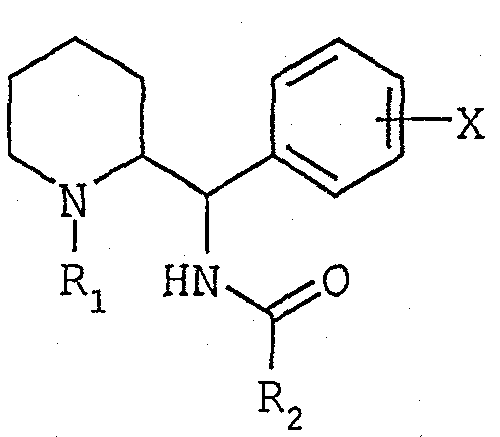
Соединения согласно изобретению отвечают общей формуле (I):
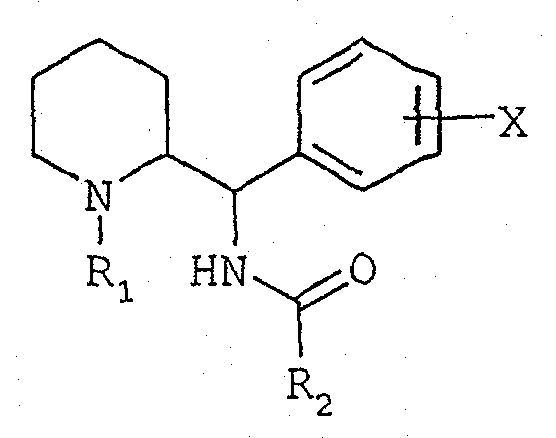
в которой:
R1 обозначает либо атом водорода, либо прямую или разветвленную группу (С1-С7)алкил, возможно замещенную одним или несколькими атомами фтора, либо группу (С3-С7)циклоалкил, либо группу (С3-С7)циклоалкил(С1-С3)алкил, либо группу фенил(С1-С3)алкил, возможно замещенную одной или двумя группами метокси, либо группу (С2-С4)алкенил, либо группу (С2-С4)алкинил,
Х обозначает атом водорода или один или несколько заместителей, выбранных из атомов галогена и групп: трифторметил, прямые или разветвленные (С1-С4)алкилы и (С1-С4)алкоксилы,
R2 обозначает группу, выбранную из групп нафталенил, пиридинил, пиримидинил, пиразинил, пиридазинил, триазинил, инданил, инденил, хинолеинил, изохинолеинил, хиназолинил, хиноксалинил, фталазинил, тиенил, фурил, пирролил, имидазолил, пиразолил, оксазолил, тиазолил, изоксазолил, изотиазолил, тиадиазолил, оксадиазолил, триазолил, бензотиенил, бензофурил, бензимидазолил, бензотиазолил, индолил, изоиндолил, индазолил, бензоксазолил, бензизоксазолил, бензотриазолил, бензизотиазолил, дигидроиндолил, пирролопиридинил, фуропиридинил, тиенопиридинил, имидазопиридинил, оксазолопиридинил, тиазолопиридинил, пиразолопиридинил, изоксазолопиридинил, изотиазолопиридинил, тетрагидрохинолеинил, тетрагидроизохинолеинил, возможно замещенных одним или несколькими заместителями, выбранными из атомов галогена и групп: (С1-С4)алкилы, (С1-С4)алкоксилы, тио(С1-С4)алкилы или фенилы, возможно замещенные одним или несколькими заместителями, выбранными из атомов галогена и групп: трифторметил, (С1-С4)алкилы и (С1-С4)алкоксилы.
Соединения общей формулы (I) могут существовать в форме рацемата конфигурации трео (1R,2R, 1S,2S) или в форме энантиомеров (1R,2R) или (1S,2S); они могут находиться в форме свободных оснований или аддитивных солей с кислотами.
Соединения, имеющие структуру, аналогичную структуре соединений согласно изобретению, описаны в патенте США 5254569 в качестве анальгетических, диуретических, антиконвульсивных, анестезирующих, седативных, церебропротекторных средств благодаря механизму, воздействующему на опиатные рецепторы. Другие соединения аналогичной структуры описаны в заявке на патент ЕР-0499995 в качестве антагонистов 5-НТ3, пригодных для лечения психотических расстройств, неврологических болезней, желудочных симптомов, тошноты и рвоты.
Предпочтительные соединения согласно изобретению не имеют активности в отношении опиатных рецепторов или рецепторов 5-НТ3, но обладают конкретной активностью - они являются специфическими ингибиторами переносчиков глицина glyt 1 и/или glyt 2.
Соединения общей формулы (I), в которой R1 не является атомом водорода, могут быть получены способом, представленным на нижеследующей схеме 1.
Осуществляют реакцию сочетания диамина общей формулы (II), в которой R1 и Х имеют значения, описанные выше (при условии, что R1 не является атомом водорода), с активированной кислотой или с хлорангидридом кислоты общей формулы (III), в которой Y обозначает активированную группу ОН или атом хлора и R2 имеет значения, описанные выше, с использованием методов, известных специалисту.
Схема 1
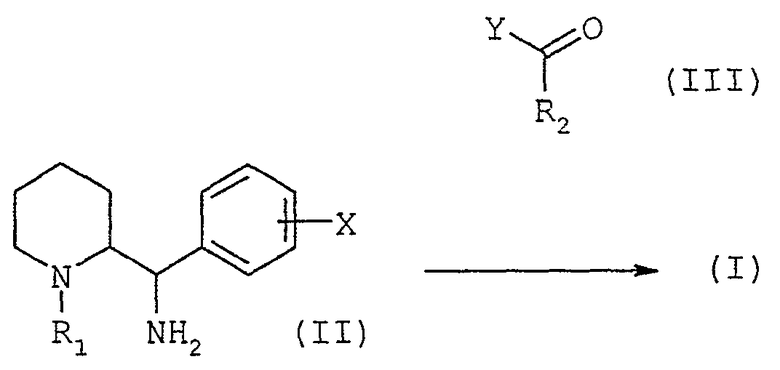
Диамин общей формулы (II) может быть получен способом, представленным на схеме 2, приведенной ниже.
Вводят во взаимодействие амид Вейнреба формулы (IV) с производным фениллития общей формулы (V), в которой Х имеет значения, описанные выше, в эфирном растворителе, таком как диэтиловый эфир, при температуре от -30°С до температуры окружающей среды; получают кетон общей формулы (VI), который восстанавливают до спирта конфигурации трео общей формулы (VII), с помощью восстановителя, такого как К-Selectride® или L-Selectride® (три-втор-бутилборгидрид калия или лития) в эфирном растворителе, таком как тетрагидрофуран, при температуре от -78°С до температуры окружающей среды. Карбамат общей формулы (VII) может быть затем восстановлен до трео-N-метиламиноспирта общей формулы (VIII) при действии смешанного гидрида, такого как двойной гидрид алюминия и лития, в эфирном растворителе,
Схема 2
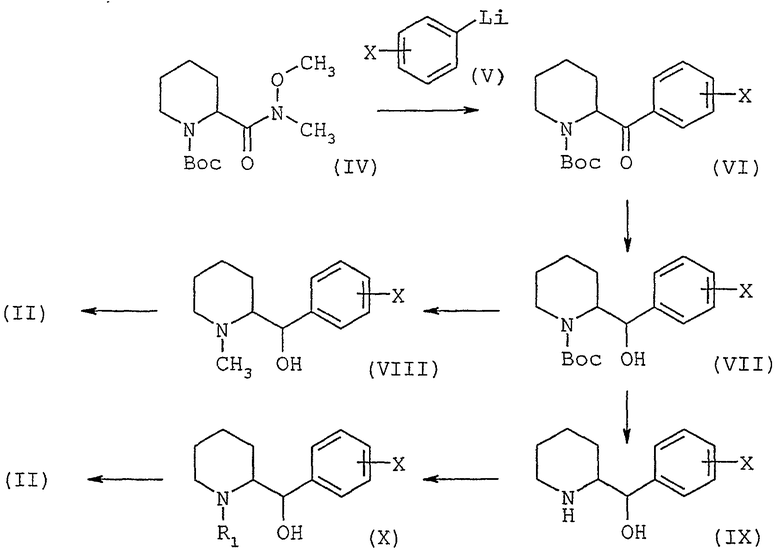
таком как тетрагидрофуран, при температуре от температуры окружающей среды до температуры кипения с обратным холодильником. Превращают затем трео-спирт общей формулы (VIII) в промежуточное трео-соединение общей формулы (II), в которой R1 обозначает группу метил, в две стадии: сначала превращают спиртовую группу в нуклеофобную, например, в метансульфонатную, при действии метилсульфонилхлорида, в хлорсодержащем растворителе, таком как дихлорметан, и в присутствии основания, такого как триэтиламин, при температуре в интервале от 0°С до температуры окружающей среды, затем подвергают нуклеофобную группу взаимодействию с жидким аммиаком при -50°С в спирте, таком как этанол, в закрытой среде, такой как автоклав, при температуре в интервале от -50°С до температуры окружающей среды. Можно также снять защитную группу с карбамата общей формулы (VII) с помощью сильного основания, такого как водный раствор едкого калия, в спирте, таком как метанол, для получения трео-аминоспирта общей формулы (IX), затем осуществить N-алкилирование при помощи галогенпроизводного формулы R1Z, в которой R1 имеет значение, описанное выше, но не является водородом, и Z обозначает атом галогена, в присутствии основания, такого как карбонат калия, в полярном растворителе, таком как N,N-диметилформамид, при температуре от температуры окружающей среды до 100°С. Обрабатывают полученный таким образом спирт общей формулы (X) так, как описано выше в отношении спирта общей формулы (VIII).
Другой вариант способа, иллюстрированный схемой 3, представленной ниже, может быть использован в случае, когда R1 обозначает метил и Х обозначает атом водорода. Кватернизируют пиридиноксим формулы (XI), например, при действии метилтрифторметансульфоната в эфирном растворителе, таком как диэтиловый эфир, при температуре окружающей среды. Полученную соль пиридиния формулы (XII) подвергают гидрированию в атмосфере водорода, в присутствии катализатора, такого как оксид платины, в смеси спирта и водного раствора кислоты, такой как смесь этанола и 1N раствора хлористоводородной кислоты. Получают диамин общей формулы (II), в которой R1 является метилом, а Х - атомом водорода, в виде смеси двух диастереоизомеров трео/эритро 9/1. Можно из него получить соль, например, со щавелевой кислотой, затем очистить продукт путем перекристаллизации образованного оксалата из смеси спирта и эфирного растворителя, такой как смесь метанола и диэтилового эфира, для получения чистого диастереоизомера трео (1R,2R; 1S,1S).
Схема 3
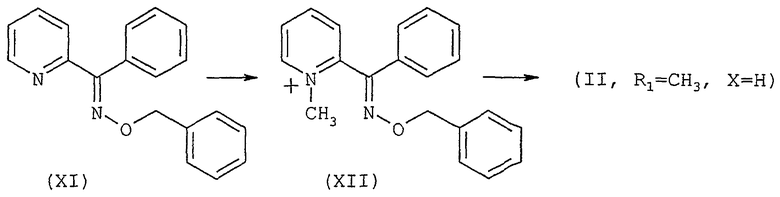
Соединения общей формулы (II), в которой R1 обозначает атом водорода, могут быть получены согласно схеме 2, с использованием соединения общей формулы (I), в которой R1 обозначает либо фенилметильную группу, возможно замещенную, с последующим снятием защитной группы с атома азота пиперидинового цикла, например, с помощью окисляющего агента или кислоты Льюиса, такой как трибромид бора, или путем гидрогенолиза, либо алкенильную группу, предпочтительно, аллильную, и снимают защитную группу с атома азота комплексом Pd0 для получения соединения общей формулы (I), в которой R1 обозначает атом водорода.
Кроме того, хиральные соединения общей формулы (I), соответствующие энантиомерам (1R,2R) или (1S,2S) диастереоизомера трео, могут быть получены путем разделения рацемических соединений путем высокоэффективной жидкостной хроматографии (ВЭЖХ) на хиральной колонке, или путем расщепления рацемического амина общей формулы (II) с использованием хиральной кислоты, такой как винная кислота, камфорсульфоновая кислота, дибензоилвинная кислота или N-ацетиллейцин, путем фракционной и предпочтительной перекристаллизации диастереоизомерной соли в спиртовом растворителе, или же путем энантиоселективного синтеза согласно схеме 2 с использованием хирального амида Вейнреба формулы (IV).
Рацемический или хиральный амид Вейнреба формулы (IV), а также кетон общей формулы (VI) могут быть получены по аналогии с методом, который описан в Eur. J. Med. Chem., 35, (2000), 979-988 J. Med. Chem., 41, (1998), 591-601. Фениллитиевое соединение общей формулы (V), в которой Х обозначает атом водорода, является коммерчески доступным продуктом. Замещенные производные этого соединения могут быть получены по аналогии с методом, который описан в Tetrahedron Lett., 57, 33, (1996), 5905-5908. Пиридиноксим общей формулы (XI) может быть получен по аналогии с методом, который описан в заявке на патент ЕР-0366006. Амин общей формулы (IX), в которой Х обозначает атом водорода, может быть получен в хиральной форме согласно методу, описанному в патенте США 2928835. Наконец, амин общей формулы (XIII) может быть получен по аналогии с методом, описанным в Chem. Pharm. Bull., 32, 12,(1984), 4893-4906 и Synthesis, (1976), 593-595.
Кислоты и хлорангидриды кислот общей формулы (III) являются коммерчески доступными продуктами, за исключением 4-[2-хлор-3-(трифторметил)фенил]-1Н-имидазол-2-карбоновой кислоты, которая может быть получена в условиях, сопоставимых с условиями, описанными в заявке на патент ЕР-0365030 и в патенте США 3336300.
Нижеописанные примеры иллюстрируют получение некоторых соединений согласно изобретению. Элементные микроанализы, ИК-спектры и ЯМР и ВЭЖХ на хиральной колонке подтверждают структуру и энантиомерную чистоту полученных соединений.
Номера, указанные в скобках в названиях примеров, соответствуют номерам 1-й колонки приведенной ниже таблицы.
ПРИМЕР 1 (СОЕДИНЕНИЕ № 4)
Гидрохлорид трео-2,5-дихлор-N-[(1-метил-2-пиперидинил)(фенил)метил]-3-тиофенкарбоксамида 1:1
1.1 Трифторметансульфонат 2-(бензилоксииминофенилметил)-1-метилпиридиния
К суспензии 35 г (120 ммоль) фенил(пиридин-2-ил)метанон-О-бензилоксима в 200 мл диэтилового эфира добавляют по каплям при 0°С 17,4 мл (120 ммоль) метилтрифторметансульфоната и перемешивают смесь при температуре окружающей среды в течение 3 часов. Отделяют образовавшийся осадок фильтрацией и сушат при пониженном давлении.
Получают 49 г продукта, который используют как таковой на следующей стадии.
1.2 Этандиоат трео-(1-метилпиперидин-2-ил)-фенилметанамина 2:1
В склянку Парра помещают 14,8 г (31,89 ммоль) трифторметансульфоната 2-(бензилоксииминофенилметил)-1-метилпиридиния и 0,74 г оксида платины в 50 мл этанола и 50 мл 1N раствора хлористоводородной кислоты и осуществляют гидрирование в течение 5 часов.
Испаряют этанол при пониженном давлении, экстрагируют остаток дихлорметаном, отделяют водную фазу, добавляют в нее раствор аммиака и экстрагируют дихлорметаном. После промывки объединенных органических фаз, сушки над сульфатом натрия, фильтрации и испарения растворителя при пониженном давлении, получают 6,7 г маслянистого продукта, содержащего 10% эритро диастереоизомера.
Получают этандиоат при растворении указанных 6,7 г продукта в метаноле путем действия двух эквивалентов этандикислоты, растворенной в минимальном количестве метанола.
Очищают полученную соль перекристаллизацией из смеси метанола и диэтилового эфира.
В заключение выделяют 4,7 г этандиоата в виде чистого трео диастереоизомера.
Т. плавления: 156-159°С.
1.3 Гидрохлорид трео-2,5-дихлор-N-[(1-метил-2-пиперидинил)(фенил)метил]тиофен-3-карбоксамида 1:1
В колбу емкостью 100 мл вводят 0,768 г (4 ммоль) 2,5-дихлортиофен-3-карбоновой кислоты, растворенной в 15 мл дихлорметана, затем в нее последовательно вводят 0,651 мл (4,7 ммоль) триэтиламина и 0,447 мл (4,7 ммоль) этилхлорформиата и перемешивают реакционную смесь при температуре окружающей среды в течение 2 часов.
Добавляют 0,80 г (3,9 ммоль) трео-(1-метилпиперидин-2-ил)фенилметанамина, растворенного в 15 мл дихлорметана, и продолжают перемешивание при температуре окружающей среды в течение 12 часов.
Обрабатывают смесь водой и экстрагируют несколько раз дихлорметаном. После промывки органических фаз водой, затем 1N водным раствором едкого натра, сушки над сульфатом натрия, фильтрации и испарения растворителя при пониженном давлении остаток очищают хроматографией на колонке из силикагеля, элюируя смесью 97/3 до 95/5 дихлорметана и метанола.
Получают 0,6 г маслянистого продукта, из которого получают гидрохлорид путем добавления 0,1N раствора хлористоводородной кислоты в пропан-2-оле.
После испарения растворителей при пониженном давлении перекристаллизовывают полученное белое твердое вещество из смеси изопропилового эфира и пропан-2-ола.
В заключение выделяют 0,474 г гидрохлорида в виде твердого вещества белого цвета.
Т. плавления: 216-217°С.
ПРИМЕР 2 (СОЕДИНЕНИЕ № 5)
Гидрохлорид 2,5-дихлор-N-[(S)-[(2S)-1-метил-2-пиперидинил](фенил)метил]тиофен-3-карбоксамида 1:1
2.1 1,1-диметилэтил-(2S)-2-бензоилпиперидин-1-карбоксилат
В колбу емкостью 500 мл вводят в атмосфере азота 11,8 г (43,3 ммоль) 1,1-диметилэтил-(2S)-2-(N-метокси-N-метилкарбамоил)пиперидин-1-карбоксилата в 100 мл безводного диэтилового эфира, охлаждают среду до -23°С, прикапывают 21,6 мл (43,2 ммоль) 1,8М раствора фениллития в смеси 70/30 циклогексана и диэтилового эфира и перемешивают смесь при температуре окружающей среды в течение 3 часов. После гидролиза насыщенным водным раствором хлорида натрия отделяют водную фазу и экстрагируют этилацетатом. Сушат органическую фазу над сульфатом натрия, фильтруют, концентрируют при пониженном давлении и очищают остаток путем хроматографии на колонке из силикагеля, элюируя смесью этилацетата и циклогексана.
Получают 4,55 г твердого продукта.
Т. плавления: 123-125°С.
[α]25 D=-25,4° (c=2,22; CH2Cl2) ее=97,2%.
2.2 1,1-диметилэтил-(1S)-2-[(2S)-гидрокси(фенил)метил]пиперидин-1-карбоксилат
В колбу емкостью 500 мл вводят в атмосфере азота 4,68 г (16,2 ммоль) 1,1-диметилэтил-(2S)-2-бензоилпиперидин-1-карбоксилата в 170 мл безводного тетрагидрофурана, охлаждают раствор до -78°С, прикапывают 48,5 мл (48,5 ммоль) 1М раствора L-Selectride® (три-втор-бутилборгидрид лития) в тетрагидрофуране и перемешивают смесь при температуре окружающей среды в течение 5 часов.
Медленно гидролизуют на холоду с помощью 34 мл воды и 34 мл 35%-ного водного раствора перекиси водорода и оставляют смесь при перемешивании на 2 часа до достижения температуры окружающей среды. Разбавляют смесь водой и этилацетатом, отделяют водную фазу и экстрагируют этилацетатом. После промывки объединенных органических фаз, сушки, сушки над сульфатом натрия, фильтрации и испарения остаток очищают хроматографией на колонке с силикагелем, элюируя смесью этилацетата и циклогексана.
Получают 4,49 г бледно-желтого масла.
[α]25 D=+63,75° (c=0,8; CH2Cl2) ее=97,8%.
2.3 (1S)-[(2S)-(1-метилпиперидин-2-ил)]фенилметанол
В колбу емкостью 200 мл вводят в атмосфере азота 2,96 г (78,1 ммоль) двойного гидрида алюминия и лития в 50 мл безводного тетрагидрофурана, кипятят смесь с обратным холодильником, добавляют 4,49 г (15,4 ммоль) раствора 1,1-диметилэтил-(1S)-2-[(2S)-гидрокси(фенил)метил]пиперидин-1-карбоксилата в 35 мл тетрагидрофурана и выдерживают смесь при кипении с обратным холодильником в течение 3,5 часов.
Смесь охлаждают, медленно гидролизуют 0,1М раствором двойного тартрата калия и натрия и оставляют смесь при перемешивании на ночь. Фильтруют и осадок промывают тетрагидрофураном, затем фильтрат концентрируют при пониженном давлении.
Получают 2,95 г бесцветного маслянистого продукта.
2.4 (1S)-[(2S)-(1-метилпиперидин-2-ил)]фенилметанамин
В колбу емкостью 250 мл вводят в атмосфере азота 2,95 г (14,4 ммоль) (1S)-[(2S)-(1-метилпиперидин-2-ил)]фенилметанола и 2 мл (14,4 ммоль) триэтиламина в 70 мл безводного дихлорметана, охлаждают среду до 0°С, добавляют 1,1 мл (14,4 ммоль) метансульфонилхлорида, оставляют смесь на 2 часа для медленного возвращения ее к температуре окружающей среды и затем концентрируют при пониженном давлении.
В автоклав, снабженный магнитной мешалкой и охлажденный до -50°С, вводят жидкий аммиак, добавляют раствор только что полученного метансульфоната в 30 мл абсолютного этанола, закрывают автоклав и перемешивают содержимое в течение 48 часов.
Выливают смесь в колбу, испаряют растворители при пониженном давлении и выделяют амин в виде маслянистого продукта, который используют как таковой на следующей стадии.
2.5 Гидрохлорид 2,5-дихлор-N-[(S)-[(2S)-1-метил-2-пиперидинил](фенил)метил]тиофен-3-карбоксамида 1:1
В колбу емкостью 250 мл вводят 0,37 г (1,88 ммоль) 2,5-дихлортиофен-3-карбоновой кислоты в 15 мл дихлорметана, последовательно добавляют 0,31 мл (2,25 ммоль) триэтиламина и 0,21 мл (2,25 ммоль) этилхлорформиата и перемешивают смесь при температуре окружающей среды в течение 1 часа.
Добавляют 0,38 г (1,88 ммоль) (1S)-[(2S)-(1-метилпиперидин-2-ил)]фенилметанамина, растворенного в 10 мл дихлорметана, и продолжают перемешивание в течение 12 часов при температуре окружающей среды.
Обрабатывают смесь водой, экстрагируют несколько раз дихлорметаном, объединяют органические фазы, промывают их 1N водным раствором едкого натра, сушат их над сульфатом натрия, фильтруют и концентрируют фильтрат при пониженном давлении.
Сырой остаток очищают хроматографией на колонке с силикагелем, элюируя смесью 98/2 дихлорметана и метанола, содержащего 0,1% гидрата окиси аммония. Получают 0,368 г маслянистого продукта, из которого получают гидрохлорид путем добавления 0,1N раствора хлористоводородной кислоты в пропан-2-оле.
После испарения растворителя при пониженном давлении твердый продукт перекристаллизовывают из смеси пропан-2-ола и изопропилового эфира.
В заключение выделяют 0,36 г гидрохлорида в виде бледно-желтого твердого продукта.
Т. плавления: 134-136°С [α]25 D=+45,6° (c=0,99); CH3ОН.
ПРИМЕР 3 (СОЕДИНЕНИЕ № 18)
Гидрохлорид трео-4-[2-хлор-3-(трифторметил)-фенил]-N-[(1-метил-2-пиперидинил)(фенил)метил]-1Н-имидазол-2-карбоксамида 1:1
В колбу емкостью 50 мл вводят 0,1 г (0,344 ммоль) 4-[2-хлор-3-(трифторметил)фенил]-1Н-имидазол-2-карбоновой кислоты, 0,066 г (0,344 ммоль) гидрохлорида 1-[3-(диметиламино)пропил]-3-этилкарбодиимида, 0,047 г (0,344 ммоль) 1-гидроксибензотриазола, растворенного в 10 мл дихлорметана, и перемешивают при температуре окружающей среды в течение 5 минут.
Добавляют 0,072 г (0,344 ммоль) трео-(1-метилпиперидин-2-ил)фенилметанамина (приготовленного согласно примеру 1.2) в нескольких мл дихлорметана и продолжают перемешивание в течение 6 часов.
Обрабатывают смесь водой, экстрагируют несколько раз дихлорметаном, промывают органические фазы водой, затем 1N водным раствором едкого натра, затем насыщенным водным раствором хлорида натрия, сушат их над сульфатом натрия и испаряют растворитель при пониженном давлении. Очищают остаток хроматографией на колонке с силикагелем, элюируя смесью дихлорметана и метанола.
Получают 91 мг продукта, из которого получают гидрохлорид путем добавления 0,1N раствора хлористоводородной кислоты в пропан-2-оле. Частично испаряют растворитель при пониженном давлении и получают после кристаллизации, 104 мг твердого соединения белого цвета.
Т. плавления: 188-195°С.
Таблица иллюстрирует химическую структуру и физические свойства некоторых соединений согласно изобретению.
В колонке “Соль“ знак “-“ обозначает соединение в форме основания и “HCl” обозначает гидрохлорид.
Вращательная способность соединения №5 составляет [α]25 D=+45,6° (c=0,99); CH3ОН.
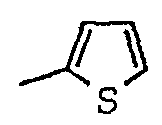
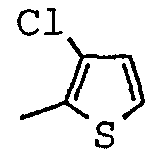
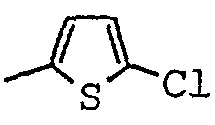
Соединения согласно изобретению были подвергнуты серии фармакологических испытаний и проявили себя интересными как вещества с терапевтической активностью.
Исследование переноса глицина в клетках SK-N-MC, экспрессирующих нативный переносчик glyt1 человека
Захват [14С]глицина изучался на клетках SK-N-MC (нейро-эпителиальные клетки человека), экспрессирующих нативный переносчик glyt 1 человека, путем измерения включенной радиоактивности в присутствии или в отсутствие исследуемого соединения. Клетки культивировали в монослое в течение 48 часов в планшетах, предварительно обработанных 0,02%-ным фибронектином. В день эксперимента культуральную среду удаляли и клетки промывали буфером Krebs-HEPES (4-(2-гидроксиэтил)пиперазин-1-этансульфоновая кислота) с рН 7,4. После 10 минут предварительной инкубации при 37°С в присутствии либо буфера (контрольная партия), либо исследуемого соединения при различных концентрациях или 10 мМ глицина (определение неспецифического захвата) вводили 10 мкМ [14С]глицина (специфическая активность 112 mCi/ммоль). Инкубирование продолжали 10 минут при 37°С, и реакцию останавливали путем 2 промывок буфером Krebs-HEPES с рН 7,4. Включенную в клетки радиоактивность измеряли после введения 100 мкл сцинтилляционной жидкости и перемешивания в течение 1 часа. Подсчет осуществляли на счетчике Microbeta Tri-luxTM. Эффективность соединения определяли по значению CI50, представляющему собой концентрацию соединения, при которой снижается на 50% специфический захват глицина, выражаемый разностью уровней радиоактивности, включенной в контрольную партию и в партию, в которой находился глицин в концентрации 10 мМ.
Соединения согласно изобретению имели в данном тесте значение CI50 порядка от 0,01 до 10 мкМ.
Изучение переноса глицина в гомогенате клеток спинного мозга мыши
Захват [14С]глицина переносчиком glyt2 изучался в гомогенате клеток спинного мозга мыши путем измерения включенной радиоактивности в присутствии или в отсутствие исследуемого соединения.
После умерщвления животных (самцы мышей породы OF1 Iffa Crédo весом 20-25 г в день эксперимента) быстро извлекали у каждого животного спинной мозг, взвешивали и хранили на льду. Образцы гомогенизировали в буфере Krebs-HEPES (4-(2-гидроксиэтил)пиперазин-1-этансульфоновая кислота) с рН 7,4 из расчета 25 мл/г ткани.
Предварительно инкубировали 50 мкл гомогената в течение 10 минут при 25°С в присутствии буфера Krebs-HEPES с рН 7,4 и исследуемого соединения при различных концентрациях, или 10 мМ глицина для определения неспецифического захвата. Затем добавляли [14C]глицин (специфическая активность = 112 mCi/ммоль) в течение 10 минут при 25°С до конечной концентрации 10 мкМ. Реакцию останавливали фильтрацией в вакууме, и радиоактивность определяли путем сцинтилляции твердой пробы и подсчета на счетчике Microbeta TriluxTM. Эффективность соединения определяли по концентрации CI50, при которой снижается на 50% специфический захват глицина, выражаемый разностью между уровнем радиоактивности, включенной в контрольную партию и в партию, в которой находился глицин в концентрации 10 мМ.
Соединения согласно изобретению в данном тесте имели значение CI50 порядка от 0,01 до 10 мкМ.
Эти результаты показывают, что соединения согласно изобретению могут быть использованы для лечения поведенческих нарушений, связанных с деменцией, психозов, в частности, шизофрении (дефицитная (негативная) форма и продуктивная форма) и острых или хронических экстрапирамидальных симптомов, наведенных нейролептиками, для лечения различных форм страха, приступов паники, фобий, компульсивных навязчивых состояний, для лечения различных форм депрессии, включая психотическую депрессию, для лечения расстройств, связанных со злоупотреблением алкоголя или с прекращением принятия алкоголя, нарушений сексуального поведения, нарушений приема пищи и для лечения мигрени.
Кроме того, соединения согласно изобретению могут быть использованы для лечения болезненных мышечных контрактур в ревматологии и в случае острых спинальных патологий, для лечения спастических контрактур мозгового и спинно-мозгового происхождения, для симптоматического лечения острых и подострых болей легкой и умеренной интенсивности, для лечения интенсивных и/или хронических болей, неврогенных и непрекращающихся болей, для лечения болезни Паркинсона и симптомов болезни Паркинсона нейродегенеративного происхождения или наведенных нейролептиками, для лечения генерализованных первичных и вторичных эпилепсий, очаговых эпилепсий с простой или сложной симптоматикой, смешанных и других эпилептических синдромов в сочетании с другим антиэпилептическим лечением, или в монотерапии, для лечения апное во сне и в качестве нейропротекторов.
В этой связи настоящее изобретение относится также к фармацевтическим композициям, содержащим эффективную дозу, по меньшей мере, одного соединения согласно изобретению, в форме фармацевтически приемлемого основания или соли или сольвата, при необходимости, в смеси с подходящими эксципиентами.
Указанные эксципиенты выбирают в зависимости от фармацевтической формы и желаемого способа введения лекарства.
Таким образом, фармацевтические композиции согласно изобретению могут быть предназначены для перорального приема, для введения под язык, для подкожного, внутримышечного, внутривенного введения, для топического нанесения, для интратрахеального, назального, трансдермального, ректального, внутриглазного введения.
Разовые вводимые формы могут быть представлены, например, в виде таблеток, желатиновых капсул, гранул, порошков, растворов или суспензий для перорального приема или для инъекций, трансдермальных наклеек (“пластырей“), свечей. Для топического нанесения можно предусмотреть помады, лосьоны и глазные капли. Указанные разовые формы выпускаются в дозах, позволяющих ежедневно принимать от 0,01 до 20 мг активного начала на кг веса тела пациента в зависимости от галеновой формы.
Для получения таблеток добавляют к активному началу, находящемуся в микронизированной или в немикронизированной форме, фармацевтический носитель, который может представлять собой соединение-разбавитель, как, например, лактоза, микрокристаллическая целлюлоза, крахмал, и вспомогательные вещества для приготовления лекарственной формы как, например, связующие (поливинилпирролидон, гидроксипропилметилцеллюлоза и т.д.), агенты для придания текучести, как, например, диоксид кремния, смазывающие агенты, как, например, стеарат магния, стеариновая кислота, трибегенат глицерина, стеарилфумарат натрия. Можно также вводить смачивающие вещества или поверхностно-активные вещества, такие как лаурилсульфат натрия.
Технологией получения может служить прямое прессование, сухое гранулирование, влажное гранулирование или горячее плавление.
Таблетки могут выпускаться без покрытия, или в виде драже, покрытого сахаром, или могут быть покрыты различными полимерами или другими соответствующими материалами. Они могут выпускаться в форме, пригодной для быстрого высвобождения активного начала, для отсроченного или пролонгированного высвобождения активного начала благодаря полимерным матрицам или специфическим полимерам, используемым для покрытия.
Для получения желатиновых капсул смешивают активное начало с фармацевтическим сухим носителем (обычное смешивание, сухое или влажное гранулирование, или горячее плавление), с жидким или полутвердым носителем.
Желатиновые капсулы могут быть твердыми или мягкими, с оболочкой или без оболочки, могут иметь быструю активность, пролонгированную или отсроченную активность (например, в случае лекарственной формы для лечения кишечника).
Композиция в форме сиропа или эликсира, или для приема в виде капель может содержать активное вещество вместе с подсластителем, предпочтительно, бескалорийным, метилпарабеном или пропилпарабеном в качестве антисептика, с вкусовым агентом и красителем.
Порошки и гранулы, диспергируемые в воде, могут содержать активное начало в смеси с диспергируемыми агентами или со смачивающими агентами, или с диспергирующими агентами, как, например, поливинилпирролидон, а также с подсластителями и корректорами вкуса. Для ректального введения используют свечи, приготовленные со связующими, плавящимися при ректальной температуре, например, масло какао или полиэтиленгликоль.
Для парентерального введения используют водные суспензии, изотонические солевые растворы или стерильные растворы для инъекций, содержащие фармакологически приемлемые диспергируемые и/или смачивающие агенты, например, пропиленгликоль или бутиленгликоль.
Активное начало может входить в состав микрокапсул, возможно, с одним или несколькими носителями или добавками, или с полимерной матрицей или с циклодекстрином (трансдермальные пластыри, формы с пролонгированным высвобождением).
Композиции для топического нанесения согласно изобретению содержат среду, совместимую с кожей. Они могут находиться, в частности, в форме водных, спиртовых или водно-спиртовых растворов, гелей, эмульсий типа вода-в-масле или масло-в-воде, имеющих вид крема или геля, микроэмульсий, аэрозолей, или же в форме везикулярных дисперсий, содержащих ионогенные и/или неионогенные липиды. Эти галеновые формы готовят согласно методам, практикующимся в соответствующей области.
Наконец, фармацевтические композиции согласно изобретению могут содержать наряду с соединением общей формулы (I) другие активные начала, которые могут быть использованы при лечении расстройств или болезней, указанных выше.
| название | год | авторы | номер документа |
|---|---|---|---|
| ПРОИЗВОДНЫЕ N-[ФЕНИЛ(ПИРРОЛИДИН-2-ИЛ)МЕТИЛ]БЕНЗАМИДА И N-[(АЗЕПАН-2-ИЛ)ФЕНИЛМЕТИЛ]БЕНЗАМИДА, СПОСОБ ИХ ПОЛУЧЕНИЯ И ИХ ПРИМЕНЕНИЕ В ТЕРАПИИ | 2004 |
|
RU2386614C2 |
| ПРОИЗВОДНЫЕ N-ГЕТЕРОЦИКЛИЛМЕТИЛБЕНЗАМИДОВ, ИХ ПОЛУЧЕНИЕ И ИХ ПРИМЕНЕНИЕ В ТЕРАПИИ | 2004 |
|
RU2346945C2 |
| ПРОИЗВОДНЫЕ N-[ФЕНИЛ(АЛКИЛПИПЕРИДИН-2-ИЛ)МЕТИЛ]БЕНЗАМИДА И ИХ ПРИМЕНЕНИЕ В ТЕРАПИИ | 2004 |
|
RU2351589C2 |
| ПРОИЗВОДНЫЕ N-[ГЕТЕРОАРИЛ(ПИПЕРИДИН-2-ИЛ)МЕТИЛ]БЕНЗАМИДА И ИХ ПРИМЕНЕНИЕ В ТЕРАПИИ | 2004 |
|
RU2351596C2 |
| (ГЕТЕРО)АРИЛЦИКЛОПРОПИЛАМИНЫ В КАЧЕСТВЕ ИНГИБИТОРОВ LSD1 | 2012 |
|
RU2681211C2 |
| ПРОИЗВОДНЫЕ БОРОНОВОЙ КИСЛОТЫ | 2018 |
|
RU2793315C2 |
| НОВЫЕ ПРОИЗВОДНЫЕ ЦИКЛОПЕНТАНА | 2010 |
|
RU2572555C2 |
| ПРОИЗВОДНЫЕ ЦИКЛОПРОПИЛАМИНА В КАЧЕСТВЕ МОДУЛЯТОРОВ H-ГИСТАМИНОВОГО РЕЦЕПТОРА | 2007 |
|
RU2449989C2 |
| НОВЫЕ КОНЪЮГАТЫ СВЯЗЫВАЮЩЕЕ СОЕДИНЕНИЕ - АКТИВНОЕ СОЕДИНЕНИЕ (ADC) И ИХ ПРИМЕНЕНИЕ | 2012 |
|
RU2610336C2 |
| ГИДРОКСИЭТИЛАМИНОСУЛЬФОНАМИДЫ, ПРОМЕЖУТОЧНЫЕ СОЕДИНЕНИЯ, ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ, СПОСОБ ИНГИБИРОВАНИЯ РЕТРОВИРУСНЫХ ПРОТЕАЗ, СПОСОБ ЛЕЧЕНИЯ РЕТРОВИРУСНЫХ ИНФЕКЦИЙ, СПОСОБ ЛЕЧЕНИЯ СПИДА | 1993 |
|
RU2173680C2 |
Изобретение относится к новым соединениям, отвечающим общей формуле (I):
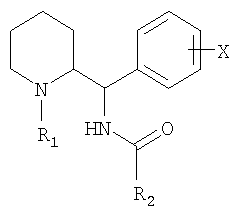
в которой: R1 обозначает прямой или разветвленный (С1-С7)алкил, X обозначает атом водорода, R2 обозначает группу, выбранную из нафталенила, пиридинила, изохинолеинила, тиенила, имидазолила, бензотиенила, бензимидазолила, индолила, бензотриазолила и возможно замещенную одним или несколькими заместителями, выбранными из атомов галогена и следующих групп: (С1-С4)алкилы, тио(С1-С4)алкилы или фенилы, возможно замещенные одним или несколькими заместителями, выбранными из атомов галогена или трифторметила, в форме свободного основания или аддитивной соли с кислотой. Изобретение также относится к лекарственному средству, к фармацевтической композиции, а также к применению. Технический результат - получение новых биологически активных соединений, обладающих активностью в отношении специфических ингибиторов переносчиков глицина glyt 1 и/или glyt 2. 5 н. и 1 з.п. ф-лы, 1 табл.
1. Соединение, отвечающее общей формуле (I):
в которой
R1 обозначает прямой или разветвленный (С1-С7)алкил, X обозначает атом водорода,
R2 обозначает группу, выбранную из нафталенила, пиридинила, изохинолеинила, тиенила, имидазолила, бензотиенила, бензимидазолила, индолила, бензотриазолила и возможно замещенную одним или несколькими заместителями, выбранными из атомов галогена и следующих групп: (С1-С4)алкилы, тио(С1-С4)алкилы или фенилы, возможно замещенные одним или несколькими заместителями, выбранными из атомов галогена или трифторметила, в форме свободного основания или аддитивной соли с кислотой.
2. Соединение по п.1, отличающееся тем, что оно выбрано из следующих соединений:
[(1-метил-пиперидин-2-ил)-фенил-метил]-амид-2,5-дихлор-тиофен-3-карбоновой кислоты;
[(1 -метил-пиперидин-2-ил)-фенил-метил]-амид-1 -бензил- 1H-индол-4-карбоновой кислоты;
[(1-метил-пиперидин-2-ил)-фенил-метил]-амид-4-бром-2-этил-5-метил-2Н-пиразол-3-карбоновой кислоты;
[(1-метил-пиперидин-2-ил)-фенил-метил]-амид-1-бензил-2-этил-5-метокси-1 Н-индол-3 -карбоновой кислоты;
[(1-метил-пиперидин-2-ил)-фенил-метил]-амид-4-(2-хлор-3-трифторметил-фенил)-1Н-имидазол-2-карбоновой кислоты;
[(1-метил-пиперидин-2-ил)-фенил-метил]-амид-4,6-дихлор-1Н-индол-2-карбоновой кислоты;
[(1-метил-пиперидин-2-ил)-фенил-метил]-амид-5-(4-хлор-фенил)-2-трифторметил-фуран-3-карбоновой кислоты;
[(1-метил-пиперидин-2-ил)-фенил-метил]-амид-2,5-дихлор-тиофен-3-карбоновой кислоты;
[(1-метил-пиперидин-2-ил)-фенил-метил]-амид-4-метил-5-фенил-тиофен-2-карбоновой кислоты;
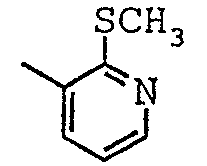
N-[(1-метил-пиперидин-2-ил)-фенил-метил]-2-метилсульфанил-никотинамид;
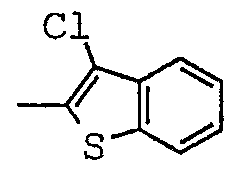
[(1-метил-пиперидин-2-ил)-фенил-метил]-амид-3-хлор-бензо[b]тиофен-2-карбоновой кислоты;
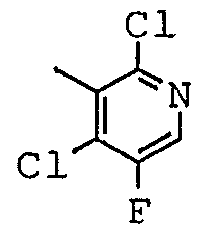
2,4-дихлор-5-фтор-N-[(1-метил-пиперидин-2-ил)-фенил-метил]-никотинамид;
[(1-метил-пиперидин-2-ил)-фенил-метил]-амид-4,5-дибромтиофен-2-карбоновой кислоты;
[(1-метил-пиперидин-2-ил)-фенил-метил]-амид-бензофуран-4-карбоновой кислоты;
[(1-метил-пиперидин-2-ил)-фенил-метил]-амид-2-метил-хинолин-4-карбоновой кислоты;
[(1-метил-пиперидин-2-ил)-фенил-метил]-амид-1-метил-1Н-индол-3-карбоновой кислоты;
[(1-метил-пиперидин-2-ил)-фенил-метил]-амид-1Н-индазол-5-карбоновой кислоты;
[(1-метил-пиперидин-2-ил)-фенил-метил]-амид-1Н-индазол-6-карбоновой кислоты;
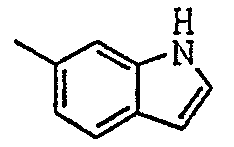
[(1-метил-пиперидин-2-ил)-фенил-метил]-амид-1Н-индол-6-карбоновой кислоты.
3. Лекарственное средство, которое обладает активностью в отношении специфических ингибиторов переносчиков глицина glyt 1 и/или glyt 2, отличающееся тем, что оно представляет собой соединение по п.1.
4. Фармацевтическая композиция, которая обладает активностью в отношении специфических ингибиторов переносчиков глицина glyt 1 и/или glyt 2, отличающаяся тем, что она включает соединение по п.1, ассоциированное с эксципиентом.
5. Применение соединения согласно п.1 для получения лекарственного средства, предназначенного для лечения поведенческих расстройств, связанных с деменцией психозов, различных форм тревог, приступов паники, фобий, компульсивных навязчивых состояний, различных форм депрессии, расстройств, связанных со злоупотреблением алкоголя или с прекращением принятия алкоголя, нарушений сексуального поведения, нарушений приема пищи и лечения мигрени.
6. Применение соединения по п.1 для получения лекарственного средства, предназначенного для лечения контрактур, боли, болезни Паркинсона и симптомов болезни Паркинсона, эпилепсий, смешанных форм и других эпилептических синдромов в сочетании с другим антиэпилептическим лечением или в монотерапии, для лечения апное во сне и в качестве нейропротектора.
| Автоматический огнетушитель | 0 |
|
SU92A1 |
| Машина для холодной стыковой сварки | 1971 |
|
SU499995A1 |
| ПРОИЗВОДНЫЕ ПИПЕРИДИНА, СПОСОБЫ ИХ ПОЛУЧЕНИЯ И ФАРМАЦЕВТИЧЕСКИЙ ПРЕПАРАТ НА ИХ ОСНОВЕ | 1995 |
|
RU2145599C1 |

