ОБЛАСТЬ, К КОТОРОЙ ОТНОСИТСЯ ИЗОБРЕТЕНИЕ
Данное изобретение относится к фармацевтическим композициям для доставки биологически активных агентов через слизистую оболочку. Более конкретно, данное изобретение относится к новому способу регуляции и стимулирования скорости и степени проникновения через слизистую оболочку и всасывания противосудорожного агента при совместном применении лекарственного средства с фармацевтически приемлемой системой сорастворителей, включающей алифатический спирт, гликоль и воду и их комбинации с биологическими поверхностно-активными веществами, такими как соли желчных кислот или лецитин. Еще более конкретно, данное изобретение относится к фармацевтическим композициям, создающим приемлемую для больного назальную систему доставки противосудорожного средства, которая применима для экстренной помощи в случае непрерывного эпилептического припадка (status epilepticus) и приступа лихорадки и при быстром и удобном способе применения.
ПРЕДПОСЫЛКИ СОЗДАНИЯ ИЗОБРЕТЕНИЯ
Статус эпилептикус (непрерывный эпилептический припадок) представляет собой критическое неврологическое состояние, при котором смертность составляет 3-35%. Основной целью лечения является быстрая ликвидация активности патологического приступа; чем позднее начнется лечение этого приступа, тем труднее его контролировать и тем больше риск хронических церебральных нарушений. Следовательно, решающим для помощи больному является четкая программа действий, включающая быстрое лечение эффективными лекарственными средствами в достаточных дозах в соответствующей фармацевтической рецептуре, а также помощь при гиповентиляции (легких) и стерильной гипотензии.
В настоящее время в лечении статус эпилептикус применяется несколько схем приема лекарственных веществ. Диазепам и лоразепам наиболее широко используемые бензодиазепины для этой цели. Внутривенное введение притивосудорожных средств является наиболее быстрым способом подавления эпилептических судорог. Однако, другие способы применения могут быть очень желательны, когда внутривенное введение неудобно и запаздывает, например, из-за технических затруднений, таких как необходимость стерильного оборудования и опытного персонала и из-за возможного развития тромбофлебита. Кроме того, внутривенные методы терапии часто связаны с гипотензией, сердечной аритмией или ослаблением функции центральной нервной системы. В связи с этим Moolenaar [Moolenaar et al., Int. J. Pharm., 5:127-137 (1986)] попытался вводить диазепам больным несколькими другими способами, такими как внутримышечная инъекция, пероральная таблетка и ректальный раствор. Было найдено, что только ректальное введение обеспечивает довольно быстрое всасывание и, следовательно, может рассматриваться как способ, альтернативный IV (в.в.) инъекции. Однако, ректальный путь очень неудобный способ введения лекарственного вещества, особенно в случае неотложной помощи. В Патенте США 4863720 (Burghardt) описывается подъязычный, впрыскиваемый фармацевтический препарат, в котором активным лекарственным веществом может быть бензодиазепин, оптимально содержащий полиэтиленгликоль (ПЭГ, PEG) и этанол, ди- и/или триглицериды жирных кислот и фармацевтически приемлемый газ-вытеснитель.
Позже оказалось, что введение через слизистую оболочку носа обеспечивает терапевтический эффект в случае многих лекарственных веществ. Назальное применение имеет то преимущество, что лекарственные вещества можно вводить легко и просто для достижения системного или локального эффекта. Однако основной проблемой при приеме лекарственного вещества через нос является то, что большинство молекул лекарственных веществ медленно и слабо диффундируют через слизистую оболочку носа и, следовательно, заданные уровни терапевтического агента не могут быть достигнуты при простом введении через нос. Дополнительное ограничение, связанное с назальным введением, - это требуемый для введения малый объем; обычно невозможно вводить в каждую ноздрю более 150 мкл; избыток попадает в глотку и проглатывается. Следовательно, требуются растворители-носители с высокой растворимостью в них лекарственного вещества и не раздражающие слизистую носа. Всасывание лекарственных веществ через нос можно увеличить совместным введением химического адъюванта или энхансеров проницаемости. Например, Lau и Slattery [Lau et al., Int. J. Pharm., 54:171-174 (1989)] пытались вводить бензодиазепин, такой как диазепам и лоразепам, растворяя эти лекарственные вещества в нескольких растворителях: триацетин, диметилсульфоксид, PEG 400, Cremophor EL, Lipal-9-LA, изопропиладипинат и Азон (Azone). Хотя многие растворители растворяли диазепам и лоразепам в нужных концентрациях, они вызывали сильное раздражение, чтобы их вводить в нос. Найдено, что Cremophor EL (кремофор) менее всех раздражает слизистую носа. Но назальное всасывание при применении этого носителя у людей достаточно медленное (Тmax=1,4 час) и максимальная концентрация низка по сравнению с концентрацией, наблюдаемой после внутривенного введения. В Патенте США 4950664 Rugby описывает назальное введение бензодиазепинового снотворного в фармацевтически приемлемом носителе. Носителем может быть водный физиологический раствор, спирт, гликоль, простой эфир гликоля или их смеси. Результаты фармакокинетических исследований на собаках показали, что время, через которое концентрация триазолама в плазме достигает максимума, составляет 18 минут после назального введения, хотя целью является срочная помощь в пределах 5 минут. Bechgaard и Hjortkjer [Bechgaard et al., J. Pharm. Pharmacol., 49: 747-750 (1997)] описали применение чистых органических растворителей, таких как гликофурол и тетраэтиленгликоль, и их комбинаций в качестве носителей для назальной доставки диазепама. Абсолютная биосовместимость, измеряемая в течение первых 30 минут после введения через нос, составляла 49-62% для наиболее перспективных изученных систем носителей. В заявке РСТ WO 95/31217 Dumex описал применение фармацевтического препарата в форме эмульсии на основе токоферола и его производных для назального введения биологически активных соединений, включая бензодиазепины.
СУЩНОСТЬ ИЗОБРЕТЕНИЯ
Данное изобретение охватывает новый способ модулируемого с помощью растворителя введения противосудорожного агента через слизистые оболочки человека и животных. Система растворителя находится в водном фармацевтическом носителе, содержащем алифатический спирт или гликоль и их комбинации с биологическим поверхностно-активным веществом, таким как соль желчной кислоты или лецитин.
Целью данного изобретения является создание фармацевтически приемлемой системы носителей, которая способна повысить проникновение и всасывание через слизистую оболочку противосудорожного агента. Ингредиенты, используемые в фармацевтической композиции, предпочтительно состоят из GRAS (общепризнанных как безопасные) веществ, так что вопрос о токсичности не стоит. Другой целью данного изобретения является создание способа регулирования доставки противосудорожного вещества через слизистую оболочку с соответствующим образом корректируемой скоростью так, чтобы достичь оптимального терапевтического эффекта и при этом избежать или снизить нежелательные побочные эффекты. Такие композиции особенно применимы для введения препаратов через нос при срочной помощи в случае статус эпилептикус и приступов лихорадки.
КРАТКОЕ ОПИСАНИЕ ФИГУР
На Фиг.1 изображен график, показывающий влияние наполнителя на in vitro проникновение препарата диазепама по изобретению через слизистую оболочку носа.
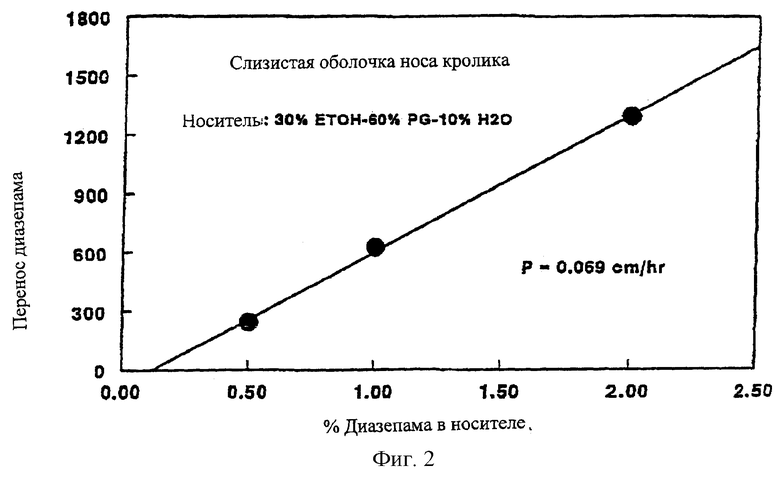
На Фиг.2 изображен график, показывающий влияние уровня концентрации лекарственного вещества на in vitro проникновение препарата диазепама из носителя по изобретению через слизистую оболочку носа.
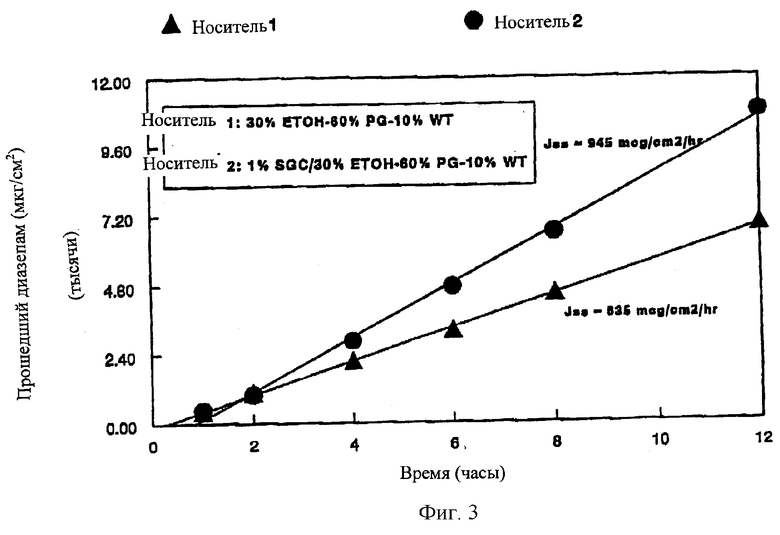
На Фиг.3 изображен график, показывающий влияние гликохолата натрия (SGC) на in vitro проникновение препарата диазепама из носителя по изобретению через слизистую оболочку носа.
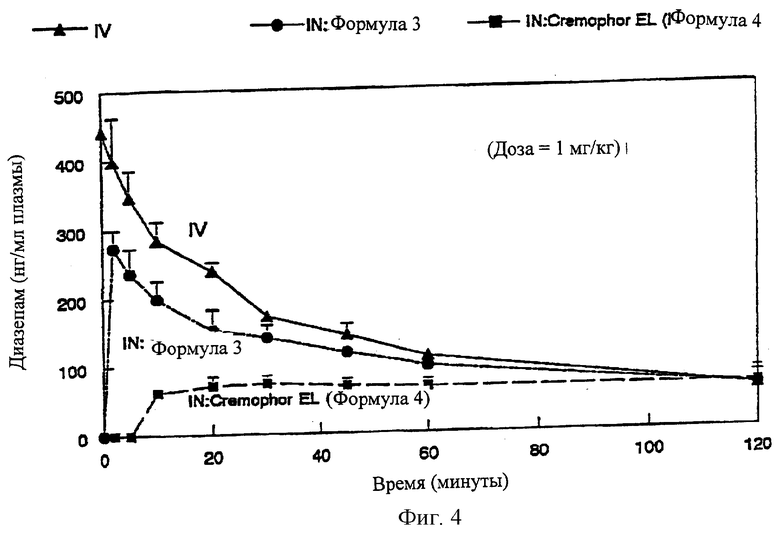
На Фиг.4 даны кривые зависимости средней концентрации диазепама от времени после внутривенного (в.в., IV) введения и интраназального введения препарата по данному изобретению (однократная доза).
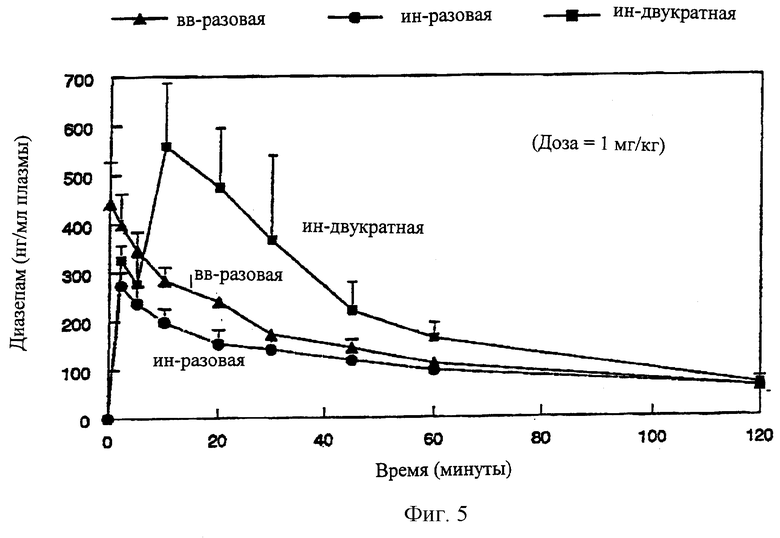
На Фиг.5 даны кривые зависимости средней концентрации диазепама в плазме от времени после внутривенного и интраназального введения препарата по изобретению (многократная доза).
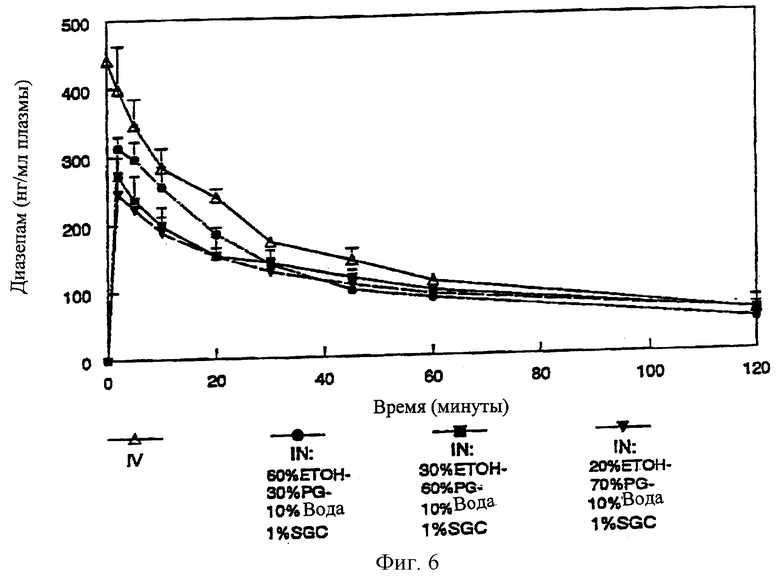
На Фиг.6 даны кривые средней концентрации диазепама в плазме - время после интраназального введения препарата как функция объемного соотношения полиэтиленгликоль, этанол в препарате по изобретению.
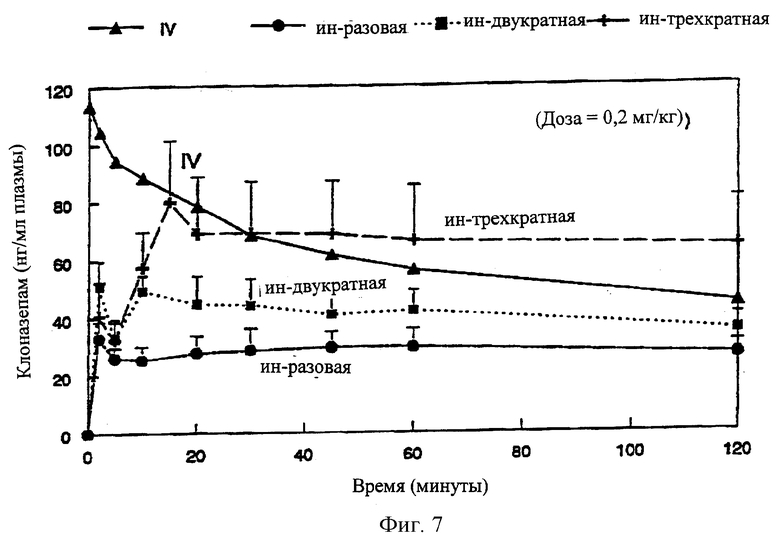
На Фиг.7 даны кривые средней концентрации клоназепама в плазме от времени после внутривенного введения и интраназального введения препарата по изобретению (однократная и многократная дозы).
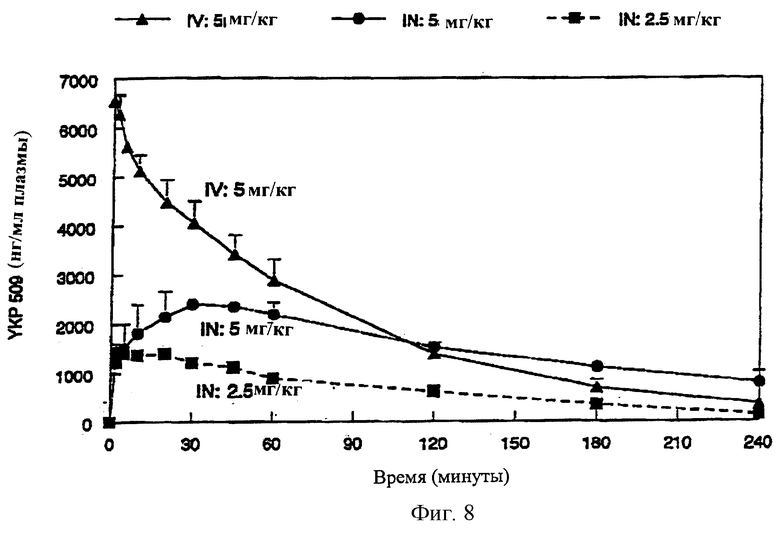
На Фиг.8 даны кривые: средняя концентрация (S)-2-карбамоилокси-1-о-хлорфенилэтанола в плазме - время после внутривенного введения и интраназального введения препарата по изобретению как функция величины дозы.
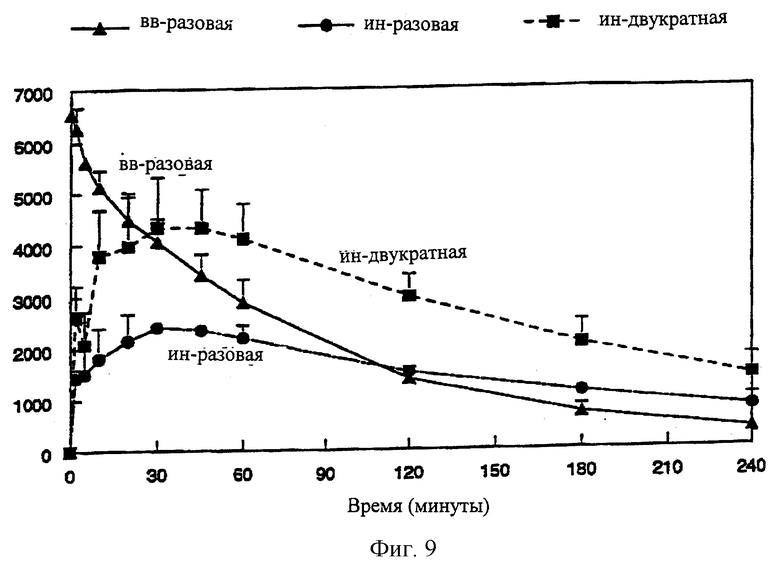
На Фиг.9 даны кривые: средняя концентрация (S)-2-карбамоилокси-1-о-хлорфенилэтанола в плазме - время после внутривенного введения и интраназального введения препарата по изобретению (однократная и многократная дозы).
ПОДРОБНОЕ ОПИСАНИЕ ИЗОБРЕТЕНИЯ
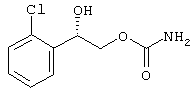
Согласно данному изобретению определенная водная система сорастворителей, содержащая один алифатический спирт, один гликоль и биологическое поверхностно-активное вещество, обеспечивает улучшенную, с регуляцией скорости, трансназальную доставку противосудорожного агента. Спирт по данному изобретению выбирают из С1-С5-алифатических спиртов; гликоль выбирают из пропиленгликоля (PG), полиэтиленгликоля (PEG, ПЭГ) 200, PEG 300 и PEG 400 и PEG 600; и биологическое поверхностно-активное вещество выбирают из солей желчных кислот, таких как холат натрия, дезоксихолат натрия, таурохолат натрия, гликохолат натрия и урсодезоксихолат натрия, или лецитин, такой как лизофосфатидилхолины, фосфатидилхолины, фосфатидилсерины, фосфатидилинозиты, фосфатидилэтаноламины и фосфатидилглицерины. Вышеописанные композиции могут применяться для лекарственных препаратов, содержащих противосудорожные вещества, подходящие для слизистой оболочки человека и животных. Более конкретно, эти препараты представляют собой композиции, которые содержат бензодиазепин, такой как диазепам, клоназепам и лоразепам, и новое противосудорожное соединение на основе монокарбамата, (S)-2-карбамоилокси-1-о-хлорфенилэтанол, представленный следующей формулой:
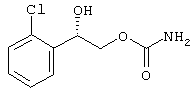
адаптированные для введения через нос, в растворе, суспензии, геле или в виде другой применимой для назального введения рецептуры. Эти назальные композиции можно применять для любой из известных терапевтических целей, для которых известны такие противосудорожные средства, включая фенитоины (фенитоин, мефенитоин и этотоин), барбитураты (фенобарбитал, мефобарбитал и примидон), иминостильбены (карбамазепин), сукцинимиды (этозуксимид), вальпроевую кислоту, оксазолидиндионы (триметадион) и другие средства против эпилептических припадков (габапентин, ламотригин, ацетазоламид, фелбамат и γ-винил GABA). Применение интраназальной рецептуры противосудорожного вещества значительно облегчает применение. По сравнению с парентеральным введением, например, простой пульверизатор или ингалятор достаточны для быстрой и удобной доставки лекарственных веществ, в частности для быстрой помощи при остром приступе эпилепсии. С клинической точки зрения интраназальное введение часто приводит к более продолжительному противосудорожному действию. По данному изобретению терапевтический эффект, с точки зрения приступа, интенсивность и длительность можно более эффективно и точно регулировать, меняя соотношение алифатического спирта и гликоля в носителе и с помощью введения в виде однократной или многократных доз препарата по изобретению. Хотя это изобретение описано относительно противосудорожного вещества как модельного соединения, понятно, что это изобретение также применимо к другим биологически активным веществам, которые применимы для слизистой оболочки человека и животных.
Изобретение далее иллюстрируется следующими примерами, которые только иллюстрируют конкретный способ практического применения изобретения и не ограничивают объем изобретения.
Пример 1
In vitro исследование проникновения (прохождения) через назальную мембрану
Слизистую оболочку носа, используемую в этих in vitro экспериментах, получают от новозеландских белых кроликов (2,5-3,0 кг). Кроликов умерщвляют с помощью (внутривенной) (IV) инъекции фенобарбитала. Носовую перегородку осторожно отделяют от кости с помощью хирургических ножниц и пилы для разрезания костей. Затем два фрагмента (две части) слизистой оболочки носа осторожно счищают с (отделяют от) перегородки, не трогая центр поверхности оболочки и промывают нормальным физиологическим раствором. Слизистую оболочку помещают между двумя половинами камеры стеклянного прибора (камеры) для изучения диффузии. Экспонируемая поверхность составляет, примерно, 0,64 см2. Испытуемый раствор или суспензию (3,5 мл) вводят со стороны слизистой оболочки мембраны в донорском отсеке, тогда как 3,5 мл 10% этанола, 40% полиэтиленгликоля и 50% изотонического раствора фосфатного буфера рН 7,4 вводят в рецепторный отсек. Всю диффузионную систему выдерживают при 37°С в течение всего эксперимента. Через определенные интервалы 100 мкл рецепторного раствора отбирают для анализа и снова заполняют тем же объемом рецепторной среды, сохраняя объем постоянным. Величину переноса в стационарном состоянии определяют по наклону прямой линии на графике совокупного количества прошедшего лекарственного вещества в зависимости от времени. Каждый эксперимент проводят по меньшей мере дважды. Этот метод используют в Примерах 2-6.
В этом исследовании применяют систему для жидкостной хроматографии высокого давления с системой-кодом нескольких (смесей) растворителей (Model 600E, Waters Associates, Milford, Mass), с автоматическим шприцом (автовводом) (Model 717 Plus, Waters Ass.), детектором в виде набора фотодиодов (Model 996 Plus, Waters Ass.), обращенно-фазовую колонку Symmetric C18 (150 мм × 3,9 мм ID, 5 мкм) и компьютерную систему с программным обеспечением Millenium 2010. Подвижные фазы и УФ-длины волн, применяемые для анализа диазепама, клоназепама и (S)-2-карбамоилокси-1-о-хлорфенилэтанола, это 70% метанола, 30% воды при 254 нм; 60% метанола, 40% воды при 252 нм; и 25% ацетонитрила и 75% воды при 262 нм соответственно.
Пример 2
В этом примере показано влияние соли желчной кислоты и лецитина, растворенных в водной среде с концентрацией 1% вес/объем на in vitro проникновение модельного лекарственного вещества диазепама через свежеиссеченную носовую мембрану. В этих исследованиях изучают ряд солей желчных кислот, таких как холат натрия, дезоксихолат натрия, таурохолат натрия и гликохолат натрия, и лецитин, такой как лизофосфатидилхолин. Скорости проникновения измеряют, применяя метод, описанный в исследовании in vitro проникновения через назальную мембрану. Средние данные трансназального переноса в стационарном состоянии, полученные таким образом, представлены в Таблице I.

Как видно из Таблицы I, соль желчной кислоты, такая как гликохолат натрия, и лецитин, такой как лизофосфатидилхолин, оказывают значительное влияние на прохождение диазепама через слизистую оболочку (мембрану) носа.
Пример 3
Данный пример иллюстрирует влияние носителя на in vitro прохождение диазепама через слизистую оболочку носа кролика при 37°С. В этом эксперименте 1% суспензию и раствор диазепама готовят, используя воду и носитель из сорастворителей, состоящий из 30% этанола (EtOH), 60% пропиленгликоля (PG) и 10% воды (WT) соответственно. Скорости проникновения определяют по методу, описанному в Примере 1. Графики трансназального прохождения (проникновения), полученные таким образом, показаны на Фиг.1.
Как видно из Фиг.1, носитель из сорастворителей, содержащий этанол, пропиленгликоль и воду, способствует почти 8-кратному повышению скорости трансназального переноса диазепама по сравнению со скоростью водной суспензии.
Пример 4
В данном примере показано влияние концентрации лекарственного вещества в донорской половине (отсеке) на прохождение (транспорт) диазепама через слизистую оболочку (мембрану) носа in vitro. В этом исследовании готовят 0,5-2% препараты диазепама, используя смесь из сорастворителей, содержащую 30% этанола, 60% пропиленгликоля и 10% воды. Скорости in vitro трансмембранного транспорта (прохождения) определяют по методу, описанному в Примере 1. Данные трансназального переноса in vitro, полученные при концентрациях диазепама в рецептурах 0,5-2%, показаны на Фиг.2.
Как видно из Фиг.2, трансназальный перенос диазепама в стационарном состоянии повышается линейно при повышении концентрации лекарственного вещества в донорской половине выше 0,5-2,0%.
Пример 5
В данном примере иллюстрируется влияние введения соли желчной кислоты в назальную рецептуру по изобретению на in vitro прохождение диазепама через слизистую оболочку носа. В этом опыте изучают эффект введения 1% гликохолата натрия в носитель, состоящий из 30% этанола, 60% пропиленгликоля и 10% воды. Готовят образцы растворов лекарственного вещества (10 мг/мл) в носителе, содержащем и не содержащем соль желчной кислоты. Скорости транспорта через мембрану измеряют по методу, описанному в Примере 1. Графики in vitro проникновения (транспорта), полученные таким образом, представлены на Фиг.3.
Как видно из Фиг.1, введение 1% гликохолата натрия значительно повышает скорость прохождения диазепама через слизистую оболочку носа. Отмечено почти 50%-ное повышение трансмембранного переноса в стационарном состоянии, когда в носитель добавляют соль желчной кислоты.
Пример 6
Данный пример иллюстрирует сравнительную трансназальную проницаемость трех модельных лекарственных веществ, таких как диазепам, клоназепам и (S)-2-карбамоилокси-1-о-хлорфенилэтанол. В этом эксперименте используют носитель из сорастворителей, состоящий из 30% этанола, 60% пропиленгликоля и 10% воды. In vitro эксперименты по проникновению (прохождению сквозь) осуществляют, используя метод анализа, описанный в Примере 1. Сравнительный коэффициент проницаемости через мембрану и данные переноса в стационарном состоянии, полученные для препаратов с начальной концентрацией лекарственных веществ 5 мг/мл, представлены в Таблице II.
Как видно из Таблицы II, трансназальная проницаемость противосудорожного вещества на основе монокарбамата, (S)-2-карбамоилокси-1-о-хлорфенилэтанола, по-видимому, в два раза выше трансназальной проницаемости диазепама.
Пример 7
Биодоступность и фармакокинетика препаратов диазепама
Характеристики биодоступности и фармакокинетики препаратов по изобретению, содержащих диазепам, определяют (анализируют) после назального введения новозеландским белым кроликам (n=3-4). Для сравнения исследуют действие инъекции диазепама (Формула 1 в Таблице III) in vivo после внутривенного введения той же дозы. В.в. Формулы 1 (10 мг/2 мл) получают из Elkins-Sinn, Inc., их готовят с пропиленгликолем (0,4 мл), спиртом (0,1 мл), бензиловым спиртом (0,015 мл), бензоатом натрия/бензойной кислоты (50 мг) и достаточным количеством воды для инъекции для получения 1 мл. Для назального применения готовят два препарата, используя систему носителей по изобретению, состоящую из 30% этанола, 60% пропиленгликоля и 10% воды с (Формула 3 в Таблице III) и без (Формула 2 в Таблице III) 1% гликохолата натрия соответственно. Другую рецептуру для носа (назальная) (Формула 4 в Таблице III) с неионным поверхностно-активным носителем, полиоксиэтилированным касторовым маслом (Cremophor EL) также испытывают после интраназального применения с целью сравнения, так как этот препарат испытывали на людях Lau и Slattery (1989). Все препараты для введения в нос готовят непосредственно перед экспериментами, растворяя 20 мг диазепама (Sigma Chemical) в 1 мл описанных выше носителях.
Непосредственно перед экспериментом кроликов (n=3-4) взвешивают и помещают в клетку для подготовки. Каждому кролику вводят 100 мкл препаратов Формулы 2 или 3 в каждую ноздрю из пульверизатора Пфайфера (Pfeiffer) за 5 сек. Кроликам, получающим в.в. (IV), вводят 1 мг/кг препарата формулы 1 в ушную вену внутривенным вливанием за 20 сек. Для изучения действия повторной дозы тот же объем препарата Формулы 3 (100 мкл) впрыскивают в каждую ноздрю через 5 минут после первой дозы. Пробы крови (1 мл) отбирают через 0, 2, 5, 10, 20, 30, 45, 60 и 120 минут после IV (в.в.) и IN-введения. Из проб крови плазму отделяют центрифугированием и хранят при -20°С до испытания. Для анализа образцы плазмы (0,5 мл) осторожно переносят в полипропиленовую пробирку для центрифуги на 1,5 мл. К образцу плазмы добавляют 0,5 мл 0,01% объем/объем перхлорной кислоты в ацетонитриле, содержащем внутренний стандарт (клоназепам 1 мкг/мл). Смесь перемешивают 30 сек и центрифугируют при 4000 об/мин в течение 10 минут. Концентрацию диазепама в плазме анализируют ВЭЖХ. Анализ осуществляют с помощью Waters ВЭЖХ, как описано в Примере 1. В этом исследовании используют колонку 3,8 мм × 150 мм × 5 мкм Symmetric C18. Подвижная фаза 50 об.% метанола:10 об.% ацетонитрила:40 об.% фосфатного буфера с рН 3,5. Скорость протекания подвижной фазы 1 мл/мин и УФ-детектирование проводят при 228,5 нм. Предел обнаружения для диазепама составляет 70 нмол/л. Площадь (AUC) под кривыми зависимости концентрация в плазме - время, от 0 мин до 120 мин, вычисляют с помощью линейного трапецеидального метода. Данные по биодоступности и фармакокинетике, полученные таким образом, приведены в Таблице III. Сравнительные фармакокинетические кривые, полученные после однократного в.в. (IV) введения (Формула 1) и однократного и двухкратного IN-введения препаратов по изобретению (Формулы 3 и 4), приведены на Фиг.1 и 5 соответственно.
(1 мг/кг × 1)
b IN (ин) Формула 2: 2% раствор диазепама в 60% PG, 30% EtOH и 10% воды
с IN (ин) Формула 3: 2% раствор диазепама в 1% SGC, 60% PG, 30% EtOH и 10% воды
d Стандартное отклонение
е Нормализованные данные, определяемые по уравнению:
F={AUCIN, 1 мг×2/2 × AUCIV, мг×1/×100}
f Время применения: t0: первая доза для назального введения
t5 минут: вторая доза для назального введения
g IN (ин) Формула 4: 2% раствор диазепама в Cremophor EL.
Как видно из Фиг.4 и Таблицы III, IN Формулы 3, приготовленный с 1% SGC, 30% EtOH, 60% PG и 10% воды, заметно повышает трансназальное всасывание по сравнению с Cremophor EL Формулы 4. Величины Сmax и AUC0-120 мин для IN Формулы 3 составляют около 69% и 76% относительно соответствующих величин для IV-введения (вв) соответственно. С другой стороны, значения Сmax и AUC0-120 мин для Cremophor EL Формулы 4 составляют около 19% и 42,6% от значений для IV-инъекции. Эти относительные результаты, по-видимому, согласуются с данными по фармакокинетике для человека, опубликованными Lau и Slattery (1989). Согласно опубликованным данным, Тmax препарата Cremophor EL составляет 1,4 час после интраназального введения человеку, а Сmax составляет только 27% по сравнению с IV-инъекцией. Достаточно неожиданным, как видно из Фиг.5 и Таблицы III, является то, что повторное интраназальное введение через 5 минут после первой дозы дает заметное увеличение интраназального всасывания диазепама. Значения Сmax и AUC точно вдвое больше после второго введения по сравнению со значениями, полученными при первом введении. Кроме того, уровень диазепама в плазме после второй дозы превосходит уровень при однократном IV за 7 минут. Эти открытия четко демонстрируют, что схему повторного введения дозы (за короткий период времени) можно эффективно использовать для срочной помощи при приступе эпилепсии, когда однократная интраназальная доза не способна дать желательный терапевтический эффект.
Пример 8
Контроль фармакокинетики при максимальных концентрациях (пиках) в плазме
Готовят два мг диазепама в 100 мкл носителя и вводят кроликам (n=3) аналогично описанному в Примере 7. Испытывают следующие носители: 60% EtOH, 30% PG и 10% воды (WT) с 1% SGC, 30% EtOH, 60% PG и 10% воды (WT) с 1% SGC, и 20% EtOH, 70% PG и 10% воды (WT) с 1% SGC. Пробы крови берут из ушной вены со следующими 5-минутными интервалами: 0, 2, 5, 10, 20, 30, 45, 60 и 120 минут. Концентрацию диазепама в плазме определяют ВЭЖХ. Фармакокинетические кривые, полученные после IV и IN-введения препаратов, представлены в Таблице IV и на Фиг.6.
(1 мг/кг × 1)
(1 мг/кг × 1)
(1 мг/кг × 1)
(1 мг/кг × 1)
b IN (ин) Формула А: 2% раствор диазепама в 60% PG, 30% EtOH и 10% воды
с IN (ин) Формула В: 2% раствор диазепама в 1% SGC, 60% PG, 30% EtOH и 10% воды
d IN (ин) Формула С: 2% раствор диазепама в 1% SGC, 70% PG, 20% EtOH и 10% воды
е Стандартное отклонение
Как видно из Таблицы IV и Фиг.6, максимальную концентрацию лекарственного вещества в плазме, наблюдаемую через 2 минуты после IN-введения, можно регулировать в зависимости от объемного соотношения EtOH/PG в исследуемом носителе. Сmax постепенно увеличивается с увеличением соотношения объемов EtOH/PG от 0,3 до 2. Кроме того, максимальная концентрация в плазме для IN-носителя, состоящего из 60% EtOH, 30% PG и 10% воды (WT) с 1% SGC, через 2 минуты составляет около 79% от соответствующего значения при IV-инъекции.
Кроме того, модулированием объемного соотношения EtOH/PG в носителях можно также регулировать график концентрация в плазме - время на стадии элиминирования.
Пример 9
Фармакологическая реакция на препарат диазепама
Фармакологическую реакцию изучают на новозеландских белых кроликах, оценивая влияние диазепама на мышечную релаксацию после IV (вв)-введения и IN (ин)-введения препаратов по изобретению при уровне дозы 1 мг/кг. Носитель назального препарата состоит из 30% этанола, 60% пропиленгликоля и 10% воды, содержащей 1% SGC. Образец препарата готовят, растворяя 2 мг диазепама в 1 мл носителя с помощью ультразвука. IV-рецептура такая же, как рецептура, применяемая в Примере 7. Фармакологический ответ измеряют у кроликов после введения 100 мкл назальной рецептуры в каждую ноздрю при положении кролика лежа, тогда как его бедро крепко прижимают пальцем. Среднее время ответа (реакции), в течение которого кролики остаются в положении лежа, при этом их задние лапы вытянуты в одну сторону, после IV (вв) и IN (ин) введения, указано в Таблице V.
Как видно из Таблицы V, назальный препарат по изобретению обеспечивает очень быстрый ответ. Время фармакологической реакции составляет 1,5 минут.
Пример 10
Биодоступность и фармакокинетика препаратов клоназепама
Интраназальную рецептуру готовят, растворяя 8,36 мг клоназепама в 2 мл носителя по изобретению, состоящего из 30% EtOH, 60% PG и 10% воды, содержащей 1% SGC. Препарат для в.в. (IV)-инъекции готовят растворением 3 мг клоназепама в 2 мл раствора - 40% PG, 30% EtOH и 30% воды и фильтрованием через стерильный фильтр в асептических условиях. Препараты вводят кроликам (n=3) при дозе 0,2 мг/кг так, как это описано в Примере 7. Также исследуют схему с повторной (многократной, двукратной, трехкратной) дозой с 5-минутными интервалами. Пробы крови берут из ушной вены в следующие временные точки: 0, 2, 5, 10, 20, 30, 45, 60 и 120 минут. Из образцов крови плазму отделяют центрифугированием и хранят при -20°С до испытания. Для анализа образцы плазмы (0,5 мл) осторожно переносят в пробирки на 1,5 мл. К образцу плазмы добавляют 10 мкл раствора внутреннего стандарта (диазепам 5 мкг/мл) и 50 мл NaOH (0,5 М). К вышеуказанной смеси добавляют 5 мл диэтилового эфира и смесь перемешивают 60 сек и центрифугируют при 4000 об/мин в течение 10 минут. Вышеуказанный эфирный раствор переносят в пробирку на 5 мл и упаривают в вакуумном испарителе при 40°С в течение 30 мин. Остаток "воспроизводят", добавляя 100 мкл подвижной фазы для ВЭЖХ-анализа, состоящей из 20% метанола, 30% ацетонитрила и 50% буферного раствора КН2PO4/Н3PO4 с рН 3,5. Концентрацию клоназепама в плазме определяют ВЭЖХ при скорости потока 1 мл/мин и используя УФ-детектирование при 254 нм. Предел обнаружения для клоназепама составляет 16 нмол/л. Данные по биодоступности и фармакокинетике, полученные после в.в. (IV) и и.н. IN-введения по схеме разовой или многократной доз, приведены в Таблице VI, а графики зависимости средней концентрации в плазме от времени представлены на Фиг.7.
(0,2 мг/кг × 1)
(0,2 мг/кг × 1)
(0,2 мг/кг × 2)
(0,2 мг/кг × 3)
b IN (ин) Формула: 0,42% раствор клоназепама в 1% SGC, 60% PG, 30% EtOH и 10% воды
c Стандартное отклонение
d Нормализованные данные, вычисленные по следующему уравнению:
F={AUCIN, 0,2 мг × 2/2 × AUCIV, 0,2 мг×1}×100
е Нормализованные данные, вычисленные по следующему уравнению:
F={AUCIN, 0,2 мг × 3/3 × AUCIV, 0,2 мг×1}×100
f Время введения: tноль: первая доза для назального введения
t5 минут: вторая доза для назального введения
t10 минут: третья доза для назального введения
Как видно из Таблицы VI и Фиг.7, первый пик концентрации в плазме наступает в течение 2 минут после первого назального введения препарата. Максимальный уровень в плазме (пик) составляет 32% от значения при IV (вв) инъекции. Однако, после третьего введения с 5-минутными интервалами максимальная концентрация в плазме (пик), наблюдаемая через 15 минут, практически идентична концентрации после однократной IV (вв)-инъекции клоназепама.
Пример 11
Фармакологический ответ препаратов клоназепама
Фармакологический ответ препаратов клоназепама изучают на новозеландских белых кроликах после введения 100 мкл 4,18 мг клоназепама/мл носителя в каждую ноздрю тем же способом, что и в Примере 9. Носитель состоит из 30% этанола, 60% пропиленгликоля и 10% воды, содержащей 1% SGC. Клоназепам растворяют в носителе с помощью ультразвука. Рецептура для IV (вв)-инъекции, используемая в исследовании, такая же, как и в Примере 10. Среднее время ответа после IV (вв) и IN (ин) введения представлено в Таблице VII.
Как показано в Таблице VII, интраназальное введение препарата клоназепама по изобретению обеспечивает более быстрый ответ (1,4 минуты), чем IV (вв)-инъекция (1,7 минут).
Пример 12
Биодоступность и фармакокинетика препаратов (S)-2-карбамоилокси-1-о-хлорфенилэтанола
Интраназальную рецептуру готовят, растворяя 50 мг или 100 мг нового противосудорожного агента на основе монокарбамата (S)-2-карбамоилокси-1-о-хлорфенилэтанола в 1 мл носителя по изобретению, состоящего из 30% EtOH, 60% PG и 10% воды, содержащей 1% SGC. Препарат для в.в. (IV)-инъекции готовят, растворяя 15 мг (S)-2-карбамоилокси-1-о-хлорфенилэтанола в 1 мл 40% PEG (ПЭГ) 400 и 60% воды и отфильтровывая через стерильную мембрану в асептических условиях. Препараты вводят кроликам (n=2-4) в виде двух различных доз 2,5 мг/кг и 5 мг/кг методом, аналогичным описанному в Примере 7. Также изучают схему многократных (повторных) доз с 5-минутными интервалами при введении в нос препарата по изобретению. Пробы крови берут из ушной вены в следующие временные точки: 0, 2, 5, 10, 20, 30, 45, 60, 120, 180 и 240 минут. Из образцов крови плазму отделяют центрифугированием и хранят при -20°С до испытания. Для анализа образцы плазмы (0,5 мл) осторожно переносят в пробирку на 15 мл. К образцу плазмы добавляют 50 мкл раствора внутреннего стандарта (2-(2,6-дихлорфенил)-2-карбамоилоксиэтил)оксокарбамид-1- мкг/мл) и 5 мл метилбутилового эфира. Смесь перемешивают 60 сек и центрифугируют при 3500 об/мин в течение 10 минут. Вышеуказанный эфирный раствор переносят в пробирку на 5 мл и упаривают в вакуумном испарителе при 40°С в течение 30 мин. Остаток разводят ("воспроизводят") в 200 мкл деионизированной воды. Концентрацию (S)-2-карбамоилокси-1-о-хлорфенилэтанола в плазме определяют методом ВЭЖХ, используя подвижную фазу, состоящую из 20% ацетонитрила и 80% воды, при скорости потока 1 мл/мин и используя УФ-детектирование при 210 нм. Предел обнаружения для (S)-2-карбамоилокси-1-о-хлорфенилэтанола составляет 23 нмол/л. Фармакокинетические параметры, определяемые после в.в. (IV) и и.н. IN-введения (S)-2-карбамоилокси-1-о-хлорфенилэтанола в виде двух различных доз, представлены в Таблице VIII. Параметры биодоступности и фармакокинетики, полученные после в.в. (IV) и и.н. IN-введения препаратов по изобретению по схеме однократной (разовой) и двукратной доз, приведены в Таблице IX. Графики зависимости: средняя концентрация в крови - время, полученные после в.в. (IV) и и.н. IN-введения препаратов (S)-2-карбамоилокси-1-о-хлорфенилэтанола в виде (по схеме) однократной и двукратной доз, представлены на Фиг.8 и 9.
(нг × мин/мл)
b IN (ин) Формула 1: 10% раствор (S)-2-карбамоилокси-1-о-хлорфенилэтанола в 1% SGC, 60% PG, 30% EtOH и 10% воды
c IN (ин) Формула 2: 5% раствор (S)-2-карбамоилокси-1-о-хлорфенилэтанола в 1% SGC, 60% PG, 30% EtOH и 10% воды
d Стандартное отклонение
(мг/кг)
(нг × мин/мл)
(5 мг/кг × 1)
(5 мг/кг × 1)
(5 мг/кг × 2)
b IN (ин) Формула 1: 10% раствор (S)-2-карбамоилокси-1-о-хлорфенилэтанола в 1% SGC, 60% PG, 30% EtOH и 10% воды
c Стандартное отклонение
d Нормализованные данные, вычисленные по следующему уравнению:
F={AUCIN, 5 мг × 2/2 × AUCIV, 5 мг×1}×100
e Время введения: tноль: первая доза для назального введения
t5 минут: вторая доза для назального введения
Как видно из Таблицы VIII, после интраназального введения начальные максимальные концентрации, наблюдаемые в пределах 5-30 минут, увеличиваются пропорционально увеличению дозы. Найдено, что биодоступность назальных препаратов составляет 73-79% от биодоступности при IV (вв)-инъекции. Фармакокинетические результаты, представленные в Таблице IX и на Фиг.9, четко демонстрируют, что второе введение назального препарата через 5 минут после первой дозы обеспечивает биодоступность, почти идентичную биодоступности, достигаемой после первой дозы. Значения Сmax и AUC0-240 минут удовлетворяются после второго интраназального введения. Кроме того, концентрация (S)-2-карбамоилокси-1-о-хлорфенилэтанола в плазме, обнаруживаемая после второй дозы, превышает уровень в плазме, получающейся при однократной IV (вв)-инъекции через 30 минут.
Пример 13
Исследование стабильности
С целью оптимизировать стабильность лекарственных веществ в фармацевтических композициях по данному изобретению проводят ускоренное исследование стабильности при температуре хранения 37°С в течение более чем 10-14-недельного периода. Образцы растворов лекарственного вещества (0,1 мг/мл) готовят, применяя носитель по изобретению, состоящий из 30% EtOH, 60% PG и 10% воды. Растворы лекарственного вещества хранят в сушильном шкафу при 37°С. С определенными временными интервалами отбирают 100 мкл образца и анализируют методом ВЭЖХ. Данные по химической стабильности, определенные в процентах вещества, представлены в Таблице X.
| название | год | авторы | номер документа |
|---|---|---|---|
| Фармацевтическая композиция для устранения судорожного синдрома на основе никетамида и препаратов бензодиазепиновой группы | 2022 |
|
RU2801050C1 |
| ТРАНСНАЗАЛЬНЫЕ МИКРОЭМУЛЬСИИ, СОДЕРЖАЩИЕ ДИАЗЕПАМ | 2004 |
|
RU2354354C2 |
| ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ И СПОСОБ ЕЕ ИНГАЛЯЦИОННОГО ВВЕДЕНИЯ | 2010 |
|
RU2445119C2 |
| ПРИМЕНЕНИЕ 1-АР(АЛК)ИЛИМИДАЗОЛИН-2-ОНОВ ДЛЯ ЛЕЧЕНИЯ СОСТОЯНИЯ СТРАХА И НАПРЯЖЕННОСТИ | 1998 |
|
RU2203055C2 |
| ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ С ПРОТИВОСУДОРОЖНОЙ АКТИВНОСТЬЮ ДЛЯ ПАРЕНТЕРАЛЬНОГО ВВЕДЕНИЯ | 2011 |
|
RU2463052C1 |
| Композиции, содержащие триптановые соединения | 2010 |
|
RU2710372C2 |
| ИНТРАНАЗАЛЬНЫЕ ФАРМАЦЕВТИЧЕСКИЕ ДОЗИРОВАННЫЕ ФОРМЫ, СОДЕРЖАЩИЕ НАЛОКСОН | 2012 |
|
RU2657441C1 |
| Композиции, содержащие триптановые соединения | 2019 |
|
RU2762725C2 |
| ЖИДКИЕ ДЕПО-ПРЕПАРАТЫ | 2005 |
|
RU2390331C2 |
| ИНТРАНАЗАЛЬНЫЕ ФАРМАЦЕВТИЧЕСКИЕ ДОЗИРОВАННЫЕ ФОРМЫ, СОДЕРЖАЩИЕ НАЛОКСОН | 2012 |
|
RU2587051C2 |
Предложено: назальные противосудорожные композиции и способ введения противосудорожных агентов. Композиции содержат терапевтически эффективное количество противосудорожного агента, растворенного или диспергированного в водном носителе, содержащем 10-80 об.% алифатического спирта, 10-80 об.% гликоля и 0,1-5 вес.% соли желчной кислоты или лецитина. Указанный носитель обеспечивает повышение проникновения в кровь и обеспечения быстрого фармакологического ответа противосудорожного агента (например, такого как бензодиазепин, в частности диазепам, клоназепам, фенитоина, мефенитоина, этотоина, фенобарбитала, карбамазепина, этосукцимида, вальпроевой кислоты, габапентина, триметадиона, ламотригина) при назальном введении. 2 н. и 9 з.п. ф-лы, 10 табл., 9 ил.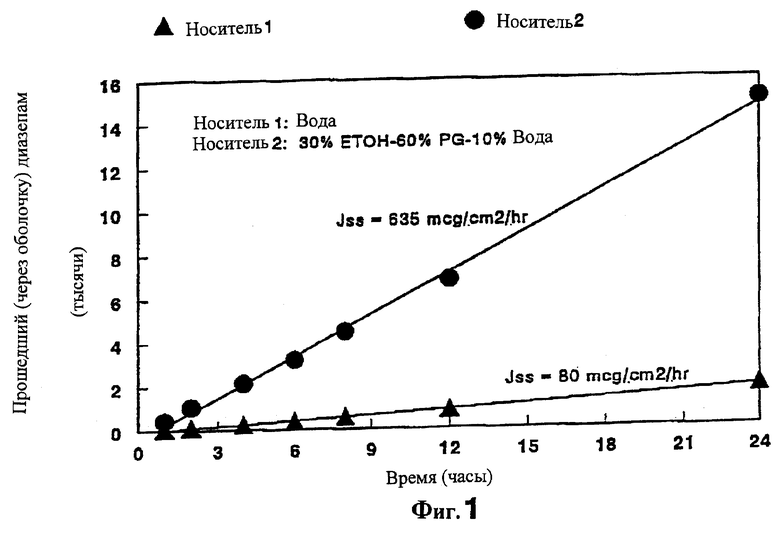
| Прибор, замыкающий сигнальную цепь при повышении температуры | 1918 |
|
SU99A1 |
| US 4863720 А 05.09.1989 | |||
| Прибор, замыкающий сигнальную цепь при повышении температуры | 1918 |
|
SU99A1 |
| СПОСОБ ЛЕЧЕНИЯ БОЛЬНЫХ ЭПИЛЕПСИЕЙ | 1995 |
|
RU2118537C1 |
| ХАРКЕВИЧ Д.А | |||
| Фармакология | |||
| М.: Медицина, 1996 с.28-30. | |||

