Изобретение относится к химико-фармацевтической промышленности, медицине и ветеринарии и описывает систему доставки 2-этил-6-метил-3-гидроксипиридина сукцината (далее ЭМГПС) для перорального применения в форме таблетки. Изобретение может быть использовано в медицине или ветеринарии в качестве лекарственного средства для перорального применения ЭМГПС для лечения и профилактики различных заболеваний и патологических состояний организма человека или животных.
Лекарственные препараты с ЭМГПС нашли эффективное применение в различных областях клинической медицины: неврологии, психиатрии, кардиологии, офтальмологии, хирургии, стоматологии, эндокринологии и др.
Препараты с ЭМГПС (торговое название - Мексидол® и др.) широко применяются в качестве антиоксидантного и антигипоксического средства, характеризующегося широким спектром фармакологического действия и высокой эффективностью (ноотропное и транквилизирующее действие - патент RU 2065299, противоишемическое и антиатеросклеротическое действие - патент RU 2144822, антиангинальное - патент RU 2168993, гепатопротекторное - патент RU 2189817, антибактериальное - Пат. RU 2157686 и другие).
ЭМГПС применяют при лечении следующих заболеваний/состояний человека:
- сердечной недостаточности;
- ишемической болезни сердца в составе комплексной терапии;
- острых нарушений мозгового кровообращения;
- последствий острых нарушений мозгового кровообращения, в т.ч. после транзиторных ишемических атак, в фазе субкомпенсации в качестве профилактических курсов;
- легких черепно-мозговых травм, последствий черепно-мозговых травм;
- энцефалопатии различного генеза (дисциркуляторные, дисметаболические, посттравматические, смешанные);
- синдрома вегетативной дистонии;
- легких когнитивных расстройств атеросклеротического генеза;
- тревожных расстройств при невротических и неврозоподобных состояниях;
- купирования абстинентного синдрома при алкоголизме с преобладанием неврозоподобных и вегетативно-сосудистых расстройств, постабстинентных расстройств;
- состояний после острой интоксикации антипсихотическими средствами;
- астенических состояний, а также для профилактики развития соматических заболеваний под воздействием экстремальных факторов и нагрузок;
- воздействия экстремальных (стрессорных) факторов;
и профилактики:
- обострений сердечной недостаточности;
- обострений ишемической болезни сердца.
Известны композиции на основе ЭМГПС в виде различных лекарственных форм: раствора для парентерального введения (Раствор Мексидола® 5% для инъекций Р N002161/01, патенты RU 2205640, 2380089, 2398583), таблеток (Мексидол® таблетки, покрытые оболочкой, 0,125 г ЛСР-002063/07), капсул (патент RU 2144822, 2145855).
Для обеспечения максимальной биодоступности действующего вещества, точной дозировки, а также интенсивной терапии пациентов в случаях необходимости быстрого создания в крови высоких концентраций лекарственного средства наиболее предпочтительны лекарственные формы для инъекционного введения. Вместе с тем общеизвестны ряд недостатков данного пути введения - риск инфицирования, необходимость присутствия квалифицированного персонала, болезненность инъекций и, как следствие, невозможность длительного (более 3-4 недель) лечения парентеральными лекарственными формами.
Кроме того, инъекционная форма не обеспечивает длительного ноотропного эффекта, при этом наблюдается изменение спектра фармакологической активности препарата, что выражается в повышении агрессивности, эмоциональной реактивности, снижении антистрессорного действия [см. описание RU 2065299].
Многогранное применение ЭМГПС требует создание лекарственных форм с различной скоростью всасывания, попадания активного вещества в кровь и поддержания его активной концентрации в крови (от максимально быстрого при экстренных ситуациях до продолжительного при длительном лечении), что напрямую ставит вопрос о необходимости получения системы доставки, обеспечивающей как высокую биодоступность активного вещества, так и создающей возможность поддержания его концентрации в плазме крови, достаточной для длительного терапевтического воздействия. Готовые лекарственные формы, известные до настоящего времени, не могут удовлетворить этот запрос. Поддержание активной концентрации в плазме в течение относительно продолжительного периода времени происходит посредством приема больших доз лекарственных форм или введением их несколько раз в сутки. Такие дозы могут создать токсичное и нежелательно высокое содержание лекарственного средства в организме больного. Введение лекарственного средства с некоторыми временными интервалами приводит к образованию колеблющегося уровня лекарственного средства, так называемого пикового и провального действия, что в свою очередь ведет к неэффективности или неудаче лечения, увеличению риска появления побочных токсических эффектов. Поэтому больным при длительной терапии некоторых заболеваний/состояний желательно иметь относительно постоянную концентрацию введенного лекарственного средства в крови.
Учитывая вышеуказанные обстоятельства, является актуальной разработка лекарственных форм для перорального введения для длительной терапии пациентов в амбулаторных условиях.
Задачей изобретения является разработка системы доставки ЭМГПС для перорального применения в форме таблетки, обеспечивающей повышенную биодоступность с одновременным продолжительным поддержанием необходимой концентрации активного вещества в плазме крови.
Авторами изобретения, описанного в патенте RU 2444359, который может быть указан в качестве ближайшего аналога, была предпринята попытка решение этой задачи. Ими был разработан состав таблеток с ЭМГПС с модифицированным высвобождением. Описанная лекарственная форма представляет собой таблетки, имеющие в своей структуре сфероиды с активным веществом, покрытые оболочкой. В зависимости от состава оболочек были созданы формы с различной растворимостью активного вещества. Для каждой группы полученных сфероидов определяли профиль растворения и на основании полученных данных рассчитывали необходимое соотношение сфероидов в готовой лекарственной форме для достижения требуемого профиля растворения. Профиль растворения получали, используя аппарат 2 фармакопеи США (USP аппарат 2): объем среды растворения 900 мл; скорость вращения мешалки 50 об/мин; температура ванны 37±0,5°С. Среда растворения 0,1 N HCl0- в течение 2 ч, остальное время - фосфатный буфер, рН 6,8.
Однако в описании патента отсутствуют сведения, позволяющие убедиться в решении поставленной задачи. Исследования, представленные в патенте, свидетельствуют лишь об изменении профиля растворения при прохождении лекарственной формы из одной среды растворения (соответствующей рН желудка) в другую (соответствующей рН кишечника). Достижение и поддержание необходимой концентрации в плазме крови, а также степень абсорбции/всасывания активного вещества в представленных исследованиях не изучались.
Лекарственные формы формируются для обеспечения и контроля требуемого высвобождения и растворения действующего вещества. Абсорбции подвергается растворенное вещество, в связи с этим процесс растворения (количество и скорость перехода ЛС в раствор), является одним из наиболее значимых факторов в обеспечении требуемой биодоступности фармакологически активного вещества из готовой лекарственной формы. Однако, помимо растворимости существуют и другие биофармацевтические факторы способные значимо модифицировать конечные свойства таблетированной микросистемы, что в конечном итоге может приводить к существенным различиям в биодоступности (см. Сеткина С.Б. и др. Биофармацевтические аспекты технологии лекарственных средств и пути модификации биодоступности, Вестник ВГМУ, 2014, том 13, №4, с. 162-172). Только при выявлении достоверной in vivo in vitro корреляции можно позволить проводить оценку поведения лекарственного средства in vivo путем изучения его кинетики растворения и, таким образом, оценивать его биодоступность (см. Раменская Г.В и др. In vivo - in vitro корреляция (ivivc): современный инструмент для оценки поведения лекарственных форм в условиях in vivo, Медицинский альманах, 2011, №1 (14) март, с. 222-226). В патенте RU 2444359 отсутствуют сведения о in vivo in vitro корреляции, в связи с этим приведенные исследования растворимости не могут свидетельствовать о повышении биодоступности ЭМГПС.
В результате проведенных нами исследований неожиданно было установлено, что биодоступность может быть повышена в результате создания системы доставки ЭМГПС, способной удерживать активное вещество более 6 часов в желудке. Также было доказано, что биодоступность при использовании лекарственных форм с пролонгированным действием сопоставима с таблетками немедленного высвобождения. Наши исследования позволили опровергнуть мнение специалистов о том, что используя пролонгированную форму с ЭМГПС можно повысить биодоступность. Составы с замедленным (пролонгированным) высвобождением начинают высвобождать ЭМГПС в разных отделах ЖКТ (начиная с желудка и заканчивая прямой кишкой), что не способствует повышению его биодоступности.
Решение задачи контролируемого всасывания активного вещества в кровь для достижения нужной концентрации и ее поддержания и/или изменения во времени достигается вспомогательными веществами, модулирующими/контролирующими высвобождение активного вещества, при этом было установлено, что повышение биодоступности возможно в том случае, если система доставки высвобождает ЭМГПС в желудке длительное время.
Настоящее изобретение обеспечивает новую технологию доставки лекарственного средства (ЭМГПС) с гастроретентивной (гастро-удерживающей) системой доставки.
Преимущества данного способа доставки лекарственного средства (ЭМГПС) по сравнению с аналогичными лекарственными препаратами, включающими ЭМГПС различного способа высвобождения, заключаются в том, что высвобождение происходит только в определенном отделе ЖКТ (большую часть в желудке), где наблюдается наибольшая биодоступность (всасывание) ЭМГПС.
В настоящее время было обнаружено, что ЭМГПС, который хорошо растворим, можно вводить перорально способом, который продлит время его высвобождения, чтобы распределить скорость его всасывания более равномерно на протяжении длительного времени. Это значительно уменьшит проблемы временной передозировки, вызванной начальным скачком концентрации, попадающей в кровоток сразу после введения и последующей недостаточной дозировки, и вместо этого контролирует дозировку до более безопасных и более эффективных уровней в течение длительного периода времени.
Кроме того, было обнаружено, что для ЭМГПС возникают проблемы из-за высвобождения его в нижнем отделе желудочно-кишечного тракта из-за низкой способности абсорбции в кровоток в нижних отделах желудочно-кишечного тракта.
Целью настоящего изобретения является создание системы доставки лекарственного средства, которая остается в желудке в течение, по меньшей мере, 6 часов.
Изобретение относится к гастроретентивной системе доставки ЭМГПС, обеспечивающей длительное время пребывания ее в желудке.
Гастроретентивные системы доставки в виде набухающих в желудке таблеток, а также способы их получения описаны в патентах ЕР 1886665, US 6340475 и международной заявке WO 03035177. Из указанных документов известны полимеры, используемые в гастроретентивных набухающих в желудке таблетках. Удержание в желудке может происходить в результате набухания и/или мукоадгезии системы доставки. Однако ранее не была описана пригодность, а тем более эффективность данных систем доставки для ЭМГПС с улучшением биодоступности.
Подбор полимеров для получения такой системы доставки, находится в области знаний специалиста и зависит от свойств полимеров, их молекулярной массы и от желаемой скорости высвобождения ЭМГПС и продолжительности удержания в желудке. Правильно подобранная комбинация полимеров может обеспечить более контролируемое высвобождение и всасывание ЭМГПС, что приводит не только к повышению биодоступности, но и сокращению количества приемов препарата. Объектом изобретения является система пероральной доставки 2-этил-6-метил-3-гидроксипиридина сукцината в виде гастроретентивной таблетки.
Агент для получения системы доставки ЭМГПС согласно изобретению содержит, по меньшей мере, один полимер, который сочетает в себе контролирующее в желудке набухание, высвобождение ЭМГПС в желудке и мукоадгезивные свойства. Согласно изобретению система может содержать один полимер с такими свойствами или комбинацию полимеров, обеспечивающих необходимые свойства. Примеры полимеров указаны в Таблице 1.
В качестве такого полимера предпочтительно могут быть использованы полиалкиленоксиды (полиэтиленоксид и полипропиленоксид; сополимеры этиленоксида и пропиленоксида). Наиболее предпочтительным является использование полиэтиленоксида (ПЭО). Этот полимер может быть скомбинирован с другими функциональными эксципиентами- полимерами, указанными в таблице 1.
Массовое соотношение 2-этил-6-метил-3-гидроксипиридина сукцината к полиэтиленоксиду предпочтительно составляет от 2,6:1 до 1,3: 1.
Также изобретение относится к удерживаемой в желудке таблетке, в которой матрица представляет собой агент доставки и ЭМГПС в терапевтически эффективном количестве. Таблетка может содержать ЭМГПС в количестве от 125 мг до 1500 мг.

Таблетка, согласно изобретению, необязательно содержит дополнительные фармацевтически приемлемые вспомогательные агенты, чтобы способствовать производству, сжимаемости, внешнему виду и вкусу препарата.
Такие агенты включают, например, разбавители или наполнители, глиданты, связующие агенты, гранулирующие агенты, антиадгезивные агенты, смазки, ароматизаторы, красители и консерванты. Другие обычные эксципиенты, известные в данной области, также могут быть включены.
Наполнитель может быть выбран из растворимых наполнителей, например, сахарозы, лактозы, в частности моногидрата лактозы, трегалоза, мальтоза, маннит и сорбит.
Смазка может быть использована для улучшения высвобождения таблетки из устройства, на котором она сформирована.
Подходящие смазочные материалы включают стеарат магния, стеарат кальция, масло канолы, глицерил пальмитостеарат, гидрогенизированное растительное масло, магний оксид, минеральное масло, полоксамер, полиэтиленгликоль, поливиниловый спирт, бензоат натрия, лаурилсульфат натрия, натрий стеарилфумарат, стеариновая кислота, стеарат цинка и тому подобное.
Состав таблеток может быть разработан с учетом технологических свойств фармацевтической субстанции, а также ее совместимости с различными вспомогательными веществами. Выбор вспомогательных компонентов и технологии производства может быть проведен экспериментальным путем.
Новая лекарственная форма обеспечивает концентрацию активного вещества в крови в диапазоне терапевтической активности, по возможности, без кратковременных пиков Cmax. Лекарственная форма, удерживая ЭМГПС в области абсорбции, показывает улучшение биодоступности благодаря медленной скорости высвобождения. Контролируемое высвобождение имеет ряд явных преимуществ перед обычными составами немедленного высвобождения. Кроме того, лекарственная форма с контролируемым высвобождением снижает максимальную концентрацию в плазме.
Действие активного вещества в желудке (гастроретенция) происходит благодаря набуханию таблетки и ее адгезивной способности (мукоадгезия). В отличие от пролонгированной формы, гастроретентивная таблетка может иметь следующую степень набухания до 243% за 6 часов, а пролонгированная 60% за 6 часов. Пролонгированная форма не обладает мукоадгезивной способностью, тогда как гастроретентивная форма может сохранять эту способность в течение 14 часов.
Также изобретение относится к способу повышения биодоступности ЭМГПС, предусматривающему введение 1 раз в сутки таблетки нуждающемуся в этом субъекту.
Повышение биодоступности и пролонгированное действие позволяет сократить частоту приема ЭМГПС до 1 раза в сутки.
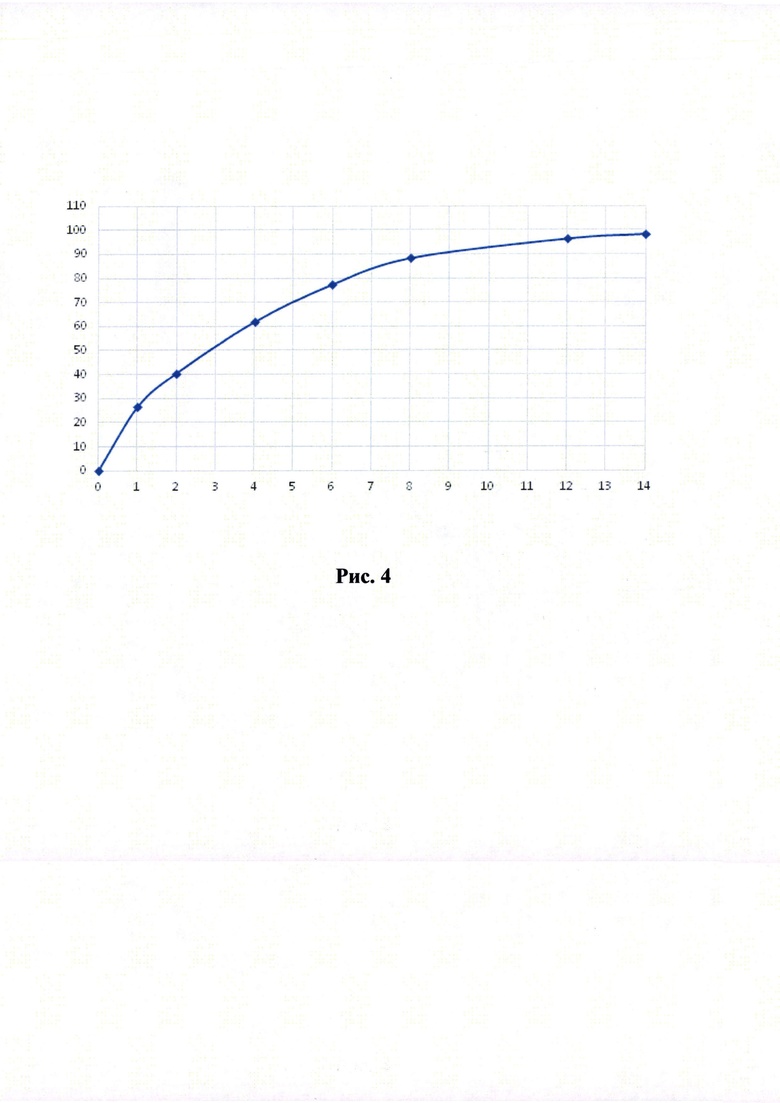
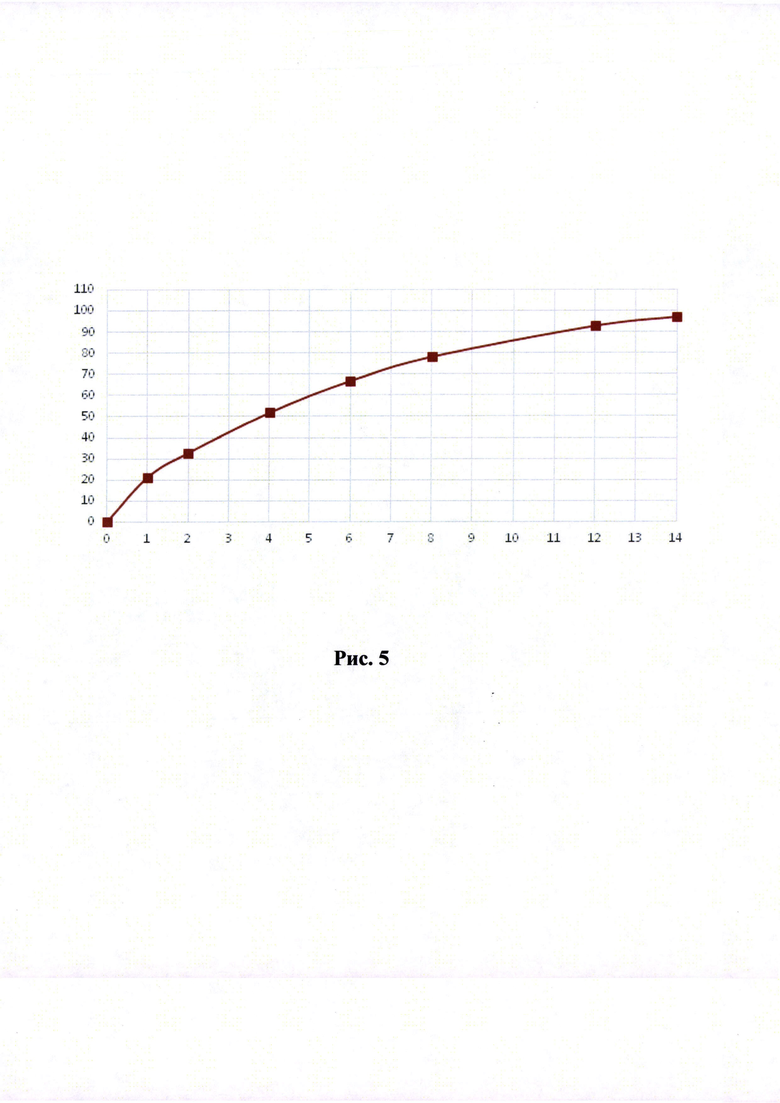
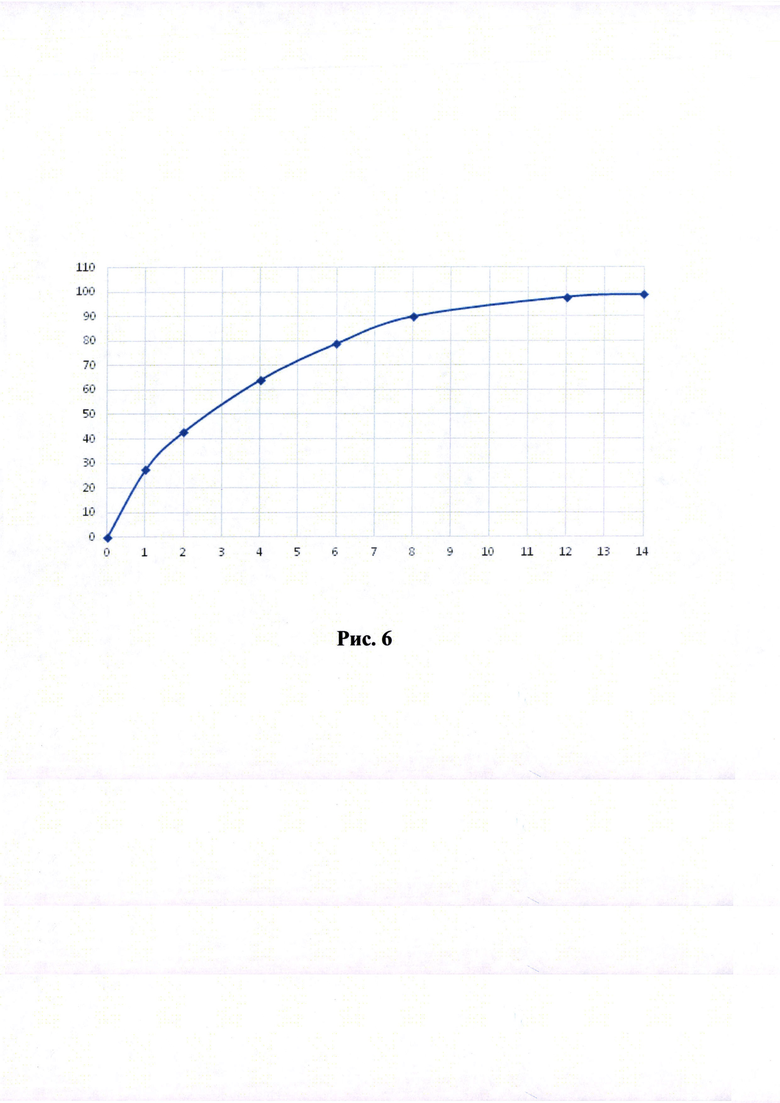
Краткое описание рисунков
Рисунки 1-6. Кривая растворимости ЭМГПС из разных составов (по оси абцисс - время в часах, по оси ординат высвобождение ЭМГПС в %).
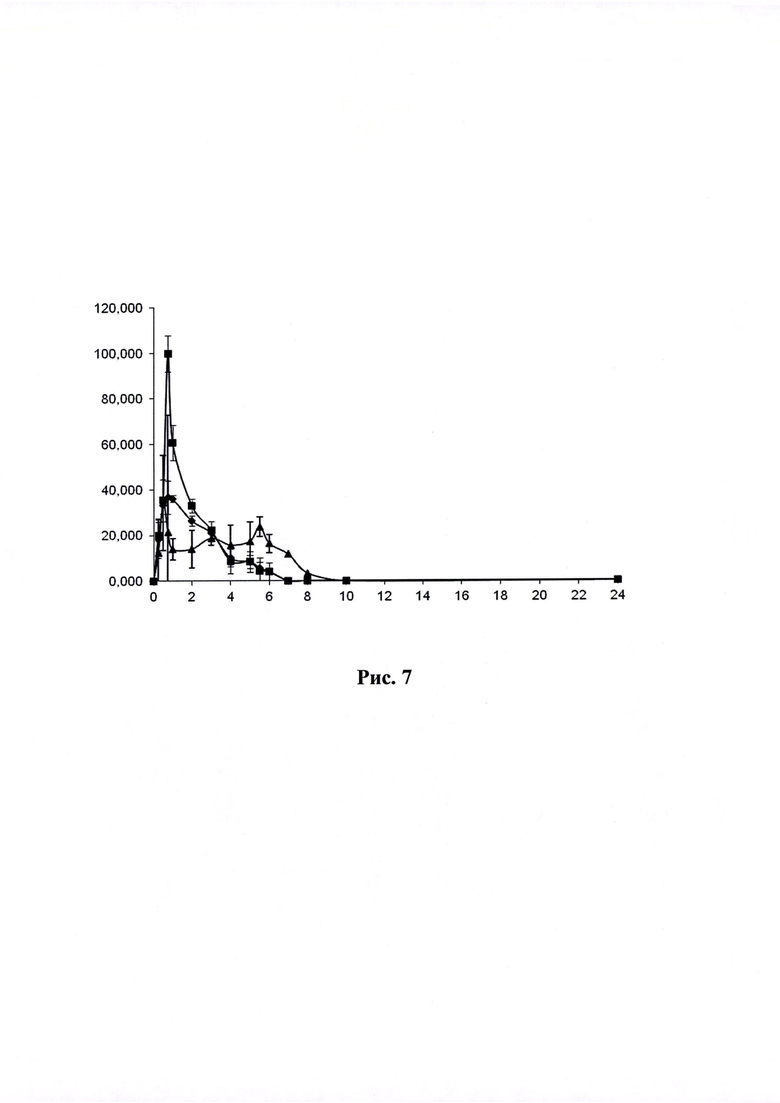
Рисунок 7. Кривая «концентрация-время» (по оси абцисс - время в часах, по оси ординат - концентрация ЭМГПС в нг/мл) после приема ГЛФ ЭМГПС карликовыми свиньями: препарат сравнения - после трехкратного перорального введения  ;
;
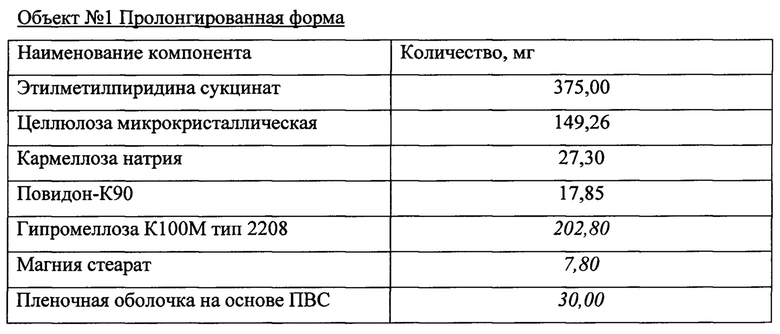
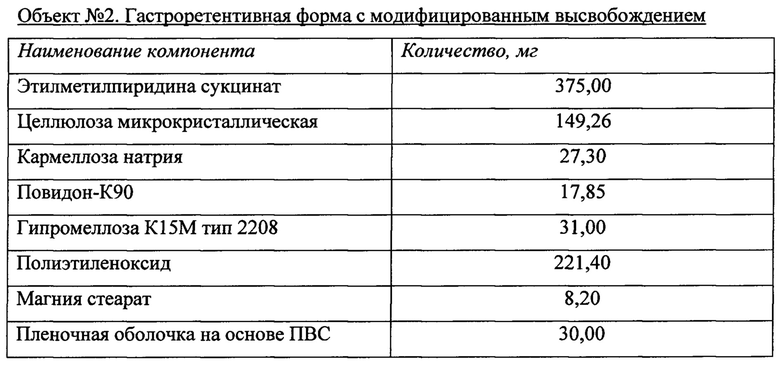
Объект №1, п/о 375 мг  и Объект 2, п/о 375 мг
и Объект 2, п/о 375 мг  после однократного перорального введения,
после однократного перорального введения, 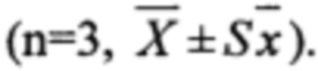
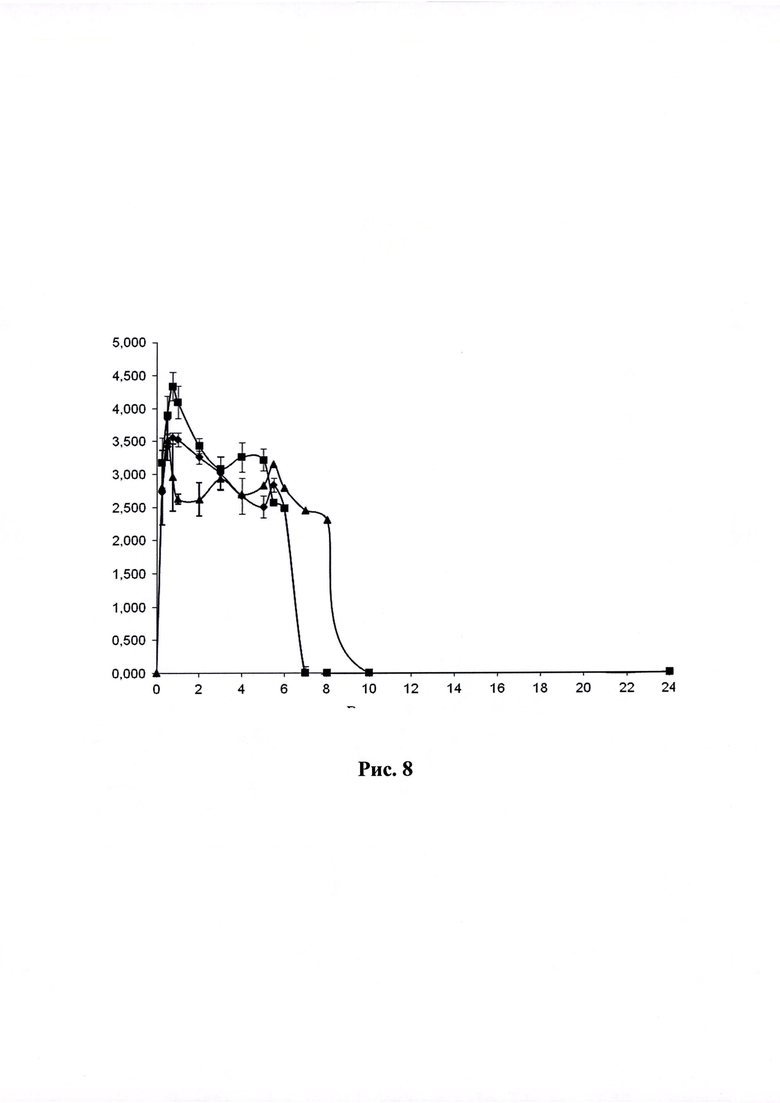
Рисунок 8. Кривая «концентрация-время» (по оси абцисс - время в часах, по оси ординат - Ln концентрации ЭМГПС) после приема ГЛФ ЭМГПС карликовыми свиньями: препарат сравнения - после трехкратного перорального введения ;
Объект №1, п/о 375 мг и Объект 2, п/о 375 мг после однократного перорального введения в полулогарифмических координатах .
Способы получения гастроретентивных таблеток не отличаются от способов получения обычных таблеток. Система доставки и таблетки могут быть получены следующими способами: технология прямого прессования, технология влажной грануляции, технология сухой грануляции и т.д.
Наиболее предпочтительными вспомогательными веществами являются следующие.
Целлюлоза микрокристаллическая
Микрокристаллическая целлюлоза 102. Данная марка целлюлозы микрокристаллической имеет средний размер частиц 130 мкм, что позволяет использовать ее для улучшения сыпучести и прессуемости.
Целлюлоза микрокристаллическая используется, как наполнитель и разбавитель, который улучшает сыпучесть и сцепление между частицами при таблетировании. Целлюлоза микрокристаллическая обеспечивает сохранение целостность формы таблеток при механической нагрузке. Содержание целлюлозы микрокристаллической будет подобрано экспериментальным путем. Содержание целлюлозы микрокристаллической ограничено, вследствие большой геометрии таблеток.
Кармеллоза натрия
Кармеллоза натрия марка CMC 7H4F в концентрации была выбрана на основании рекомендаций компании Ashland для применения в таблетках замедленного высвобождения методом влажной грануляции. Кармеллоза натрия выступает в роли связующего агента при влажном гранулировании. Кармеллоза натрия бладает мукоадгезивными (слизисто-адгезивными) свойствами.
Повидон
Повидон известное и доступное высокоэффективное связующее. В зависимости от концентрации в рецептуре может использоваться: 1) для образования комплексов с субстанцией, с целью улучшения его растворения; 2) для связывания компонентов, с целью их агломерации (получение гранул); 3) для придания ядрам таблеток технологических свойств (твердость и прочность таблеток на истирание).
Повидон является универсальным компонентом и применяется в технологиях производства лекарственных средств методом прямого прессования, сухого гранулирования, влажной грануляции, компактирования. Марка 90 F была выбрана на основании рекомендаций производителя, компании BASF, как связующего агента для влажной грануляции.
Гипромеллоза
Данный компонент внесен в рецептуру лекарственного препарата в качестве связующего агента, с целью придания таблеткам устойчивости к истиранию и сколам для возможности нанесения пленочной оболочки на ядра таблеток, а также в увеличении степени набухания в комбинации с полиэтиленоксидом. Гипромеллоза обеспечивает необходимый профиль растворения.
Полиэтиленоксид
Водорастворимые полимеры POLYOX™ (марки NF) представляют собой неионные поли (этиленоксид) полимеры. Они представляют собой гидрофильные полимеры, доступные в широком диапазоне молекулярных масс, и поставляются в виде белых сыпучих порошков. POLYOX обеспечивает быстрое смачивание и набухание для использования в технологиях осмотического насоса, а также образует гель в гидрофильной матрице. Полиэтиленоксид примененяется как гидрофильная матрица в гастроретентивных лекарственных формах и обладает мукоадгезивными (слизисто-адгезивными) свойствами.
Магния стеарат
Магния стеарат был выбран вследствие большого размера таблетки, который имеет высокий торец таблетки, что может вызывать высокое усилие выталкивания при таблетировании на высокой скорости в промышленном масштабе. Магния стеарат по природе является гидрофобным лубрикантом, используется для предотвращения прилипания смесей к прессе-инструменту.
Пленочная оболочка
В лекарственном препарате предложена оболочка компании Colorcon марки Opadry II на основе поливинилового спирта.
Пленочная оболочка на основе поливинилового спирта быстро и равномерно распределяется по поверхности таблеток, а также снижает риск образования сколов из-за большей адгезии и более низкой вязкости, чем пленочная оболочка на основе гипромеллозы. Также оболочка на основе поливинилового спирта обеспечивает легкое проглатывание таблеток, что актуально для больших таблеток лекарственного препарата.
Для получения таблеток предложены две технологии производства: влажная грануляция и прямое прессование. Использование технологии влажной грануляции позволяет:
• предотвращать расслоение многокомпонентной смеси;
• улучшать сыпучесть смеси;
• обеспечить необходимую насыпную плотность;
• обеспечить точность дозирования.
Фармацевтическая субстанция ЭМГПС обладает неудовлетворительными технологическими свойствами, что осложняет использование технологии прямого прессования.
В ходе фармацевтической разработки были получены лабораторные образцы по технологии прямого прессования. Данная концепция соответствовала показателям качества «Количественное определение» и «Растворение», но имела следующие недостатки:
• недостаточная прочность таблеток-ядер;
• высокая истираемость таблеток-ядер;
• налипание таблеток-ядер на пресс-инструмент в процессе таблетирования.
Использование метода прямого прессования при производстве будет осложнено неудовлетворительным физико-химическими и технологическими свойствами самой субстанции, которые могут оказывать влияние на критические показатели качества лекарственного препарата. Наиболее предпочтительно использовать технологию влажной грануляции.
В частности, один из вариантов гастроретентивных таблеток может быть получен описанным ниже способом.
Описание производственного процесса
Подготовка сырья
Просев сырья
ЭМГПС просеивают вручную через сито 1,0 мм. Остальное сырье просеивают на вибросите CIS A RP 200N через сито 0,5 мм.
Взвешивание сырья
Каждое наименование сырья взвешивают в отдельные чистые емкости.
Приготовление раствора увлажнителя
В емкость с водой очищенной при постоянном перемешивании вносят все количество Повидона К90. Перемешивание проводят до полного растворения (контроль визуальный). Концентрация раствора увлажнителя 8,0%.
Приготовление смеси для таблетирования
Смешивание
В модульную установку с псевдоожиженным слоем загружают ЭМГПС просеянный, МКЦ просеянный, полиэтиленоксид и/или кармеллозу натрия просеянную. Проводят перемешивание компонентов до однородного состояния (контроль визуальный).
Гранулирование
При постоянном перемешивании смеси в псевдоожиженный слой вносят раствор увлажнителя. Проводят перемешивание до образования мелкого рассыпчатого гранулята (контроль визуальный).
Сушка гранулята
Гранулят сушат в сушилке с псевдоожиженным слоем до остаточной влажности не более +0,5% к исходной при температуре 50-55°С.
Калибровка гранулята
Гранулят просеивают на Вибросите CISA RP 200N через сито 1,0 мм. Отсев гранулята протирают вручную через сито 1,0 мм.
Опудривание
В смеситель вносят гранулят, Гипромеллозу 2208 или Полиэтиленоксид и Гипромеллозу 2208. Проводят перемешивание до однородного состояния (контроль визуальный).
В смеситель загружают магний стеарат просеянный и проводят перемешивание компонентов до однородного состояния (контроль визуальный).
Смесь для таблетирования выгружают в маркированную емкость.
Таблетирование
Процесс таблетирования проводят на таблеточном прессе Fasttab 3000. Процесс таблетирования проводят на пресс-инструменте с геометрией 19×8 (18×10) мм.
Проводят настройку таблеточного пресса на производство средней массой.
После настройки средней массы таблеток, проводят настройку таблеточного пресса по остальным показателям:
• твердость;
• истираемость;
• высота;
• отклонения от средней массы таблеток.
После настройки таблеточного пресса проводят процесс таблетирования до полного расходования смесей для таблетирования. В процессе таблетирования, не реже одного раза за 5 минут проводят контроль:
• средней массы таблеток;
• твердость таблеток.
В течение процесса таблетирования таблетки собирают в маркированные емкости.
Покрытие таблеток-ядер оболочкой
Приготовление пленочной суспензии
В процессе нанесения пленочной оболочки используют 18% пленочную суспензию.
Устанавливают перемешивание воды, очищенной так, чтобы образовалась воронка на поверхности воды. Вносят постепенно сухую пленочную оболочку, не допуская образования агломератов на поверхности раствора, затем после снижают скорость перемешивания так, чтобы не происходило пенообразование, и оставляют раствор перемешиваться 45 минут.
Нанесение оболочки
Перед проведением процесса нанесения оболочки нагревают таблетки до 37-39°С при периодическом перемешивании в барабане коатера.
По достижении температуры таблеток начинают распыление пленочной суспензии. В процессе контролируют внешний вид таблеток и привес оболочки.
Процесс нанесения оболочки проводят до получения средней массы таблеток.
При достижении заданной массы таблеток процесс останавливают и охлаждают таблетки до температуры не выше 33°С. После окончания процесса, емкости закрывают.
Таблеточная композиция с гастро-удерживающими свойствами может представлять собой следующие составы:







Состав и соотношение компонентов (а также степень набухания и мукоадгезивность) помогают достичь необходимого профиля высвобождения в желудке (in-vitro тест). Были проведены эксперименты, в которых были получены кривые растворения ЭМГПС следующих составов таблеток:
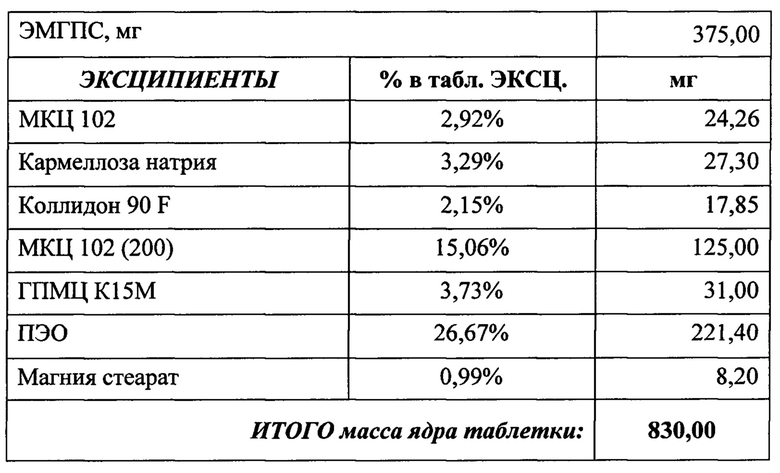

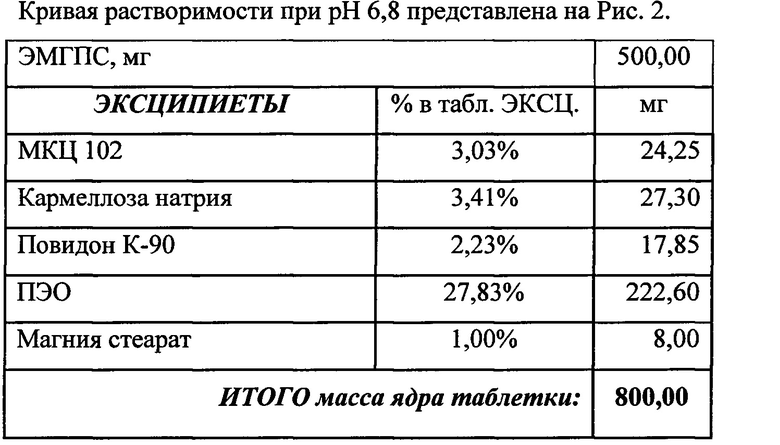
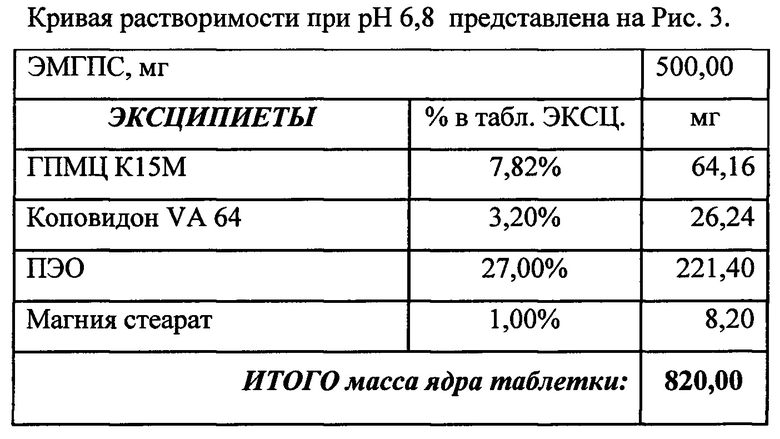
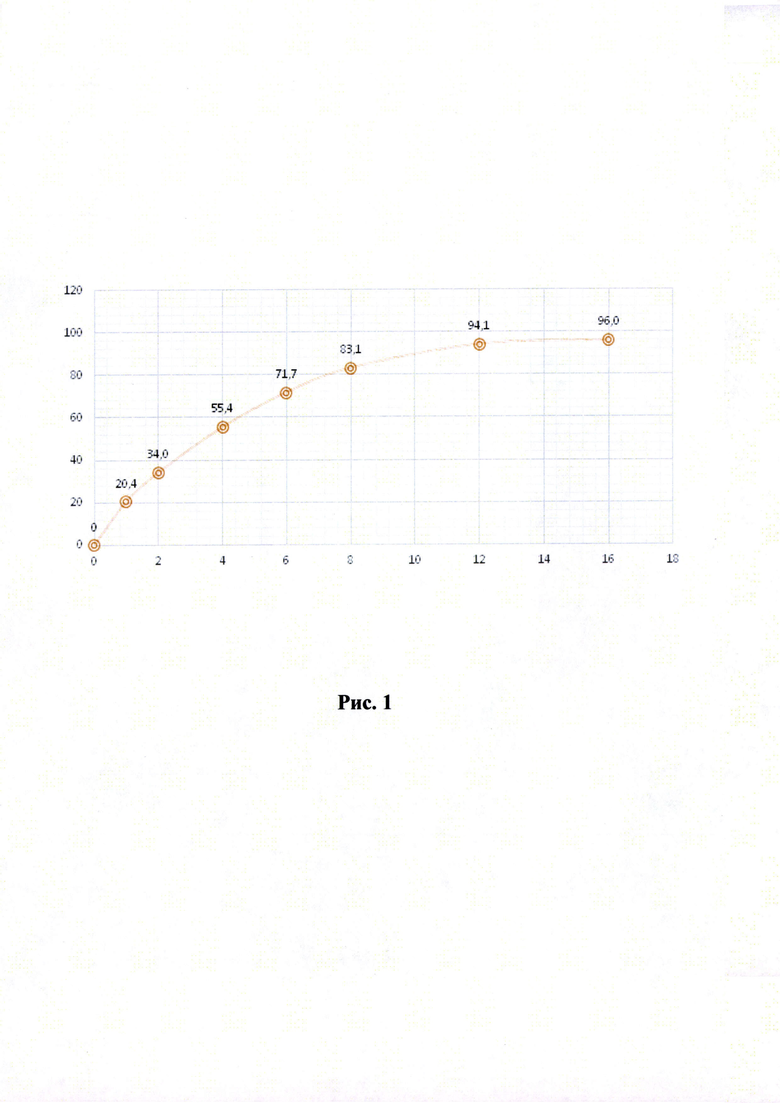
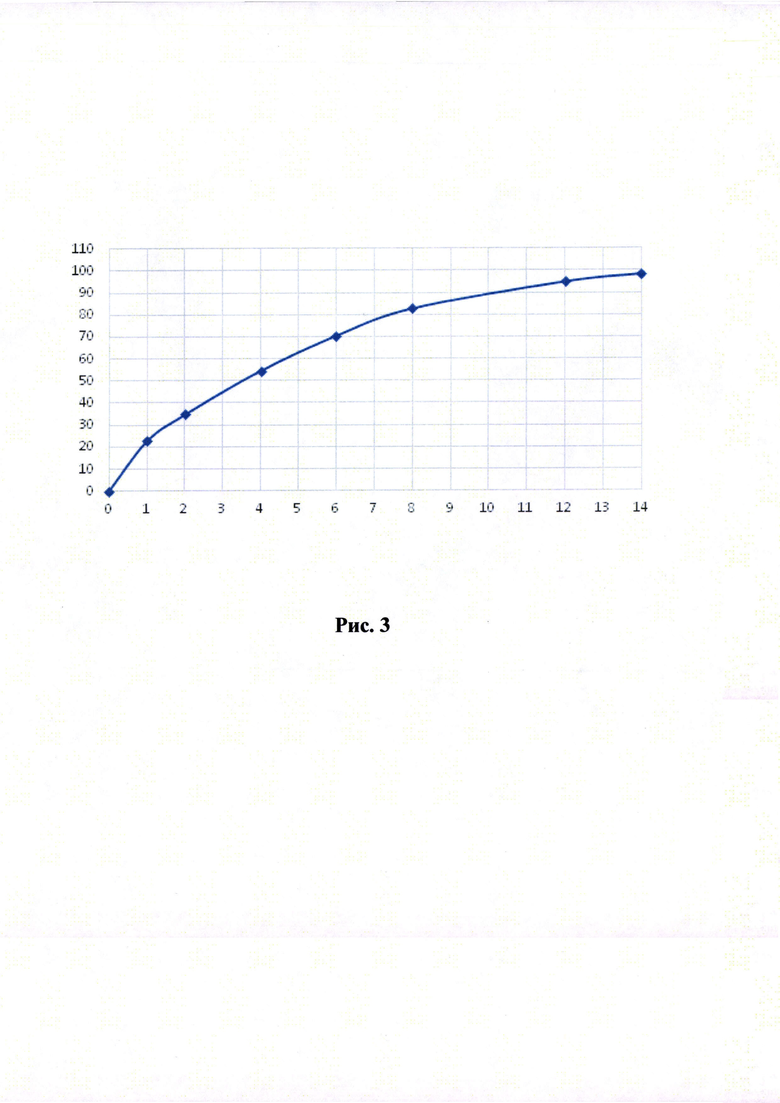
Кривые растворимости при рН 1,2 и 6,8 представлены на Рисунках 4 и 5 соответственно.
Было изучено, что в среде растворения желудка (рН 1,2 in-vitro) высвобождение действующего вещества происходит быстрее. После изменения состава была получена кривая высвобождения для следующего состава.
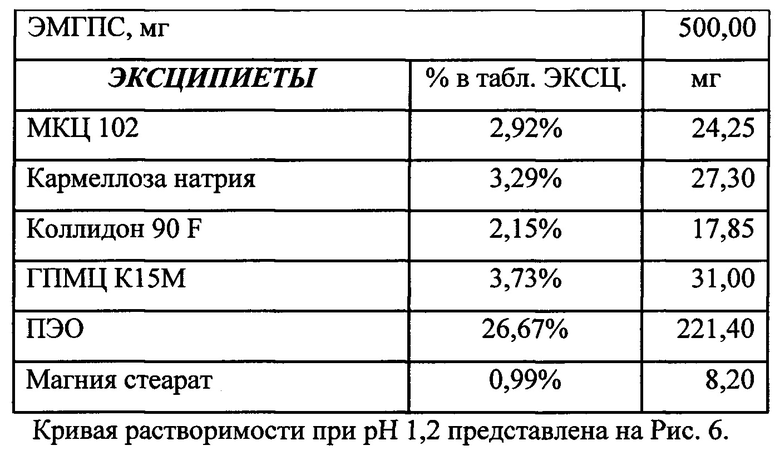
Данный состав позволяет получить необходимый профиль высвобождения и необходимые дополнительные параметры (степень набухания, мукоадгезивность) для гастроретентивной таблетки, а также удовлетворительные технологические свойства (сыпучесть смеси, прочность и истираемость таблетки). Технология влажной грануляции делает процесс таблетирования стабильным и контролируемым.
Исследование сравнительной фармакокинетики трех экспериментальных готовых лекарственных форм ЭМГПС на карликовых свиньях.
Объекты исследования:
Препарат сравнения: Мексидол, следующего состава:
ЭМГПС 125 мг, вспомогательные вещества: лактоза моногидрат - 97,5 мг, повидон - 25,0 мг, магния стеарат - 2,50 мг. Оболочка: опадрай II белый - 7,5 мг.
Материалы и методы.
Исследование проводили на половозрелых самцах карликовых свиней, поскольку им возможно введение исследуемых объектов в виде таблеток без разрушения целостности готовой лекарственной формы. Карликовые свиньи являются стандартным объектом фармакокинетических исследований (Руководство по содержанию и использованию лабораторных животных. Восьмое издание / пер. с англ. Под ред. И.В. Белозерцевой, Д.В. Блинова, М.С. Красилыциковой. - М.: ИРБИС, 2017. - 362 с).
Лабораторных животных до начала исследования содержали в вольерах для адаптации.
Длительность адаптационного периода для животных составила 5 дней. Во время периода адаптации у животных каждый день контролировали клиническое состояние путем визуального осмотра. Отклонений выявлено не было.
Распределение животных по группам проведено не было, т.к. в эксперименте было задействовано только 3 животных. Введение объектов №1, 2 осуществлялось последовательно после введения объекта сравнения: тестируемый объект №1 был введен через 2 дня после введения объекта сравнения; тестируемый объект №2 - через 4 дня после введения тестируемого объекта №1.
Животных содержали в стандартных условиях в соответствии с Директивой 2010/63/EU Европейского парламента и совета Европейского союза от 22 сентября 2010 г. по охране животных, используемых в научных целях.
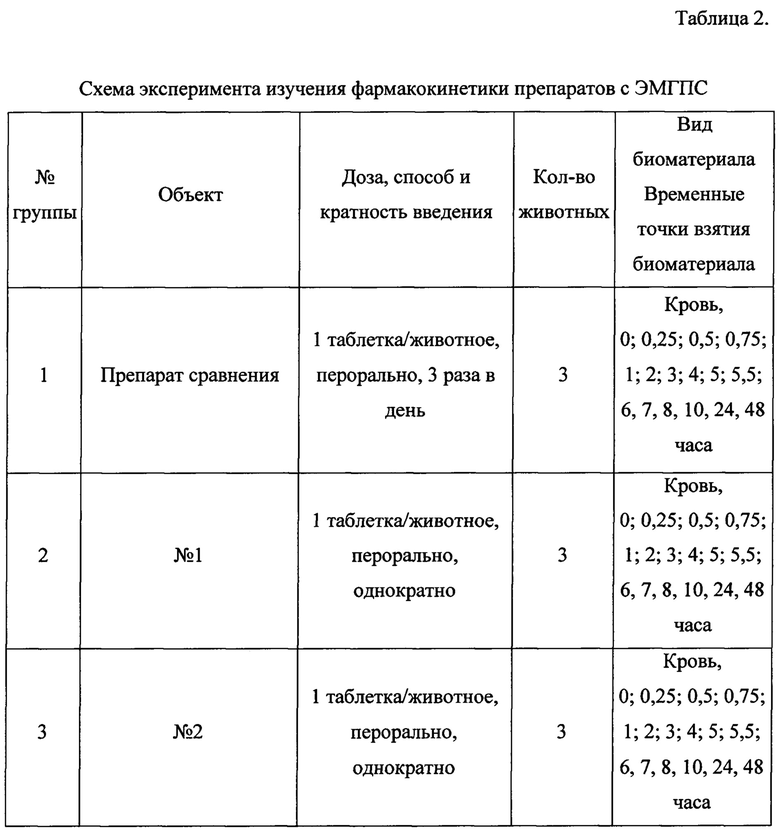
Схема эксперимента приведена в Таблице 2.
Для человека рекомендовано применение двух ГЛФ препарата с ЭМГПС, являющихся формами с модифицированным высвобождением ЭМГПС, в лекарственной форме таблетка, покрытая оболочкой, - 1 таблетка 1 раз в день. То есть по ЭМГПС 1×375 мг=375 мг/день.
Таким образом, высшая терапевтическая доза (ВТД) ЭМГПС для человека массой тела 60 кг составляет 375/60=6,25 мг/кг.
Для карликовых свиней эквивалентная доза составляет 6,25×1,1 (коэффициент пересчета доз для карликовых свиней Guidance for industry. Estimating the maximum safe starting dose for initial clinical trials for therapeutics in adult healthy volunteers. US Department of health and human services. FDA. CDER. - 2005)=6,88 мг/кг.
При введении карликовой свинье массой 20 кг 1 таблетки препарата ЭМГПС доза составит 375 мг/20 кг=18,75 мг/кг, что соответствует примерно 2,7 ВТД.
Тестируемые объекты представляют собой лекарственную форму пролонгированного действия, что предполагает минимальное ее разрушение при введении в тест-систему в ходе исследования и не допускает измельчения лекарственной формы. Поэтому в данном исследовании предусмотрено введение препарата ЭМГПС без разрушения целостности лекарственной формы. Таким образом, особенности лекарственной формы препарата ЭМГПС обуславливают выбранную дозу, 1 таблетка, как минимальную и наиболее близкую из возможных к ВТД в пересчете на карликовых свиней с учетом межвидовых коэффициентов.
Выбранная доза позволила оценить основные параметры фармакокинетики ЭМГПС при однократном введении тестируемых объектов.
Тестируемые объекты вводили карликовым свиньям в неизмененном виде перорально, однократно, до утренней раздачи корма в дозе 1 таблетка/животное с помощью таблеткодавателя.
Препарат сравнения вводили 3 раза в день по 1 таблетке с интервалом 2,5 часа, каждому животному, что эквивалентно дозе введения для тестируемых объектов (препарата ЭМГПС). Первая таблетка - до утренней раздачи корма, 2 и 3 - не зависимо от приема пищи.
На экспериментальном этапе выполняли осмотр животных в вольере для выявления отклонений поведения животных и смертности.
Забор биоматериала
Каждому животному устанавливали внутривенный катетер 22G (KD Medical GmbH Hospital Products, Германия) в ушную вену, посредством которого забирали образцы крови в объеме 2,0 мл на точку экспозиции.
В точке 0 часов биоматериал забирали перед введением препаратов. Затем проводили введение исследуемых препаратов и далее проводили забор биоматериала в соответствующих временных точках.
Методика количественного определения ЭМГПС в биологических образцах
В пластиковую центрифужную пробирку вместимостью 2 мл (типа «Эппендорф») помещали 0,2 мл плазмы крови, прибавляли 0,6 мл этилацетата, интенсивно встряхивали 3 минуты, после расслоения фаз отбирали органический слой, растворитель удаляли, для растворения аналита к сухому остатку прибавляли 0,2 мл 0,03% раствора трифторуксусной кислоты, вортексировали в течение 30 секунд, пробу переносили в виалу для автосамплера и дозировали в ВЭЖХ-систему.
Условия ВЭЖХ-анализа
Анализ выполнен на жидкостном хроматографе высокого давления фирмы Shimadzu (Япония) с диодно-матричным детектором и колонкой Luna С18 (2) 4,6×150 мм (размер частиц сорбента 5 мкм) и предколонкой (3 мм) заполненной тем же сорбентом (Phenomenex, США) в изократическом режиме элюирования смесью 0,03% раствора трифторуксусной кислоты и ацетонитрила в соотношении 95:5, скорость подачи элюента 1 мл/мин, дозируемый объем проб 20 мкл, длина волны детектирования 295 нм.
Регистрация и обработка хроматограмм выполнена с помощью программного обеспечения LabSolutions LCSolution Version 1.25 (Shimadzu, Япония).
Статистическая обработка данных
В Таблицах 3-5 приведены средние арифметические значения 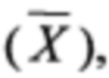 соответствующие им стандартные отклонения (SD), стандартные ошибки среднего значения
соответствующие им стандартные отклонения (SD), стандартные ошибки среднего значения 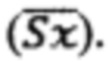
Параметры фармакокинетики (максимальная концентрация Cmax, Время достижения максимальной концентрации Tmax, площадь под кривой «концентрация-время» AUC, среднее время удерживания MRT, период полувыведения Т1/2 и показатель скорости всасывания Cmax/AUCt) рассчитаны внемодельным методом статистических моментов (Пиотровский В.К. Метод статистических моментов и интегральные модельно-независимые параметры фармакокинетики // Фармакология и токсикология. - 1986. - Т. 49, №5. - С. 118-127).
Для статистической оценки различий между фармакокинетическими параметрами был применен двухвыборочный t-тест для средних (оценку проводили при уровне достоверности 95%).
В нулевой точке (перед вводом препарата) исследование проб плазмы крови не выявило наличия аналита.
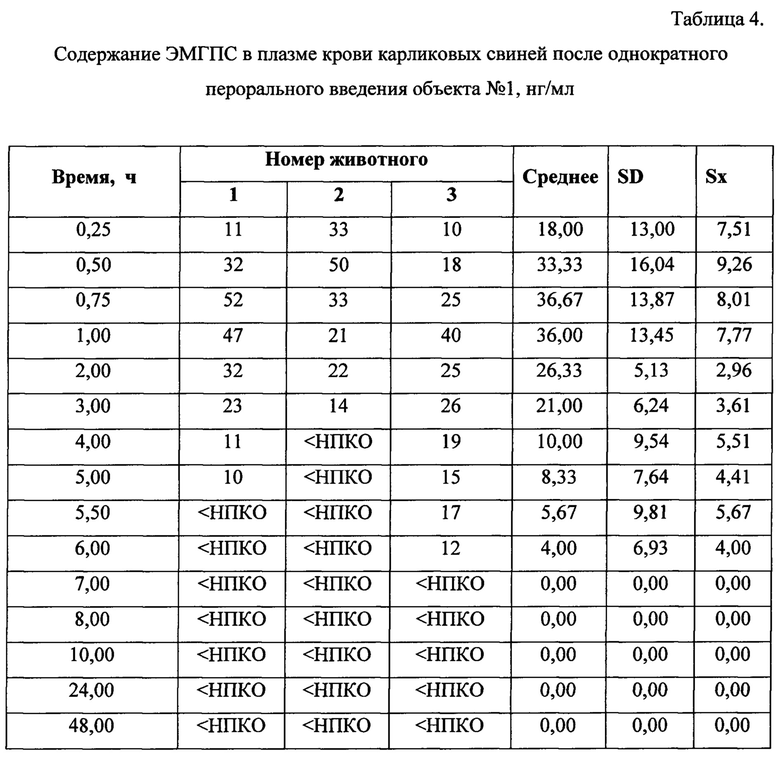
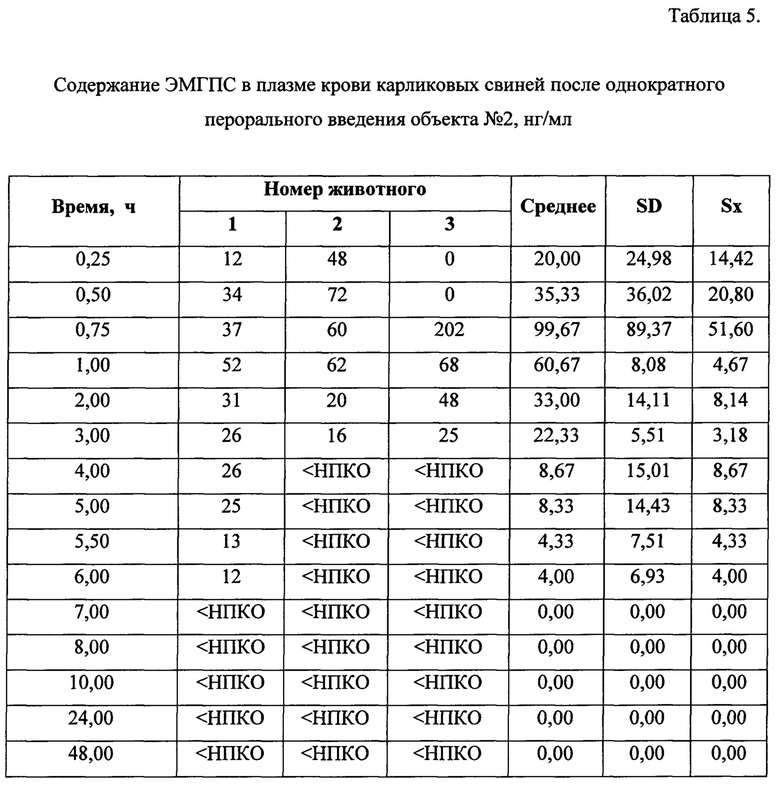
Динамика изменения концентраций ЭМГПС в плазме крови животных представлена в Таблицах 3-5. Усредненные фармакокинетические кривые в линейных и полулогарифмических координатах приведены на Рисунках 7 и 8.

Кинетика обнаружения ЭМГПС в плазме крови различалась по форме кривых, но характеризовалась наличием аналита в крови в течении 6-8 часов.
После трехкратного введения препарата сравнения, наблюдали повышение уровня ЭМГПС в крови через 0,5 часа (34,33±6,66 нг/мл); 3 часа (18,67±0,58 нг/мл) и 5,5 часов (23,67±4,16 нг/мл) после первого введения препарата, что соответствует максимальной концентрации аналита в крови через 0,5 часа после каждого введения препарата. После каждого увеличения концентрации ЭМГПС в крови наблюдалось постепенное снижение его концентрации. К 10 часу эксперимента после введения препаратов в плазме крови аналит не был обнаружен.
Максимальная концентрация ЭМГПС в плазме крови после однократного введения объектов №1,2 наблюдалась в интервале 0,75-1,0 часа: 36,67±13,87 нг/мл для объекта №1 и 99,67±89,37 нг/мл для объекта №2. Далее наблюдалось постепенное снижение концентрации ЭМГПС в плазме крови. К 8 часу эксперимента после введения препаратов в плазме крови аналит не был обнаружен.
Фармакокинетические профили фармакологического средства в крови при его пероральном введении должны быть охарактеризованы такими параметрами, как максимальная концентрация (Cmax), время достижения максимальной концентрации (Tmax), площадь под кривой «концентрация-время» (AUC), среднее время удерживания (MRT), период полувыведения (Т1/2) и показатель скорости всасывания Cmax/AUCt [13]. Параметры фармакокинетики рассчитаны внемодельным методом - методом статистических моментов (Пиотровский В.К. Метод статистических моментов и интегральные модельно-независимые параметры фармакокинетики // Фармакология и токсикология. - 1986. - Т. 49, №5. - С. 118-127), т.к. при камерном подходе могут получаться различные фармакокинетические параметры, что в значительной степени может повлиять на результаты исследований (Жердев В.П., Колыванов Г.Б., Литвин А.А. и др. Гармонизация проведения исследований биоэквивалентности лекарственных препаратов: вопросы и их возможное решение // Экспериментальная и клиническая фармакология. - 2003. - Т. 66, №2. - С. 60-64).
Таблицы 6-8 содержат индивидуальные и средние значения основных фармакокинетических параметров, характеризующих биодоступность ЭМГПС.
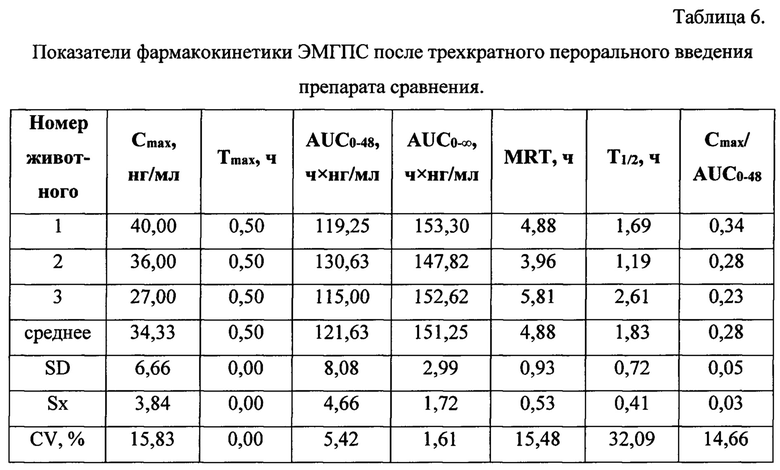

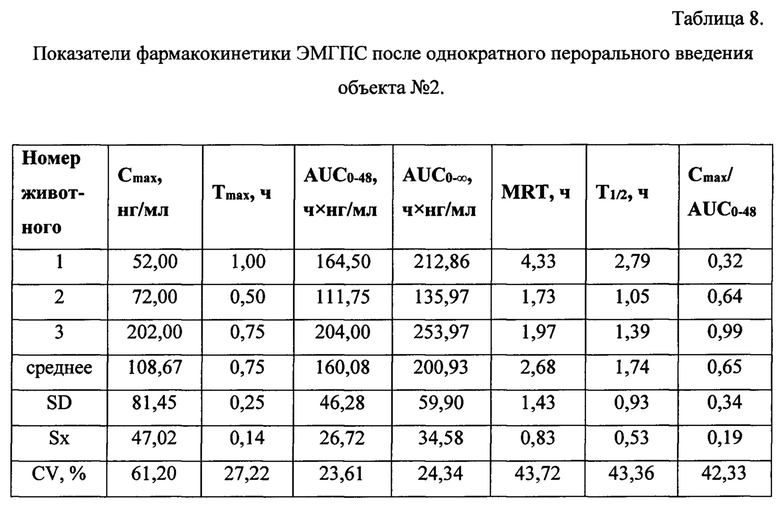
Анализ основных фармакокинетических данных ЭМГПС показал, что значения максимальной концентрации (Cmax) в плазме крови, рассчитанные как среднее значение наибольших измеренных значений у каждого животного, составили: 34,33±6,66 нг/мл для препарата сравнения; 47,33±6,43 нг/мл для объекта №1 и 108,67±81,45 нг/мл для объекта №2. Коэффициенты вариации по данному параметру составили 16, 11 и 61% для препарата сравнения, объектов №1 и 2, соответственно.
Значения основного параметра, характеризующего степень биологической доступности препарата, AUC0-48, для препарата сравнения, объектов №1 и 2 были близки и составили 121,63±4,66 ч×нг/мл; 108,75±32,77 ч×нг/мл и 160,08±46,28 ч×нг/мл, соответственно. Коэффициенты вариации по данному параметру составили 5,25 и 24% для препарата сравнения, объектов №1 и 2, соответственно.
Для параметра Tmax значения составили 0,5 часа для препарата сравнения и 0,75 часа для объектов №1 и 2. Коэффициенты вариации данного параметра не превысили 28%.
Значения показателя среднего времени удержания ЭМГПС (MRT) для препарата сравнения, объектов №1 и 2 составили 4,88±0,93; 3,59±1,43 и 2,68±1,43 часа, соответственно. Коэффициенты вариации данного параметра не превысили 44%.
Значения параметра периода полувыведения (T1/2) для препарата сравнения, объектов №1 и 2 составили 1,83±0,72; 2,25±0,97 и 1,74±0,93, соответственно. Коэффициенты вариации данного параметра составили 32-43%.
Значения показателя Cmax/AUC0-24, характеризующего скорость всасывания, составили: 0,28±0,05; 0,48±0,20 и 0,65±0,34, соответственно, для препарата сравнения, объектов №1 и 2.
Проведенные исследования показали более высокую биодоступность ЭМГПС при пероральном введении гастроретентивных таблеток по сравнению с таблетками пролонгированного действия. Было установлено повышение растворимости ЭМГПС при рН, соответствующей среде желудка. Исследования позволяют предположить наличие in vivo - in vitro корреляции растворимости ЭМГПС для гастроретентивных таблеток.
| название | год | авторы | номер документа |
|---|---|---|---|
| Фармацевтическая композиция на основе макозинона для лечения туберкулеза, включая его формы с множественной и широкой лекарственной устойчивостью. | 2020 |
|
RU2751163C1 |
| ЛЕКАРСТВЕННАЯ ФОРМА С МОДИФИЦИРОВАННЫМ ВЫСВОБОЖДЕНИЕМ 6-МЕТИЛ-2-ЭТИЛ-3-ГИДРОКСИПИРИДИНА СУКЦИНАТА | 2008 |
|
RU2411035C2 |
| ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ С 2-ЭТИЛ-6-МЕТИЛ-3-ГИДРОКСИПИРИДИНА СУКЦИНАТА ДЛЯ ПЕРОРАЛЬНОГО ПРИМЕНЕНИЯ И СПОСОБ ЕЕ ПОЛУЧЕНИЯ | 2010 |
|
RU2444359C1 |
| ТВЁРДАЯ ПЕРОРАЛЬНАЯ ЛЕКАРСТВЕННАЯ ФОРМА, ПРОЛОНГИРОВАННО ВЫСВОБОЖДАЮЩАЯ АКТИВНУЮ ФАРМАЦЕВТИЧЕСКУЮ СУБСТАНЦИЮ (АФС) В ВЕРХНЕЙ ЧАСТИ ЖЕЛУДКА | 2021 |
|
RU2786063C2 |
| СПОСОБ ПОЛУЧЕНИЯ ФАРМАЦЕВТИЧЕСКОЙ КОМПОЗИЦИИ, ОБЛАДАЮЩЕЙ НЕЙРОПРОТЕКТОРНОЙ АКТИВНОСТЬЮ ПРИ ХРОНИЧЕСКОЙ ИШЕМИИ ГОЛОВНОГО МОЗГА | 2023 |
|
RU2816359C1 |
| ГАСТРОРЕТЕНТИВНЫЕ ФАРМАЦЕВТИЧЕСКИЕ КОМПОЗИЦИИ ДЛЯ ПЕРОРАЛЬНОГО ВВЕДЕНИЯ | 2014 |
|
RU2666996C1 |
| ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ В ФОРМЕ ТАБЛЕТКИ, ПОКРЫТОЙ ОБОЛОЧКОЙ | 2022 |
|
RU2786073C1 |
| ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ ПРОКСОДОЛОЛА С КОНТРОЛИРУЕМЫМ ВЫСВОБОЖДЕНИЕМ | 2007 |
|
RU2356532C2 |
| РЕЦЕПТУРА С КОНТРОЛИРУЕМЫМ ВЫСВОБОЖДЕНИЕМ | 2006 |
|
RU2385712C2 |
| ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ МЕТФОРМИНА С ДЛИТЕЛЬНЫМ ВЫСВОБОЖДЕНИЕМ И СПОСОБ ЕЕ ПОЛУЧЕНИЯ | 2006 |
|
RU2433821C2 |

Группа изобретений относится к медицин, а именно к применению по меньшей мере одного контролирующего высвобождение в желудке полимера с мукоадгезивными свойствами для получения системы пероральной доставки 2-этил-6-метил-3-гидроксипиридина сукцината в форме гастроретентивной таблетки. Для этого по меньшей мере один полимер выбран из полиалкиленоксида. Группа изобретений также относится к гастроретентивной таблетке, содержащей систему пероральной доставки и 2-этил-6-метил-3-гидроксипиридина сукцинат, а также относится к способу повышения биодоступности 2-этил-6-метил-3-гидроксипиридина сукцината. Изобретение обеспечивает повышение биодоступности активного вещества, а также сокращение частоты приема лекарственного средства. 3 н. и 11 з.п. ф-лы, 8 ил., 8 табл.
1. Применение по меньшей мере одного контролирующего высвобождение в желудке полимера с мукоадгезивными свойствами, где по меньшей мере один полимер выбран из полиалкиленоксида, для получения системы пероральной доставки 2-этил-6-метил-3-гидроксипиридина сукцината в форме гастроретентивной таблетки.
2. Применение по п. 1, где по меньшей мере один указанный полиалкиленоксид выбран из полиэтиленоксида, полипропиленоксида или сополимера этиленоксида и пропиленоксида.
3. Применение по п. 2, где массовое соотношение 2-этил-6-метил-3-гидроксипиридина сукцината к полиэтиленоксиду составляет от 2,6:1 до 1,3:1.
4. Применение по п. 1 или 2, где указанная гастроретентивная таблетка дополнительно содержит полимер, выбранный из группы, включающей кармеллозу натрия, гидроксипропилметилцеллюлозу, целлюлозу микрокристаллическую, поливинилпирролидон или их смеси.
5. Гастроретентивная таблетка, содержащая систему пероральной доставки по любому из пп. 1-4 и 2-этил-6-метил-3-гидроксипиридина сукцинат в терапевтически эффективном количестве.
6. Таблетка по п. 5, отличающаяся тем, что покрыта пленочной оболочкой.
7. Таблетка по п. 5, отличающаяся тем, что дополнительно содержит лубрикант.
8. Таблетка по п. 7, отличающаяся тем, что в качестве лубриканта содержит стеарат магния.
9. Таблетка по п. 5, отличающаяся тем, что содержит 2-этил-6-метил-3-гидроксипиридина сукцинат в количестве от 125 мг до 1500 мг.
10. Таблетка по п. 5, отличающаяся тем, что содержит компоненты в следующем соотношении, мас. %:
11. Таблетка по п. 5, отличающаяся тем, что содержит компоненты в следующем соотношении, мас. %:
12. Таблетка по п. 5, отличающаяся тем, что содержит компоненты в следующем соотношении, мас. %:
или
или
или
13. Таблетка по любому из пп. 5-12, отличающаяся тем, что предназначена для антиоксидантного и/или антигипоксантного и/или мембранопротекторного действия.
14. Способ повышения биодоступности 2-этил-6-метил-3-гидроксипиридина сукцината, характеризующийся тем, что его вводят перорально в виде гастроретентивной таблетки по любому из пп. 5-12 нуждающемуся в этом субъекту 1 раз в сутки.
| ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ ДЛЯ ПРОФИЛАКТИКИ И ЛЕЧЕНИЯ СЕРДЕЧНО-СОСУДИСТЫХ ЗАБОЛЕВАНИЙ | 2011 |
|
RU2491070C2 |
| БЕНЗИЛАМИНОВЫЕ ПРОИЗВОДНЫЕ | 2014 |
|
RU2660421C2 |
| ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ С 2-ЭТИЛ-6-МЕТИЛ-3-ГИДРОКСИПИРИДИНА СУКЦИНАТА ДЛЯ ПЕРОРАЛЬНОГО ПРИМЕНЕНИЯ И СПОСОБ ЕЕ ПОЛУЧЕНИЯ | 2010 |
|
RU2444359C1 |

