Настоящее изобретение относится к новому способу получения 1-(4--хлорфенил)-3-(1,2,4-триазол-1-ил-метил)-4,4-диметилпентан-3-ола (общепринятое наименование - "тебуконазол")
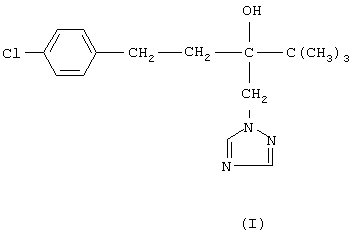
действующего вещества фунгицидных препаратов Folicur®* и Raxil®* (EP 0040345, USPat. №4723984, DE-OS 3018865, DE-OS 3018866, DE-OS 3333411).
Известно, что 1-(4-хлорфенил)-3-(1,2,4-триазол-1-ил-метил)-4,4-диметил-пентан-3-ол (I) может быть получен при взаимодействии 1,2,4-триазола с 1-(4-хлорфенил-этил)-2-трет-бутилоксираном (II) в присутствии щелочи. Известно также, что оксиран (II) получают при взаимодействии 4,4-диметил-1-(4-хлорфенил)пентан-3-она (III) с триметилсульфоний метилсульфатом (IV) в присутствии щелочи. Для получения триметилсульфоний метилсульфата (IV) используют реакцию диметилсульфида с диметилсульфатом.
Процесс получения тебуконазола (I) с использованием в качестве исходного продукта кетона (III) проводят в две стадии по следующей схеме:
1. Стадия получения 2-(4-хлорфенил-этил)-2-трет-бутилоксирана (II)
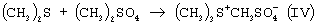

Вторую стадию осуществляют после выделения из реакционной смеси оксирана (II).
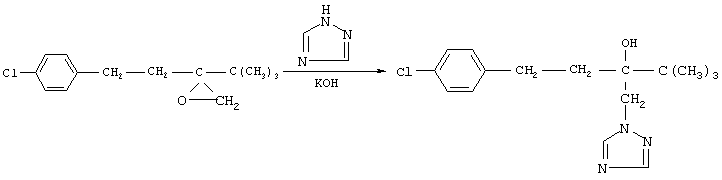
2. Стадия получения 1-(4-хлорфенил)-3-(1,2,4-триазол-1-ил-метил)-4,4-диметил-пентан-3-ола (тебуконазола) (I)
Из источников, относящихся к способу получения оксирана (II), известно, что 2-(4-хлорфенил-этил)-2-трет-бутилоксиран (II) можно получить при проведении реакции диметилсульфида и диметилсульфата в органическом растворителе с последующим добавлением к образовавшемуся триметилсульфоний метилсульфату (IV) 1-(4-хлорфенил)-4,4-диметилпентанона (III) и щелочного агента, например КОН или КОС(СН3)3. В качестве растворителей используют ацетонитрил, толуол, трет-бутиловый спирт, а также диметилсульфид, причем последний вводят в реакцию в количестве, многократно превышающем необходимое для образования триметилсульфоний метилсульфата (DE-OS 3315619, DE-OS 3315681, USPat. №4898954).
Недостатком вышеуказанных способов является использование постороннего органического растворителя, необходимость его регенерации и утилизации. В случае с диметилсульфидом имеют место сложности, связанные с его резким неприятным запахом и повышенной пожароопасностью, а также с увеличением объемов аппаратов. При проведении процесса в трет-бутиловом спирте используют на 1 моль исходного кетона 2 моля трет-бутилата калия (USPat. №4898954), для приготовления которого необходим металлический калий, то есть производство становиться особо пожароопасным.
В Европейском патенте №0219799 и DE-OS 3537817 описан способ получения 2-(4-хлорфенил-этил)-2-третбутилоксирана (II) в среде диметилсульфида с добавлением диметилсульфоксида, что ведет к существенному удорожанию процесса и делает его малопригодным для промышленных масштабов.
В заявке DE-OS 3733755 описан способ получения 2-(4-хлорфенил-этил)-2-трет-бутилоксирана (II) в среде диметилсульфида с добавлением воды в количестве 1 моль на 1 моль исходного кетона. Процесс проводят при температуре не выше 38°С, поэтому протекает он медленно (~14 часов.)
Из источников, относящихся к стадии получения тебуконазола (I) известно, что 1-(4-хлорфенил)-3-(1,2,4-триазол-1-ил-метил)-4,4-диметилпентан-3-ол может быть получен при взаимодействии 2-(4-хлорфенил-этил)-2-трет-бутилоксирана с 1,2,4-триазолом в растворе этанола или н-пропанола в присутствии КОН или NaOH, а также в диметилсульфоксиде в присутствии Nа2СО3 (ЕР 0040435, DE-OS 3315681 и ЕР 0219799). Недостатком этих способов является невысокий выход продукта и образование значительного количества примесей.
Известен также способ получения 1-(4-хлорфенил)-3-(1,2,4-триазол-1-ил-метил)-4,4-диметилпентан-3-ола (I) из соответствующих оксирана (II) и триазола в растворе N-метилпирролидона в присутствии NaOH (DE-OS 3342692). Несмотря на высокий выход продукта применение способа в промышленном масштабе нецелесообразно из-за высокой стоимости использования N-метилпирролидона.
Наиболее близким к заявляемому является способ получения 1-(4-хлорфенил)-3-(1,2,4-триазол-1-ил-метил)-4,4-диметилпентан-3-ола (I) при взаимодействии 2-(4-хлорфенил-этил)-2-трет-бутилоксирана (II) с 1,2,4-триазолом в присутствии КОН и н-бутанола при температуре от 80°С до 145°С. При этом на 1 моль 2-(4-хлорфенил-этил)-2-трет-бутилоксирана (II) в реакцию вводят от 1,0 до 1,1 молей 1,2,4-триазола, от 0,1 до 0,25 молей КОН и от 0,8 до 2,5 молей н-бутанола. Судя по приведенным в описании примерам, процесс завершается в течение 6-7 часов без учета времени, необходимого для выделения и очистки целевого продукта, выход которого составляет от 79,6% до 83,4%, считая на оксиран (II) (DE-OS 3733754).
Во всех известных способах получения 1-(4-хлорфенил)-3-(1,2,4-триазол-1-ил-метил)-4,4-диметилпентан-3-ола (I) используют выделенный из реакционной смеси и очищенный (-97% содержания основного вещества) 2-(4-хлорфенил-этил)-2-трет-бутилоксиран (II).
При разработке технологических процессов такой степени сложности, как процесс получения 1-(4-хлорфенил)-3-(1,2,4-триазол-1-ил-метил)-4,4-диметил-пентан-3-ола ("тебуконазола") одной из важных задач является достижение максимальной простоты и компактности процесса, т.е. сокращения количества операций, необходимой аппаратуры, времени проведения процесса при сохранении качества и выхода конечного продукта.
Эту задачу удалось решить путем совмещения стадий получения 2-(4-хлорфенил-этил)-2-трет-бутилоксирана (II) и 1-(4-хлорфенил)-3-(1,2,4-триазол-1-ил-метил)-4,4-диметилпентан-3-ола (I) в одном производственном цикле. Такое совмещение оказалось возможным в результате проведения процесса получения 2-(4-хлорфенил-этил)-2-трет-бутилоксирана (II) в среде н-бутилового спирта, т.е. того же растворителя, который используют в известном способе получения 1-(4-хлорфенил)-3-(1,2,4-триазол-1-ил-метил)-4,4-диметил-пентан-3-ола (DE-OS 3733754) и в разработанном ранее заявителем способе получения 1-(4-хлорфенил)-4,4-диметилпентан-3-она (III) (исходного продукта для оксирана (II) (заявка №2002126238/04 (027830) от 02.10.2002 г.)).
Оказалось, что в среде бутилового спирта реакция протекает полностью в присутствии порошкообразного КОН, который добавляют в количестве не более 3 молей на 1 моль кетона. После перемешивания реакционной смеси в течение 3,5-4 часов при 50-55°С осуществляется превращение 1-(4-хлорфенил)-4,4-диметилпентан-3-она в 2-(4-хлорфенил-этил)-2-трет-бутилоксиран со степенью конверсии не ниже 97% (по данным ГЖХ). Образовавшийся оксиран находится в растворе н-бутанола с примесью диметилсульфида. Кроме того, в реакционной смеси присутствует подвижный осадок, состоящий из смеси различных солей калия и избыточной щелочи, которая необходима для дальнейшего превращения оксирана в целевой продукт (I). К этой реакционной смеси добавляют 1,2,4-триазол в количестве, не превышающем 1,1 моля на 1 моль введенного в реакцию 1-(4-хлорфенил)-4,4-диметилпентан-3-она (III). Во время проведения реакции происходит также отгонка избытка диметилсульфида, воды и других возможных легколетучих примесей. Твердые компоненты осадка, содержащиеся в реакционной смеси, как оказалось, не препятствуют нормальному течению процесса, в результате которого получают 1-(4-хлорфенил)-3-(1,2,4-триазол-1-ил-метил)-4,4-диметилпентан-3-ол ("тебуконазол") (I) 98-99% чистоты по ВЭЖХ с выходом 80-82%, считая на 1-(4-хлорфенил)-4,4-диметилпентан-3-он (III). Эти данные вполне сопоставимы или даже превосходят результаты по прототипу (DE-OS 3733754), в котором используют предварительно выделенный и очищенный оксиран (II) и где выход рассчитан на оксиран (II), а не на предшествующий ему кетон (III).
Кроме того, на стадии выделения и очистки целевого продукта (I) оказалось возможным и достаточным использовать в отличие от прототипа кристаллизацию из петролейного эфира без охлаждения смеси ниже комнатной температуры.
Таким образом, очевидные преимущества заявляемого способа состоят в совмещении в одном процессе двух стадий - получения 2-(4-хлорфенил-этил)-2-трет-бутилоксирана (II) и 1-(4-хлорфенил)-3-(1,2,4-триазол-1-ил-метил)-4,4-диметил-пентан-3-ола (I) в результате использования на стадии получения оксирана в качестве растворителя н-бутанола и небольшого избытка КОН, который работает на стадии получения "тебуконазола". При этом уменьшается количество операций, в том числе устраняются операции по выделению и очистке оксирана (II), а также операция по регенерации "постороннего" растворителя, поскольку на всем протяжении процесса используется один растворитель - н-бутанол. Одновременно сокращается также время проведения процесса, упрощается его аппаратурное оформление при сохранении выхода конечного продукта (I) и его качества.
Как показали опыты, наилучшие результаты могут быть достигнуты, если на 1 моль 1-(4-хлорфенил)-4,4-диметилпентан-3-она (III) используют 1,5-1,6 моль триметилсульфоний метилсульфата (из эквимольных количеств диметилсульфида и диметилсульфата с небольшим избытком диметилсульфида (~0,015 М)); 2,5-3 моля КОН; 2,2-2,5 моля н-бутанола и 1,0-1,1 моля 1,2,4-триазола.
Далее следуют примеры, иллюстрирующие, но не ограничивающие изобретение.
Способ получения
1-(4-хлорфенил)-4,4-диметил-3-(1,2,4-триазол-1-ил-метил)пентан-4-ола
Пример №1
К раствору 1,13 кг (18,2 М) диметилсульфида, 2,70 кг (11,8 М) 98% 1-(4-хлорфенил)-4,4-диметилпентан-3-она в 2,5 л (26,4 М) н-бутанола при перемешивании в течение 30 мин прибавляют 2,26 кг (17,9 М) диметилсульфата. Затем реакционную смесь перемешивают в течение 1 часа при 50°С, после чего охлаждают до 30°С. Далее при перемешивании к реакционной смеси в течение 20 минут прибавляют 2,28 кг (34,5 М) 85% порошкообразного КОН. Затем реакционную смесь перемешивают 3,5-4,0 часа при 50-55°С. По истечении этого времени осуществляется превращение 1-(4-хлорфенил)-4,4-диметилпентан-3-она в 2-(4-хлорфенил-этил)-2-трет-бутилоксиран со степенью конверсии 98,8% (по данным ГЖХ). Далее к полученной смеси, содержащей бутанольный раствор 2-(4-хлорфенил-этил)-2-трет-бутилоксирана, прибавляют 0,84 кг (12,2 М) 1,2,4-триазола. Реакционную смесь нагревают до 130-140°С при этой температуре 3 часа. При этом отгоняются легколетучие компоненты. Далее смесь охлаждают до 60°С и фильтруют, после чего органический раствор промывают 800 мл воды. Органический слой отделяют и из полученного раствора при 40-90°С и остаточном давлении 20-50 мм рт.ст. удаляют н-бутанол. К оставшейся массе при перемешивании при 60°С добавляют 1,4 кг петролейного эфира и выдерживают смесь при 20°С в течение 6 часов. Выпавший осадок отфильтровывают, промывают 0,9 кг петролейного эфира и сушат 1 час при 60°С. Получают 3,15 кг 1-(4-хлорфенил)-3-(1,2,4-триазол-1-ил-метил)-4,4-диметил-пентан-3-ола (99,2% чистоты по ВЭЖХ). Выход - 82% от теоретического, считая на 1-(4-хлорфенил)-4,4-диметилпентан-3-он.
Пример №2
К раствору 3,28 кг (26,0 М) диметилсульфата, 3,89 кг (17,0 М) 98% 1-(4-хлорфенил)-4,4-диметилпентан-3-она в 3,7 л (39,1 М) н-бутанола в течение 30 минут прибавляют 1,63 кг (26,3 М) диметилсульфида. Затем реакционную смесь перемешивают 1 час при 50°С, после чего охлаждают до 30°С. Далее при перемешивании к реакционной смеси в течение 30 мин прибавляют 2,81 кг (42,5 М) 85% порошкообразного КОН. Затем реакционную смесь перемешивают 4 часа при 50-55°С. По истечении этого времени осуществляется превращение 1-(4-хлорфенил)-4,4-диметилпентан-3-она в 2-(4-хлорфенил-этил)-2-трет-бутилоксиран со степенью конверсии 97,6% (по данным ГЖХ). Далее к полученной смеси, содержащей бутанольный раствор 2-(4-хлорфенил-этил)-2-трет-бутилоксирана, прибавляют 1,29 кг (18,7 М) 1,2,4-триазола. Реакционную смесь нагревают до 130-140°С и перемешивают при этой температуре 3,5 часа. При этом отгоняются легколетучие компоненты. Далее смесь охлаждают до 70°С и фильтруют, после чего органический раствор промывают 1,2 л воды. Органический слой отделяют и из полученного раствора при пониженном давлении отгоняют н-бутанол. Оставшуюся маслообразную жидкость при перемешивании прибавляют к нагретому до 50°С петролейному эфиру в количестве 2,0 кг, и выдерживают смесь при 20°С в течение 6 часов. Выпавший осадок отфильтровывают, промывают 1,3 кг петролейного эфира и сушат 1 час при 60°С. Получают 4,48 кг 1-(4-хлорфенил)-4,4-диметил-3-(1,2,4-триазол-1-ил-метил)пентан-4-ола (98,0% чистоты по ВЭЖХ). Выход - 80%, считая на 1-(4-хлорфенил)-4,4-диметилпентан-3-он.
Пример №3
К раствору 176 кг (768 М) диметилсульфида в 104 л (1100 М) н-бутанола при перемешивании в течение 1 часа прибавляют 96 кг (760 М) диметилсульфата. Затем реакционную смесь перемешивают в течение 1,5 часа при 50°С, после чего охлаждают до 35°С. Далее при перемешивании к реакционной смеси прибавляют 114 кг (500 М) 98%-го 1-(4-хлорфенил)-4,4-диметилпентан-3-она и порциями 96 кг (1450 М) 85%-го порошкообразного КОН. Затем реакционную смесь перемешивают 4 часа при 50-55°С. По истечении этого времени осуществляется превращение 1-(4-хлорфенил)-4,4-диметилпентан-3-она в 2-(4-хлорфенил-этил)-2-трет-бутилоксиран со степенью конверсии 98,2% (по данным ГЖХ). Далее к полученной смеси, содержащей бутанольный раствор 2-(4-хлорфенил-этил)-2-трет-бутилоксирана, прибавляют 36 кг (525 М) 1,2,4-триазола. Реакционную смесь нагревают до 130-140°С при одновременной отгонке легколетучих компонентов, причем время перемешивания при 135-140°С составляет 4 часа. Далее смесь охлаждают до 80°С и промывают 30 л воды. Органический слой отделяют и из полученного раствора при пониженном давлении отгоняют н-бутанол. Оставшуюся маслообразную жидкость при перемешивании прибавляют к нагретому до 60°С петролейному эфиру в количестве 50 кг, и выдерживают смесь при 20 С в течение 8 часов. Выпавший осадок отфильтровывают, промывают 23 кг петролейного эфира и сушат 3 часа при 60°С. Получают 133 кг 1-(4-хлорфенил)-4,4-диметил-3-(1,2,4-триазол-1-ил-метил)пентан-4-ола (99,1% чистоты по ВЭЖХ). Выход - 82%, считая на 1-(4-хлорфенил)-4,4-диметилпентан-3-он.
| название | год | авторы | номер документа |
|---|---|---|---|
| СПОСОБ ПОЛУЧЕНИЯ 4,4-ДИМЕТИЛ-1-(П-ХЛОРФЕНИЛ)-ПЕНТАН-3-ОНА | 2002 |
|
RU2228327C1 |
| ФУНГИЦИДНАЯ КОМПОЗИЦИЯ, СПОСОБ БОРЬБЫ С ФИТОПАТОГЕННЫМИ ГРИБАМИ | 1994 |
|
RU2129371C1 |
| ФУНГИЦИДНОЕ СРЕДСТВО НА ОСНОВЕ ПРОИЗВОДНОГО АЗОЛА И СПОСОБ ТОРМОЖЕНИЯ КРИСТАЛЛИЗАЦИИ 1-(4-ХЛОРФЕНИЛ)-4,4-ДИМЕТИЛ-3-(1,2,4-ТРИАЗОЛ-1-ИЛМЕТИЛ)ПЕНТАН-3-ОЛА ИЗ ВОДНОГО ПРЕПАРАТА | 1994 |
|
RU2155482C2 |
| СПОСОБ ПОЛУЧЕНИЯ ЦИС-2-(1Н-1,2,4-ТРИАЗОЛ-1-ИЛ-МЕТИЛ)-2-(ГАЛОГЕНФЕНИЛ)-3-(ГАЛОГЕНФЕНИЛ)-ОКСИРАНА | 1990 |
|
RU2071473C1 |
| Замещенные 1-(1Н-1,2,4-триазол-1-илметил)-циклогексанолы, обладающие фунгицидной активностью, замещенные оксираны в качестве промежуточных продуктов для синтеза замещенных 1-(1Н-1,2,4-триазол-1-илметил)-циклогексанолов, обладающих фунгицидной активностью | 1991 |
|
SU1838304A3 |
| Замещенные 4-(азол-1-илметил)-1-фенил-5,5-диалкилспиро-[2.5]октан-4-олы, способ их получения (варианты), фунгицидная и рострегуляторная композиции на их основе | 2016 |
|
RU2648240C1 |
| ФУНГИЦИДНОЕ СРЕДСТВО | 1996 |
|
RU2143804C1 |
| ЗАМЕЩЕННЫЕ 1-(ПИРИДИНИЛ-3)-2-ФЕНОКСИЭТАНОЛЫ-1, ИХ СПОСОБ ПОЛУЧЕНИЯ И ФУНГИЦИДНАЯ КОМПОЗИЦИЯ НА ИХ ОСНОВЕ | 2003 |
|
RU2248351C1 |
| СПОСОБ БОРЬБЫ С ГРИБНЫМИ БОЛЕЗНЯМИ РАСТЕНИЙ | 1991 |
|
RU2028052C1 |
| ФУНГИЦИДНОЕ СРЕДСТВО И СПОСОБ БОРЬБЫ С ГРИБКАМИ | 1992 |
|
RU2098962C1 |
Описывается улучшенный способ получения 1-(4-хлорфенил)-4,4-диметил-3-(1,2,4-триазол-1 -ил-метил)пентан-3-ола, заключающийся во взаимодействии 1,2,4-триазола с 2-(4-хлорфенил-этил)-2-трет-бутилоксираном в присутствии гидроокиси калия в среде н-бутанола. Отличие состоит в том, что процесс проводят непосредственно в реакционной смеси, содержащей 2-(4-хлорфенил-этил)-2-трет-бутилоксиран, полученный в результате взаимодействия 4,4-диметил-1-(4-хлорфенил)пентан-3-она с триметилсульфоний метилсульфатом в среде н-бутанола при 50-55°С, без предварительного выделения оксирана и его очистки. Способ позволяет сократить количество операций, времени проведения процесса с сохранением высокого выхода продукта. 3 з.п. ф-лы.
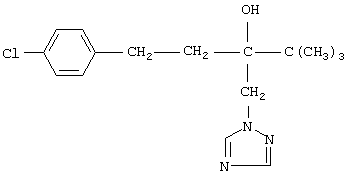
заключающийся во взаимодействии 1,2,4-триазола с 2-(4-хлорфенил-этил)-2-трет-бутилоксираном (II)
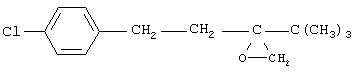
в присутствии гидроокиси калия в среде н-бутанола при температуре 130-140°С, отличающийся тем, что 1,2,4-триазол вводят непосредственно в реакционную смесь, полученную в результате взаимодействия 4,4-диметил-1-(4-хлорфенил)пентан-3-она (III)

с триметилсульфонийметилсульфатом (СH3)3S+CH3SO4 - в среде н-бутанола при температуре 50-55°С в присутствии гидроокиси калия.
| DE 3733754 А, 20.04.1989 | |||
| DE 3342692 A, 05.06.1985 | |||
| ПРОИЗВОДНЫЕ 1,2,4-ТРИАЗОЛА ИЛИ ИХ ФАРМАЦЕВТИЧЕСКИ ПРИЕМЛЕМЫЕ СОЛИ, ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ И ПРОМЕЖУТОЧНЫЕ СОЕДИНЕНИЯ | 1992 |
|
RU2095358C1 |

