ОБЛАСТЬ ТЕХНИКИ
[1] Данное изобретение относится к новым производным катехола или его фармацевтически приемлемой соли, способу их получения и содержащей их фармацевтической композиции. Более конкретно, данное изобретение относится к новому производному катехола или его фармацевтически приемлемой соли, имеющему алкильный фрагмент, замещенный алкиламино, и/или N-алкил-замещенный тиофен-(тио)карбоксамидный фрагмент, способу их получения и содержащей их фармацевтической композиции. Производное катехола или фармацевтически приемлемая соль согласно данному изобретению имеет превосходную активность по индуцированию аутофагии.
УРОВЕНЬ ТЕХНИКИ
[2] Аутофагия, также называемая аутофагоцитозом, представляет собой естественный регулируемый механизм клетки, который разбирает ненужные или дисфункциональные компоненты. Она обеспечивает упорядоченный распад и переработку клеточных компонентов. В процессе аутофагии расходуемые компоненты цитоплазмы изолируются от остальной части клетки в двухмембранном пузырьке, известном как аутофагосома. Далее аутофагосома сливается с доступной лизосомой, и в результате содержимое пузырька распадается и перерабатывается. Как правило, описывают три формы аутофагии: макроаутофагия, микроаутофагия и шаперон-опосредованная аутофагия (CMA). При заболевании аутофагию рассматривают как адаптивный ответ на стресс, способствующий выживанию клетки; но в других случаях она, по-видимому, способствует гибели клеток и осложнениям. При остром клеточном голодании распад клеточных компонентов способствует выживанию клеток, поддерживая уровень энергии клетки.
[3] В то же время при уменьшении аутофагии различные заболевания могут быть возникать из-за накопления неправильно свернутых белков и так далее. Например, сообщалось, что индуцирование аутофагии может лечить нейродегенеративные заболевания, такие как болезнь Хантингтона (HD), болезнь Паркинсона (PD), болезнь Альцгеймера (AD), прионная болезнь, рассеянный склероз и боковой амиотрофический склероз (болезнь Лу Герига) (например, патент Кореи № 10-1731908). Также сообщалось, что индуцирование аутофагии может лечить заболевания печени, такие как фиброз печени, цирроз печени, гепатит и жировая болезнь печени (например, публикация Кореи № 10-2017-0022790, опубликованная для всеобщего ознакомления). Помимо этого, сообщалось, что индуцирование аутофагии может лечить метаболические заболевания, такие как диабет, гиперлипидемия, ожирение и воспаление (например, публикация Кореи № 10-2018-0007307, опубликованная для всеобщего ознакомления). Кроме того, сообщалось, что индуцирование аутофагии может ингибировать чрезмерные иммунные ответы, связанные с сепсисом (например, публикация Кореи № 10-2012-0131401, опубликованная для всеобщего ознакомления).
[4] Следовательно, предполагается, что соединение, индуцирующее аутофагию, может быть успешно применено для профилактики, облегчения или лечения различных заболеваний, связанных с аутофагией, таких как нейродегенеративные заболевания, заболевания печени, метаболические заболевания, сепсис и так далее.
ПОДРОБНОЕ РАСКРЫТИЕ ИЗОБРЕТЕНИЯ
ТЕХНИЧЕСКАЯ ЗАДАЧА
[5] Авторы настоящего изобретения обнаружили, что определенное производное соединение катехола или его фармацевтически приемлемая соль, имеющее алкильный фрагмент, замещенный алкиламино, и/или N-алкил-замещенный тиофен- (тио) карбоксамидный фрагмент, имеет превосходную активность по индуцированию аутофагии и, следовательно, может быть эффективно применено для профилактики, облегчения или лечения различных заболеваний, связанных с аутофагией.
[6] Данное изобретение предлагает указанное производное соединение катехола или его фармацевтически приемлемую соль, способ их получения и содержащую их фармацевтическую композицию.
ТЕХНИЧЕСКОЕ РЕШЕНИЕ
[7] Одним объектом данного изобретения является новое производное соединение катехола или его фармацевтически приемлемая соль.
[8] Другим объектом данного изобретения является способ получения указанного производного соединения катехола или его фармацевтически приемлемой соли.
[9] Еще одним объектом данного изобретения является фармацевтическая композиция, содержащая указанное производное соединение катехола или его фармацевтически приемлемую соль в качестве активного ингредиента.
[10] Еще одним объектом данного изобретения является способ лечения заболевания, связанного с аутофагией, у млекопитающего, нуждающегося в данном лечении, включающий введение млекопитающему эффективного количества указанного производного соединения катехола или его фармацевтически приемлемой соли.
[11] Еще одним объектом данного изобретения является применение упомянутого производного соединения катехола или его фармацевтически приемлемой соли для производства лекарственного средства для профилактики, облегчения или лечения заболевания, связанного с аутофагией.
ТЕХНИЧЕСКИЕ РЕЗУЛЬТАТЫ ИЗОБРЕТЕНИЯ
[12] Соединение согласно данному изобретению, то есть производное соединение катехола (далее по тексту может быть названо как производное или соединение) или его фармацевтически приемлемая соль, имеющее алкильный фрагмент, замещенный алкиламино, и/или N-алкил-замещенный тиофен-(тио)карбоксамидный фрагмент, имеет превосходную активность по индуцированию аутофагии. Следовательно, соединение или его фармацевтически приемлемая соль согласно данному изобретению может быть успешно применено для профилактики, облегчения или лечения различных заболеваний, связанных с аутофагией, включая нейродегенеративные заболевания, заболевания печени, метаболические заболевания, сепсис и так далее. В частности, производное катехола или его фармацевтически приемлемая соль в соответствии с настоящим изобретением имеет молекулярную структуру, способную проникать через гематоэнцефалический барьер, то есть алкилзамещенный аминный фрагмент, такая структура позволяет применять его для профилактики, облегчения или лечения заболеваний, связанных с церебральным кровотоком, например, нейродегенеративных заболеваний, таких как болезнь Хантингтона (HD), болезнь Паркинсона (PD), болезнь Альцгеймера (AD), прионная болезнь, множественный склероз, болезнь Лу Герига и подобные.
ОПИСАНИЕ ФИГУР
[13] На ФИГ. 1 показаны результаты применения экспериментальных способов согласно Тестовому примеру 2 для оценки активности по улучшению функции печени путем перорального введения на модели повреждения печени.
[14] На ФИГ. 2 показаны результаты применения экспериментальных способов согласно Тестовому примеру 3 для оценки активности по улучшению функции печени путем перорального введения на модели повреждения печени.
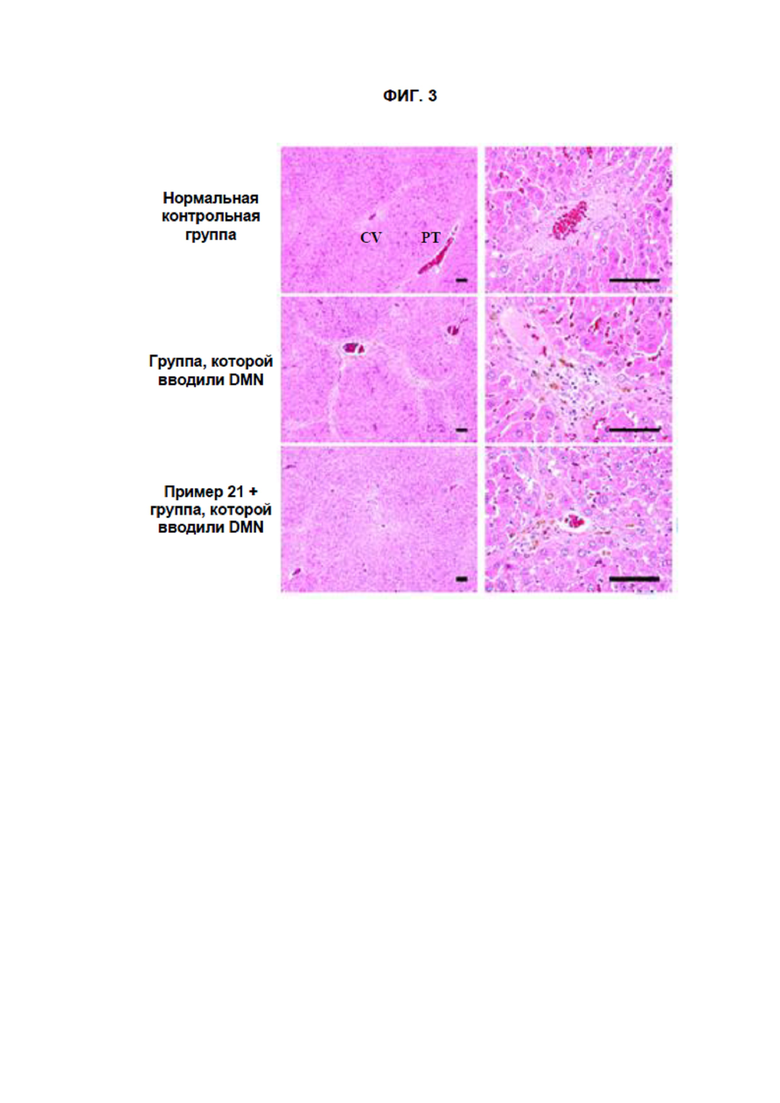
[15] На ФИГ. 3 показаны результаты, полученные при окрашивании гематоксилином и эозином (H&E) образцов ткани, полученных в Тестовом примере 3. Масштабная метка: левая черная линия 100 мкм, правая черная линия 100 мкм.
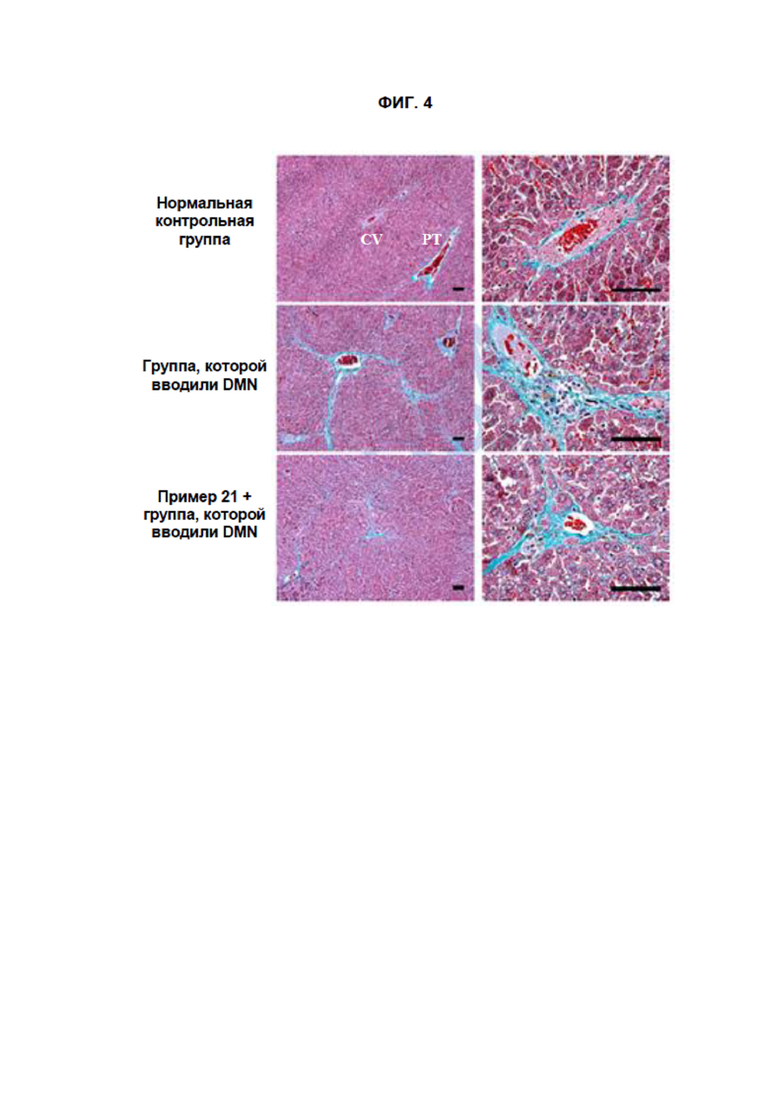
[16] На ФИГ. 4 показаны результаты, полученные путем окрашивания трихромом по Массону образцов ткани, полученных в Тестовом примере 3. Масштабная метка: левая черная линия 100 мкм, правая черная линия 100 мкм.
ЛУЧШИЙ ПРИМЕР ОСУЩЕСТВЛЕНИЯ ИЗОБРЕТЕНИЯ
[17] Используемый здесь термин «алкил» относится к линейному или разветвленному алифатическому углеводородному радикалу. Например, C1-C6 алкил означает линейный или разветвленный алифатический углеводород, содержащий от 1 до 6 атомов углерода, такой как метил, этил, пропил, н-бутил, н-пентил, н-гексил, изопропил, изобутил, втор-бутил, трет-бутил, неопентил и изопентил.
[18] Термин «гидрокси» относится к радикалу -ОН. Термин «алкокси» относится к радикалу, образованному замещением атома водорода гидроксигруппы алкилом. Например, C1-C6 алкокси содержит метокси, этокси, пропокси, н-бутокси, н-пентилокси, изопропокси, втор-бутокси, трет-бутокси, неопентилокси и изопентилокси.
[19] Термин «галоген» относится к радикалу фтора, брома, хлора или йода.
[20] Термин «амино» относится к радикалу -NH2. Термин «алкиламино» относится к амино, образованному замещением атома(ов) водорода аминогруппы моно- или диалкилом. Например, C1-C6 алкиламино содержит амино, замещенный моно- или ди-C1-6 алкилом.
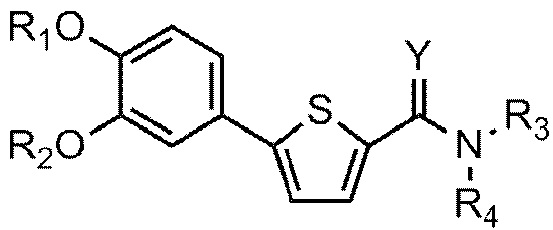
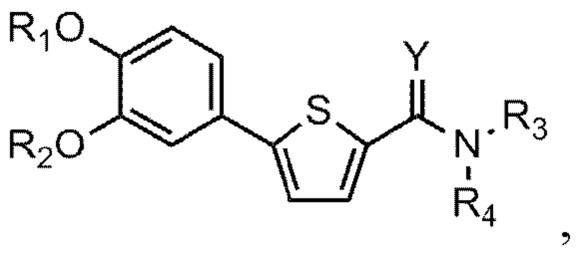
[21] Данное изобретение относится к соединению или его соли, имеющему превосходную активность по индуцированию аутофагии, то есть соединению Формулы 1 или его фармацевтически приемлемой соли:
[22] Формула 1
[23]
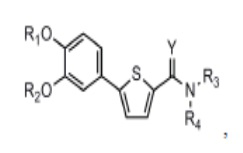
[24] причем
[25] Y - это O или S,
[26] (1) если Y - O,
[27] R1 представляет собой водород или C1-C4 алкильную группу, замещенную моно- или ди-C1-C5 алкиламино,
[28] R2 - C1-C6 алкильная группа,
[29] R3 - водород, и R4 - (4- (диметиламино) тетрагидро-2H-пиран-4-ил)метильная группа; C1-C6 алкильная группа; C1-C4 алкильная группа, замещенная моно- или ди-C1-C5 алкиламино; C1-C4 алкильная группа, замещенная азотсодержащим циклическим кольцом (причем азотсодержащее циклическое кольцо необязательно замещено C1-C4 алкилом); или пиперидинильная группа, необязательно замещенная C1-C4 алкилом, или
[30] R3 и R4 соединены друг с другом атомом азота, к которому они присоединены для образования пиперазинового кольца (причем пиперазиновое кольцо необязательно замещено C1-C4 алкилом),
[31] (2) если Y – это S,
[32] R1 и R2 независимо друг от друга представляют собой водород; C1-C6 алкильную группу или C1-C4 алкильную группа, замещенную моно- или ди-C1-C5 алкиламино,
[33] R3 - водород, а R4 - C1-C6 алкильная группа или C1-C4 алкильная группа, замещенная моно- или ди-C1-C5 алкиламино, или
[34] R3 и R4 соединены друг с другом атомом азота, к которому они присоединены для образования пиперазинового кольца, причем пиперазиновое кольцо необязательно замещено C1-C4 алкилом.
[35] В соединении или его фармацевтически приемлемой соли согласно данному изобретению Y может быть O. Предпочтительно, если Y является O; R1 может быть водородом или диэтиламиноэтильной группой, а R2 может быть метильной группой. Также азотсодержащее циклическое кольцо может быть морфолином, пиперидином или пирролидином.
[36] В одном из примеров осуществления изобретения предложено соединение или его фармацевтически приемлемая соль, причем:
[37] Y - это O,
[38] R1 - водород или диэтиламиноэтильная группа,
[39] R2 - метильная группа,
[40] R3 - водород, а
[41] R4 - (4- (диметиламино) тетрагидро-2H-пиран-4-ил) метильная группа; изопропильная группа, диметиламиноэтильная группа, диэтиламиноэтильная группа, диизопропиламиноэтильная группа, морфолиноэтильная группа, необязательно замещенная C1-C4 алкилом, пиперидиноэтильная группа, необязательно замещенная C1-C4 алкилом, пирролидиноэтильная группа, необязательно замещенная C1-C4 алкилом, или пиперидинильная группа, необязательно замещенная C1-C4 алкилом.
[42] В другом примере осуществления изобретения предложено соединение или его фармацевтически приемлемая соль, причем:
[43] Y - это O,
[44] R1 - водород или диэтиламиноэтильная группа,
[45] R2 - метильная группа,
[46] R3 - водород, а
[47] R4 - (4- (диметиламино) тетрагидро-2H-пиран-4-ил)метильная группа.
[48] В еще одном примере осуществления изобретения предложено соединение или его фармацевтически приемлемая соль, причем:
[49] Y - O,
[50] R1 - водород или диэтиламиноэтильная группа,
[51] R2 - метильная группа, и
[52] R3 и R4 соединены друг с другом атомом азота, к которому они присоединены для образования пиперазинового кольца, необязательно замещенного C1-C4 алкилом.
[53] В соединении или его фармацевтически приемлемой соли согласно данному изобретению Y может представлять собой S. Предпочтительно, когда Y - S, R1 может быть водородом или диэтиламиноэтильной группой, а R2 - метильной группой.
[54] В одном из примеров осуществления изобретения предложено соединение или его фармацевтически приемлемая соль, причем:
[55] Y - это S,
[56] R1 - водород или диэтиламиноэтильная группа,
[57] R2 - метильная группа,
[58] R3 - водород, а
[59] R4 - изопропильная группа или диизопропиламиноэтильная группа.
[60] В другом примере осуществления изобретения предложено соединение или его фармацевтически приемлемая соль, причем:
[61] Y - это S,
[62] R1 - водород или диэтиламиноэтильная группа,
[63] R2 - метильная группа, и
[64] R3 и R4 соединены друг с другом атомом азота, к которому они присоединены для образования пиперазинового кольца, необязательно замещенного C1-C4 алкилом.
[65] Предпочтительно, чтобы соединение или его фармацевтически приемлемая соль согласно данному изобретению могли быть одним или несколькими соединениями, выбранными из группы, включающей:
[66] N-(4-метилпиперазино)-5-(4-гидрокси-3-метоксифенил)тиофен-2-карбоксамид гидрохлорид,
[67] N-(4-метилпиперазино)-5-(4-(2-диэтиламино)этокси-3-метоксифе-нил)тиофен-2-карбоксамид гидрохлорид,
[68] N-(4-метилпиперазино)-5-(4-гидрокси-3-метоксифенил)тиофен-2-тиокарбоксамид гидрохлорид,
[69] N-(4-метилпиперазино)-5-(4-(2-диэтиламино)этокси-3-метоксифе-нил)тиофен-2-тиокарбоксамид гидрохлорид,
[70] N-(2-(диизопропиламино) этил)-5-(4-гидрокси-3-метоксифенил) тиофен-2-карбоксамид гидрохлорид,
[71] N-(2-(диизопропиламино)этил)-5-(4-(2-диэтиламино)этокси-3-метоксифенил)тиофен-2-карбоксамид гидрохлорид,
[72] N-(2-(диизопропиламино)этил)-5-(4-гидрокси-3-метоксифенил)тио-фен-2-тиокарбоксамид гидрохлорид,
[73] N-(2-(диизопропиламино)этил)-5-(4-(2-диэтиламино)этокси-3-метоксифенил)тиофен-2-тиокарбоксамид гидрохлорид,
[74] N-изопропил-5-(4-(2-диэтиламино)этокси-3-метоксифенил)тиофен-2-карбоксамид гидрохлорид,
[75] N-изопропил-5-(4-(2-диэтиламино)этокси-3-метоксифенил)тиофен-2-тиокарбоксамид гидрохлорид,
[76] N-изопропил-5-(4-гидрокси-3-метоксифенил)тиофен-2-тиокарбокса-мид,
[77] N-(2-(диэтиламино)этил)-5-(4-гидрокси-3-метоксифенил)тиофен-2-карбоксамид гидрохлорид,
[78] N-(2-(диэтиламино)этил)-5-(4-(2-диэтиламино)этокси-3-метоксифе-нил)тиофен-2-карбоксамид гидрохлорид,
[79] N-(2-(диметиламино)этил)-5-(4-гидрокси-3-метоксифенил)тиофен-2-карбоксамид гидрохлорид,
[80] N-(2-(диметиламино)этил)-5-(4-(2-диэтиламино)этокси-3-метокси-фенил)тиофен-2-карбоксамид гидрохлорид,
[81] N-(2-(4-морфолино)этил)-5-(4-гидрокси-3-метоксифенил)тиофен-2-карбоксамид гидрохлорид,
[82] N-(2-(1-пиперидино)этил)-5-(4-гидрокси-3-метоксифенил)тиофен-2-карбоксамид гидрохлорид,
[83] N-(2-(1-пиперидино)этил)-5-(4-(2-диэтиламино)этокси-3-метокси-фенил)тиофен-2-карбоксамид гидрохлорид,
[84] N-(2-(1-пирролидино)этил)-5-(4-гидрокси-3-метоксифенил)тиофен-2-карбоксамид гидрохлорид,
[85] N-((4-(диметиламино)тетрагидро-2H-пиран-4-ил)метил)-5-(4-гидрокси-3-метоксифенил)тиофен-2-карбоксамид гидрохлорид,
[86] N-((4-(диметиламино)тетрагидро-2H-пиран-4-ил)метил)-5-(4-(2-диэтиламино) этокси-3-метоксифенил)тиофен-2-карбоксамид гидрохлорид,
[87] N-(4-(1-метил)пиперидинил)-5-(4-гидрокси-3-метоксифенил)тио-фен-2-карбоксамид гидрохлорид,
[88] N-(2-(2-(1-метил)пирролидино)этил)-5-(4-гидрокси-3-метоксифе-нил)тиофен-2-карбоксамид гидрохлорид,
[89] N-(2-(2-(1-метил)пирролидино)этил)-5-(4-(2-диэтиламино)этокси-3-метоксифенил)тиофен-2-карбоксамид гидрохлорид,
[90] N-((S)-2-(1-этил)пирролидинометил)-5-(4-гидрокси-3-метоксифе-нил)тиофен-2-карбоксамид гидрохлорид и
[91] N-((S)-2-(1-этил)пирролидинометил)-5-(4-(2-диэтиламино)этокси-3-метоксифенил) тиофен-2-карбоксамид гидрохлорид.
[92] Более предпочтительно, чтобы соединение согласно данному изобретению представляло собой N-((4-(диметиламино)тетрагидро-2H-пиран-4-ил)метил)-5-(4-гидрокси-3-метоксифенил)тиофен-2-карбоксамид или его фармацевтически приемлемую соль (например, гидрохлорид); или представляло собой N-((4-(диметиламино)тетрагидро-2H-пиран-4-ил)метил)-5-(4-(2-диэтиламино)этокси-3-метоксифенил)тиофен-2-карбоксамид или его фармацевтически приемлемую соль (например, гидрохлорид).
[93] Соединение формулы 1 согласно данному изобретению может быть представлено в форме фармацевтически приемлемой соли, например, в форме кислотно-аддитивной соли. В частности, соединение согласно данному изобретению имеет алкилзамещенный аминный фрагмент и, следовательно, может быть легко изолировано в форме кислотно-аддитивной соли (например, в форме гидрохлорида) в отличие от обычных соединений (например, соединений, описанных в публикации Кореи № 10-2017-0022790). То есть соединение согласно данному изобретению в форме кислотно-аддитивной соли может быть легко получено процессами кислотно-щелочной обработки и может быть легко применено для способов равномерного увеличения без проведения процессов колоночной хроматографии в отличие от обычных соединений (например, соединений, описанных в публикации Кореи № 10-2017-0022790). Также соединение согласно данному изобретению в форме кислотно-аддитивной соли имеет превосходную растворимость в воде и, следовательно, может быть легко приготовлено, а также обеспечивает превосходную биодоступность при пероральном введении. Кислотно-аддитивная соль может быть производной неорганической кислоты или органической кислоты, такой кислоты как соляная кислота, бромистоводородная кислота, йодистоводородная кислота, азотная кислота, серная кислота, фосфорная кислота, уксусная кислота, молочная кислота, лимонная кислота, винная кислота, янтарная кислота, малеиновая кислота, малоновая кислота, щавелевая кислота, фумаровая кислота, глюконовая кислота, сахарная кислота, бензойная кислота, метансульфоновая кислота, этансульфоновая кислота, бензолсульфоновая кислота, п-толуолсульфоновая кислота, памоевая кислота и т.д., без ограничения вышеперечисленным. Кислотно-аддитивная соль может быть получена в ходе реакции соединения Формулы 1 с неорганической кислотой или органической кислотой в обычном растворителе, например воде, спирте, тетрагидрофуране, ацетоне или их смеси.
[94] Соединение Формулы 1 или его фармацевтически приемлемая соль может иметь заместитель(и), содержащий асимметричный углерод, и, следовательно, может находиться в форме рацемической смеси (RS) или в формах оптических изомеров, таких как (R) или (S) изомер. Следовательно, если не указано иное, соединение Формулы 1 или его фармацевтически приемлемая соль содержит как рацемическую смесь (RS), так и оптические изомеры, такие как (R) или (S) изомер. Также соединение Формулы 1 или его фармацевтически приемлемая соль может быть в форме цис- или транс-геометрического изомера в зависимости от заместителя(ей). Следовательно, если не указано иное, соединение Формулы 1 или его фармацевтически приемлемая соль содержит как цис-, так и транс-геометрические изомеры. Также соединение Формулы 1 или его фармацевтически приемлемая соль может быть в форме одного или нескольких диастереомерных изомеров или их смеси. Следовательно, если не указано иное, соединение Формулы 1 или его фармацевтически приемлемая соль содержит как диастереомерный изомер(ы), так и их смесь.
[95] Соединение Формулы 1 или его фармацевтически приемлемая соль согласно данному изобретению может быть в безводной форме, в форме гидрата или в форме сольвата. Помимо этого, соединение Формулы 1 или его фармацевтически приемлемая соль согласно данному изобретению может быть в аморфной или кристаллической формах. Указанные аморфные или кристаллические формы также могут быть в форме гидрата или в форме сольвата. Гидрат или сольват может содержать воду или органический растворитель в стехиометрическом или нестехиометрическом количестве по отношению к соединению Формулы 1 или его фармацевтически приемлемой соли.
[96] Данное изобретение также включает способ получения соединения Формулы 1 или его фармацевтически приемлемой соли.
[97] Например, соединение Формулы 1 или его фармацевтически приемлемая соль, где Y представляет собой O (то есть соединение Формулы 1a или его фармацевтически приемлемая соль), может быть получено ацилированием соединения Формулы 4 соединением Формулы 5 для получения соединения Формулы 2; и связыванием соединения Формулы 2 с соединением Формулы 3 для получения соединения Формулы 1a, как показано на следующей схеме реакции 1:
[98] Схема реакции 1
[99]
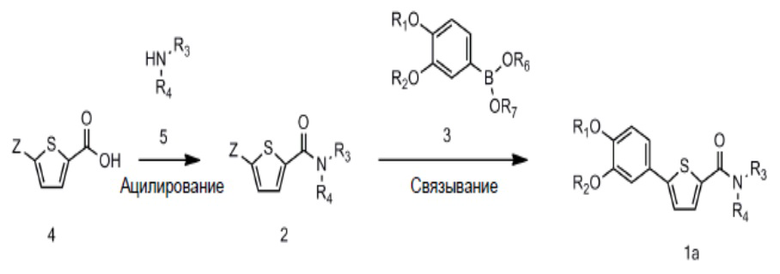
[100] На схеме реакции 1 R1, R2, R3 и R4 такие же, как определено выше, Z - галоген, а R6 и R7 представляют собой водород; или соединены друг с другом атомом бора, к которому они присоединены, для образования 4,4,5,5-тетраметил-1,3,2-диоксаборолана.
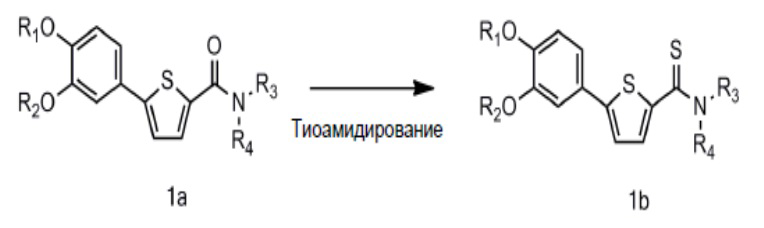
[101] Соединения Формул 4 и 5, которые являются известными соединениями, коммерчески доступны. Ацилирование может быть проведено с использованием ацилирующего агента, такого как 1-этил-3-(3-диметиламинопропил)карбодиимид (EDC), дициклогексилкарбодиимид (DCC), 1,1'-карбонилдиимидазол (CDI), N-этоксикарбонил-2-этокси-1,2-дигидрохинолин (EEDQ). Ацилирование также может быть проведено через реакцию соединения Формулы 4 с тионилхлоридом, оксалилхлоридом, хлоридом фосфора и т.д. для получения ацилхлорида с последующим вступлением в реакцию с соединением Формулы 5. Ацилирование может быть проведено в обычном органическом растворителе, например дихлорметане.
[102] Связывание может быть выполнено в присутствии катализатора (например, тетракис (трифенилфосфин)палладия (0)) и основания (например, карбоната натрия). Реакция соединения Формулы 2 с соединением Формулы 3 может быть проведена при молярном соотношении, находящемся в диапазоне от 1:2 до 2:1, предпочтительно в молярном соотношении приблизительно 1:1. Связывание может быть выполнено в воде, спирте C1-C4, тетрагидрофуране, 1,2-диметоксиэтане или их смеси.
[103] Помимо этого, соединение Формулы 1 или его фармацевтически приемлемая соль, где Y представляет собой S (то есть соединение Формулы 1b или его фармацевтически приемлемая соль), может быть получено выполнением тиоамидирования соединения Формулы 2 для получения соединения Формулы 6 и связыванием соединения Формулы 6 с соединением Формулы 3 для получения соединения Формулы 1b, как показано на следующей схеме реакции 2:
[104] Схема реакции 2
[105]
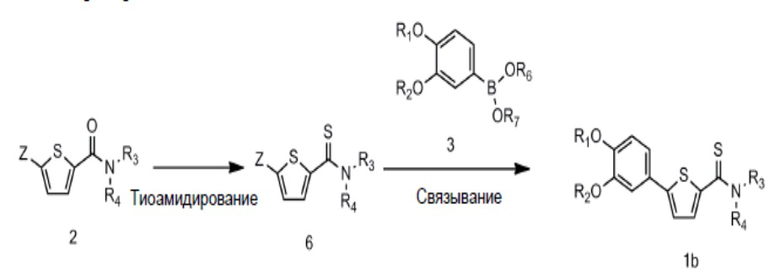
[106] На схеме реакции 2 R1, R2, R3, R4, Z, R6 и R7 такие же, как определено выше.
[107] В схеме реакции 2 соединение Формулы 2 может быть получено, как описано для схемы реакции 1. Тиоамидирование может быть проведено через реакцию соединения Формулы 2 с P4S10, бис(трициклогексилтин) сульфидом или реагентом Лавессона. Реакция тиоамидирования может быть проведена в толуоле, дихлорметане, тетрагидрофуране или их смешанном растворителе. Помимо этого, реакция связывания может быть проведена, как описано на схеме реакции 1, с использованием соединения Формулы 6 вместо соединения Формулы 2.
[108] А также, соединение Формулы 1 или его фармацевтически приемлемая соль, где Y представляет собой S (то есть соединение Формулы 1b или его фармацевтически приемлемая соль), может быть получено тиоамидированием соединения Формулы 1a для получения соединения Формулы 1b, как показано на следующей схеме реакции 3:
[109] Схема реакции 3
[110]
[111] На схеме реакции 3 R1, R2, R3, и R4 такие же, как определено выше.
[112] На схеме реакции 3 соединение Формулы 1a может быть получено, как описано на схеме реакции 1. Помимо этого, тиоамидирование может быть выполнено, как описано на схеме реакции 2, с использованием соединения Формулы 1a вместо соединения Формулы 2.
[113] Производное катехола согласно данному изобретению, то есть соединение Формулы 1 или его фармацевтически приемлемая соль, имеет превосходную активность по индуцированию аутофагии. Следовательно, соединение Формулы 1 или его фармацевтически приемлемая соль согласно данному изобретению может быть успешно применено для профилактики, облегчения или лечения различных заболеваний, связанных с аутофагией, включая нейродегенеративные заболевания, заболевания печени, метаболические заболевания, сепсис и так далее.
[114] Следовательно, данное изобретение содержит фармацевтическую композицию для индуцирования аутофагии, содержащую терапевтически эффективное количество соединения Формулы 1 или его фармацевтически приемлемой соли в качестве активного ингредиента.
[115] Заболевания, связанные с аутофагией, включают без ограничения различные заболевания, которые можно предотвратить, облегчить или излечить путем индуцирования аутофагии. Например, фармацевтическая композиция согласно данному изобретению может быть фармацевтической композицией для профилактики, облегчения или лечения нейродегенеративных заболеваний, выбранных из группы, содержащей болезнь Хантингтона, болезнь Паркинсона, болезнь Альцгеймера, прионную болезнь, рассеянный склероз и болезнь Лу Герига; заболевания печени, выбранные из группы, включающей фиброз печени, цирроз печени, гепатит и жировую болезнь печени; метаболические заболевания, выбранные из группы, включающей диабет, гиперлипидемию, ожирение и воспаление, или сепсис.
[116] Фармацевтическая композиция согласно данному изобретению может содержать фармацевтически приемлемый носитель, такой как разбавители, разрыхлители, подсластители, смазывающие вещества или вкусоароматические агенты. Фармацевтическая композиция может быть приготовлена в форме, предназначенной для приема внутрь, такой как таблетки, капсулы, порошки, гранулы, суспензии, эмульсии или сиропы; или в лекарственной форме для парентерального применения, такой как внешние растворы, внешние суспензии, внешние эмульсии, гели (например, мази), ингаляторы, спреи, инъекции, в соответствии с традиционными способами. Лекарственная форма может представлять собой различные формы, например лекарственные формы для однократного или для многократного применения.
[117] Фармацевтическая композиция согласно данному изобретению может содержать, например, разбавитель (например, лактозу, кукурузный крахмал и т.д.), смазывающее вещество (например, стеарат магния), эмульгирующий агент, суспендирующий агент, стабилизатор и/или изотонический агент. При необходимости композиция дополнительно содержит подсластители и/или вкусоароматические агенты.
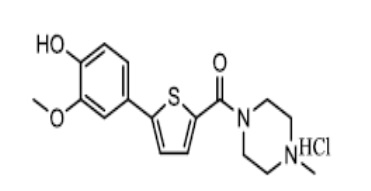
[118] Композицию согласно данному изобретению можно применять внутрь или парентерально, в том числе путем ингалируемого, внутривенного, интраперитонеального, подкожного, интрацеребровентрикулярного, ректального и местного способа применения. Таким образом, композицию согласно данному изобретению можно включать в различные формы, такие как таблетки, капсулы, водные растворы или суспензии. В случае таблеток для приема внутрь обычно используют такие носители, как лактоза, кукурузный крахмал и смазывающие вещества, такие как стеарат магния. В случае капсул для приема внутрь в качестве разбавителя можно использовать лактозу и/или высушенный кукурузный крахмал. Если для приема внутрь требуется водная суспензия, активный ингредиент можно объединять с эмульгирующими и/или суспендирующими агентами. При необходимости можно использовать некоторые подсластители и/или вкусоароматические агенты. Для внутримышечного, интраперитонеального, подкожного и внутривенного введения обычно готовят стерильные растворы активного ингредиента; при этом необходимо соответствующим образом регулировать pH этих растворов и буферизовать их. Для придания препарату изотоничности необходимо контролировать общую концентрацию растворенных веществ для внутривенного введения. Композиция согласно данному изобретению может иметь форму водного раствора, содержащего фармацевтически приемлемые носители, например физиологический раствор, уровень pH которого составляет 7,4. Эти растворы можно вводить в кровоток пациента внутримышечно путем местной болюсной инъекции.
[119] Производное катехола согласно данному изобретению, то есть соединение Формулы 1 или его фармацевтически приемлемая соль, может быть введено пациенту в терапевтически эффективном количестве в диапазоне от примерно 0,0001 мг/кг до примерно 100 мг/кг в сутки, предпочтительно от примерно 0,001 мг/кг до примерно 100 мг/кг в сутки. Введение может быть выполнено перорально или парентерально один или несколько раз в сутки. Безусловно, доза может быть изменена в зависимости от возраста пациента, состояния, весы тела, восприимчивости, степени заболевания, способа введения, продолжительности введения и т.п. В зависимости от способа введения фармацевтическая композиция согласно данному изобретению может содержать соединение Формулы 1 или его фармацевтически приемлемую соль в количестве от 0,001 до 99% по массе, предпочтительно от 0,01 до 60% по массе.
[120] Данное изобретение также включает способ индуцирования аутофагии у млекопитающего, нуждающегося в этом, предусматривающий введение млекопитающему терапевтически эффективного количества соединения Формулы 1 или его фармацевтически приемлемой соли. Например, данное изобретение включает способ для профилактики, облегчения или лечения нейродегенеративных заболеваний, выбранных из группы, содержащей болезнь Хантингтона, болезнь Паркинсона, болезнь Альцгеймера, прионную болезнь, рассеянный склероз и болезнь Лу Герига; заболевания печени, выбранные из группы, содержащей фиброз печени, цирроз печени, гепатит и жировую болезнь печени; метаболические заболевания, выбранные из группы, содержащей диабет, гиперлипидемию, ожирение и воспаление; или сепсис.
[121] Данное изобретение также содержит применение соединения Формулы 1 или его фармацевтически приемлемой соли для производства лекарственного средства для индуцирования аутофагии у млекопитающего, нуждающегося в этом. Например, данное изобретение включает применение соединения Формулы 1 или его фармацевтически приемлемой соли для производства лекарственного средства для профилактики, облегчения или лечения нейродегенеративных заболеваний, выбранных из группы, содержащей болезнь Хантингтона, болезнь Паркинсона, болезнь Альцгеймера, прионную болезнь, рассеянный склероз и болезнь Лу Герига; заболевания печени, выбранные из группы, содержащей фиброз печени, цирроз печени, гепатит и жировую болезнь печени; метаболические заболевания, выбранные из группы, содержащей диабет, гиперлипидемию, ожирение и воспаление; или сепсис.
[122] Приведенные ниже примеры и тестовые примеры представлены только в целях иллюстрации и не предназначены для ограничения объема изобретения.
[123] Анализ соединений, полученных в приведенных ниже примерах, проводили, как описано ниже: анализ спектра ядерного магнитного резонанса (ЯМР) проводили на спектрометре Bruker 400 МГц и результаты анализа химических сдвигов выражены в мкг/г (ppm). Колоночную хроматографию проводили на силикагеле (Merck, 70-230 меш). Если не указано иное, все исходные материалы были приобретены на коммерческой основе и использовались без дополнительной очистки. Все реакции и хроматографические фракции анализировали посредством тонкослойной хроматографии (ТLC) на пластине силикагеля 250 нм и визуализировали с помощью окрашивания ультрафиолетом или йодом (I2).
[124]
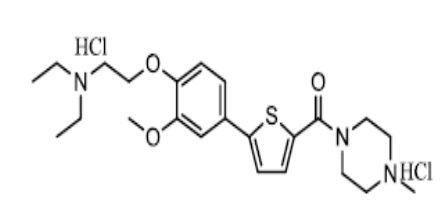
[125] Пример 1: Получение N-(4-метилпиперазино)-5-(4-гидрокси-3-метоксифенил)тиофен-2-карбоксамид гидрохлорида
[126]
[127] Смесь 5-бромтиофен-2-карбоновой кислоты (6,21 г) с дихлорметаном (30 мл) и диметилформамидом (0,25 мл) перемешивали в течение 10 минут. К смеси добавили тионилхлорид (2,64 мл), после чего смесь нагревали в колбе с обратным холодильником при перемешивании в течение 3 часов. Реакционную смесь охладили до комнатной температуры и затем сконцентрировали при пониженном давлении. К полученному концентрату добавили дихлорметан (45 мл), который затем охладили до 0 ~ 10 °C. К реакционной смеси добавили K2CO3 (4,62 г), после чего смесь перемешивали в течение 20 минут. К смеси добавили N-метилпиперазин (3,30 мл). Реакционную смесь перемешивали при комнатной температуре в течение 2 часов, после чего к ней добавили очищенную воду (45 мл). Полученную промывочную воду экстрагировали дихлорметаном (30 мл). Экстракт объединили с реакционной смесью. Полученную смесь промыли очищенной водой (30 мл), высушили над безводным сульфатом натрия, сконцентрировали в вакууме для получения 8,66 г промежуточного соединения, то есть N-(4-метилпиперазино) -5-бром-тиофен-2-карбоксамида (выход: 100 %).
[128] К N-(4-метилпиперазино)-5-бром-тиофен-2-карбоксамиду (8,66 г) добавили тетракис(трифенилфосфин)палладий(0) (3,48 г) и 1,2-диметоксиэтан (51 мл). К смеси добавили раствор карбоната натрия (9,36 г) в очищенной воде (51 мл), после чего смесь перемешивали при комнатной температуре в течение 30 минут. К реакционной смеси добавили раствор (4-гидрокси-3-метоксифенил)-(тетраметил-1,3-окса)боролана (8,25 г) в этаноле (51 мл), после чего смесь перемешивали при температуре примерно 80 °C в течение 5 часов. Реакционную смесь охладили до комнатной температуры, после чего отфильтровали для удаления нерастворимого материала. Полученный нерастворимый материал промыли этанолом (40 мл), после чего полученный промывочный раствор объединили с фильтратом. Полученный фильтрат сконцентрировали при пониженном давлении для удаления растворителя. К полученному остатку при перемешивании добавили очищенную воду (200 мл) и 6N соляную кислоту (10 мл). Полученный раствор дважды промыли хлороформом (100 мл и 50 мл соответственно), после чего его pH довели до pH 8 ~ 9, используя гидроксид натрия (около 3,0 г). Раствор дважды экстрагировали хлороформом (100 мл и 50 мл соответственно). Объединенный экстракт высушили над безводным сульфатом натрия, после чего сконцентрировали в вакууме. К полученному концентрату добавили ацетон (30 мл). Смесь перемешивали в течение 1 часа, после чего отфильтровали. Полученное твердое вещество промыли ацетоном (5 мл), высушили в вакууме при 30 °C в течение 3 часов для получения N-(4-метилпиперазино)-5-(4-гидрокси-3-метоксифенил)тиофен-2-карбоксамида (8,68 г, выход: 87,0%). Высушенное твердое вещество (1,50 г) растворили в смешанном растворителе из метанола (10 мл) и хлороформа (3 мл), после чего к нему добавили 2N раствор соляной кислоты в этаноле (1,5 мл). Смесь сконцентрировали при пониженном давлении. К полученному концентрату добавили ацетон (10 мл). Смесь перемешивали в течение 1 часа, после чего отфильтровали. Полученное твердое вещество промыли ацетоном (5 мл), после чего высушили в вакууме для получения 1,52 г указанного в заголовке соединения (выход: 91,6%, общий выход: 80,0%).
[129] TLC Rf = 0,20 в 10% MeOH в хлороформе.
[130] 1H NMR (400MHz, MeOH-d4) δ 7,46 (d, 1H, J = 4,0 Гц), 7,31 (d, 1H, J = 4,0 Гц), 7,22 (d, 1H, J = 2,0 Гц), 7,16 (dd, 1H, J = 2,0 Гц, 8,0 Гц), 6,98 (d, 1H, J = 8,0 Гц), 4,72 ~ 4,60 (m, 2H), 3,93 (s, 3H), 3,70 ~ 3,48 (m, 4H), 3,30~3,20 (m, 2H), 2,99 (s, 3H)
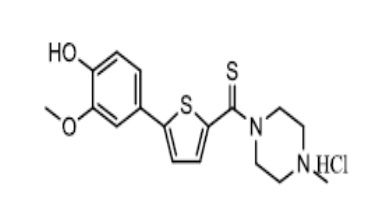
[131]
[132] Пример 2: Получение N-(4-метилпиперазино)-5-(4-(2-диэтиламино)этокси-3-метоксифенил)тиофен-2-карбоксамид гидрохлорида
[133]
[134] К смеси N-(4-метилпиперазино)-5-(4-гидрокси-3-метокси-фенил)тиофен-2-карбоксамида (3,32 г), полученного посредством тех же процедур, что и в Примере 1, с 2-диэтиламиноэтилхлорид гидрохлоридом (1,72 г) и толуолом (50 мл) добавили гидроксид натрия (0,80 г). Реакционную смесь перемешивали при 85 °C в течение 4 часов, после чего охладили до комнатной температуры. К реакционной смеси добавили очищенную воду (50 мл), после чего смесь перемешивали в течение 30 минут. Отделенный органический слой промыли насыщенным раствором карбоната натрия, после чего экстрагировали 1N соляной кислотой (50 мл). Экстракт промыли этилацетатом (20 мл), после чего довели его pH до pH 7~8, используя гидроксид натрия (около 2,0 г). Раствор дважды экстрагировали дихлорметаном (50 мл и 30 мл соответственно). Полученный экстракт высушили над безводным сульфатом натрия, после чего сконцентрировали в вакууме для получения 4,0 г неочищенного продукта, то есть N-(4-метилпиперазино)-5-(4-(2-диэтиламино)этокси-3-метоксифенил)тио-фен-2-карбоксамида (выход: 89,5%). К полученному остатку добавили 4N раствор соляной кислоты в этаноле (6,0 мл) и ацетоне (60 мл). Смесь перемешивали при комнатной температуре в течение 1 часа, после чего отфильтровали. Полученное твердое вещество промыли ацетоном (5 мл), после чего высушили в вакууме для получения 4,08 г указанного в заголовке соединения (выход: 87,4%, общий выход: 78,2%).
[135] TLC Rf = 0,21 в 10% MeOH в хлороформе.
[136] 1H NMR (400 мГц, MeOH-d4) δ 7.50 (d, 1H, J = 4,0 Гц), 7.41 (d, 1H, J = 4,0 Гц), 7,33 (d, 1H, J = 2,0 Гц), 7,30 (dd, 1H, J = 2,0 Гц, 8,4 Гц), 7,13 (d, 1H, J = 8,4 Гц), 4,72 ~ 4,60 (m, 2H), 4,42 (t, 2H, J = 4,8 Гц), 3,97 (s, 3H), 3,70 ~ 3,60 (m, 6H), 3,50~3,38 (m, 4H), 3,30~3,20 (m, 2H), 2,99 (s, 3H), 1,42 (t, 6H, J = 7,2 Гц).
[137]
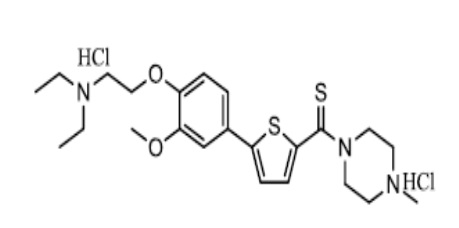
[138] Пример 3: Получение N-(4-метилпиперазино)-5-(4-гидрокси-3-метоксифенил)тиофен-2-тиокарбоксамид гидрохлорида
[139]
[140] Смесь N-(4-метилпиперазино)-5-бром-тиофен-2-карбоксамида (2,92 г), полученного по тем же процедурами, что и в Примере 1, с толуолом (15 мл) и тетрагидрофураном (15 мл) перемешивали в течение 10 минут. Реагент Лавессона (4,25 г) добавили к смеси, которую затем перемешивали при 50 °C в течение 5 часов. Реакционную смесь охладили до комнатной температуры, после чего отфильтровали для удаления нерастворимого материала. К полученному фильтрату добавили этилацетат (200 мл), после чего экстрагировали смешанным раствором очищенной воды (200 мл) и 2N соляной кислоты (25 мл) и смешанным раствором очищенной воды (200 мл) и 2N соляной кислоты (15 мл) соответственно. Объединенный экстракт промыли этилацетатом (200 мл), после чего довели его pH до pH 9~10, используя гидроксид натрия (около 4,3 г). Раствор трижды экстрагировали хлороформом (100 мл, 50 мл и 50 мл соответственно). Полученный экстракт высушили над безводным сульфатом натрия, после чего сконцентрировали в вакууме для получения 2,47 г промежуточного соединения, то есть N-(4-метилпиперазино)-5-бром-тиофен-2-тиокарбоксамида (выход: 80,0 %).
[141] К N-(4-метилпиперазино)-5-бром-тиофен-2-карбоксамиду (2,47 г) добавили тетракис(трифенилфосфин)палладий(0) (1,04 г) и 1,2-диметоксиэтан (15 мл). К смеси добавили раствор карбоната натрия (2,81 г) в очищенной воде (15 мл), после чего смесь перемешивали при комнатной температуре в течение 30 минут. К реакционной смеси добавили раствор (4-гидрокси-3-метоксифенил)- (тетраметил-1,3-окса)боролана (2,48 г) в этаноле (15 мл), после чего смесь перемешивали при температуре примерно 80 °C в течение 5 часов. Реакционную смесь охладили до комнатной температуры, после чего отфильтровали для удаления нерастворимого материала. Полученный фильтрат промыли этанолом (15 мл), после чего сконцентрировали при пониженном давлении для удаления растворителя. К полученному остатку при перемешивании добавили очищенную воду (60 мл) и 6N соляную кислоту (3 мл). Полученный раствор дважды промыли хлороформом (30 мл и 15 мл соответственно), после чего его pH довели до pH 8 ~ 9, используя гидроксид натрия (около 1,0 г). Раствор дважды экстрагировали хлороформом (30 мл и 50 мл соответственно). Полученный экстракт высушили над безводным сульфатом натрия, после чего сконцентрировали в вакууме. К полученному концентрату добавили ацетон (10 мл). Смесь перемешивали в течение 1 часа, после чего отфильтровали. Полученное твердое вещество (то есть N-(4-метилпиперазино)-5-(4-гидрокси-3-метоксифенил)тиофен-2-тиокарбоксамид) растворили в смешанном растворителе из метанола (50 мл) и хлороформа (15 мл), после чего к нему добавляли 2N раствор соляной кислоты в этаноле (5 мл). Смесь сконцентрировали при пониженном давлении. К полученному концентрату добавили ацетон (50 мл). Смесь перемешивали в течение 1 часа, после чего отфильтровали. Полученное твердое вещество промыли ацетоном (5 мл), после чего высушили в вакууме для получения 2,49 г указанного в заголовке соединения (выход: 80,0 %).
[142] TLC Rf = 0,33 в 10% MeOH в хлороформе.
[143] 1H NMR (400 мГц, MeOH-d4) δ 7,30 (d, 1H, J = 3,6 Гц), 7,21~7,15 (m, 3H), 6,98 (d, 1H, J = 8,0 Гц), 5,20 ~ 5,10 (m, 2H), 3,93 (s, 3H), 3,85 ~ 3,65 (m, 4H), 3,32 ~ 3,25(m, 2H), 3,00 (s, 3H)
[144]
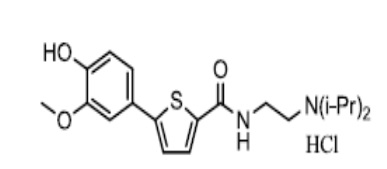
[145] Пример 4: Получение N-(4-метилпиперазино)-5-(4-(2-диэтиламино)этокси-3-метоксифенил)тиофен-2-тиокарбоксамид гидрохлорида
[146]
[147] Смесь N-(4-метилпиперазино)-5-(4-(2-диэтиламино)этокси-3-метоксифенил)тиофен-2-карбоксамида (4,0 г), полученного по тем же процедурам, что и в Примере 2, с толуолом (15 мл) и тетрагидрофураном (15 мл) перемешивали в течение 10 минут. Реагент Лавессона (4,04 г) добавили к реакционной смеси, которую затем перемешивали при 50 °C в течение 5 часов. Реакционную смесь охладили до комнатной температуры, после чего отфильтровали для удаления нерастворимого материала. К полученному фильтрату добавили хлороформ (100 мл), после чего экстрагировали раствором очищенной воды (60 мл) и 2N соляной кислоты (40 мл). Полученный экстракт промыли хлороформом (30 мл), после чего довели его pH до pH 8~9, используя гидроксид натрия (около 4,4 г). Раствор трижды экстрагировали хлороформом (100 мл, 50 мл и 50 мл соответственно). Полученный экстракт высушили над безводным сульфатом натрия, после чего сконцентрировали в вакууме. К полученному остатку добавили 4N раствор соляной кислоты в этаноле (6,0 мл) и ацетоне (60 мл). Смесь перемешивали при комнатной температуре в течение 1 часа, после чего отфильтровали. Полученное твердое вещество промыли ацетоном (5 мл), после чего высушили в вакууме для получения 4,0 г указанного в заголовке соединения (выход: 83.0 %).
[148] TLC Rf = 0,29 в 10% MeOH в хлороформе.
[149] 1H NMR (400 мГц, MeOH-d4) δ 7,35 (d, 1H, J = 3,6 Гц), 7,32 ~ 7,26 (m, 3H), 7,12 (d, 1H, J = 8,4 Гц), 5,25 ~ 5,15 (m, 2H), 4,41 (t, 2H, J = 4,8 Гц), 3,97 (s, 3H), 3,85 ~ 3,75 (m, 2H), 3,66 (t, 4H, J = 4,8 Гц), 3,50 ~ 3,35 (m, 6H), 3,01 (s, 3H), 1,42 (t, 3H, J = 7,2 Гц)
[150]
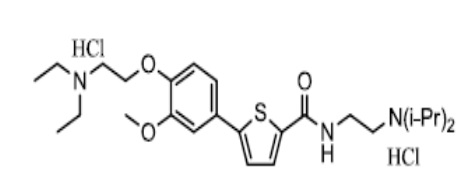
[151] Пример 5: Получение N-(2-(диизопропиламино)этил)-5-(4-гидрокси-3-метоксифенил)тиофен-2-карбоксамид гидрохлорида
[152]
[153] Указанное в заголовке соединение получили по тем же процедурам, что и в Примере 1, используя 2-(диизопропиламино)этиламина вместо N-метилпиперазина (выход: 80,0 %).
[154] TLC Rf = 0,23 в 10% MeOH в хлороформе.
[155] 1H NMR (400 мГц, MeOH-d4) δ 7,70 (d, 1H, J = 4,0 Гц), 7,34 (d, 1H, J = 4,0 Гц), 7,23 (d, 1H, J = 2,0 Гц), 7,18 (dd, 1H, J = 2,0 Гц, 8,0 Гц), 6,86 (d, 1H, J = 8,4 Гц), 3,94 (s, 3H), 3,85 (p, 2H, J = 6,4 Гц), 3,73 (t, 2H, J = 6.4 Гц), 3,38 (t, 2H, J = 6.4 Гц), 1,47 ~ 1,42 (m, 12H)
[156]
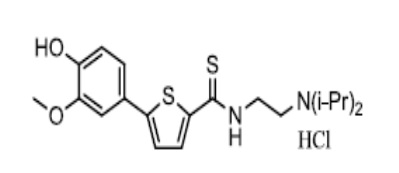
[157] Пример 6: Получение N-(2-(диизопропиламино)этил)-5-(4-(2-диэтиламино)этокси-3-метоксифенил)тиофен-2-карбоксамид гидрохлорида
[158]
[159] К смеси N-(2-(диизопропиламино)этил)-5-бром-тиофен-2-карбоксамида (4,00 г) (промежуточное соединение, полученное по тем же процедурам, что и в Примере 5), с тетракис(трифенилфосфин)палладием(0) (1,39 г) и 1,2-диметоксиэтаном (20 мл) добавили раствор карбоната натрия (3,80 г) в очищенной воде (20 мл). Полученную смесь перемешивали при комнатной температуре в течение 30 минут, после чего к ней добавили раствор (4-(2-диэтиламино)этокси-3-метоксифенил)-(тетраметил-1,3-окса)боролана (4,19 г) в этаноле (20 мл). Реакционную смесь перемешивали при температуре около 80 °C в течение 5 часов, охладили до комнатной температуры, после чего отфильтровали для удаления нерастворимого материала. Полученный фильтрат промыли этанолом (40 мл), после чего сконцентрировали при пониженном давлении для удаления растворителя. К полученному остатку при перемешивании добавили 2N соляную кислоту (70 мл). Полученный раствор дважды промыли этилацетатом (50 мл), после чего его pH довели до pH 8 ~ 9, используя гидроксид натрия (около 6,0 г). Раствор дважды экстрагировали дихлорметаном (100 мл и 50 мл соответственно). Объединенный экстракт высушили над безводным сульфатом натрия, после чего сконцентрировали в вакууме. К полученному концентрату добавили 2N раствор соляной кислоты в этаноле (1,0 мл). Смесь сконцентрировали при пониженном давлении. К полученному концентрату добавили ацетон (60 мл). Смесь перемешивали в течение 1 часа, после чего отфильтровали. Полученное твердое вещество промыли ацетоном (5 мл), после чего высушили в вакууме для получения 5,53 г указанного в заголовке соединения (выход: 83.0 %).
[160] TLC Rf = 0,15 в 10% MeOH в хлороформе.
[161] 1H NMR (400 мГц, MeOH-d4) δ 7,76 (d, 1H, J = 4,0 Гц), 7.43 (d, 1H, J = 4,0 Гц), 7.33 (d, 1H, J = 2,0 Гц), 7,31 (dd, 1H, J = 2,0 Гц, 8,0 Гц), 7,13 (d, 1H, J = 8,0 Гц), 4,42 (t, 2H, J = 4,8 Гц), 3,97 (s, 3H), 3,85 (p, 2H, J = 6,4 Гц), 3,75 (t, 2H, J = 6,4 Гц), 3,66 (t, 2H, J = 4,8 Гц), 3,48 ~ 3,36 (m, 6H), 1,45 (dd, 12H, J = 2,4 Гц, 6,4 Гц), 1,42 (t, 6H, J = 7,2 Гц)
[162]
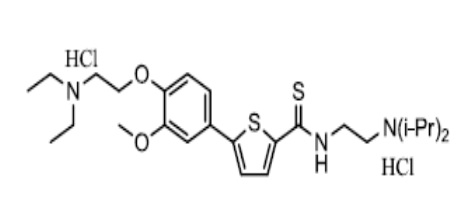
[163] Пример 7: Получение N-(2-(диизопропиламино)этил)-5-(4-гидрокси-3-метоксифенил)тиофен-2-тиокарбоксамид гидрохлорида
[164]
[165] Указанное в заголовке соединение получили по тем же процедурам, что и в Примере 4, используя N-(2-(диизопропиламино)этил)-5-(4-гидрокси-3-метоксифенил)тиофен-2-карбоксамид, полученный по тем же процедурам, что и в Примере 5 (выход: 75,0 %).
[166] TLC Rf = 0,30 в 10% MeOH в хлороформе.
[167] 1H NMR (400 мГц, MeOH-d4) δ 7,58 (d, 1H, J = 4,0 Гц), 7,24 (d, 1H, J = 4,0 Гц), 7,18 ~ 7,08 (m, 2H), 6,76 (d, 1H, J = 8,0 Гц), 4,25 (t, 2H, J = 6,4 Гц), 3,81 (p, 2H, J = 6,4 Гц), 3,96 (s, 3H), 3,38 (t, 2H, J = 6,4 Гц), 1,45 ~ 1,38 (m, 12H)
[168]
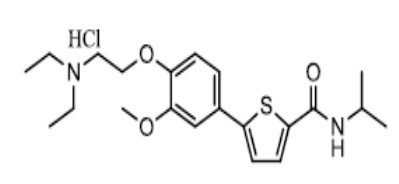
[169] Пример 8: Получение N-(2-(диизопропиламино)этил)-5-(4-(2-диэтиламино)этокси-3-метоксифенил)тиофен-2-тиокарбоксамид гидрохлорида
[170]
[171] Указанное в заголовке соединение получили по тем же процедурам, что и в Примере 4, используя N-(2-(диизопропиламино)этил)-5-(4-(2-диэтиламино)этокси-3-метоксифенил)тиофен-2-карбоксамида, полученный по тем же процедурам, что и в Примере 6 (выход: 80,0 %).
[172] TLC Rf = 0,25 в 10% MeOH в хлороформе.
[173] 1H NMR (400 мГц, MeOH-d4) δ 7,66 (d, 1H, J = 4,0 Гц), 7,40 (d, 1H, J = 4,0 Гц), 7,30 ~ 7,25 (m, 2H), 7,11 (d, 1H, J = 8,0 Гц), 4,40 (t, 2H, J = 4,8 Гц), 4,13 (t, 2H, J = 6,4 Гц), 3,96 (s, 3H), 3,83 (p, 2H, J = 6,4 Гц), 3,63 (t, 2H, J = 4,8 Гц), 3,45 ~ 3,33 (m, 6H), 1,42 (dd, 12H, J = 2,4 Гц, 6,4 Гц), 1,38 (t, 6H, J = 7,2 Гц)
[174]
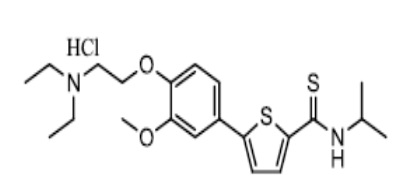
[175] Пример 9: Получение N-изопропил-5-(4-(2-диэтиламино)этокси-3-метоксифенил)тиофен-2-карбоксамид гидрохлорида
[176]
[177] Указанное в заголовке соединение получили по тем же процедурам, что и в Примере 2, используя изопропиламин вместо N-метилпиперазина (выход: 73,0 %).
[178] TLC Rf = 0,20 в 10% MeOH в хлороформе.
[179] 1H NMR (400 мГц, MeOH-d4) δ 7,68 (d, 1H, J = 4,0 Гц), 7,36 (d, 1H, J = 4,0 Гц), 7,32 (d, 1H, J = 2,4 Гц), 7,29 (dd, 1H, J = 2,4 Гц, 8,4 Hz), 7,11 (d, 1H, J = 8,4 Гц), 4,40 (t, 2H, J = 4,8 Гц), 4,20 (p, 1H, J = 6,8 Гц), 3,97 (s, 3H), 3,65 (t, 2H, J = 4,8 Гц), 3,46~3,37 (m, 4H), 1,41 (t, 6H, J = 7,2 Гц), 1,28 (d, 6H, J = 6,8 Гц)
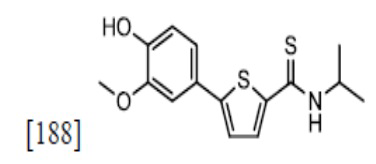
[180]
[181] Пример 10: Получение N-изопропил-5-(4-(2-диэтиламино)этокси-3-метоксифенил)тиофен-2-тиокарбоксамид гидрохлорида
[182]
[183] Указанное в заголовке соединение получили по тем же процедурам, что и в Примере 4, используя N-изопропил-5-(4-(2-диэтиламино)этокси-3-метоксифенил)тиофен-2-карбоксамид, полученный по тем же процедурам, что и в Примере 9 (выход: 70,0 %).
[184] TLC Rf = 0,26 в 10% MeOH в хлороформе.
[185] 1H NMR (400 мГц, MeOH-d4) δ 7,56 (d, 1H, J = 4,0 Гц), 7,33 (d, 1H, J = 4,0 Гц), 7,30~7,25 (m, 2H), 7,08 (d, 1H, J = 8,0 Гц), 4,80 (p, 1H, J = 6,8 Гц), 4,38 (t, 2H, J = 4,8 Гц), 3,95 (s, 3H), 3,62 (t, 2H, J = 4,8 Гц), 3,45 ~ 3,35 (m, 4H), 1,39 (t, 6H, J = 7,2 Гц), 1,33 (d, 6H, J = 6,8 Гц)
[186]
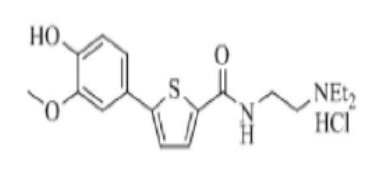
[187] Пример 11: Получение N-изопропил-5-(4-гидрокси-3-метоксифенил)тиофен-2-тиокарбоксамида
[189] Промежуточное соединение (то есть N-изопропил-5-(4-гидрокси-3-метоксифенил) тиофен-2-карбоксамид) (3,5 г) получили по тем же процедурам, что и в Примере 1, используя изопропиламин вместо N-метилпиперазина (Выход: 85,0%). К нему добавили толуол (50 мл), после чего перемешивали в течение 10 минут. Реагент Лавессона (5,0 г) добавили к смеси, которую затем перемешивали при приблизительно 80 °C в течение 5 часов. Реакционную смесь охладили до комнатной температуры, после чего отфильтровали для удаления нерастворимого материала. Полученный фильтрат сконцентрировали в вакууме. Полученный остаток очистили колоночной хроматографией (этилацетат / гексан = 1/3, об./об.), после чего сконцентрировали при пониженном давлении. Полученный концентрат перемешивали в смешанном растворителе из этилацетата и гексана (1/3 (об./об.), 10 мл) в течение 1 часа, после чего отфильтровали. Полученное твердое вещество промыли гексаном (5 мл), после чего сушили при 40 °C в течение 5 часов для получения 2,4 г указанного в заголовке соединения (выход: 65,0 %).
[190] TLC Rf = 0,35 в 10% MeOH в хлороформе.
[191] 1H NMR (400 мГц, MeOH-d4) δ 7,53 (d, 1H, J = 4,0 Гц), 7,24 (d, 1H, J = 4,0 Гц), 7,19 (d, 1H, J = 2,0 Гц), 7,14 (dd, 1H, J = 2,0 Гц, 8,4 Гц), 6,82 (d, 1H, J = 8,4 Гц), 4,80 (p, 1H, J = 6,8 Гц), 3,92 (s, 3H), 1,32 (d, 6H, J = 6,8 Гц)
[192]
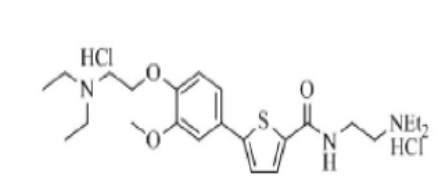
[193] Пример 12: Получение N-(2-(диэтиламино)этил)-5-(4-гидрокси-3-метоксифенил)тиофен-2-карбоксамид гидрохлорида
[194]
[195] Указанное в заголовке соединение получили по тем же процедурам, что и в Примере 1, используя 2-(диэтиламино)этиламин вместо N-метилпиперазина (выход: 34,8 %).
[196] TLC Rf = 0,13 в 10% MeOH в хлороформе.
[197] 1H NMR (400 мГц, MeOH-d4) δ 7,71 (d, 1H, J = 4,0 Гц), 7,32 (d, 1H, J = 4,0 Гц), 7,22 (d, 1H, J = 2,0 Гц), 7,17 (dd, 1H, J = 2,0 Гц, 8,0 Гц), 6,86 (d, 1H, J = 8,4 Гц), 3,94 (s, 3H), 3,76 (t, 2H, J = 6,0 Гц), 3,41 (t, 2H, J = 6,0 Гц), 3,38 ~ 3,34 (m, 4H), 1,38 (t, 6H, J = 7,2 Гц)
[198]
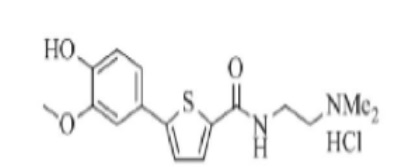
[199] Пример 13: Получение N-(2- (диэтиламино)этил)-5-(4-(2-диэтиламино)этокси-3-метоксифенил)тиофен-2-карбоксамид гидрохлорида
[200]
[201] К раствору гидроксида натрия (0,96 г) в смешанном растворителе из очищенной воды (10 мл) и тетрагидрофурана (50 мл) добавили N-(2-(диэтиламино)этил)-5-(4-гидрокси-3-метоксифенил)тиофен-2-карбоксамид (2,21 г), полученный по тем же процедурам, что и в Примере 12, и 2-диэтиламиноэтилхлорид гидрохлорида (0,99 г). Реакционную смесь перемешивали при температуре около 70°C в течение 4 часов, охладили до комнатной температуры, после чего разделили на водный слой и органический слой. Полученный органический слой сконцентрировали при пониженном давлении, после чего экстрагировали 1N соляной кислотой (20 мл). Полученный экстракт промыли этилацетатом (20 мл), после чего его pH довели до pH 8 ~ 9, используя гидроксид натрия (около 0,9 г). Раствор дважды экстрагировали дихлорметаном (50 мл и 30 мл соответственно). Полученный экстракт высушили над безводным сульфатом натрия, после чего сконцентрировали в вакууме для получения неочищенного продукта, N-(2-(диэтиламино)этил)-5-(4-(2-диэтиламино)этокси-3-метоксифенил)тиофен- 2-карбоксамида. К полученному остатку добавили этанол (15 мл). К полученному раствору добавили раствор 1N соляной кислоты в эфире (15 мл). Смесь нагревали в течение 1 часа, после чего сконцентрировали. К полученному остатку добавили ацетон (50 мл). Смесь перемешивали при комнатной температуре в течение 30 минут, после чего отфильтровали. Полученное твердое вещество промывали ацетоном (5 мл), после чего высушили в вакууме для получения 2,20 г указанного в заголовке соединения (выход: 73,3 %).
[202] TLC Rf = 0,23 в 20% MeOH в хлороформе.
[203] 1H NMR (400 мГц, MeOH-d4) δ 7,75 (d, 1H, J = 4,0 Гц), 7,42 (d, 1H, J = 4,0 Гц), 7.33 (d, 1H, J = 2,0 Гц), 7,31 (dd, 1H, J = 2,0 Гц, 8,0 Гц), 7,12 (d, 1H, J = 8,0 Гц), 4,42 (t, 2H, J = 4,8 Гц), 3,97 (s, 3H), 3,77 (t, 2H, J = 6,0 Гц), 3,66 (t, 2H, J = 6,0 Гц), 3,50 ~ 3.35 (m, 10H), 1,42 (t, 6H, J = 7,2 Гц), 1,39 (t, 6H, J = 7,2 Гц)
[204]
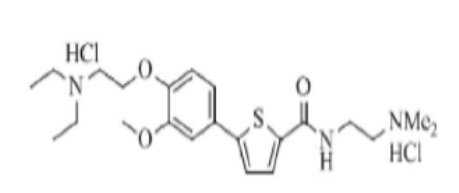
[205] Пример 14: Получение N-(2-(диметиламино)этил)-5-(4-гидрокси-3-метоксифенил)тиофен-2-карбоксамид гидрохлорида
[206]
[207] Указанное в заголовке соединение получили по тем же процедурам, что и в Примере 1, используя 2-(диметиламино)этиламин вместо N-метилпиперазина (выход: 64,5 %).
[208] TLC Rf = 0,12 в 10% MeOH в хлороформе.
[209] 1H NMR (400 мГц, MeOH-d4) δ 7,69 (d, 1H, J = 4,0 Гц), 7,32 (d, 1H, J = 4,0 Гц), 7,23 (d, 1H, J = 2,0 Гц), 7,17 (dd, 1H, J = 2,0 Гц, 8,0 Гц), 6,86 (d, 1H, J = 8,0 Гц), 3,94 (s, 3H), 3,76 (t, 2H, J = 6,0 Гц), 3,40 (t, 2H, J = 6,0 Гц), 3,01 (s, 6H)
[210]
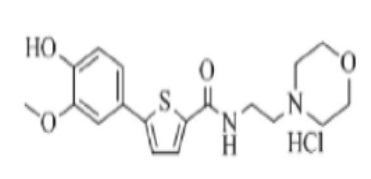
[211] Пример 15: Получение N-(2- (диметиламино)этил)-5-(4-(2-диэтиламино)этокси-3-метоксифенил)тиофен-2-карбоксамид гидрохлорида
[212]
[213] Указанное в заголовке соединение получили по тем же процедурам, что и в Примере 13, используя N-(2-(диметиламино)этил)-5-(4-гидрокси-3-метоксифенил)тиофен-2-карбоксамид, полученный по тем же процедурами, что и в Примере 14, вместо N-(2-(диэтиламино)этил)-5-(4-гидрокси-3-метоксифенил)тиофен-2-карбоксамида (выход: 54,7 %).
[214] TLC Rf = 0,34 в 20% MeOH в хлороформе.
[215] 1H NMR (400 мГц, MeOH-d4) δ 7,77 (d, 1H, J = 4,0 Гц), 7,41 (d, 1H, J = 4,0 Гц), 7.33 (d, 1H, J = 2,0 Гц), 7,30 (dd, 1H, J = 2,0 Hz, 8,0 Гц), 7,12 (d, 1H, J = 8,4 Гц), 4,42 (t, 2H, J = 4,8 Гц), 3,97 (s, 3H), 3,78 (t, 2H, J = 6,0 Гц), 3,66 (t, 2H, J = 6,0 Гц), 3,50 ~ 3,35 (m, 6H), 3,01 (s, 6H), 1,42 (t, 6H, J = 7,2 Гц)
[216]
[217] Пример 16: Получение N-(2-(4-морфолино)этил)-5-(4-гидрокси-3-метоксифенил)тиофен-2-карбоксамид гидрохлорида
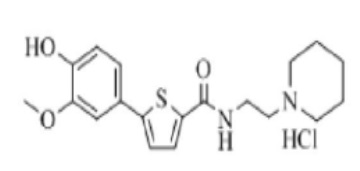
[218]
[219] Указанное в заголовке соединение получили по тем же процедурам, что и в Примере 1, используя 4-(2-аминоэтил)морфолин вместо N-метилпиперазина (выход: 16,8 %).
[220] TLC Rf = 0,32 в 10% MeOH в хлороформе.
[221] 1H NMR (400 мГц, MeOH-d4) δ 7,71 (d, 1H, J = 4,0 Гц), 7,33 (d, 1H, J = 4,0 Гц), 7,23 (d, 1H, J = 2,0 Гц), 7,17 (dd, 1H, J = 2,0 Гц, 8,0 Гц), 6,86 (d, 1H, J = 8,0 Гц), 4,14 ~ 4,09 (m, 2H), 3,94 (s, 3H), 3,87 ~ 3,77 (m, 4H), 3,74 ~ 3,69 (m, 2H), 3,43 (t, 2H, J = 5,6 Гц), 3,28 ~ 3,23 (m, 2H)
[222]
[223] Пример 17: Получение N-(2-(1-пиперидино)этил)-5-(4-гидрокси-3-метоксифенил)тиофен-2-карбоксамид гидрохлорида
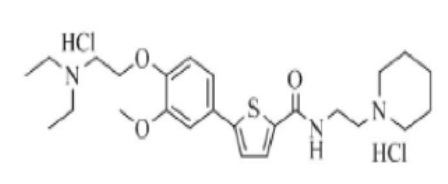
[224]
[225] Указанное в заголовке соединение получили по тем же процедурам, что и в Примере 1, используя 1-(2-аминоэтил)пиперидин вместо N-метилпиперазина (выход: 52,7 %).
[226] TLC Rf = 0,15 в 10% MeOH в хлороформе.
[227] 1H NMR (400 мГц, MeOH-d4) δ 7,69 (d, 1H, J = 4,0 Гц), 7,33 (d, 1H, J = 4,0 Гц), 7,23 (d, 1H, J = 2,0 Гц), 7,17 (dd, 1H, J = 2,0 Гц, 8,0 Гц), 6,86 (d, 1H, J = 8,4 Гц), 3,94 (s, 3H), 3,76 (t, 2H, J = 6,0 Гц), 3,75 ~ 3,67 (m, 2H), 3,38 ~ 3,35 (m, 2H), 3,02 (t, 2H, J = 12 Гц), 2,03 ~ 1,95 (m, 2H), 1,92 ~ 1,77 (m, 3H), 1,63 ~ 1,52 (m, 1H)
[228]
[229] Пример 18: Получение N-(2-(1-пиперидино)этил)-5-(4-(2-диэтиламино)этокси-3-метоксифенил)тиофен-2-карбоксамид гидрохлорида
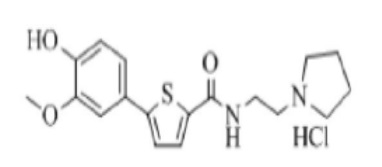
[230]
[231] Указанное в заголовке соединение получили по тем же процедурам, что и в Примере 13, используя N-(2-(1-пиперидино)этил)-5-(4-гидрокси-3-метоксифенил)тиофен-2-карбоксамид, полученный по тем же процедурам, что и в Примере 17, вместо N-(2-(диэтиламино)этил)-5-(4-гидрокси-3-метоксифенил)тиофен-2-карбоксамида (выход: 82,6 %).
[232] TLC Rf = 0,27 в 20% MeOH в хлороформе.
[233] 1H NMR (400 мГц, MeOH-d4) δ 7,75 (d, 1H, J = 4,0 Гц), 7,42 (d, 1H, J = 4,0 Гц), 7,33 (d, 1H, J = 2,0 Гц), 7,31 (dd, 1H, J = 2,0 Гц, 8,0 Гц), 7,12 (d, 1H, J = 8,4 Гц), 4.42 (t, 2H, J = 4,8 Гц), 3,97 (s, 3H), 3,78 (t, 2H, J = 6,0 Гц), 3,75 ~ 3,67 (m, 2H), 3,70 (t, 2H, J = 11 Гц), 3,52 ~ 3,35 (m, 6H), 3,02 (m, 2H), 2,03 ~ 1,95 (m, 2H), 1,91 ~ 1,79 (m, 3H), 1,62 ~ 1,55 (m, 1H), 1,42 (t, 6H, J = 7,2 Гц)
[234]
[235] Пример 19: Получение N-(2-(1-пирролидино)этил)-5-(4-гидрокси-3-метоксифенил)тиофен-2-карбоксамид гидрохлорида
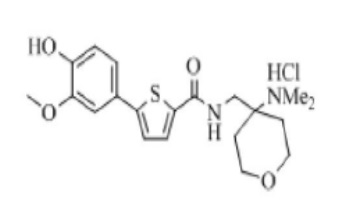
[236]
[237] Указанное в заголовке соединение получили по тем же процедурам, что и в Примере 1, используя 1-(2-аминоэтил)пирролидин вместо N-метилпиперазина (выход: 20,9 %).
[238] TLC Rf = 0,08 в 10% MeOH в хлороформе.
[239] 1H NMR (400 мГц, MeOH-d4) δ 7,69 (d, 1H, J = 4,0 Гц), 7,32 (d, 1H, J = 4,0 Гц), 7,23 (d, 1H, J = 2,0 Гц), 7,17 (dd, 1H, J = 2,0 Гц, 8,0 Гц), 6,86 (d, 1H, J = 8,0 Гц), 3,94 (s, 3H), 3,76 (t, 2H, J = 6,0 Гц), 3,85 ~ 3,78 (m, 2H), 3,75 (t, 2H, J = 6,0 Гц), 3,46 (t, 2H, J = 6,0 Гц), 3,22 ~ 3,15 (m, 2H), 2,28 ~ 2,13 (m, 2H), 2,13 ~ 2,00 (m, 2H)
[240]
[241] Пример 20: Получение N-((4-(диметиламино)тетрагидро-2H-пиран-4-ил)метил)-5-(4-гидрокси-3-метоксифенил)тиофен-2-карбоксамид гидрохлорида
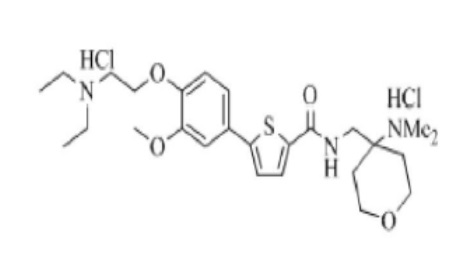
[242]
[243] Указанное в заголовке соединение получили по тем же процедурам, что и в Примере 1, используя 4-(аминометил)-N, N-диметилтетрагидро-2H-пиран-4-амин вместо N-метилпиперазина (выход: 48,4 %).
[244] TLC Rf = 0,37 в 10% MeOH в хлороформе.
[245] 1H NMR (400 мГц, MeOH-d4) δ 7,80 (d, 1H, J = 4,0 Гц), 7,34 (d, 1H, J = 4,0 Гц), 7,23 (d, 1H, J = 2,0 Гц), 7,18 (dd, 1H, J = 2,0 Гц, 8,0 Гц), 6,86 (d, 1H, J = 8,4 Гц), 4,08 ~ 4,04 (m, 2H), 4,02 (s, 2H), 3,94 (s, 3H), 3,82 ~ 3,75 (m, 2H), 2,99 (s, 6H), 2,05 ~ 1,94 (m, 4H)
[246]
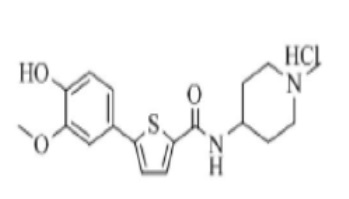
[247] Пример 21: Получение N-((4-(диметиламино)тетрагидро-2H-пиран-4-ил)метил)-5-(4-(2-диэтиламино)этокси-3-метоксифенил)тиофен-2-карбоксамид гидрохлорида
[248]
[249] Указанное в заголовке соединение получили по тем же процедурам, что и в Примере 13, используя N-((4-(диметиламино)тетрагидро-2H-пиран-4-ил)метил)-5-(4-гидрокси-3-метоксифенил)тиофен-2-карбоксамид, полученный по тем же процедурам, что и в Примере 20, вместо N-(2-(диэтиламино)этил)-5-(4-гидрокси-3-метоксифенил)тиофен-2-карбоксамида (выход: 72,3 %).
[250] TLC Rf = 0,44 в 20% MeOH в хлороформе.
[251] 1H NMR (400 мГц, MeOH-d4) δ 7,87 (d, 1H, J = 4,0 Гц), 7,43 (d, 1H, J = 4,0 Гц), 7,33 (d, 1H, J = 2,0 Гц), 7,31 (dd, 1H, J = 2,0 Гц, 8,0 Гц), 7,12 (d, 1H, J = 8,4 Гц), 4,42 (t, 2H, J = 4,8 Гц), 4,08 ~ 4,02 (m, 2H), 4,03 (s, 2H), 3,97 (s, 3H), 3,82 ~ 3,75 (m, 2H), 3,66 (t, 2H, J = 4,8 Гц), 2,99 (s, 6H), 2,04 ~ 1,94 (m, 4H), 1,42 (t, 6H, J = 7,2 Гц)
[252]
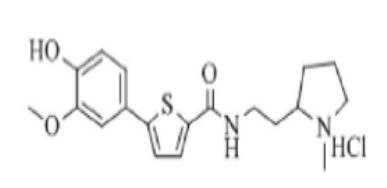
[253] Пример 22: Получение N-(4-(1-метил)пиперидинил)-5-(4-гидрокси-3-метоксифенил)тиофен-2-карбоксамид гидрохлорида
[254]
[255] Указанное в заголовке соединение получили по тем же процедурам, что и в Примере 1, используя 4-амино-1-метилпиперидин вместо N-метилпиперазина (выход: 63,4 %).
[256] TLC Rf = 0,20 в 20% MeOH в хлороформе.
[257] 1H NMR (400 мГц, MeOH-d4) δ 7,71 ~ 7,67 (m, 1H), 7,30 (d, 1H, J = 4,0 Гц), 7,22 (d, 1H, J = 2,0 Гц), 7,17 (dd, 1H, J = 2,0 Гц, 8,0 Гц), 6,86 (d, 1H, J = 8,0 Гц), 4,18 ~ 4,08 (m, 1H), 3,94 (s, 3H), 3,62 ~ 3,57 (m, 2H), 3,25 ~ 3,14 (m, 2H), 2,92 (s, 3H), 2,31 ~ 2,24 (m, 2H), 1,98 ~ 1,87 (m, 2H)
[258]
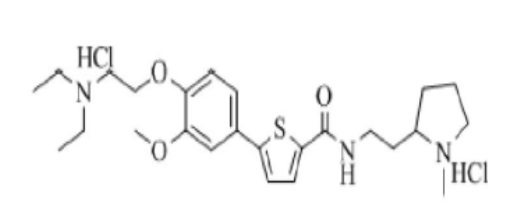
[259] Пример 23: Получение N-(2-(2-(1-метил)пирролидино)этил)-5-(4-гидрокси-3-метоксифенил)тиофен-2-карбоксамид гидрохлорида
[260]
[261] Указанное в заголовке соединение получили по тем же процедурам, что и в Примере 1, используя 2-(2-аминоэтил)-1-метилпирролидин вместо N-метилпиперазина (выход: 55.6 %).
[262] TLC Rf = 0,16 в 20% MeOH в хлороформе.
[263] 1H NMR (400 мГц, MeOH-d4) δ 7,64 (d, 1H, J = 4,0 Гц), 7,31 (d, 1H, J = 4,0 Гц), 7.22 (d, 1H, J = 2,0 Гц), 7,17 (dd, 1H, J = 2,0 Гц, 8,0 Гц), 6,86 (d, 1H, J = 8,0 Гц), 3,94 (s, 3H), 3,73 ~ 3,66 (m, 1H), 3,53 ~ 3,48 (m, 2H), 3,41 ~ 3,35 (m, 1H), 3,23 ~ 3,15 (m, 1H), 2,97 (s, 3H), 2,57 ~ 2,47 (m, 1H), 2,35 ~ 2,25 (m, 1H), 2,25 ~ 2,04 (m, 2H), 1,92 ~ 1,80 (m, 2H)
[264]
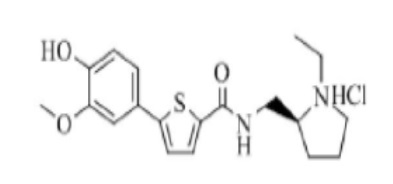
[265] Пример 24: Получение N-(2-(2-(1-метил)пирролидино)этил)-5-(4-(2-диэтиламино)этокси-3-метоксифенил)тиофен-2-карбоксамид гидрохлорида
[266]
[267] Указанное в заголовке соединение получили по тем же процедурам, что и в Примере 13, используя N-(2-(2-(1-метил)пирролидино)этил)-5-(4-гидрокси-3-метоксифенил)тиофен-2-карбоксамид, полученный по тем же процедурам, что и в Примере 23, вместо N-(2-(диэтиламино)этил)-5-(4-гидрокси-3-метоксифенил)тиофен-2-карбоксамида (выход: 60,9 %).
[268] TLC Rf = 0,12 в 20% MeOH в хлороформе.
[269] 1H NMR (400 мГц, DMSO-d6) δ 10,37 (s, 1H), 10,21 (s, 1H), 8,71 (t, 1H, J = 6,0 Гц), 7,78 (d, 1H, J = 4,0 Гц), 7,51 (d, 1H, J = 4,0 Гц), 7,30 (d, 1H, J = 2,0 Гц), 7,26 (dd, 1H, J = 2,0 Гц, 8,0 Гц), 7,10 (d, 1H, J = 8,0 Гц), 4,39 (t, 2H, J = 4,8 Гц), 3,88 (s, 3H), 3,48 ~ 3,51 (m, 3H), 3,43 ~ 3,15 (m, 5H), 3,10 ~ 3,00 (m, 1H), 2,79 (d, 3H, J = 4,8 Гц), 2,40 ~ 2,35 (m, 1H), 2,22 ~ 2,13 (m, 1H), 2,07 ~ 1,80 (m, 3H), 1,72 ~ 1,63 (m, 1H), 1,27 (t, 6H, J = 7,2 Гц)
[270]
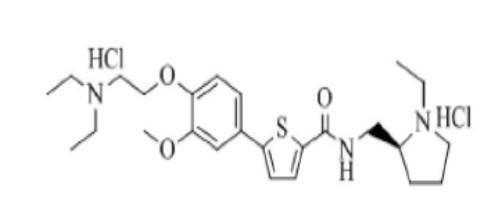
[271] Пример 25: Получение N-((S)-2-(1-этил)пирролидинометил)-5-(4-гидрокси-3-метоксифенил)тиофен-2-карбоксамид гидрохлорида
[272]
[273] Указанное в заголовке соединение получили по тем же процедурам, что и в Примере 1, используя (S)-2-(аминометил)-1-этилпирролидин вместо N-метилпиперазина (выход: 42,1 %).
[274] TLC Rf = 0,29 в 20% MeOH в хлороформе.
[275] 1H NMR (400 мГц, DMSO-d6) δ 9,85 (s, 1H), 9,44 (s, 1H), 8,97 (t, 1H, J = 5,6 Гц), 7,82 (d, 1H, J = 4,0 Гц), 7,44 (d, 1H, J = 4,0 Гц), 7,22 (d, 1H, J = 2,0 Гц), 7,13 (dd, 1H, J = 2,0 Гц, 8,0 Гц), 6,83 (d, 1H, J = 8,0 Гц), 3,85 (s, 3H), 3,72 ~ 3,65 (m, 1H), 3,65 ~ 3,53 (m, 2H), 3,48 ~ 3,38 (m, 2H), 3,15 ~ 3,05 (m, 2H), 2,18 ~ 2,08 (m, 1H), 2,03 ~ 1,93 (m, 1H), 1,93 ~ 1,76 (m, 2H), 1,28 (t, 3H, J = 7,2 Гц)
[276]
[277] Пример 26: Получение N-((S)-2-(1-этил) пирролидинометил)-5-(4-(2-диэтиламино)этокси-3-метоксифенил)тиофен-2-карбоксамид гидрохлорида
[278]
[279] Указанное в заголовке соединение получили по тем же процедурам, что и в Примере 13, используя N-((S)-2-(1-этил)пирролидинометил)-5-(4-гидрокси-3-метоксифенил)тиофен-2-карбоксамид, полученный по тем же процедурам, что и в Примере 25, вместо N-(2-(диэтиламино)этил)-5-(4-гидрокси-3-метоксифенил)тиофен-2-карбоксамида (выход: 37,6 %).
[280] TLC Rf = 0,24 в 20% MeOH в хлороформе.
[281] 1H NMR (400 мГц, DMSO-d6) δ 10,41 (s, 1H), 10,25 (s, 1H), 9,14 (t, 1H, J = 5,6 Гц), 7,92 (d, 1H, J = 4,0 Гц), 7,53 (d, 1H, J = 4,0 Гц), 7,31 (d, 1H, J = 2,0 Гц), 7,27 (dd, 1H, J = 2,0 Гц, 8,0 Гц), 7,11 (d, 1H, J = 8,0 Гц), 4,40 (t, 2H, J = 4,8 Гц), 3,88 (s, 3H), 3,82 ~ 3,72 (m, 1H), 3,67 ~ 3,50 (m, 4H), 3,48 ~ 3,40 (m, 2H), 3,40 ~ 3,18 (m, 4H), 3,18 ~ 3,03 (m, 2H), 2,18 ~ 2,08 (m, 1H), 2,03 ~ 1,93 (m, 1H), 1,93 ~ 1,75 (m, 2H), 1,45 ~ 1,35 (m, 9H)
[282]
[283] Тестовый пример 1: Активность по индуцированию аутофагии
[284] Активность по индуцированию аутофагии, согласно данному изобретению измерили в клеточных линиях Hela (корейский банк клеточных линий), используя комплект для обнаружения аутофагии (ab139484, Abcam) в соответствии с инструкциями производителя. В частности, в каждую лунку 96-луночной пластины добавили 100 мкл EBSS (сбалансированного солевого раствора Эрла, WELGENE (LB 002-03)), содержащего 10% фетальную бычью сыворотку (FBS). Клеточные линии Hela (1 × 104 клеток) добавили в каждую лунку, после чего инкубировали в течение ночи при температуре 37°С в инкубаторе CO2 для стабилизации клеток. Среду каждой лунки, содержащей стабилизированные клетки, заменили средой, полученной добавлением 10 нм или 100 нм соединений, полученных в Примерах (испытуемые соединения), к EBSS, содержащему 10% FBS. В качестве положительного контроля рапамицин растворили в диметилсульфоксиде, после чего обработали в концентрации 500 нм на лунку. После обработок клетки инкубировали при температуре 37°C в инкубаторе CO2 в течение 4 или 24 часов, после чего дважды промыли аналитическим буфером 1X (приготовленным путем добавления 9 мл дистиллированной воды к аналитическому буферу 10X). EBSS, содержащий 5% фетальной бычьей сыворотки, 1 мкл/мл проявляющего реагента Грина и 1 мкл / мл ядерного красителя, добавили к ним в количестве 100 мкл на лунку. Клетки инкубировали в течение 1 часа при температуре 37°С в инкубаторе CO2, после чего дважды промыли аналитическим буфером 1X. Значения коэффициента абсорбции измерили при 488 нм, используя планшет-ридер (Cytation 3). Испытания повторили четыре раза.
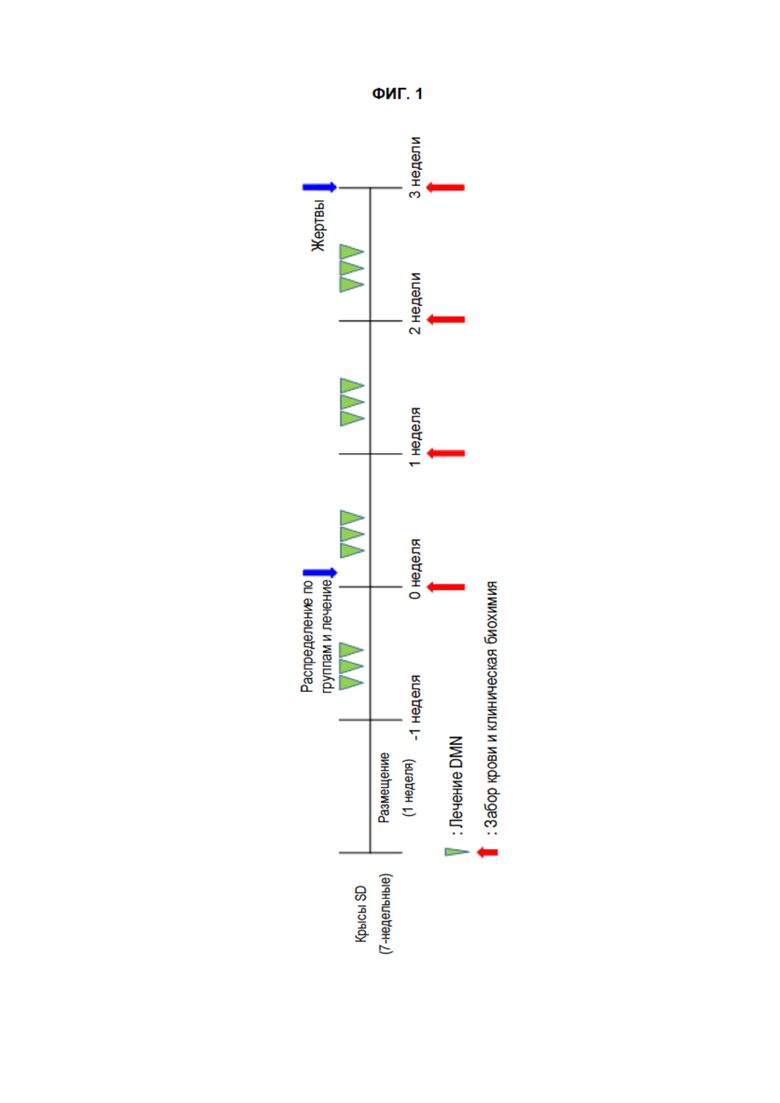
[285] Значения коэффициента абсорбции, которые были получены в результате обработки клеточных линий Hela испытуемыми соединениями (100 нм) и положительным контролем (рапамицин, 500 нм) с последующим инкубированием в течение 4 часов, как описано выше, представлены в Таблице 1 ниже. Также значения коэффициента абсорбции, которые были получены в результате обработки клеточных линий Hela испытуемыми соединениями (10 нм или 100 нм) и положительным контролем (рапамицин, 500 нМ) с последующим инкубированием в течение 24 часов, как описано выше, показаны в Таблице 2 ниже.
[286] Таблица 1
Коэффициент абсорбции (инкубация в течение 4 часов)
[287]
[288] Таблица 2
Коэффициент абсорбции (инкубация в течение 24 часов)
[289] По результатам, приведенным в Таблице 1 выше, можно видеть, что при инкубации в течение 4 часов после обработки испытуемых соединений соединения согласно данному изобретению продемонстрировали активность по индуцированию аутофагии даже при концентрации 1/5, равной или выше, чем у положительного контроля (то есть рапамицина). В частности, по результатам, приведенным в Таблице 2, можно видеть, что при инкубации в течение 24 часов после обработки испытуемых соединений соединения согласно данному изобретению продемонстрировали превосходную (то есть, по меньшей мере, в пять раз или более) активность по индуцированию аутофагии даже при концентрациях 1/50 и 1/5 по сравнению с положительным контролем (то есть рапамицином). Следовательно, соединения согласно данному изобретению демонстрируют превосходную активность по индуцированию аутофагии, благодаря чему может быть успешно применено для профилактики, облегчения или лечения различных заболеваний, связанных с аутофагией, включая нейродегенеративные заболевания, заболевания печени, метаболические заболевания, сепсис и так далее.
[290]
[291] Тестовый пример 2: Оценка активности по улучшению функции печени при пероральном применении на модели повреждения печени (1)
[292] Соединения согласно данному изобретению вводили перорально самцам крыс SD с повреждением печени, вызванным диметилнитрозамином (DMN), в течение 3 недель, чтобы оценить активность по улучшению функции печени. В частности, 7-недельных самцов крыс SD (Orient Bio, Корея) помещали в лабораторную среду при комнатной температуре на 7 дней. Наблюдали общие внешние проявления, после чего для эксперимента использовали только здоровых животных. Крыс разделили на 9 групп (n = 5 для каждой группы): нормальную контрольную группу, группу, которой вводили только DMN, и группы, которым вводили как соединение согласно данному изобретению (соединения Примеров 2, 5, 6, 9, 10, 20 или 21), так и DMN. DMN растворяли в очищенной воде, после чего вводили внутрибрюшинно в дозе 10 мг/кг в течение 3 недель подряд (3 раза в течение 4ой недели). Забор крови делали в День 3 первой недели после завершения индуцирования повреждения печени, после чего измерили значения АЛТ (аланинтрансаминазы) и АСТ (аспартаттрансаминазы), чтобы подтвердить повреждение печени. Соединения согласно данному изобретению вводили в течение 3 недель, то есть после Дня 4 после завершения индуцирования повреждения печени в течение первой недели по отношению к периоду введения DMN. Семь соединений согласно данному изобретению растворяли в очищенной воде или кукурузном масле, после чего вводили перорально с помощью перорального зонда в дозе 25 мг/кг один раз в день в течение 3 недель. Забор крови был сделан в День 0 (в 3й день первой недели после завершения индуцирования повреждения печени) и в День 7, 14 и 21 после введения испытуемых соединений. Собранные образцы крови вводили в пробирку с вакуумом, содержащую активатор сгустков, после чего выдерживали при комнатной температуре в течение примерно 20 минут для коагуляции каждого образца крови. После центрифугирования в течение 10 минут полученные сыворотки подвергали биохимическим исследованиям крови. Экспериментальные способы кратко представлены на ФИГ. 1.
[293] Значения сывороточных АЛТ и АСТ, полученные при проведении биохимического анализа крови, как описано выше, представлены в Таблицах 3 и 4 ниже.
[294] Таблица 3
[295]
[296] Таблица 4
[297] Как можно видеть по результатам, приведенным в Таблицах 3 и 4, значения сывороточных АЛТ и АСТ в группе, которой вводили DMN, увеличились соответственно примерно в 3 раза и примерно в 2 раза через 3 недели. Тем не менее, в группах, которым вводили как соединения согласно данному изобретению, так и DMN, значения АЛТ через 3 недели после введения составили 101,18 ~ 126,10 Ед / л (то есть уменьшились примерно на 19 ~ 35% по сравнению с данным значениями в группе, которой вводили DMN); а значения АСТ через 3 недели после введения составили 195,10 ~ 250,76 Ед/л (то есть уменьшились примерно на 17 ~ 35% по сравнению с данными значениями в группе, которой вводили DMN). Таким образом, можно подтвердить, что соединения согласно данному изобретению демонстрируют эффективную активность по улучшению печени при повреждениях печени, включая фиброз печени.
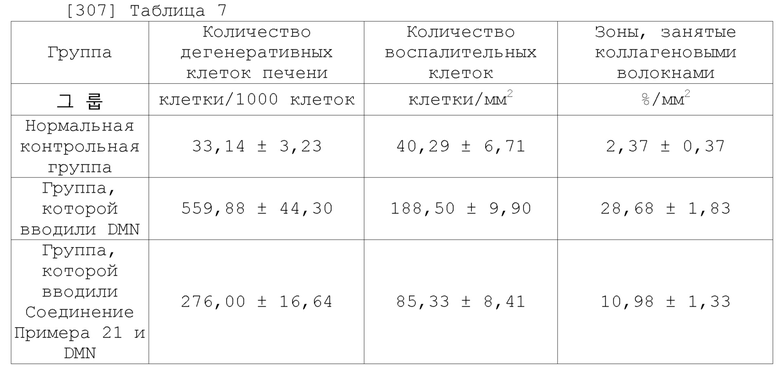
[298]
[299] Тестовый пример 3: Оценка активности по улучшению функции печени при пероральном применении на модели повреждения печени (2)
[300] Соединения согласно данному изобретению вводили перорально самцам крыс SD с повреждением печени, вызванным диметилнитрозамином (DMN), в течение 4 недель, чтобы оценить активность по улучшению функции печени. В частности, 7-недельных самцов крыс SD (Orient Bio, Корея) помещали в лабораторную среду при комнатной температуре на 7 дней. Наблюдали общие внешние признаки, после чего для эксперимента использовали только здоровых животных. Крыс разделили на 3 группы (n = 10 для каждой группы): нормальную контрольную группу, группу, которой вводили только DMN, и группы, которым вводили как соединение согласно данному изобретению (соединение Примера 21), так и DMN. DMN растворяли в очищенной воде, после чего вводили внутрибрюшинно в дозе 10 мг/кг в течение 3 недель подряд (3 раза в неделю в течение 4 недель). Забор крови делали после завершения индуцирования повреждения печени в четвертую неделю, после чего измерили значения АЛТ (аланинтрансаминазы) и АСT (аспартаттрансаминазы), чтобы подтвердить повреждение печени. Соединение согласно данному изобретению вводили в течение 4 недель, начиная со Дня 1 после завершения четырехнедельного индуцирования повреждения печени. Соединение согласно данному изобретению (соединение Примера 21) растворяли в очищенной воде, после чего вводили перорально с помощью перорального зонда в дозе 25 мг/кг один раз в день в течение 4 недель. Забор крови был сделан в День 0 (через 1 дня после завершения четырехнедельного индуцирования повреждения печени) и в День 7, 14, 21 и 28 после начала введения испытуемых соединений. Собранные образцы крови вводили в пробирку с вакуумом, содержащую активатор сгустков, после чего выдерживали при комнатной температуре в течение примерно 20 минут для коагуляции каждого образца крови. После центрифугирования в течение 10 минут полученные сыворотки подвергали биохимическим исследованиям крови. Помимо этого, через 24 часа после последнего введения было выполнено вскрытие для гепатэктомии и фиксации для подготовки их образцов ткани. После подготовки предметных стекол с образцами тканей на них провели окрашивание гематоксилином и эозином (H&E) для микроскопического наблюдения повреждений ткани печени и инфильтрации воспалительных клеток в тканях печени. Помимо этого, на данных образцах ткани также провели окрашивание трихромом по Массону для микроскопического наблюдения за отложениями коллагеновых волокон в тканях печени. Экспериментальные способы кратко представлены на ФИГ. 2.
[301] Значения сывороточных АЛТ и АСТ, полученные при проведении биохимического анализа крови, как описано выше, представлены в Таблицах 5 и 6 ниже.
[302] Таблица 5
[303]
[304] Таблица 6
[305] Как можно увидеть по результатам, показанным в Таблицах 5 и 6, значения сывороточных АЛТ и АСТ в группе, которой вводили DMN, увеличились соответственно примерно в 4-5 раза и примерно в 3,2 раза через 4 недели. Тем не менее, в группе, которой вводили как соединение согласно данному изобретению (соединение Примера 21), так и DMN, значения АЛТ через 3 и 4 недели после введения составили 44,48 и 48,33 Ед/л соответственно (то есть уменьшились на 30% и 26% по сравнению с данным значениями в группе, которой вводили DMN); а значения АСТ через 3 и 4 недели после введения составили 112,06 и 110,45 Ед/л соответственно (то есть уменьшились примерно на 22% и 30% по сравнению с данными значениями в группе, которой вводили DMN). Таким образом может быть подтверждено, что соединения согласно данному изобретению демонстрируют эффективную активность по улучшению печени при повреждениях печени, включая фиброз печени.
[306] Помимо этого, результаты, полученные путем окрашивания гематоксилином и эозином (H&E) и окрашивания трихромом по Массону, как описано выше, представлены на ФИГ. 3 и ФИГ. 4 соответственно. По результатам, показанным на ФИГ. 3, измерили степень повреждения ткани печени и степень инфильтрации воспалительных клеток в тканях печени. Также по результатам, показанным на ФИГ.4, измерили степень отложения коллагеновых волокон в тканях печени. Результаты приведены ниже в Таблице 7.
[308] Как можно увидеть по результатам, показанным на ФИГ. 3 и в Таблице 7, повреждение ткани печени и инфильтрация воспалительных клеток в тканях печени в группе, которой вводили DMN, соответственно увеличилось примерно в 17 раз и примерно в 5 раз через 4 недели по сравнению с данными значениями в нормальной контрольной группе. Тем не менее, в группе, которой вводили как соединение согласно данному изобретению (соединение Примера 21), так и DMN, повреждение ткани печени и инфильтрация воспалительных клеток в тканях печени составили 276,0 клеток/1000 клеток и 85,33 клеток/мм2 соответственно (то есть улучшение примерно на 50% и 55% по сравнению с данными значениями в группе, которой вводили DMN).
[309] Также, как можно увидеть по результатам, приведенным на ФИГ. 4 и в Таблице 7, отложения коллагеновых волокон в тканях печени в группе, которой вводили DMN, увеличились примерно в 12 раз через 4 недели по сравнению с данным значением в нормальной контрольной группе. Тем не менее, в группе, которой вводили как соединение согласно данному изобретению (соединение Примера 21), так и DMN, отложения коллагеновых волокон в тканях печени составили 10,98%/мм2 (то есть уменьшились примерно на 62% по сравнению с данным значением в группе, которой вводили DMN).
[310] Таким образом может быть подтверждено, что соединения согласно данному изобретению демонстрируют эффективную активность по улучшению печени при повреждениях печени, включая фиброз печени.
| название | год | авторы | номер документа |
|---|---|---|---|
| НОВОЕ ПРОИЗВОДНОЕ 2-АРИЛТИАЗОЛА ИЛИ ЕГО СОЛЬ, СПОСОБ ЕГО ПОЛУЧЕНИЯ И СОДЕРЖАЩАЯ ЕГО ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ | 2021 |
|
RU2822057C1 |
| НОВЫЕ СОЕДИНЕНИЯ И ФАРМАЦЕВТИЧЕСКИЙ СОСТАВ, СОДЕРЖАЩИЙ ИХ | 2019 |
|
RU2793138C2 |
| БЕНЗОТИОФЕНОВЫЕ СОЕДИНЕНИЯ ИЛИ ИХ ФАРМАЦЕВТИЧЕСКИ ПРИЕМЛЕМЫЕ СОЛИ | 1996 |
|
RU2158737C2 |
| ПРОИЗВОДНЫЕ ТИОФЕНА | 2010 |
|
RU2536865C2 |
| НОВЫЕ АРОМАТИЗИРУЮЩИЕ ВЕЩЕСТВА, МОДИФИКАТОРЫ ВКУСА, СОЕДИНЕНИЯ, ПРИДАЮЩИЕ ВКУС, УСИЛИТЕЛИ ВКУСА, СОЕДИНЕНИЯ, ПРИДАЮЩИЕ ВКУС "УМАМИ" ИЛИ СЛАДКИЙ ВКУС, И/ИЛИ УСИЛИТЕЛИ И ИХ ПРИМЕНЕНИЕ | 2004 |
|
RU2419602C2 |
| ПРОИЗВОДНЫЕ ХИНОЛИНА И СОДЕРЖАЩИЕ ИХ ИНГИБИТОРЫ MELK | 2011 |
|
RU2582610C2 |
| МОДИФИКАТОР ПРОИЗВОДНОГО ЧЕТЫРЕХЧЛЕННОГО КОЛЬЦА, СПОСОБ ЕГО ПОЛУЧЕНИЯ И ЕГО ПРИМЕНЕНИЕ | 2020 |
|
RU2833027C1 |
| 2,6-ЗАМЕЩЕННЫЕ-4-МОНОЗАМЕЩЕННЫЙ АМИНО-ПИРИМИДИНЫ КАК АНТАГОНИСТЫ РЕЦЕПТОРА ПРОСТАГЛАНДИНА D2 | 2005 |
|
RU2417990C2 |
| ИНГИБИРУЮЩИЕ JAK СОЕДИНЕНИЯ НА ОСНОВЕ ПИРАЗОЛОПИРИМИДИНА И СПОСОБЫ | 2010 |
|
RU2567238C2 |
| КАРБОКСАМИДЫ, СПОСОБ ИХ ПОЛУЧЕНИЯ И ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ | 1995 |
|
RU2163232C2 |
Изобретение относится к соединению формулы 1 или к его фармацевтически приемлемой соли, причем Y представляет собой О или S, (1) если Y - это О, R1 представляет собой водород или C1-С4 алкильную группу, замещенную моно- или ди-C1-С5 алкиламино, R2 - C1-С6 алкильная группа, R3 - водород, и R4 - (4-(диметиламино)тетрагидро-2Н-пиран-4-ил)метильная группа, C1-C4 алкильная группа, замещенная моно- или ди-С1-С5 алкиламино, C1-C4 алкильная группа, замещенная азотсодержащим циклическим кольцом, выбранным из группы, состоящей из морфолина, пиперидина или пирролидина, причем азотсодержащее циклическое кольцо необязательно замещено C1-С4 алкилом, или пиперидинильная группа, необязательно замещенная C1-С4 алкилом, или R3 и R4 соединены друг с другом атомом азота, к которому они присоединены для образования пиперазинового кольца, причем пиперазиновое кольцо необязательно замещено C1-C4 алкилом, (2) если Y - это S, R1 и R2 независимо друг от друга представляют собой водород; C1-С6 алкильную группу или C1-C4 алкильную группу, замещенную моно- или ди-С1-С5 алкиламино, R3 - водород, a R4 - C1-C4 алкильная группа, замещенная моно- или ди-С1-С5 алкиламино, или R3 и R4 соединены друг с другом атомом азота, к которому они присоединены для образования пиперазинового кольца, причем пиперазиновое кольцо необязательно замещено C1-C4 алкилом. Изобретение также относится к способам получения соединения формулы 1 и его применению. Технический результат – получены новые соединения, которые могут найти применение в медицине для профилактики, облегчения или лечения нейродегенеративных заболеваний, заболеваний печени, метаболических заболеваний, путем индуцирования аутографии. 8 н. и 8 з.п. ф-лы, 4 ил., 7 табл., 26 пр.
Формула 1
1. Соединение формулы 1 или его фармацевтически приемлемая соль:
Формула 1
причем Y представляет собой О или S,
(1) если Y - это О,
R1 представляет собой водород или C1-С4 алкильную группу, замещенную моно- или ди-C1-С5 алкиламино,
R2 - C1-С6 алкильная группа,
R3 - водород, и R4 - (4-(диметиламино)тетрагидро-2Н-пиран-4-ил)метильная группа, C1-C4 алкильная группа, замещенная моно- или ди-С1-С5 алкиламино, C1-C4 алкильная группа, замещенная азотсодержащим циклическим кольцом, выбранным из группы, состоящей из морфолина, пиперидина или пирролидина, причем азотсодержащее циклическое кольцо необязательно замещено C1-С4 алкилом, или пиперидинильная группа, необязательно замещенная C1-С4 алкилом, или
R3 и R4 соединены друг с другом атомом азота, к которому они присоединены для образования пиперазинового кольца, причем пиперазиновое кольцо необязательно замещено C1-C4 алкилом,
(2) если Y - это S,
R1 и R2 независимо друг от друга представляют собой водород; C1-С6 алкильную группу или C1-C4 алкильную группу, замещенную моно- или ди-С1-С5 алкиламино,
R3 - водород, a R4 - C1-C4 алкильная группа, замещенная моно- или ди-С1-С5 алкиламино, или
R3 и R4 соединены друг с другом атомом азота, к которому они присоединены для образования пиперазинового кольца, причем пиперазиновое кольцо необязательно замещено C1-C4 алкилом.
2. Соединение или его фармацевтически приемлемая соль по п. 1, причем
Y-O,
R1 - водород или диэтиламиноэтильная группа, а
R2 - метильная группа.
3. Соединение или его фармацевтически приемлемая соль по п. 1, причем
Y-O,
R1 - водород или диэтиламиноэтильная группа,
R2 - метильная группа,
R3 - водород, а
R4 - (4-(диметиламино)тетрагидро-2Н-пиран-4-ил)метильная группа, диметиламиноэтильная группа, диэтиламиноэтильная группа, диизопропиламиноэтильная группа, морфолиноэтильная группа, необязательно замещенная С1-С4 алкилом, пиперидиноэтильная группа, необязательно замещенная C1-C4 алкилом, пирролидиноэтильная группа, необязательно замещенная C1-C4 алкилом, или пиперидинильная группа, необязательно замещенная С1-С4 алкилом.
4. Соединение или его фармацевтически приемлемая соль по п. 1, причем
Y-O,
R1 - водород или диэтиламиноэтильная группа,
R2 - метильная группа,
R3 - водород, а
R4 - (4-(диметиламино)тетрагидро-2Н-пиран-4-ил)метильная группа.
5. Соединение или его фармацевтически приемлемая соль по п. 1, причем
Y-O,
R1 - водород или диэтиламиноэтильная группа,
R2 - метильная группа, и
R3 и R4 соединены друг с другом атомом азота, к которому они присоединены для образования пиперазинового кольца, необязательно замещенного С1-С4 алкилом.
6. Соединение или его фармацевтически приемлемая соль по п. 1, причем
Y-S,
R1 - водород или диэтиламиноэтильная группа, а
R2 - метильная группа.
7. Соединение или его фармацевтически приемлемая соль по п. 1, причем
Y-S,
R1 - водород или диэтиламиноэтильная группа,
R2 - метильная группа,
R3 - водород, и
R4 - изопропильная группа или диизопропиламиноэтильная группа.
8. Соединение или его фармацевтически приемлемая соль по п. 1, причем
Y-S,
R1 - водород или диэтиламиноэтильная группа,
R2 - метильная группа, и
R3 и R4 соединены друг с другом атомом азота, к которому они присоединены для образования пиперазинового кольца, необязательно замещенного C1-C4 алкилом.
9. Соединение или его фармацевтически приемлемая соль, которые являются одним или несколькими соединениями, выбранными из группы, включающей:
N-(4-метилпиперазино)-5-(4-гидрокси-3-метоксифенил)тиофен-2-карбоксамид гидрохлорид,
N-(4-метилпиперазино)-5-(4-(2-диэтиламино)этокси-3-метоксифенил)тиофен-2-карбоксамид гидрохлорид,
N-(4-метилпиперазино)-5-(4-гидрокси-3-метоксифенил)тиофен-2-тиокарбоксамид гидрохлорид,
N-(4-метилпиперазино)-5-(4-(2-диэтиламино)этокси-3-метоксифенил)тиофен-2-тиокарбоксамид гидрохлорид,
N-(2-(диизопропиламино)этил)-5-(4-гидрокси-3-метоксифенил)тиофен-2-карбоксамид гидрохлорид,
N-(2-(диизопропиламино)этил)-5-(4-(2-диэтиламино)этокси-3-метоксифенил)тиофен-2-карбоксамид гидрохлорид,
N-(2-(диизопропиламино)этил)-5-(4-гидрокси-3-метоксифенил)тиофен-2-тиокарбоксамид гидрохлорида,
N-(2-(диизопропиламино)этил)-5-(4-(2-диэтиламино)этокси-3-метоксифенил)тиофен-2-тиокарбоксамид гидрохлорид,
N-изопропил-5-(4-(2-диэтиламино)этокси-3-метоксифенил)тиофен-2-карбоксамид гидрохлорид,
N-изопропил-5-(4-(2-диэтиламино)этокси-3-метоксифенил)тиофен-2-тиокарбоксамид гидрохлорид,
N-изопропил-5-(4-гидрокси-3-метоксифенил)тиофен-2-тиокарбоксамид,
N-(2-(диэтиламино)этил)-5-(4-гидрокси-3-метоксифенил)тиофен-2-карбоксамид гидрохлорид,
N-(2-(диэтиламино)этил)-5-(4-(2-диэтиламино)этокси-3-метоксифенил)тиофен-2-карбоксамид гидрохлорид,
N-(2-(диметиламино)этил)-5-(4-гидрокси-3-метоксифенил)тиофен-2-карбоксамид гидрохлорид,
N-(2-(диметиламино)этил)-5-(4-(2-диэтиламино)этокси-3-метоксифенил)тиофен-2-карбоксамид гидрохлорид,
N-(2-(4-морфолино)этил)-5-(4-гидрокси-3-метоксифенил)тиофен-2-карбоксамид гидрохлорид,
N-(2-(1-пиперидино)этил)-5-(4-гидрокси-3-метоксифенил)тиофен-2-карбоксамид гидрохлорид,
N-(2-(1-пиперидино)этил)-5-(4-(2-диэтиламино)этокси-3-метоксифенил)тиофен-2-карбоксамид гидрохлорид,
N-(2-(1-пирролидино)этил)-5-(4-гидрокси-3-метоксифенил)тиофен-2-карбоксамид гидрохлорид,
N-((4-(диметиламино)тетрагидро-2Н-пиран-4-ил)метил)-5-(4-гидрокси-3-метоксифенил)тиофен-2-карбоксамид гидрохлорид,
N-((4-(диметиламино)тетрагидро-2Н-пиран-4-ил)метил)-5-(4-(2-диэтиламино)этокси-3-метоксифенил)тиофен-2-карбоксамид гидрохлорид,
N-(4-(1-метил)пиперидинил)-5-(4-гидрокси-3-метоксифенил)тиофен-2-карбоксамид гидрохлорид,
N-(2-(2-(1-метил)пирролидино)этил)-5-(4-гидрокси-3-метоксифенил)тиофен-2-карбоксамид гидрохлорид,
N-(2-(2-(1-метил)пирролидино)этил)-5-(4-(2-диэтиламино)этокси-3-метоксифенил)тиофен-2-карбоксамид гидрохлорид,
N-((S)-2-(1-этил)пирролидинометил)-5-(4-гидрокси-3-метоксифенил)тиофен-2-карбоксамид гидрохлорид и
N-((S)-2-(1-этил)пирролидинометил)-5-(4-(2-диэтиламино)этокси-3-метоксифенил)тиофен-2-карбоксамид гидрохлорид.
10. N-((4-(диметиламино)тетрагидро-2Н-пиран-4-ил)метил)-5-(4-гидрокси-3-метоксифенил)тиофен-2-карбоксамид или его фармацевтически приемлемая соль.
11. N-((4-(диметиламино)тетрагидро-2Н-пиран-4-ил)метил)-5-(4-(2-диэтиламино)этокси-3-метоксифенил)тиофен-2-карбоксамид или его фармацевтически приемлемая соль.
12. Способ получения соединения Формулы 1а или его фармацевтически приемлемой соли, включающий связывание соединения Формулы 2 с соединением Формулы 3 для получения соединения Формулы 1а:
Формула 1а:
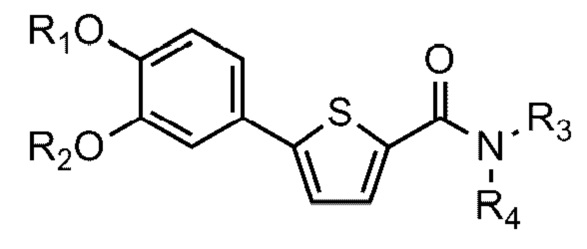
Формула 2:
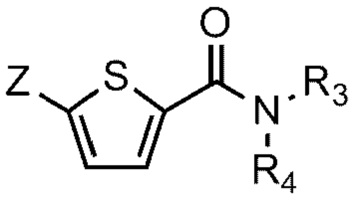
Формула 3:
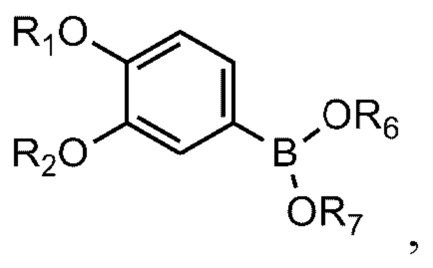
причем R1, R2, R3 и R4 такие же, как определено в п. 1, Z - галоген, a R6 и R7 представляют собой водород или соединены друг с другом атомом бора, к которому они присоединены, для образования 4,4,5,5-тетраметил-1,3,2-диоксаборолана.
13. Способ получения соединения Формулы 1b или его фармацевтически приемлемой соли, включающий связывание соединения Формулы 6 с соединением Формулы 3 для получения соединения Формулы 1b:
Формула 1b:
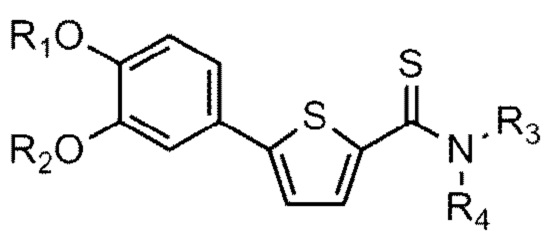
Формула 6:
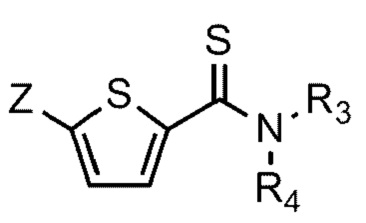
Формула 3:
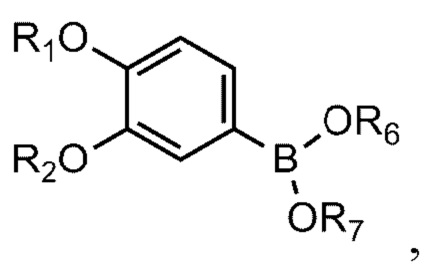
причем R1, R2, R3 и R4 такие же, как определено в п. 1, Z - галоген, a R6 и R7 представляют собой водород или соединены друг с другом атомом бора, к которому они присоединены, для образования 4,4,5,5-тетраметил-1,3,2-диоксаборолана.
14. Способ по п. 13, причем соединение Формулы 6 получают путем выполнения тиоамидирования соединения Формулы 2:
Формула 2:
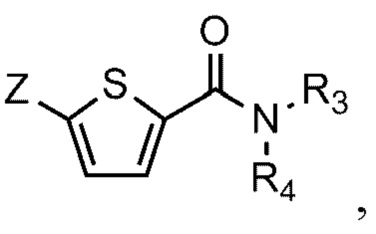
причем R3 и R4 такие же, как определено в п. 1, и Z - галоген.
15. Способ получения соединения Формулы 1b или его фармацевтически приемлемой соли, включающий выполнение тиоамидирования соединения Формулы 1а.
Формула 1а:
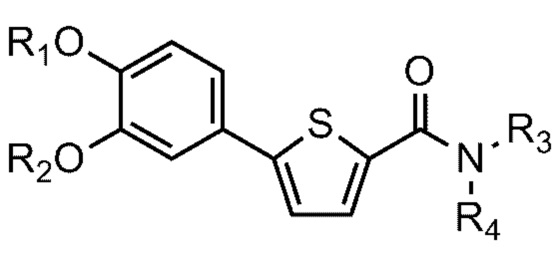
Формула 1b:
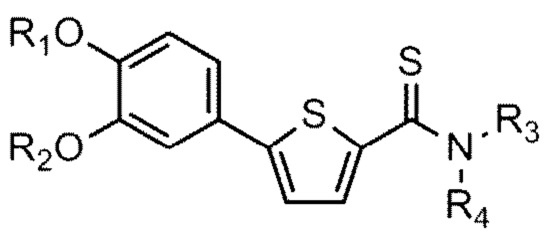
причем R1, R2, R3, и R4 такие же, как определено в п. 1.
16. Применение соединения или его фармацевтически приемлемой соли по одному из пп. 1-11 для профилактики, облегчения или лечения нейродегенеративных заболеваний, выбранных из группы, включающей болезнь Хантингтона, болезнь Паркинсона, болезнь Альцгеймера, прионную болезнь, рассеянный склероз и болезнь Лу Герига; заболевания печени, выбранные из группы, содержащей фиброз печени, цирроз печени, гепатит и жировую болезнь печени; метаболические заболевания, выбранные из группы, содержащей диабет, гиперлипидемию, ожирение и воспаление, или сепсис, путем индуцирования аутофагии.
| EP 3339299 A2, 27.06.2018 | |||
| WO 2011162267 A1, 29.12.2011 | |||
| CN 104725347 A, 24.06.2015 | |||
| WO 2003028731 A1, 10.04.2003 | |||
| RU 2006107782 A, 10.08.2006 | |||
| СОЕДИНЕНИЯ (2-КАРБОКСИАМИДО)(3-АМИНО)ТИОФЕНА | 2004 |
|
RU2339633C2 |

