Изобретение относится к способу лечения воспалительных и аллергических заболеваний с использованием норкетотифена, 10-гидрокси-кетотифена, 10-гидрокси-норкетотифена или оптических активных изомеров кетотифена, норкетотифена, 10-гидрокси-кетотифена или 10-гидрокси-норкетотифена, или их фармацевтически приемлемых солей и сольватов. Более конкретно, настоящее изобретение относится к способам лечения легочных заболеваний (таких как астма, бронхит), кожных болезней (таких как крапивница и атопический дерматит) и желудочно-кишечных расстройств (таких как раздражение желудка и энтерит) без седативных побочных эффектов и побочных эффектов, воздействующих на сердечно-сосудистую систему, ассоциированных с введением антигистаминных препаратов. Другой вариант осуществления изобретения основан на обнаружении того факта, что норкетотифен и оптически активные изомеры норкетотифена являются особенно эффективными для лечения глазных болезней, таких как конъюнктивит и кератит.
Предшествующий уровень техники
Настоящее изобретение относится, в частности, к противовоспалительным и антиаллергическим соединениям, которые могут быть использованы для лечения различных заболеваний, в частности, у пациентов, страдающих легочными заболеваниями, включая астму и бронхит; кожными болезнями, включая крапивницу и атопический дерматит; и желудочно-кишечными расстройствами, включая раздражение желудка и энтерит.
Соединениями, описанными в настоящем изобретении, являются метаболиты кетотифена (4-(1-метил-4-пиперидилин)-4Н-бензо(4,5)-циклогепта-(1,2-b)тиофен-10-он). Из-за серьезных седативных побочных эффектов, ассоциированных с кетотифеном, это соединение имеет ограниченное терапевтическое использование.
Кетотифен подвергается метаболизму в организме различными путями:

Метаболит норкетотифена (также называемый норкетотифеном) образуется путем деметилирования кетотифена:

Метаболиты 10-гидрокси-кетотифена и 10-гидрокси-норкетотифена образуются путем восстановления молекул кетотифена и норкетотифена, соответственно.
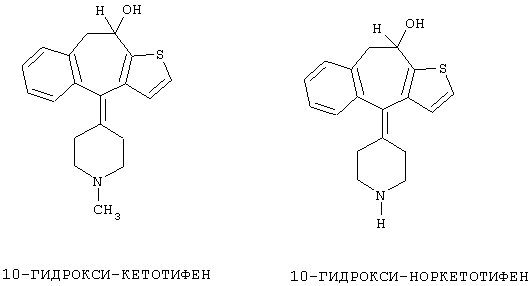
Другие метаболиты кетотифена также образуются в организме после введения кетотифена. Таким образом, молекула кетотифена может подвергаться N-глюкуронидированию и может превращаться в N-оксид кетотифена. Полученные гидроксилированные изомеры могут затем подвергаться метаболизму с образованием 10-гидрокси-глюкуронидатов. Могут быть образованы также и другие метаболиты, причем у различных видов пути метаболизма отличаются и могут даже отличаться у детей и взрослых.
Не существует каких-либо известных опубликованных фармакологических исследований рацематов или изомеров кетотифена, 10-гидрокси-кетотифена или 10-гидрокси-норкетотифена. Фармакологические свойства изомеров кетотифена были описаны Polivka et al.: 4H-benzo(4,5)cyclohepta(1,2-b)thiophenes and 9,10-dihydro derivatives-sulfonium analogues of pizotifen and ketotifen; chirality of ketotifen; synthesis of the 2-bromo derivative of ketotifen. Collect.Czech.Chem. Commun., 1989, 54, 2443-2469.
Краткое описание изобретения
Изомеры кетотифена были синтезированы и фармакологически исследованы in vitro и in vivo. Также были синтезированы и фармакологически исследованы различные метаболиты кетотифена. Было обнаружено, что антигистаминное действие рацемического норкетотифена качественно аналогично антигистаминному действию рацемического кетотифена. Таким образом, как кетотифен, так и норкетотифен являются антагонистами гистамина Н-1 с различной степенью антагонизма к гистамину Н-2. Однако, неожиданно было обнаружено, что между рацемическим кетотифеном и соединениями, описанными в настоящем изобретении, имеется важное и весьма качественное отличие: описанные здесь соединения не обладают сильной и дозоограничивающей седативной активностью кетотифена. Было также установлено, что рацемический норкетотифен, а в частности его изомер, обладают сильными противоспалительными и антигистаминными свойствами, и при этом они имеют незначительный седативный эффект или вообще не имеют такого эффекта. Аналогичным образом было также обнаружено, что, хотя оба изомера кетотифена обладают той же самой антигистаминной активностью, однако почти все эти седативные побочные эффекты присущи R(+)-кетотифену. Было также обнаружено, что метаболиты 10-гидрокси-норкетотифена и 10-гидрокси-кетотифена и изомеры обоих соединений ингибируют воспаление и блокируют рецепторы гистамина Н-1, оказывая при этом значительно меньшее седативное действие, чем кетотифен.
Подробное описание изобретения
Химический синтез кетотифена, норкетотифена, 10-гидрокси-кетотифена и 10-гидрокси-норкетотифена, их стереохимически изомерных форм и диастереомеров.
Рацемический кетотифен и норкетотифен получают методами, описанными в работе Waldvogel et al. (Helv.Chim.Acta, 59, 866-877, 1976), которая включена в настоящее описание в виде ссылки. R-(+)-кетотифен и S-(-)-кетотифен получают путем фракционированной кристаллизации солей рацемического кетотифена с (-)-O,O'-ди-(п-толуоил)-R-винной кислотой и (+)-O',O-ди(п-толуоил)-S-винной кислотой, соответственно, как описано в работе Polivka et al. (Collect.Czech.Chem.Commun., 54, 2443-2469, 1989), которая вводится в настоящее описание посредством ссылки.
Предпочтительный способ получения оптически активных изомеров норкетотифена, 10-гидроксикетотифена и 10-гидрокси-норкетотифена основан на отдельных R- и S-энантиомерах кетотифена. Заявлен новый способ синтеза кетотифена, который позволяет избежать применения жестких условий, описанных Waldvogel и др., 1976, которые могут приводить к рацемизации указанного продукта. Другими способами являются стереоселективный синтез с использованием хиральных матриц, разделение соответствующих рацематов стандартными методами, такими как фракционированная кристаллизация диастереомерных солей с хиральными кислотами, и хроматография с использованием хиральных носителей.
10-Гидроксипроизводные получают путем каталитического восстановления соответствующих энантиомеров кетотифена методами, описанными Waldvogel et al., 1976. Необходимо отметить, что 10-гидрокси-кетотифены имеют дополнительный хиральный центр, и что были получены диастереомеры соответствующих R- и S-энантиомеров кетотифена. Эти энантиомеры могут быть разделены стандартными методами кристаллизации или хроматографии, что обусловлено различиями в растворимости и в хроматографической подвижности диастереомерных изомеров. Альтернативно, восстановление R- и S-энантиомеров кетотифена хиральными восстановителями может быть осуществлено для получения только нужного диастереомерного продукта (или его высокообогащенных смесей).
Определение абсолютных конфигураций
Энантиомеры кетотифена и норкетотифена не имеют асимметрических атомов углерода, которые являются типичными для оптических изомеров. Точнее, указанные энантиомеры образуются в результате молекулярной асимметрии, обусловленной затрудненным взаимопревращением семичленного кольца. (+)-Кетотифен обозначается R и имеет конфигурацию, наблюдаемую в рентгеновской кристаллической структуре (+)-кетотифена, (-)-O,O'-ди(п-толуил)-R-тартрата, описанного Polivka и др., 1989. (-)-Кетотифен обозначен S.
У производных 10-гидроксикетотифена присутствует дополнительный хиральный центр - 10-атом углерода, а R- и S-конфигурации определяются в соответствии со стандартными правилами стереохимии. В названии диастереомеров, образующихся в результате восстановления энантиомеров кетотифена, первые буквы R или S означают конфигурацию семичленного кольца, а вторая буква R или S означает конфигурацию 10-атома углерода.
Синтез (R)- и (S)-норкетотифена и фумаратов (R)- и (S)-норкетотифена
(Эти методы синтеза систематизированы на схемах).
(R)-4-(1-(2,2,2-Трихлорэтоксикарбонил)-4-пиперидилиден)-9,10-дигидро-4Н-бензо-(4,5)-циклогепта(1,2-b)тиофен-10-он.
Безводный карбонат натрия (95,0 мг, 898 мкмоль), (+)-(R)-кетотифен (278 мг, 898 мкмоль) и хлорид бензилтриэтиламмония (ВТЕАС) (205 мг, 898 мкмоль) объединяли в сухой колбе и помещали в высокий вакуум на два дня. К полученной смеси добавляли свежедистиллированный дихлорметан (2 мл) в атмосфере N2, при комнатной температуре и при перемешивании, а затем добавляли 2,2,2-трихлорэтилхлорформиат (556 мкл, 4,04 ммоль). Двухфазную реакционную смесь кипятили с обратным холодильником в течение одного часа и после охлаждения до комнатной температуры гасили путем добавления насыщенного водного раствора карбоната натрия (7 мл). Полученную смесь разбавляли дихлорметаном (50 мл) и органическую фазу отделяли, сушили над сульфатом натрия, фильтровали, выпаривали и хроматографировали (SiO2, смесь этилацетата-петролейного эфира,  ). Фракции, соответствующие промежуточному трихлорэтилкарбамату, собирали, выпаривали и помещали в высокий вакуум с получением 279 мг 66% указанного в заголовке соединения.
). Фракции, соответствующие промежуточному трихлорэтилкарбамату, собирали, выпаривали и помещали в высокий вакуум с получением 279 мг 66% указанного в заголовке соединения.
1H ЯМР (400 МГц, CDCl3) δ 7,58 (с, 1Н), 7,37-7,30 (м, 1Н), 7,23-7,19 (м, 2Н), 7,18-7,11 (м, 1Н), 7,03-7,00 (м, 1Н), 4,88-4,69 (м, 2Н), 4,20 (д, 1Н, J=13,0 Гц), 4,03-3,84 (м, 2Н), 3,78 (д, 1Н, J=13,0 Гц), 3,35-3,09 (м, 2Н), 2,75-2,64 (м, 1Н), 2,62-2,57 (м, 1Н), 2,50-2,39 (м, 2Н).
(R)-4-(4-Пиперидилиден)-9,10-дигидро-4Н-бензо-(4,5)-циклопента(1,2-b)тиофен-10-он ((R)-норкетотифен).
Кадмиевую пыль (178 мг, 1,59 ммоль) и 10% смесь кадмия/свинца (356 мг, 3,174 ммоль) добавляли к интенсивно перемешиваемой смеси трихлорэтилкарбамата (279 мг, 592 мкмоль) в ТГФ (5,5 мл) и водного ацетата аммония (1М, 3,0 мл) в атмосфере азота. Через 2 часа смесь фильтровали через целит и промывали большим количеством дихлорметана и воды. Фильтрат подщелачивали насыщенным водным карбонатом натрия и органический слой отделяли, сушили над сульфатом натрия, фильтровали и выпаривали с получением неочищенного (R)-норкетотифена (157 мг, 90%).
1H ЯМР (400 МГц, CDCl3) δ 7,54 (д, 1Н, J=5,2 Гц), 7,34-7,29 (м, 1Н), 7,23-7,13 (м, 3Н), 7,02 (д, 1Н, J=5,2 Гц), 4,20 (д, 1Н, J=13,6 Гц), 3,76 (м, 1Н, J=13,6 Гц), 3,21-3,14 (м, 1Н), 3,13-3,05 (м, 1Н), 2,86-2,76 (м, 1Н), 2,75-2,58 (м, 3Н), 2,47-2,39 (м, 2Н).
Энантиомерную чистоту определяли с помощью аналитической хиральной ВЭЖХ (5 мкм Merck Chiradex, подвижная фаза смесь фосфата натрия-ацетонитрила, 95:5, рН 4,0, скорость потока 1 мл/мин, скорость движения диаграммной бумаги 0,3 см/мин, 254 нм): Rt 13,0 мин, 95% э.и.
СХЕМА
Синтез фумарата (R)-норкетотифена иа (+)-(R)-кетотифвна

Синтез фумарата (S)-норкетотифена из (-)-(S)-кетотифена

Фумарат (R)-норкетотифена.
Неочищенный (R)-норкетотифен (157 мг, 533 мкмоль) растворяли в дихлорметане (1 мл) и добавляли к раствору фумаровой кислоты (61,8 мг, 533 мкмоль) в этаноле (2 мл). После выпаривания 50% растворителя кристаллы фумарата (R)-норкетотифена собирали и промывали этанолом, а затем, после сушки в высоком вакууме, получали 117 мг продукта.
1H ЯМР (ДМСО-d6) δ 7,99(м, 1Н), 7,40-7,31 (м, 1Н), 7,30-7,11 (м, 4Н), 6,43 (с, 2Н), 4,35 (д, 1Н, J=13,2 Гц), 3,65 (д, 1Н, J=13,2 Гц), 3,25-3,08 (м, 2Н), 2,99-2,89 (м, 1Н), 2,83-2,74 (м, 1Н), 2,70-2,54 (м, 2Н), 2,50-2,40 (м, 1Н), 2,38-2,28 (м, 1Н).
Энантиомерную чистоту определяли с помощью аналитической хиральной ВЭЖХ (5 мкм Merck Chiradex, подвижная фаза смесь фосфата натрия-ацетонитрила, 95:5, рН 4,0, скорость потока 1 мл/мин, скорость движения диаграммной бумаги 0,3 см/мин, 254 нм): Rt 13,0 мин, 95% э.и.
(S)-4-(1-(2,2,2-трихлорэтоксикарбонил)-4-пиперидилиден)-9,10-дигидро-4Н-бензо-(4,5)-циклогепта(1,2-b)-тиофен-10-он.
В соответствии с процедурой, описанной выше для (R)-энантиомера, получали (S)-энантиомер с аналогичным выходом:
1H ЯМР (400 МГц, CDCl3) δ 7,58 (с, 1Н), 7,37-7,30 (м, 1Н), 7,23-7,19 (м, 2Н), 7,18-7,11 (м, 1Н), 7,03-7,00 (м, 1Н), 4,88-4,69 (м, 2Н), 4,20 (д, 1Н, J=13,0 Гц), 4,03-3,84 (м, 2Н), 3,78 (д, 1Н, J=13,0 Гц), 3,35-3,09 (м, 2Н), 2,75-2,64 (м, 1Н), 2,62-2,57 (м, 1Н), 2,50-2,39 (м, 2Н).
(S)-4-(4-пиперидилиден)-9,10-дигидро-4Н-бензо-(4,5)-циклогепта-(1,2-b)тиофен-10-он.
В соответствии с процедурой, описанной выше для (R)-энантиомера, получали (S)-энантиомер с аналогичным выходом:
1H ЯМР (400 МГц, CDCl3) δ 7,54 (д, 1Н, J=5,2 Гц), 7,34-7,29 (м, 1Н), 7,23-7,13 (м, 3Н), 7,02 (д, 1Н, J=5,2 Гц), 4,20 (д, 1Н, J=13,6 Гц), 3,76 (м, 1Н, J=13,6 Гц), 3,21-3,14 (м, 1Н), 3,13-3,05 (м, 1Н), 2,86-2,76 (м, 1Н), 2,75-2,58 (м, 3Н), 2,47-2,39 (м, 2Н).
Энантиомерную чистоту определяли с помощью аналитической хиральной ВЭЖХ (5 мкм Merck Chiradex, подвижная фаза смесь фосфата натрия-ацетонитрила, 95:5, рН 4,0, скорость потока 1 мл/мин, скорость движения диаграммной бумаги 0,3 см/мин, 254 нм): Rt 7,5 мин, 95% э.и.
Фумарат (S)-норкетотифена.
В соответствии с процедурой, описанной выше для (R)-энантиомера, получали (S)-энантиомер с аналогичным выходом:
1H ЯМР (400 МГц, ДМСО-d6) δ 7,99 (м, 1Н), 7,40-7,31 (м, 1Н), 7,30-7,11 (м, 4Н), 6,43 (с, 2Н), 4,35 (д, 1Н, J=13,2 Гц), 3,65 (д, 1Н, J=13,2 Гц), 3,25-3,08 (м, 2Н), 2,99-2,89 (м, 1Н), 2,83-2,74 (м, 1Н), 2,70-2,54 (м, 2Н), 2,50-2,40 (м, 1Н), 2,38-2,28 (м, 1Н).
Энантиомерную чистоту определяли с помощью аналитической хиральной ВЭЖХ (5 мкм Merck Chiradex, подвижная фаза смесь фосфата натрия-ацетонитрила, 95:5, рН 4,0, скорость потока 1 мл/мин, скорость движения диаграммной бумаги 0,3 см/мин, 254 нм): Rt 7,5 мин, 95% э.и.
Фармакологические исследования кетотифена, норкетотифена, 10-ОН-кетотифена или 10-ОН-норкетотифена и их оптически активных изомеров
Как обсуждалось выше, было показано, что S-изомер кетотифена и рацематы и изомерные формы норкетотифена, 10-ОН-кетотифена или 10-ОН-норкетотифена обладают благоприятным фармакологическим действием, которое может быть использовано для лечения таких заболеваний, как, например, аллергические заболевания, легочные заболевания, кожные болезни и желудочно-кишечные расстройства, но, при этом, они не имеют значительных седативных побочных эффектов кетотифена. Были установлены неожиданные факты, которые описаны в нижеследующих примерах.
Метод испытаний 1. Связывание с гистаминеррическиии рецепторами
Аффинности рацемических и изомерных тестируемых соединений по отношению к рецептору гистамина Hi оценивали с использованием анализа на связывание с 3H-пириламином, как описано Dini et al. (Agents and Actions, 1991, 33: 181-184). Для этого мембраны, полученные из мозжечка морских свинок, инкубировали с 3H-пириламином и с различными концентрациями тестируемого(ых) соединения(й). Специфическое связывание радиоактивного лиганда с рецептором определяли в присутствии избыточного количества немеченного лиганда. Результаты выражали как процент специфического связывания в присутствии соединений. Величины IC50 (концентрация, необходимая для 50%-ного ингибирования специфического связывания) и коэффициенты Хилла (nH) определяли с помощью нелинейного регрессионного анализа кривой конкурентного связывания. Эти параметры были получены исходя из кривой, построенной по уравнению Хилла с использованием программного обеспечения Sigmaplot™.
Метод испытаний 2. Антигистаминные эффекты in vitro
Полоски бронхиальной ткани или ткани других гладких мышц иссекали из тела самцов морских свинок весом 400-600 г. Эти ткани суспендировали в окисленном буфере, имеющем состав (мМ): NaCl, 133; KCl, 4,7; CaCl2, 2,5; MgSO4, 0,6; NaH2PO4, 1,3; NaHCO3, 16,3; и глюкоза, 7,7, или в растворе с аналогичным составом. Этот раствор поддерживали при 37,5°С. Сокращения регистрировали с помощью изометрических датчиков (Model FT-10) на полиграфе Грасса.
Для оценки жизнеспособности каждой ткани и для использования ее в качестве системы отсчета сначала регистрировали сокращение каждой полоски ткани в ответ на воздействие окисленного буфера, в котором NaCl был заменен на KCl с получением концентрации 137,7 мМ KCl. Затем возвращались к стандартному окисленному буферу, а после этого обрабатывали возрастающими в соответствующей прогрессии концентрациями гистамина, причем отдельные обработки каждой концентрацией осуществляли до тех пор, пока не будет зарегистрирован максимальный ответ. Затем, оставляя одну полоску живой ткани необработанной, каждую из остальных полосок ткани обрабатывали в течение предварительно определенного интервала времени одной концентрацией антагониста. И наконец, ответы на возрастающие концентрации гистамина с последующей обработкой 137,7 мМ KCl регистрировали во второй раз.
Метод испытаний 3. Связывание с мускариновыми рецепторами
Эти эксперименты проводили на мембранах, полученных из клеток SF9, инфицированных бакуловирусом, для экспрессии подтипов человеческого рекомбинантного мускаринового рецептора. После инкубирования с тестируемым материалом и соответствующим радиолигандом и после промывки определяли связанную радиоактивность на жидкостном сцинтилляционном счетчике с использованием коммерчески доступной сцинтилляционной смеси. Специфическое связывание радиолиганда с каждым рецептором определяли как разность между общим связыванием и неспецифическим связыванием, определенным в присутствии избыточного количества немеченного лиганда. Величины IC50 (концентрация, необходимая для 50%-го ингибирования специфического связывания) определяли с помощью нелинейного регрессионного анализа кривой конкурентного связывания.
Метод испытаний 4. Ингибирование бронхиальной аккумуляции эозинофилов
Ингибирование аккумуляции эозинофилов в легких определяли у сенсибилизированных морских свинок (400-600 граммов) после внутрибрюшинной инъекции аллергена, такого как, например, PAF (фактора агрегации тромбоцитов) или альбумин бычьей сыворотки. Через заранее определенный промежуток времени животных умерщвляли путем введения барбитурата. Трахею вынимали и каннюлировали. После этого последовательно вводили 6×10 мл аликвот забуференного модифицированного раствора Тироде (состав: NaHCO3, 11,9; NaCl, 136,9; KCl 2,7; Na2HPO4, 0,4; глюкоза, 5,6; EDTA 19,8; желатин, 0,1% масс./об; рН 7,4) и отсасывали путем мягкого сдавливания легких. Общее выделение жидкости обычно превышало 80%. Клеточные суспензии концентрировали путем центрифугирования на низкой скорости (200 G в течение 10 минут) и полученный клеточный осадок ресуспендировали в 1 мл модифицированного раствора Тироде. Общее число клеток определяли путем разведения 10 мкл клеточной суспензии в 90 мкл жидкости Тэрка. Лейкоцитарную формулу определяли из мазков, фиксированных в метаноле (100%) и окрашивали красителем Лейшмана. Общее количество по крайней мере 500 клеток на мазок подсчитывали при 1000-кратном увеличении для дифференциации типа клеток. Лекарственные средства вводили в виде пролонгированной подкожной инфузии из имплантированного мининасоса Alza (Alzet 2001 или аналогичного) или путем повторных пероральных или повторных парентеральных инъекций.
Метод испытаний 5. Противовоспалительное действие на кожу
Противовоспалительное действие на кожу тестировали методом индуцирования воспаления нанесением кретонового масла на уши мышей. Этот метод испытаний описан Tarrida J. et al., Meth. Find.Exp.Clin.Pharmacol., (1996) 18(4):233-234 и Blazzo et al., Prostaglandins, (1995) 50:161-168. Вкратце, самцов мышей (25-30 г) обрабатывали 5 мл/кг 2 мг/мл раствора каждого тестируемого соединения в физиологическом растворе путем внутрибрюшинной инъекции. Через тридцать минут после инъекции на оба уха каждой мыши местно наносили кротоновое масло (0,20 мкл 1,0% кротонового масла в ацетоне) или ацетон (контроль). Во время нанесения кротонового масла или ацетона животные были закреплены, а затем их освобождали и помещали в клетку. До нанесения кротонового масла или ацетона и в заранее определенные промежутки времени после их нанесения группы животных анестезировали галотаном и подвергали эвтаназии путем смещения шейных позвонков. Уши удаляли и взвешивали. Затем строили график зависимости среднего веса уха от времени. Всего, на каждый момент времени, тестировали 4 животных для каждого соединения и контрольного, носителя.
Метод испытаний 6. Исследования на седативные побочные эффекты
В исследованиях настоящего изобретения используется тест на индуцированный физостигмином летальный исход. Этот тест представляет собой модифицированный анализ на седативный эффект, описанный VILLANI et al., в патенте США №4659716. Вкратце, физостигмин (1,9 мг/кг s.c.) при его введении мышам, разделенным на группы по 10 животных в каждой пластиковой клетке (11 см × 26 см × 13 см), в 90-100% случаев приводил к летальному исходу. Мыши, которым до введения физостигмина вводили седативное средство, такое как, например, седативный антигистамин, были защищены от гибели и выживали. В настоящих исследованиях тестируемые соединения вводили перорально в течение 60 минут перед введением физостигмина. Число выживших особей подсчитывали через 30 минут после введения физостигмина.
Фармацевтические композиции
Термины "фармацевтически приемлемые соли" или "его фармацевтически приемлемая соль" означают соли или сольваты, полученные из фармацевтически приемлемых нетоксичных кислот. Подходящими фармацевтически приемлемыми кислотно-аддитивными солями соединений настоящего изобретения являются соли уксусной, бензолсульфоновой (безилат), бензойной, камфорсульфоновой, лимонной, этансульфоновой, фумаровой, глюконовой, глутаминовой, бромистоводородной, хлористо-водородной, изетионовой, молочной, малеиновой, яблочной, миндальной, метансульфоновой, слизевой, азотной, памовой, пантотеновой, фосфорной, янтарной, серной, винной, п-толуолсульфоновой кислот и т.п. Особенно предпочтительным является бифумарат.
Настоящее изобретение относится к фармацевтическим композициям, содержащим одно или несколько соединений настоящего изобретения вместе с одним или несколькими фармацевтически приемлемыми носителями. Указанные фармацевтические композиции могут быть специально приготовлены для перорального введения, инстилляции в конъюктиву, подъязычного введения, парентерального введения, чрезкожного введения, ректального введения, трансбуккального введения или местного введения, либо для введения путем ингаляции или инсуффляции порошка или аэрозоля.
Фармацевтические композиции настоящего изобретения могут быть введены человеку или другим млекопитающим перорально, подъязычно, парентерально, подкожно, чрезкожно, ректально, трансбуккально, местно, путем инстилляции в конъюктиву или в виде перорального или интраназального спрея или аэрозоля. Термин "парентеральное" введение означает внутривенную, внутриартериальную, внутримышечную, внутрибрюшинную, чрезкожную, подкожную или внутрисуставную инъекцию или инфузию. Термин "чрезкожный" относится к использованию различных приспособлений ("пластырей" и т.п.), которые могут облегчать или модифицировать транспорт или абсорбцию лекарственного средства через кожу. Термин "местный" относится к нанесению композиции, содержащей лекарственное средство, на кожу и на слизистые оболочки.
Формы для перорального введения
Фармацевтическими композициями по настоящему изобретению для перорального введения твердых лекарственных форм являются капсулы, гранулы, драже, порошки и таблетки. В указанных твердых лекарственных формах активное соединение может быть смешано с одним или несколькими фармацевтическими эксципиентами или носителями (например, цитратом натрия, дифосфатом кальция), наполнителями или добавками, придающими объем (например, крахмалом, лактозой, сахарозой, глюкозой, маннитом, кремниевой кислотой), связующими (например, карбоксиметилцеллюлозой, альгинатами, желатином, поливинилпирролидоном, сахарозой, аравийской камедью), увлажнителями (например, глицерином), агентами, замедляющими растворение (например, парафином), разрыхлителями (например, агар-агаром, карбонатом кальция, крахмалом, альгиновой кислотой, силикатами, карбонатом натрия), ускорителями абсорбции (например, четвертичными аммониевыми соединениями), смачивающими агентами (например, цетиловым спиртом, моностеаратом глицерина), абсорбентами (например, каолином, бентонитом), замасливателями (например, тальком, стеаратом кальция, стеаратом магния, полиэтиленгликолями, лаурилсульфатом натрия) и/или забуферивающими агентами.
Твердые формы, а именно: капсулы, драже, гранулы, пилюли и таблетки могут иметь покрытие и/или оболочки (например, энтеросолюбильные покрытия), известные специалистам. Указанные композиции могут быть также приготовлены таким образом, чтобы активный(е) ингредиент(ы) высвобождался в определенной части желудочно-кишечного тракта путем регулируемого высвобождения, замедленного высвобождения или пролонгированного высвобождения.
Указанные композиции могут быть также приготовлены таким образом, чтобы осуществлялась лимфатическая абсорбция активного ингредиента(ов).
Указанное активное соединение(я) может быть также микроинкапсулировано с одним или несколькими вышеуказанными наполнителями.
Жидкими лекарственными формами для перорального введения являются фармацевтически приемлемые эмульсии, растворы, суспензии, сиропы и эликсиры. Жидкая лекарственная форма может также содержать общеизвестные разбавители (например, воду, другие растворители, солюбилизирующие агенты), эмульгаторы (например, этанол, изопропиловый спирт, этилкарбонат, этилацетат, бензиловый спирт, бензилбензоат, пропиленгликоль, бутиленгликоль, диметилформамид, масла, олеиновую кислоту, глицерин, полиэтиленгликоли, сложные эфиры сорбитана и жирных кислот и их смеси).
Помимо инертных разбавителей пероральные композиции могут также включать адъюванты, такие как смачивающие агенты, эмульгаторы, суспендирующие агенты, подслащивающие агенты или ароматизаторы.
Суспензии могут содержать один или несколько суспендирующих агентов, известных специалистам в области фармацевтики.
Формы для местного введения
Композициями для местного введения соединений настоящего изобретения являются растворы, суспензии, капли, спреи, мази, кремы и порошки. Композиции, предназначенные для нанесения на кожу, могут содержать стимуляторы проницаемости и другие агенты, известные специалистам. Для нанесения на кожу могут быть также использованы пластыри, повязки и т.п.
Помимо терапевтически активных ингредиентов композиции настоящего изобретения для местного введения, включая различные препараты для местного офтальмического введения, могут, кроме того, включать различные ингредиенты лекарственных препаратов, такие как противомикробные консерванты и агенты, регулирующие изотоничность. Примерами подходящих противомикробных консервантов являются хлорид бензалкония, тимерозал, хлорбутанол, метилпарабен, пропилпарабен, фенилэтиловый спирт, динатрийэдетат, сорбиновая кислота, ONAMER М и другие агенты, известные специалистам. Указанные консерванты, если они используются, могут быть введены в количестве от 0,001 до 1,0 мас.%. Примерами подходящих агентов, которые могут быть использованы для придания данной композиции изотоничности или осмомолярности, являются хлорид натрия, хлорид калия, маннит, декстроза, глицерин и пропиленгликоль. Указанные агенты, если они используются, могут быть введены в количестве от 0,1% до 10,0 % по массе (мас.%). Указанные композиции, предпочтительно, являются водными и имеют рН в пределах от 3,5 до 8,0.
Как очевидно для каждого специалиста, композиции могут быть изготовлены в виде различных лекарственных форм, подходящих для местной офтальмической доставки, включая растворы, суспензии, эмульсии, гели и разлагаемые твердые офтальмические прокладки.
Парентеральные формы для введения
Фармацевтические композиции для парентеральных инъекций включают фармацевтически приемлемые стерильные водные или безводные растворы, дисперсии, суспензии, эмульсии и стерильные порошки для приготовления стерильных инъецируемых растворов или дисперсий непосредственно перед использованием. Могут быть использованы различные водные или безводные носители, разбавители, растворители и носители (например, вода, этанол, глицерин, гликоль), а также растительные масла (например, оливковое масло) и органические сложные эфиры (например, этилолеат) или их смеси. Текучесть может поддерживаться с использованием материала для покрытий, такого как лецитин, с помощью ограничения размера частиц и с использованием поверхностно-активных веществ.
Указанные композиции могут также содержать адъюванты, такие как консерванты, смачивающие агенты, эмульгаторы, диспергирующие агенты, антибактериальные агенты, противогрибковые агенты, агенты, придающие изотоничность и/или агенты, замедляющие абсорбцию. Эффекты пролонгирования абсорбции или замедления абсорбции могут быть достигнуты путем инъекции кристаллической или аморфной суспензии с низкой водорастворимостью. Замедленная абсорбция может быть также достигнута посредством растворения или суспендирования указанного лекарственного средства в масляном носителе, либо посредством использования инъецируемых депоформ (например, микроинкапсулированных матриц лекарственного средства в биологически разлагаемых полимерах, таких как полилактид-полигликолид, полиортоэфиры, полиангидриды) или использования различных типов липосом или микроэмульсий, содержащих лекарственное средство. Композиции для инъекций могут быть стерилизованы различными методами.
Ведение путем ингаляции
Соединения настоящего изобретения, например, норкетотифен, могут быть введены путем ингаляции, которая может оказаться предпочтительным путем введения для некоторых пациентов, например пациентов, страдающих астмой. Могут быть использованы различные устройства для ингаляции, такие как, например, ингаляторы с дозирующим клапаном, ингаляторы для распыления сухого порошка и аэрозольные ингаляторы, известные специалистам.
Формы для ректального введения
Композициями для ректального введения, предпочтительно, являются суппозитории.
Формы для буккального введения
Композициями для буккального введения, предпочтительно, являются зубные пасты, жидкости для полоскания рта, препараты для подъязычного введения, жевательные резинки и т.п.
Формы для подъязычного введения
Могут быть использованы различные галеновы препараты, а именно концентрированные растворы или суспензии лекарственных средств, которые могут быть введены подъязычно с помощью различных капельных устройств; для распыления лекарственного средства на слизистые оболочки рта могут быть использованы различные аэрозольные устройства; а для быстрой доставки полной дозы могут быть также использованы специально разработанные таблетки, капсулы или порошки быстрого растворения.
Формы для чрезкожного введения
Композициями для чрезкожного введения соединений настоящего изобретения являются различные известные пластыри, повязки и т.п.
Аэрозольный препарат или капли для перорального (буккального) или интраназального введения
Композиции для пероральных или интраназальных аэрозолей или капель могут быть приготовлены в форме растворов, суспензий или сухих, порошков и могут быть разработаны для интраназальной, трансбуккальной, бронхиальной/легочной и/или желудочной абсорбции лекарственного средства.
Терапевтические дозы
Поскольку соединения настоящего изобретения не обладают сильными и дозоограничивающими седативными побочными эффектами кетотифена, то они могут быть введены в более высоких дозах, чем обычно используемые дозы кетотифена.
Фактические дозы активных ингредиентов в фармацевтических композициях настоящего изобретения могут варьироваться для достижения нужного терапевтического эффекта. Таким образом, количество используемого лекарственного средства может варьироваться в зависимости от таких факторов, как форма введения, тяжесть заболевания, частота введения дозы и т.п. Для введения в качестве лекарственного средства пациентам, страдающим легкими расстройствами дыхательных путей или бронхиальными расстройствами (такими как астма, бронхит и т.п.), пероральные дозы соединения настоящего изобретения вводят в количествах от 0,5 мг до около 200 мг, а предпочтительно от 1 мг до 20 мг, от одного до четырех раз в день в расчете на пациента весом 60 кг. Ежедневная доза может быть увеличена или снижена в зависимости от различных факторов, таких как вес и серьезность заболевания пациента.
Примерами использования лекарственного средства для лечения пациентов, страдающих сезонными аллергическими состояниями, такими как, например, аллергический ринит, является введение пероральных доз соединения настоящего изобретения в количествах от 0,1 мг до около 100 мг, и предпочтительно от 1 мг до 20 мг, от одного до четырех раз в день в расчете на пациента весом 60 кг. Для пациентов, страдающих сезонным аллергическим конъюнктивитом, концентрация раствора, содержащего соединения настоящего изобретения для инстилляции в конъюктивальный мешок, может составлять от 0,01% до 4,0%, и предпочтительно от 0,02% до 2,0%, активного ингредиента. Пациентам, страдающим астмой, соединения настоящего изобретения вводят в виде пероральной дозы от 2 мг до 100 мг, и предпочтительно от 2 мг до 20 мг, от одного до четырех раз в день в расчете на пациента весом 60 кг. Частота введения и количество дозы могут быть определены врачом-клиницистом с учетом различных клинических факторов, таких как, например, масса тела пациента и тяжесть его заболевания. Офтальмическое применение обычно предусматривает местное введение от одной до двух капель (или определенное количество твердой или полутвердой лекарственной формы) в пораженный глаз от одного до четырех раз в день. Кожное применение обычно предусматривает нанесение на кожу мази, содержащей от 0,1% до 10,0% соединения настоящего изобретения.
Пероральные лекарственные композиции
Примеры изготовления таблеток
Активный ингредиент (в примере вышеуказанным соединением является рацемический норкетотифен) смешивают с лактозой и целлюлозой до образования однородной смеси. Затем добавляют лак и снова смешивают. И, наконец, подмешивают стеарат кальция и полученную смесь прессуют в таблетки с использованием 9/32-дюймового (7 мм) плоско-вогнутого штампа. Таблетки с другой концентрацией лекарственного средства могут быть получены путем изменения отношения количества активного ингредиента к количеству наполнителей или к конечной массе таблетки. Помимо ингредиентов, указанных в этом примере, нужные композиции для перорального введения могут содержать другие или дополнительные ингредиенты, описанные выше в разделе "Формы для перорального введения".
Терапевтические показания
Соединения по настоящему изобретению могут быть использованы для различных терапевтических показаний, включая такие показания, для которых может быть использован кетотифен. Соединения по настоящему изобретению обладают фармакологическим действием, аналогичным действию кетотифена, но соединения настоящего изобретения не имеют значительных и дозоограничивающих седативных побочных эффектов исходного соединения (кетотифена), который используется в терапевтических целях.
Настоящее изобретение относится к способам лечения и/или профилактики всех форм аллергических расстройств, включая, но не ограничиваются ими, аллергический ринит, поливалентные аллергии, кожные и глазные аллергии. Эти способы предусматривают введение млекопитающему лекарственного средства по настоящему изобретению либо в виде одного изомера, либо в виде смеси изомеров, либо в виде его фармацевтически приемлемой соли или сольвата.
Настоящее изобретение также относится к способам лечения и/или профилактики бронхиальных и легочных заболеваний, включая, но не ограничиваясь ими, астму, бронхит, бронхиальную гиперреактивность, кашель и хроническую обструктивную болезнь легких (ХОБЛ). Эти способы предусматривают введение млекопитающему лекарственного средства настоящего изобретения либо в виде одного изомера, либо в виде смеси изомеров, либо в виде его фармацевтически приемлемой соли или сольвата.
Настоящее изобретение также относится к способам лечения и/или профилактики кожных болезней, включая, но не ограничиваясь ими, атопический дерматит, крапивницу, другие виды зуда или воспалительных состояний и псориаз. Эти способы предусматривают введение млекопитающему лекарственного средства настоящего изобретения либо в виде одного изомера, либо в виде смеси изомеров, либо в виде его фармацевтически приемлемой соли или сольвата.
Настоящее изобретение относится к способам лечения и/или профилактики некоторых форм глазных болезней, таких как конъюнктивит, кератит, блефарит, эписклерит, склерит, передний увеит, задний увеит, эндофтальмит, неврит зрительного нерва, краниальный артериит, симпатическая офтальмия у млекопитающих, таких как человек, где указанные способы позволяют избежать раздражения глаз, а также седативных и других токсических проявлений кетотифена и стероидов. Эти способы предусматривают введение млекопитающему лекарственного средства по настоящему изобретению либо в виде одного изомера, либо в виде смеси изомеров, либо в виде его фармацевтически приемлемой соли или сольвата.
Настоящее изобретение относится к способам лечения и/или профилактики определенных форм желудочно-кишечных заболеваний, таких как, например, синдромы гиперактивности или гиперсекреции, включая синдром Золлингера-Эллисона, раздражение желудка, энтерит, язву желудка или двенадцатиперстной кишки, гиперхлоргидрию, изжогу, нарушение перистальтики, желудочный рефлюкс или нежелательную секрецию желудочного сока. Эти способы предусматривают введение млекопитающему лекарственного средства настоящего изобретения либо в виде одного изомера, либо в виде смеси изомеров, либо в виде его фармацевтически приемлемой соли или сольвата.
Совместное введение
Настоящее изобретение также относится к способам введения одного или нескольких соединений настоящего изобретения вместе с адренергическими агонистами, включая, но не ограничиваясь ими, альбутерол, тербуталин, фенотерол, формотерол или сальметерол, что позволяет исключить или ослабить побочные эффекты, которые могут быть индуцированы указанной терапией с использованием бета-агонистов.
Настоящее изобретение также относится к способам совместного введения одного или нескольких соединений настоящего изобретения с агентами или лекарственными средствами, вызывающими бронхиальную гиперреактивность, включая, но не ограничиваясь ими, агенты, блокирующие адренергические бета-рецепторы, или ингибиторы циклооксигеназы, что позволяет исключить или снизить бронхиальную гиперреактивность, индуцированную указанной терапией. Настоящее изобретение также относится к способам совместного введения одного или нескольких соединений настоящего изобретения по крайней мере с одним из лекарственных средств следующих классов: агентов, снижающих глазное давление, адренергических антагонистов, антибактериальных агентов, противовирусных агентов, стероидов, ингибиторов циклооксигеназы, антагонистов лейкотриена, ингибиторов липоксигеназы, местных анестезирующих средств и офтальмических терапевтических лекарственных средств. Настоящее изобретение также относится к совместному введению соединений настоящего изобретения с противоотечными средствами, такими как, например, фенилэфедрин, нафазолин, тетрагидрозолин, или с антибактериальными агентами, такими как бацитрацин, неомицин и полимиксин.
Эквиваленты
Многие эквиваленты описанных здесь конкретных вариантов осуществления изобретения очевидны или могут быть получены каждым специалистом посредством лишь рутинного экспериментирования. Такими эквивалентами являются терапевтическое использование одного изомера и композиции, содержащей указанный(е) изомер(ы), не обладающий(е) побочными эффектами, присущими соответствующему(им) изомеру(ам) или исходному соединению (кетотифену) или его изомеру. Такими эквивалентами являются также множество фармацевтически приемлемых солевых форм, например, сульфата, гидробромида, гидрохлорида, дигидрохлорида, метансульфоната, фумарата, гидроксинафтоата, или, если это возможно, одна или другая из гидратных форм этих солей, см. Merck Index llth edition (1989) items 9089, 209, 3927, 4628, 8223, 5053, 5836, 8142, 2347, 7765, 1840, 9720, 7461, 1317, 4159 и 963, а также цитируемые в них работы, и Am.Rev.Resp.Dis. 1988, 137: (4;2/2) 32. Эквивалентами также является совместное введение по крайней мере одного соединения настоящего изобретения с любым другим лекарственным средством, используемым для лечения заболеваний у млекопитающих, упомянутых в настоящем документе. Для каждого специалиста-медика очевидно, что может также оказаться предпочтительным использование более высоких или более низких доз, чем те, которые были указаны в настоящем описании, и эти дозы могут быть введены с большей или меньшей частотой, чем частота, указанная в настоящем описании.
Специалистам-фармакологам известно, что бронхиальная гиперреактивность обычно наблюдается у пациентов, страдающих различными легочными расстройствами, такими как, например, астма. Кроме того, специалистам также известно, что бронхиальная гиперреактивность может быть индуцирована лекарственными средствами, такими как, например, агонисты адренергических бета-рецепторов.
Соединения настоящего изобретения, обладающие определенными фармакологическими свойствами (такими как ингибирующая активность, направленная на различные типы гистаминовых рецепторов, ФАТ-антагонистическая активность, воздействие на (окись азота)-синтазу, активность, стабилизирующая тучные клетки и т.п.) могут быть использованы для других показаний, чем те, которые были указаны в настоящей заявке. Такими показаниями являются эквиваленты описанных здесь конкретных вариантов осуществления изобретения.
Использование одного изомера соединения по настоящему изобретению позволяет избежать побочных эффектов, присущих соответствующему диастереомеру. Такими побочными эффектами могут быть, например, побочные эффекты, действующие на сердечно-сосудистую систему, такие как, например, сердечная недостаточность и сердечная аритмия, побочные эффекты, действующие на желудочно-кишечный тракт, такие как, например, раздражение желудка, или побочные эффекты, действующие на ЦНС, такие как, например, седативный эффект или сонливость. Все эквиваленты входят в объем настоящего изобретения.
| название | год | авторы | номер документа |
|---|---|---|---|
| БЕНЗОЦИКЛОГЕПТАТИОФЕНОВЫЕ СОЕДИНЕНИЯ, СПОСОБЫ ПРОФИЛАКТИКИ ИЛИ ЛЕЧЕНИЯ, ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ | 1998 |
|
RU2193557C2 |
| ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ ДЛЯ ЛЕЧЕНИЯ АЛЛЕРГИЧЕСКИХ ЗАБОЛЕВАНИЙ, СПОСОБ АНТИГИСТАМИННОГО ЛЕЧЕНИЯ, ПРИМЕНЕНИЕ КОМПОЗИЦИИ ДЛЯ ПОЛУЧЕНИЯ ЛЕКАРСТВЕННОГО ПРЕПАРАТА | 1993 |
|
RU2167657C2 |
| ТЕРАПЕВТИЧЕСКИЕ АГЕНТЫ | 2008 |
|
RU2468025C2 |
| КОМПОЗИЦИИ, СОДЕРЖАЩИЕ ИНГИБИТОРЫ ОБРАТНОГО ЗАХВАТА ДОПАМИНА, И СПОСОБЫ ИХ ПРИМЕНЕНИЯ | 2004 |
|
RU2358719C2 |
| КОМПОЗИЦИИ, СОДЕРЖАЩИЕ ИНГИБИТОРЫ ОБРАТНОГО ЗАХВАТА ДОПАМИНА, И СПОСОБЫ ИХ ПРИМЕНЕНИЯ | 1999 |
|
RU2238084C2 |
| ПРОИЗВОДНЫЕ ИМИДАЗОЛА, АКТИВНЫЕ В ОТНОШЕНИИ РЕЦЕПТОРА СВ1 | 2005 |
|
RU2377238C2 |
| ИЗОИНДОЛИНОНОВЫЕ ИНГИБИТОРЫ ВЗАИМОДЕЙСТВИЯ MDM2-P53, ОБЛАДАЮЩИЕ ПРОТИВОРАКОВОЙ АКТИВНОСТЬЮ | 2016 |
|
RU2794333C1 |
| КОМПОЗИЦИИ (-)-E-10-OH-NT И СПОСОБЫ ИХ СИНТЕЗА И ПРИМЕНЕНИЯ | 2008 |
|
RU2469715C2 |
| 3,3-ДИФТОРПИПЕРИДИНКАРБАМАТНЫЕ ГЕТЕРОЦИКЛИЧЕСКИЕ СОЕДИНЕНИЯ В КАЧЕСТВЕ АНТАГОНИСТОВ NR2B NMDA-РЕЦЕПТОРА | 2016 |
|
RU2735277C2 |
| ФАРМАЦЕВТИЧЕСКИЕ КОМПОЗИЦИИ | 2011 |
|
RU2568882C2 |
Изобретение относится к области химико-фармацевтической промышленности и касается фармацевтической композиции, содержащей S-изомер соединения формулы I, или его фармацевтически приемлемые соли и сольваты вместе с фармацевтически приемлемым носителем; способа синтеза соединения S-изомера формулы I, а также способа лечения заболевания, выбранного из группы, включающей респираторные заболевания, аллергические заболевания, кожные заболевания, желудочно-кишечные заболевания и глазные болезни. Композиция позволяет избежать побочных седативных эффектов при лечении указанных заболеваний. 3 н. и 11 з.п. ф-лы.


где R представляет собой Н,
или его фармацевтически приемлемые соли и сольваты вместе с фармацевтически приемлемым носителем.
Приоритет по признакам:
US 60/153566 от 13.09.1999 - относится к рацемическим метаболитам кетотифена, охватывает рацемический норкетотифен;. US 60/197905 от 15.04.2000 - относится к R-норкетотифену;. US 60/197363 от 15.04.2000 - относится к S-норкетотифену;. US 60/197906 от 15.04.2000 - относится к R-кетотифену;. US 60/197985 от 15.04.2000 - относится к S-кетотифену.
| WO 9856381 A1, 17.12.1998 | |||
| WO 9843640 A1, 08.10.1998 | |||
| Автореферат АБД STN Polivka, Zdenek "4H-Benzo[4,5-cyclohepta[1,2-b]thiophenes and 9,10-dihydroderivatives | |||
| Sulfonium analogs of pizotifen and ketotifen | |||
| Chirality of ketotifen | |||
| Аппарат для очищения воды при помощи химических реактивов | 1917 |
|
SU2A1 |
| Inst | |||
| Pharm | |||
| Biochem., Prague, 130 60 | |||
| Collection of | |||

