Изобретение относится к новым соединениям, к способу их получения, их применению и фармацевтическим композициям, содержащим эти новые соединения. Эти новые соединения являются полезными в терапии и, в частности, для лечения боли.
Предшествующий уровень техники
δ-Рецептор идентифицирован как рецептор, играющий роль во многих функциях организма, таких как сердечно-сосудистая и лимфатическая и болевая системы. Следовательно, лиганды для δ-рецептора могут находить потенциальное применение в качестве анальгетиков и/или в качестве гипотензивных агентов. Показано также, что лиганды для δ-рецептора обладают иммуномодулирующей активностью.
Идентификация по меньшей мере трех различных групп опиоидных рецепторов (μ, δ и κ) в настоящее время четко установлена, и очевидно, что все три находятся как в центральной, так и в периферической нервных системах многих видов, включая человека. Аналгезию наблюдали в различных животных моделях при активации одного или более чем одного из этих рецепторов.
За редким исключением, имеющиеся в настоящее время селективные δ-опиоидные лиганды являются пептидными по природе и не подходят для введения посредством системных путей. Одним из примеров не пептидного δ-агониста является SNC80 (Bilsky E.J. et al., Journal of Pharmacology and Experimental Therapeutics, 273 (1), pp.359-366 (1995)). Однако все еще существует необходимость в селективных δ-агонистах, обладающих не только повышенной селективностью, но также улучшенным профилем побочных эффектов.
Таким образом, задачей, лежащей в основе настоящего изобретения, являлся поиск новых анальгетиков, обладающих улучшенными болеутоляющими эффектами, но также с улучшенным профилем побочных эффектов по сравнению с современными μ-агонистами, а также обладающих улучшенной системной эффективностью.
Анальгетики, которые идентифицированы и имеются в уровне техники, имеют много недостатков, заключающихся в том, что они обладают неудовлетворительной фармакокинетикой и не оказывают болеутоляющего действия при введении посредством системных путей. Также документально подтверждено, что предпочтительные соединения δ-агонисты, описанные в данном уровне техники, проявляют значительные судорожные эффекты при системном введении.
Авторами изобретения в настоящее время обнаружено, что некоторые соединения, конкретно не раскрытые, но включенные в объем WO 98/28270, демонстрируют неожиданно улучшенные δ-агонистические свойства и эффективность in vivo относительно соединений, раскрытых в WO 98/28270, при системном введении. Соединения по настоящему изобретению демонстрируют значительные и неожиданно повышенные уровни агонизма по отношению к дельта-рецептору и метаболической стабильности.
Краткое изложение сущности изобретения
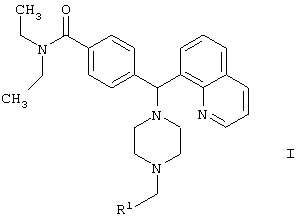
Новые соединения по настоящему изобретению определены формулой 1
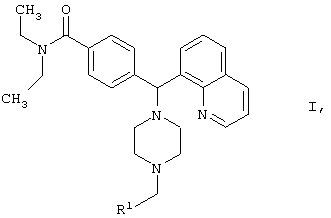
где
R1 выбран из
(1) фенила;
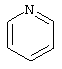
(2) пиридинила 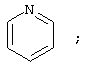
(3) тиофенила 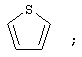
(4) фуранила 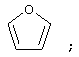
(5) имидазолила 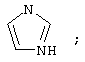
(6) триазолила 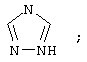
где каждое фенильное кольцо R1 и гетероароматическое кольцо R1 может быть возможно и независимо дополнительно замещено 1, 2 или 3 заместителями, выбранными из прямого и разветвленного C1-С6алкила, NO2, CF3, C1-С6алкокси, хлоро, фторо, бромо и йодо. Замещения на фенильном кольце и на гетероароматическом кольце могут иметь место в любом положении на указанных кольцевых системах.
В объем настоящего изобретения также входят фармацевтически приемлемые соли соединений формулы I, а также их изомеры.
В предпочтительном воплощении данного изобретения соединения формулы 1 находятся в виде (+)-энантиомера или в виде (-)-энантиомера.
Под «изомерами» авторы изобретения подразумевают соединения формулы I, которые различаются по положению их функциональной группы и/или ориентации. Под «ориентацией» авторы изобретения подразумевают стереоизомеры, диастереоизомеры, региоизомеры и энантиомеры.
Новые соединения по настоящему изобретению являются полезными в терапии, особенно для лечения различных болевых состояний, таких как хроническая боль, невропатическая боль, острая боль, раковая боль, боль, вызванная ревматоидным артритом, мигрень, висцеральная боль и так далее. Этот перечень, однако, не следует интерпретировать как исчерпывающий.
Соединения по настоящему изобретению являются полезными в качестве иммуномодуляторов, особенно при аутоиммунных заболеваниях, таких как артрит, для кожных трансплантатов, трансплантатов органов и подобных хирургических нужд, при коллагенозах, различных аллергиях, для применения в качестве противоопухолевых агентов и противовирусных агентов.
Соединения по настоящему изобретению являются полезными при болезненных состояниях, при которых имеется дегенерация или дисфункция опиоидных рецепторов, либо дегенерация или дисфункция опиоидных рецепторов вовлечена в процесс. В диагностические методики и в применения, связанные с визуализацией, такие как позитронная эмиссионная томография (ПЭТ), может быть также вовлечено использование меченых изотопами вариантов соединений по настоящему изобретению.
Соединения по настоящему изобретению являются полезными для лечения диареи, депрессии, тревоги, недержания мочи, различных психических заболеваний, кашля, отека легких, различных желудочно-кишечных расстройств, повреждения спинного мозга и привыкания к чрезмерному употреблению лекарств, включая лечение злоупотребления алкоголем, никотином, опиоидами и другими лекарствами, а также для лечения расстройств симпатической нервной системы, например гипертензии.
Соединения по настоящему изобретению являются полезными в качестве анальгетического агента для применения во время общей анестезии и контролируемой коррекции анестезии. Комбинации агентов с различными свойствами часто применяют для достижения баланса эффектов, необходимого для поддержания состояния анестезии (например, амнезии, аналгезии, мышечной релаксации и седативного эффекта). В данную комбинацию включены ингаляционные анестетики, снотворные средства, анксиолитики, нейромышечные блокаторы и опиоиды.
В объем настоящего изобретения также входит применение любого из соединений формулы I, указанной выше, для производства лекарства для лечения любого из состояний, обсуждаемых выше.
Следующий аспект настоящего изобретения представляет собой способ лечения субъекта, страдающего от любого из состояний, обсуждаемых выше, при котором пациенту, нуждающемуся в таком лечении, вводят эффективное количество соединения формулы I, указанной выше. Также в объем настоящего изобретения включено любое новое промежуточное соединение, как описано в приведенной ниже Схеме I, полезное в синтезе соединений формулы I, указанной выше.
Способы получения
Соединения по настоящему изобретению можно получить, следуя любой из методик, описанных в Схемах I, II, III и IV. Эти известные методики описаны в J. March, Advanced Organic Chemistry, 4th Edition, John Wiley and sons (1992); Katritsky, A.R., Lan, X. Chem. Soc. Rev., pp.363-373 (1994), которые включены в данное описание изобретения путем ссылки.
СХЕМА I
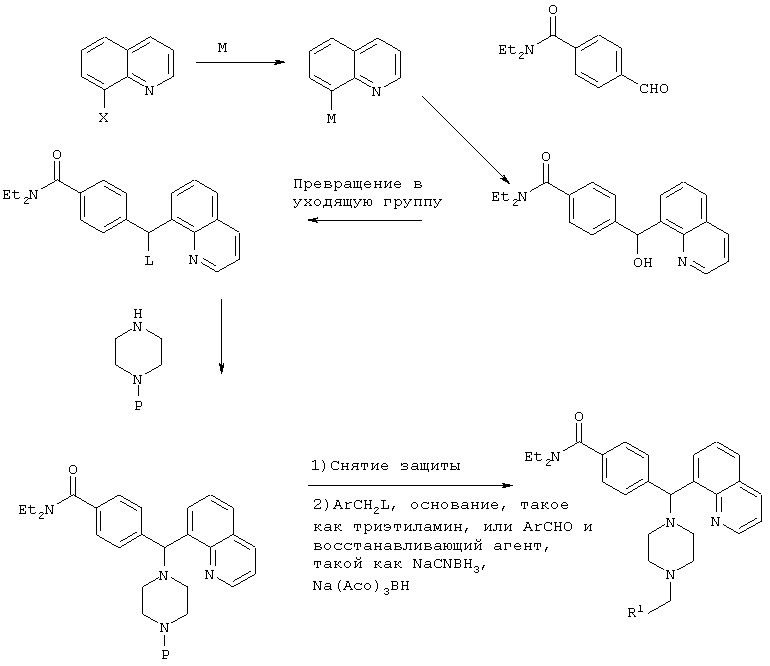
Р=защитная группа, такая как Bn, Boc, CBz
М=Li, Mg, Zn
Х=Br, I
L=Cl, Br, OMs, OTs, I
R1=как определено в формуле (I), указанной выше.
СХЕМА II
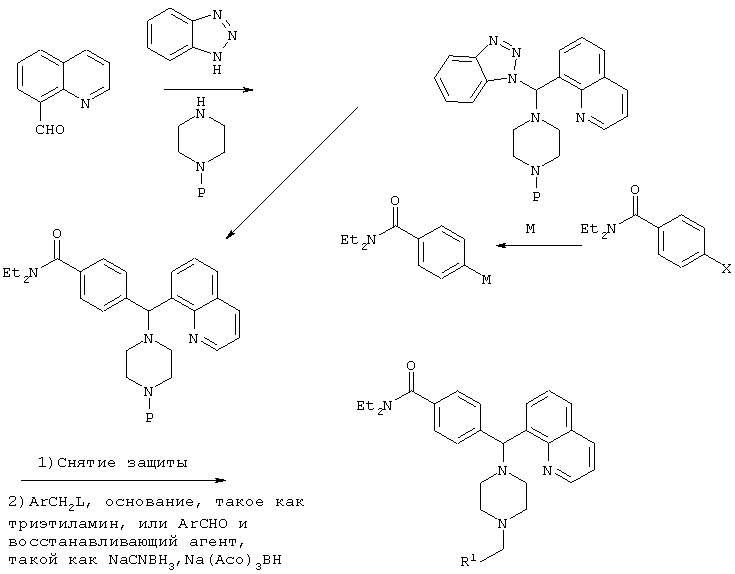
Р=защитная группа, такая как Bn, Boc, CBz
М=Li, Mg, Zn
Х=Br, I
L=Cl, Br, OMs, OTs, I
R1=как определено в формуле (I), указанной выше.
СХЕМА III
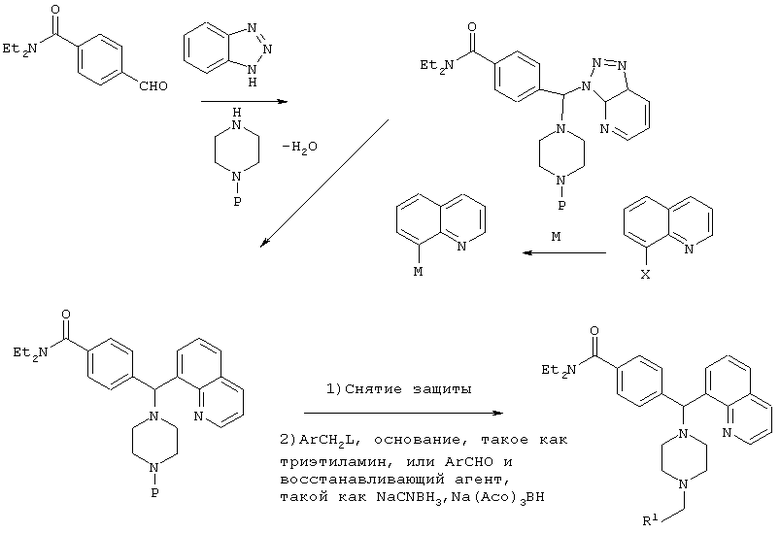
Р=защитная группа, такая как Bn, Boc, CBz
М=Li, Mg, Zn
Х=Br, I
L=Cl, Br, OMs, OTs, I
R1=как определено в формуле (I), указанной выше.
СХЕМА IV
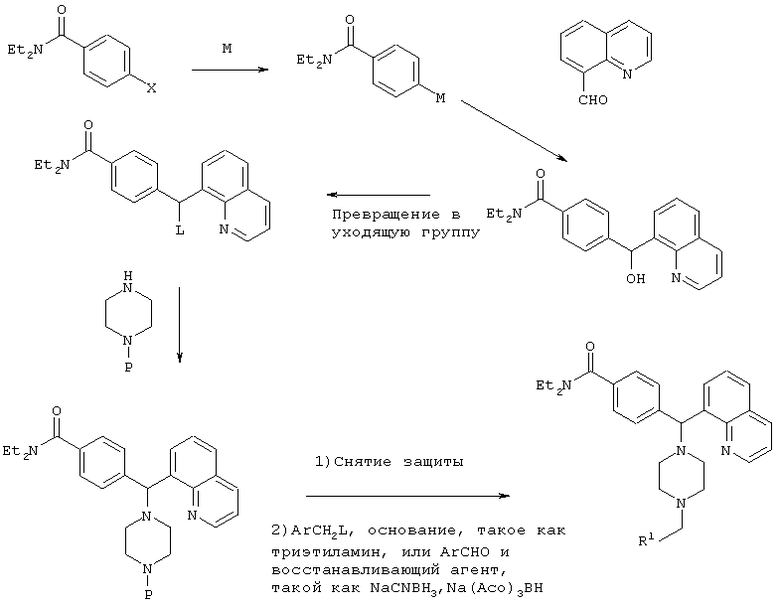
Р=защитная группа, такая как Bn, Boc, CBz
М=Li, Mg, Zn
Х=Br, I
L=Cl, Br, OMs, OTs, I
R1=как определено в формуле (I), указанной выше.
Примеры
Далее изобретение будет описано более подробно с помощью следующих примеров, которые не следует истолковывать как ограничивающие данное изобретение.
Пример 1
Получение 4-[(4-бензил-1-пиперазинил)(8-хинолинил)метил]-N,N-диэтилбензамида дигидрохлорида (соединение 2)
Соединение 2, указанное в заголовке, получили, следуя методике синтеза Схемы 1, приведенной ниже.
Схема 1
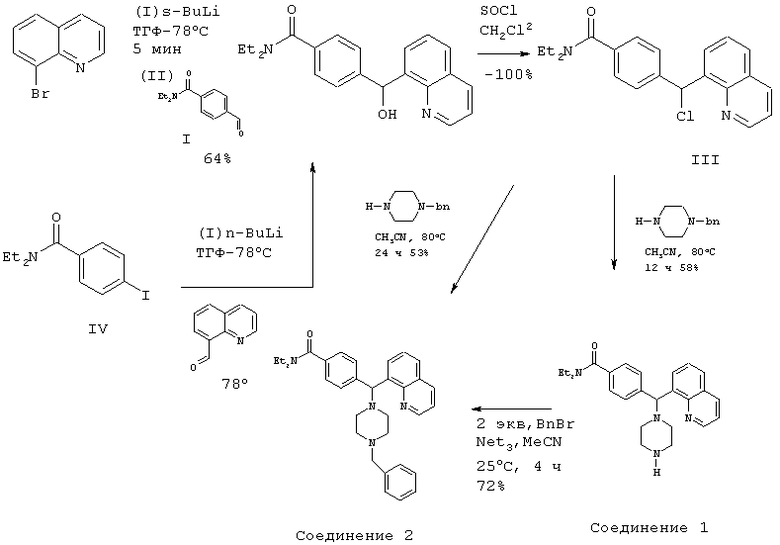
(1) Получение N,N-диэтил-4-формилбензамида (соединение I)
4-Формилбензойную кислоту (11,2 г, 74,6 ммоль) и триэтиламин (10,4 мл, 75 ммоль) растворяли в тетрагидрофуране (ТГФ) (100 мл) и охлаждали до -10°С. Добавляли изо-бутилхлорформиат (10,3 мл, 78 ммоль), и перемешивание продолжали в течение 10 минут при -10°С, после чего добавляли диэтиламин (9,7 мл, 94 ммоль), и раствор оставили нагреваться до достижения 25°С. После концентрирования, водной обработки и хроматографии на силикагеле (0-100% ЕЮАс в гептане) получили суммарно 7,4 г (50%) соединения I.
(2) Получение N,N-диэтил-4-[гидрокси(8-хинолинил)метил]бензамида (соединение II)
8-Бромхинолин (3,0 г, 14,4 ммоль) растворяли в безводном ТГФ (150 мл) и охлаждали до -78°С в атмосфере азота. Добавляли по каплям втор-бутиллитий (emop-BuLi) (11,1 мл, 1,3 М в пентане, 14,4 ммоль) в течение 5 мин (Preparation and reactions with 8-lithioquinoline: Suggs, J. Org. Chem. 1980, 45, 1514). Еще через 5 мин добавляли N,N-диэтил-4-формилбензамид (3,5 г, 17,0 ммоль), растворенный в ТГФ (5 мл). Этот раствор перемешивали в течение 1 ч, затем добавляли NH4Cl (водный). После концентрирования, водной обработки и хроматографии на силикагеле (0-100% EtOAc в гептане) получили суммарно 3,5 г (70%) соединения II.
МС: 334, 262, 234, 215, 204, 178, 156, 129.
Альтернативный путь получения соединения II из N,N-диэтил-4-йодбензамида (соединение IV)
Соединение IV (0,67 г, 2,2 ммоль) растворяли в сухом ТГФ (25 мл) и охлаждали до -78°С в атмосфере азота. Добавляли по каплям н-BuLi (1,3 мл, 1,6 М в гексане, 2,2 ммоль) в течение 5 мин. Еще через 10 мин добавляли 8-формилхинолин (0,17 г, 1,1 ммоль) (8-формилхинолин получили из 8-метилхинолина путем окисления диоксидом селена при 150-155°С в течение 12 ч (Kingsbury, J. Med. Chem. 1993, 3308)), растворенный в ТГФ (1 мл). Этот раствор перемешивали в течение 1 ч, затем добавляли NH4Cl (водный). После концентрирования, водной обработки и хроматографии на силикагеле (0-100% EtOAc в гептане) получили суммарно 0,29 г (78%) соединения II.
(3) Получение 4-[хлор(8-хинолинил)метил]-N,N-диэтилбензамида (соединение III).
Соединение II (2,0 г, 6,6 ммоль) растворяли в безводном СН2Cl2 (25 мл) и добавляли SOCl2 (0,53 мл, 7,3 ммоль). Этот раствор перемешивали при 25°С в течение 30 мин, и растворитель выпаривали в вакууме. Соединение III получили в виде масла (-100%) и использовали в следующей реакции без дальнейшей очистки.
МС: 348, 333, 233, 215, 204, 156.
(4) Получение N,N-диэтил-4-[1-пиперазинил(8-хинолинил)метил]бензамида (соединение 1).
Неочищенный продукт соединение III (˜6,6 ммоль) и пиперазин (2,3 г, 26 ммоль) растворяли в безводном MeCN (50 мл) и нагревали с обратным холодильником в течение 12 ч. Растворитель удаляли в вакууме, остаток растворяли в CH2Cl2 и промывали водой, и органическую фазу высушивали (К2СО3) и выпаривали в вакууме. После хроматографии на силикагеле (0-20% МеОН в CH2Cl2, 1% NH4OH) получили суммарно 1,8 г (68%, 2 стадии) соединения 1. Дальнейшей очистки можно достичь путем обращенно-фазовой хроматографии (LiChroprep RP-18, 10-50% MeCN в воде, 0,1% ТФУ) с получением 1,2 г бесцветного продукта. Соль дигидрохлорид получили путем обработки 2 экв. HCl в эфире.
Т.пл.:180-90°С.
ИК (KBr, νmax) 3297, 2982, 2716, 2474, 1611, 1434, 1380, 1288, 1098 см-1.
МС (амин): 402, 318, 246, 217, 109.
1H ЯМР (амин, CDCl3): δ 1.2, 1.1 (2s, 6H), 2.94, 2.51 (2m, 8H), 3.5-3.1 (m, 5H), 6.05 (s, 1H), 8.94-7.20 (m, 10H).
Анализ (C25H30N4Ox3,2 CF3СО2Н) С, N; Н: вычислено 4,36; обнаружено 3,90.
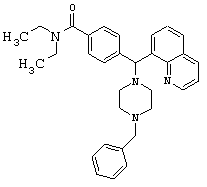
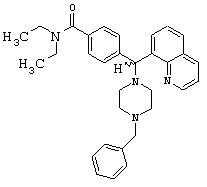
(5) Получение 4-[(4-бензил-1-пиперазинил)(8-хинолинил)метил]-N,N-диэтилбензамида дигидрохлорида (соединение 2, указанное в заголовке).
Соединение 1 (1,3 г, 3,2 ммоль) и триэтиламин (0,90 мл, 6,4 ммоль) растворяли в MeCN (10 мл). Бензилбромид (0,77 мл, 6,4 ммоль) добавляли при перемешивании при 25°С. Через 4 ч этот раствор концентрировали и очищали путем хроматографии на силикагеле (0-5% МеОН в CH2Cl2) или путем обращенно-фазовой хроматографии (LiChroprep RP-18, 20-80% MeCN в воде, 0,1% ТФУ). Суммарно получили 2,2 г (72%) соединения 2, указанного в заголовке.
Путем обработки 2 экв. HCl (водной) и лиофилизации получили соль дигидрохлорид (3,6 г).
ИК (2Х HCI, KBr): 2388, 1606, 1434, 1356, 1287 см-1.
1H ЯМР (свободный амин, CDCl3): δ=1.05 (m, 6H), 2.5 (m, 8H), 3.1-3.6 (m, 6H), 6.04 (s, 1H), 7.18-8.98 (m, 15H).
Анализ (С32Н38Cl2N4O) С, Н, N. Альтернативная методика получения соединения 2, указанного в заголовке, из соединения III
Неочищенный продукт соединение III (˜13,2 ммоль), триэтиламин (2,0 мл, 14,5 ммоль) и N-бензил-пиперазин (2,6 г, 14,5 ммоль) растворяли в безводном MeCN (50 мл) и нагревали с обратным холодильником в течение 12 ч. Добавляли дополнительное количество N-бензил-пиперазина (0,5 г, 2,8 ммоль) и нагревание продолжали в течение 12 ч. Растворитель удаляли в вакууме, остаток растворяли в CH2Cl2 и промывали водой; и органическую фазу высушивали (К2СО3) и выпаривали в вакууме. После хроматографии на силикагеле (0-10% МеОН в СН2Cl2 суммарно получили 3,5 г (53%) соединения 2, указанного в заголовке.
Примеры 2 и 3
Разделение энантиомеров соединения 2 (соединения 3 и 4)
Препаративное разделение данного соединения проводили на колонке Chiralcel OD (50 мм × 50 см), используя смесь гексан/EtOH/диэтиламин в соотношении 85:15:0,1 в качестве подвижной фазы. Обнаружили, что на колонке Chiralcel OD (+)-изомер элюируется первым.
Пример 2 (-)4-[(4-бензил-1-пиперазинил)(8-хинолинил)метил]-N,N-диэтилбензамид (соединение 3)
[α]D 25: -130° (с 0,78, МеОН)
1H ЯМР: (CD3OD): δ=1.05 (m, 6H), 3.0-3.6 (m, 14H), 5.90 (s, 1H), 7.22-8.20 (m, 13H), 8.78 (m, 1H), 9.50 (m, 1H).
АНАЛИЗ: Вычисленная масса 3,1 Н2O, С: 61.85, Н: 7.17, N: 9.02. Обнаружено С: 61.84, Н: 6.60, N: 8.89.
Пример 3
(+)4-[(4-бензил-1-пиперазинил)(8-хинолинил)метил]-N,N-диэтилбензамид (соединение 4)
[α]D 25: +130° (c 0,69, MeOH)
1H ЯМР: (CD3OD): δ=1.05 (m, 6Н), 3.0-3.6 (m, 14H), 5.90 (s, 1H), 7.22-8.20 (m, 13H), 8.78 (m, 1H), 9.50 (m, 1H).
АНАЛИЗ: Вычисленная масса 3,2 Н2O, С: 61.67, Н: 7.18, N: 8.99. Обнаружено С: 61.70, Н: 6.46, N: 8.84.
Пример 4
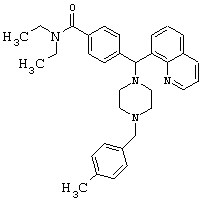
Получение N,N-диэтил-4-[[4-(4-метилбензил)-1-пиперазинил](8-хинолинил)метил]бензамида (соединение 5)
Соединение 5, указанное в заголовке, получили, следуя методике синтеза Схемы 2, приведенной ниже.
Схема 2
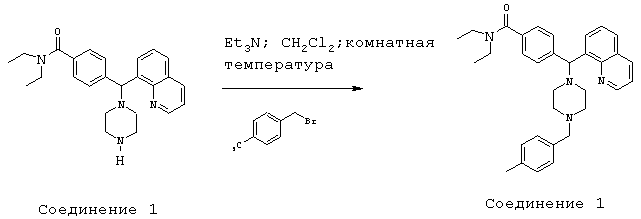
К раствору соединения 1 (0,80 г, 1,99 ммоль) в СН2Cl2 (20 мл) добавляли Et3N (0,83 мл, 5,97 ммоль), затем пара-метилбензилбромид (773 мг, 4,18 ммоль). Эту реакционную смесь перемешивали в течение ночи, а затем концентрировали при пониженном давлении. Очистка путем обращенно-фазовой хроматографии, используя 10%-30% СН3CN/N2О.
(М+1) вычисленная: 507,70, (М+1) наблюдаемая: 507,20.
ИК (NaCl, свободный амин) 2969, 2807, 2360, 1628, 1455, 1425, 1286, 1134, 1095 (см-1).
1H ЯМР: (CDCl3, свободный амин): δ=1.0, 1.1 (2m, 6H, амид-Ме), 2.31 (s, 3H, Ar-Me), 2.5 (m, 8H, пиперазин-Н), 3.2, 3.5 (2m, амид-СН2), 3.49 (s, 2H, ArCH2N), 6.03 (s, 1H, Ar2CH), 7.06-7.68 (m, 11H, Ar-Н), 8.01-8.12 (m, 2H, Ar-H), 8.93 (m, 1H, Ar-H).
Анализ (C32H38Cl2N4O) С, Н, N.
Примеры 5 и 6
Разделение энантиомеров соединения 5 с получением соединений 6 и 7
Препаративное разделение данного соединения проводили на полупрепаративной колонке Chiralcel AD (21 мм × 25 см), используя смесь гексан/EtOH/диэтиламин в соотношении 80:20:0,1 в качестве подвижной фазы. Обнаружили, что на колонке Chiralcel AD (-)-изомер элюируется первым.
Пример 5
(-)4-[(4-метилбензил-1-пиперазинил)(8-хинолинил)метил]-N,N-диэтилбензамид (соединение 6)
[α]D 25: -131o (c 1,0, MeOH)
Пример 6
(+)4-[(4-метилбензил-1-пиперазинил)(8-хинолинил)метил]-N,N-диэтилбензамид (соединение 7)
[α]D 25: +124o (c 1,4, МеОН)
Пример 7
Получение 4-[{4-[4-(трет-бутил)бензил]-1-пиперазинил}(8-хинолинил)метил]-N,N-диэтилбензамида дигидрохлорида (соединение 8)
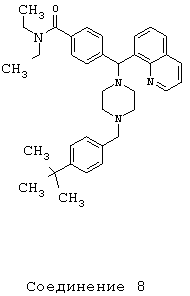
С помощью методики, аналогичной методике получения соединения 2, получили соединение 8, указанное в заголовке. Алкилирование осуществляли 4-трет-бутилбензилбромидом.
(МС) (ES): 549,53 (МН+).
ИК (NaCl, свободный амин) 2963, 2807, 2360, 1631, 1456, 1425, 1285, 1135, 1094, 1001 (см-1).
1H ЯМР; (CDCl3, свободный амин): δ=1.0, 1.2 (2m, 6H), 1.29 (s, 9H), 2.50 (m, 8H), 3.2, 3.5 (2m), 3.50 (s, 2H), 6.04 (s, 1H), 7.16-7.68 (m, 11H), 7.98-8.10 (m, 2H), 8.92 (m, 1H).
Анализ (С36Н46Cl2N4O) С, Н, N.
Пример 8
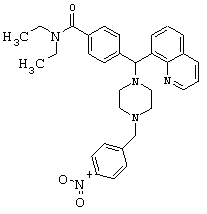
Получение N,N-диэтил-4-[[4-(4-нитробензил)1-пиперазинил](8-хинолинил)метил]бензамида дигидрохлорида (соединение 9)
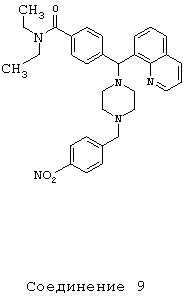
С помощью методики, аналогичной методике получения соединения 2, описанной выше, получили соединение 9, указанное в заголовке. Алкилирование осуществляли 4-нитробензилбромидом.
(МС) (ES): 538,04 (МН+).
ИК (NaCl, свободный амин) 2969, 2809, 2360, 1626, 1518, 1456, 1426, 1343, 1286, 1134, 1095, 1001 (см-1).
1H ЯМР: (CDCl3, свободный амин): δ=1.0, 1.2 (2m, 6H), 2.50 (m, 8H), 3.2, 3.5 (2m), 3.60 (s, 2H), 6.05 (s, 1H), 7.18-8.16 (m, 13H), 8.94 (m, 1H).
Анализ (С32Н37Cl2N5O3) С, Н, N.
Пример 9
Получение 4-[{4-[2,4-бис(трифторметил)бензил]1-1-пиперазинил}(8-хинолинил)метил]-N,N-диэтилбензамида дигидрохлорида (соединение 10)
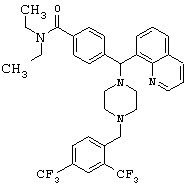
Соединение 10
Следуя методике, аналогичной методике получения соединения 2, описанной выше, получили соединение 10, указанное в заголовке. Алкилирование осуществляли 2,4-бис(трифторметил)бензилбромидом.
(МС) (ES): 629,08 (МН+).
ИК (NaCl, свободный амин) 2970, 2811, 2360, 1628, 1456, 1426, 1346, 1275, 1170, 1128 (см-1).
1H ЯМР: (CDCl3, свободный амин): δ=1.0, 1.2 (2m, 6H), 2.48 (m, 8H), 3.2, 3.5 (2m), 3.71 (s, 2H), 6.06 (s, 1H), 7.20-8.14 (m, 12H), 8.95 (m, 1H).
Анализ (C34H36Cl2F6N4O) С, Н, N.
Пример 10
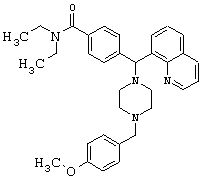
Получение N,N-диэтил-4-[[4-(4-метоксибензил)-1-пиперазинил](8-хинолинил)метил]бензамида дигидрохлорида (соединение 11)
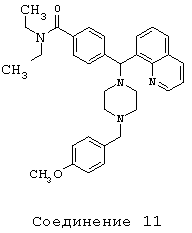
С помощью методики, аналогичной методике получения соединения 2, описанной выше, получили соединение 11, указанное в заголовке. Алкилирование осуществляли 4-метоксибензилхлоридом.
(МС) (ES): 523,45 (МН+).
ИК (NaCl, свободный амин) 2966, 2806, 2360, 1627, 1510, 1456, 1426, 1286,1246, 1134, 1095 (см-1).
1H ЯМР: (CDCl3, свободный амин): δ=1.0, 1.2 (2m, 6H), 2.48 (m, 8H), 3.2, 3.5 (2m), 3.47 (s, 2H), 3.78 (s, 3Н), 6.03 (s, 1H), 6.80-7.68 (m, 11H), 8.01-8.12 (m, 2H), 8.93 (m, 1H).
Анализ (С33Н40Cl2N4O2) С, Н, N.
Пример 11
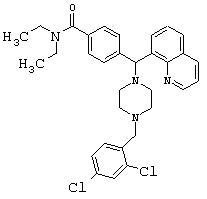
Получение 4-[[4-(2,4-дихлорбензил)-1-пиперазинил](8-хинолинил)метил]-N,N-диэтилбензамида дигидрохлорида (соединение 12)
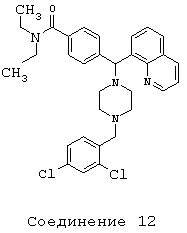
Следуя методике, аналогичной методике получения соединения 2, описанной выше, получили соединение 12, указанное в заголовке. Алкилирование осуществляли 2,4-дихлорбензилбромидом.
(МС) (ES): 562,45 (МН+).
ИК (NaCl, свободный амин) 2968, 2810, 2360, 2341, 1627, 1470, 1426, 1285, 1134, 1095 (см-1).
1H ЯМР: (CDCl3, свободный амин): δ=1.0, 1.1 (2m, 6H), 2.5 (m, 8H), 3.2, 3.5 (2m), 3.58 (s, 2H), 6.05 (s, 1H), 7.14-7.70 (m, 10H), 8.06 (m, 2H), 8.94 (m, 1H).
Анализ (С32Н36Cl4N4O) С, Н, N.
Пример 12
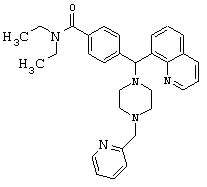
Получение N,N-диэтил-4-[[4-(2-пиридинилметил)-1-пиперазинил](8-хинолинил)метил]бензамида дигидрохлорида (соединение 13)
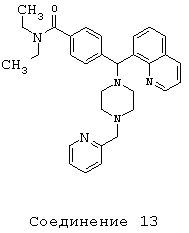
Соединение 1 (80 мг, 0,20 ммоль) растворяли в МеОН (2 мл) с 2-пиридилкарбоксальдегидом (39 мкл, 0,40 ммоль) и НОАс (1 мкл, 0,02 ммоль). Добавляли цианоборгидрид натрия (26 мг, 0,40 ммоль), и перемешивание продолжали в течение 48 ч. Растворитель выпаривали и остаток очищали путем хроматографии на силикагеле (0-10% МеОН в СН2Cl2). Получили 38 мг (39%) продукта.
(MC)(ES):494,19(MH+).
ИК (NaCl, свободный амин) 2968, 2809, 2360, 1626, 1455, 1428, 1286. 1134, 1094, 1001 (см-1).
1H ЯМР: (CDCl3, свободный амин): δ=1.0, 1.2 (2m, 6H), 2.50 (m, 8H), 3.2, 3.5 (2m), 3.69 (s, 2H), 6.05 (s, 1H), 7.12-7.70 (m, 10Н), 8.08 (m, 2H), 8.54 (m, 1H),8.94(m, 1H).
Анализ (С31Н37Cl2N5O) С, Н, N.
Пример 13
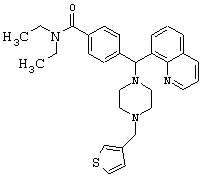
Получение N,N-диэтил-4-[[4-(3-тиенилметил)-1-пиперазинил](8-хинолинил)метил]бензамида (соединение 14)
Соединение 14, указанное в заголовке, получили, следуя методике синтеза Схемы 3, приведенной ниже.
Схема 3
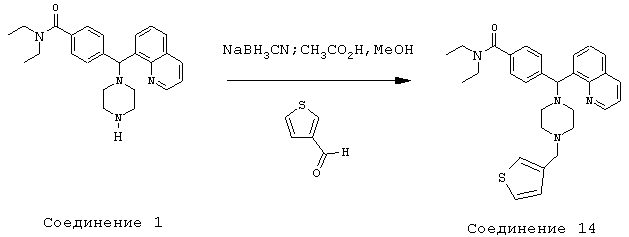
К раствору соединения 1 (500 мг, 0,99 ммоль) в метаноле (10 мл) добавляли тиофен-3-карбоксальдегид (104 мкл, 1,19 ммоль), а затем уксусную кислоту (0,1 мл, 1%) и цианоборгидрид натрия (186,6 мг, 2,97 ммоль). Эту реакционную смесь перемешивали в течение ночи, затем добавляли 2 н гидроксид натрия и эту смесь экстрагировали метиленхлоридом (3х). Объединенные метиленхлоридные экстракты высушивали над Na2SO4, фильтровали и концентрировали при пониженном давлении. С помощью очистки путем обращенно-фазовой хроматографии, используя 10%-30% СН3CN/Н2О (ТФУ в качестве буфера), получили 258 мг желаемого продукта (соль ТФУ).
Степень очистки по ВЭЖХ: >99% (215 нм); >95% (254 нм)
(М+1) вычисленная: 499,25, (М+1) наблюдаемая: 499,46.
Анализ: вычислено для (С30Н34N4OS × 2.80 С2HO2F3 × 1.80 Н2O): С: 50,28%; Н: 4,79%; N: 6,59%; О: 15,80%; S: 3,77%; F: 18,77%. Обнаружено: С: 50,28%; Н: 4,83%; N: 6,53%.
1H ЯМР: 8.95 (dd, 1H, J=4.4, 2.0 Гц), 8.38 (dd, 1H, J=8.0, 2.0 Гц), 8.00 (dd, 1H, J=7.2, 1.6 Гц), 7.84 (dd, 1H, J=8.0, 1.6 Гц), 7.52-7.62 (m, 5H), 7.45 (dd, 1H, J=4.8, 2.8 Гц), 7.20 (dd, 2H, J=8.8, 2.2 Гц), 7.11 (dd, 1H, J=4.8, 1.6 Гц), 5.96 (s, 1H), 4.27 (s, 2H), 3.34-3.44 (m, 2H), 3.22-3.28 (m, 4H), 3.04-3.14 (m, 2H), 2.66-2.88 (m, 4H), 1.04-1.14 (m, 3Н), 0.88-0.98 (m, 3H).
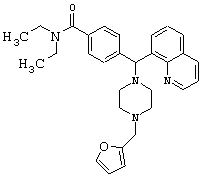
Пример 14
Получение N,N-диэтил-4-[[4-(2-фуранилметил)-1-пиперазинил](8-хинолинил)метил]бензамида (соединение 15)
Соединение 15, указанное в заголовке, получили, следуя методике синтеза Схемы 4, приведенной ниже.
Схема 4
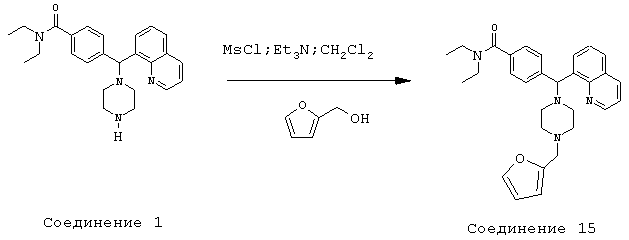
К раствору фурфурилового спирта (0,19 мл, 2,24 ммоль) и триэтиламина (0,52 мл, 3,73 ммоль) в метиленхлориде (4 мл) при 0°С добавляли метансульфонилхлорид (0,17 мл, 2,24 ммоль). Эту смесь перемешивали в течение 1 ч при 0°С, затем добавляли соединение 1 (300 мг, 0,75 ммоль). Эту реакционную смесь оставляли нагреваться до комнатной температуры и перемешивали в течение ночи, затем нагревали до 45°С и перемешивали в течение 1,5 часов. Эту реакционную смесь оставляли охлаждаться до комнатной температуры и добавляли 2 н NaOH, пока рН не стал щелочным. Эту смесь экстрагировали метиленхлоридом (3х). Объединенные метиленхлоридные экстракты высушивали над Na2SO4, фильтровали и концентрировали при пониженном давлении. С помощью очистки путем обращенно-фазовой хроматографии, используя 10%-25% СН3CN/H2О (ТФУ в качестве буфера), получили 197 мг желаемого продукта (соль ТФУ).
Степень очистки по ВЭЖХ: >99% (215 нм, 254 нм и 280 нм)
(М+1) вычисленная: 483,63, (М+1) наблюдаемая: 483,30.
1H ЯМР: 8.89 (dd, 1Н, J=4.4, 1.6 Гц), 8.29 (dd, 1H, J=8.0, 1.6 Гц), 7.97 (dd, 1Н, J=7.2, 1.6 Гц), 7.79 (d, 1H, J=7.2 Гц), 7.61 (d, 2H, J=8.0 Гц), 7.52-7.58 (m, 2H), 7.48 (dd, 1H, J=8.0, 4.4 Гц), 7.19 (d, 2H, J=8.0 Гц), 6.56 (d, 1H, J=3.2 Гц), 6.40 (dd, 1H, J=3.2, 2.4 Гц), 6.02 (s, 1H), 4.26 (s, 2H), 3.34-3.44 (m, 2H), 3.16-3.26 (m, 4H), 3.04-3.14 (m, 2H), 2.68-2.86 (m, 4H), 1.06-1.14 (m, 3Н), 0.90-0.98 (m, 3H).
Пример 15
Получение N,N-диэтил-4-[[4-(3-фуранилметил)-1-пиперазинил](8-хинолинил)метил]бензамида (соединение 16)
Соединение 16, указанное в заголовке, получили, следуя методике синтеза Схемы 5, приведенной ниже.
Схема 5
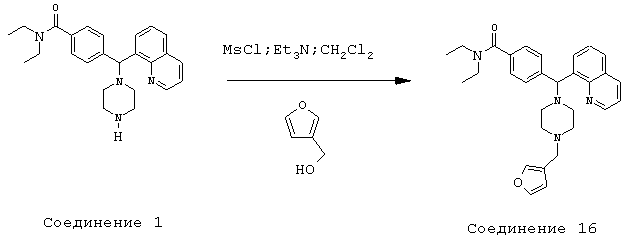
К раствору 3-фуранметанола (220 мг, 2,24 ммоль) и триэтиламина (0,52 мл, 3,73 ммоль) в метиленхлориде (4 мл) при 0°С добавляли метансульфонилхлорид (0,17 мл, 2,24 ммоль). Эту смесь перемешивали в течение 1 ч при 0°С, затем добавляли соединение 1 (300 мг, 0,75 ммоль). Эту реакционную смесь оставляли нагреваться до комнатной температуры и перемешивали в течение ночи, затем нагревали до 45°С и перемешивали в течение 3,5 часов. Эту реакционную смесь оставляли охлаждаться до комнатной температуры и добавляли 2 н NaOH, пока рН не стал щелочным. Эту смесь экстрагировали метиленхлоридом (3х). Объединенные метиленхлоридные экстракты высушивали над HO2SO4, фильтровали и концентрировали при пониженном давлении. С помощью очистки путем обращенно-фазовой хроматографии, используя 10%-25% СН3CN/Н2О (ТФУ в качестве буфера), получили 293 мг желаемого продукта (соль ТФУ).
Степень очистки по ВЭЖХ: >98% (215 нм и 280 нм); >99% (254 нм).
(М+1) вычисленная: 483,63, (М+1) наблюдаемая: 483,34.
Анализ: вычислено для (С30Н34N4О2 × 3.10 С2HO2F3 × 1.70 Н20): С: 50,17%; Н: 4,71%; N: 6,46%; О: 18,27%; F: 20,39%. Обнаружено: С: 50,14%; Н: 4,76%; N; 6,38%.
1H ЯМР: 8.93 (dd, 1H, J=4.4, 2.0 Гц), 8.36 (dd, 1H, J=8.6, 2.0 Гц), 8.00 (dd, 1H, J=7.4, 1.2 Гц), 7.82 (dd, 1H, J=7.6, 1.2 Гц), 7.48-7.66 (m, 6H), 7.19 (d, 2H, J=8.0 Гц), 6.46 (s, 1H), 5.97 (s, 1H), 4.13 (s, 2H), 3.32-3.44 (m, 2H), 3.20-3.28 (m, 4H), 3.04-3.14 (m, 2H), 2.66-2.86 (m, 4H), 1.04-1.14 (m, 3H), 0.88-0.98 (m, 3H).
Пример 16
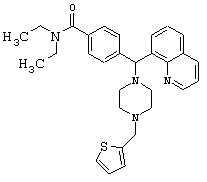
Получение N,N-диэтил-4-[[4-(2-тиофенилметил)-1-пиперазинил](8-хинолинил)метил]бензамида дигидрохлорида (соединение 17)
Соединение 17, указанное в заголовке, получили, следуя методике синтеза Схемы 6, приведенной ниже.
Схема 6
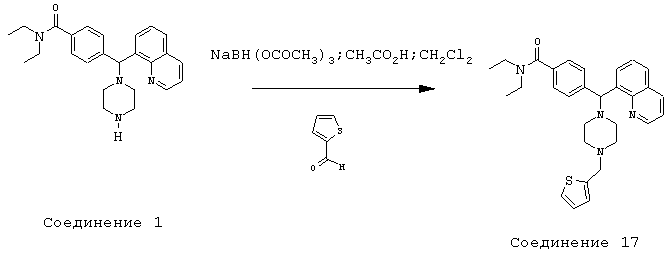
К раствору соединения 1 (0,99 ммоль) в метиленхлориде (10 мл) добавляли 2-тиофенкарбоксальдегид (190 мкл, 1,98 ммоль), а затем уксусную кислоту (0,1 мл, 1%). Эту смесь перемешивали в течение 30 минут, затем добавляли триацетоксиборгидрид натрия (0,63 г, 2,97 ммоль), и эту реакционную смесь перемешивали в течение ночи. Эту реакционную смесь нейтрализовали 2 н гидроксидом натрия и экстрагировали метиленхлоридом (3х). Объединенные метиленхлоридные экстракты высушивали над Na2SO4, фильтровали и концентрировали при пониженном давлении. С помощью очистки путем обращенно-фазовой хроматографии, используя 10%-30% CH3CN/H2O (ТФУ в качестве буфера), получили 115 мг желаемого продукта (соль ТФУ).
Степень очистки по ВЭЖХ; >99% (215 нм); >96% (254 нм).
(М+1) вычисленная: 499,25, (М+1) наблюдаемая: 499,33.
Анализ: вычислено для (C30H34N4OS × 2.50 С2HO2F3 × 0.10 H2O): С: 53,51%; Н: 4,71%; N: 7,13%; О: 12,42%; S: 4,08%; F: 18,14%. Обнаружено: С: 53,49%; Н: 4,63%; N: 7,49%.
1H ЯМР: 8.91 (dd, 1H, J=4.0, 1.6 Гц), 8.30 (dd, 1H, J=8.8, 1.6 Гц), 7.96 (dd, 1H, J=7.4, 1.4 Гц), 7.81 (d, 1H, J=7.2 Гц), 7.62 (d, 2H, J=8.0 Гц), 7.46-7.58 (m, 3H), 7.20 (d, 2H, J=8.0 Гц), 7.14-7.22 (m, 1H), 7.00 (dd, 1H, J=5.2, 3.6 Гц), 6.03 (s, 1H), 4.38 (s, 2H), 3.34-3.44 (m, 2H), 3.14-3.22 (m, 4H), 3.06-3.12 (m, 2H), 2.74-2.88 (m, 4H), 1.04-1.14 (m, 3H), 0.88-0.98 (m, 3H).
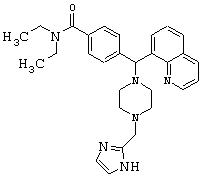
Пример 17
Получение N,N-диэтил-4-[[4-(2-имидазолилметил)-1-пиперазинил](8-хинолинил)метил]бензамида дигидрохлорида (соединение 18)
Соединение 18, указанное в заголовке, получили, следуя методике синтеза Схемы 7, приведенной ниже.
Схема 7
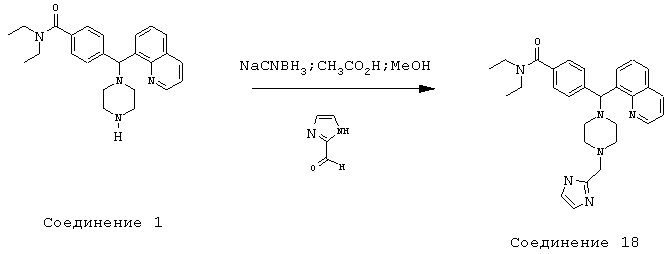
К раствору соединения 1 (0,99 ммоль) в метаноле (10 мл) добавляли 2-имидазолкарбоксальдегид (114 мг, 1,19 ммоль), а затем уксусную кислоту (0,5 мл, 5%). Эту смесь перемешивали в течение 3 часов, затем добавляли цианоборгидрид натрия (186,6 мг, 2,97 ммоль), и эту реакционную смесь перемешивали в течение ночи. Эту реакционную смесь нейтрализовали 2 н гидроксидом натрия и экстрагировали метиленхлоридом (3х). Объединенные метиленхлоридные экстракты высушивали над Na2SO4, фильтровали и концентрировали при пониженном давлении. С помощью очистки путем обращенно-фазовой хроматографии, используя 10%-30% CH3CN/H2O (ТФУ в качестве буфера), получили соль ТФУ. Соль HCl получили, используя HCl/эфир. Выход: 60,3 мг желаемого продукта (соль HCl).
Степень очистки по ВЭЖХ: >95% (215 нм); >93% (254 нм).
(М+1) вычисленная: 483,29, (М+1) наблюдаемая: 483,19.
1H ЯМР: 9.12-9.22 (m, 1Н), 8.54-8.62 (m, 1H), 8.08-8.16 (m, 1H), 7.98-8.04 (m, 1H), 7.60-7.86 (m, 4H), 7.38-7.46 (m, 2H), 7.22-7.32 (m, 2H), 6.32 (s, 1H), 4.11 (s, 2H), 2.94-3.40 (m, 12H), 0.88-1.12 (m, 6H).
Пример 18
Получение N,N-диэтил-4-[[4-(4-имидазолилметил)-1-пиперазинил](8-хинолинил)метил]бензамида дигидрохлорида (соединение 19)
Соединение 19, указанное в заголовке, получили, следуя методике синтеза Схемы 8, приведенной ниже.
Схема 8
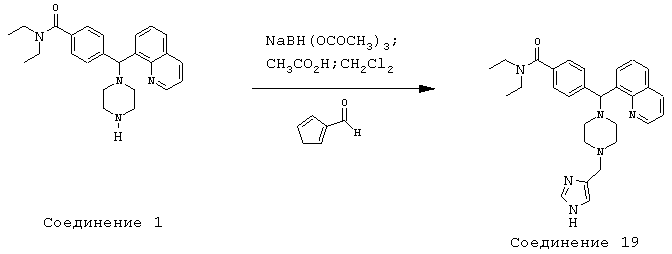
К раствору 434 (400 мг, 0,99 ммоль) и 4-имидазолкарбоксальдегида (95,5 мг, 0,99 ммоль) в метиленхлориде (10 мл) при комнатной температуре добавляли уксусную кислоту (0,1 мл). Эту смесь перемешивали в течение 5 часов, затем добавляли триацетоксиборгидрид натрия (632 мг, 2,98 ммоль). Эту реакционную смесь перемешивали в течение ночи и нейтрализовали 2 н гидроксидом натрия. Эту смесь экстрагировали метиленхлоридом (3х). Объединенные метиленхлоридные экстракты высушивали над Na2SO4, фильтровали и концентрировали при пониженном давлении. С помощью очистки путем обращенно-фазовой хроматографии, используя 15% СН3CN/H2О (ТФУ в качестве буфера), получили 103 мг желаемого продукта (соль ТФУ).
Степень очистки по ВЭЖХ: >99% (215 нм, 254 нм и 280 нм).
(М+1) вычисленная: 483,28, (М+1) наблюдаемая: 482,96.
Анализ: вычислено для (C29H34N6O × 3.80 С2HO2F3 × 0.80 H2O): С: 47,25%; Н: 4,27%; N: 9,03%; О: 16,17%; F: 23,28%. Обнаружено: С: 47,31%; Н: 4,40%; N: 8,87%.
1H ЯМР: 8.99 (dd, 1H, J=4.4, 1.2 Гц), 8.76 (d, 1H, J=1.2 Гц), 8.39 (dd, 1H, J=8.8, 1.2 Гц), 7.93 (dd, 1H, J=7.2, 1.6 Гц), 7.86 (dd, 1H, J=8.0, 1.6 Гц), 7.71 (d, 2H, J=8.8 Гц), 7.60 (dd, 1H, J=8.8, 4.4 Гц), 7.56 (dd, 1H, J=8.0, 7.2 Гц), 7.40 (s, 1H), 7.27 (d, 2H, J=8.8 Гц), 6.12 (s, 1H), 3.74 (s, 2H), 3.38 (q, 2H, J=6.4 Гц), 3.10-3.25 (m, 6H), 3.06 (q, 2H, J=7.2 Гц), 2.75-2.90 (m, 2H), 1.08 (t, 3H, J=6.4 Гц), 0.92 (t, 3H, J=7.2 Гц).
Пример 19
Получение N,N-диэтил-4-[[4-(3-триазолилметил)-1-пиперазинил](8-хинолинил)метил]бензамида дигидрохлорида (соединение 20)
Соединение 20, указанное в заголовке, получили, следуя методике синтеза Схемы 9, приведенной ниже.
Схема 9
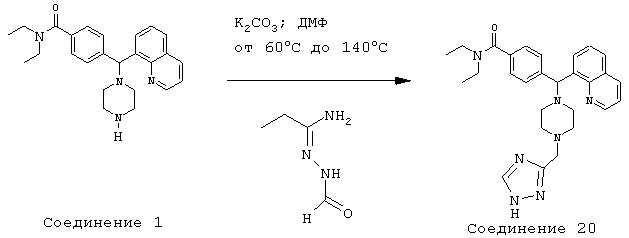
К раствору 434 (200 мг, 0,50 ммоль) в диметилформамиде (10 мл) при комнатной температуре добавляли карбонат калия (275 мг, 1,99 ммоль), а затем N-формамидо-2-(хлорметил)ацетамидин (170 мг, 1,24 ммоль). Эту реакционную смесь нагревали до 60°С и перемешивали в течение 2 суток, затем температуру повышали до 140°С и перемешивали в течение 3 часов. Эту реакционную смесь оставляли охлаждаться до комнатной температуры и добавляли воду. Эту смесь экстрагировали этилацетатом (3х). Объединенные этилацетатные экстракты высушивали над Na2SO4, фильтровали и концентрировали при пониженном давлении. С помощью очистки путем обращенно-фазовой хроматографии, используя 20% CH3CN/H2O (ТФУ в качестве буфера), получили 21 мг желаемого продукта (соль ТФУ).
Степень очистки по ВЭЖХ: >99% (215 нм, 254 нм и 280 нм).
(М+1) вычисленная: 484,28, (М+1) наблюдаемая: 483,92.
Анализ: вычислено для (С28Н33N7О × 3.30 С2HO2F3 × 3.30 Н2O): С: 45,20%; Н: 4,70%; N: 10,66%; О: 18,97%; F: 20,46%. Обнаружено: С: 45,12%; Н: 4,60%; N: 10,84.
1H ЯМР: 8.94 (dd, 1H, J=4.4, 1.6 Гц), 8.38 (s, 1H), 8.33 (dd, 1H, J=8.0, 1.2 Гц), 7.93 (d, 1H, J=7.2 Гц), 7.85 (d, 1H, J=7.2 Гц), 7.65 (d, 2H, J=8.8 Гц), 7.51-7.58 (m, 2H), 7.23 (d, 2H, J=8.8 Гц), 6.15 (s, 1H), 4.21 (s, 2H), 3.40-3.50 (m, 2H), 3.10-3.30 (m, 8H), 2.90-3.10 (m, 2H), 0.90-1.30 (m, 6H).
Фармацевтические композиции
Новые соединения по настоящему изобретению можно вводить перорально, внутримышечно, подкожно, местно, интраназально, внутрибрюшинно, интраторакально, внутривенно, эпидурально, внутриоболочечно, интрацеребровентрикулярно и путем инъекции в суставы.
Предпочтительным путем введения является пероральный, внутривенный или внутримышечный.
Дозировка будет зависеть от пути введения, тяжести заболевания, возраста и массы пациента и других факторов, обычно учитываемых лечащим врачом при определении индивидуального режима и уровня дозировки как наиболее подходящих для конкретного пациента.
Для изготовления фармацевтических композиций из соединений по данному изобретению инертные фармацевтически приемлемые носители могут быть либо твердыми, либо жидкими. Препараты в виде твердых форм включают в себя порошки, таблетки, диспергируемые гранулы, капсулы, крахмальные капсулы и суппозитории.
Твердым носителем может быть одно или более чем одно вещество, которые могут также служить в качестве разбавителей, корригентов, солюбилизирующих агентов, смазывающих веществ, суспендирующих агентов, связывающих веществ или разрыхляющих агентов для таблеток; он также может являться инкапсулирующим материалом.
В порошках носитель представляет собой тонко измельченное твердое вещество, которое находится в смеси с тонко измельченным активным компонентом. В таблетках активный компонент смешан с носителем, обладающим необходимыми связующими свойствами, в подходящих пропорциях и спрессован в желаемую форму желаемого размера.
Для приготовления композиций в виде суппозиториев сначала плавят легкоплавкий воск, такой как смесь глицеридов жирных кислот и масла какао, и диспергируют в нем активный ингредиент путем, например, перемешивания. Затем эту расплавленную однородную смесь заливают в формы подходящего размера и оставляют охлаждаться и затвердевать.
Подходящими носителями являются карбонат магния, стеарат магния, тальк, лактоза, сахар, пектин, декстрин, крахмал, трагакант, метилцеллюлоза, натрийкарбоксиметилцеллюлоза, легкоплавкий воск, масло какао и тому подобное.
Фармацевтически приемлемыми солями являются ацетат, бензолсульфонат, бензоат, бикарбонат, битартрат, бромид, ацетат кальция, камсилат, карбонат, хлорид, цитрат, дигидрохлорид, эдетат, эдисилат, эстолат, эзилат, фумарат, глюкаптат, глюконат, глутамат, гликолиларсанилат, гексилрезорцинат, гидрабамин, гидробромид, гидрохлорид, гидроксинафтоат, йодид, изэтионат, лактат, лактобионат, малат, малеат, манделат, мезилат, метилбромид, метилнитрат, метилсульфат, мукат, напсилат, нитрат, памоат (эмбонат), пантотенат, фосфат/дифосфат, полигалактуронат, салицилат, стеарат, субацетат, сукцинат, сульфат, таннат, тартрат, теоклат, триэтиодид, соли бензатина, хлорпрокаина, холина, диэтаноламина, этилендиамина, меглумина, прокаина, алюминия, кальция, лития, магния, калия, натрия и цинка. Предпочтительными фармацевтически приемлемыми солями являются гидрохлориды и битартраты. Соли гидрохлориды являются особенно предпочтительными.
В термин «композиция» следует включать препарат активного компонента с инкапсулирующим материалом в качестве носителя, образующим капсулу, в которой активный компонент (с другими носителями или без них) окружен носителем, который, таким образом, находится в связи с ним. Подобным образом включены крахмальные капсулы.
Таблетки, порошки, крахмальные капсулы и капсулы можно применять в качестве твердых лекарственных форм, подходящих для перорального введения.
Композиции в виде жидких форм включают в себя растворы, суспензии и эмульсии. Растворы активных соединений в стерильной воде или в воде-пропиленгликоле можно упомянуть в качестве примера жидких препаратов, подходящих для парентерального введения. Жидкие композиции можно также изготавливать в виде раствора препарата в растворе водного полиэтиленгликоля.
Водные растворы для перорального введения можно изготавливать путем растворения активного компонента в воде и добавления подходящих красителей, корригентов, стабилизаторов и загущающих агентов по желанию. Водные суспензии для перорального применения можно изготавливать путем диспергирования тонко измельченного активного компонента в воде вместе с вязким материалом, таким как природные и синтетические смолы, камеди, метилцеллюлоза, натрийкарбоксиметилцеллюлоза и другие суспендирующие агенты, известные в области фармацевтических препаратов.
Предпочтительно фармацевтические композиции находятся в стандартной лекарственной форме. В такой форме композиция разделена на стандартные дозы, содержащие подходящие количества активного компонента. Стандартная лекарственная форма может представлять собой упакованный препарат, причем упаковка содержит дискретные количества препаратов, например упакованных таблеток, капсул и порошков во флаконах или в ампулах. Стандартная лекарственная форма может также представлять собой капсулу, крахмальную капсулу или таблетку саму по себе, либо может представлять собой подходящее количество любых из этих упакованных форм.
БИОЛОГИЧЕСКАЯ ОЦЕНКА
Модель in vitro
Культура клеток
Клетки человека 293S, экспрессирующие клонированные μ-, δ- и κ-рецепторы человека и устойчивость к неомицину, выращивали в суспензии при 37°С и 5% CO2 в качалочных колбах, содержащих свободную от кальция модифицированную Дульбекко среду Игла (DMEM), 10% FBS, 5% BCS, 0,1% плюроник (Pluronic) F-68 и 600 мкг/мл генетицина.
Препарат мембран
Клетки осаждали и ресуспендировали в буфере для лизиса (50 мМ Трис, рН 7,0, 2,5 мМ ЭДТА с фенилметилсульфонилфторидом (PMSF), добавленным непосредственно перед использованием до 0,1 мМ из 0,1 М исходного раствора в этаноле), инкубировали на льду в течение 15 мин, затем гомогенизировали, используя Polytron, в течение 30 сек. Суспензию центрифугировали при 1000 g (максимум) в течение 10 мин при 4°С. Супернатант хранили на льду, а осадки ресуспендировали и центрифугировали, как описано выше. Супернатанты от обоих центрифугирований объединяли и центрифугировали при 46000 g (максимум) в течение 30 мин. Осадки ресуспендировали в холодном Трис-буфере (50 мМ Трис/Cl, рН 7,0) и снова центрифугировали. Конечные осадки ресуспендировали в буфере для мембран (50 мМ Трис, 0,32 М сахароза, рН 7,0). Аликвоты (1 мл) в полипропиленовых пробирках замораживали в сухом льду/этаноле и хранили при -70°С до использования. Концентрации белка определяли модифицированным анализом по Лоури с додецилсульфатом натрия (SDS).
Анализы на связывание
Мембраны подвергали оттаиванию при 37°С, охлаждали на льду, пропускали 3 раза через иглу 25 размера и разбавляли в связывающем буфере (50 мМ Трис, 3 мМ MgCl2, 1 мг/мл бычьего сывороточного альбумина (BSA) (Sigma A-7888), рН 7,4, который хранили при 4°С после фильтрования через 0,22 м фильтр и к которому были добавлены свежеприготовленные 5 мкг/мл апротинина, 10 мкМ бестатина, 10 мкМ дипротина А (без DTT). Аликвоты по 100 мкл (на мкг белка, см. Таблицу 1) добавляли в охлажденные на льду полипропиленовые пробирки 12×75 мм, содержащие 100 мкл соответствующего радиоактивного лиганда (см. Таблицу 1) и 100 мкл тестируемых пептидов в различных концентрациях. Общее (ОС) и неспецифическое (НС) связывание определяли в отсутствие и в присутствии 10 мкМ налоксона, соответственно. Пробирки встряхивали и инкубировали при 25°С в течение 60-75 мин, и через это время содержимое быстро фильтровали в вакууме и промывали примерно 12 мл/пробирку охлажденного на льду буфера для отмывки (50 мМ Трис, рН 7,0, 3 мМ MgCl2) через фильтры GF/B (Whatman), предварительно замоченные в течение по меньшей мере 2 ч в 0,1% полиэтиленимине. Радиоактивность (число распадов в минуту, dpm), оставшуюся на фильтрах, измеряли с использованием бета-счетчика после замачивания фильтров в течение по меньшей мере 12 ч в мини-флаконах, содержащих 6-7 мл сцинтилляционной жидкости. Если анализ ставили в 96-луночных планшетах с глубокими лунками, фильтрование осуществляли через 96-луночные стандартные фильтры, замоченные в PEI, которые промывали 3×1 мл буфера для отмывки и высушивали в печи при 55°С в течение 2 ч. Фильтровальные планшеты подсчитывали в TopCount (Packard) после добавления 50 мкл сцинтилляционной жидкости MS-20 на лунку.
Анализ данных
Специфичное связывание (СС) вычисляли как ОС-НС, и СС в присутствии различных тестируемых пептидов выражали в процентах от контрольного СС. Значения IC50 и коэффициента Хилла (Hill) (nH) для лигандов по замещению специфично связанного радиоактивного лиганда вычисляли на основании графиков в логарифмическом масштабе или программ для вычерчивания кривой по точкам, таких как Ligand, GraphPad Prism, SigmaPlot или ReceptorFit. Значения Ki вычисляли на основании уравнения Cheng-Prussoff. Значения среднее ± средняя квадратичная ошибка (СКО) IC50, Кi и nH представляли для тестируемых лигандов по меньшей мере в трех кривых замещения. Биологические данные представлены ниже в Таблице 1.
Эксперименты по насыщению рецептора
Значения Кδ радиоактивных лигандов определяли путем проведения анализов на связывание на клеточных мембранах с соответствующими радиоактивными лигандами в концентрациях, находящихся в диапазоне от 0,2 до 5-кратных от оцененного значения Кδ (вплоть до 10-кратных, если необходимые количества радиоактивного лиганда доступны). Специфичное связывание радиоактивного лиганда выражали в пмоль/мг мембранного белка. Значения Кδ и Вmax из индивидуальных экспериментов получили на основании нелинейных соответствий специфично связанного (СС) против нМ свободного (С) радиоактивного лиганда из индивидуального эксперимента согласно однопозиционной модели.
ОПРЕДЕЛЕНИЕ МЕХАНИЧЕСКОЙ АЛЛОДИНИИ С ИСПОЛЬЗОВАНИЕМ ТЕСТИРОВАНИЯ ПО VON FREY
Тестирование проводили между 08:00 и 16:00 ч, используя способ, описанный Chaplan et al. (1994). Крыс помещали в плексигласовые клетки на верхнюю поверхность дна из проволочной сетки, которая обеспечивала доступ к лапе, и оставляли для привыкания на 10-15 мин. Тестируемой областью была середина подошвы левой задней лапы, избегая менее чувствительных подушечек стопы. На лапу воздействовали серией из 8 волосков Von Frey логарифмически возрастающей жесткости (0,41, 0,69, 1,20, 2,04, 3,63, 5,50, 8,51 и 15,14 грамм; Stoelting, III, USA). Волосок Von Frey прикладывали снизу сетчатого пола перпендикулярно поверхности подошвы с достаточной силой, чтобы вызвать небольшой прогиб против лапы, и держали в течение примерно 6-8 секунд. Положительный ответ отмечали, если лапа резко отдергивалась. Вздрагивание немедленно после удаления волоска также считали положительным ответом. Передвижение считали неопределенным ответом, и в таких случаях стимул повторяли.
ПРОТОКОЛ ТЕСТИРОВАНИЯ
Животных тестировали на 1-е сутки после операции для группы, обработанной FCA. 50%-ное пороговое значение отдергивания определяли, используя прямой способ Dixon (1980). Тестирование начинали с 2,04 г волоска, в середине серии. Стимулы всегда давали последовательным образом, либо восходящим, либо нисходящим. В отсутствие ответа в виде отдергивания лапы на исходно выбранный волосок давали более сильный стимул; в случае отдергивания лапы выбирали следующий более слабый стимул. Для вычисления оптимального порогового значения данным способом необходимо 6 ответов в непосредственной близости от 50%-ного порогового значения, и подсчет этих 6 ответов начинали, когда появлялось первое изменение ответа, то есть, когда пороговое значение впервые пересекалось. В случаях, когда пороговые значения оказывались вне диапазона стимулов, соответственно устанавливались значения 15,14 (нормальная чувствительность) или 0,41 (максимальная аллодиния). Полученную в результате картину положительных и отрицательных ответов вносили в таблицу, используя условные обозначения: Х=отсутствие отдергивания; О=отдергивание, и 50%-ное пороговое значение отдергивания интерполировали, используя формулу:
50% г пороговое значение = 10(Xf+k δ )/10000
где Xf=значение последнего использованного волоска Von Frey (логарифмические единицы); k=табличное значение (из Chaplan et al. (1994)) для картины положительных/отрицательных ответов; и δ=значение разности между стимулами (логарифмические единицы). Здесь δ=0,224.
Пороговые значения Von Frey переводили в процент от максимального возможного ответа (% МВО) согласно Chaplan et al., 1994. Для компьютерного вычисления % МВО использовали следующее уравнение:
% МВО=Порог лекарственной обработки (г) - порог аллодинии (г) × 100
Контрольный порог (г) - порог аллодинии (г)
ВВЕДЕНИЕ ТЕСТИРУЕМОГО ВЕЩЕСТВА
Перед тестированием по Von Frey крысам инъецировали (подкожно, внутрибрюшинно, внутривенно или перорально) тестируемое вещество, время между введением тестируемого соединения и тестом Von Frey зависело от природы тестируемого соединения.
ТЕСТ WRITHING
Уксусная кислота при введении мышам внутрибрюшинно будет вызывать сокращения живота. Затем их тело будет типичным образом вытягиваться. Когда вводят обезболивающие лекарства, это описанное движение наблюдают менее часто, и это лекарство выбирают в качестве потенциального хорошего кандидата.
Рефлекс Writhing считают полным и типичным только тогда, когда присутствуют следующие элементы: животное не находится в движении; нижняя часть спины слегка опущена; видна подошвенная сторона обеих лап.
(1) Приготовление растворов
Уксусная кислота (АсОН): 120 мкл уксусной кислоты добавляют к 19,88 мл дистиллированной воды до получения конечного объема 20 мл при конечной концентрации 0,6% АсОН. Затем этот раствор перемешивают (встряхиванием), и он готов для инъекции.
Соединение (лекарство). Каждое соединение получают и растворяют в наиболее подходящем носителе согласно стандартным методикам.
(2) Введение растворов
Соединение (лекарство) вводят перорально, внутрибрюшинно (i.p.), подкожно (s.c.) или внутривенно (i.v.) в дозе 10 мл/кг (учитывая среднюю массу тела мышей) за 20, 30 или 40 минут (соответственно классу соединения и его характеристикам) до тестирования. Когда соединение доставляют центрально: интравентрикулярно (i.c.v.) или внутриоболочечно (i.t), вводят объем 5 мкл.
АсОН вводят внутрибрюшинно (i.p.) в двух местах в дозе 10 мл/кг (учитывая среднюю массу тела мышей) непосредственно перед тестированием.
(3) Тестирование
Животное (мышь) наблюдают в течение периода 20 минут и в конце эксперимента отмечают и компилируют число случаев (рефлекс Writhing). Мышей держат в индивидуальных клетках «обувная коробка» с контактной подстилкой. Обычно наблюдают всего 4 мышей одновременно: один контроль и три дозы лекарства.
Протокол для определения GTP в мозге крыс
Культура клеток
А. Клетки человека 293S, экспрессирующие клонированные κ-, δ- и μ-рецепторы человека и резистентные к неомицину, выращивали в суспензии при 37°С и 5% CO2 в качалочных колбах, содержащих модифицированную Дульбекко среду Игла (DMEM) без кальция, 10% фетальную сыворотку коровы (FBS), 5% BCS, 0,1% плюроник F-68 и 600 мкг/мл генетицина.
Б. Мозги крыс взвешивали и промывали в ледяном фосфатно-солевом буферном растворе (PBS) (содержащем 2,5 мМ ЭДТА (этилендиаминтетрауксусной кислоты), рН 7,4). Мозги гомогенизировали с помощью Politron в течение 30 секунд (крысы) в ледяном буфере для лизиса (50 мМ Tris (трис-(гидроксиметил)-аминометан), рН 7,0, 2,5 мМ ЭДТА с 0,5 мМ фенилметилсульфонилфторидом (PMSF), который добавляют непосредственно перед применением, приготавливая его из 0,5 М исходного раствора в смеси ДМСО (диметилсульфоксид):этанол).
Получение мембран
Клетки осадили центрифугированием и ресуспендировали в буфере для лизиса (50 мМ Tris, рН 7,0, 2,5 мМ ЭДТА с 0,1 мМ PMSF, который добавляют непосредственно перед применением, приготавливая его из 0,1 М исходного раствора в этаноле), инкубировали на льду в течение 15 минут, затем гомогенизировали с помощью Politron в течение 30 секунд. Суспензию центрифугировали при 1000 g (макс) в течение 10 минут при 4°С. Надосадочную жидкость сохраняли на льду и осадки ресуспендировали и центрифугировали, как описано выше. Надосадочные жидкости от двух центрифугирований объединили и центрифугировали при 46000 g (макс) в течение 30 минут. Осадки ресуспендировали в холодном Tris-буфере (50 мМ Tris/Cl, рН 7,0) и снова центрифугировали. Конечные осадки ресуспендировали в буфере для мембран (50 мМ Tris, 0,32 М сахароза, рН 7,0). Аликвоты (1 мл) в полипропиленовых пробирках заморозили в смеси сухой лед/этанол и хранили при -70°С до применения. Концентрации белка определяли с помощью модифицированного анализа по Лоури с додецилсульфатом натрия.
Тесты на связывание
Мембраны разморозили при 37°С, охладили на льду, пропустили 3 раза через иглу 25-го размера и разбавили в буфере для связывания (50 мМ Tris, 3 мМ MgCl2, 1 мг/мл бычий сывороточный альбумин (BSA) (Sigma A-7888), рН 7,4, который хранили при 4°С после фильтрования через 0,22 мкм фильтр и к которому перед экспериментом добавили 5 мкг/мл апротинина, 10 мкМ бестатина, 10 мкМ дипротина A (diprotin А), без DTT (дитиотрейтол). Аликвоты по 100 мкл добавили к охлажденным льдом полипропиленовым пробиркам (12×75 мм), содержащим 100 мкл соответствующего радиолиганда и 100 мкл тестируемого соединения в различных концентрациях. Общее (ТВ) и неспецифическое (NS) связывание определяли в отсутствие и в присутствии 10 мкМ налоксона, соответственно. Пробирки встряхивали и инкубировали при 25°С в течение 60-75 минут, после чего содержимое быстро отфильтровали в вакууме и промыли охлажденным льдом буфером для промывания в количестве примерно 12 мл/на пробирку (50 мМ Tris, рН 7,0, 3 мМ MgCl2) через GF/B фильтры (Whatman), предварительно вымоченные в течение по меньшей мере 2 часов в 0,1%-ном полиэтиленимине. Радиоактивность (число распадов в минуту), оставшуюся на фильтрах, измеряли с помощью счетчика бета-частиц после вымачивания фильтров в течение по меньшей мере 12 часов в мини-флаконах, содержащих 6-7 мл сцинтилляционной жидкости. Если тест проводили в 96-луночных планшетах с глубокими лунками, то фильтрацию осуществляют через 96-луночные универсальные фильтры, вымоченные в полиэтиленимине, которые промывали буфером для промывания (3×1 мл) и сушили в сушильном шкафу при 55°С в течение 2 часов. Радиоактивность фильтр-планшетов измеряли с помощью TopCount (Packard) после добавления 50 мкл на лунку сцинтилляционной жидкости MS-20.
Функциональные тесты
Агонистическую активность соединений измеряют определением степени, с которой комплекс рецептор-соединение активирует связывание GTP (гуанозинтрифосфата) с G-белками, с которыми связаны рецепторы. В тесте на связывание GTP GTP[γ]35S объединяют с тестируемыми соединениями и мембранами из клеток HEK-293S, экспрессирующих клонированные опиоидные рецепторы человека, или из гомогенизированного мозга крыс и мышей. Агонисты стимулируют связывание GTP[γ]35S в этих мембранах. Значения ЕС50 (концентрация соединения, при которой наблюдается эффект, равный 50% от максимального) и Emax (максимальный эффект, который может вызвать соединение) соединений определяют из кривых доза-ответ. Сдвиги вправо кривой доза-ответ дельта-антагонистом налтриндолом (naltrindole) осуществляют, чтобы подтвердить, что агонистическая активность опосредована через δ-рецепторы. Для функциональных тестов с δ-рецепторами человека ЕС50 (низкие) измеряют, когда δ-рецепторы человека, используемые в тесте, экспрессировались на низких уровнях по сравнению с таковыми, используемыми при определении ЕС50 (высокие). Значения Emax определяли по отношению к стандартному δ-агонисту SNC80, то есть, значение выше 100% имеет соединение, которое обладает лучшей эффективностью, чем SNC80.
Процедура для GTP в мозге крыс
Мембраны мозга крыс размораживают при 37°С, пропускают 3 раза через иглу с тупым концом 25-го размера и разбавляют в буфере для связывания GTPγS (50 мМ N-2-гидроксиэтилпиперазин-N'-2-этансульфоновой кислоты (Hepes), 20 мМ NaOH, 100 мМ NaCl, 1 мМ ЭДТА, 5 мМ MgCl2, pH 7,4, добавляют свежий 1 мМ DTT, 0,1% BSA (дитиотрейтол). В конце к растворам мембран добавляют 120 мкМ гуанозиндифосфата (GDP). ЕС50 и Emax соединений оценивают по 10-точечным кривым доза-ответ, составленным в 300 мкл с подходящим количеством мембранного белка (20 мкг/лунка) и 100000-130000 число распадов в минуту GTPγ35S на лунку (0,11-0,14 нМ). Базальное и максимальное стимулированное связывание определяют в отсутствие и в присутствии 3 мкМ SNC80.
Анализ данных
Специфическое связывание (SB) вычисляли как TB-NS, и SB в присутствии различных тестируемых соединений выражали как процент от контроля SB. Значения IC50 и коэффициент Хилла (nH) для лигандов при замещении специфически связанного радиолиганда вычисляли из логит-графиков или с помощью строящих кривую по точкам программ, таких как Ligand, GraphPad Prism, SigmaPlot или ReceptorFit. Значения К (константы ингибирования) вычисляли по уравнению Cheng-Prussoff. Среднее значение ± стандартная ошибка значений IC50, Ki и nH представлены для лигандов, тестированных по меньшей мере на трех кривых замещения.
| название | год | авторы | номер документа |
|---|---|---|---|
| ПРОИЗВОДНЫЕ ПИПЕРАЗИНА ИЛИ ПИПЕРИДИНА И ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ | 1996 |
|
RU2194702C2 |
| СПОСОБ ВОССТАНОВЛЕНИЯ НАРУШЕННЫХ ФУНКЦИЙ ОПИОИДНЫХ РЕЦЕПТОРОВ | 1996 |
|
RU2307833C2 |
| НОВЫЕ СОЕДИНЕНИЯ КОНДЕНСИРОВАННОГО ИМИДАЗОЛА, ОБЛАДАЮЩИЕ СВОЙСТВАМИ АГОНИСТОВ РЕЦЕПТОРА СВ2 | 2002 |
|
RU2312864C2 |
| ПРОИЗВОДНЫЕ 4-(ФЕНИЛ-ПИПЕРАЗИНИЛ-МЕТИЛ)-БЕНЗАМИДА, СПОСОБ ИХ ПОЛУЧЕНИЯ И ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ НА ИХ ОСНОВЕ | 2002 |
|
RU2297412C2 |
| ДИАРИЛМЕТИЛПИПЕРАЗИНОВЫЕ ПРОИЗВОДНЫЕ, ИХ ПОЛУЧЕНИЕ И ПРИМЕНЕНИЕ | 2005 |
|
RU2404172C2 |
| НОВЫЕ СОЕДИНЕНИЯ | 2000 |
|
RU2258703C2 |
| ПРОИЗВОДНЫЕ ХИНОЛИН-2-ОНА В КАЧЕСТВЕ ИНГИБИТОРОВ КИНАЗЫ C-KIT | 2016 |
|
RU2754858C2 |
| ПРОИЗВОДНЫЕ ФУРО[3,2-В]- И ТИЕНО[3,2-В]ПИРИДИНА В КАЧЕСТВЕ ИНГИБИТОРОВ TBK1 И IKKε | 2013 |
|
RU2622034C2 |
| ПРОИЗВОДНЫЕ ХИНОЛИНА И СОДЕРЖАЩИЕ ИХ ИНГИБИТОРЫ MELK | 2011 |
|
RU2582610C2 |
| Соединения, активные по отношению к бромодоменам | 2015 |
|
RU2743074C2 |
Настоящее изобретение относится к новым 4-(пиперазинил(8-хинолинил)метил)бензамидам общей формулы I
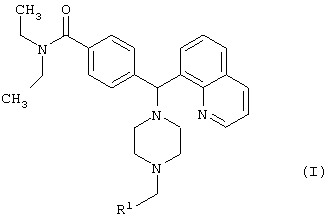
где R1 выбран из фенила, пиридинила, тиофенила, фуранила и имидазолила; причем каждое фенильное кольцо и гетероароматическое кольцо возможно и независимо дополнительно замещено 1, 2 или 3 заместителями, выбранными из прямого и разветвленного С1-С6алкила, NO2, CF3, С1-С6алкокси, хлоро, фторо, бромо и йодо, а также их фармацевтически приемлемые соли. Соединения могут быть использованы в терапии, в частности при устранении боли. Описаны также фармацевтическая композиция на основе соединений I и способ лечения боли. 3 н. и 9 з.п. ф-лы, 3 табл.
где R1 выбран из
(1) фенила;
(2) пиридинила
(3) тиофенила
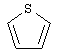
(4) фуранила
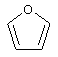
(5) имидазолила
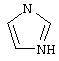
и
(6) триазолила
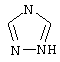
где каждое фенильное кольцо может быть возможно и независимо дополнительно замещено 1, 2 или 3 заместителями, выбранными из прямого и разветвленного С1-С6алкила, NO2, CF3, C1-С6алкокси, хлоро, фторо, бромо и йодо; а также их фармацевтически приемлемые соли и их изомеры.
4-[(4-бензил-1-пиперазинил)(8-хинолинил)метил]-N,N-диэтилбензамида дигидрохлорида (соединение 2);
N,N-диэтил-4-[[4-(4-метилбензил)-1-пиперазинил](8-хинолинил)метил]бензамида (соединение 5);
4-[{4-[4-(трет-бутил)бензил]-1-пиперазинил}(8-хинолинил)метил]-N,N-диэтилбензамида дигидрохлорида (соединение 8);
N,N-диэтил-4-[[4-(4-нитробензил)-1-пиперазинил](8-хинолинил)метил]бензамида дигидрохлорида (соединение 9);
4-[{4-[2,4-бис(трифторметил)бензил]-1-пиперазинил}(8-хинолинил)метил]-N,N-диэтилбензамида дигидрохлорида (соединение 10);
N,N-диэтил-4-[[4-(4-метоксибензил)-1-пиперазинил](8-хинолинил)метил]бензамида дигидрохлорида (соединение 11);
4-[[4-(2,4-дихлорбензил)-1-пиперазинил](8-хинолинил)метил]-N,N-диэтилбензамида дигидрохлорида (соединение 12);
N,N-диэтил-4-[[4-(2-пиридинилметил)-1-пиперазинил](8-хинолинил)метил]бензамида дигидрохлорида (соединение 13);
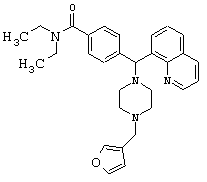
N,N-диэтил-4-[[4-(3-тиенилметил)-1-пиперазинил](8-хинолинил)метил]бензамида (соединение 14);
N,N-диэтил-4-[[4-(2-фуранилметил)-1-пиперазинил](8-хинолинил)метил]бензамида (соединение 15);
N,N-диэтил-4-[[4-(3-фуранилметил)-1-пиперазинил](8-хинолинил)метил]бензамида (соединение 16);
N,N-диэтил-4-[[4-(2-тиофенилметил)-1-пиперазинил](8-хинолинил)метил]бензамида дигидрохлорида (соединение 17);
N,N-диэтил-4-[[4-(2-имидазолилметил)-1-пиперазинил](8-хинолинил)метил]бензамида дигидрохлорида (соединение 18);
N,N-диэтил-4-[[4-(4-имидазолилметил)-1-пиперазинил](8-хинолинил)метил]бензамида дигидрохлорида (соединение 19) и
N,N-диэтил-4-[[4-(3-триазолилметил)-1-пиперазинил](8-хинолинил)метил]бензамида дигидрохлорида (соединение 20).
| RU, 2127732, C1, 20.03.1999 | |||
| WO, 97/23466, A1, 03.07.1997 | |||
| WO, 99/33806, A1, 08.07.1999. |

