Данное изобретение относится к способу получения пиримидинонового соединения и его фармацевтически приемлемых солей. Более конкретно, данное изобретение относится к удобному и высокопродуктивному способу получения пиримидинонового соединения следующей ниже формулы 1 и его фармацевтически приемлемых солей, которые пригодны для лечения сердечно-сосудистых заболеваний, обусловленных связыванием ангиотензина II с его рецепторами, благодаря антагонистической активности против рецепторов ангиотензина II.
Формула 1
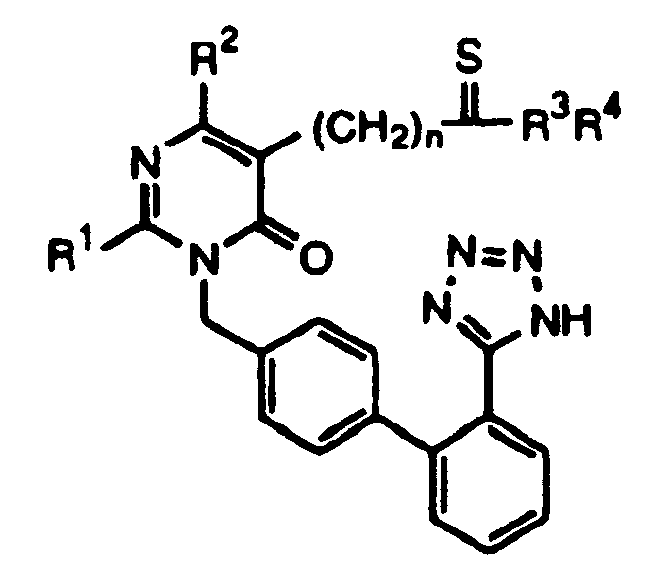
где R1 представляет С1-С4алкил с линейной или разветвленной цепью, циклоалкил, С1-С4алкилалкокси или С1-С4алкилмеркапто;
R2 представляет Н, галоген, С1-С4алкил с линейной или разветвленной цепью или арилалкил;
R3, R4, которые являются одинаковыми или различными, представляют Н, С1-С4алкил с линейной или разветвленной цепью, циклоалкил, карбоциклический арил, арилалкил, арилкарбонил, С1-С4алкоксикарбонил или аминокарбонил, которые необязательно замещены Н, галогеном, гидрокси, С1-С4алкокси, амино, алкиламино или диалкиламино (где каждый алкильный остаток имеет С1-С5), С1-С4алкоксикарбонилом или карбоксилом;
R3 и R4 вместе с атомом N образуют 4-8-членное гетероциклическое кольцо, которое может быть дополнительно замещено одним или двумя заместителями, выбранными из группы, состоящей из циклоалкила, карбоциклического арила или арилалкила, галогена, гидроксила, С1-С4алкокси, амино, алкиламино или диалкиламино (где каждый алкильный остаток имеет С1-С5), С1-С4алкоксикарбонила, карбокси или аминокарбонила и С1-С4алкила с линейной или разветвленной цепью, где указанное гетероциклическое кольцо может дополнительно включать -О-, -S-, -SO-, -SO2- или >N-R5; R5 представляет Н, С1-С4алкил, карбоциклический арил, арилалкил, замещенный алкенил, пиридил, пиримидил, С1-С4алкил или арилкарбонил, С1-С4алкоксикарбонил, аминокарбонил, CN или SO2NR3R4;
n является целым числом, выбранным из 1, 2, 3, 4, 5 и 6.
Недавно были проведены многочисленные исследования по непептидному антагонисту ангиотензина II (патенты США 4207324; 4340598; 4576958; 4582847 и 4880804; Европейские открытые публикации патентов №№ (European Laid-Open Patent Publication Nos) 028834; 245637; 253310; 291969; 323841 и 324377 и т.д.). Среди представленного выше в Европейских открытых публикациях патентов № 028834 и 253310 описаны имидазольные производные, замещенные бифенильной группой (например, лозартан), и в Европейской открытой публикации патента № 245637 описаны имидазолпиридиновые производные (например, L158809). Эти соединения, как заявлено, обладают сильным антагонистическим эффектом в отношении ангиотензина II. Кроме того, в Европейских открытых публикациях патентов №407342, 419048 и 445811 описано пиримидиноновое соединение, которое представляет собой 6-членное гетероциклическое соединение, содержащее дополнительный атом азота, по сравнению с 5-членным имидазольным производным, указанным выше. Однако эти пиримидиноновые соединения проявляют меньшую активность, чем имидазольные производные.
В результате интенсивного изучения пиримидиноновых соединений было создано новое пиримидиноновое соединение, имеющее в основе другую структуру по сравнению с указанными выше пиримидиноновыми соединениями, обладающее в то же время превосходящей активностью, то есть в 50 раз более высокой, чем активность указанных выше имидазольных производных [in vitro (аорта кролика) степень подавления равна 60-70% при концентрации 10-8 ˜ 10-9 моль], и поэтому зарегистрированы патентные заявки (международные патентные заявки №№ РСТ/KR95/00121, поданная 15 сентября 1995 и РСТ/KR 9900198, поданная 26 апреля 1999). Разработанные пиримидиноновые соединения формулы 1 были получены, как представлено на следующей схеме реакций 1, которая описана в обеих из приведенных выше заявок.
Схема реакций 1
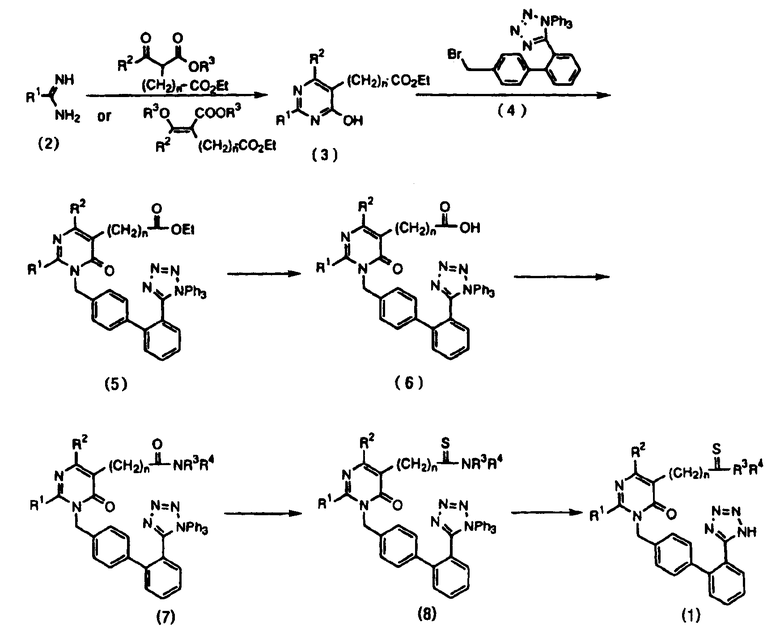
где R1, R2, R3, R4, R5 и n имеют значения, указанные выше.
Однако приведенный выше способ, состоящий из множества стадий, отчасти весьма сложен, а также очень трудно выделить соединение формулы 5 из-за неселективных реакций N, О-алкилированных соединений, которые неизбежно вызывает неудобства, связанные с колоночной очисткой, и убыточность, связанную с очень низким выходом (1,28%).
Данное изобретение разрешает проблемы предшествующих прототипов путем представления нового способа получения пиримидинонового соединения формулы 1, где данный способ включает стадии: индукции селективной реакции N-алкилирования добавлением основания к соединениям формулы 3 и формулы 4 в смешанном органическом растворителе, так что получают приведенное выше соединение формулы 5; одновременного гидролиза и амидирования соединения формулы 5 с получением соединения формулы 7; проведения реакции тиоамидирования соединения формулы 7 с использованием реагента Лавессона без обработки кислотой и обработки полученного продукта спиртовым реагентом.
Поэтому объектом данного изобретения является создание способа получения пиримидинонового соединения и его солей относительно простым и высокопродуктивным способом.
Кроме того, другим объектом данного изобретения является получение гидратов пиримидинонового соединения, полученного приведенным выше способом, и его фармацевтически приемлемых солей.
Другие объекты данного изобретения будут ясно поняты из представленного с этой целью следующего ниже подробного описания.
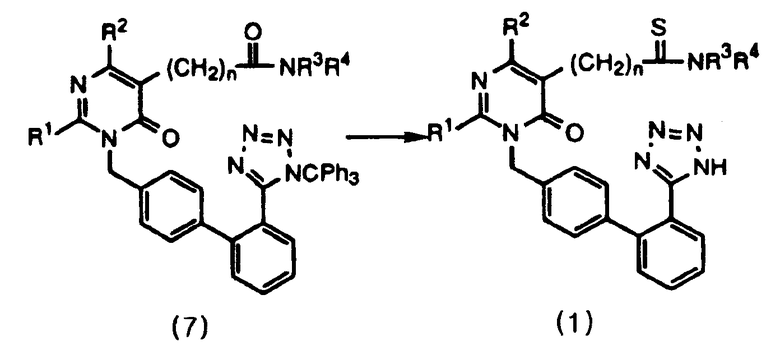
Данное изобретение относится к способу получения соединений формулы 1 и их солей, как проиллюстрировано на следующей ниже схеме реакций 2, включающему стадии тиоамидирования соединения формулы 7 с использованием реагента Лавессона с последующей обработкой продукта спиртовым реагентом.
Схема реакций 2
где R1 представляет С1-С4алкил с линейной или разветвленной цепью, циклоалкил, С1-С4алкилалкокси или С1-С4алкилмеркапто;
R2 представляет Н, галоген, С1-С4алкил с линейной или разветвленной цепью или арилалкил;
R3, R4, которые являются одинаковыми или различными, представляют Н, С1-С4алкил с линейной или разветвленной цепью, циклоалкил, карбоциклический арил, арилалкил, арилкарбонил, С1-С4алкоксикарбонил или аминокарбонил, которые необязательно замещены Н, галогеном, гидрокси, С1-С4алкокси, амино, алкиламино или диалкиламино (где каждый алкильный остаток имеет С1-С5), С1-С4алкоксикарбонилом или карбоксилом;
R3 и R4 вместе с атомом N образуют 4-8-членное гетероциклическое кольцо, которое может быть дополнительно замещено одним или двумя заместителями, выбранными из группы, состоящей из циклоалкила, карбоциклического арила или арилалкила, галогена, гидроксила, С1-С4алкокси, амино, алкиламино или диалкиламино (где каждый алкильный остаток имеет С1-С5), С1-С4алкоксикарбонила, карбокси или аминокарбонила и С1-С4алкила с линейной или разветвленной цепью, где указанное гетероциклическое кольцо может дополнительно включать -О-, -S-, -SO-, -SO2- или >N-R5; R5 представляет Н, С1-С4алкил, карбоциклический арил, арилалкил, замещенный алкенил, пиридил, пиримидил, С1-С4алкил или арилкарбонил, С1-С4алкоксикарбонил, аминокарбонил, CN или SO2NR3R4;
n является целым числом, выбранным из 1, 2, 3, 4, 5 и 6.
Далее способ получения по данному изобретению объясняется более подробно.
Данное изобретение представляет способ получения пиримидинонового соединения формулы 1 и его солей, включающий стадии тиоамидирования соединения формулы 7 путем использования реагента Лавессона и затем обработки продукта спиртовым реагентом. В соответствии с данным способом пиримидиноновое соединение формулы 1 может быть удобно получено в одну стадию с высоким выходом путем проведения реакции тиоамидирования. Его фармацевтически приемлемые соли получают добавлением соли к полученному пиримидиноновому соединению в соответствии с общепринятыми методиками. Реагент Лавессона во время тиоамидирования используют в количестве 0,5-2,0 экв., предпочтительно в количестве 1,0 экв. Затем защитные группы обрабатывают спиртовым реагентом, предпочтительно выбранным из метанола, этанола, пропанола и их смесей. В качестве соли предпочтительно используют соль калия или натрия.
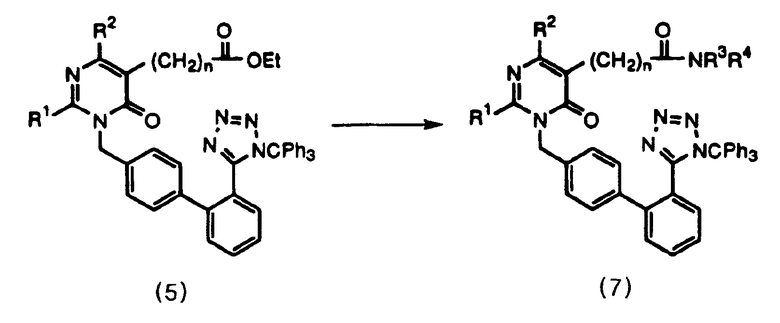
Соединение формулы 7 получают одновременным гидролизом и амидированием соединения формулы 5, как представлено в общих чертах на следующей схеме реакций 3, где гидролиз проводят, используя гидроксид натрия, и амидирование проводят, используя амин и N-гидроксибензотриазол, N-метилморфолин и дициклогексилкарбодиимид в количестве 1 экв. в хлороформе.
Схема реакций 3
Соединение формулы 5 можно легко получить селективным образованием N-алкилированного соединения добавлением основания к соединениям формулы 3 и 4 в смешанном органическом растворителе.
Схема реакций 4
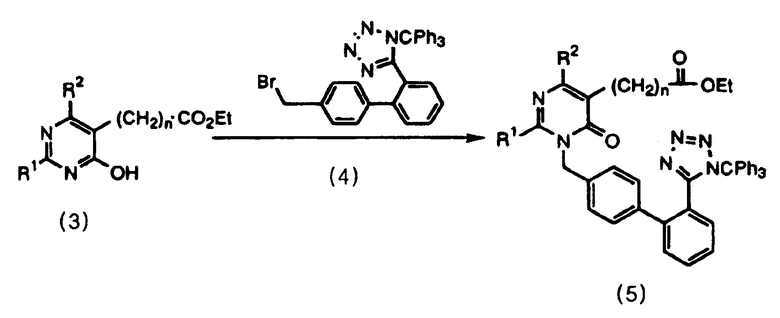
В качестве смешанного органического растворителя используют смесь диметилформамида и этилацетата, предпочтительно при отношении смешивания 1˜50:50˜1, и более предпочтительно 1˜10:10˜1. Кроме того, в качестве основания могут быть приведены гидриды щелочных металлов или органические соли щелочных металлов, и из гидридов щелочных металлов наиболее предпочтительно использовать гидрид лития.
В результате в соответствии со способом данного изобретения возможно получение N-алкилированного соединения формулы 5 при высокой селективности, которая далее делает возможным удобный режим работы на стадиях выделения и очистки. Кроме того, возможно получение пиримидинонового соединения формулы 1 со значительно высоким выходом (28,2%).
Данное изобретение обеспечивает также гидраты пиримидинонового соединения, получаемого с помощью приведенного выше способа, и его фармацевтически приемлемых солей. Как описано выше, пиримидиноновое соединение формулы 1 описано в международных заявках №№ PCT/KR95/00121, поданной 15 сентября 1995, и PCT/KR 9900198, поданной 26 апреля 1999. Однако в случае, когда указанное выше соединение находится в безводной форме, оно имеет склонность абсорбировать влагу из воздуха. Таким образом, если лекарственный препарат производят из указанного выше безводного пиримидинонового соединения, он становится нестабильным из-за влаги, присутствующей в воздухе.
Данное изобретение будет дополнительно проиллюстрировано следующими ниже примерами. Специалистам в данной области будет очевидно, что эти примеры даны только для более полной иллюстрации данного изобретения, и изобретение не ограничивается представленными примерами.
Пример получения 1: Получение 2-н-бутил-5-этоксикарбонилметил-4-гидрокси-6-метилпиримидина
1,41 кг валероамидина и 1,41 кг диэтилацетилсукцината растворяли в 4,5 л метанола и добавляли 1,16 кг гидроксида калия. Смесь перемешивали в течение 15 часов при комнатной температуре и затем при перемешивании добавляли 14 л воды. Полученное твердое вещество отфильтровывали, сушили и растворяли в 6 л этанола. Затем по каплям добавляли 840 г тионилхлорида в течение 2 часов и перемешивали в течение 12 часов при 60°С. К полученной смеси добавляли раствор 1,22 кг бикарбоната натрия в 15 л воды, полученное твердое вещество отфильтровывали и сушили с получением 1,35 кг (47,8% выход) указанного в заголовке соединения.
ИК (KBr) см-1: 1740, 1665, 1620.
1H ЯМР (ДМСО-d6): δ 0,89 (т, 3H), 1,12 (т, 3H), 1,20˜1,40 (м, 2H), 1,52-1,70 (м, 2H), 2,18 (с, 3H), 2,50 (т, 2H), 3,45 (с, 2H), 4,08 (кв, 2H), 12,38 (шир.с, 1Н).
Пример 1: Получение 2-н-бутил-5-этоксикарбонилметил-6-метил-3-[[2'-(N-трифенилметилтетразол-5-ил)бифенил-4-ил]метил]пиримидин-4(3Н)-она
1,35 кг соединения примера получения 1 растворяли в 18 л смешанного раствора диметилформамида и этилацетата (соотношение компонентов 1:8) и охлаждали до 0°С. К полученной смеси добавляли 47 г гидрида лития и перемешивали в течение 30 минут. Затем к полученному перемешиваемому раствору добавляли 4,26 кг 4-[2'-(N-трифенилметилтетразол-5-ил)фенил]бензилбромида и перемешивали в течение 90 часов при 55°С. Продукт отфильтровывали и сушили с получением 3,54 кг (90% выход) селективно N-алкилированного указанного в заголовке соединения.
ИК (чистый) cm-1: 1740, 1665, 1600.
1H ЯМР (CDCl3): δ 0,86 (т, 3H), 1,25(т, 3H), 1,52-1,70 (м, 4H), 2,30 (с, 3H), 2,52 (т, 2H), 3,63 (с, 2H), 4,18 (кв, 2H), 5,19 (с, 2H), 6,85˜6,98 (м, 7H), 7,05˜7,12 (м, 3H), 7,20˜7,40 (м, 9H), 7,40˜7,50 (м, 3H), 7,90˜7,95 (дд, 1H).
Пример 2: Получение 2-н-бутил-5-диметиламинокарбонилметил-6-метил-3-[[2'-(N-трифенилметилтетразол-5-ил)бифенил-4-ил]метил]пиримидин-4(3Н)-она
2,75 кг соединения примера 1 растворяли в 8 л смешанного раствора метанола и тетрагидрофурана (отношение компонентов 1:3) и охлаждали до 0°С. К полученной смеси добавляли 2 л 10% раствора гидроксида натрия в течение двух часов и затем перемешивали в течение 4 часов. Добавляли 1,2 л 4 N хлористоводородной кислоты для нейтрализации раствора и затем концентрировали в вакууме, чтобы затем экстрагировать 10 л хлороформа.
Хлороформ концентрировали до 6 л, затем к последовательно добавляли 330 г диметиламина гидрохлорида, 550 г N-гидроксибензотриазола, 900 мл N-метилморфолина и 920 г дициклогексилкарбодиимида при 0°С и перемешивали в течение 15 часов при комнатной температуре. Полученное твердое вещество отфильтровывали, затем фильтрат промывали 2 л воды и 2 л насыщенного раствора бикарбоната натрия и концентрировали в вакууме. Остаток растворяли в 5 л этилацетата и по каплям добавляли 10 л гексана с получением твердого продукта, который далее отфильтровывали и сушили с получением 1,97 кг (82%) указанного в заголовке соединения.
ИК (KBr) cm-1: 1660, 1620, 1555.
1H ЯМР (CDCl3): δ 0,87 (т, 3H), 1,25-1,40 (м, 2H), 1,55-1,75 (м, 2H), 2,20 (с, 3H), 2,58 (т, 2H), 2,87 (с, 3H), 3,11 (с, 3H), 3,56 (с, 2H), 5,10 (с, 2H), 6,85˜6,98 (м, 8H), 7,11 (дд, 2H), 7,22˜7,38 (м, 10H), 7,48 (м, 2H), 7,98 (дд, 1H).
Пример 3: Получение 2-н-бутил-5-диметиламинотиокарбонилметил-6-метил-3-[[2'-(1Н-тетразол-5-ил)бифенил-4-ил]метил]пиримидин-4(3Н)-она
1,97 кг соединения примера 2 растворяли в 8 л толуола и к полученной смеси добавляли реагент Лавессона (1,1 кг) при комнатной температуре. В образовавшемся мутном растворе проводили взаимодействие в течение 6 часов при 80°С и затем охлаждали до комнатной температуры, чтобы отфильтровать ненужное твердое вещество. Затем проводили концентрирование в вакууме, к остатку добавляли 6 л метанола, нагревали с обратным холодильником в течение 3 часов и концентрировали. Полученный концентрат растворяли в 4,5 л этилацетата. К полученному раствору добавляли 4,5 л воды, так чтобы получить твердый продукт. Полученный твердый продукт отфильтровывали и промывали 1 л воды, 1 л этилацетата и 3 л изопропилового эфира, отдельно, и сушили в течение 24 часов при 60°С с получением 1,1 кг (80%) указанного в заголовке соединения.
Температура плавления: 96,8-101,8°С
ТСХ Rf: 0,28 (5% МеОН в CHCl3)
1H ЯМР (CDCl3): δ 0,89 (т, 3H), 1,28˜1,45 (м, 2H), 1,58˜1,74 (м, 2H), 2,26 (с, 3H), 2,63 (т, 2H), 3,44 (с, 3H), 3,46 (с, 3H), 3,77 (с, 2H). 5,22 (с, 2H), 7,07 (с, 5H), 7,33˜7,60 (м, 3H), 7,94 (дд, 1H).
Пример 4: Стабильность 2-н-бутил-5-диметиламинотиокарбонилметил-6-метил-3-[[2'-(1Н-тетразол-5-ил)бифенил-4-ил]метил]пиримидин-4(3Н)-она
Время (часы)
60% КВ
75% КВ
Как видно из таблицы 1, содержание влаги в соединении примера 3 измеряли каждый час. В результате содержание влаги повышалось с течением времени. Однако по прошествии определенного срока содержание больше не повышалось. В результате открыто, что гидрат пиримидинонового соединения на воздухе более стабилен, чем ангидрид. Данные элементного анализа в отношении указанных выше гидратов иллюстрируется в следующей таблице 2.
Как подтверждено в приведенной выше таблице, пиримидиноновое соединение поглощает влагу, превращается в тригидратный тип и считается стабильным.
Как описано выше, данное изобретение представляет собой способ получения пиримидинонового соединения формулы 1 и его солей, включающий стадии тиоамидирования соединения формулы 7 с использованием реагента Лавессона и последующей обработки продукта спиртовым реагентом. Как представлено в приведенном выше примере 1, соединение формулы 5 может быть получено по методике только одной реакции селективного N-алкилирования, которая в свою очередь не требует процедуры очистки с использованием колонки в непрерывном способе. Поэтому в соответствии со способом данного изобретения пиримидиноновое соединение и его соли получают со значительно более высоким выходом по сравнению с общепринятым способом.
| название | год | авторы | номер документа |
|---|---|---|---|
| ПИРИМИДИНОНОВЫЕ СОЕДИНЕНИЯ, СПОСОБ ИХ ПОЛУЧЕНИЯ И ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ | 1999 |
|
RU2233278C9 |
| 2-[(ДИГИДРО)ПИРАЗОЛИЛ-3'-ОКСИМЕТИЛЕН]АНИЛИДЫ, СПОСОБЫ ИХ ПОЛУЧЕНИЯ, ПРОМЕЖУТОЧНЫЕ СОЕДИНЕНИЯ, СРЕДСТВО БОРЬБЫ С СЕЛЬСКОХОЗЯЙСТВЕННЫМИ ВРЕДИТЕЛЯМИ И ВРЕДОНОСНЫМИ ГРИБАМИ И СПОСОБЫ БОРЬБЫ | 1995 |
|
RU2151142C1 |
| ПРОИЗВОДНЫЕ ФЕНИЛУКСУСНОЙ КИСЛОТЫ, ПРОМЕЖУТОЧНЫЕ ПРОДУКТЫ ДЛЯ ИХ ПОЛУЧЕНИЯ И СОДЕРЖАЩИЕ ИХ СРЕДСТВА | 1995 |
|
RU2165411C2 |
| ПРОИЗВОДНЫЕ ФЕНИЛУКСУСНОЙ КИСЛОТЫ И СРЕДСТВО БОРЬБЫ ПРОТИВ НАСЕКОМЫХ И ПАУКООБРАЗНЫХ И ПРОТИВ ВРЕДОНОСНЫХ ГРИБОВ | 1995 |
|
RU2162075C2 |
| НОВЫЕ ПРОИЗВОДНЫЕ КАТЕХОЛА ИЛИ ИХ СОЛЬ, СПОСОБЫ ИХ ПОЛУЧЕНИЯ И СОДЕРЖАЩИЕ ИХ ФАРМАЦЕВТИЧЕСКИЕ КОМПОЗИЦИИ | 2019 |
|
RU2795227C2 |
| СПОСОБ ПОЛУЧЕНИЯ ЦИКЛИЧЕСКИХ ДИКЕТОНОВ | 2003 |
|
RU2316544C2 |
| НОВЫЕ СОЕДИНЕНИЯ МИМЕТИКИ ОБРАТНОГО ПОВОРОТА И ИХ ПРИМЕНЕНИЕ | 2008 |
|
RU2457210C2 |
| СПОСОБ ПОЛУЧЕНИЯ ЦИКЛИЧЕСКИХ ДИКЕТОНОВ | 2005 |
|
RU2384562C2 |
| ИНГИБИТОРЫ ТИРОЗИНФОСФАТАЗЫ БЕЛКА ЧЕЛОВЕКА И СПОСОБЫ ПРИМЕНЕНИЯ | 2007 |
|
RU2435763C2 |
| СПОСОБ ПОЛУЧЕНИЯ 4-АМИНО-5-ФТОР-3-ГАЛОГЕН-6-(ЗАМЕЩЕННЫХ)ПИКОЛИНАТОВ | 2012 |
|
RU2542985C1 |
Настоящее изобретение относится к способу получения пиримидинонового соединения формулы 1 и его солей, который включает стадию тиоамидирования соединения формулы 7 с использованием реагента Лавессона с последующей обработкой продукта спиртовым реагентом, как проиллюстрировано на следующей схеме реакций 2:
Схема реакции 2
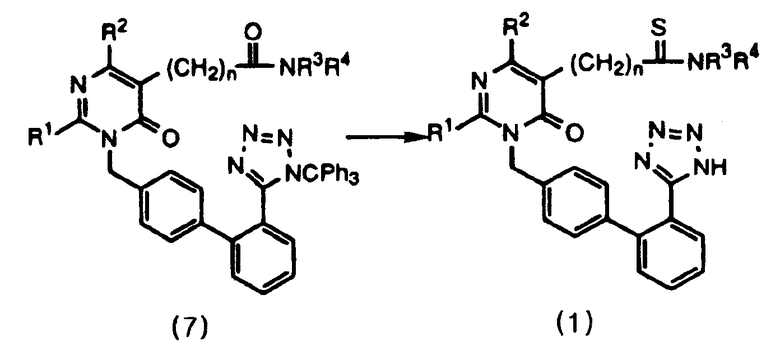
где R1 представляет С1-С4алкил с линейной или разветвленной цепью; R2 представляет С1-С4алкил с линейной или разветвленной цепью; R3, R4 представляют С1-С4алкил с линейной или разветвленной цепью; n является целым числом, выбранным из 1, 2, 3, 4, 5 и 6. Настоящее изобретение позволяет получить пиримидиноновое соединение формулы (1) относительно простым и высокопродуктивным способом. Другим объектом изобретения является тригитрат пиримидинонового соединения формулы (1). 2 н. и 8 з.п.ф-лы, 2 табл.
Схема реакции 2
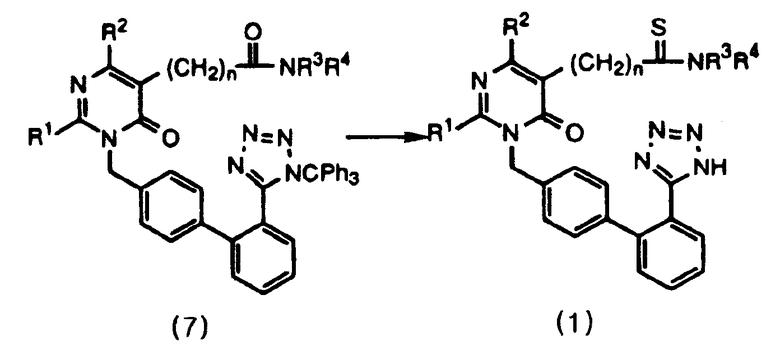
где R1 представляет С1-С4алкил с линейной или разветвленной цепью;
R2 представляет С1-С4алкил с линейной или разветвленной цепью;
R3, R4 представляют С1-С4алкил с линейной или разветвленной цепью;
n является целым числом, выбранным из 1, 2, 3, 4, 5 и 6.
Схема реакции 3
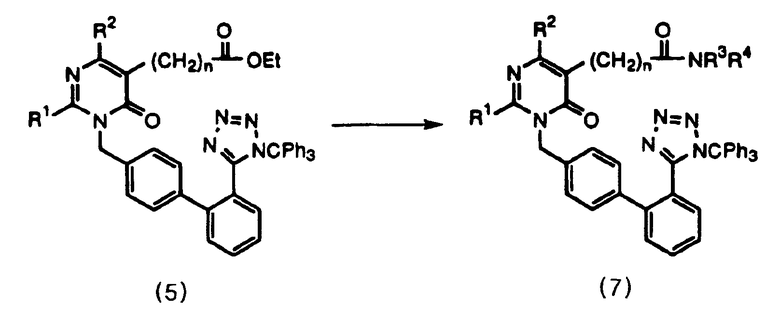
где R1, R2, R3, R4, R5 и n имеют значения, указанные в п.1.
Схема реакции 4
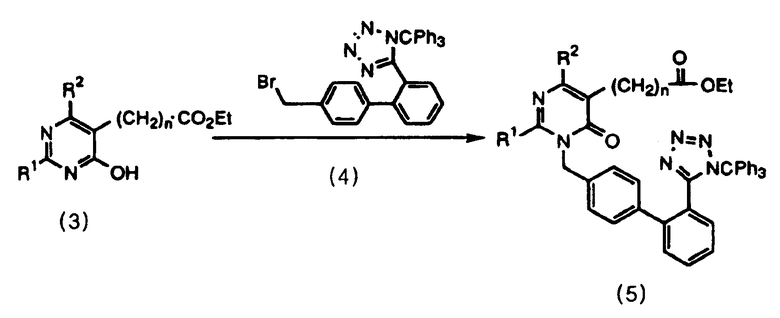
где R1, R2 и n имеют значения, указанные в п.1.
где R1 представляет С1-С4алкил с линейной или разветвленной цепью;
R2 представляет С1-С4алкил с линейной или разветвленной цепью;
R3, R4 представляют С1-С4алкил с линейной или разветвленной цепью;
n является целым числом, выбранным из 1, 2, 3, 4, 5 и 6.
| WO 9955681 A, 04.11.1999.WO 9608476 A, 21.03.1996.RU 2099331 C1, 20.12.1997. |

