Изобретение относится к новым указанным в заглавии химическим соединением, которые представлены ниже, и к способам лечение болезненных состояний, модулируемых аллергическими, воспалительными или холинергическими активностями у млекопитающих, с использованием новых упомянутых указанных в заглавии химических веществ.
Соединения данного изобретения включают указанные в заглавии химические соединения следующей формулы:
где -А-В - фрагмент, имеющий формулу
-СО-СН2- (а)
-СН2-СО- (b)
-СН2-СН2 (с)
-СНОН-СН2- (d)
-СНОН-СНОН- (e)
-СН2-СНОН- (f)
или
-СО-СО- (g)
где R - гидроксиалкильный или карбоксиалкилоксиалкильный фрагмент, и их фармацевтически приемлемые соли и оптически активные изомеры рацемических соединений.
Соединения данного изобретения обладают фармакологическими свойствами, что делает указанные соединения полезными при профилактике и лечении аллергических заболевании, воспалений, глазных заболеваний различных типов и различных типов повышенной активности гладких мышц (такой как повышенная бронхиальная и маточная активность, включая повышенную активность, вызванную лекарствами).
Более точно, данное изобретение относится к новым указанным в заглавии химическим соединениям и к способам лечения аллергических расстройств (таких как, например, аллергический ринит), легочных расстройств (таких как, например, астма, бронхит, кашель и повышенная бронхиальная реактивность), кожных расстройств (таких как, например, крапивница, псориаз и атопический дерматит), желудочно-кишечных расстройств (таких как синдромы повышенной секреции, включая синдром Золлингера - Эллисона, желудочное раздражение и энтерит) и других воспалительных заболеваний и/или аллергических расстройств (таких как, например, глазной конъюнктивит и глазной кератит), без побочных эффектов (таких как побочное седативное действие, сердечная аритмия и глазное раздражение) с использованием упомянутых новых химических соединений, указанных в заглавии.
ОПИСАНИЕ ПРЕДШЕСТВУЮЩЕГО УРОВНЯ
Данное изобретение относится, в частности, к противовоспалительным и противоаллергическим соединениям, имеющим терапевтическое применение при различных заболеваниях, что наиболее важно для пациентов, страдающих повышенной реактивностью дыхательных путей и/или обструктивными (закупоривающими) заболеваниями дыхательных путей, включая астму и бронхит, кожными расстройствами и аллергическими заболеваниями, включая крапивницу, атопический дерматит, аллергический ринит и ретинопатию, другими заболеваниями небольших сосудов, связанными с сахарным диабетом, или глазными расстройствами, включая конъюнктивит и кератит.
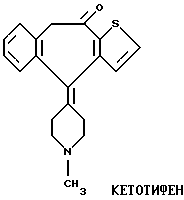
Данные соединения по химическому строению схожи с кетотифеном (Zaditen ®) и ранее не были известны заявителям. Побочное седативное действие сильно ограничило терапевтическую полезность кетотифена, и такие побочные эффекты могут быть снижены или устранены посредством применения соединений данного изобретения.
Фармакология, токсикология, метаболизм и клиническое испытание кетотифена обобщены в публикации Sorkin et al., Focus on Ketotifen. Ed. E.M. Sorkin. In Drugs, Sept. 1990, vol. 40, No. 3, pp. 412 - 448.
КРАТКОЕ ОПИСАНИЕ ИЗОБРЕТЕНИЯ
Данное изобретение относится к некоторым указанным в названии химическим соединениям, описанным ниже, способам применения указанных химических соединений для терапевтических целей и композициям, включающим один или большее количество фармацевтически приемлемых инертных носителей, и в качестве активного ингредиента, терапевтически эффективное количество по меньшей мере одного соединения, его фармацевтически приемлемых кислотно-аддитивных солей и его стереохимически изомерных форм, имеющего формулу:
где R выбран из группы, включающей гидрокси-С2-6алкил или карбокси-С1-6алкилокси-С1-6алкил и
-А-В- представляет фрагмент, имеющий формулу
-CO-CH2- (а)
-СН2-СО- (b)
-СН2-СН2 (c)
-СНОН-СН2- (d)
-СНОН-СНОН- (е)
-СН2-СНОН- (f)
или
-СО-СО- (д)
Соединения данного изобретения были синтезированы и фармакологически изучены. Были обнаружены значительные фармакологические различия между соединениями данного изобретениями и кетотифеном. Так кетотифен обладает глубоким побочным седативным действием, в то время как данные соединения, как было обнаружено, обладают пониженной седативной активностью или не обладают ею. Было также установлено, что новые соединения обладают антигистаминергическими и противовоспалительными свойствами. Важно, что новые соединения обладают сильным противовоспалительным действием на легкие и что они сильно ингибируют повышенную реактивность гладких бронхиальных мышц.
Так как воспаление дыхательных путей легких и повышенная реактивность бронхиальных гладких мышц являются отличительными признаками астмы, следовательно, новые соединения помимо того, что являются мощными антигистаминными средствами, будут полезны для клинического лечения астмы и бронхита без сопутствующего побочного седативного действия.
ПОДРОБНОЕ ОПИСАНИЕ ИЗОБРЕТЕНИЯ
БИОЛОГИЧЕСКИЕ ИСПЫТАНИЯ СОЕДИНЕНИЙ ДАННОГО ИЗОБРЕТЕНИЯ
Как обсуждалось выше, в настоящее время показано, что соединения данного изобретения обладают преимущественными фармакологическими действиями, полезными при лечении различных расстройств, таких как астма, аллергии и глазные расстройства. Новые результаты описываются в следующих далее биологических испытаниях.
1. Связывание с гистаминергическими рецепторами.
Сродство испытываемых соединений к гистаминным H1-рецепторам оценивают с использованием опыта [3Н]пириламинового связывания, модифицированного впоследствии (Chang et al. Heterogeneity of Histamine H1-Receptors. J. Neurochem. 1979, 32: 1653 - 1663). Кратко, мембраны, полученные из мозжечка коровы, выдерживают с [3Н] пириламином и испытываемым соединением при концентрациях. Специфическое связывание радиоактивного лиганда с рецептором определяют как разность между общим связыванием и неспецифическим связыванием, определенным в присутствии избытка немеченого лиганда. Значения IC50 (концентрация, необходимая для ингибирования 50% специфического связывания [3Н] пириламина) определяют c помощью нелинейного регрессионного анализах конкурентных кривых (таблица 1).
2. Связывание с мускариновыми рецепторами.
Сродство испытываемых соединений к мускариновым M1-рецепторам оценивают с использованием опыта [3Н] пирензепинового связывания, модифицированного впоследствии (Luthm et al. [3Н] Pirenzepine and [3H]QNB binding to brain nuscarinic cholinergic receptors. Molec. Pharmac. 1984, 26: 164-169). Кратко, опыты проводят на стриарных мембранах коровы, экспрессирующих мускариновые М1-рецепторы. После инкубации с испытываемым соединением и подходящим радиолигандом и промывки связанную радиоактивность определяют с помощью сцинтилляционного счетчика, используя коммерческий сцинтилляционный коктейль. Специфическое радиолигандное связывание с каждым рецептором определяют как разность между общим связыванием и неспецифическим связыванием, определенным в присутствии избытка немеченого лиганда. Значения IC50 (концентрации, необходимые для ингибирования 50% специфического связывания) определяют с помощью нелинейного регрессионного анализа конкурентных кривых (таблица 2).
3. Исследование побочного седативного действия.
Тест физостигмин-индуцированной летальности, используемый в этих опытах, представляет собой модификацию метода оценки седативного действия, описанного в публикации COLLIER et al., in Br. J. Pharmac., 1968, 32: 295-310. Кратко, физостигмин (1,9 мг/кг, подкожно) дает 100% летальность при введении мышам, которые разбиты на группы по 10 особей в каждом пластиковом садке (приблизительно 11•26•13 см). Мыши, которым перед введением физостигмина вводят обладающий седативным действием антигистамин, являются защищенными и выживают. В данном исследовании испытываемые соединения вводят перорально за 60 минут до введения физостигмина. Количество выживших мышей подсчитывает спустя 30 минут после введения физостигмина. Дозы в опыте ЦНС составляют половину молекулярного веса испытываемых соединений, выраженную в мг/кг веса тела (таблица 3).
4. Противовоспалительное действие (ингибирование бронхиальной эозинофильной аккумуляции)
Ингибирование эозинофильной аккумуляции в легком определяют на морских свинках (от 400 до 600 граммов) после интраперитонеальной инъекции 10 мкг РАЕ (фактора агрегации тромбоцитов) в 0,25% растворе сывороточного альбумина коровы в физиологическом растворе. Спустя двадцать четыре часа животных умерщвляют с использованием барбитурата. Трахею экспонируют и канюлируют 6•10 мл аликвотами буферного модифицированного раствора Тирода (Tyrode's solution) (состав: NaHCO3 11,9, NaCl 136,9, KCl 2,7, Na2HPО4 0,4, глюкоза 5,6, EDTA 19,8, желатин 0,1% мас./объем, BSA 0,5% мас./объем; рН 7,4) вводят последовательно и аспирируют с помощью мягкой компрессии легких. Общее выделение жидкости обычно превышает 80%. Клеточные суспензии концентрируют с помощью низкоскоростного центрифугирования и образующийся клеточный пеллет повторно суспендируют в 1 мл раствора Тирода. Подсчет всех клеток производят разбавлением 10 мкл клеточной суспензии в 90 мкл раствора Турка. Подсчет дифференциальных клеток проводят из мазков, смешанных в метаноле (100%) и окрашенных в красителе Лейсхмана. Всего, по меньшей мере, 500 клеток на мазок подсчитывают при 1000-кратном увеличении для того, чтобы дифференцировать типы клеток. Лекарственные средства вводят в течение 7 дней в виде длительной подкожной инфузии из имплантированного мини-насоса Альза, так что экспозиция в PAF имеет место только после пятидневного периода предобработки испытываемым(и) соединением(ями).
5. Исследование влияний на желудок.
Влияния соединений данного изобретения исследуют на брадикинининдуцированных сокращениях выделенной подвздошной кишки морской свинки. Ткань предварительно обрабатывают испытываемыми соединениями в различных концентрациях перед сократительными ответами на брадикинин (в отсутствии или в присутствии атропина с концентрацией 1 мкМ).
Влияния соединений на образование язв желудка исследуют на крысах. Образование язв желудка производят с помощью подкожной инъекции 30 мг/кг индометацина.
Изучаемым группам дают испытываемые соединения в количестве 100 мкг/100 г массы тела перорально за 30 минут до и через 5 часов после введения индометацина. Измеряют уменьшение площади язвы (мм2).
ХИМИЧЕСКИЙ СИНТЕЗ НОВЫХ СОЕДИНЕНИЙ. ПРИМЕРЫ
Синтез кетотифена, нор-кетотифена и (RS)-10-OH-кетотифена описан в публикации Waldvogel et al., Helv Chem. Acta, 1976, 59: 866 - 877, содержание которой введено в описание в виде ссылки.
Новые соединения данного изобретения являются соединениями общей формулы, представленной в Таблице 1, ниже.
Исходные соединения для этих синтезов получают в соответствии с публикацией Waldvogel et al.:
Соединение (1) 4-(4-пиперидилиден)-9,10-дигидро-4Н-бензо[4,5]циклогепта] [1,2-b]тиофен-9-он.
Соединение (2) 4-(4-пиперидилиден)-9,10-дигидро-4Н-бензо[4,5]циклогепта[1,2-b]тиофен-10-он.
Соединение (3) 4-(4-пиперидилиден)-9,10-дигидро-4Н-бензо[4,5]циклогепта[1,2-b]тиофен.
Соединение (4) 4-(4-пиперидилиден)-9,10-дигидро-4Н-бензо[4,5]циклогепта[1,2-b]тиофен-9,10-дион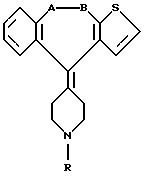
где R выбран из группы, включающей гидрокси-С1-6алкил или карбокси-С1-6алкилокси-С1-6алкил и
-А-В - представляет фрагмент, имеющий формулу
-СО-СН2- (а)
-СН2-СО- (b)
-СН2-СН2 (с)
-СНОН-СН2- (d)
-СНОН-СНОН- (е)
-СН2-СНОН- (f)
или
-СО-СО- (g)
Таблица 1. Новые соединения данного изобретения
Пример 1
Соединение Таблицы 1, где R представляет -(СН2)nОН, -А-В- имеет формулу -CO-CH2- и n = 2, получают обработкой исходного соединения (1) 2-галогенэтанолом, таким как 2-бромэтанол или 2-хлорэтанол, в присутствии основного катализатора, такого как карбонат калия, в растворителе, таком как N,N-диметилформамид (DMF), при перемешивании с нагревом или без него для инициирования реакции. После выпаривания растворителя остаток смешивают с водой и экстрагируют органическим растворителем, таким как хлороформ, метиленхлорид или этилацетат. После выпаривания органического растворителя продукт может быть очищен кристаллизацией из растворителя, такого как метанол или этанол. Аналогичные соединения, в которых n=3-6, могут быть получены таким же способом, но с использованием ω-галогенспиртов, Х-(СН2)3-6-ОН, хлор или бром.
Продукты могут быть превращены в гидрохлоридные соли посредством растворения в смеси растворителей, такой как хлороформ/диэтиловый эфир, и добавлением раствора хлористого водорода в диоксане. Выпаривание растворителей приводит к получению продукта в виде гидрохлорида.
Пример 2
Соединение Таблицы 1, где R представляет -(СН2)nOH, -А-В- имеет формулу -СН2-СО- и n=2, получают взаимодействием одного грамма соединения (2) с 2-хлорэтанолом (3 эквивалента), карбонатом калия (3 эквивалента) и йодидом калия (0,4 эквивалента) в 10 мл диметилформамида. После перемешивания в течение 4 дней при комнатной температуре растворитель выпаривают в вакууме, остаток растворяют в хлороформе (50 мл), раствор промывают водой и сушат сульфатом магния. Растворитель отгоняют и неочищенный (технический) продукт очищают хроматографией на силикагеле, используя в качестве элюента 5% метанол в хлороформе. Продукт растворяют в смеси хлороформ/диэтиловый эфир и добавляют раствор хлористого водорода в диоксане. Растворители выпаривают в вакууме, получают Пример 2 (n = 2) в виде гидрохлорида. Выход: 0,87 грамма. Данные протонного ЯМР согласуются с предполагаемой структурой.
Пример 3
Соединение Таблицы 1, где R представляет -(СН2)nОН, А-В- имеет формулу -CH2-CH2- и n= 2, получают обработкой исходного соединения (3) 2-галогенэтанолом, таким как 2-бромэтанол или 2-хлорэтанол, в присутствии основного катализатора, такого как карбонат калия, в растворителе, таком как N,N-диметилформамид (DMF), при перемешивании с нагревом или без него для осуществления реакции. После выпаривания растворителя остаток смешивают с водой и экстрагируют органическим растворителем, таким как хлороформ, метиленхлорид или этилацетат. После выпаривания органического растворителя продукт может быть очищен кристаллизацией из растворителя, такого как метанол или этанол. Аналогичные соединения, где n=3-6, могут быть получены таким же способом, но с ω-галогенспиртами, Х-(СН2)3-6-ОН, где Х - хлор или бром. Продукты могут быть превращены в хлористоводородные соли посредством растворения в смеси растворителей, такой как хлороформ/диэтиловый эфир, и добавления раствора хлористого водорода в диоксане. Выпаривание растворителей приводит к получению продукта в виде гидрохлорида.
Пример 4
Соединение Таблицы 1, где R представляет - (СН2)n-О-СН2СООН, -А-В- имеет формулу -CO-CH2- и n=2, получают из соединения Таблицы 1, где R представляет -(СН2)nОН, -А-В- имеет формулу -СО-СН2- и n=2, обработкой галогенуксусной кислотой, Х-СН2СООН, где Х = хлор или бром, в присутствии основного катализатора, такого как карбонат калия, в растворителе, таком как DMF, при перемешивании с нагреванием или без него для проведения реакции. После выпаривания растворителя остаток смешивают с водой, раствор нейтрализуют до рН 5 - 6 и водный раствор экстрагируют органическим растворителем, таким как хлороформ, метиленхлорид или этилацетат. После выпаривания органического растворителя продукт может быть очищен кристаллизацией из растворителя, такого как метанол или этанол. Аналогичные соединения, где n=3-6, могут быть получены таким же способом из соединений Таблицы 1, где R представляет - (СН2)n-О-CH2COOH, -А-В- имеет формулу -СО-СН2- и n=3-6. Альтернативно соединения Таблицы 1, где R представляет -(СН2)n-О-СН2-СООН, -А-В- имеет формулу -СО-СН2- и n=2-6, могут быть синтезированы в соответствии с общим способом, описанным в Примере 5 ниже.
Пример 5
Соединение Таблицы 1, где R представляет -(CH2)n-О-CH2- СООН, -А-В- имеет формулу -СН2-СО- и n=2, получают в две стадии. На первой стадии 1,9 грамма cоединения (2) обрабатывают (2-хлор-этокси)ацетонитрилом (2 эквивалента) и карбонатом калия (2,4 эквивалента) в 10 мл диметилформамида. После перемешивания в течение четырех дней при комнатной температуре растворитель выпаривают в вакууме и к остатку добавляют 10 мл воды.
Полученную суспензию экстрагируют хлороформом (3•10 мл), соединенные экстракты подвергают выпариванию и технический продукт очищают хроматографией на силикагеле, используя в качестве элюента 2% метанол в хлороформе. Промежуточный продукт - N-[(2-цианометокси)этил]норкетотифен (0,75 грамма) нагревают в концентрированной соляной кислоте и выдерживают в этих условиях в течение двенадцати часов. Растворитель выпаривают в вакууме, остаток розового цвета растворяют в метаноле, дважды обрабатывают активированным углем, фильтруют, растворитель выпаривают в вакууме, получают гидрохлорид Примера 5 (n=2) в виде розовой пены. Выход - 0,71 грамма. Данные протонного ЯМР согласуются с предполагаемой структурой.
Пример 6
Соединение Таблицы 1, где R представляет -(СН2)n-О-СН2-СООН, -А-В- имеет формулу -СН2-СН2- и n=2, получают из соединения Таблицы 1, где R представляет -(CH2)nOH, -А-В- имеет формулу -СН2-СН2- и n=2, обработкой галогенуксусной кислотой, Х-СН2СООН, где Х = хлор или бром, в присутствии основного катализатора, такого как карбонат калия, в растворителе, таком как DMF, при перемешивании с нагревом или без него для проведения реакции. После выпаривания растворителя остаток смешивают с водой, раствор нейтрализуют до рН 5 - 6 и водный раствор экстрагируют органическим растворителем, таким как хлороформ, метиленхлорид или этилацетат. После выпаривания органического растворителя продукт может быть очищен кристаллизацией из растворителя, такого как метанол или этанол. Аналогичные соединения, где n=3-6, могут быть получены таким же способом из соединений Таблицы 1, где R представляет -(СН2)nСН, -А-В- имеет формулу -СН2-СН2- и n=3-6. Альтернативно соединения Таблицы 1, где R представляет - (CH2)n-О-СН2-СООН, -А-В- имеет формулу -CH2-CH2- и n=2-6, могут быть синтезированы в соответствии с общим способом, описанным в Примере 5.
Пример 7
Соединение Таблицы 1, где R представляет -(СН2)nОН, -А-В- имеет формулу -СНОН-СН2- и n= 2, получают из соединения Таблицы 1, где R представляет -(CH2)nOH -А-В- имеет формулу -СО-СН2- и n=2, обработкой боргидридом натрия в растворителе, таком как этанол, при комнатной температуре. После разложения избытка реагента ацетоном растворители выпаривают и остаток кристаллизуют из растворителя, такого как метанол или этанол, с добавлением или без диэтилового эфира. Аналогичные соединения, где n=3-6, могут быть получены таким же способом из соединений Таблицы 1, где R представляет -(СН2)nОН, -А-В- имеет формулу -СО-СН2 и где n=3-6. Продукты могут быть превращены в хлористоводородные соли растворением в смеси растворителей, такой как хлороформ/диэтиловый эфир, и добавлением раствора хлористого водорода в диоксане. Выпаривание растворителей приводит к получению продукта в виде гидрохлорида.
Пример 8
Соединение Таблицы 1, где R представляет - (CH2)n-O-CH2-COOH, -А-В- имеет формулу -СНОН-СН2- и n=2, получают из соединения Таблицы 1, где R представляет -(СН2)nОН, -А-В- имеет формулу -СO-СН2- и n=2, обработкой боргидридом натрия в растворителе, таком как этанол, при комнатной температуре. После разложения избыточного реагента ацетоном растворители выпаривают и остаток смешивают с водой, раствор нейтрализуют кислотой, такой как разбавленная соляная кислота, и экстрагируют растворителем, таким как этилацетат. После выпаривания органического растворителя остаток кристаллизуют из растворителя, такого как метанол или этанол, с добавлением или без диэтилового эфира. Аналогичные соединения, где n=3-6, могут быть получены этим же способом из соединений Таблицы 1, где R представляет -(СН2)nОН, -А-В- имеет формулу -СО-СН2- и n=3-6.
Альтернативно соединения Таблицы 1, где R представляет - (СН2)n-О-СН2СООН, -А-В- имеет формулу -СНОН-СН2- и n=2-6, могут быть синтезированы в соответствии с общим способом, описанным в Примере 5.
Пример 9
Соединение Таблицы 1, где R представляет -(СН2)nОН, -А-В- имеет формулу -СН2-СНOН- и n= 2, получают из соединения Таблицы 1, где R представляет -(CH2)nOH, -А-В- имеет формулу -СН2-СО- и n=2, обработкой боргидрида натрия в растворителе, таком как этанол, при комнатной температуре. После разложения избыточного реагента ацетоном растворители выпаривают и остаток кристаллизуют из растворителя, такого как метанол или этанол, с добавлением или без диэтилового эфира. Аналогичные соединения, где n=3-6, могут быть получены таким же способом из соединений Таблицы 1, где R представляет -(СН2)nОН, -А-В- имеет формулу -СН2-СО- и n=3-6. Продукты могут быть превращены в хлористоводородные соли путем растворения в смеси растворителей, такой как хлороформ/диэтиловый эфир, и добавлением раствора хлористого водорода в диоксане. Выпаривание растворителей приводит к получению продукта в виде гидрохлорида.
Пример 10
Соединение Таблицы 1, где R представляет -(СН2)n-О-СН2-СООН, -А-В- имеет формулу -СH2СНОН- и n = 2, получают из соединения Таблицы 1, где R представляет - (СН2)n-O-CH2COOH, -А-В- имеет формулу -СН2-СО- и n=2, обработкой боргидридом натрия в растворителе, таком как этанол, при комнатной температуре. После разложения избыточного реагента ацетоном растворители выпаривают и остаток смешивают с водой, раствор нейтрализуют кислотой, такой как разбавленная соляная кислота, и экстрагируют растворителем, таким как этилацетат. После выпаривания органического растворителя остаток кристаллизую из растворителя, такого как метанол или этанол, с добавлением или без диэтилового эфира. Аналогичные соединения, где n=3-6, могут быть получены таким же способом из соединений Таблицы 1, где R представляет -(СН2)n-СН2-СООН, -А-В- имеет формулу -СН2-СО- и n=3-6.
Альтернативно соединения Таблицы 1, где R представляет -(СН2)n-O-СН2СООН, -А-В- имеет формулу -СН2-СНОН- и n=2-6, могут быть синтезированы в соответствии с общим способом, описанным в Примере 5.
Пример 11
Соединение Таблицы 1, где R представляет -(CH2)nOH, -А-В- имеет формулу -СО-СО- и n= 2, получают обработкой исходного соединения (4) 2-галогенэтанолом, таким как 2-бромэтанол или 2-хлорэтанол, в присутствии основного катализатора, такого как карбонат калия, в растворителе, таком как N,N-диметилформамид (DMF), при перемешивании с нагреванием для инициирования реакции или без него. После выпаривания растворителя остаток смешивают с водой и экстрагируют органическим растворителем, таким как хлороформ, метиленхлорид или этилацетат. После выпаривания органического растворителя продукт может быть очищен кристаллизацией из растворителя, такого как метанол или этанол. Аналогичные соединения, в которых n=3-6, могут быть получены этим же способом, но с ω-галогенспиртами, Х-(СН2)3-6ОН, где Х представляет хлор или бром.
Продукты могут быть превращены в хлористоводородные соли путем растворения в смеси растворителей, такой как хлороформ/диэтиловый эфир, и добавлением раствора хлористого водорода в диоксане. Выпаривание растворителей приводит к получению продукта в виде гидрохлорида.
Пример 12
Соединение Таблицы 1, где R представляет -(СН2)n-О-СН2СООН, -А-В- имеет формулу -СО-СО- и n=2, получают из соединения Таблицы 3, где R представляет -(CH2)nOH, -А-В- имеет формулу -СO-СО- и n=2, посредством обработки галогенуксусной кислотой, Х-СН2СООН, где Х= хлор или бром, в присутствии основного катализатора, такого как карбонат калия, в растворителе, таком как DMF, при перемешивании с нагреванием или без него для проведения реакции. После выпаривания растворителя остаток смешивают с водой, раствор нейтрализуют до рН 5 - 6 и водный раствор экстрагируют органическим растворителем, таким как хлороформ, метиленхлорид или этилацетат. После выпаривания органического растворителя продукт может быть очищен кристаллизацией из растворителя, такого как метанол или этанол. Аналогичные соединения, в которых n=3-6, могут быть получены таким же способом из соединений таблицы 1, где R представляет -(СН2)nОН, -А-В- имеет формулу -СО-СО- и n=3-6.
Альтернативно соединения Таблицы 1, где R представляет - (CH2)n-О-CH2-COOH, -А-В- имеет формулу -СО-СО- и n=2-6, могут быть синтезированы в соответствии с общим способом, описанным в Примере 5.
Пример 13
Соединение Таблицы 1, где R представляет -(CH2)nOH, -А-В- имеет формулу -СНОН-СНОН- и n=2, получают из соединения Таблицы 1, где R представляет -(СН2)nОН, -А-В- имеет формулу -СО-СО- и n=2, обработкой боргидридом натрия в растворителе, таком как этанол, при комнатной температуре. После разложения избыточного реагента ацетоном растворители выпаривают и остаток кристаллизуют из растворителя, такого как метанол или этанол с добавлением или без диэтилового эфира. Аналогичные соединения, в которых n=3-6, могут быть получены таким же способом из соединений Таблицы 1, где R представляет -(СН2 )nОН, -А-В- имеет формулу -СО-СО-.
Продукты могут быть превращены в хлористоводородные соли растворением в смеси растворителей, такой как хлороформ/диэтиловый эфир, и добавлением раствора хлористого водорода в диоксане. Выпаривание растворителей приводит к получению продукта в виде гидрохлорида.
Пример 14
Соединение Таблицы 1, где R представляет -(СН2)n-O-СН2-СООН, -А-В- имеет формулу -СНОН-СНОН- и n=2, получают из соединения Таблицы 1, где R представляет - (СН2)n-О-СН2-СООН, -А-В- имеет формулу -СО-СО- и n=2, обработкой боргидридом натрия в растворителе, таком как этанол, при комнатной температуре. После разложения избыточного реагента ацетоном растворители выпаривают, остаток смешивают с водой, раствор нейтрализуют кислотой, такой как разбавленная соляная кислота, и экстрагируют растворителем, таким как этилацетат. После выпаривания органического растворителя остаток кристаллизуют из растворителя, такого как метанол или этанол, с добавлением или без диэтиленового эфира. Аналогичные соединения, в которых n=3-6, могут быть получены этим же способом из соединений Таблицы 1, где R представляет - (СН2)n-О-СН2-СООН, -А-В- имеет формулу -СО-СО-.
Альтернативно соединения Таблицы 1, где R представляет -(СН2)n-О-СН2-СООН, -А-В- имеет формулу -СНОН-СНОН- и n=2-6, могут быть синтезированы в соответствии с общим методом, описанным в Примере 5.
Данное изобретение предоставляет соединения, описанные выше, включая изомеры рацемических соединений, и фармацевтически приемлемые кислотно-аддитивные соли и сольваты новых соединений.
Оптически активные изомеры соединений данного изобретения могут быть получены разделением рацемата с использованием традиционных способов, таких как фракционная кристаллизация диастереомерных солей с хиральными кислотами. Другие стандартные способы разделения, известные квалифицированному специалисту данной области техники, включают, но не ограничиваются только ими, кристаллизацию и хроматографию на хиральном субстрате и также могут быть использованы. Оптически активные изомеры данного изобретения также могут быть получены с помощью стереоселективного синтеза.
Термины "фармацевтически приемлемые соли" или "его фармацевтически приемлемая соль" относятся к солям, полученным из фармацевтически приемлемых нетоксичных кислот. Подходящие фармацевтически приемлемые кислотно-аддитивные соли для соединений данного изобретения включают соли уксусной, бензолсульфоновой (бузилат), бензойной, камфорсульфоновой, лимонной, этансульфоновой, фумаровой, глюконовой, глутаминовой, бромистоводородной, соляной, изотионовой, молочной, малеиновой, яблочной, манделовой, метансульфоновой, слизевой, азотной, памоиновой, патотеновой, фосфорной, п-толуолсульфоновой, сукциновой, серной, винной и т.п. кислот. Гидрофумарат является особенно предпочтительным.
Данное изобретение предоставляет также фармацевтические композиции, которые включают одно или большее количество соединений изобретения, введенных в рецептуру вместе с одним или большим количеством фармацевтически приемлемых носителей. Фармацевтические композиции могут быть специально приготовлены для перорального введения, конъюнктивальной инстилляции (закапывания), подъязычного введения, парентерального введения, трансдермального (чрескожного) введения, ректального введения, трансбуккального введения, для местного введения или для ведения ингаляцией, инсуффляцией (вдуванием) порошка или аэрозоля.
Фармацевтические композиции данного изобретения могут быть введены людям и другим млекопитающим перорально, подъязычно, парентерально, подкожно, трансдермально, ректально, трансбуккально, местно, конъюнктивальной истилляцией или в виде аэрозоля для перорального или назального введения.
Термин "парентеральное" введение включает внутривенную, внутриартериальную, внутримышечную, интраперитонеальную, внутрикожную, подкожную, внутримышечную, интраперитонеальную инъекцию и инфузию. Термин "трансдермальное" включает применение различных приспособлений ("бляшки" и т.д.), которые могут облегчить или модифицировать транспорт или абсорбцию лекарственного средства через кожу.
Формы для перорального введения
Фармацевтические композиции данного изобретения для перорального введения твердых дозированных форм включают капсулы, гранулы, пилюли, порошки и таблетки. В таких твердых дозированных формах активное соединение может быть смешано с одним или большим количеством фармацевтически приемлемых разбавителей или носителей (например, цитрат натрия, дикальций фосфат), наполнителей (например, крахмал, лактоза, сахароза, глюкоза, маннит, кремниевая кислота), связующих веществ (например, карбоксиметилцеллюлоза, альгинаты, желатин, поливинилпирролидон, сахароза), акация увлажнителей (например, глицерин), добавками, стабилизирующими раствор (например, парафин), дизинтегрирующими средствами (например, агар-агар, карбонат кальция, крахмал, альгиновая кислота, силикаты, карбонат натрия), ускорителями абсорбции (например, четвертичные аммониевые соединения), смачивающими агентами (например, цетиловый спирт, глицерин моностеарат), абсорбентами (например, каолин, бентонитовая глина), смазками (например, тальк, стеарат кальция, стеарат магния, полиэтиленгликоли, натрийлаурилсульфат) и/или буферными средствами.
Твердые формы в виде капсул, драже, гранул, пилюль и таблеток могут иметь покрытия и/или оболочки (например, энтеросолюбильные покрытия), известные в области получения фармацевтических препаратов. Композиции могут также быть разработаны для высвобождения активного(ых) ингредиента(ов) в определенной части желудочно-кишечного тракта или с контролируемым высвобождением, медленным высвобождением или длительным высвобождением действующего вещества.
Композиция может также разрабатываться для лимфатической абсорбции активного(ных) ингредиента(ов).
Активное(ые) соединение(ия) также могут быть микроинкапсулированными с одним или большим количеством указанных выше наполнителей.
Жидкие дозированные формы для перорального введения включают фармацевтически приемлемые эмульсии, растворы, суспензии, сиропы и эликсиры. Жидкая дозированная форма может также содержать обычно известные разбавители (например, воду, другие растворители, солюбилизирующие компоненты), эмульгаторы (например, этанол, изопропиловый спирт, этилкарбонат, этилацетат, бензиловый спирт, бензилбензоат, пропиленгликоль, бутиленгликоль, диметилформамид, масла, олеиновая кислота, глицерин, полиэтиленгликоли, сложные жирные сорбитан эфиры и их смеси).
Помимо инертных разбавителей композиции для перорального введения могут также включать адъюванты, такие как смачивающие, эмульгирующие, суспендирующие подслащивающие или вкусовые агенты.
Суспензии могут содержать один или большее количество суспендирующих агентов, известных в области технологии приготовления фармацевтических препаратов.
ФОРМЫ ДЛЯ МЕСТНОГО ВВЕДЕНИЯ
(включая формы для конъюнктивальной инстилляции)
Композиции для местного введения соединений данного изобретения включают растворы, суспензии, капли, спреи, мази и порошки.
Кроме терапевтически активных ингредиентов композиция данного изобретения для местного глазного или конъюнктивального введения может дополнительно включать различные рецептурные ингредиенты, такие как антимикробные консерванты и средства, регулирующие тоничность раствора. Примеры подходящих антимикробных консервантов включают: бензалконий хлорид, тимеросал, хлорбутанол, метилпарабен, пропилпарабен, фенилэтиловый спирт, эдетат динатрий, сорбиновая кислота, ONAMER М и другие компоненты, известные специалисту данной области. Такие консерванты, если используются, будут обычно применяться в количестве от 0,001% до 1,0% из расчета на массу (мас.%). Примеры подходящих средств, которые могут использоваться для регулирования тоничности или осмотического давления рецептур, включают хлорид натрия, хлорид калия, маннит, декстрозглицерин и пропиленгликоль. Такие добавки, если используются, будут применяться в количестве от 0,1% до 10,0% из расчета на массу (мас.%). Композиции предпочтительно являются водными и имеют рН в интервале значений от 3,5 до 8,0 и осмотическое давление в интервале значений от 280 до 320 миллимолей на литр.
Квалифицированному специалисту понятно, что композиции могут приготавливаться в различных дозированных формах, подходящих для местного глазного введения, включая растворы, суспензии, эмульсии, гели и твердые разлагаемые глазные вставки.
ФОРМЫ ДЛЯ ПАРЕНТЕРАЛЬНОГО ВВЕДЕНИЯ
Фармацевтические композиции для парентеральных инъекций включают фармацевтически приемлемые стерильные водные или неводные растворы, дисперсии, суспензии, эмульсии и стерильные порошки для превращения перед применением в стерильные инъекционные растворы или дисперсии. Могут использоваться различные водные и неводные носители, разбавители, растворители и наполнители (например, вода, этанол, глицерин, гликоль), а также растительные масла (например, оливковое масло), органические сложные эфиры (например, этилолеат) или их смеси. Текучесть может сохраняться посредством применения материалов для покрытий, таких как лецитин, посредством ограничения размера частиц или посредством применения поверхностно-активных веществ.
Композиции также могут содержать адъюванты, такие как консерванты, смачивающие агенты, эмульгаторы, дисперсанты, антибактериальные средства, противогрибковые средства, изотонические добавки и/или компоненты, замедляющие адсорбцию. Увеличение продолжительности абсорбции или замедление абсорбции могут достигаться посредством инъекции кристаллической или аморфной суспензии с низкой растворимостью в воде. Замедленная абсорбция может также быть получена путем растворения или суспендирования лекарственного средства в масляном носителе, или посредством применения базовых форм для инъекций (например, микроинкапсулированных матриц лекарственного средства в полимерах, способных разлагаться биологическим способом, таких как полиактидполигликолид, сложные полиортоэфиры, полиангидриды), или посредством применения липосом различных типов или микроэмульсий для удерживания лекарственного средства. Рецептуры для инъекции могут подвергаться стерилизации различными способами.
ФОРМЫ ДЛЯ РЕКТАЛЬНОГО ВВЕДЕНИЯ
Композиции для ректального введения предпочтительно представляют собой свечи.
ФОРМЫ ДЛЯ ТРАНСБУККАЛЬНОГО ВВЕДЕНИЯ
Композиции для трансбуккального введения предпочтительно представляют собой зубные пасты, жидкости для полоскания рта, препараты для подъязычного введения, жевательные резинки и т.д.
ФОРМЫ ДЛЯ ПОДЪЯЗЫЧНОГО ВВЕДЕНИЯ
Могут использоваться различные галеновые рецептуры: концентрированные растворы или суспензии лекарственного средства могут быть применены подъязычно с помощью различных капельных приспособлений; различные аэрозольные устройства могут быть использованы для распыления лекарства на слизистую оболочку рта; специально разработанные быстро растворяющиеся таблетки, капсулы или порошки могут также использоваться для быстрой доставки полной дозы.
ФОРМЫ ДЛЯ ТРАНСДЕРМАЛЬНОГО ВВЕДЕНИЯ
Композиции для трансдермального введения соединений данного изобретения включают различные известные бляшки, бандажи и т.д.
ОРАЛЬНОЕ ИЛИ НАЗАЛЬНОЕ РАСПЫЛЕНИЕ ИЛИ КАПЕЛЬНОЕ ВВЕДЕНИЕ
Композиции для оральных или назальных спреев или капель могут быть в форме раствора, суспензий или сухих порошков, могут разрабатываться для назальной, трансбуккальной, бронхиальной/легочной и/или желудочной абсорбции лекарственного средства.
ТЕРАПЕВТИЧЕСКИЕ ДОЗЫ
Фактические уровни доз активных ингредиентов в фармацевтических композициях данного изобретения могут изменяться для достижения нужного терапевтического действия. Следовательно, используемое количество лекарственного средства изменяется и может зависеть от различных факторов, таких как форма введения, тяжесть заболевания, частота дозирования и т.д. При применении в качестве лекарственного средства пациентами, страдающими доброкачественными расстройствами дыхательных путей или бронхиальными расстройствами (такими как астма, бронхит и т.д.), пероральные дозы соединения данного изобретения находятся в интервале от 0,5 мг до приблизительно 200 мг, предпочтительно от 0,5 мг до 10 мг при применении от одного до четырех раз в день пациентом с массой до 60 кг. Ежедневная доза может быть увеличена или снижена в зависимости от различных факторов, например массы и состояния болезни пациента.
Например, для применения в качестве лечения для пациентов, страдающих аллергическим конъюнктивитом, используются пероральные дозы соединения данного изобретения в интервале от 0,1 мг до приблизительно 100 мг, предпочтительно от 0,2 мг до 10 мг при приеме от одного до четырех раз в день для пациента массой до 60 кг. Для пациентов, страдающих сезонным аллергическим конъюнктивитом, концентрация нор-кетотифенового раствора для инстилляции в конъюнктивальный мешок находится в интервале от 0,01% до 2,0%, предпочтительно от 0,02% до 1,0%. Частота и количество дозировки будет определяться клинически на основании различных клинических факторов, таких как, например, масса и тяжесть заболевания пациента. Применение будет, обычно, включать местное введение одной - двух капель (или количество твердой или полутвердой дозированной формы) для воздействия на глаз от одного до четырех раз в день.
РЕЦЕПТУРА СТАНДАРТНОЙ ДОЗЫ ДЛЯ ПЕРОРАЛЬНОГО ВВЕДЕНИЯ
ПРИМЕР 15. РЕЦЕПТУРЫ ТАБЛЕТОК (таблица 4)
Активный ингредиент (в данном примере - соединение Примера 5, где n=2) смешивают с лактозой и целлюлозой до тех пор, пока не образуется однородная смесь. Добавляют краситель (lake) и дополнительно смешивают. Наконец, в смесь с перемешиванием добавляют стеарат кальция и полученную смесь прессуют в таблетки, используя вогнутый поверхностный перфоратор (g/32 дюйма, 7 мм). Таблетки с другим содержанием активного ингредиента могут быть получены посредством изменения соотношения активного ингредиента и наполнителей или конечной массы таблетки.
Изобретение предоставляет способы лечения и/или профилактики всех форм бронхиальной астмы, аллергического бронхита, мультисистемных аллергий, аллергического ринита и аллергических кожных расстройств у млекопитающих, таких как человек, при отсутствии побочного седативного действия и других токсических проявлений кетотифена. Эти способы включают введение млекопитающему, нуждающемуся в таком лечении и/или профилактике, эффективных количеств по меньшей мере одного соединения изобретения или его фармацевтически приемлемой соли.
Данное изобретение предоставляет также способы совместного введения одного или большего количества соединений данного изобретения с агонистом адренергического бета-рецептора, включая, но не ограничивая только этим перечнем, альбутерол, тербуталин, фенотерол, формотерол или сальметерол, устраняя или снижая таким образом повышенную бронхиальную реактивность, которая может быть индуцирована указанной терапией бета-агониста.
Изобретение предоставляет также способы совместного введения соединения данного изобретения с другими веществами или лекарственными средствами, вызывающими повышенную бронхиальную реактивность, включая, но не ограничиваясь только этим перечнем, блокирующие средства адренергического бета-рецептора или ингибиторы циклооксигеназы, при устранении или снижении таким образом повышенной бронхиальной реактивности, которая индуцируется такой терапией.
Данное изобретение предоставляет способы лечения и/или профилактики глазных заболеваний, таких как аллергический конъюнктивит или аллергический кератит, и воспалительных заболеваний, таких как блефарит, конъюнктивит, эписклерит, склерит, кератит, передний увеит (anterior uveitis), задний увеит (posterior uveitis), эндофтальмит, зрительный неврит, краниальный артериит, симпатическая офтальмия, у млекопитающих, таких как человек, при устранении глазного раздражения, побочного седативного действия и других токсических проявлений кетотифена и стероидов. Эти способы включают введение млекопитающему, нуждающемуся в таком лечении и/или профилактике, эффективных количеств соединения данного изобретения или его фармацевтически приемлемых солей.
Данное изобретение предоставляет также способы совместного введения соединения данного изобретения с по меньшей мере одним лекарственным средством из следующих классов: глазные антигипертензивные средства, адренергические агонисты или антагонисты, антибактериальные средства, антивирусные средства, стероиды, ингибиторы циклооксигеназы, антагонисты лейкотриена, ингибиторы липоксигеназы и другие глазные терапевтические лекарственные средства. В частности, данное изобретение обеспечивает способы совместного введения соединения данного изобретения с глазными противоотечными лекарственными средствами, такими как, например, фенилэфедрин, нафазолин, тетрагидрозолин, или с антибактериальными средствами, такими как бацитрацин, неомицин (neomycin) и полимиксин.
Изобретение также предоставляет способы введения соединения данного изобретения в сочетании с хирургическими операциями для снижения до минимума воспаления или раздражения и улучшения процесса послеоперационного выздоровления.
Данное изобретение предоставляет также способы лечения или профилактики различных форм гастроэнтерологических заболеваний, таких как синдромы повышенной секреции, включая синдром Золингера-Эллисона, желудочное раздражение, энтерит, желудочную или дуоденальную язвы, изжогу (acid indigestion) или нежелательную секреция кислоты в желудке. Эти способы включают введение млекопитающему, нуждающемуся в таком лечении и/или профилактике, эффективных количеств соединения данного изобретения или его фармацевтически приемлемых солей.
ЭКВИВАЛЕНТЫ
Квалифицированному специалисту будет понятно или он способен установить, используя только стандартный эксперимент, большое количество эквивалентов для конкретных воплощений изобретения, описанных здесь. Такие эквиваленты включают применение единственного изомера и композиции, содержащей этот же изомер, при отсутствии побочных эффектов, свойственных соответствующему(им) изомеру(ам). Такие эквиваленты также включают многочисленные фармацевтически приемлемые солевые формы, например сульфат, гидробромид, гидрохлорид, дигидрохлорид, фумарат, метансульфонат, гидроксинафтоат или соответствуют одной или другой из их гидратных форм (см. Merck Index 11th edition (1989) items 9089, 209, 3927, 4628, 8223, 5053, 5836, 8142, 2347, 7765, 1840, 9720, 7461, 1317, 4159, 963 и ссылки, приведенные в ней и Am. Rev. Resp. Dis. 1988, 137: (4; 2/2) 32). Такие эквиваленты также включают совместное введение по меньшей мере одного соединения данного изобретения с любым другим лекарственным средством, которое используется для подавления указанных в данном документе заболеваний у млекопитающих. Квалифицированный специалист в области медицины также понимает, что более высокие или более низкие дозы, чем указанные в данном описании, могут быть предпочтительными, и дозы могут приниматься более или менее часто, чем предложено в описании. Квалифицированный специалист в области фармакологии может представлять, что соединения данного изобретения обладают некоторыми фармакологическими свойствами (такими как антигистаминная активность в отношении различных типов рецепторов, PAF-антагонистической активностью, стабилизирующей активностью в отношении мастоцитов и т.д.), могут быть полезными для других показаний, которые не приведены в описании. Такие показания являются эквивалентами конкретных воплощений изобретения, описанных здесь.
Квалифицированный специалист в данной области будет представлять, что посредством применения единственного изомера (евтомера) любых рацемических соединений данного изобретения, нор-кетотифена или 10-ОН-норкетотифена можно избежать побочных эффектов, свойственных другому изомеру. Такие побочные эффекты могут включать, например, сердечно-сосудистые побочные эффекты, такие как, например, кардиодепрессия, побочные воздействия на центральную нервную систему, такие как, например, побочное седативное действие. Все эквиваленты, как подразумеваются, включены в данное изобретение.
Биологические исследования соединений по настоящему изобретению
Как упомянуто выше, исследования были предприняты с целью изучения фармакологических профилей новых соединений. Особую важность имеют исследования, касающиеся седативных побочных эффектов, антигистаминной активности и противовоспалительных эффектов.
Седативные эффекты in vivo
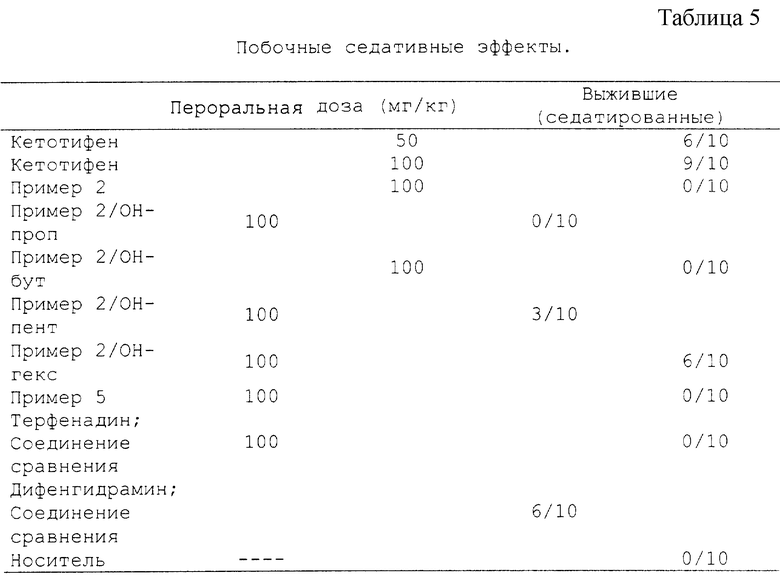
Седативные эффекты были исследованы с использованием методологии, описанной Виллару и др., в патенте США N 4659716. Данная методология в настоящее время считается "промышленным стандартом" по отношению к измерениям седативного эффекта на лабораторных животных, поскольку существует отличная корреляция между результатами, полученными в этом тесте, и клинической степенью седативности (иногда называемой "сонливостью"). Если коротко, обнаружено, что физостигмин (1,0 мг/кг внутримышечно) дает 100% летальность при введении группам мышей (10 животных в группе). Мыши, которым ввели седативный препарат перед физостигмином, надежно защищены и выживают. В приведенных здесь испытаниях тестируемые соединения давали перорально за 60 минут до физостигмина. Наши испытания включали исследования, в которых время до введения изменялось от 15 мин до 24 часов; результаты этих исследований согласуются с приведенными здесь. Количество седатированных (выживших) мышей определялось через 30 мин после введения физостигмина. Дозы тестированных веществ приведены далее в Таблице 5.
Результаты испытаний (седативность; повторное испытание)
"Пример 2/ОН-проп" означает соединение, имеющее структуру в соответствии с Примером 2, где заместителем в пиперидине является гидроксипропил. "Пример 2/ОН-бут" означает соединение, имеющее структуру в соответствии с Примером 2, где заместителем в пиперидине является гидроксибутил. "Пример 2/ОН-пент" означает соединение, имеющее структуру в соответствии с Примером 2, где заместителем в пиперидине является гидроксипентил. "Пример 2/ОН-гекс" означает соединение, имеющее структуру в соответствии с Примером 2, где заместителем в пиперидине является гидроксигексил.
Соединение сравнения терфенадин является неседативным антигистаминным препаратом, в то время как дифенгидоамин является антигистаминным препаратом с побочным седативным эффектом.
Выводы (седативность): результаты подтверждают клинические эффекты соединений сравнения, подтверждая таким образом объективность методологии. Кетотифен имеет больший седативный эффект, чем даже дифенгидрамин, тем самым подтверждая побочные седативные эффекты этого соединения. За исключение соединений, обозначенных Пример 2/ОН-пент и Пример 2/ОН-гекс, все новые соединения не имеют седативного эффекта.
Противовоспалительные эффекты: легочные воспаления
Сенсибилизированным мужским особям морских свинок Данкин-Хартли весом 400-600 г вводили интраперитонеально 10 мкг PAF (фактор агрегации тромбоцитов) в 0,25% растворе сывороточного альбумина коровы (BSA) в физиологическом растворе. Через двадцать четыре часа животных умерщвляют внутрибрюшинной инъекцией барбитурата и трахею экспонируют и канюлируют. Последовательно вводили аликвоты буферного модифицированного раствора Тирода (мМ: NаНСО3 11,9; Na2HPO4 0,4; KCl 2,7; NaCl 136,9; ЭДТА 19,8; глюкоза 5,6; желатин 0,1% вес/объем; BSA 0,5% вес/объем; рН до 7,4) и аспирировали с помощью мягкой компрессии легких. Общее выделение жидкости обычно превышает 80%. Клеточные суспензии концентрировали с помощью низкоскоростного центрифугирования (200 г в течение 10 мин) и полученный клеточный осадок повторно суспендируют в 1 мл модифицированного раствора Тирода. Подсчет всех клеток производят разбавлением 10 мкл клеточной суспензии в 90 мкл раствора Турка. Дифференциальный подсчет клеток проводят из мазков, фиксированных в метаноле (100%) и окрашенных в красителе Лейхсмана. Всего, по меньшей мере, 500 клеток на мазок подсчитывают при 1000-кратном увеличении для того, чтобы дифференцировать типы клеток. Лекарственные средства вводят в течение 6 дней в виде подкожной инфузии из имплантированного мини-насоса Альза. Экспозиция PAF имеет место после пятидневного периода обработки испытуемыми соединениями.
Результаты испытаний (Легочное воспаление): (Таблица 6)
Выводы (легочное воспаление):
Накопление воспалительных клеток в легких и дыхательных путях пациентов является хорошо установленным признаком астмы. В настоящем испытании накопление легочных эозинофилов было достигнуто внутрибрюшинными инъекциями PAF морским свинкам. Количество эозинофильных клеток увеличивается до примерно 250% от нормального (BSA) уровня. Когда животным вводили кетотифен (1 мкг/кг/день подкожно в течение 6 дней с помощью осмотического мини-насоса), количество эозинофильных клеток снижается до контрольного уровня. Введение соединения из Примера 2 снижает количество эозинофилов даже более эффективно, чем в случае кетотифена, тогда как соединение из Примера 5 в данном испытании не проявляет сколько-нибудь существенного противовоспалительного действия.
Противовоспалительные эффекты: Кожное воспаление
Уши мужских особей мышей обрабатывали 20 мкл 1,0% раствора кретонового масла в ацетоне. Группы мышей усыпляли через установленные промежутки времени после внутрибрюшинного введения исследуемых соединений (10 мкг/кг) и уши взвешивали. Вес ушей записывали и рассчитывали параметры, представленные далее в Таблице 7.
Результаты (Кожное воспаление):
Средний вес ушей контрольных мышей составил 32±2 мкг. Вес ушей животных, обработанных носителем (контроль) или испытуемыми соединениями, представлен ниже в таблице 7.
"Пример 2/ОН-проп" означает соединение структуры, указанной в Примере 2, где заместителем пиперидина является гидроксипропил.
Выводы (Кожное воспаление):
Кетотифен, Пример 2 и "Пример 2/ОН-проп" одинаково активны в качестве противовоспалительных агентов в испытаниях на кожное воспаление. Пример 5 в этом испытании не проявляет каких-либо заметных противовоспалительных эффектов.
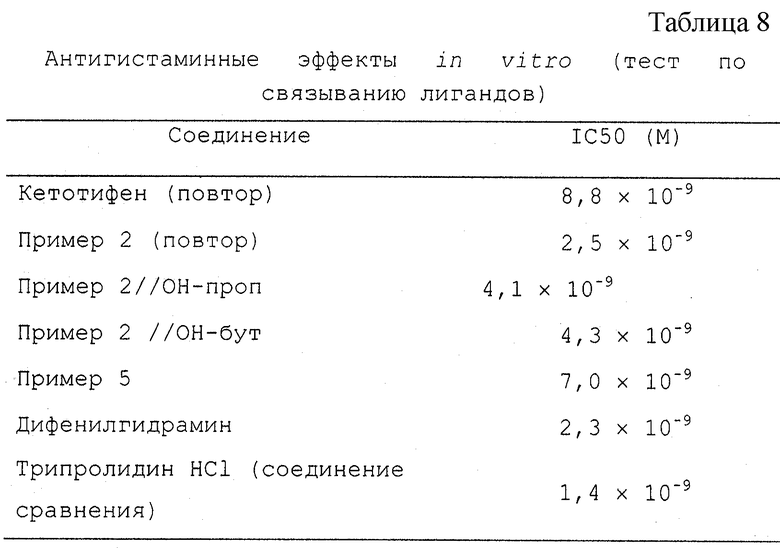
Антигистаминная активность in vitro
Сродство тестируемых соединений по отношению к H1-рецепторам гистамина оценивали с использованием анализа связывания [3Н] пириламина, модифицированного по Чангу и сотр. (Heterogeneity of Histamine H1-Receptors. J.Neurochem. 1979, 32:1653-1663). Кратко, мембраны из коровьего мозжечка инкубировали с [3Н]пириламином и тестируемыми соединениями при увеличении концентраций. Специфическое связывание радиоактивных лигандов с рецептором определяли как разницу между общим связыванием и неспецифическим связыванием, измеренным в присутствии избытка немеченного лиганда. Величины IС50 (концентрация, необходимая для ингибирования 50% специфического связывания [3Н]пириламина) определяли нелинейным регрессионным анализом конкурентных кривых.
Результаты (Антигистаминная активность in vitro): (таблица 8)
"Пример 2/ОН-проп" означает соединение структуры, указанной в Примере 2, где заместителем пиперидина является гидроксипропил. "Пример 2/ОН-бут" означает соединение структуры, указанной в Примере 2, где заместителем пиперидина является гидроксибутил.
Выводы (Антигистаминные активность in vitro):
Все новые соединения ингибируют сайты Н-1 рецепторов. Новые соединения немного менее активны, чем кетотифен, и более активны, чем дифенгидрамин.
Антигистаминная активность in vivo
Спинные волосы мужских особей крыс весом 150-200 г выстригали и депилировали. Примерно через двадцать четыре часа после депиляции животным предварительно вводили перорально (2,0 мл/кг веса тела) испытуемое вещество, носитель или соединение сравнения. Через шестьдесят (60) минут производили две подкожных инъекции дифосфата гистамина (инъекция 50 мкл из 0,6 мкг/мл раствора гистамина ди-НСl). Также производили две подкожные инъекции носителя для раствора гистамина на депилированную поверхность спины. Внутривенно вводили краситель голубой Эванса (20 мкг/кг при 1 мл/кг) за одну минуту до завершения времени предварительной обработки (= одной минуте перед двумя подкожными инъекциями гистамина и двумя подкожными инъекциями носителя гистамина). Двадцать минут отводилось для полного развития отклика на краситель, после чего животных быстро усыпляли асфиксией СO2. Вдоль спины делали разрез и отделяли спинные кожные ткани, содержащие внутрикожный краситель. Площадь участков голубого цвета измеряли в квадратных миллиметрах, оклик из двух опытов усредняли.
В случае животных, обработанных носителем, площадь голубых пятен в данном испытании увеличивалась гистамином от примерно 20 мм2 до примерно 120 мм2. Разница названа "гистаминным эффектом". В случае животных, предварительно обработанных активными исследуемыми веществами, эффект гистамина ниже, чем в случае животных, предварительно обработанных носителем.
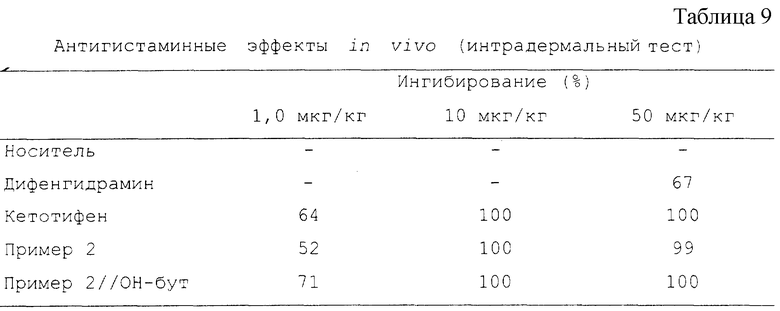
Ингибирование рассчитано в процентах. Результаты представлены в Таблице 9.
"Пример 2/ОН-бут" означает соединение структуры, описанной в Примере 2, где пиперидин замещен гидроксибутилом.
Выводы (Антигистаминная активность in vivo):
Кетотифен, Пример 2 и Пример 2//ОН-бут хорошо абсорбировались после перорального введения и в этом испытании продемонстрировали свойства потенциальных антигистаминных соединений. Новые гидроксилированные соединения в этом исследовании проявили одинаковую активность и были примерно в 50 раз активнее, чем дифенгидрамин.
Антигистаминовая активность производных бензоциклогептатиофена in vivo.
Методология. Самцов крыс весом 160-200 г, имеющий волосяной покров на спине, усыпляли и подвергали депиляции коммерческим агентом для депиляции. Четыре опытные площади на спине были помечены несмываемыми чернилами, тщательно избегая при этом области вблизи позвоночника. Все животные были лишены пищи в течение всей ночи и примерно 12 часов после депиляции, животных предварительно обработали перорально (2,0 мл/кг веса тела) испытываемым веществом, наполнителем или контрольным соединением. Таким образом, испытываемые вещества вводили только один раз каждому животному. Спустя 60 минут, две внутрикожные инъекции дифосфата гистамина (50 мкл инъекции 0,6 мг/мл гистамина дифосфата) были введены. Две внутрикожные инъекции наполнителя для гистаминового раствора были также осуществлены. Голубой краситель Эванса (20 мг/кг при 1,0 мл/кг) вводили внутривенно за одну минуту до окончания времени предобработки (= 1 минуте перед двумя внутрикожными инъекциями гистамина и двумя внутрикожными инъекциями наполнителя для гистамина). Двадцать минут давали для на полное развитие ответной реакции в виде пузырьков, затем животных быстро умерщвляли асфиксией двуокисью углерода. Делали разрез вдоль спины и спинную кожу, содержащую внутрикожные пузырьки, отгибали. Помеченные голубым площади измеряли в кв. мм и двукратно измеренные ответы усредняли.
У обработанных наполнителем животных площадь голубого пятна была увеличена гистамином от приблизительно 20 мм2 до приблизительно 120 мм2 в этой экспериментальной системе. Разница была названа как "эффект гистамина". У животных, предобработанных активным исследуемым веществом, эффект гистамина был меньше, чем у животных предобработанных наполнителем. Ингибирование было вычислено в процентах. Испытанные соединения представляли собой контрольные соединения дифенгидрамин (Benadril®) и кетотифен (Zaditen®), производное бензоциклогептатиофена по пункту 1, где -А-В- представляет -СН3-СО- и где R представляет гидроксиэтил (Пример 2) или карбоксиметоксиэтил (Пример 5) (таблица 10).
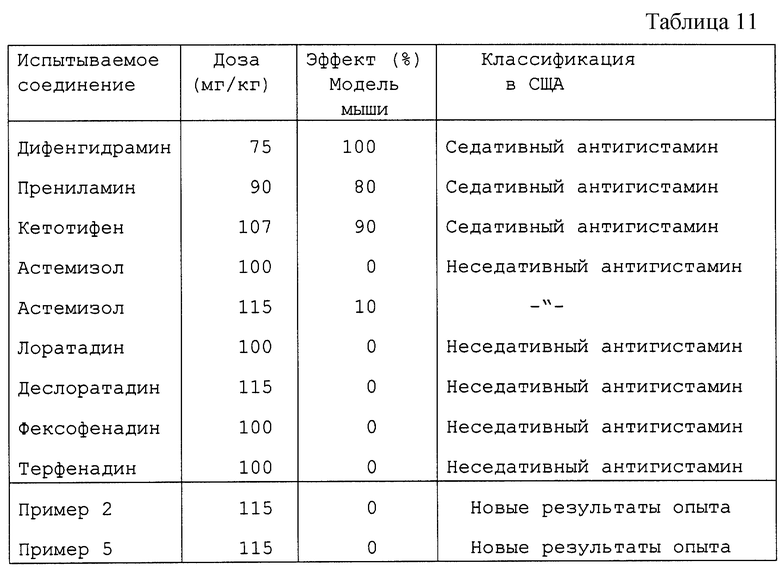
Выводы:
Контрольные соединения дифенгидрамин и кетотифен, новые соединения Примера 2 и Примера 5 все поглощались после перорального введения. Соединения настоящего изобретения (Пример 2 и Пример 5) являются эффективными антигистаминовыми соединениями в этой экспериментальной системе, где воздействия соединений испытывали через 80 минут после перорального введения.
Соединения настоящего изобретения по активности равны кетотифену (наиболее сильному антигистаминовому соединению среди производимых и продаваемых) и были примерно в 50 раз более активными, чем дифенгидрамин (таблица 11).







Патентуются N-замещенные гидроксиалкильные или карбоксиалкилоксиалкильные аналоги 9- и/или 10-оксо-4Н-бензо[4,5]циклогепта [1,2-b]тиофеновых соединений или их 9-ОН- и/или 10-ОН-замещенные аналоги формулы I, где R - гидрокси С2-6, алкил, карбокси C1-6 алкокси-С1-6 алкил, которые обладают антигистаминными и антиастматическими свойствами с пониженными седативными побочными действиями. Описываются также их оптически активные изомеры и фармацевтически приемлемые соли. Соединения, как было установлено, оказывают также профилактическое действие на повышенную реактивность гладких мышц.

-А-В-= -СО-СН2- (а)
-А-В-= -СН2-СО- (b)
-А-В-= -СН2-СН2- (с)
-А-В-= -СНОН-СН2- (d)
-А-В-= -СНОН-СНОН- (e)
-А-В-= -СН2-СНОН- (f)
-А-В-= -СО-СО- (g)
8 с. и 12 з.п. ф-лы, 11 табл.

включая их стереохимически изомерные формы и их фармацевтически приемлемые соли, где R выбран из группы, включающей гидрокси-С2-6алкил и карбокси-С1-6алкилокси-С1-6алкил, и
-А-В- представляет фрагмент, имеющий формулу
-CO-CH2-; (а)
-СН2-СО-; (b)
-СН2-СН2-; (с)
-СНОН-СН2-; (d)
-СНОН-СНОН-; (е)
-СН2-СНОН- или (f)
-СО-СО-. (g)
2. Соединение по п. 1, в котором -А-В- имеет формулу -СН2-СО-, или его фармацевтически приемлемая соль.
| US 4255036, 1982 | |||
| US 5250681, 1993 | |||
| МЕТИЛОВЫЙ ЭФИР 4-ЙОД-2-[N-(N-АЛКИЛАМИНОКАРБОНИЛ)АМИНОСУЛЬФОНИЛ]-БЕНЗОЙНОЙ КИСЛОТЫ, ЕГО ПРОИЗВОДНЫЕ И СПОСОБ ИХ ПОЛУЧЕНИЯ | 1997 |
|
RU2309948C2 |

