Настоящее изобретение относится к применению перфторалкилсодержащих комплексов металлов, характеризующихся критической концентрацией мицеллообразования менее 10-3 молей/л, гидродинамическим диаметром мицелл (2Rh) более 1 нм и релаксационностью протонов в плазме (R1) более 10 л/ммоль-с, в качестве контрастных веществ в магнитно-резонансной (МР) томографии для визуализации бляшек, лимфатических узлов, инфарцированной и некротической ткани, а также для независимой визуализации некротической ткани и опухолевой ткани. При создании изобретения было установлено, что перфторалкилсодержащие комплексы металлов, обладающие указанными выше свойствами, наиболее пригодны для независимой визуализации бляшек, опухолей и некрозов с помощью МР-томографии и одновременно позволяют охватить играющую важную роль в диагностике инфарктов и некрозов область томографии.
Артериосклероз представляет собой имеющее наиболее важное значение и наиболее часто встречающееся болезненное изменение артерий, сопровождающееся уплотнением и утолщением их стенок, утратой их стенками эластичности и стенозом. Артериосклероз является наиболее частой причиной смерти в западных промышленно развитых странах. Изменения стенок сосудов в результате отложения липидов, разрастания соединительной ткани и кальциноза с неравномерным распределением приводят к нестабильности стенок сосудов, их сужению и к отложению сгустков. Подобное заболевание обусловлено самыми различными экзогенными и эндогенными факторами, соответственно болезнями, например гипертонией, гиперлипидемией, гиперфибриногенемией, сахарным диабетом, токсинами, никотином, комплексами антиген-антитело, воспалительными процессами, гипоксией, психическим стрессом, возрастом и проблемами в семейных отношениях. Под влиянием всех подобных факторов нарушается целостность внутренней оболочки сосудов, нарушается контроль за ростом гладкомышечных клеток стенок сосудов, а также нарушается процесс разложения компонентов состарившихся клеток. Собственно артериосклероз в принципе не поддается лечению, и поэтому все врачебные усилия преимущественно сводятся к предупреждению этого заболевания за счет уменьшения факторов риска его возникновения, например за счет использования средств, понижающих уровень липидов в крови.
Для диагностики артериосклероза в настоящее время в клинической практике преимущественно используется ангиография, которая в этой области является своего рода "золотым стандартом". Однако все методы, основанные на измерении степени уменьшения просвета сосуда, ограничены диагностикой той ранней стадии развития заболевания, для которой характерно утолщение стенки сосуда при нормальном его просвете (см. Glagov S., Zarins С.К., Quantitating atherosclerosis, Clinical Diagnosis of Atherosclerosis. Quantitative Methods of Evaluation, под ред. Bond M.G., Insull W., Glagov S., Chandler А.В., Cornhill J.F., изд-во Springer Verlag, New York 1983, cc.11-35). Другим методом, позволяющим в диагностических целях оценить состояние стенки сосуда и его просвет, является внутрисосудистое или чрескожное ультразвуковое исследование.
Томография, основанная на методе ядерного магнитного резонанса (ЯМР-томография или МРТ), является современным, неинвазивным радиологическим методом, позволяющим с исключительно высоким пространственным и временным разрешением визуализировать физиологические и патофизиологические структуры. Применение же при подобной томографии особых контрастных веществ, способных селективно накапливаться в определенных тканях и органах, позволяет существенно повысить ее ценность как метода диагностического исследования. Очевидно, что композиции на основе контрастных веществ, способных селективно накапливаться в атеросклеротических бляшках, позволили бы определять в более ранний момент времени местонахождение очага заболевания и степень его развития и тем самым обеспечили бы возможность целенаправленного лечения и профилактики того или иного заболевания, и поэтому поиск пригодных для этой цели контрастных веществ ведется уже достаточно давно.
Так, например, в патенте US 4577636 было предложено использовать гематопорфириновые производные в качестве контрастных веществ для выявления атеросклеротических бляшек. В качестве методов исследования в указанном патенте названы сцинтиграфия, рентгенография, основанные на флюоресценции методы, а для парамагнитных металлопорфиринов - также ЯМР-спектрометрия. В качестве парамагнитных ионов в указанной публикации упомянуты Gd, Cr, Co, Ni, Ag и Eu. Недостаток всех этих соединений состоит в том, что порфирины откладываются в коже и изменяют ее цвет, каковой эффект может сохраняться в течение нескольких недель. Помимо этого такие соединения вызывают фотосенсибилизацию. Кроме того, при длительном нахождении этих соединений in vivo существует опасность "потери" металла металлопорфирином.
В заявке WO 95/09856 описаны металлопорфирины (дейтеропорфирины), предназначенные для диагностики и терапии бляшек. В качестве диагностического метода в этой публикации упомянута МРТ. Однако и относящиеся к этому типу порфирины также изменяют цвет кожи.
В заявке WO 95/09013 описаны конъюгаты из специфически связывающихся полипептидов и комплексов металлов. Согласно указанной заявке эти соединения должны также связываться с бляшками и тем самым обеспечивать возможность их диагностики и терапии. В качестве диагностических методов в этой публикации упомянуты сцинтиграфия, компьютерная томография и МРТ. В этой заявке экспериментально подтвержденные данные приведены только для сцинтиграфии, тогда как для МРТ какие-либо данные отсутствуют.
В патенте US 5807536 описаны меченые фикоцианины, предназначенные для применения в качестве контрастных веществ для визуализации бляшек. В качестве диагностических методов в этой публикации упомянуты рентгенография, компьютерная томография, сцинтиграфия, однофотонная эмиссионная компьютерная томография (ОФЭКТ) и МРТ. Экспериментально подтвержденные данные приведены в указанном патенте только для сцинтиграфии.
Из литературы известны многочисленные контрастные вещества для визуализации инфарктов и некрозов. Попытки повысить эффективность локализации, т.е. определения местонахождения, инфарктов и некрозов за счет применения контрастных веществ при неинвазивных методах, таких как сцинтиграфия или ЯМР-томография, предпринимались уже достаточно давно. При этом большое число опубликованных работ посвящено экспериментальным исследованиям по использованию порфиринов для визуализации некрозов. Однако полученные в ходе подобных исследований результаты носят противоречивый характер. Так, в частности, в работе Winkelman и Hoyes, опубликованной в Nature, 200 (1967), с.903, говорится о селективном накоплении марганец-5,10,15,20-тетракис(4-сульфонатофенил)порфирина (ТФПС) в некротической области опухоли.
В отличие от этого в работе Lyon и др. (Magn. Res. Med. 4 (1987), с.24) говорится о том наблюдавшемся этими авторами эффекте, что марганец-ТФПС распределяется по существу по всему организму, а именно, накапливается в почках, печени, опухоли и лишь незначительно в мышечных тканях. Особый интерес при этом представляет тот факт, что концентрация указанного вещества в опухоли достигает максимума только на 4-й день после введения, и то лишь после повышения дозы с 0,12 ммолей/кг до 0,2 ммолей/кг. Поэтому авторами делается также вывод о неспецифическом накоплении ТФПС в опухолевой ткани. В работе Bockhurst и др., опубликованной в Acta Neurochir. 60 (1994, дополн.), с.347, вновь говорится о селективном связывании Mn-ТФПС с опухолевыми клетками.
В свою очередь по результатам исследований, проводившихся Foster и др. (J. Nucl. Med. 26 (1985), с.756), было установлено, что 11In-5,10,15,20-тетракис(4-N-метилпиридиний)порфирин (ТМПиП) накапливается не в некротической области, а в окружающих ее живых краевых слоях. На основании этого можно было бы сделать очевидный вывод о наличии взаимодействия между порфирином и тканью, что, однако, не обязательно соответствует действительности.
В работе Ni и др., опубликованной в Circulation, т.90, №4, часть 2, с.1468, Реферат №2512 (1994), сообщалось о возможности визуализации затронутых инфарктом областей с помощью марганец-тетрафенилпорфирина (Mn-ТФП) и гадолиний-мезопорфирина (Gd-МП). Согласно заявке WO 95/31219 оба этих вещества использовались для визуализации инфарктов и некрозов. В этой заявке, авторами которой являются Marchal и Ni, говорится (см. пример 3), что при использовании соединения Gd-МП содержание металла в пораженной инфарктом почке находилось на том же уровне, что и в неинфарцированном органе, тогда как в миокарде содержание металла в инфарцированной ткани (см. пример 1) в девять раз превышало его содержание в неинфарцированной ткани. Неожиданным является при этом тот факт, что при МРТ соотношение между интенсивностью сигнала, формируемого инфарцированной тканью, и интенсивностью сигнала, формируемого здоровой тканью, в обоих случаях находилось на сравнительно высоком уровне и составляло 2,10 и 2,19 соответственно. Другие металлопорфирины описаны в заявке DE 19835082 (на имя Schering AG).
Порфирины обладают тенденцией накапливаться в коже, что приводит к ее фотосенсибилизации. Подобная сенсибилизация может сохраняться в течение нескольких дней, а иногда и в течение нескольких недель. В этом заключается нежелательный побочный эффект, проявляющийся при применение порфиринов в качестве диагностикумов. Помимо этого порфирины имеют лишь исключительно низкий терапевтический индекс, поскольку, например, в случае Mn-ТФПС его действие проявляется только при его использовании в дозе 0,2 ммоля/кг, тогда как его летальная доза ЛД50 составляет уже 0,5 ммоля/кг.
Другие контрастные вещества, не являющиеся производными порфиринового каркаса и предназначенные для визуализации некрозов и инфарктов, описаны в заявках DE 19744003 (на имя Schering AG), DE 19744004 (на имя Schering AG) и WO 99/17809 (на имя EPIX).
Так, в частности, в заявке DE 19744003 описаны олигомерные соединения, состоящие из ядра, с которым связано от 1 до 3 комплексов металлов.
В заявке DE 19744004 описаны липофильные комплексы металлов для визуализации некрозов и инфарктов. К таким соединениям относятся металлические комплексы полиаминополикарбоновых кислот, полиаминополифосфоновых кислот, порфиринов, тексафиринов, сапфиринов, пептидов.
В поданной на имя EPIX заявке WO 99/17809 описано применение производных ДТПК (диэтилентриаминпентауксусной кислоты) для визуализации некрозов. Наиболее значимым соединением при этом является гадолиниевый комплекс фосфодиэфира гидроксиметил-ДТПК (MS-325).
Известно также применение перфторалкилсодержащих комплексов металлов в качестве контрастных веществ в МР-томографии. Так, в частности, в заявках WO 97/26017 (на имя Schering) и WO 99/01161 (на имя Schering) описано применение перфторалкилсодержащих комплексов металлов в качестве контрастных веществ в лимфографии. В заявке WO 99/01161 говорится, кроме того, о пригодности этих соединений для визуализации внутрисосудистой полости (контрастные вещества для визуализации пулов крови).
В литературе описано также применение контрастных веществ для индивидуальной визуализации опухолей и некрозов с помощью МР-томографии.
Так, в частности, в заявке ЕР 417870 А1 описаны соединения, предназначенные для диагностики и терапии опухолей. В этой заявке говорится также о возможности визуализации инфарктов и ишемий. Однако в указанной заявке не представлено никаких экспериментально полученных данных, которые подтверждали бы подобную возможность. Описанные в этой заявке соединения представляют собой хелатные комплексы типов N2S2 и N3S с радиоизотопами. В качестве диагностического метода в этой заявке упомянута сцинтиграфия.
Сцинтиграфия используется в качестве диагностического метода и в заявке DE 19646762. В этой публикации описаны хелатные комплексы металлов, применяемые в качестве радиосенсибилизаторов для терапии обусловленных гипоксией опухолей и для диагностики обусловленных гипоксией состояний и некрозов. В описательной части этой заявки в качестве диагностических методов упоминаются ЯМР-диагностика, рентгенодиагностика и радионуклидная диагностика.
В заявке DE 19824653 описаны порфирины в качестве обладающих аффинностью по отношению к некрозам веществ, предназначенные для терапии опухолей. В этой заявке говорится, в частности, что эти соединения накапливаются в некротических и гипоксических областях опухолей. Такие соединения могут применяться в диагностических целях в виде их металлических производных с парамагнитными ионами, соответственно радиоизотопами.
Общим для обеих указанных заявок - DE 19646762 и DE 19824653 - является тот факт, что в них не предусмотрена возможность визуализации некрозов независимо от визуализации опухолей (или наоборот), а возможна только визуализация некроза как части опухоли.
В основу настоящего изобретения была положена задача предложить пригодные для МР-томографии контрастные вещества, которые позволяли бы не только визуализировать бляшки, лимфатические узлы, инфарцированную и некротическую ткань, но и позволяли бы визуализировать некрозы независимо от визуализации опухолей (или наоборот).
При создании изобретения неожиданно было установлено, что перфторалкилсодержащие комплексы металлов, характеризующиеся критической концентрацией мицеллообразования менее 10-3 молей/л, гидродинамическим диаметром мицелл (2Rh) более 1 нм и релаксационностью протонов в плазме (R1) более 10 л/ммоль-с, в высшей степени пригодны для применения в качестве контрастных веществ в МР-томографии для визуализации бляшек. Помимо этого такие соединения могут использоваться также для визуализации лимфатических узлов, инфарцированной и некротической ткани, а также для независимой визуализации некротической и опухолевой тканей.
Под пригодными для предусмотренного изобретением применения перфторалкилсодержащими комплексами металлов подразумеваются амфифильные соединения, в молекуле которых в качестве неполярного фрагмента имеется перфторалкильная боковая цепь, которая необязательно связана со всей молекулой липофильным линкером. Полярный фрагмент предлагаемых в изобретении соединений образован одним или несколькими металлическими комплексами и необязательно присутствующими другими полярными группами.
В водных системах подобные амфифильные молекулы проявляют характерные для традиционных ПАВ (таких, например, как додецилсульфат натрия, ДСН) свойства. Так, например, эти молекулы снижают поверхностное натяжение воды. С помощью тензиометрии можно определить так называемую критическую концентрацию мицеллообразования (ККМ, в молях/л). С этой целью определяют поверхностное натяжение в зависимости от концентрации анализируемого вещества. ККМ можно вычислить на основании характеристики изменения полученной функции поверхностного натяжения (с). Критическая концентрация мицеллообразования у предлагаемых в изобретении соединений должна быть меньше 10-3 молей/л, предпочтительно меньше 10-4 молей /л.
Предлагаемые в изобретении амфифильные соединения ассоциированы в растворе и представлены в виде агрегатов. Размер (2Rh) подобных агрегатов (имеющих, например, форму мицелл, палочек, облаток и т.п.) можно определять с помощью фотонно-корреляционной спектроскопии (ФКС).
В соответствии с этим вторым критерием является гидродинамический диаметр мицелл 2Rh, который должен быть больше 1 нм. Более предпочтительными являются согласно изобретению те перфторалкилсодержащие комплексы металлов, величина 2Rh которых равна или превышает 3 нм, наиболее предпочтительно превышает 4 нм.
Методика определения ККМ, а также фотонно-корреляционная спектроскопия описаны у H.-D. Dörfler, "Grenzflächen- und Kolloidchemie", изд-во VSH, Weinheim, New York, Basel, Cambridge, Tokyo, 1994.
Третьим критерием служит релаксационность протонов в плазме (R1) при 40°С и напряженности поля 0,47 тесла. Релаксационность, выражаемая в [л/ммоль-с], является количественной мерой, характеризующей уменьшение времени релаксации T1 протонов. Для предусмотренного изобретением применения такая релаксационность должна быть предельно высокой и должна составлять более 10 л/ммоль-с, предпочтительно более 13 л/ммоль-с, наиболее предпочтительно более 15 л/ммоль-с.
Релаксационность R1 [л/ммоль-с] предлагаемых в изобретении контрастных веществ для МР-томографии определяли с помощью прибора Minispec P 20 фирмы Bruker. Измерения проводили при 40°С и напряженности поля 0,47 тесла. Для каждой T1-последовательности: 180°-TI-90°, режим "инверсия-восстановление", измерения проводили в 8 точках. В качестве среды при измерениях использовали бычью плазму, поставляемую фирмой Kraeber. Концентрация контрастного вещества в анализируемых смесях составляла от 0,30 до 1,16 ммолей/л.
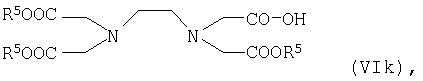
В одном из вариантов осуществления настоящего изобретения предпочтительными для применения являются соединения общей формулы I согласно п.п.8-11 формулы изобретения. При этом речь идет об известных соединениях, описанных в заявке WO 97/26017. Способ получения таких соединений также описан в этой заявке. Неожиданно было установлено, что эти соединения пригодны и для применения в качестве контрастных веществ при МРТ для визуализации бляшек. Наиболее предпочтительно при этом применять соединения, представляющие собой металлические комплексы I-IV, VI и XI-XIII (см. также таблицу 1).
В другом варианте осуществления настоящего изобретения предпочтительными для применения являются соединения общей формулы Ia согласно п.п.12-21. Эти соединения известны и описаны в заявке WO 99/01161. Однако возможность применения таких соединений в качестве контрастных веществ при МРТ для визуализации бляшек до настоящего времени в литературе описано не было. Среди этих соединений особенно предпочтительно применять металлический комплекс XIV (см. таблицу 1).
Согласно еще одному предпочтительному варианту осуществления изобретения можно применять макроциклические перфторалкильные соединения общей формулы Ib
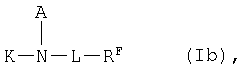
в которой
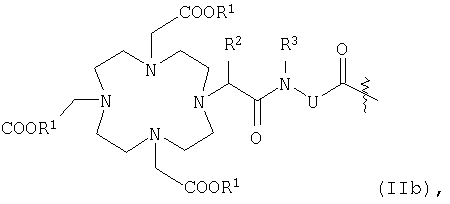
К представляет собой комплексообразователь или металлический комплекс общей формулы IIb

где
R1 обозначает атом водорода или эквивалент иона металла с порядковым номером 23-29, 42-46 или 58-70,
R2 и R3 обозначают атом водорода, С1-С7алкильную группу, бензильную группу, фенильную группу, -СН2ОН или -СН2-ОСН3 и
U2 представляет собой остаток L1, при этом L1 и U2 независимо друг от друга могут иметь идентичные или различные значения,
А1 представляет собой атом водорода, линейную или разветвленную С1-С30алкильную группу, которая необязательно прервана 1-15 атомами кислорода и/или необязательно замещена 1-10 гидроксигруппами, 1-2 СООН-группами, фенильной группой, бензильной группой и/или 1-5-ORg-группами, где Rg обозначает атом водорода либо С1-С7алкильный остаток, или -L1-RF,
L1 представляет собой линейную или разветвленную С1-С30алкиленовую группу, которая необязательно прервана 1-10 атомами кислорода, 1-5 -NH-CO-группами, 1-5 -CO-NH-группами, необязательно замещенной СООН-группой фениленовой группой, 1-3 атомами серы, 1-2 -N(B1)-SO2-группами и/или 1-2 -SO2-N(B1)-группами, где В1 имеет указанные для А1 значения или обозначает NHCO-группу, CONH-группу, N(B1)-SO2-группу либо -SO2-N(B1)-группу и/или необязательно замещена остатком RF, и
RF представляет собой линейный или разветвленный перфорированный алкильный остаток формулы СnF2nЕ, где
n обозначает числа от 4 до 30, а
Е обозначает концевой атом фтора, хлора, брома, йода или водорода, при этом необязательно присутствующие кислотные группы при определенных условиях могут быть представлены в виде солей органических и/или неорганических оснований или аминокислот либо амидов аминокислот.
Поскольку предлагаемые в изобретении соединения предназначены для их применения при ЯМР-диагностике, ион металла формирующей сигнал группы должен быть парамагнитным. К подобным ионам относятся прежде всего двух- и трехвалентные ионы элементов порядковых номеров 23-29, 42-46 и 58-70. В качестве примера пригодных для применения в указанных целях ионов можно назвать ионы хрома(III), железа(II), кобальта(II), никеля(II), меди(II), празеодима(III), неодима(III), самария(III) и иттербия(III). Наиболее предпочтительны при этом с учетом их высокого магнитного момента ионы гадолиния(III), тербия(III), диспрозия(III), гольмия(III), эрбия(III), железа(III) и марганца(II).
Предпочтительны ионы марганца(II), железа(II), железа(III), празеодима(III), неодима(III), самария(III), гадолиния(III) и иттербия(III), прежде всего диспрозия(III).
Алкильные группы, указанные в качестве значений для R2, R3, Rg, могут иметь прямую или разветвленную цепь. В качестве примеров таких групп можно назвать метил, этил, пропил, изопропил, н-бутил, 1-метилпропил, 2-метилпропил, н-пентил, 1-метилбутил, 2-метилбутил, 3-метилбутил и 1,2-диметилпропил.
Предпочтительными значениями R2, R3, и Rg являются водород и С1-С4алкильные группы, а наиболее предпочтительными являются водород и метильная группа.
Бензильная и фенильная группа, указанные в качестве значений для R2, A1 и В1, могут быть замещены в фенильном кольце. В качестве заместителя при этом рассматривается СООН-группа.
Если в соединении формулы Ib одновременно присутствуют остатки L1 и U2, то каждый из этих остатков L1 и U2 может иметь значение, отличное от значения другого остатка.
С1-С30алкиленовые группы, указанные в качестве значений для U2, могут иметь прямую или разветвленную цепь. В качестве примера таких групп можно назвать метилен, этилен, пропилен, изопропилен, н-бутилен, 1-метилпропилен, 2-метилпропилен, н-пентилен, 1-метилбутилен, 2-метилбутилен, 3-метилбутилен и 1,2-диметилпропилен.
Предпочтительными значениями для U2, когда он представляет собой алкилен, являются C1-С10алкиленовые группы, наиболее предпочтительно С1-С4алкиленовые группы.
С1-С30алкильные группы, указанные в качестве значений для А1, могут иметь прямую или разветвленную цепь. В качестве примера таких групп можно назвать метил, этил, пропил, изопропил, н-бутил, 1-метилпропил, 2-метилпропил, н-пентил, 1-метилбутил, 2-метилбутил, 3-метилбутил, 1,2-диметилпропил и н-гексил.
Помимо этого С1-С30алкильные группы, указанные в качестве значений для А1, могут быть прерваны 1-15 атомами кислорода и/или могут быть замещены 1-10 гидроксигруппами, 1-5 алкоксигруппами или 1-2 СООН-группами, и в соответствии с этим могут прдставлять собой, например С2Н4-O-СН3, С3Н6-O-СН3, С2Н4-O-(С2Н4-O)t-С2Н4-ОН, С2Н4-O-(С2Н4-O)t-С2Н4-ОСН3, где t=0-13, С2Н4OH, С3Н6ОН, C4H8OH, С3Н10ОН, C6H12OH, C7H14OH, а также их разветвленные изомеры, СН(ОН)СН2OH, СН(ОН)СН(ОН)СН2OH, СН2[СН(ОН)]u1СН2OH, где u1=1-10, СН[СН2(ОН)]СН(ОН)СН2OH, С2Н4СН(ОН)СН2OH, (CH2)sCOOH, где s=1-15, С2Н4-O-(С2Н4-O)t-СН2СООН, где t=0-13, С2Н4-O-(С2Н4-O)t-С2Н4-СnF2nЕ, где t=0-13, n=4-20, а Е обозначает атом фтора, водорода, хлора, брома или йода.
Предпочтительными значениями А1 являются водород, C1-С10алкил, С2Н4-O-СН3, С3Н6-O-СН3, С2Н4-O-(С2Н4-O)х-С2Н4-ОН, С2Н4-O-(С2Н4O)х-С2Н4-ОСН3, где х=0-5, С2Н4OH, С3Н6ОН, СН2[СН(ОН)]уСН2OH, где y=1-6, СН[СН2(ОН)]СН(ОН)СН2OH, (CH2)wCOOH, где w=1-10, С2Н4-O-(С2Н4-O)х-СН2СООН, где х=0-5, C2H4-O-(C2H4-O)x-C2H4-CnF2nE, где х=0-5, n=4-15, а Е обозначает атом фтора.
Если в соединении общей формулы Ib присутствуют два остатка L1-RF, то они могут иметь различные, не зависящие друг от друга значения.
В качестве примера значения остатка L можно назвать следующие, при этом α обозначает связь с атомом азота, а β обозначает связь с остатком RF:
α-(СН2)s-β, где s=1-15, α-СН2-СН2-(O-СН2-СН2-)у-β, где y=1-6,
α-СН2-(O-СН2-СН2-)у-β, где y=1-6, α-CH2-NH-CO-β, α-CH2-CH2-NH-SO2-β,
α-CH2-NH-CO-CH2-N(CH2COOH)-SO2-β, α-CH2-NH-CO-CH2-N(C2H5)-SO2-β,
α-CH2-NH-CO-CH2-N(C10Н21)-SO2-β, α-CH2-NH-CO-CH2-N(C6H13)-SO2-β,
α-CH2-NH-CO-(CH2)10-N(C2H5 )-SO2-β, α-CH2-NH-CO-CH2-N(-CH2-C6H5)-SO2-β,
α-CH2-NH-CO-CH2-N(-CH2-CH2-OH)SO2-β, α-CH2-NHCO-(CH2)10-S-CH2CH2-β,
α-СН2NHCOCH2-O-СН2СН2-β, α-СН2-СН2NHCOCH2-O-СН2СН2-β,
α-СН2-(СН2-СН2-O)у-(СН2)3NHCO-СН2-O-СН2СН2-β, где y=1-6,
α-CH2NHCO(CH2)10-O-CH2CH2-β, α-CH2CH2NHCO(CH2)10-O-CH2CH2-β,
α-СН2-С6Н4-O-СН2СН2-β, при этом фениленовая группа присоединена в положении 1,4 или 1,3, α-СН2-O-СН2-С(СН2-ОСН2СН2-С6F13)2-СН2-ОСН2-СН2-β,
α-CH2-NHCOCH2CH2CON-CH2CH2NHCOCH2N(C2H5)SO2C8F17-β,
α-CH2-CH2NHCOCH2N(C2H5)-SO2-β,
α-CH2-O-CH2-CH(OC10Н21)-CH2-O-CH2CH2-β, α-(CH2NHCO)4-CH2O-CH2CH2-β,
α-(СН2NHCO)3-СН2O-СН2СН2-β, α-СН2-ОСН2С(СН2OH)2-СН2-O-СН2СН2-β,

α-CH2NHCOCH2N(C6H5)-SO2-β, α-NHCO-CH2-CH2-β, α-NHCO-CH2-O-CH2CH2-β,
α-NH-CO-β, α-NH-CO-CH2-N(CH2COOH)-SO2-β, α-NH-CO-CH2-N(C2H5)-SO2-β,
α-NH-CO-CH2-N(C10Н21)-SO2-β, α-NH-CO-CH2-N(C6H13)-SO2-β,
α-NH-CO-(CH2)10-N(C2H5)-SO2-β, α-NH-CO-CH2-N(-CH2-C6H5)-SO2-β,
α-NH-CO-CH2-N(-CH2-CH2-OH)SO2-β, α-NH-CO-CH2-β,
α-СН2-O-С6Н4-O-СН2-СН2-β, α-СН2-С6Н4-O-СН2-СН2-β, α-N(С2Н5)-SO2-β,
α-N(C6H5)-SO2-β, α-N(C10Н21)-SO2-β, α-N(C6H13)-SO2-β, α-N(C2H4OH)-SO2-β,
α-N(CH2COOH)-SO2-β, α-N(CH2C6H5)-SO2-β, α-N-[CH(CH2OH)2]-SO2-β,
α-N-[CH(CH2OH)CH(OH)(CH2OH)]-SO2-β.
Предпочтительны, в частности, следующие остатки: α-СН2-О-СН2СН2-β,
α-СН2-СН2-(O-СН2-СН2-)у-β, где y=1-6, α-СН2-(O-СН2-СН2-)у-β, где y=1-6,
α-CH2-CH2-NH-SO2-β, α-CH2NHCOCH2-O-CH2CH2-β,
α-CH2-CH2NHCOCH2-O-CH2CH2-β,
α-СН2-(СН2-СН2-O)у-(СН2)3NHCO-СН2-O-СН2СН2-β, где y=1-6,
α-CH2NHCO(CH2)10-O-CH2CH2-β, α-CH2CH2NHCO(CH2)10-O-CH2CH2-β,
α-СН2-O-СН2-СН(ОС10Н21)-СН2-O-СН2СН2-β, α-СН2-O-С6Н4-O-СН2-СН2-β,
α-СН2-С6Н4-O-СН2-СН2-β.
Согласно изобретению наиболее предпочтительны остатки L1, указанные в приведенных ниже примерах соединений.
К значениям U2 относятся приведенные выше остатки в качестве значений L1 и указанные как предпочтительные и наиболее предпочтительные остатки, а также приведенные выше для значения "алкилен" и при определенных условиях указанные в качестве предпочтительных и наиболее предпочтительных остатки, при условии, что в α-положении не должен присутствовать атом азота, а в концевом положении (β-положении) не должна присутствовать SO2- или СО-группа.
Предпочтительными значениями остатка В1 являются водород, линейные или разветвленные С1-С10алкильные остатки, которые необязательно прерваны 1-5 атомами кислорода и/или необязательно замещены 1-5 гидроксигруппами, 1-2 СООН-группами, необязательно замещенной СООН-группой, фенильной группой, бензильной группой и/или 1-5 -ORg-группами, где Rg представляет собой атом водорода или С1-С3алкильный остаток.
Предпочтительными значениями остатка RF являются линейные или разветвленные перфторированные алкильные остатки формулы CnF2nE, в которой n обозначает числа от 4 до 15, а Е представляет собой концевой атом фтора.
Предлагаемые в изобретении соединения общей формулы Ib

в которой К представляет собой комплексообразователь или комплекс металла общей формулы IIb
можно получать рассмотренными ниже методами.
Метод А
Карбоновая кислота формулы IIIb уже содержит эквивалент иона металла R1.

Необязательно активированную in situ карбоновую кислоту формулы IIIb, в которой R1 представляет собой эквивалент иона металла, подвергают взаимодействию в условиях реакции сочетания с амином формулы IVb с получением амида формулы Ib.
Этот метод получения металлических комплексов амидов карбоновых кислот известен из DE 19652386.
Используемую в реакции сочетания смесь, состоящую из металлического комплекса карбоновой кислоты IIIb, присутствующие в которой при определенных условиях карбокси- и/или гидроксигруппы представлены в защищенной форме, и по меньшей мере одного способствующего растворению вещества в количестве до 5, предпочтительно от 0,5 до 2, молярных эквивалентов в пересчете на металлический комплекс карбоновой кислоты, либо можно получать на предшествующей реакционной стадии, выделять (например, путем упаривания, сублимационной сушки или распылительной сушки водного или смешивающегося с водой раствора компонентов или же путем осаждения из подобного раствора органическим растворителем) и затем подвергать взаимодействию в ДМСО с дегидратирующим реагентом (осушителем) и необязательно со вспомогательным агентом сочетания, либо можно получать in situ необязательно за счет добавления к суспензии металлического комплекса карбоновой кислоты в ДМСО способствующего(-их) растворению вещества(веществ), дегидратирующего реагента и необязательно вспомогательного агента сочетания.
Полученный одним из этих методов реакционный раствор для его предварительной обработки (активирования кислоты) в течение 1-24 ч, предпочтительно 3-12 ч, выдерживают при температуре от 0 до 50°С, предпочтительно при комнатной температуре.
После этого в реакционный раствор добавляют амин общей формулы IVb
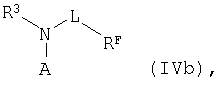
в которой остатки R3, L1, RF и А1 имеют указанные выше значения, который используют без растворителя или в растворенном виде, например в диметилсульфоксиде, спиртах, таких как метанол, этанол, изопропанол или их смеси, формамид, диметилформамид, вода либо смеси указанных растворителей, предпочтительно в диметилсульфоксиде, в воде или в смешанных с водой растворителях. Затем для образования амида полученный таким путем реакционный раствор выдерживают для проведения реакции сочетания при температуре от 0 до 70°С, предпочтительно от 30 до 60°С, в течение 1-48 ч, предпочтительно 8-24 ч.
В некоторых случаях, как было установлено, амин предпочтительно использовать в реакции в виде его соли, например в виде гидробромида или гидрохлорида. Для высвобождения амина добавляют основание, такое, например, как триэтиламин, диизопропилэтиламин, N-метилморфолин, пиридин, трипропиламин, трибутиламин, гидроксид лития, карбонат лития, гидроксид натрия или карбонат натрия.
После этого все еще присутствующие при определенных условиях защитные группы отщепляют.
Реакционный продукт выделяют известными методами, предпочтительно осаждением органическими растворителями, предпочтительно ацетоном, 2-бутаноном, диэтиловым эфиром, этиловым эфиром уксусной кислоты, метил-трет-бутиловым эфиром, изопропанолом или их смесями. Последующую очистку можно проводить, например, путем хроматографии, кристаллизации или ультрафильтрации.
Для применения в качестве способоствующих растворению веществ пригодны соли щелочных металлов, соли щелочноземельных металлов, соли триалкиламмония, соли тетраалкиламмония, мочевины, N-гидроксиимиды, гидроксиарилтриазолы, замещенные фенолы и соли гетероциклических аминов. При этом в качестве конкретных примеров можно назвать хлорид лития, бромид лития, йодид лития, бромид натрия, йодид натрия, метансульфонат лития, метансульфонат натрия, n-толуолсульфонат лития, n-толуолсульфонат натрия, бромид калия, йодид калия, хлорид натрия, бромид магния, хлорид магния, йодид магния, n-толуолсульфонат тетраэтиламмония, n-толуолсульфонат тетраметиламмония, n-толуолсульфонат пиридиния, n-толуолсульфонат триэтиламмония, 2-морфолиноэтилсульфоновую кислоту, 4-нитрофенол, 3,5-динитрофенол, 2,4-дихлорфенол, N-гидроксисукцинимид, N-гидроксифталимид, мочевину, тетраметилмочевину, N-метилпирролидон, формамид, а также циклические мочевины, при этом среди указанных выше пять первых из них предпочтительны.
В качестве дегидратирующих реагентов можно использовать все известные специалистам средства. При этом в качестве примера можно назвать карбодиимиды и ониевые реагенты, такие как дициклогексилкарбодиимид (ДЦКД), 1-этил-3-(3-диметиламинопропил)карбодиимидгидроксихлорид (ЭДК), гексафторфосфат бензотриазол-1-илокситрис(диметиламино)фосфония (БОФ) и гексафторфосфат O-(бензотриазол-1-ил)-1,1,3,3-тетраметилурония (ГБТУ), предпочтительно ДЦКД.
Ниже в качестве примера представлены литературные источники, в которых описаны приемлемые методы;
- активирование карбоновых кислот: обзорная информация в Houben-Weyl, Methoden der Organischen Chemie, том XV/2, изд-во Georg Thieme Verlag Stuttgart, 1974 (и J. Chem. Research (S) 1996, с.302);
- активирование карбодиимидами: R.Schwyzer и Н.Kappeler, Helv. 46 (1963), с.1550;
- E.Wünsch и др., В. 100 (1967), с.173;
- активирование карбодиимидами/гидроксисукцинимидом: J. Am. Chem. Soc. 86 (1964), с.1839, а также J. Org. Chem. 53 (1988), с.3583, Synthesis 453 (1972);
- ангидридный метод, 2-этокси-1-этоксикарбонил-1,2-дигидрохинолин: В.Belleau и др., J. Am. Chem. Soc., 90 (1986), с.1651, Н.Kunz и др., Int. J.Pept. Prot. Res., 26 (1985), с.493, и J.R.Voughn, Am. Soc. 73 (1951), с.3547;
- имидазолидный метод: B.F.Gisin, R.B.Menifield, D.C.Tosteon, Am. Soc. 91 (1969), с.2691;
- методы с хлорангидридами кислот, тионилхлорид: Helv., 42 (1959), с.1653;
- оксалилхлорид: J. Org. Chem., 29 (1964), с.843.
Для применения в качестве необязательно используемых вспомогательных агентов сочетания пригодны все известные специалистам агенты (Houben-Weyl, Methoden der organischen Chemie, т. XV/2, изд-во Georg Thieme Verlag, Stuttgart, 1974). В качестве примера при этом можно назвать 4-нитрофенол, N-гидроксисукцинимид, 1-гидроксибензотриазол, 1-гидрокси-7-азабензотриазол, 3,5-динитрофенол и пентафторфенол. Среди этих соединений предпочтительны 4-нитрофенол и N-гидроксисукцинимид, а наиболее предпочтительным является первый из указанных агентов.
Защитные группы отщепляют по известным методам, например путем гидролиза, гидрогенолиза, щелочного омыления сложных эфиров щелочью в водно-спиртовом растворе при температуре от 0 до 50°С, кислотного омыления минеральными кислотами или - в случае, например, сложных трет-бутиловых эфиров - с помощью трифторуксусной кислоты [Protective Groups in Organic Synthesis, 2-е изд., T.W.Greene и P.G.M.Wuts, изд-во John Wiley and Sons, Inc. New York, 1991], а в случае простых бензиловых эфиров - с помощью водорода в присутствии палладия на угле.
Способ получения исходного материала, т.е. соединений формулы IIIb

известен из DE 19652386.
Амины общей формулы IVb
представляют собой имеющиеся в продаже продукты (Fluorochem, ABCR) или их можно получать следующим методом из соединений общей формулы Vb взаимодействием с амином общей формулы VIb и последующим восстановлением соединений общей формулы VIIb:

где
RF, А1, L1 и R3 имеют указанные выше значения,
L' имеет указанные для группы L1 значения, в которой еще отсутствует α-CH2-группа, и
Rg обозначает водород или метильную группу.
Кислоту формулы Vb перед ее взаимодействием с амином формулы VIb активируют по методам, уже рассмотренным выше для активирования карбоновой кислоты формулы IIIb и описанным в литературе. Если Rg обозначает метильную группу, то проводят аминолиз.
Соединения общей формулы Vb представляют собой имеющиеся в продаже продукты (Fluorochem, ABCR) или их получают по способу, описанному в DE 19603033.
Соединения формулы VIb представляют собой имеющиеся в продаже продукты (Fluorochem, ABCR) или их можно получать по методу, описанному в Houben-Weyl, Methoden der organischen Chemie, т. XI/2, раздел "Stickstoffverbindungen", изд-во Georg Thieme Verlag, Stuttgart, 1957, с.680;.E. Rickman и Т. Atkins, Am. Chem. Soc., 96 (1974), с.2268; F. Chavez и A.D. Sherry, J. Org. Chem., 54 (1989), с.2990.
Соединения общей формулы IVb получают известным методом [Helv. Chim. Acta, 77 (1994), с.23] восстановлением соединений общей формулы VII, например, дибораном или алюмогидридом лития и последующим отщеплением защитных групп.
Метод Б
Исходным материалом служит карбоновая кислота формулы IIIx, где R1 обозначает водород, которая еще не содержит эквивалент иона металла R1. Карбоксильные группы защищают известными методами с получением соединения формулы IIIy, где R5 представляет собой любую приемлемую защитную группу, а R5' представляет собой ее предшественник.
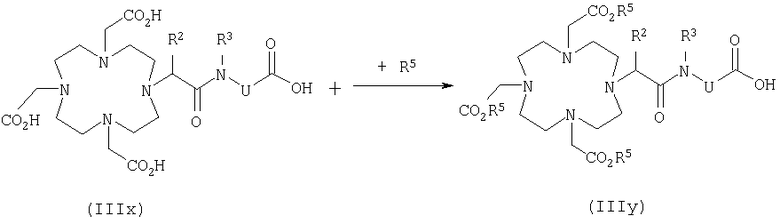
В качестве карбоксизащитных групп могут рассматриваться, например, линейные или разветвленные С1-С6алкильные, арильные и аралкильные группы, например метильная, этильная, пропильная, бутильная, фенильная, бензильная, дифенилметильная, трифенилметильная, бис(n-нитрофенил)метильная, а также триалкилсилильная группы. Предпочтительна трет-бутильная группа.

Взаимодействие защищенной карбоновой кислоты формулы IIIy с амином формулы IVb и отщепление защитных групп проводят по описанной для метода А методике и на последующей стадии полученную карбоновую кислоту формулы Ix дополнительно подвергают взаимодействию по меньшей мере с одним оксидом металла или одной солью металла, требуемого с порядковым номером, как это описано, например, в DE 19525924.
Если полученный по методу А или методу Б комплекс металла все еще содержит свободные СООН-группы, то эти группы могут быть также представлены в виде солей физиологически приемлемых неорганических или органических оснований.
Возможно еще присутствующие свободные карбоксигруппы в этом случае нейтрализуют с помощью неорганических оснований (например, гидроксидов, карбонатов или бикарбонатов), например, натрия, калия, лития, магния или кальция, и/или органических оснований, в частности первичных, вторичных и третичных аминов, таких, например, как этаноламин, морфолин, глюкамин, N-метил- и N,N-диметилглюкамин, а также основных аминокислот, таких, например, как лизин, аргинин и орнитин, или амидов первоначально нейтральных или кислых аминокислот.
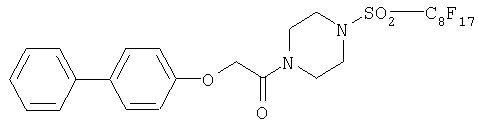
Наиболее предпочтительными для предусмотренного изобретением применения являются металлические комплексы V, VII, VIII, IX и Х (см. таблицу 1).
Эти соединения общей формулы Ib при их применении в качестве контрастных веществ в МРТ особо пригодны для визуализации бляшек.
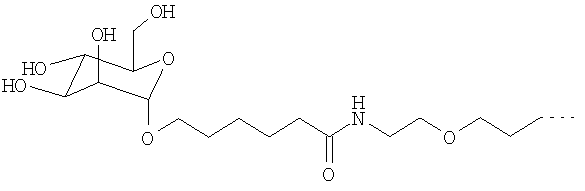
Согласно другому предпочтительному варианту осуществления изобретения можно использовать перфторалкилсодержащие комплексы с остатками сахаров общей формулы Ic
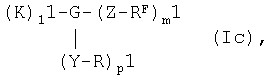
в которой
R обозначает присоединенный через 1-ОН- или 1-SH-положение моно- или олигосахаридный остаток,
RF обозначает перфторированную, прямую или разветвленную углеродную цепь формулы -CnF2nE, где
Е представляет собой концевой атом фтора, хлора, брома, йода или водорода, а
n обозначает числа 4-30,
К обозначает металлический комплекс общей формулы IIc
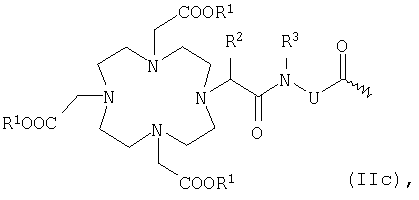
в которой
R1 представляет собой атом водорода или эквивалент иона металла с порядковым номером 23-29, 42-46 или 58-70, при условии, что по меньшей мере два радикала R1 обозначают эквиваленты ионов металлов,
R2 и R3 независимо друг от друга обозначают водород, С1-С7алкил, бензил, фенил, -СН2OH или -СН2ОСН3 и
U представляет собой -С6H4-О-СН2-ω-, -(СН2)1-5-ω-, фениленовую группу,-СН2-NHCO-СН2-СН(СН2СООН)-С6Н4-ω-, -С6Н4-(ОСН2СН2)0-1-N(СН2СООН)-СН2-ω- или необязательно прерванную одним или несколькими атомами кислорода, 1-3 -NHCO-группами, 1-3 -CONH-группами и/или замещенную 1-3 -(СН2)0-5СООН-группами С1-С12алкиленовую или С7-С12-С6Н4-O-группу, при этом ω обозначает место присоединения к -СО-, или общей формулы IIIc

в которой
R1 имеет вышеуказанные значения,
R4 обозначает водород или указанный для R1 эквивалент иона металла и
U1 обозначает -С6Н4-O-СН2-ω-, где ω обозначает место присоединения к -СО-,
или общей формулы IVc

в которой R1 и R2 имеют указанные выше значения,
или общей формулы VcA либо VcB

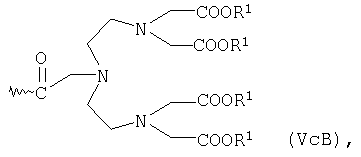
в которой R1 имеет указанные выше значения,
или общей формулы VIc

в которой R1 имеет указанные выше значения,
или общей формулы VIIc
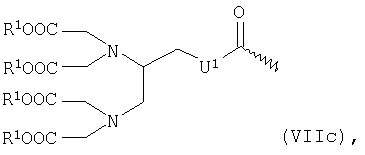
в которой
R1 имеет указанные выше значения, а
U1 обозначает -С6Н4-О-СН2-ω-, где ω обозначает место присоединения к -СО-,
или общей формулы VIIIc

в которой R1 имеет указанные выше значения,
при этом необязательно присутствующие в остатке К свободные кислотные группы необязательно могут быть представлены в виде солей органических и/или неорганических оснований или аминокислот либо в виде амидов аминокислот,
G в том случае, если К обозначает металлический комплекс IIc-VIIc, представляет собой по меньшей мере трехкратно функционализованный остаток, выбранный из следующих остатков a)-j):


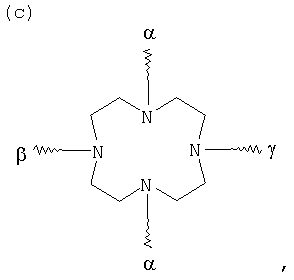

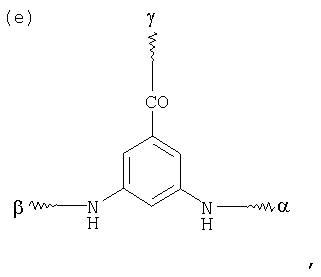

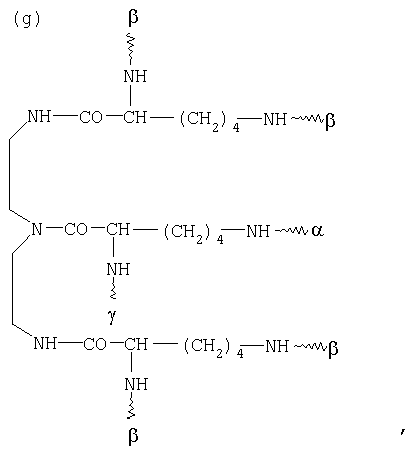


 , и
, и
G в том случае, если К обозначает металлический комплекс VIIIc, представляет собой по меньшей мере трехкратно функционализованный остаток, выбранный из остатков k) и I):

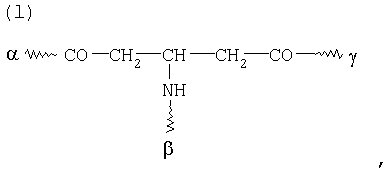
при этом α обозначает место присоединения G к комплексу К, β обозначает место присоединения G к остатку Y, а γ обозначает место присоединения G к остатку Z,
Y обозначает -СН2-, δ-(CH2)1-5CO-β, δ-СН2-СНОН-СО-β или δ-СН(СНОН-СН2OH)-СНОН-СНОН-СО-β, где δ обозначает место присоединения к остатку сахара R, а β обозначает место присоединения к остатку G,
Z обозначает группу

γ-COCH2-N(C2H5)-SO2-ε,
γ-COCH2-O-(CH2)2-SO2-ε,
 или
или
γ-NHCH2CH2-O-CH2CH2-ε,
где γ обозначает место присоединения Z к остатку G, а ε обозначает место присоединения Z к перфторированному остатку RF,
l1, m1 независимо друг от друга обозначают целые числа 1 или 2 и
р1 обозначает целые числа от 1 до 4.
Поскольку предлагаемые в изобретении соединения предназначены для их применения при ЯМР-диагностике, ион металла формирующей сигнал группы должен быть парамагнитным. К подобным ионам относятся прежде всего двух- и трехвалентные ионы элементов порядковых номеров 23-29, 42-46 и 58-70. В качестве примера пригодных для применения в указанных целях ионов можно назвать ионы хрома(III), железа(II), кобальта(II), никеля(II), меди(II), празеодима(III), неодима(III), самария(III) и иттербия(III). Наиболее предпочтительны при этом с учетом их высокого магнитного момента ионы гадолиния(III), тербия(III), диспрозия(III), гольмия(III), эрбия(III), железа(III) и марганца(II).
Предпочтительны ионы марганца(II), железа(II), железа(III), празеодима(III), неодима(III), самария(III), гадолиния(III) и иттербия(III), прежде всего диспрозия(III).
Присутствующие при определенных условиях в R1 кислотные атомы водорода, т.е. атомы, которые не замещены центральным ионом, необязательно могут быть полностью или частично заменены на катионы неорганических и/или органических оснований или аминокислот либо амидов аминокислот.
В качестве примера приемлемых неорганических катионов можно назвать ион лития, ион калия, ион кальция и прежде всего ион натрия. Приемлемыми катионами органических оснований являются, в частности, таковые первичных, вторичных или третичных аминов, таких, например, как этаноламин, диэтаноламин, морфолин, глюкамин, N,N-диметилглюкамин и прежде всего N-метилглюкамин. В качестве примера приемлемых катионов аминокислот можно назвать катионы лизина, аргинина и орнитина, а также амиды в остальном кислых или нейтральных аминокислот.
К наиболее предпочтительным соединениям общей формулы I с относятся соединения, содержащие макроцикл К общей формулы IIc.
Остаток U в металлическом комплексе К предпочтительно обозначает -СН2- или С6Н4-O-СН2-ω, где со представляет собой место присоединения к -СО-.
Указанные в качестве значений R2 и R3 алкильные группы в макроцикле общей формулы IIc могут иметь прямую или разветвленную цепь. При этом в качестве примеров можно назвать метил, этил, пропил, изопропил, н-бутил, 1-метилпропил, 2-метилпропил, н-пентил, 1-метилбутил, 2-метилбутил, 3-метилбутил, 1,2-диметилпропил. Предпочтительно R2 и R3 независимо друг от друга обозначают водород или С1-С4алкил.
В одном из особо предпочтительных вариантов R2 обозначает метил, а R3 обозначает водород.
Указанная в качестве значения R2 или R3 бензильная группа или фенильная группа в макроцикле К общей формулы IIc может быть также замещена в кольце.
Остаток R в общей формуле I с обозначает присоединенный через 1-ОН- или 1-SH-положение моно- или олигосахаридный остаток либо остаток тиосахара, причем в этом случае согласно изобретению речь может также идти о дезоксисахарах, которые вместо одной или более ОН-групп содержат Н-атом. В одном из предпочтительных вариантов осуществления изобретения R обозначает моносахаридный остаток с 5 или 6 С-атомами, предпочтительно глюкозу, маннозу, галактозу, рибозу, арабинозу или ксилозу либо их дезоксисахара, такие, например, как 6-дезоксигалактоза (фукоза) или 6-дезоксиманноза (рамноза), либо их тиосахара, при этом наиболее предпочтительны глюкоза, манноза и галактоза.
Среди предлагаемых в изобретении соединений общей формулы Ic предпочтительны далее соединения, в которых RF обозначает -CnF2n+1. При этом n предпочтительно обозначает числа от 4 до 15. Наиболее предпочтительны остатки -C4F9, -С6F13, -C8F17, -C12F25 и -C14F29, а также остатки рассмотренных в примерах соединений.
По меньшей мере трехкратно функционализованный остаток G в общей формуле Ic, который является "скелетом", в одном из предпочтительных вариантов осуществления изобретения обозначает остаток лизина (а) или (b).
Y и Z представляют собой указанный для общей формулы I с линкер, при этом независимо друг от друга Z предпочтительно обозначает группу
а Y предпочтительно обозначает группу δ-СН2СО-β.
Способ получения перфторалкилсодержащих комплексов металлов с остатками сахаров общей формулы Ic
в которой К обозначает один из металлических комплексов общих формул IIc-VIIc, a G представляет собой одну из групп формул a)-j), при этом Y, Z, R, RF,
m1, р1 и l1 имеют указанные выше значения, заключается в том, что карбоновую кислоту общей формулы IIi

в которой R5 обозначает эквивалент иона металла с порядковым номером 23-29, 42-46 или 58-70 или карбоксизащитную группу, а R2, R3 и U имеют указанные выше значения, или карбоновую кислоту общей формулы IIIi

в которой R4, R5 и U1 имеют указанные выше значения, или карбоновую кислоту общей формулы IVi
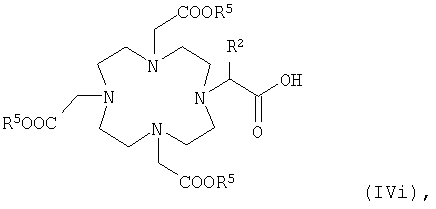
в которой R5 и R2 имеют указанные выше значения, или карбоновую кислоту общей формулы Vi или Vii
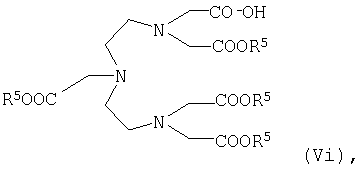
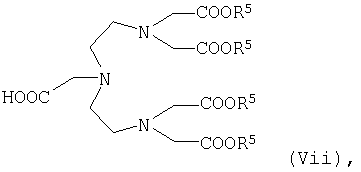
где R5 имеет указанные выше значения, или карбоновую кислоту общей формулы VIi

в которой R5 имеет указанные выше значения, или карбоновую кислоту общей формулы VIIi

в которой R5 и U1 имеют указанные выше значения, необязательно в активированной форме подвергают по известной методике взаимодействию в условиях реакции сочетания с амином общей формулы IXc
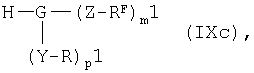
в которой G обозначает одну из групп формул a)-j), а и R, RF, Y, Z, m1 и р1 имеют указанные выше значения, и затем при необходимости отщепляют присутствующие при определенных условиях защитные группы с получением в результате комплекса металла общей формулы Ic либо, если R5 представляет собой защитную группу, после отщепления таких защитных групп на следующей стадии подвергают взаимодействию по известной методике по меньшей мере с одним оксидом металла или солью металла с порядковым номером 23-29, 42-46 или 58-70, после чего при необходимости присутствующие при определенных условиях кислотные атомы водорода замещают катионами неорганических и/или органических оснований, аминокислот либо амидов аминокислот.
Способ получения предлагаемых в изобретении соединений общей формулы Ic, в которой К обозначает комплекс металла общей формулы VIIIc, a G представляет собой группу формулы k) или l), состоит в том, что амин общей формулы VIIIi

в которой R5 обозначает эквивалент иона металла с порядковым номером 23-29, 42-46 или 58-70 или карбоксизащитную группу, подвергают по известной методике взаимодействию в условиях реакции сочетания с необязательно активированной карбоновой кислотой общей формулы Хс

в которой G обозначает группу формулы k) или l), а R, RF, Y, Z, m1 и р1 имеют указанные выше значения, и затем при необходимости отщепляют присутствующие при определенных условиях защитные группы с получением в результате комплекса металла общей формулы Ic либо, если R5 представляет собой защитную группу, после отщепления таких защитных групп на следующей стадии подвергают взаимодействию по известной методике по меньшей мере с одним оксидом металла или солью металла с порядковым номером 23-29, 42-46 или 58-70, после чего при необходимости присутствующие при определенных условиях кислотные атомы водорода замещают катионами неорганических и/или органических оснований, аминокислот либо амидов аминокислот.
Используемые в описанных выше реакциях карбоновые кислоты общих формул IIi-VIIi либо являются известными соединениями, либо их получают по описанным в примерах методам. Так, в частности, способ получения карбоновых кислот общей формулы IIi известен из DE 19652386. Способ получения карбоновых кислот общей формулы IVi описан в DE 19728954.
Предшественником соединений общей формулы VcA является N3-(2,6-диоксоморфолиноэтил)-N6-(этоксикарбонилметил)-3,6-диазапробковая кислота, которая описана в ЕР 263059.
Соединения общей формулы VcB являются производными изомерной диэтилентриаминпентауксусной кислоты (ДТПК), которая присоединена через находящуюся у центрального N-атома уксусную кислоту. Эта ДТПК описана в DE 19507819 и DE 19508058.
Соединения общей формулы VIc являются производными N-(карбоксиметил)-N-[2-(2,6-диоксо-4-морфолинил)этил]глицина, способ получения которого описан в J. Am. Oil. Chem. Soc., 59 (2) (1982), cc.104-107.
Соединения общей формулы VIIc являются производными 1-(4-карбоксиметоксибензил)этилендиаминтетрауксусной кислоты, которая описана в патенте US 4622420.
Используемые в качестве исходных веществ пербензилированные сахарные кислоты можно получать аналогично методу, описанному у Lockhoff в Angew. Chem., 110, №24 (1998), сс.3634 и далее. Так, например, 1-O-уксусную кислоту получают из пербензилглюкозы в две стадии, а именно, через трихлорацетимидат и взаимодействие с этиловым эфиром гидроксиуксусной кислоты, при катализе с помощью BF3 в ТГФ с последующим омылением с помощью NaOH в МеОН/ТГФ.
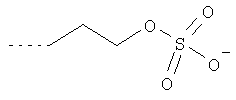
В одном из предпочтительных вариантов используемые в качестве исходных веществ пербензилированные сахарные кислоты можно также получать растворением пербензилированного 1-ОН-сахара в не смешивающемся с водой органическом растворителе с последующим взаимодействием с алкилирующим агентом общей формулы XIc
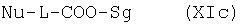
где Nu представляет собой нуклеофоб, L обозначает -(CH2)(1-5)-, -СН2-СНОН-, -СН(СНОН-СН2OH)-СНОН-СНОН-, a Sg представляет собой защитную группу, в присутствии основания и необязательно межфазного катализатора. Алкилирующий агент общей формулы XIc может содержать в качестве нуклеофоба, например, -Cl, -Br, -J, -OTs, -OMs, -OSO2CF3, -OSO2C4F9 или -OSO2C8F17. Под защитной группой подразумевается обычно используемая в подобных целях кислотозащитная группа. Такие защитные группы хорошо известны специалистам (см., например, Protective Groups in Organic Syntheses, T.W. Greene и P.G.M. Wuts, 2-е изд., изд-во John Wiley & Sons Inc., New York 1991).
Рассмотренное выше взаимодействие можно проводить при температуре от 0 до 50°С, предпочтительно от 0°С до комнатной температуры. Продолжительность реакции составляет от 10 мин до 24 ч, преимущественно от 20 мин до 12 ч.
Основание добавляют либо в твердом виде, предпочтительно в виде тонкоизмельченного порошка, либо в виде 10-70%-ного, предпочтительно 30-50%-ного водного раствора. Предпочтительными основаними при этом являются NaOH и КОН.
В качестве органического не смешивающегося с водой растворителя в предлагаемом в изобретении методе алкилирования можно использовать, например, толуол, бензол, CF3-бензол, гексан, циклогексан, диэтиловый эфир, тетрагидрофуран, дихлорметан, МТБ или их смеси.
В качестве межфазных катализаторов в предлагаемом в изобретении способе используют известные по их применению в этих целях соли четвертичного аммониевого основания либо фосфония или же краун-эфиры, такие, например, как [15]-краун-5 или [18]-краун-6. Предпочтительны при этом соли четвертичного аммониевого основания с четырьмя идентичными или различными углеводородными группами у катиона, выбранными из метила, этила, пропила, изопропила, бутила и изобутила. Углеводородные группы у катиона должны быть достаточно большими для того, чтобы обеспечить хорошую растворимость алкилирующего агента в органическом растворителе. Согласно изобретению наиболее предпочтительно при этом использовать N(бутил)4 +-Cl-, N(бутил)4 +-HSO4 -, а также N(метил)4 +-Cl-.
Среди соединений общей формулы I с наиболее предпочтительно использовать согласно изобретению металлический комплекс XV, представленный в таблице 1 (пример 1).
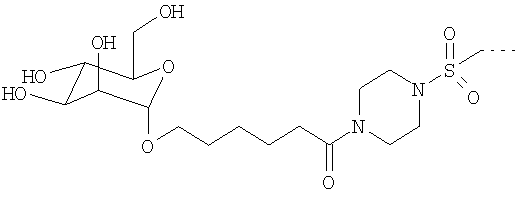
Согласно еще одному предпочтительному варианту осуществления изобретения предлагается использовать перфторалкилсодержащие комплексы с полярными остатками общей формулы Id

в которой
RF обозначает перфторированную, прямую или разветвленную углеродную цепь формулы -CnF2nE, где
Е представляет собой концевой атом фтора, хлора, брома, йода или водорода, а
n обозначает числа 4-30,
К обозначает металлический комплекс общей формулы IId

в которой
R1 обозначает атом водорода или эквивалент иона металла с порядковым номером 23-29, 42-46 или 58-70,
при условии, что по меньшей мере два радикала R1 обозначают эквиваленты ионов металлов,
R2 и R3 независимо друг от друга обозначают водород, С1-С7алкил, бензил, фенил, -СН2ОН или -СН2ОСН3 и
U обозначает -С6Н4-O-СН2-ω-, -(CH2)1-5-ω-, фениленовую группу,
-CH2-NHCO-CH2-CH(CH2COOH)-С6Н4-ω-,
-С6Н4-(ОСН2СН2)0-1-N(СН2СООН)-СН2-ω;- или необязательно прерванную одним или несколькими атомами кислорода, 1-3 -NHCO-группами, 1-3 -CONH-группами и/или замещенную 1-3 -(СН2)0-5COOH-группами С1-С12алкиленовую или С7-С12-С6Н4-O-группу, где ω обозначает место присоединения к -СО-, или общей формулы IIId

в которой
R1 имеет указанные выше значения,
R4 обозначает водород или указанный для R1 эквивалент иона металла и
U1 обозначает -С6Н4-О-СН3-ω-, где ω обозначает место присоединения к -СО-,
или общей формулы IVd

в которой R1 и R2 имеют указанные выше значения,
или общей формулы VdA или VdB

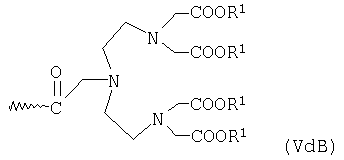
где R1 имеет указанные выше значения,
или общей формулы VId

где R1 имеет указанные выше значения,
или общей формулы VIId

в которой
R1 имеет указанные выше значения, а
U1 обозначает -С6Н4-O-СН2-ω-, где ω обозначает место присоединения к -СО-,
при этом необязательно присутствующие в остатке К свободные кислотные группы необязательно могут быть представлены в виде солей органических и/или неорганических оснований или аминокислот либо в виде амидов аминокислот,
G обозначает по меньшей мере трехкратно функционализованный остаток, выбранный из следующих остатков a)-i):




где α обозначает место присоединения G к комплексу К, β обозначает место присоединения G к остатку R, а γ обозначает место присоединения G к остатку Z,
Z обозначает группу
γ-С(O)СН2O(СН3)2-ε,
где γ обозначает место присоединения Z к остатку G, а ε обозначает место присоединения Z к перфторированному остатку RF,
R представляет собой полярный остаток, выбранный из комплексов К общих формул IId-VIId, причем в этом случае R1 обозначает атом водорода или эквивалент иона металла с порядковым номером 20, 23-29, 42-46 или 58-70, а остатки R2, R3, R4, U и U1 имеют указанные выше значения, или представляет собой остаток фолиевой кислоты
или присоединенную через -СО-, SO2- или прямую связь к остатку G углеродную цепь с 2-30 С-атомами, которая является прямой или разветвленной, насыщенной или ненасыщенной и которая необязательно прервана 1-10 атомами кислорода, 1-5 -NHCO-группами, 1-5-CONH-группами, 1-2 атомами серы, 1-5 -NH-группами или 1-2 фениленовыми группами, которые необязательно могут быть замещены 1-2 ОН-группами, 1-2 NH2-группами, 1-2 -СООН-группами или 1-2 -SO3H-группами, или необязательно замещена 1-8 ОН-группами, 1-5 -СООН-группами, 1-2 SO3Н-группами, 1-5 NH2-группами, 1-5 С1-С4алкоксигруппами, и
l1, m1, p2 независимо друг от друга обозначают целые числа 1 или 2.
Поскольку предлагаемые в изобретении соединения предназначены для их применения при ЯМР-диагностике, ион металла формирующей сигнал группы должен быть парамагнитным. К подобным ионам относятся прежде всего двух- и трехвалентные ионы элементов порядковых номеров 23-29, 42-46 и 58-70. В качестве примера пригодных для применения в указанных целях ионов можно назвать ионы хрома(III), железа(II), кобальта(II), никеля(II), меди(II), празеодима(III), неодима(III), самария(III) и иттербия(III). Наиболее предпочтительны при этом с учетом их высокого магнитного момента ионы гадолиния(III), тербия(III), диспрозия(III), гольмия(III), эрбия(III), железа(III) и марганца(II).
Предпочтительны ионы марганца(II), железа(II), железа(III), празеодима(III), неодима(III), самария(III), гадолиния(III) и иттербия(III), прежде всего диспрозия(III).
Присутствующие при определенных условиях в R1 кислотные атомы водорода, т.е. атомы, которые не замещены центральным ионом, необязательно могут быть полностью или частично заменены на катионы неорганических и/или органических оснований или аминокислот либо амидов аминокислот.
В качестве примера приемлемых неорганических катионов можно назвать ион лития, ион калия, ион кальция и прежде всего ион натрия. Приемлемыми катионами органических оснований являются, в частности, таковые первичных, вторичных или третичных аминов, таких, например, как этаноламин, диэтаноламин, морфолин, глюкамин, N,N-диметилглюкамин и прежде всего N-метилглюкамин. В качестве примера приемлемых катионов аминокислот можно назвать катионы лизина, аргинина и орнитина, а также амиды в остальном кислых или нейтральных аминокислот.
К наиболее предпочтительным соединениям общей формулы Id относятся соединения, содержащие макроцикл К общей формулы IId, IIId, VdB или VIId.
Остаток U в металлическом комплексе К предпочтительно обозначает -СН2- или С6Н4-O-СН2-ω, где ω представляет собой место присоединения к -СО-.
Указанные в качестве значений R2 и R3 алкильные группы в макроцикле общей формулы IId могут иметь прямую или разветвленную цепь. При этом в качестве примеров можно назвать метил, этил, пропил, изопропил, н-бутил, 1-метилпропил, 2-метилпропил, н-пентил, 1-метилбутил, 2-метилбутил, 3-метилбутил, 1,2-диметилпропил. Предпочтительно R2 и R3 независимо друг от друга обозначают водород или С1-С4алкил.
В одном из особо предпочтительных вариантов R2 обозначает метил, а R3 обозначает водород.
Указанная в качестве значения R2 или R3 бензильная группа или фенильная группа в макроцикле К общей формулы IId может быть также замещена в кольце.
Полярный остаток R в общей формуле Id в одном из предпочтительных вариантов представляет собой комплекс К, которым предпочтительно помимо Gd3+- или Mn2+-комплекса может также являться Са2+-комплекс. Наиболее предпочтительны в качестве полярных остатков R комплексы К общих формул IId, IIId, VdA и VIId. В качестве R1 они в особо предпочтительном варианте содержат эквивалент иона металла с порядковым номером 20, 25, 39 или 64.
Согласно еще одному предпочтительному варианту полярный остаток R имеет следующие значения: -С(O)СН2СН2SO3Н,
-С(O)СН2OCH2СН2OCH2СН2OH,-С(O)СН2OCH2СН2OH,
-C(O)CH2OCH2CH(OH)CH2OH,-C(O)CH2NH-C(O)CH2COOH,
-С(O)СН2СН(ОН)СН2OH, -С(O)СН2OCH2СООН, -SO2CH2CH2COOH,
-С(O)-С6Н3-(м-СООН)2,-С(O)СН2O(СН2)2-С6Н3-(м(-СООН)2, -С(O)СН2O-С6Н4-м-SO3H, -C(O)CH2NHC(O)CH2NHC(O)CH2OCH2COOH,
-С(O)СН2OCH2СН2OCH2СООН,-С(O)СН2OCH2СН(ОН)СН2O-СН2СН2OH,
-С(O)СН2OCH2СН(ОН)СН2OCH2-СН(ОН)-СН2OH,-С(O)СН2SO3Н,
-С(O)СН2СН2СООН, -С(O)СН(ОН)СН(ОН)СН2OH, -С(O)СН2O[(СН2)2O]1-9-СН3,
-С(O)СН2O[(СН2)2O]1-9-Н, -С(O)СН2OCH(СН2OH)2,
-С(O)СН2OCH(СН2OCH2СООН)2,-С(O)-С6Н3-(м-ОСН2СООН)2,
-СО-СН2O-(СН2)2O(СН2)2O-(СН2)2O(СН2)2OCH3, предпочтительно
-С(O)СН2O[(СН2)2O]4-СН3.
В другом предпочтительном варианте полярный остаток R представляет собой остаток фолиевой кислоты.
Среди предлагаемых в изобретении соединений общей формулы Id предпочтительны далее соединения, в которых RF обозначает -CnF2n+1. n предпочтительно обозначает числа от 4 до 15. Наиболее предпочтительны остатки -С4F9, -С6F13, -C8F17, -C12F25 и -С14F29.
По меньшей мере трехкратно функционализованный остаток G в общей формуле Id, который является "скелетом", в одном из предпочтительных вариантов осуществления изобретения обозначает остаток лизина (а) или (b).
Z обозначает указанный для общей формулы Id линкер, при этом предпочтительна группа

Способ получения перфторалкилсодержащих комплексов металлов с полярными остатками общей формулы Id
в которой К, G, R, Z, RF, l1, m1 и р2 имеют указанные выше значения, состоит в том, что карбоновую кислоту общей формулы IIk
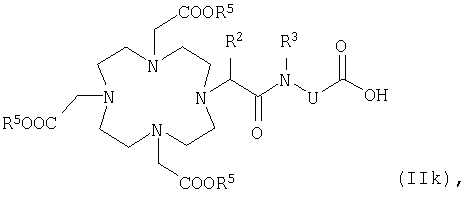
в которой R5 представляет собой эквивалент иона металла с порядковым номером 23-29, 42-46 или 58-70 либо карбоксизащитную группу, а R2, R3 и U имеют указанные выше значения, или карбоновую кислоту общей формулы IIIk

в которой R4, R5 и U1 имеют указанные выше значения, или карбоновую кислоту общей формулы IVk
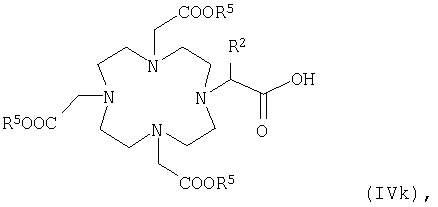
в которой R5 и R2 имеют указанные выше значения, или карбоновую кислоту общей формулы Vk или Vm


где R5 имеет указанные выше значения, или карбоновую кислоту общей формулы VIk
в которой R5 имеет указанные выше значения, или карбоновую кислоту общей формулы VIIk

в которой R5 и U1 имеют указанные выше значения, необязательно в активированной форме подвергают по известной методике взаимодействию в условиях реакции сочетания с амином общей формулы VIIId

в которой G, R, Z, RF, m1 и р2 имеют указанные выше значения, и затем при необходимости отщепляют присутствующие при определенных условиях защитные группы с получением в результате комплекса металла общей формулы Id либо, если R5 представляет собой защитную группу, после отщепления указанных защитных групп на следующей стадии подвергают взаимодействию по известной методике по меньшей мере с одним оксидом металла или солью металла с порядковым номером 23-29, 42-46 или 58-70, после чего при необходимости присутствующие при определенных условиях кислотные атомы водорода замещают катионами неорганических и/или органических оснований, аминокислот либо амидов аминокислот.
Используемые в описанных выше реакциях карбоновые кислоты общих формул IIk-VIIk либо являются известными соединениями, либо их получают по описанным в примерах методам. Так, в частности, способ получения карбоновых кислот общей формулы IIk известен из DE 19652386. Способ получения карбоновых кислот общей формулы IVk описан в DE 19728954.
Предшественником соединений общей формулы VdA является N3-(2,6-диоксоморфолиноэтил)-N6-(этоксикарбонилметил)-3,6-диазапробковая кислота, которая описана в ЕР 263059.
Соединения общей формулы VdB являются производными изомерной диэтилентриаминпентауксусной кислоты (ДТПК), которая присоединена через находящуюся у центрального N-атома уксусную кислоту. Эта ДТПК описана в DE 19507819 и DE 19508058.
Соединения общей формулы VId являются производными N-(карбоксиметил)-N-[2-(2,6-диоксо-4-морфолинил)этил]глицина, способ получения которого описан в J. Am. Oil. Chem. Soc., 59 (2) (1982), сс.104-107.
Соединения общей формулы VIId являются производными 1-(4-карбоксиметоксибензил)этилендиаминтетрауксусной кислоты, которая описана в патенте US 4622420.
Среди соединений общей формулы Id наиболее предпочтительно применять согласно изобретению металлический комплекс XVI, представленный ниже а таблице 1.
В другом предпочтительном варианте осуществления изобретения можно применять галеновы композиции, которые содержат парамагнитные и диамагнитные перфторалкилсодержащие вещества. Такие парамагнитные перфторалкилсодержащие вещества предпочтительно применять в виде раствора в водном растворителе.
В качестве парамагнитных перфторалкилсодержащих соединений в таких композициях могут применяться согласно изобретению все вышеуказанные комплексы металлов общих формул I, Ia, Ib, Ic и/или Id.
Диамагнитные перфторалкилсодержащие вещества представляют собой соединения общей формулы XX:

в которой RF обозначает линейный или разветвленный перфторалкильный остаток с 4-30 атомами углерода, L2 обозначает линкер, а В2 обозначает гидрофильную группу. Линкер L2 представляет собой прямую связь, -SO2-группу или прямую либо разветвленную углеродную цепь, которая содержит до 20 атомов углерода и которая может быть замещена одной или несколькими группами -ОН, -СОО-, -SO3 и/или необязательно содержит одну или несколько -O-, -S-, -СО-, -CONH-, -NHCO-, -CONR9-, -NR9CO-, -SO2-, -PO4 --, -NH-, -NR9-групп, арильное кольцо или пиперазин, при этом R9 обозначает С1-С20алкильный остаток, который в свою очередь может содержать один или несколько O-атомов и/или может быть замещен -СОО-- или SO3-группами.
Гидрофильная группа В2 представляет собой моно- либо дисахарид, одну либо несколько смежных -СОО-- либо -SO3 -групп, дикарбоновую кислоту, изофталевую кислоту, пиколиновую кислоту, бензолсульфоновую кислоту, тетрагидропирандикарбоновую кислоту, 2,6-пиридиндикарбоновую кислоту, ион четвертичного аммония, аминополикарбоновую кислоту, аминодиполиэтиленгликольсульфоновую кислоту, аминополиэтиленгликольную группу, SO2-(СН2)2-ОН-группу, полигидроксиалкильную цепь по меньшей мере с двумя гидроксильными группами или одну либо несколько полиэтиленгликольных цепей по меньшей мере с двумя гликольными звеньями, при этом полиэтиленгликольные звенья оканчиваются группой -ОН или -ОСН3. Подобные вещества уже частично известны, а частично были вновь синтезированы для получения предлагаемых в изобретении композиций. Известные перфторалкилсодержащие вещества и их получение описаны в следующих публикациях:
J.G.Riess, Journal of Drug Targeting, 1994, т.2, сс.455-468;
J.В.Nivet и др., Eur. J. Med. Chem., 1991, т.26, сс.953-960;
M.-P.Krafft и др., Angew. Chem., 1994, т.106, №10, сс.1146-1148;
M.Lanier и др., Tetrahedron Letters, 1995, т.36, №14, сс.2491-2492;
F.Guillod и др., Carbohydrate Research, 1994, т.261, сс.37-55;
S.Achilefu и др., Journal of Fluorine Chemistry, 1995, т.70, сс.19-26;
L.Clary и др., Tetrahedron, 1995, т.51, №47, сс.13073-13088;
F.Szoni и др., Journal of Fluorine Chemistry, 1989, т.42, сс.59-68;
Н.Wu и др., Supramolecular Chemistry, 1994, т.3, сс.175-180;
F.Guileri и др., Angew. Chem. 1994, т.106, №14, сс.1583-1585;
M.-P. Krafft и др., Eur. J. Med. Chem., 1991, т.26, сс.545-550;
J.Greiner и др., Journal of Fluorine Chemistry, 1992, т.56, сс.285-293;
A.Milius и др., Carbohydrate Research, 1992, т.229, сс.323-336;
J.Riess и др., Colloids and Surfaces A, 1994, т.84, сс.33-48;
G.Merhi и др., J. Med. Chem., 1996, т.39, сс.4483-4488;
V.Cirkva и др., Jounal of Fluorine Chemistry, 1997, т.83, сс.151-158;
A.Ould Amanetoullah и др., Journal of Fluorine Chemistry, 1997, т.84, cc.149-153;
J.Chen и др., Inorg. Chem., 1996, т.35, сс.1590-161;
L.Clary и др., Tetrahedron Letters, 1995, т.36, №4, сс.539-542;
M.M. Chaabouni и др., Journal of Fluorine Chemistry, 1990, т.46, сс.307-315;
A.Milius и др., New J. Chem., 1991, т.15, сс.337-344;
М.-Р. Krafft и др., New J. Chem., 1990, т.14, сс.869-875;
J.-B. Nivet и др., New J. Chem., 1994, т.18, сс.861-869;
С.Santaella и др., New J. Chem., 1991, т.15, сс.685-692;
С.Santaella и др., New J. Chem., 1992, т.16, сс.399-404;
A.Milius и др., New J. Chem., 1992, т.16, сс.771-773;
F.Szonyi и др., Journal of Fluorine Chemistry, 1991, т.55, сс.85-92;
С.Santaella и др., Angew. Chem., 1991, т.103, №5, сс.584-586;
М.-Р.Krafft и др., Angew. Chem., 1993, т.105, №5, сс.783-785;
ЕР 0548096 В1.
Получение новых перфторалкилсодержащих веществ аналогично получению вышеуказанных известных из литературы соединений и описано в примерах. При этом речь идет о веществах общей формулы XXI

в которой
RF обозначает линейный или разветвленный перфторалкильный остаток с 4-30 атомами углерода, а
Х1 обозначает остаток, выбранный из группы следующих остатков (при этом n обозначает число от 1 до 10);



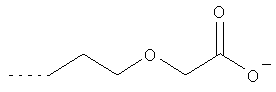


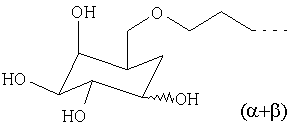



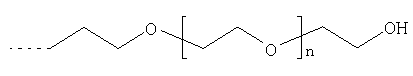





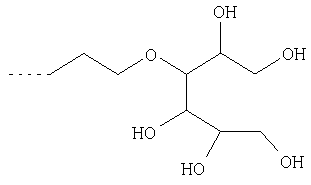




Предпочтительными диамагнитными перфторалкилсодержащими веществами являются таковые, которые содержат моносахарид в качестве гидрофильной группы В2.
Особо предпочтительные диамагнитные перфторалкилсодержащие соединения содержат перфторалкильный остаток Rf с 6-12 атомами углерода, линкер L2, которым является -SO2-группа либо прямая или разветвленная углеродная цепь, число атомов углерода в которой составляет до 20 и которая в свою очередь содержит одну или несколько -O-, -СО-, -CONH-, -NHCO-, -CONR-, -NRCO-, -SO2-групп или пиперазин, где R имеет указанные выше значения, и моносахарид в качестве гидрофильной группы В2.
Другими приемлемыми диамагнитными перфторалкилсодержащими соединениями являются конъюгаты циклодекстрина и перфторалкилсодержащих соединений. Такие конъюгаты состоят из α-, β- или γ-циклодекстрина и соединений общей формулы XXII

в которой А1 обозначает молекулу адамантана, бифенила или антрацена, L3 обозначает линкер, а RF обозначает линейный или разветвленный перфторалкильный остаток с 4-30 атомами углерода. Линкер L3 представляет собой при этом прямую углеводородную цепь с 1-20 атомами углерода, которая может быть прервана одним или несколькими атомами кислорода, одной или несколькими СО-, SO2-, CONH-, NHCO-, CONR-, NRCO-, NH-, NR-группами или пиперазином, при этом R представляет собой С1-С5алкильный остаток. Предпочтительными являются, в частности, следующие соединения:







Соотношение в смеси между парамагнитными и диамагнитными перфторалкилсодержащими соединениями в составе предлагаемых в изобретении галеновых композиций составляет от 5:95 до 95:5. Более предпочтительно, чтобы соотношение в смеси между обоими веществами составляло от 40:60 до 60:40. Оба этих вещества используют в миллимолярных концентрациях. При этом их концентрация составляет от 0,5 до 1000 ммолей/л растворителя. В качестве растворителя предпочтительно использовать воду. Концентрация металла или металлов в композициях составляет от 50 до 250 ммолей/л.
Предпочтительны смеси из парамагнитных и диамагнитных перфторалкилсодержащих соединений, у которых длина перфторалкильных цепей составляет от 6 до 12 атомов углерода. Более предпочтительны смеси, в которых и парамагнитные, и диамагнитные перфторалкилсодержащие соединения имеют перфторалкильную цепь с 8 атомами углерода.
Технология получения галеновых композиций состоит в том, что парамагнитные перфторалкилсодержащие соединения (компонент А) и диамагнитные перфторалкилсодержащие вещества (компонент Б) смешивают между собой с таким расчетом, чтобы мольная доля одного из компонентов А или Б составляла от 0,05 до 0,95, и растворяют в пригодном для этой цели растворителе. Наиболее предпочтительно при этом использовать воду в качестве растворителя. К этому раствору затем в избытке добавляют обычно применяемые в галеновых формах добавки, такие, например, как буферные растворы, и Са-соль комплексообразователя. Далее раствор интенсивно перемешивают при температуре от 10 до 100°С. Альтернативно раствор можно подвергать при температуре от 10 до 100°С обработке ультразвуком. Согласно еще одному варианту раствор можно подвергать обработке микроволновым излучением.
К веществам, которые как отдельные компоненты нерастворимы в воде, предпочтительно добавлять гидротропный солюбилизатор, такой как спирт (например, метанол или этанол), либо иной смешивающийся с водой растворитель и затем медленно отгонять его. Подобную отгонку можно проводить в вакууме. После этого остаток растворяют в воде и полученный раствор фильтруют. Существует также возможность по отдельности растворять каждый из компонентов в соответствующем растворителе, а затем объединять полученные растворы и подвергать их дальнейшей обработке по описанной выше методике. Было установлено, что целесообразно сначала приготавливать сравнительно высококонцентрированный раствор (более 100 ммолей) комплекса металла (компонент А), а затем в чистом виде добавлять к этому раствору компонент Б и после этого по описанной выше методике перемешивать полученный раствор либо обрабатывать его ультразвуком, соответственно микроволновым излучением.
Резюмируя все сказанное выше, следует отметить, что предусмотренным настоящим изобретением критериям удовлетворяют представленные ниже в таблице 1 гадолиниевые комплексы I-XVI, которые являются наиболее предпочтительными для предлагаемого в изобретении применения соединениями. Физические характеристики этих металлических комплексов I-XVI приведены в таблице 2.
Следует также отметить, что предлагаемые в изобретении парамагнитные соединения общих формул I, Ia, Ib, Ic и Id, равно как и предлагаемые в изобретении композиции, содержащие парамагнитные и диамагнитные перфторалкилсодержащие вещества, в высшей степени пригодны для применения в качестве контрастных веществ в МР-томографии для визуализации бляшек, опухолей и некрозов.
Наиболее предпочтительные для предусмотренного изобретением применения металлические комплексы
Физико-химические характеристики применяемых согласно изобретению комплексов, представленных в таблице 1
Примечание:
ККМ обозначает критическую концентрацию мицеллообразования;
2Rh обозначает гидродинамический диаметр мицелл;
R1 обозначает релаксационность.
Примеры осуществления изобретения
Пример 1
а) N-(2-метокси)этиламид 2Н,2Н,4Н,4Н,5Н,5Н-3-оксаперфтортридекановой кислоты
К 30 г (57,45 ммоля) 2Н,2Н,4Н,4Н,5Н,5Н-3-оксаперфтортридекановой кислоты в 300 мл дихлорметан добавляют 8,90 г (70 ммолей) оксалилхлорида и перемешивают в течение 12 ч при комнатной температуре. Смесь упаривают в вакууме досуха. Остаток растворяют в 100 мл дихлорметана и при 0°С по каплям добавляют к раствору 4,51 г (60 ммолей) 2-метоксиэтиламина и 6,07 г (60 ммолей) триэтиламина в 200 мл дихлорметана. Смесь перемешивают в течение 3 ч при 0°С, а затем в течение 6 ч при комнатной температуре. Далее добавляют 300 мл 5%-ной водной соляной кислоты и тщательно перемешивают в течение 15 мин. Органическую фазу отделяют, сушат над сульфатом магния и досуха упаривают в вакууме. Остаток хроматографируют на силикагеле (элюент:дихлорметан/ацетон в соотношении 20:1).
Выход: 30,28 г (91% от теории) бесцветного твердого вещества.
Элементный анализ:
б) N-(2-метоксиэтил)-N-(1H,1H,2H,2H,4H,4H,5H,5H-3-окса)перфтортридециламин
30 г (51,79 ммоля) соединения, указанного в заголовке примера 1а, растворяют в 300 мл тетрагидрофурана и добавляют 31 мл 10 М борандиметилсульфида (в тетрагидрофуране). Далее в течение 16 ч кипятят с обратным холодильником. После этого охлаждают до 0°С и по каплям добавляют 200 мл метанол, после чего досуха упаривают в вакууме. Остаток растворяют в смеси из 300 мл этанола, 50 мл 10%-ной водной соляной кислоты и в течение 8 ч перемешивают при 40°С. Смесь досуха упаривают в вакууме, остаток растворяют в 300 мл 5%-ного водного едкого натра и трижды экстрагируют дихлорметаном порциями по 300 мл. Органические фазы сушат над сульфатом магния, досуха упаривают в вакууме и остаток хроматографируют на силикагеле (элюент:дихлорметан/2-пропанол в соотношении 20:1).
Выход: 26,93 г (92% от теории) бесцветного твердого вещества.
Элементный анализ (в пересчете на безводное вещество):
в) 1,4,7-трис(карбоксилатометил)-10-{(3-аза-4-оксогексан-5-ил)-кислота-[N-(2-метоксиэтил)-N-(1Н,1Н,2Н,2Н,4Н,4Н,5Н,5Н-3-окса)перфтортридецил]амид}-1,4,7,10-тетраазациклододекан, гадолиниевый комплекс (металлический комплекс X)
10 г (15,88 ммоля) гадолиниевого комплекса 10-[1-(карбоксиметилкарбамоил)этил]-1,4,7,10-тетраазациклододекан-1,4,7-триуксусной кислоты и 1,35 г (31,76 ммоля) хлорида лития при 60°С растворяют в 100 мл диметилсульфоксида. Далее охлаждают до 15°С и добавляют 8,98 г (15,88 ммоля) соединения, указанного в заголовке примера 16. Смесь перемешивают в течение 10 мин, а затем добавляют 7,42 г (30 ммолей) 2-этокси-1-этоксикарбонил-1,2-дигидрохинолина. После этого перемешивают в течение 12 ч при комнатной температуре. Раствор сливают в смесь из 200 мл ацетона и 1300 мл диэтилового эфира и перемешивают в течение 2 ч при комнатной температуре. Выпавший осадок отфильтровывают, растворяют в смеси из небольшого количества этанола и воды и хроматографируют на силикагеле RP-18 (элюент:градиент тетрагидрофурана/ацетонитрила/воды).
Выход: 15,14 г (81% от теории) бесцветного аморфного порошка.
Содержание воды: 5,7%.
Элементный анализ (в пересчете на безводное вещество):
Пример 2
а) N-(2,3-дигидроксипропил)амид 2Н,2Н,4Н,4Н,5Н,5Н-3-оксаперфтортридекановой кислоты
К 30 г (57,45 ммоля) 2Н,2Н,4Н,4Н,5Н,5Н-3-оксаперфтортридекановой кислоты в 300 мл дихлорметана добавляют 8,90 г (70 ммолей) оксалилхлорида и перемешивают в течение 12 ч при комнатной температуре. Смесь упаривают в вакууме досуха. Остаток растворяют в 100 мл дихлорметана и при 0°С добавляют по каплям к раствору из 5,47 г (60 ммолей) 2,3-дигидроксипропиламина и 6,07 г (60 ммолей) триэтиламина в 200 мл дихлорметана. Смесь перемешивают в течение 3 ч при 0°С, а затем в течение 6 ч при комнатной температуре. Далее добавляют 300 мл 5%-ной водной соляной кислоты и тщательно перемешивают в течение 15 мин. Органическую фазу отделяют, сушат над сульфатом магния и досуха упаривают в вакууме. Остаток хроматографируют на силикагеле (элюент:дихлорметан/этанол в соотношении 15:1).
Выход: 29,70 г (87% от теории) бесцветного твердого вещества.
Элементный анализ:
б) N-(2,3-дигидроксипропил)-N-(1Н,1Н,2Н,2Н,4Н,4Н,5Н,5Н-3-оксаперфтортридецил)амин
30 г (48,8 ммоля) соединения, указанного в заголовке примера 2а, растворяют в 300 мл тетрагидрофурана и добавляют 50 мл 10 М борандиметилсульфида (в тетрагидрофуране). Далее в течение 16 ч кипятят с обратным холодильником. После этого охлаждают до 0°С и по каплям добавляют 300 мл метанола и затем досуха упаривают в вакууме. Остаток растворяют в смеси из 300 мл этанола и 50 мл 10%-ной водной соляной кислоты и в течение 8 ч перемешивают при 60°С. Смесь досуха упаривают в вакууме, остаток растворяют в 300 мл 5%-ного водного едкого натра и трижды экстрагируют дихлорметаном порциями по 300 мл. Органические фазы сушат над сульфатом магния, досуха упаривают в вакууме и остаток хроматографируют на силикагеле (элюент:дихлорметан/метанол в соотношении 15:1).
Выход: 24,07 г (85% от теории) бесцветного твердого вещества.
Элементный анализ:
в) 1,4,7-трис(карбоксилатометил)-10-[(3-аза-4-оксогексан-5-ил)-кислота-N-(2,3-дигидроксипропил)-N-(1Н,1Н,2Н,2Н,4Н,4Н,5Н,5Н-3-окса)перфтортридецил)амид]-1,4,7,10-тетраазациклододекан, гадолиниевый комплекс (металлический комплекс V)
10 г (15,88 ммоля) гадолиниевого комплекса 10-[1-(карбоксиметилкарбамоил)этил]-1,4,7,10-тетраазациклододекан-1,4,7-триуксусной кислоты и 1,35 г (31,76 ммоля) хлорида лития растворяют при 60°С в 100 мл диметилсульфоксида. Далее охлаждают до 15°С и добавляют 9,21 г (15,88 ммоля) соединения, указанного в заголовке примера 26. Смесь перемешивают в течение 10 мин и затем добавляют 7,42 г (30 ммолей) 2-этокси-1-этоксикарбонил-1,2-дигидрохинолина. После этого перемешивают в течение 12 ч при комнатной температуре. Раствор сливают в смесь из 200 мл ацетона и 1300 мл диэтилового эфира и перемешивают в течение 2 ч при комнатной температуре. Выпавший осадок отфильтровывают, растворяют в смеси из небольшого количества этанола/воды и хроматографируют на силикагеле RP-18 (элюент:градиент тетрагидрофурана/ацетонитрила/воды).
Выход: 16,09 г (85% от теории) бесцветного аморфного порошка.
Содержание воды: 6,3%.
Элементный анализ (в пересчете на безводное вещество):
Пример 3
а) N-(5-гидрокси-3-оксапентил)амид 2Н,2Н,4Н,4Н,5Н,5Н-3-оксаперфтортридекановой кислоты
К 30 г (57,45 ммоля) 2Н,2Н,4Н,4Н,5Н,5Н-3-оксаперфтортридекановой кислоты в 300 мл дихлорметана добавляют 8,90 г (70 ммолей) оксалилхлорида и перемешивают в течение 12 ч при комнатной температуре. Смесь упаривают в вакууме досуха. Остаток растворяют в 100 мл дихлорметана и при 0°С по каплям добавляют к раствору из 6,25 г (60 ммолей) 5-гидрокси-3-оксапентиламина и 6,07 г (60 ммолей) триэтиламина в 200 мл дихлорметана. Смесь перемешивают в течение 3 ч при 0°С, а затем в течение 6 ч при комнатной температуре. Далее добавляют 300 мл 5%-ной водной соляной кислоты и тщательно перемешивают в течение 15 мин. Органическую фазу отделяют, сушат над сульфатом магния и досуха упаривают в вакууме. Остаток хроматографируют на силикагеле (элюент:дихлорметан/ацетон в соотношении 15:1).
Выход: 32,20 г (92% от теории) бесцветного твердого вещества.
Элементный анализ:
б) N-(5-гидрокси-3-оксапентил)-N-(1Н,1Н,2Н,2Н,4Н,4Н,5Н,5Н-3-оксаперфтортридецил)амин
30 г (49,24 ммоля) соединения, указанного в заголовке примера 3а, растворяют в 300 мл тетрагидрофурана и добавляют 31 мл 10 М борандиметилсульфида (в тетрагидрофуране). Далее в течение 16 ч кипятят с обратным холодильником. После этого охлаждают до 0°С, по каплям добавляют 200 мл метанола и затем досуха упаривают в вакууме. Остаток растворяют в смеси из 300 мл этанола и 50 мл 10%-ной водной соляной кислоты и в течение 10 ч перемешивают при 50°С. Смесь досуха упаривают в вакууме, остаток растворяют в 300 мл 5%-ного водного едкого натра и трижды экстрагируют дихлорметаном порциями по 300 мл. Органические фазы сушат над сульфатом магния, досуха упаривают в вакууме и остаток хроматографируют на силикагеле (элюент:дихлорметан/2-пропанол в соотношении 20:1).
Выход: 26,09 г (89% от теории) бесцветного твердого вещества.
Элементный анализ:
в) 1,4,7-трис(карбоксилатометил)-10-[(3-аза-4-оксогексан-5-ил)-кислота-N-(5-гидрокси-3-оксапентил)-N-(1Н,1Н,2Н,2Н,4Н,4Н,5Н,5Н-3-окса)перфтортридецил)амид]-1,4,7,10-тетраазациклододекан, гадолиниевый комплекс (металлический комплекс VIII)
10 г (15,88 ммоля) гадолиниевого комплекса 10-[1-(карбоксиметилкарбамоил)этил]-1,4,7,10-тетраазациклододекан-1,4,7-триуксусной кислоты и 1,35 г (31,76 ммоля) хлорида лития при 60°С растворяют в 100 мл диметилсульфоксида. Далее охлаждают до 15°С и добавляют 9,45 г (15,88 ммоля) соединения, указанного в заголовке примера 3б. Смесь перемешивают в течение 10 мин и затем добавляют 7,42 г (30 ммолей) 2-этокси-1-этоксикарбонил-1,2-дигидрохинолина. После этого перемешивают в течение 12 ч при комнатной температуре. Раствор сливают в смесь из 200 мл ацетона и 1300 мл диэтилового эфира и перемешивают в течение 2 ч при комнатной температуре. Выпавший осадок отфильтровывают, растворяют в смеси из небольшого количества этанола и воды и хроматографируют на силикагеле RP-18 (элюент:градиент тетрагидрофурана/ацетонитрила/воды).
Выход: 16,10 г (84% от теории) бесцветного аморфного порошка.
Содержание воды: 5,7%.
Элементный анализ (в пересчете на безводное вещество):
Пример 4
а) N-(2-гидроксиэтил)амид 2Н,2Н,4Н,4Н,5Н,5Н-3-оксаперфтортридекановой кислоты
К 30 г (57,45 ммоля) 2Н,2Н,4Н,4Н,5Н,5Н-3-оксаперфтортридекановой кислоты в 300 мл дихлорметана добавляют 8,90 г (70 ммолей) оксалилхлорида и перемешивают в течение 12 ч при комнатной температуре. Смесь упаривают в вакууме досуха. Остаток растворяют в 100 мл дихлорметана и при 0°С по каплям добавляют к раствору из 3,66 г (60 ммолей) 2-аминоэтанола и 6,07 г (60 ммолей) триэтиламина в 200 мл дихлорметана. Смесь перемешивают в течение 3 ч при 0°С, а затем в течение 6 ч при комнатной температуре. Далее добавляют 300 мл 5%-ной водной соляной кислоты и тщательно перемешивают в течение 15 мин. Органическую фазу отделяют, сушат над сульфатом магния и досуха упаривают в вакууме. Остаток хроматографируют на силикагеле (элюент:дихлорметан/ацетон в соотношении 20:1).
Выход: 28,90 г (89% от теории).
Элементный анализ:
б) N-(2-гидроксиэтил)-N-(1Н,1Н,2Н,2Н,4Н,4Н,5Н,5Н-3-оксаперфтортридецил)амин
28 г (49,54 ммоля) соединения, указанного в заголовке примера 4а, растворяют в 300 мл тетрагидрофурана и добавляют 31 мл 10 М борандиметилсульфида (в тетрагидрофуране). Далее в течение 16 ч кипятят с обратным холодильником. После этого охлаждают до 0°С, по каплям добавляют 200 мл метанола и затем досуха упаривают в вакууме. Остаток растворяют в смеси из 300 мл этанола и 50 мл 10%-ной водной соляной кислоты и в течение 10 ч перемешивают при 50°С. Смесь досуха упаривают в вакууме, остаток растворяют в 300 мл 5%-ного водного едкого натра и трижды экстрагируют дихлорметаном порциями по 300 мл. Органические фазы сушат над сульфатом магния, досуха упаривают в вакууме и остаток хроматографируют на силикагеле (элюент:дихлорметан/2-пропанол в соотношении 15:1).
Выход: 25,12 г (92% от теории) бесцветного твердого вещества.
Элементный анализ (в пересчете на безводное вещество):
в) 1,4,7-трис(карбоксилатометил)-10-[(3-аза-4-оксогексан-5-ил)-кислота-N-(2-гидроксиэтил)-N-(1Н,1Н,2Н,2Н,4Н,4Н,5Н,5Н-3-окса)перфтортридецил)аминамид]-1,4,7,10-тетраазациклододекан, гадолиниевый комплекс
10 г (15,88 ммоля) гадолиниевого комплекса 10-[1-(карбоксиметилкарбамоил)этил]-1,4,7,10-тетраазациклододекан-1,4,7-триуксусной кислоты и 1,35 г (31,76 ммоля) хлорида лития при 60°С растворяют в 100 мл диметилсульфоксида. Далее охлаждают до 15°С и добавляют 8,75 г (15,88 ммоля) соединения, указанного в заголовке примера 4б. Смесь перемешивают в течение 10 мин и затем добавляют 7,42 г (30 ммолей) 2-этокси-1-этоксикарбонил-1,2-дигидрохинолина. После этого перемешивают в течение 12 ч при комнатной температуре. Раствор сливают в смесь из 200 мл ацетона и 1300 мл диэтилового эфира и перемешивают в течение 2 ч при комнатной температуре. Выпавший осадок отфильтровывают, растворяют в смеси из небольшого количества этанола и воды и хроматографируют на силикагеле RP-18 (элюент:градиент тетрагидрофурана/ацетонитрила/воды).
Выход: 16,81 г (91% от теории) бесцветного аморфного порошка.
Содержание воды: 7,2%.
Элементный анализ (в пересчете на безводное вещество):
Пример 5
а) Амид 2Н,2Н,4Н,4Н,5Н,5Н-3-оксаперфтортридекановой кислоты
К 30 г (57,45 ммоля) 2Н,2Н,4Н,4Н,5Н,5Н-3-оксаперфтортридекановой кислоты в 300 мл дихлорметана добавляют 8,90 г (70 ммолей) оксалилхлорида и перемешивают в течение 12 ч при комнатной температуре. Смесь упаривают в вакууме досуха. Остаток растворяют в 200 мл дихлорметана. Затем раствор при 0°С барботируют газообразным аммиаком в течение примерно 2 ч. После этого смесь перемешивают в течение 4 ч при 0°С, а затем в течение 2 ч при комнатной температуре. Далее добавляют 300 мл 5%-ной водной соляной кислоты и тщательно перемешивают в течение 15 мин. Органическую фазу отделяют, сушат над сульфатом магния и досуха упаривают в вакууме. Остаток хроматографируют на силикагеле (элюент:дихлорметан/ацетон в соотношении 20:1).
Выход: 27,85 г (93% от теории).
Элементный анализ:
б) 1Н,1Н,2Н,2Н,4Н,4Н,5Н,5Н-3-оксаперфтортридециламин, гидрохлорид
27 г (51,8 ммоля) соединения, указанного в заголовке примера 5а, растворяют в 300 мл тетрагидрофурана и добавляют 31 мл 10 М борандиметилсульфида (в тетрагидрофуране). Далее в течение 16 ч кипятят с обратным холодильником. После этого охлаждают до 0°С, по каплям добавляют 200 мл метанола и затем досуха упаривают в вакууме. Остаток растворяют в смеси из 400 мл этанола и 100 мл 10%-ной водной соляной кислоты и перемешивают в течение 8 ч при 60°С. Смесь досуха упаривают в вакууме и остаток перекристаллизовывают из небольшого количества этанола/диэтилового эфира.
Выход: 26,75 г (95% от теории) бесцветного, кристаллического твердого вещества.
Элементный анализ:
в) N-(1Н,1Н,2Н,2Н,4Н,4Н,5Н,5Н-3-оксаперфтортридецил)амид 3,6,9,12,15-пентаоксагексадекановой кислоты
К 26,5 г (48,74 ммоля) указанного в заголовке примера 5б соединения и 14,8 г (146,2 ммоля) триэтиламина, растворенных в 300 мл дихлорметана, при 0°С добавляют 14,24 г (50 ммолей) хлорангидрида 3,6,9,12,15-пентаоксагексадекановой кислоты и перемешивают в течение 3 ч при 0°С. Далее добавляют 300 мл 5%-ной водной соляной кислоты и тщательно перемешивают в течение 30 мин. Органическую фазу отделяют, сушат над сульфатом магния и досуха упаривают в вакууме. Остаток хроматографируют на силикагеле (элюент:дихлорметан/ацетон в соотношении 20:1).
Выход: 32,03 г (87% от теории) бесцветного масла.
Элементный анализ:
г) N-(3,6,9,12,15-пентаоксагексадецил)-N-(1Н,1Н,2Н,2Н,4Н,4Н-3-оксаперфтортридецил)амин
31 г (41,03 ммоля) соединения, указанного в заголовке примера 5в, растворяют в 300 мл тетрагидрофурана и добавляют 25 мл 10 М борандиметилсульфида (в тетрагидрофуране). Далее в течение 16 ч кипятят с обратным холодильником. После этого охлаждают до 0°С, по каплям добавляют 200 мл метанола и затем досуха упаривают в вакууме. Остаток растворяют в смеси из 300 мл этанола и 50 мл 10%-ной водной соляной кислоты и в течение 8 ч перемешивают при 40°С. Смесь досуха упаривают в вакууме, остаток растворяют в 300 мл 5%-ного водного едкого натра и трижды экстрагируют дихлорметаном порциями по 300 мл. Органические фазы сушат над сульфатом магния, досуха упаривают в вакууме и остаток хроматографируют на силикагеле (элюент:дихлорметан/2-пропанол в соотношении 15:1).
Выход: 27,68 г (91% от теории).
Элементный анализ:
д) 1,4,7-трис(карбоксилатометил)-10-{(3-аза-4-оксогексан-5-ил)-кислота-[N-(3,6,9,12,15-пентаоксагексадецил)-N-(1Н,1Н,2Н,2Н,4Н,4Н,5Н,5Н-3-окса)перфтортридецил]амид}-1,4,7,10-тетраазациклододекан, гадолиниевый комплекс (металлический комплекс VII)
10 г (15,88 ммоля) гадолиниевого комплекса 10-[1-(карбоксиметилкарбамоил)этил]-1,4,7,10-тетраазациклододекан-1,4,7-триуксусной кислоты и 1,35 г (31,76 ммоля) хлорида лития при 60°С растворяют в 100 мл диметилсульфоксида. Далее охлаждают до 15°С и добавляют 11,77 г (15,88 ммоля) соединения, указанного в заголовке примера 5г. Смесь перемешивают в течение 10 мин и затем добавляют 7,42 г (30 ммолей) 2-этокси-1-этоксикарбонил-1,2-дигидрохинолина. После этого перемешивают в течение 12 ч при комнатной температуре. Раствор сливают в смесь из 200 мл ацетона и 1300 мл диэтилового эфира и перемешивают в течение 2 ч при комнатной температуре. Выпавший осадок отфильтровывают, растворяют в смеси из небольшого количества этанола и воды и хроматографируют на силикагеле RP-18 (элюент:градиент тетрагидрофурана/ацетонитрила/воды).
Выход: 18,05 г (84% от теории) бесцветного аморфного порошка.
Содержание воды: 6,2%.
Элементный анализ (в пересчете на безводное вещество):
Пример 6
а) 1,4,7-трис(карбоксилатометил)-10-[(3-аза-4-оксогексан-5-ил)-кислота-N-(12-амино-3,6,9-триоксадодецил)амид]-1,4,7,10-тетраазациклододекан, гадолиниевый комплекс
10 г (15,88 ммоля) гадолиниевого комплекса 10-[1-(карбоксиметилкарбамоил)этил]-1,4,7,10-тетраазациклододекан-1,4,7-триуксусной кислоты, 1,35 г (31,76 ммоля) хлорида лития и 3,66 г (31,76 ммоля) N-гидроксисукцинимида при 60°С растворяют в 100 мл диметилсульфоксида. Далее охлаждают до 15°С и добавляют 3,51 г (17 ммолей) N,N'-дициклогексилкарбодиимида и перемешивают в течение 5 ч при 15°С. Для отделения мочевины раствор фильтруют. К фильтрату добавляют 14,66 г (60 ммолей) 1,12-диамино-3,6,9-триоксадодекана и 2,02 г (20 ммолей) триэтиламина и перемешивают в течение 12 ч при комнатной температуре. Раствор сливают в смесь из 1500 мл диэтилового эфира и 50 мл н-бутанола и перемешивают в течение 30 мин. Выпавшее в осадок твердое вещество отфильтровывают и хроматографируют на силикагеле RP-18 (элюент:градиент тетрагидрофурана/ацетонитрила/воды).
Выход: 12,66 г (69% от теории) бесцветного аморфного порошка.
Содержание воды: 3,5%.
Элементный анализ (в пересчете на безводное вещество):
б) 1,4,7-трис(карбоксилатометил)-10-{(3-аза-4-оксогексан-5-ил)-кислота-[N-(3,6,9,16-тетраокса-13-аза-14-оксо-С19-С26гептадекафтор)гексакозил]амид}-1,4,7,10-тетраазациклододекан, гадолиниевый комплекс (металлический комплекс IX)
11,3 г (21,64 ммоля) 2Н,2Н,4Н,4Н,5Н,5Н-3-оксаперфтортридекановой кислоты, 0,85 г (20 ммолей) хлорида лития и 4,95 г (43 ммоля) N-гидроксисукцинимида растворяют при 25°С в 150 мл диметилсульфоксида. Далее охлаждают до 15°С, добавляют 6,19 г (30 ммолей) N,N'-дициклогексилкарбодиимида и перемешивают в течение 5 ч при 15°С. Для отделения мочевины раствор фильтруют. К фильтрату добавляют 12,5 г (10,82 ммоля) соединения, указанного в заголовке примера 6а, и 3,29 г (32,47 ммоля) триэтиламина и перемешивают в течение 12 ч при комнатной температуре. Раствор сливают в смесь из 1300 мл диэтилового эфира и 100 мл ацетона и перемешивают в течение 30 мин. Выпавшее в осадок твердое вещество отфильтровывают и хроматографируют на силикагеле RP-18 (элюент:градиент тетрагидрофурана/ацетонитрила/воды).
Выход: 13,01 г (90% от теории).
Содержание воды: 6,7%.
Элементный анализ (в пересчете на безводное вещество):
Пример 7
1,4,7-трис(карбоксилатометил)-10-[(3-аза-4-оксогексан-5-ил)-кислота-N-(1Н,1Н,2Н,2Н,4Н,4Н,5Н,5Н-3-оксаперфтортридецил)амид]-1,4,7,10-тетраазациклододекан, гадолиниевый комплекс
10 г (15,88 ммоля) гадолиниевого комплекса 10-[1-(карбоксиметилкарбамоил)этил]-1,4,7,10-тетраазациклододекан-1,4,7-триуксусной кислоты, 1,35 г (31,76 ммоля) хлорида лития и 3,66 г (31,76 ммоля) N-гидроксисукцинимида при 60°С растворяют в 100 мл диметилсульфоксида. Далее охлаждают до 15°С и добавляют 3,51 г (17 ммолей) N,N'-дициклогексилкарбодиимида и перемешивают в течение 5 ч при 15°С. Для отделения мочевины раствор фильтруют. К фильтрату добавляют 8,63 г (15,88 ммоля) соединения, указанного в заголовке примера 5б, и 5,06 г (50 ммолей) триэтиламина и перемешивают в течение 12 ч при комнатной температуре. Раствор сливают в смесь из 1500 мл диэтилового эфира и 100 мл ацетона и перемешивают в течение 30 мин. Выпавшее в осадок твердое вещество отфильтровывают и хроматографируют на силикагеле RP-18 (элюент:градиент тетрагидрофурана/ацетонитрила/воды).
Выход: 13,86 г (78% от теории) бесцветного аморфного порошка.
Содержание воды: 9,3%.
Элементный анализ (в пересчете на безводное вещество):
Пример 8
а) N-(2,3,4,5,6-пентагидрокси)гексиламид 2Н,2Н,4Н,4Н,5Н,5Н-3-оксаперфтортридекановой кислоты
К 30 г (57,45 ммоля) 2Н,2Н,4Н,4Н,5Н,5Н-3-оксаперфтортридекановой кислоты в 300 мл дихлорметана добавляют 8,90 г (70 ммолей) оксалилхлорида и перемешивают в течение 12 ч при комнатной температуре. Смесь упаривают в вакууме досуха. Остаток растворяют в 100 мл дихлорметана и при 0°С по каплям добавляют к раствору из 10,87 г (60 ммолей) глюкамина и 6,07 г (60 ммолей) триэтиламина в смеси из 150 мл дихлорметана и 150 диоксана. Смесь перемешивают в течение 3 ч при 0°С, а затем в течение 8 ч при комнатной температуре. Далее добавляют 400 мл 5%-ной водной соляной кислоты и тщательно перемешивают в течение 15 мин. Органическую фазу отделяют, сушат над сульфатом магния и досуха упаривают в вакууме. Остаток хроматографируют на силикагеле (элюент:дихлорметан/метанол в соотношении 5:1).
Выход: 30,71 г (78% от теории).
Элементный анализ:
б) N-(2,3,4,5,6-пентагидроксигексил)-N-1Н,1Н,2Н,2Н,4Н,4Н,5Н,5Н-3-оксаперфтортридециламин
30 г (43,77 ммоля) соединения, указанного в заголовке примера 8а, растворяют в 300 мл тетрагидрофурана и добавляют 50 мл 10 М борандиметилсульфида (в тетрагидрофуране). Далее в течение 48 ч кипятят с обратным холодильником. После этого охлаждают до 0°С, по каплям добавляют 500 мл метанола и затем досуха упаривают в вакууме. Остаток растворяют в смеси из 500 мл этанола и 100 мл 10%-ной водной соляной кислоты и в течение 15 ч перемешивают при 60°С. Смесь досуха упаривают в вакууме, остаток растворяют в 400 мл 5%-ного водного едкого натра и пятикратно экстрагируют хлороформом порциями по 400 мл. Органические фазы сушат над сульфатом магния, досуха упаривают в вакууме и остаток хроматографируют на силикагеле (элюент:дихлорметан/метанол в соотношении 3:1).
Выход: 19,69 г (67% от теории) бесцветного твердого вещества.
Элементный анализ:
в) 1,4,7-трис(карбоксилатометил)-10-{(3-аза-4-оксогексан-5-ил)-кислота-N-[-(2,3,5,6-пентагидрокси)гексил-N-(1Н,1Н,2Н,2Н,4Н,4Н,5Н,5Н-3-оксаперфтортридецил]амид}-1,4,7,10-тетраазациклододекан, гадолиниевый комплекс
10 г (15,88 ммоля) гадолиниевого комплекса 10-[1-(карбоксиметилкарбамоил)этил]-1,4,7,10-тетраазациклододекан-1,4,7-триуксусной кислоты и 1,35 г (31,76 ммоля) хлорида лития при 60°С растворяют в 100 мл диметилсульфоксида. Далее охлаждают до 15°С и добавляют 15,88 г (15,88 ммоля) соединения, указанного в заголовке примера 8б. Смесь перемешивают в течение 10 мин и затем добавляют 7,42 г (30 ммолей) 2-этокси-1-этоксикарбонил-1,2-дигидрохинолина. После этого перемешивают в течение 12 ч при комнатной температуре. Раствор сливают в смесь из 200 мл ацетона и 1300 мл диэтилового эфира и перемешивают в течение 2 ч при комнатной температуре. Выпавший осадок отфильтровывают, растворяют в смеси из небольшого количества этанола и воды и хроматографируют на силикагеле RP-18 (элюент:градиент тетрагидрофурана/ацетонитрила/воды).
Выход: 16,10 г (79% от теории) бесцветного аморфного порошка.
Содержание воды: 6,3%.
Элементный анализ (в пересчете на безводное вещество):
Пример 9
а) N-(2,2-диметил-5-гидрокси-1,3-диоксепан-6-ил)амид 2Н,2Н,4Н,4Н,5Н,5Н-3-оксаперфтортридекановой кислоты
К 30 г (57,45 ммоля) 2Н,2Н,4Н,4Н,5Н,5Н-3-оксаперфтортридекановой кислоты в 300 мл дихлорметана добавляют 8,90 г (70 ммолей) оксалилхлорида и перемешивают в течение 12 ч при комнатной температуре. Смесь упаривают в вакууме досуха. Остаток растворяют в 100 мл дихлорметана и при 0°С по каплям добавляют к раствору из 9,67 г (60 ммолей) 5-амино-2,2-диметил-1,3-диоксепан-6-ола и 6,07 г (60 ммолей) триэтиламина в 200 мл дихлорметана. Смесь перемешивают в течение 3 ч при 0°С, а затем в течение 5 ч при комнатной температуре. Далее добавляют 300 мл воды и тщательно перемешивают в течение 15 мин. Органическую фазу отделяют, сушат над сульфатом магния и досуха упаривают в вакууме. Остаток хроматографируют на силикагеле (элюент:дихлорметан/ацетон в соотношении 15:1).
Выход: 27,62 г (85% от теории).
Элементный анализ:
б) N-(1-гидроксиметил-2,3-дигидроксипропил)-N-(1Н,1Н,2Н,2Н,4Н,4Н,5Н,5Н-3-оксаперфтортридецил)амин
27 г (40,58 ммоля) соединения, указанного в заголовке примера 9а, растворяют в 300 мл тетрагидрофурана и добавляют 26 мл 10 М борандиметилсульфида (в тетрагидрофуране). Далее в течение 20 ч кипятят с обратным холодильником. После этого охлаждают до 0°С, по каплям добавляют 300 мл метанола и затем досуха упаривают в вакууме. Остаток растворяют в смеси из 300 мл этанола и 100 мл 10%-ной водной соляной кислоты и в течение 6 ч перемешивают при 60°С. Смесь досуха упаривают в вакууме, остаток растворяют в 400 мл 5%-ного водного едкого натра и пятикратно экстрагируют хлороформом порциями по 250 мл. Органические фазы сушат над сульфатом магния, досуха упаривают в вакууме и остаток хроматографируют на силикагеле (элюент:дихлорметан/метанол в соотношении 6:1).
Выход: 20,09 г (81% от теории) бесцветного твердого вещества.
Элементный анализ:
в) 1,4,7-трис(карбоксилатометил)-10-{(3-аза-4-оксогексан-5-ил)-кислота-[N-(1-гидроксиметил-2,3-дигидроксипропил)-N-(1Н,1Н,2Н,2Н,4Н,4Н,5Н,5Н-3-оксаперфтортридецил]амид}-1,4,7,10-тетраазациклододекан, гадолиниевый комплекс
10 г (15,88 ммоля) гадолиниевого комплекса 10-[1-(карбоксиметилкарбамоил)этил]-1,4,7,10-тетраазациклододекан-1,4,7-триуксусной кислоты и 1,35 г (31,76 ммоля) хлорида лития при 60°С растворяют в 100 мл диметилсульфоксида. Далее охлаждают до 15°С и добавляют 9,71 г (15,88 ммоля) соединения, указанного в заголовке примера 9б. Смесь перемешивают в течение 10 мин и затем добавляют 7,42 г (30 ммолей) 2-этокси-1-этоксикарбонил-1,2-дигидрохинолина. После этого перемешивают в течение 12 ч при комнатной температуре. Раствор сливают в смесь аиз 200 мл ацетона и 1300 мл диэтилового эфира и перемешивают в течение 2 ч при комнатной температуре. Выпавший осадок отфильтровывают, растворяют в смеси из небольшого количества этанола и воды и хроматографируют на силикагеле RP-18 (элюент:градиент тетрагидрофурана/ацетонитрила/воды).
Выход: 13,40 г (69% от теории) бесцветного аморфного порошка.
Содержание воды: 9,1%.
Элементный анализ (в пересчете на безводное вещество):
Пример 10
а) N-[(2-бензилоксикарбониламино)этил]амид перфтороктилсульфоновой кислоты
40 г (173,4 ммоля) гидрохлорида 1-бензилоксикарбониламино-2-аминоэтана, 87,1 г (173,4 ммоля) перфтороктилсульфофторида и 35,42 г (350 ммолей) триэтиламина нагревают до 80°С и выдерживают при этой температуре в течение 10 ч. Далее охлаждают до комнатной температуры и непосредственно вносят в силикагелевую колонку для хроматографической очистки (элюент:дихлорметан/ацетон в соотношении 20:1).
Выход: 42,22 г (36% от теории) бесцветного твердого вещества.
Элементный анализ:
б) N-[(2-амино)этил]амид перфтороктилсульфоновой кислоты
30 г (44,36 ммоля) соединения, указанного в заголовке примера 10а, растворяют в 300 мл метанола, добавляют 5 г палладиевого катализатора (10%-ный Pd/C) и гидрируют в течение ночи при комнатной температуре. После этого катализатор отфильтровывают и фильтрат досуха упаривают в вакууме.
Выход: 24,05 г (количественный) бесцветного твердого вещества.
Элементный анализ:
в) 1,4,7-трис(карбоксилатометил)-10-{(3-аза-4-оксогексан-5-ил)-кислота-N-[(2-перфтороктилсульфониламино)этил]амид}-1,4,7,10-тетраазациклододекан, гадолиниевый комплекс
10 г (15,88 ммоля) гадолиниевого комплекса 10-[1-(карбоксиметилкарбамоил)этил]-1,4,7,10-тетраазациклододекан-1,4,7-триуксусной кислоты, 1,35 г (31,76 ммоля) хлорида лития и 3,66 г (31,76 ммоля) N-гидроксисукцинимида при 60°С растворяют в 100 мл диметилсульфоксида. Далее охлаждают до 15°С, добавляют 3,51 г (17 ммолей) N,N'-дициклогексилкарбодиимида и перемешивают в течение 5 ч при 15°С. Для отделения мочевины раствор фильтруют. К фильтрату добавляют 8,61 г (15,88 ммоля) соединения, указанного в заголовке примера 10б, и 2,02 г (20 ммолей) триэтиламина и перемешивают в течение 12 ч при комнатной температуре. Раствор сливают в смесь из 1500 мл диэтилового эфира и 100 мл ацетона и перемешивают в течение 30 мин. Выпавшее в осадок твердое вещество отфильтровывают и хроматографируют на силикагеле RP-18 (элюент:градиент тетрагидрофурана/ацетонитрила/воды).
Выход: 15,76 г (86% от теории) бесцветного аморфного порошка.
Содержание воды: 6,5%.
Элементный анализ (в пересчете на безводное вещество):
Пример 11
а) N-(2-бензилоксикарбоксиаминоэтил)амид 2Н,2Н,4Н,4Н,5Н,5Н-3-оксаперфтортридекановой кислоты
К 30 г (57,45 ммоля) 2Н,2Н,4Н,4Н,5Н,5Н-3-оксаперфтортридекановой кислоты в 300 мл дихлорметана добавляют 8,90 г (70 ммолей) оксалилхлорида и перемешивают в течение 12 ч при комнатной температуре. Смесь упаривают в вакууме досуха. Остаток растворяют в 100 мл дихлорметана и при 0°С по каплям добавляют к раствору из 13,84 г (60 ммолей) гидрохлорида 1-бензилоксикарбониламин-2-аминоэтана и 12,14 г (120 ммолей) триэтиламина в 200 мл дихлорметана. Смесь перемешивают в течение 3 ч при 0°С, а затем в течение 5 ч при комнатной температуре. Далее добавляют 300 мл 5%-ной водной соляной кислоты и тщательно перемешивают в течение 15 мин. Органическую фазу отделяют, сушат над сульфатом магния и досуха упаривают в вакууме. Остаток хроматографируют на силикагеле (элюент:дихлорметан/ацетон в соотношении 20:1).
Выход: 33,30 г (83% от теории) бесцветного твердого вещества.
Элементный анализ:
б) N-[(2-амино)этил]амид 2Н,2Н,4Н,4Н,5Н,5Н-3-оксаперфтортридекановой кислоты
30 г (42,96 ммоля) соединения, указанного в заголовке примера 11а, растворяют в 500 мл метанола, добавляют 5 г палладиевого катализатора (10%-ного Pd/C) и гидрируют в течение ночи при комнатной температуре. После этого катализатор отфильтровывают и фильтрат досуха упаривают в вакууме.
Выход: 24,24 г (количественный) бесцветного твердого вещества.
Элементный анализ:
в) 1,4,7-трис(карбоксилатометил)-10-{(3-аза-4-оксогексан-5-ил)-кислота-N-[3-аза-6-окса-4-оксо-(С9-С16гептадекафтор)гексадецил]амид}-1,4,7,10-тетраазациклододекан, гадолиниевый комплекс
10 г (15,88 ммоля) гадолиниевого комплекса 10-[1-(карбоксиметилкарбамоил)этил]-1,4,7,10-тетраазациклододекан-1,4,7-триуксусной кислоты, 1,35 г (31,76 ммоля) хлорида лития и 3,66 г (31,76 ммоля) N-гидроксисукцинимида при 60°С растворяют в 100 мл диметилсульфоксида. Далее охлаждают до 15°С, добавляют 3,51 г (17 ммолей) N,N'-дициклогексилкарбодиимида и перемешивают в течение 5 ч при 15°С. Для отделения мочевины раствор фильтруют. К фильтрату добавляют 8,96 г (15,88 ммоля) соединения, указанного в заголовке примера 11б, и 2,02 г (20 ммолей) триэтиламина и перемешивают в течение 12 ч при комнатной температуре. Раствор сливают в смесь из 1500 мл диэтилового эфира и 100 мл ацетона и перемешивают в течение 30 мин. Выпавшее в осадок твердое вещество отфильтровывают и хроматографируют на силикагеле RP-18 (элюент:градиент тетрагидрофурана/ацетонитрила/воды).
Выход: 15,31 г (82% от теории) бесцветного аморфного порошка.
Содержание воды: 6,3%.
Элементный анализ (в пересчете на безводное вещество):
Пример 12
а) N-(2-гидрокси)этил]амид 2Н,2Н,4Н,4Н,5Н,5Н-3-оксаперфторундекановой кислоты
К 24,25 г (57,45 ммоля) 2Н,2Н,4Н,4Н,5Н,5Н-3-оксаперфтортридекановой кислоты в 300 мл дихлорметана добавляют 8,90 г (70 ммолей) оксалилхлорида и перемешивают в течение 12 ч при комнатной температуре. Смесь упаривают в вакууме досуха. Остаток растворяют в 100 мл дихлорметана и при 0°С по каплям добавляют к раствору из 3,66 г (60 ммолей) этаноламина и 6,07 г (60 ммолей) триэтиламина в 200 мл дихлорметана. Смесь перемешивают в течение 3 ч при 0°С, а затем в течение 6 ч при комнатной температуре. Далее добавляют 300 мл 5%-ной водной соляной кислоты и тщательно перемешивают в течение 15 мин. Органическую фазу отделяют, сушат над сульфатом магния и досуха упаривают в вакууме. Остаток хроматографируют на силикагеле (элюент:дихлорметан/ацетон в соотношении 20:1).
Выход: 24,86 г (93% от теории) бесцветного твердого вещества.
Элементный анализ:
б) N-(2-гидроксиэтил)-N-1H,1Н,2Н,2Н,4Н,4Н,5Н,5Н-3-оксаперфторундецил)амин
24 г (51,59 ммоля) соединения, указанного в заголовке примера 12а, растворяют в 300 мл тетрагидрофуранаа и добавляют 31 мл 10 М борандиметилсульфида (в тетрагидрофуране). Далее в течение 12 ч кипятят с обратным холодильником. После этого охлаждают до 0°С, по каплям добавляют 200 мл метанола и затем досуха упаривают в вакууме. Остаток растворяют в смеси из 300 мл этанола и 50 мл 10%-ной водной соляной кислоты и перемешивают в течение 8 ч при 40°С. Смесь досуха упаривают в вакууме, остаток растворяют в 300 мл 5%-ного водного едкого натра и трижды экстрагируют дихлорметаном порциями по 300 мл. Органические фазы сушат над сульфатом магния, досуха упаривают в вакууме и остаток хроматографируют на силикагеле (элюент:дихлорметан/2-пропанол в соотношении 20:1).
Выход: 20,95 г (90% от теории) бесцветного твердого вещества.
Элементный анализ:
в) 1,4,7-трис(карбоксилатометил)-10-{(3-аза-4-оксогексан-5-ил)-кислота-N-[(2-гидрокси)этил-N-(1Н,1Н,2Н,2Н,4Н,4Н,5Н,5Н-3-окса)перфторундецил]амид}-1,4,7,10-тетраазациклододекан, гадолиниевый комплекс
10 г (15,88 ммоля) гадолиниевого комплекса 10-[1-(карбоксиметилкарбамоил)этил]-1,4,7,10-тетраазациклододекан-1,4,7-триуксусной кислоты и 1,35 г (31,76 ммоля) хлорида лития при 60°С растворяют в 100 мл диметилсульфоксида. Далее охлаждают до 15°С и добавляют 8,98 г (15,88 ммоля) соединения, указанного в заголовке примера 12б. Смесь перемешивают в течение 10 мин и затем добавляют 7,42 г (30 ммолей) 2-этокси-1-этоксикарбонил-1,2-дигидрохинолина. После этого перемешивают в течение 12 ч при комнатной температуре. Раствор сливают в смесь из 200 мл ацетона и 1300 мл диэтилового эфира и перемешивают в течение 2 ч при комнатной температуре. Выпавший осадок отфильтровывают, растворяют в смеси из небольшого количества этанола и воды и хроматографируют на силикагеле RP-18 (элюент:градиент тетрагидрофурана/ацетонитрила/воды).
Выход: 14,01 г (83% от теории) бесцветного аморфного порошка.
Элементный анализ:
Пример 13
а) N-(3,6,9,12-тетраоксатридецил)амид 2Н,2Н,4Н,4Н,5Н,5Н-3-оксаперфторундекановой кислоты
К 24,25 г (57,45 ммоля) 2Н,2Н,4Н,4Н,5Н,5Н-3-оксаперфторундекановой кислоты в 300 мл дихлорметана добавляют 8,90 г (70 ммолей) оксалилхлорида и перемешивают в течение 12 ч при комнатной температуре. Смесь упаривают в вакууме досуха. Остаток растворяют в 100 мл дихлорметана и при 0°С по каплям добавляют к раствору из 12,44 г (60 ммолей) 3,6,9,12-тетраоксатридециламина и 6,07 г (60 ммолей) триэтиламина в 200 мл дихлорметана. Смесь перемешивают в течение 3 ч при 0°С, а затем в течение 6 ч при комнатной температуре. Далее добавляют 300 мл 5%-ной водной соляной кислоты и тщательно перемешивают в течение 15 мин. Органическую фазу отделяют, сушат над сульфатом магния и досуха упаривают в вакууме. Остаток хроматографируют на силикагеле (элюент:дихлорметан/ацетон в соотношении 15:1).
Выход: 31,61 г (90% от теории) бесцветного твердого вещества.
Элементный анализ:
б) N-(3,6,9,12-тетраоксатридецил)-N-(1Н,1Н,2Н,2Н,4Н,4Н,5Н,5Н-3-оксаперфторундецил)амин
31 г (50,7 ммоля) соединения, указанного в заголовке примера 13а, растворяют в 300 мл тетрагидрофурана и добавляют 32 мл 10 М борандиметилсульфида (в тетрагидрофуране). Далее в течение 16 ч кипятят с обратным холодильником. После этого охлаждают до 0°С, по каплям добавляют 200 мл метанола и затем досуха упаривают в вакууме. Остаток растворяют в смеси из 300 мл этанола и 50 мл 10%-ной водной соляной кислоты и перемешивают в течение 8 ч при 40°С. Смесь досуха упаривают в вакууме, остаток растворяют в 300 мл 5%-ного водного едкого натра и трижды экстрагируют дихлорметаном порциями по 300 мл. Органические фазы сушат над сульфатом магния, досуха упаривают в вакууме и остаток хроматографируют на силикагеле (элюент:дихлорметан/2-пропанол в соотношении 20:1).
Выход: 28,17 г (93% от теории) бесцветного твердого вещества.
Элементный анализ (в пересчете на безводное вещество):
в) 1,4,7-трис(карбоксилатометил)-10-{(3-аза-4-оксогексан-5-ил)-кислота-N-[(3,6,9,12-тетраокса)тридецил-N-(1Н,1Н,2Н,2Н,4Н,4Н,5Н,5Н-3-окса)перфторундецил]амид}-1,4,7,10-тетраазациклододекан, гадолиниевый комплекс
10 г (15,88 ммоля) гадолиниевого комплекса 10-[1-(карбоксиметилкарбамоил)этил]-1,4,7,10-тетраазациклододекан-1,4,7-триуксусной кислоты и 1,35 г (31,76 ммоля) хлорида лития при 60°С растворяют в 100 мл диметилсульфоксида. Далее охлаждают до 15°С и добавляют 9,49 г (15,88 ммоля) соединения, указанного в заголовке примера 13б. Смесь перемешивают в течение 10 мин, а затем добавляют 7,42 г (30 ммолей) 2-этокси-1-этоксикарбонил-1,2-дигидрохинолина. После этого перемешивают в течение 12 ч при комнатной температуре. Раствор сливают в смесь из 200 мл ацетона и 1300 мл диэтилового эфира и перемешивают в течение 2 ч при комнатной температуре. Выпавший осадок отфильтровывают, растворяют в смеси из небольшого количества этанола и воды и хроматографируют на силикагеле RP-18 (элюент:градиент тетрагидрофурана/ацетонитрила/воды).
Выход: 16,13 г (84% от теории).
Элементный анализ:
Пример 14
а) трет-Бутиловый эфир 2-N-(1H,1H,2H,2H,4H,4H,5H,5H-3-оксаперфтортридецил)аминоуксусной кислоты
К 32,0 г (58,65 ммоля) соединения, указанного в заголовке примера 5б, и 24,89 г (180 ммолей) карбоната калия в 300 мл ацетонитрила при 50°С по каплям добавляют 6,523 г (40 ммолей) трет-бутилового эфира бромуксусной кислоты и перемешивают в течение 3 ч при этой температуре. Далее добавляют 300 мл дихлорметана, выпавшие в осадок соли отфильтровывают и фильтрат досуха упаривают в вакууме. Остаток хроматографируют на силикагеле (элюент:дихлорметан/2-пропанол в соотношении 20:1).
Выход: 28,11 г (57% от теории) бесцветного твердого вещества.
Элементный анализ:
б) 1,4,7-трис(карбоксилатометил)-10-{(3-аза-4-оксогексан-5-ил)-кислота-N-[(трет-бутилоксикарбонилметил)-N-(1Н, Н,2Н,2Н,4Н,4Н,5Н,5Н-3-окса)перфтортридецил]амид}-1,4,7,10-тетраазациклододекан, гадолиниевый комплекс
10 г (15,88 ммоля) гадолиниевого комплекса 10-[1-(карбоксиметилкарбамоил)этил]-1,4,7,10-тетраазациклододекан-1,4,7-триуксусной кислоты и 1,35 г (31,76 ммоля) хлорида лития при 60°С растворяют в 100 мл диметилсульфоксида. Далее охлаждают до 15°С и добавляют 9,87 г (15,88 ммоля) соединения, указанного в заголовке примера 14а. Смесь перемешивают в течение 10 мин и затем добавляют 7,42 г (30 ммолей) 2-этокси-1-этоксикарбонил-1,2-дигидрохинолина. После этого перемешивают в течение 12 ч при комнатной температуре. Раствор сливают в смесь из 200 мл ацетона и 1300 мл диэтилового эфира и перемешивают в течение 2 ч при комнатной температуре. Выпавший осадок отфильтровывают, растворяют в смеси из небольшого количества этанола и воды и хроматографируют на силикагеле RP-18 (элюент:градиент тетрагидрофурана/ацетонитрила/воды).
Выход: 16,64 г (85% от теории).
Элементный анализ:
в) 1,4,7-трис-(карбоксилатометил)-10-{(3-аза-4-оксогексан-5-ил)-кислота-N-[(карбоксиметил)-N-(1Н,1Н,2Н,2Н,4Н,4Н,5Н,5Н-3-окса)перфтортридецил]амид}-1,4,7,10-тетраазациклододекан, гадолиниевый комплекс
10 г (8,11 ммоля) соединения, указанного в заголовке примера 14б, растворяют в 50 мл трифторуксусной кислоты и в течение 5 ч перемешивают при комнатной температуре. Смесь досуха упаривают в вакууме и остаток хроматографируют на силикагеле RP-18 (элюент:градиент тетрагидрофурана/ацетонитрила/воды). После упаривания содержащих продукт фракций остаток растворяют в воде и значение рН устанавливают на 7,2 с помощью 5%-ного водного едкого натра. Раствор фильтруют и фильтрат лиофилизуют.
Выход: 10,48 г (91% от теории).
Элементный анализ (в пересчете на безводное вещество):
Пример 15
а) N-(2-гидроксиэтил)амид 2Н,2Н,4Н,4Н,5Н,5Н-3-оксаперфтортридекановой кислоты
К 32 г (56,61 ммоля) соединения, указанного в заголовке примера 4а, добавляют 2,96 г (74 ммоля) гидрида натрия (из 60%-ного гидрида натрия в парафиновом масле) в 300 мл тетрагидрофурана и перемешивают в течение 3 ч при комнатной температуре в атмосфере азота. Далее по каплям добавляют 7,67 г (74 ммоля) трет-бутилового эфира бромуксусной кислоты, растворенного в 20 мл тетрагидрофурана, и перемешивают в течение 5 ч при 50°С. Далее добавляют 50 мл метанола и досуха упаривают в вакууме. Остаток хроматографируют на силикагеле (элюент:дихлорметан/2-пропанол в соотношении 20:1).
Выход: 23,46 г (61% от теории).
Элементный анализ:
б) N-(1Н,1Н,2Н,2Н,4Н,4Н,5Н,5Н-3-оксаперфтортридецил)-N-[4-трет-бутилоксикарбонил-3-окса)бутил]амин
35,0 г (51,52 ммоля) соединения, указанного в заголовке примера 15а, растворяют в 300 мл тетрагидрофурана и добавляют 31 мл 10 М борандиметилсульфида (в тетрагидрофуране). Далее в течение 16 ч кипятят с обратным холодильником. После этого охлаждают до 0°С, по каплям добавляют 200 мл метанола и затем досуха упаривают в вакууме. Остаток растворяют в смеси из 300 мл этанола и 50 мл 10%-ной водной соляной кислоты и перемешивают в течение 8 ч при 40°С. Смесь досуха упаривают в вакууме, остаток растворяют в 300 мл 5%-ного водного едкого натра и трижды экстрагируют дихлорметаном порциями по 300 мл. Органические фазы сушат над сульфатом магния, досуха упаривают в вакууме и остаток хроматографируют на силикагеле (элюент:дихлорметан/2-пропанол в соотношении 20:1).
Выход: 31,88 г (93% от теории).
Элементный анализ:
в) 1,4,7-трис(карбоксилатометил)-10-{(3-аза-4-оксогексан-5-ил)-кислота-N-[(4-трет-бутилоксикарбонил-3-окса)бутил)-N-(1Н,1Н,2Н,2Н,4Н,4Н,5Н,5Н-3-окса)перфтортридецил]амид}-1,4,7,10-тетраазациклододекан, гадолиниевый комплекс
10 г (15,88 ммоля) гадолиниевого комплекса 10-[1-(карбоксиметилкарбамоил)этил]-1,4,7,10-тетраазациклододекан-1,4,7-триуксусной кислоты и 1,35 г (31,76 ммоля) хлорида лития при 60°С растворяют в 100 мл диметилсульфоксида. Далее охлаждают до 15°С и добавляют 10,57 г (15,88 ммоля) соединения, указанного в заголовке примера 15б. Смесь перемешивают в течение 10 мин и затем добавляют 7,42 г (30 ммолей) 2-этокси-1-этоксикарбонил-1,2-дигидрохинолина. После этого перемешивают в течение 12 ч при комнатной температуре. Раствор сливают в смесь из 200 мл ацетона и 1300 мл диэтилового эфира и перемешивают в течение 2 ч при комнатной температуре. Выпавший осадок отфильтровывают, растворяют в смеси из небольшого количества этанола и воды и хроматографируют на силикагеле RP-18 (элюент:градиент тетрагидрофурана/ацетонитрила/воды).
Выход: 16,63 г (82% от теории).
Элементный анализ:
г) 1,4,7-трис(карбоксилатометил)-10-{(3-аза-4-оксогексан-5-ил)-кислота-[N-(4-карбокси-3-окса)бутил)-N-(1Н,1Н,2Н,2Н,4Н,4Н,5Н,5Н-3-окса)перфтортридецил]амид}-1,4,7,10-тетраазациклододекан, гадолиниевый комплекс
12 г (9,40 ммоля) соединения, указанного в заголовке примера 15в, растворяют в 50 мл трифторуксусной кислоты и в течение 5 ч перемешивают при комнатной температуре. Смесь досуха упаривают в вакууме и остаток хроматографируют на силикагеле RP-18 (элюент:градиент тетрагидрофурана/ацетонитрила/воды). После упаривания содержащих продукт фракций остаток растворяют в воде и значение рН устанавливают на 7,2 с помощью 5%-ного водного едкого натра. Раствор фильтруют и фильтрат лиофилизуют.
Выход: 11,41 г (92% от теории).
Содержание воды: 5,8%.
Элементный анализ (в пересчете на безводное вещество):
Пример 16
а) N-(2Н,2Н,4Н,4Н,5Н,5Н-3-оксаперфтортридецил)амид 2Н,2Н,4Н,4Н,5Н,5Н-3-оксаперфтортридекановой кислоты
К 30 г (57,45 ммоля) 2Н,2Н,4Н,4Н,5Н,5Н-3-оксаперфтортридекановой кислоты в 300 мл дихлорметана добавляют 8,90 г (70 ммолей) оксалилхлорида и перемешивают в течение 12 ч при комнатной температуре. Смесь упаривают в вакууме досуха. Остаток растворяют в 100 мл дихлорметана и при 0°С по каплям добавляют к раствору из 32,62 г (60 ммолей) соединения, указанного в заголовке примера 5б, и 6,07 г (60 ммолей) триэтиламина в 200 мл дихлорметана. Смесь перемешивают в течение 3 ч при 0°С, а затем в течение 6 ч при комнатной температуре. Далее добавляют 300 мл 5%-ной водной соляной кислоты и тщательно перемешивают в течение 15 мин. Органическую фазу отделяют, сушат над сульфатом магния и досуха упаривают в вакууме. Остаток хроматографируют на силикагеле (элюент:дихлорметан/ацетон в соотношении 15:1).
Выход: 52,87 г (91% от теории).
Элементный анализ:
б) N-бис[(1Н,1Н,2Н,2Н,4Н,4Н,5Н,5Н-3-окса)перфтортридецил]амин
52 г (51,42 ммоля) соединения, указанного в заголовке примера 16а, растворяют в 500 мл тетрагидрофурана и добавляют 31 мл 10 М борандиметилсульфида (в тетрагидрофуране). Далее в течение 16 ч кипятят с обратным холодильником. После этого охлаждают до 0°С, по каплям добавляют 200 мл метанола и затем досуха упаривают в вакууме. Остаток растворяют в смеси из 400 мл этанола и 70 мл 10%-ной водной соляной кислоты и перемешивают в течение 8 ч при 40°С. Смесь досуха упаривают в вакууме, остаток растворяют в 400 мл 5%-ного водного едкого натра и трижды экстрагируют дихлорметаном порциями по 400 мл. Органические фазы сушат над сульфатом магния, досуха упаривают в вакууме и остаток хроматографируют на силикагеле (элюент:дихлорметан/2-пропанол в соотношении 20:1).
Выход: 47,18 г (92% от теории) бесцветного твердого вещества.
Элементный анализ:
в) 1,4,7-трис(карбоксилатометил)-10-[(3-аза-4-оксогексан-5-ил)-кислота-N-бис(1Н,1Н,2Н,2Н,4Н,4Н,5Н,5Н-3-оксаперфтортридецил)амид]-1,4,7,10-тетраазациклододекан, гадолиниевый комплекс
10 г (15,88 ммоля) гадолиниевого комплекса 10-[1-(карбоксиметилкарбамоил)этил]-1,4,7,10-тетраазациклододекан-1,4,7-триуксусной кислоты и 1,35 г (31,76 ммоля) хлорида лития при 60°С растворяют в 100 мл диметилсульфоксида. Далее охлаждают до 15°С и добавляют 15,84 г (15,88 ммоля) соединения, указанного в заголовке примера 16б. Смесь перемешивают в течение 10 мин и затем добавляют 7,42 г (30 ммолей) 2-этокси-1-этоксикарбонил-1,2-дигидрохинолина. После этого перемешивают в течение 12 ч при комнатной температуре. Раствор сливают в смесь из 200 мл ацетона и 1300 мл диэтилового эфира и перемешивают в течение 2 ч при комнатной температуре. Выпавший осадок отфильтровывают, растворяют в смеси из небольшого количества этанола и воды и хроматографируют на силикагеле RP-18 (элюент:градиент тетрагидрофурана/ацетонитрила/воды).
Выход: 20,95 г (82% от теории).
Элементный анализ:
Пример 17
а) N-(5-гидрокси-3-оксапентил)амид 2Н,2Н,4Н,4Н,5Н,5Н-3-оксаперфтортридекановой кислоты
К 32 г (52,52 ммоля) соединения, указанного в заголовке примера 3а, добавляют 2,80 г (70 ммолей) гидрида натрия (из 60%-ного гидрида натрия в парафиновом масле) в 300 мл тетрагидрофурана и перемешивают в течение 3 ч при комнатной температуре в атмосфере азота. Затем по каплям добавляют 9,68 г (70 ммолей) трет-бутилокого эфира бромуксусной кислоты в 20 мл тетрагидрофурана и перемешивают в течение 5 ч при 50°С. Далее добавляют 50 мл метанола и досуха упаривают в вакууме. Остаток хроматографируют на силикагеле (элюент:дихлорметан/2-пропанол в соотношении 20:1).
Выход: 19,31 г (59% от теории).
Элементный анализ:
б) N-(3,6-диоксагептил)-N-(1H,1H,2H,2H,4H,4H,5H,5H-3-оксаперфтортридецил)амин
32 г (51,34 ммоля) соединения, указанного в заголовке примера 17а, растворяют в 300 мл тетрагидрофуранаа и добавляют 31 мл 10 М борандиметилсульфида (в тетрагидрофуране). Далее в течение 16 ч кипятят с обратным холодильником. После этого охлаждают до 0°С, по каплям добавляют 200 мл метанола и затем досуха упаривают в вакууме. Остаток растворяют в смеси из 300 мл этанола и 50 мл 10%-ной водной соляной кислоты и перемешивают в течение 8 ч при 40°С. Смесь досуха упаривают в вакууме, остаток растворяют в 300 мл 5%-ного водного едкого натра и трижды экстрагируют дихлорметаном порциями по 300 мл. Органические фазы сушат над сульфатом магния, досуха упаривают в вакууме и остаток хроматографируют на силикагеле (элюент:дихлорметан/2-пропанол в соотношении 20:1).
Выход: 28,47 г (91% от теории).
Элементный анализ:
в) 1,4,7-трис(карбоксилатометил)-10-[(3-аза-4-оксогексан-5-ил)-кислота-N-(3,6-диокса)гептил-N-(1Н,1Н,2Н,2Н,4Н,4Н,5Н,5Н-3-оксаперфтортридецил)амид]-1,4,7,10-тетраазациклододекан, гадолиниевый комплекс
10 г (15,88 ммоля) гадолиниевого комплекса 10-[1-(карбоксиметилкарбамоил)этил]-1,4,7,10-тетраазациклододекан-1,4,7-триуксусной кислоты и 1,35 г (31,76 ммоля) хлорида лития при 60°С растворяют в 100 мл диметилсульфоксида. Далее охлаждают до 15°С и добавляют 9,68 г (15,88 ммоля) соединения, указанного в заголовке примера 17б. Смесь перемешивают в течение 10 мин и затем добавляют 7,42 г (30 ммолей) 2-этокси-1-этоксикарбонил-1,2-дигидрохинолина. После этого перемешивают в течение 12 ч при комнатной температуре. Раствор сливают в смесь из 200 мл ацетон и 1300 мл диэтилового эфира и перемешивают в течение 2 ч при комнатной температуре. Выпавший осадок отфильтровывают, растворяют в смеси из небольшого количества этанола и воды и хроматографируют на силикагеле RP-18 (элюент:градиент тетрагидрофурана/ацетонитрила/воды).
Выход: 16,09 г (83% от теории).
Элементный анализ:
Пример 18
а) N-(гексил)амид 2Н,2Н,4Н,4Н,5Н,5Н-3-оксаперфтортридекановой кислоты
К 30 г (57,45 ммоля) 2Н,2Н,4Н,4Н,5Н,5Н-3-оксаперфтортридекановой кислоты в 300 мл дихлорметана добавляют 8,90 г (70 ммолей) оксалилхлорида и перемешивают в течение 12 ч при комнатной температуре. Смесь упаривают в вакууме досуха. Остаток растворяют в 100 мл дихлорметана и при 0°С по каплям добавляют к раствору из 6,07 г (60 ммолей) н-гексиламина и 6,07 г (60 ммолей) триэтиламина в 200 мл дихлорметана. Смесь перемешивают в течение 3 ч при 0°С, а затем в течение 6 ч при комнатной температуре. Далее добавляют 300 мл 5%-ной водной соляной кислоты и тщательно перемешивают в течение 15 мин. Органическую фазу отделяют, сушат над сульфатом магния и досуха упаривают в вакууме. Остаток хроматографируют на силикагеле (элюент:дихлорметан/ацетон в соотношении 20:1).
Выход: 30,95 г (89% от теории).
Элементный анализ:
б) N-(гексил)-N-(1Н,1Н,2Н,2Н,4Н,4Н,5Н,5Н-3-оксаперфтортридецил)амин
31 г (51,21 ммоля) соединения, указанного в заголовке примера 18а, растворяют в 300 мл тетрагидрофурана и добавляют 31 мл 10 М борандиметилсульфида (в тетрагидрофуране). Далее в течение 16 ч кипятят с обратным холодильником. После этого охлаждают до 0°С, по каплям добавляют 200 мл метанола и затем досуха упаривают в вакууме. Остаток растворяют в смеси из 300 мл этанола и 50 мл 10%-ной водной соляной кислоты и перемешивают в течение 8 ч при 40°С. Смесь досуха упаривают в вакууме, остаток растворяют в 300 мл 5%-ного водного едкого натра и трижды экстрагируют дихлорметаном порциями по 300 мл. Органические фазы сушат над сульфатом магния, досуха упаривают в вакууме и остаток хроматографируют на силикагеле (элюент:дихлорметан/2-пропанол в соотношении 20:1).
Выход: 28,16 г (93% от теории).
Элементный анализ:
в) 1,4,7-трис(карбоксилатометил)-10-{(3-аза-4-оксогексан-5-ил)-кислота-N-(гексил)-N-(1Н,1Н,2Н,2Н,4Н,4Н,5Н,5Н-3-оксаперфтортридецил]амид}-1,4,7,10-тетраазациклододекан, гадолиниевый комплекс
10 г (15,88 ммоля) гадолиниевого комплекса 10-[1-(карбоксиметилкарбамоил)этил]-1,4,7,10-тетраазациклододекан-1,4,7-триуксусной кислоты и 1,35 г (31,76 ммоля) хлорида лития при 60°С растворяют в 100 мл диметилсульфоксида. Далее охлаждают до 15°С и добавляют 10,98 г (15,88 ммоля) соединения, указанного в заголовке примера 18б. Смесь перемешивают в течение 10 мин и затем добавляют 7,42 г (30 ммолей) 2-этокси-1-этоксикарбонил-1,2-дигидрохинолина. После этого перемешивают в течение 12 ч при комнатной температуре. Раствор сливают в смесь из 200 мл ацетона и 1300 мл диэтилового эфира и перемешивают в течение 2 ч при комнатной температуре. Выпавший осадок отфильтровывают, растворяют в смеси из небольшого количества этанола и воды и хроматографируют на силикагеле RP-18 (элюент:градиент тетрагидрофурана/ацетонитрила/воды).
Выход: 16,29 г (84% от теории).
Элементный анализ:
Пример 19
а) N-[(10-трет-бутилоксикарбонил)децил]амид 2Н,2Н,4Н,4Н,5Н,5Н-3-оксаперфтортридекановой кислоты
К 30 г (57,45 ммоля) 2Н,2Н,4Н,4Н,5Н,5Н-3-оксаперфтортридекановой кислоты в 300 мл дихлорметана добавляют 8,90 г (70 ммолей) оксалилхлорида и перемешивают в течение 12 ч при комнатной температуре. Смесь упаривают в вакууме досуха. Остаток растворяют в 100 мл дихлорметана и при 0°С по каплям добавляют к раствору из 15,45 г (60 ммолей) трет-бутилового эфира 11-аминоундекановой кислоты и 6,07 г (60 ммолей) триэтиламина в 200 мл дихлорметана. Смесь перемешивают в течение 3 ч при 0°С, а затем в течение 6 ч при комнатной температуре. Далее добавляют 300 мл 5%-ной водной соляной кислоты и тщательно перемешивают в течение 15 мин. Органическую фазу отделяют, сушат над сульфатом магния и досуха упаривают в вакууме. Остаток хроматографируют на силикагеле (элюент:дихлорметан/ацетон в соотношении 20:1).
Выход: 42,04 г (92% от теории).
Элементный анализ:
б) N-(10-трет-бутилоксикарбонилдецил)-N-(1Н, 1Н,2Н,2Н,4Н,4Н,5Н,5Н-3-оксаперфтортридецил)амин
39 г (51,21 ммоля) соединения, указанного в заголовке примера 19а, растворяют в 300 мл тетрагидрофурана и добавляют 31 мл 10 М борандиметилсульфида (в тетрагидрофуране). Далее в течение 16 ч кипятят с обратным холодильником. После этого охлаждают до 0°С, по каплям добавляют 200 мл метанола и затем досуха упаривают в вакууме. Остаток растворяют в смеси из 400 мл этанола и 70 мл 10%-ной водной соляной кислоты и перемешивают в течение 8 ч при 40°С. Смесь досуха упаривают в вакууме, остаток растворяют в 350 мл 5%-ного водного едкого натра и трижды экстрагируют дихлорметаном порциями по 400 мл. Органические фазы сушат над сульфатом магния, досуха упаривают в вакууме и остаток хроматографируют на силикагеле (элюент:дихлорметан/2-пропанол в соотношении 20:1).
Выход: 34,84 г (91% от теории).
Элементный анализ:
в) 1,4,7-трис(карбоксилатометил)-10-{(3-аза-4-оксогексан-5-ил)-кислота-N-(10-трет-бутилоксикарбонил)децил-N-(1Н,1Н,2Н,2Н,4Н,4Н,5Н,5Н-3-оксаперфтортридецил]амид}-1,4,7,10-тетраазациклододекан, гадолиниевый комплекс
10 г (15,88 ммоля) гадолиниевого комплекса 10-[1-(карбоксиметилкарбамоил)этил]-1,4,7,10-тетраазациклододекан-1,4,7-триуксусной кислоты и 1,35 г (31,76 ммоля) хлорида лития при 60°С растворяют в 100 мл диметилсульфоксида. Далее охлаждают до 15°С и добавляют 11,87 г (15,88 ммоля) соединения, указанного в заголовке примера 19б. Смесь перемешивают в течение 10 мин и затем добавляют 7,42 г (30 ммолей) 2-этокси-1-этоксикарбонил-1,2-дигидрохинолина. После этого перемешивают в течение 12 ч при комнатной температуре. Раствор сливают в смесь из 200 мл ацетона и 1300 мл диэтилового эфира и перемешивают в течение 2 ч при комнатной температуре. Выпавший осадок отфильтровывают, растворяют в смеси из небольшого количества этанола и воды и хроматографируют на силикагеле RP-18 (элюент:градиент тетрагидрофурана/ацетонитрила/воды).
Выход: 17,92 г (83% от теории).
Элементный анализ:
г) 1,4,7-трис(карбоксилатометил)-10-{(3-аза-4-оксогексан-5-ил)-кислота-[N-(10-карбокси)децил-N-(1Н,1Н,2Н,2Н,4Н,4Н,5Н,5Н-3-окса)перфтортридецил]амид}-1,4,7,10-тетраазациклододекан, гадолиниевый комплекс, натриевая соль
12 г (8,83 ммоля) соединения, указанного в заголовке примера 19в, растворяют в 50 мл трифторуксусной кислоты и в течение 5 ч перемешивают при комнатной температуре. Смесь досуха упаривают в вакууме и остаток хроматографируют на силикагеле RP-18 (элюент:градиент тетрагидрофурана/ацетонитрила/воды). После упаривания содержащих продукт фракций остаток растворяют в воде и значение рН устанавливают на 7,2 с помощью 5%-ного водного едкого натра. Раствор фильтруют и фильтрат лиофилизуют.
Выход: 12,48 г (92% от теории).
Содержание воды: 6,2%.
Элементный анализ (в пересчете на безводное вещество):
Пример 20
а) N-(1Н,1Н,2Н,2Н,4Н,4Н,5Н,5Н-3-окса)перфтортридецил)амид 15-бензил-3,6,9,12,15-пентаоксагексадециловой кислоты
К 19,67 г (57,45 ммоля) 15-бензил-3,6,9,12,15-пентаоксагексадециловой кислоты в 250 мл дихлорметана добавляют 8,90 г (70 ммолей) оксалилхлорида и перемешивают в течение 12 ч при комнатной температуре. Смесь упаривают в вакууме досуха. Остаток растворяют в 100 мл дихлорметана и при 0°С по каплям добавляют к раствору из 32,62 г (60 ммолей) гидрохлорида 1Н,1Н,2Н,2Н,4Н,4Н,5Н,5Н-3-оксаперфтортридециламина и 6,07 г (60 ммолей) триэтиламина в 200 мл дихлорметана. Смесь перемешивают в течение 3 ч при 0°С, а затем в течение 6 ч при комнатной температуре. Далее добавляют 300 мл 5%-ной водной соляной кислоты и тщательно перемешивают в течение 15 мин. Органическую фазу отделяют, сушат над сульфатом магния и досуха упаривают в вакууме. Остаток хроматографируют на силикагеле (элюент:дихлорметан/ацетон в соотношении 20:1).
Выход: 44,91 г (94% от теории) бесцветного твердого вещества.
Элементный анализ:
б) N-15-бензил-3,6,9,12,15-пентаоксагексадецил)-N-(1Н, 1Н,2Н,2Н,4Н,4Н,5Н,5Н-3-окса)перфтортридецил)амин
43 г (51,72 ммоля) соединения, указанного в заголовке примера 20а, растворяют в 400 мл тетрагидрофурана и добавляют 31 мл 10 М борандиметилсульфида (в тетрагидрофуране). Далее в течение 16 ч кипятят с обратным холодильником. После этого охлаждают до 0°С, по каплям добавляют 200 мл метанола и затем досуха упаривают в вакууме. Остаток растворяют в смеси из 400 мл этанола и 50 мл 10%-ной водной соляной кислоты и перемешивают в течение 8 ч при 40°С. Смесь досуха упаривают в вакууме, остаток растворяют в 350 мл 5%-ного водного едкого натра и трижды экстрагируют дихлорметаном порциями по 400 мл. Органические фазы сушат над сульфатом магния, досуха упаривают в вакууме и остаток хроматографируют на силикагеле (элюент:дихлорметан/2-пропанол в соотношении 20:1).
Выход: 39,32 г (93% от теории).
Элементный анализ:
в) 1,4,7-трис(карбоксилатометил)-10-{(3-аза-4-оксогексан-5-ил)-кислота-[N-(15-бензил-3,6,9,12,15-пентаокса)гексадецил-N-(1Н,1Н,2Н,2Н,4Н,4Н,5Н,5Н-3-окса)тридецил] амид} -1,4,7,10-тетраазациклододекан, гадолиниевый комплекс
10 г (15,88 ммоля) гадолиниевого комплекса 10-[1-(карбоксиметилкарбамоил)этил]-1,4,7,10-тетраазациклододекан-1,4,7-триуксусной кислоты и 1,35 г (31,76 ммоля) хлорида лития при 60°С растворяют в 100 мл диметилсульфоксида. Далее охлаждают до 15°С и добавляют 12,98 г (15,88 ммоля) соединения, указанного в заголовке примера 20б. Смесь перемешивают в течение 10 мин и затем добавляют 7,42 г (30 ммолей) 2-этокси-1-этоксикарбонил-1,2-дигидрохинолина. После этого перемешивают в течение 12 ч при комнатной температуре. Раствор сливают в смесь из 200 мл ацетона и 1300 мл диэтилового эфира и перемешивают в течение 2 ч при комнатной температуре. Выпавший осадок отфильтровывают, растворяют в смеси из небольшого количества этанола и воды и хроматографируют на силикагеле RP-18 (элюент:градиент тетрагидрофурана/ацетонитрила/воды).
Выход: 18,84 г (83% от теории).
Элементный анализ:
г) 1,4,7-трис(карбоксилатометил)-10-{(3-аза-4-оксогексан-5-ил)-кислота-[N-(14-гидрокси-3,6,9,12-тетраокса)тетрадецил-N-(1Н,1Н,2Н,2Н,4Н,4Н,5Н,5Н-3-окса)перфтортридецил]амид} -1,4,7,10-тетраазациклододекан, гадолиниевый комплекс
12 г (8,40 ммоля) соединения, указанного в заголовке примера 20в, растворяют в 150 мл метанола, добавляют 1,0 г палладиевого катализатора (10%-ного Pd/C) и гидрируют в течение ночи при комнатной температуре. После этого катализатор отфильтровывают и фильтрат досуха упаривают в вакууме.
Выход: 10,13 г (95% от теории).
Элементный анализ:
Пример 21
а) 2-N-трифторацетил-6-N-бензилоксикарбонил-L-лизин
100,0 г (356,7 ммоля) 6-N-бензилоксикарбонил-L-лизина растворяют в смеси из 1000 мл этилового эфира трифторуксусной кислоты и 500 мл этанола и в течение 24 ч перемешивают при комнатной температуре. После этого упаривают досуха и остаток кристаллизуют из диизопропилового эфира.
Выход: 128,9 г (96% от теории) бесцветного кристаллического порошка.
Температура плавления: 98,5°С.
Элементный анализ:
б) [1-(4-перфтороктилсульфонил)пиперазин]амид 2-N-трифторацетил-6-N-бензилоксикарбонил-L-лизина
К 125,0 г (332,0 ммоля) соединения, указанного в заголовке примера 21 а, и 188,7 г (332,0 ммоля) 1-перфтороктилсульфонилпиперазина (полученного согласно DE 19603033) в 750 мл тетрагидрофурана при 0°С добавляют 164,2 г (0,664 ммоля) ЭЭДХ (этиловый эфир 2-этокси-1,2-дигидрохинолин-1-карбоновой кислоты) и перемешивают в течение ночи при комнатной температуре. После этого досуха упаривают в вакууме и хроматографируют на силикагеле (элюент:дихлорметан/метанол в соотношении 20:1).
Выход: 286,0 г (93% от теории) бесцветного твердого вещества.
Температура плавления: 92°С.
Элементный анализ:
в) [1-(4-перфтороктилсульфонил)пиперазин]амид 6-N-бензилоксикарбонил-L-лизина
Раствор из 280,0 г (302,2 моля) соединения, указанного в заголовке примера 21б, в 2000 мл этанола при 0°С в течение одного часа барботируют газообразным аммиаком. Затем смесь перемешивают в течение 4 ч при 0°С. После этого упаривают досуха и остаток выделяют из воды путем перемешивания. Твердое вещество отфильтровывают и сушат в вакууме при 50°С.
Выход: 243,5 г (97% от теории) аморфного твердого вещества.
Элементный анализ:
г) [1-(4-перфтороктилсульфонил)пиперазин]амид 6-N-бензилоксикарбонил-2-N-[1-O-α-D-карбонилметил(2,3,4,6-тетра-O-бензилманнопираноза]-L-лизина
К раствору из 100,0 г (120,4 моля) соединения, указанного в заголовке примера 21в, 72,1 г (120,4 моля) 1-O-α-D-карбоксиметил-2,3,4,6-тетра-O-бензилманнопиранозы и 13,86 г (120,4 моля) N-гидроксисукцинимида в 500 мл диметилформамида при 0°С добавляют 41,27 г (200,0 ммолей) N,N-дициклогексилкарбодиимида. Смесь перемешивают в течение 3 ч при 0°С, а затем в течение ночи при комнатной температуре. Выпавшую в осадок мочевину отфильтровывают, фильтрат досуха упаривают в вакууме и хроматографируют на силикагеле (элюент:дихлорметан/этанол в соотношении 20:1).
Выход: 136,1 г (87% от теории) вязкого масла.
Элементный анализ:
д) 1-[(4-перфтороктилсульфонил)пиперазин]амид 2-N-[1-O-α-D-карбонилметилманнопираноза]-L-лизина
130,0 г (100,0 ммолей) соединения, указанного в заголовке примера 21г, растворяют в 2000 мл этанола и добавляют 10,0 г палладиевого катализатора (10%-ного Pd/C). После этого гидрируют в течение 12 ч при комнатной температуре. Затем катализатор отфильтровывают и фильтрат досуха упаривают в вакууме.
Выход: 91,7 г (количественный) бесцветного твердого вещества.
Элементный анализ:
е) [1-(4-перфтороктилсульфонил)пиперазин]амид 6-N-[1,4,7-трис(карбоксилатометил)-1,4,7,10-тетраазациклододекан-10-N-(пентаноил-3-аза-4-оксо-5-метил-5-ил)]-2-N-[1-O-α-D-карбонилметилманнопираноза]-L-лизина, Gd-комплекс (металлический комплекс XV)
50,0 г (54,55 ммоля) соединения, указанного в заголовке примера 21д, 6,28 г (54,55 ммоля) N-гидроксисукцинимида, 4,62 г (109,0 молей) хлорида лития и 34,35 г (54,55 моля) Gd-комплекса 1,4,7-трис(карбоксилатометил)-10-(карбокси-3-аза-4-оксо-5-метилпент-5-ил)-1,4,7,10-тетраазациклододекана растворяют при умеренном нагревании в 400 мл диметилсульфоксида. Далее при 10°С добавляют 16,88 г (81,8 моля) N,N-дициклогексилкарбодиимида и затем перемешивают в течение ночи при комнатной температуре. Раствор сливают в 3000 мл ацетона и перемешивают в течение 10 мин. Выпавшее в осадок твердое вещество отфильтровывают и затем очищают хроматографией (RP-18, элюент:градиент воды/этанола/ацетонитрила).
Выход: 75,9 г (91,0% от теории) бесцветного твердого вещества.
Содержание воды: 8,6%.
Элементный анализ (в пересчете на безводное вещество):
Пример 22
а) [1-(4-перфтороктилсульфонил)пиперазин]амид 6-N-[1,4,7-трис(карбоксилатометил)-1,4,7,10-тетраазациклододекан-10-N-(пентаноил-3-аза-4-оксо-5-метил-5-ил)]-2-N-[1-O-α-D-карбонилметилманнопираноза]-L-лизина, Gd-комплекс
50,0 г (54,55 ммоля) соединения, указанного в заголовке примера 21д, 6,28 г (54,55 ммоля) N-гидроксисукцинимида, 4,62 г (109,0 молей) хлорида лития и 34,35 г (54,55 моля) Gd-комплекса 1,4,7-трис(карбоксилатометил)-10-(карбокси-3-аза-4-оксо-5-метилпент-5-ил)-1,4,7,10-тетраазациклододекана растворяют при умеренном нагревании в 400 мл диметилсульфоксида. Далее при 10°С добавляют 16,88 г (81,8 ммоля) N,N-дициклогексилкарбодиимида и затем перемешивают в течение ночи при комнатной температуре. Раствор сливают в 3000 мл ацетона и перемешивают в течение 10 мин. Выпавшее в осадок твердое вещество отфильтровывают и затем очищают хроматографией (RP-18, элюент:градиент воды/этанола/ацетонитрила).
Выход: 76,0 г (92,0% от теории) бесцветного твердого вещества.
Содержание воды: 6,88%.
Элементный анализ (в пересчете на безводное вещество):
Пример 23
а) Метиловый эфир 2-[4-(3-оксаэтилпропионат)]фенилуксусной кислоты
К 200,0 г (1204,0 ммоля) метилового эфира 4-гидроксифенилуксусной кислоты и 212,0 г (2000,0 ммолей) карбоната натрия в 2000 мл ацетона добавляют 233,8 г (1400,0 ммолей) этилового эфира 2-бромуксусной кислоты и в течение 5 ч кипятят с обратным холодильником. Твердое вещество отфильтровывают и досуха упаривают в вакууме. Остаток хроматографируют на силикагеле (элюент:н-гексан/этиловый эфир уксусной кислоты в соотношении 15:1).
Выход: 288,5 г (95,0% от теории) бесцветного масла.
Элементный анализ:
б) Метиловый эфир 2-[4-(3-оксаэтилпропионат)]фенил-2-бромуксусной кислоты
К 285,0 г (1130,0 ммолей) указанного в заголовке примера 23а соединения, растворенного в 2000 мл четыреххлористого углерода, добавляют 201,0 г (1130,0 ммолей) N-бромсукцинимида и 100,0 мг дибензоилпероксида и в течение 8 ч кипятят с обратным холодильником. Далее охлаждают на ледяной бане, выпавший в осадок сукцинимид отфильтровывают и фильтрат досуха упаривают в вакууме. Остаток очищают на силикагеле (элюент:н-гексан/ацетон в соотношении 15:1).
Выход: 359,2 г (96,0% от теории) бесцветного вязкого масла.
Элементный анализ:
в) Метиловый эфир 2-[4-(3-оксаэтилпропионат)]фенил-2-[1-(1,4,7,10-тетраазациклододекан-1-ил]уксусной кислоты
К 603,0 г (3500,0 ммолей) 1,4,7,10-тетраазациклододекана в 6000 мл хлороформа добавляют 350,0 г (1057,0 ммолей) соединения, указанного в заголовке примера 23б, и перемешивают в течение ночи при комнатной температуре. Далее трижды экстрагируют водой порциями по 3000 мл, органическую фазу сушат над сульфатом магния и досуха упаривают в вакууме. Остаток без дополнительной очистки используют в последующей реакции (пример 23 г).
Выход: 448,0 г (количественный) вязкого масла.
Элементный анализ:
г) 2-[4-(3-оксапропионовая кислота)]фенил-2-[1,4,7-трис(карбоксиметил)-1,4,7,10-тетраазациклододекан-10-ил]уксусная кислота
445,0 г (1053,0 ммоля) соединения, указанного в заголовке примера 23в, и 496,0 г (5270,0 ммолей) хлоруксусной кислоты растворяют в 4000 мл воды. Значение рН с помощью 30%-ного водного едкого натра устанавливают на 10 и перемешивают в течение 8 ч при 70°С. После этого значение рН реакционного раствора устанавливают на 13 смешением с 30%-ным водным едким натром и в течение 30 мин кипятят с обратным холодильником. Затем раствор охлаждают на ледяной бане и добавлением концентрированной соляной кислоты значение рН устанавливают на 1. После этого досуха упаривают в вакууме. Остаток растворяют в 4000 мл метанола и в течение часа перемешивают при комнатной температуре. Выпавшую в осадок поваренную соль отфильтровывают, фильтрат упаривают досуха и остаток очищают на С RP-18 (элюент:градиент воды/этанола/ацетонитрила).
Выход: 403,0 г (69,0% от теории) бесцветного твердого вещества.
Содержание воды: 10,2%.
Элементный анализ (в пересчете на безводное вещество):
д) 2-[4-(3-оксапропионовая кислота)]фенил-2-[1,4,7-трис(карбоксилатометил)-1,4,7,10-тетраазациклододекан-10-ил]уксусная кислота, Gd-комплекс
К 400 г (721,3 ммоля) соединения, указанного в заголовке примера 23 г, в 2000 мл воды добавляют 130,73 г (360,65 ммоля) оксида гадолиния и перемешивают в течение 5 ч при 80°С. Раствор фильтруют и фильтрат лиофилизуют.
Выход: 511 г (количественный) аморфного твердого вещества.
Содержание воды: 11,0%.
Элементный анализ (в пересчете на безводное вещество):
e) [1-(4-перфтороктилсульфонил)пиперазин]амид 6-N-[2-[4-(3-оксапропионил)фенил]-2-[1,4,7-трис(карбоксилатометил)-1,4,7,10-тетраазациклододекан-10-ил]уксусная кислота)]-2-N-(1-O-α-D-карбонилметилманнопираноза)-L-лизина, Gd-комплекс, натриевая соль
50,0 г (54,55 ммоля) соединения, указанного в заголовке примера 21д, 6,28 г (54,55 ммоля) N-гидроксисукцинимида, 4,62 г (109,0 ммолей) хлорида лития и 38,66 г (54,55 ммоля) соединения, указанного в заголовке примера 23д, растворяют при умеренном нагревании в 400 мл диметилсульфоксида. Далее при 10°С добавляют 16,88 г (81,8 ммоля) N,N-дициклогексилкарбодиимида и затем перемешивают в течение ночи при комнатной температуре. Раствор сливают в 3000 мл ацетона и перемешивают в течение 10 мин. Выпавшее в осадок твердое вещество отфильтровывают и затем очищают хроматографией (RP-18, элюент:градиент воды/этанола/ацетонитрила). Полученный продукт растворяют в небольшом количестве воды и значение рН раствора устанавливают на 7,4 с помощью водного едкого натра. После этого содержащий продукт раствор лиофилизуют.
Выход: 79,1 г (89% от теории) бесцветного твердого вещества.
Содержание воды: 10,3%.
Элементный анализ (в пересчете на безводное вещество):
Пример 24
а) [1-(4-перфтороктилсульфонил)пиперазин]амид 6-N-[1,4,7-трис(трет-бутилоксикарбонилметил)-10-карбоксиметил-1,4,7,10-тетраазациклододекан-10-карбонилметил]-2-N-(1-O-α-D-карбонилметилманнопираноза)-L-лизина
15,0 г (26,19 ммоля) 1,4,7-трис(трет-бутилоксикарбонилметил)-10-карбоксиметил-1,4,7,10-тетраазациклододекана, 24,0 г (26,19 ммоля) соединения, указанного в заголовке примера 21д, и 3,01 г (26,19 ммоля) N-гидроксисукцинимида растворяют в 150 мл диметилформамида и при 0°С добавляют 8,25 г (40,0 ммолей) N,N-дициклогексилкарбодиимида. Смесь перемешивают в течение ночи при комнатной температуре. Выпавшую в осадок мочевину отфильтровывают и фильтрат досуха упаривают в вакууме. Остаток хроматографируют на силикагеле (элюент:дихлорметан/метанол в соотношении 20:1).
Выход: 35,45 г (92,0% от теории) бесцветного твердого вещества.
Элементный анализ:
б) [1-(4-перфтороктилсульфонил)пиперазин]амид 6-N-[1,4,7-трис(карбоксилатометил)-1,4,7,10-тетраазациклододекан-10-карбонилметил]-2-N-[1-O-α-D-карбонилметилманнопираноза]-L-лизина, Gd-комплекс
30,0 г (20,39 ммоля) соединения, указанного в заголовке примера 24а, растворяют в 50 мл хлороформа и добавляют 300 мл трифторуксусной кислоты. Смесь перемешивают в течение 10 мин при комнатной температуре. После этого досуха упаривают в вакууме и остаток растворяют в 300 мл воды. Далее добавляют 3,69 г (10,19 ммоля) оксида гадолиния и перемешивают в течение 5 ч при 80°С. Раствор досуха упаривают в вакууме и очищают на силикагеле (RP-18, элюент:градиент воды/этанола/ацетонитрила).
Выход: 11,0 г (37,0% от теории) бесцветного и аморфного твердого вещества.
Содержание воды: 11,3%.
Элементный анализ (в пересчете на безводное вещество):
Пример 25
а) [1-(4-перфтороктилсульфонил)пиперазин]амид 6-N-[3,6,9-трис(карбоксиметил)-3,6,9-триазанонандикарбоновая кислота-1-карбокси-11-оил]-2-N-[1-O-α-D-карбонилметилманнопираноза]-L-лизина
К 24,0 г (26,19 ммоля) указанного в заголовке примера 21д соединения, растворенного в смеси из 100 мл диметилформамида и 30 мл пиридина, добавляют 12,10 г (30,0 ммолей) 3-N-(2,6-диоксоморфолиноэтил)-6-N-(этоксикарбонилметил)-3,6-диазапробковой кислоты и перемешивают в течение 5 ч при 50°С. После этого досуха упаривают в вакууме. Остаток растворяют в 200 мл воды и значение рН полученного раствора устанавливают на 13 добавлением 20%-ного водного едкого натра. Смесь перемешивают в течение 8 ч при 22°С и значении рН, равном 13. Далее значение рН раствора устанавливают на 7,2 добавлением концентрированной соляной кислоты и затем досуха упаривают в вакууме. Остаток хроматографируют на силикагеле RP-18 (элюент:градиент воды/этанола/ацетонитрила).
Выход: 17,26 г (51,0% от теории) бесцветного твердого вещества.
Содержание воды: 9,3%.
Элементный анализ (в пересчете на безводное вещество):
б) [1-(4-перфтороктилсульфонил)пиперазин]амид 6-N-[3,6,9-трис(карбоксилатометил)-3,6,9-триазанонандикарбоновая кислота-1-карбокси-11-оил]-2-N-[1-O-α-D-карбонилметилманнопираноза]-L-лизина, Gd-комплекс, натриевая соль
К 10,0 г (7,74 ммоля) соединения, указанного в заголовке примера 25а, в 100 мл воды добавляют 1,40 г (3,87 ммоля) оксида гадолиния и перемешивают в течение 2 ч при 70°С. После этого раствор фильтруют. Значение рН фильтрата устанавливают на 7,4 с помощью 2н. едкого натра и лиофилизуют.
Выход: 11,36 г (количественный) аморфного твердого вещества.
Содержание воды: 10,5%.
Элементный анализ (в пересчете на безводное вещество):
Пример 26
а) [1-(4-перфтороктилсульфонил)пиперазин]амид 6-N-бензилоксикарбонил-2-N-[1,4,7-трис(карбоксилатометил)-1,4,7,10-тетраазациклододекан-10-(пентаноил-3-аза-4-оксо-5-метил-5-ил)]-L-лизина, Gd-комплекс
50,0 г (60,20 ммоля) соединения, указанного в заголовке примера 21в, 6,93 г (60,20 ммоля) N-гидроксисукцинимида, 5,09 г (120,0 ммолей) хлорида лития и 37,91 г (60,20 ммоля) Gd-комплекса 1,4,7-трис(карбоксилатометил)-1,4,7,10-тетраазациклододекан-10-(пентаноил-3-аза-4-оксо-5-метил-5-ила) растворяют при умеренном нагревании в 400 мл диметилсульфоксида. Далее при 10°С добавляют 20,63 г (100,0 ммолей) N,N-дициклогексилкарбодиимида и затем перемешивают в течение ночи при комнатной температуре. Раствор сливают в 3000 мл ацетона и перемешивают в течение 10 мин. Выпавшее в осадок твердое вещество отфильтровывают и затем очищают хроматографией (силикагель RP-18, элюент:градиент воды/этанола/ацетонитрила).
Выход: 75,53 г (87,0% от теории) бесцветного твердого вещества.
Содержание воды: 10,1%.
Элементный анализ (в пересчете на безводное вещество):
б) [1-(4-перфтороктилсульфонил)пиперазин]амид 2-N-[1,4,7-трис(карбоксилатометил)-1,4,7,10-тетраазациклододекан-10-(пентаноил-3-аза-4-оксо-5-метил-5-ил)]-L-лизина, Gd-комплекс
70,0 г (48,53 ммоля) соединения, указанного в заголовке примера 21г, растворяют в смеси из 500 мл воды и 100 мл этанола, смешивают с 5,0 г палладиевого катализатора (10%-ного Pd/C) и гидрируют при комнатной температуре в атмосфере водорода (1 атм) до тех пор, пока не прекратится поглощение водорода. После отделения катализатора вакуум-фильтрацией тщательно промывают этанолом (дважды порциями по 75 мл) и досуха концентрируют в вакууме. Таким путем получают указанное в заголовке соединение в виде высоковязкого и бесцветного масла.
Выход: 63,5 г (количественный).
Содержание воды: 9,8%.
Элементный анализ (в пересчете на безводное вещество):
в) [1-(4-перфтороктилсульфонил)пиперазин]амид 6-N-(1-O-α-D-карбонилметил-2,3,4,6-тетра-O-бензилманнопираноза)-2-N-[1,4,7-трис(карбоксилатометил)-1,4,7,10-тетраазациклододекан-10-(пентаноил-3-аза-4оксо-5-метил-5-ил)]-L-лизина, Gd-комплекс
50,0 г (38,22 ммоля) соединения, указанного в заголовке примера 26б, 4,40 г (38,22 ммоля) N-гидроксисукцинимида, 3,39 г (80,0 ммолей) хлорида лития и 22,88 г (38,22 ммоля) 1-O-α-D-карбоксиметил-2,3,4,6-тетра-O-бензил-маннопиранозы растворяют при умеренном нагревании (30-40°С) в 400 мл диметилсульфоксида. Далее при 10°С добавляют 10,32 г (50,0 ммолей) N,N-дициклогексилкарбодиимида и затем перемешивают в течение ночи при комнатной температуре. После этого раствор сливают в 3000 мл ацетона и перемешивают в течение 10 мин. Выпавшее в осадок твердое вещество отфильтровывают и затем очищают хроматографией (силикагель RP-18, элюент:
градиент воды/этанола/ацетонитрила).
Выход: 64,25 г (89,0% от теории) бесцветного твердого вещества.
Содержание воды: 10,9%.
Элементный анализ (в пересчете на безводное вещество):
г) [1-(4-перфтороктилсульфонил)пиперазин]амид 6-N-(1-O-α-D-карбонилметил-2,3,4,6-тетра-O-бензилманнопираноза)-2-N-[1,4,7-трис(карбоксилатометил)-1,4,8,10-тетраазациклододекан-10-(пентаноил-3-аза-4-оксо-5-метил-5-ил)]-L-лизина, Gd-комплекс
60,0 г (31,77 ммоля) соединения, указанного в заголовке примера 26в, растворяют в 500 мл этанола и смешивают с 6,0 г палладиевого катализатора (10%-ного Pd/C). После этого гидрируют при комнатной температуре в атмосфере водорода (1 атм) до тех пор, пока не прекратится поглощение водорода. После этого катализатор удаляют вакуум-фильтрацией, тщательно промывают этанолом (дважды порциями по 150 мл) и досуха концентрируют в вакууме.
Выход: 48,55 г (количественный) бесцветного твердого вещества.
Содержание воды: 3,9%.
Элементный анализ (в пересчете на безводное вещество):
Пример 27
а) 1,7-бис(бензилоксикарбонил)-4-[2-(N-этил-N-перфтороктилсульфонил)амино]ацетил-1,4,7,10-тетраазациклододекан
К 50,0 г (113,5 ммоля) 1,7-бис(бензилоксикарбонил)-1,4,7,10-тетраазациклододекана и 66,42 г (113,5 ммоля) 2-(N-этил-N-перфтороктилсульфонил)аминоуксусной кислоты (полученной согласно DE 19603033) в 300 мл тетрагидрофурана при 0°С добавляют 49,46 г (200,0 ммолей) ЭЭДХ (этиловый эфир 2-этокси-1,2-дигидрохинолин-1-карбоновой кислоты) и перемешивают в течение ночи при комнатной температуре. После этого досуха упаривают в вакууме и хроматографируют на силикагеле (элюент:дихлорметан/метанол в соотношении 20:1).
Выход: 65,2 г (57% от теории) бесцветного твердого вещества.
Элементный анализ:
б) 1,7-бис(бензилокси)-4-[2-(N-этил-N-перфтороктилсульфонил)амино]ацетил-10-[1-O-α-D-карбонилметил-2,3,4,6-тетра-O-бензилманнопираноза]-1,4,7,10-тетраазациклододекан
К 60,0 г (59,53 ммоля) соединения, указанного в заголовке примера 27а, и 35,64 г (59,53 ммоля) 1-O-α-D-карбоксиметил-2,3,4,6-тетра-O-бензилманнопиранозы, полученной согласно DE 19728954, в 300 мл тетрагидрофурана при 0°С добавляют 24,73 г (100 ммолей) ЭЭДХ (этиловый эфир 2-этокси-1,2-дигидрохинолин-1-карбоновой кислоты) и перемешивают в течение ночи при комнатной температуре. После этого досуха упаривают в вакууме и хроматографируют на силикагеле (элюент:дихлорметан/метанол в соотношении 20:1).
Выход: 76,6 г (81,0% от теории) бесцветного твердого вещества.
Элементный анализ:
в) 1-[2-(N-этил-N-перфтороктилсульфонил)амино]ацетил-7-(1-O-α-D-карбонилметилманнопираноза)-1,4,7,10-тетраазациклододекан
70 г (44,07 ммоля) соединения, указанного в заголовке примера 27б, растворяют в 800 мл этанола и добавляют 8 г палладиевого катализатора (10%-ного Pd/C). После этого гидрируют при комнатной температуре. Затем катализатор отфильтровывают и фильтрат досуха упаривают в вакууме.
Выход: 42,3 г (количественный) бесцветного твердого вещества.
Элементный анализ:
г) 1,7-бис[1,4,7-трис(карбоксилатометил)-1,4,7,10-тетраазациклододекан-10-(пентаноил-3-аза-4-оксо-5-метил-5-ил)-4-[2-(N-этил-N-перфтороктилсульфонил)амино]ацетил-10-(1-O-α-D-карбонилметилманнопираноза)-1,4,7,10-тетраазациклододекан, Gd-комплекс
20 г (20,84 ммоля) соединения, указанного в заголовке примера 27в, 5,09 г (120 ммолей) хлорида лития и 37,78 г (60 ммолей) Gd-комплекса 1,4,7-трис(карбоксилатометил)-10-(пентаноил-3-аза-4-оксо-5-метил-5-ил)-1,4,7,10-тетраазациклододекана растворяют при умеренном нагревании в 400 мл диметилсульфоксида. Далее при 10°С добавляют 29,67 г (120 ммолей) ЭЭДХ и затем перемешивают в течение ночи при комнатной температуре. Раствор сливают в 3000 мл ацетона и перемешивают в течение 10 мин. Выпавшее в осадок твердое вещество отфильтровывают и затем очищают хроматографией (силикагель RP-18, элюент:градиент воды/этанола/ацетонитрила).
Выход: 13,2 г (29,0% от теории) бесцветного твердого вещества.
Содержание воды: 11,8%.
Элементный анализ (в пересчете на безводное вещество):
Пример 28
а) 1,7-бис(бензилоксикарбонил)-4-[2-(N-этил-N-перфтороктилсульфонил)амино]ацетил-10-[пентаноил-3-аза-4-оксо-5-метил-5-ил-[1,4,7-трис(карбоксилатометил)-Gd-комплекс-1,4,7,10-тетраазациклододекан-10-ил]-1,4,7,10-тетраазациклододекан
50,0 г (49,61 ммоля) соединения, указанного в заголовке примера 27а, 5,71 г (49,61 ммоля) N-гидроксисукцинимида, 4,24 г (100 ммолей) хлорида лития и 31,24 г (49,61 ммоля) Gd-комплекса 1,4,7-трис(карбоксилатометил)-10-(пентаноил-3-азаоксо-5-метил-5-ил)-1,4,7,10-тетраазациклододекана растворяют при умеренном нагревании в 350 мл диметилсульфоксида. Далее при 10°С добавляют 15,47 г (75 ммолей) N,N-дициклогексилкарбодиимида и затем перемешивают в течение ночи при комнатной температуре. Раствор сливают в 2000 мл ацетона и перемешивают в течение 10 мин. Выпавшее в осадок твердое вещество отфильтровывают и затем очищают хроматографией (силикагель RP-18, элюент:градиент воды/этанола/ацетонитрила).
Выход: 65,1 г (81,0% от теории) бесцветного твердого вещества.
Содержание воды: 7,9%.
Элементный анализ (в пересчете на безводное вещество):
б) 1-[2-(N-этил-N-перфтороктилсульфонил)амино]ацетил-7-{(пентаноил-3-аза-4-оксо-5-метил-5-ил)-10-[1,4,7-трис(карбоксилатометил)-1,4,7,10-тетраазациклододекан,Gd-комплекс]}-1,4,7,10-тетраазациклододекан
60,0 г (37,05 ммоля) соединения, указанного в заголовке примера 28а, растворяют в 600 мл этанола и добавляют 6,0 г палладиевого катализатора (10%-ного Pd/C). После этого гидрируют при комнатной температуре. Затем катализатор отфильтровывают и фильтрат досуха упаривают в вакууме.
Выход: 50,06 г (количественный) бесцветного твердого вещества.
Содержание воды: 3,9%.
Элементный анализ (в пересчете на безводное вещество):
в) 1-[2-(N-этил-N-перфтороктилсульфонил)амино]ацетил-4,10-бис[1-O-α-D-карбонилметил-2,3,4,6-тетра-O-бензилманнопираноза]-7-{(пентаноил-3-аза-4-оксо-5-метил-5-ил)-[1,4,7-трис(карбоксилатометил)-1,4,7,10-тетраазациклододекан-10-ил]-Gd-комплекс}-1,4,7,10-тетраазациклододекан
40,0 г (29,60 ммоля) соединения, указанного в заголовке примера 28б, 2,54 г (60,0 ммолей) хлорида лития и 44,9 г (75,0 ммолей) 1-O-α-D-карбоксиметил-2,3,4,6-тетра-О-бензилманнопиранозы растворяют при умеренном нагревании в 300 мл диметилсульфоксида. Далее при 10°С добавляют 24,73 г (100,0 ммолей) ЭЭДХ и затем перемешивают в течение ночи при комнатной температуре. Раствор сливают в 3000 мл ацетона и перемешивают в течение 10 мин. Выпавшее в осадок твердое вещество отфильтровывают и затем очищают хроматографией (силикагель RP-18, элюент:градиент воды/этанола/ацетонитрила).
Выход: 31,98 г (43,0% от теории) бесцветного твердого вещества.
Содержание воды: 3,5%.
Элементный анализ (в пересчете на безводное вещество):
г) 1-[2-(N-этил-N-перфтороктилсульфонил)амино]ацетил-4,10-бис[1-O-α-D-карбонилметил-2,3,4,6-тетра-O-бензилманнопираноза]-7-{(пентаноил-3-аза-4-оксо-5-метил-5-ил)-[1,4,7-трис(карбоксилатометил)-1,4,7,10-тетраазациклододекан-10-ил]-Gd-комплекс}-1,4,7,10-тетраазациклододекан
30,0 г (11,94 ммоля) соединения, указанного в заголовке примера 28в, растворяют в смеси из 300 мл этанола и 30 мл воды и добавляют 4,0 г палладиевого катализатора (10%-ного Pd/C). После этого гидрируют при комнатной температуре, катализатор отфильтровывают и фильтрат досуха упаривают в вакууме.
Выход: 21,39 г (количественный) бесцветного твердого вещества.
Содержание воды: 3,4%.
Элементный анализ (в пересчете на безводное вещество):
Пример 29
а) [1-(4-перфтороктилсульфонил)пиперазин]амид 6-N-[3,6-бис(карбоксиметил)октан-1,8-дикарбоновая кислота-1-карбокси-8-оил]-2-N-(1-O-α-D-карбоксиметилманнопираноза)лизина
К 27,5 г (30,0 ммолей) указанного в заголовке примера 21д соединения, растворенного в смеси из 300 мл диметилформамида и 100 мл пиридина, добавляют 25,62 г (100,0 ммолей) диангидрида этилендиамин-N,N,N',N'-тетрауксусной кислоты и перемешивают в течение 5 ч при 50°С. После этого досуха упаривают в вакууме. Остаток растворяют в 300 мл воды, значение рН добавлением 20%-ного водного едкого натра устанавливают на 10, затем значение рН щелочного содержащего продукт раствора добавлением концентрированной соляной кислоты устанавливают на 3 и досуха упаривают в вакууме. Остаток хроматографируют на силикагеле RP-18 (элюент:градиент воды/этанола/ацетонитрила).
Выход: 18,22 г (51,0% от теории) бесцветного твердого вещества.
Содержание воды: 7,9%.
Элементный анализ (в пересчете на безводное вещество):
б) [1-(4-перфтороктилсульфонил)пиперазин]амид 6-N-[3,6-бис(карбоксилатометил)октан-1,8-дикарбоновая кислота-1-карбоксилато-8-оил]-2-N-(1-O-α-D-карбоксиметилманнопираноза)-L-лизина, Mn-комплекс, натриевая соль
10 г (8,397 ммоля) соединения, указанного в заголовке примера 29а, растворяют в 200 мл воды. Далее добавляют 965 мг (8,397 ммоля) карбоната марганца(II) и перемешивают в течение 3 ч при 60°С. Затем значение рН раствора устанавливают на 7,4 с помощью 5%-ного водного едкого натра, фильтруют и после этого лиофилизуют.
Выход: 10,52 г (99,0% от теории) бесцветного твердого вещества.
Содержание воды: 7,8%.
Элементный анализ (в пересчете на безводное вещество):
Пример 30
а) 1,2,3,4,6-пента-O-ацетил-α,β-D-маннопираноза
Аналогично методу, описанному в литературе [M.L.Wolfrom и A.Thompson, "Methods in Carbohydrate Chemistry" (под ред. R.L.Whistler, M.L.Wolfrom и J.N.BeMiller), изд-во Academic Press, New York, т.II, 53, сс.211-215 (1963)], в результате взаимодействия 150 г (832,5 ммоля) α,β-D-маннопиранозы со смесью из 1500 мл абсолютного пиридина и 1500 мл уксусного ангидрида после переработки получают 315 г (96,7%) указанного выше в заголовке соединения в виде сырого продукта в форме вязкого и бесцветного масла. Соотношение между обоими α- и β-аномерами в полученном таким путем указанном в заголовке соединении составляет 4:1 по данным 1Н-ЯМР-спектроскопического анализа. Для проведения последующих реакций указанное выше в заголовке соединение можно не разделять на отдельные α,β-аномеры.
Элементный анализ:
б) 1-O-α-D-(5-этоксикарбонил)пентил-2,3,4,6-тетра-O-ацетилманнопираноза
Аналогично методу, описанному в литературе для синтеза арилгликопиранозидов [J.Conchie и G.A.Levvy, "Methods in Carbohydrate Chemistry" (под ред. R.L.Whistler, M.L.Wolfrom и J.N.BeMiller), изд-во Academic Press, New York, т.II, 90, сс.345-347 (1963)], в результате взаимодействия 156,2 г (400 ммолей) указанного в заголовке примера 21а соединения в виде смеси α,β-аномеров с 67 мл (400 ммолей) этилового эфира 6-гидроксигексановой кислоты и 60,8 мл (520 ммолей) хлорида олова(IV) в 1,2-дихлорэтане общим количеством 600 мл после очистки колоночной хроматографией (элюент:гексан/этиловый эфир уксусной кислоты в соотношении 2:1) получают 100,05 г (51% от теории) указанного выше в заголовке соединения в виде бесцветного и вязкого масла. Данные 1Н-ЯМР-спектроскопического анализа полученного таким путем указанного в заголовке соединения свидетельствуют о том, что это соединение представляет собой только чистый α-аномер.
Элементный анализ:
в) 1-O-α-D-(5-карбокси)пентил-2,3,4,6-тетра-O-бензилманнопираноза
Перемешанную суспензию 141,0 г (289 ммоля) соединения, указанного в заголовке примера 30б, в 200 мл диоксана при комнатной температуре и при одновременно интенсивном перемешивании порциями смешивают с высокодисперсным порошковым гидроксидом калия общим количеством 238,5 г (4,26 моля). Реакционную смесь для повышения ее размешиваемости смешивают еще с 200 мл диоксана, полученную в результате суспензию затем нагревают до температуры кипения и при этой температуре в течение двух часов по каплям смешивают с бензилбромидом общим количеством 372 мл (3,128 моля). После проведения реакции в течение 4 ч при 110°С, а затем в течение 12 ч при комнатной температуре реакционную смесь для ее переработки медленно сливают в смесь воды со льдом общим количеством 2,5 л и водную фазу затем полностью экстрагируют диэтиловым эфиром. После промывки полученной таким путем эфирной фазы и последующей ее сушки над сульфатом натрия соль отделяют вакуум-фильтрацией и диэтиловый эфир удаляют в вакууме. После этого избыток бензилбромида количественно отгоняют из реакционной смеси в вакууме, создаваемом масляным насосом, при температуре масляной бани 180°С. Полученный таким путем смолянисто-маслянистый остаток очищают на силикагеле с использованием этилового эфира уксусной кислоты/гексана (в соотношении 1:10) в качестве элюента.
Выход: 172,2 г (91,0% от теории) указанного выше в заголовке соединения в виде бесцветного и высоковязкого масла.
Элементный анализ:
г) [1-(4-перфтороктилсульфонил)пиперазин]амид 6-N-бензилоксикарбонил-2-N-[1-O-α-D-(5-карбонил)пентил-2,3,4,6-тетра-O-бензилманнопираноза]-L-лизина
100,0 г (134,0 ммоля) полученной в примере 30в карбоновой кислоты и 32,4 г (281,4 ммоля) N-гидроксисукцинимида растворяют в 500 мл диметилформамида, при 0°С порциями смешивают с N,N'-дициклогексилкарбодиимидом общим количеством 58,0 г (281,4 ммоля) и затем перемешивают в течение 3 ч при этой температуре. К полученному таким путем раствору активированного сложного эфира по каплям добавляют охлажденный до 0°С раствор 111,3 г (134,0 ммоля) указанного в заголовке примера 21в соединения в 300 мл диметилформамида и перемешивают в течение 2 ч при 0°С, а затем в течение 12 ч при комнатной температуре. Для переработки выпавшую в осадок дициклогексилмочевину отфильтровывают и после этого отгоняют растворитель до получения сухого остатка. Полученный таким путем остаток затем хроматографируют на силикагеле (элюент:дихлорметан/этанол в соотношении 20:1, хроматографию проводят с использованием градиента растворителя, непрерывно увеличивая долю этанола).
Выход: 132,5 г (67,4% от теории) указанного в заголовке соединения в виде бесцветного и высоковязкого масла.
Элементный анализ:
д) [1-(4-перфтороктилсульфонил)пиперазин]амид 2-N-[1-O-α-D-(5-карбонил)пентилманнопираноза]-L-лизина
120,0 г (81,77 ммоля) полученного в примере 30г соединения растворяют в 800 мл этанола, смешивают с 4,5 г катализатора Перлмана (20%-ный Pd/C) и гидрируют при комнатной температуре в атмосфере водорода (1 атм) до тех пор, пока не прекратится поглощение водорода (примерно в течение 8 ч). После этого катализатор удаляют вакуум-фильтрацией, затем тщательно промывают этанолом (примерно 200 мл) и досуха концентрируют в вакууме. Таким путем получают указанное в заголовке соединение в виде высоковязкого и бесцветного масла.
Выход: 78,5 г (98,7% от теории).
Элементный анализ:
е) [1-(4-перфтороктилсульфонил)пиперазин]амид 2-N-[1-O-α-D-(5-карбонил)пентилманнопираноза]-6-N-[1,4,7-трис(карбоксилатометил)-10-(3-аза-4-оксо-5-метил-5-илпентаноил)-1,4,7,10-тетраазациклододекан]-L-лизина, Gd-комплекс
99,8 г (158,4 ммоля, 2,2 молярных эквивалента в пересчете на количество используемого аминового компонента из примера 30д) описанного в заявке DE 19728954 С1 в примере 31h Gd-комплекса 10-(4-карбокси-1-метил-2-оксо-3-азабутил)-1,4,7,10-тетраазациклододекан-1,4,7-триуксусной кислоты и 6,7 г безводного хлорида лития (158,4 ммоля) при 40°С растворяют при перемешивании в 800 мл абсолютного диметилсульфоксида. Далее при этой температуре смешивают с N-гидроксисукцинимидом общим количеством 18,2 г (158,4 ммоля) и 70,0 г (71,96 ммоля) указанного в заголовке примера 30д соединения, растворенного в 250 мл абсолютного диметилсульфоксида. После охлаждения до комнатной температуры реакционный раствор смешивают с 32,7 г (158,4 ммоля) N,N'-дициклогексилкарбодиимида и перемешивают в течение 12 ч при комнатной температуре. Затем полученную суспензию смешивают с достаточным количеством ацетона до полного осаждения указанного выше в заголовке соединения, осадок отделяют вакуум-фильтрацией, сушат, растворяют в воде, отфильтровывают нерастворившуюся дициклогексилмочевину и фильтрат для его обессоливания и очистки от низкомолекулярных компонентов пропускают через ультрафильтрационную мембрану AMICON® YM-3 (предельная проницаемость: 3000 Да). Ретентат (вещество, не прошедшее через фильтрационную мембрану) в завершение лиофилизуют.
Выход: 93,0 г (81,6% от теории) в виде бесцветного лиофилизата.
Н2О-содержание (при использовании реактива Карла Фишера): 9,53%.
Элементный анализ (в пересчете на безводное вещество):
Пример 31
а) [1-(4-перфтороктилсульфонил)пиперазин]амид 2-N-[1-O-α-D-(5-карбонил)пентилманнопираноза]-6-N-{2-[4-(3-оксапропионил)фенил]-2-[1,4,7-трис(карбоксилатометил)-1,4,7,10-тетраазациклододекан-10-ил]уксусная кислота} -L-лизина, Gd-комплекс, натриевая соль
Перемешанную суспензию 5,0 г (9,06 ммоля) соединения, указанного в заголовке примера 23д, в 15 мл абсолютного диметилсульфоксида при 70°С смешивают с 0,68 г (15,9 ммоля) хлорида лития. После 30-минутного перемешивания при 70°С прозрачный реакционный раствор порциями смешивают с N-гидроксисукцинимидом общим количеством 1,83 г (15,9 ммоля) и реакционную смесь выдерживают еще в течение 1 ч при этой температуре. После охлаждения до 0°С смешивают с 4,52 г (23,85 ммоля) дициклогексилкарбодиимида и реакционный раствор перемешивают еще в течение 1 ч при 0°С, а затем в течение 12 ч при 22°С. Полученный таким путем реакционный раствор N-гидроксисукцинимидоэфира соединения, указанного в заголовке примера 23д, при 22°С по каплям смешивают с раствором 4,0 г (4,12 ммоля) указанного в заголовке примера 30д соединения в 15 мл абсолютного диметилсульфоксида и перемешивают еще в течение 12 ч при комнатной температуре. Для переработки реакционный раствор при 22°С по каплям добавляют к 900 мл ацетона, при этом указанное в заголовке соединение выпадает в виде бесцветного осадка. Этот осадок отделяют вакуум-фильтрацией, растворяют в 200 мл дистиллированной воды и затем значение рН этого раствора устанавливают точно на 7,2 с помощью 1-молярного едкого натра. Полученный таким путем водный содержащий продукт раствор для обессоливания и для отделения низкомолекулярных компонентов трижды подвергают ультрафильтрации через ультрафильтрационную мембрану YM-3 (AMICON®, предельная проницаемость: 3000 Да). Полученный в результате ретентат в завершение лиофилизуют.
Выход: 6,33 г (92,4% от теории, в пересчете на количество используемого аминового компонента) в виде бесцветного лиофилизата с содержанием воды 7,38%.
Элементный анализ (в пересчете на безводное вещество):
Пример 32
а) N-(3-окса-1Н,1Н,2Н,2Н,4Н,4Н,5Н,5Н-перфтортридецил)амид 3,5-бисбензилоксикарбониламинобензойной кислоты
20 г (47,5 ммоля) 3,5-бисбензилоксикарбониламинобензойной кислоты (синтез согласно методу, описанному у Skulnick Harvey I., Johnson Paul D., Aristoff Paul A., Morris Jeanette K., Lovasz Kristine D. и др., J. Med. Chem., 40, 7, (1997), сс.1149-1164), а также 4,78 г (47,5 ммоля) триэтиламина растворяют в смеси растворителей, состоящей из 125 мл сухого тетрагидрофурана и 125 мл сухого диоксана. После охлаждения до -15°С при перемешивании по каплям медленно добавляют раствор 6,56 г (48 ммолей) изобутилового эфира хлормуравьиной кислоты в 30 мл сухого тетрагидрофурана, при этом внутреннюю температуру следует поддерживать ниже -10°С. После проведения реакции в течение 15 мин при -15°С по каплям при -20°С добавляют раствор 58,6 г (47,5 ммоля) 1-амино-1Н,1Н,2Н,2Н,4Н,4Н,5Н,5Н-3-оксоперфтортридекана и 4,78 г (47,5 ммоля) триэтиламина в 100 мл сухого тетрагидрофурана. После проведения реакции в течение одного часа при -15°С, а затем в течение двух часов при комнатной температуре реакционный раствор досуха концентрируют в вакууме. Полученный остаток растворяют в 300 мл этилового эфира уксусной кислоты и дважды промывают насыщенным раствором гидрокарбоната натрия порциями по 200 мл и однократно водой порцией 300 мл. После сушки органической фазы над сульфатом натрия соль отделяют вакуум-фильтрацией и этиловый эфир уксусной кислоты отгоняют в вакууме. Полученный маслянистый остаток очищают на силикагеле с использованием дихлорметана/гексана/2-пропанола (в соотношении 10:5:1) в качестве элюента.
Выход: 36,2 г (82,5% от теории) указанного в заголовке соединения в виде бесцветного масла.
Элементный анализ:
б) N-(3-окса-1Н,1Н,2Н,2Н,4Н,4Н,5Н,5Н-перфтортридецил)амид 3,5-диаминобензойной кислоты
30,0 г (32,4 ммоля) полученного в примере 32а амида растворяют в 300 мл этанола и смешивают с 1,2 г катализатора Перлмана (20%-ный Pd/C). После этого гидрируют при комнатной температуре в атмосфере водорода (1 атм) до тех пор, пока не прекратится поглощение водорода. После этого катализатор удаляют вакуум-фильтрацией, тщательно промывают этанолом (примерно 300 мл) и досуха концентрируют в вакууме. Таким путем получают указанное в заголовке соединение в виде высоковязкого желтоватого масла.
Выход: 20,12 г (94,8% от теории).
Элементный анализ:
в) N-(3-окса-1Н,1Н,2Н,2Н,4Н,4Н,5Н,5Н-перфтортридецил)амид 3-N-(1-O-α-D-карбонилметил-2,3,4,6-тетра-O-бензилманнопираноза)-5-аминобензойной кислоты
10,95 г (18,30 ммоля) 1-карбоксиметилокси-2,3,4,6-тетра-O-бензил-α-D-маннопиранозида [получение согласно патенту DE 19728954 С1] растворяют в 150 мл диметилформамида и смешивают с N-гидроксисукцинимидом общим количеством 2,09 г (18,3 ммоля). Далее охлаждают до 0°С и добавляют 3,78 г (18,3 ммоля) дициклогексилкарбодиимида. После этого перемешивают в течение часа при 0°С, а затем в течение 4 ч при комнатной температуре. Далее охлаждают до 0°С и в течение 3 ч по каплям медленно добавляют раствор 24,0 г (36,6 ммоля, 2 молярных эквивалента в пересчете на используемое количество карбоновой кислоты) полученного в примере 32б диаминосоединения в 350 мл диметилформамида. Далее перемешивают еще в течение часа при 0°С, а затем в течение ночи при комнатной температуре. Смесь упаривают в вакууме досуха и остаток растворяют в 300 мл этилового эфира уксусной кислоты. Выпавшую в осадок мочевину отфильтровывают и фильтрат дважды промывают 5%-ным водным раствором соды порциями по 100 мл. Органическую фазу сушат над сульфатом магния и досуха упаривают в вакууме. Остаток хроматографируют на силикагеле (элюент:н-гексан/изопропанол в соотношении 13:1). Таким путем получают 16,8 г (74,3% от теории, в пересчете на количество используемой карбоновой кислоты) указанного в заголовке соединения в виде бесцветного масла. За счет повышения полярности элюента изменением соотношения между н-гексаном и изопропанолом до 5:1 в последующих хроматографических фракциях рекуперируют в целом 10,15 г непрореагировавшего диаминосоединения из примера 32б, которое вновь можно использовать в реакции в соответствии с рассмотренной выше методикой.
Элементный анализ:
г) N-(3-окса-1Н,1Н,2Н,2Н,4Н,4Н,5Н,5Н-перфтортридецил)амид 3-N-(1-O-α-D-карбонилметилманнопираноза)-5-аминобензойной кислоты
Аналогично методу синтеза соединения, указанного в заголовке примера 32б, в результате гидрогенолиза 12,0 г (9,70 ммоля) соединения, указанного в заголовке примера 32в, с использованием 0,5 г катализатора Перлмана (20%-ный Pd/C) в смеси этанола и воды (в соотношении 9:1) после переработки получают 8,08 г (96,7% от теории) указанного выше в заголовке соединения в виде вязкого масла желтого цвета.
Элементный анализ:
д) N-(3-окса-1Н,1Н,2Н,2Н,4Н,4Н,5Н,5Н-перфтортридецил)амид 3-N-(1-O-α-D-карбонилметилманнопираноза)-5-N-{2-[4-(3-оксапропионил)фенил]-2-[1,4,7-трис(карбоксилатометил)-1,4,7,10-тетраазациклододекан-10-ил]уксусная кислота} бензойной кислоты, Gd-комплекс, натриевая соль
13,6 г (19,2 ммоля, 2,2 молярных эквивалента в пересчете на количество используемого аминового компонента из примера 32г) описанного в примере 23д Gd-комплекса и 0,81 г безводного хлорида лития (19,2 ммоля) при 40°С растворяют при перемешивании в 100 мл абсолютного диметилсульфоксида и при этой температуре смешивают с N-гидроксисукцинимидом общим количеством 2,2 г (19,2 ммоля) и с 7,5 г (8,7 ммоля) указанного в заголовке примера 32г соединения, растворенного в 50 мл абсолютного диметилсульфоксида. После охлаждения до комнатной температуры реакционный раствор смешивают с 3,96 г (19,2 ммоля) N,N'-дициклогексилкарбодиимида и перемешивают в течение 12 ч при комнатной температуре. Полученную суспензию затем смешивают с достаточным количеством ацетона до полного осаждения указанного выше в заголовке соединения, осадок отделяют вакуум-фильтрацией, сушат, растворяют в воде, отфильтровывают нерастворившуюся дициклогексилмочевину и фильтрат для обессоливания и очистки от низкомолекулярных компонентов пропускают через ультрафильтрационную мембрану AMICON® YM-3 (предельная проницаемость: 3000 Да). Ретентат в завершение лиофилизуют.
Выход: 11,51 г (84,5% от теории) в виде бесцветного лиофилизата.
Н2O-содержание (при использовании реактива Карла Фишера): 6,77%.
Элементный анализ (в пересчете на безводное вещество):
Пример 33
а) 3,5-бис(бензилоксикарбониламино)-1-{N-[1-(4-перфтороктилсульфонил)пиперазин]}бензамид
10 г (23,75 ммоля) 3,5-бисбензилоксикарбониламинобензойной кислоты (синтез согласно методу, описанному у Skulnick Harvey I., Johnson Paul D., Aristoff Paul A., Morris Jeanette K., Lovasz Kristine D. и др., J. Med. Chem., 40, 7 (1997), сс.1149-1164), а также 2,39 г (23,75 ммоля) триэтиламина растворяют в смеси растворителей, состоящей из 60 мл сухого тетрагидрофурана и 70 мл сухого диоксана. После охлаждения до -15°С при перемешивании по каплям медленно добавляют раствор 3,28 г (24 ммоля) изобутилового эфира хлормуравьиной кислоты в 20 мл сухого тетрагидрофурана, при этом внутренняя температура не превышает -10°С. После проведения реакции в течение 15 мин при -15°С по каплям при -20°С добавляют раствор 23,0 г (23,75 ммоля) перфтороктилсульфонилпиперазина и 2,39 г (23,75 ммоля) триэтиламина в 50 мл сухого тетрагидрофурана. После проведения реакции в течение часа при -15°С, а затем в течение двух часов при комнатной температуре реакционный раствор досуха концентрируют в вакууме. Полученный остаток растворяют в 200 мл этилового эфира уксусной кислоты и дважды промывают насыщенным раствором гидрокарбоната натрия порциями по 100 мл и однократно водой порцией 300 мл. После сушки органической фазы над сульфатом натрия соль отделяют вакуум-фильтрацией и этиловый эфир уксусной кислоты отгоняют в вакууме. Полученный маслянистый остаток очищают на силикагеле с использованием дихлорметана/гексана/2-пропанола (в соотношении 15:5:1) в качестве элюента.
Выход: 18,35 г (79,6% от теории) указанного в заголовке соединения в виде бесцветного масла.
Элементный анализ:
б) 3,5-диамино-1-{N[1-(4-перфтороктилсульфонил)пиперазин]}бензамид
9,70 г (10,0 ммолей) полученного в примере 33а амида растворяют в 100 мл этанола и смешивают с 0,4 г катализатора Перлмана (20%-ный Pd/C). После этого гидрируют при комнатной температуре в атмосфере водорода (1 атм) до тех пор, пока не прекратится поглощение водорода. После этого катализатор удаляют вакуум-фильтрацией, тщательно промывают этанолом (примерно 150 мл) и досуха концентрируют в вакууме. Таким путем получают указанное в заголовке соединение в виде высоковязкого желтоватого масла.
Выход: 6,9 г (98,2% от теории).
Элементный анализ:
в) N-[1-(4-перфтороктилсульфонил)пиперазин]амид 5-амино-3-N-(1-O-α-D-карбонилметил-2,3,4,6-тетра-O-бензилманнопираноза)бензойной кислоты
5,48 г (9,15 ммоля) 1-карбоксиметилокси-2,3,4-тетра-O-бензил-α-D-маннопиранозида [получение по методу, описанному в патенте DE 19728954 С1] растворяют в 100 мл диметилформамида и смешивают с N-гидроксисукцинимидом общим количеством 1,04 г (9,15 ммоля). Далее охлаждают до 0°С и добавляют 1,89 г (9,15 ммоля) дициклогексилкарбодиимида. После этого перемешивают в течение часа при 0°С, а затем в течение 4 ч при комнатной температуре. После повторного охлаждения до 0°С в течение 3 ч по каплям медленно добавляют раствор из 12,85 г (18,30 ммоля, 2 молярных эквивалента в пересчете на количество используемой карбоновой кислоты) описанного в примере 33б диаминосоединения в 250 мл диметилформамида. После этого перемешивают еще в течение часа при 0°С, а затем в течение ночи при комнатной температуре. Смесь упаривают в вакууме досуха и остаток растворяют в 100 мл этилового эфира уксусной кислоты. Выпавшую в осадок мочевину отфильтровывают и фильтрат промывают дважды 5%-ным водным раствором соды порциями по 100 мл. Органическую фазу сушат над сульфатом магния и досуха упаривают в вакууме. Остаток хроматографируют на силикагеле (элюент:н-гексан/изопропанол в соотношении 13:1). Таким путем получают 8,14 г (69,4% от теории, в пересчете на количество используемой карбоновой кислоты) указанного в заголовке соединения в виде бесцветного масла. За счет повышения полярности элюента изменением в процессе хроматографии соотношения между его компонентами (н-гексаном и изопропанолом) до 6:1 в последующих хроматографических фракциях рекуперируют в целом еще 4,36 г непрореагировавшего диаминосоединения из примера 33б, которое вновь можно использовать в реакции в соответствии с рассмотренной выше методикой.
Элементный анализ:
г) N-[1-(4-перфтороктилсульфонил)пиперазин]амид 5-амино-3-N-(1-O-α-D-карбонилметилманнопираноза)бензойной кислоты
Аналогично методу, описанному для синтеза указанного в заголовке примера 33б соединения, в результате гидрогенолиза 6,4 г (5,0 ммолей) указанного в заголовке примера 33в соединения с использованием 0,3 г катализатора Перлмана (20%-ный Pd/C) в смеси этанола и воды (в соотношении 8:1) после переработки получают 4,43 г (96,2% от теории) указанного выше в заголовке соединения в виде вязкого масла желтого цвета.
Элементный анализ:
д) N-[1-(4-перфтороктилсульфонил)пиперазин]амид 3-N-(1-O-α-D-карбонилметилманнопираноза)-5-N-[1,4,7-трис(карбоксилатометил)-10-(3-аза-4-оксо-5-метил-5-илпентаноил)-1,4,7,10-тетраазациклододекан]бензойной кислоты, Gd-комплекс
5,54 г (8,8 ммоля, 2,2 молярных эквивалента в пересчете на количество используемого аминового компонента из примера 33г) описанного в заявке DE 19728954 С1 в примере 31h Gd-комплекса 10-(4-карбокси-1-метил-2-оксо-3-азабутил)-1,4,7,10-тетраазациклододекан-1,4,7-триуксусной кислоты и 0,37 г безводного хлорида лития (8,8 ммоля) при 40°С растворяют при перемешивании в 60 мл абсолютного диметилсульфоксида и при этой температуре смешивают с N-гидроксисукцинимидом общим количеством 1,01 г (8,8 ммоля) и с 3,7 г (4,0 ммоля) указанного в заголовке примера 33г соединения, растворенного в 40 мл абсолютного диметилсульфоксида. После охлаждения до комнатной температуры реакционный раствор смешивают с 1,82 г (8,8 ммоля) N,N'-дициклогексилкарбодиимида и перемешивают в течение 12 ч при комнатной температуре. Полученную суспензию затем смешивают с достаточным количеством ацетона до полного осаждения указанного выше в заголовке соединения, осадок отделяют вакуум-фильтрацией, сушат, растворяют в воде, отфильтровывают нерастворившуюся дициклогексилмочевину и фильтрат для обессоливания и очистки от низкомолекулярных компонентов пропускают через ультрафильтрационную мембрану AMICON® YM-3 (предельная проницаемость: 3000 Да). Ретентат в завершение лиофилизуют.
Выход: 5,36 г (87,4% от теории) в виде бесцветного лиофилизата.
Н2O-содержание (при использовании реактива Карла Фишера): 6,77%.
Элементный анализ (в пересчете на безводное вещество):
Пример 34
а) Диамид 1,4,7-триазагептан-1,7-бис(2-N-трифторацетил-6-N-бензилоксикарбонил-L-лизина)
100 г (107,9 ммоля) полученной в примере 21а карбоновой кислоты и 26,1 г (226,59 ммоля) N-гидроксисукцинимида растворяют в 500 мл диметилформамида, при 0°С порциями смешивают с N,N'-дициклогексилкарбодиимидом общим количеством 46,7 г (226,59 ммоля) и затем перемешивают в течение 3 ч при этой температуре. К полученному таким путем раствору активированного сложного эфира по каплям добавляют охлажденный до 0°С раствор 5,57 г (53,95 ммоля) диэтилентриамина в 60 мл диметилформамида и перемешивают в течение 2 ч при 0°С, а затем в течение 12 ч при комнатной температуре. Для переработки выпавшую в осадок дициклогексилмочевину отфильтровывают и затем отгоняют растворитель до получения сухого остатка. Полученный таким путем остаток после этого хроматографируют на силикагеле (элюент:дихлорметан/этанол в соотношении 15:1; хроматографию проводят с использованием градиента растворителя, непрерывно увеличивая долю этанола).
Выход: 26,0 г (58,8% от теории, в пересчете на количество используемого аминового компонента) указанного в заголовке соединения в виде бесцветного и высоковязкого масла.
Элементный анализ:
б) 1,7-бис(2-N-трифторацетил-6-N-бензилоксикарбонил-L-лизин)диамид-4-[2-(N-этил-N-перфтороктилсульфонил)амино]ацетил-1,4,7-триазагептан
К раствору из 20 г (24,4 ммоля) полученного в примере 34а диамида в смеси, содержащей 150 мл тетрагидрофурана и 15 мл хлороформа, при 0°С в атмосфере азота добавляют 16,18 г (27,0 ммолей) 2-(N-этил-N-перфтороктилсульфонил)аминоуксусной кислоты (получение согласно DE 19603033), растворенной в 50 мл тетрагидрофурана. После этого при 0°С порциями добавляют в целом 18,0 г (36,6 ммоля) ЭЭДХ [2-этокси-1-этоксикарбонил-1,2-дигидрохинолин], оставляют на ночь для перемешивания при комнатной температуре и затем концентрируют в вакууме. Полученное масло хроматографируют на силикагеле (элюент:н-гексан/изопропанол в соотношении 15:1). Таким путем получают 24,74 г (72,4% от теории, в пересчете на количество используемого вторичного амина) указанного в заголовке соединения в виде бесцветного масла.
Элементный анализ:
в) 1,7-бис(6-N-бензилоксикарбонил-L-лизин)диамид-4-[2-(N-этил-N-перфтороктилсульфонил)амино]ацетил-1,4,7-триазагептан
22,0 г (15,7 ммоля) полученного в примере 34б соединения растворяют в 100 мл этанола и при 0°С этот раствор в течение 40 мин барботируют газообразным аммиаком. После этого перемешивают в течение 4 ч при 0°С, а затем в течение 3 ч при комнатной температуре и досуха концентрируют в вакууме при температуре бани 40°С. Полученный маслянистый остаток очищают на силикагеле с использованием дихлорметана/гексана/2-пропанола (в соотношении 20:10:1) в качестве элюента.
Выход: 12,92 г (98,4% от теории) указанного выше в заголовке соединения в виде бесцветного и высоковязкого масла.
Элементный анализ:
г) 1,7-бис[6-N-бензилоксикарбонил-2-N-(1-O-α-D-карбонилметил-2,3,4,6-тетра-O-бензилманнопираноза)-L-лизин]диамид-4-[2-(N-этил-N-перфтороктилсульфонил)амино]ацетил-1,4,7-триазагептан
5,47 г (9,15 ммоля) 1-карбоксиметилокси-2,3,4,6-тетра-O-бензил-α-D-маннопиранозида [получение по методу, описанному в патенте DE 19728954 С1] растворяют в 80 мл диметилформамида и смешивают с N-гидроксисукцинимидом общим количеством 1,05 г (9,15 ммоля). Далее охлаждают до 0°С и добавляют 1,9 г (9,15 ммоля) дициклогексилкарбодиимида. После этого перемешивают в течение часа при 0°С, а затем в течение 4 ч при комнатной температуре. Далее охлаждают до 0°С и в течение 3 ч по каплям медленно добавляют раствор 7,65 г (9,15 ммоля) полученного в примере 34в аминосоединения в 50 мл диметилформамида. После этого перемешивают в течение часа при 0°С, а затем в течение ночи при комнатной температуре. Смесь упаривают в вакууме досуха и остаток растворяют в 100 мл этилового эфира уксусной кислоты. Выпавшую в осадок мочевину отфильтровывают и фильтрат дважды промывают 5%-ным водным раствором соды порциями по 50 мл. Органическую фазу сушат над сульфатом магния и досуха упаривают в вакууме. Остаток хроматографируют на силикагеле (элюент:н-гексан/изопропанол в соотношении 20:1). Таким путем получают 17,01 г (78,9% от теории, в пересчете на количество используемой карбоновой кислоты) указанного в заголовке соединения в виде бесцветного масла.
Элементный анализ:
д) 1,7-бис[2-N-(1-O-α-D-карбонилметилманнопираноза)-L-лизин]диамид-4-[2-(N-этил-N-перфтороктилсульфонил)амино]ацетил-1,4,7-триазагептан
15,0 г (6,36 ммоля) полученного в примере 34г амида растворяют в 150 мл этанола и смешивают с 0,5 г катализатора Перлмана (20%-ный Pd/C). После этого гидрируют при комнатной температуре в атмосфере водорода (1 атм) до тех пор, пока не прекратится поглощение водорода. После этого катализатор удаляют вакуум-фильтрацией, тщательно промывают этанолом (примерно 100 мл) и досуха концентрируют в вакууме. Таким путем получают указанное в заголовке соединение в виде высоковязкого, желтоватого масла.
Выход: 8,54 г (97,2% от теории).
Элементный анализ:
e) 1,7-бис[2-N-(1-O-α-D-карбонилметилманнопираноза)-6-N-[1,4,7-трис(карбоксилатометил)-10-(3-аза-4-оксо-5-метил-5-илпентаноил)-1,4,7,10-тетраазациклододекан]-L-лизин]диамид-4-[2-(N-этил-N-перфтороктилсульфонил)амино]ацетил-1,4,7-триазагептан, дигадолиниевый комплекс
Перемешанную суспензию 5,7 г (9,06 ммоля) описанного в заявке DE 19728954 С1 в примере 31h Gd-комплекса 10-(4-карбокси-1-метил-2-оксо-3-азабутил)-1,4,7,10-тетраазациклододекан-1,4,7-триуксусной кислоты в 75 мл абсолютного диметилсульфоксида при 70°С смешивают с 0,68 г (15,9 ммоля) хлорида лития. После 30-минутного перемешивания при 70°С прозрачный реакционный раствор порциями смешивают с N-гидроксисукцинимидом общим количеством 1,83 г (15,9 ммоля) и реакционную смесь выдерживают еще в течение 1 ч при этой температуре. После охлаждения до 0°С смешивают с 4,52 г (23,85 ммоля) дициклогексилкарбодиимида и реакционный раствор перемешивают еще в течение 1 ч при 0°С, а затем в течение 12 ч при 22°С. Полученный таким путем реакционный раствор N-гидроксисукцинимидоэфира Gd-комплекса 10-(4-карбокси-1-метил-2-оксо-3-азабутил)-1,4,7,10-тетраазациклододекан-1,4,7-триуксусной кислоты при 22°С по каплям смешивают с раствором 2,84 г (2,06 ммоля) указанного в заголовке примера 34д соединения в 15 мл абсолютного диметилсульфоксида и затем перемешивают в течение 12 ч при комнатной температуре. Для переработки реакционный раствор по каплям добавляют при 22°С к 500 мл ацетона, при этом указанное в заголовке соединение выпадает в виде бесцветного осадка. Этот осадок отделяют вакуум-фильтрацией, растворяют в 200 мл дистиллированной воды и для обессоливания и для отделения низкомолекулярных компонентов трижды подвергают ультрафильтрации через ультрафильтрационную мембрану YM-3 (AMICON®, предельная проницаемость: 3000 Да). Полученный в результате ретентат в завершение лиофилизуют.
Выход: 4,80 г (89,6% от теории, в пересчете на количество используемого аминового компонента) в виде бесцветного лиофилизата с содержанием воды 8,98%.
Элементный анализ (в пересчете на безводное вещество):
Пример 35
а) 1,7-бис(бензилоксикарбонил)-4-{3-оксапентан-1,5-дикарбоновая кислота-1-оил-5-[1-(4-перфтороктилсульфонил)пиперазин]амид}-1,4,7,10-тетраазациклододекан
К раствору 10,75 г (24,4 ммоля) 1,7-бис[бензилоксикарбонил]-1,4,7,10-тетраазациклододекана в смеси, состоящей из 150 мл тетрагидрофурана и 15 мл хлороформа, при 0°С в атмосфере азота добавляют 16,56 г (24,4 ммоля) указанного в заголовке примера 35д соединения, растворенного в 150 мл тетрагидрофурана. После этого при 0°С порциями добавляют в целом 18,0 г (36,6 ммоля) ЭЭДХ [2-этокси-1-этоксикарбонил-1,2-дигидрохинолин], оставляют на ночь при комнатной температуре для перемешивания и затем концентрируют в вакууме. Полученное масло хроматографируют на силикагеле (элюент:н-гексан/изопропанол в соотношении 12:1). Таким путем получают 17,22 г (64,3% от теории, в пересчете на количество используемого вторичного амина) моноамида, а также 3,8 г (8,8% от теории) диамида в качестве побочного продукта. Указанное в заголовке соединение выделяют в виде бесцветного масла.
Элементный анализ:
б) 1,7-бис(бензилоксикарбонил)-4-{3-оксапентан-1,5-дикарбоновая кислота-1-оил-5-[1-(4-перфтороктилсульфонил)пиперазин]амид}-10-[1-O-α-D-(5-карбонил)пентил-2,3,4,6-тетра-О-бензилманнопираноза]-1,4,7,10-тетраазациклододекан
10,0 г (13,4 ммоля) полученной в примере 30в карбоновой кислоты и 3,24 г (28,1 ммоля) N-гидроксисукцинимида растворяют в 100 мл диметилформамида, при 0°С порциями смешивают с N,N'-дициклогексилкарбодиимидом общим количеством 5,8 г (28,1 ммоля) и затем перемешивают в течение 3 ч при этой температуре. К полученному таким путем раствору активированного сложного эфира по каплям добавляют охлажденный до 0°С раствор 14,83 г (13,4 ммоля) указанного в заголовке примера 35а соединения в 100 мл диметилформамида и перемешивают в течение 2 ч при 0°С, а затем в течение 12 ч при комнатной температуре. Для переработки выпавшую в осадок дициклогексилмочевину отфильтровывают и затем отгоняют растворитель до получения сухого остатка. Полученный таким путем остаток затем хроматографируют на силикагеле (элюент:дихлорметан/этиловый эфир уксусной кислоты в соотношении 20:1; хроматографию проводят с использованием градиента растворителя, непрерывно увеличивая долю этилового эфира уксусной кислоты).
Выход: 18,3 г (78,2% от теории) указанного в заголовке соединения в виде бесцветного и высоковязкого масла.
Элементный анализ:
в) 1-{3-оксапентан-1,5-дикарбоновая кислота- 1-оил-5-[1 -(4-перфтороктилсульфонил)пиперазин]амид}-7-[1-O-α-D-(5-карбонил)пентилманнопираноза]-1,4,7,10-тетраазациклододекан
17,0 г (9,75 ммоля) полученного в примере 34б соединения растворяют в 150 мл этанола, смешивают с 1,0 г катализатора Перлмана (20%-ный Pd/C) и гидрируют при комнатной температуре в атмосфере водорода (1 атм) до тех пор, пока не прекратится поглощение водорода. После этого катализатор удаляют вакуум-фильтрацией, тщательно промывают этанолом (дважды порциями по 75 мл) и досуха концентрируют в вакууме. Таким путем получают указанное в заголовке соединение в виде высоковязкого и бесцветного масла.
Выход: 10,76 г (99,0% от теории).
Элементный анализ:
г) 1,7-бис[1,4,7-трис(карбоксилатометил)-1,4,7,10-тетраазациклододекан-10-(пентаноил-3-аза-4-оксо-5-метил-5-ил)-4-[2-(N-этил-N-перфтороктилсульфонил)амино]ацетил-2-оксаацетил]-10-[1-O-α-D-6-карбонилпентилманнопираноза]-1,4,7,10-тетраазациклододекан, Gd-комплекс
24,86 г (39,46 ммоля, 4,4 молярных эквивалента в пересчете на количество используемого аминового компонента из примера 35в) описанного в заявке DE 19728954 С1 в примере 31h Gd-комплекса 10-(4-карбокси-1-метил-2-оксо-3-азабутил)-1,4,7,10-тетраазациклододекан-1,4,7-триуксусной кислоты и 1,67 г безводного хлорида лития (39,46 ммоля) при 40°С растворяют при перемешивании в 200 мл абсолютного диметилсульфоксида и при этой температуре смешивают с N-гидроксисукцинимидом общим количеством 4,53 г (39,46 ммоля) и с 10,0 г (8,97 ммоля) указанного в заголовке примера 34в соединения, растворенного в 100 мл абсолютного диметилсульфоксида. После охлаждения до комнатной температуры реакционный раствор смешивают с 8,14 г (39,46 ммоля) N,N'-дициклогексилкарбодиимида и перемешивают в течение 12 ч при комнатной температуре. Полученную суспензию затем смешивают с достаточным количеством ацетона до полного осаждения указанного выше в заголовке соединения, осадок отделяют вакуум-фильтрацией, сушат, растворяют в воде, отфильтровывают нерастворившуюся дициклогексилмочевину и фильтрат для обессоливания и для очистки от низкомолекулярных компонентов пропускают через ультрафильтрационную мембрану AMICON® YM-3 (предельная проницаемость: 3000 Да). Ретентат в завершение лиофилизуют.
Выход: 16,37 г (79,3% от теории) в виде бесцветного лиофилизата.
Н2О-содержание (при использовании реактива Карла Фишера): 7,65%.
Элементный анализ (в пересчете на безводное вещество):
д) Моно[1-(4-перфтороктилсульфонил)пиперазин]амид 3-оксапентан-1,5-дикарбоновой кислоты
25 г (44,0 ммоля) 1-перфтороктилсульфонилпиперазина растворяют в 150 мл тетрагидрофурана, смешивают при комнатной температуре с ангидридом дигликолевой кислоты общим количеством 5,1 г (44,0 ммоля) и полученный таким путем реакционный раствор в течение 12 ч кипятят с обратным холодильником. После охлаждения до комнатной температуры концентрируют досуха и полученный маслянистый остаток очищают на силикагеле с использованием дихлорметана/2-пропанола (в соотношении 16:1) в качестве элюента
Выход: 27,94 г (92,8% от теории) указанного выше в заголовке соединения в виде бесцветного и вязкого масла.
Элементный анализ:
Пример 36
а) 1,7-бис(бензилоксикарбонил)-4-{3-оксапентан-1,5-дикарбоновая кислота-1-оил-5-[1-(4-перфтороктилсульфонил)пиперазин]амид}-(10-[1-O-β-D-6-карбонилпентил-2,3,4,6-тетра-О-бензилглюкопираноза]-1,4,7,10-тетраазациклододекан)
В 750 мл сухого тетрагидрофурана растворяют 68,5 г (91,79 ммоля) 1-карбоксиметилокси-2,3,4,6-тетра-O-бензил-α-D-маннопиранозида [получение по методу, описанному в патенте DE 19728954 С1] и затем добавляют 9,25 г (91,79 ммоля) триэтиламина. После охлаждения реакционного раствора до температуры в пределах от -15 до -20°С при этой температуре и при перемешивании по каплям медленно добавляют раствор 12,64 г (92,5 ммоля) изобутилового эфира хлормуравьиной кислоты в 150 мл сухого тетрагидрофурана, при этом скорость капельного добавления должна быть такой, чтобы внутренняя температура не превышала -10°С. После проведения реакции в течение 15 мин при -15°С по каплям при -20°С медленно добавляют раствор 101,6 г (91,79 ммоля) указанного в заголовке примера 35а соединения и 9,25 г (91,79 ммоля) триэтиламина в 500 мл сухого тетрагидрофурана. После проведения реакции в течение часа при -15°С, а затем в течение двух часов при комнатной температуре реакционный раствор досуха концентрируют в вакууме. Полученный остаток растворяют в 450 мл этилового эфира уксусной кислоты и дважды промывают насыщенным раствором гидрокарбоната натрия порциями по 300 мл и однократно водой порцией 400 мл. После сушки органической фазы над сульфатом натрия соль отделяют вакуум-фильтрацией и этиловый эфир уксусной кислоты отгоняют в вакууме. Полученный маслянистый остаток очищают на силикагеле с использованием дихлорметана/гексана/2-пропанола (в соотношении 10:20:1) в качестве элюента.
Выход: 130,6 г (81,6% от теории) указанного выше в заголовке соединения в виде бесцветного и высоковязкого масла.
Элементный анализ:
б) 1-{3-оксапентан-1,5-дикарбоновая кислота-1-оил-5-[1-(4-перфтороктилсульфонил)пиперазин]амид}-7-[1-O-α-D-(5-карбонил)пентилманнопираноза]-1,4,7,10-тетраазациклододекан
110,0 г (63,08 ммоля) полученного в примере 36а соединения растворяют в 1000 мл этанола, смешивают с 5,0 г катализатора Перлмана (20%-ный Pd/C) и гидрируют до количественного поглощения водорода. После этого катализатор удаляют вакуум-фильтрацией, после чего промывают этанолом и досуха концентрируют в вакууме. Таким путем получают указанное в заголовке соединение в виде вязкого и бесцветного масла.
Выход: 92,61 г (99,5% от теории).
Элементный анализ:
в) 1,7-бис[1,4,7-трис(карбоксилатометил)-10-(3-аза-4-оксо-5-метил-5-илпентаноил)-4-{3-оксапентан-1,5-дикарбоновая кислота-1-оил-5-[1-(4-перфтороктилсульфонил)пиперазин]амид}-10-[1-O-α-D-(5-карбонил)пентилманнопираноза]-1,4,7,10-тетраазациклододекан, дигадолиниевый комплекс
55,4 г (88,0 ммолей, 4,4 молярных эквивалента в пересчете на количество используемого диаминового компонента из примера 33 г) описанного в заявке DE 19728954 С1 в примере 31h Gd-комплекса 10-(4-карбокси-1-метил-2-оксо-3-азабутил)-1,4,7,10-тетраазациклододекан-1,4,7-триуксусной кислоты и 3,7 г безводного хлорида лития (88,0 ммолей) при 40°С растворяют при перемешивании в 500 мл абсолютного диметилсульфоксида и при этой температуре смешивают с N-гидроксисукцинимидом общим количеством 10,1 г (88,0 ммолей) и с 29,5 г (20,0 ммолей) указанного в заголовке примера 36б соединения, растворенного в 200 мл абсолютного диметилсульфоксида. После охлаждения до комнатной температуры реакционный раствор смешивают с 18,2 г (88,0 ммолей) N,N'-дициклогексилкарбодиимида и перемешивают в течение 12 ч при комнатной температуре. Полученную суспензию затем смешивают с достаточным количеством ацетона до полного осаждения указанного выше в заголовке соединения, осадок отделяют вакуум-фильтрацией, сушат, растворяют в воде, отфильтровывают нерастворившуюся дициклогексилмочевину и фильтрат для обессоливания и для очистки от низкомолекулярных компонентов пропускают через ультрафильтрационную мембрану AMICON® YM-3 (предельная проницаемость: 3000 Да). Ретентат в завершение лиофилизуют.
Выход: 35,96 г (76,9% от теории) в виде бесцветного лиофилизата.
Н2О-содержание (при использовании реактива Карла Фишера): 5,98%.
Элементный анализ (в пересчете на безводное вещество):
Пример 37
а) 5-(этоксикарбонил)пентил-2,3,4,6-тетра-O-ацетил-α-D-маннопиранозид
Аналогично методу, описанному в литературе для синтеза арилгликопиранозидов [J.Conchie и G.A.Levvy, "Methods in Carbohydrate Chemistry (под ред. R.L.Whistler, M.L.Wolfrom и J.N.BeMiller), изд-во Academic Press, New York, т.II, 90, сс.345-347 (1963)] в результате взаимодействия 156,2 г (400 ммолей) пентаацетата D-маннозы в виде смеси α,β-аномеров (соотношение между α и β составляет 4:1) [синтез 1,2,3,4,6-пента-O-ацетил-α,β-D-маннопиранозы см. у M.L.Wolfrom и A.Thompson, "Methods in Carbohydrate Chemistry" (под ред. R.L.Whistler, M.L.Wolfrom и J.N.BeMiller), изд-во Academic Press, New York, т.II, 53, сс.211-215 (1963)] с 67 мл (400 ммолей) этилового эфира 6-гидроксигексановой кислоты и 60,8 мл (520 ммолей) хлорида олова(IV) в 1,2-дихлорэтане общим количеством 600 мл после очистки колоночной хроматографией (элюент:гексан/этиловый эфир уксусной кислоты в соотношении 2:1) получают 100,05 г (51% от теории) указанного выше в заголовке соединения в виде бесцветного и вязкого масла. Данные 1H-ЯМР-спектроскопического анализа полученного таким путем указанного в заголовке соединения свидетельствуют о том, что это соединение представляет собой только чистый α-аномер.
Элементный анализ:
б) 5-(карбокси)пентил-2,3,4,6-тетра-O-бензил-α-D-маннопиранозид
Перемешанную суспензию 141,0 г (289 ммолей) указанного в заголовке примера 37а соединения в 200 мл диоксана при комнатной температуре и одновременном интенсивном перемешивании порциями смешивают с высокодисперсным порошковым гидроксидом калия общим количеством 238,5 г (4,26 моля). Реакционную смесь для повышения ее размешиваемости смешивают еще с 200 мл диоксана и полученную в результате суспензию затем нагревают до температуры кипения и при этой температуре в течение двух часов по каплям смешивают с бензилбромидом общим количеством 372 мл (3,128 моля). После проведения реакции в течение 4 ч при 110°С, а затем в течение 12 ч при комнатной температуре реакционную смесь для переработки медленно сливают в смесь воды со льдом общим количеством 2,5 л и водную фазу затем полностью экстрагируют диэтиловым эфиром. После промывки полученной таким путем эфирной фазы и последующей ее сушки над сульфатом натрия соль отделяют вакуум-фильтрацией и диэтиловый эфир отгоняют в вакууме. После этого избыток бензилбромида количественно отгоняют из реакционной смеси в вакууме, создаваемом масляным насосом, при температуре масляной бани 180°С. Полученный таким путем смолянисто-маслянистый остаток очищают на силикагеле с использованием этилового эфира уксусной кислоты/гексана (в соотношении 1:10) в качестве элюента.
Выход: 172,2 г (91,0% от теории) указанного выше в заголовке соединения в виде бесцветного и высоковязкого масла.
Элементный анализ:
в) 5-[(карбокси)пентил-2,3,4,6-тетра-O-бензил-α-D-маннопиранозид]-N-гидроксисукцинимидоэфир
60,0 г (91,5 ммоля) соединения, указанного в заголовке примера 37б, растворяют в 750 мл диметилформамида и смешивают с N-гидроксисукцинимидом общим количеством 10,4 г (91,5 ммоля). Далее охлаждают до 0°С и добавляют 18,9 г (91,5 ммоля) дициклогексилкарбодиимида. После этого перемешивают в течение часа при 0°С, а затем в течение 4 ч при комнатной температуре. Растворитель отгоняют в вакууме, полученный остаток смешивают с 100 мл этилового эфира уксусной кислоты и охлаждают до 0°С. Выпавшую в осадок мочевину отфильтровывают и полученный фильтрат досуха концентрируют в вакууме. Полученный таким путем смолянисто-маслянистый остаток очищают на силикагеле с использованием этилового эфира уксусной кислоты/гексана (в соотношении 1:20) в качестве элюента.
Выход: 61,23 г (89,0% от теории) указанного выше в заголовке соединения в виде бесцветного и вязкого масла.
Элементный анализ:
г) Метиловый эфир 2,6-бис{6-Nε-2-Nα-[1-O-α-D-6-карбонилпентил-(2,3,4,6-тетра-O-бензил)маннопираноза]-L-лизина}
К охлажденному до 0°С раствору из 4,26 г (18,30 ммоля, 0,5 молярных эквивалента в пересчете на количество используемой карбоновой кислоты) дигидрохлорида метилового эфира L-лизина (поставляется фирмой Bachem) и 4,05 г (40,26 ммоля) триэтиламина в 100 мл диметилформамида по каплям добавляют раствор 27,51 г (36,6 ммоля) указанного в заголовке примера 37в соединения в 150 мл диметилформамида. По завершении этого добавления перемешивают еще в течение часа при 0°С, а затем в течение ночи при комнатной температуре. Смесь упаривают в вакууме досуха и остаток растворяют в 300 мл этилового эфира уксусной кислоты. Выпавшую в осадок мочевину отфильтровывают и фильтрат дважды промывают 5%-ным водным раствором соды порциями по 100 мл. Органическую фазу сушат над сульфатом магния и досуха упаривают в вакууме. Остаток хроматографируют на силикагеле (элюент:н-гексан/изопропанол в соотношении 25:1). Таким путем получают 39,56 г (75,4% от теории) указанного в заголовке соединения в виде бесцветного масла.
Элементный анализ:
д) 2,6-бис[6-Nε-2-Nα-[1-O-α-D-6-карбонилпентил-(2,3,4,6-тетра-O-бензил)маннопираноза]]-L-лизин
В 150 мл этанола растворяют 30,0 г (20,92 ммоля) полученного в примере 37 г соединения. Далее добавляют раствор 4 г (100,0 ммолей) гидроксида натрия в 10 мл дистиллированной воды и перемешивают в течение 3 ч при 50°С. Согласно хроматограмме тонкослойной хроматографии омыление является количественным. После этого смесь досуха концентрируют в вакууме, полученный остаток растворяют в 300 мл этилового эфира уксусной кислоты и органическую фазу дважды экстрагируют разбавленным водным раствором лимонной кислоты порциями по 100 мл. После сушки над сульфатом натрия фильтруют и досуха концентрируют в вакууме. Остаток хроматографируют на силикагеле (элюент:н-гексан/изопропанол в соотношении 13:1). Таким путем получают 25,56 г (88,5% от теории) указанного в заголовке соединения в виде бесцветного масла.
Элементный анализ:
е) N-гидроксисукцинимидоэфир 2,6-бис[6-Nε-2-Nα-[1-O-α-D-6-карбонилпентил-(2,3,4,6-тетра-O-бензил)маннопираноза]-L-лизина]
14,0 г (9,15 ммоля) соединения, указанного в заголовке примера 37д, растворяют в 100 мл диметилформамида и смешивают с N-гидроксисукцинимидом общим количеством 1,04 г (9,15 ммоля). Далее охлаждают до 0°С и добавляют 1,89 г (9,15 ммоля) дициклогексилкарбодиимида. После этого перемешивают в течение часа при 0°С, а затем в течение 4 ч при комнатной температуре. Далее растворитель отгоняют в вакууме, полученный остаток смешивают с 100 мл этилового эфира уксусной кислоты и охлаждают до 0°С. Выпавшую в осадок мочевину отфильтровывают и полученный фильтрат досуха концентрируют в вакууме. Полученный таким путем смолянисто-маслянистый остаток очищают на силикагеле с использованием этилового эфира уксусной кислоты/н-гексана (в соотношении 1:20) в качестве элюента.
Выход: 12,94 г (92,4% от теории) указанного выше в заголовке соединения в виде бесцветного и вязкого масла.
Элементный анализ:
ж) 1,7-(1,4,7-триазагептан)диамид 2,6-N,N'-бис[1-O-α-D-(6-карбонил)пентил-2,3,4,6-тетра-O-бензилманнопираноза]-L-лизина
К охлажденному до 0°С раствору 0,47 г (4,57 ммоля) диэтилентриамина в 25 мл диметилформамида по каплям медленно добавляют раствор 14,0 г (9,15 ммоля; 2 молярных эквивалента в пересчете на количество используемого амина) указанного в заголовке примера 37е соединения в 100 мл диметилформамида. По завершении этого добавления перемешивают еще в течение часа при 0°С, а затем в течение ночи при комнатной температуре. Смесь упаривают в вакууме досуха и остаток растворяют в 200 мл этилового эфира уксусной кислоты. Выпавшую в осадок мочевину отфильтровывают и фильтрат дважды промывают 5%-ным водным раствором соды порциями по 50 мл. Органическую фазу сушат над сульфатом магния и досуха упаривают в вакууме. Остаток хроматографируют на силикагеле (элюент:н-гексан/изопропанол в соотношении 25:1). Таким путем получают 9,53 г (71,4% от теории) указанного в заголовке соединения в виде бесцветного масла.
Элементный анализ:
з) Метиловый эфир 2-N-[2-(N-этил-N-перфтороктилсульфонил)амино]ацетил-6-N-(бензилоксикарбонил)-L-лизина
20,8 г (35,6 ммоля) 2-(N-этил-N-перфтороктилсульфонил)аминоуксусной кислоты, а также 3,60 г (35,6 ммоля) триэтиламина растворяют в 200 мл диметилформамида и добавляют 4,09 г (35,6 моля) N-гидроксисукцинимида. Далее охлаждают до 0°С и добавляют 7,34 г (35,6 ммоля) дициклогексилкарбодиимида. После этого перемешивают в течение часа при 0°С, а затем в течение 4 ч при комнатной температуре. Далее охлаждают до 0°С и в течение 10 мин по каплям добавляют раствор 11,77 г (35,6 ммоля) гидрохлорида метилового эфира 6-N-бензилоксикарбонил-L-лизина и 4,0 г (40,0 ммолей) триэтиламина в 100 мл диметилформамида. Смесь перемешивают в течение часа при 0°С, а затем в течение ночи при комнатной температуре. Смесь упаривают в вакууме досуха и остаток растворяют в 100 мл этилового эфира уксусной кислоты. Выпавшую в осадок мочевину отфильтровывают и фильтрат дважды промывают 5%-ным водным раствором соды порциями по 100 мл. Органическую фазу сушат над сульфатом магния и досуха упаривают в вакууме. Остаток хроматографируют на силикагеле (элюент:н-гексан/этиловый эфир уксусной кислоты в соотношении 20:1). Таким путем получают 27,43 г (88,0% от теории) бесцветного масла.
Элементный анализ:
и) 2-Nα-{[2-(N-этил-N-перфтороктилсульфонил)амино]ацетил}-6-Nε-(бензилоксикарбонил)-L-лизин
В 150 мл этанола растворяют 25,0 г (28,55 ммоля) полученного в примере 37з соединения. Далее добавляют раствор 4 г (100,0 ммолей) гидроксида натрия в 10 мл дистиллированной воды и перемешивают в течение 3 ч при 50°С. Согласно хроматограмме тонкослойной хроматографии омыление является количественным. После этого досуха концентрируют в вакууме, полученный остаток растворяют в 300 мл этилового эфира уксусной кислоты и органическую фазу дважды экстрагируют разбавленным водным раствором лимонной кислоты порциями по 100 мл. После сушки над сульфатом натрия фильтруют и досуха концентрируют в вакууме. Остаток хроматографируют на силикагеле (элюент:н-гексан/изопропанол в соотношении 10:1). Таким путем получают 22,73 г (92,4% от теории) указанного в заголовке соединения в виде бесцветного масла.
Элементный анализ:
к) 1,4,7-триазагептан-4-{2-N-[2-(N-этил-N-перфтороктилсульфонил)амино]ацетил-6-N-бензилоксикарбонил}-L-лизинамид-1,7-бис{2,6-N,N'-бис[1-O-α-D-(5-карбонил)пентил-2,3,4,6-тетра-O-бензилманнопираноза]-L-лизиндиамид}
15,33 г (17,8 ммоля) указанного в заголовке примера 37и соединения, а также 1,80 г (17,8 ммоля) триэтиламина растворяют в 250 мл сухого тетрагидрофурана. После охлаждения реакционного раствора до температуры в интервале от -15 до -20°С при этой температуре по каплям при перемешивании медленно добавляют раствор 4,92 г (35,6 ммоля) изобутилового эфира хлормуравьиной кислоты в 50 мл сухого тетрагидрофурана, при этом скорость капельного добавления должна быть такой, чтобы внутренняя температура не превышала -10°С. После проведения реакции в течение 15 мин при -15°С по каплям при -20°С медленно добавляют раствор 52,0 г (17,8 ммоля) указанного в заголовке примера 37ж соединения и 1,80 г (17,8 ммоля) триэтиламина в 300 мл сухого тетрагидрофурана. После проведения реакции в течение часа при -15°С, а затем в течение двух часов при комнатной температуре реакционный раствор досуха концентрируют в вакууме. Полученный остаток растворяют в 500 мл этилового эфира уксусной кислоты и дважды промывают насыщенным раствором гидрокарбоната натрия порциями по 200 мл и однократно водой порцией 200 мл. После сушки органической фазы над сульфатом натрия соль отделяют вакуум-фильтрацией и этиловый эфир уксусной кислоты отгоняют в вакууме. Полученный маслянистый остаток очищают на силикагеле с использованием этилового эфира уксусной кислоты/н-гексана (в соотношении 1:20) в качестве элюента.
Выход: 54,6 г (81,6% от теории) указанного выше в заголовке соединения в виде бесцветного и высоковязкого масла.
Элементный анализ:
л) 1,4,7-триазагептан-4-{2-N-[2-(N-этил-N-перфтороктилсульфонил)амино]ацетил}-L-лизинамид-1,7-бис{2,6-N,N'-бис[1-O-α-D-(5-карбонил)пентилманнопираноза]-L-лизиндиамид}
50,0 г (13,28 ммоля) полученного в примере 37к соединения растворяют в 500 мл этанола, смешивают с 4,0 г катализатора Перлмана (20%-ный Pd/C) и гидрируют при комнатной температуре в атмосфере водорода (1 атм) до тех пор, пока не прекратится поглощение водорода. После этого катализатор удаляют вакуум-фильтрацией, тщательно промывают этанолом (примерно 400 мл) и досуха концентрируют в вакууме. Таким путем получают указанное в заголовке соединение в виде высоковязкого и бесцветного масла.
Выход: 26,85 г (93,0% от теории).
Элементный анализ:
м) 1,4,7-триазагептан-4-{2-N-[2-(N-этил-N-перфтороктилсульфонил)амино]ацетил-6-N-[1,4,7-трис(карбоксилатометил)-10-(3-аза-4-оксо-5-метил-5-илпентаноил)-1,4,7,10-тетраазациклододекан}-L-лизинамид-1,7-бис{2,6-N,N'-бис[1-O-α-D-(5-карбонил)пентилманнопираноза]-L-лизиндиамид}, гадолиниевый комплекс
5,54 г (8,8 ммоля, 2,2 молярных эквивалента в пересчете на количество используемого аминового компонента из примера 37л) описанного в заявке DE 19728954 С1 в примере 31h Gd-комплекса 10-(4-карбокси-1-метил-2-оксо-3-азабутил)-1,4,7,10-тетраазациклододекан-1,4,7-триуксусной кислоты и 0,37 г безводного хлорида лития (8,8 ммоля) при 40°С растворяют при перемешивании в 60 мл абсолютного диметилсульфоксида и при этой температуре смешивают с N-гидроксисукцинимидом общим количеством 1,01 г (8,8 ммоля) и с 1,84 г (4,0 ммоля) указанного в заголовке примера 37л соединения, растворенного в 40 мл абсолютного диметилсульфоксида. После охлаждения до комнатной температуры реакционный раствор смешивают с 1,82 г (8,8 ммоля) N,N'-дициклогексилкарбодиимида и перемешивают в течение 12 ч при комнатной температуре. Полученную суспензию затем смешивают с достаточным количеством ацетона до полного осаждения указанного выше в заголовке соединения, осадок отделяют вакуум-фильтрацией, сушат, растворяют в воде, отфильтровывают нерастворившуюся дициклогексилмочевину и фильтрат для обессоливания и для очистки от низкомолекулярных компонентов пропускают через ультрафильтрационную мембрану AMICON® YM-3 (предельная проницаемость: 3000 Да). Ретентат в завершение лиофилизуют.
Выход: 8,77 г (78,7% от теории) в виде бесцветного лиофилизата.
Н2О-содержание (при использовании реактива Карла Фишера): 4,43%.
Элементный анализ (в пересчете на безводное вещество):
Пример 38
а) Метиловый эфир 2-Nα-6-Nε-бис[1-O-α-D-карбонилметил-2,3,4,6-тетра-O-бензилманнопираноза]-L-лизина]
10,95 г (18,30 ммоля) 1-карбоксиметилокси-2,3,4,6-тетра-O-бензил-α-D-маннопиранозида [получение по методу, описанному в патенте DE 19728954 С1] растворяют в 150 мл диметилформамида и смешивают с N-гидроксисукцинимидом общим количеством 2,09 г (18,3 ммоля). Далее охлаждают до 0°С и добавляют 3,78 г (18,3 ммоля) дициклогексилкарбодиимида. После этого перемешивают в течение часа при 0°С, а затем в течение 4 ч при комнатной температуре. Далее охлаждают до 0°С и в течение часа по каплям добавляют раствор 2,13 г (9,15 ммоля, 0,5 молярных эквивалента в пересчете на количество используемой карбоновой кислоты) дигидрохлорида метилового эфира L-лизина (поставляется фирмой Bachem) и 2,02 г (20,13 ммоля) триэтиламина в 70 мл диметилформамида. По завершении этого добавления перемешивают еще в течение часа при 0°С, а затем в течение ночи при комнатной температуре. Смесь упаривают в вакууме досуха и остаток растворяют в 300 мл этилового эфира уксусной кислоты. Выпавшую в осадок мочевину отфильтровывают и фильтрат дважды промывают 5%-ным водным раствором соды порциями по 100 мл. Органическую фазу сушат над сульфатом магния и досуха упаривают в вакууме. Остаток хроматографируют на силикагеле (элюент:н-гексан/изопропанол в соотношении 25:1). Таким путем получают 10,05 г (82,3% от теории) указанного в заголовке соединения в виде бесцветного масла.
Элементный анализ:
б) 2-Nα-6-Nε-бис[1-O-α-D-карбонилметил-2,3,4,6-тетра-O-бензилманнопираноза]-L-лизин
Аналогично методу, описанному в примере 37д для синтеза указанного в его заголовке соединения, в результате омыления 15 г (11,23 ммоля) метилового эфира соединения, указанного в заголовке примера 38а, получают 13,89 г (93,6% от теории) указанного выше в заголовке соединения в виде бесцветного и вязкого масла.
Элементный анализ:
в) N-гидроксисукцинимидоэфир 2-Nα-6-Nε-бис[1-O-α-D-карбонилметил-2,3,4,6-тетра-O-бензилманнопираноза]-L-лизина
12,09 г (9,15 ммоля) соединения, указанного в заголовке примера 38б, растворяют в 100 мл диметилформамида и смешивают с N-гидроксисукцинимидом общим количеством 1,04 г (9,15 ммоля). Далее охлаждают до 0°С и добавляют 1,89 г (9,15 ммоля) дициклогексилкарбодиимида. После этого перемешивают в течение часа при 0°С, а затем в течение 4 ч при комнатной температуре. Далее растворитель отгоняют в вакууме, полученный остаток смешивают с 100 мл этилового эфира уксусной кислоты и охлаждают до 0°С. Выпавшую в осадок мочевину отфильтровывают и полученный фильтрат досуха концентрируют в вакууме. Полученный таким путем смолянисто-маслянистый остаток очищают на силикагеле с использованием этилового эфира уксусной кислоты/н-гексан (в соотношении 1:20) в качестве элюента.
Выход: 12,24 г (94,4% от теории) указанного выше в заголовке соединения в виде бесцветного и вязкого масла.
Элементный анализ:
г) [1-(4-перфтороктилсульфонил)пиперазин]амид 6-N-бензилоксикарбонил-2-N-{[2,6-N,N'-бис(1-O-α-D-карбонилметил-2,3,4,6-тетра-O-бензилманнопираноза)]-L-лизил}-L-лизина
19,0 г (13,4 ммоля) полученного в примере 38в N-гидроксисукцинимидоэфира карбоновой кислоты растворяют в 75 мл диметилформамида и при 0°С по каплям смешивают с охлажденным до 0°С раствором 11,13 г (13,4 ммоля) указанного в заголовке примера 21в соединения в 50,0 мл диметилформамида. Полученный реакционный раствор перемешивают еще в течение 2 ч при 0°С, а затем в течение 12 ч при комнатной температуре. Для переработки выпавшую в осадок дициклогексилмочевину отфильтровывают и затем растворитель отгоняют в вакууме до получения сухого остатка. Полученный таким путем остаток хроматографируют на силикагеле [элюент:дихлорметан/этанол в соотношении 28:1, при этом хроматографию проводят с использованием градиента растворителя, непрерывно увеличивая в процессе хроматографии долю используемого в элюенте его полярного компонента (в данном случае этанола)].
Выход: 25,28 г (88,4% от теории) указанного в заголовке соединения в виде бесцветного и высоковязкого масла.
Элементный анализ:
д) [1-(4-перфтороктилсульфонил)пиперазин]амид 2-N-{[2,6-N,N'-бис(1-O-α-D-карбонилметилманнопираноза)]-L-лизил-L-лизина
20,0 г (9,37 ммоля) полученного в примере 38г соединения растворяют в 200 мл этанола, смешивают с 1,5 г катализатора Перлмана (20%-ный Pd/C) и гидрируют при комнатной температуре в атмосфере водорода (1 атм) до тех пор, пока не прекратится поглощение водорода. После этого катализатор удаляют вакуум-фильтрацией, тщательно промывают этанолом (дважды порциями примерно по 100 мл) и досуха концентрируют в вакууме. Таким путем получают указанное в заголовке соединение в виде высоковязкого и бесцветного масла.
Выход: 11,62 г (97,0% от теории).
Элементный анализ:
e) [1-(4-перфтороктилсульфонил)пиперазин]амид 6-N-[1,4,7-трис(карбоксилатометил)-10-(3-аза-4-оксо-5-метил-5-илпентаноил)-1,4,7,10-тетраазациклододекан)-2-N-{[2,6-N,N'-бис(1-O-α-D-карбонилметилманнопираноза)]-L-лизил}-L-лизина, Gd-комплекс
9,98 г (15,84 ммоля, 2,2 молярных эквивалента в пересчете на количество используемого аминового компонента из примера 38д) описанного в заявке DE 19728954 С1 в примере 31h Gd-комплекса 10-(4-карбокси-1-метил-2-оксо-3-азабутил)-1,4,7,10-тетраазациклододекан-1,4,7-триуксусной кислоты и 0,67 г безводного хлорида лития (15,84 ммоля) при 40°С растворяют при перемешивании в 100 мл абсолютного диметилсульфоксида и при этой температуре смешивают с N-гидроксисукцинимидом общим количеством 1,82 г (15,84 ммоля) и с 9,19 г (7,19 ммоля) указанного в заголовке примера 38д соединения, растворенного в 50 мл абсолютного диметилсульфоксида. После охлаждения до комнатной температуры реакционный раствор смешивают с 3,27 г (15,84 ммоля) N,N'-дициклогексилкарбодиимида и перемешивают в течение 12 ч при комнатной температуре. Полученную суспензию затем смешивают с достаточным количеством ацетона до полного осаждения указанного выше в заголовке соединения, осадок отделяют вакуум-фильтрацией, сушат, растворяют в воде, отфильтровывают нерастворившуюся дициклогексилмочевину и фильтрат для обессоливания и для одновременной очистки от возможно еще присутствующих в нем низкомолекулярных компонентов подвергают ультрафильтрации через ультрафильтрационную мембрану AMICON® YM-3 (предельная проницаемость: 3000 Да). Ретентат в завершение лиофилизуют.
Выход: 11,85 г (87,2% от теории) в виде бесцветного лиофилизата.
Н2О-содержание (при использовании реактива Карла Фишера): 5,54%.
Элементный анализ (в пересчете на безводное вещество):
Пример 39
а) 1,7-бис(бензилоксикарбонил)-4-(3-окса-2Н,2Н,4Н,4Н,5Н,5Н-перфтортридеканоил)-1,4,7,10-тетраазациклододекан
К раствору 10,75 г (24,4 ммоля) 1,7-бис[бензилоксикарбонил]-1,4,7,10-тетраазациклододекана в смеси из 150 мл тетрагидрофурана и 15 мл хлороформа при 0°С в атмосфере азота добавляют 12,74 г (24,4 ммоля) указанного в заголовке примера 39ж соединения, растворенного в 150 мл тетрагидрофурана. После этого при 0°С порциями добавляют в целом 18,0 г (36,6 ммоля) ЭЭДХ [2-этокси-1-этоксикарбонил-1,2-дигидрохинолин], оставляют на ночь для перемешивания при комнатной температуре и затем концентрируют в вакууме. Полученное масло хроматографируют на силикагеле (элюент:н-гексан/изопропанол в соотношении 16:1). Таким путем получают 15,89 г (69,0% от теории, в пересчете на количество используемого вторичного амина) моноамида, а также 3,8 г (8,8% от теории) диамида в качестве побочного продукта. Указанное в заголовке соединение выделяют в виде бесцветного масла.
Элементный анализ:
б) 1,7-бис(бензилоксикарбонил)-4-(3-окса-2Н,2Н,4Н,4Н,5Н,5Н-перфтортридеканоил)-10-[1-S-α-D-(2-карбонил)этил-2,3,4,6-тетра-O-ацетилманнопираноза]-1,4,7,10-тетраазациклододекан
7,09 г (13,4 ммоля) N-гидроксисукцинимидоэфира 3-(2,3,4,6-тетра-O-ацетил-1-тио-α-D-маннопиранозил)пропионовой кислоты (получение согласно методу, описанному у J.Haensler и др., Bioconjugate Chem. 4, 85 (1993); Chipowsky S. и Lee Y.C., Synthesis of 1-thio-aldosides, Carbohydrate Research 31(1973), cc.339-346) растворяют в 100 мл диметилформамида и при 0°С по каплям смешивают с предварительно охлажденным до 0°С раствором 12,65 г (13,4 ммоля) указанного в заголовке примера 39а соединения в 100 мл диметилформамида. Смесь перемешивают в течение 2 ч при 0°С, а затем в течение 12 ч при комнатной температуре. Для переработки растворитель отгоняют в вакууме до получения сухого остатка, который затем хроматографируют на силикагеле (элюент:дихлорметан/этиловый эфир уксусной кислоты в соотношении 20:1; хроматографию проводят с использованием градиента растворителя, непрерывно увеличивая долю этилового эфира уксусной кислоты).
Выход: 16,23 г (88,9% от теории) указанного в заголовке соединения в виде бесцветного и высоковязкого масла.
Элементный анализ:
в) 1-(3-окса-2Н,2Н,4Н,4Н,5Н,5Н-перфтортридеканоил)-7-[1-S-α-O-(2-карбонил)этил-2,3,4,6-тетра-O-ацетилманнопираноза]-1,4,7,10-тетраазациклододекан
15,0 г (11,0 ммолей) полученного в примере 39б соединения растворяют в 150 мл этанола, смешивают с 1,0 г катализатора Перлмана (20%-ный Pd/C) и гидрируют при комнатной температуре в атмосфере водорода (1 атм) до тех пор, пока не прекратится поглощение водорода. После этого катализатор удаляют вакуум-фильтрацией, тщательно промывают этанолом (дважды порциями по 75 мл) и досуха концентрируют в вакууме. Таким путем получают указанное в заголовке соединение в виде высоковязкого и бесцветного масла.
Выход: 11,56 г (96,0% от теории).
Элементный анализ:
г) 1-(3-окса-2Н,2Н,4Н,4Н,5Н,5Н-перфтортридеканоил)-7-[1-S-α-D-(2-карбонил)этилманнопираноза]-1,4,7,10-тетраазациклододекан
10,0 г (9,13 ммоля) соединения, указанного в заголовке примера 39в, суспендируют в 100 мл абсолютного метанола и при 5°С смешивают с каталитическим количеством метанолята натрия. После проведения реакции в течение 3 ч при комнатной температуре данные контроля за протеканием реакции тонкойслойной хроматографией (элюент:хлороформ/метанол в соотношении 4:1) указывают уже на количественное превращение. Для переработки прозрачный реакционный раствор нейтрализуют смешением с катионообменной смолой Amberlite IR 120 (Н+-форма), ионит удаляют вакуум-фильтрацией, после чего промывают метанолом и полученный таким путем метанольный фильтрат перегоняют в вакууме до получения сухого остатка. Полученный маслянистый остаток очищают колоночной хроматографией на силикагеле (элюент:дихлорметан/н-гексан/этиловый эфир уксусной кислоты в соотношении 15:20:1; хроматографию проводят с использованием градиента растворителя, непрерывно увеличивая долю этилового эфира уксусной кислоты). Данные 1Н-ЯМР-спектроскопического анализа указанного в заголовке соединения позволяют на основании величины констант взаимодействия J1,2=0,9 Гц сделать однозначный вывод о наличии α-конфигурации у аномерного центра D-маннопиранозы. Подобная α-конфигурация является единственной присутствующей у центра аномерии конфигурацией, т.е. количество возможно образовавшегося аномера указанного в заголовке соединения с β-конфигурацией лежит тем самым ниже порога обнаружения при 1Н-ЯМР-спектроскопическом анализе. В соответствии с этим указанное выше в заголовке соединение получают только в виде чистого аномера с α-конфигурацией.
Выход: 8,28 г (98,0% от теории) указанного в заголовке соединения в виде бесцветного и высоковязкого масла.
Элементный анализ:
д) 1-(3-окса-2Н,2Н,4Н,4Н,5Н,5Н-перфтортридеканоил)-7-[1-S-α-D-(2-карбонил)этилманнопираноза]-4,10-бис[1,4,7-трис(карбоксилатометил)-10-(3-аза-4-оксо-5-метил-5-илпентаноил)]-1,4,7,10-тетраазациклододекан, дигадолиниевый комплекс
2,48 г (3,94 ммоля, 4,4 молярных эквивалента в пересчете на количество используемого диаминового компонента из примера 39г) описанного в заявке DE 19728954 С1 в примере 31h Gd-комплекса 10-(4-карбокси-1-метил-2-оксо-3-азабутил)-1,4,7,10-тетраазациклододекан-1,4,7-триуксусной кислоты и 167 мг безводного хлорида лития (3,94 ммоля) при 40°С растворяют при перемешивании в 40 мл абсолютного диметилсульфоксида и при этой температуре смешивают с N-гидроксисукцинимидом общим количеством 453 мг (3,94 ммоля) и с 980 мг (0,895 ммоля) указанного в заголовке примера 39г соединения, растворенного в 10 мл абсолютного диметилсульфоксида. После охлаждения до комнатной температуры реакционный раствор смешивают с 814 мг (3,946 ммоля) N,N'-дициклогексилкарбодиимида и перемешивают в течение 12 ч при комнатной температуре. Полученную суспензию затем смешивают с достаточным количеством ацетона до полного осаждения указанного выше в заголовке соединения, осадок отделяют вакуум-фильтрацией, сушат, растворяют в воде, отфильтровывают нерастворившуюся дициклогексилмочевину и фильтрат для обессоливания и для очистки от низкомолекулярных компонентов пропускают через ультрафильтрационную мембрану AMICON® YM-3 (предельная проницаемость: 3000 Да). Ретентат в завершение лиофилизуют.
Выход: 1,32 г (69,1% от теории) в виде бесцветного лиофилизата.
Н2О-содержание (при использовании реактива Карла Фишера): 7,65%.
Элементный анализ (в пересчете на безводное вещество):
е) трет-Бутиловый эфир 3-окса-2Н,2Н,4Н,4Н,5Н,5Н-перфтортридекановой кислоты
25,0 г (53,8 ммоля) 1Н,1Н,2Н,2Н-перфтор-1-деканола [поставляемого фирмой Lancaster] растворяют в 250 мл абсолютного толуола и при комнатной температуре смешивают с каталитическим количеством (примерно 0,75 г) гидросульфата тетра-н-бутиламмония. После этого при 0°С добавляют в целом 7,55 г (134,6 ммоля, 2,5 эквивалента в пересчете на количество используемого спиртового компонента) высокодисперсного порошкового гидроксида калия, а затем 15,73 г (80,7 ммоля, 1,5 эквивалента в пересчете на используемое количество спиртового компонента) трет-бутиловото эфира бромуксусной кислоты и перемешивают еще в течение 2 ч при 0°С. Полученный таким путем реакционный раствор перемешивают в течение 12 ч при комнатной температуре и затем для переработки смешивают с этиловым эфиром уксусной кислоты общим количеством 500 мл и с 250 мл воды. Органическую фазу отделяют и дважды промывают водой. После сушки органической фазы над сульфатом натрия соль отделяют вакуум-фильтрацией и растворитель отгоняют в вакууме. Полученный маслянистый остаток очищают на силикагеле с использованием этилового эфира уксусной кислоты/гексана (в соотношении 1:10) в качестве элюента.
Выход: 26,3 г (84,6% от теории) указанного выше в заголовке соединения в виде бесцветного и высоковязкого масла.
Элементный анализ:
ж) 3-окса-2Н,2Н,4Н,4Н,5Н,5Н-перфтортридеканкарбоновая кислота
20,0 г (34,58 ммоля) соединения, указанного в заголовке примера 39е, при комнатной температуре суспендируют при перемешивании в 200 мл смеси, состоящей из метанола и 0,5-молярного едкого натра в пропорции 2:1, и затем нагревают до 60°С. После проведения реакции в течение 12 ч при 60°С прозрачную реакционную смесь для ее переработки нейтрализуют смешением с катионообменной смолой Amberlite IR 120 (Н+-форма), ионит удаляют вакуум-фильтрацией и полученный в результате метанольно-водный фильтрат перегоняют в вакууме до получения сухого остатка. Полученный аморфно-маслянистый остаток очищают на силикагеле с использованием этилового эфира уксусной кислоты/н-гексана (в соотношении 1:3) в качестве элюента.
Выход: 16,0 г (88,6% от теории) указанного выше в заголовке соединения в виде бесцветного и высоковязкого масла.
Элементный анализ:
Пример 40
а) Метиловый эфир 6-бензилоксикарбонил-2-[2-(N-этил-N-перфтороктилсульфонил)амино]ацетил-L-лизина
К 8,0 г (24,4 ммоля) гидрохлорида метилового эфира ε-карбонилоксибензил-L-лизина (поставляется фирмой Bachem), растворенного в смеси из 150 мл тетрагидрофурана, 15 мл хлороформа и 2,62 г (26,0 ммолей) триэтиламина, при 0°С в атмосфере азота по каплям добавляют 16,18 г (27,0 ммолей) 2-(N-этил-N-перфтороктилсульфонил)аминоуксусной кислоты (получение согласно DE 19603033), растворенной в 50 мл тетрагидрофурана. После этого при 0°С порциями добавляют в целом 18,0 г (36,6 ммоля) ЭЭДХ [2-этокси-1-этоксикарбонил-1,2-дигидрохинолин] и оставляют на ночь для перемешивания при комнатной температуре. После этого концентрируют в вакууме и полученное масло хроматографируют на силикагеле (элюент:н-гексан/изопропанол в соотношении 15:1). Таким путем получают 17,0 г (79,6% от теории, в пересчете на количество используемого первичного амина) указанного в заголовке соединения в виде бесцветного масла.
Элементный анализ:
б) Метиловый эфир 2-[2-(N-этил-N-перфтороктилсульфонил)амино]ацетил-L-лизина
15,0 г (20,23 ммоля) полученного в примере 40а соединения растворяют в 200 мл этанола, смешивают с 800 мг катализатора Перлмана (20%-ный Pd на активированном угле) и гидрируют до поглощения расчетного количества водорода. После этого катализатор удаляют вакуум-фильтрацией, тщательно промывают этанолом и досуха концентрируют в вакууме. Таким путем получают указанное в заголовке соединение в виде бесцветного масла.
Выход: 14,68 г (97,9% от теории)
Элементный анализ:
в) Метиловый эфир 6-(1-O-α-D-карбонилметил-2,3,4,6-тетра-O-бензилманнопираноза)-2-[2-(N-этил-N-перфтороктилсульфонил)амино]ацетил-L-лизина
В 500 мл сухого тетрагидрофурана растворяют 21,31 г (35,6 ммоля) 1-карбоксиметилокси-2,3,4,6-тетра-O-бензил-α-D-маннопиранозида [получение по методу, описанному в патенте DE 19728954 С1], а также 3,60 г (35,6 ммоля) триэтиламина. После охлаждения реакционного раствора до температуры в интервале от -15 до -20°С при этой температуре по каплям при перемешивании медленно добавляют раствор 4,92 г (35,6 ммоля) изобутилового эфира хлормуравьиной кислоты в 75 мл сухого тетрагидрофурана, при этом скорость капельного добавления должна быть такой, чтобы внутренняя температура не превышала -10°С. После проведения реакции в течение 15 мин при -15°С по каплям при -20°С медленно добавляют раствор 26,39 г (35,6 ммоля) указанного в заголовке примера 40б соединения и 3,60 г (35,6 ммоля) триэтиламина в 100 мл сухого тетрагидрофурана. После проведения реакции в течение часа при -15°С, а затем в течение двух часов при комнатной температуре реакционный раствор досуха концентрируют в вакууме. Полученный остаток растворяют в 250 мл этилового эфира уксусной кислоты и дважды промывают насыщенным раствором гидрокарбоната натрия порциями по 100 мл и однократно водой порцией 200 мл. После сушки органической фазы над сульфатом натрия соль отделяют вакуум-фильтрацией и этиловый эфир уксусной кислоты отгоняют в вакууме. Полученный маслянистый остаток очищают на силикагеле с использованием этилового эфира уксусной кислоты/н-гексана (в соотношении 1:10) в качестве элюента.
Выход: 38,12 г (81,0% от теории) указанного выше в заголовке соединения в виде бесцветного и высоковязкого масла.
Элементный анализ:
г) 6-(1-O-α-D-карбонилметил-2,3,4,6-тетра-O-бензилманнопираноза)-2-[2-(N-этил-N-перфтороктилсульфонил)амино]ацетил-L-лизин
27,65 г (20,92 ммоля) полученного в примере 40в соединения растворяют в 250 мл метанола. Далее добавляют раствор 4,0 г (100,0 ммолей) гидроксида натрия в 10 мл дистиллированной воды и перемешивают в течение 3 ч при 50°С. Контроль за протеканием реакции тонкослойной хроматографией указывает на уже количественное омыление метилового эфира. После этого смесь досуха концентрируют в вакууме, полученный остаток растворяют в 300 мл этилового эфира уксусной кислоты и органическую фазу дважды экстрагируют разбавленным водным раствором лимонной кислоты порциями по 100 мл. После сушки над сульфатом натрия фильтруют и досуха концентрируют в вакууме. Полученный остаток хроматографируют на силикагеле (элюент:н-гексан/хлороформ/изопропанол в соотношении 15:10:1). Таким путем получают 24,31 г (88,9% от теории) указанного в заголовке соединения в виде бесцветного и вязкого масла.
Элементный анализ:
д) 6-(1-O-α-D-карбонилметилманнопираноза)-2-[2-(N-этил-N-перфтороктилсульфонил)амино]ацетил-L-лизин
20,0 г (15,30 ммоля) соединения, указанного в заголовке примера 40г, растворяют в смеси из 250 мл 2-пропанола и 25 мл воды и смешивают с 1,0 г палладиевого катализатора (10%-ный Pd на активированном угле). После этого гидрируют в течение 12 ч при комнатной температуре и давлении водорода одна атмосфера. Далее катализатор отфильтровывают и фильтрат досуха упаривают в вакууме. Остаток растворяют в 200 мл метанола и реакционный продукт осаждают смешением с диэтиловым эфиром общим количеством 800 мл. После отделения полученного таким путем твердого вещества вакуум-фильтрацией его сушат в вакууме при 50°С.
Выход: 14,32 г (99,0% от теории) аморфного твердого вещества.
Элементный анализ:
е) N-{2-гидроксипроп-3-ил-[1,4,7-трис(карбоксилатометил)-1,4,7,10-тетраазациклододекан-10-ил]}амид 6-(1-O-α-D-карбонилметилманнопираноза)-2-[2-(N-этил-N-перфтороктилсульфонил)амино]ацетил-L-лизина, Gd-комплекс
7,48 г (7,91 ммоля) соединения, указанного в заголовке примера 40д, при 40°С растворяют в 50 мл диметилсульфоксида и добавляют 1,00 г (8,70 моля) N-гидроксисукцинимида. Далее охлаждают до 20°С и добавляют 1,795 г (8,7 ммоля) дициклогексилкарбодиимида. После этого перемешивают в течение часа при 20°С, а затем в течение 4 ч при 40°С. Далее при этой температуре в течение 10 мин по каплям добавляют раствор 4,53 г (7,91 ммоля) гадолиниевого комплекса 10-(2-гидрокси-3-аминопропил)-4,7,10-трис(карбоксиметил)-1,4,7,10-тетраазациклододеканина [получение см. в WO 97/02051] в 20 мл диметилсульфоксида. Смесь перемешивают в течение часа при 40°С, а затем в течение ночи при комнатной температуре. Полученную таким путем суспензию затем смешивают с достаточным количеством ацетона до полного осаждения указанного выше в заголовке соединения, осадок отделяют вакуум-фильтрацией, сушат, растворяют в воде, отфильтровывают нерастворившуюся дициклогексилмочевину и фильтрат для обессоливания и для очистки от низкомолекулярных компонентов пропускают через ультрафильтрационную мембрану AMICON® YM-3 (предельная проницаемость: 3000 Да). Ретентат в завершение лиофилизуют.
Выход: 9,71 г (81,7% от теории) в виде бесцветного лиофилизата.
Н2О-содержание (при использовании реактива Карла Фишера): 3,97%.
Элементный анализ (в пересчете на безводное вещество):
Пример 41
а) Метиловый эфир 6-N-[1-O-α-D-(5-карбонил)пентил-2,3,4,6-тетра-O-бензилманнопираноза]-2N-[2-(N-этил-N-перфтороктилсульфонил)амино]ацетил-L-лизина
5,23 г (8,0 ммолей) описанного в примере 30в 5-(карбокси)пентил-2,3,4,6-тетра-О-бензил-α-D-маннопиранозида, 1,3 г (8,0 ммолей) 1-гидроксибензотриазола и 2,6 г (8,0 ммолей) тетрафторбората 2-(1Н-бензотриазол-1-ил)-1,1,3,3-тетраметилурония (ТБТУ; фирма Peboc Limited, Великобритания) растворяют в 75 мл ДМФ и перемешивают в течение 15 мин. Этот раствор затем смешивают с 5,16 мл (30 ммолей) N-этилдиизопропиламина и с 5,93 г (8,0 ммолей) описанного в примере 40б амина и перемешивают в течение 1,5 дней при комнатной температуре. Для переработки растворитель отгоняют в вакууме до получения сухого остатка, который затем хроматографируют на силикагеле (элюент:дихлорметан/этиловый эфир уксусной кислоты в соотношении 30:1; хроматографию проводят с использованием градиента растворителя, непрерывно увеличивая долю этилового эфира уксусной кислоты).
Выход: 9,70 г (88,0% от теории) указанного в заголовке соединения в виде бесцветного и высоковязкого масла.
Элементный анализ:
б) 6-N-[1-O-α-D-(5-карбонил)пентил-2,3,4,6-тетра-O-бензилманнопираноза]-2N-[2-(N-этил-N-перфтороктилсульфонил)амино]ацетил-L-лизин
9,0 г (12,40 ммоля) полученного в примере 41а соединения растворяют в 150 мл метанола. Далее добавляют раствор 2,48 г (62,0 ммоля) гидроксида натрия в 15 мл дистиллированной воды и перемешивают в течение 3 ч при 50°С. Контроль за протеканием реакции тонкослойной хроматографией указывает уже на количественное омыление метилового эфира по истечении указанного выше времени реакции. Затем смесь досуха концентрируют в вакууме, полученный остаток растворяют в 300 мл этилового эфира уксусной кислоты и органическую фазу дважды экстрагируют разбавленным водным раствором лимонной кислоты порциями по 100 мл. После сушки над сульфатом натрия фильтруют и досуха концентрируют в вакууме. Полученный остаток хроматографируют на силикагеле (элюент:н-гексан/хлороформ/изопропанол в соотношении 25:10:1). Таким путем получают 15,88 г (93,9% от теории) указанного в заголовке соединения в виде бесцветного и высоковязкого масла.
Элементный анализ:
в) 6-N-[1-O-α-D-(5-карбонил)пентилманнопираноза]-2N-[2-(N-этил-N-перфтороктилсульфонил)амино]ацетил-L-лизин
13,0 г (9,52 ммоля) соединения, указанного в заголовке примера 41б, растворяют в смеси из 150 мл 2-пропанола и 25 мл воды и добавляют 1,0 г палладиевого катализатора (10%-ного Pd на активированном угле). После этого гидрируют в течение 12 ч при давлении водорода 1 атмосфера и комнатной температуре. После этого катализатор отфильтровывают и фильтрат досуха упаривают в вакууме. Полученный остаток хроматографируют на силикагеле (элюент:н-гексан/хлороформ/изопропанол в соотношении 15:10:1). Таким путем получают 9,09 г (95,1% от теории) указанного в заголовке соединения в виде бесцветного и высоковязкого масла.
Элементный анализ:
г) N-{2-гидроксипроп-3-ил-[1,4,7-трис(карбоксилатометил)-1,4,7,10-тетраазациклододекан-10-ил]}амид 6-N-[1-O-α-D-(5-карбонил)пентилманнопираноза]-2N-[2-(N-этил-N-перфтороктилсульфонил)амино]ацетил-L-лизина, Gd-комплекс
7,93 г (7,91 ммоля) соединения, указанного в заголовке примера 41в, растворяют при 40°С в 75 мл диметилсульфоксида и смешивают с 1,00 г (8,70 моля) N-гидроксисукцинимида. Далее охлаждают до комнатной температуры и добавляют в целом 1,795 г (8,7 ммоля) дициклогексилкарбодиимида. После этого перемешивают в течение часа при 20°С, а затем в течение 4 ч при 40°С. К этому раствору активированного сложного эфира соединения, указанного в заголовке примера 41в, затем при 40°С в течение 10 мин по каплям добавляют раствор 4,53 г (7,91 ммоля) гадолиниевого комплекса 10-(2-гидрокси-3-аминопропил)-4,7,10-трис(карбоксиметил)-1,4,7,10-тетраазациклододеканина [получение см. в WO 97/02051] в 20 мл диметилсульфоксида. Смесь перемешивают в течение часа при 40°С, а затем в течение ночи при комнатной температуре. Полученную таким путем суспензию затем смешивают с достаточным количеством смеси ацетон/2-пропанол (в соотношении 2:1) до полного осаждения указанного выше в заголовке соединения, осадок отделяют вакуум-фильтрацией, после чего промывают этиловым эфиром уксусной кислоты, сушат, растворяют в воде, нерастворившуюся дициклогексилмочевину отфильтровывают и фильтрат для обессоливания и для очистки от низкомолекулярных компонентов пропускают через ультрафильтрационную мембрану AMICON® YM-3 (предельная проницаемость: 3000 Да). Ретентат в завершение лиофилизуют.
Выход: 9,71 г (78,8% от теории) в виде бесцветного лиофилизата.
Н2О-содержание (при использовании реактива Карла Фишера): 6,65%.
Элементный анализ (в пересчете на безводное вещество):
Пример 42
а) [1-(4-перфтороктилсульфонил)пиперазин]амид 6-N-{4-[2,3-бис(N,N-бис(трет-бутилоксикарбонилметил)амино)пропил]фенил}-3-оксапропионил-2-N-(1-α-D-карбонилметилманнопираноза)-L-лизина
5,25 г (7,72 ммоля) тетра-трет-бутилового эфира "Tyr-ЭДТК-карбоновой кислоты" и 781 мг (7,72 ммоля) триэтиламина растворяют в 50 мл метиленхлорида. Далее в течение 5 мин при -15°С по каплям добавляют раствор 1,16 г (8,5 ммоля) изобутилового эфира хлормуравьиной кислоты в 10 мл метилхлорида и перемешивают еще в течение 20 мин при -15°С. После этого раствор охлаждают до -25°С, в течение 30 мин по каплям добавляют раствор 7,07 г (7,72 ммоля) указанного в заголовке примера 30д соединения и 2,12 г (21,0 ммоль) триэтиламина в 70 мл тетрагидрофурана и затем перемешивают еще в течение 30 мин при -15°С, а затем в течение ночи при комнатной температуре. Для переработки растворитель отгоняют в вакууме и полученный маслянистый остаток растворяют в 250 мл хлороформа. Хлороформную фазу дважды экстрагируют 10%-ным водным раствором хлорида аммония порциями по 100 мл, органическую фазу сушат над сульфатом магния и досуха упаривают в вакууме. Остаток хроматографируют на силикагеле (элюент:метиленхлорид/этанол в соотношении 20:1).
Выход: 9,60 г (79,0% от теории) бесцветного и высоковязкого масла.
Элементный анализ:
б) [1-(4-перфтороктилсульфонил)пиперазин]амид 6-N-{4-[2,3-бис(N,N-бис(карбоксиметил)амино)пропил]фенил}-3-оксапропионил-2-N-(1-α-D-карбонилметилманнопираноза)-L-лизина
9,0 г (5,70 ммоля) полученного в примере 42а соединения растворяют в 150 мл метанола. Далее добавляют раствор 4,0 г (100,0 ммолей) гидроксида натрия в 25 мл дистиллированной воды и перемешивают в течение 6 ч при 60°С. Контроль за протеканием реакции тонкослойной хроматографией указывает на уже количественное омыление тетра-трет-бутилового эфира по истечении указанного выше времени реакции. После этого смесь досуха концентрируют в вакууме, полученный остаток растворяют в теплых условиях в 50 мл диметилсульфоксида и затем смешивают с достаточным количеством смеси ацетона с этиловым эфиром уксусной кислоты (в соотношении 1:1) до полного осаждения указанного выше в заголовке соединения, после чего полученный таким путем осадок отделяют вакуум-фильтрацией, тщательно промывают этиловым эфиром уксусной кислоты, сушат, растворяют в воде, значение рН содержащего продукт раствора устанавливают на 3,5 с помощью 1-молярной соляной кислоты, отфильтровывают возможно присутствующие нерастворимые компоненты и фильтрат для обессоливания и для очистки от низкомолекулярных компонентов пропускают через ультрафильтрационную мембрану AMICON® YM-3 (предельная проницаемость: 3000 Да). Ретентат в завершение лиофилизуют.
Выход: 6,76 г (87,6% от теории) в виде бесцветного лиофилизата.
Н2О-содержание (при использовании реактива Карла Фишера): 3,30%.
Элементный анализ (в пересчете на безводное вещество):
в) [1-(4-перфтороктилсульфонил)пиперазин]амид 6-N-{4-[2,3-бис(N,N-бис(карбоксилатометил)амино)пропил]фенил}-3-оксапропионил-2-N-(1-α-D-карбонилметилманнопираноза)-L-лизина, Mn-комплекс, динатриевая соль
3,0 г (2,22 ммоля) соединения, указанного в заголовке примера 42б, растворяют при температуре кипения в 150 мл смеси воды с этанолом (в соотношении 3:1) и при 80°С порциями смешивают с 0,25 г (2,22 ммоля) карбоната марганца(II). После этого полученный таким путем реакционный раствор в течение 5 ч кипятят с обратным холодильником. После охлаждения до комнатной температуры смесь растворителей полностью отгоняют в вакууме и полученный остаток растворяют в смеси 200 мл дистиллированной воды с н-бутанолом (в соотношении 1:1). Далее значение рН при интенсивном перемешивании устанавливают на 7,2 смешением с 1н. едким натром. После полной отгонки н-бутанола в вакууме оставшуюся водную фазу для обессоливания и для очистки от низкомолекулярных компонентов пропускают через ультрафильтрационную мембрану AMICON® YM-3 (предельная проницаемость: 3000 Да). Ретентат в завершение лиофилизуют.
Выход: 3,19 г (99,0% от теории) в виде бесцветного лиофилизата.
Н2О-содержание (при использовании реактива Карла Фишера): 5,08%.
Элементный анализ (в пересчете на безводное вещество):
Пример 43
а) [1-(4-перфтороктилсульфонил)пиперазин]моноамид 3-бензилоксикарбониламиноглутаровой кислоты
Перемешанный раствор 25,0 г (94,96 ммоля) ангидрида 3-N-(бензилоксикарбонил)глутаровой кислоты [синтез согласно Hatanaka Minoru, Yamamoto Yu-ichi, Nitta Hajime, Ishimaru Toshiyasu, TELEAY; Tetrahedron Lett., EN, 22, 39 (1981), сс.3883-3886] в 150 мл абсолютного тетрагидрофурана при перемешивании по каплям смешивают с раствором 53,97 г (95,0 ммолей) 1-перфтороктилсульфонилпиперазина в 150 мл тетрагидрофурана и полученный таким путем реакционный раствор в течение 12 ч кипятят с обратным холодильником. После охлаждения до комнатной температуры концентрируют досуха и полученный маслянистый остаток очищают на силикагеле с использованием дихлорметана/2-пропанола (в соотношении 20:1) в качестве элюента.
Выход: 75,80 г (96,0% от теории) указанного выше в заголовке соединения в виде бесцветного и вязкого масла.
Элементный анализ:
б) [1-(4-перфтороктилсульфонил)пиперазин]моноамид 3-аминоглутаровой кислоты
31,50 г (37,88 ммоля) полученного согласно примеру 43б соединения растворяют в 300 мл этанола, смешивают с 2,5 г катализатора Перлмана (20%-ный Pd/C) и гидрируют до количественного поглощения водорода при давлении водорода 1 атмосфера. После этого катализатор удаляют вакуум-фильтрацией, промывают этанолом и досуха концентрируют в вакууме. Таким путем получают указанное в заголовке соединение в виде бледно-желтого вязкого масла.
Выход: 25,22 г (95,5% от теории).
Элементный анализ:
в) [1-(4-перфтороктилсульфонил)пиперазин]моноамид 3-N-(1-α-D-карбонилметил-2,3,4,6-тетра-O-бензилманнопираноза)глутаровой кислоты
21,52 г (18,96 ммоля) 1-карбоксиметилокси-2,3,4,6-тетра-O-бензил-α-D-маннопиранозида [получение по методу, описанному в патенте DE 19728954 С1] растворяют при комнатной температуре в 100 мл абсолютного диметилформамида и при 0°С смешивают с 2,56 г (22,2 ммоля) N-гидроксисукцинимида, а затем с 4,55 г (22,2 ммоля) дициклогексилкарбодиимида. После проведения реакции в течение 60 мин при 0°С и в течение 3 ч при 22°С нерастворившуюся дициклогексилмочевину отфильтровывают и полученный таким путем прозрачный раствор активированного эфира указанного выше в заголовке соединения при 0°С по каплям медленно добавляют к перемешанному раствору 13,22 г (18,96 ммоля) соединения из примера 43б в 100 мл диметилформамида. После проведения реакции в течение 12 ч при комнатной температуре растворитель отгоняют в вакууме и полученный остаток растворяют в 300 мл этилового эфира уксусной кислоты, после чего мочевину отфильтровывают и органический фильтрат дважды промывают насыщенным раствором гидрокарбоната натрия порциями по 100 мл, однократно 100 мл 10%-ного водного растворя лимонной кислоты и однократно 200 мл воды. После сушки органической фазы над сульфатом натрия соль отделяют вакуум-фильтрацией и этиловый эфир уксусной кислоты отгоняют в вакууме. Полученный маслянистый остаток очищают на силикагеле с использованием этилового эфира уксусной кислоты/н-гексана (в соотношении 1:15) в качестве элюента.
Выход: 21,39 г (88,3% от теории) указанного выше в заголовке соединения в виде бесцветного и высоковязкого масла.
Элементный анализ:
г) [1-(4-перфтороктилсульфонил)пиперазин]моноамид 3-N-(1-α-D-карбонилметилманнопираноза)глутаровой кислоты
19,55 г (15,30 ммоля) соединения, указанного в заголовке примера 43в, растворяют в смеси из 250 мл 2-пропанола и 25 мл воды и смешивают с 1,5 г палладиевого катализатора (10%-ного Pd на активированном угле). После этого гидрируют в течение 12 ч при комнатной температуре и давлении водорода одна атмосфера. Затем катализатор отфильтровывают и фильтрат досуха упаривают в вакууме. Остаток растворяют в 200 мл метанола и реакционный продукт осаждают смешением с диэтиловым эфиром общим количеством 800 мл. После отделения полученного таким путем твердого вещества вакуум-фильтрацией его сушат в вакууме при 40°С.
Выход: 17,49 г (97,5% от теории) аморфного твердого вещества.
Элементный анализ:
д) 3-N-(1-α-O-карбонилметилманнопираноза)глутаровая кислота-[1-(4-перфтороктилсульфонил)пиперазин]амид-5-N-{2-гидроксипроп-3-ил-[1,4,7-трис(карбоксилатометил)-1,4,7,10-тетраазациклододекан-10-ил]} амид, Gd-комплекс
14,43 г (15,84 ммоля) соединения, указанного в заголовке примера 43г, и 0,67 г безводного хлорида лития (15,84 ммоля) при 40°С растворяют при перемешивании в 100 мл абсолютного диметилсульфоксида и при этой температуре смешивают с N-гидроксисукцинимидом общим количеством 1,82 г (15,84 ммоля) и с раствором 9,08 г (15,84 ммоля) гадолиниевого комплекса 10-(2-гидрокси-3-аминопропил)-4,7,10-трис(карбоксиметил)-1,4,7,10-тетраазациклододеканина [получение см. в WO 97/02051] в 50 мл диметилсульфоксида. После охлаждения до комнатной температуры реакционный раствор смешивают с 3,27 г (15,84 ммоля) N,N'-дициклогексилкарбодиимида и перемешивают в течение 12 ч при комнатной температуре. Полученную суспензию затем смешивают с достаточным количеством ацетона до полного осаждения указанного выше в заголовке соединения, осадок отделяют вакуум-фильтрацией, сушат, растворяют в воде, отфильтровывают нерастворившуюся дициклогексилмочевину и фильтрат для обессоливания и для одновременной очистки от возможно еще присутствующих в нем низкомолекулярных компонентов подвергают ультрафильтрации через ультрафильтрационную мембрану AMICON® YM-3 (предельная проницаемость: 3000 Да). Ретентат в завершение лиофилизуют.
Выход: 18,71 г (80,2% от теории) в виде бесцветного лиофилизата.
Н2О-содержание (при использовании реактива Карла Фишера): 4,87%.
Элементный анализ (в пересчете на безводное вещество):
Пример 44
а) 1,7-бис(бензилоксикарбонил)-4-{3-оксапентан-1,5-дикарбоновая кислота-1-оил-5-[1-(4-перфтороктилсульфонил)пиперазин]амид}-10-[2,6-N,N'-бис(1-O-α-D-карбонилметил-2,3,4,6-тетра-O-бензилманнопираноза)]-L-лизил-1,4,7,10-тетраазациклододекан
К раствору 27,0 г (24,4 ммоля) полученного в примере 35а вторичного амина в смеси из 150 мл тетрагидрофурана и 15 мл хлороформа при 0°С в атмосфере азота добавляют 33,04 г (25,0 ммолей) указанного в заголовке примера 38в соединения, растворенного в 250 мл тетрагидрофурана. После этого при 0°С порциями добавляют в целом 18,0 г (36,6 ммоля) ЭЭДХ [2-этокси-1-этоксикарбонил-1,2-дигидрохинолин] и оставляют на ночь для перемешивания при комнатной температуре. Затем смесь досуха концентрируют в вакууме и полученное масло хроматографируют на силикагеле (элюент:н-гексан/изопропанол в соотношении 25:1). Таким путем получают 45,87 г (78,0% от теории, в пересчете на количество используемого вторичного амина) указанного в заголовке соединения в виде бесцветного масла.
Элементный анализ:
б) 1-{3-оксапентан-1,5-дикарбоновая кислота-1-оил-5-[1-(4-перфтороктилсульфонил)пиперазин]амид}-7-[2,6-N,N'-бис(1-O-α-D-карбонилметилманнопираноза)]-L-лизил-1,4,7,10-тетраазациклододекан
24,1 г (10,0 ммолей) полученного в примере 44а и указанного в его заголовке соединения растворяют в 250 мл этанола и смешивают с 1,4 г катализатора Перлмана (20%-ный Pd/C). После этого гидрируют до количественного поглощения водорода, после чего катализатор удаляют вакуум-фильтрацией, тщательно промывают этанолом и досуха концентрируют в вакууме. Продукт получают в виде высоковязкого масла желтого цвета.
Выход: 12,80 г (90,1% от теории).
Элементный анализ:
в) 1-{3-оксапентан-1,5-дикарбоновая кислота-1-оил-5-[1-(4-перфтороктилсульфонил)пиперазин]амид}-7-[2,6-N,N-бис(1-O-α-D-карбонилметилманнопираноза)]-L-лизил-4,10-бис[1,4,7-трис(карбоксилатометил)-10-(3-аза-4-оксо-5-метил-5-илпентаноил)]-1,4,7,10-тетраазациклододекан, дигадолиниевый комплекс
5,54 г (8,8 ммоля, 2,2 молярных эквивалента в пересчете на количество используемого аминового компонента из примера 44б) описанного в заявке DE 19728954 С1 в примере 31h Gd-комплекса 10-(4-карбокси-1-метил-2-оксо-3-азабутил)-1,4,7,10-тетраазациклододекан-1,4,7-триуксусной кислоты и 0,37 г (8,8 ммоля) безводного хлорида лития при 40°С растворяют при перемешивании в 60 мл абсолютного диметилсульфоксида и при этой температуре смешивают с N-гидроксисукцинимидом общим количеством 1,01 г (8,8 ммоля) и с 5,68 г (4,0 ммоля) указанного в заголовке примера 44б соединения, растворенного в 40 мл абсолютного диметилсульфоксида. После охлаждения до комнатной температуры реакционный раствор смешивают с 1,82 г (8,8 ммоля) N,N'-дициклогексилкарбодиимида и перемешивают в течение 12 ч при комнатной температуре. Полученную суспензию затем смешивают с достаточным количеством ацетона до полного осаждения указанного выше в заголовке соединения, осадок отделяют вакуум-фильтрацией, сушат, растворяют в воде, отфильтровывают нерастворившуюся дициклогексилмочевину и фильтрат для обессоливания и для очистки от низкомолекулярных компонентов пропускают через ультрафильтрационную мембрану AMICON® YM-3 (предельная проницаемость: 3000 Да). Ретентат в завершение лиофилизуют.
Выход: 8,52 г (80,6% от теории; в пересчете на количество используемого диаминового компонента) в виде бесцветного лиофилизата.
H2О-содержание (при использовании реактива Карла Фишера): 6,09%.
Элементный анализ (в пересчете на безводное вещество):
Пример 45
а) 1,7-бис(бензилоксикарбонил)-4-{3-оксапентан-1,5-дикарбоновая кислота-1-оил-5-[1-(4-перфтороктилсульфонил)пиперазин]амид}-10-{2,6-N,N'-бис(1-O-α-D-(5-карбонил)пентил-2,3,4,6-тетра-О-бензилманнопираноза)}-L-лизил-1,4,7,10-тетраазациклододекан
К раствору 27,0 г (24,4 ммоля) полученного в примере 35а вторичного амина в смеси из 150 мл тетрагидрофурана и 15 мл хлороформа при 0°С в атмосфере азота добавляют 35,80 г (25,0 ммолей) указанного в заголовке примера 37д соединения, растворенного в 250 мл тетрагидрофурана. После этого при 0°С порциями добавляют в целом 18,0 г (36,6 ммоля) ЭЭДХ [2-этокси-1-этоксикарбонил-1,2-дигидрохинолин] и оставляют на ночь для перемешивания при комнатной температуре. Затем смесь досуха концентрируют в вакууме и полученное масло хроматографируют на силикагеле (элюент:н-гексан/изопропанол в соотношении 20:1). Таким путем получают 49,48 г (80,4% от теории, в пересчете на количество используемого вторичного амина) указанного в заголовке соединения в виде бесцветного масла.
Элементный анализ:
б) 1-{3-оксапентан-1,5-дикарбоновая кислота- 1-оил-5-[1-(4-перфтороктилсульфонил)пиперазин]амид}-7-[2,6-N,N'-бис(1-O-α-D-(5-карбонил)пентилманнопираноза)]-L-лизил-1,4,7,10-тетраазациклододекан
25,2 г (10,0 ммолей) полученного в примере 45а и указанного в его заголовке соединения растворяют в 250 мл этанола и смешивают с 1,8 г катализатора Перлмана (20%-ный Pd/C). После этого гидрируют до количественного поглощения водорода, после чего катализатор удаляют вакуум-фильтрацией, тщательно промывают этанолом и досуха концентрируют в вакууме. Продукт получают в виде высоковязкого масла желтого цвета.
Выход: 14,11 г (92,5% от теории).
Элементный анализ:
в) 1-{3-оксапентан-1,5-дикарбоновая кислота-1-оил-5-[1-(4-перфтороктилсульфонил)пиперазин]амид}-7-[2,6-N,N'-бис(1-O-α-D-(5-карбонил)пентилманнопираноза)]-L-лизил-4,10-бис[1,4,7-трис(карбоксилатометил)-10-(3-аза-4-оксо-5-метил-5-илпентаноил)]-1,4,7,10-тетраазациклододекан, дигадолиниевый комплекс
5,54 г (8,8 ммоля, 2,2 молярных эквивалента в пересчете на количество используемого аминового компонента из примера 45б) описанного в заявке DE 19728954 С1 в примере 31h Gd-комплекса 10-(4-карбокси-1-метил-2-оксо-3-азабутил)-1,4,7,10-тетраазациклододекан-1,4,7-триуксусной кислоты и 0,37 г (8,8 ммоля) безводного хлорида лития при 40°С растворяют при перемешивании в 60 мл абсолютного диметилсульфоксида и при этой температуре смешивают с N-гидроксисукцинимидом общим количеством 1,01 г (8,8 ммоля) и с 6,10 г (4,0 ммоля) указанного в заголовке примера 45б соединения, растворенного в 40 мл абсолютного диметилсульфоксида. После охлаждения до комнатной температуры реакционный раствор смешивают с 1,82 г (8,8 ммоля) N,N'-дициклогексилкарбодиимида и перемешивают в течение 12 ч при комнатной температуре. Полученную суспензию затем смешивают с достаточным количеством ацетона до полного осаждения указанного выше в заголовке соединения, осадок отделяют вакуум-фильтрацией, сушат, растворяют в воде, отфильтровывают нерастворившуюся дициклогексилмочевину и фильтрат для обессоливания и для очистки от низкомолекулярных компонентов пропускают через ультрафильтрационную мембрану AMICON® YM-3 (предельная проницаемость: 3000 Да). Ретентат в завершение лиофилизуют.
Выход: 9,26 г (84,0% от теории; в пересчете на количество используемого диаминового компонента) в виде бесцветного лиофилизата.
Н2О-содержание (при использовании реактива Карла Фишера): 5,89%.
Элементный анализ (в пересчете на безводное вещество):
Пример 46
а) [1-(4-перфтороктилсульфонил)пиперазин]амид 6-N-трет-бутилоксикарбонил-2-N-бензилоксикарбонил-L-лизина
19,02 г (50,0 ммолей) α-N-(бензилоксикарбонил)-ε-N'-(трет-бутилоксикарбонил)-L-лизина (поставляется фирмой Bachem) растворяют в 150 мл абсолютного тетрагидрофурана. Далее при 0°С по каплям добавляют 8,31 г (50,0 ммолей) карбонилдиимидазола и 5,03 г (50,0 ммолей) триэтиламина, растворенных в 75 мл сухого тетрагидрофурана, и в течение 10 мин перемешивают при этой температуре. Затем при 0°С по каплям добавляют раствор 48,42 г (50,0 ммолей) перфтороктилсульфонилпиперазина и 5,03 г (50,0 ммолей) триэтиламина в 250 мл сухого тетрагидрофурана. После перемешивания в течение ночи тетрагидрофуран отгоняют в вакууме и полученное масло хроматографируют на силикагеле (элюент:н-гексан/изопропанол в соотношении 15:1). Таким путем получают 49,48 г (80,4% от теории, в пересчете на количество используемого вторичного амина) указанного в заголовке соединения в виде бесцветного масла.
Элементный анализ (в пересчете на безводное вещество):
б) [1-(4-перфтороктилсульфонил)пиперазин]амид 6-N-трет-бутилоксикарбонил-L-лизина
30,0 г (32,2 ммоля) соединения, указанного в заголовке примера 46а, растворяют в 300 мл изопропанола и смешивают с 1,5 г катализатора Перлмана (20%-ный гидроксид палладия на угле). После этого гидрируют в течение 10 ч при комнатной температуре, при этом контроль за протеканием реакции тонкослойной хроматографией указывает уже на количественное гидрогенолитическое отщепление бензилоксикарбонилзащитной группы по истечении указанного выше времени реакции. Затем катализатор отфильтровывают и фильтрат досуха концентрируют в вакууме. Полученный остаток хроматографируют на силикагеле (элюент:н-гексан/изопропанол в соотношении 25:1). Таким путем получают 25,13 г (98,0% от теории) указанного в заголовке соединения в виде бесцветного масла.
Элементный анализ:
в) [1-(4-перфтороктилсульфонил)пиперазин]амид 6-N-трет-бутилоксикарбонил-2-N-[1-S-α-D-(2-карбонил)этил-2,3,4,6-тетра-O-ацетилманнопираноза]-L-лизина
15,53 г (35,60 ммолей) 3-(2,3,4,6-тетра-O-ацетил-1-тио-α-D-маннопиранозил)пропионовой кислоты (получение согласно J. Haensler и др., Bioconjugate Chem. 4 (1993), с.85; Chipowsky S. и Lee Y.C, Synthesis of 1-thio-aldosides. Carbohydrate Research 31 (1973), сс.339-346), а также 3,60 г (35,60 ммоля) триэтиламина растворяют в 300 мл сухого тетрагидрофурана. После охлаждения реакционного раствора до температуры в пределах от -15 до -20°С при этой температуре по каплям при перемешивании медленно добавляют раствор 4,92 г (35,60 ммоля) изобутилового эфира хлормуравьиной кислоты в 75 мл сухого тетрагидрофурана, при этом скорость капельного добавления должна быть такой, чтобы внутренняя температура не превышала -10°С. После проведения реакции в течение 15 мин при -15°С по каплям при 20°С медленно добавляют раствор 28,35 г (35,60 ммоля) указанного в заголовке примера 42б соединения и 3,60 г (35,60 ммоля) триэтиламина в 200 мл сухого тетрагидрофурана. После проведения реакции в течение часа при -15°С, а затем в течение двух часов при комнатной температуре реакционный раствор досуха концентрируют в вакууме. Полученный остаток растворяют в 250 мл этилового эфира уксусной кислоты и дважды промывают насыщенным раствором гидрокарбоната натрия порциями по 100 мл и однократно водой порцией 200 мл. После сушки органической фазы над сульфатом натрия соль отделяют вакуум-фильтрацией и этиловый эфир уксусной кислоты отгоняют в вакууме. Полученный маслянистый остаток хроматографируют на силикагеле с использованием этилового эфира уксусной кислоты/н-гексана (в соотношении 1:25) в качестве элюента.
Выход: 34,21 г (79,1% от теории) указанного выше в заголовке соединения в виде бесцветного и высоковязкого масла.
Элементный анализ:
г) [1-(4-перфтороктилсульфонил)пиперазин]амид 6-N-трет-бутилоксикарбонил-2-N-[1-S-α-D-(2-карбонил)этилманнопираноза]-L-лизина
29,93 г (24,64 ммоля) соединения, указанного в заголовке примера 46в, суспендируют в 400 мл абсолютного метанола и при 5°С смешивают с каталитическим количеством метанолята натрия. После проведения реакции в течение 3 ч при комнатной температуре контроль за протеканием реакции тонкослойной хроматографией (элюент:хлороформ/метанол в соотношении 9:1) указывает на уже количественное превращение. Для переработки прозрачный реакционный раствор нейтрализуют смешением с катионообменной смолой Amberlite® IR 120 (Н+-форма), ионит удаляют вакуум-фильтрацией и полученный таким путем метанольный фильтрат отгоняют в вакууме до получения сухого остатка. Полученный аморфный остаток очищают хроматографией на силикагеле с использованием 2-пропанола/этилового эфира уксусной кислоты/н-гексана (в соотношении 1:1:15) в качестве элюента.
Выход: 23,42 г (90,8% от теории) бесцветного вязкого масла.
Элементный анализ:
д) [1-(4-перфтороктилсульфонил)пиперазин]амид 2-N-[1-S-α-D-(2-карбонил)этилманнопираноза]-L-лизина
20,93 г (20,0 ммолей) указанного в заголовке примера 46г соединения при 0°С растворяют при интенсивном перемешивании в смеси из 50 мл трифторуксусной кислоты и 100 мл дихлорметана и перемешивают в течение 10 мин при этой температуре. После этого смесь досуха упаривают в вакууме и остаток растворяют в 150 мл воды. Значение рН этого водного, содержащего продукт раствора устанавливают на 9,5, добавляя по каплям 2-молярный водный едкий натр. Далее водный, содержащий продукт раствор для обессоливания и для одноврменной очистки от возможно еще присутствующих в нем низкомолекулярных компонентов подвергают ультрафильтрации через ультрафильтрационную мембрану AMICON® YM-3 (предельная проницаемость: 3000 Да). Ретентат в завершение лиофилизуют.
Выход: 17,79 г (94,2% от теории) свободного амина в виде бесцветного лиофилизата.
Н2О-содержание (при использовании реактива Карла Фишера): 3,09%.
Элементный анализ (в пересчете на безводное вещество):
e) [1-(4-перфтороктилсульфонил)пиперазин]амид 2-N-[1-S-α-D-(2-карбонил)этилманнопираноза]-6-N-[1,4,7-трис(карбоксилатометил)-10-(3-аза-4-оксо-5-метил-5-илпентаноил)-1,4,7,10-тетраазациклододекан]-L-лизина, гадолиниевый комплекс
5,54 г (8,8 ммоля, 2,2 молярных эквивалента в пересчете на количество используемого аминового компонента из примера 46д) описанного в заявке DE 19728954 С1 в примере 31h Gd-комплекса 10-(4-карбокси-1-метил-2-оксо-3-азабутил)-1,4,7,10-тетраазациклододекан-1,4,7-триуксусной кислоты и 0,37 г безводного хлорида лития (8,8 ммоля) при 40°С растворяют при перемешивании в 60 мл абсолютного диметилсульфоксида и при этой температуре смешивают с N-гидроксисукцинимидом общим количеством 1,01 г (8,8 ммоля) и с 3,78 г (4,0 ммоля) указанного в заголовке примера 46д соединения, растворенного в 40 мл абсолютного диметилсульфоксида. После охлаждения до комнатной температуры реакционный раствор смешивают с 1,82 г (8,8 ммоля) N,N'-дициклогексилкарбодиимида и перемешивают в течение 12 ч при комнатной температуре. Полученную суспензию затем смешивают с достаточным количеством ацетона до полного осаждения указанного выше в заголовке соединения, осадок отделяют вакуум-фильтрацией, растворяют в воде, отфильтровывают нерастворившуюся дициклогексилмочевину и фильтрат для обессоливания и для очистки от низкомолекулярных компонентов пропускают через ультрафильтрационную мембрану AMICON® YM-3 (предельная проницаемость: 3000 Да). Ретентат в завершение лиофилизуют.
Выход: 5,17 г (83,0% от теории) в виде бесцветного лиофилизата.
Н2О-содержание (при использовании реактива Карла Фишера): 4,43%.
Элементный анализ (в пересчете на безводное вещество):
Пример 47
а) [1-(4-перфтороктилсульфонил)пиперазин]амид 6-N-бензилоксикарбонил-2-N-(1-O-β-D-карбонилметил-2,3,4,6-тетра-O-бензилглюкопираноза)-L-лизина
8,02 г (13,4 ммоля) соединения, указанного в заголовке описанного в заявке DE 19728954 С1 примера 46а [1-карбоксиметилокси-2,3,4,6-тетра-O-бензил-β-D-глюкопиранозид] и 3,24 г (28,14 ммоля) N-гидроксисукцинимида растворяют в 100 мл диметилформамида и при 0°С порциями смешивают с N,N'-дициклогексилкарбодиимидом общим количеством 5,80 г (28,14 ммоля). Затем перемешивают в течение 3 ч при этой температуре. К полученному таким путем раствору активированного сложного эфира по каплям добавляют охлажденный до 0°С раствор 11,13 г (13,4 ммоля) указанного в заголовке примера 21в соединения, растворенного в 50 мл диметилформамида, и перемешивают в течение 2 ч при 0°С, а затем в течение 12 ч при комнатной температуре. Для переработки выпавшую в осадок дициклогексилмочевину отфильтровывают и затем отгоняют растворитель до получения сухого остатка. Полученный таким путем остаток затем хроматографируют на силикагеле (элюент:дихлорметан/этанол в соотношении 20:1; хроматографию проводят с использованием градиента растворителя, непрерывно увеличивая долю этанола).
Выход: 12,67 г (67,0% от теории) указанного в заголовке соединения в виде бесцветного и высоковязкого масла.
Элементный анализ:
б) [1-(4-перфтороктилсульфонил)пиперазин]амид 2-N-(1-O-β-D-карбонилметилглюкопираноза)-L-лизина
11,52 г (8,17 ммоля) полученного в примере 47а соединения растворяют в 100 мл этанола, смешивают с 0,5 г катализатора Перлмана (20%-ный Pd/C) и гидрируют при комнатной температуре в атмосфере водорода (1 атм) до тех пор, пока не прекратится поглощение водорода. После этого катализатор удаляют вакуум-фильтрацией, тщательно промывают этанолом (трижды порциями примерно по 40 мл) и досуха концентрируют в вакууме. Таким путем получают указанное в заголовке соединение в виде высоковязкого и бесцветного масла.
Выход: 7,36 г (98,4% от теории).
Элементный анализ:
в) [1-(4-перфтороктилсульфонил)пиперазин]амид 2-N-(1-O-β-D-карбонилметилглюкопираноза)-6-N-[1,4,7-трис(карбоксилатометил)-10-(аза-4-оксо-5-метил-5-илпентаноил)-1,4,7,10-тетраазациклододекан]-L-лизина, Gd-комплекс
9,98 г (15,84 ммоля, 2,2 молярных эквивалента в пересчете на количество используемого аминового компонента из примера 47б) описанного в заявке DE 19728954 С1 в примере 31h Gd-комплекса 10-(4-карбокси-1-метил-2-оксо-3-азабутил)-1,4,7,10-тетраазациклододекан-1,4,7-триуксусной кислоты и 0,67 г (15,84 ммоля) безводного хлорида лития при 40°С растворяют при перемешивании в 80 мл абсолютного диметилсульфоксида и при этой температуре смешивают с N-гидроксисукцинимидом общим количеством 1,82 г (15,84 ммоля) и с 7,25 г (7,19 ммоля) указанного в заголовке примера 47б соединения, растворенного в 30 мл абсолютного диметилсульфоксида. После охлаждения до комнатной температуры реакционный раствор смешивают с 3,27 г (15,84 ммоля) N,N'-дициклогексилкарбодиимида и перемешивают в течение 12 ч при комнатной температуре. Полученную суспензию затем смешивают с достаточным количеством ацетона до полного осаждения указанного выше в заголовке соединения, осадок отделяют вакуум-фильтрацией, растворяют в воде, отфильтровывают нерастворившуюся дициклогексилмочевину и фильтрат для обессоливания и для очистки от низкомолекулярных компонентов пропускают через ультрафильтрационную мембрану AMICON® YM-3 (предельная проницаемость: 3000 Да). Ретентат в завершение лиофилизуют.
Выход: 9,11 г (83,0% от теории) в виде бесцветного лиофилизата.
Н2О-содержание (при использовании реактива Карла Фишера): 4,02%.
Элементный анализ (в пересчете на безводное вещество):
Пример 48
а) [1-(4-перфтороктилсульфонил)пиперазин]амид 2-N-трифторацетил-L-лизина
10,0 г (11,46 ммоля) полученного в примере 21б соединения растворяют в 100 мл этанола, смешивают с 1,0 г катализатора Перлмана (20%-ный Pd/C) и гидрируют до количественного поглощения водорода. После этого катализатор удаляют вакуум-фильтрацией, промывают этанолом и досуха концентрируют в вакууме. Таким путем получают указанное в заголовке соединение в виде вязкого и бесцветного масла.
Выход: 8,85 г (97,5% от теории).
Элементный анализ:
б) [1-(4-перфтороктилсульфонил)пиперазин]амид 2-N-трифторацетил-6-N-[1-O-α-D-(5-карбонил)пентил-2,3,4,6-тетра-O-бензилманнопираноза]-L-лизина
К охлажденному до 0°С раствору 29,0 г (36,6 ммоля) соединения, указанного в заголовке примера 48а, и 4,05 г (40,26 ммоля) триэтиламина в 100 мл диметилформамида по каплям добавляют раствор 27,51 г (36,6 ммоля) указанного в заголовке примера 37в соединения в 150 мл диметилформамида. По завершении этого добавления перемешивают еще в течение часа при 0°С, а затем в течение ночи при комнатной температуре. Смесь упаривают в вакууме досуха и остаток растворяют в 300 мл этилового эфира уксусной кислоты. Нерастворившиеся компоненты отфильтровывают и фильтрат дважды промывают 5%-ным водным раствором соды порциями по 100 мл. Органическую фазу сушат над сульфатом магния и досуха упаривают в вакууме. Остаток хроматографируют на силикагеле (элюент:н-гексан/изопропанол в соотношении 25:1). Таким путем получают 42,05 г (80,4% от теории) указанного в заголовке соединения в виде бесцветного масла.
Элементный анализ;
в) [1-(4-перфтороктилсульфонил)пиперазин]амид 6-N-[1-O-α-D-(5-карбонил)пентил-2,3,4,6-тетра-O-бензилманнопираноза]-L-лизина
20,0 г (14,0 ммолей) полученного в примере 48б соединения растворяют в 150 мл этанола. Далее добавляют раствор 2,8 г (70,0 ммолей) гидроксида натрия в 25 мл дистиллированной воды и перемешивают в течение 0,5 ч при 50°С. Согласно хроматограмме тонкослойной хроматографии к этому моменту времени происходит количественное отщепление защитной группы. Затем смесь досуха концентрируют в вакууме и путем многократной совместной перегонки с этанолом удаляют следы воды. Остаток хроматографируют на силикагеле (элюент:н-гексан/изопропанол в соотношении 20:1). Таким путем получают 16,66 г (89,3% от теории) указанного в заголовке соединения в виде бесцветного масла.
Элементный анализ:
г) [1-(4-перфтороктилсульфонил)пиперазин]амид 6-N-[1-O-α-D-(5-карбонил)пентилманнопираноза]-L-лизина
15,0 г (11,25 ммоля) полученного в примере 48в соединения растворяют в 150 мл смеси этанола и воды в соотношении 10:1 и смешивают с 1,0 г катализатора Перлмана (20%-ный Pd/C). После этого гидрируют до количественного поглощения водорода при комнатной температуре и при давлениии водорода одна атмосфера. После этого катализатор удаляют вакуум-фильтрацией, остаток промывают смесью этанола и воды (в соотношении 10:1) и досуха концентрируют в вакууме. Таким путем получают указанное в заголовке соединение в виде вязкого и бесцветного масла.
Выход: 10,77 г (98,4% от теории).
Элементный анализ:
д) [1-(4-перфтороктилсульфонил)пиперазин]амид 6-N-[1-O-α-D-(5-карбонил)пентилманнопираноза]-2-N-[1,4,7-трис(карбоксилатометил)-10-(3-аза-4-оксо-5-метил-5-илпентаноил)-1,4,7,10-тетраазациклододекан]-L-лизина, Gd-комплекс
5,54 г (8,8 ммоля, 2,2 молярных эквивалента в пересчете на количество используемого аминового компонента из примера 48г) описанного в заявке DE 19728954 С1 в примере 31h Gd-комплекса 10-(4-карбокси-1-метил-2-оксо-3-азабутил)-1,4,7,10-тетраазациклододекан-1,4,7-триуксусной кислоты и 0,37 г (8,8 ммоля) безводного хлорида лития при 40°С растворяют при перемешивании в 60 мл абсолютного диметилсульфоксида и при этой температуре смешивают с N-гидроксисукцинимидом общим количеством 1,01 г (8,8 ммоля) и с 3,89 г (4,0 ммоля) указанного в заголовке примера 48г соединения, растворенного в 60 мл абсолютного диметилсульфоксида. После охлаждения до комнатной температуры реакционный раствор смешивают с 1,82 г (8,8 ммоля) N,N'-дициклогексилкарбодиимида и перемешивают в течение 12 ч при комнатной температуре. Полученную суспензию затем смешивают с достаточным количеством ацетона до полного осаждения указанного выше в заголовке соединения, осадок отделяют вакуум-фильтрацией, растворяют в воде, отфильтровывают нерастворившуюся дициклогексилмочевину и фильтрат для обессоливания и для очистки от низкомолекулярных компонентов пропускают через ультрафильтрационную мембрану AMICON® YM-3 (предельная проницаемость: 3000 Да). Ретентат в завершение лиофилизуют.
Выход: 4,81 г (75,9% от теории) в виде бесцветного лиофилизата.
Н2О-содержание (при использовании реактива Карла Фишера): 8,98%.
Элементный анализ (в пересчете на безводное вещество):
Пример 49
а) 1,7-бис(бензилоксикарбонил)-4-(1-O-β-D-карбонилметил-2,3,4,6-тетра-O-бензилгалактопираноза)-10-{3-оксапентан-1,5-дикарбоновая кислота-1-оил-5-[1-(4-перфтороктилсульфонил)пиперазин]амид}-1,4,7,10-тетраазациклододекан
К раствору 27,0 г (24,4 ммоля) полученного в примере 35а вторичного амина в смеси из 150 мл тетрагидрофурана и 15 мл хлороформа при 0°С в атмосфере азота добавляют 35,80 г (25,0 ммолей) указанного в заголовке примера 37д соединения, растворенного в 250 мл тетрагидрофурана. После этого при 0°С порциями добавляют в целом 18,0 г (36,6 ммоля) ЭЭДХ [2-этокси-1-этоксикарбонил-1,2-дигидрохинолин] и оставляют на ночь для перемешивания при комнатной температуре. Затем досуха концентрируют в вакууме и полученное масло хроматографируют на силикагеле (элюент:н-гексан/изопропанол в соотношении 20:1). Таким путем получают 32,11 г (78,0% от теории, в пересчете на количество используемого вторичного амина) указанного в заголовке соединения в виде бесцветного масла.
Элементный анализ:
б) 1-(1-O-β-D-карбонилметилгалактопираноза)-7-{3-оксапентан-1,5-дикарбоновая кислота-1-оил-5-[1-(4-перфтороктилсульфонил)пиперазин]амид}-1,4,7,10-тетраазациклододекан
30,0 г (17,77 ммоля) полученного в примере 49а и указанного в его заголовке соединения растворяют в 250 мл этанола и смешивают с 3,0 г катализатора Перлмана (20%-ного Pd/C). После этого гидрируют до количественного поглощения водорода, после чего катализатор удаляют вакуум-фильтрацией, тщательно промывают этанолом и досуха концентрируют в вакууме. Продукт получают в виде высоковязкого масла желтого цвета.
Выход: 17,89 г (95,1% от теории).
Элементный анализ:
в) 1-(1-O-β-D-карбонилметилгалактопираноза)-7-{3-оксапентан-1,5-дикарбоновая кислота-1-оил-5-[1-(4-перфтороктилсульфонил)пиперазин] амид}-4,10-бис[1,4,7-трис(карбоксилатометил)-10-(3-аза-4-оксо-5-метил-5-илпентаноил)-1,4,7,10-тетраазациклододекан]-1,4,7,10-тетраазациклододекан, дигадолиниевый комплекс
5,54 г (8,8 ммоля, 4,4 молярных эквивалента в пересчете на количество используемого аминового компонента из примера 49б) описанного в заявке DE 19728954 С1 в примере 31h Gd-комплекса 10-(4-карбокси-1-метил-2-оксо-3-азабутил)-1,4,7,10-тетраазациклододекан-1,4,7-триуксусной кислоты и 0,37 г (8,8 ммоля) безводного хлорида лития при 40°С растворяют при перемешивании в 60 мл абсолютного диметилсульфоксида и при этой температуре смешивают с N-гидроксисукцинимидом общим количеством 1,01 г (8,8 ммоля) и с 2,11 г (2,0 ммоля) указанного в заголовке примера 49б соединения, растворенного в 25 мл абсолютного диметилсульфоксида. После охлаждения до комнатной температуры реакционный раствор смешивают с 1,82 г (8,8 ммоля) N,N'-дициклогексилкарбодиимида и перемешивают в течение 12 ч при комнатной температуре. Полученную суспензию затем смешивают с достаточным количеством ацетона до полного осаждения указанного выше в заголовке соединения, осадок отделяют вакуум-фильтрацией, сушат, растворяют в воде, отфильтровывают нерастворившуюся дициклогексилмочевину и фильтрат для обессоливания и для очистки от низкомолекулярных компонентов пропускают через ультрафильтрационную мембрану AMICON® YM-3 (предельная проницаемость: 3000 Да). Ретентат в завершение лиофилизуют.
Выход: 3,29 г (72,2% от теории; в пересчете на количество используемого аминового компонента) в виде бесцветного лиофилизата.
Н2О-содержание (при использовании реактива Карла Фишера): 5,99%.
Элементный анализ (в пересчете на безводное вещество):
Пример 50
а) Метиловый эфир 3-(1-O-α-D-2,3,4,6-тетра-O-бензилманнопираноза)-2-N-бензилоксикарбонил-L-серина
В 500 мл сухого ацетонитрила растворяют 21,42 г (39,61 ммоля) 2,3,4,6-тетра-О-бензил-α-D-маннопиранозы (получение согласно F. Kong и др., J. Carbohydr. Chem., 16, 6 (1997), сс.877-890). После охлаждения реакционного раствора до 5°С при этой температуре по каплям при перемешивании медленно добавляют раствор 13,23 г (59,52 ммоля) триметилсилилового эфира трифторметансульфоновой кислоты в 30 мл ацетонитрила, а затем раствор 20,06 г (79,21 ммоля) метилового эфира N-бензилоксикарбонил-L-серина (поставляется фирмой Bachem) в 50 мл ацетонитрила, при этом скорость капельного добавления должна быть такой, чтобы внутренняя температура не превышала 10°С. После проведения реакции в течение 15 ч при комнатной температуре реакционный раствор досуха концентрируют в вакууме. Полученный остаток растворяют в 250 мл этилового эфира уксусной кислоты и дважды промывают насыщенным раствором гидрокарбоната натрия порциями по 100 мл и однократно водой порцией 200 мл. После сушки органической фазы над сульфатом натрия соль отделяют вакуум-фильтрацией и этиловый эфир уксусной кислоты отгоняют в вакууме. Полученный маслянистый остаток очищают на силикагеле с использованием этилового эфира уксусной кислоты/н-гексана (в соотношении 1:5) в качестве элюента.
Выход: 23,60 г (76,8% от теории) указанного выше в заголовке соединения в виде бесцветного масла.
Элементный анализ:
б) 3-(1-O-α-D-2,3,4,6-тетра-O-бензилманнопираноза)-2-N-бензилоксикарбонил-L-серин
10,0 г (12,90 ммоля) полученного в примере 50а соединения растворяют в смеси из 20 мл метанола, 20 мл воды и 50 мл тетрагидрофурана. Далее при комнатной температуре добавляют 0,47 г (19,35 ммоля) гидроксида лития, растворенного в 25 мл дистиллированной воды, и затем перемешивают в течение 6 ч при 60°С. Контроль за протеканием реакции тонкослойной хроматографией (элюент:метиленхлорид/метанол в соотношении 10:1) указывает на уже количественное омыление метилового эфира из примера 50а по истечении указанного выше времени реакции. Для переработки содержащий продукт раствор досуха концентрируют в вакууме и полученный остаток в теплых условиях (примерно 60°С) растворяют в 250 мл этилового эфира уксусной кислоты. После этого полученную таким путем этилацетатную фазу дважды промывают 15%-ной водной соляной кислотой порциями по 50 мл и однократно дистиллированной водой порцией 100 мл. Органическую фазу сушат над сульфатом магния, фильтруют и досуха упаривают в вакууме. Остаток хроматографируют на силикагеле (элюент:н-гексан/этиловый эфир уксусной кислоты в соотношении 5:1). Таким путем получают 8,40 г (85,7% от теории) указанного в заголовке соединения в виде бесцветного масла.
Элементный анализ:
в) [1-(4-перфтороктилсульфонил)пиперазин]амид 3-(1-O-α-D-2,3,4,6-тетра-O-бензилманнопираноза)-2-N-бензилоксикарбонил-L-серина
К 13,86 г (24,40 ммоля) 1-перфтороктилсульфонилпиперазина (полученного согласно DE 19603033), растворенного в смеси из 150 мл тетрагидрофурана и 15 мл хлороформа, при 0°С в атмосфере азота по каплям добавляют 20,57 г (27,0 ммоля) полученной согласно примеру 50б карбоновой кислоты, растворенной в 50 мл тетрагидрофурана. После этого при 0°С порциями добавляют в целом 18,0 г (36,60 ммолей) ЭЭДХ [2-этокси-1-этоксикарбонил-1,2-дигидрохинолин] и оставляют на ночь для перемешивания при комнатной температуре. Для переработки реакционный раствор концентрируют в вакууме и полученное исключительно вязкое масло хроматографируют на силикагеле с использованием смеси н-гексан/изопропанол (в соотношении 15:1) в качестве элюента. Таким путем получают 17,0 г (79,6% от теории, в пересчете на количество используемого первичного амина) указанного в заголовке соединения в виде бесцветного вязкого масла.
Элементный анализ:
г) [1-(4-перфтороктилсульфонил)пиперазин]амид 3-(1-O-α-D-маннопираноза)-L-серина
15,0 г (11,41 ммоля) полученного согласно примеру 50в соединения растворяют в 200 мл этанола и смешивают с 1,5 г катализатора Перлмана (20%-ный Pd/C). После этого реакционный раствор гидрируют при комнатной температуре в атмосфере водорода (1 атм) до тех пор, пока не прекратится поглощение водорода (примерно в течение 8 ч). Для переработки катализатор удаляют вакуум-фильтрацией, тщательно промывают этанолом (дважды порциями примерно по 100 мл) и содержащий продукт этанольный фильтрат досуха концентрируют в вакууме. Таким путем получают указанное в заголовке соединение в виде высоковязкого и бесцветного масла.
Выход: 8,79 г (94,0% от теории).
Элементный анализ:
д) [1-(4-перфтороктилсульфонил)пиперазин]амид 3-(1-O-α-D-маннопираноза)-2-N-[1,4,7-трис(карбоксилатометил)-10-(3-аза-4-оксо-5-метил-5-илпентаноил)-1,4,7,10-тетраазациклододекан]-L-серина, Gd-комплекс
Перемешанную суспензию 5,7 г (9,06 ммоля, соответствует 1,5 молярных эквивалентов в пересчете на количество используемого соединения, указанного в заголовке примера 50г (первичного амина)) описанного в заявке DE 19728954 С1 в примере 31h Gd-комплекса 10-(4-карбокси-1-метил-2-оксо-3-азабутил)-1,4,7,10-тетраазациклододекан-1,4,7-триуксусной кислоты в 75 мл абсолютного диметилсульфоксида при 70°С смешивают с 0,68 г (15,9 ммоля) хлорида лития. После 30-минутного перемешивания при 70°С прозрачный реакционный раствор порциями смешивают с N-гидроксисукцинимидом общим количеством 1,83 г (15,9 ммоля) и реакционную смесь выдерживают еще в течение 1 ч при 70°С. После охлаждения до 10°С смешивают с 4,52 г (23,85 ммоля) дициклогексилкарбодиимида и реакционный раствор перемешивают еще в течение 1 ч при 0°С, а затем в течение 12 ч при 22°С. Полученный таким путем раствор N-гидроксисукцинимидоэфира Gd-комплекса 10-(4-карбокси-1-метил-2-оксо-3-азабутил)-1,4,7,10-тетраазациклододекан-1,4,7-триуксусной кислоты затем по каплям смешивают при 22°С с раствором 4,94 г (6,03 ммоля) указанного в заголовке примера 50г соединения в 15 мл абсолютного диметилсульфоксида и после этого перемешивают еще в течение 12 ч при комнатной температуре. Для переработки реакционный раствор при 22°С по каплям медленно добавляют к смеси растворителей, состоящей из 250 мл ацетона и 250 мл 2-пропанола, при этом указанное в заголовке соединение в течение 12 ч полностью выпадает в осадок при 10°С в виде масла светло-желтого цвета. Надосадочный элюент осторожно декантируют и маслянистый продукт растворяют в 200 мл дистиллированной воды, при этом такой продукт полностью переходит в раствор с образованием имеющего светло-желтую окраску водного раствора указанного выше в заголовке соединения. Далее этот водный, содержащий продукт раствор сначала фильтруют через мембранный фильтр, а затем для обессоливания и для отделения низкомолекулярных компонентов его трижды подвергают ультрафильтрации через ультрафильтрационную мембрану YM3 (AMICON®, предельная проницаемость: 3000 Да). Полученный в результате ретентат в завершение лиофилизуют.
Выход: 8,63 г (80,2% от теории, в пересчете на количество используемого соединения, указанного в заголовке примера 50г) в виде бесцветного лиофилизата с содержанием воды 7,65%.
Элементный анализ (в пересчете на безводное вещество):
Пример 51
а) [1-(4-перфтороктилсульфонил)пиперазин]амид 6-N-бензилоксикарбонил-2-N-[O-β-D-галактопиранозил-(1→4)-глюконозил]-L-лизина
К перемешанному раствору 4,98 г (6,0 ммолей) соединения, указанного в заголовке примера 21в, в 40 мл абсолютного диметилсульфоксида при комнатной температуре по каплям добавляют раствор 13,3 г (37,2 ммоля) O-β-D-галактопиранозил-(1→4)-D-глюконо-1,5-лактона [лактобионолактон; получение согласно (a) Williams T.J., Plessas N.R., Goldstein I.J., Carbohydr. Res. 67, Cl. (1978); (б) Kobayashi К., Sumitomo H., Ina Y., Polym. J. 17 (1985), с.567; (в) Hiromi Kitano, Katsuko Sohda и Ayako Kosaka, Bioconjugate Chem. 6 (1995), сс.131-134] в 40 мл абсолютного диметилсульфоксида. Полученный таким путем реакционный раствор затем перемешивают в течение 14 ч при 40°С. Для переработки этот раствор смешивают при комнатной температуре с 500 мл абсолютного 2-пропанола, полученный бесцветный осадок отделяют вакуум-фильтрацией с помощью фритты G4 и затем тщательно промывают абсолютным 2-пропанолом общим количеством 250 мл. Полученное таким путем твердое вещество далее растворяют в 300 мл дистиллированной воды и трижды подвергают ультрафильтрации через ультрафильтрационную мембрану YM3 (AMICON®, предельная проницаемость: 3000 Да). Подобная трехкратная ультрафильтрация позволяет отделить от целевого продукта избыток лактобионолактона, а также возможно еще присутствующие низкомолекулярные компоненты. Осадок на ультрафильтрационной мембране в завершение полностью растворяют в 300 мл дистиллированной воды и лиофилизуют.
Выход: 6,51 г (92,7% от теории) в виде бесцветного лиофилизата.
Содержание воды: 10,03%.
Элементный анализ (в пересчете на безводное вещество):
б) [1-(4-перфтороктилсульфонил)пиперазин]амид 2-N-[O-β-D-галактопиранозил-(1→4)-глюконозил]-L-лизина
5,0 г (4,27 ммоля) полученного в примере 51а соединения растворяют в 100 мл этанола, смешивают с 0,5 г катализатора Перлмана (20%-ный Pd/C) и гидрируют до количественного поглощения водорода при давлении водорода 1 атмосфера. После этого катализатор удаляют вакуум-фильтрацией, затем промывают этанолом и досуха концентрируют в вакууме. Таким путем получают указанное в заголовке соединение в виде бесцветного и вязкого масла.
Выход: 4,36 г (98,5% от теории).
Элементный анализ:
в) [1-(4-перфтороктилсульфонил)пиперазин]амид 2-N-[O-β-D-галактопиранозил-(1→4)-глюконозил]-6-N-[1,4,7-трис(карбоксилатометил)-10-(3-аза-4-оксо-5-метил-5-илпентаноил)-1,4,7,10-тетраазациклододекан]-L-лизина, Gd-комплекс
5,54 г (8,8 ммоля, 2,2 молярных эквивалента в пересчете на количество используемого аминового компонента из примера 51б) описанного в заявке DE 19728954 С1 в примере 31h Gd-комплекса 10-(4-карбокси-1-метил-2-оксо-3-азабутил)-1,4,7,10-тетраазациклододекан-1,4,7-триуксусной кислоты и 0,37 г (8,8 ммоля) безводного хлорида лития при 40°С растворяют при перемешивании в 60 мл абсолютного диметилсульфоксида и при этой температуре смешивают с N-гидроксисукцинимидом общим количеством 1,01 г (8,8 ммоля) и с 3,85 г (4,0 ммоля) указанного в заголовке примера 51б соединения, растворенного в 60 мл абсолютного диметилсульфоксида. После охлаждения до комнатной температуры реакционный раствор смешивают с 1,82 г (8,8 ммоля) N,N'-дициклогексилкарбодиимида и перемешивают в течение 12 ч при комнатной температуре. Полученную суспензию затем смешивают с достаточным количеством ацетона/2-пропанола (в соотношении 1:1) до полного осаждения указанного выше в заголовке соединения и осадок отделяют вакуум-фильтрацией. Полученный таким путем осадок затем растворяют в 300 мл воды и отфильтровывают нерастворившуюся дициклогексилмочевину. Фильтрат трижды подвергают ультрафильтрации через ультрафильтрационную мембрану AMICON® YM-3 (предельная проницаемость: 3000 Да). Подобная трехкратная ультрафильтрация позволяет отделить от целевого продукта избыток Gd-комплекса, а также возможно еще присутствующие низкомолекулярные компоненты. Осадок на ультрафильтрационной мембране в завершение полностью растворяют в 500 мл дистиллированной воды и лиофилизуют.
Выход: 4,64 г (70,4% от теории) в виде бесцветного лиофилизата.
Н2О-содержание (при использовании реактива Карла Фишера): 10,08%.
Элементный анализ (в пересчете на безводное вещество):
Пример 52
а) 2-N-трифторацетил-6-Н-бензилоксикарбониллизин
100 г (356,7 ммоля) 6-N-бензилоксикарбониллизина растворяют в смеси из 1000 мл этилового эфира трифторуксусной кислоты и 500 мл этанола и в течение 24 ч перемешивают при комнатной температуре. После этого упаривают досуха и остаток кристаллизуют из диизопропилового эфира.
Выход: 128,9 г (96% от теории) бесцветного кристаллического порошка.
Элементный анализ:
б) [1-(4-перфтороктилсульфонил)пиперазин]амид 2-N-трифторацетил-6-N-бензилоксикарбониллизина
К 125 г (332 ммоля) соединения, указанного в заголовке примера 52а, и 188,7 г (332 ммоля) 1-перфтороктилсульфонилпиперазина (полученного согласно DE 19603033) в 800 мл тетрагидрофурана при 0°С добавляют 164,2 г (0,664 ммоля) ЭЭДХ (этиловый эфир 2-этокси-1,2-дигидрохинолин-1-карбоновой кислоты) и перемешивают в течение ночи при комнатной температуре. После этого досуха упаривают в вакууме и хроматографируют на силикагеле (элюент:дихлорметан/метанол в соотношении 20:1).
Выход: 286 г (93% от теории) бесцветного твердого вещества.
Элементный анализ:
в) [1-(4-перфтороктилсульфонил)пиперазин]амид 6-N-бензилоксикарбониллизина
Раствор 280 г (302,2 ммоля) соединения, указанного в заголовке примера 52б, в 2000 мл этанола при 0°С барботируют в течение часа газообразным аммиаком. Затем смесь перемешивают в течение 4 ч при 0°С. После этого упаривают досуха и остаток выделяют из воды путем перемешивания. Твердое вещество отфильтровывают и сушат в вакууме (50°С).
Выход: 243,5 г (97% от теории) аморфного твердого вещества.
Элементный анализ:
г) [1-(4-перфтороктилсульфонил)пиперазин]амид 6-N-бензилоксикарбонил-2-N-(3,6,9,12,15-пентаоксагексадеканоил)лизина
К 50 г (60,20 ммоля) соединения, указанного в заголовке примера 52в, и 7,10 г (70 ммолей) триэтиламина в 350 мл дихлорметана при 0°С по каплям добавляют раствор 19,93 г (70 ммолей) хлорангидрида 3,6,9,12,15-пентаоксагексадекановой кислоты [полученного согласно Liebigs Ann. Chem. (6), (1980), сс.852-862] в 50 мл дихлорметана и перемешивают в течение 3 ч при 0°С. Далее добавляют 200 мл 5%-ной водной соляной кислоты и перемешивают в течение 5 мин при комнатной температуре. Органическую фазу отделяют, сушат над сульфатом магния и досуха упаривают в вакууме. Остаток хроматографируют на силикагеле (элюент:дихлорметан/ацетон в соотношении 15:1)
Выход: 53,7 г (93% от теории) бесцветного вязкого масла.
Элементный анализ:
д) [1-(4-перфтороктилсульфонил)пиперазин]амид 2-N-(3,6,9,12,15-пентаоксагексадеканоил)лизина
50 г (52,15 ммоля) соединения, указанного в заголовке примера 52г, растворяют в 500 мл этанола и добавляют 6 г палладиевого катализатора (10%-ный Pd/C). После этого гидрируют при комнатной температуре. Затем катализатор отфильтровывают и фильтрат досуха упаривают в вакууме.
Выход: 43,0 г (количественный) бесцветного твердого вещества.
Элементный анализ:
е) [1-(4-перфтороктилсульфонил)пиперазин]амид 6-N-[1,4,7-трис(карбоксилатометил)-1,4,7,10-тетраазациклододекан-10-(пентаноил-3-аза-4-оксо-5-метил-5-ил)]-2-N-(3,6,9,12,15-пентаоксагексадеканоил)лизина, Gd-комплекс
20 г (24,25 ммоля) соединения, указанного в заголовке примера 52д, 2,79 г (24,25 ммоля) N-гидроксисукцинимида, 2,12 г (50 ммолей) хлорида лития и 15,27 г (24,25 ммоля) Gd-комплекса 1,4,7-трис(карбоксилатометил)-10-[(3-аза-4-оксо-5-метил-5-ил)пентановая кислота]-1,4,7,10-тетраазациклододекана растворяют при умеренном нагревании в 200 мл диметилсульфоксида. Далее при 10°С добавляют 8,25 г (40 ммолей) N,N-дициклогексилкарбодиимида и затем перемешивают в течение ночи при комнатной температуре. Раствор сливают в 3000 мл ацетона и перемешивают в течение 10 мин. Выпавшее в осадок твердое вещество отфильтровывают и затем очищают хроматографией (силикагель RP-18, элюент:градиент воды/этанола/ацетонитрила).
Выход: 28,21 г (81% от теории) бесцветного твердого вещества.
Содержание воды: 11,0%.
Элементный анализ (в пересчете на безводное вещество):
Пример 53
а) [1-(4-перфтороктилсульфонил)пиперазин]амид 6-N-[3,9-бис(трет-бутилоксикарбонилметил)-3,6,9-триазаундекан-1,11-дикарбоновая кислота-бис(трет-бутиловый эфир)-6-карбонилметил]-2-N-(3,6,9,12,15-пентаоксагексадеканоил)лизина
К раствору 20 г (24,08 ммоля) соединения, указанного в заголовке примера 52д, 14,88 г (24,08 ммоля) бис(трет-бутилового эфира) 3,9-бис(трет-бутилоксикарбонилметил-3,6,9-триазаундекан-1,11-дикарбоновой кислоты и 2,77 г (24,08 ммоля) N-гидроксисукцинимида в 150 мл диметилформамида при 0°С добавляют 8,25 г (40 ммолей) N,N-дициклогексилкарбодиимида. Смесь перемешивают в течение 3 ч при 0°С, а затем в течение ночи при комнатной температуре. Выпавшую в осадок мочевину отфильтровывают, фильтрат досуха упаривают в вакууме и хроматографируют на силикагеле (элюент:дихлорметан/этанол в соотношении 20:1).
Выход: 31,61 г (91% от теории) вязкого масла.
Элементный анализ:
б) [1-(4-перфтороктилсульфонил)пиперазин]амид 6-N-[6-карбонилметил-3,9-бис(карбоксилатометил)-3,6,9-триазаундекандикарбоновая кислота-1-карбокси-11-карбоксилато]-2-N-(3,6,9,12,15-пентаоксагексадеканоил)лизина, Gd-комплекс, натриевая соль
30 г (20,8 ммоля) соединения, указанного в заголовке примера 53а, растворяют в 300 мл трифторуксусной кислоты и в течение 5 ч перемешивают при комнатной температуре. После этого упаривают досуха, остаток растворяют в 300 мл воды и значение рН устанавливают на 2,5 с помощью 10%-ного водного NaOH. Далее добавляют 3,77 г (10,4 ммоля) оксида гадолиния и перемешивают в течение 3 ч при 60°С. После этого смеси дают охладиться до комнатной температуры и значение рН устанавливают на 7,4 с помощью едкого натра. Затем упаривают досуха и остаток очищают на силикагеле RP-18 (элюент:градиент воды/ацетонитрила).
Выход: 19,18 г (67% от теории) бесцветного аморфного твердого вещества.
Содержание воды 9,8%.
Элементный анализ (в пересчете на безводное вещество):
Пример 54
а) [1-(4-перфтороктилсульфонилпиперазин]амид лизина
20 г (24,08 ммоля) соединения, указанного в заголовке примера 52в, растворяют в 300 мл этанола и добавляют 4 г палладиевого катализатора (10%-ный Pd/C). После этого гидрируют при комнатной температуре. Затем катализатор отфильтровывают и фильтрат досуха упаривают в вакууме.
Выход: 16,77 г (количественный) бесцветного твердого вещества.
Элементный анализ:
б) [1-(4-перфтороктилсульфонилпиперазин]амид 2,6-N,N'-бис[1,4,7-трис(карбоксилатометил)-1,4,7,10-тетраазациклододекан-10-(пентаноил-3-аза-4-оксо-5-метил-5-ил)]лизина, Gd-комплекс (металлический комплекс XVI)
10 г (14,36 ммоля) соединения, указанного в заголовке примера 54а, 3,34 г (29 ммолей) N-гидроксисукцинимида, 2,54 г (ммоля) хлорида лития и 18,26 г (29 ммолей) Gd-комплекса 1,4,7-трис(карбоксилатометил)-10-(3-аза-4-оксо-5-метил-5-ил)-1,4,7,10-тетраазациклододекана растворяют при умеренном нагревании в 200 мл диметилсульфоксида. Далее при 10°С добавляют 12,38 г (60 ммолей) N,N-дициклогексилкарбодиимида и затем перемешивают в течение ночи при комнатной температуре. Раствор сливают в 3000 мл ацетона и перемешивают в течение 10 мин. Выпавшее в осадок твердое вещество отфильтровывают и затем очищают хроматографией (силикагель RP-18, элюент:градиент воды/этанола/ацетонитрила).
Выход: 19,02 г (69% от теории) бесцветного твердого вещества.
Содержание воды: 11,3%.
Элементный анализ (в пересчете на безводное вещество):
Пример 55
а) Метиловый эфир 2-[4-(3-оксаэтилпропионат)]фенилуксусной кислоты
К 200 г (1,204 моля) метилового эфира 4-гидроксифенилуксусной кислоты и 212 г (2 моля) карбоната натрия в 2000 мл ацетона добавляют 233,8 г (1,4 моля) этилового эфира 2-бромуксусной кислоты и в течение 5 ч кипятят с обратным холодильником. Твердое вещество отфильтровывают и досуха упаривают в вакууме. Остаток хроматографируют на силикагеле (элюент:н-гексан/этиловый эфир уксусной кислоты в соотношении 15:1).
Выход: 288,5 г (95% от теории) бесцветного масла.
Элементный анализ:
б) Метиловый эфир 2-[4-(3-оксаэтилпропионат)]фенил-2-бромуксусной кислоты
К 285 г (1,13 моля) указанного в заголовке примера 55а соединения, растворенного в 2000 мл четыреххлористого углерода, добавляют 201 г (1,13 моля) N-бромсукцинимида и 100 мг дибензилпероксида и в течение 8 ч кипятят с обратным холодильником. Далее охлаждают на ледяной бане, выпавший в осадок сукцинимид отфильтровывают и фильтрат досуха упаривают в вакууме. Остаток очищают на силикагеле (элюент:н-гексан/ацетон в соотношении 15:1).
Выход: 359,2 г (96% от теории) бесцветного вязкого масла.
Элементный анализ:
в) Метиловый эфир 2-[4-(3-оксаэтилпропионат)]фенил-2-[1-(1,4,7,10-тетраазациклододекан-7-ил]уксусной кислоты
К 603 г (3,5 моля) 1,4,7,10-тетраазациклододекана в 6000 мл хлороформа добавляют 350 г (1,057 моля) соединения, указанного в заголовке примера 55б, и перемешивают в течение ночи при комнатной температуре. Далее трижды экстрагируют 3000 мл воды, органическую фазу сушат над сульфатом магния и досуха упаривают в вакууме. Остаток без дополнительной очистки используют в последующей реакции (пример 55г).
Выход: 448 г (количественный) вязкого масла.
Элементный анализ:
г) 2-[4-(3-оксапропионовая кислота)]фенил-2-[1,4,7-трис(карбоксиметил)-1,4,7,10-тетраазациклододекан-10-ил]уксусная кислота
445 г (1,053 моля) соединения, указанного в заголовке примера 55в, и 496 г (5,27 моля) хлоруксусной кислоты растворяют в 4000 мл воды. Затем значение рН устанавливают на 10 с помощью 30%-ного водного едкого натра. Далее смесь нагревают до 70°С и значение рН добавлением 30%-ного водного едкого натра поддерживают на уровне 10. Смесь перемешивают в течение 8 ч при 70°С. После этого значение рН устанавливают на 13 и в течение 30 мин кипятят с обратным холодильником. Затем раствор охлаждают на ледяной бане и добавлением концентрированной соляной кислоты значение рН устанавливают на 1. После этого досуха упаривают в вакууме. Остаток растворяют в 4000 мл метанола и перемешивают в течение часа при комнатной температуре. Выпавшую в осадок поваренную соль отфильтровывают, фильтрат упаривают досуха и остаток очищают на силикагеле RP-18 (элюент:градиент воды/этанола/ацетонитрила).
Выход: 403 г (69% от теории) бесцветного твердого вещества.
Содержание воды: 10,2%.
Элементный анализ (в пересчете на безводное вещество):
д) 2-[4-(3-оксапропионовая кислота)]фенил-2-[1,4,7-трис(карбоксиметил)-1,4,7,10-тетраазациклододекан-10-ил]уксусная кислота, Gd-комплекс
К 400 г (721,3 ммоля) соединения, указанного в заголовке примера 55г, в 2000 мл воды добавляют 130,73 г (360,65 ммоля) оксида гадолиния и перемешивают в течение 5 ч при 80°С. Раствор фильтруют и фильтрат лиофилизуют.
Выход: 511 г (количественный) аморфного твердого вещества.
Содержание воды: 11,0%.
Элементный анализ (в пересчете на безводное вещество):
е) [4-перфтороктилсульфонил)пиперазин]амид 2,6-N,N'-бис{2-[4-(3-оксапропионил)фенил]-2-[1,4,7-трис(карбоксилатометил)-1,4,7,10-тетраазациклододекан-10-ил]уксусная кислота]лизина, дигадолиниевый комплекс, динатриевая соль
10 г (14,36 ммоля) соединения, указанного в заголовке примера 54а, 3,45 г (30 ммолей) N-гидроксисукцинимида, 2,54 г (60 ммолей) хлорида лития и 21,26 г (30 ммолей) соединения, указанного в заголовке примера 55д, растворяют при умеренном нагревании в 250 мл диметилсульфоксида. Далее при 10°С добавляют 16,51 г (80 ммолей) N,N-дициклогексилкарбодиимида и затем перемешивают в течение ночи при комнатной температуре. Раствор сливают в 2000 мл ацетона и перемешивают в течение 10 мин. Выпавшее в осадок твердое вещество отфильтровывают и затем очищают хроматографией (силикагель RP-18, элюент:градиент воды/этанола/ацетонитрила). Это вещество растворяют в небольшом количестве воды, значение рН устанавливают на 7,4 с помощью едкого натра и лиофилизуют.
Выход: 21,02 г (69% от теории) бесцветного твердого вещества.
Содержание воды: 11,2%.
Элементный анализ (в пересчете на безводное вещество):
Пример 56
а) [1-(4-перфтороктилсульфонил)пиперазин]амид 2,6-N,N'-бис[6-карбонилметил-3,9-бис(трет-бутилоксикарбонилметил)-3,6,9-триазаундекан-1,11-дикарбоновая кислота-бис(трет-бутиловый эфир)]лизина
К раствору 10 г (14,36 ммоля) соединения, указанного в заголовке примера 54а, 18,53 г (30 ммолей) бис(трет-бутилового эфира) 3,9-бис(трет-бутилоксикарбонилметил)-6-карбоксиметил-3,6,9-триазаундекан-1,11-дикарбоновой кислоты и 3,45 г (30 молей) N-гидроксисукцинимида в 150 мл диметилформамида при 0°С добавляют 10,32 г (50 ммолей) N,N-дициклогексилкарбодиимида. Смесь перемешивают в течение 3 ч при 0°С, а затем в течение ночи при комнатной температуре. Выпавшую в осадок мочевину отфильтровывают, фильтрат досуха упаривают в вакууме и хроматографируют на силикагеле (элюент:дихлорметан/этанол в соотношении 20:1).
Выход: 19,60 г (72% от теории) вязкого масла.
Элементный анализ:
б) 2,6-N,N-бис[6-карбонилметил-3,9-бис(карбоксилатометил)-3,6,9-триазаундекандикарбоновая кислота-1-карбокси-11-карбоксилатолизин-[1-(4-перфтороктилсульфонил)пиперазин]амид, Gd-комплекс, натриевая соль]
15 г (7,91 моля) соединения, указанного в заголовке примера 56а, растворяют в 50 мл хлороформа и добавляют 200 мл трифторуксусной кислоты. Смесь перемешивают в течение 10 мин при комнатной температуре. После этого досуха упаривают в вакууме и остаток растворяют в 150 мл воды. Далее добавляют 2,87 г (7,91 ммоля) оксида гадолиния и перемешивают в течение 5 ч при 80°С. Затем смеси дают охладиться до комнатной температуры и значение рН устанавливают на 7,4 с помощью 2н. едкого натра. Раствор досуха упаривают в вакууме и очищают на RP-18 (элюент:градиент воды/этанола/ацетонитрила).
Выход: 8,11 г (57% от теории) бесцветного аморфного твердого вещества.
Содержание воды: 9,6%.
Элементный анализ (в пересчете на безводное вещество):
Пример 57
а) [1-(4-перфтороктилсульфонил)пиперазин]амид 6-N-бензилоксикарбонил-2-N-[6-карбоксиметил-3,9-бис(трет-бутилоксикарбонилметил)-3,6,9-триазаундекан-1,11-дикарбоновая кислота-бис(трет-бутиловый эфир)]лизина
К раствору 20 г (24,08 ммоля) соединения, указанного в заголовке примера 52в, 14,88 г (24,08 ммоля) бис(трет-бутилового эфира) 3,9-бис(трет-бутилоксикарбонилметил)-6-карбоксиметил-3,6,9-триазаундекан-1,11-дикарбоновой кислоты и 2,88 г (25 ммолей) N-гидроксисукцинимида в 100 мл диметилформамида при 0°С добавляют 8,25 г (40 молей) N,N-дициклогексилкарбодиимида. Смесь перемешивают в течение 3 ч при 0°С, а затем в течение ночи при комнатной температуре. Выпавшую в осадок мочевину отфильтровывают, фильтрат досуха упаривают в вакууме и хроматографируют на силикагеле (элюент:дихлорметан/этанол в соотношении 20:1).
Выход: 27,21 г (79% от теории) вязкого масла.
Элементный анализ:
б) [1-(4-перфтороктилсульфонил)пиперазин]амид 2-N-[карбонилметил-3,9-бис(трет-бутилоксикарбонилметил)-3,6,9-триазаундекан-1,11-дикарбоновая кислота-бис(трет-бутиловый эфир)]лизина
25 г (17,48 ммоля) соединения, указанного в заголовке примера 57а, растворяют в 350 мл этанола и добавляют 5 г палладиевого катализатора (10%-ный Pd/C). После этого гидрируют при комнатной температуре. Затем катализатор отфильтровывают и фильтрат досуха упаривают в вакууме.
Выход: 22,66 г (количественный) бесцветного твердого вещества.
Элементный анализ:
в) [1-(4-перфтороктилсульфонил)пиперазин]амид 6-N-[1,4,7-трис(карбоксилатометил)-1,4,7,10-тетраазациклододекан-10-(пентаноил-3-аза-4-оксо-5-метил-5-ил)]-2-N-[6-карбонилметил-3,9-бис(трет-бутилоксикарбонилметил)-3,6,9-триазаундекан-1,11-дикарбоновая кислота-бис(трет-бутиловый эфир)]лизина, Gd-комплекс
20 г (15,43 ммоля) соединения, указанного в заголовке примера 57б, 1,78 г (15,43 ммоля) N-гидроксисукцинимида, 1,48 г (35 ммолей) хлорида лития и 9,72 г (15,43 ммоля) Gd-комплекса 1,4,7-трис(карбоксилатометил)-10-(3-аза-4-оксо-5-метил-5-ил)пентановая кислота-1,4,7,10-тетраазациклододекана растворяют при умеренном нагревании в 150 мл диметилсульфоксида. Далее при 10°С добавляют 5,16 г (25 ммолей) N,N-дициклогексилкарбодиимида и затем перемешивают в течение ночи при комнатной температуре. Раствор сливают в 2500 мл ацетона и перемешивают в течение 10 мин. Выпавшее в осадок твердое вещество отфильтровывают и затем очищают хроматографией (силикагель RP-18, элюент:градиент воды/этанола/ацетонитрила).
Выход: 22,94 г (78% от теории) бесцветного твердого вещества.
Содержание воды: 7,9%.
Элементный анализ (в пересчете на безводное вещество):
г) [1-(4-перфтороктилсульфонил)пиперазин]амид 6-N-[1,4,7-трис(карбоксилатометил)-1,4,7,10-тетраазациклододекан-10-(3-аза-4-оксо-5-метил-5-илпентаноил)]-2-N-[6-карбонилметил-3,9-бис(карбоксилатометил)-3,6,9-триазаундекан-дикарбоновая кислота-карбокси-11-карбоксилато-z]лизина, дигадолиниевый комплекс, натриевая соль
20 г (10,49 ммоля) соединения, указанного в заголовке примера 57в, растворяют в 200 мл трифторуксусной кислоты. Смесь перемешивают в течение 60 мин при комнатной температуре. Далее досуха упаривают в вакууме и остаток растворяют в 150 мл воды. Затем добавляют 1,90 г (5,25 ммоля) оксида гадолиния и перемешивают в течение 5 ч при 80°С. Смеси дают охладиться до комнатной температуры и значение рН устанавливают на 7,4 с помощью едкого натра. Раствор досуха упаривают в вакууме и очищают на силикагеле RP-18 (элюент:градиент воды/этанола/ацетонитрила).
Выход: 11,89 г (61% от теории) бесцветного аморфного твердого вещества.
Содержание воды: 10,2%.
Элементный анализ (в пересчете на безводное вещество):
Пример 58
а) трет-Бутиловый эфир 5,6-бис(бензилокси)-3-оксагексановой кислоты
100 г (376,2 ммоля) 1,2-ди-О-бензилглицерина [полученного согласно Chem. Phys. Lipids, 43(2) (1987), сс.113-127] и 5 г гидросульфата тетрабутиламмония растворяют в смеси из 400 мл толуола и 200 мл 50%-ного водного едкого натра. Далее при 0°С в течение 30 мин по каплям добавляют 78 г (400 ммолей) трет-бутилового эфира 2-бромуксусной кислоты и затем перемешивают в течение 3 ч при 0°С. Органическую фазу отделяют, сушат над сульфатом магния и досуха упаривают в вакууме. Остаток хроматографируют на силикагеле (элюент:N-гексан/ацетон в соотношении 20:1).
Выход: 133,4 г (94% от теории) бесцветного масла.
Элементный анализ:
б) 5,6-бис(бензилокси)-3-оксагексановая кислота
130 г (336,4 ммоля) соединения, указанного в заголовке примера 58а, растворяют в 200 мл дихлорметана и при 0°С добавляют 100 мл трифторуксусной кислоты. Смесь перемешивают в течение 4 ч при комнатной температуре и затем упаривают досуха. Остаток кристаллизуют из пентана/диэтилового эфира.
Выход: 102,2 г (92% от теории) воскоподобного твердого вещества.
Элементный анализ:
в) [1-(4-перфтороктилсульфонил)пиперазин]амид 6-N-бензилоксикарбонил-2-N-[1,4,7-трис(карбоксилатометил)-1,4,7,10-тетраазациклододекан-10-(пентаноил-3-аза-4-оксо-5-метил-5-ил)]лизина, Gd-комплекс
50 г (60,20 ммоля) соединения, указанного в заголовке примера 52в, 6,93 г (60,20 ммоля) N-гидроксисукцинимида, 5,09 г (120 ммолей) хлорида лития и 37,91 г (60,20 ммоля) Gd-комплекса 1,4,7-трис(карбоксилатометил)-1,4,7,10-тетраазациклододекан-10-пентаноил-3-аза-4-оксо-5-метил-5-ила растворяют при умеренном нагревании в 400 мл диметилсульфоксида. Далее при 10°С добавляют 20,63 г (100 ммолей) N,N-дициклогексилкарбодиимида и затем перемешивают в течение ночи при комнатной температуре. Раствор сливают в 3000 мл ацетона и перемешивают в течение 10 мин. Выпавшее в осадок твердое вещество отфильтровывают и затем очищают хроматографией (силикагель RP-18, элюент:градиент воды/этанола/ацетонитрила).
Выход: 75,53 г (87% от теории) бесцветного твердого вещества.
Содержание воды: 10,1%.
Элементный анализ (в пересчете на безводное вещество):
г) [1-(4-перфтороктилсульфонил)пиперазин]амид 2-N-[1,4,7-трис(карбоксилатометил)-1,4,7,10-тетраазациклододекан-10-(пентаноил-3 -аза-4-оксо-5 метил-5-ил]лизина, Gd-комплекс
70 г (48,53 ммоля) соединения, указанного в заголовке примера 58в, растворяют в смеси из 500 мл воды и 100 мл этанола и добавляют 5 г палладиевого катализатора (10%-ный Pd/C). После этого гидрируют при комнатной температуре. Затем катализатор отфильтровывают и фильтрат досуха упаривают в вакууме.
Выход: 63,5 г (количественный) бесцветного твердого вещества.
Содержание воды: 9,8%.
Элементный анализ (в пересчете на безводное вещество):
д) [1-(4-перфтороктилсульфонил)пиперазин]амид 6-N-[5,6-бис(бензилокси)-3-оксагексаноил]-2-N-[1,4,7-трис(карбоксилатометил)-1,4,7,10-тетраазациклододекан-10-(пентаноил-3-аза-4-оксо-5-метил-5-ил)]лизина, Gd-комплекс
10 г (7,64 ммоля) соединения, указанного в заголовке примера 58г, 3,30 г (10 ммолей) соединения, указанного в заголовке примера 58б, 0,85 г (20 ммолей) хлорида лития и 1,15 г (10 ммолей) N-гидроксисукцинимида растворяют при умеренном нагревании в 150 мл диметилсульфоксида. Далее при 10°С добавляют 3,10 г (15 ммолей) N,N'-дициклогексилкарбодиимида и перемешивают в течение 8 ч при комнатной температуре. Реакционный раствор сливают в 2000 мл ацетона и выделяют выпавший осадок. Указанное в заголовке соединение очищают на силикагеле RP-18 (элюент:градиент воды/этанола/ацетонитрила).
Выход: 11,14 г (90% от теории) бесцветного аморфного твердого вещества.
Содержание воды: 4,3%.
Элементный анализ (в пересчете на безводное вещество):
е) [1-(4-перфтороктилсульфонил)пиперазин]амид 6-N-(5,6,-дигидрокси-3-оксагексаноил)-2-N-[1,4,7-трискарбоксилатометил)-1,4,7,10-тетраазациклододекан-10-пентаноил-3-аза-4-оксо-5-метил-5-ил)]лизина, Gd-комплекс
10 г (6,17 ммоля) соединения, указанного в заголовке примера 58д, растворяют в 200 мл этанола и добавляют 3 г палладиевого катализатора (10%-ный Pd/C). После этого гидрируют при комнатной температуре. Затем катализатор отфильтровывают и фильтрат досуха упаривают в вакууме.
Выход: 8,89 г (количественный) бесцветного твердого вещества.
Содержание воды: 3,1%.
Элементный анализ (в пересчете на безводное вещество):
Пример 59
а) [1-(4-перфтороктилсульфонил)пиперазин]амид 6-N-бензилоксикарбонил-2-N-[5,6-бис(бензилокси)-3-оксагексаноил]лизина
К раствору из 20 г (24,08 ммоля) соединения, указанного в заголовке примера 52в, 9,91 г (30 ммолей) соединения, указанного в заголовке примера 58б, и 3,45 г (30 ммолей) N-гидроксисукцинимида в 100 мл диметилформамида при 0°С добавляют 9,28 г (45 ммолей) N,N-дициклогексилкарбодиимида. Смесь перемешивают в течение 3 ч при 0°С, а затем в течение ночи при комнатной температуре. Выпавшую в осадок мочевину отфильтровывают, фильтрат досуха упаривают в вакууме и хроматографируют на силикагеле (элюент:дихлорметан/этанол в соотношении 20:1).
Выход:24,50 г (89% от теории) вязкого масла.
Элементный анализ:
б) [1-(4-перфтороктилсульфонил)пиперазин]амид 2-N-(5,6-дигидрокси-3-оксагексаноил)лизина
20 г (17,5 ммоля) соединения, указанного в заголовке примера 59а, растворяют в 300 мл этанола и добавляют 5 г палладиевого катализатора (10%-ный Pd/C). После этого гидрируют при комнатной температуре. Затем катализатор отфильтровывают и фильтрат досуха упаривают в вакууме.
Выход: 17,65 г (количественный) бесцветного твердого вещества.
Элементный анализ:
в) [1-(4-перфтороктилсульфонил)пиперазин]амид 6-N-[1,4,7-трис(карбоксилатометил)-1,4,7,10-тетраазациклододекан-10-(пентаноил-3-аза-4-оксо-5-метил-5-ил)]лизина, Gd-комплекс
15 г (14,87 ммоля) соединения, указанного в заголовке примера 59б, 1,73 г (15 ммолей) N-гидроксисукцинимида, 1,27 г (30 ммолей) хлорида лития и 9,48 г (15 ммолей) Gd-комплекса 1,4,7-трис(карбоксилатометил)-10-(3-аза-4-оксо-5-метил-5-ил)пентановая кислота-1,4,7,10-тетраазациклододекана растворяют при умеренном нагревании в 100 мл диметилсульфоксида. Далее при 10°С добавляют 5,16 г (25 моля) N,N-дициклогексилкарбодиимида и затем перемешивают в течение ночи при комнатной температуре. Раствор сливают в 1500 мл ацетона и перемешивают в течение 10 мин. Выпавшее в осадок твердое вещество отфильтровывают и затем очищают хроматографией (силикагель RP-18, элюент:градиент воды/этанола/ацетонитрила).
Выход: 19,28 г (80% от теории) бесцветного твердого вещества.
Содержание воды: 10,3%.
Элементный анализ (в пересчете на безводное вещество):
Пример 60
а) [1-(4-перфтороктилсульфонил)пиперазин]амид 6-N-бензилоксикарбонил-2-N-[2,6-N,N'-бис(бензилоксикарбонил)лизил]лизина
20 г (24,08 ммоля) соединения, указанного в заголовке примера 52в, и 2,53 г (25 ммолей) триэтиламина растворяют в 200 мл тетрагидрофурана (ТГФ) и добавляют 14,46 г (27 ммолей) ди-N,N'-паранитрофенолового эфира Z-лизина. Смесь перемешивают в течение 5 ч при 50°С. После этого досуха упаривают в вакууме и остаток хроматографируют на силикагеле (элюент:дихлорметан/метанол в соотношении 20:1).
Выход: 28,07 г (95% от теории) бесцветного твердого вещества.
Элементный анализ:
б) [1-(4-перфтороктилсульфонил)пиперазин]амид 2-N-(лизил)лизина, тригидробромид
К 25 г (20,37 ммоля) соединения, указанного в заголовке примера 60а, добавляют 100 мл бромистоводородной кислоты в ледяной уксусной кислоте (48%-ной) и перемешивают в течение 2 ч при 40°С. Далее охлаждают до 0°С, по каплям добавляют 1500 мл диэтилового эфира и выпавшее в осадок твердое вещество отфильтровывают. После сушки в вакууме (60°С) получают 21,52 г (99% от теории) кристаллического твердого вещества светло-желтого цвета.
Элементный анализ:
в) [1-(4-перфтороктилсульфонил)пиперазин]амид 6-N-[1,4,7-трис(карбоксилатометил)-1,4,7,10-тетраазациклододекан-10-(пентаноил-3-аза-4-оксо-5-метил-5-ил)]-2-N-[2,6-N,N'-бис[1,4,7-трис(карбоксилатометил)-1,4,7,10-тетраазациклододекан-10-(пентаноил-3-аза-4-оксо-5-метил-5-ил)]лизил]лизина, тригадолиниевый комплекс
31,49 г (50 ммолей) Gd-комплекса 1,4,7-трис(карбоксилатометил)-10-(3-аза-4-оксо-5-метил)-5-ил)пентановой кислоты, 6,91 г (60 ммолей) N-гидроксисукцинимида и 4,24 г (100 ммолей) хлорида лития растворяют при умеренном нагревании в 350 мл диметилсульфоксида. Далее при 10°С добавляют 16,51 г (80 ммолей) N,N-дициклогексилкарбодиимида и перемешивают в течение 5 ч при 10°С. К этой смеси добавляют 10 г (9,37 ммоля) соединения, указанного в заголовке примера 60б, и 3,03 г (30 ммолей) триэтиламина и перемешивают в течение 12 ч при 60°С. Далее смеси дают охладиться до комнатной температуры и после этого ее сливают в 3000 мл ацетона. Выпавший осадок отфильтровывают и очищают на силикагеле RP-18 (элюент:градиент воды/этанола/ацетонитрила).
Выход: 16,7 г (67% от теории) бесцветного твердого вещества.
Содержание воды: 7,9%.
Элементный анализ (в пересчете на безводное вещество):
Пример 61
а) 1,7-бис(бензилоксикарбонил)-4-(3,6,9,12,15-пентаоксагексадеканоил)-1,4,7,10-тетраазациклододекан
К 18,13 г (68,1 ммоля) 3,6,9,12,15-пентаоксагексадекановой кислоты и 30 г (68,1 ммоля) 1,7-ди-Z-циклена, полученного согласно Z.Kovacs и A.D.Sherry, J. Chem. Soc. chem. Commun., 2 (1995), с.185, в 300 мл тетрагидрофурана при 0°С добавляют 24,73 г (100 ммолей) ЭЭДХ (этиловый эфир 2-этокси-1,2-дигидрохинолин-1-карбоновой кислоты) и перемешивают в течение ночи при комнатной температуре. После этого досуха упаривают в вакууме и хроматографируют на силикагеле (элюент:дихлорметан/метанол в соотношении 20:1).
Выход: 19,13 г (42% от теории) бесцветного твердого вещества.
Элементный анализ:
б) 1,7-бис(бензилоксикарбонил)-4-(3,6,9,12,15-пентаоксагексадеканоил)-10-(2Н,2Н,4Н,5Н,5Н-3-оксаперфтортридеканоил)-1,4,7,10-тетраазациклододекан
К 18 г (26,91 ммоля) соединения, указанного в заголовке примера 61a, и 14,05 г (26,91 ммоля) 2Н,2Н,4Н,4Н,5Н,5Н-3-оксаперфтортридекановой кислоты, полученной согласно DE 19603033, в 300 мл тетрагидрофурана при 0°С добавляют 12,36 г (50 ммолей) ЭЭДХ (этиловый эфир 2-этокси-1,2-дигидрохинолин-1-карбоновой кислоты) и перемешивают в течение ночи при комнатной температуре. После этого досуха упаривают в вакууме и хроматографируют на силикагеле (элюент:дихлорметан/метанол в соотношении 20:1).
Выход: 21,51 г (67% от теории) бесцветного твердого вещества.
Элементный анализ:
в) 1-(3,6,9,12,15-пентаоксагексадеканоил)-7-(2Н,2Н,4Н,4Н,5Н,5Н-3-оксаперфтортридеканоил)-1,4,7,10-тетраазациклододекан
20 г (16,77 ммоля) соединения, указанного в заголовке примера 52г, растворяют в 200 мл этанола и добавляют 2,5 г палладиевого катализатора (10%-ный Pd/C). После этого гидрируют при комнатной температуре. Затем катализатор отфильтровывают и фильтрат досуха упаривают в вакууме.
Выход: 15,5 г (количественный) бесцветного твердого вещества.
Элементный анализ:
г) 1,7-бис(1,4,7-трис(карбоксилатометил)-1,4,7,10-тетраазациклододекан-10-(пентаноил-3-аза-4-оксо-5-метил-5-ил)]-4-(3,6,9,12,15-пентаоксагексадеканоил)-10-(2Н,2Н,4Н,4Н,5Н,5Н-3-оксаперфтортридеканоил)-1,4,7,10-тетраазациклододекан, Gd-комплекс
15 г (16,22 ммоля) соединения, указанного в заголовке примера 61в, 4,60 г (40 ммолей) N-гидроксисукцинимида, 3,39 г (80 ммолей) хлорида лития и 25,19 г (40 ммолей) Gd-комплекса 1,4,7-трис(карбоксилатометил)-10-(3-аза-4-оксо-5-метил-5-ил)пентановой кислоты растворяют при умеренном нагревании в 300 мл диметилсульфоксида. Далее при 10°С добавляют 24,73 г (100 ммолей) ЭЭДХ и затем перемешивают в течение ночи при комнатной температуре. Раствор сливают в 3000 мл ацетона и перемешивают в течение 10 мин. Выпавшее в осадок твердое вещество отфильтровывают и затем очищают хроматографией (силикагель RP-18, элюент:градиент воды/этанола/ацетонитрила).
Выход: 19,86 г (57% от теории) бесцветного твердого вещества.
Содержание воды: 11,3%.
Элементный анализ (в пересчете на безводное вещество):
Пример 62
а) 1-[(4-перфтороктилсульфонил)пиперазин]амид 3,5-динитробензойной кислоты
К 20 г (35,2 ммоля) перфтороктилсульфонилпиперазина и 8,1 г (80 ммолей) триэтиламина, растворенных в 200 мл дихлорметана, при 0°С по каплям добавляют раствор 8,76 г (38 ммолей) 3,5-динитробензоилхлорида в 55 мл дихлорметана и перемешивают в течение 3 ч при 0°С. Далее добавляют 200 мл 5%-ной водной соляной кислоты и перемешивают в течение 5 мин при комнатной температуре. Органическую фазу отделяют, сушат над сульфатом магния и досуха упаривают в вакууме. Остаток хроматографируют на силикагеле (элюент:дихлорметан/ацетон в соотношении 15:1).
Выход: 24,96 г (93% от теории) бесцветного твердого вещества.
Элементный анализ:
б) [1-(4-перфтороктилсульфонил)пиперазин]амид 3,5-диаминобензойной кислоты
20 г (26,23 ммоля) соединения, указанного в заголовке примера 62а, растворяют в 400 мл этанола и добавляют 6 г палладиевого катализатора (10%-ный Pd/C). После этого гидрируют при комнатной температуре. Затем катализатор отфильтровывают и фильтрат досуха упаривают в вакууме.
Выход: 18,43 г (количественный) твердого вещества кремового цвета.
Элементный анализ:
в) [1-(4-перфтороктилсульфонил)пиперазин]амид 3,5-N,N'-бис[1,4,7-трис(карбоксилатометил)-1,4,7,10-тетраазациклододекан-10-(пентаноил-3-аза-4-оксо-5-метил-5-ил)]бензойной кислоты, Gd-комплекс
10 г (14,24 ммоля) соединения, указанного в заголовке примера 62б, 3,45 г (30 ммолей) N-гидроксисукцинимида, 2,54 г (60 молей) хлорида лития и 18,89 г (30 ммолей) Gd-комплекса 1,4,7-трис(карбоксилатометил)-10-(3-аза-4-оксо-5-метил-5-ил)пентановой кислоты растворяют при умеренном нагревании в 200 мл диметилсульфоксида. Далее при 10°С добавляют 10,32 г (50 ммолей) N,N-дициклогексилкарбодиимида и затем перемешивают в течение ночи при комнатной температуре. Раствор сливают в 2000 мл ацетона и перемешивают в течение 10 мин. Выпавшее в осадок твердое вещество отфильтровывают и затем очищают хроматографией (силикагель RP-18, элюент:градиент воды/этанола/ацетонитрила).
Выход: 19,74 г (72% от теории) бесцветного твердого вещества.
Содержание воды: 11,8%.
Элементный анализ (в пересчете на безводное вещество):
Пример 63
а) трет-Бутиловый эфир 3-окса-2Н,2Н,4Н,4Н,5Н,5Н-перфтортридеканкарбоновой кислоты
25,0 г (53,8 ммоля) 1Н,1Н,2Н,2Н-перфтор-1-деканола [поставляется фирмой Lancaster] растворяют в 250 мл абсолютного толуола и при комнатной температуре смешивают с каталитическим количеством (примерно 0,75 г) гидросульфата тетра-и-бутиламмония. Далее при 0°С добавляют в целом 7,55 г (134,6 ммоля, 2,5 эквивалента в пересчете на количество используемого спиртового компонента) высокодисперсного порошкового гидроксида калия, а затем 15,73 г (80,7 ммоля, 1,5 эквивалента в пересчете на количество используемого спиртового компонента) трет-бутилового эфира бромуксусной кислоты и оставляют на 2 ч для перемешивания при 0°С. Полученный таким путем реакционный раствор затем перемешивают в течение 12 ч при комнатной температуре и для переработки смешивают с этиловым эфиром уксусной кислоты общим количеством 500 мл и 250 мл воды. Органическую фазу отделяют и дважды промывают водой. После сушки органической фазы над сульфатом натрия соль отделяют вакуум-фильтрацией и растворитель отгоняют в вакууме. Полученный маслянистый остаток очищают на силикагеле с использованием этилового эфира уксусной кислоты/гексана (в соотношении 1:10) в качестве элюента.
Выход: 26,3 г (84,6% от теории) указанного выше в заголовке соединения в виде бесцветного и высоковязкого масла.
Элементный анализ:
б) 3-окса-2Н,2Н,4Н,4Н,5Н,5Н-перфтортридеканкарбоновая кислота
20,0 г (34,58 ммоля) соединения, указанного в заголовке примера 63а, при перемешивании суспендируют при комнатной температуре в 200 мл смеси, состоящей из метанола и 0,5-молярного едкого натра в соотношении 2:1, и затем нагревают до 60°С. После проведения реакции в течение 12 ч при 60°С прозрачную реакционную смесь для переработки нейтрализуют смешением с катионообменной смолой Amberlite® IR 120 (Н+-форма), ионит удаляют вакуум-фильтрацией и полученный таким путем метанольно-водный фильтрат перегоняют в вакууме до получения сухого остатка. Полученный аморфно-маслянистый остаток очищают на силикагеле с использованием этилового эфира уксусной кислоты/н-гексана (в соотношении 1:3) в качестве элюента.
Выход: 16,0 г (88,6% от теории) указанного выше в заголовке соединения в виде бесцветного и высоковязкого масла.
Элементный анализ:
в) 1,7-бис {[1,4,7-трис(карбоксилатометил)-10-(3-аза-4-оксо-5-метил-5-илпентаноил)]-1,4,7,10-тетраазациклододекан}диэтилентриамин, дигадолиниевый комплекс
2,48 г (3,94 ммоля, 2,05 молярных эквивалента в пересчете на количество используемого диэтилентриамина) описанного в заявке DE 19728954 С1 в примере 31h Gd-комплекса 10-(4-карбокси-1-метил-2-оксо-3-азабутил)-1,4,7,10-тетраазациклододекан-1,4,7-триуксусной кислоты и 167 мг безводного хлорида лития (3,94 ммоля) при 40°С растворяют при перемешивании в 40 мл абсолютного диметилсульфоксида и при этой температуре смешивают с N-гидроксисукцинимидом общим количеством 453 мг (3,94 ммоля). После охлаждения до комнатной температуры полученный таким путем реакционный раствор смешивают с 814 мг (3,946 ммоля) N,N'-дициклогексилкарбодиимида и в течение 2 ч перемешивают при комнатной температуре. Полученную суспензию активированного эфира затем смешивают с 198,3 мг (1,92 ммоля) диэтилентриамина, растворенного в 5 мл абсолютного диметилсульфоксида, и перемешивают в течение 12 ч при комнатной температуре. Для переработки реакционную смесь смешивают с достаточным количеством ацетона до полного осаждения указанного выше в заголовке соединения, осадок отделяют вакуум-фильтрацией, сушат, растворяют в воде, отфильтровывают нерастворившуюся дициклогексилмочевину и фильтрат для обессоливания и для очистки от низкомолекулярных компонентов пропускают через ультрафильтрационную мембрану AMICON® YM-3 (предельная проницаемость 3000 Да). Ретентат в завершение лиофилизуют.
Выход: 1,85 г (72,7% от теории) в виде бесцветного лиофилизата.
Н2О-содержание (при использовании реактива Карла Фишера): 3,89%.
Элементный анализ (в пересчете на безводное вещество):
г) 1,7-бис{[1,4,7-трис(карбоксилатометил)-10-(3-аза-4-оксо-5-метил-5-илпентаноил)]-1,4,7,10-тетраазациклододекан}-4-(3-окса-2Н,2Н,4Н,4Н,5Н,5Н-перфтортридеканоил)диэтилентриамин, дигадолиниевый комплекс
К раствору 3,23 г (2,44 ммоля) соединения, указанного в заголовке примера 63в, в смеси из 30 мл диметилсульфоксида и 3 мл тетрагидрофурана при 50°С в атмосфере азота по каплям добавляют 1,27 г (2,44 ммоля) указанного в заголовке примера 63б соединения, растворенного в смеси из 15 мл тетрагидрофурана и 15 мл диметилсульфоксида. После этого при 0°С порциями добавляют в целом 1,80 г (3,66 ммоля) ЭЭДХ [2-этокси-1-этоксикарбонил-1,2-дигидрохинолин] и оставляют на ночь для перемешивания при комнатной температуре. Полученный реакционный раствор затем смешивают с достаточным количеством ацетона до полного осаждения указанного выше в заголовке соединения, осадок отделяют вакуум-фильтрацией, сушат, растворяют в воде, отфильтровывают нерастворившиеся компоненты и фильтрат подвергают ультрафильтрации через ультрафильтрационную мембрану AMICON® YM-3 (предельная проницаемость: 3000 Да), что обеспечивает полное обессоливание указанного в заголовке соединения и одновременно его очистку от низкомолекулярных компонентов. Ретентат в завершение лиофилизуют.
Выход: 3,54 г (79,4% от теории) в виде бесцветного лиофилизата.
Н2О-содержание (при использовании реактива Карла Фишера): 5,87%.
Элементный анализ (в пересчете на безводное вещество):
Пример 64
а) 2-N-трифторацетил-6-N-бензилоксикарбонил-L-лизин
100,0 г (356,7 ммоля) 6-N-бензилоксикарбонил-L-лизина растворяют в смеси из 1000 мл этилового эфира трифторуксусной кислоты и 500 мл этанола и в течение 24 ч перемешивают при комнатной температуре. После этого упаривают досуха и остаток кристаллизуют из диизопропилового эфира.
Выход: 128,9 г (96% от теории) бесцветного кристаллического порошка.
Температура плавления: 98,5°С.
Элементный анализ:
б) [1-(4-перфтороктилсульфонил)пиперазин]амид 2-N-трифторацетил-6-N-бензилоксикарбонил-L-лизина
К 125,0 г (332,0 ммоля) соединения, указанного в заголовке примера 52а, и 188,7 г (332,0 ммоля) 1-перфтороктилсульфонилпиперазина (полученного согласно DE 19603033) в 750 мл тетрагидрофурана при 0°С добавляют 164,2 г (0,664 ммоля) ЭЭДХ (этиловый эфир 2-этокси-1,2-дигидрохинолин-1-карбоновой кислоты) и перемешивают в течение ночи при комнатной температуре. После этого досуха упаривают в вакууме и хроматографируют на силикагеле (элюент:дихлорметан/метанол в соотношении 20:1).
Выход: 286,0 г (93% от теории) бесцветного твердого вещества.
Температура плавления: 92°С.
Элементный анализ:
в) [1-(4-перфтороктилсульфонил)пиперазин]амид 6-N-бензилоксикарбонил-L-лизина
Раствор 280,0 г (302,2 моля) соединения, указанного в заголовке примера 52б, в 2000 мл этанола при 0°С барботируют в течение часа газообразным аммиаком. После этого смесь перемешивают в течение 4 ч при 0°С. Далее упаривают досуха и остаток выделяют из воды путем перемешивания. Твердое вещество отфильтровывают и сушат в вакууме при 50°С.
Выход: 243,5 г (97,0% от теории) аморфного твердого вещества.
Элементный анализ:
г) [1-(4-перфтороктилсульфонил)пиперазин]амид L-лизина
200,0 г (240,8 ммоля) полученного в примере 64в соединения растворяют в 1000 мл этанола, смешивают с 5,0 г катализатора Перлмана (20%-ный Pd/C) и гидрируют при комнатной температуре в атмосфере водорода (1 атм) до тех пор, пока не прекратится поглощение водорода. После этого катализатор удаляют вакуум-фильтрацией, тщательно промывают этанолом (трижды порциями примерно по 100 мл) и досуха концентрируют в вакууме. Таким путем получают указанное в заголовке соединение в виде высоковязкого масла желтого цвета.
Выход: 162,5 г (96,9% от теории).
Элементный анализ:
д) [1-(4-перфтороктилсульфонил)пиперазин]амид 6N-2N-бис{4-[2,3-бис(N,N-бис(трет-бутилоксикарбонилметил)амино)пропил]фенил}-3-оксапропионил-L-лизина
5,25 г (7,72 ммоля) 4-[2,3-бис-(N,N-бис(трет-бутилоксикарбонилметил)амино)пропил]фенил}-3-оксапропионовой кислоты и 781,0 мг (7,72 ммоля) триэтиламина растворяют в 50 мл метиленхлорида. Далее при -15°С по каплям в течение 5 мин добавляют раствор 1,16 г (8,5 ммоля) изобутилового эфира хлормуравьиной кислоты в 10 мл метилхлорида и перемешивают еще в течение 20 мин при -15°С. Далее раствор охлаждают до -25°С, в течение 30 мин по каплям добавляют раствор из 2,68 г (3,86 ммоля) указанного в заголовке примера 64г соединения и 2,12 г (21,0 ммоль) триэтиламина в 70 мл тетрагидрофурана, после чего перемешивают еще в течение 30 мин при -15°С, а затем в течение ночи при комнатной температуре. Для переработки растворитель отгоняют в вакууме и полученный маслянистый остаток растворяют в 250 мл хлороформа. Хлороформную фазу дважды экстрагируют 10%-ным водным раствором хлорида аммония порциями по 100 мл, органическую фазу сушат над сульфатом магния и досуха упаривают в вакууме. Остаток хроматографируют на силикагеле (элюент:метиленхлорид/этанол в соотношении 20:1).
Выход: 5,37 г (68,8% от теории) бесцветного и высоковязкого масла.
Элементный анализ:
е) [1-(4-перфтороктилсульфонил)пиперазин]амид 6N-2N-бис{4-[2,3-бис(N,N-бис(карбоксилатометил)амино)пропил]фенил}-3-оксапропионил-L-лизина, октанатриевая соль
5,0 г (2,47 ммоля) соединения, указанного в заголовке примера 64д, растворяют в 60 мл абсолютного дихлорметана. После этого полученный раствор при 0°С по каплям смешивают с трифторуксусной кислотой общим количеством 75 мл. После проведения реакции в течение 12 ч при комнатной температуре смесь досуха концентрируют в вакууме. Полученный остаток смешивают с 100 мл воды и вновь перегоняют в вакууме до получения сухого остатка. Полученный таким путем остаток растворяют в 200 мл дистиллированной воды и водный, содержащий продукт раствор указанного выше в заголовке соединения дважды экстрагируют диэтиловым эфиром порциями по 60 мл. Общий объем полученного водного содержащего продукт раствора доводят смешением с водой до 300 мл, отфильтровывают нерастворившиеся компоненты и фильтрат подвергают ультрафильтрации через ультрафильтрационную мембрану AMICON® YM-3 (предельная проницаемость: 3000 Да), что обеспечивает полное обессоливание указанного в заголовке соединения и одновременно его очистку от низкомолекулярных компонентов. Общий объем ретентата смешением с водой доводят до 200 мл и затем значение рН этого раствора устанавливают на 10,0 с помощью 15%-ного едкого натра. Щелочной водный содержащий продукт раствор в завершение лиофилизуют.
Таким путем получают 4,0 г (92,8% от теории) указанного в заголовке соединения в виде октанатриевой соли в форме аморфного лиофилизата.
Содержание воды: 5,37%.
Элементный анализ (в пересчете на безводное вещество):
ж) [1-(4-перфтороктилсульфонил)пиперазин]амид 6N-2N-бис{4-[2,3-бис(N,N-бис(карбоксиметил)амино)пропил]фенил}-3-оксапропионил-L-лизина, димарганцевый комплекс, тетранатриевая соль
1,94 г (1,11 ммоля) соединения, указанного в заголовке примера 64е, растворяют в 100 мл дистиллированной воды и значение рН полученного раствора устанавливают на 4,0 смешением с 1-молярной водной соляной кислотой. Далее при 80°С порциями смешивают с 0,25 г (2,22 ммоля) карбоната марганца(II). Полученный таким путем реакционный раствор затем в течение 5 ч кипятят с обратным холодильником. После охлаждения до комнатной температуры значение рН водного содержащего продукт раствора устнавливают при интенсивном перемешивании на 7,2 смешением с 1н. едким натром и для обессоливания и для очистки от низкомолекулярных компонентов пропускают через ультрафильтрационную мембрану AMICON® YM-3 (предельная проницаемость: 3000 Да). Ретентат в завершение лиофилизуют.
Выход: 1,80 г (92,0% от теории) указанного в заголовке соединения в виде бесцветного лиофилизата.
Н2O-содержание (при использовании реактива Карла Фишера): 7,28%.
Элементный анализ (в пересчете на безводное вещество):
Пример 65
а) [1-(4-перфтороктилсульфонил)пиперазин]амид 6-N-(бензилоксикарбонил)-2-N-[(N-птероил)-L-глутаминил]лизина
20 г (45,31 ммоля) фолиевой кислоты растворяют в 300 мл диметилсульфоксида и при 10°С добавляют 9,49 г (46 ммолей) N,N-дициклогексилкарбодиимида. Смесь перемешивают в течение ночи при комнатной температуре. Далее к этой смеси добавляют 29,1 г (35 ммолей) соединения, указанного в заголовке примера 52в, и 20 мл пиридина и перемешивают в течение 3 ч при 50°С. Затем охлаждают до комнатной температуры и добавляют смесь из 1500 мл диэтилового эфира и 1500 мл ацетона. Выпавший осадок отфильтровывают и очищают на силикагеле RP-18 (элюент:градиент воды/этанола/тетрагидрофурана).
Выход: 21,59 г (38% от теории) желтого твердого вещества.
Содержание воды: 2,1%.
Элементный анализ (в пересчете на безводное вещество):
б) [1-(4-перфтороктилсульфонил)пиперазин]амид 2-N-[(N-птероил)-L-глутаминил]лизина
К 20 г (15,95 ммоля) соединения, указанного в заголовке примера 65а, добавляют 200 мл бромистоводородной кислоты в ледяной уксусной кислоте (48%-ной) и перемешивают в течение 2 ч при 40°С. Далее охлаждают до 0°С, по каплям добавляют 2000 мл диэтилового эфира и выпавшее в осадок твердое вещество отфильтровывают. После сушки в вакууме (60°С) получают 18,96 г (99% от теории) кристаллического твердого вещества желтого цвета.
Элементный анализ:
в) [1-(4-перфтороктилсульфонил)пиперазин]амид 6-N-[1,4,7-трис(карбоксилатометил)-1,4,7,10-тетраазациклододекан-10-(пентаноил-3-аза-4-оксо-5-метил-5-ил]-2-N-[(N-птероил)-L-глутаминил]лизина, Gd-комплекс
0,92 г (8 ммолей) N-гидроксисукцинимида, 0,68 г (16 молей) хлорида лития и 5,04 г (8 ммолей) Gd-комплекса 1,4,7-трис(карбоксилатометил)-10-(3-аза-4-оксо-5-метил-5-ил)-1,4,7-10-тетраазациклододекана растворяют при умеренном нагревании в 80 мл диметилсульфоксида. Далее при 10°С добавляют 2,06 г (10 молей) N,N-дициклогексилкарбодиимида и затем перемешивают в течение 3 ч при комнатной температуре. К этому реакционному раствору добавляют 5 г (4,16 ммоля) соединения, указанного в заголовке примера 65б, и 10 мл пиридина. Смесь перемешивают в течение ночи при комнатной температуре. Затем раствор сливают в 1000 мл ацетона и перемешивают в течение 10 мин. Выпавшее в осадок твердое вещество отфильтровывают и после этого очищают хроматографией (силикагель RP-18, элюент:градиент воды/этанола/ацетонитрила). Продукт растворяют в небольшом количестве воды, значение рН устанавливают на 7,4 с помощью едкого натра и лиофилизуют.
Выход: 3,87 г (53% от теории) желтого твердого вещества.
Содержание воды: 5,8%.
Элементный анализ (в пересчете на безводное вещество):
Пример 66
а) N-(2,3-дигидроксипропил)амид 2Н,2Н,4Н,4Н,5Н,5Н-3-окса)перфтортридекановой кислоты
К 30 г (57,45 ммоля) 2Н,2Н,4Н,4Н,5Н,5Н-3-оксаперфтортридекановой кислоты в 300 мл дихлорметана добавляют 8,90 г (70 ммолей) оксалилхлорида и перемешивают в течение 12 ч при комнатной температуре. Смесь упаривают в вакууме досуха. Остаток растворяют в 100 мл дихлорметана и при 0°С по каплям добавляют к раствору из 5,47 г (60 ммолей) 2,3-дигидроксипропиламина и 6,07 г (60 ммолей) триэтиламина в 200 мл дихлорметана. Смесь перемешивают в течение 3 ч при 0°С, а затем в течение 6 ч при комнатной температуре. Далее добавляют 300 мл 5%-ной водной соляной кислоты и тщательно перемешивают в течение 15 мин. Органическую фазу отделяют, сушат над сульфатом магния и досуха упаривают в вакууме. Остаток хроматографируют на силикагеле (элюент:дихлорметан/этанол в соотношении 15:1).
Выход: 29,70 г (87% от теории) бесцветного твердого вещества.
Элементный анализ:
б) N-(2,3-дигидроксипропил)-N-(1Н,1Н,2Н,2Н,4Н,4Н,5Н,5Н-3-оксаперфтортридецил)амин
30 г (48,8 ммоля) соединения, указанного в заголовке примера 66а, растворяют в 300 мл тетрагидрофурана и добавляют 50 мл 10 М борандиметилсульфида (в тетрагидрофуране). Далее в течение 16 ч кипятят с обратным холодильником. После этого охлаждают до 0°С, по каплям добавляют 300 мл метанола и затем досуха упаривают в вакууме. Остаток растворяют в смеси из 300 мл этанола и 50 мл 10%-ной водной соляной кислоты и перемешивают в течение 8 ч при 60°С. Смесь досуха упаривают в вакууме, остаток растворяют в 300 мл 5%-ного водного едкого натра и трижды экстрагируют дихлорметаном порциями по 300 мл. Органические фазы сушат над сульфатом магния, досуха упаривают в вакууме и остаток хроматографируют на силикагеле (элюент:дихлорметан/метанол в соотношении 15:1).
Выход: 24,07 г (85% от теории) бесцветного твердого вещества.
Элементный анализ:
в) 1,4,7-трис(карбоксилатометил)-10-[(3-аза-4-оксогексан-5-ил)-кислота-N-(2,3-дигидроксипропил)-N-(1Н,1Н,2Н,2Н,4Н,4Н,5Н,5Н-3-окса)перфтортридецил)амид]-1,4,7,10-тетраазациклододекан, гадолиниевый комплекс
10 г (15,88 ммоля) гадолиниевого комплекса 10-[1-(карбоксиметилкарбамоил)этил]-1,4,7,10-тетраазациклододекан-1,4,7-триуксусной кислоты и 1,35 г (31,76 ммоля) хлорида лития при 60°С растворяют в 100 мл диметилсульфоксида. Далее охлаждают до 15°С и добавляют 9,21 г (15,88 ммоля) соединения, указанного в заголовке примера 66б. Смесь перемешивают в течение 10 мин и затем добавляют 7,42 г (30 ммолей) 2-этокси-1-этоксикарбонил-1,2-дигидрохинолина. После этого перемешивают в течение 12 ч при комнатной температуре. Раствор сливают в смесь из 200 мл ацетона и 1300 мл диэтилового эфира и перемешивают в течение 2 ч при комнатной температуре. Выпавший осадок отфильтровывают, растворяют в смеси из небольшого количества этанола и воды и хроматографируют на силикагеле RP-18 (элюент:градиент тетрагидрофурана/ацетонитрила/воды).
Выход: 16,09 г (85% от теории) бесцветного аморфного порошка.
Содержание воды: 6,3%.
Элементный анализ (в пересчете на безводное вещество):
Пример 67
1,4,7-трис(карбоксилатометил)-10-[(3-аза-4-оксогексан-5-ил)-кислота-N-(1Н,1Н,2Н,2Н,4Н,4Н,5Н,5Н-3-оксаперфтортридецил)амид]-1,4,7,10-тетраазациклододекан, гадолиниевый комплекс
10 г (15,88 ммоля) гадолиниевого комплекса 10-[1-(карбоксиметилкарбамоил)этил]-1,4,7,10-тетраазациклододекан-1,4,7-триуксусной кислоты, 1,35 г (31,76 ммоля) хлорида лития и 3,66 г (31,76 ммоля) N-гидроксисукцинимида при 60°С растворяют в 100 мл диметилсульфоксида. Далее охлаждают до 15°С, добавляют 3,51 г (17 ммолей) N,N'-дициклогексилкарбодиимида и перемешивают в течение 5 ч при 15°С. Для отделения мочевины раствор фильтруют. К фильтрату добавляют 8,63 г (15,88 ммоля) соединения, указанного в заголовке примера 68б, и 5,06 г (50 ммолей) триэтиламина и перемешивают в течение 12 ч при комнатной температуре. Раствор сливают в смесь из 1500 мл диэтилового эфира и 100 мл ацетона и перемешивают в течение 30 мин. Выпавшее в осадок твердое вещество отфильтровывают и хроматографируют на силикагеле RP-18 (элюент:градиент тетрагидрофурана/ацетонитрила/воды).
Выход: 13,86 г (78% от теории) бесцветного аморфного порошка.
Содержание воды: 9,3%.
Элементный анализ (в пересчете на безводное вещество):
Пример 68
а) Амид 2Н,2Н,4Н,4Н,5Н,5Н-3-оксаперфтортридекановой кислоты
К 30 г (57,45 ммоля) 2Н,2Н,4Н,4Н,5Н,5Н-3-оксаперфтортридекановой кислоты в 300 мл дихлорметана добавляют 8,90 г (70 ммолей) оксалилхлорида и перемешивают в течение 12 ч при комнатной температуре. Смесь упаривают в вакууме досуха. Остаток растворяют в 200 мл дихлорметана. Затем раствор при 0°С барботируют газообразным аммиаком в течение примерно 2 ч. После этого смесь перемешивают в течение 4 ч при 0°С, а затем в течение 2 ч при комнатной температуре. Далее добавляют 300 мл 5%-ной водной соляной кислоты и тщательно перемешивают в течение 15 мин. Органическую фазу отделяют, сушат над сульфатом магния и досуха упаривают в вакууме. Остаток хроматографируют на силикагеле (элюент:дихлорметан/ацетон в соотношении 20:1).
Выход: 27,85 г (93% от теории).
Элементный анализ:
б) 1Н,1Н,2Н,2Н,4Н,4Н,5Н,5Н-3-оксаперфтортридециламин, гидрохлорид
27 г (51,8 ммоля) соединения, указанного в заголовке примера 68а, растворяют в 300 мл тетрагидрофурана и добавляют 31 мл 10 М борандиметилсульфида (в тетрагидрофуране). Далее в течение 16 ч кипятят с обратным холодильником. После этого охлаждают до 0°С, по каплям добавляют 200 мл метанола и затем досуха упаривают в вакууме. Остаток растворяют в смеси из 400 мл этанола и 100 мл 10%-ной водной соляной кислоты и перемешивают в течение 8 ч при 60°С. Смесь досуха упаривают в вакууме и остаток перекристаллизовывают из небольшого количества этанола/диэтилового эфира.
Выход: 26,75 г (95% от теории) бесцветного кристаллического твердого вещества.
Элементный анализ:
в) N-(1H,1Н,2Н,2Н,4Н,4Н,5Н,5Н-3-окса)перфтортридецил)амид 3,6,9,12,15-пентаоксагексадекановой кислоты
К 26,5 г (48,74 ммоля) соединения, указанного в заголовке примера 68б, и 14,8 г (146,2 ммоля) триэтиламина, растворенных в 300 мл дихлорметана, при 0°С добавляют 14,24 г (50 ммолей) хлорангидрида 3,6,9,12,15-пентаоксагексадекановой кислоты и перемешивают в течение 3 ч при 0°С. Далее добавляют 300 мл 5%-ной водной соляной кислоты и тщательно перемешивают в течение 30 мин. Органическую фазу отделяют, сушат над сульфатом магния и досуха упаривают в вакууме. Остаток хроматографируют на силикагеле (элюент:дихлорметан/ацетон в соотношении 20:1).
Выход: 32,03 г (87% от теории) бесцветного масла.
Элементный анализ:
г) N-(3,6,9,12,15-пентаоксагексадецил)-N-(1Н,1Н,2Н,2Н,4Н,4Н-3-окса)перфтортридецил)амид
31 г (41,03 ммоля) соединения, указанного в заголовке примера 68в, растворяют в 300 мл тетрагидрофурана и добавляют 25 мл 10 М борандиметилсульфида (в тетрагидрофуране). Далее в течение 16 ч кипятят с обратным холодильником. После этого охлаждают до 0°С, по каплям добавляют 200 мл метанола и затем досуха упаривают в вакууме. Остаток растворяют в смеси из 300 мл этанола и 50 мл 10%-ной водной соляной кислоты и перемешивают в течение 8 ч при 40°С. Смесь досуха упаривают в вакууме, остаток растворяют в 300 мл 5%-ного водного едкого натра и трижды экстрагируют дихлорметаном порциями по 300 мл. Органические фазы сушат над сульфатом магния, досуха упаривают в вакууме и остаток хроматографируют на силикагеле (элюент:дихлорметан/2-пропанол в соотношении 15:1).
Выход: 27,68 г (91% от теории).
Элементный анализ:
д) 1,4,7-трис(карбоксилатометил)-10-{(3-аза-4-оксогексан-5-ил)-кислота-[N-(3,6,9,12,15-пентаоксагексадецил)-N-(1Н,1Н,2Н,2Н,4Н,4Н,5Н,5Н-3-окса)перфтортридецил]амид}-1,4,7,10-тетраазациклододекан, гадолиниевый комплекс
10 г (15,88 ммоля) гадолиниевого комплекса 10-[1-(карбоксиметилкарбамоил)этил]-1,4,7,10-тетраазациклододекан-1,4,7-триуксусной кислоты и 1,35 г (31,76 ммоля) хлорида лития при 60°С растворяют в 100 мл диметилсульфоксида. Далее охлаждают до 15°С и добавляют 11,77 г (15,88 ммоля) соединения, указанного в заголовке примера 68г. Смесь перемешивают в течение 10 мин и затем добавляют 7,42 г (30 ммолей) 2-этокси-1-этоксикарбонил-1,2-дигидрохинолина. Смесь перемешивают в течение 12 ч при комнатной температуре. Раствор сливают в смесь из 200 мл ацетона и 1300 мл диэтилового эфира и перемешивают в течение 2 ч при комнатной температуре. Выпавший осадок отфильтровывают, растворяют в смеси из небольшого количества этанола и воды и хроматографируют на силикагеле RP-18 (элюент:градиент тетрагидрофурана/ацетонитрила/воды).
Выход: 18,05 г (84% от теории) бесцветного аморфного порошка.
Содержание воды: 6,2%.
Элементный анализ (в пересчете на безводное вещество):
Пример 69
а) N-(5-гидрокси-3-оксапентил)амид 2Н,2Н,4Н,4Н,5Н,5Н-3-оксаперфтортридекановой кислоты
К 30 г (57,45 ммоля) 2Н,2Н,4Н,4Н,5Н,5Н-3-оксаперфтортридекановой кислоты в 300 мл дихлорметана добавляют 8,90 г (70 ммолей) оксалилхлорида и перемешивают в течение 12 ч при комнатной температуре. Смесь упаривают в вакууме досуха. Остаток растворяют в 100 мл дихлорметана и при 0°С по каплям добавляют к раствору из 6,25 г (60 ммолей) 5-гидрокси-3-оксапентиламина и 6,07 г (60 ммолей) триэтиламина в 200 мл дихлорметана. Смесь перемешивают в течение 3 ч при 0°С, а затем в течение 6 ч при комнатной температуре. Далее добавляют 300 мл 5%-ной водной соляной кислоты и тщательно перемешивают в течение 15 мин. Органическую фазу отделяют, сушат над сульфатом магния и досуха упаривают в вакууме. Остаток хроматографируют на силикагеле (элюент:дихлорметан/ацетон в соотношении 15:1).
Выход: 32,20 г (92% от теории) бесцветного твердого вещества.
Элементный анализ:
б) N-(5-гидрокси-3-оксапентил)-N-(1Н,1Н,2Н,2Н,4Н,4Н,5Н,5Н-3-оксаперфтортридецил)амин
30 г (49,24 ммоля) соединения, указанного в заголовке примера 69а, растворяют в 300 мл тетрагидрофурана и добавляют 31 мл 10 М борандиметилсульфида (в тетрагидрофуране). Далее в течение 16 ч кипятят с обратным холодильником. После этого охлаждают до 0°С, по каплям добавляют 200 мл метанола и затем досуха упаривают в вакууме. Остаток растворяют в смеси из 300 мл этанола и 50 мл 10%-ной водной соляной кислоты и перемешивают в течение 10 ч при 50°С. Смесь досуха упаривают в вакууме, остаток растворяют в 300 мл 5%-ного водного едкого натра и трижды экстрагируют дихлорметаном порциями по 300 мл. Органические фазы сушат над сульфатом магния, досуха упаривают в вакууме и остаток хроматографируют на силикагеле (элюент:дихлорметан/2-пропанол в соотношении 20:1).
Выход: 26,09 г (89% от теории) бесцветного твердого вещества.
Элементный анализ:
в) 1,4,7-трис(карбоксилатометил)-10-[(3-аза-4-оксогексан-5-ил)-кислота-N-(5-гидрокси-3-окса-пентил)-N-(1Н,1Н,2Н,2Н,4Н,4Н,5Н,5Н-3-окса)перфтортридецил)амид]-1,4,7,10-тетраазациклододекан, гадолиниевый комплекс
10 г (15,88 ммоля) гадолиниевого комплекса 10-[1-(карбоксиметилкарбамоил)этил]-1,4,7,10-тетраазациклододекан-1,4,7-триуксусной кислоты и 1,35 г (31,76 ммоля) хлорида лития при 60°С растворяют в 100 мл диметилсульфоксида. Далее охлаждают до 15°С и добавляют 9,45 г (15,88 ммоля) соединения, указанного в заголовке примера 69б. Смесь перемешивают в течение 10 мин и затем добавляют 7,42 г (30 ммолей) 2-этокси-1-этоксикарбонил-1,2-дигидрохинолина. Смесь перемешивают в течение 12 ч при комнатной температуре. Раствор сливают в смесь из 200 мл ацетона и 1300 мл диэтилового эфира и перемешивают в течение 2 ч при комнатной температуре. Выпавший осадок отфильтровывают, растворяют в смеси из небольшого количества этанола и воды и хроматографируют на силикагеле RP-18 (элюент:градиент тетрагидрофурана/ацетонитрила/воды).
Выход: 16,10 г (84% от теории) бесцветного аморфного порошка.
Содержание воды: 5,7%.
Элементный анализ (в пересчете на безводное вещество):
Пример 70
а) 1,2,3,4,6-пента-O-ацетил-α,β-D-маннопираноза
Аналогично описанному в литературе методу [M.L.Wolfrom и A.Thompson, "Methods in Carbohydrate Chemistry" (под ред. R.L.Whistler, M.L.Wolfrom и J.N.BeMiller), изд-во Academic Press, New York, т.II, 53, сс.211-215 (1963)] в результате взаимодействия 150 г (832,5 ммоля) α,β-D-маннопиранозы со смесью из 1500 мл абсолютного пиридина и 1500 мл уксусного ангидрида после переработки получают 315 г (96,7%) указанного выше в заголовке соединения в виде сырого продукта в форме вязкого и бесцветного масла. Соотношение между обоими α- и β-аномерами в полученном таким путем указанном в заголовке соединении составляет 4:1 по данным 1Н-ЯМР-спектроскопического анализа. Для проведения последующих реакций указанное выше в заголовке соединение можно не разделять на отдельные α,β-аномеры.
Элементный анализ:
б) Этиловый эфир 6-[1-O-α-(2,3,4,6-тетра-O-ацетил-D-маннопиранозил)]гексановой кислоты
Аналогично методу, описанному в литературе для синтеза арилгликопиранозидов [J.Conchie и G.A.Levvy, "Methods in Carbohydrate Chemistry" (под ред. R.L. Whistler, M.L. Wolfrom и J.N. BeMiller), изд-во Academic Press, New York, т.II, 90, сс.345-347 (1963)] в результате взаимодействия 156,2 г (400 ммолей) указанного в заголовке примера 70а в виде смеси α,β-аномеров с 67 мл (400 ммолей) этилового эфира 6-гидроксигексановой кислоты и 60,8 мл (520 ммолей) хлорида олова(IV) в 1,2-дихлорэтане общим количеством 600 мл после очистки колоночной хроматографией (элюент:гексан/этиловый эфир уксусной кислоты в соотношении 2:1) получают 100,05 г (51% от теории) указанного выше в заголовке соединения в виде бесцветного и вязкого масла. Данные 1Н-ЯМР-спектроскопического анализа полученного таким путем указанного в заголовке соединения свидетельствуют о том, что это указанное в заголовке соединение представляет собой только чистый α-аномер.
Элементный анализ:
в) 6-[1-O-α-(2,3,4,6-тетра-O-бензил-D-маннопиранозил)гексановая кислота
Перемешанную суспензию из 141,0 г (289 ммолей) указанного в заголовке примера 70б соединения в 200 мл диоксана порциями смешивают при комнатной температуре и одновременно интенсивном перемешивании с высокодисперсным порошковым гидроксидом калия общим количеством 238,5 г (4,26 моля). Реакционную смесь для повышения ее размешиваемости смешивают еще с 200 мл диоксана, полученную в результате суспензию затем нагревают до температуры кипения и при этой температуре в течение двух часов по каплям смешивают с бензилбромидом общим количеством 372 мл (3,128 моля). После проведения реакции в течение 4 ч при 110°С, а затем в течение 12 ч при комнатной температуре реакционную смесь для переработки медленно сливают в смесь воды со льдом общим количеством 2,5 л и водную фазу затем полностью экстрагируют диэтиловым эфиром. После промывки полученной таким путем эфирной фазы и ее последующей сушки над сульфатом натрия соль отделяют вакуум-фильтрацией и диэтиловый эфир отгоняют в вакууме. После этого избыток бензилбромида количественно отгоняют из реакционной смеси в вакууме, создаваемом масляным насосом, при температуре масляной бани 180°С. Полученный таким путем смолянисто-маслянистый остаток очищают на силикагеле с использованием этилового эфира уксусной кислоты/гексана (в соотношении 1:10) в качестве элюента.
Выход: 172,2 г (91,0% от теории) указанного выше в заголовке соединения в виде бесцветного и высоковязкого масла.
Элементный анализ:
г) N-(3-окса-1Н,1Н,2Н,2Н,4Н,4Н,5Н,5Н-перфтортридецил)амид 6-[1-O-α-(2,3,4,6-тетра-O-бензил-D-маннопиранозил)гексановой кислоты
100 г (134 ммоля) описанной в примере 70в кислоты, а также 13,5 г (134 ммоля) триэтиламина растворяют в 1200 мл сухого тетрагидрофурана. После охлаждения до -15°С по каплям при перемешивании медленно добавляют раствор 18,45 г (135 ммолей) изобутилового эфира хлормуравьиной кислоты в 200 мл сухого тетрагидрофурана, при этом внутренняя температура не должна превышать -10°С. После проведения реакции в течение 15 мин при -15°С по каплям при -20°С добавляют раствор 165,5 г (134 ммоля) 1-амино-1Н,1Н,2Н,2Н-перфтордекана и 13,5 г (134 ммоля) триэтиламина в 250 мл сухого тетрагидрофурана. После проведения реакции в течение часа при -15°С, а затем в течение двух часов при комнатной температуре реакционный раствор досуха концентрируют в вакууме. Полученный остаток растворяют в 300 мл этилового эфира уксусной кислоты и дважды промывают 400 мл насыщенным раствором гидрокарбоната натрия порциями по 400 мл и однократно водой порцией 500 мл. После сушки органической фазы над сульфатом натрия соль отделяют вакуум-фильтрацией и этиловый эфир уксусной кислоты отгоняют в вакууме. Полученный маслянистый остаток очищают на силикагеле с использованием дихлорметана/гексана/2-пропанола (в соотношении 10:5:1) в качестве элюента.
Выход: 143,8 г (86,9% от теории).
Элементный анализ:
д) N-(3-окса-1Н,1Н,2Н,2Н,4Н,4Н,5Н,5Н-перфтортридецил)амид 6-[1-O-α-D-маннопиранозил)гексановой кислоты
40,0 г (32,38 ммоля) соединения, указанного в заголовке примера 70г, растворяют в 750 мл 2-пропанола и смешивают с 2,0 г палладиевого катализатора (10%-ный Pd/C). Затем реакционный раствор гидрируют в течение 12 ч при 22°С и давлении водорода 1 атмосфера. После этого катализатор отфильтровывают и фильтрат концентрируют досуха. Полученный остаток растворяют в 300 мл диметилсульфоксида и из полученного таким путем содержащего продукт раствора смешением с диэтиловым эфиром общим количеством 1000 мл и после отделения выпавшего в осадок твердого вещества вакуум-фильтрацией получают 21,52 г (88,0% от теории) указанного выше в заголовке соединения в виде бесцветного кристаллического порошка с температурой плавления 88,5°С (с разложением).
Элементный анализ:
е) Получение композиции, содержащей металлический комплекс I и N-(3-окса-1Н,1Н,2Н,2Н,4Н,4Н,5Н,5Н-перфтортридецил)амид 6-[1-O-α-D-маннопиранозил)гексановой кислоты
К 35 мл раствора металлического комплекса I (280 ммолей/л) в 0,45%-ном водном растворе поваренной соли (рН 7,4, 0,25 мг/л CaNa3ДТПА) добавляют 3,17 г (4,2 ммоля) соединения, указанного в заголовке примера 70д, и объем с помощью 0,9%-ного водного раствора поваренной соли доводят до 98 мл. Далее выдерживают в течение 2 ч при 60°С, подвергая смесь воздействию ультразвука. Раствор охлаждают до комнатной температуры и значение рН устанавливают на 7,4 с помощью водного 2н. едкого натра. После этого фильтруют через фильтр с размером пор 0,2 мкм и фильтрат расфасовывают во флаконы. Полученный таким путем раствор можно непосредственно использовать для проведения биологических экспериментов. (Концентрация Gd составляет 100 ммолей/л).
Пример 71
а) 1-O-α-D-[(1-перфтороктилсульфонилпиперазин-4-карбонил)пентил-5]-2,3,4,6-тетра-О-бензилманнопираноза
В 800 мл смеси из тетрагидрофурана и ацетонитрила (в соотношении 7:3) растворяют 74,59 г (100 ммолей) описанной в примере 71в кислоты, а также 10,11 г (100 ммолей) триэтиламина. После этого при комнатной температуре по каплям смешивают с 500 мл раствора 58,0 г (102,0 ммоля) 1-перфтороктилсульфонилпиперазина в тетрагидрофуране, 10,11 г (100 ммолей) триэтиламина и 16,84 г (110 ммолей) 1-гидроксибензотриазола. Полученный таким путем реакционный раствор смешивают при -5°С с раствором 22,7 г (110 ммолей) дициклогексилкарбодиимида в 100 мл тетрагидрофурана и затем перемешивают при -5°С в течение двух последующих часов. После оттаивания реакционного раствора его перемешивают при комнатной температуре еще в течение 12 ч, выпавшую в осадок дициклогексилмочевину отфильтровывают и полученный фильтрат досуха концентрируют в вакууме. Полученный остаток растворяют в 600 мл этилового эфира уксусной кислоты и дважды промывают насыщенным раствором гидрокарбоната натрия порциями по 300 мл и дважды водой порциями по 300 мл. После сушки органической фазы над сульфатом натрия соль отделяют вакуум-фильтрацией и этиловый эфир уксусной кислоты отгоняют в вакууме. Полученный маслянистый остаток очищают на силикагеле с использованием дихлорметана/ацетона/2-пропанола (в соотношении 16:2:1) в качестве элюента
Выход: 113,01 г (79,8% от теории) бесцветного и вязкого масла.
Элементный анализ:
б) 1-O-α-D-[(1-перфтороктилсульфонилпиперазин-4-карбонил)пентил-5]-маннопираноза
50 г (35,30 ммоля) соединения, указанного в заголовке примера 71а, растворяют в смеси из 500 мл 2-пропанола и 50 мл воды и добавляют 2 г палладиевого катализатора (10%-ный Pd на активированном угле). После этого гидрируют в течение 12 ч при комнатной температуре. Затем катализатор отфильтровывают и фильтрат досуха упаривают в вакууме. Остаток растворяют в 200 мл метанола и реакционный продукт осаждают смешением с диэтиловым эфиром общим количеством 800 мл. После отделения полученного таким путем твердого вещества вакуум-фильтрацией его сушат в вакууме при 50°С.
Выход: 29,51 г (99% от теории) аморфного твердого вещества.
Элементный анализ:
в) Получение композиции, содержащей металлический комплекс II и 1-O-α-D-[(1-перфтороктилсульфонилпиперазин-4-карбонил)пентил-5]маннопиранозу
К 47 мл раствора металлического комплекса II (250 ммолей/л) в 0,45%-ном водном растворе хлорида натрия добавляют 9,92 г (11,75 ммоля) соединения, указанного в заголовке примера 71б, и в течение 10 мин нагревают в микроволновой печи. Раствор охлаждают до комнатной температуры, фильтруют через фильтр с размером пор 0,2 мкм и фильтрат расфасовывают во флаконы. Полученный таким путем раствор можно непосредственно использовать для проведения биологических экспериментов. (Концентрация Gd составляет 250 ммолей/л.)
Пример 72
а) 2-ацетамидо-2-дезокси-1,3,4,6-(тетра-O-бензил)-α,β-D-глюкопираноза
К перемешанной суспензии 20,16 г (700 ммолей) гидрида натрия (80%-ный в минеральном масле) в 150 мл диметилсульфоксида при комнатной температуре по каплям добавляют в целом 24,0 г (108,5 ммоля) 2-ацетамидо-2-дезокси-α,β-D-глюкопиранозы, растворенной в 500 мл абсолютного диметилсульфоксида. После этого на 120 мин оставляют для перемешивания при комнатной температуре и затем по каплям добавляют 159,5 г (1,26 моля) бензилхлорида. Полученный таким путем реакционный раствор затем перемешивают еще в течение 12 ч при комнатной температуре. Для переработки реакционный раствор медленно сливают в 1,5 л смеси воды со льдом и после этого практически полностью экстрагируют диэтиловым эфиром. Объединенные диэтилэфирные фазы затем дважды промывают насыщенным раствором гидрокарбоната натрия порциями по 600 мл и дважды водой порциями по 800 мл. После сушки органической фазы над сульфатом натрия соль отделяют вакуум-фильтрацией и растворитель отгоняют в вакууме. Полученный маслянистый остаток очищают на силикагеле с использованием этилового эфира уксусной кислоты/гексана (в соотношении 1:5) в качестве элюента.
Выход: 48,68 г (73,6% от теории) указанного выше в заголовке соединения в виде вязкого и бесцветного масла.
Элементный анализ:
б) 1-O-бензил-3,4,6-три-O-бензил-2-амино-2-дезокси-α,β-D-глюкопираноза
30,0 г (49,2 ммоля) соединения, указанного в заголовке примера 72а, суспендируют в смеси из 750 мл метанола и 215 мл воды и при комнатной температуре по каплям смешивают с 0,112-молярным водным раствором перхлорной кислоты общим количеством 440 мл (49,2 ммоля). По завершении этого добавления реакционный раствор перемешивают еще в течение 10 мин при комнатной температуре и полученный в результате гомогенный реакционный раствор затем досуха концентрируют в вакууме. Полученный маслянистый остаток кристаллизуют его смешением со смесью гексана и дихлорметана, взятыми в равной пропорции. Кристаллический реакционный продукт отделяют вакуум-фильтрацией, промывают гексаном и сушат в вакууме при комнатной температуре.
Выход: 27,08 г (86% от теории) указанного выше в заголовке соединения в виде его перхлората, представляющего собой бесцветное кристаллическое соединение.
Температура плавления: 180,5-181,5°С.
Элементный анализ:
в) 1,3,4,6-тетра-O-бензил-2-дезокси-2-[ацетил-(2-амино-N-этил-N-перфтороктилсульфонил)амино]-1-α,β-D-глюкопираноза
20,8 г (35,6 ммоля) 2-[N-этил-N-перфтороктилсульфонил)аминоуксусной кислоты и 3,60 г (35,6 ммоля) триэтиламина растворяют в 350 мл сухого тетрагидрофурана. После охлаждения реакционного раствора до температуры в пределах от -15 до -20°С при этой температуре по каплям при перемешивании медленно добавляют раствор 4,92 г (35,6 ммоля) изобутилового эфира хлормуравьиной кислоты в 75 мл сухого тетрагидрофурана, при этом скорость капельного добавления должна быть такой, чтобы внутренняя температура не превышала -10°С. После проведения реакции в течение 15 мин при -15°С по каплям при -20°С медленно добавляют раствор 22,78 г (35,6 ммоля) перхлората указанного в заголовке примера 72б соединения и 3,60 г (35,6 ммоля) триэтиламина в 100 мл сухого тетрагидрофурана. После проведения реакции в течение часа при -15°С, а затем в течение двух часов при комнатной температуре реакционный раствор досуха концентрируют в вакууме. Полученный остаток растворяют в 250 мл этилового эфира уксусной кислоты и дважды промывают насыщенным раствором гидрокарбоната натрия порциями по 100 мл и однократно водой порцией 200 мл. После сушки органической фазы над сульфатом натрия соль отделяют вакуум-фильтрацией и этиловый эфир уксусной кислоты отгоняют в вакууме. Полученный маслянистый остаток очищают на силикагеле с использованием этилового эфира уксусной кислоты/гексана (в соотношении 1:5) в качестве элюента.
Выход: 33,3 г (84,6% от теории) указанного выше в заголовке соединения в виде бесцветного и высоковязкого масла.
Элементный анализ:
г) 2-дезокси-2-[ацетил-(2-амино-N-этил-N-перфтороктилсульфонил)амино]-1-α,β-D-глюкопираноза
20,0 г (18,06 ммоля) соединения, указанного в заголовке примера 72в, растворяют в 250 мл 2-пропанола и смешивают с 1,5 г палладиевого катализатора (10%-ный Pd/C). Реакционный раствор гидрируют в течение 12 ч при 22°С и давлении водорода 1 атмосфера. После этого катализатор отфильтровывают и фильтрат концентрируют досуха. Полученный остаток растворяют в 300 мл диметилсульфоксида и из полученного таким путем содержащего продукт раствора смешением с 750 мл смеси диэтилового эфира и этилового эфира уксусной кислоты, взятыми в равных пропорциях, и после отделения выпавшего в осадок твердого вещества вакуум-фильтрацией получают 12,65 г (93,8% от теории) указанного выше в заголовке соединения в виде бесцветного кристаллического порошка. Это указанное выше в заголовке соединение представлено в виде смеси α/β-аномеров, при этом соотношение между обоими возможными аномерами составляет по данным 1Н-ЯМР-спектроскопического анализа примерно 1:1,2. В соответствии с этим указанное в заголовке соединение представляет собой смесь α/β-аномеров с практическим одинаковым соотношением между ними.
Температура плавления: 132,5-133°С.
Элементный анализ:
д) Получение композиции, содержащей металлический комплекс XIV и 2-дезокси-2-[ацетил-(2-амино-N-этил-N-перфтороктилсульфонил)амино]-1-α,β-D-глюкопиранозу
К 51 мл раствора металлического комплекса XIV (300 ммолей/л) в 0,45%-ном растворе хлорида натрия (рН 7,4; 0,25 мг/л CaN3ДТПА) добавляют раствор 4,90 г (6,57 ммоля) указанного в заголовке примера 3г соединения в 200 мл этанола и перемешивают в течение 2 ч при 50°С. Далее раствор досуха концентрируют в вакууме и объем остатка добавлением дистиллированной воды доводят до 153 мл. Смесь перемешивают в течение 10 мин при 40°С и фильтруют через фильтр с размером пор 0,2 мкм. Фильтрат расфасовывают во флаконы. Полученный таким путем раствор можно непосредственно использовать для проведения биологических экспериментов. (Концентрация Gd составляет 100 ммолей/л.)
Пример 73
а) 1,2,3,4,6-пента-O-ацетил-α-D-глюкопираноза
Аналогично методу, описанному для синтеза указанного в заголовке примера 70а соединения, в результате взаимодействия 100 г (555,0 ммолей) α-D-глюкопиранозы со смесью из 1000 мл абсолютного пиридина и 1000 мл уксусного ангидрида после переработки и перекристаллизации из 95%-ного водного этанола получают 190,6 г (88,0%) указанного выше в заголовке соединения в виде бесцветного кристаллического соединения. В полученном таким путем указанном в заголовке соединении соотношение между обоими возможными α- и β-аномерами составляет по данным 1Н-ЯМР-спектроскопического анализа примерно 98:2. В соответствии с этим указанное в заголовке соединение представлено только в виде аномера с α-конфигурацией.
Температура плавления: 110,5°С.
Элементный анализ:
б) 5-(этоксикарбонил)пентил-2,3,4,6-тетра-O-ацетил-α-D-глюкопиранозид
Аналогично методу, описанному для синтеза указанного в заголовке примера 70б соединения, в результате взаимодействия 130,0 г (332,8 ммоля) соединения, указанного в заголовке примера 4а, с 55,8 мл (332,8 ммоля) этилового эфира 6-гидроксигексановой кислоты и 50,6 мл (520 ммолей) хлорида олова(IV) в 500 мл 1,2-дихлорэтана после очистки колоночной хроматографией (элюент:гексан/этиловый эфир уксусной кислоты в соотношении 2:1) получают 101,85 г (62,4% от теории) указанного выше в заголовке соединения в виде бесцветного и вязкого масла. Данные 1Н-ЯМР-спектроскопического анализа указанного в заголовке соединения позволяют на основании величины констант взаимодействия J1,2=8,8 Гц сделать однозначный вывод о наличии β-конфигурации у аномерного центра, которая, кроме того, является единственной присутствующей у центра аномерии конфигурацией. В соответствии с этим указанное выше в заголовке соединение можно получить только в виде аномера с β-конфигурацией.
Элементный анализ:
в) 5-(карбокси)пентил-2,3,4,6-тетра-O-бензил-α-D-глюкопиранозид
Перемешанную суспензию 100,0 г (204,96 ммоля) соединения, указанного в заголовке примера 73б, в 150 мл диоксана при комнатной температуре и одновременном интенсивном перемешивании порциями смешивают с общим количеством 169,14 г (3,02 моля) высокодисперсного порошкового гидроксида калия. Реакционную смесь для повышения ее размешиваемости смешивают еще с 150 мл диоксана, полученную в результате суспензию затем нагревают до температуры кипения и при этой температуре в течение двух часов по каплям смешивают с бензилбромидом общим количеством 264 мл (2,218 моля). После проведения реакции в течение 4 ч при 110°С, а затем в течение 12 ч при комнатной температуре реакционную смесь для переработки медленно сливают в смесь воды со льдом общим количеством 2,0 л и водную фазу затем полностью экстрагируют диэтиловым эфиром. После промывки полученной таким путем эфирной фазы и последующей сушки органической фазы над сульфатом натрия соль отделяют вакуум-фильтрацией и диэтиловый эфир отгоняют в вакууме. После этого избыток бензилбромида количественно отгоняют из реакционной смеси в вакууме, создаваемом масляным насосом, при температуре масляной бани 180°С. Полученный таким путем маслянистый остаток очищают на силикагеле с использованием этилового эфира уксусной кислоты/гексана (в соотношении 1:10) в качестве элюента.
Выход: 128,8 г (84,3% от теории) указанного выше в заголовке соединения в виде бесцветного и высоковязкого масла.
Элементный анализ:
г) 2,3,4,6-тетра-O-бензил-1-O-β-D-[6-гексановая кислота-N-(3-окса-1Н,1Н,2Н,2Н,4Н,4Н,5Н,5Н-перфтортридецил)амид]глюкопираноза
68,5 г (91,79 ммоля) описанной в примере 73в кислоты, а также 9,25 г (91,79 ммоля) триэтиламина растворяют в 825 мл сухого тетрагидрофурана. После охлаждения реакционного раствора до температуры в пределах от -15 до -20°С при этой температуре по каплям при перемешивании медленно добавляют раствор 12,64 г (92,5 ммоля) изобутилового эфира хлормуравьиной кислоты в 150 мл сухого тетрагидрофурана, при этом скорость капельного добавления должна быть такой, чтобы внутренняя температура не превышала -10°С. После проведения реакции в течение 15 мин при -15°С по каплям при -20°С медленно добавляют раствор 46,40 г (91,79 ммоля) 1Н,1Н,2Н,2Н-гептадекафторо-1-(2-аминоэтокси)декана и 9,25 г (91,79 ммоля) триэтиламина в 200 мл сухого тетрагидрофурана. После проведения реакции в течение часа при -15°С, а затем в течение двух часов при комнатной температуре реакционный раствор досуха концентрируют в вакууме. Полученный остаток растворяют в 250 мл этилового эфира уксусной кислоты и дважды промывают насыщенным раствором гидрокарбоната натрия порциями по 300 мл и однократно водой порцией 400 мл. После сушки органической фазы над сульфатом натрия соль отделяют вакуум-фильтрацией и этиловый эфир уксусной кислоты отгоняют в вакууме. Полученный маслянистый остаток хроматографируют на силикагеле с использованием дихлорметана/гексана/2-пропанола (в соотношении 10:5:1) в качестве элюента.
Выход: 104,7 г (92,4% от теории) указанного выше в заголовке соединения в виде бесцветного и высоковязкого масла.
Элементный анализ:
д) 1-O-β-D-[6-гексановая кислота-N-(3-окса-1Н,1Н,2Н,2Н,4Н,4Н,5Н,5Н-перфтортридецил)амид]глюкопираноза
40,0 г (32,38 ммоля) соединения, указанного в заголовке примера 73г, растворяют в 750 мл 2-пропанола и смешивают с 2,0 г палладиевого катализатора (10%-ный Pd/C). Реакционный раствор гидрируют в течение 12 ч при 22°С и давлении водорода 1 атмосфера. Затем катализатор отфильтровывают и фильтрат концентрируют досуха. Полученный остаток растворяют в 300 мл диметилсульфоксида и из полученного таким путем содержащего продукт раствора смешением с диэтиловым эфиром общим количеством 1000 мл и последующего отделения выпавшего в осадок твердого вещества вакуум-фильтрацией получают 22,05 г (90,2% от теории) указанного в заголовке соединения в виде бесцветного кристаллического порошка с температурой плавления 122-124°С (с разложением).
Элементный анализ:
е) Получение композиции, содержащей указанное в заголовке примера 12 в заявке WO 99/01161 соединение (1,4,7-трис{1,4,7-трис(N-карбоксилатометил)-10-(N-1-метил-3-аза-2,5-диоксопентан-1,5-диил]-1,4,7,10-тетраазациклододекан, Gd-комплекс}-10-[2-(N-этил-N-перфтороктилсульфонил)амино]ацетил-1,4,7,10-тетраазациклододекан) и 1-O-β-D-[6-гексановая кислота-N-(3-окса-1Н,1Н,2Н,2Н,4Н,4Н,5Н,5Н-перфтортридецил)амид]глюкопиранозу
К 37 мл раствора 1,4,7-трис{1,4,7-трис(N-карбоксилатометил)-10-(N-1-метил-3-аза-2,5-диоксопентан-1,5-диил]-1,4,7,10-тетраазациклододекан, Gd-комплекс}-10-[2-(N-этил-N-перфтороктилсульфонил)амино]ацетил-1,4,7,10-тетраазациклододекана (300 ммолей/л) в 0,45%-ном водном растворе поваренной соли (рН 7,4; 0,25 мг/л CaNa3ДТПА) добавляют 20,29 г (25,9 ммоля) соединения, указанного в заголовке примера 73д, и объем с помощью 0,9%-ного водного раствора поваренной соли доводят до 111 мл. Далее выдерживают в течение 2 ч при 60°С, подвергая смесь воздействию ультразвука. Раствор охлаждают до комнатной температуры и значение рН устанавливают на 7,4 с помощью водного 2н. едкого натра. После этого фильтруют через фильтр с размером пор 0,2 мкм и фильтрат расфасовывают во флаконы. Полученный таким путем раствор можно непосредственно использовать для проведения биологических экспериментов. (Концентрация Gd составляет 100 ммолей/л.)
Пример 74
а) 1-O-(1Н,1Н,2Н,2Н-перфтородецил)-(2,3,4,6-тетра-O-ацетил)-α-D-маннопираноза
Взаимодействием 50 г (128,09 ммоля) указанного в заголовке примера 70а соединения, которое используют в виде смеси α,β-аномеров в соотношении между ними 4:1, с раствором 75,84 г (128,1 ммоля) 1-гидрокси-1Н,1Н,2Н,2Н-перфтородекана в 150 мл 1,2-дихлорэтана, а также с хлоридом олова(IV) общим количеством 19,47 г (166,53 ммоля) аналогично методу, описанному для синтеза указанных в заголовках примеров 1б и 4б соединений, после переработки и очистки колоночной хроматографией (элюент:гексан/этиловый эфир уксусной кислоты в соотношении 2:1) получают 74,2 г (63,4% от теории) указанного выше в заголовке соединения в виде вязкого бесцветного масла. Данные 1H-ЯМР-спектроскопического анализа указанного в заголовке соединения позволяют на основании величины констант взаимодействия J1,2=1,3 Гц сделать однозначный вывод о наличии α-конфигурации у аномерного центра, которая, кроме того, является единственной присутствующей у центра аномерии конфигурацией, и поэтому указанное выше в заголовке соединение можно получить только в виде чистого аномера с α-конфигурацией.
Элементный анализ:
б) 1-O-(1Н,1Н,2Н,2Н-перфтородецил)-α-D-маннопираноза
25 г (27,33 ммоля) соединения, указанного в заголовке примера 74а, суспендируют в 400 мл абсолютного метанола и при 5°С смешивают с каталитическим количеством метанолята натрия. После проведения реакции в течение 3 ч при комнатной температуре данные контроля за протеканием реакции тонкослойной хроматографией (элюент:хлороформ/метанол в соотношении 9:1) указывают уже на количественное превращение. Для переработки прозрачный реакционный раствор нейтрализуют смешением с катионообменной смолой Amberlite IR 120 (Н+-форма), ионит удаляют вакуум-фильтрацией и полученный таким путем метанольный фильтрат отгоняют в вакууме до получения сухого остатка. Полученный кристаллический остаток очищают двукратной перекристаллизацией из этанола. Данные 1Н-ЯМР-спектроскопического анализа указанного в заголовке соединения позволяют на основании величины констант взаимодействия J1,2=1,0 Гц сделать однозначный вывод о наличии α-конфигурации у аномерного центра. Подобная α-конфигурация является единственной присутствующей у центра аномерии конфигурацией, т.е. количество возможно образовавшегося аномера указанного в заголовке соединения с β-конфигурацией лежит ниже порога обнаружения при 1Н-ЯМР-спектроскопическом анализе. В соответствии с этим указанное выше в заголовке соединение получают только в виде чистого аномера с α-конфигурацией.
Выход: 16,2 г (94,6% от теории) бесцветного кристаллического твердого вещества.
Температура плавления: 172-174°С (с разложением).
Элементный анализ:
в) Получение композиции, содержащей металлический комплекс II и 1-O-(1Н, 1Н,2Н,2Н-перфтородецил)-α-D-маннопиранозу
К 50 мл раствора металлического комплекса II (150 ммолей/л) в 0,45%-ном растворе хлорида натрия (рН 7,4; 0,25 мг/л CaNa3ДТПА), добавляют раствор 2,01 г (3,21 ммоля) указанного в заголовке примера 74б соединения в 200 мл этанола и перемешивают в течение 2 ч при 50°С. Далее раствор досуха концентрируют в вакууме и объем остатка добавлением дистиллированной воды доводят до 75 мл. Смесь перемешивают в течение 10 мин при 40°С и фильтруют через фильтр с размером пор 0,2 мкм. Фильтрат расфасовывают во флаконы. Полученный таким путем раствор можно непосредственно использовать для проведения биологических экспериментов. (Концентрация Gd составляет 100 ммолей/л.)
Пример 75
а) 1-O-(1Н,1Н,2Н,2Н-перфторододецил)-2,3,4,6-тетра-O-ацетил-α-D-маннопираноза
Взаимодействием 35 г (89,66 ммоля) указанного в заголовке примера 70а соединения, которое используют в виде смеси α,β-аномеров в соотношении между ними 4:1, с раствором 50,60 г (89,7 ммоля) 1-гидрокси-1Н,1Н,2Н,2Н-перфторододекана в 100 мл 1,2-дихлорэтана, а также с хлоридом олова(IV) общим количеством 13,63 г (16,61 ммоля) аналогично методу, описанному для синтеза указанных в заголовках примеров 1б, 4б и 5б соединений, и после переработки и очистки колоночной хроматографией (элюент:гексан/этиловый эфир уксусной кислоты в соотношении 2:1) получают 62,49 г (68,7% от теории) указанного выше в заголовке соединения в виде вязкого бесцветного масла. Данные 1Н-ЯМР-спектроскопического анализа указанного в заголовке соединения позволяют на основании величины констант взаимодействия J1,2=1,4 Гц сделать однозначный вывод о наличии α-конфигурации у аномерного центра, которая, кроме того, является единственной присутствующей у центра аномерии конфигурацией, и поэтому указанное выше в заголовке соединение можно получить только в виде чистого аномера с α-конфигурацией.
Элементный анализ:
б) 1-O-(1Н,1Н,2Н,2Н-перфторододецил)-α-D-маннопираноза
25 г (24,64 ммоля) соединения, указанного в заголовке примера 75а, суспендируют в 400 мл абсолютного метанола и при 5°С смешивают с каталитическим количеством метанолята натрия. После проведения реакции в течение 3 ч при комнатной температуре данные контроля за протеканием реакции тонкослойной хроматографией (элюент:хлороформ/метанол в соотношении 9:1) указывают уже на количественное превращение. Для переработки прозрачный реакционный раствор нейтрализуют смешением с катионообменной смолой Amberlite IR 120 (Н+ -форма), ионит удаляют вакуум-фильтрацией и полученный таким путем метанольный фильтрат отгоняют в вакууме до получения сухого остатка. Полученный кристаллический остаток очищают двукратной перекристаллизацией из смеси 2-пропанола с этанолом (в соотношении 1:1). Данные 1Н-ЯМР-спектроскопического анализа указанного в заголовке соединения позволяют на основании величины констант взаимодействия J1,2=0,9 Гц сделать однозначный вывод о наличии α-конфигурации у аномерного центра. Подобная α-конфигурация является единственной присутствующей у центра аномерии конфигурацией, т.е. количество возможно образовавшегося аномера указанного в заголовке соединения с β-конфигурацией лежит ниже порога обнаружения при 1Н-ЯМР-спектроскопическом анализе. В соответствии с этим указанное выше в заголовке соединение получают только в виде чистого аномера с α-конфигурацией.
Выход: 16,96 г (90,8% от теории) бесцветного кристаллического твердого вещества.
Температура плавления: 187-188°С (с разложением).
Элементный анализ:
в) Получение композиции, содержащей металлический комплекс VI и 1-O-(1Н,1Н,2Н,2Н-перфторододецил)-α-D-маннопиранозу
К 52 мл раствора металлического комплекса VI (180 ммолей/л) в 0,45%-ном водном растворе хлорида натрия добавляют 1,70 г (2,34 ммоля) соединения, указанного в заголовке примера 75б, и в течение 10 мин нагревают в микроволновой печи. Раствор охлаждают до комнатной температуры, фильтруют через фильтр с размером пор 0,2 мкм и фильтрат расфасовывают во флаконы. Полученный таким путем раствор можно непосредственно использовать для проведения биологических экспериментов. (Концентрация Gd составляет 180 ммолей/л.)
Пример 76
а) 2,3,4,6-тетра-O-ацетил-1-O-α-D-[3,6,9-триокса(С12-С19гептадекафтор)нонадецил]маннопираноза
Взаимодействием 20 г (51,23 ммоля) указанного в заголовке примера 70а соединения, которое используют в виде смеси α,β-аномеров в соотношении между ними 4:1, с раствором 30,54 г (51,23 ммоля) 1-гидрокси-трис-(1Н,1Н,2Н,2Н-O)-1Н,1Н,2Н,2Н-перфтородекана в 100 мл 1,2-дихлорэтана, а также с хлоридом олова(IV) общим количеством 5,98 г (51,23 ммоля) аналогично методу, описанному для синтеза указанных в заголовках примеров 1б, 4б и 5б соединений, после переработки и очистки колоночной хроматографией (элюент:гексан/этиловый эфир уксусной кислоты в соотношении 1:1) получают 34,22 г (72,1% от теории) указанного выше в заголовке соединения в виде вязкого бесцветного масла. Данные 1Н-ЯМР-спектроскопического анализа указанного в заголовке соединения позволяют на основании величины констант взаимодействия J1,2=1,1 Гц сделать однозначный вывод о наличии α-конфигурации у аномерного центра, которая, кроме того, является единственной присутствующей у центра аномерии конфигурацией, и поэтому указанное выше в заголовке соединение можно получить только в виде чистого аномера с α-конфигурацией.
Элементный анализ:
б) 1-O-α-D-[3,6,9-триокса(С12-С19гептадекафтор)нонадецил]маннопираноза
20 г (21,58 ммоля) соединения, указанного в заголовке примера 76а, суспендируют в 350 мл абсолютного метанола и при 5°С смешивают с каталитическим количеством метанолята натрия. После проведения реакции в течение 3 ч при комнатной температуре данные контроля за протеканием реакции тонкослойной хроматографией (элюент:хлороформ/метанол в соотношении 6:1) указывают уже на количественное превращение. Для переработки прозрачный реакционный раствор нейтрализуют смешением с катионообменной смолой Amberlite IR 120 (Н+ -форма), ионит удаляют вакуум-фильтрацией и полученный таким путем метанольный фильтрат отгоняют в вакууме до получения сухого остатка. Полученный кристаллический остаток очищают двукратной перекристаллизацией из смеси этилового эфира уксусной кислоты с 2-пропанолом и этанолом (в соотношении 1:0,5:1). Данные 1Н-ЯМР-спектроскопического анализа указанного в заголовке соединения позволяют на основании величины констант взаимодействия J1,2=1,0 Гц сделать однозначный вывод о наличии α-конфигурации у аномерного центра. Подобная α-конфигурация является единственной присутствующей у центра аномерии конфигурацией, т.е. количество возможно образовавшегося аномера указанного в заголовке соединения с β-конфигурацией лежит ниже порога обнаружения при 1Н-ЯМР-спектроскопическом анализе. В соответствии с этим указанное выше в заголовке соединение получают только в виде чистого аномера с α-конфигурацией.
Выход: 15,20 г (92,9% от теории) бесцветного кристаллического твердого вещества.
Температура плавления: 141°С.
Элементный анализ:
в) Получение композиции, содержащей указанное в заголовке примера 68 соединение и 1-O-α-D-[3,6,9-триокса(С12-С19гептадекафтор)нонадецил]маннопиранозу
К 38 мл раствора указанного в заголовке примера 68 соединения (300 ммолей/л) в 0,45%-ном водном растворе поваренной соли (рН 7,4; 0,25 мг/л CaNa3ДТПА) добавляют 3,71 г (4,89 ммоля) соединения, указанного в заголовке примера 76б, и объем с помощью 0,9%-ного водного раствора поваренной соли доводят до 114 мл. Далее выдерживают в течение 2 ч при 60°С, подвергая смесь воздействию ультразвука. Раствор охлаждают до комнатной температуры и значение рН устанавливают на 7,4 с помощью водного 2н. едкого натра. После этого фильтруют через фильтр с размером пор 0,2 мкм и фильтрат расфасовывают во флаконы. Полученный таким путем раствор можно непосредственно использовать для проведения биологических экспериментов. (Концентрация Gd составляет 100 ммолей/л.)
Пример 77
а) 2,3,4,6-тетра-O-ацетил-1-α-D-[3-тиопропионовая кислота-N-(3-окса-1Н,1Н,2Н,2Н,4Н,4Н,5Н,5Н-перфтортридецил)амид]маннопираноза
В 500 мл сухого тетрагидрофурана растворяют 25,0 г (57,28 ммоля) 3-(тетра-О-ацетил-α-D-маннопиранозилмеркапто)пропионовой кислоты [получение согласно Ponpipom Mitree M., Bugianesi Robert L., Robbins James C., Doebber T.W., Shen T.Y., J. Med. Chem., 24, 12 (1981), сс.1388-1395], а также 5,77 г (57,28 ммоля) триэтиламина. После охлаждения реакционного раствора до температуры в пределах от -15 до -20°С при этой температуре по каплям при перемешивании медленно добавляют раствор 7,82 г (57,28 ммоля) изобутилового эфира хлормуравьиной кислоты в 100 мл сухого тетрагидрофурана, при этом скорость капельного добавления должна быть такой, чтобы внутренняя температура не превышала -10°С. После проведения реакции в течение 15 мин при -15°С по каплям при -20°С медленно добавляют раствор 29,05 г (57,28 ммоля) 1Н,1Н,2Н,2Н-гептадекафтор-1-(2-аминоэтокси)декана и 5,77 г (57,28 ммоля) триэтиламина в 200 мл сухого тетрагидрофурана. После проведения реакции в течение часа при -15°С, а затем в течение двух часов при комнатной температуре реакционный раствор досуха концентрируют в вакууме. Полученный остаток растворяют в 250 мл этилового эфира уксусной кислоты и дважды промывают насыщенным раствором гидрокарбоната натрия порциями по 200 мл и однократно водой порцией 300 мл. После сушки органической фазы над сульфатом натрия соль отделяют вакуум-фильтрацией и этиловый эфир уксусной кислоты отгоняют в вакууме. Полученный маслянистый остаток очищают на силикагеле с использованием дихлорметана/гексана/2-пропанола (в соотношении 8:5:1) в качестве элюента.
Выход: 44,90 г (84,7% от теории) указанного выше в заголовке соединения в виде бесцветного и высоковязкого масла.
Элементный анализ:
б) 1-α-D-[3-тиопропионовая кислота-N-(3-окса-1Н,1Н,2Н,2Н,4Н,4Н,5Н,5Н-перфтортридецил)амид]маннопираноза
30 г (32,41 ммоля) соединения, указанного в заголовке примера 77а, суспендируют в 400 мл абсолютного метанола и при 5°С смешивают с каталитическим количеством метанолята натрия. После проведения реакции в течение 3 ч при комнатной температуре данные контроля за протеканием реакции тонкослойной хроматографией (элюент:хлороформ/метанол в соотношении 9:1) указывают уже на количественное превращение. Для переработки прозрачный реакционный раствор нейтрализуют смешением с катионообменной смолой Amberlite IR 120 (Н+-форма), ионит удаляют вакуум-фильтрацией и полученный таким путем метанольный фильтрат отгоняют в вакууме до получения сухого остатка. Полученный кристаллический остаток очищают перекристаллизацией из смеси этилового эфира уксусной кислоты с метанолом (в соотношении 0,5:1). Данные 1H-ЯМР-спектроскопического анализа указанного в заголовке соединения позволяют на основании величины констант взаимодействия J1,2=1,1 Гц сделать однозначный вывод о наличии α-конфигурации у аномерного центра. Подобная α-конфигурация является единственной присутствующей у центра аномерии конфигурацией, т.е. количество возможно образовавшегося аномера указанного в заголовке соединения с β-конфигурацией лежит ниже порога обнаружения при 1Н-ЯМР-спектроскопическом анализе. В соответствии с этим указанное выше в заголовке соединение получают только в виде чистого аномера с α-конфигурацией.
Выход: 23,76 г (96,8% от теории) бесцветного кристаллического твердого вещества.
Температура плавления: 113-114,5°С.
Элементный анализ:
в) Получение композиции, содержащей указанное в заголовке примера 66 соединение и 1-α-D-[3-тиопропионовая кислота-N-(3-окса-1Н,1Н,2Н,2Н,4Н,4Н,5Н,5Н-перфтортридецил)амид]маннопиранозу
К 47 мл раствора указанного в заголовке примера 66 соединения (330 ммолей/л) в 0,45%-ном растворе хлорида натрия (рН 7,4; 0,25 мг/л CaNa3ДТПА) добавляют раствор 27,41 г (36,19 ммоля) указанного в заголовке примера 77б соединения в 200 мл этанола и перемешивают в течение 2 ч при 50°С. Далее раствор досуха концентрируют в вакууме и объем остатка добавлением дистиллированной воды доводят до 155 мл. Смесь перемешивают в течение 10 мин при 40°С и фильтруют через фильтр с размером пор 0,2 мкм. Фильтрат расфасовывают во флаконы. Полученный таким путем раствор можно непосредственно использовать для проведения биологических экспериментов. (Концентрация Gd составляет 100 ммолей/л.)
Пример 78
а) 2,3,4,6-тетра-O-ацетил-1-β-D-[3,6,9-триокса(С12-С19гептадекафтор)нонадецил]глюкопиранозилуроновая кислота
20,2 г (50,85 ммоля) метил(1-бром-2,3,4-три-O-ацетил-α-D-глюкопиранозид)уроната [получение согласно Pelzer, Hoppe-Seyler's Z. Physiol. Chem., 314 (1949), сс.234, 237, а также Goebel, Babers, J. Biol. Chem., Ill (1935), cc.347, 350, и Bollenback и др., J. Amer. Chem. Soc., 77 (1955), сс.3310, 3313] и 60,64 г (101,7 ммоля) 3,6,9-триокса(С12-С19гептадекафтор)нонадекан-1-ола растворяют в 250 мл безводного ацетонитрила и при комнатной температуре смешивают с 13,0 г свежеосажденного оксида серебра. После проведения реакции в течение 12 ч при комнатной температуре нарастворившиеся соли серебра отфильтровывают, после чего соли тщательно промывают дихлорметаном и полученный таким путем фильтрат отгоняют в вакууме до получения сухого остатка. Полученный остаток очищают колоночной хроматографией (элюент:гексан/этиловый эфир уксусной кислоты в соотношении 3:1).
Выход: 22,99 г (53,3% от теории) указанного выше в заголовке соединения в виде бесцветного, высоковязкого масла.
Элементный анализ:
б) 1-O-β-D-[3,6,9-триокса(С12-С19гептадекафтор)нонадецил]глюкопиранозилуроновая кислота
10,0 г (11,78 ммоля) соединения, указанного в заголовке примера 78а, при комнатной температуре суспендируют при перемешивании в 200 мл смеси, состоящей из метанола и 0,5-молярного едкого натра в соотношении 2:1. После проведения реакции в течение 12 ч при комнатной температуре прозрачную реакционную смесь для ее переработки нейтрализуют смешением с катионообменной смолой Amberlite IR 120 (Н+-форма), ионит удаляют вакуум-фильтрацией и полученный таким путем метанольно-водный фильтрат отгоняют в вакууме до получения сухого остатка. Полученный кристаллический остаток очищают перекристаллизацией из смеси этилового эфира уксусной кислоты с метанолом (в соотношении 0,25:1). Данные 1Н-ЯМР-спектроскопического анализа указанного в заголовке соединения позволяют на основании величины констант взаимодействия J1,2=9,2 Гц сделать однозначный вывод о наличии β-конфигурации у аномерного центра. Подобная β-конфигурация является единственной присутствующей у центра аномерии конфигурацией, т.е. количество возможно образовавшегося аномера указанного в заголовке соединения с α-конфигурацией лежит ниже порога обнаружения при 1Н-ЯМР-спектроскопическом анализе. В соответствии с этим указанное выше в заголовке соединение получают только в виде чистого аномера с β-конфигурацией.
Температура плавления: 78,5°С.
Элементный анализ:
в) Получение композиции, содержащей металлический комплекс I и 1-O-β-D-[3,6,9-триокса(C12-C19гептадекафтор)нонадецил]глюкопиранозилуроновую кислоту
К 38 мл раствора металлического комплекса I (280 ммолей/л) в 0,45%-ном водном растворе поваренной соли (рН 7,4; 0,25 мг/л CaNa3ДТПА) добавляют 19,18 г (24,83 ммоля) соединения, указанного в заголовке примера 78б, и объем с помощью 0,9%-ного водного раствора поваренной соли доводят до 53,2 мл. Далее выдерживают в течение 2 ч при 60°С, подвергая смесь воздействию ультразвука. Раствор охлаждают до комнатной температуры и значение рН устанавливают на 7,4 с помощью водного 2н. едкого натра. После этого фильтруют через фильтр с размером пор 0,2 мкм и фильтрат расфасовывают во флаконы. Полученный таким путем раствор можно непосредственно использовать для проведения биологических экспериментов. (Концентрация Gd составляет 200 ммолей/л.)
Пример 79
a) 6-(2-окса-1H,1H,3H,3H,4H,4H-перфтордецил)-O1,O2,O3,O4-диизопропилиден-α-D-галактопираноза
К перемешанной суспензии 2,01 г (70,0 ммолей) гидрида натрия (80%-ный в минеральном масле) в 25 мл диметилформамида при комнатной температуре по каплям добавляют в целом 12,15 г (46,66 ммоля) O1,O2,O3,O4-диизопропилиден-α-галактопиранозы [получение согласно Levene, Meyer, J. Biol. Chem., 64 (1925), с.473, а также McCreath, Smith, J. Chem. Soc. 1939, cc.387, 389 и Freudenberg, Hixon, Chem. Ber., 56 (1923), cc.2119, 2122], растворенной в 200 мл абсолютного диметилформамида. После этого оставляют еще на 120 мин для перемешивания при комнатной температуре и затем по каплям медленно добавляют в целом 30,09 г (48,0 ммолей) 1-бром-1Н,1Н,2Н,2Н-перфторододекана, растворенного в 150 мл абсолютного диметилформамида. Затем полученный таким путем реакционный раствор перемешивают в течение последующих 12 ч при комнатной температуре. Для переработки реакционный раствор медленно сливают в 1 л смеси воды со льдом, после чего практически полностью экстрагируют диэтиловым эфиром. Объединенные органические фазы затем дважды промывают насыщенным раствором гидрокарбоната натрия порциями по 200 мл и дважды водой порциями по 200 мл. После сушки органической фазы над сульфатом натрия соль отделяют вакуум-фильтрацией и растворитель отгоняют в вакууме. Полученный маслянистый остаток очищают на силикагеле с использованием этилового эфира уксусной кислоты/гексана (в соотношении 1:10) в качестве элюента.
Выход: 29,8 г (79,3% от теории) указанного выше в заголовке соединения в виде вязкого бесцветного масла.
Элементный анализ:
б) 6-(2-окса-1Н, 1Н,3H,3H,4Н,4Н-перфтордецил)-α-D-галактопираноза
20 г (24,8 ммоля) соединения, указанного в заголовке примера 79а, смешивают с 300 мл 1%-ного водного раствора серной кислоты и перемешивают в течение 3 ч при 80°С. После охлаждения до комнатной температуры нейтрализуют смешением с водным раствором гидроксида бария, после чего выпавший в осадок сульфат бария отфильтровывают и полученный таким путем прозрачный водный содержащий продукт раствор лиофилизуют. Данные 1Н-ЯМР-спектроскопического анализа указанного в заголовке соединения позволяют сделать однозначный вывод о наличии обеих возможных конфигураций у аномерного центра, при этом соотношение между присутствующими у центра аномерии α/β-конфигурациями составляет согласно данным 1Н-ЯМР-спектроскопического анализа 1:1,4 (α:β). В соответствии с этим указанное выше в заголовке соединение выделяют только в виде смеси α- и β-аномеров при соотношении между ними 1:1,4, т.е. полученное соединение не разделяют на отдельные аномеры.
Выход: 15,28 г (98,4% от теории) указанного выше в заголовке соединения в виде бесцветного лиофилизата.
Элементный анализ (в пересчете на безводное вещество):
в) Получение композиции, содержащей указанное в заголовке примера 67 соединения и 6-(2-окса-1Н,1Н, 3H,3H,4Н,4Н-перфтордецил)-α-D-галактопиранозу
К 43 мл раствора указанного в заголовке примера 67 соединения (250 ммолей/л) в 0,45%-ном растворе хлорида натрия (рН 7,4; 0,25 мг/л CaNa3ДТПА) добавляют раствор 1,68 г (2,69 ммоля) указанного в заголовке примера 79б соединения в 200 мл этанола и перемешивают в течение 2 ч при 50°С. Далее раствор досуха концентрируют в вакууме и объем остатка добавлением дистиллированной воды доводят до 107,5 мл. Смесь перемешивают в течение 10 мин при 40°С и фильтруют через фильтр с размером пор 0,2 мкм. Фильтрат расфасовывают во флаконы. Полученный таким путем раствор можно непосредственно использовать для проведения биологических экспериментов. (Концентрация Gd составляет 100 ммолей/л.)
Пример 80
а) 1-O-α-D-[(1-перфтороктилсульфонилпиперазин-4-карбонил)метил]маннопираноза
30 г (52,8 ммоля) 1-перфтороктилсульфонилпиперазина (получение описано в DE 19603033) и 31,73 г (53 ммоля) 2,3,4,6-тетра-O-бензил-α-D-карбоксиметилманнопиранозы (получение описано в DE 19728954) растворяют в 300 мл тетрагидрофурана. Далее при 0°С добавляют 24,73 г (100 ммолей) ЭЭДХ (этиловый эфир 1,2-дигидро-2-этоксихинолин-1-карбоновой кислоты) и перемешивают в течение 3 ч при 0°С, а затем в течение 6 ч при комнатной температуре. Далее раствор досуха упаривают в вакууме и остаток очищают экспресс-хроматографией на силикагеле (элюент:гексан/этиловый эфир уксусной кислоты в соотношении 10:1). Содержащие продукт фракции упаривают досуха, остаток растворяют в смеси из 200 мл метанола и 150 мл дихлорметана и в течение 8 ч гидрируют над палладием на угле (2 г 10%-ного Pd/C). После этого катализатор гидрирования отфильтровывают и фильтрат упаривают досуха. Остаток перекристаллизовывают из ацетона/диэтилового эфира.
Выход: 30,39 г (73% от теории) воскоподобного бесцветного твердого вещества.
Элементный анализ:
б) Получение композиции, содержащей металлический комплекс I и 1-O-α-D-[(1-перфтороктилсульфонилпиперазин-4-карбонил)метил]маннопиранозу
К 32 мл раствора металлического комплекса I (280 ммолей/л) в 0,45%-ном водном растворе поваренной соли (рН 7,4; 0,25 мг/л CaNa3ДТПА) добавляют 4,71 г (5,97 ммоля) соединения, указанного в заголовке примера 80а, и объем с помощью 0,9%-ного водного раствора поваренной соли доводят до 55 мл. Далее выдерживают в течение 2 ч при 60°С, подвергая смесь воздействию ультразвука. Раствор охлаждают до комнатной температуры и значение рН устанавливают на 7,4 с помощью водного 2н. едкого натра. После этого фильтруют через фильтр с размером пор 0,2 мкм и фильтрат расфасовывают во флаконы. Полученный таким путем раствор можно непосредственно использовать для проведения биологических экспериментов. (Концентрация Gd составляет 200 ммолей/л.)
Пример 81
а) 3-окса-2Н,2Н,4Н,4Н,5Н,5Н-перфтортридекановая кислота, натриевая соль
20 г (38,3 ммоля) 3-окса-2Н,2Н,4Н,4Н,5Н,5Н-перфтортридекановой кислоты (получение описано в DE 19603033) растворяют в 300 мл этанола и добавляют 7,7 мл 5н. водного едкого натра. После этого упаривают досуха и остаток сушат в вакуумном сушильном шкафу (8 ч, 60°С).
Выход: 20,85 г (количественный) бесцветного кристаллического порошка.
Элементный анализ:
б) Получение композиции, содержащей металлический комплекс I и натриевую соль 3-окса-2Н,2Н,4Н,4Н,5Н,5Н-перфтортридекановой кислоты
К 32 мл раствора металлического комплекса I (280 ммолей/л) в 0,45%-ном водном растворе поваренной соли (рН 7,4; 0,25 мг/л CaNa3ДТПА) добавляют 2,09 г (3,84 ммоля) соединения, указанного в заголовке примера 81а, и объем с помощью 0,9%-ного водного раствора поваренной соли доводят до 90 мл. Далее выдерживают в течение 2 ч при 60°С, подвергая смесь воздействию ультразвука. Раствор охлаждают до комнатной температуры и значение рН устанавливают на 7,4 с помощью водного 2н. едкого натра. После этого фильтруют через фильтр с размером пор 0,2 мкм и фильтрат расфасовывают во флаконы. Полученный таким путем раствор можно непосредственно использовать для проведения биологических экспериментов. (Концентрация Gd составляет 100 ммолей/л.)
в) Получение композиции, содержащей металлический комплекс I и натриевую соль 3-окса-2Н,2Н,4Н,4Н,5Н,5Н-перфтортридекановой кислоты
К 32 мл раствора металлического комплекса I (280 ммолей/л) в 0,45%-ном водном растворе поваренной соли (рН 7,4; 0,25 мг/л CaNa3ДТПА) добавляют 1,00 г (1,84 ммоля) соединения, указанного в заголовке примера 81а, и объем с помощью 0,9%-ного водного раствора поваренной соли доводят до 90 мл. Далее выдерживают в течение 2 ч при 60°С, подвергая смесь воздействию ультразвука. Раствор охлаждают до комнатной температуры и значение рН устанавливают на 7,4 с помощью водного 2н. едкого натра. После этого фильтруют через фильтр с размером пор 0,2 мкм и фильтрат расфасовывают во флаконы. Полученный таким путем раствор можно непосредственно использовать для проведения биологических экспериментов. (Концентрация Gd составляет 100 ммолей/л.)
г) Получение композиции, содержащей металлический комплекс I и натриевую соль 3-окса-2Н,2Н,4Н,4Н,5Н,5Н-перфтортридекановой кислоты
К 32 мл раствора металлического комплекса I (280 ммолей/л) в 0,45%-ном водном растворе поваренной соли (рН 7,4; 0,25 мг/л CaNa3ДТПА) добавляют 0,54 г (1,0 ммоль) соединения, указанного в заголовке примера 81а, и объем с помощью 0,9%-ного водного раствора поваренной соли доводят до 90 мл. Далее выдерживают в течение 2 ч при 60°С, подвергая смесь воздействию ультразвука. Раствор охлаждают до комнатной температуры и значение рН устанавливают на 7,4 с помощью водного 2н. едкого натра. После этого фильтруют через фильтр с размером пор 0,2 мкм и фильтрат расфасовывают во флаконы. Полученный таким путем раствор можно непосредственно использовать для проведения биологических экспериментов. (Концентрация Gd составляет 100 ммолей/л.)
Пример 82
а) 1-перфтороктилсульфонил-4-(3,6,9,12,15-пентаоксагексадеканоил)пиперазин
20 г (35,2 ммоля) перфтороктилсульфонилпиперазина (см. пример 80а) растворяют в 300 мл дихлорметана и добавляют 5,06 г (50 ммолей) триэтиламина. Далее охлаждают до 0°С, после чего по каплям в течение 20 мин добавляют 14,24 г (50 ммолей) хлорангидрида 3,6,9,12,15-пентаоксагексановой кислоты и перемешивают в течение 3 ч при 0°С. Далее добавляют 400 мл 5%-ной водной соляной кислоты и тщательно перемешивают. Органическую фазу отделяют, сушат над сульфатом магния и досуха упаривают в вакууме. Остаток хроматографируют на силикагеле (элюент:дихлорметан/метанол в соотношении 15:1).
Выход: 26,44 (92% от теории) воскоподобного твердого вещества.
Элементный анализ:
б) Получение композиции, содержащей металлический комплекс I и 1-перфтороктилсульфонил-4-(3,6,9,12,15-пентаоксагексадеканоил)пиперазин
К 47 мл раствора металлического комплекса I (280 ммолей/л) в 0,45%-ном водном растворе поваренной соли (рН 7,4; 0,25 мг/л CaNa3ДТПА) добавляют 4,61 г (5,64 ммоля) соединения, указанного в заголовке примера 82а, и объем с помощью 0,9%-ного водного раствора поваренной соли доводят до 66 мл. Далее выдерживают в течение 2 ч при 60°С, подвергая смесь воздействию ультразвука. Раствор охлаждают до комнатной температуры и значение рН устанавливают на 7,4 с помощью водного 2н. едкого натра. После этого фильтруют через фильтр с размером пор 0,2 мкм и фильтрат расфасовывают во флаконы. Полученный таким путем раствор можно непосредственно использовать для проведения биологических экспериментов. (Концентрация Gd составляет 200 ммолей/л.)
Пример 83
а) Эфир 1Н,1Н,2Н,2Н-перфтордецил-n-толуолсульфоновой кислоты
20 г (43,1 ммоля) 1Н,1Н,2Н,2Н-перфтордеканола растворяют в 200 мл пиридина и при 0°С порциями добавляют 9,53 г (50 ммолей) хлорангидрида n-толуолсульфоновой кислоты. Смесь перемешивают в течение 5 ч при комнатной температуре. Далее раствор сливают в 1000 мл смеси воды со льдом и перемешивают в течение 10 мин. Осадок отфильтровывают, промывают большим количеством воды и после этого перекристаллизовывают из ацетона.
Выход: 22,04 г (97% от теории) бесцветного кристаллического твердого вещества.
Элементный анализ:
б) С18-С25 гептадекафтор-3,6,9,12,15-пентаоксапентакозан-1-ол
20 г (37,94 ммоля) соединения, указанного в заголовке примера 83а, 35,74 г (150 ммолей) пентаэтиленгликоля и 1 г краун-эфира 18-краун-6 растворяют в 300 мл тетрагидрофурана и добавляют 10,1 г (180 ммолей) высокодисперсного порошкового гидроксида калия. Смесь перемешивают в течение 10 ч при комнатной температуре. После этого твердое вещество отфильтровывают и фильтрат досуха концентрируют в вакууме. Остаток хроматографируют на силикагеле (элюент:дихлорметан/метанол в соотношении 15:1).
Выход: 5,45 г (21% от теории) бесцветного вязкого масла.
Элементный анализ:
в) Получение композиции, содержащей указанное в заголовке примера 69 соединение и С18-С25гептадекафтор-3,6,9,12,15-пентаоксапентакозан-1-ол
К 53 мл раствора указанного в заголовке примера 69 соединения (310 ммолей/л) в 0,45%-ном водном растворе хлорида натрия добавляют 44,98 г (65,72 ммоля) соединения, указанного в заголовке примера 83б, и в течение 10 мин нагревают в микроволновой печи. Раствор охлаждают до комнатной температуры, фильтруют через фильтр с размером пор 0,2 мкм и фильтрат расфасовывают во флаконы. Полученный таким путем раствор можно непосредственно использовать для проведения биологических экспериментов. (Концентрация Gd составляет 310 ммолей/л.)
Пример 84
а) Амид N,N-бис(8-гидрокси-3,6-диоксаоктил)перфтороктилсульфоновой кислоты
15 г (29,23 ммоля) амида перфтороктилсульфоновой кислоты и 22,16 г (87,7 мл) 9-(тетрагидропиран-2-ил)-3,6,9-триоксанонилхлорида растворяют в 200 мл ацетонитрила. Далее добавляют 41,46 г (300 ммолей) карбоната калия и 1 г (6 ммолей) йодида калия и в течение 10 ч кипятят с обратным холодильником. Твердое вещество отфильтровывают и фильтрат досуха упаривают в вакууме. Остаток растворяют в 400 мл этанола и добавляют 30 мл 10%-ной водной соляной кислоты. Смесь перемешивают в течение 2 ч при комнатной температуре. Далее значение рН устанавливают на 7 с помощью едкого натра и раствор концентрируют в вакууме. Остаток хроматографируют на силикагеле (элюент:дихлорметан/метанол в соотношении 10:1).
Выход: 11,38 г (51% от теории) бесцветного вязкого масла.
Элементный анализ:
б) Получение композиции, содержащей металлический комплекс I и амид N,N-бис(8-гидрокси-3,6,-диоксаоктил)перфтороктилсульфоновой кислоты
К 37 мл раствора металлического комплекса I (280 ммолей/л) в 0,45%-ном водном растворе поваренной соли (рН 7,4; 0,25 мг/л CaNa3ДТПА) добавляют 7,91 г (10,36 ммоля) соединения, указанного в заголовке примера 84а, и объем с помощью 0,9%-ного водного раствора поваренной соли доводят до 104 мл. Далее выдерживают в течение 2 ч при 60°С, подвергая смесь воздействию ультразвука. Раствор охлаждают до комнатной температуры и значение рН устанавливают на 7,4 с помощью водного 2н. едкого натра. После этого фильтруют через фильтр с размером пор 0,2 мкм и фильтрат расфасовывают во флаконы. Полученный таким путем раствор можно непосредственно использовать для проведения биологических экспериментов. (Концентрация Gd составляет 100 ммолей/л.)
Пример 85
а) Амид N,N-бис(трет-бутилоксикарбонилметил)перфтороктилсульфоновой кислоты
20 г (38,97 ммоля) амида перфтороктилсульфоновой кислоты и 20,73 г (150 моля) карбоната калия суспендируют в 200 мл ацетона и добавляют 17,56 г (90 ммолей) трет-бутилового эфира бромуксусной кислоты. Далее в течение 3 ч кипятят с обратным холодильником. Твердое вещество отфильтровывают и фильтрат досуха упаривают в вакууме. Остаток хроматографируют на силикагеле (элюент:н-гексан/этиловый эфир уксусной кислоты в соотношении 10:1).
Выход: 23,53 г (83% от теории) бесцветного воскоподобного твердого вещества.
Элементный анализ:
б) Амид N,N-бис(карбоксиметил)перфтороктилсульфоновой кислоты, динатриевая соль
23 г (31,62 ммоля) соединения, указанного в заголовке примера 85а, растворяют в 300 мл трифторуксусной кислоты и в течение 5 ч перемешивают при комнатной температуре. Смесь досуха упаривают в вакууме и остаток перекристаллизовывают из ацетона. Кристаллы отделяют вакуум-фильтрацией и сушат в вакууме при 50°С.
Выход: 17,7 г (91% от теории) бесцветного кристаллического порошка.
17 г (27,63 ммоля) полученной таким путем дикислоты растворяют в смеси из 100 мл воды и 300 мл этанола и добавляют 9,2 мл 3 н. водного едкого натра. Смесь перемешивают в течение 20 мин при комнатной температуре и затем досуха упаривают в вакууме. Остаток сушат в вакууме (60°С, 8 ч).
Выход; 18,2 г бесцветного кристаллического порошка.
Элементный анализ:
в) Получение композиции, содержащей металлический комплекс II и динатриевую соль амида N,N-бис(карбоксиметил)перфтороктилсульфоновой кислоты
К 41 мл раствора металлического комплекса II (250 ммолей/л) в 0,45%-ном водном растворе поваренной соли (рН 7,4; 0,25 мг/л CaNa3ДТПА) добавляют 2,89 г (4,39 ммоля) соединения, указанного в заголовке примера 85б, и объем с помощью 0,9%-ного водного раствора поваренной соли доводят до 52 мл. Далее выдерживают в течение 2 ч при 60°С, подвергая смесь воздействию ультразвука. Раствор охлаждают до комнатной температуры и значение рН устанавливают на 7,4 с помощью водного 2н. едкого натра. После этого фильтруют через фильтр с размером пор 0,2 мкм и фильтрат расфасовывают во флаконы. Полученный таким путем раствор можно непосредственно использовать для проведения биологических экспериментов. (Концентрация Gd составляет 200 ммолей/л.)
Пример 86
а) Моноэфир 1Н,1Н,2Н,2Н-перфтордодецилсерной кислоты, натриевая соль
10 г (17,73 ммоля) 1Н,1Н,2Н,2Н-перфтордодеканола растворяют в 300 мл хлороформа и при 0°С добавляют 2,82 г (17,73 ммоля) комплекса из триоксида серы и пиридина. Смесь перемешивают в течение часа при 0°С и затем досуха упаривают в вакууме. Остаток растворяют в 300 мл этанола и смешивают с 17,8 мл 1н. водного едкого натра. Раствор упаривают досуха и остаток сушат в вакууме (60°С, 2 ч).
Выход: 11,81 г (количественный).
Элементный анализ:
б) Получение композиции, содержащей металлический комплекс VI и натриевую соль моноэфира 1Н,1Н,2Н,2Н-перфтордодецилсерной кислоты
К 38 мл раствора металлического комплекса VI (290 ммолей/л) в 0,45%-ном водном растворе хлорида натрия добавляют 4,90 г (7,35 ммоля) соединения, указанного в заголовке примера 86а, и в течение 10 мин нагревают в микроволновой печи. Раствор охлаждают до комнатной температуры, фильтруют через фильтр с размером пор 0,2 мкм и фильтрат расфасовывают во флаконы. Полученный таким путем раствор можно непосредственно использовать для проведения биологических экспериментов. (Концентрация Gd составляет 290 ммолей/л.)
Пример 87
а) 2Н,2Н,4Н,4Н,5Н,5Н-3-оксаперфторпентадекановая кислота, натриевая соль
10 г (16,07 ммоля) 2Н,2Н,4Н,4Н,5Н,5Н-3-оксаперфторпентадекановой кислоты растворяют в 300 мл этанола и смешивают с 16,1 мл 1н. водного едкого натра. Раствор упаривают досуха и остаток сушат в вакууме (60°С, 2 ч).
Выход: 10,35 г (количественный) бесцветного аморфного порошка.
Элементный анализ:
б) Получение композиции, содержащей указанное в заголовке примера 66 соединение и натриевую соль 2Н,2Н,4Н,4Н,5Н,5Н-3-оксаперфторпентадекановой кислоты
К 45 мл раствора указанного в заголовке примера 66 соединения (270 ммолей/л) в 0,45%-ном растворе хлорида натрия (рН 7,4; 0,25 мг/л CaNa3ДТПА) добавляют раствор 3,36 г (5,21 ммоля) указанного в заголовке примера 87а соединения в 200 мл этанола и перемешивают в течение 2 ч при 50°С. Далее раствор досуха концентрируют в вакууме и объем остатка добавлением дистиллированной воды доводят до 122 мл. Смесь перемешивают в течение 10 мин при 40°С и фильтруют через фильтр с размером пор 0,2 мкм. Фильтрат расфасовывают во флаконы. Полученный таким путем раствор можно непосредственно использовать для проведения биологических экспериментов. (Концентрация Gd составляет 100 ммолей/л.)
Пример 88
a) N-(1Н, 1Н,2Н,2Н,4Н,4Н,5Н,5Н-3-оксаперфтортридецил)моноамид этилендиамин-N,N-тетрауксусной кислоты
К 30 г (117,1 ммоля) бисангидрида ЭДТК, суспендированного в 200 мл диметилформамида и 50 мл пиридина, при 50°С порциями добавляют 10,14 г (20 ммолей) 1Н,1Н,2Н,2Н,4Н,4Н,5Н,5Н-3-оксаперфтортридециламина и перемешивают в течение 6 ч при 50°С. Далее добавляют 10 мл воды, перемешивают в течение 10 мин при 50°С и остаток упаривают досуха. Далее остаток растворяют в небольшом количестве воды и значение рН устанавливают на 4 с помощью ледяной уксусной кислоты. Нерастворившийся осадок отфильтровывают и хроматографируют на RP-18 (элюент:градиент ацетонитрила/воды).
Выход: 9,58 г (61% от теории) бесцветного твердого вещества.
Содержание воды: 8%.
Элементный анализ:
б) N-(1Н,1Н,2Н,2Н,4Н,4Н,5Н,5Н-3-оксаперфтортридецил)моноамид этилендиамин-N,N-тетрауксусной кислоты, кальциевая соль, натриевая соль
9,0 г (11,46 ммоля) указанного в заголовке примера 88а соединения суспендируют в 300 мл воды и добавляют 11,4 мл 1 н. водного едкого натра. После этого добавляют 1,15 г (11,46 ммоля) карбоната кальция и перемешивают в течение 5 ч при 50°С. Раствор фильтруют и фильтрат лиофилизуют.
Выход: 9,7 г (100% от теории) бесцветного аморфного твердого вещества.
Содержание воды: 7,5%.
Элементный анализ:
в) Получение композиции, содержащей металлический комплекс I и кальциевую соль и натриевую соль N-(1H,1H,2H,2H,4H,4H,5H,5H-3-оксаперфтортридецил)моноамида этилендиамин-N,N-тетрауксусной кислоты
К 43 мл раствора металлического комплекса I (280 ммолей/л) в 0,45%-ном водном растворе поваренной соли (рН 7,4; 0,25 мг/л CaNa3ДТПА) добавляют 2,54 г (3,01 ммоля) соединения, указанного в заголовке примера 88б, и объем с помощью 0,9%-ного водного раствора поваренной соли доводят до 121 мл. Далее выдерживают в течение 2 ч при 60°С, подвергая смесь воздействию ультразвука. Раствор охлаждают до комнатной температуры и значение рН устанавливают на 7,4 с помощью водного 2н. едкого натра. После этого фильтруют через фильтр с размером пор 0,2 мкм и фильтрат расфасовывают во флаконы. Полученный таким путем раствор можно непосредственно использовать для проведения биологических экспериментов. (Концентрация Gd составляет 100 ммолей/л.)
Пример 89
а) Простой 1Н,1Н,2Н,2Н-перфтордецил(2,2-диметил-5-гидрокси-1,3-диоксепан-6-иловый) эфир
30 г (64,64 ммоля) 1Н,1Н,2Н,2Н-перфтордеканола растворяют в 200 мл тетрагидрофурана и при 0°С добавляют 1,68 г (70 ммолей) гидрида натрия. Смесь перемешивают в течение 2 ч при комнатной температуре, а затем в течение 4 ч при 60°С. Далее раствор помещают в металлический автоклав, после чего добавляют 9,31 г (64,64 ммоля) 2,2-диметил-1,3,6-триоксабицикло[5.1.0]октана и затем в течение 10 ч выдерживают при 150°С. После этого реакционный раствор сливают в смесь воды со льдом и дважды экстрагируют диэтиловым эфиром. Объединенные органические фазы упаривают досуха и остаток хроматографируют на силикагеле (элюент:дихлорметан/ацетон в соотношении 10:1)
Выход: 16,12 г (41% от теории) бесцветного твердого вещества.
Элементный анализ:
б) Простой 1Н,1Н,2Н,2Н-перфтордецил(1-гидроксиметил-2,3-дигидроксипропиловый) эфир
15 г (24,66 ммоля) соединения, указанного в заголовке примера 89а, растворяют в 300 мл этанола и добавляют 30 мл 10%-ной водной соляной кислоты. Далее в течение 5 ч кипятят с обратным холодильником. Затем значение рН устанавливают на 7 с помощью едкого натра, после чего концентрируют досуха и остаток хроматографируют на RP-18 (элюент:градиент ацетонитрила/воды).
Выход: 12,75 г (91% от теории) бесцветного твердого вещества.
Содержание воды: 4,5%.
Элементный анализ:
в) Получение композиции, содержащей указанное в заголовке примера 12 в WO 99/01161 соединение (1,4,7-трис{1,4,7-трис(N-карбоксилатометил)-10-(N-1-метил-3-аза-2,5-диоксопентан-1,5-диил]-1,4,7,10-тетраазациклододекан, Gd-комплекс}-10-[2-(N-этил-N-перфтороктилсульфонил)амино]ацетил-1,4,7,10-тетраазациклододекан) и простой 1Н,1Н,2Н,2Н-перфтордецил(1-гидроксиметил-2,3-дигидроксипропиловый) эфир
К 37 мл раствора 1,4,7-трис{1,4,7-трис(N-карбоксилатометил)-10-(N-1-метил-3-аза-2,5-диоксопентан-1,5-диил]-1,4,7,10-тетраазациклододекан, Gd-комплекс}-10-[2-(N-этил-N-перфтороктилсульфонил)амино]ацетил-1,4,7,10-тетраазациклододекана (300 ммолей/л) в 0,45%-ном водном растворе поваренной соли (рН 7,4; 0,25 мг/л CaNa3ДТПА) добавляют 9,46 г (16,65 ммоля) соединения, указанного в заголовке примера 89б, и объем с помощью 0,9%-ного водного раствора поваренной соли доводят до 111 мл. Далее выдерживают в течение 2 ч при 60°С, подвергая смесь воздействию ультразвука. Раствор охлаждают до комнатной температуры и значение рН устанавливают на 7,4 с помощью водного 2н. едкого натра. После этого фильтруют через фильтр с размером пор 0,2 мкм и фильтрат расфасовывают во флаконы. Полученный таким путем раствор можно непосредственно использовать для проведения биологических экспериментов. (Концентрация Gd составляет 100 ммолей/л.)
Пример 90
а) Простой 1Н, 1Н,2Н,2Н-перфтордецил[1,2-бис(2,2-диметил-1,3-диоксолан-4-ил)-2-гидроксиэтиловый] эфир
30 г (64,64 ммоля) 1Н,1Н,2Н,2Н-перфтордеканола растворяют в 200 мл тетрагидрофурана и при 0°С добавляют 1,68 г (70 ммолей) гидрида натрия. Смесь перемешивают в течение 2 ч при комнатной температуре, а затем в течение 4 ч при 60°С. Далее раствор помещают в металлический автоклав, после чего добавляют 15,78 г (64,64 ммоля) 1,2-бис-(2,2-диметил-1,3-диоксолан-4-ил)оксирана и затем в течение 10 ч выдерживают при 150°С. После этого реакционный раствор сливают в смесь воды со льдом и дважды экстрагируют диэтиловым эфиром. Объединенные органические фазы упаривают досуха и остаток хроматографируют на силикагеле (элюент:дихлорметан/ацетон в соотношении 10:1)
Выход: 14,2 г (31% от теории) бесцветного твердого вещества.
Элементный анализ:
б) Простой 1Н,1Н,2Н,2Н-перфтордецил[1,2-бис(1,2-дигидроксиэтил)-2-гидроксиэтиловый]эфир
14 г (19,76 ммоля) соединения, указанного в заголовке примера 90а, растворяют в 300 мл этанола и добавляют 30 мл 10%-ной водной соляной кислоты. Далее в течение 5 ч кипятят с обратным холодильником. Затем значение рН устанавливают на 7 с помощью едкого натра, после чего концентрируют досуха и остаток хроматографируют на RP-18 (элюент:градиент ацетонитрила/воды).
Выход: 10,55 г (85% от теории) бесцветного твердого вещества.
Содержание воды: 3,2%.
Элементный анализ:
в) Получение композиции, содержащей металлический комплекс II и простой 1Н,1Н,2Н,2Н-перфтордецил[1,2-бис(1,2-дигидроксиэтил)-2-гидроксиэтиловый] эфир
К 41 мл раствора металлического комплекса II (300 ммолей/л) в 0,45%-ном водном растворе поваренной соли (рН 7,4; 0,25 мг/л CaNa3ДТПА) добавляют 11,98 г (19,07 ммоля) соединения, указанного в заголовке примера 90б, и объем с помощью 0,9%-ного водного раствора поваренной соли доводят до 64 мл. Далее выдерживают в течение 2 ч при 60°С, подвергая смесь воздействию ультразвука. Раствор охлаждают до комнатной температуры и значение рН устанавливают на 7,4 с помощью водного 2н. едкого натра. После этого фильтруют через фильтр с размером пор 0,2 мкм и фильтрат расфасовывают во флаконы. Полученный таким путем раствор можно непосредственно использовать для проведения биологических экспериментов. (Концентрация Gd составляет 200 ммолей/л.)
Пример 91
а) N,N-бис[(8-серная кислота-моноэфир, натриевая соль)-3,6-диоксаоктил]амид перфтороктилсульфоновой кислоты
13,54 г (17,73 ммоля) соединения, указанного в заголовке примера 84а, растворяют в 300 мл хлороформа и при 0°С добавляют 2,82 г (17,73 ммоля) комплекса триоксида серы с пиридином. Смесь перемешивают в течение часа при 0°С и затем досуха упаривают в вакууме. Остаток растворяют в 300 мл этанола и смешивают с 17,8 мл 1н. водного едкого натра. Раствор упаривают досуха и остаток сушат в вакууме (60°С, 2 ч).
Выход: 17,15 г (количественный).
Элементный анализ:
б) Получение композиции, содержащей металлический комплекс I и N,N-бис[(8-серная кислота-моноэфир, натриевая соль)-3,6-диоксаоктил]амид перфтороктилсульфоновой кислоты
К 43 мл раствора металлического комплекса I (380 ммолей/л) в 0,45%-ном водном растворе поваренной соли (рН 7,4; 0,25 мг/л CaNa3ДТПА) добавляют 142,29 г (147,06 ммоля) соединения, указанного в заголовке примера 91а, и объем с помощью 0,9%-ного водного раствора поваренной соли доводят до 164 мл. Далее выдерживают в течение 2 ч при 60°С, подвергая смесь воздействию ультразвука. Раствор охлаждают до комнатной температуры и значение рН устанавливают на 7,4 с помощью водного 2н. едкого натра. После этого фильтруют через фильтр с размером пор 0,2 мкм и фильтрат расфасовывают во флаконы. Полученный таким путем раствор можно непосредственно использовать для проведения биологических экспериментов. (Концентрация Gd составляет 100 ммолей/л.)
Пример 92
а) Диэтиловый эфир 2-(2Н,2Н,3H,3H,5,5Н,6Н,6Н-1,4-диоксаперфтортетрадец-1-ил)янтарной кислоты
30 г (59,03 ммоля) 1Н,1Н,2Н,2Н,4Н,4Н,5Н,5Н-3-оксаперфтортридеканола растворяют в 300 мл тетрагидрофурана и при 0°С добавляют 1,68 г (70 ммолей) гидрида натрия. Далее перемешивают в течение часа при 0°С, а затем в течение 5 ч при 40°С. Затем к этому нагретому до 40°С раствору по каплям в течение 10 мин добавляют 20,25 г (80 ммолей) диэтилового эфира 2-бромянтарной кислоты, после чего перемешивают в течение 12 ч при этой температуре. Далее добавляют 500 мл смеси воды со льдом и дважды экстрагируют 300 мл диэтилового эфира. Объединенные органические фазы досуха упаривают в вакууме и остаток хроматографируют на силикагеле (элюент:н-гексан/этанол в соотношении 20:1).
Выход: 12,05 г (30% от теории).
Элементный анализ:
б) 2-(2Н,2Н,3H,3H,5Н,5Н,6Н,6Н-1,4-диоксаперфтортетрадец-1-ил)янтарная кислота, динатриевая соль
К 11,5 г (16,90 ммоля) указанного в заголовке примера 92а соединения, растворенного в 300 мл метанола, добавляют 50 мл 3 н. водного едкого натра и в течение 8 ч кипятят с обратным холодильником. После этого упаривают досуха и остаток растворяют в 300 мл воды. Далее водные фазы дважды экстрагируют 300 мл диэтилового эфира. После этого водные фазы подкисляют концентрированной соляной кислоты до рН 1 и дважды экстрагируют 300 мл хлороформа. Объединенные хлороформные фазы сушат над сульфатом магния и упаривают досуха. Остаток растворяют в 300 мл воды и значение рН устанавливают на 7,4 с помощью 5%-ного водного едкого натра. В завершение лиофилизуют.
Выход: 10,50 г (93% от теории) бесцветного аморфного твердого вещества.
Содержание воды: 5,7%.
Элементный анализ:
в) Получение композиции, содержащей металлический комплекс II и динатриевую соль 2-(2Н,2Н,3H,3H,5Н,5Н,6Н,6Н-1,4-диоксаперфтортетрадец-1-ил)янтарной кислоты
К 57 мл раствора металлического комплекса II (300 ммолей/л) в 0,45%-ном водном растворе поваренной соли (рН 7,4; 0,25 мг/л CaNa3ДТПА) добавляют 1,14 г (1,71 ммоля) соединения, указанного в заголовке примера 92б, и объем с помощью 0,9%-ного водного раствора поваренной соли доводят до 154 мл. Далее выдерживают в течение 2 ч при 60°С, подвергая смесь воздействию ультразвука. Раствор охлаждают до комнатной температуры и значение рН устанавливают на 7,4 с помощью водного 2н. едкого натра. После этого фильтруют через фильтр с размером пор 0,2 мкм и фильтрат расфасовывают во флаконы. Полученный таким путем раствор можно непосредственно использовать для проведения биологических экспериментов. (Концентрация Gd составляет 100 ммолей/л.)
Пример 93
а) N-(сукцин-2-ил)амид 2Н,2Н,4Н,4Н,5Н,5Н-3-оксаперфтортридекановой кислоты
К 20 г (38,30 ммоля) 2Н,2Н,4Н,4Н,5Н,5Н-3-оксаперфтортридекановой кислоты и 9,21 г (80 ммолей) N-гидроксисукцинимида, растворенным в 150 мл диметилформамида, при 0°С добавляют 16,51 г (80 ммолей) N,N'-дициклогексилкарбодиимида и перемешивают в течение 3 ч при этой температуре. К полученному таким путем раствору активированного сложного эфира добавляют охлажденный до 0°С раствор 5,10 г (38,30 ммоля) L-аспарагиновой кислоты в 300 мл 5%-ного водного раствора карбоната натрия и перемешивают в течение 2 ч при 0°С. Далее сливают в 500 мл смеси воды со льдом, выпавшую в осадок дициклогексилмочевину отфильтровывают и значение рН устанавливают на 1 с помощью концентрированной соляной кислоты. После этого трижды экстрагируют 300 мл хлороформа. Объединенные органические фазы концентрируют досуха и остаток хроматографируют на RP-18 (элюент:градиент ацетонитрила/воды). Полученную таким путем дикислоту растворяют в 400 мл воды и значение рН устанавливают на 7,4 с помощью 1н. водного едкого натра. После этого фильтруют и фильтрат лиофилизуют.
Содержание воды: 6,3%.
Выход: 21,13 г (81% от теории) бесцветного аморфного порошка.
Элементный анализ:
б) Получение композиции, содержащей металлический комплекс XIV и N-(сукцин-2-ил)амид 2Н,2Н,4Н,4Н,5Н,5Н-3-оксаперфтортридекановой кислоты
К 37 мл раствора металлического комплекса XIV (300 ммолей/л) в 0,45%-ном водном растворе поваренной соли (рН 7,4; 0,25 мг/л CaNa3ДТПА) добавляют 422 мг (0,62 ммоля) соединения, указанного в заголовке примера 93а, и объем с помощью 0,9%-ного водного раствора поваренной соли доводят до 111 мл. Далее выдерживают в течение 2 ч при 60°С, подвергая смесь воздействию ультразвука. Раствор охлаждают до комнатной температуры и значение рН устанавливают на 7,4 с помощью водного 2н. едкого натра. После этого фильтруют через фильтр с размером пор 0,2 мкм и фильтрат расфасовывают во флаконы. Полученный таким путем раствор можно непосредственно использовать для проведения биологических экспериментов. (Концентрация Gd составляет 100 ммолей/л.)
Пример 94
Получение композиции, содержащей указанное в заголовке примера 67 соединение и натриевую соль перфтороктансульфоновой кислоты
К 43 мл раствора указанного в заголовке примера 67 соединения (250 ммолей/л) в 0,45%-ном растворе хлорида натрия (рН 7,4; 0,25 мг/л CaNa3ДТПА) добавляют раствор 1,34 г (2,69 ммоля) натриевой соли перфтороктансульфоновой кислоты в 200 мл этанола и перемешивают в течение 2 ч при 50°С. Далее раствор досуха концентрируют в вакууме и объем остатка добавлением дистиллированной воды доводят до 108 мл. Смесь перемешивают в течение 10 мин при 40°С и фильтруют через фильтр с размером пор 0,2 мкм. Фильтрат расфасовывают во флаконы. Полученный таким путем раствор можно непосредственно использовать для проведения биологических экспериментов. (Концентрация Gd составляет 100 ммолей/л.)
Пример 95
Получение композиции, содержащей указанное в заголовке примера 68 соединение и натриевую соль перфтордекансульфоновой кислоты
К 49 мл раствора указанного в заголовке примера 68 соединения (310 ммолей/л) в 0,45%-ном водном растворе хлорида натрия добавляют 3,03 г (5,06 ммоля) натриевой соли перфтордекансульфоновой кислоты и в течение 10 мин нагревают в микроволновой печи. Раствор охлаждают до комнатной температуры, фильтруют через фильтр с размером пор 0,2 мкм и фильтрат расфасовывают во флаконы. Полученный таким путем раствор можно непосредственно использовать для проведения биологических экспериментов. (Концентрация Gd составляет 310 ммолей/л.)
Пример 96
а) Простой (1Н,1Н,2Н,2Н-перфтордецил)-5-[(1,3-дикарбокси, динатриевая соль)фениловый] эфир
К 20 г (80,62 ммоля) тринатриевой соли 5-гидроксиизофталевой кислоты в 300 мл диметилформамида добавляют 42,5 г (80,62 ммоля) соединения, указанного в заголовке примера 14а, и перемешивают в течение 10 ч при 60°С. После этого сливают в 1500 мл смеси воды со льдом и значение рН устанавливают на 1 с помощью концентрированной соляной кислоты. Далее трижды экстрагируют 300 мл хлороформа. Объединенные органические фазы упаривают и остаток хроматографируют на RP-18 (элюент:градиент ацетонитрила/воды). Очищенную таким путем дикислоту растворяют в 400 мл воды и значение рН устанавливают на 7,4 с помощью 1н. водного едкого натра. В завершение фильтруют и фильтрат лиофилизуют.
Выход: 20,05 г (37% от теории) бесцветного аморфного твердого вещества.
Содержание воды: 5,0%.
Элементный анализ:
б) Получение композиции, содержащей указанное в заголовке примера 12 в WO 99/01161 соединение (1,4,7-трис{1,4,7-трис(N-карбоксилатометил)-10-(N-1-метил-3-аза-2,5-диоксопентан-1,5-диил]-1,4,7,10-тетраазациклододекан, Gd-комплекс}-10-[2-(N-этил-N-перфтороктилсульфонил)амино] ацетил-1,4,7,10-тетраазациклододекан) и простой (1Н,1Н,2Н,2Н-перфтордецил)-5-[(1,3-дикарбокси, динатриевая соль)фениловый] эфир
К 51 мл раствора 1,4,7-трис{1,4,7-трис(N-карбоксилатометил)-10-(N-1-метил-3-аза-2,5-диоксопентан-1,5-диил]-1,4,7,10-тетраазациклододекан, Gd-комплекс}-10-[2-(N-этил-N-перфтороктилсульфонил)амино] ацетил-1,4,7,10-тетраазациклододекана (300 ммолей/л) в 0,45%-ном водном растворе поваренной соли (рН 7,4; 0,25 мг/л CaNa3ДТПА) добавляют 6,86 г (10,2 ммоля) соединения, указанного в заголовке примера 96а, и объем с помощью 0,9%-ного водного раствора поваренной соли доводят до 153 мл. Далее выдерживают в течение 2 ч при 60°С, подвергая смесь воздействию ультразвука. Раствор охлаждают до комнатной температуры и значение рН устанавливают на 7,4 с помощью водного 2 н. едкого натра. После этого фильтруют через фильтр с размером пор 0,2 мкм и фильтрат расфасовывают во флаконы. Полученный таким путем раствор можно непосредственно использовать для проведения биологических экспериментов. (Концентрация Gd составляет 100 ммолей/л.)
Пример 97
Получение композиции, содержащей металлический комплекс XIV и натриевую соль 3-окса-2Н,2Н,4Н,4Н,5Н,5Н-перфтортридекановой кислоты
К 4 мл раствора металлического комплекса XIV (320 ммолей/л) в 0,45%-ном водном растворе поваренной соли (рН 7,4; 0,25 мг/л CaNa3ДТПА) добавляют 434 мг (0,55 ммоля) соединения, указанного в заголовке примера 80а, и объем с помощью 0,9%-ного водного раствора поваренной соли доводят до 12,8 мл. Далее выдерживают в течение 2 ч при 60°С, подвергая смесь воздействию ультразвука. Раствор охлаждают до комнатной температуры и значение рН устанавливают на 7,4 с помощью водного 2 н. едкого натра. После этого фильтруют через фильтр с размером пор 0,2 мкм и фильтрат расфасовывают во флаконы. Полученный таким путем раствор можно непосредственно использовать для проведения биологических экспериментов. (Концентрация Gd составляет 100 ммолей/л.)
Пример 98
а) трет-Бутиловий эфир (адамант-1-ил)-3-оксапропионовой кислоты
К 15,22 г (100 ммолей) 1-адамантанола в 300 мл 50%-ного водного едкого кали и 200 мл толуола при 0°С добавляют 29,26 г (150 ммолей) трет-бутилового эфира бромуксусной кислоты и интенсивно перемешивают в течение 2 ч. После этого сливают в 1500 мл воды и дважды экстрагируют 300 мл диэтилового эфира. Объединенные органические фазы сушат над сульфатом магния и досуха упаривают в вакууме. Остаток хроматографируют на силикагеле (элюент:н-гексан/диэтиловый эфир в соотношении 20:1).
Выход: 21,58 г (81% от теории) вязкого бесцветного масла.
Элементный анализ:
б) (Адамант-1-ил)-3-оксапропионовая кислота
20 г (75 ммолей) соединения, указанного в заголовке примера 98а, при 0°С растворяют в 200 мл трифторуксусной кислоты и в течение 8 ч перемешивают при комнатной температуре. После этого упаривают досуха и остаток перекристаллизовывают из диизопропилового эфира.
Выход: 14,68 г (93% от теории) бесцветного вещества в виде чешуек или хлопьев.
Элементный анализ:
в) 1-(перфтороктилсульфонил)-4-[(адамант-1-ил)оксапропионил]пиперазин
14 г (66,6 ммоля) соединения, указанного в заголовке примера 98б, и 37,50 г (66,6 ммоля) 1-перфтороктилсульфонилпиперазина растворяют в 300 мл тетрагидрофурана и при 0°С добавляют 32,15 г (130 ммолей) этилового эфира 1,2-дигидро-2-этоксихинолин-1-карбоновой кислоты (ЭЭДХ). Смесь перемешивают в течение 5 ч при комнатной температуре. Затем раствор досуха упаривают в вакууме и остаток хроматографируют на силикагеле (элюент:дихлорметан/диэтиловый эфир в соотношении 30:1).
Выход: 43,05 г (85% от теории) бесцветного твердого вещества.
Элементный анализ:
г) Композиция, содержащая 0,5 части металлического комплекса I и 0,5 части соединения включения в виде гидрата β-циклодекстрина и 1-(перфтороктилсульфонил)-4-[(адамант-1-ил)оксапропионил]пиперазина
К 32 мл раствора металлического комплекса I (280 ммолей/л) в 0,45%-ном водном растворе поваренной соли (рН 7,4; 0,25 мг/л CaNa3ДТПА) добавляют 6,81 г (8,96 ммоля) указанного в заголовке примера 98в соединения и 10,33 г (8,96 ммоля) моногидрата β-циклодекстрина и объем с помощью 0,9%-ного водного раствора поваренной соли доводят до 98 мл. Далее выдерживают в течение 2 ч при 60°С, подвергая смесь воздействию ультразвука. Раствор охлаждают до комнатной температуры и значение рН устанавливают на 7,4 с помощью водного 2н. едкого натра. После этого фильтруют через фильтр с размером пор 0,2 мкм и фильтрат расфасовывают во флаконы. Полученный таким путем раствор можно непосредственно использовать для проведения биологических экспериментов. (Концентрация Gd составляет 100 ммолей/л.)
Пример 99
а) N-(1-адамантил)амид 3-окса-2Н,2Н,4Н,4Н,5Н,5Н-перфтортридекановой кислоты
К 15,12 г (100 ммолей) 1-аминоадамантана, 52,21 г (100 ммолей) 3-окса-2Н,2Н,4Н,4Н,5Н,5Н-перфтортридекановой кислоты и 11,51 г (100 ммолей) N-гидроксисукцинимида, растворенных в 300 мл тетрагидрофурана, при 0°С добавляют 30,95 г (150 ммолей) N,N-дициклогексилкарбодиимида. Смесь перемешивают в течение 2 ч при 0°С, а затем в течение 6 ч при комнатной температуре. Выпавшую в осадок мочевину отфильтровывают, фильтрат упаривают досуха и остаток хроматографируют на силикагеле (элюент:дихлорметан/ацетон в соотношении 30:1).
Выход: 54,4 г (83% от теории) воскоподобного твердого вещества.
Элементный анализ:
б) Композиция, содержащая 0,6 части металлического комплекса II и 0,4 части соединения включения в виде гидрата β-циклодекстрина и N-(1-адамантил)амида 3-окса-2Н,2Н,4Н,4Н,5Н,5Н-перфтортридекановой кислоты
К 41 мл раствора металлического комплекса II (250 ммолей/л) в 0,45%-ном водном растворе поваренной соли (рН 7,4; 0,25 мг/л CaNa3ДТПА) добавляют 4,48 г (6,83 ммоля) соединения, указанного в заголовке примера 99а, и 7,87 г (6,83 ммоля) моногидрата (3-циклодекстрина и объем с помощью 0,9%-ного водного раствора поваренной соли доводят до 103 мл. Далее выдерживают в течение 2 ч при 60°С, подвергая смесь воздействию ультразвука. Раствор охлаждают до комнатной температуры и значение рН устанавливают на 7,4 с помощью водного 2н. едкого натра. После этого фильтруют через фильтр с размером пор 0,2 мкм и фильтрат расфасовывают во флаконы. Полученный таким путем раствор можно непосредственно использовать для проведения биологических экспериментов. (Концентрация Gd составляет 100 ммолей/л.)
Пример 100
а) N-(адамантил)амид 2-[N-(этил)-N-(перфтороктилсульфонил)амино]уксусной кислоты
К 15,12 г (100 ммолей) 1-аминоадамантана, 58,52 г (100 ммолей) N-(этил)-N-(перфтороктилсульфонил)аминоуксусной кислоты и 11,51 г (100 ммолей) N-гидроксисукцинимида, растворенных в 300 мл тетрагидрофурана, при 0°С добавляют 30,95 г (150 ммолей) N,N-дициклогексилкарбодиимида. Смесь перемешивают в течение 2 ч при 0°С, а затем в течение 6 ч при комнатной температуре. Выпавшую в осадок мочевину отфильтровывают, фильтрат упаривают досуха и остаток хроматографируют на силикагеле (элюент:дихлорметан/ацетон в соотношении 30:1).
Выход: 55,65 г (79% от теории) аморфного твердого вещества.
Элементный анализ:
б) Композиция, содержащая 0,6 части металлического комплекса I и 0,4 части соединения включения в виде гидрата β-циклодекстрина и N-(1-адамантил)амида 2-N-(этил)-N-(перфтороктилсульфонил)амино]уксусной кислоты
К 32 мл раствора металлического комплекса I (280 ммолей/л) в 0,45%-ном водном растворе поваренной соли (рН 7,4; 0,25 мг/л CaNa3ДТПА) добавляют 4,20 г (5,97 ммоля) соединения, указанного в заголовке примера 100а, и 6,88 г (5,97 ммоля) моногидрата β-циклодекстрина и объем с помощью 0,9%-ного водного раствора поваренной соли доводят до 90 мл. Далее выдерживают в течение 2 ч при 60°С, подвергая смесь воздействию ультразвука. Раствор охлаждают до комнатной температуры и значение рН устанавливают на 7,4 с помощью водного 2н. едкого натра. После этого фильтруют через фильтр с размером пор 0,2 мкм и фильтрат расфасовывают во флаконы. Полученный таким путем раствор можно непосредственно использовать для проведения биологических экспериментов. (Концентрация Gd составляет 100 ммолей/л.)
Пример 101
а) [1-(4-перфтороктилсульфонил)пиперазин]амид 6-N-бензилоксикарбонил-2-N-(3,6,9,12-тетраоксатридеканоил)лизина
К 50 г (60,20 ммоля) указанного в заголовке примера 52в соединения и 7,10 г (70 ммолей) триэтиламина, растворенных в 350 мл дихлорметана, при 0°С по каплям добавляют раствор 16,85 г (70 ммолей) хлорангидрида 3,6,9,12-тетраоксатридекановой кислоты в 50 мл дихлорметана и перемешивают в течение 3 ч при 0°С. Далее добавляют 200 мл 5%-ной водной соляной кислоты и перемешивают в течение 5 мин при комнатной температуре. Органическую фазу отделяют, сушат над сульфатом магния и досуха упаривают в вакууме. Остаток хроматографируют на силикагеле (элюент:дихлорметан/ацетон в соотношении 15:1)
Выход: 30,94 г (92% от теории) бесцветного, вязкого масла.
Элементный анализ:
б) [1-(4-перфтороктилсульфонил)пиперазин]амид 2-N-(3,6,9,12-тетраоксатридеканоил)лизина
53,96 г (52,15 ммоля) соединения, указанного в заголовке примера 101а, растворяют в 500 мл этанола и добавляют 6 г палладиевого катализатора (10%-ный Pd/C). После этого гидрируют при комнатной температуре. Затем катализатор отфильтровывают и фильтрат досуха упаривают в вакууме.
Выход: 43,0 г (количественный) бесцветного твердого вещества.
Элементный анализ:
в) [1-(4-перфтороктилсульфонил)пиперазин]амид 6-N-[1,4,7-трис(карбоксилатометил)-1,4,7,10-тетраазациклододекан-10-(пентаноил-3-аза-4-оксо-5-метил-5-ил)]-2-N-(3,6,9,12-тетраоксатридеканоил)лизина, Gd-комплекс
21,84 (24,25 ммоля) соединения, указанного в заголовке примера 101б, 2,79 г (24,25 ммоля) N-гидроксисукцинимида, 2,12 г (50 ммолей) хлорида лития и 15,27 г (24,25 ммоля) Gd-комплекса 1,4,7-трис(карбоксилатометил)-10-[(3-аза-4-оксо-5-метил-5-ил)пентановая кислота]-1,4,7,10-тетраазациклододекана растворяют при умеренном нагревании в 200 мл диметилсульфоксида. Далее при 10°С добавляют 8,25 г (40 ммолей) N,N-дициклогексилкарбодиимида и затем перемешивают в течение ночи при комнатной температуре. Раствор сливают в 3000 мл ацетона и перемешивают в течение 10 мин. Выпавшее в осадок твердое вещество отфильтровывают и затем очищают хроматографией (силикагель RP-18, элюент:градиент воды/этанола/ацетонитрила).
Выход: 28,21 г (81% от теории) бесцветного твердого вещества.
Содержание воды: 11,0%.
Элементный анализ (в пересчете на безводное вещество):
Пример 102
а) [1-(4-перфтороктилсульфонил)пиперазин]амид 6-N-бензилоксикарбонил-2-N-(пропил-3-сульфоновая кислота)лизина
К 50 г (60,20 ммоля) соединения, указанного в заголовке примера 52в, и 7,10 г (70 ммолей) триэтиламина, растворенных в 250 мл сухого тетрагидрофурана, при 50°С по каплям добавляют раствор 7,33 г (60 молей) пропансультона в 50 мл тетрагидрофурана и перемешивают в течение 3 ч при 60°С. Далее добавляют 200 мл 5%-ной водной соляной кислоты и перемешивают в течение 5 мин при комнатной температуре. Органическую фазу отделяют, сушат над сульфатом магния и досуха упаривают в вакууме. Остаток хроматографируют на силикагеле (элюент:дихлорметан/ацетон в соотношении 15:1).
Выход: 45,16 г (79% от теории) бесцветного вязкого масла.
Элементный анализ:
б) [1-(4-перфтороктилсульфонил)пиперазин]амид 2-N-(пропил-3-сульфоновая кислота)лизина
49,68 г (52,15 ммоля) соединения, указанного в заголовке примера 102а, растворяют в 500 мл этанола и добавляют 6 г палладиевого катализатора (10%-ный Pd/C). После этого гидрируют при комнатной температуре. Затем катализатор отфильтровывают и фильтрат досуха упаривают в вакууме.
Выход: 42,69 г (количественный) бесцветного твердого вещества.
Элементный анализ:
в) [1-(4-перфтороктилсульфонил)пиперазин]амид 6-N-[1,4,7-трис(карбоксилатометил)-1,4,7,10-тетраазациклододекан-10-(пентаноил-3-аза-4-оксо-5-метил-5-ил)]-2-N-(пропил-3-сульфоновая кислота)лизина, Gd-комплекс
19,85 г (24,25 ммоля) соединения, указанного в заголовке примера 102б, 2,79 г (24,25 ммоля) N-гидроксисукцинимида, 2,12 г (50 ммолей) хлорида лития и 15,27 г (24,25 ммоля) Gd-комплекса 1,4,7-трис(карбоксилатометил)-10-[(3-аза-4-оксо-5-метил-5-ил)пентановая кислота]-1,4,7,10-тетраазациклододекана растворяют при умеренном нагревании в 200 мл диметилсульфоксида. Далее при 10°С добавляют 8,25 г (40 ммолей) N,N-дициклогексилкарбодиимида и затем перемешивают в течение ночи при комнатной температуре. Раствор сливают в 3000 мл ацетона и перемешивают в течение 10 мин. Выпавшее в осадок твердое вещество отфильтровывают и затем очищают хроматографией (силикагель RP-18, элюент:градиент воды/этанола/ацетонитрила).
Выход: 28,13 г (81% от теории) бесцветного твердого вещества.
Содержание воды: 11,0%.
Элементный анализ (в пересчете на безводное вещество)
Пример 103
а) [1-(4-перфтороктилсульфонил)пиперазин]амид 6-N-бензилоксикарбонил-2-N,N-бис(пропил-3-сульфоновая кислота)лизина
К 50 г (60,20 ммоля) соединения, указанного в заголовке примера 52в, и 12,14 г (120 ммолей) триэтиламина, растворенных в 250 мл сухого тетрагидрофурана, при 50°С по каплям добавляют раствор 14,65 г (120 ммолей) 1,3-пропансультона в 100 мл тетрагидрофурана и перемешивают в течение 3 ч при 60°С. Далее добавляют 400 мл 5%-ной водной соляной кислоты, перемешивают в течение 5 мин при комнатной температуре, смешивают с хлоридом натрия, органическую фазу отделяют, сушат ее над сульфатом магния и досуха упаривают в вакууме. Остаток хроматографируют на силикагеле (элюент:дихлорметан/ацетон в соотношении 15:1).
Выход: 52,24 г (81% от теории) бесцветного вязкого масла.
Элементный анализ:
б) [1-(4-перфтороктилсульфонил)пиперазин]амид 2-N,N-бис(пропил-3-сульфоновая кислота)лизина
53,74 г (52,15 ммоля) соединения, указанного в заголовке примера 103а, растворяют в 500 мл этанола и добавляют 6 г палладиевого катализатора (10%-ный Pd/C). После этого гидрируют при комнатной температуре. Затем катализатор отфильтровывают и фильтрат досуха упаривают в вакууме.
Выход: 49,06 г (количественный) бесцветного твердого вещества.
Элементный анализ:
в) [1-(4-перфтороктилсульфонил)пиперазин]амид 6-N-[1,4,7-трис(карбоксилатометил)-1,4,7,10-тетраазациклододекан-10-(пентаноил-3-аза-4-оксо-5-метил-5-ил)]-2-N,N-бис(пропил-3-сульфоновая кислота)лизина, Gd-комплекс, динатриевая соль
38,76 г (24,25 ммоля) соединения, указанного в заголовке примера 103б, 2,79 г (24,25 ммоля) N-гидроксисукцинимида, 2,12 г (50 ммолей) хлорида лития и 15,27 г (24,25 ммоля) Gd-комплекса 1,4,7-трис(карбоксилатометил)-10-[(3-аза-4-оксо-5-метил-5-ил)пентановая кислота]-1,4,7,10-тетраазациклододекана растворяют при умеренном нагревании в 200 мл диметилсульфоксида. Далее при 10°С добавляют 8,25 г (40 ммолей) N,N-дициклогексилкарбодиимида и затем перемешивают в течение ночи при комнатной температуре. Раствор сливают в 3000 мл ацетона и перемешивают в течение 10 мин. Выпавшее в осадок твердое вещество отфильтровывают и затем очищают хроматографией (силикагель RP-18, элюент:градиент воды/этанола/ацетонитрила).
Выход: 31,63 г (81% от теории) бесцветного твердого вещества.
Содержание воды: 11,0%.
Элементный анализ (в пересчете на безводное вещество):
Пример 104
а) 5-бензиловый эфир N-трифторацетил-L-глутаминовой кислоты
100 г (421,5 ммоля) 5-бензилового эфира L-глутаминовой кислоты растворяют в смеси из 1000 мл этилового эфира трифторуксусной кислоты и 500 мл этанола и перемешивают в течение 24 ч при комнатной температуре. После этого упаривают досуха и остаток кристаллизуют из диизопропилового эфира.
Выход: 140,47 г (96% от теории) бесцветного кристаллического порошка.
Элементный анализ:
б) N-бис(2-гидроксиэтил)амид 5-бензилового эфира 2-N-трифторацетил-L-глутаминовой кислоты
К раствору 24,9 г (24,08 ммоля) соединения, указанного в заголовке примера 104а, 2,53 г (24,08 ммоля) диэтаноламина и 2,77 г (24,08 ммоля) N-гидроксисукцинимида в 150 мл диметилформамида при 0°С добавляют 8,25 г (40 ммолей) N,N-дициклогексилкарбодиимида. Смесь перемешивают в течение 3 ч при 0°С, а затем в течение ночи при комнатной температуре. Выпавшую в осадок мочевину отфильтровывают, фильтрат досуха упаривают в вакууме и хроматографируют на силикагеле (элюент:дихлорметан/этанол в соотношении 20:1).
Выход: 9,11 г (90% от теории) вязкого масла.
Элементный анализ:
в) N-бис(2-гидроксиэтил)моноамид N-трифторацетил-L-глутаминовой кислоты
21,92 г (52,15 ммоля) соединения, указанного в заголовке примера 104б, растворяют в 500 мл этанола и добавляют 3 г палладиевого катализатора (10%-ный Pd/C). После этого гидрируют при комнатной температуре. Затем катализатор отфильтровывают и фильтрат досуха упаривают в вакууме.
Выход: 43,0 г (количественный) бесцветного твердого вещества.
Элементный анализ:
г) Трифторацетил-L-глутаминовая кислота-N-бис(2-гидроксиэтил)амид-5-[1-(4-перфтороктилсульфонил)пиперазин]амид
К 10,96 г (33,2 ммоля) соединения, указанного в заголовке примера 104а, и 18,87 г (33,2 ммоля) 1-перфтороктилсульфонилпиперазина (полученного согласно DE 19603033) в 80 мл тетрагидрофурана при 0°С добавляют 16,42 г (66,4 ммоля) ЭЭДХ (этиловый эфир 2-этокси-1,2-дигидрохинолин-1-карбоновой кислоты) и перемешивают в течение ночи при комнатной температуре. После этого досуха упаривают в вакууме и хроматографируют на силикагеле (элюент:дихлорметан/метанол в соотношении 20:1).
Выход: 30,93 г (93% от теории) бесцветного твердого вещества.
Элементный анализ:
д) L-глутаминовая кислота-N-бис(2-гидроксиэтил)амид-5-[1-(4-перфтороктилсульфонил)пиперазин]амид
Раствор 30,24 г (30,22 ммоля) указанного в заголовке примера 104б в 200 мл этанола при 0°С в течение часа барботируют газообразным аммиаком. Смесь затем перемешивают в течение 4 ч при 0°С. После этого упаривают досуха и остаток выделяют из воды путем перемешивания. Твердое вещество отфильтровывают и сушат в вакууме (50°С).
Выход: 26,55 г (97% от теории) аморфного твердого вещества.
Элементный анализ:
e) N-[1,4,7-трис(карбоксилатометил)-1,4,7,10-тетраазациклододекан-10-(пентаноил-3-аза-4-оксо-5-метил-5-ил)]-L-глутаминовая кислота-N-бис(2-гидроксиэтил)амид-5-[1-(4-перфтороктилсульфонил)пиперазин]амид, Gd-комплекс
211,96 г (24,25 ммоля) соединения, указанного в заголовке примера 104д, 2,79 г (24,25 ммоля) N-гидроксисукцинимида, 2,12 г (50 ммолей) хлорида лития и 15,27 г (24,25 ммоля) Gd-комплекса 1,4,7-трис(карбоксилатометил)-10-[(3-аза-4-оксо-5-метил-5-ил)пентановая кислота]-1,4,7,10-тетраазациклододекана растворяют при умеренном нагревании в 200 мл диметилсульфоксида. Далее при 10°С добавляют 8,25 г (40 ммолей) N,N-дициклогексилкарбодиимида и затем перемешивают в течение ночи при комнатной температуре. Раствор сливают в 3000 мл ацетона и перемешивают в течение 10 мин. Выпавшее в осадок твердое вещество отфильтровывают и затем очищают хроматографией (силикагель RP-18, элюент:градиент воды/этанола/ацетонитрила).
Выход: 27,43 г (81% от теории) бесцветного твердого вещества.
Содержание воды: 11,0%.
Элементный анализ (в пересчете на безводное вещество):
Пример 105
а) N-диметилбис(1,1-дигидроксиметил)амид 5-бензилового эфира N-трифторацетил-L-глутаминовой кислоты
К раствору 8,03 г (24,08 ммоля) соединения, указанного в заголовке примера 104а, 3,98 г (24,08 ммоля) диметилбис(1,1-дигидроксиметил)амина и 2,77 г (24,08 ммоля) N-гидроксисукцинимида в 150 мл диметилформамида при 0°С добавляют 8,25 г (40 ммолей) N,N-дициклогексилкарбодиимида. Смесь перемешивают в течение 3 ч при 0°С, а затем в течение ночи при комнатной температуре. Выпавшую в осадок мочевину отфильтровывают, фильтрат досуха упаривают в вакууме и хроматографируют на силикагеле (элюент:дихлорметан/этанол в соотношении 20:1).
Выход: 110,53 г (91% от теории) вязкого масла.
Элементный анализ:
б) [1-(4-перфтороктилсульфонил)пиперазин]амид 5-бензилового эфира N-трифторацетил-L-глутаминовой кислоты
25,05 г (52,15 ммоля) соединения, указанного в заголовке примера 105а, растворяют в 500 мл этанола и добавляют 6 г палладиевого катализатора (10%-ный Pd/C). После этого гидрируют при комнатной температуре. Затем катализатор отфильтровывают и фильтрат досуха упаривают в вакууме.
Выход: 20,36 г (количественный) бесцветного твердого вещества.
Элементный анализ:
в) N-трифторацетил-L-глутаминовая кислота-N-диметилбис(1,1-дигидроксиметил)амид-5-[1-(4-перфтороктилсульфонил)пиперазин]амид
К 12,96 г (33,2 ммоля) соединения, указанного в заголовке примера 105б, и 18,87 г (33,2 ммоля) 1-перфтороктилсульфонилпиперазина (полученного согласно DE 19603033) в 800 мл тетрагидрофурана при 0°С добавляют 16,42 г (66,4 ммоля) ЭЭДХ (этиловый эфир 2-этокси-1,2-дигидрохинолин-1-карбоновой кислоты) и перемешивают в течение ночи при комнатной температуре. После этого досуха упаривают в вакууме и хроматографируют на силикагеле (элюент:дихлорметан/метанол в соотношении 20:1).
Выход: 28,42 г (91% от теории) бесцветного твердого вещества.
Элементный анализ:
г) L-глутаминовая кислота-N-[диметилбис(1,1-дигидроксиметил)]амид-5-[1-(4-перфтороктилсульфонил)пиперазин]амид
Раствор 28,41 г (30,2 ммоля) соединения, указанного в заголовке примера 105в, в 200 мл этанола при 0°С в течение часа барботируют газообразным аммиаком. Смесь затем перемешивают в течение 4 ч при 0°С. После этого упаривают досуха и остаток выделяют из воды путем перемешивания. Твердое вещество отфильтровывают и сушат в вакууме (50°С).
Выход: 24,74 г (97% от теории) аморфного твердого вещества.
Элементный анализ:
д) 2-N-[1,4,7-трис(карбоксилатометил)-1,4,7,10-тетраазациклододекан-10-(пентаноил-3-аза-4-оксо-5-метил-5-ил)]-L-глутаминовая кислота-N-[диметилбис(1,1-дигидроксиметил)амид]-5-[1-(4-перфтороктилсульфонил)пиперазин]амид, Gd-комплекс
20,48 г (24,25 ммоля) соединения, указанного в заголовке примера 105г, 2,79 г (24,25 ммоля) N-гидроксисукцинимида, 2,12 г (50 ммолей) хлорида лития и 15,27 г (24,25 ммоля) Gd-комплекса 1,4,7-трис(карбоксилатометил)-10-[(3-аза-4-оксо-5-метил-5-ил)пентановая кислота]-1,4,7,10-тетраазациклододекана растворяют при умеренном нагревании в 200 мл диметилсульфоксида. Далее при 10°С добавляют 8,25 г (40 ммолей) N,N-дициклогексилкарбодиимида и затем перемешивают в течение ночи при комнатной температуре. Раствор сливают в 3000 мл ацетона и перемешивают в течение 10 мин. Выпавшее в осадок твердое вещество отфильтровывают и затем очищают хроматографией (силикагель RP-18, элюент:градиент воды/этанолы/ацетонитрила).
Выход: 29,05 г (83% от теории) бесцветного твердого вещества.
Содержание воды: 11,0%.
Элементный анализ (в пересчете на безводное вещество):
Пример 106
а) [1-(4-перфтороктилсульфонил)пиперазин]амид 5-бензилового эфира N-трифторметилацетил-L-глутаминовой кислоты
К 11,06 г (33,2 ммоля) соединения, указанного в заголовке примера 104а, и 18,87 г (33,2 ммоля) 1-перфтороктилсульфонилпиперазина (полученного согласно DE 19603033) в 80 мл тетрагидрофурана при 0°С добавляют 16,42 г (66,4 ммоля) ЭЭДХ (этиловый эфир 2-этокси-1,2-дигидрохинолин-1-карбоновой кислоты) и перемешивают в течение ночи при комнатной температуре. После этого досуха упаривают в вакууме и хроматографируют на силикагеле (элюент:дихлорметан/метанол в соотношении 20:1).
Выход: 27,28 г (93% от теории) бесцветного твердого вещества.
Элементный анализ:
б) 5-[1-[4-перфтороктилсульфонил)пиперазин]амид N-трифторацетил-L-глутаминовой кислоты
21,92 г (52,15 ммоля) соединения, указанного в заголовке примера 106а, растворяют в 500 мл этанола и добавляют 3 г палладиевого катализатора (10%-ный Pd/C). После этого гидрируют при комнатной температуре. Затем катализатор отфильтровывают и фильтрат досуха упаривают в вакууме.
Выход: 41,37 г (количественный) бесцветного твердого вещества.
Элементный анализ:
в) N-трифторацетил-L-глутаминовая кислота-N-бис(2-гидроксиэтил)амид-5-[1-(4-перфтороктилсульфонил)пиперазин]амид
К раствору 24,9 г (24,08 ммоля) соединения, указанного в заголовке примера 106а, 2,53 г (24,08 ммоля) диэтаноламина и 2,77 г (24,08 ммоля) N-гидроксисукцинимида в 150 мл диметилформамида при 0°С добавляют 8,25 г (40 ммолей) N,N-дициклогексилкарбодиимида. Смесь перемешивают в течение 3 ч при 0°С, а затем в течение ночи при комнатной температуре. Выпавшую в осадок мочевину отфильтровывают, фильтрат досуха упаривают в вакууме и хроматографируют на силикагеле (элюент:дихлорметан/этанол в соотношении 20:1).
Выход: 9,11 г (90% от теории) вязкого масла.
Элементный анализ:
г) L-глутаминовая кислота-N-бис(2-гидроксиэтил)амид-5-[1-(4-перфтороктилсульфонил)пиперазин]амид
Раствор 26,61 г (30,22 ммоля) соединения, указанного в заголовке примера 106в, в 200 мл этанола при 0°С в течение часа барботируют газообразным аммиаком. Смесь затем перемешивают в течение 4 ч при 0°С. После этого упаривают досуха и остаток выделяют из воды путем перемешивания. Твердое вещество отфильтровывают и сушат в вакууме (50°С).
Выход: 23,93 г (97% от теории) аморфного твердого вещества.
Элементный анализ:
д) N-[1,4,7-трис(карбоксилатометил)-1,4,7,10-тетраазациклододекан-10-(пентаноил-3-аза-4-оксо-5-метил-5-ил)]-L-глутаминовая кислота-N-бис(2-гидроксиэтил)амид-5-[1-(4-перфтороктилсульфонил)пиперазин]амид, Gd-комплекс
16,43 г (24,25 ммоля) соединения, указанного в заголовке примера 106г, 2,79 г (24,25 ммоля) N-гидроксисукцинимида, 2,12 г (50 ммолей) хлорида лития и 15,27 г (24,25 ммоля) Gd-комплекса 1,4,7-трис(карбоксилатометил)-10-[(3-аза-4-оксо-5-метил-5-ил)пентановая кислота]-1,4,7,10-тетраазациклододекана растворяют при умеренном нагревании в 200 мл диметилсульфоксида. Далее при 10°С добавляют 8,25 г (40 ммолей) N,N-дициклогексилкарбодиимида и затем перемешивают в течение ночи при комнатной температуре. Раствор сливают в 3000 мл ацетона и перемешивают в течение 10 мин. Выпавшее в осадок твердое вещество отфильтровывают и затем очищают хроматографией (силикагель RP-18, элюент:градиент воды/этанола/ацетонитрила).
Выход: 28,10 г (83% от теории) бесцветного твердого вещества.
Содержание воды: 11,0%.
Элементный анализ (в пересчете на безводное вещество)
Пример 107
а) [1-(4-перфтороктилсульфонил)пиперазин]амид 5-бензилового эфира N-трифторацетилглутаминовой кислоты
К 11,06 г (33,2 ммоля) соединения, указанного в заголовке примера 104а, и 18,87 г (33,2 ммоля) 1-перфтороктилсульфонилпиперазина (полученного согласно DE 19603033) в 80 мл тетрагидрофурана при 0°С добавляют 16,42 г (66,4 ммоля) ЭЭДХ (этиловый эфир 2-этокси-1,2-дигидрохинолин-1-карбоновой кислоты) и перемешивают в течение ночи при комнатной температуре. После этого досуха упаривают в вакууме и хроматографируют на силикагеле (элюент:дихлорметан/метанол в соотношении 20:1).
Выход: 27,28 г (93% от теории) бесцветного твердого вещества.
Элементный анализ:
б) 5-[1-[4-перфтороктилсульфонил)пиперазин]амид N-трифторацетил-L-глутаминовой кислоты
21,92 г (52,15 ммоля) соединения, указанного в заголовке примера 107а, растворяют в 500 мл этанола и добавляют 3 г палладиевого катализатора (10%-ный Pd/C). После этого гидрируют при комнатной температуре. Затем катализатор отфильтровывают и фильтрат досуха упаривают в вакууме.
Выход: 41,37 г (количественный) бесцветного твердого вещества.
Элементный анализ:
Пример 108
а) [1-(4-перфтороктилсульфонил)пиперазин]амид 6-N-бензилоксикарбонил-2-N-(2,3,4,5-пентагидроксигексаноил)-L-лизина
К раствору 100,0 г (120,4 моля) соединения, указанного в заголовке примера 21в, в 500 мл сухого тетрагидрофуран при 50°С по каплям добавляют раствор 21,45 г (120,4 моля) 5-глюконолактона в 50 мл тетрагидрофурана. Смесь перемешивают в течение 3 ч при 60°С, а затем в течение ночи при комнатной температуре. После этого досуха упаривают в вакууме и остаток хроматографируют на силикагеле (элюент:дихлорметан/этанол в соотношении 20:1).
Выход: 98,37 г (82% от теории) вязкого масла.
Элементный анализ:
б) 1-[(4-перфтороктилсульфонил)пиперазин]амид 2-N-(2,3,4,5-пентагидроксигексаноил)-L-лизина
100,9 г (100,0 ммолей) соединения, указанного в заголовке примера 108а, растворяют в 2000 мл этанола и добавляют 10,0 г палладиевого катализатора (10%-ный Pd/C). После этого гидрируют в течение 12 ч при комнатной температуре. Затем катализатор отфильтровывают и фильтрат досуха упаривают в вакууме.
Выход: 87,46 г (количественный) бесцветного твердого вещества.
Элементный анализ:
в) [1-(4-перфтороктилсульфонил)пиперазин]амид 6-N-[1,4,7-трис(карбоксилатометил)]-1,4,7,10-тетраазациклододекан-10-N-(пентаноил-3-аза-4-оксо-5-метил-5-ил)]-2-N-[1-O-α-D-карбонилметилманнопираноза]-L-лизина, Gd-комплекс
50,0 г (54,55 ммоля) соединения, указанного в заголовке примера 21д, 6,28 г (54,55 ммоля) N-гидроксисукцинимида, 4,62 г (109,0 моля) хлорида лития и 34,35 г (54,55 моля) Gd-комплекса 1,4,7-трис(карбоксилатометил)-10-(карбокси-3-аза-4-оксо-5-метилпент-5-ил)-1,4,7,10-тетраазациклододекана растворяют при умеренном нагревании в 400 мл диметилсульфоксида. Далее при 10°С добавляют 16,88 г (81,8 моля) N,N-дициклогексилкарбодиимида и затем перемешивают в течение ночи при комнатной температуре. Раствор сливают в 3000 мл ацетона и перемешивают в течение 10 мин. Выпавшее в осадок твердое вещество отфильтровывают и затем очищают хроматографией (RP-18, элюент:градиент воды/этанола/ацетонитрила).
Выход: 75,9 г (91,0% от теории) бесцветного твердого вещества.
Содержание воды: 8,6%.
Элементный анализ (в пересчете на безводное вещество):
Пример 109
а) [1-(4-перфтороктилсульфонил)пиперазин]амид 6-N-бензилоксикарбонил-2-N-(2,3,4,5-пентагидроксигексаноил)-L-лизина
К раствору 100,0 г (120,4 ммоля) соединения, указанного в заголовке примера 21в, и 12,18 г (120,4 ммоля) триэтиламина в 500 мл сухого тетрагидрофурана при 50°С по каплям добавляют раствор 21,45 г (120,4 моля) 5-глюконолактона в 50 мл тетрагидрофурана. Смесь перемешивают в течение 3 ч при 60°С, а затем в течение ночи при комнатной температуре. После этого добавляют 400 мл 5%-ной водной соляной кислоты, перемешивают в течение 5 мин при комнатной температуре, смешивают с хлоридом натрия, органическую фазу отделяют, сушат ее над сульфатом магния, досуха упаривают в вакууме и остаток хроматографируют на силикагеле (элюент:дихлорметан/этанол в соотношении 20:1).
Выход: 100,97 г (82% от теории) вязкого масла.
Элементный анализ:
б) 1-[(4-перфтороктилсульфонил)пиперазин]амид 2-N-(2,3,4,5-пентагидроксигексаноил)-L-лизина
100,9 г (100,0 ммолей) соединения, указанного в заголовке примера 108а, растворяют в 2000 мл этанола и добавляют 10,0 г палладиевого катализатора (10%-ный Pd/C). После этого гидрируют в течение 12 ч при комнатной температуре. Затем катализатор отфильтровывают и фильтрат досуха упаривают в вакууме.
Выход: 87,46 г (количественный) бесцветного твердого вещества.
Элементный анализ:
в) [1-(4-перфтороктилсульфонил)пиперазин]амид 6-N-[1,4,7-трис(карбоксилатометил)]-1,4,7,10-тетраазациклододекан-10-N-(пентаноил-3-аза-4-оксо-5-метил-5-ил)]-2-N-[1-O-α-D-карбонилметилманнопираноза]-L-лизина, Gd-комплекс
50,0 г (54,55 ммоля) соединения, указанного в заголовке примера 21д, 6,28 г (54,55 ммоля) N-гидроксисукцинимида, 4,62 г (109,0 моля) хлорида лития и 34,35 г (54,55 моля) Gd-комплекса 1,4,7-трис(карбоксилатометил)-10-(карбокси-3-аза-4-оксо-5-метилпент-5-ил)-1,4,7,10-тетраазациклододекана растворяют при умеренном нагревании в 400 мл диметилсульфоксида. Далее при 10°С добавляют 16,88 г (81,8 моля) N,N-дициклогексилкарбодиимида и затем перемешивают в течение ночи при комнатной температуре. Раствор сливают в 3000 мл ацетона и перемешивают в течение 10 мин. Выпавшее в осадок твердое вещество отфильтровывают и затем очищают хроматографией (RP-18, элюент:градиент воды/этанола/ацетонитрила).
Выход: 75,9 г (91,0% от теории) бесцветного твердого вещества.
Содержание воды: 8,6%.
Элементный анализ (в пересчете на безводное вещество):
Пример 110
а) [1-(4-перфтороктилсульфонил)пиперазин]амид 6-N-бензилоксикарбонил-2-N-[1-O-α-D-карбонилметил-(2,3,4,6-тетра-O-бензилглюкопираноза)]-L-лизина
К раствору 100,0 г (120,4 моля) соединения, указанного в заголовке примера 21в, 72,1 г (120,4 моля) 1-O-α-D-карбоксиметил-2,3,4,6-тетра-O-бензилглюкопиранозы и 13,86 г (120,4 моля) N-гидроксисукцинимида в 500 мл диметилформамида при 0°С добавляют 41,27 г (200,0 ммолей) N,N-дициклогексилкарбодиимида. Смесь перемешивают в течение 3 ч при 0°С, а затем в течение ночи при комнатной температуре. Выпавшую в осадок мочевину отфильтровывают, фильтрат досуха упаривают в вакууме и хроматографируют на силикагеле (элюент:дихлорметан/этанол в соотношении 20:1).
Выход: 136,1 г (87% от теории) вязкого масла.
Элементный анализ:
б) 1-[(4-перфтороктилсульфонил)пиперазин]амид 2-N-[1-O-α-D-карбонилметилглюкопираноза]-L-лизина
130,0 г (100,0 ммолей) соединения, указанного в заголовке примера 110а, растворяют в 2000 мл этанола и добавляют 10,0 г палладиевого катализатора (10%-ный Pd/C). После этого гидрируют в течение 12 ч при комнатной температуре. Затем катализатор отфильтровывают и фильтрат досуха упаривают в вакууме.
Выход: 91,7 г (количественный) бесцветного твердого вещества.
Элементный анализ:
в) [1-(4-перфтороктилсульфонил)пиперазин]амид 6-N-[1,4,7-трис(карбоксилатометил)]-1,4,7,10-тетраазациклододекан-10-N-(пентаноил-3-аза-4-оксо-5-метил-5-ил)]-2-N-[1-O-α-D-карбонилметилглюкопираноза]-L-лизина, Gd-комплекс
50,0 г (54,55 ммоля) соединения, указанного в заголовке примера 110б, 6,28 г (54,55 ммоля) N-гидроксисукцинимида, 4,62 г (109,0 моля) хлорида лития и 34,35 г (54,55 моля) Gd-комплекса 1,4,7-трис(карбоксилатометил)-10-(карбокси-3-аза-4-оксо-5-метилпент-5-ил)-1,4,7,10-тетраазациклододекана растворяют при умеренном нагревании в 400 мл диметилсульфоксида. Далее при 10°С добавляют 16,88 г (81,8 моля) N,N-дициклогексилкарбодиимида и затем перемешивают в течение ночи при комнатной температуре. Раствор сливают в 3000 мл ацетона и перемешивают в течение 10 мин. Выпавшее в осадок твердое вещество отфильтровывают и затем очищают хроматографией (RP-18, элюент:градиент воды/этанола/ацетонитрила).
Выход: 75,9 г (91,0% от теории) бесцветного твердого вещества.
Содержание воды: 8,6%.
Элементный анализ (в пересчете на безводное вещество):
Пример 111
а) [1-(4-перфтороктилсульфонил)пиперазин]амид 6-N-бензилоксикарбонил-2-N-[1-O-α-D-карбонилметил-(2,3,4,6-тетра-O-бензилгалактопираноза)]-L-лизина
К раствору 50,0 г (60,2 моля) соединения, указанного в заголовке примера 21в, 36,05 г (60,2 ммоля) 1-O-α-D-карбоксиметил-2,3,4,6-тетра-O-бензилгалактопиранозы и 6,93 г (60,2 ммоля) N-гидроксисукцинимида в 500 мл диметилформамида при 0°С добавляют 20,64 г (100,0 ммолей) N,N-дициклогексилкарбодиимида. Смесь перемешивают в течение 3 ч при 0°С, а затем в течение ночи при комнатной температуре. Выпавшую в осадок мочевину отфильтровывают, фильтрат досуха упаривают в вакууме и хроматографируют на силикагеле (элюент:дихлорметан/этанол в соотношении 20:1).
Выход: 68,1 г (87% от теории) вязкого масла.
Элементный анализ:
б) 1-[(4-перфтороктилсульфонил)пиперазин]амид 2-N-[1-O-α-D-карбонилметилгалактопираноза]-L-лизина
65,0 г (50,0 ммолей) соединения, указанного в заголовке примера 111а, растворяют в 1000 мл этанола и добавляют 5,0 г палладиевого катализатора (10%-ный Pd/C). После этого гидрируют в течение 12 ч при комнатной температуре. Затем катализатор отфильтровывают и фильтрат досуха упаривают в вакууме.
Выход: 45,85 г (количественный) бесцветного твердого вещества.
Элементный анализ:
в) [1-(4-перфтороктилсульфонил)пиперазин]амид 6-N-[1,4,7-трис(карбоксилатометил)]-1,4,7,10-тетраазациклододекан-10-N-(пентаноил-3-аза-4-оксо-5-метил-5-ил)]-2-N-[1-O-α-D-карбонилметилгалактопираноза]-L-лизина, Gd-комплекс
50,0 г (54,55 ммоля) соединения, указанного в заголовке примера 111б, 6,28 г (54,55 ммоля) N-гидроксисукцинимида, 4,62 г (109,0 моля) хлорида лития и 34,35 г (54,55 моля) Gd-комплекса 1,4,7-трис(карбоксилатометил)-10-(карбокси-3-аза-4-оксо-5-метилпент-5-ил)-1,4,7,10-тетраазациклододекана растворяют при умеренном нагревании в 400 мл диметилсульфоксида. Далее при 10°С добавляют 16,88 г (81,8 моля) N,N-дициклогексилкарбодиимида и затем перемешивают в течение ночи при комнатной температуре. Раствор сливают в 3000 мл ацетона и перемешивают в течение 10 мин. Выпавшее в осадок твердое вещество отфильтровывают и затем очищают хроматографией (RP-18, элюент:градиент воды/этанола/ацетонитрила).
Выход: 37,95 г (91,0% от теории) бесцветного твердого вещества.
Содержание воды: 8,6%.
Элементный анализ (в пересчете на безводное вещество):
Пример 112
а) Монобензиловый эфир N-трифторацетил-L-глутаминовой кислоты
100 г (421,5 ммоля) монобензилового эфира L-глутаминовой кислоты растворяют в смеси из 1000 мл этилового эфира трифторуксусной кислоты и 500 мл этанола и в течение 24 ч перемешивают при комнатной температуре. После этого упаривают досуха и остаток кристаллизуют из диизопропилового эфира.
Выход: 140,47 г (96% от теории) бесцветного кристаллического порошка.
Элементный анализ:
б) 5-N-(метил)-N-(2,3,4,5,6-пентагидроксигексил)амид монобензилового эфира 2-N-трифторацетил-L-глутаминовой кислоты
К раствору 24,9 г (24,08 ммоля) соединения, указанного в заголовке примера 112а, 2х г (24,08 ммоля) N-метилглюкамина и 2,77 г (24,08 ммоля) N-гидроксисукцинимида в 150 мл диметилформамида при 0°С добавляют 8,25 г (40 ммолей) N,N-дициклогексилкарбодиимида. Смесь перемешивают в течение 3 ч при 0°С, а затем в течение ночи при комнатной температуре. Выпавшую в осадок мочевину отфильтровывают, фильтрат досуха упаривают в вакууме и хроматографируют на силикагеле (элюент:дихлорметан/этанол в соотношении 20:1).
Выход: 109,40 г (89% от теории) вязкого масла.
Элементный анализ:
в) N-(метил)-N-(2,3,4,5,6-пентагидроксигексил)амид N-трифторацетил-L-глутаминовой кислоты
21,9хх г (52,15 ммоля) соединения, указанного в заголовке примера 112б, растворяют в 500 мл этанола и добавляют 3 г палладиевого катализатора (10%-ный Pd/C). После этого гидрируют при комнатной температуре. Затем катализатор отфильтровывают и фильтрат досуха упаривают в вакууме.
Выход: 43,0 г (количественный) бесцветного твердого вещества.
Элементный анализ:
г) Трифторацетил-L-глутаминовая кислота-5-N-(метил)-N-(2,3,4,5,6-пентагидроксигексил)амид-[1-(4-перфтороктилсульфонил)пиперазин]амидпиперазин]амид
К 10,96 г (33,2 ммоля) соединения, указанного в заголовке примера 112в, и 18,87 г (33,2 ммоля) 1-перфтороктилсульфонилпиперазина (полученного согласно DE 19603033) в 80 мл тетрагидрофурана при 0°С добавляют 16,42 г (66,4 ммоля) ЭЭДХ (этиловый эфир 2-этокси-1,2-дигидрохинолин-1-карбоновой кислоты) и перемешивают в течение ночи при комнатной температуре. После этого досуха упаривают в вакууме и хроматографируют на силикагеле (элюент:дихлорметан/метанол в соотношении 20:1).
Выход: 28,67 г (92% от теории) бесцветного твердого вещества.
Элементный анализ:
д) L-глутаминовая кислота-5-N-(метил)-N-(2,3,4,5,6-пентагидроксигексил)амид-[1-(4-перфтороктилсульфонил)пиперазин]амид
Раствор 28,36 г (30,22 ммоля) соединения, указанного в заголовке примера 112г, в 200 мл этанола при 0°С в течение часа барботируют газообразным аммиаком. Смесь затем перемешивают в течение 4 ч при 0°С. После этого упаривают досуха и остаток выделяют из воды путем перемешивания. Твердое вещество отфильтровывают и сушат в вакууме (50°С).
Выход: 24,19 г (95% от теории) аморфного твердого вещества.
Элементный анализ:
e) N-[1,4,7-трис(карбоксилатометил)-1,4,7,10-тетраазациклододекан-10-(пентаноил-3-аза-4-оксо-5-метил-5-ил)]-L-глутаминовая кислота-5-N-(метил)-N-(2,3,4,5,6-пентагидроксигексил)амид-5-[1-(4-перфтороктилсульфонил)пиперазин]амид, Gd-комплекс
20,43 г (24,25 ммоля) соединения, указанного в заголовке примера 112д, 2,79 г (24,25 ммоля) N-гидроксисукцинимида, 2,12 г (50 ммолей) хлорида лития и 15,27 г (24,25 ммоля) Gd-комплекса 1,4,7-трис(карбоксилатометил)-10-[(3-аза-4-оксо-5-метил-5-ил)пентановая кислота]-1,4,7,10-тетраазациклододекана растворяют при умеренном нагревании в 200 мл диметилсульфоксида. Далее при 10°С добавляют 8,25 г (40 ммолей) N,N-дициклогексилкарбодиимида и затем перемешивают в течение ночи при комнатной температуре. Раствор сливают в 3000 мл ацетона и перемешивают в течение 10 мин. Выпавшее в осадок твердое вещество отфильтровывают и затем очищают хроматографией (силикагель RP-18, элюент:градиент воды/этанола/ацетонитрила).
Выход: 28,45 г (79% от теории) бесцветного твердого вещества.
Содержание воды: 11,0%.
Элементный анализ (в пересчете на безводное вещество):
Пример 113
а) [1-(4-перфтороктилсульфонил)пиперазин]амид 6-N-бензилоксикарбонил-2-N-[1-O-α-D-карбонилметил(2,3,4-три-O-бензилглюкуроновая кислота-бензиловый эфир)]-L-лизина
К раствору 100,0 г (120,4 моля) соединения, указанного в заголовке примера 21в, 73,77 г (120,4 моля) бензилового эфира 1-O-α-D-карбоксиметил-2,3,4-три-О-бензилглюкуроновой кислоты и 13,86 г (120,4 моля) N-гидроксисукцинимида в 500 мл диметилформамида при 0°С добавляют 41,27 г (200,0 ммолей) N,N-дициклогексилкарбодиимида. Смесь перемешивают в течение 3 ч при 0°С, а затем в течение ночи при комнатной температуре. Выпавшую в осадок мочевину отфильтровывают, фильтрат досуха упаривают в вакууме и хроматографируют на силикагеле (элюент:дихлорметан/этанол в соотношении 20:1).
Выход: 147,58 г (86% от теории) вязкого масла.
Элементный анализ:
б) 1-[(4-перфтороктилсульфонил)пиперазин]амид 2-N-[1-O-α-D-карбонилметилглюкуроновая кислота]-L-лизина
142,52 г (100,0 ммолей) соединения, указанного в заголовке примера 113а, растворяют в 2000 мл этанола и добавляют 10,0 г палладиевого катализатора (10%-ный Pd/C). После этого гидрируют в течение 12 ч при комнатной температуре. Затем катализатор отфильтровывают и фильтрат досуха упаривают в вакууме.
Выход: 93,06 г (количественный) бесцветного твердого вещества.
Элементный анализ:
в) [1-(4-перфтороктилсульфонил)пиперазин]амид 6-N-[1,4,7-трис(карбоксилатометил)]-1,4,7,10-тетраазациклододекан-10-N-(пентаноил-3-аза-4-оксо-5-метил-5-ил)]-2-N-[1-O-α-D-карбонилметилглюкуроновая кислота]-L-лизина, Gd-комплекс, натриевая соль
50,76 г (54,55 ммоля) соединения, указанного в заголовке примера 113б, 6,28 г (54,55 ммоля) N-гидроксисукцинимида, 4,62 г (109,0 моля) хлорида лития и 34,35 г (54,55 моля) Gd-комплекса 1,4,7-трис(карбоксилатометил)-10-(карбокси-3-аза-4-оксо-5-метилпент-5-ил)-1,4,7,10-тетраазациклододекана растворяют при умеренном нагревании в 400 мл диметилсульфоксида. Далее при 10°С добавляют 16,88 г (81,8 моля) N,N-дициклогексилкарбодиимида и затем перемешивают в течение ночи при комнатной температуре. Раствор сливают в 3000 мл ацетона и перемешивают в течение 10 мин. Выпавшее в осадок твердое вещество отфильтровывают и затем очищают хроматографией (RP-18, элюент:градиент воды/этанола/ацетонитрила).
Выход; 75,14,9 г (88,0% от теории) бесцветного твердого вещества.
Содержание воды: 8,6%.
Элементный анализ (в пересчете на безводное вещество):
Пример 114
a) 6-N-бензилоксикарбонил-2-[2-(N-этил-N-перфтороктилсульфонил]амино]ацетил-L-лизин
К раствору 31,82 г (113,5 ммоля) 6-N-бензилоксикарбонил-L-лизина и 66,42 г (113,5 ммоля) 2-(N-этил-N-перфтороктилсульфонил)аминоуксусной кислоты (полученной согласно DE 19603033) в 300 мл тетрагидрофурана при 0°С добавляют 49,46 г (200,0 ммолей) ЭЭДХ (этиловый эфир 2-этокси-1,2-дигидрохинолин-1-карбоновой кислоты) и перемешивают в течение ночи при комнатной температуре. После этого досуха упаривают в вакууме и хроматографируют на силикагеле (элюент:дихлорметан/метанол в соотношении 20:1).
Выход: 55,79 г (58% от теории) бесцветного твердого вещества.
Элементный анализ:
б) N-метил-N-(2,3,4,5,6-пентагидроксигексил)амид 6-N-бензилоксикарбонил-2-N-[2-(N-этил-N-перфтороктилсульфонил)амино]ацетил-L-лизина
К раствору 51,02 г (60,2 моля) соединения, указанного в заголовке примера 114а, 11,75 г (60,2 моля) N-метилглюкамина и 6,93 г (60,2 моля) N-гидроксисукцинимида в 250 мл диметилформамида при 0°С добавляют 20,64 г (100,0 ммолей) N,N-дициклогексилкарбодиимида. Смесь перемешивают в течение 3 ч при 0°С, а затем в течение ночи при комнатной температуре. Выпавшую в осадок мочевину отфильтровывают, фильтрат досуха упаривают в вакууме и хроматографируют на силикагеле (элюент:дихлорметан/этанол в соотношении 20:1).
Выход: 53,05 г (86% от теории) вязкого масла.
Элементный анализ:
в) N-метил-N-(2,3,4,5,6-пентагидроксигексил)амид 2-N-[2-(N-этил-N-перфтороктилсульфонил)амино]ацетил-L-лизина
102,48 г (100,0 ммолей) соединения, указанного в заголовке примера 114б, растворяют в 2000 мл этанола и добавляют 10,0 г палладиевого катализатора (10%-ный Pd/C). После этого гидрируют в течение 12 ч при комнатной температуре. Затем катализатор отфильтровывают и фильтрат досуха упаривают в вакууме.
Выход: 89,06 г (количественный) бесцветного твердого вещества,
Элементный анализ:
г) N-метил-N-(2,3,4,5,6-пентагидроксигексил)амид 6-N-[1,4,7-трис(карбоксилатометил)]-1,4,7,10-тетраазациклододекан-10-N-(пентаноил-3-аза-4-оксо-5-метил-5-ил)]-2-N-[2-(N-этил-N-перфтороктилсульфонил)амино]ацетил-L-лизина, Gd-комплекс
48,58 г (54,55 ммоля) соединения, указанного в заголовке примера 114в, 6,28 г (54,55 ммоля) N-гидроксисукцинимида, 4,62 г (109,0 моля) хлорида лития и 34,35 г (54,55 моля) Gd-комплекса 1,4,7-трис(карбоксилатометил)-10-(карбокси-3-аза-4-оксо-5-метилпент-5-ил)-1,4,7,10-тетраазациклододекана растворяют при умеренном нагревании в 400 мл диметилсульфоксида. Далее при 10°С добавляют 16,88 г (81,8 моля) N,N-дициклогексилкарбодиимида и затем перемешивают в течение ночи при комнатной температуре. Раствор сливают в 3000 мл ацетона и перемешивают в течение 10 мин. Выпавшее в осадок твердое вещество отфильтровывают и затем очищают хроматографией (RP-18, элюент:градиент воды/этанола/ацетонитрила).
Выход: 73,27 г (89,4% от теории) бесцветного твердого вещества.
Содержание воды: 8,6%.
Элементный анализ (в пересчете на безводное вещество):
Пример 115: Визуализация атеросклеротических бляшек с помощью МРТ после внутривенного введения предлагаемых в изобретении металлических комплексов
У кроликов с генетически индуцированным артериосклерозом (кролики Ватанабе) через 5-60 мин, а также через 24 ч и 48 ч после внутривенного (в.в.) введения предлагаемых в изобретении соединений с концентрацией Gd 25 мкмолей/кг веса тела на T1-взвешенных изображениях, полученных на основе градиентного эха (TR 11,1 мс, ТЕ 4,3 мс, угол поворота спинов α 15°), наблюдалось существенное увеличение интенсивности сигнала, формируемого атеросклеротическими бляшками. В отличие от этого контрастное вещество не накапливалось вовсе или накапливалось лишь в незначительной степени в стенках здоровых сосудов, для которых поэтому на T1-взвешенных изображениях не было зафиксировано увеличение интенсивности сигнала или было зафиксировано лишь незначительное увеличение интенсивности сигнала. Благодаря ярко выраженному контрасту между формирующей интенсивный сигнал бляшкой и формирующей слабый сигнал стенкой здорового сосуда удалось диагностировать атеросклеротические изменения стенки сосуда.
На фиг.1 показаны полученные при МР-томографии изображения аорты до введения, а также через 24 ч и 48 ч после внутривенного введения кроликам Ватанабе (с генетически индуцированным артериосклерозом) металлического комплекса XV из расчета 25 мкмолей Gd на кг веса тела. На T1-взвешенных изображениях, полученных на основе градиентного эха (1,5 Тл; TR: 11,1 мс, ТЕ: 4,3 мс; NA: 2; матрица: 213×256; толщина слоя: 1,0 мм), видно существенное увеличение интенсивности сигнала, формируемого атеросклеротической бляшкой. Местонахождение бляшек, прежде всего в дуге аорты, а также в местах ответвления сосудов, подтверждали окрашиванием суданом-Ш.
Пример 116: Визуализация атеросклеротических бляшек с помощью МРТ после внутривенного введения кроликам Ватанабе металлическиого комплекса XV, а также корреляция с посмертным изображением, полученным при окрашивании суданом-III
На фиг.2 показаны полученные при МР-томографии изображения аорты до введения, а также через 35 мин, 60 мин и 24 ч после внутривенного введения кроликам Ватанабе (с генетически индуцированным аретериосклерозом) гадолиниевого металлического комплекса XV из расчета 10 мкмолей Gd на кг веса тела. На T1-взвешенных изображениях, полученных на основе градиентного эха (MPRange; 1,5 Тл; TR: 11,1 мс, ТЕ: 4,3 мс; NA: 2; матрица: 213×256; толщина слоя: 1,0 мм), видно существенное увеличение интенсивности сигнала, формируемого атеросклеротической бляшкой. Местонахождение бляшек, прежде всего в дуге аорты, а также в местах ответвления сосудов, подтверждали окрашиванием суданом-III. После этого вновь исследовали помещенный в агар препарат, получая его МР-изображения на основе T1-взвешенной последовательности градиентного эха (MPRange; 1,5 Тл; TR 11,1 мс, ТЕ 4,3 мс, угол поворота спинов α 15°; NA: 2; матрица: 213×256), а также последовательности спинового эха (1,5 Тл; TR: 400 мс, ТЕ: 15 мс; NA: 16; матрица: 256×256) (посмертные изображения). При этом была выявлена исключительно высокая корреляция между результатами, полученными для участков аорты, в которых после введения металлического комплекса наблюдалось значительное увеличение интенсивности формируемого ими сигнала, и результатами, полученными при окрашивании бляшек гистологическим красителем, что свидетельствует о накоплении предлагаемых в изобретении соединений в атеросклеротических бляшках.
Пример 117: Визуализация инфаркта (МРТ) после внутривенного введения крысам металлического комплекса XV
На фиг.3 показаны полученные при МР-томографии изображения сердца (in vivo и посмертные) через 24 ч после внутривенного введения крысам с индуцированным острым инфарктом миокарда металлического комплекса XV из расчета 100 мкмолей Gd на кг веса тела. На T1-взвешенных изображениях, полученных на основе спинового эха (1,5 Тл; TR: 400 мс, ТЕ: 6 мс; NA: 4; матрица: 128×128; толщина слоя: 2,5 мм), видно существенное увеличение интенсивности сигнала в затронутой инфарктом области. Успешную индукцию острого инфаркта миокарда подтверждали окрашиванием NBT.
Пример 118: Визуализация инфаркта (МРТ) после внутривенного введения крысам металлического комплекса I
На фиг.4 показаны полученные при МР-томографии изображения сердца (in vivo и посмертные) через 24 ч после внутривенного введения крысам с индуцированным острым инфарктом миокарда металлического комплекса I из расчета 100 мкмолей Gd на кг веса тела. На T1-взвешенных изображениях, полученных на основе спинового эха (1,5 Тл; TR: 400 мс, ТЕ: 6 мс; NA: 4; матрица: 128×128; толщина слоя: 2,5 мм), видно существенное увеличение интенсивности сигнала в затронутой инфарктом области. Успешную индукцию острого инфаркта миокарда подтверждали окрашиванием NBT.
Пример 119: Визуализация лимфатических узлов (МРТ) после внутривенного введения металлического комплекса XV кроликам с опухолевыми клетками VX2
На фиг.5 показаны полученные при МР-томографии изображения подвздошных лимфатических узлов, зарегистрированные до введения контрастного вещества, а также в различные моменты времени в течение 24-часового периода после внутривенного введения кроликам с внутримышечно (в.м.) имплантированными опухолевыми клетками VX2 металлического комплекса XV из расчета 200 мкмолей Gd на кг веса тела. На Т1-взвешенных изображениях, полученных на основе градиентного эха (1,5 Тл; последовательность: MPRange; TR 11,1 мс, ТЕ 4,3 мс, α 15°), видно значительное увеличение интенсивности сигнала, формируемого здоровыми тканями лимфатических узлов. Те области в пределах лимфатического узла, в которых интенсивность сигнала не увеличивалась, были диагностированы как метастазы, что подтверждали гистологически (окрашивание срезов лимфатических узлов с помощью Н/Е). Однако в более поздние моменты времени после введения контрастного вещества (через 24 ч) неожиданно было обнаружено обратное изменение интенсивности сигнала. Иными словами, в здоровой ткани лимфатических узлов интенсивность формируемого ими сигнала снизилась, тогда как в метастазе, наоборот, наблюдалось значительное увеличение интенсивности формируемого им сигнала.
Следует отметить тот неожиданный факт, что уже сразу же после введения контрастного вещества наблюдалось явное увеличение интенсивновти сигнала, формируемого первичной опухолью (прежде всего в ее периферии). В более поздние моменты времени (через 24 ч после введения) интенсивность сигнала постепенно увеличивалась и в направлении к центру опухоли.
Пример 120: Визуализация опухолей (МРТ) после внутривенного введения металлического комплекса I кроликам с опухолевыми клетками VX2
На фиг.6 показаны полученные при МР-томографии изображения подвздошного лимфатического узла и первичной опухоли (внутримышечно имплантированные опухолевые клетки VX2), зарегистрированные до введения контрастного вещества, а также через 60 мин и через 20 ч после внутривенного введения кроликам металлического комплекса I с из расчета 100 мкмолей Gd на кг веса тела. На Т1-взвешенных изображениях, полученных на основе градиентного эха (1,5 Тл; последовательность: MPRange; TR 11,1 мс, ТЕ 4,3 мс, α 15°), видно значительное увеличение интенсивности сигнала, формируемого здоровыми тканями лимфатического узла. В более ранний момент времени после введения (через 60 мин после введения) наблюдалось явное увеличение интенсивности сигнала, формируемого первичной опухолью (прежде всего в ее периферии). В более поздний момент времени (через 20 ч после введения) интенсивность сигнала постепенно увеличивалась и в направлении к центру опухоли.
Особо следует отметить увеличение интенсивности сигнала, формируемого патологической структурой (вероятно вторичной опухолью или некрозом) на контралатеральной стороне, что стало заметно лишь на полученном в более поздний момент времени изображении ("позднее увеличение интенсивности").
Металлический комплекс Х
Пример 121: Визуализация инфаркта (МРТ) после внутривенного введения контрастного вещества крысам
На фиг.7 показаны полученные при МР-томографии изображения сердца (in vivo и посмертные) через 6 ч после внутривенного введения крысам с острым индуцированным инфарктом миокарда полярного Gd-хелата с перфторированной боковой цепью (металлический комплекс X) из расчета 100 мкмолей Gd на кг веса тела. На T1-взвешенных, ЭКГ-иницированных изображениях, полученных на основе спинового эха (1,5 Тл; TR (эффективный): 400 мс, ТЕ: 12 мс; NA: 4; матрица: 128×128; толщина слоя: 2,5 мм), видно существенное увеличение интенсивности сигнала в затронутой инфарктом области. Успешную индукцию острого инфаркта миокарда подтверждали окрашиванием с помощью NBT.
Пример 122: Распределение контрастного вещества в органах (включая накопление в лимфатических узлах) после его внутривенного введения крысам
После внутривенного введения крысам полярного Gd-хелата с перфторированной боковой цепью (металлический комплекс X) из расчета 100 мкмолей общего гадолиния на кг веса тела через 24 ч после введения определяли содержание металла в различных органах, а также в лимфатических узлах (данные по которым сгруппированы в виде данных для брызжеечных и периферических лимфатических узлов) (в таблице 3 указаны средние значения, n=2).
Пример 123: Визуализация лимфатических узлов (МРТ) после внутривенного введения контрастного вещества крысам
На фиг.8 в качестве примера показаны полученные при МР-томографии изображения подколенных лимфатических узлов, зарегистрированные до введения контрастного вещества, а также в различные моменты времени в течение 60-минутного периода времени после внутривенного введения крысам металлического комплекса Х из расчета 100 мкмолей Gd на кг веса тела. На Т1 взвешенных изображениях, полученных на основе градиентного эха (1,5 Тл; последовательность: MPRange; TR 11,1 мс, ТЕ 4,3 мс, α 15°), видно значительное увеличение интенсивности сигнала в здоровой ткани лимфатического узла, наблюдаемое уже через короткий интервал времени после инъекции. Так, в частности, интенсивность сигнала через 15 мин после введения составила 263% от исходного уровня, а через 60 мин после введения составила 254% от исходного уровня.
Пример 124: Визуализация лимфатических узлов (МРТ) после внутривенного введения контрастного вещества кроликам с опухолевыми клетками VX2
На фиг.9 в качестве примера показаны полученные при МР-томографии изображения подвздошных лимфатических узлов, зарегистрированные до введения контрастного вещества, а также в различные моменты времени в течение 60-минутного периода времени после внутривенного введения кроликам с внутримышечно имплантированными опухолевыми клетками VX2 металлического комплекса Х из расчета 200 мкмолей Gd на кг веса тела. На Т1 взвешенных изображениях, полученных на основе градиентного эха (1,5 Тл; последовательность: MPRange; TR 11,1 мс, ТЕ 4,3 мс, α 15°), видно значительное увеличение интенсивности сигнала в здоровой ткани лимфатического узла. Интенсивность сигнала, формируемого здоровой тканью лимфатического узла, через 15 мин после введения составила 382% от исходного уровня, а через 60 мин после введения составила 419% от исходного уровня. Те области в пределах лимфатического узла, в которых интенсивность сигнала не увеличивалась, были диагностированы как метастазы, что подтверждали гистологически (окрашивание срезов лимфатических узлов с помощью Н/Е). Соотношение между интенсивностью сигнала, формируемого здоровой тканью лимфатического узла, и интенсивностью сигнала, формируемого метастазом, составило 3,0 через 15 мин после введения и 3,4 через 60 мин после введения.
Следует отметить тот неожиданный факт, что уже сразу же после введения контрастного вещества наблюдалось явное увеличение интенсивновти сигнала, формируемого не только лимфатическим узлом, но и первичной опухолью (прежде всего в ее периферии) (через 15 мин после введения интенсивность сигнала составила 277% от исходного уровня). В более поздние моменты времени (в течение 24 ч после введения) интенсивность сигнала постепенно увеличивалась и в направлении к центру опухоли (через 24 ч после введения интенсивность сигнала составила 217% от исходного уровня).
Металлический комплекс V
Пример 125: Визуализация инфаркта (МРТ) после внутривенного введения контрастного вещества крысам
На фиг.10 показаны полученные при МР-томографии изображения сердца (in vivo и посмертные) через 24 ч после внутривенного введения крысам с острым индуцированным инфарктом миокарда полярного Gd-хелата с перфторированной боковой цепью (металлический комплекс V) из расчета 100 мкмолей Gd на кг веса тела. На T1-взвешенных, ЭКГ-инициированных изображениях, полученных на основе спинового эха (1,5 Тл; TR (эффективный): 400 мс, ТЕ: 12 мс; NA: 4; матрица: 128×128; толщина слоя: 2,5 мм), видно существенное увеличение интенсивности сигнала в затронутой инфарктом области. Успешую индукцию острого инфаркта миокарда подтверждали окрашиванием с помощью NBT.
Пример 126: Распределение контрастного вещества в органах (включая накопление в лимфатических узлах) после его внутривенного введения крысам
После внутривенного введения крысам полярного Gd-хелата с перфторированной боковой цепью (металлический комплекс V) из расчета 200 мкмолей общего гадолиния на кг веса тела через 24 ч после введения определяли содержание металла в различных органах, а также в лимфатических узлах (данные по которым сгруппированы в виде данных для брызжеечных и периферических лимфатических узлов) (в таблице 4 указаны средние значения, n=2).
Пример 127: Визуализация лимфатических узлов (МРТ) после внутривенного введения контрастного вещества крысам
На фиг.11 в качестве примера показаны полученные при МР-томографии изображения подколенных лимфатических узлов, зарегистрированные до введения контрастного вещества, а также в различные моменты времени в течение 60-минутного периода времени после внутривенного введения крысам металлического комплекса V из расчета 200 мкмолей Gd на кг веса тела. На Т1 взвешенных изображениях, полученных на основе градиентного эха (1,5 Тл; последовательность: MPRange; TR 11,1 мс, ТЕ 4,3 мс, α 15°), видно значительное увеличение интенсивности сигнала в здоровой ткани лимфатического узла, наблюдаемое уже через короткий интервал времени после инъекции. Так, в частности, интенсивность сигнала через 15 мин после введения составила 147% от исходного уровня, а через 60 мин после введения составила 230% от исходного уровня.
Пример 128: Визуализация лимфатических узлов (МРТ) после внутривенного введения контрастного вещества кроликам с опухолевыми клетками VX2
На фиг.12 в качестве примера показаны полученные при МР-томографии изображения подвздошных лимфатических узлов, зарегистрированные до введения контрастного вещества, а также в различные моменты времени в течение 60-минутного периода времени после внутривенного введения кроликам с внутримышечно имплантированными опухолевыми клетками VX2 металлического комплекса V из расчета 200 мкмолей Gd на кг веса тела. На T1-взвешенных изображениях, полученных на основе градиентного эха (1,5 Тл; последовательность: MPRange; TR 11,1 мс, ТЕ 4,3 мс, α 15°), видно значительное увеличение интенсивности сигнала в здоровой ткани лимфатического узла. Интенсивность сигнала, формируемого здоровой тканью лимфатического узла, через 15 мин после введения составила 246% от исходного уровня, а через 60 мин после введения составила 282% от исходного уровня. Те области в пределах лимфатического узла, в которых интенсивность сигнала не увеличивалась, были диагностированы как метастазы, что подтверждали гистологически (окрашивание срезов лимфатического узла с помощью Н/Е). Соотношение между интенсивностью сигнала, формируемого здоровой тканью лимфатического узла, и интенсивностью сигнала, формируемого метастазом, составило 2,5 через 15 мин после введения и 1,7 через 60 мин после введения.
Следует отметить тот неожиданный факт, что уже сразу же после введения контрастного вещества наблюдалось явное увеличение интенсивности сигнала, формируемого не только лимфатическим узлом, но и первичной опухолью (прежде всего в ее периферии) (через 15 мин после введения интенсивность сигнала составила 350% от исходного уровня). В более поздние моменты времени (в течение 24 ч после введения) интенсивность сигнала постепенно увеличивалась и в направлении к центру опухоли (через 24 ч после введения интенсивность сигнала составила 106% от исходного уровня).
Металлический комплекс XIV
Пример 129: Визуализация инфаркта (МРТ) после внутривенного введения контрастного вещества крысам
На фиг.13 показаны полученные при МР-томографии изображения сердца (in vivo и посмертные) через 3 ч после внутривенного введения крысам с острым индуцированным инфарктом миокарда полярного Gd-хелата с перфторированной боковой цепью (металлический комплекс XIV) из расчета 100 мкмолей Gd на кг веса тела. На Т1-взвешенных, ЭКГ-инициированных изображениях, полученных на основе спинового эха (1,5 Тл; TR (эффективный): 400 мс, ТЕ: 12 мс; NA: 4; матрица: 128×128; толщина слоя: 2,5 мм), видно существенное увеличение интенсивности сигнала в затронутой инфарктом области. Успешую индукцию острого инфаркта миокарда подтверждали окрашиванием с помощью NBT.
Пример 130: Распределение контрастного вещества в органах (включая накопление в опухоли и лимфатических узлах) после его внутривенного введения крысам с раком предстательной железы
После внутривенного введения крысам (копулятивный инбридинг, за 12 дней до проведения опытов животным внутримышечно имплантировали клетки рака предстательной железы линии Dunning R3327 MAT-Lu) полярного Gd-хелата с перфторированной боковой цепью (металлический комплекс XIV) из расчета 200 мкмолей общего гадолиния на кг веса тела через 10 мин, 1 ч и 24 ч после введения (п.в.) определяли содержание металла в различных органах, в опухоли, а также в лимфатических узлах (данные по которым сгруппированы в виде данных для брызжеечных и периферических лимфатических узлов) (в таблице 5 указаны средние значения ± среднеквадратичное отклонение, n=3).
Пример 131: Визуализация лимфатических узлов (МРТ) после внутривенного введения контрастного вещества морским свинкам
На фиг.14 в качестве примера показаны полученные при МР-томографии изображения подвздошных и паховых лимфатических узлов, зарегистрированные до введения контрастного вещества, а также в различные моменты времени в течение 24-часового периода времени после внутривенного введения морским свинкам со стимулированными лимфатическими узлами (адъювантом Фрейнда) металлического комплекса XIV из расчета 200 мкмолей Gd на кг веса тела. На T1-взвешенных изображениях, полученных на основе градиентного эха (2,0 Тл; TR 10 мс, ТЕ 5 мс, α 40°), видно значительное увеличение интенсивности сигнала в здоровой ткани лимфатических узлов, наблюдаемое уже через короткий интервал времени после инъекции. Так, в частности, интенсивность сигнала через 60 мин после введения составила 127% от исходного уровня.
Пример 132: Визуализация лимфатических узлов (МРТ) после внутривенного введения контрастного вещества кроликам с опухолевыми клетками VX2
На фиг.15 в качестве примера показаны полученные при МР-томографии изображения подвздошных лимфатических узлов, зарегистрированные до введения контрастного вещества, а также в различные моменты времени в течение 23-часового периода времени после внутривенного введения кроликам с внутримышечно имплантированными опухолевыми клетками VX2 металлического комплекса XIV из расчета 200 мкмолей Gd на кг веса тела. На T1-взвешенных изображениях, полученных на основе градиентного эха (1,5 Тл; последовательность: MPRange; TR 11,1 мс, ТЕ 4,3 мс, α 15°), видно значительное увеличение интенсивности сигнала в здоровой ткани лимфатического узла. Интенсивность сигнала, формируемого здоровой тканью лимфатического узла, через 10 мин после введения составила 297% от исходного уровня, а через 60 мин после введения составила 269% от исходного уровня. Те области в пределах лимфатического узла, в которых интенсивность сигнала не увеличивалась, были диагностированы как метастазы, что подтверждали гистологически (окрашивание срезов лимфатического узла с помощью Н/Е). Соотношение между интенсивностью сигнала, формируемого здоровой тканью лимфатического узла, и интенсивностью сигнала, формируемого метастазом, составило 5,1 через 10 мин после введения и 1,9 через 60 мин после введения.
Следует отметить тот неожиданный факт, что уже сразу же после введения контрастного вещества наблюдалось явное увеличение интенсивности сигнала, формируемого не только лимфатическими узлами, но и первичной опухолью (прежде всего в ее периферии) (через 15 мин после введения интенсивность сигнала составила 594% от исходного уровня). В более поздние моменты времени (в течение 24 ч после введения) интенсивность сигнала постепенно увеличивалась и в направлении к центру опухоли (через 120 мин после введения интенсивность сигнала составила 162% от исходного уровня).
Металлический комплекс III
Пример 133: Визуализация инфаркта (МРТ) после внутривенного введения контрастного вещества крысам
На фиг.16 показаны полученные при МР-томографии изображения сердца (in vivo и посмертные) через 22 ч после внутривенного введения крысам с острым индуцированным инфарктом миокарда полярного Gd-хелата с перфторированной боковой цепью (металлический комплекс III) из расчета 100 мкмолей Gd на кг веса тела. На T1-взвешенных, ЭКГ-инициированных изображениях, полученных на основе спинового эха (1,5 Тл; TR (эффективный): 400 мс, ТЕ: 12 мс; NA: 4; матрица: 128×128; толщина слоя: 2,5 мм), видно существенное увеличение интенсивности сигнала в затронутой инфарктом области. Успешую индукцию острого инфаркта миокарда подтверждали окрашиванием с помощью NBT.
Пример 134: Распределение контрастного вещества в органах (включая накопление в лимфатических узлах) после его внутривенного введения крысам
После внутривенного введения крысам полярного Gd-хелата с перфторированной боковой цепью (металлический комплекс III) из расчета 200 мкмолей общего гадолиния на кг веса тела через 24 ч после введения определяли содержание металла в различных органах, а также в лимфатических узлах (данные по которым сгруппированы в виде данных для брызжеечных и периферических лимфатических узлов) (в таблице указаны средние значения, n=2).
Пример 135: Визуализация лимфатических узлов (МРТ) после внутривенного введения контрастного вещества крысам
На фиг.17 в качестве примера показаны полученные при МР-томографии изображения подвздошных лимфатических узлов, зарегистрированные до введения контрастного вещества, а также в различные моменты времени в течение 60-минутного периода времени после внутривенного введения крысам металлического комплекса III из расчета 200 мкмолей Gd на кг веса тела. На T1-взвешенных изображениях, полученных на основе градиентного эха (1,5 Тл; последовательность: MPRange; TR 11,1 мс, ТЕ 4,3 мс, α 15°), видно значительное увеличение интенсивности сигнала в здоровой ткани лимфатического узла, наблюдаемое уже через короткий интервал времени после инъекции. Так, в частности, интенсивность сигнала через 15 мин после введения составила 320% от исходного уровня, а через 60 мин после введения составила 401% от исходного уровня.
Пример 136: Визуализация лимфатических узлов (МРТ) после внутривенного введения контрастного вещества кроликам с опухолевыми клетками VX2
На фиг.18 в качестве примера показаны полученные при МР-томографии изображения подвздошных лимфатических узлов, зарегистрированные до введения контрастного вещества, а также в различные моменты времени в течение 60-минутного периода времени после внутривенного введения кроликам с внутримышечно имплантированными опухолевыми клетками VX2 металлического комплекса III из расчета 200 мкмолей Gd на кг веса тела. На T1-взвешенных изображениях, полученных на основе градиентного эха (1,5 Тл; последовательность: MPRange; TR 11,1 мс, ТЕ 4,3 мс, α 15°), видно значительное увеличение интенсивности сигнала в здоровой ткани лимфатического узла. Интенсивность сигнала, формируемого здоровой тканью лимфатического узла, через 15 мин после введения составила 195% от исходного уровня, а через 60 мин после введения составила 233% от исходного уровня. Те области в пределах лимфатического узла, в которых интенсивность сигнала не увеличивалась, были диагностированы как метастазы, что подтверждали гистологически (окрашивание срезов лимфатического узла с помощью Н/Е). Соотношение между интенсивностью сигнала, формируемого здоровой тканью лимфатического узла, и интенсивностью сигнала, формируемого метастазом, составило 1,9 через 15 мин после введения и 1,8 через 60 мин после введения.
Следует отметить тот неожиданный факт, что уже сразу же после введения контрастного вещества наблюдалось явное увеличение интенсивности сигнала, формируемого не только лимфатическим узлом, но и первичной опухолью (прежде всего в ее периферии) (через 15 мин после введения интенсивность сигнала составила 232% от исходного уровня).
| название | год | авторы | номер документа |
|---|---|---|---|
| ПЕРФТОРАЛКИЛСОДЕРЖАЩИЕ КОМПЛЕКСЫ С ОСТАТКАМИ САХАРОВ, СПОСОБЫ ИХ ПОЛУЧЕНИЯ | 2001 |
|
RU2280644C2 |
| ПЕРФТОРАЛКИЛСОДЕРЖАЩИЕ КОПЛЕКСЫ С ПОЛЯРНЫМИ ОСТАТКАМИ, СПОСОБ ИХ ПОЛУЧЕНИЯ, ФАРМАЦЕВТИЧЕСКОЕ СРЕДСТВО | 2001 |
|
RU2289579C2 |
| ПЕРФТОРАЛКИЛСОДЕРЖАЩИЕ СОЕДИНЕНИЯ, СПОСОБ ИХ ПОЛУЧЕНИЯ (ВАРИАНТЫ) И ФАРМАЦЕВТИЧЕСКОЕ СРЕДСТВО, СОДЕРЖАЩЕЕ УКАЗАННЫЕ СОЕДИНЕНИЯ | 1997 |
|
RU2242477C2 |
| ХЕЛАТЫ МЕТАЛЛОВ, ИМЕЮЩИЕ ПЕРФТОРИРОВАННЫЙ ПЭГ РАДИКАЛ, СПОСОБЫ ИХ ПОЛУЧЕНИЯ И ИХ ПРИМЕНЕНИЕ | 2007 |
|
RU2470014C2 |
| СОЕДИНЕНИЕ, СПОСОБ ЕГО ПОЛУЧЕНИЯ | 2002 |
|
RU2305096C2 |
| ПРИМЕНЕНИЕ ПЕРФТОРАЛКИЛСОДЕРЖАЩИХ КОМПЛЕКСОВ МЕТАЛЛОВ В КАЧЕСТВЕ КОНТРАСТНЫХ ВЕЩЕСТВ ПРИ МАГНИТНО-РЕЗОНАНСНОЙ ТОМОГРАФИИ ДЛЯ ВИЗУАЛИЗАЦИИ ВНУТРИСОСУДИСТЫХ ТРОМБОВ | 2003 |
|
RU2328310C2 |
| НОВЫЕ ХЕЛАТНЫЕ СОЕДИНЕНИЯ ГАДОЛИНИЯ С ВЫСОКОЙ РЕЛАКСИВНОСТЬЮ ДЛЯ ПРИМЕНЕНИЯ В МАГНИТНО-РЕЗОНАНСНОЙ ВИЗУАЛИЗАЦИИ | 2017 |
|
RU2755181C2 |
| КОМПЛЕКСЫ КАСКАДНЫХ ПОЛИМЕРОВ, СОДЕРЖАЩЕЕ ИХ ДИАГНОСТИЧЕСКОЕ КОНТРАСТНОЕ СРЕДСТВО И ПРОМЕЖУТОЧНЫЕ СОЕДИНЕНИЯ | 1996 |
|
RU2197495C2 |
| КАСКАДНЫЕ ПОЛИМЕРНЫЕ КОМПЛЕКСЫ, ИСХОДНЫЕ СОЕДИНЕНИЯ И ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ | 1996 |
|
RU2166501C2 |
| N-МЕТИЛГОМОЦИСТЕИНЫ, ИХ ПРИМЕНЕНИЕ И СПОСОБ ИХ ПОЛУЧЕНИЯ | 2002 |
|
RU2305678C2 |



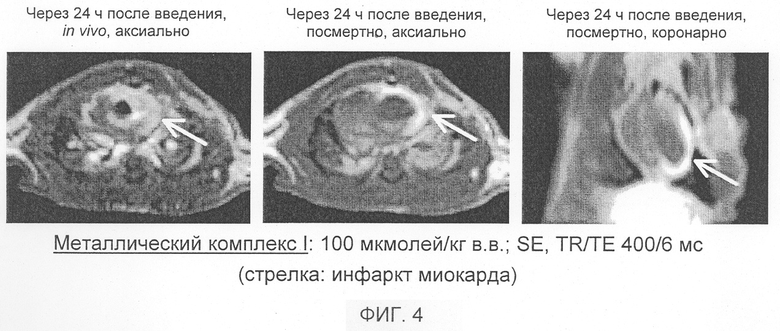
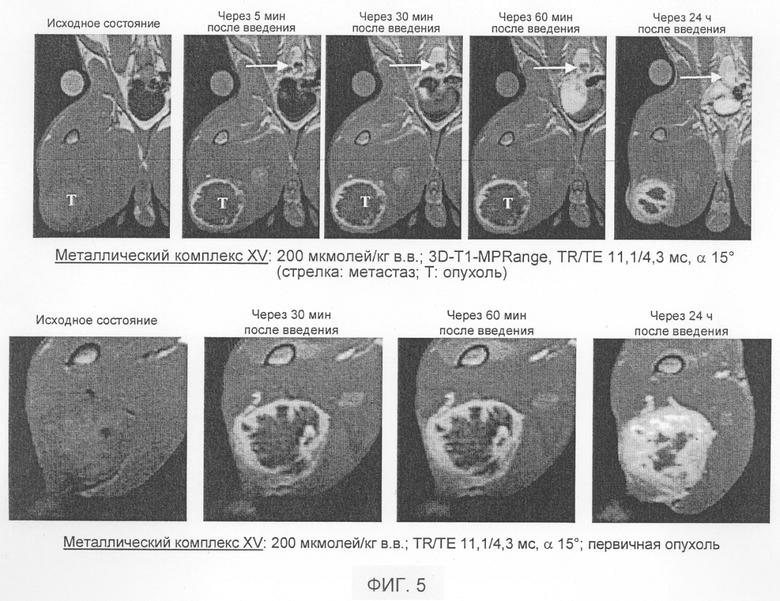



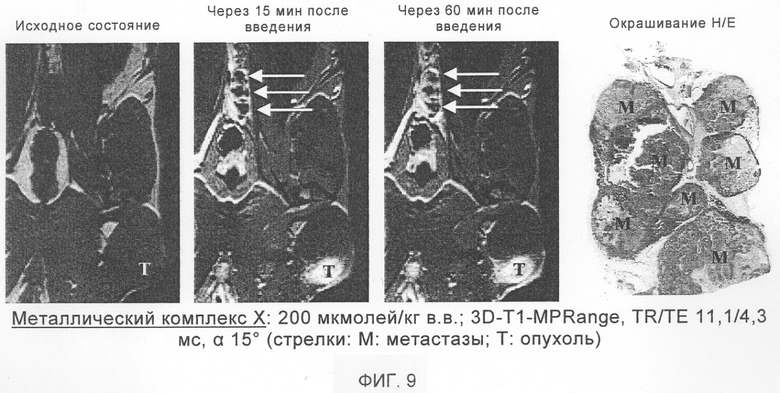
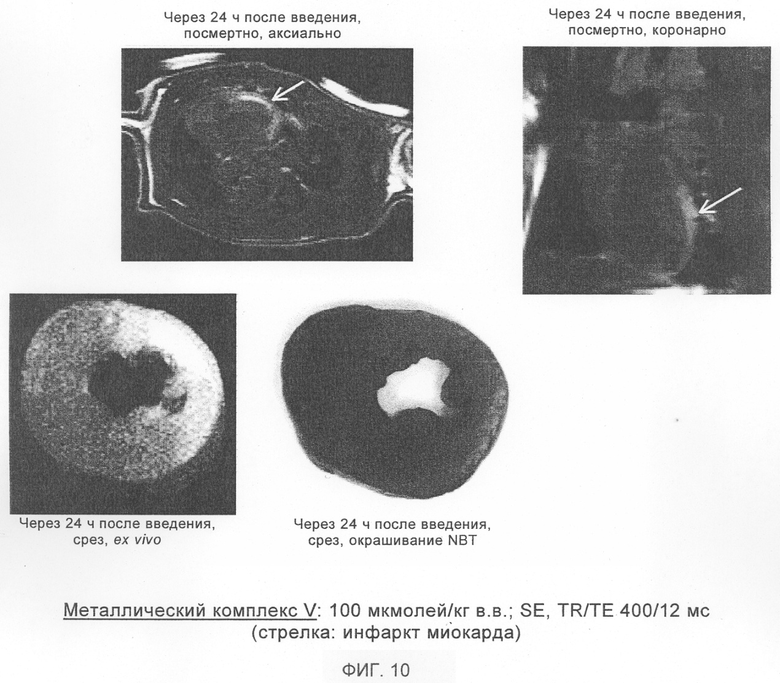






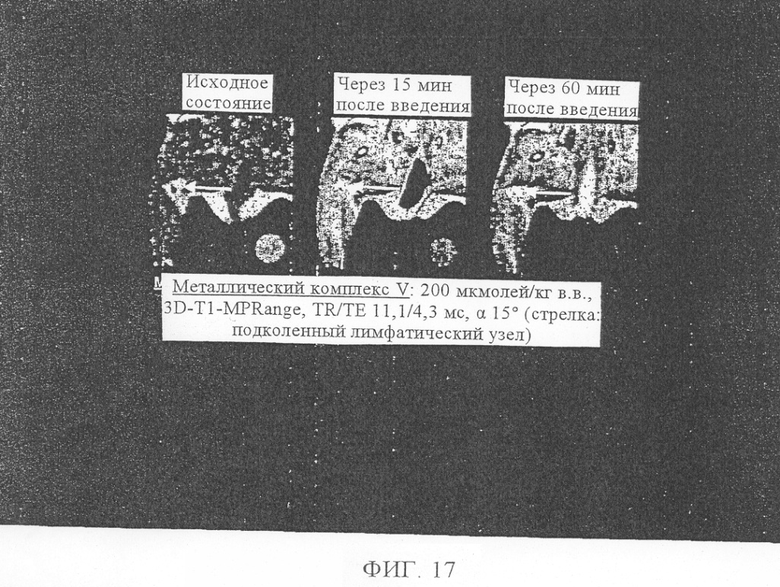

Изобретение относится к области медицины и касается применения гадолиниевого комплекса [1-(4-перфтороктилсульфонил)пиперазин]амид 6-N-[1,4,7-трис(карбоксилатометил)-1,4,7,10-тетраазациклододекан-10-N-(пентаноил-3-аза-4-оксо-5-метил-5-ил)]-2-N-[1-0-α-D-карбонилметилманнопираноза]-L-лизина в качестве контрастного вещества при магнитно-резонансной томографии (МРТ) для визуализации бляшек. Изобретение повышает точность диагностики. 4 табл., 18 ил.
Применение гадолиниевого комплекса [1-(4-перфтороктилсульфонил)пиперазин]амид 6-N-[1,4,7-трис(карбоксилатометил)-1,4,7,10-тетраазациклододекан-10-N-(пентаноил-3-аза-4-оксо-5-метил-5-ил)]-2-N-[1-O-α-D-карбонилметилманнопираноза]-L-лизина в качестве контрастного вещества при магнитно-резонансной томографии (МРТ) для визуализации бляшек.
| Бесколесный шариковый ход для железнодорожных вагонов | 1917 |
|
SU97A1 |
| Прибор для очистки паром от сажи дымогарных трубок в паровозных котлах | 1913 |
|
SU95A1 |
| Прибор, замыкающий сигнальную цепь при повышении температуры | 1918 |
|
SU99A1 |
| RU 96122883, A, 20.01.1999. | |||

