Изобретение относится к медицине, в частности к фармации, конкретно к фармацевтическим композициям, обладающим анксиолитическим действием. Изобретение обеспечивает получение фармацевтической композиции, обладающей пролонгированным высвобождением активного фармацевтического ингредиента 5-этокси-2-[2-(морфолино)-этилтио] бензимидазола дигидрохлорида (торговое название «Афобазол», международное непатентованное наименование - Фабомотизол). Пролонгированное действие активного компонента обеспечивается предложенным составом фармацевтической композиции, а именно, включением в нее комбинации модификаторов высвобождения, состоящей, по меньшей мере, из одного гидрофильного модификатора высвобождения и, по меньшей мере, из одного замедлителя высвобождения. Комбинированное взаимодействие и количественное взаимоотношение данных вспомогательных веществ, где замедлители высвобождения используются в пределах от 50,5 до 70 мас. %, а модификаторы высвобождения - от 10 до 30 мас. %, в составе фармацевтической композиции на основе 5-этокси-2-[2-(морфолино)-этилтио] бензимидазола дигидрохлорида обеспечивают получение нового технического результата, недостижимого ранее известным аналогом в этом уровне техники, а именно: увеличение длительности нахождения лекарственного средства в крови в концентрации, необходимой и достаточной для достижения терапевтического эффекта на протяжении 8 часов после его однократного приема, повышение условий комфортности пациентов за счет сокращения числа приемов лекарственного средства.
Уровень техники. 5-этокси-2-[2-(морфолино)-этилтио] бензимидазола дигидрохлорид - оригинальное лекарственное средство (патент РФ 2061686). Оно, не относясь к классу агонистов бензодиазепиновых рецепторов, обладает анксиолитическим действием с активирующим компонентом, не сопровождающимся гипноседативными эффектами при отсутствии миорелаксантных свойств и негативного влияния на показатели памяти и внимания. Известно о наличии активирующего компонента действия 5-этокси-2-[2-(морфолино)-этилтио] бензимидазола дигидрохлорида, оказывающих позитивное влияние на психофизиологические характеристики человека (сенсомоторное реагирование, координация, внимание, кратковременная зрительная память), хотя свойствами психомоторного стимулятора препарат не обладает [Незнамов Г.Г., Сюняков С.А., Чумаков Д.В. Новый селективный анксиолитик афобазол // Журнал неврологии и психиатрии им. С.С.Корсакова. 2005. Т. 105, №4. С.48- 54].
При исследовании эффектов 5-этокси-2-[2-(морфолино)-этилтио] бензимидазола дигидрохлорида по защите ишемизированного мозга выявлена способность соединения восстанавливать структуру мозговой ткани и ограничивать зону ишемического поражения как при предварительном введении, так и в пределах терапевтического окна; показана способность соединения активировать энергетический обмен мозговой ткани повышением активности сукцинатдегидрогеназы; также доказана его способность устранять анксиогенное воздействие локальной ишемии и предотвращать ослабление когнитивных свойств и памяти при окклюзии средней мозговой артерии [Середенин С.Б., Крайнева В.А. Нейропротекторные свойства афобазола при экспериментальном моделировании геморрагического инсульта // Эксперим. и клинич. фармакология. М., 2009, Т. 72, №1. С. 24-28].
Известны твердые лекарственные формы фармацевтической композиции на основе 5-этокси-2-[2-(морфолино)-этилтио] бензимидазола дигидрохлорида («Фармацевтическая композиция на основе афобазола»; Патент №2289403). Основным недостатком указанной композиции в ее таблетированной лекарственной форме является неконтролируемое и практически моментальное полное высвобождение действующего начала - Афобазола в ЖКТ при пероральном приеме и короткий период полувыведения (около часа). Фармакодинамическая активность Афобазола - 5-этокси-2-[2-(морфолино)-этилтио] бензимидазола дигидрохлорида проявляется как куполообразная зависимость эффекта от дозы, и поэтому на практике эффективная концентрация препарата (обусловленная его фармакокинетикой) поддерживается при оральном приеме нескольких доз, разнесенных по времени в течение всего дня, а не при разовом приеме одной большой дозы, что увеличивает риск пропуска дозы.
Предпочтительно использование фармацевтических композиций, характеризующихся контролируемым пролонгированным высвобождением действующего вещества, что позволяет стабилизировать концентрацию препарата в организме человека в течение суток (предупреждает чрезмерное падение концентрации в конце интервала дозирования и резкий рост после приема последующих доз препарата), обеспечивает создание его терапевтически эффективных концентраций в системном кровотоке и, соответственно, более длительный терапевтический эффект после однократного приема лекарственной формы, содержащей необходимую дозу активного вещества.
Преимущества пролонгированной лекарственной формы очевидны: это более упорядоченный прием лекарства, уменьшение количества приемов в день и соответственно меньший риск пропустить время приема (в тех случаях, когда это важно), уменьшенный риск, связанный с побочными эффектами, более равномерное и длительное действие лекарства. Однако необходимость поддерживать нужную скорость высвобождения в течение длительного времени предъявляет к лекарственной форме более жесткие требования в плане качества активной субстанции, вспомогательных веществ и технологического процесса [Трофимов С.В. Высокомолекулярные эфиры целлюлозы. Механизмы действия в матричных таблетках пролонгирующего действия. Зависимость профиля высвобождения активной субстанции от молекулярной массы и гидрофильных свойств полимера // Фармация и фармакология. Т. 5. №(12), 2015. С. 18-25].
Известны фармацевтические композиции Афобазола - 5-этокси-2-[2-(морфолино)-этилтио] бензимидазола дигидрохлорида с измененным высвобождением [Сизяков С.А. Разработка составов и технологии таблеток афобазола: дис… кандидата фарм. наук: 15.00.01, М., 2009 (далее, Сизяков, 2009); Полковникова Ю.А. Разработка состава, технологические исследования и определение норм качества капсулированных лекарственных форм афобазола: дис… кандидата фарм. наук: 14.04.01 М., 2011], где пролонгирование высвобождения действующего начала достигается путем постепенного растворения лекарственного средства из матрицы.
В работе Сизякова С.А. изучались таблетки с пролонгированным высвобождением Афобазола на основе таких матриц, как Kollidon SR, ГПМЦ (гидроксипропилметилцеллюлоза), Карбопол 71 G, композиционный полимерный носитель (КПН) в соотношениях (1:1, 1:2, 1:3, 1:4), дополнительно в систему в качестве смазывающего вещества вводился стеарат магния. Полученные фармацевтические составы на основе КПН, не укладывались в нормы ОФС «Растворение» (не более 10% растворения действующего вещества в кислой среде за 120 минут). Причем наиболее оптимальный из изученных составов по рассматриваемому эффекту состав, соотношение Афобазола к КПН в котором равнялось 1:4, обеспечивал наименьшее выделение Афобазола в кислой среде, имитирующей среду кишечника, в количестве 36,41% за 120 мин и превосходил все изученные составы на основе ГПМЦ [Сизяков, 2009, стр. 19, 1 абз.].
Наиболее длительное высвобождение наблюдалось при использовании таблеток, полученных методом прямого прессования, следующего состава: Афобазол - 0,03 г, Kollidon SR - 0,1185 г, магния стеарат - 0,0015 г. Однако подобный состав также не обеспечивал соблюдение нормы «Растворение» в кислой среде, что привело к необходимости покрывать ядро таблетки кишечнорастворимой оболочкой (AcrylEZE). Предложенные автором составы способны замедлять высвобождение действующего вещества, однако, несмотря на использованную максимально возможную дозировку, они, не могли обеспечить создание терапевтически эффективных концентраций действующего вещества в системном кровотоке, а использование кишечнорастворимой оболочкой, способствовало еще более медленному высвобождению свободной активной субстанции быстро элиминирующимися незначительными порциями, что также не могло обеспечить терапевтически эффективных концентраций Афобазола - 5-этокси-2-[2-(морфолино)-этилтио] бензимидазола дигидрохлорида в системном кровотоке.
В работе Полковниковой Ю.А. предложен вариант формы пролонгированного высвобождения Афобазола - микрокапсулы без матрицы, покрытые кишечнорастворимой оболочкой. Содержание компонентов на 100 капсул (г): микрокапсулы Афобазола - 3,0, желатин - 6,0, вода очищенная - 8,0; оболочка: Колликут МАЕ 30 ДП - 1,5, пропиленгликоль - 0,4, вода - 3,2, титана диоксид - 0,075, тальк - 11,2. Профиль высвобождения действующего вещества этой композиция схож с профилем композиции, предложенной Сизяковым С.А. Увеличение времени высвобождения лекарственного вещества до 8 часов достигалось за счет использования полимерной матрицы и кислотоустойчивого покрытия: из капсул с микрокапсулами, покрытыми Колликут МАЕ 30, однако через 2 часа высвобождалось всего 8,9% Афобазола, что не создавало требуемых терапевтических концентрации вещества в кровотоке.
Высокие показатели скорости элиминации Афобазола и куполообразная зависимость эффекта от дозы, несмотря на пролонгирование его высвобождения, не обуславливают пролонгирование терапевтического действия по причине быстрого его удаления из системного кровотока.
Недостатками описанных выше технических решений при создании пролонгированной фармацевтической композиции Афобазола явилась их неспособность обеспечить создание терапевтически эффективных концентраций действующего вещества в системном кровотоке на протяжении не менее 8 часов из-за недостаточности высвобождения Афобазола в единицу времени и/или его быстрого выведения из кровеносного русла, что в итоге не обеспечивало пролонгации фармакологической активности Афобазола.
Поскольку Афобазол - 5-этокси-2-[2-(морфолино)-этилтио] бензимидазола дигидрохлорид, как соединение, характеризуется низкими значениями биодоступности, куполообразной зависимостью доза-эффект, имеет «окно всасывания» в области тонкого кишечника (кислотность рН=7,2÷7,5) и короткое время «пребывания» в организме (за счет прямого вывода из организма и ускоренного метаболирования), лечебное средство на его основе (в таблетированной форме) нуждается в особом подходе к разработке состава и технологии производства соответствующей эффективной фармацевтической композиции.
Одним из существенных моментов создания лекарственной формы, соответствующей заданным характеристикам, является разработка оптимального качественного и количественного соотношения составляющих фармацевтической композиции. Исторически вспомогательным веществам отводилась роль абсолютно инертных веществ, необходимых для формирования лекарственной формы. Однако стремительное развитие фармацевтической технологии привело к значимому повышению роли вспомогательных веществ в возможности реализации надлежащего терапевтического эффекта. При этом влияние вспомогательных веществ на терапевтический эффект также может быть как крайне положительным, так и крайне отрицательным и реализуется большей частью на уровне высвобождения, растворения и всасывания действующего вещества. Каждое используемое в фармацевтической промышленности вспомогательное вещество обладает совокупностью специфических технологических и биофармацевтических характеристик, оказывающих в конечном итоге значимое влияние на параметры готовой лекарственной формы и высвобождение действующего вещества, причем один и тот же с химической точки зрения наполнитель может обладать совершенно различными технологическими свойствами [Воскобойникова И.В., Авакян С.Б., Сокольская Т.А. и соавт. Современные вспомогательные вещества в производстве таблеток. Использование высокомолекулярных соединения для совершенствования лекарственных форм и оптимизации технологического процесса // Хим.-фарм. журнал. 2005; Т. 2005(1), С. 22-28]. Одни и те же вспомогательные вещества могут усилить или ослабить действие лекарственных веществ, изменить характер действия под влиянием различных причин: комплексообразования, молекулярных реакций и т.д. [Фармацевтическая технология. Изготовление лекарственных препаратов: учебник / А.С. Гаврилов. 2010. 624 с.; Хоружая Т.Г., Чучалин B.C. Биофармация - научное направление в разработке и совершенствовании лекарственных препаратов: Учебное пособие. Томск: Лаб. оперативной полиграфии СибГМУ, 2006. 75 с.].
В ряде случаев вспомогательные вещества могут изменять профиль высвобождения лекарственного средства в нежелательную сторону. Наиболее распространенные причины этого: формирование плохо растворимых комплексов, рост примесных соединений, снижение количественного содержания непосредственно лекарственного средства, создание неблагоприятной среды для сохранения стабильности лекарственного средства, вступление в реакцию химического взаимодействия, а также формирование готовой формы с ненадлежащими свойствами (замедление разрушения, высвобождения действующего вещества). Одним из классических примеров является случай замены наполнителя в таблетированной форме фенитоина натрия с кальция сульфата дигидрата на лактозу. В первом случае фенитоин образовывал плохо растворимые комплексы с кальцием, что приводило к снижению всасывания и биодоступности. После изменения вспомогательного вещества на лактозу, всасывание фенитоина резко повысилось, и у пациентов с эпилепсией, ранее стабилизированных на прежней технологической форме, на той же дозе фенитоина развились признаки фенитоиновой токсичности [Aulton ЕМ. Aulton's Pharmaceutics: the Design and Manufacturing of Medicines / Churchill Livingstone Elsevier, 2007. 717 p.].
Отмеченные факты могут не только изменить содержание действующего вещества в готовой фармацевтической композиции, но и может изменить профиль переносимости терапии.
В производстве твердых лекарственных форм (таблеток, капсул и гранул) микрокристаллическая целлюлоза (МКЦ) применяется в качестве наполнителя, стабилизатора и эмульгатора, проявляющего связующие и улучшающие скольжение свойства [Егошина Ю.А., Поцелуева Л.А. Современные вспомогательные вещества в таблеточном производстве // Успехи современного естествознания. 2009. №10. С. 30-33]. Выявлены некоторые специфические особенности препаратов, получаемых с использованием МКЦ [Аутлов С.А., Базарнова Н.Г., Кушнир Е.Ю. Микрокристаллическая целлюлоза: структура, свойства и области применения (обзор) // Химия растительного сырья. 2013. №3. С. 33-41]. Так, известно, что в процессе приготовления лекарственных форм (в особенности с применением механоактивации) между МКЦ и молекулами лекарственных веществ, содержащими разнообразные функциональные группы, имеет место физико-химическое взаимодействие достаточно сложного характера, которое приводит к диспергированию и распределению активного вещества в матрице носителя с образованием «привитых комплексов», нанокристаллических или аморфных композитов, увеличивая тем самым удельную поверхность активного ингредиента. Совместная механическая обработка смесей лекарственных веществ с МКЦ позволяет стабилизировать образующиеся метастабильные состояния, способствует увеличению скорости растворения и растворимости трудно растворимых лекарственных веществ, повышению их биологической доступности. В то же время межмолекулярные взаимодействия между молекулами лекарственного вещества и матрицы носителя - МКЦ могут приводить к замедлению высвобождения активного ингредиента и пролонгированию действия лекарственного средства [Аутлов С.А., Базарнова Н.Г., Кушнир Е.Ю. Микрокристаллическая целлюлоза: структура, свойства и области применения (обзор) // Химия растительного сырья. 2013. №3. С. 33-41].. Таким образом, одно и то же вспомогательное вещество, в данном случае МКЦ, может способствовать как увеличению растворимости лекарственных веществ, повышению их биологической доступности, так и приводить к замедлению высвобождения активного ингредиента.
Пероксидные примеси в повидоне, выступающем в роли связующего, и кросповидоне, выступающем в роли дезинтегранта, могут приводить к деградации структуры лекарственного средства, как это случилось при создании композиции ралоксифена гидрохлорида [Kollocoat IR - связующее с уникальными свойствами. http://www.pharmtech-expo.ru/www_pharmtech/files/fc/fcf050ec-6d27-4a00-8324-b9954fd65087.pdf].
Следует отметить, что определение оптимального состава вспомогательных веществ на этапе фармацевтической разработки имеет различную значимость для лекарственных средств с различными показателями растворимости и биодоступности, и наиболее значимым, вплоть до потери терапевтической эффективности и/или изменения профиля безопасности, данное влияние может являться для следующих групп лекарственных веществ (Сеткина С.Б., Хишова О.М. Биофармацевтические аспекты технологии лекарственных средств и пути модификации биодоступности // Вестник Витебского государственного медицинского университета. 2014. Т. 13. №4. С. 162-172):
- лекарственные средства с низкой растворимостью и/или биодоступностью;
- лекарственные средства, имеющие особенности всасывания (всасывание на определенном участке желудочно-кишечного тракта, «окно всасывания»);
- соединения, обладающие свойствами физико-химической нестабильности в кислой либо щелочной среде;
- лекарственные средства с узким терапевтическим интервалом, для которых незначительные изменения биодоступности могут привести к существенному изменению терапевтического эффекта.
Исходя из данных соображений, 5-этокси-2-[2-(морфолино)-этилтио] бензимидазола дигидрохлорид, как соединение, характеризующееся низкими значениями биодоступности, куполообразной зависимостью доза-эффект и преимущественным всасыванием в области тонкого кишечника, нуждается в оптимизации подбора состава и технологии производства фармацевтической композиции на его основе, так как использование общих подходов может не привести к должному терапевтическому эффекту.
Включение в систему 5-этокси-2-[2-(морфолино)-этилтио] бензимидазола дигидрохлорида только гидрофильных полимеров в качестве модификаторов высвобождения могло быть нецелесообразным в силу их хорошей растворимости в воде и неспособности обеспечить требуемые характеристики высвобождения для данного технического решения. Стандартный профиль высвобождения систем на основе гидрофильных полимеров характеризуется высоким процентом флуктуаций относительного начального периода высвобождения и плоским участком кривой с неполным высвобождением ближе к концу. Для контролирования такого рода флуктуаций высвобождения применяют гидрофобные полимеры или покрытия лекарственных форм. Использование же только замедлителей высвобождения могло привести к высокому начальному флуктуационному уровню высвобождения, или, в случае биофармацевтических особенностей системы, характерной для соединений с физико-химической лабильностью, наоборот, к замедлению высвобождения до субтерапевтических концентраций. Подобное наблюдалось в работе Сизякова С.А. [Сизяков С.А. Разработка составов и технологии таблеток афобазола: дис… кандидата фарм. наук: 15.00.01, М., 2009], где использование только замедлителя высвобождения (например, Kollidon SR, ГПМЦ, Карбопол 71 G, КПН) в количестве до 80 мас. % от массы фармацевтической композиции не обеспечило создание терапевтически эффективных концентраций действующего вещества.
Таким образом, использование только одного компонента: гидрофильного модификатора высвобождения или замедлителя высвобождения, при создании фармацевтической композиции на основе Афобазола - 5-этокси-2-[2-(морфолино)-этилтио] бензимидазола дигидрохлорида могло не только не обеспечить достижение технического результата - пролонгированного действия, но и привести к потере терапевтической эффективности соединения.
Данный аспект привел к следующему решению - использованию комбинации из гидрофильных модификаторов высвобождения и замедлителей высвобождения.
Под комбинацией модификаторов высвобождения и ее использовании понимается одновременное и обязательное включение двух функциональных составляющих (каждое из которых может включать в себя одни или несколько компонентов) вспомогательных веществ - замедлителя высвобождения и модификатора высвобождения в определенных количествах в состав новой фармацевтической композиции, приводящей к пролонгированному терапевтическому действию Афобазола (5-этокси-2-[2-(морфолино)-этилтио] бензимидазола дигидрохлорида за счет постепенного его высвобождения и поддержания концентрации препарата в крови на должном уровне длительное время.
Существенность качественного состава (комбинации модификаторов высвобождения) фармацевтической композиции неотрывно от количественного соотношения этих вспомогательных веществ. Так, известно, что гидроксипропилцеллюлоза в концентрации 15-35 мас. % может быть использована для получения таблеток с пролонгированным высвобождением лекарственного средства как составляющее матрицы с пролонгированным высвобождением [Raymond С Rowe, Paul J Sheskey, Marian E Quinn, Handbook of Pharmaceutical excipients, шестое издание, опубликовано в 2009 г., стр. 328], а также, что производные акриловой кислоты могут применяться в количестве 5-50 мас. % в качестве агента контролирующего высвобождение [там же, стр. 110].
Проведенные предварительные опыты при создании фармацевтической композиции на основе 5-этокси-2-[2-(морфолино)-этилтио] бензимидазола дигидрохлорида (Афобазола) с преимущественным использованием одного из компонентов комбинации высвобождения - гидрофильного модификатора высвобождения (например: МКЦ, гидроксипропилцеллюлоза) в количестве 30-60 мас. % и замедлителя высвобождения (например: КПН, смеси повидона и поливинилацетата) в количестве 30-90 мас. %, при отсутствии или незначительном количестве второго компонента приводило к созданию композиций, которые характеризовались наличием флуктуаций (пиков) в кинетике высвобождения, что нежелательно для лекарственных форм с пролонгированным высвобождением, и/или демонстрировали недостаточный уровень высвобождения активного вещества даже после 8 часов, что в сочетании с невысокими значениями биодоступности Афобазола указывало на невозможность достижения технического результата.
Неожиданно было установлено, что использование комбинации из хотя бы одного замедлителя высвобождения в количественном соотношении от приблизительно 50,5 мас. % до приблизительно 70 мас. % и хотя бы одного модификатора высвобождения в количественном соотношении от приблизительно 10 мас. % до приблизительно 30 мас. % при создании фармацевтической композиции на основе 5-этокси-2-[2-(морфолино)-этилтио] бензимидазола дигидрохлорида (Афобазола) приводит к желаемому техническому результату.
Техническим результатом, на достижение которого направлено заявляемое изобретение, является повышение терапевтического эффекта, за счет придания композиции свойства отсроченного замедленного высвобождения Афобазола, что обеспечивает создание и поддержание в системном кровотоке практически постоянной терапевтически эффективной концентрации активного вещества в течение периода, превышающего 8 часов. Также техническим результатом является повышение условий комфортности пациентов при использовании композиции в лечебных целях за счет сокращения числа приемов лекарственного средства.
Задачей настоящего изобретения явилось создание улучшенной фармацевтической композиции и лекарственных форм на ее основе, содержащих в качестве действующего вещества 5-этокси-2-[2-(морфолино)-этилтио] бензимидазола дигидрохлорид (торговое название «Афобазол», или Фабомотизол (МНН)), обеспечивающих замедленное высвобождение и создание в системном кровотоке терапевтически эффективной концентрации Афобазола в течение периода, превышающего 8 часов при условии эквивалентности доз действующего вещества, и снижение, вследствие этого, необходимости частого приема препарата.
В качестве решения поставленной задачи предлагается отвечающая всем требованиям Государственной Фармакопеи XII издания выполненная в виде твердых лекарственных форм фармацевтическая композиция модифицированного пролонгированного высвобождения Афобазола - 5-этокси-2-[2-(морфолино)-этилтио] бензимидазола дигидрохлорида в терапевтически эффективном количестве, включающая кроме него комбинацию модификаторов высвобождения, состоящую из по меньшей мере одного гидрофильного модификатора высвобождения и по меньшей мере одного замедлителя высвобождения для контролируемого растворения и выхода из системы поддерживающих доз активного вещества, при определенном их количественном соотношении, и другие фармацевтически приемлемые вспомогательные вещества - наполнитель, смазывающее, скользящее, связывающее и биоразлагаемую пленочную оболочку при следующем соотношении компонентов, мас. %:
5-этокси-2-[2-(морфолино)-этилтио] бензимидазола
В качестве фармацевтически приемлемых вспомогательных веществ (смазывающее/скользящее, связующее, наполнитель, скользящее) в заявляемом техническом решении могут быть использованы следующие компоненты:
- в качестве наполнителя: производные целлюлозы, дисахариды, полисахариды, желатин, а также соли магния и кальция, коллоидный диоксид кремния в качестве целевых примесей к основному наполнителю и др. Предпочтительно применять наполнитель в количестве, близком к нижнему пределу интервала массы наполнителя;
- в качестве связующего: крахмальный клейстер, сахарный сироп, растворы: карбоксиметилцеллюлозы (КМЦ), оксиэтилцеллюлозы (ОЭЦ), оксипропилметилцеллюлозы (ОПМЦ); поливинилового спирта (ПВС), альгиновой кислоты, натрия альгината, желатина, метилцеллюлозы, гидроксипропилметилцеллюлозы, крахмала и др.; где растворители - вода очищенная, спирт этиловый, спирт изопропиловый; смеси вышеуказанных веществ. Предпочтительными связывающими веществами являются целлюлоза или производные целлюлозы, такие как натриевая карбоксиметилцеллюлоза, гидроксипропилцеллюлоза, гидроксипропилметилцеллюлоза или метилцеллюлоза, этилцеллюлоза, гидроксиэтилцеллюлоза, крахмал;
- в качестве смазывающего/скользящего вещества: стеариновая кислота и/или ее соли, аэросил, тальк, гидрированное растительное масло, воск из карнауба и др.
В то же время в качестве дополнительных фармацевтически приемлемых компонентов, сообщающих лекарственному средству на основе заявляемой композиции новые потребительские качества, могут быть использованы:
- в качестве замедлителя высвобождения: повидон и/или поливинилацетат, или их смеси, производные акриловой кислоты: редкосшитые акриловые полимеры, метакриловая кислота, композиционный полимерный носитель (КПН); причем, замедлитель высвобождения присутствует в количестве от приблизительно 50,5 мас. % до приблизительно 70,0 мас. % в расчете на массу композиции;
- в качестве модификатора высвобождения: производные целлюлозы, в частности, метилцеллюлоза, этилцеллюлоза, гидроксипропилцеллюлоза, гидроксипропилметилцеллюлоза, гидроксиэтилцеллюлоза, а также, кальция фосфат, кальция дигидрофосфат, кальция гидрофосфат дигидрат, кальция сульфат дигидрат, кальция карбонат основной, эфиры целлюлозы, микрокристаллическую целлюлозу, причем, микрокристаллическая целлюлоза и смеси, содержащие ее и один или несколько дополнительных компонентов, например, предварительно прежелатинизированный крахмал - особенно предпочтительны. Модификаторы высвобождения присутствует в количестве от приблизительно 10,0 мас. % до приблизительно 30,0 мас. % в расчете на массу композиции;
- пленочная оболочка, в которой в качестве пленкообразующего могут быть использованы полиэтиленгликоль, поливиниловый спирт, гидроксипропилцеллюлоза, гидроксиметилцеллюлоза и гидроксипропилметилцеллюлоза, оксипропилметилцеллюлозы фталат, ацетилцеллюлозы фталат, поливинилацетата фталат, метилцеллюлозы фталат, сополимер метакриловой кислоты или сложные метиловые эфиры метакриловой кислоты, этилцеллюлоза, ацетилцеллюлоза, сополимеры поливинилового спирта и малеинового ангидрида, полиэтиленгликоль, в частности, полиэтиленгликоль 6000, триэтилцитрат, диэтилфталат, пропиленгликоль, глицерин, бутилфталат, двуокись титана и красители, окись железа, алюминиевые красители, пленочные покрытия с сополимерами метакриловой кислоты, сополимеры этилакрилата и метилметакрилата. Пленочная оболочка может составлять от 0,0 до 3,0 масс. % от массы всей таблетки
Твердые дозированные лекарственные формы 5-этокси-2-[2-(морфолино)-этилтио] бензимидазола дигидрохлорида могут быть изготовлены из предлагаемых в данном изобретении композиций методами прямого прессования, предварительного сухого гранулирования и влажного гранулирования. При получении предлагаемой фармацевтической композиции в виде порошка лекарственное вещество и вспомогательные вещества предпочтительно пропускают через сито размером 30-40 меш.
В варианте осуществления способа с этапом влажного гранулирования, действующее и вспомогательные вещества гранулируют водой или раствором связующего вещества, и полученные гранулы после этого сушат. Остальные добавки, которые не использованы для включения в гранулы, затем смешивают с высушенными гранулами с получением в результате композиции, пригодной для капсулирования, таблетирования или др. В другом варианте приготовления лекарственной формы активное вещество и наполнители гранулируют в псевдоожиженном слое или путем размола брикетов. Полученные таким образом гранулы после просушки затем смешивают с любым оставшимся наполнителем и другими добавками, после чего из них можно получать таблетки, капсулы или другие лекарственные формы.
Для обеспечения длительного срока годности гранулят, полученный влажным гранулированием или в псевдоожиженном слое, либо другим методом на водной основе, должен быть практически полностью высушен. Процесс сушки обычно осуществляют в лотковой сушилке, или сушкой в псевдоожиженном слое. Сушку обычно проводят при температуре на входе около 50°C и относительной влажности менее 50%. Полученные гранулы затем смешивают с другими вспомогательными веществами: связывающим, смазывающими веществами и т.д. Композиции, полученные любым из вышеуказанных способов, формуют в необходимую лекарственную форму таблетированием, включением в твердую желатиновую капсулу и т.д. Из предлагаемых композиций можно изготавливать лекарственные препараты известными методами с обеспечением унифицированных доз заявляемого соединения для перорального приема в виде капсул, таблеток и тому подобное. Предварительно на данные гранулы может быть нанесена пленочная оболочка, и/или нанесение оболочки производят на модельные ядра, полученные прессованием данных гранул. Нанесение оболочки обычно осуществляют методом псевдоожижение или в установках барабанного типа.
Для получения гранул возможно использование технологии влажного гранулирования с этапом увлажнения раствором связывающего вещества и брикетирования. Высушенные гранулы пропускают через сито 18-20 меш для обеспечения надлежащего смешивания их со вспомогательными веществами. Массовое отношение воды (предпочтительно очищенной воды или воды для инъекции) к твердым материалам, предпочтительно находится в области от приблизительно 25% до приблизительно 80%. На гранулы может быть нанесено пленочное покрытие. Композиции данного изобретения могут использоваться также для получения гранулированных препаратов, гранул для дисперсии или капсул, последние, например, наполняют порошком или указанным выше гранулами. Также из указанных порошков и гранул могут быть получены путем прессования таблетированные формы, на которые также при необходимости может быть нанесена пленочная оболочка.
Пленочное покрытие таблетки при использовании водного пленкообразующего состава желательно осуществлять при температуре ядер таблеток от 30 до 50°C, температуре на входе от 50 до 80°C и относительной влажности менее 50%.
Для обеспечения высокой стабильностью при хранении лекарственного препарата важно, чтобы лекарственная форма с пленочным покрытием была высушена до влагосодержания не более 4% и предпочтительно не более 3%.
Следующие примеры иллюстрируют настоящее изобретение и не ограничивают его объем. Количество ингредиентов фармацевтической композиции, использованных в примерах, представлено в мас. %.
Пример №1.
Получение состава 1. Предварительно все ингредиенты просеиваются. Смешиваются 5-этокси-2-[2-(морфолино)-этилтио] бензимидазола дигидрохлорид (15,0) с наполнителем (лактоза) (11,5); гидрофильным модификатором высвобождения (гидроксипропилметилцеллюлоза Methocel® К4М) (12,5), замедлителем высвобождения: смесь поливинилацетата и повидона (Kollidon SR) (60,0) до получения однородной смеси. Затем таблеточная масса поступает на стадию опудривания смазывающим веществом (магния стеарат) (1,0) и таблетируется. Модельные таблетки поступают на стадию нанесения оболочки Opadry 85 F (Opadray II 85 F (18422) в состав которой входят спирт поливиниловый частично гидролизованный, титана диоксид, макрогол 4000, тальк (3,0% от массы таблетки).
Пример №2.
Получение состава 2. Предварительно все ингредиенты просеиваются. Смешиваются 5-этокси-2-[2-(морфолино)-этилтио] бензимидазола дигидрохлорид (16,5) с наполнителем (кальция карбонат основной) (14,5); гидрофильным модификатором высвобождения (гидроксипропилцеллюлоза) (15,5), связывающим веществом (гидроксипропилметилцеллюлоза) (2,0) и замедлителем высвобождения: смесь повидона и поливинилацетата (50,5) до получения однородной смеси. Затем таблеточная масса поступает на стадию опудривания смазывающим веществом (магния стеарат) (1,0) и таблетируется.
Пример №3.
Получение состава 3. Предварительно все ингредиенты просеиваются. Смешиваются 5-этокси-2-[2-(морфолино)-этилтио] бензимидазола дигидрохлорид (22,0) с наполнителем (маннит) (11,0); гидрофильным модификатором высвобождения (этилцеллюлоза) (13,3), связывающим веществом (оксипропилметилцеллюлоза) (0,7) и замедлителем высвобождения КПН (52,0) до получения однородной смеси. Затем таблеточная масса поступает на стадию опудривания смазывающим веществом (магния стеарат) (1,0) и таблетируется.
Пример №4.
Получение состава 4. Все ингредиенты просеиваются. Смешиваются часть наполнителя (лактоза) (12,0) и гидрофильный модификатор высвобождения (кальция гидрофосфат дигидрат - 11,2) до получения однородной смеси. Полученную порошковую смесь просеивают для достижения однородности смешивания и подвергают влажному гранулированию. В качестве связывающего используется раствор поливинилового спирта (5,0) с растворенным в нем 5-этокси-2-[2-(морфолино)-этилтио] бензимидазола дигидрохлоридом (10,8). Гранулят высушивают при температуре от 30 до 40°C до постоянной влажности, не превышающей 3%. Высушенный гранулят смешивают с оставшейся частью наполнителя - алюмометосиликат магния (Neusilin US2) (7,0) и замедлителем высвобождения КПН (53,0). Затем таблеточная масса поступает на стадию опудривания смазывающим веществом (1,0) и таблетируется.
Пример №5.
Получение состава 5. Все ингредиенты просеиваются. Смешиваются часть наполнителя (натриевая соль карбоксиметилцеллюлозы) (8,0) и гидрофильный модификатор высвобождения - эфир целлюлозы (Methocel® К100 LV) (23,0) или гидроксипропилметилцеллюлоза с такими же молекулярными массами и значением вязкости водного раствора до получения однородной смеси. Полученную порошковую смесь просеивают для достижения однородности смешивания и подвергают влажному гранулированию. В качестве связывающего используется раствор поливинилового спирта (1,0) с растворенным в нем 5-этокси-2-[2-(морфолино)-этилтио] бензимидазола дигидрохлоридом (5,0). Гранулят высушивают при температуре 30-40°C до постоянной влажности, не превышающей 3%. Высушенный гранулят смешивают с оставшейся частью наполнителя - алюмометосиликат магния (Neusilin US2) (7,0) и замедлителем высвобождения (КПН) (55,0). Затем таблеточная масса поступает на стадию опудривания смазывающим веществом (1,0) и таблетируется. Полученная таблетка покрывается пленочной оболочкой из гидроксипропилцеллюлозы (3,0 от массы таблетки).
Пример №6.
Получение состава 6. Предварительно все ингредиенты просеиваются. Смешиваются 5-этокси-2-[2-(морфолино)-этилтио] бензимидазола дигидрохлорид (7,5) с наполнителем (лактоза) (11,5); гидрофильным модификатором высвобождения - гидроксипропилметилцеллюлоза (Methocel® К4М) (10,0), замедлителем высвобождения - смесь поливинилацетата и повидона (Kollidon SR) (70,0) до получения однородной смеси. Затем таблеточная масса поступает на стадию опудривания смазывающим веществом (магния стеарат) (1,0) и таблетируется. Модельные таблетки поступают на стадию нанесения оболочки Opadry 85 F (3,0% от массы таблетки).
Пример №7.
Получение состава 7. Предварительно все ингредиенты просеиваются. Смешиваются 5-этокси-2-[2-(морфолино)-этилтио] бензимидазола дигидрохлорид (9,5) с наполнителем (лактоза) (9,0); гидрофильным модификатором высвобождения: гидроксипропилметилцеллюлоза (Methocel® К4М) (30,0), замедлителем высвобождения (Kollidon SR) (50,5) до получения однородной смеси. Затем таблеточная масса поступает на стадию опудривания смазывающим веществом (магния стеарат) (1,0) и таблетируется. Модельные таблетки поступают на стадию нанесения оболочки Opadry 85 F (3,0% от массы таблетки).
Пример №8.
Получение состава 8. Предварительно все ингредиенты просеиваются. Смешиваются 5-этокси-2-[2-(морфолино)-этилтио] бензимидазола дигидрохлорид (30,0) с наполнителем (лактоза) (5,0); гидрофильным модификатором высвобождения: гидроксипропилметилцеллюлоза (Methocel® К4М) (13,0), замедлителем высвобождения (Kollidon SR) (51,0) до получения однородной смеси. Затем таблеточная масса поступает на стадию опудривания смазывающим веществом (магния стеарат) (1,0) и таблетируется. Модельные таблетки поступают на стадию нанесения оболочки Opadry 85 F (3,0% от массы таблетки).
Для фармацевтических композиций по приведенным примерам были проведены испытания модельных смесей и таблеток (Табл. 1).
Как видно из приведенных в таблице 1 данных, все композиции характеризовались отсутствием флуктуаций (пиков) в кинетике высвобождения, что желательно для лекарственных форм с пролонгированным высвобождением. При этом все составы показали высокий уровень высвобождения терапевтического агента из модельных таблеток. Полученные данные позволяют сделать вывод о рациональном подборе компонентов фармацевтической композиции, что демонстрируется хорошим технологическим показателями в сочетании с оптимальной для лекарственных средств с низкими показателями биодоступности кинетикой высвобождения.
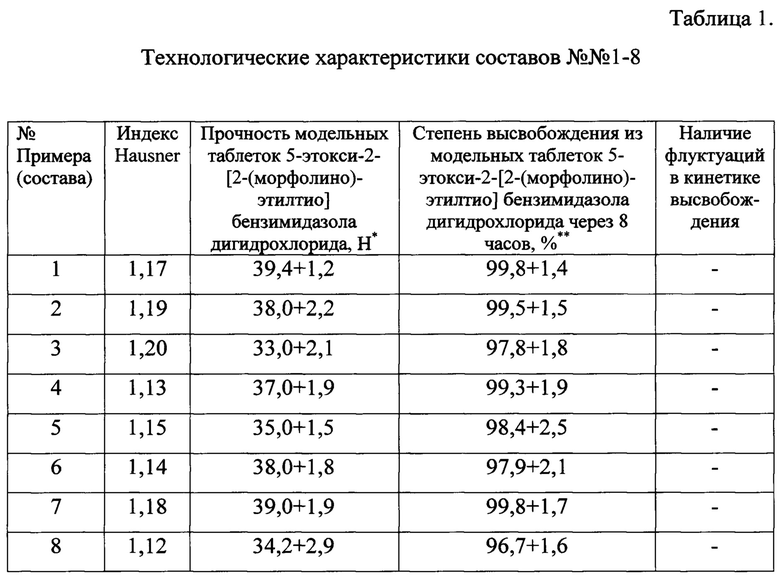
* - прочность таблеток оценивалась по ГФ XIII, ОФС 1.4.2.0011.15 для таблеток диаметром 8 мм;
** - степень высвобождения оценивалась по тесту «Растворение», проводившемуся согласно ГФ XIII, ОФС 1.4.2.0014.15.
Модификация высвобождения 5-этокси-2-[2-(морфолино)-этилтио] бензимидазола дигидрохлорида из предлагаемой фармацевтической композиции, установленная в результате фармакокинетических и биофармацевтических исследований, обуславливает пролонгирование действия активного вещества, показанное в исследованиях выраженности и продолжительности фармакодинамической активности, что проиллюстрировано следующими примерами: сравнительные фармакодинамические и фармакокинетические исследования изготовленных согласно приведенной в изобретении рецептуре и технологии твердых лекарственных форм и таблетированной формы Афобазола, выпускаемой промышленностью.
Объектами исследования являлись таблетированные лекарственные формы 5-этокси-2-[2-(морфолино)-этилтио] бензимидазола дигидрохлорида (Афобазола): таблетки - Т-1, выпускаемые промышленностью с немодифицированным высвобождением действующего вещества, и таблетки Т-2, приготовленные согласно предложенному изобретению (состав 1), с модифицированным высвобождением действующего вещества.
Пример №9.
Сравнительное фармакокинетическое исследование фармацевтических композиций на основе 5-этокси-2-[2-(морфолино)-этилтио] бензимидазола дигидрохлорида (Афобазола). Фармакокинетику Афобазола изучали после перорального введения таблеток беспородным кроликам-самцам (2,0-3,1 кг). Кроликов содержали в стандартных условиях вивария ФГБНУ "НИИ фармакологии имени В.В. Закусова" при 12-ти часовом световом режиме и стандартном рационе (комбикорм, вода). За 12 часов до начала эксперимента животных лишали пищи, оставляя свободный доступ к воде. Исследования выполняли согласно «Правилам лабораторной практики» (Приказ Минздравсоцразвития Российской Федерации №708н от 23 августа 2010 г.).
Животным в случайном порядке вводили попеременно таблетки Т-1 и Т-2 в пересчете на дозу Афобазола 40 мг/кг. Во всех случаях использовали перекрестную рандомизированную схему введения. Интервал времени между введением таблеток Афобазола разных модификаций составлял 2 недели. Фармакокинетические кривые 5-этокси-2-[2-(морфолино)-этилтио] бензимидазола гидрохлорида после введения кроликам таблеток Т-1 и Т-2 представлены на фиг. 1.
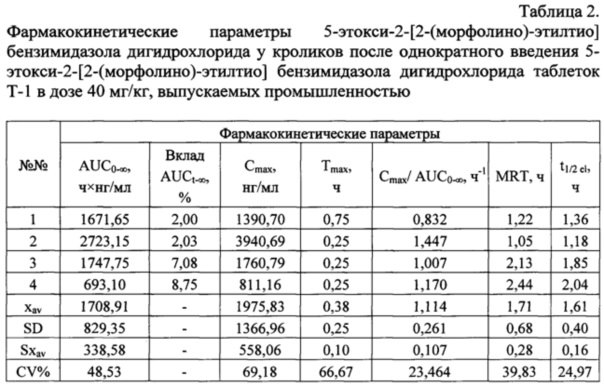
После введения кроликам таблеток 5-этокси-2-[2-(морфолино)-этилтио] бензимидазола дигидрохлорида - Т-2, действующее вещество определяется в плазме крови животных в течение 24 ч. В случае введения таблеток Т-1 - в течение 8 ч. Фармакокинетические параметры, рассчитанные после введения кроликам соединения Т-1 представлены в таблице 2, а после введения соединения Т-2 - в таблице 3.
Используемые в таблице 2 и 3 сокращения:
AUC(0→∞) (нг×ч/мл) - площадь под фармакокинетической кривой «концентрация лекарственного вещества - время»; AUC0→∞ - рассчитывается от момента введения до бесконечности;
Tmax (ч) - время достижения максимальной концентрации лекарственного вещества в плазме крови;
Cmax (нг/мл) - максимальная концентрация лекарственного вещества в плазме крови;
T1/2 el (ч) - период полувыведения - период, за который выводится половина введенной и всосавшейся дозы лекарственного вещества; в таблице;
MRT (ч) - среднее время удержания лекарственного вещества в организме;
Cmax/AUC(0→∞) (ч-1) - параметр, характеризующий скорость всасывания препарата в системный кровоток;
AUC (нг×ч/мл) - площадь под кривой зависимости «концентрация/время».
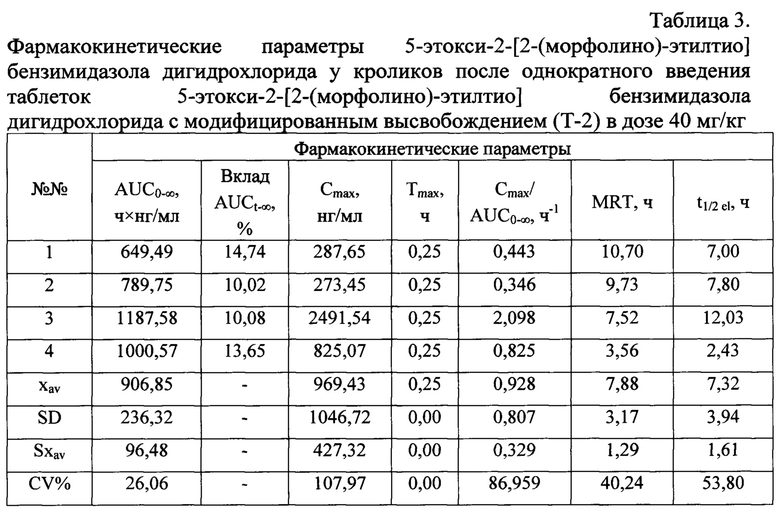
Несмотря на то, что Cmax 5-этокси-2-[2-(морфолино)-этилтио] бензимидазола гидрохлорида после введения животным Т-1 - 1975,83±558,06 нг/мл в 2,0 раза выше величины Cmax, полученной после введения Т-2 - 969,43±427,32 нг/мл, при сравнении максимальных концентраций достоверные различия не были выявлены (фиг. 2б). При сравнении значений Cmax/AUC0→∞ видно, что в случае таблеток Т-1 и Т-2 Афобазол всасывался в системный кровоток примерно с одинаковой скоростью (1,114±0,107 и 0,928±0,329 ч-1; фиг. 2в).
Среднее значение Tmax для таблеток Т-2 составило 0,25 ч. Тот же параметр для таблеток Т-1 в среднем составил 0,38±0,10 ч. Различия Tmax для сравниваемых таблетированных форм статистически не значимы (фиг. 2г).
Таким образом, сравнение таких фармакокинетических параметров, как Cmax/AUC0→∞ и Tmax для таблеток 5-этокси-2-[2-(морфолино)-этилтио] бензимидазола дигидрохлорида, изготовленных по разным технологиям, показало, что Афобазол одинаковое время всасывался из таблеток Т-1 и Т-2. Величина t1/2el для таблеток Т-2 выросла в 4,5 раза и составила в среднем 7,32±1,61 ч (фиг. 2д). Различия значений приведенных параметров являются статистически значимыми при выбранном уровне достоверности.
Значение MRT для таблеток Т-2 также возросло и составило в среднем 7,88±1,29 ч, в то время как аналогичный параметр для таблеток Т-1 составил лишь 1,71±0,28 ч (фиг. 2е). Различия значений приведенных параметров являются статистически значимыми.
Значения относительной биодоступности (F) 5-этокси-2-[2-(морфолино)-этилтио] бензимидазола дигидрохлорида из таблеток Т-2 представлены в таблице 4.

Таким образом, относительная биодоступность 5-этокси-2-[2-(морфолино)-этилтио] бензимидазола дигидрохлорида из таблеток Т-2 составила 70,04±26,12%.
Сравнение таких фармакокинетических параметров, как t1/2el и MRT для таблеток 5-этокси-2-[2-(морфолино)-этилтио] бензимидазола дигидрохлорида, разных модификаций, показало, что афобазол более длительное время определялся в плазме крови животных и медленнее выводился из организма в случае использования таблеток с модифицированным высвобождением (Т-2), что, в совокупности с повышением показателей биодоступности благодаря сочетанию быстрого и поддерживаемого пролонгированного высвобождения при сопоставлении с немодифицированной формой (таблетками, выпускаемыми промышленностью (Т-1)) оптимизирует фармакокинетический профиль препарата.
Пример №10.
10.1. Для подтверждения заявленных преимуществ предлагаемой композиции, состоящих в увеличении продолжительности фармакологического эффекта при однократном применении лекарственных форм было проведено изучение фармацевтических композиций на основе 5-этокси-2-[2-(морфолино)-этилтио] бензимидазола дигидрохлорида в таблетированной лекарственной форме с немодифицированным высвобождением (выпускаемые промышленностью) - Т-1, и модифицированным высвобождением, изготовленные согласно предложенному изобретению (состав №1) - Т-2, на поведение самцов крыс линии Вистар в тесте "приподнятый крестообразный лабиринт" (ПКЛ).
Метод ПКЛ является общепринятым для оценки анксиолитического действия [Pellow S, Chopin Р, File SE & Briley M. Validation of open: closed arm entries in an elevated plus-maze as a measure of anxiety in the rat. J. Neurosci. Methods. 1985. №14, p. 149-167; Waif AA and Frye CA. The use of the elevated plus maze as an assay of anxiety-related behavior in rodents. Nature Protocols. 2007. №2, P. 322-328]. Сущность метода заключается в анализе соотношения реакции страха животных в незнакомом пространстве и высоты с одной стороны и поисковой активности в новой обстановке с другой.
Установка ПКЛ представляет собой две взаимопересекающиеся под прямым углом горизонтальные дорожки 100×14 см. Два противоположных отсека имеют непрозрачные вертикальные стенки высотой 30 см. Лабиринт приподнят от пола на 55 см. В месте перекрестья плоскостей находится открытая центральная платформа размером 14×14 см. Эксперимент проводился в условиях дневного освещения. В начале тестирования животное помещалось в центр лабиринта носом к открытому рукаву, что давало возможность переместиться в темные, либо светлые рукава лабиринта - в зависимости от преобладания тревоги (боязнь высоты, открытого пространства) или исследовательской активности (что побуждало животное выходить из «защищенных» рукавов). Фиксировали следующие показатели поведения в ПКЛ в течение 300 сек: время нахождения в открытых рукавах, время нахождения в закрытых рукавах, время нахождения на центральной площадке, число заходов в открытые рукава, число заходов в закрытые рукава.
В соответствии с характерными для лабораторных крыс формами поведения реакция страха определяла стремление животных находиться в закрытых рукавах лабиринта, снижение двигательной активности. Поисковая (анксиолитическая) активность определяет пребывание животных в открытых рукавах, увеличение двигательной активности.
В исследовании использованы крысы-самцы линии Вистар, масса которых на момент эксперимента составляла 320-340 г. Животные были адаптированы в лаборатории в течение 7 дней до начала исследования. Животные содержались в контролируемых условиях окружающей среды (20-22°C и 30-70% относительная влажность, 10-ти кратная смены объема воздуха комнаты в час), световом цикле - 12 часов светлый и 12 часов темный периоды, в пластмассовых клетках с верхней крышкой из нержавеющей стали с подстилкой, обеспыленной из деревянной стружки, по 10 крыс в каждой клетке, при постоянном доступе к корму и воде при использовании полного рациона экструдированного брикетированного корма (ГОСТ на корм Р 50258-92). Содержание животных соответствовало правилам лабораторной практики (GLP) и нормативным документам - «Санитарные правила по устройству, оборудованию и содержанию вивариев», утвержденным Главным Государственным санитарным врачом 06.04.1973 г. №1045-73 и Приказом Министерства здравоохранения и социального развития Российской Федерации от 23 августа 2010 г. №708н «Об утверждении Правил лабораторной практики».
За 5 суток до эксперимента, животных распределяли по группам рандомизированно, используя в качестве критерия массу тела, так, чтобы индивидуальное значение массы не отклонялось от среднего значения в пределах всех групп более чем на ±3%. Число животных в каждой группе составляло 8 крыс. Тестируемые соединения вводились однократно интрагастрально через зонд в виде готовых таблетированных лекарственных форм. Диаметр таблетки составлял 3 мм, средняя масса - 0,02 г. В таблетках 5-этокси-2-[2-(морфолино)-этилтио] бензимидазола гидрохлорида немодифицированного высвобождения с составом, идентичным выпускаемым промышленностью - Т-1, и в таблетках, выполненных согласно предлагаемой фармацевтической композиции (состав 1) - Т-2, содержалось по 3 мг 5-этокси-2-[2-(морфолино)-этилтио] бензимидазола дигидрохлорида (Афобазола). Доза 5-этокси-2-[2-(морфолино)-этилтио] бензимидазола дигидрохлорида (Афобазола) для каждого животного рассчитанная в соответствии с массой составляла 10,0±0,3 мг/кг. В состав таблеток Т-0 (группа «Плацебо») входили только вспомогательные вещества.
Статистическую обработку полученных результатов проводили, используя однофакторный дисперсионный анализ (Anova, критерий Краскела-Уоллиса) и непараметрический анализ для независимых переменных (U-критерий Манна-Уитни). Данные в таблицах представлены в виде М (SD), где М-среднее арифметическое, SD-стандартное отклонение.
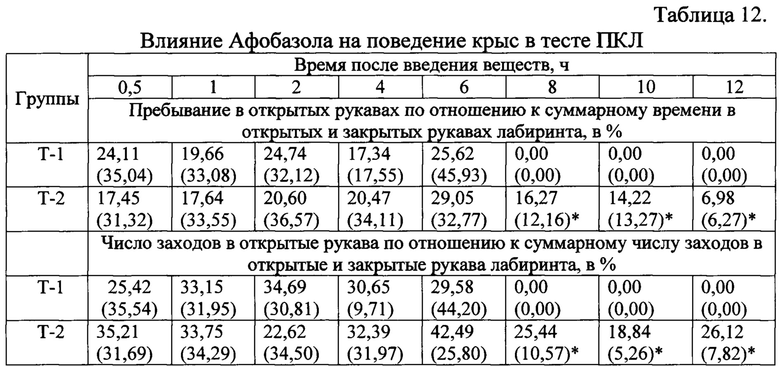
Анализ поведения крыс Вистар в ПКЛ через час после введения таблетированных лекарственных форм показал наличие выраженного анксиолитического эффекта у животных группы «Афобазол». Значимо увеличилось время пребывания и число заходов в открытые рукава лабиринта по сравнению с группой «Плацебо (Т-0)» и «Т-2» (Табл. 5). У животных входящих в группу «Т-2» отмечалось значимое по сравнению с группой «Плацебо» снижение времени пребывания в закрытых рукавах лабиринта и увеличение времени нахождения на центральной площадке, усиление двигательной активности, что можно интерпретировать как активацию исследовательского поведения и снижение уровня тревожности.
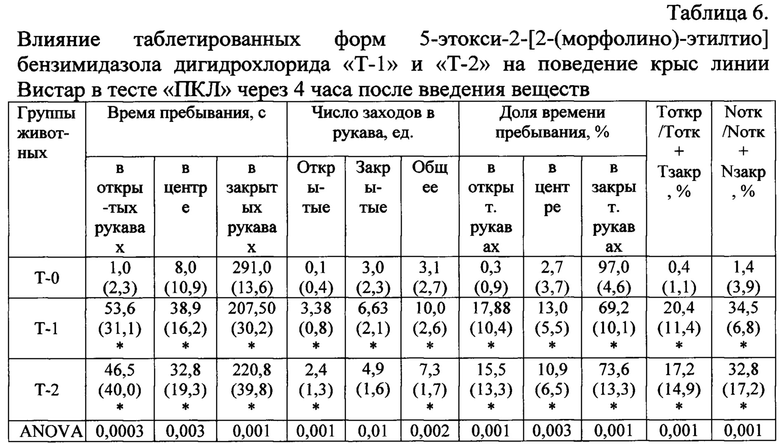
Результаты статистического анализа полученных данных через 4 часа после введения таблетированных лекарственных форм 5-этокси-2-[2-(морфолино)-этилтио] бензимидазола дигидрохлорида демонстрируют выраженное влияние лекарственных форм «Т-1» и «Т-2» на все измеряемые показатели поведения крыс: общее число заходов в рукава лабиринта, число заходов и время нахождения в открытых рукавах (Табл. 6). Значимо уменьшилось время пребывания крыс в закрытых рукавах лабиринта. Достоверное изменение изученных параметров в сравнении с группой «Плацебо» носит характер анксиолитического действия, сопровождающегося активацией исследовательского поведения.
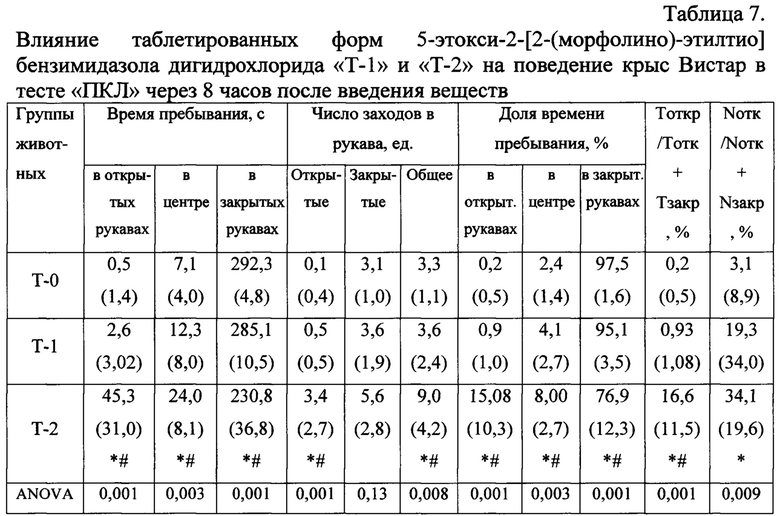
Данные поведения крыс в ПКЛ через 8 часов после введения свидетельствуют о наличии анксиолитического действия только у таблетированной лекарственной формы «Т-2» в сравнении с группами, получавшими «Плацебо» и «Афобазол». Значимо увеличилось время пребывания в открытых рукавах лабиринта, более чем в 15 раз вырос процент пребывания в открытых рукавах по отношению к суммарному времени в открытых и закрытых рукавах, что считается наиболее адекватным критерием анксиолитического действия (Табл. 7).
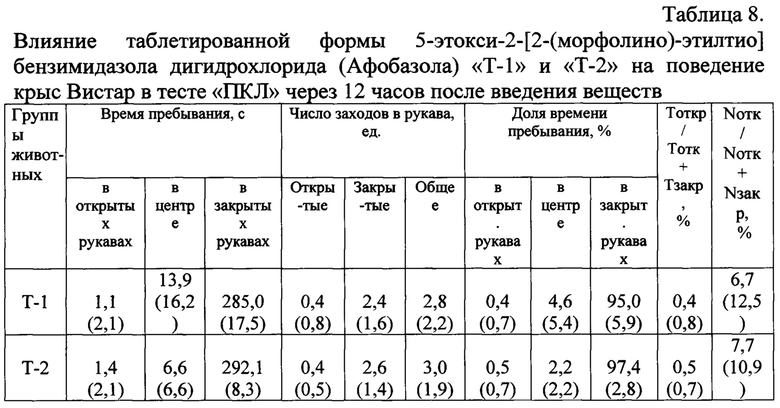
Поведение крыс в ПКЛ через 12 часов после введения таблеток «Т-2» не изменилось по сравнению с животным получившими «Плацебо» (Табл. 8).
Таким образом, на основании анализа результатов экспериментов по изучению транквилизирующей (анксиолитической) активности у соединений установлено наличие анксиолитического действия у таблетированной лекарственной формы Афобазола, выпускаемой промышленностью - Т1 во временном диапазоне: 1 час - 4 часа, при этом у модифицированной таблетированной лекарственной формы Афобазола - «Т-2», полученной согласно изобретению, продолжительность терапевтического действия отмечалась в диапазоне: 1 час - 8 часов после введения, что подтверждает увеличение продолжительности фармакодинамической активности при использовании фармацевтической композиции с модифицированным высвобождением, включающим в себя стадию быстрого создания терапевтической концентрации действующего вещества в крови и стадию поддержания данной концентрации путем замедленного высвобождения терапевтического агента.
Используемые сокращения в таблицах 5-8:
Р (Anova) - функция однофакторного дисперсионного анализа.
Тоткр / Тотк + Тзакр, % - длительность нахождения в открытых рукавах = 100 × (время в открытых рукавах) / (время в открытых рукавах + время в закрытых рукавах), %;
Nотк / Nотк + Nзакр, % - число заходов в открытые рукава = 100 × (число заходов в открытые рукава) / (число заходов в открытые рукава + число заходов в закрытые рукава), %.
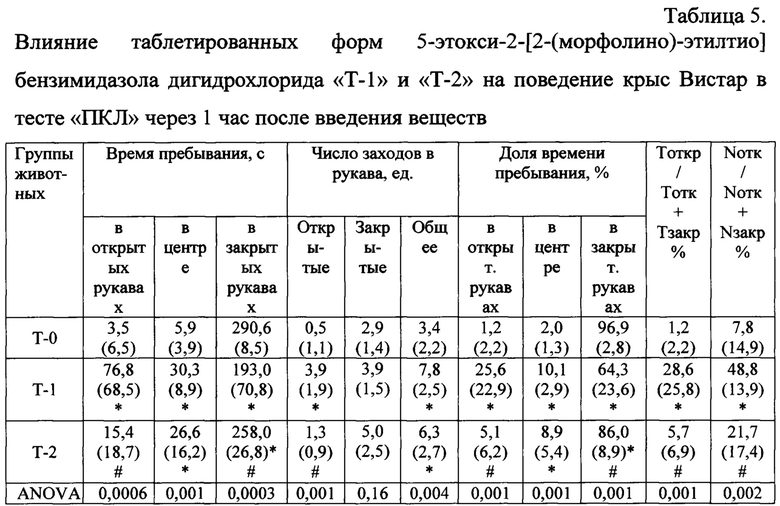
Примечания: * - статистически значимые различия (p<0,05) по сравнению с группой «Плацебо» по критерию Манна-Уитни;
# - статистически значимые различия (p<0,05) по сравнению с группой «Афобазол - Т1» по критерию Манна-Уитни.
Примечания: * - статистически значимые различия (p<0,05) по сравнению с группой «Плацебо» согласно критерию Манна-Уитни.
Примечания: * - статистически значимые различия (p<0,05) по сравнению с группой «Плацебо» по критерию Манна-Уитни; # - статистически значимые различия (p<0,05) по сравнению с группой «Афобазол - Т1» по критерию Манна-Уитни.
10.2. Детальное изучение продолжительности специфического действия новой фармацевтической композиции на основе 5-этокси-2-[2-(морфолино)-этилтио] бензимидазола дигидрохлорида (Т-2) в сравнении с Афобазолом, выпускаемым промышленностью (Т-1), на поведение крыс линии Вистар в тесте ПКЛ через 30 минут, 1, 2, 4, 6, 8, 10 и 12 часов после однократного интрагастрального введения веществ в дозе 10 мг/кг.
Анализ поведения крыс линии Вистар в ПКЛ через 30 минут, 1, 2 и 4 часа после введения таблеток Афобазола: Т-1 и Т-2 в дозе 10,0 мг/кг интрагастрально (внутрь) показал наличие выраженного анксиолитического эффекта у животных обеих групп. На фоне Т-1 и Т-2 значимо увеличилось время пребывания и число заходов в открытые рукава, сократилось время пребывания в закрытых рукавах лабиринта по сравнению с группой «Плацебо» - Т-0 (Таблица 9). Различий между поведением крыс в ПКЛ, получавших Афобазол, выпускаемый промышленностью (Т1) и Афобазол с модифицированным высвобождением (Т-2), через 30 минут, 1, 2 и 4 часа после введения препаратов не выявлено.
Данные поведения крыс в ПКЛ через 6 часов после введения Афобазола (Т-1) и Афобазол с модифицированным высвобождением (Т-2) в дозе 10,0 мг/кг свидетельствуют о наличии анксиолитического действия только у Т-2: на фоне его введения отмечалось достоверное увеличение продолжительности нахождения крыс в открытых рукавах лабиринта и числа заходов в них по сравнению с группой «Плацебо» - Т-0 (Таблица 9).
 Примечания к таблице 9: количество животных в каждой группе-8; Тоткр / Тотк + Тзакр, %, - 100 × (время в открытых рукавах) / (время в открытых рукавах + время в закрытых рукавах); Nотк / Nотк + Nзакр, % - 100 × (число заходов в открытые рукава) / (число заходов в открытые рукава + число заходов в закрытые рукава); данные представлены в виде M(SD), где М - среднее арифметическое, SD - стандартное отклонение; *, **, *** - p<0.05, 0,01 или 0,001 - статистически значимые различия по сравнению с группой «Плацебо» согласно критерию Манна-Уитни; #, ##, ### - p<0.05, 0,01, 0,001 - статистически значимые различия по сравнению с группой «Афобазол» согласно критерию Манна-Уитни.
Примечания к таблице 9: количество животных в каждой группе-8; Тоткр / Тотк + Тзакр, %, - 100 × (время в открытых рукавах) / (время в открытых рукавах + время в закрытых рукавах); Nотк / Nотк + Nзакр, % - 100 × (число заходов в открытые рукава) / (число заходов в открытые рукава + число заходов в закрытые рукава); данные представлены в виде M(SD), где М - среднее арифметическое, SD - стандартное отклонение; *, **, *** - p<0.05, 0,01 или 0,001 - статистически значимые различия по сравнению с группой «Плацебо» согласно критерию Манна-Уитни; #, ##, ### - p<0.05, 0,01, 0,001 - статистически значимые различия по сравнению с группой «Афобазол» согласно критерию Манна-Уитни.
Через 8, 10 и 12 часов после введения Афобазола - 5-этокси-2-[2-(морфолино)-этилтио] бензимидазола дигидрохлорида с модифицированным высвобождением, выявлено выраженное активирующее влияние препарата на поведение крыс в ПКЛ: увеличилось число заходов в рукава лабиринта, число заходов и время нахождения в открытых рукавах лабиринта и интегральные показатели по сравнению с группой «Плацебо» и группой «Афобазол» (Таблица 9).
Таким образом, установлено наличие анксиолитического действия у новой таблетированной лекарственной формы Афобазола с модифицированным высвобождением - Т-2 во временном диапазоне от 30 минут до 12 часов после введения, а у таблетированной лекарственной формы Афобазола, выпускаемой промышленностью «Афобазол» - в диапазоне от 30 мин до 4 часов после введения.
10.3 Сопоставление фармакодинамических и фармакокинетических данных для оценки продолжительности эффекта исследуемых лекарственных форм Афобазола Т-1 и Т-2.
Изучение фармакокинетики Афобазола в плазме крови крыс проводили после однократного интрагастрального введения таблеток с пролонгированным высвобождением (Т-2) и таблеток Афобазола с обычным высвобождением (Т-1) в дозе 10 мг/кг.
Фармакокинетические параметры исследуемого соединения в плазме крови животных после однократного интрагастрального введения анализируемых лекарственных форм представлены в таблице 10.
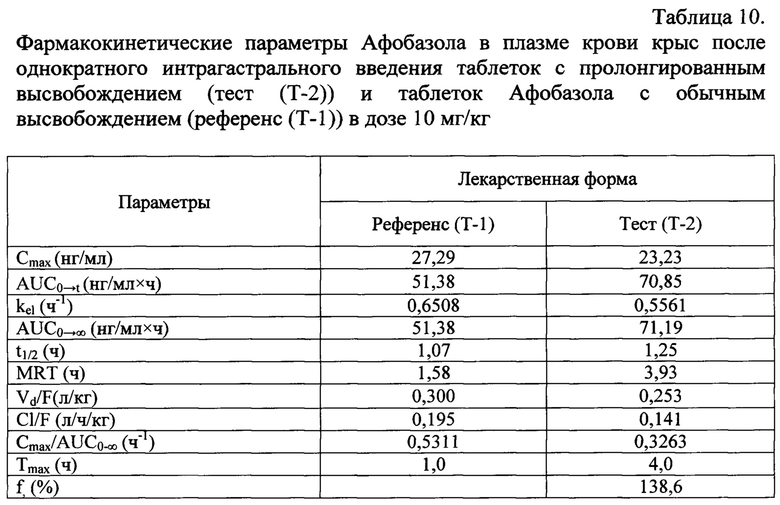
Модельно-независимым методом рассчитаны такие фармакокинетические параметры, как максимальная концентрация Афобазола в плазме крови (Cmax), время ее достижения (Tmax), константа скорости элиминации (kel), площадь под кривой «концентрация Афобазола в плазме крови - время» (AUC), период полувыведения (t1/2) Афобазола из плазмы крови, среднее время удерживания (MRT) Афобазола в организме, кажущийся клиренс (Cl/F), кажущийся объем распределения (Vd/F), относительная биодоступность (f%).
Сравнительный анализ основных фармакокинетических параметров 5-этокси-2-[2-(морфолино)-этилтио] бензимидазола дигидрохлорида (Афобазола) в плазме крови крыс (таблица 10) показал, что изучаемое соединение всасывается из ЖКТ в 1,6 раз быстрее, чем из таблеток Афобазола с обычным высвобождением (Референс). Величина Cmax/AUC0→∞ для данной лекарственной формы составила 0,5311 ч-1 по сравнению с введением таблеток Афобазола с пролонгированным высвобождением (Тест), составившее 0,3263 ч-1. Время достижения максимальной концентрации (Tmax) Афобазола в плазме крови после введения Референс-препарата составило 1,0 ч, а величина максимальной концентрации (Cmax) - 27,29 нг/мл. В то же время, Tmax для Тест-препарата наступало в 4 раза медленнее и составило 4,0 ч, а величина Cmax - 23,23 нг/мл, Следует отметить, что усредненные значения Cmax Афобазола после введения Тест- и Референс-препарата были близки.
Площадь под фармакокинетической кривой Афобазола (AUC0→∞) после введения Тест-препарата (Т-2) составила 71,19 нг/мл×ч, что больше в 1,4 раза значения AUC0→∞. Референс-препарата (51,38 нг/мл×ч). Относительная биодоступность (f%) таблеток Афобазола с пролонгированным высвобождением по отношению к таблеткам Афобазола с обычным высвобождением составила 138,6%.
Изменение степени биодоступности Афобазола можно объяснить влиянием вспомогательных веществ, входящих в состав новой лекарственной формы Тест-препарата, которое заключается в модификации структур мембран клеток кишечника и, как следствие, изменении скорости и степени всасывания лекарств из ЖКТ [Goole J, et, al, // Int, J, Pharm, 2010, Vol, 393, №, 1, P, 17-31; Vilar G, et, al, // Curr, Drug Deliver, 2012, Vol, 9, №, 4, P, 367-394].
Анализ фармакокинетических параметров позволяет заключить, что Афобазол быстрее выводится из организма крыс, после введения Афобазола в виде таблеток с обычным высвобождением (Т-1) в сравнении с таблетками Афобазола с пролонгированным высвобождением (Т-2), на что указывают значения констант скорости элиминации из плазмы крови (kel), которые составили 0,6508 ч-1 и 0,5561 ч-1, Среднее время удерживания (MRT) Афобазола после введения Тест-препарата (Т-2), увеличилось в 2,5 раза в сравнении с введением Референс-препарата (Т-1) - 1,58 ч и составило 3,93 ч. Период полувыведения анксиолитика из организма (t1/2 el) после введения Тест-препарата составил 1,25 ч и после введения Референс-препарата - 1,07 ч, соответственно.
Количественную оценку элиминируемого вещества дает кажущийся клиренс (CL/F) - объем крови, очищаемой от препарата за единицу времени. Значение CL/F Афобазола у крыс после введения Тест-препарата в 1,4 раза меньше аналогичного параметра, полученного после введения Референс-препарата (таблица 10). Таким образом, Афобазол более длительное время пребывает в организме крыс в случае введения таблеток с пролонгированным высвобождением. Величины кажущихся объемов распределения (Vd/F) Афобазола после введения Тест- и Референс-препарата были близки и составили 2,53 и 3,00 л/кг. Необходимо отметить, что рассчитанные значения Vd/F Афобазола (таблица 3) превышают реальный объем жидкости в организме крысы, который составляет 0,67 л/кг [Davies В,, Morris Т, // Pharm, Res, 1993, Vol, 10, №, 7, Р, 1093-1095]. Высокие значения кажущихся объемов распределения свидетельствуют о том, что Афобазол активно проникает в биологические жидкости и ткани экспериментальных животных.
10.4 Сопоставление фармакодинамических и фармакокинетических показателей таблеток Афобазола с пролонгированным (Т-2) и обычным высвобождением (Т-1)
С целью выявления возможных корреляционных зависимостей (степени выраженности связи) между фармакодинамическими и фармакокинетическими показателями проведено сопоставление поведенческих характеристик и концентраций Афобазола в плазме крови крыс после однократного интрагастрального введения таблеток Афобазола с пролонгированным (Тест-препарат (Т-2)) и обычным высвобождением (Референс-препарат (Т-1)).
Концентрации Афобазола в плазме крови крыс после введения исследуемых препаратов измеряли в дискретные временные интервалы: 0,5, 1, 2, 4, 6, 8, 10, 12 ч (Таблица 11). Оценку поведенческих показателей проводили в тесте «ПКЛ» в те же временные интервалы. В частности, оценивали пребывание (в %) животных в открытых рукавах по отношению к суммарному времени в открытых и закрытых рукавах лабиринта и число заходов (в %) в открытые рукава по отношению к суммарному числу заходов в открытые и закрытые рукава лабиринта (Таблица 12).
Показано, что между концентрациями Афобазола в плазме крови крыс и эффектом пребыванием крыс в открытых рукавах по отношению к суммарному времени в открытых и закрытых рукавах лабиринта и числом заходов в открытые рукава по отношению к суммарному числу заходов в открытые и закрытые рукава лабиринта после однократного введения Тест- и Референс-препаратов существуют причинно-следственные связи (корреляционные зависимости). Так, после введения Референс-препарата (Т-1) коэффициенты корреляции Пирсона линейного регрессионного уравнения равнялись 0,8374 и 0,8327, а после введения Тест-препарата (Т-2) - 0,6333 и 0,2913. Поскольку рассчитанные значения коэффициентов корреляции r>0, это означает, что с увеличением концентрации Афобазола в плазме крови крыс его эффекты усиливаются. По мере снижения концентрации Афобазола в плазме крови наступает момент, когда создавшийся уровень Афобазола в плазме крови не вызывает анксиолитический эффект. По-видимому, концентрации Афобазола в плазме крови крыс выше 0,19 нг/мл обеспечивают анксиолитический эффект. Более низкие значения коэффициента корреляции в случае введения Тест-препарата (Т-2) можно объяснить отклонениями от кинетики первого порядка на фазе всасывания Афобазола, что отразилось, в частности, на значении Tmax, которое в среднем составило 4 ч, тогда как для Референс-препарата (Т-1) этот параметр равнялся 1,0 ч.

Примечания: количество животных в каждой группе 8; данные представлены в виде M(SD), где М - среднее арифметическое, SD - стандартное отклонение; * - p<0.05 статистически значимые различия по сравнению с группой «Афобазол» согласно критерию Манна-Уитни.
Примечание: n=8; данные представлены в виде M(SD), где М - среднее арифметическое, SD - стандартное отклонение; * - p<0.05 статистически значимые различия по сравнению с группой «Афобазол» согласно критерию Манна-Уитни.
Таким образом, Афобазол в новой лекарственной форме более длительное время определяется в плазме крови и медленнее выводится из организма животных в сравнении с таблетками Афобазола, выпускаемыми промышленностью. Между концентрациями Афобазола в плазме крови крыс и выраженностью анксиолитического эффекта выявлена корреляционная зависимость.
Пример 11.
Изучение влияния пролонгированной лекарственной формы Афобазола (Т-2) на поведение крыс линии Вистар в тесте конфликтной ситуации или наказуемого взятия воды по Vogel в сравнении с Афобазолом с немодифицированным высвобождением (Т-1)
Конфликтная ситуация, создаваемая у грызунов путем подавления болевым электрическим раздражителем питьевого рефлекса при потреблении ими воды из трубки-поилки основывается на столкновении двух мотиваций - питьевой и оборонительной (страха наказания при попытке удовлетворения питьевой потребности) [Vogel JR, Beer В, Clody DE // А simple and reliable conflict procedure for testing anti-anxiety agents, Psychopharmacologia (Berl.), 1971, V. 21, С, 1-7].
Для моделирования тревожного состояния по методу Vogel у крыс использовали установку фирмы Panlab HARVARD apparatus (Испания), которая представляет собой бокс размером 50×23×14 см с электродным полом и поилкой, расположенной на боковой стенке бокса на высоте 5 см от пола, шокера (LE 100-25 Vogel Shoker), соединенного с поилкой. Регистрация числа наказуемых взятий воды (ЧНВВ) происходит автоматически и обрабатывается программным обеспечением PACWIN.
Перед тестированием крыс лишают воды на 48 часов, не ограничивая доступа к пище. Затем вырабатывают навык взятия воды из поилки, помещая крысу в бокс на 10 минут. Затем крысу возвращают в домашнюю клетку, где она получает только корм. Через 24 часа крысу помещают в бокс установки и каждое взятие воды сопровождается электроболевым раздражением, подаваемым на поилку (сила тока 0,5 мА, продолжительность 0,2 секунды). Регистрируют ЧНВВ в течение 10-ти минут. Критерием наличия анксиолитического эффекта считают увеличение ЧНВВ относительно показателей животных без терапии.
Препараты (Т-1 и Т-2) и плацебо (Т-0) вводили в таблетированной лекарственной форме однократно перорально (внутрижелудочно) за 1, 4, 6, 8 и 10 часов до теста.
Установлено, что в контрольных группах животных, получавших плацебо (Т-0) за 1, 4, 6, 8 и 10 часов до предъявления теста, ЧНВВ составляло от 27 до 44, что свидетельствует о развитии у животных анксиогенного состояния. При этом латентное время первого наказуемого взятия воды было вариабельным и находилось в диапазоне от 26 до 130 секунд на группу.
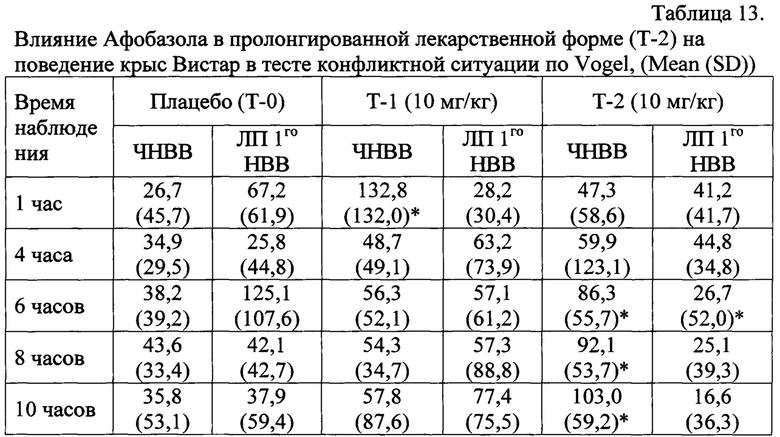
Афобазол (10 мг/кг) в обычной таблетированной лекарственной форме (Т-1) через 1 час после введения в 5 раз статистически достоверно увеличивал ЧНВВ в сравнении с группой «Плацебо» и снижал время первого взятия воды с 67 до 28 секунд. Через 4, 6, 8 и 10 часов после введения Т-1 достоверного влияния на анксиогенное состояние животных не оказывал, что выражалось в отсутствии увеличения ЧНВВ относительно соответствующих значений контрольных животных, получавших плацебо (Таблица 13).
Примечания: число животных в группе: 9-11.
ЧНВВ - число наказуемых взятий воды; ЛП 1го НВВ - латентный период до первого наказуемого взятия воды; * p≤0,05 - статистически значимые различия по сравнению с группой «Плацебо» согласно критерию Манна-Уитни.
Афобазол в пролонгированной таблетированной лекарственной форме (Т-2) в дозе 10 мг/кг увеличивал ЧНВВ во всех точках наблюдения, что было особенно выражено через 6-10 часов после его введения. Так, через 6 часов после введения Т-2 отмечалось увеличение в 2,2 раза ЧНВВ (p<0,05) и уменьшение в 4,7 раза латентного времени первого подхода к «опасной» поилке (p<0,05). Через 8 часов после введения Т-2 выявлено увеличение в 2,1 раза ЧНВВ (p<0,05) и снижение в 1,7 раза ЛП подхода к «опасной» поилке (Таблица 13). Через 10 часов после введения Афобазола в пролонгированной форме (Т-2) у препарата продолжала сохраняться высокая анксиолитическая активность, регистрируемая по ЧНВВ, превосходящих в 2,9 раза контрольные значения (Таблица 13).
Таким образом, Афобазол (10 мг/кг) при однократном пероральном введении в обычной таблетированной лекарственной форме (Т-1) достоверно увеличивает ЧНВВ, являющихся мерой анксиолитического эффекта, только через 1 час после его введения и не оказывает достоверного эффекта при тестировании через 4-10 часов после введения. Анксиолитический эффект таблетированной пролонгированной лекарственной формы Афобазола (Т-2) в дозе 10 мг/кг статистически значимо проявляется через 6 часов после однократного перорального введения и сохраняется на протяжении 10 и более часов.
Пример 12.
Изучение влияния Афобазола в пролонгированной лекарственной форме (Т-2) на поведение крыс линии Вистар в тесте «социальное взаимодействие» с оценкой продолжительности эффекта
Процедура: крысы были поделены на 2 группы по 8 особей в каждой. Крысы первой группы (резиденты) перед тестированием проходят 5-и дневную изоляцию в индивидуальных клетках со свободным доступом к корму и воде. Вторая группа крыс (интрудеры) не подвергается социальной изоляции и находится в клетках группового содержания. Эксперимент по взаимодействию резидент-интрудер проводится на нейтральной арене. В течение 10 мин регистрируют два показателя поведения: количество контактов (обнюхиваний) и их продолжительность. Анксиолитики, введенные интрудеру, увеличивают число и продолжительность контактов.
Препараты: Афобазол с пролонгированным высвобождением (Т-2) в дозе 10 мг/кг, Афобазол с обычным высвобождением (Т-1) в дозе 10 мг/кг и плацебо (Т-0) вводили в таблетированной лекарственной форме однократно перорально (внутрижелудочно) за 1, 4, 6, 8 и 10 часов до теста.
Оценка влияния Афобазола: Т-1 и Т-2 на поведение крыс линии Вистар в тесте «Социальное взаимодействие» показала, что время контактов крыс после введения таблеток Афобазола, выпускаемых промышленностью - Т-1 статистически значимо увеличилось по сравнению с группой «Плацебо» (Т-0) при введении препарата за 1-4 часа до тестирования (Таблица 14).

Примечание: Данные представлены в виде M(SD), где М - среднее арифметическое, SD - стандартное отклонение;
* p≤0,05 - статистически значимые различия по сравнению с группой «Плацебо» согласно критерию Манна-Уитни.
# - p≤0,05 - статистически значимые различия по сравнению с группой «Т-2» согласно критерию Манна-Уитни.
Увеличение времени контактов при использовании Афобазола с модифицированным высвобождением (Т-2) в дозе 10 мг/кг происходило на протяжении от 1 до 10 часов после введения, по сравнению с группой «Плацебо» (Т-0). Кроме того, На фоне введения Т-2 отмечалось межгрупповые различия с группой Т-1 на протяжении всех интервалов наблюдения (Табл. 14).
Количество контактов через 1-8 часов после введения Т-2 также было больше по сравнению с группой Т-0.
Таким образом, для Афобазола с модифицированным высвобождением (Т-2) в дозе 10 мг/кг установлена более выраженная и длительная (до 10 часов) анксиолитическая активность в тесте «Социальное взаимодействие» в сравнении с Афобазолом в дозе 10 мг/кг, продолжительность действия которого наблюдалась на протяжении 4 часов.
Оценка анксиолитического действия новой лекарственной формы Афобазола - 5-этокси-2-[2-(морфолино)-этилтио] бензимидазола дигидрохлорида с пролонгированным высвобождением активного фармацевтического ингредиента в сравнении с лекарственной формой Афобазола, выпускаемой промышленностью, проведена с использованием трех методик с определением продолжительности основного эффекта: тест ПКЛ, наказуемое поведение в тесте конфликтная ситуация по Vogel и в тесте «социальное взаимодействие». Проведено сопоставление фармакодинамических и фармакокинетических данных для оценки продолжительности эффекта исследуемых лекарственных форм Афобазола.
В тесте конфликтная ситуация, анксиолитический эффект таблетированной пролонгированной лекарственной формы Афобазола 5-этокси-2-[2-(морфолино)-этилтио] бензимидазола дигидрохлорида (Т-2) в дозе 10 мг/кг сохраняется на протяжении 10 часов после введения препарата, тогда как на фоне таблеток Афобазола в дозе 10 мг/кг, выпускаемых промышленностью (Т-1), специфическое действие препарата наблюдалось только через 1 час после его введения.
В тесте «Социальное взаимодействие» для Афобазола с модифицированным высвобождением в дозе 10,0 мг/кг установлена более выраженная и длительная (до 10 часов) анксиолитическая активность, проявляющаяся с первых часов после введения, в сравнении с таблетками Афобазола, выпускаемыми промышленностью, в аналогичной дозе (продолжительность эффекта - 4 часа).
Анксиолитические свойства Афобазола были установлены в тесте «приподнятого крестообразного лабиринта», для новой защищаемой таблетированной лекарственной формы Афобазола с модифицированным высвобождением в диапазоне 30 минут - 12 часов, а у таблетированной лекарственной формы Афобазола, выпускаемой промышленностью «Афобазол» 30 мин - 4 часа после введения.
Сравнительное фармакокинетическое исследование показало, что Афобазол - 5-этокси-2-[2-(морфолино)-этилтио] бензимидазола дигидрохлорида (Афобазола) в новой лекарственной форме, содержащей комбинацию модификаторов высвобождения, значительно дольше определяется в плазме крови и медленнее выводится из организма животных в сравнении с таблетками Афобазола, выпускаемыми промышленностью, что свидетельствует об эффекте пролонгирования новой лекарственной формы. Между концентрацией Афобазола в плазме крови крыс и выраженностью анксиолитического эффекта в тесте «ПКЛ» выявлена корреляционная зависимость, что подтверждает связь между продолжительностью терапевтического действия препарата и поддержания определенной концентрации действующего вещества (5-этокси-2-[2-(морфолино)-этилтио] бензимидазола дигидрохлорид) в крови в течение длительного времени (до 12 часов), что достигается при использовании заявляемой фармацевтической композиции 5-этокси-2-[2-(морфолино)-этилтио] бензимидазола дигидрохлорида (Афобазола).
Описание чертежей
Фиг. 1 Усредненные фармакокинетические кривые 5-этокси-2-[2-(морфолино)-этилтио] бензимидазола гидрохлорида в плазме крови кроликов после однократного введения таблеток 5-этокси-2-[2-(морфолино)-этилтио] бензимидазола гидрохлорида дигидрохлорида, приготовленных по различным технологиям (n=4; xav, dSxav).
По оси абсцисс отложено t - время (ч).
По оси ординат отложены значения натурального логарифма концентрации 5-этокси-2-[2-(морфолино)-этилтио] бензимидазола гидрохлорида в плазме крови кроликов в размерности нг/мл.
Фиг. 2 Значения AUC0-∞ (а), Cmax (б), Cmax/AUC0-∞ (в), Tmax (г), t1/2el (д) и MRT (е) 5-этокси-2-[2-(морфолино)-этилтио] бензимидазола гидрохлорида в плазме крови кроликов после однократного введения таблетированных форм (Т-1 и Т-2) 5-этокси-2-[2-(морфолино)-этилтио] бензимидазола гидрохлорида в пересчете на дозу 40 мг/кг (xav, dSxav; n=4; р=0,05) * - достоверные различия в сравнении с Т-1.
По оси абсцисс фиг. 2а-е приведены таблетированные формы.
По оси ординат отложены:
Фиг. 2а - AUC0-∞, 5-этокси-2-[2-(морфолино)-этилтио] бензимидазола гидрохлорида в плазме крови кроликов, нг/мл*ч.
Фиг. 2б - Cmax 5-этокси-2-[2-(морфолино)-этилтио] бензимидазола гидрохлорида в плазме крови кроликов, нг/мл.
ФИГ. 2в - Cmax/AUC0→∞, 1/ч.
Фиг. 2г - Tmax 5-этокси-2-[2-(морфолино)-этилтио] бензимидазола гидрохлорида в плазме крови кроликов, ч.
Фиг. 2д - t1/2el 5-этокси-2-[2-(морфолино)-этилтио] бензимидазола гидрохлорида в плазме крови кроликов, ч.
Фиг. 2е - MRT 5-этокси-2-[2-(морфолино)-этилтио] бензимидазола гидрохлорида в плазме крови кроликов, ч.
| название | год | авторы | номер документа |
|---|---|---|---|
| ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ ФАБОМОТИЗОЛА ДИГИДРОХЛОРИДА | 2022 |
|
RU2828872C2 |
| ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ НА ОСНОВЕ АФОБАЗОЛА | 2004 |
|
RU2289403C2 |
| СРЕДСТВО ДЛЯ СНИЖЕНИЯ ФУНКЦИОНАЛЬНОЙ АКТИВНОСТИ И ЭКСПРЕССИИ ГЛИКОПРОТЕИНА-Р | 2017 |
|
RU2649134C1 |
| СРЕДСТВО ДЛЯ КОРРЕКЦИИ РАССТРОЙСТВ АУТИСТИЧЕСКОГО СПЕКТРА | 2017 |
|
RU2666598C1 |
| ЗАМЕЩЕННЫЕ 2-[2-(3-ОКСОМОРФОЛИН-4-ИЛ)ЭТИЛТИО]БЕНЗИМИДАЗОЛЫ, ОБЛАДАЮЩИЕ АНКСИОЛИТИЧЕСКОЙ АКТИВНОСТЬЮ | 2007 |
|
RU2373202C2 |
| ПРОИЗВОДНЫЕ 2-МЕРКАПТОБЕНЗИМИДАЗОЛА, ОБЛАДАЮЩИЕ СЕЛЕКТИВНОЙ АНКСИОЛИТИЧЕСКОЙ АКТИВНОСТЬЮ | 1994 |
|
RU2061686C1 |
| Фармацевтическая композиция на основе N-бензил-N-метил-1-фенилпирроло [1,2-a] пиразин-3-карбоксамида | 2017 |
|
RU2689396C2 |
| Фармацевтическая композиция на основе N-бутил-N-метил-1-фенилпирроло[1,2-а]пиразин-3-карбоксамида | 2023 |
|
RU2811453C1 |
| СРЕДСТВО ДЛЯ КУПИРОВАНИЯ АБСТИНЕНТНОГО СИНДРОМА ПРИ ЗАВИСИМОСТИ ОТ ОПИАТОВ | 2012 |
|
RU2485954C1 |
| СРЕДСТВО ДЛЯ ЛЕЧЕНИЯ ИШЕМИЧЕСКИХ ПОРАЖЕНИЙ МОЗГА | 2004 |
|
RU2288714C2 |


Изобретение относится к медицине, в частности к фармацевтической композиции, обладающей анксиолитическим действием и содержащей в качестве лекарственного вещества терапевтически эффективное количество Афобазола - 5-этокси-2-[2-(морфолино)-этилтио]бензимидазола дигидрохлорида и/или основания, а в качестве дополнительных фармацевтически приемлемых компонентов, сообщающих лекарственному средству на основе заявляемой композиции новые потребительские качества: комбинацию модификаторов высвобождения, а именно замедлитель высвобождения и гидрофильный модификатор высвобождения, при определенном их количественном соотношении, а также группы веществ, обеспечивающих достаточную массу лекарственной формы, связывающие вещества, скользящие/смазывающие вещества, и пленочную оболочку. Лекарственные формы на основе Афобазола могут быть представлены как в виде таблеток, так и капсул, гранул, порошка, обеспечивающих продолжительность действия лекарственного средства не менее 8 часов. Осуществление изобретения обеспечивает получение фармацевтической композиции, обладающей пролонгированным высвобождением активного фармацевтического ингредиента, что позволяет стабилизировать концентрацию препарата в организме человека в течение суток и за счет этого уменьшить кратность приема препарата в течение суток для достижения необходимого терапевтического эффекта. 5 з.п. ф-лы, 14 табл., 2 ил.
1. Фармацевтическая композиция анксиолитического действия, содержащая 5-этокси-2-[2-(морфолино)-этилтио] бензимидазола дигидрохлорид в терапевтически эффективном количестве с замедленным высвобождением действующего вещества, отличающаяся тем, что включает комбинацию модификаторов высвобождения, состоящую из по меньшей мере одного гидрофильного модификатора высвобождения и по меньшей мере одного замедлителя высвобождения при определенном их количественном соотношении, и включающая другие обычно применяемые фармацевтически приемлемые вспомогательные вещества при следующем соотношении компонентов, мас. %:
где замедлителем высвобождения является смесь поливинилацетата и повидона;
где гидрофильным модификатором высвобождения является
гидроксипропилметилцеллюлоза.
2. Фармацевтическая композиция по п. 1, в которой в качестве смеси поливинилацетата и повидона используется Kollidon SR.
3. Фармацевтическая композиция по п. 1, в которой в качестве гидроксипропилметилцеллюлозы используется Methocel® К4М.
4. Фармацевтическая композиция по п. 1, которая в качестве скользящих/смазывающих может содержать стеариновую кислоту и/или ее соли, аэросил, тальк, гидрированное растительное масло, воск из карнауба, натрия лаурилсульфат; в качестве наполнителя может содержать производные целлюлозы, дисахариды, полисахариды, желатин, а также соли магния и кальция, коллоидный диоксид кремния в качестве целевых примесей к основному наполнителю.
5. Фармацевтическая композиция по п. 1, которая может содержать пленочную оболочку в количестве до 3,0 мас. % от массы таблетки, в которой в качестве пленкообразующего могут быть использованы: полиэтиленгликоль, поливиниловый спирт, гидроксипропилцеллюлоза, гидроксиметилцеллюлоза и гидроксипропилметилцеллюлоза, оксипропилметилцеллюлозы фталат, ацетилцеллюлозы фталат, поливинилацетата фталат, метилцеллюлозы фталат, сополимер метакриловой кислоты или сложные метиловые эфиры метакриловой кислоты, этилцеллюлоза, ацетилцеллюлоза, сополимеры поливинилового спирта и малеинового ангидрида, полиэтиленгликоль, в частности полиэтиленгликоль 6000, триэтилцитрат, диэтилфталат, пропиленгликоль, глицерин, бутилфталат, двуокись титана и красители, окись железа, алюминиевые красители, пленочные покрытия с сополимерами метакриловой кислоты, сополимеры этилакрилата и метилметакрилата.
6. Фармацевтическая композиция по п. 1, выполненная в виде твердых лекарственных форм: твердых желатиновых капсул, таблеток, гранул, порошка.
| ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ НА ОСНОВЕ АФОБАЗОЛА | 2004 |
|
RU2289403C2 |
| ЛЕКАРСТВЕННАЯ ФОРМА С ЗАДЕРЖКОЙ ЛЕКАРСТВ ДЛЯ ЛЕЧЕНИЯ БЕССОННИЦЫ | 2005 |
|
RU2458685C2 |
| Сизяков С.А | |||
| Разработка составов и технологии таблеток афобазола // афтореф | |||
| диссер | |||
| на соиск | |||
| уч.ст | |||
| канд | |||
| фармацевтических наук | |||
| - Москва | |||
| Переносная печь для варки пищи и отопления в окопах, походных помещениях и т.п. | 1921 |
|
SU3A1 |
| Устройство для электрической сигнализации | 1918 |
|
SU16A1 |

