Область изобретения
Настоящее изобретение относится к новым соединениям и их солям, представляющим собой агонисты рецептора СВ2. Эти соединения полезны в лечении, в частности для лечения боли. Настоящее изобретение также относится к способам получения новых соединений, содержащим их фармацевтическим композициям и к применению этих соединений в лечении, в частности для лечения боли.
Предшествующий уровень техники
Известны два типа каннабиноидных рецепторов: один экспрессируется преимущественно в центральной нервной системе (СВ1), тогда как другой расположен на периферии и в основном ограничен клетками и тканями, имеющими происхождение от иммунной системы (СВ2) (Abood and Martin Int. Rev. of Neurobio. 39, 197-221 (1996)).
Хотя агонисты рецептора СВ1 и смешанные агонисты высокоэффективны в моделях антиноцицепции у животных, оказалось, что невозможно в какой-либо значительной степени отделить желаемые аналгезирующие действия от нежелательных побочных эффектов в отношении центральной нервной системы (ЦНС). Известно, что эти нежелательные побочные действия в отношении ЦНС опосредованы рецептором СВ1.
Множество сообщений указывают на важную роль СВ2 в патофизиологии. В частности, Munro et. al. f Nature 365 61-65 (1993)] обнаружил, что экспрессия рецептора СВ2 индуцируется в условиях активации клеток иммунной системы. Hanus et. al. [PNAS 96, 14228-14233 (1999)] недавно привел доказательство того, что агонист СВ2 проявляет противовоспалительное и периферическое аналгезирующее действие. Кроме того, Mazzari et. al. [Soc. Neurosci. Abstr. 23 652 (1995)] продемонстрировал, что активация СВ2 ингибирует механическую гипералгезию, ассоциированную с повреждением нервов. Эти результаты указывают на то, рецептор СВ2 представляет собой вызывающую интерес мишень для разработки новых аналгезирующих средств, которые, как можно ожидать, лишены опосредованных СВ1 побочных действий, ассоциированных с обычными каннабиноидными агонистами, например, тетрагидроканнабинолом (ТГК). Кроме того, поскольку расположение рецепторов СВ2 ограничено периферией, можно ожидать, что селективные агонисты СВ2 уменьшают боль без психоактивных побочных действий и обычно осознаваемой возможности злоупотребления, характерных для действующих в отношении центральной нервной системы каннабимиметиков (СВ1) или опиатных лекарств.
Аналгезирующие средства, которые были выявлены и известны из предшествующего уровня техники, обладают множеством недостатков, среди которых можно выделить плохую фармакокинетику и потерю аналгезирующего действия при системном введении.
Подробное описание изобретения
Соединения по данному изобретению определены формулой I, и их фармацевтически приемлемая соль, и диастереоизомеры, и энантиомеры и их смеси:

где
R1 выбран из группы, состоящей из -(С1-С8)алкила, -(С2-С8)алкенила, Р4 2N(C1-C6)алкила-, R4 2NC(=O)(C1-C6)алкила-, R4O(С1-C6)алкила-, R4OC(=O)(C1-C6)алкила-, R4C(=О)(С1-C6)алкила-, R4C(=O)NR4(C1-C6)алкила-, R4 2NSO2(С1-C6)алкила-, R4CSO2NR4(C1-C6)алкила-, R4 2NC(=O)NR4(C1-C6)алкила-, R4 2NSO2NR4(C1-C6)алкила-, арил(С1-С6)алкила-, радикалов ароил(С1-С6)алкил, гетероарил(С1-С6)алкил, гетероароил(С1-С6)алкил, гетероциклоалкил(С1-С6)алкил, бициклический гетероарил(С1-С6)алкил и бициклический гетероароил(С1-С6)алкил;
группировки R1 включают в себя незамещенный -(С2-С8)алкенил и -(С1-С8)алкил, незамещенный или замещенный одной или более чем одной группировкой, независимо выбранной из группы, состоящей из галогена, циано, ацетоксиметила и нитро;
Ar представляет собой возможно замещенную арильную группировку;
R2 представляет собой -(С1-С6)алкил, незамещенный или замещенный (по 1-6 атомам углерода) одним или более чем одним заместителем фтором, или (С3-С6)циклоалкил;
R3 выбран из группы, состоящей из:
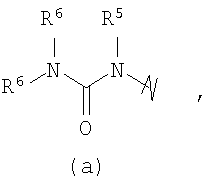
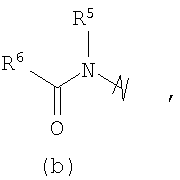
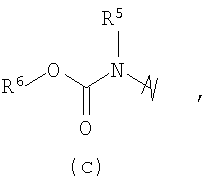
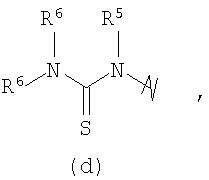
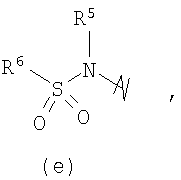
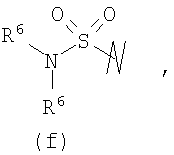
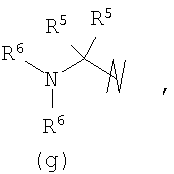
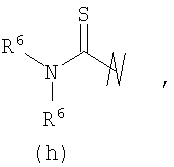
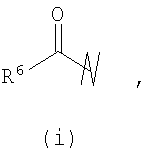
 и
и 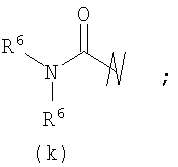
R4 представляет собой группировку, независимо выбранную из группы, состоящей из -Н, -(С1-С6)алкила, -(С2-С6)алкенила и -(С2-С6)алкинила;
NR4 2 включает в себя соединения, где NR4 2 образует гетероциклильную кольцевую систему, например пиррол, пиперидин, пиперазин или пирролидинон;
группировки R5 независимо выбраны из группы, состоящей из -Н, -(С1-С6)алкила, -(С2-С6)алкенила и гетероциклила;
NR5 2 включает в себя соединения, где NR5 2 образует гетероциклильную кольцевую систему, например пиррол, пиперидин, пиперазин или пирролидинон;
группировки R6 независимо выбраны из группы, состоящей из -Н, -(C1-С6)алкила, -(С2-С6)алкенила и -(С1-С6)алканоила, гетероциклила, радикала гетероциклил(С1-С3)алкил, арила, радикала арил(С1-С3)алкил, гетероарила, радикалов гетероарил(С1-С3)алкил, бициклический гетероарил и бициклический гетероарил(С1-С3)алкил;
R5 и R6 могут объединяться с образованием 5-7-членного гетероцикла, например пиррола, пиперидина, пиперазина, пирролидинона, гомопиперазина или гексаметиленимина;
Х выбран из группы, состоящей из -C(R5)2-, -NR5-, C(=O)-, -CH2-CH2-, -СН=СН-, -О-, -C(R)(R')- и -S(O)n- (где n равно 0, 1 или 2), где R и R' представляют собой (С1-С6)алкил, OR" или Н, и R" представляет собой Н или (С1-С6)алкил; и
Y представляет собой С или N.
Используемый здесь термин "алкил" включает в себя заместители с прямой цепью, с разветвленной цепью и циклические заместители, например, метил, этил, н-пропил, изопропил, н-бутил, изобутил, трет-бутил, циклопропилметил и циклопентил, и группировки на алкильных цепях могут находиться в любом месте цепи, так что амино(С1-С6)алкил включает в себя 1-аминопропил и 2-аминопропил.
Используемый здесь термин "галоген" включает в себя фтор, хлор, бром и йод.
Термин "арильная группировка" включает в себя ароматические карбоциклы, пятичленные гетероароматические кольцевые системы, шестичленные гетероароматические кольцевые системы и бициклические гетероароматические кольцевые системы.
Ароматический карбоцикл включает в себя фенил и нафтил.
Пятичленная гетероароматическая кольцевая система представляет собой моноциклическую ароматическую кольцевую систему, имеющую пять атомов в кольце, где 1, 2 или 3 атома кольца независимо выбраны из N, О и S.
Предпочтительно пятичленные гетероароматические кольцевые системы выбраны из группы, состоящей из тиенила, фурила, пирролила, имидазолила, тиазолила, оксазолила, пиразолила, изотиазолила, изоксазолила, 1,2,3-триазолила, тетразолила, 1,2,3-тиадиазолила, 1,2,3-оксадиазолила, 1,2,4-триазолила, 1,2,4-тиадиазолила, 1,2,4-оксадиазолила, 1,3,4-триазолила, 1,3,4-тиадиазолила и 1,3,4-оксадиазолила.
Шестичленная гетероароматическая кольцевая система представляет собой моноциклическую ароматическую кольцевую систему, имеющую шесть атомов в кольце, где 1, 2 или 3 атома кольца представляют собой N.
Предпочтительно шестичленные гетероароматические кольцевые системы выбраны из группы, состоящей из пиридила, пиразинила, пиримидинила, триазинила и пиридазинила.
Бициклическая гетероароматическая кольцевая система представляет собой кольцевую систему, имеющую два пяти- или шестичленных гетероароматических кольца, или фенил и пяти- или шестичленное гетероароматическое кольцо, или фенил и гетероциклильное кольцо, или пяти- или шестичленное гетероароматическое кольцо и гетероциклильное кольцо; соединенные посредством слияния колец, причем указанная бициклическая гетероароматическая кольцевая система включает в себя от 8 до 12 атомов в кольце, где 1, 2 или 3 атома кольца независимо выбраны из N, О и S.
Бициклические гетероароматические кольцевые системы предпочтительно выбраны из группы, состоящей из индола, индолина, хинолина, тетрагидрохинолина, изохинолина, тетрагидроизохинолина, 1,4-бензодиоксана, кумарина, дигидрокумарина, бензофурана, 2,3-дигидробензофурана, 1,2-бензизоксазола, бензотиофена, бензоксазола, бензотиазола, бензимидазола, бензотриазола, пирролизидина и хинолизидина.
Гетероциклильная или гетероциклическая группировка представляет собой насыщенную или частично насыщенную кольцевую систему, имеющую от 3 до 7 атомов в кольце, где 1, 2 или 3 атома кольца независимо выбраны из N, О и S.
Гетероциклильные группировки предпочтительно выбраны из группы, состоящей из азиридина, оксирана, тиирана, азетидина, оксетана, тиетана, пирролидина, пирролина, имидазолидина, пиразолидина, диоксолана, сульфолана 2,3-дигидрофуранила, 2,5-дигидрофуранила, тетрагидрофуранила, тиофанила, пиперидина, пиперазина, морфолина, 2,3-дигидропиранила, тетрагидропиранила, 1,4-дигидропиридинила, 1,4-диоксанила, 1,3-диоксанила, диоксанила, гомопиперидинила, гомопиперазинила, 1,3-диоксепана, 4,7-дигидро-1,3-диоксепина и оксида гексаметилена.
Ароматические, гетероароматические, гетероциклильные и бициклические гетероароматические группировки являются незамещенными или замещенными по атомам углерода кольца, предпочтительно группировками, независимо выбранными из группы, состоящей из: галогена, трифторметила, циано, нитро, гидрокси, -NR4 2, -C(=O)OR4, -C(=O)R4, -C(=O)NR4 2, -NR4C(=O)R4, -(С1-С6)алкила, -(С2-С6)алкенила, -(С2-С6)алкинила, -OR4, -SR4, -SO2R4, оксо(=O), имино(=NR4), тио(=S) и оксимино(=N-OR4).
Атомы азота кольца пятичленных гетероароматических, гетероциклильных или бициклических гетероароматических кольцевых систем являются незамещенными или замещенными, если такое замещение химически возможно, без кватернизации указанного атома азота кольца, предпочтительно группировками, независимо выбранными из группы, состоящей из -(С1-С6)алкила и -C(=O)R4.
Заместители -(С1-С6)алкил, -(С2-С6)алкенил и -(С1-С6)алканоил являются незамещенными или замещенными по одному или более чем одному атому углерода группировками, независимо выбранными из группы, состоящей из галогена, гидрокси, -OR4 и -NR4 2.
Понятно, что когда соединения по настоящему изобретению содержат один или более чем один хиральный центр, соединения по данному изобретению могут существовать и могут быть выделены в виде энантиомерных или диастереоизомерных форм или в виде рацемической смеси. Настоящее изобретение включает в себя любые возможные энантиомеры, диастереоизомеры, рацематы или их смеси соединения формулы I, которое действует как агонист СВ2. Синтез оптически активных форм может быть осуществлен с помощью стандартных способов органической химии, хорошо известных из области техники, например путем хирального хроматографического разделения рацемата, путем синтеза из оптически активных исходных веществ или путем асимметрического синтеза.
Также понятно, что некоторые соединения по настоящему изобретению могут существовать в виде геометрических изомеров, например Е- и Z-изомеров алкенов. Настоящее изобретение включает в себя любой геометрический изомер соединения формулы I, которое действует как агонист СВ2. Понятно, что настоящее изобретение охватывает таутомеры соединений формулы I.
Также понятно, что некоторые соединения по настоящему изобретению могут существовать в виде сольватированных, например гидратированных, а также несольватированных форм. Понятно, что настоящее изобретение охватывает все такие сольватированные формы соединений формулы I, которые действуют как агонисты СВ2.
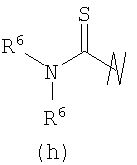
В предпочтительных воплощениях формулы I, если R1 представляет собой R4 2N(C1-C6)алкил-, где оба R4 представляют собой -(С1-С6)алкил, то R3 выбран из группировок (как указано выше) (а), (с), (d), (e), (f), (g), (h) и (k) и возможно (b) и (i) (за исключением того, где R3 представляет собой -(C1-С6)алкил (в особенности, метил, например Рз представляет собой ацетил или ацетамидо)) и (j) (за исключением того, где R5 в обоих случаях представляет собой Н, например R3 представляет собой первичный амин).
Предпочтительные соединения по настоящему изобретению представляют собой соединения формулы I, где:
R1 выбран из группы, состоящей из -(С1-С8)алкила, -(С2-С8)алкенила, радикала арил(С1-С6)алкил, R4 2N(C1-C6)алкила-, R4O(C1-C6)алкила-, радикалов -гетероциклоалкил(С1-С6)алкил (4-8 членного) и гетероарил(С1-С6)алкил;
где арильные и гетероарильные группировки R1 не замещены или замещены -(С1-С6)алкилом, ацетоксиметилом или галогеном;
R2 выбран из группы, состоящей из -СН3, -СН2СН3, -СН(СН3)2, (С3-С5)циклоалкила и CF3;
R3 выбран из группы, состоящей из:
 и
и
Ar представляет собой арильную группировку, незамещенную или замещенную одной или более чем одной группировкой, независимо выбранной из группы, состоящей из (С1-С6)алкила, галогена, трифторметила, циано, нитро, гидрокси и -OR4;
Х выбран из группы, состоящей из -СН2-, -СН2СН2-, -С(=O)-, -S-, -O-, -C(R)(R')- и -N(R)-, где R и R' представляют собой (С1-С6)алкил, OR" или Н, и R" представляет собой Н или (С1-С6)алкил;
когда Ar представляет собой фенил или шестичленную гетероароматическую кольцевую систему, Х расположен на кольце Ar в положении 1, 4 по отношению к группе -O-R2;
когда Ar представляет собой 5-членную гетероароматическую кольцевую систему, Х расположен на кольце Ar в положении 1,3 по отношению к группе -O-R2;
R4 независимо выбран из группы, состоящей из -Н и -(С1-С6)алкила;
R5 независимо выбран из группы, состоящей из -Н, -(С1-С6)алкила и -(С2-С6)алкенила; и
R6 независимо выбран из группы, состоящей из -Н, -(С1-С6)алкила, -(С2-С6)алкенила и гетероарила;
где указанный гетероарил является незамещенным или замещен -(С1-С6)алкилом.
Более предпочтительные соединения по настоящему изобретению представляют собой соединения формулы I, где;
R1 выбран из группы, состоящей из циклопропилметила, этила, пропила, аллила, изопентила, бензила, метоксиэтила, диметиламиноэтила, 4-пиридилметила, 2-пиридилметила, 1-пирролилэтила, 1-морфолиноэтила, циклогексилметила, 2-пирролидилметила, N-метил-2-пирролидилметила, 2-пиперидилметила, N-метил-2-пиперидилметила, 3-тиенилметила, 2-тетрагидрофуранилметила, (2-нитротиофен-5-ил)метила, (1-метил-1Н-имидазол-2-ил)метила, (5-(ацетоксиметил)-2-фуранил)метила, (2,3-дигидро-1 Н-изоиндол-1-ил)метила и 5-(2-метилтиазолила);
R2 выбран из группы, состоящей из -СН3, -СН2СН3, -СН(СН3)2 и CF3;
R4 представляет собой -(С1-С6)алкил;
R5 выбран из группы, состоящей из -Н, -СН3, -СН2СН3, -СН=СН2 и -СН2-СН=СН2;
R6 выбран из группы, состоящей из -СН3, -СН2СН3, -СН=СН2, -CH2-СН=СН2, -СН2-СН2-СН=СН2, -СН2СН(СН3)2 и 5-метил-3-изоксазола;
Ar представляет собой фенил или шестичленную гетероароматическую кольцевую систему, каждый из которых может быть незамещенным или замещенным одной или более чем одной группировкой, независимо выбранной из группы, состоящей из (С1-С6)алкила, галогена, трифторметила, циано, нитро, гидрокси и -OR4;
Х выбран из группы, состоящей из -СН2-, -СН2СН2-, -S-, -O-, -С(=O)-, -C(R)(R')- и -N(R)-, где R и R' представляют собой (С1-С6)алкил, OR" или Н, и R" представляет собой Н или (С1-С6)алкил; и
Х расположен на кольце Ar в положении 1, 4 относительно группы -O-R2.
Наиболее предпочтительные соединения по настоящему изобретению представляют собой соединения формулы I, где:
R2 представляет собой -СН2СН3;
Ar представляет собой незамещенный фенил или пиридил;
Х выбран из группы, состоящей из -СН2-, -СН2СН2-, -S-, -O-, -C(R)(R')- и -N(R)-, где R и R' представляют собой (С1-С6)алкил, OR" или Н, и R" представляет собой Н или (С1-С6)алкил;
Х расположен на кольце Ar в положении 1, 4 относительно группы -O-R2;
и
R4 представляет собой метил.
В некоторых предпочтительных соединениях Х выбран из группы, состоящей из СН(СН3)-, -С(СН3)2-, -СН(ОН)-, -NH- и -N(СН3)-; наиболее предпочтительно СН(СН3).
Авторы изобретения обнаружили, что соединения по настоящему изобретению проявляют избирательное действие в отношении сайта рецептора СВ2 и полезны в облегчении боли, в частности хронической боли, например хронической воспалительной боли, невропатической боли, боли в спине, боли при раке и висцеральной боли. Соединения по настоящему изобретению также полезны в лечении острой боли. В дополнение, соединения по настоящему изобретению полезны при других болезненных состояниях, при которых присутствует или в которые вовлечена дегенерация или дисфункция рецепторов СВ2.
В объем данного изобретения также включены соли соединений формулы I. Как правило, фармацевтически приемлемые соли соединений по настоящему изобретению могут быть получены с использованием стандартных способов, хорошо известных из уровня техники, например путем взаимодействия достаточного количества основного соединения, например алкиламина, с подходящей кислотой, например HCl или уксусной кислотой, с получением физиологически приемлемого аниона. Также возможно получить соответствующую соль щелочного металла (такого как натрий, калий или литий) или щелочноземельного металла (такого как кальций) путем обработки соединения по настоящему изобретению, имеющего подходящий кислотный протон, такого как карбоновая кислота или фенол, одним эквивалентом гидроксида или алкоксида (такого как этоксид или метоксид) щелочного или щелочноземельного металла или подходящего основного органического амина (такого как холин или меглюмин) в водной среде с последующими обычными способами очистки.
Также в объем настоящего изобретения включено применение соединений по настоящему изобретению в лечении.
Новые соединения по настоящему изобретению полезны в лечении, в частности в лечении различных болевых состояний, включающих в себя острую боль, хроническую боль, невропатическую боль, боль в спине, боль при раке и висцеральную боль, но не ограничивающихся ими.
При использовании для лечения у теплокровного животного, такого как человек, агонист СВ2, как правило, вводят в форме обычной фармацевтической композиции, и, как правило, эта композиция может быть в форме, подходящей для перорального или подъязычного введения, например в виде таблетки или капсулы, для парентеральной инъекции (включая внутривенную, подкожную, внутримышечную, внутрисосудистую инъекцию или инфузию), например в виде стерильного раствора, суспензии или эмульсии, для местного введения, например в виде мази или крема, или для ректального введения, например в виде суппозитория. Как правило, вышеописанные композиции могут быть приготовлены обычным образом с использованием обычных носителей. Композиции по настоящему изобретению преимущественно представлены в виде стандартной лекарственной формы.
Терапевтически эффективное количество для практического применения настоящего изобретения может быть определено с использованием известных критериев, включающих в себя возраст, массу и реакцию отдельного пациента, и интерпретировано с учетом заболевания, которое лечат или предотвращают, специалистом в данной области техники.
Также в объем данного изобретения включено применение любого соединения формулы I, указанной выше, для изготовления лекарственного средства для лечения боли.
Дополнительно предложено применение любого соединения формулы I для изготовления лекарственного средства для лечения различных болевых состояний, включающих в себя: острую боль, хроническую боль, невропатическую боль, боль в спине, боль при раке и висцеральную боль, но не ограничивающихся ими
Еще один аспект данного изобретения представляет собой способ лечения субъекта, страдающего от любого из состояний, рассмотренных выше, при котором пациенту, нуждающемуся в таком лечении, вводят эффективное количество соединения формулы I, указанной выше. Кроме того, предложена фармацевтическая композиция, включающая в себя соединение формулы I или его фармацевтически приемлемую соль совместно с фармацевтически приемлемым носителем.
В частности, предложена фармацевтическая композиция, включающая в себя соединение формулы I или его фармацевтически приемлемую соль совместно с фармацевтически приемлемым носителем, для лечения, более конкретно для лечения боли.
Кроме того, предложено применение фармацевтической композиции, включающей в себя соединение формулы I или его фармацевтически приемлемую соль совместно с фармацевтически приемлемым носителем, при любом из состояний, рассмотренных выше. Термин "лечение" в контексте настоящего изобретения означает введение эффективного количества соединения по настоящему изобретению для облегчения или уже существующего болезненного состояния, острого или хронического, или повторяющегося состояния. Это определение также охватывает профилактическое лечение для предотвращения повторяющихся состояний и продолжительное лечение хронических расстройств.
Фармацевтические композиции
Новые соединения по настоящему изобретению можно вводить перорально, подъязычно, внутримышечно, подкожно, местно, интраназально, интраперитонеально, интраторакально, внутривенно, эпидурально, интратекально, интрацеребровентрикулярно и путем инъекции в суставы.
Предпочтительными путями введения являются пероральный, внутривенный или внутримышечный.
Дозировка зависит от пути введения, тяжести заболевания, возраста и массы пациента и других факторов, которые учитываются в норме штатным врачом больницы при определении индивидуального режима и уровня дозировки, наиболее подходящих для отдельного пациента.
Для приготовления фармацевтических композиций из соединений по данному изобретению инертные фармацевтически приемлемые носители могут быть и твердыми и жидкими. Препараты в виде твердой формы включают в себя порошки, таблетки, диспергируемые гранулы, капсулы, крахмальные облатки и суппозитории.
Твердый носитель может представлять собой одно или более чем одно вещество, которое может также действовать в качестве разбавителей, корригентов, солюбилизаторов, смазывающих веществ, суспендирующих агентов, связывающих агентов или разрыхлителей; также это может быть вещество для инкапсуляции.
В порошках носитель представляет собой тонкоизмельченное твердое вещество, которое находится в смеси с тонкоизмельченным активным компонентом. В таблетках активный компонент смешан с носителем, обладающим необходимыми связывающими свойствами, в подходящих пропорциях и спрессован в желаемую форму и размер.
Для приготовления композиций суппозиториев легкоплавкий воск, такой как смесь глицеридов жирных кислот и масла какао, сначала плавят и диспергируют в нем активный ингредиент, например, путем перемешивания. Расплавленную гомогенную смесь затем выливают в формы подходящих размеров и оставляют охлаждаться и затвердевать.
Подходящие носители включают в себя карбонат магния, стеарат магния, тальк, лактозу, сахар, пектин, декстрин, крахмал, трагакант, метилцеллюлозу, натрий-карбоксиметилцеллюлозу, легкоплавкий воск, масло какао и тому подобное.
Соли включают в себя фармацевтически приемлемые соли, но не ограничиваются ими. Примеры фармацевтически приемлемых солей в объеме настоящего изобретения включают в себя: ацетат, бензолсульфонат, бензоат, бикарбонат, битартрат, карбонат, цитрат, фумарат, глюконат, глутамат, гидробромид, гидрохлорид, лактат, малеат, манделат, мезилат, фосфат/дифосфат, салицилат, сукцинат, сульфат, тартрат, холин, диэтаноламин, этилендиамин, меглюмин, алюминий, кальций, магний, калий, натрий и цинк.
Примеры фармацевтически неприемлемых солей в объеме настоящего изобретения включают в себя: гидроиодид, перхлорат, тетрафторборат, литий.
Предпочтительные фармацевтически приемлемые соли представляют собой гидрохлориды, сульфаты и битартраты.
Особенно предпочтительными являются соли гидрохлорид и сульфат.
Под термином "композиция" подразумевают препарат активного компонента с веществом для инкапсуляции в качестве носителя, обеспечивающего капсулу, в которой активный компонент (вместе или без других носителей) окружен носителем, который, таким образом, находится с ним в ассоциации. Подобным образом, включены крахмальные облатки.
Таблетки, порошки, крахмальные облатки и капсулы могут быть использованы в виде твердых лекарственных форм, подходящих для перорального введения.
Композиции в виде жидкой формы включают в себя растворы, суспензии и эмульсии. Стерильный водный раствор или раствор вода-пропиленгликоль активных соединений могут быть упомянуты в качестве примера жидких препаратов, подходящих для парентерального введения. Жидкие композиции также могут быть приготовлены в виде раствора в водном растворе полиэтиленгликоля.
Водные растворы для перорального введения могут быть приготовлены путем растворения активного компонента в воде и добавления походящих красителей, корригентов, стабилизаторов и загустителей в соответствии с тем, как требуется. Водные суспензии для перорального применения могут быть приготовлены путем диспергирования тонкоизмельченного активного компонента в воде вместе с вязким веществом, таким как натуральные синтетические смолы, камеди, метилцеллюлоза, натрий-карбоксиметилцеллюлоза и другие суспендирующие агенты, известные из фармацевтической области.
Предпочтительно фармацевтические композиции находятся в виде стандартной лекарственной формы. В такой форме композиция разделена на стандартные дозы, содержащие подходящие количества активного компонента. Стандартная лекарственная форма может представлять собой упакованный препарат, упаковку, содержащую определенные количества препаратов, например упакованные таблетки, капсулы и порошки в пузырьках или ампулах. Стандартная лекарственная форма также может представлять собой капсулу, крахмальную облатку, таблетку саму по себе или может представлять собой подходящее количество любой из этих упакованных форм.
Способы получения
Способ А1
Способ А1 для получения соединений общей формулы I,
при котором осуществляют следующие стадии:
соединения общей формулы II,
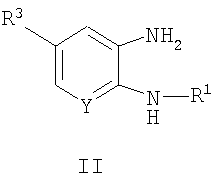
где R1, R3 и Y являются такими, как определено для формулы I, можно подвергать взаимодействию с соединениями общей формулы III,
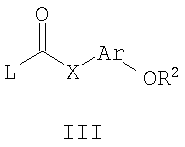
где R2, Ar и Х являются такими, как определено для формулы I, и L представляет собой -ОН или уходящую группу, такую как галогенид, O-тозил или O-мезил. Удобно проводить эту реакцию в инертном растворителе, таком как толуол, при температуре окружающей среды в течение 20 минут. После этого добавляют каталитическое количество концентрированной HCl и смесь нагревают в течение 12 часов при 85°С. Обработку осуществляют посредством водной экстракции, а очистку продукта осуществляют посредством нормальной или обращенно-фазовой хроматографии.
Способ А2
Способ А2 для получения соединений общей формулы II, при котором осуществляют следующие стадии:
соединения общей формулы IV,
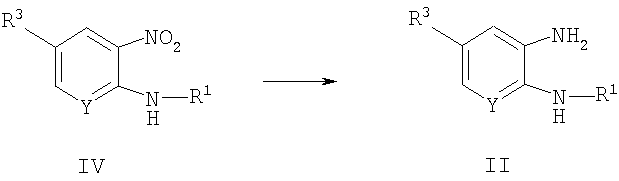
где R1, R3 и Y являются такими, как определено для формулы I, можно восстановить до соответствующего анилина (формулы II) путем взаимодействия с водородом при давлении 10-50 ф./кв. дюйм (68,95 кПа -344,75 кПа). Эту реакцию удобно проводить в инертном растворителе, таком как этанол, метанол или тетрагидрофуран, при температуре окружающей среды. Реакцию катализируют с помощью катализатора на основе переходного металла, удобно с помощью 5-10% палладия на тонкоизмельченном углероде.
Способ A3
Способ A3 для получения соединений общей формулы IV, при котором осуществляют следующие стадии:
соединения общей формулы V:
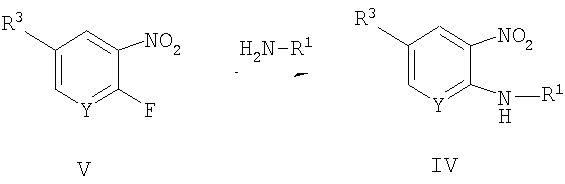
где R1, R3 и Y являются такими, как определено для формулы I, подвергают взаимодействию с первичным амином. Удобно проводить эту реакцию в протонном растворителе, таком как 80% этанол, при температуре 50-100°С. Обработку удобно осуществляют путем водной экстракции, и очистку удобно проводят посредством нормально-фазовой хроматографии.
Способ А4
Способ А4 для получения соединений общей формулы III, при котором осуществляют следующие стадии:
соединения общей формулы VI:
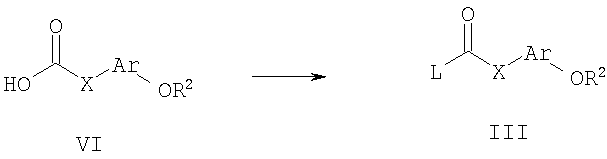
где R2, Ar и Х являются такими, как определено для формулы I, и L представляет собой -ОН или уходящую группу, такую как галогенид, O-тозил или O-мезил, подвергают взаимодействию с галогенирующим агентом, таким как тионилхлорид. Удобно проводить эту реакцию в инертном растворителе, таком как бензол или толуол, при температуре 25-100°С. Обработку удобно осуществляют путем удаления растворителя при пониженном давлении, а очистку удобно производят путем перегонки.
Способ А5
Способ А5 для получения соединений общей формулы VIII, при котором осуществляют следующие стадии:
используя КОН, гидролизуют соединения общей формулы VII (полученные способом А1),
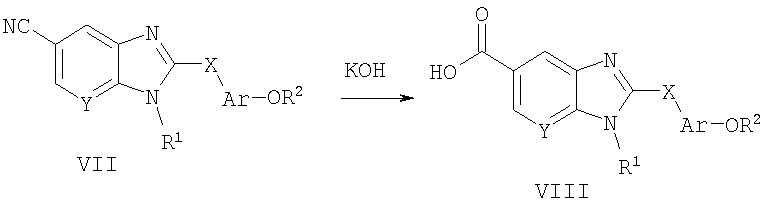
где R1, R2, Ar, Х и Y являются такими, как определено для формулы I, и полученные общими способами, описанными выше. Эту реакцию легко осуществляют в водной смеси растворителей, такой как 1:1 этанол/вода, при температуре дефлегмации. После нейтрализации продукт удобно выделяют путем фильтрации охлажденной реакционной смеси.
Способ А6
В качестве альтернативы, способ А6 для получения соединений общей формулы VIII, при котором осуществляют следующие стадии:
соединения общей формулы Х (полученные способом А1),

где R1, R2, Ar, Х и Y являются такими, как определено для формулы I, и полученные общими способами, описанными выше, превращают в соответствующую карбоновую кислоту VIII путем гидролиза эфира Х с использованием водного основания, такого как гидроксид натрия, до соответствующей карбоновой кислоты.
Способ А7
Способ А7 для получения соединений общей формулы IX, при котором осуществляют следующие стадии:
соединения формулы VIII,
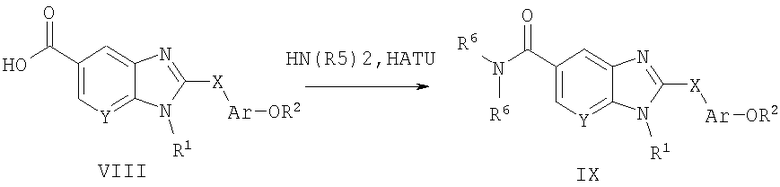
где R1, R2, Ar, Х и Y являются такими, как определено для формулы I, и полученные общими способами, описанными выше, превращают в соответствующий амид путем взаимодействия с первичным или вторичным амином в присутствии кислотного активирующего агента, такого как HATU [гексафторфосфат O-(7-азабензотриазол-1-ил)-N,N,N',N'-тетраметилурония], HBTU [гексафторфосфат O-бензотриазол-1-ил)-N,N,N',N'-тетраметилурония] или TBTU [тетрафторборат O-(1Н-бензотриазол-1-ил)-N,N,N',N'-пентаметиленурония]. Эту реакцию легко проводят в полярном апротонном растворителе, таком как диметилформамид (ДМФ), при температуре окружающей среды совместно с третичным амином, таким как триэтиламин или диизопропилэтиламин, который служит в качестве акцептора кислоты. Продукт без труда выделяют путем водной экстракции и очищают путем нормально-фазовой хроматографии.
Способ А8
Способ А8 для получения соединений общей формулы XI, при котором осуществляют следующие стадии:
соединения формулы IX (полученные способом А1 или А7),
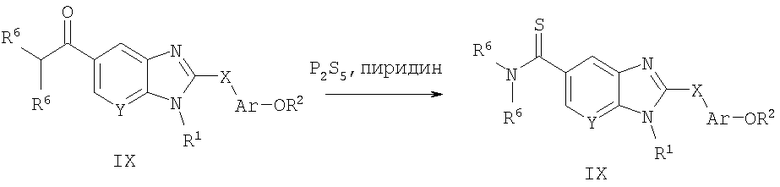
где R1, R2, R6, Ar, Х и Y являются такими, как определено для формулы I, и полученные общими способами, описанными выше, превращают в соответствующий тиоамид (формула XI) путем взаимодействия с P2S5. Эту реакцию легко проводят в пиридине при температуре 100°С. Продукт без труда выделяют путем водной экстракции декантированной части реакционной смеси и очищают путем нормально-фазовой хроматографии.
Способ А9
Способ А9 для получения соединений общей формулы XII, при котором осуществляют следующие стадии:
соединения формулы VII (полученные способом А1),
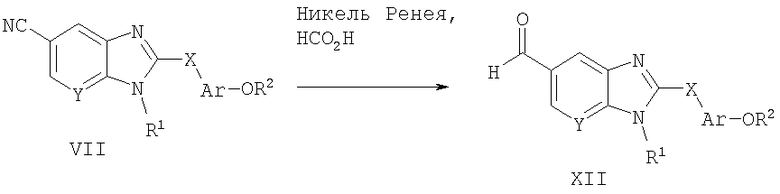
где R1, R2, Ar, Х и Y являются такими, как определено для формулы I, и полученные общими способами, описанными выше, каталитически восстанавливают, используя каталитическое количество никеля Ренея в 50% водной муравьиной кислоте. Эту реакцию легко проводят в кислой водной смеси растворителей, такой как 50% водная муравьиная кислота, при 90°С. Продукт удобно выделяют путем фильтрации охлажденной реакционной смеси через подушку из диатомовой земли, концентрирования и очистки посредством нормально-фазовой хроматографии.
Способ А10
Способ А10 для получения соединений общей формулы XIII, при котором осуществляют следующие стадии:
соединения формулы XII,
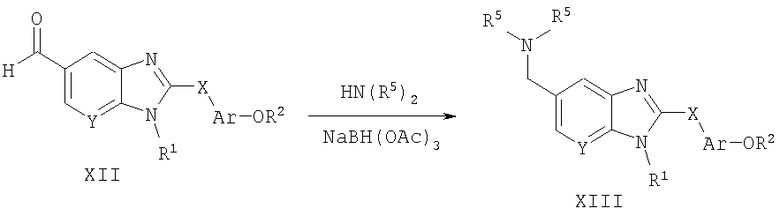
где R1, R2, R5, Ar, Х и Y являются такими, как определено для формулы I, и полученные общими способами, описанными выше, подвергают восстановительному аминированию с использованием первичного или вторичного амина в присутствии подходящего восстановителя, такого как триацетоксиборогидрид натрия. Эту реакцию удобно проводят в тетрагидрофуране с 1-1,5 эквивалентами уксусной кислоты и 1-1,5 эквивалентами триацетоксиборогидрида натрия при температуре окружающей среды. Продукт удобно выделяют путем разрушения боратного эфирного промежуточного соединения 1н. HCl с последующей водной экстракцией. Концентрирование органического экстракта позволила получить неочищенный продукт, и очистку осуществляют путем нормально-фазовой хроматографии.
Способ А11
Способ А11 для получения соединений общей формулы XIV, при котором осуществляют следующие стадии:
соединения формулы XII,
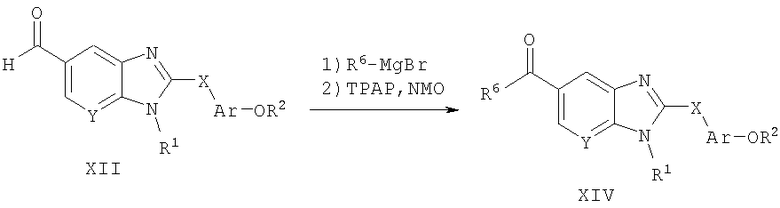
где R1, R2, R6, Ar и Х являются такими, как определено для формулы I, и полученные общими способами, описанными выше, соединяют с металлоорганическим агентом, таким как реактив Гриньяра, с последующим окислением промежуточного спирта до кетона. Реакцию Гриньяра удобно осуществляют в тетрагидрофуране с шестью эквивалентами магнийорганического галогенида, такого как метилмагнийбромид, при 0°С. Продукт удобно выделяют путем разрушения излишка металлоорганического реактива путем добавления воды с последующей водной экстракцией и концентрированием органического экстракта. Окисление этого промежуточного спирта осуществляют путем взаимодействия с каталитическим количеством (приблизительно 5 мол.%) перрутената тетрапропиламмония (ТРАР) и 1-1,5 эквивалентами N-метилморфолин-N-оксида (NMO) в присутствии молекулярных сит 4A. Эту реакцию удобно осуществляют в дихлорметане при температуре окружающей среды. Продукт удобно выделяют путем концентрирования реакционной смеси и очистки ее путем нормально-фазовой хроматографии.
Способ А12
Способ А12 для получения соединений общей формулы XVI, при котором осуществляют следующие стадии:
соединения формулы XV (полученные способом А1),
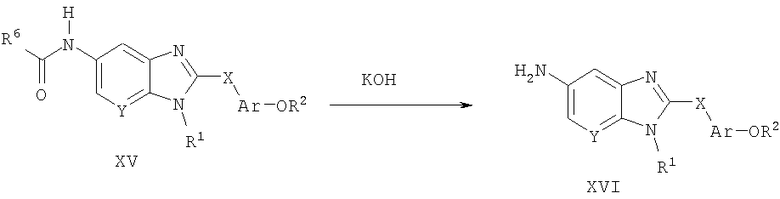
где R1, R2, R6, Ar, Х и Y являются такими, как определено для формулы I, и полученные общими способами, описанными выше, превращают в соответствующий анилин путем гидролиза с использованием основания, такого как гидроксид калия, в водном растворителе, таком как 50% водный этанол. Реакцию удобно проводят при температуре дефлегмации в течение продолжительного периода времени (8-16 часов). Продукт удобно выделяют путем того, что реакционную смесь оставляют охлаждаться до температуры окружающей среды, подкисляют смесь 1 н. HCl и собирают осажденный продукт путем фильтрования.
Способ А13
Способ А13 для получения соединений общей формулы XVII, при котором осуществляют следующие стадии:
соединения формулы XVI,
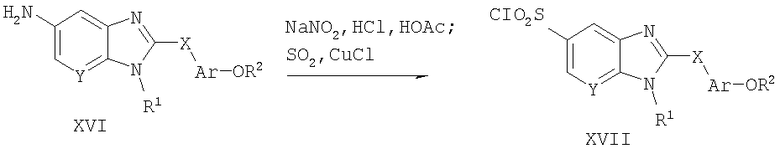
где R1, R2, Ar, Х и Y являются такими, как определено для формулы I, и полученные общими способами, описанными выше, превращают в соответствующий сульфонилхлорид путем взаимодействия с нитритом натрия в водной HCl и уксусной кислоте с получением промежуточной соли диазония. Реакцию удобно проводят при температуре менее -10°С. Промежуточную соль диазония немедленно превращают в сульфонилхлорид путем добавления по каплям свежеприготовленного насыщенного раствора диоксида серы в уксусной кислоте в присутствии хлорида меди (I). Эту реакцию удобно проводят при температуре от -10 до -5°С. Продукт удобно выделяют путем выливания реакционной смеси в ледяную воду и экстрагирования дихлорметаном и концентрирования в вакууме. Сульфонилхлориды формулы XVII обычно используют без дополнительной очистки.
Способ А14
Способ А14 для получения соединений общей формулы XVIII, при котором осуществляют следующие стадии:
соединения формулы XVII,
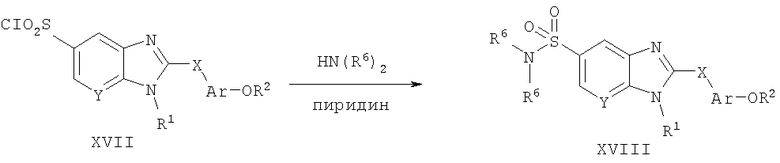
где R1, R2, R6, Ar и Х являются такими, как определено для формулы I, и полученные общими способами, описанными выше, превращают в соответствующий сульфонамид путем взаимодействия с первичным или вторичным амином. Реакцию удобно проводят в неполярном растворителе, таком как дихлорметан, при температуре окружающей среды в присутствии акцептора кислоты, такого как пиридин. Продукт удобно выделяют путем водной экстракции и очищают путем нормально-фазовой хроматографии.
Способ А15
Способ А15 для получения соединений общей формулы XX, при котором осуществляют следующие стадии:
Соединения формулы XIX,
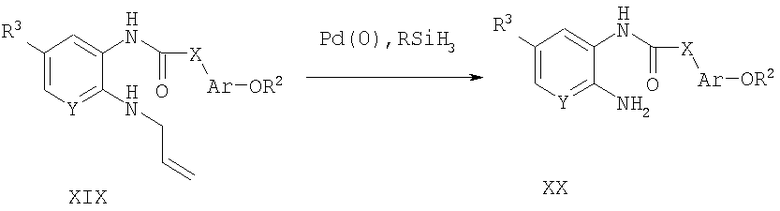
где R2, R3, Ar, Х и Y являются такими, как определено для формулы I, и полученные описанным выше способом А1, превращают в соединения формулы XX путем реакции деаллилирования, опосредованной палладием (0), в присутствии акцептора катионов, такого как фенилсилан. Продукт удобно выделяют путем водной экстракции и очищают путем нормально-фазовой хроматографии.
Способ А16
Способ А16 для получения соединений общей формулы XX, при котором осуществляют следующие стадии:
соединения формулы XXI,
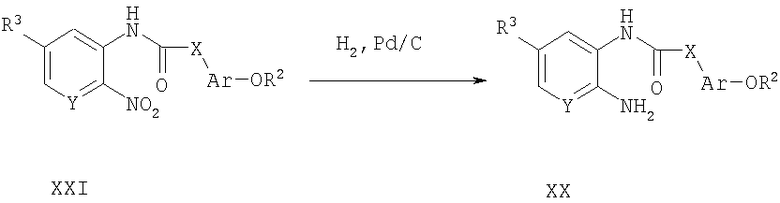
где R2, R3, Ar, X и Y являются такими, как определено для формулы I, и полученные описанным выше способом А2, превращают в соединения формулы XX в условиях катализируемого палладием гидрирования. Продукт удобно выделяют путем фильтрации и очищают путем нормально-фазовой хроматографии или используют непосредственно без хроматографической очистки.
Способ А17
Способ А17 для получения соединений общей формулы I, при котором осуществляют следующие стадии:
соединения формулы XX,
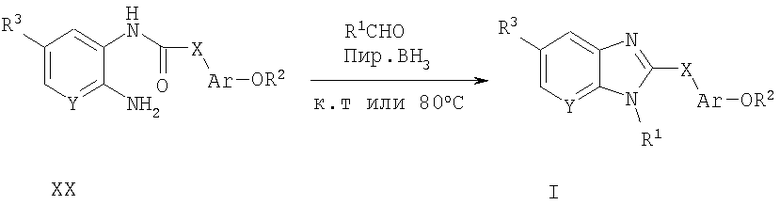
где R1, R2, R3, Ar, X и Y являются такими, как определено для формулы I, превращают в соответствующие соединения общей формулы I путем взаимодействия с альдегидом с последующим восстановлением борана в одну стадию. Эту реакцию удобно осуществляют в смешанном растворителе, таком как 1,2-дихлорэтан и уксусная кислота, при температуре окружающей среды или повышенной температуре. Продукт удобно выделяют путем водной экстракции и очищают путем нормально-фазовой или обращенно-фазовой хроматографии.
Способ А18
Еще один аспект данного изобретения представляет собой способ избирательного восстановления группировки нитро, находящейся в положении орто относительно заместителя амино на фенильном или пиридильном кольце соединения, в присутствии группировки нитро, находящейся в положении пара относительно заместителя амино, при котором обрабатывают раствор соединения в растворителе, предпочтительно неполярном растворителе, таком как этилацетат, палладиевым катализатором, таким как Pd/C, в присутствии водорода, возможно, под давлением (например, 1-10 атмосфер (101325-1013250 Па), предпочтительно 1-5 атмосфер (101325-506625 Па), еще более предпочтительно 2-4 атмосферы (202650-405300 Па)). В некоторых воплощениях оба заместителя нитро находятся в положении пара относительно атома азота пиридинового кольца. Пример протокола описан ниже в примере 58Б.
Еще один аспект настоящего изобретения представляют собой промежуточные соединения общих формул VIII, Х и XIII, представленные ниже, и их применение в синтезе соединений формулы I.

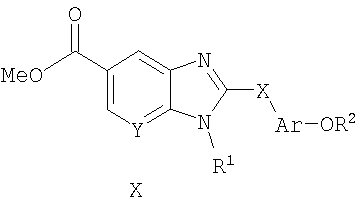
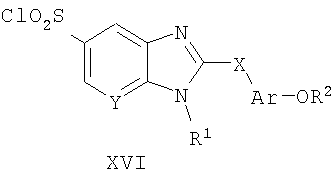
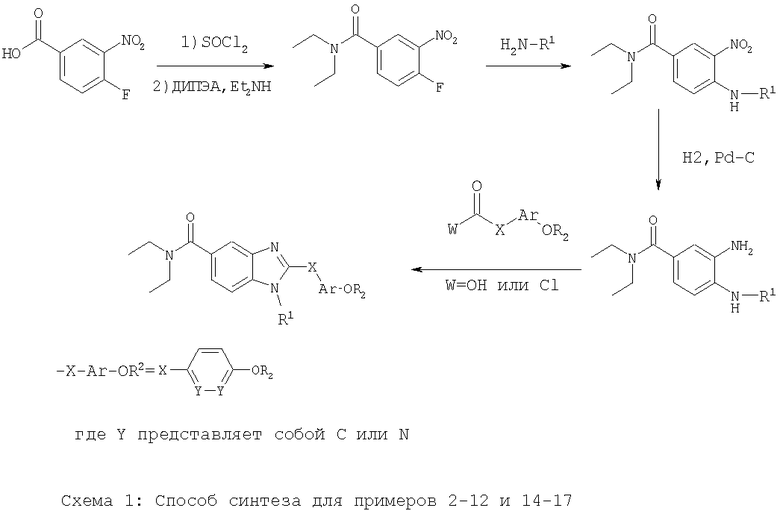
Соединения по настоящему изобретению могут быть синтезированы в соответствии со способами, описанными ниже на схемах 1-15, находящихся на следующих страницах.
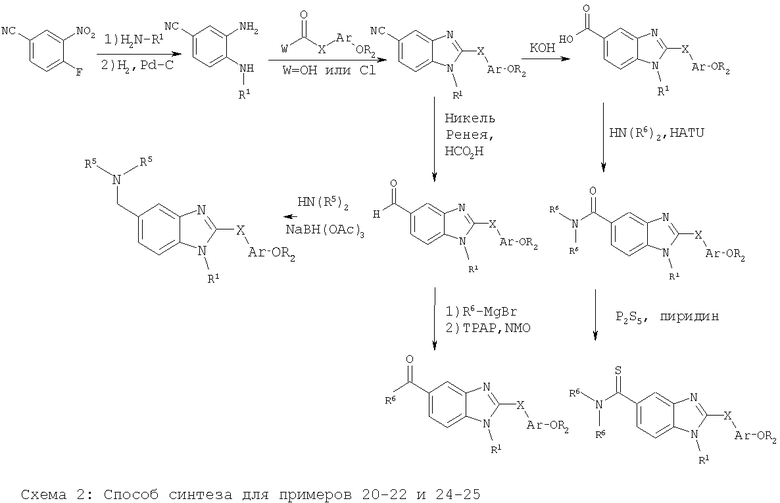
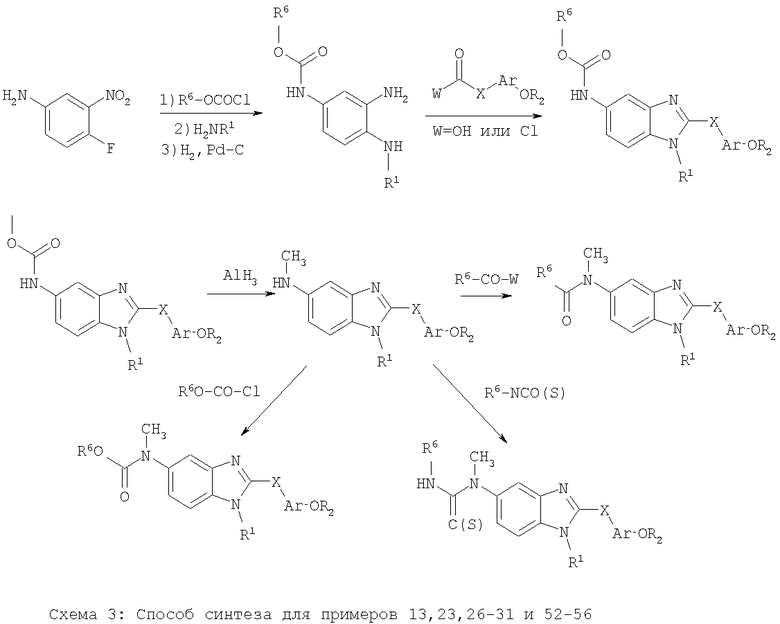
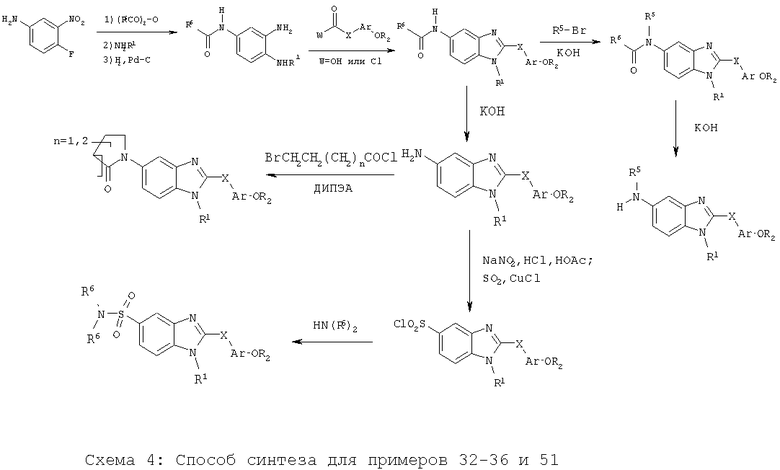
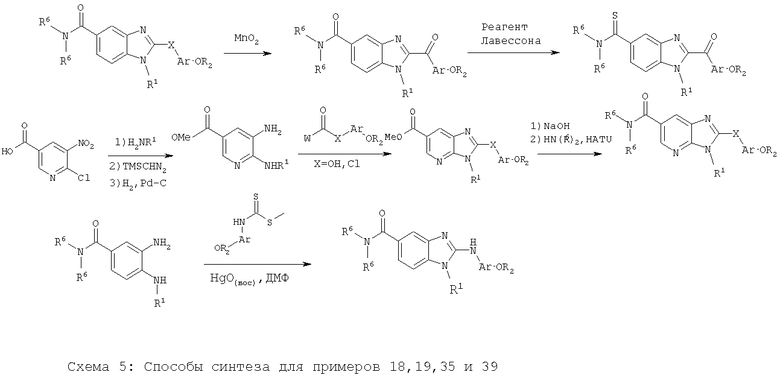
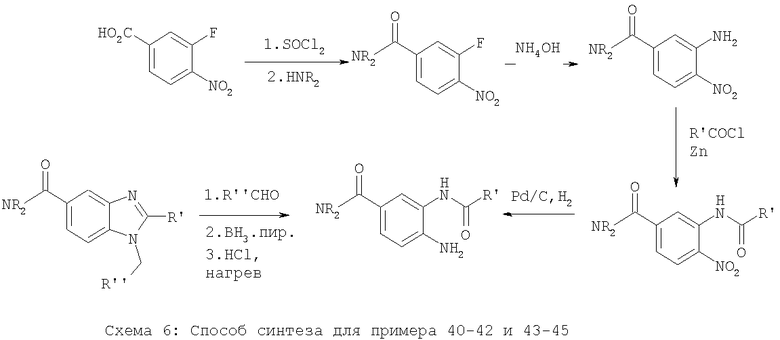
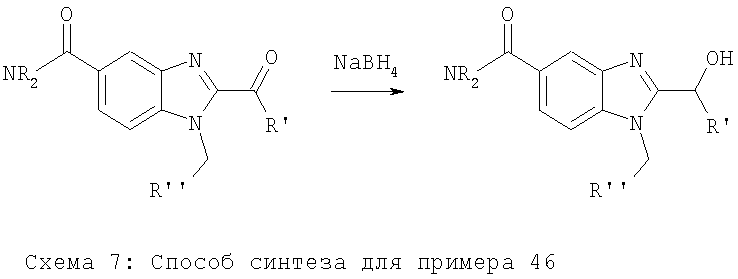
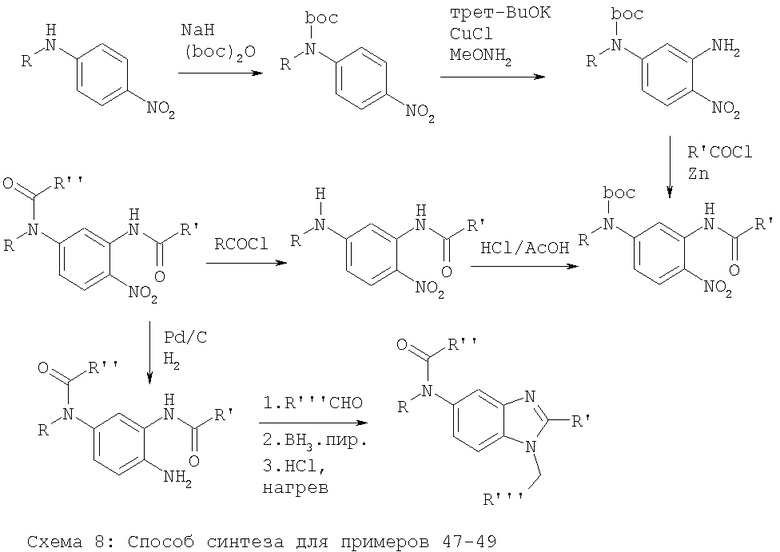
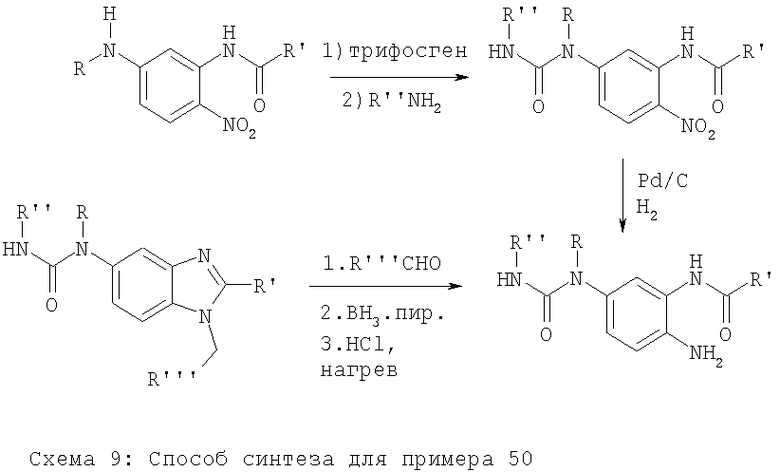
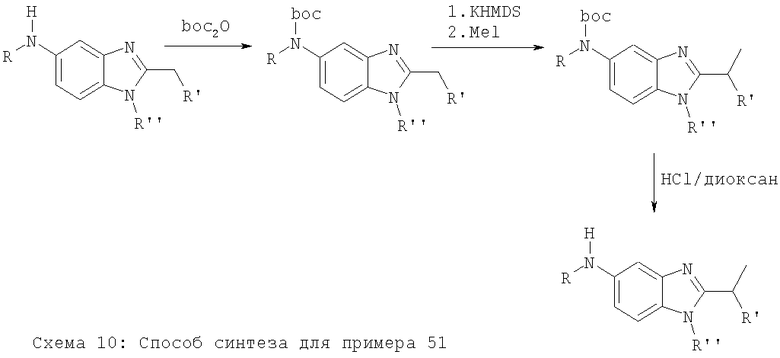
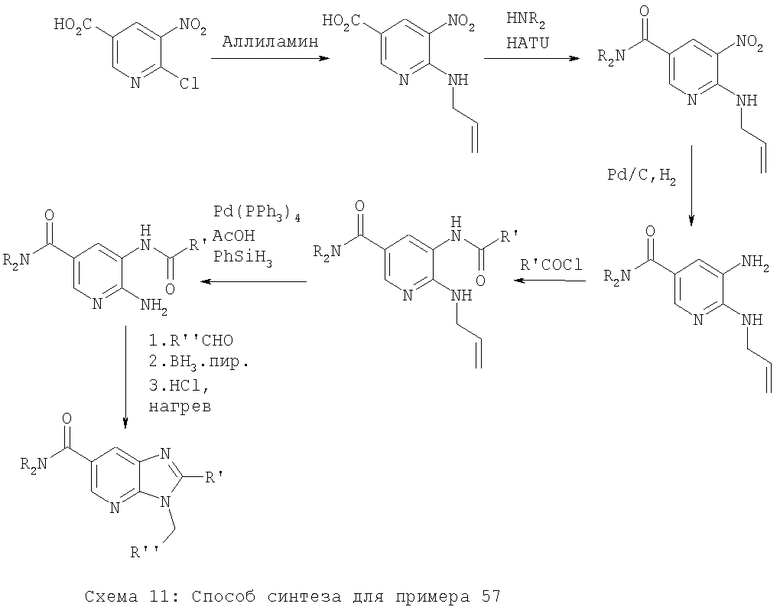
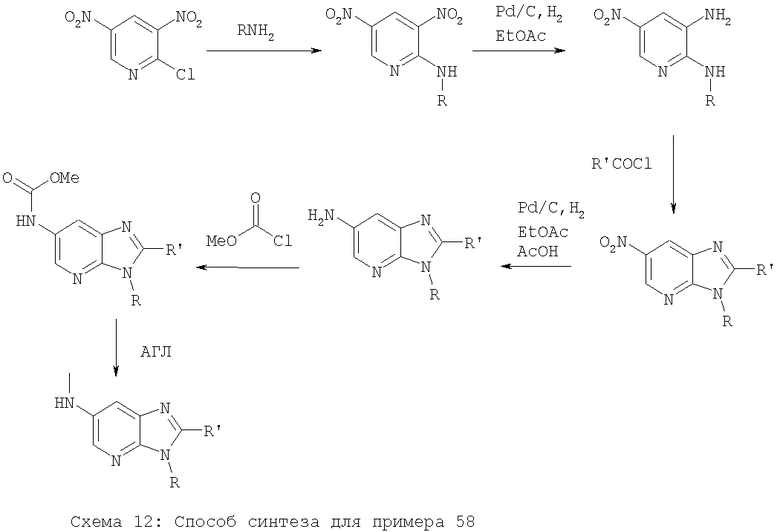
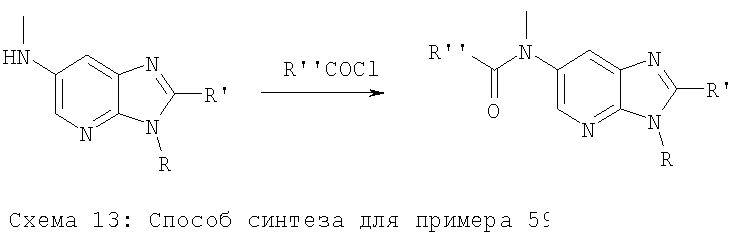
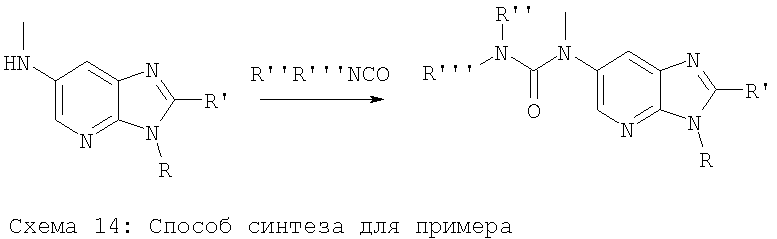
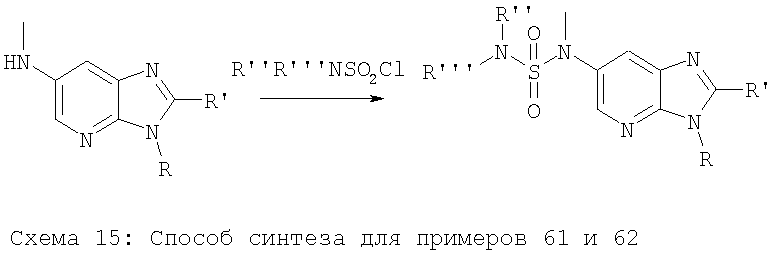
Примеры
Данное изобретение далее описано более подробно следующими примерами, которые раскрывают способы, посредством которых соединения по настоящему изобретению могут быть получены, очищены, проанализированы и протестированы на наличие биологической активности, но эти примеры не следует рассматривать как ограничивающие данное изобретение.
Пример 1: Определение биологической активности
Связывание с рецептором hCВ1 и hСВ2
Мембраны человека СВ1 (полученные от Receptor Biology) или СВ2 (полученные от BioSignal) размораживают при 37°С, 3 раза пропускают через иглу 25-го размера с тупым концом, разбавляют буфером для связывания каннабиноидов (50 мМ трис(гидроксиметил)метиламин (Tris), 2,5 мМ этилендиаминтетрауксусная кислота (ЭДТА), 5 мМ MgCl2, и 0,5 мг/мл бычий сывороточный альбумин (БСА) без жирных кислот, рН 7,4), и аликвоты, содержащие подходящее количество белка, распределяют на 96-луночных планшетах. ИК50 соединений в отношении hСВ1 и hСВ2 оценивают по 10-точечным кривым зависимости "доза-ответ", полученным с использованием 3H-СР55,940 при 20000-25000 распадов в минуту на лунку (0,17-0,21 нМ) в конечном объеме 300 мкл. Общее и неспецифическое связывание определяют соответственно при отсутствии и в присутствии 0,2 мкМ HU210. Планшеты встряхивают и инкубируют в течение 60 минут при комнатной температуре, фильтруют через фильтры Unifilter GF/B (предварительно вымоченные в 0,1% полиэтиленимине) с использованием сборщика Tomtec или Packard и 3 мл буфера для промывки (50 мМ Tris, 5 мМ MgCl2, 0,5 мг БСА, рН 7,0). Фильтры сушат в течение 1 часа при 55°С. Радиоактивность (имп./мин) подсчитывают в TopCount (Packard) после добавления 65 мкл/лунку сцинтилляционной жидкости MS-20.
Связывание гуанозинтрифосфата ГТФγS с hСВ1 и hСВ2
Мембраны человека СВ1 (Receptor Biology) или СВ2 (BioSignal) размораживают при 37°С, 3 раза пропускают через иглу 25-го размера с тупым концом и разбавляют буфером для связывания ГТФγS (50 мМ N-2-гидроксиэтилпиперазин-N'-2-этансульфоновая кислота (Hepes), 20 мМ NaOH, 100 мМ NaCl, 1 мМ ЭДТА, 5 мМ MgCl2, рН 7,4, 0,1% БСА). ЕС50 и Емакс. соединений оценивают по 10-точечным кривым "доза-ответ", полученным на 300 мкл с использованием подходящего количества белка мембран и 100000-130000 распадов в минуту ГТФg35S на лунку (0,11-0,14 нМ). Основное и максимальное стимулированное связывание определяют соответственно при отсутствии и в присутствии 1 мкМ (СВ2) или 10 мкМ (СВ1) Win 55,212-2. Мембраны предварительно инкубируют в течение 5 минут с 56,25 мкМ (СВ2) или 112,5 мкМ (СВ1) гуанозиндифосфата (ГДФ) перед распределением на планшетах (конечная концентрация ГДФ 15 мкМ (СВ2) или 30 мкМ (СВ1)). Планшеты встряхивают и инкубируют в течение 60 минут при комнатной температуре, фильтруют через фильтры Unifilter GF/B (предварительно вымоченные в воде) с использованием сборщика Tomtec или Packard и 3 мл буфера для промывки (50 мМ Tris, 5 мМ MgCl2, 50 мМ NaCl, pH 7,0). Фильтры сушат в течение 1 часа при 55°С. Радиоактивность (имп./мин) подсчитывают в TopCount (Packard) после добавления 65 мкл/лунку сцинтилляционной жидкости MS-20. Исследования с обратимым действием антагониста осуществляют тем же способом за исключением того, что (а) кривую "доза-ответ" для агониста получают в присутствии постоянной концентрации антагониста, или (б) кривую "доза-ответ" для антагониста получают в присутствии постоянной концентрации агониста. Биологические данные для избранных соединений приведены в Таблице 1 ниже.
Биологические данные для избранных соединений примеров 1-39
Пример 2:
2-(4-Метоксибензил)-N,N-диэтил-1-[2-(4-морфолинил)этил]-1Н-бензимидазол-5-карбоксамид
2А: N,N-диэтил-4-фтор-3-нитробензамид:
Перемешиваемый раствор 4-фтор-3-нитробензойной кислоты (25,0 г, 135 ммоль) и тионилхлорида (40,0 мл, 548 ммоль) в CH2Cl2 (40 мл) кипятили с обратным холодильником до тех пор, пока исходные вещества не израсходовались. Растворитель выпаривали в вакууме, совместно выпаривая с толуолом (2×20 мл). Полученный таким образом хлорангидрид карбоновой кислоты растворяли в CH2Cl2 (60 мл) и охлаждали до 0°С перед добавлением диизопропилэтиламина (ДИПЭА) (28,2 мл, 162 ммоль) и диэтиламина (14,0 мл, 135 ммоль) при энергичном перемешивании. Через 2 часа при комнатной температуре растворитель выпаривали в вакууме и получившееся в результате масло растворяли в Et2O (200 мл), промывали 5% NaOH (3×100 мл), 5% KHSO4 (100 мл) и рассолом (100 мл). Органический слой сушили над MgSO4, фильтровали и выпаривали в вакууме. Неочищенное масло растворяли в EtOAc (10 мл) и после выдерживания в течение ночи при -20°С, ярко-оранжевое твердое вещество фильтровали, промывали охлажденным EtOAc (5 мл) и охлажденными гексанами (10 мл) с получением указанного в заголовке соединения (19,6 г). Фильтрат выпаривали и подвергали кристаллизации аналогично в EtOAc (2 мл) с получением еще 6,7 г указанного в заголовке соединения (80,5%) в виде ярко-оранжевого твердого вещества. 1Н-ЯМР (CDCl3): δ 8,11 (dd, J=7,6 Гц, J=1,8 Гц, 1Н), 7,71-7,67 (m, 1H), 7,36 (dd, J=10,8 Гц, J=8,4 Гц, 1Н), 3,56 (brs, 2H), 3,28 (brs, 2H), 1,22 (brs, 6H).
2Б: N,N-Диэтил-4-{[2-(4-морфолинил)этил]амино}-3-нитробензамид
Смесь N,N-диэтил-4-фтор-3-нитробензамида (1,00 г, 4,16 ммоль), 2-(4-морфолинил)этанамина (0,600 мл, 4,58 ммоль) в 80% водн. EtOH (50 мл) кипятили с обратным холодильником в течение 4 часов. Реакционную смесь затем концентрировали в вакууме и остаток растворяли в EtOAc (40 мл). Органическую фазу промывали насыщенным NaHCO3 (2×10 мл) и объединенные водные фазы экстрагировали дополнительным количеством EtOAc (2×20 мл). Объединенные органические фазы сушили над MgSO4, фильтровали и концентрировали в вакууме с получением указанного в заголовке соединения. Неочищенный продукт очищали путем колоночной хроматографии на силикагеле (100% EtOAc до 5% триэтиламин/EtOAc) с получением указанного в заголовке соединения (1,12 г, 77%) в виде ярко-желтого твердого вещества. 1Н-ЯМР (CDCl3): δ 8,67 (s, 1H), 8,28 (d, J=1,2 Гц, 1 Н), 7,56 (dd, J=8,8 Гц, J=1,6 Гц, 1Н), 6,85 (d, J=8,8 Гц, 1Н), 3,77 (t, J=5,2 Гц, 4Н), 3,55-3,20 (brm, 6H), 2,74 (t, J=6,4 Гц, 2H), 2,53 (brs, 4H), 1,22 (t, J=6,8 Гц, 6H). Масс-спектроскопия (МС) (электрогидродинамическая ионизация (ЭГДИ)) (М+Н)+=351.
2В: 3-Амино-N,N-диэтил-4-{[2-(4-морфолинил)этил]амино}бензамид
Смесь N,N-диэтил-4-{[2-(4-морфолинил)этил]амино}-3-нитробензамида (1,10 г, 3,14 ммоль) и 10% Pd/C в МеОН (50 мл) гидрировали в течение 2 часов при 40 ф./кв. дюйм (275,8 кПа). После завершения реакции реакционную смесь фильтровали через диатомовую землю. Удаление растворителя позволило получить указанное в заголовке соединение (0,958 г, 95%), которое использовали без дополнительной очистки. МС (ЭГДИ) (М+Н)+=321.
2Г: 2-(4-Метоксибензил)-N,N-диэтил-1-[2-(4-морфолинил)этил]-1Н-бензимидазол-5-карбоксамид
(4-Метоксифенил)ацетилхлорид (0,063 г, 0,343 ммоль) добавляли к 3-амино-N,N-диэтил-4-{[2-(4-морфолинил)этил]амино}бензамиду (0,100 г, 0,312 ммоль) в уксусной кислоте (2 мл) и смесь перемешивали при 95°С в течение ночи. Реакционную смесь затем концентрировали в вакууме и остаток растворяли в EtOAc (15 мл). Органическую фазу промывали 1н. NaOH (2×8 мл) и объединенные водные фазы экстрагировали дополнительным количеством EtOAc (2×15 мл). Объединенные органические фазы сушили над MgSO4, фильтровали и концентрировали в вакууме. Неочищенный продукт очищали путем обращенно-фазовой хроматографии высокого давления (HPLC) с получением указанного в заголовке соединения в виде соли с трифторуксусной кислотой (ТФУ) (0,132 г, 53%). 1Н-ЯМР (Диметилсульфоксид (ДМСО)-d6): δ 7,74 (d, J=8,0 Гц, 1Н), 7,57 (s, 1H), 7,34 (d, J=8,0 Гц, 1Н), 7,25 (dd, J=8,0 Гц, 2Н), 6,91 (d, J=8,0 Гц, 2Н), 4,64 (t, J=7,6 Гц, 2Н), 4,36 (s, 2Н), 3,76 (brs, 2Н), 3,71 (s, 3Н), 3,35 (brs, 4H), 3,20 (brs, 6H), 1,07 (brs, 6H). МС (ЭГДИ) (M+H)+=451. Аналитически рассчитано для С26H34N4O3+3,0 ТФУ+0,6 Н2O: С, 47,84; Н, 4,79; N, 6,97. Обнаружено: С, 47,83; Н, 4,83; N, 6,96.
Пример 3:
2-(4-Этоксибензил)-N,N-диэтил-1-(2-метоксиэтил)-1Н-бензимидазол-5-карбоксамид
3А: N,N-Диэтил-4-[(2-метоксиэтил)амино]-3-нитробензамид
В соответствии с общим способом 2Б смесь N,N-диэтил-4-фтор-3-нитробензамида (0,200 г, 0,833 ммоль), 2-метоксиэтанамина (0,065 мл, 0,757 ммоль) в 80% водн. EtOH (5 мл) нагревали при 90°С в течение ночи. После обработки неочищенный продукт очищали путем колоночной хроматографии на силикагеле (33% EtOAc/гексаны до 50% EtOAc/гексаны) с получением указанного в заголовке соединения (0,191 г, 85%) в виде ярко-желтого твердого вещества. 1Н-ЯМР (CDCl3): δ 8,34 (s, 1Н), 8,28 (d, J=2,8 Гц, 1Н), 7,56 (dd, J=8,4 Гц, J=1,6 Гц, 1Н), 6,91 (d, J=9,2 Гц, 1Н), 3,69 (t, J=5,6 Гц, 2Н), 3,53 (q, J=5,6 Гц, 2Н), 3,44 (s, перекрывающийся с brs, 7Н), 1,22 (t, J=6,4 Гц, 6Н). МС (ЭГДИ) (М+H)+=296.
3Б: 3-Амино-N,N-диэтил-4-[(2-метоксиэтил)амино]
В соответствии с общим способом 2В смесь N,N-диэтил-4-[(2-метоксиэтил)амино}-3-нитробензамида (0,150 г, 0,508 ммоль) и 10% Pd/C в EtOAc (15 мл) гидрировали в течение ночи при 40 ф./кв. дюйм (275,8 кПа). Обычная обработка позволила получить указанное в заголовке соединение (0,159 г), которое использовали без дополнительной очистки. 1H-ЯМР (CDCl3): δ 6,86 (dd, J=8,4 Гц, J=2,0 Гц, 1Н), 6,80 (d, J=1,6 Гц, 1Н), 6,60 (d, J=8,0 Гц, 1Н), 3,66 (t, J=5,6 Гц, 2Н), 3,41 (s, перекрывающийся с brs, 7H), 3,31 (t, J=5,6 Гц, 2Н), 1,18 (t, J=7,6 Гц, 6Н). МС (ЭГДИ) (М+H)+=266.
3В: (4-Этоксифенил)ацетилхлорид
К перемешиваемому раствору (4-этоксифенил)уксусной кислоты (10,0 г, 55,5 ммоль) в бензоле (100 мл) добавляли тионилхлорид (50 мл, 68,5 ммоль) и реакционную смесь перемешивали при 80°С в течение 2 часов. Растворитель выпаривали в вакууме и неочищенный продукт очищали путем перегонки (температура кипения 86°С, 0,4 торр (53,3 Па)) с получением указанного в заголовке соединения (10,39 г, 94%) в виде желтого масла. МС метилового эфира: МС (ЭГДИ) (M+H)+=194.
3Г: 2-(4-Этоксибензил)-N,N-диэтил-1-(2-метоксиэтил)-1H-бензимидазол-5-карбоксамид
В соответствии с общим способом 2Г (4-Этоксифенил)ацетилхлорид (0,107 г, 0,539 ммоль) добавляли к 3-амино-N,N-диэтил-4-[(2-метоксиэтил)амино]бензамиду (0,130 г, 0,490 ммоль) в толуоле (2,5 мл). Через 20 минут добавляли одну каплю конц. HCl и смесь нагревали при 85°С в течение 12 часов. После обычной обработки неочищенный продукт очищали путем обращенно-фазовой HPLC с получением указанного в заголовке соединения в виде соли с ТФУ (0,120 г, 36%) в виде масла. 1H-ЯМР (ДМСО-d6): δ 7,86 (d, J=9,2 Гц, 1Н), 7,63 (s, 1H), 7,43 (d, J=8,4 Гц, 1Н), 7,25 (d, J=8,4 Гц, 2Н), 6,90 (d, J=9,2 Гц, 2Н), 4,60 (t, J=4,8 Гц, 2Н), 4,47 (s, 2H), 3,97 (q, J=7,6 Гц, 2Н), 3,57 (m, 2H), 3,40 (brs, 2H), 3,20 (brs, 2H), 3,15 (s, 3H), 1,28 (t, J=7,6 Гц, ЗН), 1,07 (br s, 6H). МС (ЭГДИ) (М+Н)+=411. Аналитически рассчитано для С24Н31N3O3+2,2 ТФУ+0,6 H2O: С, 50,82; Н, 5,17; N, 6,26. Обнаружено: С, 50,85; Н, 5,30; N, 6,06.
Пример 4:
1-[2-(Ацетиламино)этил]-2-(4-этоксибензил)-N,N-диэтил-1Н-бензимидазол-5-карбоксамид
4А: 4-{[2-(Ацетиламино)этил]амино}-N,N-диэтил-3-нитробензамид
В соответствии с общим способом 2Б смесь N,N-диэтил-4-фтор-3-нитробензамида (0,200 г, 0,833 ммоль), N-(2-аминоэтил)ацетамида (0,077 г, 0,757 ммоль) в 80% водн. EtOH (5 мл) нагревали при 90°С в течение ночи. После обработки неочищенный продукт очищали путем колоночной хроматографии на силикагеле (100% EtOAc до 5% МеОН/EtOAc) с получением указанного в заголовке соединения (0,152 г, 63%) в виде ярко-желтого твердого вещества. 1Н-ЯМР (CDCl3): δ 8,27 (d перекрывающийся с brs, J=2,0 Гц, 2Н), 7,55 (dd, J=8,4 Гц, J=2,0 Гц, 1Н), 6,99 (d, J=9,2 Гц, 1Н), 6,06 (brs, 1H), 3,58-3,50 (m, 4Н), 3,44 (brs, 4H), 2,02 (s, 3H), 1,22 (t, J=7,2 Гц, 6H). МС (ЭГДИ) (M+H)+=323.
4Б: 4-{[2-(Ацетиламино)этил]амино}-3-амино-N,N-диэтилбензамид
В соответствии с общим способом 2В смесь 4-{[2-(ацетиламино)этил]амино}-N,N-диэтил-3-нитробензамида (0,150 г, 0,465 ммоль) и 10% Pd/C в EtOAc (15 мл) гидрировали в течение ночи при 40 ф./кв. дюйм (275,8 кПа). Обычная обработка позволила получить указанное в заголовке соединение (0,164 г, 100%), которое использовали без дополнительной очистки. 1H-ЯМР (CDCl3): δ 6,83 (dd, J=8,0 Гц, J=2,0 Гц, 1Н), 6,76 (d, J=2,0 Гц, 1Н), 6,53 (d, J=8,4 Гц, 1H), 6,26 (brt, 1H), 3,53 (q, J=5,6 Гц, 2Н), 3,43 (brs, 4H), 3,23 (t, J=5,6 Гц, 2Н), 1,99 (s, 3H), 1,17 (t, J=6,4 Гц, 6Н). МС (ЭГДИ) {M+H)+=293.
4В: 1-[2-(Ацетиламино)этил]-2-(4-этоксибензил)-N,N-диэтил-1Н-бензимидазол-5-карбоксамид
В соответствии с общим способом 2Г (4-Этоксифенил)ацетилхлорид (0,097 г, 0,490 ммоль) добавляли к 4-{[2-(ацетиламино)этил]амино}-3-амино-N,N-диэтилбензамиду (0,130 г, 0,445 ммоль) в толуоле (2,5 мл). Через 20 минут добавляли одну каплю конц. HCl и смесь нагревали при 85°С в течение 12 часов. После обычной обработки неочищенный продукт очищали путем обращенно-фазовой HPLC с получением указанного в заголовке соединения в виде соли с ТФУ (0,042 г, 14%) в виде желтовато-коричневого твердого вещества. 1H-ЯМР (ДМСО-d6): δ 8,01 (t, J=5,6 Гц, 1Н), 7,81 (d, J=8,4 Гц, 1Н), 7,63 (s, 1Н), 7,45 (d, J=8,4 Гц, 1Н), 7,26 (d, J=8,4 Гц, 2Н), 6,90 (t, J=8,4 Гц, 2Н), 4,43 (m, 4Н), 3,97 (q, J=7,2 Гц, 2Н), 3,40 (brs, 2H), 3,38 (t, J=4,8 Гц, 2Н), 3,20 (brs, 2H), 3,15 (s, 3Н), 1,61 (s, 3Н), 1,28 (t, J=7,2 Гц, 3Н), 1,06 (brs, 6H). MC (ЭГДИ) (M+H)+=437. Аналитически рассчитано для C25H32N4O3+2,1 ТФУ+0,8 H2O: С, 50,80; Н, 5,21; N, 8,11. Обнаружено: С, 50,87; Н, 5,87; N, 8,09. Пример 5:
Метил-3-[5-[(диэтиламино)карбонил]-2-(4-этоксибензил)-1Н-бензимидазол-1-ил]пропаноат
5А: Этил-3-{4-[(диэтиламино)карбонил]-2-нитроанилино}пропаноат
В соответствии с общим способом 2Б смесь N,N-диэтил-4-фтор-3-нитробензамида (0,200 г, 0,833 ммоль), этил-3-аминопропаноата (0,116 г, 0,757 ммоль) в 80% водн. EtOH (5 мл) нагревали при 90°С в течение ночи. После обработки неочищенный продукт очищали путем колоночной хроматографии на силикагеле (50% EtOAc/гексаны до 100% EtOAc) с получением указанного в заголовке соединения (0,162 г, 63%). 1Н-ЯМР (CDCl3): δ 8,34 (brt, J=5,6 Гц, 1Н), 8,29 (d, J=2,0 Гц, 1Н), 7,58 (dd, J=8,4 Гц, J=2,0 Гц, 1Н), 6,92 (d, J=9,2 Гц, 1Н), 4,21 (q, J=7,2 Гц, 2H), 3,69 (q, J=6,4 Гц, 2H), 3,44 (brs, 4H), 2,72 (t, J=6,4 Гц, 2H), 1,30 (t, J=6,4 Гц, 3Н), 1,22 (t, J=6,8 Гц, 6H). MC (ЭГДИ) (М+H)+=338.
5Б: Этил-3-{2-амино-4-[(диэтиламино)карбонил]анилино}пропаноат
В соответствии с общим способом 2В смесь этил-3-{4-[(диэтиламино)карбонил]-2-нитроанилино}пропаноата (0,150 г, 0,445 ммоль) и 10% Pd/C в EtOAc (15 мл) гидрировали в течение ночи при 40 ф./кв. дюйм 275,8 кПа). Обычная обработка позволила получить указанное в заголовке соединение (0,158 г), которое использовали без дополнительной очистки. 1H-ЯМР (CDCl3): δ 6,86 (dd, J=8,4 Гц, J=2,0 Гц, 1 Н), 6,80 (d, J=2,0 Гц, 1 Н), 6,62 (d, J=8,4 Гц, 1Н), 4,18 (q, J=7,2 Гц, 2H), 3,46 (t и перекрывающийся brs, J=5,6 Гц, 6H), 2,66 (t, J=6,8 Гц, 2H), 1,28 (t, J=7,2 Гц, 3Н), 1,18 (t, J=6,8 Гц, 6H). MC (ЭГДИ) (M+H)+=308.
5В: Метил-3-[5-[(диэтиламино)карбонил]-2-(4-этоксибензил)-1H-бензимидазол-1-ил]пропаноат
В соответствии с общим способом 2Г (4-этоксифенил)ацетилхлорид (0,092 г, 0,465 ммоль) добавляли к этил-3-{2-амино-4-[(диэтиламино)карбонил]анилино}пропаноату (0,130 г, 0,423 ммоль) в толуоле (2,5 мл). Через 20 минут добавляли одну каплю конц. HCl и смесь нагревали при 85°С в течение 12 часов. После обработки неочищенный продукт очищали путем обращенно-фазовой HPLC (которую сопровождали транс-этерификацией с использованием МеОН) с получением указанного в заголовке соединения в виде соли с ТФУ (0,084 г, 30%) в виде масла. 1H-ЯМР (ДМСО-d6): δ 7,88 (d, J=9,2 Гц, 1Н), 7,62 (s, 1H), 7,41 (d, J=8,4 Гц, 1Н), 7,25 (d, J=8,0 Гц, 2Н), 6,90 (d, J=8,4 Гц, 2Н), 4,59 (t, J=6,4 Гц, 2Н), 4,49 (s, 2H), 3,97 (q, J=6,4 Гц, 2Н), 3,55 (s, 3Н), 3,40 (brs, 2H), 3,16 (brs, 2H), 2,77 (t, J=6,4 Гц, 2H), 1,28 (t, J=7,6 Гц, 3Н), 1,07 (brs, 6H). МС (ЭГДИ) (M+H)+=438. Аналитически рассчитано для С25Н31N3O4+1,8 ТФУ+0,8 H2O: С, 52,27; Н, 5,28; N, 6,39. Обнаружено: С, 52,31;H, 5,22; N, 6,37.
Пример 6:
1-(2-Аминоэтил)-2-(4-этоксибензил)-N,N-диэтил-1H-бензимидазол-5-карбоксамид
6А: Трет-бутил-2-{4-[(диэтиламино)карбонил]-2-нитроанилино}этилкарбамат
В соответствии с общим способом 2Б смесь N,N-диэтил-4-фтор-3-нитробензамида (0,200 г, 0,833 ммоль), трет-бутил-2-аминоэтилкарбамата (0,121 г, 0,757 ммоль) в 80% водн. этаноле (3 мл) нагревали при 85°С в течение ночи. После обработки неочищенный продукт очищали путем перекристаллизации из EtOAc с получением указанного в заголовке соединения (0,165 г, 57%) в виде ярко-желтого твердого вещества. 1Н-ЯМР (CDCl3): δ 8,31 (brs, 1H), 8,29 (d, J=1,6 Гц, 1H), 7,57 (dd, J=9,6 Гц, J=2,0 Гц, 1H), 6,97 (d, J=9,6 Гц, 1H), 4,83 (brs, 1H), 3,55-3,40 (m, 8H), 1,47 (s, 9H), 1,22 (t, J=7,6 Гц, 6H), МС (ЭГДИ) (М+Н)+=381.
6Б: Трет-бутил-2-{2-амино-4-[(диэтиламино)карбонил]анилино}этилкарбамат
В соответствии с общим способом 2В смесь трет-бутил-2-{4-[(диэтиламино)карбонил]-2-нитроанилино}этилкарбамата (0,155 г, 0,407 ммоль) и 10% Pd/C в EtOAc (15 мл) гидрировали в течение ночи при 40 ф./кв. дюйм (275,8 кПа). Обычная обработка позволила получить указанное в заголовке соединение (0,164 г), которое использовали без дополнительной очистки. 1H-ЯМР (CDCl3): δ 6,85 (dd, J=8,4 Гц, J=1,6 Гц, 1Н), 6,80 (d, J=2,0 Гц, 1Н), 6,57 (d, J=7,6 Гц, 1H), 4,87 (brs, 1H), 3,48-3,38 (brs, 6H), 3,27 (t, J=5,6 Гц, 2Н), 1,46 (s, 9H), 1,18 (t, J=6,8 Гц, 6H), МС (ЭГДИ) (M+H)+=351.
6В: 1-(2-Аминоэтил)-2-(4-этоксибензил)-N,N-диэтил-1H-бензимидазол-5-карбоксамид
В соответствии с общим способом 2Г (4-этоксифенил)ацетилхлорид (0,085 г, 0,424 ммоль) добавляли к трет-бутил-2-{2-амино-4-[(диэтиламино)карбонил]анилино}этилкарбамату (0,135 г, 0,385 ммоль) в толуоле (2,5 мл). Через 20 минут добавляли одну каплю конц. HCl и смесь нагревали при 85°С в течение 12 часов. После обработки неочищенный продукт очищали путем обращенно-фазовой HPLC с получением указанного в заголовке соединения в виде соли с ТФУ (0,123 г, 44%) в виде белого твердого вещества. 1H-ЯМР (CD3OD): δ 7,91 (d, J=8,4 Гц, 1H), 7,73 (s, 1H), 7,57 (dd, J=8,4 Гц, J=2,0 Гц, 2Н), 7,30 (d, J=8,4 Гц, 2H), 6,97 (d, J=8,4 Гц, 2Н), 4,75 (t, J=6,8 Гц, 2Н), 4,55 (s, 2Н), 4,03 (q, J=6,8 Гц, 2H), 3,58 (brs, 2H), 3,36 (t, J=6,4 Гц, 2H), 3,29 (brs, 2H), 1,37 (t, J=6,4 Гц, 3H), 1,27 (brs, 3H), 1,12 (brs, 3Н), МС (ЭГДИ) (M+H)+=395, Аналитически рассчитано для С23Н30N4O2 + 2,8 ТФУ + 0,8 Н2О: С, 47,17; Н, 4,76; N, 7,69. Обнаружено: С, 47,16; Н, 4,80; N, 7,52.
Пример 7:
1-{2-[Ацетил(метил)амино]этил}-2-(4-этоксибензил)-N,N-диэтил-1Н-бензимидазол-5-карбоксамид
К раствору продукта стадии 6В, соли 1-(2-аминоэтил)-2-(4-этоксибензил)-N,N-диэтил-1H-бензимидазол-5-карбоксамида с ТФУ (0,085 г, 0,117 ммоль), триэтиламину (0,070 мл, 0,50 ммоль) в СН2Cl2 (3 мл) добавляли ацетилхлорид (0,015 г, 0,200 ммоль) и получившуюся в результате смесь перемешивали при комнатной температуре в течение 15 минут. Смесь промывали насыщенным водным раствором NaHCO3, рассолом и органическую фазу сушили над MgSO4, фильтровали и концентрировали в вакууме.
Указанный выше неочищенный продукт растворяли в диметилформамиде (ДМФ) (2 мл), добавляли NaH (60% дисперсия в масле, 0,005 г, 0,217 ммоль) и смесь перемешивали при комнатной температуре в течение 20 минут. Растворитель выпаривали в вакууме и остаток растворяли в EtOAc. Органическую фазу промывали насыщенным водным NaHCO3, рассолом, сушили над MgSO4, фильтровали и концентрировали в вакууме. Неочищенный продукт очищали путем обращенно-фазовой HPLC с получением указанного в заголовке соединения в виде соли с ТФУ (0,017 г, 26%) в виде белого твердого вещества. 1H-ЯМР (CD3OD): δ 7,97 (d, J=8,4 Гц, 1Н), 7,69 (s, 1H), 7,59 (d, J=7,2 Гц, 1Н), 7,33 (d, J=8,4 Гц, 2Н), 6,96 (d, J=8,4 Гц, 2Н), 4,66 (t, J=6,0 Гц, 2Н), 4,60 (s, 2Н), 4,02 (q, J=7,1 Гц, 2H), 3,72 (t, J=6,0 Гц, 2Н), 3,56 (br, 2Н), 3,28 (br, 4H), 3,06 (s, 3Н), 1,79 (s, 3H), 1,36 (t, J=8,6 Гц, 3Н), 1,25 (br, 3H), 1,10 (br, 3Н), МС (ЭГДИ) (M+H)+=451.
Пример 8:
2-(4-Этоксибензил)-N,N-диэтил-1-метил-1H-бензимидазол-5-карбоксамид
8А: N,N-Диэтил-4-(метиламино)-3-нитробензамид
В соответствии с общим способом 2Б смесь N,N-диэтил-4-фтор-3-нитробензамида (0,125 г, 0,521 ммоль), гидрохлорида метиламина (0,035 г, 0,521 ммоль) в 80% водн. EtOH (3 мл) нагревали при 85°С в течение 4 часов. Обычная обработка позволила получить указанное в заголовке соединение (0,130 г, 100%), которое использовали без дополнительной очистки. МС (ЭГДИ) (M+H)+=252.
8Б: 3-Амино-N,N-диэтил-4-(метиламино)бензамид
В соответствии с общим способом 2В смесь N,N-диэтил-4-(метиламино)-3-нитробензамида (0,130 г, 0,517 ммоль) и 10% Pd/C в EtOAc (10 мл) гидрировали при 40 ф./кв. дюйм (275,8 кПа). Обычная обработка позволила получить указанное в заголовке соединение (0,124 г), которое использовали без дополнительной очистки. МС (ЭГДИ) (M+H)+=222.
8В: 2-(4-Этоксибензил)-N,N-диэтил-1-метил-1H-бензимидазол-5-карбоксамид
В соответствии с общим способом 2Г (4-этоксифенил)ацетилхлорид (0,119 г, 0,596 ммоль) добавляли к 3-амино-N,N-диэтил-4-(метиламино)бензамиду (0,120 g, 0,542 ммоль) в уксусной кислоте (2 мл) и смесь перемешивали при 90°С в течение ночи. После обработки неочищенный продукт очищали путем обращенно-фазовой HPLC с получением указанного в заголовке соединения в виде соли с ТФУ (0,087 г, 26%) в виде красного масла. 1H-ЯМР (ДМСО-d6): δ 7,90 (d, J=8,4 Гц, 1Н), 7,70 (s, 1H), 7,49 (d, J=8,4 Гц, 1Н), 7,29 (d, J=8,4 Гц, 2H), 6,92 (d, J=8,4 Гц, 2Н), 4,50 (s, 2Н), 3,98 (q, J=7,6 Гц, 2Н), 3,93 (s, 3Н), 3,42 (brs, 2H), 3,14 (brs, 2H), 1,28 (t, J=6,8 Гц, 3H), 1,11 (brs, 3H), 1,04 (brs, 3H), MC (ЭГДИ) (M+H)+=366. Аналитически рассчитано для С22Н27N3O2+2,1 ТФУ+0,2 Н2O: С, 51,71; Н, 4,89; N, 6,91. Обнаружено: С, 51,73; Н, 4,82; N, 6,93.
Пример 9:
2-(4-Этоксибензил)-N,N-диэтил-1-(2-фенилэтил)-1H-бензимидазол-5-карбоксамид
9А: N,N-Диэтил-3-нитро-4-[(2-фенилэтил)амино]бензамид
В соответствии с общим способом 2Б смесь N,N-диэтил-4-фтор-3-нитробензамида (0,125 г, 0,521 ммоль), 2-фенилэтанамина (0,065 мл, 0,521 ммоль) в 80% водн. EtOH (3 мл) нагревали при 85°С в течение 4 часов. Обычная обработка позволила получить указанное в заголовке соединение (0,170 г, 96%), которое использовали без дополнительной очистки. MC (ЭГДИ) (M+H)+=342.
9Б: 3-Амино-N,N-диэтил-4-[(2-фенилэтил)амино]бензамид
В соответствии с общим способом 2В смесь N,N-диэтил-3-нитро-4-[(2-фенилэтил)амино]бензамида (0,170 g, 0,498 ммоль) и 10% Pd/C в EtOAc (10 мл) гидрировали при 40 ф./кв. дюйм (275,8 кПа). Обычная обработка позволила получить указанное в заголовке соединение (0,156 г), которое использовали без дополнительной очистки. MC (ЭГДИ) (М+Н)+=312.
9В: 2-(4-Этоксибензил)-N,N-диэтил-1-(2-фенилэтил)-1H-бензимидазол-5-карбоксамид
В соответствии с общим способом 2Д (4-этоксифенил)ацетилхлорид (0,105 г, 0,530 ммоль) добавляли к 3-амино-N,N-диэтил-4-[(2-фенилэтил)амино]бензамиду (0,150 г, 0,482 ммоль) в уксусной кислоте (2 мл) и смесь перемешивали при 90°С в течение ночи. После обработки неочищенный продукт очищали путем обращенно-фазовой HPLC с получением указанного в заголовке соединения в виде соли с ТФУ (0,100 г, 36%) в виде фиолетового твердого вещества. 1H-ЯМР (ДМСО-d6): δ 7,74 (d, J=8,4 Гц, 1Н), 7,62 (s, 1H), 7,34 (d, J=7,2 Гц, 1H), 7,24-7,19 (m, 5H), 7,10 (d, J=8,4 Гц, 2Н), 6,91 (d, J=8,4 Гц, 2Н), 4,56 (t, J=7,2 Гц, 2Н), 4,23 (s, 2Н), 3,97 (q, J=6,8 Гц, 2H), 3,40 (brs, 2H), 3,19 (brs, 2H), 2,86 (t, J=7,2 Гц, 2H), 1,28 (t, J=6,8 Гц, 3Н), 1,08 (brs, 6H), MC (ЭГДИ) (M+H)+=456. Аналитически рассчитано для С29Н33N3O2+1,0 ТФУ+0,2 H2O: С, 64,96; Н, 6,05; N, 7,33. Обнаружено: С, 65,05; Н, 6,09; N, 7,29.
Пример 10:
2-(4-Этоксибензил)-N,N-диэтил-1-[2-(1-пиперидинил)этил]-1Н-бензимидазол-5-карбоксамид
10А: N,N-Диэтил-3-нитро-4-{[2-(1-пиперидинил)этил]амино}бензамид
В соответствии с общим способом 2Б смесь N,N-диэтил-4-фтор-3-нитробензамида (1,0 г, 4,2 ммоль), 2-(1-пиперидинил)этанамина (0,564 мл, 3,96 ммоль) в 80% водн. EtOH (30 мл) нагревали при 85°С в течение 10 часов. После обычной обработки неочищенную смесь растворяли в 1н. HCl (40 мл) и промывали CH2Cl2 (2×10 мл). Водный слой подщелачивали 5 н NaOH (10 мл) и экстрагировали СН2Cl2 (3×10 мл). Объединенные органические фазы сушили над Na2SO4, фильтровали и концентрировали в вакууме с получением указанного в заголовке соединения (0,800 г, 57%) в виде ярко-желтого твердого вещества, которое использовали без дополнительнйо очистки. 1H-ЯМР (CDCl3): δ 8,64 (brs, 1H), 8,25 (d, J=2,4 Гц, 1H), 7,53 (d, J=8,8 Гц, 1H), 6,82 (d, J=8,8 Гц, 1H), 3,41 (brs, 4H), 3,35 (brs, 2H), 2,64 (brs, 2H), 2,53 (brs, 4H), 1,61 (brs, 4H), 1,44 (brs, 2H), 1,19 (t, J=7,0 Гц, 6Н).
10Б: 3-Амино-N,N-диэтил-4-{[2-(1-пиперидинил)этил]амино}бензамид
В соответствии с общим способом 2В смесь N,N-диэтил-3-нитро-4-{[2-(1-пиперидинил)этил]амино}бензамида (0,800 g, 2,30 ммоль) и 10% Pd/C в EtOAc (30 мл) гидрировали в течение 24 часов при 30 ф./кв. дюйм (206,85 кПа). Обычная обработка позволила получить указанное в заголовке соединение (0,724 г, 99%), которое использовали без дополнительной очистки.
10В: 2-(4-Этоксибензил)-N,N-диэтил-1-[2-(1-пиперидинил)этил]-1Н-бензимидазол-5-карбоксамид
В соответствии с общим способом 2Г раствор 1М (4-этоксифенил)ацетилхлорида в толуоле (0,095 мл, 0,095 ммоль) добавляли к 3-амино-N,N-диэтил-4-{[2-(1-пиперидинил)этил]амино}бензамиду (0,027 г, 0,856 ммоль) в толуоле (1,0 мл). Через 20 минут добавляли одну каплю конц. HCl и смесь нагревали при 85°С в течение ночи. После обработки неочищенный продукт очищали путем обращенно-фазовой HPLC с получением указанного в заголовке соединения в виде соли с ТФУ (0,026 г, 39%). 1H-ЯМР (CD3OD): δ 7,89 (d, J=8,0 Гц, 1Н), 7,70 (s, 1H), 7,52 (d, J=8,4 Гц, 1Н), 7,27 (d, J=7,6 Гц, 2Н), 6,96 (d, J=7,2 Гц, 2Н), 4,52 (s, 2Н), 4,02 (q, J=7,6 Гц, 2H), 3,56 (brs, 4H), 3,34 (t, J=7,6 Гц, 2H), 3,28 (brs, 4H), 3,00 (brs, 2H), 1,88 (brs, 4H), 1,53 (brs, 2H), 1,36 (t, J=6,4 Гц, 3Н), 1,25 (brs, 3Н), 1,11 (brs, 3H). МС (ЭГДИ) (М+Н)+=463. Аналитически рассчитано для С28Н38N4O2+2,6 ТФУ+0,5 W: С, 51,92; Н, 5,46; N, 7,29. Обнаружено: С, 51,94; Н, 5,43; N, 7,25.
Пример 11:
1-[2-(Диметиламино)-1-метилэтил]-2-(4-этоксибензил)-N,N-диэтил-1Н-бензимидазол-5-карбоксамид
11А: 4-{[2-(Диметиламино)-1-метилэтил]амино}-N,N-диэтил-3-нитробензамид
Смесь N,N-диэтил-4-фтор-3-нитробензамида (0,500 г, 2,08 ммоль), N',N'-диметил-1,2-пропандиамина (0,636 г, 6,24 ммоль), ДИПЭА (2,2 мл, 12,5 ммоль) и ДМФ (12 мл) перемешивали в течение 4 часов при комнатной темпеаратуре. Реакционную смесь затем концентрировали в вакууме, остаток растворяли в 2 н. NaOH (30 мл) и экстрагировали CH2Cl2 (3×40 мл). Объединенные органические фазы промывали рассолом (10 мл), сушили над MgSO4, фильтровали и концентрировали в вакууме. Неочищенную реакционную смесь очищали путем колоночной хроматографии на силикагеле (100% CH2Cl2 до 5% MeOH/CH2Cl2) с получением указанного в заголовке соединения (0,621 g, 93%) в виде ярко-желтого твердого вещества. 1Н-ЯМР (CDCl2): δ 8,33 (d, J=6,4 Гц, 1H), 8,27 (d, J=2,0 Гц, 1Н), 7,55 (dd, J=8,8 Гц, J=2,0 Гц, 1Н), 6,91 (d, J=9,6 Гц, 1Н), 3,79 (гептет, J=7,2 Гц, 1Н), 3,45 (brd, J=5,2 Гц, 4Н), 2,55 (dd, J=12,0 Гц, J=7,6 Гц, 1Н), 2,39 (dd, J=12,4 Гц, J=5,8 Гц, 1Н), 2,29 (s, 6H), 1,69 (brs, 1H), 1,31 (d, J=6,8 Гц, 2Н), 1,22 (t, J=7,4 Гц, 6H). MC (ЭГДИ) (М+Н)+=323.
11Б: 3-Амино-4-{[2-(диметиламино)-1-метилэтил]амино}-N,N-диэтилбензамид
В соответствии с общим способом 2В смесь 4-{[2-(диметиламино)-1-метилэтил]амино}-N,N-диэтил-3-нитробензамида (0,516 g, 1,60 ммоль) и 10% Pd/C в EtOAc (20 мл) гидрировали в течение ночи при 35 ср./кв. дюйм (241,3 кПа). Обычная обработка позволила получить указанное в заголовке соединение (0,408 г, 87%), которое использовали без дополнительной очистки. 1H-ЯМР (CDCl3): δ 6,82 (dd, J=8,0 Гц, J=2,0 Гц,1Н), 6,77 (d, J=2,4 Гц, 1H), 6,62 (d, J=8,0 Гц, 1H), 3,52-336 (m, 6H), 2,52 (dd, J=12,0 Гц, J=9,6 Гц, 2Н), 2,28-2,19 (m, 7Н), 1,17 (m, 8H). MC (ЭГДИ) (M+H)+=293.
11В: 1-[2-(Диметиламино)-1-метилэтил]-2-(4-этоксибензил)-N,N-диэтил-1Н-бензимидазол-5-карбоксамид
В соответствии с общим способом 2Г раствор 1М (4-этоксифенил)ацетилхлорида в толуоле (0,095 мл, 0,095 ммоль) добавляли к 3-амино-4-{[2-(диметиламино)-1-метилэтил]амино}-N,N-диэтилбензамиду (0,025 г, 0,0856 ммоль) в толуоле (1,0 мл). Через 20 минут добавляли одну каплю конц. HCl и смесь нагревали при 85°С в течение ночи. После обработки неочищенный продукт очищали путем обращенно-фазовой HPLC с получением указанного в заголовке соединения в виде соли с ТФУ (0,024 г, 38%). 1Н-ЯМР (CD3OD): δ 7,96 (d, J=8,4 Гц, 1H), 7,72 (s, 1H), 7,47 (d, J=7,6 Гц, 2Н), 7,24 (m, 2H), 6,94 (m, 2H), 5,20 (brs, 1H), 4,50 (s, 2H), 4,03-3,95 (m, 3Н), 3,77 (dd, J=14,0 Гц, J=5,6 Гц, 1H), 3,56 (brs, 2H), 3,28 (m, 2H), 2,81 (s, 6H), 1,56 (d, J=6,8 Гц, 3Н), 1,35 (t, J=6,4 Гц, 3Н), 1,25 (brs, 3Н), 1,14 (brs, 3Н). MC (ЭГДИ) (М+Н)+=437. Аналитически рассчитано для С26Н36N4O2+2,5 ТФУ+0,7 Н2O: С, 50,71; Н, 5,48; N, 7,63. Обнаружено: С, 50,76; Н, 5,46; N, 7,61.
Пример 12:
1-(Циклопропилметил)-2-(4-этоксибензил)-N,N-диэтил-1Н-бензимидазол-5-карбоксамид
12А: 4-[(Циклопропилметил)амино]-N,N-диэтил-3-нитробензамид
В соответствии с общим способом 2Б N,N-диэтил-4-фтор-3-нитробензамид (0,823 г, 3,42ммоль) и циклопропилметанамин (0,39 мл, 4,50 ммоль) в 80% водном этаноле (17 мл) перемешивали при 85°С в течение 16 часов. Неочищенный продукт (1,00 г) в виде оранжевого твердого вещества использовали на последующих стадиях. 1H-ЯМР (CD3OD): δ 8,23 (d, J=2,0 Гц, 1Н), 7,55 (dd, J=9,2 Гц, J=2,0 Гц, 1H), 7,09 (d, J=9,2 Гц, 1Н), 3,47 (brs, 3H), 3,31 (q, J=2,0 Гц, 2Н), 3,27 (d, J=6,4 Гц, 2Н), 1,23 (перекрывающийся t и m, J=7,0 Гц, 7H), 0,68-0,60 (m, 2H), 0,40-0,34 (m, 2H). МС (ЭГДИ) (М+Н)+=292.
12Б: 3-Амино-4-[(циклопропилметил)амино]-N,N-диэтилбензамид
В соответствии с общим способом 2В 4-[(циклопропилметил)амино]-N,N-диэтил-3-нитробензамид (1,00 г) гидрировали в течение 24 ч с получением указанного в заголовке соединения (0,889 г) в виде зеленовато-коричневого твердого вещества. Неочищенный продукт использовали на последующих стадиях. 1H-ЯМР (CD3OD): δ 6,72-6,78 (m, 2H), 6,58 (d, J=8,4 Гц, 1H), 3,43 (brs, 4H), 2,99 (d, J=6,4 Гц, 2H), 1,18 (перекрывающийся brt и m, J=6,4 Гц, 7Н), 0,59-0,52 (m, 2H), 0,30-0,24 (m, 2H), МС (ЭГДИ) (M+H)+=262.
12В: 1-(Циклопропилметил)-2-(4-этоксибензил)-N,N-диэтил-1Н-бензимидазол-5-карбоксамид
В соответствии с общим способом 2Г (4-этоксифенил)ацетилхлорид (0,109 г, 0,547 ммоль) добавляли к 3-амино-4-[(циклопропилметил)амино]-N,N-диэтилбензамиду (0,130 г, 0,497 ммоль) в уксусной кислоте (2 мл) и смесь перемешивали при 90°С в течение ночи. После обработки неочищенный продукт очищали путем обращенно-фазовой HPLC с получением указанного в заголовке соединения в виде соли с ТФУ (0,042 г, 14%) в виде масла. 1H-ЯМР (ДМСО-d6): δ 7,59 (d, J=8,4 Гц, 1H), 7,30 (s, 1H), 7,08 (d, J=7,2 Гц, 1H), 6,91 (d, J=9,2 Гц, 2H), 6,53 (d, J=8,4 Гц, 2H), 4,14 (s, 2H), 3,97 (d, J=7,2 Гц, 2H), 3,60 (q, J=6,4 Гц, 2H), 3,04 (brs, 2H), 2,80 (brs, 2H), 0,90 (t, J=6,4 Гц, 3H), 0,78 (brm, 7H), 0,10-0,03 (m, 4H), MS (ЭГДИ) (М+Н)+=406. Аналитически рассчитано для С25Н31N3O2+1,9 ТФУ: С, 55,60; Н, 5,33; N, 6,75. Обнаружено: С, 55,51; Н, 5,25; N, 6,74.
Пример 13:
1-(Циклопропилметил)-2-[(6-этокси-3-пиридинил)метил]-N,N-диэтил-1Н-бензимидазол-5-карбоксамид
13А: (6-Этокси-З-пиридинил)уксусная кислота
К перемешиваемому раствору (6-хлор-3-пиридинил)уксусной кислоты (0,094 г, 0,545 ммоль) в EtOH (1,6 мл) добавляли 3,1 М EtONa в EtOH (0,7 мл, 2,17 ммоль) и реакционную смесь перемешивали при 100 °С в течение 24 часов, после чего добавляли избыток NaH (60% дисперсия в масле) вместе с 1 мл EtOH и нагревание при 100°С продолжали в течение 96 ч. Растворитель выпаривали в вакууме и остаток растворяли в диэтиловом эфире (5 мл). Органическую фазу промывали разбавленной HCl (2×2 мл) и рассолом (2 мл), сушили над MgSO4, фильтровали и концентрировали в вакууме с получением указанного в заголовке соединения (0,081 г, 82%) в виде бледно-коричневого твердого вещества, которое использовали без дополнительной очистки. 1H-ЯМР (CDCl3): δ 10,06 (brs, 1H), 8,06 (s, 1H), 7,55 (d, J=9,2 Гц, 1Н), 6,72 (d, J=8,4 Гц, 1H), 4,30 (q, J=6,1 Гц, 2H), 3,58 (s, 2H) 1,38 (t, J=8,0 Гц, 3Н). МС (ЭГДИ) (M+H)+=182.
13Б: 1-(Циклопропилметил)-2-[(6-этокси-3-пиридинил)метил]-N,N-диэтил-1Н-бензимидазол-5-карбоксамид
К перемешиваемому раствору (6-этокси-3-пиридинил)уксусной кислоты (0,081 г, 0,448 ммоль) в ДМФ (2 мл) добавляли гексафторфосфат O-(7-азабензотриазол-1-ил)-N,N,N',N'-тетраметилурония (HATU) (0,178 г, 0,468 ммоль) и ДИПЭА (0,156 мл, 0,895 ммоль) и реакционную смесь перемешивали при комнатной температуре в течение 10 мин. Добавляли 3-амино-4-[(циклопропилметил)амино]-N,N-диэтилбензамид (0,111 г, 0,448 ммоль) и получающуюся в результате смесь перемешивали в течение 1 часа при комнатной температуре. Смесь разбавляли EtOAc (15 мл), промывали насыщенным NaHCO3 (8 мл) и затем рассолом (8 мл), сушили над MgSO4 и концентрировали в вакууме с получением неочищенного амида, который использовали без дополнительной очистки.
Вышеописанное неочищенное промежуточное соединение растворяли в уксусной кислоте (5 мл) и смесь нагревали при 90°С в течение ночи. Реакционную смесь концентрировали и остаток растворяли в EtOAc (15 мл). Органическую фазу промывали 1н. NaOH (2×8 мл) и затем рассолом (8 мл), сушили над MgSO4 и концентрировали в вакууме. Неочищенный продукт очищали путем колоночной хроматографии на силикагеле (2% МеОН/EtOAc) с получением указанного в заголовке соединения (0,111 г, 65%) в виде бледно-коричневого твердого вещества. 1H-ЯМР (CDCl3): δ 8,05 (s, 1H), 7,75 (s, 1H), 7,49 (d, J=8,4 Гц, 1H), 7,38-7,30 (m, 2H), 6,68 (d, J=8,4 Гц, 1H), 4,32 (qd, J=7,2 Гц, J=2,2 Гц, 2H), 4,25 (s, 2H), 3,55 (brs, 2H), 3,38 (brs, 2H), 1,38 (td, J=6,8 Гц, J=2,0 Гц, 3H), 1,22 (brs, 6H), 1,12-1,03 (m, 1H), 0,60-0,54 (m, 2H), 0,33-0,28 (m, 2H), МС (ЭГДИ) (М+Н)+=407. Аналитически рассчитано для C24H28N4O2+ 0,2Н2O: С, 70,63; Н, 7,01; N, 13,73. Обнаружено: С, 70,83; Н, 7,50; N, 13,67.
Пример 14:
1-[2-(Диметиламино)этил]-2-[2-(4-этоксифенил)этил]-N,N-диэтил-1Н-бензимидазол-5-карбоксамид
14А: 4-{[2-(Диметиламино)этил]амино}-N,N-диэтил-3-нитробензамид
В соответствии с общим способом 2Б N,N-диэтил-4-фтор-3-нитробензамид (0,534 г, 2,22 ммоль), N1,N1-диметил-1,2-этандиамин (0,22 мл, 2,02 ммоль) перемешивали при 85°С в течение 14 часов. Неочищенный продукт очищали путем растворения в 1 н. HCl и экстракции Et2O (2×15 мл). Водную фазу доводили до рН 11 5 н. NaOH и затем экстрагировали CH2Cl2 (4×15 мл). Объединенные СН2Cl2-фазы сушили над MgSO4, фильтровали и концентрировали в вакууме с получением указанного в заголовке соединения (0,560 г, 82%) в виде оранжевого масла. 1Н-ЯМР (CD3OD): δ 8,22 (d, J=2,8 Гц, 1H), 7,56 (dd, J=8,4 Гц, J=2,8 Гц, 1H), 7,09 (d, J=8,4 Гц, 1Н), 3,53-3,40 (перекрывающийся t, J=6,4 Гц и brs, 6H), 2,67 (t, J=6,4 Гц, 2H), 2,33 (s, 6H), 1,22 (t, J=7,6 Гц, 6H). МС (ЭГДИ) (M+H)+=309.
14Б: 3-Амино-4-{[2-(диметиламино)этил]амино}-N,N-диэтилбензамид
В соответствии с общим способом 2В 4-{[2-(диметиламино)этил]амино}-N,N-диэтил-3-нитробензамид (0,560 г, 1,82 ммоль) гидрировали в течение 3 ч с получением указанного в заголовке соединения (0,531 г) в виде зеленовато-коричневого твердого вещества. Неочищенный продукт использовали на последующих стадиях. 1Н-ЯМР (CD3OD): δ 6,77, 6,76 (перекрывающийся s и dd, J=6,4 Гц, J=2,0 Гц, 2Н), 6,61 (d, J=8,4 Гц, 1H), 3,45 (brs, 4H), 3,29 (t, J=6,4 Гц, 2H), 2,66 (t, J=6,4 Гц, 2Н), 2,34 (s, 6Н), 1,19 (brt, J=6,8 Гц, 6H). МС (ЭГДИ) (M+H)+=279.
14В: 1-[2-(Диметиламино)этил]-2-[2-(4-этоксифенил)этил]-N,N-диэтил-1H-бензимидазол-5-карбоксамид
В соответствии со способом 13Б объединяли 3-(4-этоксифенил)пропановую кислоту (0,0386 г, 0,198 ммоль), HATU (0,0756 г, 0,199 ммоль), ДИПЭА (0,047 мл, 0,27 ммоль) и 4-{[2-(диметиламино)этил]амино}-N,N-диэтил-3-аминобензамид (0,0503 г, 0,180 ммоль). Неочищенный продукт очищали путем колоночной хроматографии (9:1 CH2Cl2:MeOH) с получением указанного в заголовке соединения (0,0284 г, 36%). 1Н-ЯМР (CD3OD): δ 7,64 (s, 1H), 7,53 (d, J=8,4 Гц, 1H), 7,29 (d, J=8,4 Гц, 1H), 7,06 (d, J=8,4 Гц, 2H), 6,78 (d, J=8,4 Гц, 2H), 4,14 (t, J=8,0 Гц, 2H), 3,96 (q, J=7,2 Гц, 2H), 3,57 (brs, 2H), 3,35 (brs, 2H), 3,21-3,10 (m, 4H), 2,43 (t, J=7,6 Гц, 2H), 2,26 (s, 6H), 1,34 (t, J=7,2 Гц, 3Н), 1,26, 1,16 (2 перекрывающиеся brs, 6H). 13С-ЯМР (CD3OD): δ 174,03, 159,16, 158,09, 142,67, 136,55, 133,78, 132,24, 130,54, 122,10, 117,34, 115,65, 111,44, 64,43, 58,74, 45,80, 45,16, 42,64, 41,00, 34,46, 30,53, 15,18, 14,43, 13,12. МС (ЭГДИ) (М+Н)+=437. Соль с HCl получали с использованием HCl в Et2O. После лиофилизации получали белое твердое вещество. Аналитически рассчитано для С26Н36N4O2·2,8 HCl·2,2 H2O: С, 54,00; Н, 7,53; N, 9,69. Обнаружено: С, 54,12; Н, 7,53; N, 9,36.
Пример 15:
1-(Циклопропилметил)-2-[2-(4-этоксифенил)этил]-N,N-диэтил-1Н-бензимидазол-5-карбоксамид
В соответствии с общим способом 13Б объединяли 3-(4-этоксифенил)пропановую кислоту (0,0409 г, 0,211 ммоль), HATU (0,0801 г, 0,211 ммоль), ДИПЭА (0,050 мл, 0,29 ммоль) и 3-амино-4-[(циклопропилметил)амино]-N,N-диэтилбензамид (0,0500 г, 0,191 ммоль). Неочищенный продукт очищали путем колоночной хроматографии (19:1 CH2Cl2:MeOH) с получением указанного в заголовке соединения (0,0457 г, 57%). 1Н-ЯМР (CD3OD): δ 7,64 (s, 1H), 7,55 (d, J=8,4 Гц, 1H), 7,27 (d, J=8,4 Гц, 1H), 7,08 (d, J=8,4 Гц, 2H), 6,77 (d, J=8,4 Гц, 2H), 3,97 (d, J=7,2 Гц, 2H), 3,95 (q, J=6,8 Гц, 2H), 3,57 (brs, 2H), 3,34 (brs, 2H), 3,23-3,07 (m, 4H), 1,34 (t, J=6,4 Гц, ЗН), 1,36-1,02 (перекрывающиеся 2 x brs и m, 7H), 0,55-0,46 (m, 2H), 0,40-0,32 (m, 2H). 13С-ЯМР (CD3OD): δ 174,08, 159,08, 157,72, 142,60, 136,95, 133,70, 132,03, 130,46, 121,97, 117,21, 115,59, 111,83, 64,41, 48,69, 45,15, 40,98, 34,38, 30,54, 15,18, 14,43, 13,12, 12,15, 4,54. МС (ЭГДИ) (М+Н)+=420. Соль с HCl получали с использованием HCl в Et2O. После лиофилизации получали белое твердое вещество. Аналитически рассчитано для С26Н33N3O2·1,0 HCl·0,8 Н2O: С, 66,38; Н, 7,63; N, 8,93. Обнаружено: С, 66,35; Н, 7,60; N, 8,81.
Пример 16:
2-(4-Этоксибензил)-N,N-диэтил-1-изопентил-1Н-бензимидазол-5-карбоксамид
16А: N,N-Диэтил-4-(изопентиламино)-3-нитробензамид
В соответствии с общим способом 2Б N,N-диэтил-4-фтор-3-нитробензамид (1,077 г, 4,48 ммоль) и З-метил-1-бутанамин (0,68 мл, 5,83 ммоль) перемешивали при 85°С в течение 14 часов с получением указанного в заголовке соединения (1,425 г) в виде оранжевого масла. Неочищенный продукт использовали на следующих стадиях. 1H-ЯМР (CD3OD): δ 8,22 (d, J=2,0 Гц, 1Н), 7,56 (dd, J=9,2 Гц, J=2,0 Гц, 1H), 7,09 (d, J=9,2 Гц, 1Н), 3,52-3,40 (br m, 6H), 1,83-1,72 (m, 1H), 1,64 (q, J=7,6 Гц, 2H), 1,23 (t, J=7,6 Гц, 6Н), 1,01 (d, J=6,8 Гц, 6H). МС (ЭГДИ) (M+H)+=308.
16Б: 3-Амино-N,N-диэтил-4-(изопентиламино)бензамид
В соответствии с общим способом 2В N,N-диэтил-4-(изопентиламино)-3-нитробензамид (1,425 г, 4,64 ммоль) гидрировали в течение 41 ч с получением указанного в заголовке соединения (1,312 г) в виде темно-синего твердого вещества. Неочищенный продукт использовали на последующих стадиях. 1H-ЯМР (CD3OD): δ 6,78-6,74 (m, 2H), 6,58 (d, J=8,4 Гц, 1H), 3,45 (brs, 4H), 3,17 (t, J=7,6 Гц, 2H), 1,83-1,72 (m, 1H), 1,59 (q, J=7,2 Гц, 2H), 1,19 (t, J=6,4 Гц, 6H), 0,98 (d, J=6,4 Гц, 6H). МС (ЭГДИ) (М+Н)+=278.
16В: 2-(4-Этоксибензил)-N,N-диэтил-1-изопентил-1H-бензимидазол-5-карбоксамид
В соответствии с общим способом 13Б объединяли (4-этоксифенил)уксусную кислоту (0,110 г, 0,612 ммоль), HATU (0,233 г, 0,611 ммоль), ДИПЭА (0,15 мл, 0,86 ммоль) и 3-амино-N,N-диэтил-4-(изопентиламино)бензамид (0,154 г, 0,556 ммоль). Неочищенный продукт очищали путем колоночной хроматографии (19:1 CH2Cl2:MeOH) с получением указанного в заголовке соединения (0,211 г, 90%). 1H-ЯМР (CD3OD): δ 7,67 (s, 1Н), 7,50 (d, J=8,4 Гц, 1H), 7,30 (dd, J=8,4 Гц, J=2,0 Гц, 1Н), 7,16 (d, J=9,2 Гц, 2Н), 6,86 (d, J=8,4 Гц, 2Н), 4,28 (s, 2Н), 4,12 (m, 2H), 3,98 (q, J=7,6 Гц, 2Н), 3,57 (brs, 2H), 3,34 (brs, 2H), 1,62-1,48 (m, 1H), 1,34 (t, J=7,6 Гц, 3Н), 1,36-1,07 (перекрывающиеся 2×brs и m, 8H), 0,88 (d, J=6,8 Гц, 6Н). МС (ЭГДИ) (M+H)+=422. Аналитически рассчитано для C26H35N3O2-1,7 ТФУ 0,2 Н2O: С, 57,05; Н, 6,04; N, 6,79. Обнаружено: С, 57,11; Н, 6,09; N, 6,69.
Пример 17:
2-(4-Этоксибензил)-N,N-диэтил-1-(4-пиридинилметил)-1Н-бензимидазол-5-карбоксамид
17А: N,N-Диэтил-3-нитро-4-[(4-пиридинилметил)амино]бензамид
В соответствии с общим способом 2Б N,N-диэтил-4-сртор-3-нитробензамид (0,272 г, 1,13 ммоль) и 4-пиридинилметанамин (0,11 мл, 1,12 ммоль) перемешивали при комнатной температуре в течение 87 часов. Неочищенный продукт очищали путем колоночной хроматографии (100% EtOAc, затем 9:1 CH2Cl2:MeOH) с получением указанного в заголовке соединения (0,181 г, 60%). 1H-ЯМР (CD3OD): δ 8,49 (d, J=6,4 Гц, 2H), 8,27 (d, J=2,8 Гц, 1H), 7,50-7,42 (m, 3Н), 6,87 (d, J=8,4 Гц, 1H), 4,77 (s, 2H), 3,44 (brs, 4H), 1,21 (t, J=7,2 Гц, 6Н). МС (ЭГДИ) (М+Н)+=329.
17Б: 3-Амино-N,N-диэтил-4-[(4-пиридинилметил)амино]бензамид
В соответствии с общим способом 2В N,N-диэтил-3-нитро-4-[(4-пиридинилметил)амино]бензамид (0,181 г, 0,551 ммоль) гидрировали в течение 20 ч с получением указанного в заголовке соединения (0,172 г) в виде вязкого коричневого масла. Неочищенный продукт использовали на последующих стадиях. 1H-ЯМР (CD3OD): δ 8,45 (d, J=6,8 Гц, 2H), 7,46 (d, J=6,4 Гц, 2H), 6,79 (d, J=2,0 Гц, 1H), 6,64 (dd, J=7,2 Гц, J=2,0 Гц, 1H), 6,36 (d, J=8,4 Гц, 1H), 4,51 (s, 2H), 3,42 (brs, 4H), 1,17 (brt, 6H). МС (ЭГДИ) (M+H)+=299.
17В: 2-(4-Этоксибензил)-N,N-диэтил-1-(4-пиридинилметил)-1Н-бензимидазол-5-карбоксамид
В соответствии со способом 13Б объединяли (4-этоксифенил)уксусную кислоту (0,0364 г, 0,202 ммоль), HATU (0,0768 г, 0,202 ммоль), ДИПЭА (0,048 мл, 0,28 ммоль) и 3-амино-N,N-диэтил-4-[(4-пиридинилметил)амино]бензамид (0,0548 г, 0,184 ммоль). Неочищенный продукт очищали путем обращенно-фазовой жидкостной хроматографии среднего давления (MPLC) (градиент 10-50% СН2Cl2 в Н2O) с получением указанного в заголовке соединения (0,0459 г, 36%) в виде его соли с ТФУ. Это вещество лиофилизировали из смеси Н2O/диоксан с получением белого твердого вещества. 1H-ЯМР (CD3OD): δ 8,60 (brs, 2H), 7,87 (s, 1Н), 7,70 (d, J=8,4 Гц, 1H), 7,52 (dd, J=8,4 Гц, J=2,0 Гц, 1Н), 7,42 (d, J=5,6 Гц, 2H), 7,13 (d, J=8,4 Гц, 2H), 6,71 (d, J=9,6 Гц, 2H), 6,06 (s, 2H), 4,54 (s, 2H), 3,93 (q, J=7,2 Гц, 2H), 3,59 (brs, 2H), 3,31 (brs, 2H), 1,35 (t, J=6,8 Гц, 3H), 1,38-1,04 (2×brs, 6H). MC (ЭГДИ) (М+Н)+=443. Аналитически рассчитано для С27Н30N4O2·2,1 ТФУ 0,8 Н2O: С, 53,81; Н, 4,88; N, 8,04. Обнаружено: С, 53,74; Н, 4,89; N, 8,07.
Пример 18:
2-(4-Этоксибензоил)-N,N-диэтил-1-изопентил-1Н-бензимидазол-5-карбоксамид
MnO2 (0,641 г, 7,37 ммоль) добавляли к перемешиваемому раствору 2-(4-этоксибензил)-N,N-диэтил-1-изопентил-1Н-бензимидазол-5-карбоксамида (0,211 г, 0,500 ммоль) в безводном диоксане (4 мл) в атмосфере N2. Реакционную смесь нагревали до 50°С. Через 64 ч добавляли дополнительную порцию MnO2 (0,600 г, 6,90 ммоль) и нагревание продолжали при 50°С в течение еще 24 ч. Реакционную смесь охлаждали до комнатной температуры, разбавляли СН2Cl2 и фильтровали через диатомовую землю. Твердое вещество тщательно промывали дополнительным количеством CH2Cl2 и фильтрат концентрировали с получением неочищенного продукта. Очистка с использованием колоночной хроматографии (1:1 EtOAc:гептан) позволила получить указанное в заголовке соединение (0,139 г, 64%) в виде вязкого бесцветного масла. 1H-ЯМР (CD3OD): δ 8,19 (d, J=9,6 Гц, 2H), 7,83 (s, 1H), 7,72 (d, J=8,4 Гц, 1H), 7,48 (dd, J=8,4 Гц, J=1,6 Гц, 1H), 7,01 (d, J=9,2 Гц, 2H), 4,53 (dd, J=7,6 Гц, J=7,2 Гц, 2H), 4,13 (q, J=6,4 Гц, 2H), 3,58 (brs, 2H), 3,36 (brs, 2H), 1,78-1,70 (m,2H), 1,72-1,56 (m, 1H), 1,41 (t, J=7,2 Гц, 3H), 1,34-1,10 (2×brs, 6Н), 0,94 (d, J=6,8 Гц, 6Н). 13С-ЯМР (CD3OD): 5 185,72, 173,32, 165,47, 149,57, 142,00, 137,18, 134,65, 133,57, 130,19, 124,88, 119,93, 115,31, 112,75, 65,10, 45,12, 44,94, 41,00, 40,13, 27,18, 22,79, 14,99, 14,46, 13,12. MC (ЭГДИ) (М+Н)+=436.
Аналитически рассчитано для С26Н33N3О3·0,5 H2O: С, 70,24; Н, 7,71; N, 9,45. Обнаружено: С, 70,40; Н, 7,64; N, 8,97.
Пример 19:
2-(4-Этоксибензоил)-N,N-диэтил-1-изопентил-1H-бензимидазол-5-карботиоамид
Реагент Лавессона (0,0963 г, 0,238 ммоль) добавляли к перемешиваемому раствору 2-(4-этоксибензоил)-N,N-диэтил-1-изопентил-1H-бензимидазол-5-карбоксамида (0,0451 г, 0,104 ммоль) в безводном толуоле (5 мл) в атмосфере N2. Реакционную смесь нагревали до температуры дефлегмации в течение 0,5 ч и затем охлаждали до комнатной температуры и концентрировали. Остаток очищали путем колоночной хроматографии (3:1 гексаны:EtOAc) с получением указанного в заголовке соединения (0,0137 г, 29%) в виде стекловидного желтого твердого вещества. 1H-ЯМР (CD3OD): δ 8,19 (d, J=9,2 Гц, 2Н), 7,67 (d, J=8,4 Гц, 1Н), 7,64 (s, 1H), 7,36 (d, J=8,4 Гц, 1Н), 7,06 (d, J=9,2 Гц, 2Н), 4,55 (dd, J=8,4 Гц, J=7,2 Гц, 2Н), 4,17 (q, J=7,6 Гц, 4Н), 3,53 (q, J=7,6 Гц, 2Н), 1,80-1,71 (m, 2H), 1,71-1,58 (m, 1H), 1,44 (t, J=7,6 Гц, ЗН), 1,41 (t, J=7,6 Гц, 3Н), 1,17 (t, J=7,6 Гц, 3Н), 0,97 (d, J=6,4 Гц, 6Н). 13С-ЯМР (CD3OD): δ 201,23, 185,95, 165,57, 149,55, 141,87, 141,18, 136,28, 134,64, 130,31, 124,56, 118,20, 115,41, 112,36, 65,15, 47,48, 44,89, 40,18, 27,20, 22,77, 14,95, 14,07, 11,44. МС (ЭГДИ) (M+H)+=452. Аналитически рассчитано для C26H33N3O2S·0,3 Н2O: С, 68,33; Н, 7,41; N, 9,19. Обнаружено: С, 68,44; Н, 7,52; N, 9,01.
Пример 20:
N-Циклогексил-1-(циклопропилметил)-2-(4-этоксибензил)-N-метил-1Н-бензимидазол-5-карбоксамид
20А: 4-[(Циклопропилметил)амино]-3-нитробензонитрил
В соответствии с общим способом 2Б к 733 мг (4,42 ммоль) 4-фтор-3-нитробензонитрила в 80% водном этаноле (20 мл) при комнатной температуре добавляли циклопропилметиламин (377 мг, 5,3 ммоль). Смесь перемешивали при 60°С в течение 3 ч перед обработкой. Неочищенный продукт (920 мг, 96%) использовали на следующей стадии без дополнительной очистки. 1H-ЯМР (400 МГц, CDCl3): δ 8,52 (s, 1H), 8,51-8,47 (br, 1H), 7,59 (d, J=8,4 Гц, 1H), 6,90 (d, J= 8,4 Гц, 1H), 3,22-3,20 (m, 2H), 1,25-1,15 (m, 1H), 0,73-0,59 (m, 2H), 0,42-0,35 (m, 2H). МС (ЭГДИ) [2×(М+Н)+]:436.
20Б: 3-Амино-4-[(циклопропилметил)амино]бензонитрил
В соответствии с общим способом 2В неочищенный 4-[(циклопропилметил)-амино]-3-нитробензонитрил (920 мг, 4,24 ммоль) гидрировали в течение 2 ч (35 ф./кв. дюйм (241,3 кПа)) в 40 мл EtOAc с получением 750 мг неочищенного продукта (95%) и использовали на следующей стадии без дополнительной очистки. МС (ЭГДИ) (M+H)+=188.
20В: 1-(Циклопропилметил)-2-(4-этоксибензил)-1H-бензимидазол-5-карбонитрил
В соответствии с общим способом 2Г только что полученный (4-этоксифенил)ацетилхлорид (из 793 мг кислоты, 4,4 ммоль) и неочищенный 3-амино-4-[(циклопропилметил)амино]бензонитрил (750 мг, 4,01 ммоль) нагревали в уксусной кислоте (НОАс) в течение ночи. После обработки неочищенный остаток очищали путем колоночной хроматографии на силикагеле с получением чистого продукта (1,04 г, 78%) в виде твердого вещества белого цвета с желтоватым оттенком. 1Н-ЯМР (400 МГц, CDCl3): δ 8,08 (s, 1H), 7,50 (d, J=8,4 Гц, 1Н), 7,38 (d, J=8,4 Гц, 1Н), 7,14 (d, J=8,4 Гц, 2Н), 6,84 (d, J=8,4 Гц, 2Н), 4,29 (s, 2Н), 4,01 (d, J=7,6 Гц, 2Н), 3,96 (q, J=7,6 Гц, 2Н), 1.39 (t, J=7,6 Гц, 3Н), 1,04-1,00 (m, 1H), 0,58-0,53 (m, 2H), 0,29-0,25 (m, 2H). МС (ЭГДИ) (М+H)+=332. Аналитически рассчитано для C21H21N3O.0,1 H2O: С, 75,50; Н, 6,41; N, 12,61. Обнаружено: С, 75,76; Н, 6,72; N, 12,45.
20Г: 1-(Циклопропилметил)-2-(4-этоксибензил)-1Н-бензимидазол-5-карбоновая кислота
К раствору 1-(циклопропилметил)-2-(4-этоксибензил)-1Н-бензимидазол-5-карбонитрила (500 мг, 1,51 ммоль) в 1:1 EtOH:Н2O (16 мл) добавляли КОН (338 мг, 6,04 ммоль). Получившуюся в результате смесь кипятили с обратным холодильником в течение 36 ч. Смесь подкисляли 1 н. HCl до рН˜6,0, осадок собирали путем фильтрации с получением белого твердого вещества (520 мг, 99%), которое использовали на следующей стадии без дополнительной очистки. Аналитически чистое соединение получали путем перекристаллизации (из EtOH-Н2O). 1H-ЯМР (400 МГц, CD3OD): δ 8,25 (s, 1H), 7,89 (d, J=8,4 Гц, 1H), 7,47 (d, J=8,4 Гц, 1H), 7,18 (d, J=8,2 Гц, 2H), 6,82 (d, J=8,2 Гц, 2H), 4,28 (s, 2H), 4,06 (d, J=6,8 Гц, 2H), 3,89 (q, J=7,6 Гц, 2H), 1,32 (t, J=7,6 Гц, 3Н), 1,06-0,92 (m, 1H), 0,50-0,48 (m, 2H), 0,32-0,26 (m, 2H). МС (ЭГДИ) (М+Н)+=351. Аналитически рассчитано для С21Н22N2O3.0,7HCl: С, 67,09; Н, 6,09; N, 7,45. Обнаружено: С, 66,98; Н, 6,31;N, 7,09.
20Д: N-Циклогексил-1-(циклопропилметил)-2-(4-этоксибензил)-N-метил-1H-бензимидазол-5-карбоксамид
1-(Циклопропилметил)-2-(4-этоксибензил)-1Н-бензимидазол-5-карбоновую кислоту (80 мг, 0,228 ммоль) растворяли в ДМФ (2 мл) и затем добавляли HATU (104 мг, 0,274 ммоль) с последующим ДИПЭА (48 мкл, 0,274 ммоль). После перемешивания в течение 10 минут добавляли циклогексилметиламин (0,456 ммоль) и полученную в результате смесь перемешивали в течение ночи. Смесь разбавляли EtOAc (50 мл), промывали насыщенным NaHCO3 (2×10 мл) и затем Н2O (2×10 мл), сушили над MgSO4 и концентрировали с получением неочищенного амида. Неочищенный остаток очищали путем колоночной хроматографии на силикагеле с получением чистого продукта, который обрабатывали 1 М раствором HCl в диэтиловом эфире с получением соли с HCl в виде белого твердого вещества (93 мг, 78%). 1H-ЯМР (400 МГц, CD3OD): δ 7,66 (s, 1H), 7,58 (d, J=8,0 Гц, 1Н), 7,30 (d, J=8,0 Гц, 1H), 7,16 (d, J=8,6 Гц, 2Н), 6,84 (d, J=8,6 Гц, 2Н), 4,30 (s, 2H), 4,10-4,04 (m, 5H), 3,96 (q, J=7,6 Гц, 2H), 3,66-3,54 (m, 1H), 1,90-1,50 (m, 10Н), 1,33 (t, J=7,6 Гц, 3Н), 1,10-0,98 (m, 1H), 0,50-0,42 (m, 2H), 0,32-0,27 (m, 2H). МС (ЭГДИ) (М+Н)+=446. Аналитически рассчитано для С28Н35N3O2·2,2HCl: С, 63,96; Н, 7,13; N, 7,99. Обнаружено: С, 64,11; Н, 7,24; N, 7,73.
Пример 21:
1-Циклопропилметил)-2-(4-этоксибензил)-5-(1-пирролидинилкарбонил)-1Н-бензимидазол
В соответствии с общим способом 20Д 1-(циклопропилметил)-2-(4-этоксибензил)-1Н-бензимидазол-5-карбоновую кислоту (80 мг, 0,228 ммоль) растворяли в ДМФ (2 мл) и затем добавляли HATU (104 мг, 0,274 ммоль) с последующим ДИПЭА (48 мкл, 0,274 ммоль). После перемешивания в течение 10 минут добавляли пирролидин (0,456 ммоль) и полученную в результате смесь перемешивали в течение ночи. Неочищенный остаток после обработки очищали путем колоночной хроматографии на силикагеле с получением чистого продукта, который обрабатывали 1М раствором HCl в диэтиловом эфире с получением соли с HCl в виде белого твердого вещества (82 мг, 79%).
1H-ЯМР (400 МГц, CD3OD): δ 7,85 (s, 1H), 7,59 (d, J=8,6 Гц, 1Н), 7,48 (d, J=8,6 Гц, 1H), 7,18 (d, J=8,1 Гц, 2Н), 6,87 (d, J=8,1 Гц, 2Н), 4,32 (s, 2H), 4,07 (d, J=6,8 Гц, 2H), 4,00 (q, J=7,6 Гц, 2H), 3,64 (t, J=7,2 Гц, 2H), 3,53 (t, J=7,2 Гц, 2H), 2,06-1,96 (m, 2H), 1,96-1,85 (m, 2H), 1,36 (t, J=7,6 Гц, 3Н), 1,10-1,04 (m, 1H), 0,52-0,46 (m, 2H), 0,34-0,28 (m, 2H). МС (ЭГДИ) (M+H)+=404. Аналитически рассчитано для С25Н29N3O2.1,5HCl: С, 65,53; Н, 6,71; N, 9,17. Обнаружено: С, 65,48; Н, 6,77; N, 8,75.
Пример 22:
1-(Циклопропилметил)-2-(4-этоксибензил)-5-(1-пирролидинилкарботиоил)-1Н-бензимидазол
К раствору свободного основания 1-(циклопропилметил)-2-(4-этоксибензил)-5-(1-пирролидинилкарбонил)-1H-бензимидазола (пример 21, из 80 мг 1-(циклопропилметил)-2-(4-этоксибензил)-1Н-бензимидазол-5-карбоновой кислоты) в пиридине (2 мл) добавляли Р2S5 (507 мг, 1,14 ммоль). Смесь нагревали при 100°С в течение 24 часов и охлаждали до комнатной температуры. После декантирования супернатант концентрировали и остаток разбавляли EtOAc (20 мл), промывали 1н. NaOH (2×10 мл) и затем Н2O (2×10 мл). Органический слой сушили над MgSO4 и выпаривали. Неочищенный остаток очищали путем колоночной хроматографии на силикагеле с получением масла (59 мг, 62% для 2 стадий). 1Н-ЯМР (400 МГц, CD3OD): δ 7,70 (s, 1H), 7,39 (d, J=8,0 Гц, 1H), 7,28 (d, J=8,0 Гц, 1H), 7,12 (d, J=8,4 Гц, 2H), 6,81 (d, J=8,4 Гц, 2H), 4,27 (s, 2H), 4,08-3,95 (m, 4H), 3,89 (d, J=6,8 Гц, 2H), 3,57 (t, J=7,2 Гц, 2H), 2,14-2,03 (m, 2H), 2,00-1,92 (m, 2H), 1,38 (t, J=7,2 Гц, 3Н), 1,06-0,98 (m, 1H), 0,54-0,48 (m, 2H), 0,28-0,22 (m, 2H). МС (ЭГДИ) (M+H)+=420.
Пример 23:
1-(Циклопропилметил)-2-[(5-этокси-2-пиридинил)метил]-5-(1-пирролидинилкарбонил)-1Н-бензимидазол
23А: [5-(Бензилокси)-2-пиридинил]уксусная кислота
К раствору 1,45 г (6,46 ммоль) [5-(бензилокси)-2-пиридинил]ацетонитрила (полученного в 6 стадий из 6-метил-З-пиридинола, см.: W.M. Golebiewski и J.T. Wrobel, Bull. Pol. Acad. Sci., 1990, 38, 17) в МеОН (9 мл) добавляли 25% NaOH (3 мл). Реакционную смесь перемешивали при температуре дефлегмации в течение 48 ч. После охлаждения большую часть МеОН удаляли в вакууме, добавляли воду (10 мл) и водный раствор промывали диэтиловым эфиром (2×10 мл) с последующим подкислением (рН ˜6,0). Смесь экстрагировали EtOAc (3×20 мл), сушили над MgSO4 и концентрировали с получением бледно-желтых кристаллов (1,4 г, 89%). МС (ЭГДИ) (M+H)+=244.
23Б: 2-{[5-(Бензилокси)-2-пиридинил]метил}-1-(циклопропилметил)-1Н-бензимидазол-5-карбонитрил
В соответствии с общим способом 13Б 360 мг (1,48 ммоль) [5-(бензилокси)-2-пиридинил]уксусной кислоты соединяли с 3-амино-4-[(циклопропилметил)амино]бензонитрилом, используя HATU. Получающееся в результате промежуточное соединение нагревали в НОАс (20 мл) при 90°С в течение ночи. После концентрирования неочищенный остаток очищали путем колоночной хроматографии на силикагеле с получением чистого продукта (230 мг, 40% для 2 стадий) в виде масла. МС (ЭГДИ) (М+H)+=395.
23В: 1-(Циклопропилметил)-2-[(5-гидрокси-2-пиридинил)метил]-1Н-бензимидазол-5-карбонитрил
К раствору 2-{[5-(бензилокси)-2-пиридинил]метил}-1-(циклопропилметил)-1H-бензимидазол-5-карбонитрила (200 мг, 0,51 ммоль) в EtOH (10 мл) добавляли 10% Pd/C (20 мг). Смесь гидрировали в течение ночи (40 ф./кв. дюйм (275,8 кПа)). После фильтрации и концентрирования неочищенный продукт получали в виде бледно-желтого масла (120 мг, 77%), которое использовали на следующей стадии без дополнительной очистки. МС (ЭГДИ) (M+H)+=305.
23Г: 1-(Циклопропилметил)-2-[(5-этокси-2-пиридинил)метил]-1Н-бензимидазол-5-карбонитрил
К раствору 1-(циклопропилметил)-2-[(5-гидрокси-2-пиридинил)метил]-1Н-бензимидазол-5-карбонитрила (120 мг, 0,394 ммоль) в ДМСО (1 мл) добавляли MeONa (25% в МеОН, 0,47 ммоль). После перемешивания при комнатной температуре в течение 30 мин МеОН удаляли в вакууме и добавляли EtI (0,47 ммоль) и получающуюся в результате смесь перемешивали при комнатной температуре в течение ночи. Раствор разбавляли H2O (10 мл), экстрагировали EtOAc (2×20 мл), промывали H2O (2×10 мл) и в заключение сушили над MgSO4. Остаток, полученный после выпаривания, очищали путем колоночной хроматографии на силикагеле с получением чистого продукта в виде масла (84 мг, 64%). 1Н-ЯМР (400 МГц, CDCl3): δ 8,19 (d, J=3,0 Гц, 1Н), 8,04 (s, 1H), 7,49 (d, J=8,4 Гц, 1H), 7,42 (d, J=8,4 Гц, 1H), 7,24 (d, J=8,8 Гц, 1H), 7,13 (dd, J=10,0 Гц, J= 2,8 Гц, 1H), 4,47 (s, 2H), 4,15 (d, J=6,8 Гц, 2Н), 4,04 (q, J=7,0 Гц, 2Н), 1,41 (t, J=7,0 Гц, 3Н), 1,14-1,08 (m, 1H), 0,60-0,54 (m, 2H), 0,36-0,30 (m, 2H). МС (ЭГДИ) (М+Н)+=333.
23Д: 1-(Циклопропилметил)-2-[(5-этокси-2-пиридинил)метил]-1Н-бензимидазол-5-карбоновая кислота
К раствору 1-(циклопропилметил)-2-[(5-этокси-2-пиридинил)метил]-1H-бензимидазол-5-карбонитрила (400 мг, 1,203 ммоль) в 1:1 EtOH:Н2О (12 мл) добавляли КОН (269 мг, 4,81 ммоль). Получающуюся в результате смесь кипятили с обратным холодильником в течение 36 ч. Смесь подкисляли 1 н. HCl до рН˜6,0. Осадок (397 мг, 94%) использовали на следующей стадии без дополнительной очистки. МС (ЭГДИ) (М+Н)+=352.
23Е: 1-(Циклопропилметил)-2-[(5-этокси-2-пиридинил)метил]-5-(1-пирролидинил-карбонил)-1Н-бензимидазол
В соответствии с общим способом 20Д 1-(циклопропилметил)-2-[(5-этокси-2-пиридинил)метил]-1Н-бензимидазол-5-карбоновую кислоту (80 мг, 0,228 ммоль) растворяли в ДМФ (2 мл) и добавляли HATU (104 мг, 0,274 ммоль) с последующим ДИПЭА (48 мкл, 0,274 ммоль). После перемешивания в течение 10 мин добавляли пирролидин и получающуюся в результате смесь перемешивали в течение ночи. Неочищенный остаток, полученный после обработки, очищали, используя обращенно-фазовую препаративную HPLC, с получением соли с ТФУ в виде белого твердого вещества (42 мг, 37%). 1H-ЯМР (400 МГц, CD3OD): δ 8,18 (d, J=2,8 Гц, 1H), 7,98 (d, J=9,2 Гц, 1H), 7,87 (s, 1H), 7,72 (dd, J=9,2 Гц, J=2,0 Гц, 1H), 7,52 (d, J=9,2 Гц, 1H), 7,46 (dd, J=9,2 Гц, J=2,8 Гц, 1H), 4,89 (s, 2H), 4,43 (d, J=6,8 Гц, 2H), 4,09 (q, J=7,2 Гц, 2H), 3,60 (t, J=7,6 Гц, 2H), 3,44 (t, J=7,6 Гц, 2H), 2,02-1,94 (m, 2H), 1,92-1,86 (m, 2H), 1,37 (t, J=7,2 Гц, 3Н), 1,30-1,22 (m, 1H), 0,64-0,58 (m, 2H), 0,48-0,44 (m, 2H). МС (ЭГДИ) (M+H)+=405. Аналитически рассчитано для С24Н28N4O2.0,8 ТФУ.0,2 Н2О: С, 55,32; Н, 5,12; N, 9,49. Обнаружено: С, 55,30; Н, 4,91; N, 9,23.
Пример 24:
1-(Циклопропилметил)-2-(4-этоксибензил)-5-(4-морфолинилметил)-1Н-бензимидазол
24А: 1-(Цикпопропилметил)-2-(4-этоксибензил)-1Н-бензимидазол-5-карбальдегид
К раствору 1-(циклопропилметил)-2-(4-этоксибензил)-1Н-бензимидазол-5-карбонитрила (0,049 г, 0,149 ммоль) в 50% водной муравьиной кислоте (1 мл) добавляли каталитическое количество никеля Ренея (50% суспензия в воде) и получающуюся в результате смесь нагревали при 90°С в течение 6 часов. Смесь фильтровали через тонкий слой диатомовой земли и промывали EtOAc. Растворитель выпаривали в вакууме и остаток растворяли в EtOAc (5 мл). Органическую фазу промывали 1н. NaOH (2×2 мл) и рассолом (2 мл), сушили над MgSO4, фильтровали и концентрировали в вакууме. Неочищенный продукт очищали путем колоночной хроматографии на силикагеле (40% EtOAc/гексаны) с получением указанного в заголовке соединения (0,041 г, 82%) в виде белого твердого вещества. 1H-ЯМР (ацетон-d6): δ 10,16 (s, 1H), 8,29 (s, 1H), 7,90 (d, J=8,4 Гц, 1H), 7,76 (d, J=8,4 Гц, 1H), 7,34 (d, J=8,4 Гц, 2Н), 6,94 (d, J=8,4 Гц, 2Н), 4,44 (s, 2H), 4,24 (d, J=6,4 Гц, 2Н) 4,07 (q, J=6,8 Гц, 2Н), 1,40 (t, J=7,0 Гц, 3Н), 1,27-1,14 (m, 1H), 0,58-0,49 (m, 2H), 0,48-0,38 (m, 2H). MC (ЭГДИ) (M+H)+=335.
24Б: 1-(Циклопропилметил)-2-(4-этоксибензил)-5-(4-морфолинилметил)-1Н-бензимидазол
К раствору 1-(циклопропилметил)-2-(4-этоксибензил)-1H-бензимидазол-5-карбальдегида (0,077 г, 0,231 ммоль) и уксусной кислоты (0,135 мл, 0,236 ммоль) в тетрагидрофуране (ТГФ) (0,25 мл) добавляли триацетоксиборогидрид натрия (0,245 г, 0,268 ммоль) и получающуюся в результате смесь перемешивали при комнатной температуре в течение б часов. Добавляли воду (2 мл) и 1 н. HCl (5 мл) и смесь перемешивали в течение 10 минут перед промыванием EtOAc (2х5 мл). Водный слой подщелачивали 5 н. NaOH (5 мл) и экстрагировали EtOAc (3×10 мл). Объединенные органические фазы промывали рассолом (5 мл), сушили над MgSO4, фильтровали и концентрировали в вакууме. Неочищенный продукт очищали путем колоночной хроматографии на силикагеле (5% МеОН/EtOAc) с получением указанного в заголовке соединения (0,0445 г, 47%), которое растворяли в 1M HCl в диэтиловом эфире с получением после выпаривания растворителя соли с HCl в виде масла. 1Н-ЯМР (свободное основание, CDCl3): δ 7,68 (s, 1H), 7,28-7,23 (m, 2Н), 7,14 (d, J=8,4 Гц, 2H), 6,81 (d, J=8,4 Гц, 2Н), 4,26 (s, 2H), 3,98 (q, J=7,2 Гц, 2H), 3,88 (d, 6,4Н), 3,72-3,65 (m, 4Н) 3,63 (s, 2H), 2,53-2,44 (m, 4Н), 1,38 (t, J=8,6 Гц, 3Н), 1,07-1,00 (m, 1H), 0,53-0,48 (m, 2H), 0,29-0,22 (m, 2H). МС (ЭГДИ) (М+Н)+=406.
Пример 25:
1-[1-(Циклопропилметил)-2-(4-этоксибензил)-1H-бензимидазол-5-ил]этанон
25А: 1-[1-(Циклопропилметил)-2-(4-этоксибензил)-1Н-бензимидазол-5-ил]этанол
К раствору 1-(циклопропилметил)-2-(4-этоксибензил)-1Н-бензимидазол-5-карбальдегида (2,32 г, 6,94 ммоль) в ТГФ (60 мл) при 0°С добавляли 3М метилмагнийбромид в ТГФ (13,9 мл, 41,7 ммоль) и получающуюся в результате смесь перемешивали при 0°С в течение 90 минут. Добавляли воду (50 мл) и смесь перемешивали в течение 10 минут перед экстракцией EtOAc (3×100 мл). Объединенные органические фазы промывали рассолом (50 мл), сушили над MgSO4, фильтровали и концентрировали в вакууме. К неочищенному продукту добавляли СН2Cl2(5 мл), получающуюся в результате суспензию фильтровали, твердое вещество промывали CH2Cl2 (2 мл) и сушили с получением указанного в заголовке соединения (1,915 г, 79%) в виде белого твердого вещества. 1H-ЯМР (CDCl2): δ 7,74 (s, 1H), 7,31 (s, 2H), 7,12 (d, J=8,4 Гц, 2H), 6,79 (d, J=8,0 Гц, 2H), 5,03 (q, J=6,3 Гц, 2H), 4,27 (s, 2H), 3,98 (q, J=6,8 Гц, 2H), 3,90 (d, J=6,4 Гц, 2H), 2,33 (brs, 1H), 1,56 (d, J=6,4 Гц, 3Н), 1,38 (t, J=7,0 Гц, 3Н), 1,05-0,99 (m, 1H), 0,53-0,48 (m, 2H), 0,26-0,23 (m, 2H). МС (ЭГДИ) (M+H)+=351. Аналитически рассчитано для C22H26N2O2: С, 75,40; Н, 7,48; N, 7,99. Обнаружено: С, 75,22; Н, 7,32; N, 7,99.
25Б: 1-[1-(Циклопропилметил)-2-(4-этоксибензил)-1H-бензимидазол-5-ил]этанон
К смеси 1-[1-(циклопропилметил)-2-(4-этоксибензил)-1Н-бензимидазол-5-ил]этанола (1,35 г, 3,86 ммоль), N-метилморфолин-N-оксида (0,497 г, 4,24 ммоль) и молекулярных сит 4 А (2,0 г) в CH2Cl2 (10 мл) добавляли перрутенат тетрапропиламмония (0,068 г, 0,193 ммоль) и получающуюся в результате смесь перемешивали при комнатной температуре в течение 90 минут. Добавляли N-метилморсролин-N-оксид (0,124 г, 1,06 ммоль) и ацетонитрил (1 мл) и смесь перемешивали при комнатной температуре в течение ночи. Растворитель концентрировали в вакууме и неочищенный продукт очищали путем колоночной хроматографии на силикагеле (30% EtOAc/гексаны до 60% EtOAc/гексаны) с получением указанного в заголовке соединения (0,880 г, 65%) в виде белого твердого вещества. 1Н-ЯМР (CDCl3): δ 8,34 (s, 1H), 7,94 (dd, J=8,4 Гц, J=2,0 Гц, 1H), 7,35 (d, J=8,4 Гц, 1H), 7,15 (d, J=8,4 Гц, 2Н), 6,91 (dd, J=6,4 Гц, J=2,0 Гц, 2H), 4,29 (s, 2H), 3,98 (q, J=7,2 Гц, 2Н) 3,41 (d, J=6,8 Гц, 2H), 2,67 (s, 3Н), 1,38 (t, J=7,0 Гц, 3Н), 1,07-1,00 (m, 1H), 0,55-0,51 (m, 2H), 0,29-0,26 (m, 2H). 13С-ЯМР (CDCl3): δ 198,71, 159,01, 156,43, 143,29, 140,01, 132,68, 130,44, 128,67, 123,50, 121,95, 115,83, 110,73, 64,38, 49,23, 34,63, 27,63, 15,76, 11,99, 5,20. МС (ЭГДИ) (M+H)+=349. Аналитически рассчитано для C22H24N2O2+0,1 Н2O: С, 75,44; Н, 6,96; N, 8,00. Обнаружено: С, 75,48; Н, 7,13; N, 8,01.
Пример 26:
Метил-1-(циклопропилметил)-2-(4-этоксибензил)-1Н-бензимидазол-5-илкарбамат
26А: Метил-4-фтор-3-нитрофенилкарбамат
К перемешиваемому раствору 4-фтор-3-нитроанилина (10,30 г, 65,97 ммоль), ДИПЭА (15,00 мл, 86,11 ммоль) в CH2Cl2 (150 мл) по каплям добавляли метилхлорформиат (6,86 г, 72,59 ммоль) при 0°С. Реакционную смесь оставляли нагреваться до комнатной температуры и затем перемешивали при комнатной температуре в течение ночи. Смесь разбавляли CH2Cl2 (100 мл), промывали 3 н. HCl (50 мл), рассолом (20 мл) и сушили над сульфатом натрия. Удаление растворителя позволило получить неочищенный карбамат в виде светло-коричневого твердого вещества (13,03 г, 92%).
26Б: Метил-4-[(циклопропилметил)амино]-3-нитрофенилкарбамат
В соответствии с общим способом 2Б к метил-4-фтор-3-нитрофенилкарбамату (6,50 г, 30,35 ммоль, с предыдущей стадии) в 80% водном этаноле (100 мл) добавляли циклопропанметиламин (5,00 мл, 57,65 ммоль) при комнатной температуре. Реакционную смесь нагревали при 60°С в течение ночи. После обычной обработки неочищенный продукт, который выпадал в осадок, собирали (8,21 г). МС (ЭГДИ) (M+H)+=266.
26В: Метил-3-амино-4-[(циклопропилметил)амино]фенилкарбамат
В соответствии с общим способом 2В метил-4-[(цикпопропилметил)амино]-3-нитрофенилкарбамат (8,21 г) гидрировали s этилацетате (100 мл), катализируя 10% Pd/C (500 мг) при давлении Н2 15-25 ф./кв. дюйм (103,4 кПа - 172,4 кПа) в течение 1 ч. Реакционную смесь фильтровали через диатомовую землю и удаление растворителя позволило получить желаемый диамин (4,0 г, 56% для двух стадий). Это вещество использовали на следующей стадии без дополнительной очистки. МС (ЭГДИ) (M+H)+=236.
26Г: Метил-1-(циклопропилметил)-2-(4-этоксибензил)-1H-бензимидазол-5-илкарбамат
В соответствии с общим способом 13Б объединяли метил-3-амино-4-[(циклопропилметил)амино]-фенилкарбамат (4,0 г, 17,0 ммоль), ДИПЭА (5 мл), 4-этоксифенилуксусную кислоту (3,06 г, 17,0 ммоль) и HATU (7,10 г, 18,7 ммоль). Желаемый продукт (3,34 г, 52%) осаждался из реакционной смеси. 1H-ЯМР (CDCl3): δ 7,65 (d, J=2,0 Гц, 1Н), 7,37 (br, 1H), 7,26 (d, J=4 Гц), 7,12 (d, J=9,0 Гц, 2Н), 6,81 (d, J=9,0 Гц, 2Н), 6,74 (br., 1H), 4,25 (s, 2Н), 3,98 (q, J=7,4 Гц, 2Н), 3,87 (d, J=7,2 Гц, 2Н), 3,79 (s, 3H), 1,38 (t, J=7,4 Гц, 3Н), 1,05-0,97 (m, 1H), 0,52-0,47 (m, 2H), 0,25-0,20 (m, 2H). МС (ЭГДИ) (М+Н)+=380.
Пример 27:
N-[1-(Циклопропилметил)-2-(4-этоксибензил)-1Н-бензимидазол-5-ил]-N,5-диметил-3-изоксазолкарбоксамид
27А: 1-(Циклопропилметил)-2-(4-этоксибензил)-N-метил-1H-бензимидазол-5-амин
Метил-1-(циклопропилметил)-2-(4-этоксибензил)-1H-бензимидазол-5-илкарбамат (1,65 г, 4,35 ммоль) порциями добавляли к охлажденному (0°С) раствору AlH3 (˜0,3 М), который готовят свежим путем добавления по каплям конц. H2SO4 (0,80 г, 8,10 ммоль) к 1М раствору UAIH4 в ТГФ (16 мл, 16 ммоль) в ТГФ (30 мл) при 0°С. Реакционную смесь перемешивали при комнатной температуре в течение ночи и затем осторожно гасили путем добавления EtOAc (5 мл), Н2О (3 мл) и Et2O (100 мл) и затем Na2SO4·5 H2O (10 г). Реакционную смесь перемешивали до получения прозрачного раствора и твердое вещество отфильтровывали. Фильтрат сушили над Na2SO4 и концентрировали в вакууме с получением желаемого соединения (1,21 г). Неочищенный продукт использовали на следующей стадии без дополнительной очистки.
27Б: N-[1-(циклопропилметил)-2-(4-этоксибензил)-1Н-бензимидазол-5-ил]-N,5-диметил-3-изоксазолкарбоксамид
К раствору 1-(циклопропилметил)-2-(4-этоксибензил)-N-метил-1H-бензимидазол-5-амина (100 мг, 0,30 ммоль) и Et2N (200 мг, 0,20 ммоль) в СН2Cl2 (5 мл) при комнатной температуре добавляли 5-метил-3-изоксазолкарбонилхлорид (0,1 мл). Реакционную смесь перемешивали при комнатной температуре в течение ночи и гасили насыщенным NaHCO3 (1 мл). Смесь разбавляли Et2O (30 мл), промывали насыщенным NaHCO3 и затем рассолом и, в заключение, сушили над Na2SO4. После концентрирования получающийся в результате остаток очищали путем препаративной HPLC с получением желаемого вещества в виде соли с ТФУ (40 мг, 24%). Соль с ТФУ растворяли в Н2О (10 мл) и нейтрализовали насыщенным NaHCO3, экстрагировали Et2O (2×20 мл). Объединенный эфирный раствор промывали рассолом, сушили над Na2SO4 и концентрировали. Свободное основание превращали в соль с HCl (20 мг). 1H-ЯМР (CD3OD): δ 7,86 (br, 1H), 7,62 (br, 1H), 7,45 (br, 1H), 7,24 (d, J=8,4 Гц, 2Н), 6,93 (d, J=8,4 Гц, 2Н), 4,53 (s, 2H), 4,34 (d, J=6,8 Гц, 2H), 4,00 (q, J=7,0 Гц, 2H), 3,50 (s, 3Н), 2,26 (s, 3Н), 1,34 (t, J=7,0 Гц, 3Н), 1,26-1,16 (m, 1H), 0,62-0,55 (m, 2H), 0,46-0,40 (m, 2H). МС (ЭГДИ) (М+Н)+=445. Аналитически рассчитано для С26Н28N403.1,15 HCl: С, 64,19; Н, 5,92; N, 11,51. Обнаружено: С, 64,59; Н, 5,74; N, 10,97.
Пример 28:
Изопропил-1-(циклопропилметил)-2-(4-этоксибензил)-1Н-бензимидазол-5-илметил)карбамат
К раствору 1-(циклопропилметил)-2-(4-этоксибензил)-N-метил-1H-бензимидазол-5-амина (67 мг, 0,20 ммоль) и Et2N (0,1 мл) в CH2Cl2 (3 мл) при комнатной температуре добавляли изопропилхлорформиат (1М в толуоле, 0,24 мл, 0,24 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение ночи и гасили насыщенным NaHCO3 (1 мл). Смесь разбавляли Et2O (30 мл), промывали насыщенным NaHCO3 и рассолом, сушили над Na2SO4 и затем концентрировали. Получающийся в результате остаток очищали путем препаративной HPLC с получением соли с ТФУ (10 мг, 9%). 1H-ЯМР (CD3OD): δ 7,87 (d, J=8,4 Гц, 1Н), 7,62 (d, J=1,6 Гц, 1Н), 7,50 (dd, J=1,6, 8,4 Гц, 1Н), 7,24 (d, J=8,4 Гц, 2Н), 6,93 (d, J=8,4 Гц, 2Н), 4,95-4,85 (m, 1H), 4,54 (s, 2H), 4,35 (d, J=7,2 Гц, 2Н), 4,01 (q, J=6,8 Гц, 2Н), 3,31 (s, 3Н), 1,343 (t, J=6,8 Гц, 3Н), 1,27-1,19 (m, 1H), 1,19 (d, J=5,6 Гц, 6Н), 0,63-0,56 (m, 2H), 0,47-0,42 (m, 2H). МС (ЭГДИ) (M+H)+=422.. Аналитически рассчитано для С25Н31N3О3.1,35 ТФУ.0,05 Н2O: С, 57,72; Н, 5,67; N, 7,29. Обнаружено: С, 57,68; Н, 5,33; N, 7,23.
Пример 29:
(25)-N-[1-(Циклопропилметил)-2-(4-этоксибензил)-1H-бензимидазол-5-ил]-N,1-диметил-2-пирролидинкарбоксамид
К перемешиваемому раствору 1-(циклопропилметил)-2-(4-этоксибензил)-N-метил-1H-бензимидазол-5-амина (80 мг, 0,24 ммоль) в ДМФ (3 мл) добавляли ДИПЭА (0,2 мл) и (2S)-1-метил-2-пирролидинкарбоновую кислоту (80 мг, 0,62 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение 5 мин и затем добавляли HATU (280 мг, 0,73 ммоль). Реакционную смесь затем перемешивали при комнатной температуре в течение ночи. Добавляли насыщенный NaHCO3 (1 мл) и H2O (20 мл) и смесь экстрагировали EtOAc (2×20 мл). Объединенные экстракты промывали насыщенным NaHCO3 (5 мл) и рассолом, сушили над Na2SO4 и концентрировали в вакууме с получением неочищенного амида, который очищали путем препаративной HPLC с получением его соли с ТФУ (18 мг, 13%). МС (ЭГДИ) (M+H)+=447.
Пример 30:
N'-трет-Бутил-N-[1-(циклопропилметил)-2-(4-этоксибензил)-1Н-бензимидазол-5-ил]-N-метилмочевина
трет-Бутилизоцианат (0,1 мл) добавляли к раствору 1-(циклопропилметил)-2-(4-этоксибензил)-N-метил-1Н-бензимидазол-5-амина (34 мг, 0,10 ммоль) в этиленхлориде (5 мл) и реакционную смесь затем нагревали при 60°С в течение ночи. Неочищенный продукт очищали путем флэш-хроматографии на силикагеле (EtOAc) с получением желаемой мочевины (26 мг, 59%). 1H-ЯМР (CDCl3): свободное основание δ 7,61 (d, J=2,0 Гц, 1H), 7,33 (d, J=8,4 Гц, 1H), 7,16 (d, J=8,4 Гц, 2H), 7,10 (dd, J=2,0, 8,4 Гц, 1H), 6,83 (d, J=8,4 Гц, 2H), 4,28 (s, 1H), 4,27 (s, 2H), 4,00 (q, J=7,0 Гц, 2H), 3,93 (d, J=6,8 Гц, 2H), 3,26 (s, 3Н), 1,40 (t, J=7,0 Гц, 3Н), 1,28-1,20 (m, 1H), 1,25 (s, 9H), 0,58-0,53 (m, 2H), 0,31-0,26 (m, 2Н). МС (ЭГДИ) (М+H)+=435. Соль с HCl получали, используя HCl в диэтиловом эфире. Аналитически рассчитано для С26Н34N4O2.1,20 HCl. 1,10 С2Н6О: С, 64,03; Н, 7,96; N, 10,59. Обнаружено: С, 64,10; Н, 7,53; N, 10,30.
Пример 31:
N-[1-(Циклопропилметил)-2-(4-этоксибензил)-1Н-бензимидазол-5-ил]-N'-(2-метоксифенил)-N-метилтиомочевина
1-Изотиоцианато-2-метоксибензол (100 мг, 0,61 ммоль) добавляли к раствору 1-(циклопропилметил)-2-(4-этоксибензил)-N-метил-1H-бензимидазол-5-амина (100 ммоль, 0,30 ммоль) в ДМФ (5 мл). Реакционную смесь нагревали при 50°С в течение ночи. Неочищенный продукт, выделенный после удаления растворителя, очищали путем хроматографии на силикагеле (EtOAc/CH2Cl2 1:1) с получением желаемого продукта (85 мг, 57%) 1H-ЯМР (CDCl3): свободное основание δ 8,26 (dd, J=2,0, 8,4 Гц, 1Н), 7,74 (d, J=2,0 Гц, 1Н), 7,50 (s, 1H), 7,43 (d, J=8,4 Гц, 1H), 7,20 (dd, J=1,6, 8,4 Гц, 1H), 7,15 (d, J=8,4 Гц, 2H), 7,08-7,02 (m, 1H), 6,98-6,92 (m, 1H), 6,83 (d, J=8,4 Гц, 2H), 6,75 (d, J=8,4 Гц, 1H), 4,29 (s, 2H), 4,00 (q, J=7,0 Гц, 2H), 3,96 (d, J=6,8 Гц, 2H), 3,80 (s, 3H), 3,58 (s, 3H), 1,37 (t, J=7,0 Гц, 3H), 1,10-1,00 (m, 1H), 0,59-0,52 (m, 2H), 0,32-0,25 (m, 2H). МС (ЭГДИ) (М+Н)+=501.
Пример 32:
N-Аллил-N-[1-(циклопропилметил)-2-(4-этоксибензил)-1Н-бензимидазол-5-ил]ацетамид
32А: N-(4-Фтор-3-нитрофенил)ацетамид
4-Фтор-З-нитроанилин (45,0 г) порциями добавляли к уксусному ангидриду (150 мл) при комнатной температуре. Реакционную смесь перемешивали при комнатной температуре в течение 2 ч. Желаемый продукт осаждался и его собирали и сушили в вакууме (42,0 г, 70%).
32Б: N-{4-[(Циклопропилметил)амино]-3-нитрофенил}ацетамид
В соответствии с общим способом 2Б к раствору N-(4-фтор-3-нитрофенил)ацетамида (15,4 г, 77,7 ммоль) в 80% водном этаноле (150 мл) добавляли циклопропанметиламин (10 мл) при комнатной температуре. Реакционную смесь нагревали до температуры дефлегмации в течение ночи. После охлаждения до комнатной температуры добавляли Н2О (300 мл) и желаемый продукт осаждался в виде оранжевого твердого вещества (18 г, 92%).
32В: N-{3-Амино-4-[(циклопропилметил)амино]фенил}ацетамид
В соответствии с общим способом 2В N-{4-[(циклопропилметил)амино]-3-нитрофенил}ацетамид (7,0 г, 28 ммоль) гидрировали в этилацетате (300 мл) с 10% Pd/C (0,8 г) при давлении H2 25 ф./кв. дюйм (172,4 кПа) в шейкере Парра. После обработки выделяли желаемый продукт (5,5 г, 89%).
32Г: N-[1-(Циклопропилметил)-2-(4-этоксибензил)-1Н-бензимидазол-5-ил]ацетамид
В соответствии со способом 13Б N-{3-амино-4-[(циклопропилметил)амино]фенил}ацетамид (5,50 г, 25,11 ммоль), ДИПЭА (6,50 мл, 37,5 ммоль), 4-этоксифенилуксусную кислоту (5,40 г, 30,0 ммоль) и HATU (11,40 г, 30,0 ммоль) в ДМФ (100 мл) перемешивали при комнатной температуре в течение ночи. Выделяли желаемый продукт (4,5 г, 86%).
1H-ЯМР (CD3OD): δ 7,89 (s, 1H), 7,44-7,36 (m, 2Н), 7,13 (d, J=8,4 Гц, 2Н), 6,83 (d, J=8,4 Гц, 2Н), 4,25 (s, 2Н), 4,03-3,94 (m, 4H), 2,14 (s, 3Н), 1,34 (t, J=7,0 Гц, 3Н), 1,06-0,96 (m, 1H), 0,48-0,41 (m, 2H), 0,29-0,22 (m, 2H). МС (ЭГДИ) (M+H)+=364. Аналитически рассчитано для C22H25N3O2.1,5 H2O: С, 67,67; Н, 7,23; N, 10,76. Обнаружено: С, 67,50; Н, 7,12; N, 10,65.
32Д: N-Аллил-N-[1-(циклопропилметил)-2-(4-этоксибензил)-1H-бензимидазол-5-ил]ацетамид
Аллилбромид (0,3 мл) добавляли к хорошо перемешиваемому двухфазному раствору 50% КОН (5 мл), N-[1-(циклопропилметил)-2-(4-этоксибензил)-1Н-бензимидазол-5-ил]ацетамида (545 мг, 1,50 ммоль) и н-Bu4NBr (64 мг, 0,2 ммоль) в СН2Cl2 (15 мл) при комнатной температуре в атмосфере азота. Реакционную смесь перемешивали при комнатной температуре в течение 2 ч и затем разбавляли H2O (30 мл), экстрагировали СН2Cl2 (2×50 мл). Экстракты объединяли и промывали насыщенным NaHCO3, рассолом, сушили над Na2SO4. Удаление растворителя позволило получить желтоватый остаток (530 мг), который очищали путем флэш-хроматографии на силикагеле (MeOH/CH2Cl2, 1:10) с получением продукта (510 мг, 84%).
1H-ЯМР (CDCl3): δ 7,54 (d, J=2,0 Гц, 1H), 7,31 (d, J=8,4 Гц, 1H), 7,16 (d, J=8,4 Гц, 2H), 7,02 (dd, J=2,0, 8,4 Гц, 1H), 6,84 (d, J=8,4 Гц, 2H), 5,98-5,84 (m, 1Н), 5,12-5,04 (m, 2H), 4,34 (d, J=5,6 Гц, 2Н), 4,27 (s, 2Н), 4,01 (q, J=7,6 Гц, 2Н), 3,93 (d, J=6,4 Гц, 2H), 1,88 (s, 3Н), 1,39 (t, J=7,6 Гц, 3Н), 1,10-1,00 (m, 1H), 0,59-0,52 (m, 2H), 0,31-0,26 (m, 2H). МС (ЭГДИ) (M+H)+=404. Соль с HCl получали, используя HCl в диэтиловом эфире. Аналитически рассчитано для С25Н29N3O2.HCl.1,5 Н2O: С, 64,24; Н, 7,07; N, 8,99. Обнаружено: С, 63,96; Н, 6,80; N, 8,84.
Пример 33:
N-Аллил-1-(циклопропилметил)-2-(4-этоксибензил)-1Н-бензимидазол-5-амин
К раствору N-аллил-N-[1-(циклопропилметил)-2-(4-этоксибензил)-1Н-бензимидазол-5-ил]ацетамида (510 мг) в EtOH (20 мл) добавляли 20% КОН (20 мл). Реакционную смесь нагревали до температуры дефлегмации в течение ночи и затем оставляли охлаждаться до комнатной температуры. Смесь концентрировали в вакууме приблизительно до 20 мл и экстрагировали CH2Cl2 (2×100 мл). Объединенные экстракты промывали рассолом, сушили над Na2SO4 и концентрировали в вакууме. Остаток очищали путем хроматографии на силикагеле (CH2Cl2:MeOH 25:1) с получением светло-желтого масла (400 мг, 87%).
1H-ЯМР (CD3OD): δ 8,03 (br, 1H), 7,64 (br, 1H), 7,54 (br, 1H), 7,28 (d, J=8,4 Гц, 2H), 6,96 (d, J=8,4 Гц, 2H), 6,03-5,92 (m, 1H), 5,43 (d, J=24,8 Гц, 1H), 5,40 (d, J=18,8 Гц, 1H), 4,59 (s, 2H), 4,41 (d, J=7,6 Гц, 2H), 4,04 (d, J=6,4 Гц, 2H), 4,03 (q, J=6,4 Гц, 2H), 1,36 (t, J=6,4 Гц, 3Н), 1,30-1,18 (m, 1H), 0,66-0,58 (m, 2H), 0,50-0,42 (m, 2H). МС (ЭГДИ) (M+H)+=362. Соль с HCl получали, используя HCl в диэтиловом эфире. Аналитически рассчитано для C23H27N3O.2 HCl.1,25 Н2O: С, 60,46; Н, 6,89; N, 9,19. Обнаружено: С, 60,53; Н, 6,64; N, 8,89.
Пример 34:
1-[1-(Циклопропилметил)-2-(4-этоксибензил)-1Н-бензимидазол-5-ил]-2-пирролидинон
34А: 1-(Циклопропилметил)-2-(4-этоксибензил)-1Н-бензимидазол-5-амин
Раствор N-[1-(циклопропилметил)-2-(4-этоксибензил)-1H-бензимидазол-5-илацетамида (727 мг, 2 ммоль) в 1:1 20% КОН:EtOH (20 мл) перемешивали при температуре дефлегмации в течение ночи. После охлаждения смесь подкисляли 1 н. HCl до рН˜6,0 и осадок собирали путем фильтрации с получением желаемого продукта в виде светло-желтого твердого вещества, 611 мг (95%). МС (ЭГДИ) (M+H)+=322.
34Б: 1-[1-(Циклопропилметил)-2-(4-этоксибензил)-1H-безимидазол-5-ил]-2-пирролидинон
К 1-(циклопропилметил)-2-(4-этоксибензил)-1H-бензимидазол-5-амину (72 мг, 0,22 ммоль) и ДИПЭА (150 мкл) в CH2Cl2 (3 мл) добавляли 4-бромбутирилхлорид (40 мкл, 0,35 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение 3 ч, затем в реакционную смесь добавляли 50% КОН (3 мл) и н-Bu4NBr (5 мг). Реакционную смесь интенсивно перемешивали в течение 4 ч, разбавляли H2O (10 мл) и экстрагировали СН2Cl2 (2×20 мл). Объединенные экстракты промывали Н2O, затем рассолом, сушили над Na2SO4 и концентрировали в вакууме с получением неочищенного лактама, который очищали путем препаративной тонкослойной хроматографии (ТСХ) и затем путем препаративной HPLC с получением желаемых продуктов в виде соли с ТФУ (11 мг, 10%). 1H-ЯМР (CD3OD): δ 8,14 (d, J=2,0 Гц, 1Н), 7,91 (d, J=9,2 Гц, 1Н), 7,80 (dd, J=2,0, 9,2 Гц, 1Н), 7,26 (d, J=9,2 Гц, 2Н), 6,95 (d, J=9,2 Гц, 2Н), 4,56 (s, 2H), 4,37 (d, J=6,4 Гц, 2Н), 4,03 (t, J=7,6 Гц, 2Н), 4,01 (q, J=6,8 Гц, 2H), 2,63 (t, J=8,4 Гц, 2H), 2,20 (tt, J=7,6, 8,4 Гц, 2H), 1,36 (t, J=6,8 Гц, 3Н), 1,28-1,18 (m, 1H), 0,64-0,57 (m, 2H), 0,49-0,42 (m, 2H). МС (ЭГДИ) (М+H)+=390. Аналитически рассчитано для С24Н27N3O2.ТФУ.0,1 Н2O: С, 61,80; Н, 5,63; N, 8,32. Обнаружено: С, 61,75; Н, 5,30; N, 7,99.
Пример 35:
3-(Циклопропилметил)-2-(4-этоксибензил)-N,N-диэтил-3Н-имидазо[4,5-b]пиридин-6-карбоксамид
35А: 6-[(Циклопропилметил)амино]-5-нитроникотиновая кислота
Циклопропанметиламин (1,70 г, 23,90 ммоль) добавляли к раствору 6-хлор-5-нитроникотиновой кислоты (1,12 г, 5,53 ммоль) в МеОН (25 мл) при комнатной температуре. Реакционную смесь перемешивали при комнатной температуре в течение 2 ч и концентрировали в вакууме. Остаток растворяли в Н2O (20 мл) и раствор подкисляли путем добавления 3 н. HCl до рН˜3. Смесь экстрагировали EtOAc и СН2Cl2 (2×100 мл) и объединенные экстракты промывали рассолом, сушили над Na2SO4, концентрировали в вакууме с получением ярко-желтого твердого вещества (1,30 г), которое использовали в следующей реакции без дополнительной очистки.
35Б: Метил-6-[(циклопропилметил)амино]-5-нитроникотинат
6-[(Циклопропилметил)амино]-5-нитроникотиновую кислоту (1,30 г) растворяли в МеОН/толуол (1:1, 60 мл) и по каплям добавляли тетраметилсилан-CHN3 (TMSCHN2) (2М в гексане, 6 мл). Реакционную смесь перемешивали при комнатной температуре в течение ночи и концентрировали в вакууме с получением неочищенного метилового эфира (1,5 г), который использовали в течение следующей стадии без очистки.
35В: Метил-5-амино-6-[(циклопропилметил)амино]никотинат
В соответствии с общим способом 2В метил-6-[(циклопропилметил)амино]-5-нитроникотинат (1,5 г) гидрировали в этилацетате (50 мл) 10% Pd/C (100 мг) при давлении Н2 20 ф./кв. дюйм (137,9 кПа) в шейкере Парра в течение 2 ч. Реакционную смесь фильтровали через диатомовую землю и растворитель удаляли с получением диамина (1,12 г), который использовали без дополнительной очистки на следующей стадии.
35Г: Метил-3-(циклопропилметил)-2-(4-этоксибензил)-3H-имидазо[4,5-b]пиридин-6-карбоксилат
В соответствии с общим способом 13Б объединяли метил-5-амино-6-[(циклопропилметил)амино]никотинат (950 мг, ˜4,3 ммоль), ДИПЭА (1,1 мл), 4-этоксифенилуксусную кислоту (850 мг, 4,72 ммоль) и HATU (1,80, 4,74 ммоль) в ДМФ (15 мл). Неочищенный продукт очищали путем флэш-хроматографии на силикагеле (EtOAc) с получением желаемого продукта (530 мг, 34%).
1H-ЯМР (CDCl3): δ 9,00 (d, J=2,0 Гц, 1Н), 8,61 (d, J=2,0 Гц, 1Н), 7,17 (d, J=8,0 Гц, 2Н), 6,84 (d, J=8,0 Гц, 2Н), 4,33 (s, 2H), 4,06 (d, J=7,2 Гц, 2Н), 4,00 (q, J=7,0 Гц, 2H), 3,97 (s, 3Н), 1,39 (t, J=7,0 Гц, 3Н), 1,16-1,06 (m, 1H), 0,52-0,45 (m, 2H), 0,44-0,38 (m, 2H).
35Д: 3-(Циклопропилметил)-2-(4-этоксибензил)-3Н-имидазо[4,5-b]пиридин-6-карбоновая кислота
К раствору метил-3-(циклопропилметил)-2-(4-этоксибензил)-3Н-имидазо[4,5-b]пиридин-6-карбоксилата (260 мг, 0,71 ммоль) в ТГФ/МеОН (1:1, 6 мл) добавляли 1 н. NaOH (3 мл, 3 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение 3 ч и концентрировали в вакууме.
Остаток растворяли в Н2О (10 мл) и подкисляли путем добавления 1 к раствора HCl до рН˜5. Твердое вещество собирали (230 мг) и фильтрат экстрагировали EtOAc (2×50 мл). Экстракты сушили над Na2SO4 и концентрировали в вакууме с получением дополнительного выхода продукта (20 мг). Желаемую кислоту получали с количественным выходом. 1H-ЯМР (CD3OD): δ 8,93 (d, J=1,6 Гц, 1Н), 8,50 (d, J=1,6 Гц, 1Н), 7,15 (d, J=9,2 Гц, 2Н), 6,82 (d, J=9,2 Гц, 2Н), 4,33 (s, 2H), 4,11 (d, J=6,4 Гц, 2Н), 3,95 (q, J=7,2 Гц, 2Н), 1,31 (t, J=7,2 Гц, 3Н), 1,16-1,03 (m, 1H), 0,43-0,32 (m, 4H).
35E: 3-(Циклопропилметил)-2-(4-этоксибензил)-N,N-диэтил-3Н-имидазо[4,5-b]пиридин-6-карбоксамид
К перемешиваемому раствору 3-(циклопропилметил)-2-(4-этоксибензил)-3Н-имидазо[4,5-b]пиридин-6-карбоновой кислоты (150 мг, 0,43 ммоль) в ДМФ (5 мл) добавляли ДИПЭА (0,12 мл, 0,69 ммоль) и диэтиламин (60 мкл, 0,58 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение 5 мин и затем добавляли HATU (200 мг, 0,53 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение ночи и затем выливали в ледяную воду (20 мл) при перемешивании. Смесь экстрагировали EtOAc (100 мл) и экстракт промывали Н2О (20 мл), сушили с использованием Na2SO4 и концентрировали в вакууме. Остаток очищали путем флэш-хроматографии на силикагеле (EtOAc/МеОН, 20:1) с получением желаемого амида (115 мг, 64%), который превращали в соответствующую соль с HCl, используя HCl в диэтиловом эфире с метанолом в качестве сорастворителя. 1H-ЯМР (CD3OD): δ 8,46 (d, J=2,0 Гц, 1H), 8,04 (d, J=2,0 Гц, 1H), 7,21 (d, J=8,4 Гц, 2H), 6,88 (d, J=8,4 Гц, 2H), 4,46 (s, 2H), 4,24 (d, J=7,2 Гц, 2H), 3,98 (q, J=7,0 Гц, 2H), 3,57 (br, 2H), 3,32 (br, 2H), 1,33 (t, J=7,0 Гц, 3Н), 1,32-1,10 (m, 7H), 0,56-0,44 (m, 4H). MC (ЭГДИ) (М+Н)+=423. Аналитически рассчитано для С24Н30N4O2 HCl·0,20 СН3ОН: С, 64,68; Н, 7,07; N, 12,48. Обнаружено: С, 65,07; Н,7,13; N, 12,13.
Пример 36:
N-Циклопентил-1-(циклопропилметил)-2-(4-этоксибензил)-1Н-бензимидазол-5-сульфонамид
36А: 1-(Циклопропилметил)-2-(4-этоксибензил)-1H-бензимидазол-5-сульфонилхлорид
К смеси конц. HCl (0,6 мл) и НОАс (0,18 мл) добавляли 1-(циклопропилметил)-2-(4-этоксибензил)-1H-бензимидазол-5-амин (193 мг, 0,6 ммоль). Получающуюся в результате смесь перемешивали и охлаждали до температуры менее -10°С и затем медленно добавляли раствор NaNO3 (44,8 мг, 0,65 ммоль) в Н2О (0,065 мл) и смесь перемешивали при температуре в диапазоне от -10°С до -5°С в течение 45 мин. В отдельной колбе в течение 30 мин барботировали SO2 в уксусную кислоту (0,6 мл). Затем добавляли CuCl (15 мг) и снова барботировали SO2 до тех пор, пока желто-зеленая суспензия не стала сине-зеленой. Смесь, содержащую медь, затем охлаждали до температуры менее 10°С и при перемешивании по каплям добавляли диазотирующую смесь. После добавления всей соли диазония смесь выливали в ледяную воду (1:1, 4 мл) и затем экстрагировали дихлорметаном (3×15 мл), промывали Н2О (2×10 мл), сушили над MgSO4 и концентрировали с получением желтого масла (190 мг, 78%), которое использовали в следующей реакции вскоре после образования и без дополнительной очистки. МС (ЭГДИ) (М+H)+=405.
36Б: N-Циклопентил-1-(циклопропилметил)-2-(4-этоксибензил)-1Н-бензимидазол-5-сульфонамид
К неочищенному раствору 1-(циклопропилметил)-2-(4-этоксибензил)-1H-бензимидазол-5-сульфонилхлорида (из 0,2 ммоль анилинового предшественника) в дихлорметане (4 мл) добавляли циклопентиламин (0,4 ммоль), а затем добавляли пиридин (0,5 мл). Получающуюся в результате смесь перемешивали при комнатной температуре в течение ночи. Добавляли воду (10 мл) и смесь экстрагировали CH2Cl2 (2×10 мл), сушили над MgSO4 и концентрировали. Остаток очищали путем колоночной хроматографии на силикагеле с получением чистого продукта, который обрабатывали 1М раствором HCl в диэтиловом эфире с образованием соли с HCl (62 мг, 81%). 1H-ЯМР (400 МГц, CD3OD): δ 8,29 (s, 1H), 7,78 (d, J=8,0 Гц, 1Н), 7,42 (d, J=8,0 Гц, 1Н), 7,16 (d, J=8,4 Гц, 2Н), 6,84 (d, J=8,4 Гц, 2Н), 4,32 (s, 2H), 4,02-3,94 (m, 4H), 3,62-3,58 (m, 1Н), 1,80-1,74 (m, 2H), 1,64-1,56 (m, 2H), 1,50-1,42 (m, 2H), 1,39 (t, J=8,2 Гц, 3Н), 1,42-1,34 (m, 2H), 1,08-1,00 (m, 1H), 0,58-0,54 (m, 2H), 0,30-0,25 (m, 2H). MC (ЭГДИ) (М+Н)+=454. Аналитически рассчитано для С25Н31N3O2 5.0,3HCl: С, 64,64; Н, 6,79; N, 9,05. Обнаружено: С, 64,45; Н, 6,97; N, 8,66.
Пример 37:
1-(Циклопропилметил)-2-(4-этоксибензил)-5-[(4-метил-1-пиперазинил)сульфонил]-1H-бензимидазол
В соответствии с общим способом 36Б использовали 1-(циклопропилметил)-2-(4-этоксибензил)-1Н-бензимидазол-5-сульфонилхлорид (из 0,2 ммоль анилинового предшественника), 1-метилпиперазин (0,4 ммоль), пиридин (0,5 мл) в CH2Cl2 (4 мл). Получающуюся в результате смесь перемешивали при комнатной температуры в течение ночи. Выделяли желаемый продукт в виде соли с HCl после колоночной хроматографии и обработки 1М раствором HCl в диэтиловом эфире (59 мг, 75%). 1H-ЯМР (400 МГц, CD3OD): δ 8,02 (s, 1H), 7,71 (d, J=8,6 Гц, 1H), 7,64 (d, J=8,6 Гц, 1H), 7,13 (d, J=8,8 Гц, 2H), 6,83 (d, J=8,8 Гц, 2H), 4,80 (s, 3Н), 4,31 (s, 2H), 4,10 (d, J=7,2 Гц, 2H), 3,95 (q, J=8,0 Гц, 2H), 3,04-2,96 (m, 4H), 2,50-2,44 (m, 4H), 1,32 (t, J=8,0 Гц, 3Н), 1,08-0,98 (m, 1H), 0,47-0,43 (m, 2H), 0,31-0,26 (m, 2H). MC (ЭГДИ) (М+H)+=469. Аналитически рассчитано для C25H32N4O3S.2,7 HCl. 0,4 Н2O: С, 52,29; Н, 6,23; N, 9,76. Обнаружено: С, 52,54; Н, 6,21; N, 8,67.
Пример 38:
1-(Циклопропилметил)-2-(4-этоксибензил)-N-этил-1Н-бензимидазол-5-сульфонамид
В соответствии с общим способом 36Б использовали 1-(циклопропилметил)-2-(4-этоксибензил)-1Н-бензимидазол-5-сульфонилхлорид (из 0,2 ммоль анилинового предшественника), этиламин (2М раствор в ТГФ, 0,4 ммоль) и пиридин (0,5 мл) в CH2Cl2 (4 мл). Получающуюся в результате смесь перемешивали при комнатной температуре в течение ночи. Выделяли желаемый продукт в виде соли с HCl после колоночной хроматографии и обработки 1М раствором HCl в диэтиловом эфире (61 мг, 87%). 1Н-ЯМР (400 МГц, CD3OD): δ 8,28 (s, 1H), 7,78 (d, J=8,0 Гц, 1H), 7,42 (d, J=8,0 Гц, 1H), 7,15 (d, J=8,8 Гц, 2Н), 6,83 (d, J=8,8 Гц, 2Н), 4,32 (s, 2H), 4,02-3,93 (m, 4H), 3,06-2,98 (m, 2Н), 1,38 (t, J=7,6 Гц, 3Н), 1,10 (t, J=8,0 Гц, 3Н), 1,07-1,00 (m, 1H), 0,55-0,52 (m, 2H), 0,30-0,24 (m, 2H). МС (ЭГДИ) (M+H)+=414. Аналитически рассчитано для С22Н27N3О3.1,1HCl: С, 58,25; Н, 6,24; N, 9,26. Обнаружено: С, 58,19; Н, 6,73; N, 8,62.
Пример 39:
2-(4-Этоксианилино)-N,N-диэтил-1-изопентил-1Н-бензимидазол-5-карбоксамид
Раствор метил-4-этоксифенилдитиокарбамата (0,0274 г, 0,121 ммоль) в ДМФ (0,2 мл) добавляли к смеси 3-амино-N,N-диэтил-4-(изопентиламино)бензамида (0,0334 г, 0,120 ммоль) и красному оксиду ртути (II) (0,0261 г, 0,121 ммоль) в ДМФ (0,2 мл). Получающуюся в результате суспензию интенсивно перемешивали в течение 5,5 ч и добавляли дополнительную порцию красного оксида ртути (II) (0,0130 г, 0,0600 ммоль). После перемешивания в течение дополнительных 16 ч реакционную смесь разбавляли смесью 9:1 CH2Cl2:MeOH, наносили на небольшой слой силикагеля и элюировали той же системой растворителя. Элюант концентрировали и остаток очищали путем обращенно-фазовой HPLC (градиент 20-70% CH3CN в Н2О) с получением указанного в заголовке соединения (0,0287 г, 45%) в виде соли с ТФУ. Это вещество лиофилизировали из смеси Н2O/диоксан с получением белого твердого вещества. 1H-ЯМР (CD3OD): δ 7,60 (d, J=8,4 Гц, 1H), 7,44-7,38 (2 перекрывающихся d, 3Н), 7,35 (s, 1H), 7,11 (d, J=9,2 Гц, 2H), 4,32 (brt, J=7,2 Гц, 2H), 4,12 (q, J=7,6 Гц, 2H), 3,56 (brs, 2H), 3,32 (brs, 2H), 1,85-1,76 (brm, 3Н), 1,43 (t, J=7,6 Гц, 3Н), 1,25 (brs, 3Н), 1,14 (brs, 3Н), 1,08 (d, J=6,4 Гц, 6Н). 13С-ЯМР (CD3OD): δ 172,51, 160,57, 151,15, 134,20, 132,87, 130,29, 128,70, 128,44, 123,46, 117,18, 111,54, 111,23, 65,02, 45,07, 43,16, 41,13, 37,49, 27,26, 22,76, 15,07, 14,35, 13,07. Аналитически рассчитано для МС (ЭГДИ) (M+H)+=423.
Биологические данные для соединений из примеров 40-62
Для соединений из примеров 61 и 62 Kи измеряли исходя из 2 соединений на планшет.
Пример 40
2-[(4-Этоксифенил)метил]-N,N-диэтил-1-(3-тиенилметил)-1Н-бензимидазол-5-карбоксамид
40А: N,N-Диэтил-3-фтор-4-нитро-бензамид
3-Фтор-4-нитробензойную кислоту (5,0 г, 27,0 ммоль) кипятили с обратным холодильником в смеси 2:1 CH2Cl2/SOCl2 (150 мл) в течение ночи. Растворитель концентрировали и остаток растворяли в CH2Cl2 (50 мл). Затем по каплям в охлажденный перемешиваемый раствор (0°С) хлорангидрида добавляли дополнительный раствор CH2Cl2 (50 мл) диэтиламина (3,35 мл, 1,2 экв.) и триэтиламина (7,5 мл, 2 экв.). Раствор перемешивали при комнатной температуре (к.т.) в течение 1 ч. Раствор затем промывали 5% раствором KHSO4, насыщенным раствором NaHCO3, рассолом и сушили над безводным MgSO4. Неочищенный продукт очищали путем флэш-хроматографии, используя 2:1/гексан:EtOAc на силикагеле, с получением указанного в заголовке соединения (5,10 г, выход 79%); 1H-ЯМР (400 МГц, CD3Cl3) δ 8,12 (m, 1H), 7,29 (m, 2H), 3,56 (br d, 2H), 3,23 (br d, 2H), 1,27 (brs, 3Н), 1,15 (br s, 3Н).
40Б: 3-Амино-N,N-диэтил-4-нитро-бензамид
N,N-Диэтил-3-фтор-4-нитро-бензамид (5,1 г, 21,2 ммоль) кипятили с обратным холодильником в смеси 2:1 NH4OH/EtOH (150 мл) в течение 48 ч. Раствор охлаждали до к.т. и растворитель концентрировали. Раствор затем экстрагировали (3×) EtOAc. Объединенные органические фазы промывали рассолом и сушили над безводном MgSO4. Неочищенный продукт кристаллизовался из смеси EtOAc/гексаны с получением указанного в заголовке соединения (4,35 г, выход 86%). 1Н-ЯМР (400 МГц, CD3OD) δ 8,12 (d, J=8,8 Гц, 1H), 6,92 (s, 1H), 6,56 (d, J=8,8 Гц, 1H), 3,51 (q, J=7,2 Гц, 2H), 3,28 (m, 2H), 1,23 (t, J=7,2 Гц, 3Н), 1,13 (t, J=7,2 Гц, 3Н).
40В: N-[5-[(Диэтиламино)карбонил]-2-нитрофенил]-4-этокси-бензолацетамид
К перемешиваемому толуоловому раствору (100 мл) 3-амино-N,N-диэтил-4-нитро-бензамида (1,00 г, 4,21 ммоль) добавляли 4-этоксифенилацетилхлорид (1,25 г, 1,5 экв.) и цинковую пыль (415 мг, 1,5 экв.). Раствор перемешивали при к.т. в течение ночи. Затем добавляли еще 0,5 экв. хлорангидрида и цинковой пыли и раствор перемешивали при к.т. в течение еще 24 ч. Раствор затем фильтровали через целит и промывали EtOAc. Органическую фазу промывали насыщенным раствором NaHCO3, рассолом и сушили над безводным MgSO4. Неочищенный продукт очищали путем флэш-хроматографии на силикагеле, используя смесь 1:1/гексан:EtOAc, для получения желаемого продукта (1,52 г, выход 90%). 1H-ЯМР (400 МГц, CDCl3) δ 8,81 (s, 1H), 8,20 (d, J=8,8 Гц, 1Н), 7,26 (m, 3Н), 7,15 (d, J=8,8 Гц, 1H), 6,94 (d, J=8,4 Гц, 2Н), 4,06 (q, J=7,2 Гц, 2Н), 3,75 (s, 2Н), 3,54 (q, J=6,8 Гц, 2Н), 3,24 (q, J=6,8 Гц, 2Н), 1,59 (brs, 1H), 1,42 (t, J=6,8 Гц, 3Н), 1,25 (t, J=6,8 Гц, 3Н), 1,16 (t, J=6,8 Гц, 3Н).
40Г: N-[2-Амино-5-[(Диэтиламино)карбонил]фенил]-4-этокси-бензолацетамид
N-[5-[(Диэтиламино)карбонил]-2-нитрофенил]-4-этокси-бензолацетамид (1,00 г, 2,50 ммоль) растворяли в EtOAc (50 мл), содержащем каталитическое количество 10% Pd/C. Раствор перемешивали в атмосфере Н2 (35 ф./кв. дюйм (241,3 кПа)) при к.т. в течение ночи. Раствор фильтровали через целит и растворитель концентрировали. Анализ с помощью ЖХ/МС (жидкостная хроматография/масс-спектрометрия) показал, что указанное в заголовке соединение достаточно чистое (более 95%) и может непосредственно использоваться на следующей стадии. Выход: 927 мг (99%); 1H-ЯМР (400 МГц, CDCl3) δ 7,54 (s, 1H), 7,25 (d, J=7,6 Гц, 2Н), 7,07 (s, 1H), 7,01 (d, J=8,0 Гц, 1H), 6,91 (d, J=7,2 Гц, 2Н), 6,66 (d, J=8,4 Гц, 1H), 4,04 (q, J=6,8 Гц, 2Н), 3,69 (s, 2H), 3,39 (brs, 4Н), 1,42 (t, J=6,8 Гц, 3Н), 1,15 (brs, 6H).
40Д: 2-[(4-Этоксифенил)метил]-N,N-диэтил-1-(3-тиенилметил)-1Н-бензимидазол-5-карбоксамид
N-[2-Амино-5-[(диэтиламино)карбонил]фенил]-4-этокси-бензолацетамид (100 мг, 0,271 ммоль) растворяли в 3 мл смеси 2:1 дихлорэтана (ДХЭ):АсОН. Добавляли тиофен-3-карбоксальдегид (37 мл, 1,5 экв.) и раствор перемешивали при к.т. в течение 15 мин. Добавляли раствор комплекса боран/пиридин (55 мл, 2 экв.) и раствор перемешивали при к.т. в течение 1 ч. Добавляли несколько капель концентрированной HCl (5 капель) и раствор перемешивали при 85°С в течение 3 ч. Раствор охлаждали до к.т. и растворитель концентрировали. Неочищенный продукт непосредственно очищали путем обращенно-фазовой хроматографии (колонка С-18), используя градиент 15-65% СН3CN/Н2О, и затем лиофилизировали. Указанный в заголовке продукт выделяли в виде соответствующей соли с ТФУ. Выход: 118 мг (78%). 1H-ЯМР (400 МГц, CD3OD) 5 7,78 (d, J=8,4 Гц, 1Н), 7,70 (s, 1H), 7,48 (d, J=8,4 Гц 1H), 7,38 (m, 1H), 7,23 (s, 1H), 7,18 (d, J=7,2 Гц, 2Н), 6,84 (m, 3Н), 5,69 (s, 2H), 4,53 (s, 2H), 3,97 (q, J=7,2 Гц, 2H), 3,54 (brs, 2H), 3,27 (brs, 2H), 1,34 (t, J=7,2 Гц, 3Н), 1,23 (brs, 3H), 1,10 (brs, 3Н); МС (ЭГДИ) (M+H)+=448,32; Аналитически рассчитано для С26Н29N3O2S+1,7 ТФУ+0,1 Н2O0: С, 54,90; Н, 4,84; N, 6,53. Обнаружено: С, 54,88; Н, 4,86; N, 6,53.
Пример 41
2-[(4-Этоксифенил)метил]-N,N-диэтил-1-[(2R)-2-пирролидинилметил]-1Н-бензимидазол-5-карбоксамид
В соответствии со способом 40Д использовали N-[2-амино-5-[(диэтиламино)карбонил]фенил]-4-этокси-бензолацетамид (100 мг, 0,270 ммоль) и N-(трет-бутоксикарбонил)-D-пролинал (prolinal) (76 мл, 1,5 экв.). Неочищенный продукт непосредственно очищали путем обращенно-фазовой хроматографии (колонка С-18), используя градиент 5-50% СН3CN/H2О, и затем лиофилизировали. Указанный в заголовке продукт выделяли в виде соответствующей соли с ТФУ. Выход: 86 мг (78%). 1Н-ЯМР (400 МГц, CD3OD) δ 7,94 (d, J=8,8 Гц, 1H), 7,70 (s, 1H), 7,55 (d, J=8,4 Гц, 1H), 7,28 (d, J=8,8 Гц, 2H), 6,94 (d, J=8,8 Гц, 2H), 4,83 (d, J=7,2 Гц, 2H), 4,55 (s, 2H), 4,02 (q, J=7,2 Гц, 2H), 3,96 (m, 1H), 3,56 (brs, 2H), 3,48 (m, 1H), 3,27 (m, 3Н), 2,24 (m, 1H), 2,14 (m, 1H), 2,04 (m, 1H), 1,87 (m, 1H), 1,36 (t, J=7,2 Гц, 3Н), 1,26 (brs, 3Н), 1,11 (brs, 3H); МС (ЭГДИ) (М+Н)+=435,45; Аналитически рассчитано для С26Н34N4O2+2,2 ТФУ+1,6 Н2O: С, 51,12; Н, 5,56; N, 7,84. Обнаружено: С, 51,12; Н, 5,62; N, 7,82.
Пример 42
2-[(4-Этоксифенил)метил]-N,N-диэтил-1-[[(25)-тетрагидро-2-фуранил]метил]-1Н-бензимидазол-5-карбоксамид
Смесь N,N-диэтил-4-фтор-3-нитробензамида (120 мг, 0,500 ммоль), триэтиламина (0,105 мл, 1,5 экв.) и 8-(+)-тетрагидрофуриламина (55 мг, 1,1 экв.) перемешивали в 3 мл EtOH при 85°С в течение 3 ч. Раствор охлаждали до к.т. и растворитель концентрировали. Остаток растворяли в EtOAc и промывали насыщенным раствором NaHCO3, рассолом и сушили над безводным MgSO4. Аддукт очищали путем флэш-хроматографии на силикагеле, используя смесь 3:1/гексан:EtOAc. Выход: 147 мг (92%). 1H-ЯМР (400 МГц, CDCl3) δ 8,35 (s, 1H), 8,25 (s, 1Н), 7,53 (d, J=8,8 Гц, 1Н), 6,89 (d, J=8,8 Гц, 1Н), 4,17 (m, 1H), 3,92 (m, 1Н), 3,82 (m, 1H), 2,08 (m, 5H), 2,05 (m, 1H), 1,93 (m, 2H), 1,69 (m, 2H), 1,19 (t, J=6,8 Гц, 3Н).
Это нитросоединение (125 мг, 0,389 ммоль) затем растворяли в 20 мл EtOAc, содержащем каталитическое количество 10% Pd/C. Раствор перемешивали в атмосфере Н2 (40 ф./кв. дюйм (275,8 кПа)) при к.т. в течение 6 ч. Раствор фильтровали через целит и растворитель концентрировали с получением желаемого анилина. Выход: 113 мг (99%); МС (ЭГДИ) (М+Н)+=292,31.
Этот анилин (113 мг, 0,388 ммоль) вместе с 4-этоксифенилацетилхлоридом (85 мг, 1,1 экв.) перемешивали в 2 мл дихлорэтана при к.т. в течение 30 мин. Добавляли несколько капель концентрированной HCl (5 капель) и раствор перемешивали при 85°С в течение 3 ч. Раствор охлаждали до к.т. и растворитель концентрировали. Неочищенный продукт непосредственно очищали путем обращенно-фазовой хроматографии (колонка С-18), используя градиент 15-65% СН2CN/Н2О, и затем лиофилизировали с получением указанного в заголовке соединения. Продукт выделяли в виде соответствующей соли с ТФУ. Выход: 148 мг (69%); 1H-ЯМР (400 МГц, CD3OD) δ 7,99 (d, J=8,4 Гц, 1H), 7,68 (s, 1H), 7,58 (d, J=8,8 Гц, 1H), 7,28 (d, J=8,8 Гц, 2H), 6,97 (d, J=8,4 Гц, 2H), 4,70 (d, J=14,8 Гц, 1H), 4,60 (s, 2H), 4,51 (m, 1H), 4,21 (m, 1H), 4,02 (q, J=7,2 Гц, 2H), 3,89 (m, 1H), 3,73 (m, 1H), 3,57 (brd, 2H), 3,27 (brs, 2H), 2,18 (m, 1H), 1,97 (m, 2H), 1,76 (m, 1H), 1,36 (t, J=7,2 Гц, 3Н), 1,26 (brs, 3Н), 1,11 (brs, 3Н); МС (ЭГДИ) (M+H)=436,43;
Аналитически рассчитано для С26Н33N3О3+2,5 ТФУ+0,4 Н2O: С, 52,64; Н, 5,23; N, 6,06. Обнаружено: С, 52,59; Н, 5,08; N, 6,33.
Пример 43
2-[(4-Этоксифенил)метил]-N,N-диэтил-1-[[(2R)-1-метил-2-пирролидинил]метил]-1Н-бензимидазол-5-карбоксамид
N-[2-Амино-5-[(Диэтиламино)карбонил]фенил]-4-этокси-бензолацетамид (85 мг, 0,230 ммоль) и N-(трет-бутоксикарбонил)-D-пролинал (0,06 мл, 1,5 экв.) соединяли и подвергали циклизации в соответствии со способом в примере 40Д растворитель затем концентрировали. Остаток растворяли в МеОН и добавляли 37% формальдегид в воде (формалин) (несколько капель, избыток) с последующим добавлением цианоборогидрида натрия (43 мг, 3 экв.). Раствор затем перемешивали при к.т. в течение 1 ч. Неочищенный продукт непосредственно очищали путем обращенно-фазовой хроматографии (колонка С-18), используя градиент 15-65% СН2CN/H2О, и затем лиофилизировали с получением указанного в заголовке соединения (46 мг, выход 36%) в виде соответствующей соли с ТФУ; 1H-ЯМР (400 МГц, CD3OD) δ 7,80 (d, J=8,8 Гц, 1H), 7,69 (s, 1H), 7,48 (d, J=8,4 Гц, 1Н), 7,22 (d, J=8,8 Гц, 2Н), 6,92 (d, J=8,4 Гц, 2Н), 4,85 (m, 1H), 4,66 (m, 1H), 4,49 (s, 2H), 4,01 (q, J=7,2 Гц, 2Н), 3,78 (brs, 2Н), 3,56 (brs, 2H), 3,27 (brs, 3H), 2,95 (s, 3H), 2,02 (m, 3H), 1,85 (m, 1H), 1,35 (t, J=7,2 Гц, 3H), 1,25 (brs, 3H), 1,11 (brs, 3H); MC (ЭГДИ) (M+H)+=449,45; Аналитически рассчитано для C27H36N4O2+2,4 ТФУ+1,1 Н2O: С, 51,47; Н, 5,51; N, 7,55. Обнаружено: С, 51,46; Н, 5,43; N, 7,67.
Примеры 44 и 45
2-[(4-Этоксифенил)метил]-N,N-диэтил-1-[[(2R)-1-метил-2-пиперидинил]метил]-1Н-бензимидазол-5-карбоксамид и 2-[(4-этоксифенил)метил]-N,N-диэтил-1-[[(25)-1-метил-2-пиперидинил]метил]-1Н-бензимидазол-5-карбоксамид
В соответствии со способом из примера 40Д использовали N-[2-Амино-5-[(диэтиламино)карбонил]фенил]-4-этокси-бензолацетамид (70 мг, 0,189 ммоль) и 2-N-(трет-бутоксикарбонил)-1-пиперидинкарбоксальдегид (52 мг, 1,5 экв.). Неочищенный продукт затем растворяли в МеОН (3 мл), содержащем несколько капель ледяной уксусной кислоты. Добавляли избыток 37% НСНО/Н2O с последующим добавлением NaCNBH2 (24 мг, 2 экв.). Раствор перемешивали при к.т. в течение 30 мин. Растворитель концентрировали и неочищенный продукт непосредственно очищали путем обращенно-фазовой хроматографии (колонка С-18), используя градиент 5-50% СН3CN/Н2O, и затем лиофилизировали с получением желаемого продукта в виде смеси двух энантиомеров. Продукт выделяли в виде соответствующей соли с ТФУ. Выход: 53 мг (50%). Два энантиомера разделяли путем хиральной хроматографии, используя калонку Chiral AD с изократическим элюентом 30% изо-PrOH/гексаны, содержащим 0,1% диэтиламин, с получением двух энантиомеров 2-[(4-этоксифенил)метил]-N,N-диэтил-1-[[(2R)-1-метил-2-пиперидинил]метил]-1Н-бензимидазол-5-карбоксамида (20 мг, 50%) и 2-[(4-этоксифенил)метил]-N,N-диэтил-1-[[(25)-1-метил-2-пиперидинил]метил]-1Н-бензимидазол-5-карбоксамида (20 мг, 50%).
Энантиомер: 2-[(4-этоксифенил)метил]-N,N-диэтил-1-[[(2R)-1-метил-2-пиперидинил]метил]-1Н-бензимидазол-5-карбоксамид: 1H-ЯМР (400 МГц, CD3OD) δ 7,71 (brs, 1Н), 7,68 (s, 1H), 7,43 (d, J=8,4 Гц, 1Н), 7,21 (d, J=8,6 Гц, 2Н), 6,92 (d, J=8,6 Гц, 2Н), 4,85 (s, 2Н), 4,42 (s, 2H), 3,97 (q, J=7,2 Гц, 2Н), 3,55 (brs, 2H), 3,27 (m, 5H), 3,03 (s, 3Н), 2,98 (brs, 1H), 1,85 (brs, 1H), 1,74 (m, 3H), 1,50 (brs, 1H), 1,34 (t, J=7,2 Гц, 3Н), 1,25 (brs, 3Н), 1,11 (brs, 3Н). МС (ЭГДИ) (M+H)+=463,51; Аналитически рассчитано для С28Н38N4O2+2,1 ТФУ+0,6 Н2О: С, 54,25; Н, 5,84; N, 7,86. Обнаружено: С, 54,24; Н, 5,69; N, 8,17; HPLC k': 1,99 (колонка: Chiral AD, градиент: 30% Б в течение 25 мин, скорость потока 1 мл/мин, 25°С; Растворитель А: 0,1% диэтиламин (ДЭА) в гексанах, Растворитель Б: 0,1% ДЭА в изо-PrOH).
Энантиомер: 2-[(4-этоксифенил)метил]-N,N-диэтил-1-[[(2R)-1-метил-2-пиперидинил]метил]-1H-бензимидазол-5-карбоксамид: 1H-ЯМР, МС и элементный анализ идентичны его энантиомеру; HPLC k': 4,97 (колонка: Chiral AD, градиент: 30% Б в течение 25 мин, скорость потока 1 мл/мин, 25°С; Растворитель А: 0,1% ДЭА в гексанах, Растворитель Б: 0,1% ДЭА в изо-PrOH).
Пример 46
2-[{4-Этоксифенил)гидроксиметил]-N,N-диэтил-1-(3-метилбутил)-1Н-бензимидазол-5-карбоксамид
2-(4-Этоксибензоил)-N,N-диэтил-1-(3-метилбутил)-1Н-бензимидазол-5-карбоксамид (110 мг, 0,252 ммоль) растворяли в 3 мл EtOH. Добавляли NaBH4 (12 мг, 1,2 экв.) и раствор перемешивали при к.т. в течение 1 ч. Растворитель концентрировали и остаток растворяли в EtOAc. Органическую фазу промывали насыщенным раствором NaHCO3, рассолом и сушили над безводным MgSO4. Неочищенный продукт непосредственно очищали путем обращенно-фазовой хроматографии (колонка С-18), используя градиент 10-65% СН2Cl2/Н2O, и затем лиофилизировали с получением указанного в заголовке соединения. Продукт выделяли в виде соответствующей соли с ТФУ. Выход: 102 мг (73%); 1H-ЯМР (400 МГц, CD3OD) δ 7,85 (br s, 2H), 7,62 (br s, 1H), 7,40 (br s, 2H), 7,01 (br s, 2H), 6,33 (br s, 1H), 4,29 (br s, 2H), 4,06 (br s, 2H), 3,62 (br s, 2H), 3,32 (br s, 2H), 1,55 (br, 2H), 1,38 (br s, 3H), 1,30 (br s, 3H), 1,17 (br s, 2H), 0,90 (s, 3H), 0,83 (s, 3H); МС (ЭГДИ) (М+Н)+=438,30; Аналитически рассчитано для C26H35N3O3+1,2 ТФУ: С, 59,38; Н, 6,35; N, 7,32. Обнаружено: С, 59,36; Н, 5,97; N, 7,37.
Пример 47
N-[2-[(4-Этоксифенил)метил]-1-(3-тиенилметил)-1Н-бензимидазол-5-ил]-N,3-диметил-бутанамид
47А: 1,1-Диметилэтиловый эфир метил-(4-нитрофенил)-карбаминовой кислоты
К перемешиваемому раствору NaH (1,15 г, 1,5 экв., 60% в масле) в безводном ДМФ (100 мл) добавляли раствор N-метил-4-нитроанилина (3,00 г, 19,7 ммоль) в ДМФ (25 мл) при 0°С. Раствор затем перемешивали при 0°С в течение 15 мин. Затем добавляли раствор ди-трет-бутилдикарбоната (4,30 г, 1,2 экв.) в ДМФ (50 мл) и раствор интенсивно перемешивали при к.т. в течение 3 ч. Раствор гасили путем добавления насыщенного раствора NH4Cl и растворитель концентрировали. Остаток растворяли в EtOAc и промывали насыщенным раствором NaHCO3, рассолом и сушили над безводным MgSO4. Неочищенный продукт очищали путем флэш-хроматографии на силикагеле, используя смесь 4:1/гексан:EtOAc, с получением 4,50 г (выход 90%) желаемого продукта, представляющего собой 1,1-диметилэтиловый эфир метил-(4-нитрофенил)-карбаминовой кислоты. 1Н-ЯМР (400 МГц, CDCl3) δ 8,19 (d, J=8,8 Гц, 2Н), 7,46 (d, J=8,8 Гц, 2Н), 3,35 (s, 3Н), 1,51 (s, 9H).
47Б: 1,1-Диметилэтиловый эфир (3-амино-4-нитрофенил)метил-карбаминовой кислоты
К охлажденному раствору (0°С) ДМФ (50 мл) трет-BuOK (9,0 г, 4,5 экв.) и CuCl (176 мг, 0,1 экв.) в атмосфере N2 добавляли раствор 1,1-диметилэтилового эфира метил-(4-нитрофенил)-карбаминовой кислоты (4,49 г, 17,8 ммоль) и метоксиламингидрохлорида (1,85 г, 1,25 экв.) в ДМФ (100 мл). Раствор оставляли нагреваться до к.т. в течение 1 ч. Реакционную смесь затем гасили насыщенным NH4Cl и растворитель концентрировали. Остаток разбавляли водой и экстрагировали EtOAc. Органическую фазу промывали рассолом и сушили над безводным MgSO4. Неочищенный продукт очищали путем флэш-хроматографии на силикагеле, используя смесь 2:1/гексан:EtOAc, с получением желаемого продукта 1,1-диметилэтилового эфира (3-амино-4-нитрофенил)метил-карбаминовой кислоты (1,10 г, выход 24%). 1H-ЯМР (400 МГц, CDCl3) δ 8,06 (d, J=9,2 Гц, 1Н,), 6,78 (s, 1H), 6,67 (d, J=9,2 Гц 1Н), 6,10 (brs, 2H), 3,28 (s, 3H), 1,50(s, 9H).
47В: 1,1-Диметилэтиловый эфир [3-[[(4-этоксифенил)ацетил]амино]-4-нитрофенил]метил-карбаминовой кислоты
К перемешиваемому раствору 1,1-диметилэтилового эфира (3-амино-4-нитрофенил)метил-карбаминовой кислоты (1,10 г, 4,12 ммоль) в толуоле (100 мл) добавляли 4-этоксифенилацетилхлорид (980 мг, 1,2 экв.) и цинковую пыль (400 мг, 1,5 экв.). Раствор перемешивали при к.т. в течение ночи. Затем добавляли еще 0,5 экв. хлорангидрида и цинковую пыль и раствор перемешивали при к.т. в течение дополнительных 24 ч. Раствор затем фильтровали через целит и промывали EtOAc. Органическую фазу промывали насыщенным раствором NaHCO3, рассолом и сушили над безводным MgSO4. Неочищенный продукт очищали путем флэш-хроматографии на силикагеле, используя смесь 2:1/гексан:EtOAc, с получением желаемого продукта 1,1-диметилэтилового эфира [3-[[(4-этоксифенил)ацетил]амино]-4-нитрофенил]метил-карбаминовой кислоты (1,18 г, выход 67%); 1H-ЯМР (400 МГц, CDCl3) δ 8,77 (s, 1H), 8,11 (d, J=9,2 Гц, 1H), 7,22 (m, 3H), 6,93 (d, J=9,2 Гц, 2H), 4,40 (m, 2H), 3,75 (s, 2H), 3,33 (s, 3H), 1,50 (s, 9H), 1,42 (t, J=6,8 Гц, 3Н).
47Г: 4-Этокси-N-[5-(метиламино)-2-нитрофенил]-бензолацетамид
1,1-Диметилэтиловый эфир [3-[[(4-этоксифенил)ацетил]амино]-4-нитрофенил]метил-карбаминовой кислоты (1,10 г, 2,56 ммоль) перемешивали в 15 мл 1M HCl/AcOH при к.т. в течение 2 ч. Растворитель концентрировали. Остаток растворяли в EtOAc и промывали насыщенным раствором NaHCO3, рассолом и сушили над безводным MgSO4. Продукт непосредственно использовали на следующей стадии. Выход 845 мг (99%); 1H-ЯМР (400 МГц, CDCl3) δ 8,80 (d, J=9,2 Гц, 1H), 8,02 (s, 1H), 7,25 (d, J=6,8 Гц, 2H), 6,91 (d, J=8,4 Гц, 2H), 6,22 (d, J=9,2 Гц, 1H), 4,74 (brd, 1H), 4,04 (q, J=7,2 Гц, 2H), 3,73 (s, 2H), 2,94 (s, 3Н), 1,41 (t, J=7,2 Гц, 3Н).
47Д: 4-Этокси-N-[5-[метил(3-метил-1-оксобутил)амино]-2-нитрофенил]-бензолацетамид
К раствору 4-этокси-N-[5-(метиламино)-2-нитрофенил]-бензолацетамида (845 мг, 2,56 ммоль) и 4-диметиламинофенола (ДМАФ) (470 мг, 1,5 экв.) в смеси 1:1/ДХЭ:СН3CN (50 мл) добавляли изовалерилхлорид (0,47 мл, 1,5 экв.).
Раствор перемешивали при к.т. в течение 48 ч. Раствор затем промывали 5% KHSO4, насыщенным раствором NaHCO3, рассолом и сушили над безводным MgSO4. Неочищенный продукт очищали путем флэш-хроматографии, используя смесь 7:1/СН2Cl2:эфир, с получением желаемого продукта 4-этокси-N-[5-[метил(3-метил-1-оксобутил)амино]-2-нитрофенил]-бензолацетамида (1,06 г, выход 99%); 1H-ЯМР (400 МГц, CDCl3) δ 8,72 (s, 1H), 8,21 (d, J=8,8 Гц, 1Н), 7,26 (m, 3Н), 6,96 (m, 2H), 4,06 (m, 2Н), 3,76 (s, 2H), 3,31 (s, 3Н), 2,15 (m, 3H), 1,42 (t, J=7,2 Гц, 3Н), 0,88 (d, J=6,8 Гц, 6Н).
47Е: 4-Этокси-N-[2-амино-5-[метил(3-метил-1-оксобутил)амино]фенил]-бензолацетамид
4-Этокси-N-[5-[метил(3-метил-1-оксобутил)амино]-2-нитрофенил]-бензолацетамид (1,05 г, 2,53 ммоль) растворяли в EtOAc (50 мл), содержащем каталитическое количество 10% Pd/C. Раствор встряхивали в атмосфере Н2 (35 ф./кв. дюйм (241,3 кПа)) при к.т. в течение ночи. Раствор фильтровали через целит и растворитель концентрировали. Анализ посредством ЖХ/МС продемонстрировал, что указанное в заголовке соединения является чистым (более 95%) и может непосредственно использоваться на следующей стадии. Выход: 965 мг (99%). 1H-ЯМР (400 МГц, CDCl3) δ 7,23 (d, J=8,8 Гц, 2H), 7,12 (s, 1H), 7,00 (s, 1H), 6,93 (d, J=8,8 Гц, 1H), 6,78 (m, 2H), 4,01 (q, J=7,0 Гц, 2H), 3,69 (s, 2H), 3,13 (s, 3Н), 2,05 (m, 1H), 1,93 (d, J=7,2 Гц, 2H), 1,39 (t, J=7,2 Гц, 3Н), 0,78(d,J=6,8 Гц, 6H).
47Ж: N-[2-[(4-Этоксифенил)метил]-1-(3-тиенилметил)-1H-бензимидазол-5-ил]-N,3-диметил-бутанамид
В соответствии со способом из примера 40Д использовали 4-этокси-N-[2-амино-5-[метил(3-метил-1-оксобутил)амино]фенил]-бензолацетамид (75 мг, 0,196 ммоль) и тиофен-3-карбоксальдегид (26 мл, 1,5 экв.). Неочищенный продукт непосредственно очищали путем обращенно-фазовой хроматографии (колонка С-18), используя градиент 15-65% СН3CN, и затем лиофилизировали. Указанное в заголовке соединение выделяли в виде соответствующей соли с ТФУ. Выход: 55 мг (50%); 1H-ЯМР (400 МГц, CD3OD) δ 7,54 (m, 2H), 7,30 (d, J=4,8 Гц, 1H), 7,17 (d, J=8,4 Гц, 1H), 7,12 (d, J=8,4 Гц, 2H), 7,05 (s, 1H), 6,80 (d, J=8,8 Гц, 2H), 6,70 (d, J=5,2 Гц, 1H), 5,48 (s, 2H), 4,35 (s, 2H), 3,95 (q, J=7,2 Гц, 2H), 3,23 (s, 3Н), 2,03 (brs, 1Н), 1,94 (brs, 2H), 1,31 (t, J=7,2 Гц, 3Н), 0,76 (brs, 6H); МС (ЭГДИ) (M+H)+=462,43; Аналитически рассчитано для С27Н31N3O2S+0,7 ТФУ+0,5 Н2O: С, 61,97; Н, 5,99; N, 7,63. Обнаружено: С, 61,88; Н, 6,03; N, 7,62.
Пример 48
N-[2-[(4-Этоксифенил)метил]-1-[(2R)-2-пирролидинилметил]-1Н-бензимидазол-5-ил]-N,3-диметил-бутанамид
В соответствии со способом из примера 40Д использовали 4-этокси-N-[2-амино-5-[метил(3-метил-1-оксобутил)амино]фенил]-бензолацетамид (75 мг, 0,196 ммоль) и N-(трет-бутоксикарбонил)-O-пролинал (55 мл, 1,5 экв.). Неочищенный продукт непосредственно очищали путем обращенно-фазовой хроматографии (колонка С-18), используя градиент 5-50% СН3CN/H2О, и затем лиофилизировали. Указанное в заголовке соединение выделяли в виде соответствующей соли с ТФУ. Выход: 81 мг (73%); 1H-ЯМР (CD3OD) δ 7,91 (d, J=8,8 Гц, 1Н), 7,59 (s, 1Н), 7,43 (d, J=8,8 Гц, 1Н), 7,26 (d, J=8,8 Гц, 2Н), 6,93 (d, J=8,8 Гц, 2Н), 4,79 (d, J=7,2 Гц, 2Н), 4,53 (s, 2H), 3,98 (q, J=7,2 Гц, 2Н), 3,88 (m, 1Н), 3,44 (m, 1H), 3,23 (s, 3Н), 2,18 (m, 1H), 2,13 (m, 1H), 2,05 (m, 2H), 1,98 (m, 3Н), 1,81 (m, 1H), 1,33 (t, J=7,2 Гц, 3Н), 0,78 (brs, 6H); МС (ЭГДИ) (М+Н)+=449,50; Аналитически рассчитано для С27Н36N4O2+2,3 ТФУ+1,8 Н2О: С, 51,06; Н, 5,68; N, 7,54. Обнаружено: С, 51,06; Н, 5,73; N, 7,26.
Пример 49
N-[1-[[5-[(Ацетилокси)метил]-2-фуранил]метил]-2-[(4-этоксифенил)метил]-1Н-бензимидазол-5-ил]-N,3-диметил-бутанамид
В соответствии со способом из примера 40Д использовали 4-этокси-N-[2-амино-5-[метил(3-метил-1-оксобутил)амино]фенил]-бензолацетамид (75 мг, 0,196 ммоль) и 5-[(ацетилокси)метил]-2-фуранкарбоксальдегид (50 мг, 1,5 экв.). Неочищенный продукт непосредственно очищали путем обращенно-фазовой хроматографии (колонка С-18), используя градиент 15-65% СН2CN/H2О, и затем лиофилизировали. Указанное в заголовке соединение выделяли в виде соответствующей соли с ТФУ. Выход: 63 мг (51%); 1Н-ЯМР (400 МГц, CD3OD) δ 8,01 (d, J=8,8 Гц, 1H), 7,64 (s, 1H), 7,49 (d, J=8,8 Гц, 1H), 7,27 (d, J=8,4 Гц, 2H), 6,94 (d, J=8,4 Гц, 2H), 6,50 (d, J=3,2 Гц, 1H), 6,39 (d, J=3,2 Гц, 1H), 5,75 (s, 2H), 4,92 (s, 2H), 4,68 (s, 2H), 4,00 (q, J=7,2 Гц, 2H), 3,27 (s, 3Н), 2,02 (m, 1H), 1,96 (brs, 5H), 1,36 (t, J=7,2 Гц, 3Н), 0,79 (s, 6H); МС (ЭГДИ) (М+Н)+=518,49; Аналитически рассчитано для С30Н35N3O5+1,6 ТФУ+0,6 Н2О: С, 56,10; Н, 5,36; N, 5,91. Обнаружено: С, 56,14; Н, 5,40; N, 5,95.
Пример 50
N-[2-(4-Этоксибензил)-1-[(2R)-2-пирролидинилметил]-1Н-бензимидазол-5-ил]-N'-N-(1-изопропил)мочевина
50А: N-[5-[Метил[[изопропиламино]карбонил]амино]-2-нитрофенил]-4-этокси-бензолацетамид
Смесь 4-этокси-N-[5-(метиламино)-2-нитрофенил]-бензолацетамида (2,20 г, 6,69 ммоль) в 1,2-дихлорэтане (100 мл) перемешивали при комнатной температуре в атмосфере азота в виде трифосгена (1,99 г, 6,69 ммоль) и добавляли ТЭА (0,93 мл, 6,69 ммоль). Через 30 минут добавляли ДМАФ (817 мг, 6,69 ммоль) и изопропиламин (3,42 мл, 40,0 ммоль) и содержимое перемешивали в течение 16 часов при 45°С. Смесь гасили насыщенным водным NaHCO3 (40 мл) и слои разделяли. Водную фазу экстрагировали дихлорметаном (2×50 мл) и органические слои объединяли и промывали рассолом (50 мл) и сушили с использованием Na2SO4. Твердое вещество отфильтровывали и фильтрат концентрировали в вакууме до остатка, который очищали путем колоночной хроматографии (75% EtOAc, 25% гептан на силикагеле) с получением указанного в заголовке соединения (2,44 г, 88%). 1H-ЯМР (400 МГц, CDCl3): δ 1,14 (d, J=6,4, 6Н), 1,38 (t, J=7,1 Гц, 3Н), 3,29 (s, 3H), 3,71 (s, 2H), 4,01 (q, J=7,1 Гц, 2Н), 4,70 (s, 1Н), 6,90 (d, J=8,8 Гц, 2Н), 7,00 (dd, J=2,5, 9,2 Гц, 1H),7,22(d,J=8,8 Гц, 2H), 8,13 (d, J=9,2 Гц, 1H),8,61 (d, J=2,5 Гц, 1Н). МС (ЭГДИ) (М+Н)+=415.
50Б: N-[2-Амино-5-[метил[[изопропиламино]карбонил]амино]фенил]-4-этокси-бензолацетамид
Смесь N-[5-[метил[[изопропиламино]карбонил]амино]-2-нитрофенил]-4-этокси-бензолацетамида (190 мг, 0,46 ммоль) и 10% Pd/C в EtOAc (5,0 мл) гидрировали в течение 4 часов при давлении 30 ф./кв. дюйм (206,85 кПа). Содержимое фильтровали через целит и целит промывали EtOAc (2×10,0 мл). Растворитель удаляли в вакууме с получением указанного в заголовке соединения (170 мг, 99%), которое использовали без дополнительной очистки. 1H-ЯМР (400 МГц, CDCl3): δ 0,98 (d, J=6,5 Гц, 6Н), 1,38 (t, J=7,1 Гц, 3Н), 3,09 (s, 3Н), 3,69 (s, 2H), 3,86 (spt, J=6,7 Гц, 1Н), 3,99 (q, J=7,0 Гц, 2H), 4,11-4,13 (m, 1Н), 6,71 (d, J=8,4 Гц, 1Н), 6,82 (dd, J=2,4 Гц, J=8,4 Гц, 1Н), 6,87 (d, J=8,6 Гц, 2Н), 6.96 (d, J=2,3 Гц, 1Н), 7,24 (d, J=8,6 Гц, 2Н), 7,58 (s, 1H). МС (ЭГДИ) (М+H)+=385.
50В: N-[2-(4-Этоксибензил)-1-[(25)-2-пирролидинилметил]-1Н-бензимидазол-5-ил]-N'-N-(1-изопропил)мочевина
В соответствии со способом из примера 40Д смесь N-[2-амино-5-[метил[[изопропиламино]карбонил]амино]фенил]-4-этокси-бензолацетамида (83 мг, 0,22 ммоль) и 2-(3)-N-Вос-пролинала (56 мкл, 0,30 ммоль) в смеси 1,2-дихлорэтан/АсОН (2:1, 7,5 мл) перемешивали при комнатной температуре в течение 2,5 часов. Посредством шприца добавляли комплекс ВН3 · пиридин (43 мкл, 0,42 ммоль) и содержимое перемешивали в течение 16 часов. Смесь нагревали до температуры дефлегмации в течение 6 часов. Обычная обработка позволила получить остаток, который очищали путем обращенно-фазовой жидкостной хроматографии высокого давления (HPLC, колонка С-18) с получением указанного в заголовке соединения в виде соли с трифторуксусной кислотой (ТФУ) (25 мг, 26%). 1H-ЯМР (400 МГц, MeOH-d6): δ 1,08 (d, J=6,6 Гц, 6Н), 1,36 (t, J=6,9 Гц, 3Н), 1,75-1,85 (m, 1H), 1,96-2,05 (m, 1H), 2,10-2,20 (m, 2Н), 3,26 (s, 3Н), 3,44 (m, 1H), 3,85-3,93 (m, 2Н), 4,01 (q, J=7,1 Гц, 2Н), 4,47 (s, 2H), 4,70 (d, J=7,2 Гц, 2H), 6,93 (d, J=8,6 Гц, 2H), 7,24 (d, J=8,6 Гц, 2H), 7,38 (dd, J=2,0, J=8,8 Гц, 1Н), 7,57 (d, J=2,0 Гц), 7,77 (d, J=8,6 Гц). МС (ЭГДИ) (М+H)+=450.
Пример 51
1-(Циклопропилметил)-2-[1-(4-этоксифенил)этил]-N-метил-1Н-бензимидазол-5-амин
Способ А:
51АА: 1-(Циклопропилметил)-2-[(4-этоксифенил)метил]-N-метил-1Н-бензимидазол-5-амин
К суспензии метил-[1-(циклопропилметил)-2-[(4-этоксифенил)метил]-1Н-бензимидазол-5-ил]карбамата (0,781 г, 2,06 ммоль) в ТГФ добавляли алюмогидрид лития (АГЛ) (0,32 г, 8,43 ммоль) при 0°С. Смесь перемешивали в течение 1 ч при 0°С и 2,5 ч при температуре дефлегмации, охлаждали до -78°С и гасили МеОН (5 мл) и Н2О (5 мл). После добавления Na2SO4 (10 г) получающуюся в результате смесь перемешивали в течение 1 ч при комнатной температуре. После фильтрации и концентрирования остаток очищали путем жидкостной хроматографии среднего давления на силикагеле, используя EtOAc, с получением 641,7 мг (93%) указанного в заголовке соединения в виде твердого вещества белого цвета с желтоватым оттенком. 1H-ЯМР (CDCl3): S 0,22 (m, 2H), 0,48 (m, 2Н), 1,26 (m, 1H), 1,38 (t, J=7,0 Гц, 3Н), 2,89 (s, 3H), 3,83 (d, J=6,6 Гц, 2H), 3,98 (q, J=7,0 Гц, 2H), 4,23 (s, 2H), 6,64 (dd, J=8,6, 2,2 Гц, 1H), 6,80 (d, J=8,6 Гц, 2H), 6,99 (d, J=2,1 Гц, 1 Н), 7,13 (m, 3Н). МС (ЭГДИ) (M+H)+=336,42.
51АБ: 1,1-Диметилэтил-[1-(циклопропилметил)-2-[(4-этоксифенил)метил]-1Н-бензимидазол-5-ил]метилкарбамат
Раствор 1-(циклопропилметил)-2-[(4-этоксифенил)метил]-N-метил-1Н-бензимидазол-5-амина (816,9 мг, 2,44 моль) и Вос2О (930,1 мг, 4,26 ммоль) в ТГФ (60 мл) перемешивали в течение выходных при комнатной температуре. После выпаривания растворителя остаток очищали путем MPLC на силикагеле, используя гексан/EtOAc (1:1), с получением 917,7 мг (87%) указанного в заголовке соединения в виде белого твердого вещества. МС (ЭГДИ) (М+Н)+=436,49.
51АВ: 1,1-Диметилэтил-[1-(циклопропилметил)-2-[1-(4-этоксифенил)этил]-1H-бензимидазол-5-ил]метилкарбамат
К раствору 1,1-диметилэтил-[1-(циклопропилметил)-2-[(4-этоксифенил)метил]-1Н-бензимидазол-5-ил]метилкарбамата (654,3 мг, 1,50 ммоль) в ТГФ добавляли KHMDS (0,5M, 4 мл, 2,0 ммоль) при -78°С. После пеемешивания в течение 40 мин добавляли Mel (283,9 мг, 2,0 ммоль). Смесь перемешивали в течение ночи при комнатной температуре, гасили МеОН (0,5 мл) и насыщ. NaHCO3 (10 мл). Две фазы разделяли. Водную фазу экстрагировали EtOAc (3×30 мл). Объединенные органические фазы промывали рассолом (2×30 мл) и сушили Na2SO4. После концентрирования остаток очищали путем MPLC на силикагеле, используя Гексан/EtOAc (1:1), с получением 541,3 мг (80%) желаемого продукта в виде желтого сиропа. МС (ЭГДИ) (М+Н)+=450,44.
51АГ: 1 -(Циклопропилметил)-2-[1-(4-этоксифенил)этил]-N-метил-1Н-бензимидазол-5-амин
Смесь 1,1-диметилэтил-[1-(циклопропилметил)-2-[1-(4-этоксифенил)этил]-1H-бензимидазол-5-ил]метилкарбамата (541,3 мг, 1,20 ммоль) в смеси 4 н. HCl/диоксан (20 мл) перемешивали в течение 2,5 ч при комнатной температуре. После выпаривания растворителя остаток растворяли в воде (30 мл), нейтрализовали 2 н. NaOH и экстрагировали EtOAc (4×30 мл). Объединенные органические фазы промывали насыщенным NaHCO3 и сушили над Na2SO4. После фильтрации и концентрирования получали 523,0 мг (100%) указанного в заголовке соединения в виде сиропа. МС (ЭГДИ) (М+Н)+=350,35.
Способ Б:
51 БА: Этил-4-этоксифенил-2-пропионат
К раствору 4-гидроксифенил-2-пропионовой кислоты (5,83 г, 35,1 ммоль) в ДМФ (100 мл) добавляли К2СО3 (12,12 г, 87,7 ммоль) при 0°С. После перемешивания в течение 1 ч добавляли Etl (7,0 мл, 13,68 г, 87,7 ммоль). Смесь перемешивали в течение выходных при комнатной температуре, разбавляли водой (400 мл) и экстрагировали EtOAc (4×100 мл). Объединенные органические фазы промывали водой (2×100 мл) и рассолом (100 мл) и сушили с использованием Na2SO4. После фильтрации и концентрирования остаток очищали путем MPLC на силикагеле, используя смесь гексан/EtOAc (4:1), с получением 7,61 г (98%) указанного в заголовке соединения в виде бесцветной жидкости. 1H-ЯМР (400 МГц, CDCl3): δ 1,18 (t, J=7,2 Гц, 3Н), 1,38 (t, J=7,0 Гц, 3Н), 1,44 (d, J=7,0 Гц, 3Н), 3,62 (q, J=7,0 Гц, 2Н), 3,99 (q, J=7,1 Гц, 2Н), 4,1 (m, 1H), 6,8 (m, 2H), 7,2 (m, 2H).
51 ББ: 4-Этоксифенил-2-пропионовая кислота
К раствору этил-4-этоксифенил-2-пропионата (1,33 г, 5,98 ммоль) в 30 мл ТГФ-Н20 (7:3) добавляли LiOH (0,29 г, 12,0 ммоль). Смесь нагревали в течение 24 ч при 40°С и разбавляли водой (20 мл). Водную фазу экстрагировали дизтиловым эфиром, подкисляли 2 н. HCl и затем экстрагировали диэтиловым эфиром (4×20 мл). Объединенные органические фазы промывали рассолом и сушили с использованием Na2SO4. После фильтрации и концентрирования получали 1,14 г (98%) указанного в заголовке соединения в виде белого твердого вещества. 1H-ЯМР (400 МГц, CDCl3): δ 1,38 (t, J=7,0 Гц, 3Н), 1,47 (d, J=7,2 Гц, 3 Н), 3,66 (q, J=7,2 Гц, 1Н), 3,99 (q, J=7,0 Гц, 2Н), 6,8 (m, 2H), 7,2 (m, 2H).
51 БВ: N-[1-(Циклопропилметил)-2-[1-(4-этоксифенил)этил]-1Н-бензимидазол-5-ил]ацетамид
К раствору N-[3-амино-4-[(циклопропилметил)амино]фенил]ацетамида (1,28 г, 5,82 ммоль) и 4-этоксифенил-2-пропионовой кислоты (1,13 г, 5,82 ммоль) в ДМФ (40 мл) добавляли ДИПЭА (1,52 мл, 1,13 г, 8,73 ммоль) при комнатной температуре. После перемешивания в течение 10 мин добавляли одну порцию HATU (2,66 г, 6,98 ммоль). Смесь перемешивали в течение ночи при комнатной температуре, концентрировали до небольшого объема (10 мл) с последующим добавлением Н2О (100 мл), затем ее экстрагировали EtOAc (4×50 мл). Объединенные органические фазы промывали рассолом и сушили с использованием Na2SO4. После фильтрования и выпаривания остаток растворяли в уксусной кислоте (40 мл) и затем нагревали в течение 20 ч при 100°С. После выпаривания растворителя остаток растворяли в EtOAc (200 мл) и промывали 10% Na2CO3 и сушили с использованием Na2SO4. После концентрирования неочищенный продукт очищали путем MPLC, используя EtOAc, с получением 2,14 г (97%) указанного в заголовке соединения в виде светло-желтого твердого вещества. 1H-ЯМР (400 МГц, CDCl3): δ 0,11 (m, 1H), 0,27 (m, 1H), 0,47 (m, 2H), 0,94 (m, 1H), 1,38 (t, J=7,0 Гц, 3Н), 1,81 (d, J=7,0 Гц, 3Н),2.21 (s, 3Н), 3,76 (dd, J=15,0, 6,8 Гц, 1H), 3,86 (dd, J=15,0, 6,5 Гц, 1Н),3,97 (q, J=6,8 Гц, 2H), 4,28 (q, J=7,0 Гц, 1H), 6,79 (d, J=8,6 Гц, 2H), 7,10 (d, J=8,6 Гц, 2Н), 7,23 (d, J=8,6 Гц, 1Н), 7,43 (s, 1Н), 7,53 (dd, J=8,6, 1,9 Гц, 1Н), 7,78 (d, J=2,0 Гц, 1H). МС (ЭГДИ) (M+H)+=378,38.
51 БГ: N-[1-(Циклопропилметил)-2-[1-(4-этоксифенил)этил]-1Н-бензимидазол-5-ил]-N-метилацетамид
К раствору N-[1-(циклопропилметил)-2-[1-(4-этоксифенил)этил]-1Н-бензимидазол-5-ил]-ацетамида (2,13 г, 5,64 ммоль) в ТГФ (100 мл) добавляли NaH (0,45 г, 11,28 ммоль) при 0°С. После перемешивания в течение 2 ч при комнатной температуре добавляли йодметан (1,60 г, 11,28 ммоль). Смесь перемешивали в течение ночи, гасили МеОН (2 мл) и разбавляли диэтиловым эфиром (200 мл), затем промывали насыщенным раствором хлорида аммония (100 мл) и сушили с использованием Na2SO4. После фильтрации и выпаривания получали указанное в заголовке соединение (2,43 г, 100%) в виде неочищенного продукта. МС (ЭГДИ) (M+H)+=392,40.
51 БД: 1-(Циклопропилметил)-2-[1-(4-этоксифенил)этил]-N-метил-1Н-бензимидазол-5-амин
Смесь N-[1-(циклопропилметил)-2-[1-(4-этоксифенил)этил]-1Н-бензимидазол-5-ил]-N-метилацетамида (2,43 г, 5,64 ммоль) и 40% КОН (50 мл) в EtOH нагревали в течение 20 ч при температуре дефлегмации. После охлаждения смесь концентрировали до небольшого объема (50 мл) и разбавляли Н2О (50 мл), затем ее экстрагировали дихлорметаном (4×50 мл). Объединенные органические фазы сушили с использованием Na2SO4. После фильтрации и выпаривания растворителя остаток очищали путем MPLC, используя EtOAc на силикагеле, с получением 1,93 г (99%) указанного в заголовке соединения в виде твердого вещества белого цвета с желтоватым оттенком. МС (ЭГДИ) (М+Н)+=350,37.
Пример 52
N-[1-(Циклопропилметил)-2-[1-(4-этоксифенил)этил]-1H-бензимидазол-5-ил]-N,3-диметилбутанамид
К раствору 1-(циклопропилметил)-2-[1-(4-этоксифенил)этил]-N-метил-1Н-бензимидазол-5-амин (349,5 мг, 1,0 ммоль) в MeCN (25 мл) добавляли ДИПЭА (258,5 мг, 2,0 ммоль), ДМАФ (10 мг) и изовалерилхлорид (180,9 мг, 1,5 ммоль) при 0°С. Реакционную смесь перемешивали в течение ночи при комнатной температуре, затем ее гасили Н2O (150 мл) и экстрагировали EtOAc (4×50 мл). Объединенные органические фазы промывали рассолом и сушили с использованием Na2SO4. После фильтрации и выпаривания растворителя остаток очищали путем MPLC, используя EtOAc на силикагеле, с получением 431,9 мг (99%) желаемого продукта в виде бесцветного сиропа, который превращали в соль с ТФУ в виде белого твердого вещества. 1H-ЯМР (400 МГц, CD3OD): δ 0,22 (m, 1Н), 0,45 (m, 2H), 0,57 (m, 1Н), 0,86 (d, J=5,9 Гц, 6Н), 1,02 (m, 1H), 1,38 (t, J=7,0 Гц, 3Н), 1,83 (d, J=7,0 Гц, 3Н), 2,06 (m,2H), 2,10 (m, 1Н), 3,34 (s, 3Н), 4,02 (m, 3Н), 4,21 (dd, J=15,0, 6,6 Гц, 1Н), 4,65 (q, J=7,0 Гц, 1Н), 6,90 (d, J=8,6 Гц, 2H), 7,21 (d, J=8,6 Гц, 2H), 7,28 (d, J=8,4 Гц, 1Н), 7,62 (s, 1H), 7,72 (d, J=8,6 Гц, 1H). МС (ЭГДИ) (М+Н)+=434,42 (М+1)+ Аналитически рассчитано для С27Н35N3O2+0,80 ТФУ+0,90 МеОН: С, 64,00; Н, 7,17; N, 7,54. Обнаружено: С, 63,98; Н, 7,02; N, 7,34.
Примеры 53, 54 и 55
(рац)-N-[1-(Циклопропилметил)-2-[1-(4-этоксифенил)этил]-1Н-бензимидазол-5-ил]-N-метил-N-(1-метилэтил)-мочевина, (-)-Н-[N-(циклопропилметил)-2-[1-(4-этоксифенил)этил]-1H-бензимидазол-5-ил]-N-метил-N'-(1-метилэтил)-мочевина и (+)-N-[1-(Циклопропилметил)-2-[1-(4-этоксифенил)этил]-1Н-бензимидазол-5-yl]-N-метил-N'-(1-метилэтил)-мочевина
К раствору 1-(циклопропилметил)-2-[1-(4-этоксифенил)этил]-N-метил-1Н-бензимидазол-5-амина (523,0 мг, 1,20 ммоль) в 1,2-дихлорэтане (25 мл) добавляли изопропилизоцианат (1,02 г, 12 ммоль) при комнатной температуре. Смесь нагревали в течение 14 ч при 60°С. После концентрирования остаток очищали путем MPLC, используя EtOAc на силикагеле, с получением 477,9 мг (92%) желаемого продукта (рацемического) в виде светло-желтого твердого вещества, которое превращали в соль ТФУ в виде белого твердого вещества. 1H-ЯМР (400 МГц, CD3OD): δ 0,27 (m, 1Н), 0,51 (m, 2H), 0,60 (m, 1H), 1,08 (m, 1H), 1,15 (d, J=6,4 Гц, 6Н), 1,39 (t, J=7,0 Гц, 3Н), 1,90 (d, J=7,1 Гц, 3Н), 3,35 (s, 3Н), 3,97 (сеп., J=6,7 Гц, 1H), 4,04 (q, J=7,0 Гц, 2H), 4,26 (dd, J=14,8, 7,0 Гц, 1H), 4,38 (dd, J=15,0, 7,2 Гц, 1H), 4,89 (q, J=7,0 Гц, 1H), 6,97 (d, J=8,8 Гц, 2H), 7,26 (d, J=8,6 Гц, 2H), 7,52 (dd, J=8,6, 1,8 Гц, 1Н), 7,71 (s, 1Н), 7,91 (d, J=9,0 Гц, 1H). МС (ЭГДИ) (M+H)+=435,44 (М+1)+ Аналитически рассчитано для С26Н34N4O2+1,20 ТФУ+0,10 H2O: С, 59,51; Н, 6,22; N, 9,77. Обнаружено: С, 59,64; Н, 6,22; N, 9,53.
Рацемическую смесь (рац)-N-[1-(циклопропилметил)-2-[1-(4-этоксифенил)этил]-1Н-бензимидазол-5-ил]-N-метил-N'-(1-метилэтил)-мочевину разделяли на колонке AD-chiral, используя гексан/узо-PrOH (9:1).
Энантиомер: (-)-N-[1-(циклопропилметил)-2-[1-(4-этоксифенил)этил]-1Н-бензимидазол-5-ил]-N-метил-N'-(1-метилэтил)-мочевина: 221,2 мг (40%), соль с ТФУ, белое твердое вещество. [α]D - 11,7° (с 0,25, ErOH). 1Н-ЯМР(400 МГц, CD3OD): δ 0,27 (m, 1H), 0,51 (m, 2H), 0,60 (m, 1H), 1,08 (m, 1H), 1,15 (dd, J=6,6, 1,6 Гц, 6 Н), 1,39 (t, J=7,0 Гц, 3Н), 1,90 (d, J=7,2 Гц, 3Н), 3,35 (s, 3Н), 3,97 (сеп., J=6,6 Гц, 1Н), 4,04 (q, J=7,0 Гц, 2H), 4,26 (dd, J=15,2, 7,2 Гц, 1Н), 4,39 (dd, J=15,0, 6,8 Гц, 1Н), 4,90 (q, J=7,0 Гц, 1Н), 6,97 (m, 2H), 7,25 (m, 2H), 7,51 (dd, J=8,8, 1,8 Гц, 1Н), 7,71 (d, J=1,8 Гц, 1Н), 7,91 (d, J=9,0 Гц, 1Н). МС (ЭГДИ) (М+Н)+=435,47 (М+1)+. Аналитически рассчитано для С26H34N4O2+1,10 ТФУ+0,10 Н2O: С, 60,29; Н, 6,33; N, 9,97. Обнаружено: С, 60,33; Н, 6,33; N, 10.02.
Энантиомер: (+)-N-[1-(Циклопропилметил)-2-[1-(4-этоксифенил)этил]-1Н-бензимидазол-5-ил]-N-метил-N'-(1-метилэтил)-мочевина: 186,4 мг (34%), соль с ТФУ, белое твердое вещество. [α]D +12.2° (с 0,27, ErOH). 1Н-ЯМР (400 МГц, CD3OD): δ 0,27 (m, 1H),0,51 (m, 2H), 0,60 (m, 1H), 1,08 (m, 1Н), 1,15 (dd, J=6,6, 1,6 Гц, 6Н), 1,39 (t, J=7,0 Гц, 3Н), 1,90 (d, J=7,2 Гц, 3Н), 3,35 (s, 3Н), 3,97 (сеп., J=6,6 Гц, 1Н), 4,04 (q, J=7,0 Гц, 2H), 4,26 (dd, J=15,0, 7,2 Гц, 1Н), 4,39 (dd, J=15,0, 6,8 Гц, 1H), 4,90 (q, J=7,2 Гц, 1H), 6,97 (m, 2H), 7,26 (m, 2H), 7,51 (dd, J=9,0, 2,0 Гц, 1H), 7,71 (d, J=1,8, 1H), 7,91 (d, J=9,0 Гц, 1H). МС (ЭГДИ) (М+Н)+=435,46 (М+1)+ Аналитически рассчитано для С26Н34N4O2+1,10 ТФУ+0,20 Н2O: С, 60,10; Н, 6,35; N, 9,94. Обнаружено: С, 60,02; Н, 6,28; N, 10,07.
Пример 56
N-[1-(Циклогексилметил)-2-[1-(4-этоксифенил)этил]-1Н-бензимидазол-5-ил]-N-метил-N'-(1-метилэтил)-мочевина
56А: [4-[(Циклогексилметил)амино]-3-нитрофенил]-метиловый эфир карбаминовой кислоты
К перемешиваемой смеси метил-4-фтор-3-нитрофенилкарбамата (3 г, 14 ммоль) в смеси этанол:вода 4:1 (40 мл+10 мл) добавляли циклогексилметиламин (3,6 мл, 28 ммоль) при комнатной температуре. Реакционную смесь нагревали при 60°С в течение ночи, охлаждали до комнатной температуры. К смеси добавляли воду (20 мл), темно-оранжевое твердое вещество осаждалось из раствора, и его собирали в виде желаемого продукта (6 г, 100%). МС (ЭГДИ) (М+Н)+=308,32 (М+1)+
56Б: [3-Амино-4-[(Циклогексилметил)амино]фенил]-метиловый эфир карбаминовой кислоты
[4-[(Циклогексилметил)амино]-3-нитрофенил]-метиловый эфир карбаминовой кислоты гидрировали в этилацетате, катализируя 10% Pd/C при давлении 30-40 ф./кв. дюйм (206,85 кПа - 275,8кПа) в течение 6 часов. Реакционную смесь фильтровали через целит и растворитель удаляли с получением желаемого продукта в виде темно-пурпурного твердого вещества (2,02 г, 37%). МС (ЭГДИ) (М+Н)+=278,30 (М+1)+.
56В: [1-(Циклогексилметил)-2-[1-(4-этоксифенил)этил]-1Н-бензимидазол-5-ил]-метиловый эфир карбаминовой кислоты
К перемешиваемому раствору [3-амино-4-[(циклогексилметил)амино]фенил]-метилового эфира карбаминовой кислоты (2,02 г, 7,3 ммоль), 4-этокси-а-метилбензолуксусной кислоты (1,4 г, 7,3 ммоль) и диизопропилэтиламина (2,2 мл, 12,4 ммоль) в безводном ДМФ (24 мл) одной порцией при комнатной температуре добавляли HATU (3,1 г, 8,1 ммоль). Раствор распределяли между CH2Cl2 и водой и экстрагировали (3×30 мл). Объединенные органические слои промывали рассолом (30 мл), сушили над Na2SO4 и подвергали фильтрации и концентрированию. Остаток растворяли в уксусной кислоте (180 мл) и нагревали при 80°С в течение ночи. Растворитель выпаривали, остаток растворяли в EtOAc (200 мл), промывали 2 н. NaOH, рассолом и сушили над Na2SO4. Смесь подвергали фильтрации и концентрированию с получением указанного в заголовке соединения в виде желтой пены (2,44 г, 77%). МС (ЭГДИ) (M+H)+=436,42 (М+1)+.
56Г: 1-(Циклогексилметил)-2-[1-(4-этоксифенил)этил]-N-метил-1Н-бензимидазол-5-амин
К раствору [1-(циклогексилметил)-2-(1-(4-этоксифенил)этил]-1Н-бензимидазол-5-ил]-метилового эфира карбаминовой кислоты (2,44 г, 5,6 ммоль) в H2О-МеОН (100 мл, количество МеОН должно быть достаточным для того, чтобы только растворить соединение) по каплям добавляли 1М раствор HCl в Et2O до тер пор, пока не образовывался дополнительный осадок (6 мл). Растворитель удаляли в вакууме. Соль с HCl растворяли в безводном Et2O (120 мл) и ТГФ (60 мл), смесь охлаждали до 0°С и одной порцией добавляли HATU (986 мг, 25,2 ммоль). Раствор медленно нагревали до комнатной температуры и перемешивали в течение ночи с последующим охлаждением до -78°С, гасили МеОН (12 мл) и водой (12 мл), нагревали до комнатной температуры, добавляли Na2SO4 (25 г) и перемешивали при комнатной температуре в течение 3 часов. Фильтрование через целит, промывка EtOAc и концентрирование позволили получить указанное в заголовке соединение в виде коричневой пены (2,1 г, 96%). МС (ЭГДИ) (M+H)+=392,46 (М+1)+
56Д: N-[1-(Циклогексилметил)-2-[1-(4-этоксифенил)этил]-1Н-бензимидазол-5-ил]-N-метил-N'-(1-метилэтил)-мочевина
К раствору 1-(циклогексилметил)-2-[1-(4-этоксифенил)этил]-N-метил-1Н-бензимидазол-5-амина (2,1 г, 5,4 ммоль) в 1,2-дихлорэтане (270 мл) добавляли изопропилизоцианат (1,1 мл, 10,8 ммоль) при комнатной температуре и реакционную смесь нагревали при 60°С в течение ночи. Раствор охлаждали до комнатной температуры, растворитель выпаривали и коричневый остаток очищали путем MPLC (EtOAc на силикагеле) с получением указанного в заголовке соединения в виде бежевой пены (1,5 г, 58%). 1H-ЯМР (400 МГц, CD3OD) δ 0,95-1,18 (m, 11H), 1,36 (t, J=7,0 Гц, 3Н), 1,41-1,68 (m, 6H), 1,82 (d, J=7,0 Гц, 2H), 3,31 (s, 3Н), 3,89-3,95 (m, 1H), 4,01 (apq, J=7,0 Гц, 2Н), 4,13-4,18 (m, 1H), 4,68 (q, J=7,2 Гц, 1H), 7,17-7,20 (m, 2H), 7,35 (dd, J=2,0, 8,8 Гц, 1H), 7,63 (d, J=2,0 Гц, 1H), 7,71 (d, J=8,8 Гц, 1H). МС (ЭГДИ) (M+H)+=477,48 (М+1)+ Аналитически рассчитано для С29Н40N4O2+0,5 HCl+0,8 СН3ОН: С, 68,76; Н, 8,46; N, 10,26. Обнаружено: С, 68,75; Н, 8,61; N, 10,98.
Пример 57
2-[(4-Этоксифенил)метил]-N,N-диэтил-3-[(5-нитро-2-тиенил)метил]-3H-имидазо[4,5-b]пиридин-6-карбоксамид
57А: N,N-Диэтил-5-нитро-6-(2-пропениламино)-3-пиридинкарбоксамид
К раствору 6-хлор-5-нитро-З-пиридинкарбоновой кислоты (4,72 г, 23,30 ммоль) в метаноле (70 мл) при комнатной температуре добавляли аллиламин (5,25 мл, 70,0 ммоль) и смесь перемешивали в течение ночи. Затем реакционную смесь концентрировали в вакууме и остаток растворяли в воде (100 мл). Раствор доводили до рН 3 путем добавления водного раствора 1М HCl. Получающуюся в результате суспензию экстрагировали EtOAc (50 мл). Водную фазу экстрагировали дополнительным количеством EtOAc (2×50 мл). Органические фазы объединяли, сушили с использованием MgSO4 и фильтровали. Раствор концентрировали в вакууме с получением 5-нитро-6-(2-пропениламино)-3-пиридинкарбоновой кислоты, которую использовали без дополнительной очистки.
Этот остаток растворяли в диэтиламине (100 мл). К этому раствору добавляли HATU (9,30 г, 24,47 ммоль). Смесь перемешивали в течение 48 часов при комнатной температуре. Реакционную смесь затем концентрировали в вакууме и остаток растворяли в насыщенном водном растворе NaHCO3 (100 мл) и EtOAc (100 мл). Фазы разделяли и водную фазу экстрагировали дополнительным количеством EtOAc (2×50 мл). Органические фазы объединяли, сушили с использованием MgSO4, фильтровали и концентрировали в вакууме. Остаток очищали, используя флэш-хроматографию на силикагеле ([2% МеОН+1% NH4OH водн.] в СН2Cl2, с получением 4,83 г указанного в заголовке соединения N,N-диэтил-5-нитро-6-(2-пропениламино)-3-пиридинкарбоксамида (выход 74,5% по отношению к 6-хлор-5-нитро-3-пиридинкарбоновой кислоте). 1H-ЯМР (400 МГц, CDCl3): δ 1,26 (m, 6Н), 3,28 (brs, 1Н), 3,47 (m, 4Н), 4,26 (m, 2Н), 5,22 (m, 1Н), 5,30 (m, 1Н), 7,40 (d, J=8,2 Гц, 1Н), 7,70 (d, J=8,2 Гц, 1Н). МС (ЭГДИ) (М+Н)+=279.
57Б: 5-Амино-N,N-диэтил-6-(2-пропениламино)-3-пиридинкарбоксамид
К раствору N,N-диэтил-5-нитро-6-(2-пропениламино)-3-пиридинкарбоксамида (4,83 г, 17,25 ммоль) в ДМФ (50 мл) при комнатной температуре добавляли дигидрат дихлорида олова (8,56 г, 37,95 ммоль). Смесь перемешивали в течение ночи при 80°С. Реакционную смесь доводили до комнатной температуры и концентрировали в вакууме. Остаток растворяли в насыщенном водном растворе NaHCO3 (100 мл) и EtOAc (100 мл). Суспензию фильтровали и фазы разделяли. Водную фазу экстрагировали дополнительным количеством EtOAc (100 мл). Органические фазы объединяли, сушили с использованием MgSO4, фильтровали и концентрировали в вакууме. Остаток очищали, используя флэш-хроматографию на силикагеле ([5% МеОН+1% NH4OH водн.] в CH2Cl2), с получением 2,10 г указанного в заголовке соединения 5-амино-N,N-диэтил-6-(2-пропениламино)-3-пиридинкарбоксамида (выход 49,0% по отношению к N,N-диэтил-5-нитро-6-(2-пропениламино)-3-пиридинкарбоксамиду). 1H-ЯМР (400 МГц, CDCl3): δ 1,20 (t, J=7,2 Гц, 6Н), 3,23 (brs, 1Н), 3,45 (m, 4H), 4,12 (m, 2H), 4,44 (brs, 2H), 5,17 (d, J=12,5 Гц, 1Н), 5,26 (d, J=17,2 Гц, 1Н), 6,04 (m, 1Н), 7,00 (d, J=2,0 Гц, 1Н), 7,83 (d, J=2,0 Гц, 1Н). МС (ЭГДИ) (М+Н)+=2,49.
57В: 5-[[(4-Этоксифенил)ацетил]амино]-N,n-диэтил-6-(2-пропениламино)-3-пиридинкарбоксамид
К раствору 5-амино-N,N-диэтил-6-(2-пропениламино)-3-пиридинкарбоксамида (1,61 г, 6,50 ммоль) в дихлорметане (50 мл) при 0°С добавляли 4-этокси-бензолацетилхлорид (1,35 г, 6,80 ммоль). Смесь перемешивали в течение 3 часов при комнатной температуре. Реакционную смесь затем гасили насыщенным водным раствором NaHCO3 (100 мл) и экстрагировали дихлорметаном (50 мл). Водную фазу экстрагировали дополнительным количеством дихлорметана (2×50 мл). Органические фазы объединяли, сушили с использованием MgSO4, фильтровали и концентрировали в вакууме. Остаток очищали, используя флэш-хроматографию на силикагеле ([2% МеОН + 0,5% NH4OH водн.] в CH2Cl2), с получением 1,69 г указанного в заголовке соединения 5-[[(4-этоксифенил)ацетил]амино]-N,N-диэтил-6-(2-пропениламино)-3-пиридинкарбоксамида (выход 63,3% по отношению к 5-амино-N,N-диэтил-6-(2-пропениламино)-3-пиридинкарбоксамиду). 1Н-ЯМР (400 МГц, CDCl3): δ 1,18 (t, J=7,2 Гц, 6Н), 1,41 (t, J=7,8H 4,3H), 3,42 (brs, 1H), 3,69 (s, 2Н), 4,02 (m, 8Н), 5,13 (m, 2Н), 5,92 (m, 1H), 6,90 (m, 2H), 7,27 (m, 2H), 7,34 (m, 1H), 8,00 (m, 1H). МС (ЭГДИ)(М+H)+.
57Г: 6-Амино-5-[[(4-этоксифенил)ацетил]амино]-N,N-диэтил-3-пиридинкарбоксамид
К дегазированному раствору 5-[[(4-этоксифенил)ацетил]амино]-N,N-диэтил-6-(2-пропениламино)-3-пиридинкарбоксамида (1,04 г, 2,53 ммоль) в дихлорметане (20 мл) при комнатной температуре добавляли тетракиспалладий (117 мг, 0,10 ммоль) с последующим добавлением уксусной кислоты (580 мкл, 10,14 ммоль) и фенилсилана (625 мкл, 5,07 ммоль). Смесь перемешивали в течение 6 часов. Реакционную смесь затем гасили насыщенным водным раствором NaHCO3 (50 мл) и экстрагировали EtOAc (20 мл). Водную фазу экстрагировали дополнительным количеством дихлорметана (2×20 мл). Органические фазы объединяли, сушили с использованием MgSO4, фильтровали и концентрировали в вакууме. Остаток очищали, используя флэш-хроматографию на силикагеле ([3% МеОН+1% NH4OH водн.] в СН2Cl2), с получением 705 мг указанного в заголовке соединения 6-амино-5-[[(4-этоксифенил)ацетил]амино]-N,N-диэтил-3-пиридинкарбоксамида (выход 75,2% по отношению к 5-[[(4-этоксифенил)ацетил]амино]-N,N-диэтил-6-(2-пропениламино)-3-пиридинкарбоксамида). 1H-ЯМР (400 МГц, CDCl3): δ 1,18 (brs, 6Н), 1,43 (t, J=7,0 Гц, 3Н), 3,42 (brs, 4H), 3,69 (s, 2H), 4,04 (q, J=7,0 Гц, 2Н), 4,73 (s, 2Н), 6,91 (d, J=8,6 Гц, 2Н), 7,26 (d, J=8,6 Гц, 2Н), 7,38 (m, 1H), 7,78 (s, 1Н), 7,95 (m, 1H). МС (ЭГДИ) (М+Н)+=372.
57Д: 2-[(4-Этоксифенил)метил]-N,N-диэтил-3-[(5-нитро-2-тиенил)метил]-3H-имидазо[4,5-b]пиридин-6-карбоксамид
К раствору 6-амино-5-[[(4-этоксифенил)ацетил]амино]-N,N-диэтил-3-пиридинкарбоксамида (30 мг, 0,08 ммоль) в дихлорэтане (0,5 мл) и уксусной кислоты (0,5 мл) при комнатной температуре добавляли 5-нитро-2-тиофенкарбоксальдегид (19 мг, 0,12 ммоль). Смесь перемешивали в течение 4,5 часов. Добавляли ВН3×пиридин (8,2 мкл, 0,08 ммоль) и реакционную смесь доводили до 84°С. После перемешивания в течение ночи смесь охлаждали комнатной температуры и добавляли 5-нитро-2-тиофенкарбоксальдегид (19 мг, 0,12 ммоль). Смесь перемешивали в течение 5 часов. Добавляли ВН3·пиридин (8,2 мкл, 0,08 ммоль) и реакционную смесь доводили до 84°С. После перемешивания в течение ночи реакционную смесь гасили водным раствором 1M NaOH (20 мл) и экстрагировали EtOAc (20 мл). Водную фазу экстрагировали дополнительным количеством дихлорметана (2×20 мл). Органические фазы объединяли, сушили с использованием MgSO4, фильтровали и концентрировали в вакууме. Остаток очищали, используя флэш-хроматографию на силикагеле ([3% МеОН+0,5% NH4OH водн.] в СН2Cl2, с получением указанного в заголовке соединения 2-[(4-этоксифенил)метил]-N,N-диэтил-3-[(5-нитро-2-тиенил)метил]-3H-имидазо[4,5-b]пиридин-6-карбоксамида (чистота: более 84% при 215 нм, более 81% при 254 нм, более 68% при 280 нм). 1H-ЯМР (400 МГц, CDCl3): δ 1,23 (m, 6H), 1,40 (t, J=6,8 Гц, 3Н), 3,36 (brs, 2H), 3,60 (brs, 2H), 3,98 (q, J=6,8 Гц, 2H), 4,29 (s, 2H), 4,84 (s, 2H), 6,80 (d, J=8,6 Гц, 2H), 7,07 (d, J=8,6 Гц, 2H), 7,66 (d, J=4,1 Гц, 1H), 7,77 (d, J=4,1 Гц, 1H), 8,03 (d, J=2,0 Гц, 1H), 8,03 (d, J=2,0 Гц, 1H). МС (ЭГДИ) (М+Н)+=494.
Пример 58
3-(Циклопропилметил)-2-[(4-этоксифенил)метил]-N-метил-3Н-имидазо[4,5-b]пиридин-6-амин
58А: N-(Циклопропилметил)-3,5-динитро-2-пиридинамин
К раствору 2-хлор-3,5-динитро-пиридина (1,00 г, 4,91 ммоль) в дихлорметане (10 мл), поддерживаемому при комнатной температуре при помощи водяной бани, по каплям добавляли циклопропилметиламин (852 мкл, 9,83 ммоль). После добавления сразу наблюдали оранжевое окрашивание и обнаруживали осадки. Смесь перемешивали в течение 1 часа. Реакционную смесь гасили водным раствором 1М NaOH (10 мл) и экстрагировали EtOAc (20 мл). Органическую фазу промывали рассолом, сушили с использованием MgSO4, фильтровали и концентрировали в вакууме. Способ позволил получить 1,117 г указанного в заголовке соединения N-(циклопропилметил)-3,5-динитро-2-пиридинамина (выход 96% по отношению к 2-хлор-3,5-динитропиридину) с чистотой более 96% в соответствии с анализом путем HPLC. Остаток использовали без дополнительной очистки. 1Н-ЯМР (400 МГц, CDCl3): δ 0,36 (m, 2H), 0,66 (m, 2H), 1,18 (m, 1H), 3,61 (dd, J=5,3, 7,2 Гц, 2Н), 8,85 (m, 1Н), 9,23 (m, 2Н). МС (ЭГДИ) (М+Н)+=239.
58Б: N-(Циклопропилметил-нитро-3-пиридиндиамин
К раствору N-(циклопропилметил)-3,5-динитро-2-пиридинамина (1,17 г, 4,92 ммоль) в EtOAc (50 мл) при комнатной температуре добавляли Pd/C (394 мг, 0,25 ммоль, содержание палладия 10%). Смесь помещали в аппарат Парра при давлении H2 35 ф./кв. дюйм (241,3 кПа). Смесь перемешивали в течение ночи. Реакционную смесь фильтровали через целит и концентрировали в вакууме. Остаток очищали, используя флэш-хроматографию на силикагеле ([2,5% МеОН+1% NH4OH водн.] в CH2Cl2), с получением 792 мг указанного в заголовке соединения N2-(циклопропилметил-5-нитро-3-пиридиндиамина (выход 77,3% по отношению к N-(циклопропилметил)-3,5-динитро-2-пиридинамину). 1Н-ЯМР (400 МГц, CDCl3): δ 0,31 (m, 2H), 0,61 (m, 2H), 1,14 (m, 1 Н), 3,34 (brs, 2H), 3,42 (dd, J=5,3, 7,2 Гц, 2H), 5,15 (brs, 1 Н), 7,62 (d, J=2,5 Гц, 1H), 8,73 (d, J=2,5 Гц, 2H). МС (ЭГДИ) (М+Н)+=209.
58В: 3-(Циклопропилметил)-2-[(4-этоксифенил)метил]-6-нитро-3H-имидазо[4,5-b]пиридин
К раствору N2-(циклопропилметил-5-нитро-3-пиридиндиамина (792 мг, 3,80 ммоль) в дихлорметане (50 мл) при комнатной температуре добавляли 4-этокси-бензолацетилхлорид (795 мг, 4,00 ммоль). Смесь перемешивали в течение 10 часов. Смесь концентрировали в вакууме и остаток растворяли в дихлорэтане (25 мл) и уксусной кислоте (25 мл). Смесь перемешивали в течение 48 часов при 85°С. Реакционную смесь охлаждали до комнатной температуры и доводили до рН 10 путем добавления водного раствора 1М NaOH. Смесь экстрагировали EtOAc (50 мл). Водную фазу экстрагировали дополнительным количеством EtOAc (2×50 мл). Органические фазы объединяли, сушили с использованием MgSO4, фильтровали и концентрировали в вакууме. Остаток очищали, используя флэш-хроматографию на силикагеле ([2,5% МеОН+0,5% NH4OH водн.] в CH2Cl2), с получением 1,02 г указанного в заголовке соединения 3-(циклопропилметил)-2-[(4-этоксифенил)метил]-6-нитро-3Н-имидазо[4,5-b]пиридина (выход 76,0% по отношению к N2-циклопропилметил-5-нитро-3-пиридиндиамину). 1H-ЯМР (400 МГц, CDCl3): δ 0,44 (m, 2Н), 0,52 (m, 2H), 1,10 (m, 1H), 1,40 (t, J=6,8 Гц, 3Н), 4,01 (q, J=6,8 Гц, 2H), 4,10 (d, J=7,2 Гц, 2H), 4,35 (s, 2H), 6,86 (d, J=8,8 Гц, 2H), 7,18 (d, J=8,8 Гц, 2H), 8,80 (d, J=2,3 Гц, 1H), 9,26 (d, J=2,3 Гц, 1H). МС (ЭГДИ) (М+Н)+=353.
58Г: 3-(Циклопропилметил)-2-[(4-этоксифенил)метил]-3Н-имидазо[4,5-b]пиридин-6-амин
К раствору 3-(циклопропилметил)-2-[(4-этоксифенил)метил]-6-нитро-3H-имидазо[4,5-b]пиридина (1,35 г, 3,84 ммоль) в EtOAc (20 мл) и уксусной кислоте (1 мл) при комнатной температуре добавляли Pd/C (308 мг, содержание палладия 10%). Смесь помещали в аппарат Парра при давлении Н2 35 ф./кв. дюйм (241,3 кПа). Смесь перемешивали в течение 72 часов. Реакционную смесь фильтровали через Целит. Фильтрат растворяли в водном растворе 1М NaOH (25 мл) и EtOAc (50 мл) и фазы разделяли. Водную фазу экстрагировали дополнительным количеством EtOAc (2×50 мл). Органические фазы объединяли, сушили с использованием MgSO4, фильтровали и концентрировали в вакууме. Способ позволил получить 1,18 г указанного в заголовке соединения 3-(циклопропилметил)-2-[(4-этоксифенил)метил]-3H-имидазо[4,5-b]пиридин-6-амина (выход 95% по отношению к 3-(циклопропилметил)-2-[(4-этоксифенил)метил]-6-нитро-3H-имидазо[4,5-b]пиридину), с чистотой более 95% в соответствии с анализом путем HPLC. Остаток использовали без дополнительной очистки. 1H-ЯМР (400 МГц, CDCl3): δ 0,35 (m, 2H), 0,46 (m, 2H), 1,11 (m, 1 Н), 1,39 (t, J=6,8 Гц, 3Н), 3,26 (brs, 2H), 3,98 (m, 4H), 4,25 (s, 2H), 6,83 (d, J=8,8 Гц, 2H), 7,16 (d, J=8,8 Гц, 2H), 7,34 (d, J=2,3 Гц, 1H), 7,87 (d, J=2,3 Гц, 1H). МС (ЭГДИ) (М+Н)+=323.
58Д: Метиловый эфир [3-(циклопропилметил)-2-[(4-этоксифенил)метил]-3Н-имидазо[4,5-b]пиридин-6-ил]карбаминовой кислоты
К раствору 3-(циклопропилметил)-2-[(4-этоксифенил)метил]-3H-имидазо[4,5-b]пиридин-6-амина (1,18 г, 3,66 ммоль) в дихлорметане (20 мл) при 0°С добавляли основание Хунига (869 мкл, 4,99 ммоль) с последующим добавлением метилхлорформиата (326 мкл, 4,22 ммоль). Смесь доводили до комнатной температуры и перемешивали в течение ночи. Реакционную смесь гасили насыщенным водным раствором NH4Cl и экстрагировали дихлорметаном (50 мл).
Водную фазу экстрагировали дополнительным количеством дихлорметана (2×50 мл). Органические фазы объединяли, сушили с использованием MgSO4, фильтровали и концентрировали в вакууме. Остаток очищали, используя флэш-хроматографию на силикагеле ([от 3 до 7% МеОН+1% NH4OH водн.] в CH2Cl2) с получением 1,02 г указанного в заголовке соединения метилового эфира [3-(циклопропилметил)-2-[(4-этоксифенил)метил]-3H-имидазо[4,5-b]пиридин-6-ил]-карбаминовой кислоты (выход 73,3% по отношению к 3-(циклопропилметил)-2-[(4-этоксифенил)метил]-3Н-имидазо[4,5-b]пиридин-6-амину). 1Н-ЯМР (400 МГц, CDCl3): δ 0,38 (m, 2H), 0,45 (m, 2H), 1,11 (m, 1H), 1,39 (t, J=7,0 Гц, 3Н), 3,81 (s, 3H), 4,00 (m, 4H), 4,29 (s, 2H), 6,69 (brs, 1H), 6,84 (d, J=8,6 Гц, 2H), 7,15 (d, J=8,6 Гц, 2H), 8,13 (brs, 1H), 8,26 (brs, 1H). МС (ЭГДИ) (М+Н)+=381.
58Е: 3-(Циклопропилметил)-2-[(4-этоксифенил)метил]-N-метил-3H-имидазо[4,5-b]пиридин-6-амин
К раствору метилового эфира [3-(циклопропилметил)-2-[(4-этоксифенил)метил]-3Н-имидазо[4,5-b]пиридин-6-ил]-карбаминовой кислоты (550 мг, 1,46 ммоль) в дихлорметане (20 мл) при комнатной температуре добавляли 1M HCl в растворе диэтилового эфира (3 мл). Смесь перемешивали в течение 10 минут и концентрировали в вакууме. Остаток растворяли в ТГФ (15 мл) и дизтиловом эфире (30 мл) и охлаждали до 0°С. К раствору добавляли АГЛ (137 мг, 3,61 ммоль). Смесь перемешивали в течение ночи. Затем реакционную смесь охлаждали to -78°C и гасили МеОН (3,20 мл) и водой (3,20 мл). К смеси, которой дали возможность медленно нагреться до комнатной температуры, добавляли Na2SO4. Смесь фильтровали через Целит и концентрировали в вакууме. Остаток очищали, используя флэш-хроматографию на силикагеле ([3,5% МеОН+0,5% NH4OH водн.] в СН2Cl2), с получением 445 мг указанного в заголовке соединения 3-(циклопропилметил)-2-[(4-этоксифенил)метил]-N-метил-3Н-имидазо[4,5-b]пиридин-6-амина (выход 90,6% по отношению к метиловому эфиру [3-(циклопропилметил)-2-[(4-этоксифенил)метил]-3H-имидазо[4,5-6]пиридин-6-ил]-карбаминовой кислоты). 1H-ЯМР (400 МГц, CDOCl3): δ 0,35 (m, 2H), 0,46 (m, 2H), 1,11 (m, 1H), 1,39 (t, J=7,0 Гц, 3Н), 2,90 (s, 3Н), 3,98 (m, 4H), 4,25 (s, 2H), 6,83 (d, J=8,8 Гц, 2H), 7,15 (d, J=8,8 Гц, 2H), 7,23 (d, J=2,5 Гц, 1Н), 7,83 (d, J=2,5 Гц, 1Н). МС (ЭГДИ) (М+Н)+:337.
Пример 59
N-[3-(Циклопропилметил)-2-[(4-этоксифенил)метил]-3Н-имидазо[4,5-6]пиридин-6-ил]-N,3-диметил-бутанамид
К раствору 3-(циклопропилметил)-2-[(4-этоксифенил)метил]-N-метил-3H-имидазо[4,5-b]пиридин-6-амина (143,6 мг, 0,43 ммоль) в дихлорэтане (17 мл) при комнатной температуре добавляли триэтиламин (300 мкл, 2,15 ммоль) с последующим добавлением изовалерилхлорида (156 мкл, 1,28 ммоль). Смесь перемешивали в течение ночи. Реакционную смесь гасили насыщенным водным раствором Na2СО3 (10 мл) и экстрагировали EtOAc (25 мл). Водную фазу экстрагировали дополнительным количеством EtOAc (25 мл). Органические фазы объединяли, сушили с использованием MgSO4, фильтровали и концентрировали в вакууме. Остаток очищали, используя препаративную HPLC (колонка С-18, от 10 до 70% [0,1% ТФУ в растворе AcCN] в 0,1% водном растворе ТФУ), с получением 95,5 мг соли с ТФУ указанного в заголовке соединения N-[3-(циклопропилметил)-2-[(4-этоксифенил)метил]-3H-имидазо[4,5-b]пиридин-6-ил]-N,3-диметил-бутанамида (выход 41,5% по отношению к 3-(циклопропилметил)-2-[(4-этоксифенил)метил]-N-метил-3H-имидазо[4,5-b]пиридин-6-амина). 1Н-ЯМР (400 МГц, d6-MeOD): δ 0,45 (m, 4H), 0,82 (m, 6H), 1,17 (m, 1H), 1,36 (t, J=7,0 Гц, 3Н), 2,00 (m, 2H), 3,32 (s, 3Н), 4,00 (t, J=7,0 Гц, 2H), 4,22 (d, J=7,2 Гц, 2H), 4,44 (s, 2H), 6,90 (d, J=8,6 Гц, 2H), 7,23 (d, J=8,6 Гц, 2H), 7,97 (d, J=2,15 Гц, 1H), 8,33 (brs, 1H). МС (ЭГДИ) (М+Н)+:421.
Пример 60
N-[3-(Циклопропилметил)-2-[(4-этоксифенил)метил]-3Н-имидазо[4,5-b]пиридин-6-ил]-N-метил-N'-(1-метилэтил)-мочевина
К раствору 3-(циклопропилметил)-2-[(4-этоксифенил)метил]-N-метил-3H-имидазо[4,5-b]пиридин-6-амина (163,4 мг, 0,49 ммоль) в ДМФ (20 мл) при комнатной температуре добавляли изопропилизоцианат (48 мкл, 0,49 ммоль). Смесь перемешивали в течение 72 часов при 50°С. Анализ реакционной смеси путем HPLC указал на присутствие исходного вещества. Поэтому добавляли дополнительное количество изопропилизоцианата (144 мкл, 1,47 ммоль). Получающуюся в результате смесь перемешивали в течение 12 часов при 50°С. Реакционную смесь растворяли в воде (150 мл) и EtOAc (150 мл). Водную фазу экстрагировали дополнительным количеством EtOAc (100 мл). Органические фазы объединяли, сушили с использованием MgSO4, фильтровали и концентрировали в вакууме. Остаток очищали, используя препаративную HPLC (колонка С-18, от 10 до 70% [0,1% ТФУ в растворе AcCN] в 0,1% водном растворе ТФУ), с получением 92 мг соли с ТФУ указанного в заголовке соединения N-[3-(циклопропилметил)-2-[(4-этоксифенил)метил]-3H-имидазо[4,5-b]пиридин-6-ил]-N-метил-N'-(1-метилэтил)-мочевины (выход 35,0% по отношению к 3-(циклопропилметил)-2-[(4-этоксифенил)метил]-N-метил-3Н-имидазо[4,5-b]пиридин-6-амину). 1Н-ЯМР (400 МГц, d6-MeOD): δ 0,46 (m, 4H), 1,09 (d, J=6,6 Гц, 6Н), 1,18 (m, 1Н), 1,36 (t, J=7,0 Гц, 3Н), 3,30 (s, 3Н), 3,92 (m, 1Н), 4,00 (t, J=7,0 Гц, 2Н), 4,22 (d, J=7,2 Гц, 2Н), 4,44 (s, 2Н), 6,90 (d, J=8,6 Гц, 2Н), 7,22 (d, J=8,6 Гц, 2Н), 7,93 (d, J=2,2 Гц, 1Н), 8,34 (d, J=2,2 Гц, 1Н). МС (ЭГДИ) (М+Н)+=422.
Пример 61
N-[3-(Циклопропилметил)-2-[(4-этоксифенил)метил]-3Н-имидазо[4,5-b]пиридин-6-ил]-N-метил-бензолсульфонамид
К раствору 3-(циклопропилметил)-2-[(4-этоксифенил)метил]-N-метил-3H-имидазо[4,5-b]пиридин-6-амина (237,0 мг, 0,70 ммоль) в ацетонитриле (5 мл) при комнатной температуре добавляли триэтиламин (196 мкл, 1,41 ммоль) с последующим добавлением бензолсульфонилхлорида (156 мкл, 0,92 ммоль). Смесь перемешивали в течение ночи. Реакционную смесь концентрировали в вакууме. Остаток растворяли в EtOAc (10 мл) и насыщенном водном растворе Na2СО3 (10 мл). Фазы разделяли и водную фазу экстрагировали дополнительным количеством EtOAc (10 мл). Органические фазы объединяли, сушили с использованием MgSO4, фильтровали и концентрировали в вакууме. Остаток очищали, используя препаративную HPLC (колонка С-18, от 20 до 80% [0,1% АсОН в растворе AcCN] в 0,1% водном растворе АсОН), с получением 63 мг соли с АсОН указанного в заголовке соединения N-[3-(циклопропилметил)-2-[(4-этоксифенил)метил]-3H-имидазо[4,5-b]пиридин-6-ил]-N-метил-бензолсульфонамида (выход 17% по отношению к 3-(циклопропилметил)-2-[(4-этоксифенил)метил]-N-метил-3Н-имидазо[4,5-b]пиридин-6-амина). 1H-ЯМР (400 МГц, d6-MeOD): δ 0,37 (m, 2H), 0,42 (m, 2Н), 1,10 (m, 1H), 1,35 (t, J=7,0 Гц, 3Н), 3.31 (s, 3Н), 3,99 (t, J=7,0 Гц, 2H), 4,10 (d, J=7,2 Гц, 2H), 4,32 (s, 2H), 6,86 (d, J=8,6 Гц, 2H), 7,17 (d, J=8,6 Гц, 2H), 7,55 (m, 2H), 7,57 (m, 2H), 7,59 (m, 1H), 7,70 (m, 1H),8,11 (d,J=2,2 Гц, 1H). МС (ЭГДИ) (М+Н)+:477.
Пример 62
N-[3-(Циклопропилметил)-2-[(4-этоксифенил)метил]-3Н-имидазо[4,5-b]пиридин-6-ил]-N-метил-2-тиофенсульфонамид
К раствору 3-(циклопропилметил)-2-[(4-этоксифенил)метил]-N-метил-3H-имидазо[4,5-b]пиридин-6-амина (250,0 мг, 0,74 ммоль) в ацетонитриле (5 мл) при комнатной температуре добавляли триэтиламин (207 мкл, 1,49 ммоль) с последующим добавлением 2-тиофенсульфонилхлорида (176 мкл, 0,96 ммоль). Смесь перемешивали в течение ночи. Реакционную смесь концентрировали в вакууме. Остаток растворяли в EtOAc (10 мл) и насыщенном водном растворе Na2СО3 (10 мл). Фазы разделяли и водную фазу экстрагировали дополнительным количеством EtOAc (10 мл). Органические фазы объединяли, сушили с использованием MgSO4, фильтровали и концентрировали в вакууме. Остаток очищали, используя препративную HPLC (колонка С-18, от 20 до 80% [0,1% АсОН в растворе AcCN] в 0,1% водном растворе АсОН), с получением 72 мг соли с АсОН указанного в заголовке соединения N-[3-(циклопропилметил)-2-[(4-этоксифенил)метил]-3H-имидазо[4,5-b]пиридин-6-ил]-N-метил-2-тиофенсульфонамида (выход 18% по отношению к 3-(циклопропилметил)-2-[(4-этоксифенил)метил]-N-метил-3H-имидазо[4,5-b]пиридин-6-амину). 1-ЯМР (400 МГц, d6-MeOD): δ 0,37 (m, 2H), 0,43 (m, 2H), 1,10 (m, 1H), 1,35 (t, J=7,0 Гц, 3Н), 3.32 (s, 3Н), 3,99 (t, J=7,0 Гц, 2H), 4,10 (d, J=7,2 Гц, 2H), 4,32 (s, 2H), 6,86 (d, J=8,8 Гц, 2Н), 7,17 (d, J=8,8 Гц, 2Н), 7,20 (m, 1H), 7,44 (d, J=5,3 Гц, 1Н), 7,61 (d, J=2,3H 4,1H), 7,85 (d, J= 5,1 Гц, 1H), 8,15 (d, J=2,3 Гц, 1H). МС (ЭГДИ) (М+Н)+=483.
Другие соединения по изобретению
Пример 110
N-[1-(Циклопропилметил)-2-[(4-этоксифенил)метил]-1Н-бензимидазол-5-ил]-N-метил-1-пиперидинкарбоксамид
В раствор 1-(циклопропилметил)-2-[1-(4-этоксифенил)метил]-N-метил-1H-бензимидазол-5-амина (67,2 мг, 0,2 ммоль), который получали в соответствии со способом, описанным в примере 51АА), ДИПЭА (51,7 мг, 0,4 ммоль) и ДМАФ (5 мг) в MeCN (8 мл) добавляли пиперидинкарбонилхлорид (44,3 мг, 0,3 ммоль) при комнатной температуре. Реакционную смесь нагревали в течение 24 ч при температуре дефлегмации, гасили Н2O (50 мл) и экстрагировали EtOAc (4×20 мл). Объединенные органические фазы промывали водным раствором NaCl и сушили над Na2SO4. После фильтрации и выпаривания растворителя остаток очищали путем MPLC, используя EtOAc на силикагеле, с получением 75,2 мг (84%) бесцветного сиропа, который превращали в соль с ТФУ в виде белого твердого вещества. 1Н-ЯМР (CD3OD): δ 0,51 (m, 2H), 0,66 (m, 2Н), 1,28 (m, 1H), 1,41 (m, 7H), 1,55 (m, 2H), 3,28 (s, 3Н), 3,34 (m, 4H), 4,07 (q, J=7,1 Гц, 2H), 4,39 (d, J=7,2 Гц, 2H), 4,57 (s, 2H), 7,01 (m, 2H), 7,30 (m, 2H), 7,39 (dd, J=9,0, 2,1 Гц, 1H), 7,44 (d, J=2,0 Гц, 1H), 7,90 (d, J=9,0 Гц, 1H). МС (ЭГДИ) (M+H)+=447,36. Аналитически рассчитано для С27Н34N4O2+0,90 ТФУ+0,60 Н2O: С, 61,77; Н, 6,50; N, 10,00. Обнаружено: С, 61,90; Н, 6,65; N, 9,79.
Пример 111
N-[1-(Циклопропилметил)-2-[(4-этоксифенил)метил]-1Н-бензимидазол-5-ил]-N-метил-1-пирролидинкарбоксамид
В раствор 1-(циклопропилметил)-2-[1-(4-этоксифенил)метил]-N-метил-1Н-бензимидазол-5-амина (67,2 мг, 0,2 ммоль), который получали в соответствии со способом, описанном в примере 51АА), ДИПЭА (51,7 мг, 0,4 ммоль) и ДМАФ (5 мг) в MeCN (8 мл) добавляли пирролидинкарбонилхлорид (40,1 мг, 0,3 ммоль) при комнатной температуре. Реакционную смесь нагревали в течение 24 ч при температуре дефлегмации, гасили Н2O (50 мл) и экстрагировали EtOAc (4×20 мл). Объединенные органические фазы промывали водным раствором NaCl и сушили над Na2SO4. После фильтрации и выпаривания растворителя остаток очищали путем MPLC, используя EtOAc на силикагеле, с получением 79,4 мг (92%) бесцветного сиропа, который превращали в соль с ТФУ, представляющую собой белое твердое вещество. 1H-ЯМР (CD3OD): δ 0,50 (m, 2Н), 0,65 (m, 2H), 1,29 (m, 1H), 1,41 (t, J=7,2 Гц, 3Н), 1,75 (m, 4H), 3,28 (s, 3H), 3,34 (m, 4H), 4,07 (q, J=7,0 Гц, 2H), 4,39 (d, J=7,2 Гц, 2H), 4,58 (s, 2H), 7,01 (m, 2H), 7,31 (m, 2H), 7,43 (dd, J=9,0, 2,1 Гц, 1H), 7,48 (m, 1H), 7,91 (d, J=8,8 Гц, 1H). MC (ЭГДИ) (M+H)+=433,34. Аналитически рассчитано для C26H32N4O2+0,90 ТФУ+0,20 Н2O: С, 61,97; Н, 6,23; N, 10,40. Обнаружено: С, 62,01; Н, 6,20; N, 10,04.
Пример фармацевтической композиции
Таблетки
Таблетки готовят следующим образом: соединение по изобретению, лактозу, кукурузный крахмал, две трети мелкокристаллической целлюлозы и половину стеарата магния смешивают и гранулируют. К гранулам добавляют оставшиеся микрокристаллическую целлюлозу и стеарат магния и все вместе прессуют в таблетки в таблеточной машине.
| название | год | авторы | номер документа |
|---|---|---|---|
| ПРОИЗВОДНЫЕ БЕНЗИМИДАЗОЛА, СОДЕРЖАЩИЕ ИХ КОМПОЗИЦИИ, ИХ ПОЛУЧЕНИЕ И ПРИМЕНЕНИЕ | 2004 |
|
RU2346938C2 |
| (АЗА)ИНДОЛ-, БЕНЗОТИОФЕН- И БЕНЗОФУРАН-3-СУЛЬФОНАМИДЫ | 2017 |
|
RU2767904C2 |
| ХИНОЛИНОВЫЕ ПРОИЗВОДНЫЕ В КАЧЕСТВЕ ИНГИБИТОРОВ SMO | 2015 |
|
RU2695815C2 |
| Композиции для лечения гипертензии и/или фиброза | 2014 |
|
RU2661878C2 |
| ПРОИЗВОДНЫЕ ПИРИДАЗИНОН-АМИДОВ | 2014 |
|
RU2666899C2 |
| ГЕТЕРОЦИКЛИЧЕСКИЕ СОЕДИНЕНИЯ, ИХ ПРИМЕНЕНИЕ И ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ ДЛЯ ЛЕЧЕНИЯ СОСТОЯНИЙ, ОПОСРЕДОВАННЫХ СХСR4 И CCR5 | 2001 |
|
RU2277092C2 |
| ГЕТЕРОЦИКЛИЧЕСКИЕ СОЕДИНЕНИЯ С ПОВЫШЕННОЙ ЭФФЕКТИВНОСТЬЮ, СВЯЗЫВАЮЩИЕСЯ С РЕЦЕПТОРОМ ХЕМОКИНА | 2002 |
|
RU2325387C2 |
| МАКРОЦИКЛИЧЕСКИЕ АНТИБИОТИКИ ШИРОКОГО СПЕКТРА ДЕЙСТВИЯ | 2016 |
|
RU2766543C2 |
| ПИРИДИНИЛ- И ПИРАЗИНИЛ(АЗА)ИНДОЛСУЛЬФОНАМИДЫ | 2019 |
|
RU2799321C2 |
| ПРОИЗВОДНЫЕ ТЕТРАГИДРОХИНОЛИНА, ДЕМОНСТРИРУЮЩИЕ ЗАЩИТНОЕ ОТ ВИЧ-ИНФЕКЦИИ ДЕЙСТВИЕ | 2005 |
|
RU2350604C2 |
Изобретение относится к новым соединениям формулы I или его фармацевтически приемлемым солям, которые обладают свойствами агонистов рецептора СВ2 и могут быть использованы для приготовления лекарственных средств, обладающих аналгезирующим действием, в частности для лечении боли. В соединении формулы I
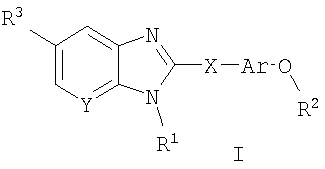
R1 выбран из группы, состоящей из -(С1-С6)алкила, -(С3-С6)циклоалкила, -(С3-С6)циклоалкил(С1-С6)алкила, -(С2-С6)алкенила, R4 2N(C1-C6)алкила-, R4 2NC(=O)(C1-С6)алкила-, R4O(C1-C6)алкила-, R4OC(=O)(C1-C6)алкила-, R4С(=O)(С1-С6)алкила-, R4C(=O)NR4(C1-C6)алкила-, R4 2NSO2(C1-C6)алкила-, R4 2NC(=O)NR4(C1-C6)алкила-, радикалов фенил(С1-С6)алкил, гетероарил(С1-С6)алкил, гетероциклоалкил(С1-С6)алкил, бициклический гетероарил(С1-С6)алкил; Ar представляет собой фенил или пиридил; R2 представляет собой -(С1-С6)алкил, незамещенный или замещенный по 1-6 атомам углерода одним или более чем одним заместителем фтора, или (С3-С6)циклоалкил; R3 выбран из группы, состоящей из:
 ,
,  ,
,  ,
,
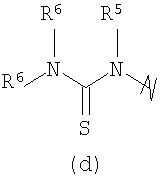 ,
, 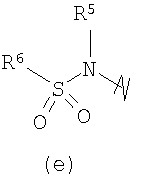 ,
, 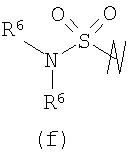 ,
, 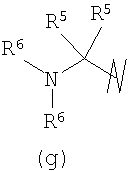 ,
,
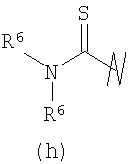 ,
, 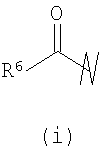 ,
,
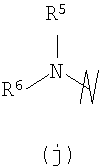 и
и 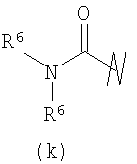 ;
;
R4 представляет собой группировку, независимо выбранную из группы, состоящей из -H, -(С1-С6)алкила, -(С2-С6)алкенила; группировки R5 независимо выбраны из группы, состоящей из -Н, -(С1-С6)алкила, -(С2-С6)алкенила; группировки R6 независимо выбраны из группы, состоящей из: -Н, -(С1-С6)алкила, -(С3-С6)циклоалкила, -(С2-С6)алкенила, гетероциклила, радикала гетероциклил(С1-С3)алкил, фенила, радикала фенил(С1-С3)алкил, гетероарила, радикалов гетероарил(С1-С3)алкил, бициклический гетероарил и бициклический гетероарил(С1-С3)алкил; R5 и R6 могут объединяться с образованием 5-7-членного гетероцикла; Х выбран из группы, состоящей из -C(R5)2-, -NR5-, С(=O)-, -СН2-СН2-, -СН=СН- и -C(R)(R')-, где R и R' представляют собой (С1-С6)алкил, OR'' или Н, и R'' представляет собой Н; и Y представляет собой СН или азот; при этом гетероциклил или гетероциклоалкил представляет собой насыщенное 5-6-членное кольцо, содержащее от 1 до 2 гетероатомов, выбранных из N и О, которое является незамещенным или замещено -(С1-С6)алкилом; гетероарил представляет собой гетероароматическое 5-6-членное кольцо, содержащее от 1 до 2 гетероатомов, выбранных из N, О и S, которое является незамещенным или замещено группировкой, выбранной из группы, состоящей из -(С1-С6)алкила, нитро, галогена и ацетоксиметила; бициклический гетероарил представляет собой 5-6-членное азотсодержащее кольцо, конденсированное с бензольным кольцом. Изобретение также относится к фармацевтической композиции и способу лечения боли. 4 н. и 5 з.п. ф-лы, 3 табл.
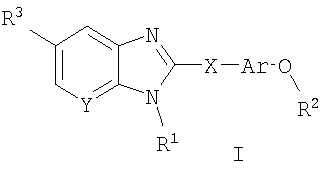
где R1 выбран из группы, состоящей из -(С1-С6)алкила, -(С3-С6)циклоалкила, -(С3-С6)циклоалкил(С1-С6)алкила, -(С2-С6)алкенила, R4 2N(C1-C6)алкила-, R4 2NC(=0)(C1-C6)алкила-, R4O(C1-C6)алкила-, R4OC(=O)(C1-C6)алкила-, R4C(=O)(C1-C6)алкила-, R4C(=O)NR4(C1-С6)алкила-, R4 2NSO2(C1-C6)алкила-, R4 2NC(=O)NR4(C1-C6)алкила-, радикалов фенил(С1-С6)алкил, гетероарил(С1-С6)алкил, гетероциклоалкил(С1-С6)алкил, бициклический гетероарил(С1-С6)алкил;
Ar представляет собой фенил или пиридил;
R2 представляет собой -(С1-С6)алкил, незамещенный или замещенный по 1-6 атомам углерода одним или более чем одним заместителем фтора, или (С3-С6)циклоалкил;
R3 выбран из группы, состоящей из
 ,
,  ,
,  ,
,  ,
,
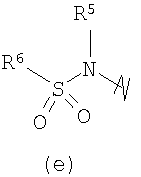 ,
, 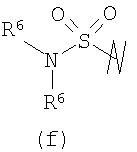 ,
, 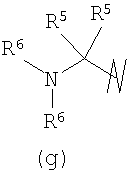 ,
, 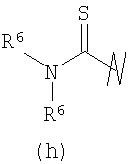 ,
, 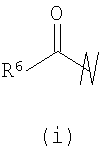 ,
,
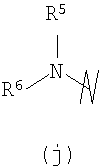 и
и  ;
;
R4 представляет собой группировку, независимо выбранную из группы, состоящей из -Н, -(С1-С6)алкила, -(С2-С6)алкенила;
группировки R5 независимо выбраны из группы, состоящей из -Н, -(С1-С6)алкила, -(С2-С6)алкенила;
группировки R6 независимо выбраны из группы, состоящей из -Н, -(С1-С6)алкила, -(С3-С6)циклоалкила, -(С2-С6)алкенила, гетероциклила, радикала гетероциклил(С1-С3)алкил, фенила, радикала фенил(С1-С3)алкил, гетероарила, радикалов гетероарил(С1-С3)алкил, бициклический гетероарил и бициклический гетероарил(С1-С3)алкил;
R5 и R6 могут объединяться с образованием 5-7-членного гетероцикла;
Х выбран из группы, состоящей из -C(R5)2-, -NR5-, С(=O)-, -СН2-СН2-,
-СН=СН- и -C(R)(R')-, где R и R' представляют собой (С1-С6)алкил, OR'' или Н, и R'' представляет собой Н; и
Y представляет собой CH или азот;
где, если R1 представляет собой R4 2N(С1-С6)алкил-, где оба R4 представляют собой -(C1-С6)алкил, то R3 не представляет собой ацетил, -NH2 или ацетамидо;
где гетероциклил или гетероциклоалкил представляет собой насыщенное 5-6-членное кольцо, содержащее от 1 до 2 гетероатомов, выбранных из N и О, которое является незамещенным или замещено -(С1-С6)алкилом;
где гетероарил представляет собой гетероароматическое 5-6-членное кольцо, содержащее от 1 до 2 гетероатомов, выбранных из N, О и S, которое является незамещенным или замещено группировкой, выбранной из группы, состоящей из -(С1-С6)алкила, нитро, галогена и ацетоксиметила; и
где бициклический гетероарил представляет собой 5-6-членное азотсодержащее кольцо, конденсированное с бензольным кольцом;
при условии, что соединение не является 1-[2-(N,N-диэтиламино)этил]-2-(п-этоксибензил)-5-[N-(трет-бутоксикарбонил)амино]бензимидазолом.
где R1 выбран из группы, состоящей из -(С1-С6)алкила, -(С2-С6)алкенила, радикала фенил(С1-С6)алкил, R4 2N(C1-C6)алкила-, R4O(C1-C6)алкила-, радикалов -гетероциклоалкил(С1-С6)алкил и гетероарил(С1-С6)алкил;
где гетероарильные группировки R1 являются незамещенными или замещены -(C1-С6)алкилом или галогеном;
R2 выбран из группы, состоящей из -СН3, -CH2СН3, -СН(СН3)2 и CF3;
R3 выбран из группы, состоящей из
, , , ,
, и ;
Ar представляет собой фенил или пиридил;
Х выбран из группы, состоящей из -CH2-, -СН(СН3)-, -СН(ОН)-, -NH-, -CH2CH2-, -С(=O)-;
Х расположен на кольце Ar в положении 1,4 по отношению к группе -O-R2;
R4 независимо выбран из группы, состоящей из -Н и -(С1-С6)алкила;
R5 независимо выбран из группы, состоящей из -Н, -(С1-С6)алкила и -(С2-С6)алкенила; и
R6 независимо выбран из группы, состоящей из -Н, -(С1-С6)алкила, -(С2-С6)алкенила и гетероарила;
где указанный гетероарил является незамещенным или замещен -(С1-С6)алкилом.
где R1 выбран из группы, состоящей из циклопропилметила, аллила, изопентила, метоксиэтила, диметиламиноэтила, 4-пиридилметила, 2-пиридилметила, 1-морфолиноэтила, циклогексилметила, 2-пирролидилметила, N-метил-2-пирролидилметила, N-метил-2-пиперидилметила, 3-тиенилметила, 2-тетрагидрофуранилметила, (2-нитротиофен-5-ил)метила и (5-(ацетоксиметил)-2-фуранил)метила;
R2 выбран из группы, состоящей из -СН3, -CH2CH3, -СН(СН3)2 и CF3;
R4 представляет собой -(С1-С6)алкил;
R5 выбран из группы, состоящей из -Н, -СН3, -CH2CH3, -СН=СН2 и -CH2-CH=CH2;
R6 выбран из группы, состоящей из -СН3, -СН2СН3, -СН2-СН=СН2, -СН2-СН2-СН=СН2, -СН2СН(СН3)2 и 5-метил-3-изоксазола;
Ar представляет собой фенил или пиридинил;
Х выбран из группы, состоящей из -СН2-, -СН2СН2-, -СН(СН3)-, СН(ОН)-, -NH-, и -С(=O)-; и
Х расположен на кольце Ar в положении 1,4 относительно группы -O-R2.
где R2 представляет собой -СН2СН3;
Ar представляет собой незамещенный фенил или пиридил;
Х выбран из группы, состоящей из -СН2-, -СН2СН2-, -СН(СН3)-, -СН(ОН)-, -NH-;
Х расположен на кольце Ar в положении 1,4 относительно группы -O-R2;и
R4 представляет собой метил.
| GRELLA GIUSEPPE et al | |||
| Переносная печь для варки пищи и отопления в окопах, походных помещениях и т.п. | 1921 |
|
SU3A1 |
| IGNACZAK WOJCIECH et al | |||
| Печь для непрерывного получения сернистого натрия | 1921 |
|
SU1A1 |
| US 5620978 A, 15.04.1997 | |||
| Прибор для очистки паром от сажи дымогарных трубок в паровозных котлах | 1913 |
|
SU95A1 |
| CAI SUI XIONG et al. | |||

