-4-(3,5-дифтор-4-гидроксифенил)спиро[2Н-1-бензопиран-2,4’-пиперидина] (1,7 г, получен по методу, описанному на стадии А) суспендируют в растворе хлористого водорода в этиловом спирте (120 см3) и смесь нагревают при температуре кипения с обратным холодильником в течение 16 часов. При охлаждении летучие материалы удаляют в вакууме с получением твердого остатка, который распределяют между этилацетатом (100 см3) и смесью воды (100 см3) и насыщенного водного раствора бикарбоната калия (30 см3). Водный слой экстрагируют этилацетатом (2×50 см3), после чего объединенные экстракты промывают водой (2×50 см3) и сушат (Na2SO4). Неочищенный материал фильтруют через силикагель, используя в качестве элюента этилацетат, который затем выпаривают с получением твердого продукта (1,54 г).
Стадия С: Этил-(3,5-дифтор-4-этоксифенил)спиро[2Н-1-бензопиран-2,4’-пиперидин]-1’-ацетат
К этил (3,5-дифтор-4-гидроксифенил)спиро[2Н-1-бензопиран-2,4’-пиперидин]-1’-ацетату (350 мг), карбонату цезия (412 мг) и иодиду натрия (13 мг) добавляют N,N-диметилформамид (9 см3) и затем йодэтан (1,7 молярных эквивалента). Полученную смесь нагревают при перемешивании до 65°С в течение 3 часов. При охлаждении реакционную смесь разбавляют этилацетатом (90 см3) и затем промывают водой (5×35 см3) и сушат (Na2SO4). Указанный раствор неочищенного материала фильтруют затем через слой силикагеля, после чего растворитель удаляют в вакууме с получением гомогенного продукта.
Стадия D: Из этил (3,5-дифтор-4-этоксифенил)спиро[2Н-1-бензопиран-2,4’-пиперидин]-1’-ацетата в соответствии с процедурой, описанной в примере 1, получают гидрохлорид 1’-карбоксиметил-4-(3,5-дифтор-4-этоксифенил)спиро[2H-1-бензопиран-2,4’-пиперидина]: положительный ион ESI (M+H)+ 416,5.
Указанным методом получают также следующие соединения:
Пример 43: Гидрохлорид 1’-карбоксиметил-4-(3,5-дифтор-4-н-пропоксифенил)спиро[2Н-1-бензопиран-2,4’-пиперидина]: положительный ион ESI (M+H)+ 430,3.
Пример 44: Гидрохлорид 1’-карбоксиметил-4-(3,5-дифтор-4-н-бутоксифенил)спиро[2Н-1-бензопиран-2,4’-пиперидина]: положительный ион ESI (M+H)+ 444,4.
Пример 45: Гидрохлорид 1’-карбоксиметил-4-(4-бензилокси-3,5-дифторфенил)спиро[2Н-1-бензопиран-2,4’-пиперидина]: положительный ион ESI (M+H)+ 478/0.
Пример 46: Гидрохлорид 1’-карбоксиметил-4-(3,5-дифтор-4-изо-пентилоксифенил)спиро[2Н-1-бензопиран-2,4’-пиперидина]: Тпл. 211-215°С; положительный ион ESI (M+H)+ 458,5.
Пример 47: Гидрохлорид 1’-карбоксиметил-4-(4-этокси-3-фторфенил)спиро[2Н-1-бензопиран-2,4’-пиперидина]: положительный ион ESI (M+H)+ 398,2.
Пример 48: Гидрохлорид 1’-карбоксиметил-4-(3-фтор-4-н-пропоксифенил)спиро[2Н-1-бензопиран-2,4’-пиперидина]: положительный ион ESI (M+H)+ 412,0.
Пример 49: Гидрохлорид 1’-карбоксиметил-4-(3-фтор-4-н-бутоксифенил)спиро[2Н-1-бензопиран-2,4’-пиперидина]: положительный ион ESI (M+H)+ 426,1.
Пример 50: Гидрохлорид 1’-карбоксиметил-4-(4-бензилокси-3-фторфенил)спиро[2Н-1-бензопиран-2,4’-пиперидина]: положительный ион ESI (M+H)+ 460,3.
Пример 51: Гидрохлорид 1’-карбоксиметил-4-(3-фтор-4-изо-пентилоксифенил)спиро-[2Н-1-бензопиран-2,4’-пиперидина]: положительный ион ESI (M+H)+ 440,2.
Пример 52: Гидрохлорид 1’-карбоксиметил-4-(3-фтор-4-метоксиэтоксифенил)спиро[2Н-1-бензопиран-2,4’-пиперидина]: положительный ион ESI (М+Н)+ 428,2.
Пример 53: Гидрохлорид 1’-карбоксиметил-4-(3-фтор-4-изо-бутилоксифенил)спиро[2Н-1-бензопиран-2,4’-пиперидина]: положительный ион ESI (M+H)+ 425,8.
Пример 54: Гидрохлорид 1’-карбоксиметил-4-[3-фтор-4-метоксибензилокси)фенил]спиро[2Н-1-бензопиран-2,4’-пиперидина]: положительный ион ESI (M+H)+ 490,0.
Пример 55: Гидрохлорид 4-(4-аллилокси-3,5-дифторфенил)-1’-карбоксиметил)спиро[2Н-1-бензопиран-2,4’-пиперидина]:
Стадия А: N-бензил-4-(3,5-дифтор-4-аллилоксифенил)спиро-[2H-1-бензопиран-2,4’-пиперидин]
К смеси N-бензил-4-(3,5-дифтор-4-гидроксифенил)спиро-[2Н-1-бензопиран-2,4’-пиперидина] (750 мг, 1,79 ммоль; получен в соответствии с процедурами, описанными в примерах 1 и 42), карбоната цезия (1,1 молярных эквивалента) и безводного N,N-диметилформамида (15 см3) добавляют по каплям аллилбромид (1,1 молярных эквивалента). После перемешивания в течение 2 часов при комнатной температуре не остается исходного материала. Реакционную смесь разбавляют этилацетатом (50 см3) и обрабатывают водой (100 см3), после чего водный слой отделяют и повторно экстрагируют этилацетатом (2×25 см3). Объединенные органические экстракты промывают водой (4×100 см3) и затем сушат (Na2SO4) и удаляют летучие материалы в вакууме с получением требуемого материала высокой степени чистоты (60%).
Стадия В: 4-(3,5-дифтор-4-аллилоксифенил)спиро[2Н-1-бензопиран-2,4’’-пиперидин]
Проводят дебензилирование в соответствии с процедурой стадии С примера 1 со следующими изменениями: в качестве растворителя используют дихлорметан (который специально не сушат) и реакцию не ведут в инертной атмосфере. В некоторых примерах разложение промежуточного карбамата оказывается неэффективным при нагревании при температуре кипения с обратным холодильником в присутствии метилового спирта. В таких случаях требуется добавление избытка 10н водного гидроксида калия с нагреванием в течение ночи при температуре кипения с обратным холодильником.
Далее амин превращают в конечный продукт в соответствии с процедурами, описанными в примере 1: Тпл. 221-225°С; положительный ион ESI (М+Н)+ 428,2.
Пример 56: Гидрохлорид 1’-карбоксиметил-4-(3,5-дифтор-4-изо-пропилоксифенил)спиро[2Н-1-бензопиран-2,4’-пиперидина]: Тпл. 241-243°С; положительный ион ESI (М+Н)+ 430,3.
Пример 57: Гидрохлорид 1’-карбоксиметил-4-(3,5-дифтор-4-пропаргилоксифенил)спиро[2Н-1-бензопиран-2,4’-пиперидина]: Тпл. 211-217°С; положительный ион ESI (М+Н)+ 426,1.
Пример 58: Гидрохлорид 1’-карбоксиметил-4-(3,5-дифтор-4-циклопропилметоксифенил)спиро[2Н-1-бензопиран-2,4’-пиперидина]: Тпл. 225-231°С; положительный ион ESI (M+H)+ 442,0.
Пример 59: Гидрохлорид 1’-карбоксиметил-4-(3,5-дифтор-4-трифторэтоксифенил)спиро[2Н-1-бензопиран-2,4’-пиперидина]: Тпл.244-251°С; положительный ион ESI (М+Н)+ 470,2.
Пример 60: Гидрохлорид 1’-карбоксиметил-4-(3-фтор-4-изо-пропилоксифенил)спиро[2Н-1-бензопиран-2,4’-пиперидина]: Тпл.>260°С; положительный ион ESI (М+Н)+ 412,4.
Пример 61: Гидрохлорид 1’-карбоксиметил-4-(3-фтор-4-трифторэтоксифенил)спиро[2Н-1-бензопиран-2,4’-пиперидина]: Тпл. 230-235°С; положительный ион ESI (М+Н)+ 452,2.
Пример 62: Гидрохлорид 1’-карбоксиметил-4-(3-фтор-4-феноксифенил)спиро[2Н-1-бензопиран-2,4’-пиперидина]: Тпл. 160-180°С; положительный ион ESI (M+H)+ 446,0.
Пример 63: 4-фенилспиро[2Н-1-бензопиран-2,4’-пиперидин]-1’-ацетат лития: Тпл. 280-281°С; положительный ион ESI (М+Н)+ 336,2.
Пример 64: 4-(4-трифторметилфенил)спиро[2Н-1-бензопиран-2,4’-пиперидин]-1’-ацетат лития: положительный ион ESI (М-Li+2H)+ 404,4.
Пример 65: Этил 4-(4-этилфенил)спиро[2Н-1-бензопиран-2,4’-пиперидин]-1’-ацетат: Тпл. 119-121°С; положительный ион ESI (М+Н)+ 392,3.
Пример 66: Фенил 4-(4-этилфенил)спиро[2Н-1-бензопиран-2,4’-пиперидин]-1’-ацетат: Тпл. 104-105°С; положительный ион ESI (M+H)+ 440,3.
Пример 67: Гидрохлорид 1’-карбоксиметил-4-(4-изопентилоксифенил)спиро[2Н-1-бензопиран-2,4’-пиперидина]: положительный ион ESI (M+H)+ 422,0.
Пример 68: [4-(2-пиридинометилокси)фенил]спиро[2Н-1-бензопиран-2,4’-пиперидин]-1’-ацетат натрия; получают в соответствии с процедурой примера 42 с использованием гидрохлорида 2-пиколилхлорида. Заключительный гидролиз проводят в соответствии с процедурой примера 1 за исключением того, что используют гидроксид натрия; положительный ион ESI (M+H)+ 443,4.
Пример 69: Гидрохлорид 1’-карбоксиметил-4-[4-(4-метилфенилсульфонилокси)фенил]спиро[2Н-1-бензопиран-2,4’-пиперидина]:
Стадия А: Этиловый эфир 1’-карбоксиметил-4-[4-(4-метилфенилсульфонилокси)фенил]спиро[2Н-1-бензопиран-2,4’’-пиперидина]
К перемешиваемой при -5°С смеси этил (4-гидроксифенил)спиро[2Н-1-бензопиран-2,4’-пиперидин]-1’-ацетата (182 мг, 0,5 ммоль; получен по процедуре примера 42), дихлорметана (13 см3) и пиридина (158 мг, 2,0 см3) осторожно добавляют раствор п-толуолсульфонилхлорида (288 мг, 1,5 ммоль) в дихлорметане (13 см3). Затем реакционной смеси позволяют нагреться до температуры окружающей среды, перемешивают в течение еще 2 часов, после чего раствор оставляют стоять в течение ночи. К смеси добавляют воду (7 см3) и после перемешивания в течение 10 минут выпаривают растворитель с получением продукта в виде камеди, которая затвердевает при стоянии. Указанный продукт разбивают шпателем, отфильтровывают и промывают водой (20 см3). Полученную лепешку отсасывают почти досуха и затем сушат в вакууме при 65°С с получением продукта (300 мг); (М+Н)+ 534,2 m/z.
Указанный сложный эфир гидролизуют в соответствии с процедурой, описанной в примере 1, за исключением того, что гидролиз проводят в н-бутаноле: положительный ион ESI (М+Н)+ 506,2.
Пример 70: Гидрохлорид 1’-карбоксиметил-8-фтор-4-фенилспиро[2Н-1-бензопиран-2,4’-пиперидина]
В том случае, когда нужный исходный 2’-гидроксиацетофенон коммерчески не доступен, он может быть получен указанным ниже способом:
Стадия А: (2-фторфенил)ацетат
К перемешиваемой смеси 2-фторфенола (60,0 г, 535 ммоль) и водного раствора гидроксида натрия (4 н, 214 см3, 856 ммоль) при 0°С добавляют уксусный ангидрид (81 см3, 856 ммоль). После перемешивания реакционной смеси в течение 30 минут водный слой отделяют от органической части и промывают дихлорметаном (150 см3). Объединенные органические экстракты промывают водным раствором гидроксида натрия (4 N, 150 см3) и затем насыщенным раствором соли (100 см3). Органическую фазу сушат (Na2SO4) и растворитель выпаривают в вакууме с получением указанного в заголовке соединения (84,9 г, >100%.
Стадия В: 1’-гидрокси-3’-фторацетофенон
Мелкоизмельченный трихлорид алюминия (32,0 г, 239 ммоль) добавляют к (2-фторфенил)ацетату (23,0 г, 149 ммоль) и смесь нагревают до 180°С в течение 1 часа. При охлаждении до комнатной температуры реакционную смесь осторожно вливают в смесь льда/воды и продукт экстрагируют дихлорметаном (150 см3). Отделяют органический слой, промывают его насыщенным раствором соли (150 см3), сушат (Na2SO4) и растворитель удаляют в вакууме. Неочищенную(сырую) реакционную смесь очищают хроматографией на колонке (силикагель, дихлорметанметиловый спирт 99:1) с получением указанного в заголовке соединения (3,1 г, 14%).
Далее ацетофенон превращают в конечный продукт с использованием процедуры, описанной в примере 1: Тпл.180-182°С; положительный ион ESI (М+Н)+ 354,0.
В соответствии с указанным способом получают также следующие соединения:
Пример 71: Гидрохлорид 1’-карбоксиметил-7-хлор-6-фтор-4-фенилспиро[2Н-1-бензопиран-2,4’-пиперидина]; Тпл. 275-293°С; положительный ион ESI (M+Н)+ 387,7.
Пример 72: Гидрохлорид 1’-карбоксиметил-5-фтор-4-фенилспиро[2Н-1-бензопиран-2,4’-пиперидина]
Нужный исходный 2’-гидроксиацетофенон коммерчески недоступен, исходя из чего его получают описанным ниже способом:
Стадия А: 2’-фтор-6’-метоксиацетофенон
Смесь 2-фтор-6-метоксибензонитрила (15,94 г, 105,5 ммоль) и раствора метилиодида магния в диэтиловом эфире (3 М, 46,0 см3, 137 ммоль) нагревают до 100°С в течение 18 часов. При охлаждении до комнатной температуры добавляют водный раствор соляной кислоты (3 М, 94 см3) и смесь нагревают до температуры кипения с обратным холодильником в течение 4 часов. Как только реакционная смесь охладится до комнатной температуры, отделяют органический слой и водную фазу экстрагируют этилацетатом (100 см3). Объединенные органические экстракты промывают насыщенным раствором соли (100 см3) и затем сушат (Na2SO4). Растворитель удаляют в вакууме с получением указанного в заголовке соединения в виде масла (117,7 г, 100%).
Стадия В: 2’-фтор-6’-гидроксиацетофенон
Раствор трибромида бора в дихлорметане (69,0 г, 69,6 ммоль) добавляют по каплям в течение 25 минут при температуре -78°С к раствору 2’-фтор-6’метоксиафетофенона (17,72 г, 105,5 ммоль) в дихлорметане (150 см3). После повышения температуры реакционной смеси до 0°С реакцию гасят добавлением воды (100 см3) и продукт экстрагируют дихлорметаном (100 см3). Органический раствор промывают водой (100 см3), насыщенным раствором соли (150 см3) и затем сушат (Na2SO4). Выпаривание растворителя в вакууме дает неочищенный продукт, который используют далее без дополнительной очистки.
В дальнейшем ацетофенон превращают в конечный продукт с помощью процедур, описанных в примере 1: Тпл. 252-254°С; положительный ион ESI (M+H)+ 354,2.
Пример 73: Гидрохлорид 1’-карбоксиметил-4-[4-(2-этоксиморфолино)фенил]спиро[2Н-1-бензопиран-2,4’-пиперидина]. Получают с использованием методов, описанных в примере 42, посредством алкилирования соответствующего фенола гидрохлоридом 4-(2-хлорэтил)морфолина; положительный ион ESI (М+2Н)+ 465,2.
Пример 74: Гидрохлорид 1’-карбоксиметил-6-гидрокси-4-фенилспиро[2Н-1-бензопиран-2,4’-пиперидина]. Получают с использованием методов, описанных в примерах 1 и 42, положительный ион ESI (М+Н)+ 352,2.
Пример 75: Гидрохлорид 1’-карбоксиметил-7-метилтио-4-фенилспиро[2Н-1-бензопиран-2,4’-пиперидина].
Стадия А: 7-фтор-N-метилспиро[2Н-1-бензопиран-2,4’-пиперидина]-4(3Н)-он
Перемешиваемый раствор 4’-фтор-2’-гидроксиацетофенона (15,97 г), 1-метил-4-пиперидона (12,74 см3) и пирролидина (4,325 см3) в метиловом спирте (250 см3) нагревают до температуры кипения с обратным холодильником в атмосфере азота. Через 0,5 часа добавляют еще порцию пирролидина (4,33 см3), затем еще одну порцию через следующие 0,5 часа и раствору дают охладиться. После этого реакционную смесь выпаривают при пониженном давлении с получением масла, которое растворяют в дихлорметане (400 см3). Раствор промывают водой (3×400 см3), сушат (Na2SO4) и выпаривают с получением масла (25,7 г). Флэш-хроматография на силикагеле (при элюировании смесью дихлорметан:метиловый спирт:33% водный аммиак = 380:20:1) дает очищенный продукт в виде масла (14,41 г.
Стадия В: 7-метилтио-N-метилспиро[2Н-1-бензопиран-2,4’’пиперидин]-4(3Н)-он
К перемешиваемому раствору 7-фтор-N-метилспиро [2Н-1-бензопиран-2,4’-пиперидин]-4 (3Н)-она (4,54 г) в диметилформамиде (20 см3) в атмосфере азота добавляют тиометоксид натрия (1,40 г). Смесь перемешивают при комнатной температуре в течение 1,25 часа, при 60°С - в течение 4,5 часа и затем позволяют охладиться и выдерживают в течение ночи при комнатной температуре. Далее указанную смесь перемешивают и нагревают при 130°С в течение 2,5 часов, затем позволяют охладиться и выдерживают в течение ночи при комнатной температуре, после чего выливают в воду при перемешивании (140 см3). Твердый продукт отфильтровывают, промывают водой и растворяют в дихлорметане. Раствор сушат (Na2SO4) и выпаривают с получением продукта в виде камеди (4,44 г).
Далее 7-метилтио-N-метилспиро[2Н-1-бензопиран-2,4'-пиперидин]-4(3Н)-он превращают в конечное соединение с использованием процедуры, описанной в примере 1, в которую внесены следующие изменения: при проведении стадии деметилирования целевой вторичный амин неожиданно превращается в менее полярное соединение, которое обрабатывают водным раствором гидроксида калия (10 н) в метиловом спирте при температуре кипения с обратным холодильником с образованием целевого амина; (М+H)+ = 382,0.
Пример 76: Гидрохлорид 1’-карбоксиметил-4-(4-трифтор-метоксифенил)спиро[2Н-1-бензопиран-2,4’-пиперидина]
Стадия А: Сложный эфир трифторметансульфоновой кислоты-4-[1’-фенилметилспиро(2Н-1-бензопиран-2,4’-пиперидина)]
К перемешиваемому охлажденному раствору (-78°С) N-бензилспиро[2Н-1-бензопиран-2,4’-пиперидин]-4-(3Н)-она (1,54 г, 5 ммоль; получен по методу примера 1) в сухом тетрагидрофуране (40 см3) добавляют гексаметилдисилазид лития (1 М раствор в гексане, 7,5 см3, 7,5 ммоль) по каплям в течение примерно 5 минут. После перемешивания при указанной температуре в течение 1 часа добавляют сульфонамид N-фенилтрифторметана (2,68 г, 7,5 ммоль) в виде одной порции и полученную реакционную смесь оставляют перемешиваться и медленно нагревают в течение ночи. Реакцию гасят водой (10 см3) и экстрагируют этилацетатом (2×50 см3). Органическую часть промывают насыщенным водным раствором хлорида аммония (50 см3), насыщенным водным раствором хлорида натрия (50 см3) и водой (50 см3) и затем сушат (Na2SO4) и концентрируют. Продукт очищают колоночной хроматографией на силикагеле (3:1, гептан-этилацетат) с получением требуемого материала (1,75 г, 80%), который либо сразу же используют, либо хранят в инертной атмосфере при -20°С.
Стадия В: N-бензил-4-(4-трифторметоксифенил)спиро[2Н-1-бензопиран-2,4’-пиперидин]-4(3Н)-он
Смесь {N-бензилспиро[2Н-1-бензопиран-2,4’-пиперидин]}-4-(фенил-4-трифторметилсульфоната) (1,69 г, 3,86 ммоль), 4-трифторметоксибензолбороновой кислоты (1,25 экв.), диметоксиэтана (40 см3), хлорида лития (2,5 экв.), тетракис(трифенилфосфин)палладия (0) (2,5 моль%) и водного 2 н раствора карбоната натрия (2 экв.) нагревают при температуре кипения с обратным холодильником в течение 12 часов. При охлаждении смесь обрабатывают водой (75 см3) и этилацетатом (75 см3). После встряхивания органический слой отделяют и промывают водой (2×100 см3), сушат (Na2SO4) и растворитель удаляют с получением целевого продукта (96%), который используют на следующей стадии без дополнительной очистки.
В дальнейшем N-бензил-4-(4-трифторметоксифенил)спиро [2Н-1-бензопиран-2,4’-пиперидин]-4 (3Н)-он превращают в конечное соединение с использованием процедуры, описанной в примере 1: Тпл. 233-237°С; (М+H)+ = 420,2.
Пример 77: Гидрохлорид 1’-карбоксиметил-4-(4-метилтио-фенил)спиро[2Н-1-бензопиран-2,4’-пиперидина]. Получают с использованием методики, описанной в примере 76; положительный ион ESI (М+Н)+ 352,2.
Пример 78: Гидрохлорид 1’-карбоксиметил-4-[4-(N-метил-N-метилсульфонамидо)фенил]спиро[2Н-1-бензопиран-2,4’-пиперидина]
Стадия А: этил 4-(4-гидроксифенил)спиро[2Н-1-бензопиран-2,4’-пиперидин]–1’-ацетат
К смеси 1’-карбоксиметил-4-(4-гидроксифенил)спиро[2Н-1-бензопиран-2,4’-пиперидин]-1’ ацетата (432 мг, 1 ммоль, получен по процедуре примера 42), гидрокарбоната натрия (176 мг, 2,1 ммоль) в сухом N,N-диметилформамиде (25 см3) добавляют этилиодид (0,088 см3, 1,1 ммоль) и смесь нагревают до 80°С в течение 2 часов. При охлаждении указанную смесь разбавляют этилацетатом (100 см3) и промывают водой (5×100 см3). Затем органический раствор сушат над сульфатом натрия, после чего растворитель удаляют в вакууме. Далее неочищенный материал пропускают через небольшой слой силикагеля с использованием вначале дихлорметана (который затем отбрасывают) и затем этилацетата. Растворитель удаляют с получением требуемого продукта (194 мг, 51%).
Стадия В: Сложный этиловый эфир трифторметансульфоновой кислоты-{1’-карбоксиметил-4-фенилспиро[2Н-1-бензопиран-2,4’-пиперидина]
К охлажденной перемешиваемой (-20°С) суспензии этил 4-(4-гидроксифенил)спиро[2Н-1-бензопиран-2,4’-пиперидин]-1’-ацетата (158 мг, 4,16×10-4 моль) в безводном дихлорметане (10 см3) в инертной атмосфере добавляют триэтиламин (0,065 см3, 4,6×10-4 моль). Через 10 минут добавляют по каплям примерно в течение 5 минут раствор трифторметансульфонового ангидрида (0,077 см3, 4,6×10-4 моль) в дихлорметане (7,7 см3). В течение примерно 20 следующих минут твердый материал исчезает. Однако анализ с использованием тонкослойной хроматографии показывает, что реакция еще не завершена, так что добавляют еще порцию трифторметансульфонового ангидрида (0,25 экв.). Примерно через 20 минут реакционную смесь разбавляют этилацетатом (60 см3), промывают водой (3×60 см3), после чего сушат (Na2SO4) и растворитель удаляют в вакууме. Неочищенный продукт очищают хроматографией на колонке с силикагелем (дихлорметан: этилацетат, 3:1) с получением требуемого материала (154 мг, 75%).
Стадия С: Этиловый эфир 4-[4-(дифенилимино)фенил]-спиро[2Н-1-бензопиран-2,4’-пиперидин]-1-уксусной кислоты
Указанное в заголовке соединение получают с помощью модифицированной методики Бухвальда с соавт. (Buchwald et al.. Tetrahedron Letters, 1997, 38, 6367).
К раствору трифторметансульфоновой кислоты-{1’-карбоксиметил-4-фенилспиро[2Н-1-бензопиран-2,4’-пиперидин]-этиловый эфир} (421 мг, 8,43×10-4 моль) в сухом тетрагидрофуране (25 см3) в ходе перемешивания при комнатной температуре в атмосфере сухого азота добавляют имин бензофенона (1,2 экв., 0,283 см3). Затем добавляют ацетат палладия (1,25 моль%), (R)-(+)-2,2’-бис(дифенилфосфино)-1,1’-бинафтил (3,75 моль%) и карбонат цезия (1,40 экв., 385 мг). Смесь нагревают до температуры кипения с обратным холодильником в течение 24 часов и затем позволяют ей охладиться до комнатной температуры. Далее смесь разбавляют диэтиловым эфиром (не сухим) (250 см3), фильтруют через фильтровальную подушку и удаляют в вакууме летучие продукты. Остаток очищают колоночной хроматографией на силикагеле (гептан:этилацетат, 1:1) с получением требуемого материала (547 мг, с примесью имина бензофенона).
Стадия D: этил 4-(4-аминофенил)спиро[2Н-1-бензопиран-2,4’-пиперидин]-1’-ацетат
Указанное в заголовке соединение получают по методу
Бухвальда с соавт. (Buchwald et al., Tetrahedron Letters, 1997, 38, 6367).
К перемешиваемому раствору этил 4-(4-бензофенониминофенил)спиро[2Н-1-бензопиран-2,4’-пиперидин]-1’-ацетата (542 мг, 1 ммоль) в метиловом спирте (25 см3) добавляют ацетат натрия (198 мг, 2,4 ммоль) и затем гидрохлорид гидроксиламина (125 мг, 1,8 экв.) и реакционную смесь перемешивают при комнатной температуре в течение 30 минут. Метиловый спирт удаляют в вакууме, после чего остаток распределяют между дихлорметаном (25 см3) и водным раствором гидроксида натрия (0,1 М, 25 см3). Отделяют органический слой и летучие продукты удаляют в вакууме, после чего продукт очищают колоночной хроматографией (силикагель, этилацетат) (220 мг, 69% из трифлата).
Стадия Е: этил 4-(метил 4-фенилсульфонамид)спиро[2Н-1-бензопиран-2,4’-пиперидин]-1’-ацетат
Указанное в заголовке соединение получают по методу Сандберга с соавт. (R.J. Sundberg et al., Journal of Organic chemistry, 1984, 49, 249).
Колбу, содержащую смесь этил 4-(4-аминофенил)спиро[2Н-1-бензопиран-2,4’-пиперидин]-1’-ацетата (214 мг, 0,566 ммоль), пиридина (3 экв., 0,137 см3) и безводного дихлорметана (10 см3) перемешивают в бане, содержащей лед/метиловый спирт. Через 10 минут при указанной температуре добавляют по каплям в течение примерно 5 минут раствор метансульфонилхлорида (1,5 экв., 0,066 см3) в сухом дихлорметане (0,66 см3). После перемешивания в течение 2 часов реакционную смесь выливают в насыщенный водный раствор гидрокарбоната калия (10 см3). Органический слой промывают водой (3×50 см3) и растворитель удаляют в вакууме. Добавляют толуол (30 см3) и затем удаляют его в вакууме (указанную процедуру повторяют до тех пор, пока исчезнет запах пиридина) и затем добавляют метиловый спирт (30 см3) и удаляют его. Остаток представляет собой гомогенный материал (234 мг, 91%).
Стадия F: этил 4-(метил 4-фенил-N-метилсульфо-намид)спиро[2Н-1-бензопиран-2,4’-пиперидин]-1’-ацетат
К раствору этил 4-(метил 4-фенилсульфонамид)спиро[2Н-1-бензопиран-2,’пиперидин]-1’-ацетата (108 мг, 0,236 ммоль) в метиловом спирте (10 см3) добавляют без перемешивания раствор триметилсилилдиазометана (2 М в гексанах, 5 см3). Сразу же при добавлении отмечается выделение азота. После выдерживания в течение 1 часа при комнатной температуре летучие продукты удаляют в вакууме, после чего остаток очищают хроматографией на колонке (силикагель, этилацетат) с получением гомогенного продукта (74 мг, 67%).
Пример 79 Гидрохлорид 1’-карбоксиметил 4-(N-метил 4-фенил-N-метилсульфонамид)спиро[2Н-1-бензопиран-2,4’-пиперидина]
В дальнейшем синтез проводят по методу, описанному в примере 1; положительный ион ESI (М+Н)+ = 443,2
Пример 80: Дигидрохлорид 1’-карбоксиметил-4-(4-амино-3,5-дифтофенил)спиро[2H-1-бензопиран-2,4’-пиперидина] (4-бром-2,5-дифторфенил)-2,5-диметилпиррол получают, следуя методике Брукельмана с соавт. (Bruekelmann et al., J. Chem. Soc., Perkin Trans. 1, 1984, 2801).
Указанный материал превращают в соответствующий реагент Гриньяра и проводят реакцию с N-метилспиро[2Н-1-бензопиран-2,4’-пиперидин]-4(3Н)-оном по методу примера 1, за исключением того, что получают неразделенную смесь пиролла и освобожденного от защитной группы анилина. Указанную смесь подвергают N-дезалкилированию, как описано в примере 1 (вместе с проведением сопутствующего снятия защиты анилиновой группы) с получением 4-(4-амино-3,5-дифторфенил)спиро-[2Н-1-бензопиран-2,4’-пиперидина]. Далее полученный амин алкилируют этилбромацетатом и образовавшийся сложный эфир гидролизуют по методу примера 1; положительный ион ESI (M+H)+ 387,1.
Пример 81: Гидрохлорид 1’-карбоксиметил-3,4-дигидро-4-(4-метилфенил)спиро[2Н-1-бензопиран-2,4’-пиперидина]
Стадия А: этил 3,4-дигидро-4-(4-метилфенил)спиро[2Н-1-бензопиран-2,4’-пиперидин]-1’-ацетат
К раствору этил 4-(4-метилфенил)спиро[2Н-1-бензопиран-2,4’-пиперидин]-1’-ацетата (730 мг, 0,194 ммоль) в этиловом спирте (125 см3) добавляют 10% палладий на угле. Указанную смесь нагревают до 40°С в течение 7 часов в атмосфере водорода (4 бара) и затем фильтруют в теплом состоянии и образовавшуюся фильтровальную лепешку промывают горячим этиловым спиртом (3×50 см3). Растворитель удаляют в вакууме и полученное соединение повторно кристаллизуют из этилового спирта (67%). Указанный сложный эфир гидролизуют по процедуре примера 1; Тпл. 247-249°С; (М+Н)+ = 352,2.
Указанным методом получают также:
Пример 82: 3,4-дигидро-4-(4-фторфенил)спиро[2Н-1-бензопиран-2,4’-пиперидин]-1’-ацетат лития: Тпл. 274-278°С (разлож.); отрицательный ион ESI (M-Li)- 354,40.
Пример 83:
Метод определения поглощения глицина в СНО клетках, гетерологично экспрессирующих переносчик GlyT-1b человека.
А: Клонирование: кДНК получают посредством ПЦР по методу, описанному Ким с соавт, (Kirn K.-M. et al., Mol. Pharmacol. 1994, 45, 608-617). Последовательность подтверждают методом дидезоксисеквенирования с использованием секвенатора ДНК ALF DNA sequencer™ (Pharmacia) и клонируют в экспрессирующий конструкт pcDNA3 (Invitrogen).
В: Трансфекция: Трансфекцию hGlyT-1b в СНО клетки проводят по стандартной кальций-фосфатной методике, описанной Самбруком с соавт. (Sumbrook, J. et al., (1989) in Molecular Cloning: A Laboratory Manual, Cold Spring Harbor Laboratory, Cold Spring Harbor, NY).
С: Селекция: Проводят селекцию стабильно трансфицированных клеток в течение 1 недели в ростовой среде, содержащей 1 мг/см3 генетицина. Отбирают индивидуальные клоны для дальнейшего анализа и проводят позитивные пассажи, как описано далее.
D: Условия культивирования: Клетки, стабильно экспрессирующие ген hGlyT-1b, культивируют при 37°С в атмосфере с содержанием 5% СO2 в среде DMEM-NUT.MIX.F12 и с Glutamax-1 (Gibco), содержащей генетицин (0,5 мг/см3, Gibco) и с добавкой 10% Fetalclone II (Hyclone). Культуру поддерживают в стандартных проветриваемых колбах 80 см2 (2×10-6 м фильтр, Nunc) и клетки при достижении состояния слияния субкультивируют с трипсином (Sigma).
Е: Процедура анализа: Клетки, используемые в исследованиях поглощения, помещают в 96-луночные планшеты (17000 клеток на лунку) без генетицина и культивируют в течение 48 часов перед использованием. Для определения уровня транспорта глицина клетки промывают дважды сбалансированным солевым раствором Хэнкса (HBSS), предварительно нагретым до 37°С, и затем избыток жидкости удаляют с последующим внесением исследуемых соединений, растворенных в 0,200 см3 HBSS. Планшеты инкубируют при 37°С в течение 5 минут, после чего добавляют [3H]глицин (0,050 см3, 150×10-6 М, 248 Бк/нмоль1, NEN) и продолжают инкубацию еще в течение 10 минут. Поглощение останавливают посредством промывания клеток охлажденной ледяной средой HBSS, после чего удаляют избыток жидкости и к каждой лунке добавляют 0,200 см3 сцинтиляционного коктейля. Планшеты закрывают липкой пленкой, встряхивают до достижения гомогенности образцов и проводят сцинтиляционный счет в счетчике для планшетов.
F: Анализ данных: Полученные данные анализируют посредством построения стандартных кривых для вычисления значения plC50 (где рlС50 обозначает отрицательный логарифм концентрации исследуемого соединения, вызывающей 50% ингибирование поглощения).
G: Результаты: Соединения согласно настоящему изобретению селективно ингибируют транспорт глицина переносчиком GlyT-1b человека в сравнении с переносчиком GlyT-2 человека (молекулярное клонирование и функциональная экспрессия переносчика GlyT-2 человека описаны Морроу с соавт. (Morrow, J.A. et al., FEBS letters 1998, 439, 334-340).
В Таблице 1 приведены значения plC50 для ряда соединений согласно настоящему изобретению


| название | год | авторы | номер документа |
|---|---|---|---|
| АРОИЛАМИНО- И ГЕТЕРОАРОИЛАМИНО-ЗАМЕЩЕННЫЕ ПИПЕРИДИНЫ В КАЧЕСТВЕ ИНГИБИТОРОВ GLYT-1 | 2010 |
|
RU2517701C2 |
| Соединения формул (I) и (A), фармацевтическая композиция, лекарственное средство, применение и способ получения соединения формулы (I) | 2018 |
|
RU2822758C2 |
| НОВОЕ ПРОИЗВОДНОЕ КУМАРИНА, ОБЛАДАЮЩЕЕ ПРОТИВООПУХОЛЕВОЙ АКТИВНОСТЬЮ | 2007 |
|
RU2428420C2 |
| ПРОИЗВОДНЫЕ N-ФЕНИЛ(ПИПЕРИДИН-2-ИЛ)МЕТИЛ-БЕНЗАМИДА И ИХ ПРИМЕНЕНИЕ В ТЕРАПИИ | 2004 |
|
RU2351588C2 |
| ЗАМЕЩЕННЫЕ ТРИЦИКЛИЧЕСКИЕ СОЕДИНЕНИЯ И СПОСОБ ПРИМЕНЕНИЯ | 2016 |
|
RU2744766C2 |
| Полусинтетические производные гелиомицина, ингибирующие опухолевый рост | 2016 |
|
RU2644780C2 |
| НОВЫЕ БЕНЗОДИОКСОЛЫ | 2003 |
|
RU2304580C2 |
| АЗОТСОДЕРЖАЩИЕ ГЕТЕРОЦИКЛИЧЕСКИЕ ПРОИЗВОДНЫЕ И ИХ ПРИМЕНЕНИЕ В ЛЕКАРСТВЕННЫХ ПРЕПАРАТАХ | 2014 |
|
RU2694254C1 |
| ЗАМЕЩЕННЫЕ (2R,3R,5R)-3-ГИДРОКСИ-(5-ПИРИМИДИН-1-ИЛ)ТЕТРАГИДРОФУРАН-2-ИЛМЕТИЛ АРИЛ ФОСФОРАМИДАТЫ | 2013 |
|
RU2553996C1 |
| ПОЛИЦИКЛИЧЕСКИЕ СОЕДИНЕНИЯ И СПОСОБЫ ЦЕЛЕНАПРАВЛЕННОЙ ДЕГРАДАЦИИ ПОЛИПЕПТИДОВ БЫСТРО УСКОРЕННОЙ ФИБРОСАРКОМЫ | 2019 |
|
RU2830173C2 |
Настоящее изобретение относится к новым производным спиро[2Н-1-бензопиран-2,4’-пиперидина], имеющим общую формулу (I)
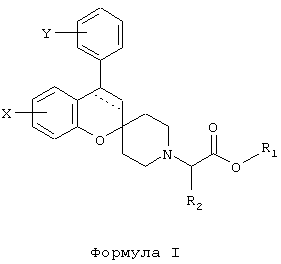
где пунктирная линия обозначает необязательную связь;
Y обозначает 1-4 заместителя, независимо выбираемые из водорода, галогена, (С1-6)алкила (необязательно замещенного одним или более галогенами), (C1-6)алкилокси (необязательно замещенного одним или более галогенами или (С3-6)циклоалкилом), (С2-6)алкенилокси, (С2-6)алкинилокси, (С3-6)циклоалкилокси, (С6-12)арилокси, арилалкилокси, пиридилметокси, SR3, NR3R4, OSO2R5 и NR3SO2R4;
2 заместителя Y могут вместе образовывать O-(СН2)n-O или O-(CF2)n-O, где n принимает значение 1 или 2; или Y обозначает конденсированную (С5-6)арильную группу;
X обозначает 1-3 заместителя, независимо выбираемые из водорода, галогена, гидрокси, (C1-6)алкилокси, и (С1-4)алкила;
r1 обозначает водород, (С1-4)алкил или (C6-12)арил;
r2, r3 и R4 обозначают независимо водород или (C1-4)алкил;
R5 обозначает (С6-12)арил; или его фармацевтически приемлемой соли. Изобретение также относится к фармацевтическим композициям, включающим указанные производные, обладающим активностью в отношении ЦНС. Технический результат - получение новых соединений с ценным фармакологическим действием. 2 н. и 7 з.п. ф-лы, 1 табл.
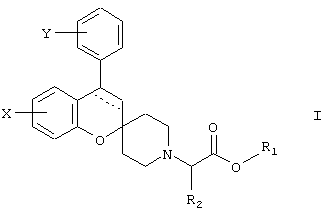
где пунктирная линия обозначает необязательную связь;
Y обозначает 1-4 заместителя, независимо выбираемых из водорода, галогена, (С1-6)алкила (необязательно замещенного одним или более галогенами), (C1-6)алкилокси (необязательно замещенного одним или более галогенами или (С3-6)циклоалкилом), (С2-6)алкенилокси, (С2-6)алкинилокси, (С3-6)циклоалкилокси, (С6-12)арилокси, арилалкилокси, пиридилметокси, SR3, NR3R4, OSO2R5 и NR3SO2R4;
2 заместителя Y могут вместе образовывать O-(СН2)n-О или O-(CF2)n-O, где n принимает значение 1 или 2; или Y обозначает конденсированную (С5-6)арильную группу;
Х обозначает 1-3 заместителя, независимо выбираемых из водорода, галогена, гидрокси, (C1-6)алкилокси, и (С1-4)алкила;
R1 обозначает водород, (С1-4)алкил или (С6-12)арил;
R2, R3 и R4 обозначают независимо водород или (C1-4)алкил;
R5 обозначает (С1-4)алкил (необязательно замещенный одним или более галогенами) или (С6-12)арил (необязательно замещенный (С1-4)алкилом); или его фармацевтически приемлемая соль.
| СПОСОБ ПОЛУЧЕНИЯ ФАРМАЦЕВТИЧЕСКОЙ КОМПОЗИЦИИ ДЛЯ ЛЕЧЕНИЯ СОСТОЯНИЙ БЕСПОКОЙСТВА, ПРОИЗВОДНЫЕ ПИПЕРИДИНА, ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ ДЛЯ ЛЕЧЕНИЯ СОСТОЯНИЙ БЕСПОКОЙСТВА ИЛИ ЭПИЛЕПСИИ | 1992 |
|
RU2142952C1 |
| WO 9522548 А, 24.08.1995 | |||
| WO 9429317 А, 22.12.1994 | |||
| WO 9413678 А, 23.06.1994 | |||
| WO 9418204 А, 18.08.1994. | |||

