Настоящее изобретение относится к фармацевтически полезному новому гетероциклическому производному или его соли и к фармацевтической композиции, содержащей их в качестве активного ингредиента.
В качестве анальгетика используют наркотический анальгетик (такой как морфин), ненаркотический анальгетик (такой как аспирин или индометацин) или наркотическо-антагонистический анальгетик (такой как пентазоцин). Наркотический анальгетик оказывает анальгетическое действие главным образом путем ингибирования центральной болевой передачи возбуждения. Ненаркотический анальгетик оказывает анальгетическое действие главным образом благодаря ингибированию продуцирования периферического вызывающего боль вещества. Наркотическо-антагонистический анальгетик оказывает анальгетическое действие по механизму, аналогичному механизму действия наркотического анальгетика.
Однако отсутствует анальгетик, который является эффективным против хронической боли, которая не подавляется морфином, при аллодинии, сопровождающейся опоясывающим лишаем, или при повышенной болевой чувствительности и было бы желательно разработать превосходный анальгетик.
Ноцицептин представляет собой нейропептид, относящийся к разнообразной нервной активности, включая болевую чувствительность in vivo. В не прошедшей экспертизу патентной публикации Японии №10-212290 описано, что агонист и/или антагонист ноцицептина может оказаться эффективным при лечении психического расстройства, невропатии и физиологических нарушений и особенно эффективен для снижения беспокойства и стресса, депрессии, травматического нарушения, амнезии вследствие болезни Альцгеймера или другого слабоумия, симптомов эпилепсии и спазма, острой и/или хронической боли, симптомов при прекращении приема наркотиков, для контроля за балансом воды, для выделения Na+, нарушения артериального давления крови и при расстройстве питания, таком как ожирение.
В качестве непептидного соединения, воздействующего на ноцицептиновый рецептор, известны лофентанил, бензоилгидразон налоксона и производное 2-оксоимидазола (Международная публикация WO 9854168). Однако данные соединения все еще находятся на стадии фундаментального исследования и ни одно из них не является коммерчески доступным.
В качестве соединения, аналогичного производному хиназолина в гетероциклических производных соединений в соответствии с настоящим изобретением, известны различные соединения (Международная Публикация WO 9307124, публикация прошедшего экспертизу патента Японии №2923742, 9720821, Международная Публикация WO 9850370, Международная Публикация WO 9909986, не прошедшая экспертизу патентная публикация Японии №47-2927, Международная Публикация WO 9817267 и тому подобные). Среди них в Международной Публикации WO 9720821 описано, что производное 2-ациламинохиназолина обладает ингибирующим действием на подтип-Y5 нейропептидного Y(NPY) рецептора и является эффективным для уменьшения интенсивности повышенной чувствительности к боли или амнезии.
Задачей настоящего изобретения является разработка нового соединения, обладающего превосходным анальгетическим действием. Более конкретно настоящее изобретение относится к новому анальгетику (болеутоляющему средству), обладающему анальгетическим действием, который очень эффективен против хронической боли или аллодинии, сопровождающейся опоясывающим лишаем, путем воздействия на ноцицептиновый (болевой) рецептор.
Задача настоящего изобретения достигается соединением, представленным следующей общей формулой (1), являющимся агонистом и/или антагонистом ноцицептинового рецептора и обладающим превосходным анальгетическим действием.
Соответственно настоящее изобретение относится к соединению, представленному следующей общей формулой (1):

или его соли.
В формуле Х и Y являются одинаковыми или различными, и каждый представляет собой атом азота или СН;
R1 представляет собой атом водорода или алкил;
А1 и А2 являются одинаковыми или различными, и каждый представляет собой (1) одинарную связь или (2) двухвалентную алифатическую углеводородную группу, которая может быть замещенной и может включать 1-3 ненасыщенные связи в любом положении (такая алифатическая углеводородная группа может содержать один гетероатом, выбранный из группы, состоящей из -NH-, О и S);
Q представляет собой (1) одинарную связь, (2) необязательно замещенную 3-8-членную циклоалкиленовую группу, (3) необязательно замещенную фениленовую группу или (4) необязательно замещенную 4-8-членную двухвалентную гетероциклическую группу;
R2А, R2С и R2D являются одинаковыми или различными, и каждый представляет собой атом водорода, алкил или фенил, R2B представляет собой атом водорода, алкил, циано, нитро или фенил, или два атома азота гуанидино-группы образуют цикл вместе с одним или двумя ее заместителями R2B, R2C и R2D с образованием насыщенного или ненасыщенного 5-или 6-членного кольца;
или взяты вместе в виде -N(R1)-A1-Q-A2-N(R2A)- с образованием 5-7-членного кольца;
Е представляет собой (1) этенилен, (2) -NRCO-, (3)
-NRCONH-, (4) -CONR-, (5) этинилен, (6) -NRSO2- или (7) аминоалкилен (где R представляет собой водород или необязательно замещенный алкил);
R3 представляет собой необязательно замещенную фенильную группу или гетероциклическую группу;
R4 и R5 (1) являются одинаковыми или различными, и каждый представляет собой атом водорода, алкил, алкокси, аралкилокси, галоген, нитро, гидрокси, алкоксикарбонил, -NR6R7, -NR6COR7, -NR6SO2R7, -CONR6R7 (в которых R6 и R7 являются одинаковыми или различными, и каждый представляет собой атом водорода или алкил), или (2) когда они являются смежными друг с другом, то взятые вместе образуют -O(СН2)nО (где n представляет собой целое число 1 или 2) или -СН=СН-СН=СН-.
Предпочтительно в формуле (1) каждый Х и Y представляет собой атом азота или СН;
R1 представляет собой атом водорода или алкил;
А1 и А2 являются одинаковыми или различными, и каждый представляет собой (1) одинарную связь или (2) алкилен, который может быть замещен алкилом, карбамоилом, моноалкилкарбамоилом, диалкилкарбамоилом, гидрокси, алкокси или трифторметилом и может содержать 1-3 ненасыщенные связи в любых положениях;
Q представляет собой (1) одинарную связь, (2) 3-8-членную циклоалкиленовую группу, которая может быть замещена алкилом, алкоксикарбонилом, карбамоилом, моноалкилкарбамоилом, диалкилкарбамоилом или алкокси, (3) фениленовую группу, которая может быть замещена алкилом, алкокси, алкоксикарбонилом, карбамоилом, моноалкилкарбамоилом, диалкилкарбамоилом, сульфамоилом, моноалкилсульфамоилом, диалкилсульфамоилом, амино, моноалкиламино, диалкиламино, нитро, галогеном, циано или трифторметилом, или (4) 4-8-членную двухвалентную гетероциклическую группу, которая может быть замещена алкилом, алкокси, алкоксикарбонилом, карбамоилом, моноалкилкарбамоилом, диалкилкарбамоилом, амино, моноалкиламино или диалкиламино;
R2A, R2C и R2D являются одинаковыми или различными, и каждый представляет собой атом водорода, алкил или фенил, R2B представляет собой атом водорода, алкил, цианогруппу, нитрогруппу или фенил, или два атома азота гуанидино-группы циклизованы вместе с одним или двумя ее заместителями R2B, R2C и R2D с образованием насыщенного или ненасыщенного 5-или 6-членного кольца;
или взяты вместе в виде -N(R1)-A1-Q-A2-N(R2A)- с образованием 5-7-членного кольца;
Е представляет собой (1) этенилен, (2) -NRCO-, (3) -NRCONH-, (4) -CONR-, (5) этинилен, (6) -NRSO2- или (7) аминоалкилен (в котором R представляет собой водород или необязательно замещенный алкил);
R3 представляет собой необязательно замещенную фенильную группу или гетероциклическую группу, которая может быть замещена алкилом, алкокси, алкоксикарбонилом, карбамоилом, моноалкилкарбамоилом, диалкилкарбамоилом, сульфамоилом, моноалкилсульфамоилом, диалкилсульфамоилом, алкилсульфониламино, N-(алкил)алкилсульфониламино, амино, моноалкиламино, диалкиламино, нитро, галогеном, циано, гидрокси или трифторметилом, и
R4 и R5 (1) являются одинаковыми или различными, и каждый представляет собой атом водорода, алкил, алкокси, аралкилокси, галоген, нитро, гидрокси, алкоксикарбонил, -NR6R7, -NR6COR7, -NR6SO2R7, -CONR6R7 (в которых R6 и R7 являются одинаковыми или различными, и каждый представляет собой атом водорода или алкил) или (2) когда они являются смежными друг с другом, то взятые вместе образуют -O(СН2)nО (где n представляет собой целое число 1 или 2) или -СН=СН-СН=СН-.
Более предпочтительное соединение представлено общей формулой (1), где каждый Х и Y представляет собой атом азота, R1 представляет собой атом водорода или алкил, А1 и А2 являются одинаковыми или различными, и каждый представляет собой (1) одинарную связь или (2) необязательно замещенный алкилен, Q представляет собой (1) одинарную связь, (2) необязательно замещенную 4-8-членную циклоалкиленовую группу, (3) необязательно замещенную фениленовую группу или (4) необязательно замещенную 5-7-членную двухвалентную гетероциклическую группу, R2A, R2B, R2C и R2D являются одинаковыми или различными, и каждый представляет собой атом водорода, алкил или фенил, или взятые вместе в виде -N-(R1)-A1-Q-A2-N-(R2A)- образуют 5-7-членное кольцо, Е представляет собой (1) этенилен, (2) -NRCO- или (3) -CONR-, и R4 и R5 (1) являются одинаковыми или различными, и каждый представляет собой атом водорода, алкил, алкокси, аралкилокси, галоген, нитро, гидрокси или алкоксикарбонил, или (2) когда они являются смежными друг с другом, то взятые вместе образуют -O(СН2)nО- (где n представляет собой целое число 1 или 2) или -СН=СН-СН=СН-.
Дополнительное предпочтительное соединение представлено общей формулой (1), где каждый Х и Y представляет собой атом азота, R1 является водородом. А1 и А2 являются одинаковыми или различными, и каждый представляет собой (1) одинарную связь или (2) необязательно замещенный алкилен, Q представляет собой (1) одинарную связь, (2) необязательно замещенную 5-7-членную циклоалкиленовую группу, (3) необязательно замещенную фениленовую группу, R2A, R2B, R2C и R2D являются одинаковыми или различными, и каждый представляет собой атом водорода, алкил или фенил, Е представляет собой (1) этенилен, (2) -NRCO-, и R4 и R5 являются одинаковыми или различными, и каждый представляет собой атом водорода, алкил, алкокси, аралкилокси, галоген или нитро.
Кроме того, настоящее изобретение относится к фармацевтической композиции, включающей любое из вышеуказанных соединений или его соль, и более конкретно к анальгетику.
Структурными признаками соединения настоящего изобретения являются следующие: присутствие гуанидиновой группы на конце заместителя, -N(R1)-A1-Q-A2-, в 4-положении хиназолиновой или хинолиновой структуры или в 1-положении изохинолиновой структуры; или циклизация двух NH групп в гуанидиновой группе вместе с заместителем после этого.
Соединение согласно настоящему изобретению, которое имеет описанную выше характерную особенность, представляет собой новое соединение, которое не было раскрыто в ссылках. Соединение согласно настоящему изобретению действует на ноцицептиновый рецептор, оказывая тем самым превосходное анальгетическое действие.
Далее настоящее изобретение будет описано подробно.
Примеры «алкила» в настоящем изобретении могут включать линейный или разветвленный алкил, имеющий от 1 до 6 атомов углерода, например метил, этил, н-пропил, изопропил, н-бутил, изобутил, втор-бутил, трет-бутил, н-пентил, 5-изопентил, н-гексил, изогексил и тому подобные. В частности, предпочтительным является алкил, содержащий от 1 до 4 атомов углерода.
Примеры «алкокси» могут включать линейную или разветвленную алкокси-группу, содержащую от 1 до 6 атомов углерода, например метокси, этокси н-пропокси, изопропокси, н-бутокси, изобутокси, втор-бутокси, трет-бутокси, н-пентилокси, изопентилокси, н-гексилокси, изогексилокси и тому подобные. В частности, предпочтительным является алкокси, содержащий от 1 до 4 атомов углерода.
Примеры «аралкилокси» могут включать аралкилокси, содержащий от 7 до 10 атомов углерода, например бензилокси, фенэтилокси и тому подобные. В частности, предпочтительным является бензилокси.
Примеры «двухвалентной алифатической углеводородной группы» могут включать линейный или разветвленный алкилен, содержащий от 1 до 6 атомов углерода (например, метилен, этилен, триметилен, тетраметилен, пентаметилен, гексаметилен, 2-этилтриметилен и 1-метилтетраметилен), линейный или разветвленный алкенилен, содержащий от 2 до 6 атомов углерода (например, винилен или пропенилен) или линейный или разветвленный алкинилен, содержащий от 2 до 6 атомов углерода (например, этинилен). Такая алифатическая углеводородная группа может содержать один гетероатом, выбранный из группы, состоящей из NH, атома кислорода и атома серы.
Примеры алкилена в «аминоалкилене» могут включать алкилен, перечисленный в «двухвалентной алифатической углеводородной группе».
Примеры «циклоалкилена» могут включать циклоалкилен, содержащий от 3 до 8 атомов углерода, например циклопропилен, циклобутилен, циклопентилен, циклогексилен, циклогептилен и циклооктилен. Такой циклоалкилен может содержать 1-2 заместителя, и примеры таких заместителей могут включать алкил, алкоксикарбонил, карбамоил, моноалкилкарбамоил, диалкилкарбамоил или алкокси. Он также может содержать ненасыщенную связь, и примеры циклоалкилена, содержащего такую ненасыщенную связь, включают циклогексенилен, циклогептенилен, циклооктенилен и тому подобные.
Примеры «галогена» могут включать атомы фтора, хлора, брома и иода.
Примеры гетероциклического кольца в «гетероциклической группе» и «двухвалентной гетероциклической группе» могут включать 4-8-членное моноциклическое или конденсированное кольцо, которое содержит 1-2 гетероатома, выбранные из группы, состоящей из NH, атома кислорода и атома серы, и которое может содержать 1-4 ненасыщенные связи. Примеры R3 могут включать 2-пиридил, 3-пиридил, 4-пиридил, 2-пиримидинил, 4-пиримидинил, 5-пиримидинил, 3-пиридазинил, 4-пиридазинолил, 2-пиразинил и 3-пиразинил. Такая гетероциклическая группа может содержать 1-2 заместителя и примеры заместителей могут включать алкил, алкокси, алкоксикарбонил, карбамоил, моноалкилкарбамоил, диалкилкарбамоил, сульфамоил, моноалкилсульфамоил, диалкилсульфамоил, алкилсульфониламино, N-(алкил) алкилсульфониламино, амино, моноалкиламино, диалкиламино, нитро, галоген, циано, гидрокси или трифторметокси. Примеры гетероциклического кольца в гетероциклической группе Q могут включать пиридин, пиримидин, пиперазин, гомопиперазин, фуран, тиофен и тому подобные. Гетероциклическая группа Q может содержать 1-2 заместителя, и примеры таких заместителей могут включать алкил, алкокси, алкоксикарбонил, карбамоил, моноалкилкарбамоил, диалкилкарбамоил, амино, моноалкиламино или диалкиламино.
«Фениленовая группа» может содержать 1-2 заместителя, и примеры таких заместителей могут включать алкил, алкокси, алкоксикарбонил, карбамоил, моноалкилкарбамоил, диалкилкарбамоил, сульфамоил, моноалкилсульфамоил, диалкилсульфамоил, амино, моноалкиламино, диалкиламино, гидрокси, нитро, галоген, циано и трифторметил.
Пример кольца, представленного -N(R1)-A1-Q-A2-N(R2A)- могут включать 5-7-членное насыщенное кольцо, такое как пиперазино или гомопиперазино.
Примеры «соли» соединения (1), охватываемые настоящим изобретением, могут включать соль неорганической кислоты, такой как хлористоводородная кислота, серная кислота, азотная кислота, фосфорная кислота, фтористоводородная кислота или бромистоводородная кислота, или соль органической кислоты, такой как уксусная кислота, винная кислота, молочная кислота, лимонная кислота, фумаровая кислота, малеиновая кислота, янтарная кислота, метансульфоновая кислота, этансульфоновая кислота, бензолсульфоновая кислота, толуолсульфоновая кислота, нафталинсульфоновая кислота или камфорсульфоновая кислота.
Примеры особенно предпочтительного соединения могут включать дигидрохлорид (1S,2R)-N-амидино-2-{[2(4-хлорбензоиламино)-6-метоксихиназолин-4-ил]амино} циклогексиламина, тригидрохлорид N-амидино-2-[6-метокси-4-{2-[2-(2-пиридил)этенил]хиназолин-4-ил)аминоэтил]фенилэтиламина, тригидрохлорид цис-4-гуанидинометил-цис-2-метил-N-{6-метокси-2-[2-{2-пиридил)этенил]хиназолин-4-ил}циклогексиламина, тригидрохлорид N-амидино-N'-{6-метил-2-[2-(2-пиридил)этенил] хиназолин-4-ил}-1,6-гександиамина, дигидрохлорид (1S,2R)-цис-N-амидино-2-{[2-(4-хлорстирил)-6-метоксихиназолин-4-ил]амино} циклогексиламина и тригидрохлорид N-амидино-N'-{6-метокси-2-[2-(2-пиридил)этенил]хиназолин-4-ил}-1,6-гександиамина.
Соединение согласно настоящему изобретению может существовать в виде цис (Z-форма) изомера или транс (Е форма) изомера, и каждый изомер и их смесь также включены в настоящее изобретение.
Некоторые соединения согласно настоящему изобретению могут иметь асимметрические атомы углерода и каждый оптический изомер и их рацематы также включены в настоящее изобретение. Оптический изомер можно получать, например, исходя из рацемата, полученного как описано выше, с использованием его основных свойств, используя оптически активную кислоту (винную кислоту, дибензоилвинную кислоту, миндальную кислоту, 10-камфорсульфоновую кислоту и тому подобные) известным способом для осуществления оптического разделения, или исходя из предварительно полученного оптически активного соединения.
Соединение (1) согласно настоящему изобретению может быть получено, например, следующими способами.
Способ получения А

(где X, Y, R1, R2A R2B, R2c, R2D, R3, R4, R5, A1, A2, Q и Е имеют описанные выше значения. R2b и R2c являются одинаковыми или разными, и каждый представляет собой атом водорода, алкил, фенил, циано, нитро или защитную группу. L представляет собой уходящую группу.)
Примеры защитной группы могут включать трет-бутоксикарбонил, бензилоксикарбонил, бензил и тому подобные. Примеры уходящей группы могут включать пиразол-1-ил, метилтио, метокси, галоген и тому подобные. Соединение (2) подвергают взаимодействию с соединением (3) в количестве от одного эквивалента до избыточного количества в углеводородах, таких как бензол и толуол, простых эфирах, таких как диоксан и тетрагидрофуран, галогенированных углеводородах, таких как хлороформ, хлористый метилен и 1,2-дихлорэтан, или в N,N-диметилформамиде при температуре от 0°С до температуры кипения используемого растворителя в течение от нескольких часов до нескольких дней с последующим снятием защитных групп R2b и R2c, когда они присутствуют в качестве защитных групп, с использованием известного самого по себе способа, получая таким образом соединение (1). Особенно предпочтительно использовать пиразол-1-ил в качестве уходящей группы L в соединении (3) и трет-бутоксикарбонил в качестве защитной группы, 1,2-дихлорэтан в качестве растворителя и проводить реакцию при комнатной температуре в течение от 1 до 48 часов с последующим снятием защиты хлористоводородной кислотой.
Способ получения В (где R2B в соединении (1) представляет собой атом водорода)
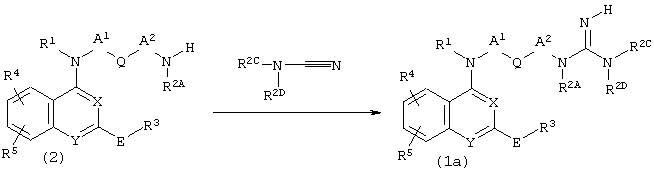
(где X, Y, R1, R2A, R2B, R2C, R2D, R4, R5, A1, A2, Q и Е имеют описанные выше значения.
Соединение (1a) может быть получено взаимодействием соединения (2) с R2CR2DN-CN по известному способу (J. Med. Chem., 18, 90, 1975 и тому подобные).
Способ получения С (где R2D в соединении (1) представляет собой атом водорода)

(где X, Y, R1, R2A, R2B, R2C, R3, R4, R5, A1, A2, Q и Е имеют описанные выше значения.
Соединение (1b) может быть получено взаимодействием соединения (2) с R2B-N=C=N-R2C по известному способу (J. Am.Chem. Soc., 3673, 1962 и тому подобные).
Способ получения D

(где X, Y, R1, R2A, R2B, R2C, R2D, R3, R4, R5, A1, A2, Q и Е имеют описанные выше значения. R8 представляет собой алкил).
Соединение (1) может быть получено из соединения (120) известным способом (Synthesis, 6, 460, 1988 и тому подобные). Алкил в качестве R8 представляет собой алкил, имеющий от 1 до 4 атомов углерода и предпочтительно представляет собой метил.
Способ получения Е
Соединение (1А), которое представляет собой соединение (1), где Е представляет собой этенилен, оба Х и Y представляют собой N, может быть получено по представленной ниже реакции:
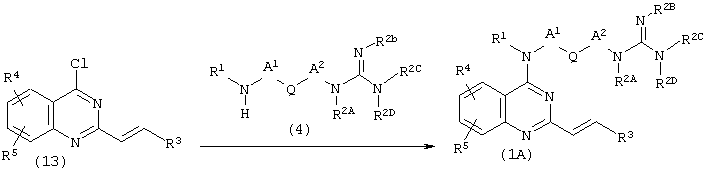
(где R1, R2A, R2B, R2C, R2D, R3, R4, R5, A1, A2, R2b, R2c и Q имеют описанные выше значения).
Соединение (13) подвергают взаимодействию с амином (4) в количестве от одного эквивалента до избыточного количества в присутствии основания, такого как гидрид натрия или N,N-диизопропилэтиламин, в растворителе, имеющем высокую температуру кипения, таком как 1-пентанол, N,N-диметилформамид или фенол, при температуре от 50°С до температуры кипения используемого растворителя в течение от нескольких часов до нескольких суток с последующим снятием защитных групп R2b и R2c, когда они присутствуют в качестве защитных групп, с использованием известного способа, получая таким образом соединение (1А). Предпочтительно взаимодействие проводят в феноле при 150-180°С в течение 5-24 часов с последующим снятием защиты с использованием хлористоводородной кислоты, получая соединение (1А).
Способ получения F
Соединение (1Z), которое представляет собой соединение (1), где Е представляет собой -NRCO-, и оба Х и Y представляют собой N, также может быть получено представленным ниже реакционным способом:

(где R, R1, R2A, R2B, R2C, R2D, R3, R4 R5, A1, A2, R2b, R2c и Q имеют описанные выше значения).
Соединение (37В) подвергают взаимодействию с соединением (3) в количестве от одного эквивалента до избыточного количества в углеводородах, таких как бензол и толуол, простых эфирах, таких как диоксан и тетрагидрофуран, галогенированных углеводородах, таких как хлороформ, хлористый метилен и 1,2-дихлорэтан, или в N,N-диметилформамиде при температуре от 0°С до температуры кипения используемого растворителя в течение от нескольких часов до нескольких дней, получая таким образом соединение (37А). Особенно предпочтительно использовать пиразол-1-ил в качестве уходящей группы L в соединении (3), трет-бутоксикарбонил в качестве защитной группы и 1,2-дихлорэтан в качестве растворителя.
Соединение (37А) подвергают взаимодействию с хлорангидридом кислоты в количестве от одного эквивалента до избыточного количества в углеводородах, таких как бензол и толуол, простых эфирах, таких как диоксан и тетрагидрофуран, галогенированных углеводородах, таких как хлористый метилен, 1,2-дихлорэтан и хлороформ, в присутствии основания, такого как триэтиламин, N,N-диизопропилэтиламин или пиридин, с использованием при необходимости катализатора, такого как 4-диметиламинопиридин, при температуре от комнатной до температуры кипения используемого растворителя в течение от нескольких часов до нескольких дней с последующим снятием защитных групп R2b и R2c, когда они присутствуют в качестве защитных групп, с использованием известного способа, получая таким образом соединение (1Z).
Полученное таким образом соединение (1) может быть выделено и очищено известными методами, такими как концентрирование, преобразование жидкой фазы, разделение, экстракция растворителем, кристаллизация, перекристаллизация, фракционная перегонка или хроматография.
Исходное соединение (2) может быть получено в соответствии со следующей реакционной схемой.
(а) Когда Е представляет собой этенилен, и оба Х и Y представляют собой N в соединении (2):
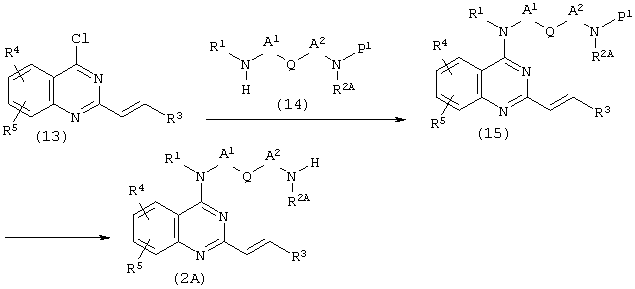
(где R1, R2, R3, R4, R5, А1, А2 и Q имеют описанные выше значения. Р1 представляет собой защитную группу).
Примеры защитной группы могут включать трет-бутоксикарбонил, бензилоксикарбонил и тому подобные.
Соединение (13) (полученное аналогично описанному на стр.13-15 в Международной Публикации WO 9909986) подвергают взаимодействию с амином (14) в количестве от одного эквивалента до избыточного количества в углеводородах, таких как бензол и толуол, простых эфирах, таких как диоксан и тетрагидрофуран, спиртах, таких как этанол и изопропанол, или в органическом растворителе, таком как N,N-диметилформамид, при необходимости в присутствии основания, такого как триэтиламин или N,N-диизопропилэтиламин, при температуре от комнатной температуры до температуры кипения используемого растворителя в течение от нескольких часов до нескольких дней с последующим снятием защиты хлористоводородной кислотой, трифторуксусной кислотой или путем гидрирования с использованием палладия-на-углероде, получая таким образом соединение (2А). Особенно предпочтительно, чтобы соединение (13) подвергали взаимодействию с 1-2 эквивалентами амина (14), в котором Р1 представляет собой трет-бутоксикарбонил, в толуоле в качестве растворителя в присутствии триэтиламина (ТЭА) при 100-130°С в течение 24-48 часов, а затем снятие защиты осуществляли хлористоводородной кислотой.
(b) Когда Е представляет собой этенилен, Х представляет собой СН, Y представляет собой N в соединении (2):
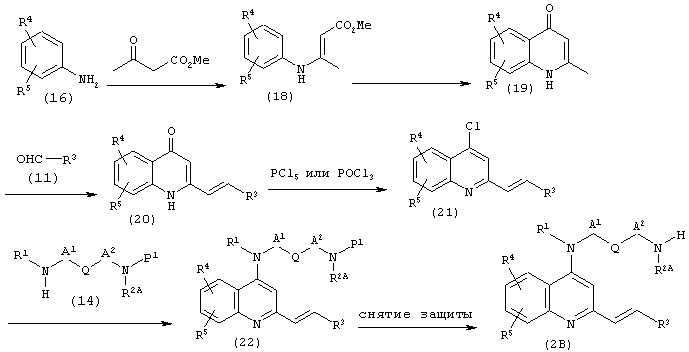
(где R1, R2A, R3, R4, R5, А1, A2, Q и Р1 имеют описанные выше значения).
Для получения соединения (19) используют известный способ (JACS 70, 4065 (1948); JACS 70, 2402 (1948); JOC 12, 456 (1947) и тому подобные), исходя из соединения (16).
Соединение (19) подвергают взаимодействию с альдегидом (11) в растворителе, таком как уксусный ангидрид, уксусная кислота или трифторуксусная кислота при температуре от комнатной температуры до температуры кипения используемого растворителя в течение 1-48 часов, предпочтительно в уксусном ангидриде в качестве растворителя при 80-100°С в течение 5-24 часов, получая таким образом соединение (20). Альдегид (11) может быть коммерчески доступным или может быть получен известным способом.
Соединение (20) подвергают взаимодействию с хлорирующим агентом, таким как оксихлорид фосфора или пентахлорид фосфора, без использования какого-либо растворителя или в растворителе, таком как толуол, ксилол или 1,2-дихлорэтан, при температуре от комнатной температуры до температуры кипения используемого растворителя или при температуре от комнатной температуры до температуры кипения используемого хлорирующего агента в том случае, когда растворитель не используется, в течение 1-24 часов, получая таким образом соединение (21). При такой методике при необходимости может присутствовать третичный амин, такой как диметиланилин или триэтиламин.
Соединение (21) подвергают взаимодействию с амином (14) в количестве от одного эквивалента до избыточного количества, так как описано выше в (а), а затем, при необходимости, проводят снятие защиты с использованием известного способа, получая соединение (2В). Особенно предпочтительно, чтобы соединение (21) взаимодействовало с 1-2 эквивалентами амина (14), в котором Р1 представляет собой трет-бутоксикарбонил, в толуоле в качестве растворителя в присутствии триэтиламина при 100-130°С в течение 24-48 часов, давая соединение (22), в котором затем снимают защиту с помощью трифторуксусной кислоты в хлористом метилене.
(с) Когда Е представляет собой этенилен, Х представляет собой N, и Y представляет собой СН в соединении (2):
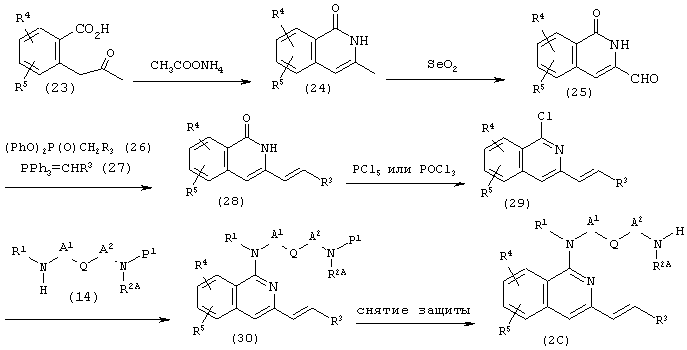
(где R1, R2A, R3, R4, R5, А1, A2, Q и Р1 имеют описанные выше значения).
Исходя из соединения 23, используя известный способ (J. Chem. Soc. Perkin Trans., 1, 1990, 1770), получают соединение (24).
Соединение (24) подвергают взаимодействию с 1-3 эквивалентами диоксида селена в простых эфирах, таких как диоксан и тетрагидрофуран, или спиртах, таких как этанол и изопропанол, при температуре от комнатной температуры до температуры кипения используемого растворителя в течение от нескольких часов до нескольких дней, предпочтительно в диоксане при 50-100°С в течение 5-48 часов, получая таким образом соединение (25).
Соединение (25) подвергают взаимодействию с соединением (26) или соединением (27) в растворителе, который не участвует в реакции, таком как диоксан или тетрагидрофуран, в присутствии основания, такого как н-бутиллитий, гидрид натрия или гексаметилдисилазид натрия, при температуре от -78°С до температуры кипения используемого растворителя в течение от нескольких часов до нескольких дней, предпочтительно в тетрагидрофуране при температуре от -20°С до комнатной температуры в течение 1-5 часов, получая таким образом соединение (28).
Аналогично методике описанной выше в части (b) соединение (28) взаимодействует с хлорирующим агентом, таким как оксихлорид фосфора или пентахлорид фосфора, в течение 1-24 часов, давая соединение (29). Соединение (29) взаимодействует с амином (14) в количестве от одного эквивалента до избыточного количества, как описано выше в части (а), а затем при необходимости проводят снятие защитных групп известным самим по себе способом, получая соединение (2С).
(d) Когда Е представляет собой -NRCO-, и оба Х и Y представляют собой N в соединении (2):
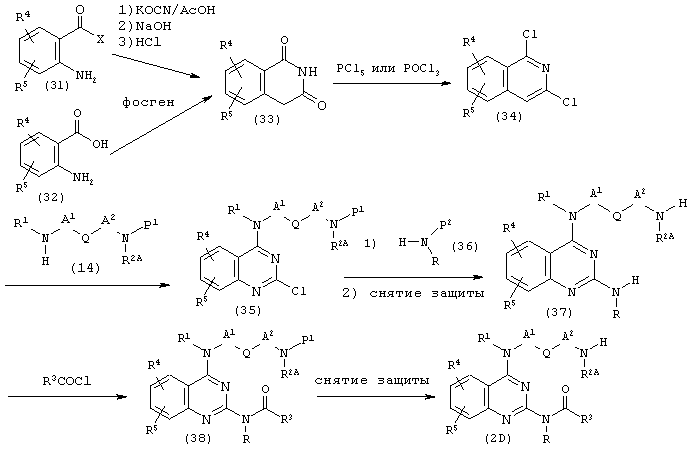
(где R, R1, R2A, R3, R4, R5, A1, A2, Q и Р1 имеют описанные выше значения. X представляет собой гидроксильную группу или аминогруппу. Р2 представляет собой атом водорода или защитную группу, такую как бензил или 4-метоксибензил).
Соединение (34) может быть получено из соединений (31) и (32) в соответствии с известным способом (патент Японии №2923742).
Соединение (34) подвергают взаимодействию с амином (14) в количестве от одного эквивалента до избыточного количества в таком же растворителе, как в описанной выше части (а), при необходимости в присутствии основания, такого как триэтиламин или N,N-диизопропилэтиламин, при температуре от 0°С до температуры кипения используемого растворителя в течение от нескольких часов до нескольких дней, предпочтительно в присутствии триэтиламина при комнатной температуре в течение 5-48 часов, получая при этом соединение (35).
Соединение (35) подвергают взаимодействию с амином (36) в количестве от одного эквивалента до избыточного количества в растворителе, имеющем высокую температуру кипения, таком как фенол или дифениловый простой эфир, при необходимости в присутствии основания, такого как триэтиламин или N,N-диизопропилэтиламин, при температуре от комнатной температуры до температуры кипения используемого растворителя в течение от нескольких часов до нескольких дней, или в углеводородах, таких как бензол, толуол и ксилол, простых эфирах, таких как диоксан и тетрагидрофуран, в присутствии металлического катализатора, такого как ацетат палладия, лиганда, такого как 2,2'-бис(дифенилфосфино)-1,1'-бинафтил, и основания, такого как трет-бутоксид натрия, при температуре от комнатной температуры до температуры кипения используемого растворителя в течение от нескольких часов до нескольких дней с последующим снятием защитной группы Р2, когда она присутствует в качестве защитной группы, с использованием способа, не оказывающего действия на Р1, при этом получая соединение (37).
Соединение (37) подвергают взаимодействию с хлорангидридом кислоты в количестве от одного эквивалента до избыточного количества в углеводородах, таких как бензол и толуол, простых эфирах, таких как диоксан и тетрагидрофуран, галогенированных углеводородах, таких как хлористый метилен, 1,2-дихлорэтан и хлороформ, в присутствии основания, такого как триэтиламин, N,N-диизопропилэтиламин или пиридин, при необходимости с использованием катализатора, такого как 4-диметиламинопиридин, при температуре от комнатной температуры до температуры кипения используемого растворителя в течение от нескольких часов до нескольких дней, получая таким образом соединение (38). Для получения соединения (2D) в соединение (38) снимают защитную группу с помощью известного самого по себе способа. Особенно предпочтительно проводить взаимодействие с использованием хлористого метилена в качестве растворителя в присутствии триэтиламина, используя каталитическое количество 4-диметиламинопиридина, при комнатной температуре в течение 24-48 часов. Хлорангидрид кислоты может быть коммерчески доступным или может быть получен известным способом.
(е) Когда Е представляет собой -NHCO-, и оба Х и Y представляют собой N в соединении (2):


(где R1, R2A, R3, R4, R5, A1, A2, Q и Р1 имеют описанные выше значения).
Исходя из соединения (39), для получения соединения (41) используют известный способ (см. JOC, 27, 4672 (1962).
Соединение (41) подвергают взаимодействию с амином (14) в количестве от одного эквивалента до избыточного количества аналогично описанному выше в части (а), получая соединение (42). Особенно предпочтительно проводить взаимодействие соединения (41) с 1-2 эквивалентами амина (14) в толуоле в качестве растворителя в присутствии триэтиламина (ТЭА) при 100-130°С в течение 24-48 часов.
Гидролизом соединения (42) известным способом получают соединение (43). Особенно предпочтительно проводить взаимодействие соединения (42) в этаноле в присутствии 1н. водного раствора гидроксида натрия при температуре от комнатной температуры до 60°С в течение 1-3 часов.
Соединение (43) подвергают взаимодействию с дифенилфосфорилазидом (DPPA) в спиртах, таких как этанол и бензиловый спирт, в присутствии основания, такого как триэтиламин или N,N-диизопропилэтиламин, при температуре от комнатной температуры до температуры кипения используемого растворителя в течение от нескольких часов до нескольких дней, получая таким образом соединение (44). Особенно предпочтительно проводить взаимодействие соединения (43) в кипящем с обратным холодильником этаноле в присутствии триэтиламина в течение 24-48 часов.
Соединение (44) гидролизуют известным способом, который не оказывает воздействия на Р1, получая таким образом соединение (45). Особенно предпочтительно проводить взаимодействие соединения (44) в метаноле в присутствии гидроксида калия при температуре от комнатной температуры до 60°С в течение 1-3 часов.
Соединение (45) подвергают взаимодействию с хлорангидридом кислоты в количестве от одного эквивалента до избыточного количества в углеводородах, таких как бензол и толуол, простых эфирах, таких как диоксан и тетрагидрофуран, галогенированных углеводородах, таких как хлористый метилен и 1,2-дихлорэтан, в присутствии основания, такого как триэтиламин, N,N-диизопропилэтиламин или пиридин, при необходимости с использованием катализатора, такого как 4-диметиламинопиридин, при температуре от комнатной температуры до температуры кипения используемого растворителя в течение от нескольких часов до нескольких дней, получая таким образом соединение (46). Особенно предпочтительно проводить взаимодействие с использованием хлористого метилена в качестве растворителя в присутствии триэтиламина, используя каталитическое количество 4-диметиламинопиридина, при комнатной температуре в течение 24-48 часов.
В соединении (46) известным способом снимают защитные группы, получая соединение (2Е). Когда Р1 представляет собой трет-бутоксикарбонил, предпочтительным является проводить взаимодействие с трифторуксусной кислотой в хлористом метилене при комнатной температуре в течение 1-5 часов. Когда Р1 представляет собой бензилоксикарбонил, является предпочтительным осуществлять гидрирование в метаноле в присутствии 5%-ного палладия-на-углероде при комнатной температуре при атмосферном давлении.
(f) Когда Е представляет собой этинилен, и оба Х и Y представляют собой N в соединении (2):
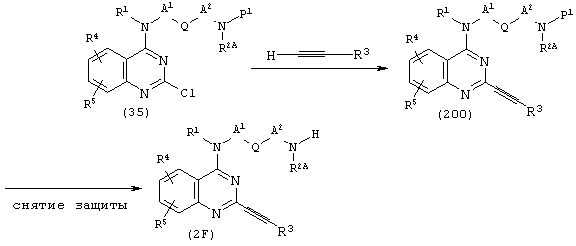
(где R1, R2A,R3, R4, R5, А1, A2, Q и Р1 имеют описанные выше значения).
Исходя из соединения (35), используют известный сам по себе способ (см. Heterocycles, 24, 2311 (1986) и тому подобные) для получения соединения (200). В соединении (200) снимают защитные группы известным способом, получая соединение (2F).
(g) Когда Е представляет собой -CONR-, и оба Х и Y представляют собой N в соединении (2):
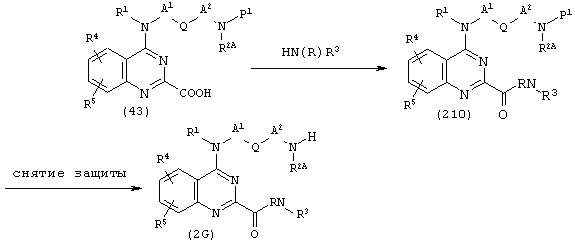
(где R, R1, R2A, R3, R4, R5, A1, A2, Q и Р1 имеют описанные выше значения).
Исходя из соединения (43), используют известный сам по себе способ амидирования для получения соединения (210). В соединении (210) затем снимают защитные группы известным способом, получая соединение (2G).
(h) Когда Е представляет собой -NRSO2-, и оба Х и Y представляют собой N в соединении (2):
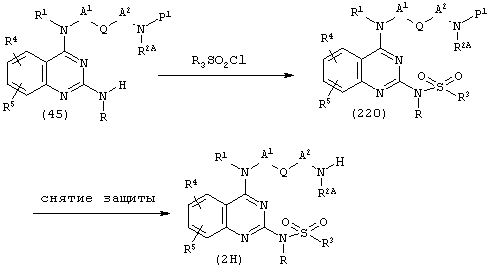
(где R, R1, R2A, R3, R4, R5, A1, А2, Q и Р1 имеют описанные выше значения).
Соединение (220) может быть получено, исходя из соединения (45), в соответствии с известным способом сульфонамидирования. В соединении (220) затем снимают защитные группы известным способом, получая соединение (2Н).
(i) Когда Е представляет собой -NRCONH-, и оба Х и Y представляют собой N в соединении (2):

(где R, R1, R2A R3, R4, R5, A1, A2, Q и Р1 имеют описанные выше значения).
Исходя из соединения (45), изоцианат подвергают взаимодействию известным способом, получая соединение (230). В соединении (230) затем снимают защитные группы известным способом, получая соединение (21).
Исходное соединение (3) может быть получено в соответствии с известным способом (J. Org. Chem., 34, 616, 1969; Synthesis 6, 460, 1988 и тому подобные).
Исходное соединение (4) может быть получено в соответствии со следующей реакционной схемой:

(где R1, R2a R2b, R2C, R2D, A1, A2, Q, L и Р1 имеют описанные выше значения).
Соединение (100) подвергают взаимодействию аналогично описанному выше способу получения А, получая исходное соединение (4). Данное исходное соединение (100) может быть коммерчески доступным или может быть получено с помощью известного способа.
Исходное соединение (120) может быть получено в соответствии со следующей реакционной схемой.

(где R1, R2A, R2B, R3, R4, R5, A1, A2, E, Q, X и Y имеют описанные выше значения).
Соединение (110) подвергают взаимодействию с R2BN=C=S в количестве от одного эквивалента до избыточного количества в растворителе, аналогично описанному выше в способе получения А, при необходимости в присутствии основания, такого как триэтиламин или N,N-диизопропилэтиламин, при температуре от комнатной температуры до температуры кипения используемого растворителя в течение от нескольких часов до нескольких дней, получая исходное соединение (120). Особенно предпочтительно проводить взаимодействие в хлористом метилене при комнатной температуре в течение 1-24 часов.
В описанном выше способе получения аминогруппа или гидроксильная группа при необходимости могут быть защищены обычно используемыми защитными группами, и после проведения взаимодействия защитная группа может быть снята на подходящей стадии способом, который известен сам по себе, таким как обработка кислотой или щелочью, или каталитическое гидрирование. Примеры аминозащитной группы могут включать бензил, бензилоксикарбонил, трет-бутоксикарбонил и трифторацетил. Примеры гидроксилзащитной группы могут включать метоксиметил, 2-метоксиэтоксиметил, метилтиометил, тетрагидропиранил, трет-бутил, бензил, триметилсилил, трет-бутилдиметилсилил и тому подобные.
Соль соединения (1) настоящего изобретения может быть получена известным способом. Например, гидрохлорид соединения (1) настоящего изобретения может быть получен обработкой соединения (1) настоящего изобретения раствором хлористого водорода в спирте или простом этиловом эфире с последующим выделением выпавших в осадок кристаллов фильтрованием, или в том случае, когда кристаллы не выпадают в осадок, путем концентрирования раствора до осаждения кристаллов, которые затем выделяют фильтрованием.
Поскольку соединение согласно настоящему изобретению, представленное формулой (1), как показано в описанных далее примерах испытаний, связывается с ноцицептиновым рецептором, оказывая агонистическое или антагонистическое действие, оно полезно в качестве анальгетика, противовоспалительного агента, диуретика, анестетика, анти-гипертензивного агента, подавляющего беспокойство агента, агента против ожирения, регулятора слуха, антидепрессанта, агента против слабоумия, агента, преодолевающего наркотическо-анальгетическую устойчивость.
Когда соединение настоящего изобретения вводят в качестве лекарственного средства, его можно вводить млекопитающему, включая человека, само по себе или в смеси с фармацевтически приемлемым нетоксичным, инертным носителем, например в виде фармацевтической композиции, содержащей соединение на уровне от 0,1% до 99,5%, предпочтительно от 0,5% до 90%.
В качестве носителя можно использовать один или несколько вспомогательных агентов для составления рецептуры, таких как твердый, полужидкий и жидкий разбавитель. Желательно, чтобы фармацевтическую композицию вводили в виде единичной препаративной лекарственной формы. Поскольку соединение настоящего изобретения является водорастворимым, его можно использовать не только в виде твердой рецептуры, но также в виде жидкой рецептуры (например, состав для внутривенных инъекций, вливания в мочевой пузырь, сироп для перорального введения). Фармацевтическую композицию можно вводить в ткани или перорально, местно (чрескожно) или ректально. Само собой разумеется, что используют препаративную лекарственную форму, подходящую для любого из описанных выше способов ведения. Например, предпочтительным является пероральное или внутривенное введение.
Хотя желательно, чтобы доза в качестве анальгетика могла быть подобрана в зависимости от состояния пациентов, включая возраст, вес тела, природу и степень боли, а также путь введения, обычно дневная доза в качестве активного ингредиента для взрослого человека составляет от 1 мг до 1000 мг для взрослого человека, предпочтительно от 1 мг до 500 мг для взрослого человека при пероральном введении, и обычно от 1 мг до 100 мг на взрослого человека, предпочтительно от 1 мг до 50 мг на взрослого человека при внутривенном введении. В некоторых случаях может оказаться достаточной более низкая доза или может потребоваться более высокая доза. Обычно дозу вводят один раз или несколько раз, разделенную на порции, или вводят внутривенно и непрерывно в течение периода от 1 до 24 часов в сутки.
Введение в ткань можно осуществлять при использовании жидкой единичной препаративной лекарственной формы, например в виде раствора или суспензии, состава для подкожной, внутримышечной, внутрь мочевого пузыря или внутривенной инъекции. Любой из данных составов может быть получен путем суспендирования или растворения определенного количества соединения в нетоксичном жидком носителе, таком как водная или масляная среда, совместимом с целью инъекции, с последующей стерилизацией указанной суспензии или раствора. Альтернативно определенное количество соединения помещают в ампулу, которую затем стерилизуют вместе с ее содержимым и затем запаивают. Для восстановления влагосодержания или смешивания непосредственно перед применением порошкообразный или лиофильно высушенный активный ингредиент снабжают дополнительной ампулой или носителем. Также для получения изотоничного раствора для инъекции возможно добавлять нетоксичную соль или раствор соли. Также можно применять стабилизатор, консервант, эмульгатор и тому подобное.
Пероральное введение может быть осуществлено в виде твердой или жидкой лекарственной формы, такой как частица, порошок, таблетка, покрытая сахаром таблетка, капсула, гранула, суспензия, жидкость, сироп, капли, буккальная рецептура, суппозиторий или другая препаративная лекарственная форма. Частицы получают распылением активного ингредиента в частицы подходящего размера. Порошок может быть получен распылением активного ингредиента в частицы подходящего размера с последующим смешиванием с фармацевтическим носителем, таким как пищевой углевод, включая крахмалы или маннит, которые также были распылены в частицы подходящего размера. При необходимости могут быть добавлены вкусовые добавки, консерванты, диспергирующие агенты, красители, ароматизаторы и тому подобное.
Капсула может быть получена путем заполнения капсулы, такой как желатиновая капсула, частицами или порошком, которые предварительно были распылены, как описано выше, или гранулами, полученными, как описано в разделе для таблетки. Также возможно перед процедурой заполнения смешать с распыленным веществом добавку, такую как смазочное вещество, сжижающий агент, такой как коллоидный диоксид кремния, тальк, стеарат магния, стеарат кальция или твердый полиэтиленгликоль. С целью усиления доступности лекарственного средства при проглатывании капсулы может быть добавлен дезинтегрирующий или солюбилизирующий агент, такой как карбоксиметилцеллюлоза, кальцийкарбоксиметилцеллюлоза, низкозамещенная гидроксипропилцеллюлоза, кросскармелоза натрия, карбоксикрахмал натрия, карбонат кальция или карбонат натрия.
Мелкораспыленный порошок может быть суспендирован и диспергирован в растительном масле, полиэтиленгликоле, глицерине и поверхностно-активном веществе, а затем инкапсулирован в желатиновую оболочку с получением таким образом мягкой капсулы. Таблетку получают путем приготовления порошковой смеси, превращения ее в гранулу или кусочки неправильной формы, добавления дезинтегранта или смазочного вещества, а затем прессования в таблетку. Порошковую смесь получают смешиванием распыленного подходящим образом вещества с разбавителем или основой, описанными выше, при необходимости вместе со связующим веществом (например, карбоксиметилцеллюлозой натрия, гидроксипропилцеллюлозой, метилцеллюлозой, гидроксипропилметилцеллюлозой, желатином, поливинилпирролидоном, поливиниловым спиртом и тому подобным), замедлителем разбавления (например, парафином, воском, отвержденным касторовым маслом и тому подобным), промотором ресорбции (например, четвертичной солью) или адсорбентом (например, бентонитом, каолином, дифосфатом кальция и тому подобным). Порошковая смесь может быть гранулирована путем смачивания связующим веществом, таким как сироп, крахмальный клей, аравийская камедь, раствор целлюлозы или раствор полимера, а затем принудительно пропущена через сито. Вместо описанной выше процедуры гранулирования порошка можно использовать другой способ, в котором смесь первоначально подвергают воздействию таблетирующего устройства с образованием морфологически незавершенных кусочков, которые затем измельчают.
Полученные таким образом гранулы для предотвращения какого-либо слипания друг с другом могут содержать в качестве смазочного вещества стеариновую кислоту, стеараты, тальк, минеральное масло и тому подобное. Смазанная таким образом смесь затем прессуется в таблетки.
Полученная таким образом плоская таблетка может быть покрыта пленкой или покрыта сахаром.
Активный ингредиент может быть смешан с жидким инертным носителем, а затем непосредственно подвергнут прессованию в таблетки без использования описанных выше способа гранулирования или преобразования в куски. Также можно использовать прозрачную или полупрозрачную защитную пленку в виде шеллаковой уплотняющей пленки, пленку сахара или полимерного материала и блестящую пленку воска.
Другие пероральные препаративные лекарственные формы, такие как раствор, сироп и эликсир, могут быть рецептурированы в виде единичных препаративных лекарственных форм, определенное количество которых содержит определенное количество лекарственного средства. Сироп получают растворением соединения в ароматизированном водном растворе, тогда как эликсир получают с использованием нетоксичного спиртового носителя. Суспензию получают путем диспергирования соединения в нетоксичном носителе. При необходимости могут быть добавлены вспомогательные вещества, такие как солюбилизирующий агент, эмульгатор (например, этоксилированные изостеариловые спирты, сложные эфиры полиоксиэтиленсорбита), консервант и вкусовая добавка (например, мятное масло, сахарин).
При желании, пероральная единичная препаративная лекарственная рецептура также может быть инкапсулирована. Такая рецептура может быть покрыта пленкой или внедрена в полимер или воск для получения пролонгированной активности или замедленного высвобождения активного ингредиента.
Ректальное введение можно осуществлять с использованием суппозитория, полученного смешиванием соединения с водорастворимым или не растворимым в воде твердым веществом, имеющим низкую температуру плавления, таким как полиэтиленгликоль, масло какао, высшие сложные эфиры (например, миристил пальмилат), а также их смесями.
Лучший способ осуществления изобретения
Далее настоящее изобретение будет описано более подробно со ссылкой на примеры получения типичных исходных веществ (Ссылочные примеры), примеры получения соединения согласно настоящему изобретению (Примеры), примеры композиций и примеры испытаний, которыми оно не ограничивается. Отметим, что оптическое вращение измеряли при 20°С.
Ссылочный пример 1
N-трет-Бутоксикарбонил-1,6-гександиамин
Раствор 5,9 г 1,6-гександиамина в 30 мл тетрагидрофурана объединяли с 30 мл 2%-ного водного раствора гидроксида натрия и охлаждали до 0°С. Данную смесь обрабатывали по каплям раствором 4,46 г ди-трет-бутилдикарбоната в 30 мл тетрагидрофурана и перемешивали в течение 15 часов, постепенно нагревая до комнатной температуры. Реакционный раствор объединяли с водой, экстрагировали этилацетатом, сушили над сульфатом натрия и затем концентрировали. Остаток очищали хроматографией на силикагеле, получая 3,1 г целевого соединения.
Следующие соединения получали способом, аналогичным способу, использованному в ссылочном примере 1:
N-трет-Бутоксикарбонил-1,2-этилендиамин
N-трет-Бутоксикарбонил-1,3-пропандиамин
N-трет-Бутоксикарбонил-1,4-бутандиамин
N-трет-Бутоксикарбонил-1,5-пентандиамин
N-трет-Бутоксикарбонил-1,7-гептандиамин
N-трет-Бутоксикарбонил-1,8-октандиамин
N-трет-Бутоксикарбонилпиперазин
Цис-N-трет-Бутоксикарбонил-1,2-циклогександиамин
Транс-N-трет-Бутоксикарбонил-1,2-циклогександиамин
Цис-N-трет-Бутоксикарбонил-1,3-циклогександиамин
Транс-N-трет-Бутоксикарбонил-1,3-циклогександиамин
Цис-N-трет-Бутоксикарбонил-1,4-циклогександиамин
Транс-N-трет-Бутоксикарбонил-1,4-циклогександиамин
Ссылочный пример 2
(1S,2R)-2-трет-Бутоксикарбониламино-циклогексиламин
Стадия 1
(1R,2R)-N-трет-Бутоксикарбонил-2-бензилоксициклогексиламин
Раствор 3,0 г (1R,2R)-2-бензилоксициклогексиламина в 30 мл тетрагидрофурана объединяли с 30 мл 2%-ного водного раствора гидроксида натрия и охлаждали до 0°С. Эту смесь обрабатывали по каплям раствором 4,46 г ди-трет-бутилдикарбоната в 30 мл тетрагидрофурана и перемешивали в течение 15 часов, постепенно нагревая до комнатной температуры. Реакционный раствор объединяли с водой и экстрагировали этилацетатом. После сушки над сульфатом магния путем концентрирования получали 4,45 г целевого соединения.
Стадия 2
(1R,2R)-N-трет-Бутоксикарбонил-2-гидроксициклогексиламин
Раствор 3,0 г (1R,2R)-N-трет-бутоксикарбонил-2-бензилоксициклогексиламина в 100 мл метанола объединяли с 300 мг 5%-ного палладия-на-углероде и гидрировали при комнатной температуре при атмосферном давлении. Через 48 часов палладий-на-углероде отфильтровывали и фильтрат концентрировали. Остаток подвергали хроматографии на силикагеле (н-гексан:этилацетат = 2:1), получая 2,0 г целевого соединения.
Стадия 3
(1R,2S)-N-трет-Бутоксикарбонил-2-фталиминоциклогексиламин
В атмосфере аргона раствор 500 мг (1R,2R)-N-трет-бутоксикарбонил-2-гидроксициклогексиламина в 20 мл безводного тетрагидрофурана объединяли с 913 мг трифенилфосфина и 513 мг фталимида, обрабатывали по каплям 1,58 мл 40%-ного раствора диэтилового эфира азодикарбоновой кислоты в толуоле при охлаждении льдом и перемешивали в течение 24 часов, давая возможность постепенно нагреваться до комнатной температуры. После отгонки растворителя остаток очищали колоночной хроматографией на силикагеле(н-гексан:этилацетат = 2:1), получая 550 мг целевого соединения.
Стадия 4
(1S,2R)-2-трет-Бутоксикарбониламиноциклогексиламин
Раствор 2,50 г (1R,2S)-N-трет-бутоксикарбонил-2-фталиминоциклогексиламина в 80 мл этанола объединяли с 1,82 г гидразингидрата и нагревали при кипении с обратным холодильником в течение 3 часов. После отгонки растворителя остаток объединяли с 10%-ным водным раствором гидроксида натрия и экстрагировали хлороформом. После концентрирования остаток очищали колоночной хроматографией на силикагеле (хлороформ:метанол = 10:1), получая 1,60 г целевого соединения.
Следующие соединения получали способом, аналогичным способу ссылочного примера 2.
(1R,2S)-2-трет-Бутоксикарбониламиноциклогексиламин
(1R,2S)-2-трет-Бутоксикарбониламиноциклопентиламин
(1S,2R)-2-трет-Бутоксикарбониламиноциклопентиламин
4-Амино-N-трет-бутоксикарбонилпиперидин
Ссылочный пример 3
1-трет-Бутоксикарбониламино-6-аминогептан
Стадия 1
6-трет-Бутоксикарбониламино-1-гексанол
Раствор 5,1 г 6-амино-1-гексанола в 100 мл хлороформа обрабатывали по каплям 10,4 г ди-трет-бутилдикарбоната и перемешивали в течение 12 часов. Реакционный раствор концентрировали и остаток промывали н-гексаном, получая 9,40 г целевого соединения в виде белых кристаллов.
Стадия 2
6-трет-Бутоксикарбониламиногексаналь
Раствор 1,0 г б-трет-бутоксикарбониламино-1-гексанола в 20 мл хлористого метилена объединяли с 3 г молекулярных сит емкостью 4 ангстрема, 808 мг N-метилморфолин-N-оксида и каталитическим количеством перрутената тетрапропиламмония и перемешивали в течение 24 часов. Реакционный раствор фильтровали через целит и концентрировали. Остаток очищали колоночной хроматографией на силикагеле (н-гексан:этилацетат = 2:1), получая 660 мг целевого соединения.
Стадия 3
1-трет-Бутоксикарбониламино-6-гидроксигептан
В атмосфере аргона раствор 650 мг 6-трет-бутоксикарбониламиногексаналя в 10 мл безводного тетрагидрофурана охлаждали до -78°С и обрабатывали по каплям 6,8 мл метилмагнийбромида (1,0 М раствор в тетрагидрофуране). Через 2 часа реакционный раствор объединяли с водой, экстрагировали этилацетатом и сушили. После концентрирования остаток очищали колоночной хроматографией на силикагеле (н-гексан:этилацетат = 2:1), получая 280 мг целевого соединения.
Стадия 4
1-трет-Бутоксикарбониламино-6-фталиминогептан
В атмосфере аргона раствор 270 мг 1-трет-бутоксикарбониламино-6-гидроксигептана в 7 мл безводного тетрагидрофурана объединяли с 367 мг трифенилфосфина и 258 мг фталимида, обрабатывали по каплям 0,80 мл 40%-ного раствора диэтилового эфира азодикарбоновой кислоты в толуоле при охлаждении льдом и перемешивали в течение 24 часов, давая возможность постепенно нагреваться до комнатной температуры. После отгонки растворителя остаток очищали колоночной хроматографией на силикагеле (н-гексан: этилацетат = 4:1), получая 321 мг целевого соединения.
Стадия 5
1-трет-Бутоксикарбониламино-6-аминогептан
Раствор 321 мг 1-трет-бутоксикарбониламино-6-фталимидогептана в 10 мл этанола объединяли с 89 мг гидразингидрата и нагревали при кипении с обратным холодильником в течение 4 часов. После отгонки растворителя остаток объединяли с 10%-ным водным раствором гидроксида натрия и экстрагировали хлороформом. После сушки над сульфатом натрия с последующим концентрированием остаток очищали колоночной хроматографией на силикагеле (хлороформ:метанол = 10:1), получая 202 мг целевого соединения.
Ссылочный пример 4
цис-4-Трифторацетиламинометилциклогексиламин
Стадия 1
Гидрохлорид метилового эфира цис-4-аминоциклогексанкарбоновой кислоты
Раствор 2,0 г цис-4-аминоциклогексанкарбоновой кислоты в 20 мл метанола объединяли с 3,57 мл хлористого тионила и перемешивали в течение 3 часов. Реакционный раствор концентрировали и остаток промывали простым этиловым эфиром, получая 2,64 г целевого соединения в виде бесцветных кристаллов.
Стадия 2
Метиловый эфир цис-4-(трет-бутоксикарбониламино) циклогексанкарбоновой кислоты
Раствор 2,64 г гидрохлорида метилового эфира цис-4-аминоциклогексанкарбоновой кислоты в 30 мл хлороформа объединяли с 1,52 г триэтиламина и к данному раствору добавляли по каплям 3,27 г ди-трет-бутилдикарбоната. Через 3 часа реакционный раствор объединяли с водой, экстрагировали хлороформом и затем сушили над сульфатом магния. Растворитель отгоняли и остаток очищали колоночной хроматографией на силикагеле (хлороформ), получая 3,62 г целевого соединения.
Стадия 3
цис-N-(трет-Бутоксикарбонил)-4-гидроксиметилциклогексиламин
В атмосфере аргона суспензию 1,29 г литийалюминийгидрида в 40 мл безводного этилового эфира при охлаждении льдом обрабатывали по каплям раствором 5,80 г метилового эфира цис-4-(трет-бутоксикарбониламино)циклогексанкарбоновой кислоты в 20 мл безводного этилового эфира и перемешивали в течение 3 часов, давая постепенно возможность нагреваться до комнатной температуры. Реакционный раствор охлаждали до 0°С, объединяли с небольшим количеством воды для разложения избытка литийалюминийгидрида. Нерастворимые вещества отфильтровывали через целит и фильтрат концентрировали, затем остаток промывали н-гексаном, получая 3,60 г целевого соединения в виде бесцветных кристаллов.
Стадия 4
цис-N-(трет-Бутоксикарбонил)-4-фталиминометилциклогексиламин
В атмосфере аргона раствор 3,60 г цис-N-(трет-бутоксикарбонил)-4-гидроксиметилциклогексиламина в 50 мл безводного тетрагидрофурана объединяли с 4,12 г трифенилфосфина и 2,31 г фталимида, обрабатывали по каплям 6,84 мл 40%-ного раствора диэтилового эфира азодикарбоновой кислоты в толуоле при охлаждении льдом и перемешивали в течение 15 часов, давая возможность постепенно нагреваться до комнатной температуры. Растворитель отгоняли и остаток очищали колоночной хроматографией на силикагеле (хлороформ), получая 3,45 г целевого соединения.
Стадия 5
цис-N-(трет-Бутоксикарбонил)-4-трифторацетиламинометилциклогексиламин
Раствор 3,45 г цис-N-(трет-бутоксикарбонил)-4-фталиминометилциклогексиламина в 35 мл этанола объединяли с 0,72 г гидразингидрата и нагревали при кипении с обратным холодильником в течение 5 часов. Растворитель отгоняли и остаток объединяли с 10%-ным водным раствором гидроксида натрия и экстрагировали хлороформом. После концентрирования раствор остатка в 25 мл метанола объединяли с 1,17 г триэтиламина и 1,64 г этилового эфира трифторуксусной кислоты и перемешивали в течение 15 часов. После концентрирования реакционного раствора остаток очищали колоночной хроматографией на силикагеле (хлороформ:метанол = 40:1), получая 2,50 г целевого соединения.
Стадия 6
цис-4-Трифторацетиламинометилциклогексиламин
Раствор 0,53 г цис-N-(трет-бутоксикарбонил)-4-трифторацетиламинометилциклогексиламина в хлористом метилене объединяли с 2 мл трифторуксусной кислоты и перемешивали в течение 2 часов. Реакционный раствор подщелачивали добавлением насыщенного раствора гидрокарбоната натрия и затем экстрагировали хлороформом. После сушки над сульфатом натрия получали 0,19 г целевого соединения в виде бледно-желтого масла.
Следующее соединение получали способом, аналогичным описанному в ссылочном примере 4.
цис-2-Трифторацетиламинометилциклогексиламин
Ссылочный пример 5
транс-N-трет-Бутоксикарбонил-1,4-бис(аминометил)циклогексан
Стадия 1
Диметиловый эфир транс-1,4-циклогександикарбоновой кислоты
При охлаждении льдом 25 мл метанола обрабатывали по каплям 6 мл хлористого тионила и перемешивали в течение 1 часа. Данный раствор объединяли с 3,44 г транс-1,4-циклогександикарбоновой кислоты и перемешивали при комнатной температуре в течение 20 часов. После концентрирования реакционного раствора остаток объединяли со льдом и подщелачивали добавлением 10%-ного водного раствора гидроксида натрия. Смесь экстрагировали хлороформом, сушили и затем концентрировали. Остаток промывали н-гексаном, получая 3,9 г целевого соединения.
Стадия 2
транс-1,4-Бис(гидроксиметил)циклогексан
В токе аргона суспензию 2,96 г литийалюминийгидрида в 100 мл безводного тетрагидрофурана обрабатывали по каплям раствором 3,9 г диметилового эфира транс-1,4-циклогександикарбоновой кислоты в безводном тетрагидрофуране при -20°С и перемешивали в течение 2,5 часов. Реакционный раствор объединяли с ледяной водой для завершения реакции и нерастворимые вещества отфильтровывали через целит. После сушки над сульфатом магния остаток концентрировали, получая 2,80 г целевого соединения.
Стадия 3
транс-1,4-Бис(фталиминометил)циклогексан
Раствор 1,60 г транс-1,4-бис(гидроксиметил)циклогексана в 200 мл толуола объединяли с 6,98 г трифенилфосфина, обрабатывали по каплям 3,92 г фталимида и 11,58 мл 40%-ного раствора диэтилового эфира азодикарбоновой кислоты в толуоле при охлаждении льдом и перемешивали в течение 18 часов. Остаток объединяли с водой, экстрагировали хлороформом, сушили и затем концентрировали. Остаток промывали этиловым эфиром и метанолом, получая 3,53 г целевого соединения.
Стадия 4
транс-N,N-Ди-трет-бутоксикарбонил-1,4-бис(аминометил) циклогексан
Суспензию 3,50 г транс-1,4-бис(фталиминометил) циклогексана в 50 мл этанола объединяли с 4,35 г гидразингидрата и нагревали при кипении с обратным холодильником в течение 2 часов. Реакционный раствор концентрировали, объединяли с 20 мл 10%-ного водного раствора гидроксида натрия и 30 мл 1,4-диоксана, обрабатывали по каплям 6,50 г ди-трет-бутилдикарбоната при охлаждении льдом и затем перемешивали при комнатной температуре в течение 2 часов. После экстрагирования хлороформом экстракт сушили и концентрировали. Остаток промывали н-гексаном и сушили, получая 2,80 г целевого соединения.
Стадия 5
транс-N-трет-Бутоксикарбонилбис-1,4-(аминометил)циклогексан
Раствор 2,75 г транс-N,N'-ди-трет-бутоксикарбонил-1,4-бис(аминометил)циклогексана в 40 мл хлористого метилена объединяли с 5 мл 4н. раствора хлористого водорода в этилацетате, перемешивали при комнатной температуре в течение 2 часов и затем реакционный раствор концентрировали. Полученный остаток объединяли с 40 мл 10%-ного водного раствора гидроксида натрия и 20 мл 1,4-диоксана, обрабатывали по каплям 0,90 г ди-трет-бутилдикарбоната при охлаждении льдом и перемешивали при комнатной температуре в течение 2 часов. После экстрагирования хлороформом экстракт сушили и концентрировали. Остаток очищали колоночной хроматографией на силикагеле (хлороформ:метанол = 20:1), получая 0,25 г целевого соединения.
Следующее соединение получали способом, аналогичным описанному в ссылочном примере 5.
цис-N-трет-Бутоксикарбонил-1,4-бис(аминометил)циклогексан
Пример 1
Дигидрохлорид цис-N-Амидино-2-[2-(4-хлорстирил)-6-метоксихиназолин-4-ил]аминоциклогексиламина
Стадия 1
цис-N-трет-Бутоксикарбонил-2-[2-(4-хлорстирил)-6-метоксихиназолин-4-ил]аминоциклогексиламин
Раствор 391 мг 4-хлор-2-(4-хлорстирил)-6-метоксихиназолина, 379 мг цис-2-(трет-бутоксикарбонил) аминоциклогексиламина и 358 мг триэтиламина в 20 мл толуола объединяли с каталитическим количеством 4-диметиламинопиридина и нагревали при кипении с обратным холодильником в течение 20 часов. После отгонки реакционного раствора остаток объединяли с водой, экстрагировали хлороформом, сушили над сульфатом магния и концентрировали. Остаток очищали колоночной хроматографией на силикагеле (хлороформ:метанол = 50:1), получая 580 мг целевого соединения.
Стадия 2
цис-2-[2-(4-Хлорстирил)-6-метоксихиназолин-4-ил] аминоциклогексиламин
Раствор 520 мг цис-N-трет-бутоксикарбонил-2-[2(4-хлорстирил)-6-метоксихиназолин-4-ил]аминоциклогексиламина в 10 мл метанола объединяли с 5 мл 4н. раствора хлористого водорода в этилацетате и проводили взаимодействие при 50°С в течение 24 часов. После отгонки растворителя остаток подщелачивали добавлением 10%-ного водного раствора гидроксида натрия и экстрагировали хлороформом. После концентрирования остаток очищали колоночной хроматографией на силикагеле (хлороформ:метанол = 10:1), получая 378 мг целевого соединения.
Стадия 3
цис-N-[N,N'-Бис(трет-бутоксикарбонил)]амидино-2-[2-(4-хлорстирил)-6-метоксихиназолин-4-ил]аминоциклогексиламин
Раствор 400 мг цис-2-[2-(4-хлорстирил)-6-метоксихиназолин-4-ил]аминоциклогексиламина в 5 мл дихлорэтана и 1 мл N,N-диметилформамида объединяли с 273 мг N,N'-бис(трет-бутоксикарбонил)-1Н-пиразол-1-карбоксиамидина и перемешивали при комнатной температуре в течение 15 часов. Реакционный раствор объединяли с водой и экстрагировали этилацетатом. Органический слой затем промывали водой и насыщенным раствором соли и сушили над сульфатом магния. После концентрирования остаток очищали колоночной хроматографией на силикагеле (хлороформ:метанол = 30:1), получая 580 мг целевого соединения.
Стадия 4
Дигидрохлорид цис-N-амидино-2-[2-(4-хлорстирил)-6-метоксихиназолин-4-ил]аминоциклогексиламина
Раствор 570 мг цис-N-[N,N'-бис(трет-бутоксикарбонил)] амидино-2-[2-(4-хлорстирил)-6-метоксихиназолин-4-ил] аминоциклогексиламина в 8 мл метанола и 8 мл хлороформа объединяли с 5 мл 4н. раствора хлористого водорода в этилацетате и осуществляли взаимодействие при 50°С в течение 48 часов. После концентрирования проводили кристаллизацию из этилацетата, получая 310 мг целевого соединения в виде бледно-желтых кристаллов.
Катион FAB-масс-спектр (бомбардировка быстрыми атомами) m/z: 451[M+H]+.
Данные элементного анализа (для C24H27N6ClO·2HCl·H2O
Вычисленное значение (%) С: 51,48 Н: 5,94 N: 15,01
Найденное значение (%) С: 51,75 Н: 5,64 N: 15,01
Пример 2
Дигидрохлорид цис-N-амидино-2-[2-(4-хдорбензоиламино)-6-метоксихиназолин-4-ил]аминоциклогексиламин
Стадия 1
цис-N-трет-Бутоксикарбонил-2-(2-этоксикарбонил-6-метоксихиназолин-4-ил]аминоциклогексиламин
Раствор 1,96 г 4-хлор-2-этоксикарбонил-6-метоксихиназолина в 70 мл толуола объединяли с 1,58 г цис-2-трет-бутоксикарбониламиноциклогексиламина и 0,74 г триэтиламина и нагревали при кипении с обратным холодильником в течение 15 часов. После концентрирования смесь объединяли с водой, экстрагировали хлороформом и сушили. После отгонки растворителя остаток очищали колоночной хроматографией на силикагеле (хлороформ), получая 2,70 г целевого соединения.
Стадия 2
цис-N-трет-Бутоксикарбонил-2-(2-карбокси-6-метоксихиназолин-4-ил]аминоциклогексиламин
Раствор 1,70 г цис-N-трет-бутоксикарбонил-2(2-этоксикарбонил-6-метоксихиназолин-4-ил)аминоциклогексиламина в 5 мл метанола объединяли с 5 мл 1н. водного раствора гидроксида натрия и перемешивали при комнатной температуре в течение 3 часов. После доведения значения рН до 5 путем добавления к реакционному раствору 1н. водного раствора гидросульфата калия, его затем экстрагировали хлороформом и сушили. После отгонки растворителя остаток очищали колоночной хроматографией на силикагеле (хлороформ:метанол = 10:1), получая 1,10 г целевого соединения.
Стадия 3
цис-N-трет-Бутоксикарбонил-2(2-этоксикарбониламино-6-метоксихиназолин-4-ил)аминоциклогексиламин
Раствор 1,03 г цис-М-трет-бутоксикарбонил-2-(2-карбокси-6-метоксихиназолин-4-ил)аминоциклогексиламина в 10 мл тетрагидрофурана объединяли с 0,82 г дифенилфосфорилазида, 1,14 г этанола и 0,3 г триэтиламина и проводили взаимодействие при 80°С в течение 72 часов. Реакционный раствор концентрировали и затем объединяли с водой, экстрагировали хлороформом и сушили. После отгонки растворителя остаток очищали колоночной хроматографией на силикагеле (хлороформ:метанол = 50:1), получая 800 мг целевого соединения.
Стадия 4
цис-2-(2-Амино-6-метоксихиназолин-4-ил)амино-N-трет-бутоксикарбонилциклогексиламин
Раствор 300 мг цис-N-трет-бутоксикарбонил-2(2-этоксикарбониламино-6-метоксихиназолин-4-ил) аминоциклогексиламина в 10 мл метанола объединяли с 50 мг порошкообразного гидроксида калия и перемешивали при комнатной температуре в течение 3 часов. Реакционный раствор нейтрализовали добавлением насыщенного водного раствора хлорида аммония и затем экстрагировали хлороформом и сушили. После отгонки растворителя остаток очищали колоночной хроматографией на силикагеле (хлороформ:метанол = 10:1), получая 250 мг целевого соединения.
Стадия 5
цис-N-трет-Бутоксикарбонил-2-[2-(4-хлорбензоиламино)-6-метоксихиназолин-4-ил]аминоциклогексиламин
Раствор 68 мг 4-хлорбензоилхлорида и 200 мг диизопропилэтиламина в 8 мл хлористого метилена объединяли с 150 мг цис-2-(2-амино-6-метоксихиназолин-4-ил)амино-N-трет-бутоксикарбонилциклогексиламина и перемешивали при комнатной температуре в течение 15 часов. Реакционный раствор объединяли с водой, экстрагировали хлороформом и сушили. После отгонки растворителя остаток очищали колоночной хроматографией на силикагеле (хлороформ:метанол = 30:1), получая 120 мг целевого соединения.
Стадия 6
цис-2-[2-(4-Хлорбензоиламино)-6-метоксихиназолин-4-ил]аминоциклогексиламин
Раствор 120 мг цис-N-трет-бутоксикарбонил-2-[2-(4-хлорбензоиламино)-6-метоксихиназолин-4-ил] аминоциклогексиламина в 5 мл хлористого метилена объединяли с 2 мл трифторуксусной кислоты и осуществляли взаимодействие при комнатной температуре в течение 1 часа. Реакционный раствор подщелачивали добавлением насыщенного раствора гидрокарбоната натрия, экстрагировали хлороформом и сушили. После отгонки растворителя остаток очищали колоночной хроматографией на силикагеле (хлороформ:метанол = 30:1), получая 80 мг целевого соединения.
Стадия 7
цис-N-[N,N'-Бис(трет-бутоксикарбонил)]амидино-2-[2(4-хлорбензоиламино)-6-метоксихиназолин-4-ил]аминоциклогексиламин
Раствор 80 мг цис-2-[2-(4-хлорбензоиламино)-6-метоксихиназолин-4-ил]аминоциклогексиламина в 5 мл дихлорэтана и 1 мл N,N-диметилформамида объединяли с 58 мг N,N'-бис(трет-бутоксикарбонил) -1Н-пиразол-1-карбоксиамидина и перемешивали при комнатной температуре в течение 15 часов. Реакционный раствор объединяли с водой и экстрагировали этилацетатом. Органический слой промывали водой и насыщенным раствором соли и затем сушили над сульфатом магния. После концентрирования остаток очищали колоночной хроматографией на силикагеле (хлороформ:метанол = 30:1), получая 120 мг целевого соединения.
Стадия 8
Дигидрохлорид цис-N-амидино-2[2(4-хлорбензоиламино)-6-метоксихиназолин-4-ил]аминоциклогексиламин
Раствор 120 мг цис-N-[N,N'-бис(трет-бутоксикарбонил) ] амидино-2-[2-(4-хлорбензоиламино)-б-метоксихиназолин-4-ил] аминоциклогексиламина в 5 мл метанола и 5 мл хлороформа объединяли с 3 мл 4н. раствора хлористого водорода в этилацетате и проводили взаимодействие при 50°С в течение 72 часов. После концентрирования проводили обработку смесью метанол-этиловый эфир, получая 22 мг целевого соединения в виде бледно-желтого порошка.
Катион FAB-Macc-спектр m/z: 468[M+H]+.
Данные элементного анализа (для С23Н26ClN7O2·2НСl·3Н2O)
Вычисленное значение (%) С: 46,43 Н: 5,76 N: 16,48
Найденное значение (%) С: 46,45 Н: 5,55 N: 16,25
Пример 3
Дигидрохлорид цис-N-ацетимид-2-[2-(4-хлорстирил-6-метоксихиназолин-4-ил]аминоциклогексиламин
Раствор 50 мг цис-2-[2-(4-хлорстирил)-6-метоксихиназолин-4-ил]аминоциклогексиламина в 8 мл этанола объединяли с 76 мг этилацетимидата и 123 мг триэтиламина и нагревали при кипении с обратным холодильником в течение 3 часов. После концентрирования реакционного раствора остаток очищали колоночной хроматографией на силикагеле (хлороформ:метанол:водный аммиак = 100:10:1). Раствор 50 мг полученного цис-N-ацетимид-2-[2-(4-хлорстирил)-6-метоксихиназолин-4-ил]аминоциклогексиламина в 3 мл метанола объединяли с 1 мл 4н. раствора хлористоводородной кислоты в этилацетате и перемешивали в течение 30 минут. После концентрирования проводили кристаллизацию в простом этиловом эфире, получая 45 мг целевого соединения в виде бледно-желтых кристаллов.
Данные элементного анализа (для C25H28ClN5O·2HCl·1,5H2O)
Катион FAB-масс-спектр m/z: 450 [М+Н]+
Вычисленное значение (%) С: 65,28 Н: 6,14 N: 12,18
Найденное значение (%) С: 65,23 Н: 5,92 N: 12,12
Пример 4
Тригидрохлорид цис-4-гуанидинометил-N-{2[2(2-пиридил)этенил]-6-метоксихиназолин-4-ил}циклогексиламина
Стадия 1
цис-4-Трифторацетиламинометил-N-{2-[2-(2-пиридил)этенил]-6-метоксихиназолин-4-ил}циклогексиламин
Раствор 140 мг цис-4-трифторацетиламинометилциклогексиламина в 15 мл толуола объединяли с 180 мг 4-хлор-6-метокси-2-[2-(2-пиридил)этенил] хиназолина, 500 мг триэтиламина и 20 мг 4-диметиламинопиридина и нагревали при кипении с обратным холодильником в течение 15 часов. После отгонки реакционного раствора остаток объединяли с водой и экстрагировали хлороформом. После сушки над сульфатом магния и концентрирования остаток очищали колоночной хроматографией на силикагеле (хлороформ:метанол = 30:1), получая 140 мг целевого соединения.
Стадия 2
цис-4-Аминометил-N-{2-[2-(2-пиридил)этенил]-6-метоксихиназолин-4-ил}циклогексиламин
Раствор цис-4-трифторацетиламинометил-N-{2-[2-(2-пиридил) этенил]-6-метоксихиназолин-4-ил}циклогексиламина в 45 мл метанола и 5 мл воды объединяли с 414 мг карбоната калия и перемешивали при комнатной температуре в течение 15 часов. Реакционный раствор объединяли с водой, экстрагировали хлороформом и затем сушили над сульфатом натрия и концентрировали. Остаток очищали колоночной хроматографией на силикагеле (хлороформ:метанол:водный аммиак = 100:10:1), получая 120 мг целевого соединения.
Стадия 3
Тригидрохлорид цис-4-гуанидинометил-N-{2-[2-(2-пиридил)этенил-6-метоксихиназолин-4-ил}циклогексиламина
Раствор 120 мг цис-4-аминометил-N-{2[2(2-пиридил) этенил]-6-метоксихиназолин-4-ил}циклогексиламина в 15 мл дихлорэтана и 3 мл N,N-диметилформамида объединяли с 150 мг N,N'-бис(трет-бутоксикарбонил)-1Н-пиразол-1-карбоксиамидина и перемешивали при комнатной температуре в течение 15 часов. Реакционный раствор объединяли с водой и экстрагировали этилацетатом. Органический слой промывали водой и насыщенным раствором соли и затем сушили над сульфатом магния. После концентрирования остаток очищали колоночной хроматографией на силикагеле (хлороформ:метанол = 30:1). Полученный продукт растворяли в 3 мл метанола, объединяли с 3 мл 4н. раствора хлористого водорода в этилацетате и проводили взаимодействие при 50°С в течение 24 часов. Реакционный раствор концентрировали и затем обрабатывали смесью метанол-этиловый простой эфир, получая 84 мг целевого соединения.
Катион FAB-масс-спектр m/z: 416 [М+Н]+.
Внешний вид: желтый порошок.
Пример 5
Дигидрохлорид N-2-(2-имидазолинил)-N'-[2-(4-хлорстирил)-6-метилхиназолин-4-ил]-1,4-циклогександиамина
100 мг N-[2-(4-хлорстирил)-6-метилхиназолин-4-ил]-1,4-циклогександиамин в 10 мл метанола объединяли с 82 мг 1-трет-бутоксикарбонил-2-метилтио-2-имидазолина и нагревали при кипении с обратным холодильником в течение 10 часов. Реакционный раствор концентрировали и остаток очищали колоночной хроматографией на силикагеле (хлороформ: метанол = 20:1) и полученный при очистке продукт объединяли с 7 мл 4н. раствора хлористого водорода в этилацетате и проводили взаимодействие при 50°С в течение 24 часов. После концентрирования проводили обработку смесью метанол-этиловый простой эфир, получая 80 мг целевого соединения. Внешний вид: белый порошок
Данные элементного анализа (для C26H29ClN6·3HCl·5H2O)
Вычисленное значение (%) С: 50,75 Н: 6,06 N: 13,66
Найденное значение (%) С: 51,15 Н: 5,70 N: 13,47
Соединения следующих примеров 6-48, 52-59, 61 и 64-68 получали способом, аналогичным описанному в примере 1.
Пример 6
Дигидрохлорид N-амидино-N'-[2-(4-хлорстирил)хиназолин-4-ил]-1.4-бутандиамина
Катион FAB-масс-спектр m/z: 395 [М+Н]+
Внешний вид: бледно-желтые кристаллы
Пример 7
Дигидрохлорид N-амидино-N'-[2-(4-хлорстирил)хиназолин-4-ил]-1.5-пентандиамина
Катион FAB-масс-спектр m/z: 409 [М+Н]+
Внешний вид: бледно-желтый порошок
Данные элементного анализа (для C22H25ClN6·2HCl·1,5H2O)
Вычисленное значение (%) С: 51,93 Н: 5,94 N: 16,52
Найденное значение (%) С: 51,99 Н: 5,76 N: 16,25
Пример 8
Дигидрохлорид N-амидино-N'-[2-(4-хлорстирил)-6-метилхиназолин-4-ил]-1,6-гександиамина
Катион FAB-масс-спектр m/z: 437[M+H]+
Внешний вид: белый порошок
Пример 9
Дигидрохлорид N-амидино-N'-[2(4-хлорстирил)хиназолин-4-ил]-1,3-пропандиамина
Катион FAB-масс-спектр m/z: 381[М+Н]+
Внешний вид: бледно-красный порошок
Данные элементного анализа (для C20H21ClN6·2HCl·H2O)
Вычисленное значение (%) С: 50,91 Н: 5,34 N: 17,82
Найденное значение (%) С: 50,79 H: 5,07 N: 18,30
Пример 10
Дигидрохлорид N-амидино-N'-[2-(4-хлорстирил)-6-метилхиназолин-4-ил]-1,4-бутандиамина
Катион FAB-масс-спектр m/z: 409[М+Н]+
Внешний вид: бледно-желтый порошок
Пример 11
Дигидрохлорид N-амидино-N'-[2(4-хлорстирил)-6-метилхиназолин-4-ил]-1,5-пентандиамина
Катион FAB-масс-спектр m/z: 423[М+Н]+
Внешний вид: белый порошок
Пример 12
Дигидрохлорид цис-N-амидино-2-[2-(4-хлорстирил)-6-метилхиназолин-4-ил]аминоциклогексиламина
Катион FAB-масс-спектр m/z: 435[M+H]+
Внешний вид: желтый порошок
Данные элементного анализа (для С24Н27ClN6·3НСl)
Вычисленное значение (%) С: 52,96 Н: 5,55 N: 15,44
Найденное значение (%) С: 52,60 Н: 5,73 N: 15,77
Пример 13
Дигидрохлорид (1R,2S)-N-амидино-2[2(4-хлорстирил)-6-метилхиназолин-4-ил]аминоциклогексиламина
Катион FAB-масс-спектр m/z: 435[М+Н]+
Данные элементного анализа (для С24Н27ClN6·6НСl·2,5Н2О)
Вычисленное значение (%) С: 52,13 Н: 6,20 N: 15,20
Найденное значение (%) С: 52,40 Н: 5,80 N: 15,43
Оптическое вращение [α]20 D=+87,6° (с=1,0, метанол)
Пример 14
Дигидрохлорид (1S,2R)-N-амидино-2-[2-(4-хлорстирил)-6-метилхиназолин-4-ил]аминоциклогексиламина
Катион FAB-масс-спектр m/z: 435 [М+Н]+
Данные элементного анализа (для C24H27N6Cl·2HCl·2H2O)
Вычисленное значение (%) С: 53,00 Н: 6,12 N: 15,45
Найденное значение (%) С: 52,95 Н: 5,95 N: 15,40
Оптическое вращение [α]20 D=-86,7° (с=1,1, метанол)
Пример 15
Дигидрохлорид N-амино-N'-[6-трет-бутил-2-(4-хлорстирил)хиназолин-4-ил]-1,6-гександиамина
Внешний вид: бледно-красный порошок
Пример 16
Дигидрохлорид N-амидино-N'-[2(4-хлорстирил)-6-метоксихиназолин-4-ил]-1,6-гександиамина
Катион FAB-масс-спектр m/z: 453[М+Н]+
Внешний вид: бледно-желтый порошок
Данные элементного анализа (для C24H29ClN6O·2HCl·H2O)
Вычисленное значение (%) С: 53,00 Н: 6,12 N: 15,45
Найденное значение (%) С: 52,73 Н: 5,99 N: 15,64
Пример 17
Дигидрохлорид N-амидино-N'-[2-(4-хлорстирил)-6,7-диметилхиназолин-4-ил]-1,5-пентандиамина
Катион FAB-масс-спектр m/z: 437[М+Н]+
Данные элементного анализа (для C24H29ClN6·3HCl)
Вычисленное значение (%) С: 52,76 Н: 5,90 N: 15,38
Найденное значение (%) С: 52,45 Н: 6,12 N: 15,10
Пример 18
Дигидрохлорид N-амидино-N'-[2-(4-хлорстирил)-6-изопропилхиназолин-4-ил]-1,6-гександиамина
Катион FAB-масс-спектр m/z: 465 [М]+
Данные элементного анализа (для С26Н33ClN6·2НСl·1,4Н2О)
Вычисленное значение (%) С: 55,45 Н: 6,77 N: 14,92
Найденное значение (%) С: 55,50 Н: 6,61 N: 14,82
Пример 19
Дигидрохлорид цис-N-амидино-N'-[2-(4-хлорстирил)-6-метилхиназолин-4-ил]-1,4-циклогександиамина
Катион FAB-масс-спектр m/z: 435[М+Н]+
Данные элементного анализа (для С24Н27ClN6·2НСl·2Н2O)
Вычисленное значение (%) С: 53,00 Н: 6,11 N: 15,45
Найденное значение (%) С: 53,50 Н: 6,08 N: 14,92
Пример 20
Дигидрохлорид цис-N-амидино-2-[2-(4-хлорстирил)-6,7-диметилхиназолин-4-ил]аминоциклогексиламина
Катион FAB-масс-спектр m/z: 449[M+H]+
Пример 21
Дигидрохлорид цис-N-амидино-3-[2-(4-хлорстирил)-6-метилхиназолин-4-ил]аминоциклогексиламина
Катион FAB-масс-спектр m/z: 435[М+Н]+
Данные элементного анализа (для С24Н27ClN6·2НСl·2Н2O)
Вычисленное значение (%) С: 53,00 Н: 6,12 N: 15,45
Найденное значение (%) С: 52,52 Н: 5,79 N: 15,23
Пример 22
Дигидрохлорид транс-N-амидино-3-[2-(4-хлорстирил)-6-метилхиназолин-4-ил]аминоциклогексиламина
Катион FAB-масс-спектр m/z: 435[М+Н]+
Пример 23
Дигидрохлорид N-амидино-N'-{6-метокси-2-[2-(2-пиридил)этенил]хиназолин-4-ил}-1,6-гександиамина
Внешний вид: желтые кристаллы
Катион FAB-масс-спектр m/z: 420[М+Н]+
Данные элементного анализа (для C23H29N7O·3HCl·3H2O)
Вычисленное значение (%) С: 47,39 Н: 6,57 N: 16,82
Найденное значение (%) С: 47,32 Н: 6,29 N: 16,89
Пример 24
Дигидрохлорид (1R,2S)-N-амидино-2[2(4-хлорстирил)-6-метоксихиназолин-4-ил]аминоциклогексиламина
Внешний вид: бледно-желтый порошок
Данные элементного анализа (для С24Н27ClN6O·2НСl·0,5Н2O)
Вычисленное значение (%) С: 54,09 Н: 5,67 N: 15,77
Найденное значение (%) С: 53,71 Н: 5,60 N: 15,65
Оптическое вращение [α]20 D=+81,7° (с=1,1, метанол)
Пример 25
Дигидрохлорид (1S,2R)-N-амидино-2-[2-(4-хлорстирил)-6-метоксихиназолин-4-ил]аминоциклогексиламина
Внешний вид: бледно-желтый порошок
Катион FAB-масс-спектр m/z: 451 [М+Н]+
Данные элементного анализа (для C24H27ClN6O·2HCl·H2O)
Вычисленное значение (%) С: 53,19 Н: 5,77 N: 15,51
Найденное значение (%) С: 53,37 Н: 5,54 N: 15,61
Оптическое вращение [α]20 D=-77,6° (с=0,6, метанол)
Пример 26
Дигидрохлорид N-амидино-N'-[2(4-хлорстирил)хиназолин-4-ил]-1,4-бис(аминометил)циклогексана
Внешний вид: бледно-красный порошок
Катион FAB-масс-спектр m/z: 449[М+Н]+
Данные элементного анализа (для С25Н29ClN6·2НСl·Н2O)
Вычисленное значение (%) С: 55,62 Н: 6,16 N: 15,56
Найденное значение (%) С: 55,64 Н: 6,16 N: 15,02
Пример 27
Дигидрохлорид N-амидино-N'-[2-(4-хлорстирил)бензо[g]хиназолин-4-ил]-1,6-гександиамина
Катион FAB-масс-спектр m/z: 473[М+Н]+
Внешний вид: оранжевый порошок
Данные элементного анализа (для С27Н29ClN6·3НСl·0,5Н2O)
Вычисленное значение (%) С: 54,84 Н: 5,62 N: 14,21
Найденное значение (%) С: 55,11 Н: 5,65 N: 14,37
Пример 28
Дигидрохлорид цис-N-амидино-2-[2-(4-хлорстирил)-6-изопропилхиназолин-4-ил]аминоциклогексиламина
Катион FAB-масс-спектр m/z: 464 [М+Н]+
Данные элементного анализа (для С26Н31ClN6·2,0НСl·2Н2O)
Вычисленное значение (%) С: 54,60 Н: 6,52 N: 14,69
Найденное значение (%) С: 54,82 Н: 6,20 N: 14,85
Пример 29
Дигидрохлорид N-амидино-N'-[2(4-хлорстирил)-6-метилхиназолин-4-ил]-1,4-бис(аминометил)циклогексана
Катион FAB-масс-спектр m/z: 463[М+Н]+
Данные элементного анализа (для С26Н31ClN6·2НСl·1,5Н2O)
Вычисленное значение (%) С: 55,47 Н: 6,45 N: 14,93
Найденное значение (%) С: 55,81 Н: 6,52 N: 14,72
Пример 30
Дигидрохлорид цис-N-амидино-2-[2-(4-хлорстирил)-6-гидроксихиназолин-4-ил]аминоциклогексиламина
Внешний вид: бледно-зеленый порошок
Катион FAB-масс-спектр m/z: 437[M+H]+
Данные элементного анализа (для С23Н25ClN6O·3HСl·1,5Н2O)
Вычисленное значение (%) С: 48,40 Н: 5,30 N: 14,37
Найденное значение (%) С: 48,18 Н: 5,45 N: 14,66
Пример 31
Тригидрохлорид цис-N-амидино-2-{6-метил-2-[2-(4-пиридил; этенил]хиназолин-4-ил}аминоциклогексиламина
Катион FAB-масс-спектр m/z: 402[М+Н]+
Данные элементного анализа (для C23H27N7·3HCl·6H2O)
Вычисленное значение (%) С: 44,63 Н: 6,84 N: 15,84
Найденное значение (%) С: 45,00 Н: 6,59 N: 15,65
Пример 32
Дигидрохлорид цис-N-Амидино-2-[2-(4-хлорбензоиламино)-6-метилхиназолин-4-ил]аминоциклогексиламина
Катион FAB-масс-спектр m/z:452[М+Н]+
Пример 33
Дигидрохлорид цис-N-амидино-2-[2-(4-хлорстирил)-6-этоксихиназолин-4-ил]аминоциклогексиламина
Внешний вид: бледно-желтый порошок
Катион FAB-масс-спектр m/z: 465[М+Н]+
Данные элементного анализа (для C25H29ClN6O·3HCl·H2O)
Вычисленное значение (%) С: 51,19 Н: 5,86 N: 13,90
Найденное значение (%) С: 50,69 Н: 5,79 N: 14,19
Пример 34
Тригидрохлорид цис-N-амидино-2-(6-метокси-2-[2-(3-пиридил)этенил]хиназолин-4-ил]аминоциклогексиламина
Катион FAB-масс-спектр т/z: 418 [М+Н]+
Данные элементного анализа (для C23H27N7 O·3HCl·H2O)
Вычисленное значение (%) С: 50,70 Н: 5,92 N: 18,00
Найденное значение (%) С: 50,57 Н: 5,85 N: 17,98
Пример 35
Тригидрохлорид (1R,2S)-цис-N-амидино-2{6-метокси-2[2(3-пиридил)этенил]хиназолин-4-ил}аминоциклогексиламина
Катион FAB-масс-спектр m/z: 418[М+Н]+
Пример 36
Тригидрохлорид (1S,2R)-цис-N-амидино-2-(6-метокси-2-[2-(3-пиридил)этенил]хиназолин-4-ил]аминоциклогексиламина
Катион FAB-масс-спектр m/z: 418[М+Н]+
Пример 37
Дигидрохлорид (1R,2S)-N-амидино-2[2(4-хлорстирил)-6-метоксихиназолин-4-ил]аминоциклопентиламина
Катион FAB-масс-спектр m/z: 437[М+Н]+
Данные элементного анализа (для С23Н25ClN6O·2НСl·Н2O)
Вычисленное значение (%) С: 52,33 Н: 5,54 N: 15,92
Найденное значение (%) С: 52,72 Н: 5,24 N: 16,07
Оптическое вращение [α]20 D=-52,3° (с=1,0, метанол)
Пример 38
Дигидрохлорид (1S,2R)-N-амидино-2-[2-(4-хлорстирил)-6-метоксихиназолин-4-ил]аминоциклопентиламина
Катион FAB-масс-спектр m/z: 437[M+H]4'
Данные элементного анализа (для С23Н25ClN6O·2НСl·Н2O)
Вычисленное значение (%) С: 52,33 Н: 5,54 N: 15,92
Найденное значение (%) С: 52,42 Н: 5,34 N: 15,98
Пример 39
Тригидрохлорид цис-N-амидино-2-(6-метокси-2-[2-(2-пиридил)этенил]хиназолин-4-ил]аминоциклогексиламина
Катион FAB-масс-спектр m/z: 418[М+Н]+
Данные элементного анализа (для C23H27N7O·3HCl·H2O)
Вычисленное значение (%) С: 50,70 Н: 5,92 N: 18,00
Найденное значение (%) С: 50,58 Н: 5,75 N: 18,10
Пример 40
Тригидрохлорид (1R,2S)-цис-N-амидино-2-{6-метокси-2[2(2-пиридил)этенил]хиназолин-4-ил}аминоциклогексиламина
Катион FAB-масс-спектр m/z: 418[М+Н]+
Пример 41
Тригидрохлорид (1S,2R)-цис-N-амидино-2-{6-метокси-2-[2-(2-пиридил)этенил]хиназолин-4-ил}аминоциклогексиламина
Катион FAB-масс-спектр m/z: 418 [М+Н]+
Пример 42
Тригидрохлорид цис-N-амидино-2-{6-метокси-2[2(4-пиридил)этенил]хиназолин-4-ил}аминоциклогексиламина
Катион FAB-масс-спектр m/z: 418 [М+Н]+
Пример 43
Дигидрохлорид цис-N-амидино-2-[6-метокси-2-(2-метоксистирил)хиназолин-4-ил]аминоциклогексиламина
Катион FAB-масс-спектр m/z: 447[M+H]+
Данные элементного анализа (для С25Н30N6O2·3НСl)
Вычисленное значение (%) С: 54,01 Н: 5,98 N: 15,12
Найденное значение (%) С: 54,11 Н: 6,22 N: 15,14
Пример 44
Тригидрохлорид цис-N-амидино-N'(6-метокси-2[2(2-пиридил)этенил]хиназолин-4-ил]-1,4-бис(аминометил)циклогексана
Внешний вид: оранжевый порошок
Катион FAB-масс-спектр m/z: 446[М+Н]+
Пример 45
Тригидрохлорид транс-N-амидино-N'-{6-метокси-2-[2-(2-пиридиол)этенил]хиназолин-4-ил}-1,4-бис(аминометил)циклогексана
Внешний вид: оранжевый порошок
Катион FAB-масс-спектр m/z: 446[М+Н]+
Пример 46
Тригидрохлорид N-амидино-N'-{6-метил-2-[2-(2-пиридил)этенил]хиназолин-4-ил}-1,6-гександиамина
Катион FAB-масс-спектр m/z: 404 [М+Н]+
Данные элементного анализа (для С23Н29N7·3НСl·2Н2O)
Вычисленное значение (%) С: 50,33 Н: 6,61 N: 17,86
Найденное значение (%) С: 50,93 Н: 6,69 N: 17,26
Пример 47
Тригидрохлорид N-амидино-N'-{6-метил-2[2(2-пиридил)этенил]хиназолин-4-ил}-1,8-октандиамина
Внешний вид: желтый порошок
Катион FAB-масс-спектр m/z: 432[М+Н]+
Данные элементного анализа (для С25Н33N7·3НСl·Н2O)
Вычисленное значение (%) С: 53,72 Н: 6,85 N: 17,54
Найденное значение (%) С: 53,83 Н: 7,03 N: 17,03
Пример 48
Тригидрохлорид N-амидино-6-{6-метокси-2-[2-(2-пиридил)этенил]хиназолин-4-ил}аминогептиламина
Внешний вид: желтый порошок
Катион FAB-масс-спектр m/z: 434 [М+Н]+
Данные элементного анализа (для C24H31N7O·3HCl·1,5H2O)
Вычисленное значение (%) С: 65,28 Н: 6,14 N: 12,18
Найденное значение (%) С: 65,23 Н: 5,92 N: 12,12
Пример 49
Дигидрохлорид N-[2-(4-хлорстирил)хиназолин-4-ил]-N'-(2-пиримидил)пиперазина
Указанное в заголовке соединение получали в виде бледно-желтого порошка способом, аналогичным способу примера 3.
Катион FAB-масс-спектр m/z: 429[М+Н]+
Данные элементного анализа (для C24H21ClN6·3HCl)
Вычисленное значение (%) С: 56,33 Н: 6,47 N: 11,43
Найденное значение (%) С: 56,05 Н: 6,31 N: 11,36
Пример 50
Дигидрохлорид цис-N-[2-(4-хлорстирил)6-метоксихиназолин-4-ил]-2-гуанидинометилциклогексиламина
Указанное в заголовке соединение получали способом, аналогичным способу примера 4.
Катион FAB-масс-спектр m/z: 465[М+Н]+
Данные элементного анализа (для C25H29ClN6O·3HCl)
Вычисленное значение (%) С: 52,28Н: 5,62 N: 14,63
Найденное значение (%) С: 52,24 Н: 5,66 N: 14,27
Пример 51
Дигидрохлорид N-2-(2-имидазолинил)-N' [2-(4-хлорстирил)-6-метилхиназолин-4-ил]-1,6-гександиамина
Указанное в заголовке соединение получали в виде белого порошка способом, аналогичным способу примера 5.
Температура плавления: 305°С
Пример 52
Дигидрохлорид N-[2-(4-хлорстирил)хиназолин-4-ил]-1,2-этандиамина
Внешний вид: бледно-красный порошок
Катион FAB-масс-спектр m/z: 367[М+Н]+
Данные элементного анализа (для С19Н19ClN6·2НСl·Н2О)
Вычисленное значение (%) С: 49,85 Н: 5,06 N: 18,36
Найденное значение (%) С: 49,97 Н: 4,97 N: 18,26
Пример 53
Дигидрохлорид N-амидино-N'-[2-(4-хлорстирил)хиназолин-4-ил]-1,6-гександиамина
Внешний вид: белый порошок
Катион FAB-масс-спектр m/z: 423[M+H]+
Пример 54
Дигидрохлорид N-амидино-N'-[2-(4-хлорстирил)-6-метилхинолин-4-ил]-1,6-гександиамина
Внешний вид: бледно-желтый порошок
Катион FAB-масс-спектр m/z: 436 [М+Н]+
Данные элементного анализа (для С25Н30ClN5·2НСl·0,7Н2O)
Вычисленное значение (%) С: 57,58 Н: 6,45 N: 13,43
Найденное значение (%) С: 57,48 Н: 6,32 N: 13,34
Пример 55
Дигидрохлорид N-амидино-N'-[2-(4-хлорстирил)хинолин-4-ил]-1,6-гександиамина
Внешний вид: бледно-желтый порошок
Катион FAB-масс-спектр m/z: 422 [М+Н]+
Пример 56
Дигидрохлорид цис-N-амидино-2-[2-(4-хлорстирил)-6-метилхинолин-4-ил]аминоциклогексиламина
Внешний вид: белый порошок
Катион FAB-масс-спектр m/z: 434 [М+Н]+
Данные элементного анализа (для C25H28ClN5·2HCl·1,5H2O)
Вычисленное значение (%) С: 56,24 Н: 6,23 N: 13,12
Найденное значение (%) С: 56,14 Н: 6,02 N: 13,08
Пример 57
Дигидрохлорид цис-N-амидино-2-[2-(4-хлорстирил)-6-метоксихинолин-4-ил]аминоциклогексиламина
Внешний вид: бледно-желтый порошок
Катион FAB-масс-спектр m/z: 450[М+Н]+
Данные элементного анализа (для C25H28ClN5O·3HCl·H2O)
Вычисленное значение (%) С: 52,01 Н: 5,76 N: 12,13
Найденное значение (%) С: 52,00 Н: 5,59 N: 12,01
Пример 58
Дигидрохлорид цис-N-амидино-2-[3-(4-хлорстирил)изохинолин-1-ил]аминоциклогексиламина
Внешний вид: бледно-желтый порошок
Катион FAB-масс-спектр m/z: 420 [М+Н]+
Пример 59
Тригидрохлорид N-амидино-4-{6-метил-2-[2-(2-пиридил)этенил] хиназолин-4-ил]аминометил-бензиламина
Внешний вид: белый порошок
Катион FAB-масс-спектр m/z: 424 [М+Н]+
Данные элементного анализа (для C25H25N7·3HCl·2H2O)
Вычисленное значение (%) С: 52,78 Н: 5,66 N: 17,23
Найденное значение (%) С: 53,28 Н: 5,36 N: 17,09
Пример 60
Тригидрохлорид N-(N-изобутил-N'-фенил)амидино-N'-{6-метокси-2-[2-(2-пиридил)этенил]хиназолин-4-ил}-1,6-гександиамина
Стадия 1
N-[6-{6-Метокси-2-[2-(2-пиридил)этенил]хиназолин-4-ил}амино]гексил-N'-фенил-тиомочевина
Раствор 135 мг N-{6-метокси-2-[2-(2-пиридил)этенил]хиназолин-4-ил}-1,6-гександиамина в хлористом метилене объединяли с 58 мг фенилизоцианата и перемешивали при комнатной температуре в течение 2 часов. После отгонки реакционного раствора остаток очищали колоночной хроматографией на силикагеле (хлороформ:метанол = 50:1), получая 174 мг целевого соединения.
Стадия 2
N-[6-{6-Метокси-2-[2-(2-пиридил)этенил]хиназолин-4-ил}амино]гексил-N'-фенил-S-метилизотиомочевина
Раствор 10 мг соединения со стадии 1 в 3 мл хлористого метилена объединяли с избытком йодистого метила и перемешивали в течение 15 часов. После отгонки реакционного раствора остаток очищали колоночной хроматографией на силикагеле (хлороформ:метанол:водный аммиак = 10:1:0,1), получая 11 мг целевого соединения.
Стадия 3
Тригидрохлорид N(N-изобутил-N'-фенил)амидино-N'-{6-метокси-2-[2-(2-пиридил)этенил]хиназолин-4-ил}-1,6-гександиамина
Раствор 11 мг соединения со стадии 2 в 3 мл этанола объединяли с избытком изобутиламина и нагревали при кипении с обратным холодильником в течение 15 часов. После отгонки летучих компонентов из реакционного раствора остаток очищали колоночной хроматографией на силикагеле (хлороформ: метанол: водный аммиак =10:1:0,1) и затем обрабатывали 4н. раствором хлористого водорода в этилацетате, получая 13 мг целевого соединения.
Катион FAB-масс-спектр m/z: 552 [М+Н]+
Соединения следующих примера 62 и примера 63 получали способом, аналогичным способу примера 60.
Пример 61
Тригидрохлорид 2-гуанидиноэтокси-N-{6-метил-2-[2-(2-пиридил)этенил]хиназолин-4-ил}этиламина
Внешний вид: бледно-желтый порошок
Катион FAB-масс-спектр m/z: 392 [М+Н]+
Пример 62
Тригидрохлорид N-(N-метил-N'-фенил)амидино-N'-{6-метокси-2-[2-(2-пиридил)этенил]хиназолин-4-ил}-1,6-гександиамина
Внешний вид: бледно-желтый порошок
Катион FAB-масс-спектр m/z: 510[М+Н]+
Пример 63
Тригидрохлорид N-(N-этил-N'-метил)амидино-N'-{6-метокси-2-[2-(2-пиридил)этенил]хиназолин-4-ил)-1,6-гександиамина
Катион FAB-масс-спектр m/z: 462[M+Н]+
Пример 64
Тригидрохлорид 2-гуанидинопропилокси-N-{6-метил-2[2(2-пиридил)этенил]хиназолин-4-ил}этиламин
Внешний вид: желтый порошок
Катион FAB-масс-спектр m/z: 406[M+H]+
Пример 65
Тригидрохлорид 3-гуанидиноэтокси-N-{6-метил-2[2(2-пиридил)этенил]хиназолин-4-ил}пропиламина
Внешний вид: бледно-желтый порошок
Катион FAB-масс-спектр m/z: 406 [М+Н]+
Пример 66
Тригидрохлорид N-амидино-2-{6-метокси-2-[2-(2-пиридил)этенил]хиназолин-4-ил}аминоэтил-фенилэтиламина
Внешний вид: желтый порошок
Катион FAB-масс-спектр m/z: 468[М+Н]+
Данные элементного анализа (для C27H29N7O·3HCl)
Вычисленное значение (%) С: 56,21 Н: 5,59 N: 16,99
Найденное значение (%) С: 55,92 Н: 5,59 N: 16,28
Пример 67
Тригидрохлорид транс-4-гуанидинометил-цис-2-метил-N-{6-метокси-2-[2-(2-пиридил)этенил]хиназолин-4-ил}циклогексиламин
Внешний вид: желтый порошок
Катион FAB-масс-спектр m/z: 446[М+Н]+
Данные элементного анализа (для С25Н31N7O·3HСl·Н2O)
Вычисленное значение (%) С: 49,31 Н: 6,62 N: 16,10
Найденное значение (%) С: 49,60 Н: 6,42 N: 16,01
Пример 68
Тригидрохлорид цис-4-гуанидинометил-цис-2-метил-N-{6-метокси-2-[2-(2-пиридил)этенил]хиназолин-4-ил}циклогексиламина
Внешний вид: желтый порошок
Катион FAB-масс-спектр m/z: 446[M+H]+
Данные элементного анализа (для С25Н31N7O·3НСl·2,5Н2O)
Вычисленное значение (%) С: 50,05 Н: 6,55 N: 16,34
Найденное значение (%) С: 49,87 Н: 6,30 N: 16,22
Пример 69
Дигидрохлорид (1R,2S)-N-амидино-2(2(4-хлорбензоиламино)-6-метоксихиназолин-4-ил)аминоциклогексиламина
Стадия 1
(1R,2S)-N-трет-Бутоксикарбонил-2(2-хлор-6-метоксихиназолин-4-ил)аминоциклогексиламин
Раствор 710 мг 2,4-дихлор-6-метоксихиназолина в 20 мл хлористого метилена объединяли с 471 мг триэтиламина и 750 мг (1S,2R)-2-трет-бутоксикарбониламиноциклогексиламина и перемешивали при комнатной температуре в течение 48 часов. После концентрирования смесь объединяли с водой, экстрагировали хлористым метиленом и сушили. После отгонки растворителя остаток очищали колоночной хроматографией на силикагеле (хлороформ:метанол = 20:1), получая 1,20 г целевого соединения.
Стадия 2
(1R,2S)-N-трет-Бутоксикарбонил-2[2(4-метоксибензиламино)-6-метоксихиназолин-4-ил]аминоциклогексиламин
В атмосфере аргона раствор 1,75 г (1R,2S)-N-трет-бутоксикарбонил-2-(2-хлор-6-метоксихиназолин-4-ил) аминоциклогексиламина и 1,47 г 4-метоксибензиламина в 100 мл безводного толуола объединяли с 97 мг ацетата палладия, 268 мг 2,2'-бис(дифенилфосфино)-1,1'-бинафтила и 1,03 г трет-бутоксида натрия и перемешивали при 70°С в течение 5 часов. Реакционный раствор концентрировали и затем объединяли с водой, экстрагировали хлороформом и сушили. После отгонки растворителя остаток очищали колоночной хроматографией на силикагеле (хлороформ:метанол = 20:1), получая 1,62 г целевого соединения.
Стадия 3
(1R,2S)-N-[N,N'-Бис(трет-бутоксикарбонил)]амидино-2-[2-(4-метоксибензиламино)-6-метоксихиназолин-4-ил]аминоциклогексиламин
Раствор 1,70 г (1R,2S)-N-трет-бутоксикарбонил-2-[2(4-метоксибензиламино)-6-метоксихиназолин-4-ил] аминоциклогексиламина в 30 мл хлористого метилена объединяли с 10 мл трифторуксусной кислоты при охлаждении льдом и перемешивали в течение 2 часов. Реакционный раствор нейтрализовали насыщенным раствором гидрокарбоната натрия, экстрагировали хлористым метиленом и сушили. После отгонки растворителя раствор остатка в 30 мл хлористого метилена объединяли с 1,18 г N,N'-бис(трет-бутоксикарбонил)-1H-пиразол-1-карбоксиамидина и перемешивали при комнатной температуре в течение 15 часов. Реакционный раствор объединяли с водой, экстрагировали хлористым метиленом и сушили. После отгонки растворителя остаток очищали колоночной хроматографией на силикагеле (хлороформ:метанол = 20:1), получая 1,86 г целевого соединения.
Стадия 4
(1R,2S)-N-[N,N'-Бис(трет-бутоксикарбонил)]амидино-2-(2-амино-6-метоксихиназолин-4-ил)аминоциклогексиламин
Раствор 1,76 г (1R, 2S)-N-[N,N'-бис(трет-бутоксикарбонил)]амидино-2-[2-(4-метоксибензиламино)-6-метоксихиназолин-4-ил] аминоциклогексиламина в 80 мл хлористого метилена объединяли с 3,17 г N-метилморфолин-N-оксида и 95 мг перрутената тетрапропиламмония и перемешивали в течение 9 часов. Реакционный раствор объединяли с водой, экстрагировали хлористым метиленом и сушили. После отгонки растворителя остаток очищали колоночной хроматографией на силикагеле (хлороформ:метанол = 10:1), получая 0,95 г целевого соединения.
Стадия 5
(1R,2S)-N-[N,N'-Бис(трет-бутоксикарбонил)]амидино-2-[2-(4-хлорбензоиламино)-6-метоксихиназолин-4-ил]аминоциклогексиламин
Раствор 366 мг N,N-диизопропилэтиламина в 10 мл хлористого метилена объединяли с 60 мг 4-диметиламинопиридина и 0,156 мл 4-хлорбензоилхлорида. Эту смесь обрабатывали по каплям раствором 500 мг (1R,2S)-N-[N,N'-бис(трет-бутоксикарбонил)]амидино-2-(2-амино-6-метоксихиназолин-4-ил) аминоциклогексиламина в 10 мл хлористого метилена и перемешивали при комнатной температуре в течение 3 часов. Реакционный раствор объединяли с водой, экстрагировали хлористым метиленом и сушили. После отгонки растворителя остаток очищали колоночной хроматографией на силикагеле (хлороформ:метанол = 30:1), получая 560 мг целевого соединения.
Стадия 6
Дигидрохлорид (1R,2S)-N-амидино-2{[2-(4-хлорбензоиламино)-6-метоксихиназолин-4-ил]амино}циклогексиламина
Раствор 450 мг (1R,2S)-N-[N,N'-бис(трет-бутоксикарбонил)]амидино-2-[2-(4-хлорбензоиламино)-6-метоксихиназолин-4-ил] аминоциклогексиламина в 5 мл метанола и 5 мл хлороформа объединяли с 5 мл 4н. раствора хлористого водорода в этилацетате и проводили взаимодействие при 50°С в течение 72 часов. После концентрирования проводили обработку смесью метанол-простой этиловый эфир, получая 260 мг целевого соединения в виде бесцветного порошка.
Катион FAB-масс-спектр m/z: 468[М+Н]+
Данные элементного анализа (для С23Н26ClN7O2·2НСl·1,5Н2O)
Вычисленное значение (%) С: 48,64 Н: 5,50 N: 17,26
Найденное значение (%) С: 48,87 Н: 5,38 N: 17,29
Оптическое вращение [α]20 D=+64,97 (с=1,0, метанол)
Соединение следующего примера 70 получали способом, аналогичным способу примера 69.
Пример 70
Дигидрохлорид (1S,2R)-N-амидино-2-{[2-(4-хлорбензоиламино)-6-метоксихиназолин-4-ил]амино}циклогексиламина
Катион FAB-масс-спектр m/z: 468[M+H]+
Данные элементного анализа (для С23Н26ClN7O2•2НСl•3Н2O)
Вычисленное значение (%) С: 46,43 Н: 5,76 N: 16,48
Найденное значение (%) С: 46,41 Н: 5,56 N: 16,50
Оптическое вращение [α]20 D=+65,98° (с=1,0, метанол)
Пример испытания 1
Анализ связывания ноцицептинового рецептора
Суспензию клеточных мембран, полученную от экспрессирующей ноцицептин клетки человека, получали таким образом, что она содержала от 5 до 10 мкг/мл мембранного белка в Tris-буфере (50 мм Tris-HCl (pH 7,8), 5 мМ MgCl2, 1 мМ EGTA, 0,1% БСА). К данной суспензии добавляли [3Н]ноцицептин (разбавленный до конечной концентрации 0,08 нМ Tris-буфером) и исследуемое вещество и смесь инкубировали при 25°С в течение 60 минут. С помощью клеточного харвестера и промывочного раствора [50 мМ Tris-HCl (pH 7,8), 4°С] мембраны выделяли на GF/B фильтре, который предварительно обрабатывали 0,3% PEI и затем дополнительно промывали 4 раза. Фильтр переносили в ампулу, в которую был добавлен сцинтиллятор, и измеряли радиоактивность с использованием жидкостного сцинтилляционного счетчика. Отметим, что неспецифическое связывание рассматривали как связывание в присутствии 10 мкМ ноцицептина, а специфическое связывание получали, вычитая неспецифическое связывание из общего связывания. Из соотношения ингибирования связывания в присутствии исследуемого вещества получали значение IC50 и затем использовали вместе со значением Кd для [3Н]ноцицептина для расчета значения Ki для исследуемого вещества. Результаты представлены в таблице 1.
(Пример №)
Пример испытания 2
Анализ связывания μ-рецептора
Препарат экспрессирующей μ-рецептор клеточной мембраны человека (Receptor Biology) получали таким образом, что он содержал 8,5 мкг/мл мембранного белка в Tris-буфере (50 мМ Tris-HCl (рН 7,8), 5 мМ MgCl2, 1 мМ EGTA, 0,1% БСА). К данной суспензии добавляли [3Н] дипренорфин (разбавленный до конечной концентрации 0,13 нМ Tris-буфером) и исследуемое вещество и смесь инкубировали при 25°С в течение 90 минут. С помощью клеточного харвестера и промывочного раствора [50 мМ Tris-HCl (рН 7,8), 4°С] мембраны выделяли на GF/B фильтре, который предварительно был обработан 0,3% PEI, затем дополнительно промывали 4 раза. Фильтр переносили в ампулу, в которую был добавлен сцинтиллятор, и измеряли радиоактивность с использованием жидкостного сцинтилляционного счетчика. Отметим, что неспецифическое связывание рассматривали как связывание в присутствии 100 мкМ налоксона, а специфическое связывание получали, вычитая неспецифическое связывание из общего связывания. Из соотношения ингибирования связывания в присутствии исследуемого вещества получали значение IC50 и затем использовали вместе со значением Кd для [3Н] дипренорфина для расчета значения Ki для исследуемого вещества. Результаты представлены в таблице 2.
Как видно из таблицы 1 и таблицы 2, каждое соединение настоящего изобретения обладало превосходным связывающим действием в отношении ноцицептинового рецептора.
Пример испытаний 3
Тест болевых судорог с помощью уксусной кислоты у мышей
Десять самцов мышей (Slc:ddY, возраст 4-5 недель) предписывали к каждой группе. Шкуру на спине каждой мыши надрезали вбок на длину приблизительно 3 см и после акклиматизации в течение 30 минут или дольше вводили в положение вокруг L3-L4 иглу 27G, закрепленную на верхушке силиконовой трубочки, связанной с микрошприцем, через которую вводили 5 мкл раствора лекарственного средства, проводя таким образом введение в спинальное подпаутинное пространство. Исследуемое вещество растворяли в физиологическом растворе и вводили из расчета 10 нмоль/животное. В контрольной группе аналогичным образом вводили физиологический раствор.
Мышь, которой не давали еды за день до эксперимента, помещали в камеру наблюдения (20×20×15 см), где ей давали возможность акклиматизироваться в течение 30 минут или дольше, а затем ей вводили внутрибрюшинно с помощью иглы 27G 100 мкл на 10 г веса тела 0,6% раствор уксусной кислоты. Число судорожных реакций с растяжением брюшной полости подсчитывали в течение 20 минут после введения уксусной кислоты и полученные данные представляли как среднее значение ± стандартная ошибка. Только отобранные данные подвергали тесту на значимое различие с помощью t-теста между двумя группами контроля и группой обработки или путем одностороннего анализа расхождения среди различных групп с последующим тестом множественного сравнения Даннетта (Dunnett), и значимое различие рассматривали как имеющее место, когда р<0,05. Результаты представлены в таблице 3.
Таблица 3
Тест болевых судорог с помощью уксусной кислоты у мышей
(Число судорог)
Животное №
10 нмоль
10 нмоль
10 нмоль
10 нмоль
Как видно из таблицы 3, было выявлено, что каждое соединение настоящего изобретения снижает число значительных судорожных реакций, проявляя таким образом анальгетическое (обезболивающее) действие.
Пример состава 1
100 г Соединения примера 70, 292 г D-маннита, 120 г кукурузного крахмала и 28 г низкозамещенной гидроксипропилцеллюлозы помещали в гранулятор кипящего слоя (STREA; PAUREC) и гранулировали с распылением определенного количества 5%-ного водного раствора гидроксипропилцеллюлозы. После сушки, а затем измельчения с помощью измельчающего/размалывающего устройства (COMIL, PAULEC) смешивали с определенным количеством стеарата магния с помощью смесителя (BOHRE контейнерный смеситель модель МС20, KOTOBUKI-GIKEN) и смесь подвергали действию роторной таблетирующей прессующей машины (CORRECT 12HUK; KIKUSUI) для формования таблеток диаметром 7 мм каждая, весом 140 мг на таблетку, получая таким образом таблетку, содержащую 25 мг соединения по настоящему изобретению.
Пример состава 2
75 г Соединения примера 70, 180 г лактозы, 75 г кукурузного крахмала и 18 г кроскармелозы кальция помещали в перемешивающий гранулятор (вертикальная модель гранулятора VG-01), объединяли с определенным количеством 5%-ного водного раствора гидроксипропилметилцеллюлозы и гранулировали, затем сушили с помощью гранулирующей сушки в кипящем слое (STREA; PAUREC), а затем измельчали с помощью измельчающего/размалывающего устройства (COMIL, производство PAULEC). 120 мг Измельченного материала заполняли капсулу №3 с использованием устройства для наполнения капсул (капсульное наполняющее устройство; SHIONOGI QUALICAPS), получая таким образом капсулу, содержащую 25 мг соединения по настоящему изобретению.
Пример состава 3
Отвешивали 2,5 г соединения примера 70 и 4,5 г хлорида натрия, объединяли с 450 мл воды для инъекций и перемешивали растворяли и доводили до рН 6,5 с использованием растворов 0,1 моль/л соляной кислоты и 0,1 моль/л гидроксида натрия. Затем добавляли воду для инъекций для достижения общего количества 500 мл. Полученный таким образом раствор фильтровали под давлением через мембранный фильтр (размер пор: 0,22 мкм), затем коричневую ампулу емкостью 5 мл заполняли 5,3 мл состава в асептических условиях, получая таким образом состав для инъекций, содержащий 25 мг соединения по настоящему изобретению. Процедуру для получения посредством заполнения проводили в асептических условиях.
Пример состава 4
99,75 г UITEPSOL Н-15 (производства HIRTH) растворяли при 45°С и объединяли с 0,25 г соединения примера 70 и диспергировали с использованием перемешивания. Эту смесь, пока она оставалась горячей, осторожно для предотвращения седиментации вводили в форму для 1 г суппозитория, отверждали и вынимали из формы, получая таким образом суппозиторий, содержащий 25 мг соединения по настоящему изобретению.
Промышленная применимость
Поскольку соединение по настоящему изобретению обладает превосходной способностью связывания с ноцицептиновым рецептором, оно может благополучно использоваться в течение продолжительного периода времени против вызывающих боль заболеваний, таких как боль, мигрень, ревматоидный артрит и невралгия, и в качестве агента для преодоления устойчивости к морфину или тому подобному.
| название | год | авторы | номер документа |
|---|---|---|---|
| ПРОИЗВОДНЫЕ ХИНАЗОЛИНА В КАЧЕСТВЕ ИНГИБИТОРОВ ВАСКУЛЯРНОГО ЭНДОТЕЛИАЛЬНОГО ФАКТОРА РОСТА (VEGF) | 2000 |
|
RU2291868C2 |
| ПРОИЗВОДНЫЕ N-АЦИЛ- α -АМИНОКИСЛОТЫ ИЛИ ИХ ФИЗИОЛОГИЧЕСКИ ПРИЕМЛЕМЫЕ СОЛИ, ПРОСТЫЕ ИЛИ СЛОЖНЫЕ ЭФИРЫ, АМИДЫ ИЛИ ГИДРАТЫ И КОМПОЗИЦИЯ ИНГИБИРУЮЩАЯ СВЯЗЫВАНИЕ АДГЕЗИВНЫХ ПРОТЕИНОВ С ТРОМБОЦИТАМИ И АГРЕГАЦИЮ ТРОМБОЦИТОВ | 1992 |
|
RU2097378C1 |
| ХИНАЗОЛИНОВЫЕ СОЕДИНЕНИЯ | 2003 |
|
RU2365588C2 |
| ПРОИЗВОДНЫЕ ХИНАЗОЛИНА, СПОСОБ ИХ ПОЛУЧЕНИЯ И ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ | 2001 |
|
RU2267489C2 |
| ПРОИЗВОДНЫЕ ХИНАЗОЛИНА В КАЧЕСТВЕ ИНГИБИТОРОВ АНГИОГЕНЕЗА | 2000 |
|
RU2262935C2 |
| ПРОИЗВОДНЫЕ ЭТИЛЕНДИАМИНА И СОДЕРЖАЩИЕ ИХ ИНГИБИТОР FXa И АНТИКОАГУЛЯНТ | 2001 |
|
RU2268259C2 |
| ИНГИБИТОР СЕРИНОВЫХ ПРОТЕАЗ И ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ НА ЕГО ОСНОВЕ | 1999 |
|
RU2232760C2 |
| ЗАМЕЩЕННЫЕ АМИНОМАСЛЯНЫЕ ПРОИЗВОДНЫЕ В КАЧЕСТВЕ ИНГИБИТОРОВ НЕПРИЛИЗИНА | 2012 |
|
RU2604522C2 |
| ТАКСАНЫ, ФАРМАЦЕВТИЧЕСКИЕ КОМПОЗИЦИИ, СПОСОБЫ ИНГИБИРОВАНИЯ | 2001 |
|
RU2265019C2 |
| ПИРИДОПИРИМИДИНОВЫЕ ИНГИБИТОРЫ CDK2/4/6 | 2017 |
|
RU2726115C1 |
Предложено анальгетическое средство, обладающее анальгетическим действием, эффективное против боли, путем действия на ноцицептиновый рецептор. Анальгетическое средство в качестве активного ингредиента содержит соединение, представленное общей формулой (1) или его соль. X, Y, Е, Q, А1, A2, R1, R3, R4, R5, R2A, R2C, R2D, R2B определены в формуле изобретения. 3 табл.

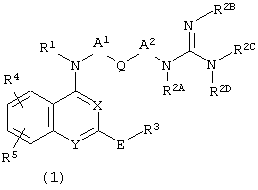
где Х и Y являются одинаковыми или различными, и каждый представляет собой атом азота или СН;
R1 представляет собой атом водорода или алкил;
А1 и А2 являются одинаковыми или различными, и каждый представляет собой (1) одинарную связь или (2) двухвалентную алифатическую углеводородную группу, которая может быть замещенной и может содержать 1-3 ненасыщенные связи в любом положении (такая алифатическая углеводородная группа может содержать один гетероатом, выбранный из группы, состоящей из -NH- и О);
Q представляет собой (1) одинарную связь, (2) необязательно замещенную 3-8-членную циклоалкиленовую группу или (3) необязательно замещенную фениленовую группу;
R2A, R2C и R2D являются одинаковыми или различными, и каждый представляет собой атом водорода, алкил или фенил, R2B представляет собой атом водорода, алкил, циано, нитро или фенил, или два атома азота гуанидино-группы образуют цикл вместе с одним или двумя ее заместителями R2B, R2C и R2D с образованием насыщенного или ненасыщенного 5- или 6-членного кольца;
или взяты вместе в виде -N(R1)-A1-Q-A2-N(R2A)- с образованием 5-7-членного кольца;
Е представляет собой (1) этенилен, (2) -NRCO-, (3) -NRCONH-, (4) -CONR-, (5) этинилен или (7) аминоалкилен (где R представляет собой водород или необязательно замещенный алкил);
R3 представляет собой необязательно замещенную фенильную группу или 4-8-членное моноциклическое или конденсированное кольцо, содержащее 1-2 гетероатома, выбранные из группы, состоящей из NH, кислорода и серы, и может иметь 1-4 ненасыщенные связи, и
R4 и R5 (1) являются одинаковыми или различными, и каждый представляет собой атом водорода, алкил, алкокси, галоген, нитро, гидрокси, алкоксикарбонил, -NR6R7, -NR6COR7, -CONR6R7 (в которых R6 и R7 являются одинаковыми или различными, и каждый представляет собой атом водорода или алкил), или (2) когда они являются смежными друг с другом, то взятые вместе образуют -O(СН2)nО- (где n представляет собой целое число 1 или 2) или -СН=СН-СН=СН-,
или его соль.
Приоритет по признакам радикалов R2a, R2D, R2c, R2B - представляющие фенил - от 02.11.2000. Приоритет по признакам радикала R2B, представляющего CN, NO2; R4 и R5 -NR6COR7, при R6=водород - от 31.03.2000 и 02.11.2000, при других значениях радикалов - от 30.03.2001. Приоритет по признакам Е, представляющего -NRCONH-, CONR-, этинилен, аминоалкилен, -NRCO-, в котором R представляет необязательно замещенный алкил - от 30.03.2001. Приоритет по признакам -NRCO-, где R=H- от 31.03.2000 и 02.11.2000. Приоритет по признакам остальных радикалов - от 31.03.2000 и 02.11.2000.
| WO 9720821 A1, 12.06.1997 | |||
| Горизонтальный вибрационный смеситель | 1977 |
|
SU856514A1 |
| WO 9948492 A1, 30.09.1999 | |||
| RU 98114078 А, 27.03.2000. | |||

