Объектом настоящего изобретения являются низкомолекулярные производные пептидов, которые способны действовать как ингибиторы взаимодействия между ламинином и нидогеном (ламинин/нидоген взаимодействие), способ их получения, приготовленные на их основе фармацевтические композиции и их использование для изготовления фармацевтических препаратов и для идентификации ингибиторов взаимодействия ламинина/нидогена.
Ассоциация ламинина (гликопротеин размером 80 кДа) и нидогена (гликопротеин размером 1-60 кДа) рассматривается как важнейший биомолекулярный механизм, участвующий в синтезе и стабилизации базальных мембран (Mayer, U. и Timpl, R., 1994, Extracellular Matrix Assembly and Structure (P.D. Yurchenco, D. Birk и R.P. Mecham, Ed.), 389-416, Academic Press, Orlando, FL). Способность нидогена образовывать трехкомпонентные комплексы со всеми основными ингредиентами базальной мембраны, такими как, например, изоформы, содержащие γ1-ламинин (информацию по номенклатуре см.: Burgeson, R.E., Chiquet, M., Deutzmann, R., Ekblom, P., Engel, J., Kleinmann, H., Martin, G.R., Meneguzzi, G., Paulsson, M., Sanes, J., Timpl, R., Tryggvasson, K., Yamada, Y., Yurchenco, P.D., 1994, Matrix Biology, 14, 209-211), коллаген IV, перлекан и фибулин, а также ассоциированные структуры, образованные на основе каждой из них, означает, что он выполняет функции линкера, который соединяет, пространственно организует и стабилизирует независимо действующие макромолекулы (Fox, J.W., Mayer, U., Nischt, R., Aumailley, M., Reinhardt, D., Wiedemann, H., Mann, K., Timpl, R., Krieg, Т., Engel, J. и Chu, M.-L., 1991, EMBO J., 10, 3137-3146).
Базальные мембраны представляют собой высокоспециализированные внеклеточные структуры, с которыми связывают реализацию важных функций в контроле клеточной и тканевой активности, структуры тканей, клеточного роста, клеточной трансформации, миграции клеток и тканеспецифичной экспрессии генов (Adams, J.C. и Watt, F.M., 1993, Development, 117, 1183-1198). Эксперименты, проведенные с использованием моноклональных антител против ламинина, предоставили очевидные доказательства центральной роли взаимодействия ламинина/нидогена в процессе синтеза функционирующей базальной мембраны. Указанные антитела получают при иммунизации кроликов ламинином Р1 или полученными рекомбинантными методами нидогенсвязывающим доменом ламинина (γ1 III 3-5). После концентрирования антител с помощью аффинной хроматографии на ламинине Р1 или ламинине γ1 III 3-5 матрицы демонстрируют в тестах на ингибирование полное ингибирование ассоциации типа ламинин/нидоген. Такой эффект основан на стерической блокаде антителами доступа нидогена к ламинину, регионы связывания которых находятся в непосредственной близости от нидоген-связывающей последовательности ламинина (Mayer, U., Nischt, R., Е., Mann, К., Fukuda, К., Gerl, M., Yamada, Y., Timpl, R., 1993, EMBO J., 12, 1879-1885).
В культурах эмбриональных органов указанные антитела ингибируют развитие почечных канальцев, образование легочных альвеол, а также морфогенез слюнных желез у эмбрионов. Все три указанные модели относятся к тем программам онтогенеза, для которых имеет существенное значение беспрепятственный синтез базальной мембраны (Ekblom, P., Ekblom, M., Fecker, L., Klein, G., Zhang, H.-Y., Kadoya, Y., Chu, M.-L., Mayer, U., Timpl, R., 1994, Development, 120, 2003-2014).
Антитела, действующие против γ1 цепи последовательности ламинина в регионе, ответственном за связывание с нидогеном, также способны ингибировать ассоциацию ламинин/нидоген. Однако указанное ингибирование носит конкурентный характер в отличие от действия указанных выше антител против ламинина, поскольку в данном случае они конкурируют непосредственно с нидогеном за сайт связывания на ламинине (WO 98/31709).
Моноклональное антитело из подкласса IgM (против ламинина P1 A6/2/4 - DSM ACC2327, см. WO 98/31709) ингибирует in vitro взаимодействие ламинина/нидогена со значением ИК50, равным 30 нМ. Как и описанный выше препарат поликлональных антител против ламинина, оно препятствует морфогенезу эмбриональных слюнных желез в культуре органов. Этот факт подчеркивает специфичность взаимодействия ламинина/нидогена и важность LE-4 модуля, а также идентифицированной последовательности региона в домене ламинина γ1 III 4 в осуществлении указанного взаимодействия.
Нидоген-связывающий домен ламинина был вполне определенно идентифицирован и охарактеризован с точки зрения его локализации, первичной последовательности и пространственной структуры (проведен рентгеноструктурный анализ кристаллической решетки и ЯМР анализ первичной структуры) (Gerl, M., Mann, К., Aumailley, M., Timpl, R., 1991, Eur. J. Biochem., 202, 167-174; Mayer, U., Nischt, R., Pöschl, E., Mann, K., Fukuda, K., Gerl, M., Yamada, Y., Timpl, R., 1993, EMBO J., 12, 1879-1885; Baumgartner, R., Czisch, M., Mayer, U., Pöschl, E., Huber, R., Timpl, R., Holak, T.A., 1996, J. Mol. Biol., 257, 658-668; Stetefeld, J., Mayer, U., Timpl, R., Huber, R., 1996, J. Mol. Biol., 257, 644-657). Указанный домен расположен в LE-модуле (эпидермальном ростоподобном факторе ламининового типа) короткого плеча γ1 цепи ламинина в домене γ1 III 4. LE-модули представляют собой структурный мотив, состоящий из 50-60 аминокислот с характерной сложной складчатой структурой, аналогичной ЭФР (эпидермального фактора роста), с 4 дисульфидными мостиками (Bairoch, A., 1995, Nomenclature of extracellular domains. The SWISS-PROT Protein sequence data bank release, 310; Engel, J., 1989, FEBS Letters, 251, 1-7).
Высокая аффиность по связыванию нидогена с комплементарным ламининовым доменом была обнаружена для ламинина Р1 из EHS опухоли мыши, для ламинина 2 и ламинина 4 из человеческой плаценты и для ламинина дрозофилы. Причина такого межвидового перекрывания специфичности связывания кроется в чрезвычайно высокой идентичности последовательностей γ1 III 4 домена в отношении исследованных видов. Она составляет 97% для последовательностей человека и мыши, 61% - для мыши и дрозофилы и, как ни странно, 51% - между мышью и Caenorhabditis elegans, относительно всего домена (Pikkarinen, Т., Kallunki, Т., Tryggvasson, К., 1987, J. Biol. Chem., 263, 6751-6758; Chi, H.-C., Hui, C.-F., 1989, J. Biol. Chem., 264, 1543-1550; Wilson, R. et al., 1994, Nature, 368, 32-38; Pöschl, E., Mayer, U., Stetefeld, J., Baumgartner, R., Holak, T.A., Huber, R., Timpl, R., 1996, EMBO J., 15, 5154-5159).
Исследования выявили не только наличие зависимости процесса связывания нидогена от интактной трехмерной структуры, но также привели к идентификации четко установленных последовательностей регионов, расположенных в стабилизированных с помощью S-S связей петлях а и с домена γ1 III 4. При этом были определены пять незаменимых аминокислот, четыре из которых расположены внутри участка петли а, состоящего из 7 аминокислот, и одна из них - тирозин - в боковой цепи петли с (Mann, К., Deutzmann, R., Timpl, R., 1988, Eur. J. Biochem., 178, 71-80). Синтетические пептиды, которые могут быть получены на основе соответствующих регионов γ1 III 4 домена и которые способны ингибировать связывание ламинина/нидогена в специфических тестах на связывание, раскрыты в патенте Фокса и Тимпла (J.W. Fox and R. Timpl, патент США No. 5493008).
По всей видимости, для достижения высокой аффиности связывания с сайтом связывания нидогена на молекуле ламинина требуется взаимодействие с тирозином или гистидином на петле (петле с), соседней с действующей связывающей последовательностью. Указанное взаимодействие с ароматическими аминокислотами рассматривается как предварительное условие ингибирования в диапазоне значений ИК50<500 нМ, с учетом размера γ1 III 3-5, равного 3 Да, и на основе известного характера связи структура-функция, который описан в патенте США No. 5493008. Однако до сих пор остается неясным вопрос, взаимодействует ли петля с непосредственно с нидогеном или она лишь участвует в стабилизации подходящей пространственной структуры NIDPNAV последовательности соответствующего региона (Pöschl, Е., Fox, J.W., Block, D., Mayer, U., Timpl, R., 1994, EMBO J., 13, 3741-3747; Baumgartner. R., Czisch, M., Mayer, U., Pöschl, E., Huber, R., Timpl, R., Holak, T.A., 1996, J. Mol. Biol., 257, 658-668; Stetefeld, J., Mayer, U., Timpl, R., Huber, R., 1996, J. Mol. Biol., 257, 644-657).
На взаимодействие типа ламинин/нидоген оказывает сильное воздействие конформационный компонент (Mayer, U., Nischt, R., Pöschl, E., Mann, К., Fukuda, К., Gerl, M., Yamada, Y., Timpl, R., 1993, EMBO J., 12, 1879-1885; Mann, К., Deutzmann, R., Timpl, R., 1988, Eur. J. Biochem., 178, 71-80). Синтетические пептиды, которые могут быть получены на основе нидоген-связывающего сайта ламинина, не способны образовывать дисульфидную связку, такую как присутствует в LE-модулях, но они демонстрируют активность в тестах на ингибирование, которая примерно в 400-10000 раз слабее, чем активность интактного ламинина Р1 или ламинина γ1 III 3-5 (Pöschl, Е., Fox, J.W., Block, D., Mayer, U., Timpl, R., 1994, EMBO J., 13, 3741-3747; J.W. Fox и R. Timpl, патент США No. 5493008). Такое снижение активности выглядит достаточно необычно, поскольку известно, что в водном растворе пептиды могут принимать большое множество различных конформаций и лишь небольшой процент пептидов находится в биологически активной конформации. Наиболее активный пептид из числа описанных к настоящему времени (со значением ИК50, равным 22 нМ) имеет молекулярный вес примерно 2700 Да (т.е. примерно 50% соответствуют LE-модулю). Его структура включает интактную S-S петлю, которая предположительно стабилизирует структуру последовательности из незаменимых аминокислот NIDPNAV в соответствующем регионе (Poschl, E., Fox, J.W., Block, D., Mayer, U., Timpl, R., 1994, EMBO J., 13, 3741-3747; J.W. Fox и R. Timpl, патент США No. 5493008).
Химическая формула последовательности NIDPNAV (Asn-Ile-Asp-Pro-Asn-Ala-Val) имеет следующий вид:
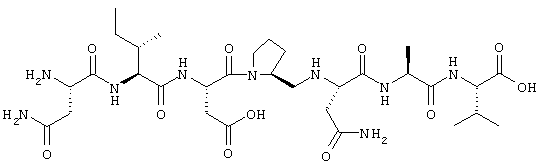
Ингибиторы взаимодействия ламинин/нидоген могут найти применение для изготовления фармацевтических средств, употребляемых при заболеваниях, связанных с повышенным или нежелательным синтезом базальных мембран.
Такие заболевания включают, в частности, все типы диабета, которые сопровождаются утолщением базальных мембран (особенно в почках, глазах, сосудистой системе), фиброз печени, особенно фиброз печени, вызванный алкогольной интоксикацией, который характеризуется синтезом непрерывной базальной мембраны в синусоидах и соответствующей капилляризацией, все фиброзы (хронические или ятрогенные), при которых наблюдается повышенный синтез базальной мембраны или ее компонентов (фиброз почки, легкого, кожи), атеросклероз, при котором имеет место ограничение регуляции липидного метаболизма и которое может быть вызвано в числе других также нарушенной фильтрацией липопротеинов через частично капилляризованные синусоиды печени (патологические изменения в сосудистой системе, наблюдаемые при атеросклерозе, также могут вносить определенный вклад в модификацию состава и структуры базальных мембран сосудов), заболевания, при которых ангиогенез усугубляет тяжесть клинической картины, например при раке, когда для роста опухоли требуется неоваскуляризация, а также диабетическая ретинопатия, ретролентальная фиброплазия, болезни с выраженным воспалительным компонентом (например, ревматоидный артрит, остеоартрит, васкулит), гемангиома, псориаз и многие другие.
Однако использование пептидов, описанных в патенте США 5493008, в качестве лекарственных препаратов ограничено в значительной мере их конформационной гибкостью, нестабильностью в отношении протеаз, плохой биодоступностью и низкой фармакодинамикой (Milner-White, E.J., 1989, Trends Pharmacol, Sci., 10, 70-74; Verber, D.F., Freidinger, R.M., 1985, Trends. Neurosci., 8, 392-396; Hruby, V.J,, 1994, Peptides, Proc. Thirteenth American Symposium (Hodges, R.S., Smith, J.A., Ed.), 3-17, ESCOM, Leiden, Netherlands).
3адачей настоящего изобретения является выявление низкомолекулярных пептидных производных, которые способны специфически взаимодействовать с нидоген-связывающим сайтом на ламинине и таким образом конкурентно ингибировать в низких концентрациях ассоциацию между ламинином и нидогеном.
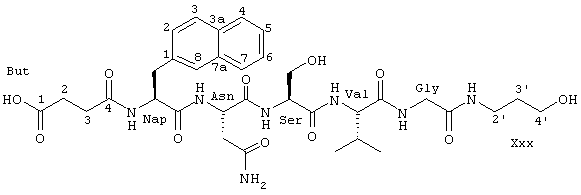
В этой связи объектом настоящего изобретения является соединение формулы I:
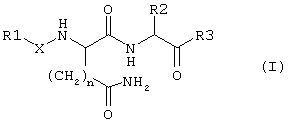
в которой
R1 обозначает группу формулы
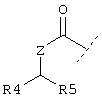 или
или 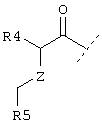 или
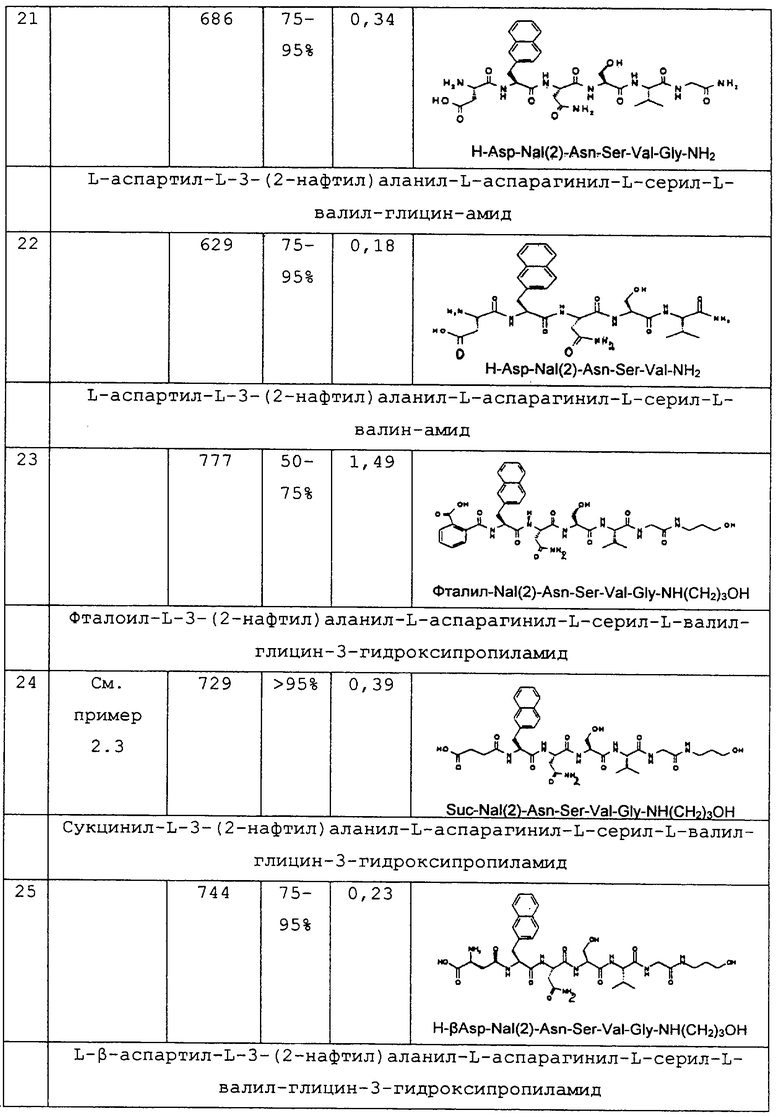
или 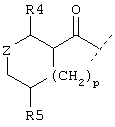 или
или
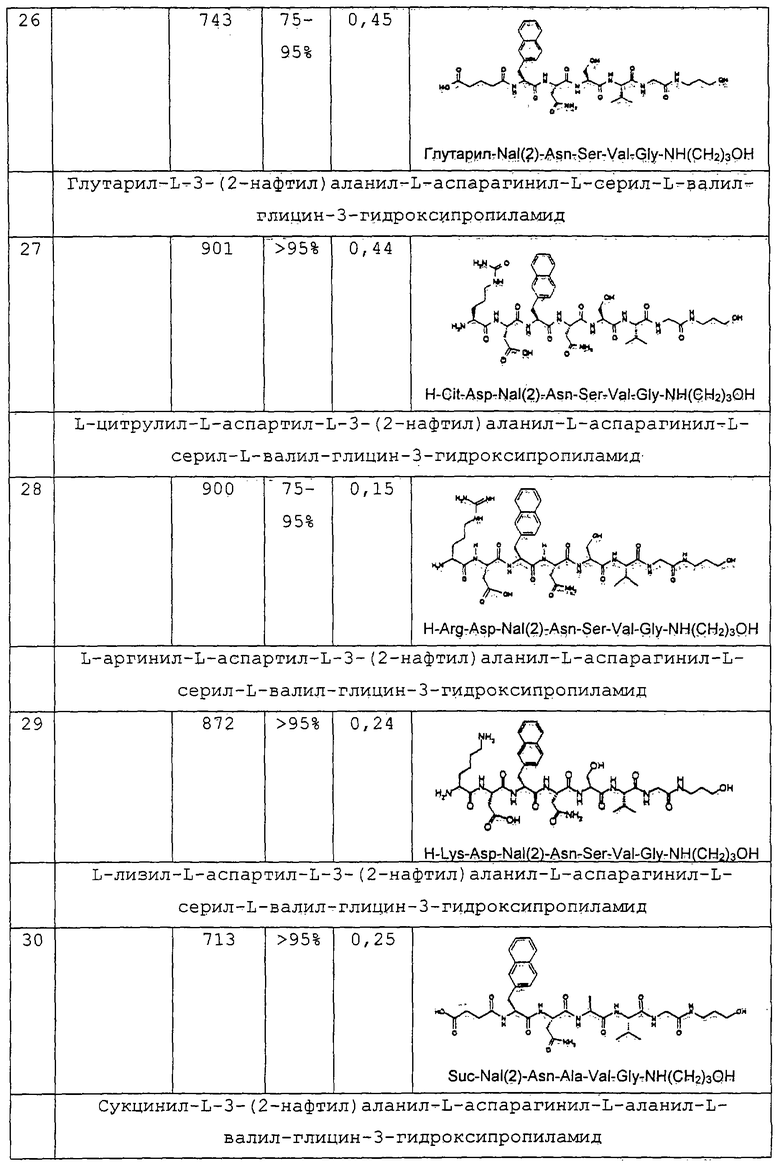
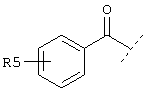 или
или 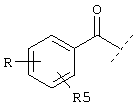 или
или 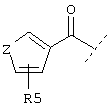
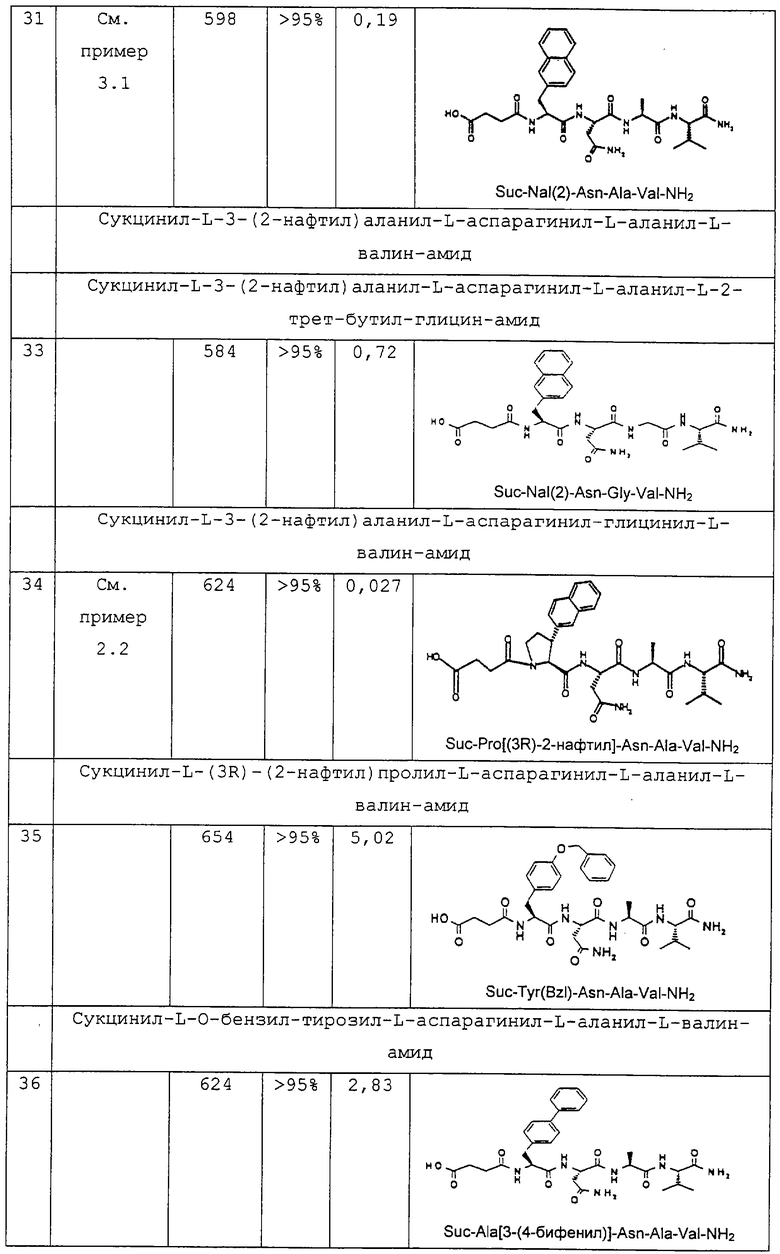
или 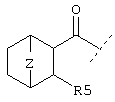 или
или 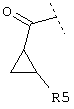
где R4 обозначает -A, -NH2, -NHR, -NR2, А2, -NHR1,
 или
или  или
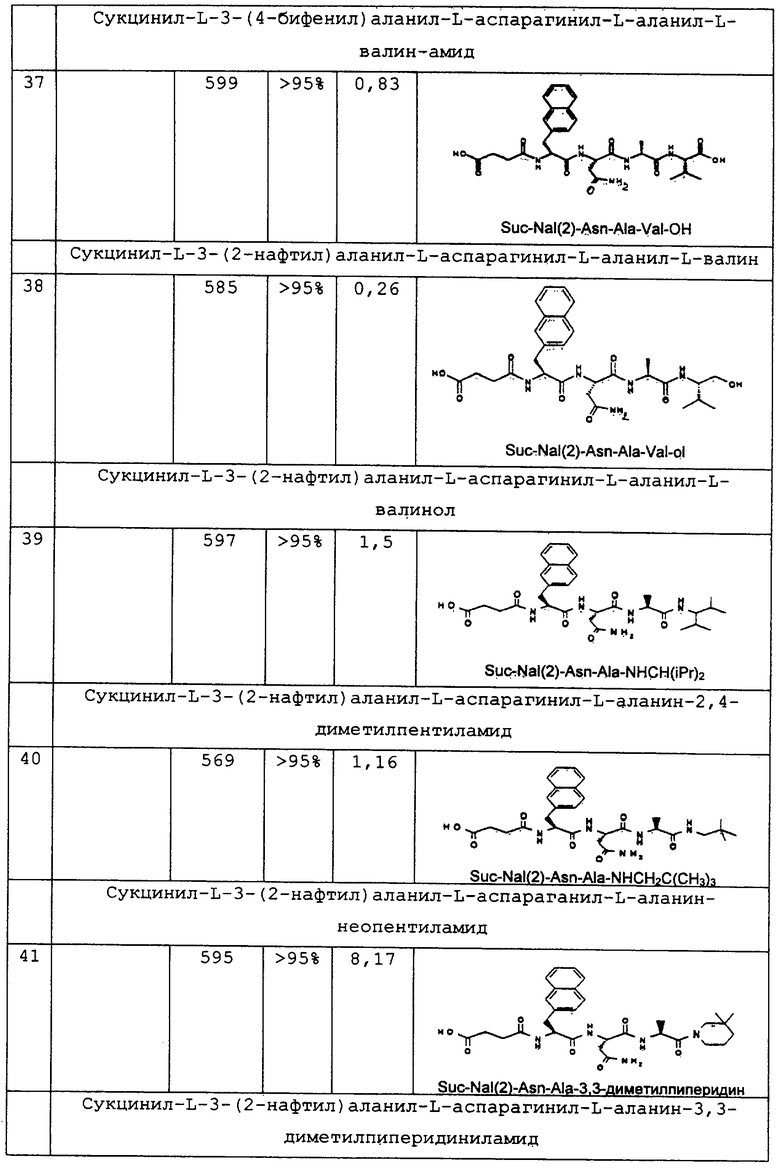
или 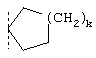
и R5 обозначает -(CH2)1COOA, - (СН2)1CONH2, -(CH2)1NH2 или -(СН2)1-SO3Н,
 или
или
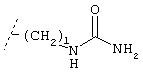
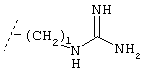
Х обозначает группу, описываемую одной из следующих формул
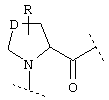 или
или 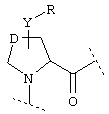 или
или 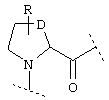 или
или
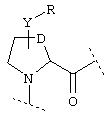 или
или 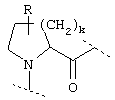 или
или 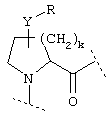 или
или
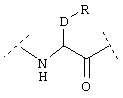 или
или 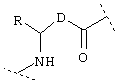
где Y обозначает О, S, -N(A)-CO- или -(СН2)r-,
D обозначает (СН2)r, О, S, NH, NR, (CH2)r-O, (CH2)r-S, (CH2)r-NH или (CH2)r-NR и
R2 обозначает -А, -Е-ОН, -Е-СООН или -E-CONH2,
где Е обозначает линейную или разветвленную C1-С10-алкильную цепь, которая может быть не замещена или замещена группами -А, -(СН2)2mОН, -(CH2)m-СООН, -(СН2)m-С(О)NA2 или С5-С10-циклоалкильной группой,
или Е обозначает C5-C10-циклоалкил, который может быть не замещен или замещен группами -А, -(СН2)m-ОН, -(СН2)m-СООН, - (CH2)m-C(О)NA2 или C5-C10-циклоалкильной группой,
и R3 представляет собой группу одной из нижеприведенных формул:
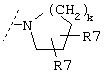 или
или 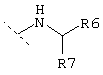
или 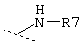
где R6 обозначает -Н, -СООН, -CONH2, -CONHR, -CONR2, -CH2OH или
 или
или
 или
или 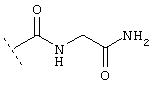
и где R7 обозначает линейную или разветвленную C1-С10-алкильную группу, которая может быть не замещена или замещена группами -А, -(СН2)m-ОН, -(СН2)m-СООН, -(СН2)m-С(О)NA2 или C5-С10-циклоалкильной группой,
или R7 обозначает С5-С10-циклоалкильную группу, которая может быть не замещена или замещена группами -А, -(СН2)m-ОН, -(СН2)m-СООН, -(CH2)m-C(O)NA2 или С5-С10-циклоалкильной группой,
и R обозначает разветвленный или неразветвленный C1-C6-алкил, С2-С6-алкенил, С2-С6-алкинил, С5-С10-циклоалкил, Het или Ar, которые необязательно могут быть замещены одним или более атомами галогена, C1-C6-алкилокси, разветвленной или неразветвленной C1-С6-алкильной, С2-С6-алкенильной, С2-С6-алкинильной или С5-С10-циклоалкильной группами, или -C1-C6-алкил-Het, -C1-C6-алкил-Ar, -О-С1-С6-алкил-Het, -O-C1-С6-алкил-Ar, Het или Ar,
где Het обозначает моноциклическое или бициклическое, 5-10-членное ароматическое или неароматическое кольцо, содержащее 1 или 2 одинаковых или разных гетероатома в качестве членов указанного кольца, выбранных из группы, состоящей из азота, кислорода и серы, которое может быть незамещенным или замещенным одной или более гидрокси- или карбоксигруппами, и где
Ar обозначает моноциклическое или бициклическое 5-10-членное ароматическое кольцо, которое может быть незамещенным или замещенным одной или более гидрокси- или карбоксигруппами, и
Z обозначает (СН2)m, О, S, NH, NR, N-C(O)-R или NSO2R,
А обозначает Н или С1-С4-алкил и
l, m и r обозначают целые числа от 0 до 3,
n и k обозначают целые числа от 1 до 2,
р обозначает целое число от 0 до 1 и
q обозначает целое число от 1 до 3
и все их стереоизомерные формы и их смеси во всех соотношениях, включая все физиологически толерантные соли.
Физиологически толерантные соли включают, например, соли неорганических и органических кислот, например соляной кислоты, серной кислоты, уксусной кислоты, лимонной кислоты или п-толуолсульфоновой кислоты, или соли неорганических и органических оснований, таких как NH4OH, NaOH, KOH, Са(ОН)2, Mg(ОН)2, диэтаноламин или этилендиамин, или соли аминокислот, таких как аргинин, лизин, лизил-лизин или глютаминовая кислота.
В одном предпочтительном варианте реализации настоящее изобретение относится к соединению формулы I, в которой n равно 1.
Еще один предпочтительный вариант реализации относится к соединению формулы I, в которой R в группе Х обозначает Het или Ar, которые могут быть необязательно замещены -C1-C6-алкил-Het, -C1-C6-алкил-Ar, -O-C1-C6-алкил-Het, -O-C1-С6-алкил-Ar, Het или Ar. Более предпочтительно R в группе Х обозначает Het. Например, Het может обозначать:
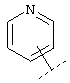
или
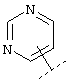
или
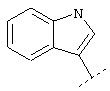
или
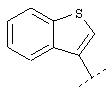
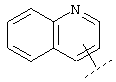
или
Предпочтительный вариант осуществления настоящего изобретения включает также соединение формулы I, в которой R в группе Х обозначает Ar, который может быть необязательно замещен -C1-С6-алкил-Ar, -O-C1-C6-алкил-Ar или Ar. Предпочтительно R в группе Х обозначает Ar.
Например, Ar может обозначать:
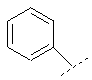
или
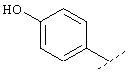
или
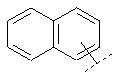
или
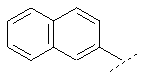
Предпочтительный вариант осуществления настоящего изобретения включает также соединение формулы I, в которой R в группе Х обозначает:
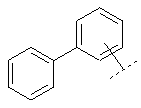
или
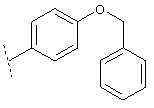
В соединении формулы I X предпочтительно обозначает группу следующей формулы:
Предпочтительно Y обозначает -(СН2)r, где r предпочтительно имеет значение 0 или 1 и k предпочтительно имеет значение 1 или 2.
Еще один предпочтительный вариант осуществления настоящего изобретения относится к соединению формулы 1, в которой группа Х имеет следующую формулу:
причем D предпочтительно обозначает -(СН2)r-, где r принимает значение 0 или 1.
Также предпочтительным является вариант осуществления настоящего изобретения, относящийся к соединению формулы I, в которой R1 обозначает группу следующей формулы:
где Z обозначает предпочтительно (СН2)m и m имеет значение 0 или 1. Предпочтительно R5 обозначает -(CH2)1-COOA, где А обозначает предпочтительно Н, или R5 обозначает -(CH2)1-COONH2, где 1 имеет значение 0. Предпочтительно R4 обозначает -NH2 или -А, где А предпочтительно обозначает Н, или предпочтительно R4 обозначает -NHR1, где -NHR1 предпочтительно обозначает:
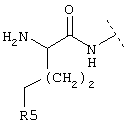
и где R5 в группе -NHR1 предпочтительно обозначает
и 1 предпочтительно имеет значение 0, или R5 в группе -NHR1 предпочтительно обозначает
и 1 предпочтительно имеет значение 0, или R5 в группе -NHR1 предпочтительно обозначает (CH2)1-NH2, и 1 предпочтительно обозначает 0.
Еще один предпочтительный вариант реализации настоящего изобретения относится к соединению А формулы I, где R1 обозначает группу следующей формулы:
где Z обозначает предпочтительно -(CH2)m- и m предпочтительно имеет значение 1, R4 предпочтительно обозначает -NH2 и R5 предпочтительно обозначает -(CH2)1-COOA, где 1 предпочтительно имеет значение 0, и А обозначает предпочтительно Н.
Еще один предпочтительный вариант реализации настоящего изобретения относится к соединению А формулы I, где R1 обозначает группу следующей формулы:
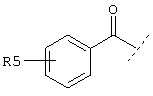
где R5 предпочтительно обозначает -(СН2)1-COOA, где 1 предпочтительно имеет значение 0, и А обозначает предпочтительно Н.
Еще один предпочтительный вариант реализации настоящего изобретения относится к соединению формулы I, где R2 обозначает А, причем А предпочтительно обозначает группу -СН3, или R2 обозначает группу -Е-СООН, предпочтительно -СН2-СООН, или R2 обозначает группу -Е-ОН, предпочтительно -СН2-ОН.
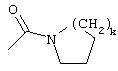
Еще один предпочтительный вариант реализации настоящего изобретения относится к соединению формулы I, где R3 обозначает группу следующей формулы:
где k предпочтительно обозначает 2.
Еще один предпочтительный вариант реализации настоящего изобретения относится к соединению формулы I, где R3 обозначает группу следующей формулы:
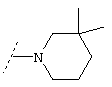
Еще один предпочтительный вариант реализации настоящего изобретения относится к соединению формулы I, где R3 обозначает группу следующей формулы:
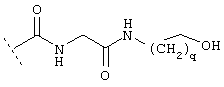
где R7 предпочтительно обозначает разветвленную C1-С10-алкильную группу, предпочтительно -СН(СН3)2, -С(СН3)3, -СН(СН3)СН2-СН3 или -СН2-СН(СН3)2 и где R6 предпочтительно обозначает -Н, -СООН, -CONH2, -СН2OH, -CON(СН3)2, или более предпочтительно R6 обозначает:
где q обозначает предпочтительно 2.
Еще один предпочтительный вариант реализации настоящего изобретения относится к соединению формулы I, где R3 обозначает группу следующей формулы:
где R7 предпочтительно обозначает -СН(СН(СН3)2)2 или -СН2С(СН3)3.
Соединения согласно настоящему изобретению являются неприродными (т.е. они не встречаются в природе) низкомолекулярными производными пептидов, которые способны ингибировать взаимодействие ламинин/нидоген в нМ диапазоне концентраций. Неожиданно было обнаружено, что низкомолекулярные структуры оказались способными осуществлять высокоаффинное связывание с сайтами связывания нидогена на молекуле ламинина, обходясь без взаимодействия с тирозином или гистидином из петли (петли с), соседней с действующей связывающей последовательностью.
Еще более неожиданным стало обнаружение того факта, что рассматриваемые в настоящем изобретении низкомолекулярные производные пептидов со значениями молекулярного веса от 550 до 800 Да демонстрируют ингбирование того же порядка, что и наиболее активные из числа известных на настоящий момент пептидов (ИК50 примерно 22 нМ), которые имеют молекулярный вес примерно 2700 Да (т.е. примерно 50% от значения, характерного для LE-модуля) и включают интактную S-S петлю, которая преимущественно стабилизирует структуру необходимого региона с последовательностью NIDPNAV (J.W. Fox и R. Timpl, патент США 5493008).
Целевое соединение было получено путем специфического синтеза, на основе знания природы взаимодействия структура/функция и известной по литературным данным трехмерной структуры связывающего нидоген сайта, в виде пептидных производных на смоле, использованной в качестве подложки. Строительные блоки, применявшиеся для пептидного синтеза, варьировали в соответствии с заданными критериями для достижения нужных структурных различий и интеграции неприродных блоков. Для тестирования наличия у получаемых пептидных производных ингибирующей активности и последующего сравнения их между собой был использован чувствительный метод скрининг-анализа, который применялся после отщепления производных от смолы-подложки.
Соединения согласно настоящему изобретению могут использоваться для получения фармацевтических препаратов, применяемых при лечении заболеваний, связанных с повышенным или нежелательным синтезом базальных мембран.
В соответствии с вышесказанным, возможными областями терапевтического применения настоящих пептидных производных и/или их физиологически толерантных солей являются:
1. Все типы поздних осложнений диабета, связанные с утолщением базальных мембран (особенно в почках, глазах, сосудистой системе).
2. Цирроз печени, особенно алкогольный цирроз печени, характеризующийся синтезом непрерывной мембраны в синусоидах и вызванной этим процессом капилляризацией.
3. Все фиброзы (хронические или ятрогенные), при которых может наблюдаться повышенный синтез базальных мембран или их компонентов (почка, легкое, кожа).
4. Атеросклероз, который характеризуется ограниченной регуляцией липидного метаболизма, что может быть вызвано в том числе нарушенной фильтрацией липопротеинов через частично капилляризованные синусоиды печени. К тому же патологические изменения в сосудистой системе, которые могут иметь место при атеросклерозе, также вносят определенный вклад в модификацию состава и структуры базальных мембран сосудов.
5. 3аболевания, при которых ангиогенез способствует ухудшению клинической картины, например в случае рака, когда для роста опухоли требуется неоваскуляризация, в случае диабетической ретинопатии, ретролентальной фиброплазии, болезней с выраженным воспалительным компонентом (например, ревматоидный артрит, остеоартрит, васкулит), гемангиома, псориаз и многие другие.
Таким образом, соединения согласно настоящему изобретению и/или их физиологически толерантные соли могут применяться в качестве фармацевтических средств. В этой связи еще одним объектом настоящего изобретения является фармацевтическая композиция, содержащая, по меньшей мере, одно соединение согласно настоящему изобретению и/или его физиологически толерантные соли.
Соединения формулы I и их физиологически толерантные соли и производные могут вводиться согласно настоящему изобретению животным, предпочтительно млекопитающим, в частности людям, для лечения или профилактики. Они могут вводиться per se, в виде смесей друг с другом или в виде фармацевтических препаратов, пригодных для энтерального или парентерального введения, и, будучи активными составляющими, содержат эффективную дозу, по меньшей мере, одного соединения формулы I и/или его физиологически толерантных солей и производных, а также могут объединяться с традиционными фармацевтически безвредными наполнителями и/или добавками.
Указанные фармацевтические препараты могут вводиться системно или местно. Так, например, они могут употребляться в виде пилюль, таблеток, таблеток, покрытых оболочкой, таблеток, покрытых сахарной пленкой, гранул, твердых и мягких желатиновых капсул, порошков, растворов, сиропов, эмульсий, суспензий или других фармацевтических форм. Кроме того, введение может осуществляться внутривагинально или ректально, например, в виде суппозиторий, или парентерально или путем имплантирования, например, в виде растворов для инъекций или для инфузии, микрокапсул или стержневидных форм, и кроме того, введение может быть местным или чрескожным, например, в виде мазей, растворов или настоек, или с использованием любого другого способа, например в виде назального распылителя или аэрозольных смесей, или в виде вдыхаемых препаратов сухих порошков. В случае парентерального введения растворов может быть выбран, например, внутривенный, внутримышечный, подкожный, интраартикулярный, внутрисуставной или другой способ, например путем ингаляции влажных аэрозолей или сухих порошковых препаратов.
Фармацевтические препараты согласно настоящему изобретению получают с помощью известных в технике способов, позволяющих использовать фармацевтически инертные неорганические и/или органические носители в сочетании с соединением(ями) формулы I и/или его/их физиологически толерантными солями и производными. Для получения пилюль, таблеток, таблеток, покрытых сахарной пленкой, и твердых желатиновых капсул могут использоваться, например, лактоза, кукурузный крахмал или его производные, тальк, стеариновая кислота или ее соли и др. Функции носителей в случае мягких желатиновых капсул и суппозиториев могут выполнять, например, жиры, воски, полутвердые и жидкие полиолы, полиэтиленгликоли, натуральные или отвержденные масла и др. Приемлемыми наполнителями при изготовлении растворов, например растворов для инъекций, или эмульсий или сиропов могут быть, например, вода, спирты, глицерин, диолы, полиолы, сахароза, инвертный сахар, глюкоза, растительные масла и др. Приемлемые носители для получения микрокапсул, имплантатов или стержневидных форм включают, например, сополимеры гликолевой кислоты и молочной кислоты. В норме фармацевтические препараты содержат примерно от 0,5 до 90 вес.% соединений формулы I и/или их физиологически толерантных солей и производных.
Кроме активных соединений и носителей фармацевтические препараты могут также включать вспомогательные средства или добавки, такие как, например, наполнители, средства, способствующие разложению, связующие вещества, замасливатели, увлажняющие средства, стабилизаторы, эмульгаторы, консерванты, подсластители, красители, вкусовые вещества или ароматизаторы, загустители, разбавители, забуферивающие вещества, растворители или солюбилизирующие агенты, средства для достижения депо-эффекта, соли, воздействующие на осмотическое давление, средства, применяемые для нанесения покрывающей оболочки, или антиоксиданты. Они также могут содержать два или более соединений формулы I и/или их физиологически толерантных солей и производных. Кроме того, указанные препараты могут включать один или более терапевтически или профилактически активных веществ в дополнение, по меньшей мере, к одному соединению формулы I и/или его физиологически толерантным солям и производным. В норме фармацевтические препараты могут содержать от 0,2 до 500 мг, предпочтительно от 1 до 100 мг активного соединения формулы I и/или его физиологически толерантных солей и производных в расчете на дозу.
Если соединения формулы I или содержащие их фармацевтические препараты применяются в виде аэрозолей, например в виде назальных аэрозолей или влажных аэрозолей, или при ингаляции сухого порошка, то указанное введение может осуществляться с помощью распылителя, атомайзера, насосного атомайзера, устройства для ингаляции, ингалятора с отмеряемой дозой или ингалятора для сухого порошка соответственно. Фармацевтические формы, используемые для введения соединений формулы I в виде аэрозоля, могут быть приготовлены с помощью способа, известного на достигнутом уровне техники любому специалисту в данной области. Так, для получения растворов или дисперсий соединений формулы I в воде могут использоваться в смесях вода-спирт или подходящих солевых растворах обычные добавки, например бензиловый спирт или другие подходящие консерванты, усилители абсорбции с целью повышения биодоступности, солюбилизирующие агенты, средства, способствующие диспергированию, и другие, а в случае соответствующих форм - традиционные пропелленты, например хлорфторуглеводороды и/или фторуглеводороды, тогда как порошковые формы препаратов на основе соединений формулы I и/или их физиологически толерантных солей могут быть получены при лиофильной сушке или предпочтительно при распылительном высушивании водных растворов соединений формулы I и/или их физиологически толерантных солей и приемлемых водорастворимых добавок, таких как сахара или производные сахаров и аминокислоты.
Доза соединений формулы I в фармацевтических композициях может варьировать в широких пределах и как обычно определяется лечащим врачом в каждом конкретном случае при учете специфических индивидуальных условий. Так, например, она зависит от природы и тяжести заболевания, которое предстоит лечить, от конкретного используемого соединения, от того, в острой или хронической форме предстоит лечить заболевание или применять препарат с профилактической целью, а также от того, вводятся ли в сочетании с соединением формулы I в составе композиции другие активные соединения. В целом в случае перорального введения подходящей для достижения эффективных результатов у взрослых людей является дневная доза, составляющая примерно от 0,01 до 100 мг/кг, предпочтительно от 0,1 до 10 мг/кг, в частности от 0,3 до 2 мг/кг (в каждом случае имеется в виду доза на кг веса тела). В случае внутривенного введения дневная доза составляет в основном примерно от 0,01 до 50 мг/кг, предпочтительно от 0,01 до 10 мг/кг веса тела. В частности, при введении относительно больших количеств дневная доза может быть разделена на несколько приемов, например на 2, 3 или 4 отдельных введения. В ряде случаев при наличии соответствующих условий может возникнуть потребность повысить или снизить указанную дозировку.
Кроме того, соединения формулы I и их соли согласно настоящему изобретению могут использоваться в качестве промежуточных продуктов для изготовления других соединений, в частности других фармацевтически активных соединений, получаемых из соединений формулы I, например, при соответствующей модификации радикалов или при введении в них нужных групп, например в ходе реакций этерификации, восстановления, окисления или других видов превращения функциональных групп.
Пептидные производные согласно настоящему изобретению могут, с одной стороны, непосредственно использоваться как терапевтическое средство, но могут также представлять собой и основу для создания родственных структур, которые также могут применяться в качестве лекарственного средства при лечении заболеваний, связанных с повышенным или нежелательным синтезом базальных мембран.
Другим объектом настоящего изобретения является способ идентификации соединения, которое способно ингибировать взаимодействие нидогена и ламинина, заключающийся в том, что указанное соединение согласно настоящему изобретению используется как конкурентный ингибитор. Указанный способ может также включать композицию идентифицированного соединения в фармацевтически приемлемой форме.
Объектом настоящего изобретения является также способ получения фармацевтической композиции, предназначенной для идентификации соединения, ингибирующего взаимодействие нидогена и ламинина, заключающийся в том, что соединение согласно настоящему изобретению, используемое как конкурентный ингибитор, и/или его физиологически толерантные соли, смешивают с фармацевтически приемлемым носителем.
Объектом настоящего изобретения является также способ получения соединения формулы I согласно настоящему изобретению.
Соединение формулы I:
согласно настоящему изобретению получают посредством фрагментарной конденсации соединения формулы II:
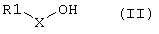
с соединением формулы III
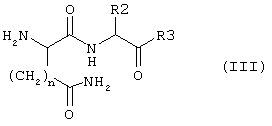
где переменные R1, X, n, R2 и R3 имеют указанные выше значения, и в этой связи соединения формул II и III могут быть защищены на определенных выше функциональных группах с использованием методов, известных в пептидной химии (см., например, Houben-Weyl, Methoden der Organischen Chemie, vol. 15/1 и 15/2, Georg Thieme Verlag, Stuttgart, 1974). Подходящие методы конденсации хорошо известны в технике (Houben-Weyl, Methoden der Organischen Chemie, vol. 15/1 и 15/2, Georg Thieme Verlag, Stuttgart, 1974). Приемлемые конденсирующие или связывающие реагенты представляют собой, например, карбонилдиимидазолы, карбодиимиды, такие как дициклогексилкарбодиимид или диизопропилкарбодиимид, или О-((циано(этоксикарбонил)-метилен)амино)-N,N,N',N'-тетра-метил-урония тетрафторборат (TOTU) или ангидрид пирофосфорной кислоты (РРА). Реакции конденсации проводят в стандартных условиях. Как правило, при пептидной конденсации возникает, необходимость в защите аминогрупп, которые не участвуют в реакции связывания, с помощью защитных групп, которые затем легко удаляются в условиях, отличных от тех, что применяют для реакции связывания. То же самое относится к карбоксигруппам, которые не участвуют в реакции связывания и которые предпочтительно защищают в виде C1-С6-алкиловых эфиров, бензиловых эфиров или трет-бутиловых эфиров в ходе реакции связывания. 3ащита аминогрупп не является необходимой в том случае, если аминогруппы присутствуют также в виде предшественников аминогрупп, например в форме нитро- или цианогрупп. При этом аминогруппы получают затем на стадии гидратации после реакции конденсации. По завершении стадии конденсации защитные группы удаляют с помощью известных и приемлемых методов, например бензилоксикарбонильные и бензильные группы могут быть удалены при гидратации в бензиловых эфирах, а защитные трет-бутильные группы в основном расщепляются в кислых условиях, тогда как 9-флуоренилметилоксикарбонильный остаток удаляется во вторичных аминах.
Получение соединения формулы I согласно настоящему изобретению может также осуществляться при постадийном добавлении соответствующих компонентов, например природных и неприродных аминокислот, а также их производных в твердую фазу, при этом указанные компоненты могут добавляться в разные последовательности.
С целью получения соединения формулы I может быть также полезно проводить не прямое связывание компонентов формул I и II при фрагментарной конденсации, а проводить указанную реакцию с соответствующими их предшественниками, получая нужные промежуточные соединения, которые затем уже могут быть преобразованы в соединение формулы I, например, посредством соответствующих реакций.
Описанный выше способ введения в молекулу функциональных групп не непосредственно, а через соответствующие предшественники с целью получения промежуточных соединений, из которых затем может быть легко получен конечный продукт при трансформации групп предшественника в соответствующие функциональные группы с последующей реакцией конденсации, может применяться в приложении к разным частям молекулы соединения формулы I, т.е. применительно к боковой цепи соединения формулы I: R1- или R1-X соответственно.
Примеры
Приведенные ниже сокращения имеют следующие значения:
Агенты и растворители:
Химические группы:
1. Скрининг библиотеки ингибиторов взаимодействия ламинина/нидогена
Библиотека была создана для поиска более мелких, более мощных и метаболически более стабильных пептидов относительно известного на достигнутом уровне гептапептида NIDPNAV (Pöschl, Е., Fox, J.W., Block, D., Mayer, U., Timpl, R. (1994) EMBO J., 13, 3741-3747, Pöschl, E., Mayer, U., Stetefeld, J., Baumgartner, R., Holak, T.A., Huber, R., Timpl, R. (1996) EMBO J., 15, 5154-5159; Baumgartner, R., Czisch, M., Mayer, U., Pöschl, E., Huber, R., Timpl, R., Holak, T.A. (1996) J. Mol. Biol. 257, 658-668). Синтез и скрининг библиотеки проводили в виде трех подбиблиотек: пентамеров, гексамеров и гептамеров. Ниже приведено описание стратегии скрининга пентамерной подбиблиотеки. Указанный метод является репрезентативным для методов, используемых в отношении двух других подбиблиотек, за исключением того, что при скрининге гексамеров на первой стадии используют примерно 50 шариков на ячейку, а при скрининге гептамеров используют примерно 100 шариков на ячейку.
1.1. Скрининг пентамерной библиотеки
Пентамерная библиотека содержит 2160 различных соединений.
1) Примерно 8800 отдельных шариков суспендируют в 0,1% HCl и распределяют по семи фильтрам на дне 96-гнездного микротитрационного планшета примерно по 14 шариков на ячейку.
2) Шарики промывают два раза 200 мкл деионизированной воды и затем добавляют 50 мкл 500 мМ HEPES, рН 7,0. Используемый в библиотеке линкер высвобождает одну аликвоту соединения при повышении рН до 7,0, и указанную стадию расщепления осуществляют в течение всей ночи.
3) Планшеты складывают вершинами фильтровальных пластин с дном U-образной формы и центрифугируют. Смеси соединений, высвобождаемых из шариков, собирают на дне планшетов, тогда как соответствующие шарики остаются на исходном фильтре планшета.
4) Добавляют к шарикам 25 мкл ДМСО для вымывания оставшегося свободного соединения из шариков, и планшеты снова центрифугируют для отделения соединений в растворе от шариков. Полученный препарат для хранения представляет собой преимущественно 27 мкМ концентрацию на соединение в 333 мМ HEPES, 33% ДМСО.
5) Проводят предварительную инкубацию раствора для хранения, содержащего соединение, с нидогеном (10 мкл соединения из раствора для хранения с 90 мкл раствора нидогена) и проводят анализ, как описано в приложенном протоколе исследования, с достижением конечной концентрации при скрининге в 2,7 мкМ на соединение.
6) В 25 ячейках для анализа, где достигается воспроизводимое ингибирование ≥62%, соответствующие шарики из исходных фильтровальных пластин суспендируют в смеси 0,05% HCl и 0,1% Твин-20 и пипеткой переносят на пять новых фильтровальных пластин в количестве 1 шарик на ячейку. К каждой пластине в качестве контролей добавляют два контрольных шарика с родительским соединением на том же самом линкере.
7) Шарики промывают два раза 200 мкл деионизированной воды и затем к каждой ячейке добавляют 25 мкл 50 мМ NaOH. Линкер, используемый в библиотеке, высвобождает вторую эквимолярную аликвоту соединения при повышении рН от значения 7,0 до 10,0 или более. Стадию расщепления проводят в течение 3 часов.
8) Планшеты складывают наверху фильтровальных пластин с дном U-образной формы и центрифугируют. Соединения, высвобождаемые из шариков, собирают на дне планшетов, тогда как соответствующие шарики остаются на исходном фильтре планшета.
9) Шарики промывают 20 мкл 50 мМ HEPES (исходный рН 7,0) при добавлении 50 мМ HCl, после чего раствор центрифугируют в нижнюю пластину и объединяют с первой высвобожденной пробой.
10) Шарики промывают третий раз 25 мкл ДМСО для уравновешивания с шариками в течение 10 минут перед центрифугированием.
11) Полученные высвобожденные пробы анализируют в 1/10 объема, как указано на стадии 5.
12) Растворы, которые ингибируют так же или лучше, чем контрольные шарики (примерно 50%-ный уровень ингибирования), рассматриваются как лучшие. Восстанавливают 23 шарика из числа лучших, тогда как два других потенциально работающих шарика определяются как дополнительные слабые ингибиторы в отдельных ячейках.
13) Наиболее активные растворы подвергают масс-спектрофотометрическому анализу для определения их молекулярного веса.
14) Соответствующие отдельные активные шарики подвергают деградации Эдмана для определения пептидных последовательностей.
15) Анализируют объединенные MS данные и результаты анализа по Эдману для идентификации структур активных соединений.
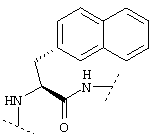
Ниже показаны структуры и частота их восстановления. G-Нора обозначает гидроксипропиламид глицина, линкерный элемент.

Легенда:
Na12=L-3-(2-нафтил)аланил:
G-Hopa=глицин-3-гидроксипропиламид:
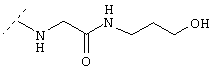
D=Asp (аспартил), Р=Pro (пролил), N=Asn (аспарагинил), А=Ala (аланил), V=Val (валинил), S=Ser (серил), I=Ile (изолейцил).
1.2. Процедуры: Получение пептидной библиотеки
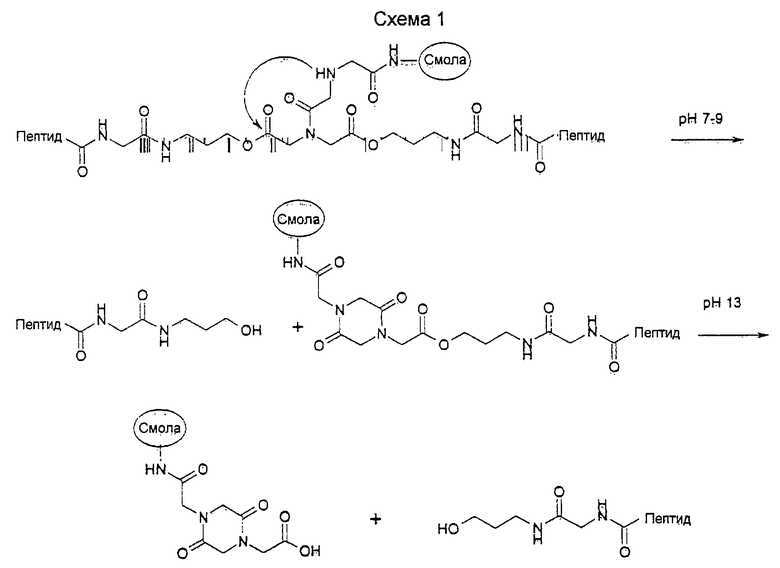
Пептидные библиотеки синтезируют с использованием техники расщепления/смешивания (Lam, K.S., Salmon, S.E., Hersh, E.M., Hruby, V.J., Kazmierski, W.M., и Knapp, R.J. (1991) Nature 354, 82; Furka, A., Sebestyen, F., Asgedom, M., и Dibo, G. (1991) Int. J. Pept. Protein Res. 37, 487) с использованием методов твердофазнои химии синтеза пептидов с помощью Fmoc (Stewart, J.M., и Young, J.D. (1984) Solid Phase Peptide Synthesis. Pierce Chemical Co., Rockford, IL., Atherton, E., и Sheppard, R.C. (1989) Solid Phase Peptide Synthesis. IRL Press Oxford). В каждом цикле связывания каждый смоляной шарик подвергается воздействию только одной активированной аминокислоты. В этой связи при завершении синтеза библиотеки каждый смоляной шарик представляет собой только одну пептидную форму. Поскольку не представлялось возможным отдельно тестировать все соединения, авторы на каждом смоляном шарике построили в двух копиях одинаковые структуры с помощью дифференциально расщепляемого линкера, фиг.1 (Kocis, P., Krchnak., V., и Lebl, М. (1993) Tetr. Lett. 34, 7251; Lebl, M., Krchnak, V., Salmon, S.E., и Lam, K.S. (1994) A Companion to Methods in Enzymology 6, 381). 3атем может быть достигнуто высвобождение пептида из смоляного шарика в ходе ряда последовательных стадий с использованием разных механизмов расщепления. Высвобождение первой порции пептида в виде гидроксипропиламида проводят в буфере при рН 7-9. Высвобождение второй порции пептида активируется при использовании более высоких значений рН (Схема 1).
В пептидных библиотеках используют полистирольные шарики с привитым полиэтиленгликолем или TentaGel®S NH2. Фактически приемлемы для использования любые смоляные шарики, совместимые с условиями пептидного синтеза и скрининга в водных средах.
Пента-, гекса- и гептамерную библиотеки получают с одной фиксированной позицией (L-аспарагин). Гидроксипропиламид глицина на С-конце представляет собой часть линкера;
H-X4X3-Asn-X2X1-Gly-NH(CH2)3OH (2160 пептидов)
H-X5X4X3-Asn-X2X1-Gly-NH(CH2)3OH (25920 пептидов)
H-X6X5X4X3-Asn-X2X1-Gly-NH(CH2)3OH (311040 пептидов)
X1: N-Fmoc-L-аминокислоты (9), используемые в первой рандомизации: валин, изолейцин, треонин, фенилаланин, β(2-нафтил)аланин, 2-азетидинкарбоновая кислота, пролин, циклогексилглицин, фенилглицин.
Х2: N-Fmoc-L-аминокислоты (4), используемые во второй рандомизации: аланин, глицин, серин, аспарагиновая кислота.
Х3=Х5=Х6: N-Fmoc-L-аминокислоты (12), используемые в третьей, пятой и шестой рандомизации: пипеколиновая кислота, Р(2-нафтил)аланин, глютаминовая кислота, лизин, 2-азетидинкарбоновая кислота, треонин, пролин, аспарагин, изолейцин, 3,5-дийодтирозин, цитруллин, аргинин.
Х4: N-Fmoc-L-аминокислоты (5), используемые в четвертой рандомизации: аспарагиновая кислота, глютаминовая кислота, 2-аминоадипиновая кислота, O-сульфат тирозина, γ-карбокси-глютаминовая кислота.
Смола (PEG-PS·HCl, Millipore®, 20 г, нагрузка 0,58 ммоль/г, средний размер частиц 220 мкм) набухает в N,N-диметилформамиде в течение 2 часов, после чего ее нейтрализуют 10% N,N-диизопропилэтиламином в дихлорметане. Далее смолу промывают дихлорметаном и N,N-диметилформамидом. Связывают с линкером (фиг.1, 3 экв.) с использованием 1,3-диизопропилкарбодиимида и 1-гидроксибензотриазола (каждого по 3 экв.) в N,N-диметилформамиде при комнатной температуре в течение 12 часов. Ход реакции отслеживают по тесту с бромфеноловым синим (Krchnak, V., Vagner, J., Safar, P., и Lebl, M. (1988) Collec. Czech. Cem. Commun. 53, 2542). Окончание реакции связывания определяют по нингидриновому тесту (Kaiser, E., Colescott, R.L., Bossinger, C.D., и Cook, P.I. (1969) Anal. Biochem. 34, 595). После промывания N,N-диметилформамидом удаляют Fmoc-защитную группу при обработке 50% пиперидином в N,N-диметилформамиде в течение 15 минут. 3атем смолу промывают N,N-диметилформамидом и определяют количество высвобожденного фульвен-пиперидинового аддукта на УФ спектрофотометре (302 нм). Стабильный уровень нагрузки смолы (ммоль/г), определяемый этим методом в ходе всего процесса синтеза библиотеки, является одним из контролируемых параметров качества.
Смолу разделяют на 9 равных порций. 3атем в каждую аликвоту смолы добавляют отдельно девять Fmoc-защищенных аминокислот (X1) и в течение 2 часов проводят связывание по описанной выше процедуре. Далее смолу собирают в цилиндрическом стеклянном сосуде, прикрепленном фриттой к дну. Смолу барботируют сухим азотом с целью перемешивания. 3атем удаляют Fmoc-защитные группы, как описано выше.
После этого смолу разделяют на 4 равные порции. В каждую аликвоту смолы добавляют отдельно четыре Fmoc-защищенных аминокислоты (Х2) и проводят связывание по той же процедуре, что и ранее. Удаляют Fmoc-защитную группу и определяют уровень нагрузки смолы. В следующем цикле связывают L-аспарагин по указанной процедуре. 3атем смолу разделяют на аликвоты для использования в другом цикле связывания. После завершения всех стадий рандомизации удаляют Fmoc-защитные группы, и защитные группы боковой цепи расщепляют в течение 2,5 часа смесью трифторуксусной кислоты (82,5%), анизола (5%), воды (5%), тиоанизола (5%) и этандитиола (2,5%). Далее смолу промывают трифторуксусной кислотой, дихлорметаном, N,N-диметилформамидом и метанолом. Полученные библиотеки хранят при температуре 4°С.
Для подтверждения качества библиотеки проводят выборочный анализ нескольких шариков в рамках деградации Эдмана и масс-спектрофотометрического анализа.
1.3. Результаты (см. также фиг.2-12)
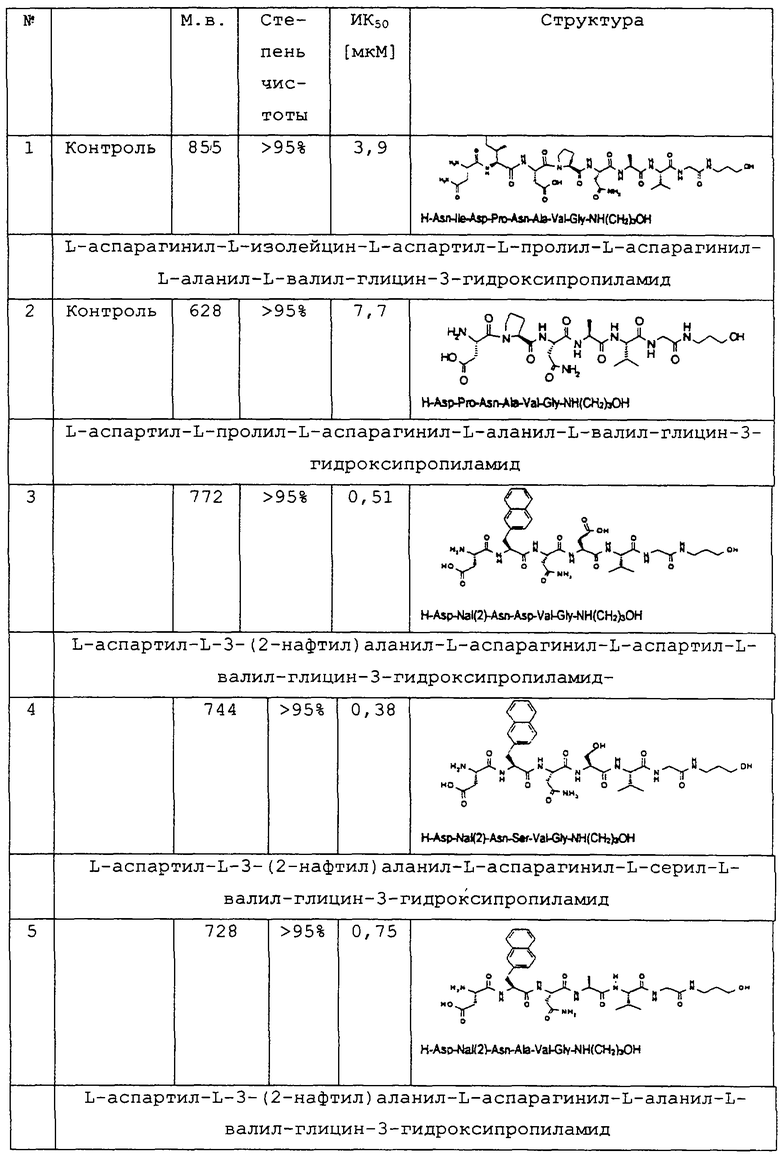
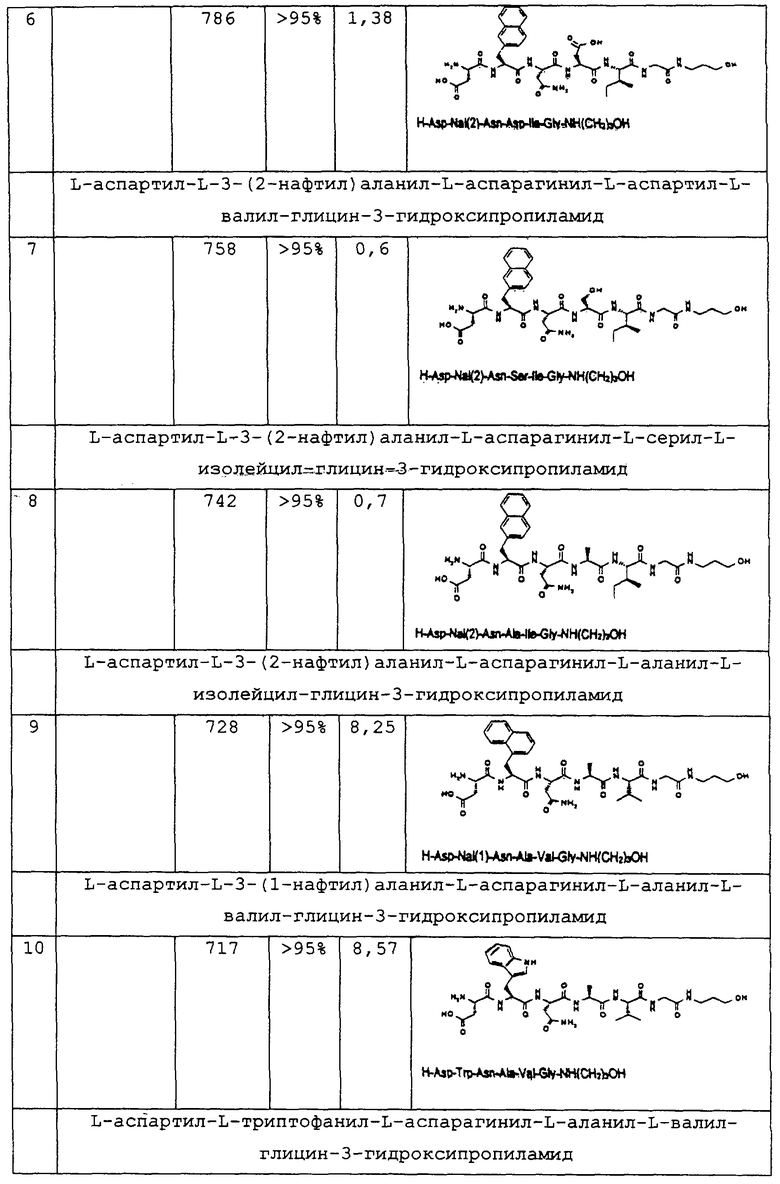
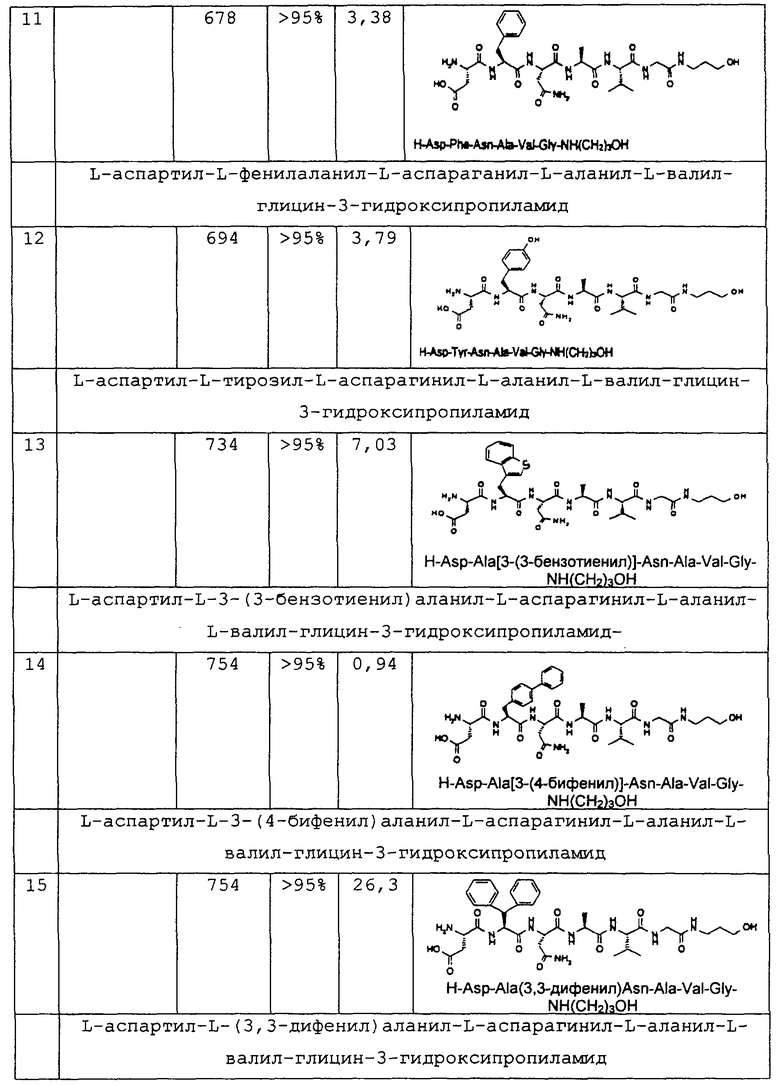
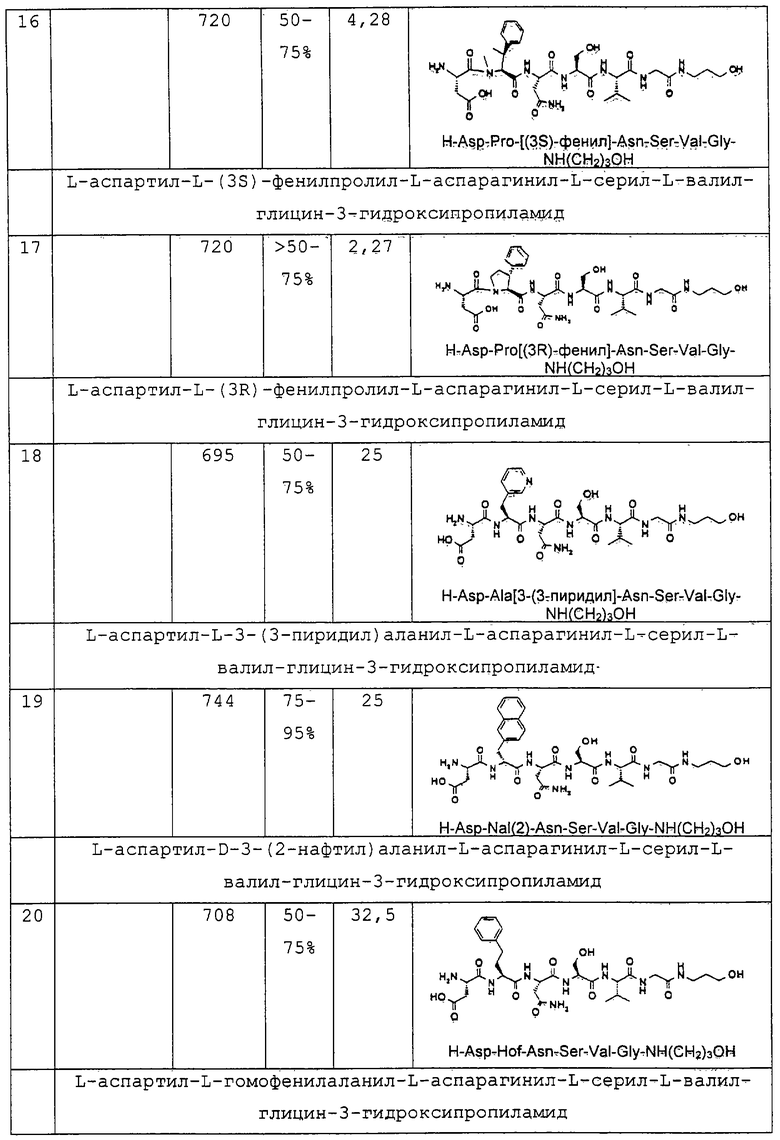
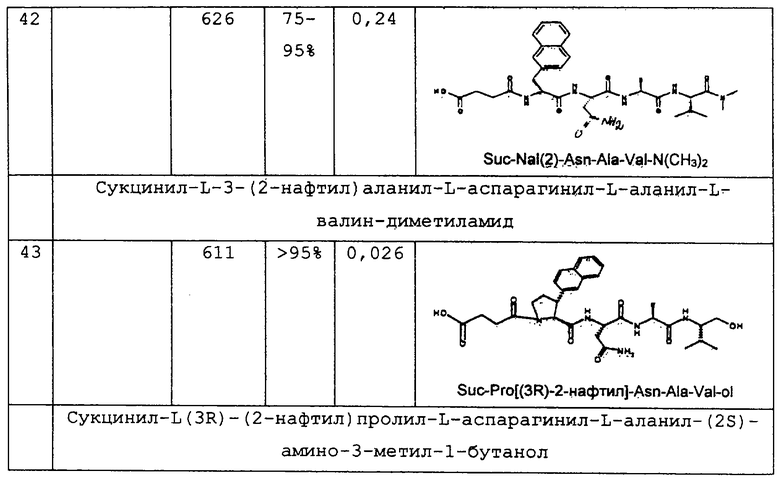
2. Синтез в больших масштабах
2.1. Синтез N-Fmoc-транс-3-(2'-нафтил)-L-пролина (А8)
Краткое описание: N-Fmoc-транс-3-(2'-нафтил)-L-пролин (А8) получают по способу, включающему 10 стадий:
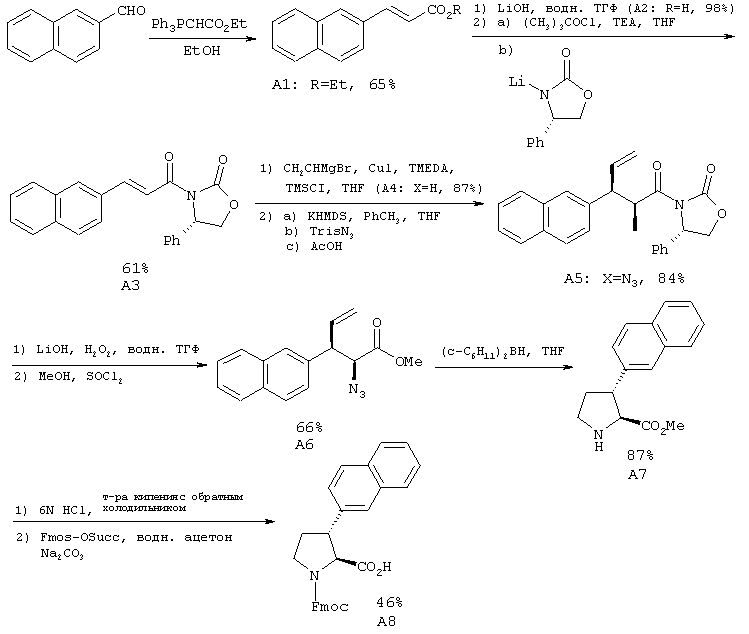
2.1.1. Этил транс-3-(2'-нафтил)-пропеноат (А1)
К перемешанному раствору 2-нафтальдегида (7,8 г, 50 ммоль) в 50 мл этанола добавляют (карбэтоксиметилен)трифенил-фосфоран (18,3 г, 52,5 ммоль). Отмечается слабая экзотермическая реакция. При перемешивании смеси в течение ночи образуется осадок. Далее реакционную смесь разбавляют Et2O (500 мл) и промывают 1М Н3РО4 (2×100 мл), далее насыщенным раствором NaHCO3 (1×100 мл), водой (100 мл) и солевым раствором (100 мл). Органическую фракцию сушат (MgSO4) и концентрируют под пониженным давлением. Остаток пропускают через прокладку с SiO2 и элюируют смесью гексан:EtOAc (9:1). После концентрирования под вакуумом получают почти количественный выход продукта в виде смеси в соотношении 85:15 геометрических изомеров (скорее всего в транс-форме по данным ЯМР). Указанный материал повторно кристаллизуют из смеси гексан/EtOAc (при обогащении гексаном) с получением 4,5 г целевого продукта в виде смеси изомеров в соотношении 97:3 (по данным ЯМР). Маточную жидкость концентрируют и повторно кристаллизуют, как было описано ранее, с получением еще 2,9 г (всего 7,4 г, 33 ммоль, 65% выход). ЯМР (CDCl3) δ 7,93 (с, 1Н), 7,88-7,83 (с, 4Н), 7,67 (дд, 1Н, J=1,6, 8,6 Гц), 7,53-7,50 (с, 2Н), 6,55 (д, 1Н, J=16,0 Гц), 4,30 (кв, 2Н, J=7,1 Гц), 1,42 (т, 3Н, J=7,1 Гц).
2.1.2. Транс-3-(2'-нафтил) пропеноевая кислота (А2)
К раствору сложного эфира А1 (4,24 г, 18,8 ммоль) в ТГФ (75 мл) добавляют LiOH·Н2О (2,36 г, 56,3 ммоль) в воде (19 мл). Изначально гетерогенную смесь энергично перемешивают в течение ночи, после чего она становится гомогенной. Далее реакционную смесь подкисляют концентрированной HCl (рН≈2), в связи с чем образуется осадок. Указанную гетерогенную смесь переносят в делительную воронку и экстрагируют EtOAc (3×150 мл). Объединенные экстракты сушат (MgSO4) и концентрируют под вакуумом с получением карбоновой кислоты в виде белого твердого вещества (3,66 г, 98% выход). ЯМР (CDCl3) δ 7,97 (д, 1Н, J=15,7 Гц), 7,90 (д, 1Н, J=15,3 Гц), 7,90-7,83 (с, 3Н), 7,70 (дд, 1Н, J=1,6; 8,6 Гц), 7,57-7,50 (с, 2Н), 6,58 (д, 1Н, J=16,0 Гц).
2.1.3. Транс-(4S)-3-(3'-(2"-нафтил)-пропеноил)-4-фенил-2-оксазолидинон (A3)
Раствор карбоновой кислоты А2 (3,66 г, 18,5 ммоль) и триэтиламина (1,87 г, 2,56 мл, 18,5 ммоль) в безводном ТГФ (74 мл) охлаждают до -78°С. В течение 2 минут добавляют пивалоилхлорид (2,35 г, 2,40 мл, 19,4 ммоль), что сопровождается образованием белого осадка. Через 10 минут колбу помещают в баню с температурой 0°С на 10 минут, после чего колбу вновь охлаждают до -78°С на 1,5 часа. В отдельной колбе оксазолидинон, полученный из L-фенилглицинола (3,31 г, 20,3 ммоль), в безводном ТГФ (74 мл) охлаждают до -78°С. После этого добавляют раствор n-BuLi (1,6 М в гексане, 11,6 мл, 18,5 ммоль) и перемешивание продолжают в течение примерно 1 часа, что сопровождается осаждением металлированного оксазолидинона из раствора ТГФ/гексан. Смешанный ангидрид добавляют через канюлю к металлированному оксазолидинону, и реакционную смесь помещают в баню с температурой 0°С. Через 1 час баню убирают, и смесь нагревают до комнатной температуры в течение ночи. Далее реакцию гасят добавлением 50 мл насыщенного NH4Cl. ТГФ удаляют под пониженным давлением и после переноса в делительную воронку смесь экстрагируют СН2Cl2 (3×75 мл). Объединенные органические фракции промывают 1М NaOH (2×50 мл), сушат (MgSO4) и концентрируют. Остаток повторно кристаллизуют из смеси EtOAc/гексан с получением белого твердого вещества (3,87 г, 11,2 ммоль, 61% выход). ЯМР (CDCl3) δ 8,05 (д, 1Н, J=15,7 Гц), 7,94 (д, 1Н, J=15,4 Гц), 7,87-7,81 (с, 3Н), 7,76 (дд, 1Н, J=1,5; 8,6 Гц), 7,53-7,47 (с, 2Н), 7,41-7,34 (с, 5Н), 5,58 (дд, 1Н, J=8,7; 3,9 Гц), 4,76 (т, 1Н, J=8,7 Гц), 4,33 (дд, 1Н, J=8,8; 3,9 Гц).
2.1.4. (3'R4S)-3-(3'-(2"-нафтил)-4'-пентеноил)-4-фенил-2-оксазолидинон (А4)
К раствору CuI (3,96 г, 20,9 ммоль) и ТМЭДА (2,66 г, 3,46 мл, 22,9 ммоль) в безводном ТГФ (92 мл) при -78°С добавляют винилбромид магния (1,0 М в ТГФ, 20,9 мл, 20,9 ммоль). Смесь перемешивают в течение 15 минут. В отдельной колбе к раствору ненасыщенного имида A3 (3,87 г, 11,3 ммоль) в безводном ТГФ (42 мл) добавляют триметилсилилхлорид (5,69 г, 6,64 мл, 52,2 ммоль). В связи с нерастворимостью имида мембранный элемент колбы, содержащей купратный реагент, убирают и добавляют в виде одной порции кашицу имида с последующим быстрым промыванием большим количеством ТГФ. Температуру бани повышают до -30°С и перемешивание продолжают еще в течение 1 часа. Далее реакционную смесь вливают в 250 мл смеси насыщенный NH4Cl/концентрированный NH4OH в соотношении 3:2 соответственно. Разделяют слои, и водную фракцию экстрагируют EtOAc (3×200 мл). Объединенные органические фракции промывают последовательно насыщенным NH4Cl (1×100 мл) и водой (1×100 мл). Органическую фракцию сушат (MgSO4) и концентрируют под пониженным давлением. Остаток очищают при пропускании через прокладку с SiO2 и элюируют смесью гексан:EtOAc 4:1. Элюат концентрируют под вакуумом с получением белого твердого вещества (3,64 г, 9,81 ммоль, 87% выход). ЯМР (CDCl3) δ 7,87-7,82 (с, 3Н), 7,72 (с, 1Н), 7,54-7,27 (с, 8Н), 6,11 (ддд, 1Н, J=6,7; 10,4, 17,0 Гц), 5,34 (дд, 1H, J=8,6, 3,5 Гц), 5,10 (д, 1Н, J=8,2, Гц), 5,08 (д, 1Н, J=17,2 Гц), 4,56 (т, 1H, J=8,8 Гц), 4,26 (дд, 1H, J=8,8, 3,5 Гц), 4,16 (ддд, 1H, J=8,1; 7,0; 6,9 Гц), 3,68 (дд, 1H, J=8,4; 16,5 Гц), 3,50 (дд, 1H, J=6,5% 16,5 Гц).
2.1.5. (2'S3'R4S)-3-(2'-азидо-3'-(2"-нафтил)-4'-пентеноил)-4-фенил-2-оксазолидинон (А5)
К безводному ТГФ (34 мл) при -78°С добавляют в виде одной порции гексаметилдисилазид калия (0,5 М в толуоле, 25,5 мл, 12,8 ммоль). Делают кашицу из имида А4 (3,64 г, 9,81 ммоль) в ТГФ (34 мл) и вводят ее через канюлю, промывая ТГФ (2×11 мл) для достижения более полного переноса. Через 30 минут трисилазид (4,40 г, 14,2 ммоль) растворяют в ТГФ (34 мл), охлаждают до -78°С и добавляют через канюлю. Через тридцать минут добавляют АсОН (1,41 г, 1,34 мл, 23,4 ммоль) для гашения реакции. После этого смесь перемешивают при комнатной температуре в течение ночи. Далее смесь распределяют между СН2Cl2 (300 мл) и разбавленным солевым раствором (150 мл). Слои разделяют, и водную фазу экстрагируют CH2Cl2 (3×150 мл). Объединенные органические фракции сушат (MgSO4) и концентрируют под пониженным давлением. Остаток очищают флэш-хроматографией с получением продукта (3,41 г, 8,28 ммоль, 84% выход). ЯМР (CDCl3) δ 7,85-7,82 (с, 3Н), 7,72 (с, 1H), 7,53-7,47 (с, 2Н), 7,42 (дд, 1H, J=1,7; 8,5 Гц), 7,37-7,31 (с, 3Н), 7,18-7,15 (с, 2Н), 6,28 (ддд, 1H, J=8,2; 10,2; 17,1 Гц), 5,63 (д, 1H, J=10,2 Гц), 5,37 (д, 1H, J=17,0 Гц), 5,34 (д, 1H, J=10,2 Гц), 4,83 (дд, 1H, J=3,0, 8,3 Гц), 4,14 (т, 1H, J=7,2 ГЦ), 4,07 (дд, 1H, J=9,3; 17,9 Гц), 3,94 (дд, 1Н, J=3,0; 5,8 Гц), 3,68 (т, 1Н, J=8,6 Гц).
2.1.6. Метил-(2S3R)-2-азидо-3-(2'-нафтил)-4-пентеноат (А6)
К раствору имида А5 (3,41 г, 8,28 ммоль) в ТГФ (62 мл) добавляют воду (21 мл), 35% Н2O2 (2,7 мл) и LiOH·H2O (695 мг, 16,6 ммоль). Через 2 часа добавляют Na2SO3 (4,17 г, 33,1 ммоль) в виде водного раствора (41 мл). Указанную смесь перемешивают в течение 15 минут, и ТГФ удаляют под пониженным давлением. Водный раствор подкисляют HCl и экстрагируют EtOAc (2×150 мл). Объединенные экстракты сушат (MgSO4) и концентрируют под пониженным давлением. Полученный остаток пропускают через колонку с прокладкой SiO2, элюируя смесью гексан:EtOAc (1:1), с получением после концентрирования белого твердого вещества, которое представляет собой преимущественно смесь карбоновой кислоты и хирального вспомогательного реагента. Повторная кристаллизация из смеси гексан:EtOAc дает хиральное вспомогательное вещество в виде игольчатых кристаллов. Маточную жидкость концентрируют и переносят на стадию этерификации. Остаток, содержащий неочищенную карбоновую кислоту, растворяют в безводном МеОН (46 мл) и охлаждают до 0°С. 3атем добавляют тионилхлорид (1,18 г, 725 мкл, 9,94 ммоль) и через 10 минут смесь нагревают при температуре кипения с обратным холодильником в течение 2 часов. К указанной смеси добавляют воду (1,0 мл), перемешивают все в течение 10 минут, и содержимое колбы концентрируют под пониженным давлением. Остаток распределяют между EtOAc (150 мл) и солевым раствором (100 мл). Разделяют слои, и органическую фракцию сушат (MgSO4) и концентрируют под пониженным давлением. Остаток очищают флэш-хроматографией (гексан:EtOAc, 19:1) с получением метилового эфира (1,54 г, 5,48 ммоль, 66% выход). ЯМР (CDCl3) δ 7,84-7,80 (с, 3Н), 7,71 (с, 1Н), 7,50-7,46 (с, 2Н), 7,39 (дд, 1Н, J=1,8; 8,5 Гц), 6,23 (ддд, 1Н, J=8,3; 10,9; 17,6 Гц), 5,30 (д, 1Н, J=9,9 Гц), 5,28 (д, 1Н, J=17,7 Гц), 4,22 (д, 1Н, J=7,5 Гц), 4,06 (т, 1Н, J=7,9 Гц).
2.1.7. Метиловый эфир транс-3-(2'-нафтил)-L-пролина (А7)
Боран-метилсульфидный комплекс (2,0 М в ТГФ, 6,57 мл, 13,1 ммоль) разбавляют безводным ТГФ (26 мл) и охлаждают до 0°С. Осторожно шприцем добавляют циклогексен (2,16 г, 2,66 мл, 26,3 ммоль). Через 30 минут образуется белый осадок. Перемешивание продолжают еще в течение трех часов. Далее содержимое колбы концентрируют под вакуумом. Реагент разводят в CH2Cl2 до состояния кашицы (36 мл) и охлаждают до 0°С. Растворяют в СН2Cl2 (9 мл) винилазид А6 (1,23 г, 4,38 ммоль) и добавляют его через канюлю. Реакционная смесь становится бледно-желтой и начинается заметное выделение газа. Далее смесь нагревают до комнатной температуры в течение ночи. Добавляют МеОН (26 мл) и перемешивают еще в течение 15 минут. 3атем смесь концентрируют под пониженным давлением. Полученный остаток отбирают в Et2O (25 мл) и экстрагируют 0,1М HCl (5×25 мл). Водные экстракты подщелачивают насыщенным NaHCO3 и экстрагируют CH2Cl2 (3×100 мл). Органические экстракты сушат (MgSO4) и концентрируют под вакуумом с получением циклического продукта вместе с некоторым количеством примесей в виде производных дициклогексилборана (974 мг, 3,82 ммоль, 87% выход неочищенного материала), ЯМР (CDCl3) δ 7,84-7,78 (с, 3Н), 7,71 (с, 1Н), 7,49-7,41 (с, 3Н), 3,91 (д, 1Н, J=6,9 Гц), 3,69 (с, 3Н), 3,63 (м, 1Н), 3,48 (дд, 1Н, J=8,2; 15,4, Гц), 3,27 (д, 1Н, J=7,8 Гц), 3,25 (д, 1Н, J=7,8 Гц), 2,33 (м, 1Н), 2,09 (м, 1Н).
2.1.8. N-Fmoc-транс-3-(2'-нафтил)-L-пролин (А8)
510 мг (2 ммоль) метилового эфира (А7) в 12 мл 6 н. HCl нагревают при 100°С в течение 10 часов. 3атем реакционный раствор концентрируют под пониженным давлением, и полученный твердый остаток суспендируют в 15 мл ацетона. Далее в суспензии доводят значение рН до 9-10 с использованием 2 н. раствора Na2CO3. После этого медленно добавляют 742 мг (2,2 ммоль) Fmoc-O-сукцинимида. 3начение рН затем снова доводят до 9-10, и смесь перемешивают при комнатной температуре в течение 4 часов, после чего выдерживают при комнатной температуре в течение ночи. 3атем значение рН доводят до 2 с помощью концентрированной HCl, и полученную смесь перемешивают с этилацетатом. Отфильтровывают с отсасыванием 560 мг осажденного продукта. Водную фазу экстрагируют три раза этилацетатом и затем смешивают с метиленхлоридом. Указанная процедура приводит к получению еще 185 мг продукта в виде осадка. Выход составляет 745 мг (80,4%). ЯМР (d6-ДМСО) δ 7,95-7,80 (с, 6Н), 7,68 (д, 1Н, J=7,3 Гц), 7,60 (д, 1Н, J=7,4 Гц), 7,50-7,34 (с, 6Н), 7,25 (м, 1Н), 4,39-4,15 (с, 4Н), 3,70-3,48 (с, 3Н), 2,29 (м, 1Н), 2,14 (м, 1Н).
2.2. N-сукцинил-транс-3-(2'-нафтил)-L-пролил-L-аспарагинил-L-аланил-L-валин-амид (34)
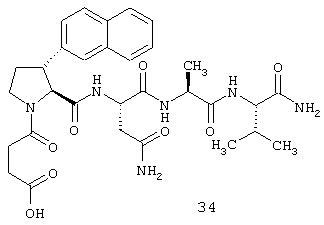
2.2.1. N-Fmoc-транс-3-(2'-нафтил)-L-пролин-L-аспарагин-L-аланин-L-валин-амид (B1)
463,5 мг (1 ммоль) N-Fmoc-транс-3-(2'-нафтил)-L-пролина (А8), 338 мг H-Asn-Ala-Val-NH2 гидрохлорида (полученного с использованием обычных методов пептидной химии) и 135 мг НОВТ растворяют в 20 мл ДМФ. При 0°С добавляют 0,13 мл N-этилморфолина и 220 мг ДЦК. Смесь перемешивают при 0°С в течение 1 часа и затем при комнатной температуре в течение 3 часов, после чего выдерживают при комнатной температуре в течение ночи. Осадок отфильтровывают с отсасыванием, и полученный раствор концентрируют под высоким вакуумом. Образовавшийся остаток распределяют между пентанолом и раствором NaHCO3. Пентанольную фазу промывают раствором KHSO4 и раствором H2O/NaCl. Осадок отфильтровывают с отсасыванием, и полученный материал тщательно растирают с диэтиловым эфиром. Указанная процедура приводит к получению 473 мг продукта. Пентанольную фазу сушат с помощью Na2SO4 и концентрируют. Остаток растирают дважды с диэтиловым эфиром. Эта процедура дает еще 257 мг продукта.
Выход: 730 мг (97,7%)
2.2.2. Транс-3-(2'-нафтил)-L-пролин-L-аспарагин-L-аланин-L-валин-амид (В2)
248 мг (0,332 ммоль) N-Fmoc-транс-3-(2'-нафтил)-L-пролин-L-аспарагин-L-аланин-L-валин-амида В1 отбирают в 5 мл ДМФ. Добавляют 0,35 мл (3,32 ммоль) диэтидамина, и смесь перемешивают при комнатной температуре в течение 15 минут. Далее смесь отфильтровывают с отсасыванием через осветляющий слой и концентрируют под высоким вакуумом. Твердый остаток растирают с диэтиловым эфиром и отфильтровывают с отсасыванием.
Выход: 141 мг (81%)
2.2.3. Метил трет-бутилсукцинат (В3).
В атмосфере аргона суспендируют 13,2 г (100 ммоль) монометилсукцината в 500 мл метиленхлорида. В течение 30 минут добавляют по каплям 12,9 мл (150 ммоль) оксалилхлорида, и затем смесь перемешивают при комнатной температуре в течение 6 часов. Примерно через 3,5 часа образуется прозрачный раствор. Далее добавляют по каплям 300 мл трет-бутанола. Указанную смесь выдерживают при комнатной температуре в течение 21 часа и концентрируют прозрачный раствор. Полученный остаток растворяют в этилацетате и промывают Н2O, раствором NaHCO3 и снова Н2О. 3атем указанный раствор сушат с использованием Na2SO4 и концентрируют.
Выход: 21,6 г (неочищенный маслянистый продукт)
2.2.4. Моно-трет-бутилсукцинат (В4)
9,4 г (50 ммоль) метил трет-бутилсукцината (В3) растворяют в 115 мл 1,4-диоксана. После этого добавляют 110 мл 0,5 н. NaOH. Указанную смесь выдерживают при комнатной температуре, при этом продукт выпадает в осадок. Далее смесь выдерживают при комнатной температуре в течение недели и затем концентрируют. Водный раствор экстрагируют диэтиловым эфиром. Водную фазу охлаждают до 0°С и подкисляют до рН 4 с помощью 2 н. H2SO4. 3атем смесь экстрагируют пять раз диэтиловым эфиром. Органические фазы объединяют, промывают Н2O, сушат с помощью Na2SO4 и концентрируют. Выход: 5,62 г масла (64,5%).
2.2.5. N-трет-бутил-сукцинил-транс-3-(2'-нафтил)-L-пролин-L-аспарагин-L-аланин-L-валин-амид (В5)
262 мг (0,5 ммоль) транс-3-(2'-нафтил)-L-пролин-L-аспарагин-L-аланин-L-валин-амида (В2), 87,1 мг (0,5 ммоль) моно-трет-бутилсукцината (В4) и 67,5 мг HOBt растворяют в 5 мл ДМФ. При 0°С добавляют 110 мг ДЦК, и смесь перемешивают при 0°С в течение 1 часа и затем при комнатной температуре в течение 2 часов, после чего выдерживают при комнатной температуре в течение ночи. Полученный осадок отфильтровывают с отсасыванием, и фильтрат концентрируют под высоким вакуумом. Остаток растирают с раствором NaHCO3, отфильтровывают с отсасыванием, промывают водой и сушат в эксикаторе.
Выход: 169 мг (49,6%).
2.2.6. N-сукцинил-транс-3-(2'-нафтил)-L-пролин-L-аспарагин-L-аланин-L-валин-амид (34)
316 мг N-трет-бутилсукцинил-транс-3-(2'-нафтил)-L-пролин-L-аспарагин-L-аланин-L-валин-амида (В5) растворяют в 2 мл 90%-трифторуксусной кислоты и выдерживают при комнатной температуре в течение 1 часа. После этого смесь фильтруют через осветляющий слой и концентрируют. Остаток растирают с диэтиловым эфиром и отфильтровывают с отсасыванием. Указанная процедура дает 159 мг неочищенного продукта. Для очистки вещество хроматографируют на сефадексе (Sephadex® LH20) с использованием смеси бутанол/ледяная уксусная кислота/вода.
Выход: 27,5 мг (9,5%),
m/z 625,298949 (М+Н)+(масс-спектр высокого разрешения)
Данные ЯМР для соединения 34:
Химические сдвиги для соединения 34 в ДМСО при 300 К:
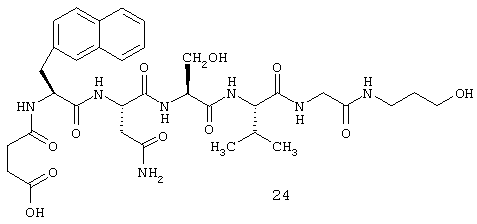
2.3. N-сукцинил-L-(2-нафтил) аланинил-L-аспарагинил-L-серинил-L-валинил-глицин-3-гидроксипропиламид (24)
2.3.1. Бензилоксикарбонил-глицин-(3-пропанол)амид (С1)
627 г (30 ммоль) Gly-OH, 2,45 мл 3-амино-1-пропанола и 4,05 г HOBt растворяют в 60 мл ДМФ. Далее при 0°С добавляют 6,6 г ДЦК. Смесь перемешивают в течение 1 часа при 0°С и в течение 3 часов при комнатной температуре, после чего выдерживают при комнатной температуре в течение ночи. Образовавшийся осадок отфильтровывают с отсасыванием, и фильтрат концентрируют под высоким вакуумом. Остаток распределяют между этилацетатом и раствором NaHCO3. 3атем органическую фазу промывают раствором NaHCO3 и смесью H2O/NaCl, сушат с использованием Na2SO4 и концентрируют. Остаток растирают с диэтиловым эфиром.
Выход: 7,05 г (88,2%).
2.3.2. Бензилоксикарбонил-глицин-(3-пропанол трет-бутиловый эфир)амид (С2)
7 г (26,28 ммоль) бензилоксикарбонил-глицин-(3-пропанол)амида (С1) растворяют в 60 мл диоксана. При низкой температуре (жидкий СО2) медленно добавляют 6 мл Н2SO4. После этого добавляют 60 мл конденсированного изобутилена. Указанную смесь встряхивают в автоклаве при комнатной температуре под давлением азота примерно 20 бар в течение 3 дней. 3атем смесь перемешивают с диэтиловым эфиром и экстрагируют три раза 2 н. раствором Na2CO3. Водный раствор промывают диэтиловым эфиром. Далее органические фазы объединяют, промывают водой, сушат с использованием Na2SO4 и концентрируют.
Выход: 7,98 г (94,2%)
2.3.3. Глицин-(3-пропанол трет-бутиловый эфир) амида гидрохлорид (С3)
7,98 г (24,75 ммоль) бензилоксикарбонил-глицин-(3-пропанол трет-бутиловый эфир) амида (С2) растворяют в 80 мл МеОН, смешивают с Pd на угле и гидрогенизируют в автотитраторе с использованием метанольного раствора HCl и H2. 3атем катализатор отфильтровывают с отсасыванием и фильтрат концентрируют. Образовавшийся остаток сушат под высоким вакуумом.
Выход: 4,7 г (84,5%).
2.3.4 Бензилоксикарбонил-L-валин-глицин-(3-пропанол трет-бутиловый эфир)амид (С4)
5,13 г (20,43 ммоль) бензилоксикарбонил-Val-OH, 4,59 г (20,43 ммоль) глицин-(3-пропанол трет-бутиловый эфир) амида гидрохлорида (С3) и 2,75 г HOBt растворяют в 60 мл ДМФ. Далее добавляют при 0°С 2,65 г N-этилморфолина и 4,5 г ДЦК. Полученную смесь перемешивают при 0°С в течение 1 часа и затем при комнатной температуре еще в течение 2 часов. Далее смесь выдерживают при комнатной температуре в течение ночи, после чего концентрируют под высоким вакуумом. Остаток распределяют между ледяной уксусной кислотой и раствором NaHCO3. 3атем фазу ледяной уксусной кислоты промывают раствором NaHCO3, раствором KHSO4 и смесью H2O/NaCl, сушат с использованием Na2SO4 и концентрируют. Твердый остаток растирают с диэтиловым эфиром и отфильтровывают с отсасыванием.
Выход: 7,32 г (85%).
2.3.5. L-валин-глицин-(3-пропанол трет-бутиловый эфир) амида гидрохлорид (С5)
7,29 г (17,3 ммоль) бензилоксикарбонил-L-валин-глицин-(3-пропанол трет-бутиловый эфир) амида (С4) растворяют в 90 мл МеОН, смешивают с Pd на угле и гидрогенизируют в автотитраторе с использованием метанольного раствора HCl. 3атем катализатор отфильтровывают с отсасыванием, и фильтрат концентрируют. Остаток (аморфный) сушат под высоким вакуумом, растирают с диэтиловым эфиром и фильтруют с отсасыванием.
Выход: 5,22 г (93,2%).
2.3.6. Бензилоксикарбонил-L-серин(трет-бутиловый эфир)-L-валин-глицин-(3-пропанол трет-бутиловый эфир)амид (С6)
5,46 г (18,5 ммоль) Z-Ser(But)ОН, 6 г (18,5 ммоль) L-валин-глицин-(3-пропанол трет-бутиловый эфир) амида гидрохлорида (С5) и 2,5 г HOBt растворяют в 60 мл ДМФ. 3атем при 0°С добавляют 2,4 мл N-этилморфолина и 4,07 г ДЦК. Полученную смесь перемешивают при 0°С в течение 1 часа и затем при комнатной температуре еще в течение 3 часов. Далее указанную смесь выдерживают при комнатной температуре в течение ночи, после чего концентрируют под высоким вакуумом. Полученный твердый остаток распределяют между ледяной уксусной кислотой и раствором NaHCO3. 3атем фазу ледяной уксусной кислоты промывают раствором NaHCO3, раствором KHSO4 и смесью H2O/NaCl, после чего сушат с использованием Na2SO4 и концентрируют, Остаток растирают с диэтиловым эфиром и отфильтровывают с отсасыванием.
Выход: 9,74 (93,2%)
2.3.7. L-серин(трет-бутиловый эфир)-L-валин-глицин-(3-пропанол трет-бутиловый эфир) амида гидрохлорид (С7)
9,74 г (17,25 ммоль) бензилоксакарбонил-L-серин(трет-бутиловый эфир)-L-валин-глицин-(3-пропанол трет-бутиловый эфир) амида (С6) растворяют примерно в 100 мл МеОН, смешивают с Pd на угле и гидрогенизируют в автотитраторе с использованием метанольного раствора HCl. После этого катализатор отфильтровывают с отсасыванием, и фильтрат концентрируют. Остаток (аморфный) сушат под высоким вакуумом, растирают с диэтиловым эфиром и отфильтровывают с отсасыванием.
Выход: 8,02 г (99,6%).
2.3.8. Бензилоксикарбонил-L-аспарагин-L-серин(трет-бутиловый эфир)-L-валин-глицин-(3-пропанол трет-бутиловый эфир)амид (С8)
4,53 г (17 ммоль) Z-Asn-OH, 7,94 г L-серин(трет-бутиловый эфир)-L-валин-глицин-(3-пропанол трет-бутиловый эфир) амида гидрохлорида (С7) и 2,3 г HOBt растворяют в 60 мл ДМФ. При 0°С добавляют 2,21 мл N-этилморфолина и 3,74 г ДЦК. Полученную смесь перемешивают при 0°С в течение 1 часа и затем при комнатной температуре еще в течение 3 часов, после чего концентрируют под высоким вакуумом. Полученный остаток распределяют между пентанолом и раствором NaHCO3. Пентанольную фазу промывают раствором NaHCO3, раствором KHSO4 и смесью H2O/NaCl, после чего сушат с помощью Na2SO4 и отфильтровывают с отсасыванием, и полученный фильтрат концентрируют под высоким вакуумом. Остаток растирают с диэтиловым эфиром, охлаждают и отфильтровывают с отсасыванием. Продукт сушат в эксикаторе над Р2O5.
Выход: 10,8 г, (93,6%).
2.3.9. L-аспарагин-L-серин(трет-бутиловый эфир)-L-валин-глицин-(3-пропанол трет-бутиловый эфир) амида гидрохлорид (С9)
10,8 г (15,9 ммоль) бензилоксикарбонил-L-аспарагин-L-серин(трет-бутиловый эфир)-L-валин-глицин-(3-пропанол трет-бутиловый эфир) амида (С8) растворяют примерно в 160 мл теплого МеОН, смешивают с Pd на угле и гидрогенизируют в автотитраторе с использованием метанольного раствора HCl. 3атем катализатор отфильтровывают с отсасыванием, и фильтрат концентрируют. Полученный аморфный остаток сушат под высоким вакуумом, растирают с диэтиловым эфиром, охлаждают и фильтруют с отсасыванием.
Выход: 8,96 г (97%).
2.3.10. Бензилоксикарбонил-L-2-нафтилаланин-L-аспарагин-L-серин(трет-бутиловый эфир)-L-валин-глицин-(3-пропанол трет-бутиловый эфир)амид (С10)
5,24 г (15 ммоль) бензилоксикарбонил-2-Nal-OH, 8,72 г (15 ммоль) L-аспарагин-L-серин(трет-бутиловый эфир)-L-валин-глицин-(3-пропанол трет-бутиловый эфир) амида гидрохлорида (С9) и 2,04 г HOBt растворяют в 60 мл ДМФ. 3атем при 0°С добавляют 1,95 мл N-этилморфолина и 3,3 г ДЦК. Полученную смесь перемешивают при 0°С в течение 1 часа и затем при комнатной температуре еще в течение 3 часов. Далее указанную смесь выдерживают при комнатной температуре в течение ночи, разбавляют ДМФ и немного нагревают. Образовавшийся осадок отфильтровывают с отсасыванием, и фильтрат концентрируют под высоким вакуумом. Полученный остаток растирают с раствором NaHCO3 и фильтруют с отсасыванием, после чего растирают с раствором KHSO4, фильтруют с отсасыванием, растирают с Н2O, фильтруют с отсасыванием, промывают Н2О и сушат в эксикаторе над P2O5.
Выход: 13,25 г (>99%).
2.3.11. L-2-нафтилаланин-L-аспарагин-L-серин(трет-бутиловый эфир)-L-валин-глицин-(3-пропанол-трет-бутиловый эфир)амида гидрохлорид (С11)
8,85 г (10,1 ммоль) бензилоксикарбонил-L-2-нафтилаланин-L-аспарагин-L-серин(трет-бутиловый эфир)-L-валин-глицин-(3-пропанол трет-бутиловый эфир) амида (С10) частично растворяют в 270 мл МеОН, смешивают с Pd на угле и гидрогенизируют в автотитраторе с использованием метанольного раствора HCl. Полученную суспензию разбавляют ДМФ. Примерно через 6 часов смесь концентрируют до половины исходного объема. Весь материал при этом растворяется. Полученную смесь выдерживают при комнатной температуре в течение ночи. После этого катализатор отфильтровывают с отсасыванием, и фильтрат разбавляют тем же самым количеством МеОН, перемешивают с новым катализатором (Pd на угле) и снова гидрогенизируют на автотитраторе. Через 7 часов смесь помещают на ночь в условия комнатной температуры. Указанную смесь гидрогенизируют еще в течение 4 часов, катализатор отфильтровывают с отсасыванием, и фильтрат концентрируют. Остаток (аморфный) сущат под высоким вакуумом, растирают с диэтиловым эфиром и отфильтровывают с отсасыванием.
Выход: 7,56 г (96,2%).
2.3.12. N-трет-бутил-сукцинил-L-2-нафтилаланин-L-аспарагин-L-серин-L-валин-глицин-(3-пропанол)амид (С12)
523 мг (3 ммоль) L-2-нафтилаланин-L-аспарагин-L-серин(трет-бутиловый эфир)-L-валин-глицин-(3-пропанол трет-бутиловый эфир) амида гидрохлорида (С11), 2,33 г моно-трет-бутилсукцината (В4) и 405 мг HOBt растворяют в 20 мл ДМФ. При 0°С добавляют 0,39 мл N-этилморфолина и 660 мг ДЦК. Полученную смесь перемешивают при 0°С в течение 1 часа и при комнатной температуре в течение 2 часов, после чего выдерживают при комнатной температуре в течение ночи. Указанную смесь концентрируют под высоким вакуумом, твердый остаток растирают с раствором NaHCO3 и отфильтровывают с отсасыванием. 3атем полученный продукт растирают с раствором KHSO4, отфильтровывают с отсасыванием, промывают Н2О и сушат в эксикаторе над Р2O5.
Выход: 3,04 г (неочищенный продукт).
2.3.13. N-сукцинил-L-2-нафтилаланин-L-аспарагин-L-серин-L-валин-глицин-(3-пропанол)амид (С24)
3 г (неочищенный продукт) N-трет-бутил-сукцинил-L-2-нафтилаланин-L-аспарагин-L-серин-L-валин-глицин-(3-пропанол) амида (С12) растворяют в 30 мл 90% трифторуксусной кислоты и все выдерживают при комнатной температуре в течение 1 часа. 3атем указанную смесь концентрируют, и полученный остаток растирают с диэтиловым эфиром и отфильтровывают с отсасыванием. Указанная процедура приводит к получения 2,6 г неочищенного продукта. Для очистки 250 мг указанного продукта растворяют в теплой ледяной уксусной кислоте и хроматографируют на сефадексе (Sephadex® LH20) с использованием смеси бутанол/ледяная уксусная кислота/вода.
Выход: 103 мг
m/z 730,341246 (М+Н)+ (масс-спектр высокого разрешения)
Данные ЯМР для соединения 24:
Химические сдвиги для соединения 24 в ДМСО при 300 К:
3. Ингибирование взаимодействия ламинин/нидоген и биологическая активность
Если особо не оговорено иное, используемые химические реактивы приобретаются от компании Мерк (Merk (Дармштадт)), Сигма (Sigma (Мюнхен)), или Ридель де Хайн (Riedel de Haen (Seelze)).
Выделение лиминина Р1 из плаценты человека, человеческого нидогена из трансфицированных НЕК-293 клеток и мышиного ламинина γ1 III 3-5 из НЕК-293 клеток описано в документе WO 98/31709.
Пример 3.1
Тест на ингибирование - обнаружение ингибирования связывания ламинина/нидогена пептидными производными
3.1.1. HTS-скрининг (высокочувствительный вариант теста):
Метод флуоресцентного анализа процесса растворения с течением времени
Нанесение покрытия на исследуемые пробирки
Микротитрационные планшеты (например, FluoroNunc®) покрывают 75 мкл 0,1 мкг/мл раствором ламинина Р1 (в 0,159 г Na2CO3, 0,293 г NaHCO3, 0,02 г NaN3/л, pH 9,2) при комнатной температуре в течение 1 часа. 3атем раствор раскапывают, и свободные сайты связывания блокируют при инкубации с 0,5% БСА (в среде, содержащей 7,9 г NaCl, 1,2 г Na2HPO4, 0,31 г KCl, 0,23 г NaH2PO4, 0,04% Твин 20/л, рН 7,2) при комнатной температуре в течение 0,5 часа. По завершении реакции блокирования проводят декантацию раствора и промывают планшеты один раз 250 мкл промывочного буфера (ФБР/0,04% Твин).
Последовательное ингибирование
Параллельно с нанесением проб на планшет проводят предварительную инкубацию 85-100 мкл 0,25 нМ раствора нидогена (полученного рекомбинантным методом из человеческого нидогена) с ингибитором или стандартом в отдельном реакционном сосуде (при комнатной температуре в течение 1 часа в среде, содержащей 7,9 г NaCl, 1,2 г Na2HPO4, 0,31 г KCl, 0,23 г NaH2PO4, 0,04% Твин 20/л, 0,5% БСА, рН 7,2).
75 мкл Преинкубационной среды (нидоген +ингибитор или стандарт) переносят на заполненные ячейки микротитрационного планшета и инкубируют при комнатной температуре в течение 1 часа. После этого проводят дважды промывку с использованием ФБР/0,04% Твин.
Обнаружение связанного нидогена проводят при инкубации (при комнатной температуре) в течение 1 часа с 75 мкл специфического препарата антитела, полученного из желтка куриных яиц, иммунизированных человеческим нидогеном. Фракция IgY используется в разбавлении 1:500 в смеси ФБР/0,04% Твин. Комплекс нидогена и специфически связанных антител после промывки дважды ФБР/0,04% Твин выявляют при добавлении антикуриного IgY-биотина (75 мкл в разбавлении 1:2500; Promega, Madison, WI 53711, 608-274-4330). С этой целью после инкубации в течение 1 часа и промывки дважды смесью ФБР/0,04% Твин проводят инкубацию со стрептовидин-европием (Wallac, 1 час при комнатной температуре) и дважды промывают смесью ФБР/0,04% Твин. В итоге становится возможным после добавления 100 мкл усиливающего раствора (Wallac) и взбалтывания в течение 5 минут измерить флуоресцентный сигнал на счетчике Victor для подсчета множественных меток с использованием протокола обнаружения европия. Определяют связь между количеством связанного нидогена в растворах с ингибитором и нидогена без добавления ингибитора.
3.1.2. Трехдневный анализ на достижение равновесия
Выбранные ингибиторы исследуют на их ингибирующую активность в указанном варианте анализа. Анализ описан в патенте США 5493008.
Приведенная ниже таблица дает сравнительные значения ИК50 выбранных веществ вместе с результатами HTS-скрининга. Из них видно, что 3-дневный анализ дает несколько меньшее значение и, как и ожидалось, он более чувствителен, чем скрининг-тест. Однако, что также ясно из приведенного сравнения, ингибирующие структуры могут быть надежно идентифицированы при проведении скрининг-анализа, разработанного авторами.
Пример 3.2 (гипотетический)
Тестирование биологической активности пептидных производных
Несколько моделей, которые были детально описаны в литературе, могут использоваться для определения биологической активности пептидных производных.
Ниже приведены некоторые репрезентативные работы:
Formation of tubuli in cultures of embryonic kidneys (Образование канальцев в культурах эмбриональных почек).
Grobstein, С. (1956) Exp. Cell Res. 10: 424-440.
Ekblom, P. et al. (1994) Development 120: 2003-2014.
Branching morphology in embryonic lungs. (Разветвленная морфология эмбриональных легких).
Grobstein, С. (1953) J. Exp. Zool. 124: 383-413.
Kadoya, Y. et al., (1997) Development 124: 683-691.
Basement membrane assembly in a organotypic skin culture. (Сборка базальной мембраны в органотипической культуре кожи).
Smola, H., Stark, H.-J, Thiekötter, G., Mirancea, N., Krieg, Т., Fusenig, N.E. (1998) Exp. Cell. Res. 239: 399-410.
Reconstitution of hydra from disintegrated cells. (Восстановление гидры из дезинтегрированных клеток).
Yang, Y.G., Mayura, К., Spainhour, С.В., Edwards, Jr., J.F., Phillips, T.D. (1993) Toxicology 85: 179-198.
Thickening of basement membranes in hydra after culturing at increased glucose concentration.(Утолщение базаальных мембран у гидры после культивирования с повышенной концентрацией глюкозы).
Zhang, X.; Huff, J.K.; Hudson, B.G.; Sarras Jr.; M.P. (1990) Diabetologia 33: 704-707.
Все виды количественного анализа ангиогенеза обобщены в обзорной статье Jain, R.K. et al., Nature Medicine (1997) Vol.3, No.11, например:
Induction of Haemangiomes in mice by implantation of cells from spontaneous hemangioendotheliomes,
O'Reilly, M.S., Brem, M.S., Folkman, J. (1995) J. Pediatr. Surg. 30:2, 325-329.
Growth of micro-vessels in a serum-free culture of rat aorta,
Nicosia, R.F., Ottinetti, A. (1990) Lab. Invest Vol.63, No.1, 115-122.
Formation of capillaries of endothelic cells on micro-carriers after imbedding into a fibrin gel
Nehls, V.; Drenckhahn, D. (1995) Microvascular Research 50: 311-322.
| название | год | авторы | номер документа |
|---|---|---|---|
| НИЗКОМОЛЕКУЛЯРНЫЕ ПРОИЗВОДНЫЕ ПЕПТИДОВ КАК ИНГИБИТОРЫ ВЗАИМОДЕЙСТВИЯ ЛАМИНИНА/НИДОГЕНА | 2000 |
|
RU2380371C2 |
| ПРОИЗВОДНЫЕ ГИДРАЗИНА ИЛИ ИХ СОЛИ, ФАРМАЦЕВТИЧЕСКОЕ СРЕДСТВО, АМИНОАЛКИЛГИДРАЗИНЫ ИЛИ ИХ СОЛИ | 1992 |
|
RU2092492C1 |
| НОВЫЕ АНАЛОГИ ГЛЮКАГОН-ПОДОБНОГО ПЕПТИДА, КОМПОЗИЦИЯ И СПОСОБ ПРИМЕНЕНИЯ | 2010 |
|
RU2557301C2 |
| АЗОТСОДЕРЖАЩИЕ ГЕТЕРОАРИЛЬНЫЕ СОЕДИНЕНИЯ И ИХ ПРИМЕНЕНИЕ ПРИ ПОВЫШЕНИИ ЭНДОГЕННОГО ЭРИТРОПОЭТИНА | 2004 |
|
RU2379291C2 |
| ПРОТИВОВИРУСНЫЕ ЭФИРЫ ИЗОСТЕРОВ СУБСТРАТОВ АСПАРТАТПРОТЕАЗЫ ИЛИ ИХ СОЛИ, СПОСОБ ИХ ПОЛУЧЕНИЯ, ФАРМАЦЕВТИЧЕСКИЙ ПРЕПАРАТ И КОМПОЗИЦИЯ | 1995 |
|
RU2164229C2 |
| СПОСОБ ПОЛУЧЕНИЯ ОПТИЧЕСКИ АКТИВНЫХ АЛЬФА-АМИНОАЦЕТАЛЕЙ | 2009 |
|
RU2507194C2 |
| СПОСОБ ПОЛУЧЕНИЯ N-КАРБОКСИАНГИДРИДОВ ИЛИ N-ТИОКАРБОКСИАНГИДРИДОВ УРЕТАНЗАЩИЩЕННЫХ АМИНОКИСЛОТ | 1989 |
|
RU2007396C1 |
| ЗАМЕЩЕННЫЕ ПИРАЗОЛЫ, ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ И СПОСОБ ИНГИБИРОВАНИЯ АКТИВНОСТИ КАТЕПСИНА S | 2001 |
|
RU2278863C2 |
| ПРОИЗВОДНЫЕ 5-АМИНОЦИКЛИЛМЕТИЛОКСАЗОЛИДИН-2-ОНА | 2008 |
|
RU2492169C2 |
| ПРИМЕНЕНИЕ ПЕРФТОРАЛКИЛСОДЕРЖАЩИХ КОМПЛЕКСОВ МЕТАЛЛОВ В КАЧЕСТВЕ КОНТРАСТНЫХ ВЕЩЕСТВ В МАГНИТНО-РЕЗОНАНСНОЙ ТОМОГРАФИИ ДЛЯ ВИЗУАЛИЗАЦИИ БЛЯШЕК, ОПУХОЛЕЙ И НЕКРОЗОВ | 2001 |
|
RU2290206C2 |
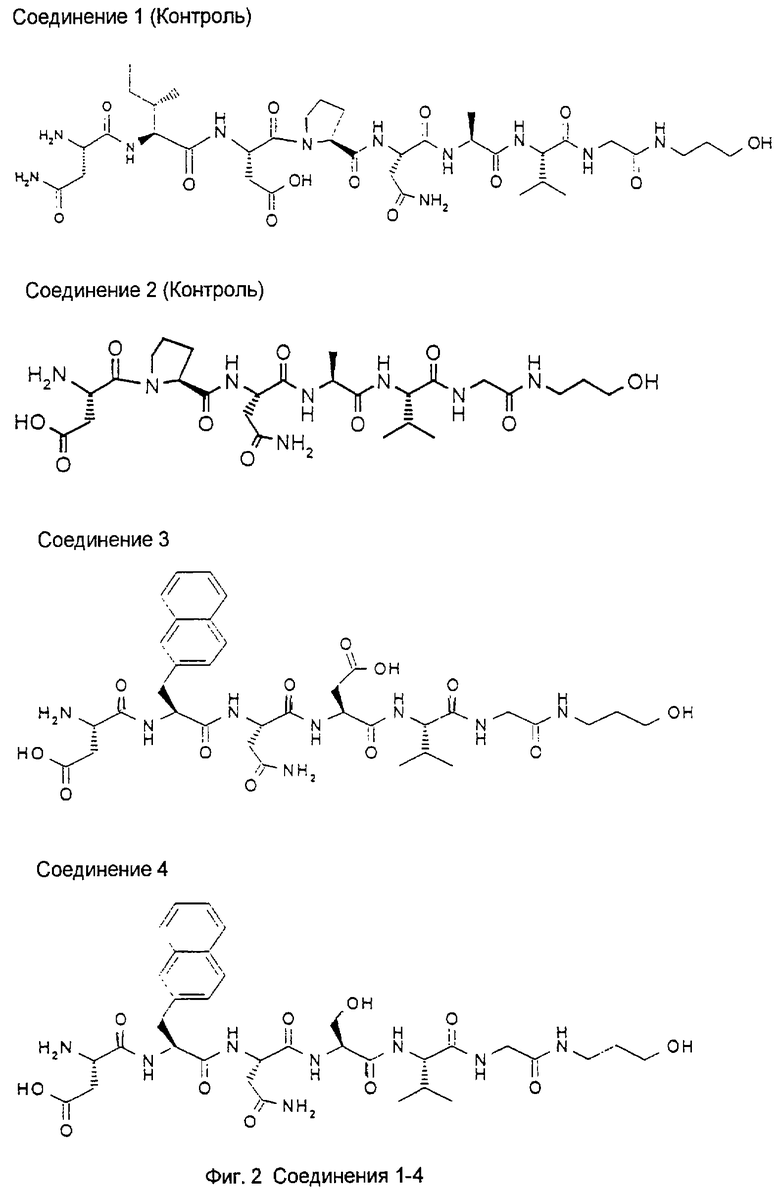
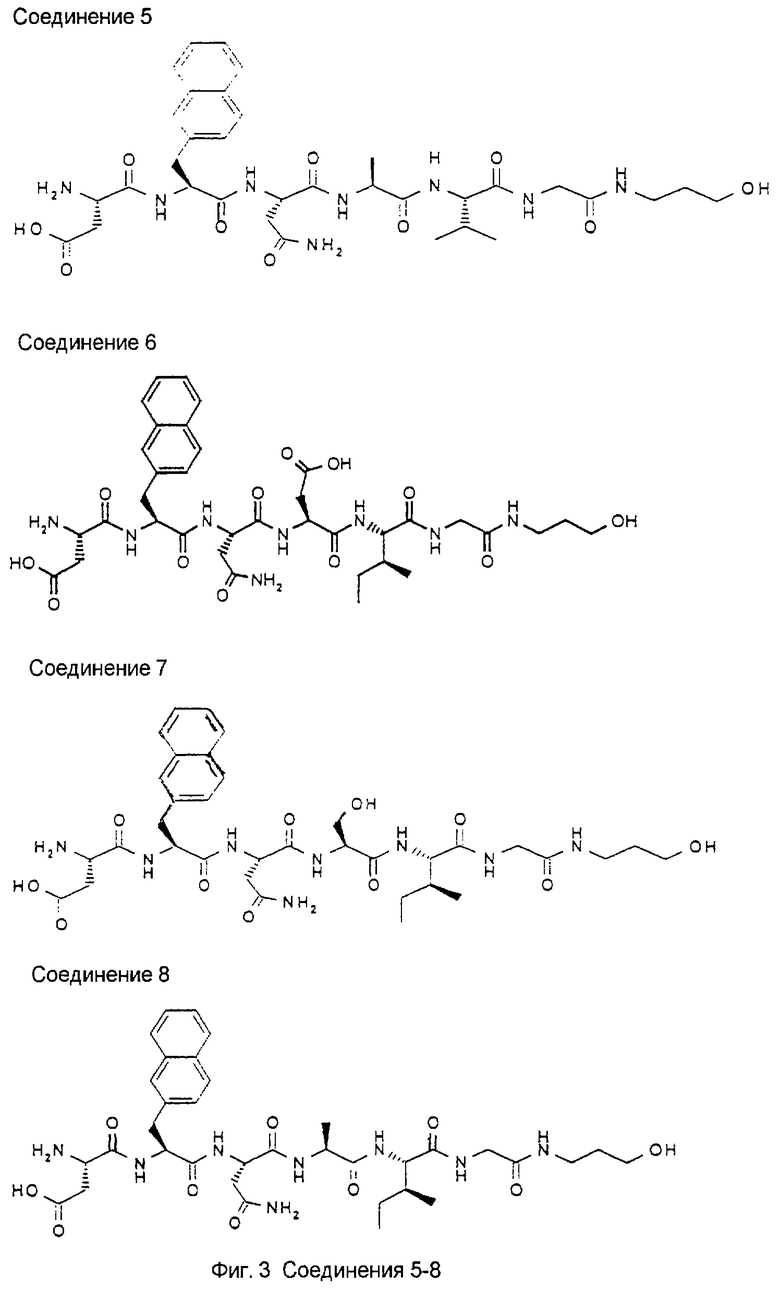
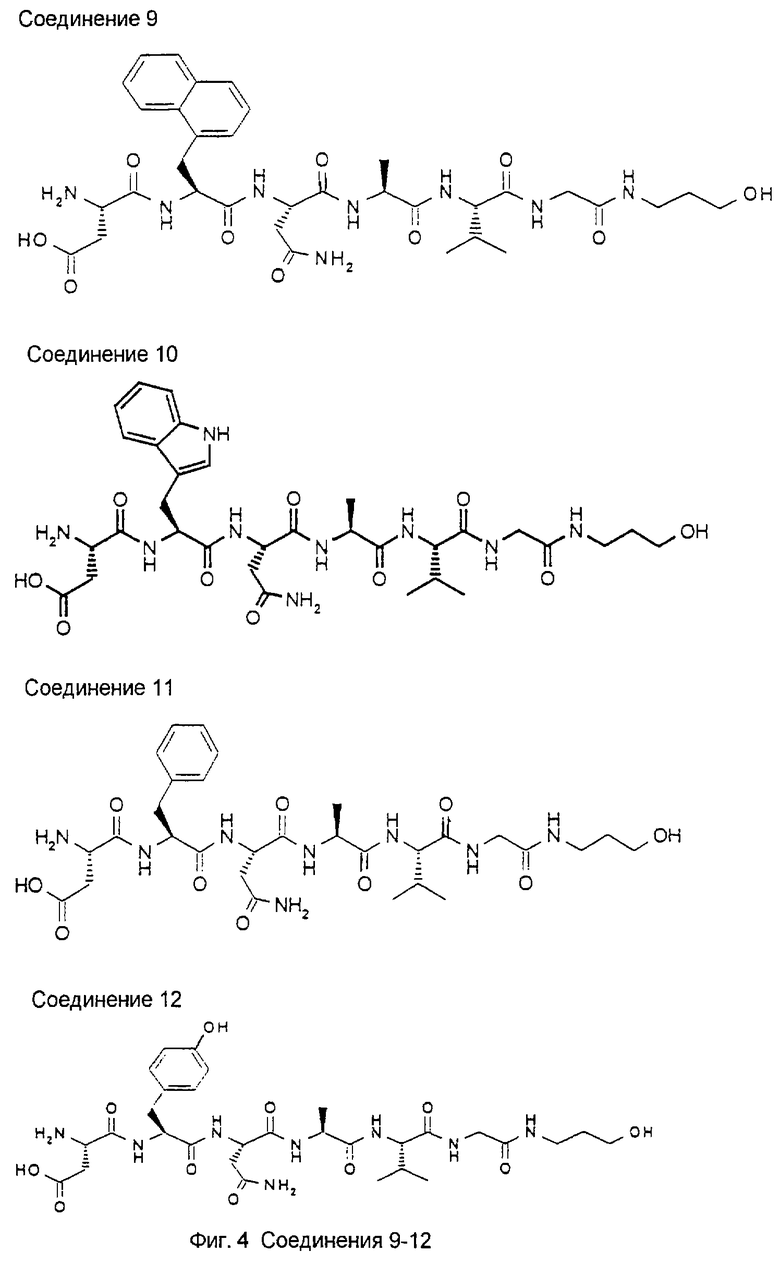
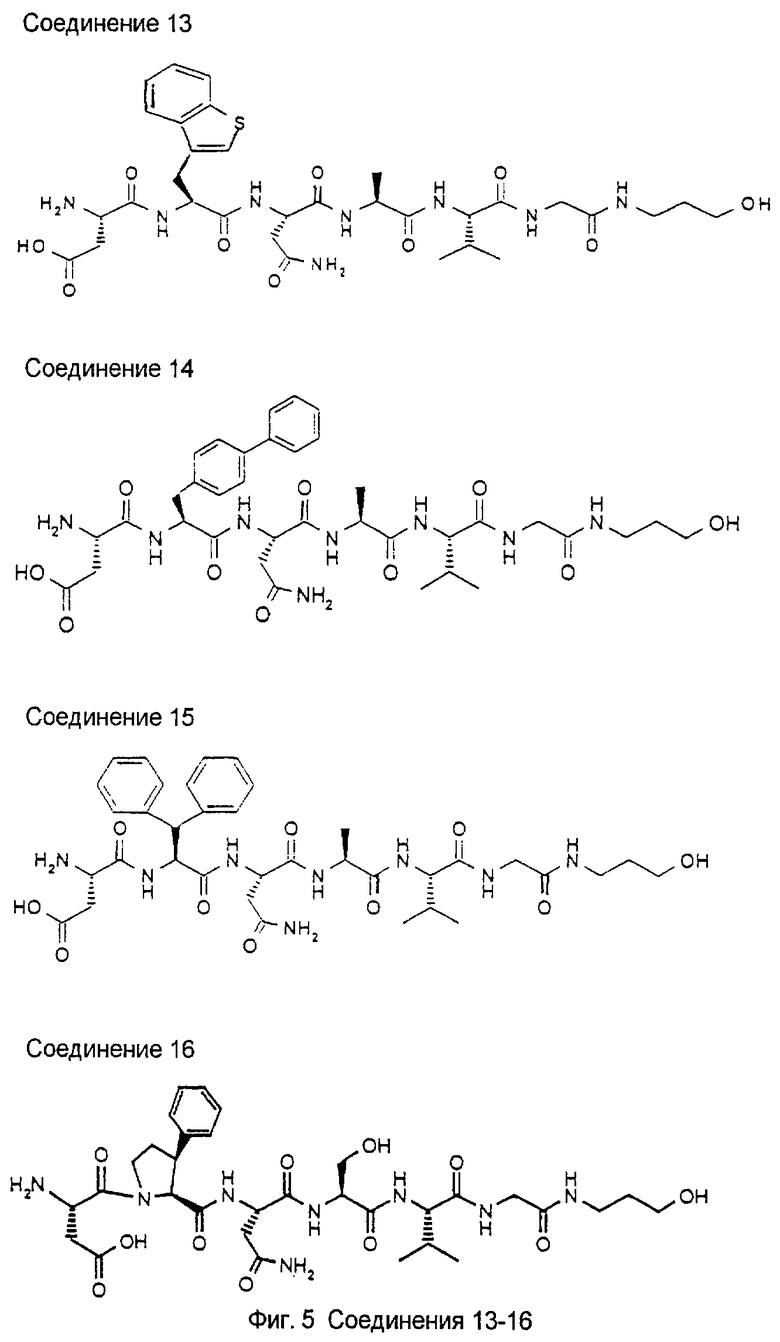
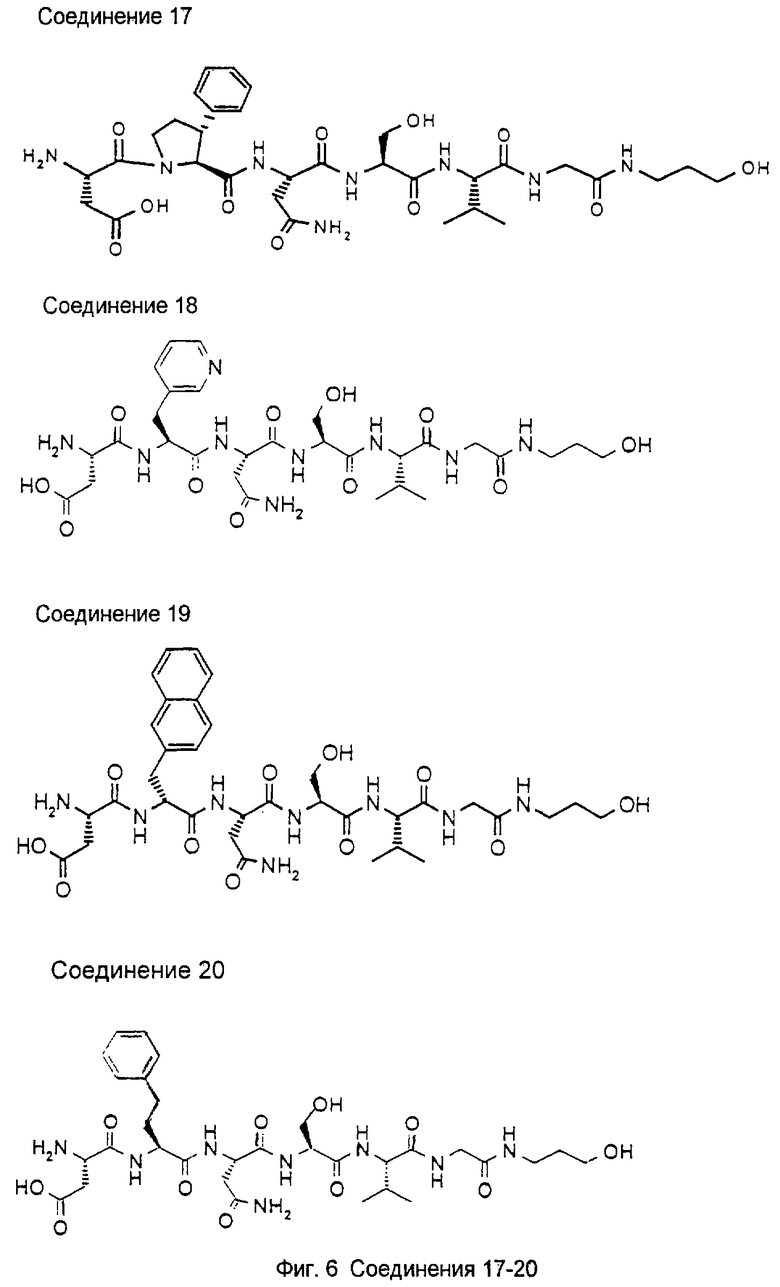
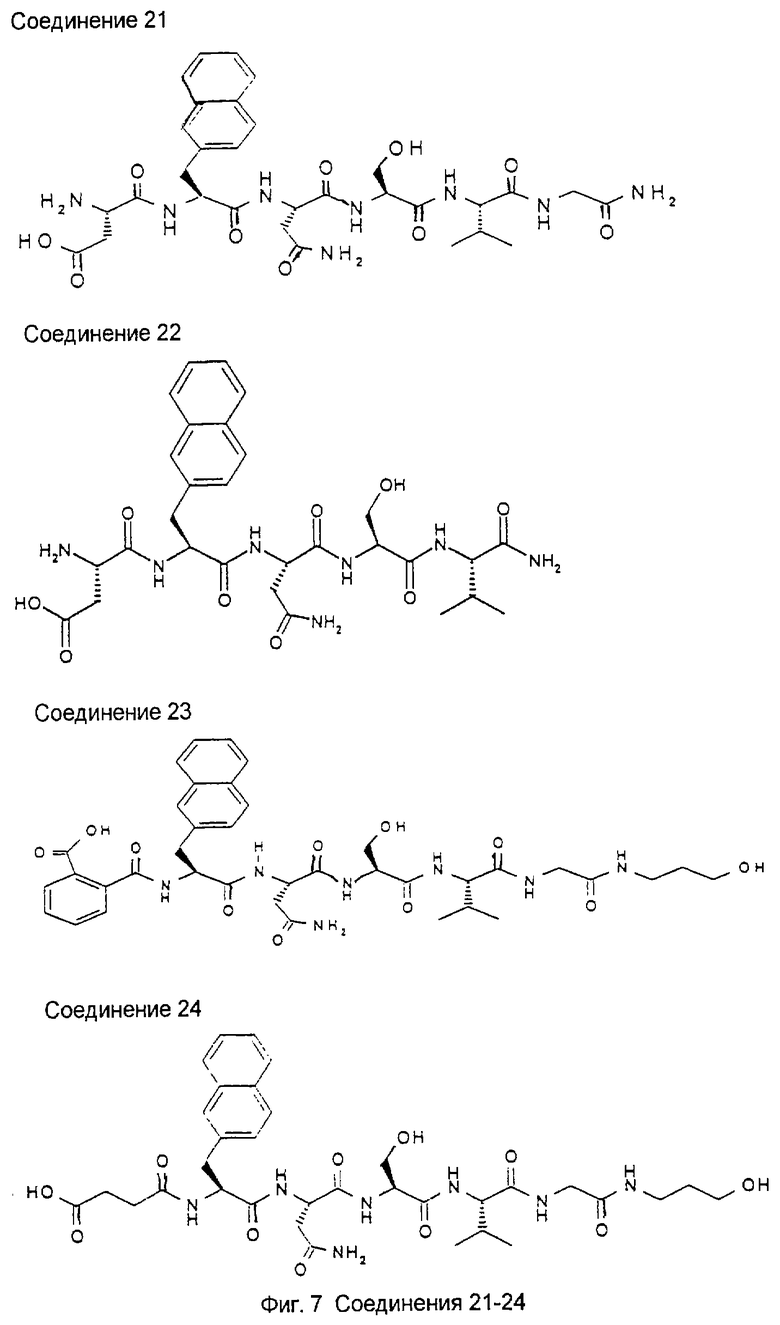
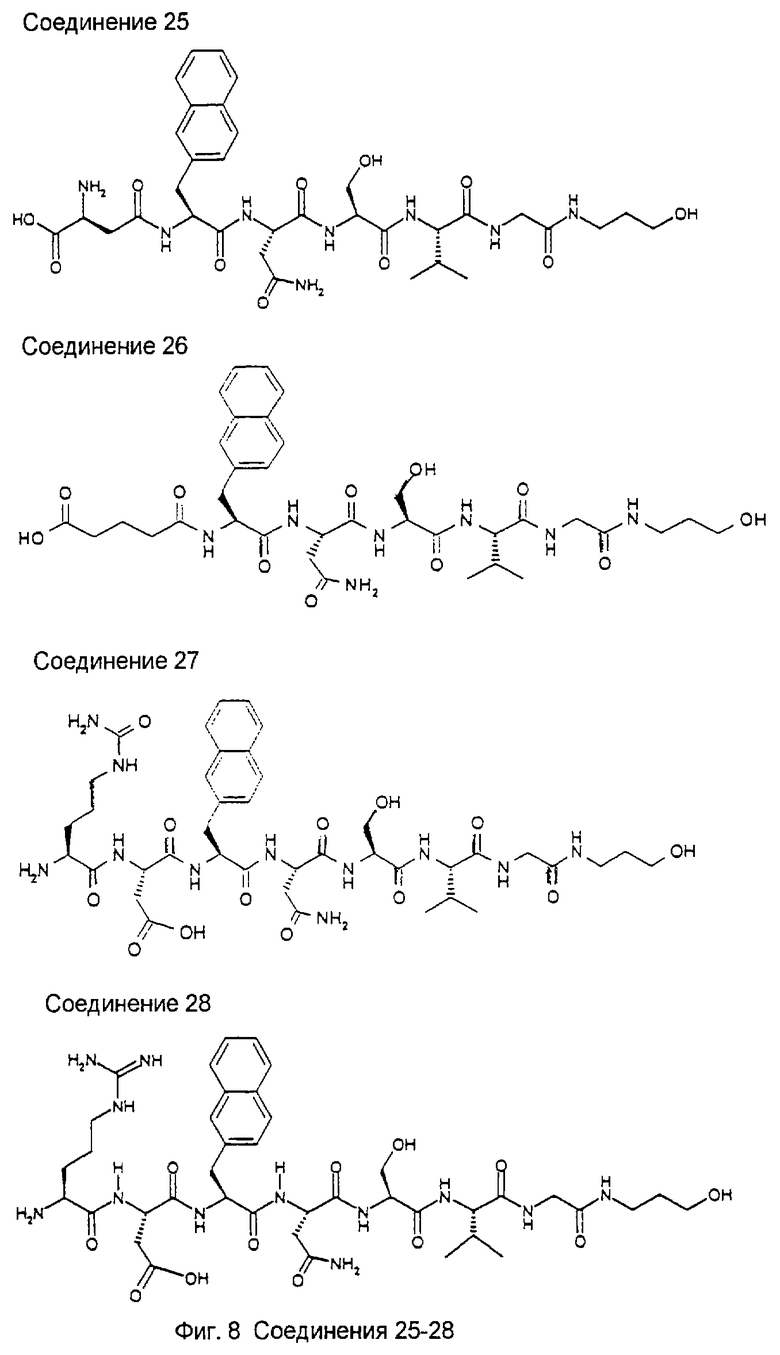
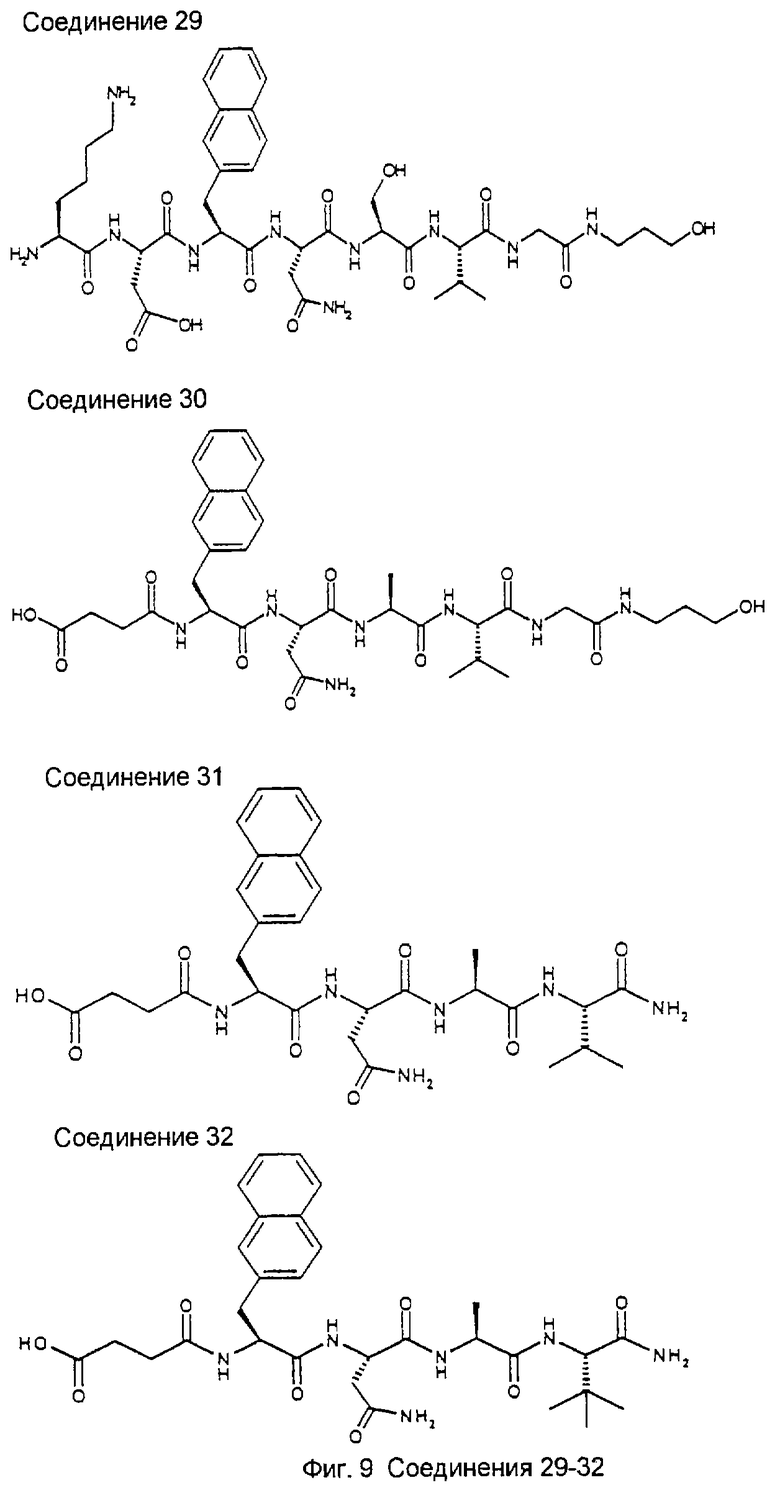
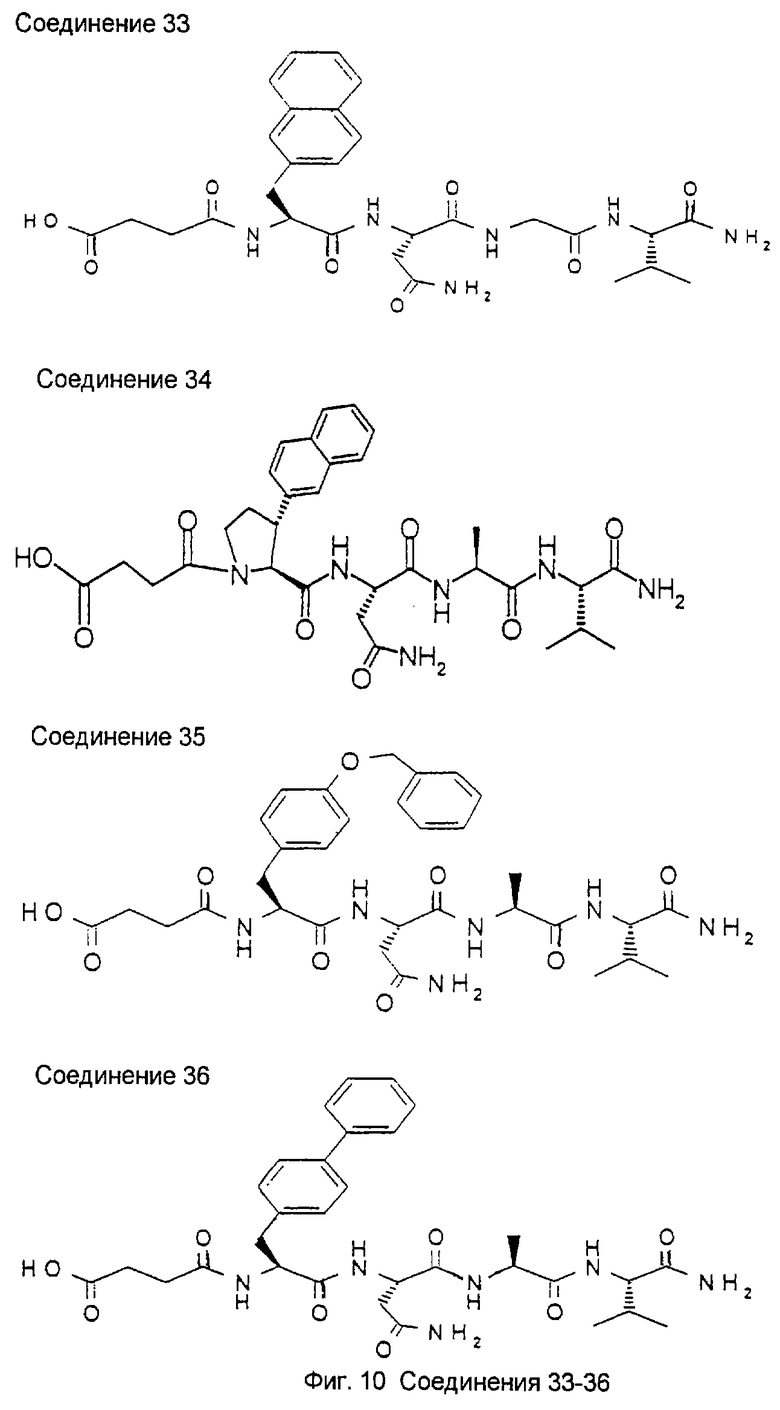
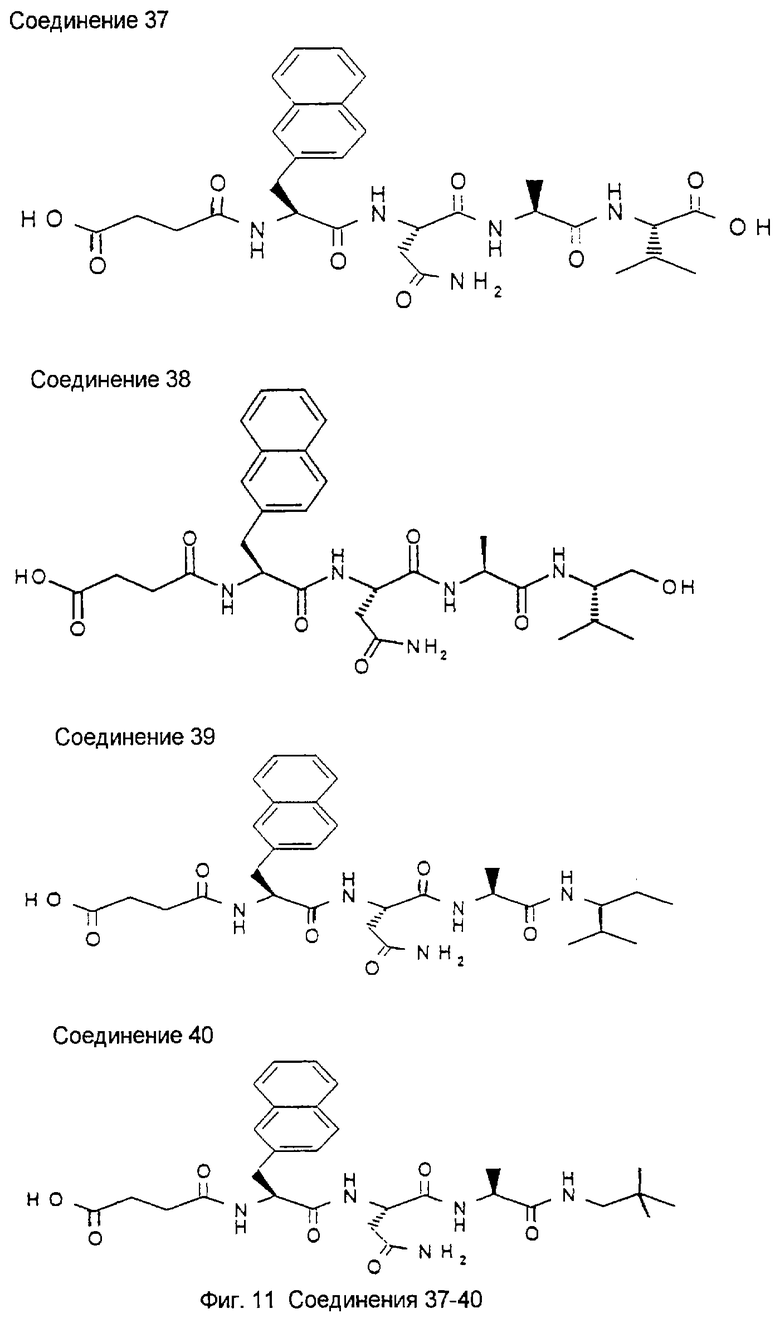
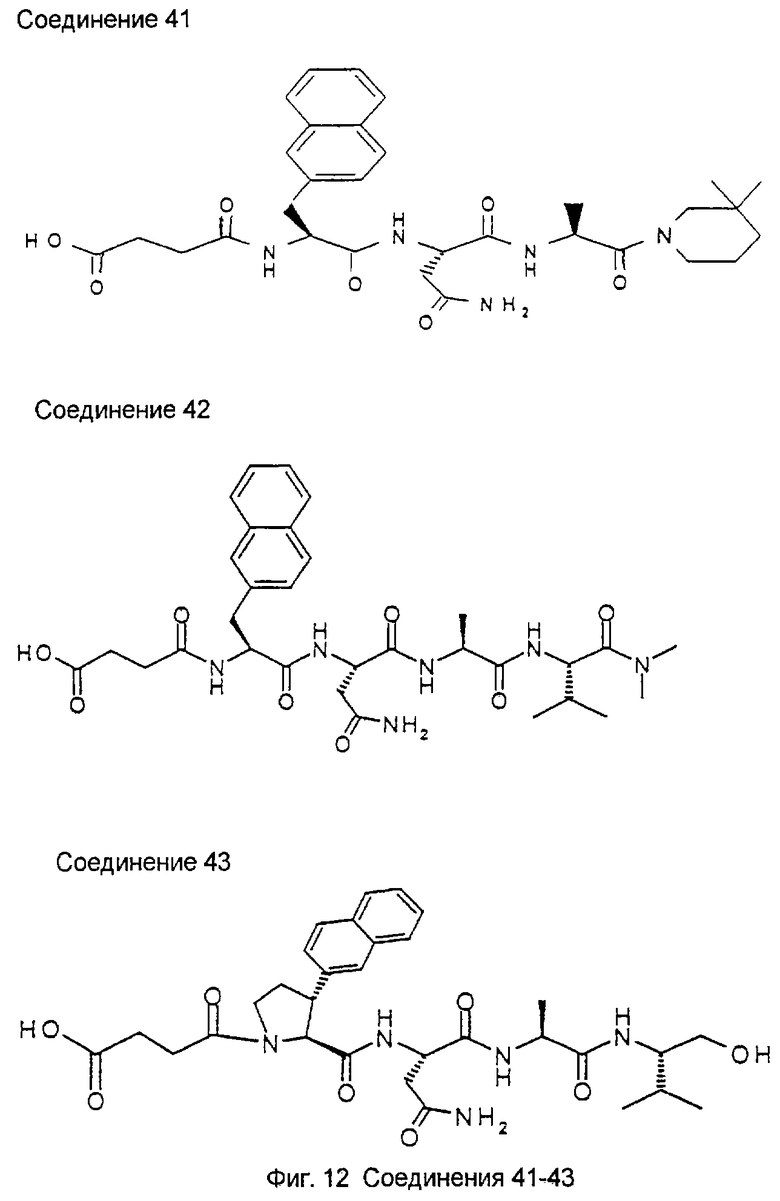
Объектом настоящего изобретения являются низкомолекулярные производные пептидов, которые способны действовать как ингибиторы взаимодействия между ламинином и нидогеном (взаимодействия ламинин/нидоген), способ их получения, приготовленные на их основе фармацевтические композиции и их использование для получения фармацевтических средств и для идентификации ингибиторов взаимодействия ламинин/нидоген. 3 н. и 2 з.п.ф-лы, 12 ил.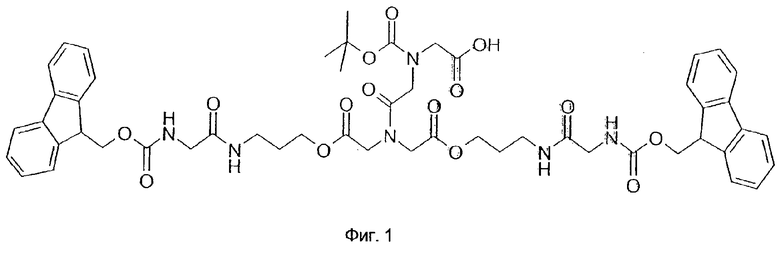
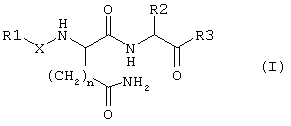
в которой R1 обозначает группу одной из нижеприведенных формул
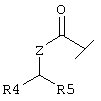 или
или 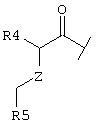 или
или 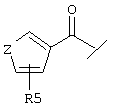
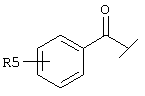
где Z обозначает -(СН2)m;
R4 обозначает -A, -NH2,
и R5 обозначает -(CH2)1СООА, (CH2)1CONH2, -(CH2)1NH2 или
 или
или
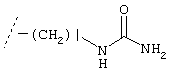
Х обозначает группу, описываемую одной из следующих формул:
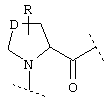
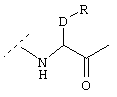
где D обозначает (CH2)r и
R2 обозначает -А, -Е-ОН, где Е обозначает линейную или разветвленную C1-С10-алкильную цепь,
и R3 представляет собой группу одной из нижеприведенных формул
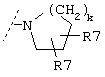 или
или 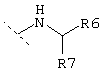
или 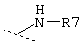
где R6 обозначает
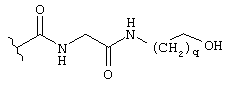
и где R7 обозначает линейную или разветвленную C1-С10-алкильную группу, которая может быть незамещенной или замещенной группами -А, -(СН2)mОН,
и R обозначает Het или Ar, которые необязательно могут быть замещены -O-С1-С6-алкил-Ar или Ar, где Het обозначает моноциклическое или бициклическое, 5-10-членное ароматическое или неароматическое кольцо, содержащее 1 или 2 одинаковых или разных гетероатома в качестве членов указанного кольца, выбранных из группы, состоящей из азота и серы, и где
Ar обозначает моноциклическое или бициклическое, 5-10-членное ароматическое кольцо,
Z обозначает (СН2)m;
А обозначает Н или С1-С4-алкил;
l, m и r обозначают целые числа от 0 до 3;
n и k обозначают целые числа от 1 до 2;
р обозначает целое число от 0 до 1;
q обозначает целое число от 1 до 3,
включая их физиологически толерантные соли.
| ПРОИЗВОДНЫЕ ПЕПТИДОВ, ОБЛАДАЮЩИЕ СПОСОБНОСТЬЮ РЕГУЛИРОВАТЬ ПРОЛИФЕРАЦИЮ КЛЕТОК, И СПОСОБЫ ИНГИБИРОВАНИЯ КЛЕТОЧНОЙ ПРОЛИФЕРАЦИИ ЧЕЛОВЕКА И ЖИВОТНОГО (ВАРИАНТЫ) | 1989 |
|
RU2079509C1 |
| RU 97109041 A 10.05.1999 | |||
| НЕУПОРЯДОЧЕННАЯ БИБЛИОТЕКА ПЕПТИДОВ, СПОСОБ ЕЕ ПОЛУЧЕНИЯ И СПОСОБ ИДЕНТИФИКАЦИИ ПЕПТИДА, СИНТЕЗИРОВАННОГО ТВЕРДОФАЗНЫМ СИНТЕЗОМ | 1991 |
|
RU2145233C1 |
| US 5493008 A 20.02.1996 | |||
| MAYER U | |||
| et al | |||
| «A single EGF-like motif of laminine is responsible for high affinity NIDOGEN Binding» EMBO journal, 1993, v.12, №5, p.1879-85. | |||

