Настоящее изобретение относится к кристаллической форме соединения
(S)-2-этокси-3-[4-(2-{4-метансульфонилоксифенил}этокси)фенил]пропановая кислота, представленного формулой I ниже

или его фармацевтически приемлемой соли и их сольватов. Данное изобретение также относится к способам лечения одного или более чем одного состояния, связанного с нарушением обмена веществ, в частности состояний, ассоциированных с синдромом инсулинорезистентности, и к применению кристаллической формы данного соединения или его фармацевтически приемлемой соли, или их сольвата в приготовлении лекарства для терапевтического использования в случае одного или более чем одного из указанных нарушений обмена веществ.
Данное изобретение также относится к фармацевтическим композициям, содержащим кристаллическую форму данного соединения, или его фармацевтически приемлемой соли, или их сольвата в качестве активного ингредиента, а также к способам получения кристаллической формы данного соединения или его фармацевтически приемлемой соли, или их сольвата.
При приготовлении лекарственных композиций важно, чтобы лекарственное вещество находилось в такой форме, в которой им было бы легко манипулировать и обрабатывать. Это важно не только с точки зрения получения коммерчески осуществимого способа получения, но также с позиции последующего приготовления фармацевтических препаратов, содержащих активное соединение.
Химическая стабильность, стабильность твердого состояния и срок хранения активных ингредиентов также представляют собой очень важные факторы. Лекарственное вещество и композиции, содержащие его, должны быть способны эффективно храниться в течение значительных периодов времени без существенного изменения физико-химических свойств активного компонента (например его химического состава, плотности, гигроскопичности и растворимости).
Кроме того, также важно, чтобы можно было получать лекарственное средство в такой форме, которая является настолько химически чистой, насколько это возможно.
Аморфные вещества могут представлять существенные проблемы в этом отношении. Например, такие вещества, как правило, сложнее обрабатывать и готовить в виде фармацевтического препарата, чем кристаллическое вещество, они имеют ненадежную растворимость и, как это часто обнаруживается, являются нестабильными и не являются химически чистыми.
Специалисту понятно, что если лекарственное средство может быть легко получено в виде стабильной кристаллической формы, то вышеперечисленные проблемы могут быть решены.
Таким образом, при приготовлении коммерчески осуществимых и фармацевтически приемлемых лекарственных композиций желательно, когда это возможно, получать лекарственное средство в по существу кристаллической и стабильной форме.
Тем не менее, следует отметить, что эта цель не всегда достижима. Действительно, как правило, невозможно предсказать, исходя из одной только молекулярной структуры, каково будет кристаллизационное поведение соединения, это обычно может быть определено лишь эмпирически.
Вышеуказанное соединение предназначено для терапевтического использования при синдроме инсулинорезистентности (IRS, Insulin Resistance Syndrome), который относится к группе проявлений, включающих в себя резистентность к инсулину с сопутствующей гиперинсулинемией, возможно сахарным диабетом 2-го типа, артериальной гипертензией, общим (висцеральным) ожирением, дислипидемией, наблюдаемой в виде нарушенных уровней липопротеинов, как правило, характеризующихся повышенными концентрациями липопротеинов очень низкой плотности (ЛПОНП) и пониженными концентрациями липопротеинов высокой плотности (ЛПВП) и пониженным фибринолизом.
Недавнее эпидемиологическое исследование засвидетельствовало, что индивидуумы с инсулинорезистентностью подвержены значительно большему риску сердечно-сосудистых заболеваний и смертности от них, особенно они страдают от инфаркта миокарда и инсульта. При сахарном диабете 2-го типа связанные с атеросклерозом состояния являются причиной до 80% всех смертельных исходов.
В клинической медицине существует осознание необходимости увеличения чувствительности к инсулину у пациентов, страдающих IRS, и, соответственно, коррекции дислипидемии, которая, как предполагают, вызывает ускоренное развитие атеросклероза. Однако это заболевание не является универсально четко определенным.
Настоящее изобретение относится к кристаллической твердой форме соединения формулы I. Значительные преимущества могут иметь место в случае, когда соединение формулы I может быть выделено в кристаллической форме, например, при получении соединения с уровнями чистоты и однородностью, соответствующими требованиям органов контроля, и для облегчения приготовления и однородности препарата.
Авторы изобретения выделили соединение формулы I в виде кристаллического твердого вещества. Конкретная выделенная кристаллическая форма существует в форме, которая практически или по существу не содержит растворителя (далее именуемая как "безводная форма"). В качестве альтернативы может быть получена сольватированная форма, например гидратированная форма.
Авторы в качестве объекта данного изобретения представляют кристаллическую форму соединения формулы I или его сольвата. В качестве альтернативного объекта данного изобретения авторы представляют кристаллическую форму фармацевтически приемлемой соли соединения формулы I или ее сольвата.
В используемый термин "сольватированный" авторы данного изобретения также включают понятие «гидратированный».
Под используемым термином "кристаллическая форма" авторы изобретения подразумевают любую и каждую возможную кристаллическую форму соединения формулы I, предпочтительно безводную форму.
Кристаллическая форма соединения формулы I может быть определена путем указания его точки плавления, картины дифракции рентгеновских лучей на порошке и данных рентгеновского анализа монокристалла.
Точка плавления кристаллической формы соединения формулы I, как правило, зависит от уровня чистоты и может быть определена обычными способами, хорошо известными из уровня техники, например посредством дифференциальной сканирующей калориметрии (ДСК). Как правило, безводная форма имеет точку плавления, находящуюся в диапазоне 82-92°С, например приблизительно 85-89°С.
Безводная форма имеет картину дифракции рентгеновских лучей на порошке, содержащую характерные пики высокой интенсивности при 6,2, 4,47 и 4,15 Å. Дополнительные характерные пики с более низкой интенсивностью относительно первых пиков находятся при 4,69, 3,64, 3,60 и 3,45 Å.
Кристаллическая форма соединения формулы I может быть получена из некристаллической формы соединения формулы I путем кристаллизации из подходящего растворителя (включая органические растворители, водные растворы и их смеси), такого как толуол и этилацетат, или из смеси растворителей, такой как смесь этанол/вода, изопропанол/вода или толуол/изооктан. Для инициации кристаллизации может потребоваться затравка кристаллическим соединением формулы I. Кристаллизация соединения из подходящей системы растворителей может быть осуществлена путем достижения перенасыщения, например путем охлаждения, путем выпаривания растворителя и/или путем добавления антирастворителя (растворителя, в котором соединение формулы I плохо растворимо, примеры подходящих антирастворителей включают в себя гептан или изооктан). Температуры и время кристаллизации будут варьировать в зависимости от концентрации соединения формулы I в растворе, используемой системы растворителей и выбранного способа кристаллизации.
Кристаллическая форма соединения формулы I может быть выделена с использованием способов, хорошо известных специалистам в данной области техники, например путем декантирования, фильтрации или центрифугирования. Также кристаллическая форма может быть высушена в соответствии с хорошо известными способами.
Возможная стадия(ии) перекристаллизации может быть осуществлена с использованием тех же самых или других систем растворителей для уменьшения дополнительных загрязнений, таких как аморфное вещество, химические примеси, или для превращения кристаллической формы в сольватированную/гидратированную форму или в безводную форму.
Предпочтительно кристаллизацию осуществляют непосредственно из реакционного раствора. В качестве альтернативы кристаллизацию осуществляют из последующего раствора.
Дополнительным объектом данного изобретения является способ получения кристаллической формы соединения формулы I, при котором кристаллизуют соединение формулы I.
При использовании термина "безводная форма" авторы изобретения не исключают присутствия внутри структуры кристаллической решетки некоторого количества растворителя, включая воду. Растворитель, включая воду, может также присутствовать снаружи структуры кристаллической решетки.
Аспектом данного изобретения является кристаллическая форма соединения формулы I, как описано выше, для применения в терапевтическом лечении.
В соответствии с еще одним аспектом данного изобретения предложена фармацевтическая композиция, которая содержит кристаллическую форму соединения формулы I, как описано выше, в сочетании с фармацевтически приемлемым разбавителем или носителем. Применение кристаллической формы соединения формулы I, как описано выше, в приготовлении фармацевтической композиции путем объединения кристаллической формы соединения формулы 1 с фармацевтически приемлемым разбавителем или носителем.
Композиция может находиться в форме, пригодной для перорального применения, например в форме таблетки, капсулы, водного или масляного раствора, суспензии или эмульсии; для местного применения, например в форме крема, мази, геля или водного или масляного раствора или суспензии; для назального применения, например в форме лекарства для вдыхания через нос, назального спрея или назальных капель; для вагинального или ректального применения, например в форме суппозитория; для введения путем ингаляции, например в виде тонкоизмельченного порошка, такого как сухой порошок, микрокристаллическая форма или жидкий аэрозоль; для сублингвального или трансбуккального применения, например в форме таблетки или капсулы; или для парентерального применения (включая внутривенное, подкожное, внутримышечное, внутрисосудистое введение или вливание), например в форме стерильного водного или масляного раствора или суспензии.
В общем вышеуказанные композиции могут быть приготовлены обычным образом с использованием традиционных эксципиентов.
Количество кристаллической формы соединения формулы I, как описано выше, которое объединяют с одним или более чем одним эксципиентом с получением разовой лекарственной формы, будет непременно варьировать в зависимости от пациента, которого лечат, и конкретного пути введения.
Например, препарат, предназначенный для перорального введения людям, как правило, содержит, например, от 0,001 мг до 50 мг активного агента, смешанного с соответствующим и подходящим количеством эксципиента(ов), которое может варьировать от приблизительно 10 до приблизительно 99,9999 процентов по массе от массы всей композиции.
Данное изобретение также включает в себя применение кристаллического соединения по данному изобретению, как описано выше, в приготовлении лекарства для использования в:
(1) лечении дислипидемии;
(2) лечении сахарного диабета 2-го типа;
(3) лечении гипергликемии;
(4) лечении гиперинсулинемии;
(5) лечении гиперлипемии;
(6) лечении артериальной гипертензии и/или
(7) лечении абдоминального ожирения.
Данное изобретение также включает в себя способ получения эффекта, как определено выше, или лечения заболевания или расстройства, как определено выше, при котором теплокровному животному, предпочтительно человеку, нуждающемуся в таком лечении, вводят эффективное количество кристаллической формы соединения формулы I, как описано выше.
Размер дозы кристаллической формы соединения формулы I для терапевтических или профилактических целей будет, естественно, варьировать в соответствии с природой и тяжестью медицинского состояния, возрастом и полом животного или пациента, которого лечат, и путем введения в соответствии с хорошо известными постулатами медицины.
Пригодные суточные дозы соединений по данному изобретению при терапевтическом лечении людей составляют приблизительно 0,001-50 мг/кг массы тела, предпочтительно 0,01-10 мг/кг массы тела.
Кристаллическая форма соединения формулы I может вводиться в качестве монотерапии, либо она может вводиться в комбинации с другими фармакологически активными агентами, такими как антидиабетический, антигипертензивный, диуретический или антигиперлипемический агент.
Кристаллические формы, приготовленные в соответствии с Примером(ами), приведенным(и) ниже, демонстрируют по существу одни и те же картины дифракции рентгеновских лучей на порошке и/или термограммы ДСК. При сравнении релевантных картин/термограмм (с возможностью экспериментальной ошибки) было ясно, что образовывалась одна и та же кристаллическая форма. Начальные температуры ДСК могут варьировать в диапазоне ±5°С (например ±2°С), и значения периодов на картинах дифракции рентгеновских лучей на порошке могут варьировать в диапазоне ±5 в последнем десятичном разряде.
Сокращения
EtOAc = этилацетат
ЖХВД = жидкостная хроматография высокого давления
i-PrOAc = изопропилацетат
NMP = N-метил-2-пирролидинон
THF = тетрагидрофуран
Синтез (S)-2-этокси-3-[4-(2-{4-метансульфонилоксифенил}этокси)фенил]пропановой кислоты
1) Этил (S)-2-этокси-3-(4-гидроксифенил)пропаноат
а) Получение этил 2-этоксиэтаноата
Раствор 2-хлоруксусной кислоты (50 г, 529 ммоль, 1,0 экв.) в абсолютном этаноле (110 мл, 2,2 об., где об. далее обозначает объемный эквивалент) вводят в этанольный раствор этоксида натрия (494 мл, 90 г, 1,32 моль, 2,5 об.). Температуру во время введения поддерживают на уровне 15-25°С. Когда введение завершается, температуру поднимают до 50°С. Реакционную смесь охлаждают до 15°С, когда достигается степень превращения свыше 95%. Затем вводят HCl (г) до тех пор, пока рН смеси не станет меньше 1. Когда степень превращения становится свыше 95%, суспензию охлаждают до 15°С и нейтрализуют до рН 5-7 раствором этоксида натрия (приблизительно 5-20% от изначально введенного количества). После нейтрализации суспензию охлаждают до 5°С и вводят этилацетат (150 мл, 3 об.). Образующийся в реакции хлорид натрия затем отфильтровывают и промывают этилацетатом. Раствор затем выпаривают. Максимально остаточный этанол составляет 20 об.%.
Общий выход указанного в подзаголовке соединения составляет 58% от теоретического значения (потеря при выпаривании). Химическая чистота составляет свыше 99%.
б) Получение этил 2-этокси-3-(4-метоксифенил)пропеноата
4-Метоксибензальдегид (100 г, 734 ммоль, 1,0 экв.) и этил 2-этоксиэтаноат (116 г, 881 ммоль, 1,2 экв.) растворяют в THF (600 мл, 6 об.) в атмосфере азота. Раствор охлаждают до -20°С. В получающийся в результате раствор медленно вводят раствор трет-бутоксида калия (98,8 г, 880 ммоль, 1,2 экв.) в THF (704 мл, 7,1 об., соответствующие трет-бутоксиду калия), поддерживая температуру ниже -10°С. После завершения введения реакционную смесь перемешивают в течение 1 часа при температуре от -15°С до -10°С. В суспензию затем вводят ледяную уксусную кислоту (53 г, 1,24 моль, 1,7 экв.), поддерживая температуру ниже +5°С. THF затем выпаривают до тех пор, пока не останется приблизительно 1/3. Добавляют толуол (824 мл, 8,24 об.) и остаток THF выпаривают. К толуольной суспензии добавляют воду (200 мл, 2 об.) и метансульфоновую кислоту (50 мл, 0,5 об.) с получением рН в водном слое 2-3. Водный слой отделяют. Толуольный слой затем выпаривают для удаления остаточной воды. К толуольному раствору добавляют метансульфоновую кислоту (2,11 г, 22 ммоль, 0,03 экв.). Толуольный раствор кипятят с присоединенным устройством Дина-Старка (Dean-Starke) до достижения полного превращения. Раствор охлаждают до 25°С. Раствор затем промывают гидроксидом натрия (водн., 48%) (1,83 г, 22 ммоль, 0,03 экв.), разбавленным водой (15 мл).
Общий выход указанного в подзаголовке соединения приблизительно составляет 52% от теоретического значения.
в) Получение 2-этокси-3-(4-метоксифенил)пропеновой кислоты
NaOH (водн., 48%) (122 г, 1,46 моль, 2,0 экв.), воду (244 мл, 2,44 об.) и EtOH (90 мл, 0,9 об.) вводят в толуольный раствор этил 2-этокси-3-(4-метоксифенил)пропеноата (приблизительно 96 г, 382 ммоль, 0,52 экв.). Реакционную смесь нагревают до 50°С и перемешивают до достижения полного превращения. После завершения реакции толуольный слой отделяют и водный слой затем промывают толуолом (100 мл, 1 об.). После разделения водный слой охлаждают до +5°С и подкисляют конц. HCl (приблизительно 173 мл, 2,1 моль, 2,9 экв.). В процессе введения кислоты температуру поддерживают ниже 10°С. К кислотной водной суспензии добавляют EtOAc (100 мл, 1 об.). После экстракции фазы разделяют. EtOAc раствор выпаривают и добавляют толуол (288 мл, 3 об.).
В толуольный раствор вводят затравку 2-этокси-3-(4-метоксифенил)пропеновой кислоты и охлаждают до 0°С. После кристаллизации вещество отфильтровывают. Влажное вещество используют без сушки на следующей стадии.
Общий выход указанного в подзаголовке соединения составляет 42% от теоретического значения для стадий (б) и (в) вместе. Химическая чистота составляет 99,7%.
г) Получение 2-этокси-3-(4-метоксифенил)пропановой кислоты
Палладий на угле (5%, 60%-ная влажность) (13,2 г, 0,26 г Pd, 2,44 ммоль Pd, 0,0054 экв.) вводят в раствор 2-этокси-3-(4-метоксифенил)пропеновой кислоты (100 г, 450 ммоль, 1,0 экв.) в этаноле (800 мл, 8 об.) в атмосфере азота. В сосуде затем создают с помощью водорода общее давление 4 бара (0,4 МПа). Гидрирование продолжают до достижения полного превращения. Катализатор отфильтровывают и этанол выпаривают в вакууме. Добавляют толуол (500 мл, 5 об.) и затем выпаривают. Остаток растворяют в толуоле (500 мл, 5 об.) и упаривают до объема 260 мл. Раствор нагревают до 50°С и добавляют изооктан (800 мл, 8 об.). Раствор охлаждают до 35°С и затем вводят затравку 2-этокси-3-(4-метоксифенил)пропановой кислоты. Температуру поддерживают на уровне 35°С в течение 30 мин. Разбавленную суспензию затем охлаждают со скоростью 10°С/час до температуры +5°С, которую поддерживают в течение ночи. Кристаллы затем отфильтровывают и промывают изооктаном (220 мл, 2,2 об.). Кристаллы сушат в вакууме при 30°С.
Выход указанного в подзаголовке соединения составляет 88% от теоретического значения. Химическая чистота составляет 99,8%.
д) Получение (1S)-1-(1-нафтил)-1-этанаминия (2S)-2-этокси-3-(4-метоксифенил)пропаноата
Раствор 2-этокси-3-(4-метоксифенил)пропионовой кислоты (100 г, 446 ммоль, 1,0 экв.) в i-PrOAc (2000 мл, 20 об.) перемешивают при 0-5°С в атмосфере азота. К получающемуся в результате раствору добавляют (S)-1-(1-нафтил)этиламин (45,8 г, 268 ммоль, 0,6 экв.). Получающуюся в результате суспензию нагревают до 75-80°С для растворения всех частиц, посредством чего получают раствор. Этот раствор затем охлаждают и вводят затравку соли (S)-1-(1-нафтил)этиламина и (2S)-2-этокси-3-(4-метоксифенил)пропановой кислоты. Желаемую диастереомерную соль собирают путем фильтрации. Кристаллы промывают i-PrOAc.
Полученную соль (1S)-1-(1-нафтил)этиламина и (2S)-2-этокси-3-(4-метоксифенил)пропановой кислоты (67 г, 169 ммоль, 1,0 экв.) растворяют путем нагревания до 75-80°С в i-PrOAc (1340 мл, 20 об.). Полученный продукт собирают путем фильтрации, промывают i-PrOAc и сушат в вакууме при 40°С до постоянной массы.
Общий выход в течение двух стадий кристаллизации составляет 74% от теоретического значения. Химическая чистота составляет свыше 99%. Энантиомерный избыток (э.и.) составляет 97,8%.
е) Получение (S)-2-этокси-3-(4-гидроксифенил)пропановой кислоты
Соль (1S)-1-(1-нафтил)этиламина и (2S)-2-этокси-3-(4-метоксифенил)пропановой кислоты (100 г, 253 ммоль, 1,0 экв.) суспендируют в толуоле. Смесь затем обрабатывают NaOH (11,1 г, 278 ммоль, 1,1 экв.) в воде (280 мл, 5 об.). Верхний толуольный слой, содержащий хиральный амин, отделяют. Нижний водн. слой промывают еще двумя частями толуола (280 мл, 5 об.). Нижний водн. слой подкисляют до рН=1 водн. 37%-ной HCl (30 г, 304 ммоль, 1,2 экв.). Водный раствор, содержащий (S)-2-этокси-3-(4-метоксифенил)пропановую кислоту, экстрагируют двумя порциями EtOAc (280 мл, 5 об.). Объединенный EtOAc экстракт промывают одной порцией воды (280 мл, 5 об.). Растворитель заменяют на NMP при пониженном давлении.
NaOH (гранулы) (45,5 г, 1,14 моль, 4,5 экв.) и октантиол (129 г, 154 мл, 884 ммоль, 3,5 экв.) вводят в раствор (S)-2-этокси-3-(4-метоксифенил)пропановой кислоты (приблизительно 56,6 г, 253 ммоль, 1,0 экв.) в NMP (680 мл, 12 об.) в атмосфере азота. Реакционную смесь нагревают до 120°С и поддерживают при 115-125°С до завершения реакции, что определяют с помощью ЖХВД.
Реакционную смесь охлаждают до 60°С и затем гасят водой. рН затем доводят до 2-3 с помощью конц. HCl. Температуру поддерживают на уровне 60-70°С. Образуются два слоя, из которых верхний слой содержит в основном октантиол и соответствующий метиловый эфир (образующийся в ходе реакции). Слои разделяют и слой, содержащий воду и NMP, концентрируют до 3-4 объемов в вакууме при внутренней температуре 80-100°С.
Остаток экстрагируют смесью H2O:EtOAc. EtOAc раствор затем промывают 3 раза 15%-ным раствором NaCl.
EtOAc выпаривают и остаток непосредственно используют на следующей стадии, или он также может кристаллизоваться из толуола с получением белого твердого вещества.
Выход составляет 52% с использованием кристаллизации, 90% - с использованием только выпаривания. Химическая чистота составляет 99,8%. Энантиомерный избыток (э.и.) составляет 97,8%.
ж) Получение этил (S)-2-этокси-3-(4-гидроксифенил)пропаноата
(S)-2-Этокси-3-(4-гидроксифенил)пропановую кислоту (874 г, 4,16 моль, 1,0 экв.) растворяют в EtOAc (1250 мл). В этот раствор вводят этанол (3000 мл) и HCI (37%, водн.) (40 мл, 0,48 моль, 0,12 экв.). Раствор нагревают до кипения (приблизительно 72°С) и отгоняют смесь вода/EtOAc/EtOH (2000 мл). Еще одну порцию EtOH (2000 мл) вводят и еще 2000 мл отгоняют. Эту процедуру повторяют еще раз. В этот момент достигается степень превращения приблизительно 95%. Затем добавляют EtOH (99,5%, 1000 мл) и выпаривают. Это повторяют до тех пор, пока не достигается степень превращения свыше 97,5%. Раствор затем концентрируют до объема 1700-2000 мл в вакууме и затем охлаждают до 20°С.
EtOAc раствор, содержащий этил (S)-2-этокси-3-(4-гидроксифенил)пропаноат, затем при интенсивном перемешивании медленно (30-40 мин) вводят в раствор NaHCO3 (7 об.%, 3500 мл). Кристаллизация происходит через несколько минут. После введения суспензию охлаждают до 0-5°С и затем перемешивают при 0-5°С в течение по меньшей мере одного часа. Кристаллы затем отфильтровывают и сушат в вакууме.
Выход составляет приблизительно 93%. Химическая чистота составляет свыше 99%. Энантиомерный избыток (э.и.) составляет свыше 97,8%.
2) 2-(4-(Метансульфонилоксифенил)этилметансульфонат
2-(4-Гидроксифенил)этанол (356 г, 2,58 моль, 1,0 экв.) растворяют в метиленхлориде (3500 мл) и триэтиламине (653 г, 6,44 моль, 2,5 экв.). Смесь охлаждают до -20°С. Затем добавляют метансульфонилхлорид (657 г, 5,74 моль, 2,2 экв.), поддерживая температуру между -25°С и -15°С. Когда степень превращения становится выше 95%, образующуюся во время реакции соль отфильтровывают и промывают метиленхлоридом (600 мл). Органический слой промывают сначала насыщенным раствором гидрокарбоната натрия (700 мл) при 20°С, а затем водой (700 мл). Метиленхлорид выпаривают и заменяют ацетонитрилом. Ацетонитрильный раствор затем используют на следующей стадии.
3) Этил (S)-2-этокси-3-[4-(2-{4-метансульфонилоксифенил}этокси)фенил]пропаноат
Этил (S)-2-этокси-3-(4-гидроксифенил)пропаноат (325 г, 1,34 моль, 1,0 экв.) растворяют в ацетонитриле (2600 мл). Когда образуется гомогенный раствор, добавляют карбонат калия (560 г, 4,05 моль, 3,0 экв.) и сульфат магния (110 г, (0,2 г/г К2СО3)). Ацетонитрильный раствор 2-(4-(метансульфонилоксифенил)этилметансульфоната (общий объем приблизительно: 2050 мл, (0,3 г/мл, 2,21 моль, 1,65 экв.) вводят в реакционный сосуд и смеси дают возможность вступать в реакцию при кипячении с обратным холодильником, 82°С, в течение 24 часов при интенсивном перемешивании, поддерживая объем постоянным путем порционного добавления ацетонитрила. Когда степень превращения становится выше 98%, реакционную смесь охлаждают до комнатной температуры. Оставшуюся соль отфильтровывают и промывают ацетонитрилом (800 мл). Фильтрат выпаривают досуха. Остаток затем используют на следующей стадии.
4) (S)-2-Этокси-3-[4-(2-{4-метансульфонилоксифенил}этокси)фенил]пропановая кислота
К маслу этил (S)-2-этокси-3-[4-(2-{4-метансульфонилоксифенил}этокси)фенил]пропаноата (723 г (71,2% анализ), 1,18 моль, 1,0 экв.) добавляют THF (3900 мл). Когда образуется гомогенный раствор, добавляют воду (900 мл). Смесь охлаждают до +10°С. Раствор гидроксида лития (390 мл, 4 М, 1,32 экв.) добавляют в течение 1 часа. Температуру затем поднимают до +30°С и реакции дают возможность протекать при этой температуре в течение 2-3 часов. Реакцию останавливают, когда степень превращения становится выше 99%. Добавляют EtOAc (500 мл) и смесь охлаждают до комнатной температуры. Раствор перемешивают в течение приблизительно 30 минут и THF выпаривают. Когда выпарено приблизительно 80-90% THF, добавляют воду (1900 мл). Выпаривание продолжают до тех пор, пока THF не исчезнет из смеси. Щелочной водный раствор затем промывают EtOAc (1000 мл, 2×1250 мл и 950 мл). рН водного раствора (S)-2-этокси-3-[4-(2-{4-метансульфонилоксифенил}этокси)фенил]пропановой кислоты затем доводят до 2,0-2,5 с помощью HCI (водн.) (550 мл, 3,0 М). Добавляют EtOAc (2500 мл) и фазы разделяют. Этилацетатный раствор (S)-2-этокси-3-[4-(2-{4-метансульфонилоксифенил}этокси)фенил]пропановой кислоты затем промывают водой (700 мл) и после разделения выпаривают досуха. Оставшееся масло затем используют в последующей кристаллизации.
Кристаллизация (S)-2-этокси-3-[4-(2-{4-метансульфонилоксифенил}этокси)фенил]пропановой кислоты
Неочищенное вещество из 3 партий (S)-2-этокси-3-[4-(2-{4-метансульфонилоксифенил}этокси)фенил]пропановой кислоты (общая масса 1871 г, 1262 г соединения, 3,09 моль, 1,0 экв.), содержащих EtOAc (500 мл), растворяют в толуоле (5000 мл) при 50°С. Когда получается прозрачный раствор, его выпаривают для уменьшения количества присутствующего EtOAc. Объем перед выпариванием составляет 6750 мл. Добавляют еще одну порцию толуола (2500 мл), объем после добавления составляет 7750 мл, и выпаривание продолжают. Затем к раствору добавляют третью порцию толуола (2500 мл), объем перед добавлением составляет 6300 мл, объем после добавления составляет 8800 мл. Выпаривание продолжают до образования непрозрачного раствора, объем составляет 8200 мл. К раствору, который нагрет до 40°С, добавляют изооктан (1000 мл). Кристаллизацию инициируют путем введения затравки при 40°С. Смесь интенсивно перемешивают до образования суспензии. Скорость перемешивания затем уменьшают. Суспензию оставляют кристаллизоваться в течение ночи. Суспензию затем фильтруют и промывают смесью толуол: изооктан 5:1 (1800 мл). Кристаллы затем сушат при пониженном давлении при 40°С.
Перекристаллизация (S)-2-этокси-3-[4-(2-{4-метансульфонилоксифенил}этокси)фенил]пропановой кислоты
(S)-2-Этокси-3-[4-(2-{4-метансульфонилоксифенил}этокси)фенил]пропановую кислоту (1040 г (96,4% анализ), 2,45 моль, 1,0 экв.) растворяют в толуоле (7000 мл) при температуре 60°С. Когда образуется прозрачный раствор, к нему добавляют изооктан (1720 мл). Раствор затем фильтруют через Силикагель 60 (Silica 60 gel). Раствор затем охлаждают с 50°С до 45°С, причем при этой температуре происходит кристаллизация. Суспензию охлаждают до 20°С. Твердое вещество затем отфильтровывают и промывают смесью толуол: изооктан 5:1 (1500 мл). Кристаллы сушат при пониженном давлении при 40°С.
Определение точки плавления
Дифференциальная сканирующая калориметрия (ДСК) была осуществлена с использованием прибора Mettler DSC820 в соответствии со стандартными способами, например, описанными в: Höhne, G. W et al (1996), Differential Scanning Calorimetry, Springer, Berlin.
ДСК безводной формы продемонстрировала эндотерму с экстраполированной начальной температурой, составляющей приблизительно 87°С (приблизительно 102 Дж/г).
Определение картины дифракции рентгеновских лучей на порошке
Порошковые дифрактограммы рентгеновских лучей (ПДРЛ) были определены с использованием рентгеновского дифрактометра Siemens D5000 и/или ренгеновского дифрактометра Philips X'Pert. ПДРЛ получали на образцах, приготовленных в соответствии со стандартными способами, примеры которых описаны в: Giacovazzo, С. et al (1995), Fundamentals of Crystallography, Oxford University Press; Jenkins, R. and Snyder, R. L. (1996), Introduction to X-Ray Powder Diffractometry, John Wiley and Sons, New York; Bunn, C. W. (1948), Chemical Crystallography, Clarendon Press, London; или Klug, H. P. and Alexander, L. E. (1974), X-Ray Diffraction Procedures, John Wiley and Sons, New York.
Порошковые дифрактограммы рентгеновских лучей типичного образца безводной кристаллической формы соединения формулы I представлены на Фиг.1.
Кристаллы безводной формы были проанализированы посредством ПДРЛ, результаты представлены ниже в Таблице I (где ОИ представляет собой относительную интенсивность) и изображены на Фиг.1. Дифрактограмму измеряли с переменными щелями и без внутреннего стандарта. Интенсивности основаны на интенсивностях, наблюдаемых при измерении с переменными щелями без вычитания фона. Относительные интенсивности являются менее достоверными и вместо числовых величин используют следующие определения:
Некоторые дополнительные слабые или очень слабые пики, обнаруженные на дифрактограмме, были исключены из Таблицы.
Таблица. Данные по дифракции рентгеновских лучей на порошке для безводной формы кристаллической формы соединения формулы I, представленной на Фиг.1.
Понятно, что d-значения картин дифракции рентгеновских лучей на порошке могут слегка варьировать от одного инструмента к другому и поэтому приведенные значения не следует интерпретировать как абсолютные. Следует полагать, что кристаллическая форма соединения формулы I является той, которая описана здесь, если d-значения находятся в пределах ±5 в последнем десятичном знаке, особенно в пределах ±2 в последнем десятичном знаке.
Определение картины дифракции рентгеновских лучей от монокристалла
Элементарную ячейку определяли из рентгеновских данных для монокристалла безводной формы. Она является орторомбической с симметрией P212121, Z=4 и имеет следующие показатели: а=5,762Å, b=14,426Å, с=24,785Å, α=β=γ=90° и V=2060,2Å3.
Исследование стабильности кристаллов (S)-2-этокси-3-[4-(2-{4-метансульфонилоксифенил}этокси)фенил]пропановой кислоты
Были протестированы три партии кристаллов безводной формы на стабильность после 12-месячного хранения при 25°С/60% относительной влажности в двойных полиэтиленовых пакетах (стандартные условия для тестирования стабильности коммерческих препаратов). Никаких изменений в кристаллах при тестировании с помощью дифракции рентгеновских лучей на порошке или инфракрасной спектроскопии не было обнаружено при сравнении со спектрами исходного эталонного вещества.
| название | год | авторы | номер документа |
|---|---|---|---|
| ИЗМЕЛЬЧЕННАЯ ФОРМА (S)-2-ЭТОКСИ-3-[4-(2-{4-МЕТАНСУЛЬФОНИЛОКСИФЕНИЛ}ЭТОКСИ)ФЕНИЛ]ПРОПАНОВОЙ КИСЛОТЫ | 2000 |
|
RU2248966C2 |
| ПОЛИМОРФЫ С-MET/HGFR ИНГИБИТОРА | 2006 |
|
RU2387650C2 |
| НОВЫЕ ТИЕНОПИРИМИДИНОВЫЕ ПРОИЗВОДНЫЕ, СПОСОБ ИХ ПОЛУЧЕНИЯ И ФАРМАЦЕВТИЧЕСКИЕ КОМПОЗИЦИИ, СОДЕРЖАЩИЕ ИХ | 2014 |
|
RU2605403C2 |
| НОВЫЕ БИЦИКЛИЧЕСКИЕ ПРОИЗВОДНЫЕ, СПОСОБ ИХ ПОЛУЧЕНИЯ И ФАРМАЦЕВТИЧЕСКИЕ КОМПОЗИЦИИ, СОДЕРЖАЩИЕ ИХ | 2016 |
|
RU2760554C1 |
| СПОСОБ ПОЛУЧЕНИЯ (4S)-4-(4-ЦИАНО-2-МЕТОКСИФЕНИЛ)-5-ЭТОКСИ-2,8-ДИМЕТИЛ-1,4-ДИГИДРО-1,6-НАФТИРИДИН-3-КАРБОКСАМИДА И ВЫДЕЛЕНИЯ (4S)-4-(4-ЦИАНО-2-МЕТОКСИФЕНИЛ)-5-ЭТОКСИ-2,8-ДИМЕТИЛ-1,4-ДИГИДРО-1,6-НАФТИРИДИН-3-КАРБОКСАМИДА ПОСРЕДСТВОМ ЭЛЕКТРОХИМИЧЕСКИХ МЕТОДОВ | 2016 |
|
RU2715226C2 |
| (2S,4R)-5-(5'-ХЛОР-2'-ФТОРБИФЕНИЛ-4-ИЛ)-4-(ЭТОКСИОКСАЛИЛАМИНО)-2-ГИДРОКСИМЕТИЛ-2-МЕТИЛПЕНТАНОВАЯ КИСЛОТА | 2016 |
|
RU2726623C2 |
| НОВЫЕ ПРОИЗВОДНЫЕ АММОНИЯ, СПОСОБ ИХ ПОЛУЧЕНИЯ И ФАРМАЦЕВТИЧЕСКИЕ КОМПОЗИЦИИ, СОДЕРЖАЩИЕ ИХ | 2016 |
|
RU2743098C2 |
| СПОСОБ ПОЛУЧЕНИЯ (4S)-4-(4-ЦИАНО-2-МЕТОКСИФЕНИЛ)-5-ЭТОКСИ-2,8-ДИМЕТИЛ-1-4-ДИГИДРО-1,6-НАФТИРИДИН-3-КАРБОКСАМИДА И ЕГО ОЧИСТКИ ДЛЯ ПРИМЕНЕНИЯ В КАЧЕСТВЕ ФАРМАЦЕВТИЧЕСКОГО АКТИВНОГО ИНГРЕДИЕНТА | 2015 |
|
RU2729998C2 |
| СПОСОБЫ ПОЛУЧЕНИЯ (S)-1-(3-ЭТОКСИ-4-МЕТОКСИФЕНИЛ)-2- МЕТАНСУЛЬФОНИЛЭТИЛАМИНА | 2013 |
|
RU2632875C2 |
| КРИСТАЛЛИЧЕСКАЯ ФУМАРАТНАЯ СОЛЬ (S)-[3,4-ДИФТОР-2-(2-ФТОР-4-ЙОДФЕНИЛАМИНО)ФЕНИЛ][3-ГИДРОКСИ-3-(ПИПЕРИДИН-2-ИЛ)АЗЕТИДИН-1-ИЛ]МЕТАНОНА | 2016 |
|
RU2762181C2 |
Настоящее изобретение относится к новой кристаллической форме соединения (S)-2-этокси-3-[4-(2-{4-метансульфонилоксифенил} этокси)фенил] пропановая кислота, представленного формулой (I).

Соединение может быть использовано при лечении состояний, связанных с нарушением обмена веществ, в частности состояний, ассоциированных с синдромом инсулинорезистентности. Кристаллическая форма соединения формулы I способствует облегчению приготовления и однородности препарата. 2 з.п. ф-лы, 1 табл., 1 ил.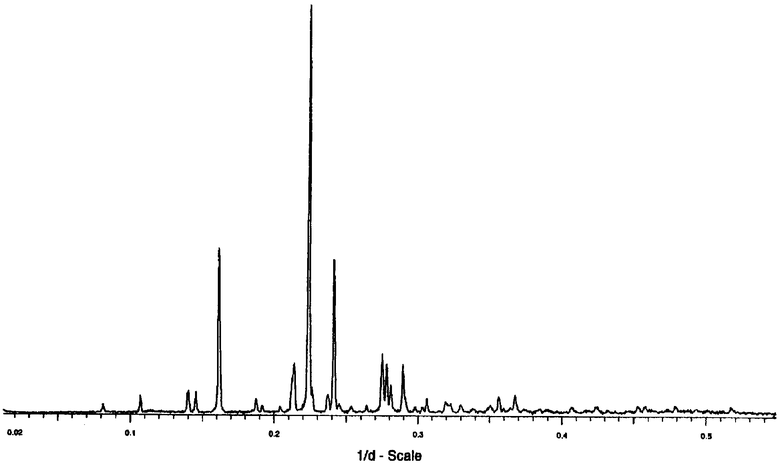

имеющая точку плавления (87±5)°С и имеющая картину дифракции рентгеновских лучей на порошке, содержащую характерные пики высокой интенсивности при 6,2, 4,47 и 4,15 Å.
| СПОСОБ ПОЛУЧЕНИЯ СЛОЖНЫХ ЭФИРОВ а-МЕТИЛОЛБЕНЗОИНСУЛЬФОКИСЛОТЫ | 0 |
|
SU335831A1 |
| Бесколесный шариковый ход для железнодорожных вагонов | 1917 |
|
SU97A1 |

