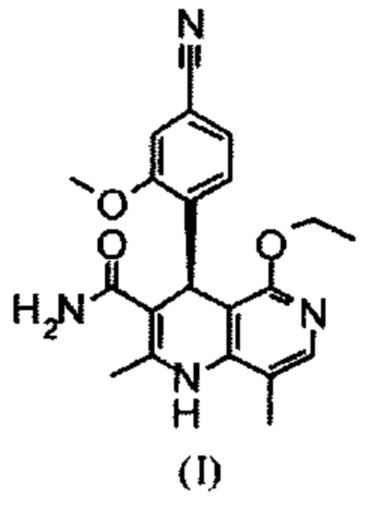
Настоящее изобретение относится к новому и улучшенному способу получения (4S)-4-(4-циано-2-метоксифенил)-5-этокси-2,8-диметил-1,4-дигидро-1,6-нафтиридин-3-карбоксамида формулы (I)
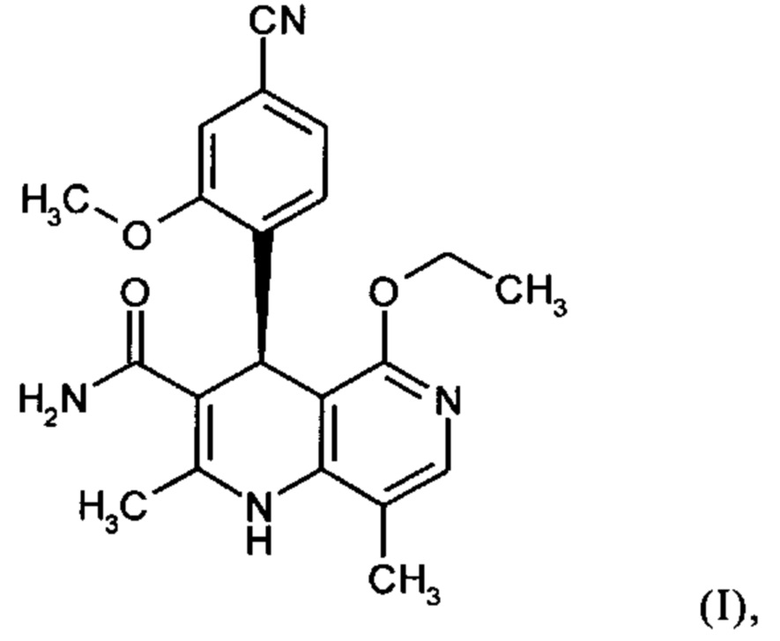
а также к получению и применению кристаллического полиморфа I (4S)-4-(4-циано-2-метоксифенил)-5-этокси-2,8-диметил-1,4-дигидро-1,6-нафтиридин-3-карбоксамида формулы (I).
Соединение формулы (I) действует как нестероидный антагонист минералокортикоидного рецептора и может быть использовано, например, в качестве средства для профилактики и/или лечения сердечно-сосудистых и почечных нарушений, таких как сердечная недостаточность и диабетическая нефропатия.
Соединение формулы (I) и способ его получения описаны в WO 2008/104306 и ChemMedChem 2012, 7, 1385, где, в обеих публикациях, раскрыто подробное описание исследования синтеза. Недостатком описанного в них синтеза является тот факт, что этот синтез непригоден для дальнейшего крупномасштабного процесса, поскольку многие стадии протекают при очень высоком разбавлении с очень большим избытком реагентов и, следовательно, обеспечивают относительно низкий общий выход. Кроме того, требуется много промежуточных хроматографических очисток, которые технически обычно очень трудоемки и требуют высокого расхода растворителей, которые являются дорогостоящими и поэтому их следует избегать, если это возможно. Некоторые стадии недоступны из-за трудностей с безопасностью и технологическими процессами.
Таким образом, существует необходимость в промышленно осуществимом синтезе, который обеспечивает воспроизводимость соединения формулы (I) с высоким общим выходом, низкими издержками производства и высокой чистотой и отвечает всем нормативным требованиям для обеспечения клинических испытаний с активным ингредиентом и использоваться для последующей заявки на регистрацию.
В рамках настоящего изобретения был найден очень эффективный синтез, который позволяет удовлетворить упомянутые выше требования.
В публикации ChemMedChem 2012, 7, 1385, где описан синтез соединения формулы (I) в масштабе исследования, где соединение формулы (I) получают в 10 стадий, исходя из ванилина с общим выходом 3,76% от теоретического. Соединение формулы (I) получали упариванием хроматографических фракций в виде аморфного твердого вещества; определенный способ кристаллизации для конечной стадии образования полиморфа до сих пор не был описан.
Следующая схема 1 демонстрирует известный способ получения соединения формулы (I).
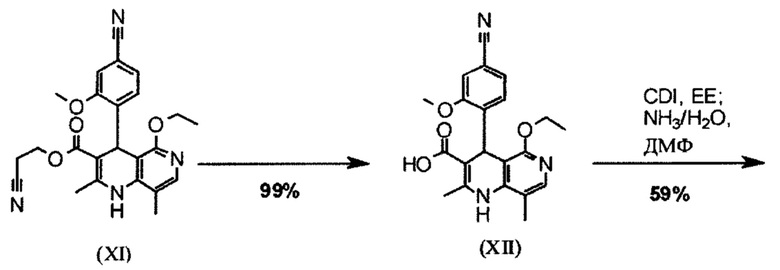
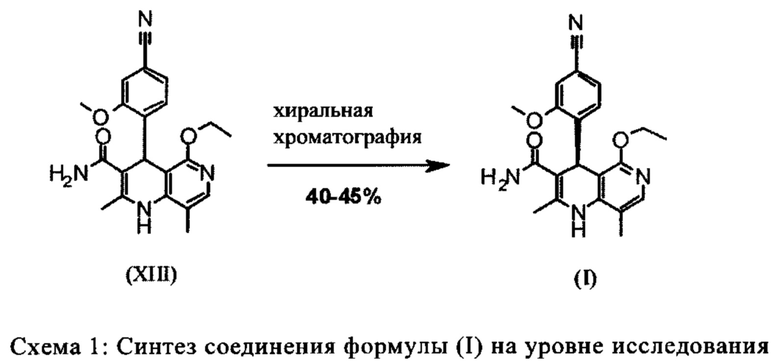
Используют три хроматографические очистки, а также стадию хиральной хроматографии для разделения энантиомеров рацемата формулы (XIII). Некоторые из стадий проходят при очень высоком разбавлении и использовании очень больших количеств реагента.
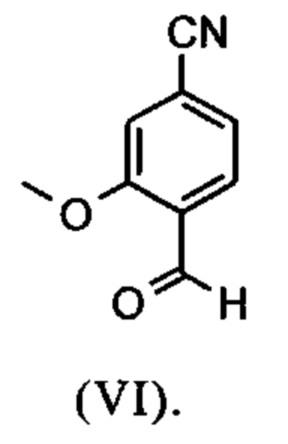
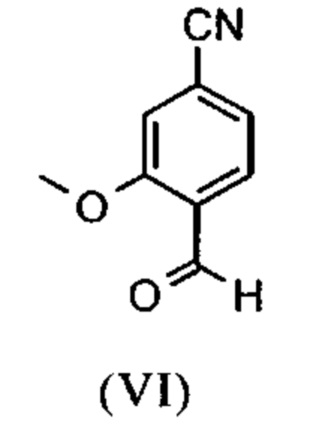
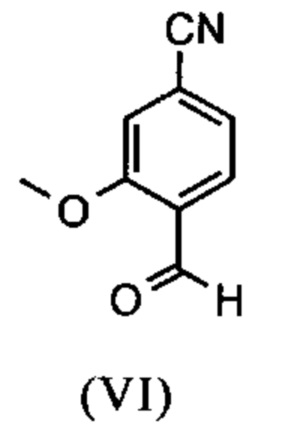
Например, последовательность получения нитрил-альдегидного промежуточного соединения (VI), в частности, которая играет центральную роль в этом синтезе, неприемлема с точки зрения экономии атомов.
Кроме того, этот способ не может быть перенесен в промышленный масштаб, поскольку прежде всего используются очень дорогие реагенты, такие как ангидрид трифторметансульфокислоты  и избытки трет-бутилакрилата. При проведении реакции Хека
и избытки трет-бутилакрилата. При проведении реакции Хека  в промышленных масштабах, в резервуаре образуется остаток, подобный пластику, который возникает в результате полимеризации трет-бутилакрилата, используемого в избытке. Это неприемлемо в промышленной методике, поскольку существует опасность того, что это может вызвать разрушение мешалки и приведет к образованию остатков в механизме мешалки, которые слишком трудно удалить.
в промышленных масштабах, в резервуаре образуется остаток, подобный пластику, который возникает в результате полимеризации трет-бутилакрилата, используемого в избытке. Это неприемлемо в промышленной методике, поскольку существует опасность того, что это может вызвать разрушение мешалки и приведет к образованию остатков в механизме мешалки, которые слишком трудно удалить.
Последующего расщепления двойной связи периодатом натрия и высокотоксичным тетроксидом осмия также следует избегать, так как в описанных экспериментальных условиях происходит задержка реакции, которая приводит к сильной экзотермичности и, следовательно, связана с нерегулируемой реакцией.
Схема 2 иллюстрирует новый способ в соответствии с изобретением, который дает соединение формулы (I) в 9 стадий с общим выходом 27,7% от теоретического без хроматографической очистки промежуточных продуктов.
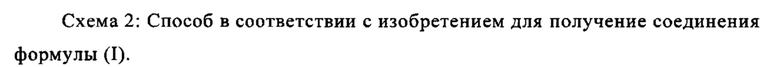
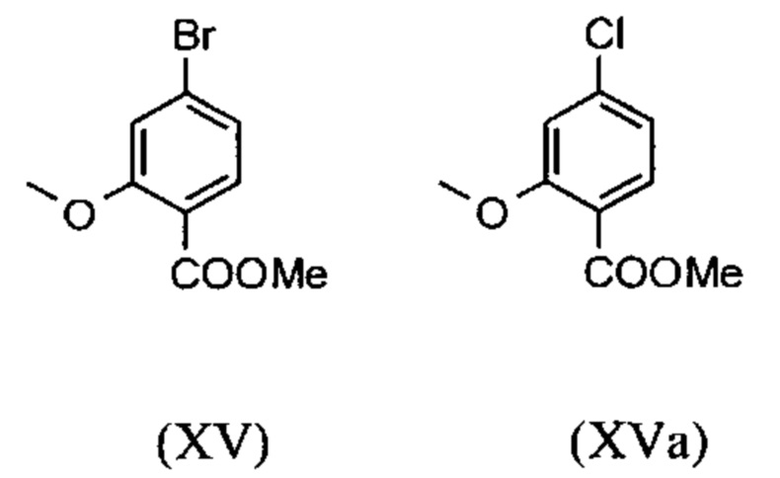
Сложный метиловый эфир (XV) и альдегид (XVI) не выделяются, но не подвергаются далее реакции непосредственно в раствор, что приводит только к 7 стадиям для выделения. Метод препаративной хиральной ВЭЖХ (например, SMB Technology, Varicol) применяют для разделения энантиомеров.
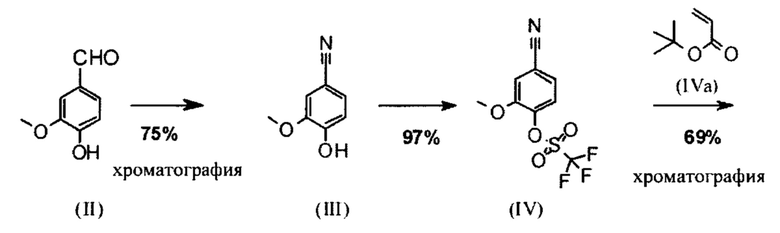
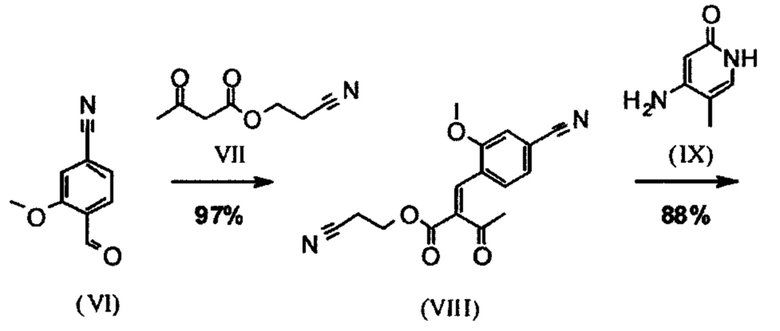
Альдегид (VI) является известным из литературы (J. Med. Chem. 2007, 50, 2468-2485) и является важным промежуточным соединением в этом синтезе. Между тем, есть также возможность приобрести соединение коммерчески.
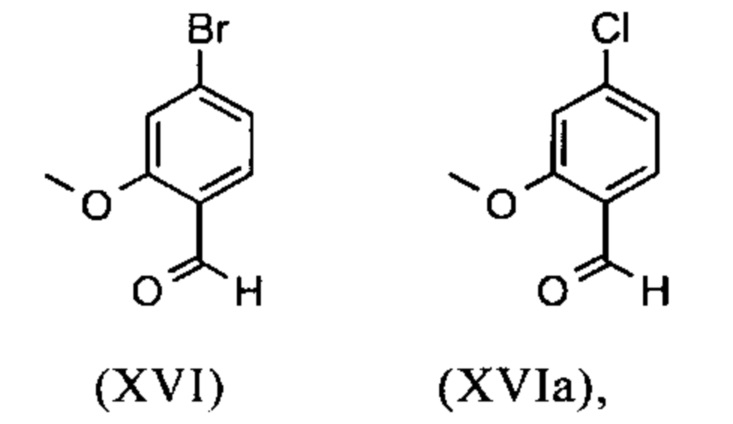
Исходя из 4-циано-2-метокситолуола (VIa) получают дибромид (VIb) с использованием NBS, который подвергают реакции в этаноле с 2,46 экв. нитрата серебра (в воде) для получения целевого альдегида (VI). Этот синтез, описанный в литературе, и способ, описанный в синтезе на уровне исследования, совершенно непригодны для масштабирования до многотонной шкалы, так что существует большая потребность в новом, более эффективном и экономически более жизнеспособном синтезе.
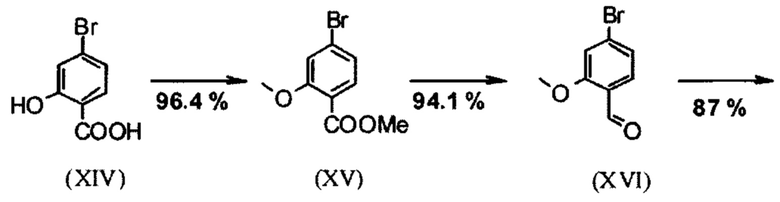
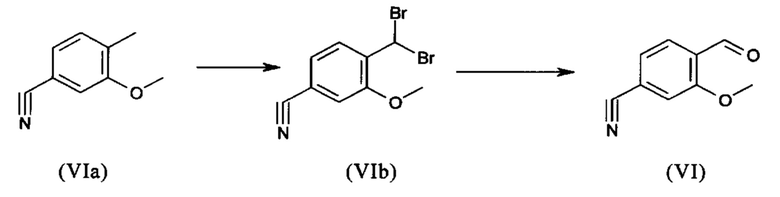
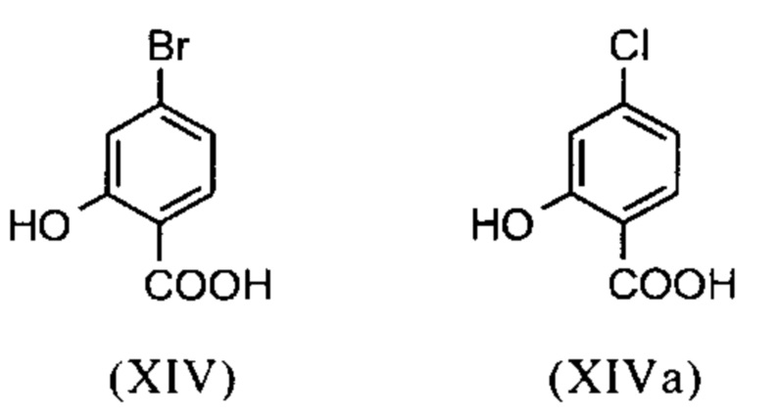
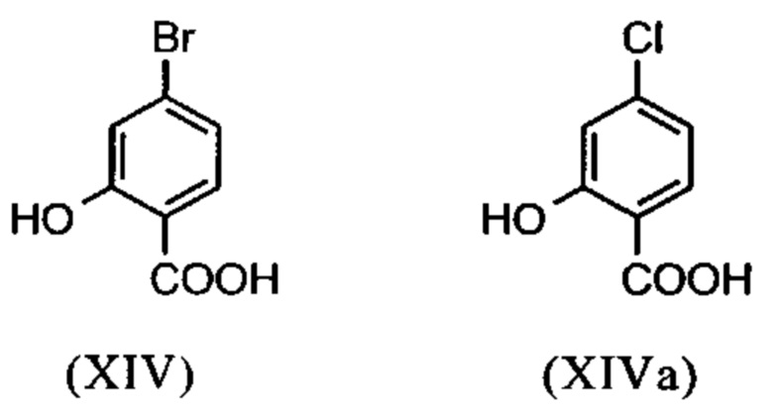
Галогензамещенные кислоты (XIV) и (XIVa)
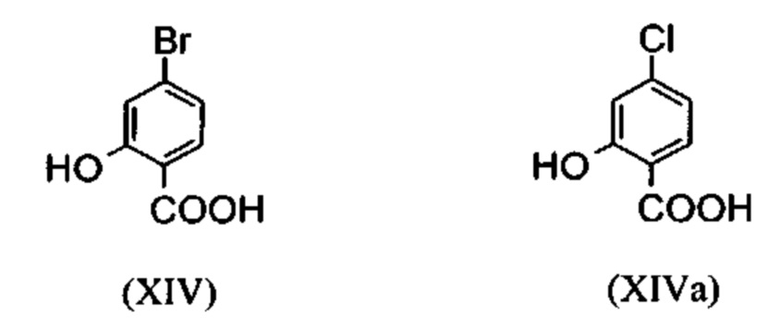
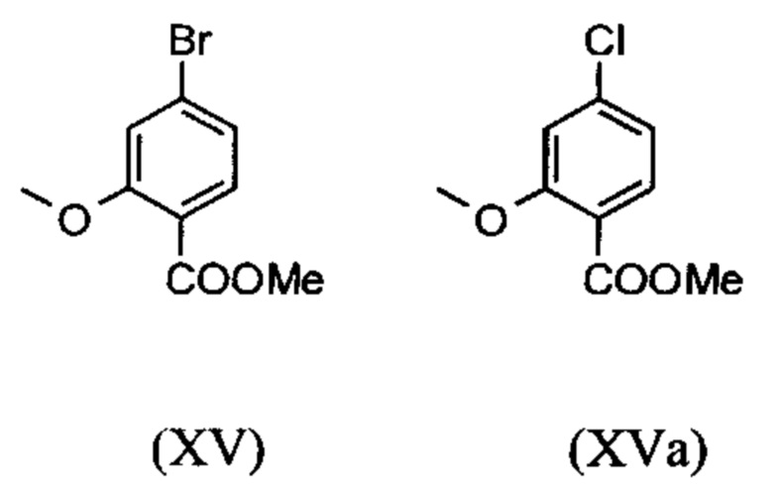
являются доступными в относительно больших количествах. Был создан очень эффективный и более дешевый способ, в котором промежуточные соединения (XV) и (XVI)
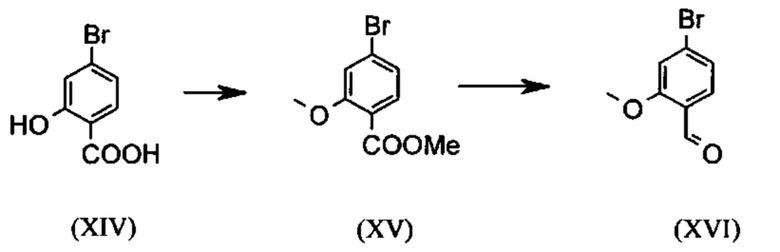
не выделяются, но подвергаются далее реакции, растворенными в растворе. Это возможно только потому, что выход и чистота в каждой реакции очень высоки (>95% от теоретического). Метиловый простой эфир - сложный эфир (XV) является известным из литературы (Journal of Medicinal Chemistry, 1992, vol. 35, p. 734-740) и его получают путем реакции с использованием сильно летучего, вредного для здоровья и дорогостоящего йодида металла.
С новым способом в соответствии с изобретением было возможно показать, что нелетучий, менее дорогой диметилсульфат можно использовать по аналогии. Исходя из кислоты (XIV), указанную кислоту вводят в реакцию в растворителе, таком как ацетон, 2-бутанон, ТГФ, 2-метил-ТГФ, ДМФА, DMA или NMP, с диметилсульфатом с помощью вспомогательного основания, такого как карбонат калия, карбонат натрия, карбонат кальция, карбонат лития, N-метилимидазол. триэтиламин, пиридин или 2,6-лутидин, при температурах 50-100°С с получением метилового простого эфира-сложного эфира (XV). Методы, известные специалисту в данной области техники, представляют собой эстерификация кислот и этерификация фенолов (Tetrahedron, 2013, vol. 69, p. 2807-2815, Journal of American Chemical Society, 2013, vol. 135, p. 5656-5668). Оказалось, что реакция в ацетоне при нагревании с обратным холодильником (56°С) с использованием диметилсульфата и карбоната калия является особенно предпочтительной. В данном случае, диметилсульфат добавляют к кипящей реакционной смеси в течение 4 часов. Ацетон отгоняют и заменяют его на толуол (повторная перегонка). Для обработки с целью выделения продукта, добавляют воду (разложение излишка диметилсульфата), толуольную фазу отделяют и промывают водой и раствором насыщенного хлорида натрия и толуольный раствор потом отгоняют до определенного объема (служит азеотропной сушкой, т.е. удаление воды для последующей стадии). Определение содержания раствора показывает практически полное превращение (>96% от теоретического). Вместо соединения брома, можно по аналогии использовать соединение хлора, для которого полученные превращения являются идентичными соединению брома.
Получение альдегида (XVI) описано в литературе, примеры которой включают: Glaxo Group Limited US 2008/312209 A1, 2008, European Journal of Medicinal Chemistry, 1986, vol. 21, p. 397-402, Journal of Medicinal Chemistry, 1992, vol. 35, p. 734-740, Journal of Materials Chemistry, 2011, vol. 21, p. 9523-9531. Однако, используемые в реакциях исходные вещества являются очень дорогостоящими и их невозможно получить в больших количествах, поэтому был создан новый способ, исходя из метилового простого эфира-сложный эфир (XV). Превращение соединения (XV) в альдегид (XVI) возможно при использовании REDAL (натрия бис(2-метоксиэтокси)алюминия дигидрида) в толуоле путем добавления N-метилпиперазина. Этот метод описан в литературе (Синтез 2003, №6, 823-828 и Tetrahedron 57 (2001) 2701-2710). Если реакцию проводят аналогично стехиометрии, описанной в литературе, дополнительное соединение обнаруживают в смеси при добавлении к альдегиду. Было показано, что оно соответствует бензиловому спирту, который образовывается с помощью сверхвосстановления вплоть до 10%. Было продемонстрировано, что очень важно установить стехиометрию REDAL и N-метилпиперазина до точных 1.21 экв. REDAL + 1.28 экв. N-метилпиперазина, делая возможным уменьшить этот побочный продукт, который нарушает кристаллизацию на следующей стадии, до <1%. С этой целью, загружают 65% раствор REDAL в толуоле при 0-5°С (предпочтительно 1.21 экв.) и добавляют 1.28 экв. N-метилпиперазина. Таким образом полученный раствор REDAL с N-метилпиперазином добавляют в течение прибл. 30 минут к раствору бромметилового сложного эфира (XIV), загруженного в толуол, и потом смесь перемешивают в течение одного часа при 0°С. Реакционный раствор гасят в смеси вода/кислота, предпочтительно водной серной кислоте, и толуольную фазу отделяют и промывают водой и раствором насыщенного хлорида натрия. Толуол отгоняют и повторно отгоняют в ДМФА (растворитель для следующей стадии). Выход реакционной смеси составляет обычно >94% от теоретического. Соответствующую реакцию с соединением хлора проводят по аналогии и выходы являются эквивалентными. Раствор ДМФА используют непосредственно в следующей реакции.
В ходе дальнейшего синтеза, бромальдегид (XVI) превращают в нитрил способом, известным per se с помощью методов, известных специалисту в данной области техники (Synth. Commun. 1994, 887-890, Angew. Chemie 2003, 1700-1703, Tetrahedron Lett. 2007, 2555-2557, Tetrahedron Lett. 2004, 1441-1444, JACS 2003, 125, 2890-2891, Journal of Organometallic Chemistry 689 (2004), 4576-4583), где в данном случае получают нитрильный альдегид (VI). Было доказано, что особенно предпочтительным в случае соединения брома проводить палладия-катализированную реакцию с гексацианоферратом калия * 3 Н2О в качестве источника цианида (Tetrahedron Lett. 48 (2007), 1087-1090). С этой целью, бромальдегид (XVI) загружают в ДМФА (8-10-кратно), загружают 0.22 экв. гексацианоферрата калия * 3 Н2О и 1 экв. карбоната натрия и затем добавляют 0.005 экв. ацетата палладия. Смесь нагревают до 120°С в течение 3 часов. Раствор охлаждают до 20°С, затем добавляют воду и этилацетат. Этилацетатную фазу отделяют, водную фазу снова промывают этилацетатом и объединенные этилацетатные фазы затем повторно отгоняют в изопропаноле. Продукт осаждается путем осаждения воды при кипящей температуре. После выделения, продукт сушат в вакууме. В некоторых случаях, продукт осаждали непосредственно путем добавления воды к ДМФА и использовали непосредственно на следующей стадии после выделения и сушки. Выходы реакционной смеси обычно составляют >85% от теоретического. Ацетат палладия не является достаточным для превращения соединения хлора и было доказано, что является предпочтительным в данном случае применение катализаторов палладия, известных специалисту в данной области техники, так как это описано в Tetrahedron Lett. 48 (2007), 1087-1090, где выходы являются несколько более низкими, чем в случае с соединением брома, обычно 80-85% от теоретического.
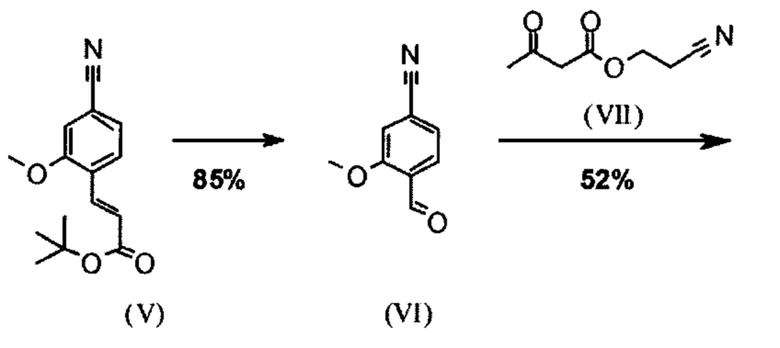
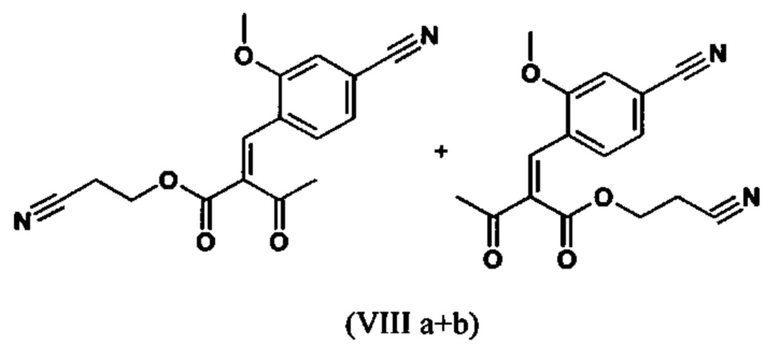
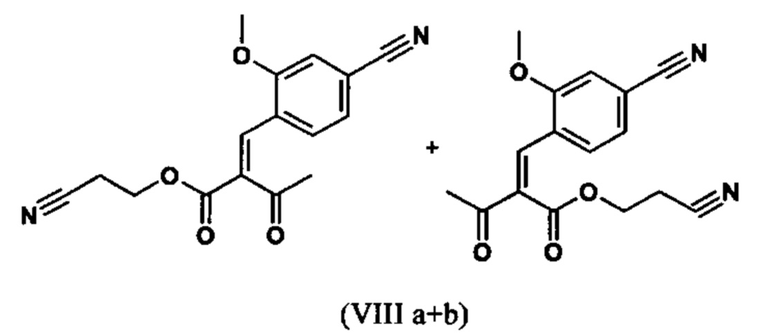
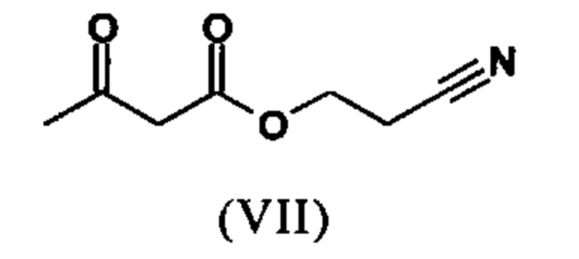
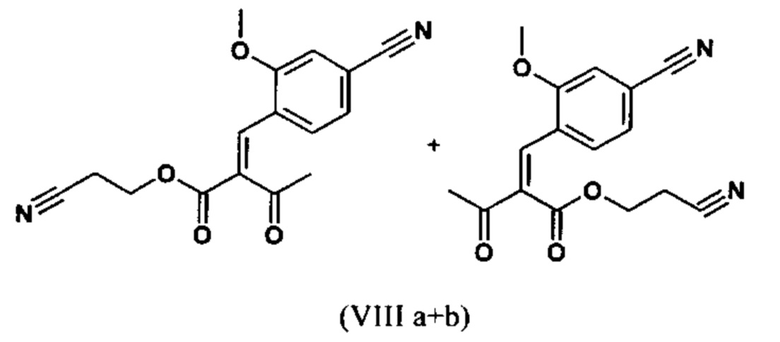
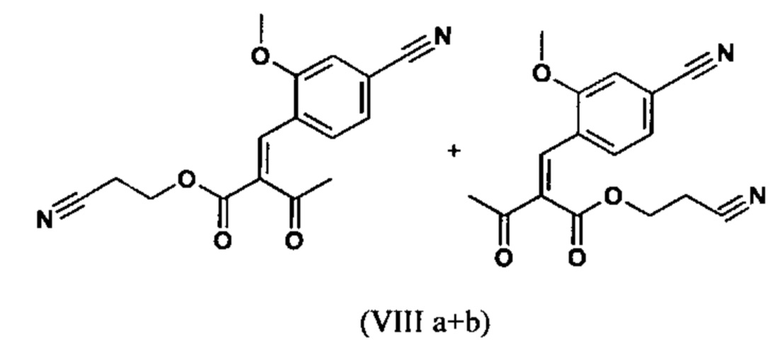
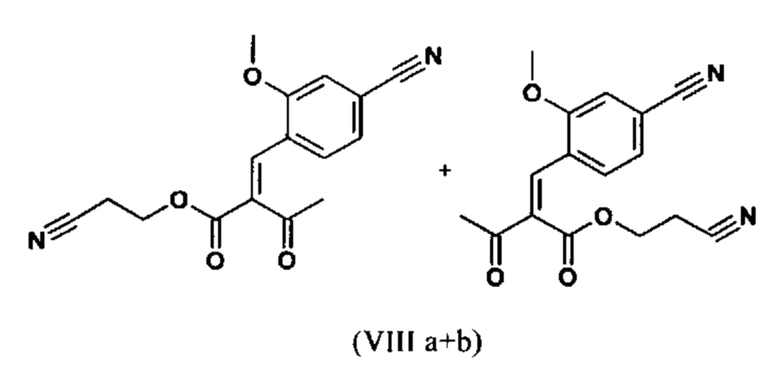
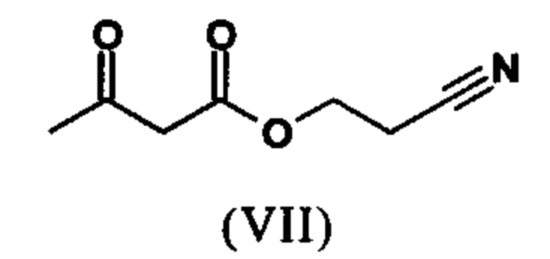
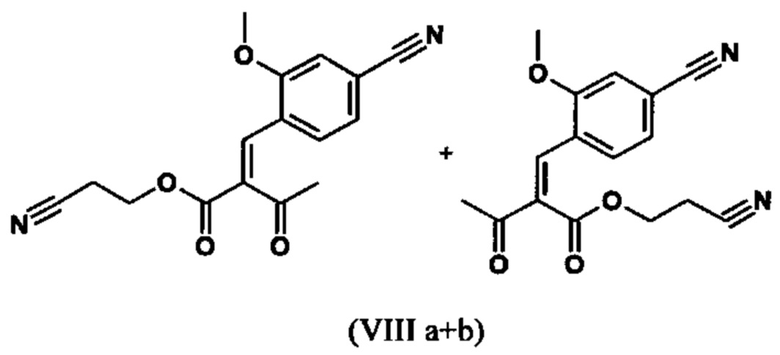
Сложный эфир коричной кислоты (VIII а,b) получают в виде смеси E/Z, исходя из альдегида формулы (VI) путем реакции Кневенагеля со сложным цианоэфиром (VIII):

В ходе исследования, дихлорметан и 0.2 экв. пиперидина/0.2 экв. ледяной уксусной кислоты 16.6-кратно нагревают в течение 20 часов на водоотделителе. После водной обработки с целью выделения продукта, продукт кристаллизуют из метанола после упаривания растворителя, при этом получая целевое соединение в количестве 52% от теоретического.
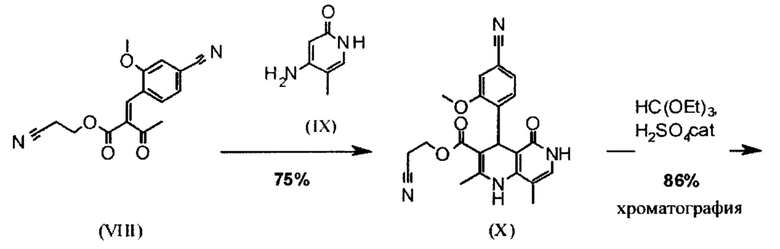
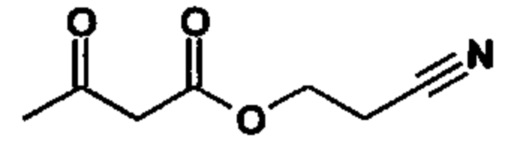
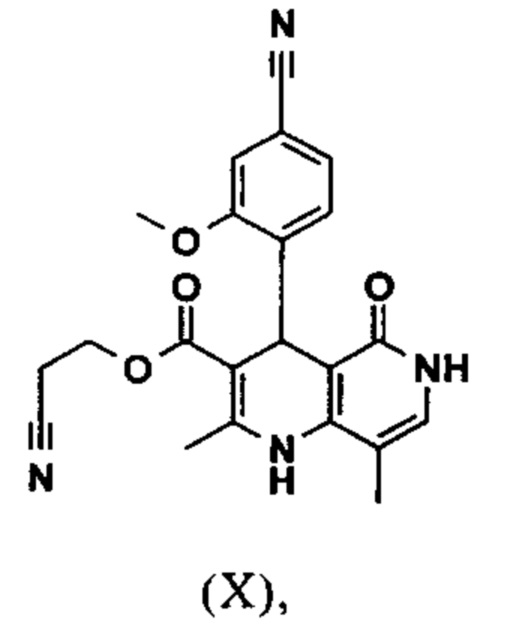
Реакцию продолжают предпочтительно в кипящем дихлорметане (10-кратно) путем добавления 5-20 мол. % пиперидина, предпочтительно 10 мол. % и 5-20 мол. % ледяной уксусной кислоты, предпочтительно 5-10 мол. %, на водоотделителе. Время реакции составляет 4-12 ч, но предпочтительно 5-6 ч, особенно предпочтительно 6 ч. Добавляют 1.0-1.5 экв., однако предпочтительно 1.1-1.35 экв. или 1.25 экв. - 1.35 экв. сложного цианоэфира (VII). Особенно предпочтительно добавляют 1.1 экв.. Получение сложного цианоэфира (VII) описано в Pharmazie, 2000, vol. 55, p. 747-750 и Bioorg. Med. Chem. Lett. 16, 798-802 (2006). После завершения, реакционную смесь охлаждают до 20°С и органическую фазу промывают дважды водой. Органическую промывку повторно отгоняют в 2-бутаноле и смесь E/Z сложных эфиров коричной кислоты (VIII а+b) используют непосредственно без выделения промежуточного соединения в следующей реакции с гетероциклом (IX) с получением дигидропиридина (X):
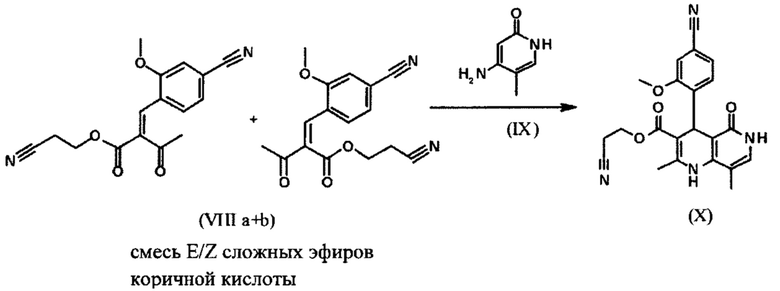
Для дальнейшей реакции в синтезе на уровне исследования, смесь нагревали с обратным холодильником с помощью гетероцикла (IX) в изопропаноле в течение 40 часов.
Было обнаружено, что реакцию можно проводить, предпочтительно во вторичном спирте, таком как изопропанол, изобутанол, 2-амиловый спирт или циклогексанол при температурах 80-160°С, при атмосферном давлении, а также в автоклаве (2-10 бар), при времени реакции 8-40 ч, но предпочтительно в течение 20-25 ч в кипящем 2-бутаноле при атмосферном давлении или еще в изопропаноле в автоклаве (100°С, 2-10 бар, предпочтительно 3-5 бар, 8-24 ч). Для обработки с целью выделения продукта, смесь охлаждают до 0°С - 20°С, кристаллы отфильтровывают и промывают изопропанолом и затем сушат (в вакууме, 60°С).
Если применение дихлорметана следует избежать из экологических соображений, было доказано, что достаточно предпочтительно получать сложный эфир коричной кислоты (VIII а,b) в изопропаноле, в случае чего альдегид (VI) загружают в изопропанол (3-9-кратно, предпочтительно 5-7-кратно) и добавляют 5-20 мол. % пиперидина, предпочтительно 5-10 мол. %, 10 мол. % и 5-20 мол. % ледяной уксусной кислоты, предпочтительно 5-10 мол. % или 10 мол. %. При 30°С, 1.0-1.5 экв., предпочтительно 1.1-1.35 экв. или 1.35 экв.., особенно предпочтительно 1.1 экв. сложного цианоэфира (VII) добавляют в течение 3 часов, необязательно растворяют в литре изопропанола, и смесь перемешивают при 30°С в течение 1 час. Сложный эфир коричной кислоты (VIII a, b) выкристаллизовывается во время реакции. Потом продукт отфильтровывают, необязательно после охлаждения, предпочтительно при 0°С, промывают литром изопропанола (охлаждают до 0°С) и используют влажным в последующей реакции, как описано выше. Выход составляет >96% от теоретического. Последующую реакцию предпочтительно осуществляют 10-15-кратно (в отношении альдегида (VI)), предпочтительно 11-12-кратно - изопропанола в течение 20-24 часов при 100°С под давлением. После завершения реакции и охлаждения, продукт выделяют путем фильтрации или центрифугирования. Продукт потом сушат при 40-90°С в вакууме. Поскольку превращение в сложный эфир коричной кислоты продолжается практически количественно, способ для последующей стадии может быть легко стандартизирован без необходимости установления количество гетероцикла (IX) в каждом случае, так как продукт можно использовать влажным от изопропанола. Выходы составляют >87% от теоретического. Гетероцикл (IX) можно получить известными в литературе методами, как описано, например, в Synthesis 1984, 765-766.
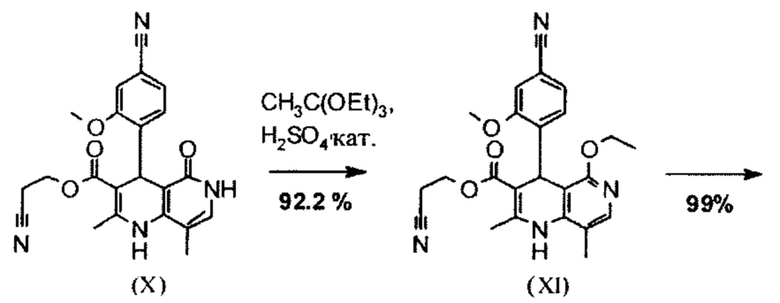
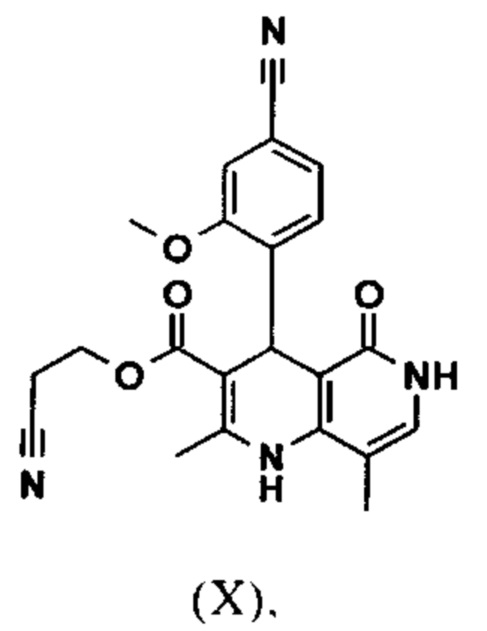
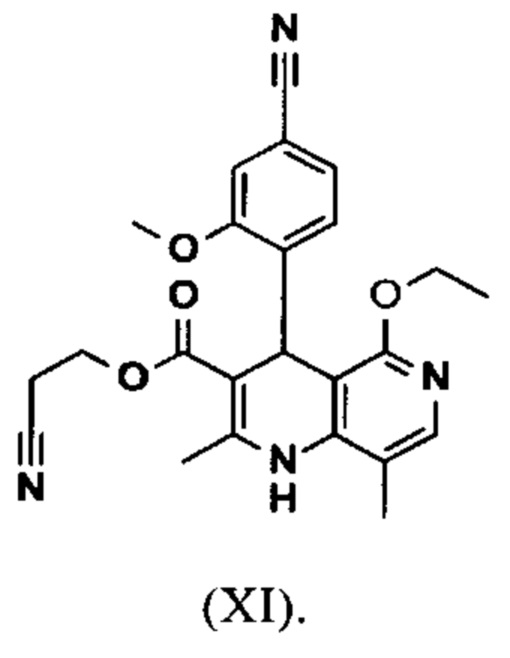
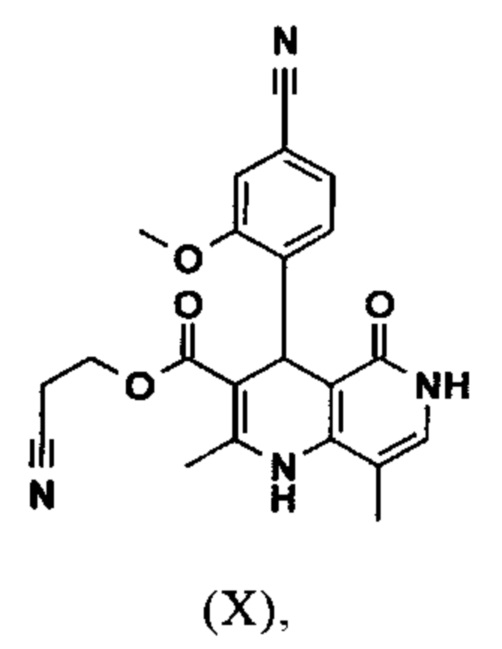
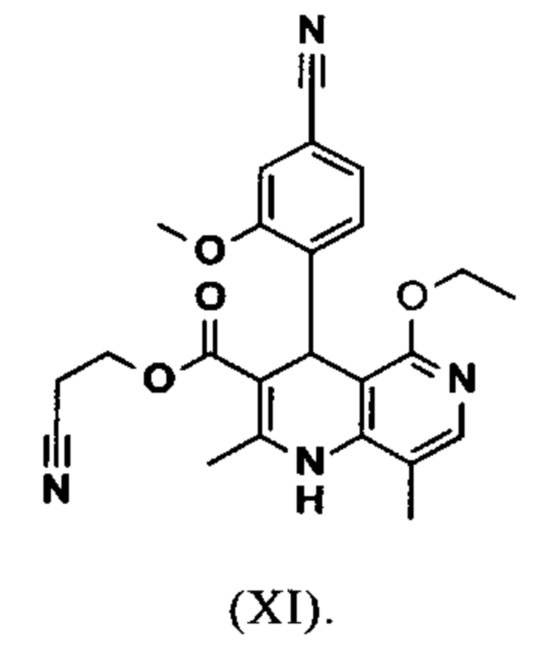
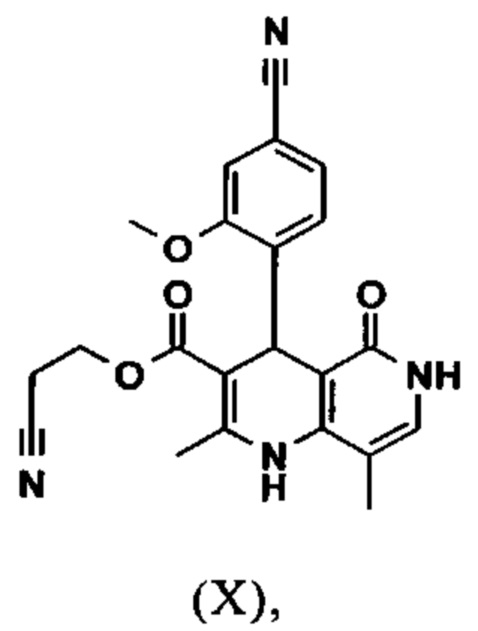
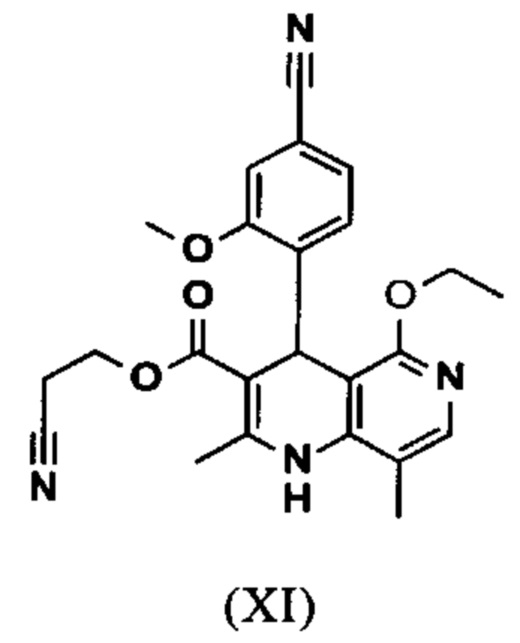
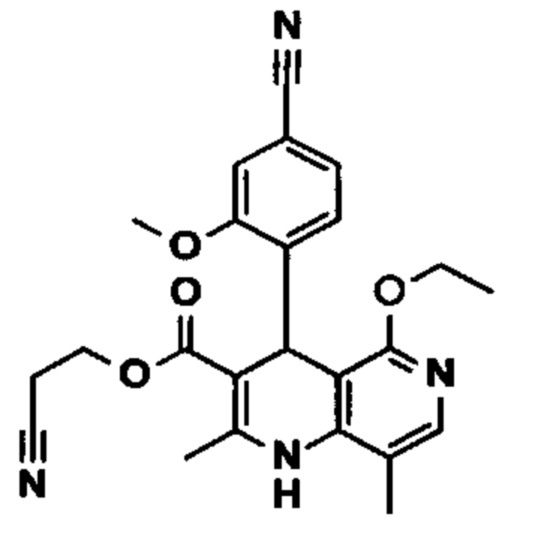
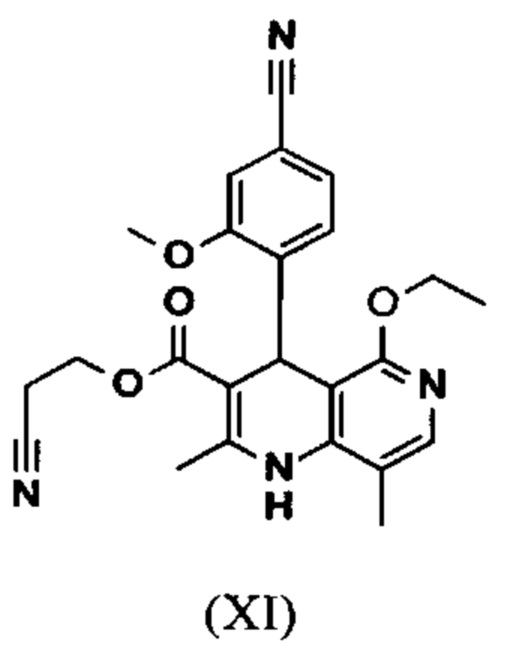
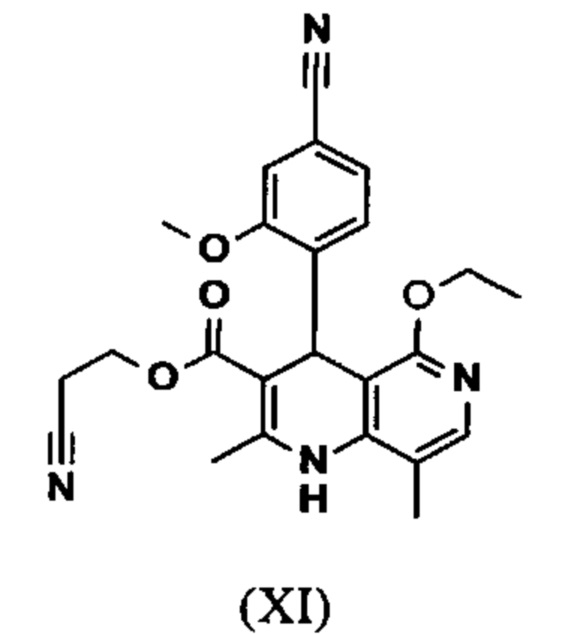
Исходя из дигидропиридина (X), получают этиловый эфир (XI) путем реакции под кислотным катализатором с ортоэфиром, где R представляет собой -Н или -метил:
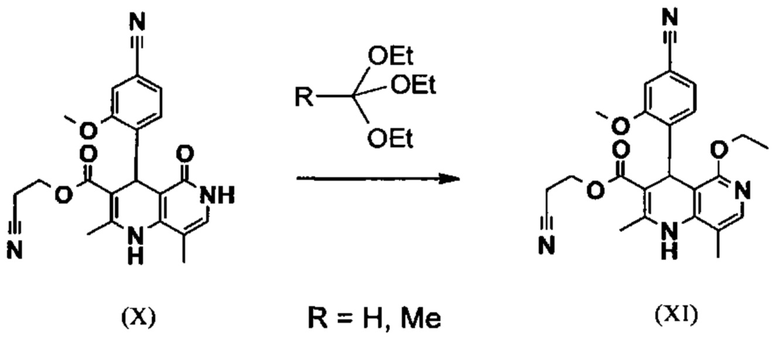
В синтезе на уровне исследования, реакцию проводили в 25-кратном ДМФА с 20.2 экв. триэтилортоформиата и каталитическими количествами конц. серной кислоты при 135°С. Смесь концентрировали досуха и остаток очищали с помощью хроматографии с выходом 86% от теоретического. Этот метод не является пригодным как техническая методика, из-за высокого разведения и применения триэтилортоформиата, его высокой воспламеняемости при низкой температуре, который используют в очень большом избытке, и последующей хроматографии.
Неожиданно было обнаружено, что реакцию можно проводить при высокой концентрации (вплоть до 1.5 г растворителя на 1 г реагента) в растворителях, таких как диметилацетамид, NMP (1-метил-2-пирролидон) или ДМФА (диметилформамид) путем добавления 4-10% по массе, предпочтительно 6-8% по массе, конц. серной кислоты. Реакцию продолжают, неожиданно, даже с 2.5-5 экв. или 5 экв. сложного ортоэфира. Было обнаружено, что намного более удобно применять соответствующий триэтилортоацетат в реакции, поскольку он, с одной стороны, реагирует намного чище, и является намного менее воспламеняемым, делая особенно подходящими условия технической процедуры. Реакцию предпочтительно проводят в DMA (диметилацетамид) и/или NMP (1-метил-2-пирролидон), при температурах 100-120°С, предпочтительно 115°С. Перед началом самой реакции, было доказано, что предпочтительно отогнать некоторое количество растворителя (DMA и/или NMP) при повышенной температуре (100-120°С в вакууме) для того, чтобы удалить любые остатки изопропанола, присутствующего от предшественника, так как в обратном случае присутствуют нежелательные побочные продукты. Реакция: Перемешивание в течение 1.5-3 часов, предпочтительно 2 часов. Для обработки с целью выделения продукта, воду добавляют непосредственно к смеси, где продукт выкристаллизовывается. С целью получения особенно стабильного и репродуктивного процесса, вначале добавляют порцию воды (например, 1/3), затем в нее вносят затравку, и добавляют оставшееся количество воды. Эта процедура гарантирует, что всегда получают тот же самый кристаллический полиморф, что показывает оптимальные характеристики выделения. Продукт промывают водой и сушат. Выходы составляют >92% от теоретического.
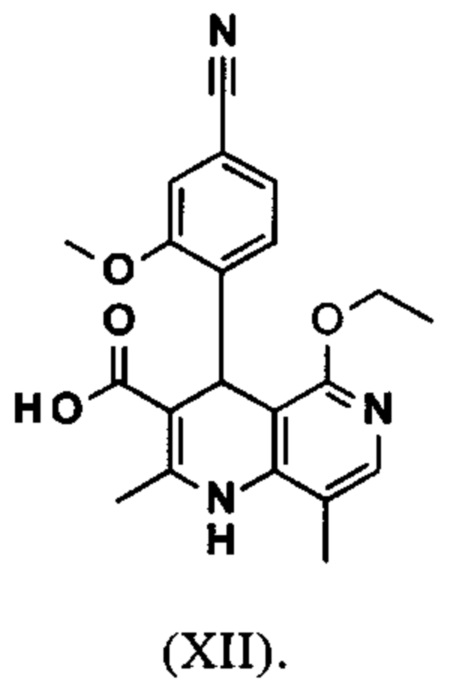
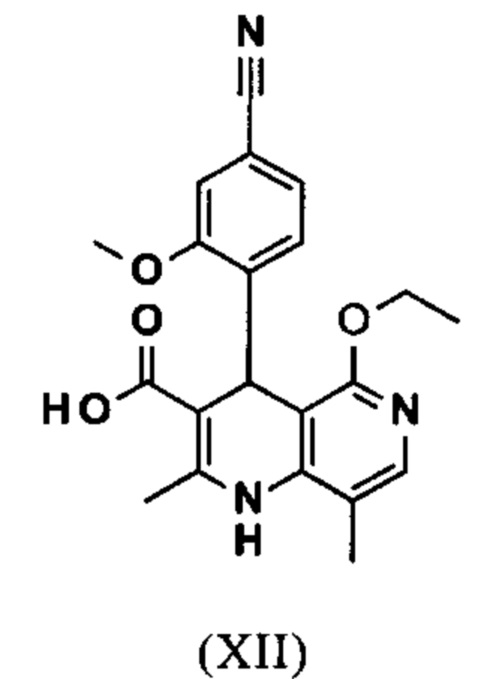
Исходя из этилового эфира (XI), получают кислоту (XII) путем щелочного омыления и последующей кислотной обработки с целью выделения продукта.
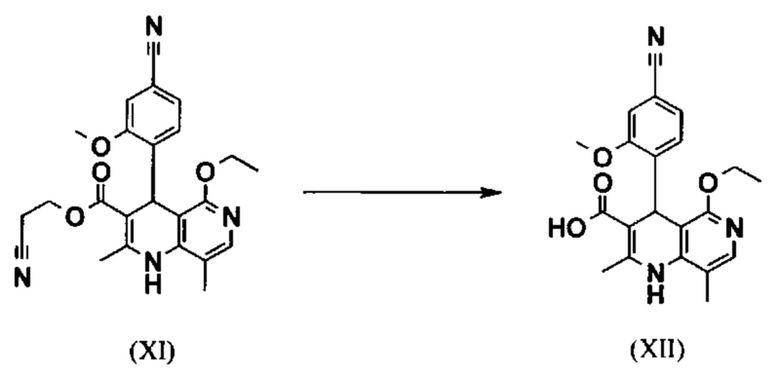
В синтезе на уровне исследования, омыление проводили при высоком разведении (33.9-кратно) в смеси DME/вода 3:1. В данном случае, было необходимо прежде всего повысить пропускную способность и заменить используемый DME (диметоксиэтан), который имеет очень низкую температуру воспламенения и, следовательно, считается особо критичным для широкомасштабного использования. Неожиданно было обнаружено, что реакцию также можно также проводить очень легко очень концентрированно в смеси ТГФ/вода. Для этого, реакцию предпочтительно осуществляют в смеси ТГФ/вода 2:1 (9-кратно), добавляют водный раствор гидроксида натрия при 0-5°С, затем смесь перемешивают при 0-5°С в течение 1-2 часов. Водный раствор гидроксида калия также можно использовать, но предпочтительно используют NaO. Для обработки с целью выделения продукта, смесь экстрагируют МТВЕ (метил трет-бутиловым эфиром) и этилацетатом и для выделения рН устанавливают с помощью минеральной кислоты, такой как соляная кислота, серная кислота или фосфокислота, но предпочтительно соляной кислоты, на значение рН 6.5-7.0 или рН 7. Смесь затем смешивают с насыщенным раствором аммониевой соли соответствующей кислоты, но предпочтительно с раствором хлорида аммония, где продукт количественно выкристаллизовывается. После выделения, продукт промывают водой и этилацетатом или ацетонитрилом или ацетоном, но предпочтительно ацетонитрилом, и сушат в вакууме при 40-50°С. Выход по сути подсчитывают количественно (99%). Альтернативная предпочтительная обработка с целью выделения продукта: в качестве альтернативной обработки с целью выделения продукта, толуол добавляют к смеси, добавляют ацетат натрия и смесь перемешивают при 20°С, фазы затем разделяют и водную фазу устанавливают при 0°С с использованием 10% водной соляной кислоты на значение рН 6.5-7.0 (можно необязательно вносить затравку при рН 9.5-10). Смесь недолго перемешивают и продукт отфильтровывают, промывают небольшим количеством воды и толуола и сушат при 40-50°С в вакууме. Полученные выходы также подсчитывают количественно в данном случае.
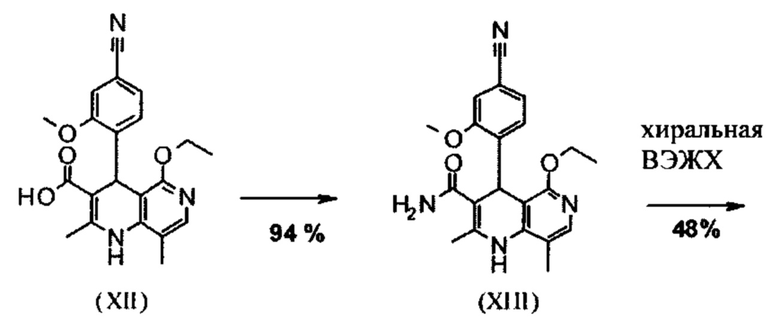
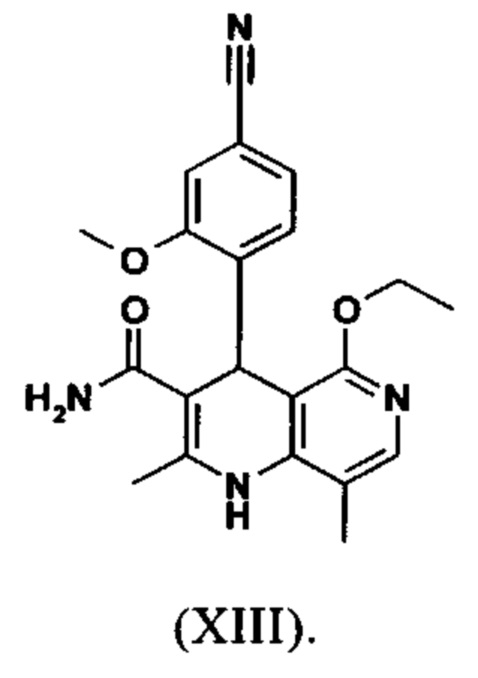
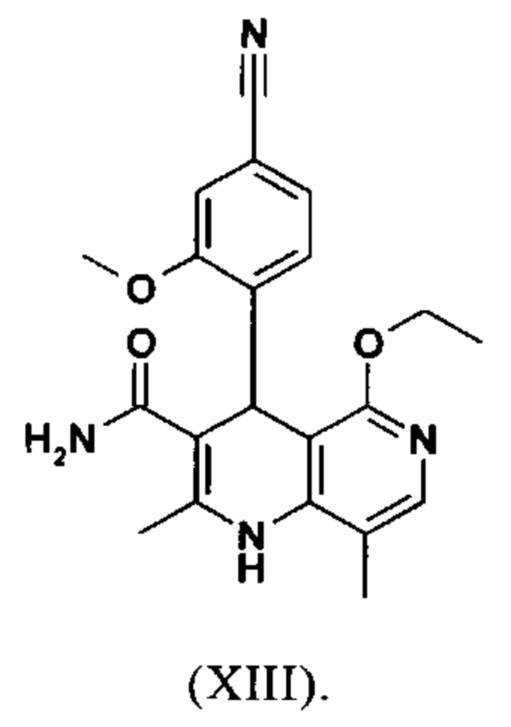
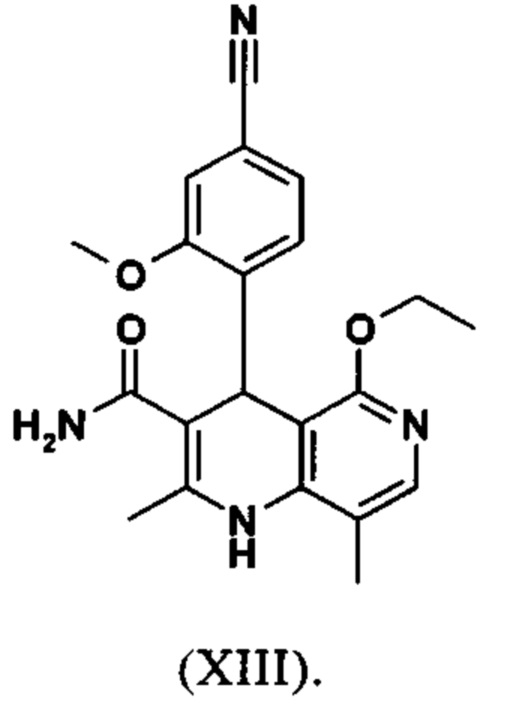
Последующее превращение кислоты в амид (XIII) проводили в на стадии исследования, как указано ниже: кислоту (XII) растворяли прибл. 10-кратно в ДМФА, добавляли 1.25 экв. 1,1'-карбодиимидазола и 0.1 экв. DMAP (4-(диметиламино)пиридин) и смесь перемешивали при комнатной температуре в течение 4 часов. Потом добавляли 20 экв. аммиака в виде водного 25% раствора и эту смесь переносили на масляную баню, предварительно нагретую до 110°С. В данной методике, мгновенно образуются относительно большие количества газообразного аммиака, который выходит из системы, и в добавление обеспечивает резкое повышение давления. Эту смесь добавляли прибл. 90-кратно к воде и устанавливали на значение рН 7 путем добавления ацетата натрия. Осажденный продукт отфильтровывали и сушили (выход: 59% от теоретического). Дополнительную порцию выделяли из основного раствора путем исчерпывающего экстрагирования (прибл. 100-кратно этилацетатом), которую перемешивали с легковоспламеняющимся диэтиловым эфиром и содержала прибл. 14% ДМФА. Совершенно ясно, что такой метод не может быть достигнут таким способом в рамках методики, а поэтому существует высокая потребность в альтернативной процедуре. Усилия, необходимые для выделения этой порции, непропорциональны количеству выделенного в этом случае.
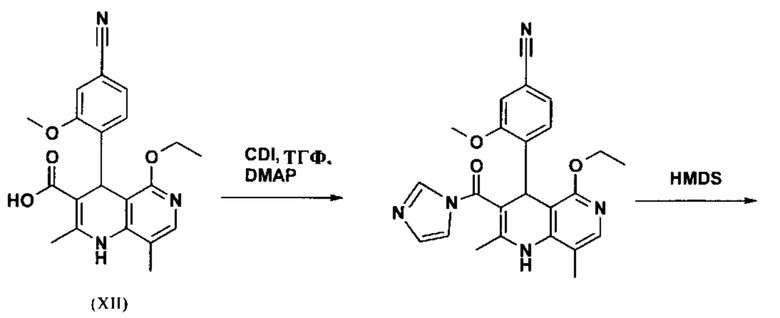
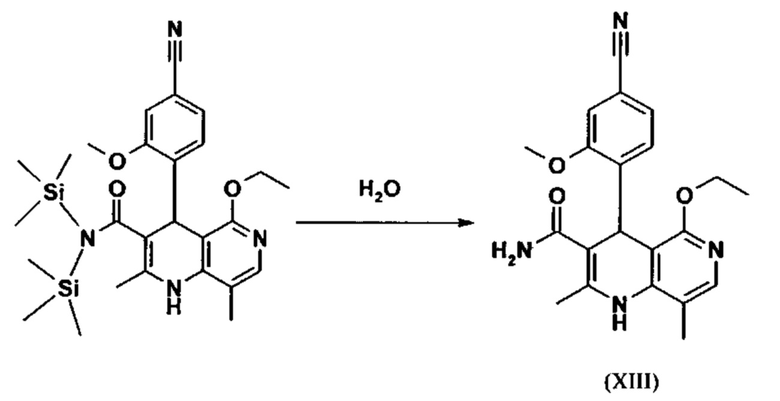
Неожиданно было обнаружено, что в реакции кислоты (XII) в ТГФ, амид (XIII) выкристаллизовывается непосредственно из раствора и может быть получен при высоких выходе и чистоте. С этой целью, карбоновую кислоту (XII) в ТГФ вводят в реакцию с 1.1-1.6 экв., предпочтительно 1.3-1.4 экв. 1,1'-карбодиимидазола в условиях катализа DMAP (5-15 мол. %, предпочтительно 10 мол. %) с получением имидазолида, который происходит при температурах между 20-50°С, предпочтительным подходом оказался вначале тот, где начинали при 20°С, затем перемешивали 1-2 часа при этой температуре и затем еще перемешивали при 50°С в течение 2-3 часов. После завершения активизации, добавляют 3-8 экв., предпочтительно 4.5 экв. гексаметилдисилазана и смесь кипятят с обратным холодильником в течение 16-24 часов, но предпочтительно 16 часов. Полученное в результате дисилиламидное соединение в данном случае можно необязательно выделить, но оказалось, что предпочтительно продолжить реакцию, путем реакции, которую проводят в одном реакционном сосуде без выделения промежуточных соединений. Поэтому, после завершения реакции, смесь охлаждают до 0-3°С и добавляют смесь воды/или в смеси с ТГФ, при этом оказалось, что предпочтительно применять количество воды 0.5-0.7-кратно (относительно реагента), особенно предпочтительно количество воды 0.52-кратно. Воду можно добавлять непосредственно или в виде смеси с приблизительно эквивалентом вплоть до двойного количества ТГФ по объему. После окончания гашения, смесь нагревают с обратным холодильником в течение 1-3 часов в целом, предпочтительно 1 час. Смесь охлаждают до 0°С и перемешивают в течение 1-5 часов, предпочтительно 3 часов, при этой температуре, затем продукт выделяют путем фильтрации или центрифугирования. Продукт промывают ТГФ и водой и сушат в вакууме при повышенной температуре (30-100°С, предпочтительно при 60°С - 90°С или при 40°С - 70°С). Выходы являются очень высокими и обычно составляют >93% от теоретического. Чистота обычно составляет >99% (ВЭЖХ, 100% метод). Соединение (XIII) можно также получить непосредственно с помощью реакции с аммиачным газом в автоклаве (прибл. 25-30 бар). С этой целью, осуществляется предварительная активация, описанная выше, и реакционную смесь нагревают под давлением в условиях газообразного аммиака. После завершения реакции, реакционную смесь охлаждают и продукт отфильтровывают. Выходы и чистоты, достигнутые таким образом, можно сравнивать.
Для получения соединения формулы (I), рацемическую смесь амидов (XIII) необходимо разделить на антиподы. В опубликованном синтезе на уровне исследования с этой целью использовали специфически синтезированную хиральную фазу (продукт собственного производства), которая включала N-(дициклопропилметил)-N2-метакрилоил-D-лейцинамид в качестве хирального селектора. Этот селектор получали многостадийным способом и затем полимеризировали на специальном силикагеле. Смесь метанол/этилацетат служила как элюент. Основным недостатком данного метода была очень низкая загрузка, 30 мг в перерасчете на разделение на хроматографической колонке 500*63 мм, так что была большая необходимость найти как можно наиболее эффективный метод разделения, который позволит осуществить разделение антиподов в многотонном диапазоне. Неожиданно было обнаружено, что разделение можно осуществлять на легкодоступной фазе. Это фаза Chiralpak AS-V, 20 мкм. Используемым элюентом была смесь метанол/ацетонитрил 60:40. Эта смесь имеет то основное преимущество, что она может быть восстановлена как элюент после дистилляционной обработки с целью выделения продукта, имея идентичный состав (60:40 соответствует азеотропу. Таким образом достигают очень эффективного способа, при котором выход разделения составляет >47% от теоретического (50% теоретически возможно). Оптическая чистота в данном случае составляет >93% е.е., но предпочтительно >98.5% е.е. В данном случае, хроматографию можно осуществлять на обычной хроматографической колонке, но предпочтительно используют методики, известные специалисту в данной области техники, такие как SMB или Varicol (Computers and Chemical Engineering 27 (2003) 1883-1901). Например, прибл. 500 кг рацемического амида (XIII) разделяли с использованием системы SMB, при этом достигая выхода 48%. Продукт получают в виде 3-8%, предпочтительно 5-7% раствора в смеси метанол/ацетонитрил 60:40 и его непосредственно можно использовать в "конечной обработке". Другие соотношения смеси растворителей ацетонитрил к метанолу также возможны (90:10 - 10:90). Альтернативно, другие смеси растворителей можно также использовать, однако, для разделения SMB, такие как ацетонитрил/этанол соотношения в смеси 10:90 - 90:10. Особое соотношение растворителей частично зависит от технических свойств системы SMB и должны быть установлены, при необходимости (например, изменяющаяся скорость потока, повторное использование растворителя на тонкопленочном испарителе).
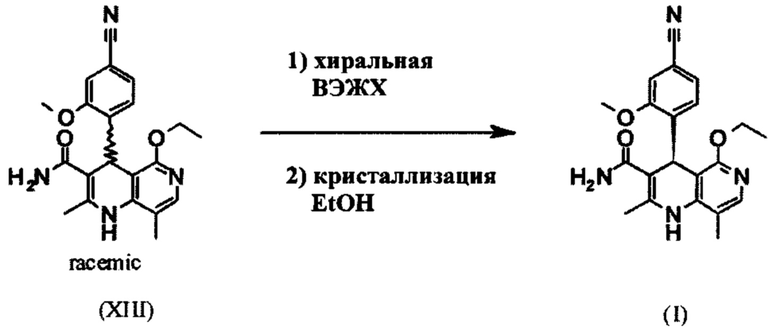
Так как соединение формулы (I) было разработано в форме таблетки, существует большая потребность в том, чтобы выделенное соединение формулы (I) выделялось в определенной кристаллической форме воспроизводимым образом, так чтобы обеспечивалась воспроизводимая биодоступность. Неожиданно было обнаружено, что соединение формулы (I) можно кристаллизовать из метанола, этанола, ТГФ, ацетонитрила, а также их смесей с водой, где воспроизводимо формируется только один полиморф I, имеющий определенную температуру плавления 252°С. Преимущественно используют этанол или денатурированный этанол.
Процесс конечной кристаллизации: с этой целью прибл. 5-7% раствор продукта в смеси метанол/ацетонитрил 60:40 (или, если использовали смесь этанол/ацетонитрил, прибл. 3-4% раствор смеси этанол/ацетонитрил 50:50), полученный в результате хроматографии, вначале подвергают фильтрации частиц по техническим причинам GMP и потом заменяют растворитель этанолом, предпочтительно с использованием этанола, денатурированного толуолом. С этой целью раствор неоднократно повторно отгоняют, концентрируют и каждый раз добавляют свежий этанол. После замены, добавляют как можно больше этанола до тех пор, пока не пройдет фаза раствора при температуре кипения и затем ее концентрируют при атмосферном давлении или при слегка пониженном давлении до прибл. 3-4-кратного по объему, после чего продукт выкристаллизовывается. Полученное охлаждают до 0°С и затем выделяют кристаллы и сушат при 40-50°С в вакууме. Выходы обычно составляют >90% от теоретического. Достигнутая химическая чистота составляет >99.8% и содержание ~ 100% соответствуют критерию для коммерческих продуктов в соответствии с Руководящими принципами Международной конференции по гармонизации и нормами надлежащей клинической практики. Остаточный растворитель, в случае этанола, составляет <0.02%. Оптическая чистота составляет >>99% е.е.
Настоящее изобретение обеспечивает соединение формулы (I) в кристаллической форме полиморфа I

которое отличается тем, что рентгеновская дифрактограмма соединения показывает максимальные значения пиков угла 2 тета на 8.5, 14.1, 17.2, 19.0, 20.5, 25.6. 26.5.
Настоящее изобретение также обеспечивает соединение формулы (I) в кристаллической форме полиморфа I

которое отличается тем, что ИК-спектр (IR-ATR) соединения показывает максимальные значения полос на 3475, 2230, 1681, 1658, 1606, 1572, 1485, 1255, 1136 и 1031 см-1.
Настоящее изобретение также обеспечивает соединение формулы (I) в кристаллической форме полиморфа I
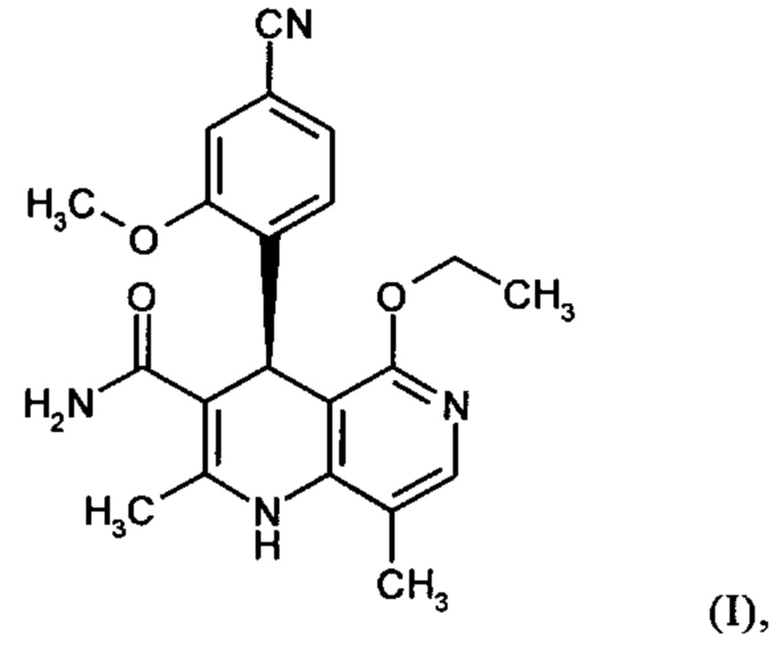
которое отличается тем, что спектр Рамана соединения показывает максимальные значения полос на 3074, 2920, 2231, 1601, 1577, 1443, 1327, 1267, 827 и 155 см-1.
Настоящее изобретение также обеспечивает способ получения соединения формулы (I) в кристаллической форме полиморфа I, который отличается тем, что соединение формулы (I), которое присутствует в виде одного или нескольких полиморфов или в виде сольвата в инертном растворителе, перемешивают при температуре 20°С - 120°С, и соединение формулы (I) выделяют в виде кристаллического полиморфа I.
Предпочтительными растворителями для способа получения соединения формулы (I) в кристаллической форме полиморфа I являются метанол, этанол, ТГФ, ацетонитрил, а также их смеси. Особенно предпочтительным является этанол или денатурированный этанол.
Предпочтительным диапазоном температур для способа получения соединения формулы (I) в кристаллической форме полиморфа I является 20°С - 90°С.
Настоящее изобретение также обеспечивает соединение формулы (I) в кристаллической форме полиморфа (I), как описано выше, для лечения нарушений.
Настоящее изобретение также обеспечивает лекарственное средство, которое содержит, соединение формулы (I) в кристаллической форме полиморфа (I), как описано выше, и не большую пропорциональную часть какой-либо другой формы соединения формулы (I) в кристаллической форме полиморфа (I), как описано выше. Настоящее изобретение также обеспечивает лекарственное средство, которое содержит соединение формулы (I) в кристаллической форме полиморфа (I), как описано выше, в количестве более 90 масс. % в перерасчете на общее количество соединения формулы (I), присутствующего в кристаллической форме полиморфа (I), как описано выше.
Настоящее изобретение также обеспечивает применение соединения формулы (I) в кристаллической форме полиморфа I, как описано выше, для изготовления лекарственного средства для лечения сердечно-сосудистых нарушений.
Настоящее изобретение также обеспечивает способ лечения сердечнососудистых нарушений, который включает введение эффективного количества соединения формулы (I) в кристаллической форме полиморфа (I), как описано выше.
Настоящее изобретение также обеспечивает способ получения соединения (I), который отличается тем, что соединение формулы (XIV) или формулы (XIVa)
вводят в реакцию путем добавления диметилсульфата с получением соединения формулы (XV) или (XVa)
и невыделенные метиловые сложные эфиры формулы (XV) или (XVa) восстанавливают с помощью 1.21 экв. REDAL (натрия бис(2-метоксиэтокси)алюминия дигидрида) и 1.28 экв. N-метилпиперазина с получением альдегида формулы (XVI) или (XVIa)
и альдегид (XVI) или (XVIa) вводят далее в реакцию без выделения с получением нитрила формулы (VI)
Настоящее изобретение также обеспечивает способ получения соединения формулы (I), который отличается тем, что соединение формулы (VI)
растворенное в изопропаноле (3-7-кратно), 5-10 мол. % пиперидина и 5-10 мол. % ледяной уксусной кислоты при 30°С вводят в реакцию с соединением формулы (VII)
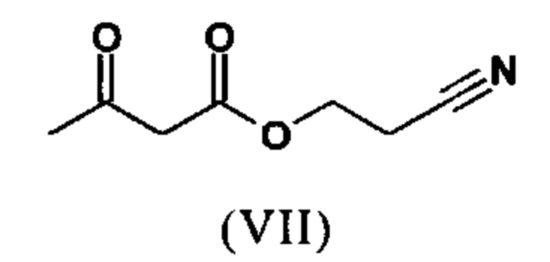
с получением соединения (VIIIa+b).
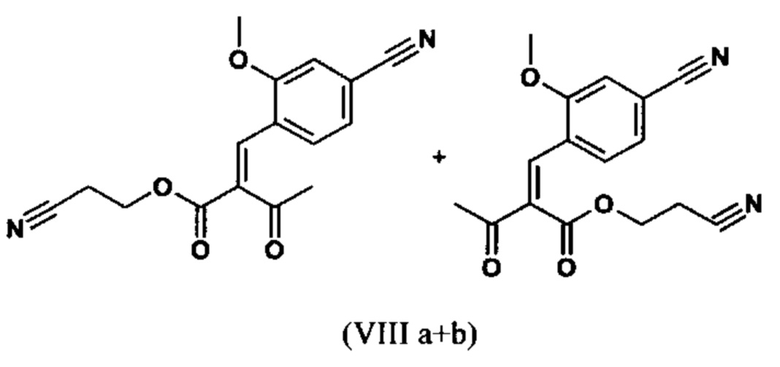
Настоящее изобретение также обеспечивает способ получения соединения формулы (I), который отличается тем, что соединение формулы (X)
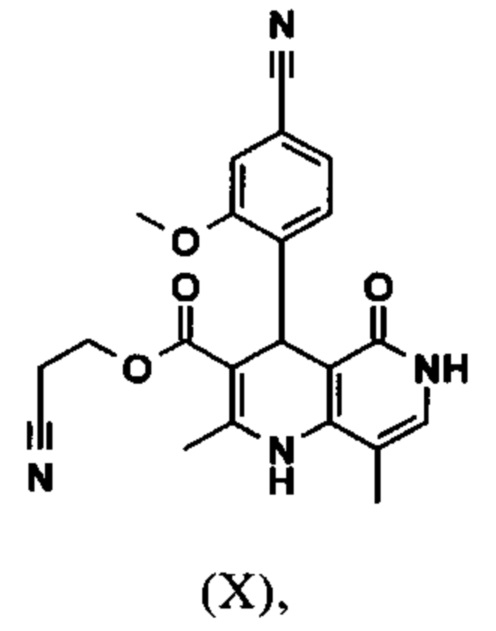
вводят в реакцию при перемешивании с 2.5-5 экв. триэтилортоацетата в диметилацетамиде при 100-120°С в течение 1.5-3 часов с получением соединения формулы (XI)
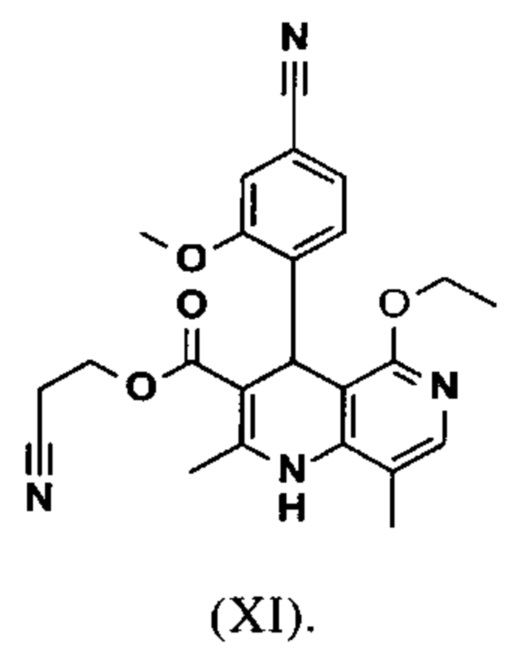
Настоящее изобретение также обеспечивает способ получения соединения формулы (I), который отличается тем, что соединение формулы (XI)
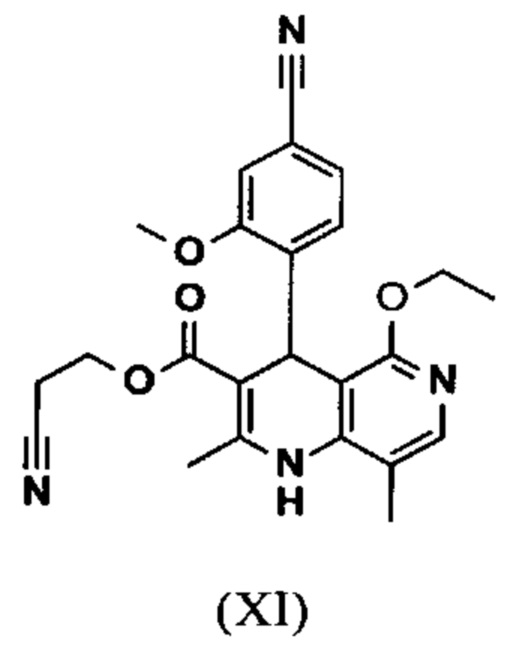
омыляют в смеси ТГФ/вода (2:1, 9-кратно) водным раствором гидроксида натрия с получением соединения формулы (XII)
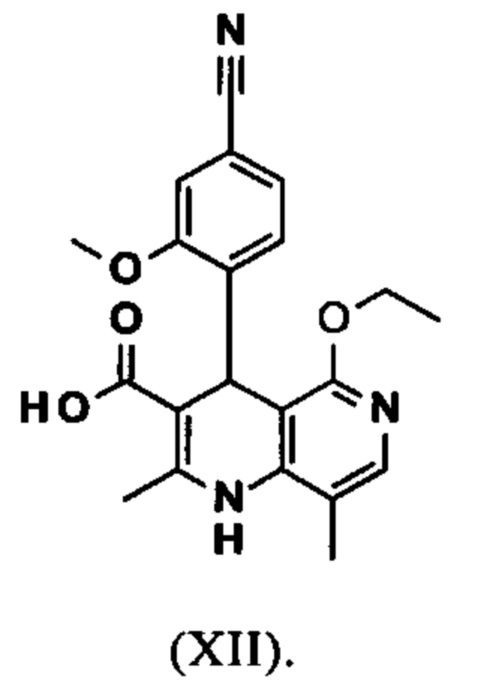
Настоящее изобретение также обеспечивает способ получения соединения формулы (I), который отличается тем, что соединение формулы (XII)
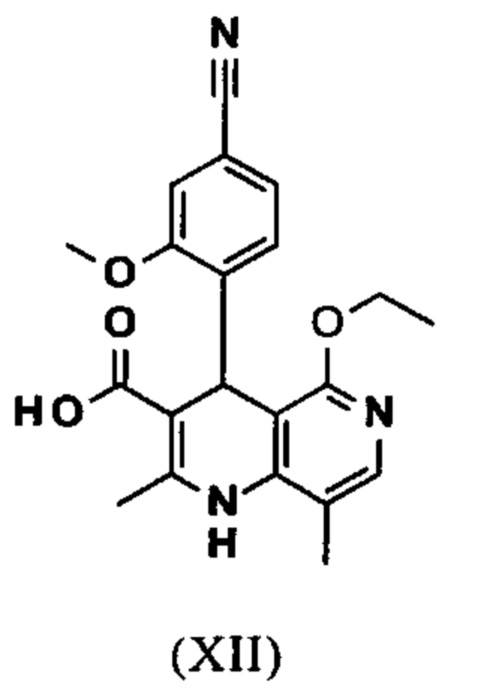
вводят в реакцию, которую проводят в одном реакционном сосуде без выделения промежуточных соединений, в ТГФ вначале с карбодиимидазолом и каталитическими количествами 4-(диметиламино)пиридина, на второй стадии нагревают с обратным холодильником вместе с гексаметилдисилазаном в течение 16-24 часов, и на третьей стадии гидролизуют в воде с использованием ТГФ или воды с получением соединения формулы (XIII)
Настоящее изобретение также обеспечивает способ получения соединения формулы (I), который отличается тем, что соединение формулы (XIV) или формулы (XIVa)
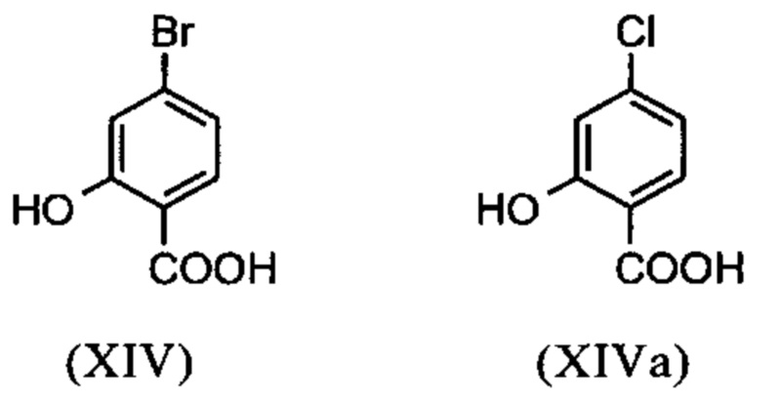
вводят в реакцию путем добавления диметилсульфата с получением соединения формулы (XV) или (XVa)
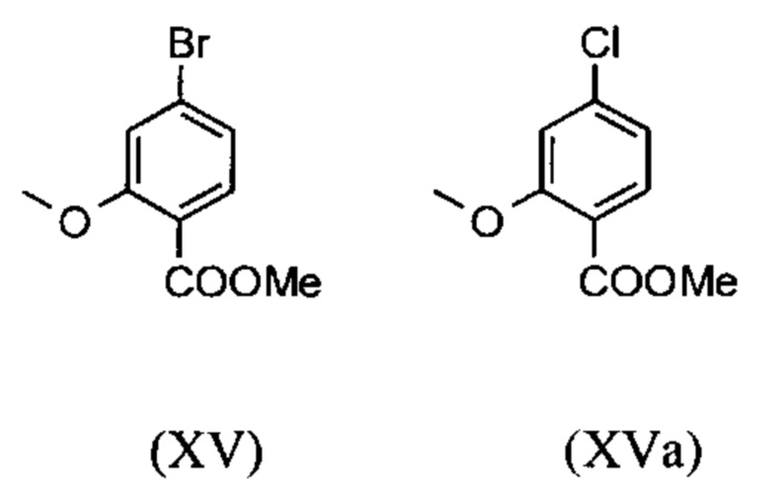
и невыделенные метиловые сложные эфиры формулы (XV) или (XVa) восстанавливают с помощью 1.21 экв. REDAL (натрия бис(2-метоксиэтокси)алюминия дигидрида) и 1.28 экв. N-метилпиперазина с получением альдегида формулы (XVI) или (XVIa)
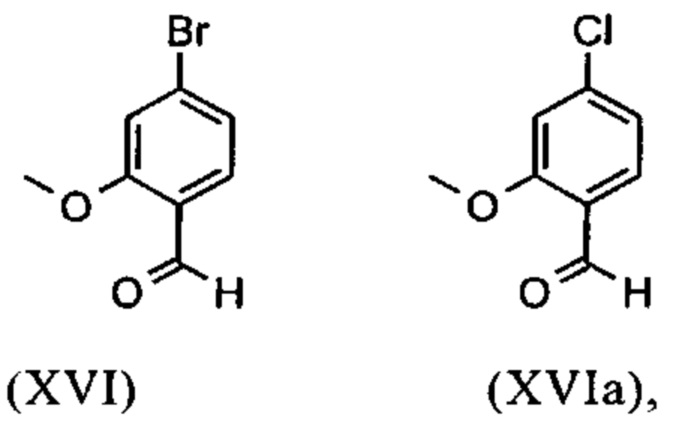
и альдегид (XVI) или (XVIa) вводят далее в реакцию без выделения с получением нитрила формулы (VI)
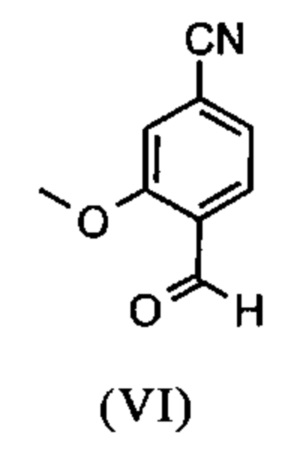
и соединение формулы (VI)
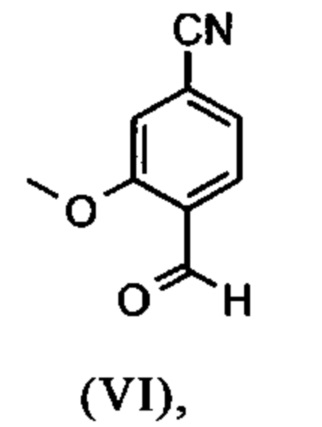
растворенное в изопропаноле (3-7-кратно), 5-10 мол. % пиперидина и 5-10 мол. % ледяной уксусной кислоты при 30°С вводят в реакцию с соединением формулы (VII)
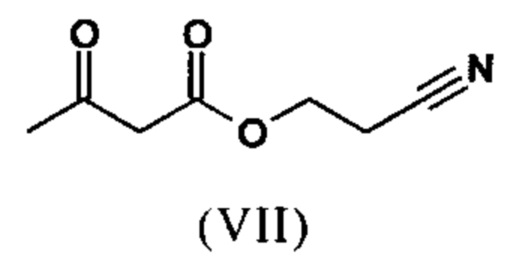
с получением соединения (VIIIa+b).

Настоящее изобретение также обеспечивает способ получения соединения формулы (I), который отличается тем, что соединение формулы (VI)
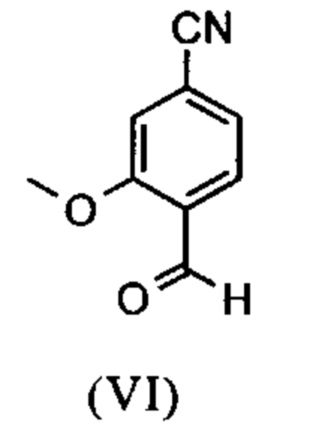
растворенное в изопропаноле (3-7-кратно), 5-10 мол. % пиперидина и 5-10 мол. % ледяной уксусной кислоты при 30°С вводят в реакцию с соединением формулы (VII)
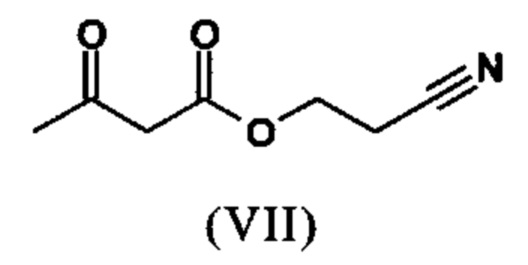
с получением соединения (VIIIa+b)
и что соединение формулы (X)
вводят в реакцию при перемешивании с 2.5-5 экв. триэтилортоацетата в диметилацетамиде при 100-120°С в течение 1.5-3 часов с получением соединения формулы (XI)
Настоящее изобретение также обеспечивает способ получения соединения формулы (I), который отличается тем, что соединение формулы (X)

вводят в реакцию при перемешивании с 2.5-5 экв. триэтилортоацетата в диметилацетамиде при 100-120°С в течение 1.5-3 часов с получением соединения формулы (XI)
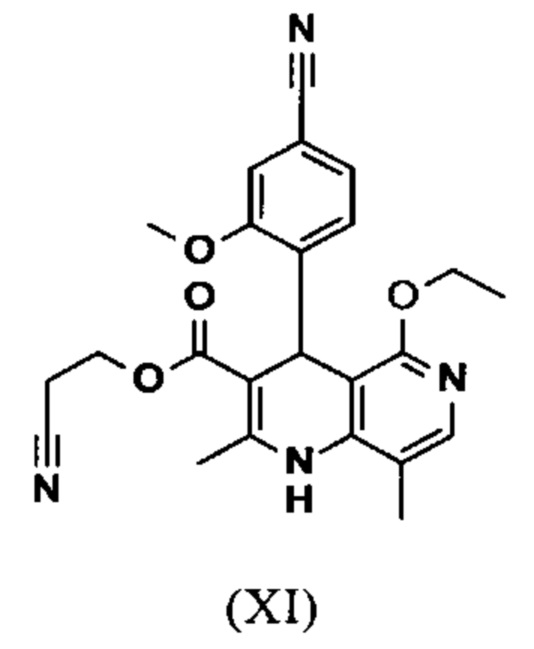
и что соединение формулы (XI)
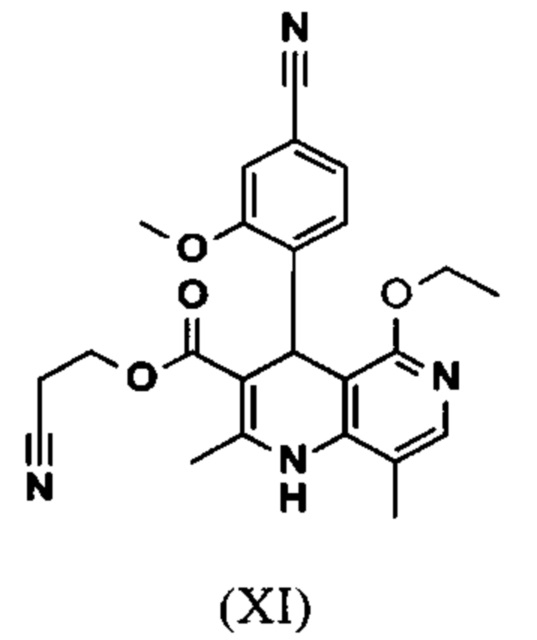
омыляют в смеси ТГФ/вода (2:1, 9-кратно) водным раствором гидроксида натрия с получением соединения формулы (XII)
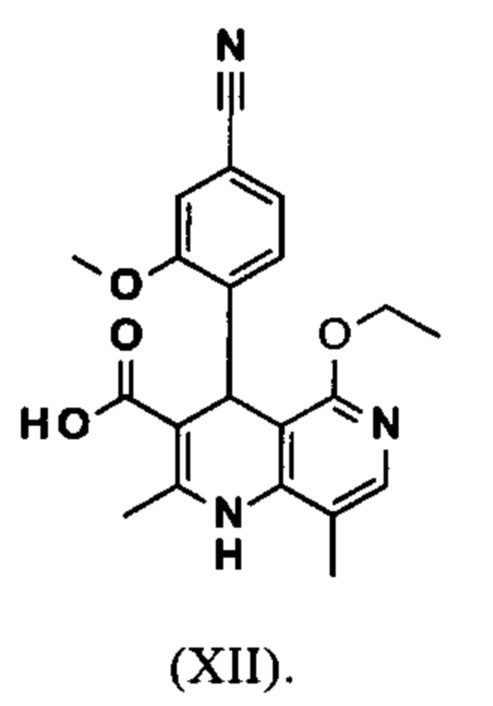
Настоящее изобретение также обеспечивает способ получения соединения формулы (I), который отличается тем, что соединение формулы (XI)
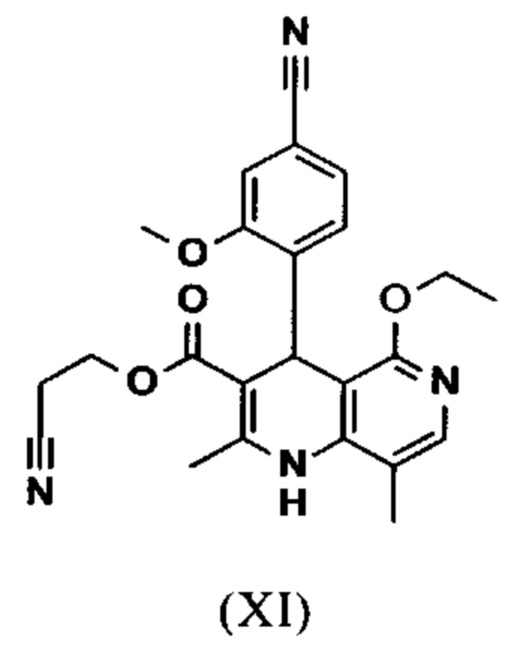
омыляют в смеси ТГФ/вода (2:1, 9-кратно) водным раствором гидроксида натрия с получением соединения формулы (XII)
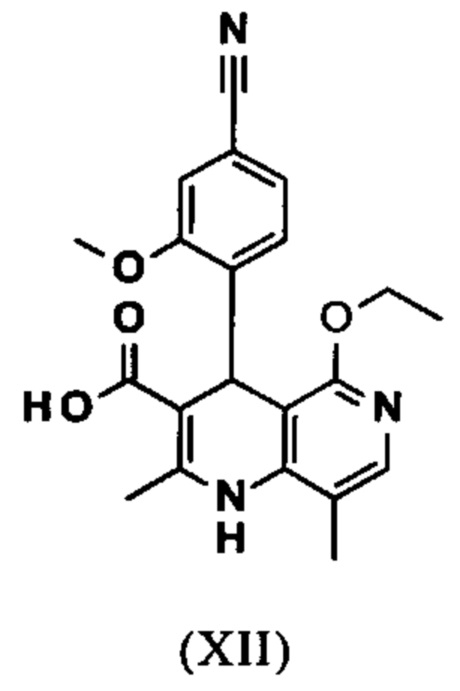
и что соединение формулы (XII)
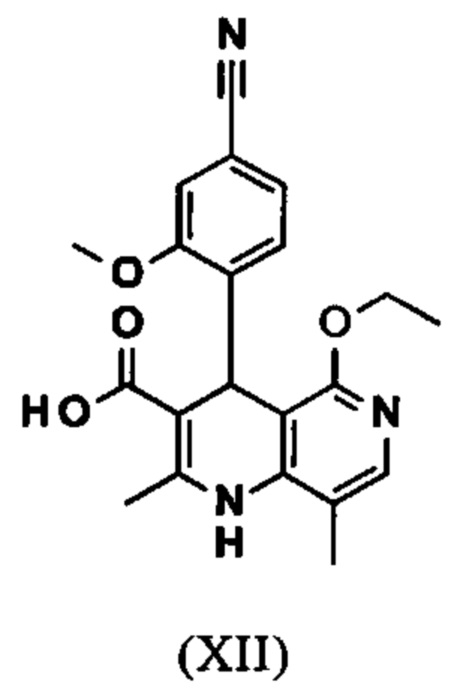
вводят в реакцию, которую проводят в одном реакционном сосуде без выделения промежуточных соединений, в ТГФ вначале с карбодиимидазолом и каталитическими количествами 4-(диметиламино)пиридина, на второй стадии нагревают с обратным холодильником вместе с гексаметилдисилазаном в течение 16-24 часов, и на третьей стадии гидролизуют в воде с использованием ТГФ или воды с получением соединения формулы (XIII)
Настоящее изобретение также обеспечивает способ получения соединения формулы (I), который отличается тем, что соединение формулы (XIV) или формулы (XIVa)
вводят в реакцию путем добавления диметилсульфата с получением соединения формулы (XV) или (XVa)
и невыделенные метиловые сложные эфиры формулы (XV) или (XVa) восстанавливают с помощью 1.21 экв. REDAL (натрия бис(2-метоксиэтокси)алюминия дигидрида) и 1.28 экв. N-метилпиперазина с получением альдегида формулы (XVI) или (XVIa)
и альдегид (XVI) или (XVIa) вводят далее в реакцию без выделения с получением нитрила формулы (VI)
и соединение формулы (VI)
растворенное в изопропаноле (3-7-кратно), 5-10 мол. % пиперидина и 5-10 мол. % ледяной уксусной кислоты при 30°С, вводят в реакцию с соединением формулы (VII)

с получением соединения (VIIIa+b)
и что соединение формулы (X)
вводят в реакцию при перемешивании с 2.5-5 экв. триэтилортоацетата в диметилацетамиде при 100-120°С в течение 1.5-3 часов с получением соединения формулы (XI)
Настоящее изобретение также обеспечивает способ получения соединения формулы (I), который отличается тем, что соединение формулы (VI)
растворенное в изопропаноле (3-7-кратно), 5-10 мол. % пиперидина и 5-10 мол. % ледяной уксусной кислоты при 30°С, вводят в реакцию с соединением формулы (VII)
с получением соединения (VIIIa+b)
и что соединение формулы (X)
вводят в реакцию при перемешивании с 2.5-5 экв. триэтилортоацетата в диметилацетамиде при 100-120°С в течение 1.5-3 часов с получением соединения формулы (XI)
и что соединение формулы (XI)

омыляют в смеси ТГФ/вода (2:1, 9-кратно) водным раствором гидроксида натрия с получением соединения формулы (XII)
Настоящее изобретение также обеспечивает способ получения соединения формулы (I), который отличается тем, что соединение формулы (X)
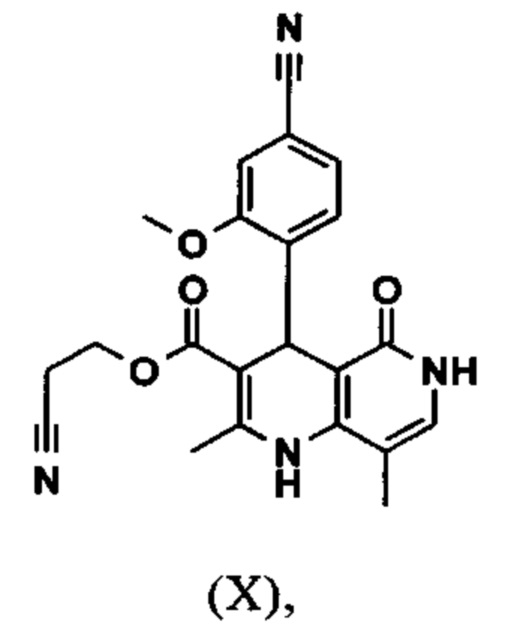
вводят в реакцию при перемешивании с 2.5-5 экв. триэтилортоацетата в диметилацетамиде при 100-120°С в течение 1.5-3 часов с получением соединения формулы (XI)
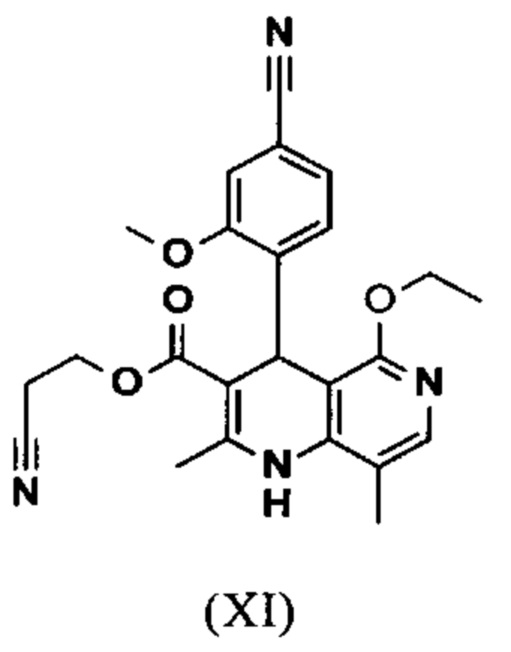
и что соединение формулы (XI)

омыляют в смеси ТГФ/вода (2:1, 9-кратно) водным раствором гидроксида натрия с получением соединения формулы (XII)

и что соединение формулы (XII)
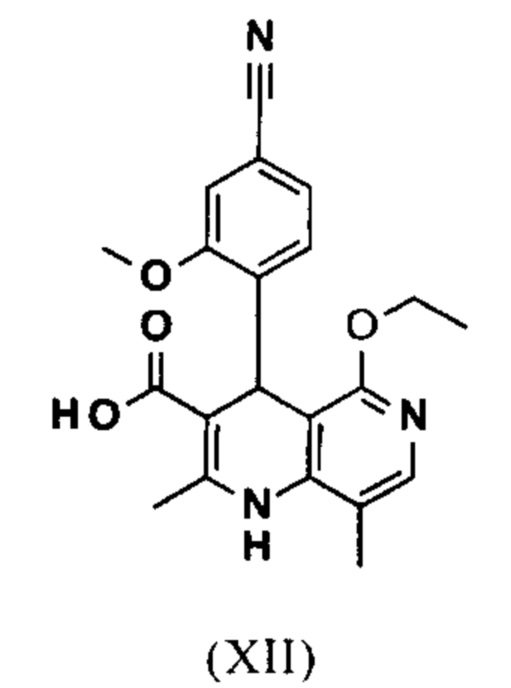
вводят в реакцию, которую проводят в одном реакционном сосуде без выделения промежуточных соединений, в ТГФ вначале с карбодиимидазолом и каталитическими количествами 4-(диметиламино)пиридина, на второй стадии нагревают с обратным холодильником вместе с гексаметилдисилазаном в течение 16-24 часов, и на третьей стадии гидролизуют в воде с использованием ТГФ или воды с получением соединения формулы (XIII)

Настоящее изобретение также обеспечивает способ получения соединения формулы (I), который отличается тем, что соединение формулы (XIV) или формулы (XIVa)
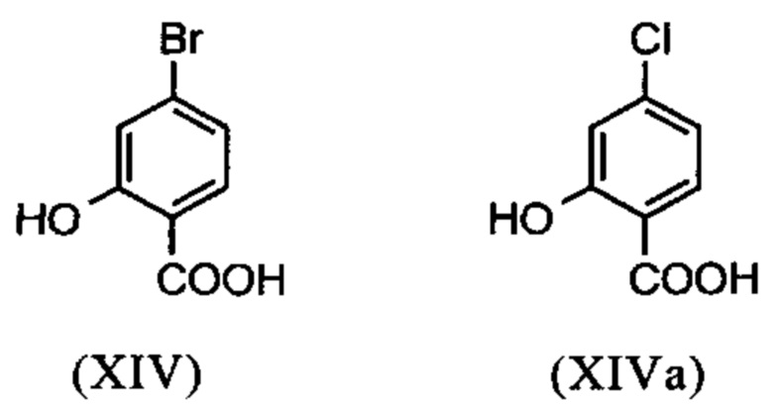
вводят в реакцию путем добавления диметилсульфата с получением соединения формулы (XV) или (XVa)
и невыделенные метиловые сложные эфиры формулы (XV) или (XVa) восстанавливают с помощью 1.21 экв. REDAL (натрия бис(2-метоксиэтокси)алюминия дигидрида) и 1.28 экв. N-метилпиперазина с получением альдегида формулы (XVI) или (XVIa)
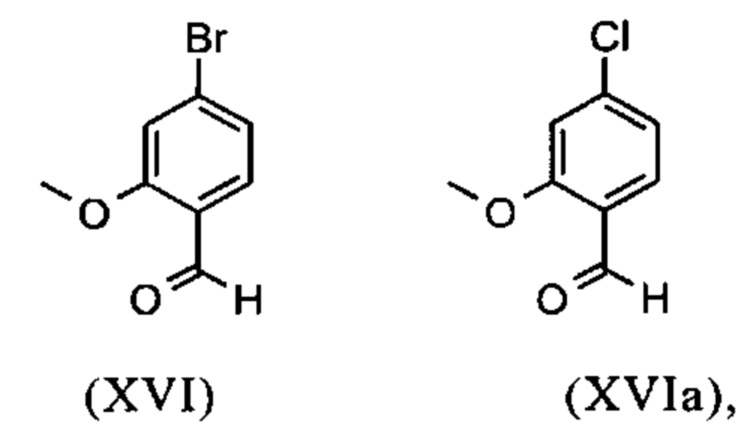
и альдегид (XVI) или (XVIa) вводят далее в реакцию без выделения с получением нитрила формулы (VI)
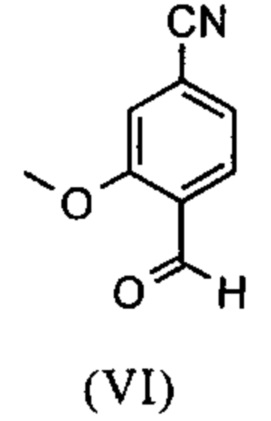
и соединение формулы (VI)
растворенное в изопропаноле (3-7-кратно), 5-10 мол. % пиперидина и 5-10 мол. % ледяной уксусной кислоты при 30°С, вводят в реакцию с соединением формулы (VII)
с получением соединения (VIIIa+b)
и что соединение формулы (X)
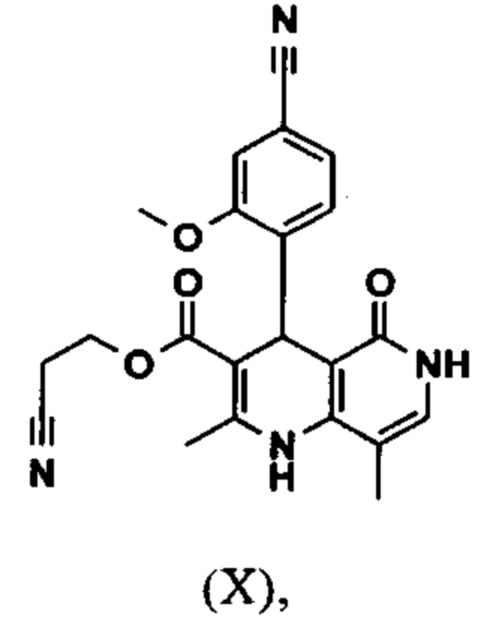
вводят в реакцию при перемешивании с 2.5-5 экв. триэтилортоацетата в диметилацетамиде при 100-120°C в течение 1.5-3 часов с получением соединения формулы (XI)
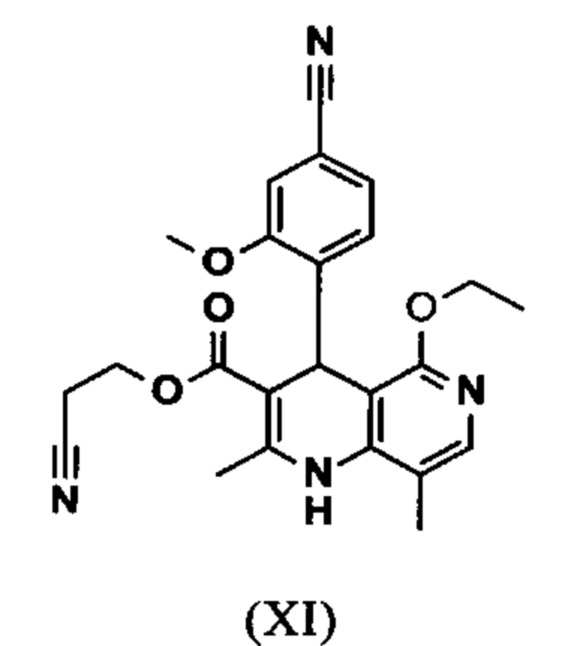
и что соединение формулы (XI)

омыляют в смеси ТГФ/вода (2:1, 9-кратно) водным раствором гидроксида натрия с получением соединения формулы (XII)
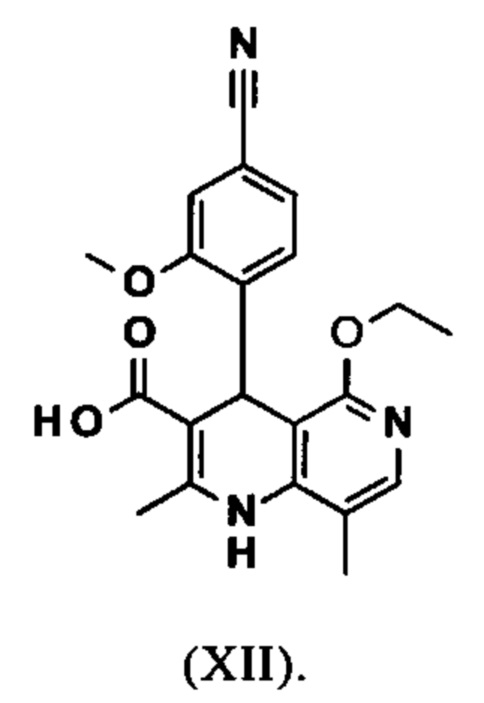
Настоящее изобретение также обеспечивает способ получения соединения формулы (I), который отличается тем, что соединение формулы (VI)
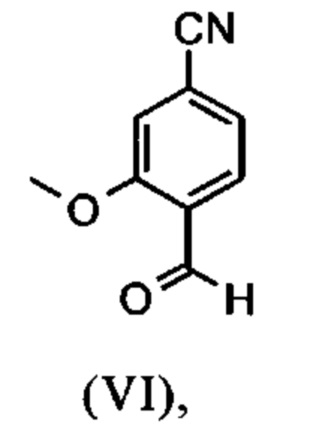
растворенное в изопропаноле (3-7-кратно), 5-10 мол. % пиперидина и 5-10 мол. % ледяной уксусной кислоты при 30°C, вводят в реакцию с соединением формулы (VII)
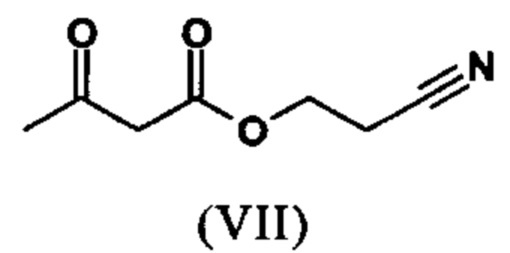
с получением соединения (VIIIa+b)
и что соединение формулы (X)
вводят в реакцию при перемешивании с 2.5-5 экв. триэтилортоацетата в диметилацетамиде при 100-120°C в течение 1.5-3 часов с получением соединения формулы (XI)

и что соединение формулы (XI)
омыляют в смеси ТГФ/вода (2:1, 9-кратно) водным раствором гидроксида натрия с получением соединения формулы (XII)
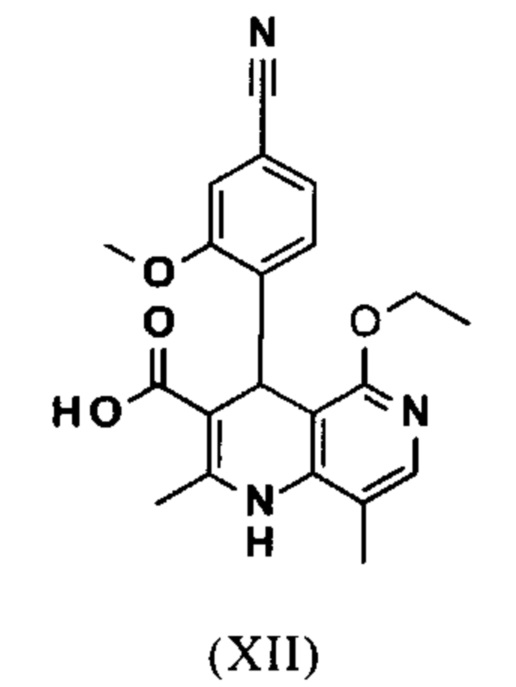
и что соединение формулы (XII)
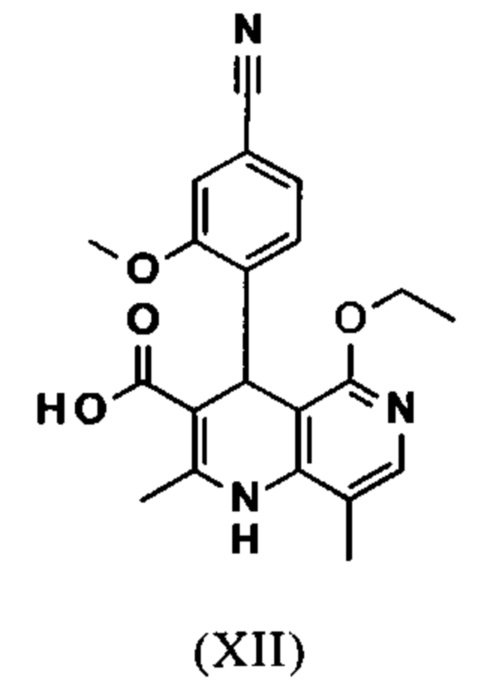
вводят в реакцию, которую проводят в одном реакционном сосуде без выделения промежуточных соединений, в ТГФ вначале с карбодиимидазолом и каталитическими количествами 4-(диметиламино)пиридина, на второй стадии нагревают с обратным холодильником вместе с гексаметилдисилазаном в течение 16-24 часов, и на третьей стадии гидролизуют в воде с использованием ТГФ или воды с получением соединения формулы (XIII)
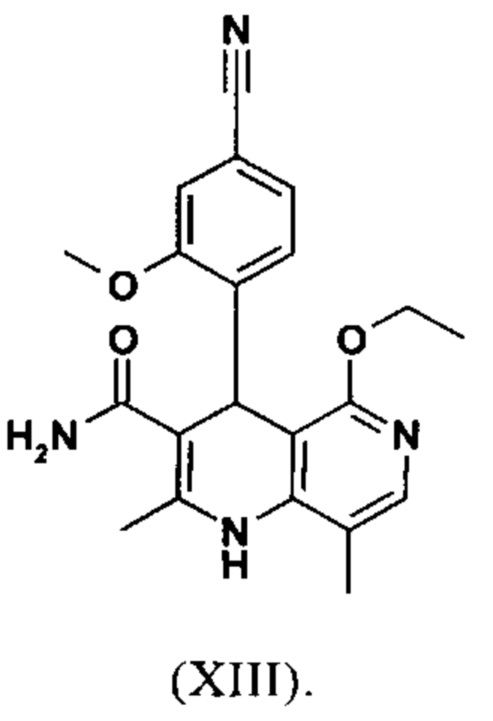
Настоящее изобретение также обеспечивает способ получения соединения формулы (I), который отличается тем, что соединение формулы (XIV) или формулы (XIVa)
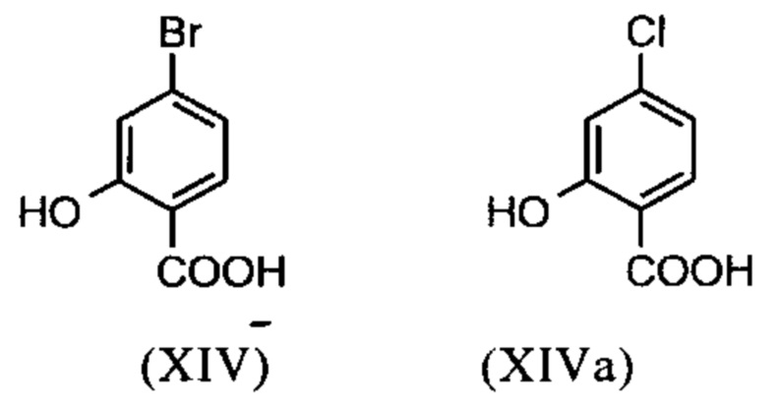
вводят в реакцию путем добавления диметилсульфата с получением соединения формулы (XV) или (XVa)
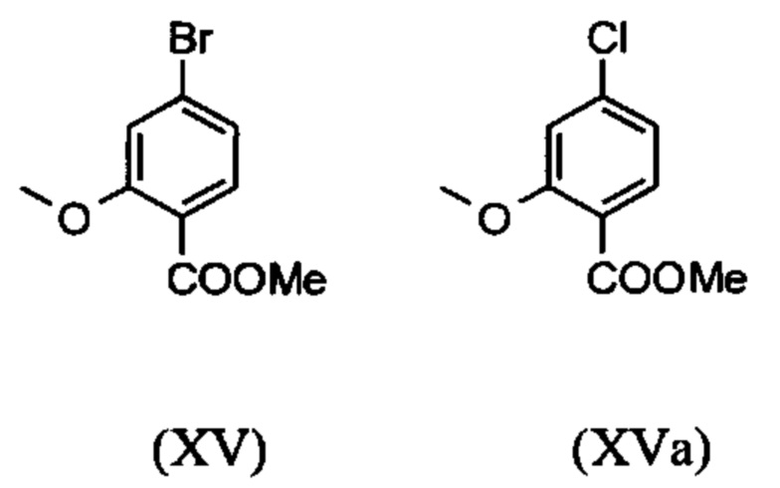
и невыделенные метиловые сложные эфиры формулы (XV) или (XVa) восстанавливают с помощью 1.21 экв. REDAL (натрия бис(2-метоксиэтокси)алюминия дигидрида) и 1.28 экв. N-метилпиперазина с получением альдегида формулы (XVI) или (XVIa)
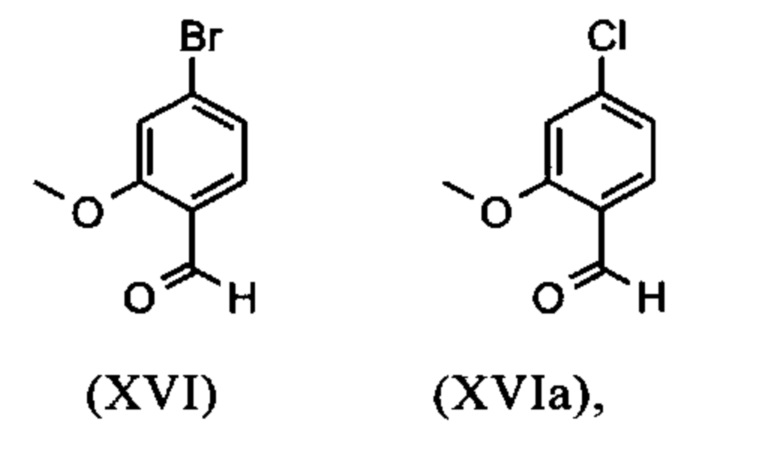
и альдегид (XVI) или (XVIa) вводят далее в реакцию без выделения с получением нитрила формулы (VI)
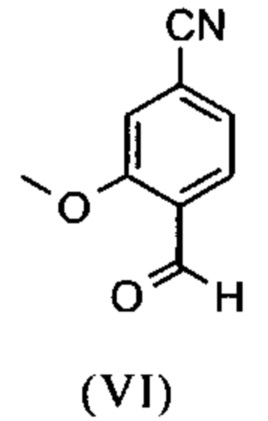
и соединение формулы (VI)
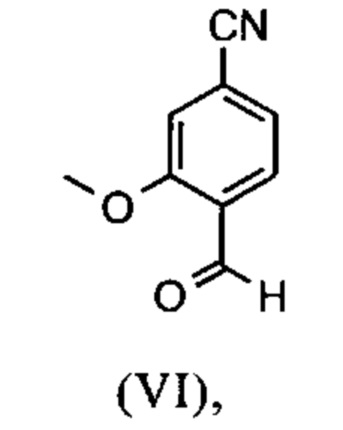
растворенное в изопропаноле (3-7-кратно), 5-10 мол. % пиперидина и 5-10 мол. % ледяной уксусной кислоты при 30°С, вводят в реакцию с соединением формулы (VII)
с получением соединения (VIIIa+b)
и что соединение формулы (X)

вводят в реакцию при перемешивании с 2.5-5 экв. триэтилортоацетата в диметилацетамиде при 100-120°С в течение 1.5-3 часов с получением соединения формулы (XI)
и что соединение формулы (XI)
омыляют в смеси ТГФ/вода (2:1, 9-кратно) водным раствором гидроксида натрия с получением соединения формулы (XII)

и что соединение формулы (XII)
вводят в реакцию, которую проводят в одном реакционном сосуде без выделения промежуточных соединений, в ТГФ вначале с карбодиимидазолом и каталитическими количествами 4-(диметиламино)пиридина, на второй стадии нагревают с обратным холодильником вместе с гексаметилдисилазаном в течение 16-24 часов, и на третьей стадии гидролизуют в воде с использованием ТГФ или воды с получением соединения формулы (XIII)
Процесс кристаллизации очень мощный и дает соединение формулы I в кристаллической форме полиморфа I воспроизводимым образом (температура плавления 252°C). Неожиданно, но также можно использовать вещество с более низкой оптической чистотой в процессе кристаллизации, и было показано, что даже материал с 93% е.е. все еще возрастает после кристаллизации до >99% е.е.
Соединение формулы (I) обычно тонко измельчается и вводится в состав фармацевтического препарата. Обнаружено, что соединение формулы (I) в кристаллической форме полиморфа I обладает очень хорошими стабильными свойствами (даже при высокой атмосферной влажности) и может храниться без каких-либо проблем в течение более 2 лет.
В соответствии с новым синтезом согласно изобретению можно получить соединение формулы (I) очень эффективным способом. Этот способ предлагает значительные преимущества по сравнению с уровнем техники, относящимся к масштабируемости и техническим характеристикам. Общий выход значительно выше по сравнению с опубликованными данными, и также достигается отличная чистота активного ингредиента. Новый способ обеспечивает воспроизводимое, экономичное получение определенного соединения формулы (I) в кристаллической форме полиморфа I, о существовании которого в уровне техники нигде не было описано.
Используя представленный здесь способ в соответствии с изобретением, уже успешно было получено 200 кг материала для клинических испытаний.
Соединения согласно изобретению, соединение формулы (I), и из которых соединение формулы (I) в кристаллической форме полиморфа I действуют как антагонисты минералокортикоидного рецептора и проявляют непредвиденный, полезный спектр фармакологической активности. Поэтому они пригодны для использования в качестве лекарственных средств для лечения и/или профилактики расстройств у людей и животных.
Соединения согласно изобретению пригодны для профилактики и/или лечения различных расстройств и заболеваний, в частности заболеваний, характеризующихся либо увеличением концентрации альдостерона в плазме, либо изменением концентрации в плазме альдостерона по отношению к концентрации в плазме ренина или связанных с этими изменениями. Примеры включают: идиопатический первичный гиперальдостеронизм, гиперальдостеронизм, связанный с гиперплазией надпочечников, аденомами надпочечников и/или карциномами надпочечников, гиперальдостеронизм, связанный с циррозом печени, гиперальдостеронизм, связанный с сердечной недостаточностью, и (относительный) гиперальдостеронизм, связанный с гипертонической болезнью.
Соединения согласно изобретению также пригодны из-за их механизма действия для профилактики внезапной сердечной смерти у пациентов с повышенным риском смерти от внезапной сердечной смерти. В частности, это пациенты, страдающие, например, от любого из следующих нарушений: первичной и вторичной гипертонии, гипертонической болезни сердца с или без застойной сердечной недостаточности, резистентной к терапии гипертензии, острой и хронической сердечной недостаточности, ишемической болезни сердца, стабильной и нестабильной стенокардии, ишемии миокарда, инфаркта миокарда, дилатационных кардиомиопатий, наследственных первичных кардиомиопатий, например синдрома Бругады, кардиомиопатий, вызванных болезнью Шагаса, шока, артериосклероза, предсердной и желудочковой аритмии, транзиторных и ишемических атак, инсульта, воспалительных сердечно-сосудистых нарушений, периферических сердечно-сосудистых расстройств, нарушений периферического кровотока, артериальных окклюзионных расстройств, таких как перемежающаяся хромота, бессимптомная левожелудочковая дисфункция, миокардит, гипертрофические изменения в сердце, легочная гипертензия, спазмы коронарных артерий и периферических артерий, тромбозы, тромбоэмболические расстройства, и васкулит.
Соединения согласно изобретению могут также использоваться для профилактики и/или лечения образования отеков, например отека легких, отека почек или отека, связанного с сердечной недостаточностью, и рестенозов, таких как терапия тромболизиса, чрескожная транслюминальная ангиопластика (ЧТА) и чрескожная транслюминальная коронарная ангиопластика (ЧТКА), трансплантация сердца и операции шунтирования.
Соединения согласно изобретению также пригодны для использования в качестве калийсберегающего мочегонного средства и для электролитных нарушений, например, гиперкальциемии, гипернатриемии или гипокалиемии.
Соединения согласно изобретению в равной степени пригодны для лечения почечных нарушений, таких как острая и хроническая почечная недостаточность, гипертоническая болезнь почек, атеросклеротический нефрит (хронический и интерстициальный), нефросклероз, хроническая почечная недостаточность и кистозная почечная недостаточность, для профилактики почечного повреждения, которое может быть вызвано, например, иммунодепрессантами, такими как циклоспорин А в случае трансплантации органов, и для рака почек.
Соединения настоящего изобретения могут дополнительно использоваться для профилактики и/или лечения сахарного диабета и диабетических осложнений, например невропатии и нефропатии.
Соединения настоящего изобретения могут также использоваться для профилактики и/или лечения микроальбуминурии, например, вызванной сахарным диабетом или высоким кровяным давлением, и протеинурии.
Соединения согласно изобретению пригодны также для профилактики и/или лечения расстройств, связанных либо с повышением концентрации глюкокортикоидов в плазме, либо с локальным увеличением концентрации глюкокортикоидов в ткани (например, сердца). Примеры включают: дисфункции надпочечников, приводящие к сверхпроизводству глюкокортикоидов (синдром Кушинга), адренокортикальные опухоли с сверхпроизводством глюкокортикоидов и опухоли гипофиза, которые автономно производят АКТГ (адренокортикотропный гормон) и, таким образом, приводят к гиперплазии надпочечников с последующей болезнью Кушинга.
Соединения согласно изобретению могут дополнительно использоваться для профилактики и/или лечения ожирения, метаболического синдрома и обструктивного апноэ во сне.
Соединения согласно изобретению могут также использоваться для профилактики и/или лечения воспалительных заболеваний, вызванных, например, вирусами, спирохетами, грибами, бактериями или микобактериями, а также воспалительных заболеваний неизвестной этиологии, таких как полиартрит, красная волчанка, пери- или полиартериит, дерматомиозит, склеродермия и саркоидоз.
Соединения согласно изобретению могут также быть использованы для лечения центральных нервных расстройств, таких как депрессия, состояния тревоги и хронической боли, особенно мигрени, и для нейродегенеративных расстройств, таких как болезнь Альцгеймера и синдром Паркинсона.
Соединения согласно изобретению также пригодны для профилактики и/или лечения поражения сосудов, например, таких процедур, как чрескожная транслюминальная коронарная ангиопластика (ЧТКА), имплантация стентов, коронарная ангиоскопия, реокклюзия или рестеноз после операций шунтирования и эндотелиальная дисфункция, при болезни Рейно, для облитерирующего тромбангиита (синдром Бюргера) и для синдрома звона в ушах.
Настоящее изобретение также предусматривает применение соединений согласно изобретению для лечения и/или профилактики расстройств, особенно вышеупомянутых расстройств.
Настоящее изобретение также предусматривает применение соединений согласно изобретению для получения лекарственного средства для лечения и/или профилактики расстройств, в частности, расстройств, упомянутых выше.
Настоящее изобретение также относится к способу лечения и/или профилактики расстройств, в частности расстройств, упомянутых выше, с использованием эффективного количества, по меньшей мере, одного из соединений согласно изобретению.
Соединения согласно изобретению могут использоваться по отдельности или, при необходимости, в комбинации с другими активными соединениями. Настоящее изобретение, кроме того, относится к лекарственным средствам, содержащим по меньшей мере одно из соединений согласно изобретению и одно или несколько дополнительных активных соединений, в частности для лечения и/или профилактики вышеупомянутых заболеваний. Предпочтительные примеры активных соединений, подходящих для комбинаций, включают:
• Активные соединения, которые снижают кровяное давление, например, и предпочтительно из группы антагонистов кальция, антагонистов ангиотензина AII, ингибиторов АСЕ, антагонистов эндотелина, ингибиторов ренина, блокаторов альфа-рецепторов, бета-рецепторных блокаторов и ингибиторов Rho-киназы;
• диуретики, особенно петлевые диуретики, тиазиды и тиазидоподобные диуретики;
• антитромботические агенты, в качестве примера и предпочтительно из группы ингибиторов агрегации тромбоцитов, антикоагулянтов или профибринолитических веществ;
• активные соединения, изменяющие липидный обмен, например, и предпочтительно, из группы агонистов тиреоидных рецепторов, ингибиторов синтеза холестерина, таких как, например, предпочтительно ингибиторы HMG-СоА-редуктазы или ингибиторы синтеза сквалена, ингибиторы АСАТ, ингибиторы СЕТР, ингибиторы МТР, агонисты PPAR-альфа, PPAR-гамма и/или PPAR-дельта, ингибиторы абсорбции холестерина, ингибиторы липазы, адсорбенты полимерной желчной кислоты, ингибиторы реабсорбции желчной кислоты и антагонисты липопротеина(а);
• органические нитраты и NO-доноры, например нитропруссид натрия, нитроглицерин, изосорбид мононитрат, изосорбид динитрат, молсидомин или SIN-1 и ингалированный NO;
• соединения, имеющие положительный инотропный эффект, например сердечные гликозиды (дигоксин), бета-адренергические и допаминергические агонисты, такие как изопротеренол, адреналин, норадреналин, допамин и добутамин;
• соединения, которые ингибируют разложение циклического гуанозинмонофосфата (цГМФ) и/или циклического аденозинмонофосфата (цАМФ), например ингибиторы фосфодиэстераз (ФДЭ) 1, 2, 3, 4 и/или 5, особенно ингибиторы ФДЭ 5, такие как силденафил, варденафил и тадалафил и ингибиторы ФДЭ 3, такие как амринон и милринон;
• натрийуретические пептиды, например «предсердный натрийуретический пептид» (ANP, анаритид), «натрийуретический пептид В-типа» или «мозговой натрийуретический пептид» (BNP, несиритид), «натрийуретический пептид С-типа» и уродилат;
• сенсибилизаторы кальция, предпочтительным примером является левозимендан;
• NO-независимые, но гем-зависимые стимуляторы гуанилатциклазы, такие как, в частности, соединения, описанные в WO 00/06568, WO 00/06569, WO 02/42301 и WO 03/095451;
• NO- и гем-независимые активаторы гуанилатциклазы, такие как, в частности, соединения, описанные в WO 01/19355, WO 01/19776, WO 01/19778, WO 01/19780, WO 02/070462 и WO 02/070510;
• ингибиторы нейтрофильной эластазы человека (HNE), например, сивелестат или DX-890 (Reltran);
• соединения, которые ингибируют каскад сигнальной трансдукции, например, ингибиторы тирозинкиназы, особенно сорафениб, иматиниб, гефитиниб и эрлотиниб; и/или
• соединения, которые влияют на энергетический метаболизм сердца, предпочтительными примерами являются этомоксир, дихлорацетат, ранолазин или триметазидин.
В предпочтительном варианте осуществления изобретения соединения согласно изобретению вводят в комбинации с диуретиком, в качестве примера и предпочтительно, с фуросемидом, буметанидом, торсемидом, бендрофлюметиазидом, хлортиазидом, гидрохлортиазидом, гидрофлуметиазидом, метиклотиазидом, политиазидом, трихлорметиазидом, хлорталидоном, индапамидом, метолазоном, квиназоном, ацетазоламидом, дихлорфенамидом, метазоламидом, глицерином, изосорбидом, маннитом, амилоридом или триамтереном.
Под веществами, которые понижают кровяное давление, предпочтительно следует понимать соединения из группы антагонистов кальция, антагонистов ангиотензина AII, ингибиторов АПФ, антагонистов эндотелина, ингибиторов ренина, блокаторов альфа-рецепторов, блокаторов бета-рецепторов, ингибиторов Rho-киназы и диуретиков.
В предпочтительном варианте осуществления изобретения соединения согласно изобретению вводят в комбинации с антагонистом кальция, в качестве примера и предпочтительно, с нифедипином, амлодипином, верапамилом или дилтиаземом.
В предпочтительном варианте осуществления изобретения соединения согласно изобретению вводят в комбинации с антагонистом ангиотензина АН, предпочтительными примерами являются лозартан, кандесартан, валсартан, телмисартан или эмбсусартан.
В предпочтительном варианте осуществления изобретения соединения согласно изобретению вводят в комбинации с ингибитором АПФ в качестве примера и предпочтительно, с эналаприлом, каптоприлом, лизиноприлом, рамиприлом, делаприлом, фозиноприлом, хиноприлом, периндоприлом или трандоприлом.
В предпочтительном варианте осуществления изобретения соединения согласно изобретению вводят в комбинации с антагонистом эндотелина, в качестве примера и предпочтительно, с босентаном, дарусентаном, амбрисентаном или ситаксентаном.
В предпочтительном варианте осуществления изобретения соединения согласно изобретению вводят в комбинации с ингибитором ренина, предпочтительными примерами являются алискирен, SPP-600, SPP-635, SPP-676, SPP-800 или SPP-1148.
В предпочтительном варианте осуществления изобретения соединения в соответствии с изобретением вводят в комбинации с блокатором альфа-1-рецептора, в качестве примера и предпочтительно с празозином.
В предпочтительном варианте осуществления изобретения соединения согласно изобретению вводят в комбинации с бета-рецепторным блокатором, в качестве примера и предпочтительно с пропранололом, атенололом, тимололом, пиндололом, алпренололом, окспренололом, пенбутололом, бупранололом, метипранололом, надололом, мепиндололом, каразалолом, соталолом, метопрололом, бетаксололом, целипрололом, бисопрололом, картеололом, эсмололом, лабеталолом, карведилолом, адаптололом, ландиололом, небивололом, эпанололом или буциндололом.
В предпочтительном варианте осуществления изобретения соединения согласно изобретению вводят в комбинации с ингибитором rho-киназы, в качестве примера и предпочтительно, с фасудилом, Y-27632, SLx-2119, BF-66851, BF-66852, BF -66853, KI-23095 или ВА-1049.
Антитромботические агенты (антитромботические средства) предпочтительно следует понимать как соединения из группы ингибиторов агрегации тромбоцитов, антикоагулянтов или профиринолитических веществ.
В предпочтительном варианте осуществления изобретения соединения согласно изобретению вводят в комбинации с ингибитором агрегации тромбоцитов, например, предпочтительно, с аспирином, клопидогрелем, тиклопидином или дипиридамолом.
В предпочтительном варианте осуществления изобретения соединения согласно изобретению вводят в комбинации с ингибитором тромбина, в качестве примера и предпочтительно, с ксимелагатраном, мелагатраном, бивалирудином или клексаном.
В предпочтительном варианте осуществления изобретения соединения согласно изобретению вводят в комбинации с антагонистом GPIIb/IIIa, в качестве примера, предпочтительно, с тирофибаном или абциксимабом.
В предпочтительном варианте осуществления изобретения соединения согласно изобретению вводят в комбинации с ингибитором фактора Ха в качестве примера и предпочтительно, с ривароксабаном (BAY 59-7939), DU-176b, апиксабаном, отамиксабаном, фидексабаном, разаксабаном, фондапаринуксом, идрапаринуксом, PMD-3112, YM-150, KFA-1982, EMD-503982, МСМ-17, MLN-1021, DX 9065а, DPC 906, JTV 803, SSR-126512 или SSR-128428.
В предпочтительном варианте осуществления изобретения соединения согласно изобретению вводят в комбинации с гепарином или с низкомолекулярным (LMW) производным гепарина.
В предпочтительном варианте осуществления изобретения соединения согласно изобретению вводят в комбинации с антагонистом витамина К, в качестве примера и предпочтительно с кумарином.
Предпочтительно, модификаторы метаболизма липидов означают соединения из группы ингибиторов СЕТР, агонистов тиреоидного рецептора, ингибиторов синтеза холестерина, таких как ингибиторы HMG-CoA-редуктазы или ингибиторы синтеза сквалена, ингибиторы АСАТ, ингибиторы МТР, PPAR-альфа, PPAR-гамма И/или агонисты PPAR-дельта, ингибиторы абсорбции холестерина, адсорбенты полимерной желчной кислоты, ингибиторы реабсорбции желчной кислоты, ингибиторы липазы и антагонисты липопротеина(а).
В предпочтительном варианте осуществления изобретения соединения согласно изобретению вводят в комбинации с ингибитором СЕТР в качестве примера и предпочтительно с торцетрапибом (СР-529 414), JJT-705, BAY 60-5521, BAY 78- 7499 или СЕТР вакциной (Avant).
В предпочтительном варианте осуществления изобретения соединения согласно изобретению вводят в комбинации с агонистом рецептора щитовидной железы в качестве примера и предпочтительно с D-тироксином, 3,5,3'-трийодтиронином (Т3), CGS 23425 или акситиромом (CGS 26214).
В предпочтительном варианте осуществления изобретения соединения согласно изобретению вводят в комбинации с ингибитором HMG-CoA-редуктазы из класса статинов, в качестве примера и предпочтительно с ловастатином, симвастатином, правастатином, флувастатином, аторвастатином, розувастатином, церивастатином или питавастатином.
В предпочтительном варианте осуществления изобретения соединения согласно изобретению вводят в комбинации с ингибитором синтеза сквалена, в качестве примера и предпочтительно с BMS-188494 или TAK-475.
В предпочтительном варианте осуществления изобретения соединения согласно изобретению вводят в комбинации с ингибитором АСАТ в качестве примера и предпочтительно с авасимибом, мелинамидом, пактимибом, эфлюцимибом или SMP-797.
В предпочтительном варианте осуществления изобретения соединения согласно изобретению вводят в комбинации с ингибитором МТР, в качестве примера и предпочтительно с имплитапидом, BMS-201038, R-103757 или JTT-130.
В предпочтительном варианте осуществления изобретения соединения согласно изобретению вводят в комбинации с агонистом PPAR-гамма, в качестве примера и предпочтительно с пиоглитазоном или розиглитазоном.
В предпочтительном варианте осуществления изобретения соединения согласно изобретению вводят в комбинации с агонистом PPAR-дельта, предпочтительными примерами являются GW-501516 или BAY 68-5042.
В предпочтительном варианте осуществления изобретения соединения в соответствии с изобретением вводят в комбинации с ингибитором абсорбции холестерина, например, предпочтительно, с эзетимибом, тикстидом или памаквесидом.
В предпочтительном варианте осуществления изобретения соединения согласно изобретению вводят в комбинации с ингибитором липазы, в качестве примера и предпочтительно, с орлистатом.
В предпочтительном варианте осуществления изобретения соединения согласно изобретению вводят в комбинации с полимерным адсорбентом желчной кислоты, например, предпочтительно, с холестирамином, колестиполом, колесолвамом, CholestaGel или колестимидом.
В предпочтительном варианте осуществления изобретения соединения согласно изобретению вводят в комбинации с ингибитором реабсорбции желчных кислот, в качестве примера и предпочтительно, с ингибиторами ASBT (=IBAT), например, AZD-7806, S-8921, AK-105, BARI-1741, SC-435 или SC-635.
В предпочтительном варианте осуществления изобретения соединения в соответствии с изобретением вводят в комбинации с антагонистом липопротеина(а), в качестве примера и предпочтительно с гемкабеновым кальцием (CI-1027) или никотиновой кислотой.
Настоящее изобретение также относится к лекарственным средствам, которые содержат по меньшей мере одно соединение согласно изобретению, обычно вместе с одним или несколькими инертными нетоксичными фармацевтически приемлемыми вспомогательными веществами и к их применению для вышеуказанных целей.
Соединения согласно изобретению могут действовать системно и/или локально. Для этой цели их можно вводить подходящим способом, например, пероральным, парентеральным, легочным, назальным, подъязычным, лингвальным, трансбуккальным, ректальным, дермальным, трансдермальным, конъюнктивальным или отическим путем или в качестве имплантата или стента.
Соединения в соответствии с изобретением можно вводить в подходящих формах введения для этих путей введения.
Приемлемыми формами для перорального введения являются те, которые работают согласно известному уровню техники и высвобождают соединения согласно изобретению быстро и/или модифицированным образом и которые содержат соединения согласно изобретению в кристаллической и/или аморфной и/или растворенной форме, например таблетки (таблетки без покрытия или таблетки с покрытием, например, с устойчивым к желудочному соку или с замедленным растворением или нерастворимые покрытия, которые контролируют высвобождение соединения в соответствии с изобретением), таблетки или пленки/наполнители, которые быстро разлагаются в полости рта, пленки/лиофилизаты, капсулы (например, твердые или мягкие желатиновые капсулы), таблетки с сахарным покрытием, гранулы, пеллеты, порошки, эмульсии, суспензии, аэрозоли или растворы.
Парентеральное введение можно осуществить, избегая стадии ресорбции (например, внутривенным, внутриартериальным, внутрисердечным, внутриспинальным или внутрилумбарным путем) или с ресорбцией (например, внутримышечным, подкожным, внутрикожным, чрескожным или внутрибрюшинным путем). Формы введения, подходящие для парентерального введения, включают препараты для инъекций и инфузий в виде растворов, суспензий, эмульсий, лиофилизатов или стерильных порошков.
Для других путей введения подходящими примерами являются ингаляционные лекарственные формы (включая порошковые ингаляторы, небулайзеры), капли для носа, растворы или спреи, таблетки, пленки/наполнители или капсулы для лингвального, подъязычного или трансбуккального введения, суппозитории, препараты для ушей или глаз, вагинальные капсулы, водные суспензии (лосьоны, встряхиваемые смеси), липофильные суспензии, мази, кремы, чрескожные терапевтические системы (например, пластыри), молоко, пасты, пены, разбрызгивающие порошки, имплантаты или стенты.
Пероральное и парентеральное введение являются предпочтительными, особенно пероральное и внутривенное введение.
Соединения согласно изобретению могут быть преобразованы в указанные формы введения. Это можно осуществить известным способом путем смешивания с инертными, нетоксичными фармацевтически приемлемыми вспомогательными веществами. Эти вспомогательные вещества включают носители (например, микрокристаллическую целлюлозу, лактозу, маннит), растворители (например, жидкие полиэтиленгликоли), эмульгаторы и диспергирующие или смачивающие агенты (например, додецилсульфат натрия, полиоксисорбитан олеат), связывающие вещества (например, поливинилпирролидон), синтетические и природные полимеры (например, альбумин), стабилизаторы (например, антиоксиданты, например аскорбиновая кислота), красители (например, неорганические пигменты, например оксиды железа) и корректоры вкуса и/или запаха.
В общем, было обнаружено, что в случае парентерального введения предпочтительно вводить количества от приблизительно 0,001 до 1 мг/кг, предпочтительно от приблизительно 0,01 до 0,5 мг/кг массы тела, для достижения эффективных результатов. В случае перорального введения дозировка составляет приблизительно от 0,01 до 100 мг/кг, предпочтительно от приблизительно 0,01 до 20 мг/кг и наиболее предпочтительно от 0,1 до 10 мг/кг массы тела.
Тем не менее, при необходимости может потребоваться отклонение от заявленных количеств, в частности, в зависимости от веса тела, способа введения, индивидуальной реакции на активное соединение, характера препарата и времени или интервала, в течение которого происходит введение. Таким образом, в некоторых случаях может быть недостаточно меньше упомянутой минимальной дозы, в то время как в других случаях вышеуказанный верхний предел должен быть превышен. В случае введения больших количеств, возможно, целесообразно разделить их на несколько индивидуальных доз в течение дня.
Приведенные ниже рабочие примеры иллюстрируют изобретение. Изобретение не ограничивается примерами.
Если не указано иначе, проценты в тестах и примерах, которые приведены ниже, являются массовыми процентами; части - это массовые части. Соотношения растворителей, соотношения разбавления и данные о концентрациях для жидкости/жидких растворов в каждом случае зависят от объема.
Экспериментальная часть
Сокращения и аббревиатуры;
МС: масса от масс-спектрометрии
ВЭЖХ: высокоэффективная жидкостная хроматография
ДМФА: диметилформамид
Red-Al раствор в толуоле: натрия бис(2-метоксиэтокси)алюминия дигидрид в толуоле
ТГФ: тетрагидрофуран
Водн. HCl: водный раствор соляной кислоты
DMAP: 4-(диметиламино)пиридин
Примеры
Пример 1
Метил 4-бром-2-метоксибензоат (XV)
3.06 кг (22.12 моль) карбоната калия в начале загружают в 3.6 л ацетона и нагревают с обратным холодильником. К этой суспензии добавляют 1.2 кг 4-бром-2-гидроксибензойной кислоты (5.53 моль), суспендируют в 7.8 л ацетона и дополнительно прополаскивают 0.6 л ацетона. Суспензию нагревают с обратным холодильником в течение 1 часа (сильное выделение газа!). Затем добавляют 2.65 кг (21.01 моль) диметилсульфата в течение 4 часов при кипячении. Потом смесь перемешивают при нагревании с обратным холодильником в течение 2.5 часов. Растворитель отгоняют в значительной степени (до состояния возможности перемешивания) и добавляют 12 л толуола и затем остаточный ацетон отгоняют при 110°С. Приблизительно 3 л дистиллята отгоняют, при этом дополнительно добавляя еще 3 л толуола к смеси. Смеси дают охладиться до 20°С и добавляют 10.8 л воды и энергично ее перемешивают. Органическую фазу отделяют и водную фазу экстрагируют еще раз 6.1 л толуола. Объединенные органические фазы промывают 3 л насыщенного раствора хлорида натрия и фазу толуола концентрируют до прибл. 4 л. Определение содержания путем упаривания порции приводит к выходу в 1.306 кг (96.4% от теоретического). Раствор используют непосредственно на следующей стадии без дополнительной очистки.
ВЭЖХ метод А: В.у. прибл. 11.9 мин.
МС (EIполож.): m/z=245 [М+Н]+
1Н ЯМР (400 МГц, CD2Cl2): δ=3.84 (s, 3Н), 3.90 (s, 3Н), 7.12-7.20 (m, 2Н), 7.62 (d, 1Н).
Пример 2
4-Бром-2-метоксибензальдегид (XVI)
В 1.936 кг (6.22 моль) 65% раствора Red-A1 в толуоле загружают 1.25 л толуола при -5°С. К этому раствору добавляют 0.66 кг (6.59 моль) 1-метилпиперазина, который прополосчен 150 мл толуола, поддерживая температуру между -7 и -5°С. Затем смеси дают перемешаться при 0°С в течение 30 минут. Этот раствор затем добавляют к раствору 1.261 кг (5.147 моль) метил 4-бром-2-метоксибензоата (XV), растворенному в 4 л толуола, поддерживая температуру при -8-0°С. После дополнительного ополаскивания дважды 0.7 л толуола, смесь затем перемешивают при 0°С в течение 1.5 часов. Для обработки с целью выделения продукта, раствор добавляют к холодному водному раствору серной кислоты при 0°С (12.5 л вода +1.4 кг конц. серной кислоты). Температура повыситься до максимум 10°С (медленное добавление). Значение рН устанавливают на рН 1, при необходимости, путем добавления еще серной кислоты. Органическую фазу отделяют и водный фазу экстрагируют 7.6 л толуола. Объединенные органические фазы промывают 5.1 л воды и затем в значительной степени концентрируют и остаток вносят в 10 л ДМФА. Раствор снова концентрируют до объема прибл. 5 л. Определение содержания путем упаривания порции приводит к выходу в 1.041 кг (94.1% от теоретического). Раствор используют непосредственно на следующей стадии без дополнительной очистки.
ВЭЖХ метод А: В.у. прибл. 12.1 мин.
МС (EIполож.): m/z=162 [М+Н]+
1Н-ЯМР (CDCl3, 400 МГц): δ=3.93 (3Н, s), 7.17 (2Н, m), 7.68 (1Н, d), 10.40 (1H, s)
Пример 3
4-Формил-3-метоксибензонитрил (VI)
В 719 г (3.34 моль) 4-бром-2-метоксибензальдегида (XVI) в виде раствора в 4.5 л ДМФА загружают 313 г (0.74 моль) гексацианоферрата калия (K4[Fe(CN)6]) и 354 г (3.34 моль) карбоната натрия и добавляют еще 1.2 л ДМФА и 3.8 г (0.017 моль) ацетата палладия. Смесь перемешивают при 120°С в течение 3 часов. Смеси дают охладиться до 20°С и добавляют к смеси 5.7 л воды. Смесь экстрагируют 17 л этилацетата и водную фазу промывают еще раз 17 л этилацетата. Органические фазы объединяют и в значительной степени концентрируют, вносят в 5 л изопропанола и концентрируют до прибл. 2 л. Смесь нагревают до кипячения и по каплям добавляют 2 л воды. Смеси дают охладиться до 50°С и опять добавляют 2 л воды. Смесь охлаждают до 3°С и перемешивают при этой температуре в течение одного часа. Продукт отфильтровывают и промывают водой (2 раза 1.2 л). Продукт сушат при 40°С в вакууме.
Выход: 469 г (87% от теоретического) бежевого твердого вещества.
ВЭЖХ метод А: В.у. прибл. 8.3 мин.
МС (EIполож.): m/z=162 [М+Н]+
1Н-ЯМР (300 МГц, ДМСО-d6): δ=3.98 (s, 3Н), 7.53 (d, 1Н), 7.80 (s, 1H), 7.81 (d, 1Н), 10.37 (s, 1Н).
Пример 4
2-Цианоэтил 4-(4-циано-2-метоксифенил)-2,8-диметил-5-оксо-1,4,5,6-тетрагидро-1,6-нафтиридин-3-карбоксилат (X)
Вариант А
1.035 кг (6.422 моль) 4-формил-3-метоксибензонитрила (VI), 1.246 кг (8.028 моль) 2-цианоэтил 3-оксобутаноата, 54.6 г (0.642 моль) пиперидина и 38.5 г (0.642 моль) ледяной уксусной кислоты нагревали с обратным холодильником в 10 л дихлорметана в течение 6.5 часов на водоотделителе. Смеси дают охладиться до комнатной температуры и органическую фазу дважды промывают 5 л воды каждый раз. Фазу дихлорметана затем концентрируют при атмосферном давлении и все еще перемешиваемый остаток вносят в 15.47 кг 2-бутанола и добавляют 0.717 кг (5.78 моль) 4-амино-5-метилпиридона. Остаточный дихлорметан отгоняют до достижения внутренней температуры 98°С. Смесь потом нагревают с обратным холодильником в течение 20 часов. Смесь охлаждают до 0°С, позволяют перемешаться при этой температуре в течение 4 часов и продукт отфильтровывают. Продукт сушат при 40°С в вакууме в атмосфере уносящего газа.
Выход: 2.049 кг (87.6% от теоретического в перерасчете на 4-амино-5-метилпиридон, поскольку этот компонент используется субстехиометрически) светло-желтого твердого вещества.
ВЭЖХ метод А: В.у. прибл. 9.7 мин.
МС (EIполож.): m/z=405 [М+Н]+
1Н-ЯМР (300 МГц, ДМСО-d6): δ=2.03 (s, 3Н), 2.35 (s, 3Н), 2.80 (m, 2Н), 3.74 (s, 3Н), 4.04 (m, 1H), 4.11 (m, 1Н), 5.20 (s, 1Н), 6.95 (s, 1H), 7.23 (dd, 1H), 7.28-7.33 (m, 2Н), 8.18 (s, 1Н), 10.76 (s, 1Н).
Вариант В
1.344 кг (8.34 моль) 4-формил-3-метоксибензонитрила (VI), 71 г (0.834 моль) пиперидина и 50.1 г (0.834 моль) ледяной уксусной кислоты загружают в 6 л изопропанола и при 30°С раствор 1.747 кг (11.26 моль) 2-цианоэтил 3-оксобутаноата в 670 мл изопропанола добавляют в течение 3 часов. Смесь затем перемешивают при 30°С в течение одного часа. Смесь охлаждают до 0-3°С и перемешивают в течение 0.5 часов. Продукт отфильтровывают и промывают дважды 450 мл холодного изопропанола каждый раз. Для определения выхода, продукт сушат при 50°С в вакууме (2.413 кг, 97% от теоретического); однако, благодаря высокому выходу, увлажненный изопропанолом продукт обычно непосредственно далее обрабатывают. Для этого, продукт вносят в 29 л изопропанола и добавляют 1.277 кг (7.92 моль) 4-амино-5-метилпиридона и затем смесь нагревают до внутренней температуры 100°С при положительном давлении прибл. 1.4 бар в течение 24 ч в закрытом сосуде. Смесь затем охлаждают до 0°C с помощью градиента в течение 5 ч и затем перемешивают при 0°С в течение 3 часов. Продукт затем отфильтровывают и промывают 2.1 л холодного изопропанола. Продукт сушат при 60°С в вакууме.
Выход: 2.819 кг (88% от теоретического в перерасчете на 4-амино-5-метилпиридон, поскольку этот компонент используют субстехиометрически) светло-желтого твердого вещества.
ВЭЖХ метод А: В.у. прибл. 9.7 мин.
МС (EIполож.): m/z=405 [М+Н]+
1Н-ЯМР (300 МГц, ДМСО-d6): δ=2.03 (s, 3Н), 2.35 (s, 3Н), 2.80 (m, 2Н), 3.74 (s, 3Н), 4.04 (m, 1H), 4.11 (m, 1Н), 5.20 (s, 1Н), 6.95 (s, 1H), 7.23 (dd, 1H), 7.28-7.33 (m, 2Н), 8.18 (s, 1H), 10.76 (s, 1Н).
Пример 5
2-Цианоэтил 4-(4-циано-2-метоксифенил)-5-этокси-2,8-диметил- 1,4-дигидро-1,6-нафтиридин-3-карбоксилат (XI)
2.142 кг (5.3 моль) 2-цианоэтил 4-(4-циано-2-метоксифенил)-2,8-диметил-5-оксо-1,4,5,6-тетрагидро-1,6-нафтиридин-3-карбоксилата (X) и 4.70 кг (29 моль) триэтилортоацетата растворяют в 12.15 л диметилацетамида и добавляют 157.5 г концентрированной серной кислоты. Смесь нагревают при 115°С в течение 1.5 часов и затем охлаждают до 50°С. При 50°С, 12.15 л воды по каплям добавляют в течение 30 минут. После завершения добавления, смесь вносят затравку 10 г указанного в заголовке соединения (XI) и затем по каплям добавляют еще 12.15 л воды в течение 30 минут при 50°С. Смесь охлаждают до 0°С (градиент, 2 часов) и перемешивают при 0°С в течение двух часов. Продукт отфильтровывают, промывают дважды 7.7 л воды каждый раз и сушат при 50°С в вакууме.
Выход: 2114.2 г (92.2% от теоретического) светло-желтого твердого вещества.
ВЭЖХ метод В: В.у. прибл. 10.2 мин.
МС (EIполож.): m/z=433 [М+Н]+
1Н-ЯМР (300 МГц, ДМСО-d6): δ=1.11 (t, 3Н), 2.16 (s, 3Н), 2.42 (s, 3Н), 2.78 (m, 2Н), 3.77 (s, 3Н), 4.01-4.13 (m, 4Н), 5.37 (s, 1Н), 7.25 (d, 1H), 7.28-7.33 (m, 2Н), 7.60 (s, 1H), 8.35 (s, 1Н).
В качестве альтернативы, реакцию можно проводить в NMP (1-метил-2-пирролидон)
2-Цианоэтил 4-(4-циано-2-метоксифенил)-5-этокси-2,8-диметил-1,4-дигидро-1,6-нафтиридин-3-карбоксилат (XI)
2.142 кг (5.3 моль) 2-цианоэтил 4-(4-циано-2-метоксифенил)-2,8-диметил-5-оксо-1,4,5,6-тетрагидро-1,6-нафтиридин-3-карбоксилата (X) и 2.35 кг (14.5 моль) триэтилортоацетата растворяют в 3.21 кг NMP (1-метил-2-пирролидона) и добавляют 157.5 г концентрированной серной кислоты. Смесь нагревают при 115°С в течение 1.5 часов и затем охлаждают до 50°С. При 50°С, по каплям добавляют 2.2 л воды в течение 30 минут. После завершения добавления, в смесь вносят затравку 10 г указанного в заголовке соединения (XI) и затем по каплям добавляют еще 4.4 л воды в течение 30 минут при 50°С. Смесь охлаждают до 0°С (градиент, 2 часов) и затем перемешивают при 0°С в течение двух часов. Продукт отфильтровывают, промывают дважды 4 л воды каждый раз и сушат при 50°С в вакууме.
Выход: 2180.7 г (95.1% от теоретического) светло-желтого твердого вещества.
ВЭЖХ метод В: В.у. прибл. 10.2 мин.
Пример 6
4-(4-Циано-2-метоксифенил)-5-этокси-2,8-диметил-1,4-дигидро-1,6-нафтиридин-3-карбоновая кислота (XII)
2.00 кг (4.624 моль) 2-цианоэтил 4-(4-циано-2-метоксифенил)-5-этокси-2,8-диметил-1,4-дигидро-1,6-нафтиридин-3-карбоксилата (XI) растворяют в смеси 12 л ТГФ и 6 л воды и охлаждают до 0°С. К этому раствору при 0°С по каплям добавляют в течение 15 минут водный раствор гидроксида натрия (полученного из 0.82 кг 45% водн. NaOH (9.248 моль) и 4.23 л воды и смесь затем перемешивают при 0°С в течение 1.5 часа. Смесь экстрагируют дважды 4.8 л метил-трет-бутилового эфира каждый раз и один раз 4.8 л этилацетата. Водный раствор при 0°С устанавливают на значение рН 7 с помощью разбавленной соляной кислоты (полученной из 0.371 кг 37% HCl и 1.51 л воды). Раствору дают нагреться до 20°С и добавляют водный раствор 2.05 кг хлорида аммония в 5.54 л воды. Раствор перемешивают при 20°С в течение 1 часа, продукт фильтруют и промывают дважды 1.5 л воды каждый раз и один раз 4 л ацетонитрила. продукт сушат при 40°С в атмосфере уносящего газа.
Выход: 1736.9 г (99% от теоретического) почти бесцветного порошка (очень легкий желтый оттенок).
ВЭЖХ метод С: В.у.: прибл. 6.8 мин.
МС (EIполож.): m/z=380 [М+Н]+
1Н-ЯМР (300 МГц, ДМСО-d6): δ=1.14 (t, 3Н), 2.14 (s, 3Н), 2.37 (s, 3Н). 3.73 (s, 3Н), 4.04 (m, 2Н), 5.33 (s, 1Н), 7.26 (m, 2Н), 7.32 (s, 1Н), 7.57 (s, 1H), 8.16 (s, 1H), 11.43 (br. s, 1Н).
Альтернативная обработка с целью выделения продукта с использованием толуола для экстрагирования:
4-(4-Циано-2-метоксифенил)-5-этокси-2,8-диметил-1,4-дигидро-1,6-нафтиридин-3-карбоновая кислота (XII)
2.00 кг (4.624 моль) 2-цианоэтил 4-(4-циано-2-метоксифенил)-5-этокси-2,8-диметил-1,4-дигидро-1,6-нафтиридин-3-карбоксилата (XI) растворяют в смеси 12 л ТГФ и 6 л воды и охлаждают до 0°С. К этому раствору при 0°С по каплям добавляют в течение 15 минут водный раствор гидроксида натрия (полученный из 0.82 кг 45% водн. NaOH (9.248 моль) и 4.23 л воды и смесь затем перемешивают при 0°С в течение 1.5 часа. 5 л толуола и 381.3 г натрия ацетат добавляют и энергично перемешивают. Фазам дают осадиться и органическую фазу отделяют. Водную фазу устанавливают на значение рН 6.9 с помощью 10% соляной кислоты (при прибл. значении рН 9.5 в раствор вносят затравку 10 г указанного в заголовке соединения). После завершения осаждения продукта, смесь перемешивают при 0°С в течение одного часа и затем фильтруют и промывают дважды 4 л воды каждый раз и дважды 153 мл толуола каждый раз. Продукт сушат при 40°С в вакууме в атмосфере уносящего газа (азот, 200 мбар. Выход: 1719.5 г (98% от теоретического) почти бесцветного порошка (очень легкий желтый оттенок).
ВЭЖХ метод С: В.у.: прибл. 6.8 мин.
Пример 7
4-(4-Циано-2-метоксифенил)-5-этокси-2,8-диметил-1,4-дигидро-1,6-нафтиридин-3-карбоксамид (XIII)
1.60 кг (4.22 моль) 4-(4-циано-2-метоксифенил)-5-этокси-2,8-диметил-1,4-дигидро-1,6-нафтиридин-3-карбоновой кислоты (XII) и 958 г (5.91 моль) 1,1-карбодиимидазола загружают в 8 л ТГФ и добавляют 51 г (0.417 моль) DMAP при 20°С. Смесь перемешивают при 20°С (выделение газа!) в течение одного часа и затем нагревают до 50°С в течение 2.5 часов. К этому раствору добавляют 2.973 кг (18.42 моль) гексаметилдисилазана и кипятят с обратным холодильником в течение 22 часов. Добавляют еще 1.8 л ТГФ и смесь охлаждают до 5°С. Смесь 1.17 л ТГФ и 835 г воды добавляют в течение 3 часов так, что температура остается между 5 и 20°С. Потом смесь кипятят с обратным холодильником в течение одного часа, затем охлаждают с помощью градиента (3 часов) до 0°С и перемешивают при этой температуре в течение одного часа. Продукт отфильтровывают и промывают дважды 2.4 л ТГФ каждый раз и дважды 3.2 л воды каждый раз. Продукт сушат при 70°С в вакууме в атмосфере уносящего газа.
Выход: 1.501 кг (94% от теоретического) почти бесцветного порошка (очень легкий желтый оттенок).
ВЭЖХ метод В: В.у. прибл. 6.7 мин.
МС (EIполож.): m/z=379 [М+Н]+
1Н-ЯМР (300 МГц, ДМСО-d6): δ=1.05 (t, 3Н), 2.12 (s, 3Н), 2.18 (s, 3Н), 3.82 (s, 3Н), 3.99-4.07 (m, 2Н), 5.37 (s, 1H), 6.60-6.84 (m, 2Н), 7.14 (d, 1Н), 7.28 (dd, 1H), 7.37 (d, 1Н), 7.55 (s, 1H), 7.69 (s, 1Н).
Пример 8
(4S)-4-(4-Циано-2-метоксифенил)-5-этокси-2,8-диметил-1,4-дигидро-1,6-нафтиридин-3-карбоксамид (I) в виде раствора в смеси ацетонитрил/метанол 40:60
Разделение энантиомеров на системе SMB
Исходным раствором является раствор, состоящий из 50 г рацемического 4-(4-циано-2-метоксифенил)-5-этокси-2,8-диметил-1,4-дигидро-1,6-нафтиридин-3-карбоксамида (XIII), растворенного в 1 литре смеси метанол/ацетонитрил 60:40.
Раствор хроматографируют с помощью системы SMB на неподвижной фазе: Chiralpak AS-V, 20 мкм. Давление составляет 30 бар и смесь метанол/ацетонитрил 60:40 используют в качестве элюента.
9.00 кг 4-(4-циано-2-метоксифенил)-5-этокси-2,8-диметил-1,4-дигидро-1,6-нафтиридин-3-карбоксамида (XII) растворяют в 180 л смеси, состоящей из метанола/ацетонитрила 60:40 и хроматографируют с помощью SMB. После концентрации продукт-содержащих фракций, 69.68 литров 6.2% раствора (которые соответствуют 4.32 кг (4S)-4-(4-циано-2-метоксифенил)-5-этокси-2,8-диметил-1,4-дигидро-1,6-нафтиридин-3-карбоксамида (I)) получают в виде раствора в смеси ацетонитрил/метанол 40:60).
Выход: 4.32 кг (48% от теоретического), в виде бесцветной фракции растворенной в 69.68 литрах смеси ацетонитрил/метанол 40:60.
Энантиомерная чистота: >98.5% е.е. (ВЭЖХ, метод D)
Образец концентрируют в вакууме и получают:
МС (EIполож.): m/z=379 [М+Н]+
1Н-ЯМР (300 МГц, ДМСО-d6): δ=1.05 (t, 3Н), 2.12 (s, 3Н), 2.18 (s, 3Н), 3.82 (s, 3Н), 3.99-4.07 (m, 2Н), 5.37 (s, 1Н), 6.60-6.84 (m, 2Н), 7.14 (d, 1Н), 7.28 (dd, 1H), 7.37 (d, 1Н), 7.55 (s, 1Н), 7.69 (s, 1Н).
Пример 9
(4S)-4-(4-Циано-2-метоксифенил)-5-этокси-2,8-диметил-1,4-дигидро-1,6-нафтиридин-3-карбоксамид (I)
Кристаллизация и образование полиморфа
64.52 литров 6.2% раствора из Примера 8 в смеси ацетонитрил/метанол 40:60 (соответствующие 4.00 кг соединения 1) фильтруют через фильтрующий картридж (1.2 мкм) и потом в достаточной мере концентрируют при 250 мбар так, что раствор все еще можно перемешивать. Добавляют 48 л этанола, денатурированного толуолом, и и снова дистиллируют при 250 мбар вплоть до ограничения перемешиваемости (повторная перегонка в этаноле). Добавляют еще 48 л этанола, денатурированного толуолом, и затем отгоняют при атмосферном давлении до получения общего объема прибл. 14 л (температура в рубашке 98°С). Смесь охлаждают градиентом (4 часов) до 0°С, перемешивают при 0°С в течение 2 часов и продукт отфильтровывают. Продукт промывают дважды 4 л холодного этанола каждый раз и затем сушат при 50°С в вакууме.
Выход: 3.64 кг (91% от теоретического) бесцветного кристаллического порошка.
Энантиомерная чистота: >>99% е.е. (ВЭЖХ метод D); Относительное время удержания/о.в.у.: (4S)-4-(4-циано-2-метоксифенил)-5-этокси-2,8-диметил-1,4-дигидро-1,6-нафтиридин-3-карбоксамид (1) - прибл. 11 мин. о.в.у.: 1.00; (4R)-4-(4-циано-2-метоксифенил)-5-этокси-2,8-диметил-1,4-дигидро-1,6-нафтиридин-3-карбоксамид (I) - прибл. 9 мин. о.в.у.: 0.82
Чистота: >99.8% (ВЭЖХ метод В), В.у.: прибл. 6.7 мин.
Содержание: 99.9% (относительно внешнего стандарта)
удельное вращение (хлороформ, 589 нм, 19.7°С, с=0.38600 г / 100 мл): - 148.8°.
МС (EIполож.): m/z=379 [М+Н]+
1Н ЯМР (300 МГц, ДМСО-d6): δ=1.05 (t, 3Н). 2.12 (s, 3Н), 2.18 (s, 3Н), 3.82 (s, 3Н), 3.99-4.07 (m, 2Н), 5.37 (s, 1Н), 6.60-6.84 (m, 2Н), 7.14 (d, 1Н), 7.28 (dd, 1H), 7.37 (d, 1Н), 7.55 (s, 1Н), 7.69 (s, 1Н).
Температура плавления: 252°С (соединение формулы (I) в кристаллической форме полиморфа I)
Физико-химическая характеристика соединения формулы (I) в кристаллической форме полиморфа I
Соединение формулы (I) в кристаллической форме полиморфа I плавится при 252°С, ΔН=95-113 Дж⋅г-1 (скорость нагревания 20 Kмин-1, Фигура 1).
Ослабление температуры плавления наблюдали в зависимости от скорости нагревания.
Температура плавления понижается при более низкой скорости нагревания (например, 2 Кмин-1) поскольку происходить разложение.
Никакие другие перемещения фаз не наблюдались. Наблюдали потерю массы прибл. 0.1% вплоть до температуры 175°С.
Стабильность и абсорбция влаги
Образцы соединения формулы (I) в кристаллической форме полиморфа I хранили при относ, влажности 85% и 97% (25°С). Образцы оценивали через 12 месяцев с помощью DSC, TGA и XRPD. Через 12 месяцев наблюдалось изменение массы <0.1% в обоих случаях. Это означает, что соединение формулы (I) в кристаллической форме полиморфа I не демонстрирует значительной абсорбции воды в таких условиях хранения. В соответствии с DSC, TGA и XRPD, не появилось никакой разницы в соединении формулы (I) в кристаллической форме полиморфа I.
Фармацевтический состав (4S)-4-(4-циано-2-метоксифенил)-5-этокси-2,8-диметил-1,4-дигидро-1,6-наФтиридин-3-карбоксамида формулы (I)
Гранулированный раствор соединения формулы (I) в кристаллической форме полиморфа I в тонкоизмельченной форме, гипромеллозы 5 сР и лаурилсульфата натрия приготавливали в очищенной воде.
Микрокристаллическую целлюлозу, моногидрат лактозы и кроскармеллозу натрия смешивали (премикс) в контейнере или грануляторе с псевдоожиженным слоем.
Премикс и гранулированный раствор гранулировали в грануляторе с псевдоожиженным слоем.
Добавляли смазывающее вещество стеарат магния, после чего гранулят сушили и просеивали. Таким образом получали готовую прессованную смесь.
Готовую прессованную смесь спрессовывали с получением таблеток, используя роторный таблеточный пресс.
Однородную суспензию для покрытия получали из гипромеллозы, талька, диоксида титана, желтого оксида железа, красного оксида железа и очищенной воды. Суспензию для покрытия наносили распылением на таблетки в подходящем устройстве для нанесения покрытия.
ВЭЖХ условия/методы
Метод А
YMC Hydrosphere CI8
150*4.6 мм, 3.0 мкм
25°C, 1 мл/мин, 270 нм, 4 нм
0': 70% ТФУ 0.1%*; 30% ацетонитрил
17': 20% ТФУ 0.1%*; 80% ацетонитрил
18': 70% ТФУ 0.1%*; 30% ацетонитрил
*: ТФУ в воде
Метод В
YMC Hydrosphere С18
150*4.6 мм, 3.0 мкм
25°С, 1 мл/мин., 255 нм, 6 нм
0': 90% ТФУ 0.1%; 10% ацетонитрил
20': 10% ТФУ 0.1%; 90% ацетонитрил
18': 10% ТФУ 0.1%; 90% ацетонитрил
Метод С
Nucleodur Gravity С18
150*2 мм, 3.0 мкм
35°С; 0.22 мл/мин., 255 нм, 6 нм
Раствор А: 0.58 г гидрофосфат аммония и 0.66 г дигидрофосфат аммония в 1 л воды (буферный раствор аммония фосфат, рН 7.2)
Раствор В:
ацетонитрил 0': 30% В; 70% А
15': 80% В; 20% А
25': 80% В; 20% А
Метод D
Длина колонки: 25 см
Внутренний диаметр: 4.6 мм
Упаковка: Chiralpak IA, 5 мкм
Реагенты: 1. Ацетонитрил чистоты для ВЭЖХ
2. Метил трет-бутил эфир (МТВЕ), ч.д.а.
Образец тестового раствора растворяют при концентрации 1.0 мг/мл в ацетонитриле.
(например, прибл. 25 мг образца, точно взвешенного, растворенного в ацетонитриле до 25.0 мл).
Элюент А. ацетонитрил
В. Метил-трет-бутиловый эфир (МТВЕ), ч.д.а.
Скорость потока 0.8 мл/мин
Температура термостата колонок: 25°С
Измерянная длина волны: 255 нм
Ширина полосы: 6 нм
Объемы инжекции: 5 мкл
Состав смеси элюентов А и B в соотношении по объему 90:10
Время работы хроматограммы: 30 мин
Относительное время удержания/О.В.У.:
(4S)-4-(4-циано-2-метоксифенил)-5-этокси-2,8-диметил-1,4-дигидро-1,6-нафтиридин-3-карбоксамид (1) - прибл. 11 мин. О.В.У.: 1.00
(4R)-4-(4-циано-2-метоксифенил)-5-этокси-2,8-диметил-1,4-дигидро-1,6-нафтиридин-3-карбоксамид (1) - прибл. 9 мин. О.В.У.: 0.82
Параметры решетки соединения формулы (I) в кристаллической форме полиморфа I
Полиморф I
Ортотриметрическая кристаллическая система
Пространственная группа Р2(1)2(1)2(1)
Молекулы на элементарную ячейку 4
Длина оси а  7.8610(3)
7.8610(3)
Длина оси b 11.7797(6)
Длина оси с 20.1792(8)
α [°] 90
β [°] 90
γ [°] 90
Рассчитанная плотность при 100 K [г⋅см-3] 1.345
Измерительные параметры рентгеновской дифрактометрии для измерения соединения формулы (I) в кристаллической форме полиморфа I



Измерительные условия для ИК-спектроскопии и спектроскопии Рамана для измерения соединения формулы (I) в кристаллической форме полиморфа I:
ИК-Спектроскопия:
Спектроскопия Рамана:

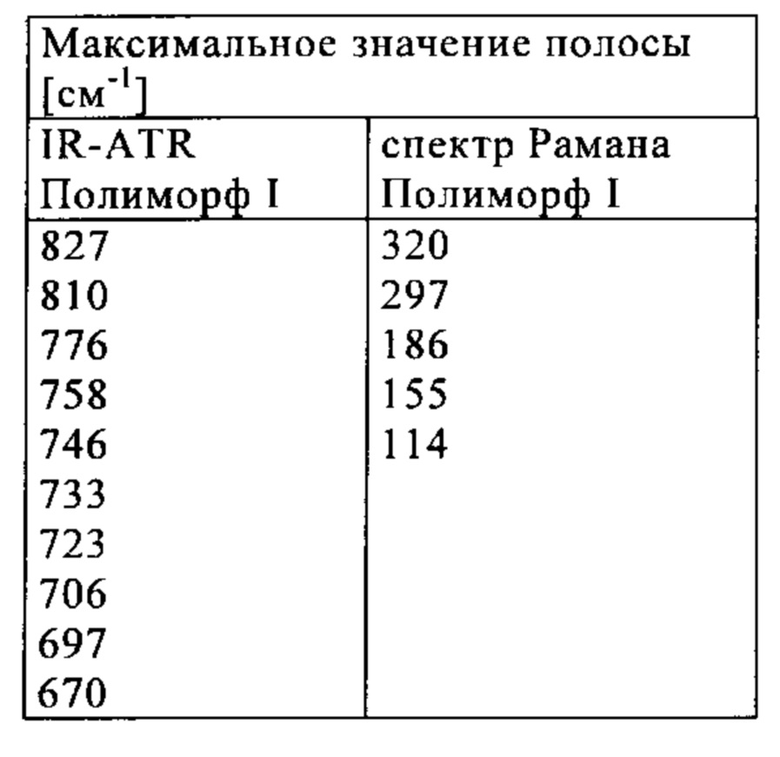
Описание фигур:
Фигура 1: DSC (20 Kмин-1) и TGA соединения формулы (I) в кристаллической форме полиморфа I
Фигура 2: рентгеновский луч единичного кристалла полиморфа 1 (4S)-4-(4-циано-2-метоксифенил)-5-этокси-2,8-диметил-1,4-дигидро-1,6-нафтиридин-3-карбоксамида (1)
Фигура 3: рентгеновская дифрактограмма соединения формулы (I) в кристаллической форме полиморфа I
Фигура 4: спектр Рамана соединения формулы (I) в кристаллической форме полиморфа I
Фигура 5: спектр, соединения формулы (I) в кристаллической форме полиморфа I, полученный инфракрасной (IR) спектроскопией с Фурье-преобразованием (KBr)
Фигура 6: спектр, соединения формулы (I) в кристаллической форме полиморфа I, полученный инфракрасной (IR) спектроскопией с Фурье-преобразованием (ATR)
Фигура 7: спектр соединения формулы (I) в кристаллической форме полиморфа I в ближнем инфракрасном диапазоне (NIR), полученный инфракрасной спектроскопией с Фурье-преобразованием
Фигура 8: спектр соединения формулы (I) в кристаллической форме полиморфа I в дальнем инфракрасном диапазоне (FIR), полученный инфракрасной спектроскопией с Фурье-преобразованием
Фигура 9: 13С-ЯМР спектр соединения формулы (I) в твердом состоянии в кристаллической форме полиморфа I
Фигура 10: стабильность соединения формулы (I) в кристаллической форме полиморфа I во влажности воздуха (x - ось относительная влажность в % / y - ось изменение массы в %)
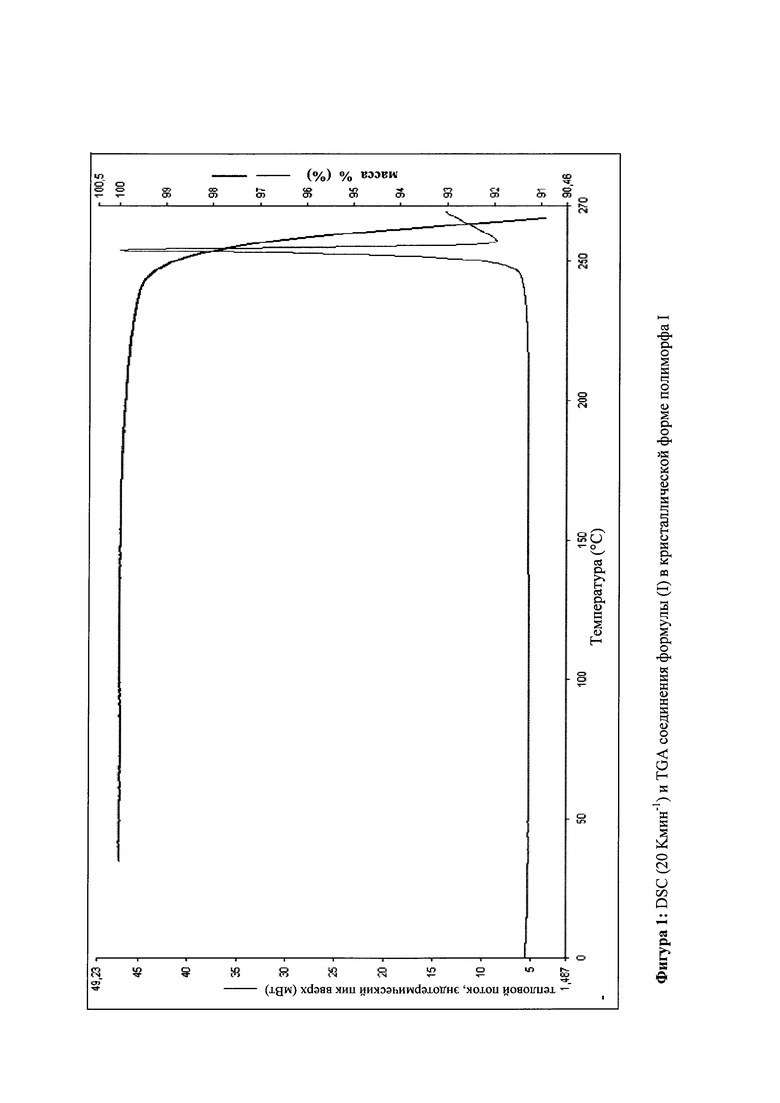
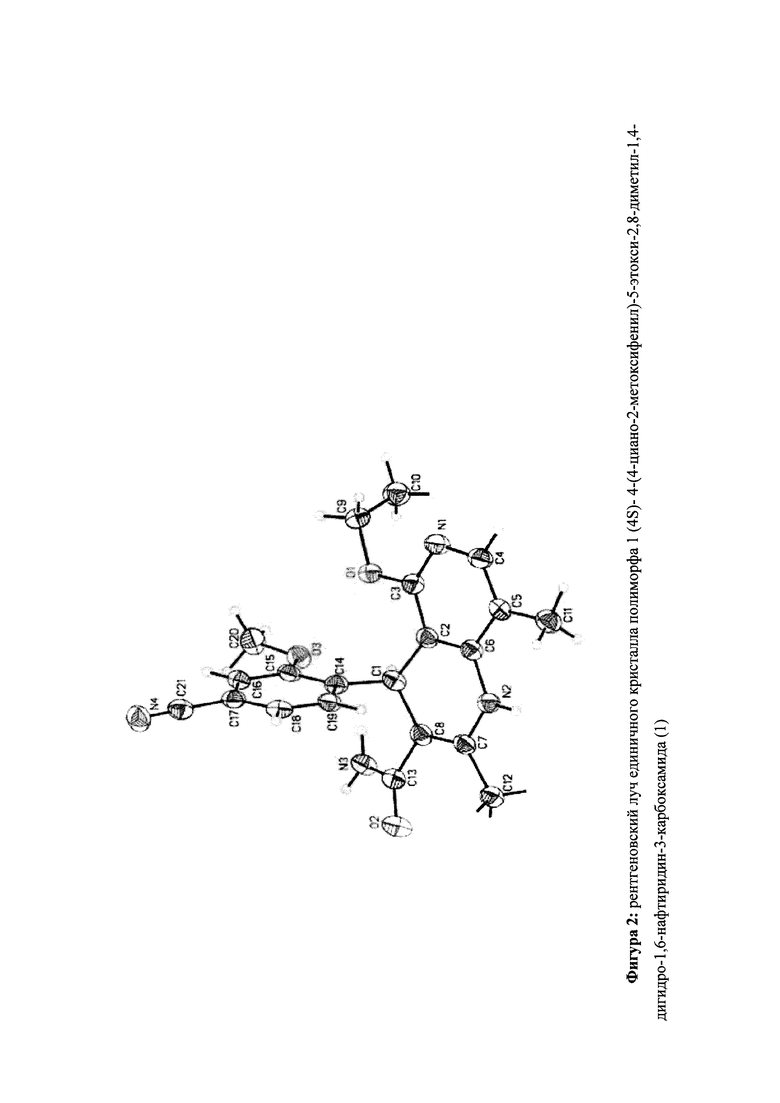
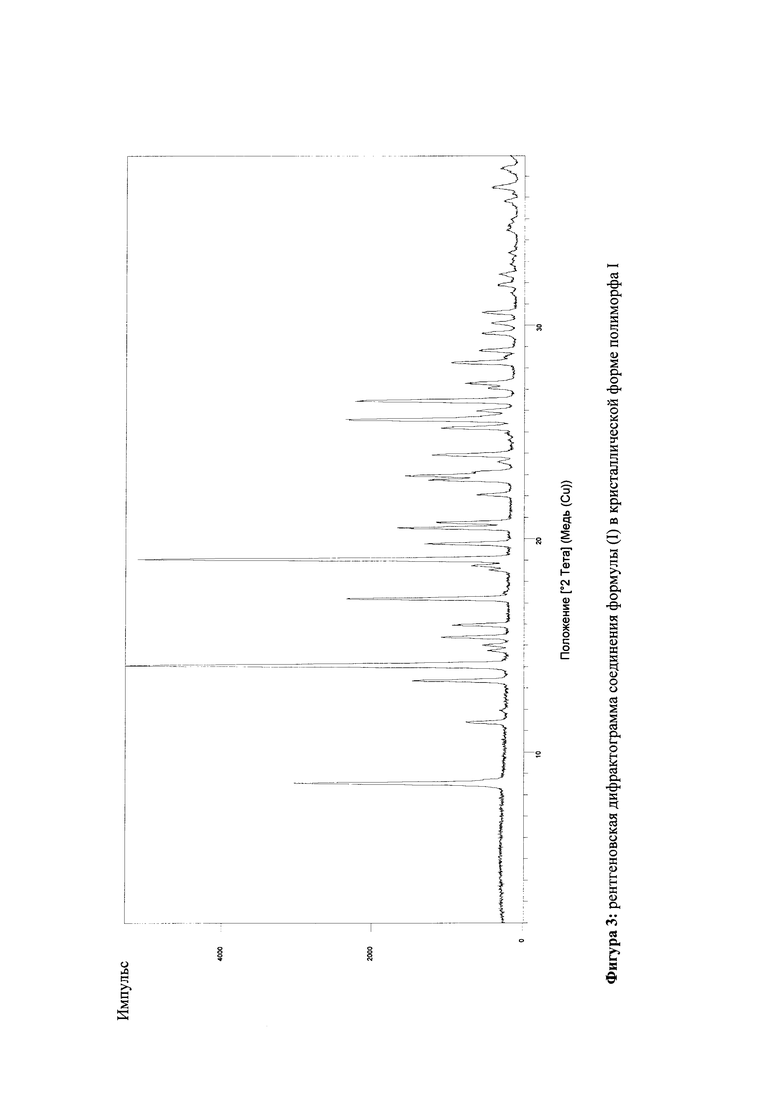
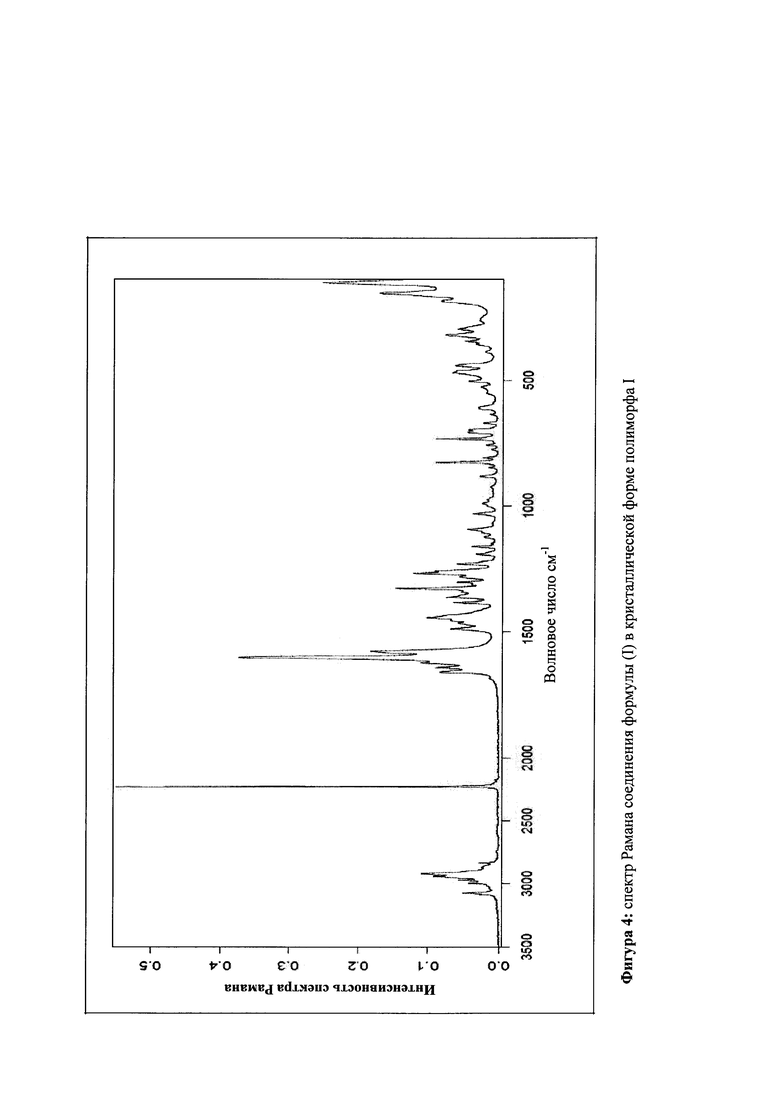

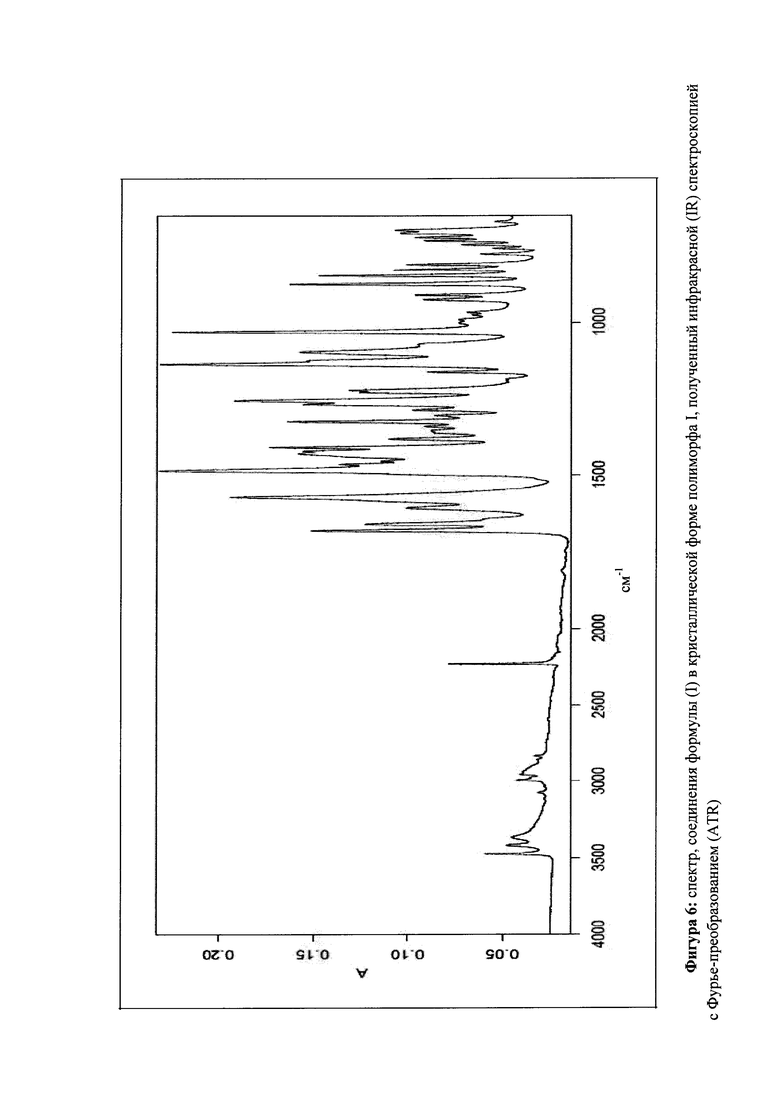


Изобретение относится к области органической химии, а именно к способу получения (4S)-4-(4-циано-2-метоксифенил)-5-этокси-2,8-диметил-1,4-дигидро-1,6-нафтиридин-3-карбоксамида формулы (I), который включает взаимодействие соединения формулы (XIV) или (XIVa) с диметилсульфатом с получением соединения формулы (XV) или (XVa), которые без выделения направляют на восстановление с помощью 1.21 экв. REDAL (натрия бис(2-метоксиэтокси)алюминия дигидрида) и 1.28 экв. N-метилпиперазина с получением альдегида формулы (XVI) или (XVIa), который вводят далее в реакцию без выделения с получением нитрила формулы (VI), которое растворяют в 3-7-кратном количестве изопропанола, 5-10 мол. % пиперидина и 5-10 мол. % ледяной уксусной кислоты при 30°С, вводят далее в реакцию с соединением формулы (VII) с получением соединения (VIIIa+b), которое затем реагирует с 4-амино-5-метилпиридоном с получением соединения (X), которое вводят в реакцию при перемешивании с 2.5-5 экв. триэтилортоацетата в диметилацетамиде при 100-120°С в течение 1.5-3 часов с получением соединения формулы (XI), которое омыляют в 9-кратном количестве смеси ТГФ/вода, имеющей соотношение 2:1, водным раствором гидроксида натрия с получением соединения формулы (XII), которое затем без выделения промежуточных соединений вводят в реакцию вначале в ТГФ с карбодиимидазолом и каталитическими количествами 4-(диметиламино)пиридина, на второй стадии нагревают с обратным холодильником вместе с гексаметилдисилазаном в течение 16-24 часов, и на третьей стадии гидролизуют в воде с использованием ТГФ или воды с получением соединения формулы (XIII), которое используют на стадии хиральной хроматографии для получения соединения формулы (I) в виде раствора в инертном растворителе. Также изобретение относится к способу получения кристаллической формы соединения формулы (I) и способу получения лекарственного средства на основе соединения формулы (I) в кристаллической форме. Технический результат: разработан улучшенный способ получения, который дает соединение формулы (I) в 9 стадий с общим выходом 27,7% от теоретического без хроматографической очистки промежуточных продуктов. 3 н.п. ф-лы, 10 ил., 8 пр.
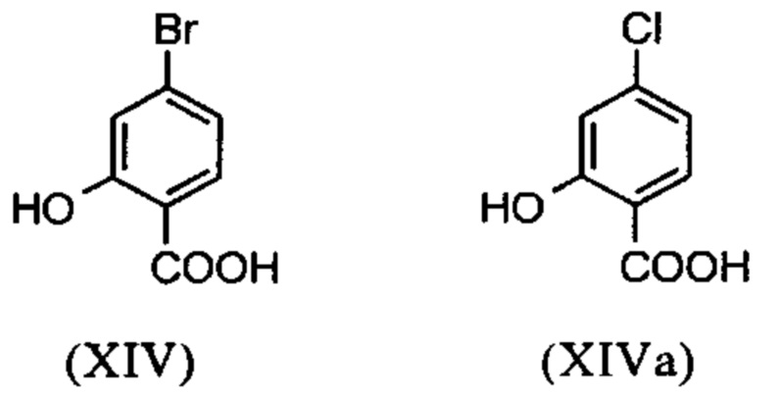
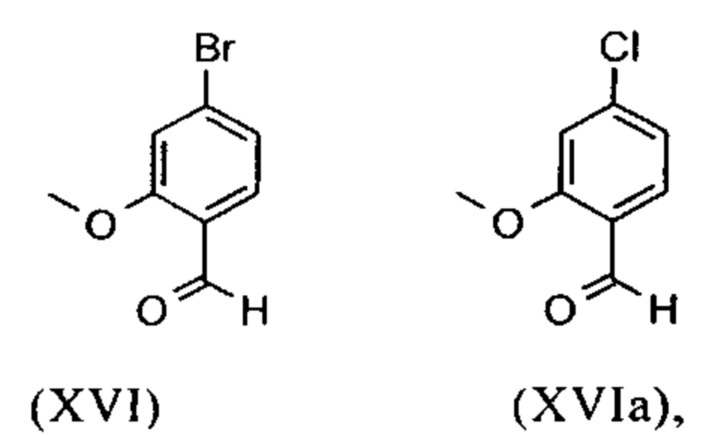
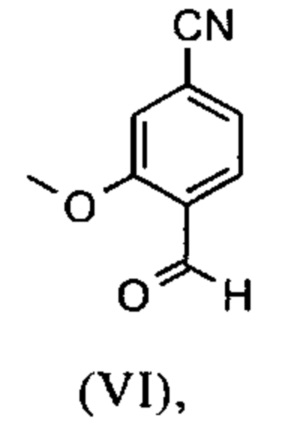
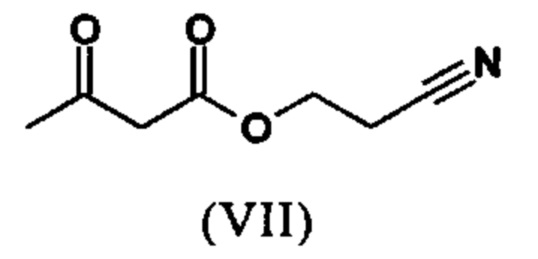

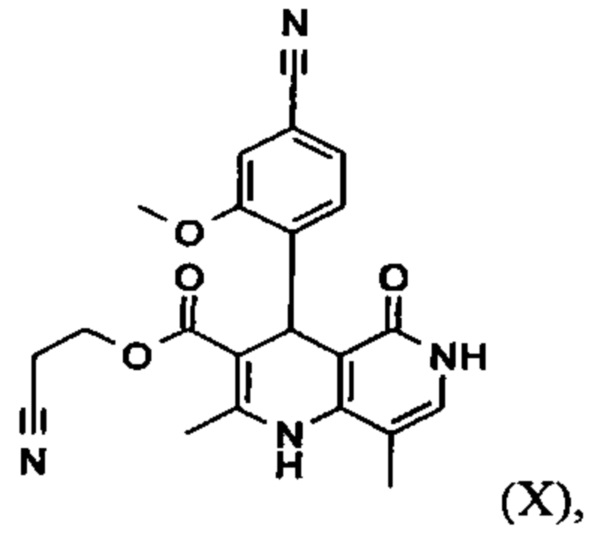
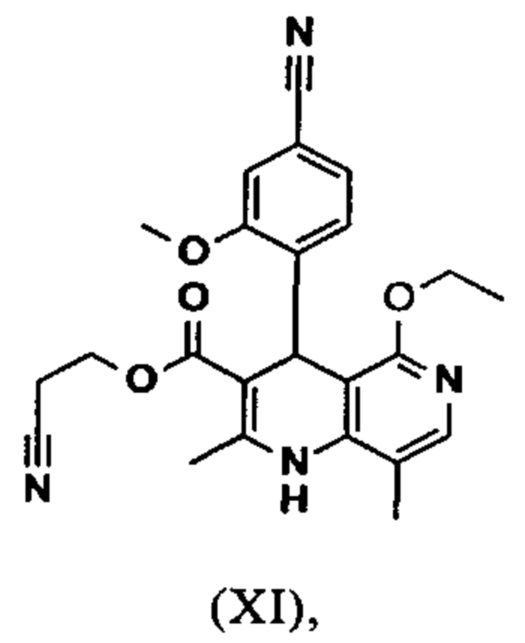
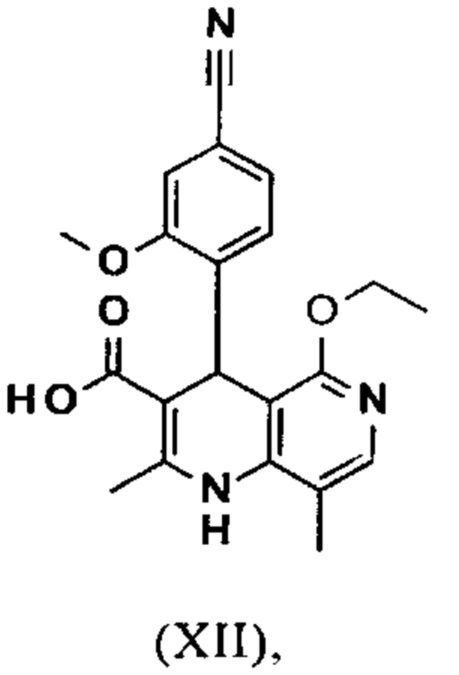
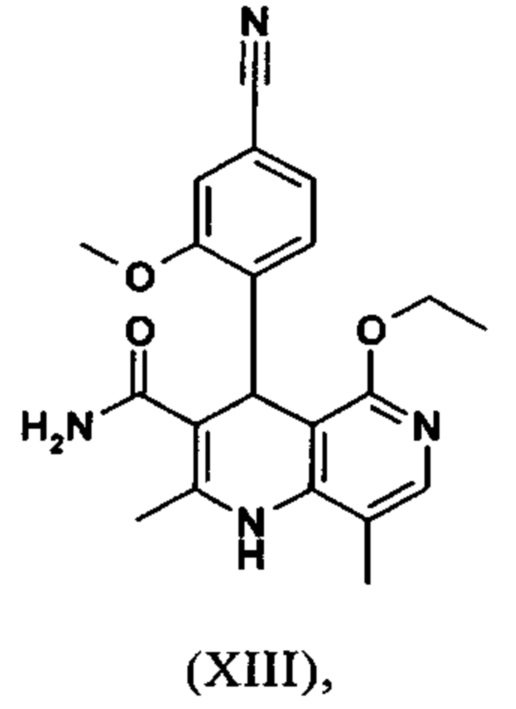
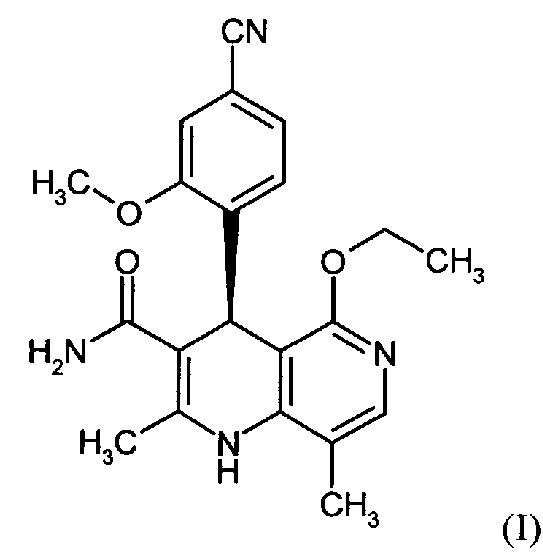
1. Способ получения соединения формулы (I)
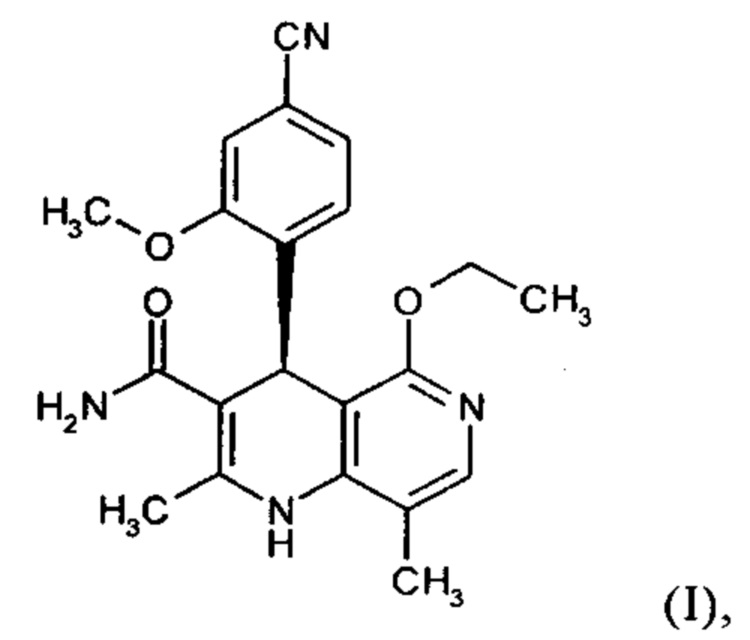
который отличается тем, что соединение формулы (XIV) или формулы (XIVa)
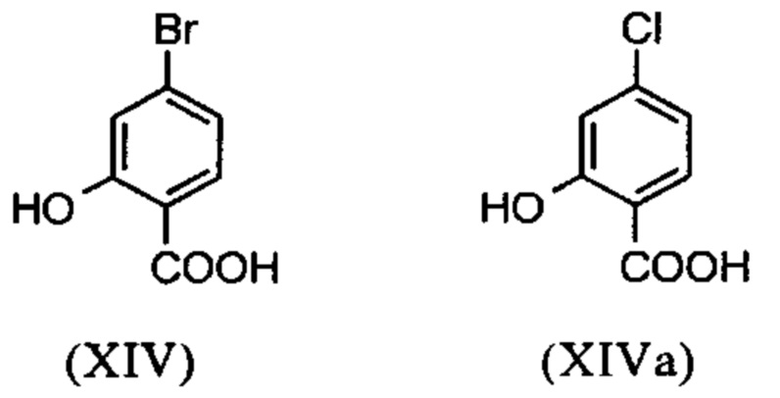
вводят в реакцию путем добавления диметилсульфата с получением соединения формулы (XV) или (XVa)
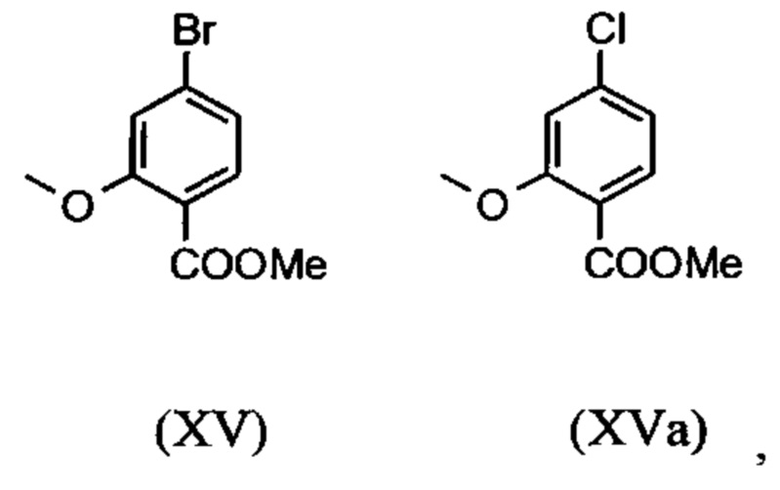
и невыделенные метиловые сложные эфиры формулы (XV) или (XVa) восстанавливают с помощью 1.21 экв. REDAL (натрия бис(2-метоксиэтокси)алюминия дигидрида) и 1.28 экв. N-метилпиперазина с получением альдегида формулы (XVI) или (XVIa)
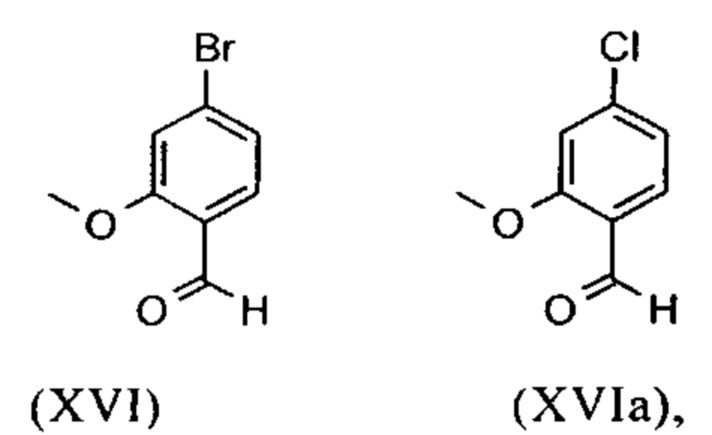
и альдегид (XVI) или (XVIa) вводят далее в реакцию без выделения с получением нитрила формулы (VI)
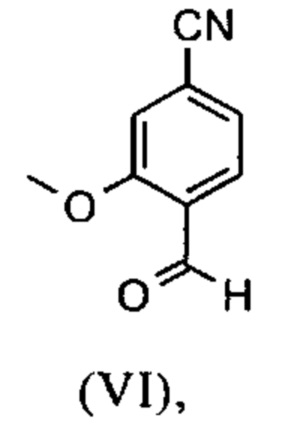
и соединение формулы (VI), растворенное в 3-7-кратном количестве изопропанола, 5-10 мол. % пиперидина и 5-10 мол. % ледяной уксусной кислоты при 30°С, вводят далее в реакцию с соединением формулы (VII)
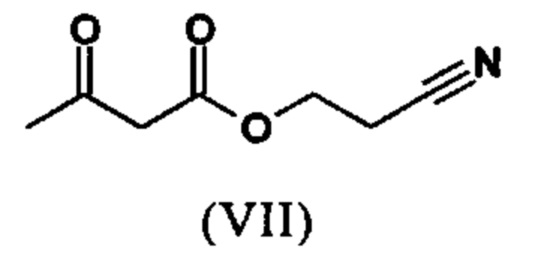
с получением соединения (VIIIa+b)
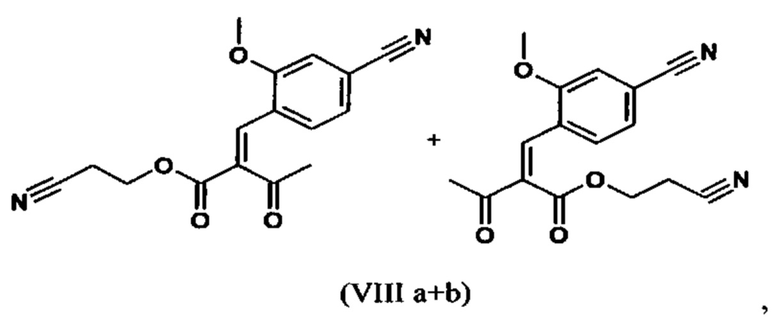
и соединение (VIIIa+b) реагирует с 4-амино-5-метилпиридоном с получением соединения (X)

и что соединение формулы (X)
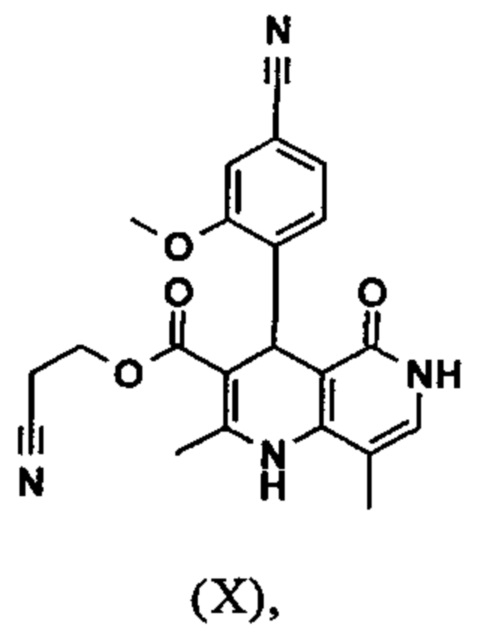
вводят в реакцию при перемешивании с 2.5-5 экв. триэтилортоацетата в диметилацетамиде при 100-120°С в течение 1.5-3 часов с получением соединения формулы (XI)
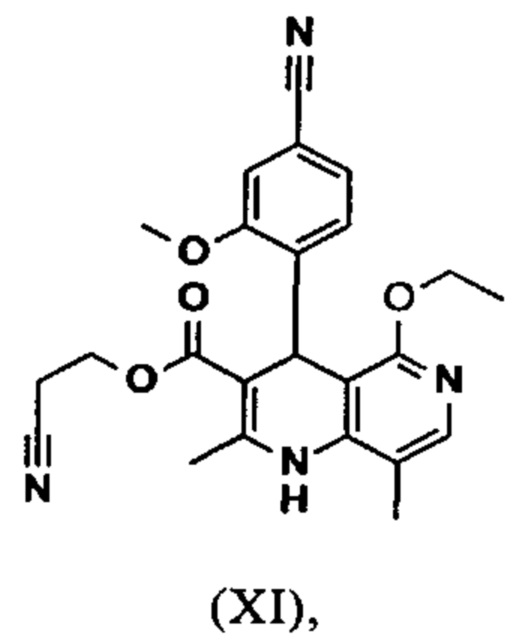
и что соединение формулы (XI) омыляют в 9-кратном количестве смеси ТГФ/вода, имеющей соотношение 2:1, водным раствором гидроксида натрия с получением соединения формулы (XII)
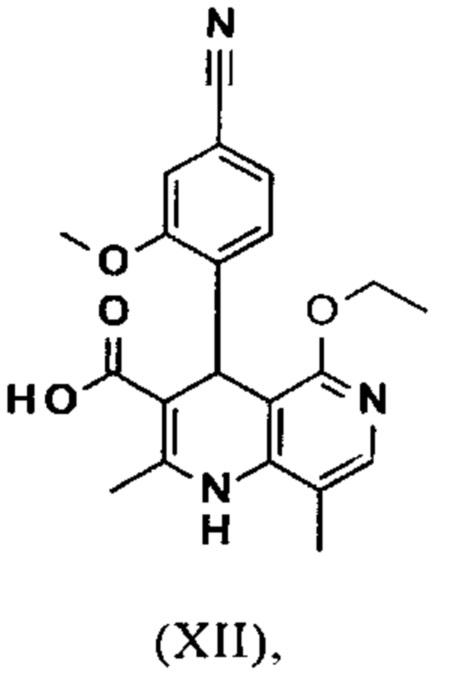
и что соединение формулы (XII) вводят в реакцию, которую проводят в одном реакционном сосуде без выделения промежуточных соединений, в ТГФ вначале с карбодиимидазолом и каталитическими количествами 4-(диметиламино)пиридина, на второй стадии нагревают с обратным холодильником вместе с гексаметилдисилазаном в течение 16-24 часов, и на третьей стадии гидролизуют в воде с использованием ТГФ или воды с получением соединения формулы (XIII)
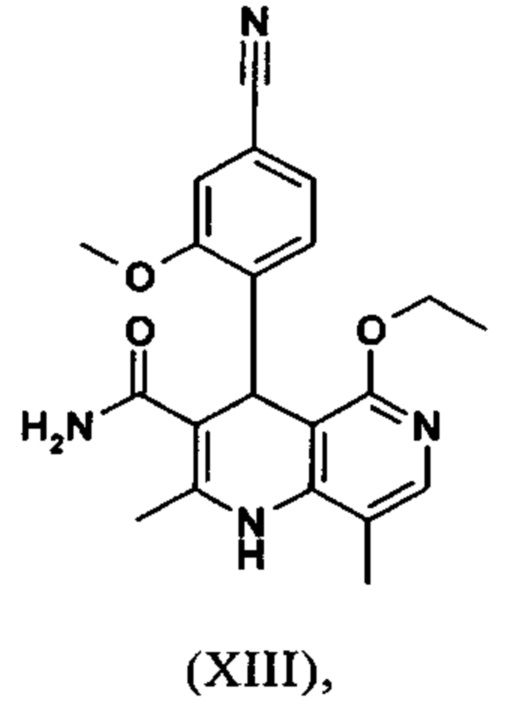
которое используют на стадии хиральной хроматографии для получения соединения формулы (I) в виде раствора в инертном растворителе.
2. Способ получения соединения формулы (I) в кристаллической форме полиморфа I, который отличается тем, что соединение формулы (I), полученное способом по п. 1, которое присутствует в виде раствора в инертном растворителе, перемешивают при температуре 20-120°С, и соединение формулы (I) выделяют в виде кристаллического полиморфа I, которое характеризуется рентгеновской дифрактограммой, измеренной с использованием источника излучения Cu-K Альфа 1, которая показывает максимальные значения пиков угла 2 тета на 8.5, 14.1, 17.2, 19.0, 20.5, 25.6, 26.5.
3. Способ получения лекарственного средства, включающий получение соединения формулы (I) в кристаллической форме полиморфа I согласно стадиям способа по п. 2 и смешивание полученного соединения с одним или несколькими инертными, нетоксичными, фармацевтически приемлемыми вспомогательными веществами.
| Станок для изготовления деревянных ниточных катушек из цилиндрических, снабженных осевым отверстием, заготовок | 1923 |
|
SU2008A1 |
| Mino R | |||
| Caira: "Crystalline polymorphism of organic compounds", Topics in Current Chemistry, 198, стр.163-208, 1998 | |||
| Способ получения производных 1,6-нафтиридина в виде рацематов или энантиомеров,или их кислотно-аддитивных солей | 1985 |
|
SU1395143A3 |

