ОБЛАСТЬ ТЕХНИКИ
Настоящее изобретение касается новой пероральной галеновой формы молсидомина с пролонгированным высвобождением действующего вещества, предназначенной для лечения грудной жабы во всех ее разновидностях (стенокардия напряжения или стенокардия покоя, нестабильная стенокардия).
ПРЕДШЕСТВУЮЩИЙ УРОВЕНЬ ТЕХНИКИ
Известно, что молсидомин (N-(этоксикарбонил)-3(4-морфолинил)сиднонимин) - это первый представитель нового семейства антиангинных препаратов - сиднониминов, который был описан в специальном патенте на лекарственное средство №6734.
Это соединение применимо, в частности, в превентивном лечении приступа стенокардии во всех проявлениях, поскольку оно вызывает расслабление гладких мышечных волокон кровеносных сосудов и подавление ранних стадий образования атеросклеротических бляшек.
Действие этого соединения основано на его способности в ходе своей биологической трансформации напрямую высвобождать радикал NO.
Точнее говоря, молсидомин является пролекарством - предшественником действующего вещества.
После перорального введения молсидомин полностью всасывается и подвергается в печени ферментативному изменению (гидролизу и декарбоксилированию). При этом образуется SIN-1, который, в свою очередь, преобразуется в крови без участия ферментов в SIN-1A. SIN-1 и SIN-1A являются активными метаболитами молсидомина.
Затем SIN-1A посредством окисления распадается до неактивного SIN-1С с высвобождением NO.
Вслед за тем SIN-1С, в свою очередь, подвергается обмену в печени, как описано в работе Bernd Rosenkranz and al., Clinical Pharmacokinetics of Molsidomine, Clin. Pharmacokinet. 1996, May; 30 (5) 372-384.
В настоящее время молсидомин является коммерческим препаратом и в основном представлен в виде таблеток в дозе 2 мг и 4 мг, обыкновенно назначаемых трижды в день при лечении стенокардии напряжения и четырежды в день при лечении стенокардии покоя и стенокардии напряжения в тяжелой форме.
Позже была предложена новая галенова форма молсидомина с пролонгированным высвобождением дозой 8 мг, предназначенная для введения два раза в день для профилактического и длительного лечения грудной жабы - продаваемый в Бельгии препарат под названием CORVATARD™ (см. Compendium 2003, 21éme édition, Association Générale de I'lndustrie du Médicament).
В этой лекарственной форме максимальная концентрация молсидомина в плазме крови наблюдается между первым и третьим часами после введения.
Обычно молсидомин действует в течение 4-5 часов, будучи назначен в дозе 4 мг, и в течение 10-12 часов, будучи назначен в дозе 8 мг.
В общем случае, с точки зрения удобства для пациента, более предпочтительны лекарственные формы с более длительным терапевтическим действием, что позволяет сократить количество приемов лекарства в день, а сокращение числа приемов более удобно для пациента.
В то же время в фармацевтике известно, что продление терапевтического действия вызывает значимое уменьшение максимальной концентрации в плазме и более позднее поступление в терапевтическую зону.
СУЩНОСТЬ ИЗОБРЕТЕНИЯ
Было обнаружено, и это составляет основу настоящего изобретения, что в отношении молсидомина и его активных метаболитов продление терапевтического действия может быть достигнуто без значимого уменьшения максимальной концентрации в плазме крови, при поступлении в терапевтическую зону, сравнимом с их поступлением из лекарственных форм дозой 4 мг и 8 мг.
Таким образом, форма галенова препарата (галенова форма) в соответствии с настоящим изобретением, обладая пролонгированным действием, высвобождает достаточное и выверенное количество активного вещества в кислой среде, главным образом в желудке, что обеспечивает быстрое поступление (около 30 мин натощак и 1 ч 30 мин при назначении после еды) в терапевтическую зону (5-10 нг/мл) и пиковое содержание в плазме крови (33-40 нг/мл), которое соответствует максимальному содержанию препарата в плазме крови, измеренному для форм галеновых препаратов немедленного высвобождения.
Вклад обмена в желудке или, иными словами, в кислой среде удалось показать благодаря измерению соотношений in vivo - in vitro. Именно в среде 0,1 н. HCl наблюдается наибольшее соотношение между долей высвобождения in vitro и долей всасывания in vivo, причем для всех форм молсидомина.
Таким образом, прежде всего, объектом настоящего изобретения является твердая пероральная форма галенова препарата с пролонгированным высвобождением молсидомина, отличающаяся тем, что она содержит терапевтически эффективное количество молсидомина или одного из его активных метаболитов и тем, что она имеет следующую скорость растворения активного вещества in vitro (измеренную согласно методике, описанной в Pharmacopée Européenne, 3 изд. (или USP XXIV) при 50 об/мин в 500 мл 0,1 н. HCl при 37°С):
15-25% молсидомина, высвобожденного через 1 час,
20-35% молсидомина, высвобожденного через 2 часа,
50-65% молсидомина, высвобожденного через 6 часов,
75-95% молсидомина, высвобожденного через 12 часов,
>85% молсидомина, высвобожденного через 18 часов,
>90% молсидомина, высвобожденного через 24 часа,
причем пиковая концентрация молсидомина в плазме крови достигается in vivo через 2,5-5 часов, предпочтительно через 3-4 часа после введения указанной формы, и имеет значение от 25 до 40 нг/мл плазмы.
В настоящем описании «пиковая концентрация молсидомина в плазме крови... in vitro» соответствует средней максимальной концентрации молсидомина, обнаруживаемой в плазме по меньшей мере 10-ти здоровых добровольцев.
Выражение «терапевтически эффективное количество», применяемое в рамках настоящего изобретения, означает количество молсидомина, достаточное для обеспечения его концентрации в плазме крови, равной по меньшей мере 5 нг/мл, предпочтительно - не менее 10 нг/мл, в течение срока около 24 часов.
Как правило, форма галенова препарата согласно настоящему изобретению может содержать 14-34 мг, предпочтительно 16-20 мг молсидомина на единицу дозы, причем в настоящее время предпочтение отдается форме, содержащей 16 мг молсидомина.
Выражение «активные метаболиты» молсидомина имеет целью объединить, в частности, соединения SIN-1 и SIN-1A, образующиеся в результате биологического превращения, которому подвергается молсидомин после его введения.
Новая форма галенова препарата согласно настоящему изобретению имеет много преимуществ по сравнению с существующими в настоящее время коммерческими галеновыми формами молсидомина.
Прежде всего, она очень удобна для пациента, поскольку для достижения желаемого терапевтического действия достаточно одного приема препарата в день. Соответствие запросам пациента при этом возрастает.
Кроме того, поддержание повышенной максимальной концентрации в плазме крови обеспечивает оптимальную эффективность в течение первых часов после введения при очень быстром (30 мин натощак и 1 час 30 мин после еды) поступлении в терапевтическую зону (5-10 нг/мл).
По этой причине предложенная новая галенова форма позволяет избежать
- с одной стороны, каких-либо промежутков времени, в течение которых пациент был бы не защищен (характеризуемых концентрацией ниже 5-10 нг/мл) и,
- с другой стороны, побочных эффектов, вызванных наведением нескольких пиковых концентраций в плазме крови в день, связанных с несколькими ежедневными приемами препарата.
Кроме того, предварительные клинические испытания совершенно неожиданно показали, что пациенты, состояние которых стабилизировалось в результате лечения молсидомином (таблетки пролонгированного действия дозой 8 мг), назначавшимся 2 раза в день в сочетании с подъязычным приемом органических нитропроизводных в момент приступа, после последующего лечения галеновой формой согласно настоящему изобретению (в дозировке, например, 16 мг) испытывали значительное уменьшение числа приступов грудной жабы, в результате чего уменьшалось потребление органических нитропроизводных.
Новая форма галенова препарата в соответствии с изобретением может иметь форму, например, таблеток, порошка или сфероидов (шариков), причем форма таблеток является предпочтительной.
Для достижения поставленной задачи молсидомин встраивается в систему высвобождения, позволяющую достичь тех скоростей растворения действующего вещества in vitro, что определены выше.
Такая система высвобождения действующего вещества может, в частности, состоять в использовании носителя («матрицы») с пролонгированным высвобождением или же в применении молсидомина в традиционной форме, покрытой защитной оболочкой, обеспечивающей его пролонгированное высвобождение.
В соответствии с одним из примеров осуществления настоящего изобретения, такая система высвобождения действующего вещества состоит из активного носителя, содержащего смешанные с молсидомином либо с одним из его активных метаболитов полимерный материал, сильно разбухающий при соприкосновении с водой или водными растворами, или гелеобразующий полимерный материал, причем вышеупомянутые полимерные материалы могут представлять собой один и тот же полимерный материал, одновременно обладающий свойствами набухания и гелеобразования; при этом вышеупомянутый носитель может дополнительно содержать разнообразные обычные вспомогательные вещества (адъюванты), в частности имеющие целью придать ему подходящие для таблетирования характеристики.
Такими вспомогательными веществами могут быть, в частности, растворимые вещества, такие как лактоза, смазывающие вещества, такие как стеарат магния, гранулирующие агенты, такие как поливинилпирролидон, агенты, улучшающие текучесть, такие как, например, коллоидная окись кремния и красящие вещества, такие как окись железа.
Эти вспомогательные вещества могут быть включены в указанный носитель в количестве 25-60 мас.% по отношению к общему весу носителя.
Полимерными материалами, которые имеют повышенное свойство набухания и которые могут быть использованы в рамках настоящего изобретения, являются, например, карбоксиметилцеллюлоза сетчатой структуры в натриевой форме, гидроксипропилцеллюлоза сетчатой структуры, гидроксиметилпропилцеллюлоза большого молекулярного веса, полиметилметакрилат, поливинилпирролидон сетчатой структуры или высокомолекулярный поливиниловый спирт.
Подходящими для использования в рамках настоящего изобретения гелеобразующими полимерами являются, например, метилцеллюлоза, карбоксиметилцеллюлоза, низкомолекулярная гидроксипропилметилцеллюлоза, низкомолекулярный поливиниловый спирт, полиоксиэтиленгликоль или поливинилпирролидон несетчатой структуры.
В рамках настоящего изобретения предпочтительно используется один полимерный материал, обладающий одновременно свойствами набухания и гелеобразования. Таким веществом является высокомолекулярная гидроксипропилметилцеллюлоза, такая как препарат, известный под торговым названием METHOCEL® К100М, причем это соединение дополнительно сообщает конечной смеси превосходную вязкость.
В общем случае полимерный материал, имеющий повышенную способность к набуханию, и гелеобразующий полимерный материал составляют в сумме от 40% до 60%, предпочтительно 49,0 масс.% от общей массы указанного носителя.
Массовое соотношение между полимерным материалом, обладающим повышенной способностью к набуханию, и гелеобразующим полимерным материалом может изменяться в широких пределах.
В некоторых случаях для достижения желаемых скоростей растворения in vitro может оказаться необходимым включить в носитель липофильное вещество, предназначенное для регулирования скорости высвобождения молсидомина.
Таким липофильным веществом может быть гидрофобное липидное соединение, такое как гидрогенизированное касторовое масло (Cutina®), стеариловый, цетостеариловый, цетиловый спирты, моно-, ди-, триглицериды, такие как глицерилпальмитостеарат, глицерилмоноолеат, твердый парафин.
В рамках настоящего изобретения предпочтительно будет применяться глицерилбегенат, в частности препарат, известный под торговым названием COMPRITOL® 888 АТО, поскольку это соединение позволяет превосходно регулировать проницаемость матрицы носителя.
Упомянутое липофильное вещество может присутствовать в составе матрицы носителя в количестве порядка 12-20 мас.% по отношению к общей массе матрицы носителя.
Система высвобождения молсидомина, используемая в рамках настоящего изобретения, может быть приготовлена традиционными способами, известными профессионалам, включающими стадии смешивания, просеивания, гранулирования, высушивания и таблетирования.
Для достижения желаемого профиля высвобождения может оказаться целесообразным придать матрице носителя геометрическую форму, обеспечивающую продление высвобождения действующего вещества вплоть до 24 часов.
Так, система высвобождения действующего вещества, применимая в рамках настоящего изобретения, может представлять собой многослойную матрицу носителя, состоящую из по меньшей мере одного «активного» слоя, содержащего молсидомин, в сочетании с по меньшей мере одним «неактивным» слоем, предпочтительно состоящим главным образом из тех же веществ, что и активный слой, но не содержащим молсидомина.
В соответствии с предпочтительным вариантом осуществления изобретения, форма галенова препарата согласно настоящему изобретению изготавливается в виде таблетки, в которой активный слой находится между двумя неактивными слоями.
Более подробно настоящее изобретение поясняют следующие примеры, приведенные только в разъяснительных целях.
СВЕДЕНИЯ, ПОДТВЕРЖДАЮЩИЕ ВОЗМОЖНОСТЬ
ОСУЩЕСТВЛЕНИЯ ИЗОБРЕТЕНИЯ
ПРИМЕР 1
Приготовление формы галенова препарата согласно настоящему изобретению в виде многослойных таблеток дозировкой 16 мг
Форма галенова препарата согласно настоящему изобретению выполнена в виде таблетки, содержащей активный слой, расположенный в промежутке между двумя неактивными слоями, имеющей следующие размеры:
- диаметр таблетки: 8,0 мм
- толщина промежуточного активного слоя: около 2,1 мм
- толщина каждого неактивного слоя: около 1,55 и 1,95 мм
Каждый из этих слоев был приготовлен из веществ, главным образом идентичных, взятых в количествах, приведенных в Таблице I, в расчете на одну таблетку.
Активный слой готовили следующим образом.
Смешивали до однородности молсидомин, полимерный материал (METHOCEL® K100 М), липофильное вещество (COMPRITOL® 888 АТО), гидрофильный наполнитель (MANNITOL® 60) и гранулирующий агент (PLASDONE® K29-32) в подходящем смесителе.
Кроме того, готовили 95%-ный раствор этанола, применяемый для смачивания вышеописанной пылевидной смеси.
Полученную таким образом однородную массу подвергали гранулированию и высушиванию над сжиженным воздухом для получения, после калибровки, гранулята.
Полученный таким образом гомогенный гранулят смешивали с противоприлипателем (веществом, улучшающим текучесть - AEROSIL® 200) и смазывающим веществом (магния стеарат), затем таблетировали.
Неактивные слои получали по той же прописи, что приведена выше для активного слоя, при этом силу сдавливания на всех стадиях таблетирования выбирали так, чтобы получить совершенно однородную таблетку (давление около 1000 кг/см2).
ПРИМЕР 2
Определение профиля растворения in vitro галеновой формы согласно настоящему изобретению
Доли растворения in vitro галеновой формы согласно настоящему изобретению, в частности галеновой формы, приготовленной в Примере 1, измеряли с помощью методики, описанной в Pharmacopée Européenne, 3 изд. (или USP XXIV).
Измерения осуществляли в следующих экспериментальных условиях:
Прибор Sotax AT7 с колонками
Скорость вращения: 50 об/мин
Температура среды: 37°С
Фильтрация: фильтр Wathman GF-D
Дозирование: УФ-спектрофотометрия при длине волны около 286 или 311 нм
Спектроскопия: Hitachi U-3000 с кварцевой кюветой толщиной 1 см
Среда для растворения: 500 мл 0,1 н. HCl (кислый рН).
При этом были получены следующие результаты:
18% высвобожденного молсидомина через 1 час
27% высвобожденного молсидомина через 2 часа
57% высвобожденного молсидомина через 6 часов
88% высвобожденного молсидомина через 12 часов
96% высвобожденного молсидомина через 18 часов
100% высвобожденного молсидомина через 24 часа
ПРИМЕР 3
Сравнительное изучение основных фармакокинетических характеристик лекарственных форм на основе молсидомина
Для доказательства преимуществ галеновой формы согласно настоящему изобретению по сравнению с ранее известными галеновыми формами молсидомина были измерены основные фармакокинетические характеристики трех следующих лекарственных форм.
Лекарственная форма на основе молсидомина дозировкой 4 мг, соответствующая бельгийскому коммерческому препарату CORVATON® 4 mg.
Лекарственная форма на основе молсидомина дозировкой 8 мг, соответствующая бельгийскому коммерческому препарату CORVATARD®.
Лекарственная форма в соответствии с настоящим изобретением дозировкой 16 мг (полученная в соответствии с Примером 1).
При использовании экспериментальных процедур, хорошо знакомых специалистам, для каждой из этих лекарственных форм были измерены следующие параметры:
Cmax: максимальная концентрация в плазме крови.
Tmax: время, в течение которого наблюдалась величина Cmax.
AUC 0-t: площадь под кривой между значениями времени 0 и t.
T1/2: период полужизни препарата.
MRT: среднее время присутствия вещества в организме.
В случае лекарственной формы согласно настоящему изобретению эти фармакокинетические характеристики определялись у здоровых молодых добровольцев натощак, а затем после еды.
Полученные таким образом результаты сведены в Таблицу II
Кроме того, на Фиг.1 представлены графики изменения концентрации каждого из испытуемых препаратов в плазме крови в зависимости от времени.
Полученные результаты показывают, что лекарственная форма дозировкой 4 мг обеспечивает присутствие активного вещества в плазме крови в продолжении около 4-5 часов, лекарственная форма дозировкой 8 мг - в продолжении около 10-12 часов, а лекарственная форма согласно настоящему изобретению дозировкой 16 мг - в продолжении около 24 часов.
Можно утверждать, что максимальная концентрация в плазме крови относительно эквивалентна для всех трех лекарственных форм и составляет от 33 до 40 нг/мл.
Результат, полученный для лекарственной формы согласно настоящему изобретению, оказался совершенно неожиданным, поскольку продление терапевтического действия не приводит здесь к существенному уменьшению Cmax. Не существует статистически значимого различия по этому признаку между галеновой формой согласно настоящему изобретению и традиционными формами (дисперсионный анализ с последующим исследованием post-hoc по Бонферонни).
Кроме того, полученные результаты показывают, что лекарственное средство в соответствии с настоящим изобретением обеспечивает эффективность, сопоставимую с эффективностью известных лекарственных средств, даже в первые часы после приема, при быстром поступлении в терапевтическую зону (в течение около 30 минут натощак и 90 минут после приема пищи).
ПРИМЕР 4
Сравнительное изучение корреляций между кинетикой высвобождения in vitro в различной среде и кинетикой усвоения in vivo
при рН 5,0 в продолжении 30 минут
при рН 6,3 в продолжении 3 часов, при рН 7,0 в продолжении оставшегося времени
Все коэффициенты корреляции значимы (р<0,01; тест Пирсона). Наилучшая корреляция (0,958) получена для формы немедленного действия (молсидомин 4 мг), что представляется весьма логичным. В самом деле известно, что чем более усложняется галенова форма, тем сложнее найти зависимость между высвобождением in vitro и усвоением in vivo. Для молсидомина 8 мг коэффициент корреляции остается очень высок (0,855). Для молсидомина 16 мг наилучшая корреляция (0,812) наблюдается, когда учитывается кинетика после принятия пищи и растворение в кислой среде. В общем случае, для форм с пролонгированным высвобождением действующего вещества корреляция всегда лучше, когда высвобождение осуществляется в кислой среде. Это объясняется тем обстоятельством, что большая часть кинетики молсидомина зависит от всасывания в желудке.
ПРИМЕР 5
Исследование концентрации препарата в плазме крови при назначении лекарственной формы молсидомина согласно настоящему изобретению в зависимости от времени у пожилых пациентов, страдающих стенокардией.
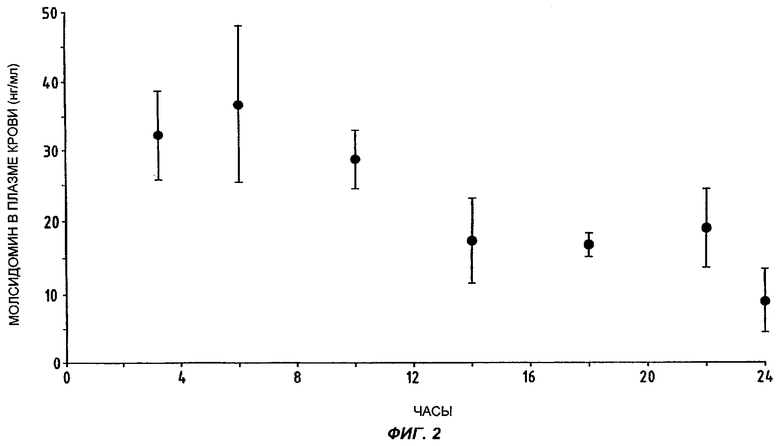
Тридцать три коронарных больных со стабильной стенокардией получили однократную дозу молсидомина 16 мг (лекарственная форма согласно Примеру 1), принятую в 8 часов утра с завтраком. Выборка была представлена 22 мужчинами и 11 женщинами со средним возрастом 62,6±1,3 года (крайние значения 49 и 73 года). Разделенные на 7 групп пациенты сдавали кровь на содержание молсидомина соответственно через 3, 6, 10, 14, 18, 22 и 24 часа после принятия препарата.
На Фиг.2 представлена средняя концентрация молсидомина в плазме крови и стандартная ошибка средней для каждой из 7 групп испытуемых пациентов.
Как показано на Фиг.2, кинетика, определенная у пожилых пациентов, сильно коррелирует с кинетикой, определенной у здоровых молодых добровольцев и представленной на Фиг.1. Кроме того, и величины Cmax, Tmax и время полужизни сравнимы в двух испытуемых группах.
Получены также следующие данные:
- средняя максимальная концентрация составляет 36,0±10,8 нг/мл;
- наивысшая средняя концентрация определена в группе 2, то есть через 6 часов после приема молсидомина;
- концентрация снижается медленно, на 50% в течение 8 часов;
- концентрация молсидомина в плазме крови поддерживается приблизительно на одном уровне в течение 8 часов, между 14-м и 22-м часами после приема и колеблется между 16,5 и 18,1 нг/мл;
- остаточная концентрация 8,5±4,3 нг/мл наблюдается еще через 24 часа после назначения однократной дозы молсидомина 16 мг.
ПРИМЕР 6
Изучение корреляции между клинической эффективностью и концентрацией в плазме крови лекарственной формы молсидомина согласно настоящему изобретению.
Десять коронарных больных, страдающих стабильной стенокардией, получали комплексное лечение грудной жабы (нитропроизводные пролонгированного действия, молсидомин, антагонисты кальция и/или бета-блокаторы) по меньшей мере в продолжении трех дней, при этом в случае приема бета-блокаторов лечение было более длительным; некоторым был прописан в течение этого периода прием изосорбид-тринитрата 5 мг в таблетках под язык или применение нитроглицерина в аэрозоле.
Затем испытуемые пациенты получали однократно молсидомин 16 мг (в лекарственной форме согласно Примеру 1) или плацебо в соответствии с техникой двойного слепого рандомизированного плацебо-контролируемого эксперимента, включающего период наблюдения по меньшей мере в продолжении двух суток.
Выборка состояла из 8 мужчин и 2 женщин со средним возрастом 61,3±3,1 года (крайние значения 49 лет и 73 года).
Будучи разделенные на 7 групп, пациенты после нагрузки на велоэргометре сдавали кровь на содержание молсидомина спустя соответственно 3, 6, 10, 14, 18, 22 и 24 часа по приеме лекарства или плацебо.
Испытание на велоэргометре состояло из начальной нагрузки в 30 Вт с повышением на 30 Вт каждые 3 минуты до момента остановки испытания при возникновении симптомов грудной жабы, достижения максимальной теоретической частоты сердечных сокращений, мышечной усталости или из соображений безопасности (нарушение ритма или внутрисердечной проводимости, скачок давления более чем на 20 мм рт.ст., подавление участка ST≥3 мм). Как в покое, так и в продолжении всего испытания нагрузкой снимались ЭКГ и показания артериального давления.
Клиническая эффективность лекарственной формы согласно настоящему изобретению определялась количественно по следующим показателям:
а) разница в общей длительности упражнения, в секундном выражении, под действием плацебо и под действием молсидомина;
б) разница в объеме выполненной работы, выраженная как сумма ватт·мин, произведенных на каждой ступени нагрузки, под действием плацебо и под действием молсидомина.
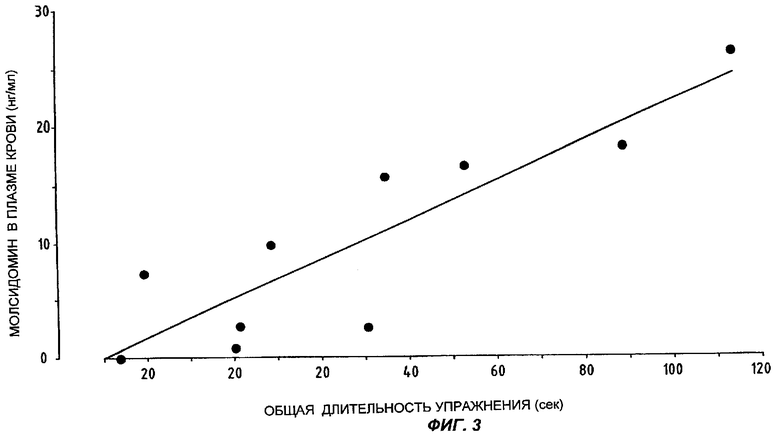
На Фиг.3 представлены данные о концентрациях молсидомина в плазме крови, выявленные у каждого из 10 пациентов, и соответствующие различия в общей длительности упражнения.
Уравнение линейной регрессии между переменной Х (разница между общей длительностью упражнения под действием плацебо и под действием молсидомина) и переменной У (концентрация молсидомина в плазме крови) таково: Y=0,18Х+5,35.
Коэффициент корреляции Пирсона г равен 0,88 (Р<0,001), что соответствует коэффициенту детерминации r2=0,77, то есть 77% вариационного ряда данных о клинической эффективности может быть объяснено за счет присутствия молсидомина в плазме крови пациентов.
Ни квадратическая, ни кубическая модели не улучшают коэффициента корреляции.
Если считать клинически значимой разницу в 30 секунд между испытаниями под нагрузкой под действием плацебо и молсидомина, то оказывается, что необходимая для такого уровня эффективности концентрация молсидомина в плазме крови составляет 10,75 нг/мл - величина, практически достижимая даже спустя 24 часа после однократного приема молсидомина 16 мг согласно настоящему изобретению (см. Фиг.2).
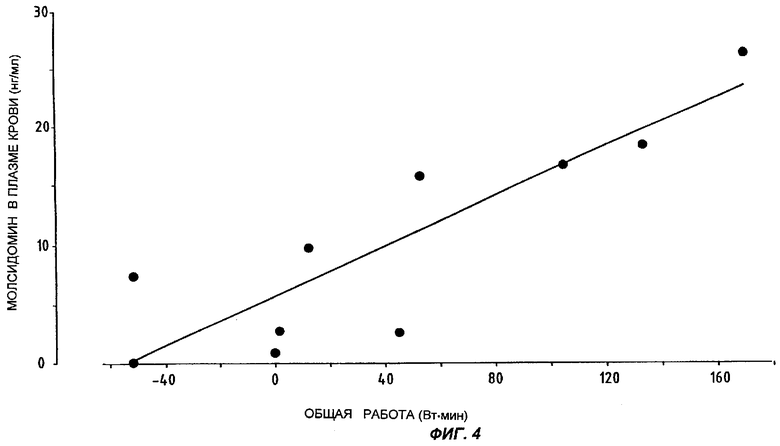
На Фиг.4 представлены данные о концентрациях молсидомина в плазме крови у каждого из 10 пациентов и разница в произведенной общей работе.
Уравнение линейной регрессии между переменной Х (разница между общей произведенной работой под действием плацебо и под действием молсидомина) и переменной У (концентрация молсидомина в плазме крови) таково: Y=0,11Х+5,90.
Коэффициент корреляции Пирсона r составляет 0,86 (Р=0,002), что соответствует коэффициенту детерминации r2=0,74, т.е. 74% вариационного ряда показателей клинической эффективности может быть объяснено за счет присутствия молсидомина в плазме крови пациентов.
Ни квадратическая, ни кубическая модели не улучшают коэффициента корреляции.
Если считать клинически значимой разницу в 50 Вт·мин между испытаниями под нагрузкой под действием плацебо и молсидомина, то оказывается, что необходимая для такого уровня эффективности концентрация молсидомина в плазме крови составляет 11,40 нг/мл - величина, практически достижимая даже спустя 24 часа после однократного приема молсидомина 16 мг согласно настоящему изобретению (см. Фиг.2).
ПРИМЕР 7
Исследование клинической эффективности лекарственной формы молсидомина согласно настоящему изобретению
Двести двадцать два пациента со стабильной стенокардией напряжения участвовали в международном многоцентровом рандомизированном двойном слепом плацебо-контролируемом исследовании.
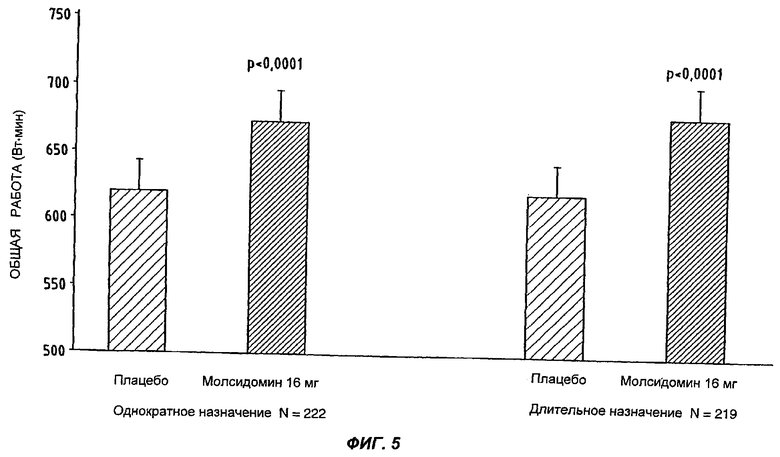
После однократного назначения разовой дозы молсидомина 16 мг (лекарственная форма согласно Примеру 1) или плацебо пациенты проходили испытание под нагрузкой на велоэргометре, отсроченное на временной промежуток от 2-х до 24-х часов после приема медикамента. Объем нагрузки (общая произведенная работа, выраженная в Вт·мин) под действием молсидомина оказался значимо больше, чем под действием плацебо как статистически (р<0,001), так и клинически (среднее улучшение составляет 53 Вт·мин).
Введение той же однократной ежедневной дозы в 16 мг молсидомина (в лекарственной форме согласно Примеру 1) в течение 2-х недель вызывает улучшение физического состояния, сравнимое с улучшением, достигнутым при однократном введении, значимое как статистически (р<0,001), так и клинически (среднее улучшение составляет 58 Вт·мин).
Эти результаты свидетельствуют об отсутствии привыкания к молсидомину 16 мг после длительного лечения (Фиг.5). Они указывают также на то, что терапевтический эффект растягивается на 24 часа.
Заметим, что улучшение клинической картины, наблюдаемое в этом исследовании, находится в хорошем соответствии с клинически эффективными концентрациями молсидомина в плазме крови, выведенными из корреляции общая работа / молсидомин в плазме, представленной на Фиг.4.
Результаты, полученные в вышеизложенных Примерах 5, 6 и 7, доказывают оригинальность новой галеновой формы молсидомина согласно изобретению.
Основные преимущества этой формы в сравнении с существующими формами могут быть изложены следующим образом.
Поддержание на относительно неизменном уровне повышенной концентрации молсидомина в плазме крови (16,5-18,1 нг/мл) в течение 8 часов (от 14-го до 22-го часа после приема препарата). Относительно неизменный уровень концентрации в плазме крови, обеспечивающий значительную клиническую эффективность у коронарных больных со стабильной стенокардией, о чем говорит тесная корреляция между повышением объема нагрузки и концентрацией молсидомина в крови, причем указанное улучшение сохраняется в продолжение 24 часов после введения препарата.
Отсутствие привыкания к молсидомину, причем значимая клиническая эффективность сохраняется после двухнедельного лечения коронарных больных со стабильной стенокардией.
| название | год | авторы | номер документа |
|---|---|---|---|
| ПЕРОРАЛЬНАЯ КОМПОЗИЦИЯ С ПРОЛОНГИРОВАННЫМ ВЫСВОБОЖДЕНИЕМ МОЛСИДОМИНА ДЛЯ ЛЕЧЕНИЯ АТЕРОСКЛЕРОЗА | 2005 |
|
RU2388475C2 |
| ПРЕПАРАТЫ РАНОЛАЗИНА С ПРОЛОНГИРОВАННЫМ ДЕЙСТВИЕМ | 1999 |
|
RU2214233C2 |
| СПОСОБ ЛЕЧЕНИЯ СЕРДЕЧНО-СОСУДИСТЫХ ЗАБОЛЕВАНИЙ | 1999 |
|
RU2207856C2 |
| Фармацевтическая композиция пролонгированного действия на основе 5-этокси-2-[2-(морфолино)-этилтио] бензимидазола дигидрохлорида и/или основания (Афобазола) | 2017 |
|
RU2694837C2 |
| Система доставки 2-этил-6-метил-3-гидроксипиридина сукцината для перорального применения в форме гастроретентивной таблетки | 2019 |
|
RU2734970C1 |
| СОСТАВЫ ЛОРАЗЕПАМА С ЗАМЕДЛЕННЫМ ВЫСВОБОЖДЕНИЕМ | 2014 |
|
RU2678324C2 |
| ФАРМАЦЕВТИЧЕСКИЕ КОМПОЗИЦИИ ДЛЯ ПРОЛОНГИРОВАННОГО ВЫСВОБОЖДЕНИЯ СЕБАКОИЛ-ДИНАЛБУФИНОВОГО ЭФИРА | 2016 |
|
RU2718900C2 |
| ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ ДЛЯ ПРОЛОНГИРОВАННОГО ВЫСВОБОЖДЕНИЯ ТРИМЕТАЗИДИНА | 2013 |
|
RU2621128C2 |
| Лекарственное средство пролонгированного действия на основе анастрозола | 2017 |
|
RU2659689C1 |
| КОМПОЗИЦИИ В ВИДЕ МНОЖЕСТВА ЧАСТИЦ С МОДИФИЦИРОВАННЫМ ВЫСВОБОЖДЕНИЕМ | 1999 |
|
RU2236847C2 |
Настоящее изобретение касается новой пероральной формы галенова препарата молсидомина с пролонгированным высвобождением действующего вещества, предназначенной для лечения приступа грудной жабы во всех ее разновидностях (стенокардия напряжения, спастическая стенокардия, нестабильная стенокардия). Согласно настоящему изобретению, эта новая форма галенова препарата содержит терапевтически эффективное количество молсидомина или одного из его активных метаболитов и характеризуется следующей скоростью растворения in vitro [измеренной спектрофотометрически при длине волны 286 или 311 нм согласно способу, изложенному в Pharmacopee Europeenne, 3-е изд. (или USP XXIV) при 50 об/мин в 500 мл среды, состоящей из 0,1 н. HCl при 37°C]: 15-25% молсидомина, высвобожденного через 1 час, 20-35% молсидомина, высвобожденного через 2 часа, 50-65% молсидомина, высвобожденного через 6 часов, 75-95% молсидомина, высвобожденного через 12 часов, >85% молсидомина, высвобожденного через 18 часов, >90% молсидомина, высвобожденного через 24 часа, причем пик содержания молсидомина в плазме крови in vivo появляется через 2,5-5 часов, предпочтительно через 3-4 часа, после приема вышеупомянутой формы и имеет значение от 25 до 40 нг/мл плазмы. Изобретение позволяет сократить количество приемов лекарства в день, что более удобно для пациента. 13 з.п. ф-лы, 5 ил., 2 табл.
15-25% молсидомина, высвобожденного через 1 ч,
20-35% молсидомина, высвобожденного через 2 ч,
50-65% молсидомина, высвобожденного через 6 ч,
75-95% молсидомина, высвобожденного через 12 ч,
>85% молсидомина, высвобожденного через 18 ч,
>90% молсидомина, высвобожденного через 24 ч,
причем пик содержания молсидомина в плазме крови in vivo появляется через 2,5-5 ч, предпочтительно через 3-4 ч, после приема вышеупомянутой формы и имеет значение от 25 до 40 нг/мл плазмы, при этом она включает в себя систему высвобождения молсидомина, состоящую из матрицы пролонгированного высвобождения или из обычной лекарственной формы с защитной оболочкой, обеспечивающей пролонгированное высвобождение молсидомина.
| Огнетушитель | 0 |
|
SU91A1 |
| Способ сборки электродинамических громкоговорителей | 1978 |
|
SU714661A1 |
| Устройство для контроля цифровых устройств | 1977 |
|
SU624370A1 |
| КОМПОЗИЦИЯ ДЛЯ ПОКРЫТИЯ ПРЕПАРАТА С РЕГУЛИРУЕМЫМ ВЫДЕЛЕНИЕМ АКТИВНОГО ВЕЩЕСТВА | 1991 |
|
RU2072836C1 |

