Область изобретения
Настоящее изобретение относится к соединениям, которые воздействуют на холинергические рецепторы, особенно, мускариновые рецепторы. Настоящее изобретение предлагает соединения, которые являются агонистами холинергических рецепторов, включая мускариновые рецепторы, особенно подтипы m1 и m4 мускариновых рецепторов. Изобретение предлагает также способы использования предложенных соединений для модуляции состояний, связанных с холинергическими рецепторами, особенно для лечения или ослабления симптомов болезненных состояний, связанных с мускариновыми рецепторами, например подтипами m1 или m4 рецепторов.
Предпосылки создания изобретения
Мускариновые холинергические рецепторы опосредуют действия нейротрансмиттера ацетилхолина в центральной и периферической нервных системах, желудочно-кишечной системе, сердце, железах внутренней секреции, легких и других тканях. Мускариновые рецепторы играют центральную роль в центральной нервной системе в отношении высших познавательных функций, а также в периферической парасимпатической нервной системе. Идентифицировано пять различных подтипов мускариновых рецепторов, m1-m5. Подтип m1 является преобладающим подтипом, обнаруженным в коре головного мозга, и полагают, что этот подтип принимает участие в регулировании познавательных функций; m2 является преобладающим подтипом, обнаруженным в сердце, и полагают, что этот подтип принимает участие в регулировании частоты сердечных сокращений; m3, как полагают, принимает участие в стимуляции желудочно-кишечного тракта и мочевых путей, а также в потоотделении и слюноотделении; m4 присутствует в головном мозге и может принимать участие в способности к передвижению, и m5 присутствует в головном мозге и может принимать участие в некоторых функциях центральной нервной системы, связанных с допаминергической системой.
Состояния, связанные с познавательной недостаточностью, такие как болезнь Альцгеймера, сопровождаются снижением (потерей) уровня ацетилхолина в головном мозге. Считается, что это является результатом дегенерации холинергических нейронов в базальном переднем мозге, которые иннервируют участки ассоциативной зоны коры головного мозга и гиппокампа, который принимает участие в высших процессах.
Попытки повысить уровни ацетилхолина были сфокусированы на повышении уровней холина, предшественника синтеза ацетилхолина, и на блокировании ацетилхолинэстеразы (AChE), фермента, который метаболизирует ацетилхолин. Введение холина или фосфатидилхолина было не очень успешным. Ингибиторы AChE проявляли некоторую терапевтическую эффективность, но могли вызвать холинергические побочные эффекты вследствие стимуляции периферического ацетилхолина, включая абдоминальные спазмы, тошноту, рвоту, диарею, анорексию, потерю массы, миопатию и депрессию. Желудочно-кишечные побочные эффекты наблюдали почти у трети пациентов, подвергаемых лечению. Кроме того, некоторые ингибиторы AChE, такие как такрин, как было также обнаружено, вызывают значительную гепатотоксичность с повышением уровня трансаминаз печени, наблюдаемым приблизительно у 30% пациентов. Побочные действия ингибиторов AChE ограничивают их клиническую эффективность.
Обнаружено, что известные мускариновые агонисты m1, такие как ареколин, являются слабыми агонистами подтипа m2, а также m3, и не являются очень эффективными при лечении познавательной недостаточности, наиболее вероятно, вследствие ограничивающих дозу побочных действий.
Существует потребность в соединениях, которые повышают передачу сигнала ацетилхолином или действуют в головном мозге. Конкретно, существует потребность в мускариновых агонистах, которые являются активными в отношении различных подтипов мускариновых рецепторов в центральной и периферической нервной системе. Кроме того, существует потребность в более высокоселективных мускариновых агонистах, таких как m1- или m4-селективные агенты, как в качестве фармакологических средств, так и в качестве терапевтических агентов.
Краткое изложение сущности изобретения
Настоящее изобретение относится к соединениям, которые воздействуют на холинергические, особенно, мускариновые рецепторы, которые обладают агонистической активностью у подтипов m1 или m4 мускариновых рецепторов или у обоих. Соединения изобретения являются соединениями общей формулы (I):
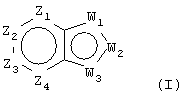
где:
Z1 представляет CR1 или N, Z2 представляет CR2 или N, Z3 представляет CR3 или N и Z4 представляет CR4 или N, где не более чем два из Z1, Z2, Z3 и Z4 являются N;
W1 представляет О, S или NR5, один из W2 и W3 представляет N или CR6 и другой из W2 и W3 представляет CG; W1 представляет NG, W2 представляет CR5 или N и W3 представляет CR6 или N; или W1 и W3 представляют N и W2 представляет NG;
G представляет собой формулу (II):
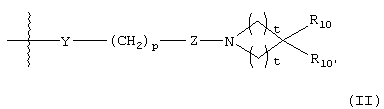
Y представляет О, S, CHOH, -NHC(O)-, -C(O)NH-, -C(O)-, -OC(O)-, -(O)CO-, -NR7-, -CH=N- или отсутствует;
р равно 1, 2, 3, 4 или 5;
Z представляет CR8R9 или отсутствует;
каждый t равно 1, 2 или 3;
каждый R1, R2, R3 и R4, независимо, представляет Н, амино, гидроксил, галоген или неразветвленный или разветвленный С1-6-алкил, С2-6-алкенил, С2-6-алкинил, С1-6-гетероалкил, С1-6-галогеналкил, -CN, -CF3, -OR11, -COR11, -NO2, -SR11, -NHC(O)R11, -C(O)NR12R13, -NR12R13, -NR11C(O)NR12R13, -SO2NR12R13, -OC(O)R11, -O(CH2)qNR12R13 или -(CH2)qNR12R13, где q равно целому числу от 2 до 6, или R1 и R2 вместе образуют -NH-N=N- или R3 и R4 вместе образуют -NH-N=N-;
каждый из R5, R6 и R7, независимо, представляет Н, С1-6-алкил; формил; С3-6-циклоалкил; С5-6-арил, необязательно замещенный галогеном или С1-6-алкилом; или С5-6-гетероарил, необязательно замещенный галогеном или С1-6-алкилом;
каждый из R8 и R9, независимо, представляет Н или С1-8-алкил с неразветвленной или разветвленной цепью;
R10 представляет неразветвленный или разветвленный С1-8-алкил, С2-8-алкенил, С2-8-алкинил, С1-8-алкилиден, С1-8-алкокси, С1-8-гетероалкил, С1-8-аминоалкил, С1-8-галогеналкил, С1-8-алкоксикарбонил, С1-8-гидроксиалкокси, С1-8-гидроксиалкил, -SH, С1-8-алкилтио, -О-СН2-С5-6-арил, -С(О)-С5-6-арил (замещенный С1-3-алкилом или галогеном), С5-6-арил, С5-6-циклоалкил, С5-6-гетероарил, С5-6-гетероциклоалкил, -NR12R13, -C(O)NR12R13, -NR11C(O)NR12R13, -CR11R12R13, -OC(O)R11, -(O)(CH2)sNR12R13 или -(CH2)sNR12R13, причем s равно целому числу от 2 до 8;
R'10 представляет Н, неразветвленный или разветвленный С1-8-алкил, С2-8-алкенил, С2-8-алкинил, С1-8-алкилиден, С1-8-алкокси, С1-8-гетероалкил, С1-8-аминоалкил, С1-8-галогеналкил, С1-8-алкоксикарбонил, С1-8-гидроксиалкокси, С1-8-гидроксиалкил или С1-8-алкилтио;
каждый R11, независимо, представляет Н, неразветвленный или разветвленный С1-8-алкил, С2-8-алкенил, С2-8-алкинил, С2-8-гетероалкил, С2-8-аминоалкил, С2-8-галогеналкил, С1-8-алкоксикарбонил, С2-8-гидроксиалкил, -С(О)-С5-6-арил (замещенный С1-3-алкилом или галогеном), С5-6-арил, С5-6-гетероарил, С5-6-циклоалкил, С5-6-гетероциклоалкил, -C(O)NR12R13, -CR5R12R13, -(CH2)tNR12R13, причем t равно целому числу от 2 до 8; и
каждый R12 и R13, независимо, представляет Н, С1-6-алкил; С3-6-циклоалкил; С5-6-арил, необязательно замещенный галогеном или С1-6-алкилом, или С5-6-гетероарил, необязательно замещенный галогеном или С1-6-алкилом, или R12 и R13 вместе образуют циклическую структуру;
или их фармацевтически приемлемыми солями, эфирами и пролекарствами.
Настоящее изобретение далее относится к фармацевтическим композициям, включающим эффективное количество соединения формулы (I) или его фармацевтически приемлемых солей, эфиров или пролекарств.
Изобретение относится также к способам повышения активности холинергического рецептора, включающим контактирование холинергического рецептора или системы, содержащей холинергический рецептор, с эффективным количеством соединения формулы (I), а также наборам для осуществления способа. Рецептор, предпочтительно, является мускариновым рецептором подтипа m1 или m4. Рецептор может быть расположен в центральной нервной системе, периферической нервной системе, желудочно-кишечной системе, сердце, железах внутренней секреции или легких, и рецептор может быть усеченным, мутированным или модифицированным холинергическим рецептором.
Кроме того, настоящее изобретение относится к способу активации холинергического рецептора, включающему контактирование холинергического рецептора или системы, содержащей холинергический рецептор, с эффективным количеством, по меньшей мере, одного соединения формулы (I), а также к наборам для проведения способа. В предпочтительном осуществлении соединение является селективным в отношении подтипа m1 или m4 мускаринового рецептора или для обоих подтипов. В другом предпочтительном осуществлении соединение оказывает слабое действие или не оказывает его на активность m2 или m3.
Другой аспект настоящего изобретения относится к способу лечения болезненного состояния, связанного с холинергическим рецептором, включающий введение субъекту, нуждающемуся в таком лечении, эффективного количества, по меньшей мере, одного из соединений изобретения. Изобретение относится также к набору для проведения способа. Болезненные состояния, которые лечат, включают, но не ограничиваются перечисленным, состояния дисфункции познавательной способности, забывчивость, спутанность сознания, потерю памяти, дефицит внимания, дефицит визуального восприятия, депрессию, боль, нарушения сна и психоз. Болезненные состояния включают также, но не ограничиваются перечисленным, болезнь Альцгеймера, болезнь Паркинсона, хорею Хантингтона, атаксию Фридериха, синдром Жиля де ла Туретта, синдром Дауна, болезнь Пика, деменцию pugilistica, клиническую депрессию, возрастное отклонение в познавательной способности, нарушение, связанное с дефицитом внимания, и синдром внезапной смерти внешне здоровых младенцев.
Изобретение далее относится к способам лечения симптомов заболевания или состояния, связанного с пониженными уровнями ацетилхолина, включающим введение эффективного количества, по меньшей мере, одного соединения изобретения.
В другом воплощении настоящее изобретение относится к способу лечения болезни Альцгеймера. Способ включает введение субъекту, нуждающемуся в таком лечении, эффективного количества, по меньшей мере, одного соединения изобретения.
Еще в одном воплощении настоящее изобретение относится к способу лечения глаукомы. Способ включает введение эффективного количества, по меньшей мере, одного соединения изобретения.
Другим аспектом настоящего изобретения является способ идентификации предрасположенности к генетическому полиморфизму субъекта, который восприимчив к соединению изобретения. Способ включает введение субъекту терапевтически эффективного количества соединения; измерение ответной реакции указанного субъекта на соединение (посредством чего идентифицируют восприимчивого субъекта, имеющего ослабленное (клинически не проявляющееся) болезненное состояние, связанное с холинергическим рецептором), и идентификацию генетического полиморфизма у восприимчивого субъекта, где указанный генетический полиморфизм приводит к предрасположенности субъекта к восприимчивости к соединению.
Настоящее изобретение относится также к способу идентификации субъекта, подходящего для лечения соединением изобретения, и наборам для такой идентификации. Способ включает обнаружение присутствия полиморфизма у субъекта, где полиморфизм приводит к предрасположенности указанного субъекта к восприимчивости к соединению и где присутствие полиморфизма указывает на то, что субъект подходит для лечения соединением.
Подробное описание изобретения
Определения
Для целей настоящего описания следующие определения во всей их полноте будут использованы для определения технических терминов и для определения объема химических соединений, защиту которых определяют в формуле изобретения.
Термин "рецептор" включает любую молекулу, присутствующую внутри или на поверхности клетки, которая может воздействовать на клеточную физиологию, когда его ингибируют или стимулируют лигандом. В типичном случае рецептор включает внеклеточный домен со связывающим лиганд свойствами, трансмембранный домен, который закрепляет рецептор в клеточной мембране, и цитоплазматический домен, который генерирует клеточный сигнал в ответ на связывание лиганда ("сигнальная трансдукция"). Рецептор включает также любую молекулу, имеющую характерную структуру рецептора, но без идентифицируемого лиганда. Кроме того, рецептор включает усеченный, модифицированный, мутированный рецептор или любую молекулу, включающую частично или все последовательности рецептора.
"Лиганд" означает любое вещество, которое взаимодействует с рецептором.
"Агонист" определяют как соединение, которое повышает активность рецептора при взаимодействии его с рецептором.
"Рецептор m1" определяют как рецептор, имеющий активность, соответствующую активности подтипа m1 мускаринового рецептора, характеризующейся посредством молекулярного клонирования и фармакологии.
"Селективный" или "селективность" определяют как способность соединения вызывать нужную реакцию определенного типа, подтипа, класса или подкласса рецептора при меньшей или незначительной реакции со стороны других типов рецептора. "Селективный" или "селективность" соединения, являющегося мускариновым рецептором m1 или m4, означает способность соединения повышать активность мускаринового рецептора m1 или m4, соответственно, вызывая незначительную активность или не вызывая активность других подтипов, включая подтипы m3 и m5, предпочтительно, подтипа m2. Соединения настоящего изобретения могут также проявлять селективность по отношению к рецепторам как m1, так и m4, т.е. повышать активность мускариновых рецепторов как m1, так и m4, при незначительной активности или отсутствии активности других подтипов, включая подтипы m3 и m5, предпочтительно, подтипа m2.
Термин "субъект" относится к животному, предпочтительно, млекопитающему или человеку, который является объектом лечения, наблюдения или эксперимента.
Используемый здесь термин "совместное введение" фармакологически активных соединений относится к доставке двух или более отдельных химических агентов, независимо от того, вводят ли их in vitro или in vivo. Совместное введение означает одновременную доставку отдельных агентов, одновременную доставку смеси агентов; а также доставку одного агента после доставки второго агента или дополнительных агентов. Агенты, которые вводят совместно, предназначаются в типичном случае для действия в сочетании друг с другом.
Термин "эффективное количество", используемый здесь, означает количество активного соединения или фармацевтического агента, который вызывает биологическую или медицинскую реакцию в ткани, системе, организме животного или человека, которое определяется исследователем, ветеринаром, врачом или другим клиницистом, и которое включает облегчение или ослабление симптомов заболевания, подвергаемого лечению.
"Алкил" означает группу алкана с неразветвленной или разветвленной цепью с 1-6 атомами углерода в цепи, например, метил, этил, пропил, изопропил, н-бутил, втор-бутил, трет-бутил и т.д. Термин "гетероалкил" предназначен для обозначения группы алкана, содержащей 1 или 2 гетероатома, выбранных из О, S или N.
"Алкенил" означает группу алкена с неразветвленной или разветвленной цепью с 2-6 атомами углерода в цепи, термин "алкинил" предназначен для обозначения группы алкина с неразветвленной или разветвленной цепью с 2-6 атомами углерода в цепи.
Термины "арил" и "циклоалкил", предпочтительно, относится к структурам моно- или бициклических колец, включающих 5-12 атомов углерода, более предпочтительно, моноциклическим кольцам, включающим 5 или 6 атомов углерода. Когда такие кольца включают один или несколько гетероатомов, выбранных из N, S и О (т.е. гетероциклические или гетероарильные кольца), такие кольца включают всего от 5 до 12 атомов, более предпочтительно, 5 или 6 атомов. Гетероциклические кольца включают, но не ограничиваются перечисленным, фурил, пирролил, пиразолил, тиенил, имидазолил, индолил, бензофуранил, бензотиофенил, индазолил, бензоимидазолил, бензотиазолил, изоксазолил, оксазолил, тиазолил, изотиазолил, пиридил, пиперидинил, пиперазинил, пиридазинил, пиримидинил, пиразинил, морфолинил, оксадиазолил, тиадиазолил, имидазолинил, имидазолидинил и тому подобное. Кольцо может быть замещено одной или несколькими группами, включенными в приведенное выше определение R2. Понятно, что заместители С1-6-алкил, С2-6-алкенил, С2-6-алкинил, С1-6-алкокси, С1-6-гетероалкил, С1-6-аминоалкил, С1-6-галогеналкил или С1-6-алкоксикарбонил, если они присутствуют, могут быть замещены одним или несколькими гидроксилами, С1-4-алкокси, галогенами, циано, амино или нитро.
Используемый здесь термин "галоген" включает хлор, фтор, которые являются предпочтительными, и иод и бром.
Настоящее изобретение относится к соединениям, которые являются агонистами холинергических рецепторов, включая мускариновые рецепторы. Настоящее изобретение, особенно, относится к соединениям, которые являются селективными для подтипа m1 или m4 мускаринового рецептора или для обоих типов. Соединения, предложенные настоящим изобретением, обладают терапевтическим действием и их можно использовать для лечения болезненных состояний, ассоциированных с холинергическими рецепторами, например, недостаточности познавательной способности при болезни Альцгеймера, глаукомы, боли или шизофрении.
В соответствии с одним воплощением, настоящее изобретение относится к соединениям формулы I:
где:
Z1 представляет CR1 или N, Z2 представляет CR2 или N, Z3 представляет CR3 или N и Z4 представляет CR4 или N, но не более чем два из Z1, Z2, Z3 и Z4 являются N;
W1 представляет О, S или NR5, один из W2 и W3 представляет N или CR6 и другой из W2 и W3 представляет CG; W1 представляет NG, W2 представляет CR5 или N и W3 представляет CR6 или N; или W1 представляет N, W2 представляют NG и W3 представляет N;
G представляет собой формулу (II):
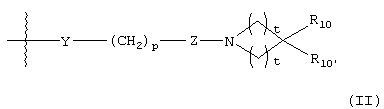
Y представляет О, S, CHOH, -NHC(O)-, -C(O)NH-, -C(O)-, -OC(O)-, -(O)CO-, -NR7-, -CH=N- или отсутствует;
р равно 1, 2, 3, 4 или 5;
Z представляет CR8R9 или отсутствует;
каждый t равен 1, 2 или 3;
каждый R1, R2, R3 и R4, независимо, представляет Н, амино, гидроксил, галоген или неразветвленный или разветвленный С1-6-алкил, С2-6-алкенил, С2-6-алкинил, С1-6-гетероалкил, С1-6-галогеналкил, -CN, -CF3, -OR11, COR11, -NO2, -SR11, -NHC(O)R11, -C(O)NR12R13, -NR12R13, -NR11C(O)NR12R13, -SO2NR12R13, -OC(O)R11, -O(CH2)qNR12R13 или -(CH2)qNR12R13, где q равно целому числу от 2 до 6, или R1 и R2 вместе образуют -NH-N=N- или R3 и R4 вместе образуют -NH-N=N-;
каждый из R5, R6 и R7, независимо, представляет Н, С1-6-алкил; формил; С3-6-циклоалкил; С5-6-арил, необязательно замещенный галогеном или С1-6-алкилом; или С5-6-гетероарил, необязательно замещенный галогеном или С1-6-алкилом;
каждый из R8 и R9, независимо, представляет Н или С1-8-алкил с неразветвленной или разветвленной цепью;
R10 представляет неразветвленный или разветвленный С1-8-алкил, С2-8-алкенил, С2-8-алкинил, С1-8-алкилиден, С1-8-алкокси, С1-8-гетероалкил, С1-8-аминоалкил, С1-8-галогеналкил, С1-8-алкоксикарбонил, С1-8-гидроксиалкокси, С1-8-гидроксиалкил, -SH, С1-8-алкилтио, -О-СН2-С5-6-арил, -С(О)-С5-6-арил (замещенный С1-3-алкилом или галогеном), С5-6-арил, С5-6-циклоалкил, С5-6-гетероарил, С5-6-гетероциклоалкил, -NR12R13, -C(O)NR12R13, -NR11C(O)NR12R13, -CR11R12R13, -OC(O)R11, -(O)(CH2)sNR12R13 или -(CH2)sNR12R13, причем s равно целому числу от 2 до 8;
R'10 представляет Н, неразветвленный или разветвленный С1-8-алкил, С2-8-алкенил, С2-8-алкинил, С1-8-алкилиден, С1-8-алкокси, С1-8-гетероалкил, С1-8-аминоалкил, С1-8-галогеналкил, С1-8-алкоксикарбонил, С1-8-гидроксиалкокси, С1-8-гидроксиалкил или С1-8-алкилтио;
каждый R11, независимо, представляет H, неразветвленный или разветвленный С1-8-алкил, С2-8-алкенил, С2-8-алкинил, С2-8-гетероалкил, С2-8-аминоалкил, С2-8-галогеналкил, С1-8-алкоксикарбонил, С2-8 -гидроксиалкил, -С(О)-С5-6 -арил (замещенный С1-3-алкилом или галогеном), С5-6-арил, С5-6-гетероарил, С5-6-циклоалкил, С5-6-гетероциклоалкил, -C(O)NR12R13, -CR5R12R13, (CH2)tNR12R13, причем t равно целому числу от 2 до 8; и
каждый R12 и R13, независимо, представляет Н, С1-6-алкил; С3-6-циклоалкил; С5-6-арил, необязательно замещенный галогеном или С1-6-алкилом, или С5-6-гетероарил, необязательно замещенный галогеном или С1-6-алкилом, или R12 и R13 вместе образуют циклическую структуру;
или их фармацевтически приемлемым солям, эфирам и пролекарствам.
В соответствии с предпочтительным воплощением, t равно 2 и R'10 представляет Н.
В соответствии с предпочтительным рядом воплощений, Y представляет -С(О)-, -NHC(O)-, S, O, -OC(O)- или отсутствует. В другом R10 представляет алкил и Z1 представляет CR1 или N, Z2 представляет CR2, Z3 представляет CR3 или N и Z4 представляет CR4. В одном воплощении р равно 2. В другом R5 представляет Н или С1-6-алкил.
В одном предпочтительном воплощении каждый из R1, R2, R3 и R4, независимо, представляет Н, галоген, -NO2 или неразветвленный или разветвленный С1-6-алкил или R1 и R2 вместе образуют -NH-N=N- или R3 и R4 вместе образуют -NH-N=N-.
Конкретные соединения изобретения включают:
3-[3-(4-метоксипиперидин)-1-илпропил]-1Н-индол;
3-[3-(4-этоксипиперидин)-1-илпропил]-1Н-индол;
3-[3-(4-пропоксипиперидин)-1-илпропил]-1Н-индол;
3-[3-(4-бутоксипиперидин)-1-илпропил]-1Н-индол;
3-[3-(4-метоксиметилпиперидин)-1-илпропил]-1Н-индол;
3-[3-(4-этоксиметилпиперидин)-1-илпропил]-1Н-индол;
3-[3-(4-пропоксиметилпиперидин)-1-илпропил]-1Н-индол;
3-[3-(4-метилпиперидин)-1-илпропил]-1Н-индол;
3-[3-(4-этилпиперидин)-1-илпропил]-1Н-индол;
3-[3-(4-н-пропилпиперидин)-1-илпропил]-1Н-индол;
3-[3-(4-н-бутилпиперидин)-1-илпропил]-1Н-индол;
3-[2-(4-метоксипиперидин)-1-илэтил]-1Н-индол;
3-[2-(4-этоксипиперидин)-1-илэтил]-1Н-индол;
3-[2-(4-пропоксипиперидин)-1-илэтил]-1Н-индол;
3-[2-(4-бутоксипиперидин)-1-илэтил]-1Н-индол;
3-[2-(4-метоксиметилпиперидин)-1-илэтил]-1Н-индол;
3-[2-(4-этоксиметилпиперидин)-1-илэтил]-1Н-индол;
3-[2-(4-пропоксиметилпиперидин)-1-илэтил]-1Н-индол;
3-[2-(4-метилпиперидин)-1-илэтил]-1Н-индол;
3-[2-(4-этилпиперидин)-1-илэтил]-1Н-индол;
3-[2-(4-н-пропилпиперидин)-1-илэтил]-1Н-индол;
3-[2-(4-н-бутилпиперидин)-1-илэтил]-1Н-индол;
3-[2-(4-метоксипиперидин)-1-илэтил]бензо[d]изоксазол;
3-[2-(4-бутоксипиперидин)-1-илэтил]бензо[d]изоксазол;
3-[2-(4-метоксипиперидин)-1-илпропил]бензо[d]изоксазол;
3-[3-(4-бутоксипиперидин)-1-илпропил]бензо[d]изоксазол;
3-[4-(4-метоксипиперидин)-1-илбутил]бензо[d]изоксазол;
3-[4-(4-бутоксипиперидин)-1-илбутил]бензо[d]изоксазол;
1-[3-(4-метоксипиперидин)-1-илпропил]-1Н-индол;
1-[3-(4-этоксипиперидин)-1-илпропил]-1Н-индол;
1-[3-(4-пропоксипиперидин)-1-илпропил]-1Н-индол;
1-[3-(4-бутоксипиперидин)-1-илпропил]-1Н-индол;
1-[3-(4-метоксиметилпиперидин)-1-илпропил]-1Н-индол;
1-[3-(4-этоксиметилпиперидин)-1-илпропил]-1Н-индол;
1-[3-(4-пропоксиметилпиперидин)-1-илпропил]-1Н-индол;
1-[3-(4-метилпиперидин)-1-илпропил]-1Н-индол;
1-[3-(4-этилпиперидин)-1-илпропил]-1Н-индол;
1-[3-(4-н-пропилпиперидин)-1-илпропил]-1Н-индол;
1-[3-(4-н-бутилпиперидин)-1-илпропил]-1Н-индол;
1-[2-(4-метоксипиперидин)-1-илэтил]-1Н-индол;
1-[2-(4-этоксипиперидин)-1-илэтил]-1Н-индол;
1-[2-(4-пропоксипиперидин)-1-илэтил]-1Н-индол;
1-[2-(4-бутоксипиперидин)-1-илэтил]-1Н-индол;
1-[2-(4-метоксиметилпиперидин)-1-илэтил]-1Н-индол;
1-[2-(4-этоксиметилпиперидин)-1-илэтил]-1Н-индол;
1-[2-(4-пропоксиметилпиперидин)-1-илэтил]-1Н-индол;
1-[2-(4-метилпиперидин)-1-илэтил]-1Н-индол;
1-[2-(4-этилпиперидин)-1-илэтил]-1Н-индол;
1-[2-(4-н-пропилпиперидин)-1-илэтил]-1Н-индол;
1-[2-(4-н-бутилпиперидин)-1-илэтил]-1Н-индол;
1-[3-(4-метоксипиперидин)-1-илпропил]-1Н-бензотриазол;
1-[3-(4-этоксипиперидин)-1-илпропил]-1Н-бензотриазол;
1-[3-(4-пропоксипиперидин)-1-илпропил]-1Н-бензотриазол;
1-[3-(4-бутоксипиперидин)-1-илпропил]-1Н-бензотриазол;
1-[3-(4-метоксиметилпиперидин)-1-илпропил]-1Н-бензотриазол;
1-[3-(4-этоксиметилпиперидин)-1-илпропил]-1Н-бензотриазол;
1-[3-(4-пропоксиметилпиперидин)-1-илпропил]-1Н-бензотриазол;
1-[3-(4-метилпиперидин)-1-илпропил]-1Н-бензотриазол;
1-[3-(4-этилпиперидин)-1-илпропил]-1Н-бензотриазол;
1-[3-(4-н-пропилпиперидин)-1-илпропил]-1Н-бензотриазол;
1-[3-(4-н-бутилпиперидин)-1-илпропил]-1Н-бензотриазол;
1-[2-(4-метоксипиперидин)-1-илэтил]-1Н-бензотриазол;
1-[2-(4-этоксипиперидин)-1-илэтил]-1Н-бензотриазол;
1-[2-(4-пропоксипиперидин)-1-илэтил]-1Н-бензотриазол;
1-[2-(4-бутоксипиперидин)-1-илэтил]-1Н-бензотриазол;
1-[2-(4-метоксиметилпиперидин)-1-илэтил]-1Н-бензотриазол;
1-[2-(4-этоксиметилпиперидин)-1-илэтил]-1Н-бензотриазол;
1-[2-(4-пропоксиметилпиперидин)-1-илэтил]-1Н-бензотриазол;
1-[2-(4-метилпиперидин)-1-илэтил]-1Н-бензотриазол;
1-[2-(4-этилпиперидин)-1-илэтил]-1Н-бензотриазол;
1-[2-(4-н-пропилпиперидин)-1-илэтил]-1Н-бензотриазол;
1-[2-(4-н-бутилпиперидин)-1-илэтил]-1Н-бензотриазол;
1-[4-(4-метоксипиперидин)-1-илбутил]-1Н-бензотриазол;
1-[4-(4-этоксипиперидин)-1-илбутил]-1Н-бензотриазол;
1-[4-(4-пропоксипиперидин)-1-илбутил]-1Н-бензотриазол;
1-[4-(4-бутоксипиперидин)-1-илбутил]-1Н-бензотриазол;
1-[4-(4-метоксиметилпиперидин)-1-илбутил]-1Н-бензотриазол;
1-[4-(4-этоксиметилпиперидин)-1-илбутил]-1Н-бензотриазол;
1-[4-(4-пропоксиметилпиперидин)-1-илбутил]-1Н-бензотриазол;
1-[4-(4-метилпиперидин)-1-илбутил]-1Н-бензотриазол;
1-[4-(4-этилпиперидин)-1-илбутил]-1Н-бензотриазол;
1-[4-(4-н-пропилпиперидин)-1-илбутил]-1Н-бензотриазол;
1-[4-(4-н-бутилпиперидин)-1-илбутил]-1Н-бензотриазол;
2-[4-(4-метилпиперидин)-1-илбутил]-1Н-бензотриазол;
2-[4-(4-этилпиперидин)-1-илбутил]-1Н-бензотриазол;
2-[4-(4-н-пропилпиперидин)-1-илбутил]-1Н-бензотриазол;
2-[4-(4-н-бутилпиперидин)-1-илбутил]-1Н-бензотриазол;
2-[3-(4-метилпиперидин)-1-илпропил]-1Н-бензоимидазол;
2-[3-(4-этилпиперидин)-1-илпропил]-1Н-бензоимидазол;
2-[3-(4-н-пропилпиперидин)-1-илпропил]-1Н-бензоимидазол;
2-[3-(4-н-бутилпиперидин)-1-илпропил]-1Н-бензоимидазол;
2-[2-(4-метилпиперидин)-1-илэтил]-1Н-бензоимидазол;
2-[2-(4-этилпиперидин)-1-илэтил]-1Н-бензоимидазол;
2-[2-(4-н-пропилпиперидин)-1-илэтил]-1Н-бензоимидазол;
2-[2-(4-н-бутилпиперидин)-1-илэтил]-1Н-бензоимидазол;
1-(1Н-бензоимидазол-2-ил)-4-(4-метилпиперидин)бутанон;
1-(1Н-бензоимидазол-2-ил)-4-(4-этилпиперидин)бутанон;
1-(1Н-бензоимидазол-2-ил)-4-(4-н-пропилпиперидин)бутанон;
1-(1Н-бензоимидазол-2-ил)-4-(4-н-бутилпиперидин)бутанон;
1-(1Н-бензоимидазол-2-ил)-3-(4-метилпиперидин)пропанон;
1-(1Н-бензоимидазол-2-ил)-3-(4-этилпиперидин)пропанон;
1-(1Н-бензоимидазол-2-ил)-3-(4-н-пропилпиперидин)пропанон;
1-(1Н-бензоимидазол-2-ил)-3-(4-н-бутилпиперидин)пропанон;
3-[3-(4-метилпиперидин)-1-илпропил]-1Н-индазол;
3-[3-(4-этилпиперидин)-1-илпропил]-1Н-индазол;
3-[3-(4-н-пропилпиперидин)-1-илпропил]-1Н-индазол;
3-[3-(4-н-бутилпиперидин)-1-илпропил]-1Н-индазол;
1-(3-бензофуран-3-илпропил)-4-метилпиперидин;
1-(3-бензофуран-3-илпропил)-4-этилпиперидин;
1-(3-бензофуран-3-илпропил)-4-н-пропилпиперидин;
1-(3-бензофуран-3-илпропил)-4-н-бутилпиперидин;
3-(3-(4-метилпиперидин)-1-илпропил)бензо[d]изотиазол;
3-(3-(4-этилпиперидин)-1-илпропил)бензо[d]изотиазол;
3-(3-(4-н-пропилпиперидин)-1-илпропил)бензо[d]изотиазол;
3-(3-(4-н-бутилпиперидин)-1-илпропил)бензо[d]изотиазол;
1-[3-(4-метилпиперидин)-1-илпропил]-1Н-бензоимидазол;
1-[3-(4-этилпиперидин)-1-илпропил]-1Н-бензоимидазол;
1-[3-(4-н-пропилпиперидин)-1-илпропил]-1Н-бензоимидазол;
1-[3-(4-н-бутилпиперидин)-1-илпропил]-1Н-бензоимидазол;
1-[2-(4-метилпиперидин)-1-илэтил]-1Н-бензоимидазол;
1-[2-(4-этилпиперидин)-1-илэтил]-1Н-бензоимидазол;
1-[2-(4-н-пропилпиперидин)-1-илэтил]-1Н-бензоимидазол;
1-[2-(4-н-бутилпиперидин)-1-илэтил]-1Н-бензоимидазол;
1-[3-(4-метилпиперидин)-1-илпропил]-1Н-бензоимидазол;
1-[3-(4-этилпиперидин)-1-илпропил]-1Н-индазол;
1-[3-(4-н-пропилпиперидин)-1-илпропил]-1Н-индазол;
1-[3-(4-н-бутилпиперидин)-1-илпропил]-1Н-индазол;
2-[4-(4-метилпиперидин)-1-илбутил]-1Н-бензотиазол;
2-[4-(4-этилпиперидин)-1-илбутил]-1Н-бензотиазол;
2-[4-(4-н-пропилпиперидин)-1-илбутил]-1Н-бензотиазол;
2-[4-(4-н-бутилпиперидин)-1-илбутил]-1Н-бензотиазол;
2-[3-(4-метилпиперидин)-1-илпропил]-1Н-бензотиазол;
2-[3-(4-этилпиперидин)-1-илпропил]-1Н-бензотиазол;
2-[3-(4-н-пропилпиперидин)-1-илпропил]-1Н-бензотиазол;
2-[3-(4-н-бутилпиперидин)-1-илпропил]-1Н-бензотиазол;
2-[2-(4-метилпиперидин)-1-илэтил]-1Н-бензотиазол;
2-[2-(4-этилпиперидин)-1-илэтил]-1Н-бензотиазол;
2-[2-(4-н-пропилпиперидин)-1-илэтил]-1Н-бензотиазол;
2-[2-(4-н-бутилпиперидин)-1-илэтил]-1Н-бензотиазол;
2-[3-(4-метилпиперидин)-1-илпропил]бензоксазол;
2-[3-(4-этилпиперидин)-1-илпропил]бензоксазол;
2-[3-(4-н-пропилпиперидин)-1-илпропил]бензоксазол;
2-[3-(4-н-бутилпиперидин)-1-илпропил]бензоксазол;
2-[2-(4-метилпиперидин)-1-илэтил]бензоксазол;
2-[2-(4-этилпиперидин)-1-илэтил]бензоксазол;
2-[2-(4-н-пропилпиперидин)-1-илэтил]бензоксазол;
2-[2-(4-н-бутилпиперидин)-1-илэтил]бензоксазол;
2-[4-(4-метилпиперидин)-1-илбутил]бензоксазол;
2-[4-(4-этилпиперидин)-1-илбутил]бензоксазол;
2-[4-(4-н-пропилпиперидин)-1-илбутил]бензоксазол;
2-[4-(4-н-бутилпиперидин)-1-илбутил]бензоксазол;
4,5-дифтор-2-(3-(4-н-бутилпиперидин-1-ил)пропил)-1Н-бензоимидазол;
6-фтор-5-нитро-2-(3-(4-н-бутилпиперидин-1-ил)пропил)-1Н-бензоимидазол;
5-трет-бутил-2-(3-(4-н-бутилпиперидин-1-ил)пропил)-1Н-бензоимидазол;
5-хлор-6-метил-2-(3-(4-н-бутилпиперидин-1-ил)пропил)-1Н-бензоимидазол;
4,6-дифтор-2-(3-(4-н-бутилпиперидин-1-ил)пропил)-1Н-бензоимидазол;
2-(3-(4-н-бутилпиперидин-1-ил)пропил)-1Н-имидазо[4,5-c]пиридин;
8-(3-(4-н-бутилпиперидин)-1-илпропил)-9Н-пурин;
7-(3-(4-н-бутилпиперидин)-1-илпропил)-3,8-дигидроимидазо[4',5':3,4]бензо[1,2-d][1,2,3]триазол;
2-(3-(4-н-бутилпиперидин-1-ил)пропил)-3а,4,5,6,7,7а-гексагидро-1Н-бензоимидазол;
3-метил-1-(3-(4-н-бутилпиперидин)-1-илпропил)-1Н-индол;
5-бром-1-(3-(4-н-бутилпиперидин)-1-илпропил)-1Н-индол;
3-формил-1-(3-(4-н-бутилпиперидин)-1-илпропил)-1Н-индол;
7-бром-1-(3-(4-н-бутилпиперидин)-1-илпропил)-1Н-индол;
3-(3-(4-н-бутилпиперидин)-1-илпропил)бензо[d]изоксазол;
4-нитро-2-(3-(4-н-бутилпиперидин)-1-илпропил)-1Н-бензоимидазол;
5-нитро-2-(3-(4-н-бутилпиперидин)-1-илпропил)-1Н-бензоимидазол;
4-гидрокси-2-(3-(4-н-бутилпиперидин)-1-илпропил)-1Н-бензоимидазол;
4-метил-2-(3-(4-н-бутилпиперидин)-1-илпропил)-1Н-бензоимидазол;
3-(2-(4-н-бутилпиперидин)этокси)-7-метилбензо[d]изоксазол;
1-(3-(4-метилпиперидин)-1-илпропил)-1Н-индазол;
1-(3-(4-пентилпиперидин)-1-илпропил)-1Н-индазол;
1-(3-(4-пропилпиперидин)-1-илпропил)-1Н-;
1-(3-(4-(3-метилбутил)пиперидин)-1-илпропил)-1Н-индазол;
1-(3-(4-пентилиденпиперидин)-1-илпропил)-1Н-индазол;
1-(3-(4-пропилиденпиперидин)-1-илпропил)-1Н-индазол;
1-бензо[b]тиофен-2-ил-4-(4-бутилпиперидин-1-ил)бутан-1-он;
4-(4-бутилпиперидин-1-ил)-1-(3-метилбензофуран-2-ил)бутан-1-он;
4-(4-бутилпиперидин-1-ил)-1-(5-фтор-3-метилбензо[b]тиофен-2-ил)бутан-1-он;
1-бензофуран-2-ил-4-(4-бутилпиперидин-1-ил)бутан-1-он;
1-(3-бромбензо[b]тиофен-2-ил)-4-(4-бутилпиперидин-1-ил)бутан-1-он;
1-(3-бензо[b]тиофен-2-илпропил)-4-бутилпиперидин;
1-(3-бензофуран-2-илпропил)-4-бутилпиперидин;
4-бутил-1-[3-(3-метилбензофуран-2-ил)пропил]пиперидин;
4-бутил-1-[3-(5-фтор-3-метилбензо[b]тиофен-2-ил)пропил] пиперидин;
2-(3-иодпропил)бензо[b]тиофен;
1-(3-бензо[b]тиофен-2-илпропил)-4-метилпиперидин;
1-(3-бензо[b]тиофен-2-илпропил)-4-бензилпиперидин;
1-(3-бензо[b]тиофен-2-илпропил)-4-(2-метоксифенил)пиперидин;
2-(3-бромпропил)-2Н-бензотриазол;
2-[3-(4-бутилпиперидин-1-ил)пропил]-2Н-бензотриазол;
1-(3-бромпропил)-1Н-бензотриазол;
1-[3-(4-бутилпиперидин-1-ил)пропил]-1Н-бензотриазол;
1-[3-(4-бутилпиперидин-1-ил)пропил]-1Н-индол-3-карбальдегид;
{1-[3-(4-бутилпиперидин-1-ил)пропил]-1Н-индол-3-ил}метанол;
1-[3-(4-бутилпиперидин-1-ил)пропил]-2-фенил-1Н-бензоимидазол;
1-[3-(4-бутилпиперидин-1-ил)пропил]-3-хлор-1Н-индазол;
1-[3-(4-бутилпиперидин-1-ил)пропил]-6-нитро-1Н-индазол;
Бензо[d]изоксазол-3-ол;
3-(2-хлорэтокси)бензо[d]изоксазол;
3-[2-(4-Бутилпиперидин-1-ил)этокси]бензо[d]изоксазол;
3-(1Н-индол-3-ил)пропан-1-ол;
гидрохлорид 3-[3-(4-бутилпиперидин-1-ил)пропил]-1Н-индола;
метиловый эфир 4-(4-бутилпиперидин-1-ил)масляной кислоты;
2-[3-(4-бутилпиперидин-1-ил)пропил]-1-метил-1Н-бензимидазол;
(2-(4-бутилпиперидин)-1-илэтил)амид 1Н-индазол-3-карбоновой кислоты;
1-[3-(4-бутилпиперидин-1-ил)пропил]-5-нитро-1Н-индазол;
2-[3-(4-бутилпиперидин-1-ил)пропил]-5-нитро-2Н-индазол;
1-[3-(4-бутилпиперидин-1-ил)пропил]-2-метил-1Н-индол;
1-{1-[3-(4-бутилпиперидин-1-ил)пропил]-1Н-индол-3-ил}этанон;
{1-[3-(4-бутилпиперидин-1-ил)пропил]-1Н-индол-3-ил}ацетонитрил;
1-[3-(4-бутилпиперидин-1-ил)пропил]-1Н-индол-3-карбонитрил;
1-[3-(4-бутилпиперидин-1-ил)пропил]-5,6-диметил-1Н-бензоимидазол;
1-[3-(4-бутилпиперидин-1-ил)пропил]-5(6)-диметил-1Н-бензоимидазол;
1-[3-(4-бутилпиперидин-1-ил)пропил]-5-метокси-1Н-бензоимидазол;
{1-[3-(4-бутилпиперидин-1-ил)пропил]-1Н-бензоимидазол-2-ил} метанол;
1-[3-(4-бутилпиперидин-1-ил)пропил]-2-трифторметил-1Н-бензоимидазол;
трет-бутиловый эфир (2-триметилстаннилфенил)карбаминовой кислоты;
трет-бутиловый эфир [2-(4-хлорбутирил)фенил]карбаминовой кислоты;
трет-бутиловый эфир {2-[4-(4-бутилпиперидин-1-ил)бутирил] фенил}карбаминовой кислоты;
3-[3-(4-бутилпиперидин-1-ил)пропил]-1Н-индазол, HCl;
3-[3-(4-бутилпиперидин-1-ил)пропил]-5-нитро-1Н-индазол;
3-[3-(4-бутилпиперидин-1-ил)пропил]-5,7-динитро-1Н-индазол;
4-(4-бутилпиперидин-1-ил)-1-(2-метилсульфанилфенил)бутан-1-он;
3-[3-(4-бутилпиперидин-1-ил)пропил]бензо[d]изотиазол;
3-[3-(4-бутилпиперидин-1-ил)пропил]-5-метокси-1Н-индазол;
3-[3-(4-бутилпиперидин-1-ил)пропил]-4-метокси-1Н-индазол;
3-[3-(4-бутилпиперидин-1-ил)пропил]-6-метокси-1Н-индазол;
3-[3-(4-бутилпиперидин-1-ил)пропил]-1Н-индазол-4-ол (53MF51);
3-[3-(4-бутилпиперидин-1-ил)пропил]-1Н-индазол-6-ол (53MF52) и
3-[3-(4-бутилпиперидин-1-ил)пропил]-1Н-индазол-5-ол.
Настоящее изобретение также относится к фармацевтическим композициям, включающим эффективное количество, по меньшей мере, одного соединения изобретения, включая все соединения в диапазоне формулы (I).
В общем, соединения настоящего изобретения являются активными в отношении холинергических, особенно, мускариновых рецепторов. Предпочтительные соединения обладают общим свойством действия в качестве агонистов подтипа m1 или m4 мускариновых рецепторов или обоих типов. В предпочтительном осуществлении соединения настоящего изобретения являются селективными по отношению к m1, m4 или обоим подтипам мускариновых рецепторов из m1 и m4, т.е. соединения обладают слабым или не обладают по существу действием на другие типы мускариновых рецепторов. В типичном случае селективные для m1 и/или m4 соединения изобретения не оказывают действия на другие родственные рецепторы, включая G-белок-связанные рецепторы, например, серотонин, гистамин, допамин или адренергические рецепторы. Изобретение относится к соединениям, которые являются селективными в качестве агонистов подтипов либо m1 либо m4, а также соединениям, которые являются агонистами подтипов рецепторов как m1, так и m4. В одном воплощении соединения настоящего изобретения обладают меньшим или не обладают действием на подтипы мускариновых рецепторов m2 и m3. В другом воплощении соединения настоящего изобретения обладают меньшим или не обладают действием на подтипы мускариновых рецепторов m2, m3, m4 и m5.
Соединения настоящего изобретения в типичном случае обладают терапевтическими действиями, их можно использовать для лечения или ослабления симптомов болезненных состояний, связанных с холинергическими рецепторами, такими как недостаточность познавательной способности, забывчивость, спутанность сознания, потеря памяти, дефицит внимания, дефицит визуального восприятия, депрессия, боль, нарушения сна, психоз, галлюцинации, агрессивность, паранойя и повышенное внутриглазное давление. Болезненное состояние может быть результатом дисфункции, повышенной активности, модификации, мутации, усечения или потери холинергических рецепторов, особенно мускариновых рецепторов, а также сниженных уровней ацетилхолина.
Соединения настоящего изобретения можно также использовать для лечения заболеваний, например возрастные снижения познавательной способности, болезни Альцгеймера, болезни Паркинсона, хореи Хантингтона, атаксии Фридериха, синдрома Жиля де ла Туретта, синдрома Дауна, болезни Пика, деменции, клинической депрессии, нарушения, связанного с дефицитом внимания, синдрома внезапной смерти внешне здорового младенца и глаукомы.
Соединения настоящего изобретения обладают способностью повышать активность холинергического рецептора или активировать холинергические рецепторы. Активность холинергических рецепторов включает активность в передаче сигнала или любую другую активность, которая непосредственно или косвенно относится к холинергической передаче сигнала или активации. Холинергические рецепторы включают мускариновые рецепторы, особенно подтип m1 или m4 мускариновых рецепторов. Мускариновый рецептор может быть, например, в центральной нервной системе, периферической нервной системе, желудочно-кишечной системе, сердце, железах внутренней системы или легких. Мускариновый рецептор может быть мускариновым рецептором дикого типа, усеченным, мутированным или модифицированным холинергическим рецептором. Наборы, включающие соединения настоящего изобретения для повышения активности холинергических рецепторов или активации холинергических рецепторов также рассматриваются настоящим изобретением.
Системой, содержащей холинергический рецептор, может быть, например, субъект, такой как млекопитающее, не являющийся человеком примат или человек. Система может быть экспериментальной моделью in vivo или in vitro, такой как система модели клеточной культуры, которая экспрессирует холинергический рецептор, не содержащий клетки ее экстракт, который содержит холинергический рецептор или очищенный рецептор. Неограничивающими примерами таких систем являются клетки тканевой культуры, экспрессирующие рецептор, или их экстракты, или лизаты. Клетки, которые можно использовать в настоящем способе, включают любые клетки, способные опосредовать сигнальную трансдукцию посредством холинергических рецепторов, особенно мускаринового рецептора m1, либо посредством эндогенной экспрессии этого рецептора (некоторые типы линий нейронных клеток, например, нативно экспрессируют рецептор m1) либо, например, после введения экзогенного гена в клетку, например, трансфекцией клеток плазмидами, содержащими ген рецептора. Такие клетки в типичном случае являются клетками млекопитающих (или другими эукариотическими клетками, такими как клетки насекомых или ооциты Xenopus), поскольку у клеток низших форм жизни обычно отсутствуют пути подходящей сигнальной трансдукции для настоящей цели. Примеры подходящих клеток включают: мышиную фибробластную клеточную линию (NIH 3T3 (ATCC CRL (1658), которая реагирует на трансфицированные рецепторы m1 усиленным ростом; клетки RAT 1 (Pace et al., Proc. Natl. Acad. Sci. USA 88:7031-35 (1991)) и клетки гипофиза (Vallar et al., Nature 330:556-58 (1987)). Другие пригодные клетки млекопитающих для настоящего способа включают, но не ограничиваются перечисленным, клетки HEK 293, клетки СНО и клетки COS.
Соединения настоящего изобретения обладают также способностью снижать внутриглазное давление и, следовательно, их можно использовать при лечении таких заболеваний, как глаукома. Глаукома является заболеванием, при котором аномалию наблюдают в механизме регулирования циркуляции внутриглазной жидкости, заполняющей переднюю камеру глазного яблока, т.е. пространство, образованное между роговицей и хрусталиком. Это приводит к повышению объема внутриглазной жидкости и повышению внутриглазного давления, впоследствии приводящие к нарушению поля зрения и даже к потери зрения вследствие сжатия и сокращения сосочка зрительного нерва.
Настоящее изобретение относится также к области прогностической медицины, в которой для целей прогноза используют фармакогеномику. Фармакогеномика имеет дело с клинически значительными наследственными вариациями в реакции на лекарственные средства вследствие измененного распределения лекарственного средства и аномального действия у пораженных индивидуумов (см., например, Eichelbaum, Clin. Exp. Pharmacol. Physiol. 23:983-985 (1996) и Linder, Clin. Chem. 43:254-66 (1977)). В общем, можно различать два типа фармакогенетических состояний: генетические состояния, связанные с отдельными факторами, изменяющими путь действия лекарственных средств на организм (измененное действие лекарственного средства), или генетические состояния, связанные с отдельными факторами, изменяющими путь воздействия организма на лекарственные средства (измененный метаболизм лекарственного средства). Эти фармакогенетические состояния могут иметь место как встречающиеся в природе полиморфизмы.
Один подход фармакогеномики к идентификации генов, который предсказывает реакцию на лекарственное средство, известный как "ассоциация по широкому исследованию генома", основан в основном на карте высокого разрешения генома человека, состоящей из известных, относящихся к гену маркеров (например, карте "биаллельных" генных маркеров, которая состоит из 60000-100000 полиморфных или вариабельных сайтов генома человека, каждый из которых имеет два варианта). Такую генетическую карту высокого разрешения можно сравнить с картой генома любого статистически значимого числа пациентов, принимающих участие в испытании лекарственного средства в фазе II/III, для идентификации маркеров, связанных с конкретно наблюдаемой реакцией на лекарственное средство или побочным действием. В альтернативном случае такую карту высокого разрешения можно получить из комбинации приблизительно десяти миллионов известных единичных нуклеотидных полиморфизмов (SNPs) в геноме человека. Используемый здесь "SNP" является общим изменением, которое имеет место в одном нуклеотидном основании в участке ДНК. Например, SNP может встречаться один раз на каждые 1000 оснований ДНК. SNP может принимать участие в процессе заболевания, хотя значительное большинство может быть не связано с заболеванием. Данную генную карту индивидуумов, основанную на присутствии таких SNPs, можно сгруппировать по генетическим категориям в зависимости от конкретной картины SNPs в их индивидуальном геноме. Таким способом можно специально разработать лечебные схемы для групп генетически подобных индивидуумов, принимая во внимание особенности, которые могут быть общими у таких генетически подобных индивидуумов.
По другому варианту для идентификации генов, которые предсказывают реакцию на лекарственное средство, можно использовать способ, названный "подход по гену-кандидату". В соответствии с этим способом, если ген, который кодирует мишень лекарственного средства, известен (например, белок или рецептор по настоящему изобретению), все обычные варианты этого гена можно идентифицировать в популяции. Его можно легко определить стандартными способами, конкретная версия связана с реакцией гена на определенное лекарственное средство.
В альтернативном случае для идентификации генов, которые предсказывают реакцию на лекарственное средство, можно использовать способ, названный "анализом профиля экспрессии гена". Например, экспрессия гена животного, которому вводили дозы лекарственного средства (например, соединения или композиции настоящего изобретения), может дать указание на то, включались ли генные пути, связанные с токсичностью.
Информацию, полученную из более чем одного из вышеуказанных подходов фармакогеномики, можно использовать для определения подходящей дозы и лечебных схем для профилактического или терапевтического лечения индивидуума. Это знание, когда его применяют для выбора дозы или лекарственного средства, может предотвратить побочные реакции или терапевтическую недостаточность и, таким образом, повысить терапевтическую или профилактическую эффективность при лечении субъекта соединением или композицией настоящего изобретения, такое как модулятор, идентифицированный одним из описанных здесь примерных анализов скрининга. Эти подходы можно также использовать для идентификации нового рецептора-кандидата или других генов, подходящих для дальнейшей фармакологической характеристики in vitro и in vivo.
В соответствии с этим, другой аспект настоящего изобретения описывает способы и наборы для идентификации генетического полиморфизма, ведущего к предрасположенности субъекта к восприимчивости к описанному здесь соединению. Способ включает введение субъекту эффективного количества соединения; идентификацию восприимчивого субъекта, имеющего сниженную интенсивность симптомов болезненного состояния (клинически не выраженную), связанного с холинергическим рецептором, и идентификацию генетического полиморфизма у восприимчивого субъекта, где генетический полиморфизм ведет к предрасположенности субъекта к восприимчивости к соединению. Идентификацию генетического полиморфизма у восприимчивого субъекта можно проводить любыми способами, известными в данной области, в том числе способами, описанными выше. Кроме того, набор, который используют для идентификации генетического полиморфизма, ведущего к предрасположенности субъекта к восприимчивости к соединению, по изобретению, включает соединение настоящего изобретения и, предпочтительно, реагенты и инструкции для проведения теста на генетический полиморфизм.
В одном воплощении субъект можно подвергнуть тестированию на известный полиморфизм, который ведет к предрасположенности субъекта к восприимчивости к соединению настоящего изобретения. Присутствие полиморфизма указывает на то, что субъект является пригодным для лечения.
В предпочтительных воплощениях соединения настоящего изобретения можно представить, как показано в формулах (IIIa-e):
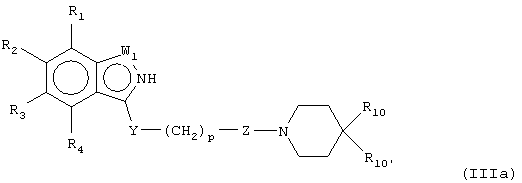
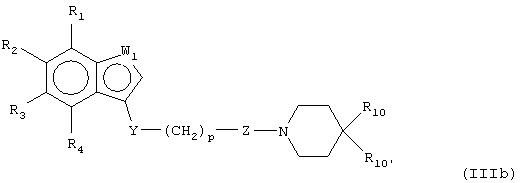
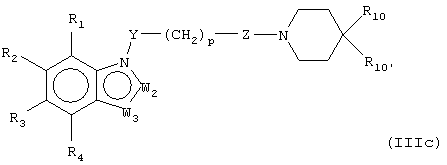
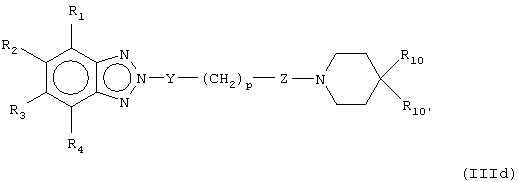
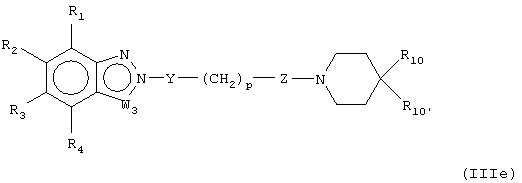
где W1 представляет О, S или NR5, W2 представляет CR5 или N и W3 представляет CR5 или N или где W3 представляет NR5, S или О, или их фармацевтически приемлемой солью, эфиром или пролекарством.
Соединения настоящего изобретения можно получить способами, аналогичными способам, описанным в патенте Великобритании №1142143 и патенте США №3816433, каждый из которых включен здесь в качестве ссылки. Пути модификации этих способов для включения других реагентов и т.д. должны быть очевидны для специалистов в данной области. Так, например, соединения формулы III (например, IIIb, где W1 представляет NR5) можно получить, как указано в следующей реакционной схеме.
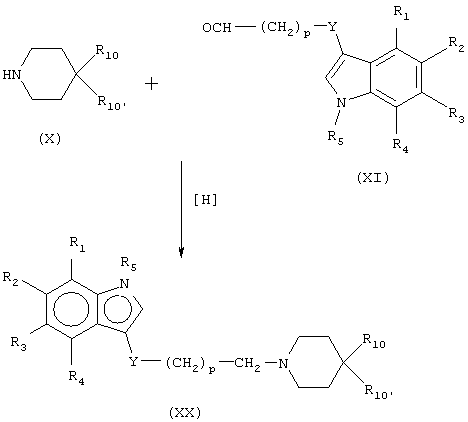
Исходное соединение, имеющее формулу (Х), можно получить общими способами органических синтезов. Для общих способов получения соединений формулы (Х) дается ссылка на Fuller, et al., J. Med. Chem. 14:322-325 (1971); Foye, et al., J. Pharm. Sci. 68:591-595 (1979); Bossier, et al., Chem. Abstr. 66:46195h and 67:21527a (1967); Aldous, J. Med. Chem. 17:1100-1111 (1974); Fuller, et al., J. Pharm. Pharmacol. 25:828-829 (1973); Fuller, et al., Neuropharmacology 14:739-746 (1975); Conde, et al., J. Med. Chem. 21:978-981 (1978); Lukovits, et al., Int. J. Quantum Chem. 20:429-438 (1981); and Law, Cromatog. 407:1-18 (1987), описание которых включается здесь в качестве ссылки во всей их полноте. Соединения формулы XI получают, например, как описано в Darbre, et al., Helv. Chim. Acta, 67:1040-1052 (1984) или Ihara, et al, Heterocycles, 20:421-424 (1983), также включенных здесь в качестве ссылки. Меченые радиоактивным изотопом производные, имеющие формулу (ХХ), можно получить, например, с использованием тритированного восстанавливающего агента для проведения восстановительного аминирования или с использованием 14С-меченого исходного материала.
Для получения соединений формулы (I) можно использовать соединения формулы (XXII). Соединения формулы (XXII) получают, например, как описано в Ishii, et al, J. Org. Chem. 61:3088-3092 (1996) или Britton, et al. Bioorg. Med. Chem. Lett. 9:475-480 (1999), также включенных здесь в качестве ссылки. Когда исходное соединение включает карбонильную группу, соединение, имеющее формулу (XXII), можно восстановить, например, AlH3, системой диборан:метилсульфид или другими стандартными восстанавливающими карбонильными реагентами для получения лиганда, имеющего формулу (ХХХ).
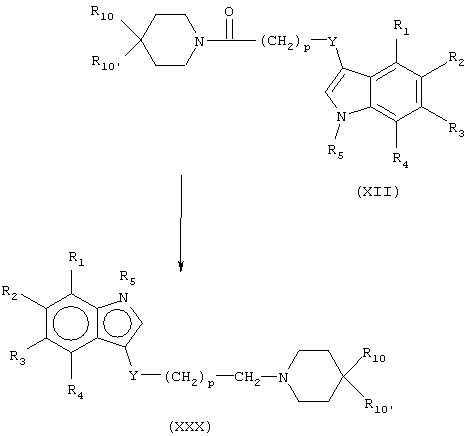
Лиганды рецепторов, имеющие формулу (XXXII), можно получить нуклеофильным замещением подходящего нуклеофуга (Е) аминопроизводным (XXXI). Примеры нуклеофугов, которые можно использовать для этой цели, включают галогениды, такие как I, Cl, Br, или тозилат или мезилат.
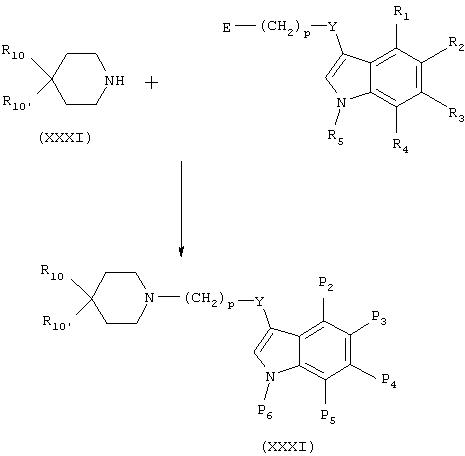
Когда Y в формуле (ХХХ) представляет -С(О)-, это соединение можно получить окислением вторичного спирта, например, хлорхроматом пиридиния, N-хлорсукцинимидом, CrO3-H2SO4, или никелем посредством методик Сверна или Десс-Мартина.
Когда Y в формуле (ХХХ) представляет -О-, это соединение можно получить арилированием спирта арилгалогенидами, например, при катализе с Cu.
Когда Y в формуле (ХХХ) представляет -S-, это соединение можно получить арилированием тиола арилгалогенидами, например, при катализе с Cu.
Когда Y в формуле (ХХХ) представляет -СНОН-, это соединение можно получить восстановлением соответствующего кетона каталитическим гидрированием или используя NaBH4 или LiAlH4.
Подходящие фармацевтически приемлемые соли соединений данного изобретения включают кислотно-аддитивные соли, которые, например, могут быть образованы смешиванием раствора соединения по изобретению с раствором фармацевтически приемлемой кислоты, такой как хлористоводородная кислота, серная кислота, фумаровая кислота, малеиновая кислота, янтарная кислота, уксусная кислота, бензойная кислота, щавелевая кислота, лимонная кислота, винная кислота, угольная кислота или фосфорная кислота. Кроме того, когда соединения изобретения имеют кислотную часть, их подходящие фармацевтически приемлемые соли включают соли щелочных металлов, например, соли натрия или калия; соли щелочноземельных металлов, например соли кальция или магния, и соли, образованные с подходящими органическими лигандами, например четвертичные аммониевые соли. Примеры фармацевтически приемлемых солей включают соль ацетат, бензолсульфонат, бензоат, бикарбонат, бисульфат, битартрат, борат, бромид, кальций, карбонат, хлорид, клавуланат, цитрат, дигидрохлорид, фумарат, глюконат, глутамат, гидробромид, гидрохлорид, гидроксинафтоат, иодид, изотионат, лактат, лактобионат, лаурат, малеат, манделат, мезилат, метилбромид, метилнитрат, метилсульфат, нитрат, аммониевую соль N-метилглутамина, олеат, оксалат, фосфат/дифосфат, салицилат, стеарат, сульфат, сукцинат, таннат, тартрат, тозилат, триэтиодид и валерат.
Настоящее изобретение включает в свой объем пролекарства соединений данного изобретения. В общем, такие пролекарства являются производными соединений данного изобретения, которые легко превращаются in vivo в нужное соединение. Общепринятые процедуры отбора и получения подходящих пролекарственных производных описаны, например, в Design of Prodrugs (Bundgaard, ed. Elsevier, 1985). Метаболиты этих соединений включают активные виды, продуцированные при введении соединений данного изобретения в биологическую среду.
Когда соединения по изобретению имеют, по меньшей мере, один хиральный центр, они могут существовать в виде рацематов или энантиомеров. Следует отметить, что все такие изомеры и их смеси включены в объем настоящего изобретения. Кроме того, некоторые кристаллические формы соединений настоящего изобретения могут существовать в виде полиморфов и как таковые, как предполагается, включены в настоящее изобретение. Кроме того, некоторые из соединений настоящего изобретения могут образовывать сольваты с водой (т.е. гидраты) или обычными органическими растворителями. Такие сольваты также включены в объем данного изобретения.
Когда способы получения соединений по изобретению дают смесь стереоизомеров, такие изомеры можно разделить общепринятыми способами, такими как препаративная хиральная хроматография. Соединения можно получить в рацемической форме или индивидуальные энантиомеры можно получить либо стереоселективным синтезом или разделением на энантиомеры. Соединения можно, например, разделить на их энантиомерные компоненты стандартными способами, такими как образование диастереомерной парой соли с оптически активной кислотой, такой как (-)-ди-п-толуоил-d-винная кислота и/или (+)-ди-п-толуоил-l-винная кислота, с последующей фракционной кристаллизацией и образованием свободного основания. Соединения можно также разделить образованием диастереомерных сложных эфиров или амидов с последующим хроматографическим разделением и удалением хирального вспомогательного вещества.
В ходе любого из способов получения соединений настоящего изобретения может быть необходимо и/или желательно защитить чувствительные или реакционноспособные группы на любой из участвующих в реакции молекул. Это можно достичь с помощью общепринятых защитных групп, таких как группы, описанные в Protective Groups in Organic Chemistry (McOmie ed., Plenum Press, 1973) и Greene & Wuts, Protective Groups in Organic Synthesis (John Wiley and Sons, 1991). Защитные группы можно удалить на приемлемой последующей стадии с использованием способов, известных в данной области.
Соединения настоящего изобретения можно вводить в любой из приводимых композиций и в соответствии со схемами приема лекарственного средства, установленными в данной области, всякий раз, когда требуется определенная фармакологическая модификация активности мускариновых рецепторов.
Настоящее изобретение относится также к фармацевтическим композициям, включающим одно или несколько соединений изобретения вместе с фармацевтически приемлемым разбавителем или эксципиентом. Такие композиции, предпочтительно, являются стандартными дозированными лекарственными формами, такими как таблетки, пилюли, капсулы (в том числе готовые препаративные формы с длительным или замедленным высвобождением), порошки, гранулы, эликсиры, настойки, сиропы и эмульсии, стерильные парентеральные растворы или суспензии, аэрозольные или жидкие спреи, капли, ампулы, устройства-автоинжекторы или суппозитории; для перорального, парентерального (например, внутривенного, внутримышечного или подкожного), интраназального, сублингвального или ректального введения, или для введения ингаляцией или инсуффляцией и могут быть изготовлены подходящим способом и в соответствии с общепринятыми методиками, такими, как практики, описанные в Remington's Pharmaceutical Sciences (Gennaro, ed., Mack Publishing Co., Easton PA, 1990). В альтернативном случае композиции могут быть в форме с замедленным высвобождением, пригодной для введения один раз в неделю или один раз в месяц; например, нерастворимую соль активного соединения, такую как деканоатную соль, можно приспособить для получения препарата-депо для внутримышечной инъекции. Настоящее изобретение рассматривает также подходящие местные готовые препаративные формы для введения, например, в глаза, нанесения на кожу или слизистую оболочку.
Для перорального введения, например, в форме таблетки или капсулы активный лекарственный компонент можно комбинировать с пероральным, нетоксичным фармацевтически приемлемым инертным носителем, таким как этанол, глицерин, вода и тому подобное. Кроме того, когда желательно или необходимо, в смесь можно также ввести подходящие связующие, смазывающие вещества, дезинтегрирующие агенты, корригенты и красящие агенты. Подходящие связующие включают, без ограничения, крахмал, желатин, натуральные сахара, такие как глюкоза или бета-лактоза, природные и синтетические камеди, такие как аравийская камедь, трагакант или альгинат натрия, карбоксиметилцеллюлоза, полиэтиленгликоль, воски и тому подобное. Смазывающие вещества, используемые в этих лекарственных формах, включают, без ограничения, олеат натрия, стеарат натрия, стеарат магния, бензоат натрия, ацетат натрия, хлорид натрия и тому подобное. Дезинтеграторы включают, без ограничения, крахмал, метилцеллюлозу, агар, бентонит, ксантановую камедь и тому подобное.
Для получения твердых композиций, таких как таблетки, активный ингредиент смешивают с подходящим фармацевтическим эксципиентом, таким как эксципиенты, описанные выше, и другими фармацевтическими разбавителями, например, водой, с образованием композиции в виде твердой готовой препаративной формы, содержащей гомогенную смесь соединения настоящего изобретения или его фармацевтически приемлемой соли. Термин "гомогенная" означает, что активный ингредиент равномерно диспергирован на всем объеме композиции, так что композицию можно легко разделить на равно эффективные стандартные лекарственные формы, такие как таблетки, пилюли и капсулы. Твердую предварительно приготовленную композицию можно затем разделить на стандартные лекарственные формы типа, описанного выше, содержащие приблизительно от 0,01 до приблизительно 50 мг активного ингредиента настоящего изобретения. Таблетки или пилюли настоящего изобретения можно покрыть оболочкой или составить иным путем для получения лекарственной формы, обеспечивающей преимущества пролонгированного действия. Например, таблетка или пилюля может включать внутреннее ядро, содержащее активное соединение, и внешний слой, как покрытие, окружающее ядро. Внешний слой может быть энтеросолюбильным слоем, который служит для препятствия дезинтеграции в желудке и позволяет внутреннему ядру проходить интактным в двенадцатиперстную кишку или замедлять высвобождение. Для таких энтеросолюбильных слоев или покрытий можно использовать различные материалы, причем такие материалы включают различные полимерные кислоты и смеси полимерных кислот с общепринятыми материалами, такими как шеллак, цетиловый спирт и ацетат целлюлозы.
Жидкие формы, в которые настоящие композиции можно включить для введения перорально или инъекцией, включают водные растворы, сиропы, подходящим образом обработанные для изменения вкуса и запаха, водные или масляные суспензии и эмульсии, обработанные для изменения вкуса и запаха пищевыми маслами, такими как хлопковое масло, кунжутное масло, кокосовое масло или арахисовое масло, а также эликсиры и аналогичные фармацевтические носители. Подходящие диспергирующие или суспендирующие агенты для водных суспензий включают синтетические и природные камеди, такие как трагакант, аравийская камедь, альгинат, декстран, натриевая соль карбоксиметилцеллюлозы, желатин, метилцеллюлоза и поливинилпирролидон. Другие диспергирующие агенты, которые можно использовать, включают глицерин и тому подобное. Для парентерального введения желательны стерильные суспензии и растворы. Изотонические препараты, которые обычно содержат подходящие консерванты, применяют, когда желательно внутривенное введение. Композиции можно также изготовить в виде офтальмического раствора или суспензии, т.е. глазных капель, для глазного введения.
Соединения настоящего изобретения можно ввести в виде одной суточной дозы или общую суточную дозу можно ввести в виде разделенных доз два, три или четыре раза в день. Кроме того, соединения настоящего изобретения можно ввести в интраназальной форме посредством местного использования подходящих интраназальных наполнителей или через трансдермальные пути с использованием, например, форм чрескожных пластырей на кожу, которые хорошо известны специалистам в данной области. Для введения в форме чрескожной системы доставки введение дозы должно быть непрерывным, а не прерывистым, на протяжении схемы приема лекарственного средства.
Схему приема лекарственного средства, использующую соединения настоящего изобретения, выбирают в соответствии с различными факторами, включающими тип, виды, возраст, массу, пол и медицинское состояние пациента; серьезность состояния, которое лечат; путь введения, функцию почек и печени пациента и применяемого конкретного соединения. Врач и ветеринар средней квалификации может легко определить и прописать эффективное количество лекарственного средства, требуемого для профилактики, противодействия или остановки развития подвергаемого лечению заболевания или нарушения.
Суточная доза продуктов может варьировать в широком диапазоне от 0,01 до 100 мг для взрослого человека в день. Для перорального введения композиции, предпочтительно, предоставляют в форме таблеток, содержащих 0,01, 0,05, 0,1, 0,5, 1,0, 2,5, 5,0, 10,0, 15,0, 25,0 или 50,0 мг активного ингредиента для симптоматической установки дозы для подвергаемого лечению пациента. Стандартная доза обычно содержит приблизительно от 0,001 мг до приблизительно 50 мг активного ингредиента, предпочтительно, приблизительно от 1 мг до приблизительно 10 мг активного ингредиента. Эффективное количество лекарственного средства обычно применяют при уровне доз приблизительно от 0,0001 мг/кг до приблизительно 25 мг/кг массы тела в день. Диапазон, предпочтительно, составляет приблизительно от 0,001 до 10 мг/кг массы тела в день и, особенно, приблизительно от 0,001 мг/кг до 1 мг/кг массы тела в день. Соединения можно вводить, например, при режиме от 1 до 4 раз в день.
Соединения по настоящему изобретению можно использовать, как взятые отдельно, при подходящих дозах, определяемых обычным тестированием для достижения оптимального фармакологического действия на мускариновый рецептор, особенно подтип m1 или m4 мускаринового рецептора, при минимизации любого потенциального токсического или других нежелательных действий. Кроме того, в некоторых случаях может быть желательно совместное введение или последовательное введение других агентов, которые повышают действие соединения.
Фармакологические свойства и селективность соединений данного изобретения для определенных подтипов мускариновых рецепторов можно продемонстрировать различными способами анализа с использованием, например, рекомбинантных подтипов рецептора, предпочтительно, рецепторов человека в качестве доступного, например, стандартного второго мессенджера, или анализов на связывание. Особенно пригодной системой функционального анализа является выбор рецептора и анализ на амплификацию, описанный в патенте США №5707798, в котором описывается способ отбора биоактивных соединений посредством использования способности клеток, трансфицированных рецепторной ДНК, например, кодирующей различные мускариновые подтипы, амплифицироваться в присутствии лиганда рецептора. Размножение клеток определяют по повышенным уровням маркера, также экспрессированного клетками.
Изобретение далее описывается подробно в следующих примерах, которые ни в коем случае не предназначаются для ограничения объема заявленного изобретения.
ПРИМЕРЫ
Способы получения
Соединения в соответствии с настоящим изобретением можно синтезировать способами, описанными ниже, или модификацией этих способов. Способы модификации методик включают, например, изменения температур, растворителей, реагентов и т.д., они должны быть очевидны для специалистов в данной области.
Общая процедура ЖХ-МС: Все спектры получали с использованием прибора HP1100 LC/MSD. Использовали установку с двойным насосом, автопробоотборником, термостатом для колонок, диодным матричным детектором и интерфейс ионизацию электронораспылением. Использовали колонку с обращенной фазой (С18 Luna, размер частиц 3 мм, 7,5 см×4,6 мм, внутр. диаметр) с системой защитного картриджа. Колонку выдерживали при температуре 30°C. Подвижной фазой была система ацетонитрил/8 мМ водный ацетат аммония. Использовали 15 минутную градиентную программу, которая начинается при 70% ацетонитриле, достигает на протяжении 12 минут 95% ацетонитрила, на протяжении 1 минуты возвращается обратно к 70% ацетонитрила и остается такой в течение 2 минут. Скорость потока была 0,6 мл/мин. Величины tr, указанные в конкретных примерах ниже, были получены с использованием этой процедуры.
2-(3-(4-н-Бутилпиперидин-1-ил)пропил)бензотиазол (5). 1-Бензил-4-н-бутилиденпиперидин (2). В 3-горлую колбу на 500 мл, снабженную мешалкой, загружают гидрид натрия (1,61 г, 67 ммоль) и ДМСО (мл). Образовавшуюся суспензию нагревают до 90°C в течение 30 минут до прекращения выделения водорода. Суспензию охлаждают на ледяной бане в течение 20 минут с последующим добавлением суспензии бромида бутилтрифенилфосфония (26,6 г, 67 ммоль) в ДМСО (70 мл). Красную смесь перемешивают в течение 15 мин при комнатной температуре. Медленно в течение 30 мин добавляют 1-бензил-4-пиперидон 1 (14,0 г, 74 ммоль) и смесь перемешивают при комнатной температуре в течение ночи. К реакционной смеси добавляют Н2О (200 мл) с последующей экстракцией гептаном (4×100 мл) и этилацетатом (2×100 мл). Объединенные органические фазы сушат и выпаривают досуха, получая при этом 38,1 г желтого масла. Масло перегоняют с получением 14,9 г (88%) соединения 2, т.кип. 101-105°С (0,1 мм Hg). 1ЯМР (CDCl3) δ 0,90-0,95 (т, 3Н), 1,25-1,41 (м, 2H), 1,90-2,20 (м, 2Н), 2,18-2,30 (м, 4Н), 2,40-2,45 (м, 4H), 2,50 (с, 2H), 5,17 (т, 1Н), 7,20-7,42 (м, 5Н).
4-н-Бутилпиперидин (3). В колбу на 500 мл, снабженную мешалкой, добавляют суспензию 2 (13,2 г, 58 ммоль) и 10% палладий на угле (1,2 г) в этаноле (70 мл) с последующим добавлением концентрированной хлористоводородной кислоты (1,5 мл). Из реакционной колбы выкачивают воздух и через реакционную колбу добавляют водород. Всего поглощается 2,5 дм3 водорода. Реакционную смесь фильтруют и упаривают и остаток растворяют в Н2О (40 мл) и NaOH (20 мл, 2 М) с последующей экстракцией этилацетатом (3×100 мл). Объединенные органические фазы промывают насыщенным раствором соли (30 мл) и упаривают досуха с получением 7,1 г сырого продукта 3. Сырой продукт подвергают колоночной хроматографии (элюент: гептан:EtOAc (4:1)), получая при этом чистый продукт 3 (2,7 г, 33%). 1H ЯМР (CDCl3) δ 0,85 (т, 3H), 1,0-1,38 (м, 9H), 1,65 (дд, 2H), 2,38 (с, 1H), 2,55 (дт, 2H), 3,04 (дт, 2H).
Метиловый эфир 4-(4-н-бутилпиперидин-1-ил)масляной кислоты (4). В колбу на 50 мл загружают смесь соединения 3 (2,7 г, 15 ммоль), метилового эфира 4-броммасляной кислоты (9,9 г, 55 ммоль) и карбоната калия (8,6 г, 62 ммоль) в ацетонитриле (25 мл). Смесь перемешивают при комнатной температуре в течение 72 часов с последующим упариванием досуха. Сырой продукт подвергают колоночной хроматографии (элюент: СН2Cl2:СН3ОН (96:4)) с получением чистого продукта 4 (3,4 г, 94%). 1H ЯМР (CDCl3) δ 0,89 (т, 3H), 1,20-1,39 (м, 9H), 1,69 (д, 2H), 1,89 (кв., 2H), 1,98 (т, 2H), 2,36 (т, 2H), 2,43 (т, 2H), 3,99 (д, 2H), 3,67 (с, 3H).
Общая процедура получения 2-(3-(4-н-бутилпиперидин-1-ил) пропил)гетероароматических соединений (5, 6, 7, 8, 9, 10, 11, 12, 13). Небольшую запаянную ампулу, снабженную магнитной мешалкой и загруженную соединением 4 (121 мг, 0,50 ммоль), подходящими бенздиаминами (как указано под каждым из соединений) (0,55 ммоль) и полифосфорной кислотой (2,1 г), нагревают до 150°C в течение 2 часов. Реакционную смесь выливают в воду и нейтрализуют бикарбонатом натрия и фильтруют. Дальнейшей обработкой фильтрата 2М NaOH получают дополнительные кристаллы, которые отфильтровывают и объединяют с первой порцией с последующим промыванием, сушкой и перекристаллизацией из эфира.
Пример 1. 2-(3-(4-н-Бутилпиперидин-1-ил)пропил)бензотиазол (5) (34JJ15). 2-Аминобензолтиол используют в качестве исходного материала и общую методику применяют для получения чистого соединения 5 (70 мг, 43%). 1H ЯМР (CDCl3) δ 0,88 (т, 3H), 1,08-1,20 (м, 2H), 1,50 (м, 2Н), 1,55-1,70 (м, 7H), 1,72 (кв., 2H), 1,73-1,75 (м, 2H), 2,35-2,39 (м, 2H), 2,41 (т, 2H), 2,61 (т, 2H), 7,39(дт, 2H), 7,89(дд, 2H).
Пример 2. 2-(3-(4-н-Бутилпиперидин-1-ил)пропил)бензоксазол (6) (34JJ17). 2-Аминофенол используют в качестве исходного материала и общую методику применяют для получения чистого соединения 6 (137 мг, 83%). 1H ЯМР (CDCl3) δ 0,88 (т, 3H), 1,18-1,32 (м, 10H), 1,65 (д, 2H), 1,95 (т, 2H), 2,12 (кв., 2H), 2,49 (т, 2H), 2,92-3,00 (м, 3H), 7,28-7,32 (м, 2H), 7,45-7,50 (м, 1H), 7,64-7,68 (м, 1H).
Пример 3. 4,5-Дифтор-2-(3-(4-н-бутилпиперидин-1-ил)пропил)-1Н-бензоимидазол (7) (34JJ21). 3,4-Дифтор-1,2-диаминобензол используют в качестве исходного материала и общую методику применяют для получения чистого соединения 7 (55 мг, 30%). 1H ЯМР (CDCl3) δ 0,93 (т, 3H), 1,30-1,44 (м, 9H), 1,82 (д, 2H), 1,98 (кв., 2H), 2,09 (т, 2H), 2,63 (дт, 2H), 3,07 (д, 2H), 3,14 (дт, 2H), 6,95-7,03 (м, 1H), 7,16-7,21 (м, 1H).
Пример 4. 4-Фтор-5-нитро-2-(3-(4-н-бутилпиперидин-1-ил) пропил)-1Н-бензоимидазол (8) (34JJ13). 4-Фтор-5-нитро-1,2-диаминобензол используют в качестве исходного материала и общую методику применяют для получения чистого соединения 8 (12 мг, 6%). 1H ЯМР (CDCl3) δ 0,93 (т, 3H), 1,30-1,54 (м, 7H), 1,60 (к., 2H), 1,93 (д, 2H), 2,22 (кв., 2H), 2,42 (т, 2H), 2,82 (т, 2H), 3,24 (т, 2H), 3,31 (д, 2H), 7,34 (д, 1H), 8,29 (д, 1H).
Пример 5. 5-трет-Бутил-2-(3-(4-н-бутилпиперидин-1-ил)пропил)-1Н-бензоимидазол (9) (23JJ83). 4-трет-Бутил-1,2-диаминобензол используют в качестве исходного материала и общую методику применяют для получения чистого соединения 9 (74 мг, 38%). 1H ЯМР (CDCl3) δ 0,93 (т, 3H), 1,30-1,42 (м, 18H), 1,81 (д, 2H), 1,96 (кв., 2H), 2,04 (т, 2H), 2,55 (т, 2H), 3,02 (д, 2H), 3,07 (т, 2H), 7,26 (дд, 1H), 7,45 (д, 1H), 7,53 (д, 1H).
Пример 6. 5-Хлор-6-метил-2-(3-(4-н-бутилпиперидин-1-ил) пропил)-1Н-бензоимидазол (10) (23JJ93). 4-Хлор-5-метил-1,2-диаминобензол используют в качестве исходного материала и общую методику применяют для получения чистого соединения 10 (7 мг, 3%). 1H ЯМР (CDCl3) δ 0,94 (т, 3H), 1,30-1,41 (м, 9H), 1,83 (д, 2H), 1,95 (кв., 2H), 2,08 (т, 2H), 2,46 (с, 3H), 2,57 (т, 2H), 3,04 (д, 2H), 3,09 (т, 2H), 7,32 (с, 1H), 7,50 (с, 1H).
Пример 7. 4,6-Дифтор-2-(3-(4-н-бутилпиперидин-1-ил)пропил)-1Н-бензоимидазол (11) (23JJ77). 3,5-Дифтор-1,2-диаминобензол используют в качестве исходного материала и общую методику применяют для получения чистого соединения 11 (50 мг, 27%). 1H ЯМР (CDCl3) δ 0,92 (т, 3H), 1,22-1,43 (м, 7H), 1,56 (к., 2H), 1,87 (д, 2H), 2,13 (кв., 2H), 2,38 (т, 2H), 2,87 (т, 2H), 3,19 (т, 2H), 2,29 (д, 2H), 6,69 (дт, 1H), 7,02 (дд, 1H).
Пример 8. 2-(3-(4-н-Бутилпиперидин)-1-илпропил)-1Н-имидазо [4,5-c]пиридин (12) (23JJ81). Пиридин-3,4-диамин используют в качестве исходного материала и общую методику применяют для получения чистого соединения 12 (18 мг, 11%). 1H ЯМР (CDCl3) δ 0,94 (т, 3H), 1,30-1,42 (м, 9H), 1,87 (д, 2H), 2,01 (кв., 2H), 2,13 (т, 2H), 2,64 (т, 2H), 3,08 (д, 2H), 3,17 (т, 2H), 7,41 (д, 1H), 8,35 (д, 1H), 8,90 (с, 1H).
Пример 9. 8-(3-(4-н-Бутилпиперидин)-1-илпропил)-9Н-пурин (13) (34JJ27). Пиримидин-4,5-диамин используют в качестве исходного материала и общую методику применяют для получения чистого соединения 13 (94 мг, 57%). 1H ЯМР (MeOD) δ 0,92 (т, 3H), 1,29-1,39 (м, 6H), 1,43-1,60 (м, 3H), 2,00 (д, 2H), 2,43 (кв., 2H), 3,00 (т, 2H), 3,21-3,35 (м, 4H), 3,64 (д, 2H), 9,25 (с, 1H), 9,38 (с, 1H).
Пример 10. 7-(3-(4-н-Бутилпиперидин)-1-илпропил)-3,8-дигидроимидазо[4',5':3,4]бензо[1,2-d][1,2,3]триазол (14) (34JJ39). 1Н-Бензотриазол-4,5-диамин используют в качестве исходного материала и общую методику применяют для получения чистого соединения 14 (24 мг, 13%). 1H ЯМР (DMSO) δ 0,83 (т, 3H), 1,00-1,28 (м, 9H), 1,57 (д, 2H), 1,80 (т, 2H), 1,94 (кв., 2H), 2,32 (т, 2H), 2,82 (д, 2H), 2,88 (т, 2H), 7,49 (д, 1H), 7,62 (д, 1H).
Пример 11. 2-(3-(4-н-Бутилпиперидин)-1-илпропил)-3а,4,5,6,7,7а-гексагидро-1Н-бензоимидазол (15). Циклогексан-1,2-диамин используют в качестве исходного материала и общую методику применяют для получения чистого соединения 15 (79 мг, 47%). 1H ЯМР (CDCl3) δ 0,80-1,05 (м, 11H), 1,27-1,75 (м, 17H), 2,57 (т, 2H), 2,66 (т, 2H), 3,57 (к., 1H), 4,48 (к., 1H).
Общая процедура получения производных замещенного индола (16, 17, 18, 19, 20 и 21). 1,3-Дибромпропан (205 мкл, 2,0 ммоль) в 5 мл ДМФА помещают в 50 мл колбу. Соответствующий индол (2,0 ммоль) и КОН (280 мг, 5,0 ммоль) частично растворяют в 5 мл ДМФА и добавляют при перемешивании. Образовавшуюся суспензию перемешивают в течение ночи при комнатной температуре. Добавляют 4-бутилпиперидин (3) (178 мг, 1,0 ммоль) в 5 мл ДМФА и смесь перемешивают в течение ночи при комнатной температуре. Добавляют этилацетат (20 мл) и воду (20 мл). Фазы разделяют и водную фазу снова экстрагируют этилацетатом (20 мл). Объединенные органические фазы промывают насыщенным раствором соли, сушат над сульфатом магния и упаривают досуха, получая при этом сырой продукт. Сырой продукт очищают колоночной хроматографией (0-5% метанол:дихлорметан) с получением чистых продуктов.
Пример 12. 1-(3-(4-н-Бутилпиперидин)-1-илпропил)-1Н-индол (16) (35AKU-15). 1Н-Индол используют в качестве исходного материала и общую методику применяют для получения чистого соединения 16 (69 мг, 23%). 1H ЯМР (CDCl3) δ 0,9 (т, 3H), 1,2-1,3 (м, 7H), 1,5 (к., 2H), 1,75 (д, 2H), 2,1-2,3 (м, 4H), 2,5 (т, 2H), 3,1 (д, 2H), 4,25 (т, 2H), 6,5 (д, 1H), 7,1 (м, 2H), 7,2 (т, 1H), 7,35 (д, 1H), 7,6 (д, 1H).
Пример 13. 1-(3-(4-н-Бутилпиперидин)-1-илпропил)-1Н-бензоимидазол (17) (35AKU-16). 1Н-Бензоимидазол используют в качестве исходного материала и общую методику применяют для получения чистого соединения 17 (69 мг, 23%). 1H ЯМР (CDCl3) δ 0,9 (т, 3H), 1,2-1,3 (м, 7H), 1,5 (к., 2H), 1,75 (д, 2H), 2,25 (м, 4H), 2,6 (т, 2H), 3,1 (д, 2H), 4,3 (т, 2H), 7,2-7,3 (м, 2H), 7,45 (д, 1H), 7,75 (д, 1H), 8,0 (с, 1H).
Пример 14. 3-Метил-1-(3-(4-н-бутилпиперидин)-1-илпропил)-1Н-индол (18) (35AKU-22). 3-Метил-1Н-индол используют в качестве исходного материала и общую методику применяют для получения чистого соединения 18. 1H ЯМР (CDCl3) δ 0,9 (т, 3H), 1,2-1,3 (м, 9H), 1,65 (д, 2H), 1,9 (т, 2H), 2,0 (м, 2H), 2,25 (м, 2H), 2,3 (с, 3H), 2,85 (д, 2H), 4,1 (т, 2H), 6,85 (с, 1H), 7,1 (т, 1H), 7,2 (т, 1H), 7,55 (д, 1H).
Пример 15. 5-Бром-1-(3-(4-н-бутилпиперидин)-1-илпропил)-1Н-индол (19) (35AKU-23). 5-Бром-1Н-индол используют в качестве исходного материала и общую методику применяют для получения чистого соединения 19. 1H ЯМР (CDCl3) δ 0,9 (т, 3H), 1,2-1,3 (м, 9H), 1,65 (д, 2H), 1,85 (т, 2H), 2,0 (т, 2H), 2,2 (т, 2H), 2,8 (д, 2H), 4,15 (т, 2H), 6,4 (д, 1H), 7,1 (д, 1H), 7,25 (м, 2H), 7,75 (с, 1H).
Пример 16. 3-Формил-1-(3-(4-н-бутилпиперидин)-1-илпропил)-1Н-индол (20) (35AKU-24). 3-Формил-1Н-индол используют в качестве исходного материала и общую методику применяют для получения чистого соединения 20. 1H ЯМР (CDCl3) δ 0,9 (т, 3H), 1,2-1,3 (м, 9H), 1,7 (д, 2H), 1,95 (т, 2H), 2,1 (м, 2H), 2,3 (т, 2H), 2,9 (д, 2H), 4,3 (т, 2H), 7,3-7,5 (м, 3H), 8,3 (м, 1H), 10,0 (с, 1H).
Пример 17. 7-Бром-1-(3-(4-н-бутилпиперидин)-1-илпропил)-1Н-индол (21) (35AKU-25). 7-Бром-1Н-индол используют в качестве исходного материала и общую методику применяют для получения чистого соединения 21. 1H ЯМР (CDCl3) δ 0,9 (т, 3H), 1,2-1,3 (м, 9H), 1,65 (д, 2H), 1,9 (т, 2H), 2,05 (м, 2H), 2,3 (т, 2H), 2,9 (д, 2H), 4,55 (т, 2H), 6,45 (д, 1H), 6,9 (т, 1H), 7,1 (д, 1H), 7,35 (д, 1Н), 7,55 (д, 1H).
Пример 18. 1-(3-Бромпропил)-1Н-индазол (22). 1,3-Дибромпропан (508 мкл, 5,0 ммоль) растворяют в 10 мл ДМФА и помещают в 100 мл колбу. Добавляют индазол (592 мг, 5,0 ммоль) и КОН (282 мг, 5,0 ммоль) и суспензию перемешивают в течение ночи при комнатной температуре. Добавляют этилацетат (50 мл) и воду (50 мл). Фазы разделяют и водную фазу снова экстрагируют этилацетатом (50 мл). Объединенные органические фазы промывают насыщенным раствором соли, сушат над сульфатом магния и упаривают досуха, получая при этом 751 мг желтого масла. Сырой продукт далее очищают колоночной хроматографией (0-10% метанол:дихлорметан), получая при этом чистое соединение 22 (169 мг, 14%).
Пример 19. 1-(3-(4-н-Бутилпиперидин)-1-илпропил)-1Н-индазол (23) (35AKU-21). В 50 мл колбу добавляют соединение 22 (169 мг, 0,7 ммоль) и 10 мл ДМФА. 4-Бутилпиперидин (3) (142 мг, 1,0 ммоль) и КОН (113 мг, 2,0 ммоль) частично растворяют в ДМФА (5 мл) и добавляют. Суспензию перемешивают в течение ночи при комнатной температуре. Добавляют этилацетат (20 мл) и воду (20 мл). Фазы разделяют и водную фазу снова экстрагируют этилацетатом (20 мл). Объединенные органические фазы промывают насыщенным раствором соли, сушат над сульфатом магния и упаривают досуха, получая при этом 192 мг светло-коричневого масла. Сырой продукт очищают колоночной хроматографией (0-10% метанол:дихлорметан), получая при этом чистый продукт 23 (61 мг, 29%). Оксалатную соль получают из щавелевой кислоты (1,1 экв.) в смеси метанол/диэтиловый эфир. 1H ЯМР (CDCl3) δ 0,9 (т, 3H), 1,2-1,3 (м, 9H), 1,65 (д, 2H), 1,9 (т, 2H), 2,15 (м, 2H), 2,3 (т, 2H), 2,85 (д, 2H), 4,45 (т, 2H), 7,1 (т, 1H), 7,35 (т, 1H), 7,5 (д, 1H), 7,7 (д, 1H), 8,0 (с, 1H).
Пример 20. Оксим 1-(2-гидроксифенил)этанона (24). Хлорид гидроксиламмония (6,96 г, 100 ммоль) и ацетат натрия·3Н2О (13,6 г, 100 ммоль) растворяют в 150 мл смеси этанол:вода (7:3) и добавляют к раствору 2-гидроксиацетофенона (6,81 г, 50 ммоль) в 50 мл смеси этанол:вода (7:3). рН регулируют до 4-5 4 н HCl (˜10 мл) и реакционную смесь затем нагревают при кипении с обратным холодильником (100°C) в течение 1 часа. Масляную баню удаляют, и смесь оставляют на ночь при перемешивании. Этанол частично удаляют выпариванием, и водную фазу экстрагируют этилацетатом два раза. Объединенные органические фазы сушат над сульфатом магния и упаривают досуха с получением 7,55 г чистого соединения 24.
Пример 21. 3-Метилбензо[d]изоксазол (25). В колбу на 100 мл с соединением 24 (7,55 г, 50 ммоль) добавляют уксусный ангидрид (7,1 мл, 75 ммоль). Смесь нагревают до 60°C в течение 3 часов с последующим упариванием досуха. Карбонат калия (8,7 г, 63 ммоль) частично растворяют в 40 мл ДМФА и добавляют к смеси. Смесь перемешивают при комнатной температуре в течение ночи и, наконец, нагревают до 100°C в течение 30 минут. Добавляют этилацетат и воду. Фазы разделяют, и водную фазу экстрагируют этилацетатом и дихлорметаном. Объединенные органические фазы сушат над сульфатом магния и упаривают досуха, получая при этом 5,6 г желтого масла. Сырой продукт очищают колоночной хроматографией (100% дихлорметан), получая при этом чистое соединение 25 (4,6 г). 1H ЯМР (CDCl3) δ 2,6 (с, 3H), 7,3 (м, 1H), 7,55 (м, 2H), 7,65 (м, 1H).
Пример 22. 3-Бут-3-енилбензо[d]изоксазол (26). Сухой ТГФ в количестве 3,0 мл добавляют в высушенную в сушильном шкафу колбу на 25 мл и охлаждают до -78°C на бане сухой лед/изопропанол. Добавляют диизопропиламин (840 мкл, 6,0 ммоль), затем н-BuLi (3,8 мл, 1,6 М, 6,0 ммоль). Раствор LDA, который был получен, оставляют при комнатной температуре. Соединение 25 (666 мг, 5,0 ммоль) растворяют в 10 мл сухого ТГФ и добавляют в высушенную в сушильном шкафу колбу на 50 мл, затем добавляют аллилбромид (476 мкл, 5,5 ммоль). Свежеприготовленный раствор LDA медленно добавляют при -78°C и смесь выдерживают при комнатной температуре в течение 30 мин. Добавляют этилацетат и воду. Фазы разделяют и водную фазу экстрагируют этилацетатом. Объединенные органические фазы сушат над сульфатом магния и упаривают досуха, получая при этом 893 мг светло-коричневого масла. Сырой продукт очищают колоночной хроматографией (гептан:этилацетат; 9:1, изократный), получая при этом чистое соединение 26 (355 мг, 41%).
Пример 23. 3-(Бензо[d]изоксазол-3-ил)пропиональдегид (27). Соединение 26 (549 мг, 3,2 ммоль), воду (5 мл), 1,4-диоксан (15 мл) и тетраоксид осмия (15 мг, 0,06 ммоль) перемешивают в течение 5 мин в небольшой колбе. На протяжении 30 мин добавляют метапериодат натрия (1,56 г, 7,3 ммоль) и суспензию затем перемешивают в течение 1 часа. Добавляют этилацетат и воду. Фазы разделяют, и водную фазу экстрагируют этилацетатом и дихлорметаном. Объединенные органические фазы сушат над сульфатом магния и упаривают досуха, получая при этом 784 мг сырого соединения 27, который используют непосредственно, без дальнейшей очистки, в синтезе соединения 28.
Пример 24. 3-(3-(4-н-Бутилпиперидин)-1-илпропил)бензо[d]изоксазол (28) (35AKU-2). Соединение 27 (˜500 мг, 2-3 ммоль) растворяют в 5 мл метанола. 4-Бутилпиперидин·HCl 3 (260 мг, 1,5 ммоль) растворяют в 10 мл метанола и добавляют. Добавляют цианоборогидрид натрия (188 мг, 3,0 ммоль) в 10 мл метанола, получая при этом темно-коричневый раствор, который перемешивают в течение ночи. Добавляют воду, и метанол частично удаляют выпариванием. Водную фазу экстрагируют этилацетатом и дихлорметаном. Объединенные органические фазы сушат над сульфатом магния и упаривают досуха. Сырой продукт далее очищают препаративной ВЭЖХ (подвижная фаза 0-80% ацетонитрил в воде (0,1% ТФУ)), получая при этом соединение 28 (244 мг, 54%). Соль HCl получают из 2 М HCl в диэтиловом эфире. Кристаллы отфильтровывают и промывают диэтиловым эфиром. 1H ЯМР (CDCl3) δ 0,9 (т, 3H), 1,2-1,3 (м, 9H), 1,65 (д, 2H), 1,9 (т, 2H), 2,05 (м, 2H), 2,45 (т, 2H), 2,9 (д, 2H), 3,0 (т, 2H), 7,3 (м, 1H), 7,55 (м, 2H), 7,7 (д, 1H).
Пример 25. 3-(1Н-Индол-3-ил)пропан-1-ол (29). Суспензию литийалюминийгидрида (4,68 г, 126 ммоль) в 230 мл безводного диэтилового эфира интенсивно перемешивают. 3-Индолпропионовую кислоту (10,0 г, 53 ммоль), растворенную в 460 мл безводного диэтилового эфира, переносят в капельную воронку и добавляют при такой скорости, чтобы поддерживалось слабое кипение с обратным холодильником. Реакционную смесь оставляют для перемешивания при температуре флегмы в течение 2 час, затем при комнатной температуре в течение ночи. Затем кипячение с обратным холодильником продолжают в течение 2 час перед охлаждением до комнатной температуры. Медленно добавляют 25 мл Н2О, затем 70 мл Н2О/H2SO4 (1:3 Н2 О:H2SO4). Образовавшуюся прозрачную смесь экстрагируют 110 мл диэтилового эфира три раза. Объединенные органические фазы промывают насыщенным раствором соли, сушат Na2SO4, фильтруют и концентрируют до светлого масла, которое используют без дополнительной очистки.
Пример 26. 3-(1Н-Индол-3-ил)пропиловый эфир метансульфоновой кислоты (30). Соединение 29 (1,8 г, 5,44 ммоль) переносят в высушенную в пламени колбу с аргоном и растворяют в безводном ТГФ, затем охлаждают до -40°С. Шприцем добавляют триэтиламин (0,72 г, 7,07 ммоль), затем MeSO2Cl (0,75 г, 6,53 ммоль). Реакционную смесь оставляют для повышения температуры до комнатной температуры (10-15 минут) до того, как ее быстро фильтруют и концентрируют. Сырое масло растворяют в СН2Cl2 и промывают Н2О. Органическую фазу сушат MgSO4, фильтруют и концентрируют в вакууме до темного, коричневого масла. Сырой продукт используют сразу в следующей стадии.
Пример 27. 3-(3-(4-н-Бутилпиперидин)-1-илпропил)-1Н-индол (31) (39MF34). Na2CO3 (1,28 г, 11,97 ммоль) добавляют к раствору гидрохлорида 4-бутилпиперидина 3 (967 мг, 5,44 ммоль) в безводном DME. Образовавшуюся суспензию перемешивают в течение 30 мин. Соединение 30 растворяют в безводном DME и добавляют к суспензии. Образовавшуюся смесь перемешивают в атмосфере аргона при 82°С в течение ночи. Смесь охлаждают, добавляют EtOAc и Н2О, разделяют две фазы, и воду экстрагируют EtOAc три раза. Объединенные органические фазы промывают насыщенным раствором соли, сушат Na2SO4, фильтруют и концентрируют в вакууме. Сырое масло растворяют в безводном СН2Cl2 и добавляют HCl в диоксане (4М, 2 мл). Продукт (31) выделяют в виде белых кристаллов перекристаллизацией из смеси МеОН/диэтиловый эфир. 1H ЯМР (CDCl3) δ 0,93 (т, 3H), 1,32-1,58 (м, 7H), 1,60 (к., 2H), 1,93 (д, 2H), 2,22 (кв., 2H), 2,42 (т, 2H), 2,82 (т, 2H), 3,24 (т, 2H), 3,31 (д, 2H), 6,91-7,10 (м, 2H), 7,34 (д, 1H), 7,53 (д, 1H).
Пример 28. 4-Нитро-2-(3-(4-н-бутилпиперидин)-1-илпропил)-1Н-бензоимидазол (32) (29MF03). В колбу на 25 мл, снабженную холодильником и магнитной мешалкой, загружают 1,2-диамино-3-нитробензол (0,251 г, 1,64 ммоль) и метиловый эфир 4-(4-н-бутилпиперидин-1-ил)масляной кислоты (4) (0,395 г, 1,64 ммоль) в 5 мл 4М HCl. Реакционную смесь кипятят с обратным холодильником в течение 24 час с последующим добавлением 2,0 М NaOH для создания основных условий, перемешивают при комнатной температуре в течение 1 часа и экстрагируют этилацетатом (5×50 мл). Объединенные органические фазы промывают 15 мл насыщенного раствора соли, затем сушат над MgSO4 и упаривают досуха, получая при этом 0,45 г сырого продукта. Сырой материал подвергают колоночной хроматографии (элюент: СН2Cl2:МеОН (20:1)), получая при этом чистое, указанное в заголовке соединение (32) (0,03 г, 5%). 1H ЯМР (CDCl3) δ 0,92 (т, 3H), 1,25-1,42 (м, 9H), 1,55-1,64 (м, 2H), 1,75-1,82 (м, 2H), 2,10-2,23 (м, 2H), 2,24-2,31 (м, 2H), 2,67-2,77 (м, 2H), 3,17-3,22 (м, 4H), 7,25-7,35 (м, 1H), 7,97-8,04 (м, 1H), 8,08-8,13 (м, 1H).
Пример 29. 5-Нитро-2-(3-(4-н-бутилпиперидин)-1-илпропил)-1Н-бензоимидазол (33) (29MF04). В колбу на 25 мл, снабженную холодильником и магнитной мешалкой, загружают 1,2-диамино-4-нитробензол (0,259 г, 1,69 ммоль) и метиловый эфир 4-(4-н-бутилпиперидин-1-ил)масляной кислоты (4) (0,408 г, 1,69 ммоль) в 5 мл 4М HCl. Реакционную смесь кипятят с обратным холодильником в течение 24 час с последующим добавлением 2,0 М NaOH для создания основных условий, затем перемешивают при комнатной температуре в течение 1 часа и экстрагируют этилацетатом (5×50 мл). Объединенные органические фазы промывают 15 мл насыщенного раствора соли, затем сушат над MgSO4 и упаривают досуха, получая при этом 0,27 г сырого материала. Сырой материал подвергают колоночной хроматографии (элюент: СН2Cl2:МеОН (20:1)), получая при этом конечное соединение (122 мг). Этот материал выделяют и растворяют в 2,0 М растворе HCl в эфире с последующим упариванием досуха, получая при этом чистое, указанное в заголовке соединения (33) (80 мг, 10%). 1H ЯМР (CD3OD) 0,92 (т, 3H), 1,34 (м, 6H), 1,55 (м, 3H), 2,00 (д, 2H), 2,45 (м, 2H), 3,01 (т, 2H), 3,29-3,37 (дт, 4H), 3,64 (д, 2H), 7,94 (д, 1H), 8,43 (дд, 1H), 8,65 (д, 1H).
Пример 30. 4-Гидрокси-2-(3-(4-н-бутилпиперидин)-1-илпропил)-1Н-бензоимидазол (34) (29MF07). В колбу на 25 мл, снабженную холодильником и магнитной мешалкой, загружают 1,2-диамино-4-гидроксибензол (0,177 г, 1,43 ммоль) и метиловый эфир 4-(4-н-бутилпиперидин-1-ил)масляной кислоты (4) (0,345 г, 1,43 ммоль) в 5 мл 4 М HCl. Реакционную смесь кипятят с обратным холодильником в течение 20 час с последующим добавлением 2,0 М NaOH для создания основных условий. Смесь упаривают досуха на 10 мл диоксида кремния и подвергают колоночной хроматографии (элюент: СН2Cl2:МеОН (20:1), получая при этом сырой продукт (0,145 г). Сырой продукт подвергают препаративной ВЭЖХ (элюент: буфер А: 0,1% ТФУ; буфер В:80% CH3CN+0,1% ТФУ) и выделенный продукт упаривают с 1,0 М ТФУ в эфире, получая при этом чистое, указанное в заголовке соединение 34 (74 мг, 16%) в виде соли трифторуксусной кислоты. 1H ЯМР (CD3OD) 0,98 (т, 3H), 1,32-1,45 (м, 6H), 1,51-1,69 (м, 3H), 1,97-2,08 (д, 2H), 2,37-2,47 (м, 2H), 2,95-3,12 (м, 2H), 3,26-3,41 (м, 4H), 3,58-3,72 (м, 2H), 6,91-6,97 (д, 1H), 7,19-7,25 (д, 1H), 7,35-7,43 (т, 1H).
Пример 31. 2-(3-(4-н-Бутилпиперидин)-1-илпропил)-1Н-бензоимидазол (35) (21MF25). В колбу на 25 мл, снабженную холодильником и магнитной мешалкой, загружают 1,2-диаминобензол (0,201 г, 18,6 ммоль) и метиловый эфир 4-(4-н-бутилпиперидин-1-ил)масляной кислоты (4) (0,50 г, 2,1 ммоль) в 6 мл 4 М HCl. Реакционную смесь кипятят с обратным холодильником в течение 20 час с последующим добавлением 2,0 М NaOH для создания основных условий. Осадок отфильтровывают и сушат в вакууме с последующей колоночной хроматографией (элюент: СН2Cl2:МеОН (10:1)), получая при этом чистое, указанное в заголовке соединение 35 (0,40 г, 73%). Т.пл. 78-79°С. 1H ЯМР (CDCl3) 0,92 (т, 3H), 1,33 (м, 6H), 1,50 (м, 3H), 1,80-1,95 (м, 2H), 2,02-2,15 (м, 2H), 2,16-2,24 (м, 2H), 2,62-2,75 (м, 2H), 3,17-3,21 (м, 4H), 7,20-7,23 (м, 2H), 7,52-7,59 (м, 2H).
Пример 32. 4-Метил-2-(3-(4-н-бутилпиперидин)-1-илпропил)-1Н-бензоимидазол (36) (29MF08). В колбу на 25 мл, снабженную холодильником и магнитной мешалкой, загружают 1,2-диамино-3-метилбензол (0,168 г, 1,37 ммоль) и метиловый эфир 4-(4-н-бутилпиперидин-1-ил)масляной кислоты (4) (0,331 г, 1,37 ммоль) в 5 мл 4 М HCl. Реакционную смесь кипятят с обратным холодильником в течение 48 час с последующим добавлением 4,0 М NaOH. Реакционную смесь экстрагируют дихлорметаном (4×25 мл). Объединенные органические фазы сушат над MgSO4 и упаривают, получая при этом 0,40 г сырого продукта. Сырой материал подвергают колоночной хроматографии (элюент: СН2Cl2:МеОН (20:1) и выделенный продукт упаривают досуха с 1,0 М HCl в эфире, получая при этом чистое, указанное в заголовке соединение 36 (0,210 г, 44%). 1H ЯМР (CD3OD) 0,92 (т, 3H), 1,33 (м, 6H), 1,54 (м, 3H), 1,99 (д, 2H), 2,43 (м, 2H), 2,65 (м, 2H), 3,00 (м, 2H), 3,28 (м, 2H), 3,63 (м, 2H), 7,38 (д, 1H), 7,47 (т, 1H), 7,59 (д, 1H).
Пример 33. 3-(2-(4-н-Бутилпиперидин)-1-илэтил)-1Н-индол (37). В колбу на 25 мл, снабженную магнитной мешалкой, загружают гидрохлорид 4-н-бутилпиперидина 3 (0,256 г, 1,4 ммоль) и карбонат калия (0,5 г, 3,6 ммоль) в диоксане (5 мл). Смесь перемешивают при комнатной температуре в течение 2 час с последующим добавлением 3-(2-бромэтил)индола (0,30 г, 1,3 ммоль), растворенного в диоксане (5 мл). Смесь затем перемешивают при 50°С в течение 24 час. Добавляют воду (15 мл) с последующей экстракцией этилацетатом (3×50 мл). Объединенные органические фазы сушат над MgSO4 и упаривают, получая при этом 1,02 г сырого продукта. Сырой продукт подвергают колоночной хроматографии (элюент: СН2Cl2:МеОН (20:1)), получая при этом чистое, указанное в заголовке соединение 37 (0,08 г, 21%). 1H ЯМР (CDCl3) 0,90 (т, 3H), 1,25-1,49 (м, 9H), 1,72-1,79 (м, 2H), 2,77 (т, 2H), 3,06 (т, 2H), 3,16 (д, 2H), 7,03 (с, 1H), 7,11 (т, 1H), 7,19 (т, 1H), 7,36 (д, 1H), 7,61 (д, 1H), 8,09-8,16 (с, 1H).
Пример 34. трет-Бутиловый эфир (2-(4-хлорбутан-1-он)фенил) карбаминовой кислоты (38). В сухую 100 мл одногорлую колбу, снабженную холодильником, магнитной мешалкой и вводным отверстием для аргона, добавляют 4-хлорбутаноилхлорид (624 мг, 44 ммоль) м бис(ацетонитрил)дихлорпалладий (34 мг) в 10 мл сухого толуола. К смеси добавляют трет-бутиловый эфир (2-триметилстаннилфенил)карбаминовой кислоты (1,5 г, 42 ммоль) (Bioorg. Med. Chem., 6:811 (1998)), растворенный в 15 мл сухого толуола. Смесь затем кипятят с обратным холодильником в течение 1 часа и затем перемешивают при комнатной температуре в течение 17 час. Реакционную смесь упаривают досуха, что дает сырой продукт (1,6 г), который подвергают колоночной хроматографии (элюент:гептан:EtOAc, 10:1), получая при этом чистое, указанное в заголовке соединение 38 (1,15 г, 92%). 1H ЯМР (CDCl3) 1,52 (т, 9H), 2,22 (м, 2H), 3,22 (т, 2H), 3,68 (т, 2H), 7,03 (т, 1H), 7,51 (т, 1H), 7,91 (д, 1H), 8,48 (д, 1H), 10,90 (с, 1H).
Пример 35. Трет-Бутиловый эфир (2-(3-(4-н-бутилпиперидин)-1-илпропил)фенил)карбаминовой кислоты (39). В сухую колбу на 5 мл, снабженную магнитной мешалкой и вводом для аргона, добавляют 38 (0,5 г, 1,7 ммоль) и 4-н-бутилпиперидин 3 (1,5 г, 10,6 ммоль) и оставляют для перемешивания при 60°C в течение 70 час. Сырую реакционную смесь подвергают колоночной хроматографии (элюент: СН2Cl2:МеОН, 20:1), получая при этом чистое соединение 39 (0,49 г, 72%). 1H ЯМР (CDCl3) 0,87 (т, 3H), 1,18-1,27 (м, 9H), 1,52 (с, 9H), 1,64 (м, 2H), 1,94 (м, 4H), 2,41 (т, 2H), 2,91 (д, 2H), 3,03 (т, 2H), 7,00, (т, 1H), 7,49 (т, 1H), 7,91 (д, 1H), 8,46 (д, 1H), 10,97 (с, 1H).
Пример 36. 3-(3-(4-н-Бутилпиперидин)-1-илпропил)-1Н-индазол (40) (39MF34). Соединение 39 (0,06 г, 0,15 ммоль), растворенное в 2 мл 4,0М HCl в диоксане, добавляют в 5 мл колбу и перемешивают при комнатной температуре в течение 1 часа. Смесь упаривают досуха и затем снова растворяют в 1 мл концентрированной HCl и температуру регулируют до 0°C при помощи бани лед/вода. К охлажденной смеси добавляют нитрит натрия (0,010 г, 0,15 ммоль), растворенный в 2 мл воды, и реакционную смесь выдерживают при 0°C в течение 1,5 часа с последующим добавлением дихлорида олова (0,08 г, 0,36 ммоль), растворенного в 2 мл концентрированной HCl. После выдерживания 1,5 часа при 0°C образуются кристаллы. Кристаллы отфильтровывают и промывают водой, получая при этом сырой продукт (0,07 г). Сырой продукт подвергают колоночной хроматографии (элюент: СН2Cl2:МеОН, 20:1), получая при этом чистое соединение 40 (9,0 мг, 20%). 1H ЯМР (CDCl3) 0,88 (т, 3H), 1,19-1,33 (м, 9H), 1,67 (д, 2H), 1,95 (т, 2H), 2,08 (м, 2H), 2,50 (т, 2H), 2,93-3,20 (м, 4H), 7,12 (т, 1H), 7,36 (т, 1H), 7,43 (д, 1H), 7,71 (д, 1H), 9,87-10,05 (с, 1H).
Пример 37. 3-(2-Хлорэтокси)-7-метилбензо[d]изоксазол (41). 1-Бром-2-хлорэтан (168 мкл, 2,0 ммоль) добавляют к 5 мл ДМФ в 50 мл колбе. Добавляют 7-метилбензо[d]изоксазол-3-ол (298 мг, 2,0 ммоль), карбонат калия (276 мг, 2,0 ммоль) и дополнительный ДМФ (5 мл) и смесь перемешивают в течение 12 час. Добавляют этилацетат (50 мл) и Н2О (50 мл). Две фазы разделяют и водную фазу экстрагируют этилацетатом. Объединенные органические фазы промывают насыщенным раствором соли, сушат над MgSO4 и упаривают досуха, получая при этом 420 мг сырого продукта. Сырой продукт подвергают колоночной хроматографии (0-5% метанол в дихлорметане) с получением чистого, указанного в заголовке соединения 41 (290 мг, 70%). 1H ЯМР (CDCl3) 2,5 (с, 3H), 3,9 (т, 2H), 4,7 (т, 2H), 7,2 (т, 1H), 7,3 (д, 1H), 7,5 (д, 2H).
Пример 38. 3-(2-(4-н-Бутилпиперидин)этокси)-7-метилбензо[d]изоксазол (42) (35AKU-41). Соединение 41 (294 мг, 1,4 ммоль) растворяют в ДМФА (5 мл) в 50 мл колбе с последующим добавлением смеси 4-н-бутилпиперидина (284 мг; 1,6 ммоль) и карбоната калия (442 мг; 3,2 ммоль), растворенной в ДМФА (15 мл). Смесь перемешивают в течение 2 дней при 80°C. Добавляют этилацетат (50 мл) и Н2О (50 мл), фазы разделяют и водную фазу экстрагируют этилацетатом (3×50 мл). Объединенные органические фазы промывают насыщенным раствором соли, сушат над MgSO4 и упаривают досуха с получением сырого продукта (454 мг). Сырой продукт подвергают колоночной хроматографии (0-5% метанол в дихлорметане), получая при этом чистое, указанное в заголовке соединение 42 (131 мг, 30%). Оксалатную соль получают из щавелевой кислоты (1,1 экв.) в смеси метанол/диэтиловый эфир. 1H ЯМР (CDCl3) 0,9 (т, 3H), 1,2-1,3 (м, 9H), 1,7 (д, 2H), 2,1 (т, 2H), 2,5 (с, 3H), 2,9 (т, 2H), 3,0 (д, 2H), 4,6 (т, 2H), 7,15 (т, 1H), 7,3 (д, 1H), 7,45 (д, 1H).
Пример 39. 1-(3-(4-Метилпиперидин)-1-илпропил)-1Н-индазол (43) (46RO13.48). Твердый К2СО3 (70 мг, 0,5 ммоль) добавляют к смеси 7-бром-1-(3-(4-н-бутилпиперидин)-1-илпропил)-1Н-индола (96 мг, 0,4 ммоль) и 4-метилпиперидина (30 мг, 0,3 ммоль) в CH3CN (2 мл). Образовавшуюся суспензию перемешивают при 50°C в течение 48 час и затем охлаждают до температуры окружающей среды. Суспензию затем выливают в воду (10 мл) и обрабатывают следующим образом: экстрагируют этилацетатом (3×10 мл), промывают собранные органические фазы последовательно водой (3×5 мл) и насыщенным раствором соли с последующей сушкой над MgSO4 и удалением растворителя роторным испарением. Остаток очищают на ISOLUTE SCX, получая при этом соединение 43 (25 мг, 24%). Оксалатную соль получают из щавелевой кислоты (1,1 экв.) в смеси метанол/диэтиловый эфир. 1H ЯМР (CD3OD) δ 0,9 (т, 3H), 1,2 (м, 2H), 1,6 (м, 1H), 1,8 (д, 2H), 2,15 (м, 2H), 2,8 (м, 2H), 3,0 (м, 2H), 3,4 (м, 2H), 4,45 (т, 2H), 7,1 (т, 1H), 7,35 (т, 1H), 7,5 (д, 1H), 7,7 (д, 1H), 8,0 (с, 1H).
Пример 40. 1-(3-(4-Пентилпиперидин)-1-илпропил)-1Н-индазол (44) (46RO13.57). Твердый К2СО3 (35 мг, 0,25 ммоль) добавляют к смеси 7-бром-1-(3-(4-н-бутилпиперидин)-1-илпропил)-1Н-индола (48 мг, 0,4 ммоль) и 4-пентилпиперидина (23 мг, 0,15 ммоль) в CH3CN (2 мл). Образовавшуюся суспензию перемешивают при 50°C в течение 48 час и затем охлаждают до температуры окружающей среды. Суспензию затем выливают в воду (10 мл) и обрабатывают следующим образом: экстрагируют этилацетатом (3×10 мл), промывают собранные органические фазы последовательно водой (3×5 мл) и насыщенным раствором соли с последующей сушкой над MgSO4 и удалением растворителя роторным испарением. Остаток очищают на ISOLUTE SCX, получая при этом соединение 44 (25 мг, 40%). Оксалатную соль получают из щавелевой кислоты (1,1 экв.) в смеси метанол/диэтиловый эфир. 1H ЯМР (CD3OD) δ 0,9 (т, 3H), 1,2 (м, 12H), 1,6 (м, 1H), 1,8 (д, 2H), 2,15 (м, 2H), 2,8 (м, 2H), 3,0 (м, 2H), 3,4 (м, 2H), 4,45 (т, 2H), 7,1 (т, 1H), 7,35 (т, 1H), 7,5 (д, 1H), 7,7 (д, 1H), 8,0 (с, 1H).
Пример 41. 1-(3-(4-Пропилпиперидин)-1-илпропил)-1Н-индазол (45) (46RO13.55LH). Твердый К2СО3 (35 мг, 0,25 ммоль) добавляют к смеси 7-бром-1-(3-(4-н-бутилпиперидин)-1-илпропил)-1Н-индола (48 мг, 0,2 ммоль) и 4-пропилпиперидина (19 мг, 0,15 ммоль) в CH3CN (2 мл). Образовавшуюся суспензию перемешивают при 50°C в течение 48 час и затем охлаждают до температуры окружающей среды. Суспензию затем выливают в воду (10 мл) и обрабатывают следующим образом: экстрагируют этилацетатом (3×10 мл), промывают собранные органические фазы последовательно водой (3×5 мл) и насыщенным раствором соли с последующей сушкой над MgSO4 и удалением растворителя роторным испарением. Остаток очищают на ISOLUTE SCX, получая при этом указанное в заголовке соединение 45 (16 мг, 28%). Оксалатную соль получают из щавелевой кислоты (1,1 экв.) в смеси метанол/диэтиловый эфир. 1H ЯМР (CD3OD) δ 0,9 (т, 3H), 1,2 (м, 6H), 1,6 (м, 1H), 1,8 (д, 2H), 2,15 (м, 2H), 2,8 (м, 2H), 3,0 (м, 2H), 3,4 (м, 2H), 4,45 (т, 2H), 7,1 (т, 1H), 7,35 (т, 1H), 7,5 (д, 1H), 7,7 (д, 1H), 8,0 (с, 1H).
Пример 42. 1-(3-(4-(3-Метилбутил)пиперидин)-1-илпропил)-1Н-индазол (46) (46RO13.58). Твердый К2СО3 (35 мг, 0,25 ммоль) добавляют к смеси 7-бром-1-(3-(4-н-бутилпиперидин)-1-илпропил)-1Н-индола (48 мг, 0,2 ммоль) и 4-(3-метилбутил) пиперидина (23 мг, 0,15 ммоль) в CH3CN (2 мл). Образовавшуюся суспензию перемешивают при 50°C в течение 48 час и затем охлаждают до температуры окружающей среды. Суспензию затем выливают в воду (10 мл) и обрабатывают следующим образом: экстрагируют этилацетатом (3×10 мл), промывают собранные органические фазы последовательно водой (3×5 мл) и насыщенным раствором соли с последующей сушкой над MgSO4 и удалением растворителя роторным испарением. Остаток очищают на ISOLUTE SCX, получая при этом указанное в заголовке соединение 46 (18 мг, 30%). Оксалатную соль получают из щавелевой кислоты (1,1 экв.) в смеси метанол/диэтиловый эфир. 1H ЯМР (CD3OD) δ 0,9 (т, 6H), 1,2-1,5 (м, 8H), 1,8 (д, 2H), 2,15 (м, 2H), 2,8 (м, 2H), 3,0 (м, 2H), 3,4 (м, 2H), 4,45 (т, 2H), 7,1 (т, 1H), 7,35 (т, 1H), 7,5 (д, 1H), 7,7 (д, 1H), 8,0 (с, 1H).
Пример 43. 1-(3-(4-Пентилиденпиперидин)-1-илпропил)-1Н-индазол (47) (46RO13.46). Твердый К2СО3 (35 мг, 0,25 ммоль) добавляют к смеси 7-бром-1-(3-(4-н-бутилпиперидин)-1-илпропил)-1Н-индола (48 мг, 0,2 ммоль) и 4-пентилиденпиперидина (23 мг, 0,15 ммоль) в CH3CN (2 мл). Образовавшуюся суспензию перемешивают при 50°C в течение 48 час и затем охлаждают до температуры окружающей среды. Суспензию выливают в воду (10 мл) и обрабатывают следующим образом: экстрагируют этилацетатом (3×10 мл), промывают собранные органические фазы последовательно водой (3×5 мл) и насыщенным раствором соли с последующей сушкой над MgSO4 и удалением растворителя роторным испарением. Остаток очищают на ISOLUTE SCX, получая при этом указанное в заголовке соединение 47 (3 мг, 5%). Оксалатную соль получают из щавелевой кислоты (1,1 экв.) в смеси метанол/диэтиловый эфир. 1H ЯМР (CD3OD) δ 0,9 (т, 3H), 1,3 (м, 4H), 2,0 (м, 2H), 2,3 (м, 3H), 2,35 (д, 2H), 2,7 (м, 2H), 3,1 (м, 3H), 3,4 (м, 2H), 4,45 (т, 2H), 5,3 (м, 1H), 7,1 (т, 1H), 7,35 (т, 1H), 7,5 (д, 1H), 7,7 (д, 1H), 8,0 (с, 1H).
Пример 44. 1-(3-(4-Пропилиденпиперидин)-1-илпропил)-1Н-индазол (48) (46RO13.45). Твердый К2СО3 (35 мг, 0,25 ммоль) добавляют к смеси 7-бром-1-(3-(4-н-бутилпиперидин)-1-илпропил)-1Н-индола (48 мг, 0,2 ммоль) и 4-пропилиденпиперидина (18 мг, 0,15 ммоль) в CH3CN (2 мл). Образовавшуюся суспензию перемешивают при 50°C в течение 48 час и затем охлаждают до температуры окружающей среды. Суспензию затем выливают в воду (10 мл) и обрабатывают следующим образом: экстрагируют этилацетатом (3×10 мл), промывают собранные органические фазы последовательно водой (3×5 мл) и насыщенным раствором соли с последующей сушкой над MgSO4 и удалением растворителя роторным испарением. Остаток очищают на ISOLUTE SCX, получая при этом указанное в заголовке соединение 48 (10 мг, 25%). Оксалатную соль получают из щавелевой кислоты (1,1 экв.) в смеси метанол/диэтиловый эфир. 1H ЯМР (CD3OD) δ 0,9 (т, 3H), 2,0 (т, 2H), 2,4 (м, 6H), 3,1 (м, 4H), 3,4 (м, 2H), 4,45 (т, 2H), 5,35 (т, 1H), 7,1 (т, 1H), 7,35 (т, 1H), 7,5 (д, 1H), 7,7 (д, 1H), 8,0 (с, 1H).
Пример 45. 1-Бензо[b]тиофен-2-ил-4-(4-бутилпиперидин-1-ил) бутан-1-он (49) (45NK99/оксалат). н-BuLi в гептанах (0,77 мл, 1,0 ммоль, 1,3 М) добавляют по каплям к бензо[b]тиофену (134 мг, 1,0 ммоль) в ТГФ (4 мл) при -78°C в атмосфере аргона. Реакционную смесь перемешивают при -78°C в течение 15 мин, затем добавляют 4-(4-бутилпиперидин-1-ил)-N-метокси-N-метилбутирамид (135 мг, 0,5 ммоль) в ТГФ (1 мл). Реакционную смесь перемешивают при -78°C в течение 30 мин, затем добавляют NH4Cl (насыщ., водн., 1 мл) и реакционную смесь нагревают до комнатной температуры. Продукт экстрагируют этилацетатом (2×20 мл) и органический слой промывают водой (10 мл), сушат (К2СО3), фильтруют и концентрируют в вакууме. Продукт очищают колоночной хроматографией (0-25% этилацетат в гептанах + 0,1% Et3N) Выход 94 мг (55%). Оксалатную соль получают добавлением щавелевой кислоты в смеси диэтиловый эфир:метанол (10:1), получая при этом белый осадок, который отфильтровывают и сушат. 1H ЯМР (DMSO): δ 0,91 (т, 3H), 1,24-1,56 (м, 9H), 1,87 (шир.д, 2H), 2,08 (м, 2H), 2,93 (м, 2H), 3,14 (м, 2H), 3,24 (м, 2H), 3,47 (м, 2H), 7,46-7,59 (м, 2H), 8,05 (м, 2H), 8,36 (с, 1H).
Пример 46. 4-(4-Бутилпиперидин-1-ил)-1-(3-метилбензофуран-2-ил)бутан-1-он (50) (45NK100/оксалат). н-BuLi в гептанах (0,85 мл, 1,1 ммоль, 1,3 М) добавляют по каплям к 3-метилбензофурану (132 мг, 1,0 ммоль) в ТГФ (4 мл) при -78°C в атмосфере аргона. Реакционную смесь перемешивают при -78°C в течение 20 мин, затем добавляют 4-(4-бутилпиперидин-1-ил)-N-метокси-N-метилбутирамид (135 мг, 0,5 ммоль) в ТГФ (1 мл). Реакционную смесь перемешивают при -78°C в течение 45 мин, затем добавляют NH4Cl (насыщ., водн., 1 мл) и реакционную смесь нагревают до комнатной температуры. Продукт экстрагируют этилацетатом (2×20 мл) и органический слой промывают водой (10 мл), сушат (К2СО3), фильтруют и концентрируют в вакууме. Продукт очищают колоночной хроматографией (0-20% этилацетат в гептанах + 0,1% Et3N). Выход 38 мг (22%). Оксалатную соль получают добавлением щавелевой кислоты в смеси диэтиловый эфир:метанол (10:1), получая при этом белый осадок, который отфильтровывают и сушат. 1H ЯМР (CD3OD): δ 0,91 (т, 3H), 1,32 (м, 6H), 1,42-1,64 (м, 3H), 1,89 (шир.д, 2H), 2,15 (тт, 2H), 2,58 (с, 3H) 2,96 (м, 2H), 3,17 (м, 4H), 3,60 (м, 2H), 7,33 (м, 1H), 7,52 (м, 2H), 7,71 (м, 1H).
Пример 47. 4-(4-Бутилпиперидин-1-ил)-1-(5-фтор-3-метилбензо[b] тиофен-2-ил)бутан-1-он (51) (45NK105). н-BuLi в гептанах (0,50 мл, 0,8 ммоль, 1,6М) добавляют по каплям к 5-фтор-3-метилбензо[b]тиофену (166 мг, 1,0 ммоль) в ТГФ (4 мл) при -40°C в атмосфере аргона. Реакционную смесь перемешивают при -40°C в течение 40 мин, затем добавляют 4-(4-бутилпиперидин-1-ил)-N-метокси-N-метилбутирамид (135 мг, 0,5 ммоль) в ТГФ (1 мл). Реакционную смесь перемешивают при -40°C в течение 30 мин, затем добавляют NH4Cl (насыщ., водн., 1 мл) и реакционную смесь нагревают до комнатной температуры. Продукт экстрагируют этилацетатом (2×20 мл) и органический слой промывают водой (10 мл), сушат (К2СО3), фильтруют и концентрируют в вакууме. Продукт очищают на Isco CombiFlash Sq 16x (колонка с 4,1 г диоксида кремния, элюирование гептанами (5 мин), 0-15% этилацетатом в гептанах (20 мин), 15% этилацетатом в гептанах (15 мин), всеми растворителями + 0,1% Et3N). Выход 39 мг (21%). Гидрохлоридную соль получают добавлением HCl (4М в диоксане) и перекристаллизовывают из смеси метанол-диэтиловый эфир, получая при этом белый осадок, который отфильтровывают и сушат. 1H ЯМР (свободное основание CDCl3): δ 0,87 (т, 3H), 1,10-1,35 (м, 9H), 1,62 (шир.д, 2H), 1,96 (м, 4H), 2,42 (т, 2H), 2,71 (с, 3H), 2,93 (м, 4H), 7,34 (дт, 1H), 7,49 (дд, 1H), 7,76 (дд, 1H).
Пример 48. 1-Бензофуран-2-ил-4-(4-бутилпиперидин-1-ил)бутан-1-он (52) (45NK106). н-BuLi в гептанах (0,50 мл, 0,8 ммоль, 1,6М) добавляют по каплям к бензофурану (118 мг, 1,0 ммоль) в ТГФ (4 мл) при -40°C в атмосфере аргона. Реакционную смесь перемешивают при -40°C в течение 40 мин, затем добавляют 4-(4-бутилпиперидин-1-ил)-N-метокси-N-метилбутирамид (135 мг, 0,5 ммоль) в ТГФ (1 мл). Реакционную смесь перемешивают при -40°C в течение 30 мин, затем добавляют NH4Cl (насыщ., водн., 1 мл) и реакционную смесь нагревают до комнатной температуры. Продукт экстрагируют этилацетатом (2×20 мл) и органический слой промывают водой (10 мл), сушат (К2СО3), фильтруют и концентрируют в вакууме. Продукт очищают на Isco CombiFlash Sq 16x (колонка с 4,1 г диоксида кремния, элюирование гептанами (5 мин), 0-15% этилацетатом в гептанах (20 мин), 15% этилацетатом в гептанах (15 мин), всеми растворителями + 0,1% Et3N). Выход 61 мг (50%). Гидрохлоридную соль получают добавлением HCl (4М в диоксане) и перекристаллизовывают из смеси метанол-диэтиловый эфир, получая при этом белый осадок, который отфильтровывают и сушат. 1H ЯМР (свободное основание CDCl3): δ 0,87 (т, 3H), 1,10-1,30 (м, 9H), 1,59 (шир.д, 2H), 1,93 (м, 2H), 1,99 (тт, 2H), 2,40 (т, 2H), 2,87 (м, 2H), 2,96 (т, 2H), 7,30 (м, 1H), 7,45 (м, 1H), 7,48 (м, 1H), 7,57 (м, 1H), 7,69 (м, 1H).
Пример 49. 1-(3-Бромбензо[b]тиофен-2-ил)-4-(4-бутилпиперидин-1-ил)бутан-1-он (53) (45NK108). н-BuLi в пентанах (0,48 мл, 0,8 ммоль, 1,7М) добавляют по каплям к 3-бромбензо[b]тиофену (213 мг, 1,0 ммоль) в ТГФ (4 мл) при -78°C в атмосфере аргона. Реакционную смесь перемешивают при -78°C в течение 40 мин, затем добавляют 4-(4-бутилпиперидин-1-ил)-N-метокси-N-метилбутирамид (135 мг, 0,5 ммоль) в ТГФ (1 мл). Реакционную смесь перемешивают при -78°C в течение 30 мин, затем добавляют NH4Cl (насыщ., водн., 1 мл) и реакционную смесь нагревают до комнатной температуры. Продукт экстрагируют этилацетатом (2×20 мл) и органический слой промывают водой (10 мл), сушат (К2СО3), фильтруют и концентрируют в вакууме. Продукт очищают на Isco CombiFlash Sq 16x (колонка с 4,1 г диоксида кремния, элюирование гептанами (5 мин), 0-15% этилацетатом в гептанах (20 мин), 15% этилацетатом в гептанах (15 мин), всеми растворителями + 0,1% Et3N). Выход 18 мг (4%). Гидрохлоридную соль получают добавлением HCl (4 М в диоксане) и перекристаллизовывают из смеси метанол-диэтиловый эфир, получая при этом белый осадок, который отфильтровывают и сушат. 1H ЯМР (свободное основание CDCl3): δ 0,88 (т, 3H), 1,12-1,28 (м, 9H), 1,62 (шир.д, 2H), 1,94 (м, 2H), 2,02 (тт, 2H), 2,45 (т, 2H), 2,92 (шир.д, 2H), 3,18 (т, 2H), 7,51 (м, 2H), 7,83 (м, 1H), 7,98 (м, 1H).
Пример 50. 1-(3-Бензо[b]тиофен-2-илпропил)-4-бутилпиперидин (54) (45NK124). Н-BuLi в гептанах (0,75 мл, 1,2 ммоль, 1,6 М) добавляют по каплям к бензо[b]тиофену (134 мг, 1,0 ммоль) в ТГФ (4 мл) при -5°C в атмосфере аргона. Реакционную смесь перемешивают при -5°C в течение 15 мин, затем добавляют 1-хлор-3-иодпропан (151 мкл, 1,2 ммоль) и иодид меди (I) (19 мг, 0,1 ммоль). Реакционную смесь перемешивают при -5°C в течение 1 часа, затем при комнатной температуре в течение 0,5 часа. Добавляют воду (5 мл), продукт экстрагируют диэтиловым эфиром (2×10 мл) и органический слой сушат (К2СО3), фильтруют и концентрируют в вакууме. Продукт очищают колоночной хроматографией (0-2% этилацетат в гептанах), получая при этом 2-(3-хлорпропил)бензо[b]тиофен (93 мг, 44%). 1H ЯМР (CDCl3): δ 2,22 (тт, 2H), 3,10 (дт, 2H), 3,61 (т, 2H), 7,06 (м, 1H), 7,30 (м, 2H), 7,69 (м, 1H), 7,78 (м, 1H).
2-(3-Хлорпропил)бензо[b]тиофен (53 мг, 0,25 ммоль), 4-бутилпиперидин (36 мг, 0,25 ммоль), иодид натрия (75 мг, 0,5 ммоль) и карбонат натрия (53 мг, 0,5 ммоль) в ацетонитриле (2 мл) встряхивают при 80°C в течение 18 час, затем реакционную смесь охлаждают до комнатной температуры. Добавляют воду (5 мл) и продукт экстрагируют этилацетатом (2×10 мл), сушат (К2СО3), фильтруют и концентрируют в вакууме. Продукт очищают колоночной хроматографией (0-15% этилацетат в гептанах + 0,1% Et3N), получая при этом указанное в заголовке соединение 54. Выход 29 мг (37%). Гидрохлоридную соль получают добавлением HCl (4М в диоксане) и перекристаллизовывают из смеси метанол-диэтиловый эфир с получением белого осадка, который отфильтровывают и сушат. 1H ЯМР (CD3OD): δ 0,91 (т, 3H), 1,32 (м, 6H), 1,39 (м, 2H), 1,55 (м, 1H), 1,96 (шир.д, 2H), 2,19 (тт, 2H), 2,93 (м, 2H), 3,04 (т, 2H), 3,14 (м, 2H), 3,53 (м, 2H), 7,14 (шир.с, 1H), 7,26 (м, 1H), 7,31 (м, 1H), 7,68 (м, 1H), 7,77 (м, 1H).
Пример 51. 1-(3-Бензофуран-2-илпропил)-4-бутилпиперидин (55) (56NK03). н-BuLi в гептанах (1,5 мл, 2,4 ммоль, 1,6М) добавляют по каплям к бензофурану (236 мг, 2,0 ммоль) в ТГФ (5 мл) при -20°C в атмосфере аргона. Реакционную смесь перемешивают при -15°C в течение 30 мин, затем добавляют 1-хлор-3-иодпропан (322 мкл, 3,0 ммоль) и иодид меди (I) (38 мг, 0,2 ммоль). Реакционную смесь перемешивают при -15°C в течение 1 часа, затем добавляют NH4Cl (насыщ. водн., 5 мл). Продукт экстрагируют диэтиловым эфиром (2×30 мл) и органический слой промывают насыщенным раствором соли (10 мл), сушат (К2СО3), фильтруют и концентрируют в вакууме. Продукт очищают колоночной хроматографией (0-1% диэтиловый эфир в гептанах), получая при этом 2-(3-хлорпропил)бензофуран (101 мг, 26%). 1H ЯМР (CDCl3): δ 2,23 (тт, 2H), 2,97 (дт, 2H), 3,62 (т, 2H), 6,45 (к., 1H), 7,21 (м, 2H), 7,42 (м, 1H), 7,50 (м, 1H).
2-(3-Хлорпропил)бензофуран (101 мг, 0,52 ммоль), 4-бутилпиперидин (74 мг, 0,52 ммоль), иодид натрия (156 мг, 1,04 ммоль) и карбонат натрия (110 мг, 1,04 ммоль) в ацетонитриле (2 мл) встряхивают при 80°C в течение 18 час, затем реакционную смесь охлаждают до комнатной температуры. Добавляют воду (1 мл), продукт экстрагируют этилацетатом (2×2 мл) и органический слой загружают в ионообменную колонку Varian SCX. Колонку промывают метанолом (2 объема колонки) и продукт элюируют из колонки с использованием 10% гидроксида аммония в метаноле (2 объема колонки). Сорбат концентрируют в вакууме, растворяют в ацетоне, сушат (К2СО3) и концентрируют в вакууме. Продукт очищают колоночной хроматографией (0-12% этилацетат в гептанах + 0,1% Et3N), получая при этом указанное в заголовке соединение 55. Выход 86 мг (55%). Гидрохлоридную соль получают добавлением HCl (4М в диоксане) и перекристаллизовывают из смеси метанол-диэтиловый эфир с получением белого чешуйчатого твердого вещества, которое отфильтровывают и сушат. 1H ЯМР (CD3OD): δ 0,90 (т, 3H), 1,30 (м, 6H), 1,48 (м, 3H), 1,95 (шир.д, 2H), 2,21 (м, 4H), 2,91 (м, 4H), 3,16 (м, 2H), 3,55 (шир.д, 2H), 6,57 (с, 1H), 7,17 (м, 2H), 7,38 (м, 2H), 7,48 (м, 1H).
Пример 52. 4-Бутил-1-[3-(3-метилбензофуран-2-илпропил] пиперидин (56) (56NK04). н-BuLi в гептанах (1,5 мл, 2,4 ммоль, 1,6М) добавляют по каплям к 3-метилбензофурану (264 мг, 2,0 ммоль) в ТГФ (5 мл) при -20°C в атмосфере аргона. Реакционную смесь перемешивают при -15°C в течение 30 мин, затем добавляют 1-хлор-3-иодпропан (322 мкл, 3,0 ммоль) и иодид меди (I) (38 мг, 0,2 ммоль). Реакционную смесь перемешивают при -15°C в течение 1 часа, затем добавляют NH4Cl (насыщ. водн., 5 мл). Продукт экстрагируют диэтиловым эфиром (2×30 мл) и органический слой промывают насыщенным раствором соли (10 мл), сушат (К2СО3), фильтруют и концентрируют в вакууме. Продукт очищают колоночной хроматографией (0-1% диэтиловый эфир в гептанах), получая при этом 2-(3-хлорпропил)-3-метилбензофуран (25 мг, 6%). 1H ЯМР (CDCl3): δ: 2,19 (тт, 2H), 2,22 (с, 3H), 2,94 (т, 2H), 3,57 (т, 2H), 7,22 (м, 2H), 7,38 (м, 1H), 7,44 (м, 1H).
2-(3-Хлорпропил)-3-метилбензофуран (25 мг, 0,12 ммоль), 4-бутилпиперидин (17 мг, 0,12 ммоль), иодид натрия (35 мг, 0,24 ммоль) и карбонат натрия (25 мг, 0,24 ммоль) в ацетонитриле (2 мл) встряхивают при 80°C в течение 18 час, затем реакционную смесь охлаждают до комнатной температуры. Добавляют воду (1 мл), продукт экстрагируют этилацетатом (2×2 мл) и органический слой загружают в ионообменную колонку Varian SCX. Колонку промывают метанолом (2 объема колонки), затем продукт элюируют из колонки с использованием 10% гидроксида аммония в метаноле (2 объема колонки). Сорбат концентрируют в вакууме, растворяют в ацетоне, сушат (К2СО3) и концентрируют в вакууме. Продукт очищают колоночной хроматографией (0-12% этилацетат в гептанах + 0,1% Et3N), получая при этом указанное в заголовке соединение 56. Выход 14 мг (38%). Гидрохлоридную соль получают добавлением HCl (4М в диоксане) и перекристаллизовывают из смеси метанол-диэтиловый эфир с получением белого твердого вещества, которое отфильтровывают и сушат. 1H ЯМР (CD3OD): δ 0,91 (т, 3H), 1,28-1,45 (м, 8H), 1,55 (м, 1H), 1,96 (шир.д, 2H), 2,17 (м, 2H), 2,22 (с, 3H), 2,89 (т, 2H), 2,94 (м, 2H), 3,14 (м, 2H), 3,54 (м, 2H), 7,20 (м, 2H), 7,34 (м, 1H), 7,45 (м, 1H).
Пример 53. 4-Бутил-1-[3-(5-фтор-3-метилбензо[b]тиофен-2-ил) пропил]бутилпиперидин (57) (56NK05). н-BuLi в гептанах (1,5 мл, 2,4 ммоль, 1,6М) добавляют по каплям к 5-фтор-3-метилбензо[b]тиофену (332 мг, 2,0 ммоль) в ТГФ (5 мл) при -20°C в атмосфере аргона. Реакционную смесь перемешивают при -15°C в течение 30 мин, затем добавляют 1-хлор-3-иодпропан (322 мкл, 3,0 ммоль) и иодид меди (I) (38 мг, 0,2 ммоль). Реакционную смесь перемешивают при -15°C в течение 1 часа, затем добавляют NH4Cl (насыщ. водн., 5 мл). Продукт экстрагируют диэтиловым эфиром (2×30 мл) и органический слой промывают насыщенным раствором соли (10 мл), сушат (К2СО3), фильтруют и концентрируют в вакууме. Продукт очищают колоночной хроматографией (0-1% диэтиловый эфир в гептанах), получая при этом 2-(3-хлорпропил)-5-фтор-3-метилбензо[b]тиофен (180 мг, 37%). 1H ЯМР (CDCl3): δ 2,19 (тт, 2H), 2,22 (с, 3H), 2,94 (т, 2H), 3,57 (т, 2H), 7,04 (дт, 1H), 7,28 (дд, 1H), 7,66 (дд, 1H).
2-(3-Хлорпропил)-5-фтор-3-метилбензо[b]тиофен (180 мг, 0,74 ммоль), 4-бутилпиперидин (212 мг, 0,74 ммоль), иодид натрия (225 мг, 1,48 ммоль) и карбонат натрия (159 мг, 1,48 ммоль) в ацетонитриле (2 мл) встряхивают при 80°C в течение 18 час, затем реакционную смесь охлаждают до комнатной температуры. Добавляют воду (1 мл), продукт экстрагируют этилацетатом (2×2 мл) и органический слой загружают в ионообменную колонку Varian SCX. Колонку промывают метанолом (2 объема колонки) и продукт элюируют из колонки с использованием 10% гидроксида аммония в метаноле (2 объема колонки). Сорбат концентрируют в вакууме, растворяют в ацетоне, сушат (К2СО3) и концентрируют в вакууме. Продукт очищают колоночной хроматографией (0-12% этилацетат в гептанах + 0,1% Et3N), получая при этом указанное в заголовке соединение 57. Выход 185 мг (72%). Гидрохлоридную соль получают добавлением HCl (4М в диоксане) и перекристаллизовывают из смеси метанол-диэтиловый эфир с получением белых кристаллов, которые отфильтровывают и сушат. 1H ЯМР (CD3OD): δ 0,90 (т, 3H), 1,31 (м, 6H), 1,37-1,62 (м, 3H), 1,94 (шир.д, 2H), 2,15 (м, 2H), 2,31 (с, 3H), 2,92 (шир.т, 2H), 3,01 (тм, 2H), 3,14 (м, 2H), 3,54 (шир.д, 2H), 7,06 (дт, 2H), 7,34 (дд, 1H), 7,73 (дд, 1H).
Пример 54. 2-(3-Иодпропил)бензо[b]тиофен (58). Смесь 2-(3-Хлорпропил)бензо[b]тиофен (902 мг, 4,28 ммоль) и иодид натрия (1,29 г, 8,6 ммоль) нагревают до 50°C в ацетоне (5 мл) в течение 72 час, затем охлаждают до комнатной температуры. Добавляют водный тиосульфат натрия (1М, 10 мл) и продукт экстрагируют диэтиловым эфиром (2×20 мл). Органический слой сушат (К2СО3), фильтруют и концентрируют в вакууме, получая при этом белое твердое вещество, которое фильтруют через целит и элюируют гептанами. Фильтрат концентрируют в вакууме с получением белого твердого вещества. Выход 1,038 г (80%). 1H ЯМР (CDCl3): δ 2,24 (тт, 2H), 3,04 (дт, 2H), 3,27 (т, 2H), 7,07 (кв, 1H), 7,28 (м, 2H), 7,68 (м, 1H), 7,77 (м, 1H).
Общая процедура алкилирования аминов
2-(3-Иодпропил)бензо[b]тиофен (33 мг, 0,11 ммоль) в DCM (240 мкл) добавляют к амину (0,10 ммоль) в DCM (200 мкл) и реакционную смесь встряхивают при комнатной температуре в течение 18 час. Добавляют DCM (1 мл) с последующей загрузкой макропористого триэтиламмонийметилполистиролкарбоната (50 мг, 3,06 ммоль/г, Agronaut Technologies) и реакционную смесь встряхивают при комнатной температуре в течение 1 часа. Добавляют полистиролметилизоцианат (60 мг, 1,25 ммоль/г, Argonaut Technologies) и реакционную смесь встряхивают при комнатной температуре в течение 2 час. Реакционную смесь затем загружают в ионообменную колонку Varian SCX. Колонку промывают метанолом (2 объема колонки) и продукт элюируют из колонки с использованием 10% гидроксида аммония в метаноле (2 объема колонки). Сорбат концентрируют в вакууме, растворяют в ацетоне, сушат (К2СО3) и концентрируют в вакууме.
Пример 55. 1-(3-Бензо[b]тиофен-2-илпропил)-4-метилпиперидин (59) (56NK38). Взаимодействие проводят по общей процедуре с использованием 4-метилпиперидина (17 мг, 0,10 ммоль), получая при этом 14 мг (53%) 1-(3-бензо[b]тиофен-2-илпропил)-4-метилпиперидина. 1H ЯМР (CD3OD): δ 0,92 (д, 3H), 1,27 (м, 2H), 1,34 (м, 1H), 1,63 (м, 2H), 1,94 (м, 4H), 2,40 (т, 2H), 2,91 (м, 4H), 7,00 (д, 1H), 7,28 (м, 2H), 7,66 (м, 1H), 7,76 (м, 1H).
Пример 56. 1-(3-Бензо[b]тиофен-2-илпропил)-4-бензилпиперидин (60) (56NK40). Взаимодействие проводят по общей процедуре с использованием 4-бензилпиперидина (17 мг, 0,10 ммоль), получая при этом 16 мг (45%) 1-(3-бензо[b]тиофен-2-илпропил)-4-бензилпиперидина. 1H ЯМР (CD3OD): δ 1,29 (м, 2H), 1,47-1,67 (м, 4H), 1,92 (м, 4H), 2,38 (м, 2H), 2,52 (м, 3H), 2,88 (м, 4H), 7,03 (м, 1H), 7,10-7,15 (м, 3H), 7,18-7,28 (м, 4H), 7,63 (м, 1H), 7,72 (м, 1H).
Пример 57. 1-(3-Бензо[b]тиофен-2-илпропил)-4-(2-метоксифенил) пиперидин (61) (56NK42). Взаимодействие проводят по общей процедуре с использованием 4-(2-метоксифенил)пиперидина (17 мг, 0,10 ммоль), получая при этом 17 мг (47%) 1-(3-бензо [b]тиофен-2-илпропил)-4-(2-метоксифенил)пиперидина. 1H ЯМР (CD3OD): δ 1,77 (м, 4H), 1,98 (м, 2H), 2,10 (м, 2H), 2,46 (м, 2H), 2,94 (м, 3H), 3,04 (м, 2H), 3,79 (с, 3H), 6,88 (м, 2H), 7,06 (шир.с, 1H), 7,13 (м, 2H), 7,26 (м, 2H), 7,65 (м, 1H), 7,73 (м, 1H).
Пример 58. 2-(3-Бромпропил)-2Н-бензотриазол (35AKU-17-2) (62). К раствору 1,3-дибромпропана (510 мкл, 5,0 ммоль) в диметилформамиде (10 мл) добавляют бензотриазол (600 мг, 5,0 ммоль) и КОН (430 мг, 7,7 ммоль). После перемешивания в течение 20 час при комнатной температуре добавляют воду (10 мл) и этилацетат (10 мл). Фазы разделяют и водную фазу снова экстрагируют этилацетатом (3×15 мл). Объединенные органические фазы сушат над MgSO4 и концентрируют в вакууме, получая при этом 1,44 г сырого материала. Сырой продукт очищают флэш-хроматографией (0-10% метанол в DCM) с получением 274 мг (23%) указанного в заголовке соединения 62. ТСХ (5% метанол в DCM): Rf=0,7. 1H ЯМР (400 МГц, CDCl3): δ=7,88-7,83 (2H, м); 7,41-7,36 (2H, м); 4,91 (2H, т); 3,44 (2H, т); 2,66 (2H, м).
Пример 59. 2-[3-(4-Бутилпиперидин-1-ил)пропил]-2Н-бензотриазол (63)(35AKU-18). К раствору 2-(3-бромпропил)-2Н-бензотриазола (274 мг, 1,14 ммоль) в диметилформамиде (5 мл) добавляют раствор 4-бутилпиперидина (142 мг, 1,0 ммоль) и КОН (125 мг, 2,2 ммоль) в диметилформамиде (5 мл). Смесь перемешивают в течение 20 час при комнатной температуре и затем добавляют этилацетат (10 мл) и воду (10 мл). Фазы разделяют и водную фазу снова экстрагируют этилацетатом (3×20 мл). Объединенные органические фазы сушат над MgSO4 и концентрируют в вакууме с получением 383 мг сырого материала. Сырой продукт очищают флэш-хроматографией (0-10% метанол в DCM), получая при этом 232 мг (77%) указанного в заголовке соединения 63. Оксалатную соль получают из щавелевой кислоты (1,1 экв.) в диэтиловом эфире. ТСХ (10% метанол в DCM): Rf=0,4. ВЭЖХ-МС (способ А): М+=301,2 (УФ/МС(%)=100/89). 1H ЯМР (400 МГц, CDCl3): δ=7,86 (2H, м); 7,37 (2H, м); 4,78 (2H, т); 2,93 (2H,д); 2,45 (2H, д); 2,34 (2H, м); 1,94 (2H, т); 1,61 (2H, д); 1,32-1,13 (9H, м); 0,88 (3H, т).
Пример 60. 1-(3-Бромпропил)-1Н-бензотриазол (35AKU-17-1) (64). К раствору 1,3-дибромпропана (510 мкл, 5,0 ммоль) в диметилформамиде (10 мл) добавляют бензотриазол (600 мг, 5,0 ммоль) и КОН (430 мг, 7,7 ммоль). После перемешивания в течение 20 час при комнатной температуре добавляют воду (15 мл) и этилацетат (15 мл). Фазы разделяют и водную фазу снова экстрагируют этилацетатом (3×20 мл). Объединенные органические фазы сушат над MgSO4 и концентрируют с получением 1,44 г сырого продукта. Сырой продукт очищают флэш-хроматографией (0-10% метанол в DCM), получая при этом 705 мг (59%) указанного в заголовке соединения 64. ТСХ (5% метанол в DCM): Rf=0,4. ВЭЖХ-МС (способ А): М+=239,9 (УФ/МС(%)=52/58).
Пример 61. 1-[3-(4-Бутилпиперидин-1-ил)пропил]-1Н-бензотриазол (65)(35AKU-19). К раствору 1-(3-бромпропил)-1Н-бензотриазола (705 мг, 1,6 ммоль) в диметилформамиде (5 мл) добавляют раствор 4-бутилпиперидина (140 мг, 1,0 ммоль) и КОН (240 мг, 4,3 ммоль) в диметилформамиде (5 мл). Смесь перемешивают в течение 20 час при комнатной температуре. Затем добавляют этилацетат (10 мл) и воду (10 мл). Фазы разделяют и водную фазу снова экстрагируют этилацетатом (3×15 мл). Объединенные органические фазы промывают насыщенным раствором соли, сушат над MgSO4 и упаривают досуха с получением 776 мг сырого материала. Сырой продукт очищают флэш-хроматографией (0-10% метанол в DCM), получая при этом 146 мг (49%) указанного в заголовке соединения 65. Оксалатную соль получают из щавелевой кислоты (1,1 экв.) в диэтиловом эфире. ТСХ (10% метанол в DCM): Rf=0,4. ВЭЖХ-МС (способ А): М+=301,2 (УФ/МС(%)=100/99). 1H ЯМР (400 МГц, CDCl3): δ=8,05 (1H, м); 7,62-7,33 (3H, м); 4,71 (2H, т); 2,85 (2H, д); 2,34 (2H, м); 2,22 (2H, м); 1,90 (2H, т); 1,67 (2H, д); 1,33-1,16 (9H, м); 0,89 (3H, т).
Пример 62. 1-[3-(4-Бутилпиперидин-1-ил)пропил]-1Н-индол-3-карбальдегид (66)(35AKU-24). К раствору 1,3-дибромпропана (410 мкл, 4,0 ммоль) в диметилформамиде (5 мл) добавляют раствор 1Н-индол-3-карбоксальдегида (582 мг, 4,0 ммоль) и КОН (456 мг, 8,1 ммоль) в диметилформамиде (5 мл). После перемешивания в течение 24 час добавляют 4-бутилпиперидин (359 мг, 2,0 ммоль) и дополнительный КОН (200 мг, 3,6 ммоль). После перемешивания в течение 20 час добавляют воду и этилацетат. Фазы разделяют и водную фазу снова экстрагируют этилацетатом (3×15 мл). Объединенные органические фазы промывают насыщенным раствором соли, сушат над MgSO4 и упаривают досуха с получением 1,04 г сырого продукта. Сырой продукт очищают флэш-хроматографией (0-10% метанол в DCM), получая при этом 252 мг (39%) указанного в заголовке соединения 66. ТСХ (10% метанол в DCM): Rf=0,5. ВЭЖХ-МС (способ А): М+=327,2 (УФ/МС(%)=99/96).
Пример 63. {1-[3-(4-Бутилпиперидин-1-ил)пропил]-1Н-индол-3-ил} метанол (67) (35AKU-26). К раствору 1-[3-(4-бутилпиперидин-1-ил)пропил]-1Н-индол-3-карбальдегида (120 мг, 0,37 ммоль) в метаноле (2 мл) медленно добавляют раствор NaBH4 (9,2 мг, 0,24 ммоль) в 20 мкл 2 М NaOH/1 мл воды. Смесь затем перемешивают в течение 20 час при комнатной температуре. Добавляют дополнительный NaBH4 (12 мг, 0,32 ммоль) и смесь перемешивают в течение дополнительных 2 час. Добавляют другую порцию NaBH4 (14 мг, 0,37 ммоль) и смесь перемешивают в течение ночи. Метанол частично удаляют с использованием Rotavap и добавляют этилацетат (10 мл) и воду (10 мл). Фазы разделяют и водную фазу снова экстрагируют этилацетатом (3×15 мл). Объединенные органические фазы сушат над MgSO4 и упаривают досуха, получая при этом 93 мг (71%) указанного в заголовке соединения 67. ТСХ (10% метанол в DCM): Rf=0,4. ВЭЖХ-МС (способ А): М+=329,2 (УФ/МС(%)=98/79). 1H ЯМР (400 МГц, CDCl3): δ=7,72 (1H, д); 7,36 (1H, д); 7,25-7,10 (3H, м); 4,86 (1H, с); 4,15 (2H, т); 2,84 (2H, д); 2,26 (2H, т); 1,99 (2H, м); 1,86 (2H, т); 1,71-1,62 (4H, м); 1,34-1,16 (9H, м); 0,90 (3H, т).
Пример 64. 1-[3-(4-Бутилпиперидин-1-ил)пропил]-2-фенил-1Н-бензоимидазол (68)(35AKU-28). К раствору 1,3-дибромпропана 205 мкл, 2,0 ммоль) в диметилформамиде (5 мл) добавляют 2-фенилбензимидазол (389 мг, 2,0 ммоль) и КОН (266 мг, 4,7 ммоль). После перемешивания в течение 16 час при комнатной температуре добавляют гидрохлорид 4-бутилпиперидина (176 мг, 1,0 ммоль). После 24 час перемешивания добавляют дополнительный КОН (270 мг, 4,8 ммоль) и смесь нагревают при 90°C в течение 3 час. После охлаждения добавляют воду (10 мл) и этилацетат (10 мл). Фазы разделяют и водную фазу снова экстрагируют этилацетатом (3×15 мл). Объединенные органические фазы промывают насыщенным раствором соли, сушат над MgSO4 и концентрируют в вакууме с получением 643 мг сырого продукта. Сырой продукт очищают флэш-хроматографией (0-10% метанол в DCM), получая при этом 71 мг (19%) указанного в заголовке соединения 68. ТСХ (10% метанол в DCM): Rf=0,7. ВЭЖХ-МС (способ А): М+=376,3 (УФ/МС(%)=100/100). 1H ЯМР (400 МГц, CDCl3): δ=7,85-7,27 (9H, м); 4,32 (2H, т); 2,73 (2H, д); 2,25 (2H, т); 1,95 (2H, м); 1,81 (2H, т); 1,62 (2H, д); 1,33-1,08 (9H, м); 0,90 (3H, т).
Пример 65. 1-[3-(4-Бутилпиперидин-1-ил)пропил]-3-хлор-1Н-индазол (69)(35AKU-34). К раствору 1,3-дибромропана (205 мкл, 2,0 ммоль) в диметилформамиде (5 мл) добавляют 3-хлориндазол (306 мг, 2,0 ммоль) и КОН (400 мг, 7,1 ммоль). После перемешивания суспензии в течение 16 час добавляют гидрохлорид 4-бутилпиперидина (180 мг, 1,0 ммоль) и диметилформамид (2 мл). После перемешивания 20 час добавляют воду (10 мл) и этилацетат (10 мл). Фазы разделяют и водную фазу снова экстрагируют этилацетатом (3×15 мл). Объединенные органические фазы промывают насыщенным раствором соли, сушат над MgSO4 и концентрируют в вакууме с получением 500 мг сырого продукта. Сырой продукт очищают флэш-хроматографией (0-10% метанол в DCM), получая при этом 121 мг (36%) указанного в заголовке соединения 69. Оксалатную соль получают из щавелевой кислоты (1,1 экв.) в диэтиловом эфире. ТСХ (10% метанол в DCM): Rf=0,5. ВЭЖХ-МС (способ А): М+=334,1 (УФ/МС(%)=100/100). 1H-ЯМР (400 МГц, CDCl3): δ=7,68-7,16 (4H, м); 4,46 (2Н, т); 3,13 (2H, д); 2,62 (2H, т); 2,35 (2H, м); 2,22 (2H, т); 1,76 (2H, д); 1,61-1,46 (2H, м); 1,36-1,24 (7H, м), 0,89 (3H, т).
Пример 66. 1-[3-(4-Бутилпиперидин-1-ил)пропил]-6-нитро-1Н-индазол (70)(35AKU-40). К раствору 1,3-дибромпропана (205 мкл, 2,0 ммоль) в диметилформамиде (20 мл) добавляют 6-нитроиндазол (325 мг, 2,0 ммоль) и К2СО3 (590 мг, 4,3 ммоль). После перемешивания суспензии в течение 20 час добавляют гидрохлорид 4-бутилпиперидина (178 мг, 1,0 ммоль) и диметилформамид (5 мл). После перемешивания 20 час добавляют воду (15 мл) и этилацетат (15 мл). Фазы разделяют и водную фазу снова экстрагируют этилацетатом (3×20 мл). Объединенные органические фазы промывают насыщенным раствором соли, сушат над MgSO4 и концентрируют в вакууме с получением 511 мг сырого продукта. Сырой продукт очищают ионообменной хроматографией (вымывают 10% водным NH4OH (25%) в метаноле) и флэш-хроматографией (0-10% метанол в DCM), получая при этом 21 мг (6%) указанного в заголовке соединения 70. Оксалатную соль получают из щавелевой кислоты (1,1 экв.) в диэтиловом эфире. ТСХ (10% метанол в DCM): Rf=0,4. ВЭЖХ-МС (способ А): М+=345,1 (УФ/МС(%)=97/96). 1H-ЯМР (400 МГц, CDCl3): δ=8,70 (1H, м); 8,07 (1H, м); 7,90 (1H, м); 7,75 (1H, м); 4,56 (2H, т); 2,86 (2H, д); 2,32 (2H, т); 2,24 (2H, м); 1,92 (2H, т); 1,68 (2H, м); 1,35-1,16 (9H, м); 0,89 (3H, т).
Пример 67. Бензо[d]изоксазол-3-ол (35AKU-44) (71). К раствору салицилгидроксамовой кислоты (1,53 г, 10 ммоль) в ТГФ (40 мл) добавляют раствор карбонилдиимидазола (1,62 г, 20 ммоль) в тетрагидрофуране (20 мл). Смесь перемешивают при кипячении с обратным холодильником в течение 4 час перед упариванием досуха. Добавляют воду (20 мл) и конц. HCl (водн.) (5 мл) и раствор охлаждают (5°C) в течение 30 мин. Образовавшийся осадок собирают фильтрованием и промывают 2 М HCl. Твердый материал растворяют в метаноле и концентрируют в вакууме, получая при этом 725 мг (54%) указанного в заголовке соединения 71. ТСХ (10% метанол в DCM): Rf=0,2. ВЭЖХ-МС (способ А): М+=136,1 (УФ/МС(%)=94/100). 1H-ЯМР (400 МГц, CDCl3, MeOD): δ=7,73 (1H, м); 7,56 (1H, м); 7,38 (1H, м); 7,28 (1H, м); 3,87 (1H, с).
Пример 68. 3-(2-Хлорэтокси)бензо[d]изоксазол (35AKU-45 (72)). К раствору 1-бром-2-хлорэтана (250 мкл, 3,0 ммоль) в диметилформамиде (10 мл) добавляют бензо[d]изоксазол-3-ол (400 мг, 3,0 ммоль) и К2СО3 (440 мг, 3,2 ммоль). Смесь перемешивают в течение 20 час и затем нагревают при 80°C в течение 1 часа. Добавляют этилацетат (10 мл) и воду (10 мл). Фазы разделяют и водную фазу снова экстрагируют этилацетатом (3×15 мл). Объединенные органические фазы промывают насыщенным раствором соли, сушат над MgSO4 и концентрируют в вакууме с получением 543 мг сырого продукта. Сырой продукт очищают флэш-хроматографией (0-10% метанол в DCM), получая при этом 378 мг (64%) указанного в заголовке соединения 72. ТСХ (10% метанол в DCM): Rf=0,8. 1H-ЯМР (400 МГц, CDCl3): δ=7,68 (1H, д); 7,55 (1H, т); 7,44 (1H, д); 7,28 (1Н, т); 4,72 (2H, т); 3,94 (2H, т).
Пример 69. 3-[2-(4-Бутилпиперидин-1-ил)этокси]бензо[d] изоксазол (73) (35AKU-46). Раствор 3-(2-хлорэтокси)бензо[d] изоксазол (378 мг, 1,9 ммоль), гидрохлорида 4-бутилпиперидина (270 мг, 1,5 ммоль) и К2СО3 (537 мг, 3,9 ммоль) в диметилформамиде (15 мл) нагревают до 80°C и перемешивают в течение 24 час. После охлаждения до комнатной температуры добавляют воду (15 мл) и этилацетат (15 мл). Фазы разделяют и водную фазу снова экстрагируют этилацетатом (3×20 мл). Объединенные органические фазы промывают насыщенным раствором соли, сушат над MgSO4 и концентрируют в вакууме с получением 586 мг сырого материала. Сырой продукт очищают флэш-хроматографией (0-5% метанол в DCM), получая при этом 157 мг (35%) указанного в заголовке соединения 73. Оксалатную соль получают из щавелевой кислоты (1,1 экв.) в диэтиловом эфире. ТСХ (5% метанол в DCM): Rf=0,3. ВЭЖХ-МС (способ А): М+=303,1 (УФ/МС(%)=100/100). 1H-ЯМР (400 МГц, CDCl3): δ=7,69-7,22 (4H, м); 4,57 (2H, т); 2,99 (2H, д); 2,88 (2H, т); 2,11 (2H, т); 1,68 (2H, м); 1,32-1,18 (9H, м); 0,89(3H, т).
Пример 70. 3-(1Н-Индол-3-ил)пропан-1-ол (74) (32HS28). Суспензию литийалюминийгидрида (4,68 г, 126 ммоль) в безводном диэтиловом эфире (230 мл) энергично перемешивают. 3-Индолпропионовую кислоту (10,0 г, 53 ммоль) растворяют в безводном диэтиловом эфире и добавляют по каплям при кипячении реакционной смеси с обратным холодильником. Реакционную смесь далее кипятят с обратным холодильником в течение 2 час и затем перемешивают при комнатной температуре (кт) в течение ночи. Медленно добавляют воду (25 мл), затем водный раствор H2SO4 (1:3 Н2О/конц. H2SO4) (20 мл). Образовавшуюся прозрачную смесь экстрагируют диэтиловым эфиром (3×110 мл) и объединенные органические фазы промывают насыщенным раствором соли, сушат (Na2SO4), фильтруют и концентрируют в вакууме, получая при этом сырое масло указанного в заголовке соединения (74) (1,8 г). Сырой продукт используют без дальнейшей очистки.
Пример 71. Гидрохлорид 3-[3-(4-Бутилпиперидин-1-ил)пропил]-1Н-индола (75) (32HS34). Сырой 3-(1Н-индол-3-ил)пропан-1-ол (1,8 г) растворяют в безводном ТГФ и охлаждают до -40°C. Шприцем добавляют триэтиламин (720 мг, 7,1 ммоль) с последующим добавление метансульфонилхлорида (750 мг, 6,5 ммоль). Смесь оставляют для нагревания до 20°C и затем фильтруют и концентрируют в вакууме, получая при этом сырой продукт, который снова растворяют в DCM и промывают водой. Органическую фазу сушат над MgSO4, фильтруют и концентрируют в вакууме с получением коричневого масла. Данный материал используют сразу без дополнительной очистки.
Гидрохлорид 4-н-бутилпиперидина (967 мг, 5,4 ммоль) и Na2CO3 (1,28 г, 12 ммоль) суспендируют в DME, перемешивают при кт в течение 30 мин и затем добавляют к сырому материалу в DME. Образовавшуюся смесь перемешивают при 82°C в течение ночи. Смесь охлаждают перед добавлением этилацетата (15 мл) и воды (15 мл), экстрагируют этилацетатом (3×20 мл). Объединенные органические фазы промывают насыщенным раствором соли, сушат (Na2SO4) и концентрируют в вакууме. Очистка препаративной ВЭЖХ с последующей обработкой HCl в диоксане (4М, 2 мл) дает указанное в заголовке соединение (75) в виде белых кристаллов после промывания DCM. Выход: 130 мг, 0,3% (общий). ВЭЖХ-МС (способ А): М+=298,3 (УФ/МС(%)=100/100). 1H-ЯМР (400 МГц, CD3OD): δ 7,55 (д, 1Н); 7,34 (д, 1Н), 7,09 (м, 2H), 7,01 (т, 1H), 3,46 (м, 2H), 3,09 (м, 2H), 2,87 (м, 5H), 2,14 (м, 2H), 1,91 (2, 2H), 1,58-1,24 (м, 9H), 0,90 (т, 3H).
Пример 72. Метиловый эфир 4-(4-бутилпиперидин-1-ил)масляной кислоты (76) (40-LH-58). К раствору метилового эфира 4-броммасляной кислоты (1,35 г, 7,5 ммоль) в сухом ацетонитриле (10 мл) добавляют 4-бутилпиперидин (1,00 г, 7,1 ммоль) и К2СО3 (1,10 г, 7,8 ммоль). После перемешивания при кт в течение 12 час реакционную смесь упаривают досуха с последующим добавлением воды (15 мл). Водную фазу экстрагируют этилацетатом (3×20 мл) и объединенные органические фазы сушат (Na2SO4) и концентрируют в вакууме с получением 1,71 г сырого указанного в заголовке соединения 76. Сырой продукт очищают флэш-хроматографией (МеОН:этилацетат, 2:8), получая при этом чистое, указанное в заголовке соединение. Выход 1,27 г (74%). 1H-ЯМР (CD3OD): δ 3,65 (с, 3H), 2,93 (д, 2H), 2,33 (к., 4H), 1,98 (т, 2H), 1,81 (кв., 2H), 1,69 (д, 2H), 1,35-1,18 (м, 9H), 0,90 (т, 3H).
Пример 73. 2-[3-(4-Бутилпиперидин-1-ил)пропил]-1-метил-1Н-бензимидазол (77) (40-LH-59В). Смесь N-метилбензол-1,2-диамина (68 мг, 0,56 ммоль) и метилового эфира 4-(4-бутилпиперидин-1-ил)масляной кислоты (130 мг, 0,54 ммоль) в полифосфорной кислоте (1 мл) нагревают и встряхивают в запаянной ампуле при 150°C в течение 1,5 часа. Реакционную смесь выливают в охлаждаемую льдом баню (NaOH (4 н): лед, 1:1) при перемешивании, после чего образуется серый осадок. Серый осадок отфильтровывают и промывают холодным эфиром. Оксалатную соль получают из щавелевой кислоты (1,1 экв.) в диэтиловом эфире. Выход 141 мг (92%). 1H-ЯМР (CDCl3): δ 7,71 (м, 1H), 7,30-7,19 (м, 3H), 3,74 (с, 3H), 2,90 (к., 4H), 2,43 (т, 2H), 2,06 (кв., 2H), 1,89 (т, 2H), 1,65 (д, 2H), 1,31-1,14 (м, 9H), 0,89 (т, 3H).
Пример 74. (2-(4-Бутилпиперидин)-1-илэтил)амид 1Н-индазол-3-карбоновой кислоты (78) (40-LH-70-17В). К встряхиваемому раствору 1Н-индазол-3-карбоновой кислоты (49 мг, 0,30 ммоль) и N-гидроксисукцинимида (36 мг, 0,31 ммоль) в сухом ДМФА (2 мл) добавляют раствор дициклогексилкарбодиимида (62 мг, 0,30 ммоль) в сухом ДМФА (1 мл). Смесь встряхивают в течение 16 час с последующим добавлением 2-(4-бутилпиперидин-1-ил)этиламина (28 мг, 0,15 ммоль). Реакционную смесь далее встряхивают в течение еще 24 час с последующим фильтрованием. Органическую фазу загружают в ионообменную колонку Varian SCX. Колонку промывают последовательно метанолом (5 мл), изопропанолом (5 мл) и метанолом (5 мл). Продукт элюируют из колонки с использованием 5% аммиака в метаноле (5 мл). Сорбат концентрируют в вакууме, растворяют в ацетоне, сушат (К2СО3) и концентрируют в вакууме, получая при этом указанное в заголовке соединение 78. Выход 47 мг (95%). 1H-ЯМР (CD3OD): δ 8,21 (д, 1H), 7,56 (д, 1H), 7,40 (дт, 1H), 7,24 (дт, 1H), 3,59 (т, 2H), 3,01 (шир.д, 2H), 2,63 (т, 2H), 2,08 (т, 2H), 1,71 (д, 2H), 1,34-1,21 (м, 9H), 0,90 (т, 3H).
Пример 75. 1-[3-(4-Бутилпиперидин-1-ил)пропил]-5-нитро-1Н-индазол (79) (64LHY29-1). Пример 76. 2-[3-(4-Бутилпиперидин-1-ил)пропил]-5-нитро-2Н-индазол (80) (64LHY29-2). К охлажденному раствору (-78°C) 5-нитроиндазола (41,20 мг, 0,25 ммоль) в ТГФ (1 мл) добавляют раствор н-бутиллития в гексане (1,5М, 0,17 мл, 0,25 ммоль) с последующим добавлением 1-бром-3-иодпропана (27 мкл, 0,25 ммоль). После выдерживания 16 час при кт смесь концентрируют в вакууме. Добавляют метилэтилкетон (1 мл) и 4-бутилпиперидин (35,3 мг, 0,25 ммоль). Реакционную смесь встряхивают в течение 16 час при 60°C с последующим фильтрованием и органический слой затем упаривают досуха. Твердое вещество растворяют в метаноле (1 мл) перед загрузкой в колонку с ионообменной смолой Varian SCX. Колонку промывают метанолом (3×6 мл) и продукт элюируют 10% NH3 в метаноле (5 мл). Сорбат концентрируют в вакууме. Образуется два изомера при отношении 1:1 по данным анализа ЖХ-МС сырой смеси. Два изомера выделяют после очистки препаративной ВЭЖХ. 79 (64LHY12-1): 1H-ЯМР (CDCl3): δ 0,88 (т, 3H), 1,l8-1,33 (м, 9H), 1,73-1,64 (шир.д, 2H), 1,92 (шир.т, 2H), 2,21 (ддд, 2H), 2,30 (дд, 2H), 2,85 (шир.д, 2H), 4,55 (т, 2H), 7,75 (ддд, 1H), 8,10 (дд, 1H), 8,24 (д, 1H), 8,73 (дд, 1H); 13С-ЯМР (CDCl3): δ=14,0, 22,8, 27,3, 28,9 (2C), 32,3, 35,6, 36,1, 52,1, 53,9 (2C), 54,7, 118,2, 119,2, 119,9, 120,1, 127,3, 143,0, 149,8; LC-MS: (М+H)+ 445,2, tr 3,69 min, 80 (64LHY29-2): 1H-ЯМР (CDCl3): (0,90 (т, 3H), 1,14-1,38 (м, 9H), 1,62 (шир.д, 2H), 1,86 (шир.дд, 2H), 2,16 (ддд, 2H), 2,21 (дд, 2H), 2,75 (шир.д, 2H), 4,50 (т, 2H), 7,59 (ддд, 1H), 8,21 (д, 1H), 8,25 (дд, 1H), 8,73 (дд, 1H); 13C-ЯМР (CDCl3): (14,3, 23,1, 27,3, 29,2, 32,8 (2C), 36,0, 36,5, 47,2, 54,2 (2C), 55,2, 110,0, 119,1, 121,3, 123,0, 136,0, 141,8, 142,5; LC-MS: (М+H)+ 445,2, tr 5,30 min.
Общая процедура получения производных индола
Индол (1,20 ммоль) растворяют в сухом ДМФА (3 мл) перед добавлением гидрида натрия (2,50 ммоль) при кт с последующим добавлением 3-хлор-1-иодпропана (0,20 г, 1,0 ммоль). Реакционную смесь встряхивают в запаянной ампуле при кт. в течение 16 час. Добавляют 4-бутилпиперидин (130 мг, 0,9 ммоль) и реакционную смесь далее встряхивают при 50°C в течение 72 час. Смесь фильтруют и фильтрат загружают в ионообменную колонку Varian SCX. Колонку промывают метанолом (10 мл, 2 объема колонки) и продукт элюируют из колонки с использованием 5% гидроксида аммония в метаноле (5 мл, 1 объем колонки). Сорбат концентрируют в вакууме, получая при этом указанное в заголовке соединение (79, 80).
Пример 77. 1-[3-(4-Бутилпиперидин-1-ил)пропил]-2-метил-1Н-индол (81) (55-LH-1-1-(1402). Взаимодействие проводят по общей процедуре с использованием 2-метил-1Н-индола (157 мг, 1,20 ммоль). Сырой продукт далее очищают флэш-хроматографией (МеОН:этилацетат, 1:4), получая при этом указанное в заголовке соединение 81. Выход 19 мг (21%). (УФ/МС(%)=98/89); 1H-ЯМР (CDCl3): δ 7,50 (д, 1H), 7,31 (д, 1H), 7,12 (дт, 1H), 7,04 (дт, 1H), 6,23 (с, 1H), 4,12 (т, 2H), 2,87 (д, 2H), 2,44 (с, 3H), 2,31 (т, 2H), 1,98-1,83 (м, 4H), 1,67 (д, 2H), 1,32-1,19 (м, 9H), 0,89 (т, 3H).
Пример 78. 1-{1-[3-(4-Бутилпиперидин-1-ил)пропил]-1Н-индол-3-ил}этанон (82) (55-LH-1-2-(1403). Взаимодействие проводят по общей процедуре с использованием 1-(1Н-индол-3-ил)этанона (191 мг, 1,20 ммоль), получая при этом указанное в заголовке соединение 82. Выход 33 мг (32%). (УФ/МС(%)=99/91); 1H-ЯМР (CDCl3): δ 8,39-8,34 (м, 1H), 7,80 (с, 1H), 7,41-7,36 (м, 1H), 7,30-7,25 (м, 2H), 4,24 (т, 2H), 2,80 (д, 2H), 2,21 (т, 2H), 2,01 (кв., 2H), 1,86 (т, 2H), 1,69 (д, 2H), 1,32-1,19 (м, 9H), 0,89 (т, 3H).
Пример 79. {1-[3-(4-Бутилпиперидин-1-ил)пропил]-1Н-индол-3-ил}ацетонитрил (83) (55-LH-1-3-(1404). Взаимодействие проводят по общей процедуре с использованием (1Н-индол-3-ил)ацетонитрила (187 мг, 1,20 ммоль), получая при этом указанное в заголовке соединение 83. Выход 33 мг (11%). (УФ/МС(%)=99/92); 1H-ЯМР (CDCl3): δ 7,55 (д, 1H), 7,38 (д, 1H), 7,24 (т, 1H), 7,15 (т, 1H), 4,17 (т, 2H), 3,82 (с, 2H), 2,82 (д, 2H), 2,23 (т, 2H), 1,98 (кв., 2H), 1,85 (т, 2H), 1,67 (д, 2H), 1,33-1,17 (м, 9H), 0,89 (т, 3H).
Пример 80. 1-[3-(4-Бутилпиперидин-1-ил)пропил]-1Н-индол-3-карбонитрил (84) (55-LH-1-4-(1405). Взаимодействие проводят по общей процедуре с использованием 1Н-индол-3-карбонитрила (170 мг, 1,20 ммоль), получая при этом указанное в заголовке соединение 84. Выход 30 мг (31%). (УФ/МС(%)=99/96); 1H-ЯМР (CDCl3): δ 7,75 (д, 1H), 7,63 (с, 1H), 7,45 (д, 1H), 7,35-7,25 (м, 2H), 4,25 (т, 2H), 2,79 (д, 2H), 2,20 (т, 2H), 1,99 (кв., 2H), 1,86 (т, 2H), 1,68 (д, 2Н), 1,33-1,18 (м, 9H), 0,89 (т, 3H).
Общая процедура получения производных бензимидазола
Бензимидазол (0,60 ммоль) растворяют в сухом ТГФ (1 мл) перед добавлением к нему по каплям н-BuLi (1,6М в гексане) (413 мкл, 0,66 ммоль) при кт. Смесь перемешивают в течение 15 мин с последующим добавлением 1,3-дибромпропана (100 мг, 0,50 ммоль) и затем выдерживают при кт в течение 16 час. Добавляют 4-бутилпиперидин (64 мг, 0,45 ммоль) и реакционную смесь встряхивают при 60°C в течение 72 час. Смесь фильтруют и фильтрат концентрируют в вакууме перед очисткой препаративной ВЭЖХ.
Пример 81. 1-[3-(4-Бутилпиперидин-1-ил)пропил]-5,6-диметил-1Н-бензоимидазол (85) (55-LH-8-2 (1387)). Взаимодействие проводят по общей процедуре с использованием 5,6-диметилбензоимидазола (88 мг, 0,60 ммоль), получая при этом указанное в заголовке соединение 85. Выход 20 мг (14%). (МС(%)=100); 1H-ЯМР (CDCl3): δ 7,78 (с, 1H), 7,55 (с, 1H), 7,18 (д, 1H), 4,20 (т, 2H), 2,81 (д, 2H), 2,39 (с, 3H), 2,37 (с, 3H), 2,22 (т, 2H), 2,00 (кв., 2H), 1,85 (т, 2H), 1,68 (д, 2H), 1,33-1,18 (м, 9H), 0,90 (т, 3H).
Пример 82. 1-[3-(4-Бутилпиперидин-1-ил)пропил]-5(6)-диметил-1Н-бензоимидазол (86) (55-LH-8-3 (1388)). Взаимодействие проводят по общей процедуре с использованием 5-метилбензоимидазола (79 мг, 0,60 ммоль), получая при этом указанное в заголовке соединение (86) в виде смеси 50/50 двух региоизомеров по данным 1Н ЯМР. Выход 42 мг (30%). (УФ/МС(%)=100/100).
Пример 83. 1-[3-(4-Бутилпиперидин-1-ил)пропил]-5-метокси-1Н-бензоимидазол (87) (55-LH-8-6(1393)). Взаимодействие проводят по общей процедуре с использованием 5-метоксибензоимидазола (89 мг, 0,60 ммоль), получая при этом указанное в заголовке соединение (87) в виде смеси 50/50 двух региоизомеров по данным 1Н ЯМР. Выход 62 мг (42%). (УФ/МС(%)=100/100).
Пример 84. {1-[3-(4-Бутилпиперидин-1-ил)пропил]-1H-бензоимидазол-2-ил}метанол (88) (55-LH-8-9(1400)). Взаимодействие проводят по общей процедуре с использованием (1Н-бензоимидазол-2-ил)метанола (89 мг, 0,60 ммоль), получая при этом указанное в заголовке соединение (88). Выход 56 мг (38%). (УФ/МС(%)=95/85). 1H-ЯМР (CDCl3): δ 7,69-7,65 (м, 1H), 7,32-7,28 (м, 1H), 7,21-7,18 (м, 2H), 4,88 (с, 2H), 4,38 (т, 2H), 2,70 (д, 2H), 2,18-2,06 (м, 4H), 1,74 (т, 2H), 1,58 (д, 2H), 1,24-1,14 (м, 9H), 0,81 (т, 3H).
Пример 85. 1-[3-(4-Бутилпиперидин-1-ил)пропил]-2-трифторметил-1Н-бензоимидазол (89) (55-LH-8-10 (1401)). Взаимодействие проводят по общей процедуре с использованием 2-трифторметил-1Н-бензоимидазола (112 мг, 0,60 ммоль), получая при этом указанное в заголовке соединение (89). Выход 48 мг (29%). (УФ/МС(%)=100/95). 1H-ЯМР (CDCl3): δ 7,78 (д, 1H), 7,74 (д, 1H), 7,49 (т, 1H), 7,41 (т, 1H), 4,48 (т, 2H), 2,86 (д, 2H), 2,41 (т, 2H), 2,08 (кв., 2H), 1,92 (т, 2H), 1,67 (д, 2H), 1,31-1,15 (м, 9H), 0,89 (т, 3H).
Пример 86. трет-Бутиловый эфир (2-триметилстаннилфенил) карбаминовой кислоты (90) (53MF36). К раствору трет-бутилового эфира фенилкарбаминовой кислоты (10,02 г, 52 ммоль) в сухом ДМФА (150 мл) добавляют по каплям трет-BuLi (1,7 М в гексане) (80 мл, 0,14 моль) при -70°C. Реакционную смесь перемешивают в течение 30 мин при -70°C и 2 часа при -20°C перед добавлением раствора триметилоловохлорида в сухом ТГФ (1М) (77,0 мл, 78 моль). Реакционную смесь далее перемешивают при -20°C в течение 1 часа с последующим добавлением водного раствора хлорида аммония (15%) (100 мл). Смесь экстрагируют диэтиловым эфиром (3×300 мл) и объединенные органические фазы сушат над MgSO4 и упаривают в вакууме, получая при этом сырое, указанное в заголовке соединение (90) (17,0 г), которое используют в следующей реакции без дополнительной очистки.
Пример 87. трет-Бутиловый эфир [2-(4-хлорбутирил)фенил] карбаминовой кислоты (91) (53MF37). К раствору трет-бутилового эфира (2-триметилстаннилфенил)карбаминовой кислоты (17,0 г, 36 ммоль) в сухом толуоле (300 мл) добавляют 4-хлорбутирилхлорид (5,3 г, 38 ммоль) и дихлорбис(ацетонитрил) палладий(II) (300 мг, 1,2 ммоль). Реакционную смесь нагревают для кипячения с обратным холодильником и оставляют на 12 час с последующим упариванием досуха и колоночной хроматографией (гептан:этилацетат, 10:1), получая при этом указанное в заголовке соединение 91. Выход 7,2 г (47% от трет-бутилового эфира фенилкарбаминовой кислоты).
Пример 88. трет-Бутиловый эфир {2-[4-(4-бутилпиперидин-1-ил)бутирил]фенил}карбаминовой кислоты (92)(53MF38). В колбу загружают трет-бутиловый эфир [2-(4-хлорбутирил)фенил] карбаминовой кислоты (2,1 г, 7,1 ммоль) и 4-бутилпиперидин (1,2 г, 8,5 ммоль) перед добавлением пиридина (5 мл). К реакционной смеси добавляют карбонат калия (1,17 г, 8,5 ммоль) и смесь перемешивают при 100°C в течение 12 час. Добавляют воду (50 мл) с последующей экстракцией этилацетатом (3×150 мл). Объединенные органические фазы сушат над MgSO4, фильтруют и упаривают досуха. Сырой материал подвергают колоночной хроматографии (DCM:метанол; 20:1), которая дает чистое, указанное в заголовке соединение (92) (1,48 г, 52%).
Пример 89. 3-[3-(4-Бутилпиперидин-1-ил)пропил]-1Н-индазол, HCl (93) (53MF39). Раствор трет-бутилового эфира {2-[4-(4-бутилпиперидин-1-ил)бутирил]фенил}карбаминовой кислоты (1,48 г, 3,7 ммоль) в растворе HCl в диоксане (4 н) (20 мл) перемешивают при кт в течение 1 часа перед выпариванием досуха. Остаток растворяют в HCl (конц.) (15 мл) перед добавлением раствора нитрита натрия (255 мг, 3,7 ммоль), растворенного в воде (3 мл). Смесь перемешивают при 0°C в течение 1 часа перед добавлением станнилхлорида (1,7 г, 7,4 ммоль) и затем далее перемешивают при кт. в течение 3 час. рН реакционной смеси регулируют NaOH (2 н) до достижения основного значения с последующей экстракцией этилацетатом (3×400 мл). Объединенные органические фазы сушат над MgSO4, фильтруют и упаривают в вакууме. Сырой материал подвергают колоночной хроматографии (DCM:метанол; 20:1), которая дает сырое, указанное в заголовке соединение 93. Сырое соединение растворяют в диэтиловом эфире с последующим добавлением HCl в эфире (1,0М) и перемешивают в течение 0,5 часа. Раствор упаривают досуха и твердый материал перекристаллизовывают дважды из смеси DCM:диэтиловый эфир, получая при этом чистое, указанное в заголовке соединение. Выход 0,44 г (32%). (УФ/МС(%)=100/100); т.пл. 160,5-164,0°C; 1H-ЯМР (CD3OD): δ 7,96 (д, 1H), 7,62 (д, 2H), 7,33 (д, 1H), 3,58 (дт, 2H), 3,24-3,19 (м, 4H), 2,95 (т, 2H), 2,33 (кв., 2H), 1,97 (д, 2H), 1,65-1,28 (м, 9H), 0,91 (т, 3H). 13С-ЯМР (CD3OD): δ 143,9, 141,0, 130,3, 122,5, 120,8, 120,5, 110,9, 56,4, 53,2, 35,4, 33,6, 29,7, 28,5, 23,1, 22,7, 22,6, 13,1.
Пример 90. 3-[3-(4-Бутилпиперидин-1-ил)пропил]-5-нитро-1Н-индазол (94) (39MF43NO2).
Пример 91. 3-[3-(4-Бутилпиперидин-1-ил)пропил]-5,7-динитро-1Н-индазол (95) (39MF43DiNO2). Раствор 3-[3-(4-бутилпиперидин-1-ил)пропил]-1Н-индазола (120 мг, 0,4 ммоль) в смеси 1:1 азотной кислоты (дымящей) и серной кислоты (конц.) (2 мл) перемешивают при 0°C в течение 1,5 часа. рН смеси регулируют NaOH (8 н), в результате чего желтый маслянистый материал осаждается. Материал фильтруют и подвергают препаративной ТСХ (DCM:метанол; 10:1), которая дает два чистых, указанных в заголовке соединения. Выход: 25 мг (18%) (3-[3-(4-бутилпиперидин-1-ил)пропил]-5-нитро-1Н-индазола (94). 1H-ЯМР (CDCl3): δ 8,45 (с, 1H), 8,01 (д, 2H), 7,48 (д, 1H), 3,48 (д, 2H), 3,18-2,95 (м, 4H), 2,62 (т, 2H), 2,27 (кв., 2H), 1,82 (д, 2H), 1,58 (кв., 2H), 1,44-1,38 (м, 1H), 1,30-1,19 (м, 6H), 0,91 (т, 3H), 13С-ЯМР (CDCl3): 147,4, 143,4, 141,9, 121,6, 121,1, 117,7, 111,3, 57,4, 53,6, 35,4, 34,4, 30,0, 28,9, 24,2, 23,5, 22,9, 14,2. Выход: 10 мг (6%) (3-[3-(4-Бутилпиперидин-1-ил)пропил]-5,7-динитро-1Н-индазола (95). 1H-ЯМР (CDCl3): δ 9,18 (д, 1H), 9,05 (д, 1H), 3,18-3,10 (м, 4H), 2,68 (т, 2H), 2,25-2,l4 (м, 4H), 1,74 (д, 2H), 1,45-1,22 (м, 7H), 0,91 (т, 3H).
Пример 92. 4-(4-Бутилпиперидин-1-ил)-1-(2-метилсульфанилфенил) бутан-1-он (96) (65MF07). К перемешиваемому раствору 2-бромтиоанизола (12,85 г, 63,3 ммоль) в сухом ТГФ (60 мл) при -78°C добавляют н-BuLi (1,6 н в гексане) (41 мл, 65,3 ммоль) через шприц на протяжении 30 мин. Реакционную смесь далее перемешивают при -78°C в течение 30 мин перед добавления раствора 4-(4-бутилциклогексил)-N-метокси-N-метилбутирамида (11,41 г, 42,2 ммоль) в сухом ТГФ (10 мл). Смесь выдерживают при -78°C в течение 0,5 часа и при кт в течение 0,5 часа перед добавлением воды (100 мл) и экстракцией этилацетатом (3×150 мл). Объединенные органические фазы сушат над MgSO4, фильтруют и упаривают в вакууме, получая при этом сырое, указанное в заголовке соединение 96 (11,9 г). Чистота по анализу ЖХ-МС: УФ/МС(%)=90/91).
Пример 93. 3-[3-(4-Бутилпиперидин-1-ил)пропил]бензо[d]изотиазол (97) (65MF08). Смесь сырого 4-(4-бутилпиперидин-1-ил)-1-(2-метилсульфанилфенил)бутан-1-она (11,9 г, 36 ммоль) и гидроксиламин-О-сульфоновой кислоты (6,11 г, 54 ммоль) в уксусной кислоте (500 мл) перемешивают при кт в течение 72 час с последующим нагреванием при 100°C в течение 24 час. Реакционную смесь охлаждают до кт и рН регулируют 2 н NaOH до основного условия (рН 9) перед экстракцией этилацетатом (3×400 мл). Объединенные органические фазы сушат над MgSO4, фильтруют и упаривают в вакууме, получая при этом 12,1 г сырого продукта. Сырой продукт очищают колоночной хроматографией (DCM:MeOH; 20:1), получая при этом указанное в заголовке соединение 97. Выход 3,67 г (18,3%) от 2-бромтиоанизола. Оксалатную соль получают добавлением щавелевой кислоты и перекристаллизовывают из смеси метанол-диэтиловый эфир с получением белых кристаллов, которые фильтруют и сушат. (УФ/МС(%)=90/91), т.пл. 193,4-194,0°C. 1H-ЯМР (CDCl3): δ 7,98 (д, 1H), 7,91 (д, 1H), 7,50 (т, 1H), 7,41 (т, 1H), 3,14 (т, 2H), 2,92 (д, 2H), 2,46 (т, 2H), 2,18 (кв., 2H), 1,92 (т, 2H), 1,66 (д, 2H), 1,35-1,18 (м, 9H), 0,88 (т, 3H), 13С-ЯМР (CDCl3): 166,6, 152,5, 134,9, 127,6, 124,5, 123,6, 120,0, 58,5, 54,2, 36,4, 35,9, 32,5, 29,7, 25,5, 23,1, 14,2.
Пример 94. 3-[3-(4-Бутилпиперидин-1-ил)пропил]-5-метокси-1Н-индазол (98) (53MF35). В небольшую колбу загружают 1-(2-амино-5-метоксифенил)-4-(4-бутилпиперидин-1-ил)бутан-1-он (1,58 г, 47 ммоль) в конц. HCl (15 мл). Смесь охлаждают до 0°C с последующим добавлением нитрита натрия (0,61 г, 88 ммоль) и воды (3 мл) и перемешиванием при 0°C в течение 2 час. Добавление дигидрата хлорида олова(II) (2,68 г, 11,9 ммоль) дает осадок, который фильтруют, промывают дважды охлажденной льдом водой и сушат. Фильтрат растворяют в этилацетате (100 мл) и 1 н NaOH (150 мл) с последующей экстракцией этилацетатом (3×150 мл). Объединенные органические фазы сушат над MgSO4, фильтруют и упаривают в вакууме, получая при этом 1,30 г сырого продукта. Сырой продукт очищают колоночной хроматографией (DCM:MeOH; 20:1) с получением указанного в заголовке соединения 98. Оксалатную соль получают добавлением щавелевой кислоты и перекристаллизовывают из смеси метанол-диэтиловый эфир, получая при этом белые кристаллы, которые фильтруют и сушат. Выход 0,97 г (49%). 1H-ЯМР (CDCl3): δ 7,32 (дд, 1H), 7,03 (дд, 1H), 6,98 (д, 1H), 3,85 (с, 3H), 3,10 (д, 2H), 2,96 (т, 2H), 2,61 (т, 2H), 2,15-2,05 (м, 4H), 1,68 (д, 2H), 1,40-1,20 (м, 9H), 0,87 (т, 3H). 13С-ЯМР (CDCl3): 177,2, 154,5, 145,6, 137,4, 122,4, 119,0, 111,2, 99,6, 58,0, 55,9, 53,5, 36,1, 35,5, 31,5, 29,1, 25,2, 24,9, 23,4, 23,0, 14,2.
Пример 95. 3-[3-(4-Бутилпиперидин-1-ил)пропил]-4-метокси-1Н-индазол (99) (53MF47О). К раствору 3-(3-хлорпропил)-4-метокси-1Н-индазола (0,99 г, 4,41 ммоль) в ацетонитриле (25 мл) добавляют 4-бутилпиперидин (0,61 г, 4,41 ммоль) при кт. Реакционную смесь перемешивают при кт. в течение 3 дней перед добавлением воды (50 мл). Водную фазу экстрагируют этилацетатом (3×50 мл) и объединенные органические фазы сушат над MgSO4, фильтруют и упаривают в вакууме с получением 1,40 г сырого продукта. Сырой продукт очищают колоночной хроматографией (этилацетат:МеОН:Et3N; 10:5:3), получая при этом указанное в заголовке соединение 99. Выход 0,65 г (45%). 1H-ЯМР (CDCl3): δ 7,24 (д, 1H), 7,00 (д, 1H), 6,41 (т, 1H), 3,91 (с, 3H), 3,58 (д, 2H), 3,20-2,99 (м, 4H), 2,55 (т, 2H), 2,22 (кв., 2H), 1,81 (д, 2H), 1,61 (к., 2H), 1,41-1,08 (м, 7H), 0,87 (т, 3H). 13С-ЯМР (CDCl3): 154,9, 144,3, 143,3, 129,2, 113,1, 103,1, 99,8, 57,1, 55,4, 53,3, 35,3, 34,3, 29,5, 28,8, 25,6, 23,1, 22,8, 14,1.
Пример 96. 3-[3-(4-Бутилпиперидин-1-ил)пропил]-6-метокси-1Н-индазол (100) (53MF47Р). К раствору 3-(3-хлорпропил)-6-метокси-1Н-индазола (0,99 г, 4,41 ммоль) в ацетонитриле (25 мл) добавляют 4-бутилпиперидин (0,61 г, 4,41 ммоль) при кт. Реакционную смесь перемешивают при кт. в течение 3 дней перед добавлением воды (50 мл). Водную фазу экстрагируют этилацетатом (3×50 мл) и объединенные органические фазы сушат над MgSO4, фильтруют и упаривают в вакууме с получением 0,85 г сырого продукта. Сырой продукт очищают колоночной хроматографией (этилацетат:МеОН:Et3N; 10:5:3), получая при этом указанное в заголовке соединение 100. Выход 0,55 г (38%). 1H-ЯМР (CDCl3): δ 7,42 (д, 1H), 6,80-6,72 (3, 2H), 3,80 (с, 3H), 3,60 (д, 2H), 3,11-2,92 (м, 4H), 2,55 (т, 2H), 2,23 (кв., 2H), 1,79 (д, 2H), 1,57 (к., 2H), 1,40-1,08 (м, 7H), 0,83 (т, 3H). 13С-ЯМР (CDCl3): 160,8, 143,8, 142,6, 120,7, 116,2, 114,0, 91,2, 57,1, 55,6, 53,4, 35,3, 34,3, 29,5, 28,8, 23,7, 22,8, 22,7, 14,1.
Пример 97. 3-[3-(4-Бутилпиперидин-1-ил)пропил]-1Н-индазол-4-ол (101) (53MF51). 3-[3-(4-Бутилпиперидин-1-ил)пропил]-4-метокси-1Н-индазол (28 мг, 0,09 ммоль) растворяют в сухом DCM (1,0 мл) и охлаждают до 0°C перед добавлением 1М раствора бромтрибромида в DCM (0,50 мл, 0,50 ммоль). Реакционную смесь выдерживают при кт. в течение 12 час с последующим добавлением воды (5 мл) и 2 н NaOH (10 мл). Водную фазу экстрагируют DCM (3×25 мл) и объединенные органические фазы сушат над MgSO4, фильтруют и упаривают в вакууме, получая при этом 13 мг сырого продукта. Очистка препаративной ВЭЖХ с последующей обработкой HCl в диоксане (4М, 2 мл) дает указанное в заголовке соединение (101) в виде белых кристаллов после промывания DCM. Выход: 6,0 мг, 17%. 1H-ЯМР (CDCl3): δ 7,18 (т, 1H), 6,81 (д, 1H), 6,50 (т, 1H), 2,85 (д, 2H), 2,23 (т, 2H), 1,98 (кв., 2Н), 1,83 (т, 2H), 1,62 (д, 2H), 1,41 (д, 2H), 1,21-1,01 (м, 9H), 0,78 (т, 3H).
Пример 98. 3-[3-(4-Бутилпиперидин-1-ил)пропил]-1Н-индазол-6-ол (102) (53MF52). 3-[3-(4-Бутилпиперидин-1-ил)пропил]-6-метокси-1Н-индазол (28 мг, 0,09 ммоль) растворяют в сухом DCM (1,0 мл) и охлаждают до 0°C перед добавлением 1 М раствора бромтрибромида в DCM (0,50 мл, 0,50 ммоль). Смесь выдерживают при кт. в течение 12 час с последующим добавлением воды (5 мл) и 2 н NaOH (10 мл). Водную фазу экстрагируют DCM (3×25 мл) и объединенные органические фазы сушат над MgSO4, фильтруют и упаривают в вакууме, получая при этом 17 мг сырого продукта. Очистка препаративной ВЭЖХ с последующей обработкой HCl в диоксане (4М, 2 мл) дает указанное в заголовке соединение (102) в виде белых кристаллов после промывания DCM. Выход: 10 мг, 17%. 1H-ЯМР (CD3OD): δ 7,54 (д, 1H), 6,77 (с, 1H), 6,71 (д, 1H), 3,55 (д, 2H), 3,15 (т, 2H), 3,04 (т, 2H), 2,90 (дт, 2H), 2,22 (кв., 2H), 1,97 (д, 2H), 1,58-1,28 (м, 9H), 0,92 (т, 3H). 13С ЯМР (CD3OD): 159,0, 145,5, 144,4, 121,7, 117,2, 113,7, 94,4, 58,2, 54,3, 36,5, 34,8, 31,0, 29,7, 24,7, 24,3, 23,7, 14,3.
Пример 99. 3-[3-(4-Бутилпиперидин-1-ил)пропил]-1Н-индазол-5-ол (103) (53MF50). 3-[3-(4-Бутилпиперидин-1-ил)пропил]-5-метокси-1Н-индазол (28 мг, 0,09 ммоль) растворяют в сухом DCM (1,0 мл) и охлаждают до 0°C перед добавлением 1 М раствора бромтрибромида в DCM (0,50 мл, 0,50 ммоль). Смесь выдерживают при кт. в течение 12 час с последующим добавлением воды (5 мл) и 2 н NaOH (10 мл). Водную фазу экстрагируют DCM (3×25 мл) и объединенные органические фазы сушат над MgSO4, фильтруют и упаривают в вакууме, получая при этом 14 мг сырого продукта. Очистка препаративной ВЭЖХ с последующей обработкой HCl в диоксане (4М, 2 мл) дает указанное в заголовке соединение (103) в виде белых кристаллов после промывания DCM. Выход: 16 мг, 60%. 1H-ЯМР (CD3OD): δ 7,54 (д, 1H), 6,77 (с, 1H), 6,71 (д, 1H), 3,55 (д, 2H), 3,15 (т, 2H), 3,04 (т, 2H), 2,90 (дт, 2H), 2,22 (кв., 2H), 1,97 (д, 2H), 1,58-1,28 (м, 9H), 0,92 (т, 3H). 13С ЯМР (CD3OD): 159,0, 145,5, 144,4, 121,7, 117,2, 113,7, 94,4, 58,2, 54,3, 36,5, 34,8, 31,0, 29,7, 24,7, 24,3, 23,7, 14,3.
Пример 100. Отбор испытуемых соединений в анализе с использованием подтипов мускариновых рецепторов m1, m2, m3, m4 и m5. Подтипы мускариновых рецепторов m1, m2, m3, m4 и m5 клонировали по существу так, как описано Bonner et al., Science 273:527(1987) and Bonner et al., Neuron 1:403 (1988). Анализы R-SAT проводили по существу так, как описано в патентах США №№5707798, 5912132 и 5955281, и Braüner-Osborne & Brann, Eur. J. Pharmacol. 295:93 (1995). Клетки NIH-3T3 (доступные от American Type Culture Collection как ATCC CRL 1658) трансфицировали плазмидной ДНК, кодирующей рецепторы подтипов m1, m2, m3, m4 или m5, и плазмидной ДНК, кодирующей β-галактозидазу. Трансфицированные клетки выращивали в присутствии от 1 нМ до 40 мкМ испытуемого соединения в течение 5 дней. На день 5 клетки лизировали с использованием 0,5% Nonidet-P и экспрессию β-галактозидазы количественно определяли с использованием хромогенного субстрата о-нитрофенил-β-D-галактозида (ONGP).
Данные нормализовали относительно максимальной реакции клеток на мускариновый агонист карбахол и этим данным соответствует следующее уравнение:
реакция = минимум + (максимум - минимум)/(1+EC50/лиганд])
где [лиганд] = концентрация лиганда
% эффективности определяли как:
(максимум-минимум)/(максимальная реакция клеток на карбахол).
рЕС50=-Log (EC50).
Когда данные дают колоколобразную кривую, "максимум" определяют как самую высокую наблюдаемую реакцию.
Результаты, которые демонстрируют селективную агонистическую активность ряда соединений изобретения, представлены ниже в таблице 1.
Селективность мускариновых агонистов
pEC50: - log EC50
Нет реакции: Эффективность <25% максимальной реакции на карбахол. Этот уровень активности рассматривается как не отличающийся значительно от уровня "нет реакции".
Пример 101. Влияние соединения 35AKU-21 на внутриглазное давление у приматов. Все исследования проводили на находящихся в полном сознании самках обезьян cynomolgus (макаки-крабоеды, Macacca fascicularis), весящих 3-4 кг. Одностороннюю глазную гипертензию вызывали аргонной лазерной фотокоагуляцией срединной трабекулярной сети (Sawyer & McGuigan, Invest. Ophthalmol. Vis. Sci. 29:81 (1988).
Животных готовили так, чтобы они позволяли измерить внутриглазное давление (IOP) пвневмотонометром модели 30 Classic (Mentor O&O Co.). На всем протяжении каждого исследования обезьяны сидели в специально сконструированных креслах (Primate Products, San Francisco) и получали фрукты и соки по потребности.
Лекарственное средство вводили местным способом. Лекарственное средство изготовляли в водном растворе, таком как дистиллированная вода, физиологический раствор или цитратный буфер при рН 5-7 и применяли односторонним образом в виде 35 мкл капли; другой глаз получал равный объем физиологического раствора (или наполнителя). Два базовых измерения проводили перед введением лекарственного средства с последующими периодическими измерениями вплоть до 6 часов после введения лекарственного средства. Результаты данного исследования показаны ниже в таблице 2.
Глазное гипотензивное действие 35AKU-21 на обезьян с глаукомой
| название | год | авторы | номер документа |
|---|---|---|---|
| ПРОИЗВОДНЫЕ БЕНЗИМИДАЗОЛИДИНОНА В КАЧЕСТВЕ АГЕНТОВ МУСКАРИНОВЫХ РЕЦЕПТОРОВ | 2002 |
|
RU2288919C2 |
| БЕНЗИМИДАЗОЛЬНЫЕ, БЕНЗТИАЗОЛЬНЫЕ И БЕНЗОКСАЗОЛЬНЫЕ ПРОИЗВОДНЫЕ И ИХ ПРИМЕНЕНИЕ В КАЧЕСТВЕ МОДУЛЯТОРОВ LTA4H | 2004 |
|
RU2373204C2 |
| ГЕТЕРОЦИКЛИЧЕСКИЕ МУСКАРИНОВЫЕ АГОНИСТЫ И КОМПОЗИЦИИ, ИХ ПРИМЕНЕНИЕ И СПОСОБЫ ЛЕЧЕНИЯ | 2002 |
|
RU2292346C2 |
| АКТИВАТОРЫ ГЛЮКОКИНАЗЫ И ФАРМАЦЕВТИЧЕСКИЕ КОМПОЗИЦИИ, СОДЕРЖАЩИЕ ИХ В КАЧЕСТВЕ АКТИВНОГО ИНГРЕДИЕНТА | 2008 |
|
RU2450001C2 |
| АРИЛ- И ГЕТЕРОАРИЛЗАМЕЩЕННЫЕ ТЕТРАГИДРОИЗОХИНОЛИНЫ И ИХ ПРИМЕНЕНИЕ ДЛЯ БЛОКИРОВАНИЯ ОБРАТНОГО ЗАХВАТА НОРЭПИНЕФРИНА, ДОПАМИНА И СЕРОТОНИНА | 2005 |
|
RU2388751C2 |
| ГЕТЕРОЦИКЛИЧЕСКИЕ АЗОТИСТЫЕ ПРОИЗВОДНЫЕ ПИРРОЛА, ИХ ПОЛУЧЕНИЕ И ФАРМАЦЕВТИЧЕСКОЕ ПРИМЕНЕНИЕ | 2008 |
|
RU2473543C2 |
| КОНДЕНСИРОВАННОЕ 4-ОКСОПИРИМИДИНОВОЕ ПРОИЗВОДНОЕ | 2005 |
|
RU2358969C2 |
| ТИЕНОПИРАЗОЛЫ | 2004 |
|
RU2358978C2 |
| АГОНИСТ РЕЦЕПТОРА GPR40, СПОСОБЫ ЕГО ПОЛУЧЕНИЯ И ФАРМАЦЕВТИЧЕСКИЕ КОМПОЗИЦИИ, СОДЕРЖАЩИЕ ЕГО В КАЧЕСТВЕ АКТИВНОГО ИНГРЕДИЕНТА | 2013 |
|
RU2650506C2 |
| ПРОИЗВОДНЫЕ ИНДОЛА И ИНДАЗОЛА, ОБЛАДАЮЩИЕ КОНСЕРВИРУЮЩИМ ДЕЙСТВИЕМ ПО ОТНОШЕНИЮ К КЛЕТКАМ, ТКАНЯМ И ОРГАНАМ | 2009 |
|
RU2460525C2 |
Настоящее изобретение относится к соединениям общей формулы I
где:
Z1 представляет CR1 или N, Z2 представляет CR2, Z3 представляет CR3 или N, и Z4 представляет CR4;
W1 представляет О, S или NR5, один из W2 и W3 представляет N или CR6, и другой из W2 и W3 представляет CG; W1 представляет NG, W2 представляет CR5 или N, и W3 представляет CR6 или N; или W1 и W3 представляют N, и W2 представляет NG;
G представляет собой формулу (II):
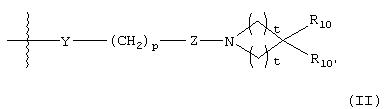
Y представляет О, -С(O)- или отсутствует;
р равно 1, 2, 3, 4 или 5; Z отсутствует; каждый t равен 2. Описаны также способ повышения активности мускаринового холинергического рецептора и способ лечения болезненных состояний, в которых модификация активности холинергических, особенно мускариновых рецепторов m1, m4 или как m1, так и m4, оказывает благотворное действие. 3 н. и 11 з.п. ф-лы, 2 табл.
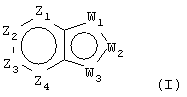
где Z1 представляет CR1 или N, Z2 представляет CR2, Z3, представляет CR3 или N, и Z4 представляет CR4;
W1 представляет О, S или NR5, один из W2 и W3 представляет N или CR6, и другой из W2 и W3 представляет CG; W1 представляет NG, W2 представляет CR5 или N, и W3 представляет CR6 или N; или W1 и W3 представляют N, и W2 представляет NG;
G представляет собой формулу (II):
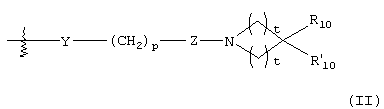
Y представляет О, -С(O)- или отсутствует;
р равно 1, 2, 3, 4 или 5;
Z отсутствует;
каждый t равен 2;
каждый R1, R2, R3 и R4, независимо, представляет Н, гидроксил, галоген, или неразветвленный или разветвленный C1-6-алкил или C1-6-галогеналкил, или -NO2;
каждый из R5 и R6, независимо, представляет Н, C1-6-алкил; формил; С3-6-циклоалкил или С5-6-арил;
R10 представляет Н, неразветвленный или разветвленный C1-8-алкил, С1-8-алкилиден, C1-8-алкокси, C1-8-алкоксикарбонил, С5-6-арил или -C(O)NR12R13;
R'10 представляет Н, неразветвленный или разветвленный C1-8-алкил, C1-8-алкилиден, C1-8-алкокси или C1-8-алкоксикарбонил;
каждый R12 и R13, независимо, представляет Н, С1-6-алкил, необязательно замещенный галогеном или C1-6-алкилом;
или его фармацевтически приемлемая соль или эфир.
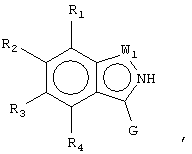
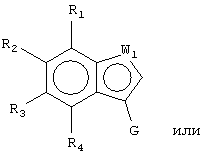
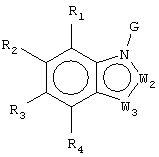
где W1 представляет O, S или NR5, W2 представляет CR5 или N и W3 представляет CR5 или N.
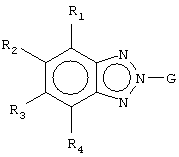 или
или 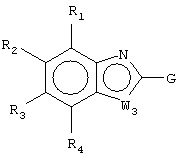
где W3 представляет NR5, S или О.
2-(3-(4-н-бутилпиперидин-1-ил)пропил)бензотиазол;
2-(3-(4-н-бутилпиперидин-1-ил)пропил)бензоксазол;
4,5-дифтор-2-(3-(4-н-бутилпиперидин-1-ил)пропил)-1Н-бензоимидазол;
6-фтор-5-нитро-2-(3-(4-н-бутилпиперидин-1-ил)пропил)-1Н-бензоимидазол;
5-трет-бутил-2-(3-(4-н-бутилпиперидин-1-ил)пропил)-1Н-бензоимидазол;
5-хлор-6-метил-2-(3-(4-н-бутилпиперидин-1-ил)пропил)-1Н-бензоимидазол;
4,6-дифтор-2-(3-(4-н-бутилпиперидин-1-ил)пропил)-1Н-бензоимидазол;
2-(3-(4-н-бутилпиперидин-1-ил)пропил)-1Н-имидазо[4,5-с]пиридин;
8-(3-(4-н-бутилпиперидин)-1-илпропил)-9Н-пурин;
7-(3-(4-н-бутилпиперидин)-1-илпропил)-3,8-дигидроимидазо[4',5':3,4]бензо[1,2-d][1,2,3]триазол;
2-(3-(4-н-бутилпиперидин-1-ил)пропил)-3а,4,5,6,7,7а-гексагидро-1Н-бензоимидазол;
1-(3-(4-н-бутилпиперидин)-1-илпропил)-1Н-индол;
1-(3-(4-н-бутилпиперидин)-1-илпропил)-1Н-бензоимидазол;
3-метил-1-(3-(4-н-бутилпиперидин)-1-илпропил)-1Н-индол;
5-бром-1-(3-(4-н-бутилпиперидин)-1-илпропил)-1Н-индол;
3-формил-1-(3-(4-н-бутилпиперидин)-1-илпропил)-1Н-индол;
7-бром-1-(3-(4-н-бутилпиперидин)-1-илпропил)-1Н-индол;
1-(3-(4-н-бутилпиперидин)-1-илпропил)-1Н-индазол;
3-(3-(4-н-бутилпиперидин)-1-илпропил)бензо[d]изоксазол;
3-(3-(4-н-бутилпиперидин)-1-илпропил)-1Н-индол;
4-нитро-2-(3-(4-н-бутилпиперидин)-1-илпропил)-1Н-бензоимидазол;
5-нитро-2-(3-(4-н-бутилпиперидин)-1-илпропил)-1Н-бензоимидазол;
4-гидрокси-2-(3-(4-н-бутилпиперидин)-1-илпропил)-1Н-бензоимидазол;
2-(3-(4-н-бутилпиперидин)-1-илпропил)-1Н-бензоимидазол;
4-метил-2-(3-(4-н-бутилпиперидин)-1-илпропил)-1Н-бензоимидазол;
3-(2-(4-н-бутилпиперидин)-1-илэтил)-1Н-индол;
3-(3-(4-н-бутилпиперидин)-1-илпропил)-1Н-индазол;
3-(2-(4-н-бутилпиперидин)этокси)-7-метилбензо[d]изоксазол;
1-(3-(4-н-метилпиперидин)-1-илпропил)-1Н-индазол;
1-(3-(4-пентилпиперидин)-1-илпропил)-1Н-индазол;
1-(3-(4-пропилпиперидин)-1-илпропил)-1Н-индазол;
1-(3-(4-(3-метилбутил)пиперидин)-1-илпропил)-1Н-индазол;
1-(3-(4-пентилиденпиперидин)-1-илпропил)-1Н-индазол;
1-(3-(4-пропилиденпиперидин)-1-илпропил)-1Н-индазол;
1-бензо[b]тиофен-2-ил-4-(4-бутилпиперидин-1-ил)бутан-1-он;
4-(4-бутилпиперидин-1-ил)-1-(3-метилбензофуран-2-ил)бутан-1-он;
4-(4-бутилпиперидин-1-ил)-1-(5-фтор-3-метилбензо[b]тиофен-2-ил)бутан-1-он;
1-бензофуран-2-ил-4-(4-бутилпиперидин-1-ил)бутан-1-он;
1-(3-бромбензо[b]тиофен-2-ил)-4-(4-бутилпиперидин-1-ил)бутан-1-он;
1-(3-бензо[b]тиофен-2-илпропил)-4-бутилпиперидин;
1-(3-бензофуран-2-илпропил)-4-бутилпиперидин;
4-бутил-1-[3-(3-метилбензофуран-2-ил)пропил]пиперидин;
4-бутил-1-[3-(5-фтор-3-метилбензо[b]тиофен-2-ил)пропил]пиперидин;
2-(3-иодпропил)бензо[b]тиофен;
1-(3-бензо[b]тиофен-2-илпропил)-4-метилпиперидин;
1-(3-бензо[b]тиофен-2-илпропил)-4-бензилпиперидин;
1-(3-бензо[b]тиофен-2-илпропил)-4-(2-метоксифенил)пиперидин;
2-(3-бромпропил)-2Н-бензотриазол;
2-[3-(4-бутилпиперидин-1-ил)пропил]-2Н-бензотриазол;
1-(3-бромпропил)-1Н-бензотриазол;
1-[3-(4-бутилпиперидин-1-ил)пропил]-1Н-бензотриазол;
1-[3-(4-бутилпиперидин-1-ил)пропил]-1Н-индол-3-карбальдегид;
{1-[3-(4-бутилпиперидин-1-ил)пропил]-1Н-индол-3-ил}метанол;
1-[3-(4-бутилпиперидин-1-ил)пропил]-2-фенил-1Н-бензоимидазол;
1-[3-(4-бутилпиперидин-1-ил)пропил]-3-хлор-1Н-индазол;
1-[3-(4-бутилпиперидин-1-ил)пропил]-6-нитро-1Н-индазол;
бензо[d]изоксазол-3-ол;
3-(2-хлорэтокси)бензо[d]изоксазол;
3-[2-(4-бутилпиперидин-1-ил)этокси]бензо[d]изоксазол;
3-(1Н-индол-3-ил)пропан-1-ол;
гидрохлорид 3-[3-(4-бутилпиперидин-1-ил)пропил]-1Н-индола;
метиловыйэфир 4-(4-бутилпиперидин-1-ил)масляной кислоты;
2-[3-(4-бутилпиперидин-1-ил)пропил]-1-метил-1Н-бензимидазол;
(2-(4-бутилпиперидин)-1-илэтил)амид 1Н-индазол-3-карбоновой кислоты;
1-[3-(4-бутилпиперидин-1-ил)пропил]-5-нитро-1Н-индазол;
2-[3-(4-бутилпиперидин-1-ил)пропил]-5-нитро-2Н-индазол;
1-[3-(4-бутилпиперидин-1-ил)пропил]-2-метил-1Н-индол;
1-{1-[3-(4-бутилпиперидин-1-ил)пропил]-1Н-индол-3-ил}этанон;
{1-[3-(4-бутилпиперидин-1-ил)пропил]-1Н-индол-3-ил}ацетонитрил;
1-[3-(4-бутилпиперидин-1-ил)пропил]-1Н-индол-3-карбонитрил;
1-[3-(4-бутилпиперидин-1-ил)пропил]-5,6-диметил-1Н-бензоимидазол;
1-[3-(4-бутилпиперидин-1-ил)пропил]-5(6)-диметил-1Н-бензоимидазол;
1-[3-(4-бутилпиперидин-1-ил)пропил]-5-метокси-1Н-бензоимидазол;
{1-[3-(4-бутилпиперидин-1-ил)пропил]-1Н-бензоимидазол-2-ил}метанол;
1-[3-(4-бутилпиперидин-1-ил)пропил]-2-трифторметил-1Н-бензоимидазол;
трет-бутиловый эфир (2-триметилстаннанилфенил)карбаминовой кислоты;
трет-бутиловый эфир [2-(4-хлорбутирил)фенил]карбаминовой кислоты;
трет-бутиловый эфир {2-[4-(4-бутилпиперидин-1-ил)бутирил]фенил} карбаминовой кислоты;
3-[3-(4-бутилпиперидин-1-ил)пропил]-1Н-индазол, HCl;
3-[3-(4-бутилпиперидин-1-ил)пропил]-5-нитро-1Н-индазол;
3-[3-(4-бутилпиперидин-1-ил)пропил]-5,7-динитро-1Н-индазол;
4-(4-бутилпиперидин-1-ил)-1-(2-метилсульфанилфенил)бутан-1-он;
3-[3-(4-бутилпиперидин-1-ил)пропил]бензо[d]изотиазол;
3-[3-(4-бутилпиперидин-1-ил)пропил]-5-метокси-1Н-индазол;
3-[3-(4-бутилпиперидин-1-ил)пропил]-4-метокси-1H-индазол;
3-[3-(4-бутилпиперидин-1-ил)пропил]-6-метокси-1Н-индазол;
3-[3-(4-бутилпиперидин-1-ил)пропил]-1Н-индазол-4-ол;
3-[3-(4-бутилпиперидин-1-ил)пропил]-1Н-индазол-6-ол или
3-[3-(4-бутилпиперидин-1-ил)пропил]-1Н-индазол-5-ол.
| ПРОИЗВОДНЫЕ ИНДОЛА, ИХ ОПТИЧЕСКИЕ ИЗОМЕРЫ И ФАРМАЦЕВТИЧЕСКИ ПРИЕМЛЕМЫЕ СОЛИ | 1991 |
|
RU2095360C1 |
| US 4870085 А, 26.09.1989 | |||
| DE 4039631 А, 17.06.1992. | |||

