Настоящее изобретение относится к производным хиназолина, способам их получения, фармацевтическим композициям, содержащим их в качестве активного ингредиента, способам лечения патологических состояний, ассоциированных с развитием кровеносных сосудов и/или повышенной васкулярной проницаемостью, к их применению в качестве лекарственных средств и к их применению при изготовлении лекарственных препаратов для использования при продуцировании антиангиогенных и/или снижающих васкулярную проницаемость действий у теплокровных животных, таких как человек.
Нормальное развитие кровеносных сосудов играет важную роль в различных процессах, включающих эмбриональное развитие, заживление ран и несколько компонентов женской репродуктивной функции. Нежелательное или патологическое развитие кровеносных сосудов было связано с патологическими состояниями, включающими диабетическую ретинопатию, псориаз, рак, ревматоидный артрит, атерому, саркому Капоши и гемангиому (Fan et al, 1995, Trends Pharmacol Sci. 16: 57-66; Folkman, 1995, Nature Medicine 1: 27-31). Полагают, что изменение васкулярной проницаемости играет роль как в нормальных, так и патологических физиологических процессах (Cullinan-Bove et al, 1993, Endocrinology 133: 829-837; Senger et al, 1993, Cancer and Metastasis Reviews, 12: 303-324). Были идентифицированы несколько полипептидов со стимулирующей in vitro рост эндотелиальных клеток активностью, включая кислотные и основные факторы роста фибробластов (aFGF и bFGF) и васкулярный эндотелиальный фактор роста (VEGF). Вследствие ограниченной экспрессии его рецепторов активность фактора роста VEGF, в противоположность активности FGFs, является относительно избирательной по отношению к эндотелиальным клеткам. Последние данные указывают на то, что VEGF является важным стимулятором как нормального, так и патологического развития кровеносных сосудов (Jakeman et al. Endocrinology, 133: 848-859; Kolch et al, 1995, Breast Cancer Research and Treatment, 36: 139-155) и васкулярной проницаемости (Connolly et al, 1989, J. Biol. Chem. 264: 20017-20024). Антагонизм действию VEGF секвестрацией VEGF антителом может привести к ингибированию роста опухоли (Kim et al, 1993, Nature 362: 841-844).
Рецепторные тирозинкиназы (RTKs) являются важными в передаче биохимических сигналов через плазматическую мембрану клеток. Эти трансмембранные молекулы характеристично состоят из внеклеточного, связывающего лиганд домена, связанного через сегмент в плазматической мембране с доменом внутриклеточной тирозинкиназы. Связывание лиганда с рецептором приводит к стимуляции активности ассоциированной с рецептором тирозинкиназы, которая приводит к фосфорилированию остатков тирозина как на рецепторе, так и других внутриклеточных молекулах. Указанные изменения в фосфорилировании тирозина инициируют каскад передачи сигнала, приводящий к различным клеточным ответным реакциям. На сегодняшний день идентифицировано, по меньшей мере, девятнадцать различных подсемейств RTK, определяемых гомологией аминокислотной последовательности. Одно из этих подсемейств в настоящее время включает fms-подобный рецептор тирозинкиназы, Flt или Flt1, рецептор, содержащий домен вставки киназы, KDR (называемый также Flk-1), и другой fms-подобный рецептор тирозинкиназы, Flt4. Было показано, что два из этих родственных RTKs, Flt и KDR, связывают VEGF с высоким сродством ((De Vries at al, 1992, Science 255: 989-991; Terman et al, 1992, Biochem. Biophys. Res. Comm. 1992, 187: 1579-1586). Связывание VEGF с указанными рецепторами, экспрессированными в гетерологичных клетках, было ассоциировано с изменениями в статусе фосфорилирования тирозина клеточных белков и кальциевых потоков.
Производные хиназолина, которые являются ингибиторами рецепторной тирозинкиназы VEGF, описаны в публикации Международных патентных заявок WO 97/30035 и WO 98/13354. В WO 97/30035 и WO 98/13354 описываются соединения, которые обладают активностью против рецепторной тирозинкиназы VEGF, в то же время обладая некоторой активностью против рецепторной тирозинкиназы EGF.
Соединения по настоящему изобретению подпадают под объем широкого общего описания WO 97/30035 и WO 98/13354. Авторами обнаружено, что соединения по настоящему изобретению обладают очень высокой ингибирующей активностью в отношении рецепторной тирозинкиназы VEGF. Соединения по настоящему изобретению, которые были испытаны, проявляют in vitro активность в отношении ряда ксенотрансплантатов опухолей у мышей. Соединения по настоящему изобретению обладают выгодным токсикологическим профилем при испытании в течение 14 дней на крысах. Соединения по настоящему изобретению обладают очень высокой ингибирующей активностью в отношении рецепторной тирозинкиназы VEGF, проявляют in vivo активность против ряда ксенотрансплантатов опухолей у мышей и обладают выгодным токсикологическим профилем при испытании в течение 14 дней на крысах.
Соединения по настоящему изобретению ингибируют действие VEGF, что является ценным свойством при лечении патологических состояний, ассоциированных с развитием кровеносных сосудов и/или повышенной васкулярной проницаемостью, таких как рак, диабет, псориаз, ревматоидный артрит, саркома Капоши, гемангиома, острые и хронические нефропатии, атерома, артериальный рестеноз, аутоиммунные заболевания, острое воспаление, избыточные образования шрамов и спаек, эндометриоз, дисфункциональное маточное кровотечение и глазные болезни с пролиферацией ретинальных сосудов.
Соединения по настоящему изобретению обладают высокой активностью в отношении рецепторной тирозинкиназы VEGF, в то же время обладая некоторой активностью в отношении рецепторной тирозинкиназы EGF. Кроме того, некоторые соединения по настоящему изобретению обладают значительно более высокой активностью в отношении рецепторной тирозинкиназы VEGF по сравнению с активностью в отношении рецепторной тирозинкиназы EGF или рецепторной тирозинкиназы EGF R1. Без какой-либо привязки к теории такие соединения могут, например, представлять интерес при лечении опухолей, которые ассоциируются с VEGF, особенно тех опухолей, рост которых зависит от VEGF. Кроме того, полагают, что указанные соединения могут представлять интерес при лечении опухолевых состояний, ассоциированных как с VEGF, так и EGF, особенно, когда пациент страдает состоянием, при котором присутствуют опухоли, рост которых зависит как от VEGF, так и EGF.
В соответствии с одним аспектом настоящего изобретения предложено производное хиназолина формулы I
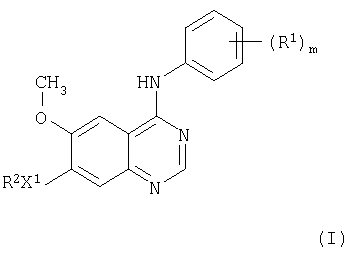
где m равно целому числу от 1 до 3;
R1 представляет галоген или C1-3 - алкил;
X1 представляет -О-;
R2 выбран из одной из следующих трех групп:
1) С1-5 - алкил-R3 (где R3 представляет пиперидин-4-ил, который имеет один или два заместителя, выбранных из гидрокси, галогена, C1-4 - алкила, C1-4 - гидроксиалкила и C1-4 - алкокси);
2) С2-5 - алкенил-R3 (где R3 имеет указанные выше значения);
3) С2-5 - алкинил-R3 (где R3 имеет указанные выше значения);
и где любая алкильная, алкенильная или алкинильная группа может иметь один или несколько заместителей, выбранных из гидрокси, галогена и амино;
или его соль, или его пролекарство.
Предпочтительно, m равно 2.
Фенильная группа, содержащая (R1)m, предпочтительно, выбрана из 2-фтор-4-метилфенила, 4-хлор-2,6-дифторфенила, 4-бром-2,6-дифторфенила, 4-хлор-2-фторфенильной группы и 4-бром-2-фторфенила.
Фенильная группа, содержащая (R1)m, более предпочтительно, выбрана из 4-хлор-2-фторфенила и 4-бром-2-фторфенила.
Фенильная группа, имеющая (R1)m, очень предпочтительно, выбрана из 4-бром-2-фторфенила.
R2, предпочтительно, представляет C1-5-алкил-R3 (где R3 имеет указанные выше значения).
R2, более предпочтительно, представляет С1-3-алкил-R3 (где R3 имеет указанные выше значения).
Конкретно, R2 представляет пиперидин-4-илметил, в котором кольцо пиперидина может иметь один или два заместителя, как указано здесь выше.
Более конкретно, R2 представляет пиперидин-4-илметил, в котором кольцо пиперидина может иметь один или два заместителя, выбранных из C1-4-алкила.
В частности, R2 представляет 1-метилпиперидин-4-илметил.
В соответствии со следующим аспектом настоящего изобретения предложено производное хиназолина формулы II
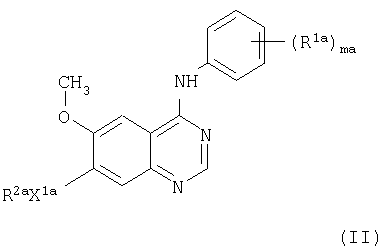
где ma равно целому числу от 1 до 3;
R1а представляет галоген или C1-3-алкил;
X1a представляет -О-;
R2a выбран из одной из следующих трех групп:
1) С1-5-алкил-R3 (где R3 имеет указанные выше значения);
2) С2-5-алкенил-R3 (где R3 имеет указанные выше значения);
3) С2-5-алкинил-R3 (где R3 имеет указанные выше значения);
или его соль, или его пролекарство.
Предпочтительно, ma равно 2.
Фенильная группа, имеющая (R1a)ma, предпочтительно, выбрана из 2-фтор-4-метилфенила, 4-хлор-2,6-дифторфенила, 4-бром-2,6-дифторфенила, 4-хлор-2-фторфенильной группы и 4-бром-2-фторфенила.
Фенильная группа, имеющая (R1a)ma, более предпочтительно, выбрана из 4-хлор-2-фторфенила и 4-бром-2-фторфенила.
Фенильная группа, имеющая (R1a)ma, очень предпочтительно, выбрана из 4-бром-2-фторфенила.
R2a, предпочтительно, представляет С1-5-алкил-R3 (где R3 имеет указанные выше значения).
R2a, более предпочтительно, представляет С1-3-алкил-R3 (где R3 имеет указанные выше значения).
Конкретно, R2a представляет пиперидин-4-илметил, в котором кольцо пиперидина может иметь один или два заместителя, как указано здесь выше.
Более конкретно, R2a представляет пиперидин-4-илметил, в котором кольцо пиперидина может иметь один или два заместителя, выбранных из C1-4-алкила.
В частности, R2a представляет 1-метилпиперидин-4-илметил. Предпочтительные соединения настоящего изобретения включают:
4-(4-хлор-2-фторанилино)-6-метокси-7-(1-метилпиперидин-4-илметокси)хиназолин,
4-(2-фтор-4-метиланилино)-6-метокси-7-(1-метилпиперидин-4-илметокси)хиназолин,
4-(4-бром-2-фторанилино)-6-метокси-7-(1-метилпиперидин-4-илметокси)хиназолин,
4-(4-хлор-2,6-дифторанилино)-6-метокси-7-(1-метилпиперидин-4-илметокси)хиназолин,
4-(4-бром-2,6-дифторанилино)-6-метокси-7-(1-метилпиперидин-4-илметокси)хиназолин,
4-(4-хлор-2-фторанилино)-6-метокси-7-(пиперидин-4-илметокси)хиназолин,
4-(2-фтор-4-метиланилино)-6-метокси-7-(пиперидин-4-илметокси)хиназолин,
4-(4-бром-2-фторанилино)-6-метокси-7-(пиперидин-4-илметокси)хиназолин,
4-(4-хлор-2,6-дифторанилино)-6-метокси-7-(пиперидин-4-илметокси)хиназолин,
4-(4-бром-2,6-дифторанилино)-6-метокси-7-(пиперидин-4-илметокси)хиназолин
и их соли, особенно их гидрохлоридные соли.
Более предпочтительные соединения настоящего изобретения включают:
4-(4-хлор-2-фторанилино)-6-метокси-7-(1-метилпиперидин-4-илметокси)хиназолин,
4-(4-бром-2-фторанилино)-6-метокси-7-(1-метилпиперидин-4-илметокси)хиназолин,
4-(4-хлор-2,6-дифторанилино)-6-метокси-7(1-метилпиперидин-4-илметокси)хиназолин,
4-(4-бром-2,6-дифторанилино)-6-метокси-7-(1-метилпиперидин-4-илметокси)хиназолин,
4-(4-хлор-2-фторанилино)-6-метокси-7-(пиперидин-4-илметокси)хиназолин,
4-(4-бром-2-фторанилино)-6-метокси-7-(пиперидин-4-илметокси)хиназолин,
4-(4-хлор-2,6-дифторанилино)-6-метокси-7-(пиперидин-4-илметокси)хиназолин,
4-(4-бром-2,6-дифторанилино)-6-метокси-7-(пиперидин-4-илметокси)хиназолин
и их соли, особенно их гидрохлоридные соли.
Особенно предпочтительные соединения настоящего изобретения включают:
4-(4-хлор-2-фторанилино)-6-метокси-7-(1-метилпиперидин-4-илметокси)хиназолин,
4-(4-бром-2-фторанилино)-6-метокси-7-(1-метилпиперидин-4-илметокси)хиназолин,
4-(4-хлор-2,6-дифторанилино)-6-метокси-7-(1-метилпиперидин-4-илметокси)хиназолин,
4-(4-бром-2,6-дифторанилино)-6-метокси-7-(1-метилпиперидин-1-илметокси)хиназолин
и их соли, особенно их гидрохлоридные соли.
Более конкретно, предпочтительные соединения настоящего изобретения включают:
4-(4-хлор-2-фторанилино)-6-метокси-7-(1-метилпиперидин-4-илметокси)хиназолин и
4-(4-бром-2-фторанилино)-6-метокси-7-(1-метилпиперидин-4-илметокси)хиназолин
и их соли, особенно их гидрохлоридные соли.
Особенно предпочтительным соединением настоящего изобретения является 4-(4-бром-2-фторанилино)-6-метокси-7-(1-метилпиперидин-4-илметокси)хиназолин и его соли, особенно гидрохлоридные соли.
Во избежание каких-либо сомнений, следует учесть, что в тех случаях, когда в данном описании группа определяется выражением «ранее здесь определяемая» или «определяемая здесь ранее», указанная группа включает первое встречающееся и самое широкое определение, а также каждое и все из предпочтительных определений для этой группы. Аналогичное условие применимо для выражения «как указано далее» или «определяемая далее».
В данном описании, если не указано иное, термин «алкил» включает алкильные группы как с неразветвленной, так и с разветвленной цепью, но ссылки на индивидуальные алкильные группы, такие как «пропил», являются конкретными только для варианта неразветвленной цепи. Аналогичное условие применимо для других родовых терминов. Если не указано иное, термин «алкил» преимущественно относится к цепям с 1-5 атомами углерода, предпочтительно, 1-3 атомами углерода. Термин «алкокси», используемый здесь, если не указано иначе, включает «алкил»-О-группы, в которых «алкил» имеет значения, указанные здесь выше. Термин «арил», используемый здесь, если не указано иначе, включает отношение к С6-10-арильной группе, которая может, при необходимости, иметь один или несколько заместителей, выбранных из галогена, алкила, алкокси, нитро, трифторметила и циано (где алкил и алкокси имеют значения, указанные здесь выше). Термин «арилокси», используемый здесь, если не указано иначе, включает «арил»-О-группы, в которых «арил» имеет значения, указанные здесь выше. Термин «сульфонилокси», используемый здесь, относится к алкилсульфонилокси- и арилсульфонилоксигруппам, в которых «алкил» и «арил» имеют значения, указанные здесь выше. Термин «алканоил», используемый здесь, если не указано иначе, включает формил и алкил-С=О-группы, в которых «алкил» имеет значения, указанные здесь выше, например С2-алканоил является этаноилом и относится к СН3С=О, C1-алканоил является формилом и относится к СНО. В данном описании, если не указано иначе, термин «алкенил» включает алкенильные группы как с неразветвленной, так и разветвленной цепью, но ссылки на индивидуальные алкенильные группы, такие как 2-бутенил, являются специфичными только для варианта неразветвленной цепи. Если не указано иначе, термин «алкенил», преимущественно, относится к цепям с 2-5 атомами, предпочтительно, 3-5 атомами углерода. В данном описании, если не указано иначе, термин «алкинил» включает алкинильные группы как с неразветвленной, так и разветвленной цепью, но ссылки на индивидуальные алкенильные группы, такие как 2-бутинил, являются специфичными только для варианта неразветвленной цепи. Если не указано иначе, термин «алкинил», преимущественно, относится к цепям с 2-5 атомами, предпочтительно, 3-5 атомами углерода.
В формуле I, как указано здесь выше, водород может присутствовать в положениях 2, 5 и 8 хиназолиновой группы.
В настоящем изобретении должно быть ясно, что соединение формулы I или его соль может проявлять феномен таутомерии и что изображения формул в данном описании могут представлять только одну из возможных таутомерных форм. Должно быть понятно, что изобретение включает любую таутомерную форму, которая ингибирует активность рецепторной тирозинкиназы VEGF, и не должно ограничиваться только любой одной таутомерной формой, используемой в изображениях формул.
Должно быть понятно также, что некоторые соединения формулы I и их соли могут существовать в сольватированных, а также несольватированных формах, таких как, например, гидратированные формы. Должно быть понятно, что изобретение включает все такие сольватированные формы, которые ингибируют активность рецепторной тирозинкиназы VEGF.
Чтобы избежать появления сомнений, следует знать, что в соединении формулы I, когда R2 представляет, например, группу формулы С2-5-алкенил-R3, он представляет С2-5-алкенильную часть, которая связана с X1, и аналогичную условность применяют для других групп. Когда R2 представляет группу 1-R3-проп-1-ен-3-ил, он является первым углеродом, к которому присоединен R3, и он является третьим углеродом, который связан с X1, аналогично, когда R2 представляет группу 2-R3-пен-3-ен-5-ил, он является вторым углеродом, к которому присоединена группа R3, и он является пятым углеродом, который связан с X1, и аналогичную условность применяют для других групп.
Соединения формулы I можно вводить в виде пролекарства, которое расщепляется в организме человека или животного с выделением соединения формулы I. Примеры пролекарств включают гидролизуемые in vivo сложные эфиры соединения формулы I.
Различные формы пролекарств известны в данной области. Примеры таких пролекарственных производных см. в публикациях:
a) Design of Prodrugs, edited by H. Bundgaard, (Elsevier, 1985) and Methods in Enzymotogy, Vol.42, p.309-396, edited by K. Widder, et al. (Academic Press, 1985);
b) A Textbook of Drug Design and Development, edited by Krogsgaard-Larsen and H.Bundgaard, Chapter 5 "Design and Application of Prodrugs", by H.Bundgaard p.113-191 (1991);
c) H.Bundgaard, Advanced Drug Delivery Reviews, 8, 1-38 (1992);
d) H.Bundgaard, et al., Journal of Pharmaceutical Sciences, 77, 285 (1988); and
e) N.Kakeya. et al., Chem Pharm Bull, 32, 692 (1984).
Гидролизуемый in vivo сложный эфир соединения формулы I, содержащего гидроксигруппу, включает неорганические эфиры, такие как фосфатные эфиры (в том числе фосфорамидные циклические эфиры), ацилоксиалкиловые простые эфиры и аналогичные соединения, которые в результате гидролиза in vivo сложного эфира расщепляются с образованием исходной гидроксигруппы(групп). Примеры ацилоксиалкиловых простых эфиров включают ацетоксиметокси и 2,2-диметилпропионилоксиметокси. Выбор групп, образующих гидролизуемые in vivo сложные эфиры для гидрокси, включает алканоил, бензоил, фенилацетил и замещенный бензоил и фенилацетил, алкоксикарбонил (для получения алкилкарбонатных эфиров), диалкилкарбамоил и N-(диалкиламиноэтил)-N-алкилкарбамоил (для получения карбаматов), диалкиламиноацетил и карбоксиацетил. Примеры заместителей бензоила включают морфолино и пиперазино, соединенные по атому азота кольца через метиленовую группу с 3- или 4-положением кольца бензоила.
Настоящее изобретение относится к соединениям формулы I, как указано здесь выше, а также их солям. Соли для использования в фармацевтических композициях должны быть фармацевтически приемлемыми солями, но другие соли могут быть пригодными при получении соединений формулы I и их фармацевтически приемлемых солей. Фармацевтически приемлемые соли по изобретению могут, например, включать кислотно-аддитивные соли соединений формулы I, как указано здесь выше, которые являются достаточно основными для образования таких солей. Такие кислотно-аддитивные соли включают, например, соли с неорганическими или органическими кислотами, образующими фармацевтически приемлемые анионы, такие как галогенводородные (особенно, хлористоводородная или бромистоводородная кислота, из которых хлористоводородная кислота особенно предпочтительна), или с серной или фосфорной кислотой, или с трифторуксусной кислотой, лимонной или малеиновой кислотой. Кроме того, когда соединения формулы I являются достаточно кислотными, можно получить фармацевтически приемлемые соли с неорганическим или органическим основанием, которое образует фармацевтически приемлемый катион. Такие соли с неорганическими или органическими основаниями включают, например, соль щелочного металла, такую как соль натрия или калия, соль щелочно-земельного металла, такую как соль кальция или магния, соль аммония или, например, соль с метиламином, диметиламином, триметиламином, пиперидином, морфолином или трис(2-гидроксиэтил)амином.
Соединение формулы I или его соль, или другие соединения изобретения (как указано здесь выше) можно получить любым способом, который, как известно, является пригодным для получения химически близких соединений. Такие способы включают, например, способы, иллюстрированные в Европейских патентных заявках, публикации №№0520722, 0566226, 0602851 и 0635498, и в публикациях Международных патентных заявок WO 97/22596, WO 97/30035, WO 97/32856 и WO 98/13354. Такие способы предложены в качестве следующих признаков изобретения и описаны ниже. Необходимые исходные материалы можно получить стандартными методиками органической химии. Получение таких исходных материалов описывается в сопровождающих неограничивающих примерах. В альтернативном случае необходимые исходные материалы можно получить методиками, аналогичными иллюстрированным методикам, которые находятся в пределах знаний химика-органика средней квалификации.
Таким образом, следующие способы от (а) до (d) и от (i) до (iv) составляют следующие признаки настоящего изобретения.
Синтез соединений формулы I
(а) Соединения формулы I и их соли можно получить взаимодействием соединения формулы III
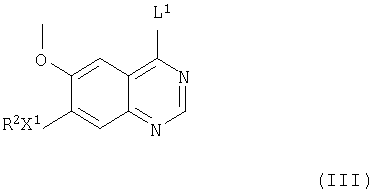
(где R2 и Х1 имеют значения, указанные выше, и L1 представляет заменяемую часть) с соединением формулы IV
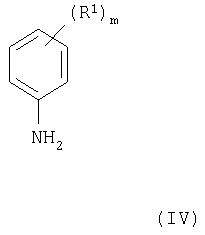
(где R1 и m имеют значения, указанные выше), при этом получают соединения формулы I и их соли. Подходящая заменяемая группа L1 представляет, например, галоген, алкокси (предпочтительно, C1-4-алкокси), арилокси или сульфонилоксигруппу, например хлор, бром, метокси, фенокси, метансульфонилокси или толуол-4-сульфонилоксигруппу.
Взаимодействие преимущественно проводят в присутствии либо кислоты, либо основания. Такой кислотой является, например, безводная неорганическая кислота, такая как хлористый водород. Таким основанием является, например, органический амин, такой как, например, пиридин, 2,6-лутидин, коллидин, 4-диметиламинопиридин, триэтиламин, морфолин, N-метилморфолин или диазабицикло[5.4.0]ундец-7-ен, или, например, карбонат или гидроксид щелочного металла или щелочно-земельного металла, например карбонат натрия, карбонат калия, карбонат кальция, гидроксид натрия или гидроксид калия. В альтернативном случае таким основанием является, например, гидрид щелочного металла, например гидрид натрия, или амид щелочного металла или щелочно-земельного металла, например амид натрия или бис(триметилсилил)амид натрия. Взаимодействие, предпочтительно, проводят в присутствии инертного растворителя или разбавителя, например алканола или сложного эфира, такого как метанол, этанол, 2-пропанол или этилацетат, галогенированного растворителя, такого как метиленхлорид, трихлорметан или тетрахлорид углерода, простого эфира, такого как тетрагидрофуран или 1,4-диоксан, ароматического углеводородного растворителя, такого как толуол, или диполярного органического растворителя, такого как N,N-диметилформамид, N,N-диметилацетамид, N-метилпирролидин-2-он или диметилсульфоксид. Взаимодействие подходящим образом проводят при температуре в диапазоне, например, от 10 до 150°С, предпочтительно, в диапазоне от 20 до 80°С.
Соединение по изобретению можно получить указанным способом в виде свободного основания или, в альтернативном случае, его можно получить в виде соли с кислотой формулы H-L1, где L1 имеет значения, указанные выше. Когда необходимо получить свободное основание из соли, соль можно обработать основанием, как указано выше, с использованием общепринятой методики.
(b) Соединения формулы I и их соли можно получить взаимодействием, подходящим образом, в присутствии основания, как указано выше, соединения формулы V
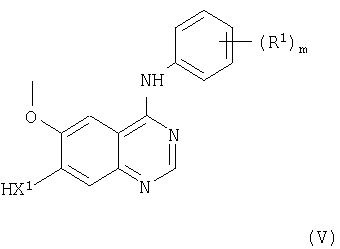
(где m, X1 и R1 имеют значения, указанные выше) с соединением формулы VI

(где R2 и L1 имеют значения, указанные выше); L1 представляет заменяемую группу, например галоген или сульфонилоксигруппу, такую как бром- или метансульфонилоксигруппа. В подходящем случае L1 представляет группу O-+P(Y)3 (где Y представляет бутил или фенил), и в таких случаях соединение формулы VI можно удобно получить in situ. Взаимодействие, предпочтительно, проводят в присутствии основания (как указано выше в способе (а)) и, преимущественно, в присутствии инертного растворителя (как указано выше в способе (а)), преимущественно, при температуре в диапазоне, например, от 10 до 150°С, в подходящем случае, приблизительно при 50°С.
(с) Соединения формулы I и их соли можно получить взаимодействием соединения формулы VII
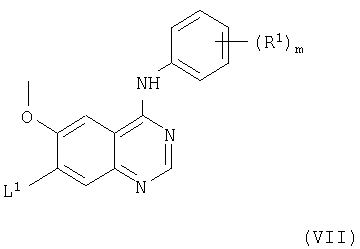
с соединением формулы VIII

(где L1, R1, R2, m и X1, все, имеют значения, указанные выше). Взаимодействие подходящим образом можно проводить в присутствии основания (как указано здесь выше в способе (а)) и, преимущественно, в присутствии инертного растворителя или разбавителя (как указано выше в способе (а)), преимущественно, при температуре в диапазоне, например, от 10 до 150°С, в подходящем случае приблизительно при 100°С.
(d) Соединения формулы I и их соли можно получить снятием защиты у соединения формулы IX
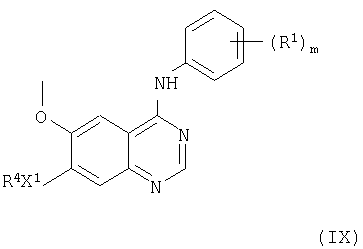
где R1, m и Х1, все, имеют указанные выше значения, и R4 представляет защищенную группу R2, где R2 имеет указанные выше значения, но дополнительно имеет одну или несколько защитных групп Р2. Выбор защитной группы находится в пределах стандартного знания химика-органика, например, знания, включенного в стандартные пособия, такие как «Protective Groups in Organic Synthesis» T.W. Greene and R.G.M. Wuts, 2nd Ed. Wiley 1991. Предпочтительно, Р2 представляет такую защитную группу, как карбамат (алкоксикарбонил) (такой как, например, трет-бутоксикарбонил, трет-амилоксикарбонил, циклобутоксикарбонил, пропоксикарбонил, метоксикарбонил, этоксикарбонил, изопропоксикарбонил, аллилоксикарбонил или бензилоксикарбонил). Более предпочтительно, Р2 представляет трет-бутоксикарбонил. Взаимодействие, предпочтительно, проводят в присутствии кислоты. Такая кислота является, например, неорганической кислотой, такой как хлористый водород, бромистый водород, или органической кислотой, такой как трифторуксусная кислота, трифторметансульфоновая кислота. Взаимодействие можно проводить в присутствии инертного растворителя, такого как метиленхлорид, трихлорметан, и в присутствии следов воды. Взаимодействие в подходящем случае проводят при температуре в диапазоне 10-100°С, предпочтительно, в диапазоне 20-80°С.
Синтез промежуточных продуктов
(1) Соединения формулы III и их соли, в которых L1 представляет галоген, можно, например, получить галогенированием соединения формулы X
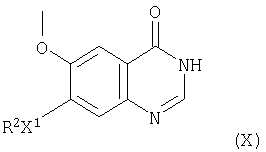
(где R2 и Х1 имеют указанные выше значения).
Подходящие галогенирующие агенты включают галогенангидриды неорганических кислот, например тионилхлорид, хлорид фосфора (III), оксихлорид фосфора (V) и хлорид фосфора (V). Реакцию галогенирования в подходящем случае проводят в присутствии инертного растворителя или разбавителя, такого как, например, галогенированный растворитель, такой как метиленхлорид, трихлорметан или тетрахлорид углерода, или ароматический углеводородный растворитель, такой как бензол или толуол. Взаимодействие подходящим образом проводят при температуре в диапазоне, например, от 10 до 150°С, предпочтительно, в диапазоне от 40 до 100°С.
Соединения формулы Х и их соли можно, например, получить взаимодействием соединения формулы XI
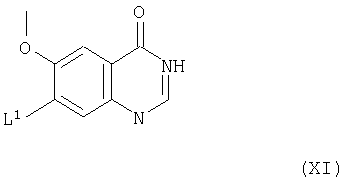
(где L1 имеет указанные выше значения) с соединением формулы VIII, как указано выше. Взаимодействие можно подходящим образом проводить в присутствии основания (как указано выше в способе (а)) и, преимущественно, в присутствии инертного растворителя или разбавителя (как указано выше в способе (а)), преимущественно, при температуре в диапазоне, например, от 10 до 150°С, в подходящем случае, приблизительно при 100°С.
Соединения формулы Х и их соли можно также получить циклизацией соединения формулы XII
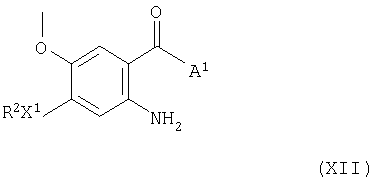
(где R2 и X1 имеют значения, указанные выше, и А1 представляет гидрокси-, алкокси-(предпочтительно, C1-4-алкокси) или аминогруппу), при этом получают соединение формулы Х или его соль. Циклизацию можно проводить взаимодействием соединения формулы XII, где А1 представляет гидрокси- или алкоксигруппу, с формамидом или его эквивалентом, эффективным для того, чтобы вызвать циклизацию, при этом получают соединение формулы Х или его соль, такое как хлорид [3-(диметиламино)-2-азапроп-2-енилиден]диметиламмония. Циклизацию в подходящем случае проводят в присутствии формамида в качестве растворителя или в присутствии инертного растворителя или разбавителя, такого как простой эфир, например 1,4-диоксан. Циклизацию в подходящем случае проводят при повышенной температуре, предпочтительно, в диапазоне от 80 до 200°С. Соединения формулы Х можно также получить циклизацией соединения формулы XII, где А1 представляет аминогруппу, с муравьиной кислотой или ее эквивалентом, эффективным для того, чтобы вызвать циклизацию, при этом получают соединение формулы Х или его соль. Эквиваленты муравьиной кислоты, эффективные для того, чтобы вызвать циклизацию, включают, например, три-С1-4-алкоксиметан, например, триэтоксиметан и триметоксиметан. Циклизацию в подходящем случае проводят в присутствии каталитического количества безводной кислоты, такой как сульфоновая кислота, например, п-толуолсульфоновая кислота, и в присутствии инертного растворителя или разбавителя, такого как, например, галогенированного растворителя, такой как метиленхлорид, трихлорметан или тетрахлорид углерода, простой эфир, такой как диэтиловый эфир или тетрагидрофуран, или ароматический углеводородный растворитель, такой как толуол. Циклизацию в подходящем случае проводят при температуре в диапазоне, например, от 10 до 100°С, предпочтительно, в диапазоне от 20 до 50°С.
Соединения формулы XII и их соли можно, например, получить восстановлением нитрогруппы в соединении формулы XIII
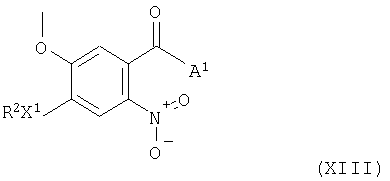
(где R2, X1 и А1 имеют указанные выше значения) с получением соединения формулы XII, как указано здесь ранее. Восстановление нитрогруппы можно в подходящем случае осуществить любой из методик, известных для такого превращения. Восстановление можно проводить, например, гидрированием раствора нитросоединения в присутствии инертного растворителя или разбавителя, как указано здесь выше, в присутствии металла, эффективного для катализа реакций гидрирования, такого как палладий или платина. Следующим восстанавливающим агентом является, например, активированный металл, такой как активированное железо (полученный, например, промыванием порошка железа разбавленным раствором кислоты, такой как хлористоводородная кислота). Так, например, восстановление можно проводить нагреванием нитросоединения и активированного металла в присутствии растворителя или разбавителя, такого как смесь воды и спирта, например, метанола или этанола, до температуры в диапазоне, например, от 50 до 150°С, в подходящем случае, приблизительно при 70°С.
Соединения формулы XIII и их соли можно например, получить взаимодействием соединения формулы XIV
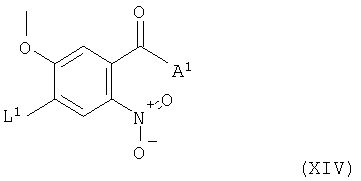
(где L1 и А1 имеют указанные выше значения) с соединением формулы VIII, как указано здесь выше, при этом получают соединение формулы XIII. Взаимодействие соединений формул XIV и VIII в подходящем случае проводят в условиях, которые описаны для указанного выше способа (с).
Соединения формулы XIII и их соли можно, например, получить также взаимодействием соединения формулы XV
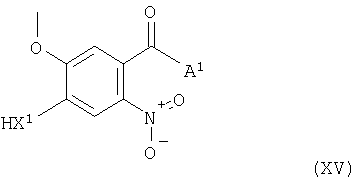
(где L1 и А1 имеют указанные выше значения) с соединением формулы VI, как указано здесь выше, при этом получают соединение формулы XIII, как указано здесь выше. Взаимодействие соединений формул XV и VI в подходящем случае проводят в условиях, которые описаны здесь выше для способа (b).
Соединения формулы III и их соли можно также получить, например, взаимодействием соединения формулы XVI
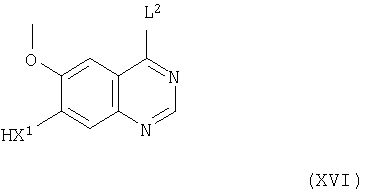
(где X1 имеет значения, указанные здесь выше, и L2 представляет заменяемую защитную часть) с соединением формулы VI, как указано здесь выше, при этом получают соединение формулы III, в которой L1 представлен L2.
Подходящим образом используют соединение формулы XVI, где L2 представляет феноксигруппу, которая может, если необходимо, содержать вплоть до 5 заместителей, предпочтительно, вплоть до 2 заместителей, выбранных из галогена, нитро и циано. Взаимодействие можно в подходящем случае проводить в условиях, описываемых для указанного выше способа (b).
Соединения формулы XVI и их соли, как указано здесь выше, можно, например, получить удалением защитной группы у соединения формулы XVII
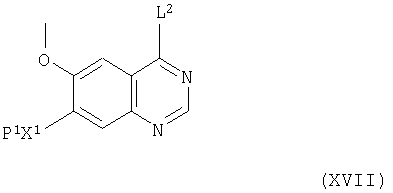
(где X1 и L2 имеют значения, указанные здесь выше, и Р1 представляет защитную группу фенольного гидроксила). Выбор защитной группы фенольного гидроксила Р1 находится в пределах стандартного знания химика-органика, например, знания, включенного в стандартные пособия, такие как «Protective Groups in Organic Synthesis» T.W.Greene and R.G.M.Wuts, 2nd Ed. Wiley 1991, и включает простые эфиры (например, метиловый, метоксиметиловый, аллиловый и бензиловый и бензиловый, замещенный заместителями вплоть до двух, выбранных из C1-4-алкокси и нитро), силиловые простые эфиры (например, трет-бутилдифенилсилиловый и трет-бутилдиметилсилиловый), сложные эфиры (например, ацетат и бензоат) и карбонаты (например, метил и бензил и бензил, замещенный заместителями вплоть до двух, выбранных из C1-4-алкокси и нитро). Снятие защиты можно осуществить методиками, хорошо известными в литературе, например, когда Р1 представляет бензильную группу, снятие защиты можно осуществить гидрогенолизом или обработкой трифторуксусной кислотой.
Удаление такой защитной группы фенольного гидроксила можно осуществить любой из методик, известных для такого превращения, включая условия взаимодействия методик, указанных в стандартных пособиях, таких как пособие, указанное здесь ранее, или относящейся к ним методикой. Условия взаимодействия, предпочтительно, являются такими, что гидроксипроизводное получают без нежелательных взаимодействий у других участков в пределах исходных соединений или соединений-продуктов. Например, когда защитная группа Р1 представляет ацетат, преобразование можно в подходящем случае осуществить обработкой производного хиназолина основанием, как указано здесь выше, включающим аммиак и его моно- и диалкилированные производные, предпочтительно, в присутствии протонного растворителя или сорастворителя, такого как вода или спирт, например метанол или этанол. Такое взаимодействие можно осуществить в присутствии дополнительного инертного растворителя или разбавителя, как указано здесь выше, и при температуре в диапазоне от 0 до 50°С, в подходящем случае, приблизительно при 20°С.
Одно соединение формулы III можно, если необходимо, преобразовать в другое соединение формулы III, в которой часть L1 является другой. Так, например, соединение формулы III, в которой L1 другой, чем галоген, например, необязательно замещенный фенокси, можно преобразовать в соединение формулы III, в которой L1 представляет галоген, гидролизом соединения формулы III (в которой L1 является другим, чем галоген) с получением соединения формулы X, как указано здесь выше, с последующим введением галогенида в соединение формулы X, таким образом полученного, как указано здесь выше, при этом получают соединение формулы III, в которой L1 представляет галоген.
(ii) Соединения формулы V, как указано выше, и их соли можно получить снятием защиты у соединения формулы XVIII
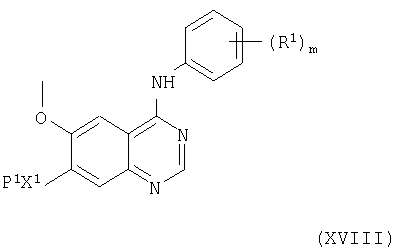
(где R1, Р1, Х1 и m имеют значения, указанные здесь выше) способом, например, описываемым выше в (i).
Соединения формулы XVIII и их соли можно получить взаимодействием соединений формул XVII и IV, как указано здесь выше, в условиях, описанных здесь выше в (а), при этом получают соединение формулы XVIII или его соль.
(iii) Соединения формулы VII и их соли, как указано здесь выше, можно получить взаимодействием соединения формулы XIX
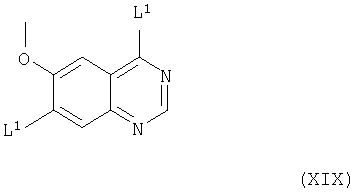
(где L1 имеет значения, указанные выше, и L1 в 4- и 7-положениях могут быть одинаковыми или разными) с соединением формулы IV, как указано здесь выше, причем взаимодействие проводят, например, способом, описываемым выше в (а).
(iv) Соединение формулы IX можно получить взаимодействием соединения формулы V, как указано здесь выше, с соединением формулы XX

где R4 и L1 имеют значения, указанные выше, в условиях, описанных в указанном выше способе (b), при этом получают соединение формулы IX или его соль. Взаимодействие, предпочтительно, проводят в присутствии основания (как указано здесь выше в способе (а)) и преимущественно в присутствии инертного растворителя или разбавителя (как указано выше в способе (а)), преимущественно, при температуре в диапазоне, например, от 10 до 150°С, в подходящем случае, в диапазоне 20-50°С.
Когда необходима фармацевтически приемлемая соль соединения формулы I, ее можно получить, например, взаимодействием указанного соединения, например, с кислотой с использованием общепринятой методики, причем кислота имеет фармацевтически приемлемый анион или ее можно получить взаимодействием указанного соединения с основанием общепринятой методикой.
Идентификация соединений, которые, возможно, ингибируют активность тирозинкиназы, ассоциированную с рецепторами VEGF, такими как Flt и/или KDR, и которые ингибируют развитие кровеносных сосудов и/или повышают васкулярную проницаемость, является необходимой и представляет предмет настоящего изобретения. Указанные свойства можно оценить, например, с использованием одной или нескольких методик, указанных ниже.
(а) Испытание ингибирования in vitro рецепторной тирозинкиназы
Данный анализ определяет способность испытуемого соединения ингибировать активность тирозинкиназы. ДНК, кодирующую цитоплазматические домены рецептора VEGF или эпидермального фактора роста (EGF), можно получить общим синтезом гена (Edwards M, International Biotechnology Lab 5(3), 19-25, 1987) или клонированием. Ее можно затем экспрессировать в подходящей системе экспрессии для получения полипептида с активностью тирозинкиназы. Было обнаружено, что, например, рецепторные цитоплазмические домены VEGF или EGF, которые были получены экспрессией рекомбинантного белка в клетках насекомых, демонстрируют свойственную им активность тирозинкиназы. В случае рецептора VEGF Flt (Genbank accession number X51602) фрагмент ДНК с 1,7 т.п.н., кодирующий большую часть цитоплазматического домена, начинающегося с метионина 783 и включающего концевой кодон, описанный Shibuya et al. (Oncogene, 1990, 5: 519-524), был выделен из кДНК и клонирован в бакуловирусный вектор-переносчик (например, pAcYM1 (см. Бакуловирусная система экспрессии: A Laboratory Guide, L.A.King and R.D.Possee, Chapman and Hall, 1992) или рАс360 или pBlueBacHis (доступен от Invitrogen Corporation). Данную рекомбинантную конструкцию котрансфекцировали в клетки насекомых (например, Spodoptera frugiperda 21(Sf21)) с вирусной ДНК (например, Pharmingen BaculoGold) для получения рекомбинантного бакуловируса. (Детали способов сборки молекул рекомбинантной ДНК, получение и использование рекомбинантного бакуловируса можно найти в стандартных монографиях, например, в Sambrook et al, 1989, Molecular cloning-A Laboratory Manual, 2nd edition, Cold Spring Harbour Laboratory Press and O'Reilly et al. 1992, Baculovirus Expression Vectors - A Laboratory Manual, W.H. Freeman and Co, New York). Для других тирозинкиназ, используемых в анализах, цитоплазматические фрагменты, начинающиеся с метионина 806 (KDR, Genbank accession number L04947) и метионина 668 (EGF receptor, Genbank accession number Х00588), можно клонировать и экспрессировать аналогичным способом.
Для экспрессии активности тирозинкиназы cFIt клетки Sf21 инфекцировали очищенным из бляшек рекомбинантным вирусом cFIt при множественности инфекции 3 и собирали спустя 48 часов. Собранные клетки промывали охлажденным льдом забуференным фосфатом солевым раствором (PBS) (10 мМ фосфат натрия, рН 7,4, 138 мМ хлорид натрия, 2,7 мМ хлорид калия), затем снова суспендировали в охлажденном льдом HNTG/PMSF (20 мМ Hepes, рН 7,5, 150 мМ хлорид натрия, 10% об./об. глицерин, 1% об./об. тритон Х100, 1,5 мМ хлорид магния, 1 мМ этиленгликольбис(β-аминоэтиловый эфир)-N,N,N'N'-тетрауксусная кислота (EGTA), 1 мМ PMSF (фенилметилсульфонилфторид); PMSF добавляют непосредственно перед использованием из свежеприготовленного 100 мМ раствора в метаноле, причем используют 1 мл HNTG/PMSF на 10 миллионов клеток. Суспензию центрифугировали в течение 10 минут при 13000 об/мин при 4°С, супернатант (исходный раствор фермента) удаляли и сохраняли в аликвотах при -70°С. Каждую новую серию исходного раствора фермента титровали в анализе разбавлением разбавителем фермента (100 мМ Hepes, рН 7,4, 0,2 мМ ортованадат натрия, 0,1% об./об. тритона Х100, 0,2 мМ дитиотреит). Для типичной серии исходный раствор фермента разбавляли 1 к 2000 разбавителем фермента и 50 мкл разбавленного фермента использовали для каждой лунки анализа.
Исходный раствор субстрата получали из статистического сополимера, содержащего тирозин, например, поли(Glu, Ala, Tyr), 6:3:1 (Sigma Р3899), сохраняли в виде исходного раствора 1 мг/мл в PBS при -20°С и разбавляли 1 к 500 PBS для покрытия планшета.
За день до анализа 100 мкл разбавленного раствора субстрата распределяли по всем лункам аналитических планшетов (Nunc maxisorb 96-well immunoplates), которые герметизировали и оставляли на ночь при 4°С.
В день анализа раствор субстрата выгружали и лунки аналитического планшета промывали один раз PBST (PBS, содержащий 0,05% об./об. твина 20) и один раз 50 мМ Hepes с рН 7,4.
Испытуемые соединения разбавляли 10% диметилсульфоксидом (ДМСО) и 25 мкл разбавленного соединения переносили в лунки промытых аналитических планшетов. «Общие» контрольные лунки содержали 10% ДМСО вместо соединения. Двадцать пять микролитров 40 мМ хлорида марганца(II), содержащего 8 мкМ аденозин-5'-трифосфат (АТФ), добавляли во все лунки для испытания, за исключением «слепых» контрольных лунок, которые содержали хлорид марганца(II) без АТФ. Для начала взаимодействий 50 мкл свежеразбавленного фермента добавляли в каждую лунку и планшеты инкубировали при комнатной температуре в течение 20 минут. Жидкость затем выгружали и лунки промывали два раза PBST. Сто микролитров мышиных IgG-антител против фосфотирозина (Upstate Biotechnology Ink. Product 05-321), разбавленных 1 к 6000 содержащим PBST 0,5% масс./об. бычьим сывороточным альбумином (BSA), добавляли к каждой лунке и планшеты инкубировали в течение 1 часа при комнатной температуре до выгрузки жидкости и промывания лунок два раза PBST. Добавляли 100 микролитров связанных с пероксидазой из хрена (HRP) овечьих антител против мышиного Ig (Amersham product NXA 931), разбавленных 1 к 500 PBST, содержащего 0,5% масс./об. BSA, и планшеты инкубировали в течение 1 час при комнатной температуре до выгрузки жидкости и промывания лунок два раза PBST. В каждую лунку добавляли 100 микролитров раствора 2,2'-азинобис-(3-этилбензтиазолин-6-сульфоновой кислоты) (ABST), свежеприготовленного с использованием одной таблетки 50 мг ABTS (Boehringer 1204 521) в 50 мл свежеполученного раствора 50 мМ фосфат-цитратного буфера, рН 5,0,+0,03% пербората натрия (получен с 1 капсулой фосфат-цитратного буфера с перборатом натрия (PCSB) (Sigma Р4922) на 100 мл дистиллированной воды). Планшеты затем инкубировали в течение 20-60 минут при комнатной температуре до тех пор, пока величина оптической плотности «общих» контрольных лунок, измеренная при 405 нм с использованием считывающего планшет спектрофотометра, не стала приблизительно 1,0. «Слепые» (без АТФ) и «общие» (без соединения) контрольные величины использовали для определения диапазона разбавления испытуемого соединения, который дает 50% ингибирование ферментативной активности.
(b) Анализ пролиферации in vitro HUVEC
Данный анализ определяет способность испытуемого соединения ингибировать стимулированную фактором роста пролиферацию эндотелиальных клеток умбиликальной вены человека (HUVEC).
Клетки HUVEC выделяли в MCDB 131 (Gibco BRL) + 7,5% об./об. фетальной телячьей сыворотки (FCS) и высевали (при пересевах от 2 до 8) в MCDB 131+2% об./об. FCS+3 мкг/мл гепарина + 1 мкг/мл гидрокортизона при концентрации 1000 клеток/лунку в 96-луночных планшетах. После не менее чем 4 дней к ним добавляли дозы соответствующего фактора роста (например, 3 нг/мл VEGF, 3 нг/мл EGF или 0,3 нг/мл b-FGF) и соединения. Культуры затем инкубировали в течение 4 дней при 37°С с 7,5% диоксида углерода. На четвертый день культуры подвергали кратковременному мечению 1 мкКи/лунку меченного литием тимидина (Amersham product TRA 61) и инкубировали в течение 4 часов. Клетки собирали с использованием харвестера 96-луночного планшета (Tomtek) и затем анализировали для определения включения трития счетчиком бета-излучения планшетов. Включение радиоактивности в клетки, выраженное как число импульсов в минуту, использовали для измерения ингибирования соединениями стимулированной фактором роста пролиферации клеток.
(с) Модель твердого опухолевого заболевания
Данное испытание измеряет способность соединений ингибировать рост твердых опухолей.
Ксенотрансплантаты опухоли CaLu-6 имплантировали в боковую часть тела самок мышей athymic Swiss nu/nu подкожной инъекцией 1x106 клеток CaLu-6/мышь в 100 мкл 50% (об./об.) раствора Matrigel в не содержащей сыворотку культуральной среде. Спустя 10 дней после клеточной имплантации мышей распределяли по 8-10 группам так, чтобы достичь сравнимые средние объемы групп. Опухоли измеряли с использованием нониусного штангенциркуля и объемы вычисляли так: (l×w)×√(l×w)×(π/6), где l представляет самый большой диаметр и w представляет диаметр, перпендикулярный самому большому диаметру. Испытуемые соединения вводили перорально один раз в день в течение минимум 21 дней, а контрольные животные получали разбавитель соединения. Размер опухолей измеряли два раза в неделю. Уровень ингибирования роста вычисляли сравнением среднего объема опухоли контрольной группы по сравнению с группой обработки, и статистическую значимость определяли с использованием t-критерия Стьюдента и/или критерия суммы рангов Манна-Уитни. Ингибирующее действие обработкой соединением считали значимым, когда р<0,05.
Токсикологический профиль соединений настоящего изобретения можно анализировать, например, с использованием исследования крыс в течение 14 дней, как описано ниже.
(d) Испытание токсичности на крысах в течение 14 дней
Данное испытание измеряет активность соединений в увеличении зоны гипертрофии в бедренных эпифезарных ростовых пластинах дистальной бедренной кости и проксимальной большеберцовой кости и позволяет оценить гистопатологические изменения в других тканях.
Развитие кровеносных сосудов является существенным событием в эндохрящевой оссификации во время удлинения длинных костей, и было предположено, что васкулярная инвазия ростовой пластины зависит от продуцирования VEGF гипертрофическими хондроцитами. Увеличение зоны гипертрофических хондроцитов и ингибирование развития кровеносных сосудов были продемонстрированы после обработки агентами, которые специфически секвестрируют VEGF, таким как, например,
(i) растворимый рецепторный химерный белок VEGF (Flt-(1-3)-IgG) у мышей (Geiber, H-P., Vu, T.H., Ryan, A.M, Kowalski, J., Werb, Z. and Ferrara, N. VEGF couples hypertrophic cartilage remodelling, ossification and angiogenesis during endochondral bone formation, Nature Med., 5: 623-628, 1999)
и (ii) рекомбинантное гуманизированное моноклональное IgG1-антитело против VEGF в организме обезьяны cynomologus (Ryan, A.M., Eppler, D.B., Hagler, K.E., Bruner, R.H., Thomford, P.J., Hall, R.L., Shopp, G.M. and O'Niell, C.A. Preclinical Safety Evaluation of rhuMAbVEGF, an antiangiogenic humanised monoclonal antibody, Tox. Path., 27: 78-86, 1999).
Ингибитор активности рецепторной тирозинкиназы VEGF, следовательно, должен также ингибировать васкулярную инвазию хряща и увеличивать зону гипертрофии в бедренных эпифезарных ростовых пластинах дистальной бедренной кости и проксимальной большеберцовой кости у растущих животных.
Соединения сначала вводили в состав композиции суспендированием в 1% (об./об.) растворе моноолеата полиоксиэтилен(20)сорбитана в деионизированной воде, дроблением в шаровой мельнице при 4°C в течение ночи (по меньшей мере, 15 часов). Соединения снова суспендировали перемешиванием непосредственно перед дозированием. Молодым крысам Alderley Park (получены из Wistar, масса 135-150 г, возраст от 4 до 8 недель, 5-6 на группу) дозированным образом вводили один раз в сутки посредством перорального кормления через зонд в течение 14 последовательных дней соединение (при 0,25 мл/100 г массы тела) или наполнитель. На день 15 животных гуманно умерщвляли с использованием повышаемой концентрации диоксида углерода и проводили вскрытие трупов. Область тканей, которые включали бедренно-тибиальные суставы, собирали и обрабатывали стандартными гистологическими способами для получения парафиновых срезов. Гистологические срезы окрашивали гематоксилином и эозином и исследовали для определения гистопатологии оптическим микроскопом. Площади бедренных эпифезарных ростовых пластин дистальной бедренной кости и проксимальной большеберцовой кости измеряли в срезах бедренной кости и большеберцовой кости с использованием морфометрического томографического анализа. Увеличение в зоне гипертрофии определяли сравнением средней площади эпифезарных ростовых пластин контрольной группы с группой обработки и статистическую значимость определяли с использованием одностороннего t-критерия Стьюдента. Ингибирующее действие обработки соединением считали значимым, когда р<0,05.
Хотя фармакологические свойства соединений формулы I изменяются со структурным изменением, в общем, активность, которой обладают соединения формулы I, можно демонстрировать при следующих концентрациях или дозах в одном или нескольких вышеуказанных испытаниях (а), (b), (с) и (d):
Испытание (а): - IC50 в диапазоне, например, <5 мкМ;
Испытание (b): - IC50 в диапазоне, например, 0,001-5 мкМ;
Испытание (с): - активность в диапазоне, например, 0,1-100 мг/кг/;
Испытание (d): - активность в диапазоне, например, 0,1-100 мг/кг.
В соответствии с одним аспектом настоящего изобретения соединения формулы I, которые оценивали в испытании на токсичность в течение 14 дней на крысах, имеют благотворный токсикологический профиль по сравнению с другими соединениями в пределах объема публикации Международной патентной заявки WO 98/13354.
В соответствии с другим аспектом настоящего изобретения соединения формулы I, которые оценивали в испытании на токсичность в течение 14 дней на крысах, имеют благотворный токсикологический профиль по сравнению с другими соединениями в пределах объема публикации Международной патентной заявки WO 97/30035.
Хотя фармакологические свойства соединений формулы I изменяются со структурным изменением и изменением видов, при дозах для крыс, предпочтительно, при дозах, которые меньше чем или равны 150 мг/кг, более предпочтительно, при дозах, которые меньше чем или равны 100 мг/кг, особенно при дозах, которые меньше чем или равны 50 мг/кг, соединения формулы I, которые вызывают статистически значимое увеличение в площади бедренных эпифезарных ростовых пластин дистальной бедренной кости и/или проксимальной большеберцовой кости, не вызывают неприемлемые гистопатологические изменения в других тканях в испытаниях (а), которые были проведены нами.
Так, в качестве примера соединение 4-(4-бром-2-фтор-анилино)-6-метокси-7-(1-метилпиперидин-4-илметокси)хиназолин (пример 2), которое испытывали по вышеуказанным тестам (а), (b), (с) и (d), дает следующие результаты:
(a) Flt - IC50 1,6 мкМ
KDR - IC50 0,04 мкМ
EGER - IC50 0,5 мкМ
(b) VEGF - IC50 0,06 мкМ
EGF - IC50 0,17 мкМ
Базальный - IC50>3 мкМ
(c) 78% ингибирование роста опухоли при 50 мг/кг; р<0,001 (критерий суммы рангов Манна-Уитни);
(d) 75% увеличение в гипертрофии эпифизарных ростовых пластин при 100 мг/кг/день у самок крыс; р<0,001 (односторонний t-критерий Стьюдента).
В соответствии со следующим аспектом изобретения предложена фармацевтическая композиция, которая содержит соединение формулы I, как указано здесь ранее, или его фармацевтически приемлемую соль в сочетании с фармацевтически приемлемым наполнителем или носителем.
Композиция может быть в виде, пригодном для перорального введения (например, в виде таблеток, лепешек, твердых или мягких капсул, водных или масляных суспензий, эмульсий, диспергируемых порошков или гранул, сиропов или эликсиров), для введения ингаляцией (например, в виде тонкоизмельченного порошка или жидкого аэрозоля), для введения инсуффляцией (например, в виде тонкоизмельченного порошка), для парентеральной инъекции (например, в виде стерильного раствора, суспензии или эмульсии для внутривенного, подкожного, внутримышечного, внутриваскулярного или инфузионного дозирования), для местного введения (например, в виде кремов, мазей, гелей или водных, или масляных растворов, или суспензий) или для ректального введения (например, в виде суппозитория). Обычно, вышеуказанные композиции можно получить общепринятым способом с использованием общепринятых эксципиентов.
Композиции по настоящему изобретению преимущественно представлены в виде стандартной лекарственной формы. Соединение обычно вводят теплокровному животному в единичной дозе в пределах 5-5000 мг на квадратный метр площади тела животного, т.е. приблизительно 0,1-100 мг/кг. Предусматривается единичная доза в диапазоне, например, 1-100 мг/кг, предпочтительно, 1-50 мг/кг, и она обычно обеспечивает терапевтически эффективную дозу. Стандартная лекарственная форма, такая как таблетка или капсула, обычно содержит, например, 1-250 мг активного ингредиента.
В соответствии со следующим аспектом настоящего изобретения здесь предложено соединение формулы I или его фармацевтически приемлемая соль, как определено здесь ранее, для использования в способе лечения организма человека или животного терапией.
Нами обнаружено, что соединения настоящего изобретения ингибируют активность рецепторной тирозинкиназы VEGF и, следовательно, представляют интерес по причине их антиангиогенных действий и/или их способности вызывать снижение в васкулярной проницаемости.
Следующим объектом настоящего изобретения является соединение формулы I или его фармацевтически приемлемая соль для использования в качестве лекарственного средства, в подходящем случае, соединение формулы I или его фармацевтически приемлемую соль для использования в качестве лекарственного средства для индуцирования антиангиогенного и/или снижающего васкулярную проницаемость действия у теплокровного животного, такого как человек.
Таким образом, в соответствии со следующим аспектом изобретения, предложено использование соединения формулы I или его фармацевтически приемлемой соли при получении лекарственного средства для использования при индуцировании антиангиогенного и/или снижающего васкулярную проницаемость действия у теплокровного животного, такого как человек.
В соответствии со следующим аспектом настоящего изобретения предложен способ индуцирования антиангиогенного и/или снижающего васкулярную проницаемость действия у теплокровного животного, такого как человек, нуждающегося в таком лечении, который включает введение указанному животному эффективного количества соединения формулы I или его фармацевтически приемлемой соли, как указано выше.
Как утверждалось выше, размер дозы, требуемой для терапевтического или профилактического лечения конкретного патологического состояния, обязательно будет изменяться в зависимости от подвергаемого лечению хозяина, пути введения и серьезности заболевания, которое лечат. Используют, предпочтительно, суточную дозу в диапазоне 1-50 мг/кг. Однако, суточная доза обязательно будет изменяться в зависимости от подвергаемого лечению хозяина, конкретного пути введения и серьезности заболевания, которое лечат. В соответствии с этим оптимальная доза может быть определена практикующим врачом, который лечит любого конкретного пациента.
Антиангиогенное и/или снижающее васкулярную проницаемость лечение, указанное здесь выше, можно использовать в качестве единственной терапии или оно может включать, кроме соединения по изобретению, одно или несколько других веществ и/или лечений. Такое совместное лечение можно осуществить посредством одновременного, последовательного или раздельного введения индивидуальных компонентов лечения. В области медицинской онкологии нормальной практикой является использование комбинации различных форм лечения при лечении каждого пациента с раковым заболеванием. В медицинской онкологии другим компонентом(ами) такого совместного лечения, кроме антиангиогенного и/или снижающего васкулярную проницаемость лечения, указанного здесь выше, могут быть хирургия, радиотерапия или химиотерапия. Такая химиотерапия может включать пять основных категорий терапевтического агента:
(i) другие антиангиогенные агенты, которые действуют по механизмам, отличным от механизма, указанного здесь ранее (например, линомид, ингибиторы функции интегрина ανβ3, ангиостатин, эндостатин, разоксин, талидомид) и включают васкулярные «прицельные» агенты (например, комбрестатин-фосфат и васкулярные поражающие агенты, описанные в публикации Международной патентной заявки WO 99/02166, причем полное описание данного документа включается здесь в качестве ссылки (например, N-ацетилколхинол-О-фосфат);
(ii) цитостатические агенты, такие как антиэстрогены (например, тамоксифен, торемифен, ралоксифен, дролоксифен, иодоксифен), прогестероны (например, мегестролацетат), ингибиторы ароматазы (например, анастрозол, летразол, воразол, эксеместан), антипрогестогены, антиандрогены (например, флутамид, нилутамид, билсалутамид, ципротеронацетат), агонисты и антагонисты LHRH (например, гозерелинацетат, люпролид, абареликс), ингибиторы тестостерон-5α-дигидроредуктазы (например, финастерид), агенты против инвазии (например, ингибиторы металлопротеиназы, подобные маримастану, и ингибиторы функции рецептора активатора урокиназного плазминогена) и ингибиторы функции фактора роста (такие факторы роста включают, например, полученный из тромбоцитов фактор роста и гепатоцитный фактор роста, такие ингибиторы включают антитела фактора роста, антитела рецептора фактора роста, ингибиторы тирозинкиназы и ингибиторы серин/треонинкиназы);
(iii) модификаторы биологических ответных реакций (например, интерферон);
(iv) антитела (например, edrecolomAb) и
(v) антипролиферативные/противоопухолевые лекарственные средства и их комбинации, используемые в медицинской онкологии, такие как антиметаболиты (например, антифолаты, подобные метотрексату, фторпиримидины, подобные аналогам 5-фторурацила, пурина и аденозина, цитозинарабинозид); противоопухолевые антибиотики (например, антрациклины, подобные доксорубицину, даунорубицину, эпирубицину и идарубицину, митомицину-С, дактиномицину, митрамицину); производные платины (например, цисплатин, карбоплатин); алкилирующие агенты (например, азотные аналоги горчичного газа, мелфалан, хлорамбуцил, бузулфан, циклофосфамид, ифосфамид, нитрозомочевины, тиотепа); антимитотические агенты (например, винкаалкалоиды, подобные винкристину, и таксоиды, подобные таксолу, таксотеру); ферменты (например, аспарагиназа); ингибиторы тимидилатсинтазы (например, ралтитрексед); ингибиторы топоизомеразы (например, эпиподофиллотоксины, подобные этопозиду и тенипозиду, амсакрин, топотекан, иринотекан).
Например, такое совместное лечение можно обеспечить посредством одновременного, последовательного или раздельного введения соединения формулы I, как указано здесь выше, такого как 4-(4-бром-2-фторанилино)-6-метокси-7-(1-метилпиперидин-4-илметокси)хиназолин или его соль, особенно его гидрохлоридную соль, и васкулярный «прицельный» агент, описанный в WO 99/02166, такой как N-ацетилколхинол-O-фосфат (пример 1 WO 99/02166).
Как утверждалось выше, соединения, указанные в настоящем изобретении, представляют интерес по причине их антиангиогенного и/или снижающего васкулярную проницаемость действия. Предполагается, что такие соединения являются полезными для широкого ряда патологических состояний, включая рак, диабет, псориаз, ревматоидный артрит, саркома Капоши, гемангиома, острая и хроническая нефропатии, атерома, артериальный рестеноз, аутоиммунные заболевания, острое воспаление, избыточные образования шрамов и спаек, эндометриоз, дисфункциональное маточное кровотечение и глазные болезни с пролиферацией ретинальных сосудов. В частности, предполагается, что такие соединения изобретения, преимущественно, замедляют рост первичных и рецидивных твердых опухолей, например толстой кишки, молочной железы, простаты, легких и кожи. Более конкретно, предполагается, что такие соединения изобретения ингибируют рост тех первичных и рецидивных опухолей, которые ассоциируются с VEGF, особенно тех опухолей, рост и распространение которых в значительной степени зависит от VEGF, включая, например, некоторые опухоли толстой кишки, молочной железы, простаты, легких, наружных половых женских органов и кожи.
В другом аспекте настоящего изобретения предполагается, что соединения формулы I ингибируют рост первичных и рецидивных опухолей, которые ассоциируются с EGF, особенно тех опухолей, рост и распространение которых в значительной степени зависит от EGF.
В другом аспекте настоящего изобретения предполагается, что соединения формулы I ингибируют рост первичных и рецидивных опухолей, которые ассоциируются как с VEGF, так и ECG, особенно тех опухолей, рост и распространение которых в значительной степени зависит от VEGF и EGF.
Кроме их использования в терапевтической медицине, соединения формулы I и их фармацевтически приемлемые соли можно также использовать в качестве средств при разработке и стандартизации in vitro и in vivo тест-систем для оценки действия ингибиторов активности рецепторной тирозинкиназы VEGF на лабораторных животных, таких как кошки, собаки, кролики, обезьяны, крысы и мыши, как части поиска новых терапевтических агентов.
Должно быть понятно, что везде, где используют термин «простой эфир» в данном описании, он относится к простому диэтиловому эфиру.
Изобретение теперь будет иллюстрировано, но не ограничено, следующими примерами, в которых, если не указано иное:
(i) выпаривание проводили роторным испарением в вакууме и процедуры обработки осуществляли после удаления остаточных твердых веществ, таких как осушающие агенты, фильтрованием;
(ii) процессы проводили при температуре окружающей среды, которая находится в диапазоне 18-25°С, и в атмосфере инертного газа, такого как аргон;
(iii) колоночную хроматографию (флэш-методика) и жидкостную хроматографию при среднем давлении (MPLC) проводили на диоксиде кремния Merck Kieselgel (Art. 9385) или диоксиде кремния с обращенной фазой Merck Lichroprep RP-18 (Art. 9303), полученном от Е. Merck, Darmstadt, Germany;
(iv) выходы даны только для иллюстрации и не обязательно являются максимально достижимыми;
(v) точки плавления являются некорректированными и были определены с использованием автоматической аппаратуры для определения точки плавления Mettler SP62, аппаратурой с масляной баней или аппаратурой с горячей пластиной Koffler;
(vi) структуры конечных продуктов формулы I подтверждали способами ядерного (обычно протонного) магнитного резонанса (ЯМР) и масс-спектральными способами; величины химических сдвигов протонного магнитного резонанса измеряли на дельта-шкале и многообразие пиков обозначают следующим образом: с, синглет; д, дублет; т, триплет; м, мультиплет; шир., широкий; к, квартет; спектры ЯМР регистрировали на приборе 400 МГц при 24°С;
(vii) промежуточные продукты обычно не были полностью характеризованы и чистоту анализировали тонкослойной хроматографией (ТСХ), высокоэффективной жидкостной хроматографией (ВЭЖХ), инфракрасным анализом (ИК) и ЯМР-анализом;
(viii) использовали следующие аббревиатуры:
ДМФ - N,N-диметилформамид, ДМСО - диметилсульфоксид, ТГФ - тетрагидрофуран, ТФУ - трифторуксусная кислота, NMP - 1-метил-2-пирролидинон.
Пример 1
ТФУ (3 мл) добавляют к суспензии 4-(4-бром-2-фторанилино)-7(1-(трет-бутоксикарбонил)пиперидин-4-илметокси)-6-метоксихиназолина (673 мг, 1,2 ммоль) в метиленхлориде (10 мл). После перемешивания в течение 1 часа при температуре окружающей среды летучие компоненты удаляют в вакууме. Остаток растирают со смесью вода/простой эфир. Органический слой отделяют. Водный слой снова промывают простым эфиром. Значение рН водного слоя устанавливают на 10 при помощи 2 н. водного гидроксида натрия. Водный слой экстрагируют метиленхлоридом. Органический слой сушат (MgSO4) и растворитель удаляют в вакууме. Твердое вещество растирают со смесью простой эфир/петролейный эфир (1/1), фильтруют, промывают простым эфиром и сушат в вакууме, при этом получают 4-(4-бром-2-фторанилино)-6-метокси-7-(пиперидин-4-илметокси)хиназолин (390 мг, 70,5%).
MS-ESI:461-463[MH]+
1Н NMR Spectrum: (DMSd6) 1,13-1,3 (m, 2Н), 1.75 (d, 2H), 1.87-2.0 (m, 1H), 2.5 (d, 2H), 3.0 (d, 2H), 3.96 (s, 3Н), 3.98 (d, 2H), 7.2 (s, 1H), 7.5 (dd, 1H), 7.55 (t, 1H), 7.68 (dd, 1H), 7.80 (s, 1H), 8.36 (s, 1H), 9.55 (br s, 1H)
Spectrum = спектр, m=м, d=д, с=с, t=т, br=шир.
Элементный анализ: Найдено С 54,5 Н4,9 N 12,1
C21H22N4O2BrF Вычислено С 54,7 Н 4,8 N 12,1%.
Исходный материал получают следующим образом:
Раствор гидрохлорида 7-бензилокси-4-хлор-6-метоксихиназолина (8,35 г, 27,8 ммоль (получен, например, как описано в WO 97/22596, пример 1) и 4-бром-2-фторанилина (5,65 г, 29,7 ммоль) в 2-пропаноле (200 мл) нагревают при кипячении с обратным холодильником в течение 4 часов. Образовавшийся осадок собирают фильтрованием, промывают 2-пропанолом и затем простым эфиром и сушат в вакууме, при этом получают гидрохлорид 7-бензилокси-4-(4-бром-2-фторанилино)-6-метоксихиназолина (9,46 г, 78%).
1Н NMR Spectrum: (DMSOd6; CD3COOD) 4.0 (s, 3H); 5.37 (s, 2H); 7.35-7.5 (m, 4H); 7.52-7.62 (m, 4H); 7.8 (d, 1H); 8.14 (9s, 1H); 8.79 (s, 1H)
MS-ESI: 456 [МН]+
Элементный анализ: Найдено С 54,0 Н 3,7 N 8,7
С22Н17N3О2BrE 0,9 HCl Вычислено С 54,2 Н 3,7 N 8,6%.
Раствор гидрохлорида 7-бензилокси-4-(4-бром-2-фторанилино-6-метоксихиназолина (9,4 г, 19,1 ммоль) в ТФУ (90 мл) нагревают при кипячении с обратным холодильником в течение 50 минут. Смеси дают возможность охладиться и выливают на лед. Образовавшийся осадок собирают фильтрованием и растворяют в метаноле (70 мл). Устанавливают рН раствора 9-10 с помощью концентрированного водного раствора аммиака. Смесь концентрируют до половины начального объема упариванием. Образовавшийся осадок собирают фильтрованием, промывают водой, затем простым эфиром и сушат в вакууме, при этом получают 4-(4-бром-2-фторанилино)-7-гидрокси-6-метоксихиназолин (5,66 г, 82%).
1Н NMR Spectrum: (DMSOd6; CD3COOD) 3.95 (s, 3Н); 7.09 (s, 1H); 7.48 (s, 1H); 7.54 (t, 1H); 7.64 (d, 1H); 7.79 (s, 1H); 8.31 (s, 1H)
MS-ESI: 366 [MH]+
Элементный анализ: Найдено С 49,5 Н 3,1 N 11,3
C15H11N3O2BrF Вычислено С 49,5 Н 3,0 N 11,5%.
Поддерживая температуру в области 0-5°С по частям добавляют раствор ди-трет-бутилдикарбоната (41,7 г, 0,19 моль) в этилацетате (75 мл) к раствору этил-4-пиперидин-карбоксилата (30 г, 0,19 моль) в этилацетате (150 мл), охлажденному до 5°С. После перемешивания в течение 48 часов при температуре окружающей среды смесь выливают в воду (300 мл). Органический слой отделяют, промывают последовательно водой (200 мл), 0,1 н. водной соляной кислотой (200 мл), насыщенным гидрокарбонатом натрия (200 мл) и насыщенным раствором соли (200 мл), сушат (MgSO4) и упаривают, при этом получают этил-4-(1-(трет-бутоксикарбонил)пиперидин)карбоксилат (48%, 98%).
1Н NMR Spectrum: (CDCl3) 1.25 (t, 3Н); 1.45 (s, 9H); 1.55-1.70 (m, 2H); 1.8-2.0 (d, 2H); 2.35-2.5 (m, 1H); 2.7-2.95 (t, 2H); 3.9-4.1 (br s, 2H); 4.15 (q, 2H)
Раствор 1 M литийалюминийгидрида в ТГФ (133 мл, 0,133 моль) добавляют частями к раствору этил-4-(1-(трет-бутоксикарбонил)пиперидин)карбоксилата (48 г, 0,19 моль) в сухом ТГФ (180 мл), охлажденном до 0°С. После перемешивания при 0°С в течение 2 часов добавляют воду (30 мл), затем 2 н. гидроксид натрия (10 мл). Осадок удаляют фильтрованием через диатомовую землю и промывают этилацетатом. Фильтрат промывают водой, насыщенным раствором соли, сушат (MgSO4) и упаривают, при этом получают 1-(трет-бутоксикарбонил)-4-гидроксиметилпиперидин (36,3 г, 89%).
MS (EI): 215 [М.]+
1H NMR Spectrum: (CDCL3) 1.05-1.2 (m, 2H); 1.35-1.55 (m, 10H); 1.6-1.8 (m, 2H); 2.6-2.8 (t, 2H); 3.4-3.6 (t, 2H); 4.0-4.2 (br s, 2H)
1,4-Диазабицикло[2.2.2]октан (42,4 г, 0,378 моль) добавляют к раствору 1-(трет-бутоксикарбонил)-4-гидроксиметилпиперидина (52,5 г, 0,244 ммоль) в трет-бутилметиловом эфире (525 мл). После перемешивания в течение 15 минут при температуре окружающей среды смесь охлаждают до 5°С и частями в течение 2 часов добавляют раствор толуолсульфонилхлорида (62,8 г, 0,33 ммоль) в трет-бутилметиловом эфире (525 мл) при поддержании температуры при 0°С. После перемешивания в течение 1 часа при температуре окружающей среды добавляют петролейный эфир (1 л). Осадок удаляют фильтрованием. Фильтрат упаривают, при этом получают твердое вещество. Твердое вещество растворяют в простом эфире и промывают последовательно 0,5 н. водной соляной кислотой (2×500 мл), водой, насыщенным гидрокарбонатом натрия и насыщенным раствором соли, сушат (MgSO4) и упаривают, при этом получают 1-(трет-бутоксикарбонил)-4-(4-метилфенилсульфонилоксиметил)пиперидин (76,7 г, 85%).
MS (ESI): 392 [MNa]+
1H NMR Spectrum: (CDCl3) 1.0-1.2 (m, 2H); 1.45 (s, 9H); 1.65 (d, 2H); 1.75-1.9 (m, 2H); 2.45 (s, 3H); 2.55-2.75 (m, 2H); 3.85 (d, 1H); 4.0-4.2 (br s, 2H); 7.35 (d, 2H); 7.8 (d, 2H)
Карбонат калия (414 мг, 3 ммоль) добавляют к суспензии 4-(4-бром-2-фторанилино)-7-гидрокси-6-метоксихиназолина (546 мг, 1,5 ммоль) в ДМФ (5 мл). После перемешивания в течение 10 минут при температуре окружающей среды добавляют L-(трет-бутоксикарбонил)-4-(4-метилфенилсульфонилоксиметил)пиперидин (636 мг, 1,72 ммоль) и смесь нагревают при 95°С в течение 2 часов. После охлаждения смесь выливают на охлажденную воду (20 мл). Осадок собирают фильтрованием, промывают водой, сушат в вакууме, при этом получают 4-(4-бром-2-фторанилино)-7-(1-(трет-бутоксикарбонил)пипе-ридин-4-илметокси)-6-метоксихиназолин (665 мг, 79%).
MS - ESI: 561-563 [МН]+
1H NMR Spectrum: (DMSOd6) 1.15-1.3 (m, 2H), 1.46 (s, 9H), 1.8 (d, 2H), 2.0-2.1 (m, 1H), 2.65-2.9 (m, 2H), 3.95 (s, 3H), 4.02 (br s, 2H), 4.05 (d, 2H), 7.2 (s, 1H), 7.48 (d, 1H), 7.55 (t, 1H), 7.65 (d, 1H), 7.8 (s, 1H), 8.35 (s, 1H), 9.55 (br s, 1H)
Пример 2а
Раствор 37% водного формальдегида (50 мкл, 0,6 ммоль), затем цианоборогидрид натрия (23 мг, 0,36 ммоль) добавляют к раствору 4(4-бром-2-фторанилино)-6-метокси-7-(пиперидин-4-илметокси)хиназолин (139 мг, 0,3 ммоль) (получен, как описано в примере 1) в смеси ТГФ/метанол (1,4 мл/1,4 мл). После перемешивания в течение 1 часа при температуре окружающей среды добавляют воду и летучие компоненты удаляют в вакууме. Остаток растирают с водой, фильтруют, промывают водой и сушат в вакууме. Твердое вещество очищают хроматографией на нейтральном оксиде алюминия, элюируя метиленхлоридом, затем смесью метиленхлорид/этилацетат (1/1), затем смесью метиленхлорид/этилацетат/метанол (50/45/5). Фракции, содержащие предполагаемый продукт, упаривают в вакууме. Образовавшееся белое твердое вещество растворяют в смеси метиленхлорид/метанол (3 мл/3 мл) и добавляют 3 н. хлорид водорода в простом эфире (0,5 мл). Летучие компоненты удаляют в вакууме. Твердое вещество растирают с простым эфиром, фильтруют, промывают простым эфиром и сушат в вакууме, при этом получают гидрохлорид 4-(4-бром-2-фторанилино)-6-метокси-7-(1-метилпиперидин-4-илметокси)хиназолина (120 мг, 69%).
MC-ESI: 475-477 [МН]+.
ЯМР-спектр протонированной формы гидрохлорида 4-(4-бром-2-фторанилино)-6-метокси-7-(1-метилпиперидин-4-илметокси)хиназолина показал присутствие 2 форм, А и В, в отношении А:В приблизительно 9:1.
1Н NMR Spectrum: (DMSOd6; CF3COOD) 1.55-1.7 (m, form A 2H); 1.85-2.0 (m, form В 4H); 2.03 (d, form A 2H); 2.08-2.14 (br s, form A 1H); 2.31-2.38 (br s, form В 1Н); 2.79 (s, form A 3H); 2.82 (s, form В 3H); 3.03 (t, form A 2H); 3.21 (br s, form В 2H); 3.30 (br s, form В 2H); 3.52 (d, form A 2H); 4.02 (s, 3H); 4.12 (d, form A 2H); 4.30 (d, form В 2H); 7.41 (s, 1H); 7.5-7.65 (m, 2H); 7.81 (d, 1H); 8.20 (s, 1H); 8.88 (s, 1H)
Элементный анализ: Найдено С 46,0 Н 5,2 N 9,6
C22H24N4О2BrF 0,3 Н2О 2,65 HCl
Вычислено С 45,8 H4,8 N 9,7%.
Пример 2b
37% Водный формальдегид (3,5 мл, 42 ммоль) добавляют к раствору 4-(4-бром-2-фторанилино)-7-(1-(трет-бутоксикарбонил)пиперидин-4-илметокси)-6-метоксихиназолина (3,49 г, 6,22 ммоль) (получен, как описано для исходного материала в примере 1) в муравьиной кислоте (35 мл). После нагревания при 95°С в течение 4 часов летучие компоненты удаляют в вакууме. Остаток суспендируют в воде и рН смеси устанавливают 10,5 с помощью медленного добавления раствора 2 н. гидроксида натрия. Суспензию экстрагируют этилацетатом. Органический слой промывают насыщенным раствором соли, сушат MgSO4 и упаривают, при этом получают 4-(4-бром-2-фторанилино)-6-метокси-7-(1-метилпиперидин-4-илметокси)хиназолин (2,61 г, 88%).
MS-ESI: 475-477 [МН]+
1H NMR Spectrum: (DMSOd6) 1.3-1.45 (m, 2H), 1.8 (d, 2H), 1.7-1.9 (m, 1H), 1.95 (t, 2H), 2.2 (s, 3H), 2.85 (d, 2H), 3.96 (s, 3H), 4.05 (d, 2H), 7.19 (s, 1H), 7.5 (d, 1H), 7.55 (t, 1H), 7.67 (d, 1H), 7.81 (s, 1H), 8.37 (s, 1H), 9.54 (s, 1H)
Элементный анализ: Найдено С 55,4 Н 5,1 N 11,6
C22H24N4О2BrF Вычислено С 55,6 Н 5, 1 N 11,8%.
Пример 2с
Суспензию 4-хлор-6-метокси-7-(1-метилпиперидин-4-илметокси)хиназолина (200 мг, 0,62 ммоль) и 4-бром-2-фторанилина (142 мг, 0,74 ммоль) в изопропаноле (3 мл), содержащую 6 н. хлорид водорода в изопропаноле (110 мкл, 0,68 мл) нагревают при кипячении с обратным холодильником в течение 1,5 часа. После охлаждения осадок собирают фильтрованием, промывают изопропанолом, затем простым эфиром и сушат в вакууме, при этом получают гидрохлорид 4-(4-бром-2-фторанилино)-6-метокси-7-(1-метилпиперидин-4-илметокси)хиназолина (304 мг, 90%).
Элементный анализ: Найдено С 47,9 Н 4,9 N 10,0
C22H24N4О2BrF 0,5 Н2О 1,8 HCl 0,08 изопропанола
Вычислено С 48,2 Н 5,8 N 10,1%.
ЯМР-спектр протонированной формы гидрохлорида 4-(4-бром-2-фторанилино)-6-метокси-7-(1-метилпиперидин-4-илметокси)хиназолина показал присутствие 2 форм, А и В, в отношении А:В приблизительно 9:1.
1H NMR Spectrum: (DMSOd6) 1.6-1.78 (m, form A 2H); 1.81-1.93 (br s, form В 4Н); 1.94-2.07 (d, form A 2H); 2.08-2.23 (br s, form A 1H); 2.29-2.37 (br s, form В 1H); 2.73 (d, form A 3H); 2.77 (d, form В 3H); 2.93-3.10 (q, form A 2H); 3.21 (br s, form В 2H); 3.27 (br s, form В 2H); 3.42-3.48 (d, form A 2H); 4.04 (s, 3H); 4.10 (d, form A 2H); 4.29 (d, form В 2H); 7.49 (s, 1H); 7.53-7.61 (m, 2H); 7.78 (d, 1H); 8.47 (s, 1H); 8.81 (s, 1H); 10.48 (br s, form A 1H); 10.79 (br s, form В 1H); 11.90 (br s, 1H)
Для снятия другого показания ЯМР некоторое количество твердого карбоната калия добавляют в раствор в ДМСО гидрохлорида 4-(4-бром-2-фторанилино)-6-метокси-7-(1-метилпиперидин-4-илметокси)хиназолина, описанного выше, чтобы выделить свободное основание в трубке ЯМР. Затем снова регистрировали ЯМР-спектр и обнаружили только одну форму, как описано ниже:
1Н NMR Spectrum: (DMSOd6; solid potassium carbonate) 1.3-1.45 (m, 2H); 1.75 (d, 2H); 1.7-1.9 (m, 1H); 1.89 (t, 2H); 2.18 (s, 3H); 2.8 (d, 2H); 3.98 (s, 3H); 4.0 (d, 2H); 7.2 (s, 1H); 7.48 (d, 1H); 7.55 (t, 1H); 7.68 (d, 1H); 7.8 (s, 1H); 8.35 (s, 1H); 9.75 (s, 1H)
1a) solid potassium carbonate=твердый карбонат калия
Образец 4-(4-бром-2-фторанилино)-6-метокси-7(1-метилпиперидин-4-илметокси)хиназолина (свободное основание) получают из гидрохлорида 4-(4-бром-2-фторанилино)-6-метокси-7-(1-метилпиперидин-4-илметокси)хиназолина (получен, как описано выше) следующим образом:
Гидрохлорид 4-(4-бром-2-фторанилино)-6-метокси-7-(1-метилпиперидин-4-илметокси)хиназолина (50 мг) суспендируют в метиленхлориде (2 мл) и промывают насыщенным гидрокарбонатом натрия. Раствор в метиленхлориде сушат (MgSO4) и летучие компоненты удаляют, упаривая, при этом получают 4-(4-бром-2-фторанилино)-6-метокси-7-(1-метилпиперидин-4-илметокси)хиназолин (свободное основание). ЯМР полученного таким образом свободного основания показал только одну форму, как описано ниже:
1Н NMR Spectrum: (DMSOd6) 1.3-1.45 (m, 2H); 1.76 (d, 2H); 1.7-1.9 (m, 1H); 1.9 (t, 2H); 2.19 (s, 3Н); 2.8 (d, 2H); 3.95 (s, 3Н); 4.02 (d, 2H); 7.2 (s, 1H); 7.48 (d, 1H); 7.55 (t, 1H); 7.68 (dd, 1H); 7.8 (s, 1H); 8.38 (s, 1H); 9.55 (br s, 1H)
Для снятия другого показания ЯМР некоторое количество CF3COOD добавляют в раствор 4-(4-бром-2-фторанилино)-6-метокси-7(1-метилпиперидин-4-илметокси)хиназолина (свободное основание) в ДМСО для ЯМР, описанный выше, и ЯМР-спектр регистрируют снова. Спектр полученной таким образом протонированной формы трифторацетатной соли 4-(4-бром-2-фторанилино)-6-метокси-7-(1-метилпиперидин-4-илметокси)хиназолина показал присутствие 2 форм, А и В, в отношении А:В приблизительно 9:1.
1Н NMR Spectrum: (DMSOd6; CF3COOD) 1.5-1.7 (m, form A 2H); 1.93 (br s, form В 4Н); 2.0-2.1 (d, form A 2H); 2.17 (br s, form A 1H); 2.35 (br s, form B1H); 2.71 (s, form A 3H); 2.73 (s, form В 3Н); 2.97-3.09 (t, form A 2H); 3.23 (br s, form В 2H); 3.34 (br s, form В 2H); 3.47-3.57 (d, form A 2H); 4.02 (s, 3H); 4.15 (d, form A 2H); 4.30 (d, form В 2Н); 72 (s, 1H); 7.3-7.5 (m, 2H); 7.6 (d, 1H); 7.9 (s, 1H); 8.7 (s, 1H)
Исходный материал получают следующим образом:
1-(трет-Бутоксикарбонил)-4-(4-метилфенилсульфонилоксиметил)пиперидин (40 г, 0,11 моль) (получен, как описано для исходного соединения в примере 1) добавляют к суспензии этил-4-гидрокси-3-метоксибензоата (19,6 г, 0,1 моль) и карбоната калия (28 г, 0,2 моль) в сухом ДМФ (200 мл). После перемешивания при 95°С в течение 2,5 часа смесь охлаждают до температуры окружающей среды и распределяют между водой и смесью этилацетат/простой эфир. Органический слой промывают водой, насыщенным раствором соли, сушат (MgSO4) и упаривают. Образовавшееся масло кристаллизуют из петролейного эфира и суспензию сохраняют в течение ночи при 5°С. Твердое вещество собирают фильтрованием, промывают петролейным эфиром и сушат в вакууме, при этом получают этил-4-(1-(трет-бутоксикарбонил)пиперидин-4-илметокси)-3-метоксибензоат (35 г, 89%).
Т.пл. 81-83°С.
MS(ESI): 416[MNa]+
1H NMR Spectrum: (CDCl3) 1.2-1.35 (m, 2H); 1.4 (t, 3Н); 1.48 (s, 9H); 1.8-1.9 (d, 2H); 2.0-2.15 (m, 2H); 2.75 (t, 2H); 3.9 (d, 2H); 3.95 (s, 3Н); 4.05-4.25 (br s, 2H); 4.35 (q, 2H); 6.85 (d, 1H); 7.55 (s, 1H); 7.65 (d, 1H)
Элементный анализ: Найдено С 63,4 H 8,0 N 3,5
C21H31NO6 0,3 H2O Вычислено С 63,2 Н 8,0 N 3,5%.
Формальдегид (12 М, 37% в воде, 35 мл, 429 ммоль) добавляют к раствору этил-4-(1-(трет-бутоксикарбонил)пиперидин-4-илметокси)-3-метоксибензоата (35 г, 89 ммоль) в муравьиной кислоте (35 мл). После перемешивания при 95°С в течение 3 часов летучие компоненты удаляют выпариванием. Остаток растворяют в метиленхлориде и добавляют 3 М хлорид водорода в простом эфире (40 мл, 120 ммоль). После разбавления простым эфиром смесь растирают до образования твердого вещества. Твердое вещество собирают фильтрованием, промывают простым эфиром и сушат в вакууме в течение ночи при 50°С, при этом получают этил-3-метокси-4-(1-метилпиперидин-4-илметокси)бензоат (36 г, количественный выход).
MS (ESI): 308 [MH]+
1HNMR Spectrum: (DMSOd6) 1.29 (1, 3H); 1.5-1.7 (m, 2H); 1.95 (d, 2H); 2.0-2.15 (br s, 1H); 2.72 (s, 3H); 2.9-3.1 (m, 2H); 3.35-3.5 (br s, 2H); 3.85 (s, 3H); 3.9-4.05 (br s, 2H); 4.3 (q, 2H); 7.1 (d, 1H); 7.48 (s, 1H); 7.6 (d, 1H)
Раствор этил-3-метокси-4-(1-метилпиперидин-4-илметокси)бензоата (30,6 г, 89 ммоль) в метиленхлориде (75 мл) охлаждают до 0-5°С. Добавляют ТФУ (37,5 мл), затем по каплям в течение 15 минут добавляют раствор дымящей 24 н. азотной кислоты (7,42 мл, 178 ммоль) в метиленхлориде (15 мл). После завершения добавления раствору дают возможность нагреться и перемешивают при температуре окружающей среды в течение 2 часов. Летучие компоненты удаляют в вакууме и остаток растворяют в метиленхлориде (50 мл). Раствор охлаждают до 0-5°С и добавляют простой эфир. Осадок собирают фильтрованием и сушат в вакууме при 50°С. Твердое вещество растворяют в метиленхлориде (500 мл) и добавляют 3 М хлорид водорода в простом эфире (30 мл), затем простой эфир (500 мл). Твердое вещество собирают фильтрованием и сушат в вакууме при 50°С, при этом получают 3-метокси-4-(1-метилпиперидин-4-илметокси)-6-нитробензоат (28,4 г, 82%).
MS (ESI): 353 [MHT]+
1H NMR Spectrum: (DMSOd6) 1.3 (t, 3H); 1.45-1.65 (m, 2H); 1.75-2.1 (m, 3H); 2.75 (s, 3H); 2.9-3.05 (m, 2H); 3.4-3.5 (d, 2H); 3.95 (s, 3H); 4.05 (d, 2H); 4.3 (q, 2H); 7.32 (s, 1H); 7.66 (s, 1H)
Суспензию этил-3-метокси-4-(1-метилпиперидин-4-илметокси)-6-нитробензоата (3,89 г, 10 ммоль) в метаноле (80 мл), содержащую 10% платины на активированном угле (50% влаги) (389 мг) гидрируют при давлении 1,8 атмосферы до прекращения поглощения водорода. Смесь фильтруют и фильтрат упаривают. Остаток растворяют в воде (30 мл) и рН устанавливают 10 при помощи насыщенного раствора гидрокарбоната натрия. Смесь разбавляют смесью этилацетат/простой эфир (1/1) и органический слой отделяют. Водный слой далее экстрагируют смесью этилацетат/простой эфир и органические слои объединяют. Органические слои промывают водой, насыщенным раствором соли, сушат (MgSO4), фильтруют и упаривают. Образовавшееся твердое вещество растирают в смеси простой эфир/петролейный эфир, фильтруют, промывают петролейным эфиром и сушат в вакууме при 60°С, при этом получают этил-6-амино-3-метокси-4-(1-метилпиперидин-4-илметокси)бензоат (2,58 г, 80%).
Т.пл. 111-112°С.
MS (ESI): 323 [MH]+
1H NMR Spectrum: (CDCl3) 1.35 (t, 3H); 1.4-1.5 (m, 2H); 1.85 (m, 3H); 1.95 (t, 2H); 2.29 (s, 3H); 2.9 (d, 2H); 3.8 (s, 3H); 3.85 (d, 2H); 4.3 (q, 2H); 5.55 (br s, 2H); 6.13 (s, 1H); 7.33 (s, 1H)
Элементный анализ: Найдено С 62,8 H 8,5 N 8,3
C17H26N2O4 0,2 H20 Вычислено С 62, 6 Н 8,2 N 8,6%.
Раствор этил-6-амино-3-метокси-4-(1-метилпиперидин-4-илметокси)бензоата (16,1 г, 50 ммоль) в 2-метоксиэтаноле (160 мл), содержащий ацетат формамидина (5,2 г, 50 ммоль) нагревают при 115°С в течение 2 часов. Ацетат формамидина (10,4 г, 100 ммоль) добавляют частями каждые 30 минут в течение 4 часов. Нагревание продолжают в течение 30 минут после последнего добавления. После охлаждения летучие компоненты удаляют в вакууме. Твердое вещество растворяют в этаноле (100 мл) и метиленхлориде (50 мл). Осадок удаляют фильтрованием и фильтрат концентрируют до конечного объема 100 мл. Суспензию охлаждают до 5°С и твердое вещество собирают фильтрованием, промывают холодным этанолом, затем простым эфиром и сушат в вакууме в течение ночи при 60°С, при этом получают 6-метокси-7-(1-метилпиперидин-4-илметокси)-3,4-дигидрохиназолин-4-он (12,7 г, 70%).
MS (ESI): 304 [МН]+
1H NMR Spectrum: (DMSOd6) 1.25-1.4 (m, 2H); 1.75 (d, 2H); 1.9 (t, 1H); 1.9 (s, 3H); 2.16 (s, 2H); 2.8 (d, 2H); 3.9 (s, 3H); 4.0 (d, 2H); 7.11 (s, 1H); 7.44 (s, 1H); 7.97 (s, 1H)
Раствор 6-метокси-7-(1-метилпиперидин-4-илметокси)-3,4-дигидрохиназолин-4-она (2,8 г, 9,24 ммоль) в тионилхлориде (28 мл), содержащий ДМФ (280 мкл), нагревают при кипячении с обратным холодильником при 85°С в течение 1 часа. После охлаждения летучие компоненты удаляют упариванием. Осадок растирают с простым эфиром, фильтруют, промывают простым эфиром и сушат в вакууме. Твердое вещество растворяют в метиленхлориде и добавляют насыщенный водный гидрокарбонат натрия. Органический слой отделяют, промывают водой, насыщенным раствором соли, сушат (MgSO4) и упаривают, при этом получают 4-хлор-6-метокси-7-(1-метилпиперидин-4-илметокси)хиназолин (2,9 г, 98%).
MS (ESI): 322 [МН]+
1H NMR Spectrum: (DMSOd6) 1.3-1.5 (m, 2H); 1.75-1.9 (m, 3H); 2.0 (t, 1H); 2.25 (s, 3H); 2.85 (d, 2H); 4.02 (s, 3H); 4.12 (d, 2H); 7.41 (s, 1H); 7.46 (s, 1H); 8.9 (s, 1H)
В альтернативном случае 6-метокси-7-(1-метилпиперидин-4-илметокси)-3,4-дигидрохиназолин-4-он можно получить следующим образом.
Гидрид натрия (1,44 г 60% суспензии в минеральном масле, 36 ммоль) добавляют частями в течение 20 минут к раствору 7-бензилокси-6-метокси-3,4-дигидрохиназолин-4-она (8,46 г, 30 ммоль) (получен, например, как описано в WO 97/22596, пример 1) в ДМФ (70 мл) и смесь перемешивают в течение 1,5 часа. Частями добавляют хлорметилпивалат (5,65 г, 37,5 ммоль) и смесь перемешивают в течение 2 часов при температуре окружающей среды. Смесь разбавляют этилацетатом (100 мл) и выливают на смесь лед/вода (400 мл) и 2 н. соляной кислоты (4 мл). Органический слой отделяют и водный слой экстрагируют этилацетатом, объединенные экстракты промывают насыщенным раствором соли, сушат (MgSO4) и растворитель удаляют выпариванием. Остаток растирают со смесью простого эфира и петролейного эфира, твердое вещество собирают фильтрованием и сушат в вакууме, при этом получают 7-бензилокси-6-метокси-3-((пивалоилокси)метил)-3,4-дигидрохиназолин-4-он (10 г, 84%).
1H NMR Spectrum: (DMSOd6) 1.11 (s, 9H); 3.89 (s, 3H); 5.3 (s, 2H); 5.9 (s, 2H); 7.27 (s, 1H); 7.35 (m, 1H); 7.47 (t, 2H); 7.49 (d, 2H); 7.51 (s, JH); 8.34 (s, 1H)
Смесь 7-бензилокси-6-метокси-3-((пивалоилокси)метил)-3,4-дигидрохиназолин-4-она (7 г, 17,7 ммоль) и катализатора, 10% палладия на угле (700 мг) в этилацетате (250 мл), ДМФ (50 мл), метаноле (50 мл) и уксусной кислоте (0,7 мл) перемешивают под атмосферным давлением водорода в течение 40 минут. Катализатор удаляют фильтрованием и растворитель удаляют из фильтрата упариванием. Остаток растирают с простым эфиром, собирают фильтрованием и сушат в вакууме, при этом получают 7-гидрокси-6-метокси-3-((пивалоилокси)метил)-3,4-дигидрохиназолин-4-он (4,36 г, 80%).
1H NMR Spectrum: (DMSOd6) 1.1 (s, 9H); 3.89 (s, 3H); 5.89 (s, 2H); 7.0 (s, 1H); 7.48 (s, 1H); 8.5 (s, 1H)
Трифенилфосфин (1,7 г, 6,5 ммоль) добавляют в атмосфере азота к суспензии 7-окси-6-метокси-3-((пивалоилокси)метил)-3,4-дигидрохиназолин-4-она (1,53 г, 5 ммоль) в метиленхлориде (20 мл) с последующим добавлением 1-(трет-бутоксикарбонил)-4-(гидроксиметил)пиперидина (1,29 г, 6 ммоль) (получен, как описано для исходного материала в примере 1) и затем раствора диэтилазодикарбоксилата (1,13 г, 6,5 ммоль) в метиленхлориде (5 мл). После перемешивания в течение 30 минут при температуре окружающей среды реакционную смесь выливают в колонку диоксида углерода и элюируют смесью этилацетат/петролейный эфир (1/1, затем 6/5, 6/4 и 7/3). Упариванием фракций, содержащих ожидаемый продукт, получают масло, которое кристаллизуется после растирания с пентаном. Твердое вещество собирают фильтрованием и сушат в вакууме, при этом получают 7-(1-(трет-бутоксикарбонил)пиперидин-4-илметокси)-6-метокси-3-((пивалоилокси)метил)-3,4-дигидрохиназолин-4-он (232 г, 92%).
MS-ESI: 526[MNa]+
1H NMR Spectrum: (CDCl3) 1.20 (s, 9H), 1.2-1.35 (m, 2H), 1.43 (s, 9H), 1.87 (d, 2H), 2.05-2.2 (m, 1H), 2.75 (t, 2H), 3.96 (d, 2H), 3.97 (s, 3Н), 4.1-4.25 (br s, 2H), 5.95 (s, 2H), 7.07 (s, 1H), 7.63 (s, 1H), 8.17 (s, 1H)
Элементный анализ: Найдено С 61,8 Н 7,5 N 8,3
C26H37N3O7 Вычислено С 62,0 Н 7,4 N 8,3%.
Раствор 7-(1-(трет-бутоксикарбонил)пиперидин-4-илметокси)-6-метокси-3-((пивалоилокси)метил)-3,4-дигидрохиназолин-4-она (2,32 г, 4,6 ммоль) в метиленхлориде (23 мл), содержащий ТФУ (5 мл), перемешивают при температуре окружающей среды в течение 1 часа. Летучие компоненты удаляют в вакууме. Остаток распределяют между этилацетатом и гидрокарбонатом натрия. Органический растворитель удаляют в вакууме и остаток фильтруют. Осадок промывают водой и сушат в вакууме. Твердый продукт подвергают азеотропной перегонке с толуолом и сушат в вакууме, при этом получают 6-метокси-7-(пиперидин-4-илметокси)-3-((пивалоилокси)метил)-3,4-дигидрохиназолин-4-он (1,7 г, 92%).
MS-ESI: 404[MH]+
1H NMR Spectrum: (DMSOd6; CF3COOD) 1.15 (s, 9H), 1.45-1.6 (m, 2H), 1.95 (d, 2H), 2.1-2.25 (m, 1H), 2.95 (t, 2H), 3.35 (d, 2H), 3.95 (s, 3H), 4.1 (d, 2H), 5.95 (s, 2H), 7.23 (s, 1H), 7.54 (s, 1H), 8.45 (s, 1H)
Водный 37% раствор формальдегида (501 мкл, 6 ммоль), затем цианоборогидрид натрия (228 мг, 3,6 ммоль) добавляют по частям к раствору 6-метокси-7-(пиперидин-4-илметокси)-3-((пивалоилокси)метил)-3,4-дигидрохиназолин-4-она (1,21 г, 3 ммоль) в смеси ТГФ/метанол (10 мл/10 мл). После перемешивания в течение 30 минут при температуре окружающей среды органические растворители удаляют в вакууме и остаток распределяют между метиленхлоридом и водой. Органический слой отделяют, промывают водой и насыщенным раствором соли, сушат (MgSO4) и летучие компоненты удаляют выпариванием. Остаток растирают с простым эфиром и образовавшееся твердое вещество собирают фильтрованием, промывают простым эфиром и сушат в вакууме, при этом получают 6-метокси-7-(1-метилпиперидин-4-илметокси)-3-((пивалоилокси)метил)-3,4-дигидрохиназолин-4-он (1,02 г, 82%).
MS-ESI: 418 [MH]+
1H NMR Spectrum: (CDCl3) 1.19 (s, 9H), 1.4-1.55 (m, 2H), 1.9 (d, 2H), 2.0 (t, 2H), 1.85-2.1 (m, 1H), 2.3 (s, 3Н), 2.92 (d, 2H), 3.96 (s, 3Н), 3.99 (d, 2H), 5.94 (s, 2H), 7.08 (s, 1H), 7.63 (s, 1H), 8.17 (s, 1H)
Насыщенный раствор аммиака в метаноле (14 мл) добавляют к раствору 6-метокси-7-(1-метилпиперидин-4-илметокси)-3-((пивалоилокси)метил)-3,4-дигидрохиназолин-4-она (1,38 г, 3,3 ммоль) в метаноле (5 мл). После перемешивания в течение 20 часов при температуре окружающей среды суспензию разбавляют метиленхлоридом (10 мл). Раствор фильтруют. Фильтрат упаривают в вакууме и остаток растирают с простым эфиром, собирают фильтрованием, промывают простым эфиром и сушат в вакууме, при этом получают 6-метокси-7-(1-метилпиперидин-4-илметокси)-3,4-дигидрохиназолин-4-он (910 мг, 83%).
MS-ESI: 304 [МН]+
1Н NMR Spectrum: (DMSOd6) 1.3-1.45 (m, 2H), 1.75 (d, 2H), 1.7-1.85 (m, 1H), 1.9 (t, 2H), 2.2 (s, 3H), 2.8 (d, 2H), 3.9 (s, 3H), 4.0 (d, 2H), 7.13 (s, 1H), 7.45 (s, 1H), 7.99 (s, 1H)
Пример 3а
Хлорид водорода (3,5 М) в этаноле (75 мкл, 0,26 ммоль) добавляют к суспензии 4-хлор-6-метокси-7-(1-метилпиперидин-4-илметокси)хиназолина (80 мг, 0,25 ммоль) (получен, как описано для исходного материала в примере 2с) в изопропаноле (3 мл), смесь нагревают до 50°С и добавляют 4-хлор-2-фторанилин (44 мг, 0,3 ммоль). Смесь нагревают при кипячении с обратным холодильником в течение 30 минут. После охлаждения смесь разбавляют простым эфиром (3 мл). Осадок собирают фильтрованием, промывают простым эфиром и сушат в вакууме, при этом получают гидрохлорид 4-(4-хлор-2-фторанилино)-6-метокси-7-(1-метилпиперидин-4-илметокси)хиназолина (105 мг, 82%).
MC-ESI: 431-433 [МН]+.
ЯМР-спектр протонированной формы гидрохлорида 4-(4-хлор-2-фторанилино)-6-метокси-7-(1-метилпиперидин-4-илметокси)хиназолина показал присутствие 2 форм, А и В, в отношении А:В приблизительно 9:1.
1Н NMR Spectrum: (DMSOd6; CF3COOD) 1.55-1.7 (m, form A 2H), 1.85-2.0 (m, form В 4H), 2.05 (d, form A 2H), 2.1-2.2 (m, form А 1Н), 2.35 (s, 3Н); 2.79 (s, form A 3H), 2.82 (s, form В 3Н), 3.03 (t, form A 2H), 3.2-3.3 (m, form В 2H); 3.3-3.4 (m, form В 2H), 3.52 (d, form A 2H), 4.02 (s, 3H), 4.13 (d, form A 2H), 4.3 (d, form В 2H), 7.41 (s, 1H), 7.47 (dd, 1H), 7.63 (t, 1H), 7.69 (dd, 1H), 8.19 (s, 1H), 8.88 (s, 1H)
Элементный анализ: Найдено С 51,8 H 5,6 N 10,9
C22H24N4O2ClF 0,4 H2O 2 HCl
Вычислено С 51,7 Н 5,3 N 11,0%.
Пример 3b
Альтернативный способ получения является следующим.
Трифенилфосфин (615 мг, 2,3 ммоль), затем диэтилазодикарбоксилат (369 мкл, 2,3 ммоль) добавляют к раствору 4-гидроксиметил-1-метилпиперидина (151 мг, 1,1 ммоль) (J. Med. Chem. 1973, 16, 156) и 4-(4-хлор-2-фторанилино)-7-гидрокси-6-метоксихиназолина (250 мг, 0,78 ммоль) (получен, как описано для исходного материала в примере 7) в метиленхлориде (5 мл). После перемешивания в течение 30 минут при температуре окружающей среды добавляют 4-гидроксиметил-1-метилпиперидин (51 мг, 0,39 ммоль), трифенилфосфин (102 мг, 0,39 ммоль) и диэтилазодикарбоксилат (61 мкл, 0,39 ммоль). После перемешивания в течение 15 минут летучие компоненты удаляют в вакууме и остаток очищают колоночной хроматографией при элюировании смесью метиленхло-рид/ацетонитрил/метанол (70/10/20, затем 75/5/20 и 80/0/20). Фракции, содержащие ожидаемый продукт, объединяют и летучие компоненты удаляют выпариванием. Остаток растворяют в смеси метиленхлорида и метанола и добавляют 5 М хлорид водорода в изопропаноле. Суспензию концентрируют и твердое вещество собирают фильтрованием, промывают простым эфиром, сушат в вакууме, при этом получают гидрохлорид 4-(4-хлор-2-фторанилино)-6-метокси-7-(1-метилпиперидин-4-илметокси)хиназолина (16 мг, 4%).
Пример 4
В атмосфере аргона гидрид натрия (60%, 372 мг, 9,3 ммоль) добавляют к раствору 4-бром-2,5-дифторанилина (1,67 г, 8,08 ммоль) в ДМФ. После перемешивания в течение 30 минут при температуре окружающей среды добавляют 4-хлор-6-метокси-7-(1-метилпиперидин-4-илметокси)хиназолин (1,3 г, 4,04 ммоль) и перемешивание продолжают в течение дополнительных 20 часов. Смесь выливают на воду (130 мл) и экстрагируют этилацетатом. Органические слои промывают водой, насыщенным раствором соли, сушат (MgSO4) и летучие компоненты удаляют выпариванием. Остаток очищают колоночной хроматографией на диоксиде кремния с элюированием смесью метиленхлорид/метанол (95/5), затем смесью метиленхлорид/метанол, содержащей аммиак (1%) (90/10). Фракции, содержащие ожидаемый продукт, объединяют и упаривают. Остаток растирают с простым эфиром, собирают фильтрованием, промывают простым эфиром и сушат в вакууме при 50°С, при этом получают 4-(4-бром-2,6-дифторанилино)-6-метокси-7-(1-метилпиперидин-4-илметокси)хиназолин (1,4 г, 70%).
MS-ESI: 493-495 [MH]+
1H NMR Spectrum: (DMSOd6) 1.3-1.45 (m, 2H), 1.8 (d, 2H), 1.7-1.9 (m, 1H), 1.9 (t, 2H), 2.17 (s, 3H), 2.8 (d, 2H), 3.95 (s, 3H), 4.02 (d. 2H), 7.2 (s, 1H), 7.63 (s, 1H), 7.6 (s, 1H), 7.82 (s, 1H), 8.35 (s, 1H)
Элементный анализ: Найдено С 53,8 Н 4,8 N 11,3
C22H23N4O2BrF2 Вычислено С 53,6 Н 4,7 N 11,4%.
Пример 5
С использованием процедуры, аналогичной процедуре, описанной в примере 4, 4-хлор-6-метокси-7-(1-метилпиперидин-4-илметокси)хиназолин (246 мг, 0,764 ммоль) (получен, как описано для исходного материала в примере 2с) подвергают взаимодействию с 4-хлор-2,6-дифторанилином (250 мг, 1,53 ммоль) (см. WO 97/30035, пример 15) в ДМФ (9 мл) и в присутствии гидрида натрия (60%, 76,5 мг, 1,9 ммоль), при этом получают 4-(4-хлор-2,6-дифторанилино)-6-метокси-7-(1-метилпиперидин-4-илметокси)хиназолин (210 мг, 61%).
MS-ESI: 449-451 [MH]+
1Н NMR Spectrum: (DMSOd6) 1.3-1.45 (m, 2H), 1.8 (d, 2Н), 1.7-1.9 (m, 1H), 1.9 (t, 2H), 2.2 (s, 3H), 2.8 (d, 2H), 3.96 (s, 3H), 4.02 (d, 2H), 7.21 (s, 1H), 7.52 (s, 1H), 7.54 (s, 1H), 7.82 (s, 1H), 8.35 (s, 1H)
Элементный анализ: Найдено С 59,0 Н 5,3 N 12,5
С22Н23N4O2ClF2 Вычислено С 58,9 Н 5,2 N 12,5%.
Исходный материал получают следующим образом.
Гидрохлорид 4-хлор-2,6-дифторанилина (см. WO 97/30035, пример 15) распределяют между метиленхлоридом и водой и добавляют гидрокарбонат натрия до тех пор, пока рН водного слоя не будет приблизительно 9. Органический слой отделяют, промывают насыщенным раствором соли, сушат (MgSO4) и упаривают, при этом получают 4-хлор-2,6-дифторанилин в виде свободного основания.
Пример 6
Суспензию 4-хлор-6-метокси-7-(1-метилпиперидин-4-илметокси)хиназолина (200 мг, 0,622 ммоль) (получен, как описано для исходного материала в примере 2с) и 2-фтор-4-метиланилина (94 мг, 0,764 ммоль) в изопропаноле (5 мл), содержащую 6,2 М хлорид водорода в изопропаноле (110 мкл) перемешивают при 80°С в течение 1,5 часа. После охлаждения осадок собирают фильтрованием, промывают изопропанолом, затем простым эфиром и сушат в вакууме. Твердое вещество очищают колоночной хроматографией с элюированием смесью метиленхлорид/метанол (90/10) и затем 5% аммиаком в смеси метанол/метиленхлорид (10/90). Упаривание фракций, содержащих ожидаемый продукт, дает 4-(2-фтор-4-метиланилино)-6-метокси-7-(1-метилпиперидин-4-илметокси)хиназолин (170 мг, 61%).
MS-ESI: 411 [МН]+
1H NMR Spectrum: (DMSOd6) 1.3-1.45 (m, 2H), 1.8 (d, 2Н), 1.7-1.9 (m, 1H), 1.9 (t, 2H), 2.2 (s, 3H), 2.35 (s, 3H), 2.8 (d, 2H), 3.95 (s, 3Н), 4.01 (d, 2H), 7.1 (d, 1H), 7.13 (d, 1H), 7.16 (s, 1H), 7.4 (t, 1H), 7.81 (s, 1H), 8.32 (s, 1H), 9.4 (s, 1H)
Элементный анализ: Найдено С 66,5 Н 6,7 N 13,7
C23H27N4O2F 0,3 Н2O Вычислено С 66,4 Н 6,7 N 13,5%.
Пример 7
1-трет-Бутоксикарбонил-4-гидроксиметилпиперидин (590 мг, 2,75 ммоль) (получен, как описано для исходного материала в примере 1), затем трифенилфосфин (1,2 г, 4,58 ммоль) и диэтилазодикарбоксилат (0,72 мл, 4,58 ммоль) добавляют к раствору 4-(4-хлор-2-фторанилино)-7-гидрокси-6-метоксихиназолина (585 мг, 1,83 ммоль) в метиленхлориде (20 мл). После перемешивания в течение 1 часа при температуре окружающей среды добавляют дополнительный трифенилфосфин (239 мг, 0,91 ммоль) и диэтилазодикарбоксилат (0,14 мл, 0,91 ммоль). После перемешивания в течение 1,5 часа летучие компоненты удаляют в вакууме и остаток очищают колоночной хроматографией с элюированием смесью этилацетат/метиленхлорид (1/1). Сырой продукт используют непосредственно в следующей стадии.
Раствор сырого продукта в метиленхлориде (15 мл), содержащий ТФУ (4,5 мл), перемешивают при температуре окружающей среды в течение 1,5 часа. Летучие компоненты удаляют в вакууме. Остаток распределяют между водой и этилацетатом. Значение рН водного слоя устанавливают 9 при помощи 2 н. гидроксида натрия. Органический слой отделяют, промывают водой, затем насыщенным раствором соли, сушат (MgSO4) и упаривают, при этом получают 4-(4-хлор-4-фторанилино)-6-метокси-7-(пиперидин-4-илметокси)хиназолин.
MS-ESI: 417-419 [МН]+
1Н NMR Spectrum: (DMSOd6) 1.1-1.3 (m, 2H), 1.75 (d, 2H), 1.85-2.0 (br s, 1H), 2.55 (d, 2H), 2.95 (d, 2H), 3.95 (s, 3H), 4.0 (d, 2H), 7.2 (s, 1H), 7.35 (dd, 1H), 7.55 (dd, 1H), 7.6 (t, 1H), 7.8 (s, 1H), 8.35 (s, 1H), 9.55 (s, 1H)
Гидрохлоридную соль получают следующим образом.
4-(4-Хлор-4-фторанилино)-6-метокси-7-(пиперидин-4-ил-метокси)хиназолин растворяют в смеси метанол/метиленхлорид и добавляют М хлорид водорода в простом эфире. Летучие компоненты удаляют в вакууме, остаток растирают с простым эфиром, собирают фильтрованием, промывают простым эфиром и сушат в вакууме, при этом получают гидрохлорид 4-(4-хлор-4-фторанилино)-6-метокси-7-(пиперидин-4-илметокси)хиназолина (390 мг, 47%) после двух стадий.
Элементный анализ: Найдено С 50,4 Н 5,2 N 11,0
C21H22O2N4ClF 2,25 HCl
Вычислено С 50,6 Н 4,9 N 11,2%.
Исходный материал получают следующим образом.
Раствор гидрохлорида 7-бензилокси-4-хлор-6-метоксихиназолина (1,2 г, 4 ммоль) (получен, как описано в WO 97/22596, пример 1) и 4-хлор-2-фторанилино (444 мкл, 4 ммоль) в 2-пропаноле (40 мл) нагревают при кипячении с обратным холодильником в течение 1,5 часа. После охлаждения осадок собирают фильтрованием, промывают 2-пропанолом, затем простым эфиром и сушат в вакууме, при этом получают гидрохлорид 7-бензилокси-4-(4-хлор-2-фторанилино)-6-метоксихиназолина (1,13 г, 64%). Т.пл. 239-242°С.
1H NMR Spectrum: (DMSOd6) 4.0 (s, 3Н); 5.36 (s, 2Н); 7.39-7.52 (m, 9Н); 8.1 (s, 1H); 8.75 (s, 1H)
MS-ESI:410[MH]+
Элементный анализ: Найдено С 59,2 Н 4,3 N 9,4
C22H17N3O2ClF 1HCl Вычислено С 59,2 Н 4,1 N 9,4%.
Раствор гидрохлорида 7-бензилокси-4-(4-хлор-2-фторанилино)-6-метоксихиназолина (892 мг, 2 ммоль) в ТФУ (10 мл) нагревают при кипячении с обратным холодильником в течение 50 минут. После охлаждения смесь выливают на лед. Осадок собирают фильтрованием, растворяют в метаноле (10 мл) и подщелачивают до рН 11 водным аммиаком. После концентрирования упариванием твердый продукт собирают фильтрованием, промывают водой, затем простым эфиром и сушат в вакууме, при этом получают 4-(4-хлор-2-фторанилино)-7-гидрокси-6-метоксихиназолин в виде желтого твердого вещества (460 мг, 72%). Т.пл. 141-143°С.
MS-ESI: 320-322 [МН]+
1H NMR Spectrum: (DMSOd6) 3.95 (s, 3Н); 7.05 (s, 1H); 7.35 (d, 1H); 7.54-7.59 (m, 2Н); 7.78 (s, 1H); 8.29 (s, 1H)
Пример 8
Суспензию 7-(1-(трет-бутоксикарбонил)пиперидин-4-илметокси)-4-(2-фтор-4-метиланилино)-6-метоксихиназолина (318 мг, 0,64 ммоль) в метиленхлориде (5 мл), содержащую ТФУ (2,5 мл), перемешивают при температуре окружающей среды в течение 2 часов. Летучие компоненты удаляют в вакууме и остаток распределяют между метиленхлоридом и водой. Значение рН водного слоя устанавливают 10-11. Органический слой отделяют, промывают водой, насыщенным раствором соли, сушат (MgSO4) и летучие компоненты удаляют упариванием, при этом получают 4-(2-фтор-4-метиланилино)-6-метокси-7-(пиперидин-4-илметокси)хиназолин (220 мг, 87%).
MS-ESI: 397[MH]+
1Н NMR Spectrum: (DMSOd6) 1.15-1.3 (m, 2Н); 1.75 (d, 2H); 1,85-2.0 (m, 1H); 2.4 (s, 3Н); 3.0 (d, 2H); 3.3-3.4 (d, 2H); 3.95 (s, 3H); 4.0 (d, 2H); 7.04 (d, 1H); 7.15 (d, 1H); 7.17 (s, 1H); 7.4 (t, 1H); 7.8 (s, 1H); 8.3 (s, 1H); 9.4 (s, 1H)
Исходный материал получают следующим образом.
С использованием процедуры, аналогичной процедуре, описанной в примере 6, гидрохлорид 7-бензилокси-4-хлор-6-метоксихиназолина (1,55 г, 5,15 ммоль) (получен, например, как описано в WO 97/22596, пример 1) подвергают взаимодействию с 2-фтор-4-метиланилином (700 мг, 5,67 ммоль) в изопропаноле (90 мл), содержащем 6,2 М хлорид водорода в изопропаноле (80 мкл, 0,51 ммоль), при этом получают гидрохлорид 7-бензилокси-4-(2-фтор-4-метиланилино)-6-метоксихиназолина (2 г, 91%).
MS-ESI: 390[MHT]+
1Н NMR Spectrum: (DMSOd6) 2.4 (s, 3Н), 4.01 (s, 3Н), 7.15 (d, 1H), 7.25 (d, 1H), 7.35-7.6 (m, 7Н), 8.3 (s, 1H), 8.78 (s, 1H)
Раствор гидрохлорида 7-бензилокси-4-(2-фтор-4-метиланилино)-6-метилхиназолина (2 г, 4,7 ммоль) в ТФУ (20 мл) нагревают при 80°С в течение 5 часов и перемешивают при температуре окружающей среды в течение ночи. Летучие компоненты удаляют в вакууме и остаток суспендируют в воде (50 мл). Твердый гидрокарбонат натрия добавляют до тех пор, пока рН не станет приблизительно 7. Осадок затем собирают фильтрованием, промывают водой и сушат в вакууме. Твердый продукт очищают колоночной хроматографией с элюированием смесью метанол/метиленхлорид (5/95). После удаления растворителя выпариванием твердое вещество растирают с простым эфиром, собирают фильтрованием, промывают простым эфиром и сушат в вакууме, при этом получают 4-(2-хлор-4-метиланилино)-7-гидрокси-6-метоксихиназолин (1,04 г, 74%).
MS-ESI: 300[MH]+
1Н NMR Spectrum: (DMSOd6) 2.4 (s, 3Н), 4.0 (s, 3H), 7.15 (d, 1H), 7.22 (s, 1H), 7.25 (d, 1H), 7.41 (t, 1H), 8.05 (s, 1H), 8.7 (s, 1H), 11.0 (s, 1H), 11.5-11.8 (br s, 1H)
Трифенилфосфин (2,19 г, 8,36 ммоль) добавляют к суспензии 4-(2-хлор-4-метиланилино)-7-гидрокси-6-метоксихиназолина (1 г, 3,34 ммоль) в метиленхлориде (10 мл), охлажденной при 0°С, затем добавляют 1-(трет-бутоксикарбонил)-4-гидроксиметилпиперидин (1,08 г, 5,01 ммоль) (получен, как описано для исходного материала в примере 1) и диэтилазодикарбоксилат (1,31 мл, 8,36 ммоль). После перемешивания в течение 2 часов при температуре окружающей среды летучие компоненты удаляют в вакууме. Остаток очищают колоночной хроматографией с элюированием смесью метиленхлорид/метанол (2/98). После удаления растворителя выпариванием остаток растирают с простым эфиром, собирают фильтрованием, промывают простым эфиром и сушат в вакууме, при этом получают 7-(1-(трет-бутоксикарбонил)пиперидин-4-илметокси)-4-(2-фтор-4-метиланилино)-6-метоксихиназолин (327 мг, 20%).
MS-ESI:497[MH]+
1H NMR Spectrum: (DMSOd6) 1.15-1.3 (m, 2H); 1.45 (s, 9H); 1.8 (d, 2H); 2.0-2.1 (m, 1H); 2.4 (s, 3Н); 2.75-2.9 (br s, 2H); 3.95 (s, 3Н); 4.0 (br s, 2H); 4.05 (d, 2H); 7.1 (d, 1H); 7.15 (d, 1H); 7.2 (s, 1H); 7.4 (t, 1H); 7.85 (t, 1H); 8.32 (s, 1H); 9.45 (s, 1H)
Пример 9
Раствор 4(4-бром-2,6-дифторанилино)-7-(1-(трет-бутоксикарбонил)пиперидин-4-илметокси)-6-метоксихиназолина (578 мг, 1 ммоль) в метиленхлориде (10 мл), содержащий ТФУ (4 мл), перемешивают при температуре окружающей среды в течение 2 часов. Летучие компоненты удаляют в вакууме и остаток суспендируют в воде. Значение рН водного слоя устанавливают приблизительно до 10 и слой экстрагируют метиленхлоридом. Органический слой промывают водой, насыщенным раствором соли, сушат (MgSO4) и летучие компоненты удаляют выпариванием. Остаток растирают с простым эфиром и сушат в вакууме, получают при этом 4-(4-бром-2,6-дифторанилино)-6-метокси-7-(пиперидин-4-илметокси)хиназолин (110 мг, 23%).
MS-ESI: 479-481[МН]+
1H NMR Spectrum: (DMSOd6) 1.15-1.3 (m, 2H); 1.75 (d, 2H); 1.85-2.0 (br s, 1H); 2.5 (d, 2H); 3.0 (d, 2H); 3.97 (s, 3Н); 4.0 (d, 2H); 7.2 (s, 1H); 7.62 (d, 2H); 7.82 (s, 1H); 8.35 (s, 1H)
1H NMR Spectrum: (DMSOd6; CF3COOD) 1.5-1.65 (m, 2H); 2.0 (d, 2H); 2.15-2.3 (br s, 1H); 3.0 (t, 2H); 3.4 (d, 2H); 4.02 (s, 3H); 4.15 (d, 2H); 7.4 (s, 1H); 7.75 (d, 2H); 8.1 (s, 1H); 8.92 (s, 1H)
Исходный материал получают следующим образом.
Гидрид натрия (60%, 612 мг, 15,3 ммоль) добавляют к раствору 4-бром-2,6-дифторанилина (2,77 г, 6,65 ммоль) в ДМФ (80 мл). После перемешивания в течение 30 минут при температуре окружающей среды добавляют 7-бензилокси-4-хлор-6-метоксихиназолин (2 г, 6,65 ммоль) (получен, например, как описано в WO 97/22596, пример 1, но свободное основание выделяют до использования) и перемешивание поддерживают в течение 4 часов. Смесь распределяют между этилацетатом и водой (200 мл). Органический слой отделяют, промывают водой, насыщенным раствором соли, сушат (MgSO4) и летучие компоненты удаляют выпариванием. Остаток растирают с изопропанолом, собирают фильтрованием, промывают простым эфиром и сушат в вакууме, при этом получают 7-бензилокси-4-(4-бром-2,6-дифторанилино)-6-метоксихиназолин (1,95 г, 62%).
MS-ESI: 472-474 [МН]+
1H NMR Spectrum: (DMSOd6) 3.94 (s, 3H), 5.3 (s, 2H), 7.3 (s, 1H), 7.4 (d, 1H), 7.45 (t, 2H), 7.5 (s, 1H), 7.55 (d, 1H), 7.65 (d, 2H), 7.85 (s, 1H), 8.35 (s, 1H), 9.4-9.6 (br s, 1H)
С использованием процедуры, аналогичной процедуре, описанной для синтеза исходного материала в примере 8, 7-бензилокси-4-(4-бром-2,6-дифторанилино)-6-метоксихиназолин (1,9 г, 4,02 ммоль) подвергают взаимодействию с ТФУ (20 мл), при этом получают 4-(4-бром-2,6-дифторанилино)-7-гидрокси-6-метоксихиназолин (1,5 г, 98%).
1H NMR Spectrum: (DMSOd6) 3.95 (s, 3H), 7.1 (s, 1H), 7.6 (s, 1H), 7.65 (s, 1H), 7.8 (s, 1H), 8.3 (s, 1H), 9.45 (br s, 1H), 10.5 (br s, 1H)
С использованием процедуры, аналогичной процедуре, описанной при получении исходного материала в примере 8, 4-(4-бром-2,6-дифторанилино)-7-гидрокси-6-метоксихиназолин (1 г, 2,62 ммоль) подвергают взаимодействию с 1-(трет-бутоксикарбонил)-4-гидроксиметилпиперидином (845 мг, 3,93 ммоль) (получен, как описано для исходного материала в примере 1), при этом получают 4-(4-бром-2,6-дифторанилино)-7-(1-(трет-бутоксикарбонил)пиперидин-4-илметокси)-6-метоксихиназолин (620 мг, 41%).
MS-ESI: 579-581 [MH]+
1H NMR Spectrum: (DMSOd6) 1.15-1.3 (m, 2H); 1.45 (s, 9H); 1.8 (d, 2H); 2.0-2.1 (m, 1H); 2.7-2.9 (m, 2H); 3.95 (s, 3H); 4.0 (br s, 2H); 4.05 (d, 2H); 7.22 (s, 1H); 7.65 (d, 2H); 7.85 (s, 1H); 8.35 (s, 1H); 9.4-9.6 (br s, 1H)
Пример 10
С использованием процедуры, аналогичной процедуре, описанной в примере 9, 7-(1-(трет-бутоксикарбонил)пиперидин-4-илметокси) -4-(4-хлор-2,6-дифторанилино)-6-метоксихи-назолин) (95 мг, 0,2 ммоль) в метиленхлориде (2 мл) обрабатывают ТФУ (800 мкл), при этом получают 4-(4-хлор-2,6-дифторанилино)-6-метокси-7-(пиперидин-4-илметокси)хиназолин (20 мг, 26%).
MS-ESI: 435-37 [МН]+
1H NMR Spectrum: (DMSOd6) 1.2-1.3 (m, 2H); 1.75 (d, 2H); 1.85-2.0 (br s, 1H); 2.5 (d, 2H); 3.0 (d, 2H); 3.97 (s, 3H); 4.0 (d, 2H), 7.2 (s, 1H); 752 (d, 2H); 7.85 (s, 1H); 8.35 (s, 1H)
Исходный материал получают следующим образом.
С использованием процедуры, аналогичной процедуре, описанной для получения исходного материала в примере 9, 7-бензилокси-4-хлор-6-метоксихиназолин (184 мг, 0,61 ммоль) (получен, например, как описано в WO 97/22596, пример 1, но свободное основание было получено до использования) подвергают взаимодействию с 4-хлор-2,6-дифторанилином (200 мг, 1,22 ммоль) в присутствии гидрида натрия (60%, 87 мг, 1,4 ммоль) в ДМФ (8 мл), при этом получают 7-бензилокси 4-(4-хлор-2,6-дифторанилино)-6-метоксихиназолин (212 мг, 74%).
MS-ESI: 428[MH]+
1H NMR Spectrum: (DMSOd6) 3.96 (s, 3H); 5.31 (s, 2H); 7.32 (s, 1H); 7.4 (d, 1H); 7.45 (t, 2H); 7.5-7.6 (m, 4H); 7.85 (s, 1H); 8.35 (br s, 1H); 9.55 (br s, 1H)
Раствор 7-бензилокси-4-(4-хлор-2,6-дифторанилино)-6-метоксихиназолина (200 мг, 0,47 ммоль) в ТФУ (3 мл) перемешивают при 80°С в течение 3 часов. После охлаждения летучие компоненты удаляют в вакууме и остаток растворяют в воде, содержащей 5% метанола. Значение рН устанавливают 8 при помощи гидрокарбоната натрия, твердое вещество собирают фильтрованием и промывают водой. Твердое вещество растворяют в смеси этилацетат/метанол/метиленхлорид (47/6/47). Летучие компоненты удаляют в вакууме, при этом получают 4-(4-хлор-2,6-дифторанилино)-7-гидрокси-6-метоксихиназолин (126 мг, 80%).
MS-ESI: 338 [MH]+
1H NMR Spectrum: (DMSO6) 3.95 (s, 3H); 7.1 (s, 1H); 7.55 (d, 2H); 7.8 (s, 1H); 8.3 (s, 1H); 9.42 (br s, 1H)
С использованием процедуры, аналогичной процедуре, описанной для получения исходного материала в примере 9, 4-(4-хлор-2,6-дифторанилино)-7-гидрокси-6-метоксихиназолин (150 мг, 0,44 ммоль) подвергают взаимодействию с 1-(трет-бутоксикарбонил)-4-гидроксиметилпиперидином (150 мг, 0,88 ммоль) (получен, как описано для исходного материала в примере 1), при этом получают 7-(1-(трет-бутоксикарбонил)пиперидин-4-илметокси)-4-(4-хлор-2,6-дифторанилино)-6-метоксихиназолин (113 мг, 59%).
MS-ESI: 535 [MH]+
1H NMR Spectrum: (DMSOd6) 1.15-1.3 (m, 2H); 1.45 (s, 9H); 1.8 (d, 2H); 2.0-2.1 (m, 1H); 2.7-2.9 (m, 2H); 3.95 (s, 3H); 4.0 (br s, 2H); 4.05 (d, 2H); 7.2 (s, 1H); 7.6 (m, 2H); 7.8 (s, 1H); 8.35 (s, 1H); 9.4-9.6 (br s, 1H)
Пример 11
Следующий пример иллюстрирует репрезентативные, применяемые в фармации лекарственные формы, содержащие соединение формулы I или его фармацевтически приемлемую соль (далее соединение X) для терапевтического или профилактического использования при лечении людей:
Примечание
Вышеуказанные готовые препаративные формы можно получить общепринятыми методиками, хорошо известными в фармацевтической области. Таблетки (а)-(с) могут быть покрыты энтеросолюбильной оболочкой общепринятыми способами, например, для получения оболочки из ацетата-фталата целлюлозы.
| название | год | авторы | номер документа |
|---|---|---|---|
| НОВОЕ АМИДНОЕ ПРОИЗВОДНОЕ ДЛЯ ИНГИБИРОВАНИЯ РОСТА РАКОВЫХ КЛЕТОК | 2008 |
|
RU2434010C2 |
| СПОСОБ ПОЛУЧЕНИЯ 4-(4-БРОМ-2-ФТОРАНИЛИНО)-6-МЕТОКСИ-7(1-МЕТИЛПИПЕРИДИН-4-ИЛМЕТОКСИ)ХИНАЗОЛИНА(ZD6474) И СПОСОБЫ ПОЛУЧЕНИЯ ПРОМЕЖУТОЧНЫХ СОЕДИНЕНИЙ ДЛЯ ЕГО ПОЛУЧЕНИЯ | 2006 |
|
RU2448102C2 |
| ПРОИЗВОДНЫЕ ХИНАЗОЛИНА | 2015 |
|
RU2704125C2 |
| ПРОИЗВОДНЫЕ ХИНАЗОЛИНА И ИХ ПРИМЕНЕНИЕ В КАЧЕСТВЕ ИНГИБИТОРОВ ФАКТОРА РОСТА ЭНДОТЕЛИЯ СОСУДОВ | 1997 |
|
RU2196137C2 |
| ПРОИЗВОДНЫЕ ХИНАЗОЛИНА, СПОСОБ ИХ ПОЛУЧЕНИЯ, ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ И СПОСОБ ДОСТИЖЕНИЯ АНТИПРОЛИФЕРАТИВНОГО ЭФФЕКТА | 1996 |
|
RU2153495C2 |
| ПРОИЗВОДНЫЕ ХИНАЗОЛИНА В КАЧЕСТВЕ ИНГИБИТОРОВ АНГИОГЕНЕЗА | 2000 |
|
RU2262935C2 |
| ПРОИЗВОДНЫЕ ХИНАЗОЛИНА, СПОСОБЫ ИХ ПОЛУЧЕНИЯ, СОДЕРЖАЩИЕ ИХ ФАРМАЦЕВТИЧЕСКИЕ КОМПОЗИЦИИ, СПОСОБ ДОСТИЖЕНИЯ АНТИАНГИОГЕННОГО И/ИЛИ ЭФФЕКТА СНИЖЕНИЯ ПРОНИЦАЕМОСТИ КРОВЕНОСНЫХ СОСУДОВ У ТЕПЛОКРОВНОГО ЖИВОТНОГО | 1997 |
|
RU2198879C2 |
| АГОНИСТ СФИНГОЗИН-1-ФОСФАТНЫХ РЕЦЕПТОРОВ, СПОСОБ ЕГО ПОЛУЧЕНИЯ И ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ, СОДЕРЖАЩАЯ ЕГО В КАЧЕСТВЕ АКТИВНОГО ИНГРЕДИЕНТА | 2020 |
|
RU2798605C1 |
| ПРОИЗВОДНЫЕ 2-АМИНО-4-ФЕНИЛ-4-ОКСОМАСЛЯНОЙ КИСЛОТЫ И ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ | 1994 |
|
RU2139850C1 |
| ПРОИЗВОДНЫЕ 6-КАРБОНИТРИЛ ДИПИРИДОПИРРОЛЫ И ИХ ПРИМЕНЕНИЕ ПРИ ЛЕЧЕНИИ РАКА | 2010 |
|
RU2575635C2 |
Изобретение относится к производным хиназолина формулы (I)
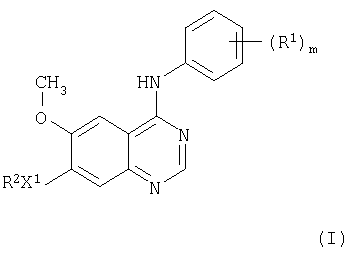
где m равно целому числу от 1 до 3; R1 представляет собой галоген или С1-3-алкил; Х1 представляет собой -O-; R2 представляет собой С1-5-алкил-R3 (где R3 представляет собой пиперидин-4-ил, который может иметь один или два заместителя, выбранных из С1-4-алкила), и их солям; способам их получения, фармацевтическим композициям, содержащим соединение формулы (I) или его фармацевтически приемлемую соль в качестве активного ингредиента. Соединения формулы (I) и их фармацевтически приемлемые соли ингибируют действия VEGF, что является ценным свойством при лечении различных патологических состояний, включающих рак и ревматоидный артрит. 9 н. и 19 з.п. ф-лы.
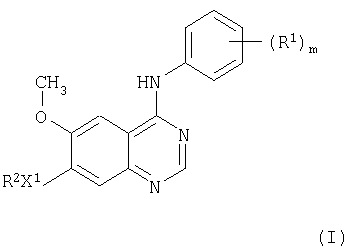
где m равно целому числу от 1 до 3;
R1 представляет собой галоген или С1-3-алкил;
X1 представляет собой -O-;
R2 представляет собой С1-5-алкил-R3 (где R3 представляет собой пиперидин-4-ил, который может иметь один или два заместителя, выбранных из С1-4-алкила);
или его соль.
4-(4-хлор-2-фторанилино)-6-метокси-7-(1-метилпиперидин-4-илметокси)хиназолина,
4-(2-фтор-4-метиланилино)-6-метокси-7-(1-метилпиперидин-4-илметокси)хиназолина,
4-(4-бром-2-фторанилино)-6-метокси-7-(1-метилпиперидин-4-илметокси)хиназолина,
4-(4-хлор-2,6-дифторанилино)-6-метокси-7-(1-метилпиперидин-4-илметокси)хиназолина,
4-(4-бром-2,6-дифторанилино)-6-метокси-7-(1-метилпиперидин-4-илметокси)хиназолина,
4-(4-хлор-2-фторанилино)-6-метокси-7-(пиперидин-4-илметокси)хиназолина,
4-(2-фтор-4-метиланилино)-6-метокси-7-(пиперидин-4-илметокси)хиназолина,
4-(4-бром-2-фторанилино)-6-метокси-7-(пиперидин-4-илметокси)хиназолина,
4-(4-хлор-2,6-дифторанилино)-6-метокси-7-(пиперидин-4-илметокси)хиназолина и
4-(4-бром-2,6-дифторанилино)-6-метокси-7-(пиперидин-4-илметокси)хиназолина
и их солей.
4-(4-хлор-2-фторанилино)-6-метокси-7-(1-метилпиперидин-4-илметокси)хиназолина,
4-(4-бром-2-фторанилино)-6-метокси-7-(1-метилпиперидин-4-илметокси)хиназолина,
4-(4-хлор-2,6-дифторанилино)-6-метокси-7-(1-метилпиперидин-4-илметокси)хиназолина и
4-(4-бром-2,6-дифторанилино)-6-метокси-7-(1-метилпиперидин-4-илметокси)хиназолина
и их солей.
4-(4-бром-2-фторанилино)-6-метокси-7-(1-метилпиперидин-4-илметокси)хиназолина и его солей.
взаимодействие соединения формулы III:
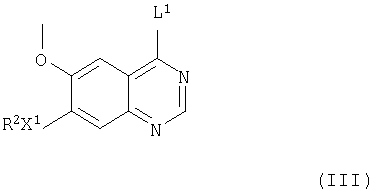
где R2 и X1 имеют значения, указанные в п.1, и L1 представляет собой заменяемую группу, с соединением формулы IV:
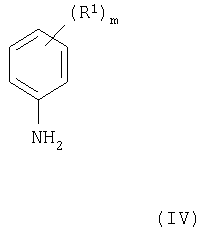
где R1 и m имеют значения, указанные в п.1,
и, когда необходима фармацевтически приемлемая соль производного хиназолина формулы I, взаимодействие полученного соединения с кислотой или основанием с получением требуемой фармацевтически приемлемой соли.
взаимодействие соединения формулы V:
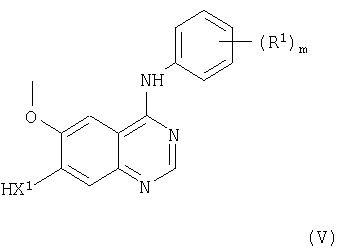
где m, X1 и R1 имеют значения, указанные в п.1, с соединением формулы VI

где R2 имеет значения, указанные в п.1, и L1 имеет значение, указанное ниже;
и, когда необходима фармацевтически приемлемая соль производного хиназолина формулы I, взаимодействие полученного соединения с кислотой или основанием с получением требуемой фармацевтически приемлемой соли.
взаимодействие соединения формулы VII
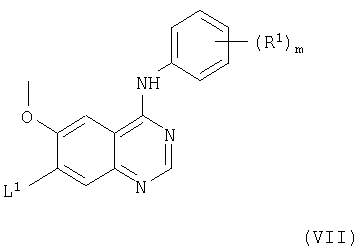
с соединением формулы VIII:

где R1, R2, m и X1 имеют значения, указанные в п.1, и L1 имеет значение, указанное ниже,
и, когда необходима фармацевтически приемлемая соль производного хиназолина формулы I, взаимодействие полученного соединения с кислотой или основанием с получением требуемой фармацевтически приемлемой соли.
удаление защиты у соединения формулы IX:
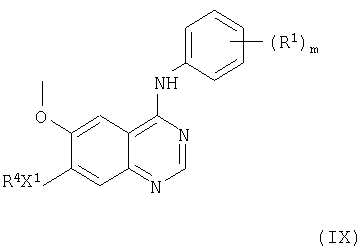
где R1, m и X1 имеют значения, указанные в п.1, и R4 представляет собой защищенную группу R2, где R2 имеет значения, указанные в п.1, но дополнительно содержит одну или несколько защитных групп Р2,
и, когда необходима фармацевтически приемлемая соль производного хиназолина формулы I, взаимодействие полученного соединения с кислотой или основанием с получением требуемой фармацевтически приемлемой соли.
(i) другого антиангиогенного агента, который действует по механизмам, отличным от механизма соединения формулы (I);
(ii) цитостатического агента;
(iii) модификатора биологических ответных реакций;
(iv) антитела; и
(v) антипролиферативного/противоопухолевого лекарственного средства или их комбинации.
(i) другого антиангиогенного агента, который действует по механизмам, отличным от механизма соединения формулы (I);
(ii) цитостатического агента;
(iii) модификатора биологических ответных реакций;
(iv) антитела; и
(v) антипролиферативного/противоопухолевого лекарственного средства или их комбинации.
| Дорожная спиртовая кухня | 1918 |
|
SU98A1 |
| Бесколесный шариковый ход для железнодорожных вагонов | 1917 |
|
SU97A1 |
| ПРОИЗВОДНЫЕ ХИНАЗОЛИНА ИЛИ ИХ ФАРМАЦЕВТИЧЕСКИ ПРИЕМЛЕМЫЕ СОЛИ, СПОСОБ ИХ ПОЛУЧЕНИЯ И ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ НА ИХ ОСНОВЕ | 1993 |
|
RU2127263C1 |

