РОДСТВЕННАЯ ЗАЯВКА США
По данной заявке испрашивается приоритет по предварительной заявке США № 61/079015, поданной 8 июля 2008 года, которая включена в настоящий документ в качестве ссылки в полном объеме.
ОБЛАСТЬ ИЗОБРЕТЕНИЯ
Настоящее изобретение относится к пероральной лекарственной форме с пролонгированным высвобождением, содержащей в качестве активного вещества такролимус или его фармацевтически активный аналог, для применения раз в сутки при иммуносупрессивном лечении пациента, нуждающегося в этом, причем эта лекарственная форма высвобождает активное вещество в течение значительно пролонгированного периода времени и обеспечивает in vivo высокую биодоступность и улучшенный фармакокинетический профиль по сравнению с традиционными лекарственными формами.
УРОВЕНЬ ТЕХНИКИ, К КОТОРОМУ ОТНОСИТСЯ ИЗОБРЕТЕНИЕ
Такролимус, также известный как FK-506 или FR-900506, представляет собой активный ингредиент Prograf®, Protopic® и Advagraf®, одобренного European Agency for the Evaluation of Medicinal Products (EMEA) 23 апреля 2007 года. В процессе разработки Advagraf®, продукт был известен как MR4. Детальные характеристики Advagraf описаны в EPAR (European Public Assessment Reports) для медицинских продуктов, разрешенных для применения у человека, в том числе в документе Scientific Discussion, который EMEA сделал общедоступным после одобрения, и Product Information (обозначение, 25/01/2008 Advagraf-H-C-712-T-03), который включен в качестве ссылки. Такролимус (Prograf®) одобрен FDA в апреле 1994 года под NDA № 050708 для профилактики отторжения органов у пациентов, получивших аллогенные трансплантаты печени. Также он одобрен в Европейском Союзе, Японии, Канаде и Швейцарии и многих других странах под тем же торговым названием. Он одобрен для профилактики отторжения органов у пациентов, получивших аллогенные трансплантаты печени, почки или сердца. Согласно оценке 72% всех реципиентов трансплантатов почки и 89% всех реципиентов трансплантатов печени получают такролимус.
Такролимус, вводимый в качестве капсул Prograf®, проявляет значительную внутри- и межиндивидуальную изменчивость всасывания и метаболизма. Вследствие этой изменчивости стандартное дозирование не является точным предсказателем концентрации. При клиническом применении часто требуется коррекция доз такролимуса, основанная на мониторинге концентраций такролимуса в крови. Такролимус имеет форму белых кристаллов или кристаллического порошка. Он практически нерастворим в воде, легко растворим в этаноле и высоко растворим в метаноле и хлороформе.
Препарат такролимуса описан в EP-A-0184162, и аналоги такролимуса описаны, например, в EP-A-0444659 и US 6387918, оба из которых включены в качестве ссылок.
Такролимус представляет собой макролидное соединение с полезной иммуносупрессивной активностью, противомикробной активностью и другими фармакологическими видами активности и является ценным для лечения или профилактики реакций отторжения при трансплантации органов или тканей, реакций "трансплантат против хозяина", аутоиммунных заболеваний и инфекционных заболеваний.
Такролимус ингибирует активацию T-лимфоцитов, хотя точный механизм его действия неизвестен. Экспериментальные данные указывают на то, что такролимус связывается с внутриклеточным белком FKBP-12. Затем образуется комплекс такролимус-FKBP-12 с кальцием, кальмодулином и кальциневрином, и фосфатазная активность кальциневрина ингибируется. Этот эффект может предотвращать дефосфорилирование и транслокацию ядерного фактора активированных T-клеток - компонента ядра, предположительно инициирующего транскрипцию генов для образования лимфокинов. Конечным результатом является ингибирование активации T-лимфоцитов, т.е. иммуносупрессия.
Такролимус в значительной степени метаболизируется изоферментом CYP3A4 в стенке пищеварительного канала и печени. Изофермент CYP3A4 присутствует или экспрессируется во всех сегментах желудочно-кишечного тракта, в том числе в толстом кишечнике. Было выявлено, что на его всасывание отрицательно влияет одновременный прием пищи. Таким образом, скорость и степень всасывания такролимуса являются наибольшими в условиях натощак.
Известно, что такролимус индуцирует значительные побочные эффекты нефро- или нейротоксического происхождения, а также побочные эффекты в желудочно-кишечном тракте и другие побочные эффекты.
Всасывание такролимуса из желудочно-кишечного тракта после перорального введения является быстрым со средним временем до максимальной концентрации (Tmax) приблизительно 1-2 часа после введения здоровым субъектам или пациентам с трансплантированными почкой или печенью, однако оно является неполным и вариабельным. Биодоступность после перорального введения, как правило, составляет не более приблизительно 20%.
Часто наблюдаемыми побочными эффектами являются рвота и тошнота, однако также наблюдают такие побочные эффекты, как тремор, головная боль, гипертензия, дисфункция почек, гиперкалиемия, гипомагниемия, гипергликемия, бессонница, диарея, запор, боль в животе, нефротоксичность и нейротоксичность.
Для перорального введения такролимус обычно изготавливают и поставляют на рынок в качестве мягких желатиновых капсул, содержащих эквивалент 0,5, 1 или 5 мг безводного такролимуса, и его поставляют на рынок под торговым названием Prograf®. Рекомендованная начальная пероральная доза для пациентов составляет приблизительно от 0,1 до 0,2 мг/кг/сутки. Доза нацелена на определенный минимальный уровень в плазме от приблизительно 5 до приблизительно 20 нг/мл. Prograf® показан для профилактики отторжения органов у пациентов, получивших аллогенные трансплантации печени или почки. Подробное описание клинической фармакологии, фармакокинетики и клинических испытаний представлено на ярлыке, одобренном FDA от 04/27/2006 для Prograf®, NDA № 50708, который включен в качестве ссылки.
Остается необходимость в новых содержащих такролимус фармацевтических композициях и/или лекарственных формах, проявляющих повышенную биодоступность и улучшенные фармакокинетические свойства. Повышенная биодоступность в комбинации с пролонгированным высвобождением состава может позволить уменьшить прием лекарственных единиц пациентом, например, вплоть до одной дозы раз в сутки без риска недостаточного клинического эффекта вследствие низких доз в последней части интервала между введениями доз. Более того, могут значительно снижаться флуктуации в профиле концентрации в плазме по отношению ко времени. Кроме того, повышенная биодоступность также может приводить к более воспроизводимому (т.е. менее изменчивому по сравнению с Prograf®) профилю высвобождения.
Составы такролимуса с замедленным высвобождением описаны в WO99/49863 (Fujisawa Pharmaceutical Co.), в частности, признанной в качестве патентов США № US 6440458, US 6576259 и US 6884433, касающихся состава, где время растворения 63,2% (величина T63,2%) такролимуса составляет от 0,7 до 15 часов. Однако состав, где 63,2% высвобождается в течение 42 минут, по-видимому, только немного отличается от общепринятого состава такролимуса с немедленным высвобождением, в случае которого в течение 30 минут высвобождается 68,4%. Утверждается, что, когда состав имеет значение T63,6 более 15 часов, высвобождение активного ингредиента является настолько замедленным, что активный ингредиент выводится из организма до достижения эффективной концентрации в крови. Наиболее предпочтительным вариантом осуществления является состав с непрерывным высвобождением с величиной T63,6, составляющей 2-5 часов. Все составы, полученные согласно примерам данной заявки, имеют величину T63,6% от 1,9 (состав с наиболее быстрым высвобождением) до 8,2 часа для состава с наиболее медленным высвобождением. Кроме того, утверждается, что такролимус превосходно всасывается, и варьирование его всасываемости снижается благодаря составам с непрерывным высвобождением. В примерах этих документов повышенной биодоступности достигают для всех протестированных составов. Величины T63,6%, описанные для этих составов, составляют 3,0, 3,3, 2,0 и 2,5, соответственно.
В патентной заявке WO 2005/020993 авторы изобретения настоящей заявки также протестировали различные составы такролимуса у собак породы бигль и у карликовых свиней, однако показали, что как таблетка с быстрым высвобождением (пример 18), так и таблетка с медленным высвобождением (пример 19) могут приводить к повышенной биодоступности по сравнению с Prograf®. Это указывает на то, что повышенная биодоступность может быть связана с наличием такролимуса в лекарственной форме в растворенном состоянии, что также следует из WO 2005/020994 тех же авторов изобретения, касающейся твердых дисперсий, содержащих такролимус. Традиционный продукт с быстрым высвобождением Prograf® содержит такролимус в физической смеси HPMC, лактозы, кроскармеллозы натрия, как описано в примере 31 WO99/49863, указанной выше и принадлежащей Fujisawa Pharmaceutical Co. (в настоящее время Asteilas), которая разработала Prograf®.
Одна из основных проблем с модифицированными лекарственными формами или лекарственными формами с пролонгированным высвобождением состоит в трудности достижения достаточного всасывания в нижней части желудочно-кишечного тракта, поскольку пероральные лекарственные формы, попадающие в толстый кишечник, могут легко экскретироваться до существенного высвобождения. Высвобождение, как правило, снижается вследствие недостатка жидкостей и физического взаимодействия лекарственных форм при возрастающем содержании твердых веществ в толстом кишечнике. Кроме того, поверхность всасывания в несколько раз меньше, чем поверхность всасывания в тонком кишечнике, и этот фактор повышает время, в течение которого высвобожденные активные вещества подвергаются возможной деградации и включению в твердые вещества, присутствующие в толстом кишечнике.
КРАТКОЕ ОПИСАНИЕ СУЩНОСТИ ИЗОБРЕТЕНИЯ
Общепризнано, что чрезмерное пролонгирование высвобождения может серьезно повлиять на биодоступность, даже в случае веществ, предположительно имеющих хорошую проникающую способность в толстом кишечнике. Для веществ, являющихся субстратами для CYP3A4, в нижнем отделе желудочно-кишечного тракта можно ожидать преимущества с точки зрения биодоступности в виде меньшей концентрации метаболизирующих ферментов. С другой стороны, относительно более высокая концентрация транспортной системы, P-гликопротеинов, в нижнем отделе желудочно-кишечного тракта обычно противодействует эффекту низкой концентрации ферментов CYP3A4, поскольку молекулы, которые проникают в энтероцит, транспортируются переносчиком обратно в просвет кишечника. Такролимус является известным субстратом для этих механизмов: как для метаболизма CYP3A4, так и для системы переносчика P-гликопротеина. Таким образом, повышенную биодоступность нельзя коррелировать с продлением высвобождения простым линейным образом. Высвобождение можно с осторожностью адаптировать до данного уровня, учитывая несколько противодействующих факторов. В толстом кишечнике эти факторы включают более низкие площади всасывания, более низкое содержание жидкости, более высокое содержание твердых веществ, бактериальную деградацию, более высокое влияние транспортной системы P-гликопротеина, более низкую подвижность, отличия в слизистом барьере и/или составе слизи и отличия в pH вдоль толстого кишечника по сравнению с тонким кишечником. Таким образом, контроль и расчет времени высвобождения in vivo пролонгированной лекарственной формы для получения предсказуемого высвобождения в различных физических условиях, присутствующих вдоль желудочно-кишечного тракта, является трудным, особенно учитывая, что иммуносупрессивное лечение у пациентов с трансплантатами требует концентраций в крови в очень узких пределах для баланса эффективности (отсутствия отторжения) и побочных эффектов (инфекции, нефротоксичность, метаболическое и сердечно-сосудистое заболевание, и т.д.). Предоставление улучшенного состава для лечения один раз в сутки, где высвобождение является пролонгированным до точного уровня, где конечные фармакокинетические параметры являются полностью оптимизированными без риска для безопасности, т.е. если изменчивость важных фармакокинетических параметров у пациента или между пациентами является высокой, если корреляция между минимальной концентрацией и биодоступностью отсутствует, является ключевым фактором при лечении лекарственным средством с узким терапевтическим индексом, таким как такролимус, где неуспех лечения тесно связан с отторжением органа, и коррекцию дозы необходимо проводить безопасным способом. Следующие факторы, снижающие вероятность клинического успеха состава для введения один раз в сутки при трансплантации органа, включают высокую частоту осложнений в желудочно-кишечном тракте, влияющих на параметры желудочно-кишечного тракта, включающие время прохождения, pH, бактериальный состав и другие функции пищеварительной системы. Эти осложнения включают тошноту, рвоту и очень часто диарею.
Таким образом, авторы настоящего изобретения неожиданно обнаружили, что лекарственная форма, которая высвобождает такролимус в течение значительно пролонгированного и контролируемого периода времени, способна доставлять такролимус in vivo таким образом, чтобы такролимус в то же время достаточно всасывался, чтобы было понятно, что такролимус не утрачивается в нижнем отделе желудочно-кишечного тракта, причем высвобождение является достаточно медленным для обеспечения очень низкой скорости всасывания, в результате чего максимальная концентрация контролируется на более низком уровне, а минимальная концентрация возрастает, обеспечивая эффективность лечения в течение полного интервала между введением доз, составляющим 24 часа. Очень важно, что минимальная концентрация, достигаемая через 24 часа после введения состава с пролонгированным высвобождением по настоящему изобретению, является высоко предсказуемой, и, таким образом, ее можно использовать в качестве маркера общей биодоступности, поскольку достигается высокая корреляция между минимальной концентрацией и фактической биодоступностью, наблюдаемой в предыдущие 24 часа перед измерением минимальной концентрации. Таким образом, минимальную концентрацию можно безопасно использовать в качестве инструмента для дозирования и коррекции в процессе лечения.
Полагают, что общепринятые способы растворения in vitro коррелируют с истинным модифицированным профилем высвобождения in vivo у человека или по меньшей мере отражают его. Таким образом, ожидается, что отличие в скорости высвобождения in vitro между двумя протестированными составами в одних и тех же условиях отражает отличие в скорости высвобождения in vivo. Однако могут существовать исключения, если, например, один состав имеет зависимое от pH высвобождение, а другой имеет независимое от pH высвобождение, и истинные значения pH для тестирования не выбраны для выявления таких отличий. Понятным примером является случай тестирования покрытого желудочно-резистентным покрытием состава при высоких pH: он обеспечивает немедленное высвобождение in vitro и замедленное высвобождение in vivo. Кроме того, при сравнении двух пролонгированных составов с различными механизмами высвобождения, например, механизмом осмотического высвобождения относительно зависимого от деградации механизма высвобождения, одинаковые профили высвобождения in vitro для двух продуктов теоретически могут привести к отличающимся профилям in vitro, однако снижение или повышение скорости растворения может отражаться in vivo для каждого продукта. Таким образом, пока не доказано обратное и предоставленные способы проводят согласно предписанию фармакопей, общепринятые способы растворения являются пригодными инструментами для дифференциации между составами и соответствующими свойствами in vivo. В соответствии с этим, настоящее изобретение относится, в его первом аспекте, к пролонгированной лекарственной форме, содержащей в качестве активного вещества такролимус или его фармацевтически активный аналог для иммуносупрессивного лечения раз в сутки пациента, нуждающегося в этом, которая высвобождает активное вещество в течение значительно пролонгированного периода времени. В следующем аспекте высвобождение характеризуется высвобождением по существу нулевого порядка для основной части высвобождения.
Общепринятые способы растворения in vitro включают способы, описанные в The United States Pharmacopeia (USP), официальном общедоступном устанавливающем стандарты авторитетном источнике для всех прописываемых и продаваемых без рецепта лекарственных средств в США, и в сходных фармакопеях Европы и Японии. Предпочтительные способы включают способ растворения I USP (корзина) и способ II (лопасть) при 50 об/мин, с использованием HPC для предотвращения прилипания лекарственного средства к оборудованию и при pH 4,5 из соображений стабильности. Поскольку такролимус не протонирован, pH не влияет на растворимость этого лекарственного средства, однако изменение pH может иметь значение в случае, когда в составе используют чувствительные к pH неактивные эксципиенты, поскольку pH 4,5 не является pH, обычно имеющемся в желудочно-кишечном тракте. Таким образом, может быть актуальным описание степени пролонгирования высвобождения с помощью альтернативных способов растворения. Кроме того, пролонгированный состав можно соответственно далее охарактеризовывать дополнительными способами растворения, среди прочих, способами с различными скоростями вращения, различными величинами pH, использованием сред для растворения, имитирующих условия желудочно-кишечного тракта (например, имитация состояния натощак и состояния сытости, среды FaSSIP и FeSSIP), применение добавок в среду для растворения, таких как SLS, для повышения смачиваемости или растворимости такролимуса, в результате чего общее измеренное время растворения снижается (скорость растворения возрастает).
Авторы изобретения открыли, что биодоступность такролимуса значительно возрастает, и фармакокинетические параметры по существу улучшаются, когда такролимус вводят млекопитающему в композиции с пролонгированным высвобождением, где высвобождение и время высвобождения активного ингредиента, т.е. профиль высвобождения in vitro и in vivo, являются пролонгированными до более чем 15 часов при измерении общепринятыми способами растворения, используемыми для лекарственных форм такролимуса, и при измерении in vivo с помощью фармакокинетических параметров, имеющих клиническое значение и значение для подтверждения пролонгирования высвобождения in vivo. Эти фармакокинетические параметры включают: значительно пролонгированное время до достижения максимальной концентрации; низкие максимальные концентрации; высокие минимальные концентрации, пролонгированное среднее время нахождения и в то же время сохранение удивительно высокой биодоступности и превосходной корреляции между минимальными концентрациями и биодоступностью.
Пролонгированное высвобождение определяют по высвобождению самое большее 63,5% содержимого активного вещества в момент времени через 12 часов, определенному по растворению in vitro и при тестировании в соответствии с тестом растворения USP II (лопасть) или тестом растворения USP I (корзина) в среде, имеющей pH 4,5 и содержащей 0,005% гидроксипропилцеллюлозы, и при вращении 50 об/мин. В другом аспекте высвобождение самое большее 63,5% активного вещества в момент времени через 12 часов сочетается с высвобождением по меньшей мере 8% через 4 часа и/или по меньшей мере 15% через 8 часов для обеспечения непрерывного высвобождения на протяжении интервала дозирования. Если на протяжении нескольких часов после введения высвобождения не происходит, пациент имеет риск, что концентрация такролимуса в крови продолжит снижаться до величины ниже желаемого терапевтического нижнего предела на несколько часов из 24-часового интервала дозирования.
В других аспектах изобретение относится к применению композиции с пролонгированным высвобождением для более безопасного иммуносупрессивного лечения вследствие улучшенного фармакокинетического профиля, достигнутого у здоровых субъектов и пациентов и продемонстрированного несколькими фармакокинетическими испытаниями однократных доз и фармакокинетических испытаний стационарного состояния по сравнению с общепринятыми коммерчески доступными лекарственными формами. Безопасное иммуносупрессивное лечение по изобретению также относится к конкретному режиму дозирования для перехода с лечения Prograf® два раза в сутки, где переход осуществляют с помощью дозировки в соотношении 1:0,66-0,80 (согласно наиболее близкой доступной дозировке таблетки). Такой режим дозирования приводит к сравнимым средним концентрациями в крови в течение интервала дозирования при измерении до и после перехода, а также к сходной с Prograf® биоэквивалентной экспозиции, исходя из других параметров, таких как AUC и минимальная концентрация.
КРАТКОЕ ОПИСАНИЕ ЧЕРТЕЖЕЙ
На фиг.1 представлено растворение состава с пролонгированным высвобождением по изобретению, тестированное способом в соответствии с тестом растворения USP II (лопасть) в среде, имеющей pH 4,5 содержащей 0,005% гидроксипропилцеллюлозы, и при вращении 50 об/мин.
На фиг.2 представлена сцинтиграфическая оценка области высвобождения при использовании состава с пролонгированным высвобождением согласно параметрам исследования 003. На фигуре показано, что высвобождение in vivo состава в соответствии с настоящим изобретением является пролонгированным до той степени, что всасывание происходит в толстом кишечнике субъекта.
На фиг.3 представлены концентрации в крови такролимуса в исследовании однократной дозы натощак у здоровых добровольцев. Закрашенные треугольники обозначают концентрацию для 5-мг состава по настоящему изобретению, звездочками обозначен 5-мг состав Prograf®; незакрашенными кругами обозначен 2-мг состав по настоящему изобретению; закрашенными прямоугольниками обозначено введение 2×2 мг в соответствии с настоящим изобретением; закрашенными ромбами обозначено введение 4×1 мг Prograf®; вертикальная линия обозначает введение 2×1 мг Prograf®. Исследование описано в таблице A настоящего документа в качестве исследования 002.
На фиг.4 представлено растворение предпочтительного состава по изобретению, имеющего композицию, сходную с композицией, описанной в примере 20. Треугольниками изображено введение 1-мг состава, прямоугольниками обозначено введение 2-мг состава. Кроме того, представлено растворение коммерческого продукта Advagraf®, используемого для сравнения в примере 20, где звездочки относятся к 0,5 мг, кресты относится к 1 мг, а круги относится к 5 мг продукта Advagraf®. Высвобождение измеряют в процентах растворенного продукта с течением времени, указанного в часах. Используемый способ растворения представляет собой способ тестирования растворения USP II (лопасть) в среде, доведенной до pH 4,5, содержащей 0,005% гидроксипропилцеллюлозы, и при вращении 50 об/мин.
На фиг.5 представлены профили в крови в стационарном состоянии, полученные до перехода (Prograf®, стационарное состояние на 7 сутки) и после перехода на 14 сутки и 21 сутки на состав с пролонгированным высвобождением в соответствии с настоящим изобретением у пациентов со стабильной печенью. Квадратами обозначен Prograf® два раза в сутки на 7 сутки, кругами обозначен LCP-Tacro раз в сутки на 17 сутки, и ромбами обозначен LCP-tacro один раз в сутки на 21 сутки. Подробное описание исследования приведено в настоящем документе в примере 19. Профили демонстрируют истинные профили после перехода на более низкую дозу состава по изобретению.
На фиг.6 представлены скорректированные по дозе профили устойчивого состояния, показанные на фиг.5.
На фиг.7 представлен PK-профиль таблеток LCP-Tacro по сравнению с капсулами Advagraf® в стационарном состоянии натощак.
На фиг.8 представлены концентрации в плазме крови после введения одной и той же единичной дозировки такролимуса в качестве 2×1-мг капсул Advagraf®, обозначенных незакрашенными кругами, и в качестве 2-мг таблеток LCT-Tacro по настоящему изобретению. Исследование описано в примере 20.
На фиг.9 описаны профили в крови в стационарном состоянии, полученные до перехода (Prograf®, стационарное состояние на 7 сутки) и после перехода на 14 сутки и 21 сутки на состав с пролонгированным высвобождением в соответствии с настоящим изобретением у пациентов со стабильной печенью. Квадратами обозначен Prograf® два раза в сутки на 7 сутки, кругами обозначен LCP-Tacro раз в сутки на 17 сутки, и ромбами обозначен LCP-tacro один раз в сутки на 21 сутки. Подробное описание исследования приведено в настоящем документе в примере 19. Профили соответствуют истинным профилям после перехода на более низкую дозу состава по изобретению.
На фиг.10 представлены скорректированные по дозе профили стационарного состояния, показанные на фиг.9.
ПОДРОБНОЕ ОПИСАНИЕ ИЗОБРЕТЕНИЯ
Определения
Как используют в настоящем документе, термин "активный ингредиент" или "активный фармацевтический ингредиент" означает любой компонент, который предназначен для обеспечения фармакологической активности или другого прямого эффекта при диагностике, устранении, смягчении, лечении или профилактике заболевания, или для влияния на структуру или любую функцию организма человека или других животных. Термин включает компоненты, которые могут претерпевать химическое изменение при изготовлении лекарственного продукта, и они присутствуют в лекарственном продукте в модифицированной форме, предназначенной для обеспечения определенной активности или эффекта.
В контексте настоящего изобретения термин "гидрофильный" описывает, что что-либо "любит воду", т.е. гидрофильная молекула или часть молекулы представляет собой молекулу, которая, как правило, является электрически поляризованной и способна образовывать водородные связи с молекулами воды, что позволяет ей растворяться более легко в воде, чем в масле или других "неполярных" растворителях.
В контексте настоящего изобретения термин "амфифильный" обозначает молекулу (такую как молекула поверхностно-активного вещества), имеющую полярную растворимую в воде группу, связанную с нерастворимой в воде углеводородной цепью. Таким образом, один конец молекулы является гидрофильным (полярным), а другой конец является гидрофобным (неполярным).
В контексте настоящего изобретения термин "гидрофобный" обозначает соединение, имеющее тенденцию к тому, чтобы быть электрически нейтральным или неполярным, и, таким образом, предпочитающее другие нейтральные и неполярные растворители или молекулярное окружение.
Как используют в настоящем документе, термин "носитель" означает любую жидкость растворителя или носителя в фармацевтическом продукте, которая не имеет фармакологической роли. Например, вода представляет собой носитель для ксилокаина, и пропиленгликоль является носителем для многих антибиотиков.
В контексте настоящего изобретения термин "твердая дисперсия" обозначает лекарственное средство или активный ингредиент или вещество, диспергированное до уровня частиц в инертном наполнителе, носителе, разбавителе или матрице в твердом состоянии, т.е. обычно дисперсию мелких частиц.
В контексте настоящего изобретения термин "твердый раствор" обозначает лекарственное средство или активный ингредиент или вещество, растворенное на молекулярном уровне в инертном наполнителе, носителе, разбавителе или матрице в твердом состоянии.
Как используют в настоящем документе, термин "аналог" обозначает химическое соединение, которое структурно напоминает другое химическое соединение.
Термин "лекарственное средство" означает соединение, предназначеное для применения в диагностике, устранении, смягчении, лечении или профилактике заболевания у человека или других животных.
В этом контексте термин "лекарственная форма" означает форму, в которой лекарственное средство доставляют пациенту. Она может представлять собой парентеральную форму, местную форму, таблетку, пероральную форму (жидкость или растворенный порошок), суппозиторий, ингаляционную форму, трансдермальную форму и т.д.
Как используют в настоящем документе, термин "биодоступность" означает степень, с которой лекарственное средство или другие вещества становятся доступными ткани-мишени после введения.
Как используют в настоящем документе, термин "биоэквивалентность" обозначает научную основу для сравнения дженериков и зарегистрированных лекарственных средств друг с другом. Например, лекарственные средства являются биоэквивалентными, если они проникают в кровоток с одинаковой скоростью при введении в сходных дозах в сходных условиях. Параметры, часто используемые в исследованиях биоэквивалентности, представляют собой Tmax, Cmax, AUC0-∞, AUC0-t. Другими важными параметрами могут быть W50, W75 и/или MRT. Таким образом, по меньшей мере один из этих параметров можно использовать при определении существования биоэквивалентности. Более того, в контексте настоящего изобретения две композиции считают биоэквивалентными, если значение используемого параметра составляет 80-125% от значения для Prograf® или сходного коммерчески доступного содержащего такролимус продукта, используемого в тесте.
В контексте настоящего изобретения Tmax обозначает время до достижения максимальной концентрации в плазме (Cmax) после введения; AUC0-∞ обозначает площадь под кривой концентрации в плазме по отношению ко времени от 0 момента времени до бесконечности; AUC0-t обозначает площадь под кривой концентрации в плазме по отношению ко времени от 0 момента времени до момента времени t; W50 обозначает время, когда концентрация в плазме составляет 50% или более от Cmax; W75 обозначает время, когда концентрация в плазме составляет 75% или более от Cmax; и MRT обозначает среднее время нахождения для такролимуса (и/или его аналога). Максимальное отклонение обозначает (Cmax-Cmin)/Cmin, и флуктуация обозначает (Cmax-Cmin)/Cсредняя. Флуктуация пригодна для сравнения лекарственных форм, обеспечивающих различную биодоступность.
В этом контексте термин "лекарственное вещество" означает соединение, используемое для лечения заболевания, повреждения или боли. Лекарственные вещества справедливо разделяют на "профилактические", т.е. для сохранения здоровья, и "терапевтические", т.е. для восстановления здоровья.
В контексте настоящего изобретения подразумевают, что термины "контролируемое высвобождение" и "модифицированное высвобождение" являются эквивалентными терминами, охватывающими любой тип высвобождения такролимуса из композиции по изобретению, который пригоден для достижения конкретного терапевтического или профилактического ответа после введения субъекту. Специалисту в данной области известно, как контролируемое высвобождение/модифицированное высвобождение отличается от высвобождения обычных таблеток или капсул. Термины "высвобождение контролируемым образом" или "высвобождение модифицированным образом" имеют то же значение, которое указано выше. Термины включают замедленное высвобождение (которое приводит к более низкой Cmax и более позднему Tmax, однако t1/2 не меняется), пролонгированное высвобождение (которое приводит к более низкой Cmax, более позднему Tmax, но кажущееся t1/2 продлевается), отсроченное высвобождение (которое приводит к неизмененной Cmax, однако лаг-период и, таким образом, Tmax замедляются, и t1/2 не меняется), а также периодическое высвобождение, импульсное высвобождение, непрерывное высвобождение, длительное высвобождение, оптимизированное по времени высвобождение, быстрое (для получения усиленного начала действия) и т.д. В эти термины также включено применение конкретных условий в организме, например, различных ферментов или изменения pH для контроля высвобождения лекарственного вещества. Термин "пролонгированное высвобождение" выбран, поскольку этот термин, как полагают, наиболее правильно охватывает высвобождение продукта in vivo.
В этом контексте термин "деградация" или "деградирующий" означает постепенное разрушение поверхности материала или структуры, например, таблетки или покрытия таблетки. Этот термин, как используют в настоящем документе, как правило, обозначает растворение полимера, ответственного за пролонгирование высвобождения, являющееся более быстрым, чем растворение активного вещества, в результате чего полимер деградирует быстрее, чем активное вещество растворяется. Иными словами высвобождение, главным образом, контролируется деградацией, а не растворением активного вещества в системе полимерной матрицы.
Настоящее изобретение относится к фармацевтическим продуктам для улучшения лечения состояний, которые отвечают на лечение такролимусом, особенно для лечения, где является желательным иммуносупрессивный эффект.
Активным ингредиентом в композициях по изобретению предпочтительно является такролимус или любой аналог или производное такролимуса, который проявляет либо фармакологическую, либо терапевтическую активность, которая по меньшей мере эквивалентна активности такролимуса (FK-506 или FR-900506). Однако в объем настоящего изобретения входит такролимус в любой физической форме (кристаллы, аморфный порошок, любые возможные полиморфы, любые возможные сольваты, включая гидраты, ангидраты, их комплексы и т.д.). Также включен любой аналог, производное или активный метаболит такролимуса, их фармацевтически приемлемые соли, сольваты, комплексы и пролекарства. Однако считают, что частицы меньших размеров в микро- и нано-диапазоне и предпочтительно молекулярный раствор могут обеспечить предсказуемое и постоянное высвобождение такролимуса in vivo.
Таким образом, в предпочтительном варианте осуществления настоящее изобретение относится к пероральной лекарственной форме с пролонгированным высвобождением, содержащей в качестве активного вещества такролимус или его фармацевтически активный аналог, для иммуносупрессивного лечения один раз в сутки пациента, нуждающегося в этом, причем эта лекарственная форма высвобождает активное вещество в течение пролонгированного периода времени, определяемого высвобождением самое большее 63,5% содержимого активного вещества в момент времени через 12 часов, определенному по растворению in vitro и при тестировании в соответствии с тестом растворения USP II (лопасть) или тестом растворения USP I (корзина) в среде, имеющей pH 4,5 и содержащей 0,005% гидроксипропилцеллюлозы, и при вращении 50 об/мин. Общепризнано, что устройство для растворения с корзиной может быть более пригодным для капсул, а устройство для растворения с лопастью является более пригодным для дезинтеграции таблеток. Однако наиболее пригодное устройство для растворения можно легко определить путем тестирования того, достигается ли наиболее высокое соответствие с помощью одного или другого устройства.
В следующем варианте осуществления самое большее 63,5% активного вещества высвобождается в момент времени через 13 часов, более предпочтительно в момент времени через 14 часов, например, в момент времени через 15 часов. В предпочтительном варианте осуществления высвобождение in vitro происходит с постоянной скоростью, в результате чего можно достигать по существу профиля высвобождения нулевого порядка в течение пролонгированного периода времени. Поскольку требуется достаточное высвобождение в момент времени, когда лекарственная форма может достигнуть толстого кишечника, такой соответствующий период, когда является желательным высвобождение нулевого порядка, можно определять по высвобождению от 8 до 15 часов при тестировании в соответствии с тестом растворения USP II (лопасть) или тестом растворения USP I (корзина) в среде, имеющей pH 4,5 и содержащей 0,005% гидроксипропилцеллюлозы, и при вращении 50 об/мин. Поскольку твердая лекарственная форма может выходить из желудка вскоре после проглатывания или может задерживаться там на несколько часов перед тем, как достигнуть желудочно-кишечного тракта, также желательно, чтобы лучше контролировалось начальное высвобождение при измерении по высвобождению in vitro, которое представляет собой по существу профиль высвобождения нулевого порядка в течение пролонгированного периода времени, который определяют по высвобождению от 2 до 10 часов при тестировании в соответствии с тестом растворения USP II (лопасть) или тестом растворения USP I (корзина) в среде, имеющей pH 4,5 и содержащей 0,005% гидроксипропилцеллюлозы, и при вращении 50 об/мин.
В другом варианте осуществления изобретения добавление поверхностно-активного вещества в среду для высвобождения обеспечивает скорость высвобождения вещества, вследствие которой высвобождение самое большее 80% активных веществ пролонгируется до периода по меньшей мере 7 часов, такого как по меньшей мере 8 часов, такого как по меньшей мере 9 часов, такого как по меньшей мере 10 часов, такого как по меньшей мере 11 часов, такого как по меньшей мере 12 часов, такого как по меньшей мере 13 часов при тестировании in vitro в соответствии с тестом растворения USP II (лопасть) или тестом растворения USP I (корзина) в среде, имеющей pH 4,5 и содержащей 0,005% гидроксипропилцеллюлозы и, кроме того, содержащей 0,5% лаурилсульфат натрия (SLS), и при вращении 50 об/мин.
Как упоминалось выше, если высвобождение является чрезмерно пролонгированным, лекарственная форма может экскретироваться до того, как она высвободиться полностью, или высвобождение происходит слишком дистально для достаточного распределения. Таким образом, содержимое лекарственной формы должно высвобождаться со скоростями, указанными в настоящем документе, однако высвобождение не должно быть пролонгированным более чем на период 24 часа, такой как самое большее 23 часа, такой как самое большее 22 часа, такой как самое большее 21 час, такой как самое большее 20 часов, такой как самое большее 19 часов, такой как самое большее 18 часов, такой как самое большее 17 часов, такой как самое большее 16 часов, вычисленный для 80% содержимого и при добавлении в среду для растворения 0,5% лаурилсульфата натрия (SLS).
Альтернативно или дополнительно лекарственная форма удовлетворяет следующему условию, согласно к которому 63,5% высвобождения активного вещества прологнируется на период самое большее 20 часов, такой как самое большее 18 часов. Также в качестве верхнего предела могут быть предпочтительными более короткие периоды растворения, такие как самое большее 16 часов, такие как самое большее 15,5 часа, также при тестировании in vitro в соответствии с тестом растворения USP II (лопасть) или тестом растворения USP I (корзина) в среде, имеющей pH 4,5 и содержащей 0,005% гидроксипропилцеллюлозы, и при вращении 50 об/мин.
Пероральная лекарственная форма с пролонгированным высвобождением, где высвобождение начинается в пределах 120 минут, например, в пределах 90 минут, например, в пределах 60 минут после помещения лекарственной формы в устройство для растворения при тестировании in vitro в соответствии с тестом растворения USP II (лопасть) или тестом растворения USP I (корзина) в среде, имеющей pH 4,5 и содержащей 0,005% гидроксипропилцеллюлозы, и при вращении 50 об/мин, указывает на композицию, которая обеспечивает предсказуемый профиль высвобождения, при условии, что высвобождение в начальный период, такой как в пределах первых 2 часов, не является слишком быстрым. Если сразу после введения не происходит никакого высвобождения, пациент имеет риск снижения концентраций. У пациента, как правило, определяют концентрации в крови, наблюдаемые непосредственно перед проглатыванием суточной дозировки, и они представляют собой минимальную концентрацию, наблюдаемую в течение суток. Задержка высвобождения приводит к более поздней неизвестной минимальной концентрации.
В объем изобретения входят следующие характеристики высвобождения:
a) Пероральная лекарственная форма с пролонгированным высвобождением, которая высвобождает самое большее приблизительно 20% масс./масс. активного вещества в пределах 1 часа, или в пределах 2 часов, или в пределах 3 часов, или в пределах 4 часов, или в пределах 5 часов, при тестировании in vitro в соответствии с тестом растворения USP II (лопасть) или тестом растворения USP I (корзина) в среде, имеющей pH 4,5 и содержащей 0,005% гидроксипропилцеллюлозы, и при вращении 50 об/мин.
b) Пероральная лекарственная форма с пролонгированным высвобождением, которая высвобождает 40% масс./масс. активного вещества в пределах от 10 до 14 часов, как, например, в пределах приблизительно от 11 до 13 часов, при тестировании in vitro в соответствии с тестом растворения USP II (лопасть) или тестом растворения USP I (корзина) в среде, имеющей pH 4,5 и содержащей 0,005% гидроксипропилцеллюлозы, и при вращении 50 об/мин.
c) Пероральная лекарственная форма с пролонгированным высвобождением, которая высвобождает 20% масс./масс. от общего количества активного вещества, высвобожденного в пределах от 6 до 10 часов, как, например, в пределах приблизительно от 7 до 9 часов, при тестировании in vitro в соответствии с тестом растворения USP II (лопасть) или тестом растворения USP I (корзина) в среде, имеющей pH 4,5 и содержащей 0,005% гидроксипропилцеллюлозы, и при вращении 50 об/мин.
d) Пероральная лекарственная форма с пролонгированным высвобождением, которая высвобождает 50% масс./масс. активного вещества в пределах от 13 до 17 часов, как, например, в пределах приблизительно от 14 до 16 часов, при тестировании in vitro в соответствии с тестом растворения USP II (лопасть) или тестом растворения USP I (корзина) в среде, имеющей pH 4,5 и содержащей 0,005% гидроксипропилцеллюлозы, и при вращении 50 об/мин.
e) Пероральная лекарственная форма с пролонгированным высвобождением, где профиль высвобождения является по существу линейным в течение периода от 4 до 8 часов, что определяют как градиент или наклон, находящийся в пределах 25% от градиента или наклона, измеренного в момент времени 6 часов, например, в пределах 15%, предпочтительно в пределах 10%.
f) Пероральная лекарственная форма с пролонгированным высвобождением, где профиль высвобождения является по существу линейным в течение периода от 6 до 10 часов, что определяют как градиент или наклон, находящийся в пределах 25% от градиента или наклона, измеренного в момент времени 8 часов, например, в пределах 15%, предпочтительно в пределах 10%.
g) Пероральная лекарственная форма с пролонгированным высвобождением, где профиль высвобождения является по существу линейным в течение периода от 8 до 12 часов, что определяют как градиент или наклон, находящийся в пределах 25% от градиента или наклона, измеренного в момент времени 10 часов, например, в пределах 15%, предпочтительно в пределах 10%.
h) Пероральная лекарственная форма с пролонгированным высвобождением, где профиль высвобождения является по существу линейным в течение периода высвобождения от момента времени, когда высвобождается 20%, до момента времени, когда высвобождается 50%, что определяют как градиент или наклон в момент времени 80%, находящийся в пределах 25% от градиента или наклона, измеренного в момент времени 20%.
i) Лекарственная форма с пролонгированным высвобождением согласно любому из вышеуказанных пунктов, где механизм пролонгирования высвобождения не обеспечивается контролирующим проникновение покрытием.
Полагают, что характеристики профиля высвобождения, определенные выше, значительно повышают биодоступность такролимуса у млекопитающих, поскольку весь активный ингредиент или его основная часть в действительности высвобождаются в желудочно-кишечном тракте таким образом, чтобы по существу предотвращался или по меньшей мере значительно снижался метаболизм посредством CYP3A4. Кроме того, предусматривается, что этот эффект коррелирует с профилем растворения in vitro фармацевтической композиции и/или лекарственных форм по изобретению или по меньшей мере отражается им, причем этот профиль легко определить проведением указанного общепринятого способа растворения композиции и/или лекарственной формы in vitro.
Желаемый профиль высвобождения фармацевтической композиции можно обеспечивать комбинированием одной или нескольких из следующих возможностей.
i) покрытие композицией желудочно-резистентным покрытием; и/или
ii) использование фармацевтической композиции, содержащей твердую дисперсию или твердый раствор активного ингредиента, т.е. такролимуса или его аналога, в гидрофильном или смешивающемся с водой носителе и одном или нескольких модифицирующих высвобождение средствах; и/или
iii) использование фармацевтической композиции, содержащей твердую дисперсию или твердый раствор активного ингредиента, т.е. такролимуса или его аналога, в гидрофобном носителе и необязательно в одном или нескольких модифицирующих высвобождение средствах.
Однако покрытый желудочно-резистентным покрытием состав может иметь недостаток в виде задержки высвобождения без пролонгирования высвобождения, и таким образом, его следует использовать в комбинации с технологией пролонгирования.
В другом варианте осуществления изобретения и более предпочтительно предусматривается содержащая такролимус фармацевтическая композиция с пролонгированным высвобождением, имеющая активный ингредиент, растворенный или диспергированный в гидрофобном носителе, как описано в настоящем документе, предпочтительно в масле, в маслянистом материале, воске или производном жирной кислоты, более предпочтительно в воске, имеющем низкую температуру плавления, например, таком как глицерилмоностеарат.
В другом варианте осуществления изобретения предусматривается содержащая такролимус фармацевтическая композиция с пролонгированным высвобождением, имеющая активный ингредиент, растворенный или диспергированный в гидрофильном или смешивающимся с водой носителе, как описано в настоящем документе, предпочтительно носителе, выбранном из полиэтиленгликоля, оксидов полиоксиэтилена, полоксамеров, стеаратов полиоксиэтилена, поли-эпсилон-капролактона, полигликолированных глицеридов, таких как Gelucire® и их смесей, более предпочтительно из полиэтиленгликоля, необязательно в смеси с полоксамером. Конкретный пример пригодной смеси представляет собой смесь 70% масс./масс. полиэтиленгликоля 6000 (PEG6000) и 30% масс./масс. полоксамера 188.
В следующем аспекте настоящее изобретение относится к фармацевтической композиции в форме частиц, содержащей такролимус и/или его аналог, вместе с одним или несколькими фармацевтически приемлемыми эксципиентами, где композиция при пероральном введении млекопитающему, нуждающемуся в этом, проявляет величину AUC/AUCPrograf® по меньшей мере приблизительно 1,3, где величины AUC определяют в сходных условиях.
Из примеров, представленных в настоящем документе, очевидно, что биодоступность, достигаемая после введения композиции по изобретению, значительно повышается. Таким образом, в конкретных вариантах осуществления величина AUC/AUCPrograf® составляет по меньшей мере приблизительно 1,25, например, приблизительно 1,5 или более, приблизительно 1,8 или более, приблизительно 1,9 или более, приблизительно 2,0 или более, причем величины AUC определяют в сходных условиях.
После перорального введения фармацевтической композиции в соответствии с настоящим изобретением предусматривается, что профиль концентрации в плазме по отношению ко времени демонстрирует пролонгированный период времени, в течение которого концентрация в плазме поддерживается в пределах терапевтического окна (т.е. концентрация в плазме приводит к терапевтическому эффекту), не приводя к тяжелым нежелательным побочным эффектам. Таким образом, также наблюдают снижение максимальной концентрации. Таким образом, изобретение относится к фармацевтической композиции в форме частиц, содержащей такролимус вместе с одним или несколькими фармацевтически приемлемыми эксципиентами, где композиция при пероральном введении млекопитающему, нуждающемся в этом, высвобождает такролимус контролируемым образом и проявляет Cmax, которая составляет самое большее приблизительно 80% от Cmax для таблеток Prograf®, например, такую как самое большее приблизительно 75%, самое большее приблизительно 70%, самое большее приблизительно 65%, самое большее приблизительно 60%, самое большее приблизительно 55%, самое большее приблизительно 50%, самое большее приблизительно 45% или самое большее приблизительно 40%.
В контексте настоящего изобретения подразумевают, что термины "контролируемое высвобождение" и "пролонгированное высвобождение" являются эквивалентными терминами, охватывающими любой тип высвобождения такролимуса из композиции по изобретению, которая пригодна для достижения конкретного терапевтического или профилактического ответа после введения субъекту. Специалисту в данной области известно, каким образом контролируемое высвобождение/пролонгированное высвобождение отличается от высвобождения обычных таблеток или капсул. Термины "высвобождение контролируемым образом" или "высвобождение пролонгированным образом" имеют то же значение, которое указано выше.
Термины контролируемое высвобождение/пролонгированное высвобождение включат медленное высвобождение (которое приводит к более низкой Cmax и более позднему Tmax, но t1/2 не меняется), пролонгированное высвобождение (которое приводит к более низкой Cmax, более позднему Tmax, но кажущееся t1/2 продлевается), отсроченное высвобождение (которое приводит к неизмененной Cmax, однако лаг-период и, таким образом, Tmax замедляются, и t1/2 не меняется), а также периодическое высвобождение, импульсное высвобождение, непрерывное высвобождение, длительное высвобождение, оптимизированное по времени высвобождение, быстрое (для обеспечения усиленного начала действия) и т.д. В эти термины также включено применение конкретных условий в организме, например, различных ферментов или изменений pH для контроля высвобождения лекарственного вещества.
Более конкретно, после перорального введения млекопитающему, в том числе человеку, фармацевтической композиции по настоящему изобретению, содержащей дозу такролимуса 5 мг, такролимус высвобождается контролируемым образом и проявляет Cmax, которая составляет самое большее приблизительно 30 нг/мл, например, такую как самое большее приблизительно 25 нг/мл или самое большее приблизительно 20 нг/мл.
Однако снижение максимальной концентрации может не приводить к снижению терапевтического эффекта, при условии, что концентрация такролимуса в плазме сохраняется в пределах терапевтического окна. Таким образом, настоящее изобретение также относится к фармацевтической композиции, где W50 составляет по меньшей мере приблизительно 2 часа, как, например, по меньшей мере приблизительно 3 часа, по меньшей мере приблизительно 4 часа, по меньшей мере приблизительно 5 часов, по меньшей мере приблизительно 6 часов, по меньшей мере приблизительно 7 часов, по меньшей мере приблизительно 8 часов, по меньшей мере приблизительно 9 часов, приблизительно 10 часов или более, приблизительно 11 часов или более, приблизительно 12 часов или более, приблизительно 13 часов или приблизительно 14 часов или более.
Кроме или более того, композиция по изобретению имеет Cdiff=[Cmax-Ct(t=12 часов)], которая является меньшей, чем для Prograf® в тех же условиях. Если Cdiff для Prograf® принять за 100, тогда Cdiff для композиции по изобретению обычно составляет 90 или менее, как, например, приблизительно 85 или менее, приблизительно 80 или менее, приблизительно 75 или менее, приблизительно 70 или менее, приблизительно 65 или менее, приблизительно 60 или менее, приблизительно 55 или менее, приблизительно 50 или менее, приблизительно 45 или менее или приблизительно 40 или менее.
Более конкретно, после перорального введения млекопитающему, в том числе человеку, фармацевтической композиции по изобретению, содержащей 5 мг такролимуса, такролимус высвобождается контролируемым образом и проявляет Cdiff приблизительно 20 нг/мл или менее, такую как, например, приблизительно 15 нг/мл или менее, приблизительно 13 нг/мл или менее или приблизительно 10 нг/мл или менее.
Фармацевтическая композиция по изобретению высвобождает такролимус контролируемым образом для пролонгирования терапевтического действия такролимуса. В одном аспекте высвобождение может быть зависимым от pH, т.е. высвобождение, главным образом, происходит после прохождения через желудок. Такое зависимое от pH высвобождение, главным образом, обеспечивают с помощью материала растворяющегося в кишечнике покрытия, как описано в настоящем документе. Высвобождение также может быть независимым от pH, например, посредством предоставления композиции с покрытием с контролируемым высвобождением, например, таким как покрытие на основе целлюлозы, например, такой как этилцеллюлоза, или посредством предоставления композиции в форме композиции в матрице, например, такой как полимерная матрица на основе гидрофильной целлюлозы, например, на основе HPMC. Также, безусловно, можно использовать комбинацию.
Как правило, изменение биодоступности и/или изменения других связанных с биодоступностью параметров обычно определяют посредством исследований in vivo в пригодной модели на животных, в которой тестируют представляющие интерес композиции вместе, например, с Prograf® или сходным коммерчески доступным содержащим такролимус продуктом. Применение модели на собаках для установления данных по биодоступности определенных составов является обычной практикой в фармацевтической промышленности.
Исследования, касающиеся такролимуса, представляют собой нерандомизированные перекрестные исследования, где каждая собака является ее собственным контролем. Обычно используют четырех собак и четыре введения. Поскольку не проводят внутривенных введений, полученная биодоступность является относительной.
Кроме того, неожиданно было выявлено, что необходимость в одновременном приеме пищи для обеспечения достаточного усвоения такролимуса значительно снижается или даже полностью устраняется.
Таким образом, фармацевтические композиции по изобретению обеспечивают значительно более высокую биодоступность такролимуса, которая может снижать количество вводимых каждые сутки дозированных единиц, и снижать или устранять необходимость во введении, связанном с приемом пищи, что обеспечивает более высокую степень свободы для реципиента фармацевтических композиций, и, следовательно, согласие и/или соблюдение режима пациентами могут значительно повышаться. Более того, композиции обеспечивают значительное снижение побочных эффектов, особенно побочных эффектов, связанных с высокой максимальной концентрацией (например, таких как нефротоксичность и нейротоксичность, диарея, запор, боль в животе, тошнота и т.д.) и обеспечивают пролонгированное высвобождение такролимуса, ведущее к улучшенной терапии.
Следующим преимуществом лекарственной формы с пролонгированным высвобождением по изобретению является возможность получения эффективного терапевтического ответа со сниженной дозировкой по сравнению с традиционным пероральным лечением. Сходная биодоступность и улучшенный профиль после введения приводят к дозе, составляющей самое большее приблизительно 85% масс./масс., например, такой как самое большее приблизительно 80% масс./масс., самое большее приблизительно 75%, самое большее приблизительно 70% масс./масс., самое большее приблизительно 65% масс./масс., самое большее приблизительно 60% масс./масс., самое большее приблизительно 55% масс./масс. или самое большее приблизительно 50% масс./масс. от введенный дозы такролимуса в форме Prograf® или сходного коммерчески доступного содержащего такролимус продукта или в форме коммерчески доступного продукта с пролонгированным высвобождением, включающего Advagraf®.
Параметры, часто используемые в исследованиях биоэквивалентности, представляют собой Tmax, Cmax, AUC0-∞, AUC0-t. Другими важными параметрами могут быть W50, W75 и/или MRT. Таким образом, при определении наличия биоэквивалентности можно использовать по меньшей мере один из этих параметров. Более того, в контексте настоящего изобретения две композиции считают биоэквивалентными, если значение используемого параметра составляет 80-125% от значения для Prograf® или сходного коммерчески доступного содержащего такролимус продукта, используемого в тесте.
В контексте настоящего изобретения "Tmax" обозначает время до достижения максимальной концентрации в плазме (Cmax) после введения; AUC0-∞ обозначает площадь под кривой концентрации в плазме по отношению ко времени от момента времени 0 до бесконечности; AUC0-t обозначает площадь под кривой концентрации в плазме по отношению ко времени от момента времени 0 до момента времени t; W50 обозначает время, когда концентрация в плазме восставляет 50% или более от Cmax; W75 обозначает, когда концентрация в плазме составляет 75% или более от Cmax; и MRT обозначает среднее время нахождения для такролимуса (и/или его аналога).
Двумя другими основными недостатками, ассоциированными с лечением или профилактикой такролимусом, является относительно высокая встречаемость побочных эффектов и относительно высокая межиндивидуальная изменчивость. Предусматривается, что композиция по изобретению может приводить к уменьшению побочных эффектов. Уменьшение может выражаться в снижении частоты или тяжести. Побочные эффекты, о которых идет речь, включают, например, нефротоксичность и нейротоксичность, диарею, запор, боль в животе, тошноту и т.д. В одном аспекте изобретение относится к фармацевтической композиции в форме частиц, содержащей такролимус или его аналог вместе с одним или несколькими фармацевтически приемлемыми эксципиентами, где композиция при пероральном введении млекопитающему, нуждающемуся в этом, высвобождает такролимус или его аналог контролируемым образом и снижает побочные эффекты по сравнению с побочными эффектами Prograf®, вводимого в тех же условиях и в дозе, которая обеспечивает эквивалентный терапевтический эффект.
Повышение биодоступности, площади под кривой (AUC), обычно приводит к снижению меж- и внутрииндивидуальной изменчивости, связанной со всасыванием лекарственного вещества. В частности, это справедливо, когда низкая и сниженная биодоступность являются следствием слабой растворимости в воде. Предусматривается, что композиции по изобретению могут обеспечить CV (коэффициент изменчивости) для данных о площади под кривой, который является значительно меньшим, чем для Prograf® и сходных продуктов.
Как упоминалось в настоящем документе, одним из основных признаков настоящего изобретения является то, что можно достигнуть повышения биодоступности пероральным введением лекарственной формы с пролонгированным высвобождением по изобретению. Обычно низкая биодоступность лекарственного вещества после перорального введения является преградой для создания композиции лекарственного вещества с контролируемым и пролонгированным высвобождением, вследствие того факта, что практически невозможно достигнуть эффективных уровней лекарственного средства на протяжении пролонгированного периода времени. Однако с помощью технологии по настоящему изобретению можно достигнуть значительно улучшенной биодоступности и, тем самым, можно создавать композиции с контролируемым, пролонгированным или замедленным высвобождением.
Такролимус экстенсивно метаболизируется изоферментом CYP3A4 в стенке пищеварительного канала и печени. Таким образом, пригодная композиция с контролируемым высвобождением может представлять собой композицию, которая предназначена для высвобождения такролимуса отсроченным образом так, чтобы предотвратить или снизить метаболизм посредством CYP3A4 в желудочно-кишечном тракте.
Замедленного высвобождения, главным образом, достигают с помощью какого-либо желудоно-резистентного покрытия. В то время как полупроницаемое покрытие может продемонстрировать некоторое замедленное высвобождение, оно не является абсолютно достаточном для "задержки" высвобождения. Кроме того, оно требует некоторого времени для высвобождения содержимого. Покрытие по этому изобретению представляет собой зависимое от pH покрытие. Этот тип покрытия является высоко устойчивым к высвобождению лекарственного средства до достижения определенного pH. В пределах совсем немногих 1/10 pH, пленка изменяет свойства и становится проницаемой. Примеры чувствительных к pH полимеров, которые являются относительно нерастворимыми и непроницаемыми при pH желудка, но которые являются более растворимыми и проницаемыми при pH тонкого кишечника и толстого кишечника, включают, но не ограничиваются ими, полиакриламиды, производные фталатов, такие как кислотные фталаты углеводов, фталат ацетата амилозы, фталат ацетата целлюлозы, другие фталаты сложных эфиров целлюлозы, другие фталаты простых эфиров целлюлозы, фталат гидроксипропилцеллюлозы, фталат гидроксипропилэтилцеллюлозы, фталат гидроксипропилметилцеллюлозы, фталат метилцеллюлозы, фталат поливинилацетата, гидрофталат поливинилацетата, фталат ацетата целлюлозы натрия, кислотный фталат крахмала, сополимер стирол-дибутилфталат малеиновой кислоты, сополимер стирол-фталат поливинилацетата малеиновой кислоты, сополимеры стирола и малеиновой кислоты, производные полиакриловой кислоты, такие как сополимеры акриловой кислоты и акрилового сложного эфира, полиметакриловая кислота и ее сложные эфиры, сополимеры акриловой и метакриловой кислот, шеллак и сополимеры винилацетата и кротоновой кислоты.
Представляющие особый интерес pH-чувствительные полимеры включают шеллак; производные фталатов, в частности, фталат ацетата целлюлозы, фталат поливинилацетата и фталат гидроксипропилметилцеллюлозы; производные полиакриловой кислоты, в частности, полиметилметакрилат, смешанный с сополимерами акриловой кислоты и акрилового сложного эфира; и сополимеры винилацетата и кротоновой кислоты.
Высвобождение активного вещества из композиции, имеющей покрытие для отсроченного высвобождения, также может представлять собой ферментативную реакцию, если в качестве материала покрытия используют, например, зеин или смеси моно/диглицеридов.
При пероральном введении млекопитающему, в том числе человеку, нуждающемуся в этом, фармацевтическая композиция с контролируемым высвобождением в соответствии с настоящим изобретением высвобождает такролимус таким образом, что в плазме достигается концентрация по меньшей мере приблизительно 5 нг/мл, например, такая как по меньшей мере приблизительно 7,5 нг/мл или по меньшей мере приблизительно 10 нг/мл в течение периода времени по меньшей мере приблизительно 24 часа. В конкретном аспекте изобретения отличия между максимальной концентрацией в плазме и концентрацией в плазме, измеренной через 24 часа после введения, составляют самое большее приблизительно 20 нг/мл, как, например, самое большее приблизительно 10 нг/мл, самое большее приблизительно 7,5 нг/мл или самое большее приблизительно 5 нг/мл.
В конкретном аспекте изобретение относится к фармацевтической композиции или твердой лекарственной форме, которая высвобождает такролимус и/или его аналог относительно быстро, чтобы обеспечить относительно быстрое появление терапевтического эффекта. В одном аспекте изобретение относится к фармацевтической композиции в форме частиц, содержащей такролимус и/или его аналог вместе с одним или несколькими фармацевтически приемлемыми эксципиентами, где композиция при пероральном введении млекопитающему, нуждающемуся в этом, контролируемым образом высвобождает по меньшей мере приблизительно 50% масс./масс. от общего количества такролимус или его аналога в пределах приблизительно 24 часов, как, например, в пределах приблизительно 22 часов, в пределах приблизительно 20 часов, в пределах приблизительно 18 часов, в пределах приблизительно 15 часов или в пределах приблизительно 12 часов.
Дополнительно или альтернативно по меньшей мере приблизительно 50% масс./масс. от общего количества такролимуса и/или его аналога высвобождается в пределах приблизительно 24 часов, в пределах приблизительно 22 часов, в пределах приблизительно 20 часов, в пределах приблизительно 18 часов, в пределах 15 часов, в пределах приблизительно 12 часов, при тестирования в тесте растворения in vitro и с использованием среды для растворения, содержащей буфер, имеющий pH 7,5. Руководство для пригодного теста растворения описано в примерах ниже, однако в объем настоящего изобретения входят варианты с точки зрения конкретного используемого способа и ингредиентов, содержащихся в среде для растворения, и т.д. Специалисту в данной области известно как проводить пригодный тест растворения, например, из руководства USP, Ph. Eur. и сходных с ними, в том числе из базы данных FDA по способам растворения лекарственных продуктов. Пригодными условиями для теста растворения in vitro являются использование теста растворения USP (способ с лопастью) и буфера с pH 7,5 содержащего 2,5% SDS и 1 г/мл панкреатина в качестве среды для растворения.
В других вариантах осуществления удовлетворяются следующие условия в отношении теста растворения in vitro:
В других вариантах осуществления удовлетворяются следующие условия в отношении теста растворения, проводимого в кислотных условиях:
i) самое большее приблизительно 30% масс./масс., как, например, самое большее приблизительно 25% масс./масс., самое большее приблизительно 20% масс./масс., самое большее приблизительно 15% масс./масс. или самое большее приблизительно 10% масс./масс. такролимуса или его аналога высвобождается в пределах 2 часов в тесте растворения in vitro с использованием среды для растворения, имеющей значение pH самое большее приблизительно 5, например, такое как самое большее приблизительно 4,5, самое большее приблизительно 4, самое большее приблизительно 3,5, самое большее приблизительно 3, самое большее приблизительно 2 или самое большее приблизительно 1,5;
ii) самое большее приблизительно 10% масс./масс., как, например, самое большее приблизительно 7,5% масс./масс., самое большее приблизительно 5% масс./масс. или самое большее приблизительно 2,5% масс./масс. такролимуса или его аналога высвобождается в пределах 2 часов в тесте растворения in vitro с использованием среды для растворения, имеющей значение pH самое большее приблизительно 5, например, такое как самое большее приблизительно 4,5, самое большее приблизительно 4, самое большее приблизительно 3,5, самое большее приблизительно 3, самое большее приблизительно 2 или самое большее приблизительно 1,5;
iii) самое большее приблизительно 60% масс./масс., как, например, самое большее приблизительно 50% масс./масс., самое большее приблизительно 40% масс./масс. или самое большее приблизительно 30% масс./масс. такролимус или его аналога высвобождается в пределах 15 часов, как, например, в пределах 12 часов, при тестировании в тесте растворения in vitro с использованием среды для растворения, имеющей значение pH самое большее приблизительно 4,5, например, такое как самое большее приблизительно 4,0, самое большее приблизительно 3,5, самое большее приблизительно 3, самое большее приблизительно 2 или самое большее приблизительно 1,5;
iv) самое большее приблизительно 40% масс./масс., как, например, самое большее приблизительно 30% масс./масс., самое большее приблизительно 25% масс./масс. или самое большее приблизительно 20% масс./масс. такролимуса или его аналога высвобождается в пределах 6 часов при тестировании в тесте растворения in vitro с использованием среды для растворения, имеющей значение pH самое большее приблизительно 4,5, такое как, например, самое большее приблизительно 4,0, самое большее приблизительно 3,5, самое большее приблизительно 3, самое большее приблизительно 2 или самое большее приблизительно 1,5, и/или
v) самое большее приблизительно 30% масс./масс., как, например, самое большее приблизительно 25% масс./масс., самое большее приблизительно 20% масс./масс. или самое большее приблизительно 15% масс./масс. такролимус или его аналога высвобождается в пределах 4 часов при тестировании в тесте растворения in vitro с использованием среды для растворения, имеющей значение pH самое большее приблизительно 4,5, например, такое как самое большее приблизительно 4,0, самое большее приблизительно 3,5, самое большее приблизительно 3, самое большее приблизительно 2 или самое большее приблизительно 1,5.
Помимо такролимуса, композиция по изобретению также может содержать дополнительное терапевтически, профилактически и/или диагностически активное вещество. Особый интерес представляют комбинации такролимуса по меньшей мере с одним из следующих активных веществ: вещества, которые показаны для применения при трансплантации органов, например, такие как стероиды, ингибиторы кальциневрина и/или антипролиферативные средства. Конкретные примеры включают преднизон, преднизолон, метилпреднизон, циклоспорин, микофенолята мофетил, азатиоприн, сиролимус, эверолимус, микофенолят натрия и FTY720 (Novartis).
Фармацевтические композиции можно получить любым удобным способом, например, таким как гранулирование, перемешивание, распылительная сушка и т.д. Особенно пригодным способом является способ, описанный в WO 03/004001. В этом документе описан способ получения материала в виде частиц способом контролируемой агломерации, т.е. способом, который обеспечивает контролируемое увеличение размера частиц. Способ вовлекает напыление первой композиции, содержащей, например, такролимус и носитель, которая является расплавленной, на второй твердый носитель. Обычно расплавляемый носитель имеет температуру плавления по меньшей мере 5°С, но ниже температуры плавления такролимуса. Температура плавления носителя может находиться в диапазоне от 10 до 150°С, например, таком как диапазон от 30 до 100°С, или наиболее предпочтительным является диапазон от 40 до 50°С.
Квалифицированный специалист способен выбрать пригодный носитель, являющийся фармацевтически приемлемым, способный растворять или по меньшей мере частично растворять такролимус и имеющий температуру плавления в желаемом диапазоне, с использованием общих знаний и общепринятого экспериментирования. Пригодные кандидаты для носителей описаны в WO 03/004001, которая включена в настоящий документ в качестве ссылки.
В контексте настоящего изобретения пригодные носители представляют собой, например, носители, упомянутые в качестве масла или маслянистого материала (как описано в настоящем документе ниже), а также носители, описанные в WO 03/004001.
Преимущество применения способа контролируемой агломерации, описанного в WO 03/004001, состоит в том, что можно наносить относительно большое количество расплава на материал в виде частиц без нежелательного увеличения размера частиц. Таким образом, в одном варианте осуществления изобретения материал фармацевтической композиции в виде частиц имеет геометрический средневзвешенный диаметр dgw≥10 мкм, например, такой как ≥20 мкм, от приблизительно 20 до приблизительно 2000, от приблизительно 30 до приблизительно 2000, от приблизительно 50 до приблизительно 2000, от приблизительно 60 до приблизительно 2000, от приблизительно 75 до приблизительно 2000, например, такой как от приблизительно 100 до приблизительно 1500 мкм, от приблизительно 100 до приблизительно 1000 мкм или от приблизительно 100 до приблизительно 700 мкм, или самое большее приблизительно 400 мкм или самое большее 300 мкм, например, такой как от приблизительно 50 до приблизительно 400 мкм, например, такой как от приблизительно 50 до приблизительно 350 мкм, от приблизительно 50 до приблизительно 300 мкм, от приблизительно 50 до приблизительно 250 мкм или от приблизительно 100 до приблизительно 300 мкм.
Материал в виде частиц, полученный указанным выше способом, имеет пригодные свойства в отношении текучести и/или прессуемости, и таким образом, он пригоден для дальнейшей переработки в фармацевтические лекарственные формы.
Твердая дисперсия и/или твердый раствор такролимуса
Твердая дисперсия или твердая дисперсия, используемая в предпочтительном варианте осуществления изобретения, содержит активный ингредиент, выбранный из такролимуса и его аналогов, который диспергирован или растворен в гидрофильном или смешивающемся с водой носителе, имеющем температуру плавления (температуру замерзания или температуру застывания) по меньшей мере 20°С, в концентрации между приблизительно 0,01% масс./масс. и приблизительно 15% масс./масс., и эта дисперсия образует твердую дисперсию или твердый раствор при температуре окружающей среды (комнатной температуре).
Концентрация активного ингредиента в гидрофильном или смешивающемся с водой носителе составляет самое большее 15% масс./масс., предпочтительно самое большее 10% масс./масс., предпочтительно самое большее 8% масс./масс., более предпочтительно самое большее 6% масс./масс., еще более предпочтительно самое большее 5% масс./масс., самое большее 4% масс./масс., особенно самое большее 3% масс./масс., в частности, сааме большее 2% масс./масс.; и/или составляет по меньшей мере приблизительно 0,05% масс./масс., предпочтительно по меньшей мере приблизительно 0,1% масс./масс., более предпочтительно по меньшей мере приблизительно 0,5% масс./масс., особенно по меньшей мере приблизительно 0,7% масс./масс., в частности, по меньшей мере приблизительно 1% масс./масс.
Физически комбинация активного ингредиента и носителя может либо образовывать твердую дисперсию, т.е. активный ингредиент диспергирован в носителе в форме частиц, либо она может образовывать твердый раствор, т.е. активный ингредиент растворен в носителе на молекулярном уровне. Активный ингредиент и носитель также могут образовывать твердую дисперсию, содержащую часть активного ингредиента, растворенную на молекулярном уровне. Физическое состояние дисперсии и/или раствора можно определять с использованием различных способов, таких как высокотемпературная микроскопия (HSM), дифференциальная сканирующая калориметрия (DSC), сканирующая электронная микроскопия (SEM), необязательно в комбинации с энергодисперсинным рентгеновским излучением (EDX) и дифракция рентгеновских лучей на порошке. В предпочтительном варианте осуществления активный ингредиент полностью растворен в носителе с образованием твердого раствора при температуре окружающей среды.
Твердая дисперсия по изобретению проявляет очень быстрое немедленное высвобождение такролимуса, когда композицию, содержащую дисперсию или раствор, тестируют в тесте растворения согласно USP с использованием водной среды для растворения, и по меньшей мере 50% масс./масс. активного фармацевтического ингредиента высвобождается в пределах приблизительно 30 минут, предпочтительно в пределах 20 минут, более предпочтительно в пределах 15 минут; например, по меньшей мере 75% масс./масс. активного фармацевтического ингредиента высвобождается в пределах приблизительно 40 минут, или более предпочтительно по меньшей мере 90% масс./масс. активного фармацевтического ингредиента высвобождается в пределах приблизительно 60 минут, предпочтительно в пределах 45 минут. Например, тест можно проводить в соответствии с любым способом и любыми указаниями, цитированными в USP. Таким образом, тест растворения можно проводить в водной среде для растворения при нейтральном или близком к нейтральному значении pH, например, при pH 6,8, или при любом кислом pH, имитирующем условия pH в желудочно-кишечном тракте. Однако в объем настоящего изобретения входят варианты с точки зрения конкретного используемого способа и ингредиентов, содержащихся в среде для растворения, и т.д. Специалисту в данной области известно, как проводить пригодный тест растворения, например, из руководства USP, Ph. Eur. и т.п. Пригодными условиями для теста растворения in vitro являются использование теста растворения USP (способ с лопастью) и буфера с pH 7,5, содержащего 2,5% SDS и 1 г/мл панкреатина, в качестве среды для растворения.
Гидрофильный или смешивающийся с водой носитель для применения в соответствии с изобретением, предпочтительно представляет собой носитель, имеющий температуру плавления (температуру замерзания или температуру застывания) по меньшей мере 20°С, более предпочтительно по меньшей мере 30°С, более предпочтительно по меньшей мере 40°С, более предпочтительно по меньшей мере 50°С, еще более предпочтительно по меньшей мере 52°С, еще более предпочтительно по меньшей мере 55°C, еще более предпочтительно по меньшей мере 59°С, особенно по меньшей мере 61°С, в частности, по меньшей мере 65°С.
Примеры пригодных гидрофильных или смешивающихся с водой носителей для применения в соответствии с этим изобретением, выбирают из группы, состоящей из полиэтиленгликолей, оксидов полиоксиэтилена, полоксамеров, стеаратов полиоксиэтилена, поли-эпсилон-капролактона, полигликолированных глицеридов, таких как Gelucire®, и их смесей.
В предпочтительном варианте осуществления изобретения носитель представляет собой полиэтиленгликоль (PEG), в частности, PEG, имеющий среднюю молекулярную массу по меньшей мере 1500, предпочтительно по меньшей мере 3000, более предпочтительно по меньшей мере 4000, особенно по меньшей мере 6000. Полиэтиленгликоль можно преимущественно смешивать с одним или несколькими гидрофильными или смешивающимися с водой носителями, например, полоксамером, предпочтительно в соотношении (в расчете на масс./масс.) от 1:3 до 10:1, предпочтительно от 1:1 до 5:1, более предпочтительно от 3:2 до 4:1, особенно от 2:1 до 3:1, в частности, приблизительно 7:3. Конкретным примером пригодной смеси является смесь PEG6000 и полоксамера 188 в соотношении 7:3.
Для полиэтиленгликолей (PEG) температура плавления (температура замерзания или температура застывания) возрастает по мере возрастания средней молекулярной массы. Например, температура плавления для PEG 400 находится в диапазоне 4-8°С, для PEG 600 она находится в диапазоне 20-25°С, для PEG1500 она находится в диапазоне 44-48°С, для PEG2000 она составляет приблизительно 52°С, для PEG4000 она составляет приблизительно 59°С, для PEG 6000 она составляет приблизительно 65°С и для PEG8000 она составляет приблизительно 61°С.
Пригодными полоксамерами (также называемыми блок-сополимерами полиоксипропилен-полиоксиэтилен) являются, например, полоксамер 188, полоксамер 237, полоксамер 338 или полоксамер 407 или другие блок-сополимеры оксида этилена и оксида пропилена, такие как серии Pluronic® и/или Tetronic®. Пригодные блок-сополимеры серии Pluronic® включают полимеры, имеющие молекулярную массу приблизительно 3000 или более, например, такую как от приблизительно 4000 до приблизительно 20000, и/или вязкость (Brookfield) от приблизительно 200 до приблизительно 4000 спз, например, такую как от приблизительно 250 до приблизительно 3000 спз. Пригодные примеры включают Pluronic® F38, P65, P68LF, P75, F77, P84, P85, F87, F88, F98, P103, P104, P105, F108, P123, F123, F127, 10R8, 17R8, 25R5, 25R8 и т.д. Пригодные блок-сополимеры серии Tetronic® включают полимеры, имеющие молекулярную массу приблизительно 8000 или более, например, такую как от приблизительно 9000 до приблизительно 35000, и/или вязкость (Brookfield) от приблизительно 500 до приблизительно 45000 спз, например, такую как от приблизительно 600 до приблизительно 40000. Вязкость, приведенную выше, определяют при 60°С для веществ, которые представляют собой пасты при комнатной температуре, и при 77°С для веществ, которые являются твердыми при комнатной температуре.
В предпочтительном варианте осуществления настоящего изобретения полоксамер представляет собой полоксамер 188, который имеет среднюю молекулярную массу приблизительно 8400 и температуру плавления приблизительно 50-54°С.
Другие пригодные гидрофильные или смешивающиеся с водой носители могут представлять собой поливинилпирролидоны, сополимеры поливинил-поливинилацетат (PVP-PVA), поливиниловый спирт (PVA), метакриловые полимеры (Eudragit RS; Eudragit RL, Eudragit NE, Eudragit E), производные целлюлозы, включающие гидроксипропилметилцеллюлозу (HPMC), гидроксипропилцеллюлозу (HPC), метилцеллюлозу, карбоксиметилцеллюлозу натрия, гидроксиэтилцеллюлозу, пектины, циклодекстрины, галактоманнаны, альгинаты, каррагенаты, ксантановые смолы и их смеси.
"Полигликолированные глицериды" означают смесь моно-, ди- и триглицеридов и сложных моно- и диэфиров полиэтиленгликоля (PEG), предпочтительно с молекулярной массой от 200 до 600, когда это целесообразно, свободного глицерина и свободного PEG, величину HLB которой корректируют посредством длины цепи PEG, и температуру плавления которой корректируют посредством длины цепей жирных кислот, PEG и степени насыщенности цепей жирных кислот, и таким образом исходного масла; примерами таких смесей являются Gelucire®. Композиции Gelucire® представляют собой инертные полутвердые восковидные материалы, которые являются амфифильными и доступны с различными физическими свойствами. Они являются поверхностно-активными и диспергируются или растворяются в водных средах, образующих мицеллы, микроскопические глобулы или везикулы. Их идентифицируют по их температуре плавления/величине HLB. Температуру плавления выражают в градусах Цельсия, а HLB (гидрофильно-липофильный баланс) представляет собой числовую шкалу от 0 до приблизительно 20. Более низкие значения HLB обозначают более липофильные и гидрофобные вещества, а более высокие значения обозначают более гидрофильные и липофобные вещества. Аффинность соединения к воде или к маслянистым веществам определяют, а их значение HLB присваивают экспериментально. Для достижения желаемых характеристик температуры плавления и/или величины HLB можно выбирать один эксципиент Gelucire® или смесь различных классов этих эксципиентов. Они представляют собой смеси сложных моноэфиров, сложных диэфиров и/или сложных триэфиров глицеридов жирных кислот длинной цепи (от C12 до C18), и сложных (моно- и/или ди-)эфиров PEG и жирных кислот длинной цепи (от C12 до C18), и они могут включать свободный PEG. Композиции Gelucire®, главным образом, описаны в качестве сложных эфиров жирных кислот и глицерина и сложных эфиров PEG или в качестве полигликолированных глицеридов. Композиции Gelucire® характеризуются широким диапазоном температур плавления от приблизительно 33°C до приблизительно 64°С и наиболее часто от приблизительно 35°С до приблизительно 55°C, и широким множеством значений HLB от приблизительно 1 до приблизительно 14, наиболее часто от приблизительно 7 до приблизительно 14. Например, Gelucire® 50/13 означает температуру плавления приблизительно 50°С и величину HLB приблизительно 13 для этого класса Gelucire®.
Фармацевтически приемлемые эксципиенты
Примеры пригодных эксципиентов для применения в композиции или твердой лекарственной форме в соответствии с настоящим изобретением включают наполнители, разбавители, дезинтегрирующие вещества, связующие вещества, смазывающие вещества и т.п. и их смеси. Поскольку композицию или твердую лекарственную форму по этому изобретению можно использовать для различных целей, выбор эксципиентов обычно проводят, учитывая такие различные применения. Другими фармацевтически приемлемыми эксципиентами, пригодными для применения, являются, например, подкислители, подщелачивающие средства, консерванты, антиоксиданты, буферные средства, хелатирующие агенты, красители, комплексообразующие средства, эмульгаторы и/или солюбилизирующие средства, вкусовые добавки и отдушки, увлажнители, подсластители, смачивающие вещества и т.п.
Примеры пригодных наполнителей, разбавителей и/или связующих веществ включают лактозу (например, высушенную распылением лактозу, α-лактозу, β-лактозу, Tabletose®, различные классы Pharmatose®, Microtose® или Fast-Roc®), микрокристаллическую целлюлозу (различные классы Avicel®, Elcema®, Vivacel®, Ming Tai® или Solka-Floc®), гидроксипропилцеллюлозу, L-гидроксипропилцеллюлозу (с низким замещением), гидроксипропилметилцеллюлозу (HPMC) (например, Methocel E, F и K, Metolose SH, Shin-Etsu, Ltd, например, такие как Methocel E и Metolose 60 SH класса 4000 спз, Methocel F и Metolose 65 SH класса 4000 спз, Methocel K класса 4000, 15000 и 100000; и Metolose 90 класса SH 4000, 15000, 39000 и 100000), полимеры метилцеллюлозы (например, такие как Methocel A, Methocel A4C, Methocel A15C, Methocel A4M), гидроксиэтилцеллюлозу, карбоксиметилцеллюлозу натрия, карбоксиметилен, карбоксиметилгидроксиэтилцеллюлозу и другие производные целлюлозы, сахарозу, агарозу, сорбит, маннит, декстрины, мальтодекстрины, крахмалы или модифицированные крахмалы (включая картофельный крахмал, кукурузный крахмал и рисовый крахмал), фосфат кальция (например, основный фосфат кальция, гидрофосфат кальция, гидрат фосфата дикальция), сульфат кальция, карбонат кальция, альгинат натрия, коллаген и т.д.
Конкретными примерами разбавителей являются, например, карбонат кальция, двухосновный фосфат кальция, трехосновный фосфат кальция, сульфат кальция, микрокристаллическая целлюлоза, порошковая целлюлоза, декстраны, декстрин, декстроза, фруктоза, каолин, лактоза, маннит, сорбит, крахмал, прежелатинизированный крахмал, сахароза, сахар и т.д.
Конкретными примерами дезинтегрирующих веществ являются, например, альгиновая кислота или альгинаты, микрокристаллическая целлюлоза, гидроксипропилцеллюлоза и другие производные целлюлозы, кроскармеллоза натрия, кросповидон, полакриллин калий, натрия крахмала гликолят, крахмал, прежелатинизированный крахмал, карбоксиметилкрахмал (например, Promogel® и Explotab®) и т.д.
Конкретными примерами связующих веществ являются, например, гуммиарабик, альгиновая кислота, агар, каррагенан кальция, карбоксиметилцеллюлоза натрия, микрокристаллическая целлюлоза, декстрин, этилцеллюлоза, желатин, жидкая глюкоза, гуаровая камедь, гидроксипропилметилцеллюлоза, метилцеллюлоза, пектин, PEG, повидон, прежелатинизированный крахмал и т.д.
Также в композицию можно включать вещества, способствующие скольжению, и смазывающие вещества. Их примеры включают стеариновую кислоту, стеарат магния, стеарат кальция или стеарат другого металла, тальк, воски и глицериды, легкое минеральное масло, PEG, глицерилбегенат, коллоидный диоксид кремния, гидрогенизированные растительные масла, кукурузный крахмал, стеарилфумарат натрия, полиэтиленгликоли, алкилсульфаты, бензоат натрия, ацетат натрия и т.д.
Другие эксципиенты, которые можно включать в композицию или твердую лекарственную форму по изобретению, представляют собой, например, вкусовые добавки, красители, корригирующие вкус средства, pH-корригирующие средства, буферные средства, консерванты, стабилизаторы, антиоксиданты, смачивающие вещества, корригирующие влажность средства, поверхностно-активные вещества, суспендирующие средства, усиливающие всасывание средства, средства для пролонгированного высвобождения и т.д.
Другие добавки в композиции или твердой лекарственной форме по изобретению могут представлять собой антиоксиданты, например, такие как аскорбиновая кислота, аскорбилпальмитат, бутилированный гидроксианизол, бутилированный гидрокситолуол, гипофосфорная кислота, монотиоглицерин, метабисульфит калия, пропилгаллат, натрия формальдегида сульфоксилат, метабисульфит натрия, тиосульфат натрия, диоксид серы, токоферол, ацетат токоферола, гемисукцинат токоферола, TPGS или другие производные токоферола и т.д. Композиция носителя также может содержать, например, стабилизаторы. Концентрация антиоксиданта и/или стабилизатора в композиции носителя обычно составляет от приблизительно 0,1% масс./масс. до приблизительно 5% масс./масс.
Композиция или твердая лекарственная форма по изобретению также может включать одно или несколько поверхностно-активных веществ или веществ, обладающих поверхностно-активными свойствами. Предусматриваются, что такие вещества вовлечены в смачивание слабо растворимого активного вещества и, таким образом, обеспечивают характеристики повышенной растворимости активного вещества.
Пригодные эксципиенты для применения в композиции или твердой лекарственной форме по изобретению представляют собой поверхностно-активные вещества, например, такие как амфифильные поверхностно-активные вещества, такие как вещества, описанные в WO 00/50007 на имя Lipocine, Inc. Примерами пригодных поверхностно-активных веществ являются
i) полиэтоксилированные жирные кислоты, например, такие как сложные моно- и диэфиры жирных кислот и полиэтиленгликоля или их смеси, например, такие как сложные моно- или диэфиры полиэтиленгликоля с лауриновой кислотой, олеиновой кислотой, стеариновой кислотой, миристиновой кислотой, рицинолеиновой кислотой, и полиэтиленгликоль может быть выбран из PEG 4, PEG 5, PEG 6, PEG 7, PEG 8, PEG 9, PEG 10, PEG 12, PEG 15, PEG 20, PEG 25, PEG 30, PEG 32, PEG 40, PEG 45, PEG 50, PEG 55, PEG 100, PEG 200, PEG 400, PEG 600, PEG 800, PEG 1000, PEG 2000, PEG 3000, PEG 4000, PEG 5000, PEG 6000, PEG 7000, PEG 8000, PEG 9000, PEG 10000, PEG 15000, PEG 20000, PEG 35000,
ii) сложные эфиры полиэтиленгликоля с жирными кислотами и глицерином, т.е. сложные эфиры, подобные указанным выше, но в форме глицерильных сложных эфиров с отдельными жирными кислотами;
iii) сложные эфиры глицерина, пропиленгликоля, этиленгликоля, PEG или сорбита, например, с растительными маслами, например, такими как гидрогенизированное касторовое масло, миндальное масло, пальмоядровое масло, касторовое масло, масло из косточек абрикоса, оливковое масло, арахисовое масло, гидрогенизированныое пальмоядровое масло и т.п.,
iv) полиглицеризованные жирные кислоты, например, такие как стеарат полиглицерина, олеат полиглицерина, рицинолеат полиглицерина, линолеат полиглицерина,
v) сложные эфиры пропиленгликоля и жирных кислот, например, такие как монолаурат пропиленгликоля, рицинолеат пропиленгликоля и т.п.,
vi) моно- и диглицериды, например, такие как глицерилмоноолеат, глицерилдиолеат, глицерилмоно- и/или диолеат, глицерилкаприлат, глицерилкапрат и т.д.;
vii) стерин и производные стерина;
viii) сложные эфиры полиэтиленгликоля сорбитана и жирных кислот (сложные эфиры PEG-сорбитана и жирных кислот), такие как сложные эфиры PEG с различными молекулярными массами, указанными выше, и различными сериями Tween®;
ix) алкильные простые эфиры полиэтиленгликоля, например, такие как олеиловый простой эфир PEG и лауриловый простой эфир PEG;
x) сложные эфиры сахаров, например, такие как монопальмитат сахарозы и монолаурат сахарозы;
xi) соединения полиэтиленгликоля с алкилфенолами, например, такие как серии Triton® X или N;
xii) блок-сополимеры полиоксиэтилен-полиоксипропилен, например, такие как серии Pluronic®, серии Synperonic®, Emkalyx®, Lutrol®, Supronic® и т.д. Общим термином для этих полимеров является термин "полоксамеры", и подходящими примерами в контексте настоящего изобретения являются полоксамер 105, 108, 122, 123, 124, 181, 182, 183, 184, 185, 188, 212, 215, 217, 231, 234, 235, 237, 238, 282, 284, 288, 331, 333, 334, 335, 338, 401, 402, 403 и 407;
xiii) сложные эфиры сорбитана и жирных кислот, такие как серия Span® или серия Ariacel®, например, такие как монолаурат сорбитана, монопальмитат сорбитана, моноолеат сорбитана, моностеарат сорбитана и т.д.;
xiv) сложные эфиры жирных кислот с низшими спиртами, например, такие как олеат, изопропилмиристат, изопропилпальмитат и т.д.;
xv) ионные поверхностно-активные вещества, включающие катионные, анионные и цвитерионные поверхностно-активные вещества, например, такие как соли жирных кислот, желчные соли, фосфолипиды, сложные эфиры фосфорной кислоты, карбоксилаты, сульфаты и сульфонаты и т.д.
Когда в композиции или твердой лекарственной форме по изобретению присутствует поверхностно-активное вещество или смесь поверхностно-активных веществ, концентрация поверхностно-активного вещества (веществ) обычно находится в диапазоне приблизительно 0,1-80% масс./масс., например, таком как от приблизительно 0,1 до приблизительно 20% масс./масс., от приблизительно 0,1 до приблизительно 15% масс./масс., от приблизительно 0,5 до приблизительно 10% масс./масс., или альтернативно от приблизительно 0,10 до приблизительно 80% масс./масс., например, таком как от приблизительно 10 до приблизительно 70% масс./масс., от приблизительно 20 до приблизительно 60% масс./масс. или от приблизительно 30 до приблизительно 50% масс./масс.
В конкретном аспекте изобретения по меньшей мере один из одного или нескольких фармацевтически приемлемых эксципиентов выбран из группы, состоящей из кремниевой кислоты или ее производного или соли, включая силикаты, диоксид кремния и их полимеры; алюмосиликат магния и/или алюмометасиликат магния, бентонит, каолин, трисалицилат магния, монтмориллонит и/или сапонит.
Такие материалы особенно пригодны в качестве сорбционного материала для масел или маслянистых материалов в фармацевтических средствах, косметических средствах и/или продуктах питания. В конкретном варианте осуществления материал используют в качестве сорбционного материала для масел или маслянистых материалов в фармацевтических препаратах. Материал, который способен выполнять функцию сорбционного материала для масел или маслянистых материалов, также называют "материалом для сорбции масел". Более того, в контексте настоящего изобретения термин "сорбция" используют для обозначения "абсорбции", а также "адсорбции". Следует понимать, что когда используют один из терминов, подразумевают, что он охватывает как явление абсорбции, так и явление адсорбции.
В частности, фармацевтически приемлемый эксципиент может содержать кремниевую кислоту или ее производное или соль, например, такие как диоксид кремния или его полимер, в качестве фармацевтически приемлемого эксципиента. В зависимости от используемого качества, диоксид кремния может представлять собой смазывающее вещество или он может представлять собой материал для сорбции масел. Качества, удовлетворяющие последней функции, кажутся наиболее важными.
В конкретном варианте осуществления композиция или твердая лекарственная форма по изобретению содержит фармацевтически приемлемый эксципиент, который представляет собой продукт диоксида кремния, который имеет свойства, соответствующие Aeroperl® 300 (доступен от Degussa, Frankfurt, Germany).
Как следует из примеров настоящего документа, высоко пригодным материалом является Aeroperl® 300 (включая материалы со свойствами, подобными или соответствующими свойствам Aeroperl® 300).
Применение материала для сорбции масла в композициях или лекарственных формах по изобретению является высоко преимущественным для приготовления фармацевтических, косметических, питательных и/или пищевых композиций, где композиция содержит масло или маслянистый материал. Одним из преимуществ является то, что можно включать относительно большое количество масла и маслянистого материала и, тем не менее, иметь твердый материал. Таким образом, с использованием сорбционного материала по изобретению можно получать твердые композиции с относительно высокой нагрузкой маслом или маслянистыми материалами. В области фармацевтики является преимущественной возможность включать относительно большое количество масла или маслянистого материала в твердую композицию, особенно в ситуации, когда активное вещество не имеет пригодных свойств в отношении растворимости в воде (например, имеет плохую растворимость в воде), стабильности в водной среде (т.е. в водной среде происходит деградация), пероральной биодоступности (например, имеет низкую биодоступность) и т.д., или в тех ситуациях, когда желательно модифицировать высвобождение активного вещества из композиции для получения активного вещества с контролируемой, отсроченной, непрерывной и/или периодической доставкой. Таким образом, в конкретном варианте осуществления их используют для изготовления фармацевтических композиций.
В важном варианте осуществления изобретения по меньшей мере часть такролимуса и/или его аналога присутствует в композиции в форме твердого раствора, включающего молекулярную дисперсию или твердую дисперсию. Обычно в композиции присутствует 10% или более, как, например, 20% или более, 30% или более, 40% или более, 50% или более, 60% или более, 70% или более, 80% или более, 90% или более, как, например, 95% или более или приблизительно 100% масс./масс. такролимуса и/или его аналога в форме твердой дисперсии.
Твердую дисперсию можно получать различными способами, например, с использованием органических растворителей или путем диспергирования или растворения активного вещества в другой пригодной среде (например, в масле или маслянистом материале, который представляет собой жидкую форму при комнатной температуре или при повышенных температурах).
Твердые дисперсии (способ растворителя), например, можно получать путем растворения физической смеси активного вещества (например, лекарственного вещества) и носителя в общем органическом растворителе, с последующим выпариванием растворителя. Носитель часто представляет собой гидрофильный полимер. Пригодные органические растворители включают фармацевтически приемлемый растворитель, в котором активное вещество является растворимым, такой как метанол, этанол, метиленхлорид, хлороформ, этилацетат, ацетон или их смеси.
Пригодные растворимые в воде носители включают полимеры, такие как полиэтиленгликоль, полоксамеры, стеараты полиоксиэтилена, поли-ε-капролактон, поливинилпирролидон (PVP), сополимер поливинилпирролидон-поливинилацетат PVP-PVA (Koilidon VA64), полиметакриловые полимеры (Eudragit® RS, Eudragit® RL, Eudragit® NE, Eudragit® E) и поливиниловый спирт (PVA), гидроксипропилцеллюлозу (HPC), гидроксипропилметилцеллюлозу (HPMC), метилцеллюлозу и полиэтиленоксид (PEO).
Полимеры, содержащие кислотные функциональные группы, могут быть пригодны для твердых дисперсий, которые высвобождают активные вещества в предпочтительном диапазоне pH, обеспечивая приемлемое всасывание в кишечнике. Такие полимеры могут представлять собой один или несколько полимеров, выбранных из группы, содержащей фталат гидроксипропилметилцеллюлозы (HMPCP), фталат поливинилацетата (PVAP), гидроксипропилметилцеллюлозы ацетата сукцинат (HPMCAS), альгинат, карбомер, карбоксиметилцеллюлозу, сополимер метакриловой кислоты (Eudragit L, Eudragit S), шеллак, фталат ацетата целлюлозы (CAP), гликолят крахмала, полакрилин, метилцеллюлозы ацетата фталат, гидроксипропилцеллюлозы ацетата фталат, целлюлозы ацетата терефталат, целлюлозы ацетата изофталат и целлюлозы ацетата триммеллитат.
Что касается количества активного вещества и полимера в твердой дисперсии, массовое соотношение активного вещества и полимера может находиться в диапазоне от приблизительно 3:1 до приблизительно 1:20. Однако также можно использовать более узкий диапазон от приблизительно 3:1 до приблизительно 1:5, например, такой как от приблизительно 1:1 до приблизительно 1:3.
Твердую дисперсию предпочтительно формируют способами распылительной сушки, контролируемой агломерации, лиофилизации или нанесения на частицы носителя, или любым других способом удаления растворителя. Высушенный продукт содержит активное вещество, присутствующее в форме твердой дисперсии, включая молекулярную дисперсию и твердый раствор.
В качестве альтернативы применению органических растворителей лекарственное средство и полимер можно совместно измельчать или экструдировать при повышенных температурах (экструзия из расплава).
Фармацевтические композиции, содержащие такролимус, по меньшей мере частично в форме твердой дисперсии или раствора, можно, в принципе, получать с использованием любого способа, пригодного для получения фармацевтических композиций, известных в данной области.
Помимо использования способа на основе органического растворителя, твердую дисперсию или твердые растворы такролимуса и/или его аналога можно получать диспергированием и/или растворением такролимуса в композиции носителя, используемой в способе контролируемой агломерации. Для обеспечения стабильности твердой дисперсии/раствора можно использовать стабилизаторы.
В другом аспекте изобретение относится к способу получения фармацевтической композиции по изобретению. Как правило, можно использовать любой пригодный способ в области фармацевтики. Однако для обеспечения включения относительно высокого количества масла или маслянистого материала особенно пригодным является способ, описанный в WO 03/004001. WO 03/004001 включена в настоящий документ в качестве ссылки. Способ включает напыление первой композиции в жидкой форме, причем указанная композиция содержит первый наполнитель или носитель и имеет температуру плавления выше 5°С, на вторую композицию, содержащую вторую подложку или носитель, причем указанная композиция, например, находится в псевдоожиженном состоянии и имеет температуру ниже температуры плавления первого наполнителя или носителя. Активное вещество может присутствовать в первой композиции наполонителя или носителя и/или во второй композиции подложки или носителя. Однако в случаях, когда такролимус и/или его аналог присутствует по меньшей мере частично в форме твердой дисперсии, преимущественно включать или растворять такролимус и/или его аналог в первой композиции наполнителя или носителя. Кроме того, в WO05020993A1 и WO05020994A1 авторов настоящего изобретения описаны композиции такролимуса, получаемые с использованием этой технологии, и его составы с замедленным высвобождением, и они включены в настоящий документ в качестве ссылок.
Твердые лекарственные формы
Фармацевтическая композиция по изобретению имеет форму частиц, и ее можно использовать как таковую. Однако во многих случаях более удобно, чтобы композиция была представлена в форме гранул, драже, микросфер, наночастиц и т.п. или в форме твердых лекарственных форм, включающих таблетки, капсулы и саше и т.п.
Твердая лекарственная форма по изобретению может представлять собой единичную лекарственную форму, или она может быть в форме лекарственной формы с множеством депо, которая содержит множество отдельных единиц, например, таких как драже, шариков и/или гранул.
Твердая лекарственная форма в соответствии с настоящим изобретением содержит фармацевтическую композицию в форме частиц, как описано выше. Детали и особенности, описанные в соответствии с этим главным аспектом изобретения, применимы с соответствующими изменениями к другим аспектам изобретения. Таким образом, свойства, связанные с повышением биодоступности, изменениями параметров биодоступности, отсутствием суточного эффекта, а также высвобождением такролимуса и/или его аналога, и т.д., описанные и/или заявленные в настоящем документе для фармацевтических композиций в форме частиц, являются алогичными для твердой лекарственной формы в соответствии с настоящим изобретением.
Рекомендованный диапазон дозировок для Prograf® составляет от 0,1 до 0,2 мг/кг/сутки, вводимые каждые 12 часов в двух разделенных дозах. Важно, что необходимо проводить мониторинг уровней в крови.
Типичный уровень для 1-3 месяцев составляет 7-20 нг/мл, и для 4-12 месяцев уровень должен составлять 5-15 нг/мл. Эти величины являются только рекомендованными, и они могут варьировать в зависимости от типов трансплантатов и этнической принадлежности.
Для пациентов с трансплантатами почки было выявлено следующее:
Ожидаемые рекомендованные дозировки для продуктов по настоящему изобретению составляют от 0,02 до 0,15 мг/кг/сутки, вводимые один раз в сутки. Пригодные лекарственные формы (дозировка) варьируют от 0,1 до 15 мг такролимуса, предпочтительно дозировка выбрана из 0,5, 1, 2 и 5 мг.
Как продемонстрировано в настоящем документе с помощью сцинтиграфии и показано на фиг.2, высвобождение в соответствии с настоящим изобретением может происходить даже в дистальной части толстого кишечника, и, тем не менее, может происходить распределение по слизистой оболочке и всасывание. Всасывание лекарственной формы с пролонгированным высвобождением по изобретению представляет собой всасывание, когда происходит высвобождение in vivo после перорального введения субъекту по существу в толстом кишечнике, такое как высвобождение в одной или нескольких областях восходящей толстой кишки, поперечной толстой кишки и нисходящей толстой кишки.
Различные варианты осуществления изобретения, касающиеся предпочтительных фармакокинетических параметров, получаемых с помощью лекарственной формы с пролонгированным высвобождением в соответствии с настоящим изобретением, приведены ниже.
В одном варианте осуществления изобретение относится к лекарственной форме с пролонгированным высвобождением, которая, при введении субъекту или группе субъектов, обеспечивает внутрииндивидуальную и/или межиндивидуальную изменчивость среднего Tmax в крови для такролимуса, которая по сравнению с изменчивостью при введении лекарственной формы коммерческого продукта Advagraf® (MR4) или биоэквивалентной лекарственной формы с пролонгированным высвобождением является сниженной по меньшей мере на 10%, например, по меньшей мере на 15%, например, по меньшей мере на 17,5%, например, по меньшей мере на 20%, например, по меньшей мере на 22,5%, например, по меньшей мере на 25%, например, по меньшей мере на 27,5%, например, по меньшей мере на 30% при определении в сходных условиях и при введении в сходных молекулярных дозировках такролимуса.
В одном варианте осуществления изобретение относится к лекарственной форме с пролонгированным высвобождением, которая, при введении субъекту или группе субъектов, обеспечивает внутрииндивидуальную и/или межиндивидуальную изменчивость средней Cmax в крови и/или AUC(0-∞) для такролимуса, которая по сравнению с изменчивостью при введении коммерческого продукта Advagraf® (MR4) или биоэквивалентной лекарственной формы с пролонгированным высвобождением является сниженной по меньшей мере на 10%, например, по меньшей мере на 15%, например, по меньшей мере на 17,5%, например, по меньшей мере на 20%, например, по меньшей мере на 22,5%, например, по меньшей мере на 25%, например, по меньшей мере на 27,5% например, по меньшей мере на 30% при определении в сходных условиях и при введении в сходных молекулярных дозировках такролимуса.
В одном варианте осуществления изобретение относится к лекарственной форме с пролонгированным высвобождением, которая при введении субъекту или группе субъектов обеспечивает сниженное значение Cmax относительно значения Cmax, получаемого при введении коммерческого продукта Advagraf® (MR4) или биоэквивалентной лекарственной формы с пролонгированным высвобождением, по меньшей мере приблизительно на 10%, или по меньшей мере приблизительно на 15%, или по меньшей мере приблизительно на 20%, или по меньшей мере приблизительно на 30%, или по меньшей мере приблизительно на 35%, или по меньшей мере приблизительно на 40 или более, или по меньшей мере приблизительно на 45%, или по меньшей мере приблизительно на 50%, или по меньшей мере приблизительно на 55%, где значения Cmax определяют в сходных условиях и введение проводят в сходных молекулярных дозировках такролимуса.
В одном варианте осуществления изобретение относится к лекарственной форме с пролонгированным высвобождением, которая при введении субъекту или группе субъектов обеспечивает повышенную биодоступность относительно биодоступности, получаемой при введения коммерческого продукта Advagraf® (MR4) или биоэквивалентной лекарственной формы с пролонгированным высвобождением, по меньшей мере приблизительно на 20%, или по меньшей мере приблизительно на 25%, или по меньшей мере приблизительно на 30%, или по меньшей мере приблизительно на 35%, или по меньшей мере приблизительно на 40%, или по меньшей мере приблизительно на 45%, или по меньшей мере приблизительно на 50%, или по меньшей мере приблизительно на 55%, или по меньшей мере приблизительно на 60%, например, по меньшей мере на 65%, где биодоступность определяют как AUC(0-∞) и в сходных условиях и введение проводят в сходных молекулярных дозировках такролимуса.
В одном варианте осуществления изобретение относится к лекарственной форме с пролонгированным высвобождением, которая, при введении субъекту или группе субъектов, обеспечивает внутрииндивидуальную и/или межиндивидуальную изменчивость среднего Tmax в крови для такролимуса, которая по сравнению с изменчивостью при введении коммерчески доступной лекарственной формы Prograf® или биоэквивалентной лекарственной формы такролимуса с немедленным высвобождением является сниженной по меньшей мере на 10%, например, по меньшей мере на 15%, например, по меньшей мере на 17,5%, например, по меньшей мере на 20%, например, по меньшей мере на 22,5%, например, по меньшей мере на 25%, например, по меньшей мере на 27,5% например, по меньшей мере на 30% при определении в сходных условиях и при введении в сходных молекулярных дозировках такролимуса.
В одном варианте осуществления изобретение относится к лекарственной форме с пролонгированным высвобождением, которая, при введении субъекту или группе субъектов обеспечивает внутрииндивидуальную и/или межиндивидуальную изменчивость среднего Cmax в крови и/или AUC(0-∞) для такролимуса, которая по сравнению с изменчивостью при введении коммерчески доступной лекарственной формы Prograf® или биоэквивалентной лекарственной формы с немедленным высвобождением является сниженной по меньшей мере на 10%, например, по меньшей мере на 15%, например, по меньшей мере на 17,5%, например, по меньшей мере на 20%, например, по меньшей мере на 22,5%, например, по меньшей мере на 25%, например, по меньшей мере на 27,5%, например, по меньшей мере на 30% при определении в сходных условиях и при введении такролимуса в сходных молекулярных дозировках.
В одном варианте осуществления изобретение относится к лекарственной форме с пролонгированным высвобождением, которая при введении субъекту или группе субъектов обеспечивает сниженное значение Cmax относительно значения Cmax, получаемого при введении коммерчески доступной лекарственной формы Prograf® или биоэквивалентной лекарственной формы с немедленным высвобождением, по меньшей мере приблизительно на 20%, или по меньшей мере приблизительно на 30%, или по меньшей мере приблизительно на 35%, или по меньшей мере приблизительно на 40%, или по меньшей мере приблизительно на 45%, или по меньшей мере приблизительно на 50% или более, или по меньшей мере приблизительно на 55%, или по меньшей мере приблизительно на 60%, или по меньшей мере приблизительно на 65% при определении в сходных условиях и при введении в сходных молекулярных дозировках такролимуса.
В одном варианте осуществления изобретение относится к лекарственной форме с пролонгированным высвобождением, которая при введении субъекту или группе субъектов обеспечивает повышенную биодоступность относительно биодоступности, получаемой при введения коммерчески доступной лекарственной формы Prograf® или биоэквивалентной лекарственной формы с немедленным высвобождением по меньшей мере приблизительно на 10%, или по меньшей мере приблизительно на 15%, или по меньшей мере приблизительно на 20%, или по меньшей мере приблизительно на 30%, или по меньшей мере приблизительно на 35%, или по меньшей мере приблизительно на 40% или более, или по меньшей мере приблизительно на 45%, или по меньшей мере приблизительно на 50%, или по меньшей мере приблизительно на 55%, где биодоступность определяют как AUC(0-∞) и в сходных условиях и введение проводят в сходных молекулярных дозировках такролимуса.
В одном варианте осуществления изобретение относится к лекарственной форме с пролонгированным высвобождением, которая при введении субъекту или группе субъектов вечером в состоянии голодания в течение по меньшей мере 4 часов, обеспечивает биодоступность, которая относительно биодоступности, получаемой после введения лекарственной формы утром в состоянии голодания в течение по меньшей мере 4 часов составляет по меньшей мере 70%, например, по меньшей мере 80%, предпочтительно по меньшей мере 85%, более предпочтительно по меньшей мере 90% и предпочтительно по меньшей мере 95% от величины, измеренной после введения утром.
В одном варианте осуществления изобретение относится к лекарственной форме с пролонгированным высвобождением, которая при введении субъекту или группе субъектов вечером в состоянии голодания в течение по меньшей мере 4 часов обеспечивает Cmax, которая относительно Cmax, получаемой после введения лекарственной формы утром в состоянии голодания в течение по меньшей мере 4 часов, составляет по меньшей мере 70%, например, по меньшей мере 80%, предпочтительно по меньшей мере 85%, более предпочтительно по меньшей мере 90% и предпочтительно по меньшей мере 95% от величины, измеренной после введения утром.
В одном варианте осуществления изобретение относится к лекарственной форме с пролонгированным высвобождением, где биодоступность является независимой от времени дозирования в течение суток, и таким образом, она пригодна для режима дозирования перед сном.
В одном варианте осуществления изобретение относится к лекарственной форме с пролонгированным высвобождением, которая, когда она содержит 5 мг такролимуса и когда ее вводят в качестве однократной дозы по меньшей мере 6 здоровым субъектам натощак, обеспечивает среднюю максимальную концентрацию (Cmax) такролимуса самое большее 15 нг/мл, например, самое большее 13 нг/мл и среднюю AUC(0-96 ч) по меньшей мере 45 мг·ч/л, такую как по меньшей мере 55 мг·ч/л, такую как по меньшей мере 60 мг·ч/л.
В одном варианте осуществления изобретение относится к лекарственной форме с пролонгированным высвобождением по изобретению, где концентрация в крови через 24 часа после введения такролимуса составляет по меньшей мере 2 нг/мл, например, по меньшей мере 3 нг/мл, например, по меньшей мере 4 нг/мл.
В одном варианте осуществления изобретение относится к лекарственной форме с пролонгированным высвобождением, для которой при введении один раз в сутки в стационарном состоянии здоровому субъекту или пациенту максимальное отклонение концентраций такролимуса в крови, измеренное как (Cmax-Cmin)/Cmin, является меньшим, чем максимальное отклонение, наблюдаемое при введении лекарственной формы Advagraf® или биоэквивалентной лекарственной формы такролимуса с пролонгированным высвобождением в режиме один раз в сутки и при определении в сходных условиях и введении в сходных молекулярных дозировках такролимуса. Снижение составляет предпочтительно по меньшей мере 10%, например, по меньшей мере 20%, предпочтительно по меньшей мере 30%, например, по меньшей мере 40%, более предпочтительно по меньшей мере 50%.
В одном варианте осуществления изобретение относится к лекарственной форме с пролонгированным высвобождением, для которой при введении один раз в сутки в стационарном состоянии здоровому субъекту или пациенту максимальное отклонение концентраций для общего такролимуса в крови, измеренное как (Cmax-Cmin)/Cmin, является меньшим, чем максимальное отклонение, наблюдаемое при введении лекарственной формы Prograf® или биоэквивалентной композиции такролимуса с немедленным высвобождением в режиме два раза в сутки и при определении в сходных условиях и введении в сходных молекулярных дозировках такролимуса. Снижение составляет предпочтительно по меньшей мере 10%, например, по меньшей мере 20%, предпочтительно по меньшей мере 30%, например, по меньшей мере 40%, более предпочтительно по меньшей мере 50%.
В одном варианте осуществления изобретение относится к лекарственной форме с пролонгированным высвобождением, для которой при введении один раз в сутки в стационарном состоянии здоровому субъекту или пациенту флуктуация концентраций для общего и/или свободного такролимуса в крови, измеренная как (Cmax-Cmin)/Cсредняя, является меньшей, чем флуктуация, наблюдаемая при введении лекарственной формы Advagraf® или биоэквивалентной композиции такролимуса с пролонгированным высвобождением в режиме один раз в сутки и при определении в сходных условиях и введении в сходных молекулярных дозировках такролимуса. Снижение составляет предпочтительно по меньшей мере 10%, например, по меньшей мере 20%, предпочтительно по меньшей мере 30%, например, по меньшей мере 40%, более предпочтительно по меньшей мере 50%.
В одном варианте осуществления изобретение относится к лекарственной форме с пролонгированным высвобождением, для которой при введении один раз в сутки в стационарном состоянии здоровому субъекту или пациенту флуктуация концентраций для общего и/или свободного такролимуса в крови, измеренная как (Cmax-Cmin)/Cсредняя, является меньшей, чем флуктуация, наблюдаемая при введении лекарственной формы Prograf® или биоэквивалентной композиции такролимуса с немедленным высвобождением в режиме два раза в сутки и при определении в сходных условиях и введении в сходных молекулярных дозировках такролимуса. Снижение составляет предпочтительно по меньшей мере 10%, например, по меньшей мере 20%, предпочтительно по меньшей мере 30%, например, по меньшей мере 40%, более предпочтительно по меньшей мере 50%.
В одном варианте осуществления изобретение относится к лекарственной форме с пролонгированным высвобождением, для которой при введении по меньшей мере 6 здоровым субъектам натощак, среднее время нахождения, MRT, такролимуса, измеренное в крови, является по меньшей мере на 10% более длительным, чем среднее время нахождения, измеренное в биоэквивалентных условиях для лекарственной формы Advagraf® или сходной лекарственной формы такролимуса с пролонгированным высвобождением. Предпочтительно MRT возрастает по меньшей мере на 20%, например, на 25%.
В одном варианте осуществления изобретение относится к лекарственной форме с пролонгированным высвобождением, для которой при введении по меньшей мере 6 здоровым субъектам натощак среднее время нахождения такролимуса, измеренное в крови, является по меньшей мере на 35% более длительным, чем среднее время нахождения, измеренное в сходных условиях для лекарственной формы Prograf® или биоэквивалентной лекарственной формы такролимуса с немедленным высвобождением.
В следующем аспекте настоящее изобретение относится к способу обеспечения иммуносупрессивного лечения пациента, нуждающегося в этом, в режиме раз в сутки путем введения состава с пролонгированным высвобождением, как описано в настоящем документе, обеспечивающего одно или несколько из сниженной Cmax, сниженного максимального отклонения, сниженной флуктуации, повышенной AUC, повышенного MRT, более длительного времени до Tmax и более высокой Cmin. Кроме того, способы обеспечивают Cmin, которая коррелирует с биодоступностью с коэффициентом корреляции от по меньшей мере 0,75 до 1, например, по меньшей мере 0,80, предпочтительно 0,85, более предпочтительно 0,90, еще более предпочтительно по меньшей мере 0,95 и еще более предпочтительно по меньшей мере 0,97.
В предпочтительном варианте осуществления отличия в биодоступности по существу не зависят от времени суток, когда вводят дозировку. Это обеспечивает возможность режима один раз в сутки перед сном или вечером, в дополнение к нормальному дозированию утром. Однако более важно, что существует риск снижения экспозиции, если пациент в действительности проглатывает лекарственную форму во время, отличное от назначенного и ожидаемого (не соблюдение пациентом режима лечения), и риск сниженной экспозиции и, таким образом повышенный риск, например, отторжения трансплантата, для пациента снижается.
В следующем аспекте изобретение относится к переходу от режима два раза в сутки с более высокой дозировкой. Одним из способов для такого перехода от, среди прочих, Prograf® на режим один раз в сутки является снижение суточной дозировки режима для такролимуса с немедленным высвобождением на от 25% до 50%, например, на от 30% до 40%, предпочтительно приблизительно на 33%. Снижение проводят до той степени, которая является возможным путем применения лекарственных форм с пролонгированным высвобождением для введения один раз в сутки, выбранных из лекарственных форм для введения один раз в сутки, составляющих 0,5, 1, 2 и 5 мг. Таким образом, соотношение для перехода составляет от 1:0,66 до 0,80 в зависимости от доступных дозировок, как упоминалось. Более того, настоящее изобретение относится к переходу от Advagraf® с соотношением от 1:0,30 до 0,75, например, от 1:0,33 до 0,7 в соответствии с доступными дозировками составов с пролонгированным высвобождением в соответствии с настоящим изобретением, выбранными из 0,5, 1, 2 и 5 мг.
Особенно важным аспектом изобретения является существенно сниженные максимальные концентрации, которые обеспечивают уменьшение связанных с максимальной концентрацией побочных эффектов. Однако такой эффект может быть трудно измерить вследствие внутрииндивидуальной и межиндивидуальной изменчивости современных способов лечения такролимуса и характера побочных эффектов, которые часто являются субъективными или могут требовать биопсии тканей для определения. Для сравнительных исследований, чтобы продемонстрировать такой эффект со значимостью, требуются тщательное анкетирование и большое число пациентов. Однако предусматривается, что лечение составом с пролонгированным высвобождением в соответствии с настоящим изобретением может снизить некоторые возможные связанные с максимальной концентрацией побочные эффекты, включая побочные эффекты неврологического происхождения, такие как тремор и головная боль.
В предпочтительном варианте осуществления снижение побочного эффекта связано с риском удлиненного интервала QTc вследствие воздействия на реполяризацию желудочков. Другими видами токсичности, которые предполагают снизить, являются развитие повреждения почек, развитие диабета, а также развитие гипертензии.
В частности, накопление, происходящее в некоторых органах, может быть ассоциировано с повреждением органа, особенно когда накопление или индивидуальная концентрация такролимуса могут не снижаться в течение периода низкой концентрации в крови в более поздние моменты времени интервала между введением доз. Эта недостаточность выведения из органа может быть связана с высокой аффинностью такролимуса к этому органу, превосходящей в ином случае ожидаемое выведение из таких высоко васкуляризированных органов. Таким образом, высокие максимальные концентрации современных доступных коммерческих лекарственных форм могут приводить к накоплению такролимуса в этих органах, которые могут содержать концентрацию, приблизительно до 30 раз превышающую концентрацию традиционных лекарственных форм в крови в стационарном состоянии.
Пациент после трансплантации de novo может быть крайне чувствительным к высоким концентрациям вследствие общего плохого состояния организма, а низкие уровни белков в плазме дополнительно повышают фракцию такролимуса, свободного для проникновения в органы. В частности, у этих пациентов высоких концентраций в крови при титровании избегают с использованием лекарственной формы по этому изобретению, которая имеет потенциал к снижению величины накопления или продлению времени до достижения концентрациями органов, где такролимус является токсичным. Таким образом, изобретение также относится к способу лечения пациентов, имеющих риск токсичности такролимуса для органов. Органы, которые, как известно, накапливают такролимус, включают надпочечники, легкое, сердце, печень, желудочно-кишечный тракт и почку. Однако, кроме того, в соответствии с настоящим изобретением полагают, что к высоким концентрациям могут быть чувствительными поджелудочная железа и, в частности, островки Лангерганса, особенно при начале введения такролимуса, тем самым риск развития диабета в последующее время может существенно возрастать. Другими органами, коррелирующими с высокой токсичностью, являются органы центральной нервной системы. Многие пациенты прекращают лечение вследствие головных болей и тремора - побочных эффектов, которые, возможно, уменьшатся при лечении в соответствии с настоящим изобретением. В частности, тремор представляет собой побочный эффект, связанный с токсическим эффектом на центральную нервную систему, вызываемым такролимусом, пересекающим гематоэнцефалический барьер. В соответствии с настоящим изобретением предусматривается, что высокие концентрации могут способствовать захвату такролимуса в центральную нервную систему, поскольку, когда концентрации являются высокими в системном кровотоке, существует более высокое отличие в концентрациях по разные стороны гематоэнцефатического барьера, что может приводить к повышенному транспорту в головной мозг по сравнению с ситуацией, когда обеспечивается постоянная и более низкая концентрация, как в случае состава с пролонгированным высвобождением в соответствии с настоящим изобретением.
Согласно следующему аспекту изобретения, состав с пролонгированным высвобождением в соответствии с настоящим изобретением относится к безопасному профилю лечения, позволяющему лечение с момента трансплантации, как только состояние пациента становится достаточно хорошим, чтобы его лечить пероральным составом. Таким образом, пациент не нуждается в первом введении общепринятого состава два раза в сутки, такого как Prograf®, с последующим переходом на лечение в соответствии с настоящим изобретением, а можно начинать с перорального иммуносупрессивного лечения такролимусом в составе для введения один раз в сутки в соответствии с настоящим изобретением.
В следующем варианте осуществления предусматривается лечение пациентов с трансплантатом de novo, поскольку состав в соответствии с настоящим изобретением способен поддерживать достаточную системную экспозицию даже через 24 часа после начала введения, тем самым предусматривается способ лечения, где ту же лекарственную форму можно использовать для начала перорального лечения такролимусом, а также для постоянного лечения, в том числе поддерживающего лечения. Полагают, что благодаря лечению один раз в сутки по настоящему изобретению составом таблетки уменьшенного размера по сравнению с другими доступными продуктами такролимуса количество коррекций доз может снизиться, и соблюдение пациентом режима лечения может улучшиться.
Выявлено, что применение Advagraf® в течение 24 часов обеспечивает системную экспозицию, которая приблизительно на 30% и 50% ниже по сравнению с введением состава Prograf®, вводимого de novo пациентам с трансплантатами почки и de novo пациентам с трансплантатами печени, соответственно. Общее объяснение этой сниженной биодоступности согласуется с научным обсуждением, представленном в связи с одобрением Advagraf® European Agency for the Evaluation of Medicinal Products (EMEA) 23 апреля 2007 года, где ее объяснили отсутствием суточного эффекта на всасывание такролимуса для состава Prograf®, вводимого вечером, относительно утра в первые сутки после трансплантации. Однако согласно авторам настоящего изобретения, более низкая экспозиция продукта Advagraf® может, главным образом, быть связана с относительно более высоким воздействием метаболизма в желудочно-кишечном тракте, возможно в комбинации с более низкой скоростью всасывания, которая обеспечивает более высокую фракцию выведения печенью при первом прохождении. У пациентов с трансплантатами печени метаболизм в желудочно-кишечном тракте может иметь более высокое воздействие, поскольку метаболизм в печени может снижаться сразу после трансплантации. Таким образом, путем предоставления состава с пролонгированным высвобождением в соответствии с настоящим изобретением воздействие метаболизма в желудочно-кишечном тракте является меньшим, поскольку состав, главным образом, высвобождает такролимус в нижнем отделе подвздошной кишки и в толстом кишечнике, где метаболизм является меньшим, и в то же время высвобождает такролимус так, чтобы всасывание в действительности происходило, например, путем высвобождения такролимуса улучшенным образом, таким как в молекулярной форме или по существу в молекулярной форме.
Таким образом, предусматривается способ начального лечения пациента с трансплантатом печени de novo такролимусом, включающий введение один раз в сутки пероральной лекарственной формы такролимуса, имеющей профиль пролонгированного высвобождения, который при измерении in vitro демонстрирует высвобождение, где через 10 часов высвобождается менее 50% от содержания такролимуса в пероральной лекарственной форме при измерении способом растворения, описанным для программы с базой данных FDA Dissolution Methods For Drug Products, и пероральная лекарственная форма обеспечивает системную экспозицию на 1 сутки, которая составляет по меньшей мере 50% от экспозиции, полученной на 1 сутки после введения той же суточной дозы, однако вводимой в качестве пероральной лекарственной формы с немедленным высвобождением два раза в сутки.
Аналогично, предусматривается способ начального лечения пациента с трансплантатом почки de novo такролимусом, включающий введение один раз в сутки пероральной лекарственной формы такролимуса, имеющей профиль пролонгированного высвобождения, который при измерении in vitro демонстрирует высвобождение, где менее 50% от содержания такролимуса в пероральной лекарственной форме высвобождается через 10 часов при измерении способом FDA для Prograf®, и пероральная лекарственная форма обеспечивает системную экспозицию на 1 сутки, которая составляет по меньшей мере 70% от экспозиции, полученной на 1 сутки после введения той же суточной дозы, но при введении в качестве пероральной лекарственной формы с немедленным высвобождением два раза в сутки.
Также способ относится к начальному лечению пациента с трансплантатом печени de novo такролимусом, включающему введение один раз в сутки пероральной лекарственной формы такролимуса, имеющей профиль пролонгированного высвобождения, который при измерении in vitro демонстрирует высвобождение, где менее 50% от содержания такролимуса в пероральной лекарственной форме высвобождается через 10 часов при измерении способом FDA для Prograf®, и пероральная лекарственная форма обеспечивает системную экспозицию на 1 сутки, которая составляет по меньшей мере 100% от экспозиции, получаемой на 1 сутки после введения той же суточной дозы, но вводимой в качестве пероральной лекарственной формы с пролонгированным высвобождением, высвобождающей более 30% такролимуса в течение 5 часов.
Аналогично способ начального лечения пациента с трансплантатом почки de novo такролимусом, включающий введение один раз в сутки пероральной лекарственной формы такролимуса, имеющей профиль пролонгированного высвобождения, который при измерении in vitro демонстрирует высвобождение, где через 10 часов менее 50% от содержания такролимуса высвобождается при измерении способом FDA для Prograf®, и пероральная лекарственная форма обеспечивает системную экспозицию на 1 сутки, которая составляет по меньшей мере 100% от экспозиции, получаемой на 1 сутки после введения той же суточной дозы, но вводимой в качестве пероральной лекарственной формы с пролонгированным высвобождением, высвобождающей более 30% такролимуса в течение 5 часов.
На твердую лекарственную форму также можно наносить покрытие для получения пригодных свойств, например, в отношении контролируемого высвобождения активного вещества. Покрытие можно наносить на единичные лекарственные формы (например, таблетки, капсулы) или его можно наносить на лекарственную форму с множеством депо или на ее отдельные единицы.
Пригодными материалами покрытия являются, например, метилцеллюлоза, гидроксипропилметилцеллюлоза, гидроксипропилцеллюлоза, актиловые полимеры, этилцеллюлоза, целлюлозы ацетата фталат, поливинилацетата фталат, гидроксипропилметилциллюлозы фталат, поливиниловый спирт, карбоксиметилцеллюлоза натрия, ацетат целлюлозы, целлюлозы ацетата фталат, желатин, сополимер метакриловой кислоты, полиэтиленгликоль, шеллак, сахароза, диоксид титана, карнаубский воск, микрокристаллический воск, глицерилмоностеарат, зеин.
В материал покрытия можно добавлять пластификаторы и другие ингредиенты. Также в материал покрытия можно добавлять то же или другое активное вещество.
В предпочтительных вариантах осуществления изобретения твердые лекарственные формы предназначены для высвобождения такролимуса и/или его аналога контролируемым образом. В контексте настоящего изобретения термин "контролируемым образом" включает все типы высвобождения, которые отличаются от высвобождения, достигаемого в случае простых таблеток. Таким образом, этот термин включает так называемое "контролируемое высвобождение", "пролонгированное высвобождение", "непрерывное высвобождение", "периодическое высвобождение", "длительное высвобождение", "импульсное высвобождение", "медленное высвобождение", "пролонгированное высвобождение", а также термины "отсроченное высвобождение" и "pH-зависимое высвобождение". Однако конкретный аспект изобретения относится к композиции или лекарственной форме с отсроченным высвобождением, которая в этом контексте означает композицию или лекарственную форму, которая высвобождает самое большее 10% масс./масс. активного вещества в течение первых 2 часов после введения и/или после начала теста растворения с использованием среды для растворения, имеющей pH самое большее приблизительно 3.
Системы с пролонгированным высвобождением
Первая система с пролонгированным высвобождение включает матричные системы, в которых такролимус погружен или диспергирован в матрице из другого материала, которая служит для замедления высвобождения такролимуса в водное окружение (т.е. жидкость просвета желудочно-кишечного тракта). Когда такролимус диспергирован в матрице этого типа, высвобождение лекарственного средства происходит, главным образом, с поверхности матрицы. Таким образом, лекарственное средство высвобождается с поверхности устройства, которое включает матрицу, после того как оно диффундирует через матрицу, и когда поверхность устройства деградирует, экспонируя лекарственное средство. В некоторых вариантах осуществления оба механизма могут действовать одновременно. Матричные системы могут быть большими, т.е. размером с таблетку (приблизительно 1 см), или небольшими (<0,3 см). Система может быть единой (например, болюс), она может быть разделенной вследствие содержания нескольких субэлементов (например, несколько капсул, которые составляют одну дозу), которые вводят по существу одновременно, или она может содержать множество частиц, и в этом случае ее называют системой множества частиц. Состав системы множества частиц может иметь многочисленные применения. Например, систему множества частиц можно использовать в качестве порошка для заполнения оболочки капсулы, или ее можно использовать как таковую для смешивания с пищей для простоты приема.
В конкретном варианте осуществления матрица из множества частиц содержит множество содержащих такролимус частиц, причем каждая частица содержит такролимус и/или его аналог, например, в форме твердого раствора/дисперсии с одним или несколькими эксципиентами, выбранными для формирования матрицы, способной контролировать скорость растворения такролимуса в водной среде. Материалы матрицы, пригодные для этого варианта осуществления, как правило, представляют собой гидрофобные материалы, такие как воски, некоторые производные целлюлозы или другие гидрофобные полимеры. Если необходимо, материалы матрицы необязательно можно включать в состав с гидрофобными материалами, которые можно использовать в качестве связующих веществ или усилителей. Материалы матрицы, пригодные для изготовления этих лекарственных форм, представляют собой такие материалы, как: этилцеллюлоза, воски, такие как парафин, модифицированные растительные масла, карнаубский воск, гидрогенизированное касторовое масло, пчелиный воск и т.п., а также синтетические полимеры, такие как поливинилхлорид, поливинилацетат, сополимеры винилацетата и этилена, полистирол и т.п. Растворимые в воде или гидрофильные связующие вещества или модифицирующие высвобождение средства, которые необязательно можно включать в состав матрицы, включают гидрофильные полимеры, такие как гидроксипропилцеллюлоза (HPC), гидроксипропилметилцеллюлоза (HPMC), метилцеллюлоза, поли(N-винил-2-пирролидинон) (PVP), полиэтиленоксид (PEO), поливиниловый спирт (PVA), ксантановая смола, каррагенан и другие такие природные и синтетические материалы. Кроме того, материалы, которые функционируют в качестве модифицирующих высвобождение средств, включают растворимые в воде материалы, такие как сахара или соли. Предпочтительные растворимые в воде материалы включают лактозу, сахарозу, глюкозу и маннит, а также гидрофильные полимеры, например, такие как HPC, HPMC и PVP.
В конкретном варианте осуществления продукт из множества частиц определяют как явяляющийся переработанным контролируемой агломерацией. В этом случае такролимус растворяют или частично растворяют в пригодном плавком носителе и напыляют на частицы носителя, содержащие вещество матрицы.
Пригодные плавкие носители упомянуты в настоящем документе выше.
Альтернативно такролимус растворяют в органическом растворителе вместе с веществом матрицы и сушат распылительной сушкой или наносят на частицы носителя, смотрите ниже. Растворители, обычно используемые для этого процесса, включают ацетон, этанол, изопропанол, этилацетат и смеси двух или более из них.
После формирования матрицы из множества частиц с такролимусом можно смешивать с прессуемыми эксципиентами, такими как лактоза, микрокристаллическая целлюлоза, дикальцийфосфат и т.п., и смесь можно прессовать с образованием таблетки. Также эффективно используют дезинтегрирующие вещества, такие как натрия крахмала гликолят или поперечно-сшитый поливинилпирролидон. Таблетки, полученные этим способом, дезинтегрируют, когда их помещают в водную среду (такую как желудочно-кишечный тракт), тем самым экспонируя матрицу из множества частиц, которая высвобождает из нее такролимус.
В следующем варианте осуществления матричная система находится в форме таблетки с гидрофильной матрицей, содержащей такролимус и/или его аналог (например, в форме твердой дисперсии) в качестве продукта из множества частиц и количество гидрофильного полимера, достаточное для обеспечения пригодной степени контроля растворения такролимуса. Гидрофильные полимеры, пригодные для образования матрицы, включают гидроксипропилметилцеллюлозу (HPMC), гидроксипропилцеллюлозу (HPC), полиэтиленоксид, поливиниловый спирт, ксантановую смолу, карбонер, каррагенан и зооглан. Предпочтительным материалом является HPMC. Также можно использовать другие сходные гидрофильные полимеры. При применении гидрофильный материал набухает под действием воды и в конечном итоге растворяется в ней. Такролимус высвобождается как путем диффузии из матрицы, так и путем деградации матрицы. Скорость высвобождения такролимуса из этих таблеток с гидрофильной матрицей можно контролировать количеством, молекулярной массой и прочностью геля используемого гидрофильного полимера. Как правило, при использовании большего количества гидрофильного полимера скорость растворения снижается, также как и при использовании более высокомолекулярного полимера. При использовании более низкомолекулярного полимера скорость растворения обычно повышается. Таблетка матрицы, как правило, содержит приблизительно от 20 до 90% по массе такролимуса и приблизительно от 80 до 10% по массе полимера.
Предпочтительная матричная таблетка содержит, по массе, от приблизительно 30% до приблизительно 80% твердой дисперсии, содержащей такролимус и/или его аналог, от приблизительно 15% до приблизительно 35% формирующего матрицу вещества (например, такого как HPMC), от 0% до приблизительно 35% лактозы, от 0% до приблизительно 20% микрокристаллической целлюлозы и от приблизительно 0,25% до приблизительно 2% смазывающего вещества (например, такого как стеарат магния).
Матричные системы как класс часто проявляют непостоянное высвобождение лекарственного средства из матрицы. Этот результат может быть следствием диффузионного механизма высвобождения лекарственного средства, и можно использовать модификации геометрии лекарственных форм, преимуществом которых является достижение более постоянной скорости высвобождения лекарственного средства.
Второй класс лекарственных форм с контролируемым высвобождением такролимуса по этому изобретению включает сдерживаемые мембраной или резервуарные системы. В этом классе резервуар с такролимусом, например, в твердом растворе/дисперсии в виде продукта из множества частиц, окружен скорость-лимитирующей мембраной. Такролимус проходит через мембрану с помощью основных транспортных механизмов, известных в данной области, включая, но не ограничиваясь ими, растворение в мембране с последующей диффузией через мембрану или диффузию через заполненные жидкостью поры в мембране. Эти лекарственные формы с индивидуальными резервуарными системами могут быть большими, как в случае таблетки, содержащей один большой резервуар, или они могут представлять собой системы множества частиц, как в случае капсул или таблеток с множеством депо, содержащих множество резервуарных частиц, каждая из которых индивидуально покрыта мембраной. Покрытие может быть непористым, но все еще проницаемым для такролимуса (например, такролимус может диффундировать прямо через мембрану), или оно может быть пористым. Как в случае других вариантов осуществления этого изобретения, полагают, что конкретный механизм транспорта не является ключевым.
Для изготовления мембраны можно использовать покрытия с непрерывным высвобождением, как известно в данной области, особенно полимерные покрытия, такие как сложный эфир или простой эфир целлюлозы, акриловый полимер или смесь полимеров. Предпочтительные материалы включают этилцеллюлозу, ацетат целлюлозы и целлюлозы ацетата бутират. Полимер можно применять в качестве раствора в органическом растворителе или в качестве водной дисперсии или латекса. Нанесение покрытия можно проводить в стандартном оборудовании, таком как устройство для нанесения покрытия в псевдоожиженном слое, устройство для нанесения покрытия Wurster или вращающееся устройство для нанесения покрытия в псевдоожиженном слое.
Если желательно, проницаемость покрытия можно корректировать путем смешивания двух или более материалов. Особенно пригодный способ коррекции пористости покрытия включает добавление в раствор или дисперсию (например, в водный латекс) формирующего мембрану полимера, подлежащего применению, заданного количества тонкоизмельченного растворимого в воде материала, такого как сахара или соли или растворимые в воде полимеры. Когда лекарственную форму проглатывают, и она попадает в водную среду желудочно-кишечного тракта, эти растворимые в воде добавки в мембрану вымываются из мембраны, оставляя поры, которые способствуют высвобождению лекарственного средства. Мембранное покрытие также можно модифицировать путем добавления пластификаторов, известных в данной области.
Особенно пригодный вариант способа нанесения мембранного покрытия, в частности, включает растворение полимера покрытия в смеси растворителей, выбранной так, чтобы по мере высыхания покрытия в применяемом растворе для покрытия происходила инверсия фазы, что приводит к мембране с пористой структурой.
Как правило, подложка для механического упрочнения мембраны не требуется.
Морфология мембраны не имеет большого значения, при условии, что удовлетворяются характеристики проницаемости, перечисленные в настоящем документе. Мембрана может быть аморфной или кристаллической. Она может иметь морфологию любой категории, получаемую каким-либо конкретным способом, и она может представлять собой, например, мембрану, полимеризующуюся на границе фаз (которая содержит тонкий скорость-лимитирующий наружный слой на пористой подложке), пористую гидрофильную мембрану, пористую гидрофобную мембрану, мембрану из гидрогеля, ионную мембрану, и другие такие материалы, которые характеризуются контролируемой проницаемостью для такролимуса.
В одном варианте осуществления изобретения задачей является снижение воздействия на желудочно-кишечный тракт высоких концентраций такролимуса. Таким образом, пригодные лекарственные формы включают формы, которые обеспечивают определенную задержку перед началом контролируемого высвобождения такролимуса. Иллюстративный вариант осуществления может быть проиллюстрирован таблеткой (или материалом в виде частиц), содержащей ядро, содержащее такролимус, покрытое первым покрытием из полимерного материала типа, пригодного для непрерывного высвобождения такролимуса, и второе покрытие типа, пригодного для отсроченного высвобождения лекарственных средств после проглатывания лекарственной формы. Первое покрытие наносят на таблетку или отдельные частицы, и оно окружает их. Второе покрытие наносят на первое покрытие, и оно окружает его.
Таблетку можно получать способами, хорошо известными в данной области, и она содержит терапевтически полезное количество такролимуса и такие эксципиенты, которые необходимы для формирования таблетки такими способами.
Первое покрытие может представлять собой покрытие для непрерывного высвобождения, известное в данной области, особенно полимерные покрытия, для изготовления мембраны, как ранее описано для резервуарных систем, или оно может представлять собой матричное ядро с контролируемым высвобождением, которое покрыто второй раз материалом с замедленным высвобождением.
Материалы, пригодные для изготовления второго покрытия на таблетке, включают полимеры, известные в данной области в качестве желудочно-резистетнтных покрытий для замедленного высвобождения фармацевтических средств. Они наиболее часто представляют собой pH-чувствительные материалы, такие как целлюлозы ацетата фталат, целлюлозы ацетата тримеллитат, фталат гидроксипропилметилцеллюлозы, поли(винилацетата фталат), и акриловые сополимеры, такие как Eudragit L-100 (Rohm Pharma) и сходные материалы, как более подробно описано ниже в разделе "Отсроченное высвобождение". Толщину покрытия с отсроченным высвобождением корректируют для получения свойства желаемого высвобождения. Как правило, более толстые покрытия являются более устойчивыми к деградации и, следовательно, приводят к более длительной и более эффективной задержке. Предпочтительная толщина покрытия находится в диапазоне от приблизительно 30 мкм до приблизительно 3 мм.
При использовании гидрофобного матричного материала, такого как глицерилмоностеарат, задерживающее покрытие не обязательно. Таблетка не высвобождает такролимус до достижения области ферментативной деградации, более конкретно, области после двенадцатиперснтной кишки.
После проглатывания дважды покрытая таблетка проходит через желудок, где второе покрытие препятствует высвобождению такролимуса в кислых условиях, преобладающих там. Когда таблетка проходит желудок и попадает в тонкий кишечник, где pH выше, второе покрытие разрушается или растворяется в соответствии с физико-химическими свойствами выбранного материала. При деградации или растворении второго покрытия первое покрытие предотвращает немедленное или быстрое высвобождение такролимуса и модулирует высвобождение, чтобы предотвратить продукцию более высоких максимальных концентраций, тем самым минимизируя побочные эффекты.
Следующий предпочтительный вариант осуществления включает систему множества частиц, где каждая частица имеет двойное покрытие, как описано выше для таблеток, первое полимером, предназначенным для достижения непрерывного высвобождения такролимуса, и с последующим покрытием полимером, предназначенным для отсроченного начала высвобождения в окружающую среду желудочно-кишечного тракта после проглатывания лекарственной формы.
Скорость высвобождения такролимуса из систем множества частиц с непрерывным высвобождением (т.е. систем множества частиц перед нанесением второго покрытия для отсроченного высвобождения) и способы модификации покрытия также контролируются факторами, рассмотренными для резервуарных систем множества частиц с такролимусом.
Вторая мембрана или покрытие для систем множества частиц с двойным покрытием представляет собой покрытие с отсроченным высвобождением, которое наносят на первое покрытие с непрерывным высвобождением, как описано выше для таблеток, и они могут быть образованы из тех же материалов. Следует отметить, что использование так называемых "желудочно-резистентных материалов" для применения на практике этого варианта осуществления значительно отличается от их применения для получения общепринятых желудочно-резистентных лекарственных форм. В случае общепринятых желудочно-резистентных форм задачей является задержка высвобождения лекарственного средства, пока лекарственная форма проходит желудок, а затем достигнет двенадцатиперстной кишки. Однако введение такролимуса прямо и полностью в двенадцатиперстную кишку может быть нежелательным, вследствие стремления минимизировать или предотвратить побочные эффекты. Таким образом, при использовании общепринятых желудочно-резистентных полимеров для осуществления на практике этого варианта осуществления может быть необходимым их нанесение более толстым слоем, чем обычно, для задержки высвобождения лекарственного средства до достижения лекарственной формой нижнего отдела желудочно-кишечного тракта. Однако также можно достигать непрерывной или контролируемой доставки такролимуса после растворения или деградации покрытия с замедленным высвобождением, таким образом, преимущества этого варианта осуществления можно реализовывать с помощью надлежащей комбинации свойства отсроченного высвобождения со свойством непрерывного высвобождения, и часть для отсроченного высвобождения отдельно может соответствовать или необязательно может соответствовать критериям USP для желудочно-резистентных форм. Толщину покрытия с замедленным высвобождением корректируют для обеспечения свойства желаемого высвобождения. Как правило, более толстые покрытия являются более устойчивыми к деградации и, следовательно, приводят к более длительной задержке.
Первый вариант осуществления с отсроченным высвобождением по изобретению представляет собой "pH-зависимую покрытую лекарственную форму", например, такую как таблетка или капсула. В случае таблетки она содержит ядро таблетки, содержащее такролимус, например, в твердом растворе/дисперсии в качестве продукта из множества частиц, матрицу для контролируемого высвобождения, например, из HPMC, дезинтегрирующее средство, смазывающее средство и один или несколько фармацевтических носителей, причем такое ядро покрыто материалом, предпочтительно полимером, который является по существу нерастворимым и непроницаемым при pH желудка, и который является более растворимым и проницаемым при pH тонкого кишечника. Предпочтительно, полимер покрытия является по существу нерастворимым и непроницаемым при pH<5,0 и растворим в воде при pH>5,0. Ядро таблетки может быть покрыто полимером в количестве, достаточном, чтобы обеспечить по существу отсутствие высвобождения такролимуса из лекарственной формы до тех пор, пока лекарственная форма не выйдет из желудка и не будет находиться в тонком кишечнике в течение приблизительно 15 минут или более, предпочтительно в течение приблизительно 30 минут или более, таким образом, обеспечивая минимальное высвобождение такролимуса в двенадцатиперстной кишке. Также можно использовать смеси pH-чувствительного полимера с нерастворимым в воде полимером. Таблетки покрывают количеством полимера, составляющим от приблизительно 10% до приблизительно 80% от массы содержащего такролимус ядра таблетки. Предпочтительно таблетки покрывают количеством полимера, составляющим от приблизительно 15% до приблизительно 50% от массы ядра таблетки с такролимусом.
pH-чувствительные полимеры, которые являются высоко нерастворимыми и непроницаемыми при pH желудка, но которые являются более растворимыми и проницаемыми при pH тонкого кишечника и толстого кишечника, включают полиакриламиды, производные фталатов, такие как кислые фталаты углеводов, амилозы ацетата фталат, целлюлозы ацетата фталат, другие фталаты сложных эфиров целлюлозы, фталаты простых эфиров целлюлозы, фталат гидроксипропилцеллюлозы, фталат гидроксипропилэтилцеллюлозы, фталат гидроксипропилметилцеллюлозы, фталат метилцеллюлозы, фталат поливинилацетата, гидрофталат поливинилацетата, фталат ацетата целлюлозы натрия, кислотный фталат крахмала, сополимер стирол-дибутилфталат малеиновой кислоты, сополимер стирол-фталат малеиновой кислоты, сополимеры стирола и малеиновой кислоты, производные полиакриловой кислоты, такие как сополимеры акриловой кислоты и акрилового сложного эфира, полиметакриловую кислоту и ее сложные эфиры, сополимеры акриловой и метакриловой кислот, шеллак и сополимеры винилацетата и кротоновой кислоты.
Предпочтительные pH-чувствительные полимеры включают шеллак; производные фталата, в частности, целлюлозы ацетата фталат, поливинилацетата фталат и гидроксипропилметилцеллюлозы фталат; производные полиакриловой кислоты, в частности, полиметилметакрилат, смешанный с акриловой кислотой и сополимерами сложных эфиров акриловой кислоты; и сополимеры винилацетата и кротоновой кислоты.
Время задержки перед высвобождением такролимуса после выхода лекарственной формы в виде "таблетки с pH-зависимым покрытием" из желудка можно контролировать путем выбора относительных количеств Eudragit-L® и Eudragit-S® в покрытии и путем выбора толщины покрытия. Пленки Eudragit-L® растворяются при pH 6,0, и пленки Eudragit-S® растворяются при pH выше 7,0, и их смеси растворяются при промежуточных pH. Поскольку pH двенадцатиперстной кишки составляет приблизительно 6,0 и pH толстого кишечника составляет приблизительно 7,0, покрытия, состоящие из смесей Eudragtt-L® и Eudragit-S®, обеспечивают защиту двенадцатиперстной кишки от такролимуса. Если желательно для задержки высвобождения такролимуса до достижения содержащей такролимус "pH-зависимой покрытой таблеткой" толстого кишечника, в качестве материала покрытия можно использовать Eudragit-S®, как описано Dew et al. (Br. J. Clin. Pharmac. 14 (1982) 405-408). Для задержки высвобождения такролимуса на приблизительно 15 минут или более, предпочтительно 30 минут или более, после выхода лекарственной формы из желудка, предпочтительные покрытия включают от приблизительно 9:1 до приблизительно 1:9 Eudragit-L®/Eudragit-S®, более предпочтительно от приблизительно 9:1 до приблизительно 1:4 Eudragit-L®/Eudragit-S®. Покрытие может составлять от приблизительно 3% до приблизительно 70% от массы непокрытого ядра таблетки. Предпочтительно, покрытие составляет от приблизительно 5% до приблизительно 50% от массы ядра таблетки.
Применение
Твердую дисперсию и/или раствор можно использовать для получения твердой пероральной лекарственной формы с пролонгированным высвобождением, такой как таблетки, капсулы или саше; или для получения гранул, драже, микросфер или наночастиц.
Следующим преимуществом лекарственной формы с пролонгированным высвобождением по настоящему изобретению является возможность достижения эффективного терапевтического ответа со сниженной дозировкой по сравнению с традиционным пероральным лечением. Таким образом, предусматривается, что твердая лекарственная форма по изобретению при пероральном введении млекопитающему, нуждающемуся в этом, в дозе, которая составляет самое большее приблизительно 85% масс./масс., как, например, самое большее приблизительно 80% масс./масс., самое большее приблизительно 75%, самое большее приблизительно 70% масс./масс., самое большее приблизительно 65% масс./масс., самое большее приблизительно 60% масс./масс., самое большее приблизительно 55% масс./масс. или самое большее приблизительно 50% масс./масс. от дозы такролимуса, вводимой в форме Prograf® или сходного коммерчески доступного содержащего такролимус продукта, является по существу биоэквивалентной Prograf® или сходному коммерчески доступному содержащему такролимус продукту.
Любое из содержащих такролимус лекарственных форм, композиций, дисперсий или растворов по изобретению может улучшать лечение состояний, которые отвечают на лечение такролимусом.
Такролимус показан (или предлагается) для лечения заболеваний, например, таких как реакции отторжения при трансплантации органов или тканей, таких как сердце, почка, печень, костный мозг, кожа, роговица, легкое, поджелудочная железа, тонкий кишечник, конечность, мышца, нерв, межпозвоночный диск, трахея, миобласт, хрящ и т.д.; реакции "трансплантат против хозяина" после трансплантации костного мозга; аутоиммунные заболевания, такие как ревматоидный артрит, системная красная волчанка, тиреоидит Хашимото, рассеянный склероз, миастения, диабет I типа, и т.д.; инфекции, вызываемые патогененными микроорганизмами (например, Aspergillus fumigatus, Fusarium oxysporum, Trichophyton asteroids и т.д.); воспалительные или гиперпролиферативные заболевания кожи или кожные проявления иммунологически опосредуемых заболеваний (например, псориаз, атопический дерматит, контактный дерматит, экзематоидный дерматит, себорейный дерматит, красный плоский лишай, пемфигус, буллезный пемфигоид, буллезный эпидермолиз, крапивница, ангионевротический отек, васкулит, эритема, дермальная эозинофилия, красная волчанка, угревая сыпь и очаговая алопеция); аутоиммунные заболевания глаза (например, кератоконъюнктивит, весенний конъюнктивит, увеит, ассоциированный с болезнью бехчета, кератит, герпетический кератит, конусовидный кератит, дистрофия эпителия роговицы, кератолейкома, глазной пемфигус, разъедающая язва роговицы, склерит, офтальмопатия Грейвса, синдром Вогта-Койанаги-Харада, сухой кератоконъюнктивит (сухой глаз), фликтена, иридоциклит, саркоидоз, эндокринная офтальмопатия, и т.д.); обратимые обструктивные заболевания дыхательных путей [астма (например, бронхиальная астма, аллергическая астма, эндогенная бронхиальная астма, экзогенная бронхиальная астма и пылевая астма), в частности, хроническая или запущенная астма (например, поздняя астма и гиперреактивность дыхательных путей) бронхит и т.д.; слизистое или сосудистое воспаление (например, язва желудка, ишемическое или тромботическое сосудистое повреждение, ишемические заболевания кишечника, энтерит, некротизирующий энтероколит, повреждения кишечника, ассоциированные с термическими ожогами, опосредуемые лейкотриеном B4 заболевания); воспаление кишечника/аллергия (например, заболевания брюшной полости, проктит, эозинофильный гастроэнтерит, мастоцитоз, болезнь Крона и язвенный колит); связанные с питанием аллергические заболевания с симптоматическими проявлениями, удаленными от желудочно-кишечного тракта (например, мигрень, ринит и экзема); заболевания почек (например, интерстициальный нефрит, синдром Гудпасчера, гемолитический уремический синдром и диабетическая нефропатия); нервные заболевания (например, множественный миозит, синдром Гийена-Барре, болезнь Меньера, множественный неврит, солитарный неврит, церебральный инфаркт, болезнь Альцгеймера, болезнь Паркинсона, боковой амиотрофический склероз (ALS) и радикулопатия); церебальное ишемическое заболевание (например, травма головы, кровоизлияние в головной мозг (например, субарахноидальное кровоизлияние, внутримозговое кровоизлияние), церебральный тромбоз, эмболия головного мозга, остановка сердца, инсульт, транзиторная ишемическая атака (TIA), гипертензивная энцефалопатия, церебральный инфаркт); эндокринные заболевания (например, гипертиреоидизм и базедова болезнь); гематологические заболевания (например, истинная эритроцитарная аплазия, апластическая анемия, гипопластическая анемия, идиопатическая тромбоцитопеническая пурпура, аутоиммунная гемолитическая анемия, агранулематоз, пернициозная анемия, мегалобластная анемия и анэритроплазия); заболевания костей (например, остеопороз); респираторные заболевания (например, саркоидоз, фиброз легких и идиопатическая интерстициальная пневмония); кожные заболевания (например, дерматомиозит, лейкодерма обыкновенная, ихтиоз обыкновенный, светочувствительность и Т-клеточная лимфома кожи); циркуляторные заболевания (например, артериосклероз, атеросклероз, аортит, узелковый полиартериит кии миокардоз); коллагеновые заболевания (например, склеродермия, гранулема Вегенера и синдром Шегрена); адипоз; эозинофильный фасциит; периодонтальные заболевания (например, повреждение десны, периодонта, альвеолярной кости или плотное вещество кости); нефротический синдром (например, гломерулонефрит); алопеция по мужскому типу, старческая алопеция; мышечная дистрофия; пиодермия и синдром Сезари; ассоциированные с хромосомными нарушениями заболевания (например, синдром Дауна); болезнь Аддисона; опосредуемые активными формами кислорода заболевания [например, повреждение органов (например, ишемические нарушения кровообращения органов (например, сердца, печени, почки, пищеварительного тракта и т.д.), ассоциированное с сохранением, трансплантацией или ишемические заболевания (например, тромбоз, инфаркт миокарда и т.д.)); заболевания кишечника (например, эндотоксиновый шок, псевдомембранозный колит, и индуцируемый лекарственными средствами или облучением колит); болезни почек (например, острая ишемическая недостаточность почек, хроническая почечная недостаточность); заболевания легких (например, токсикоз, вызываемый легочным кислородом или лекарственными средствами (например, паракортом, блеомицином и т.д.), рак легкого и эмфизема легких); глазные заболевания (например, катаракта, болезни болезнь накопления железа (siderosis bulbi), пигментный ретинит, старческие бляшки, образование рубца стекловидного тела, ожог щелочью роговицы); дерматит (например, полиформная эритема, буллезный дерматит с линейным иммуноглобулином A, цементный дерматит) и другие заболевания (например, гингивит, периодонтит, сепсис, панкреатит и заболевания, вызываемые загрязнением окружающей среды (например, загрязнением воздуха), старением, канцерогенами, метастазированием карциномы и гидробаропатией)]; заболевания, вызываемые высвобождением гистамина или высвобождением лейкотриена C4; рестеноз коронарной артерии после ангиопластики и профилактики послеоперационных спаек; аутоиммунные заболевания и воспалительные состояния (например, первичный отек слизистой оболочки, аутоиммунный атрофический гастрит, преждевременная менопауза, мужское бесплодие, ювенильный сахарный диабет, пемфигус обыкновенный, пемфигоид, симпатическая офтальмия, связанный с линзами увеит, идиопатическая лейкопения, активный хронический гепатит, идиопатический цирроз, дискоидная красная волчанка, аутоиммунный орхит, артрит (например, деформирующий артрит), или полихондрит); инфекция вирусом иммунодефицита человека (HIV), СПИД; аллергический конъюнктивит; гипертрофический рубец и келоид вследствие травмы, ожога или хирургической операции.
Кроме того, трициклические макролиды, например, такие как такролимус, обладают активностью регенерации печени и/или видами активности стимуляции гипертрофии и гиперплазии гепатоцитов. Таким образом, лекарственная форма с пролонгированным высвобождением по настоящему изобретению пригодна для увеличения эффекта лечения и/или профилактики заболеваний печени [например, иммуногенных заболеваний (например, хронических аутоиммунных заболеваний печени, таких как аутоиммунные заболевания печени, первичный билиарный цирроз или склерозирующий холангит), частичной резекции печени, острого некроза печени (например, некроза, вызываемого токсинами, вирусным гепатитом, шоком или аноксией), гепатита B, не-A не-B гепатита, печеночного цирроза и печеночной недостаточности (например, молниеносного гепатита, гепатита с поздним началом и "острой на фоне хронического заболевания" печеночной недостаточности (острой печеночной недостаточности при хронических заболеваниях печени))].
Более того, лекарственная форма с пролонгированным высвобождением в соответствии с настоящим изобретением пригодна для повышения эффекта профилактики и/или лечения различных заболеваний вследствие полезной фармакологической активности трициклических макролидов, такой как активность усиления химиотерапевтического эффекта, активность против цитомегаловирусной инфекции, противовоспалительная активность, активность ингибирования пептидилпролилизомеразы или ротамазы, антималярийная активность, противоопухолевая активность и т.д.
Кроме того, настоящее изобретение иллюстрируется с помощью примеров, представленных ниже. Однако следует отметить, что эти примеры, подобно вариантам осуществления, описанным выше, являются иллюстративными и их не следует истолковывать как ограничивающие представленный объем изобретения никоим образом.
ПРИМЕРЫ
МАТЕРИАЛЫ И СПОСОБЫ
Материалы
Такролимус (поставляемый Eurotrade); партия № RD 03-111
Моногидрат лактозы 200 меш (от DMV)
Гранулированный оксид кремния, Aeroperl® 300 (Degussa)
Полиэтиленгликоль 6000, Pluracol® E6000 (от BASF)
Полоксамер 188, Pluronic® F-68 (от BASF)
Глицерилмоностеарат, Rylo® MD50 (от Danisco Cuitor), Ph.Eur; партия № 4010056276
Avicel PH200 (микрокристаллическая целлюлоза) (от FMC)
Лактоза DCL 11 (от DMV)
Стеарат магния
Кроскармеллоза натрия, Ac-Di-Sol® (от FMC)
Eudragit® L30D.55 (от Degussa)
Триэтилцитрат (от Merck)
Противопенная эмульсия (от Unikem)
Микротальк
HPMC относится к Metolose 90SH (тип 2910, 2208) или Metolose 60SH (тип 2910) от ShinEtsu, доступным в различных степенях полимеризации (вязкость 3-100000 спз).
Таблетки, капсулы или гранулы могут быть покрытыми желудочно-резистентной оболочкой из различных типов полимеров, таких как гидроксипропилметилцеллюлозы ацетата сукцинат (Aqoat), целлюлозы ацетата фталат CAP, гидроксипропилметилцеллюлозы фталат HPMCP, или сополимеры метакриловой кислоты, такие как Eudragit L30D, Eudragit 100/S, Eudragit 100/L.
Сравнение составов такролимуса уровня техники для исследований in vivo
Prograf®, твердые желатиновые капсулы, изготовленные Fujisawa Ireland Ltd.
СПОСОБЫ
Определение варьирования массы
Таблетки, полученные согласно примерам, описанным в настоящем документе, подвергали тестированию в отношении варьирования массы, проводимому согласно Ph. Eur.
Определение средней жесткости таблеток
Таблетки, полученные согласно примерам, описанным в настоящем документе, подвергали тестированию в отношении жесткости таблетки с использованием устройства Schleuniger Model 6D, и его проводили согласно общим инструкциям для устройства.
Определение времени дезинтеграции
Время дезинтеграции таблетки, т.е. распада на частицы или агломераты, определяли согласно Ph. Eur.
Определение геометрического средневзвешенного диаметра d gw
Геометрический средневзвешенный диаметр определяли с использованием способа лазерной дифракции, диспергируя полученный материал в виде частиц (или исходный материал) в воздухе. Измерения проводили при давлении диспергирования 1 бар в оборудовании Sympatec Helos, которое регистрирует распределение эквивалентного сферического диаметра. Это распределение аппроксимируется к логарифмическому нормальному распределению объем-размер.
При использовании в настоящем документе "геометрический средневзвешенный диаметр" означает средний диаметр логарифмического нормального распределения объем-размер.
Тесты растворения in vitro
Для композиций и лекарственных форм по настоящему изобретению использовали следующие способы тестирования.
Тест 1
Тест растворения in vitro согласно способу A USP, изделия с отсроченным высвобождением (способ USP с лопастью; скорость вращения 50 об/мин; 37°С; после 2 часов в кислой среде, среду заменяют фосфатным буфером, pH 6,8).
Тест 2
Тест растворения in vitro в водной среде для растворения, доведенной до pH 4,5 (900 мл воды с 0,005% HPC (гидроксипропилцеллюлоза), доведенной до pH 4,5; 37°С; способ USP с лопастью; скорость вращения 50 об/мин.).
Представленные ниже примеры предназначены для иллюстрации изобретения и не предназначены для ограничения объема настоящего изобретения.
Фармацевтические композиции и лекарственные формы, которые могут быть оптимизированными для обеспечения желаемого профиля высвобождения по изобретению, проиллюстрированы в примерах 1-16, и эти составы описаны в патентной заявке WO 2005/020993, включенной в настоящий документ в качестве ссылки. Однако профили растворения, описанные в ней и в примере 17 настоящей заявки можно далее оптимизировать для обеспечения профилей высвобождения, удовлетворяющих параметрам пролонгированного высвобождения в соответствии с настоящим изобретением. Оптимизация может включать изменения состава, включающие изменения классов эксципиентов, изменения соотношений эксципиентов, изменения производства, такие как изменение давления при штамповании, изменение жесткости или времени дезинтеграции. Поскольку молекулярный раствор является предпочтительным, значимые изменения включают применение эксципиентов, предотвращающих деградацию активного вещества, а также применение специальных эксципиентов, менее склонных к взаимодействию с активным веществом и/или продуктами его деградации.
Пример 1
Капсулы из множества частей с модифицированным высвобождением на основе набухающей гидроколлоидной матрицы из гидроксипропилцеллюлозы
Такролимус растворяли в полиэтиленгликоле 6000 и полоксамере 188 (соотношение 70:30 масс./масс.) при 70°С. Раствор напыляли на смесь 150 г лактозы и 100 г HPMC в Strea-1 с псевдоожиженным слоем. Гранулярный продукт просеивали через сито размером 0,7 мм и заполняли в твердые желатиновые капсулы (200 мг).
Пример 2
Капсулы из множества частей с модифицированным высвобождением на основе набухающей гидроколлоидной матрицы из гидроксипропилцеллюлозы
Такролимус растворяли в глицерилмоностеарате при 70°С. Раствор напыляли на смесь из 150 г лактозы и 100 г HPMC в Strea-1 с псевдоожиженным слоем. Гранулярный продукт просеивали через сито размером 0,7 мм и заполняли в твердые желатиновые капсулы (200 мг).
Пример 3
Таблетка с матрицей с пролонгированным высвобождением на основе набухающей гидроколлоидной матрицы из гидроксипропилцеллюлозы
Такролимус растворяли в полиэтиленгликоле 6000 и полоксамере 188 (соотношение 70:30 масс./масс.) при 70°С. Раствор напыляли на 250 г лактозы в Strea-1 с псевдоожиженным слоем.
Полученный гранулярный продукт просеивали через сито размером 0,7 мм и смешивали с HPMC и стеаратом магния в течение 0,5 мин в смесителе Turbula.
Смесь прессовали в 8-мм таблетки с 1 мг активного ингредиента (таблетка массой 200 мг) в форме двойной чаши.
Среднее время дезинтеграции: 20 мин. Жесткость: 45 Н.
Пример 4
Таблетка с матрицей с модифицированным высвобождением на основе липофильной матрицы из глицерилмоностеарата
Такролимус растворяли в глицерилмоностеарате при 70°С. Раствор напыляли на 250 г лактозы в Strea-1 с псевдоожиженным слоем. Гранулярный продукт просеивали через сито размером 0,7 мм и перемешивали со стеаратом магния в течение 0,5 минуты в смесителе Turbula.
Полученную смесь прессовали в 8-мм таблетки с 1 мг активный ингредиент (200 мг таблетка) в форме двойной чаши.
Среднее время дезинтеграции: 20 мин. Жесткость: 45 Н
Пример 5
Капсула с множеством депо с модифицированным высвобождением на основе липофильной матрицы из глицерилмоностеарата
Такролимус растворяли в глицерилмоностеарате при 70°С. Раствор напыляли на 250 г лактозы в Strea-1 с псевдоожиженным слоем. Гранулярный продукт просеивали через сито размером 0,7 мм и заполняли в твердые желатиновые капсулы (200 мг).
Пример 6
Таблетка с множеством депо с модифицированным высвобождением на липофильной матрице из Gelucire® 44/14
Такролимус растворяли в Gelucire® при 70°С. Раствор распыляли на 250 г Aeroperl® в Strea-1 с псевдоожиженным слоем. Гранулярный продукт просеивали через сито размером 0,7 мм и заполняли в твердые желатиновые капсулы (200 мг).
Полученный гранулят прессовали в 8-мм таблетки с 1 мг активного ингредиента (масса таблетки 200 мг). Таблеткам придавали форму чаши.
Среднее время дезинтеграции: 25 минут. Жесткость: 43 Н.
Пример 7
Желудочно-резистентное покрытие
Капсулы и таблетки согласно примерам 1, 2, 3, 5 и 6 затем покрывали представленным ниже желудочно-резистентным покрытием для достижения отсроченного высвобождения активного ингредиента после введения.
Суспензию для покрытия приготавливали смешиванием триэтилацетилцитрата, противопенной эмульсии и очищенной воды в устройстве Ultra Turrax при 9500 об/мин в течение 30 мин. Через 1 минуту добавляли тальк. Смесь пропускали через сито № 300 и перемешивали магнитной мешалкой. Eudragit пропускали через сито № 300 и добавляли к смеси, которую перемешивали в течение 5 минут.
Условия процесса нанесения покрытия были следующими: температура на входе 40°С, температура на выходе 31°С, воздух на входе 140 м3 в час и время покрытия приблизительно 50 минут (300 г материала покрытия). Покрывали приблизительно 400 г таблеток или 200 г капсул.
Покрытые пленкой таблетки и капсулы сушили в течение 48 часов при 30°С перед тестированием растворения.
Пример 8
Таблетка с матрицей с модифицированным высвобождением на основе липофильной матрицы из моностеарата глицерина
Такролимус растворяли в моностеарате глицерина при температуре выше 80°С. Раствор распыляли с помощью подающего устройства Phast FS1.7 на 60 г лактозы и 60 г HPMC в Phast FB100 с псевдоожиженным слоем. Гранулярный продукт отверждали в нагревающей печи в течение 4 часов при 50°С. Полученный гранулярный продукт просеивали через сито размером 0,71 мм и смешивали с лактозой в течение 3 минут в смесителе Turbula.
Стеарат магния и тальк просеивали через сито № 300 и смешивали в смесителе Turbula в течение 3 минут. Гранулят смешивали со смесью стеарата магния и талька (1:9) в течение 0,5 в смесителе Turbula.
Конечную смесь прессовали в 8-мм таблетки с 2 мг активного ингредиента (210-мг таблетка) в форме двойной чаши.
Среднее время дезинтеграции: 2 часа. Жесткость: 50 Н.
Пример 9
Покрытая желудочно-резистентным покрытием таблетка с ядром на основе PEG 6000/полоксамера 188 и с желудочно-резистентным покрытием на основе Eudragit L30D 55
Ядро таблетки с такролимусом получали растворением в PEG 6000 при температуре выше 80°С. Добавляли полоксамер 188 и раствор нагревали до температуры выше 80°С. Раствор напыляли с помощью подающего устройства Phast FS1.7 на 200 г моногидрата лактозы в Phast FB100 с псевдоожиженным слоем. Полученный гранулят просеивали через сито Comill 1397, 4500 об/мин и смешивали с кроскармелозой натрия в течение 3 минут в смесителе Turbula.
Стеарат магния и тальк просеивали через сито № 300 и смешивали в смесителе Turbula в течение 3 минут. Гранулят смешивали со смесью стеарата магния и талька (1:9) в течение 0,5 минуты в смесителе Turbula.
Конечную смесь прессовали в 6-мм таблетки с 2 мг активного ингредиента (100-мг таблетка) в форме двойной чаши.
Среднее время дезинтеграции: 7 минут. Жесткость: 65 Н.
Желудочно-резистентное покрытие
Желудочно-резистентное покрытие основано на акриловом полимере Eudragit L30D-55. Eudragit L30D поставляют в качестве водной латексной суспензии, создающей нерастворимую в воде пленку, когда вода испаряется в процессе нанесения покрытия. Полимер является нерастворимым при величинах pH ниже 5,0 и хорошо растворим при величинах pH более 6,0. Композиция пленочного покрытия представляет собой:
Количество наносимого полимера пленки (Eudragit) было основано на расчете мг полимера пленки на см2 поверхности таблетки. Толщина желудочно-резистентного покрытия составляла 80 мкм. Проверка толщины наносимой пленки была основана на измерении увеличения высоты таблетки с помощью цифрового микрометра. Процесс нанесения пленочного покрытия проводили в Phast FB100 с псевдоожиженным слоем, оборудованном вставкой типа Wurster. Условия процесса представляли собой: температура воздуха на входе 50°С; поток воздуха на входе 100 м3 в час; температура продукта 38°С; скорость подачи 15 г/мин.
После нанесения покрытия образование пленки требует сушки покрытых таблеток, т.е. при 30°С в течение 48 часов в печи. Альтернативно покрытые таблетки можно более эффективно сушить при 40°С в течение 24 часов.
Пример 10
Таблетка с PEG 6000/полоксамером 188 с контролируемым высвобождением на основе матрицы из HPMC
Такролимус растворяли в PEG 6000 при температуре выше 80°С. Добавляли полоксамер 188 и раствор нагревали до температуры выше 80°С. Раствор напыляли с помощью подающего устройства Phast FS1.7 на 200 г моногидрата лактозы в Phast FB100 с псевдоожиженным слоем. Гранулярный продукт просеивали через сито Comill 1397, 4500 об/мин и смешивали с гидроксипропилметилцеллюлозой в течение 3 минут в смесителе Turbula.
Стеарат магния и тальк просеивали через сито № 300 и смешивали в смесителе Turbula в течение 3 минут. Гранулят смешивали со смесью стеарата магния и талька (1:9) в течение 0,5 минуты в смесителе Turbula. Смесь прессовали в 8-мм таблетки с 2 мг активного ингредиента (165-мг таблетка в форме двойной чаши). Среднее время дезинтеграции: 2 часа 34 минуты. Жесткость: 50 Н.
Пример 11
Состав покрытой желудочно-резистентным покрытием таблетки. Влажное гранулирование и покрытые желудочно-резистентным покрытием таблетки
Изготовление таблетки было основано на влажной грануляции в смесителе с большими сдвиговыми усилиями Pellmix 1/8. 16 г микронизированного такролимуса смешивали с 640 г лактозы 125 меш и 80 г лаурилсульфата натрия в смесителе с большими сдвиговыми усилиями. 15% водный раствор связующего вещества Kollidon VA64 закачивали в смесь при скорости лопастного колеса 500 об/мин со скоростью подачи 20 г/мин, а затем перемешивали в течение 3 минут при одной скорости. Гранулят сушили в центробежной сушилке и просеивали через сито размером 0,7 мм.
Гранулят перемешивали с 240 г Avicel PH200 в течение 3 минут и в течение добавления 4 г стеарата магния и после его добавления в течение дополнительных 0,5 минуты. Смесь прессовали в таблетки на устройстве для таблетирования с одним штампом Diaf TM20.
Диаметр таблетки: 6 мм. Форма таблетки: округлая, двойная чаша.
Затем таблетки покрывали желудочно-резистентным покрытием акрилового типа, как описано в примере 9.
Количество наносимого пленочного полимера (Eudragit) должно быть основано на расчете мг пленочного полимера на см2 поверхности таблетки. Толщина желудочно-резистентного покрытия должна составлять 50-80 мкм. Проверка толщины наносимой пленки основана на измерении увеличения высоты таблетки с помощью цифрового микрометра. Процесс нанесения пленочного покрытия проводят в Stre-1 с псевдоожиженным слоем, оборудованном вставкой типа Wurster при следующих условиях процесса.
После нанесения покрытия образование пленки требует сушки покрытых таблеток, т.е. при 30°С в течение 48 часов в печи. Альтернативно покрытые таблетки можно более эффективно сушить при 40°С в течение 24 часов.
Пример 12
Состав таблетки с контролируемым высвобождением на основе деградирующей матрицы из HPMC
HPMC добавляли в качестве части экстрагранулярной фазы. Влажное гранулирование.
Изготовление таблетки было основано на влажной грануляции в смесителе с большими сдвиговыми усилиями Pellmix 1/8. 16 г микронизированного такролимуса смешивали с 640 г лактозы 125 меш и 80 г лаурилсульфата натрия в смесителе с большими сдвиговыми усилиями. 15% водный раствор связующего вещества Kollidon VA64 закачивали в смесь при скорости лопастного колеса 500 об/мин со скоростью подачи 20 г/мин, а затем перемешивали в течение 3 минут при одной скорости. Гранулят сушили в центробежной сушилке и просеивали через сито размером 0,7 мм.
Гранулят перемешивали с 240 г Avicel PH200 и 480 г гидроксипропилметилцеллюлозы Metolose SH 90 100 спз в течение 3 минут и после добавления 8 г стеарата магния в течение дополнительных 0,5 минуты. Смесь прессовали в таблетки на устройстве для таблетирования с одним штампом Diaf TM20.
Диаметр таблетки: 7 мм. Форма таблетки: округлая, двойная чаша.
Пример 13
Состав таблетки с контролируемым высвобождением на основе деградирующей матрицы из HPMC
HPMC добавляли в качестве части внутригранулярной фазы. Влажное гранулирование.
Изготовление таблетки было основано на влажной грануляции в смесителе с большими сдвиговыми усилиями Pellmix 1/8. 16 г микронизированного такролимуса смешивали с 640 г лактозы 125 меш и 80 г лаурилсульфата натрия и 640 г гидроксипропилметилцеллюлозы Metolose SH 90 15000 спз в смесителе с большими сдвиговыми усилиями. Очищенную воду закачивали в смесь при скорости лопастного колеса 500 об/мин при скорости подачи 20 г/мин, а затем перемешивали в течение 3 минут при одной скорости. Гранулят сушили в центробежной сушилке и просеивали через сито размером 0,7 мм.
Гранулят перемешивали с 480 г Avicel PH200 в течение 3 минут и после добавления 16 г стеарата магния в течение дополнительных 0,5 минуты. Смесь прессовали в таблетки на устройстве для таблетирования с одним штампом Diaf TM20.
Диаметр таблетки: 8 мм. Форма таблетки: округлая, двойная чаша.
Пример 14
Состав таблетки с контролируемым высвобождением на основе деградирующей матрицы из HPMC
HPMC добавляли в качестве части внутригранулярной фазы. Влажное гранулирование.
Изготовление таблетки было основано на влажной грануляции в смесителе с большими сдвиговыми усилиями Pellmix 1/8. 16 г микронизированного такролимуса смешивали с 640 г лактозы 125 меш и 120 г полиэтиленгликоля 6000, 48 г полоксамера 188 и 640 г гидроксипропилметилцеллюлозы Metolose SH 90 15000 спз в смесителе с большими сдвиговыми усилиями. Рубашку емкости нагревали до 80°С и смесь нагревали при скорости вращения лопастного колеса 1000 об/мин до температуры плавления PEG и полоксамера. После плавления перемешивание продолжали в течение 4 минут при 800 об/мин. Гранулированный продукт просеивали через сито размером 0,7 мм и охлаждали на лотке. Гранулят перемешивали с 480 г Avicel PH200 в течение 3 минут и после добавления 16 г стеарата магния в течение дополнительных 0,5 минуты. Смесь прессовали в таблетки на устройстве для таблетирования с одним штампом Diaf TM20. Диаметр таблетки: 8 мм. Форма таблетки: округлая, двойная чаша.
Пример 15
Состав таблетки с контролируемым высвобождением на основе деградирующей матрицы Kollidon SR в качестве части экстрагранулярной фазы
Изготовление таблетки было основано на влажной грануляции в смесителе с большими сдвиговыми усилиями Pellmix 1/8. 16 г микронизированного такролимуса смешивали с 640 г лактозы 125 меш и 80 г лаурилсульфата натрия в смесителе с большими сдвиговыми усилиями. 15% водный раствор связующего вещества Kollidon VA64 (Kollidon SR представляет собой смесь поливинилацетат и поливинилпирролидона 80:20) закачивали в смесь при скорости лопастного колеса 500 об/мин со скоростью подачи 20 г/мин, а затем перемешивали в течение 3 минут при одной скорости. Гранулят сушили в центробежной сушилке и просеивали через сито размером 0,7 мм. Гранулят перемешивали с 400 г лактозы DC Lac 14 и 480 г Kollidon SR в течение 3 минут и в течение и после добавления 8 г стеарата магния в течение дополнительных 0,5 минуты. Смесь прессовали в таблетки на устройстве для таблетирования с одним штампом Diaf TM20.
Диаметр таблетки: 8 мм. Форма таблетки: округлая, двойная чаша.
Пример 16
Состав таблетки с желудочно-резистентным покрытием (гранулирование из расплава и покрытые желудочно-резистентным покрытием таблетки)
Изготовление таблетки было основано на грануляции из расплава в смесителе с большими сдвиговыми усилиями Pellmix 1/8. 16 г микронизированного такролимуса смешивали с 640 г лактозы 125 меш и 120 г полиэтиленгликоля 6000, 48 г полоксамера 188 в смесителе с большими сдвиговыми усилиями. Рубашку емкости нагревали до 80°С и смесь нагревали при скорости вращения лопастного колеса 1000 об/мин до температуры плавления PEG и полоксамера. После плавления перемешивание продолжали в течение 4 минут при 800 об/мин. Гранулированный продукт просеивали через сито размером 0,7 мм и охлаждали на лотке. Гранулят перемешивали с 480 г Avicel PH200 в течение 3 минут и в течение и после добавления 16 г стеарата магния в течение дополнительных 0,5 минуты. Смесь прессовали в таблетки на устройстве для таблетирования с одним штампом Diaf TM20. Диаметр таблетки: 7 мм. Форма таблетки: округлая, двойная чаша. Нанесение желудочно-резистентного покрытия таблеток проводят согласно способу, описанному в примере 11.
Пример 17
Данные растворения in vitro
Композиции и лекарственные формы согласно предыдущим примерам подвергали тестам растворения in vitro с использованием двух различных сред для растворения/тестов растворения.
A. С использованием среды для растворения/теста растворения: 900 мл водной среды с 0,005% HPC (гидроксипропилцеллюлозы), доведенной до pH=4,5 (способ USP с лопастями; скорость вращения: 50 об/мин) были выявлены следующие профили растворения:
Профиль растворения для ядер таблеток примера 9 в среде для растворения: 900 мл водной среды с 0,005% HPC (гидроксипропилцеллюлозы), доведенной до pH=4,5. Способ USP с лопастями. Скорость вращения 50 об/мин.
Профиль растворения для ядер таблеток примера 9 в среде для растворения согласно способу USP A, изделия с замедленным высвобождением. Способ USP с лопастями. Скорость вращения 50 об/мин.
Пример 18
Перечень повышающих стабильность мер для улучшения и оптимизации состава с пролонгированным высвобождением, содержащего такролимус в твердом растворе
Такролимус в солюбилизированной форме подвержен деградации, и в процессе хранения может образоваться несколько продуктов деградации. Показатели повышения стабильности, протестированные в отношении повышения стабильности такролимуса, представлены в следующем списке.
Носители, антиоксидантные составы
добавление
1000 м.д. пропилгаллата
500 м.д. α-токоферола + 500 м.д. липоида
500 м.д. аскорбилпальмитата
1000 м.д. α-токоферола
500 м.д. α-токоферола
50 м.д. α-токоферола
1000 м.д. аскорбилпальмитата + 1000 м.д. α-токоферола
500 м.д. аскорбилпальмитата + 500 м.д. α-токоферола
250 м.д. аскорбилпальмитата + 250 м.д. α-токоферола
50 м.д. аскобрилпальмитата + 50 м.д. α-токоферола
Удаление примесей в полоксамере путем фильтрации через Al2O3 (Compalox)
Добавление диметикона
Добавление BHT
Добавление органических кислот, включая виннокаменную кислоту
0,01% виннокаменную кислоту, 0,05% виннокаменную кислоту, 0,10% виннокаменную кислоту, 0,20% виннокаменную кислоту, 0,40% виннокаменную кислоту, 0,50% виннокаменную кислоту, 0,60% виннокаменную кислоту, 0,75% виннокаменную кислоту, 1% виннокаменную кислоту, 5% виннокаменную кислоту
Варьирование виннокаменной кислоты в области 0,15% с 200 м.д. α-токоферола
Таблетки, высушенные с помощью N2 при
25° в течение 4 ч, высушенные с помощью N2, нагревание при низкой температуре 65°С, низкое время нагревания,
25° в течение 24 ч, высушенные с помощью N2, нагревание при низкой температуре 65°C, низкое время нагревания,
40° в течение 4 ч, высушенные с помощью N2, нагревание при низкой температуре 65°C, низкое время нагревания,
40° в течение 24 ч, высушенные с помощью N2, низкая температура нагревания 65°C низкое время нагревания
Таблетки, открытое хранение при различной контролируемой влажности
11% влажность при LiCl
32% влажность при MgCl2
48% влажность при K2CO3, 75% влажность при NaCl
89% влажность при KNO3
Пример 19
Клиническое исследование, сравнивающее состав с пролонгированным высвобождением в соответствии с настоящим изобретением с Prograf® у стабильных пациентов с трансплантатами почки и печени, соответственно
Профили концентраций в крови для стабильных пациентов с трансплантатом почки описаны на фиг.5 и 6.
Название исследования:
Открытое многоцентровое проспективное исследование с переходом фазы II у стабильных пациентов с трансплантатами почек для сравнения фармакокинетики таблеток LCP-Tacro один раз в сутки с капсулами Prograf® два раза в сутки. Таблетки LCP-Tacro 1, 2, 5 мг.
Активный ингредиент: такролимус.
Показания: такролимус (Prograf®) используют для профилактики отторжения трансплантатов печени, почки и сердца.
Схема исследования и фаза разработки: 3-серийное открытое проспективное многоцентровое испытание с переходом (фаза II).
Название исследования: открытое многоцентровое проспективное исследование с переходом фазы II у стабильных пациентов с трансплантатами почек для сравнения фармакокинетики таблеток LCP-Tacro один раз в сутки с капсулами Prograf® два раза в сутки.
Задачи: Оценить экспозицию такролимуса (AUC0-24) и минимальные уровни (C24) у стабильных реципиентов с трансплантатами почек, переведенных с капсул Prograf® (такролимус, Astellas Pharma US, Inc.) на таблетки LCP-Tacro в трехсерийной схеме исследования, и оценить безопасность LCP-Tacro по сравнению с Prograf.
Главные критерии включения: мужчины и женщины в возрасте 18-65 лет, которые стали реципиентами трансплантата почки по меньшей мере за 6 месяцев до включения в исследование.
Тестируемые продукты/исследуемые продукты и способы введения: таблетки LCP-Tacro 1, 2, 5 мг, вводимые перорально один раз в сутки утром.
Эталонный продукт, номер партии и способ введения: капсулы Prograf®, 0,5, 1, 5 мг, вводимые два раза в сутки в двух равных разделенных дозах, одна с утра и одна вечером.
Способы: трехсерийное открытое многоцентровое проспективное исследование у стабильных пациентов с трансплантатами печени для оценки и сравнения фармакокинкетики (Cmax, C24 и AUC) и безопасности таблеток LCP-Tacro (такролимус) по сравнению с капсулами Prograf® (такролимус).
В исследование включали пациентов с трансплантатами печени, которые удовлетворяли всем критериям включения/исключения (I/E), и им вводили Prograf® в течение 7 суток. После исследования в течение 24 часов на 7 сутки для определения фармакокинетики Prograf®, всех пациентов переводили на LCP-Tacro один раз в сутки (соотношение 1:0,66-0,80) в течение 7 суток, не допуская изменения доз. На 14 сутки проводили 24-часовое исследование PK LCP-Tacro. На 15 сутки допускали одно предопределенное изменение дозы при наличии изменения более чем на 25% в среднем значении для 3 минимальных уровней, измеренных на сутки 10±1, 12±1 (отделенные по меньшей мере на 48 часов от предыдущего образца) и 14 по сравнению со средними значениями для 3 минимальных уровней, измеренных на 0, 7 и 8 сутки для каждого отдельного пациента.
Пациенты оставались на дозе, установленной на 14 сутки, в течение других 7 суток без допущения изменения доз. Другое 24-часовое исследование LCP-Tacro PK проводили на 21 сутки. На 22 сутки пациентов переводили обратно на их исходную дозу Prograf® два раза в сутки на протяжении периода для наблюдения безопасности из 30 суток, кончающегося оценкой безопасности на 52 сутки. Промежуточный анализ PK проводили после завершения 10 пациентами 21 суток, перед продолжением включения в исследование.
Моменты времени взятия крови: в процессе исследования проводили следующие взятия образцов крови:
Для LCP-Tacro: моменты времени взятия образцов крови включали: 0,00 (до введения дозы), 0,50, 1,00, 1,50, 2,00, 3,00, 4,00, 6,00, 8,00, 12,00, 14,00, 16,00, 20,00 и 24,00 часов после введения дозы, на сутки 14 и 21.
Для Prograf®: моменты времени взятия образцов крови включали: 0,00 (до введения дозы), 0,50, 1,00, 1,50, 2,00, 3,00, 4,0O1 6,00, 8,00, 12,00, 12,50, 13,00, 13,50, 14,00, 15,00, 16,00, 20,00 и 24,00 часов после утренней дозы на 7 сутки.
Критерии оценки: фармакокинетический анализ проводили у 47 пациентов. Оценку безопасности проводили у всех пациентов, которым вводили по меньшей мере 1 дозу в процессе исследования.
Фармакокинетика (PK): следующие фармакокинетические параметры для такролимуса вычисляли с помощью стандартных некомпартментных способов: AUCτ (τ=24), Cmax, Cmin, Cсреднее, Tmax, % флуктуации, % максимального отклонения и Cmax/Cmin.
Статистические способы: все демографические данные, фармакокинетические параметры, лабораторные данные и AE суммировали с использованием описательной статистики. Для непрерывных данных представлено среднее значение, стандартное отклонение, медиана, минимум и максимум. Для категорийных данных представлены процент и частота.
Некомпартментные фармакокинетические параметры [AUCτ (τ=24), Cmax, Cmin, Cсреднее, Tmax] вычисляли из данных концентрация в крови-время.
Статистические способы: с использованием способов GLM в SAS, ANOVA проводили на натуральных логарифмических (ln) преобразованных параметрах AUCτ, Cmax, Cmin и Cсреднее, и непреобразованных параметрах % флуктуации, % максимального отклонения и Cmax/Cmin. Модель включала лечение в качестве фактора. Соотношение геометрических LSM вместе с 90% CI вычисляли согласно следующим трем сравнениям для AUCτ, Cmax и Cmin:
Prograf на 14 сутки по сравнению с LCP-Tacro на 7 сутки.
Prograf на 21 сутки по сравнению с LCP-Tacro на 7 сутки.
LCP-Tacro на 21 сутки по сравнению с LCP-Tacro на 14 сутки.
Параметр Tmax анализировали с использованием непараметрических способов. Использовали знаковый ранговый критерий Вилкоксона для попарных сравнений лечений. Средний сдвиг между 2 лечениями (как описано выше) оценивали путем срединной несмещенной оценки Hodges-Lehmann и 90% точного интервала достоверности.
Степень корреляции между AUCτ и Cmin количественно определяли путем вычисления коэффициента корреляции и предоставления графических представлений данных на 7, 14 и 21 сутки. Оба параметра ln-преобразовывали перед анализом корреляций. Статистический анализ расовых подгрупп (афроамериканцы по сравнению с неафроамериканцами) проводили, поскольку было получено достаточное разделение между двумя группами.
Фармакокинетические параметры такролимуса у всех пациентов
Фармакокинетические параметры такролимуса у пациентов-афроамериканцев
Оценка относительной биодоступности такролимуса для 14 суток по сравнению с 7 сутками
у всех пациентов
у всех пациентов
у всех пациентов
Заключение
Главной задачей этого исследования была оценка экспозиции такролимуса (AUCτ) в стационарном состоянии и минимальных уровней (Cmin) у стабильных реципиентов с трансплантатами почек, переведенных с Prograf® (такролимус, Astellas Pharma US, Inc.) на LCP-Tacro в трехсерийной схеме эксперимента.
После коррекции по дозе системная экспозиция такролимуса в стационарном состоянии (AUCτ) и минимальные уровни такролимуса (Cmin) у пациентов с трансплантатами почек были значительно более высокими, когда вводили таблетки LCP-Tacro раз в сутки, по сравнению с лечением капсулами Prograf® два раза в сутки. Системная экспозиция (AUCτ и Cmin) в течение 24 часов 2-мг таблеток LCP-Tacro (раз в сутки) была на ~38% и ~36% выше, чем у капсул Prograf® (два раза в сутки). Средняя концентрация лекарственного средства на протяжении интервала дозирования, Cсреднее, была значительно более высокой для терапии LCP-Tacro по сравнению с Prograf®.
Также не было статистически значимых отличий в максимальной системной экспозиции (Cmax) такролимуса между терапией Prograf® при введении LCP-Tacro на 14 и 21 сутки. Кроме того, лечение Prograf® продемонстрировало значительно более высокую степень флуктуации и максимального отклонения по сравнению с LCP-Tacro. Корреляция между AUCτ и Cmin была более высокой на 14 и 21 сутки (терапия LCP-Tacro) по сравнении с Prograf® (7 сутки); однако величина отличий не была высокой. LCP-Tacro, вводимого на 14 сутки, по сравнению с тем, когда его вводили на 21 сутки, не дало значимых отличий в общей системной экспозиции, минимальных уровнях такролимуса или степени флуктуации или максимального отклонения.
Анализ подгрупп афроамериканцев по сравнению с неафроамерикацами, которым проводили лечение Prograf® или LCP-Tacro, показывает, что существовали статистические отличия в максимальной и системной экспозиции такролимуса, а также в минимальных уровнях такролимуса, указывая на необходимость во внимательности при введении доз популяции афроамериканцев.
Результаты этого исследования демонстрируют, что после перехода с Prograf на LCP-Tacro, терапия LCP-Tacro демонстрирует значимо более высокую системную экспозицию и меньшую степень флуктуации и максимального отклонения такролимуса в стационарном состоянии у стабильных пациентов с трансплантатом почки по сравнению с тем, когда проводят терапию Prograf®. Кроме того, максимальная экспозиция такролимуса является сходной при сравнении различных способов лечения; однако средняя концентрация на протяжении интервала дозирования была выше для LCP-Tacro.
Обсуждение и общие заключения. Главной задачей этого исследования была оценка экспозиции такролимуса (AUCτ) в стационарном состоянии и минимальных уровней (Cmin) у стабильных реципиентов трансплантатов почки, переведенных с Prograf® (такролимус, Astelias Pharma US, Inc.) на LCP-Tacro в трехсерийной схеме исследования.
После коррекции по дозе системная экспозиция такролимуса в стационарном состоянии (AUCτ) и минимальные уровни такролимуса (Cmin) у пациентов с трансплантатами почки были значимо более высокими, когда вводили таблетки LCP-Tacro раз в сутки, по сравнению с лечением капсулами Prograf® два раза в сутки. Системная экспозиция (AUCτ и Cmin) для 2-мг таблеток LCP-Tacro (раз в сутки) в течение периода 24 часов была на ~38% и ~36% выше, чем экспозиция для капсул Prograf® (два раза в сутки). Средняя концентрация лекарственного средства на протяжении интервала дозирования, Cсреднее, была значительно более высокой для лечения LCP-Tacro по сравнению с Prograf®. Также не было статистически значимых отличий в максимальной системной экспозиции (Cmax) такролимуса между терапией Prograf® и введением LCP-Tacro на 14 и 21 сутки. Кроме того, введение Prograf® продемонстрировало значимо более высокую степень флуктуации и максимального отклонения по сравнению с LCP-Tacro. Существовала более высокая корреляция между AUCτ и Cmin на 14 и 21 сутки (терапия LCP-Tacro) по сравнению с Prograf® (7 сутки); однако величина отличий не была высокой. Не было значимых отличий в общей системной экспозиции, минимальных уровнях такролимуса или степени флуктуации и максимальном отклонении при сравнении с LCP-Tacro, вводимым на 14 сутки, по сравнению с введением его на 21 сутки.
Анализ подгрупп афроамериканцев по сравнению с неафроамерикацами, которым проводили лечение Prograf® или LCP-Tacro, показывает, что существовали статистические отличия в максимальной и системной экспозиции такролимуса, а также в минимальных уровнях такролимуса, указывая на необходимость во внимательности при введении доз популяции афроамериканцев.
Результаты этого исследования демонстрируют, что после перехода с Prograf на LCP-Tacro, терапия LCP-Tacro демонстрирует значимо более высокую системную экспозицию и меньшую степень флуктуации и максимального отклонения такролимуса в стационарном состоянии у стабильных пациентов с трансплантатом почки по сравнению с тем, когда проводят терапию Prograf®. Кроме того, максимальная экспозиция такролимуса является сходной при сравнении различных способов лечения; однако средняя концентрация на протяжении интервала дозирования была выше для LCP-Tacro.
Обобщенная статистика скорректированных по дозе фармакокинетических параметров такролимуса у стабильных пациентов и пациенток с трансплантатами печени на 7, 14 и 21 сутки
Среднее значение±SD
Среднее значение±SD
Среднее значение±SD
196,35 (29,88)*
185,34 (30,26)*
202,15 (36,85)*
16,87 (42,07)*
11,80 (32,66)*
12,62 (43,39)*
6,40 (30,76)*
5,91 (37,38)*
6,37 (38,44)*
1,82 (142,58)*
5,32 (55,07)*
5,05 (55,84)*
8,18 (29,88)*
7,72 (30,26)*
8,42 (36,85)*
121,09 (42,10)*
67,02 (59,47)
65,34 (53,48)*
154,73 (46,10)*
87,59 (74,14)*
86,44 (62,35)*
2,63 (29,34)*
2,00 (39,24)*
1,98(32,37)*
N=56 на 21 сутки, N=57 на 7 сутки и на 14 сутки
^ τ=24 часа
7 сутки - Капсулы Prograf® два раза в сутки перорально
14 сутки - Таблетки LCP-Tacro раз в сутки перорально
21 сутки - Таблетки LCP-Tacro раз в сутки перорально
у пациентов и пациенток между 7 сутками и 14 сутками
90% геометрический доверительный интервал с использованием log-преобразованных данных
** Вычислено с использованием геометрических средних значений по формуле:
e((тестируемое лекарственное средство)-(эталонное лекарственное средство))×100%
*** Коэффициент вариаций для log-преобразованного фармакокинетического параметра
14 сутки - Таблетки LCP-Tacro раз в сутки перорально
7 сутки - Капсулы Prograf® два раза в сутки перорально
90% геометрический доверительный интервал с использованием log-преобразованных данных
** Вычислено с использованием геометрических средних значений по формуле:
e((тестируемое лекарственное средство)-(эталонное лекарственное средство))×100%
*** Коэффициент вариаций для log-преобразованного фармакокинетического параметра
21 сутки - Таблетки LCP-Tacro раз в сутки перорально
7 сутки - Капсулы Prograf® два раза в сутки перорально
90% геометрический доверительный интервал с использованием log-преобразованных данных
** Вычислено с использованием геометрических средних значений по формуле:
e((тестируемое лекарственное средство)-(эталонное лекарственное средство))×100%
*** Коэффициент вариаций для log-преобразованного фармакокинетического параметра
21 сутки - Таблетки LCP-Tacro раз в сутки перорально
14 сутки - Таблетки LCP-Tacro раз в сутки перорально
Среднее значение±SD
Среднее значение±SD
Среднее значение±SD
34,99 (43,13)*
45,87 (43,1
46,85 (43,31)*
3,01 (49,16)
2,92 (40,31
2,92 (39,25)*
1,14 (43,57)*
1,46 (50,08)*
1,48 (52,17)*
1,82 (142,58)*
5,32 (55,07)*
5,05 (55,84)*
1,46 (43,13)*
1,91 (43,19)*
1,95 (43,31)*
121,09 (42,10)*
67,02 (59,47)*
65,34
154,73 (46,10
87,59 (74,14
86,44 (62,35)*
2,63 (29,34)*
2,00 (39,24)*
1,98 (32,37)*
^ τ=24 часа
7 сутки - Капсулы Prograf® два раза в сутки перорально
14 сутки - Таблетки LCP-Tacro раз в сутки перорально
21 сутки - Таблетки LCP-Tacro раз в сутки перорально
у пациентов и пациенток между 7 сутками и 14 сутками
* 90% геометрический доверительный интервал с использованием log-преобразованных данных
** Вычислено с использованием геометрических средних значений по формуле:
e((тестируемое лекарственное средство)-(эталонное лекарственное средство))×100%
*** Коэффициент вариаций для log-преобразованного фармакокинетического параметра
14 сутки - Таблетки LCP-Tacro раз в сутки перорально
7 сутки - Капсулы Prograf® два раза в сутки перорально
у пациентов и пациенток между 7 сутками и 21 сутками
* 90% геометрический доверительный интервал с использованием log-преобразованных данных
** Вычислено с использованием геометрических средних значений по формуле:
e((тестируемое лекарственное средство)-(эталонное лекарственное средство))×100%
*** Коэффициент вариаций для log-преобразованного фармакокинетического параметра
21 сутки - Таблетки LCP-Tacro раз в сутки перорально
7 сутки - Капсулы Prograf® два раза в сутки перорально
у пациентов и пациенток между 14 сутками и 21 сутками
* 90% геометрический доверительный интервал с использованием log-преобразованных данных
** Вычислено с использованием геометрических средних значений по формуле:
e((тестируемое лекарственное средство)-(эталонное лекарственное средство))×100%
*** Коэффициент вариаций для log-преобразованного фармакокинетического параметра
21 сутки - Таблетки LCP-Tacro раз в сутки перорально
14 сутки - Таблетки LCP-Tacro раз в сутки перорально
Из результатов для стабильных пациентов с трансплантатами печени получено следующее. Приблизительно 31% повышение биодоступности; снижение, составляющее приблизительно 30, дозы, достигающей биоэквивалентности для AUC(0-24) и C(min24); снижение соотношения Cmax:Cmin и высокая корреляция AUC:Cmin.
Пример 20
Сравнение состава по изобретению (LCP-Tacro) относительно коммерчески доступного состава такролимуса с пролонгированным высвобождением для дозирования один раз в сутки, Advagraf®
Главной задачей этого исследования является определение и сравнение скорости и степени всасывания такролимуса из тестируемого состава 2-мг таблеток такролимуса LCP-Tacro, принимаемых один раз в сутки (q.d.), относительно эталонного Advagraf®, 2×1-мг капсулы (q.d.), в условиях многократных доз натощак.
Advagraf® изготавливается Astellas Pharma GmbH Munich, Germany.
Изложение исследования
1-мг капсулы Advagraf®
Лечение A: 1 2-мг таблетка LCP-Tacro (раз в сутки)
(суточная доза лечения = 2 мг)
Лечение B: 2 1-мг капсулы Advagraf® (раз в сутки)
(суточная доза лечения = 2 мг)
Всего 50 образцов крови (по 4 мл каждый) отбирают в каждый период согласно следующему расписанию:
Сутки 1: 0,00 (до введения дозы), 0,50, 1,00,
Сутки 5, 6, 7, 8 и 9: 0,00 (до введения дозы) и 12,00 часов после введения дозы.
Сутки 10: 0,00 (до введения дозы), 0,50, 1,00, 1,50, 2,00, 3,00, 4,00, 5,00, 6,00, 7,00, 8,00, 9,00, 10,00, 12,00, 14,00, 16,00, 20,00, 24,00, 48,00, 72,00, 96,00 и 120,00 часов после введения дозы.
Образцы отбирают в процессе этого исследования следующим образом:
До исследования: при скрининговом посещении(ях)
Сутки 3 и 8: 0,00 час (до введения дозы)
Конец исследования: после взятия крови при последнем посещении
Посещение для наблюдения после исследования: между сутками 30 и 35 после последней дозы периода II.
От каждого субъекта берут по 457,5 мл крови.
PK: Параметры PK вычисляли с использованием некомпартментного анализа для такролимуса следующим образом:
1 сутки: AUC0-24, Cmax, C24 и Tmax.
10 сутки: AUCτ (τ=24), Cmax, Cmin, Cсреднее, Tmax, t1/2, Kel, % флуктуации, % максимального отклонения, AUCτ/Cmin, Cmax/Cmin и скорость накопления (R).
Статистика: описательную статистику вычисляли для концентраций в крови и для всех параметров PK. Соотношение (тест/эталон) геометрических среднеквадратических значений (LSM) и 90% доверительного интервала (CI) вычисляли для натуральных логарифмических (ln) преобразованных параметров AUC0-24, AUCτ, Cmax, C24, Cmin, Cсреднее и непреобразованных параметров % флуктуации, % максимального отклонения, R, Cmax/Cmin, AUCτ/Cmin, Kel и t1/2. Tmax анализировали с использованием непараметрических способов.
Были получены следующие результаты, демонстрирующие значительно более высокую биодоступность продукта по изобретению и в то же время имеющие профиль, демонстрирующий значительно более пролонгированное всасывание лекарственного средства практически на 50%, значительно более низкую флуктуацию концентраций в ходе введения доз и высокую концентрацию в конце интервала дозирования, в результате чего в действительности достигается эффект при введении раз в сутки, и устраняются любая токсичность или побочные эффекты, связанные с периодами высоких концентраций, до степени, возможной с помощью перорального состава один раз в сутки. Настоящее изобретение относится к составу, который при 2-мг пероральном дозировании утром обеспечивает среднюю концентрацию на протяжении полных 24 часов, находящуюся выше 4 нг/мл (согласно представленным ниже результатам для Cmin, составляющей 4,66 нг/мл), что существенно выше, чем в случае поставляемого на рынок продукта для введения один раз в сутки Advagraf® (Cmin 2,80 нг/мл) при той же вводимой дозировке.
Фармакокинетические параметры такролимуса у здоровых мужчин-европиодов для лечения A на 1 сутки
Фармакокинетические параметры такролимуса у здоровых мужчин-европиодов для лечения B на 1 сутки
Фармакокинетические параметры такролимуса у здоровых мужчин-европиодов
для лечения А на 10 сутки
Лечение A: 1 2-мг таблетка LCP-Tacro (раз в сутки)
Фармакокинетические параметры такролимуса у здоровых мужчин-европиодов
для лечения В на 10 сутки
Сравнение Tmax для такролимуса у здоровых мужчин-европиоидов между 1 и 10 сутками для лечения A и B
Медиана (диапазон)
Медиана (диапазон)
(3,00, 16,00)
(1,00, 12,00)
(-2,00, 2,00)
(1,00, 4,00)
(1,00, 6,00)
(-0,50, 0,00)
** Значение p для сравнения лечения на основе критерия Вилкоксона-Манна-Уитни.
Лечение A: 1 2-мг таблетка LCP-Tacro (раз в сутки)
Лечение B: 2 1-мг капсулы Advagraf® (раз в сутки)
Обобщенная статистика и сравнение фармакокинетических параметров такролимуса у здоровых мужчин-европиоидов между лечением A и B на 1 сутки
48,19 (26,63)*
32,92 (27,67)*
1,73(30,65)*
0,91 (38,37)*
3,46 (28,79)*
3,31 (28,78)*
** Медиана (диапазон)
Лечение A: 1 2-мг таблетка LCP-Tacro (раз в сутки)
Лечение B: 2 1-мг капсулы Advagraf® (раз в сутки)
Обобщенная статистика и сравнение фармакокинетических параметров такролимуса у здоровых мужчин-европиоидов между лечением A и B на 1 сутки
2. Вычислено с использованием геометрических средних значений по формуле: e((тестируемое лекарственное средство)-(эталонное лекарственное средство))×100%
3. Коэффициент вариаций у каждого субъекта для log-преобразованного фармакокинетического параметра
Тест: Лечение A: 1 2-мг таблетка LCP-Tacro (раз в сутки)
Эталон: Лечение B: 2 1-мг капсулы Advagraf® (раз в сутки)
Обобщенная статистика и сравнение фармакокинетических параметров такролимуса у здоровых мужчин-европиоидов, между лечением A и B на 10 сутки
133,99 (34,73)*
89,86 (29,99)*
7,93 (34,51)*
6,71 (29,21)*
4,35 (36,60)*
2,64 (34,79)*
6,03 (42,11)*
2,16 (50,40)*
5,58 (34,73)*
3,74 (29,99)*
60,92 (35,48)*
106,46 (25,58)*
78,16 (44,03)*
150,98 (30,40)*
2,77 (22,42)*
2,70 (30,85)*
1,82 (20,29)*
2,54 (18,64)*
Лечение A: 1 2-мг таблетка LCP-Tacro (раз в сутки)
Лечение B: 2 1-мг капсулы Advagraf® (раз в сутки)
Обобщенная статистика и сравнение фармакокинетических параметров такролимуса у здоровых мужчин-европиоидов между лечением A и B на 10 сутки
* 90% геометрический интервал достоверности с использованием log-преобразованных данных
** Вычислено с использованием геометрических средних значений по формуле: e((тестируемое лекарственное средство)-(эталонное лекарственное средство))×100%
*** Коэффициент вариаций у каждого субъекта для log-преобразованного фармакокинетического параметра
Лечение A: 1 2-мг таблетка LCP-Tacro (раз в сутки)
Лечение B: 2 1-мг капсулы Advagraf® (раз в сутки)
Пример 21
В таблице представлено растворение предпочтительных вариантов осуществления изобретения, имеющих состав, как показано выше в примере 20, и растворение коммерческого продукта Advagraf®, используемого для сравнения в примере 20.
Высвобождение в процентах измеряли в способе тестирования растворения способом USP II (лопасть) в среде, доведенной до pH 4,5 и содержащей 0,005% гидроксилпропилцеллюлозы, и при вращении 50 об/мин. Растворение представлено на фиг.4.
Из растворения следует, что состав с пролонгированным высвобождением в соответствии с настоящим изобретением обеспечивает значительно более длительный и более пролонгированный профиль с в значительной степени более низким высвобождением первоначально, например, демонстрирующим менее 25% высвобождения в момент времени 5 часов, несмотря на то, что высвобождение в 3 часа составляет по меньшей мере 10%. Кроме того, профиль кривой в соответствии с настоящим изобретением имеет по существу высвобождение нулевого порядка и высоко пролонгированное высвобождение. Последнее из них отчетливо демонстрируется менее чем 50% высвобождением в момент времени 12 часов, и менее чем 62% высвобождением в момент времени 15 часов. Из примера 20 следует, что фармакокинетические параметры по существу улучшены в составе с пролонгированным высвобождением по изобретению по сравнению с коммерчески доступным продуктом Advagraf®.
Кроме того, состав с пролонгированным высвобождением в соответствии с настоящим изобретением может быть предоставлен в таблетке значительно меньшего размера по сравнению с Advagraf®, как очевидно из приведенной ниже таблицы.
Пример 22
Такролимус с пролонгированным высвобождения у пациентов с трансплантатами почек de novo
Пациентов с трансплантатами почки de novo разделяли случайным образом (соотношение 1:1) в пределах 12 часов после трансплантации (0 сутки исследования) для введения либо: 1) таблеток LCP-Tacro (как приведено в примере 20, и растворение продемонстрированно в примере 21) перорально один раз в сутки утром с интервалом 24±1 часов между дозами, начиная с 0,14 мг/кг (начальная суточная доза для пациентов-афроамериканцев составляла 0,17 мг/кг), или 2) капсул Prograf в двух равно разделенных дозах, начиная с 0,1 мг/кг каждые 12 часов (общая суточная доза 0,2 мг/кг), как рекомендовано в настоящее время в U.S. Prescribing Information (Astellas Pharma US, April 2006, NDA № 050708). Оценку фармакокинетики (PK) в течение 24 часов проводили на 1, 7 и 14 сутки исследования. 1 сутки исследования определяли как сутки, на которые вводили первую дозу лекарственного средства утром (AM), что должно было произойти в пределах 48 часов после трансплантации. Результаты представлены в таблице 22.
9,52 (59,11)
20,83 (47,41)*
23,11 (63,95)*
20,97 (52,99)*
25,65 (48,51)*
19,15 (32,04)*
4,90 (64,20)*
8,27 (47,18)*
8,92 (67,90)*
9,39 (49,92)*
9,51 (40,84)*
7,45 (38,57)*
** Медиана (диапазон)
AUCR представляет собой соотношение для накопления AUCτ на представленные сутки относительно 1 суток
AUC0-τ для LCP-Tacro является значительно более низкой, чем для Prograf® в течение первых суток после трансплантации (нескорректированная по дозе LCP-Tacro AUC0-τ на 1 сутки в данном случае составила 131,11±76,78 по сравнению с 253,45±106,58 (нг·ч/мл)). Однако для снижения исходной сверхиммуносупрессии это считается преимуществом, особенно если пациентам проводят индуцирующую терапию и терапию кортикостероидами. В соответствии с настоящим изобретением, целевой минимальный уровень 5-10 нг/мл (способ иммуноанализа) считают оптимальным лечением по сравнению с составом с пролонгированным высвобождением по изобретению. Предпочтительной является комбинация с режимом микофенолята с 1 грамом два раза в сутки, вводимого в форме микофенолята мофетила (CellCept®) NDA № 050722 и 050723 или в лекарственной форме с эквивалентным количеством микофеноловой кислоты, такой как Myfortic® NDA № 050791 (включая ее генерический препарат).
для измерения минимальных уровней такролимуса
Из представленной ниже таблицы (таблица 23) следует, что в соответствии с настоящим изобретением более высокая доля пациентов достигает целевой минимальной концентрации 5-10 нг/мл.
Также в таблице показано, что приблизительно две трети пациентов, которым вводили LCP-Tacro, имеют диапазон в пределах 5-7 нг/мл после первой дозы LCP-Tacro, и к 3 суткам свыше 80% пациентов, которым вводили LCP-Tacro, имели уровень выше минимального уровня 5 нг/мл.
Доля пациентов в пределах и за пределами целевой концентрации
5-10 нг/мл (№ (%))
Пример 23
Клинические исследования с составом с пролонгированным высвобождением в соответствии с настоящим изобретением
Таблица A
Таблица A представляет собой перечень исходных клинических исследований с составом с пролонгированным высвобождением в соответствии с настоящим изобретением. Перечень включает улучшенные фармакокинетические параметры, достигнутые с помощью состава с пролонигированным высвобождением в соответствии с настоящим изобретением, заявленного в настоящем документе.
Профили в плазме в исследованиях, приведенных в таблицах A и B, представлены на фиг.3.
Сравнительное исследование PK с одной дозой (исследование 002)
Исследование 002, сравнительное фармакокинетическое исследование с однократной дозой 4 мг LCP-Tacro (таблетки 2×2 мг) по сравнению с Prograf® 4 мг (капсулы 4×1 мг) продемонстрировало сравнимую AUC для обоих продуктов при введении в условиях натощак. Однако они отличаются скоростью всасывания при введении натощак, которое является более медленным и более непрерывным для тестируемого состава LCP-Tacro. Более длительное Tmax для таблеток LCP-Tacro (тест; HPMC) по сравнению с капсулами Prograf® (такролимус; эталон) (8,55 ч по сравнению с 1,63 ч) в сочетании с более низкой Cmax (10,05 нг/мл по сравнению с 32,75 нг/мл) и более низкой AUC(0->24) (122,4 нг·ч/мл по сравнению с 157,95 нг·ч/мл) свидетельствуют в поддержку дозирования таблеток LCP-Tacro один раз в сутки по сравнению с капсулами Prograf® (такролимус).
Исследование 003
Исследование 003 было спланировано как повторное сцинтиграфическое исследование всасывания у 8 здоровых добровольцев для оценки времени прохождения, фармакокинетического профиля и области высвобождения таблеток 2-мг LCP-Tacro, радиоактивно меченных максимум 1 Мбк 153Sm. В этом исследование профиль пролонгированного высвобождения мог быть продемонстрирован с помощью Tmax ~6,7 часа. LCP-Tacro был хорошо переносим. В целом, это исследование продемонстрировало профиль пролонгированного высвобождения in vivo таблеток LCP-Tacro с высвобождением во всех частях толстого кишечника и всасыванием такролимуса из дистальных частей желудочно-кишечного тракта. Сцинтиграфические изображения представлены на фиг.2, где изображения получены через 0,02, 0,53, 4,32, 4,57, 11,23 и 23,32 часа после введения дозы соответственно.
Сравнительное исследование PK в стационарном состоянии с многократными дозами (исследование 004)
Исследование 004 представляет собой сравнительное фармакокинетическое исследование в стационарном состоянии с многократными дозами LCP-Tacro 2 мг раз в сутки по сравнению с Prograf® 2×1 мг два раза в сутки у 14 здоровых добровольцев. Это исследование отчетливо продемонстрировало профиль LCP-Tacro один раз в сутки по сравнению с Prograf® два раза в сутки. Кроме того, это исследование продемонстрировало лучшую биодоступность таблеток LCP-Tacro, вводимых один раз в сутки, по сравнению с капсулами Prograf®, вводимыми два раза в сутки. При введении 2-мг таблеток LCP-Tacro (один раз в сутки) и 1-мг капсул Prograt® (два раза в сутки) в течение 10 последовательных суток, не наблюдали значимых отличий в концентрациях утром до введения доз на 7, 8, 9 и 10 сутки, таким образом, стационарное состояние поддерживалось с 7 суток. В стационарном состоянии системная экспозиция для 2-мг таблеток LCP-Tacro (один раз в сутки) в течение 24 часов была приблизительно на 50% выше, чем системная экспозиция 1-мг капсул Prograf (два раза в сутки). Время до достижения максимальной концентрации между однократной и многократными дозами 2 мг LCP-Tacro один раз в сутки и 2×1 мг Prograf® два раза в сутки было сходным для обоих способов лечения. Как и ожидалось вследствие профиля пролонгированного высвобождения по изобретению, 2-мг таблетки LCP-Tacro (один раз в сутки) имели более высокие значения Cmin и более низкую степень флуктуации по сравнению с 1-мг капсулами Prograf® с немедленным высвобождением (два раза в сутки). Не наблюдали значимых отличий в Cmax между 2 способами лечения, как показано ниже в таблице 25.
Обобщение фармакокинетических параметров исследования 004
Арифметическое среднее значение±SD
† n=12
Перечень дополнительных исследований
Сравнительное исследование PK многократных доз в стационарном состоянии (исследование 1012)
В этом исследовании оценивали и сравнивали биодоступность такролимуса из тестируемого состава 2-мг таблеток LCP-Tacro, принимаемых один раз в сутки (q.d.) в группе A по сравнению с эталонными 1-мг капсулами Prograf, принимаемых два раза в сутки (b.i.d.) в группе B в условиях многократных доз натощак. Исследуемая выборка состояла из 30 здоровых добровольцев, которым проводили исследуемое лечение в течение 10 суток с последующим двухнедельным вымыванием и последующим перекрестным исследованием исследуемых групп. Для этого исследования проводили оценку двадцати пяти пациентов. Фармакокинетика такролимуса на 10 сутки обобщенно представлена в таблицах 26-27 ниже.
Фармакокинетические параметры для такролимуса на 10 сутки (исследование 1012)
Оценка относительной биодоступности для такролимуса на 10 сутки (исследование 1012)
Обобщение фармакокинетических параметров исследования 1013 (пропорциональные дозы, NHV, n=25)
1×5 мг
1×5 мг+1×2 мг
2×5 мг
Исследование 1014 (суточные колебания, NHV)
В этом исследовании изучали фармакокинетический профиль такролимуса после введения 2-мг LCP-Tacro утром относительно введения вечером у 26 здоровых добровольцев, мужчин и женщин, в условиях натощак. Исследование состояло из двух периодов по восемь суток, разделенных по меньшей мере двухнедельным периодом вымывания между лечениями. Средние фармакокинетические параметры обобщенно представлены в таблице 29 ниже.
Обобщение фармакокинетических параметров исследования 1014
Исследование 1014 - Суточные колебания, NHV
Исследование 1015
В этом исследовании сравнивали скорость и степень всасывания такролимуса после 1-мг таблетки LCP-Tacro по сравнению с 1-мг капсулами Prograf® (такролимус) в условиях натощак у 17 нормальных здоровых мужчин и женщин. Исследование состояло из двух периодов по восемь суток, разделенных по меньшей мере двухнедельным периодом вымывания между лечениями. Фармакокинетические результаты обобщенно представлены в таблице ниже. Более длительное Tmax для таблеток LCP-Tacro (8,78 ч по сравнению с 1,39 ч) в сочетании с более низкой Cmax (2,54 нг/мл по сравнению с 7,04 нг/мл) и более низкой AUC(0-∞) (71,82 нг·ч/мл по сравнению с 50,18 нг·ч/мл) свидетельствуют в поддержку дозирования один раз в сутки для таблеток LCP-Tacro по сравнению с капсулами Prograf® (такролимус).
Экспозиция такролимуса после таблеток LCP-Tacro является значительно более высокой, чем после капсул Prograf со сниженной флуктцацией максимального/минимального значения и отсроченным Tmax в NHV. Эти результаты согласуются с другими данными PK, полученными для состава с пролонгированным высвобождением в соответствии с настоящим изобретением.
Время высвобождения для лекарственных форм с пролонгированным высвобождением по изобретению, используемых в клинических испытаниях, описанных в настоящем документе, обеспечивает профиль пролонгированного высвобождения, где менее 63,5% высвобождается в пределах 15 часов. Соответствующий профиль высвобождения в связи с этим представлен на фиг.1 при тестировании согласно тесту растворения USP II (лопасть) в среде, имеющей pH 4,5 и содержащей 0,005% гидроксипропилцеллюлозы и при вращении 50 об/мин.
Пример 24
Проводили двухстороннее перекрестное открытое исследование биоэквивалентности фазы 1 с многократными дозами для сравнения фармакокинетики (Cmax, C24 и AUCτ) и безопасности таблеток LCP-Tacro по сравнению с капсулами Advagraf® в условиях стационарного состояния натощак.
Двадцать здоровых мужчин-добровольцев случайным образом разделяли для введения либо одной 2-мг таблетки LCP-Tacro, либо двух 1-мг капсул Advagraf® каждые сутки в течение 10 суток. После двухнедельного периода вымывания каждому субъекту проводили альтернативное лечение. Профиль PK после 10 суток каждого из лечений проиллюстрирован на фиг.7, и на ней продемонстрировано, что таблетки LCP-Tacro обеспечивают приблизительно на 50% большую биодоступность такролимуса, чем сравнимая доза Advagraf®. Профиль PK таблеток LCP-Tacro также свидетельствует в поддержку введения один раз в сутки.
Патенты, патентные заявки, публикации, описания продуктов и протоколы, которые цитированы на протяжении этой заявки, включены в настоящий документ в качестве ссылок в полном объеме.
Варианты осуществления, проиллюстрированные и рассмотренные в этой заявке, предназначены только для указания специалистам в данной области на лучший способ, известный авторам изобретения, получения и применения изобретения. Ничто в этом описании не следует истолковывать как ограничение объема настоящего изобретения. Возможны модификации и изменения описанных выше вариантов осуществления изобретения без отклонения от изобретения, как будет понятно специалистам в данной области ввиду представленных выше указаний. Таким образом, понятно, что в объеме формулы изобретения и ее эквивалентов изобретение можно осуществлять на практике иначе, чем конкретно описано.

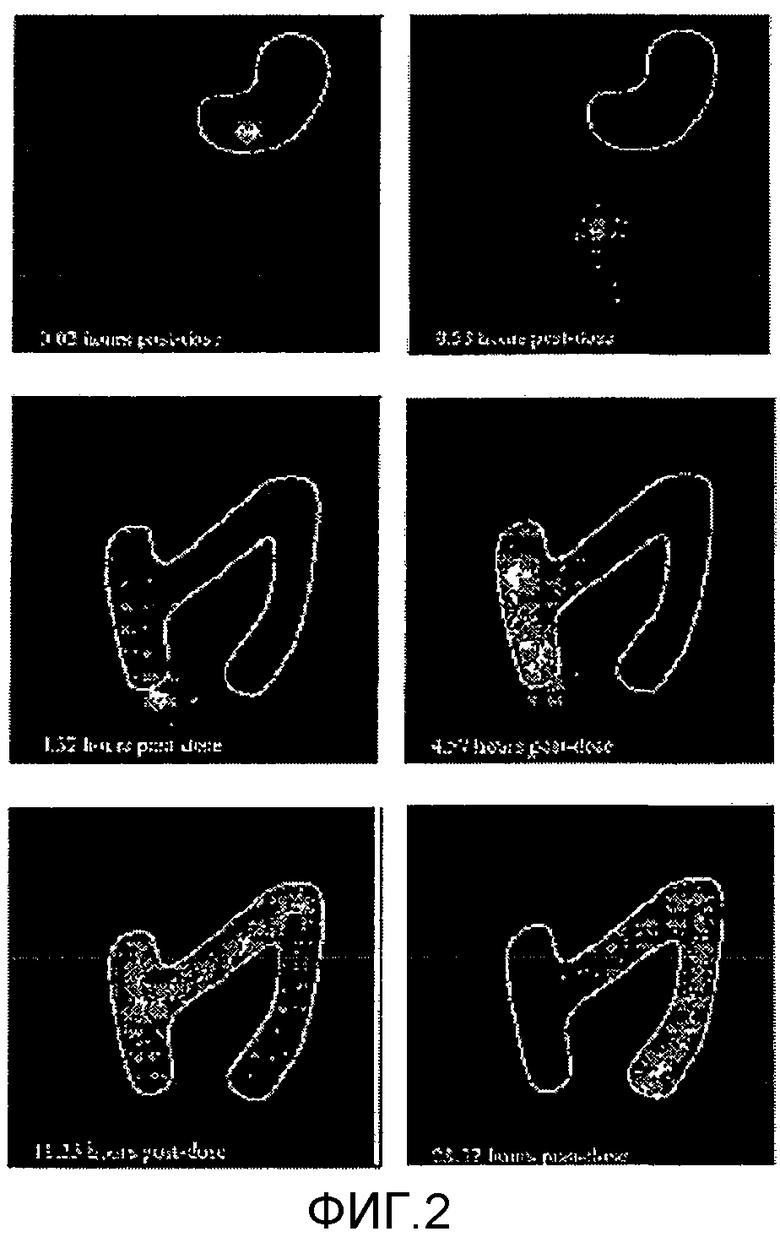
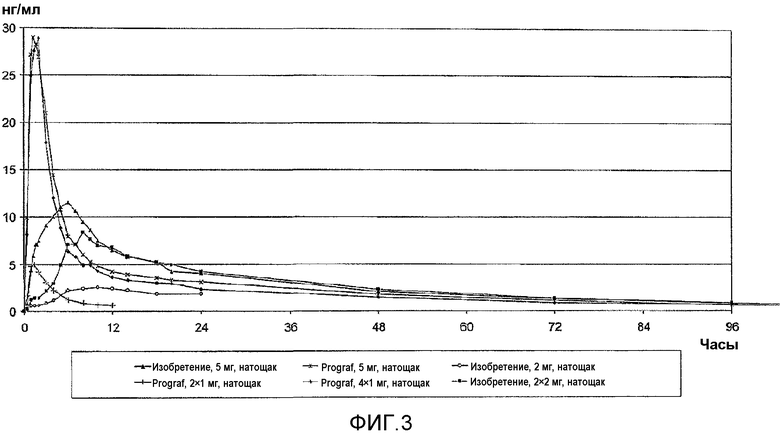
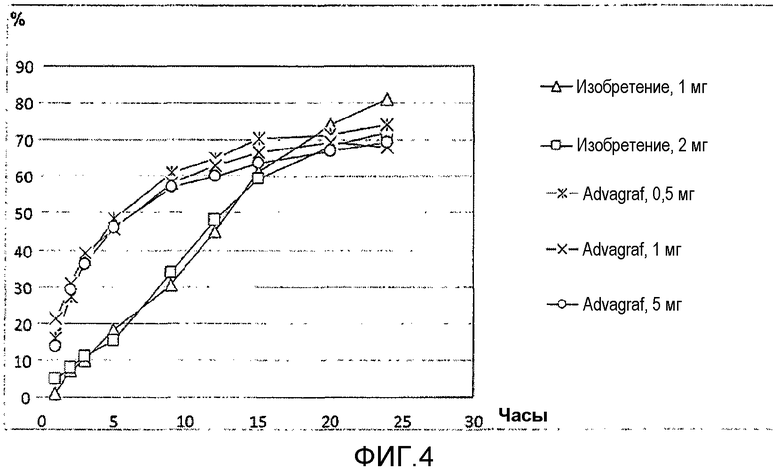
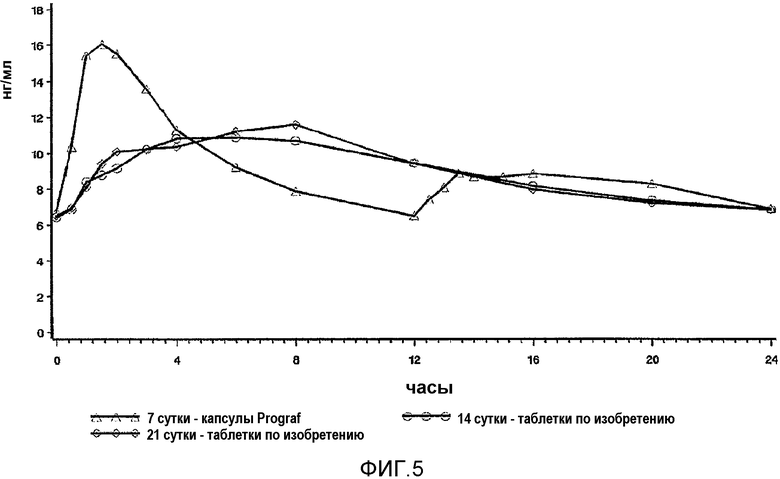
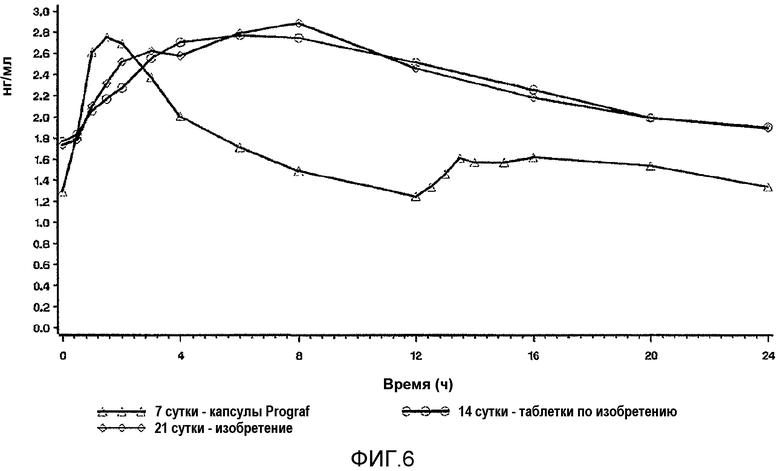
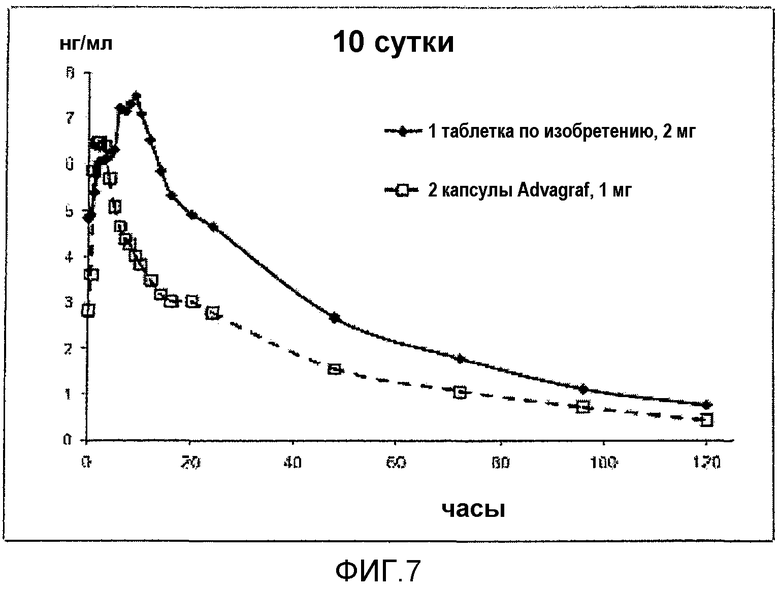
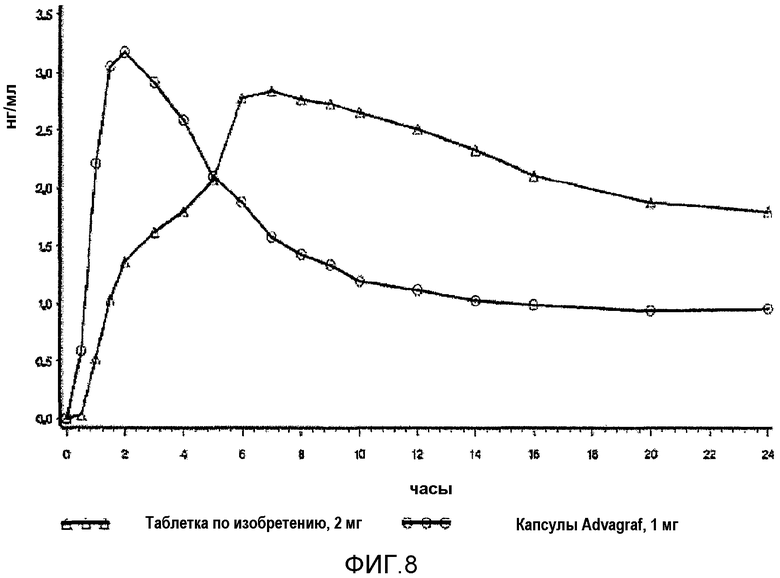
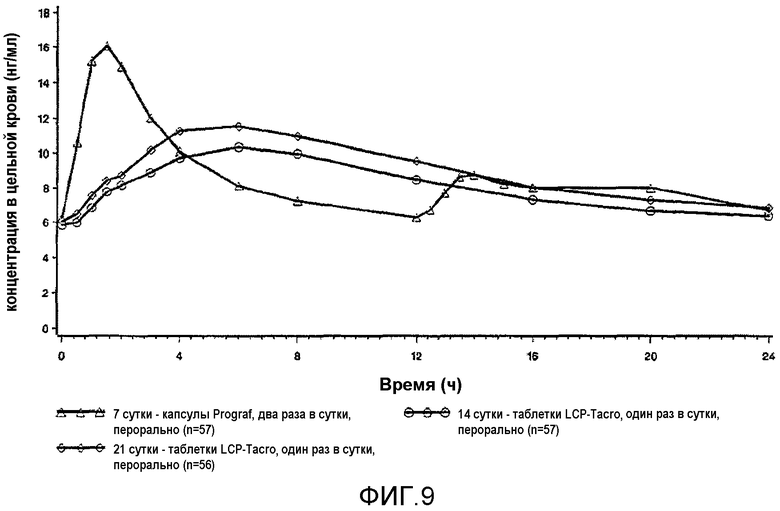
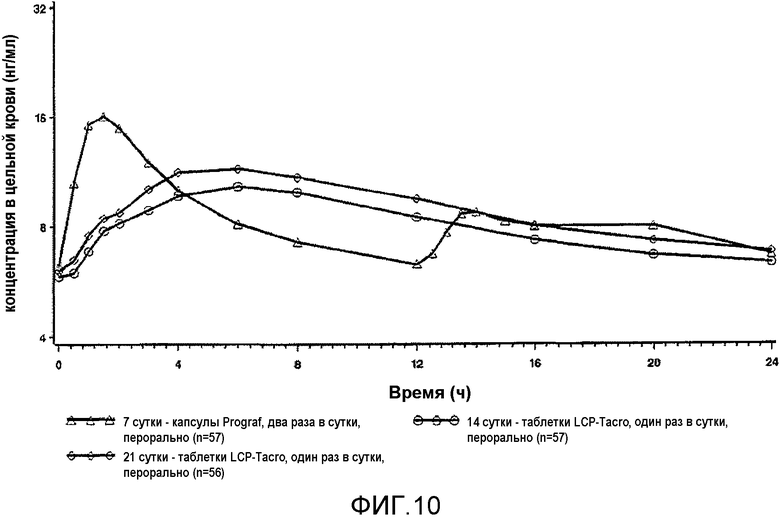
Описывается способ обеспечения иммуносупрессивного лечения пациента в режиме один раз в сутки, предпочтительно пациента с трансплантатом почки или печени, путем введения пероральной таблетки с пролонгированным высвобождением. Таблетка включает от 0,1 до 15 мг такролимуса, гидроксипропилметилцеллюлозу, моногидрат лактозы, полиэтиленгликоль 6000, полоксамер 188, стеарат магния, винную кислоту, бутилированный гидрокситолуол и диметикон 350. Таблетка такролимуса высвобождает активное вещество с линейным профилем высвобождения нулевого порядка от момента времени 8 часов до момента времени 15 часов. Также она обеспечивает улучшенные фармакокинетические параметры, включая сниженные максимальные концентрации, несмотря на повышенную биодоступность, продленные периоды времени максимальной концентрации и более высокие минимальные концентрации по сравнению с традиционными лекарственными формами такролимуса. 9 н. и 9 з.п. ф-лы, 10 ил., 30 табл., 24 пр.
1. Способ обеспечения иммуносупрессивного лечения пациента, нуждающегося в этом, в режиме один раз в сутки путем введения пероральной таблетки с пролонгированным высвобождением, включающей в качестве активного вещества от 0,1 до 15 мг такролимуса в гидрофильном или смешивающемся с водой носителе, где таблетка i) высвобождает активное вещество с профилем высвобождения по существу нулевого порядка в течение пролонгированного периода времени, определяемого по высвобождению от момента времени 8 часов до момента времени 15 часов при тестировании с помощью теста растворения USP II (лопасть) или теста растворения USP I (корзина) в среде, имеющей pH 4,5 и содержащей 0,005% гидроксипропилцеллюлозы, и при вращении 50 об/мин, причем профиль высвобождения по существу нулевого порядка определяют как линейный профиль высвобождения с отклонением не более ±15%; и ii) таблетка включает композицию, состоящую из активного вещества и приблизительно 40,5% масс./масс. гидроксипропилметилцеллюлозы (НРМС), приблизительно 26,9% масс./масс. моногидрата лактозы, приблизительно 22,1% масс./масс. полиэтиленгликоля 6000, приблизительно 9,5% масс./масс. полоксамера 188, приблизительно 1% масс./масс. стеарата магния, менее чем приблизительно 0,1% масс./масс. винной кислоты, менее чем приблизительно 0,01% масс./масс. бутилированного гидрокситолуола и менее чем приблизительно 0,01% масс./масс. диметикона 350, где проценты рассчитаны от общего количества неактивных веществ в композиции.
2. Способ по п.1, где таблетка высвобождает 40% масс./масс. активного вещества в пределах от 10 до 14 часов, при тестировании in vitro в соответствии с тестом растворения USP II (лопасть) или тестом растворения USP I (корзина) в среде, имеющей pH 4,5 и содержащей 0,005% гидроксипропилцеллюлозы, и при вращении 50 об/мин.
3. Способ по п.1, где биодоступность не зависит от времени суток дозирования.
4. Способ по п.1, где высвобождение после перорального введения субъекту частично происходит в толстом кишечнике, такое как высвобождение в одной или нескольких областях восходящей толстой кишки, поперечной толстой кишки и нисходящей толстой кишки.
5. Способ по п.1, где при введении лекарственной формы один раз в сутки в стационарном состоянии здоровому субъекту или пациенту отклонение концентраций в крови для общего и/или свободного такролимуса, измеренное как (Cmax-Cmin)/Cmin, меньше, чем отклонение, наблюдаемое при введении лекарственной формы Prograf® или биоэквивалентной композиции такролимуса с немедленным высвобождением в режиме два раза в сутки и при определении в сходных условиях, и при введении в сходных молекулярных суточных дозировках такролимуса.
6. Способ по п.1, где при введении таблетки один раз в сутки в стационарном состоянии здоровому субъекту или пациенту флуктуация концентраций в крови для общего и/или свободного такролимуса, измеренная как (Cmax-Cmin)/Ссредняя, меньше чем флуктуация, наблюдаемая при введении лекарственной формы Prograf® или биоэквивалентной композиции такролимуса с немедленным высвобождением в режиме два раза в сутки и при определении в сходных условиях, и при введении в сходных молекулярных суточных дозировках такролимуса.
7. Способ первоначального лечения пациента с трансплантатом печени de novo такролимусом, включающий введение пероральной таблетки такролимуса один раз в сутки, где i) пероральная таблетка обеспечивает системную экспозицию на 1 сутки, которая составляет по меньшей мере 50% от экспозиции, наблюдаемой в 1 сутки после введения той же суточной дозы, вводимой два раза в сутки в качестве пероральной лекарственной формы с немедленным высвобождением; и ii) таблетка является таблеткой с пролонгированным высвобождением, включающей в качестве активного вещества от 0,1 до 15 мг такролимуса в гидрофильном или смешивающемся с водой носителе, где таблетка i) высвобождает активное вещество с профилем высвобождения по существу нулевого порядка в течение пролонгированного периода времени, определяемого по высвобождению от момента времени 8 часов до момента времени 15 часов при тестировании с помощью теста растворения USP II (лопасть) или теста растворения USP I (корзина) в среде, имеющей pH 4,5 и содержащей 0,005% гидроксипропилцеллюлозы, и при вращении 50 об/мин, причем профиль высвобождения по существу нулевого порядка определяют как линейный профиль высвобождения с отклонением не более ±15%; и ii) таблетка включает композицию, состоящую из активного вещества и приблизительно 40,5% масс./масс. гидроксипропилметилцеллюлозы (НРМС), приблизительно 26,9% масс./масс. моногидрата лактозы, приблизительно 22,1% масс./масс. полиэтиленгликоля 6000, приблизительно 9,5% масс./масс. полоксамера 188, приблизительно 1% масс./масс. стеарата магния, менее чем приблизительно 0,1% масс./масс. винной кислоты, менее чем приблизительно 0,01% масс./масс. бутилированного гидрокситолуола и менее чем приблизительно 0,01% масс./масс. диметикона 350, где проценты рассчитаны от общего количества неактивных веществ в композиции.
8. Способ первоначального лечения пациента с трансплантатом почки de novo такролимусом, включающий введение пероральной таблетки такролимуса один раз в сутки, где i) таблетка обеспечивает системную экспозицию на 1 сутки, которая составляет по меньшей мере 70% от экспозиции, наблюдаемой на 1 сутки после введения той же суточной дозы, вводимой два раза в сутки в качестве пероральной лекарственной формы с немедленным высвобождением; и ii) таблетка является таблеткой с пролонгированным высвобождением, включающей в качестве активного вещества от 0,1 до 15 мг такролимуса в гидрофильном или смешивающемся с водой носителе, где таблетка i) высвобождает активное вещество с профилем высвобождения по существу нулевого порядка в течение пролонгированного периода времени, определяемого по высвобождению от момента времени 8 часов до момента времени 15 часов при тестировании с помощью теста растворения USP II (лопасть) или теста растворения USP I (корзина) в среде, имеющей pH 4,5 и содержащей 0,005% гидроксипропилцеллюлозы, и при вращении 50 об/мин, причем профиль высвобождения по существу нулевого порядка определяют как линейный профиль высвобождения с отклонением не более ±15%; и ii) таблетка включает композицию, состоящую из активного вещества и приблизительно 40,5% масс./масс. гидроксипропилметилцеллюлозы (НРМС), приблизительно 26,9% масс./масс. моногидрата лактозы, приблизительно 22,1% масс./масс. полиэтиленгликоля 6000, приблизительно 9,5% масс./масс. полоксамера 188, приблизительно 1% масс./масс. стеарата магния, менее чем приблизительно 0,1% масс./масс. винной кислоты, менее чем приблизительно 0,01% масс./масс. бутилированного гидрокситолуола и менее чем приблизительно 0,01% масс./масс. диметикона 350, где проценты рассчитаны от общего количества неактивных веществ в композиции.
9. Способ первоначального лечения пациента с трансплантатом печени de novo такролимусом, включающий введение пероральной таблетки такролимуса один раз в сутки, где i) пероральная таблетка обеспечивает системную экспозицию на 1 сутки, которая составляет по меньшей мере 100% от экспозиции, наблюдаемой на 1 сутки после введения той же суточной дозы, вводимой в качестве пероральной лекарственной формы с пролонгированным высвобождением, высвобождающей более 30% такролимуса в пределах 5 часов; и ii) таблетка является таблеткой с пролонгированным высвобождением, включающей в качестве активного вещества от 0,1 до 15 мг такролимуса в гидрофильном или смешивающемся с водой носителе, где таблетка i) высвобождает активное вещество с профилем высвобождения по существу нулевого порядка в течение пролонгированного периода времени, определяемого по высвобождению от момента времени 8 часов до момента времени 15 часов при тестировании с помощью теста растворения USP II (лопасть) или теста растворения USP I (корзина) в среде, имеющей pH 4,5 и содержащей 0,005% гидроксипропилцеллюлозы, и при вращении 50 об/мин, причем профиль высвобождения по существу нулевого порядка определяют как линейный профиль высвобождения с отклонением не более ±15%; и ii) таблетка включает композицию, состоящую из активного вещества и приблизительно 40,5% масс./масс. гидроксипропилметилцеллюлозы (НРМС), приблизительно 26,9% масс./масс. моногидрата лактозы, приблизительно 22,1% масс./масс. полиэтиленгликоля 6000, приблизительно 9,5% масс./масс. полоксамера 188, приблизительно 1% масс./масс. стеарата магния, менее чем приблизительно 0,1% масс./масс. винной кислоты, менее чем приблизительно 0,01% масс./масс. бутилированного гидрокситолуола и менее чем приблизительно 0,01% масс./масс. диметикона 350, где проценты рассчитаны от общего количества неактивных веществ в композиции.
10. Способ первоначального лечения пациента с трансплантатом почки de novo такролимусом, включающий введение пероральной таблетки такролимуса один раз в сутки, где i) таблетка обеспечивает системную экспозицию на 1 сутки, которая составляет по меньшей мере 100% от экспозиции, наблюдаемой на 1 сутки после введения той же суточной дозы, вводимой в качестве пероральной лекарственной формы с пролонгированным высвобождением, высвобождающей более 30% такролимуса в пределах 5 часов; и ii) таблетка является таблеткой с пролонгированным высвобождением, включающей в качестве активного вещества от 0,1 до 15 мг такролимуса в гидрофильном или смешивающемся с водой носителе, где таблетка i) высвобождает активное вещество с профилем высвобождения по существу нулевого порядка в течение пролонгированного периода времени, определяемого по высвобождению от момента времени 8 часов до момента времени 15 часов при тестировании с помощью теста растворения USP II (лопасть) или теста растворения USP I (корзина) в среде, имеющей pH 4,5 и содержащей 0,005% гидроксипропилцеллюлозы, и при вращении 50 об/мин, причем профиль высвобождения по существу нулевого порядка определяют как линейный профиль высвобождения с отклонением не более ±15%; и ii) таблетка включает композицию, состоящую из активного вещества и приблизительно 40,5% масс./масс. гидроксипропилметилцеллюлозы (НРМС), приблизительно 26,9% масс./масс. моногидрата лактозы, приблизительно 22,1% масс./масс. полиэтиленгликоля 6000, приблизительно 9,5% масс./масс. полоксамера 188, приблизительно 1% масс./масс. стеарата магния, менее чем приблизительно 0,1% масс./масс. винной кислоты, менее чем приблизительно 0,01% масс./масс. бутилированного гидрокситолуола и менее чем приблизительно 0,01% масс./масс. диметикона 350, где проценты рассчитаны от общего количества неактивных веществ в композиции.
11. Способ предотвращения сверхиммуносупрессии у пациента с трансплантатом почки de novo путем введения такролимуса в пределах 48 часов после трансплантации в качестве таблетки с замедленным высвобождением один раз в сутки с начальной дозой от 0,13 до 0,15 мг/кг такролимуса и целевой Cmin 5-10 нг/мл, причем таблетка с пролонгированным высвобождением обеспечивает медианное Tmax на 1 сутки лечения от 8 до 14 часов и таблетка с пролонгированным высвобождением включает в качестве активного вещества от 0,1 до 15 мг такролимуса в гидрофильном или смешивающемся с водой носителе, где таблетка i) высвобождает активное вещество с профилем высвобождения по существу нулевого порядка в течение пролонгированного периода времени, определяемого по высвобождению от момента времени 8 часов до момента времени 15 часов при тестировании с помощью теста растворения USP II (лопасть) или теста растворения USP I (корзина) в среде, имеющей pH 4,5 и содержащей 0,005% гидроксипропилцеллюлозы, и при вращении 50 об/мин, причем профиль высвобождения по существу нулевого порядка определяют как линейный профиль высвобождения с отклонением не более ±15%; и ii) таблетка включает композицию, состоящую из активного вещества и приблизительно 40,5% масс./масс. гидроксипропилметилцеллюлозы (НРМС), приблизительно 26,9% масс./масс. моногидрата лактозы, приблизительно 22,1% масс./масс. полиэтиленгликоля 6000, приблизительно 9,5% масс./масс. полоксамера 188, приблизительно 1% масс./масс. стеарата магния, менее чем приблизительно 0,1% масс./масс. винной кислоты, менее чем приблизительно 0,01% масс./масс. бутилированного гидрокситолуола и менее чем приблизительно 0,01% масс./масс. диметикона 350, где проценты рассчитаны от общего количества неактивных веществ в композиции.
12. Применение таблетки с пролонгированным высвобождением, включающей в качестве активного вещества от 0,1 до 15 мг такролимуса в гидрофильном или смешивающемся с водой носителе, где таблетка i) высвобождает активное вещество с профилем высвобождения по существу нулевого порядка в течение пролонгированного периода времени, определяемого по высвобождению от момента времени 8 часов до момента времени 15 часов при тестировании с помощью теста растворения USP II (лопасть) или теста растворения USP I (корзина) в среде, имеющей рН 4,5 и содержащей 0,005% гидроксипропилцеллюлозы, и при вращении 50 об/мин, причем профиль высвобождения по существу нулевого порядка определяют как линейный профиль высвобождения с отклонением не более ±15%; и ii) таблетка включает композицию, состоящую из активного вещества и приблизительно 40,5% масс./масс. гидроксипропилметилцеллюлозы (НРМС), приблизительно 26,9% масс./масс. моногидрата лактозы, приблизительно 22,1% масс./масс. полиэтиленгликоля 6000, приблизительно 9,5% масс./масс. полоксамера 188, приблизительно 1% масс./масс. стеарата магния, менее чем приблизительно 0,1% масс./масс. винной кислоты, менее чем приблизительно 0,01% масс./масс. бутилированного гидрокситолуола и менее чем приблизительно 0,01% масс./масс. диметикона 350, где проценты рассчитаны от общего количества неактивных веществ в композиции, для обеспечения иммуносупрессивного лечения такролимусом пациента, нуждающегося в этом, путем введения таблетки с пролонгированным высвобождением один раз в сутки.
13. Применение по п.12, где режим дозирования один раз в сутки представляет собой режим дозирования либо на ночь, либо вечером.
14. Применение по п.12, где режим дозирования один раз в сутки представляет собой режим дозирования утром.
15. Применение таблетки с пролонгированным высвобождением, включающей в качестве активного вещества от 0,1 до 15 мг такролимуса в гидрофильном или смешивающемся с водой носителе, где таблетка i) высвобождает активное вещество с профилем высвобождения по существу нулевого порядка в течение пролонгированного периода времени, определяемого по высвобождению от момента времени 8 часов до момента времени 15 часов при тестировании с помощью теста растворения USP II (лопасть) или теста растворения USP I (корзина) в среде, имеющей pH 4,5 и содержащей 0,005% гидроксипропилцеллюлозы, и при вращении 50 об/мин, причем профиль высвобождения по существу нулевого порядка определяют как линейный профиль высвобождения с отклонением не более ±15%; и ii) таблетка включает композицию, состоящую из активного вещества и приблизительно 40,5% масс./масс. гидроксипропилметилцеллюлозы (НРМС), приблизительно 26,9% масс./масс. моногидрата лактозы, приблизительно 22,1% масс./масс. полиэтиленгликоля 6000, приблизительно 9,5% масс./масс. полоксамера 188, приблизительно 1% масс./масс. стеарата магния, менее чем приблизительно 0,1% масс./масс. винной кислоты, менее чем приблизительно 0,01% масс./масс. бутилированного гидрокситолуола и менее чем приблизительно 0,01% масс./масс. диметикона 350, где проценты рассчитаны от общего количества неактивных веществ в композиции для первоначального лечения такролимусом пациента с трансплантатом почки de novo путем введения таблетки с пролонгированным высвобождением один раз в сутки в качестве лечения такролимусом.
16. Применение по п.15, где режим дозирования один раз в сутки представляет собой режим дозирования либо на ночь, либо вечером.
17. Применение по п.15, где режим дозирования один раз в сутки представляет собой режим дозирования утром.
18. Применение таблетки с пролонгированным высвобождением, включающей в качестве активного вещества от 0,1 до 15 мг такролимуса в гидрофильном или смешивающемся с водой носителе, где таблетка i) высвобождает активное вещество с профилем высвобождения по существу нулевого порядка в течение пролонгированного периода времени, определяемого по высвобождению от момента времени 8 часов до момента времени 15 часов при тестировании с помощью теста растворения USP II (лопасть) или теста растворения USP I (корзина) в среде, имеющей pH 4,5 и содержащей 0,005% гидроксипропилцеллюлозы, и при вращении 50 об/мин, причем профиль высвобождения по существу нулевого порядка определяют как линейный профиль высвобождения с отклонением не более ±15%; и ii) таблетка включает композицию, состоящую из активного вещества и приблизительно 40,5% масс./масс. гидроксипропилметилцеллюлозы (НРМС), приблизительно 26,9% масс./масс., моногидрата лактозы, приблизительно 22,1% масс./масс. полиэтиленгликоля 6000, приблизительно 9,5% масс./масс. полоксамера 188, приблизительно 1% масс./масс. стеарата магния, менее чем приблизительно 0,1% масс./масс. винной кислоты, менее чем приблизительно 0,01% масс./масс. бутилированного гидрокситолуола и менее чем приблизительно 0,01% масс./масс. диметикона 350, где проценты рассчитаны от общего количества неактивных веществ в композиции при лечении для предотвращения сверхиммуносупрессии у пациента с трансплантатом почки de novo путем введения такролимуса в пределах 48 часов после трансплантации в качестве таблетки с замедленным высвобождением один раз в сутки с начальной дозой от 0,13 до 0,15 мг/кг такролимуса и целевой Cmin 5-10 нг/мл, причем состав с пролонгированным высвобождением обеспечивает медианное Tmax на 1 сутки лечения от 8 до 14 часов.
| Пломбировальные щипцы | 1923 |
|
SU2006A1 |
| Способ обработки целлюлозных материалов, с целью тонкого измельчения или переведения в коллоидальный раствор | 1923 |
|
SU2005A1 |
| Sarah A | |||
| Cross et al | |||
| Tacrolimus Once-Daily Formulation in the Prophylaxis of Transplant Rejection in Renal or Liver Allograft Recipients | |||
| Drugs, 2007, vol.67, No.13, pp.1931-1943 | |||
| Пломбировальные щипцы | 1923 |
|
SU2006A1 |
| ЛЕКАРСТВЕННЫЕ КОМПОЗИЦИИ | 1997 |
|
RU2197226C2 |
| Станок для изготовления деревянных ниточных катушек из цилиндрических, снабженных осевым отверстием, заготовок | 1923 |
|
SU2008A1 |

