Область техники изобретения
Данное изобретение относится к способам получения фармацевтических композиций алкалоидов спорыньи с пролонгированным высвобождением и, в частности, к способам получения фармацевтических композиций алкалоидов спорыньи с пролонгированным высвобождением, характеризующихся улучшенной биодоступностью.
Предпосылки к созданию изобретения
Вообще алкалоиды спорыньи могут быть классифицированы в соответствии с их различной структурой, например эрголины, производные лизергиновой кислоты, пептидные алкалоиды спорыньи и дигидрированные пептидные алкалоиды спорыньи. Клиническое применение алкалоидов спорыньи и их производных включает в себя, помимо прочего, лечение болезни Паркинсона, головных болей, связанных с мигренью, гиперпролактинемии, цереброваскулярных нарушений.
Известно множество алкалоидов спорыньи и их производных. Например, в патенте США №3896228, выданном Richardson, обсуждается применение алкалоидов спорыньи для увеличения мочеотделения и рН мочи. В патенте США №3987173, выданном Borredon, предлагается использовать определенные смеси винкамина и алкалоидов спорыньи для лечения нарушений кровообращения. В патенте США №4229451, выданном Fehr et аl., описываются производные эргопептина, применяемые как веноконстрикторы и венотоники. В патенте США №4315937, выданном Maclay et al., рассматривается спорынья и ее применение для лечения небольших дисфункций мозга. В патенте США №4366145, выданном Stoopak et al., описывается мягкая желатиновая капсула, заполненная в центре раствором жидкого алкалоида спорыньи. В патенте США №4440722, выданном Djorjevic et al., предлагаются лекарственные препараты, содержащие соли эрготамина, эргосинина, эргокриптинина, эргокристинина и эргокорнинина, применяемые для лечения артериальной гипертензии, сердечной недостаточности, сердечной аритмии или головной боли. В патенте США №4462983, выданном Azria et al., рассматривается использование определенных пептидных алкалоидов спорыньи, приспособленных для назального или легочного введения.
Фармакологические эффекты алкалоидов спорыньи являются разнообразными и сложными, но, как оказалось, реализуются в основном в результате воздействия алкалоидов на адренергические, дофаминергические и серотонинергические рецепторы. Спектр эффектов зависит от вещества, дозы, вида, ткани и экспериментальных или физиологических условий. Вообще алкалоиды спорыньи характеризуются нестабильным всасыванием и интенсивным "метаболизмом первого прохождения" в печени с обширной биотрансформацией. Точнее всасывание алкалоидов спорыньи в желудочно-кишечном тракте оказывается низким вследствие интенсивного печеночного "метаболизма первого прохождения" и иногда нестабильным. Кроме того, изредка применение алкалоидов спорыньи может быть связано с неблагоприятными явлениями, особенно сосудистыми и сердечными явлениями. Лекарственные средства, такие как алкалоиды спорыньи, которые подвержены высокому печеночному клиренсу, могут требовать введения более высоких доз для поддержания в крови концентраций выше минимальной эффективной концентрации в течение достаточного периода времени, чтобы обеспечить желаемый фармакологический эффект. Однако если использованы обычные системы доставки лекарства, всплеск всасывания лекарственного средства, который имеет место после его введения, может индуцировать в крови концентрации, которые превосходят минимальную токсическую концентрацию. Один способ устранения описанного вредного эффекта заключается в применении более низких дозировок с чаще повторяемым приемом лекарства. Однако часто повторяемый прием лекарства не является идеальным из-за неудобства, возрастающей стоимости и повышенной вероятности, что пациент забудет принять соответствующее количество доз. Другой способ поддержания концентрации лекарственного средства на ограниченном терапевтически активном уровне заключается во введении лекарственного препарата, используя системы доставки с пролонгированным высвобождением лекарственного средства.
Системы доставки с длительным высвобождением лекарственного средства включают в себя любую систему доставки лекарственного средства, при которой успешно осуществляется медленное высвобождение лекарственного средства в течение длительного периода времени. Существуют два основных типа систем длительного высвобождения: контролируемое высвобождение и пролонгированное высвобождение. Системы контролируемого высвобождения поддерживают постоянные уровни лекарственного средства в тканях или клетках-мишенях. Системы пролонгированного высвобождения не способны поддерживать постоянный уровень лекарственного средства, но тем не менее пролонгируют присутствие терапевтического уровня лекарства в крови или ткани в течение длительного периода времени.
При создании систем доставки с длительным высвобождением следует рассматривать многие изменчивые факторы, включающие способ доставки лекарственного средства, тип системы доставки, особые свойства лекарственного средства, которое вводится, и биодоступность лекарственного средства. Системы доставки с пролонгированным высвобождением предназначены для ряда различных лекарственных средств. Например, в патенте США №4389393, выданном Schor et al., предлагаются терапевтические композиции с пролонгированным высвобождением на основе высокомолекулярной гидроксипропилметилцеллюлозы. В патенте США №5069911, выданном Züger, предлагается композиция с контролируемым высвобождением для перорального введения 9,10-дигидроалкалоида спорыньи, в патенте США №5128142, выданном Mulligan et al., описывается композиция с контролируемым высвобождением, которая включает в себя абсорбат смеси фармацевтически приемлемого активного ингредиента и неактивного вещества, абсорбированного на поперечно сшитом полимере.
В то время, как в упомянутых патентах предлагают системы доставки с пролонгированным высвобождением, которые могут обеспечить медленное высвобождение определенного лекарственного средства в течение длительного периода времени, в них не обеспечена такая система, которая также поддерживает или увеличивает биодоступность введенного лекарственного средства по сравнению с обычными системами доставки.
Краткое изложение изобретения
Поэтому аспект данного изобретения заключается в обеспечении системами доставки лекарственного средства с пролонгированным его высвобождением, которые сохраняют или увеличивают биодоступность введенного лекарственного средства по сравнению с обычными системами доставки.
Другой аспект данного изобретения заключается в обеспечении способами получения таких систем доставки.
Согласно данному изобретению, упомянутые и другие аспекты обеспечиваются посредством способа улучшения биодоступности производных спорыньи, который включает в себя комбинирование производного спорыньи или его смеси, с фармацевтически приемлемым гидрофильным набухающим агентом, или его смесью, с одним или несколькими фармацевтически приемлемыми наполнителями.
Согласно данному изобретению, биодоступность, по крайней мере, равна биодоступности производного спорыньи или его смеси, введенных, используя обычный способ доставки.
В предпочтительном аспекте производное спорыньи имеет формулу
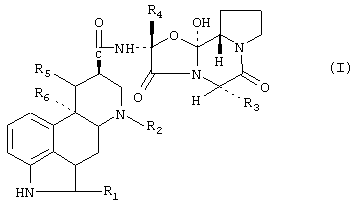
в которой:
R1 означает водород или галоген,
R2 означает водород или С1-С4алкил,
R3 означает изопропил, втор-бутил, изобутил или бензил,
R4 представляет собой метил, этил, изопропил и их смеси и простой эфир,
R5 означает водород и
R6 означает водород или метокси, или
R5 и R6 вместе представляют собой дополнительную связь, и его смеси.
Согласно данному изобретению, также предусмотрена фармацевтическая композиция. Композиция имеет биодоступность, по крайней мере, равную биодоступности производного спорыньи или его смеси, введенных с использованием обычного способа доставки.
Поэтому согласно данному изобретению способы и фармацевтические композиции могут обеспечить особенности пролонгированного высвобождения, в то же время улучшая биодоступность производных спорыньи.
Краткое описание чертежа
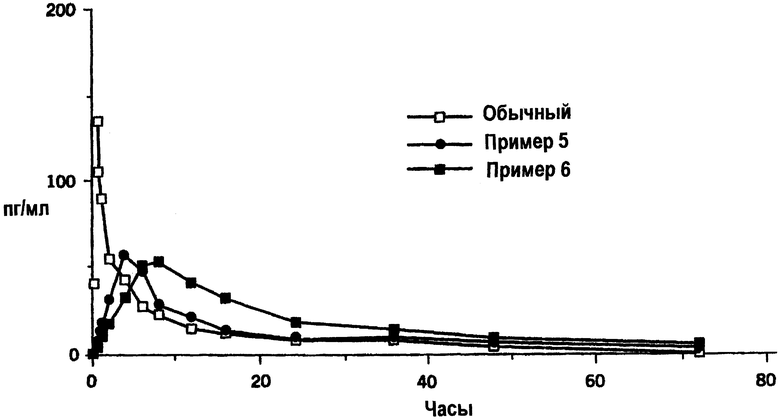
Чертеж представляет собой график концентрации в плазме α-дигидроэргокриптина в зависимости от времени после однократного перорального введения 10 мг α-дигидроэргокриптина, введенного в виде обычной таблетки или в виде таблеток с пролонгированным высвобождением, приготовленных согласно данному изобретению, как описано в примерах 5 или 6.
Подробное описание предпочтительных аспектов
Данное изобретение будет описано более подробно ниже, где представлены предпочтительные аспекты изобретения. Однако данное изобретение может быть воплощено во многих разных формах и не должно быть истолковано как ограниченное аспектами, изложенными в описании; скорее указанные аспекты представлены с тем, чтобы это изложение стало бы совершенным и полным и полностью отразило бы идею изобретения специалистам в данной области.
Данное изобретение обеспечивает композицию производных спорыньи с пролонгированным высвобождением, характеризующуюся улучшенной биодоступностью по сравнению с обычными композициями. Композиция пролонгированного действия данного изобретения содержит производное спорыньи или его смесь, фармацевтически приемлемое набухающее вещество, или его смесь, и один или несколько фармацевтически приемлемых наполнителей. Используемый в описании термин "биодоступность" определен как общее количество лекарственного средства, системно доступного в течение периода времени. Биодоступность может быть установлена посредством измерения общих системных концентраций лекарственного средства в течение периода времени после введения композиции пролонгированного действия данного изобретения и после введения композиции обычного высвобождения. Улучшенная биодоступность определяется как увеличение в "области под кривой" (AUC). AUC является интегрированным показателем системных концентраций лекарственного средства в пределах времени в единицах масса-время/объем. После введения дозы лекарства, AUC, от времени поступления лекарства в организм до момента его полного удаления из организма является показателем воздействия на пациента лекарственного средства.
Производные спорыньи данного изобретения могут представлять собой разнообразные производные спорыньи, известные специалистам в данной области. Предпочтительно производные спорыньи являются алкалоидами спорыньи. Предпочтительными алкалоидами спорыньи являются пептидные алкалоиды спорыньи и дигидрированные пептидные алкалоиды спорыньи. Особо предпочтительные алкалоиды спорыньи имеют формулу:
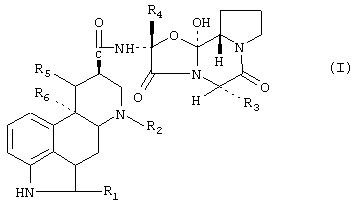
R1 означает водород или галоген,
R2 означает водород или С1-С4алкил,
R3 означает изопропил, втор-бутил, изобутил или бензил,
R4 представляет собой метил, этил, изопропил и их смеси и простой эфир,
R5 означает водород и
R6 означает водород или метокси, или
R5 и R8 вместе представляют собой дополнительную связь и его смеси или его смеси.
Если R1 означает галоген, он предпочтительно представляет собой бром.
Предпочтительными соединениями формулы I являются соединения, в которых R1, R5 и R6 означают водород, R2 означает метил, a R4 соответствует изопропилу или метилу, при условии, что R4 означает только метил, когда R3 представляет бензил.
Особо предпочтительные соединения, в которых R2 означает метил, a R1, R5 и R6 означают водород, представляют собой α-дигидроэргокриптин (R4 = изопропил, R3 = изобутил), β-дигидроэргокриптин (R4 = изопропил, R5 = втор - бутил), дигидроэргокорнин (R3 = R4 = изопропил), дигидроэргокристин (R4 = изопропил, R3 = бензил) и дигидроэрготамин (R4 = метил, R3 = бензил), вместе с их солевыми формами. Предпочтительное соединение, в котором R1 означает бром, представляет собой бромкриптин, R2 = метил, R3 = изобутил, R4 = изопропил и R5 и R6 означают вторичную связь. Приемлемые солевые формы представляют собой соли фармакологически приемлемых кислот; например метансульфонат, малеат и тартрат. Наиболее предпочтительным соединением является дигидроэргокриптин, обычно употребляемый в виде мезилата. Он предназначен для лечения болезни Паркинсона, гиперпролактинемии и мигрени. Лекарственное средство можно вводить дважды в день при ежедневной дозе приблизительно от 10 до 60 мг, предпочтительно приблизительно 20-40 мг.
Фармацевтически приемлемые набухающие вещества данного изобретения обычно представляют собой гидрофильные полимеры, такие как смолы, простые эфиры целлюлозы и материалы, полученные из белков. Предпочтительно указанные гидрофильные полимеры могут включать в себя гидроксиалкилцеллюлозы, поливиниловые спирты, полиоксиэтиленгликоли и полоксамеры. Предпочтительные гидроксиалкилцеллюлозы включают в себя метилцеллюлозу, карбоксиметилцеллюлозу, гидроксипропилцеллюлозу и гидроксипропилметилцеллюлозу.
Наиболее предпочтительным гидрофильным веществом, способствующим набуханию, является гидроксипропилцеллюлоза. Гидроксипропилметилцеллюлозы, которые могут быть использованы в данном изобретении, включают в себя Метоцел К4М® и Метоцел К15М®, обе коммерчески поставляются фирмой Colorcon of West Point, Pennsylvania. Метоцел К4М® и Метоцел К15М® содержат 19-24 мас.% метоксила и 4-12 мас.% гидроксипропила. Метоцел К4М® в 2% водном растворе имеет вязкость 4000 сП и среднюю молекулярную массу 89000, тогда как Метоцел К15М® в тех же условиях имеет вязкость 15000 сП и среднюю молекулярную массу 124000.
Композиции данного изобретения также содержат наполнители. Вообще наполнители включают в себя смазки, суспендирующие вещества, связующие вещества, разбавители, вкусовые вещества, красители, диспергаторы и смачивающие агенты, применение которых известно специалистам в данной области. В частности, приемлемыми наполнителями являются наполнители, такие как сахара, включая лактозу, сахарозу, маннит или сорбит; препараты целлюлозы, такие как, например, микрокристаллическая целлюлоза, кукурузный крахмал, пшеничный крахмал, рисовый крахмал, картофельный крахмал, желатин, трагакантовая камедь и/или поливинилпирролидон (PVP); и смазывающие вещества, такие как стеарат магния.
Композиции данного изобретения предпочтительно содержат приблизительно от 5 до 80 мг пептидных алкалоидов спорыньи. Соотношение пептидного алкалоида спорыньи к набухающему веществу предпочтительно составляет приблизительно от 1:0,5 до 1:10, более предпочтительно приблизительно от 1:2 до 1:8. Отношение дигидроэргокриптина к набухающему веществу составляет приблизительно от 1:0,5 до 1:5, более предпочтительно приблизительно от 1:1 до 1:4. Отношение пептидного алкалоида спорыньи к наполнителям предпочтительно составляет приблизительно от 1:3 до 1:100, более предпочтительно от 1:5 до 1:80 и наиболее предпочтительно приблизительно от 1:10 до 1:50.
Композиции данного изобретения обеспечивают увеличение биодоступности по сравнению с другими композициями с пролонгированным действием. Более важно, что композиции данного изобретения обеспечивают увеличение биодоступности по сравнению с обычными композициями. Предпочтительно биодоступность композиций данного изобретения выше, по крайней мере, приблизительно на 5%, более предпочтительно, по крайней мере, на 15% и наиболее предпочтительно выше, по крайней мере, приблизительно на 25%, чем биодоступность обычных композиций.
Композиции данного изобретения могут быть приготовлены в соответствии с обычными способами посредством смешивания вместе лекарственного средства и всех наполнителей, кроме смазки, чтобы получить смешанный порошок. Порошок смешивается со смазкой и полученный порошок прессуется до образования таблетки.
Следующие примеры предназначены для иллюстрации данного изобретения и не должны быть истолкованы как его ограничение. В упомянутых примерах "мг" означает миллиграмм, "нг" означает нанограмм, "рг" означает пикограмм, "мл" означает миллилитр, "мм" означает миллиметр, "С°" означает градусы Цельсия, "М" означает среднее, "SD" означает стандартное отклонение, "мПа·сек" миллипаскаль·сек, "pvp" означает поливинилпирролидон, "час" означает час и "kp" означает килопонды (kiloponds).
Примеры 1-7. Сравнение характеристик высвобождения для композиций данного изобретения с характеристиками высвобождения обычных композиций
ПРИМЕР 1
Таблетки с пролонгированным действием, содержащие 20 мг α-дигидроэргокриптина
Состав каждой таблетки
α-Дигидроэргокриптин 20,0 мг
Целлактоза® 203,0 мг1
Метоцел К15М® 25,0 мг2
Силоид 244® 1,2 мг3
Стеарат магния 0,8 мг
Примечание: 1. Содержащая 75% лактозы и 25% микрокристаллической целлюлозы, коммерчески поставляется фирмой Meggle GmbH of Wasserburg, Germany.
2. Гидроксипропилметилцеллюлоза USP тип 2208; 15000 мПа·сек, коммерчески поставляется фирмой Colorcon of West Point, Pennsylvania.
3. Двуокись кремния коммерчески поставляется фирмой W.R. Grace of Baltimore, Maryland.
Экспериментальный способ
Таблетки (250 мг), содержащие по 20 мг (8%) α-дигидроэргокриптина, получали с 80% Целлактозой® в качестве непосредственно сжимаемого наполнителя, 10% Метоцелом К15М® в качестве набухающего полимера, контролирующего высвобождение, 1,2% Силоидом 244® как свободно текущее вещество, и 0,8% стеаратом магния в качестве смазки. Лекарственное средство и все наполнители, кроме смазки, просеивали вместе по геометрическому принципу вручную с помощью сита, затем смешивали смесителем модели Turbula в течение 10 мин. После добавления стеарата магния смесь смешивали еще в течение 5 мин. Для прессования использовали роторный 8-местный лабораторный пресс с автоматической подачей порошка и контейнерными приспособлениями (12×5 мм).
Тестирование таблеток
Использовали стандартные фармацевтические способы тестирования и оборудование, чтобы определить следующее:
Твердость: средняя величина 18,6 kp (Schleuniger 4M)
Рыхлость: 0,081%
Тест растворения согласно USP XXIII, р.1792, аппарат 2, 1000 мл H2O, 50 вращений/мин:
Обычная таблеточная композиция 20 мг α-дигидроэргокриптина имела следующий состав: α-дигидроэргокриптин 20,0 мг; лактоза 148 мг; микрокристаллическая целлюлоза 70 мг; кроскармелоза 6 мг; стеарат магния 4 мг; и поливинилпирролидон 2 мг. Исследование растворения проводили в таких же условиях, как для композиции пролонгированного действия данного изобретения, описанных в данном примере, и получали в результате 96,3+/-3,6% высвобождения α-дигидроэргокриптина через 0,5 ч.
ПРИМЕР 2
Таблетки с пролонгированным действием, содержащие 20 кг α-дигидроэргокриптина
Состав каждой таблетки
α-Дигидроэргокриптин 20,0 мг
Метоцел К4М® 13,2 мг1
Натрий-СМС 26,8 мг
Лактоза 48,0 мг
PVP K30 6,7 мг
Тальк 4, 0 мг
Стеарат магния 1,3 мг
Примечание: 1. Гидроксипропилметилцеллюлоза USP тип 2208; 4000 мПа·сек, коммерчески поставлена фирмой Colorcon of West Point, Pennsylvania.
Экспериментальный способ
Таблетки (120 мг), содержащие по 20 мг (16,7%) α-дигидроэргокриптина, получали и испытывали в соответствии со способами, описанными ниже. Композицию получали с 40% лактозы как растворителем, 11% Метоцела К4М® и 22,3% натрий-СМС (вязкость среды) в качестве набухающих полимеров, контролирующих высвобождение, 5,6% PVP в качестве связывающего вещества, 3,3% талька как антисклеивающего вещества и 1,1% стеарата магния как смазки.
Получали 10% водный раствор PVP. Лекарственное вещество, разбавитель и полимеры грубо смешивали. Водный раствор PVP добавляли к порошковой смеси для образования влажной массы, которую последовательно просеивали через сито 8 меш. Влажные гранулы высушивали при 60°С, затем снова просеивали через сито 16 меш. После добавления талька и стеарата магния смесь смешивали в течение пяти минут в V-смесителе. Для прессования использовали роторный 8-местный лабораторный пресс с автоматической подачей порошка и плоскими приспособлениями, 7 мм.
Тестирование таблеток
Использовали стандартные фармацевтические способы тестирования и оборудование, чтобы определить следующее:
Твердость: средняя величина 11,2 kp (Schleuniger 4M)
Рыхлость: 0,12%
Тест растворения согласно USP XXIII, р.1792, аппарат 2, 1000 мл H2O, 50 вращении/мин:
Обычная таблеточная композиция 20 мг α-дигидроэргокриптина имела такой же состав как в примере 1. Исследование растворения проводили в таких же условиях, как для композиции пролонгированного действия данного изобретения, описанных в этом примере, и получали в результате 98,1+/-5,2% высвобождения α-дигидроэргокриптина через 0,5 час.
ПРИМЕР 3
Таблетки с пролонгированным действием, содержащие 40 мг α-дигидроэргокриптина
Состав каждой таблетки
α-Дигидроэргокриптин 40,0 мг
Лактоза DCL11® 92,5 мг1
Авицел РН101® 76,0 мг2
Метоцел К4М® 37,5 мг3
Стеарат магния 4,0 мг
Примечание: 1. Высушенная распылением лактоза поставляется фирмой Meggle GmbH of Wasserburg, Germany.
2. Микрокристаллическая целлюлоза коммерчески поставляется фирмой FMC Corporation, Pharmaceuticals Division, of Philadelphia, Pennsylvania.
3. Гидроксипропилметилцеллюлоза USP тип 2208, 4000 мПа·сек коммерчески поставляется фирмой Colorcon of West Point, Pennsylvania.
Экспериментальный способ
Таблетки (250 мг), содержащие по 40 мг (16%) α-дигидроэргокриптина, получали и испытывали в соответствии со способами, описанными ниже. Композицию получали с 37% лактозы и 30,4% микрокристаллической целлюлозы в качестве непосредственно сжимаемых наполнителей, 15% Метоцела К4М®, набухающего полимера, контролирующего высвобождение, и 1,6% стеарата магния в качестве смазки.
Лекарственное средство и все наполнители, кроме смазки, просеивали вместе по геометрическому принципу вручную с помощью сита, затем смешивали смесителем Turbula в течение 10 мин. После добавления стеарата магния смесь смешивали еще в течение 5 мин. Для прессования использовали роторный 8-местный лабораторный пресс с автоматической подачей порошка и контейнерными приспособлениями (12×5 мм).
Тестирование таблеток
Использовали стандартные фармацевтические способы тестирования и оборудование, чтобы определить следующее:
Твердость: средняя величины 14,9 kp (Schleuniger 4M)
Рыхлость: 0,072%
Тест растворения согласно USP XXIII, р.1792, аппарат 2, 1000 мл H2O, 50 вращений/мин:
Обычная таблеточная композиция, содержащая 40 мг α-дигидроэргокриптина, имела следующий состав: α-дигидроэргокриптин 40,0 мг; лактоза 128 мг; микрокристаллическая целлюлоза 70 мг; кроскармеллоза 6 мг; стеарат магния 4 мг; и поливинилпирролидон 2 мг. Исследование растворения проводили в таких же условиях, как для композиции пролонгированного действия данного изобретения, описанных в этом примере, и получали в результате 93,3+/-5,0% высвобождения α-дигидроэргокриптина через 0,5 час.
ПРИМЕР 4
Таблетки пролонгированного действия, содержащие 40 кг α-дигидроэргокриптина
Состав каждой таблетки
α-Дигидроэргокриптин 40,0 мг
Лактоза DCL11® 105,0 мг1
Авицел РН101® 76,0 мг2
Карбопол 934Р® 25,0 мг3
Стеарат магния 4,0 мг
Примечание: 1. Высушенная распылением лактоза
коммерчески поставляется фирмой Meggle GmbH of Wasserburg, Germany.
2. Микрокристаллическая целлюлоза коммерчески поставляется фирмой FMC Corporation, Pharmaceuticals Division of Philadelphia, Pennsylvania.
3. Карбомер коммерчески поставляется фирмой BF Goodrich of Cleveland, Ohio.
Экспериментальный способ
Таблетки (250 мг), содержащие по 40 мг (16%) α-дигидроэргокриптина, получали и испытывали в соответствии со способами, описанными ниже. Композицию получали с 42% лактозы и 30,4% микрокристаллической целлюлозы в качестве непосредственно сжимаемых наполнителей, 10% карбомера, набухающего полимера, контролирующего высвобождение, и 1,6% стеарата магния в качестве смазки.
Лекарственное средство и все наполнители, кроме смазки, просеивали вместе по геометрическому принципу вручную с помощью сита, затем смешивали смесителем Turbula в течение 10 мин. После добавления стеарата магния смесь смешивали в течение еще 5 мин. Для прессования использовали роторный 8-местный лабораторный пресс с автоматической подачей порошка и контейнерными приспособлениями (12×5 мм).
Тестирование таблеток
Использовали стандартные фармацевтические способы тестирования и оборудование, чтобы определить следующее:
Твердость: средняя величина 13,2 kp (Schleuniger 4M)
Рыхлость: 0,2%
Тест растворения согласно UPS XXIII, p.1792, аппарат 2, 1000 мл H2O, 50 вращении/мин:
Обычная таблеточная композиция 20 мг α-дигидроэргокриптина имела такой же состав как в примере 3. Исследование растворения проводили в таких же условиях, как для композиции пролонгированного действия данного изобретения, описанных в этом примере, и получали в результате 97,7+/-6,0% высвобождения α-дигидроэргокриптина через 0,5 час.
ПРИМЕР 5
Таблетки с пролонгированным действием, содержащие 10 мг α-дигидроэргокриптина
Состав каждой таблетки
α-Дигидроэргокриптина 10,0 мг
Целлактоза® 184,3 мг1
Метоцел К4М® 22,0 мг2
Метоцел К15М® 9,7 мг3
Натрий-СМС 2,0 мг4
Тальк 20,0 мг
Магний стеарат 2,0 мг
Примечание: 1. Составленная из 75% лактозы и 25% микрокристаллической целлюлозы, коммерчески поставляется фирмой Meggle GmbH of Wasserburg, Germany.
2. Гидроксипропилметилцеллюлоза USP типа 2208; 4000 мПа·сек, коммерчески поставляется фирмой Colorcon of West Point, Pennsylvania.
3. Гидроксипропилметилцеллюлоза USP типа 2208; 15000 мПа·сек, коммерчески поставляется фирмой Colorcon of West Point, Pennsylvania.
4. Средняя степень вязкости.
Экспериментальный способ
Таблетки (250 мг), содержащие по 10 мг (4%) α-дигидроэргокриптина, получали и испытывали в соответствии со способами, описанными ниже. Композицию получали с 73,3% Целлактозы®, в качестве непосредственно сжимаемого наполнителя, 8,8% Метоцела К4М® 3,9% Метоцела К15М® и 0,8% натрий-СМС в качестве набухающих полимеров, контролирующих высвобождение, и 8% талька как антисклеивающее вещество, и 0,8% стеарата магния в качестве смазки.
Лекарственное средство и все наполнители, кроме смазки, вместе просеивали по геометрическому принципу вручную с помощью сита, затем смешивали смесителем Turbula в течение 10 мин. После добавления стеарата магния смесь смешивали в течение еще 5 мин. Для прессования использовали роторный 8-местный лабораторный пресс с автоматической подачей порошка и контейнерными приспособлениями (12×5 мм).
Тестирование таблеток
Использовали стандартные фармацевтические способы тестирования и оборудование, чтобы определить следующее:
Твердость: средняя величина 19,1 kp (Schleuniger 4M)
Рыхлость: 0,26%
Тест растворения согласно USP XXIII, р.1792, аппарат 2, 500 мл 0,01 н. НСl, 50 вращений/мин:
Обычная таблеточная композиция 10 мг α-дигидроэргокриптина имела следующий состав: α-дигидроэргокриптин 10 мг; лактоза 158 мг; микрокристаллическая целлюлоза 70 мг; кроскармелоза 6 мг; стеарат магния 4 мг; и поливинилпирролидон 2 мг. Исследование растворения проводили в таких же условиях, как для композиции пролонгированного действия данного изобретения, описанных в этом примере, и получили в результате 96,9+/-4,8% высвобождения α-дигидроэргокриптина через 0,5 час.
ПРИМЕР 6
Таблетки пролонгированного действия, содержащие 10 мг α-дигидроэргокриптина
Состав каждой таблетки
α-Дигидроэргокриптина 10,0 мг
Целлактоза® 216,0 мг1
Метоцел К4М® 15,0 мг2
Метоцел К15М® 5,0 мг3
Натрий-СМС 2,0 мг4
Магний стеарат 2,0 мг
Примечание: 1.
Составленная из 75% лактозы и 25% микрокристаллической целлюлозы, коммерчески поставляется фирмой Meggle GmbH of Wasserburg, Germany.
2. Гидроксипропилметилцеллюлоза USP типа 2208; 4000 мПа·сек, коммерчески поставляется фирмой Colorcon of West Point, Pennsylvania.
3. Гидроксипропилметилцеллюлоза USP типа 2208; 15000 мПа·сек, коммерчески поставляется фирмой Colorcon of West Point, Pennsylvania.
4. Средняя степень вязкости.
Экспериментальный способ
Таблетки (250 мг), содержащие по 10 мг (4%) α-дигидроэргокриптина, получали и испытывали в соответствии со способами, описанными ниже. Композицию получали с 86,4% Целлактозы® в качестве непосредственно сжимаемого наполнителя, 6% Метоцела К4М®, 2% Метоцела К15М® и 0,8% натрий-СМС в качестве набухающих полимеров, контролирующих высвобождение, и 0,8% стеарата магния в качестве смазки.
Лекарственное средство и все наполнители, кроме смазки, вместе просеивали по геометрическому принципу вручную с помощью сита, затем смешивали смесителем Turbula в течение 10 мин. После добавления стеарата магния, смесь смешивали еще в течение 5 мин. Для прессования использовали роторный 8-местный лабораторный пресс с автоматической подачей порошка и контейнерными приспособлениями (12×5 мм).
Тестирование таблеток
Использовали стандартные фармацевтические способы тестирования и оборудование, чтобы определить следующее:
Твердость: средняя величина 16,1 kp (Schleuniger 4M)
Рыхлость: 0,16%
Тест растворения согласно USP XXIII, р.1792, аппарат 2, 500 мл 0,01 н. НСl, 50 вращений/мин:
Обычная таблеточная композиция 10 мг α-дигидроэргокриптина имела такой же состав, как в примере 5, и исследование растворения проводили в таких же условиях, как для композиции пролонгированного действия данного изобретения, описанных в этом примере, и получили в результате 93,7+/-3,1% высвобождения α-дигидроэргокриптина через 0,5 час.
ПРИМЕР 7
Таблетки пролонгированного действия, содержащие бромкриптин, 5 мг
Состав каждой таблетки
Бромкриптин 5,0 мг
Целлактоза® 189,3 мг1
Метоцел К4М® 22,0 мг2
Метоцел К15М® 9,7 мг3
Натрий-СМС 2,0 мг4
Тальк 20,0 мг
Стеарат магния 2,0 мг
Примечание: 1.
Составленная из 75% лактозы и 25% микрокристаллической целлюлозы, коммерчески поставляется фирмой Meggle GmbH of Wasserburg, Germany.
2. Гидроксипропилметилцеллюлоза USP типа 2208; 4000 мПа·сек, коммерчески поставляется фирмой Colorcon of West Point, Pennsylvania.
3. Гидроксипропилметилцеллюлоза USP типа 2208; 15000 мПа·сек, коммерчески поставляется фирмой Colorcon of West Point, Pennsylvania.
4. Средняя степень вязкости.
Экспериментальный способ
Таблетки (250 мг), содержащие по 5 мг (2%) бромкриптина, получали и испытывали в соответствии со способами, описанными ниже. Композицию получали с 75,7% Целлактозы® в качестве непосредственно сжимаемого наполнителя, 8,8% Метоцела К4М®, 3,9% Метоцела К15М® и 0,8% натрий-СМС в качестве набухающих полимеров, контролирующих высвобождение, 8% талька как антисклеивающего вещества и 0,8% стеарата магния в качестве смазки.
Лекарственное средство и все наполнители, кроме смазки, вместе просеивали по геометрическому принципу вручную с помощью сита, затем смешивали смесителем Turbula в течение 10 мин. После добавления стеарата магния смесь смешивали еще в течение 5 мин. Для прессования использовали роторный 8-местный лабораторный пресс с автоматической подачей порошка и контейнерными приспособлениями (12×5 мм).
Тестирование таблеток
Использовали стандартные фармацевтические способы тестирования и оборудование, чтобы определить следующее:
Твердость: средняя величина 16,4 kp (Schleuniger 4M)
Рыхлость: 0,02%
Тест растворения согласно USP XXIII, р.1792, аппарат 2, 500 мл 0,01 н. НСl, 50 вращений/мин:
Время (часы)
Обычная таблеточная композиция бромкриптина 2,5 мг, имела следующий состав: бромкриптин 2,5 мг, лактоза 115,5 мг, поливинилпирролидон 4 мг, малеиновая кислота 2 мг, стеарат магния 1,3 мг, коллоидный кремнезем 0,35 мг и кукурузный крахмал 14 мг. Исследование растворения проводили в таких же условиях, как для композиции пролонгированного действия данного изобретения, описанных в этом примере, и в результате получали 96,9+/-4,8% высвобождения бромкриптина через 0,5 час.
Пример сравнения биодоступности композиций пролонгированного действия данного изобретения с биодоступностью обычных композиций
ПРИМЕР 8
Сравнительный клинический тест
Цель исследования заключается в оценке у здоровых волонтеров фармакокинетических характеристик и биодоступности α-дигидроэргокриптина в виде пероральных таблеток пролонгированного действия согласно данному изобретению, которые описаны в примерах 5 и 6, по сравнению с обычными таблетками согласно общепринятой таблеточной композиции, описанной в примере 5. Проект изучения заключается в широкомасштабном, перекрестном, 3 цикловом исследовании. Двенадцать волонтеров, мужчин, были случайным образом определены в одну из трех схем лечения, разделенных однонедельным периодом отмывания. Лекарственное средство вводили перорально в разовой дозе, 10 мг, утром натощак (голодное состояние поддерживали в течение 4 час после приема лекарства). Образцы крови отбирали через постоянную канюлю в определенные временные точки вплоть до 72 час после введения лекарственного средства.
Концентрации в плазме во время периода наблюдения изображены на чертеже. Результаты фармакокинетического анализа плазменых концентраций представлены в таблице А (выражены в виде средних значений).
Таблица А
Представленные данные четко показывают, что обе композиции пролонгированного действия значительно снижают максимальную концентрацию и замедляют появление пика концентрации, особенно композиция пролонгированного действия, описанная в примере 6. Представленные цифры отражают низкую скорость всасывания и драматическое снижение всплеска всасывания, который обычно происходит после введения обычной композиции.
Трехкратное увеличение периода полувыведения наблюдали для композиции пролонгированного действия, описанной в примере 6, показатель пролонгированного процесса всасывания по сравнению с обычной таблеткой. Биодоступность композиций пролонгированного действия данного изобретения, измеренная посредством AUC, оказалась неожиданно выше, чем биодоступность, полученная с обычной таблеткой.
Предшествующее является иллюстрацией данного изобретения и не должно быть истолковано как его ограничение. Изобретение определено следующей формулой изобретения посредством равнозначных пунктов, включенных в него.
Приложение 1
Тесты растворения in vitro
А. Состав традиционной таблетированной композиции
α-Дигидроэргокриптина мезилат 20,0 мг
Лактоза 148,0 мг
Микрокристаллическая целлюлоза 70,0 мг
Кроскармелоза 6,0 мг
Стеарат магния 4,0 мг
Поливинилпирролидон 2,0 мг
В. Состав таблеток с пролонгированным высвобождением по настоящему изобретению
2Гидроксипропилметилцеллюлоза USP тип 2208; 4000 мПа.сек, коммерчески поставляется фирмой Colorcon из West Point, Pennsylvania.
3Гидроксипропилметилцеллюлоза USP тип 2208; 15000 мПа.сек, коммерчески поставляется фирмой Colorcon из West Point, Pennsylvania.
4Средней степени вязкости
Экспериментальный способ
Состав обрабатывали путем перемешивания в смесителе с кубическим барабаном (300 литров) в течение 15 мин, 73,72% Целлактозы® в качестве непосредственно сжимаемого агента, 8,8% Метоцела К4М® и 3,88% Метоцела К15М® и 0,8% карбоксиметилцеллюлозы натрия в качестве набухающих агентов, контролирующих высвобождение, 4% α-дигидроэргокриптина мезилата в качестве активного ингредиента и 8% талька в качестве наполнителя. После добавления 0,8% стеарата магния смесь смешивали еще в течение 15 мин. Смесь прессовали с помощью роторной таблетирующей машины (Ronchi 23N), оборудованной капсульными пункциями (punctions) ⊘12 мм.
Тестирование таблеток
Использовали стандартные фармацевтические способы тестирования и оборудование, чтобы определить следующее:
Твердость: 7,5 Кр (Schleuniger 4M)
Рыхлость: 0,081%
Средняя масса: 500 мг
Толщина: 5 мм
С. Состав таблеток с пролонгированным высвобождением по Zueger (109/7)
2Глицepиндиcтeapaт, коммерчески поставляется фирмой Giusto Favarelli SpA, Italy.
3Гидроксипропилметилцеллюлоза USP тип 2208; 4000 мПа.сек, коммерчески поставляется фирмой Colorcon из West Point, Pennsylvania.
Экспериментальный способ
Исходную смесь, состоящую из 5% α-дигидроэргокриптина мезилата в качестве активного ингредиента, 44% лактозы и 0,5% Аэросила в качестве наполнителей, просеивали через сито. К этой смеси добавляли 45% Метоцела Е4М Премиум и 5% Прецирола АТ05 и смешивали в течение 10 мин. После добавления 0,5% стеарата магния смесь перемешивали еще в течение 15 мин. Смесь затем инкапсулировали с использованием ручной инкапсулирующей машины.
D. Состав таблеток с пролонгированным высвобождением по Zueger (109/8)
2Глицериндистеарат, коммерчески поставляется фирмой Giusto Favarelli SpA, Italy.
3Гидроксипропилметилцеллюлоза USP тип 2208; 4000 мПа·сек, коммерчески поставляется фирмой Colorcon из West Point, Pennsylvania.
Экспериментальный способ
Исходную смесь, состоящую из 5% α-дигидроэргокриптина мезилата в качестве активного ингредиента, 14% лактозы и 0,5% Аэросила в качестве наполнителей, просеивали через сито. К этой смеси добавляли 30% Метоцела Е4М Премиум и 50% Прецирола АТ05 и смешивали в течение 10 мин. После добавления 0,5% стеарата магния смесь перемешивали еще в течение 15 мин. Смесь затем инкапсулировали с использованием ручной инкапсулирующей машины.
Е. Результаты тестов растворения in vitro
Тесты растворения проводили в соответствии с USP XXIII, стр.1792, прибор 2, 500 мл 0,01 н. НСl, 50 об/мин.
Традиционная таблетированная композиция
Пролонгированное высвобождение α-дигидроэргокриптина по настоящему изобретению
Пролонгированное высвобождение α-дигидроэргокриптина по Zueger (109/7)
Пролонгированное высвобождение α-дигидроэргокриптина по Zueger (109/8)
Приложение 2
Сравнительный клинический тест
Цель
Цель исследования заключается в оценке фармакокинетических характеристик и биодоступности α-дигидроэргокриптина в виде пероральных таблеток пролонгированного высвобождения согласно данному изобретению, которые описаны в Приложении 1 (далее называемых “В”), по сравнению с: обычными таблетками, имеющими традиционный для таблеток состав, описанный в Приложении 1 (далее называемых “А”), капсулами пролонгированного высвобождения, приготовленными в соответствии со способом, описанным Zueger (патент США №5069911), описанный в Приложении 1 как 109/7 (далее называемых “С”), и капсулами пролонгированного высвобождения, приготовленными в соответствии со способом, описанным Zueger (патент США №5069911), описанный в Приложении 1 как 109/8 (далее называемых “D”).
Исследование проводили в две фазы. В первой фазе фармакокинетические свойства “А” и “В” изучали в открытом, перекрестном, 2-цикловом исследовании. Двенадцать волонтеров-мужчин были случайным образом определены в одну из двух схем лечения, разделенных однонедельным периодом отмывания. Лекарственное средство вводили перорально в разовой дозе 20 мг утром натощак. Образцы крови отбирали через постоянную канюлю в определенные временные точки вплоть до 72 час после введения лекарственного средства.
Концентрации в плазме во время периода наблюдения изображены на чертеже. Результаты фармакокинетического анализа концентраций в плазме представлены в таблице 6 (выражены в виде средних значений).
Фармакокинетический анализ композиции обычного высвобождения и композиции пролонгированного высвобождения согласно данному изобретению
Во второй фазе фармакокинетические свойства “А”, “С” и “D” изучали в открытом, перекрестном, 3-цикловом исследовании. Шесть волонтеров-мужчин были случайным образом определены в одну из трех схем лечения, разделенных однонедельным периодом отмывания. Лекарственное средство вводили перорально в разовой дозе 20 мг утром натощак. Образцы крови отбирали через постоянную канюлю в определенные временные точки вплоть до 72 час после введения лекарственного средства.
Результаты фармакокинетического анализа концентраций в плазме представлены в таблице 7 (выражены в виде средних значений).
Фармакокинетический анализ композиции обычного высвобождения и композиции пролонгированного высвобождения по Zueger
Представленные данные четко показывают, что все композиции пролонгированного высвобождения, а именно “В”, “С” и “D”, значительно снижают максимальную концентрацию и замедляют появление пика концентрации. Представленные данные отражают низкую скорость всасывания и сильное снижение подъема, который обычно происходит после введения обычной композиции.
Биодоступность композиций пролонгированного высвобождения “С” и “D”, приготовленных в соответствии со способом, описанным Zueger (патент США №5069911), измеренная по AUC, сходна или слегка ниже, чем таковая обычной таблетки “А”.
Напротив, биодоступность композиций пролонгированного действия по настоящему изобретению, измеренная по AUC, неожиданно выше, чем таковая обычной таблетки.
| название | год | авторы | номер документа |
|---|---|---|---|
| КОМПОЗИЦИИ С ПРОЛОНГИРОВАННЫМ ВЫСВОБОЖДЕНИЕМ, ВКЛЮЧАЮЩИЕ ЛАМОТРИГИН | 2003 |
|
RU2325163C2 |
| Фармацевтическая композиция с модифицированным высвобождением на основе полиморфов клозапина | 2024 |
|
RU2836778C1 |
| ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ, СОДЕРЖАЩАЯ БИОТИН, И СПОСОБ ЕЕ ПОЛУЧЕНИЯ | 2017 |
|
RU2639488C1 |
| ФАРМАЦЕВТИЧЕСКИЙ СОСТАВ ПРОЛОНГИРОВАННОГО ДЕЙСТВИЯ НА ОСНОВЕ КЛОЗАПИНА ПЕРОРАЛЬНОГО ВВЕДЕНИЯ | 2009 |
|
RU2414903C1 |
| ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ ПРОКСОДОЛОЛА С КОНТРОЛИРУЕМЫМ ВЫСВОБОЖДЕНИЕМ | 2007 |
|
RU2356532C2 |
| ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ ЭНТАКАПОНА ИЛИ ЕГО СОЛЕЙ ПРОЛОНГРИРОВАННОГО ВЫСВОБОЖДЕНИЯ | 2009 |
|
RU2540465C2 |
| ТАБЛЕТКИ ТАМЗУЛОСИНА С МОДИФИЦИРОВАННЫМ ВЫСВОБОЖДЕНИЕМ | 2002 |
|
RU2335280C2 |
| ТАКРОЛИМУС ДЛЯ УЛУЧШЕННОГО ЛЕЧЕНИЯ ПАЦИЕНТОВ С ТРАНСПЛАНТАТАМИ | 2009 |
|
RU2574006C2 |
| ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ С НАБУХАЮЩИМ ПОКРЫТИЕМ | 2004 |
|
RU2375048C2 |
| ФАРМАЦЕВТИЧЕСКИЕ КОМПОЗИЦИИ РЕИНА ИЛИ ДИАЦЕРЕИНА | 2008 |
|
RU2484816C2 |
Изобретение относится к фармацевтике. Проводят объединение производного спорыньи или его смеси с фармацевтически приемлемым гидрофильным набухающим веществом, или его смеси, и одним или несколькими фармацевтически приемлемыми наполнителями. Производное спорыньи имеет формулу
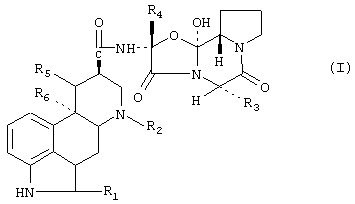
в которой R1 означает водород или галоген, R2 означает водород или C1-C4алкил, R3 представляет собой изопропил, втор-бутил, изобутил или бензил, R4 представляет собой метил, этил или изопропил, и либо R5 означает водород и R6 означает водород или метокси, либо R5 и R6, вместе соответствуют дополнительной связи. Отношение производного спорыньи к набухающему веществу составляет 1:0,5-1:10. Фармацевтическая композиция, содержащая вещество, полученное вышеуказанным способом. Она содержит от 5 до 80 мг производного спорыньи при его отношении к набухающему веществу от 1:0,5 до 1:10. Изобретение позволяет реализовать указанное назначение. 2 н. 14 з.п. ф-лы, 7 табл., 1 ил.
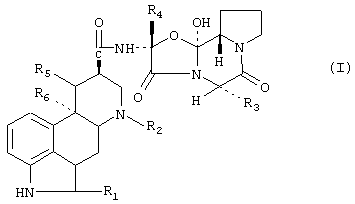
в которой R1 означает водород или галоген;
R2 означает водород или С1-С4алкил;
R3 означает изопропил, втор-бутил, изобутил или бензил;
R4 означает метил, этил или изопропил, и либо
R5 означает водород и
R6 означает водород или метокси, либо
R5 и R6 вместе соответствуют дополнительной связи,
причем указанный способ характеризуется отношением указанного производного спорыньи к указанному гидрофильному набухающему веществу от 1:0,5 до 1:10.
| ЕР 0284849 А1, 05.10.1988 | |||
| ПАТРОН ДЛЯ ЛИЧНОГО ОГНЕСТРЕЛЬНОГО ОРУЖИЯ И БРОНЕБОЙНАЯ ПУЛЯ ДЛЯ НЕГО | 2000 |
|
RU2170407C1 |
| RU 9403755 А1, 20.05.1996. | |||

