УРОВЕНЬ ТЕХНИКИ
[001] Неселективные антагонисты N-метил-D-аспартатного (NMDA) рецептора, изначально разработанные для лечения инсульта и травм головы, как недавно было показано, проявляют клиническую эффективность при лечении депрессии. Было показано, что неселективный антагонист NMDA-рецептора кетамин оказывает быстрое действие и эффективен при депрессии, устойчивой к стандартной терапии ингибиторами обратного захвата моноаминов (Mathews and Zarate, 2013, J. Clin. Psychiatry 74:516-158). Однако неселективные антагонисты NMDA-рецептора, такие как кетамин, имеют ряд нежелательных фармакологических активностей, которые ограничивают их применение для людей. В частности, для неселективных антагонистов NMDA-рецептора особенно характерны диссоциативные или психогенные побочные эффекты. Позже антагонисты NMDA-рецептора, селективно действующие в отношении подтипа NR2B, продемонстрировали возможность их широкого применения для широкого ряда клинических показаний. В частности, антагонисты NR2B также продемонстрировали активность в качестве антидепрессантов в клинических испытаниях ранней стадии (Ibrahim et al., 2012, J. Clin. Psychopharmacol. 32, 551-557; Preskorn et al., 2008, J. Clin. Psychopharmacol. 28, 631-637). Кроме того, селективные антагонисты NR2B имеют преимущества по отношению к неселективным антагонистам NMDA-рецептора, таким как кетамин, ввиду сильного уменьшения диссоциативных побочных эффектов. Однако описанные к настоящему времени антагонисты NR2B в целом демонстрировали недостатки, связанные с другими лекарственными свойствами, что ограничило возможность их применения в лекарственной терапии у людей.
КРАТКОЕ ОПИСАНИЕ
[002] Для широкого и безопасного применения у человека в отношении ряда клинических показаний, включая депрессию, необходимы селективные антагонисты подтипа NR2B. Настоящее изобретение, среди прочего, направлено на удовлетворение потребности в антагонистах рецептора NR2B, которые являются улучшенными в отношении одного или более аспектов, примерами которых являются фармакокинетические характеристики, активность при пероральном введении, безопасность в отношении сердечно-сосудистой системы и показатели терапевтической безопасности, определенные in vitro и in vivo.
[003] В некоторых вариантах реализации настоящее изобретение относится к тому, что было обнаружено, что химические соединения формулы (I):
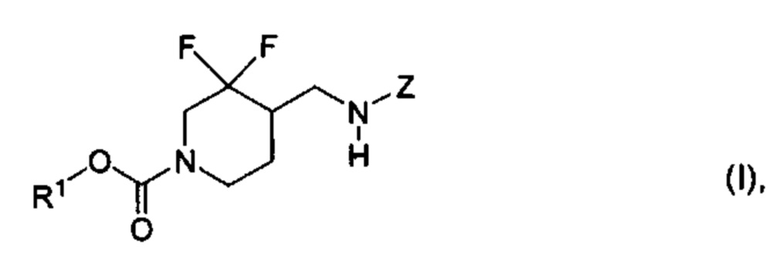
где R1 и Z определены в настоящем описании, являются селективными антагонистами подтипа рецепторов NR2B. Химические соединения формулы (I) и их фармацевтически приемлемые композиции являются подходящими для применения для лечения различных заболеваний и расстройств, связанных с антагонизмом рецепторов NR2B. Такие заболевания и расстройства включают заболевания и расстройства, описанные в настоящем описании.
КРАТКОЕ ОПИСАНИЕ ЧЕРТЕЖЕЙ
[004] На ФИГ. 1А представлены результаты описанного в примере 2.4.1 теста вынужденного плавания для мышей с применением соединения Е1-1.2. На ФИГ. 1В представлены результаты описанного в примере 2.4.2 теста вынужденного плавания для мышей с применением соединения Е1-8.2 при внутрибрюшинном введении. На ФИГ. 1С представлены результаты описанного в примере 2.4.3 теста вынужденного плавания для мышей с применением соединения Е1-21.26 при внутрибрюшинном введении. На ФИГ. 1D представлены результаты описанного в примере 2.4.4 теста вынужденного плавания для мышей с применением соединения Е1-1.2 при пероральном (п/о) введении. На ФИГ. 1D представлены результаты описанного в примере 2.4.5 теста вынужденного плавания для мышей с применением соединения Е1-1.2 при внутрибрюшинном введении. На ФИГ. 1F представлены результаты описанного в примере 2.4.6 теста вынужденного плавания для мышей с применением соединения Е1-21.26 при п/о введении. На ФИГ. 1G представлены результаты описанного в примере 2.4.7 теста вынужденного плавания для мышей с применением соединения Е1-1.2.
[005] На ФИГ. 2 представлены результаты описанного в примере 2.5.1 порогового электроконвульсивного теста (ПЭТ, англ.: electroconvulsive threshold test, ЕСТ) с применением соединения Е1-1.2.
[006] На ФИГ. 3 представлены результаты описанного в примере 2.5.2 порогового электроконвульсивного теста с применением соединения Е 1-8.2.
[007] На ФИГ. 4 представлены результаты описанного в примере 2.5.3 порогового электроконвульсивного теста с применением соединения Е1-21.26.
[008] На ФИГ. 5А представлено число животных, демонстрирующих клонические судороги в пентилентетразоловом (ПТЗ) тесте с провокацией припадка с применением соединения Е1-1.2, описанном в примере 2.6.1. На ФИГ. 5В представлено число животных, демонстрирующих тонические судороги в ПТЗ тесте с провокацией припадка с применением соединения Е1-1.2. На ФИГ. 5С представлено число животных, погибших в ПТЗ тесте с провокацией припадка с применением соединения Е1-1.2. На ФИГ. 5D представлена продолжительность латентного периода до начала клонических судорог в ПТЗ тесте с провокацией припадка с применением соединения Е1-1.2. На ФИГ. 5Е представлена продолжительность латентного периода до развития тонических судорог в ПТЗ тесте с провокацией припадка с применением соединения Е1-1.2. На ФИГ. 5F представлена продолжительность латентного периода до гибели в ПТЗ тесте с провокацией припадка с применением соединения Е1-1.2.
[009] На ФИГ. 6А представлено число животных, демонстрирующих клонические судороги в пентилентетразоловом (ПТЗ) тесте с провокацией припадка с применением соединения Е1-21.26, описанном в примере 2.6.2. На ФИГ. 6В представлено число животных, демонстрирующих тонические судороги в ПТЗ тесте с провокацией припадка с применением соединения Е1-21.26. На ФИГ. 6С представлено число животных, погибших в ПТЗ тесте с провокацией припадка с применением соединения Е1-21.26. На ФИГ. 6D представлена продолжительность латентного периода до развития клонических судорог в ПТЗ тесте с провокацией припадка с применением соединения Е1-21.26. На ФИГ. 6Е представлена продолжительность латентного периода до развития тонических судорог в ПТЗ тесте с провокацией припадка с применением соединения Е1-21.26 На ФИГ. 6F представлена продолжительность латентного периода до гибели в ПТЗ тесте с провокацией припадка с применением соединения Е1-21.26.
[010] На ФИГ. 7А представлена оценка клонических судорог передних конечностей в описанном в примере 2.7.1 тесте с провокацией припадка посредством электрошока с частотой импульсов 6 Гц для соединения Е1-1.2. На ФИГ. 7В представлено число мышей, имеющих «хвост Штрауба» в описанном в примере 2.7.1 тесте с провокацией припадка посредством электрошока с частотой импульсов 6 Гц, в случае применения соединения Е1-1.2.
[011] На ФИГ. 8А представлены результаты для соединения Е1-1.2, полученные в модели каталепсии, вызванной галоперидолом, описанной в примере 2.8.1. На ФИГ. 8В представлены результаты для амфетамина, полученные в модели каталепсии, вызванной галоперидолом. На ФИГ. 8С представлены результаты для соединения Е1-21.26, полученные в модели каталепсии, вызванной галоперидолом, описанной в примере 2.8.2.
[012] На ФИГ. 9А представлена формалиновая модель ноцицептивного поведения у крыс в фазе I для соединения Е1-1.2, описанная в примере 2.9. На ФИГ. 9В представлена формалиновая модель ноцицептивного поведения у крыс в фазе II для соединения Е1-1.2, описанная в примере 2.9.
[013] На ФИГ. 10 представлено число потенциалов постоянного тока для соединения Е1-1.2 в модели распространяющейся кортикальной депрессии (мигрени), описанной в примере 2.10.
[014] На ФИГ. 11 представлена шаростержневая модель промежуточного соединения (R)-XVIa, демонстрирующая схему нумерации, используемую в таблицах A-D.
КРАТКОЕ ОПИСАНИЕ НЕКОТОРЫХ ВАРИАНТОВ РЕАЛИЗАЦИИ
Общее описание химических соединений
[015] В некоторых вариантах реализации настоящего изобретения предложены химические соединения формулы I:
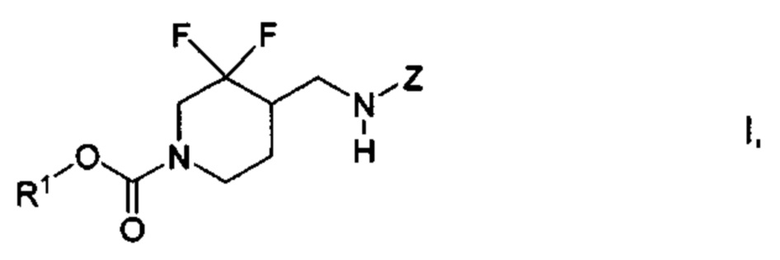
где:
R1 представляет собой водород, алкил, циклоалкил, (циклоалкил)алкил, гетероциклил, (гетероциклил)алкил, арил, (арил)алкил, гетероарил или (гетероарил)алкил,
где каждый из циклоалкила, (циклоалкил)алкила, гетероциклила, (гетероциклил)алкила, арила, (арил)алкила, гетероарила и (гетероарил)алкила независимо необязательно замещен 1-3 группами, независимо выбранными из -F, -Cl, С1-С4 алкила, циклопропила, -С≡СН, -CFH2, -CF2H, -CF3, -CF2CH3, -CH2CF3, C1-C4 алкокси, -OCFH2, -OCF2H, -OCF3, -CN, -N(R2)(R3), -NO2, C1-C4 алкилтио, C1-C4 алкилсульфонила и -S(O)2CF3;
где каждый R2 и R3 в каждом случае независимо -Н или С1-С4 алкил или -N(R2)(R3) представляет собой 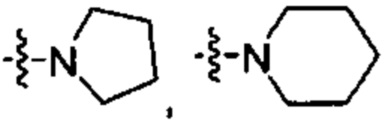 или
или 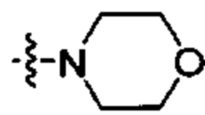 ;
;
Z представляет собой 5- или 6-членный моноциклический или 9- или 10-членный бициклический гетероарил, содержащий кольцевые атомы углерода, 1 кольцевой атом азота и 0-3 дополнительных кольцевых гетероатома, независимо выбранных из N, О и S, где указанный гетероарил необязательно замещен 1 или 2 группами Rx и необязательно замещен 1 группой Ra, где каждая Rx присоединена к кольцевому атому углерода, a Ra присоединена к кольцевому атому азота;
где:
Rx в каждом случае независимо представляет собой -F, -Cl, -СН3, -CFH2, -CF2H, -CF3, -ОН, -ОСН3, -OCF3 или -CN; и
Ra представляет собой C1-4 алкил, С3-4 циклоалкил или -S(O)2-С1-4 алкил.
[016] Если иное не указано или не следует ясно из контекста термин «химическое соединение» относится к соединению, имеющему указанную структуру, в «свободной» форме (например, форме «свободного соединения», или «свободного основания» или «свободной кислоты» в соответствующих случаях) или в форме соли, в частности в форме фармацевтически приемлемой соли, и кроме того в твердой форме или в другой форме. В некоторых вариантах реализации твердая форма представляет собой аморфную (то есть некристаллическую) форму; в некоторых вариантах реализации твердая форма представляет собой кристаллическую форму. В некоторых вариантах реализации предложена кристаллическая форма (например, полиморф, псевдогидрат или гидрат). Аналогичным образом данный термин включает соединение, представленное в твердой форме или в другой форме. Если не указано иное все сделанные в настоящем описании утверждения, касающиеся «соединений», относятся к соответствующим химическим соединениям согласно определению.
Химические соединения и определения
[017] Если не указано иное, предполагается, что слово «включать» (или любой его вариант, например «включает», «включающий» и т.д.) является «открытым» термином. Например, «А включает 1, 2 и 3» означает, что А включает, но не ограничивается ими, 1, 2 и 3.
[018] Если не указано иное, предполагается, что фраза «такой как» является открытой. Например, «А может представлять собой галоген, такой как хлор или бром» означает, что А может представлять собой, но не ограничивается ими, хлор или бром.
[019] Химические соединения согласно настоящему изобретению включают соединения, в общем описанные выше, и дополнительно проиллюстрированы классами, подклассами и видами, раскрытыми в настоящем описании. В контексте настоящего описания должны быть применены следующие определения, если не указано иное. Для целей настоящего изобретения химические элементы идентифицированы в соответствии с Периодической системой элементов, версия CAS, Handbook of Chemistry and Physics, 75е изд., с обложкой, а конкретные функциональные группы в целом определены, как описано в настоящем описании. Кроме того, общие принципы органической химии, а также конкретные функциональные фрагменты и реакционная способность описаны в источниках Thomas Sorrell, Organic Chemistry, University Science Books, Sausalito, 1999; Smith and March, March's Advanced Organic Chemistry, 5th Edition, John Wiley & Sons, Inc., New York, 2001; Larock, Comprehensive Organic Transformations, VCH Publishers, Inc., New York, 1989; и Carruthers, Some Modern Methods of Organic Synthesis, 3rd Edition, Cambridge University Press, Cambridge, 1987.
[020] Термин «алкил», используемый отдельно или в качестве части названия большего фрагмента, означает замещенную или незамещенную, линейную или разветвленную, одновалентную углеводородную цепь, которая является полностью насыщенной или содержит одну или более единиц ненасыщенности. Если не указано иное, алкильные группы содержат от 1 до 7 атомов углерода («С1-С7 алкил»). В некоторых вариантах реализации алкильные группы содержат от 1 до б атомов углерода («С1-С6 алкил»). В некоторых вариантах реализации алкильные группы содержат от 1 до 5 атомов углерода («С1-С5 алкил»). В некоторых вариантах реализации алкильные группы содержат от 1 до 4 атомов углерода («С1-С4 алкил»). В некоторых вариантах реализации алкильные группы содержат от 3 до 7 атомов углерода («С3-С7 алкил»). Примеры насыщенных алкильных групп включают метил, этил, н-пропил, изопропил, н-бутил, m-бутил, изобутил, втор-бутил, гомологи и изомеры, например, н-пентила, н-гексила, н-гептила, н-октила и тому подобное. Ненасыщенная алкильная группа представляет собой группу, имеющую одну или более двойных связей углерод-углерод или тройных связей углерод-углерод. Примеры ненасыщенных алкильных групп включают аллил, винил, 2-пропенил, кротил, 2-изопентенил, 2-(бутадиенил), 2,4-пентадиенил, 3-(1,4-пентадиенил), этинил, 1-и 3-пропинил, 3-бутинил и тому подобное. Термин «низший алкил» относится к алкильным группам, содержащим от 1 до 4 (в случае насыщенности) или от 2 до 4 (в случае ненасыщенности) атомов углерода. Иллюстративные низшие алкильные группы включают метил, этил, н-пропил, изопропил, н-бутил, втор-бутил, изобутил, т-бутил и тому подобное. Термин «алкенил» относится к алкильным группам, содержащем по меньшей мере два атома углерода и по меньшей мере одну двойную связь углерод-углерод. Термин «алкинил» относится к алкильным группам, содержащим по меньшей мере два атома углерода и по меньшей мере одну тройную связь углерод-углерод.
[021] Термин «циклоалкил», используемый отдельно или в качестве части названия большего фрагмента, например «(цикпоалкил)алкила», относится к одновалентному моноциклическому углеводороду, который является полностью насыщенным или содержит одну или более единиц ненасыщенности, но при этом не является ароматическим; или к бицикло[2.2.1]гептанилу (также называемому норборнилом) или бицикло[2.2.2]октанил. В некоторых вариантах реализации циклоалкильные группы содержат 3-8 кольцевых атомов углерода («С3-C8 циклоалкил»). Примеры циклоалкила включают циклопропил, циклобутил, циклопентил, циклогексил, 1-циклогексенил, 3-циклогексенил, циклогептил и тому подобное, а также бицикло[2.2.1]гептанил и бицикло[2.2.2]октанил.
[022] Термин «алкокси», используемый отдельно или в качестве части названия большего фрагмента, относится к группе -O-алкил.
[023] Термин «галоген» или «гало», используемый отдельно или в качестве части названия большего фрагмента, относится к фтору, хлору, брому или иоду.
[024] Термин «арил», используемый отдельно или в качестве части названия большего фрагмента, например «(арил)алкила», относится к одновалентной моноциклической или бициклической карбоциклической ароматической кольцевой системе. Если не указано иное, арильные группы содержат 6 или 10 членов колец. Примеры арила включают фенил, нафтил и тому подобное.
[025] Термин «гетероарил», используемый отдельно или в качестве части названия большего фрагмента, например «(гетероарил)алкила», относится к одновалентной моноциклической или бициклической группе, содержащей от 5 до 10 кольцевых атомов, предпочтительно 5, 6, 9 или 10 кольцевых атомов, имеющей 6, 10, или 14 π-электронов, распределенных в циклической системе, и содержащей кроме атомов углерода от одного до четырех кольцевых гетероатомов. Примеры гетероарильных групп включают тиенил, фуранил, пирролил, имидазолил, пиразолил, триазолил, тетразолил, оксазолил, изоксазолил, оксадиазолил, тиазолил, изотиазолил, тиадиазолил, пиридил, пиридазинил, пиримидинил, пиразинил, индолизинил, пуринил, нафтиридинил, птеридинил и тому подобное.
[026] Термин «гетероциклил», используемый отдельно или в качестве части названия большего фрагмента, например «(гетероциклил)алкила», относится к одновалентому стабильному 5-7-членному моноциклическому или 7-10-членному бициклическому гетероциклическому фрагмента, которое является либо насыщенным, либо частично ненасыщенным, и имеющему кроме атомов углерода от одного до четырех кольцевых гетероатомов. Примеры гетероциклильных групп включают тетрагидрофуранил, пирролидинил, тетрагидропиранил, пиперидинил, морфолинил и тому подобное.
[027] В контексте настоящего описания термин «фармацевтически приемлемая соль» относится к таким солям, которые с точки зрения медицины являются подходящими для контакта с тканями людей и более низших животных без ненадлежащей токсичности, раздражения, аллергической реакции и тому подобного и их применение соответствует целесообразному соотношению польза/риск. Фармацевтически приемлемые соли хорошо известны в данной области техники. Например, S.М. Berge с соавторами подробно описывают фармацевтически приемлемый соли в J. Pharmaceutical Sciences, 1977, 66:1-19, и данная публикация включена в настоящее описание посредством ссылки. Фармацевтически приемлемые соли соединений согласно настоящему изобретению включают соли, полученные из подходящих неорганических и органических кислот и оснований. Примерами фармацевтически приемлемых нетоксичных солей присоединения кислот являются соли, образованные по аминогруппе с неорганическими кислотами, такими как хлористоводородная кислота, бромистоводородная кислота, фосфорная кислота, серная кислота и хлорная кислота, или с органическими кислотами, такими как уксусная кислота, щавелевая кислота, малеиновая кислота, винная кислота, лимонная кислота, янтарная кислота или малоновая кислота, или путем применения других способов, используемых в данной области техники, таких как ионный обмен. Другие фармацевтически приемлемые соли включают соли адипат, альгинат, аскорбат, аспартат, бензолсульфонат, бензоат, бисульфат, борат, бутират, камфорат, камфорсульфонат, цитрат, циклопентанпропионат, диглюконат, додецилсульфат, этансульфонат, формиат, фумарат, глюкогептонат, глицерофосфат, глюконат, гемисульфат, гептаноат, гексаноат, гидроиодид, 2-гидроксиэтансульфонат, лактобионат, лактат, лаурат, лаурилсульфат, малат, малеат, малонат, метансульфонат, 2-нафталинсульфонат, никотинат, нитрат, олеат, оксалат, пальмитат, памоат, пектинат, персульфат, 3-фенилпропионат, фосфат, пивалат, пропионат, стеарат, сукцинат, сульфат, тартрат, тиоцианат, п-толуолсульфонат, ундеканоат, валерат и тому подобное.
[028] Соли, полученные из подходящих оснований, включают соли щелочных металлов, щелочно-земельных металлов, аммония и N+(C1-4 алкил)4. Иллюстративные соли щелочных или щелочно-земельных металлов включают соли натрия, лития, калия, кальция, магния и тому подобное. Другие фармацевтически приемлемые соли включают, в соответствующих случаях, соли нетоксичного аммония, четвертичного аммония и аминных катионов, полученные с противоионами, такими как галогенид, гидроксид, карбоксилат, сульфат, фосфат, нитрат, низший алкилсульфонат и арилсульфонат.
[029] В контексте настоящего описания термин «субъект» включает млекопитающее (например, человека, в некоторых вариантах реализации включая человека на внутриутрбной стадии развития). В некоторых вариантах реализации субъект страдает соответствующим заболеванием, расстройством или состоянием. В некоторых вариантах реализации субъект является восприимчивым к заболеванию, расстройству или состоянию. В некоторых вариантах реализации у субъекта проявляется один или более симптомов или характеристик заболевания, расстройства или состояния. В некоторых вариантах реализации у субъекта не проявляется никакой симптом или характеристика заболевания, расстройства или состояния. В некоторых вариантах реализации субъект имеет одну или более отличительных характеристик восприимчивости или риска заболевания, расстройства или состояния. В некоторых вариантах реализации субъект представляет собой пациента. В некоторых вариантах реализации субъект представляет собой индивидуума, которому проводят или проводили диагностику и/или терапию. В некоторых вариантах реализации субъект представляет собой плода, новорожденного, ребенка, подростка, взрослого или пожилого человека (то есть субъекта пожилого возраста, такого как субъекта старше 50). В некоторых вариантах реализации ребенок представляет собой человека от двух до 18 лет. В некоторых вариантах реализации взрослый представляет собой человека от восемнадцати и старше.
[030] Если не указано иное, предполагается, что структуры, изображенные в настоящем описании, также включают все изомерные (например, энантиомерные, диастереомерные и геометрические (или конформационные)) формы указанных структур; например R и S конфигурации для каждого центра асимметрии, Z- и Е-изомеры по двойной связи, и Z- и Е-конформационные изомеры. Таким образом, отдельные стереохимические изомеры, а также энантиомерные, диастереомерные и геометрические (или конформационные) смеси соединений согласно настоящему изобретению находятся в рамках объема настоящего изобретения. Если не указано иное, все таутомерные формы соединений согласно настоящему изобретению находятся в пределах объема настоящего изобретения. Кроме того, если не указано иное, предполагается, что структуры, изображенные в настоящем описании, также включают соединения, которые отличаются только наличием одного или более изотопно-обогащенных атомов. Например, соединения, имеющие структуры согласно настоящему изобретению, включающие замену водорода, углерода, азота, кислорода, хлора или фтора изотопами 2H, 3H, 11C, 13C, 14C, 13N, 15N, 17O, 18O, 36Cl или 18F, соответственно, находятся в рамках объема настоящего изобретения. Такие соединения являются подходящими для применения, например, в качестве аналитических инструментов, в качестве зондов в биологических исследованиях или в качестве терапевтических агентов в соответствии с настоящим изобретением. Кроме того, включение более тяжелых изотопов, таких как дейтерий (2H), может обеспечить некоторые терапевтические преимущества благодаря большей устойчивости к метаболизму, например, увеличенному периоду полувыведения in vivo, или сниженным требуемым дозировкам.
[031] Диастереомерый избыток выражается в виде % д.и., то есть для диастереомеров X и Y диастереомерный избыток X=((x-y)/(x+y))*100, где x и y представляют собой доли X и Y, соответственно.
[032] Энантиомерный избыток выражатеся как % э.и., то есть для энантиомеров X и Y энантиомерный избыток X=((x-y)/(x+y))*100, где x и y представляют собой доли X и Y, соответственно.
Иллюстративные варианты реализации химических соединений
[033] В некоторых вариантах реализации настоящего изобретения предложены химические соединения формулы (I):
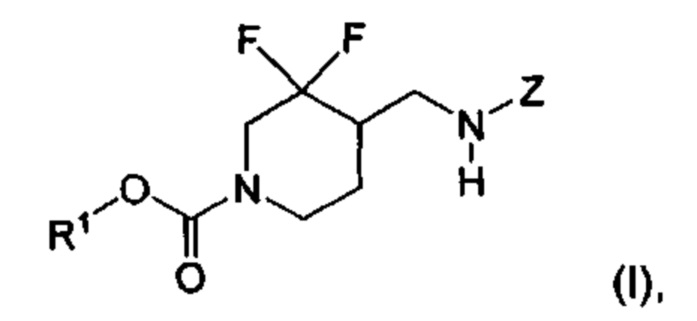
где R1 и Z такие, как описано выше.
[034] В некоторых вариантах реализации R1 представляет собой необязательно замещенный алкил.
[035] В некоторых вариантах реализации R1 представляет собой необязательно замещенный циклоалкил или необязательно замещенный (циклоалкил)алкил. В некоторых вариантах реализации R1 представляет собой необязательно замещенный циклоалкил. В некоторых вариантах реализации R1 представляет собой необязательно замещенный циклогексил. В некоторых вариантах реализации R1 представляет собой циклогексил. В некоторых вариантах реализации R1 представляет собой 4,4-дифторциклогексил. В некоторых вариантах реализации R1 представляет собой 4,4-диметилциклогексил. В некоторых вариантах реализации R1 представляет собой 4-метилциклогексил. В некоторых вариантах реализации R1 представляет собой 4-этилциклогексил. В некоторых вариантах реализации R1 представляет собой 4-циклопропилциклогексил. В некоторых вариантах реализации R1 представляет собой необязательно замещенный норборнанил. В некоторых вариантах реализации R1 представляет собой необязательно замещенный (циклоалкил)алкил. В некоторых вариантах реализации R1 представляет собой бицикло[2.2.1]гептан-2-илметил. В некоторых вариантах реализации R1 представляет собой необязательно замещенный циклогексилметил. В некоторых вариантах реализации R1 представляет собой циклогексилметил. В некоторых вариантах реализации R1 представляет собой (4,4-диметилциклогексил)метил. В некоторых вариантах реализации R1 представляет собой (4,4-дифторциклогексил)метил.
[036] В некоторых вариантах реализации R1 представляет собой необязательно замещенный гетероциклил или необязательно замещенный (гетероциклил)алкил. В некоторых вариантах реализации R1 представляет собой необязательно замещенный гетероциклил. В некоторых вариантах реализации R1 представляет собой необязательно замещенный тетрагидропиранил. В некоторых вариантах реализации R1 представляет собой тетрагидропиран-4-ил. В некоторых вариантах реализации R1 представляет собой необязательно замещенный (гетероциклил)алкил. В некоторых вариантах реализации R1 представляет собой необязательно замещенный тетрагидропиранилметил. В некоторых вариантах реализации R1 представляет собой тетрагидропиран-4-илметил.
[037] В некоторых вариантах реализации R1 представляет собой необязательно замещенный арил или необязательно замещенный (арил)алкил. В некоторых вариантах реализации R1 представляет собой необязательно замещенный (арил)алкил. В некоторых вариантах реализации R1 представляет собой необязательно замещенный бензил. В некоторых вариантах реализации R1 представляет собой 4-метилбензил. В некоторых вариантах реализации R1 представляет собой 4-этилбензил. В некоторых вариантах реализации R1 представляет собой 4-изопропилбензил. В некоторых вариантах реализации R1 представляет собой 4-(2,2,2-трифторэтил)бензил. В некоторых вариантах реализации R1 представляет собой 4-(1,1-дифторэтил)бензил. В некоторых вариантах реализации R1 представляет собой 4-m-бутилбензил. В некоторых вариантах реализации R1 представляет собой 4-хлорбензил. В некоторых вариантах реализации R1 представляет собой 4-фторбензил. В некоторых вариантах реализации R1 представляет собой 4-дифторметилбензил. В некоторых вариантах реализации R1 представляет собой 4-трифторметилбензил. В некоторых вариантах реализации R1 представляет собой 4-дифторметоксибензил. В некоторых вариантах реализации R1 представляет собой 4-трифторметоксибензил. В некоторых вариантах реализации R1 представляет собой 4-метилтиобензил. В некоторых вариантах реализации R1 представляет собой 4-этилтиобензил. В некоторых вариантах реализации R1 представляет собой 4-метилсульфонилбензил. В некоторых вариантах реализации R1 представляет собой 4-этилсульфонилбензил. В некоторых вариантах реализации R1 представляет собой 4-трифторметилсульфонилбензил.
[038] В некоторых вариантах реализации R1 представляет собой необязательно замещенный гетероарил или необязательно замещенный (гетероарил)алкил. В некоторых вариантах реализации R1 представляет собой необязательно замещенный (гетероарил)алкил. В некоторых вариантах реализации R1 представляет собой необязательно замещенный (пиридин-2-ил)метил. В некоторых вариантах реализации R1 представляет собой необязательно замещенный (5-хлорпиридин-2-ил)метил. В некоторых вариантах реализации R1 представляет собой необязательно замещенный (5-метилпиридин-2-ил)метил. В некоторых вариантах реализации R1 представляет собой необязательно замещенный (пиридин-3-ил)метил. В некоторых вариантах реализации R1 представляет собой (5-метилпиридин-3-ил)метил.
[039] В некоторых вариантах реализации Z представляет собой 5- или 6-членный моноциклический или 9- или 10-членный бициклический гетероарил, содержащий кольцевые атомы углерода, 1 кольцевой атом азота и 0-3 дополнительных кольцевых гетероатома, независимо выбранных из N, О и S, при этом указанный гетероарил необязательно замещен 1 или 2 группами Rx и необязательно замещен 1 группой Ra, где каждая Rx присоединена кольцевому атому углерода, a Ra присоединена к кольцевому атому азота.
[040] В некоторых вариантах реализации Z представляет собой 9-членную необязательно замещенную бициклическую гетероароматическую кольцевую систему, содержащую кольцевые атомы углерода, 1 кольцевой атом азота и 0-3 дополнительных кольцевых гетероатома, независимо выбранных из N, О и S.
[041] В некоторых вариантах реализации Z представляет собой 9-членную необязательно замещенную бициклическую гетероароматическую кольцевую систему, содержащую кольцевые атомы углерода, 1 кольцевой гетероатом азота и 1 кольцевой гетероатом кислорода.
[042] В некоторых вариантах реализации Z представляет собой 9-членную необязательно замещенную бициклическую гетероароматическую кольцевую систему, содержащую кольцевые атомы углерода и 2 кольцевых гетероатома азота.
[043] В некоторых вариантах реализации Z представляет собой 9-членную необязательно замещенную бициклическую гетероароматическую кольцевую систему, содержащую кольцевые атомы углерода и 3 кольцевых гетероатома азота.
[044] В некоторых вариантах реализации Z представляет собой 9-членную необязательно замещенную бициклическую гетероароматическую кольцевую систему, содержащую кольцевые атомы углерода и 4 кольцевых гетероатома азота.
[045] В некоторых вариантах реализации Z представляет собой 5- или 6-членную необязательно замещенную моноциклическую гетероароматическую кольцевую систему, содержащую кольцевые атомы углерода, 1 кольцевой атом азота и 0-2 дополнительных кольцевых гетероатома, независимо выбранных из N, О и S.
[046] В некоторых вариантах реализации Z представляет собой 6-членную необязательно замещенную моноциклическую гетероароматическую кольцевую систему, содержащую кольцевые атомы углерода, 1 кольцевой атом азота и 0 или 1 дополнительный кольцевой атом азота. В некоторых вариантах реализации Z представляет собой пиридил. В некоторых вариантах реализации Z представляет собой пиримидинил. В некоторых вариантах реализации Z представляет собой пиридазинил.
[047] В некоторых вариантах реализации Z представляет собой 6-членную необязательно замещенную моноциклическую гетероароматическую кольцевую систему, содержащую кольцевые атомы углерода и 2 кольцевых атома азота. В некоторых вариантах реализации Z представляет собой 6-членную моноциклическую гетероароматическую кольцевую систему, содержащую кольцевые атомы углерода и 2 кольцевых атома азота, при этом Z замещен 1 или 2 группами Rx. В некоторых вариантах реализации Z представляет собой 6-членную моноциклическую гетероароматическую кольцевую систему, содержащую кольцевые атомы углерода и 2 кольцевых атома азота, при этом Z замещен 1 группой Rx. Таким образом, в некоторых вариантах реализации Z представляет собой 6-членную моноциклическую гетероароматическую кольцевую систему, содержащую кольцевые атомы углерода и 2 колцьевых атома азота, при этом Z монозамещен посредством Rx. В некоторых вариантах реализации Z представляет собой пиридил, монозамещенный посредством Rx. В некоторых вариантах реализации Z представляет собой пиримидинил, монозамещенный посредством Rx. В некоторых вариантах реализации Z представляет собой пиридазинил, монозамещенный посредством Rx. В некоторых вариантах реализации Z представляет собой 5-членную необязательно замещенную моноциклическую гетероароматическую кольцевую систему, содержащую кольцевые атомы углерода, 1 кольцевой атом азота и 0-2 дополнительных кольцевых гетероатома, независимо выбранных из N, О и S.
[048] В некоторых вариантах реализации Z представляет собой 5-членную необязательно замещенную моноциклическую гетероароматическую кольцевую систему, содержащую кольцевые атомы углерода, 1 кольцевой атом азота и 0 или 1 дополнительный кольцевой гетероатом, независимо выбранный из N, О и S. В некоторых вариантах реализации Z представляет собой имидазолил или тиазолил. В некоторых вариантах реализации Z представляет собой имидазолил. В некоторых вариантах реализации Z представляет собой тиазолил.
[049] В некоторых вариантах реализации Z представляет собой 5-членную необязательно замещенную моноциклическую гетероароматическую кольцевую систему, содержащую кольцевые атомы углерода, 1 кольцевой атом азота и 0-2 дополнительных кольцевых гетероатома, независимо выбранных из N, О и S. В некоторых вариантах реализации Z представляет собой триазолил, оксадиазолил или тиадиазолил. В некоторых вариантах реализации Z представляет собой триазолил. В некоторых вариантах реализации Z представляет собой оксадиазолил. В некоторых вариантах реализации Z представляет собой тиадиазолил.
[050] В некоторых вариантах реализации Z необязательно замещен 1 или 2 группами Rx и необязательно замещен 1 группой Ra, где каждая Rx присоединена к кольцевому атому углерода, a Ra присоединена к кольцевому атому азота. В некоторых вариантах реализации каждая Rx независимо выбрана из -F, -Cl, -CH3, -CFH2, -CF2H, -CF3, -ОН, -OCH3, -OCF3 или -CN. В некоторых вариантах реализации Rx представляет собой -F или -Cl. В некоторых вариантах реализации Rx представляет собой -F, -Cl или -CN. В некоторых вариантах реализации Rx представляет собой -CH3, -CFH2, -CF2H или -CF3. В некоторых таких вариантах реализации Rx представляет собой -CFH2, -CF2H или -CF3. В некоторых вариантах реализации Rx представляет собой -CH3 или -CF3. В некоторых вариантах реализации Rx представляет собой -ОН, -OCH3 или -OCF3.
[051] В некоторых вариантах реализации каждая Ra независимо выбрана из водорода, C1-4 алкила, С3-4 циклоалкила или -S(O)2-C1-4 алкила. В некоторых вариантах реализации Ra представляет собой С1-4 алкил или С3-4 циклоалкил. В некоторых вариантах реализации Ra представляет собой С1-4 алкил. В некоторых вариантах реализации Ra представляет собой -S(O)2-C1-4 алкил.
[052] В некоторых вариантах реализации Z представляет собой одну из формул Z1-Z36, где Z необязательно замещен 1 или 2 группами Rx, где каждая Rx присоединена к кольцевому атому углерода:
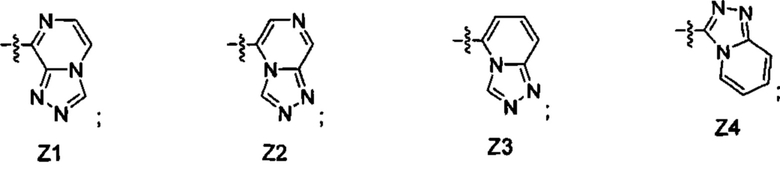
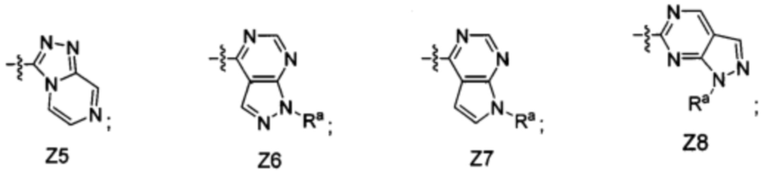

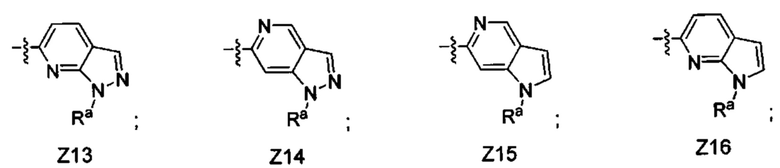
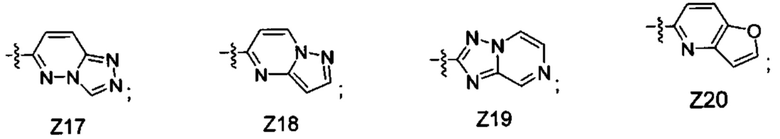
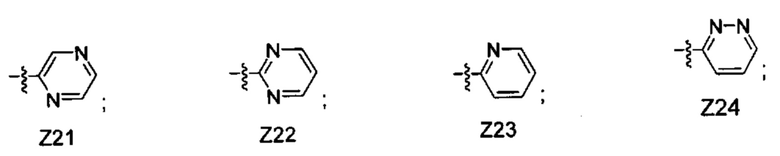
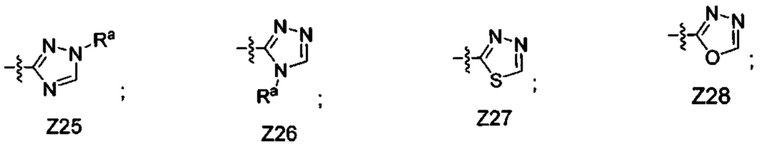

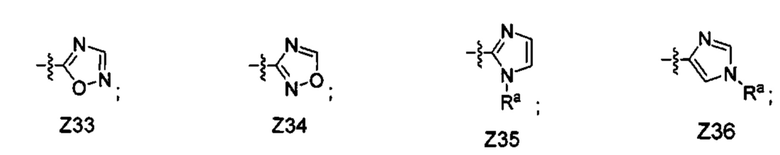
где:
в каждом случае Rx независимо представляет собой -F, -Cl, -CH3, -CFH2, -CF2H, -CF3, -ОН, -ОСН3, -OCF3 или -CN; и
Ra представляет собой водород, C1-4 алкил, С3-4 циклоалкил или -S(O)2-C1-4 алкил.
[053] В некоторых вариантах реализации Z представляет собой Z1, Z2, Z3, Z4, Z5, Z6, Z7, Z8, Z9, Z10, Z11, Z12, Z13, Z14, Z15, Z16, Z17, Z18, Z19, Z20, Z21, Z22, Z23, Z24, Z25, Z26, Z27, Z28, Z29, Z30, Z31, Z32, Z33, Z34, Z35 или Z36.
[054] В некоторых вариантах реализации Z представляет собой Z1, Z2, Z3, Z4, Z5, Z6, Z7, Z8, Z9, Z10, Z11, Z12, Z13, Z14, Z15, Z16, Z17, Z18, Z19 или Z20.
[055] В некоторых вариантах реализации Z представляет собой Z1, Z2, Z5, Z6, Z8, Z17 или Z19. В некоторых вариантах реализации Z представляет собой Z1 или Z2. В некоторых вариантах реализации Z представляет собой Z1. В некоторых вариантах реализации Z представляет собой Z2. В некоторых вариантах реализации Z представляет собой Z6 или Z8. В некоторых вариантах реализации Z представляет собой Z6. В некоторых вариантах реализации Z представляет собой Z8.
[056] В некоторых вариантах реализации Z представляет собой Z3, Z4, Z7, Z9, Z10, Z11, Z12, Z13, Z14 или Z18. В некоторых вариантах реализации Z представляет собой Z7 или Z9. В некоторых вариантах реализации Z представляет собой Z7. В некоторых вариантах реализации Z представляет собой Z9.
[057] В некоторых вариантах реализации Z представляет собой Z15, Z16 или Z20. В некоторых вариантах реализации Z представляет собой Z15 или Z16. В некоторых вариантах реализации Z представляет собой Z15. В некоторых вариантах реализации Z представляет собой Z16. В некоторых вариантах реализации Z равен 20.
[058] В некоторых вариантах реализации Z представляет собой Z21, Z22, Z23, Z24, Z25, Z26, Z27, Z28, Z29, Z30, Z31, Z32, Z33, Z34, Z35 или Z36. В некоторых вариантах реализации Z представляет собой Z21, Z22, Z23 или Z24. В некоторых вариантах реализации Z представляет собой Z25, Z26, Z27, Z28, Z29, Z30, Z31, Z32, Z33, Z34, Z35 или Z36.
[059] В некоторых вариантах реализации Z представляет собой Z23.
[060] В некоторых вариантах реализации Z представляет собой Z21, Z22, Z24, Z29, Z30, Z35 или Z36. В некоторых вариантах реализации Z представляет собой Z21, Z22, Z24, Z35 или Z36. В некоторых вариантах реализации Z представляет собой Z21 или Z22. В некоторых вариантах реализации Z представляет собой Z21. В некоторых вариантах реализации Z представляет собой Z22. В некоторых вариантах реализации Z представляет собой Z29 или Z30.
[061] В некоторых вариантах реализации Z представляет собой Z25, Z26, Z27, Z28, Z31, Z32, Z33 или Z34. В некоторых вариантах реализации Z представляет собой Z25 или Z26. В некоторых вариантах реализации Z представляет собой Z25. В некоторых вариантах реализации Z представляет собой Z26. В некоторых вариантах реализации Z представляет собой Z27, Z31 или Z32. В некоторых вариантах реализации Z представляет собой Z28, Z33 или Z34.
[062] В некоторых вариантах реализации Z представляет собой Z27, Z29, Z30, Z31 или Z32. В некоторых вариантах реализации Z представляет собой Z29 или Z30. В некоторых вариантах реализации Z представляет собой Z27, Z31 или Z32.
[063] В некоторых вариантах реализации Z представляет собой Z28, Z33 или Z34. В некоторых вариантах реализации Z представляет собой Z28.
[064] В некоторых вариантах реализации в каждом случае Rx независимо представляет собой -F, -Cl, -СН3, -CF3 или -CN. В некоторых вариантах реализации в каждом случае Rx независимо представляет собой -СН3 или -CF3.
[065] В некоторых вариантах реализации Ra представляет собой -СН3.
[066] В некоторых вариантах реализации химическое соединение формулы (I) представляет собой химическое соединение формулы (II):

где Z такой, как описано в вариантах реализации формулы (I) выше или описано в вариантах реализации в настоящем описании, как отдельно, так и в комбинации; и где R5, R6 и R7 независимо представляют собой -Н, -F, -Cl, С1-С4 алкил, циклопропил, -C≡CH, -CFH2, -CF2H, -CF3, -CF2CH3, -CH2CF3, С1-С4 алкокси, -OCFH2, -OCF2H, -OCF3, -CN, -N(R2)(R3), -NO2, C1-C4 алкилтио, С1-С4 алкилсульфонил или -S(O)2CF3;
где в каждом случае R2 и R3 независимо представляют собой -Н или С1-С4 алкил
или
-N(R2)(R3) представляет собой 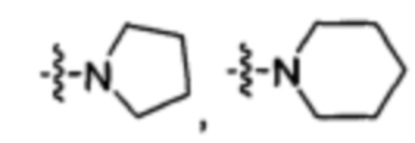 или
или 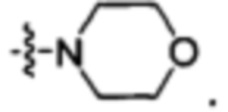
[067] В некоторых вариантах реализации Z выбран из формул Z1-Z36, где: Rx и Ra такие, как описано в вариантах реализации формул Z1-Z36 выше или описаны в вариантах реализации в настоящем описании, как отдельно, так и в комбинации.
[068] В некоторых вариантах реализации предложенное химическое соединение представляет собой химическое соединение формулы (II), где каждый из R5, R6 и R7 независимо представляет собой -Н, -F, -Cl, -СН3, -CFH2, -CF2H, -CF3, -СН2СН3, -CF2CH3, -CH2CF3, изопропил, трет-бутил, циклопропил, -OCF3, -OCF2H, -SCH3, -SCH2CH3, -S(O)2CH3, -S(O)2CH2CH3, -S(O)2CF3 или -C≡CH.
[069] В некоторых вариантах реализации предложенное химическое соединение представляет собой химическое соединение формулы (II), где каждый из R5, R6 и R7 независимо представляет собой -Н, -F, -Cl, -СН3, -CFH2, -CF2H, -CF3, -СН2СН3, -CF2CH3, -CH2CF3, циклопропил, -OCF3, -OCF2H, -SCH3, -S(O)2CH3 или -C≡CH.
[070] В некоторых вариантах реализации предложенное химическое соединение представляет собой химическое соединение формулы (II), где:
R5 представляет собой -Н, -F, -Cl, -СН3, -CFH2, -CF2H, -CF3, -CH2CH3, -CF2CH3, -CH2CF3, циклопропил, -OCF3, -OCF2H, -SCH3, -S(O)2CH3 или -С≡СН;
R6 представляет собой -H или -F; и
R7 представляет собой -Н, -F, -Cl или -СН3.
[071] В некоторых вариантах реализации предложенное химическое соединение представляет собой химическое соединение формулы (II), где каждый из R5, R6 и R7 независимо представляет собой -Н, -F, -Cl, -СН3, -CFH2, -CF2H, -CF3, -СН2СН3, -CF2CH3, -CH2CF3, изопропил, трет-бутил, циклопропил, -OCF3, -OCF2H, -SCH3, -SCH2CH3, -S(O)2CH3, -S(O)2CH2CH3, -S(O)2CF3 или -С≡СН; и Z представляет собой Z1, Z2, Z6, Z7, Z8, Z9, Z21 или Z22. В некоторых вариантах реализации Z представляет собой Z1, Z2, Z8, Z9, Z21 или Z22. В некоторых вариантах реализации Z представляет собой Z1 или Z2. В некоторых вариантах реализации Z представляет собой Z1. В некоторых вариантах реализации Z представляет собой Z2.
[072] В некоторых вариантах реализации предложенное химическое соединение представляет собой химическое соединение формулы (II), где каждый из R5, R6 и R7 независимо представляет собой -Н, -F, -Cl, -СН3, -CFH2, -CF2H, -CF3, -СН2СН3, -CF2CH3, -CH2CF3, циклопропил, -OCF3, -OCF2H, -SCH3, -S(O)2CH3 или -C≡CH; и Z представляет собой Z1, Z2, Z8, Z9, Z21 или Z22. В некоторых вариантах реализации Z представляет собой Z1 или Z2. В некоторых вариантах реализации Z представляет собой Z1. В некоторых вариантах реализации Z представляет собой Z2.
[073] В некоторых вариантах реализации предложенное химическое соединение представляет собой химическое соединение формулы (II), где:
R5 представляет собой -Н, -F, -Cl, -СН3, -CFH2, -CF2H, -CF3, -СН2СН3, -CF2CH3, -CH2CF3, циклопропил, -OCF3, -OCF2H, -SCH3, -S(O)2CH3 или -C≡CH;
R6 представляет собой -H или -F;
R7 представляет собой -Н, -F, -Cl или -СН3; и
Z представляет собой Z1, Z2, Z8, Z9, Z21 или Z22. В некоторых вариантах реализации Z представляет собой Z1 или Z2. В некоторых вариантах реализации Z представляет собой Z1. В некоторых вариантах реализации Z представляет собой Z2.
[074] Указание стереоцентра R означает, что R-изомер присутствует в большем количестве, чем соответствующий S-изомер. Например, R-изомер может присутствовать в энантиомерном избытке 50%, 60%, 70%, 80%, 85%, 90%, 92%, 94%, 96% или 98% по отношению к S-изомеру. Аналогичным образом в синтетических промежуточных соединениях, в которых может быть указано более одного стереоцентра, R-изомер может присутствовать в диастереомерном избытке 50%, 60%, 70%, 80%, 85%, 90%, 92%, 94%, 96% или 98% по отношению к S-изомеру.
[075] Указание стереоцентра как S означает, что S-изомер присутствует в большем количестве, чем соответствующий R-изомер. Например, S-изомер может присутствовать в энантиомерном избытке 50%, 60%, 70%, 80%, 85%, 90%, 92%, 94%, 96% или 98% по отношению к R-изомеру. Аналогичным образом в синтетических промежуточных соединениях, в которых может быть указано более одного стереоцентра, S-изомер может присутствовать в диастереомерном избытке 50%, 60%, 70%, 80%, 85%, 90%, 92%, 94%, 96% или 98% по отношению к R-изомеру.
[076] Указание оптического вращения химической группы означает, что указанный энантиомер присутствует в большем количестве, чем другой энантиомер. Например, (-)-изомер может присутствовать в энантиомерном избытке 50%, 60%, 70%, 80%, 85%, 90%, 92%, 94%, 96% или 98% по отношению к (+)-изомеру. Аналогичным образом (+)-изомер может присутствовать в энантиомерном избытке 50%, 60%, 70%, 80%, 85%, 90%, 92%, 94%, 96% или 98% по отношению к (-)-изомеру.
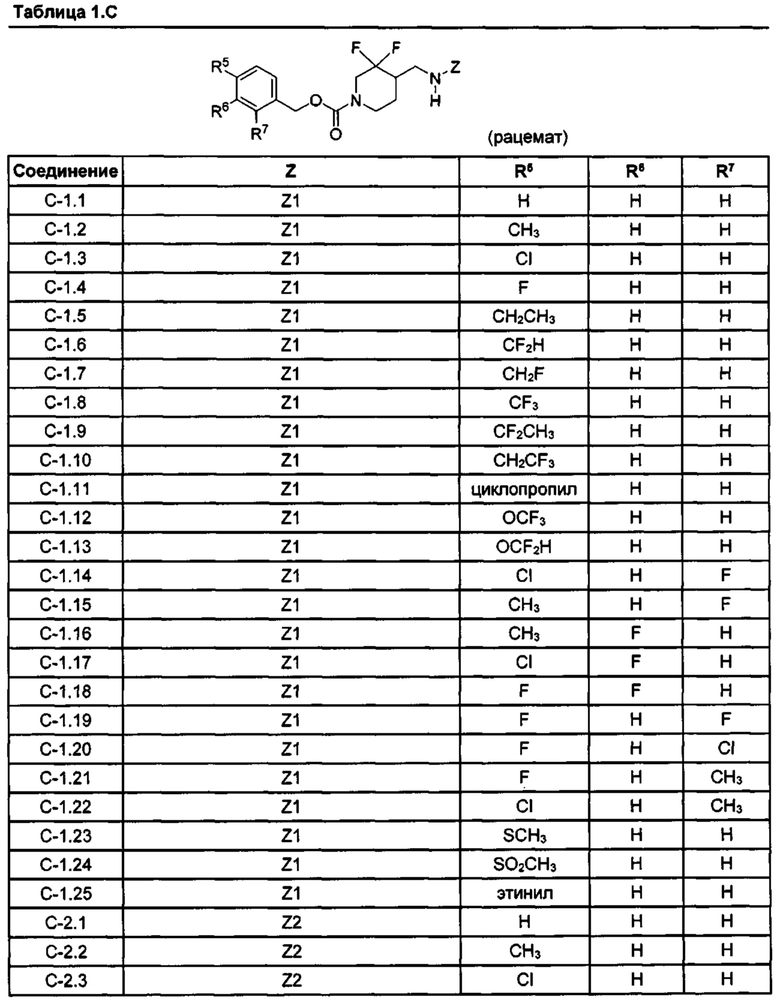
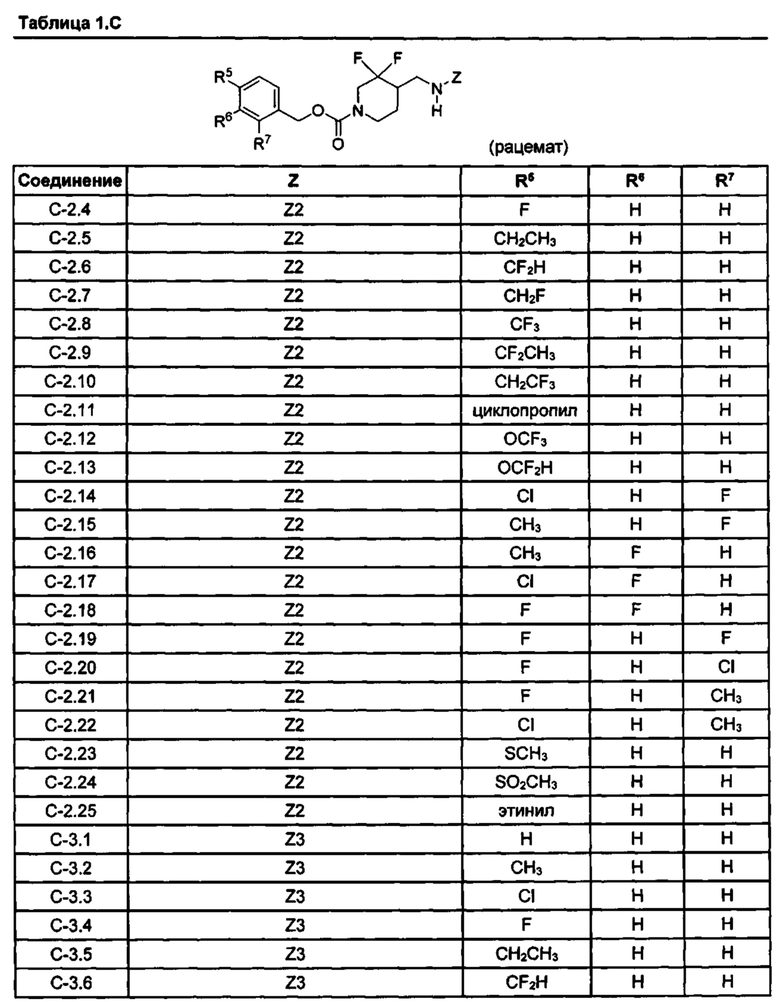
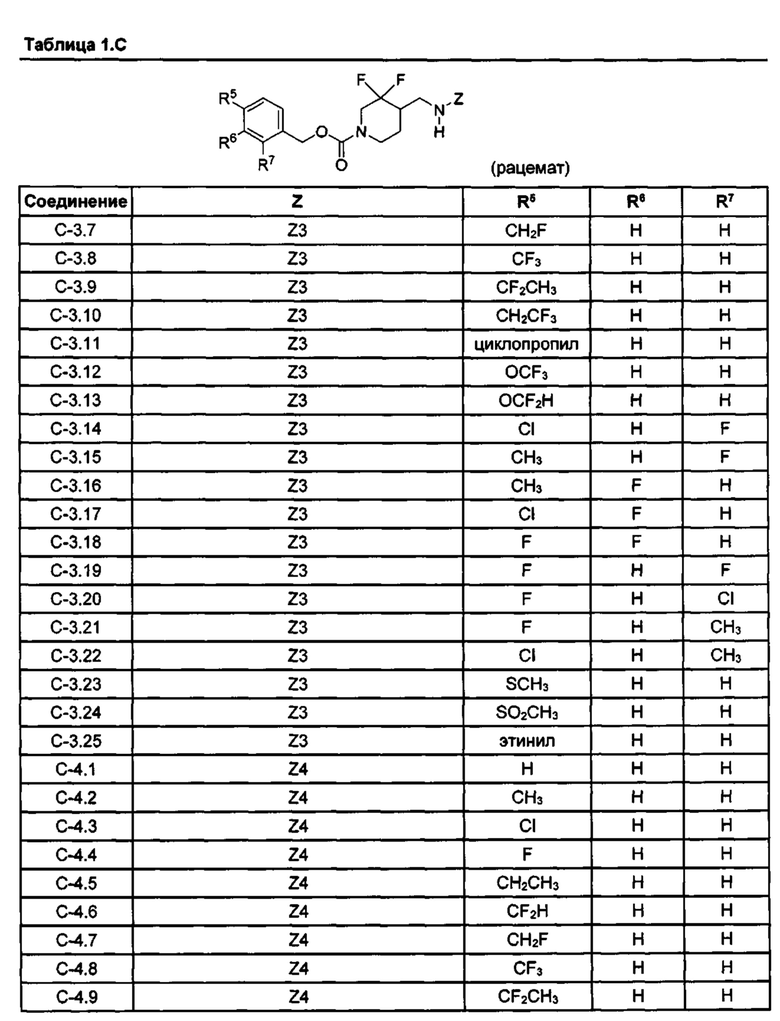
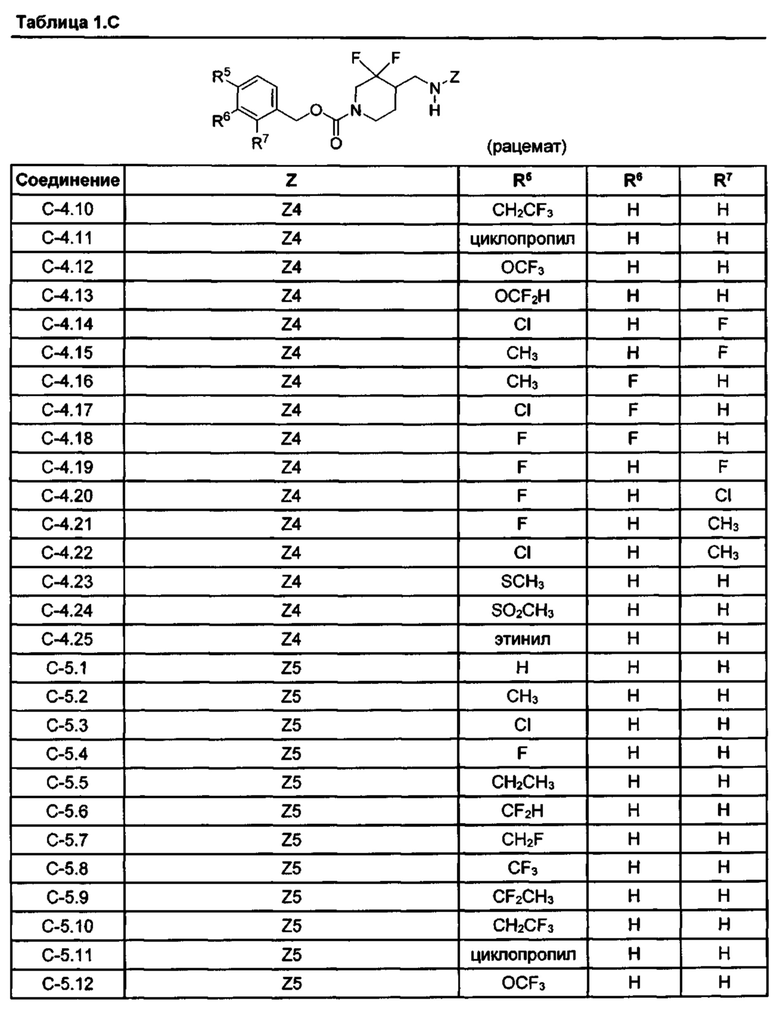
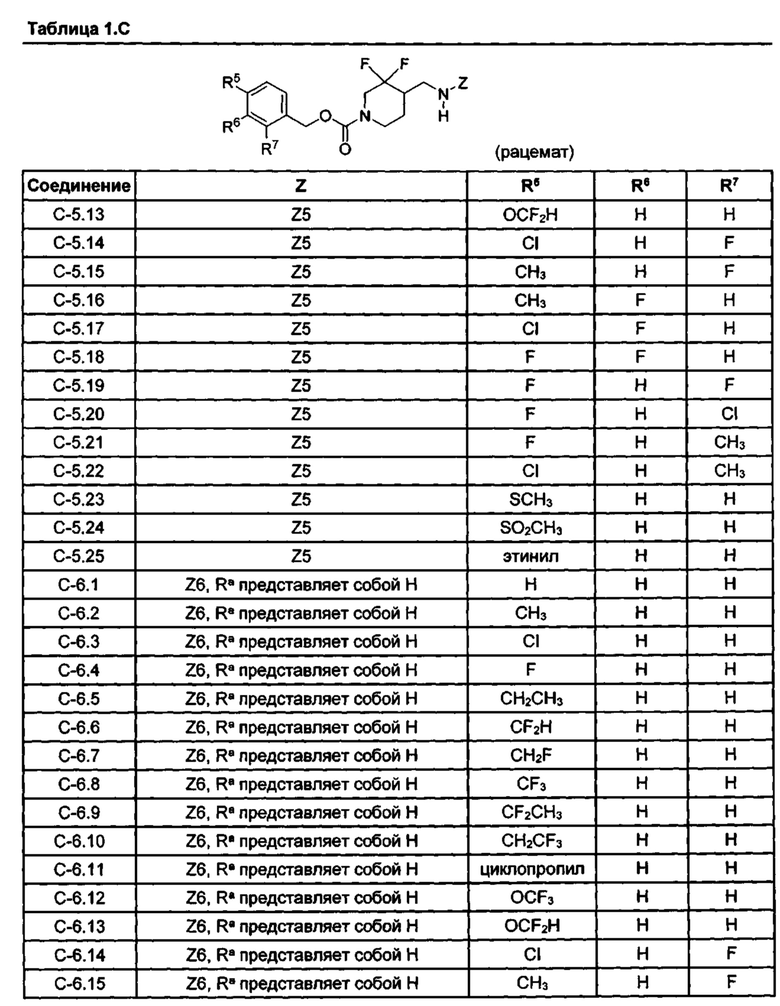
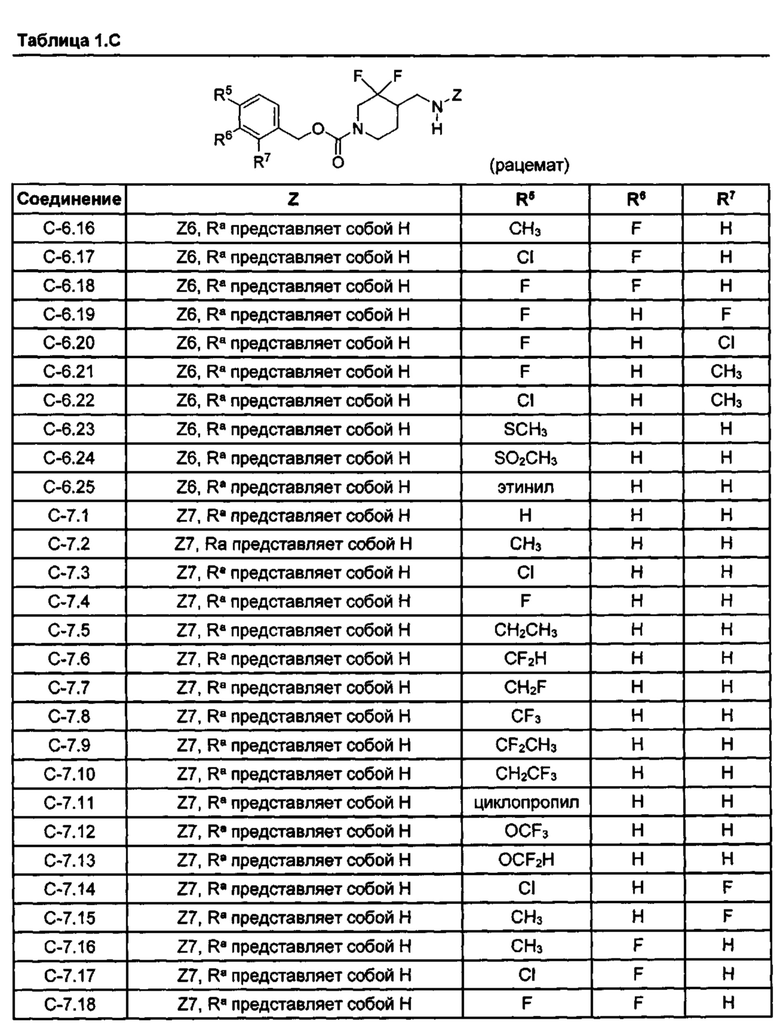
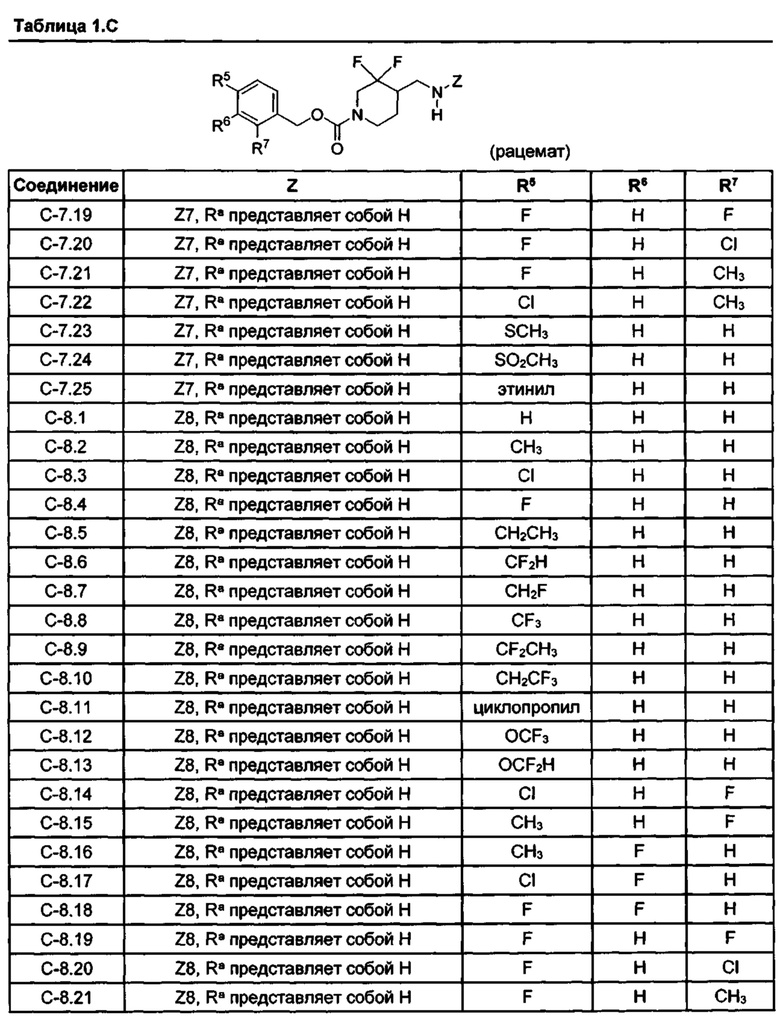
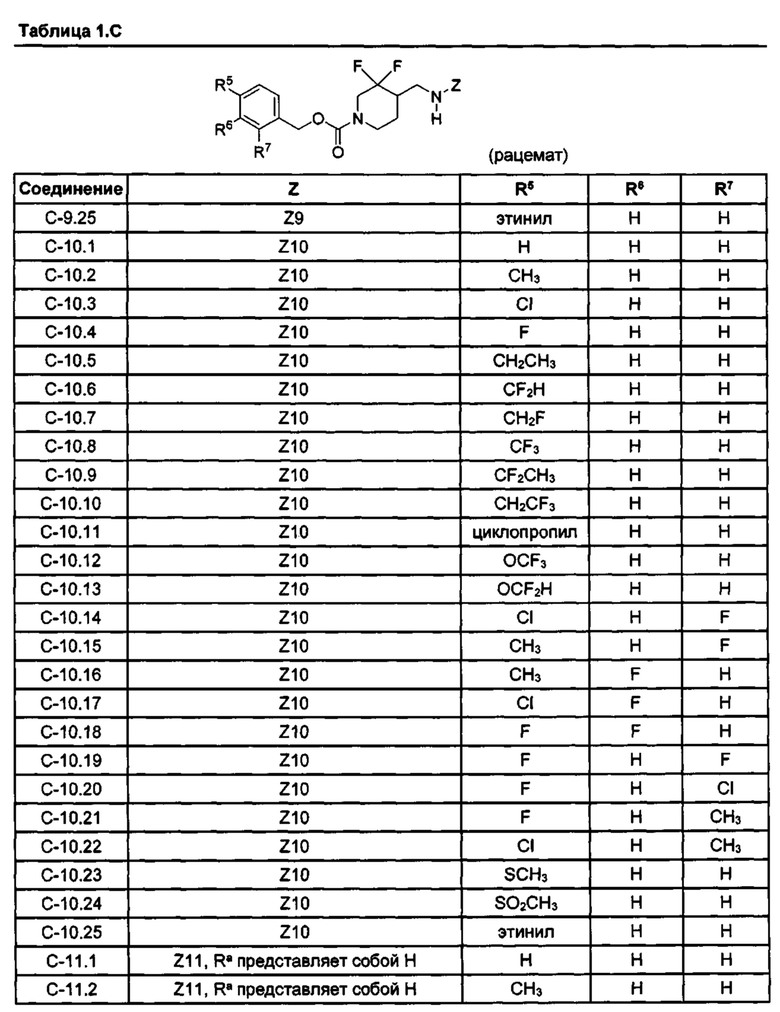
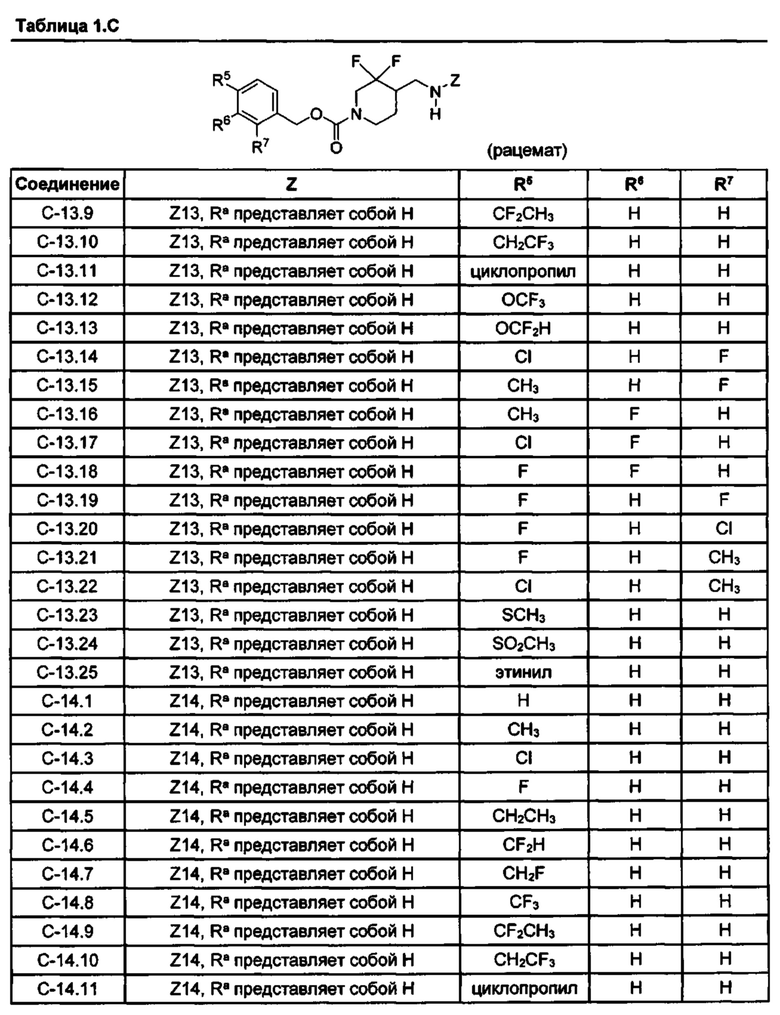
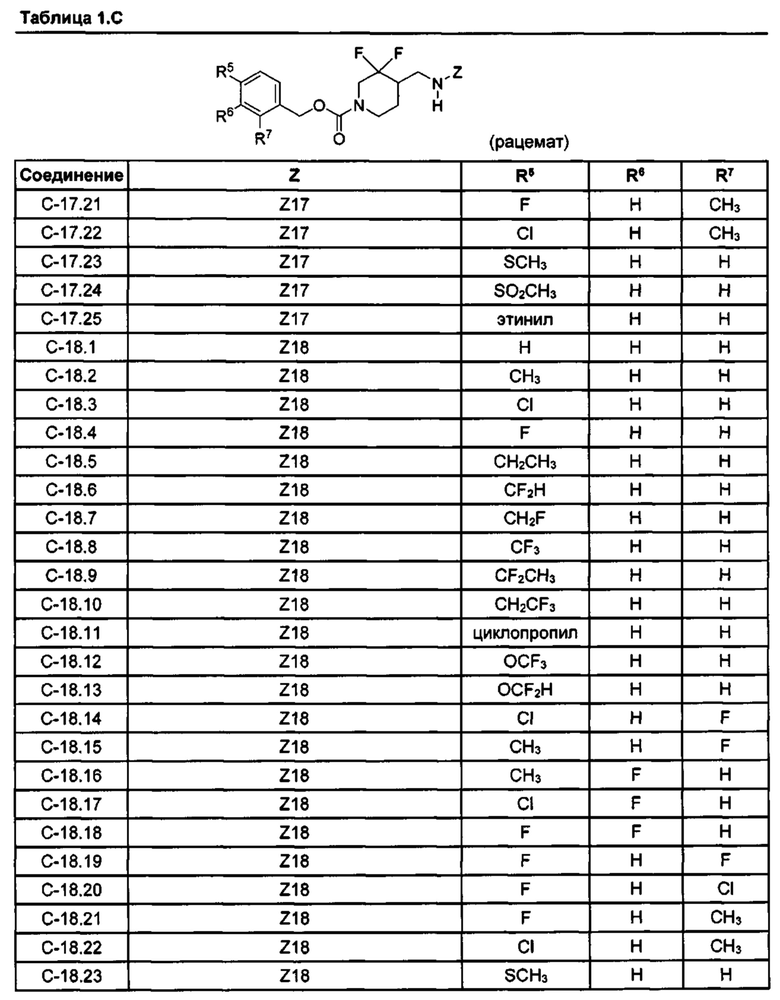
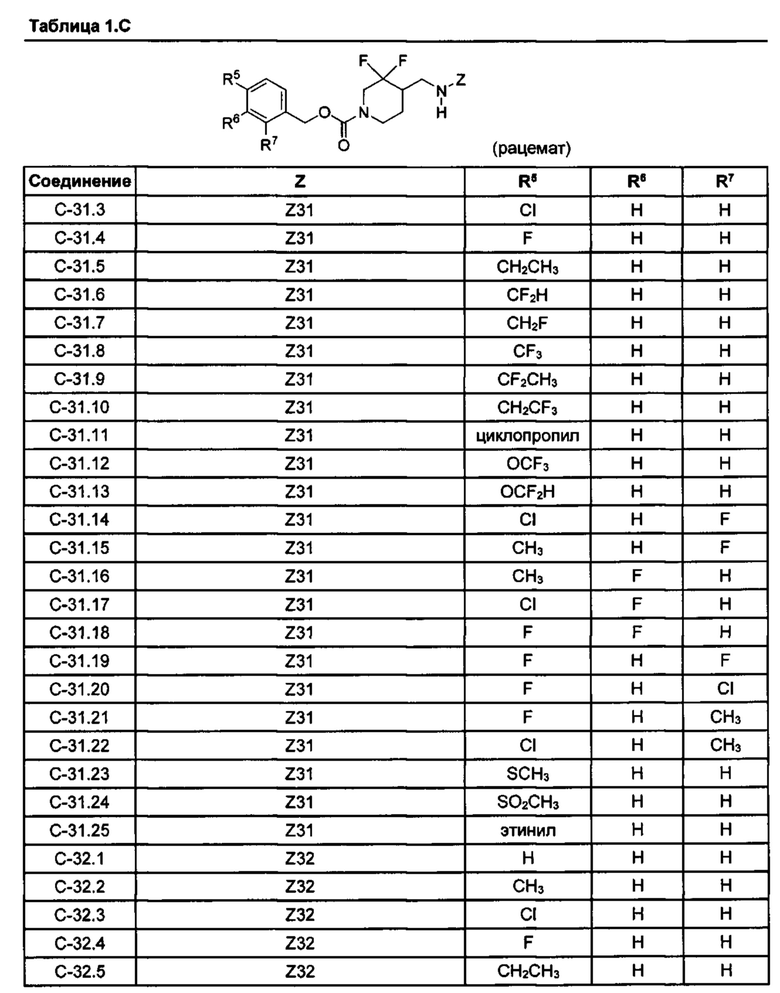
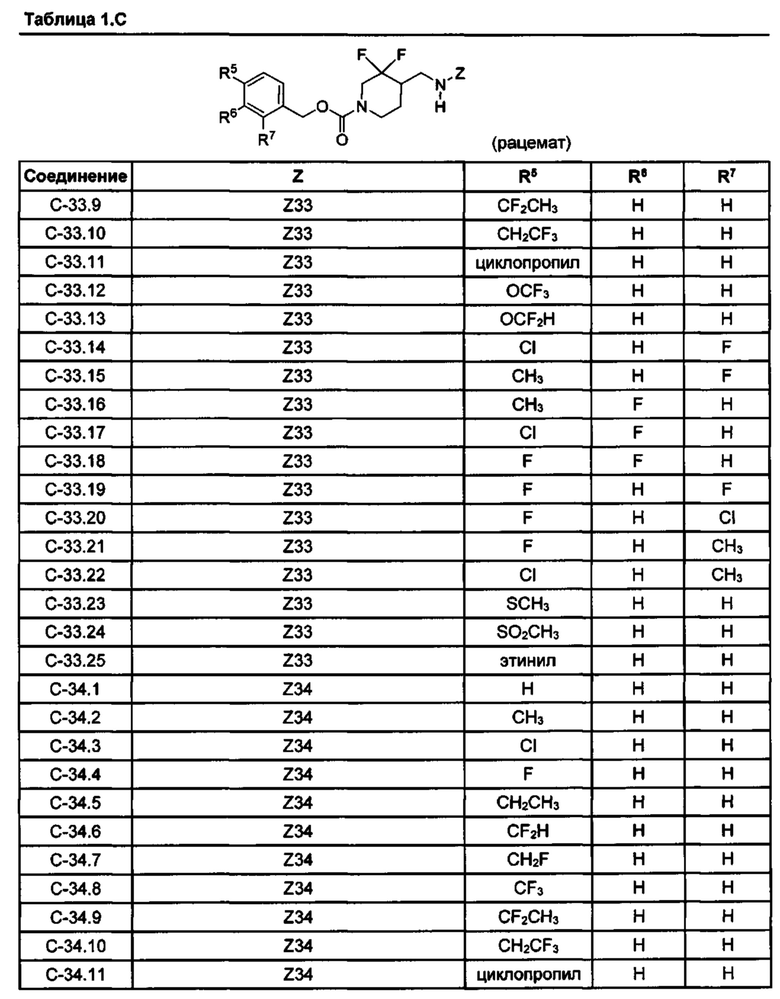
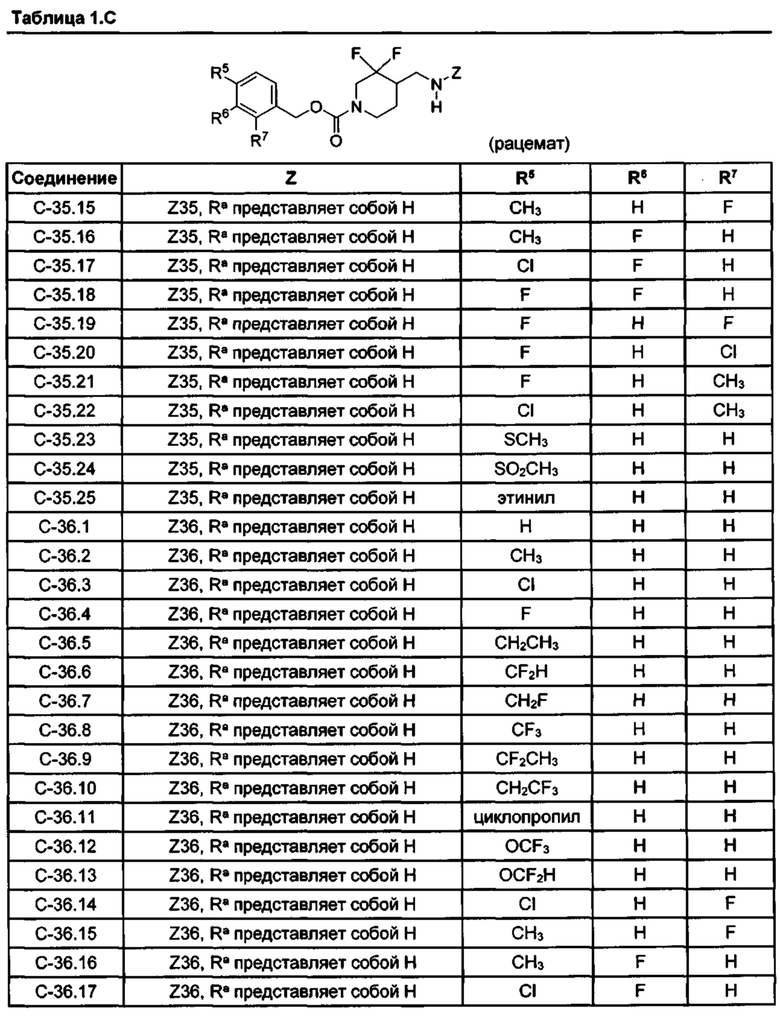
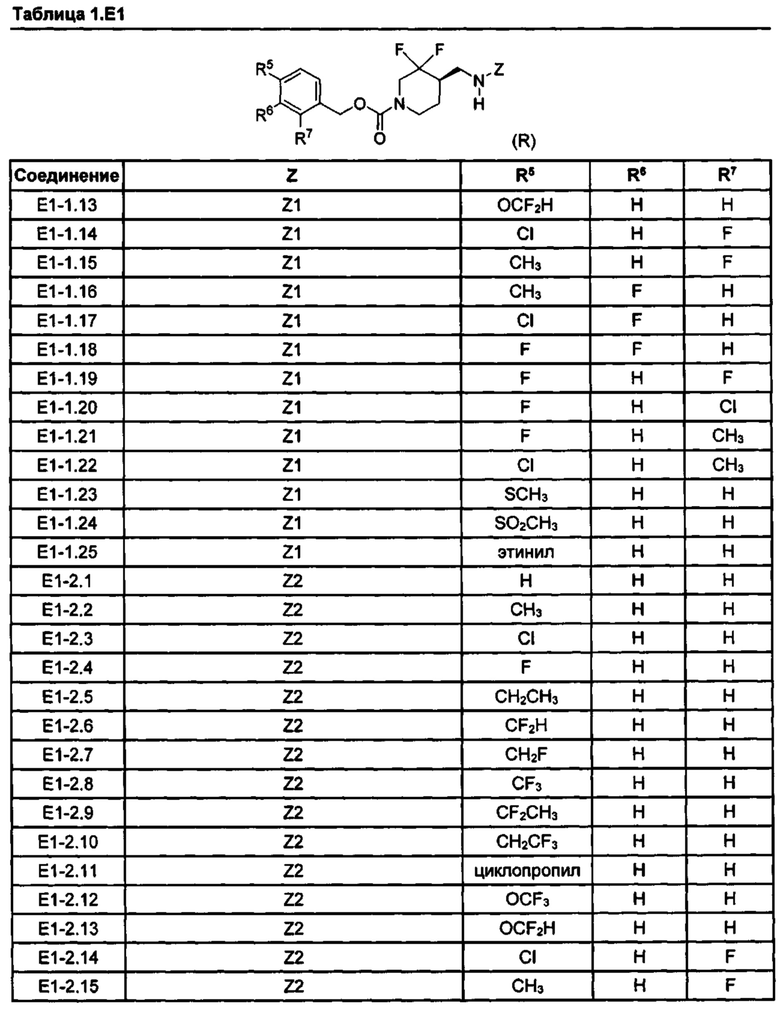
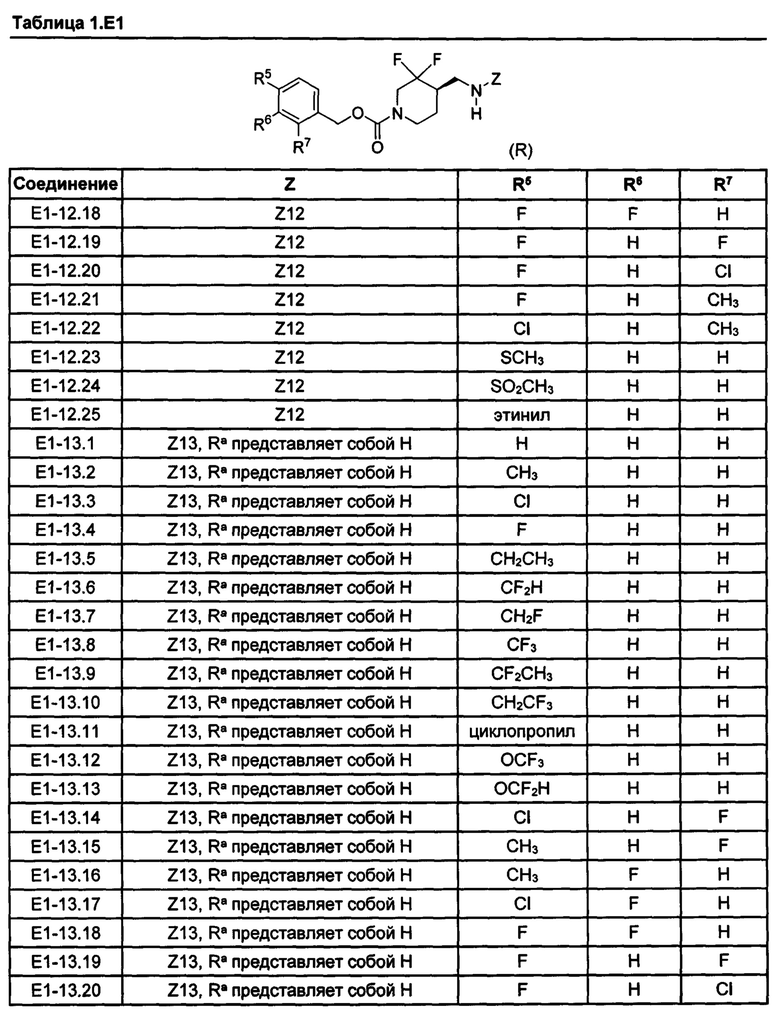
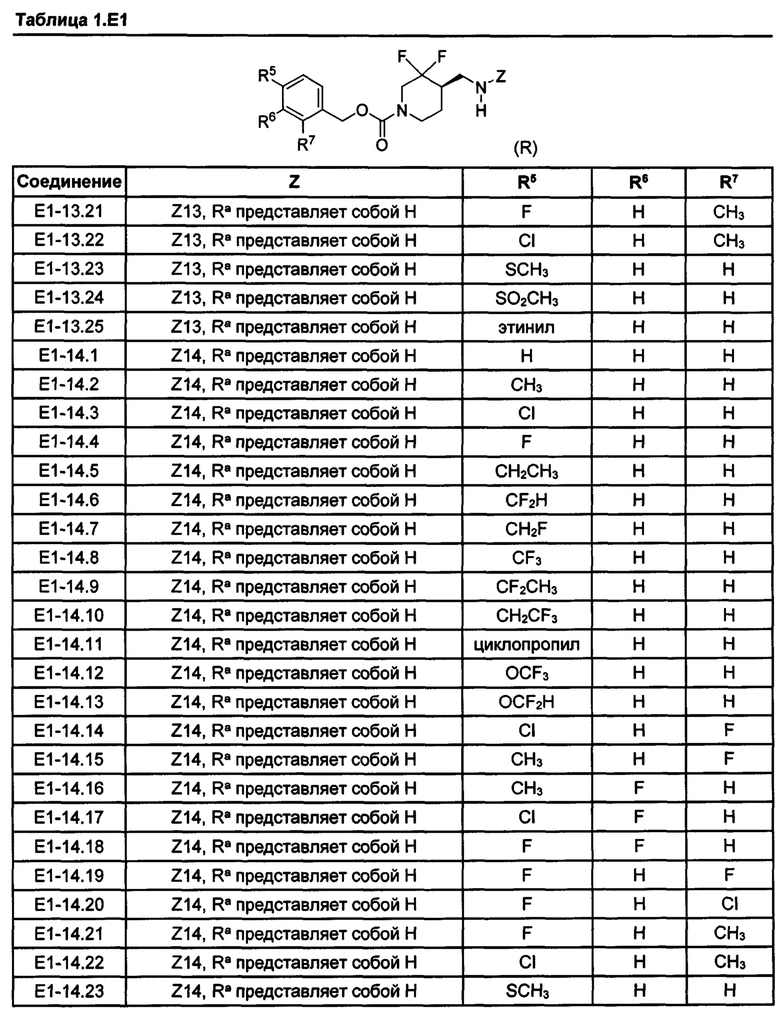
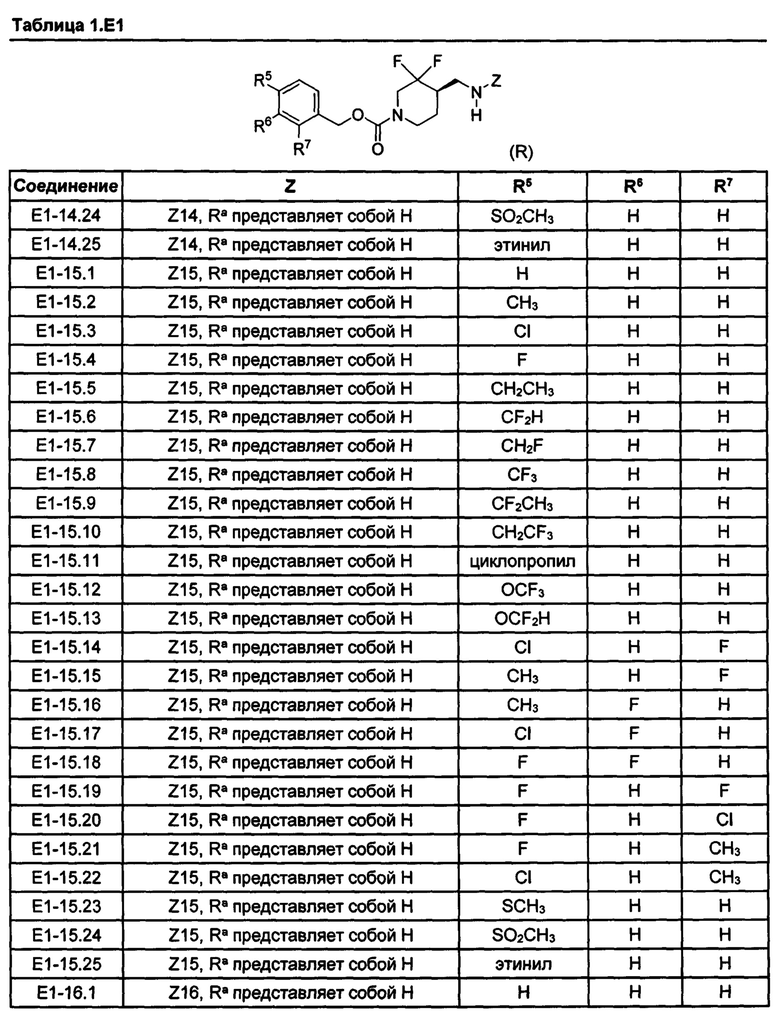
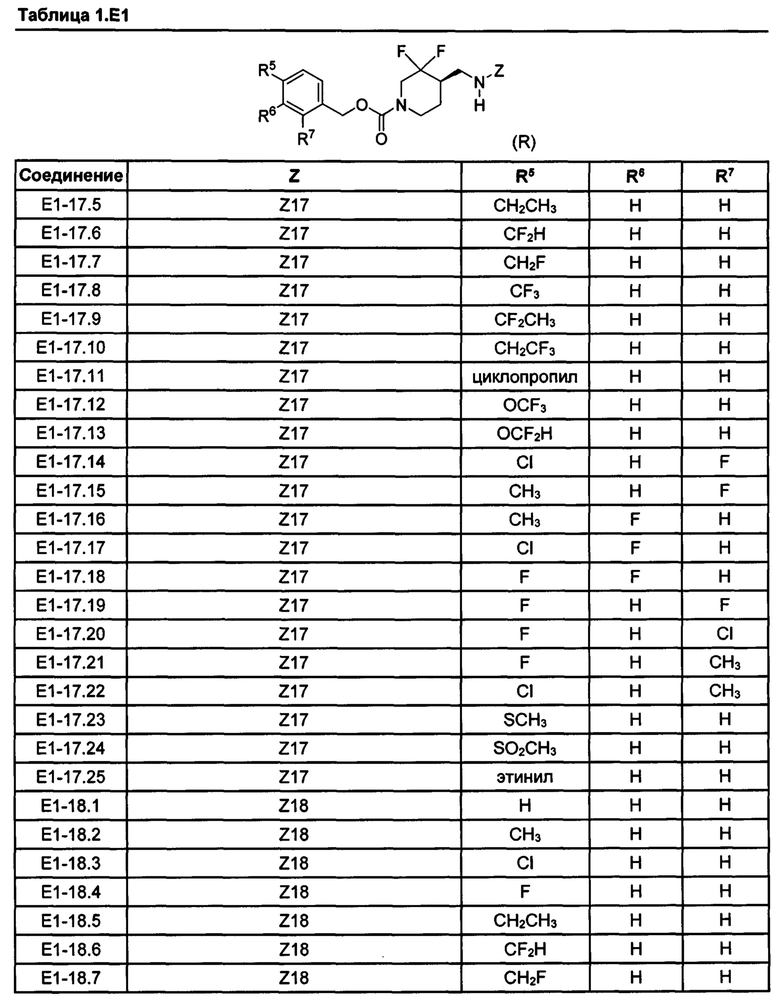
[077] Иллюстративные химические соединения формулы (I) представлены в таблицах 1.С, 1.Е1 и 1.Е2 ниже.







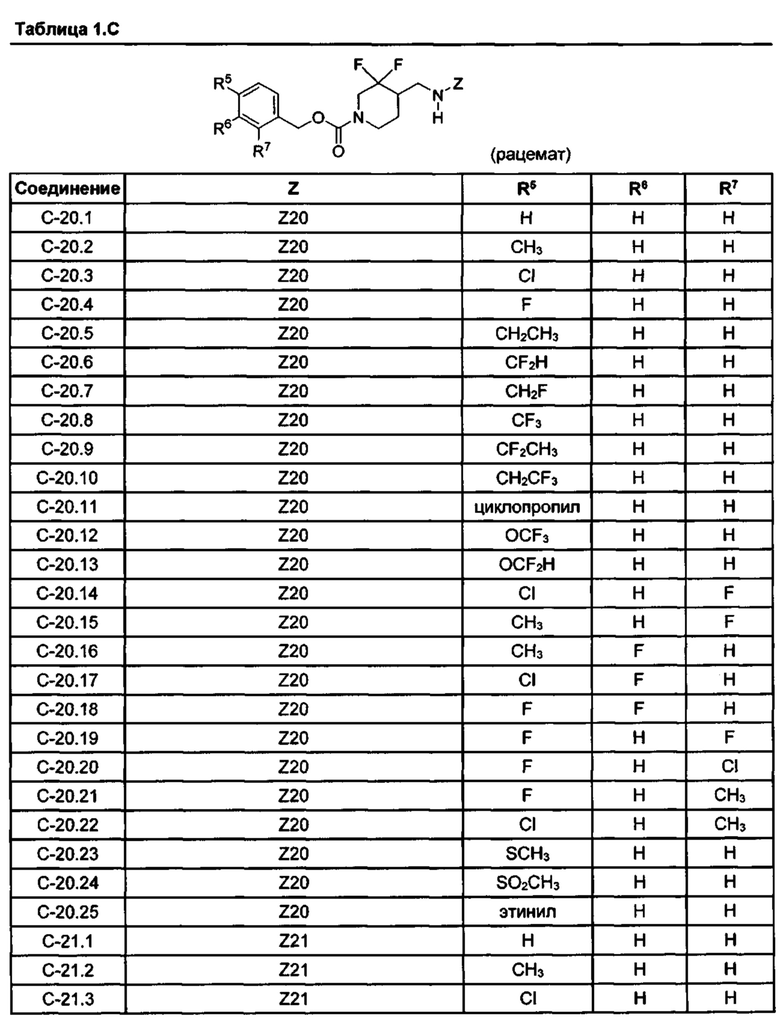


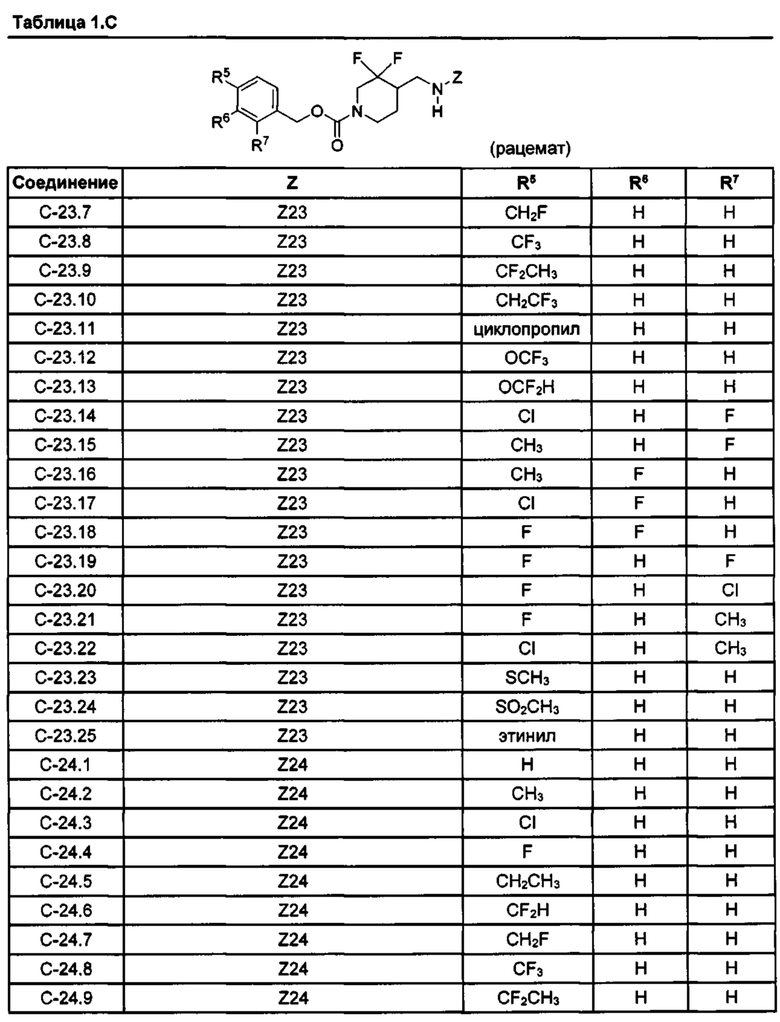
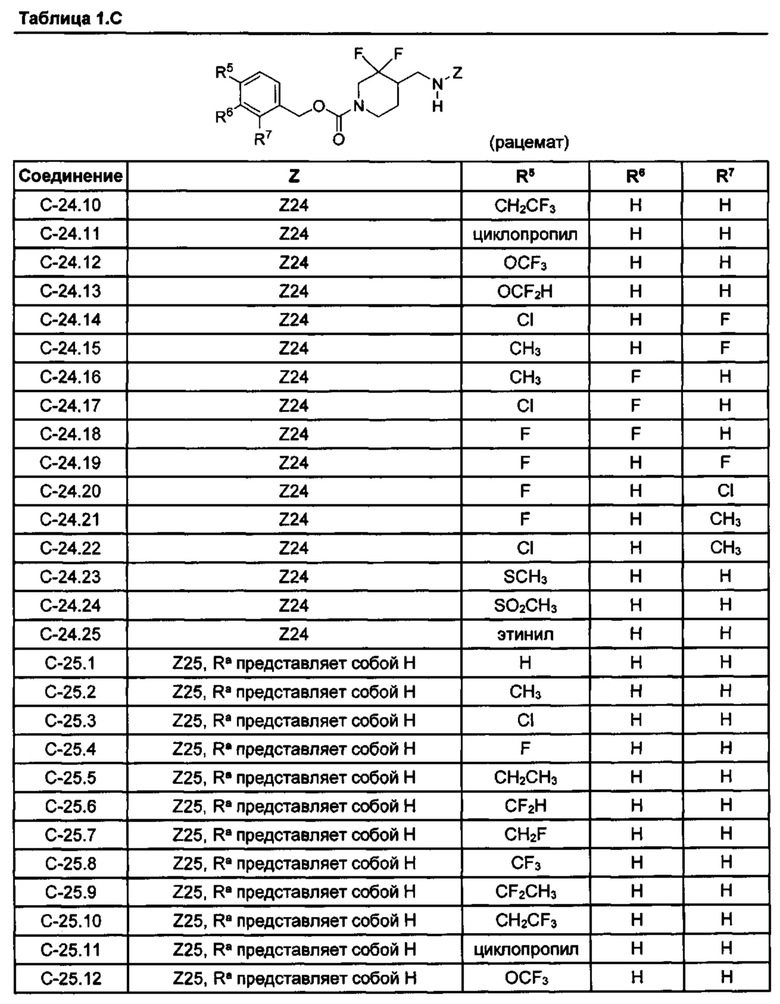



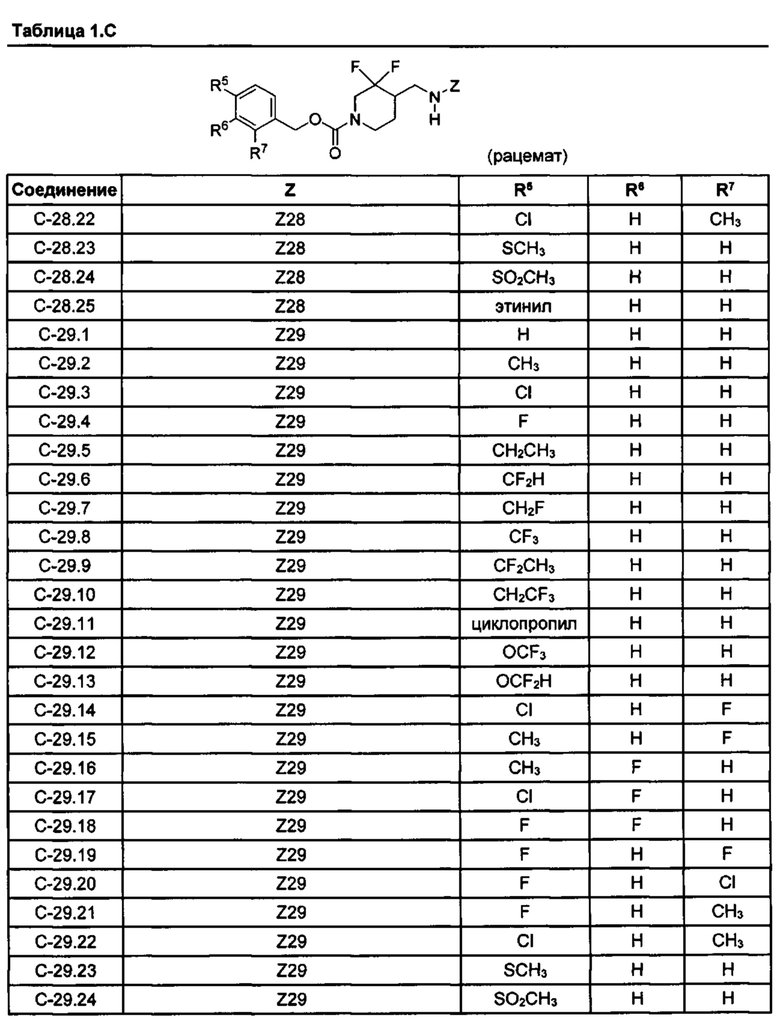



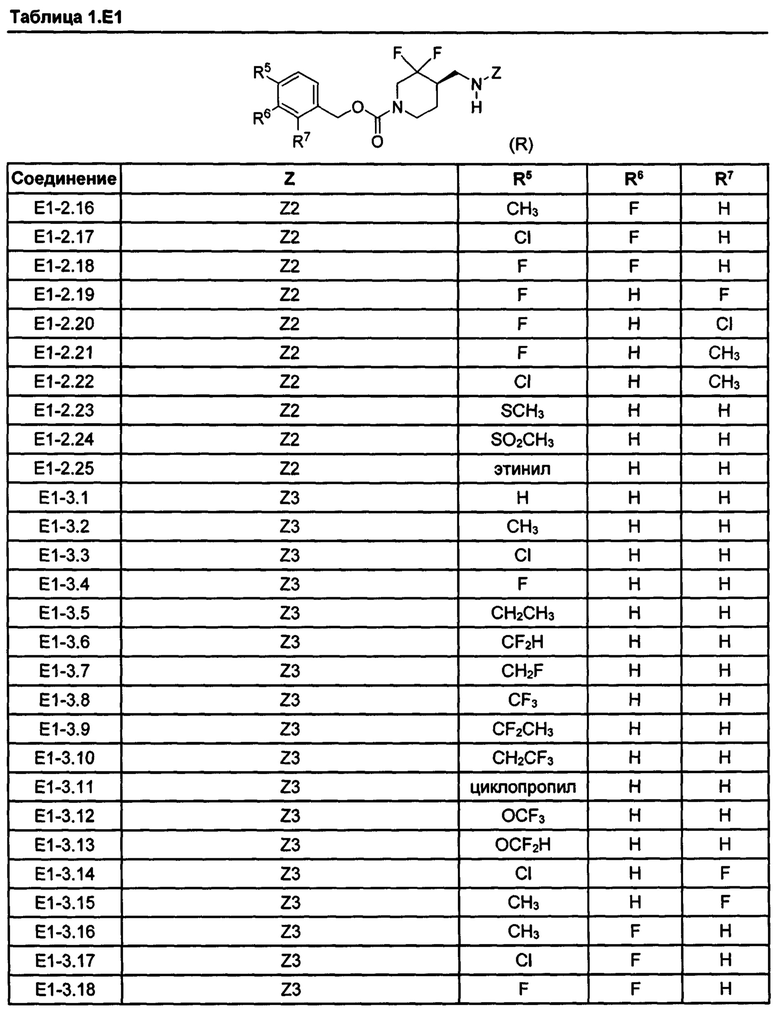



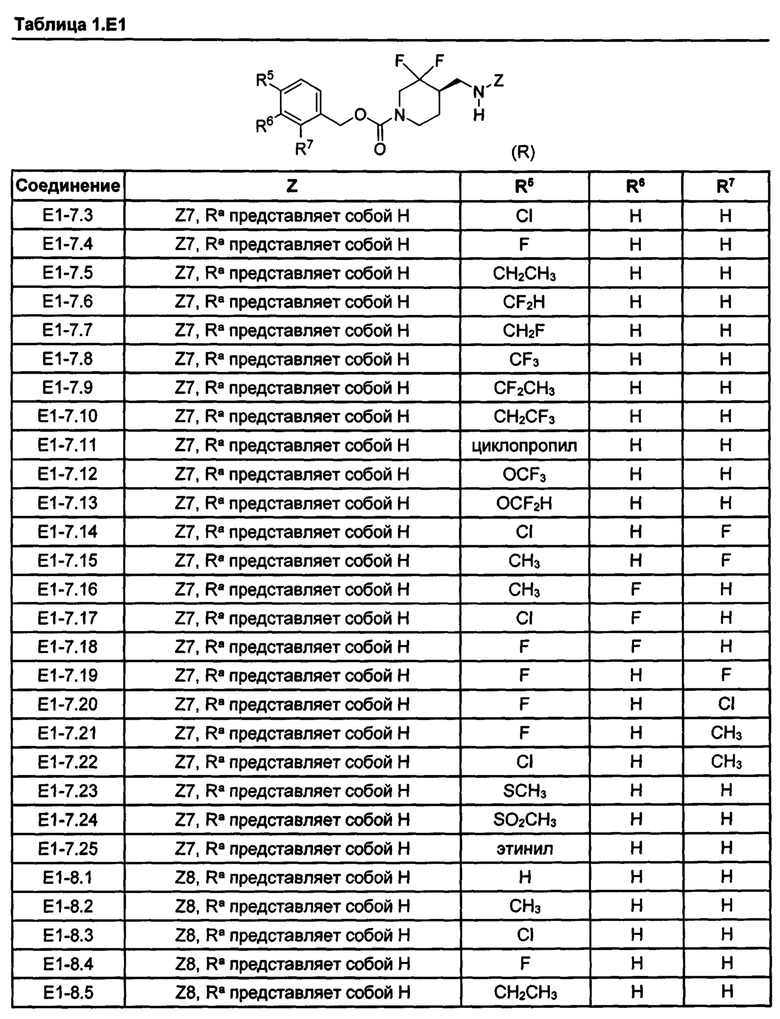

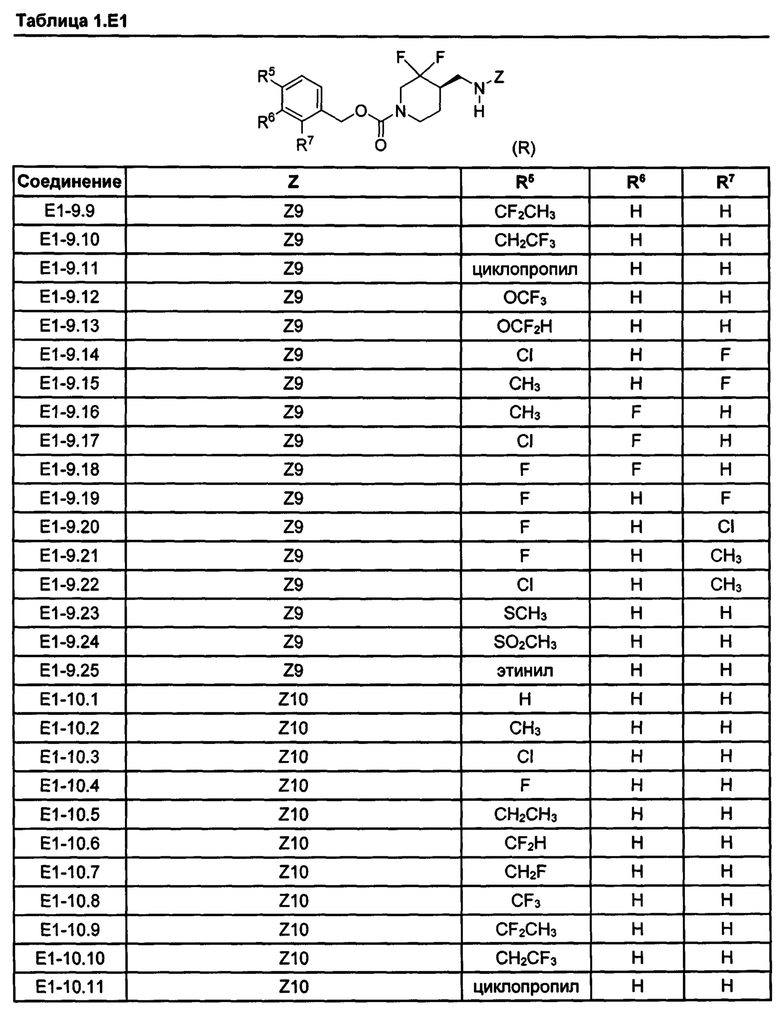

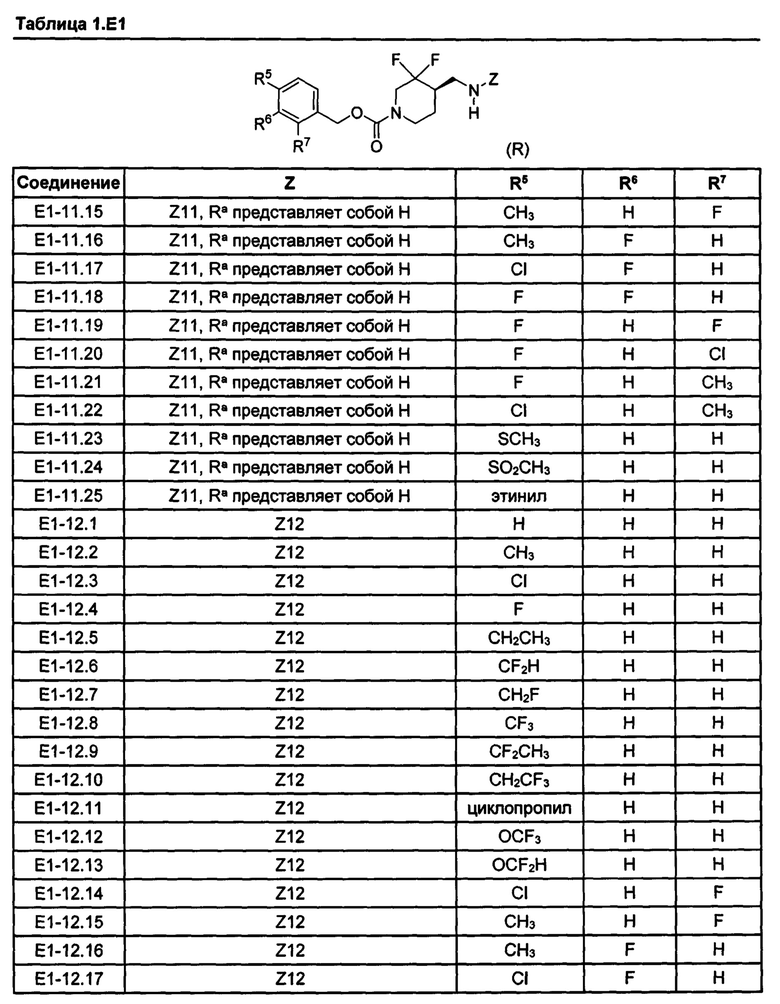





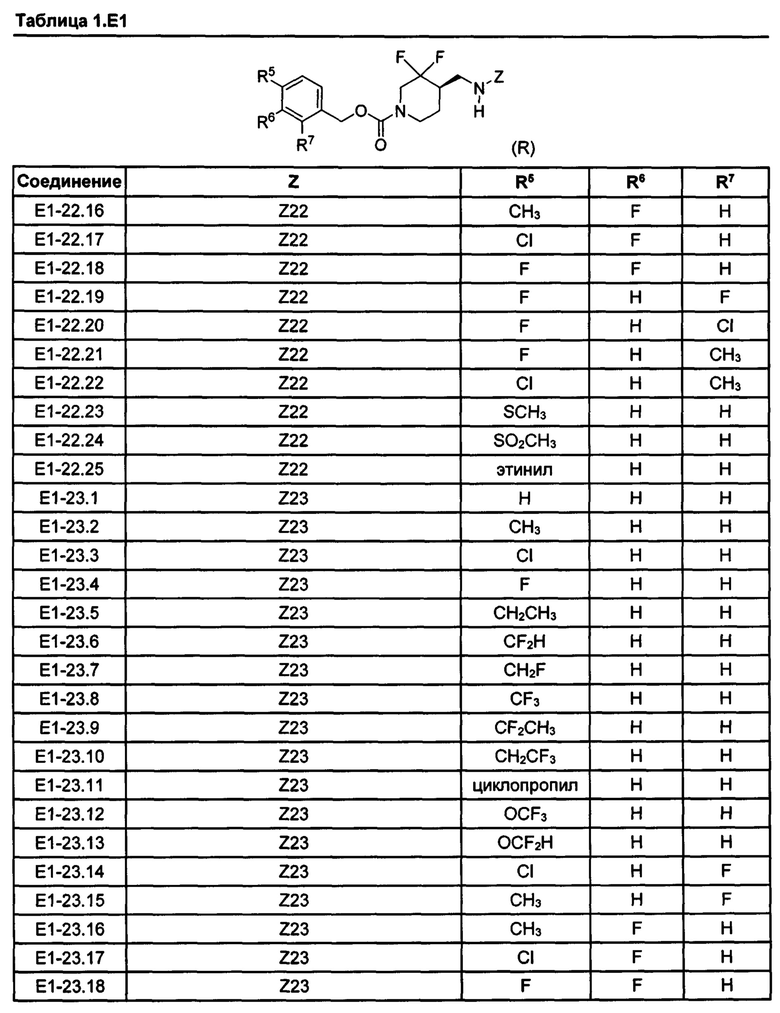
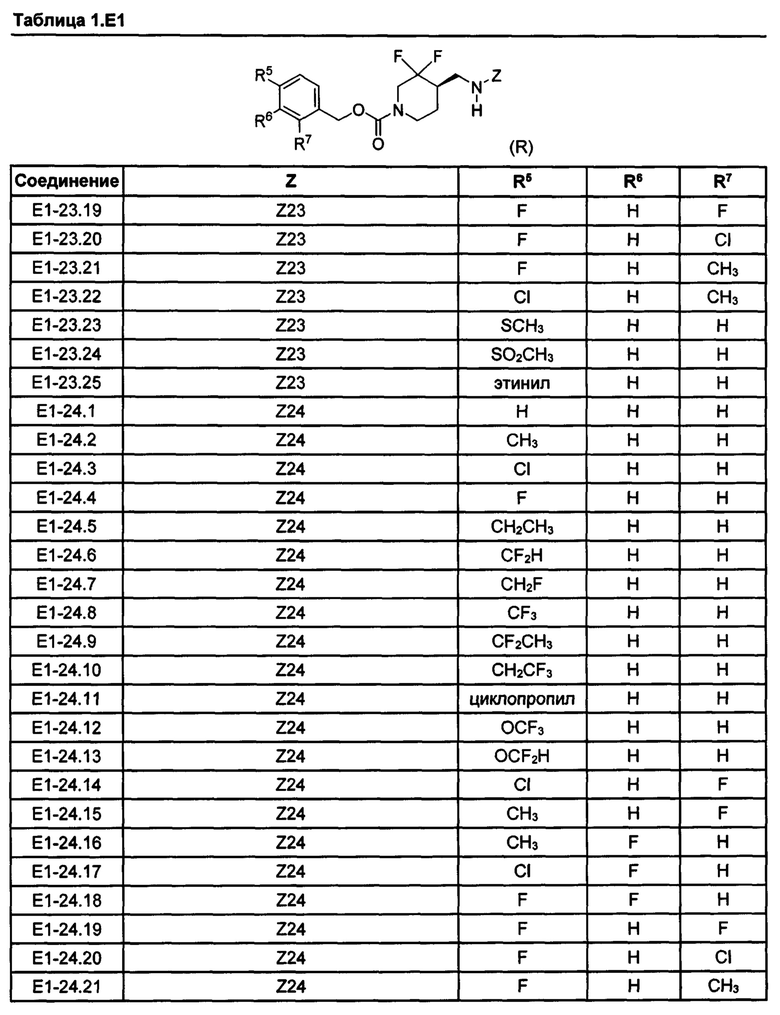

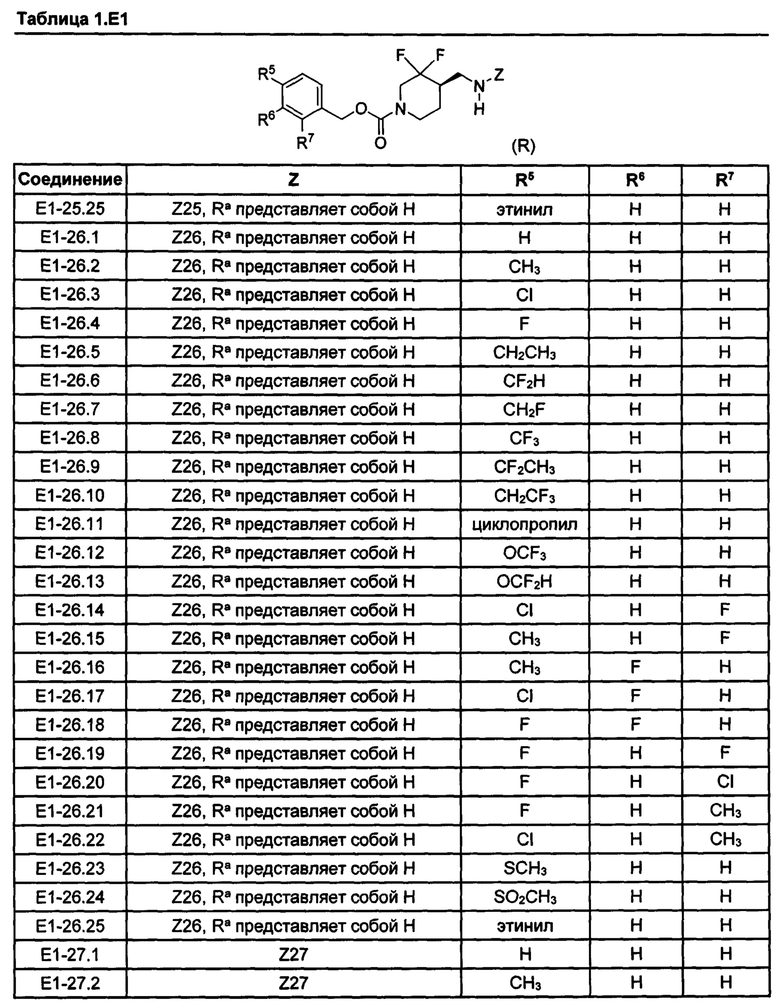

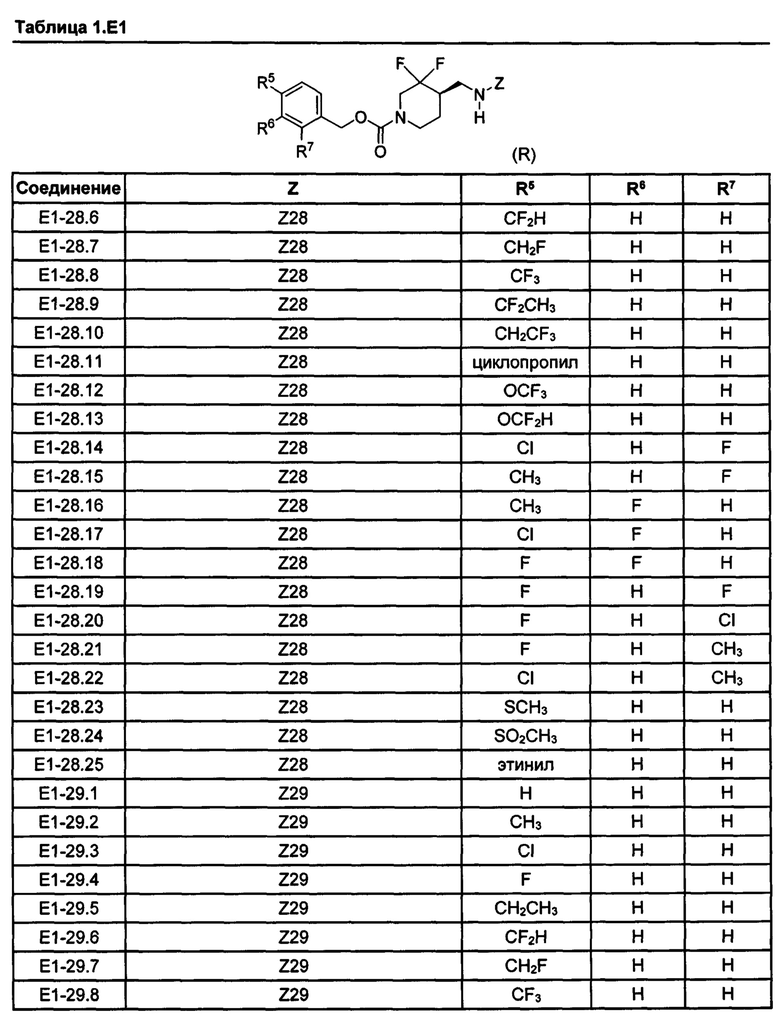
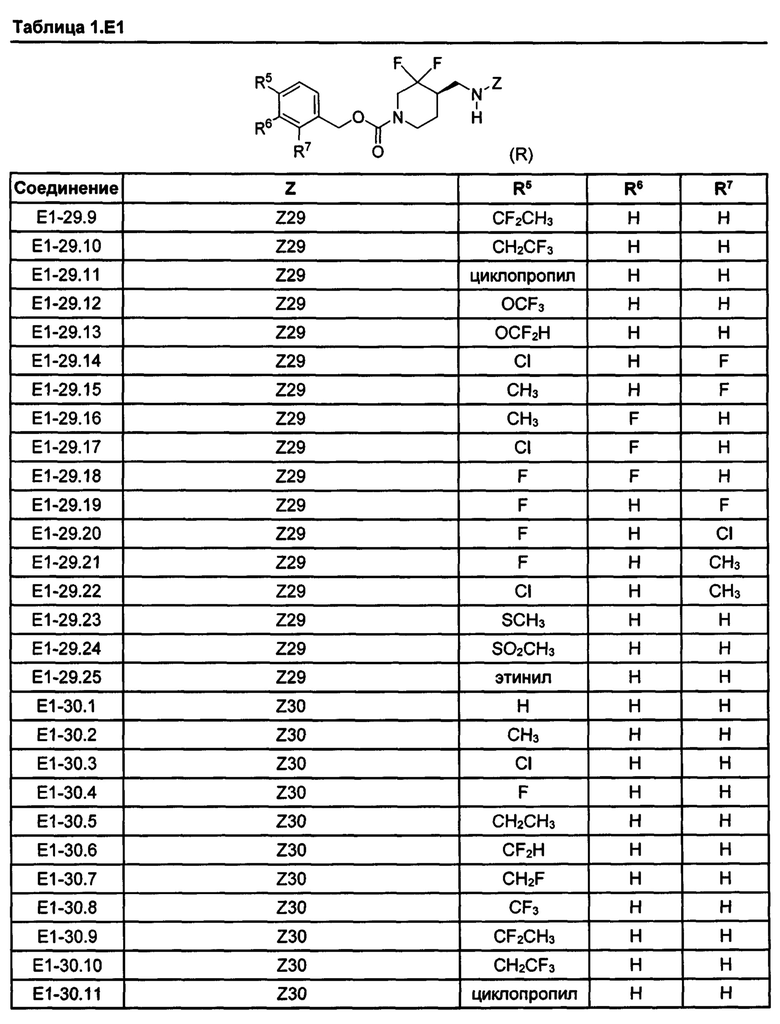

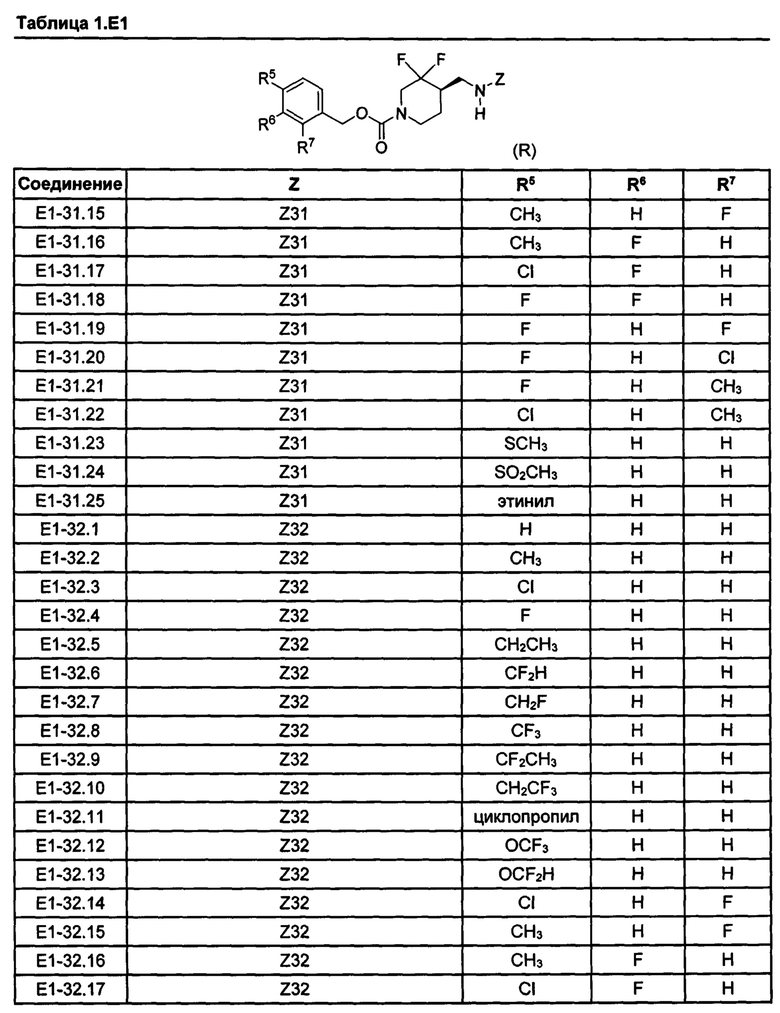
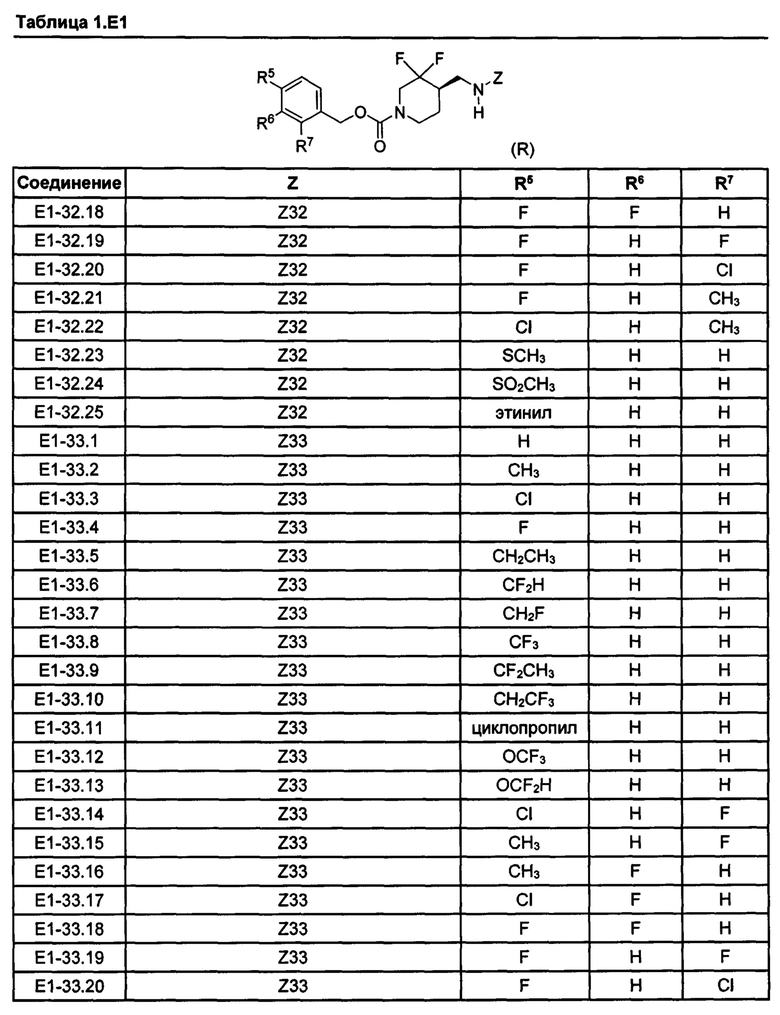
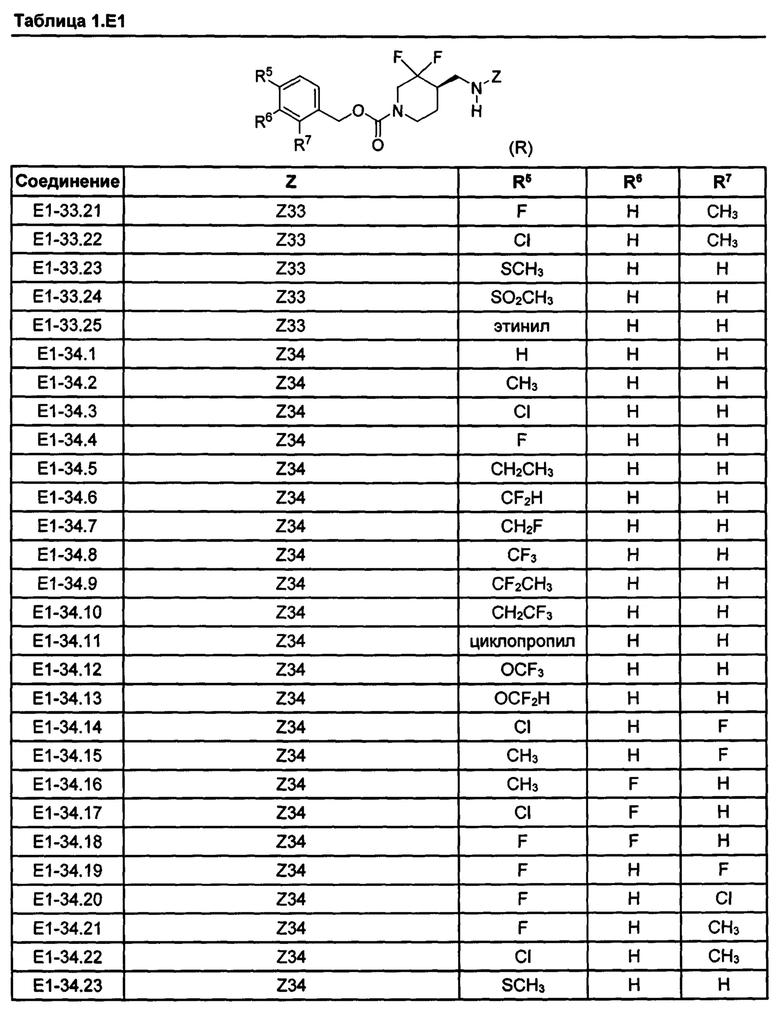


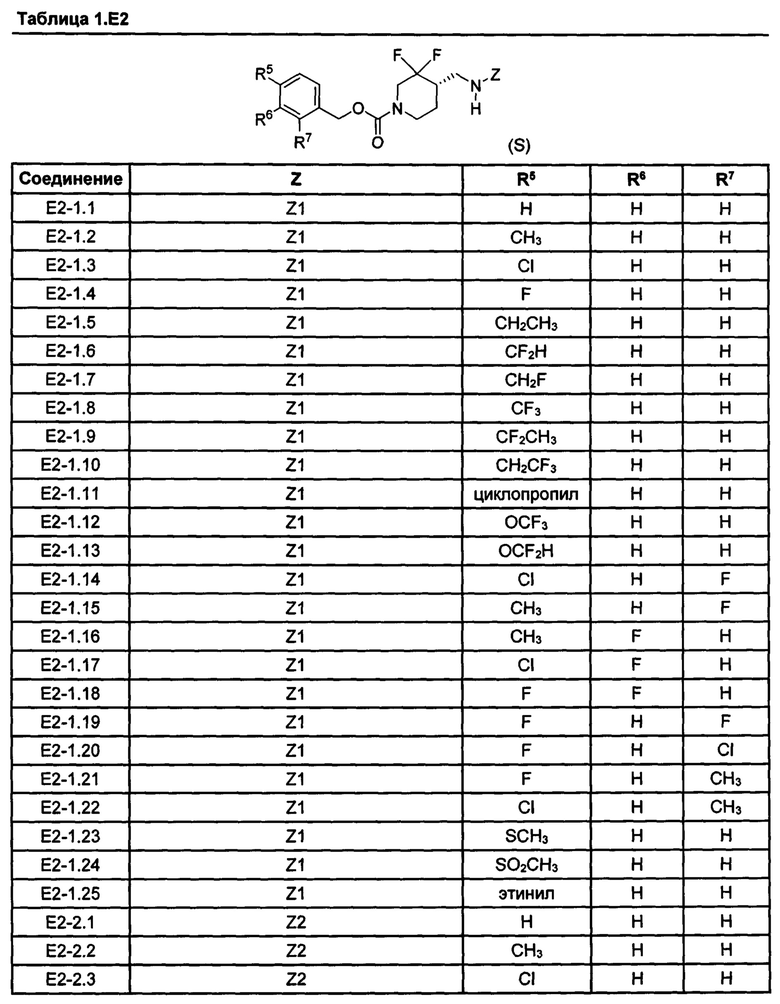
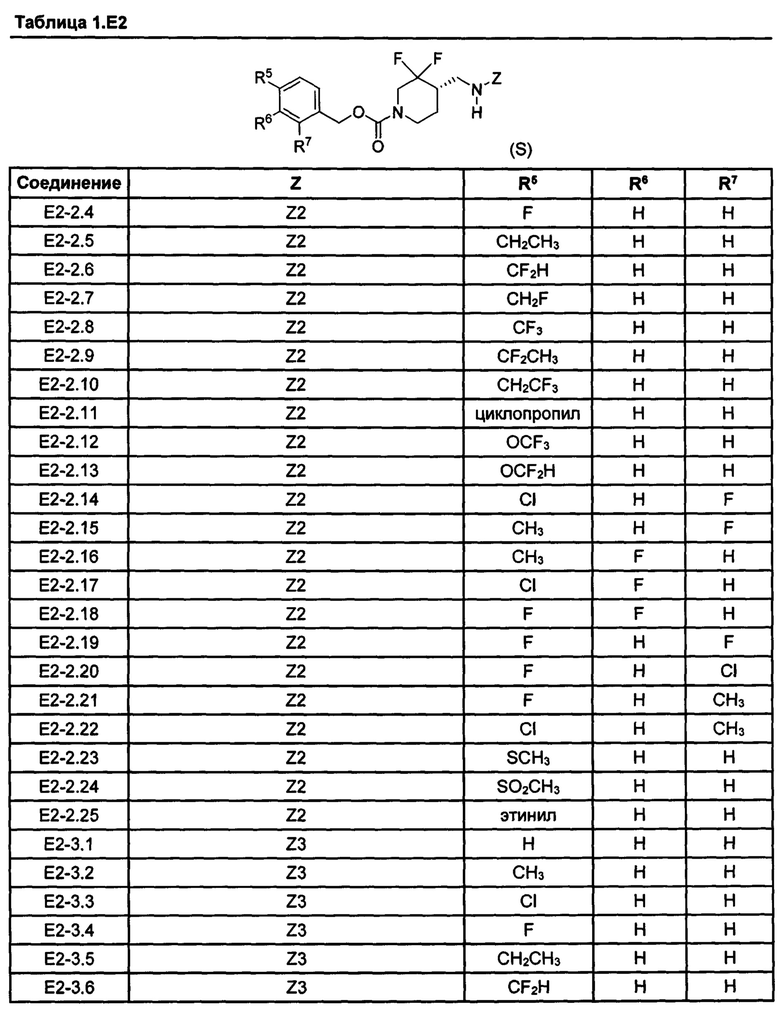

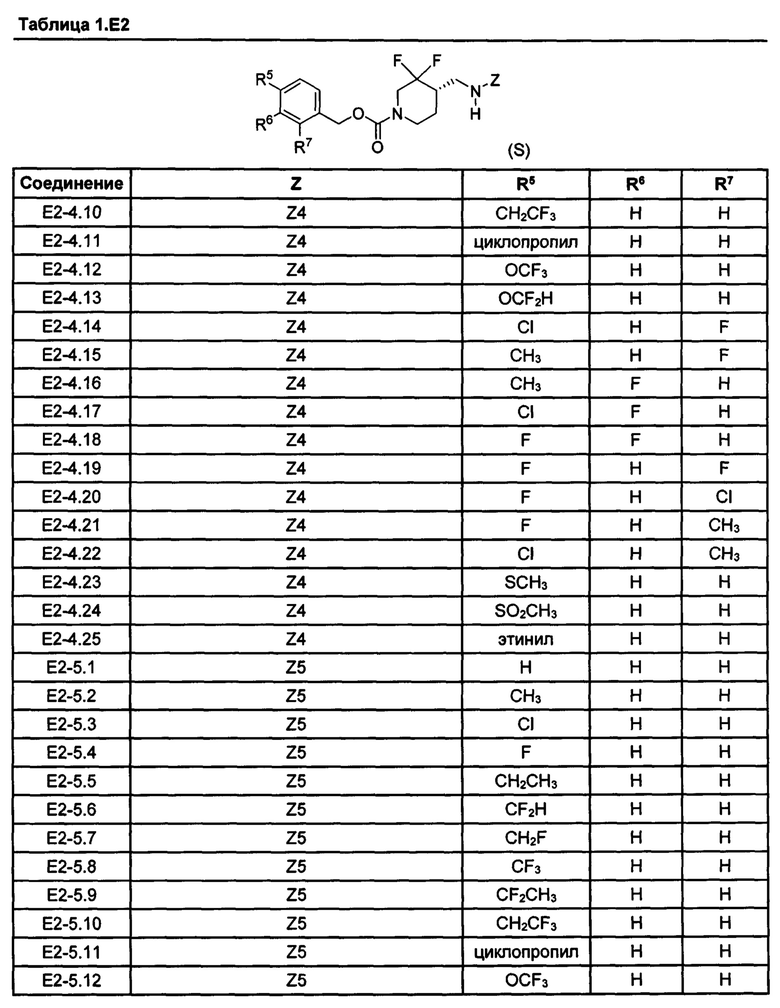
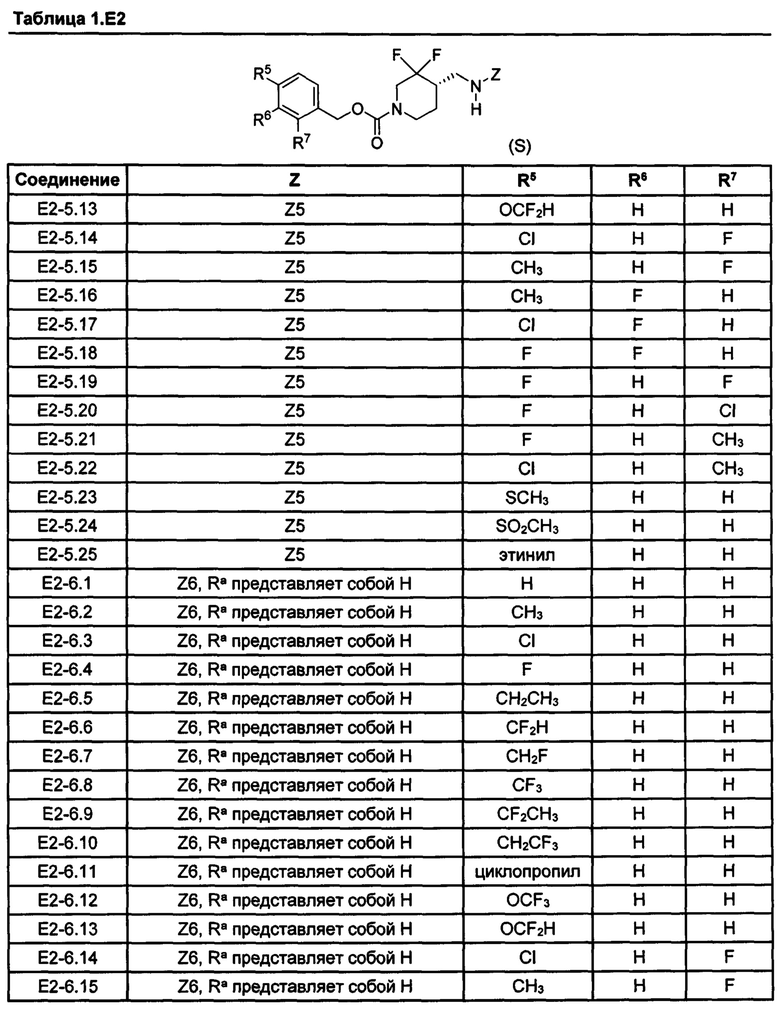
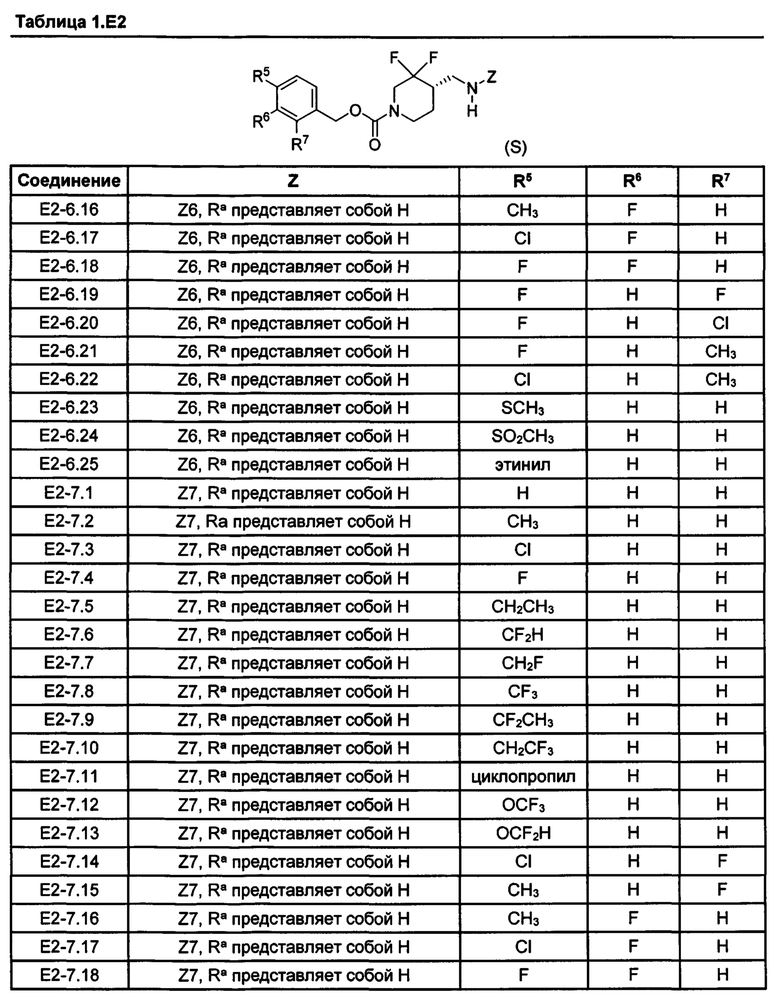
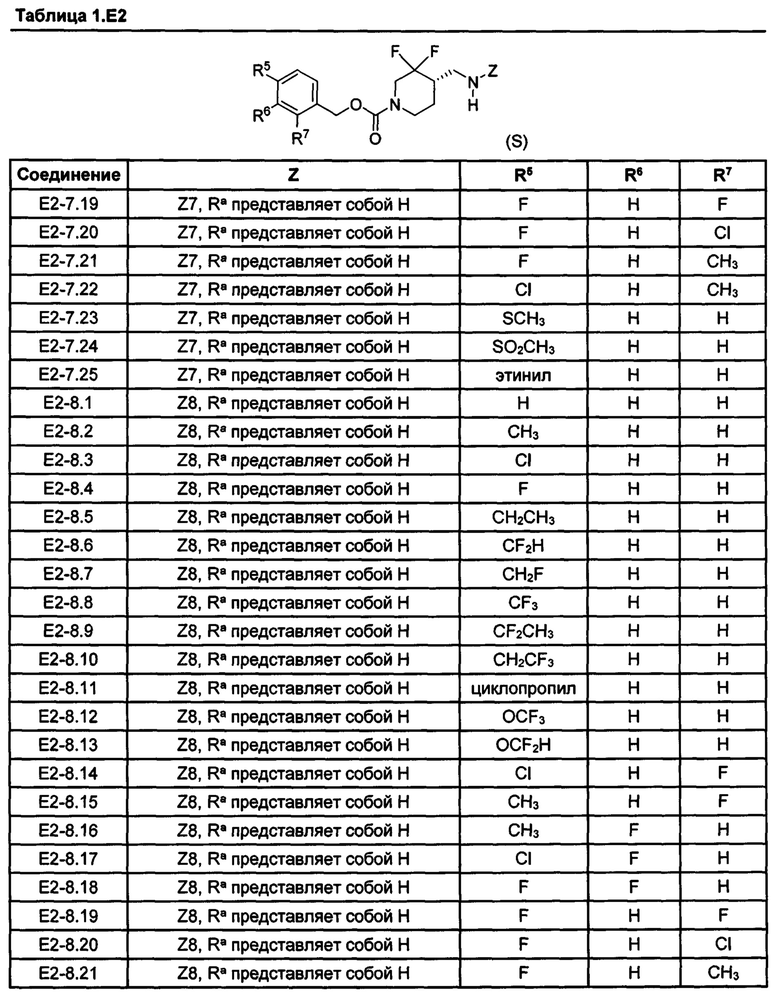
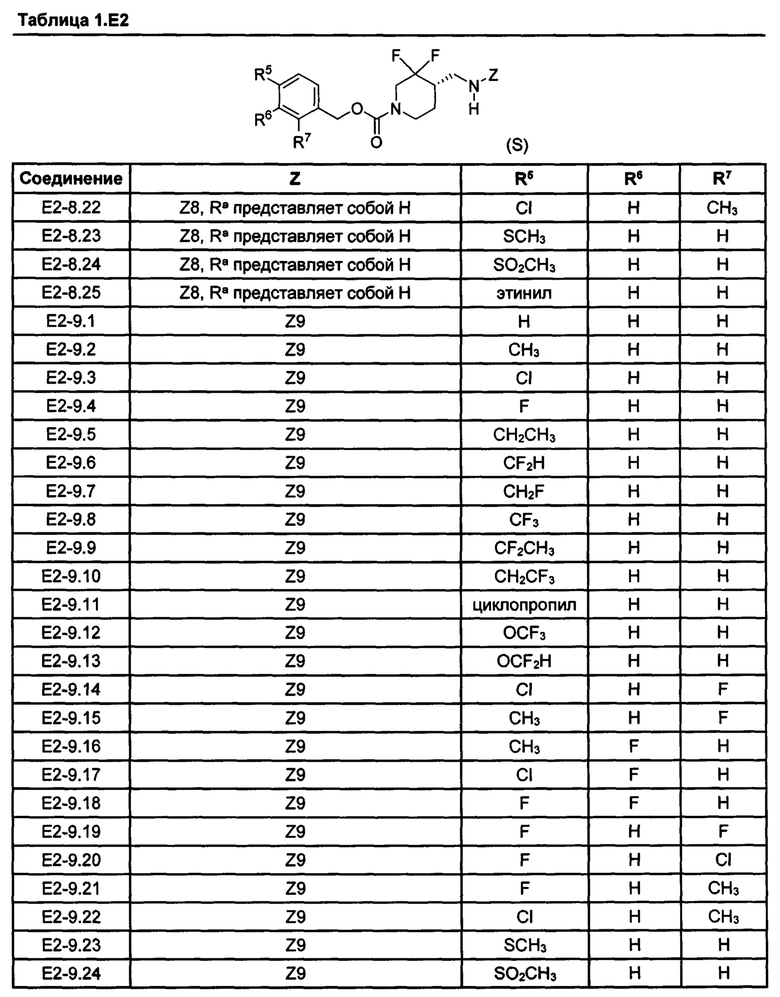
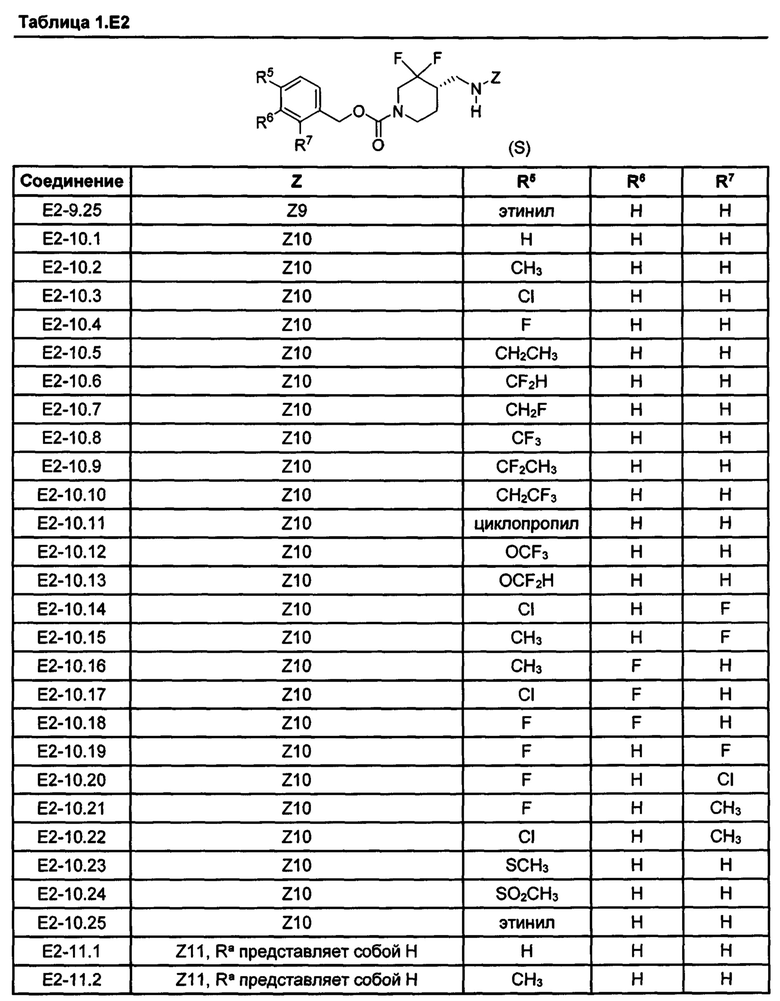
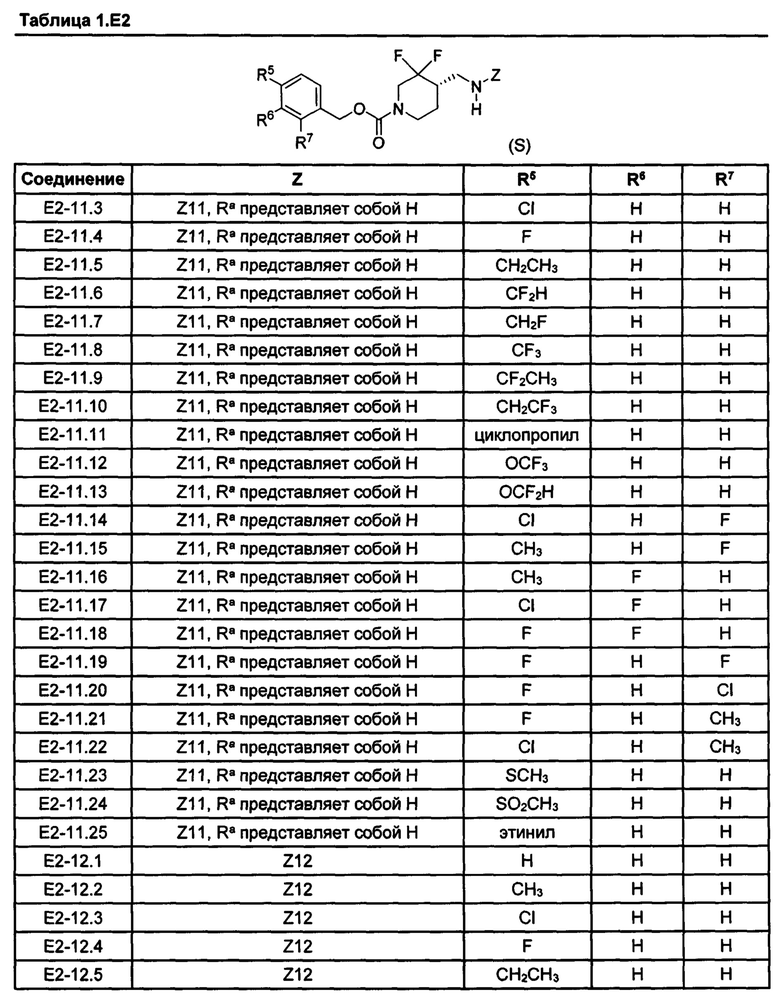



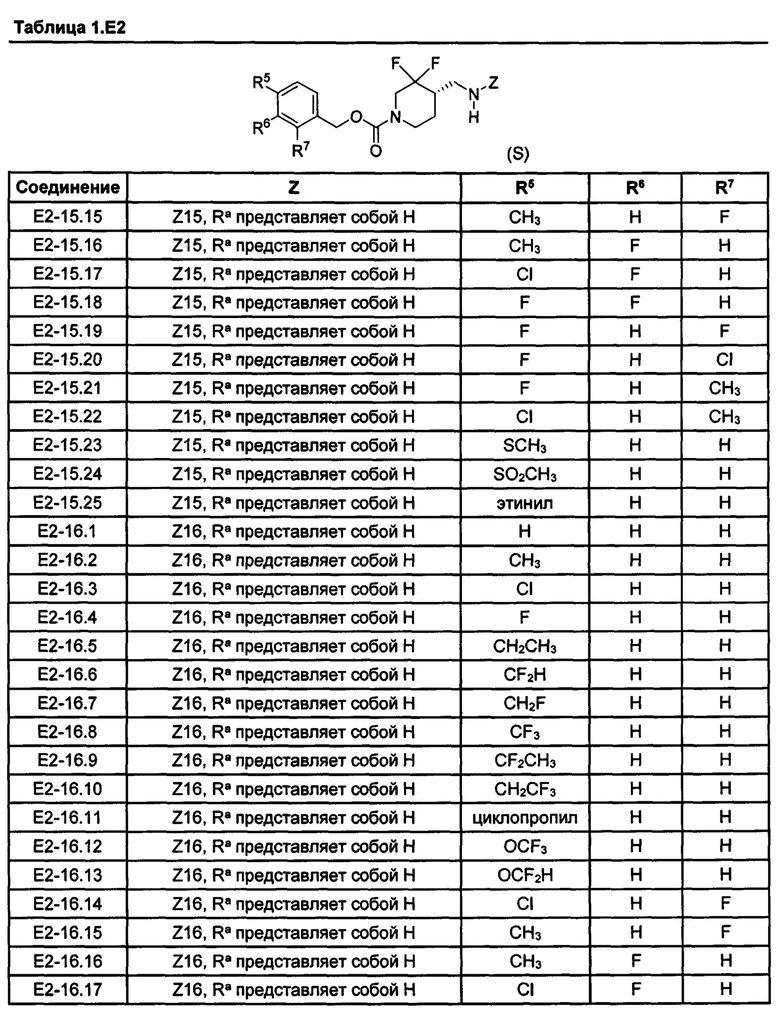

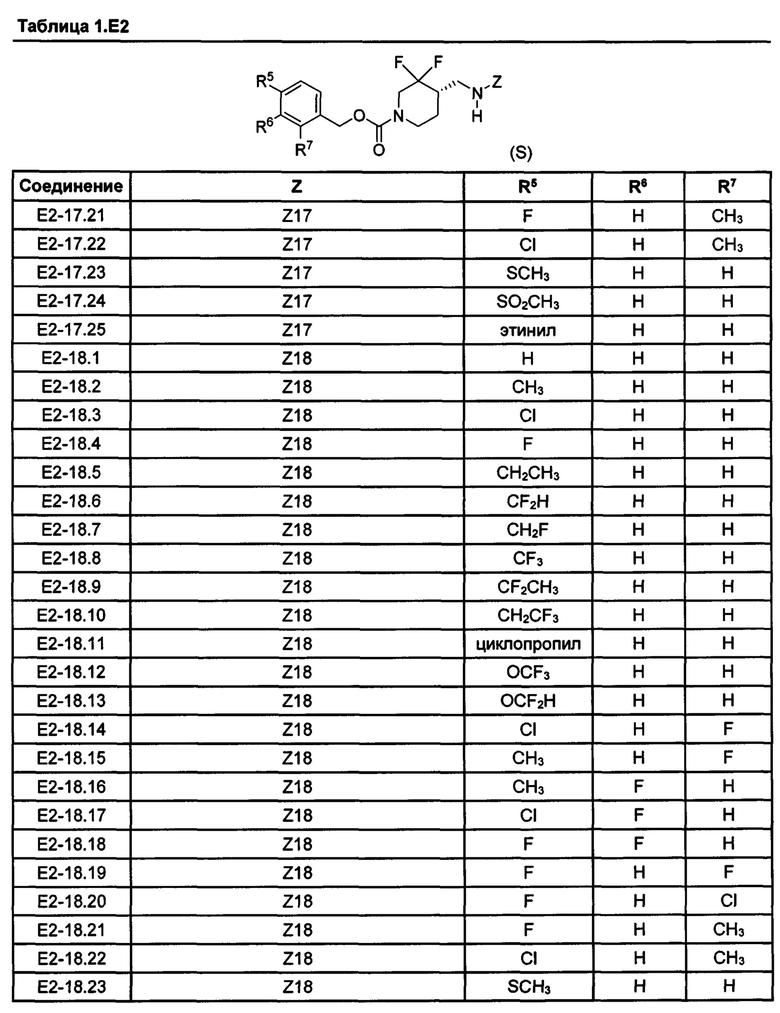

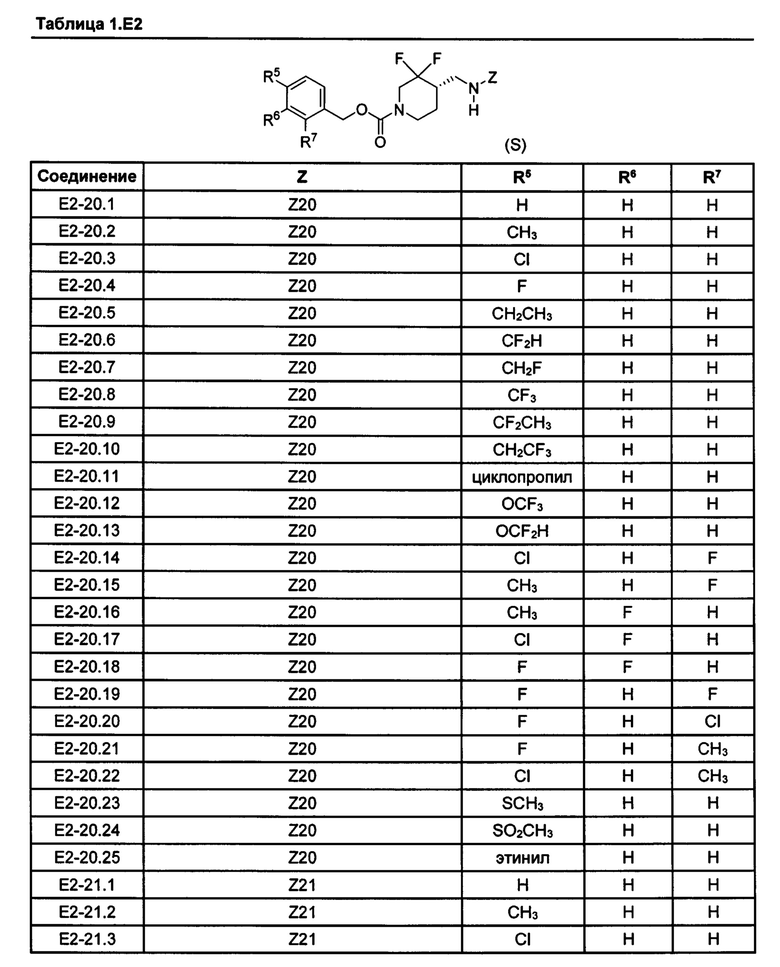
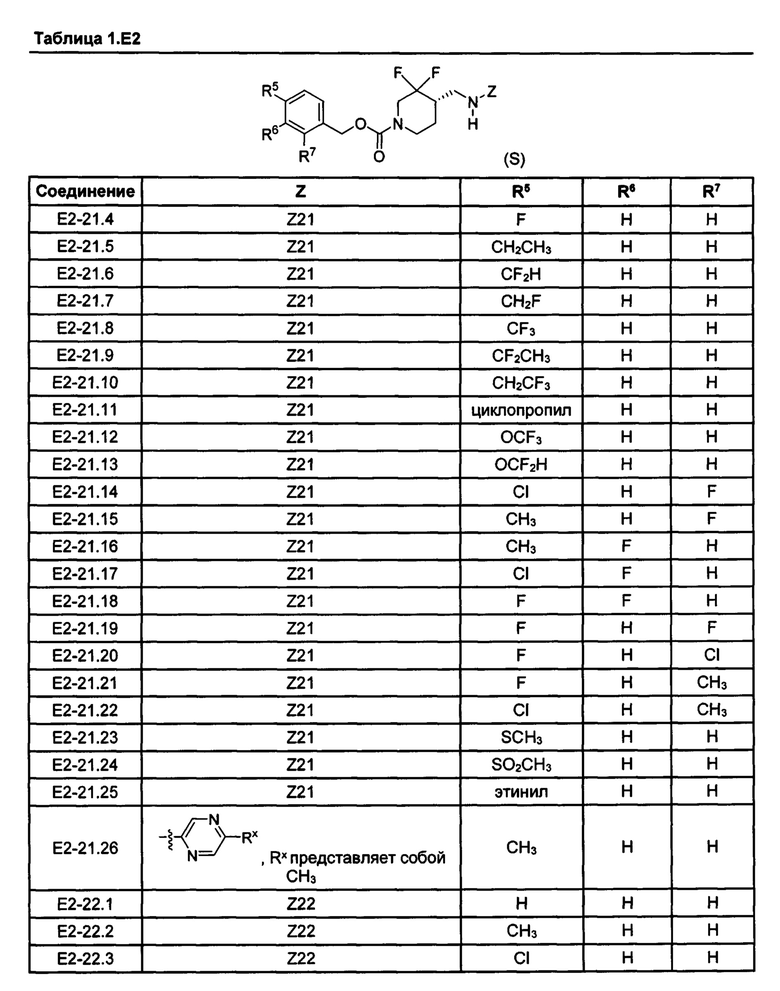


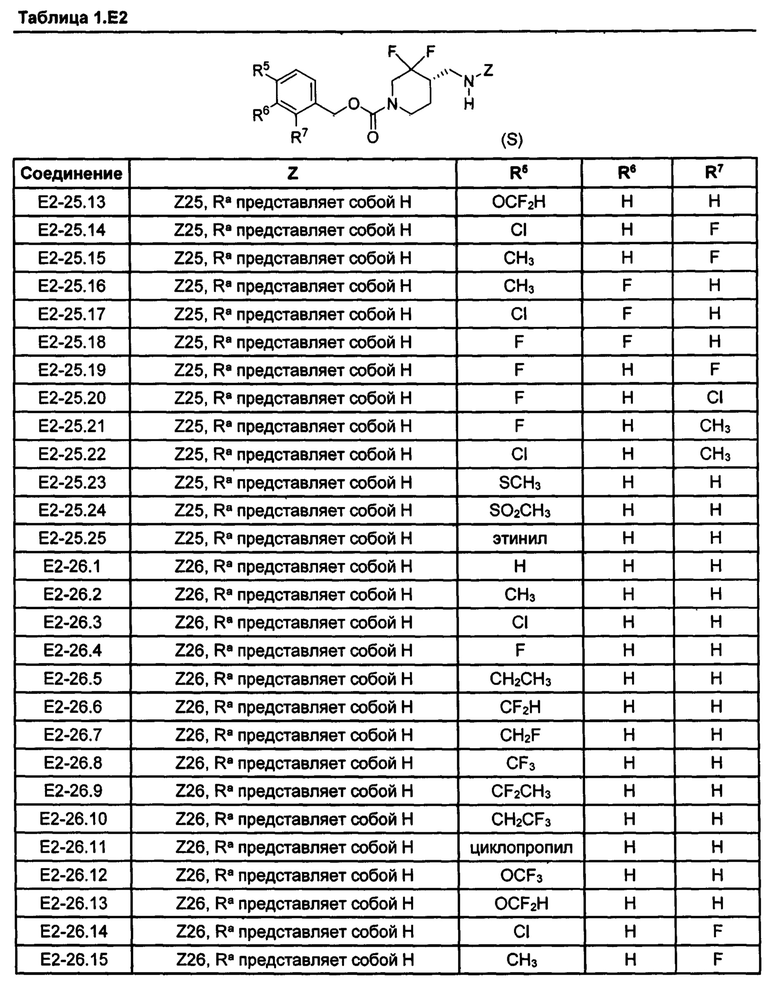




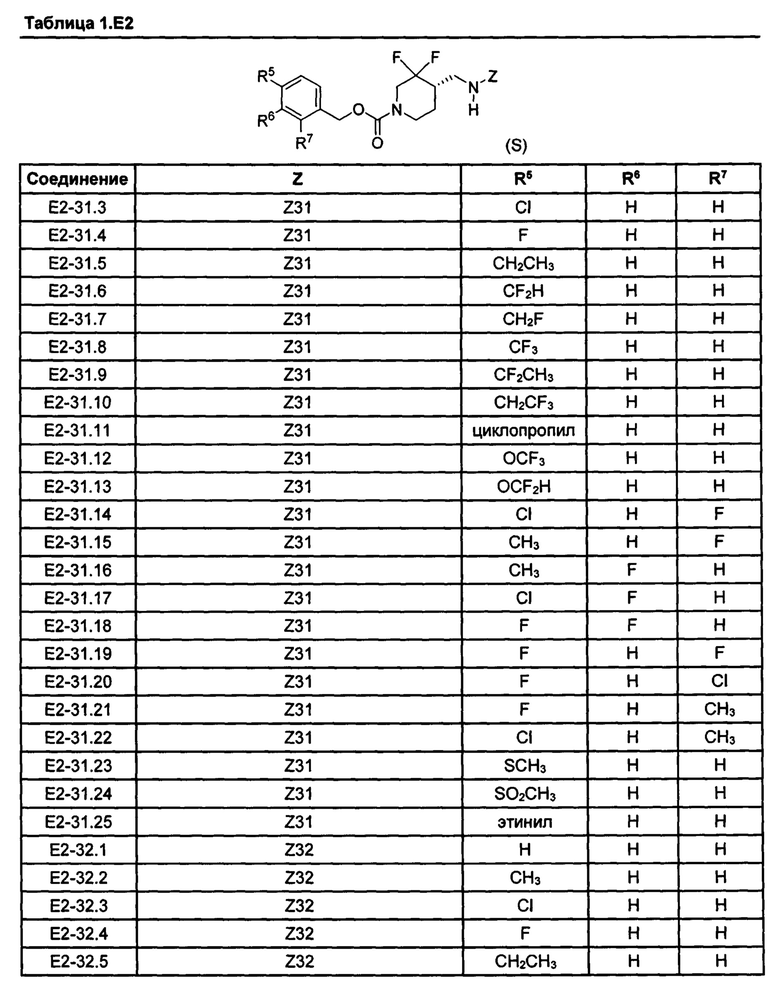
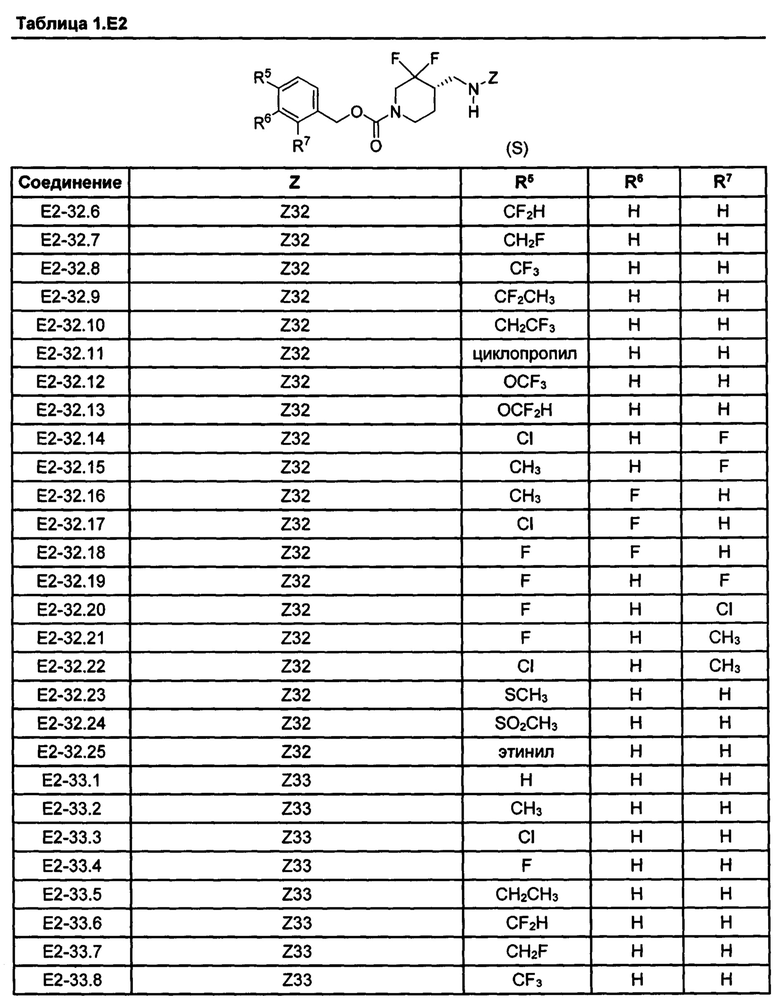
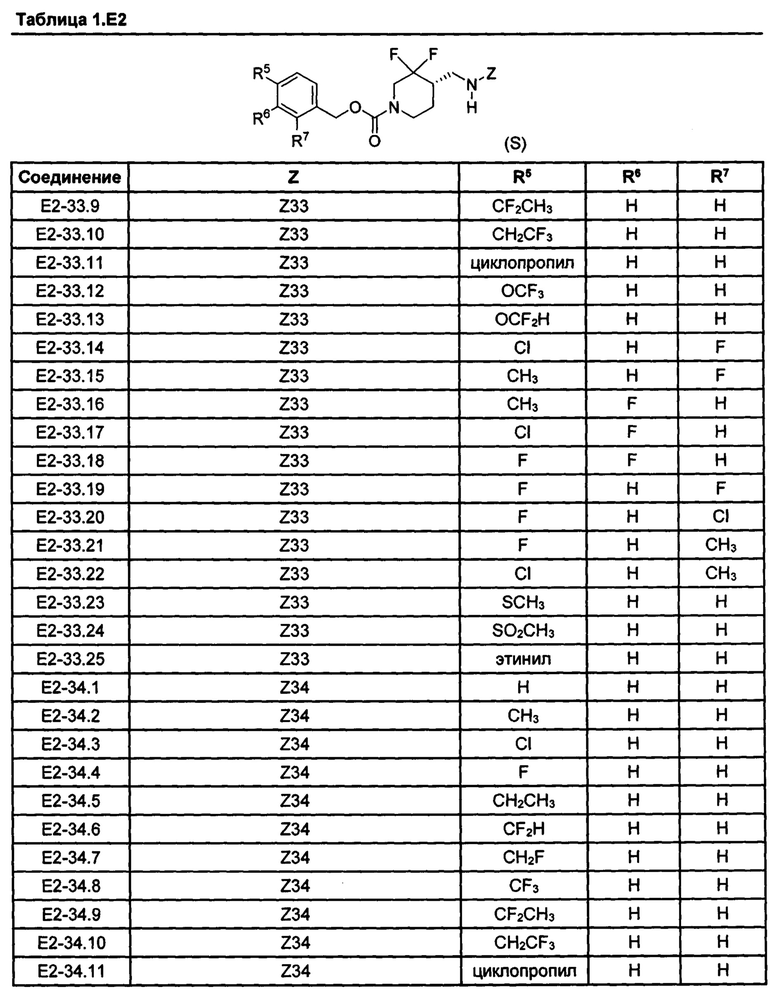
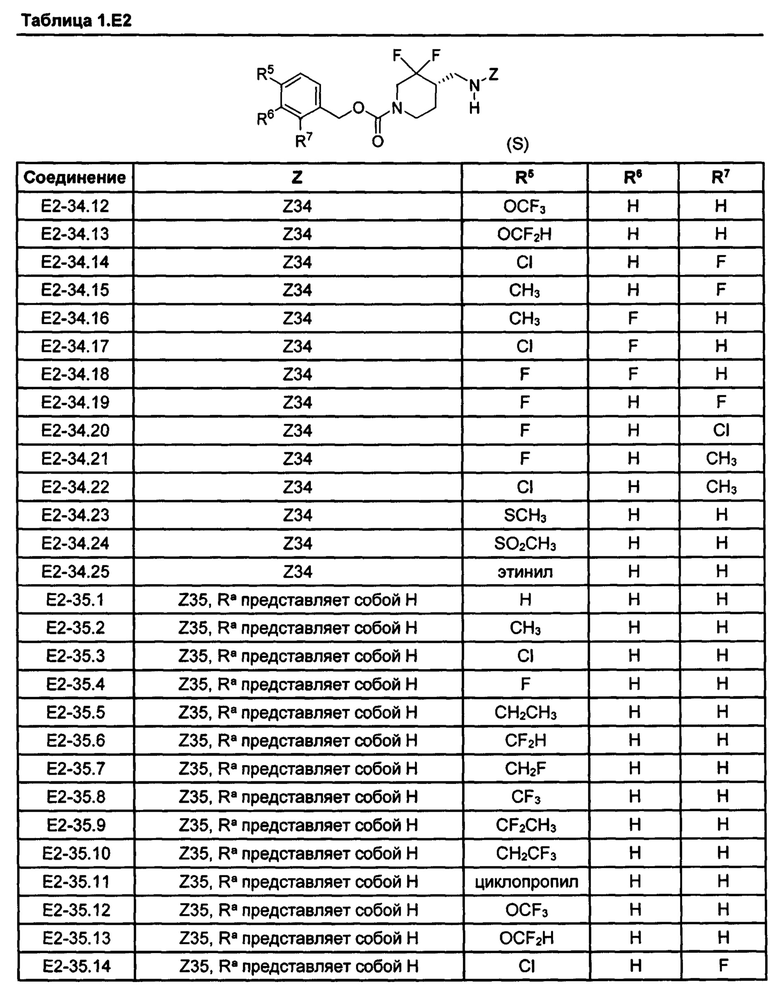

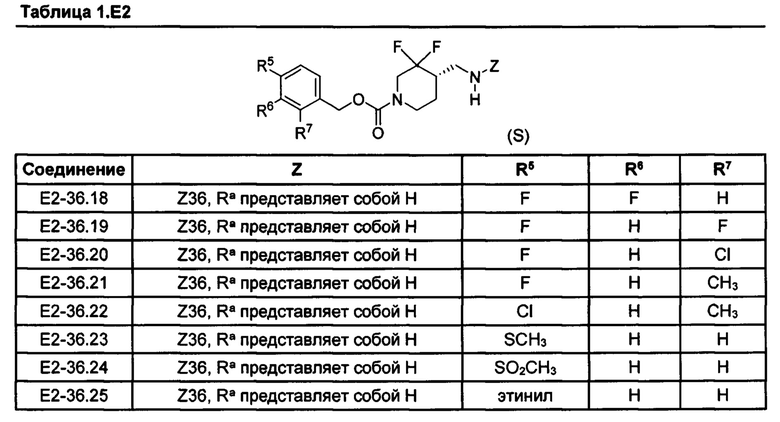
Фармакология
[078] Глутамат (GLU) представляет собой основной возбуждающий нейромедиатор в головном мозге и центральной нервной системе (ЦНС) млекопитающих. Действия этого эндогенного нейромедиатора опосредованы связыванием и активацией связывания GLU с рецепторами глутамата (GLURs), которые в широком смысле подразделяются на метаботропные связанные с G-белком (mGluRs) и лиганд-управляемые ионные каналы или ионотропные GluRs. Ионотропные GLURs с точки зрения фармакологии подразделяются на три основных вида по действию селективных агонистов рецепторов: NMDA (селективные к N-метил-D-аспартату), KA (селективные к каиновой кислоте) и АМРА (селективные к α-амино-3-гидрокси-5-метил-4-изоксазолпропионовой кислоте) рецепторы, и их структура и фармакологическая функция были недавно подробно описаны (S.F. Traynelis et al. Pharmacology Reviews, 2010, 62, 405-496). Электрофизиологические исследования показали, что NMDA-рецепторы представляют собой катионные ионные каналы, которые могут быть заблокированы в зависимости от напряжения под действием эндогенного Mg2+. Активация NMDA-рецепторов глутаматом в присутствии глицина в качестве коагониста приводит к открытию ионного канала данных рецепторов. Это в свою очередь позволяет Na+ и Са2+ проходить в клетку, в результате чего возникает возбуждающий постсинаптический потенциал (ВПСП) и активируются активируемые Са2+ сигнальные пути вторичных мессенджеров в нейронах. Ввиду проницаемости NMDA-рецепторов для Са2+ активация указанных рецепторов регулирует долговременные изменения в нейронных связях, такие как обучение и запоминание, а также синаптическая пластичность.
[079] После того, как была дана исходная фармакологическая характеристика селективным лигандам, исследования в области молекулярной биологии и клонирования позволили подробно охарактеризовать NMDA-рецепторы на молекулярном уровне (Paoletti et al., 2013, Nat. Rev. Neurosci. 14:383-400). Соответственно, NMDA-рецепторы представляют собой гетеротетрамеры, состоящие из двух субъединиц NR1 и двух субъединиц NR2. Субъединицы NR1 содержат сайт связывания коагониста глицина, а субъединицы NR2 содержат сайт связывания глутамата. Ввиду существования нескольких сплайсированных вариантов NR1 и четырех изоформ NR2 (NR2A, NR2B, NR2C и NR2D) из различных генов имеет место разнообразие молекулярных матриц и NMDA-рецепторов. Фармакологические и электрофизиологические свойства NMDA-рецепторов варьируются в зависимости от состава конкретной изоформы NR1 и подтипа NR2. Кроме того, изоформы подтипа NR2 по-разному экспрессируются в зависимости от типа клеток и областей головного мозга. Следовательно, соединения, которые селективно взаимодействуют с субъединицами NR2, могут проявлять специфические фармакологические эффекты в конкретных областях мозга и являются перспективными для лечения заболеваний ЦНС с высокой степенью специфичности и селективности (например, в отношении (vz) побочных эффектов). Например, низкая экспрессия подтипа NR2B в мозжечке по сравнению с экспрессией в других структурах мозга (Cull-Candy et al., 1998, Neuropharmacol. 37: 1369-1380) означала меньшие двигательные побочные эффекты для данного подтипа.
[080] Антагонизм NMDA-рецептора активно исследуется в отношении возможности лечения различных заболеваний ЦНС, включая инсульт, эпилепсию, боль, депрессию, болезнь Паркинсона и болезнь Альцгеймера (Paoletti et al., Nat. Rev. Neurosci 14: 383-400; Sancora, 2008, Nature Rev. Drug Disc., 7, 426-437). NMDA-рецептор позволяет предложить ряд фармакологических отправных точек для разработки ингибиторов рецепторов. Прямые блокаторы пор ионного канала NMDA-рецептора представляют собой одно из семейств антагонистических соединений, для которых могла бы быть продемонстрирована эффективность in vitro и in vivo в различных моделях заболеваний ЦНС, включая эпилепсию, боль и нейродегенерацию/инсульт. Однако соединения данного класса, представленных фенциклидином (РСР), MK-801 и кетамином, как правило, относят к неселективным в отношении разнообразных подтипов NMDA-рецепторов.

[081] У людей неселективные высокоаффинные антагонисты NMDA-рецепторов, как правило, ассоциируются с серьезными клиническими побочными эффектами, включая галлюцинации, дистрофию и нарушение координации. Тем не менее, кетамин, внутривенное лекарственное средство, изначально одобренное для применения для анестезии (Haas et. al, 1992, Anesthesia Prog., 39, 61-68), недавно продемонстрировал клиническую эффективность в качестве средства антидепрессивной терапии (Katalinic et al. 2013, Aust. N.Z. J. Psychiatry, 47, 710-727). Антидепрессивное действие при интенсивной терапии кетамином наступает практически сразу же по сравнению с примерно шестью неделями, необходимыми в случае лекарственной терапии стандартными ингибиторами обратного захвата серотонина (SSRI). Таким образом, при внутривенном введении данного лекарственного средства было продемонстрировано быстрое наступление действия и длительная эффективность, которая может сохраняться при последующих периодических введениях (Zarate et al., 2006, Arch. Gen. Psychiatry 63, 856-864). В конечном итоге было показано, что кетамин является эффективным в отношении классов депрессии, при которых имеет место устойчивость к стандартным видам лекарственной терапии (Murrough et al., 2013, American J. Psychiatry, 170, 1134-1142), в том числе в отношении биполярной депрессии (Zarate et al. 2012, Biol. Psychiatry, 71, 939-946). Однако ввиду того, что кетамин представляет собой внутривенное лекарственное средство с серьезными побочными эффектами (Gianni et. al 1985, Psychiatric Medicine, 3, 197-217; Curran et al 2000, Addiction, 95, 575-590) и потенциально продолжительным токсическим действием (Hardy et al., 2012, J. Clin. Oncol. 30: 3611-3617; Noppers et al., 2011, Pain 152: 2173-2178), терапия кетамином имеет ограниченное применение и сводится к введению в острых случаях или периодическому введению. Для расширения спектра действия и применения в качестве средства терапии депрессии и других заболеваний ЦНС необходимы перорально активные селективные антагонисты NMDA со сниженными побочными эффектами, которые можно вводить постоянно.
[082] Ифенпродил, сосудорасширяющее лекарственное средство, представляющее собой антагонист α1-адренергических рецепторов, как было определено, обладает новым механизмом действия и представляет собой аллостерический модулятор в отношении подтипа NR2B NMDA-рецептора (Reynolds et al. 1989, Mol. Pharmacol., 36, 758-765). Этот новый механизм действия открывает перспективу для нового класса лекарственных средств-антагонистов NMDA, обладающих терапевтической эффективностью без ограничивающих побочных эффектов, присущих блокаторам ионных каналов, неселективным в отношении конкретного подтипа каналов. После этого открытия были получены селективные в отношении NR2B антагонисты-аналоги ифенпродила (Borza et al., 2006, Current Topics in Medicinal Chemistry, 6, 687-695; Layton et al. Current Topics in Medicinal Chemistry, 6, 697-709), которые были оптимизированы в отношении нежелательной α1-адренергической активности, в том числе Ro-25,6981 (Fischer et al. 1997, J. Pharmacol. Exp. Ther., 283, 1285-1292) и CP-101,606, также известный как траксопродил (Chenard et al. 1995, Journal of Medicinal Chemistry, 38, 3138-3145; Menniti et al. 1998, CNS Drug Reviews., 4, 307-322). В клинических исследованиях CP-101,606 продемонстрировал антидепрессивную активность у людей после внутривенного введения с более подходящим профилем диссоциативных побочных эффектов по сравнению с неселективными антагонистами NMDA (Preskorn et al. 2008, Journal of Clinical Psychopharmacology, 28, 631-637). Однако CP-101,606 имеет неоптимальные фармакокинетические свойства и его внутривенное введение должно быть ограниченным. В случае СР-101,606 для достижения оптимальных результатов в вышеуказанном клиническом исследовании данного средства в качестве антидепрессанта был необходим протокол его медленной внутривенной инфузии (Preskorn et al. 2008, Journal of Clinical Psychopharmacology, 28, 631-637).
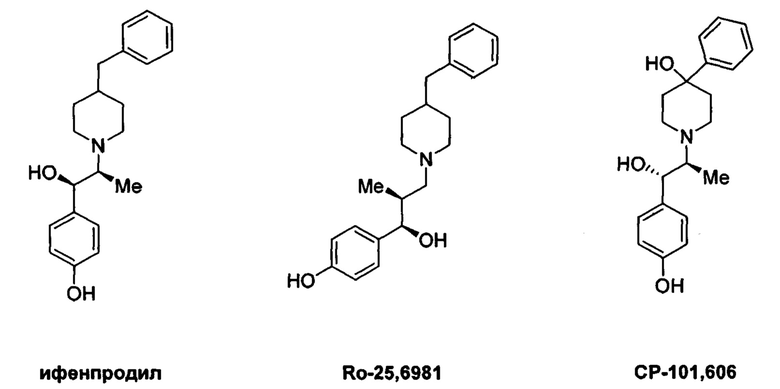
[083] Другие антагонисты NR2B, которые были описаны согласно обзору В. Ruppa с соавторами (K.В. Ruppa et al., Annual Reports in Medicinal Chemistry 2012, 47: 89-103), включают MK0657 (J.A. McCauley et al., 3rd Anglo-Swedish Medicinal Chemistry Symposium,  , Sweden, Mar. 11-14, 2007; L Mony et al., British J. of Pharmacology 2009, 157: 1301-1317; см. также публикацию международной патентной заявки №WO 2004/108705; патент США №7,592,360) и соединения формулы LX (публикация международной патентной заявки №WO 2006/113471), представленный ниже, включая специфический аналог LX-1, описанный ниже.
, Sweden, Mar. 11-14, 2007; L Mony et al., British J. of Pharmacology 2009, 157: 1301-1317; см. также публикацию международной патентной заявки №WO 2004/108705; патент США №7,592,360) и соединения формулы LX (публикация международной патентной заявки №WO 2006/113471), представленный ниже, включая специфический аналог LX-1, описанный ниже.
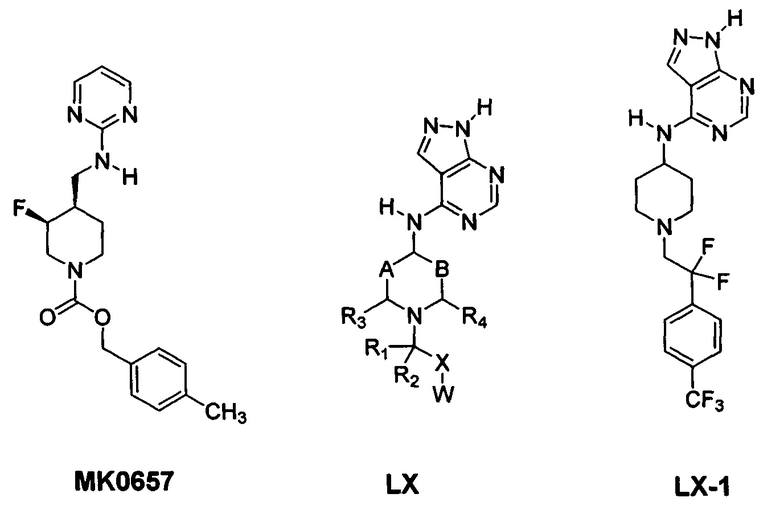
[084] Как отмечается Kawai с соавторами (М. Kawai et al., Bioorganic and Medicinal Chem. Lett. 2007, v17:5533-5536) и Brown с соавторами (Brown et al., Bioorganic and Medicinal Chem. Lett. 2011, v21:3399-3403), трудности, присущие антагонистам NR2B, имеющим основные аминные фрагменты, в отношении преодоления связанных с безопасностью недостатков, касающихся hERG и CYP2D6, при сохранении активности в отношении NR2B in vitro и in vivo, хорошо известны. Ингибирование соединением каналов hERG и связанное с этим удлинение QT на электрокардиограмме (ЭКГ) является хорошо известным серьезным риском для человека, связанным с безопасностью в отношении сердечно-сосудистой системы (Hancox et al., Molecular Pharmacology 2008, 73:1592-1595). Удлинение QT может привести к сердечной аритмии с пируэтной тахикардией (TdP), которая может перейти в желудочковую тахикардию и привести к скоропостижной смерти.
[085] Ингибирование соединением метаболических ферментов семейства цитохрома Р-450, включая CYP2D6, у человека имеет связанный с безопасностью лекарства риск, обусловленный лекарственными взаимодействиями (Drug Metabolism Handbook: Concepts and Applications, ed. Ala F. Nassar copyright 2009 Wiley & Sons, Hoboken, NJ). Таким образом, выведение лекарств, которых являются субстратами CYP2D6, может быть уменьшено соединениями, которые ингибируют CYP2D6. Результатом может быть токсичность или усиление побочных эффектов ввиду накопления определенного лекарства-субстрата CYP2D6. Лекарственные средства, действующие на ЦНС, включая антидепрессивные лекарства, занимают значительное место среди известных субстратов CYP2D6. Таким образом, ингибирование CYP2D6 является крайне нежелательным для лекарственных средств-антагонистов NR2B, особенно с учетом распространенного применения сопутствующих препаратов или избыточного применения лекарственных средств при показаниях, касающихся ЦНС, включая депрессию. Примеры субстратов CY2D6 включают антидепрессанты из класса SSRI, такие как флуоксетин, пароксетин и флувоксамин, дулоксетин, антидепрессанты из класса SSNI, различные антипсихотические средства, включая галоперидол, рисперидон и арипипразол, различные гипотензивные средства-бета-блокаторы, включая метапролол, пропранолол, тимолол и алпренолол и ингибитор холинэстеразы-лекарство от болезни Альцгеймера донепрезил (Flockhart DA (2007). "Drug Interactions: Cytochrome P450 Drug Interaction Table", Indiana University School of Medicine, accessed at <<http://medicine.iupui.edu/clinpharm/ddis/>> on May 28, 2014).
[086] MK0657 и его близкородственные аналоги (Liverton et al., J. Med. Chem. 2007, v50:807-819; являются примерами поколения антагонистов NR2B с улучшенной биодоступностью для человека при пероральном введении. Однако для MK0657 в опубликованных результатах исследований клинической эффективности, проведенных на пациентах с болезнью Паркинсона, описаны побочные эффекты в отношении сердечно-сосудистой системы, касающиеся повышения систолического, а также диастолического кровяного давления, связанного с данным лекарственным средством после его перорального введения (Addy et al., J. Clin. Pharm. 2009, v49: 856-864). Аналогичные эффекты, связанные с кровяным давлением, как сообщалось, также наблюдались после однократного введения MK0657 в исследованиях безопасности, проведенных на здоровых субъектах (Peterson et al., "A randomized, double-blind, placebo-controlled, parallel-group, three-part safety, pharmacokinetic, and pharmacodynamic study of CERC-301 in healthy subjects", National Network of Depression Centers Annual Conference, Ann Arbor, Nov. 5-6, 2015). Интересно, что MK0657 и его энантиомер (3R,4S-соединение) продемонстрировало схожую активность в отношении NR2B (MK0657=13,8 нМ; 3R,4S-энантиомер=25,5 нМ). Еще более примечательным является то, что активность цис- и транс-диастереомеров в отношении NR2B была схожей (см. Koudih et al., European J. Med. Chem. 53 (2012), 408-415).
[087] Соединение LX-1 демонстрирует биодоступность для животных при пероральном введении и не содержит фенольной группы, которая может нарушать биодоступность для людей при пероральном введении. Однако, как и другие антагонисты NR2B, содержащие основные аминные фрагменты, соединение LX-1, имеющее основный пиперидиновый атом азота, несмотря на наличие уменьшающего основность соседнего фрагмента с двумя фторами в положении бета относительно данного атома азота, демонстрирует ингибирование канала hERG у человека с IC50<10 мкМ (~4,5 мкМ), а также демонстрирует ингибирующую активность в отношении метаболического фермента CYP2D6 человека (IC50 ~ 1,0 мкМ).
[088] Как отмечается в источнике K.В. Ruppa et al., Annual Reports in Medicinal Chemistry 2012, 47: 89-103, для широкого и безопасного применения у человека необходимы улучшенные селективные антагонисты NR2B. Существует потребность в соединениях-антагонистах NR2B, которые являются улучшенными в отношении одного или более аспектов, примерами которых являются фармакокинетика, всасывание, метаболизм, выведение (ADME (всасывание, распределение, метаболизм и выведение, англ.: absorption, distribution, metabolism and excretion), например активность при пероральном введении), улучшенная эффективность, нецелевая активность, улучшенный относительный показатель терапевтической безопасности и возможность постоянной терапии с пероральным введением. Например, для MK0657 в опубликованных результатах исследования клинической эффективности, проведенных на пациентах с болезнью Паркинсона, был описан побочный эффект в отношении сердечно-сосудистой системы, касающийся повышения систолического, а также диастолического кровяного давления, связанного с данным лекарственным средством после его перорального введения (Addy et al., J. Clin. Pharm. 2009, v49: 856-864). Аналогичные эффекты, связанные с кровяным давлением, как сообщалось, также наблюдались после однократного введения MK0657 в исследованиях безопасности, проведенных на здоровых субъектах преклонного возраста.
[089] Предложенные химические соединения являются антагонистами рецептора NR2B и обладают техническими преимуществами в отношении одного или более фармацевтических лекарственных свойств, таких как биодоступность при пероральном введении, фармакокинетические параметры, характеристики ADME (например, ингибирование CYP, образование метаболитов), фармакологическая безопасность in vivo и/или in vitro.
[090] В некоторых вариантах реализации предложенное химическое соединение обладает функциональной селективностью в отношении NR2B NMDA-рецептора по сравнению с NR2A («селективностью в отношении NR2B», определяемой как отношение IC50 в отношении NR2A/IC50 в отношении NR2B, где значения IC50 определяют в соответствии с методикой из Примера 2.1) ≥ 400. В некоторых вариантах реализации предложенное химическое соединение обладает селективностью в отношении NR2B ≥ 300. В некоторых вариантах реализации предложенное химическое соединение обладает селективностью в отношении NR2B ≥ 200. В некоторых вариантах реализации предложенное химическое соединение обладает селективностью в отношении NR2B ≥ 100. В некоторых вариантах реализации предложенное химическое соединение обладает селективностью в отношении NR2B ≥ 50. В некоторых вариантах реализации предложенное химическое соединение обладает селективностью в отношении NR2B ≥ 20.
[091] В некоторых вариантах реализации предложенное химическое соединение обладает активностью в отношении hERG (определяемой как IC50 в отношении hERG, измеряемая в соответствии с методикой из Примера 2.2) ≥ 5 мкМ. В некоторых вариантах реализации предложенное химическое соединение характеризуется IC50 в отношении hERG ≥ 10 мкМ. В некоторых вариантах реализации предложенное химическое соединение характеризуется IC50 в отношении hERG ≥ 15 мкМ. В некоторых вариантах реализации предложенное химическое соединение характеризуется IC50 в отношении hERG ≥ 20 мкМ. В некоторых вариантах реализации предложенное химическое соединение характеризуется IC50 в отношении hERG ≥ 25 мкМ. В некоторых вариантах реализации предложенное химическое соединение характеризуется IC50 в отношении hERG ≥ 30 мкМ. В некоторых вариантах реализации предложенное химическое соединение характеризуется IC50 в отношении hERG ≥ 40 мкМ.
[092] В некоторых вариантах реализации предложенное химическое соединение обладает функциональной антагонистической активностью в отношении NR2B (определяемой как IC50 в отношении NR2B, измеряемая в соответствии с методикой из Примера 2.1) ≤ 200 нМ и активностью в отношении hERG (определяемой как IC50 в отношении hERG, измеряемой в соответствии с методикой из Примера 2.2) ≥ 5 мкМ. В некоторых вариантах реализации предложенное химическое соединение характеризуется IC50 в отношении NR2B ≤ 200 нМ и hERG IC50 ≥ 10 мкМ. В некоторых вариантах реализации предложенное химическое соединение характеризуется IC50 в отношении NR2B ≤ 200 нМ и IC50 в отношении hERG ≥ 15 мкМ. В некоторых вариантах реализации предложенное химическое соединение характеризуется IC50 в отношении NR2B ≤ 200 нМ и IC50 в отношении hERG ≥ 20 мкМ. В некоторых вариантах реализации предложенное химическое соединение характеризуется IC50 в отношении NR2B ≤ 200 нМ и IC50 в отношении hERG ≥ 25 мкМ. В некоторых вариантах реализации предложенное химическое соединение характеризуется IC50 в отношении NR2B ≤ 200 нМ и IC50 в отношении hERG ≥ 30 мкМ. В некоторых вариантах реализации предложенное химическое соединение характеризуется IC50 в отношении NR2B ≤ 200 нМ и IC50 в отношении hERG ≥ 40 мкМ. В некоторых вариантах реализации предложенное химическое соединение характеризуется IC50 в отношении NR2B ≤ 100 нМ и IC50 в отношении hERG ≥ 5 мкМ. В некоторых вариантах реализации предложенное химическое соединение характеризуется IC50 в отношении NR2B ≤ 100 нМ и IC50 в отношении hERG ≥ 10 мкМ. В некоторых вариантах реализации предложенное химическое соединение характеризуется IC50 в отношении NR2B ≤ 50 нМ и IC50 в отношении hERG ≥ 5 мкМ. В некоторых вариантах реализации предложенное химическое соединение характеризуется IC50 в отношении NR2B ≤ 50 нМ и IC50 в отношении hERG ≥ 10 мкМ.
[093] В некоторых вариантах реализации предложенное химическое соединение характеризуется функциональной антагонистической активностью в отношении NR2B (определяемой как IC50 в отношении NR2B, измеряемая в соответствии с методикой из Примера 2.1) ≤ 200 нМ и ингибированием CYP2D6 (определяемым как IC50 в отношении CYP2D6, измеряемая в соответствии с методикой из Примера 2.3) ≥ 2 мкМ. В некоторых вариантах реализации предложенное химическое соединение характеризуется IC50 в отношении NR2B ≤ 200 нМ и IC50 в отношении CYP2D6 ≥ 3 мкМ. В некоторых вариантах реализации предложенное химическое соединение характеризуется IC50 в отношении NR2B ≤ 200 нМ и IC50 в отношении CYP2D6 ≥ 4 мкМ. В некоторых вариантах реализации предложенное химическое соединение характеризуется IC50 в отношении NR2B ≤ 200 нМ и IC50 в отношении CYP2D6 ≥ 5 мкМ. В некоторых вариантах реализации предложенное химическое соединение характеризуется IC50 в отношении NR2B ≤ 200 нМ и IC50 в отношении CYP2D6 примерно 5-10 мкМ. В некоторых вариантах реализации предложенное химическое соединение характеризуется IC50 в отношении NR2B ≤ 200 нМ и IC50 в отношении CYP2D6 ≥ 10 мкМ. В некоторых вариантах реализации предложенное химическое соединение характеризуется IC50 в отношении NR2B ≤ 100 нМ и IC50 в отношении CYP2D6 ≥ 2 мкМ. В некоторых вариантах реализации предложенное химическое соединение характеризуется IC50 в отношении NR2B ≤ 100 нМ и IC50 в отношении CYP2D6 ≥ 3 мкМ. В некоторых вариантах реализации предложенное химическое соединение характеризуется IC50 в отношении NR2B ≤ 100 нМ и IC50 в отношении CYP2D6 ≥ 4 мкМ. В некоторых вариантах реализации предложенное химическое соединение характеризуется IC50 в отношении NR2B ≤ 100 нМ и IC50 в отношении CYP2D6 ≥ 5 мкМ. В некоторых вариантах реализации предложенное химическое соединение характеризуется IC50 в отношении NR2B ≤ 100 нМ и IC50 в отношении CYP2D6 примерно 5-10 мкМ. В некоторых вариантах реализации предложенное химическое соединение характеризуется IC50 в отношении NR2B ≤ 100 нМ и IC50 в отношении CYP2D6 ≥ 10 мкМ. В некоторых вариантах реализации предложенное химическое соединение характеризуется IC50 в отношении NR2B ≤ 50 нМ и IC50 в отношении CYP2D6 ≥ 2 мкМ. В некоторых вариантах реализации предложенное химическое соединение характеризуется IC50 в отношении NR2B ≤ 50 нМ и IC50 в отношении CYP2D6 ≥ 3 мкМ. В некоторых вариантах реализации предложенное химическое соединение характеризуется IC50 в отношении NR2B ≤ 50 нМ и IC50 в отношении CYP2D6 ≥ 4 мкМ. В некоторых вариантах реализации предложенное химическое соединение характеризуется IC50 в отношении NR2B ≤ 50 нМ и IC50 в отношении CYP2D6 ≥ 5 мкМ. В некоторых вариантах реализации предложенное химическое соединение характеризуется IC50 в отношении NR2B ≤ 50 нМ и IC50 в отношении CYP2D6 примерно 5-10 мкМ. В некоторых вариантах реализации предложенное химическое соединение характеризуется IC50 в отношении NR2B ≤ 50 нМ и IC50 в отношении CYP2D6 ≥ 10 мкМ.
Применение, приготовление и введение и фармацевтически приемлемые композиции
[094] В некоторых вариантах реализации настоящего изобретения предложена композиция, содержащая химическое соединение согласно настоящему изобретению или его фармацевтически приемлемое производное и фармацевтически приемлемый носитель, адъювант или переносящую среду. Количество химического соединения в композициях согласно настоящему изобретению таково, что оно является эффективным для определяемого ингибирования NR2B в биологическом образце или у пациента. В некоторых вариантах реализации количество химического соединения в композициях согласно настоящему изобретению таково, что оно является эффективным для определяемого ингибирования NR2B в биологическом образце или у пациента. В некоторых вариантах реализации композиция согласно настоящему изобретению приготовлена для введения пациенту, нуждающемуся в такой композиции. В некоторых вариантах реализации композиция согласно настоящему изобретению приготовлена для перорального введения пациенту.
[095] Термин «пациент» в контексте настоящего описания означает животное, предпочтительно млекопитающее и наиболее предпочтительно человека.
[096] Термин «фармацевтически приемлемый носитель, адъювант или переносящая среда» относится к нетоксичному носителю, адъюванту или переносящей среде, которая не угнетает фармакологическую активность химического соединения, в состав с которым она входит. Фармацевтически приемлемые носители, адъюванты или переносящие среды, которые могут быть применены в композициях согласно настоящему изобретению, включают ионообменные вещества, оксид алюминия, стеарат алюминия, лецитин, сывороточные белки, такие как сывороточный альбумин человека, буферные вещества, такие как фосфаты, глицин, сорбиновая кислота, сорбат калия, смеси неполных глицеридов насыщенных растительных жирных кислот, воду, соли или электролиты, такие как протаминсульфат, гидрофосфат натрия, гидрофосфат калия, хлорид натрия, соли цинка, коллоидный диоксид кремния, трисиликат магния, поливинилпирролидон, вещества на основе целлюлозы, полиэтиленгликоль, натрийкарбоксиметилцеллюлозу, полиакрилаты, воски, блок-сополимеры полиэтилена и полиоксипропилена, полиэтиленгликоль и ланолин (wool fat).
[097] «Фармацевтически приемлемое производное» означает любой нетоксичный сложный эфир, соль сложного эфира или другое производное химического соединения согласно настоящему изобретению (например, пролекарство), которое при введении получателю способно обеспечивать прямо или косвенно химическое соединение согласно настоящему изобретению или его активный в отношении ингибирования метаболит или остаток.
[098] В контексте настоящего описания термин «его активный в отношении ингибирования метаболит или остаток» означает, что метаболит или остаток указанного соединения также представляет собой ингибитор NR2B.
[099] Композиции согласно настоящему изобретению могут быть введены перорально, парентерально, с помощью спрея для ингаляций, местно, ректально, назально, буккально, вагинально или через имплантированный резервуар. Термин «парентеральный» в контексте настоящего описания включает методики подкожных, внутривенных, внутримышечных, внутрисуставных, внутрисиновиальных, внутригрудинных, интратекальных, внутрипеченочных, внутриочаговых и внутричерепных инъекций или инфузий. Предпочтительно указанные композиции вводят перорально, внутрибрюшинно или внутривенно. Стерильные инъекционные формы композиций согласно настоящему изобретению могут представлять собой водную или маслянистую суспензию. Указанные суспензии могут быть приготовлены согласно методикам, известным в данной области техники, с применением подходящих диспергирующих или смачивающих агентов и суспендирующих агентов. Стерильный препарат для инъекций также может представлять собой стерильный раствор или суспензию для инъекций в нетоксичном, подходящем для парентерального введения разбавителе или растворителе, например, находиться в виде раствора в 1,3-бутандиоле. К числу приемлемых переносящих сред и растворителей, которые могут быть применены, относятся вода, раствор Рингера и изотонический раствор хлорида натрия. Кроме того, в качестве растворителя или суспендирующей среды традиционно применяют стерильные нелетучие масла.
[0100] Для указанной цели может быть применено любое мягкое нелетучее масло, включая синтетические моно- или диглицериды. Жирные кислоты, такие как олеиновая кислота и ее глицеридные производные, подходят для применения для получения препаратов для инъекций, равно как и натуральные фармацевтически приемлемые масла, такие как оливковое масло или касторовое масло, в частности в их полиоксиэтилированных вариантах. Указанные масляные растворы или суспензии также могут содержать длинноцепочечный спиртовой разбавитель или диспергирующий агент, такой как карбоксиметилцеллюлоза или сходные диспергирующие агенты, которые широко применяются в составе фармацевтически приемлемых лекарственных форм, включая эмульсии и суспензии. Для целей приготовления также могут быть применены и другие широко используемые поверхностно-активные вещества, такие как Твины, Спаны и другие эмульгирующие агенты или агенты, улучшающие биодоступность, которые широко применяются в изготовлении фармацевтически приемлемых твердых, жидких или иных лекарственных форм.
[0101] Фармацевтически приемлемые композиции согласно настоящему изобретению могут быть перорально введены в любой перорально приемлемой лекарственной форме, включая капсулы, таблетки, водные суспензии или растворы. В случае таблеток для перорального применения широко используемые носители включают лактозу и кукурузный крахмал. Также обычно добавляют смазывающие агенты, такие как стеарат магния. В случае перорального введения в форме капсул подходящие разбавители включают лактозу и высушенный кукурузный крахмал. Когда для перорального применения требуются водные суспензии, указанный активный ингредиент объединяют с эмульгирующими и суспендирующими агентами. При необходимости также могут быть добавлены некоторые подслащивающие, ароматизирующие или окрашивающие агенты.
[0102] В качестве альтернативы фармацевтически приемлемые композиции согласно настоящему изобретению могут быть введены в форме суппозиториев для ректального введения. Указанные суппозитории могут быть получены путем смешивания указанного агента с подходящим нераздражающим вспомогательным веществом, которое является твердым при комнатной температуре, но жидким при ректальной температуре и, таким образом, будет плавиться в прямой кишке с высвобождением указанного лекарственного средства. Такие материалы включают масло какао, пчелиный воск и полиэтиленгликоли.
[0103] Фармацевтически приемлемые композиции согласно настоящему изобретению также могут быть введены местно, в частности когда мишень лечения включает области или органы, легко доступные для местного применения, включая заболевания глаз, кожи или нижнего отдела кишечного тракта. Подходящие составы для местного введения легко получить для каждой из указанных областей или органов.
[0104] Местное применение в случае нижнего отдела кишечного тракта может быть осуществлено в составе для ректальных суппозиториев (см. выше) или в подходящем составе для клизмы. Также могут быть применены местные трансдермальные пластыри.
[0105] В случае местного применения предложенные фармацевтически приемлемые композиции могут быть приготовлены в форме подходящей мази, содержащей указанный активный компонент, суспендированный или растворенный в одном или более носителях. Носители для местного введения соединений согласно настоящему изобретению включают минеральное масло, жидкий вазелин, белый вазелин, пропиленгликоль, полиоксиэтилен, полиоксипропиленовое соединение, эмульгирующий воск и воду. В качестве альтернативы предложенные фармацевтически приемлемые композиции могут быть приготовлены в форме подходящего лосьона или крема, содержащего указанные активные компоненты, суспендированные или растворенные в одном или более фармацевтически приемлемых носителях. Подходящие носители включают минеральное масло, сорбитан моностеарат, полисорбат 60, воск цетиловых эфиров, цетеариловый спирт, 2-октилдодеканол, бензиловый спирт и воду.
[0106] В случае офтальмологического применения предложенные фармацевтически приемлемые композиции могут быть приготовлены в виде микронизированных суспензий в стерильном изотоническом солевом растворе с отрегулированным рН или предпочтительно в виде растворов в стерильном изотоническом солевом растворе с отрегулированным рН с или без консерванта, такого как бензалкония хлорид. В качестве альтернативы в случае офтальмологических применений указанные фармацевтически приемлемые композиции могут быть приготовлены в форме мази, такой как вазелин.
[0107] Фармацевтически приемлемые композиции согласно настоящему изобретению также могут быть введены с помощью назального аэрозоля или ингаляции. Такие композиции получают согласно методикам, хорошо известным в области приготовления фармацевтических составов, и могут быть получены в виде растворов в солевом растворе с применением бензилового спирта или других подходящих консервантов, стимуляторов абсорбции для повышения биодоступности, фторуглеродов и/или других традиционных солюбилизирующих или диспергирующих агентов.
[0108] Наиболее предпочтительно фармацевтически приемлемые композиции согласно настоящему изобретению приготовлены для перорального введения. Такие составы могут быть введены с пищей или без нее. В некоторых вариантах реализации фармацевтически приемлемые композиции согласно настоящему изобретению вводят без пищи. В других вариантах реализации фармацевтически приемлемые композиции согласно настоящему изобретению вводят с пищей.
[0109] Количество соединений согласно настоящему изобретению, которые могут быть объединены с веществами-носителями с получением композиции в одной лекарственной форме, будет варьироваться в зависимости от целого ряда факторов, включая подвергаемого лечению субъекта и конфетный способ введения. Предпочтительно предложенные композиции должны быть приготовлены так, чтобы пациенту, получающему указанные композиции, могла быть введена доза указанного ингибитора, составляющая 0,01-100 мг/кг массы тела/сутки.
[0110] Также следует понимать, что конкретная дозировка и схема лечения для любого конкретного пациента будет зависеть от целого ряда факторов, включая активность конкретного применяемого соединения, возраст, массу тела, общее состояние здоровья, пол, рацион питания, время введения, скорость выведения, комбинацию лекарственных средств и оценку лечащего врача и тяжесть конфетного заболевания, подвергаемого лечению. Количество соединения согласно настоящему изобретению в композиции также будет зависеть от конкретного соединения в указанной композиции.
Применение химических соединений и фармацевтически приемлемых композиций
[0111] Терапевтическое применение антагонистов рецептора NR2B у человека было обобщено в обзорах Traynelis et al. (S.F. Traynelis et al., Pharmacology Reviews, 2010, 62: 405-496), Beinat et al. (C. Beinat et al., Current Medicinal Chemistry, 2010, 17:4166-4190) и Mony et al. (L. Mony et al., British J. of Pharmacology, 2009, 157:1301-1317). Антагонизм в отношении NR2B может быть применен при лечении заболеваний и расстройств, включая депрессию, боль, болезнь Паркинсона, болезнь Гентингтона, болезнь Альцгеймера, церебральную ишемию, травматическое повреждение головного мозга, судорожные расстройства (например, эпилепсию) и мигрень. (S.В. Bausch et al., Epilepsia, 2010, 51:102-105; P. Mares, Naunyn-Schmiedeberg's Arch Pharmacol, 2014, 387:753-761; E. Szczurowska et al., Brain Research Bulletin, 2015, 111:1-8).
[0112] Анализ активности химического соединения, применяемого в настоящем изобретении в качестве антагониста NR2B или лечения заболевания или расстройства центральной нервной системы (ЦНС), может быть произведен in vitro или in vivo. Оценка in vivo эффективности соединений согласно настоящему изобретению может быть выполнена с применением модели заболевания или расстройства ЦНС на животных, например модели на грызунах или приматах. Клеточные анализы могут быть выполнены с применением, например, линии клеток, выделенной из ткани, которая экспрессирует NR2B, или линии клеток, которая рекомбинантно экспрессирует NR2B. Кроме того, могут быть выполнены биохимические или основанные на механизме действия анализы, например измерение уровня цАМФ или цГМФ, нозерн-блот, полимеразная цепная реакция с обратной транскриптазой (ОТ-ПЦР) и т.д. Анализы in vitro включают анализы, которые определяют морфологию клеток, экспрессию белков и/или цитотоксичность, ингибирующую активность в отношении ферментов и/или последующие функциональные последствия обработки клеток химическими соединениями согласно настоящему изобретению. Дополнительные анализы in vitro количественно определяют способность указанного ингибитора связываться с молекулами белка или нуклеиновой кислоты внутри клетки. Связывание ингибитора может быть измерено путем введения радиоактивной метки в указанный ингибитор перед связыванием, выделения комплекса ингибитора/молекулы-мишени и определения количества связанной радиоактивной метки. В качестве альтернативы связывание ингибитора может быть определено путем выполнения эксперимента по конкурентному связыванию, в котором новые ингибиторы инкубируют с очищенными белками или нуклеиновыми кислотами, связанными с известными радиолигандами. Подробные условия проведения анализа соединения, применяемого в настоящем изобретении в качестве антагониста NR2B, приведены в примерах ниже. Вышеупомянутые анализы являются иллюстративными и не предназначены для ограничения объема настоящего изобретения. Специалисту в данной области техники следует понимать, что могут быть выполнены модификации традиционных анализов для разработки эквивалентных анализов, которые обеспечивают тот же результат.
[0113] Специалистам в данной области техники понятно, что идентификация и/или определение характеристик желаемых или эффективных соединений обычно включает оценку одного или более видов активности в модели на животных. Специалистам в данной области техники также понятно, что такие модели на животных не всегда точно воспроизводят испытания на людях (Bezard et al. Neuroscience 211:1, 2012). Среди прочего, модели на животных часто разрабатываются для имитации одного или более симптомов конкретного состояния человека, но не всегда возможно обеспечить, чтобы симптомы, наблюдаемые у животного, являлись результатом или были обусловлены тем же механизмом, который вызывает указанный симптом (симптомы) у людей, или даже определить, так ли это. Кроме того, нередко разрабатывается несколько моделей на животных для одного и того же заболевания, расстройства или состояния, каждая из которых может отражать или имитировать один или более признаков указанного заболевания, расстройства или состояния; специалистам в данной области техники понятно, что результаты в случае всех таких моделей не всегда точно согласуются между собой, и что может требоваться оценка и/или интерпретация для того, чтобы сделать выводы, достоверно прогнозирующие реакции человека. Более того, в частности в случае моделей неврологических состояний, описанных в настоящем документе, на животных, различные измерения или реакции, наблюдаемые в рамках указанной модели, могут быть более надежными, чем прочие при моделировании или прогнозировании реакции человека.
[0114] Далее приводится несколько примеров: в данной области техники известно, что доступные модели депрессии на животных имеют различные степени лицевой, конструктивной и прогностической валидности для депрессии и неодинаково способствуют нашему пониманию процессов избавления от депрессии. (Duman С.Н. Vitamins & Hormones 82:1-21, 2010).
[0115] Широко применяемые модели, релевантные относительно работы, описанной в настоящем документе, включают, например, тест принудительного плавания грызунов, который часто применяется для оценки антидепрессивной эффективности. (Can et al. J. Vis. Exp. 2012, 59:3638; Bogdanova et al. Physiol. Behav. 118:227, 2013). Для выполнения тестов принудительного плавания доступны различные протоколы. (Slattery & Cryan Nature Protocols 7:1009, 2012; Lucki et el., 2001, Psychopharmacology 155:315-322).
[0116] Модель каталепсии, вызванной галоперидолом (КВГ), применялась для оценки терапевтических агентов в отношении лечения некоторых неврологических состояний и/или симптомов. Например, модель КВГ, в частности, была рекомендована для применения для оценки терапевтических агентов в отношении их потенциальной ценности для защиты от каталепсии, связанной с болезнью Паркинсона (БП). (Steece-Collier et al. Exp. Neurol. 163:239, 2000). Кроме того, модель КВГ применялась для оценки антидепрессантов и, как сообщалось, обладала способностью выявлять различную активность среди антидепрессантов, что для сравнения могло быть выполнено в одном или более других анализах, таких как, например, тесты принудительного плавания. (Khisti et al. Indian J. Exp. Biol. 35:1297, 1997).
[0117] Сообщалось, что целый ряд различных моделей предоставляет информацию, релевантную относительно лечения эпилепсии, хотя разнообразие эпилептических синдромов и их причин может препятствовать применению какой-либо одной модели или испытания в качестве способа определения эффективности. Большинство моделей на животных, применяемых при исследовании эпилепсии, представляют собой модели эпилептического припадка, а не модели эпилепсии. Эпилепсия характеризуется периодическими спонтанными припадками, таким образом, испытание, такое как тест с провокацией припадка посредством электрошока с частотой импульсов 6 Гц (6 Hz seizure test), описанный в настоящем документе, в котором у интактного не страдающего от эпилепсии животного с помощью электрического тока индуцируют острый приступ, может не в полной мере являться моделью эпилепсии. ( Seizure 2011, 20:359-368). В то же время тест с провокацией припадка посредством электрошока с частотой импульсов 6 Гц считается потенциальным скринингом устойчивой к терапии эпилепсии. В описанном в настоящем документе анализе с применением теста с провокацией припадка посредством электрошока с частотой импульсов 6 Гц используется ток силой 44 мА, что обычно приводит к тому, что большинство противоэпилептических лекарственных средств теряет свою эффективность. Соответственно, было высказано предположение о том, тест с провокацией припадка посредством электрошока с частотой импульсов 6 Гц представляет собой подходящую модель устойчивых к терапии лимбических судорог. (Barton et al. Epilepsy Res. 47(3): 217-27, 2001).
Seizure 2011, 20:359-368). В то же время тест с провокацией припадка посредством электрошока с частотой импульсов 6 Гц считается потенциальным скринингом устойчивой к терапии эпилепсии. В описанном в настоящем документе анализе с применением теста с провокацией припадка посредством электрошока с частотой импульсов 6 Гц используется ток силой 44 мА, что обычно приводит к тому, что большинство противоэпилептических лекарственных средств теряет свою эффективность. Соответственно, было высказано предположение о том, тест с провокацией припадка посредством электрошока с частотой импульсов 6 Гц представляет собой подходящую модель устойчивых к терапии лимбических судорог. (Barton et al. Epilepsy Res. 47(3): 217-27, 2001).
[0118] Считается, что пентилентетразоловый тест (ПТЗ-тест), описанный в настоящем документе, позволяет прогнозировать активность противосудорожных лекарственных средств в отношении бессудорожных (малых или миоклонических) эпилептических припадков. Как описано в обзоре , различные противоэпилептические лекарственные средства, которые защищают от бессудорожных приступов у пациентов с эпилепсией, показали неудовлетворительные результаты в ПТЗ-тесте. ( W. Seizure 20:359-368, 2011). Таким образом, неопределенные или отрицательные данные, полученные в результате ПТЗ-теста, не обязательно означают, что испытываемая терапия (например, соединение) будет неэффективной в отношении пациентов с эпилепсией. Кроме того, такие соединения могли быть эффективными в другой модели эпилепсии на животных. Например, противоэпилептическое лекарственное средство может показать неудовлетворительные результаты в тесте с провокацией припадка посредством электрошока с частотой импульсов 6 Гц (в случае приступов, индуцированных с помощью электрического тока), но продемонстрировать эффективность в ПТЗ-тесте (в случае химически индуцированных приступов).
[0119] Специалистам в данной области техники следует понимать, что любой эффект, наблюдаемый в конкретном испытании с применением модели на животных, может иметь важное значение. Кроме того, специалистам в данной области техники следует понимать, что применение нескольких и различных моделей заболеваний на животных может положительно сказаться на оценке эффективности конкретного агента или лечения. Более того, специалистам в данной области техники следует понимать, что не требуется, чтобы каждая оценка конкретного агента в модели на животных обеспечивала убедительные доказательства активности; в некоторых случаях ясные доказательства активности в одном контексте могут быть более значительными, чем отсутствие доказательств активности в другом, в частности, поскольку понятно, что оптимизация условий реакции может выявить активность, не наблюдаемую в начальных исследованиях.
[0120] В контексте настоящего описания термины «лечение», «лечить» и «осуществление лечения» относятся к обращению вспять, ослаблению, задержке наступления или подавлению течения заболевания или расстройства или одного или более его симптомов, описанных в настоящем документе. В некоторых вариантах реализации лечение может быть проведено после развития одного или более симптомов. В других вариантах реализации лечение может быть проведено при отсутствии симптомов. Например, лечение может быть проведено в отношении восприимчивого индивидуума до появления симптомов (например, в свете симптомов в анамнезе и/или в свете генетических факторов или других факторов восприимчивости). Лечение также может быть продолжено после устранения симптомов, например, для предотвращения или отсрочки их повторения.
[0121] В соответствии со способом согласно настоящему изобретению указанные химические соединения и композиции могут быть введены с применением любого количества и любого способа введения, эффективного для лечения или уменьшения тяжести заболевания или расстройства ЦНС.
[0122] В некоторых вариантах реализации в соответствии со способом согласно настоящему изобретению указанные химические соединения и композиции могут быть введены с применением любого количества и любого способа введения, эффективного для лечения или уменьшения тяжести заболевания или расстройства, связанного с NR2B.
[0123] В некоторых вариантах реализации в соответствии со способом согласно настоящему изобретению указанные химические соединения и композиции могут быть введены с применением любого количества и любого способа введения, эффективного для лечения или уменьшения тяжести заболевания или расстройства ЦНС.
[0124] В некоторых вариантах реализации указанное заболевание или расстройство представляет собой депрессию с или без сопутствующего тревожного расстройства, например разовое и рекуррентное депрессивное расстройство, дистимическое расстройство, большое депрессивное расстройство, психотическую депрессию, предменструальное дисфорическое расстройство, послеродовую депрессию, сезонное аффективное расстройство (САР), расстройство настроения, терапевтически резистентную депрессию (ТРД, то есть большое депрессивное расстройство, при котором не было ответа на другие виды лекарственной терапии), депрессию, вызванную хроническим медицинским состоянием, таким как рак или хроническая боль, химиотерапия, хронический стресс и посттравматическое стрессовое расстройство.
[0125] В некоторых вариантах реализации указанное заболевание или расстройство представляет собой острое аффективное расстройство, например расстройство, выбранное из биполярных расстройств, включая биполярные маниакальные расстройства I и II типа.
[0126] В некоторых вариантах реализации настоящего изобретения предложен способ лечения расстройств, связанных со злоупотреблением психоактивными веществами, где лечение приводит к снижению толерантности и/или зависимости от лечения боли с помощью опиоидов, и/или лечения абстинентного синдрома, вызванного употреблением, например, алкоголя, опиоидов, героина и кокаина. В контексте настоящего описания термин «расстройства, связанные со злоупотреблением психоактивными веществами» включает зависимость от или злоупотребление психоактивными веществами с физиологической зависимостью или без нее. Психоактивные вещества, связанные с указанными расстройствами, включают алкоголь, амфетамины (или амфетаминоподобные вещества), кофеин, каннабис, кокаин, галлюциногенные вещества, летучие вещества наркотического действия, марихуану, никотин, опиоиды, фенциклидин (или фенциклидиноподобные соединения), седативно-снотворные средства или бензодиазепины и другие (или неизвестные) вещества и комбинации всех вышеперечисленных веществ.
[0127] В некоторых вариантах реализации расстройство, связанное со злоупотреблением психоактивными веществами, включает наркотические абстинентные расстройства, такие как алкогольная абстиненция с перцептивными расстройствами или без них; делирий при алкогольной абстиненции; амфетаминовая абстиненция; кокаиновая абстиненция; никотиновая абстиненция; опиоидная абстиненция; абстиненция, вызванная употреблением седативных, снотворных или анксиолитических средств, с перцептивными расстройствами или без них; делирий при абстиненции, вызванной употреблением седативных, снотворных или анксиолитических средств; и симптомы абстиненции, вызванной употреблением других веществ. Следует понимать, что указание на лечение никотиновой абстиненции включает лечение симптомов, связанных с отказом от курения. Другие расстройства, связанные со злоупотреблением психоактивными веществами, включают вызванное психоактивными веществами тревожное расстройство с возникновением во время абстиненции; вызванное психоактивными веществами расстройство настроения с возникновением во время абстиненции и вызванное психоактивными веществами нарушение сна с возникновением во время абстиненции.
[0128] В некоторых вариантах реализации указанное заболевание или расстройство представляет собой боль, например выбранную из болевых состояний, возникающих из самых разных источников, включая невропатическую боль (такую как постгерпетическая невралгия, травма/повреждение нерва, «-динии», например вульводиния, фантомная боль в ампутированной конечности, корневые авульсии, болезненная диабетическая нейропатия, компрессионная мононейропатия, ишемическая нейропатия, болезненная травматическая мононейропатия или болезненная полинейропатия), центральные болевые синдромы (потенциально вызываемые практически любым поражением на любом уровне нервной системы) и послеоперационные болевые синдромы (например, постмастэктомический синдром, постторакотомический синдром, боль в культе), боль в костях и суставах (остеоартрит, ревматоидный артрит, анкилозирующий спондилит), боль, вызванная повторяющимися движениями, туннельный синдром запястья, зубную боль, боль при раке, миофасциальную боль (мышечная травма, фибромиалгия), периоперационную боль (в общей хирургии, гинекологии), хроническую боль, дисменорею, а также боль, связанную со стенокардией, и воспалительную боль различного происхождения (например, остеоартрит, ревматоидный артрит, ревматическое заболевание, теносиновит и подагра), головную боль, мигрень и кластерную головную боль. В некоторых вариантах реализации указанное заболевание или расстройство связано с некупирующейся болью, такой как мигрень, фибромиалгия и невралгия тройничного нерва.
[0129] В некоторых вариантах реализации указанное заболевание или расстройство представляет собой мигренозное расстройство. Мигрень является одним из наиболее распространенных неврологических расстройств с распространенностью в мире, составляющей 10-15%. Треть пациентов с мигренью также страдает от кратковременных очаговых неврологических симптомов, именуемых аурой. Аура обычно начинается за период от нескольких минут до нескольких часов до головной боли, но может возникать во время фазы головной боли или даже при отсутствии головной боли. Одним из наиболее интересных аспектов ауры являются ее «растекающийся» (spreading) характер, позволяющий предполагать лежащий в основе механизм, который обеспечивает медленное распространение по прилегающей ткани мозга.
[0130] Накоплен большой объем данных, позволяющий предполагать, что распространяющаяся депрессия (РД), которая также может относиться к корковой распространяющейся депрессии (КРД), представляет собой электрофизиологический субстрат мигренозной ауры и потенциальный триггер головной боли. Иногда аура может возникать с последующей слабой головной болью или ее отсутствием. Помимо мигрени с аурой РД возникает при целом ряде значительных неврологических расстройств, например инсульте и травме головного мозга. Кроме того, РД способна активировать центральные и периферические тригеминоваскулярные ноцицептивные пути с латентным соответствием, которое возникает между аурой и головной болью у лиц, страдающих от мигрени. Наконец, что не менее важно, было продемонстрировано, что лекарственные средства для профилактики мигрени подавляют РД, из чего можно заключить, что РД представляет собой релевантную фармакологическую мишень при мигрени. (Shatillo et al. Neuropharm. 2015, 93:164-170).
[0131] РД представляет собой сильную деполяризацию нейрональных и глиальных мембран, которая распространяется в ткани головного мозга со скоростью, составляющей примерно 3 мм/мин. Волна деполяризации будет распознаваться путем регистрации постоянного тока (ПТ) на коре головного мозга у анестезированных крыс. Одновременно может быть измерено увеличение церебрального кровотока (ЦКТ) в твердой мозговой оболочке, свидетельствующее о расширении вен твердой мозговой оболочки. РД, вызываемая, когда локальные внеклеточные концентрации K+ превышают критический порог, связана с нарушением мембранных ионных градиентов, при этом сильный отток K+ и глутамата, как полагают, сначала деполяризует, а затем гиперполяризует соседние нейроны с облегчением распространения. РД может быть вызвана фармакологически путем применения глутамата, K+ и ингибиторов Na+/K+ - насоса, путем электростимуляции и путем повреждения тканей, такого как травма, кровоизлияние или ишемия. Прямые доказательства проявления РД в головном мозге человека in situ были получены с применением субдуральных электрофизиологических регистрации и интракортикальных многопараметрических электродов. (Ayata et al. Ann. Neurol. 2006, 59: 652-61).
[0132] В некоторых вариантах реализации указанное заболевание или расстройство представляет собой мигрень без ауры. В некоторых вариантах реализации указанное заболевание или расстройство представляет собой мигрень с аурой. В некоторых вариантах реализации указанное заболевание или расстройство представляет собой мигрень с типичной аурой. В некоторых вариантах реализации указанное заболевание или расстройство включает мигрень со стволовой аурой, гемиплегическую мигрень, ретинальную мигрень и/или хроническую мигрень.
[0133] В некоторых вариантах реализации мигрень может представлять собой мигрень, вызванную стрессом, «тихую» или ацефалгическую мигрень, синусовую мигрень, глазную мигрень, сезонную мигрень, синдром циклической мигрени, мигрень, сопровождающуюся гастростазом, и/или мигрень напряжения.
[0134] В некоторых вариантах реализации указанное заболевание или расстройство представляет собой первичное болевое расстройство в области головы, характеризующееся рецидивирующими головными болями, которые являются умеренными и сильными. Головная боль может затронуть одну половину головы и длиться от одного часа до нескольких суток. В некоторых вариантах реализации указанное заболевание или расстройство представляет собой головную боль напряжения и/или тригеминальную автономную цефалгию.
[0135] В некоторых вариантах реализации указанное заболевание или расстройство представляет собой кластерную головную боль. Кластерная головная боль представляет собой вид головной боли, которая повторяется в течение определенного периода времени. Как правило, пациенты с кластерными головными болями испытывают приступы от одного до трех раз в сутки в течение периода времени (кластерного периода), который может длиться от двух недель до трех месяцев. Кластерная головная боль обычно пробуждает человека от сна через один-два часа после того, как он ложится спать. Кластерные головные боли могут исчезнуть или перейти в состояние ремиссии на несколько месяцев или лет, а затем снова вернуться без предупреждения. В некоторых вариантах реализации указанная кластерная головная боль является хронической (например, кластерный период измеряется месяцами или годами, а не неделями). В некоторых вариантах реализации указанная кластерная головная боль является эпизодической.
[0136] В некоторых вариантах реализации головная боль может приводить к появлению симптомов, включающих тошноту, рвоту и повышенную чувствительность к свету, звукам или запахам. В некоторых вариантах реализации мигрень может приводить к появлению симптомов, включающих тошноту, рвоту и повышенную чувствительность к свету, звукам или запахам. В некоторых вариантах реализации осложнения мигрени включают мигренозный статус, персистирующую ауру без инфаркта, мигренозный инфаркт и/или эпилептический припадок, вызванный мигренозной аурой.
[0137] В некоторых вариантах реализации указанное заболевание или расстройство выбрано из нарушений сна и их осложнений, включая бессонницу, нарколепсию и идиопатическую гиперсомнию.
[0138] В некоторых вариантах реализации указанное заболевание или расстройство выбрано из расстройств ЦНС, характеризующихся гипервозбудимостью нейронов, таких как эпилепсия, судороги, эпилептические припадки, парциальные припадочные расстройства, генерализованные припадочные расстройства, такие как малые эпилептические припадки, атонические, миоклонические, тонические, тоникоклонические или большие («grand-mal») приступы, эпилептический статус, корковая распространяющаяся депрессия, мигренозные головные боли, церебральный паралич, синдром Отахара, синдром ломкой Х-хромосомы, эпилептические припадки у детей или вызванные генетическими причинами эпилептические припадки, такие как синдром Веста, синдром Леннокса-Гасто и синдром Ангельмана, туберозный склероз, внутричерепная гипертензия, отек центральной нервной системы, нейрональная токсичность, такая как токсичность, вызванная воздействием алкоголя, патофизиологические эсрфекты травмы головы, инсульт, ишемия, гипоксия и другие состояния, возникающие в результате или вызывающие ионный дисбаланс в центральной нервной системе или синхронные разряды популяций нейронов.
[0139] В некоторых вариантах реализации указанное заболевание или расстройство характеризуется возникновением эпилептического припадка. Приступы являются результатом неконтролируемых разрядов электрической активности в головном мозге. Приступ обычно проявляется в виде внезапных непроизвольных разрушительных и часто деструктивных сенсорных, моторных и когнитивных явлений. Приступы часто связаны с физическим повреждением тела (например, прикусыванием языка, переломом конечностей и ожогами), полной потерей сознания и недержанием. Например, типичный приступ может начинаться как спонтанное дрожание руки или ноги и прогрессировать в течение нескольких секунд или минут до ритмичного движения всего тела, потери сознания и выделения мочи или кала. Существуют как судорожные, так и бессудорожные приступы. Судорожные приступы могут представлять собой генерализованные приступы или парциальные приступы. Существует шесть основных видов генерализованных приступов: тоникоклонические, тонические, клонические, миоклонические, малые и атонические приступы. Бессудорожный приступ, например малый приступ, проявляется в виде снижения уровня сознания и обычно длится примерно 10 секунд.
[0140] В некоторых вариантах реализации указанное заболевание или расстройство представляет собой эпилепсию. Эпилепсия представляет собой нарушение в головном мозге, характеризующееся устойчивой предрасположенностью к возникновению эпилептических припадков и нейробиологическими, когнитивными, психологическими и социальными последствиями указанного состояния. (R.S. Fisher et al., Epilepsia, 2005, 46(4): 470-472). Эпилепсия может представлять собой проявление по меньшей мере одного эпилептического припадка. Эпилептический припадок представляет собой временное проявление признаков и/или симптомов вследствие аномальной повышенной или синхронной нейрональной активности в головном мозге. Эпилепсия поражает людей всех возрастов, однако чаще всего эпилепсия возникает в детском и пожилом возрасте (Institute of Medicine 2012). Точная причина эпилепсии неясна. Некоторые известные причины эпилепсии включают травму головы, инсульт, опухоли, инфекцию или аномалии головного мозга.
[0141] Эпилепсия классифицируется как идиопатическая (имеет генетическую причину) или симптоматическая (причина неизвестна) и далее подразделяется на генерализованную, затрагивающую оба полушария головного мозга, или парциальную эпилепсию, которая затрагивает одно полушарие мозга. Примеры идиопатической генерализованной эпилепсии включают детскую абсансную эпилепсию, ювенильную миоклоническую эпилепсию и эпилепсию с большими приступами. Примеры идиопатической парциальной эпилепсии включают доброкачественную фокальную эпилепсию детского возраста. Симптоматическая генерализованная эпилепсия включает синдром Веста, синдром Леннокса-Гасто и другие. Симптоматическая парциальная эпилепсия включает височную эпилепсию, лобную эпилепсию и другие.
[0142] В некоторых вариантах реализации припадочное расстройство представляет собой детское припадочное расстройство. Возможность классифицировать случай припадочного расстройства, например эпилепсии, как конкретный синдром чаще возникает в случае детей, поскольку появление приступов обычно происходит рано. Менее серьезные примеры представляют собой доброкачественную роландическую эпилепсию, детскую абсансную эпилепсию и ювенильную миокпоническую эпилепсию (A. Neligan et al., Handbook of clinical neurology 2012, 107:113-33). Другие примеры детских судорожных приступов включают фебрильные судороги, инфантильные спазмы и неонатальные судороги.
[0143] В некоторых вариантах реализации припадочное расстройство представляет собой лобную эпилепсию, ювенильную миокпоническую эпилепсию, миокпоническую эпилепсию, абсансную эпилепсию, синдром Леннокса-Гасто, синдром Ландау-Клеффнера, синдром Драве, прогрессирующие миоклонические эпилепсии, рефлекторную эпилепсию, синдром Расмуссена, височную эпилепсию, лимбическую эпилепсию, эпилептический статус, абдоминальную эпилепсию, массивный билатеральный миоклонус, катамениальную эпилепсию, джексоновскую эпилепсию, болезнь Лафоры или фотосенситивную эпилепсию или комбинацию одного или нескольких из них.
[0144] Для большинства случаев эпилепсии указанное заболевание является хроническим и требует лекарственных средств, подходящих для длительного лечения. Противоэпилептические лекарственные средства (ПЭЛ средства) в общем случае подавляют нейронную активность с помощью различных механизмов, включая изменение активности ионных каналов клеточной мембраны и склонности к генерации потенциалов действия или вспышек потенциалов действия. Указанные желаемые терапевтические эффекты часто сопровождаются нежелательным седативным побочным эффектом. Другие лекарственные средства обладают значительными побочными эффектами, не являющимися неврологическими, такими как гиперплазия десен, косметически нежелательное разрастание десен и/или утолщение черепа, как это происходит в случае фенитоина. Несмотря на то, что было доказано, что длительное употребление ПЭЛ средств является эффективным для большинства пациентов, страдающих от эпилепсии, постоянные побочные эффекты могут вызывать значительное ухудшение качества жизни пациентов. Кроме того, несмотря на доступный в настоящее время арсенал старых и новых ПЭЛ средств почти треть пациентов с эпилепсией не чувствительна (например, не поддается лечению) к любым фармакологическим схемам. (М.М. Castel-Branco et al., Methods Find Exp Clin Pharmacol, 2009, 31(2):101-106). Следовательно, существует значительная необходимость в разработке новых и более эффективных ПЭЛ средств.
[0145] В некоторых вариантах реализации припадочное расстройство не поддается лечению. Тяжелые синдромы с диффузной дисфункцией мозга, также именуемые эпилептическими энцефалопатиями, не поддаются лечению, существующему в настоящее время. Эпилептические энцефалопатии составляют группу расстройств, в которых сама эпилептическая активность, как считается, способствует тяжелым когнитивным расстройствам или тяжелому снижению когнитивных способностей, значительно превышающему то, которое можно было бы ожидать исключительно от лежащей в основе патологии. В других вариантах реализации не поддающееся лечению припадочное расстройство представляет собой расстройство, связанное с миграцией нейронов, такой как микрогирия человека. (S. Bandyopadhyay et al., Epilepsy Research, 2006, 72:127-139). Другое серьезное расстройство в подгруппе пациентов, подвергнутых хирургическому лечению фармакорезистентных приступов, представляет собой фокальную дисплазию коры головного мозга. Противосудорожная лекарственная терапия часто неэффективна в отношении пациентов с такими корковыми мальформациями. В некоторых вариантах реализации припадочное расстройство включает корковую гипервозбудимость при фокальной корковой дисплазии (мальформациях). (S. Bandyopadhyay et al., Epilepsy Research, 2006, 72:127-139).
[0146] В некоторых вариантах реализации припадочное или эпилептическое расстройство вызвано генетической аномалией. Полагают, что генетика играет большую роль в развитии различных видах эпилепсии с помощью ряда механизмов. Для некоторых из них были определены простые и сложные типы наследования. Недавние исследования секвенирования экзома и генома положили начало выявлению ряда генных мутаций «de novo», которые ответственны за некоторые эпилептические энцефалопатии, включая CHD2, и SYNGAP1, и DMN1, GABBR2, FASN и RYR3. Пациенты с эпилептической энцефалопатией, представляющей собой синдром Веста, демонстрируют ясные клинические электрофизиологические признаки, обычно проявляющиеся в период от 3 до 12 месяцев в виде кластеров инфантильных спазмов (ИС) и характерной картины электроэнцефалограммы (ЭЭГ), называемой гипсаритмией. Синдром Веста был связан с мутациями в генах ARX, CDKL5, STXBP1 и ST3GAL3, а также с различными вариациями числа копий (ВЧК, CNVs). (J.R. Lemke et al., Ann Neurol, 2014, 75(1), 147-154). Мутации в генах GRIN2A и GRIN2B, кодирующих субъединицы NR2A и NR2B NMDA-рецептора, связаны с несколькими нарушениями развития нервной системы. Мутации в гене GRIN2A недавно были обнаружены при идиопатической фокальной эпилепсии с роландическими спайками и связанными с ней эпилептическими энцефалопатиями, то есть при синдроме Ландау-Клеффнера, синдроме эпилепсии с непрерывными комплексами «спайк-волна» во время медленного сна и несиндромальной эпилепсии, связанной с умственной отсталостью (УО). В отличие от этого, ген GRIN2B до сих пор не описывался как ген эпилепсии, но неоднократно рассматривался как предполагаемый ген-«кандидат» в случае приступов, а мутации были обнаружены у пациентов с УО и шизофренией. (J.R. Lemke et al., Ann Neurol, 2014, 75(1), 147-154).
[0147] В некоторых вариантах реализации указанное заболевание или расстройство представляет собой двигательное расстройство. Двигательные расстройства включают болезнь Паркинсона, дискинезии (включая побочные эффекты, сопутствующие применению L-дигидроксифенилаланина (L-ДОФА) в обычных дозах), позднюю дискинезию, лекарственный паркинсонизм, постэнцефалитический паркинсонизм, прогрессирующий надъядерный паралич, множественную системную атрофию, кортикобазальную дегенерацию, комплекс «паркинсонизм-боковой амиотрофический склероз (БАС)-деменция», кальцификацию базальных ганглиев, акинезию, акинетико-ригидный синдром, брадикинезию, дистонию, вызванный лекарственными средствами паркинсонизм, синдром Туретта, болезнь Гентингтона, тремор, хорею, миоклонию, тикозное расстройство и дистонию.
[0148] В некоторых вариантах реализации указанное двигательное расстройство представляет собой одну или более из акинезии и акинетико-ригидных синдромов, дискинезии и вызванного лекарственными средствами паркинсонизма (такого как нейролептический паркинсонизм, злокачественный нейролептический синдром, нейролептическая острая дистония, нейролептическая острая акатизия, поздняя нейролептическая дискинезия и вызванный лекарственными средствами постуральный тремор). Примеры «акинетико-ригидных синдромов» включают болезнь Паркинсона, лекарственный паркинсонизм, постэнцефалитический паркинсонизм, прогрессирующий надъядерный паралич, множественную системную атрофию, кортикобазальную дегенерацию, комплекс «паркинсонизм-БАС-деменция» и кальцификацию базальных ганглиев. Примеры дискинезий включают тремор (включая тремор покоя, постуральный тремор и интенционный тремор), хорею (такую как хорея Сиденгама, болезнь Гентингтона, доброкачественная наследственная хорея, нейроакантоцитоз, симптоматическая хорея, лекарственная хорея и гемибаллизм), миоклонию (включая генерализованную миоклонию и фокальную миоклонию), тики (включая простые тики, сложные тики и симптоматические тики) и дистонию (включая генерализованную дистонию, такую как идиопатическая дистония, лекарственная дистония, симптоматическая дистония и пароксизмальная дистония, и фокальную дистонию, такую как блефароспазм, оромандибулярная дистония, спастическая дисфония, спастическая кривошея, аксиальная дистония, дистонический писчий спазм и гемиплегическая дистония).
[0149] В некоторых вариантах реализации указанное заболевание или расстройство представляет собой болезнь Паркинсона.
[0150] В некоторых вариантах реализации указанное заболевание или расстройство представляет собой болезнь Гентингтона.
[0151] В некоторых вариантах реализации указанное заболевание или расстройство представляет собой когнитивную дисфункцию, связанную с расстройствами, включающими шизофрению, болезнь Альцгеймера, лобно-височную деменцию, болезнь Пика, болезнь телец Леви и другие старческие деменции (например, сосудистую деменцию).
[0152] В некоторых вариантах реализации настоящего изобретения предложен способ лечения расстройства, описанного в настоящем документе, включающий введение химического соединения согласно настоящему изобретению совместно с одним или более фармацевтическими агентами. Подходящие фармацевтические агенты, которые могут быть применены в комбинации с химическими соединениями согласно настоящему изобретению, включают селективные ингибиторы обратного захвата серотонина (СИОЗС), например, при лечении депрессии; схемы дофаминзаместительной терапии и агонисты дофамина, например, при лечении болезни Паркинсона; типичные антипсихотические средства; атипичные антипсихотические средства; противосудорожные средства; стимуляторы; средства терапии болезни Альцгеймера; противомигренозные агенты и анксиолитические агенты.
[0153] Подходящие СИОЗС включают циталопрам, дапоксетин, эсциталопрам, флуоксетин, флувоксамин, индальпин, пароксетин, сертралин, вилазодон и зимелидин.
[0154] Подходящие схемы дофаминзаместительной терапии включают замену L-ДОФА на ингибитор ДОФА-декарбоксилазы, такой как карбидопа.
[0155] Подходящие агонисты дофаминовых рецепторов включают аплиндор, апоморфин, бромокриптин, каберголин, циладопа, дигидроэргокриптин, лизурид, пардопрунокс, перголид, пирибедил, прамипексол, ропинирол и ротиготин.
[0156] Подходящие типичные антипсихотические средства включают хлорпромазин, тиоридазин, мезоридазин, левомепромазин, локсапин, молиндон, перфеназин, тиотиксен, трифлуоперазин, галоперидол, флуфеназин, дроперидол, зуклопентиксол, флупентиксол и прохлорперазин.
[0157] Подходящие атипичные антипсихотические средства включают амисульприд, арипипразол, азенапин, блонансерин, клотиапин, клозапин, илоперидон, луразидон, мосапрамин, оланзапин, палиперидон, пероспирон, кветиапин, ремоксиприд, рисперидон, сертиндол, сульпирид, зипрасидон, зотепин, бисрепрунокс, пимавансерин и вабиказерин.
[0158] Подходящие противосудорожные средства включают фенитоин, карбамазепин, барбитураты, фенобарбитал, фенобарбитал, мефобарбитал, триметадион, мефенитоин, параметадион, фентенилат, фенацемид, метарбитал, бензхлорпропамид, фенсуксимид, примидон (priraidone), метсуксимид, этотоин, аминоглутетимид (aminoglutethinide), диазепам, клоназепам, клоразепат, фосфенитоин, этосуксимид, вальпроат, фелбамат, габапентин, ламотриджин, топирамат, вигабатрин (vigrabatrin), тиагабин, зиамид, клобазам, тиопентал, мидазолам, пропофол, леветирацетам, окскарбазепин, CCPene и GYKI 52466.
[0159] Подходящие стимуляторы включают Аддералл (амфетамин, смешанные соли дексамфетамина), метилфенидат, дексамфетамин, дексметилфенидат и лиздексамфетамин.
[0160] Подходящие средства терапии болезни Альцгеймера включают ингибиторы ацетилхолинэстеразы, такие как ривастигмин, донепезил, галантамин и гуперзин; агонисты альфа-7 никотиновых рецепторов, такие как энцениклин; и лекарственные средства, которые снижают уровень пептида Аβ42, такие как ингибиторы бета-секретазы (BACE), модуляторы гамма-секретазы и антитела к бета-амилоидному пептиду.
[0161] Подходящие противомигренозные лекарственные средства включают алкалоиды спорыньи (например, эрготамин и дигидроэрготамина мезилат). Другие подходящие противомигренозные лекарственные средства включают триптаны, являющиеся агонистами рецептора 5-HT1D, такие как суматриптан, алмотриптан, элетриптан, фроватриптан, наратриптан, ризатриптан и золмитриптан.
[0162] Другие подходящие агенты для применения для лечения или предотвращения головной боли (головных болей) или мигрени включают обезболивающее средство (например, аспирин, напроксен, ибупрофен и ацетаминофен), лекарственное средство от тошноты, кофеин, антигистаминные средства, изометептен мукат, дивалпрекс натрия/вальпроат натрия, топирамат, метопролол, пропранолол, тимолол, лизиноприл, кандесартан, атенолол, надолол, дилтиазем, нимодипин, верапамил, амитриптилин, нортриптилин, имипрамин, доксепин, протриптилин, пароксетин, флуоксетин, сертралин, топирамат, габапентин и дивалпрекс натрия.
[0163] В некоторых вариантах реализации один или более других подходящих агентов могут быть объединены с химическим соединением согласно настоящему изобретению для предотвращения и/или лечения головной боли. В некоторых вариантах реализации один или более других подходящих агентов могут быть объединены с химическим соединением согласно настоящему изобретению для предотвращения и/или лечения мигрени. В некоторых вариантах реализации один или более других подходящих агентов могут быть объединены с химическим соединением согласно настоящему изобретению для предотвращения и/или лечения мигрени с аурой.
[0164] В некоторых вариантах реализации один или более других подходящих агентов могут быть объединены с химическим соединением согласно настоящему изобретению для предотвращения и/или лечения кластерной головной боли. Было продемонстрировано, что кластерную головную боль лечат лекарственные средства, снижающие кровяное давление. Соответственно, в некоторых таких вариантах реализации химическое соединение согласно настоящему изобретению может быть объединено, например, с верапамилом, литием, дивалпрексом натрия, преднизоном, эрготамина тартратом, мелатонином, триптаном (например, суматриптаном), кислородом, лидокаином для интраназального введения или любым другим подходящим агентом для предотвращения и/или лечения кластерной головной боли.
[0165] Подходящие анксиолитические лекарственные средства включают модуляторы бензодиазепиновых рецепторов, такие как диазепам, алпразолам, лоразепам и клоназепам.
[0166] Другие подходящие агенты для применения совместно с химическим соединением согласно настоящему изобретению включают мемантин и модафинил.
[0167] Точное требуемое количество будет варьироваться от субъекта к субъекту в зависимости от вида, возраста и общего состояния субъекта, тяжести инфекции, конкретного агента, способа его введения и тому подобного. Химические соединения согласно настоящему изобретению предпочтительно, готовят в единичной лекарственной форме для удобства введения и постоянства дозировки. Выражение «единичная лекарственная форма» в контексте настоящего описания относится к физически дискретной единице агента, подходящей для пациента, подлежащего лечению. Однако следует понимать, что суммарное суточное потребление химических соединений и композиций согласно настоящему изобретению будет определяться лечащим врачом по результатам тщательной медицинской оценки. Конкретная величина эффективной дозы для любого конкретного пациента или организма будет зависеть от целого ряда факторов, включая расстройство, подлежащее лечению, и тяжесть указанного расстройства; активность конкретного применяемого химического соединения; конкретную применяемую композицию; возраст, массу тела, общее состояние здоровья, пол и рацион питания пациента; время введения, способ введения и скорость выведения конкретного применяемого химического соединения; продолжительность лечения; лекарственные средства, применяемые в комбинации или параллельно с конкретным применяемым химическим соединением, и другие подобные факторы, хорошо известные в области медицины. Термин «пациент» в контексте настоящего описания означает животное, предпочтительно млекопитающее и наиболее предпочтительно человека.
[0168] В некоторых вариантах реализации введение может включать дозирование, которое является дробным (например, несколько доз (то есть, по меньшей мере две дозы), разделенных во времени) и/или периодическим (например, отдельные дозы, разделенные общим периодом времени) дозированием. В некоторых вариантах реализации схема дозирования для конкретного активного агента может включать дробное или непрерывное введение, например, для обеспечения конкретного желаемого фармакокинетического профиля или другой картины воздействия в одной или более тканях или текучих средах, представляющих интерес в случае субъекта, получающего терапию. В некоторых вариантах реализации дробное дозирование может включать дозирование, которое происходит раз в сутки, раз в двое суток, раз в неделю, раз в две недели или раз в месяц.
[0169] В некоторых вариантах реализации введение может включать дозирование, при котором в течение определенного времени производится титрование доз для достижения целевой дозировки. В некоторых вариантах реализации увеличение количества доз и/или усиление режима дозирования может происходить с недельными интервалами по результатам клинической реакции и переносимости у субъекта.
[0170] В некоторых вариантах реализации одна или более особенностей схемы дозирования со временем могут быть модифицированы (например, может быть произведено увеличение или уменьшение количества активного ингредиента в любой отдельной дозе, увеличение или уменьшение временных интервалов между введением доз и т.д.), например, для оптимизации желаемого терапевтического эффекта или реакции.
[0171] В некоторых вариантах реализации фармацевтически приемлемые композиции согласно настоящему изобретению могут быть введены раз в неделю. В некоторых вариантах реализации фармацевтически приемлемые композиции согласно настоящему изобретению могут быть введены раз в сутки. В некоторых вариантах реализации фармацевтически приемлемые композиции согласно настоящему изобретению могут быть введены два раза в сутки. В некоторых вариантах реализации фармацевтически приемлемые композиции согласно настоящему изобретению могут быть введены три раза в сутки. В некоторых вариантах реализации фармацевтически приемлемые композиции согласно настоящему изобретению могут быть введены с пищей или без нее.
[0172] В некоторых вариантах реализации фармацевтически приемлемые композиции согласно настоящему изобретению могут быть введены для лечения острых состояний. Острые состояния могут начинаться внезапно. Симптомы острых состояний могут проявляться и быстро изменяться или ухудшаться. В некоторых вариантах реализации фармацевтически приемлемые композиции согласно настоящему изобретению могут быть введены для лечения хронических состояний. Хронические состояния могут представлять собой медленно развивающиеся состояния. Хроническое состояние может развиваться и ухудшаться в течение длительного периода времени.
[0173] В некоторых вариантах реализации фармацевтически приемлемые композиции согласно настоящему изобретению могут быть введены перед приступом. В некоторых вариантах реализации фармацевтически приемлемые композиции согласно настоящему изобретению могут быть введены после приступа. В некоторых вариантах реализации фармацевтически приемлемые композиции согласно настоящему изобретению могут быть введены пациенту при угрозе приступа.
[0174] Фармацевтически приемлемые композиции согласно настоящему изобретению могут быть введены людям и животным перорально, ректально, парентерально, интрацистернально, интравагинально, внутрибрюшинно, местно (в виде порошков, мазей или капель), буккально, в виде перорального или назального спрея или тому подобного в зависимости от тяжести инфекции, подвергаемой лечению. В некоторых вариантах реализации химические соединения согласно настоящему изобретению могут быть введены перорально или парентерально при уровне дозировки, составляющем от примерно 0,01 мг/кг до примерно 50 мг/кг и предпочтительно от примерно 1 мг/кг до примерно 25 мг/кг массы тела субъекта в сутки, один или несколько раз в сутки с получением желаемого терапевтического эффекта.
[0175] Жидкие лекарственные формы для перорального введения включают фармацевтически приемлемые эмульсии, микроэмульсии, растворы, суспензии, сиропы и эликсиры. Помимо активных соединений жидкие лекарственные формы могут содержать инертные разбавители, широко применяемые в данной области техники, такие как вода или другие растворители, солюбилизирующие агенты и эмульгаторы, такие как этиловый спирт, изопропиловый спирт, этилкарбонат, этилацетат, бензиловый спирт, бензилбензоат, пропиленгликоль, 1,3-бутиленгликоль, диметилформамид, масла (в частности, хлопковое, арахисовое, кукурузное, зародышевое, оливковое, касторовое и кунжутное масла), глицерин, тетрагидрофурфуриловый спирт, полиэтиленгликоли и сложные эфиры жирных кислот и сорбита, и их смеси. Помимо инертных разбавителей указанные пероральные композиции также могут содержать адъюванты, такие как смачивающие агенты, эмульгирующие и суспендирующие агенты, подслащивающие, ароматизирующие и отдушивающие агенты.
[0176] Препараты для инъекций, например стерильные водные или маслянистые суспензии для инъекций, могут быть приготовлены согласно методикам, известным в данной области техники, с применением подходящих диспергирующих или смачивающих агентов и суспендирующих агентов. Стерильный препарат для инъекций также может представлять собой стерильный раствор, суспензию или эмульсию для инъекций в нетоксичном, подходящем для парентерального введения разбавителе или растворителе, например, находиться в виде раствора в 1,3-бутандиоле. К числу приемлемых переносящих сред и растворителей, которые могут быть применены, относятся вода, раствор Рингера согласно Фармакопее США и изотонический раствор хлорида натрия. Кроме того, в качестве растворителя или суспендирующей среды традиционно применяют стерильные нелетучие масла. Для указанной цели может быть применено любое мягкое нелетучее масло, включая синтетические моно- или диглицериды. Кроме того, для получения препаратов для инъекций применяют жирные кислоты, такие как олеиновая кислота.
[0177] Составы для инъекций могут быть стерилизованы, например, путем фильтрования через фильтр, задерживающий бактерии, или путем включения стерилизующих агентов в форме стерильных твердых композиций, которые могут быть растворены или диспергированы в стерильной воде или другой стерильной среде для инъекций перед применением.
[0178] Для того чтобы продлить действие химического соединения согласно настоящему изобретению, часто желательно замедлить всасывание указанного химического соединения в результате подкожной или внутримышечной инъекции. Это может быть осуществлено путем применения жидкой суспензии кристаллического или аморфного вещества с плохой растворимостью в воде. В таком случае скорость всасывания указанного химического соединения зависит от скорости его растворения, которая, в свою очередь, может зависеть от размера кристаллов и кристаллической формы. В качестве альтернативы замедленное всасывание парентерально вводимой формы химического соединения осуществляется путем растворения или суспендирования указанного химического соединения в масляной переносящей среде. «Депо-формы» для инъекций получают путем образования микрокапсульных матриц указанного химического соединения в биоразлагаемых полимерах, таких как полилактид-полигликолид. В зависимости от соотношения химического соединения и полимера и природы конкретного применяемого полимера можно регулировать скорость высвобождения химического соединения. Примеры других биоразлагаемых полимеров включают поли(ортоэфиры) и поли(ангидриды). «Депо-составы» для инъекций также получают путем включения указанного химического соединения в липосомы или микроэмульсии, которые совместимы с тканями организма.
[0179] Композиции для ректального или вагинального введения предпочтительно представляют собой суппозитории, которые могут быть получены путем смешивания химических соединений согласно настоящему изобретению с подходящими нераздражающими вспомогательными веществами или носителями, такими как масло какао, полиэтиленгликоль или суппозиторный воск, которые являются твердыми при температуре окружающей среды, но жидкими при температуре тела и, таким образом, плавятся в прямой кишке или полости влагалища и высвобождают указанное активное химическое соединение.
[0180] Твердые лекарственные формы для перорального введения включают капсулы, таблетки, пилюли, порошки и гранулы. В таких твердых лекарственных формах указанное активное химическое соединение смешивают с по меньшей мере одним инертным, фармацевтически приемлемым носителем, таким как цитрат натрия или гидрофосфат кальция, и/или а) наполнителями или добавками (extenders), такими как крахмалы, лактоза, сахароза, глюкоза, маннитол и кремниевая кислота, b) связующими веществами, такими как карбоксиметилцеллюлоза, альгинаты, желатин, поливинилпирролидинон, сахароза и гуммиарабик, с) увлажнителями, такими как глицерин, d) разрыхляющими агентами, такими как агар-агар, карбонат кальция, картофельный или тапиоковый крахмал, альгиновая кислота, некоторые силикаты и карбонат натрия, е) агентами, замедляющими растворение, такими как парафин, f) ускорителями всасывания, такими как четвертичные аммониевые соединения, r) смачивающими агентами, такими как цетиловый спирт и моностеарат глицерина, h) абсорбентами, такими как каолин и бентонитовая глина, и i) смазывающими веществами, такими как тальк, стеарат кальция, стеарат магния, твердые полиэтиленгликоли, лаурилсульфат натрия, и их смесями. В случае капсул, таблеток и пилюль лекарственная форма также может содержать буферные агенты.
[0181] Твердые композиции подобного типа также могут быть применены в качестве наполнителей в мягких и твердых желатиновых капсулах с применением таких вспомогательных веществ, как лактоза или молочный сахар, а также высокомолекулярные полиэтиленгликоли и тому подобное. Твердые лекарственные формы, представляющие собой таблетки, драже, капсулы, пилюли и гранулы, могут быть получены с покрытиями и оболочками, такими как энтеросолюбильные покрытия и другие покрытия, хорошо известные в области приготовления фармацевтических составов. Они необязательно могут содержать замутняющие агенты и также могут иметь такой состав, что они высвобождают активный ингредиент (ингредиенты) только или преимущественно в определенной части кишечного тракта, необязательно, с отсрочкой. Примеры композиций включения, которые могут быть применены, включают полимерные вещества и воски. Твердые композиции подобного типа также могут быть применены в качестве наполнителей в мягких и твердых желатиновых капсулах с применением таких вспомогательных веществ, как лактоза или молочный сахар, а также высокомолекулярные полиэтиленгликоли и тому подобное.
[0182] Активные химические соединения также могут находиться в микрокапсулированной форме с одним или более вспомогательными веществами, указанными выше. Твердые лекарственные формы, представляющие собой таблетки, драже, капсулы, пилюли и гранулы, могут быть получены с покрытиями и оболочками, такими как энтеросолюбильные покрытия, покрытия, регулирующие высвобождение, и другие покрытия, хорошо известные в области приготовления фармацевтических составов. В таких твердых лекарственных формах указанное активное химическое соединение может быть смешано с по меньшей мере одним инертным разбавителем, таким как сахароза, лактоза или крахмал. Такие лекарственные формы также могут содержать, что является обычной практикой, дополнительные вещества, отличные от инертных разбавителей, например смазывающие вещества для получения таблеток и другие вспомогательные вещества для получения таблеток, такие как стеарат магния и микрокристаллическая целлюлоза. В случае капсул, таблеток и пилюль лекарственные формы также могут содержать буферные агенты. Они необязательно могут содержать замутняющие агенты и также могут иметь такой состав, что они высвобождают активный ингредиент (ингредиенты) только или преимущественно в определенной части кишечного тракта, необязательно, с отсрочкой. Примеры заключающих композиций, которые могут быть применены, включают полимерные вещества и воски.
[0183] Лекарственные формы для местного или чрескожного введения химического соединения согласно настоящему изобретению включают мази, пасты, кремы, лосьоны, гели, порошки, растворы, спреи, ингаляторы или пластыри. Указанное активное химическое соединение смешивают в стерильных условиях с фармацевтически приемлемым носителем и любыми необходимыми консервантами или буферами, если это потребуется. Офтальмологический состав, ушные капли и глазные капли также рассматриваются как входящие в объем настоящего изобретения. Кроме того, настоящее изобретение предполагает применение трансдермальных пластырей, которые обладают дополнительным преимуществом обеспечения регулируемой доставки химического соединения в организм. Такие лекарственные формы могут быть приготовлены путем растворения или распределения указанного химического соединения в подходящей среде. Для увеличения поступления указанного химического соединения через кожу также могут быть применены усилители всасывания. Скорость можно регулировать или путем обеспечения мембраны, регулирующей скорость, или путем диспергирования указанного химического соединения в полимерной матрице или геле.
[0184] В контексте настоящего описания термины «комбинация», «объединенный» и родственные термины относятся к одновременному или последовательному введению терапевтических агентов согласно настоящему изобретению. Например, химическое соединение согласно настоящему изобретению может быть введено с другим терапевтическим агентом одновременно или последовательно в отдельных единичных лекарственных формах или совместно в одной единичной лекарственной форме. Соответственно, в настоящем изобретении предложена одна единичная лекарственная форма, содержащая химическое соединение формулы (I), дополнительный терапевтический агент и фармацевтически приемлемый носитель, адъювант или переносящую среду.
[0185] В некоторых вариантах реализации различные агенты, вводимые в комбинации, могут быть введены с помощью различных способов доставки и/или согласно различным режимам. В качестве альтернативы или дополнительно в некоторых вариантах реализации одну или более доз первого активного агента вводят по существу одновременно с и в некоторых вариантах реализации с помощью общего способа и/или в составе одной композиции с одним или более другими активными агентами.
[0186] Количество обоих соединений, представляющих собой предложенное химическое соединение и дополнительный терапевтический агент (в композициях, которые содержат дополнительный терапевтический агент, описанный выше), которые могут быть объединены с веществами-носителями с получением одной лекарственной формы, будет варьироваться в зависимости от подвергаемого лечению носителя (host) и конкретного способа введения. Предпочтительно композиции согласно настоящему изобретению должны быть приготовлены так, чтобы могла быть введена доза предложенного химического соединения, составляющая 0,01-100 мг/кг массы тела/сутки.
[0187] В композициях, которые содержат дополнительный терапевтический агент, указанный дополнительный терапевтический агент и химическое соединение согласно настоящему изобретению могут действовать синергически. Таким образом, количество дополнительного терапевтического агента в таких композициях будет меньше, чем количество, требующееся в монотерапии с применением только указанного терапевтического агента. В составе таких композиций может быть введена доза указанного дополнительного терапевтического агента, составляющая 0,01-100 нг/кг массы тела/сутки.
[0188] Количество дополнительного терапевтического агента, присутствующего в композициях согласно настоящему изобретению, будет не больше количества, которое обычно было бы введено в составе композиции, содержащей указанный терапевтический агент в качестве единственного активного агента. Предпочтительно количество дополнительного терапевтического агента в композициях, предложенных в настоящем описании, будет составлять от примерно 50% до 100% от количества, обычно присутствующего в композиции, содержащей указанный агент в качестве единственного терапевтически активного агента.
[0189] В некоторых вариантах реализации настоящего изобретения предложено лекарственное средство, содержащее по меньшей мере одно химическое соединение формулы (I) и фармацевтически приемлемый носитель, адъювант или переносящую среду.
[0190] В некоторых вариантах реализации настоящего изобретения предложено применение химического соединения формулы (I) для получения лекарственного средства для лечения заболевания или расстройства ЦНС.
Общие способы синтеза
[0191] Химические соединения формулы (I) могут быть синтезированы согласно схеме 1 и/или с применением способов, известных в данной области техники.
Схема 1
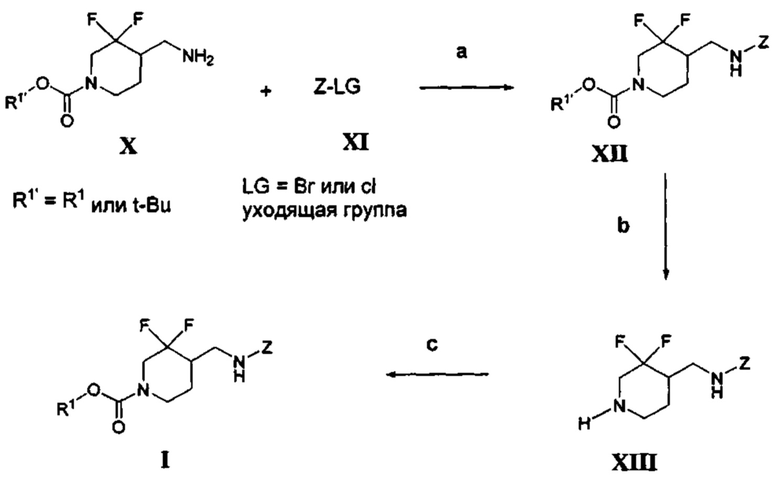
а. основание (например, диизопропилэтиламин), органический растворитель (например, н-бутанол), нагревание или условия реакции сочетания Бухвальда (например, Pd катализатор, основание, органический растворитель, нагревание) b. условия снятия защиты (например, CF3CO2H или HCl, комнатная температура, когда R1'=m-бутил) с. условия получения карбамата (например, карбонилдиимидазол, R1-ОН, ДМСО, комнатная температура).
[0192] В способе, изображенном на схеме 1, на первой стадии, соединения формулы XII могут быть получены путем сочетания промежуточных соединений формулы X, где R1'=R1 или защитная группа (например, где R1' представляет собой m-бутил, фрагмент R1'-ОС(О)- представляет собой группу Boc), с промежуточными соединениями Z-LG формулы XI. Для соединений формулы XI Z представляет собой гетероциклическую группу, определенную выше, a LG представляет собой подходящую группу для реакции сочетания, такую как хлор или бром. В некоторых случаях реакция сочетания может быть проведена в виде опосредованной основанием реакции нуклеофильного ароматического замещения. В некоторых случаях реакция сочетания может быть проведена в виде реакции Бухвальда, опосредованной катализом с помощью палладия. Реакции сочетания в виде ароматического замещения могут быть проведены в подходящем протонном (например, изопропаноле, н-бутаноле) или апротонном (например, CH2Cl2, ДМФА, ДМСО, CH3CN) растворителе при температуре от температуры окружающей среды до 160°C, например от 50°C до 120°C, с промежуточными соединениями формулы Z-Cl в присутствии подходящего основания (например, триэтиламина, диизопропилэтиламина). Реакции сочетания Бухвальда (Buchwald, S.; Muci, A. Top. Curr. Chem. 2002; 219, 133-209) могут быть проведены в подходящих органических растворителях (например, m-бутаноле, толуоле, ДМФА, ДМСО, CH3CN) в присутствии подходящего палладиевого катализатора и фосфиновой системы лигандов (например, прекатализатора Brettphos/Brettphos, BINAP/Pd2(dba)3) при температуре от 70°C до 150°C, например от 80°C до 130°C, с промежуточными соединениями формулы Z-Br в присутствии подходящего основания (например, Cs2CO3) в инертной атмосфере (например, в атмосфере азота). В случае, когда R1'=R1, соединения формулы XII эквивалентны соединениям формулы I. В случае, когда R1' представляет собой защитную группу, затем промежуточное соединение формулы XII может быть превращено в промежуточное соединение формулы XIII в условиях снятия защиты, известных в данной области техники. Например, когда R1' представляет собой m-бутильную группу (то есть когда фрагмент R1'-OC(O)- представляет собой группу Boc), промежуточные соединения формулы XII могут быть превращены в промежуточные соединения формулы XIII с применением ряда известных способов. Как правило, снятие защитной группы Boc проводят в кислых условиях либо с применением HCl (например, 1-4 н. HCl в эфире или диоксане в подходящем органическом растворителе, например дихлорметане, метаноле или ТГФ) при температуре от 0°C до 50°C, либо с применением трифторуксусной кислоты в апротонном растворителе (например, дихлорметане) при температуре от 0°C до комнатной. Последнее является особенно подходящим для соединений, чувствительных к опосредуемым хлоридом побочным реакциям. Промежуточные соединения формулы XIII могут быть превращены в соединения формулы I посредством реакции карбамоилирования с карбамоилирующим реагентом формулы R1OC(O)X, где X представляет собой подходящую уходящую группу (например, Cl, имидазолил, гидроксисукцинил). Реагенты формулы R1OC(O)X могут применяться в выделенном виде или могут быть получены in situ. Например, спирт формулы R1OH может быть обработан карбонилдиимидазолом в апротонном органическом растворителе при температуре от 0°C до комнатной для получения сначала R1OC(O)имидазолильного карбамоилирующего реагента. Реакция in situ R1OC(O)имидазолильного карбамоилирующего реагента с промежуточными соединениями формулы XIII (в форме свободного основания или кислотно-аддитивной соли при температуре от 0°C до 70°C) в апротонном растворителе (например, ДМСО) позволяет получить соединения формулы I.
[0193] Гетероарилхлоридные или гетероарилбромидные реагенты для реакции сочетания Z-LG являются либо коммерчески доступными, либо могут быть получены в соответствии с известными из литературы методиками получения конкретных соединений, либо могут быть получены с применением способов, известных в данной области техники для синтеза гетероарилхлоридов и гетероарилбромидов. Например, гетероарильное соединение Z-H может быть бромировано или хлорировано с применением способов, известных в данной области техники (например, путем обработки бромом, или N-бромсукцинимидом, или другим бромирующим реагентом или путем обработки хлорирующим реагентом, таким как сульфурилхлорид), желаемый гетероарильный реагент Z-Br или Z-Cl для реакции сочетания может быть затем выделен с помощью соответствующей методики (например, путем хроматографии, которая требуется для разделения региоизомеров). Гетероарильные реагенты Z-Cl для реакции сочетания, в которых группа хлора является частью иминохлоридной подструктуры может быть получена в стандартных условиях (например, с применением оксихлорида фосфора при повышенной температуре непосредственно в качестве растворителя или в подходящем апротонном органическом растворителе) из соответствующего исходного вещества Z-OH, которое имеет соответствующую амидную таутомерную подструктуру. Другие гетероарильные реагенты для реакции сочетания могут быть получены из соответствующего исходного вещества Z-NH2 в условиях реакции Зандмейера, которы хорошо отработаны в данной области техники (то есть реакция диазотизации с последующим хлорированием или бромированием с помощью CuCl или CuBr). В некоторых случаях необходимые исходные вещества Z-H, Z-OH или Z-NH2 могут быть получены с применением способов, известных в данной области техники для синтеза гетероарильных соединений.
[0194] Химические соединения формулы (I) могут быть получены в виде отдельных энантиомеров из рацемических смесей с применением способов, известных в данной области техники, таких как хиральная хроматография или перекристаллизация диастереомерных кислотно-аддитивных солей. В качестве альтернативы химические соединения формулы (I) могут быть получены в виде отдельных энантиомеров путем асимметрического синтеза или из соответствующих отдельных энантиомерных промежуточных соединений. Например, отдельные энантиомеры химических соединений формулы (I) могут быть получены из энантиомеров промежуточных соединений формулы X, имеющих соответствующую абсолютную стереохимическую конфигурацию по С-4 пиперидиновой кольцевой системы. Промежуточные соединения формулы X могут в свою очередь быть получены путем асимметрического синтеза, как изображено на схеме 2. В данном процессе известное исходное вещество XIV (Madaiah М. et. Al. Tempahedron Lett. 2013, 54, 1424-27) превращают в три стадии в α,β-ненасыщенное ацильное промежуточное соединение XV, замещенное оптически чистой R=(XVa) или S=(XVb) оксазолидиноновой хиральной вспомогательной системой. В случае XVa с хиральной вспомогательной системой с абсолютной стереохимией R стандартное каталитическое гидрирование с применением 10% Pd-C в этилацетате позволяет получить смесь диастереомерных продуктов (R)-XVIa (основной) и (S)-XVIa (минорный) в соотношении приблизительно 5:1. Данная смесь может быть разделена путем стандартной хроматографии на силикагеле с получением (R)-XVIa в чистом виде. Как показано на схеме 2, гидролитическое отщепление хиральной вспомогательной системы промежуточного соединения (R)-XVIa позволяет получить чистую энантиомерную кислоту - промежуточное соединение (R)-XVII. Из (R)-XVII двустадийная реакция Курциуса позволяет получить энантиомер (-)-(R)-X энантиомер. Энантиомер (+)-(S)-X может быть получен аналогичным образом при использовании в качестве исходного промежуточного соединения XVb. Чистые энантиомеры формулы X также могут быть получены из энантиомерно обогащенных или рацемических смесей посредством способов получения хиральных кислотно-аддитивных солей и перекристаллизации (например, с применением хирально чистой винной кислоты), стандартных в данной области техники.
Схема 2

а. метил (трифенилфосфоранилиден)ацетат, толуол, комнатная температура; b. NaOH, ТГФ, вода 50°C; с. триэтиламин, пивалоилхлорид, ТГФ -78°C, R-4-бензил-3-литио-2-оксазолидинон; d. 10% Pd/C, H2, этилацетат комнатная температура, разделение диастереомеров продукта с помощью колоночной хроматографии на силикагеле е. LiOH, 30% H2SO2, ТГФ, 50°C; f. дифенилфосфорилазид, PhCH2OH, триметиламин, толуол, обратный холодильник; g. 10% Pd/C, Н2, этилацетат комнатная температура; h. асимметрическое гидрирование
[0195] Энантиомерные промежуточные соединения формулы XVI могут быть также получены путем каталитического асимметрического гидрирования промежуточных соединений формулы XV, где Y образует простую ахиральную сложноэфирную группу (например, Y=ОМе, OEt). С этой целью хиральные катализаторы, содержащие хиральный фосфиновый лиганд (например, лиганды Waldphos) и переходный металл (например, Ir, Rh или Ru), могут быть применены в условиях асимметрического каталитического гидрирования при повышенных температуре и давлении с получением простых сложноэфирных промежуточных соединений XVI с высокой энантиомерной чистотой. Последние могут быть превращены в чистые энантиомеры X с применением стандартных для данной области техники способов, таких как описано выше. Гидрирование α,β-ненасыщенной кислоты и сложноэфирных систем было рассмотрено Tang с соавторами (Tang W. et al., Chem Rev. 2003, 103, 3029), а другие подходящие способы каталитического асимметрического гидрирования были описаны Krska с соавторами (Krska S.W. et al., Tempahedron 2009, 65, 8987-8994) и Tudge et al. (Tudge M. et al., Organic Process Research and Development 2010, 14, 787-798).
[0196] Чистые энантиомерные промежуточные соединения для синтеза энантиомеров соединений формулы I также могут быть получены посредством вариантов указанных выше способов с применением промежуточных соединений с альтернативными защитными группами или изомеров по двойным связям, как приведено в качестве примера на схеме 3.
Схема 3
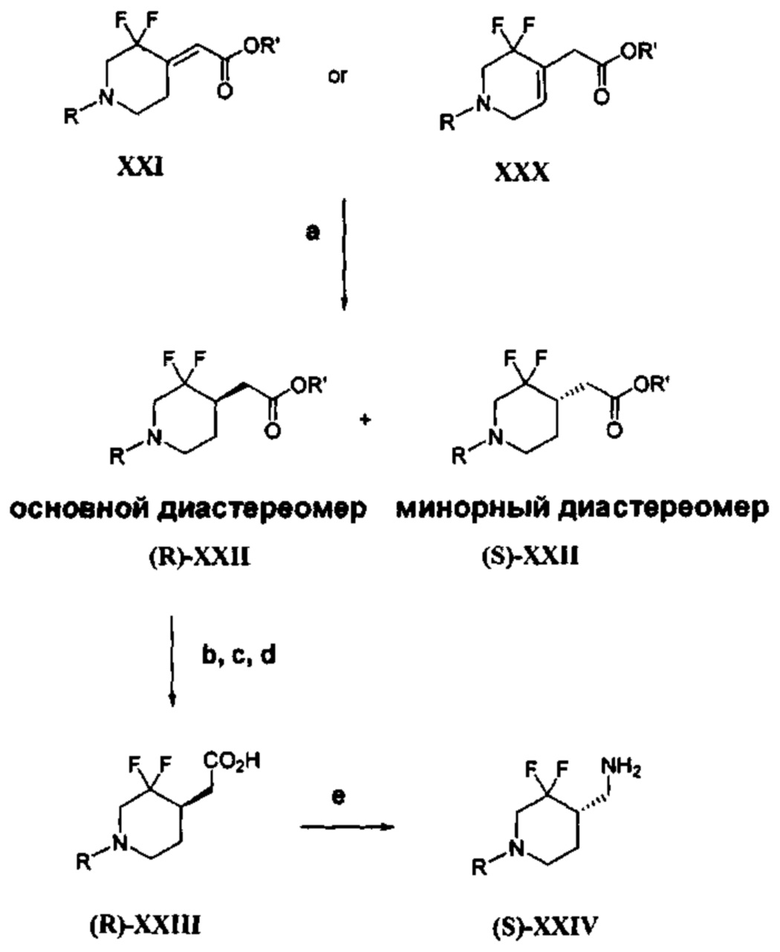
a. R = CH2Ph, R' = водород или алкил, (например, Me или Et), асимметрическое каталитическое гидрирование; b. H2 10% Pd/C для удаления бензильной защитной группы с получением R = Н; с. карбамоилирование, например карбонилдиимидазол, R1-OH, ДМСО с получением R = R1-OC(O)-; d. омыление сложного эфира, например NaOH, ТГФ; е. протокол перегруппировки Гофмана или перегруппировки Курциуса.
[0197] Например, промежуточное соединение формулы XXI, где R = бензил может быть подвергнуто вышеописанным процессам восстановления (например, каталитическому асимметрическому гидрированию с получением промежуточных соединений формулы (R)-XXII, которые являются хирально чистыми по стереоцентру С-4 пиперидина. На следующих стадиях бензильная группа может быть удалена и заменена другой защитной группой (например, Boc) или группой CO2R1, присутствующей в соединении формулы I. В качестве альтернативы та же реакционная схема может быть осуществлена путем асимметрического гидрирования изомера по двойной связи исходного вещества XXX.
ПРИМЕР 1. Химические соединения
[0198] Как показано в приведенных ниже примерах, в некоторых иллюстративных вариантах реализации химические соединения получают в соответствии со следующими методиками. Будет очевидно, что хотя в общих способах представлен синтез некоторых химических соединений согласно настоящему изобретению, ко всем химическим соединениям и подклассам и видам каждого из этих химических соединений, описанных в настоящем описании, могут быть применены следующие способы и другие способы, известные специалистам в данной области техники.
[0199] Температуры приведены в градусах Цельсия. Если не указано иное, все процедуры упаривания проводят при пониженном давлении, предпочтительно от 15 мм рт.ст. (примерно 2 кПа) до 100 мм рт.ст. (примерно 13,3 кПа). Структуры промежуточных соединений и конечных продуктов подтверждены стандартными аналитическими методами, например посредством масс-спектрометрии и ЯМР-спектроскопии. Оптические вращения измеряют по линии D натрия и приводят в градусах. Энантиомерный избыток может быть определен методами хиральной ВЭЖХ (например, с применением колонок CHIRALPAKAD-H, 4,6*150 мм, 5 мкм и при выборе подходящей подвижной фазы, например смеси гексан/изопропанол (80:20), и скорости потока, например, 1,5 мл/мин).
Сокращения:
ПРИМЕР 1.А. (-)-R-трет-бутил-4-(аминометил)-3,3-дифторпиперидин-1-карбоксилат

Стадия 1: трет-бутил-3,3-дифтор-4,4-дигидроксипиперидин-1-карбоксилат

[0200] К раствору 1-бензил-3,3-дифторпиперидин-4,4-диола (100,0 г, 412 ммоль) в этаноле (1850 мл) добавляли 10% Pd/C (10,0 г) и HCl (6,0 М, 69 мл, 414 ммоль). Полученную смесь три раза продували Н2 и гидрировали при комнатной температуре и атмосферном давлении. После того, как исходный материал был израсходован, смесь фильтровали через целит, и остаток на фильтре подвергали экстракции посредством EtOH. Объединенные фильтраты концентрировали при пониженном давлении и неочищенный продукт, содержащий 3,3-дифторпиперидин-4,4-диола гидрохлорид, непосредственно применяли на следующей стадии без очистки. Перемешанный раствор неочищенного продукта, содержащего 3,3-дифторпиперидин-4,4-диола гидрохлорид, (78 г) в воде (1000 мл) и ацетона (500 мл) подщелачивали Na2CO3 до рН 9. Затем добавляли ди-трет-бутилдикарбонат (98,9 г, 453 ммоль), и полученную смесь перемешивали при комнатной температуре в течение 4 ч. Смесь концентрировали при пониженном давлении для удаления сорастворителя-ацетона. Полученную смесь подвергали экстракции посредством EtOAc. Объединенные органические слои сушили над Na2SO4 и концентрировали, получая неочищенный продукт в виде коричневого масла. Полученное масло обрабатывали гексаном, и осажденное твердое вещество растирали и фильтровали, получая указанное в заголовке соединение в виде белого порошка (94,3 г, 90% в целом). 1Н ЯМР (400 МГц, CD3OD) δ 3,72 (t, J=11,6 Гц, 2Н), 3,56-3,46 (m, 2Н), 1,83-1,77 (m, 2Н), 1,42 (s, 9Н).
Стадия 2: трет-бутил-3,3-дифтор-4-(2-метокси-2-оксоэтилиден)пиперидин-1-карбоксилат
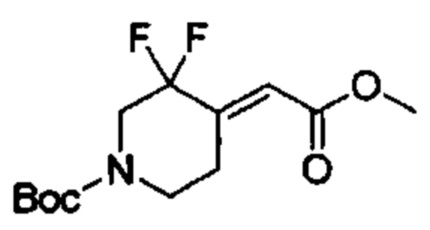
[0201] К перемешиваемому раствору трет-бутил-3,3-дифтор-4,4-дигидроксипиперидин-1-карбоксилата (80,0 г, 314 ммоль) в толуоле (1000 мл) добавляли метил-(трифенилфосфоранилиден)ацетат (126 г, 377 ммоль), и полученную реакционную смесь перемешивали при комнатной температуре в течение 4 ч. Реакционную смесь концентрировали под вакуумом и остаток очищали с помощью колоночной хроматографии на силикагеле (EtOAc/гексан = 1/5), получая указанное в заголовке соединение в виде бесцветного масла (79,6 г, 87%). 1Н ЯМР (400 МГц, CDCl3) δ 6,23 (s, 1Н), 3,83-3,71 (m, 2Н), 3,76 (s, 3Н), 3,58-3,47 (m, 2Н), 3,13-3,05 (m, 2Н), 1,48 (s, 9Н).
Стадия 3: 2-(1-(трет-бутоксикарбонил)-3,3-дифторпиперидин-4-илиден)уксусная кислота

[0202] К перемешиваемому раствору трет-бутил-3,3-дифтор-4-(2-метокси-2-оксоэтилиден)пиперидин-1-карбоксилата (590 г, 2,03 моль) в ТГФ (1,5 л) добавляли раствор NaOH (40,1 г, 1,0 моль) в воде (1,5 л) при комнатной температуре. Полученную смесь нагревали до 50°C и перемешивали в течение 1 ч. После того, как исходный материал был израсходован, концентрировали ТГФ при пониженном давлении. Водный концентрат подвергали экстракции этилацетатом и подкисляли до рН 5 4,0 М водным раствором HCl при охлаждении в ледяной бане. Водную фазу трижды подвергали экстракции этилацетатом. Объединенные органические фазы промывали водой, солевым раствором, сушили над Na2SO4 и концентрировали при пониженном давлении. Остаток растирали гексаном и суспензию фильтровали, получая указанный в заголовке продукт в виде порошка грязно-белого цвета (492 г, 87%). 1Н ЯМР (400 МГц, ДМСО-d6) δ 12,99 (s, 1Н), 6,11 (s, 1Н), 3,81 (t, J=11,6 Гц, 2Н), 3,50-3,42 (m, 2Н), 2,94-2,88 (m, 2Н), 1,42 (s, 9Н).
Стадия 4: трет-бутил-(R)-4-(2-(4-бензил-2-оксооксазолидин-3-ил)-2-оксоэтилиден)-3,3-дифторпиперидин-1-карбоксилат
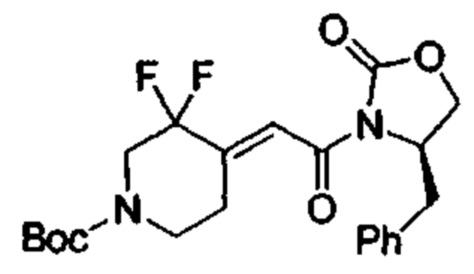
[0203] К перемешиваемому раствору 2-(1-(трет-бутоксикарбонила)-3,3-дифторпиперидин-4-илиден)уксусной кислоты (23,5 г, 84,8 ммоль) в сухом ТГФ (250 мл) добавляли Et3N (11,7 мл, 85,7 ммоль) при 0°C. По каплям добавляли пивалоилхлорид (10,5 мл, 89,0 ммоль), и полученную смесь перемешивали при 0°C в течение 2 ч. К смеси при -78°C добавляли раствор в ТГФ одного эквивалента (R)-4-бензил-3-литио-2-оксазолидинона (полученный из (R)-4-бензил-2-оксазолидинона (15,0 г, 84,7 ммоль) и n-BuLi (2,5 М в гексане, 34 мл, 85 ммоль) в ТГФ (150 мл) при -78°C). Полученной смеси позволяли нагреться до 0°C, ее перемешивали в течение 30 минут при 0°C и гасили с помощью нас. NH4Cl. Смесь подвергали экстракции этилацетатом. Объединенные органические слои промывали водой, солевым раствором, сушили над Na2SO4 и концентрировали под вакуумом. Остаток перекристаллизовывали их смеси этилацетат/гексан (60 мл/400 мл), получая указанное в заголовке соединение в виде белого порошка (24,5 г, 66%). МС (ИЭР), расчет для C22H26F2N2O5: 436,2; экспериментальное значение [459,3] [М+Na]; 1Н ЯМР (400 МГц, CDCl3) δ 7,39-7,32 (m, 3Н), 7,31-7,29 (m, 1Н), 7,22 (d, J=8,0 Гц, 2Н), 4,76-4,70 (m, 1Н), 4,27-4,19 (m, 2Н), 3,84-3,79 (m, 2Н), 3,63-3,52 (m, 2Н), 3,35 (dd, J=3,2 и 13,2 Гц, 1Н), 3,08-2,97 (m, 2Н), 2,80 (dd, J=10,0 и 13,2 Гц, 1Н), 1,49 (s, 9Н).
Стадия 5: R-трет-бутил-4-(2-((R)-4-бензил-2-оксооксазолидин-3-ил)-2-оксоэтил)-3,3-дифторпиперидин-1-карбоксилат
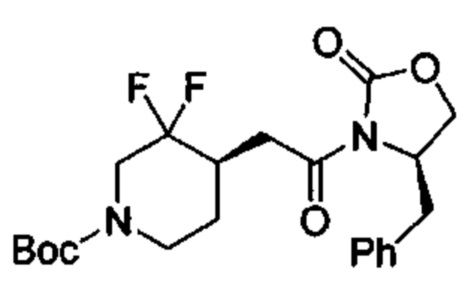
[0204] К суспензии ((R)-трет-бутил-4-(2-(4-бензил-2-оксооксазолидин-3-ил)-2-оксоэтилиден)-3,3-дифторпиперидин-1-карбоксилата (10,0 г, 22,8 ммоль) в этилацетате (150 мл) добавляли 10% палладий на угле (1,0 г). Полученную смесь гидрировали при комнатной температуре в течение 13 ч при атмосферном давлении. Смесь фильтровали через целит, и остаток на фильтре подвергали экстракции этилацетатом. Объединенные фильтраты концентрировали под вакуумом, получая 12,0 г смеси R-диастереоизомера указанного в заголовке соединения с соответствующим минорным S-диастереоизомером в соотношении приблизительно 5:1. Диастереомеры отделяли с помощью колоночной хроматографии на силикагеле (этилацетат/гексан = 1/3). Основной диастереоизомер выходил первым и был получен в виде белого порошка (5,20 г, 52%, диастереомерный избыток (д.и.) >99%) (колонка: CHIRALPAKAD-H, 4,6*150 мм, 5 мкм; подвижная фаза: А : гексан В : этанол = 70:30; t=9,07). МС (ИЭР), расчет для C22H28F2N2O5: 438,2; экспериментальное значение: 461,2 [M+Na] 1Н ЯМР (400 МГц, CDCl3) δ 7,37-7,32 (m, 2Н), 7,31-7,28 (m, 1Н), 7,20 (d, J=8,0 Гц, 2Н), 4,72-4,66 (m, 1Н), 4,48-4,00 (m, 4Н), 3,38-3,27 (m, 2Н), 3,11-2,72 (m, 4Н), 2,66-2,53 (m, 1Н), 1,93-1,89 (m, 1Н), 1,63-1,51 (m, 1Н), 1,47 (s, 9Н).
Стадия 6: R-2-(1-(трет-бутоксикарбонил)-3,3-дифторпиперидин-4-ил)уксусная кислота

[0205] К перемешиваемой смеси R-трет-бутил-4-(2-((R)-4-бензил-2-оксооксазолидин-3-ил)-2-оксоэтил)-3,3-дифторпиперидин-1-карбоксилата (5,20 г, 11,8 ммоль) в ТГФ/H2O (45 мл/45 мл) добавляли 30% водн. Н2О2 (4,9 мл, 48 ммоль) и моногидрат гидроксида лития (800 мг, 19,0 ммоль) при 0°C. После перемешивания в течение 90 мин при 0°C реакционную смесь обрабатывали 1 М раствором Na2SO3 (40 мл) и затем подвергали экстракции этилацетатом (2×100 мл) для удаления хирального вспомогательного вещества. Водную фазу подкисляли до рН=2~3 с помощью 1 М водн. HCl и затем подвергали экстракции этилацетатом (2×100 мл). Объединенные этилацетатные фазы сушили над Na2SO4 и концентрировали под вакуумом. Остаток перекристаллизовывали из смеси этилацетат/гексан (5 мл/50 мл), получая указанное в заголовке соединение в виде белого порошка (3,70 г, 79%). МС (ИЭР), расчет для C12H19F2NO4: 279,1; экспериментальное значение: 224 [М+Н - 56 (m-бутильная группа)]; 1Н ЯМР (400 МГц, CDCl3) δ 4,51-4,01 (m, 2Н), 3,10-2,70 (m, 3Н). 2,45-2,28 (m, 2Н), 1,93-1,89 (m, 1Н), 1,58-1,50 (m, 1Н), 1,46 (s, 9Н).
Стадия 7: R-трет-бутил-4-((бензилоксикарбониламино)метил)-3,3-дифторпиперидин-1-карбоксилат

[0206] К перемешиваемому раствору R-2-(1-(трет-бутоксикарбонил)-3,3-дифторпиперидин-4-ил)уксусной кислоты (6,05 г, 21,7 ммоль) в сухом толуоле (50 мл) добавляли триэтиламин (4,6 мл, 34 ммоль) и дифенилфосфорилазид (5,2 мл, 24 ммоль) в атмосфере азота. После перемешивания в течение 20 мин при 80°C добавляли бензиловый спирт (1,4 мл, 25 ммоль) и реакционную смесь перемешивали при 100°C в течение ночи. Полученной смеси давали остыть до комнатной температуры, ее концентрировали и разбавляли ДХМ (200 мл). Органическую фазу промывали 0,5 М водн. HCl (2×50 мл), водой (2×50 мл) и солевым раствором (50 мл), сушили над Na2SO4 и концентрировали под вакуумом. Остаток перекристаллизовывали из смеси этанол/H2O (30 мл/90 мл), получая указанное в заголовке соединение в виде белого порошка (6,91 г, 84%). МС (ИЭР), расчет для C19H26F2N2O4: 384,2; экспериментальное значение: 407,2 [М+Na]. 1Н ЯМР (400 МГц, CDCl3) δ 7,39-7,30 (m, 5Н), 5,10 (s, 2Н), 5,02 (ушир. s, 1Н), 4,51-3,99 (m, 2Н), 3,55-3,44 (m, 1Н), 3,39-3,26 (m, 1Н), 3,04-2,65 (m, 2Н), 2,18-2,02 (m, 1Н), 1,78-1,75 (m, 1Н), 1,58-1,48 (m, 1Н), 1,46 (s. 9Н).
Стадия 8: (-)-R-трет-бутил-4-(аминометил)-3,3-дифторпиперидин-1-карбоксилат
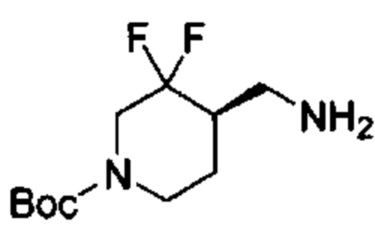
[0207] К суспензии R-трет-бутил-4-((бензилоксикарбониламино)метил)-3,3-дифторпиперидин-1-карбоксилата (7,01 г, 24,7 ммоль) в этилацетате (45 мл) добавляли 10% палладий на угле (700 мг). Смесь гидрировали при комнатной температуре под атмосферным давлением, создаваемым водородом, в течение 13 ч. Смесь фильтровали через целит, и остаток на фильтре подвергали экстракции этилацетатом. Объединенные фильтраты концентрировали под вакуумом, получая указанное в заголовке соединение в виде коричневого масла (4,60 г, 93%). [α]D=-18,3° (с=10 мг/мл, метанол, 20°C), МС (ИЭР), расчет для C11H20F2N2O2: 250,2; экспериментальное значение: 195,2 [М+Н - 56 (m-бутильная группа)]; 1Н ЯМР (400 МГц, CDCl3) δ 4,45-4,01 (m, 2Н), 3,15 (dd, J=5,2, 12,8 Гц, 1Н), 3,05-2,75 (m, 2Н), 2,68 (dd, J=6,4, 12,8 Гц, 1Н) 1,96-1,74 (m, 2Н), 1,58-1,48 (m, 1Н), 1,46 (s, 9Н), 1,40 (ушир. s, 2Н).
ПРИМЕР 1.В. 2,5-диоксопирролидин-1-ил-4-метилбензилкарбонат
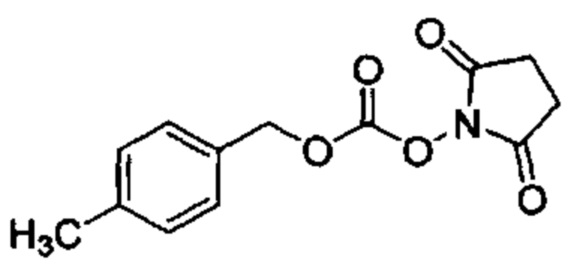
[0208] Смесь п-толилметанола (2,40 г, 19,6 ммоль) и бис(2,5-диоксопирролидин-1-ил)карбоната (5,03 г, 19,6 ммоль) в смешанном растворителе, состоящем из ацетонитрила (30 мл) и CH2Cl2 (30 мл), обрабатывали 4-диметиламинопиридином (1,20 г, 9,8 ммоль). Полученную реакционную смесь перемешивали в течение 2 ч при комнатной температуре. После того, как спирт был израсходован, смесь вливали в воду (100 мл) и отделяли органический слой, сушили над безводным сульфатом натрия и концентрировали под вакуумом. Твердое вещество, полученное таким образом, растирали с эфиром и сушили, получая указанное в заголовке соединение в виде твердого вещества белого цвета (3,40 г, 66%). 1Н ЯМР (400 МГц, CDCl3) 7,29 (d, J=8,0 Гц, 2Н), 7,20 (d, J=8,0 Гц, 2Н), 5,28 (s, 2Н), 2,82 (s, 4Н), 2,36 (s, 3Н).
ПРИМЕР 1.С. 2,5-диоксопирролидин-1-ил-4-этилбензилкарбонат

[0209] Смесь 4-этилбензилового спирта (1,0 г, 7,3 ммоль) и бис(2,5-диоксопирролидин-1-ил)-карбоната (1,88 г, 7,3 ммоль) в смеси растворителей ацетонитрила (15,0 мл) и CH2Cl2 (15,0 мл) обрабатывали 4-диметиламинопиридином (446 мг, 3,65 ммоль). Полученную реакционную смесь перемешивали в течение 2 ч при комнатной температуре. Затем смесь разбавляли этилацетатом, промывали водой, солевым раствором, сушили над безводным Na2SO4 и концентрировали при пониженном давлении, получая указанное в заголовке соединение в виде порошка грязно-белого цвета (2,0 г, 99%) который применяли без дополнительной очистки.
ПРИМЕР 1.D. (+)-S-трет-бутил-4-(аминометил)-3,3-дифторпиперидин-1-карбоксилат
[0210] Указанное в заголовке соединение может быть получено способом, аналогичным получению (-)-R-трет-бутил-4-(аминометил)-3,3-дифторпиперидин-1-карбоксилата, описанному в Примере 1.А., но с применением (S)-4-бензил-3-литио-2-оксазолидинона (полученного из (S)-4-бензил-2-оксазолидинона и n-BuLi) вместо соответствующего реагента, имеющего (R)-конфигурацию.
ПРИМЕР 1.1. (+)-R-4-метилбензил-4-(([1,2,4]триазоло[4,3-а]пиразин-8-иламино)метил)-3,3-дифторпиперидин-1-карбоксилат (Е1-1.2)

Стадия 1: R-трет-бутил-4-(([1,2,4]триазоло[4,3-а]пиразин-8-иламино)метил)-3,3-дифторпиперидин-1-карбоксилат
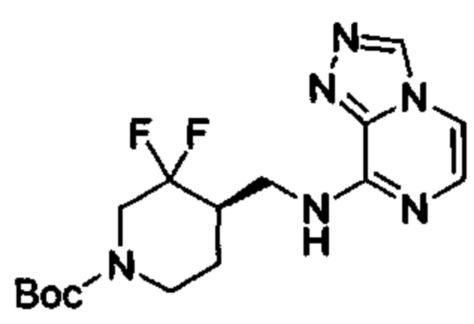
[0211] Перемешиваемую смесь (-)-R-трет-бутил-4-(аминометил)-3,3-дифторпиперидин-1-карбоксилата (230 мг, 0,92 ммоль), 8-хлор-[1,2,4]триазоло[4,3-а]пиразина (127 мг, 0,83 ммоль) и DIPEA (0,30 мл, 1,8 ммоль) в n-BuOH (5 мл) нагревали до 95°C в атмосфере азота в течение 13 ч. Полученной реакционной смеси давали остыть до комнатной температуры и проводили ее концентрирование под вакуумом. Остаток очищали с помощью колоночной хроматографии на силикагеле (этилацетат/гексан = 1/1), получая указанное в заголовке соединение в виде белого порошка (210 мг, 62%). МС (ИЭР), расчет для C16H22F2N6O2: 368,2; экспериментальное значение: 369,1 [М+Н]. 1Н ЯМР (400 МГц, CDCl3) δ 8,72 (s, 1Н), 7,41 (d, J=4,8 Гц, 1Н), 7,35 (d, J=4,8 Гц, 1Н), 6,50-6,45 (m, 1Н), 4,47-4,09 (m, 2Н), 4,02-3,96 (m, 1Н), 3,79-3,72 (m, 1Н), 3,09-2,69 (m, 2Н), 2,45-2,29 (m, 1Н), 1,92-1,87 (m, 1Н), 1,68-1,58 (m, 1Н), 1,47 (s, 9Н).
Стадия 2: R-N-((3,3-дифторпиперидин-4-ил)метил)-[1,2,4]триазоло[4,3-а]пиразин-8-амина трифторацетат
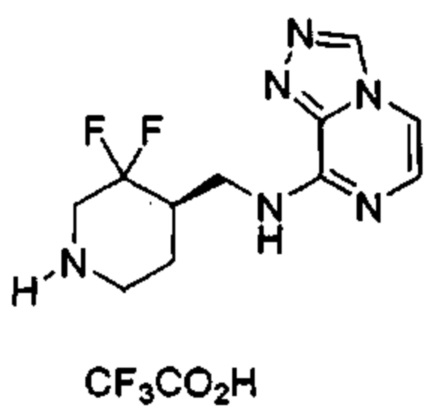
[0212] К перемешиваемой суспензии R-трет-бутил-4-(([1,2,4]триазоло[4,3-а]пиразин-8-иламино)-метил)-3,3-дифторпиперидин-1-карбоксилата (1,01 г, 2,72 ммоль) в дихлорметане (15 мл) добавляли ТФУ (3 мл). Далее полученный раствор перемешивали при температуре окружающей среды в течение 30 мин. Полученную реакционную смесь концентрировали при пониженном давлении, получая указанное в заголовке соединение в виде желтого остатка, который применяли на следующей стадии без дополнительной очистки. МС (ИЭР), расчет для C11H14F2N6: 268,1; экспериментальное значение: 269,1 [М+Н]. 1Н ЯМР (400 МГц, CD3OD) δ 9,26 (s, 1Н), 7,87 (d, J=4,8 Гц, 1Н), 7,33 (d, J=4,8 Гц, 1Н), 4,16-4,05 (m, 1Н), 3,82-3,75 (m, 2Н), 3,52-3,46 (m, 2Н), 3,20-3,13 (m, 1Н), 2,92-2,76 (m, 1Н), 2,36-2,26 (m, 1Н), 1,95-1,87 (m, 1Н).
Стадия 3: (+)-R-4-метилбензил-4-(([1,2,4]триазоло[4,3-а]пиразин-8-иламино)метил)-3,3-дифторпиперидин-1-карбоксилат

[0213] К перемешиваемому раствору R-N-((3,3-дифторпиперидин-4-ил)метил)-[1,2,4]триазоло[4,3-а]пиразин-8-амина трифторацетата с предыдущей стадии (приблизительно 2,72 ммоль) и триэтиламина (2,1 мл, 15 ммоль) в ацетонитриле (20 мл) добавляли 2,5-диоксопирролидин-1-ил-4-метилбензилкарбонат (750 мг, 2,85 ммоль). Полученную смесь перемешивали в течение 1 ч при комнатной температуре. Затем смесь разбавляли этилацетатом (100 мл), промывали водой, солевым раствором, сушили над безводным Na2SO4 и концентрировали при пониженном давлении. Остаток очищали с помощью колоночной хроматографии на силикагеле (ДХМ/МеОН = 35/1), получая указанное в заголовке соединение в виде порошка грязно-белого цвета (840 мг, 75%). [α]D=+13,5° (с=10 мг/мл, МеОН, 20°C). Хиральная чистота по ВЭЖХ: энантиомерный избыток (э.и.) >99% (CHIRALPAKAD-H 4,6*150 мм, 5 мкм; подвижная фаза: гексан : изопропанол = 80:20; комнатная температура = 6,88 мин). МС (ИЭР), расчет для C20H22F2N6O6: 416,2; экспериментальное значение: 417,5 [М+Н]. 1Н ЯМР (400 МГц, CD3OD) δ 9,11 (s, 1Н), 7,71 (d, J=4,8 Гц, 1Н), 7,34 (d, J=4,8 Гц, 1Н), 7,26 (d, J=8,0 Гц, 2Н), 7,19 (d, J=8,0 Гц, 2Н), 5,11 (s, 2Н), 4,38-4,26 (m, 1Н), 4,18-4,14 (m, 1Н), 4,01 (dd, J=5,2, 13,6 Гц, 1Н), 3,62 (dd, J=8,8, 13,6 Гц, 1Н), 3,29-3,11 (m, 1Н), 3,03-2,89 (m, 1Н), 2,65-2,50 (m, 1Н), 2,35 (s, 3Н), 2,00-1,94 (m, 1Н), 1,62-1,52 (m, 1Н).
ПРИМЕР 1.1а. R-4-метилбензил-4-(([1,2,4]триазоло[4,3-а]пиразин-8-иламино)метил)-3,3-дифторпиперидин-1-карбоксилат метансульфонат (Е1-1.2а)
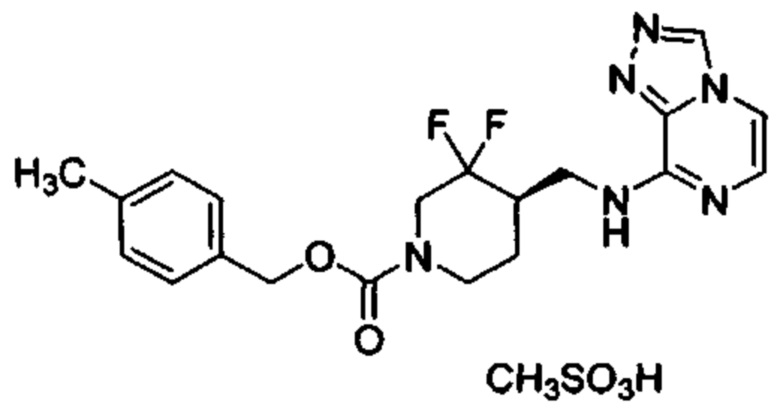
[0214] К перемешиваемому раствору (+)-R-4-метилбензил-4-(([1,2,4]триазоло[4,3-а]пиразин-8-иламино)-метил)-3,3-дифторпиперидин-1-карбоксилата (1,8 г, 4,32 ммоль) в 30 мл ДХМ/МеОН (1/1) добавляли метансульфоновую кислоту (420 мг, 4,32 ммоль). Полученный раствор перемешивали при комнатной температуре в течение 30 мин. Растворитель выпаривали, и твердое вещество, полученное таким образом, растирали с эфиром (25 мл), получая указанное в заголовке соединение в виде твердого вещества белого цвета (1,90 г, 86%). МС (ИЭР), расчет для C20H22F2N6O6: 416,2; экспериментальное значение: 417,5 [М+Н]. 1Н ЯМР (400 МГц, CD3OD) δ 9,33 (s, 1Н), 7,96 (d, J=4,8 Гц, 1Н), 7,27 (d, J=4,8 Гц, 1Н), 7,26 (d, J=8,0 Гц, 2Н), 7,19 (d, J=8,0 Гц, 2Н), 5,12 (s, 2Н), 4,47-4,33 (m, 1Н), 4,27-4,21 (m, 1Н), 4,07-3,99 (m, 1Н), 3,75-3,65 (m, 1Н), 3.30-3,14 (m, 1Н), 3,08-2,92 (m, 1Н), 2,72 (s, 3Н), 2,70-2,60 (m, 1Н), 2,35 (s, 3Н), 2,08-2,00 (m, 1Н), 1,67-1,56 (m, 1Н).
ПРИМЕР 1.2. (±)-4-метилбензил-4-(([1,2,4]триазоло[4,3-а]пиразин-8-иламино)метил)-3,3-дифторпиперидин-1-карбоксилат (С-1.2)

Стадия 1: метил-2-(1-бензил-3,3-дифторпиперидин-4-илиден)ацетат
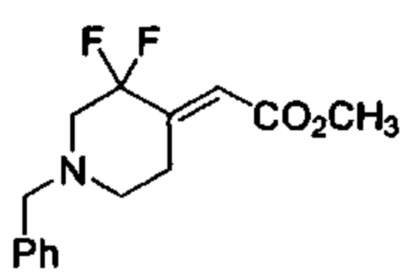
[0215] К перемешиваемому раствору 1-бензил-3,3-дифторпиперидин-4,4-диола (10,9 г, 44,8 ммоль) в толуоле (350 мл) добавляли метил-(трифенилфосфоранилиден)ацетат (18,0 г, 52 ммоль) при комнатной температуре. Полученную реакционную смесь перемешивали при комнатной температуре в течение 2 часов. Реакционную смесь концентрировали под вакуумом и очищали с помощью колоночной хроматографии на силикагеле (EtOAc/гексан = 1/6), получая указанное в заголовке соединение в виде белого порошка (10,9 г, 86%). МС (ИЭР), расчет для C15H17F2NO2: 281,1; экспериментальное значение: 282,3 [М+Н]. 1Н ЯМР (400 МГц, CDCl3) δ 7,36-7,27 (m, 5Н), 6,19 (ушир. s, 1Н), 3,74 (s, 3Н), 3,64 (s, 2Н), 3,14-3,11 (m, 2Н), 2,79 (t, J=11,2 Гц, 2Н), 2,60 (t, J=5,6 Гц, 2Н).
Стадия 2: метил-2-(3,3-дифторпиперидин-4-ил)ацетат
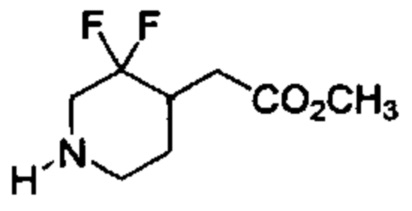
[0216] К перемешиваемому раствору метил-2-(1-бензил-3,3-дифторпиперидин-4-илиден)ацетата (10,9 г, 39 ммоль) в МеОН (200 мл) добавляли 10% Pd/C (1,0 г, 10%). Реакционную смесь трижды продували Н2 и гидрировали при комнатной температуре при атмосферном давлении. После того, как исходный материал был израсходован, смесь фильтровали через целит, и остаток на фильтре подвергали экстракции посредством МеОН. Объединенные фильтраты концентрировали и остаток очищали с помощью колоночной хроматографии на силикагеле (EtOAc/гексан = 1/6), получая указанное в заголовке соединение в виде прозрачного масла (5,30 г, 70%). МС (ИЭР), расчет для C8H13F2NO2: 193,1; экспериментальное значение: 194,4 [М+Н]. 1Н ЯМР (400 МГц, CDCl3) δ 3,67 (s, 3Н), 3,20-3,11 (m, 1Н), 3,03-2,99 (m, 1Н), 2,83-2,70 (m, 2Н), 2,66-2,56 (m, 1Н), 2,46-2,29 (m, 1Н), 2,25-2,19 (m, 1Н), 1,90-1,82 (m, 1Н), 1,64 (ушир. s, 1Н), 1,45-1,34 (m, 1Н).
Стадия 3: 4-метилбензил-3,3-дифтор-4-(2-метокси-2-оксоэтил)пиперидин-1-карбоксилат
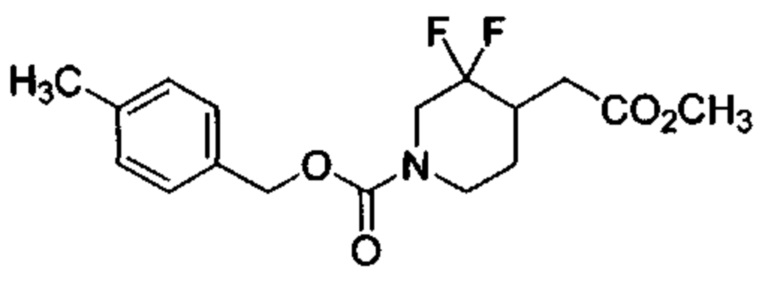
[0217] К перемешиваемому раствору п-толилметанола (1,5 г, 12 ммоль) в ДМСО (20 мл) добавляли CDI (1,9 г, 11 ммоль) при комнатной температуре в атмосфере N2. Полученную смесь перемешивали в течение 1 ч, и к ней добавляли метил-2-(3,3-дифторпиперидин-4-ил)ацетат (2,0 г, 10 ммоль) в ДМСО (10 мл) при комнатной температуре. Смесь нагревали при 50°C в течение ночи, давали остыть до комнатной температуры и разбавляли EtOAc. Органический слой промывали водой, солевым раствором, сушили над Na2SO4 и концентрировали. Полученный концентрат очищали с помощью колоночной хроматографии на силикагеле (EtOAc/гексан = 1/6), получая указанное в заголовке соединение в виде прозрачного масла (1,5 г, 43%). 1Н ЯМР (400 МГц, CDCl3) δ 7,26 (d, J=8,0 Гц, 2Н), 7,17 (d, J=8,0 Гц, 2Н), 5,09 (ушир. s, 2Н), 4,64 (d, J=5,2 Гц, 1Н), 4,54-4,06 (m, 2Н), 3,69 (d, J=12,0 Гц, 2Н), 3,12-2,73 (m, 2Н), 2,35 (s, 3Н), 2,29-2,22 (m, 1Н), 1,92-1,65 (m, 1Н), 1,59 (s, 3Н).
Стадия 4: 2-(3,3-дифтор-1-((4-метилбензилокси)карбонил)пиперидин-4-ил)уксусная кислота
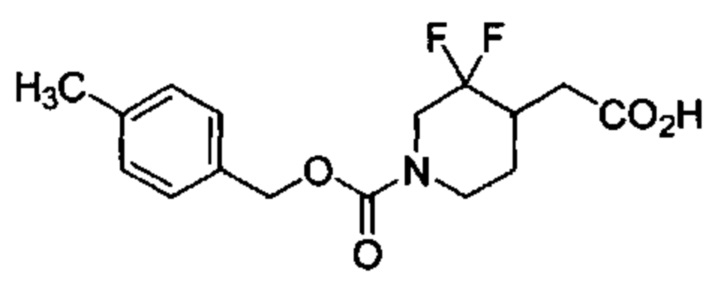
[0218] К перемешиваемому раствору 4-метилбензил-3,3-дифтор-4-(2-метокси-2-оксоэтил)пиперидин-1-карбоксилата (1,5 г, 4,4 ммоль) в ТГФ (20 мл) добавляли водный NaOH (1 М, 20 мл). Полученную реакционную смесь перемешивали при комнатной температуре в течение 5 ч и гасили 1 н. раствором HCl при охлаждении в ледяной бане. Смесь концентрировали и экстрагировали этилацетатом. Этилацетатный слой промывали водой, солевым раствором, сушили над Na2SO4 и концентрировали, получая указанное в заголовке соединение в виде твердого вещества белого цвета (650 мг, 46%). 1Н ЯМР (400 МГц, CDCl3) δ 7,25 (d, J=8,0 Гц, 2Н), 7,16 (d, J=8,0 Гц, 2Н), 5,08 (s, 2Н), 4,16 (ушир. s, 2Н), 2,91-2,71 (m, 2Н), 2,35 (s, 3Н), 2,31-2,26 (m, 1Н), 2,01-1,88 (m, 1Н), 1,80-1,69 (m, 1Н), 1,25-1,14 (m, 2Н).
Стадия 5: 4-метилбензил-4-(аминометил)-3,3-дифторпиперидин-1-карбоксилат
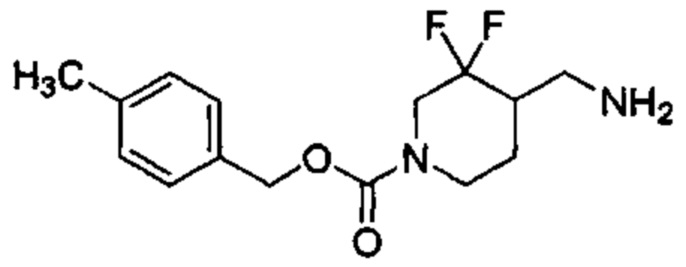
[0219] К перемешиваемому раствору 2-(3,3-дифтор-1-((4-метилбензилокси)карбонил)пиперидин-4-ил)уксусной кислоты (650 мг, 1,98 ммоль) в толуоле (3 мл) добавляли триэтиламин (93 мг, 1,0 ммоль) и DPPA (187 мг, 2,98 ммоль). Полученную реакционную смесь перемешивали при 70°C в течение 1 ч. Добавляли смесь диоксана (3 мл) и 1М водного раствора NaOH (3 мл), и реакционной смеси давали остыть до комнатной температуры. Смесь концентрировали при пониженном давлении и разбавляли EtOAc. Органический слой промывали водой, солевым раствором, сушили над Na2SO4 и концентрировали. Остаток переносили в дихлорметан и фильтровали. Фильтрат концентрировали, получая неочищенный продукт в виде желтого масла (600 мг). МС (ИЭР), расчет для C15H20F2N2O2: 298,2; экспериментальное значение: 299,2 [М+Н].
Стадия 6: (±)-4-метилбензил-4-(([1,2,4]триазоло[4,3-а]пиразин-8-иламино)метил)-3,3-дифторпиперидин-1-карбоксилат
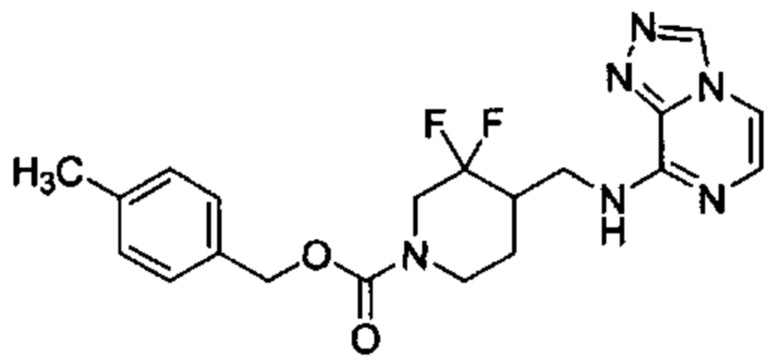
[0220] Смесь неочищенного 4-метилбензил-4-(аминометил)-3,3-дифторпиперидин-1-карбоксилата (600 мг, 2,0 ммоль), 8-хлор-[1,2,4]триазоло[4,3-а]пиразина (360 мг, 2,0 ммоль) и DIPEA (0,76 мл, 4,0 ммоль) в бутиловом спирте (10 мл) нагревали при 130°C в течение ночи. Полученной смеси давали остыть до комнатной температуры и проводили ее концентрирование при пониженном давлении. Полученный концентрат очищали с помощью колоночной хроматографии на силикагеле (100% этилацетат), получая указанное в заголовке соединение в виде светло-серого порошка (150 мг, 18%). МС (ИЭР), расчет для C20H22F2N6O2: 416,2; экспериментальное значение: 417,3 [М+Н]. 1Н ЯМР (400 МГц, CD3OD) δ 9,16 (s, 1Н), 7,77 (d, J=4,8 Гц, 1Н), 7,29 (d, J=4,8 Гц, 1Н), 7,24 (d, J=8,0 Гц, 2Н), 7,17 (d, J=8,0 Гц, 2Н), 5,09 (s, 2Н), 4,33 (ушир. s, 1Н), 4,19-4,15 (m, 1Н), 4,05-3,95 (m, 1Н), 3,72-3,58 (m, 1Н), 3,25-2,90 (m, 2Н), 2,67-2,51 (m, 1Н), 2,33 (s, 3Н), 1,99-1,93 (m, 1Н), 1,62-1,52 (m, 1Н).
ПРИМЕР 1.3. (-)-S-4-метилбензил-4-(([1,2,4]триазоло[4,3-а]пиразин-8-иламино)метил)-3,3-дифторпиперидин-1-карбоксилат (Е2-1.2)
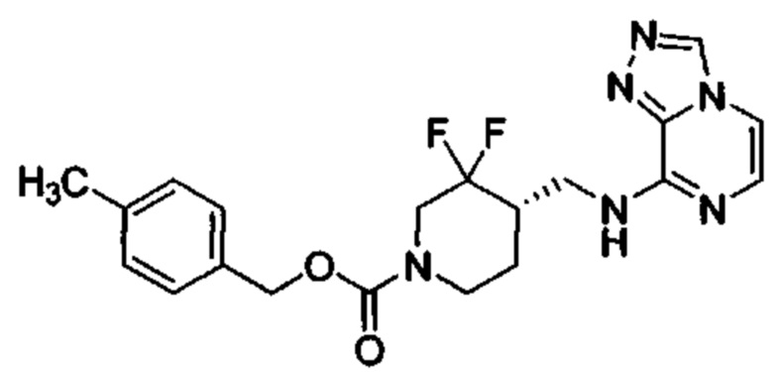
[0221] (±)-4-Метилбензил-4-(([1,2,4]триазоло[4,3-а]пиразин-8-иламино)метил)-3,3-дифтор-пиперидин-1-карбоксилат отделяли посредством хиральной ВЭЖХ [CHIRALPAKAD-H, 4,6*150 мм, 5 мкм. Подвижная фаза: А : гексан; В : (этанол/метанол = 2:1) = 70:30], получали соответствующие чистые энантиомеры, причем (+)-R-4-метилбензил-4-(([1,2,4]триазоло[4,3-а]пиразин-8-иламино)метил)-3,3-дифторпиперидин-1-карбоксилат выходил первым (время удерживания = 7,065 мин), а после него выходил (-)-S-4-метилбензил-4-(([1,2,4]триазоло[4,3-а]пиразин-8-иламино)метил)-3,3-дифторпиперидин-1-карбоксилат (время удерживания = 9,160 мин).
[0222] (+)-R-4-метилбензил-4-(([1,2,4]триазоло[4,3-а]пиразин-8-иламино)метил)-3,3-дифторпиперидин-1-карбоксилат. CHIRALPAKAD, время удерживания = 7,065 мин, α20D=+13,5° (с=10 мг/мл, МеОН).
[0223] (-)-S-4-метилбензил-4-(([1,2,4]триазоло[4,3-а]пиразин-8-иламино)метил)-3,3-дифторпиперидин-1-карбоксилат. CHIRALPAKAD, время удерживания = 9,160 мин, α20D=-12,0° (с=10 мг/мл, МеОН).
ПРИМЕР 1.4. 4-(([1,2,4]триазоло[4,3-а]пиразин-5-иламино)метил)-3,3-дифторпиперидин-1-карбоксилат (Е1-2.2)

Стадия 1: R-трет-бутил-4-(([1,2,4]триазоло[4,3-а]пиразин-5-иламино)метил)-3,3-дифторпиперидин-1-карбоксилат

[0224] Смесь R-трет-бутил-4-(аминометил)-3,3-дифторпиперидин-1-карбоксилата (300 мг, 1,20 ммоль), 5-бром-[1,2,4]триазоло[4,3-а]пиразина (356 мг, 1,80 ммоль) и DIPEA (0,42 мл, 2,40 ммоль) в NMP (9 мл) нагревали до 130°C при перемешивании в течение ночи. Полученному оранжевому раствору давали остыть до комнатной температуры и разбавляли его этилацетатом. Органическую фазу промывали водой, солевым раствором, сушили над Na2SO4 и концентрировали под вакуумом. Полученный концентрат очищали с помощью колоночной хроматографии на силикагеле (гексан/этилацетат = 1/3), получая указанное в заголовке соединение в виде порошка желтого цвета (210 мг, 48%). 1Н ЯМР (400 МГц, CDCl3) δ 9,25 (s, 1Н), 8,85 (s, 1Н), 7,27 (s, 1Н), 6,17-6,01 (m, 1Н), 4,51417 (m, 2Н), 3,93-3,84 (m, 1Н), 3,52-3,44 (m, 1Н), 3,11-2,71 (m, 2Н), 2,52-2,37 (m, 1Н), 2,03-1,92 (m, 1Н), 1,73-1,63 (m, 1Н), 1,48 (s, 9Н).
Стадия 2: R-N-((3,3-дифторпиперидин-4-ил)метил)-[1,2,4]триазоло[4,3-а]пиразин-5-амина трифторацетат

[0225] К раствору R-трет-бутил-4-(([1,2,4]триазоло[4,3-а]пиразин-5-иламино)метил)-3,3-дифторпиперидин-1-карбоксилата (256 мг, 0,69 ммоль) в дихлорметане (5 мл) добавляли ТФУ (2 мл) при комнатной температуре. После перемешивания в течение 30 мин смесь концентрировали, получая указанное в заголовке соединение в виде желтого масла, которое непосредственно применяли в виде неочищенной соли на следующей стадии. 1Н ЯМР (400 МГц, CD3OD) δ 9,38 (s, 1Н), 8,71 (s, 1Н), 7,33 (s, 1Н), 3,99-3,92 (m, 1Н), 3,84-3,75 (m, 1Н), 3,57-3,46 (m, 3Н), 3,18-3,11 (m, 1Н), 2,83-2,69 (m, 1Н), 2,40-2,33 (m, 1Н), 1,92-1,81 (m, 1Н).
Стадия 3: R-4-метилбензил-4-(([1,2,4]триазоло[4,3-а]пиразин-5-иламино)метил)-3,3-дифторпиперидин-1-карбоксилат

[0226] К раствору 2,5-диоксоциклопентил 4-метилбензилкарбоната (201 мг, 0,76 ммоль) в MeCN (5 мл) добавляли R-N-((3,3-дифторпиперидин-4-ил)метил)-[1,2,4]триазоло[4,3-а]пиразин-5-амина трифторацетат (470 мг, 0,69 ммоль) и триэтиламин (0,32 мл, 2,30 ммоль) при комнатной температуре. После перемешивания в течение 1 ч смесь разбавляли EtOAc. Органическую фазу промывали водой, солевым раствором, сушили над Na2SO4 и концентрировали под вакуумом. Полученный концентрат очищали с помощью колоночной хроматографии на силикагеле (ДХМ/МеОН = 40/1), получая указанное в заголовке соединение в виде желтого порошка (126 мг, выход 43,6% за две стадии). МС (ИЭР), расчет для C20H22F2N6O2: 416,2; экспериментальное значение: 417,5 [М+Н]. 1Н ЯМР (400 МГц, CD3OD) δ 9,33 (s, 1Н), 8,64 (s, 1Н), 7,24 (d, J=8,0 Гц, 2Н), 7,23 (s, 1Н), 7,16 (d, J=8,0 Гц, 2Н), 5,10 (s, 2Н), 4,40-4,29 (m, 1Н), 4,23-4,14 (m, 1Н), 3,86 (dd, J=4,8 и 14,4 Гц, 1Н), 3,41 (dd, J=4,8 и 14,4 Гц, 1Н), 3,30-2,15 (m, 1Н), 3,05-2,88 (m, 1Н), 2,61-2,47 (m, 1Н),2,33 (s, 3Н), 2,10-2,00 (m, 1Н), 1,65-1,53 (m, 1Н).
ПРИМЕР 1.4а. R-4-метилбензил-4-(([1,2,4]триазоло[4,3-а]пиразин-5-иламино)метил)-3,3-дифторпиперидин-1-карбоксилата метансульфонат (Е1-2.2а)
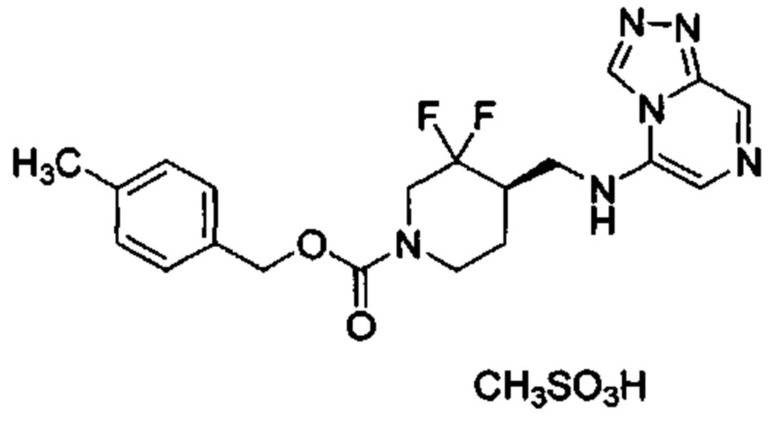
[0227] К раствору R-4-метилбензил-4-(([1,2,4]триазоло[4,3-а]пиразин-5-иламино)метил)-3,3-дифторпиперидин-1-карбоксилата (127 мг, 0,31 ммоль) в МеОН (3 мл) добавляли метансульфоновую кислоту (29 мг, 0,30 ммоль). После перемешивания в течение 1 ч при комнатной температуре смесь концентрировали, получая указанное в заголовке соединение в виде белого порошка (131 мг, 84%). МС (ИЭР), расчет для C20H22F2N6O2: 416,2; экспериментальное значение: 417,5 [М+Н]. 1Н ЯМР (400 МГц, CD3OD) δ 9,58 (s, 1Н), 8,85 (s, 1Н), 7,70 (s, 1Н),7,24 (d, J=8,0 Гц, 2Н), 7,17 (d, J=8,0 Гц, 2Н), 5,10 (s, 2Н), 4,45-4,30 (m, 1Н), 4,24-4,18 (m, 1Н), 3,95 (dd, J=5,2 и 14,0 Гц, 1Н), 3,57 (dd, J=8,0 и 14,0 Гц, 1Н), 3,26-2,90 (m, 2Н), 2,70 (s, 3Н), 2,64-2,51 (m, 1Н), 2,33 (s, 3Н), 2,11-2,02 (m, 1Н), 1,67-1,54 (m, 1Н).
ПРИМЕР 1.5. (+)-R-4-метилбензил 4-(([1,2,4]триазоло[1,5-а]пиридин-2-иламино)метил)-3,3-дифторпиперидин-1-карбоксилат (Е1-9.2)
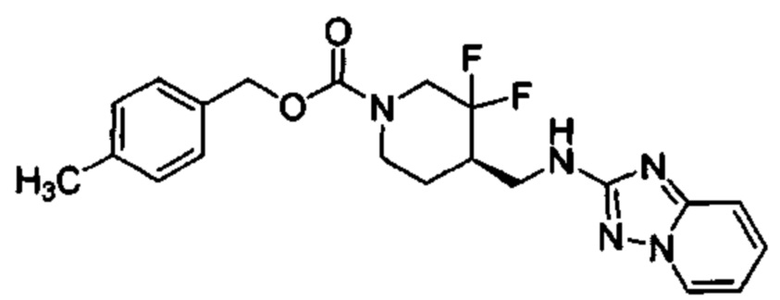
Стадия 1: R-трет-бутил-4-(([1,2,4]триазоло[1,5-а]пиридин-2-иламино)метил)-3,3-дифторпиперидин-1-карбоксилат
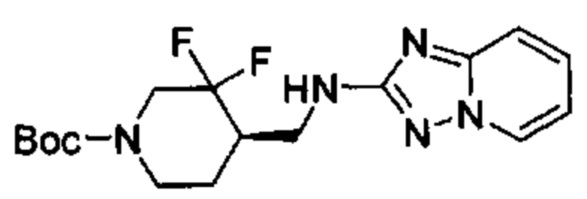
[0228] К перемешиваемой суспензии 2-бром-[1,2,4]триазоло[1,5-а]пиридина (500 мг, 2,53 ммоль) в m-бутиловом спирте (15 мл) добавляли (-)-R-трет-бутил-4-(аминометил)-3,3-дифторпиперидин-1-карбоксилат (758 мг, 3,03 ммоль), предварительный катализатор Brettphos (75 мг), Brettphos (75 мг) и Cs2CO3 (2,25 г, 5,06 ммоль) при комнатной температуре в атмосфере азота. Реакционную смесь нагревали до 100°C в течение ночи. Смеси давали остыть до комнатной температуры, разбавляли ДХМ и фильтровали через целит. Фильтрат промывали водой, сушили над Na2SO4 и концентрировали под вакуумом. Остаток очищали с помощью колоночной хроматографии на силикагеле (50% гексана в EtOAc), получая указанное в заголовке соединение в виде порошка грязно-белого цвета (298 мг, 32%). МС (ИЭР), расчет для C17H23F2N5O2: 367,2; экспериментальное значение: 368,5 [М+Н]. 1Н ЯМР (400 МГц, CD3OD) δ 8,45 (d, J=6,8 Гц, 1Н), 7,53-7,48 (m, 1Н), 7,37 (d, J=8,8 Гц, 1Н), 6,96-6,92 (m, 1Н), 4,28-4,14 (m, 1Н), 4,12-4,04 (m, 1Н), 3,77 (dd, J=14,4,8 Гц, 1Н), 3,36-3,33 (m, 1Н), 3,18-3,07 (m, 1Н), 2,92-2,84 (m, 1Н), 2,48-2,35 (m, 1Н), 1,98-1,91 (m, 1Н), 1,56-1,48 (m, 1Н), 1,47 (s, 9Н).
Стадия 2: (+)-R-4-метилбензил-4-(([1,2,4]триазоло[1,5-а]пиридин-2-иламино)метил)-3,3-дифторпиперидин-1-карбоксилат

[0229] К раствору R-трет-бутил-4-(([1,2,4]триазоло[1,5-а]пиридин-2-иламино)метил)-3,3-дифторпиперидин-1-карбоксилата (230 мг, 0,63 ммоль) в ДХМ (6 мл) добавляли ТФУ (2 мл). Полученный реакционный раствор перемешивали при комнатной температуре в течение 30 мин. Растворитель выпаривали, получая промежуточное соединение, представляющее собой соль трифторуксусной кислоты R-N-((3,3-дифторпиперидин-4-ил)метил)-[1,2,4]-триазоло[1,5-а]пиридин-2-амина в виде желтого масла (260 мг), которое применяли на следующей стадии без дополнительной очистки. К перемешиваемому раствору трифторацетатной соли R-N-((3,3-дифторпиперидин-4-ил)метил)-[1,2,4]-триазоло[1,5-а]пиридин-2-амина (260 мг) в ацетонитриле (5 мл) добавляли триэтиламин (0,26 мл) и 2,5-диоксопирролидин-1-ил-4-метилбензилкарбонат (181 мг, 0,69 ммоль). После перемешивания в течение 1 ч при комнатной температуре, реакционную смесь концентрировали под вакуумом, и остаток переносили в EtOAc. Раствор промывали водой, солевым раствором, сушили над Na2SO4 и концентрировали под вакуумом. Остаток очищали с помощью колоночной хроматографии на силикагеле (50% гексана в EtOAc), получая указанное в заголовке соединение в виде белого порошка (234 мг, 90%). [α]D=+26,2° (с=7,5 мг/мл, МеОН, 28°C). МС (ИЭР), расчет для C21H23F2N5O2: 415,2; экспериментальное значение: 416,6 [М+Н]. 1Н ЯМР (400 МГц, CD3OD) δ 8,44 (d, J=6,4 Гц, 1Н), 7,52-7,48 (m, 1Н), 7,36 (d, J=8,8 Гц, 1Н), 7,24 (d, J=8,0 Гц, 2Н), 7,17 (d, J=8,0 Гц, 2Н), 6,95-6,92 (m, 1Н), 5,12-5,06 (m, 2Н), 4,34-4,22 (m, 1Н), 4,16-4,11 (m, 1Н), 3,77 (dd, J=14,4,8 Гц, 1Н), 3,36-3,31 (m, 1Н), 3,25-3,09 (m, 1Н), 3,00-2,89 (m, 1Н), 2,51-2,38 (m, 1Н), 2,33 (s, 3Н), 2,00-1,92 (m, 1Н), 1,57-1,46 (m, 1Н).
ПРИМЕР 1.6. (+)-R-4-метилбензил-4-((1Н-пиразоло[3,4-d]пиримидин-6-иламино)метил)-3,3-дифторпиперидин-1-карбоксилат (Е1-8.2)

Стадия 1: R-трет-бутил-4-((1Н-пиразоло[3,4-d]пиримидин-6-иламино)метил)-3,3-дифторпиперидин-1-карбоксилат
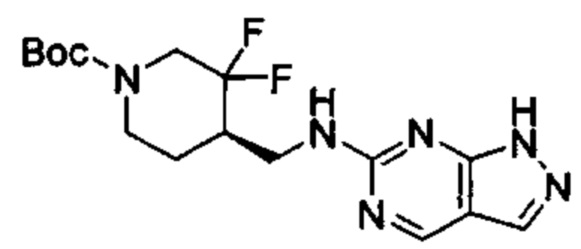
[0230] Смесь R-трет-бутил-4-(аминометил)-3,3-дифторпиперидин-1-карбоксилата (440 мг, 1,60 ммоль), 6-хлор-1Н-пиразоло[3,4-d]пиримидина (272 мг, 1,76 ммоль) и DIPEA (0,84 мл, 4,80 ммоль) в i-PrOH (10 мл) нагревали в течение ночи при 85°C. Полученной смеси давали остыть до комнатной температуры и проводили ее концентрирование при пониженном давлении. Полученный концентрат очищали с помощью колоночной хроматографии на силикагеле (гексан/этилацетат = 1/3), получая указанное в заголовке соединение в виде желтого порошка (510 мг, 86%). МС (ИЭР), расчет для C16H22F2N6O2: 368,2; экспериментальное значение: 369,4 [М+Н]. 1Н ЯМР (400 МГц, CDCl3) δ 10,88 (ушир. s, 1Н), 8,78 (s, 1Н), 7,92 (s, 1Н), 5,77-5,67 (m, 1Н), 4,50-4,00 (m, 2Н), 3,90-3,84 (m, 1Н), 3,67-3,58 (m, 1Н), 3,07-2,68 (m, 2Н), 2,38-2,23 (m, 1Н), 1,91-1,85 (m, 1Н), 1,62-1,56 (m, 1Н), 1,46 (s, 9Н).
Стадия 2: R-N-((3,3-дифторпиперидин-4-ил)метил)-1Н-пиразоло[3,4-d]пиримидин-6-амина гидрохлорид

[0231] К раствору R-трет-бутил-4-((1Н-пиразоло[3,4-d]пиримидин-6-иламино)метил)-3,3-дифторпиперидин-1-карбоксилата (510 мг, 1,39 ммоль) в ДХМ (4 мл) добавляли HCl в МеОН (10 мл, 2,0 М) при комнатной температуре. После перемешивания в течение ночи смесь концентрировали, получая указанное в заголовке соединение в виде бледно-желтого порошка (504 мг, 100%), который применяли на следующей стадии без дополнительной очистки. МС (ИЭР), расчет для C11H14F2N6: 268,1; экспериментальное значение: 269,2 [М+Н]. 1Н ЯМР (400 МГц, CD3OD) δ 9,09 (s, 1Н), 8,33 (s, 1Н), 4,04 (dd, J=5,2, 14,0 Гц, 1Н), 3,81-3,73 (m, 1Н), 3,63-3,46 (m, 3Н), 3,21-3,13 (m, 1Н), 2.83-2,69 (m, 1Н), 2,33-2,24 (m, 1Н), 1,90-1,79 (m, 1Н).
Стадия 3: (+)-R-4-метилбензил-4-((1Н-пиразоло[3,4-d]пиримидин-6-иламино)метил)-3,3-дифторпиперидин-1-карбоксилат

[0232] К раствору п-толилметанола (263 мг, 2,15 ммоль) в ДМСО (4 мл) добавляли CDI (349 мг, 2,15 ммоль) при комнатной температуре. После перемешивания в течение 1 ч добавляли R-N-((3,3-дифторпиперидин-4-ил)-метил)-1Н-пиразоло[3,4-d]пиримидин-6-амина гидрохлорид (504 мг, 1,66 ммоль). Смесь нагревали до 80°C в атмосфере N2. После перемешивания в течение ночи смесь разбавляли EtOAc. Органическую фазу промывали водой и солевым раствором, сушили над Na2SO4 и концентрировали под вакуумом. Полученный концентрат очищали с помощью колоночной хроматографии на силикагеле (ДХМ/МеОН = 40/1), получая указанное в заголовке соединение в виде белого порошка (306 мг, 49%). [α]D=+22° (с=8,5 мг/мл, 50% ДХМ в МеОН, 26°C). МС (ИЭР), расчет для C20H22F2N6O2: 416,2; экспериментальное значение: 417,4 [М+Н]. 1Н ЯМР (400 МГц. CD3OD) δ 8,77 (s, 1Н), 7,92 (s, 1Н), 7,24 (d, J=8,0 Гц, 2Н), 7,17 (d, J=8,0 Гц, 2Н), 5.09 (s, 2Н), 4.33-4.23 (m, 1Н), 4,16-4,10 (m, 1Н), 3,87 (dd, J=5,2 и 14,0 Гц, 1Н), 3,47-3,39 (m, 1Н), 3,25-3,10 (m, 1Н), 3,02-2,87 (m, 1Н), 2,54-2,40 (m, 1Н), 2,33 (s, 3Н), 1,95-1,88 (m, 1Н), 1,58-1,47 (m, 1Н).
ПРИМЕР 1.6а. (+)-R-4-метилбензил-4-((1Н-пиразоло[3,4-d]пиримидин-6-иламино)метил)-3,3-дифторпиперидин-1-карбоксилата метансульфонат (Е1-8.2а)
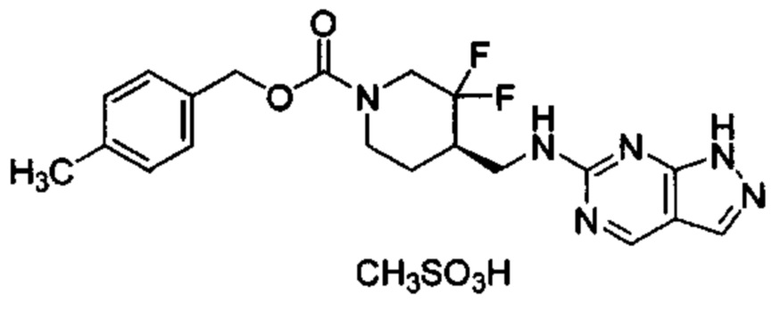
[0233] К раствору R-4-метилбензил-4-((1Н-пиразоло[3,4-d]пиримидин-6-иламино)метил)-3,3-дифторпиперидин-1-карбоксилата (184 мг, 0,44 ммоль) в ДХМ/МеОН (12 мл/4 мл) добавляли метансульфоновую кислоту (43 мг, 0,44 ммоль). После перемешивания в течение 1 ч при комнатной температуре полученную смесь концентрировали, получая указанное в заголовке соединение в виде белого порошка (191 мг, 84%). [α]D=+11,2° (с=10 мг/мл, 50% ДХМ в МеОН, 26°C). МС (ИЭР), расчет для C20H22F2N6O2: 416,2; экспериментальное значение: 417,5 [М+Н]. 1Н ЯМР (400 МГц, CD3OD) δ 9,03 (s, 1Н), 8,29 (s, 1Н), 7,24 (d, J=8,0 Гц, 2Н), 7,17 (d, J=8,0 Гц, 2Н), 5,09 (s, 2Н), 4,37-4,27 (m, 1Н), 4,20-4,13 (m, 1Н), 3,94 (dd, J=5,2 и 14,0 Гц, 1Н), 3,58-3,53 (m, 1Н), 3,24-3,10 (m, 1Н), 3,03-2,92 (m, 1Н), 2,72 (s, 3Н), 2,56-2,44 (m, 1Н), 2,33 (s, 3Н), 1,99-1,90 (m, 1Н), 1,61-1,50 (m, 1Н).
ПРИМЕР 1.7. R-4-хлорбензил-4-(([1,2,4]триазоло[4,3-а]пиразин-8-иламино)метил)-3,3-дифторпиперидин-1-карбоксилат (Е1-1.3)
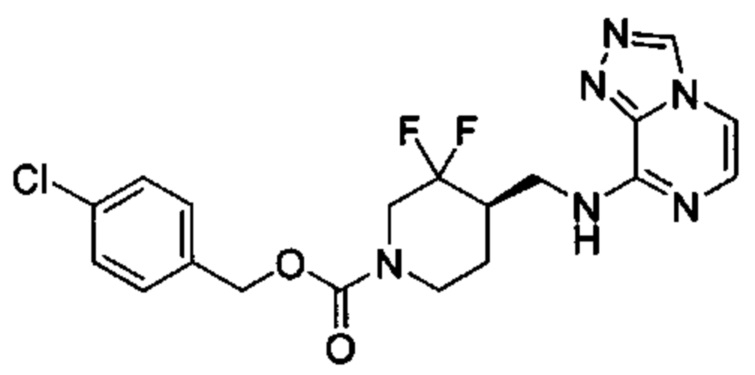
[0234] К перемешиваемому раствору 4-хлорбензилового спирта (115 мг, 0,81 ммоль) и бис-(2,5-диоксопирролидин-1-ил)карбоната (207 мг, 0,81 ммоль) в ацетонитриле (3,0 мл) и CH2Cl2 (3,0 мл) добавляли 4-диметиламинопиридин (49 мг, 0,40 ммоль), и полученную смесь перемешивали в течение 2 ч при комнатной температуре. Добавляли трифторацетатную соль N-((3,3-дифторпиперидин-4-ил)метил)-[1,2,4]триазоло[4,3-а]пиразин-8-амина (280 мг, 0,73 ммоль) в MeCN (2 мл) и ТЭА (0,3 мл, 2,2 ммоль), и полученную смесь перемешивали в течение 1 часа при комнатной температуре. Далее смесь разбавляли этилацетатом (5 мл) и органическую фазу промывали водой, солевым раствором, сушили над безводным Na2SO4 и концентрировали при пониженном давлении. Остаток очищали с помощью колоночной хроматографии на силикагеле (ДХМ/МеОН = 50/1), получая указанное в заголовке соединение в виде бледно-желтого порошка (190 мг, 59%). МС (ИЭР), расчет для C19H19ClF2N6O2: 436,1, 438,1; экспериментальное значение: 437,4, 439,4 [М+Н]. 1Н ЯМР (400 МГц, CD3OD) δ 9,08 (s, 1Н), 7,69 (d, J=4,8 Гц, 1Н), 7,39-7,33 (m, 4Н), 7,32 (d, J=4,8 Гц, 1Н), 5,16-5,09 (m, 2Н), 4,37-4,26 (m, 1Н), 4,19-4,11 (m, 1Н), 4,01-3,95 (m, 1Н), 3,65-3,57 (m, 1Н), 3,27-3,07 (m, 1Н), 3,07-2,88 (m, 1Н), 2,65-2,47 (m, 1Н), 1,99-1,91 (m, 1Н), 1,63-1,49 (m, 1Н).
ПРИМЕР 1.7а. R-4-хлорбензил-4-(([1,2,4]триазоло[4,3-а]пиразин-8-иламино)метил)-3,3-дифторпиперидин-1-карбоксилата метансульфонат (Е1-1.3а)
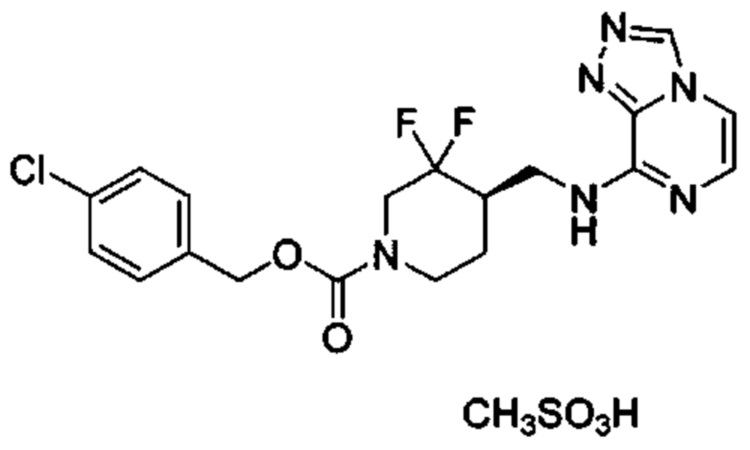
[0235] К перемешиваемому раствору R-4-хлорбензил-4-(([1,2,4]триазоло[4,3-а]пиразин-8-иламино)метил)-3,3-дифторпиперидин-1-карбоксилата (99 мг, 0,23 ммоль) в МеОН (2,0 мл) добавляли CH3SO3H (22 мг, 0,23 ммоль) при комнатной температуре. После перемешивания в течение 30 мин смесь концентрировали, получая указанное в заголовке соединение в виде порошка грязно-белого цвета (116 мг, 96%). МС (ИЭР), расчет для C19H19ClF2N6O2: 436,1, 438,4; экспериментальное значение: 437,4, 439,4 [М+Н]. 1Н ЯМР (400 МГц, CD3OD) δ 9,31 (s, 1Н), 7,94 (d, J=4,8 Гц, 1Н), 7,36 (s, 4Н), 7,25 (d, J=4,8 Гц, 1Н), 5,19-5,09 (m, 2Н), 4,44-4,33 (m, 1Н), 4,26-4,18 (m, 1Н), 4,06-3,96 (m, 1Н), 3,73-3,62 (m, 1Н), 3,29-3,14 (m, 1Н), 3,11-2,91 (m, 1Н), 2,70 (s, 3Н), 2,68-2,58 (m, 1Н), 2,07-1,99 (m, 1Н), 1,66-1,53 (m, 1Н).
ПРИМЕР 1.8. R-4-фторбензил-4-(([1,2,4]триазоло[4,3-а]пиразин-8-иламино)метил)-3,3-дифторпиперидин-1-карбоксилат (Е1-1.4)

[0236] К перемешиваемому раствору 4-фторбензилового спирта (300 мг, 2,38 ммоль) в ДХМ-MeCN (1:1 об./об., 10 мл) добавляли N,N'-дисукцинимидилкарбонат (610 мг, 2,38 ммоль) и DMAP (145 мг, 1,19 ммоль) при температуре окружающей среды. Постепенно получали прозрачный раствор и перемешивали смесь в течение 1 ч при комнатной температуре. Далее добавляли триэтиламин (1,0 мл, 7,1 ммоль), а затем соль ТФУ R-N-((3,3-дифторпиперидин-4-ил)метил)-[1,2,4]триазоло[4,3-а]пиразин-8-амина (780 мг, 2,14 ммоль) в ацетонитриле (3 мл). Полученную смесь перемешивали в течение 1 ч при комнатной температуре, и смесь концентрировали под вакуумом. Остаток растворяли в этилацетате и органическую фазу промывали водой, солевым раствором, сушили над Na2SO4 и концентрировали под вакуумом. Полученный концентрат очищали с помощью колоночной хроматографии на силикагеле (этилацетат), получая указанное в заголовке соединение в виде порошка грязно-белого цвета (446 мг, 50%). МС (ИЭР), расчет для C19H19F3N6O2: 420,2; экспериментальное значение: 421,5 [М+Н]. 1Н ЯМР (400 МГц, CDCl3) δ 8,71 (s, 1Н), 7,40 (d, J=4,8 Гц, 1Н), 7,36-7,31 (m, 3Н), 7,04 (t, J=8,4 Гц, 2Н), 6,51-6,51 (m, 1Н), 5,11 (s, 2Н), 4,54-4,10 (m, 2Н), 4,04-3,96 (m, 1Н), 3,82-3,72 (m, 1Н), 3,14-2,96 (m, 1Н), 2,94-2,78 (m, 1Н), 2,49-2,33 (m, 1Н), 1,95-1,87 (m, 1Н), 1,71-1,61 (m, 1Н).
ПРИМЕР 1.8а. R-4-фторбензил-4-(([1,2,4]триазоло[4,3-а]пиразин-8-иламино)метил)-3,3-дифторпиперидин-1-карбоксилата метансульфонат (Е1-1.4а)

[0237] К перемешиваемому раствору R-4-фторбензил-4-(([1,2,4]триазоло[4,3-а]пиразин-8-иламино)-метил)-3,3-дифторпиперидин-1-карбоксилата (446 мг, 1,06 ммоль) в ДХМ/МеОН (6 мл, 1:1) добавляли метилсульфоновую кислоту (102 мг, 1,06 ммоль) при комнатной температуре. После перемешивания в течение 30 мин, смесь концентрировали. Полученное твердое вещество промывали эфиром, получая указанное в заголовке соединение в виде твердого вещества бледно-коричневого цвета (510 мг, 93%). МС (ИЭР), расчет для C19H19F3N6O2: 420,2; экспериментальное значение: 421,5 [М+Н]. 1Н ЯМР (400 МГц, CD3OD) δ 9,31 (s, 1Н), 7,94 (d, J=5,6 Гц, 1Н), 7,43-7,37 (m, 2Н), 7,25 (d, J=5,6 Гц, 1Н), 7,09 (t, J=8,8 Гц, 2Н), 5,13 (s, 2Н), 4,43-4,32 (m, 1Н), 4,24-4,17 (m, 1Н), 4,06-3,95 (m, 1Н), 3,73-3,63 (m, 1Н), 3,32-3,10 (m, 1Н), 3,09-2,92 (m, 1Н), 2,70 (s, 3Н), 2,68-2,58 (m, 1Н), 2,06-1,98 (m, 1Н), 1,65-1,53 (m, 1Н).
ПРИМЕР 1.9. (+)-R-4-метилбензил-3,3-дифтор-4-((пиримидин-2-иламино)метил)-пиперидин-1-карбоксилат (Е1-22.2)

Стадия 1: R-трет-бутил-3,3-дифтор-4-((пиримидин-2-иламино)метил)пиперидин-1-карбоксилат
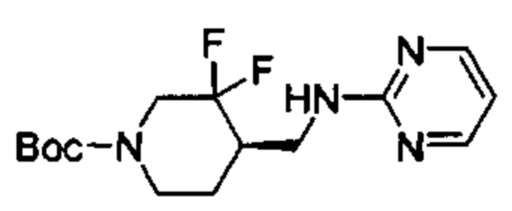
[0238] Раствор 2-хлорпиримидина (206 мг, 1,8 ммоль), R-трет-бутил-4-(аминометил)-3,3-дифторпиперидин-1-карбоксилата (500 мг, 1,8 ммоль) и DIPEA (0,63 мл, 3,6 ммоль) в n-BuOH (5 мл) нагревали до 95°C в запаянной пробирке в течение ночи при перемешивании. Полученной смеси давали возможность остыть до комнатной температуры и проводили ее концентрирование при пониженном давлении. Остаток растворяли в этилацетате и органическую фазу промывали водой и солевым раствором. Органическую фазу сушили над сульфатом натрия и концентрировали под вакуумом. Полученный концентрат обрабатывали гексаном и этилацетатом (1,5 мл+5 мл). Полученную в результате суспензию фильтровали, получая указанное в заголовке соединение в виде порошка грязно-белого цвета (420 мг, 71%). МС (ИЭР), расчет для C15H22F2N4O2: 328,2; экспериментальное значение: 329,3 [М+Н]. 1Н ЯМР (400 МГц, CDCl3) δ 8,27 (d, J=4,8 Гц, 2H), 6,55 (t, J=4,8 Гц, 1H), 5,35-5,29 (m, 1H), 4,45-4,03 (m, 2H), 3,82-3,75 (m, 1H), 3,57-3,47 (m, 1H), 3,06-2,67 (m, 2H), 2,31-2,16 (m, 1H), 1,88-1,79 (m, 1H), 1,62-1,52 (m, 1H), 1,46 (s, 9H).
Стадия 2: R-N-((3,3-дифторпиперидин-4-ил)метил)пиримидин-2-амина гидрохлорид

[0239] К раствору R-трет-бутил-3,3-дифтор-4-((пиримидин-2-иламино)метил)пиперидин-1-карбоксилата (410 мг, 1,24 ммоль) в дихлорметане (2 мл) добавляли 2 н. метанольный раствор HCl (7 мл) при комнатной температуре. После перемешивания в течение ночи при температуре окружающей среды смесь концентрировали под вакуумом, получая указанное в заголовке соединение (285 мг, 76%), которое непосредственно применяли на следующей стадии. МС (ИЭР), расчет для C10H14F2N4: 228,1; экспериментальное значение: 229,3 [М+Н]. 1Н ЯМР (400 МГц, CD3OD) δ 8,95-8,43 (m, 2Н), 7,10 (t, J=5,6 Гц, 1Н), 4,03 (dd, J=14,0, 5,6 Гц, 1Н), 3,82-3,74 (m, 1Н), 3,68 (dd, J=14,0, 5,6 Гц, 1Н), 3,59-3,48 (m, 2Н), 3,23-3,15 (m, 1Н), 2,82-2,67 (m, 1Н), 2,34-2,24 (m, 1Н), 1,91-1,78 (m, 1Н).
Стадия 3: (+)-R-4-метилбензил-3,3-дифтор-4-((пиримидин-2-иламино)метил)-пиперидин-1-карбоксилат
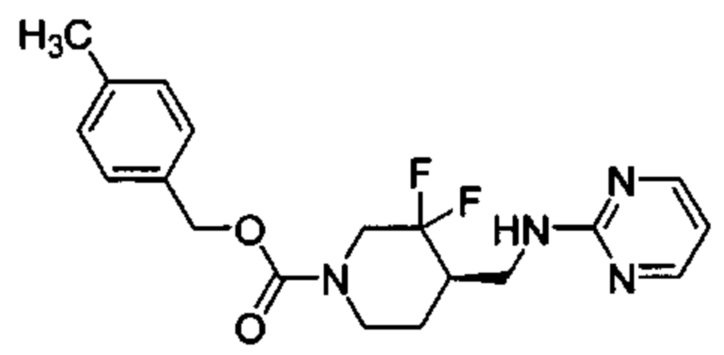
[0240] К перемешиваемому раствору 4-метилбензилового спирта (199 мг, 1,62 ммоль) в ДМСО (5 мл) добавляли CDI (263 мг, 1,62 ммоль). После перемешивания в течение 1 ч при комнатной температуре добавляли (3,3-дифторпиперидин-4-илметил)-пиримидин-2-иламина дигидрохлорид (285 мг, 0,95 ммоль), и полученную реакционную смесь перемешивали в течение ночи при 80°C. Реакционную смесь разбавляли этилацетатом, и органическую фазу промывали водой и солевым раствором. Органический слой сушили над сульфатом натрия и концентрировали под вакуумом. Полученный концентрат очищали с помощью колоночной хроматографии на силикагеле (гексан/EtOAc = 1:1), получая указанное в заголовке соединение в виде белого порошка (135 мг, 38%). %). [α]D=+10,5° (с=3,7 мг/мл, МеОН, 26°C). МС (ИЭР), расчет для C19H22F2N4O2: 376,2; экспериментальное значение: 377,4 [М+Н]. 1Н ЯМР (400 МГц, CDCl3) δ 8,27 (d, J=4,8 Гц, 2Н), 7,25 (d, J=8,0 Гц, 2Н), 7,17 (d, J=8,0 Гц, 2Н), 6,55 (t, J=4,8 Гц, 1Н), 5,37-5,26 (m, 1Н), 5,16-5,05 (m, 2Н), 4,57-4,10 (m, 2Н), 3,84-3,74 (m, 1Н), 3,59-3,47 (m, 1Н), 3,12-2,76 (m, 2Н), 2,35 (s, 3Н), 2,32-2,17 (m, 1Н), 1,92-1,78 (m, 1Н), 1,62-1,52 (m, 1Н).
ПРИМЕР 1.9а. R-4-метилбензил-3,3-дифтор-4-((пиримидин-2-иламино)метил)пиперидин-1-карбоксилата метансульфонат (Е1-22.2а)
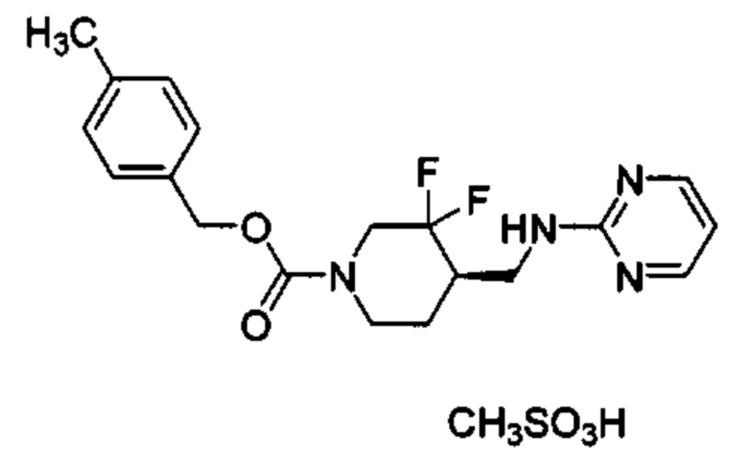
[0241] К перемешиваемому раствору (+)-R-4-метилбензил-3,3-дифтор-4-((пиримидин-2-иламино)метил)-пиперидин-1-карбоксилата (123 мг, 0,33 ммоль) в МеОН (2,0 мл) добавляли CH3SO3H (32 мг, 0,33 ммоль) при комнатной температуре. После перемешивания в течение 30 мин смесь концентрировали, получая указанное в заголовке соединение в виде порошка грязно-белого цвета (150 мг, 97%). МС (ИЭР), расчет для C19H22F2N4O2: 376,2; экспериментальное значение: 377,4 [М+Н]. 1Н ЯМР (400 МГц, CD3OD) δ 8,90-8,40 (m, 2Н), 7,24 (d, J=8,0 Гц, 2Н), 7,17 (d, J=8,0 Гц, 2Н), 7,00 (t, J=5,6 Гц, 1Н), 5,09 (s, 2Н), 4,39-4,25 (m, 1Н), 4,21-4,12 (m, 1Н), 3,91 (dd, J=5,6 и 14,0 Гц, 1Н), 3,57 (dd, J=7,6 и 14,0 Гц, 1Н), 3,27-3,08 (m, 1Н), 3,06-2,87 (m, 1Н), 2,71 (s, 3Н), 2,54-2,39 (m, 1Н), 2,33 (s, 3Н), 1,97-1,89 (m, 1Н), 1,60-1,48 (m, 1Н).
ПРИМЕР 1.10. R-4-метилбензил-3,3-дифтор-4-((пиразин-2-иламино)метил)пиперидин-1-карбоксилат (Е1-21.2)
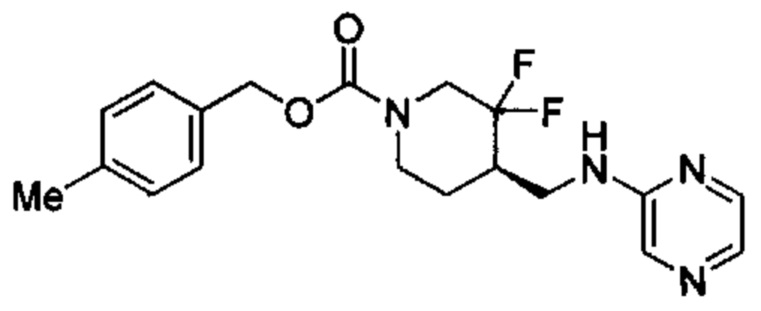
Стадия 1: R-трет-бутил-3,3-дифтор-4-((пиразин-2-иламино)метил)пиперидин-1-карбоксилат
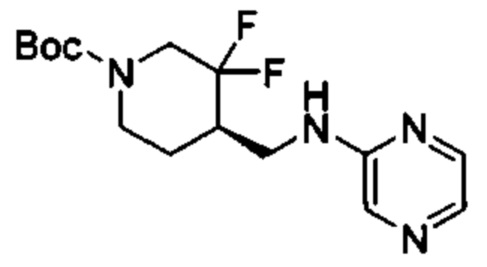
[0242] Смесь R-трет-бутил-4-(аминометил)-3,3-дифторпиперидин-1-карбоксилата (200 мг, 0,80 ммоль), 2-бромпиразина (140 мг, 0,88 ммоль) и DIPEA (0,42 мл, 2,40 ммоль) в NMP (6 мл) нагревали при перемешивании в течение ночи при 130°C. Полученной смеси давали возможность остыть до комнатной температуры и разбавляли ее этилацетатом. Органическую фазу промывали водой и солевым раствором, сушили над Na2SO4 и концентрировали под вакуумом. Полученный концентрат очищали с помощью колоночной хроматографии на силикагеле (гексан/этилацетат = 1/1), получая указанное в заголовке соединение в виде желтого масла (196 мг, 50%). МС (ИЭР), расчет для C15H22F2N4O2: 328,2; экспериментальное значение: 329,2 [М+Н]. 1Н ЯМР (400 МГц, CDCl3) δ 7,98 (dd, J=2,8 и 1,2 Гц, 1Н), 7,89 (d, J=1,2 Гц, 1Н), 7,81 (d, J=2,8 Гц, 1Н), 4,80-4,70 (m, 1Н), 4,45-4,10 (m, 2Н), 3,76-3,69 (m, 1Н), 3,57-3,49 (m, 1Н), 3,04-2,70 (m, 2Н), 2,31-2,16 (m, 1Н), 1,87-1,79 (m, 1Н), 1,63-1,53 (m, 1Н), 1,47 (s, 9Н).
Стадия 2: R-4-метилбензил-3,3-дифтор-4-((пиразин-2-иламино)метил)пиперидин-1-карбоксилат
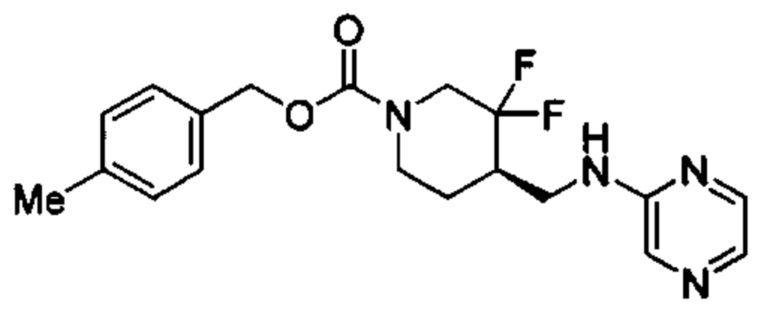
[0243] К раствору R-трет-бутил-3,3-дифтор-4-((пиразин-2-иламино)метил)-пиперидин-1-карбоксилата (196 мг, 0,59 ммоль) в ДХМ (3 мл) добавляли ТФУ (2 мл) при комнатной температуре. После перемешивания в течение 30 мин, смесь концентрировали, получая неочищенный продукт R-N-((3,3-дифторпиперидин-4-ил)метил)пиразин-2-амина трифторацетата в виде желтого масла, который непосредственно применяли на следующей стадии без дополнительной очистки. К раствору неочищенного R-N-((3,3-дифторпиперидин-4-ил)метил)пиразин-2-амина трифторацетата в MeCN (6 мл) добавляли триэтиламин (0,8 мл, 5,8 ммоль) и 2,5-диоксоциклопентил-4-метилбензилкарбонат (386 мг, 1,44 ммоль) при комнатной температуре. После перемешивания в течение 1 ч смесь разбавляли EtOAc. Органическую фазу промывали водой, солевым раствором, сушили над Na2SO4 и концентрировали под вакуумом. Полученный концентрат очищали с помощью колоночной хроматографии на силикагеле (100% EtOAc), получая указанное в заголовке соединение в виде желтого масла (167 мг, 52%). МС (ИЭР), расчет для C19H22F2N4O2: 376,2; экспериментальное значение: 377,5 [М+Н]. 1Н ЯМР (400 МГц, CD3OD) δ 7,97 (dd, J=2,8 и 1,2 Гц, 1Н), 7,88 (d, J=1,2 Гц, 1Н), 7,64 (d, J=2,8 Гц, 1Н), 7,24 (d, J=8,0 Гц, 2Н), 7,17 (d, J=8,0 Гц, 2Н), 5,09 (s, 2Н), 4,31-4,21 (m, 1Н), 4,17-4,07 (m, 1Н), 3,76 (dd, J=14,0 и 5,2 Гц, 1Н), 3,35 (dd, J=14,0 и 5,2 Гц, 1Н), 3,27-2,85 (m, 2Н), 2,44-2,34 (m, 1Н), 2,33 (s, 3Н), 1,96-1,85 (m, 1Н), 1,54-1,43 (m, 1Н).
ПРИМЕР 1.10а. R-4-метилбензил-3,3-дифтор-4-((пиразин-2-иламино)метил)пиперидин-1-карбоксилата метансульфонат (Е1-21.2а)

[0244] К раствору R-4-метилбензил-3,3-дифтор-4-((пиразин-2-иламино)метил)пиперидин-1-карбоксилата (118 мг, 0,31 ммоль) в МеОН (2 мл) добавляли метансульфоновую кислоту (27 мг, 0,28 ммоль). После перемешивания в течение 1 ч при комнатной температуре смесь концентрировали, получая указанное в заголовке соединение в виде желтого порошка (127 мг, 87%). МС (ИЭР), расчет для C19H22F2N4O2: 376,2; экспериментальное значение: 377,4 [М+Н]. 1Н ЯМР (400 МГц, CD3OD) δ 8,6 (dd, J=2,8 и 1,2 Гц, 1Н), 8,23 (d, J=1,2 Гц, 1Н), 7,85 (d, J=2,8 Гц, 1Н), 7,24 (d, J=8,0 Гц, 2Н), 7,17 (d, J=8,0 Гц, 2Н), 5,09 (s, 2H), 4,40-4,27 (m, 1H), 4,22-4,14 (m, 1H), 3,85 (dd, J=14,0 и 5,2 Гц, 1H), 3,49 (dd, J=14,0 и 5,2 Гц, 1Н), 3,30-2,90 (m, 2Н), 2,72 (s, 3Н), 2,55-2,36 (m, 1Н), 2,33 (s, 3Н), 1,99-1,89 (m, 1Н), 1,60-1,47 (m, 1Н).
ПРИМЕР 1.11. R-4-метилбензил-3,3-дифтор-4-((5-метилпиразин-2-иламино)-метил)пиперидин-1-карбоксилат (Е1-21.26)

Стадия 1: R-трет-бутил-3,3-дифтор-4-((5-метилпиразин-2-иламино)метил)-пиперидин-1-карбоксилат
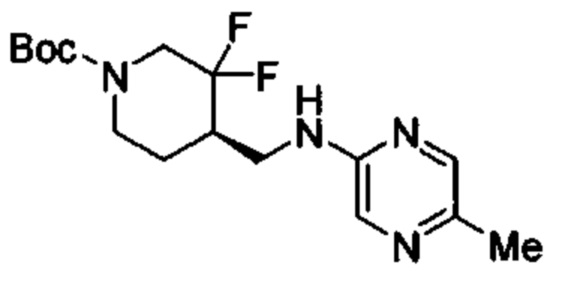
[0245] К раствору R-трет-бутил-4-(аминометил)-3,3-дифторпиперидин-1-карбоксилата (188 мг, 0,75 ммоль) в диоксане (3 мл) добавляли 2-хлор-5-метилпиразин (100 мг, 0,78 ммоль), Pd2(dba)3⋅CHCl3 (21 мг, 0,02 ммоль), Xantphos (23 мг, 0,04 ммоль) и Cs2CO3 (329 мг, 1,0 ммоль). Смесь нагревали до 90°C в атмосфере N2. После перемешивания в течение ночи реакционный раствор обрабатывали этилацетатом. Органическую фазу промывали водой, солевым раствором, сушили над Na2SO4 и концентрировали под вакуумом. Остаток очищали с помощью колоночной хроматографии на силикагеле (этилацетат/гексан = 1/1), получая указанное в заголовке соединение в виде бледно-желтого порошка (115 мг, 45%). МС (ИЭР), расчет для C16H24F2N4O2: 342,2; экспериментальное значение: 343,4 [М+Н]. 1Н ЯМР (400 МГц, CDCl3) δ 7,86 (s, 1Н), 7,83 (s, 1Н), 4,63-4,53 (m, 1Н), 4,48-4,04 (m, 2Н), 3,73-3,67 (m, 1Н), 3,52-3,45 (m, 1Н), 3,00-2,89 (m, 1Н), 2,79-2,70 (m, 1Н), 2,38 (s, 3Н), 2,28-2,15 (m, 1Н), 1,85-1,80 (m, 1Н), 1,47 (s, 9Н).
Стадия 2: R-4-метилбензил-3,3-дифтор-4-((5-метилпиразин-2-иламино)-метил)пиперидин-1-карбоксилат
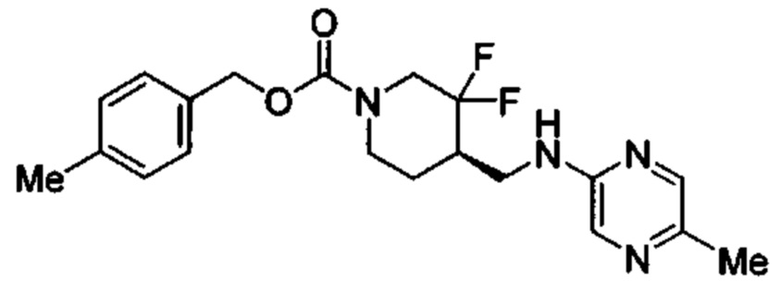
[0246] К раствору R-трет-бутил-3,3-дифтор-4-((5-метилпиразин-2-иламино)метил)пиперидин-1-карбоксилата (115 мг, 0,34 ммоль) в ДХМ (3 мл) добавляли ТФУ (1 мл) при комнатной температуре. После перемешивания в течение 30 мин, смесь концентрировали, получая R-N-((3,3-дифторпиперидин-4-ил)метил)-5-метилпиразин-2-амина трифторацетат в виде желтого масла, которое непосредственно применяли на следующей стадии без дополнительной очистки. К раствору неочищенного R-N-((3,3-дифторпиперидин-4-ил)метил)-5-метилпиразин-2-амина трифторацетата в MeCN (4 мл) добавляли триэтиламин (1 мл) и 2,5-диоксоциклопентил-4-метилбензилкарбонат (98 мг, 0,37 ммоль) при комнатной температуре. После перемешивания в течение 1 ч смесь разбавляли EtOAc. Органическую фазу промывали водой, солевым раствором, сушили над Na2SO4 и концентрировали под вакуумом. Полученный концентрат очищали с помощью колоночной хроматографии на силикагеле (этилацетат/гексан = 2/1), получая указанное в заголовке соединение в виде бледно-желтого порошка (61 мг, 46%). МС (ИЭР), расчет для C20H24F2N4O2: 390,2; экспериментальное значение: 391,2 [М+Н]. 1Н ЯМР (400 МГц, CDCl3) δ 7,86 (s, 1Н), 7,82 (s, 1Н), 7,24 (d, J=8,0 Гц, 2Н), 7,17 (d, J=8,0 Гц, 2Н), 5,10 (s, 2Н), 4,66-4,60 (m, 1Н), 4,49-4,16 (m, 2Н), 3,73-3,67 (m, 1Н), 3,51-3,44 (m, 1Н), 3,07-2,95 (m, 1Н), 2,87-2,79 (m, 1Н), 2,38 (s, 3Н), 2,35 (s, 3Н), 2,30-2,18 (m, 1Н), 1,85-1,81 (m, 1Н), 1,63-1,53 (m, 1Н).
ПРИМЕР 1.11а. R-4-метилбензил-3,3-дифтор-4-((5-метилпиразин-2-иламино)метил)-пиперидин-1-карбоксилат метансульфонат (Е1-21.26а)

[0247] К раствору R-4-метилбензил-3,3-дифтор-4-((5-метилпиразин-2-иламино)-метил)пиперидин-1-карбоксилата (54 мг, 0,138 ммоль) в ДХМ (2 мл) добавляли метилсульфоновую кислоту в МеОН (0,14 мл, 1,0 М, 0,14 ммоль). После перемешивания в течение 15 мин при комнатной температуре смесь концентрировали, получая указанное в заголовке соединение в виде порошка бледно-желтого цвета (127 мг, 87%). МС (ИЭР), расчет для C20H24F2N4O2: 390,2; экспериментальное значение: 391,2 [М+Н]. 1Н ЯМР (400 МГц, CD3OD) δ 8,22 (s, 1Н), 8,10 (s, 1Н), 7,24 (d, J=8,0 Гц, 2Н), 7,17 (d, J=8,0 Гц, 2Н), 5,09 (s, 2Н), 4,37-4,26 (m, 1Н), 4,19-4,15 (m, 1Н), 3,82 (dd, J=14,0 и 5,6 Гц, 1Н), 3,46 (dd, J=14,0 и 5,6 Гц, 1Н), 3,30-3,12 (m, 1Н), 3,02-2,91 (m, 1Н), 2,71 (s, 3Н), 2,48 (s, 3Н), 2,46-2,36 (m, 1Н), 2,33 (s, 3Н), 1,95-1,91 (m, 1Н), 1,58-1,48 (m, 1Н).
ПРИМЕР 1.12. R-4-фторбензил-4-((1Н-пиразоло[3,4-d]пиримидин-6-иламино)метил)-3,3-дифторпиперидин-1-карбоксилат (Е1-8.4)
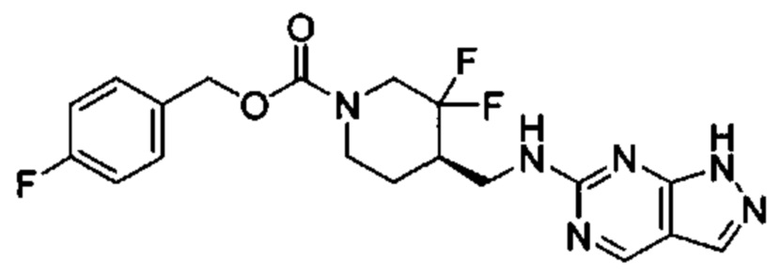
Стадия 1: R-N-((3,3-дифторпиперидин-4-ил)метил)-1Н-пиразоло[3,4-d]пиримидин-6-амина трифторацетат
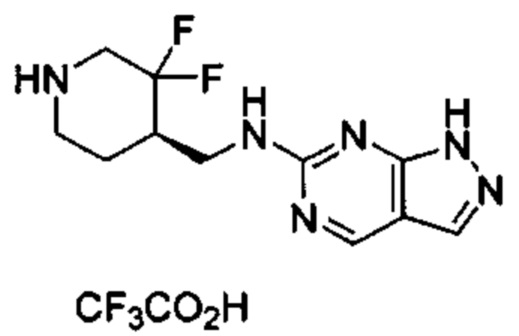
[0248] К раствору R-трет-бутил-4-((1Н-пиразоло[3,4-d]пиримидин-6-иламино)метил)-3,3-дифторпиперидин-1-карбоксилата (340 мг, 0,92 ммоль) в ДХМ (5 мл) добавляли ТФУ (3 мл) при комнатной температуре. После перемешивания в течение 30 мин, смесь концентрировали, получая указанное в заголовке соединение в виде бледно-желтого масла, которое применяли на следующей стадии без дополнительной очистки.
Стадия 2: R-4-фторбензил-4-((1Н-пиразоло[3,4-d]пиримидин-6-иламино)метил)-3,3-дифторпиперидин-1-карбоксилат

[0249] К раствору трифторацетатной соли R-N-((3,3-дифторпиперидин-4-ил)метил)-1Н-пиразоло[3,4-d]пиримидин-6-амина с предыдущей стадии (приблизительно 0,92 ммоль) в MeCN (5 мл) добавляли триэтиламин (0,6 мл, 4,6 ммоль), затем добавляли 2,5-диоксопирролидин-1-ил-4-фторбензилкарбонат (296 мг, 1,10 ммоль). Полученную смесь перемешивали в течение 1 часа при комнатной температуре. Затем смесь разбавляли этилацетатом, промывали водой, солевым раствором, сушили над безводным Na2SO4 и концентрировали при пониженном давлении. Остаток очищали с помощью колоночной хроматографии на силикагеле (этилацетат/гексан = 1/1), получая указанное в заголовке соединение в виде порошка грязно-белого цвета (160 мг, 41% за две стадии). МС (ИЭР), расчет для C19H19F3N6O2: 420,2; экспериментальное значение: 421,4 [М+Н]. 1Н ЯМР (400 МГц, CD3OD) δ 8,77 (s, 1Н), 7,92 (s, 1Н), 7,42-7,36 (m, 2Н), 7,12-7,05 (m, 2Н), 5,12 (s, 2Н), 4,33-4,21 (m, 1Н), 4,16-4,10 (m, 1Н), 3,86 (dd, J=13,6 и 4,4 Гц, 1Н), 3,50-3,43 (m, 1Н), 3,25-2,85 (m, 2Н), 2,54-2,40 (m, 1Н), 1,95-1,88 (m, 1Н), 1,58-1,47 (m, 1Н).
ПРИМЕР 1.13. R-4-метилбензил-4-((1Н-пиразоло[3,4-d]пиримидин-4-иламино)метил)-3,3-дифторпиперидин-1-карбоксилат (Е1-6.2)
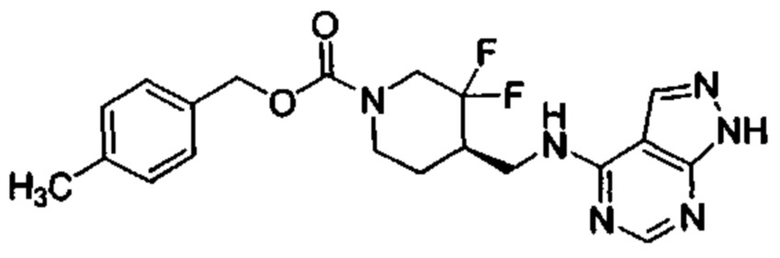
Стадия 1: R-трет-бутил-4-(([1,2,4]триазоло[4,3-а]пиразин-8-иламино)метил)-3,3-дифторпиперидин-1-карбоксилат
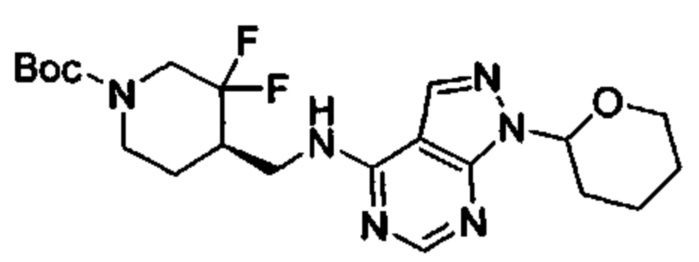
[0250] К перемешиваемому раствору R-трет-бутил-4-(аминометил)-3,3-дифторпиперидин-1-карбоксилата (600 мг, 2,4 ммоль) в n-BuOH (5 мл) добавляли 4-хлор-1-(тетрагидро-2Н-пиран-2-ил)-1Н-пиразоло[3,4-d]пиримидин (571 мг, 2,4 ммоль) и DIPEA (0,84 мл, 4,8 ммоль). Полученную смесь нагревали до 100°C в атмосфере азота в течение 13 часов. Реакционной смеси давали остыть до комнатной температуры и проводили ее концентрирование под вакуумом. Остаток растворяли в этилацетате и органическую фазу промывали водой, солевым раствором, сушили над Na2SO4 и концентрировали при пониженном давлении. Полученный концентрат очищали с помощью колоночной хроматографии на силикагеле (этилацетат/гексан = 1/1), получая указанное в заголовке соединение в виде желтого порошка (800 мг, 80%). МС (ИЭР), расчет для C21H30F2N6O3: 452,2; экспериментальное значение: 453,6 [М+Н]. 1Н ЯМР (400 МГц, CDCl3) δ 8,42 (s, 1Н), 7,95 (s, 1Н), 5,98-5,95 (m, 1Н), 5,81-5,60 (ушир. s, 1Н), 4,49-4,09 (m, 3Н), 3,99-3,87 (m, 1Н), 3,83-3,76 (m, 2Н), 3,06-2,67 (m, 2Н), 2,63-2,53 (m, 1Н), 2,40-2,23 (m, 1Н), 2,16-2,08 (m, 1Н), 1,96-1,92 (m, 1Н), 1,93-1,54 (m, 6Н), 1,47 (s, 9Н).
Стадия 2: Дигидрохлоридная соль R-N-((3,3-дифторпиперидин-4-ил)метил)-1Н-пиразоло[3,4-d]пиримидин-4-амина
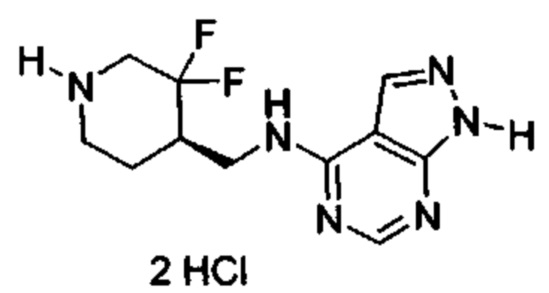
[0251] R-трет-бутил-3,3-дифтор-4-((1-(тетрагидро-2Н-пиран-2-ил)-1Н-пиразоло[3,4-d]-пиримидин-4-иламино)метил)пиперидин-1-карбоксилат (0,80 г, 1,76 ммоль) растворяли в метанольном растворе HCl (2 н., 15 мл) при комнатной температуре. После перемешивания в течение ночи полученную реакционную смесь концентрировали при пониженном давлении, получая указанное в заголовке соединение в виде твердого вещества желтого цвета (600 мг, 99%), которое применяли на следующей стадии без дополнительной очистки. МС (ИЭР), расчет для C11H14F2N6: 268,1; экспериментальное значение: 269,2 [М+Н]. 1Н ЯМР (400 МГц, CD3OD) δ 8,72 (s, 1Н), 8,64 (s, 1Н), 4,19 (dd, J=14,0 и 5,6 Гц, 1Н), 3,90 (dd, J=14,0 и 5,6 Гц, 1Н), 3,83-3,75 (m, 1Н), 3,61-3,48 (m, 2Н), 3,24-3,18 (m, 1Н), 2,93-2,78 (m, 1Н). 2,35-2,29 (m, 1Н), 1,94-1,84 (m, 1Н).
Стадия 3: R-4-метилбензил-4-((1Н-пиразоло[3,4-d]пиримидин-4-иламино)метил)-3,3-дифторпиперидин-1-карбоксилат

[0252] К перемешиваемому раствору R-N-((3,3-дифторпиперидин-4-ил)метил)-1H-пиразоло[3,4-d]пиримидин-4-амина дигидрохлорида (400 мг, 1,17 ммоль) и ТЭА (0,37 мл, 2,6 ммоль) в смеси растворителей MeCN (15 мл) и ДМФА (4 мл) добавляли 2,5-диоксопирролидин-1-ил-4-метилбензилкарбонат (307 мг, 1,17 ммоль). Полученную смесь перемешивали в течение 1 часа при комнатной температуре и затем разбавляли этилацетатом (50 мл). Органическую фазу промывали водой, солевым раствором, сушили над безводным Na2SO4 и концентрировали при пониженном давлении. Остаток очищали с помощью колоночной хроматографии на силикагеле (ДХМ/МеОН=35/1), получая указанное в заголовке соединение в виде порошка желтого цвета (250 мг, 51%). МС (ИЭР), расчет для C20H22F2N6O6: 416,2; экспериментальное значение: 417,5 [М+Н]. 1Н ЯМР (400 МГц, CD3OD) δ 8,27 (s, 1H), 8,09 (s, 1H), 7,26 (d, J=8,0 Гц, 2Н), 7,18 (d, J=8,0 Гц, 2Н), 5,11 (s, 2Н), 4,38-4,26 (ушир. s, 1Н), 4,18-4,12 (m, 1Н), 3,99-3,95 (m, 1Н), 3,67-3,61 (m, 1Н), 3,28-3,10 (m, 1Н), 3,03-2,89 (m, 1Н), 2,60-2,44 (m, 1Н), 2,35 (s, 3Н), 1,96-1,93 (m, 1Н), 1,60-1,49 (m, 1Н).
ПРИМЕР 1.13а. (+)-R-4-метилбензил-4-((1Н-пиразоло[3,4-d]пиримидин-4-иламино)метил)-3,3-дифторпиперидин-1-карбоксилата метансульфонат (Е1-6.2а)

[0253] К перемешиваемому раствору R-4-метилбензил-4-((1Н-пиразоло[3,4-d]пиримидин-4-иламино)метил)-3,3-дифторпиперидин-1-карбоксилата (230 мг, 0,55 ммоль) в ДХМ (3 мл) добавляли раствор метансульфоновой кислоты (53 мг, 0,55 ммоль) в метаноле (3 мл). Полученную смесь перемешивали при комнатной температуре в течение 30 мин. Растворитель выпаривали, и твердое вещество, полученное таким образом, растирали с эфиром (10 мл) и фильтровали, получая указанное в заголовке соединение в виде порошка желтого цвета (270 мг, 95%). [α]D=+13,5 (c=10 мг/мл, МеОН, 20°C). МС (ИЭР), расчет для C20H22F2N6O6: 416,2; экспериментальное значение: 417,5 [М+Н]. 1Н ЯМР (400 МГц, CD3OD) δ 8,55 (s, 1Н), 8,50 (s, 1Н), 7,26 (d, J=8,0 Гц, 2Н), 7,19 (d, J=8,0 Гц, 2Н), 5,11 (s, 2Н), 4,41-4,29 (m, 1Н), 4,23-4,15 (m, 1Н), 4,11-4,06 (m, 1Н), 3,84-3,78 (m, 1Н), 3,26-3,14 (m, 1Н), 3,07-2,91 (m, 1Н), 2,72 (s, 3Н), 2,63-2,50 (m, 1Н), 2,35 (s, 3Н), 2,00-1,92 (m, 1Н), 1,64-1,53 (m, 1Н).
ПРИМЕР 1.14 (+)-R-4-метилбензил-4-((7Н-пирроло[2,3-d]пиримидин-4-иламино)метил)-3,3-дифторпиперидин-1-карбоксилат (Е1-7.2)

Стадия 1: R-трет-бутил-3,3-дифтор-4-((7-тозил-7Н-пирроло[2,3-d]пиримидин-4-иламино)метил)пиперидин-1-карбоксилат

[0254] Смесь 4-хлор-7-тозил-7Н-пирроло[2,3-d]пиримидина (615 мг, 1,99 ммоль), R-трет-бутил-4-(аминометил)-3,3-дифторпиперидин-1-карбоксилата (600 мг, 2,39 ммоль) и DIPEA (0,66 мл, 3,99 ммоль) в n-BuOH (8 мл) нагревали до 130°C в атмосфере азота в течение ночи. Полученной смеси давали возможность остыть до комнатной температуры и проводили ее концентрирование под вакуумом. Полученный концентрат распределяли между этилацетатом и водой. Органический слой промывали водой, солевым раствором, сушили над Na2SO4 и концентрировали под вакуумом. Остаток очищали с помощью колоночной хроматографии на силикагеле (гексан/EtOAc=3/2), получая указанное в заголовке соединение в виде порошка грязно-белого цвета (850 мг, 82%). МС (ИЭР), расчет для C24H29F2N5O4S: 521,2; экспериментальное значение: 522,5 [М+Н]. 1Н ЯМР (400 МГц, CDCl3) δ 8,44 (s, 1H), 8,07 (d, J=8,0 Гц, 2Н), 7,47 (d, J=4,0 Гц, 1Н), 7,29 (d, J=8,0 Гц, 2H), 6,41 (d, J=4,0 Гц, 1H), 5,23-5,16 (m, 1H), 4,45-4,25 (m, 2H), 3,88-3,70 (m, 2H), 3,02-2,67 (m, 2H), 2,39 (s, 3Н), 2,35-2,19 (m, 1H), 1,85-1,77 (m, 1H), 1,62-1,50 (m, 1H), 1,46 (s, 9H).
Стадия 2: R-N-((3,3-дифторпиперидин-4-ил)метил)-7-тозил-7Н-пирроло[2,3-d]пиримидин-4-амина трифторацетат
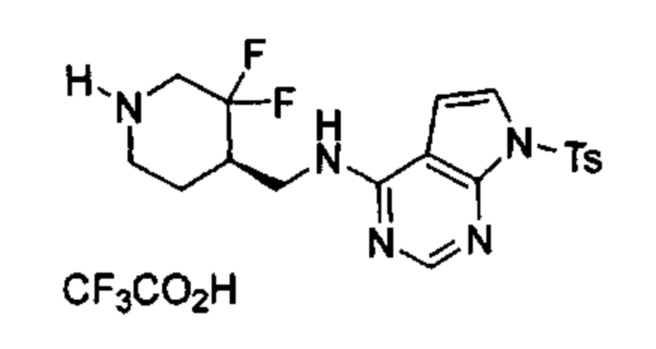
[0255] К раствору R-трет-бутил-3,3-дифтор-4-((7-тозил-7Н-пирроло[2,3-d]пиримидин-4-иламино)метил)пиперидин-1-карбоксилата (850 мг, 1,63 ммоль) в ДХМ (8 мл) добавляли ТФУ (4 мл). Полученный раствор перемешивали в течение 30 мин при комнатной температуре и затем концентрировали под вакуумом, получая указанное в заголовке соединение (2,19 г), которое непосредственно применяли на следующей стадии без дополнительной очистки. МС (ИЭР), расчет для C19H21F2N5O2S: 421,1; экспериментальное значение: 422,7 [М+Н].
Стадия 3: R-4-метилбензил-3,3-дифтор-4-((7-тозил-7Н-пирроло[2,3-d]пиримидин-4-иламино)метил)пиперидин-1-карбоксилат
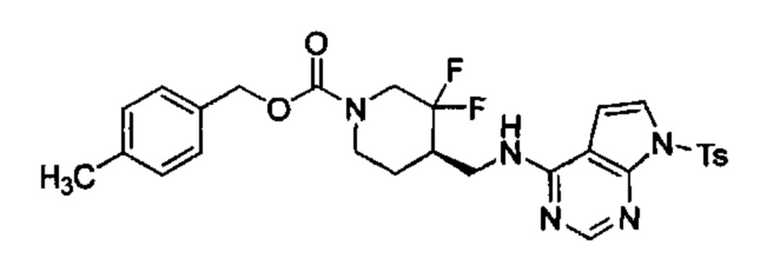
[0256] Неочищенную трифторацетатную соль R-N-((3,3-дифторпиперидин-4-ил)метил)-7-тозил-7Н-пирроло[2,3-d]пиримидин-4-амина (2,19 г, приблизительно 1,6 ммоль) растворяли в ацетонитриле (9 мл), затем добавляли триэтиламин (1,2 мл, 8,16 ммоль). Далее добавляли 2,5-диоксоциклопентил-4-метилбензилкарбонат (515 мг, 1,95 ммоль) и полученную смесь перемешивали при комнатной температуре в течение ночи. Смесь концентрировали и полученный концентрат растворяли в этилацетате. Органическую фазу промывали водой, солевым раствором, сушили над Na2SO4 и концентрировали под вакуумом. Остаток очищали с помощью колоночной хроматографии на силикагеле (гексан/EtOAc=3/2), получая указанное в заголовке соединение в виде белого порошка (847 мг, 80%). МС (ИЭР), расчет для C28H29F2N5O4S: 569,2; экспериментальное значение: 570,5 [М+Н]. 1Н ЯМР (400 МГц, CDCl3) δ 8,43 (s, 1H), 8,06 (d, J=8,0 Гц, 2Н), 7,47 (d, J=4,0 Гц, 1Н), 7,29 (d, J=8,0 Гц, 2Н), 7,24 (d, J=8,0 Гц, 2Н). 7,16 (d, J=8,0 Гц, 2Н), 6,40 (d, J=4,0 Гц, 1Н), 5,23-5,15 (m, 1Н), 5,13-5,05 (m, 2Н), 4,55-4,10 (m, 2Н), 3,87-3,71 (m, 1Н), 3,07-2,90 (m, 1Н), 2,87-2,74 (m, 1Н), 2,39 (s, 3Н), 2,35 (s, 3Н), 2,37-2,22 (m, 2Н), 1,87-1,77 (m, 1Н), 1,64-1,51 (m, 1Н).
Стадия 4: (+)-R-4-метилбензил-4-((7Н-пирроло[2,3-d]пиримидин-4-иламино)метил)-3,3-дифторпиперидин-1-карбоксилат
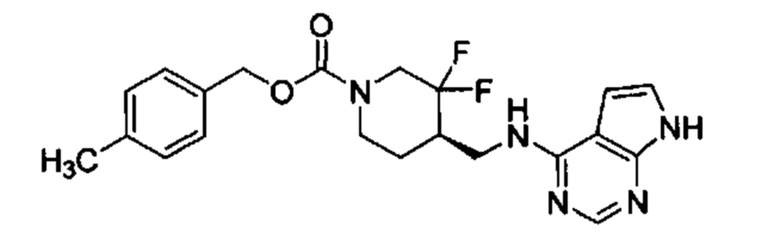
[0257] К раствору R-4-метилбензил-3,3-дифтор-4-((7-тозил-7Н-пирроло[2,3-d]пиримидин-4-иламино)метил)пиперидин-1-карбоксилата (777 мг, 1,36 ммоль) в ТГФ (8 мл) добавляли 50% водный NaOH (2 мл). Полученную смесь перемешивали в течение ночи при температуре окружающей среды и концентрировали под вакуумом для удаления растворителя, представлявшего собой ТГФ. Доводили pH оставшегося раствора до значения pH=9 с помощью HCl (6 н.) при охлаждении в ледяной бане. Водную фазу подвергали экстракции этилацетатом. Объединенные органические фазы промывали солевым раствором, сушили над Na2SO4 и концентрировали под вакуумом. Остаток очищали с помощью колоночной хроматографии на силикагеле (гексан/ацетон=1/1), получая указанное в заголовке соединение в виде порошка бледно-коричневого цвета (390 мг, 70%). [α]D=+22,5° (с=10 мг/мл, МеОН, 26°C). МС (ИЭР), расчет для C21H23F2N5O2: 415,2; экспериментальное значение: 416,5 [М+Н]. 1Н ЯМР (400 МГц, CDCl3) δ 10,08-9,98 (ушир. s, 1Н), 8,34 (s, 1Н), 7,25 (d, J=8,0 Гц, 2Н), 7,17 (d, J=8,0 Гц, 2Н), 7,07 (d. J=2,7 Гц, 1Н), 6,37 (d, J=2,7 Гц, 1Н), 5,31-5,25 (m, 1Н), 5,14-5,07 (s, 2Н), 4,56-4,10 (m, 2Н), 3,98-3,89 (m, 1Н), 3,88-3,77 (m, 1Н), 3,12-2,93 (m, 1Н), 2,91-2,77 (m, 1Н), 2,45-2,28 (m, 1Н), 2,35 (s, 3Н), 1,95-1,85 (m, 1H), 1,70-1,56 (m, 1H).
ПРИМЕР 1.15 R-4-этилбензил-4-(([1,2,4]триазоло[4,3-а]пиразин-8-иламино)метил)-3,3-дифторпиперидин-1-карбоксилат (Е1-1.5)
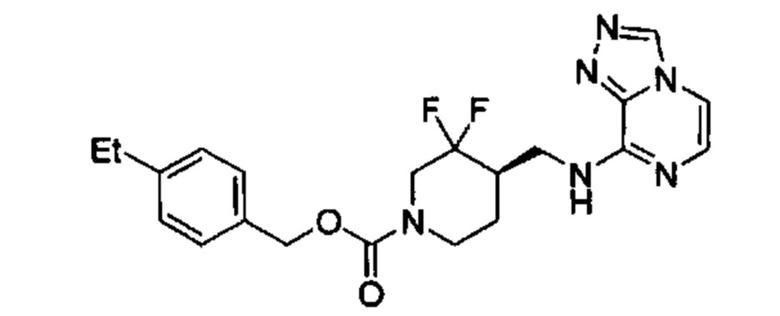
[0258] К раствору ранее описанного неочищенного N-((3,3-дифторпиперидин-4-ил)метил)-[1,2,4]триазоло[4,3-а]пиразин-8-аминтрифторацетата (373 мг, приблизительно 1,0 ммоль) в MeCN (5 мл) добавляли ТЭА (0,7 мл, 5,05 ммоль), затем добавляли 2,5-диоксопирролидин-1-ил-4-этилбензилкарбонат (335,8 мг, 1,21 ммоль). Полученную смесь перемешивали в течение 1 часа при комнатной температуре. Смесь разбавляли этилацетатом и органическую фазу промывали водой, солевым раствором, сушили над безводным Na2SO4 и концентрировали при пониженном давлении. Остаток очищали с помощью колоночной хроматографии на силикагеле (этилацетат/гексан=1/1), получая указанное в заголовке соединение в виде порошка грязно-белого цвета (302 мг). МС (ИЭР), расчет для C21H24F2N6O2: 430,2; экспериментальное значение: 431,4 [М+Н]. 1Н ЯМР (400 МГц, CD3OD) δ 9,08 (s, 1H), 7,69 (d, J=4,8 Гц, 1Н), 7,32 (d, J=4,8 Гц, 1Н), 7,26 (d, J=8,0 Гц, 2Н), 7,19 (d, J=8,0 Гц, 2Н), 5,10 (s, 2Н), 4,37-4,26 (m, 1Н), 4,19-4,11 (m, 1Н), 3,98 (dd, J=14,0 и 5,2 Гц, 1Н), 3,62 (dd, J=14,0 и 8,4 Гц, 1Н), 3,27-3,07 (m, 1Н), 3,07-2,88 (m, 1Н), 2,64 (q, J=7,6 Гц, 2Н), 2,59-2,48 (m, 1Н), 1,98-1,93 (m, 1Н), 1,61-1,51 (m, 1Н), 1,22(t, J=7,6 Гц, 3Н).
ПРИМЕР 1.15а. (+)-R-4-этилбензил-4-(([1,2,4]триазоло[4,3-а]пиразин-8-иламино)метил)-3,3-дифторпиперидин-1-карбоксилата метансульфонат (Е1-1.5а)
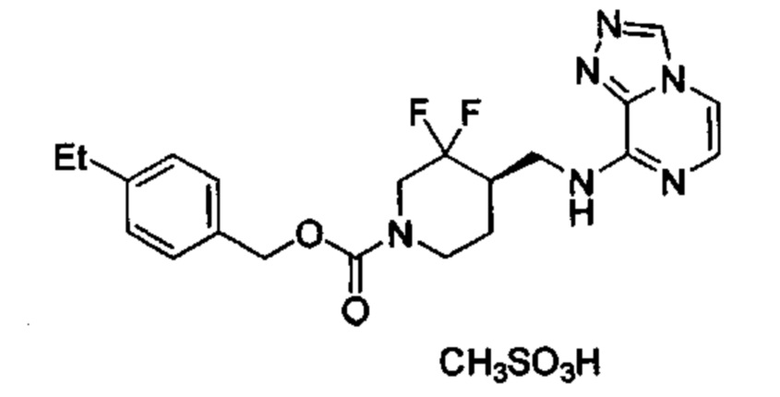
[0259] К перемешиваемому раствору R-4-этилбензил-4-(([1,2,4]триазоло[4,3-а]пиразин-8-иламино)-метил)-3,3-дифторпиперидин-1-карбоксилата (302 мг, 0,70 ммоль) в МеОН/ДХМ (4,0 мл, об./об.=1:1) добавляли раствор CH3SO3H (68 мг, 0,70 ммоль) в метаноле (1 мл) при комнатной температуре. После перемешивания в течение 30 мин смесь концентрировали, получая указанный продукт в виде порошка грязно-белого цвета (335 мг, 90,6%). [α]D=+2,4° (c=10 мг/мл, МеОН, 23°C). МС (ИЭР), расчет для C21H24F2N6O2: 430,2; экспериментальное значение: 431,5 [М+Н]. 1Н ЯМР (400 МГц, CD3OD) δ 9,32 (s, 1Н), 7,95 (d, J=5,6 Гц, 1Н), 7,27 (d, J=8,0 Гц, 2Н), 7,24 (d, J=5,6 Гц, 1Н), 7,19 (d, J=8,0 Гц, 2Н), 5,10 (s, 2Н), 4,45-4,36 (m, 1Н), 4,24-4,20 (m, 1Н), 4,04-3,99 (m, 1Н), 3,71-3,66 (m, 1Н), 3,25-2,93 (m, 2Н), 2,70 (s, 3Н), 2,68-2,60 (m, 3Н), 2,08-1,98 (m, 1Н), 1,65-1,55 (m, 1Н), 1,22 (t, J=7,6 Гц, 3Н).
ПРИМЕР 1.16 Монокристальная рентгеновская дифрактометрия (SCXRD) соединения (R)-XVIa
[0260] Результаты были получены на дифрактометре Oxford Diffraction Supernova Dual Source, CuatZero, с ПЗС-детектором Atlas, оснащенном охлаждающим устройством Oxford Cryosystems Cobra. Результаты были получены с использованием излучения CuKα. Структуры обычно расшифровывали с использованием программ SHELXS или SHELXD и уточняли с помощью программы SHELXL, входящей в состав пакета AXSSHELXTL (V6.10). Если не указано иное, атомы водорода, соединенные с углеродом, были помещены в геометрически рассчитанные положения и их изотропные параметры смещения были уточнены в модели наездника (riding). Атомы водорода, связанные с гетероатомом, были локализованы в разностных синтезах Фурье и их изотропные параметры смещения были уточнены без наложения на них каких-либо ограничений.
[0261] Образец (R)-XVIa (приблизительно 10 мг) перекристаллизовывали из 2-метил-1-пропанола (400 мкл, 40 об.) путем медленного испарения при комнатной температуре. Был выделен кристалл достаточного размера и качества для анализа методом монокристальной рентгеновской дифрактометрии с приблизительными размерами 0,20×0,15×0,10 мм.
[0262] Структуру определяли при 100 K в орторомбической системе, пространственная группа P212121 с конечным R1 [l>σ2(l)] = 4,52%. Сводная информация обо всех структурных характеристиках представлена в таблицах A-D. Соединение было идентифицировано как несольватированная форма (R)-XVIa.



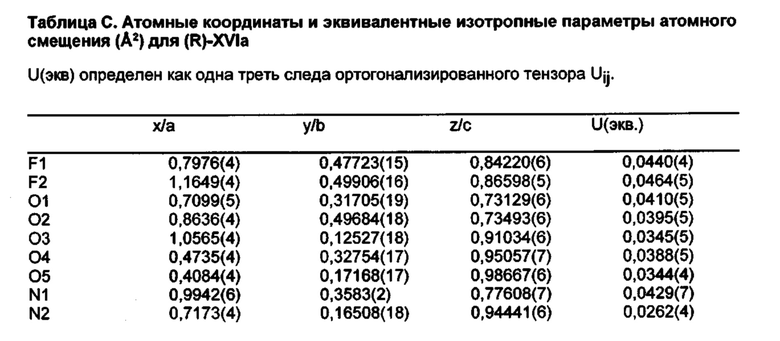

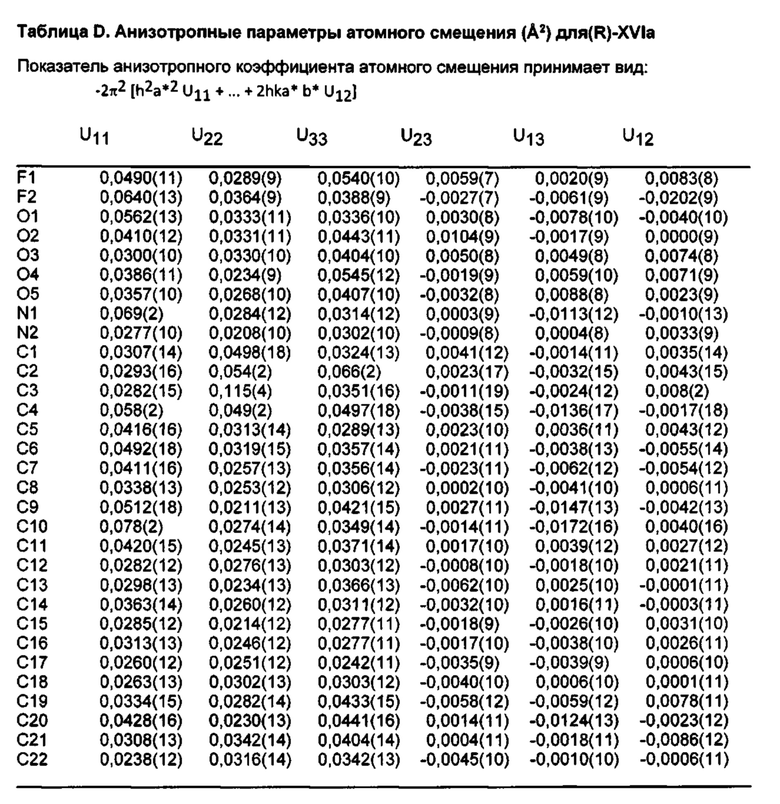
[0263] Асимметрическая единица содержит одну полностью упорядоченную молекулу (R)-XVIa. Эллипсоиды анизотропного атомного смещения атомов, отличных от атомов водорода, показаны с уровнем вероятности 50%. Атомы водорода отображены со сколь угодно малым радиусом (ФИГ. 11). Для структуры, представленной с атомами С8 и С15 в R-конфигурации, параметр Флэка (Flack)=-0,04(6) (Parsons and Flack, Acta Cryst. 2004, A60, s61). Для обращенной структуры с атомами С8 и С15 в S-конфигурации параметр Флэка=1,04(6). Определение абсолютной структуры с использованием байесовской статистики по разностям Бийво (Bijvoet) (Hooft et al., J. Appl. Cryst., 2008, 41, 96-103) показывает, что вероятность того, что абсолютная структура, соответствующая представленной, верна, составляет 1,000, в то время как вероятности того, что абсолютная структура представляет собой рацемический изомеромер или неверна, составляют 0,000. Параметр Флэка и его неопределенность, рассчитанные с помощью этой программы, составляют -0,01 (5). Расчет основывался на 1853 парах Бийво с охватом 100%. На основании параметра Флэка, байесовского статистического анализа и априорного знания можно сделать вывод, что хиральный центр при С15 имеет абсолютную конфигурацию R.
ПРИМЕР 2. Испытания
ПРИМЕР 2.1. Активность антагонистов NR2B
[0264] Линии клеток HEK293, стабильно экспрессирующие клонированные человеческие NR1/NR2B и NR1/NR2A, соответственно, были сформированы согласно стандартным способам, описанным ранее (Hansen et al., Comb. Chem High Throughput Screen. 11:304, 2008). В этих клетках активация NMDA-рецептора подтипа NR2A или NR2B глутаматом в качестве агониста и глицином в качестве коагониста приводит к притоку кальция, который можно регистрировать с помощью флюоресцентного индикатора Fluo-4. Для оценки влияния соединения на рецепторы NR2A и NR2B посредством измерения изменений флюоресценции было проведено исследование на клетках (Hansen et al., Comb. Chem High Throughput Screen. 11:304, 2008).
[0265] Клетки HEK293, стабильно экспрессирующие рецепторы NR2A или NR2B, культивировали при 37°C в увлажненном CO2-инкубаторе в DMEM с добавлением 10% эмбриональной телячьей сыворотки (FBS) (Hyclone), 10 мкМ MK801 (Sigma-Aldrich) и 50 мкМ АР-5 (Tocris). Для экспериментов клетки высевали в покрытые поли-D-лизином 96-луночные черные планшеты с прозрачным дном (Corning) при плотности ~ 50000 клеток в лунке. После культивирования в течение ночи из лунок удаляли ростовую среду и инкубировали клетки при 37°C в течение 60 минут в солевом растворе Хенкса, содержащем 4 мкМ fluo-4-АМ (Invitrogen) и 0,1% бычьего сывороточного альбумина (БСА). После окрашивания клетки три раза промывали солевым раствором Хенкса и инкубировали в течение 10 минут при комнатной температуре с различными концентрациями исследуемых соединений, приготовленных в солевом растворе Хенкса с 0,1% БСА. Планшеты с клетками помещали на флюоресцентный спектрофотометр FDSS μCell (Hamamatsu). После 20 сек считывания фоновой флюоресценции к клеткам добавляли агонист глутамат в конечной концентрации 100 мкМ и коагонист глицин в конечной концентрации 50 мкМ для активации рецептора и регистрировали и количественно оценивали полученные изменения флюоресценции. На основе изменений интенсивности флюоресценции анализировали фармакологический эффект исследуемых соединений и значения IC50, полученные путем аппроксимации концентрационно-зависимого ответа к уравнению стандартной логистической модели нелинейным методом наименьших квадратов с использованием программы Prism (Graphpad, Inc.):
Амплитуда = Макс. Амплитуда/(1+(IC50/[антагонист])n).
Результаты представлены в таблице 2.1.
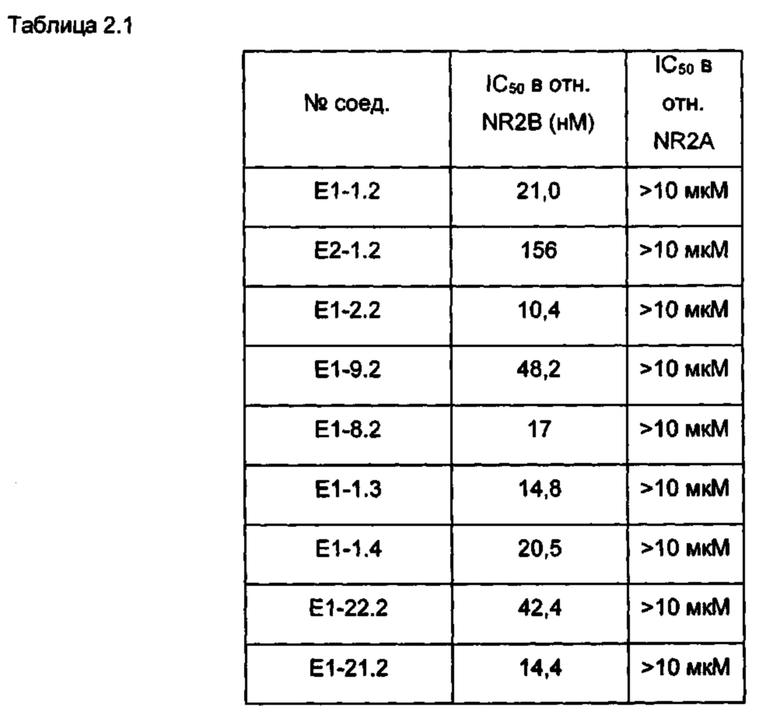
[0268] [3Н] (Е)-N1-(2-метоксибензил)-циннамидин применяли в 2 различных концентрациях, 0,5 и 30 нМ, с 30 мкг мембранных белков. Неспецифическое связывание (НС) оценивали в присутствии избытка (10 мкМ) (Е)-N1-(2-метоксибензил)-циннамидина. Был идентифицирован высокоаффинный сайт, для которого было определено значение Ki для(Е)-N1-(2-метоксибензил)-циннамидина, составляющее 0,18 нМ. Таким образом, с применением [3Н] (Е)-N1-(2-метоксибензил)-циннамидина в качестве радиолиганда рецептора NR2B (Kiss et al., Neurochemistry International. 46, p 453-464, 2005) было определено значение Ki для клонированных рецепторов NR2B, составляющее 1,0 нМ (Clairborne, С.F. Bioorganic and Medicinal Chemistry Letters, 13, 697-700, 2003), в то время как с применением [3Н]-ифенпродила в качестве радиолиганда рецептора NR2B значение Ki для клонированных рецепторов NR2B составило 0,7 нМ (Curtis N. R. et al., Bioorganic and Medicinal Chemistry Letters, 13, 693-696, 2003).
[0269] Исследуемые соединения в концентрации 10 мМ солюбилизировали в ДМСО. Затем выполняли разведения с постоянной концентрацией растворителя (1% ДМСО) в образце.
[0270] После инкубирования в течение 4,5 ч при комнатной температуре образцы фильтровали через фильтры GF/B, предварительно обработанные 0,3% (об./об.) ПЭИ, с применением системы Brandel для [3Н] MK-801 и системы Packard для [3Н] (E)-N1-(2-метоксибензил)-циннамидина. Эксперимент проводили в двух параллельных испытаниях (n=2).
[0271] Соединение Е1-1.2 проявляло только частичный эффект (40%) в отношении связывания [3Н] MK-801, соответствующий селективному связыванию с подтипом рецептора NR2B. Соединение Е1-1.2 приводило к полному перемещению [3Н] (E)-N1-(2-метоксибензил)-циннамидина на высокоаффинный сайт рецептора NR2B (96%; Ki=5,23 нМ).
[0272] Соединение Е2-1.2 проявляло только частичный эффект (36%) в отношении связывания [3Н] MK-801, соответствующий селективному связыванию с подтипом рецептора NR2B. Соединение Е2-1.2 приводило к полному перемещению [3Н] (E)-N1-(2-метоксибензил)-циннамидина на высокоаффинный сайт рецептора NR2B (98%; Ki=74,3 нМ).
[0273] Соединение Е1-1.3 проявляло только частичный эффект (41%) в отношении связывания [3Н] MK-801, соответствующий селективному связыванию с подтипом рецептора NR2B. Соединение Е1-1.3 приводило к полному перемещению [3Н] (E)-N1-(2-метоксибензил)-циннамидина на высокоаффинный сайт рецептора NR2B (97%; Ki=2,34 нМ).
[0274] Соединение Е1-1.4 проявляло только частичный эффект (32%) в отношении связывания [3Н] MK-801, соответствующий селективному связыванию с подтипом рецептора NR2B. Соединение Е1-1.4 приводило к полному перемещению [3Н] (E)-N1-(2-метоксибензил)-циннамидина на высокоаффинный сайт рецептора NR2B (98%; Ki=18,2 нМ).
[0275] Соединение Е1-1.5 проявляло только частичный эффект (48%) в отношении связывания [3Н] MK-801, соответствующий селективному связыванию с подтипом рецептора NR2B. Соединение Е1-1.5 приводило к полному перемещению [3Н] (E)-N1-(2-метоксибензил)-циннамидина на высокоаффинный сайт рецептора NR2B (97%; Ki=0,854 нМ).
[0276] Соединение Е1-8.2 проявляло только частичный эффект (33%) в отношении связывания [3Н] MK-801, соответствующий селективному связыванию с подтипом рецептора NR2B. Соединение Е1-8.2 приводило к полному перемещению [3Н] (E)-N1-(2-метоксибензил)-циннамидина на высокоаффинный сайт рецептора NR2B (95%; Ki=1,71 нМ).
[0277] Соединение Е 1-9.2 проявляло только частичный эффект (34%) в отношении связывания [3Н] MK-801, соответствующий селективному связыванию с подтипом рецептора NR2B. Соединение Е1-9.2 приводило к полному перемещению [3Н] (E)-N1-(2-метоксибензил)-циннамидина на высокоаффинный сайт рецептора NR2B (97%; Ki=11,3 нМ).
[0278] Соединение Е1-21.2 проявляло только частичный эффект (49%) в отношении связывания [3Н] MK-801, соответствующий селективному связыванию с подтипом рецептора NR2B. Соединение Е1-21.2 приводило к полному перемещению [3Н] (E)-N1-(2-метоксибензил)-циннамидина на высокоаффинный сайт рецептора NR2B (98%; Ki=0,716 нМ).
[0279] Соединение Е1-21.26 проявляло только частичный эффект (41%) в отношении связывания [3Н] MK-801, соответствующий селективному связыванию с подтипом рецептора NR2B. Соединение Е1-21.26 приводило к полному перемещению [3Н] (E)-N1-(2-метоксибензил)-циннамидина на высокоаффинный сайт рецептора NR2B (99%; Ki=1,02 нМ).
ПРИМЕР 2.2. Ингибирование канала hERG
[0280] Анализ проводили с использованием канала hERG, стабильно экспрессируемого в клетках HEK293. Клетки культивировали при 37°C в увлажненном СО2-инкубаторе в ростовой среде, состоящей из DMEM, 10% эмбриональной телячьей сыворотки и антибиотиков. Перед исследованием клетки высевали на покровное стекло диаметром 12 мм, покрытое PDL, и культивировали в чашке Петри диаметром 35 мм. После 16-40 ч культивирования покровное стекло переносили в камеру перфузионной системы OctaFlow (ALA Instrument) и при постоянном потоке внеклеточного раствора (140 мМ NaCl, 4 мМ KCl, 1 мМ MgCl2, 2 мМ CaCl2, 10 мМ HEPES, 10 мМ D-глюкозы, pH 7,35, осмолярность 290). Фиксацию потенциала цельной клетки проводили с помощью стеклянной микропипетки, заполненной внутриклеточным раствором (120 мМ KCl, 1,75 мМ MgCl2, 5,4 мМ CaCl2, 10 мМ HEPES, 10 мМ ЭГТА и 4 мМ АТФ-K2, pH 7,2, осмолярность 310). Во время испытания поддерживали гигаомный контакт. Контроль напряжения и измерение тока проводили с использованием усилителя Axon amplifier 700 В, Digidata 1440А и программного обеспечения CLAMPEX10 (Molecular Devices). Токи hERG в цельной клетке регистрировали в соответствии с протоколом Petroski: клетку выдерживали при -80 мВ, создавали скачок напряжения от -80 до 30 мВ и оставляли на 2 сек с предварительным импульсом -40 мВ в течение 20 мс. После деполяризации напряжение понижали до -40 мВ и оставляли на 2 сек, а затем возвращали напряжение -80 мВ. Исследуемое соединение вносили с помощью наконечника кварцевых капиллярных трубок (с внутренним диаметром 200 мкм) и поддерживали скорость потока на уровне 2-3 мл/мин с помощью перфузионной системы OctaFlow. Клетки обрабатывали различными концентрациями соединения в течение 5 мин, а ток hERG измеряли три раза до, во время и после обработки соединением. Данные анализировали с использованием программного обеспечения Clampfit 10 (Molecular Devices) для получения значений IC50. Результаты представлены в таблице 2.2.

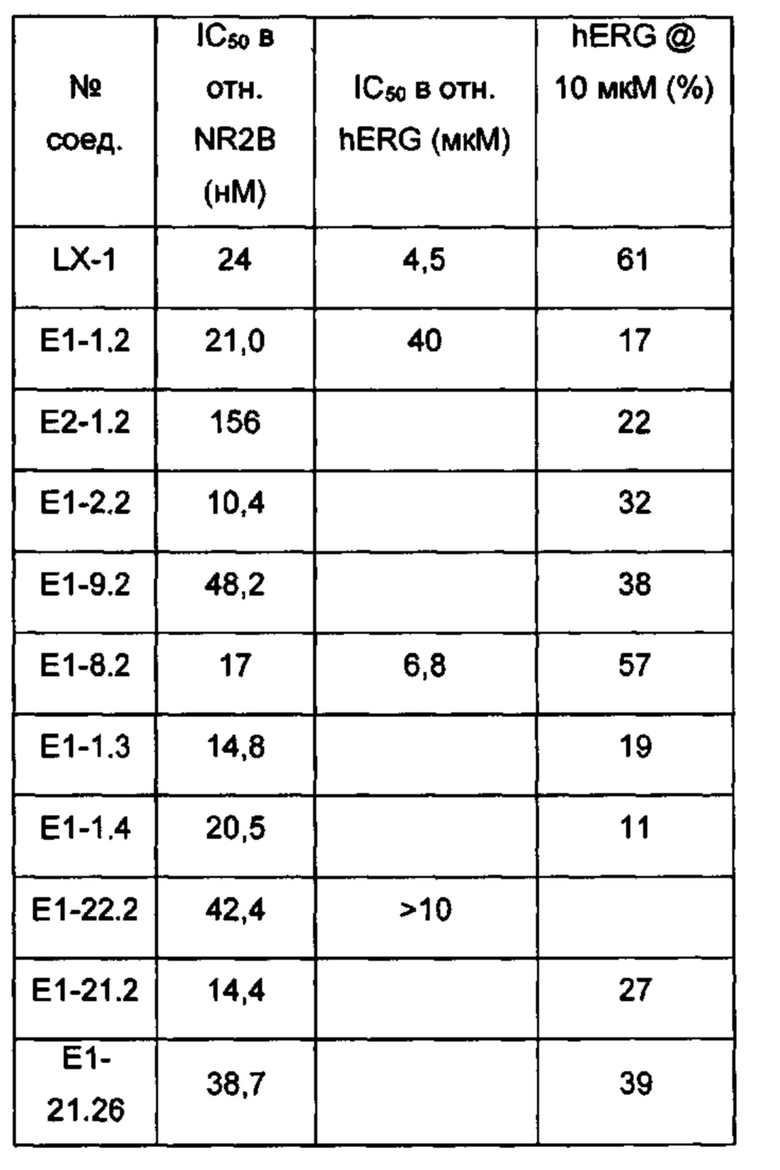
ПРИМЕР 2.3. Ингибирование фермента CYP Р450
[0281] Ингибирующую активность исследуемых соединений в отношении 5 основных изоформ CYP Р450 оценивали с использованием объединенных микросом печени человека (HLM, приобретенные у BD Gentest) и селективных субстратов для этих изоформ. Эти изоформы CYP и соответствующие им маркерные субстраты представляют собой: CYP1A2 (фенацетин, 30 мкМ), CYP2C9 (толутамид, 100 мкМ), CYP2C19 (S-мефенитоин, 40 мкМ), CYP2D6 (декстрометорфан, 5 мкМ) и CYP3A4 (мидазолам, 1 мкМ). Все маркерные субстраты использовали в концентрациях, близких к их Kms или ниже. Для эксперимента реакционную смесь исследуемого соединения в концентрации 10 мкМ или в серийном разведении, маркерного субстрата CYP, описанного выше, и 0,2 мг/мл объединенных HLM в фосфатном буферном растворе, pH 7,4, в конечном объеме, составляющем 200 мкл, предварительно инкубировали при 37°C в течение 10 минут в трех параллельных испытаниях. Реакцию инициировали добавлением НАДФН в конечной концентрации 1 мМ. Реакцию останавливали через 10 минут (CYP1A2, CYP2D6 и CYP3A4) или 30 минут (CYP2C9 и CYP2C19) посредством добавления 100 мкл охлажденного на льду ацетонитрила с внутренним стандартом (ВС). Затем образцы центрифугировали при 13000 rpm и супернатанты вводили в LC-MS/MS (Agilent Technologies) для количественного определения концентрации специфических метаболитов маркерных субстратов, образованных отдельными изоформами CYP450. Коэффициент ингибирования рассчитывают как:
(Mt-М0)/Мвода×100%,
где Mt и M0 представляют собой концентрации специфических метаболитов маркерных субстратов, образованных отдельными изоформами CYP450, в начале и в конце реакции в присутствии исследуемого соединения; в то время как Мвода представляет собой концентрацию специфического метаболита в конце реакции в отсутствие исследуемого соединения. Эксперименты по получению данных ответа, зависящего от концентрации исследуемого соединения, проводили в трех параллельных испытаниях. Средние значения IC50 CYP2D6 получали с помощью приведения данных дозозависимого ответа нелинейным методом наименьших квадратов к стандартному логистическому уравнению (Prism, GraphPad Software, Inc) для получения значений IC50 CYP2D6, представленных в таблице 2.3.
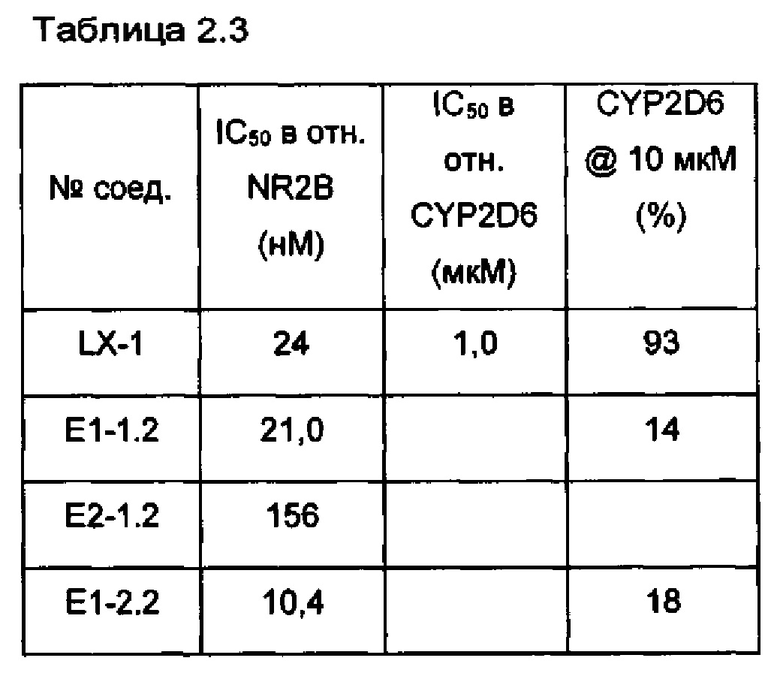

ПРИМЕР 2.4. Тест вынужденного плавания (ТВП)
[0282] Тест вынужденного плавания (ТВП), также известный как тест поведения отчаяния, использовали для оценки антидепрессивной активности (Porsolt et al., 1977 Arch. Int. Pharmacodyn. 229: 327-336, Porsolt et al., 1977, Eur. J. Pharmacol. 47:379-391). Мыши или крысы, вынужденные плавать в ситуации, из которой они не могут выйти, быстро становятся неподвижными. Лекарственные средства с антидепрессивной активностью, такие как имипрамин, уменьшают количество времени, проведенного в неподвижном состоянии. Поэтому продолжительность неподвижного состояния во время теста после введения лекарственного средства является ценным индикатором антидепрессивной активности (Lucki et al 2001 Psychopharmacology 155:315-322; Porsolt et al., 1977, Nature 266:730-732).
[0283] Исследуемые соединения E1-1.2 и E1-21.26 вводили в форме мезилатных солей (дозы в мг/кг рассчитаны на основании молекулярной массы свободного основания). Исследуемое соединение Е1-8.2 вводили в форме свободного основания.
[0284] Исследование на антидепрессивную активность проводили на крысах или мышах в соответствии с общими методиками, описанными ниже.
[0285] Мышей оценивали в одном сеансе теста плавания в течение 6 минут. Мышей помещали в прозрачный пластиковый цилиндр высотой 24 см с диаметром 13 см, содержащий 10 см воды с контролем температуры окружающей среды в целом от 22±2°C. Мышей помещали в воду на 6 минут и измеряли продолжительность неподвижного состояния в течение последних 4 минут.
[0286] Для теста на крысах использовали самцов крыс Wistar массой 197-251 г. Крыс оценивали в соответствии с методикой, состоящей из двух сеансов: первого сеанса плавания продолжительностью 15 минут в первый день эксперимента (сеанс 1) и последующего 5-минутного теста плавания через 24 часа (сеанс 2). Крыс вынуждали независимо плавать внутри вертикальных прозрачных цилиндров из плексигласа размером 40 см × 18 см, содержащих 15 см воды с поддерживаемой температурой 25°C (сеанс 1). После 15 мин пребывания в воде крыс извлекали и позволяли высохнуть в течение 15 мин в обогреваемом укрытии (32°C) перед возвращением в клетку. Крыс помещали в воду через 24 ч на 5 минут (сеанс 2) и измеряли продолжительность неподвижного состояния.
[0287] Животные находились под наблюдением «ослепленного» наблюдателя. Наблюдатель считал животное неподвижным, когда оно прекращало все виды деятельности (борьбу, плавание, прыжки и т.д.) и пассивно плавало на поверхности воды. Количество времени, проведенное каждым животным в неподвижном состоянии (и латентный период до первого эпизода неподвижности), регистрировали и использовали для статистического анализа эффекта соединения. Групповые различия оценивали с помощью t-критерия Стьюдента (эталонное вещество) или однофакторного ANOVA с последующим вычислением апостериорного критерия Даннетта (Dunnett) (исследуемые вещества).
[0288] В данном эксперименте (как в эксперименте на мышах, так и на крысах) вводили исследуемое соединение, контрольный раствор носителя и эталонное соединение имипрамин в качестве положительного контроля. Исследуемые соединения вводили в количестве одной или более доз посредством введения через желудочный зонд (п/о) или путем внутрибрюшинной инъекции (в/б) после растворения в водном растворе, содержащем 0,5% диметилсульфоксида, 4% гидроксипропил-b-циклодекстрина, в качестве носителя. Дозы исследуемого соединения составляли от 1 до 30 миллиграммов на килограмм (на сопроводительных чертежах представлены как мг на кг или мг/кг).
[0289] Контрольное соединение имипрамин растворяли в физиологическом солевом растворе. Имипрамин вводили, как обозначено в конкретных примерах.
[0290] В экспериментах как при пероральном, так и при внутрибрюшинном введении раствор дозы исследуемого соединения и контрольный раствор носителя вводили за 20 минут до помещения животных в цилиндр с водой. Имипрамин вводили, как обозначено ниже в конкретных примерах.
[0291] Для исследования использовали самцов мышей (линии NLMN) массой 25-35 г. Всех животных содержали в среде с контролируемой температурой (22-24°C) и влажностью (50-60%) со свободным доступом к пище и воде с 12-часовым циклом чередования света и темноты. Исследуемые соединения растворяли в водном растворе, содержащем 0,5% диметилсульфоксида, 4% гидроксипропил-b-циклодекстрина, для получения раствора, подходящего для введения. Лекарственные средства вводили путем внутрибрюшинной инъекции в дозе объемом 10 мл/кг. Испытание начинали через 20-60 минут после введения. Испытание на антидепрессивную активность проводили согласно описанию Darci et al. (Darci et al., 2004, Eur. J. Pharmacol. 499:135-146). Мышей помещали в белый пластмассовый цилиндр высотой 20 см с диаметром 21 см, содержащий 10 см воды с температурой 25±2°C. Мышей снимали на видео в течение 6 минут, а последние 4 минуты видеозаписи были проанализированы «ослепленным» наблюдателем в автономном режиме. Наблюдатель считал животное неподвижным, когда оно прекращало все виды деятельности (борьбу, плавание, прыжки и т.д.) и пассивно плавало на поверхности воды. Количество времени, проведенное каждым животным в неподвижном состоянии, регистрировали и использовали для статистического анализа эффекта соединения. Групповые различия оценивали с помощью t-критерия Стьюдента или однофакторного ANOVA с последующим вычислением апостериорного критерия Даннетта (Dunnett).
ПРИМЕР 2.4.1. Введение соединения Е1-1.2 мышам
[0292] Результаты представлены на ФИГ. 1А. В столбцах представлено среднее значение ± СОС продолжительности неподвижного состояния для каждой группы введения (n=10, ***: отличается от группы, получавшей носитель, р<0,001, однофакторный ANOVA, апостериорный критерий Даннетта). Дозы приведены в миллиграммах на килограмм (мг/кг). Доза имипрамина составляла 32 мг/кг.
ПРИМЕР 2.4.2. Соединение Е1-8.2, введенное мышам путем внутрибрюшинной инъекции
[0293] Результаты представлены на ФИГ. 1В. В столбцах представлено среднее значение ± СОС продолжительности неподвижного состояния для каждой группы введения (***: отличается от группы, получавшей носитель, p<0,001, однофакторный ANOVA, апостериорный критерий Даннетта). В настоящем примере соединение, применяемое в качестве положительного контроля, имипрамин (32 мг/кг в/б), введенное однократно за 30 минут до испытания, демонстрировало ожидаемую антидепрессивную активность.
ПРИМЕР 2.4.3. Соединение Е1-21.26, введенное мышам путем внутрибрюшинной инъекции
[0294] Результаты представлены на ФИГ. 1С. В столбцах представлено среднее значение ± СОС продолжительности неподвижного состояния для каждой группы введения (***: отличается от группы, получавшей носитель, р<0,001, однофакторный ANOVA, апостериорный критерий Даннетта). В настоящем примере соединение, применяемое в качестве положительного контроля, имипрамин (32 мг/кг в/б), введенное однократно за 30 минут до испытания, демонстрировало ожидаемую антидепрессивную активность.
ПРИМЕР 2.4.4. Соединение Е1-1.2, введенное мышам перорально
[0295] Результаты представлены на ФИГ. 1D. В столбцах представлено среднее значение ± СОС продолжительности неподвижного состояния для каждой группы введения (***: отличается от группы, получавшей носитель, р<0,001, однофакторный ANOVA, апостериорный критерий Даннетта). В настоящем примере соединение, применяемое в качестве положительного контроля, имипрамин (64 мг/кг п/о), введенное однократно за 60 минут до испытания, демонстрировало ожидаемую антидепрессивную активность.
ПРИМЕР 2.4.5. Соединение Е1-1.2, введенное крысам путем внутрибрюшинной инъекции
[0296] Результаты представлены на ФИГ. 1Е. В столбцах представлено среднее значение ± СОС продолжительности неподвижного состояния для каждой группы введения (***: отличается от группы, получавшей носитель, р<0,001, однофакторный ANOVA, апостериорный критерий Даннетта). В настоящем примере соединение, применяемое в качестве положительного контроля, имипрамин (32 мг/кг в/б), введенное 3 раза: за 24 ч, 4 ч и 30 мин до испытания (сеанс 2), демонстрировало ожидаемую антидепрессивную активность.
ПРИМЕР 2.4.6. Соединение Е1-21.26, введенное крысам перорально
[0297] Результаты представлены на ФИГ. 1F. В столбцах представлено среднее значение ± СОС продолжительности неподвижного состояния для каждой группы введения (***: отличается от группы, получавшей носитель, р<0,001, однофакторный ANOVA, апостериорный критерий Даннетта). В настоящем примере соединение, применяемое в качестве положительного контроля, имипрамин (64 мг/кг п/о) введенное 3 раза: за 24 ч, 4 ч и 60 мин до испытания (сеанс 2), демонстрировало ожидаемую антидепрессивную активность.
ПРИМЕР 2.4.7. Систематическое введение мышам в тесте вынужденного плавания
[0298] Мышей индивидуально помещали в цилиндр (высота = 24 см; диаметр = 13 см), содержащий 10 см воды (22°C), который они не могли покинуть. Мышей помещали в воду на 6 минут и измеряли продолжительность неподвижного состояния в течение последних 4 минут. Латентный период до первого эпизода неподвижности также регистрировали с момента начала теста.
[0299] Соединение оценивали в 2 дозах (3 и 10 мг/кг) при однократном пероральном введении за 20 минут до испытания на 7 день или ежедневном введении в течение 7 дней с последним введением за 20 минут до испытания на 7 день и сравнивали с контрольной группой, получавшей носитель. Носитель вводили ежедневно, когда введение лекарственного средства не требовалось. Имипрамин (128 мг/кг п/о), введенный однократно за 60 минут до испытания на 7 день, использовали в качестве эталонного вещества.
[0300] Результаты представлены на ФИГ. 1G. В столбцах представлено среднее значение ± СОС продолжительности неподвижного состояния для каждой группы введения (**/***: отличается от группы, получавшей носитель, р<0,01/р<0,001, однофакторный ANOVA, апостериорный критерий Даннетта). В настоящем примере соединение, применяемое в качестве положительного контроля, имипрамин (128 мг/кг п/о), введенное однократно за 60 минут до испытания на 7 день, демонстрировало ожидаемую антидепрессивную активность.
[0301] Полученные результаты показывают, что предложенные соединения проявляют антидепрессивную активность при испытании в стандартных моделях депрессии человека. Эти данные показывают, что исследуемые соединения проявляют антидепрессивную активность при систематическом введении. Кроме того, полученные результаты показывают, что предложенные соединения проявляют антидепрессивную активность при однократном и систематическом введении.
ПРИМЕР 2.5. Пороговый электроконвульсивный тест (ПЭТ)
[0302] Пороговый электроконвульсивный тест, который позволяет обнаружить проконвульсивную или противосудорожную активность, обычно проводили, как описано Swinyard et al. (J. Pharmacol. Exp. Ther., 106, 319-330, 1952). Исследуемые соединения E1-1.2 и E1-21.26 вводили в форме мезилатных солей (дозы в мг/кг рассчитаны на основании молекулярной массы свободного основания). Исследуемое соединение Е1-8.2 вводили в форме свободного основания.
[0303] Крыс подвергали воздействию электроконвульсивного шока (ЭКШ) (прямоугольный ток: ширина импульса 0,6 мс, продолжительность 1,5 с, 200 Гц) с помощью ушных электродов, соединенных с генератором импульсов постоянного тока (Ugo Basile: тип 7801). Исследуемые соединения вводили через желудочный зонд (п/о) за 1 час до испытания в дозе объемом 5 мл/кг.
[0304] Экспериментальные группы, состоящие из 20 крыс, подвергали воздействию ЭКШ следующим образом: первое животное подвергали воздействию ЭКШ 30 мА. Если у этого животного не наблюдалось судорог (тонических судорог) в течение максимум 5 секунд, животное №2 подвергали воздействию 35 мА и т.д. (с повышением на 5 мА) до тех пор, пока не наблюдали первую тоническую судорогу. Как только наблюдали первую тоническую судорогу, интенсивность ЭКШ понижали на 2 мА для следующего животного и затем понижали или повышали на 2 мА от животного к животному в зависимости от того, наблюдались ли у предыдущего животного судороги или нет. Если у первого животного наблюдались судороги (тонические судороги) в течение 5 секунд, животное №2 подвергали воздействию 25 мА и т.д. (с понижением на 5 мА) до тех пор, пока не наблюдали отсутствия тонических судорог. На данном этапе интенсивность ЭКШ повышали на 2 мА для следующего животного и затем понижали или повышали на 2 мА от животного к животному в зависимости от того, наблюдались ли у предыдущего животного судороги или нет. Минимальная применяемая сила тока составляет 5 мА, а максимальная - 95 мА. Первые 5 животных служат для определения порогового тока и не были включены в анализ. Результаты представлены как средняя сила тока, примененная к последним 15 животным из группы. Тест проводили в форме слепого испытания. Положительное процентное изменение указывает на противосудорожный эффект. Отрицательное процентное изменение указывает на проконвульсивный эффект. Исследуемое соединение оценивали в 4 дозах, как правило, с применением водного раствора, содержащего 0,5% диметилсульфоксида, 4% гидроксипропил-b-циклодекстрина, в качестве носителя, и вводили п/о за 60 минут до ЭКШ и сравнивали с контрольной группой, получавшей носитель. Диазепам (16 мг/кг п/о), введенный в тех же экспериментальных условиях, использовали в качестве эталонного вещества. В эксперимент было включено 6 групп. Результаты, полученные с применением исследуемых соединений, анализировали посредством сравнения экспериментальных групп с контрольной группой с помощью однофакторного ANOVA с последующим вычислением t-критерия Даннетта.
[0305] В примере 2.5.1 соединение, используемое в качестве положительного контроля для противосудорожной активности, диазепам (16 мг/кг п/о), демонстрировало ожидаемую противосудорожную активность. Соединение, используемое в качестве положительного контроля для проконвульсивной активности, теофиллин (128 мг/кг п/о), демонстрировало ожидаемую проконвульсивную активность. В примере 2.5.2 в исследование включали положительный контроль только для противосудорожной активности.
ПРИМЕР 2.5.1. Соединение Е1-1.2
[0306] Результаты представлены на ФИГ. 2. В столбцах представлено среднее значение ± СОС электроконвульсивного порога для каждой группы введения (n=15, ***: отличается от группы, получавшей носитель, р<0,001, однофакторный ANOVA, апостериорный критерий Даннетта). Дозы приведены в миллиграммах на килограмм (мг/кг). Доза диазепама составляла 16 мг/кг. Доза теофиллина составляла 128 мг/кг.
[0307] Соединение Е1-1.2 демонстрировало устойчивую противосудорожную активность в исследуемых дозах 3, 10 и 30 мг/кг.
ПРИМЕР 2.5.2. Соединение Е1-8.2
[0308] Результаты представлены на ФИГ. 3. В столбцах представлено среднее значение ± СОС электроконвульсивного порога для каждой группы введения (n=15, ***/*: отличается от группы, получавшей носитель, р<0,001/0,05, соответственно, однофакторный ANOVA, апостериорный критерий Даннетта). Дозы приведены в миллиграммах на килограмм (мг/кг). Доза диазепама составляла 16 мг/кг.
[0309] Соединение Е 1-8.2 демонстрировало умеренную проконвульсивную активность после введения в дозах 0,5 и 2 мг/кг и противосудорожную активность в дозах 10 и 20 мг/кг.
ПРИМЕР 2.5.3. Соединение Е1-21.26
[0310] Результаты представлены на ФИГ. 4. В столбцах представлено среднее значение ± СОС электроконвульсивного порога для каждой группы введения (n=15, ***/*: отличается от группы, получавшей носитель, р<0,001/0,05, соответственно, однофакторный ANOVA, апостериорный критерий Даннетта).
[0311] Соединение Е1-21.26 демонстрировало противосудорожную активность в дозах 3 и 10 мг/кг.
ПРИМЕР 2.6. Пентилентетразоловый (ПТЗ) тест с провокацией припадка
[0312] Метод, который позволяет обнаружить проконвульсивную или противосудорожную активность, связанную с ГАМК-эргическим механизмом, соответствует методу, описанному Krall (Epilepsia, 19, 409-428, 1978). Крысам, помещенным в отдельные макролоновые клетки (25×19×13 см), вводили пентилентетразол (ПТЗ) (100 мг/кг подкожно (п/к)). В течение 30-минутного периода отмечали продолжительность латентного периода, появление клонических и тонических судорог и гибель. В каждую группу включали по 15 крыс. Тест проводили в форме слепого испытания.
[0313] Исследуемые соединения Е1-1.2 и Е1-21.26 вводили в форме мезилатных солей (дозы в мг/кг рассчитаны на основании молекулярной массы свободного основания). Исследуемое соединение Е1-8.2 вводили в форме свободного основания.
[0314] Соединения обычно оценивали с применением водного раствора, содержащего 0,5% диметилсульфоксида, 4% гидроксипропил-b-циклодекстрина, в качестве носителя, в 1 или более дозах (например, 1, 3 и 10 мг/кг), введенных п/о за 30 минут до ПТЗ, и сравнивали с контрольной группой, получавшей носитель. Диазепам (16 мг/кг п/о), введенный за 60 минут до ПТЗ, использовали в качестве эталонного вещества. Эксперимент проводили в виде 2 отдельных частичных экспериментов с N=7-8 животных в группе и в частичном эксперименте. Количественные показатели (продолжительность латентных периодов) с применением исследуемых соединений анализировали путем сравнения экспериментальных групп с контролем с применением носителя, используя критерий Крускала-Уоллиса (Kruskal-Wallis) с последующим вычислением U-критерия Манна-Уитни (Mann-Whitney). Количественные показатели с применением эталонного соединения анализировали с использованием U-критерия Манна-Уитни. Дискретные показатели (частоты) анализировали путем сравнения экспериментальных групп с контрольными, получавшими носитель, используя критерий точной вероятности Фишера (*=р<0,05; **=р<0,01; ***=р<0,001).
ПРИМЕР 2.6.1. Соединение Е1-1.2
[0315] Результаты представлены на ФИГ. 5А, ФИГ. 5В, ФИГ. 5С, ФИГ. 5D, ФИГ. 5Е и ФИГ. 5F.
[0316] Соединение Е1-1.2 в дозе 10 мг/кг п/о снижало число крыс, демонстрирующих тонические судороги (-82%, р<0,01) и число случаев гибели (-91%, р<0,001) по сравнению с контролем с применением носителя. Соединение Е1-1.2 также увеличивало продолжительность латентных периодов до начала клонических и тонических судорог и латентного периода до гибели (+72%, р<0,05; +39%, р<0,01 и +35%, р<0,001, соответственно). Эти результаты подтверждают противосудорожную активность для соединения Е1-1.2 в дозе 10 мг/кг п/о в пентилентетразоловом тесте с провокацией припадка у крыс.
ПРИМЕР 2.6.2. Соединение Е1-21.26
[0317] Результаты представлены на ФИГ. 6А, ФИГ. 6В, ФИГ. 6С, ФИГ. 6D, ФИГ. 6Е и ФИГ 6F.
[0318] Соединение Е1-21.26 в дозе 10 мг/кг п/о снижало число крыс, демонстрирующих тонические судороги (-82%, р<0,01) и число случаев гибели (-82%, р<0,01) по сравнению с контролем с применением носителя. Соединение Е1-21.26 также увеличивало продолжительность латентных периодов до начала клонических и тонических судорог и латентного периода до гибели (+45%, р<0,05; +40%, р<0,01 и +31%, р<0,01, соответственно). Эти результаты подтверждают противосудорожную активность для соединения Е1-21.26 в дозе 10 мг/кг п/о в пентилентетразоловом тесте с провокацией припадка у крыс.
ПРИМЕР 2.7. Тест с провокацией припадка посредством электрошока с частотой импульсов 6 Гц
[0319] Тест с провокацией припадка посредством электрошока с частотой импульсов 6 Гц, который позволяет обнаружить противосудорожную активность исследуемых соединений, проводили в соответствии с методикой, описанной Brown et al. (J. Pharmacol. Exp. Ther. 107, 273-283, 1953; и Barton et al. (Epilepsy Res. 47, 217-227, 2001). Мышей подвергали действию прямоугольного тока (44 мА, прямоугольный импульс: ширина импульса 0,2 мс, продолжительность 3 с, 6 Гц) с помощью роговичных электродов, соединенных с генератором импульсов постоянного тока (Ugo Basile: type 7801). Результаты для количества припадков, отраженных в клонусе передних конечностей, регистрировали в течение первой минуты после приложения тока. Клонус передних конечностей оценивали как отсутствующий (0), умеренный (1) и сильный (2). В каждую группу включали по 15 мышей. Тест проводили в виде частично слепого испытания (исследуемое вещество против носителя). Исследуемые вещества обычно вводили с применением водного раствора, содержащего 0,5% диметилсульфоксида, 4% гидроксипропил-b-циклодекстрина, в качестве носителя, п/о за 30 минут до испытания и сравнивали с контрольной группой, получавшей носитель. Исследуемые соединения Е1-1.2 и Е1-21.26 вводили в форме мезилатных солей (дозы в мг/кг рассчитаны на основании молекулярной массы свободного основания). Исследуемое соединение Е1-8.2 вводили в форме свободного основания.
[0320] Диазепам, введенный за 60 минут до испытания, применяли в качестве эталонного вещества положительного контроля. Количественные показатели (оценки) с применением исследуемого соединения анализировали путем сравнения экспериментальных групп с контрольной, получавшей носитель, используя критерий Крускала-Уоллиса с последующим вычислением U-критерия Манна-Уитни.
ПРИМЕР 2.7.1. Соединение Е1-1.2
[0321] Результаты представлены на ФИГ. 7А. В столбцах представлено собой среднее значение ± СОС клонуса передних конечностей (произвольные единицы) для каждой группы введения (*/**/***: отличается от группы, получавшей носитель, р<0,05/0,01/0,001, однофакторный ANOVA, апостериорный критерий Даннетта). Дозы приведены в миллиграммах на килограмм (мг/кг). Доза диазепама составляла 8 мг/кг (п/о). Соединение Е1-1.2 демонстрировало дозозависимое снижение клонуса передних конечностей. (10 и 30 мг/кг) при п/о введении за 30 минут до испытания достоверно и дозозависимым образом снижали показатель приступа судорог передних конечностей по сравнению с контролем с применением носителя (-50%, р<0,05 и -70%, р<0,01, соответственно). Оно также дозозависимым образом снижало число мышей, имеющих «хвост Штрауба» со значимым эффектом в дозе 30 мг/кг (10 мг/кг: -13%, НЗ и 30 мг/кг: -40%, р<0,05) (ФИГ. 7В).
ПРИМЕР 2.8. Модель каталепсии, вызванной галоперидолом (КВГ)
[0322] Модель каталепсии, вызванной галоперидолом (КВГ), которая позволяет обнаружить антипсихотическую активность и действие селективных антагонистов NR2B (Steece-Collier et al. Exp. Neurol. 163: 239, 2000) и была основана на методе, описанном Chermat и Simon (J. Pharmacol., 6, 493-496, 1975). Способность вызывать каталепсию служит показателем предрасположенности исследуемого соединения вызывать экстрапирамидные побочные действия, в частности, паркинсонизм. Таким образом, антагонизм по отношению к каталепсии, вызванной антипсихотическими средствами, может служить для обнаружения потенциального противопаркинсонического действия.
[0323] Крысам вводили галоперидол (1 мг/кг в/б) и оценивали на наличие каталепсии с 30-минутными интервалами в период времени до 360 минут. Наличие (+) или отсутствие (-) каталепсии оценивали с помощью трех подходов: 1) принудительное скрещивание ипсилатеральных передних и задних конечностей; 2) придание животному позы Будды; 3) наклонная платформа, автоматическое устройство, которое через 5 секунд после размещения крысы вытесняет крысу из горизонтального положения в вертикальное положение и обратно, пока она цепляется за проволочную сетку передними лапами. Акинезию и каталепсию определяли в зависимости от того, двигалось ли животное до (акинезия) или во время работы платформы (каталепсия).
[0324] Для получения общей оценки каталепсии у животного учитывали 4 оценки, собранные в течение времени испытания. В каждую группу включали по 6 крыс. Тест проводили в виде слепого испытания (исследуемое вещество против носителя). Исследуемые вещества оценивали в 1 или более дозах, обычно вводимых с применением водного раствора, содержащего 0,5% диметилсульфоксида, 4% гидроксипропил-b-циклодекстрина, в качестве носителя, п/о за 15 минут до введения галоперидола (например, за 45 минут до первого измерения) и сравнивали с контрольной группой, получавшей носитель. Исследуемые соединения Е1-1.2 и Е1-21.26 вводили в форме мезилатных солей (дозы в мг/кг рассчитаны на основании молекулярной массы свободного основания).
[0325] Амфетамин (8 мг/кг п/о), введенный за 60 минут до испытания (например, за 90 минут до первого измерения), применяли в качестве эталонного соединения. Результаты с применением исследуемых соединений анализировали путем сравнения экспериментальных групп с контрольной, получавшей носитель, используя критерий Крускала-Уоллиса с последующим вычислением U-критерия Манна-Уитни. Результаты с применением эталонного вещества анализировали с использованием U-критерия Манна-Уитни.
ПРИМЕР 2.8.1. Соединение Е1-1.2
[0326] Результаты представлены на ФИГ. 8А. Дозы приведены в миллиграммах на килограмм (мг/кг). Доза амфетамина составляла 8 мг/кг (ФИГ. 8В).
[0327] Соединение, применяемое в качестве положительного контроля, амфетамин (8 мг/кг п/о), демонстрировало ожидаемую устойчивую антикаталептическую активность (ФИГ. 8В). Соединение Е1-1.2 достоверно снижало суммарную оценку каталепсии в течение 360 мин по сравнению с контролем с применением носителя (2,0, р<0,01). В дозах 1 и 3 мг/кг соединение Е1-1.2 имеет тенденцию снижать суммарную оценку каталепсии в течение 360 мин (17,0, р=0,0898 и 15,8, р=0,0526, соответственно) с достоверным эффектом через 2,5 часа в дозе 1 мг/кг (р<0,05) и через 2,5 и 3,5 часа в дозе 3 мг/кг (р<0,05 и р<0,01, соответственно). Каталепсию наблюдали между 4 и 6 часами при применении дозы 10 мг/кг. Эти результаты свидетельствуют о наличии у соединения Е1-1.2 значительной антикаталептической активности в модели каталепсии, вызванной галоперидолом, у крыс.
ПРИМЕР 2.8.2. Соединение Е1-21.26
[0328] Результаты представлены на ФИГ. 8С. Дозы приведены в миллиграммах на килограмм (мг/кг). Соединение, применяемое в качестве положительного контроля, амфетамин (8 мг/кг п/о), демонстрировало ожидаемую устойчивую антикаталептическую активность (ФИГ. 8 В). Соединение Е1-21.26 в дозах 3 мг/кг и 10 мг/кг достоверно и дозозависимым образом снижало суммарную оценку каталепсии в течение 360 мин по сравнению с контролем с применением носителя (11,7 и 1,0, соответственно, р<0,01). Каталепсию наблюдали между 1,5 и 6 часами при применении дозы 3 мг/кг. Каталепсию также наблюдали между 5 и 6 часами при применении дозы 10 мг/кг. Эти результаты свидетельствуют о наличии у соединения Е1-21.26 значительной антикаталептической активности в модели каталепсии, вызванной галоперидолом, у крыс.
ПРИМЕР 2.9. Модель с формалином на крысах
[0329] Модель с формалином на крысах представляет собой тоническую модель постоянной боли, обусловленную спонтанным ноцицептивным поведением, вызванным формалином. Инъекция формалина в лапу представляет собой общепринятую модель для оценки спонтанного ноцицептивного поведения у грызунов (Dubuisson, D. and Dennis, S.G. Pain 4:161, 1977). Подкожная инъекция формалина с подошвенной стороны приводит к двухфазному ноцицептивному поведенческому ответу у грызунов. Ранняя фаза (фаза I) продолжается в течение примерно 5-10 мин, после чего наблюдается промежуточная фаза без каких-либо заметных ноцицептивных реакций, за которой следует ноцицептивная реакция поздней фазы (фаза II), продолжающаяся от примерно 20-60 мин после инъекции формалина. Формалиновая модель представляет собой модель тонической, стойкой боли и широко используется для быстрого скрининга новых анальгетических соединений. Модель охватывает воспалительные, нейрогенные и центральные механизмы ноцицепции, а поздняя фаза, в частности, рассматривается как фармакодинамический суррогат центральной сенсибилизации. В настоящем примере эффекты исследуемого объекта оценивали в течение 0-5 минут от начала ранней фазы (фазы I) и 20-35 минут от начала поздней фазы (фаза II) ноцицептивного поведения, вызванного формалином. За 20 мин до инъекции формалина животным вводили носитель, исследуемое соединение (10, 30, 60 мг/кг в/б), как правило, с применением водного раствора, содержащего 0,5% диметилсульфоксида, 4% гидроксипропил-b-циклодекстрина, в качестве носителя и дулоксетина в качестве положительного контроля (30 мг/кг).
[0330] Животным всех групп позволяли привыкнуть к камере наблюдения в течение 15 минут непосредственно перед инъекцией формалина. Всем животным подкожно инъекционно вводили 50 мкл 5% формалина в левую заднюю лапу и затем их немедленно помещали в камеру наблюдения и непрерывно регистрировали спонтанное ноцицептивное поведение крыс, вызванное формалином, в течение 0-60 мин с использованием видеокамеры промышленного производства.
[0331] Оценка на основе записанных видеофайлов была выполнена в автономном режиме с использованием ПК наблюдателем, который был утвержден для оценки такого ноцицептивного поведения у грызунов. Общее время, проведенное в течение 5-минутного периода, регистрировали с помощью секундомера для следующего ноцицептивного поведения: подергивания, встряхивания, кусания и лизания обработанной лапы.
[0332] Исследуемое соединение Е1-1.2 вводили в форме мезилатной соли (дозы в мг/кг рассчитаны на основании молекулярной массы свободного основания).
[0333] Эффект исследуемого соединения оценивали в течение следующих периодов: 0-5 минут от начала ранней фазы (фазы I) и 20-35 минут от начала поздней фазы (фаза II).
[0334] Результаты представлены на ФИГ. 9А и 9В. В столбцах представлено среднее значение ± COC для каждой группы введения (n=8, ****: отличается от группы, получавшей носитель, р<0,0001, однофакторный ANOVA, апостериорный критерий Даннетта). Дозы приведены в миллиграммах на килограмм (мг/кг).
[0335] Соединение Е1-1.2 в каждой дозе (10, 30 и 60 мг/кг в/б) снижало суммарное время, проведенное в фазе I (0-5 мин) (ФИГ. 9А) и фазе II (20-35 мин) (ФИГ. 9В) ноцицептивного поведения.
ПРИМЕР 2.10. Распространяющаяся кортикальная депрессия (РКД), модель фазы ауры при мигрени
[0336] Модель распространяющейся кортикальной депрессии используют для исследования, влияет ли лечение с применением соединений согласно настоящему изобретению на электрофизиологические и гемодинамические события в модели мигрени у крыс.
[0337] Крыс анестезировали с применением 5% изофлурана (в 70% N2O и 30% O2, скорость потока 300 мл/мин) и помещали в стереотаксическую рамку. В течение операции и РКД концентрацию анестетика снижали до 1-1,5%. Ректальную температуру поддерживали на уровне 37,0±1,0°C с помощью гомеотермической системы покрытия. Кожу вскрывали посредством медиального разреза и сдвигали латерально. При охлаждении солевым раствором над правым полушарием просверливали три трепанационных отверстия в следующих координатах (мм от брегмы): (1) назад 4,5, латерально 2,0 (затылочная кора): место введения KCl; (2) назад 0,5, латерально 2,0 (теменная кора): место записи ЛДФ; (3) вперед 2, латерально 2 (лобная кора): место регистрации потенциала постоянного тока. Зонд для лазерной допплеровской флоуметрии (Oxyflow, Oxford Optronics, UK) для наблюдения за ЦКТ и иназивный электрод из Ag/AgCl для измерения сдвигов потенциала постоянного тока помещали в трепанационные отверстия в теменной и лобной коре на интактную твердую мозговую оболочку и в кору, соответственно. Зонд для лазерной допплеровской флоуметрии помещали в область, свободную от больших сосудов мягкой и твердой мозговых оболочек, для минимизации вклада больших сосудов в сигнал. Для измерения потенциала постоянного тока электрод сравнения закрепляли в шейке. Твердую мозговую оболочку, лежащую над затылочной корой, осторожно удаляли и принимали меры для того, чтобы избежать кровотечения. После хирургической подготовки кору оставляли для восстановления в течение 15 минут при орошении физиологическим раствором. Хлопковый шарик (диаметром 2 мм), пропитанный 1М KCl, помещали на поверхность мягкой мозговой оболочки и поддерживали во влажном состоянии посредством добавления 5 мкл раствора KCl каждые 15 минут. Исследуемое соединение или носитель вводили за десять (10) минут до начала РКД. Положительный контроль MK-801 вводили за 30 мин до начала РКД. Число эпизодов РКД, вызванных KCl, подсчитывали в течение 2 часов. ЦКТ и потенциалы постоянного тока непрерывно контролировали, начиная за 5 минут до применения KCl.
[0338] Исследуемое соединение Е1-1.2 вводили в форме мезилатной соли (дозы в мг/кг рассчитаны на основании молекулярной массы мезилатной соли).
[0339] Данные собирали с помощью программного обеспечения для сбора данных Windaq (Dataq Instruments, США) при частоте 20 кГц. Исходные данные были проанализированы в программе Clampfit (Axon Instruments, США). Сигналы были пропущены через фильтр низких частот (диапазон отсечения 5-10000 Гц), чтобы эпизоды РКД в потенциалах постоянного тока и ЦКТ могли быть подтверждены. Были проанализированы следующие параметры: i) количество, продолжительность и амплитуда потенциалов постоянного тока и ii) количество и амплитуда событий РКД.
[0340] Результаты для числа потенциалов постоянного тока представлены на ФИГ. 10. Дозы приведены в миллиграммах на килограмм (мг/кг). Доза MK-801, положительного контроля, составляла 1,25 мг/кг. В столбцах представлено среднее значение ± стандартная ошибка среднего (СОС), а различия рассматривали как статистически достоверные при уровне Р<0,05 (n=8, * отличается от группы, получавшей носитель). Статистический анализ проводили с использованием статистического программного обеспечения StatsDirect. Различия между группами анализировали с использованием однофакторного ANOVA и апостериорного критерия Даннетта.
[0341] Амплитуды потенциалов постоянного тока для соединения Е1-1.2 в различных дозах и MK-801 статистически не отличались при сравнении с носителем (данные не приведены). Продолжительность потенциалов постоянного тока для соединения Е1-1.2 в различных дозах статистически не отличалась при сравнении с носителем (данные не приведены). Продолжительность потенциалов постоянного тока для MK-801 была повышена по сравнению с носителем (р<0,05, данные не приведены).
[0342] Интенсивность ЦКТ оставалась неизменной для соединения Е1-1.2 по сравнению с носителем. Число событий ЦКТ было снижено для соединения Е1-1.2 в дозе 10 мг/кг и для MK-801 в дозе 1,25 мг/кг со статистически достоверным отличием от носителя (* р<0,05 относительно группы с применением носителя) (данные не приведены).
[0343] Соединение Е1-1.2 (в дозах 3 мг/кг, 10 мг/кг и 30 мг/кг) значительно снижало число потенциалов постоянного тока по сравнению с группой с применением носителя (* р<0,05 для всех доз относительно группы с применением носителя, данные представлены как среднее ± СОС). Настоящие результаты демонстрируют, что соединение Е1-1.2 было эффективно в модели мигрени.
| название | год | авторы | номер документа |
|---|---|---|---|
| АМИНОТРИАЗОЛОПИРИДИНЫ И ИХ ПРИМЕНЕНИЕ В КАЧЕСТВЕ ИНГИБИТОРОВ КИНАЗ | 2009 |
|
RU2552642C2 |
| ИНГИБИТОРЫ СЕРИН/ТРЕОНИН КИНАЗЫ ДЛЯ ЛЕЧЕНИЯ ГИПЕРПРОЛИФЕРАТИВНЫХ ЗАБОЛЕВАНИЙ | 2013 |
|
RU2644947C2 |
| СОЕДИНЕНИЯ ПИРИДАЗИНАМИДА И ИХ ПРИМЕНЕНИЕ В КАЧЕСТВЕ ИНГИБИТОРОВ ТИРОЗИНКИНАЗЫ СЕЛЕЗЕНКИ (SYK) | 2013 |
|
RU2627661C2 |
| ГЕТЕРОАРИЛЬНЫЕ СОЕДИНЕНИЯ И ИХ ПРИМЕНЕНИЕ | 2009 |
|
RU2734822C2 |
| СП0СОБ ПОЛУЧЕНИЯ СОЕДИНЕНИЙ ДИГИДРОИНДЕНАМИДА, ФАРМАЦЕВТИЧЕСКИЕ КОМПОЗИИ, СОДЕРЖАЩИЕ ДАННЫЕ СОЕДИНЕНИЕ И ИХ ПРИМЕНЕНИЕ В КАЧЕСТВЕ ИНГИБИТОРА ПРОТЕИНКИНАЗЫ | 2009 |
|
RU2528408C2 |
| ЗАМЕЩЕННЫЕ [1,2,4]ТРИАЗОЛО[1,5-a]ПИРИМИДИН-7-ИЛЬНЫЕ СОЕДИНЕНИЯ В КАЧЕСТВЕ ИНГИБИТОРОВ PDE2 | 2015 |
|
RU2659070C9 |
| ПРОИЗВОДНЫЕ ТИЕНОПИРРОЛА ДЛЯ ПРИМЕНЕНИЯ ДЛЯ НАЦЕЛИВАНИЯ НА БЕЛКИ, КОМПОЗИЦИИ С УКАЗАННЫМИ ПРОИЗВОДНЫМИ, СПОСОБЫ И ПРИМЕНЕНИЯ | 2017 |
|
RU2771166C2 |
| СОЕДИНЕНИЯ 8-ФТОРФТАЛАЗИН-1(2Н)-ОНА В КАЧЕСТВЕ ИНГИБИТОРОВ ТИРОЗИНКИНАЗЫ БРУТОНА | 2012 |
|
RU2622391C2 |
| ЗАМЕЩЕННЫЕ ГЕТЕРОЦИКЛИЧЕСКИЕ СУЛЬФОНАМИДНЫЕ СОЕДИНЕНИЯ, ПОЛЕЗНЫЕ В КАЧЕСТВЕ МОДУЛЯТОРОВ TRPA 1 | 2014 |
|
RU2675792C2 |
| АНТАГОНИСТЫ TLR7/8 И ИХ ПРИМЕНЕНИЕ | 2019 |
|
RU2833035C2 |
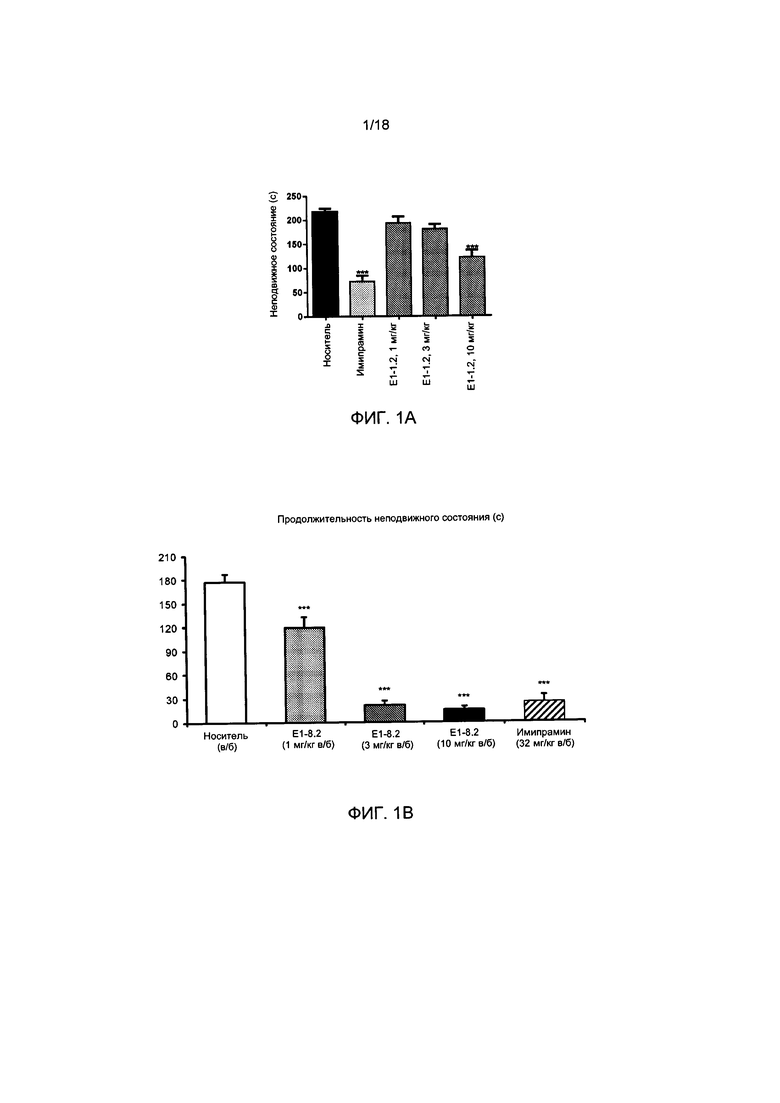
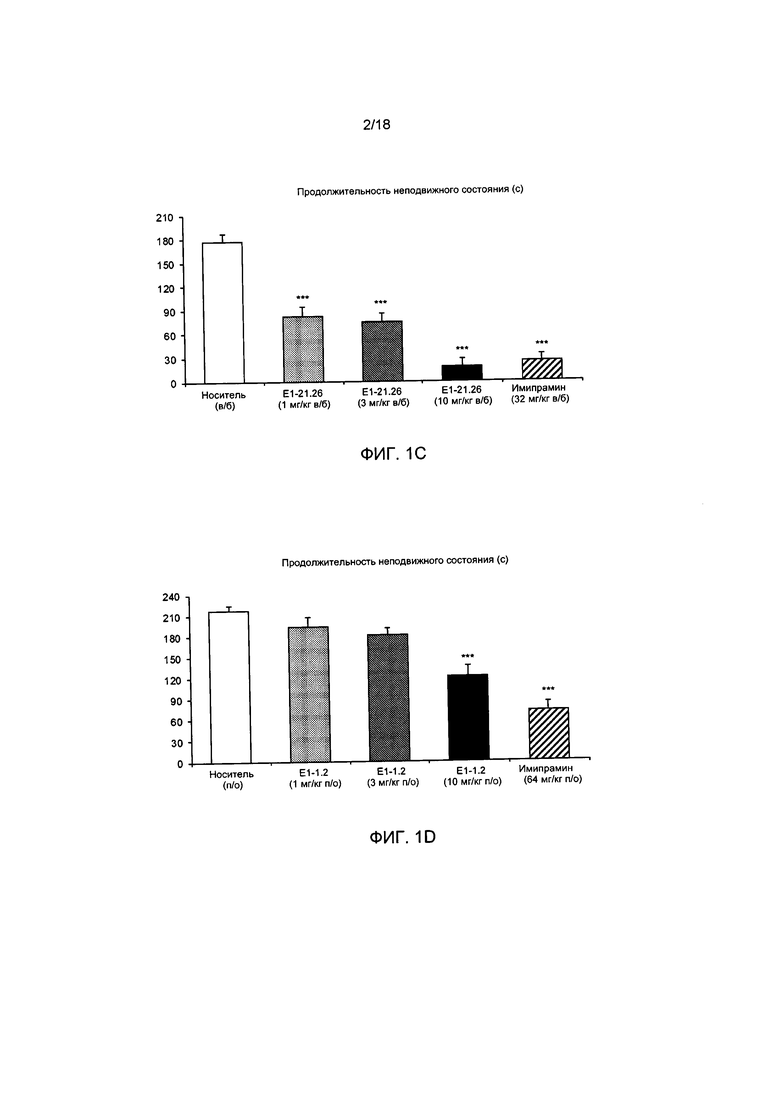
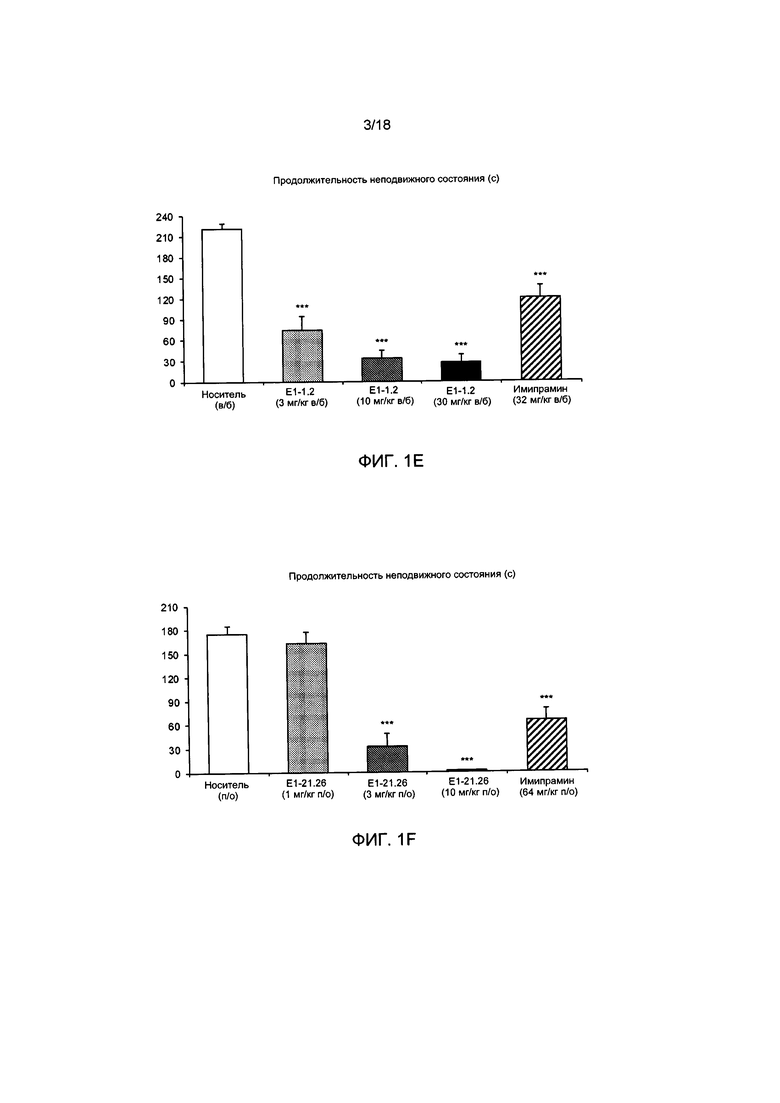
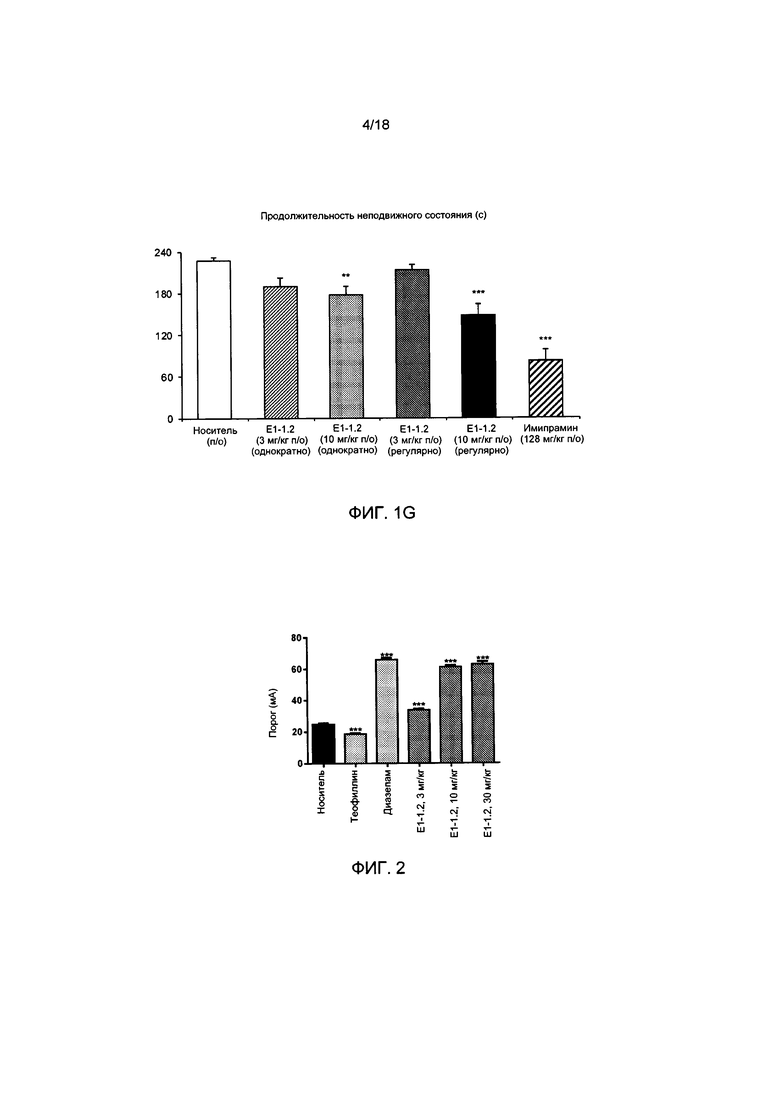

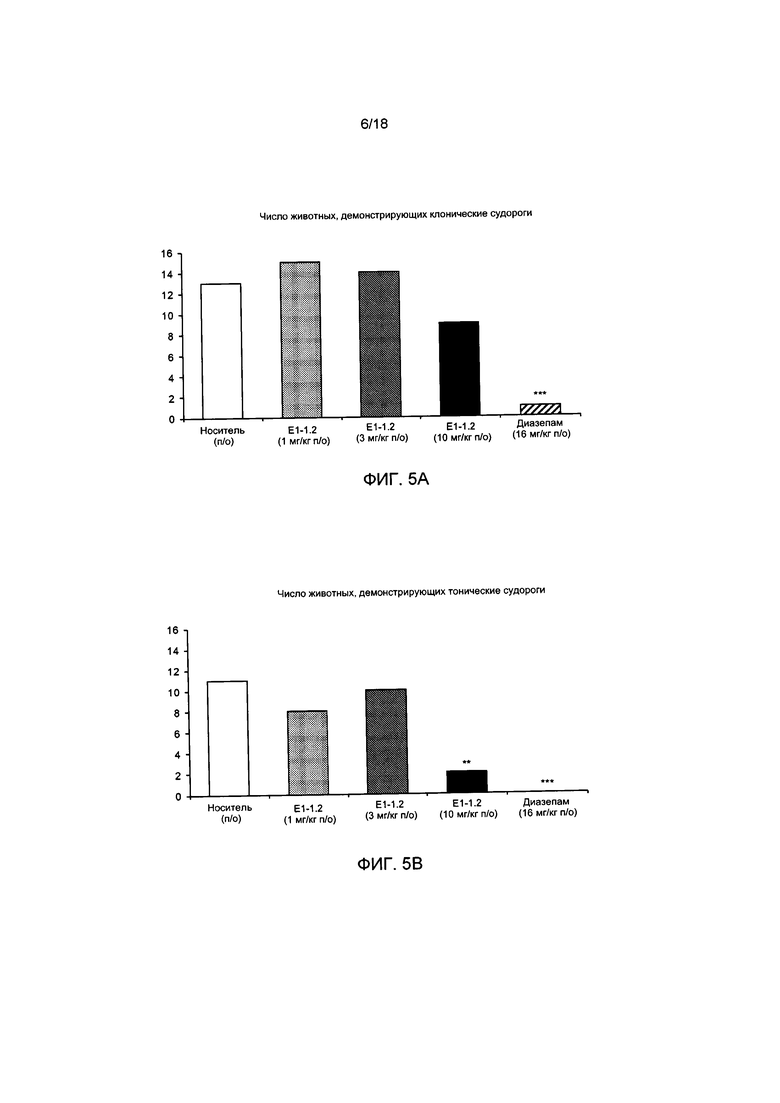
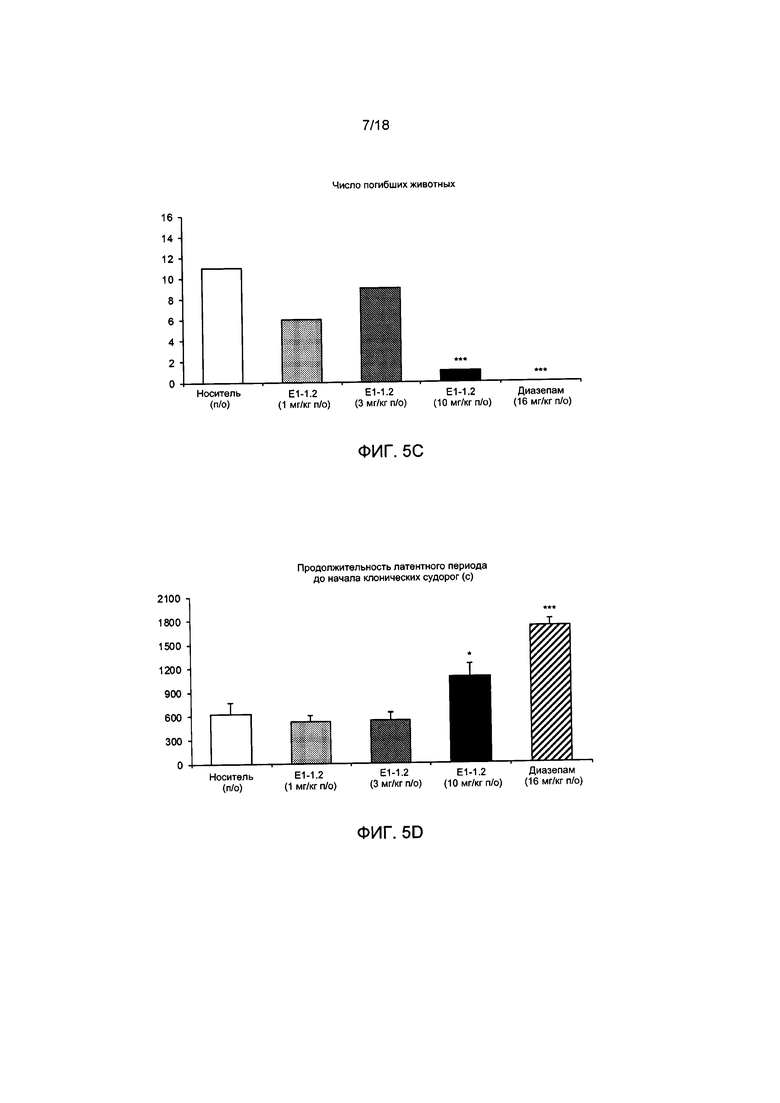




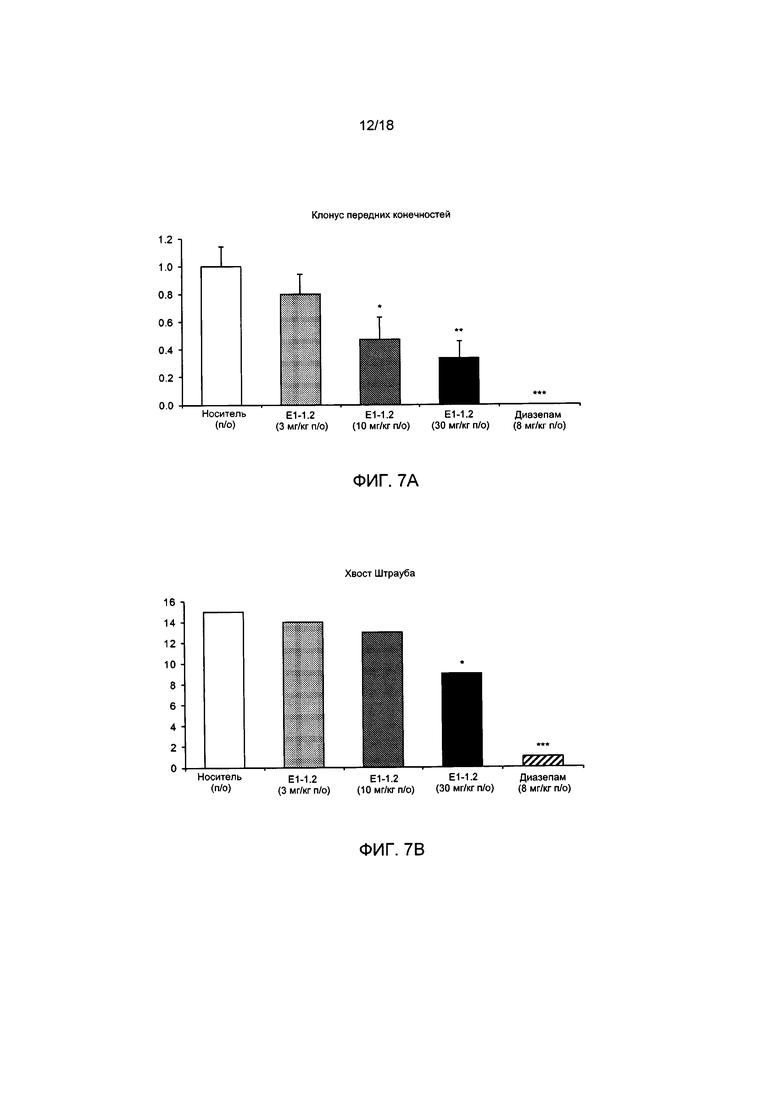
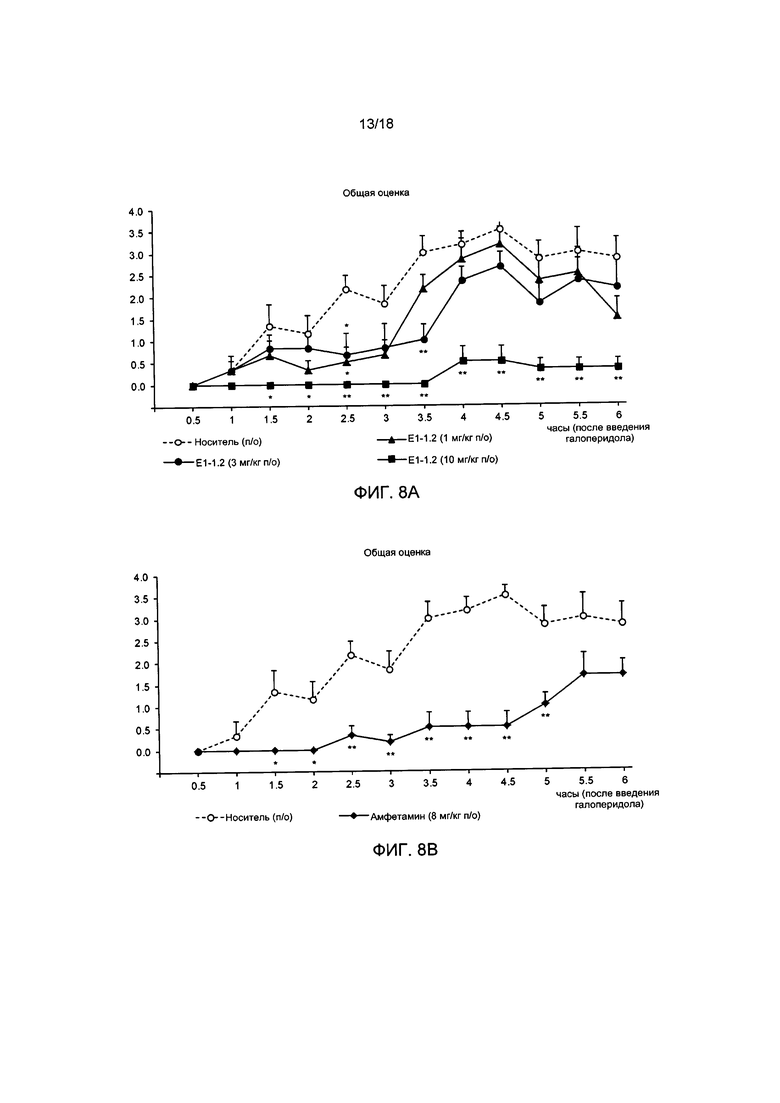
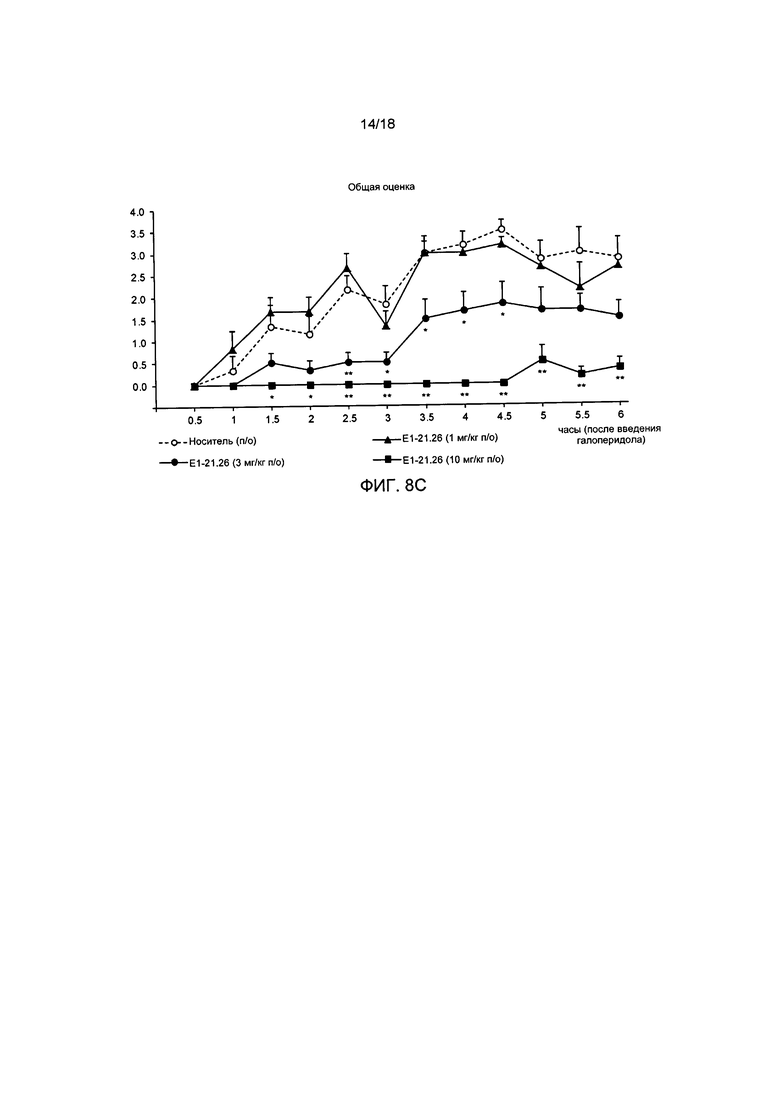
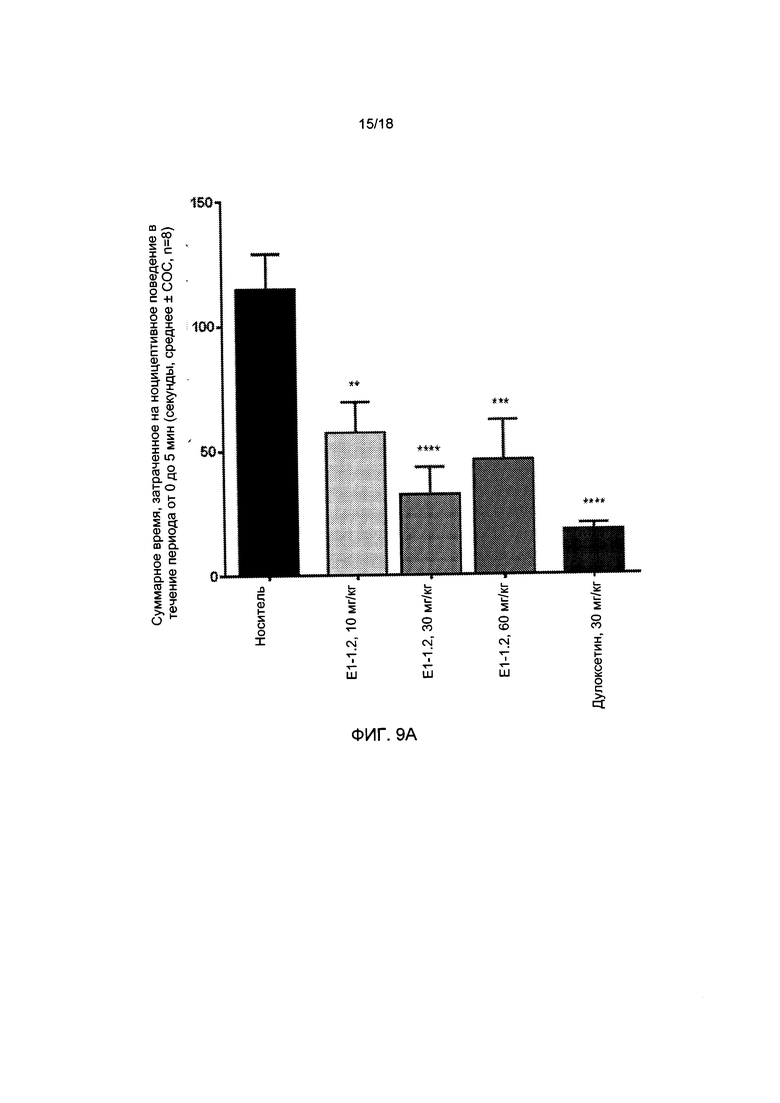

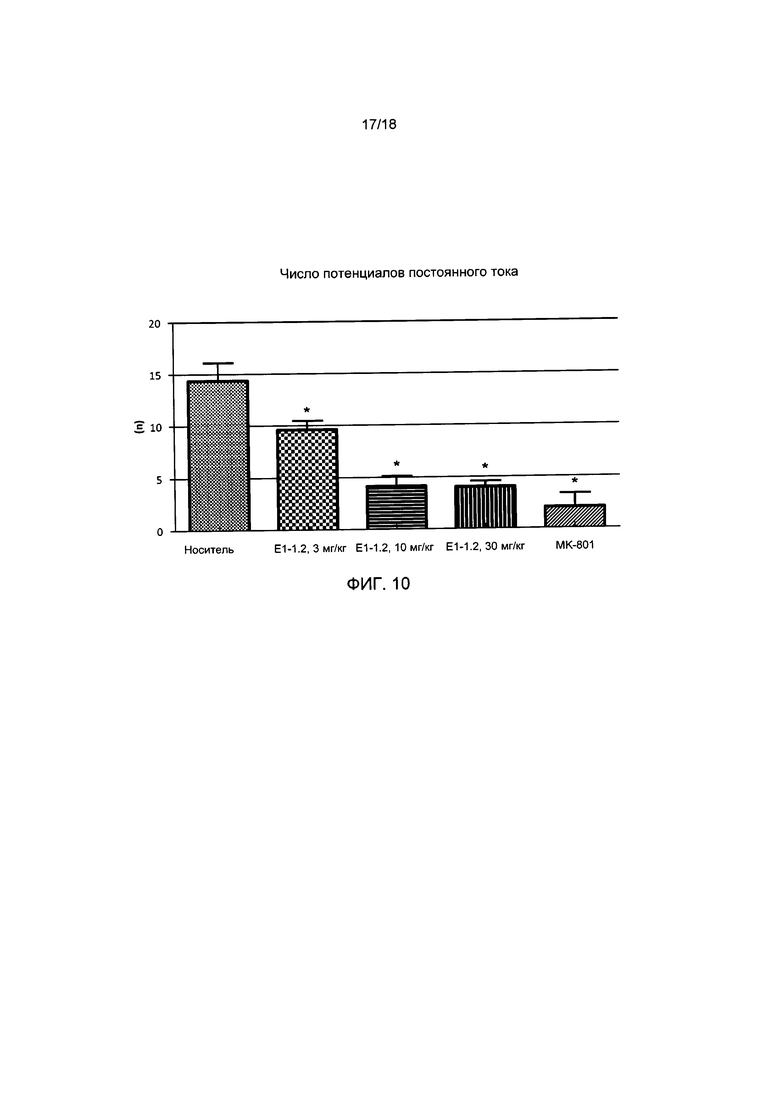

Изобретение относится к химическому соединению формулы (II), а также к фармацевтическим композициям, содержащим химическое соединение формулы (II), и к способам лечения различных заболеваний и расстройств, связанных с антагонизмом NR2B, например заболеваний и расстройств ЦНС, таких как депрессия, путем введения химического соединения формулы (II). 3 н. и 4 з.п. ф-лы, 51 пр., 10 табл., 11 ил.
1. Химическое соединение формулы (II):
 (II),
(II),
где
Z представляет собой  ;
;
R5 представляет собой -H, -F, -Cl, -СН3, -CFH2, -CF2H, -CF3, -CH2CH3, -CF2CH3, -CH2CF3, циклопропил, -OCF3, -OCF2H, -SCH3, -S(O)2CH3 или -C≡CH;
R6 представляет собой -H или -F; и
R7 представляет собой -H, -F, -Cl или -СН3.
2. Химическое соединение по п. 1, выбранное из таблицы 1.E1:

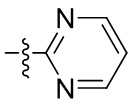
3. Фармацевтическая композиция для лечения заболевания или расстройства, отвечающего на антагонизм NR2B, содержащая эффективное количество химического соединения по любому из пп. 1 или 2 и фармацевтически приемлемый носитель.
4. Фармацевтическая композиция по п. 3, подходящая для перорального введения.
5. Способ лечения заболевания или расстройства, отвечающего на антагонизм NR2B, у субъекта, нуждающегося в таком лечении, включающий введение эффективного количества химического соединения по любому из пп. 1 или 2.
6. Способ по п. 5, где указанное заболевание или расстройство представляет собой депрессию, боль, болезнь Паркинсона, болезнь Гентингтона, болезнь Альцгеймера, церебральную ишемию, травматическое повреждение головного мозга, эпилепсию или мигрень.
7. Способ по п. 6, где указанное заболевание или расстройство представляет собой депрессию.
| US 2010105650 A1, 29.04.2010 | |||
| US 2011280808 A1, 17.11.2011 | |||
| US 2014336185 A1, 13.11.2014 | |||
| WO 2004108705 A1, 16.12.2004 | |||
| СПОСОБЫ ЛЕЧЕНИЯ НАРУШЕНИЙ С ПРИМЕНЕНИЕМ СЕЛЕКТИВНОГО АНТАГОНИСТА NR2B-ПОДТИПА NMDA РЕЦЕПТОРОВ | 2009 |
|
RU2499598C2 |

