ОБЛАСТЬ ТЕХНИКИ, К КОТОРОЙ ОТНОСИТСЯ ИЗОБРЕТЕНИЕ
Настоящее изобретение относится к композиции для консервирования клеток, тканей или органов животных, включающей в качестве активного ингредиента соединение индола или индазола формулы (1) или его фармацевтически приемлемую соль или изомер, способ консервирования путем использования этого соединения, и способ получения этого соединения. Более конкретно, соединения индола и индазола согласно настоящему изобретению являются эффективными при сохранении подвергаемых трансплантации клеток, тканей и органов животных, и предотвращают их повреждение, вызываемое в процессе транспортировки или хранении. Кроме того, соединения индола и индазола согласно настоящему изобретению предохраняют органы от повреждения тканей или органов, вызываемых реперфузией после трансплантации.
УРОВЕНЬ ТЕХНИКИ
За последнее время возросло количество случаев трансплантации органов в связи с развитием методов хирургических операций и разработкой лекарственных средств, таких как иммунодепрессанты. Многие пациенты нуждаются в трансплантатах, но многие органы хранят в несоответствующих для использования условиях, вследствие чего их отбраковывают. Поэтому, как сообщалось, все еще имеется много пациентов, ожидающих пересадку тканей или органов. Наиболее предпочтительно, чтобы орган пересаживали реципиенту, как только его извлекли из донора. Однако, на практике, многие операции по трансплантации сразу не проводят. Поэтому крайне актуальными техническими проблемами остаются осуществление возможности продолжительного хранения и улучшенное качество подвергаемых трансплантации органов в результате улучшения способа хранения.
В настоящее время, для консервации органов применяют хранение при низкой температуре (ниже 20°C, обычно ниже 4°C), в результате которого ингибируется метаболизм, но не обеспечиваются соответствующие физиологические условия in vivo. Для этого способа были разработаны и использованы в клинике большое разнообразие консервирующих растворов.
На раннем этапе использовали раствор Euro-Collin. Недавно был разработан раствор UW (University of Wisconsin, Wahlberg, J.A., et al., Transplantation, 43, pp.5-8, 1987), который может применяться в качестве консервирующего раствора для печени, кишечника и почки, а также поджелудочной железы, и который обеспечивает сохранение печени в условиях эксперимента до 24 часов. Однако при клинических испытаниях его используют для защиты пациентов в течение более короткого периода. Таким образом, крайне необходима разработка консервирующих растворов или вспомогательных средств, способных поддерживать жизнеспособность органов в течение более длительного времени и обеспечивать эффективное хранение органов.
Соединения индола и индазола согласно настоящему изобретению имеют структуру, которая хорошо подходит с точки зрения их медицинского применения, и уже опубликован ряд исследований по соединениям, имеющим строение индольного ядра. Например, достоверно доказана их активность по отношению к глюкокиназе (патентный документ WO 2006/112549), возможность применения в качестве противоопухолевого средства и ингибитора ангиогенеза сердечных сосудов (патентный документ WO 95/07276), и в качестве антибиотиков (патентный документ WO 2004/018428).
ОПИСАНИЕ ИЗОБРЕТЕНИЯ
Авторы настоящего изобретения провели интенсивные и обширные исследования по разработке соединений, которые могут ингибировать некроз различных клеток животных при консервировании различных тканей, органов или крови, в результате чего могут обеспечивать продолжительный период консервации, повышенное защитное действие и улучшенное функционирование органа после трансплантации. В результате было обнаружено, что производные индола и индазола, представленные следующей формулой (1), проявляют сильное описываемое ниже действие. Соединения индола и индазола согласно настоящему изобретению были раскрыты и заявлены в патентных документах Korean Patent Application Nos. 10-2007-0082687, 10-2008-0080519 и 10-2008-0080537, которые были выданы заявителю настоящего изобретения.
Соответственно, настоящее изобретение предлагает композицию для консервирования клеток, тканей или органов животных, включающую в качестве эффективного ингредиента соединение индола или индазола, представленное формулой (1), или его фармацевтически приемлемую соль или изомер, с фармацевтически приемлемым носителем.
Настоящее изобретение также предлагает способ получения композиции для консервирования клеток, тканей или органов животных, в частности, для предотвращения повреждения органов, выделяемых клеточных систем или тканей, вызванного хранением при низкой температуре, операцией по трансплантации или посттрансплантационной реперфузией, где указанный способ включает стадию смешения в качестве эффективного ингредиента соединения, представленного формулой (1), или его фармацевтически приемлемой соли или изомера, вместе с фармацевтически приемлемым носителем.
Настоящее изобретение также предлагает способ применения композиции согласно настоящему изобретению, включающий в качестве эффективного ингредиента соединение, представленное формулой (1), или его фармацевтически приемлемую соль или изомер, для консервирования клеток, тканей или органов животных для трансплантации.
В композиции настоящего изобретения используют в качестве эффективного ингредиента соединение индола или индазола, представленное следующей формулой (1), или его фармацевтически приемлемую соль или изомер:
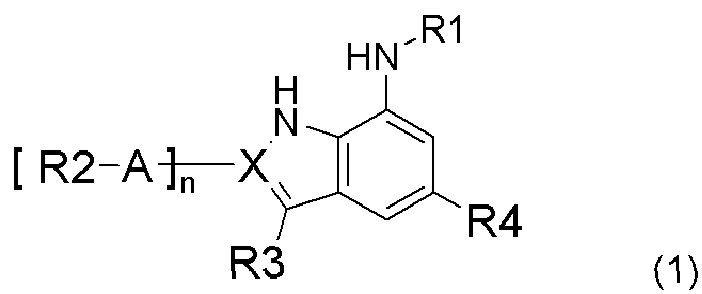
где
X представляет C или N,
n является 0 или 1, и n является 1, когда X является C, и n является 0, когда X является N,
A представляет непосредственную химическую связь, C3-C8-циклоалкил, фенил, или 5~6-членный гетероарил или гетероцикл, каждый из которых включает 1~3 гетероатомов, выбранный из атомов N, O и S,
R1 представляет водород, -C(O)-B-X'-R7 или -(CR5R6)m-B-X'-R7,
m является целым числом от 0 до 4,
каждый из R5 и R6 независимо представляет водород или C1-C5-алкил,
B представляет непосредственную химическую связь, C3-C8-циклоалкил, необязательно содержащий оксо, или 3~10-членный гетероцикл или гетероарил, каждый из которых включает 1-3 гетероатома, выбранных из атомов O, S и N,
X' представляет непосредственную химическую связь, -C(O)-, -SO2-, -CO2- или -C(О)NR5-,
R7 представляет водород, C1-C6-алкил, галоген-C1-C6-алкил, галоген, (CR5R6)m-фенил, (CR5R6)m-гидрокси или (CR5R6)m-гетероцикл, где гетероцикл необязательно содержит оксо и является 3~10-членным кольцом, включающим 1-3 гетероатома, выбранных из атомов N, O и S,
R2 представляет -(CR5R6)m-D-X''-R8,
D представляет непосредственную химическую связь или 3~10-членный гетероцикл или гетероарил, каждый из которых необязательно содержит оксо и является необязательно конденсированным, и включает 1-4 гетероатома, выбранных из атомов N, O и S,
X'' представляет непосредственную химическую связь, -C(O)-, -C(O)O-, -NR5C(O)-, -C(О)NR5- или -O-,
R8 представляет водород, галоген, C1-C6-алкил, галоген-C1-C6-алкил, три(C1-C6-алкил)силан или гидрокси-C1-C6-алкил,
R3 представляет водород, галоген, циано, нитро, арил-R9 или (CR5R6)m-D-R9,
R9 представляет водород, галоген, C1-C6-алкил, циано, нитро или C1-C6-алкокси,
R4 представляет -(CR5R6)m-Y-D-R10,
Y представляет непосредственную химическую связь, -C(O)O- или -O-, и
R10 представляет водород, нитро, галоген, C1-C6-алкил, карбокси-C1-C6-алкил, арил или -C(O)O-R5,
где каждый из указанных: алкил, алкокси, арил, циклоалкил, гетероцикл и гетероарил могут быть необязательно замещен с помощью одного или более заместителей, выбранных из группы, состоящей из гидрокси, галогена, нитрила, амино, C1-C6-алкиламина, ди(C1-C6-алкил)амина, C1-C6-алкила, галоген-C1-C6-алкила, C1-C6-алкилсульфонила, арил-C1-C6-алкокси и оксо.
При определении заместителей соединения формулы (1) согласно настоящему изобретению, термин "алкил" обозначает алифатический углеводородный радикал. Алкил может являться "насыщенным алкилом", не включающим алкенильный или алкинильный фрагмент, или "ненасыщенным алкилом", включающим, по меньшей мере, один алкенильный или алкинильный фрагмент. "Алкенил" обозначает группу, имеющую, по меньшей мере, одну двойную связь углерод-углерод, и "алкинил" обозначает группу, имеющую, по меньшей мере, одну тройную связь углерод-углерод. Алкил может являться разветвленной или линейной цепью, когда он используется сам по себе или в комбинации с алкокси.
Алкил может иметь 1-20 углеродных атомов, если здесь не определено иначе. Алкилом может являться алкил среднего размера, имеющий 1-10 углеродных атомов. Алкилом может являться низший алкил, имеющий 1-6 углеродных атомов. Типичные алкильные группы включают, но этим не ограничивая, метил, этил, пропил, изо-пропил, н-бутил, изо-бутил, трет-бутил, пентил, гексил, этенил, пропенил, бутенил, и так далее. Например, C1-C4-алкил имеет в алкильной цепи 1-4 углеродных атомов, и его выбирают из группы, состоящей из метила, этила, пропила, изо-пропила, н-бутила, изо-бутила, втор-бутила и трет-бутила.
Термин "алкокси" обозначает алкилокси, имеющую 1-10 углеродных атомов, если здесь не определено иначе.
Термин "циклоалкил" обозначает насыщенный алифатический 3~10-членный цикл, если здесь не определено иначе. Типичные циклоалкильные группы включают, но этим не ограничивая, циклопропил, циклобутил, циклопентил, циклогексил и так далее.
"Арил" включает, по меньшей мере, одно кольцо, имеющее систему с делокализованным π-электроном, например, моноциклические или конденсированные-полициклические группы (то есть кольца, совместно использующие смежные пары углеродных атомов). Иначе говоря, используемый здесь термин "арил" обозначает 4~10-членное, предпочтительно, 6~10-членное ароматическое моноциклическое или полициклическое кольцо и включает, например, фенил и нафтил.
Термин "гетероарил" обозначает 3~10-членное, предпочтительно, 4~8-членное, более предпочтительно, 5~6-членное ароматическое кольцо, которое включает 1-3 гетероатома, выбранных из атомов N, O и S, и может быть конденсировано с бензо или C3-C8 циклоалкилом, если здесь не определено иначе. Примеры моноциклического гетероарила включают, но этим не ограничивая, тиазол, оксазол, тиофен, фуран, пирол, имидазол, изоксазол, изотиазол, пиразол, триазол, триазин, тиадиазол, тетразол, оксадиазол, пиридин, пиридазин, пиримидин, пиразин и их аналоги. Примеры бициклического гетероарила включают, но этим не ограничивая, индол, индолин, бензотиофен, бензофуран, бензимидазол, бензоксазол, бензизоксазол, бензтиазол, бензтиадиазол, бензтриазол, хинолин, изохинолин, пурин, фуропиридин и их аналоги.
Термин "гетероцикл" обозначает 3-10-членное, предпочтительно 4~8-членное, и более предпочтительно, 5~6-членное кольцо, которое включает 1-3 гетероатома, выбранных из атомов N, O и S, и может быть конденсировано с бензо или C3-C8-циклоалкилалкилом, и может быть насыщенным или содержать 1 или 2 двойные связи. Примеры гетероцикла включают, но этим не ограничивая, пирролин, пирролидин, имидазолин, имидазолидин, пиразолин, пиразолидин, пиран, пиперидин, морфолин, тиоморфолин, пиперазин, гидрофуран, и так далее.
Другие используемые здесь термины и сокращения могут восприниматься специалистом в этой области в их общепринятых значениях, если здесь не определено иначе.
Предпочтительными соединениями среди соединений формулы 1 согласно настоящему изобретению являются соединения, в которых
X представляет C или N,
n является 0 или 1, и n является 1, когда X является C, и n является 0, когда X является N,
A представляет непосредственную химическую связь, фенил, или 5~6-членный гетероарил или гетероцикл, каждый из которых включает 1-3 гетероатома, выбранных из атомов N, O и S,
R1 представляет водород, -C(O)-B-X'-R7 или -(CR5R6)m-B-X'-R7,
m является целым числом от 0 до 2,
каждый из R5 и R6 независимо представляет водород или C1-C5-алкил,
B представляет непосредственную химическую связь, C4-C7-циклоалкил, необязательно содержащий оксо и необязательно замещенный с помощью галогена, или 4~8-членный гетероцикл или гетероарил, каждый из которых включает 1-3 гетероатома, выбранных из атомов O, S и N,
X' представляет непосредственную химическую связь, -C(O)-, -SO2-, -CO2- или -C(O)NH-,
R7 представляет водород, C1-C6-алкил, галоген-C1-C6-алкил, галоген, (CR5R6)m-фенил, (CR5R6)m-гидрокси или (CR5R6)m-гетероцикл, где гетероцикл необязательно содержит оксо и является 4~8-членным кольцом, включающим 1-3 гетероатома, выбранных из атомов N, O и S,
R2 представляет -(CR5R6)m-D-X''-R8,
D представляет непосредственную химическую связь или 4~8-членный гетероцикл или гетероарил, каждый из которых необязательно содержит оксо и является необязательно конденсированным, и включает 1-4 гетероатома, выбранных из атомов N, O и S,
X'' представляет -C(O)-, -C(O)O-, -NR5C(O)-, -C(O)NR5- или -O-,
R8 представляет водород, галоген, C1-C6-алкил, галоген-C1-C6-алкил, три(C1-C6-алкил)силан или гидрокси-C1-C6-алкил,
R3 представляет водород, галоген, циано, нитро, арил-R9 или (CR5R6)m-D-R9,
R9 представляет водород, галоген, C1-C6-алкил, циано, нитро или C1-C6-алкокси,
R4 представляет -(CR5R6)m-Y-D-R10,
Y представляет непосредственную химическую связь, -C(O)O- или -O-, и
R10 представляет водород, нитро, галоген, C1-C6-алкил, карбокси-C1-C6-алкил, арил или -C(O)O-R5.
В соединении формулы (1) согласно настоящему изобретению X является C или N, и структура соединения для каждого случая может быть представлена следующей формулой (1a) или (1b), соответственно.
Формула 1a
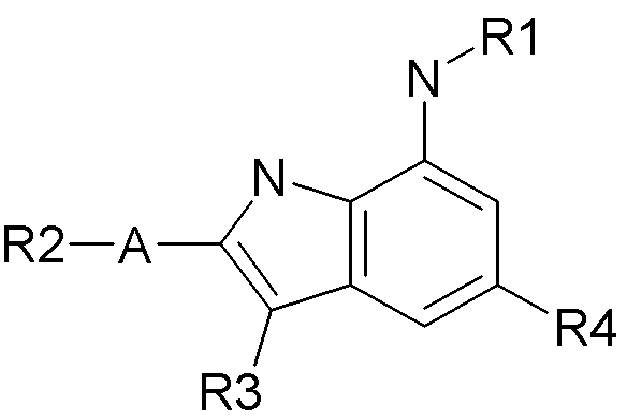
Формула 1b

В соединении формулы (1) согласно настоящему изобретению, заместитель A более предпочтительно выбирать из группы, состоящей из фенила, пиридина, 1,4-пиразина, 4,5-дигидротиазола, тиазола, 4,5-дигидрооксазола, [1,2,4]оксадиазола и [1,3,4]-оксадиазола.
Более предпочтительно, чтобы заместитель R1 представлял -C(O)-B-X'-R7 или -(CHR5)m-B-X'-R7, где m является целым числом от 0 до 2; R5 представляет C1-C3-алкил; B представляет непосредственную химическую связь, C5-C6-циклоалкил, необязательно содержащий оксо, или 5~6-членный гетероцикл или гетероарил, каждый из которых включает 1-3 гетероатома, выбранных из атомов O, S и N; X' представляет непосредственную химическую связь, -C(O)-, -SO2-, -CO2- или -C(O)NH-; и R7 представляет водород, C1-C3-алкил, галоген-C1-C3-алкил, галоген, (CH2)m-фенил, (CH2)m-гидрокси или (CH2)m-гетероцикл, где гетероцикл необязательно содержит оксо и является 5~6-членным кольцом, включающим 1-3 гетероатома, выбранных из атомов N, O и S. В заместителе R1, B наиболее предпочтительно выбирать из группы, состоящей из циклопентила, циклогексила, пиперидина, тетрагидропирана, оксоциклогексила, пирролидина, дифторциклогексила и тетрагидрофурана; и R7 наиболее предпочтительно выбирать из группы, состоящей из водорода, метила, этила, изопропила, бензила, гидроксиметила, (морфолин-4-ил)этила, тетрагидрофурана, 2,2,2-трифторэтила, гидроксиэтила, 1,1-диоксотиоморфолина, тетрагидропирана, (тетрагидропиран-4-ил)метила и трифторметила.
В заместителе R2, более предпочтительно, чтобы D представлял непосредственную химическую связь, или его выбирали из группы, состоящей из пиперазина, пирролидина, морфолина, 1,1-диоксотиоморфолина и оксопиперазина; и R8 более предпочтительно выбирать из группы, состоящей из водорода, этила, гидроксиметила, метила и фтора.
Более предпочтительно, чтобы заместитель R3 представлял водород; галоген; фенил, необязательно замещенный с помощью алкокси; или 6-членный гетероциклилметил, включающий 1~3 гетероатома, выбранных из атомов N, S и O в качестве членов кольца, и необязательно содержащий оксо. R3 наиболее предпочтительно выбирать из группы, состоящей из водорода, брома, фенила, метоксифенила, морфолин-4-илметила, оксопиперазин-4-илметила и 1,1-диоксотиоморфолин-4-илметила.
Более предпочтительно, чтобы заместитель R4 представлял -(CH2)m-Y-D-R10, где m является целым числом от 0 до 2; Y представляет непосредственную химическую связь, -C(O)O- или -O-; D представляет пиридин или 5~6-членный гетероцикл, включающий 1~3 гетероатома, выбранных из атомов N, S и O, и необязательно содержащий оксо; и R10 представляет водород, галоген, C1-C3-алкил, -(CH2)-CO2H, арил или -C(O)O-R5, где R5 представляет водород или C1-C3-алкил. В заместителе R4, D наиболее предпочтительно выбирать из группы, состоящей из 1,1-диоксо-тиоморфолина, оксопиперазина, пиридина, морфолина и 4,5-дигидро-тиазола; и R10 наиболее предпочтительно выбирать из группы, состоящей из водорода, фтора, хлора, брома, метила, этила и -(CH2)-CO2H.
Характерные соединения формулы (1) согласно настоящему изобретению включают следующие соединения:
циклопентил-[2-(4,5-дигидро-1,3-тиазол-2-ил)-1H-индол-7-ил]амин;
[2-(4,5-дигидротиазол-2-ил)-1H-индол-7-ил]-(4-метилцикло-гексил)амин;
[2-(4,5-дигидротиазол-2-ил)-1H-индол-7-ил]пиперидин-4-ил-амин;
2-5-[7-(тетрагидропиран-4-иламино)-1H-индол-2-ил]-[1,2,4]-оксадиазол-3-ил]этанол;
[(R)-2-(7-циклопентиламино-1H-индол-2-ил)-4,5-дигидро-1,3-тиазол-4-ил]метанол;
циклопентил-[2-((R)-4-пирролидин-1-илметил-4,5-дигидро-тиазол-2-ил)-1H-индол-7-ил]амин;
{(R)-2-[7-(тетрагидропиран-4-иламино)-1H-индол-2-ил]-4,5-дигидротиазол-4-ил}метанол;
[(R)-2-(7-циклопентиламино-5-фтор-1H-индол-2-ил)-4,5-дигидротиазол-4-ил]метанол;
{(R)-2-[5-фтор-7-(тетрагидропиран-4-иламино)-1H-индол-2-ил]-4,5-дигидротиазол-4-ил}метанол;
{(R)-2-[5-(пиридин-3-илокси)-7-(тетрагидропиран-4-иламино)-1H-индол-2-ил]-4,5-дигидротиазол-4-ил}метанол;
[(R)-2-(5-хлор-7-циклопентиламино-1H-индол-2-ил)-4,5-дигидротиазол-4-ил]уксусная кислота;
этиловый эфир [(R)-2-(5-хлор-7-циклопентиламино-1H-индол-2-ил)-4,5-дигидротиазол-4-ил]уксусной кислоты;
2-{(R)-2-[5-хлор-7-(тетрагидропиран-4-иламино)-1H-индол-2-ил]-4,5-дигидротиазол-4-ил}этанол;
1-[4-(2-{(R)-2-[5-хлор-7-(тетрагидропиран-4-иламино)-1H-индол-2-ил]-4,5-дигидротиазол-4-ил}этил)пиперазин-1-ил]-2-гидроксиэтанон;
1-(2-{(R)-2-[5-хлор-7-(тетрагидропиран-4-иламино)-1H-индол-2-ил]-4,5-дигидротиазол-4-ил}этил)пирролидин-3-ол;
[(R)-2-(5-бром-7-циклопентиламино-1H-индол-2-ил)-4,5-дигидротиазол-4-ил]уксусная кислота;
[(R)-2-(7-циклопентиламино-5-этокси-1H-индол-2-ил)-4,5-дигидротиазол-4-ил]уксусная кислота;
[(R)-2-(7-циклопентиламино-5-этокси-1H-индол-2-ил)-4,5-дигидротиазол-4-ил]уксусная кислота;
[2-(7-циклопентиламино-5-фенокси-1H-индол-2-ил)-4,5-дигидротиазол-4-ил]уксусная кислота;
[(R)-2-(7-циклопентиламино-5-фенокси-1H-индол-2-ил)-4,5-дигидротиазол-4-ил]уксусная кислота;
[(S)-2-(7-циклопентиламино-5-фенокси-1H-индол-2-ил)-4,5-дигидротиазол-4-ил]уксусная кислота;
этиловый эфир 3-[(R)-2-(5-хлор-7-циклопентиламино-1H-индол-2-ил)-4,5-дигидротиазол-4-ил]пропионовой кислоты;
3-[(R)-2-(5-хлор-7-циклопентиламино-1H-индол-2-ил)-4,5-дигидротиазол-4-ил]пропионовая кислота;
циклопентил-(2-пиридин-2-ил-1H-индол-7-ил)амин;
циклопентил-(2-пиразин-2-ил-1H-индол-7-ил)амин;
(2-пиразин-2-ил-1H-индол-7-ил)-(тетрагидропиран-4-ил)амин;
циклопентил-(2-тиазол-2-ил-1H-индол-7-ил)амин;
этиловый эфир 2-(7-циклопентиламино-5-метил-1H-индол-2-ил)тиазол-4-карбоновой кислоты;
2-(7-циклопентиламино-5-метил-1H-индол-2-ил)тиазол-4-карбоновая кислота;
[2-(7-циклопентиламино-5-метил-1H-индол-2-ил)тиазол-4-ил]-метанол;
[2-(7-циклопентиламино-5-метил-1H-индол-2-ил)тиазол-5-ил]-метанол;
циклопентил-(5-метил-2-[1,3,4]оксадиазол-2-ил-1H-индол-7-ил)амин;
циклопентил-(5-метил-2-пиридин-2-ил-1H-индол-7-ил)амин;
(5-метил-2-пиридин-2-ил-1H-индол-7-ил)-(тетрагидропиран-4-ил)амин;
циклогексил-(5-метил-2-пиридин-2-ил-1H-индол-7-ил)амин;
1-[4-(5-метил-2-пиридин-2-ил-1H-индол-7-иламино)пиперидин-1-ил]этанон;
(1-метилпиперидин-4-ил)-(5-метил-2-пиридин-2-ил-1H-индол-7-ил)амин;
4-(5-метил-2-пиридин-2-ил-1H-индол-7-иламино)цикло-гексанон;
(1-бензилпирролидин-3-ил)-(5-метил-2-пиридин-2-ил-1H-индол-7-ил)амин;
циклопентилметил-(5-метил-2-пиридин-2-ил-1H-индол-7-ил)-амин;
N-(5-метил-2-пиридин-2-ил-1H-индол-7-ил)бензамид;
циклопентил-(5-метил-2-пиразин-2-ил-1H-индол-7-ил)амин;
циклопентил-(5-этокси-2-пиридин-2-ил-1H-индол-7-ил)амин;
циклопентил-(5-фенокси-2-пиридин-2-ил-1H-индол-7-ил)амин;
циклопентил-(3,5-диметил-2-фенил-1H-индол-7-ил)амин;
циклопентил-(5-метил-2-фенил-1H-индол-7-ил)амин;
(2-циклогексил-5-метил-1H-индол-7-ил)циклопентиламин;
циклопентил-[5-метил-2-(6-метилпиридин-2-ил)-1H-индол-7-ил]амин;
5-метил-2-фенил-1H-индол-7-ил)-(тетрагидропиран-4-ил)-амин;
(5-метил-2-фенил-1H-индол-7-ил)-(1-метилпиперидин-4-ил)-амин;
1-[4-(5-метил-2-фенил-1H-индол-7-иламино)пиперидин-1-ил]-этанон;
гидрохлорид (5-метил-2-фенил-1H-индол-7-ил)пиперидин-4-ил-амина;
2-гидрокси-1-[4-(5-метил-2-фенил-1H-индол-7-иламино)-пиперидин-1-ил]этанон;
(1-метансульфонилпиперидин-4-ил)-(5-метил-2-фенил-1H-индол-7-ил)амин;
4-(5-метил-2-фенил-1H-индол-7-иламино)циклогексан-карбоновая кислота;
(2-морфолин-4-ил-этил)амид 4-(5-метил-2-фенил-
1H-индол-7-иламино)циклогексан-
карбоновой кислоты;
циклопентилметил-(5-метил-2-фенил-1H-индол-7-ил)амин;
(5-метил-2-фенил-1H-индол-7-ил)-(тетрагидропиран-4-илметил)-амин;
(5-хлор-2-фенил-1H-индол-7-ил)циклопентиламин;
(5-хлор-2-фенил-1H-индол-7-ил)-(тетрагидропиран-4-ил)амин;
(5-хлор-2-фенил-1H-индол-7-ил)-(1-метилпиперидин-4-ил)-амин;
(5-хлор-2-фенил-1H-индол-7-ил)циклогексиламин;
(1-бензилпирролидин-3-ил)-(5-хлор-2-фенил-1H-индол-7-ил)-амин;
метиловый эфир 4-(5-хлор-7-циклопентиламино-1H-индол-2-ил)бензойной кислоты;
4-(5-хлор-7-циклопентиламино-1H-индол-2-ил)бензойная кислота;
[4-(5-хлор-7-циклопентиламино-1H-индол-2-ил)фенил]метанол;
метиловый эфир 4-(7-циклопентиламино-5-метил-1H-индол-2-ил)бензойной кислоты;
метиловый эфир 2-(5-хлор-7-циклопентиламино-1H-индол-2-ил)бензойной кислоты;
2-(5-хлор-7-циклопентиламино-1H-индол-2-ил)бензойная кислота;
[2-(5-хлор-7-циклопентиламино-1H-индол-2-ил)фенил]метанол;
этиловый эфир 7-циклопентиламино-2-фенил-1H-индол-5-карбоновой кислоты;
7-циклопентиламино-2-фенил-1H-индол-5-карбоновая кислота;
(7-циклопентиламино-2-фенил-1H-индол-5-ил)метанол;
этиловый эфир (7-циклопентиламино-2-фенил-1H-индол-5-ил)уксусной кислоты;
(7-циклопентиламино-2-фенил-1H-индол-5-ил)уксусная кислота;
2-[(4S)-2-[5-метил-7-(оксан-4-илметиламино)-1H-индол-2-ил]-4,5-дигидро-1,3-тиазол-4-ил]уксусная кислота;
2-[(4S)-2-[5-хлор-7-(оксан-4-илметиламино)-1H-индол-2-ил]-4,5-дигидро-1,3-тиазол-4-ил]уксусная кислота;
2-[(4S)-2-[7-[(4,4-дифторциклогексил)амино]-5-метил-1H-индол-2-ил]-4,5-дигидро-1,3-тиазол-4-ил]уксусная кислота;
2-[(4S)-2-[7-(оксан-4-иламино)-5-фенокси-1H-индол-2-ил]-4,5-дигидро-l,3-тиазол-4-ил]уксусная кислота;
2-[(4R)-2-[7-(оксан-4-иламино)-5-фенокси-1H-индол-2-ил]-4,5-дигидро-1,3-тиазол-4-ил]уксусная кислота;
2-[(4R)-2-[7-(оксан-4-илметиламино)-5-фенокси-1H-индол-2-ил]-4,5-дигидро-1,3-тиазол-4-ил]уксусная кислота;
2-[(4S)-2-[7-(циклопентиламино)-1H-индол-2-ил]-4,5-дигидро-1,3-тиазол-4-ил]уксусная кислота;
2-[(4S)-2-[7-[(1-ацетилпирролидин-3-ил)амино]-5-метил-1H-индол-2-ил]-4,5-дигидро-1,3-тиазол-4-ил]уксусная кислота;
2-[(4S)-2-[7-(оксан-4-илметиламино)-1H-индол-2-ил]-4,5-дигидро-1,3-тиазол-4-ил]уксусная кислота;
2-[(4S)-2-[7-(оксан-4-иламино)-1H-индол-2-ил]-4,5-дигидро-1,3-тиазол-4-ил]уксусная кислота;
2-[(4S)-2-[7-(оксан-2-илметиламино)-1H-индол-2-ил]-4,5-дигидро-1,3-тиазол-4-ил]уксусная кислота;
2-[(4S)-2-[5-метил-7-[[1-(3,3,3-трифторпропаноил)пиперидин-4-ил]амино]-1H-индол-2-ил]-4,5-дигидро-1,3-тиазол-4-ил]уксусная кислота;
2-[(4R)-2-[7-(циклопентиламино)-5-метил-1H-индол-2-ил]-4,5-дигидро-1,3-тиазол-4-ил ]уксусная кислота;
2-[(4R)-2-[5-метил-7-(оксан-4-илметиламино)-1H-индол-2-ил]-4,5-дигидро-1,3-тиазол-4-ил]уксусная кислота;
4-[2-[(4S)-2-[5-метил-7-(оксан-4-илметиламино)-1H-индол-2-ил]-4,5-дигидро-1,3-тиазол-4-ил]этил]пиперазин-2-он;
2-[(4S)-4-[2-(1,1-диоксо-1,4-тиазинан-4-ил)этил]-4,5-дигидро-1,3-тиазол-2-ил]-5-метил-N-(оксан-4-илметил)-1H-индол-7-иламин;
N-(4,4-дифторциклогексил)-5-метил-2-[(4S)-4-(2-морфолин-4-илэтил)-4,5-дигидро-1,3-тиазол-2-ил]-1H-индол-7-иламин;
4-[2-[(4S)-2-[7-[(4,4-дифторциклогексил)амино]-5-метил-1H-индол-2-ил]-4,5-дигидро-1,3-тиазол-4-ил]этил]пиперазин-2-он;
4-[2-[(4S)-2-[7-(оксан-4-илметиламино)-5-фенокси-1H-индол-2-ил]-4,5-дигидро-1,3-тиазол-4-ил]этил]пиперазин-2-он;
2-[(4S)-4-(2-морфолин-4-илэтил)-4,5-дигидро-1,3-тиазол-2-ил]-N-(оксан-4-илметил)-5-фенокси-1H-индол-7-амин;
5-метил-2-[(4S)-4-(2-морфолин-4-илэтил)-4,5-дигидро-1,3-тиазол-2-ил]-N-(оксан-4-илметил)-1H-индол-7-амин;
1-[2-[(4S)-2-[5-метил-7-(оксан-4-илметиламино)-1H-индол-2-ил]-4,5-дигидро-1,3-тиазол-4-ил]этил]пиперидин-4-карбоксиамид;
[(2R)-1-[2-[(4S)-2-[5-метил-7-(оксан-4-илметиламино)-1H-индол-2-ил]-4,5-дигидро-1,3-тиазол-4-ил]этил]пирролидин-2-ил]метанол;
(2S)-1-[2-[(4S)-2-[5-метил-7-(оксан-4-илметиламино)-1H-индол-2-ил]-4,5-дигидро-1,3-тиазол-4-ил]этил]пирролидин-2-карбоксиамид;
4-[2-[(4R)-2-[7-(циклопентиламино)-5-метил-1H-индол-2-ил]-4,5-дигидро-1,3-тиазол-4-ил]этил]пиперазин-2-он;
2-[(4S)-2-[7-(циклопентиламино)-5-метил-1H-индол-2-ил]-4,5-дигидро-1,3-оксазол-4-ил]уксусная кислота;
{(S)-2-[5-метил-7-(тетрагидропиран-4-иламино)-1H-индол-2-ил]-4,5-дигидрооксазол-4-ил}уксусная кислота;
2-[(4S)-2-[5-метил-7-(тетрагидропиран-4-иламино)-1H-индол-2-ил]-4,5-дигидро-1,3-оксазол-4-ил]этанол;
{5-метил-2-[(S)-4-(2-морфолин-4-илэтил)-4,5-дигидро-1,3-оксазол-2-ил]-1H-индол-7-ил}-(тетрагидропиран-4-ил)амин;
4-[(5-хлор-2-фенил-1H-индол-7-ил)амино]-N-этилпиперидин-1-карбоксиамид;
[4-[(5-хлор-2-фенил-1H-индол-7-ил)амино]пиперидин-1-ил]-(оксолан-3-ил)метанон;
2-[7-(оксан-4-иламино)-2-фенил-1H-индол-5-ил]уксусная кислота;
2-[7-(циклопентилметиламино)-2-фенил-1H-индол-5-ил]уксусная кислота;
5-фтор-N-(l-метилпиперидин-4-ил)-2-фенил-1H-индол-7-амин;
2-[4-[(5-фтор-2-фенил-1H-индол-7-ил)амино]пиперидин-1-ил]этанон;
5-фтор-N-[1-(оксан-4-ил)пиперидин-4-ил]-2-фенил-1H-индол-7-амин;
N-[1-(1,1-диоксан-4-ил)пиперидин-4-ил]-5-фтор-2-фенил-1H-индол-7-амин;
N-(оксан-4-ил)-5-фенокси-2-фенил-1H-индол-7-амин;
метил 2-[(5-фтор-2-фенил-1H-индол-7-ил)амино]ацетат;
2-[(5-фтор-2-фенил-1H-индол-7-ил)амино]уксусная кислота;
метил 2-[(5-хлор-2-фенил-1H-индол-7-ил)амино]пропаноат;
2-[(5-хлор-2-фенил-1H-индол-7-ил)амино]пропановая кислота;
2-[(5-фенокси-2-фенил-1H-индол-7-ил)амино]уксусная кислота;
2-[(5-фенокси-2-фенил-1H-индол-7-ил)амино]пропановая кислота;
2-[(4S)-2-[7-(оксан-4-илметиламино)-2-фенил-1H-индол-5-ил]-4,5-дигидро-1,3-тиазол-4-ил]уксусная кислота;
2-[(4S)-2-[7-(циклопентиламино)-2-фенил-1H-индол-5-ил]-4,5-дигидро-1,3-тиазол-4-ил]уксусная кислота;
метил 2-[4-[5-хлор-7-(оксан-4-иламино)-1H-индол-2-ил]-фенил]ацетат;
метил 2-[4-[5-хлор-7-(оксан-4-илметиламино)-1H-индол-2-ил]-фенил]ацетат;
2-[4-[5-хлор-7-(оксан-4-иламино)-1H-индол-2-ил]фенил]уксусная кислота;
5-[(1,1-диоксо-1,4-тиазинан-4-ил)метил]-N-(оксан-4-ил)-2-фенил-1H-индол-7-амин;
5-[(1,1-диоксо-1,4-тиазинан-4-ил)метил]-N-(оксан-4-илметил)-2-фенил-1H-индол-7-амин;
4-[[7-(оксан-4-иламино)-2-фенил-1H-индол-5-ил]метил]-пиперазин-2-он;
5-[(1,1-диоксо-1,4-тиазинан-4-ил)метил]-2-фенил-N-пиперидин-4-ил-1H-индол-7-амин;
[4-[[5-[(1,1-диоксо-1,4-тиазинан-4-ил)метил]-2-фенил-1H-индол-7-ил]амино]пиперидин-1-ил]-(оксолан-3-ил)метанон;
N-[4-[5-[(1,1-диоксо-1,4-тиазинан-4-ил)метил]-7-(оксан-4-иламино)-1H-индол-2-ил]фенил]ацетамид;
N-[4-[7-(дициклопентиламино)-5-[(1,1-диоксо-1,4-тиазинан-4-ил)метил]-1H-индол-2-ил]фенил]ацетамид;
N-[4-[5-[(1,1-диоксо-1,4-тиазинан-4-ил)метил]-7-(оксан-4-илметиламино)-1H-индол-2-ил]фенил]ацетамид;
N-циклопентил-5-[(1,1-диоксо-1,4-тиазинан-4-ил)метил]-2-(4-метоксифенил)-1H-индол-7-амин;
5-[(1,1-диоксо-1,4-тиазинан-4-ил)метил]-2-(4-метоксифенил)-N-(оксан-4-ил)-1H-индол-7-амин;
5-[(1,1-диоксо-1,4-тиазинан-4-ил)метил]-N-(3-метоксибутил)-2-фенил-1H-индол-7-амин;
5-[(1,1-диоксо-1,4-тиазинан-4-ил)метил]-2-(3-фторфенил)-N-(оксан-4-ил)-1H-индол-7-амин;
N-циклопентил-5-[(1,1-диоксо-1,4-тиазинан-4-ил)метил]-2-(3-фторфенил)-1H-индол-7-амин;
3-бром-5-[(1,1-диоксо-1,4-тиазинан-4-ил)метил]-N-(оксан-4-ил)-2-фенил-1H-индол-7-амин;
3-бром-5-(морфолин-4-илметил)-N-(оксан-4-ил)-2-фенил-1H-индол-7-амин;
3-бром-N-циклопентил-5-[(1,1-диоксо-1,4-тиазинан-4-ил)-метил]-2-фенил-1H-индол-7-амин;
3-бром-5-[(1,1-диоксо-1,4-тиазинан-4-ил)метил]-N-(оксан-4-ил)-2-фенил-1H-индол-7-амин;
5-хлор-N-(оксан-4-ил)-3-фенил-1H-индол-7-амин;
5-хлор-N-циклопентил-3-фенил-1H-индол-7-амин;
5-хлор-N-(оксан-4-илметил)-3-фенил-1H-индол-7-амин;
5-[(1,1-диоксо-1,4-тиазинан-4-ил)метил]-N-(оксан-4-ил)-3-фенил-2-триметилсилил-1H-индол-7-амин;
4-[[5-хлор-7-(циклопентиламино)-2-фенил-1H-индол-3-ил]метил]пиперазин-2-он;
4-[[5-хлор-7-(оксан-4-иламино)-2-фенил-1H-индол-3-ил]метил]пиперазин-2-он;
4-[[5-хлор-7-(оксан-4-илметиламино)-2-фенил-1H-индол-3-ил]метил]пиперазин-2-он;
N-циклопентил-3-(4-метоксифенил)-1H-индазол-7-амин;
3-(4-метоксифенил)-N-(оксан-4-ил)-1H-индазол-7-амин;
3-(4-метоксифенил)-N-(оксан-4-илметил)-1H-индазол-7-амин и
2-(7-циклопентиламино-2-фенил-1H-индол-5-ил)этанол.
Кроме того, используемые здесь термины и сокращения имеют их исконное значение, если не определено иначе.
Соединение формулы (1) согласно настоящему изобретению может также образовывать фармацевтически приемлемые соли. Такие фармацевтически приемлемые соли включают кислотно-аддитивные соли, получаемые при взаимодействии с кислотой с образованием нетоксичной соли присоединения кислоты, имеющей фармацевтически приемлемый анион, включая, например, неорганическую кислоту, такую как хлористоводородная кислота, серная кислота, азотная кислота, фосфорная кислота, бромистоводородная кислота, йодистоводородная кислота и другие подобные кислоты; органическую кислоту, такую как винная кислота, муравьиная кислота, лимонная кислота, уксусная кислота, трихлоруксусная кислота, трифторуксусная кислота, глюконовая кислота, бензойная кислота, молочная кислота, фумаровая кислота, малеиновая кислота, салициловая кислота и другие подобные кислоты; сульфоновую кислоту, такую как метансульфоновая кислота, этансульфоновая кислота, бензолсульфоновая кислота, п-толуолсульфоновая кислота и другие подобные кислоты. Кроме того, например, фармацевтически приемлемые соли карбоновой кислоты включают соли щелочных металлов или соли щелочноземельных металлов, образованные литием, натрием, калием, кальцием, магнием и другими подобными металлами; соли аминокислот лизина, аргинина, гуанидина и других подобных аминокислот; органические соли, такие как соли дициклогексиламина, N-метил-D-глюкамина, трис(гидроксиметил)-метиламина, диэтаноламина, холина, триэтиламина и других подобных аминов. Соединение формулы (1) согласно настоящему изобретению может быть превращено в его соль при помощи традиционных методов.
Кроме того, соединение согласно настоящему изобретению может иметь центр с асимметрическим атомом (атомами) углерода, и поэтому может присутствовать в виде R или S изомерных форм, рацематов, диастереомерных смесей, и индивидуальных диастереомеров. Настоящее изобретение охватывает все эти изомерные формы и смеси.
Настоящее изобретение также предлагает способ получения соединения формулы (1). В дальнейшем, для облегчения понимания настоящего изобретения, способы получения соединения формулы (1) объясняются на основе примеров схем реакций. Однако следует иметь в виду, что специалист в этой области мог бы синтезировать соединение формулы (1) при помощи различных методов, основываясь на структуре формулы (1), и такие методы входят в объем настоящего изобретения. То есть соединение формулы (1) может быть получено, необязательно, путем объединения различных методов, раскрытых в настоящем описании и/или описанных в прототипах изобретения, которые входят в рамки настоящего изобретения, и поэтому способы получения соединения формулы (1) не ограничиваются способами, объясняемыми ниже.
Соединение формулы (1) может быть получено согласно следующей схеме реакции (1) путем восстановления нитрогруппы соединения (2) с получением соединения амина (3), и затем путем введения заместителя в образованную аминогруппу. В качестве варианта, соединение формулы (1) может быть получено согласно следующим схемам реакций (2)-(8) путем модификации заместителей R3, R5, R6, R7 соединения (4).
Схема реакции 1
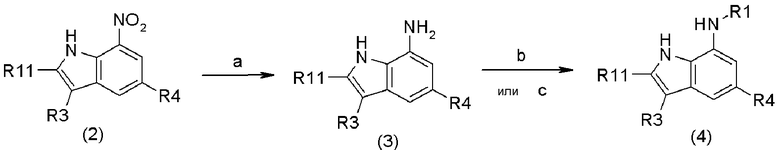
В приведенной выше схеме реакции 1:
a является Fe, Zn, или H2 (Pd/C);
b является ацилирующим реагентом в форме R7-B-CO-W, где R7 и B определены в формуле (1), W является OH или уходящей группой, такой как хлорид, бромид, йодид, смешанный ангидрид и или другая подобная группа;
c является кетоном в форме R7-B=O или соединением альдегида в форме R7-B-CHO, триацетоксиборгидридом натрия (NaBH(OAc)3) или цианоборгидридом натрия (NaBH3CN);
R3 определен в формуле (1);
R11 представляет A-R2 или CO2R12, где A и R2 определены в формуле (1), R12 представляет C1-C6алкил;
R4 определен в формуле (1); и
R1 определен в формуле (1).
Соединение (2) может быть получено при помощи способов, описанных в следующих схемах реакций (2)-(8).
Соединение (3) может быть получено путем восстановления соединения (2). Реакция восстановления может быть проведена при использовании кислотного катализатора и металла, или металлического катализатора в присутствии газообразного водорода.
В реакции восстановления при применении кислотного катализатора и металла используемой кислотой может являться, например, неорганическая кислота, такая как хлористоводородная кислота, серная кислота, азотная кислота, фосфорная кислота и другие подобные кислоты; органоугольная кислота, такая как уксусная кислота, трифторуксусная кислота и другие подобные кислоты; или соль аминокислоты, такая как хлорид аммония, и предпочтительно, хлористоводородная кислота, уксусная кислота или хлорид аммония. Используемое количество кислоты составляет обычно 0,01-10 эквивалентов, предпочтительно 0,1-5 эквивалентов, на 1 эквивалент соединения (2). Используемым металлом является, например, железо, цинк, литий, натрий, олово (обычно, хлорид олова) и другие подобные металлы, и предпочтительно железо, цинк или хлорид олова. Используемое количество металла составляет обычно 1-20 эквивалентов, предпочтительно, 1-10 эквивалентов, на 1 эквивалент соединения (2). Реакция металла в присутствии кислотного катализатора может быть проведена в инертном растворителе. Таким инертным растворителем является, например, алкиловый спирт, такой как метанол, этанол и другие подобные спирты; простой эфир, такой как тетрагидрофуран, диэтиловый эфир и другие подобные эфиры; или алкиловый сложный эфир, такой как этилацетат, и, предпочтительно, метанол, этанол, тетрагидрофуран или этилацетат. Температура реакции составляет обычно от -10 до 200°С, предпочтительно, от 25 до 120°С, и время реакции составляет предпочтительно 10 минут - 60 часов, и, предпочтительно, 10 минут - 12 часов.
В реакции восстановления в присутствии газообразного водорода используемым металлическим катализатором может являться палладий, никель, платина, рутений, родий и другие подобные металлы, и, предпочтительно, палладий, никель или другой подобный металл. Используемое количество металлического катализатора составляет обычно 0,001-2 эквивалентов, и, предпочтительно, 0,01-1 эквивалент на 1 эквивалент соединения (2). Давление газообразного водорода составляет обычно 1-10 атмосфер, и, предпочтительно, 1-3 атмосфер. Реакция может быть проведена в инертном растворителе, например, алкиловом спирте, таком как метанол, этанол и другие подобные спирты; простом эфире, таком как тетрагидрофуран, диэтиловый эфир и другие подобные эфиры; или алкилацетате, таком как метилацетат, этилацетат и другие подобные алкилацетаты, и предпочтительно, метанол, этанол или этилацетат. Температура реакции при использовании металлического катализатора составляет обычно от -10 до 200°С, и, предпочтительно, от 25 до 50°С, и время реакции обычно 10 минут - 60 часов, и, предпочтительно, 10 минут - 12 часов.
Соединение (4) может быть получено с помощью реакции ацилирования или реакции восстановительного алкилирования соединения (3).
Реакция ацилирования для аминогруппы соединения (3) может быть проведена при использовании ацилирующего реагента в присутствии основания. Используемым основанием может являться органическое основание, такое как триэтиламин, диизопропилэтиламин, пиридин, N-метилморфолин и другие подобные органические основания. Используемое количество основания составляет обычно 1-10 эквивалентов, и, предпочтительно, 1-5 эквивалентов на 1 эквивалент соединения (3). Используемое количество ацилирующего реагента обычно составляет 1-10 эквивалентов, и, предпочтительно, 1-3 эквивалента на 1 эквивалент соединения (3). Реакция может быть проведена в инертном растворителе, например простом эфире, таком как тетрагидрофуран, диэтиловый эфир и других подобных эфирах; или хлоралкане, таком как дихлорметан, хлороформ и другие подобные хлоралканы, и, предпочтительно, в дихлорметане или хлороформе. Температура реакции составляет обычно от -10 до 100°С, предпочтительно, от -10 до 50°С, и время реакции составляет обычно 10 минут - 60 часов, предпочтительно, 10 минут - 12 часов.
Восстановительное алкирирование для аминогруппы соединения (3) может быть проведено путем реакции с альдегидом или кетоном с использованием восстановителя. В случае необходимости может также быть использован кислотный катализатор. Количество альдегида или кетона составляет обычно 1-10 эквивалентов, и, предпочтительно, 1-3 эквивалента на 1 эквивалент соединения (3). Восстановителем, который может быть использован, является боргидрид натрия, цианоборгидрид натрия, триацетоксиборгидрид натрия и другие подобные восстановители. Используемое количество восстановителя составляет обычно 1-10 эквивалентов, и, предпочтительно, 1-3 эквивалента на 1 эквивалент соединения (3). Используемым кислотным катализатором может являться, например, неорганическая кислота, такая как хлористоводородная кислота, серная кислота, азотная кислота, фосфорная кислота и другие подобные кислоты; органоугольная кислота, такая как уксусная кислота, трифторуксусная кислота и другие подобные кислоты; или соль аминокислоты, такая как хлорид аммония, и, предпочтительно, хлористоводородная кислота или уксусная кислота. Используемое количество кислоты составляет обычно 0,1-10 эквивалентов, и, предпочтительно, 1-5 эквивалентов на 1 эквивалент соединения (3). Реакция может быть проведена в инертном растворителе, например, простом эфире, таком как тетрагидрофуран, диэтиловый эфир и другие подобные эфиры; или хлоралкане, таком как дихлорметан, хлороформ, дихлорэтан и другие подобные хлоралканы, и, предпочтительно, в дихлорэтане или хлороформе. Температура реакции составляет обычно от -10 до 100°С, предпочтительно, от -10 до 50°С, и время реакции составляет обычно 10 минут - 60 часов, предпочтительно, 10 минут - 12 часов.
Соединение (2) согласно настоящему изобретению может быть получено с помощью методов, конкретно приведенных в качестве примеров в следующих схемах реакций (2)-(8).
Схема реакции 2
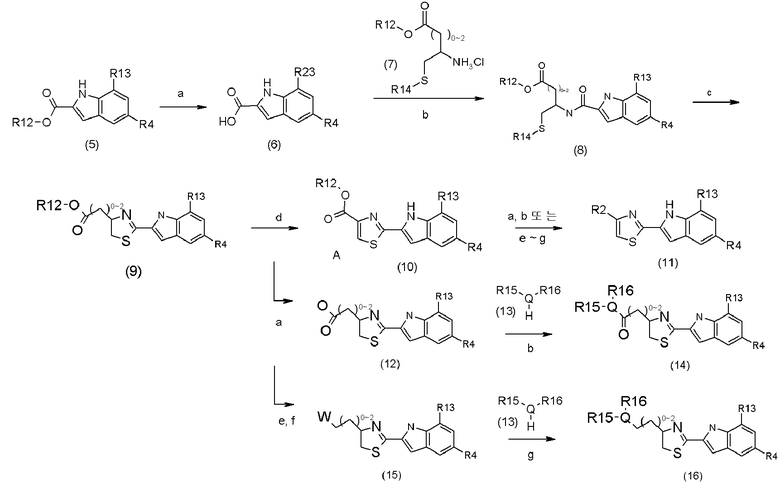
c является PCl5 или Tf2O и Ph3PO;
d является металлическим катализатором (например, Pd/C, MnO2, и так далее) или BrCCl3 или другими подобными катализаторами;
e является восстановителем (например, NaBH4, LiAlH4);
f является I2 или MsCl;
G является соединением (13);
R2 определен в формуле (1);
R4 определен в формуле (1);
R12 представляет C1-C6-алкил;
R13 представляет NO2 или R1;
R14 представляет p-MeOBn или Ph3C;
когда Q является азотом, каждый из R15 и R16 независимо представляет H, C1-C6-алкил, арил с числом членов от 6 до 12 или гетероарил с числом членов от 5 до 12; или R15 и R16 могут быть соединены друг с другом с образованием кольца с числом членов от 3 до 10;
когда Q является кислородом или серой, R15 представляет H, C1-C6-алкил, арил с числом членов от 6 до 12 или гетероарил с числом членов от 5 до 12, и R16 не присутствует; и
W является уходящей группой, например, галогеном, таким как хлорид, бромид, йодид и другие подобные группы, или сульфонилом, таким как метансульфонил, п-толуолсульфонил и другие подобные сульфонилы.
Соединение (5) может быть получено с помощью метода, описанного на схеме реакции (7) или (8).
Соединение (6) может быть получено путем реакции гидролиза соединения (5) с использованием основания. Используемым основанием может являться гидроксид лития, гидроксид натрия, гидроксид калия, бикарбонат металла, карбонат металла и другие подобные основания. Используемое количество основания составляет обычно 1-10 эквивалентов, предпочтительно, 1-5 эквивалентов на 1 эквивалент соединения (5). Реакция гидролиза может быть проведена в инертном растворителе, например, алкиловом спирте, таком как метанол, этанол и другие подобные спирты; простом эфире, таком как тетрагидрофуран, диэтиловый эфир и другие подобные эфиры. Температура реакции составляет обычно от -10 до 200°С, предпочтительно, от 25 до 120°С, и время реакции составляет обычно 10 минут - 60 часов, предпочтительно, 10 минут - 12 часов.
Соединение (7) может быть получено с помощью метода, описанного в следующей схеме реакции (9).
Соединение (8) может быть получено путем реакции сочетания группы карбоновой кислоты соединения (6) и аминогруппы соединения (7). Известные реагенты сочетания, которые могут быть использованы в реакции сочетания, включают, но этим не ограничивая, карбоимид, такой как дициклогексилкарбодиимид (DCC), 1-(3-диметиламинопропил)-3-этилкарбодиимид (EDC) или 1,1'-дикарбонилдиимидазол (CDI) в виде смеси с 1-гидрокси-бензотриазолом (HOBT) или 1-гидрокси-7-азабензотриазолом (HOAT), или бис-(2-оксо-3-оксазолидинил)фосфиновый хлорид (BOP-Cl), дифенилфосфорилазид (DPPA) или N-[диметиламино-1H-1,2,3-триазол[4,5-b]-пиридин-1-илметилен]-N-метилметанаминий (HATU). Используемое количество реагента сочетания составляет обычно 1-10 эквивалентов, предпочтительно 1-3 эквивалентов на 1 эквивалент соединения (6). Используемое количество HOBT или HOAT составляет обычно 1-10 эквивалентов, предпочтительно, 1-3 эквивалентов на 1 эквивалент соединения (5). Когда в реакции сочетания используют соль хлористоводородной кислоты и амина, кислоту следует удалять с помощью основания. Используемым основанием может являться органическое основание, такое как триэтиламин, диизопропилэтиламин и другие подобные основания. Используемое количество основания составляет обычно 1-10 эквивалентов, предпочтительно, 1-3 эквивалентов на 1 эквивалент соединения (7). Реакция сочетания может быть проведена в инертном растворителе, таком как тетрагидрофуран, диэтиловый эфир, N,N-диметилформамид, и другие подобные растворители. Температура реакции составляет обычно от -10 до 200°С, предпочтительно, от 25 до 120°С, и время реакции составляет обычно 10 минут - 60 часов, предпочтительно 10 минут - 12 часов.
Соединение (9) может быть получено путем реакции циклизации соединения (8) в соответствии с описанными в литературе методиками (Journal of Organic Chemistry, 68(24), 2003, 9506-9509; Tetrahedron, 55(34), 1999, 10271-10282, и так далее).
Когда R14 является п-метоксибензильной (p-MeOBn) группой, реакцию циклизации проводят с помощью пентахлорида фосфора (PCl5) в растворителе дихлорметане. Используемое количество PCl5 обычно 1-10 эквивалентов, предпочтительно, 1-3 эквивалентов на 1 эквивалент соединения (8). Температура реакции составляет обычно от -10 до 50°С, предпочтительно, от 0 до 25°С, и время реакции составляет обычно 10 минут - 60 часов, предпочтительно 10 минут - 12 часов.
Когда R14 является трифенилметильной (Ph3C) группой, реакцию циклизации проводят с помощью трифторметансульфонового ангидрида (Tf20) и трифенилфосфиноксида (Ph3PO) в растворителе дихлорметане. Используемое количество каждого из них составляет обычно 1-10 эквивалентов, предпочтительно, 1-3 эквивалентов на 1 эквивалент соединения (8). Температура реакции составляет обычно от -10 до 50°С, предпочтительно, 0-25°С, и время реакции составляет обычно 10 минут - 60 часов, предпочтительно 10 минут - 12 часов.
Соединение (10) может быть получено путем применения дегидрирующего реагента или металлического катализатора к соединению (9), или путем введения в это соединение уходящей группы и ее последующего удаления при помощи основания.
Дегидрирующим реагентом может являться сера, селен, различные хиноны {например, 2,3-дихлор-5,6-дицианобензохинон (DDQ)} и другие подобные реагенты, и металлическим катализатором дегидрирования может являться палладий (обычно, Pd/C), платина, никель (обычно, NiO2), марганец (обычно, MnO2) и другие подобные катализаторы. Используемое количество дегидрирующего реагента составляет обычно 1-20 эквивалентов, предпочтительно, 1-5 эквивалентов на 1 эквивалент соединения (9). Используемое количество металлического катализатора дегидрирования составляет обычно 0,001-10 эквивалентов, предпочтительно 0,1-1 эквивалент на 1 эквивалент соединение (9). Растворителем, который может быть использован, является бензол, толуол, декалин, хинолин и другие подобные растворители. Температура реакции составляет обычно от 25 до 400°С, предпочтительно, от 25 до 200°С, и время реакции составляет обычно 10 минут - 60 часов, предпочтительно 10 минут - 12 часов.
Реагент, который может быть использован при введении уходящей группы, включает бромид меди(II) (CuBr2), бромтрихлорметан (BrCCl3), N-бромсукцинимид (NBS) и другие подобные реагенты. Используемое количество составляет обычно 1-10 эквивалентов, предпочтительно, 1-5 эквивалентов на 1 эквивалент соединения (9). Основанием, которое может быть использовано, является, например, неорганическое основание, такое как карбонат натрия, бикарбонат натрия, карбонат калия и другие подобные основания; органическое основание, такое как триэтиламин, пиридин, 1,8-диазабицикло[5,4,0]ундек-7-ен (DBU) и другие подобные основания; предпочтительно, карбонат натрия или DBU. Используемое количество основания обычно составляет 0-10 эквивалентов, предпочтительно 0-3 эквивалентов на 1 эквивалент соединения (9). Растворителем, который может быть использован, является инертный растворитель, например простой эфир, такой как тетрагидрофуран, диэтиловый эфир и другие подобные эфиры; хлоралкан, такой как дихлорметан, дихлорэтан, хлороформ и другие подобные хлоралканы, и, предпочтительно, дихлорметан или хлороформ. Температура реакции составляет обычно от -10 до 200°С, предпочтительно, от 0 до 100°С, и время реакции составляет обычно 10 минут - 60 часов, предпочтительно 10 минут - 12 часов.
Соединение (11) может быть получено с помощью реакции гидролиза, реакции восстановления, реакции сочетания амина и кислоты и реакции замещения амина, и так далее, соединения (10) путем использования способов синтеза соединений (6), (15) и (16).
Соединение (12) может быть получено при помощи реакции гидролиза соединения (9) путем использования способа синтеза соединения (6).
Соединение (14) может быть получено при помощи реакции сочетания карбоновой кислоты соединения (12) и соединения (13) путем использования способа синтеза соединения (8).
Соединение (15) может быть получено путем превращения эфирной группы соединения (9) в спиртовую группу и введения уходящей группы X.
Восстановителем, который может быть использован для восстановления эфирной группы в спиртовую группу, является, например, боргидрид натрия, боргидрид лития, боран, алюмогидрид лития, гидрид диизобутилалюминия (DIBAL-H) и другие подобные восстановители. Используемое количество восстановителя составляет обычно 1-10 эквивалентов, предпочтительно, 1-3 эквивалентов на 1 эквивалент соединения (9). Реакция может быть проведена в инертном растворителе, например, спирте, таком как метанол, этанол и другие подобные спирты; простом эфире, таком как тетрагидрофуран, диэтиловый эфир и другие подобные эфиры; и предпочтительно, тетрагидрофуране или простом эфире. Температура реакции составляет обычно от -78 до 100°С, предпочтительно, от -10 до 50°С, и время реакции составляет обычно 10 минут - 60 часов, предпочтительно 10 минут - 12 часов.
Методом введения уходящей группы в спиртовую группу является реакция галогенирования или сульфонилирования. Реакция галогенирования может быть проведена путем использования такого реагента, как йод, бром, N-йодсукцинимид (NIS), N-бромсукцинимид (NBS), четыреххлористый углерод (CCl4), четырехбромистый углерод (CBr4) и другие подобные реагенты, в присутствии основания, такого как имидазол, диметиламинопиридин (DMAP), и так далее, и фосфина, такого как трифенилфосфин (Ph3P), трибутилфосфин (Bu3P), и так далее. Используемое количество каждого из галогенирующего реагента, основания и фосфинового соединения составляет обычно 1-10 эквивалентов, предпочтительно, 1-3 эквивалентов на 1 эквивалент соединения (9). Реакция может быть проведена в инертном растворителе, например, простом эфире, таком как тетрагидрофуран, диэтиловый эфир и других подобных эфирах; дихлорметане; хлороформе; ацетонитриле и других подобных растворителях. Температура реакции составляет обычно от -10 до 200°С, предпочтительно, от 0 до 50°С, и время реакции составляет обычно 10 минут - 60 часов, предпочтительно 10 минут - 12 часов.
Реакция сульфонилирования может быть проведена путем использования такого реагента, как метансульфонилхлорид, п-толуолсульфонилхлорид и другие подобные реагенты, в присутствии органического основания, такого как пиридин, триэтиламин и другие подобные основания. Используемое количество каждого из реагента сульфонилирования и основания составляет обычно 1-10 эквивалентов, предпочтительно 1-5 эквивалентов на 1 эквивалент соединения (9). Реакция может быть проведена в инертном растворителе, например, простом эфире, таком как тетрагидрофуран, диэтиловый эфир и другие подобные эфиры; хлоралкане, таком как дихлорметан, дихлорэтан, хлороформ и другие подобные хлоралканы, и предпочтительно, в дихлорметане или дихлорэтане. Температура реакции при использовании металлического катализатора составляет обычно от -10 до 200°С, предпочтительно, от 0 до 50°С, и время реакции составляет обычно 10 минут - 60 часов, предпочтительно 10 минут - 12 часов.
Соединение (16) может быть получено путем реакции сочетания соединения (13) и соединения (15) при использовании основания. Основанием, которое может быть использовано, является, например, неорганическое основание, такое как карбонат натрия, карбонат калия, карбонат цезия и другие подобные карбонаты; органическое основание, такое как триэтиламин, диизопропилэтиламин, 1,8-диазабицикло[5,4,0]ундек-7-ен (DBU) и другие подобные основания, и предпочтительно, карбонат калия, карбонат цезия или DBU. Используемое количество основания составляет обычно 1-10 эквивалентов, предпочтительно, 1-5 эквивалентов на 1 эквивалент соединения (13). Растворителем, который может быть использован, является инертный растворитель, например, простой эфир, такой как тетрагидрофуран, диэтиловый эфир и другие подобные эфиры; алкилнитрил, такой как ацетонитрил, пропионитрил и другие подобные нитрилы; амид, такой как N,N-диметилформамид, и предпочтительно, тетрагидрофуран, ацетонитрил или N,N-диметилформамид. Температура реакции составляет обычно от -10 до 200°С, предпочтительно, от 25 до 120°С, и время реакции составляет обычно 10 минут - 60 часов, предпочтительно 10 минут - 12 часов.
Схема реакции 3
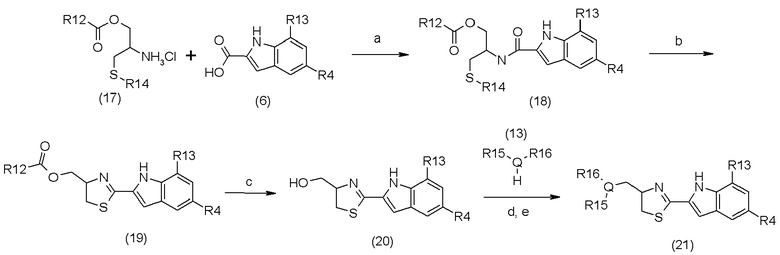
В приведенной выше схеме реакции 3:
a является реагентом сочетания (например, EDC, CDI, BOP-Cl);
b является PCl5 или Tf2O и Ph3PO;
c является гидроксидом металла (например, NaOH, LiOH);
d является I2 или MsCl или другими подобными реагентами;
e является соединением (13);
R4 определен в схеме реакции (1);
R12, R13, R14, R15, R16 и Q определены в схеме реакции (2).
Соединение (17) может быть получено при помощи способа, описанного в схеме реакции (10).
Соединение (18) может быть получено путем использования соединения (6) и соединения (17) в соответствии со способом синтеза соединения (8) в схеме реакции (2).
Соединение (19) может быть получено путем использования соединения (18) в соответствии со способом синтеза соединения (9) в схеме реакции (2).
Соединение (20) может быть получено путем использования соединения (19) в соответствии со способом синтеза соединения (6) в схеме реакции (2).
Соединение (21) может быть получено путем использования соединения (20) в соответствии со способом синтеза соединения (16) в схеме реакции (2).
Схема реакции 4
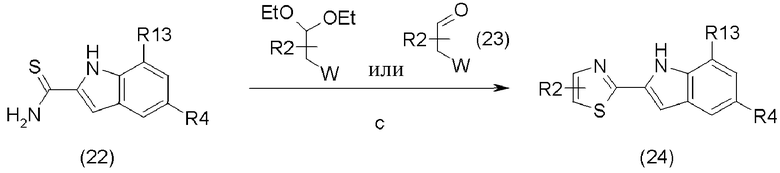
В приведенной выше схеме реакции (4),
a является SOCl2 или (COCl)2, и водным раствором NH4OH;
b является реагентом Лавессона;
c является соединением (23);
R2 определен в формуле (1);
R4 определен в схеме реакции (1);
R13 определен в схеме реакции (2);
W является уходящей группой, например, галогеном, таким как хлорид, бромид, йодид и другие подобные группы, или сульфонилом, таким как метансульфонил, п-толуолсульфонил.
Соединение (22) может быть получено путем превращения карбоновой кислоты соединения (6) в амид и затем превращения амида в тиоамид при помощи реагента Лавессона.
Метод превращения карбоновой кислоты соединения (6) в амид осуществляют путем получения хлорангидрида при помощи тионилхлорида (SOCl2) или оксалилхлорида ((COCl)2) и затем взаимодействия с водным раствор аммиака. Используемое количество хлорирующего реагента составляет обычно 1-10 эквивалентов, предпочтительно, 1-5 эквивалентов на 1 эквивалент соединения (6). Используемое количество водного раствора аммиака составляет обычно 1-5 эквивалентов. Реакция может быть проведена в инертном растворителе, таком как дихлорметан, дихлорэтан, хлороформ и другие подобные растворители. Температура реакции составляет обычно от -10 до 200°С, предпочтительно, от -10 до 100°С, и время реакции составляет обычно 10 минут - 60 часов, предпочтительно, 10 минут - 12 часов.
Тиоамидная группа может быть получена путем реакции амида с реагентом Лавессона. Используемое количество реагента Лавессона составляет обычно 1-10 эквивалентов, предпочтительно, 1-5 эквивалентов на 1 эквивалент соединения (6). Реакция может быть проведена в инертном растворителе, например, простом эфире, таком как тетрагидрофуран, диэтиловый эфир и другие подобные эфиры; хлоралкане, таком как дихлорметан, дихлорэтан, хлороформ и другие подобные хлоралканы, ароматическом углеводороде, таком как бензол, толуол и другие подобные ароматические углеводороды, и предпочтительно, тетрагидрофуране или толуоле, и так далее. Температура реакции составляет обычно от -25 до 200°С, предпочтительно, от 25 до 150°С, и время реакции составляет обычно 10 минут - 60 часов, предпочтительно 10 минут - 12 часов.
Соединение (23) выпускается в промышленности или может быть получено известными методами, например, раскрытыми в патентном документе WO 1999/02501.
Соединение (24) может быть получено путем реакции сочетания соединения (22) и соединения (23) при использовании, в случае необходимости, основания. Используемым основанием может являться, например, неорганическое основание, такое как карбонат натрия, карбонат калия, карбонат цезия и другие подобные карбонаты; органическое основание, такое как диизопропилэтиламин, DBU и другие подобные основания, и предпочтительно, карбонат калия или карбонат цезия. Используемое количество основания составляет обычно 0-10 эквивалентов, предпочтительно, 0-5 эквивалентов на 1 эквивалент соединения (22). Реакция может быть проведена в инертном растворителе, например, простом эфире, таком как тетрагидрофуран, диэтиловый эфир и другие подобные эфиры; алкилнитриле, таком как ацетонитрил, пропионитрил и другие подобные нитрилы; амиде, таком как N,N-диметилформамид, и предпочтительно, в тетрагидрофуране или N,N-диметилформамиде. Температура реакции составляет обычно от -10 до 200°С, и время реакции составляет обычно 10 минут - 60 часов, предпочтительно 10 минут - 12 часов.
Схема реакции 5
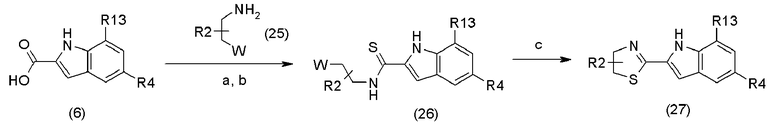
В приведенной выше схеме реакции 5:
a является соединением амина (25), реагентом сочетания (например, EDC, CDI, BOP-Cl);
b является реагентом Лавессона;
c является основанием (например, K2CO3, Cs2CO3);
R2 определен в формуле (1);
R4 определен в схеме реакции (1);
R13 определен в схеме реакции (2);
W является уходящей группой, например, галогеном, таким как хлорид, бромид, йодид и другие подобные группы, или сульфонилом, таким как метансульфонил, п-толуолсульфонил и другие подобные сульфонилы.
Соединение (25) выпускается в промышленности или может быть получено известными методами, описанными в литературе (Tetrahydron Letters, 28(48), 6068-6072, 1987; или Organic Process Research & Development 10(3), 472-480, 2006).
Соединение (26) может быть получено путем последовательного синтеза амида и синтеза тиоамида с помощью реагента Лавессона. Синтез амида путем реакции сочетания соединения (6) и соединения (25) может быть проведен в соответствии с методом получения соединения амида (8) в схеме реакции (2), а синтез соединения тиоамида (26) может быть проведен в соответствии с методом получения соединения (22) в схеме реакции (4).
Соединение (27) может быть получено путем реакции циклизации соединения (26) при использовании основания. Основанием, которое может быть использовано, является карбонат калия, карбонат цезия, диизопропилэтиламин, DBU или другие подобные основания. Используемое количество основания составляет обычно 1-10 эквивалентов, предпочтительно, 1-5 эквивалентов на 1 эквивалент соединения (26). Реакция может быть проведена в инертном растворителе, например, простом эфире, таком как тетрагидрофуран, диэтиловый эфир и другие подобные эфиры; алкилнитриле, таком как ацетонитрил, пропионитрил и другие подобные нитрилы; амиде, таком как N,N-диметилформамид, и предпочтительно, в тетрагидрофуране, ацетонитриле или N,N-диметилформамиде. Температура реакции составляет обычно от -10 до 200°С, предпочтительно, от 25 до 120°С, и время реакции составляет обычно 10 минут - 60 часов, предпочтительно 10 минут - 12 часов.
Схема реакции 6
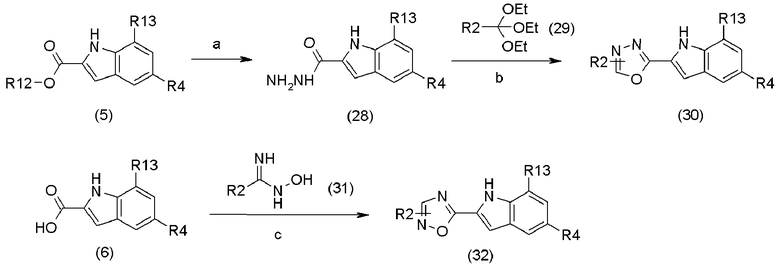
В приведенной выше схеме реакции 6,
a является H2NNH2,
b является соединением (29),
c является реагентом сочетания (например CDI, BOP-Cl) и соединением (31),
R2 определен в формуле (1),
R4 определен в схеме реакции (2), и
R12 и R13 определены в схеме реакции (2).
Соединение (28) может быть получено путем реакции соединения (5) с гидразином. Используемое количество гидразина составляет обычно 1-10 эквивалентов, предпочтительно 1-5 эквивалентов на 1 эквивалент соединения (5). Реакция может быть проведена в инертном растворителе, например, тетрагидрофуране, метаноле, этаноле и другом подобном растворителе. Температура реакции составляет обычно от -10 до 200°C, и предпочтительно, от 25 до 120°C. Время реакции составляет обычно 10 минут - 60 часов и предпочтительно 10 минут - 12 часов.
Что касается соединения (29), то используют соединение, выпускаемое промышленностью.
Соединение (30) может быть получено путем реакции сочетания соединения (28) с соединением (29). В случае необходимости, может быть использован кислотный катализатор. Используемое количество соединения (29) составляет обычно 1-10 эквивалентов, предпочтительно, 1-5 эквивалентов на 1 эквивалент соединения (28). Кислотный катализатор, который может быть использован, выбирают из неорганической кислоты, такой как хлористоводородная кислота, серная кислота, азотная кислота, фосфорная кислота и другие подобные кислоты; органической угольной кислоты, такой как уксусная кислота, трифторуксусная кислота и другие подобные кислоты; соли аминокислоты, такой как хлорид аммония; и кислоты Льюиса, такой как хлорид алюминия. Используемое количество кислоты составляет обычно 0,001-5 эквивалентов, предпочтительно, 0,01-1 эквивалент на 1 эквивалент соединения (29). Реакция может быть проведена в инертном растворителе, например, простом эфире, таком как тетрагидрофуран, диэтиловый эфир и другие подобные эфиры; ароматическом углеводороде, таком как бензол, толуол и другие подобные ароматические углеводороды; насыщенном углеводороде, таком как циклогексан, гексан и другие подобные углеводороды; или амиде, таком как N,N-диметиламид. Температура реакции составляет обычно от -10 до 200°C, и предпочтительно, от 25 до 120°C. Время реакции составляет обычно 10 минут - 60 часов и предпочтительно 10 минут - 12 часов.
Соединение (31) выпускается в промышленности или может быть получено известным методом, раскрытым в патентном документе US 2004/0019215.
Соединение (32) может быть получено путем реакции сочетания соединения (6) с соединением (31) и последующей реакции дегидратации.
Используемое количество реагента сочетания для реакции сочетания составляет обычно 1-10 эквивалентов, предпочтительно, 1-3 эквивалентов на 1 эквивалент соединения (6). Реакция может быть проведена в инертном растворителе, таком как тетрагидрофуран или N,N-диметиламид. Температура реакции составляет обычно от -10 до 200°C, и предпочтительно, от 0 до 50°C. Время реакции составляет обычно 10 минут - 60 часов и предпочтительно 10 минут - 12 часов.
Реакция дегидратации после реакции сочетания может быть проведена, необязательно, с использованием кислотного катализатора. Она может быть проведена при помощи способа для получения соединения (30).
СХЕМА РЕАКЦИИ 7
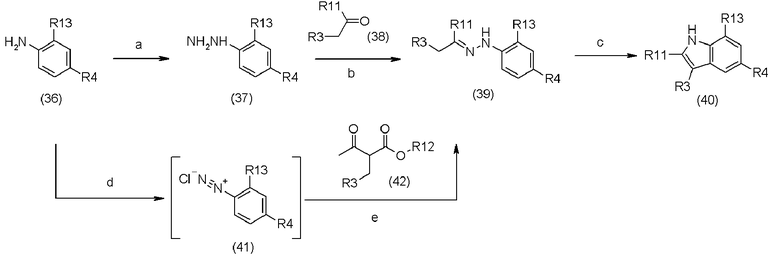
В приведенной выше схеме реакции (7),
a является нитратом натрия (NaNO2); хлоридом олова (SnCl2),
b является соединением кетона (38), основанием (например NaOAc),
c является кислотой (например, полифосфорной кислотой (PPA)),
d является NaNO2,
e является соединением (42), основанием (например NaOH),
R3 определен в формуле (1),
R11, R4 и R12 определены в схеме реакции (1), и
R13 определен в схеме реакции (2).
Соединение (36) выпускается в промышленности или может быть получено методами, описанными в литературе [Heterocycles, 68(11), 285-99, 2006, или Bioorganic & Medicinal Chemistry Letters, 14(19), 903-4906, 2004].
Соединение (37) выпускается в промышленности или может быть получено путем превращения аминогруппы соединения (36) в гидразиновую группу в соответствии с методом, описанным в литературе [Journal of the America Chemical Society, 198(48), 15374-75, 2006].
Соединение гидразина (37) может быть получено путем реакции аминогруппы с NaNO2 в присутствии хлористоводородной кислоты с получением соли диазония (42), и без отделения, путем восстановления соли диазония с помощью SnCl2. Используемое количество NaNO2 составляет обычно 1-10 эквивалентов, предпочтительно, 2-5 эквивалентов на 1 эквивалент соединения (36). Используемое количество SnCl2 составляет обычно 1-10 эквивалентов, предпочтительно, 2-5 эквивалентов на 1 эквивалент соединения (36). Реакция может быть проведена в водном растворе хлористоводородной кислоты с концентрацией 1-12N, предпочтительно, 4-8N. Температура реакции составляет обычно от -10 до 50°C. Время реакции составляет обычно 10 минут - 60 часов и предпочтительно 10 минут - 6 часов.
Что касается соединения (38), то его используют в виде соединения, выпускаемого промышленностью.
Соединение гидразона (39) может быть получено путем реакции сочетания соединения (37) с соединением кетона (38). Если соединение (37) находится в нейтральной форме, то основание не используют, но если соединение (37) находится в кислотной форме, то основание должно быть использовано для получения нейтральной формы. Основанием, которое может быть использовано, является, например, гидроксид металла, такой как гидроксид натрия, гидроксид лития, и так далее; карбонат металла, такой как бикарбонат натрия, карбонат калия, и так далее; ацетат металла, такой как ацетат натрия; или органическое основание, такое как триэтиламин, пиридин и так далее. Используемое количество основания составляет обычно 1-5 эквивалентов, предпочтительно, 1-2 эквивалентов на 1 эквивалент соединения (37). Реакция может быть проведена в инертном растворителе, таком как тетрагидрофуран, метанол, этанол и так далее. Температура реакции составляет обычно от -10 до 100°C, и время реакции составляет обычно 10 минут - 60 часов и предпочтительно 10 минут - 12 часов.
Соединение (39) может быть получено путем реакции соли диазония (41) и соединения (42) в присутствии основания в соответствии с методом перегруппировки Джэппа-Клингеманна, описанной в литературе [Organic Process Research & Development, 2, 1988, 214-220]. Используемое количество хлористоводородной кислоты для получения соли диазония (41) составляет обычно 1-10 эквивалентов, предпочтительно, 2-4 эквивалентов на 1 эквивалент соединения (36). Основанием, используемым для реакции между соединениями (41) и (42), является гидроксид натрия. Используемое количество основания составляет обычно 1-20 эквивалентов, предпочтительно, 1-10 эквивалентов на 1 эквивалент соединения (42). В качестве растворителя может быть использован 50% водный раствор этанола. Температура реакции составляет обычно от -10 до 50°C. Время реакции составляет обычно 10 минут - 60 часов и предпочтительно 10 минут - 12 часов.
Соединение (40) может быть получено путем использования кислотного катализатора и соединения (39). Кислотой, используемой в синтезе, является полифосфорная кислота, хлористоводородная кислота, п-толуолсульфоновая кислота, серная кислота, уксусная кислота, и так далее, и предпочтительно, полифосфорная кислота.
Полифосфорная кислота может быть использована или только сама по себе, или в комбинации с ароматическим углеводородом, таким как бензол, толуол и так далее. Температура реакции составляет обычно от -25 до 150°C. Время реакции составляет обычно 5 минут - 60 часов и предпочтительно 5 минут - 12 часов.
Соединение (42) выпускается в промышленности или может быть получено с помощью методов, описанных в литературе [патентные документы WO 2007040289, WO 200601079 или Organic Letters 9(3), 397-400, 2007].
Схема реакции 8
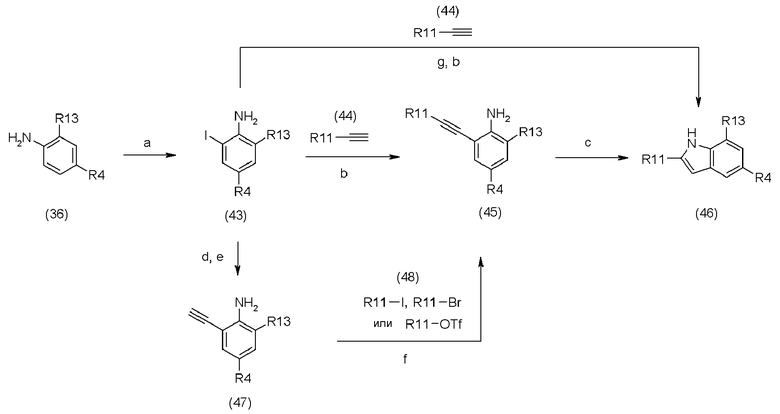
d является триметилсилилацетиленом или 2-метил-3-бутин-2-олом, Pd, Cu(I), основанием, (например, Et3N или Et2NH),
e является фторидом тетрабутиламмония (Bu4NF) или NaOH,
f является соединением (48), Pd(II), Cu(I), основанием (например, Et3N или Et2NH),
g является трифторуксусным ангидридом((CF3CO)2O),
R11 и R4 определены в схеме реакции (1), и R13 определен в схеме реакции (2).
Соединение (36) выпускается в промышленности или может быть получено с помощью методов, описанных в литературе [Heterocycles, 68(11), 285-99, 2006, или Bioorganic & Medicinal Chemistry Letters, 14(19), 903-4906, 2004].
Соединение (43) может быть получено путем реакции йодирования соединения (36). Йодирующий реагент для реакции йодирования может быть выбран из йода, монобромида йода и монохлорида йода, и иона серебра, например, нитрата серебра (AgNO3), карбоната серебра (AgCO3), сульфата серебра (Ag2SO4) и так далее, и они могут быть использованы вместе. Используемое количество йодирующего реагента составляет обычно 1-10 эквивалентов, предпочтительно, 1-3 эквивалентов на 1 эквивалент соединения (36). Используемое количество иона серебра составляет обычно 0-10 эквивалентов, предпочтительно, 0-3 эквивалентов на 1 эквивалент соединения (36). Реакция может быть проведена в инертном растворителе, например, простом эфире, таком как тетрагидрофуран, диэтиловый эфир, и так далее; алкиловом спирте, таком как метанол, этанол, и так далее; алкилнитриле, таком как ацетонитрил, пропионитрил, и так далее; или органической кислоте, такой как уксусная кислота. Температура реакции составляет обычно от -10 до 200°C и предпочтительно, от 0 до 50°C. Время реакции составляет обычно 10 минут - 60 часов и предпочтительно 10 минут - 12 часов.
Соединение (44) выпускается в промышленности или может быть получено с помощью методов, описанных в литературе [Synthesis, 59-61, 2004 или Bioorganic & Medicinal Chemistry Letters, 13, 197-209, 2003], или с помощью метода синтеза соединения (47).
Соединение (45) может быть получено путем реакции сочетания йодидной группы соединения (43) и ацетиленовой группы соединения (44) в соответствии с методом, описанном в литературе [Tetrahedron, 59, 2003, 1571-1587].
Реакция сочетания может быть проведена с использованием Pd(0) или Pd(II) катализатора [например, Pd(Ph3P)4, PdCl2(Ph3P)2)], Cu(I) катализатора (например, CuI) и основания (например, триэтиламина, диэтиламина, и так далее). Используемое количество Pd катализатора составляет обычно 0,001-5 эквивалентов, предпочтительно, 0,01-1 эквивалент на 1 эквивалент соединения (43). Используемое количество Cu(I) катализатора составляет обычно 0,001-5 эквивалентов, предпочтительно 0,01-1 эквивалент на 1 эквивалент соединения (43). Используемое количество основания составляет обычно 1-10 эквивалентов, предпочтительно, 1-5 эквивалентов на 1 эквивалент соединения (43). Реакция может быть проведена в инертном растворителе, например, простом эфире, таком как тетрагидрофуран, диэтиловый эфир, и так далее; ароматическом углеводороде, таком как бензол, толуол, и так далее; или N,N-диметилформамиде. Температура реакции составляет обычно от -10 до 200°C, и предпочтительно, от 25 до 120°C. Время реакции составляет обычно 10 минут - 60 часов и предпочтительно 10 минут - 12 часов.
Соединение (46) может быть получено путем реакции циклизации соединения (45) в соответствии с методами, описанными в литературе [патентный документ JP 2001/233855; Tetrahedron, 59, 2003, 1571-1587; Tetrahedron Letters, 47(36), 2006, 6485-6388; или Heterocycles, 64, 2004, 475-482, и так далее]. Реакция циклизации может быть проведена при использовании основания, Cu (I), Pd (II), и так далее. Основанием, которое может быть использовано, является, например, гидрид калия (KH), трет-бутилат калия (KOBut), и так далее, и используемое количество основания составляет обычно 1-10 эквивалентов, предпочтительно, 1-2 эквивалентов на 1 эквивалент соединения (45). Используемое количество каждого из Cu(I) и Pd(II) составляет обычно 0,001-5 эквивалентов, предпочтительно, 0,01-1 эквивалент на 1 эквивалент соединения (45). Реакция может быть проведена в инертном растворителе, например, простом эфире, таком как тетрагидрофуран, диэтиловый эфир, и так далее; ароматическом углеводороде, таком как бензол, толуол, и так далее; алкилнитриле, таком как ацетонитрил, пропионитрил, и так далее; N,N-диметилформамиде или N-метилпирролидиноне (NMP). Если используют основание, то предпочтительным растворителем является NMP. Если используют Cu(I) или Pd(II), то предпочтительным растворителем является ацетонитрил, толуол, N,N-диметилформамид, и так далее. Температура реакции составляет обычно от -10 до 200°C, и предпочтительно, от 0 до 120°C. Время реакции составляет обычно 10 минут - 60 часов и предпочтительно 10 минут - 12 часов.
Соединение (46) может быть также получено путем превращения аминогруппы соединения (43) в трифторацетамидную группу и затем циклизации при использовании Pd(II) в соответствии с методом, описанным в литературе [Tetrahedron, 60, 2006, 10983-10992].
Соединение (47) может быть получено путем реакции сочетания соединения (43) и ацетилена в присутствии Pd(II), Cu(I) и основания в соответствии с методом, описанным в литературе [Journal of Organic Chemistry, 71, 2006, 167-175]. Используемым ацетиленом является триметилсилилацетилен или 2-метил-3-бутин-2-ол, и используемое количество ацетилена составляет обычно 1-10 эквивалентов, предпочтительно, 1-3 эквивалентов на 1 эквивалент соединения (43). Используемое количество каждого из Cu(I) и Pd(II) составляет обычно 0,001-5 эквивалентов, предпочтительно, 0,01-1 эквивалент на 1 эквивалент соединения (46). Используемым основанием является диэтиламин, триэтиламин, диизопропилэтиламин, и так далее, и используемое количество основания составляет обычно 1-10 эквивалентов, предпочтительно, 1-5 эквивалентов на 1 эквивалент соединения (43). Реакция может быть проведена в инертном растворителе, например, простом эфире, таком как тетрагидрофуран, диэтиловый эфир, и так далее, или ароматическом углеводороде, таком как бензол, толуол, и так далее. Температура реакции составляет обычно от -10 до 200°C, и предпочтительно, от 0 до 120°C. Время реакции составляет обычно 10 минут - 60 часов и предпочтительно 10 минут - 12 часов.
Соединение (48) выпускается в промышленности или может быть получено с помощью методов, описанных в литературе [Journal of Organic Chemistry, 70, 2005, 6519-6522 или Tetrahedron, 60(48), 2004, 10983-10992].
Соединение (7) настоящего изобретения может быть получено с помощью метода, специально проиллюстрированного на следующей схеме реакции (9).
На схеме реакции (9), соединения (50), (52) и (54) соответствуют соединению (7).
Схема реакции 9
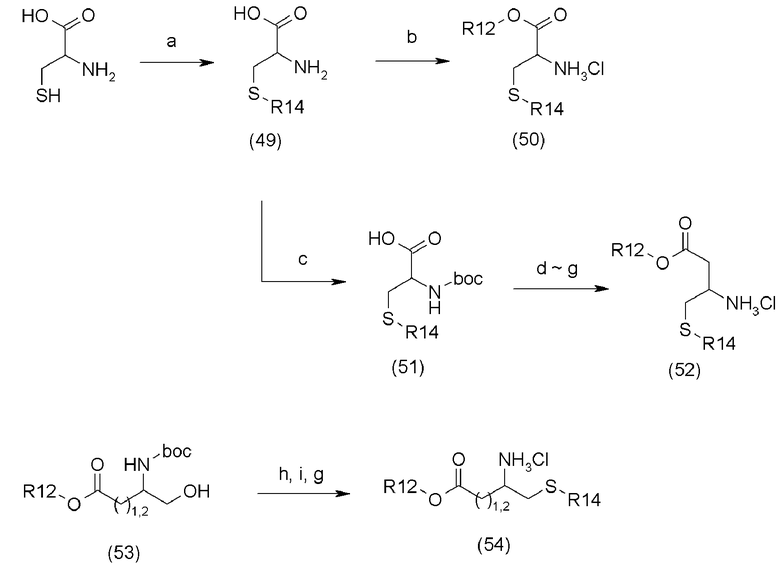
В приведенной выше схеме реакции (9),
a является п-метоксибензилхлоридом (PMBCl) или трифенилметилхлоридом (TrCl), основанием (например, NaOH),
b является алкиловым спиртом (например, метанолом, этанолом), ацетилхлоридом или тионилхлоридом,
c является ди-трет-бутилоксидикарбонилом (Boc2O), основанием (например, NaOH, K2CO3),
d является алкилхлорформиатом (например, EtOCOCl), основанием (например, N-метилморфолином),
e является диазометаном (CH2N2), основанием (например, KOH),
f является ионом серебра (например, бензоатом серебра),
g является кислотой,
h является MsCl, Et3N,
i является п-метоксибензилтиолом (PMBSH), NaH,
R12 представляет C1-C6 алкил, и
R14 представляет p-MeOBn или Ph3C.
Соединение (49) может быть получено путем защиты тиольной группы цистеина с помощью п-метоксибензилхлорида (PMBCl) или трифенилметилхлорида (TrCl) в присутствии основания.
Используемое количество PMBCl или TrCl для защиты тиольной группы составляет обычно 1-5 эквивалентов, предпочтительно, 1-2 эквивалентов на 1 эквивалент цистеина. Используемым основанием является гидроксид натрия, карбонат калия, и так далее, и его используемое количество составляет обычно 1-5 эквивалентов, предпочтительно, 1-2 эквивалентов на 1 эквивалент цистеина. Реакция может быть проведена в инертном растворителе, таком как тетрагидрофуран, метанол, этанол, вода и так далее. Температура реакции составляет обычно от -10 до 200°C, и предпочтительно, от 0 до 50°C. Время реакции составляет обычно 10 минут - 60 часов и предпочтительно 10 минут - 12 часов.
Соединение (51) может быть получено путем защиты аминогруппы соединения (49) с помощью BOC группы.
Используемое количество Boc2O для защиты аминогруппы составляет обычно 1-5 эквивалентов, предпочтительно, 1-2 эквивалентов на 1 эквивалент цистеина. Используемым основанием является, например, гидроксид, такой как гидроксид натрия, гидроксид лития и так далее; карбонат, такой как карбонат натрия, бикарбонат натрия, карбонат калия, карбонат цезия, и так далее; органическое основание, такое как диизопропилэтиламин, триэтиламин, и так далее, и предпочтительно, карбонат калия, триэтиламин, и так далее. Реакция может быть проведена в инертном растворителе, таком как тетрагидрофуран, метанол, этанол, вода, и так далее. Температура реакции составляет обычно от -10 до 200°C, и предпочтительно, от 0 до 50°C. Время реакции составляет обычно 10 минут - 60 часов и предпочтительно 10 минут - 12 часов.
Соединение (50) может быть получено путем этерификации карбоксильной группы соединения (49). Реакция этерификации может быть проведена при помощи ацетилхлорида или тионилхлорида в растворителе алкиловом спирте. Используемое количество ацетилхлорида или тионилхлорида составляет обычно 1-10 эквивалентов, предпочтительно, 1-5 эквивалентов на 1 эквивалент соединения (49). Температура реакции составляет обычно от -25 до 200°C, и предпочтительно, от 25 до 100°C. Время реакции составляет обычно 10 минут - 60 часов и предпочтительно 10 минут - 12 часов.
Соединение (52) может быть получено с помощью метода, описанного в литературе [Helvetica Chimica Acta, 87, 2004, 3131-3159].
В присутствии 1-2 эквивалентов основания (например, N-метилморфолина (NMM), триэтиламина, и так далее), 1 эквивалент соединения (51) реагирует с 1-2 эквивалентами этилхлорформиата (EtOCOCl) или изобутилхлорформиата(iBuOCOCl) в растворителе тетрагидрофуране при комнатной температуре с получением соединения ангидрида. Полученное соединение ангидрида взаимодействует с 1-5 эквивалентов диазометана и 1-5 эквивалентов водного раствора гидроксида калия в растворителе диэтиловом эфире при 0°C, и затем реагирует с ионом Ag (например, трифторацетатом серебра (CF3CO2Ag), бензоатом серебра, и так далее) при комнатной температуре в отсутствии света, с получением алкилового эфира.
Защитная BOC группа полученного соединения может реагировать с кислотой (например, хлористоводородной кислотой, трифторуксусной кислотой) в растворителе, таком как диоксан, тетрагидрофуран или дихлорметан, при комнатной температуре с целью снятия защиты, и затем может быть получено соединение (52).
Соединение (53) может быть получено с помощью метода, описанного в литературе [Synlett, 15, 2005, 2397-2399 или Journal of Organic Chemistry, 66(5), 2001, 1919-1923] при использовании в качестве исходного реагента глютаминовой кислоты или аспарагиновой кислоты.
Соединение (54) может быть получено путем превращения спиртовой группы соединения (53) в уходящую группу и затем реакции с п-метоксибензилтиолом (PMBSH), в качестве способа получения соединения (21) в схеме реакции (3).
Соединение (53) взаимодействует с 1-5 эквивалентами триэтиламина и 1-3 эквивалентами MsCl в растворителе дихлорметане при 0°C с получением соединения сульфоната. Для получения соединения (54) соединение сульфоната может взаимодействовать при температуре от 25 до 100°C с раствором PMBSNa, который получают путем добавления 2-5 эквивалентов NaH и 2-5 эквивалентов PMBSH в DMF.
Соединение (17) настоящего изобретения может быть получено с помощью способа, специально проиллюстрированного на следующей схеме реакции (10).
Схема реакции 10
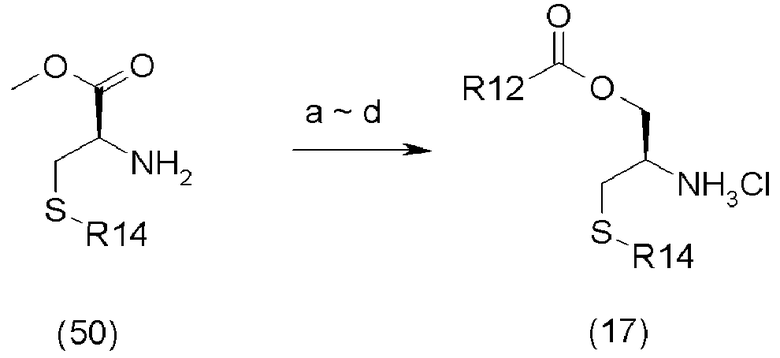
В приведенной выше схеме реакции (10),
A является Boc2O,
b является восстановителем (например, NaBH4),
c является трет-бутилкарбонилхлоридом (tBuCOCl), основанием (например, Et3N),
d является кислотой,
R12 представляет C1-C6 алкил, и
R14 представляет p-MeOBn или Ph3C.
Соединение (17) может быть последовательно получено путем защиты аминогруппы с помощью BOC группы, восстановления эфирной группы в спирт, защиты спиртовой группы с помощью эфирной группы, и снятия BOC группы.
Защита аминогруппы может быть проведена в соответствии со способом, проиллюстрированным на схеме реакции (9).
Восстановление эфирной группы может быть проведено путем взаимодействия с 2-5 эквивалентами боргидрида лития в течение 1-5 часов в растворителе тетрагидрофуране при 0°C.
Защита спиртовой группы может быть проведена путем взаимодействия с t-BuCOCl в течение 10 минут - 12 часов в присутствии 1-5 эквивалентов основания, такого как триэтиламин или пиридин и так далее, в растворителе дихлорметане при 0-25°C.
Снятие BOC группы может быть проведено путем растворения реагирующих веществ в инертном растворителе, таком как тетрагидрофуран, диоксан, этилацетат или дихлорметан, и взаимодействия с 1-10 эквивалентами хлористоводородной кислоты или уксусной кислоты в течение 10 минут - 12 часов при температуре от 0 до 50°C.
Соединения, получение которых специально не объясняется в настоящем описании, являются раскрытыми соединениями по существу или соединениями, которые могут быть получены в соответствии с уже известными синтезами или аналогичными для них синтезами.
Соединение формулы (1), полученное с помощью приведенных выше методов, может быть отделено или очищено от продуктов реакции при помощи различных методов, таких как перекристаллизация, ионный электрофорез, колоночная хроматография на силикагеле или хроматография на ионообменной смоле, и так далее.
Как уже было объяснено выше, соединения индола или индазола настоящего изобретения, исходные реагенты или промежуточные соединения, и так далее, для их получения, могут быть синтезированы с помощью различных методов.
ПРЕИМУЩЕСТВА ОСУЩЕСТВЛЕНИЯ ИЗОБРЕТЕНИЯ
Композиция настоящего изобретения, включающая в качестве активного компонента описанное выше соединение индола или индазола формулы (1), может быть использована для консервирования клеток или органов животных. Более конкретно, композиция настоящего изобретения может быть использована для предотвращения повреждения органов, выделенных клеточных систем или тканей, вызванного хранением при низких температурах, операцией по трансплантации или посттрансплантационной реперфузией. Однако применения композиции согласно настоящему изобретению не ограничиваются описанными выше применениями.
Используемый здесь термин "клетка" обозначает клетку животного, выделенную из тканей человека или не относящихся к человеку животных, и выбранную из группы, состоящей из клетки печени, клетки кожи, клетки слизистой оболочки, клетки островка Лангерганса, нервной клетки, хрящевой клетки, клетки эндотелия, эпителиальной клетки, костной клетки и мышечной клетки, или мужской половой клетки, яйцеклетки или оплодотворенной яйцеклетки животного или рыбы. "Орган" выбирают из группы, состоящей из кожи, роговицы, почки, сердца, печени, поджелудочной железы, кишечника, нерва, легкого, плаценты, пуповины и кровеносного сосуда.
Кроме того, соединение индола и индазола согласно настоящему изобретению может быть использовано для дополнительного введения его в традиционный раствор для консервирования органов. Если соединение индола и индазола согласно настоящему изобретению вводят в традиционный раствор для консервирования органов, то может быть значительно увеличен период сохранения упомянутых выше органов для трансплантации и может быть эффективно предотвращено или подвергнуто лечению повреждение органа после трансплантации.
Кроме того, в результате введения соединения индола и индазола согласно настоящему изобретению в традиционную среду для выращивания клеток или консервирующие растворы, клетки печени или клетки поджелудочной железы, и так далее, животных могут быть сохранены в течение длительного времени даже без замораживания. Таким образом, такие консервированные клетки животных могут быть использованы для применения в клеточной или тканевой инженерии для получения полезных материалов.
В случае необходимости, "фармацевтическая композиция" согласно настоящему изобретению может включать вместе с активным ингредиентом фармацевтически приемлемый носитель, разбавитель, вспомогательное вещество или их комбинацию. Такая фармацевтическая композиция облегчает введение соединения в живой организм. Существует много методов введения, включая, но этим не ограничивая, пероральное введение, инъекцию, аэрозоль, парентеральное введение и местное введение.
Используемый здесь термин "носитель" обозначает вещество, которое облегчает введение соединения в клетки или ткани. Например, диметилсульфоксид (DMSO) является типичным носителем, который используют для облегчения введения различных органических соединений в клетки или ткани живых организмов.
Используемый здесь термин "разбавитель" определяется как вещество, растворяемое в воде, которое растворяет соединение, а также которое стабилизирует данное соединение в его биологически активной форме. В технике в качестве разбавителей используют соли, растворенные в буферном растворе. Типичным буферным раствором является забуференный фосфатом физиологический раствор, который имитирует солевую форму раствора в человеческом организме. Буферные разбавители почти не изменяют биологическую активность соединения, так как буферные соли способны регулировать pH раствора при низкой концентрации.
Используемый здесь термин "фармацевтически приемлемый" обозначает свойство, которое не ослабляет биологическую активность и свойства соединения.
Соединения настоящего изобретения могут быть сформированы в различные фармацевтические лекарственные формы в соответствии с требуемой целью. Для получения фармацевтической композиции настоящего изобретения активный ингредиент, в частности соединения формулы (1), их фармацевтически приемлемая соль или изомер смешивают с различными фармацевтически приемлемыми носителями, которые могут быть выбраны в соответствии с приготавливаемой лекарственной формой. Например, в соответствии с требуемой целью, фармацевтическая композиция настоящего изобретения может быть составлена в виде препарата для инъекции, препарата для перорального введения, и так далее.
Соединения настоящего изобретения могут быть подвергнуты формированию лекарственной формы с помощью известных в технике методов, использующих известные фармацевтические носители и вспомогательные вещества, и помещены в упаковку разовой дозы лекарственной формы или в упаковку лекарственной формы для многократного приема. Формой препарата могут являться растворы, суспензии или эмульсии в масляных или водных средах, и они могут содержать типичные диспергаторы, суспендирующие средства или стабилизаторы. Кроме того, например, препарат может быть в форме сухого порошка, который перед применением предполагается растворять в стерилизованной апирогенной воде. Соединения настоящего изобретения могут также быть сформированы в суппозиторные формы, использующие типичную суппозиторную основу, такую как масло какао или другие глицериды. В качестве твердых лекарственных форм для перорального введения могут быть приготовлены капсулы, таблетки, пилюли, порошок и гранулы, и особенно являются подходящими капсулы и таблетки. Предпочтительно, чтобы таблетки и пилюли изготавливали в виде форм, покрытых энтеросолюбильной оболочкой. Твердые лекарственные формы могут быть получены путем смешения соединения настоящего изобретения вместе с носителями, такими как один или более разбавителей, таких как сахароза, лактоза, крахмал и так далее, скользящими веществами, такими как стеарат магния, разрыхлителем, связующим, и так далее.
В случае необходимости, соединение настоящего изобретения или фармацевтические композиции, содержащие это соединение, могут также быть введены в комбинации с другими активными средствами, например, другими веществами, которые могут предотвращать повреждение органов, выделенных клеточных систем или тканей, вызванное хранением при низких температурах, операцией по трансплантации или посттрансплантационной реперфузией.
Доза соединений формулы (1) зависит от предписания врача, с учетом таких факторов, как масса тела и возраст пациента, специфическая природа заболевания и тяжесть заболевания, и так далее. Однако доза, необходимая для трансплантации органа взрослому человеку, составляет обычно примерно от 1 нМ до 100 мкМ, и обычно достаточной дозой будет являться концентрация 10 мкМ или менее, но для ряда пациентов предпочтительной может являться более высокая доза.
Используемый здесь термин "лечение" означает прерывание или замедление развития заболевания при применении лечения применительно к пациенту, у которого появляются проявления симптомов начала заболевания, и термин "предотвращение" означает прерывание или замедление признака начала заболевания при применении лечения применительно к пациенту, у которого нет проявления симптомов, но существует риск появления симптомов начала заболевания.
Настоящее изобретение будет более конкретно разъяснено далее при помощи следующих синтезов и примеров. Однако следует иметь в виду, что они предназначены только для иллюстрации настоящего изобретения, и ни в коем случае не ограничивают объем настоящего изобретения. В следующих синтезах и примерах M обозначает молярную концентрацию, и N обозначает нормальную концентрацию.
ОПИСАНИЕ ЧЕРТЕЖЕЙ
На фигуре 1 показано защитное действие соединения примера 21 согласно настоящему изобретению по отношению к гепатоцитам в экспериментах с использованием первичных гепатоцитов крыс.
На фигуре 2 показано способность соединений примеров 126, 127, 137 и 138 согласно настоящему изобретению восстанавливать клетки легких LB-HEL при 37°C после холодового шока в течение 24 часов в сравнении с контрольными лекарственными средствами, IM54 и nec-1.
На фигуре 3 показано защитное действие соединения примера 126 согласно настоящему изобретению от повреждения при хранении при низких температурах на модели перфузии печени, извлеченной у крыс, в сравнении с традиционным консервирующим раствором, раствором HTK, в результате измерения LDH, AST и ALT активности.
На фигуре 4 показано защитное действие соединения примера 126 согласно настоящему изобретению от холодовой ишемии и реперфузионного повреждения при действии тепла на модели перфузии печени, извлеченной у крыс, в сравнении с традиционным консервирующим раствором, раствором HTK, в результате измерения LDH активности.
На фигуре 5 показано защитное действие соединения примера 126 согласно настоящему изобретению от холодовой ишемии/реперфузионного повреждения при действии тепла на модели перфузии печени, извлеченной у крыс, в сравнении с традиционным консервирующим раствором, раствором HTK, в результате измерения выработки желчи.
ПРЕДПОЧТИТЕЛЬНЫЙ ВАРИАНТ ОСУЩЕСТВЛЕНИЯ ИЗОБРЕТЕНИЯ
Синтез 1: Получение этилового эфира 2-[(4-фтор-2-нитрофенил)гидразоно]-пропионовой кислоты
4-Фтор-2-нитроанилин (10 г, 64 ммоль) растворяли в 6н хлористоводородной кислоте (64 мл, 0,27 моль), медленно добавляли по каплям нитрат натрия (4,4 г, 64 ммоль), растворенный в воде (50 мл), при 0°, и смесь перемешивали в течение 30 минут при 0° ~ комнатной температуре. Одновременно растворяли этил 2-метилацетоацетат (9,2 мл, 64 ммоль) и гидроксид натрия (19 г, 0,34 моль) в 80% водном растворе этанола (95 мл) и перемешивали в течение 10 минут при 0°. Полученные таким образом два раствора смешивали и перемешивали в течение 8 часов при 0° ~ комнатной температуре. К реакционной смеси добавляли воду. Нерастворившееся твердое вещество собирали, промывали водой и сушили с получением названного соединения (7,9 г, выход 46%).
1H-ЯМР (400 МГц, CDCl3); δ 10,81 (ушир.с, 1H), 8,05 (м, 1H), 7,90 (м, 1H), 7,41 (м, 1H), 4,36 (кв, 2H), 2,22 (с, 3H), 1,38 (т, 3H).
Синтез 2: Получение этилового эфира 5-фтор-7-нитро-1H-индол-2-карбоновой кислоты
Соединение, полученное синтезом 1 (8,8 г, 33 ммоль), смешивали с полифосфорной кислотой (50 мл), и смесь перемешивали в течение 7 часов при 60°. К реакционной смеси добавляли воду. Нерастворившееся твердое вещество собирали, промывали водой и сушили с получением названного соединения (3,4 г, выход 41%).
1H-ЯМР (400 МГц, DMSO-d6); δ 11,55 (ушир.с, 1H), 8,16 (м, 1H), 8,10 (м, 1H), 7,42 (с, 1H), 4,40 (кв, 2H), 1,36 (т, 3H).
Синтез 3: Получение гидрохлорида (4-хлор-2-нитрофенил)гидразина
4-Хлор-2-нитроанилин (40 г, 0,23 моль) растворяли в 12н-хлористоводородной кислоте (100 мл). Медленно добавляли по каплям при 0° нитрат натрия (16 г, 0,23 моль), растворенный в воде (50 мл), и смесь перемешивали в течение 30 минут при 0° ~ комнатной температуре. Температуру понижали до 0° и медленно добавляли по каплям хлорид олова(II) (132 г, 0,70 моль), растворенный в 12н-хлористоводородной кислоте (100 мл). Смесь перемешивали в течение 3 часов при 0° ~ комнатной температуре. Полученное желтое твердое вещество фильтровали, промывали небольшим количеством 6н-HCl, и сушили с получением названного соединения (30 г, выход, 63%).
1H-ЯМР (400 МГц, DMSO-d6); δ 9,21 (с, 1H), 7,98 (д, J=2,4 Гц, 1H), 7,66 (д, J=9,6 Гц, 1H), 7,55 (дд, J=2,4, 9,6 Гц, 1H), 4,74 (ушир.с, 2H).
Синтез 4: Получение метилового эфира 2-[(4-хлор-2-нитрофенил)гидразоно]-пропионовой кислоты
Гидразин (30 г, 0,14 моль), полученный в синтезе 3, и метилпируват (14,4 мл, 0,16 моль) растворяли в метаноле (300 мл) и добавляли к ним ацетат натрия (14,2 г, 0,17 моль). Смесь перемешивали в течение 8 часов при комнатной температуре. Полученное желтое твердое вещество фильтровали, промывали водой и метанолом, и сушили с получением названного соединения (30 г, выход 82%).
1H-ЯМР (400 МГц, CDCl3); δ 10,88 (с, 1H), 8,21 (д, J=2,4 Гц, 1H), 8,01 (д, J=9,2 Гц, 1H), 7,56 (дд, J=2,4, 9,2 Гц, 1H), 3,90 (с, 3H), 2,23 (с, 3H).
Масс-спектр (ESI, m/z): Вычислено для C10H10ClN3O4:271,04, обнаружено: 271,66.
Синтез 5: Получение метилового эфира 5-хлор-7-нитро-1H-индол-2-карбоновой кислоты
К соединению (13 г, 46 ммоль), полученному в синтезе 4, добавляли полифосфорную кислоту (100 мл), и смесь нагревали при 100° в течение 4 часов. После завершения реакции, к реакционной смеси добавляли воду. Нерастворившееся твердое вещество собирали, промывали водой и сушили с получением названного соединения (6,0 г, выход 49%).
1H-ЯМР (400 МГц, CDCl3); δ 10,32 (ушир.с, 1H), 8,29 (д, 1H), 8,03 (д, J=2,4 Гц, 1H), 7,31 (д, J=2,0 Гц, 1H), 4,01 (с, 3H).
Масс-спектр (ESI, m/z): вычислено: 254,01, обнаружено: 254,63
Синтез 6: Получение метилового эфира 5-бром-7-нитро-1H-индол-2-карбоновой кислоты
4-Бром-2-нитроанилин (15,6 г, 71,9 ммоль) подвергали взаимодействию в соответствии с такими же методиками, как в синтезах 3 - 5, с получением названного соединения (7,2 г, выход 73%).
1H-ЯМР (400 МГц, CDCl3); δ 10,33 (ушир.с, 1H), 8,41 (с, 1H), 8,18 (с, 1H), 7,30 (д, J=4,0 Гц, 1H), 4,01 (с, 3H).
Синтез 7: Получение метилового эфира 5-метил-7-нитро-1H-индол-2-карбоновой кислоты
4-Метил-2-нитроанилин (40 г, 0,26 моль) подвергали взаимодействию в соответствии с такими же методиками, как в синтезах 3 - 5, с получением названного соединения (20 г, выход 32%).
1H-ЯМР (500 МГц, DMSO-d6); δ 11,25 (ушир.с, 1H), 8,08 (с, 1H), 7,96 (с, 1H), 7,32 (с, 1H), 3,87 (с, 3H), 2,44 (с, 3H).
Синтез 8: Получение 4-этокси-2-нитрофениламина
4-Этоксианилин (40 г, 0,29 моль) и триэтиламин (61 мл, 0,44 моль) растворяли в дихлорметане (200 мл). Добавляли по каплям уксусный ангидрид (30 мл, 0,32 ммоль), и смесь перемешивали в течение 1 часа при 0° ~ комнатной температуре. Добавляли к ним 1н раствор хлористоводородной кислоты, и полученную смесь экстрагировали с помощью этилацетата, промывали насыщенным раствором хлорида натрия, и сушили над безводным сульфатом магния.
Полученное таким образом соединение ацетамида растворяли в дихлорметане (200 мл), и добавляли по каплям при 0° дымящую азотную кислоту (13 мл, 0,29 моль). Полученную смесь перемешивали в течение 1 часа при 0° ~ комнатной температуре. Добавляли насыщенный водный раствор гидрокарбоната натрия, и смесь экстрагировали с помощью этилацетата, промывали насыщенным раствором хлорида натрия и сушили над безводным сульфатом магния.
Полученное таким образом нитратное соединение растворяли в метаноле (100 мл) и тетрагидрофуране (100 мл), и добавляли по каплям 6н-гидрид натрия. Смесь перемешивали в течение 6 часов при комнатной температуре. После завершения реакции, добавляли раствор 6н-хлористоводородной кислоты для нейтрализации реакционной смеси до значения pH около 7. Смесь экстрагировали с помощью этилацетата, промывали насыщенным раствором хлорида натрия и сушили над безводным сульфатом магния с получением названного соединения (44 г, выход 83%).
Синтез 9: Получение метилового эфира 5-этокси-7-нитро-1H-индол-2-карбоновой кислоты
4-Этокси-2-нитроанилин, полученный в синтезе 8 (40 г, 0,22 моль), подвергали взаимодействию в соответствии с такими же методиками, как в синтезах 3-5, с получением названного соединения (13 г, выход 22%).
1H-ЯМР (400 МГц, DMSO-d6); δ 10,20 (ушир.с, 1H), 7,86 (с, 1H), 7,51 (с, 1H), 7,26 (с, 1H), 4,13 (м, 2H), 3,98 (с, 3H), 1,47 (м, 3H).
Синтез 10: Получение метилового эфира 7-нитро-5-фенокси-1H-индол-2-карбоновой кислоты
Фениловый эфир 4-аминофенила (20 г, 0,11 моль) подвергали взаимодействию в соответствии с такими же методиками, как в синтезе 8 и синтезах 3-5, с получением названного соединения (5 г, выход 15%).
1H-ЯМР (400 МГц, CDCl3); δ 10,26 (ушир.с, 1H), 8,05 (с, 1H), 7,69 (с, 1H), 7,39 (м, 2H), 7,26 (с, 1H), 7,15 (м, 1H), 7,01 (м, 2H), 4,00 (с, 3H).
Синтез 11: Получение этилового эфира 7-нитро-5-(пиридин-3-илокси)-1H-индол-2-карбоновой кислоты
(Стадия 1)
1-Хлор-4-нитробензол (40 г, 0,25 моль) и 3-гидроксипиридин (36 г, 0,38 моль) растворяли в N,N-диметилформамиде (100 мл). Добавляли карбонат калия (52,6 г, 0,38 моль), и смесь перемешивали в течение 20 часов при 100°С. Добавляли воду, и полученную смесь экстрагировали с помощью этилацетата, промывали насыщенным раствором хлорида натрия и сушили над безводным сульфатом магния с получением 3-(4-нитрофенокси)пиридина.
Полученное таким образом соединение растворяли с помощью воды (100 мл), тетрагидрофурана (100 мл) и метанола (100 мл). Добавляли железную пыль (103 г, 1,84 моль) и хлорид аммония (99 г, 1,84 моль), и смесь перемешивали при помощи механической мешалки в течение 3 часов при 80°. После завершения реакции, реакционную смесь фильтровали через целит, промывали с помощью метанола и концентрировали. Полученное твердое вещество фильтровали, промывали с помощью эфира и сушили с получением 4-(пиридин-3-илокси)фениламина (17 г, выход 36%).
(Стадия 2)
4-(Пиридин-3-илокси)фениламин (25 г, 0,13 моль) подвергали взаимодействию в соответствии с такими же методиками, как в синтезе 8 и синтезах 3-5 с получением названного соединения (4,2 г, выход 10%).
1H-ЯМР (400 МГц, CDCl3); δ 10,32 (ушир.с, 1H), 8,51-8,47 (м, 2H), 8,05 (д, J=2,4 Гц, 1H), 7,73 (д, J=2,0 Гц, 1H), 7,42-7,35 (м, 2H), 7,31 (д, J=2,4 Гц, 1H), 4,48 (кв, 2H), 1,47 (т, 3H).
Синтез 12: Получение 5-метил-7-нитро-2-пиридин-2-ил-1H-индола
Гидрохлорид (4-метил-2-нитрофенил)гидразина (10 г, 49 ммоль) и 2-ацетилпиридин (5,5 мл, 49 ммоль) подвергали взаимодействию в соответствии с такими же методиками, как в синтезах 4-5 с получением названного соединения (2 г, выход 16%).
1H-ЯМР (400 МГц, DMSO-d6); δ 10,89 (ушир.с, 1H), 8,70 (д, J=4,4 Гц, 1H), 8,14 (д, J=8,0 Гц, 1H), 7,98~7,92 (м, 3H), 7,42-7,39 (м, 2H), 2,49 (с, 3H).
Синтез 13: Получение 5-метил-7-нитро-2-пиразин-2-ил-1H-индола
Гидрохлорид (4-метил-2-нитрофенил)гидразина (2 г, 9,8 ммоль) и 2-ацетилпиразин (1,2 мл, 9,8 ммоль) подвергали взаимодействию в соответствии с такими же методиками, как в синтезах 4-5 с получением названного соединения (0,3 г, выход 19%).
1H-ЯМР (500 МГц, CDCl3); δ 10,64 (ушир.с, 1H), 9,06 (д, J=1,2 Гц, 1H), 8,57 (с, 1H), 8,47 (д, J=2,4 Гц, 1H), 8,00 (с, 1H), 7,76 (с, 1H), 7,12 (д, J=2,4 Гц, 1H), 2,51 (с, 3H).
Синтез 14: Получение 7-нитро-2-пиридин-2-ил-1H-индола
Гидрохлорид 2-нитрофенилгидразина (5 г, 26 ммоль) и 2-ацетилпиридин (2,5 мл, 26 ммоль) подвергали взаимодействию в соответствии с такими же методиками, как в синтезах 4-5 с получением названного соединения (1 г, выход 16%).
1H-ЯМР (400 МГц, DMSO-d6); δ 11,01 (ушир.с, 1H), 8,72 (д, J=4,0 Гц, 1H), 8,15 (м, 3H), 7,93 (м, 1H), 7,49 (с, 1H), 7,42 (м, 1H), 7,30 (м, 1H).
Синтез 15: Получение 7-нитро-2-пиразин-2-ил-1H-индола
Гидрохлорид 2-нитрофенилгидразина (3,1 г, 16 ммоль) и 2-ацетилпиразин (2,0 мл, 16 ммоль) подвергали взаимодействию в соответствии с такими же методиками, как в синтезах 4-5 с получением названного соединения (0,5 г, выход 13%).
1H-ЯМР (400 МГц, DMSO-d6); δ 11,24 (ушир.с, 1H), 9,46 (д, J=4,0 Гц, 1H), 8,76 (м, 1H), 8,65 (д, J=4,0 Гц, 1H), 8,20 (дд, J=4,0, 8,0 Гц, 1H), 7,64 (д, J=4,0 Гц, 1H), 7,34 (т, J=8,0 Гц, 1H).
Синтез 16: Получение 5-этокси-7-нитро-2-пиридин-2-ил-1H-индола
Гидрохлорид (4-этокси-2-нитрофенил)гидразина (5 г, 21 ммоль) и 2-ацетилпиридин (2,4 мл, 21 ммоль) подвергали взаимодействию в соответствии с такими же методиками, как в синтезах 4-5, с получением названного соединения (0,5 г, выход 8%).
1H-ЯМР (400 МГц, CDCl3); δ 10,97 (ушир.с, 1H), 8,60 (м, 1H), 7,79 (м, 1H), 7,73 (м, 1H), 7,61 (д, J=2,8 Гц, 1H), 7,49 (д, J=2,0 Гц, 1H), 7,24 (м, 1H), 7,02 (д, J=2,4 Гц, 1H), 4,03 (кв, 2H), 1,45 (т, 3H).
Синтез 17: Получение 7-нитро-5-фенокси-2-пиридин-2-ил-1H-индола
Гидрохлорид (4-фенокси-2-нитрофенил)гидразина (10 г, 49 ммоль) и 2-ацетилпиридин (5,5 мл, 49 ммоль) подвергали взаимодействию в соответствии с такими же методиками, как в синтезах 4-5 с получением названного соединения (2 г, выход 16%).
1H-ЯМР (400 МГц, CDCl3); δ 10,86 (ушир.с, 1H), 8,68 (д, J=4,8 Гц, 1H), 7,92 (д, J=2,0 Гц, 1H), 7,84-7,76 (м, 2H), 7,65 (д, J=2,0 Гц, 1H), 7,36 (м, 3H), 7,28 (м, 1H), 7,13 (м, 1H), 7,06 (д, J=2,0 Гц, 1H), 7,03 (с, 1H), 7,02 (с, 1H).
Синтез 18: Получение 3,5-диметил-7-нитро-2-фенил-1H-индола
Гидрохлорид (4-метил-2-нитрофенил)гидразина (1,0 г, 4,9 ммоль) и 2-пропиофенон (0,7 мл, 4,9 ммоль) подвергали взаимодействию в соответствии с такими же методиками, как в синтезах 4-5, с получением названного соединения (150 мг, выход 11%).
Синтез 19: Получение 5-метил-7-нитро-2-фенил-1H-индола
(Стадия 1)
4-Метил-2-нитроанилин (20 г, 131,5 ммоль) растворяли в этаноле (300 мл), добавляли нитрат серебра (27 г, 157,7 ммоль) и йод (40 г, 157,7 ммоль), и смесь перемешивали в течение 8 часов при комнатной температуре. После завершения реакции, реакционную смесь фильтровали через целит, промывали с помощью этилацетата (100 мл) и концентрировали. Добавляли воду, и смесь экстрагировали с помощью этилацетата, промывали насыщенным раствором хлорида натрия и сушили над безводным сульфатом магния с получением 2-йод-4-метил-6-нитрофениламина (29 г, выход 69%).
1H-ЯМР (500 МГц, CDCl3); δ 7,94 (с, 1H), 7,75 (с, 1H), 6,48 (ушир.с, 2H), 2,23 (с, 3H).
(Стадия 2)
Соединение, полученное на стадии 1 (7 г, 25,2 ммоль), и фенилацетилен (3,3 мл, 30,22 ммоль) растворяли в тетрагидрофуране (100 мл), добавляли триэтиламин (11 мл, 75,5 ммоль), дихлор(бистрифенилфосфин)палладий(II) (1,8 г, 2,52 ммоль) и йодид меди(I) (0,48 г, 2,52 ммоль), и смесь перемешивали в течение 8 часов при комнатной температуре. После завершения реакции добавляли воду. Смесь экстрагировали с помощью этилацетата, промывали насыщенным раствором хлорида натрия, сушили над безводным сульфатом магния и очищали с помощью колоночной хроматографии с получением 4-метил-2-нитро-6-фенилэтинилфениламина (4,5 г, выход 71%).
1H-ЯМР (400 МГц, CDCl3); δ 7,93 (с, 1H), 7,53 (м, 2H), 7,46 (с, 1H), 7,39 (м, 3H), 6,62 (ушир.с, 2H), 2,26 (с, 3H).
(Стадия 3)
Соединение, полученное на стадии 2 (4,5 г, 17,8 ммоль), растворяли в тетрагидрофуране (120 мл) и N-метил-пирролидиноне (30 мл). Добавляли трет-бутилат калия (4 г, 35,7 ммоль), и смесь перемешивали в течение 3 часов при комнатной температуре. После завершения реакции добавляли воду. Смесь экстрагировали с помощью этилацетата, промывали насыщенным раствором хлорида натрия, сушили над безводным сульфатом магния и очищали с помощью колоночной хроматографии с получением 5-метил-7-нитро-2-фенил-1H-индола (1,0 г, выход 22%).
1H-ЯМР (400 МГц, CDCl3); δ 11,53 (ушир.с, 1H), 8,02 (д, J-7,6 Гц, 1H), 7,94 (с, 1H), 7,87 (с, 1H), 7,50 (м, 2H), 7,41 (м, 1H), 7,09 (м, J=2,0 Гц, 1H), 2,49 (с, 3H).
Синтез 20: Получение 2-циклогексил-5-метил-7-нитро-1H-индола
6-Йод-4-метил-2-нитроанилин (500 мг, 1,8 ммоль) и циклогексилацетилен (0,23 мл, 1,8 ммоль) подвергали взаимодействию в соответствии с такой же методикой, как в синтезе 19, с получением названного соединения (290 мг, выход 62%).
1H-ЯМР (500 МГц, CDCl3); δ 9,34 (ушир.с, 1H), 7,88 (с, 1H), 7,63 (с, 1H), 6,26 (с, 1H), 2,76 (м, 1H), 2,47 (с, 3H), 2,10 (м, 2H), 1,87 (м, 2H), 1,78 (м, 1H), 1,52-1,42 (м, 4H), 1,31 (м, 1H).
Синтез 21: Получение 5-метил-2-(6-метилпиридин-2-ил)-7-нитро-1H-индола
6-Йод-4-метил-2-нитроанилин (500 мг, 1,8 ммоль) и 2-этинил-6-метилпиридин (210 мг, 1,8 ммоль) подвергали взаимодействию в соответствии с такой же методикой, как в синтезе 19, с получением названного соединения (170 мг, выход 35%).
1H-ЯМР (500 МГц, CDCl3); δ 10,77 (ушир.с, 1H), 7,98 (с, 1H), 7,73 (с, 1H), 7,64~7,58 (м, 2H), 7,08 (д, J=7,4 Гц, 1H), 6,98 (д, J=2,5 Гц, 1H), 2,62 (с, 3H), 2,50 (с, 3H).
Синтез 22: Получение гидрохлорида метилового эфира (R)-3-амино-4-(4-метоксибензилсульфанил)масляной кислоты
(Стадия 1)
К смешанному раствору диэтилового эфира (400 мл) и концентрированной хлористоводородной кислоты (400 мл) добавляли по каплям в течение 2 часов 4-метоксибензил хлорид (280 г, 1780 ммоль), растворенный в диэтиловым эфире (400 мл), и смесь перемешивали в течение 1 часа. Органический слой отделяли и добавляли к раствору, который получали растворением L-цистеина (197 г, 1625 ммоль) и 2н водного раствора гидроксида натрия (980 мл) в этаноле (1890 мл). Смесь перемешивали в течение 2 часов при комнатной температуре. После завершения реакции, смесь охлаждали до 0°, и нейтрализовали до pH 7 при помощи 3н водного раствора хлористоводородной кислоты. Полученное твердое вещество фильтровали и сушили с получением (R)-2-амино-3-(4-метокси-бензилсульфанил)пропионовой кислоты (250 г, 1035 ммоль, выход 64%).
(Стадия 2)
Соединение, полученное на стадии 1 (30,7 г, 127,3 ммоль), растворяли в тетрагидрофуране (150 мл) и воде (150 мл). Добавляли карбонат калия (26,4 г, 190 ммоль) и ди-трет-бутилоксидикарбонил (27,7 г, 127,3 ммоль), и смесь перемешивали в течение 2 часов при комнатной температуре. После завершения реакции, смесь отгоняли при пониженном давлении для удаления тетрагидрофурана. Смесь охлаждали до 0°С, и подкисляли до pH 3 при помощи 3н водного раствора хлористоводородной кислоты. Полученное твердое вещество промывали водой и сушили с получением (R)-2-трет-бутоксикарбониламино-3-(4-метоксибензил-сульфанил)пропионовой кислоты (43 г, 126 ммоль, выход 99%).
(Стадия 3)
Соединение, полученное на стадии 2 (43 г), 1-метилморфолин (14,5 мл, 132 ммоль) и этилхлорформиат (14,1 мл, 132 ммоль) растворяли в тетрагидрофуране (500 мл), и смесь перемешивали в течение 1 часа при -25°С. Одновременно, гидроксид калия (75 г, 1336 ммоль) растворяли в воде (75 мл) и диэтиловом эфире (750 мл), добавляли по каплям N-метилнитрозомочевину (26 г, 252 ммоль) в течение 2 часов при 0°С, и смесь перемешивали в течение 30 минут. Полученные таким образом два раствора смешивали и перемешивали в течение 3 часов при -25°С ~ комнатной температуре. После завершения реакции добавляли воду. Смесь промывали с помощью насыщенного водного раствора гидрокарбоната натрия и насыщенного водного раствора хлорида аммония в указанной последовательности. Органический слой концентрировали с получением трет-бутилового эфира [(R)-3-диазо-1-(4-метоксибензилсульфанилметил)-2-оксопропил]карбаминовой кислоты.
1H-ЯМР (400 МГц, CDCl3); δ 7,25 (д, J=8,8 Гц, 2H), 6,86 (д, J=8,8 Гц, 2H), 5,48 (ушир.с, 1H), 5,29 (м, 1H), 4,31 (м, 1H), 3,79 (с, 3H), 3,69 (с, 2H), 2,76 (д, J=6,0 Гц, 2H), 1,45 (с, 9H).
(Стадия 4)
Соединение, полученное на стадии 3, растворяли в метаноле (1000 мл), добавляли бензоат серебра (7,1 г, 31,1 ммоль), и смесь подвергали взаимодействию в течение 1 часа при воздействии ультразвука. После завершения реакции смесь концентрировали и очищали с помощью колоночной хроматографии с получением метилового эфира (R)-3-трет-бутоксикарбониламино-4-(4-метоксибензилсульфанил)масляной кислоты (35,2 г, 95,3 ммоль, выход 76%).
1H-ЯМР (500 МГц, CDCl3); δ 7,24 (д, J=8,6 Гц, 2H), 6,83 (д, J=8,6 Гц, 2H), 5,09 (м, 1H), 4,08 (м, 1H), 3,79 (с, 3H), 3,68 (с, 2H), 3,66 (с, 3H), 2,70-2,52 (м, 4H), 1,44 (с, 9H).
(Стадия 5)
Соединение, полученное на стадии 4 (35,2 г), растворяли в дихлорметане (70 мл), добавляли раствор 4н хлористоводородная кислота/1,4-диоксан (71 мл), и смесь перемешивали в течение 2 часов при комнатной температуре. После завершения реакции смесь концентрировали. Твердое вещество, образовавшееся при добавлении дихлорметана (30 мл) и диэтилового эфира (150 мл), фильтровали и сушили с получением названного соединения (25,5 г, 83,3 ммоль, выход 87%).
1H ЯМР (400 МГц, DMSO-d6); δ 8,21 (ушир.с, 3H), 7,25 (д, 2H), 6,83 (д, 2H), 3,78 (с, 3H), 3,68 (с, 2H), 3,65 (с, 3H), 3,29 (м, 1H), 2,51-2,48 (м, 2H), 2,35-2,31 (м, 2H).
Синтез 23: Получение гидрохлорида этилового эфира (R)-3-амино-4-(4-метокси-бензилсульфанил)масляной кислоты
L-цистеин (50 г, 0,41 моль) подвергали взаимодействию в соответствии с такой же методикой, как в синтезе 22, за исключением того, что использовали этанол вместо метанола на стадии 4 синтеза 22, с получением названного соединения (5,2 г, выход 40%).
1H ЯМР (400 МГц, CDCl3); δ 8,37 (ушир.с, 3H), 7,28 (д, J=8,0 Гц, 2H), 6,87 (д, J=8,0 Гц, 2H), 4,1 (м, 2H), 3,73 (с, 3H), 3,70 (с, 2H), 2,81-2,67 (м, 4H), 1,18 (т, 3H).
Синтез 24: Получение гидрохлорида этилового эфира (R)-4-амино-5-(4-метокси-бензилсульфанил)валериановой кислоты
(Стадия 1)
(R)-4-третбутоксикарбониламино-5-гидрокси-валериановой кислоты этиловый эфир (36 г, 137,8 ммоль), который может быть получен известным методом, и триэтиламин (38,4 мл, 275,5 моль) растворяли в дихлорметане (200 мл). Добавляли по каплям метансульфонилхлорид (11,7 мл, 151,5 ммоль), и смесь перемешивали в течение 1 часа при 0° ~ комнатной температуре. Добавляли 1н раствор хлористоводородной кислоты. Смесь экстрагировали с помощью этилацетата, промывали насыщенным раствором хлорида натрия и сушили над безводным сульфатом магния с получением этилового эфира (R)-4-трет-бутоксикарбониламино-5-метансульфонил-оксивалериановой кислоты.
(Стадия 2)
Соединение, полученное на стадии 1, и гидрид натрия (5,5 г, 137,8 ммоль) добавляли по каплям к 4-метоксибензилмеркаптану (15,4 мл, 110,2 ммоль), растворенному в N,N-диметилформамиде (150 мл), и перемешивали в течение 10 минут при 0°. Смесь перемешивали в течение 4 часов при 0°. После завершения реакции добавляли воду. Смесь экстрагировали с помощью этилацетата, промывали насыщенным раствором хлорида натрия, сушили над безводным сульфатом магния и очищали с помощью колоночной хроматографии с получением этилового эфира (R)-4-трет-бутоксикарбониламино-5-(4-метоксибензилсульфанил)валериановой кислоты (21,0 г, выход 38%).
1H-ЯМР (400 МГц, CDCl3); δ 7,25 (д, J=8,8 Гц, 2H), 6,85 (д, J=8,8 Гц, 2H), 4,56 (м, 1H), 4,12 (м, 2H), 3,79 (с, 3H), 3,69 (с, 2H), 2,53 (м, 2H), 2,33 (т, 2H), 1,93 (м, 1H), 1,70 (м, 1H), 1,44 (с, 9H), 1,25 (т, 3H).
(Стадия 3)
Соединение, полученное на стадии 2 (11 г, 62,7 ммоль), растворяли в дихлорметане (200 мл), и добавляли раствор 4н хлористоводородная кислота/этилацетат (20 мл). Смесь перемешивали в течение 2 часов при комнатной температуре. После завершения реакции смесь полностью концентрировали, и добавляли диэтиловый эфир (150 мл). Полученное твердое вещество фильтровали и сушили с получением названного соединения (20 г, выход 96%).
1H ЯМР (400 МГц, DMSO-d6); δ 8,69 (ушир.с, 3H), 7,29 (д, J=8,0 Гц, 2H), 6,89 (д, J=8,0 Гц, 2H), 4,08 (м, 2H), 3,74 (м, 5H), 3,26 (м, 1H), 2,76-2,63 (м, 2H), 2,49-2,40 (м, 2H), 1,89 (м, 2H), 1,20 (т, 3H).
Синтез 25: Получение гидрохлорида изопропилового эфира (S)-3-амино-4-(4-метокси-бензилсульфанил)масляной кислоты
Осуществляли такую же методику, как и в синтезе 24, за исключением того, что использовали изопропиловый эфир (S)-3-трет-бутоксикарбонил-амино-4-гидроксимасляной кислоты (22,0 г, 84,2 ммоль) вместо этилового эфира (R)-4-трет-бутоксикарбониламино-5-гидрокси-валериановой кислоты, с получением названного соединения (21,0 г, выход 75%).
1H ЯМР (400 МГц, MeOH-d4); δ 7,31 (д, J=8,8 Гц, 2H), 6,91 (д, J=8,8 Гц, 2H), 5,06 (м, 1H), 3,80 (с, 3H), 3,78 (с, 2H), 3,60 (м, 1H), 2,81-2,63 (м, 4H), 1,28 (дд, 6H).
Синтез 26: Получение гидрохлорида этилового эфира (R)-2-амино-3-(4-метокси-бензилсульфанил)пропионовой кислоты
Соединение кислоты, полученное на стадии 1 синтеза 22 (20 г, 83 ммоль), растворяли в этаноле (100 мл). Добавляли по каплям ацетилхлорид (12 мл, 166 ммоль), и смесь перемешивали в течение 12 часов при 50°С. После завершения реакции смесь полностью концентрировали, и добавляли к ней диэтиловый эфир. Полученное твердое вещество фильтровали и сушили с получением названного соединения (16,8 г, выход 69%).
1H ЯМР (400 МГц, CDCl3); δ 8,85 (ушир.с, 3H), 7,27 (д, J=8,0 Гц, 2H), 6,80 (д, J=8,0 Гц, 2H), 4,47 (м, 1H), 3,78-3,69 (м, 8H), 3,17 (м, 2H).
Синтез 27: Получение 2-амино-3-(4-метоксибензилсульфанил)пропилового эфира (R)-2,2-диметилпропионовой кислоты
(Стадия 1)
Соединение, полученное на стадии 1 синтеза 22 (50 г, 207,2 ммоль), растворяли в метаноле (300 мл). Добавляли по каплям ацетилхлорид (21 мл, 207,2 ммоль), и смесь перемешивали в течение 12 часов при 50°С. После завершения реакции смесь полностью концентрировали, и добавляли к ней диэтиловый эфир. Полученное твердое вещество фильтровали и сушили с получением метилового эфира (R)-2-амино-3-(4-метоксибензилсульфанил)пропионовой кислоты.
1H ЯМР (400 МГц, DMSO-d6, HCl соль); δ 8,81 (ушир.с, 3H), 7,29 (д, J=8,4 Гц, 2H), 6,91 (д, J=8,4 Гц, 2H), 4,28 (м, 1H), 3,18 (ушир.с, 8H), 2,95 (м, 2H).
(Стадия 2)
К соединению, полученному на стадии 1, добавляли для растворения тетрагидрофуран (200 мл) и воду (200 мл). Добавляли триэтиламин (87 мл, 621,6 ммоль), и добавляли по каплям при перемешивании ди-трет-бутилоксидикарбонил (43,0 г, 196,8 ммоль), растворенный в тетрагидрофуране (100 мл). Смесь перемешивали в течение 8 часов при комнатной температуре. После завершения реакции добавляли воду. Смесь экстрагировали с помощью этилацетата, промывали насыщенным раствором хлорида натрия и сушили над безводным сульфатом магния с получением метилового эфира (R)-2-трет-бутоксикарбониламино-3-(4-метоксибензилсульфанил)пропионовой кислоты.
(Стадия 3)
Соединение, полученное на стадии 2, растворяли в тетрагидрофуране (300 мл). Добавляли боргидрид лития (9,0 г, 414,4 ммоль), и смесь перемешивали в течение 3 часов при 0°С. После завершения реакции добавляли воду. Смесь экстрагировали с помощью этилацетата, промывали насыщенным раствором хлорида натрия и сушили над безводным сульфатом магния с получением трет-бутилового эфира [(R)-2-гидрокси-1-(4-метоксибензилсульфанилметил)этил]карбаминовой кислоты.
1H ЯМР (500 МГц, DMSO-d6); δ 7,24 (д, J=8,6 Гц, 2H), 6,84 (д, J-8,6 Гц, 2H), 4,96 (ушир.с, 1H), 3,78 (с, 3H), 3,76 (ушир.с, 1H), 3,70 (с, 2H), 3,7-3,66 (м, 3H), 2,58 (м, 2H), 1,44 (с, 9H).
(Стадия 4)
Соединение спирта, полученное на стадии 3, растворяли в дихлорметана (300 мл). Добавляли триэтиламин (58 мл, 414,4 ммоль) и триметилацетилхлорид (28 мл, 227,9 ммоль), и смесь перемешивали в течение 6 часов при 0°С. После завершения реакции добавляли воду. Смесь экстрагировали с помощью этилацетата, промывали насыщенным раствором хлорида натрия, сушили над безводным сульфатом магния и очищали с помощью колоночной хроматографии с получением (R)-2-трет-бутоксикарбониламино-3-(4-метоксибензилсульфанил)пропилового эфира 2,2-диметилпропионовой кислоты (81,0 г, выход 95%).
1H ЯМР (400 МГц, CDCl3); δ 7,25 (д, J=8,8 Гц, 2H), 6,85 (д, J=8,8 Гц, 2H), 4,71 (м, 1H), 4,11 (м, 2H), 3,79 (с, 3H), 3,70 (с, 2H), 2,55 (д, J=6,4 Гц, 2H), 1,52 (с, 1H), 1,27 (с, 9H).
(Стадия 5)
Соединение триметилацетата, полученное на стадии 4 (81 г, 196 ммоль), растворяли в дихлорметане (300 мл). Добавляли раствор 4н-хлористоводородная кислота/1,4-диоксан (100 мл), и смесь перемешивали в течение 8 часов при комнатной температуре. После завершения реакции смесь полностью концентрировали, и добавляли к ней диэтиловый эфир. Полученное твердое вещество фильтровали и сушили с получением названного соединения (68 г, выход 95%).
1H ЯМР (400 МГц, DMSO-d6, свободная форма); δ 7,24 (д, J=12,0 Гц, 2H), 6,85 (дд, J=4,0, 8,0 Гц, 2H), 4,04 (м, 1H), 3,95 (м, 1H), 3,80 (с, 3H), 3,68 (с, 2H), 3,10 (м, 1H), 2,60 (м, 1H), 2,36 (м, 1H), 1,18 (с, 9H).
Синтез 28: Получение 2-(4,5-дигидротиазол-2-ил)-1H-индол-7-иламина
(Стадия 1)
Этил 7-нитроиндол-2-карбоксилат (500 мг, 2,14 ммоль) растворяли в смеси растворителей тетрагидрофурана и воды (1:1, 20 мл), и добавляли гидрат гидроксида лития (448 мг, 10,7 ммоль). Смесь перемешивали в течение 8 часов при комнатной температуре, и добавляли к ней 1н раствор хлористоводородной кислоты. Смесь экстрагировали с помощью этилацетата. Экстракт сушили над безводным сульфатом магния и фильтровали. Фильтрат отгоняли при пониженном давлении с получением 7-нитро-1H-индол-2-карбоновой кислоты.
(Стадия 2)
Соединение, полученное на стадии 1, и гидрохлорид 2-хлорэтиламина (371 мг, 3,2 ммоль) растворяли в N,N-диметилформамиде (10 мл). Добавляли триэтиламин (0,6 мл, 4,3 ммоль), EDC (614 мг, 3,2 ммоль) и HOBT (433 мг, 3,2 ммоль). Смесь перемешивали в течение 8 часов при комнатной температуре, и добавляли 1н раствор хлористоводородной кислоты. Смесь экстрагировали с помощью этилацетата, промывали с помощью насыщенного раствора бикарбоната натрия, сушили над безводным сульфатом магния и фильтровали. Фильтрат отгоняли при пониженном давлении с получением (2-хлорэтил)амида 7-нитро-1H-индол-2-карбоновой кислоты.
1H ЯМР (400 МГц, CDCl3); δ 10,51 (ушир.с, 1H), 8,28 (д, J=6,4 Гц, 1H), 8,02 (д, J=6,4 Гц, 1H), 7,27 (т, 1H), 7,03 (с, 1H), 6,62 (ушир.с, 1H), 3,86 (м, 2H), 3,77 (м, 2H).
(Стадия 3)
Соединение, полученное на стадии 2, растворяли в дихлорэтане (10 мл) и толуоле (10 мл), и добавляли реагент Лавессона (1,29 г, 3,2 ммоль). Смесь кипятили с обратным холодильником в течение 4 часов, и отгоняли при пониженном давлении. Добавляли к остатку воду, и смесь экстрагировали с помощью этилацетата, сушили над безводным сульфатом магния и фильтровали. Фильтрат отгоняли при пониженном давлении, и концентрат очищали с помощью колоночной хроматографии с получением продукта реакции циклизации, то есть соединения 2-(4,5-дигидро-тиазол-2-ил)-7-нитро-1H-индол (100 мг, выход 22%).
1H-ЯМР (400 МГц, CDCl3); δ 10,49 (ушир.с, 1H), 8,24 (д, J=8,0 Hz 1H), 7,98 (д, J=7,6 Гц, 1H), 7,23 (т, 1H), 7,02 (с, 1H), 4,47 (т, 2H), 3,51 (т, 2H).
(Стадия 4)
Соединение тиазолина, полученное на стадии 3, растворяли в метаноле (50 мл). Добавляли 10% Pd/C, и смесь перемешивали в течение 8 часов в атмосфере газообразного водорода. После завершения реакции смесь фильтровали через целит, отгоняли при пониженном давлении и очищали с помощью колоночной хроматографии с получением названного соединения (80 мг, выход 91%).
1H-ЯМР (400 МГц, CDCl3); δ 9,78 (ушир.с, 1H), 7,15 (д, J=8,0 Гц, 1H), 6,95 (т, J=8,0 Гц, 1H), 6,90 (д, J=1,6 Гц, 1H), 6,60 (дд, 1H), 4,44 (дд, 2H), 3,45 (дд, 2H).
Масс-спектр (ESI, m/z): Вычислено: 217,07, обнаружено: 217,29.
Пример 1: Получение циклопентил-[2-(4,5-дигидро-1,3-тиазол-2-ил)-1H-индол-7-ил]амина
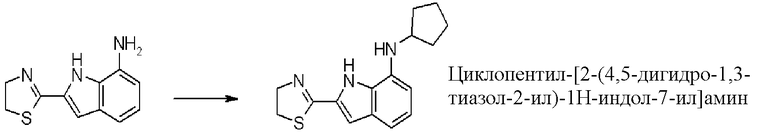
Соединение, полученное синтезом 28 (15 мг, 0,07 ммоль), растворяли в 1,2-дихлорэтане (10 мл). Добавляли циклопентанон (12 мг, 0,14 ммоль) и триацетоксиборгидрид натрия (29 мг, 0,14 ммоль), и смесь перемешивали в течение 3 часов при комнатной температуре. После завершения реакции добавляли воду. Смесь экстрагировали с помощью этилацетата, сушили над безводным сульфатом магния и фильтровали. Фильтрат отгоняли при пониженном давлении, и остаток очищали с помощью колоночной хроматографии с получением названного соединения (6,7 мг, выход 34%).
1H-ЯМР (400 МГц, CDCl3); δ 10,27 (с, 1H), 7,06 (д, J=8,0 Гц, 1H), 7,00 (т, J=7,6 Гц, 1H), 6,92 (с, 1H), 6,52 (д, J=7,2 Гц, 1H), 4,42 (м, 2H), 4,38 (м, 1H), 4,35 (м, 2H), 2,00 (м, 2H), 1,64 (м, 4H), 1,46 (м, 2H).
Масс-спектр (ESI. m/z): Вычислено: 285,13, обнаружено: 285,41.
Пример 2: Получение [2-(4,5-дигидротиазол-2-ил)-1H-индол-7-ил]-(4-метилциклогексил)амина

Соединение, полученное синтезом 28 (19 мг, 0,09 ммоль), и 4-метилциклогексанон подвергали взаимодействию в соответствие с таким же методом, как в примере 1, с получением двух диастереомеров в количестве 9,1 мг и 7,4 мг (суммарный выход 60%), соответственно.
1Н-ЯМР (400 МГц, CDCl3);
Соединение 1: δ 10,26 (с, 1H), 7,05-6,97 (м, 2H), 6,92 (с,1H), 6,51 (д, J=7,6 Гц, 1H), 4,40 (м, 2H), 3,65 (м, 1H), 3,46 (м, 2H), 1,72 (м, 3H), 1,52 (м, 3H), 1,22 (м, 3H), 0,82 (д, J=8,0 Гц, 3H).
Соединение 2: δ 10,24 (с, 1H), 7,05-6,96 (м, 2H), 6,92 (с, 1H), 6,50 (д, J=6,8 Гц, 1H), 4,44 (м, 2H), 3,47 (м, 2H), 3,25 (м, 1H), 2,11 (м, 2H), 1,74 (м, 2H), 1,36 (м, 1H), 1,10 (м, 4H), 0,90 (д, J=6,4 Гц, 1H).
Масс-спектр (RSI, m/z): вычислено: 313,16, обнаружено: 313,47.
Пример 3: Получение [2-(4,5-дигидротиазол-2-ил)-1H-индол-7-ил]пиперидин-4-иламина

К соединению, полученному из соединения синтеза 28 (20 мг, 0,09 ммоль), и 1-(трет-бутилкарбонил)-4-пиперидина в соответствии с таким же методом как в примере 1, добавляли дихлорметан и раствор трифторкарбоновой кислоты (5:1, объемное отношение, 5 мл). Смесь перемешивали в течение 1 часа при комнатной температуре и отгоняли при пониженном давлении. Добавляли насыщенный раствор бикарбоната натрия, и смесь экстрагировали с помощью этилацетата, сушили над безводным сульфатом магния и фильтровали. Фильтрат отгоняли при пониженном давлении и очищали с помощью колоночной хроматографии с получением названного соединения (4,9 мг, выход 18%).
1H-ЯМР (400 МГц, CDCl3, MeOH-d4); δ 7,39 (с, 1H), 7,07 (д, J=8,0 Гц, 1H), 6,99 (т, J=8,0 Гц, 1H), 6,93 (с, 1H), 6,47 (д, J=7,6 Гц, 1H), 4,41 (м, 2H), 3,77 (м, 1H), 3,48 (м, 4H), 3,11 (м, 2H), 2,29 (м, 2H), 1,87 (м, 2H).
Масс-спектр (ESI, m/z): вычислено: 300,14, обнаружено: 300,43.
Синтез 29: Получение 7-(тетрагидропиран-4-иламино)-1H-индол-2-карбоновой кислоты
Этил 7-нитроиндол-2-карбоксилат (2,5 г, 10,7 ммоль) растворяли в метаноле (50 мл). Добавляли 10% Pd/C (200 мг), и смесь перемешивали в течение 1 часа в атмосфере газообразного водорода. Смесь фильтровали через целит, и фильтрат отгоняли при пониженном давлении. Дистиллят растворяли в 1,2-дихлорэтане (50 мл), и добавляли тетрагидро-4H-пиран-4-он (1,3 мл, 12,8 ммоль) и триацетоксиборгидрид натрия (3,4 г, 16,1 ммоль). Смесь перемешивали в течение 8 часов при комнатной температуре. После завершения реакции добавляли воду. Смесь экстрагировали с помощью этилацетата, сушили над безводным сульфатом магния и фильтровали. Фильтрат отгоняли при пониженном давлении и концентрировали. Остаток очищали с помощью колоночной хроматографии. Полученное таким образом соединение растворяли в метаноле (50 мл) и тетрагидрофуране (50 мл), добавляли 1н-гидроксид натрия (43 мл, 42,8 ммоль), и смесь перемешивали в течение 8 часов при комнатной температуре. Добавляли 1н раствор хлористоводородной кислоты, и смесь экстрагировали с помощью этилацетата, сушили над безводным сульфатом магния и фильтровали. Фильтрат отгоняли при пониженном давлении с получением названного соединения (2,1 г, выход 76%).
1H-ЯМР (400 МГц, DMSO-d6): δ 12,87 (ушир.с, 1H), 11,46 (с, 1H),6,97 (д, J=2,0 Гц, 1H), 6,86 (д, J=1,6 Гц, 1H), 6,85 (с, 1H), 6,37 (м, 1H), 5,73 (ушир.с, 1H), 3,90 (м, 2H), 3,61 (м, 1H), 3,50 (м, 2H), 2,00 (м, 2H), 1,48 (м, 2H).
Масс-спектр (ESI, m/z): вычислено: 260,12, обнаружено: 260,30.
Синтез 30: Получение этилового эфира {5-[7-(тетрагидропиран-4-иламино)-1H-индол-2-ил]-[1,2,4]оксадиазол-3-ил}уксусной кислоты
Соединение, полученное синтезом 29 (300 мг, 1,15 ммоль), растворяли в диметилформамиде (20 мл). Добавляли к нему этил 3-(гидроксиамино)-3-иминопропионат (202 мг, 1,38 ммоль), EDC (265 мг, 1,38 ммоль) и HOBT (187 мг, 1,38 ммоль). Смесь перемешивали в течение 8 часов при комнатной температуре, и добавляли насыщенный раствор бикарбоната натрия. Смесь экстрагировали с помощью этилацетата, сушили над безводным сульфатом магния и фильтровали. Фильтрат отгоняли при пониженном давлении и концентрировали. Остаток очищали с помощью колоночной хроматографии. Полученное таким образом соединение растворяли в толуоле (20 мл) и дихлорэтане (10 мл). Смесь кипятили с обратным холодильником в течение 8 часов при 120°С, отгоняли при пониженном давлении и очищали с помощью колоночной хроматографии с получением названного соединения (80 мг, выход 19%).
1H-ЯМР (500 МГц, CDCl3); δ 9,71 (ушир.с, 1H), 7,35 (д, =1,8 Гц, 1H), 7,15 (д, J=8,0 Гц, 1H), 7,06 (т, J=8,0 Гц, 1H), 6,61 (д, J=8,0 Гц, 1H), 4,25 (м, 2H), 4,10 (м, 2H), 3,92 (с, 2H), 3,69 (м, 1H), 3,59 (м, 2H), 2,10 (м, 2H), 1,64 (м, 2H), 1,29 (т, 3H).
Масс-спектр (ESI, m/z): вычислено: 370,16, обнаружено: 370,41.
Пример 4: Получение 2-5-[7-(тетрагидропиран-4-иламино)-1H-индол-2-ил]-[1,2,4]оксадиазол-3-ил}этанола

Соединение, полученное синтезом 30 (20 мг, 0,05 ммоль), растворяли в тетрагидрофуране (2 мл), и добавляли боргидрид лития (2,4 мг, 0,10 ммоль). Смесь перемешивали в течение 2 часов при комнатной температуре, и добавляли насыщенный раствор хлорида аммония. Смесь экстрагировали с помощью этилацетата. Экстракт сушили над безводным сульфатом магния и фильтровали. Фильтрат отгоняли при пониженном давлении и очищали с помощью колоночной хроматографии с получением названного соединения (4,7 мг, выход 27%).
1H-ЯМР (500 МГц, CDCl3); δ 10,40 (ушир.с, 1H), 7,27 (с, 1H), 7,08 (д, J=8,0 Гц, 1H), 7,04 (т, J=8,0 Гц, 1H), 6,55 (д, J=7,4 Гц, 1H), 4,11 (м, 4H), 3,70 (м, 1H), 3,61 (т, 2H), 3,06 (м, 2H), 2,14 (м, 2H), 1,66 (м, 2H).
Масс-спектр (ESI, a/z): вычислено: 328,15, обнаружено: 328,37.
Синтез 31: Получение (R)-2-(7-амино-1H-индол-2-ил)-4,5-дигидротиазол-4-илметилового эфира 2,2-диметилпропионовой кислоты
(Стадия 1)
Соединение 7-нитроиндолкарбоновой кислоты, полученное на стадии 1 синтеза 28 (8,2 г, 22,7 ммоль), и соединение амина, полученное синтезом 27 (13,2 г, 27,2 ммоль), растворяли в диметилформамиде (100 мл), и добавляли EDC (6,6 г, 25,0 ммоль) и HOBT (4,6 г, 25,0 ммоль). Смесь перемешивали в течение 8 часов при комнатной температуре, и добавляли насыщенный раствор бикарбоната натрия. Смесь экстрагировали с помощью этилацетата, сушили над безводным сульфатом магния и фильтровали. Фильтрат отгоняли при пониженном давлении и концентрировали. Остаток очищали с помощью колоночной хроматографии с получением 2,2-диметилпропионовой кислоты (R)-3-(4-метоксибензилсульфанил)-2-[(7-нитро-1H-индол-2-карбонил)аминопропилового эфира (8,1 г, выход 71%).
1H-ЯМР (400 МГц, CDCl3); δ 10,47 (ушир.с, 1H), 8,27 (д,J=8,0 Гц, 1H), 8,01 (д, J=8,0 Гц, 1H), 7,26 (м, 2H), 6,93 (д, J=4,0 Гц, 1H), 6,83 (м, 2H), 6,74 (д, J=8,0 Гц, 1H), 4,56 (м, 1H), 4,44 (м, 1H), 4,24 (м, 1H), 3,74 (м, 5H), 2,77 (м, 1H), 2,62 (м, 1H), 1,18 (с, 9H).
(Стадия 2)
Соединение, полученное на стадии 1 (1,6 г, 3,2 ммоль), растворяли в дихлорметане (50 мл). Добавляли пентахлорид фосфора (1,3 г, 6,4 ммоль), и смесь перемешивали в течение 5 часов при комнатной температуре. После завершения реакции добавляли насыщенный раствор бикарбоната натрия. Смесь экстрагировали с помощью этилацетата, сушили над безводным сульфатом магния и фильтровали. Фильтрат отгоняли при пониженном давлении и концентрировали. Остаток очищали с помощью колоночной хроматографии с получением 2,2-диметилпропионовой кислоты (R)-2-(7-нитро-1H-индол-2-ил)-4,5-дигидротиазол-4-илметилового эфира (0,8 г, выход 69%).
1H-ЯМР (400 МГц, CDCl3); δ 10,53 (ушир.с, 1H), 8,26 (д, J=8,0 Гц, 1H), 7,99 (д, J=8,0 Гц, 1H), 7,04 (д, J=2,0 Гц, 1H), 6,90 (д, J=7,6 Гц, 1H), 4,78 (м, 1H), 4,46 (м, 1H), 4,30 (м, 1H), 3,59 (м, 1H), 3,36 (м, 1H), 1,20 (с, 9H).
(Стадия 3)
Соединение, полученное на стадии 2 (2,7 г, 7,5 ммоль), растворяли в смеси растворителей тетрагидрофурана, метанола и воды (1:1:1, 150 мл). Добавляли железную пыль (4,2 г, 74,7 ммоль) и хлорид аммония (4,0 г, 74,7 ммоль), и смесь перемешивали при помощи механической мешалки в течение 30 минут при 60°С. После завершения реакции добавляли воду. Смесь экстрагировали с помощью этилацетата, сушили над безводным сульфатом магния и фильтровали. Фильтрат отгоняли при пониженном давлении и очищали с помощью колоночной хроматографии с получением названного соединения (2,0 г, выход 81%).
1H-ЯМР (400 МГц, CDCl3); δ 9,86 (ушир.с, 1H), 7,30 (д, J=7,6 Гц, 1H), 7,14 (д, J=8,0 Гц, 1H), 6,89 (д, J=2,0 Гц, 1H), 6,61 (дд, J=0,8, 7,2 Гц, 1H), 4,96 (м, 1H), 4,36 (м, 2H), 3,55 (м, 1H), 3,33 (м, 1H), 1,18 (с, 9H).
Синтез 32: Получение (R)-2-(7-циклопентиламино-1H-индол-2-ил)-4,5-дигидротиазол-4-илметилового эфира 2,2-диметилпропионовой кислоты
Соединение, полученное синтезом 31 (2,0 г), подвергали взаимодействию в соответствии с таким же методом, как и в примере 1, с получением названного соединения (1,3 г, выход 54%).
Пример 5: Получение [(R)-2-(7-циклопентиламино-1H-индол-2-ил)-4,5-дигидро-1,3-тиазол-4-ил]метанола

Соединение, полученное синтезом 32 (1,3 г, 3,3 ммоль), растворяли в тетрагидрофуране (10 мл), метаноле (10 мл) и воде (10 мл). Добавляли гидрат гидроксида лития (0,4 г, 9,8 ммоль). Смесь перемешивали в течение 4 часов при комнатной температуре, отгоняли при пониженном давлении и концентрировали. К остатку добавляли 1н хлористоводородную кислоту. Смесь экстрагировали с помощью этилацетата, сушили над безводным сульфатом магния и фильтровали. Фильтрат отгоняли при пониженном давлении и очищали с помощью колоночной хроматографии с получением названного соединения (820 мг, выход 80%).
1H-ЯМР (500 МГц, CDCl3); δ 11,17-11,08 (м, 1H), 7,09 (м, 1H), 6,99 (т, 1H), 6,96 (с, 1H), 6,52 (м, 1H), 4,72 (м, 1H), 4,04 (м, 1H), 3,75 (м, 1H), 3,65 (м, 1H), 3,51 (м, 1H), 3,40 (м, 1H), 1,90 (м, 2H), 1,60-1,49 (м, 4H), 1,41-1,24 (м, 2H).
Масс-спектр (ESI, b/z): вычислено: 315,14, обнаружено: 315,44.
Синтез 33: Получение (R)-2-(7-циклопентиламино-1H-индол-2-ил)-4,5-дигидротиазол-4-илметилового эфира метансульфоновой кислоты
Соединение, полученное в примере 5 (820 мг, 2,6 ммоль), растворяли в дихлорметане (50 мл). Добавляли метансульфонил-хлорид (0,24 мл, 3,1 ммоль) и триэтиламин (0,81 мл, 3,1 ммоль), которые затем перемешивали в течение 30 минут при 0°С. После завершения реакции добавляли насыщенный раствор бикарбоната натрия. Смесь экстрагировали с помощью этилацетата, сушили над безводным сульфатом магния и фильтровали. Фильтрат отгоняли при пониженном давлении и очищали с помощью колоночной хроматографии с получением названного соединения (600 мг, выход 60%).
Пример 6: Получение циклопентил-[2-((R)-4-пирролидин-1-илметил-4,5-дигидротиазол-2-ил)-1H-индол-7-ил]амина
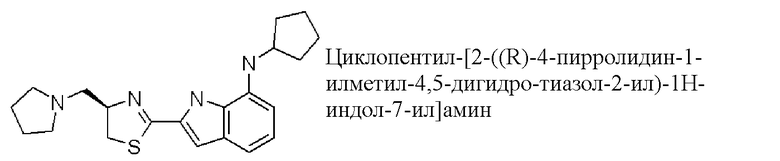
Соединение, полученное синтезом 33 (150 мг, 0,38 ммоль), растворяли в N,N-диметилформамиде (5 мл). Добавляли пирролидин (0,08 мл, 1,1 ммоль) и перемешивали в течение 4 часов при 70°С. После завершения реакции добавляли воду. Смесь экстрагировали с помощью этилацетата, сушили над безводным сульфатом магния и фильтровали. Фильтрат отгоняли при пониженном давлении и очищали с помощью колоночной хроматографии с получением названного соединения (20 мг, выход 14%).
1H-ЯМР (400 МГц, DMSO-d6); δ 11,37 (ушир.с, 1H), 6,83 (м, 1H),6,75 (д, J=2,0 Гц, 1H), 6,29 (д, J=8,0 Гц, 1H), 5,86 (д, J=8,0 Гц, 1H), 4,80 (м, 1H), 3,87 (м, 1H), 3,52 (м, 1H), 3,43 (м, 1H), 3,33 (м, 2H), 2,78 (м, 2H), 2,61 (м, 2H), 1,99 (м, 2H), 1,72 (м, 6H), 1,60 (м, 4H).
Пример 7: Получение {(R)-2-[7-(тетрагидропиран-4-иламино)-1H-индол-2-ил]-4,5-дигидротиазол-4-ил}метанола

(Стадия 1)
Соединение, полученное синтезом 31 (900 мг, 2,7 ммоль), растворяли в 1,2-дихлорэтане (100 мл). Добавляли тетрагидро-4H-пиран-4-он (0,8 мл, 8,13 ммоль), триацетоксиборгидрид натрия (1,72 г, 8,13 ммоль) и уксусную кислоту (0,47 мл, 8,13 ммоль), и перемешивали в течение 48 часов при комнатной температуре. После завершения реакции смесь разбавляли дихлорметаном, промывали с помощью насыщенного раствора бикарбоната натрия, сушили над безводным сульфатом магния и фильтровали. Фильтрат отгоняли при пониженном давлении и очищали с помощью колоночной хроматографии с получением (R)-2-[7-(тетрагидро-пиран-4-иламино)-1H-индол-2-ил]-4,5-дигидротиазол-4-илметилового эфира 2,2-диметилпропионовой кислоты.
1H-ЯМР (400 МГц, CDCl3); δ 10,91 (ушир.с, 1H), 7,01-6,91 (м, 3H), 6,48 (д, J=7,2 Гц, 1H), 4,86 (м, 1H), 4,34 (м, 2H), 4,00 (м, 2H), 3,61 (м, 1H), 3,54 (м, 3H), 3,31 (м, 1H), 2,05 (м, 2H), 1,55 (м, 2H), 1,16 (с, 9H).
(Стадия 2)
Соединение, полученное на стадии 1, растворяли в метаноле (32 мл), тетрагидрофуране (32 мл) и воде (16 мл). Добавляли 1н гидроксид натрия (7 мл) и перемешивали в течение 4 часов при комнатной температуре. После завершения реакции смесь отгоняли при пониженном давлении, экстрагировали с помощью дихлорметана, промывали насыщенным раствором хлорида натрия, сушили над безводным сульфатом магния и фильтровали. Фильтрат отгоняли при пониженном давлении и очищали с помощью колоночной хроматографии с получением названного соединения (700 мг, выход 78%).
1H-ЯМР (500 МГц, CDCl3); δ 11,04-10,95 (м, 1H), 7,11 (м, 1H), 6,99 (т, 1H), 6,96 (с, 1H), 6,52 (м, 1H), 4,74 (м, 1H), 4,02 (м, 1H), 3,92 (м, 2H), 3,68 (м, 1H), 3,46-3,30 (м, 5H), 1,91 (м, 2H), 1,28 (м, 2H).
Масс-спектр (ESI, m/z): вычислено: 331,14, обнаружено: 331,44.
Пример 8: Получение [(R)-2-(7-циклопентиламино-5-фтор-1H-индол-2-ил)-4,5-дигидротиазол-4-ил]метанола

Этил 5-фтор-7-нитро-1H-индол-2-карбоксилат, полученный в синтезе 2 (3,0 г, 11,9 ммоль), подвергали взаимодействию в соответствии с таким же методом, как и в примере 5, с получением названного соединения (600 мг, выход 15%).
1H-ЯМР (400 МГц, CDCl3); δ 10,73 (ушир.с, 1H), 6,91 (с, 1H), 6,72 (м, 1H), 6,33 (м, 1H), 4,78 (м, 1H), 4,12 (м, 1H), 3,97 (ушир.с, 1H), 3,79 (м, 1H), 3,75 (м, 1H), 3,49 (м, 2H), 2,01 (м, 2H), 1,62 (м, 4H), 1,41 (м, 2H).
Пример 9: Получение {(R)-2-[5-фтор-7-(тетрагидропиран-4-иламино)-1H-индол-2-ил]-4,5-дигидротиазол-4-ил}метанола

Этил 5-фтор-7-нитро-1H-индол-2-карбоксилат, полученный в синтезе 2 (3,0 г, 11,9 ммоль), подвергали взаимодействию в соответствии с таким же методом, как и в примере 7, с получением названного соединения (750 мг, выход 18%).
1H-ЯМР (400 МГц, CDCl3); δ 10,45 (ушир.с, 1H), 6,90 (с, 1H), 6,75 (м, 1H), 6,34 (м, 1H), 4,82 (м, 1H), 4,12 (м, 1H), 4,01 (м, 2H), 3,94 (м, 1H), 3,78 (м, 1H), 3,54-3,43 (м, 5H), 2,03 (м, 2H), 1,50 (м, 2H).
Пример 10: Получение {(R)-2-[5-(пиридин-3-илокси)-7-(тетрагидропиран-4-иламино)-1H-индол-2-ил]-4,5-дигидротиазол-4-ил}-метанола

Этиловый эфир 7-нитро-5-(пиридин-3-илокси)-1H-индол-2-карбоновой кислоты, полученный в синтезе 11 (500 мг, 1,5 ммоль), подвергали взаимодействию в соответствии с таким же способом, как и в примере 7, с получением названного соединения (40 мг, выход 6%).
1H-ЯМР (400 МГц, CDCl3); δ 10,96 (ушир.с, 1H), 8,36 (д, J=2,4 Гц, 1H), 8,26 (м, 1H), 7,27 (м, 1H), 7,19 (м, 1H), 6,83 (с, 1H), 6,63 (д, J=1,6 Гц, 1H), 6,24 (д, J=1,6 Гц, 1H), 4,81 (м, 1H), 4,01-3,94 (м, 3H), 3,75 (м, 1H), 3,47 (с, 3H), 3,48-3,29 (м, 5H), 1,93 (м, 2H), 1,52 (м, 2H).
Синтез 34: Получение 5-хлор-7-нитро-1H-индол-2-карбоновой кислоты
Соединение, полученное синтезом 5 (15,0 г, 59,1 ммоль), растворяли в тетрагидрофуране (300 мл) и метаноле (100 мл). Растворяли в воде (100 мл) гидроксид лития (7,43 г, 177 ммоль) и добавляли к реакционному раствору, который затем перемешивали в течение 3 часов при комнатной температуре. После завершения реакции удаляли путем отгонки при пониженном давлении тетрагидрофуран и метанол. Остаток нейтрализовали до значения pH около 6 при помощи 3н раствора хлористоводородной кислоты. Полученное твердое вещество фильтровали и сушили с получением названного соединения (13,1 г, выход 92%).
Синтез 35: Получение метилового эфира [(R)-2-(7-амино-5-хлор-1H-индол-2-ил)-4,5-дигидротиазол-4-ил] уксусной кислоты
(Стадия 1)
Соединение, полученное синтезом 34 (12,5 г, 52,0 ммоль), и соединение, полученное синтезом 22 (19,1 г, 62,4 ммоль), растворяли в N,N-диметилформамиде (200 мл). Добавляли триэтиламин (8,7 мл, 62,4 ммоль), HOBT (14,0 г, 104 ммоль) и EDC (16,9 г, 88,4 ммоль) и перемешивали в течение 4 часов при комнатной температуре. После завершения реакции смесь концентрировали. Остаток экстрагировали с помощью этилацетата и промывали насыщенным водным раствором гидрокарбоната натрия и насыщенным водным раствором хлорида аммония, соответственно. Органический слой концентрировали, и остаток очищали с помощью колоночной хроматографии с получением метилового эфира (R)-3-[(5-хлор-7-нитро-1H-индол-2-карбонил)амино]-4-(4-метоксибензилсульфанил)масляной кислоты (20,2 г, 41,0 ммоль, выход 79%).
1H-ЯМР (500 МГц, CDCl3); δ 10,47 (ушир.с, 1H), 8,24 (д, J=1,9 Гц, 1H), 7,96 (д, J=1,9 Гц, 1H), 7,24 (д, J=8,6 Гц, 2H), 6,89 (с, 1H), 6,81 (д, J=8,6 Гц, 2H), 4,58 (м, 1H), 3,75 (с, 3H), 3,73 (с, 2H), 3,71 (с, 3H), 2,86 (м, 1H), 2,80 (м, 1H), 2,73 (м, 1H), 2,70 (м, 1H).
(Стадия 2)
Соединение, полученное на стадии 1, растворяли в дихлорметане (200 мл). Добавляли пентахлорид фосфора (17,1 г, 82 ммоль) и перемешивали в течение 1 часа при комнатной температуре. После завершения реакции смесь концентрировали, и к остатку добавляли диэтиловый эфир (200 мл). Полученное твердое вещество фильтровали и сушили с получением метилового эфира [(R)-2-(5-хлор-7-нитро-1H-индол-2-ил)-4,5-дигидротиазол-4-ил]уксусной кислоты.
1H-ЯМР (500 МГц, CDCl3); δ 10,48 (ушир.с, 1H), 8,22 (д, J=1,8 Гц, 1H), 7,94 (д, J=1,8 Гц, 1H), 7,29 (д, J=8,6 Гц, 2H), 6,96 (д, J=2,5 Гц, 1H), 6,89 (д, J=8,6 Гц, 2H), 5,00 (м, 1H), 3,76 (с, 3H), 3,71 (м, 1H), 3,26 (м, 1H), 2,99 (м, 1H), 2,67 (м, 1H).
(Стадия 3)
Соединение, полученное на стадии 2, растворяли в тетрагидрофуране (200 мл), метаноле (200 мл) и воде (200 мл). Добавляли железную пыль (22,9 г, 410 ммоль) и хлорид аммония (21,9 г, 410 ммоль), и перемешивали при помощи механической мешалки в течение 1 часа при 60°С. После завершения реакции добавляли тетрагидрофуран (300 мл). Смесь фильтровали через целит, промывали с помощью тетрагидрофурана (100 мл), отгоняли при пониженном давлении и концентрировали. Остаток экстрагировали с помощью этилацетата, промывали насыщенным раствором хлорида натрия, сушили над безводным сульфатом магния и фильтровали. Фильтрат отгоняли при пониженном давлении и очищали с помощью колоночной хроматографии с получением названного соединения (9,0 г, выход 68%).
Синтез 36: Получение метилового эфира [(R)-2-(5-хлор-7-циклопентиламино-1H-индол-2-ил)-4,5-дигидротиазол-4-ил]уксусной кислоты
Соединение, полученное синтезом 35 (4,9 г, 15,1 ммоль), растворяли в дихлорэтане (100 мл). Добавляли циклопентанон (2,7 мл, 30,3 ммоль), ледяную уксусную кислоту (0,86 мл, 15,1 ммоль) и триацетоксиборгидрид натрия (6,42 г, 30,3 ммоль), которые затем перемешивали в течение 36 часов при комнатной температуре. После завершения реакции реакционная смесь промывали с помощью насыщенного раствора гидрокарбоната натрия (200 мл) и концентрировали. Остаток очищали с помощью колоночной хроматографии с получением названного соединения (5,15 г, выход 87%).
1H ЯМР (DMSO-d6, м.д.); δ 11,51 (с, 1H), 6,79 (с, 1H), 6,79 (с, 1H), 6,16 (с, 1H), 6,13 (д, 1H), 4,85 (м, 1H), 3,80 (м, 1H), 3,62 (м, 1H), 3,58 (с, 3H), 3,19 (м, 1H), 2,71 (м, 1H), 2,63 (м, 1H), 1,93 (м, 2H), 1,69 (м, 2H), 1,56 (м, 4H).
FAB MS(m/e) = 392.
Пример 11: Получение [(R)-2-(5-хлор-7-циклопентиламино-1H-индол-2-ил)-4,5-дигидротиазол-4-ил]уксусной кислоты

Соединение, полученное синтезом 36 (1,5 г, 3,83 ммоль), растворяли в тетрагидрофуране (100 мл) и метаноле (50 мл). Растворяли в воде (50 мл) моногидрат гидроксида лития (640 мг, 15,3 ммоль) и добавляли к реакционному раствору, который затем перемешивали в течение 4 часов при комнатной температуре. После завершения реакции удаляли тетрагидрофуран и метанол путем отгонки при пониженном давлении. Добавляли 1н раствор хлористоводородной кислоты. Смесь экстрагировали с помощью этилацетата, промывали насыщенным раствором хлорида натрия, сушили над безводным сульфатом магния и фильтровали. Фильтрат отгоняли при пониженном давлении и очищали с помощью колоночной хроматографии с получением названного соединения (13,1 г, выход 92%).
1H ЯМР (DMSO-d6, м.д.); δ 12,51 (ушир.с, 1H), 11,51 (с, 1H), 6,79 (с, 1H), 6,79 (с, 1H), 6,16 (с, 1H), 6,14 (д, 1H), 4,87 (м,1H), 3,80 (м, 1H), 3,61 (м, 1H), 3,19 (м, 1H), 2,72 (м, 1H), 2,64 (м, 1H), 1,93 (м, 2H), 1,69 (м, 2H), 1,56 (м, 4H).
FAB MS(m/e) = 378.
Пример 12: Получение этилового эфира [(R)-2-(5-хлор-7-циклопентиламино-1H-индол-2-ил)-4,5-дигидротиазол-4-ил]уксусной кислоты
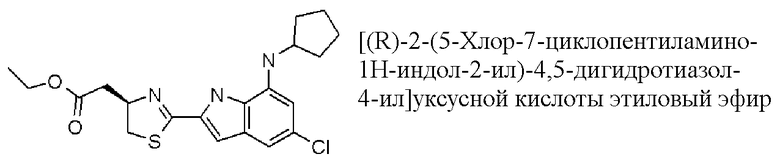
Соединение, полученное синтезом 5 (5,0 г, 19,7 ммоль), и соединение, полученное синтезом 23 (6,3 г, 19,7 ммоль), подвергали взаимодействию в соответствии с такими же методами, как и в синтезах 34-36, с получением названного соединения (840 мг, выход 11%).
Пример 13: Получение 2-{(R)-2-[5-хлор-7-(тетрагидропиран-4-иламино)-1H-индол-2-ил]-4,5-дигидротиазол-4-ил}этанола

(Стадия 1)
Соединение, полученное синтезом 35 (4,0 г, 12,4 ммоль), подвергали взаимодействию в соответствии с таким же методом, как и на стадии 1 примера 7 с получением соединения тетрагидропиран-4-иламина (4,1 г, 10,0 ммоль, выход; 81%).
1H-ЯМР (DMSO-d6, м.д.); δ 11,52 (1H, с), 6,81 (с, 1H), 6,71 (с, 1H), 6,28 (с, 1H), 6,07 (д, 1H), 4,90 (м, 1H), 3,86 (м, 2H), 3,64 (с, 3H), 3,62 (м, 2H), 3,44 (т, 2H), 2,82-2,71 (м, 2H), 1,94 (м, 2H), 1,40 (м, 2H).
FAB MS(m/e) = 408.
(Стадия 2)
Соединение, полученное на стадии 1 (2,5 г, 6,12 ммоль), подвергали взаимодействию в соответствии с таким же методом, как в примере 4, с получением названного соединения (2,19 г, 5,76 ммоль, выход 94%).
1H ЯМР (DMSO-d6, м.д.); δ 11,48 (ушир.с, 1H), 6,81 (с, 1H), 6,68 (с, 1H), 6,28 (с, 1H), 6,05 (д, 1H), 4,66 (кв, 1H), 4,54 (т, 1H), 3,87 (м, 2H), 3,61-3,54 (м, 3H), 3,44 (т, 2H), 3,15 (м, 1H), 1,99-1,93 (м, 3H), 1,73 (м, 1H), 1,40 (м, 2H), 1,20 (м, 1H).
FAB MS(m/e) = 380.
Синтез 37: Получение {5-хлор-2-[(R)-4-(2-йодэтил)-4,5-дигидротиазол-2-ил]-1H-индол-7-ил}-(тетрагидропиран-4-ил)амина
Соединение, полученное в примере 13 (3,7 г, 10,2 ммоль), растворяли в тетрагидрофуране (100 мл). Добавляли имидазол (2,1 г, 30,6 ммоль), трифенилфосфин (4,0 г, 15,3 ммоль) и йод (3,9 г, 15,3 ммоль) и перемешивали в течение 8 часов при 0°С ~ комнатной температуре. После завершения реакции добавляли этилацетат (100 мл), и реакционную смесь промывали водой (100 мл ×2). Органический слой концентрировали, и остаток очищали с помощью колоночной хроматографии с получением названного соединения (2,0 г, 4,07 ммоль, выход 40%).
Пример 14: Получение 1-[4-(2-{(R)-2-[5-хлор-7-(тетрагидропиран-4-иламино)-1H-индол-2-ил]-4,5-дигидротиазол-4-ил}этил)-пиперазин-1-ил]-2-гидроксиэтанона

(Стадия 1)
Соединение, полученное синтезом 37 (100 мг, 0,2 ммоль), и 1-трет-бутоксикарбонилпиперазин (270 мг, 1,4 ммоль) растворяли в N,N-диметилформамиде (20 мл). Добавляли карбонат калия (200 мг, 1,4 ммоль) и перемешивали в течение 4 часов при комнатной температуре. После завершения реакции добавляли воду. Смесь экстрагировали с помощью этилацетата, сушили над безводным сульфатом магния и фильтровали. Фильтрат отгоняли при пониженном давлении и концентрировали с получением трет-бутилового эфира 4-(2-{(R)-2-[5-хлор-7-(тетрагидропиран-4-иламино)-1H-индол-2-ил]-4,5-дигидротиазол-4-ил}этил)пиперазин-1-карбоновой кислоты.
(Стадия 2)
Соединение, полученное и сконцентрированное на стадии 1, растворяли в дихлорметане (10 мл). Добавляли по каплям 4н раствор хлористоводородной кислоты (0,5 мл) и перемешивали в течение 2 часов при комнатной температуре. После завершения реакции реакционную смесь концентрировали путем отгонки при пониженном давлении. Концентрат растворяли в N,N-диметилформамиде (5 мл). Добавляли гликолевую кислоту (15,1 мг, 0,2 ммоль), триэтиламин (28 мкл, 0,2 ммоль), EDC (45 мг, 0,23 ммоль) и HOBT (40 мг, 0,29 ммоль) и перемешивали в течение 8 часов при комнатной температуре. Добавляли 1н раствор хлористоводородной кислоты. Смесь экстрагировали с помощью этилацетата, сушили над безводным сульфатом магния и фильтровали. Фильтрат отгоняли при пониженном давлении и очищали с помощью колоночной хроматографии с получением названного соединения (5,1 мг, выход 5%).
1H-ЯМР (500 МГц, DMSO-d6); δ 11,48 (ушир.с, 1H), 6,81 (с, 1H),6,69 (д, J=1,8 Гц, 1H), 6,29 (с, 1H), 6,05 (д, J=7,4 Гц, 1H), 4,62 (м, 1H), 4,49 (т, 1H), 4,04 (м, 2H), 3,87 (м, 2H), 3,56 (м, 1H), 3,45 (м, 4H), 3,29 (м, 4H), 3,16 (м, 1H), 2,36 (м, 4H), 1,96 (м, 3H), 1,80 (м, 1H), 1,40 (м, 2H).
Пример 15: Получение 1-(2-{(R)-2-[5-хлор-7-(тетрагидропиран-4-иламино)-1H-индол-2-ил]-4,5-дигидротиазол-4-ил}этил)пирролидин-3-ола
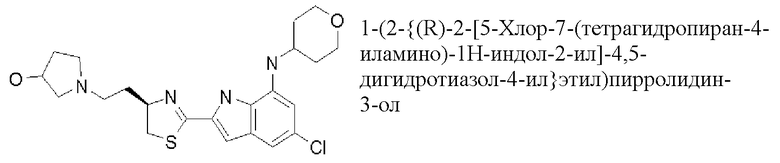
Соединение, полученное в синтезе 37 (100 мг, 0,2 ммоль), и 3-пирролидинол (0,35 мл, 4,2 ммоль) растворяли в N,N-диметилформамиде (20 мл). Добавляли карбонат калия (580 мг, 4,2 ммоль) и перемешивали в течение 4 часов при комнатной температуре. После завершения реакции добавляли воду. Реакционную смесь экстрагировали с помощью этилацетата, сушили над безводным сульфатом магния и фильтровали. Фильтрат отгоняли при пониженном давлении и концентрировали. Остаток очищали с помощью колоночной хроматографии с получением названного соединения (2 мг, выход 2%).
Пример 16: Получение [(R)-2-(5-бром-7-циклопентиламино-1H-индол-2-ил)-4,5-дигидротиазол-4-ил]уксусной кислоты

Метил 5-бром-7-нитро-1H-индол-2-карбоксилат, полученный в синтезе 6 (1,1 г, 3,7 ммоль) подвергали взаимодействию в соответствии с таким же методом, как и в примере 11 с получением названного соединения (250 мг, выход 16%).
1H-ЯМР (400 МГц, CDCl3); δ 12,50 (ушир.с, 1H), 7,10 (см 1H), 7,06 (с, 1H), 6,56 (с, 1H), 5,31 (м, 1H), 3,89 (м, 2H), 3,40 (м, 1H), 2,99 (м, 1H), 2,83 (м, 1H), 2,08 (м, 2H), 1,86 (м, 2H), 1,66 (м, 4H).
Пример 17: Получение [(R)-2-(7-циклопентиламино-5-этокси-1H-индол-2-ил)-4,5-дигидротиазол-4-ил]уксусной кислоты
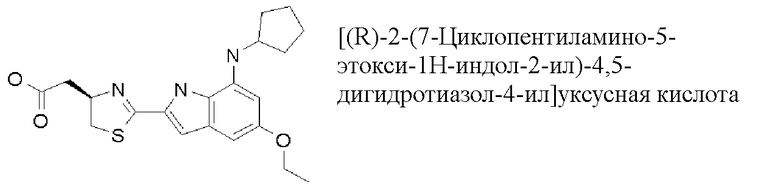
Метил 5-этокси-7-нитро-1H-индол-2-карбоксилат, полученный в синтезе 9 (1,5 г, 5,7 ммоль), подвергали взаимодействию в соответствии с таким же методом, как в примере 11, с получением названного соединения (150 мг, выход 7%).
1H-ЯМР (400 МГц, DMSO-d6); δ 11,24 (ушир.с, 1H), 6,65 (д, J=2,0 Гц, 1H), 6,26 (д, J=2,0 Гц, 1H), 5,92 (д, J=6,0 Гц, 1H), 5,88 (д, J=2,0 Гц, 1H), 4,89 (м, 1H), 3,94 (кв, 2H), 3,81 (м, 1H), 3,65 (м, 1H), 3,20 (м, 1H), 2,74 (м, 1H), 2,62 (м, 1H), 1,94 (м, 2H), 1,72 (м, 2H), 1,61 (м, 4H), 1,31 (т, 3H).
Пример 18: Получение [(S)-2-(7-циклопентиламино-5-этокси-1H-индол-2-ил)-4,5-дигидротиазол-4-ил]уксусной кислоты
(Стадия 1)
D-Цистеин (5,0 г, 31,7 ммоль) использовали вместо L-цистеина в методике синтеза 22 с получением метилового эфира (S)-3-амино-4-(4-метоксибензилсульфанил)масляной кислоты (1,2 г, выход 12%).
(Стадия 2)
5-Этокси-7-нитро-1H-индол-2-карбоксилат, полученный в синтезе 9 (1,0 г, 3,8 ммоль), и соединение, полученное на стадии 1, подвергали взаимодействию в соответствии с такими же методами, как в синтезах 34-36 и примере 11, с получением названного соединения (27 мг, выход 2%).
Пример 19: Получение [2-(7-циклопентиламино-5-фенокси-1H-индол-2-ил)-4,5-дигидротиазол-4-ил]уксусной кислоты

(Стадия 1)
Этиловый эфир 3-трет-бутоксикарбониламино-4-гидроксимасляной кислоты (15,0 г, 60,7 ммоль) использовали вместо этилового эфира (R)-4-третбутоксикарбониламино-5-гидроксивалериановой кислоты в методе синтеза 24 с получением этилового эфира 3-амино-4-(4-метоксибензилсульфанил)масляной кислоты (12,0 г, выход 61%).
(Стадия 2)
Соединение стадии 1 и метиловый эфир 7-нитро-5-фенокси-1H-индол-2-карбоновой кислоты синтеза 10 (2,0 г, 6,7 ммоль) подвергали взаимодействию в соответствии с такими же методами, как в синтезах 34-36 и примере 11 с получением названного соединения (500 мг, выход 17%).
Пример 20: Получение [(R)-2-(7-циклопентиламино-5-фенокси-1H-индол-2-ил)-4,5-дигидротиазол-4-ил]уксусной кислоты
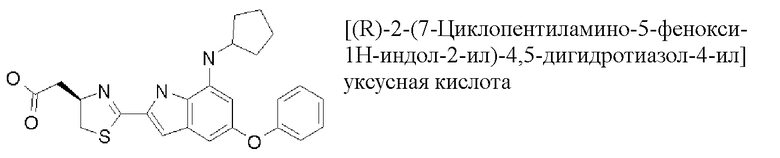
Метиловый эфир 7-нитро-5-фенокси-1H-индол-2-карбоновой кислоты синтеза 10 (101,5 г, 264,7 ммоль) и гидрохлорид этилового эфира (R)-3-амино-4-(4-метоксибензилсульфанил)масляной кислоты синтеза 23 подвергали взаимодействию в соответствии с такими же методами, как в синтезах 34-36 и примере 11, с получением названного соединения (51,0 г, выход 44%).
1H-ЯМР (400 МГц, CDCl3); δ 11,92 (ушир.с, 1H), 7,28 (м, 2H), 7,00 (м, 4H), 6,56 (с, 1H), 6,22 (с, 1H), 5,34 (ушир.с, 1H), 3,81 (ушир.с, 1H), 3,70 (м, 1H), 3,22 (д, J=12,0 Гц, 1H), 2,76-2,62 (м, 2H), 1,96 (м, 2H), 1,73 (м, 2H), 1,58 (м, 4H).
Пример 21: Получение [(S)-2-(7-циклопентиламино-5-фенокси-1H-индол-2-ил)-4,5-дигидротиазол-4-ил]уксусной кислоты
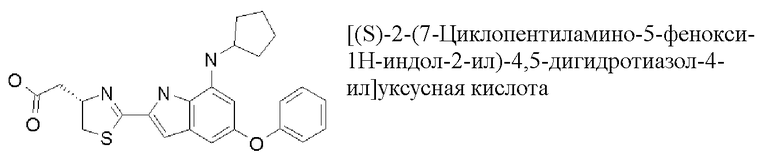
Метиловый эфир 7-нитро-5-фенокси-1H-индол-2-карбоновой кислоты синтеза 10 (55,5 г, 185,9 ммоль) и изопропиловый эфир (S)-3-амино-4-(4-метоксибензилсульфанил)масляной кислоты синтеза 25 подвергали взаимодействию в соответствии с такими же методами, как в синтезах 34-36 и примере 11 с получением названного соединения (21,0 г, выход 26%).
Синтез 38: Получение этилового эфира 3-[(R)-2-(7-амино-5-хлор-1H-индол-2-ил)-4,5-дигидротиазол-4-ил]пропионовой кислоты
Соединение кислоты, полученное в синтезе 34 (2,0 г, 8,3 ммоль), и гидрохлорид этилового эфира (R)-4-амино-5-(4-метоксибензилсульфанил)валериановой кислоты, полученный в синтезе 24 (3,4 г, 10,2 ммоль), подвергали взаимодействию в соответствии с такими же методами, как в примере 35, с получением названного соединения (0,76 г, выход 26%).
1H-ЯМР (400 МГц, CDCl3); δ 10,00 (ушир.с, 1H), 7,08 (с, 1H), 6,80 (с, 1H), 6,57 (с, 1H), 4,71 (м, 1H), 4,07 (м, 2H), 3,88 (ушир.с, 2H), 3,55 (м, 1H), 3,11 (м, 1H), 2,50 (т, 2H), 2,05 (м, 2H), 1,22 (т, 3H).
Пример 22: Получение этилового эфира 3-[(R)-2-(5-хлор-7-циклопентиламино-1H-индол-2-ил)-4,5-дигидротиазол-4-ил]пропионовой кислоты.
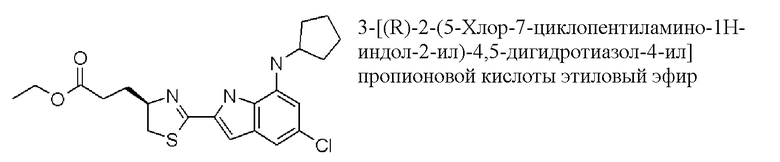
Соединение, полученное в синтезе 38 (760 мг, 2,1 ммоль), подвергали взаимодействию в соответствии с такими же методами, как в примере 1, с получением 450 мг названного соединения (выход 51%).
Пример 23: Получение 3-[(R)-2-(5-Хлор-7-циклопентиламино-1H-индол-2-ил)-4,5-дигидротиазол-4-ил]пропионовой кислоты
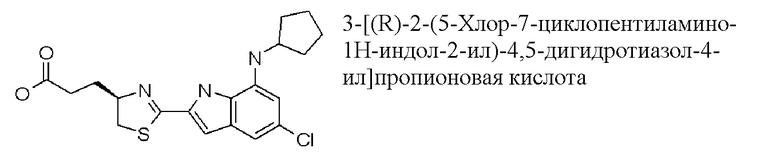
Соединение сложного эфира, полученное в примере 22 (500 мг, 1,2 ммоль), подвергали взаимодействию в соответствии с такими же методами, как в примере 11, с получением 400 мг названного соединения (выход 85%).
1H-ЯМР (400 МГц, DMSO-d6, Na соль); δ 11,69 (ушир.с, 1H), 6,82 (д, J=4,0 Гц, 1H), 6,68 (с, 1H), 6,27 (с, 1H), 6,18 (с, 1H), 4,63 (м, 1H), 3,83 (м, 1H), 3,50 (м, 1H), 3,13 (м, 1H), 2,08-1,96 (м, 6H), 1,72 (м, 2H), 1,58 (м, 4H).
Синтез 39: Получение 2-пиридин-2-ил-1H-индол-7-иламина
Соединение, полученное в синтезе 14 (1,0 г, 4,2 ммоль), подвергали взаимодействию в соответствии с такими же методами, как на стадии 3 синтеза 31, с получением 800 мг названного соединения (выход 92%).
Пример 24: Получение циклопентил(2-пиридин-2-ил-1H-индол-7-ил)амина

Соединение, полученное в синтезе 39 (150 мг, 0,7 ммоль), подвергали взаимодействию в соответствии с такими же методами, как в примере 1, с получением 55 мг названного соединения (выход 28%).
1H-ЯМР (400МГц, CDCl3); δ 10,85 (ушир.с, 1H), 8,48 (м, 1H), 7,87 (д, J=8,0 Гц, 1H), 7,74 (м, 1H), 7,12 (м, 1H), 7,08 (д, J=7,6 Гц, 1H), 7,00 (с, 1H), 6,98 (м, 1H), 6,43 (д, J=7,2 Гц, 1H), 3,81 (м, 1H), 1,89 (м, 2H), 1,49 (м, 4H), 1,26 (м, 2H).
Синтез 40: Получение 2-пиразин-2-ил-1H-индол-7-иламина
Соединение, полученное в синтезе 15 (500 мг, 2,1 ммоль), подвергали взаимодействию в соответствии с такими же методами, как на стадии 3 синтеза 31, с получением 430 мг названного соединения (выход 98%).
Пример 25: Получение циклопентил-(2-пиразин-2-ил-1H-индол-7-ил)амина

Соединение, полученное в синтезе 40 (80 мг, 0,38 ммоль), подвергали взаимодействию в соответствии с такими же методами, как в примере 1, с получением 33 мг названного соединения (выход 31%).
1H-ЯМР (400 МГц, CDCl3); δ 9,72 (ушир.с, 1H), 9,08 (д, J=1,2 Гц, 1H), 8,41 (м, 1H), 8,37 (м, 1H), 7,12 (м, 2H), 7,05 (м, 1H), 6,55 (д, J=7,6 Гц, 1H), 3,94 (м, 1H), 2,04 (м, 2H), 1,70-1,48 (м, 6H).
Пример 26: Получение (2-пиразин-2-ил-1H-индол-7-ил)-(тетрагидропиран-4-ил)амина

Соединение, полученное в синтезе 40 (80 мг, 0,38 ммоль), подвергали взаимодействию в соответствии с такими же методами, как на стадия 1 примера 7, с получением 35 мг названного соединения (выход 31%).
1H-ЯМР (400 МГц, CDCl3 & MeOH-d4); δ 9,00 (ушир.с, 1H), 8,51 (м, 1H), 8,35 (д, J=2,4 Гц, 1H), 7,13 (с, 1H), 7,10 (д, J=8,0 Гц, 1H), 7,00 (м, 1H), 6,52 (д, J=7,6 Гц, 1H), 4,05 (м, 2H), 3,68 (м, 1H), 3,61 (м, 2H),2,17 (м, 2H), 1,63 (м, 2H).
Синтез 41: Получение амида 7-нитро-1H-индол-2-тиокарбоновой кислоты
Соединение 7-нитроиндол-2-карбоновой кислоты, полученное на стадии 1 синтеза 28 (2,0 г, 9,7 ммоль) растворяли в дихлорметане (100 мл). Добавляли 2 мл (29,1 ммоль) ционилхлорида и смесь перемешивали в течение 1 часа при 60°C. В конце реакции смесь подвергали отгонке при пониженном давлении с целью концентрирования. Полученное соединение растворяли в 100 мл тетрагидрофурана и добавляли 5,0 г (14,6 ммоль) реагента Лавессона. Смесь перемешивали в течение 3 часов при 80°C. В конце реакции, смесь концентрировали. Полученное твердое вещество промывали при помощи 100 мл хлорметана с получением 1,5 г названного соединения (выход 71%).
1H-ЯМР (500 МГц, DMSO-d6); δ 10,92 (ушир.с, 1H), 10,00 (ушир.с, 1H), 9,99 (ушир.с, 1H), 8,22 (м, 2H), 7,50 (д, J=1,9 Гц, 1H), 7,30 (т, 1H).
Синтез 42: Получение 7-нитро-2-тиазол-2-ил-1H-индола
1,1 г (5,0 ммоль) соединения, полученного в синтезе 41, растворяли в 20 мл этанола и 1 мл N,N-диметилформамида. Добавляли 3,7 мл (25 ммоль) диэтилацеталя бромацетальдегида, и смесь перемешивали в течение 8 часов при 100°C. В конце реакции, добавляли воду и экстрагировали при помощи этилацетата. Экстракт сушили над безводным сульфатом магния и фильтровали. Фильтрат подвергали отгонке при пониженном давлении с целью концентрирования. Полученное твердое вещество промывали при помощи диэтилового эфира с получением 800 мг названного соединения (выход 67%).
Синтез 43: Получение 2-тиазол-2-ил-1H-индол-7-иламина
Соединение, полученное в синтезе 42 (800 мг, 3,3 ммоль), подвергали взаимодействию в соответствии с такими же методами, как на стадии 3 синтеза 31, с получением 650 мг названного соединения (выход 93%).
1H-ЯМР (500 МГц, DMSO-d6); δ 10,79 (ушир.с, 1H), 9,43 (ушир.с, 1H), 9,29 (ушир.с, 1H), 7,04 (д, J=2,5 Гц, 1H), 6,77-6,71 (м, 2H), 6,33 (м, 1H), 5,50 (ушир.с, 2H).
Пример 27: Получение циклопентил-(2-тиазол-2-ил-1H-индол-7-ил)амина

Соединение, полученное в синтезе 43 (30 мг, 3,3 ммоль), подвергали взаимодействию в соответствии с такими же методами, как в примере 1, с получением 20 мг названного соединения (выход 51%).
1H-ЯМР (400 МГц, CDCl3); δ 9,37 (ушир.с, 1H), 7,36 (ушир.с, 1H), 7,20 (ушир.с, 1H), 7,06-7,00 (м, 2H), 6,86 (д, J=2,0 Гц, 1H), 6,55 (м, 2H), 3,95 (м, 1H), 2,08 (м, 2H), 1,79 (м, 2H), 1,65 (м, 4H).
Синтез 44: Получение 5-метил-7-нитро-1H-индол-2-карбоновой кислоты
6,5 г (27,8 ммоль) соединения, полученного в синтезе 7, растворяли в 200 мл раствора смеси 1:1:1 метанола, тетрагидрофурана и воды. Добавляли к нему 3,5 г (83,3 ммоль) гидрата гидроксида лития. После перемешивания в течение 8 часов при комнатной температуре, добавляли 1н раствор HCl, и смесь экстрагировали с помощью этилацетата. Экстракт сушили над безводным сульфатом магния и фильтровали. Фильтрат подвергали отгонке при пониженном давлении с получением 5,7 г названного соединения (выход 94%).
1H-ЯМР (500 МГц, CDCl3); δ 10,26 (ушир.с, 1H), 8,17 (с, 1H), 7,87 (с, 1H), 7,38 (с, 1H), 2,59 (с, 3H).
Синтез 45: Получение этилового эфира (R)-3-(4-метоксибензилсульфанил)-2-[(5-метил-7-нитро-1H-индол-2-карбонил)амино]пропионовой кислоты
Соединение, полученное в синтезе 44 (3 г, 13,6 ммоль), и гидрохлорид этилового эфира (R)-2-амино-3-(4-метоксибензилсульфанил)пропионовой кислоты, полученный в синтезе 26 (5,8 г, 19,1 ммоль), растворяли в 100 мл диметилформамида. Добавляли к ним триэтиламин (1,9 г, 19,1 ммоль), EDC (4,4 г, 23,2 ммоль) и HOBT (3,7 г, 27,3 ммоль). После перемешивания в течение 8 часов при комнатной температуре добавляли 1н раствор HCl, и смесь экстрагировали с помощью этилацетата. Экстракт сушили над безводным сульфатом магния и фильтровали. Фильтрат подвергали отгонке при пониженном давлении и очищали при помощи колоночной хроматографии с получением 4,1 г названного соединения (выход 64%).
1H-ЯМР (400 МГц, CDCl3); δ 10,06 (ушир.с, 1H), 7,95 (с, 1H), 7,64 (с, 1H), 7,51 (д, J=8,0 Гц, 1H), 7,20 (д, J=8,0 Гц, 1H), 6,87 (с, 1H), 6,86 (д, J=4,0 Гц, 2H), 5,05 (м, 1H), 4,30 (м, 2H), 3,72 (с, 5H), 3,04 (м, 2H), 2,44 (с, 3H), 1,30 (т, 3H).
Синтез 46: Получение этилового эфира (R)-2-(7-амино-5-метил-1H-индол-2-ил)-4,5-дигидротиазол-4-карбоновой кислоты
(Стадия 1)
Соединение, полученное в синтезе 45 (4,1 г, 8,7 ммоль), растворяли в 200 мл дихлорметана, добавляли к нему 3,6 г (17,4 ммоль) пентахлорида фосфора. Смесь перемешивали в течение 4 часов при комнатной температуре. Добавляли насыщенный водный раствор бикарбоната натрия, и смесь экстрагировали с помощью этилацетата. Экстракт сушили при помощи безводного сульфата магния, фильтровали. Фильтрат подвергали отгонке при пониженном давлении с получением этилового эфира (R)-2-(5-метил-7-нитро-1H-индол-2-ил)-4,5-дигидротиазол-4-карбоновой кислоты.
(Стадия 2)
Соединение, полученное на стадии 1, растворяли в 300 мл смеси 1:1:1 воды, тетрагидрофурана и метанола. К смеси добавляли 4,6 г (86,9 ммоль) хлорида аммония и 4,9 г (86,9 ммоль) железа, и перемешивали в течение 30 минут при 60°C. Смесь фильтровали через целит и отгоняли при пониженном давлении. К концентрату добавляли насыщенный водный раствор бикарбоната натрия и экстрагировали с помощью этилацетата. Экстракт сушили при помощи безводного сульфата магния, фильтровали. Фильтрат очищали при помощи колоночной хроматографии с получением 400 мг названного соединения (выход 15%).
Синтез 47: Получение этилового эфира (R)-2-(7-циклопентиламино-5-метил-1H-индол-2-ил)-4,5-дигидротиазол-4-карбоновой кислоты
Соединение, полученное в синтезе 46 (4,0 г, 0,91 ммоль), подвергали взаимодействию в соответствии с такими же методами, как в примере 1, с получением 590 мг названного соединения (выход 12%).
1H-ЯМР (400 МГц, CDCl3); δ 9,74 (ушир.с, 1H), 6,85 (с, 1H), 6,84 (с, 1H), 6,37 (с, 1H), 5,32 (т, 1H), 4,24 (м, 2H), 3,88 (м, 1H), 3,71 (м, 2H), 2,39 (с, 3H), 2,03 (м, 2H), 1,67 (м, 2H), 1,61 (м, 2H), 1,47 (м, 2H).
Пример 28: Получение этилового эфира 2-(7-циклопентиламино-5-метил-1H-индол-2-ил)тиазол-4-карбоновой кислоты

Соединение, полученное в синтезе 47 (560 мг, 1,51 ммоль), растворяли в 20 мл дихлорметана. Добавляли бромтрихлорметан (330 мг, 1,7 ммоль) и DBU (250 мг, 1,7 ммоль). Смесь перемешивали в течение 2 часов при 0°C. В конце реакции добавляли насыщенный водный раствор бикарбоната натрия и экстрагировали с помощью этилацетата. Экстракт сушили при помощи безводного сульфата магния, фильтровали. Фильтрат подвергали отгонке при пониженном давлении и очищали при помощи колоночной хроматографии с получением 450 мг названного соединения (выход 81%).
1H-ЯМР (400 МГц, CDCl3); δ 9,36 (ушир.с, 1H), 8,07 (с, 1H), 6,91 (д, J=4,0 Гц, 1H), 6,84 (с, 1H), 6,37 (с, 1H), 4,37 (кв, 2H), 3,95 (м, 1H), 2,40 (с, 3H), 2,03 (м, 2H), 1,75 (м, 2H), 1,65 (м, 2H), 1,55 (м, 2H), 1,33 (т, 3H).
Пример 29: Получение 2-(7-циклопентиламино-5-метил-1H-индол-2-ил)тиазол-4-карбоновой кислоты

Соединение, полученное в примере 28 (100 мг, 0,27 ммоль), подвергали взаимодействию в соответствии с такими же методами, как в синтезе 44 с получением 70 мг названного соединения (выход 76%).
1H-ЯМР (400 МГц, DMSO-d6, Na соль); δ 13,20 (ушир.с, 1H), 7,89 (с, 1H), 6,81 (с, 1H), 6,65 (ушир.с, 1H), 6,59 (с, 1H), 6,13 (с, 1H), 3,87 (м, 1H), 2,31 (с, 3H), 2,01 (м, 2H), 1,75 (м, 4H), 1,55 (м, 2H).
Пример 30: Получение [2-(7-циклопентиламино-5-метил-1H-индол-2-ил)тиазол-4-ил]метанола

Соединение, полученное в пример 28 (300 мг, 0,81 ммоль), подвергали взаимодействию в соответствии с такими же методами, как в примере 4, с получением 200 мг названного соединения (выход 75%).
1H-ЯМР (400 МГц, CDCl3); δ 10,42 (ушир.с, 1H), 7,14 (с, 1H), 6,88 (д, J=4,0 Гц, 1H), 6,86 (с, 1H), 4,70 (с, 2H), 3,81 (м, 1H), 2,39 (с, 3H), 1,96 (м, 2H), 1,61 (м, 4H), 1,36 (м, 2H).
Синтез 48: Получение амида 5-метил-7-нитро-1H-индол-2-тиокарбоновой кислоты
Соединение карбоновой кислоты, полученное в синтезе 44 (3,2 г, 1,4 ммоль), подвергали взаимодействию в соответствии с такими же методами, как в синтезе 41 с получением 2,4 г названного соединения (выход 75%).
1H-ЯМР (400 МГц, DMSO-d6); δ 10,83 (ушир.с, 1H), 10,00 (ушир.с, 1H), 9,87 (ушир.с, 1H), 8,10 (с, 1H), 8,04 (с, 1H), 7,45 (с, 1H), 2,50 (с, 3H).
Синтез 49: Получение 2-{5-метил-7-нитро-1H-индол-2-ил)тиазол-5-карбальдегида
Соединение, полученное в синтезе 48 (2,5 г, 11,3 ммоль), растворяли в 50 мл N,N-диметилформамида. К раствору добавляли 2,56 г (17 ммоль) 2-броммалональдегида и перемешивали в течение 6 часов при 100°C. В конце реакции реакционную смесь добавляли в ледяную воду. Полученное твердое вещество собирали и сушили с получением 2,45 г названного соединения (выход 77%).
1H-ЯМР (400 МГц, DMSO-d6); δ 11,70 (ушир.с, 1H), 10,11 (с, 1H), 8,82 (с, 1H), 8,10 (с, 1H), 8,00 (с, 1H), 7,56 (с, 1H), 2,50 (с, 3H).
Синтез 50: Получение [2-(7-амино-5-метил-1H-индол-2-ил)-тиазол-5-ил]метанола
Соединение, полученное в синтезе 49 (1,75 г, 6,1 ммоль), растворяли в 300 мл смеси 1:1:1 воды, тетрагидрофурана и метанола. К смеси добавляли 3,26 г (60,9 ммоль) хлорида аммония и 3,4 г (60,9 ммоль) железа и перемешивали в течение 30 минут при 60°C. Смесь фильтровали через целит и отгоняли при пониженном давлении. К концентрату добавляли насыщенный водный раствор бикарбоната натрия и экстрагировали с помощью этилацетата. Экстракт сушили при помощи безводного сульфата магния, фильтровали. Фильтрат подвергали отгонке при пониженном давлении и очищали при помощи колоночной хроматографии с получением 300 мг названного соединения (выход 19%).
1H-ЯМР (400 МГц, DMSO-d6); δ 11,27 (ушир.с, 1H), 7,69 (с, 1H), 6,76 (д, J=4,0 Гц, 1H), 6,20 (с, 1H), 5,60 (т, 1H), 5,26 (ушир.с, 2H), 4,71 (д, J=8,0 Гц, 2H), 2,23 (с, 3H).
Пример 31: Получение [2-(7-циклопентиламино-5-метил-1H-индол-2-ил)тиазол-5-ил]метанола
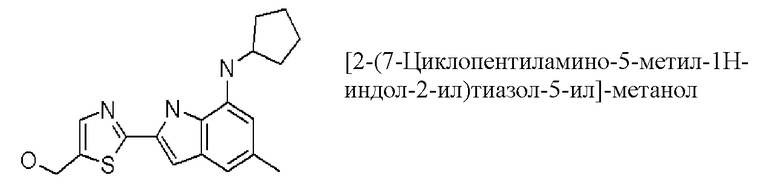
Соединение, полученное в синтезе 50 (300 мг, 1,16 ммоль), подвергали взаимодействию в соответствии с такими же методами, как в примере 1, с получением 380 мг названного соединения (выход 100%).
1H-ЯМР (400 МГц, DMSO-d6); δ 11,39 (ушир.с, 1H), 7,70 (с, 1H), 6,78 (д, J=4,0 Гц, 1H), 6,60 (с, 1H), 6,11 (с, 1H), 5,72 (д, J=8,0 Гц, 1H), 5,62 (т, 1H), 4,72 (д, J=4,0 Гц, 2H), 3,88 (м, 1H), 2,30 (с, 3H), 2,00 (м, 2H), 1,74 (м, 2H), 1,60 (м, 4H).
Синтез 51: Получение гидразида 5-метил-7-нитро-1H-индол-2-карбоновой кислоты
Соединение, полученное в синтезе 7 (5,0 г, 21,3 ммоль), растворяли в 100 мл этанола. Добавляли к нему 2 мл (40 ммоль) гидрата гидразина, и смесь перемешивали в течение 4 часов при 100°С. В конце реакции раствор охлаждали и осаждали твердое вещество. Твердое вещество собирали и промывали с помощью дихлорметана с получением 2,34 г названного соединения (выход 47%).
1H-ЯМР (400 МГц, DMSO-d6); δ 11,08 (ушир.с, 1H), 10,21 (ушир.с, 1H), 8,05 (с, 1H), 7,98 (с, 1H), 7,27 (с, 1H), 4,62 (ушир.с, 2H), 2,48 (с, 3H).
Синтез 52: Получение 5-метил-7-нитро-2-[1,3,4]оксадиазол-2-ил-1H-индола
2,3 г (9,8 ммоль) соединения, полученного в синтезе 51, объединяли с 100 мл триэтилортоформиата, и смесь перемешивали в течение 8 часов при 120°C. В конце реакции добавляли воду и экстрагировали с помощью этилацетата. Экстракт сушили над безводным сульфатом магния, фильтровали. Фильтрат подвергали отгонке при пониженном давлении с получением 600 мг названного соединения (выход 25%).
1H-ЯМР (400 МГц, DMSO-d6); δ 12,12 (ушир.с, 1H), 9,46 (с, 1H), 8,11 (с, 1H), 8,02 (с, 1H), 7,45 (с, 1H), 3,32 (с, 3H).
Пример 32: Получение циклопентил-(5-метил-2-[1,3,4]оксадиазол-2-ил-1H-индол-7-ил)амина
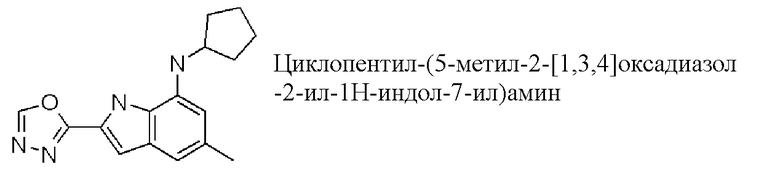
Соединение, полученное в синтезе 52 (300 мг, 1,2 ммоль), подвергали взаимодействию в соответствии с такими же методами, как в синтезе 43 и примере 1, с получением 50 мг названного соединения (выход 14%).
1H-ЯМР (400 МГц, CDCl3); δ 9,97 (ушир.с, 1H), 8,43 (с, 1H), 7,10 (д, J=4,0 Гц, 1H), 6,90 (с, 1H), 6,44 (с, 1H), 4,14 (м, 1H), 3,97 (м, 1H), 2,42 (с, 3H), 2,08 (м, 2H), 1,74 (м, 2H), 1,64 (м, 2H), 1,55 (м, 2H).
Синтез 53: Получение 5-метил-2-пиридин-2-ил-1H-индол-7-иламина
Соединение, полученное в синтезе 12 (300 мг, 1,2 ммоль), растворяли в 100 мл смеси 1:1 метанола и этилацетата. Добавляли 40 мг 10% Pd/C, и смесь перемешивали в течение 1 часа в токе газообразного водорода. В конце реакции смесь фильтровали через целит и отгоняли при пониженном давлении. Дистиллят отделяли с помощью колоночной хроматографии с получением 170 мг названного соединения (выход 64%).
1H-ЯМР (400 МГц, CDCl3); δ 9,67 (ушир.с, 1H), 8,52 (м, 1H), 7,75 (м, 1H), 7,69 (м, 1H), 7,13 (м, 1H), 6,89 (д, J=2,4 Гц, 1H), 6,89 (д, J=2,4 Гц, 1H), 6,60 (д, J=2,0 Гц, 1H), 6,27 (д, J=2,0 Гц, 1H), 4,04 (м, 2H), 1,40 (м, 3H).
Пример 33: Получение циклопентил-(5-метил-2-пиридин-2-ил-1H-индол-7-ил)амина

Соединение, полученное в синтезе 53 (50 г, 0,22 моль), подвергали взаимодействию в соответствии с такими же методами, как в примере 1, с получением 20 мг названного соединения (выход 46%).
1H-ЯМР (400МГц, CDCl3); δ 9,73 (ушир.с, 1H), 8,53 (д, J-4,8 Гц, 1H), 7,78 (д, J=8,0 Гц, 1H), 7,70 (м, 1H), 7,13 (м, 1H), 6,91 (д, J=2,0 Гц, 1H), 6,87 (с, 1H), 6,32 (с, 1H), 3,91 (м, 1H), 3,60 (ушир.с, 1H), 2,41 (с, 3H), 2,00 (м, 2H), 1,65 (м, 4H), 1,51 (м, 2H).
Пример 34: Получение (5-метил-2-пиридин-2-ил-1H-индол-7-ил)-(тетрагидропиран-4-ил)амина

Соединение, полученное в синтезе 53 (50 мг, 0,22 ммоль), подвергали взаимодействию в соответствии с такими же методами, как на стадии 1 примера 7, с получением 25 мг названного соединения (выход 36%).
1H-ЯМР (400 МГц, CDCl3); δ 10,65 (ушир.с, 1H), 8,49 (д, J=4,8 Гц, 1H), 7,81 (д, J=8,0 Гц, 1H) 7,72 (м, 1H), 7,13 (м, 1H), 6,92 (д, J=2,0 Гц, 1H), 6,88 (с, 1H), 6,27 (с, 1H), 3,92 (м, 1H), 3,49 (м, 3H), 2,39 (с, 3H), 1,95 (м, 2H), 1,30 (м, 2H).
Пример 35: Получение циклогексил-(5-метил-2-пиридин-2-ил-1H-индол-7-ил)амина
Соединение, полученное в синтезе 53 (50 мг, 0,22 ммоль), подвергали взаимодействию с получением 25 мг названного соединения (выход 36%), в соответствии с такими же методами, как в примере 1, за исключением того, что циклогексанон использовали вместо циклопентанона.
1H-ЯМР (500 МГц, CDCl3); δ 10,46 (ушир.с, 1H), 8,48 (д, J=4,9 Гц, 1H), 7,79 (д, J=8,0 Гц, 1H), 7,70 (м, 1H), 7,11 (м, 1H), 6,92 (д, J=1,8 Гц, 1H), 6,85 (с, 1H), 3,30 (м, 1H), 2,39 (с, 3H), 1,98 (м, 2H), 1,70 (м, 2H), 1,60 (м, 1H), 1,31 (м, 2H), 1,39 (м, 1H), 0,95 (м, 1H).
Пример 36: Получение 1-[4-(5-метил-2-пиридин-2-ил-1H-индол-7-иламино)пиперидин-1-ил]этанона
Соединение, полученное в синтезе 53 (30 мг, 0,13 ммоль), подвергали взаимодействию с получением 10 мг названного соединения (выход 21%), в соответствии с такими же методами, как в примере 1, за исключением того, что 1-ацетил-4-пиперидон использовали вместо циклопентанона.
1H-ЯМР (500 МГц, CDCl3 и MeOH-d4); δ 8,42 (д, J=4,9 Гц, 1H), 7,71 (д, J=8,0 Гц, 1H), 7,65 (м, 1H), 7,08 (м, 1H), 6,83 (с, 1H), 6,78 (с, 1H), 6,22 (с, 1H), 4,33 (м, 1H), 3,77 (м, 1H), 3,60 (м, 1H), 3,52 (с, 3H), 3,17 (м, 1H), 2,91 (м, 1H), 2,31 (с, 3H), 2,13 (м, 1H), 2,06 (м, 1H), 1,39 (м, 2H).
Пример 37: Получение (1-метил-пиперидин-4-ил)-(5-метил-2-пиридин-2-ил-1H-индол-7-ил)амина
Соединение, полученное в синтезе 53 (30 мг, 0,13 ммоль), подвергали взаимодействию с получением 17 мг названного соединения (выход 40%), в соответствии с такими же методами, как в примере 1, за исключением того, что 1-метил-4-пиперидон использовали вместо циклопентанона.
1H-ЯМР (400 МГц, CDCl3); δ 10,75 (ушир.с, 1H), 8,45 (д, J=4,8 Гц, 1H), 7,77 (д, J=8,0 Гц, 1H), 7,68 (м, 1H), 7,08 (м, 1H), 6,90 (д, J=2,0 Гц, 1H), 6,85 (с, 1H), 6,24 (с, 1H), 3,30 (м, 1H), 2,73 (м, 2H), 2,38 (с, 3H), 2,24 (с, 3H), 2,08 (м, 2H), 1,97 (м, 2H), 1,33 (м, 2H).
Пример 38: Получение 4-(5-метил-2-пиридин-2-ил-1H-индол-7-иламино)циклогексанона

В 10 мл ацетона и 5 мл воды растворяли соединение, полученное реакцией 30 мг (0,13 ммоль) соединения, полученного в синтезе 53, в соответствии с такими же методами, как в примере 1, за исключением того, что моноэтилен ацеталь 1,4-циклогександиона использовали вместо циклопентанона. К раствору добавляли 1 мл 1н раствора гидрохлорида, и перемешивали в течение 8 часов при 80°C. В конце реакции добавляли насыщенный раствор бикарбоната натрия и экстрагировали с помощью этилацетата. Экстракт сушили при помощи безводного сульфата магния, фильтровали, и отгоняли при пониженном давлении. Дистиллят очищали при помощи колоночной хроматографии с получением 10 мг названного соединения (выход 23%).
1H-ЯМР (400 МГц, CDCl3); δ 10,72 (ушир.с, 1H), 8,48 (д, J=4,4 Гц, 1H), 7,81 (д, J=8,0 Гц, 1H), 7,73 (м, 1H), 7,13 (м, 1H), 6,92 (д, J=2,0 Гц, 1H), 6,91 (с, 1H), 6,32 (с, 1H), 3,78 (м, 1H) 2,41 (с, 3H), 2,35 (м, 4H), 2,20 (м, 2H), 1,52 (м, 2H).
Пример 39: Получение (1-бензилпирролидин-3-ил)-(5-метил-2-пиридин-2-ил-1H-индол-7-ил)амина
Соединение, полученное в синтезе 53 (30 мг, 0,13 ммоль), подвергали взаимодействию с получением 10 мг названного соединения (выход 20%), в соответствии с такими же методами, как в примере 1, за исключением того, что 1-бензил-3-пирролидон использовали вместо циклопентанона.
1H-ЯМР (400 МГц, CDCl3); δ 10,72 (ушир.с, 1H), 8,49 (м, 1H), 7,76 (д, J=8,0 Гц, 1H), 7,66 (м, 1H), 7,31-7,21 (м, 5H), 7,08 (м, 1H), 6,89 (д, J=2,0 Гц, 1H), 6,83 (с, 1H), 6,22 (с, 1H), 4,09 (ушир.с, 1H), 3,61 (м, 2H), 2,79 (м, 1H), 2,64 (м, 1H), 2,44 (м, 2H), 2,38 (с, 3H), 2,25 (м, 1H), 1,61 (м, 1H).
Пример 40: Получение циклопентилметил-(5-метил-2-пиридин-2-ил-1H-индол-7-ил)амина

Соединение, полученное в синтезе 53 (30 мг, 0,13 ммоль), подвергали взаимодействию с получением 10 мг названного соединения (выход 20%), в соответствии с такими же методами, как в примере 1, за исключением того, что циклопентанкарбоксальдегид использовали вместо циклопентанона.
1H-ЯМР (500 МГц, CDCl3); δ 10,54 (ушир.с, 1H), 8,48 (м, 1H), 7,79 (д, J=8,0 Гц, 1H), 7,71 (м, 1H), 7,12 (м, 1H), 6,93 (д, J=2,5 Гц, 1H), 6,88 (с, 1H), 6,28 (с, 1H), 3,07 (д, J=7,3 Гц, 1H), 2,41 (с, 3H), 2,05 (м, 1H), 1,69-1,51 (м, 6H), 1,19 (м, 2H).
Пример 41: Получение N-(5-метил-2-пиридин-2-ил-1H-индол-7-ил)бензамида

Соединение, полученное в синтезе 53 (45 мг, 0,20 ммоль), растворяли в 10 мл хлорметана. Добавляли 0,2 мл триэтиламина и 0,03 мл (0,22 ммоль) бензоилхлорида, и смесь перемешивали при комнатной температуре в течение 2 часов. В конце реакции добавляли воду и экстрагировали с помощью этилацетата. Экстракт сушили над безводным сульфатом магния, фильтровали. Фильтрат подвергали отгонке при пониженном давлении и очищали при помощи колоночной хроматографии с получением 18 мг названного соединения (выход 27%).
1H-ЯМР (500 МГц, CDCl3); δ 10,98 (ушир.с, 1H), 8,61 (с, 1H), 8,50 (д, J=4,9 Гц, 1H), 7,90 (д, J=7,4 Гц, 2H), 7,77 (д, J=8,0 Гц, 1H), 7,69 (м, 1H), 7,50 (м, 1H), 7,43 (м, 2H), 7,31 (с, 1H), 7,26 (с, 1H), 7,14 (м, 1H), 6,95 (д, J=1,8 Гц, 1H), 2,41 (с, 3H).
Пример 42: Получение циклопентил-(5-метил-2-пиразин-2-ил-1H-индол-7-ил)амина
Соединение, полученное в синтезе 13 (100 мг, 0,39 ммоль), подвергали взаимодействию в соответствии с такими же методами, как в синтезе 53 и примере 1, с получением 25 мг названного соединения (выход 22%).
1H-ЯМР (400 МГц, CDCl3); δ 9,55 (ушир.с, 1H), 9,06 (с, 1H), 8,41 (д, J=4,0 Гц, 1H), 8,26 (с, 1H), 7,03 (с, 1H), 6,90 (с, 1H), 6,38 (с, 1H), 3,95 (м, 1H), 2,41 (с, 3H), 2,06 (м, 2H), 1,71-1,49 (м, 6H).
Пример 43: Получение циклопентил-(5-этокси-2-пиридин-2-ил-1H-индол-7-ил)амина
Соединение, полученное в синтезе 16 (20 мг, 0,07 ммоль), подвергали взаимодействию в соответствии с такими же методами, как в синтезе 53 и примере 1, с получением 4,5 мг названного соединения (выход 19%).
1H-ЯМР (400 МГц, CDCl3); δ 9,86 (ушир.с, 1H), 8,56 (д, J=4,8 Гц, 1H), 7,81 (д, J=8,0 Гц, 1H), 7,74 (м, 1H), 7,17 (м, 1H), 6,94 (д, J=2,0 Гц, 1H), 6,54 (д, J=2,0 Гц, 1H), 6,22 (д, J=2,0 Гц, 1H), 4,11 (кв, 2H), 3,91 (м, 1H), 2,05 (м, 2H), 1,76-1,63 (м, 4H), 1,54-1,46 (м, 5H).
Пример 44: Получение циклопентил-(5-фенокси-2-пиридин-2-ил-1H-индол-7-ил)амина
Соединение, полученное в синтезе 17 (250 мг, 0,75 ммоль), подвергали взаимодействию в соответствии с такими же методами, как в синтезе 53 и примере 1, с получением 120 мг названного соединения (выход 43%).
1H-ЯМР (400 МГц, CDCl3); δ 10,07 (ушир.с, 1H), 8,55 (д, J=4,0 Гц, 1H), 7,74 (м, 2H), 7,29 (м, 2H), 7,18 (м, 1H), 7,01 (м, 3H), 6,93 (д, J=4,0 Гц, 1H), 6,69 (д, J=4,0 Гц, 1H), 6,27 (с, 1H), 3,81 (м, 1H), 3,70 (ушир.с, 1H), 1,96 (м, 2H), 1,60 (м, 4H), 1,41 (м, 2H).
Пример 45: Получение циклопентил-(3,5-диметил-2-фенил-1H-индол-7-ил)амина

Соединение, полученное в синтезе 18 (100 мг, 0,38 ммоль), подвергали взаимодействию в соответствии с такими же методами, как в синтезе 53 и примере 1, с получением 35 мг названного соединения (выход 31%).
1H-ЯМР (400 МГц, CDCl3); δ 7,68 (ушир.с, 1H), 7,59 (м, 2H), 7,45 (м, 2H), 7,32 (м, 1H), 6,87 (с, 1H), 6,40 (с, 1H), 3,97 (м, 1H), 2,45 (с, 3H), 2,40 (с, 3H), 2,09 (м, 2H), 1,78-1,56 (м, 4H).
Пример 46: Получение циклопентил-(5-метил-2-фенил-1H-индол-7-ил)амина
Соединение, полученное в синтезе 19 (120 мг, 0,48 ммоль), подвергали взаимодействию в соответствии с такими же методами, как в синтезе 53 и примере 1 с получением 50 мг названного соединения (выход 36%).
1H-ЯМР (500 МГц, MeOH-d4) δ 7,74 (м, 2H), 7,38 (м, 2H), 7,24 (м, 1H), 6,68 (с, 1H), 6,62 (с, 1H), 6,23 (с, 1H), 3,95 (м, 1H), 2,06 (м, 2H), 1,80 (м, 2H), 1,65 (м, 4H).
Пример 47: Получение (2-циклогексил-5-метил-1H-индол-7-ил)-циклопентиламина
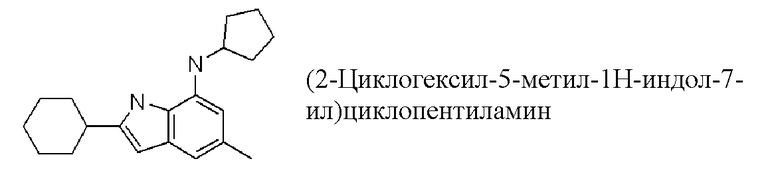
Соединение, полученное в синтезе 20 (50 мг, 0,19 ммоль), подвергали взаимодействию в соответствии с такими же методами, как в синтезе 53 и примере 1, с получением 21 мг названного соединения (выход 37%).
1H-ЯМР (400 МГц, CDCl3); δ 7,80 (ушир.с, 1H), 6,81 (с, 1H), 6,31 (с, 1H), 6,08 (с, 1H), 3,93 (м, 1H), 2,92 (м, 1H), 2,38 (с, 3H), 2,08 (м, 4H), 1,80-1,20 (м, 14H).
Пример 48: Получение циклопентил-[5-метил-2-(6-метилпиридин-2-ил)-1H-индол-7-ил]амина

Соединение, полученное в синтезе 21 (50 мг, 0,19 ммоль), подвергали взаимодействию в соответствии с такими же методами, как в синтезе 53 и примере 1, с получением 35 мг названного соединения (выход 40%).
1H-ЯМР (400 МГц, CDCl3); δ 11,11 (ушир.с, 1H), 7,59 (м, 2H), 6,94 (м, 1H), 6,88 (д, J=2,0 Гц, 1H), 6,82 (с, 1H), 6,19 (с, 1H), 3,73 (м, 1H), 2,37 (с, 3H), 2,31 (с, 3H), 1,80 (м, 2H), 1,43 (м, 4H), 1,14 (м, 2H).
Пример 49: Получение (5-метил-2-фенил-1H-индол-7-ил)-(тетрагидропиран-4-ил)амина

Соединение, полученное в синтезе 19 (70 мг, 0,28 ммоль), подвергали взаимодействию с получением 50 мг названного соединения (выход 57%), в соответствии с такими же методами, как в синтезе 53 и примере 1, за исключением того, что тетрагидро-4H-пиран-4-он использовали вместо циклопентанона.
1H-ЯМР (500 МГц, MeOH-d4); δ 7,74 (м, 2H), 7,39 (м, 2H), 7,24 (м, 1H), 6,71 (с, 1H), 6,63 (с, 1H), 6,28 (с, 1H), 4,01 (м, 1H), 3,68 (м, 1H), 3,58 (м, 2H), 2,32 (с, 3H), 2,12 (м, 2H), 1,56 (м, 2H).
Пример 50: Получение (5-метил-2-фенил-1H-индол-7-ил)-(1-метилпиперидин-4-ил)амина

Соединение, полученное в синтезе 19 (70 мг, 0,28 ммоль), подвергали взаимодействию с получением 24 мг названного соединения (выход 29%), в соответствии с такими же методами, как в синтезе 53 и примере 1, за исключением того, что 1-метил-4-пиперидон использовали вместо циклопентанона.
1H-ЯМР (500 МГц, MeOH-d4); δ 7,73 (м, 2H), 7,38 (м, 2H), 7,24 (м, 1H), 6,70 (с, 1H), 6,63 (с, 1H), 6,24 (с, 1H), 3,47 (м, 1H), 2,96 (м, 2H), 2,34 (с, 3H), 2,32 (с, 3H), 2,26 (м, 2H), 2,17 (м, 2H), 1,59 (м, 2H).
Пример 51: Получение 1-[4-(5-метил-2-фенил-1H-индол-7-иламино)пиперидин-1-ил]этанона
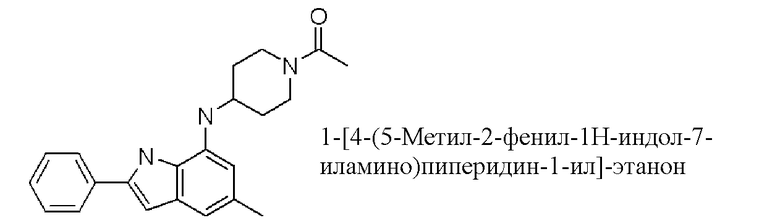
Соединение, полученное в синтезе 19 (96 мг, 0,38 ммоль), подвергали взаимодействию с получением 18 мг названного соединения (выход 13%), в соответствии с такими же методами, как в синтезе 53 и примере 1, за исключением того, что 1-ацетил-4-пиперидон использовали вместо циклопентанона.
1Н-ЯМР (500 МГц, DMSO-d6); δ 10,77 (ушир.с, 1H), 7,74 (м, 2H), 7,42 (м, 2H), 7,26 (м, 1H), 6,66 (д, J=1,85 Гц, 1H), 6,56 (с, 1H), 6,14 (с, 1H), 5,22 (д, J=7,95, 1H), 4,28 (м, 1H), 3,82 (м, 1H), 3,62 (м, 1H), 3,13 (м, 1H), 2,83 (м, 1H), 2,26 (с, 3H), 2,05 (м, 2H), 2,00 (с, 3H), 1,31 (м, 2H).
Пример 52: Получение (5-метил-2-фенил-1H-индол-7-ил)-пиперидин-4-ил)амина HCl

Соединение, полученное в синтезе 19 (140 мг, 0,56 ммоль), подвергали взаимодействию с получением 50 мг названного соединения (выход 21%), в соответствии с такими же методами, как в синтезе 53 и примере 3, за исключением того, что 1-ацетил-4-пиперидон использовали вместо циклопентанона.
Пример 53: Получение 2-гидрокси-1-[4-(5-метил-2-фенил-1H-индол-7-иламино)пиперидин-1-ил]этанона
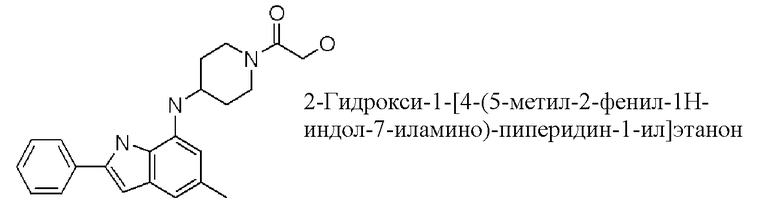
Соединение, полученное в Пример 52 (50 мг, 0,13 ммоль), растворяли в 3 мл N,N-диметилформамида. Добавляли гликолевую кислоту (15 мг, 0,20 ммоль), триэтиламин (0,06 мл, 0,43 ммоль), EDC (38 мг, 0,20 ммоль) и HOBT (27 мг, 0,20 ммоль), и смесь перемешивали при комнатной температуре в течение 8 часов. К реакционной смеси добавляли 1н раствор HCl и экстрагировали с помощью этилацетата. Экстракт сушили над безводным сульфатом магния, фильтровали. Фильтрат подвергали отгонке при пониженном давлении и очищали при помощи колоночной хроматографии с получением 20 мг названного соединения (выход 43%).
1H-ЯМР (500 МГц, MEOH-d4); δ 7,72 (м, 2H), 7,38 (м, 2H), 7,23 (м, 1H), 6,71 (2, 1H), 6,63 (с, 1H), 6,30 (с, 1H), 4,40 (м, 1H), 4,23 (м, 2H), 3,74 (м, 2H), 3,24 (м, 1H), 3,02 (м, 1H), 2,33 (с, 3H), 2,19 (м, 2H), 1,45 (м, 2H).
Пример 54: Получение (1-метансульфонилпиперидин-4-ил)-(5-метил-2-фенил-1H-индол-7-ил)амина
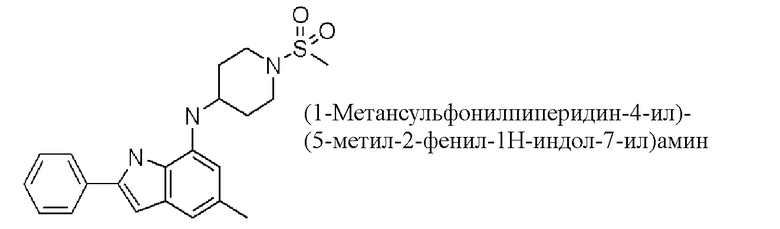
Соединение, полученное в примере 52, растворяли в 10 мл дихлорметана, и добавляли триэтиламин (0,02 мл, 0,14 ммоль), метансульфонилхлорид (10 мг, 0,09 ммоль). Смесь перемешивали при 0°C в течение 1 часа. К реакционной смеси добавляли 1н раствор HCl и экстрагировали с помощью этилацетата. Экстракт сушили над безводным сульфатом магния, фильтровали. Фильтрат подвергали отгонке при пониженном давлении и очищали при помощи колоночной хроматографии с получением 18 мг названного соединения (выход 67%).
1H-ЯМР (500 МГц, DMSO-d6); δ 10,79 (ушир.с, 1H), 7,74 (м, 2H), 7,42 (м, 2H), 7,26 (м, 1H), 6,66 (с, 1H), 6,57 (с, 1H), 6,11 (с, 1H), 5,27 (м, 1H), 3,57 (м, 2H), 3,13 (м, 1H), 2,93 (м, 1H), 2,87 (с, 3H), 2,25 (с, 3H), 2,14 (м, 2H), 1,46 (м, 2H).
Синтез 54: Получение этилового эфира 4-(5-метил-2-фенил-1H-индол-7-иламино)-циклогексанкарбоновой кислоты
Соединение, полученное в синтезе 19 (200 мг, 0,90 ммоль), подвергали взаимодействию с получением 170 мг названного соединения (выход 50%), в соответствии с такими же методами, как в синтезе 53 и примере 1, за исключением того, что этиловый эфир 4-оксо-циклогексанкарбоновой кислоты использовали вместо циклопентанона.
Пример 55: Получение 4-(5-метил-2-фенил-1H-индол-7-иламино)циклогексанкарбоновой кислоты

Соединение, полученное в синтезе 54 (170 мг, 0,45 ммоль), растворяли в смеси 5:1 тетрагидрофурана и метанола. Добавляли 0,4 мл 6н гидроксида натрия, и смесь перемешивали при комнатной температуре в течение 8 часов. В конце реакции реакционную смесь подвергали отгонке при пониженном давлении, разбавляли с помощью 1н раствора хлористоводородной кислоты и экстрагировали с помощью этилацетата. Экстракт сушили над безводным сульфатом магния, фильтровали. Фильтрат подвергали отгонке при пониженном давлении и очищали при помощи колоночной хроматографии с получением 120 мг названного соединения (выход 77%).
1H-ЯМР (500 МГц, DMSO-d6); δ 10,81 (ушир.с, 1H), 7,74 (м, 2H), 7,42 (м, 2H), 7,25 (м, 1H), 6,64 (д, J=2,45 Гц, 1H), 6,53 (с, 1H), 6,05 (с, 1H), 5,17 (м, 1H), 3,50 (м, 2H), 2,24 (с, 3H), 1,95 (м, 1H), 1,81 (м, 2H), 1,64 (м, 2H), 1,57 (м, 2H).
Пример 56: Получение (2-морфолин-4-илэтил)амида 4-(5-метил-2-фенил-1H-индол-7-иламино)циклогексанкарбоновой кислоты
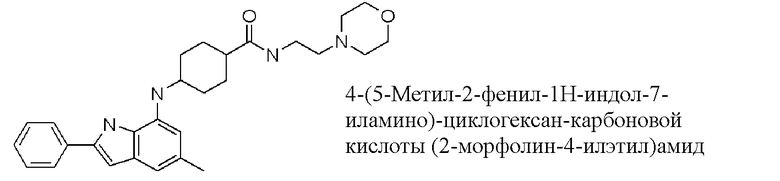
Соединение, полученное в Пример 55 (30 мг, 0,09 ммоль), растворяли в 5 мл N,N-диметилформамида. Добавляли 2-морфолин-4-илэтиламин (22 мг, 0,17 ммоль), EDC (25 мг, 0,19 ммоль) и HOBT (18 мг, 0,19 ммоль). Смесь перемешивали в течение 8 часов при комнатной температуре. Добавляли насыщенный 1н водный раствор хлорида натрия, и смесь экстрагировали с помощью этилацетата. Экстракт сушили над безводным сульфатом магния, фильтровали. Фильтрат подвергали отгонке при пониженном давлении и очищали при помощи колоночной хроматографии с получением 40 мг названного соединения (выход 100%).
1H-ЯМР (500 МГц, DMSO-d6); δ 10,91 (ушир.с, 1H), 7,75 (м, 2H), 7,65 (м, 1H), 7,41 (м, 2H), 7,26 (м, 1H), 6,64 (с, 1H), 6,53 (с, 1H), 6,02 (с, 1H), 5,20 (м, 1H), 3,57 (м, 2H), 3,51 (м, 5H), 3,14 (м, 2H), 2,29 (м, 4H), 2,24 (с, 3H), 1,87 (м, 2H), 1,78 (м, 2H), 1,68 (м, 2H), 1,53 (м, 2H).
Пример 57: Получение циклопентилметил-(5-метил-2-фенил-1H-индол-7-ил)амина
Соединение, полученное в синтезе 19 (70 мг, 0,28 ммоль), подвергали взаимодействию в соответствии с такими же методами, как в синтезе 53 и примере 1, за исключением того, что циклопентанкарбальдегид использовали вместо циклопентанона, с получением 36 мг названного соединения (выход 40%).
1H-ЯМР (500 МГц, MeOH-d4); δ 7,73 (м, 2H), 7,37 (м, 2H), 7,22 (м, 1H), 6,68 (2, 1H), 6,62 (с, 1H), 6,18 (с, 1H), 3,14 (д, J=7,35 Гц, 2H), 2,32 (с, 3H), 2,30 (м, 1H), 1,91 (м, 2H), 1,69 (м, 2H), 1,59 (м, 2H), 1,36 (м, 2H).
Пример 58: Получение (5-метил-2-фенил-1H-индол-7-ил)-(тетрагидропиран-4-илметил)амина
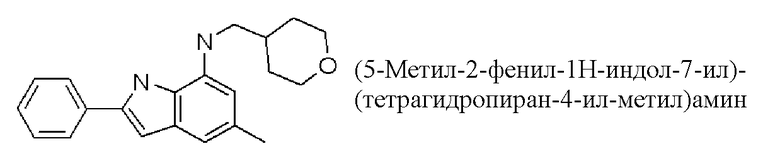
Соединение, полученное в синтезе 19 (70 мг, 0,28 ммоль), подвергали взаимодействию в соответствии с такими же методами, как в синтезе 53 и примере 1, за исключением того, что тетрагидропиран-4-карбальдегид использовали вместо циклопентанона, с получением 19 мг названного соединения (выход 21%).
1H-ЯМР (500 МГц, MeOH-d4); δ 7,73 (м, 2H), 7,38 (м, 2H), 7,23 (м, 1H), 6,68 (2, 1H), 6,63 (с, 1H), 6,19 (с, 1H), 3,97 (м, 2H), 3,44 (м, 2H), 3,16 (м, 2H), 2,32 (с, 3H), 1,99 (м, 1H), 1,83 (м, 2H), 1,40 (м, 2H).
Синтез 55: Получение 5-хлор-7-нитро-2-фенил-1H-индола
4-Хлор-2-нитроанилин (1,0 г, 3,67 ммоль) подвергали взаимодействию вместо 4-метил-2-нитроанилина в синтезе 19 с получением 750 мг названного соединения (выход 75%).
Пример 59: Получение (5-хлор-2-фенил-1H-индол-7-ил)-циклопентиламина
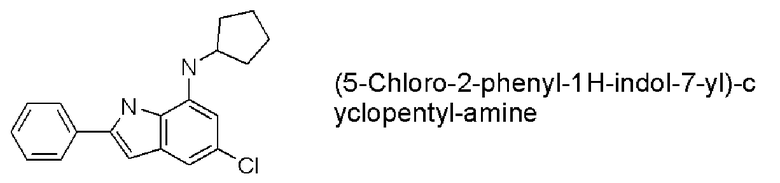
5-Хлор-7-нитро-2-фенил-1H-индол (225 мг, 0,83 ммоль) подвергали взаимодействию вместо 5-метил-7-нитро-2-фенил-1H-индолв в синтезе 46 с получением 230 мг названного соединения (выход 89%).
1H-ЯМР (500 МГц, DMSO-d6); δ 11,04 (ушир.с, 1H), 7,75 (м, 2H), 7,45 (м, 2H), 7,30 (м, 1H), 6,76 (д, J=1,8 Гц, 1H), 6,72 (д, J=2,45 Гц, 1H), 6,14 (д, J=1,8 Гц, 1H), 3,84 (м, 1H), 2,00 (м, 2H), 1,71 (м, 2H), 1,60 (м, 2H), 1,52 (м, 2H).
Пример 60: Получение (5-хлор-2-фенил-1H-индол-7-ил)-(тетрагидропиран-4-ил)амина
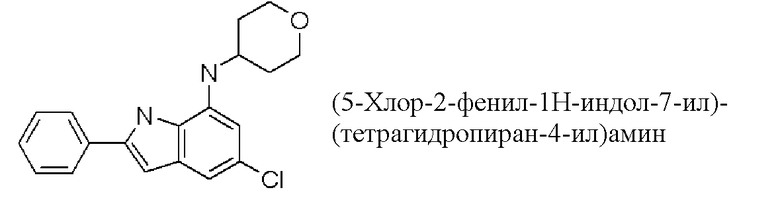
5-Хлор-7-нитро-2-фенил-1H-индол (337 мг, 1,23 ммоль) подвергали взаимодействию вместо 5-метил-7-нитро-2-фенил-1H-индола в синтезе 49 с получением 200 мг названного соединения (выход 50%).
1H-ЯМР (500 МГц, DMSO-d6); δ 11,09 (ушир.с, 1H), 7,74 (м, 2H), 7,43 (м, 2H), 7,29 (м, 1H), 6,77 (с, 1H), 6,72 (с, 1H), 6,24 (с, 1H), 5,60 (м, 1H), 4,28 (м, 1H), 3,86 (м, 2H), 3,43 (м, 2H), 1,96 (м, 2H), 1,41 (м, 2H).
Пример 61: Получение (5-хлор-2-фенил-1H-индол-7-ил)-(1-метил-пиперидин-4-ил)амина

5-Хлор-7-нитро-2-фенил-1H-индол (337 мг, 1,23 ммоль) подвергали взаимодействию вместо 5-метил-7-нитро-2-фенил-1H-индола в синтезе 50 с получением 288 мг названного соединения (выход 69%).
1H-ЯМР (500 МГц, DMSO-d6); δ 11,06 (ушир.с, 1H), 9,57 (ушир.с, 1H), 7,75 (м, 2H), 7,45 (м, 2H), 7,31 (м, 1H), 6,80 (с, 1H), 6,74 (с, 1H), 6,29 (с, 1H), 3,66 (м, 1H), 3,49 (м, 2H), 3,11 (м, 2H), 2,78 (с, 3H), 2,23 (м, 2H), 1,61 (м, 2H).
Пример 62: Получение (5-хлор-2-фенил-1H-индол-7-ил)-циклогексиламин

Соединение, полученное в синтезе 55 (170 мг, 0,62 ммоль), подвергали взаимодействию с получением 98 мг названного соединения (выход 49%), в соответствии с такими же методами, как в синтезе 53 и примере 1, за исключением того, что циклогексанон использовали вместо циклопентанона.
1H-ЯМР (500 МГц, CDCl3); δ 8,18 (ушир.с, 1H), 7,65 (м, 2H), 7,44 (м, 5H), 7,31 (м, 2H), 7,03 (с, 1H), 6,70 (с, 1H), 6,47 (д, 1H), 3,38 (м, 1H), 2,16 (м, 2H), 1,81 (м, 2H), 1,71 (м, 1H), 1,42 (м, 2H), 1,24 (м, 4H).
Пример 63: Получение (1-бензилпирролидин-3-ил)-(5-хлор-2-фенил-1H-индол-7-ил)амина
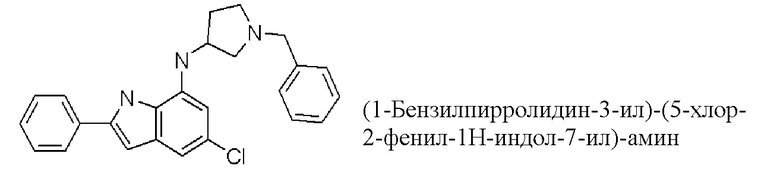
Соединение, полученное в синтезе 55 (225 мг, 0,83 ммоль), подвергали взаимодействию с получением 50 мг названного соединения (выход 15%), в соответствии с такими же методами, как в синтезе 53 и примере 1, за исключением того, что 1-бензил-пирролидин-3-он использовали вместо циклопентанона.
1H-ЯМР (500 МГц, CDCl3); δ 10,33 (ушир.с, 1H), 7,83 (м, 2H), 7,47 (м, 5H), 7,41 (м, 2H), 7,28 (м, 1H), 7,00 (д, J=1,85 Гц, 1H), 6,69 (д, J=1,85 Гц, 1H), 6,19 (д, J=1,8, 1H), 4,49 (м, 1H), 4,36 (д, J=12,8 Гц, 2H), 4,09 (д, J=12,8 Гц, 2H), 3,82 (м, 1H), 3,59 (м, 1H), 3,18 (м, 1H), 3,11 (м, 1H), 2,41 (м, 2H).
Синтез 56: Получение метилового эфира 4-этинилбензойной кислоты
(Стадия 1)
20,60 г (78,61 ммоль) метилового эфира 4-йодбензойной кислоты и 9,26 г (94,33 ммоль) этинилтриметилсилана растворяли в 200 мл тетрагидрофурана, и добавляли к ним 23,86 г (235,83 ммоль) триэтиламина, 1,65 г (2,36 ммоль) дихлор(бистрифенил-фосфин)палладия(II) и 0,45 г (2,36 ммоль) йодида меди(I). Смесь перемешивали при комнатной температуре в течение 8 часов. В конце реакции добавляли воду и экстрагировали с помощью этилацетата. Экстракт промывали с помощью насыщенного раствора хлорида натрия, сушили над безводным сульфатом магния и отделяли с помощью колоночной хроматографии с получением 18,30 г метилового эфира триметилсилилэтинилбензойной кислоты (выход 100%).
(Стадия 2)
Соединение, полученное на стадии 1 (18,30 г, 78,61 ммоль), растворяли в 300 мл смеси 2:1 метанола и дихлорметана, и добавляли 5,44 г (39,3 ммоль) карбоната калия. Смесь перемешивали при комнатной температуре в течение 1 часа. В конце реакции реакционную смесь разбавляли с помощью воды и экстрагировали с помощью этилацетата. Экстракт промывали с помощью насыщенного раствора хлорида натрия, сушили над безводным сульфатом магния и отделяли с помощью колоночной хроматографии с получением 11,90 г названного соединения (выход 95%).
Синтез 57: Получение метилового эфира 4-(5-Хлор-2-нитро-1H-индол-2-ил)-бензойной кислоты
(Стадия 1)
2-Йод-4-хлор-6-нитрофениламин (10,0 г, 33,50 ммоль) растворяли в 200 мл тетрагидрофурана и добавляли 3,97 г (50,25 ммоль) пиридина и 7,74 г (36,85 ммоль) трифторуксусного ангидрида. Смесь перемешивали в течение 9 часов при 0°C ~ комнатной температуре. В конце реакции реакционную смесь разбавляли с помощью воды и экстрагировали с помощью этилацетата. Экстракт промывали с помощью насыщенного раствора хлорида натрия, сушили над безводным сульфатом магния и отделяли с помощью колоночной хроматографии с получением 12,3 г трифторацетамида (выход 85%).
(Стадия 2)
Соединение, полученное на стадии 1 (12,3 г, 31,18 ммоль) и 6,0 г (37,42 ммоль) метилового эфира 4-этинилбензойной кислоты растворяли в 200 мл N,N-диметилформамида. Добавляли 9,47 г (93,54 ммоль) триэтиламина и 11,51 г (31,18 ммоль) йодида тетрабутиламмония, 1,09 г (1,56 ммоль) дихлор(бистрифенил-фосфин)палладия(II) и 0,30 г (1,56 ммоль) йодида меди(I). Смесь перемешивали при 80°C в течение 8 часов. В конце реакции реакционную смесь разбавляли с помощью воды и экстрагировали с помощью этилацетата. Экстракт промывали с помощью насыщенного раствора хлорида натрия, сушили над безводным сульфатом магния и отделяли с помощью колоночной хроматографии с получением 7,6 г названного соединения (выход 81%).
Пример 64: Получение метилового эфира 4-(5-хлор-7-циклопентиламино-1H-индол-2-ил)бензойной кислоты
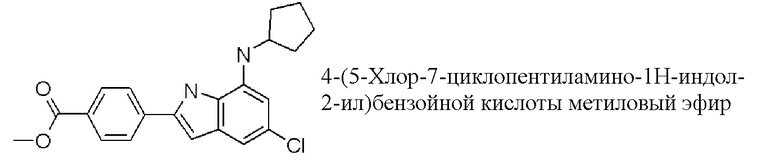
Соединение, полученное в синтезе 57 (7,60 г, 22,98 ммоль), подвергали взаимодействию вместо 5-метил-7-нитро-2-фенил-1H-индола в синтезе 46, с получением 1,25 г названного соединения (выход 15%).
1H-ЯМР (400 МГц, DMSO-d6); δ 11,25 (ушир.с, 1H), 8,07 (д, J=8 Гц, 2H), 7,94 (д, J=8 Гц, 2H), 6,95 (с, 1H), 6,84 (с, 1H), 6,22 (с, 1H), 5,77 (д, J=8 Гц, 1H), 3,89 (с, 3H), 3,88 (м, 1H), 2,05 (м, 2H), 1,75 (м, 2H), 1,65 (м, 2H), 1,63 (м, 2H).
Пример 65: Получение 4-(5-хлор-7-циклопентиламино-1H-индол-2-ил)бензойной кислоты

Соединение, полученное в примере 64 (300 мг, 0,81 ммоль), растворяли в 15 мл тетрагидрофурана, 5 мл воды и 5 мл метанола. Добавляли 100 мг (2,44 ммоль) моногидрата гидроксида лития. Смесь перемешивали при комнатной температуре в течение 21 часов. В конце реакции удаляли путем отгонки при пониженном давлении тетрагидрофуран и метанол. К дистилляту добавляли 1н раствор HCl и смесь экстрагировали с помощью этилацетата. Экстракт промывали с помощью насыщенного раствора хлорида натрия, сушили над безводным сульфатом магния, фильтровали. Фильтрат подвергали отгонке при пониженном давлении и отделяли с помощью колоночной хроматографии с получением 20 мг названного соединения (выход 7%).
1H-ЯМР (400 МГц, DMSO-d6); δ 11,59 (ушир.с, 1H), 8,01 (м, 4H), 6,94 (д, J=12 Гц, 2H), 6,31 (с, 1H), 3,91 (м, 1H), 2,01 (м, 2H), 1,77 (м, 2H), 1,62 (м, 4H).
Пример 66: Получение [4-(5-хлор-7-циклопентиламино-1H-индол-2-ил)фенил]метанола
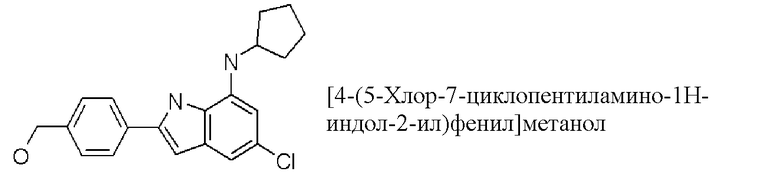
Соединение, полученное в Пример 64 (760 мг, 2,06 ммоль), растворяли в 30 мл тетрагидрофурана и добавляли 2M раствор боргидрида лития в тетрагидрофуране (2,06 мл, 4,12 ммоль). Смесь выдерживали при 80°C в течение 3 часов. Добавляли насыщенный раствор хлорида аммония, и смесь экстрагировали с помощью этилацетата. Экстракт сушили над безводным сульфатом магния, фильтровали. Фильтрат подвергали отгонке при пониженном давлении и очищали при помощи колоночной хроматографии с получением 60 мг названного соединения (выход 9%).
1H-ЯМР (400 МГц, DMSO-d6); δ 11,04 (ушир.с, 1H), 7,75 (д, J=8 Гц, 2H), 7,42 (д, J=8 Гц, 2H), 6,78 (с, 1H), 6,73 (с, 1H), 6,17 (с, 1H), 5,71 (д, J=3 Гц, 1H), 5,23 (т, 1H), 4,54 (д, J=4 Гц, 1H), 4,02 (м, 1H), 2,02 (м, 2H), 1,75 (м, 2H), 1,63 (м, 2H), 1,55 (м, 2H).
Пример 67: Получение метилового эфира 4-(7-циклопентиламино-5-метил-1H-индол-2-ил)бензойной кислоты
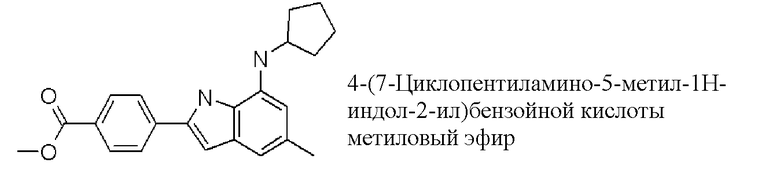
2-Йод-4-метил-6-нитрофениламин (500 мг, 1,80 ммоль) подвергали взаимодействию вместо 2-йод-4-хлор-6-нитрофениламина в соответствии с такими же методами, как в синтезе 57 и примере 64, с получением 30 мг названного соединения (выход 3%).
Синтез 58: Получение метилового эфира 2-(5-хлор-7-нитро-1H-индол-2-ил)-бензойной кислоты
Метиловый эфир 2-этинилбензойной кислоты (6,0 г 37,42 ммоль) подвергали взаимодействию вместо метилового эфира 4-этинилбензойной кислоты в синтезе 57, с получением названного 1,8 г соединения (выход 15%).
1H-ЯМР (400 МГц, CDCl3); δ 10,88 (ушир.с, 1H), 8,15 (с, 1H), 7,94 (д, 2H), 7,70 (с, 1H), 7,64 (с, 1H), 7,54 (с, 1H), 6,76 (с, 1H), 3,88 (с, 3H).
Пример 68: Получение метилового эфира 2-(5-хлор-7-циклопентиламино-1H-индол-2-ил)бензойной кислоты
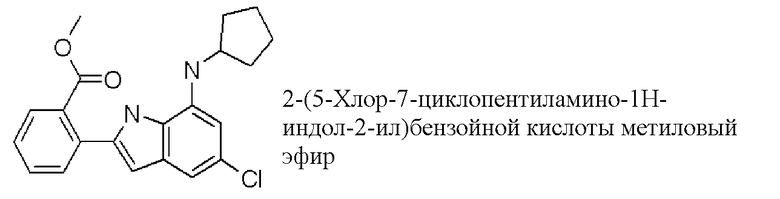
Соединения, полученные в синтезе 58 (1,80 г, 5,44 ммоль), подвергали взаимодействию вместо 5-метил-7-нитро-2-фенил-1H-индола в примере 46, с получением 800 мг названного соединения (выход 40%).
1H-ЯМР (400 МГц, CDCl3); δ 9,81 (ушир.с, 1H), 7,71 (д, J=4 Гц, 1H), 7,59 (д, J=4 Гц, 1H), 7,43 (т, 1H), 7,30 (т, 1H), 6,97 (с, 1H), 6,50 (д, J=2 Гц, 1H), 6,36 (д, J=2 Гц, 1H), 3,89 (с, 3H), 3,86 (м, 1H), 2,12 (м, 2H), 1,77 (м, 2H), 1,67 (м, 2H), 1,60 (м, 2H).
Пример 69: Получение 2-(5-хлор-7-циклопентиламино-1H-индол-2-ил)бензойной кислоты

Соединение, полученное в Пример 68 (300 мг, 0,81 ммоль), подвергали взаимодействию в соответствии с такими же методами, как в примере 65, с получением 120 мг названного соединения (выход 42%).
1H-ЯМР (400 МГц, DMSO-d6); δ 11,57 (ушир.с, 1H), 7,71 (м, 2H), 7,61 (м, 2H), 7,50 (м, 1H), 7,07 (с, 1H), 6,53 (с, 1H), 6,48 (с, 1H), 3,97 (м, 1H), 2,00 (м, 2H), 1,76 (м, 2H), 1,67 (м, 4H).
Пример 70: Получение [2-(5-хлор-7-циклопентиламино-1H-индол-2-ил)фенил]метанола
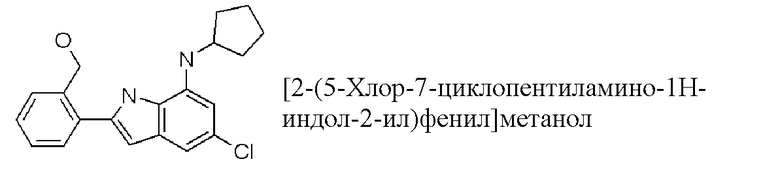
Соединение, полученное в Пример 68 (300 мг, 0,81 ммоль), подвергали взаимодействию в соответствии с такими же методами, как в примере 66, с получением 150 мг названного соединения (выход 54%).
1H-ЯМР (400 МГц, CDCl3); δ 11,37 (ушир.с, 1H), 7,69 (д, J=8 Гц, 1H), 7,47 (м, 2H), 7,42 (м, 2H), 7,21 (с, 1H), 6,69 (с, 1H), 4,78 (с, 2H), 3,97 (м, 1H), 1,96 (м, 4H), 1,86 (м, 2H), 1,56 (м, 2H).
Синтез 59: Получение этилового эфира 7-нитро-2-фенил-1H-индол-5-карбоновой кислоты
Сложный эфир 4-амино-3-нитробензойной кислоты (3,0 г, 8,9 ммоль) подвергали взаимодействию вместо 4-метил-2-нитроанилина в синтезе 19 с получением 1,6 г названного соединения (выход 58%).
1H-ЯМР (500 МГц, CDCl3); δ 10,23 (ушир.с, 1H), 8,82 (с, 1H), 8,65 (с, 1H), 7,74 (м, 2H), 7,52 (м, 2H), 7,44 (м, 1H), 7,02 (с, 1H), 4,45 (кв, 2H), 1,45 (т, 3H).
Пример 71: Получение этилового эфира 7-циклопентиламино-2-фенил-1H-индол-5-карбоновой кислоты

Соединение, полученное в синтезе 59 (600 мг, 1,9 ммоль), подвергали взаимодействию вместо 5-метил-7-нитро-2-фенил-1H-индола в примере 46, с получением 100 мг названного соединения (выход 15%).
1H-ЯМР (400 МГц, CDCl3); δ 9,47 (ушир.с, 1H), 7,90 (с, 1H), 7,67 (м, 2H), 7,34 (м, 2H), 7,24 (м, 1H), 7,18 (с, 1H), 6,83 (с, 1H), 4,41 (кв, 2H), 3,87 (м, 1H), 1,95 (м, 2H), 1,62 (м, 2H), 1,49 (м, 4H), 1,39 (т, 3H).
Пример 72: Получение 7-циклопентиламино-2-фенил-1H-индол-5-карбоновой кислоты

Соединение, полученное в примере 71 (350 мг, 1,0 ммоль), подвергали взаимодействию в соответствии с такими же способами, как в примере 65, с получением 300 мг названного соединения (выход 94%).
1H-ЯМР (400 МГц, MeOH-d4); δ 7,88 (с, 1H), 7,75 (м, 2H), 7,42 (м, 2H), 7,30 (м, 1H), 7,16 (с, 1H), 6,85 (с, 1H), 4,03 (м, 1H), 2,13 (м, 2H), 1,77 (м, 2H), 1,66 (м, 4H).
Пример 73: Получение (7-циклопентиламино-2-фенил-1H-индол-5-ил)метанола

Соединение, полученное в примере 71 (60 мг, 0,17 ммоль), подвергали взаимодействию в соответствии с такими же методами, как в примере 66, с получением 21 мг названного соединения (выход 40%).
1H-ЯМР (400 МГц, MeOH-d4); δ 7,88 (с, 1H), 7,75 (м, 2H), 7,42 (м, 2H), 7,30 (м, 1H), 7,16 (с, 1H), 6,85 (с, 1H), 4,03 (м, 1H), 2,13 (м, 2H), 1,77 (м, 2H), 1,66 (м, 4H).
Синтез 60: Получение этилового эфира (7-нитро-2-фенил-1H-индол-5-ил)уксусной кислоты
Этиловый эфир (4-амино-3-нитрофенил)уксусной кислоты (4,8 г, 13,7 ммоль) подвергали взаимодействию вместо 4-метил-2-нитроанилина в синтезе 19, с получением 3,0 г названного соединения (выход 68%).
1H-ЯМР (500 МГц, CDCl3); δ 10,03 (ушир.с, 1H), 8,05 (с, 1H), 8,00 (с, 1H), 7,72 (м, 2H), 7,49 (м, 2H), 7,49 (м, 1H), 7,39 (м, 1H), 6,87 (с, 1H), 4,19 (кв, 2H), 2,04 (с, 2H), 1,26 (т, 3H).
Пример 74: Получение этилового эфира (7-циклопентиламино-2-фенил-1H-индол-5-ил)уксусной кислоты

Соединение, полученное в синтезе 60 (3,0 г, 9,2 ммоль), подвергали взаимодействию вместо 5-метил-7-нитро-2-фенил-1H-индола в примере 46, с получением 2,0 г названного соединения (выход 60%).
1H-ЯМР (400 МГц, CDCl3); δ 8,12 (ушир.с, 1H), 7,67 (м, 2H), 7,42 (м, 2H), 7,30 (м, 1H), 6,99 (с, 1H), 6,74 (с, 1H), 6,46 (с, 1H), 4,16 (кв, 2H), 3,93 (м, 1H), 3,66 (с, 2H), 2,10 (м, 2H), 1,77 (м, 2H), 1,66 (м, 2H), 1,59 (т, 3H), 1,26 (т, 3H).
Пример 75: Получение (7-циклопентиламино-2-фенил-1H-индол-5-ил)уксусной кислоты
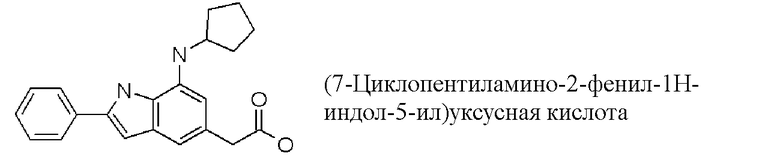
Соединение, полученное в синтезе 74 (90 мг, 0,25 ммоль), подвергали взаимодействию в соответствии с такими же методами, как в примере 65, с получением 65 мг названного соединения (выход 78%).
1H-ЯМР (400 МГц, CDCl3+MeOH-d4); δ 11,31 (с, 1H), 7,81 (м, 2H), 7,37 (м, 2H), 7,25 (м, 1H), 7,05 (с, 1H), 6,65 (с, 1H), 3,89 (м, 1H), 1,88 (м, 7H), 1,41 (м, 3H).
Пример 76: Получение 2-(7-циклопентиламино-2-фенил-1H-индол-5-ил)этанола

Соединение, полученное в синтезе 74 (50 мг, 0,14 ммоль), подвергали взаимодействию в соответствии с такими же методами, как в примере 66, с получением 40 мг названного соединения (выход 89%).
1H-ЯМР (400 МГц, CDCl3+MeOH-d4); δ 7,71 (м,2H), 7,40 (м, 2H), 7,28 (м, 1H), 6,89 (с, 1H), 6,72 (с, 1H), 6,32 (с, 1H), 3,97 (м, 1H), 3,87 (м, 2H), 3,43 (м, 1H), 2,91 (м, 2H), 2,09 (м, 2H), 1,77 (м, 2H), 1,62 (м, 4H).
Пример 77-90:
Соединения нитроиндола, полученные в синтезах 5, 7 и 10, соединения, полученные в синтезах 23 и 25, и выпускаемые в промышленности соединения кетона подвергали взаимодействию в соответствии с такими же методами, как синтезе 34, 35, 36 и примере 11, с получением следующих типичных соединений.
Пример 91-101:
Сложные эфиры, полученные в примере 21, 77, 79, 89 и 78, восстанавливали в соответствии с такими же методами, как в примере 4, и затем подвергали взаимодействию в соответствии с такими же методами, как в синтезе 37 и примере 15, с получением следующих типичных соединений.
морфолин-4-ил
разин-4-ил
моилпир-
ролидин-1-ил
Синтез 61: Получение изопропилового эфира [(S)-2-(5-метил-7-нитро-1H-индол-2-ил)-4, 5-дигидрооксазол-4-ил]уксусной кислоты
(Стадия 1)
Метиловый эфир 5-метил-7-нитро-1H-индол-2-карбоновой кислоты, полученный в синтезе 7, подвергали гидролизу с помощью LiOH в соответствии с такими же методами, как на стадии 1 синтеза 28, с получением 5-метил-7-нитро-1H-индол-2-карбоновой кислоты.
(Стадия 2)
С изопропилового эфира (S)-3-трет-бутоксикарбониламино-4-гидроксимасляной кислоты, используемого в синтезе 25, снимали защиту в соответствии с такими же методами, как на стадии 3 синтеза 24, с получением изопропилового эфира (S)-3-амино-4-гидроксимасляной кислоты.
(Стадия 3)
Соединения, полученные на стадии 1 и 2, подвергали взаимодействию в соответствии с такими же методами, как на стадия 2 синтеза 28 с получением изопропилового эфира (S)-4-гидрокси-3-[(5-метил-7-нитро-1H-индол-2-карбонил)амино]масляной кислоты.
(Стадия 4)
Соединение, полученное в Стадии 3, и метансульфонилхлорид подвергали взаимодействию в соответствии с такими же методами, как в синтезе 33, с получением изопропилового эфира (S)-4-метансульфонилокси-3-[(5-метил-7-нитро-1H-индол-2-карбонил)амино]масляной кислоты.
(Стадия 5)
Соединение, полученное на стадии 4 (890 мг, 2 ммоль), добавляли к THF (10 мл), добавляли K2CO3 (330 мг, 10 ммоль). Смесь перемешивали при 80°C в течение 2 часов. Для прерывания реакции добавляли воду. Смесь экстрагировали с помощью EtOAc, сушили над MgSO4, и удаляли растворитель при пониженном давлении. Остаток очищали при помощи колоночной хроматографии (элюент: EtOAc/n-Hex/DMC - 1/4/1) с получением названного соединения (445 мг, выход 61 %).
Пример 102: Получение [(S)-2-(7-циклопентиламино-5-метил-1H-индол-2-ил)-4,5-дигидрооксазол-4-ил]уксусной кислоты
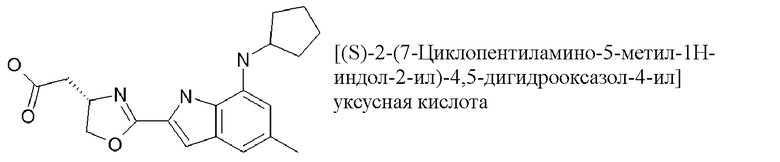
Последовательно применяли методы стадии 3 синтеза 31, синтеза 36 и стадия 1 синтеза 28 при использовании соединения, полученного в синтезе 61, и циклопентанона, с получением названного соединения.
1H-ЯМР (400 МГц, CDCl3/MeOH-d4); δ, 6,97 (с, 1H), 6,79 (с, 1H), 6,33 (с, 1H), 4,86 (м, 1H), 4,62 (дд, 1H), 4,35 (дд, 1H), 3,95 (м, 1H), 2,94 (дд, 1H), 2,67 (дд, 1H), 2,39 (с, 3H), 2,07 (м, 2H), 1,78 (м, 2H), 1,65 (м, 4H).
Пример 103: Получение [(S)-2-[5-метил-7-(тетрагидропиран-4-иламино)-1H-индол-2-ил]-4,5-дигидрооксазол-4-ил}уксусной кислоты
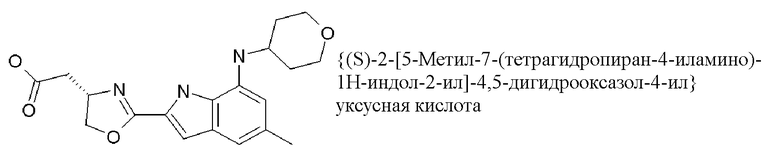
Тетрагидропиран-4-он использовали вместо циклопентанона в примере 102 с получением названного соединения.
1H-ЯМР (400 МГц, CDCl3/MeOH-d4); δ 6,94 (с, 1H), 6,81 (с, 1H), 6,32 (с, 1H), 4,87 (м, 1H), 4,59 (дд, 1H), 4,34 (дд, 1H), 4,02 (д, 1H), 3,68-3,58 (м, 3H), 2,92 (дд, 1H), 2,68 (дд, 1H), 2,38 (с, 3H), 2,13 (д, 2H), 1,57 (м, 2H).
Пример 104: Получение {(S)-2-[5-метил-7-(тетрагидропиран-4-иламино)-1H-индол-2-ил]-4,5-дигидрооксазол-4-ил}этанола
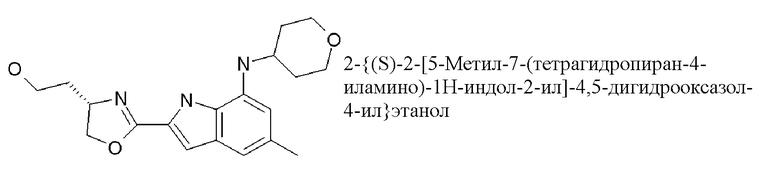
Соединение, полученное в примере 103, восстанавливали с получением названного соединения в соответствии с таким же методом, как в примере 66.
1H-ЯМР (400 МГц, CDCl3); δ 10,11 (ушир.с, 1H), 6,91 (с, 1H), 6,86 (с, 1H), 6,34 (с, 1H), 4,60 (т, 1H), 4,48 (м, 1H), 4,10-3,93 (м, 5H), 3,63-3,52 (м, 3H), 2,39 (с, 3H), 2,07 (д, 2H), 1,94 (м, 2H), 1,58 (м, 2H).
Пример 105: Получение {5-метил-2-[(S)-4-(2-морфолин-4-ил-этил)-4,5-дигидрооксазол-2-ил]-1H-индол-7-ил}-(тетрагидропиран-4-ил)амина

Соединение, полученное в примере 104, и морфолин подвергали взаимодействию в соответствии с методами, такими же, как в синтезе 33 и примере 15, с получением названного соединения.
1H-ЯМР (400 МГц, CDCl3); δ 10,11 (ушир.с, 1H), 6,91 (с, 1H), 6,86 (с, 1H), 6,34 (с, 1H), 4,60 (т, 1H), 4,48 (м, 1H), 4,10-3,93 (м, 5H), 3,63-3,52 (м, 3H), 2,39 (с, 3H), 2,07 (д, 2H), 1,94 (м, 2H), 1,58 (м, 2H).
Пример 106-125:
Следующие типичные соединения получали так же, как в синтезе 19, на стадии 3 синтеза 35 и синтезе 36.
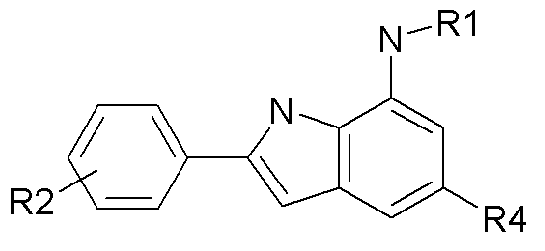
MeO2C-CH2-
Синтез 62: получение 5-(1,1-диоксо-тиоморфолин-4-илметил)-2-фенил-1H-индол-7-иламина
(Стадия 1)
Этиловый эфир 7-нитро-2-фенил-1H-индол-5-карбоновой кислоты, полученный в синтезе 59, подвергали взаимодействию в соответствии с такими же методами, как в синтезе 66, с получением (7-нитро-2-фенил-1H-индол-5-ил)метанола.
(Стадия 2)
Соединение, полученное на стадии 1, йод и 1,1-диоксотиоморфолин подвергали взаимодействию в соответствии с такими же методами, как в синтезе 37 и примере 15, с получением 5-(1,1-диоксотиоморфолин-4-илметил)-7-нитро-2-фенил-1H-индола.
(Стадия 3)
Соединение, полученное на стадии 2, и железную пыль подвергали взаимодействию в соответствии с такими же методами, как на стадии 3 синтеза 35, с получением названного соединения.
Пример 126: Получение [5-(1,1-диоксотиоморфолин-4-илметил)-2-фенил-1H-индол-7-ил]-(тетрагидропиран-4-ил)амина
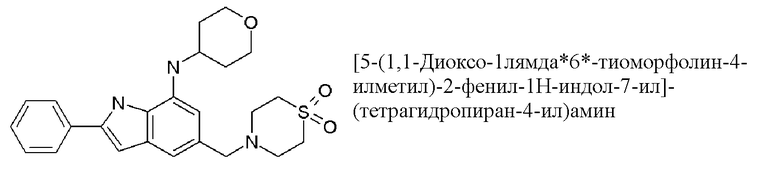
Соединение, полученное в синтезе 62, и тетрагидропиран-4-он подвергали взаимодействию в соответствии с такими же методами, как в синтезе 36, с получением названного соединения.
1H-ЯМР (500 МГц, CDCl3); δ 8,38 (ушир.с, 1H), 7,69 (д, J=7,3 Гц, 1H), 7,44 (т, J=7,3 Гц, 2H), 7,32 (т, J=7,3 Гц, 1H), 7,00 (с, 1H), 6,75 (д, J=1,85 Гц, 1H), 6,47 (д, J=1,85 Гц, 1H), 4,07 (м, 2H), 3,65 (м, 3H), 3,57 (м, 2H), 3,02 (м, 8H), 2,12 (м, 2H), 1,61 (м, 2H)
Пример 127-138:
Соединение индола, полученное методом, описанным в синтезах 59, производимые в промышленности соединения амина и карбонильные соединения подвергали взаимодействию в соответствии с такими же методами, как в синтезе 62 и примере 126, с получением типичных соединений, приведенных в таблице.
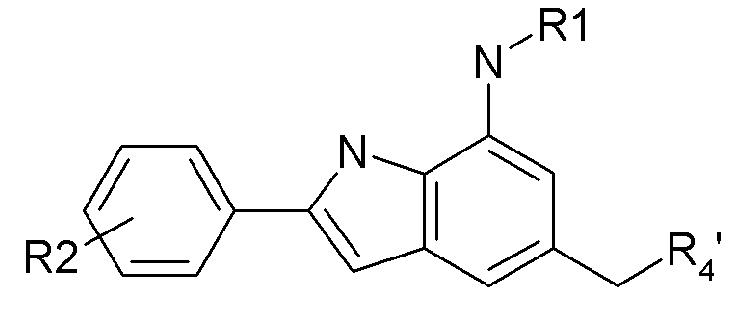
пиран-4-ил)метил
тиоморфо-
лин-4-ил
пиран-4-ил)метил
тиоморфолин-4-ил
пиран-4-ил
разин-4-ил
тиоморфо-
лин-4-ил
тиоморфо-
лин-4-ил
тиоморфо-
лин-4-ил
тиоморфо-
лин-4-ил
тиоморфолин-4-ил
пиран-4-ил
тиоморфолин-4-ил
тиоморфолин-4-ил
пиран-4-ил
тиоморфолин-4-ил
тиоморфолин-4-ил
Синтез 63: Получение 3-бром-5-(1,1-диоксотиоморфолин-4-ил)метил-7-нитро-2-фенил-1H-индола
(Стадия 1)
5-Метил-7-нитро-2-фенил-1H-индол, полученный в примере 46, и (BOC)2O подвергали взаимодействию в соответствии с такими же методами, как в синтезе 23, с получением 1-BOC-5-метил-7-нитро-2-фенилиндола.
(Стадия 2)
1-BOC-5-метил-7-нитро-2-фенилиндол (3,5 г, 10 ммоль), полученный на стадии 1, растворяли в четыреххлористом углероде (30 мл), и добавляли к нему N-бромсукцинимид (NBS, 2,3 г, 13 ммоль) и пероксид бензоила (100 мг). Смесь кипятили с обратным холодильником при перемешивании в течение 4 часов при 80°С. И затем смесь фильтровали для удаления твердых веществ, фильтрат разбавляли с помощью воды и экстрагировали с помощью DCM. Растворитель удаляли при пониженном давлении. Остаток очищали при помощи колоночной хроматографии с получением 1-BOC-3-бром-5-бромметил-7-нитро-2-фенилиндола (2,3 г, 46%).
(Стадия 3)
1-BOC-3-бром-5-бромметил-7-нитро-2-фенилиндол (1,0 г, 2 ммоль), полученный на стадии 2, растворяли в DCM (10 мл), и добавляли к нему Et3N (560 мкл, 4 ммоль) и 1,1-диоксотиоморфолин (300 мг, 3 ммоль). Смесь перемешивали в течение 12 часов при комнатной температуре. В конце реакции добавляли насыщенный водный раствор NH4Cl, экстрагировали с помощью DCM. После сушки экстракта растворитель удаляли при пониженном давлении, и остаток очищали при помощи колоночной хроматографии с получением 1-BOC-3-бром-5-(1,1-диоксотиоморфолин-4-ил)метил-7-нитро-2-фенил-1H-индола (825 мг, 78 %).
(Стадия 4)
1-BOC-3-бром-5-(1,1-диоксотиоморфолин-4-ил)метил-7-нитро-2-фенил-1H-индол, полученный на стадии 3 (825 мг, 1,6 ммоль), растворяли в диэтиловом эфире (5 мл), и затем добавляли HCl (4M раствор в диоксане, 5 мл). Реакционный раствор перемешивали в течение 2 часов при комнатной температуре. В конце реакции удаляли растворитель при пониженном давлении и сушили с получением 3-бром-5-(1,1-диоксотиоморфолин-4-ил)метил-7-нитро-2-фенил-1H-индола, который использовали на следующей стадии без дополнительной очистки.
Синтез 64: Получение 4-[(3-бром-7-нитро-2-фенил-1H-индол-5-ил)метил]морфолина
1-BOC-3-бром-5-бромметил-7-нитро-2-фенилиндол, полученный на стадии 2 синтеза 63, и морфолин подвергали взаимодействию с получением названного соединения, в соответствии с такими же методами, как на стадии 3 и 4 синтеза 63.
Пример 139: Получение 3-бром-5-(1,1-диоксотиоморфолин-4-илметил)-2-фенил-1H-индол-7-ил]-(тетрагидропиран-4-ил)амина

Соединение, полученное в синтезе 63, и тетрагидропиран-4-он подвергали взаимодействию в соответствии с такими же методами, как на стадии 3 синтеза 35 и в синтезе 36, с получением названного соединения.
1H-ЯМР (400 МГц, CDCl3); δ 8,08 (ушир.с, 1H), 7,61 (д, J= 8 Гц, 2H), 7,44 (т, 2H), 7,37 (с, 1H), 7,29 (м, 2H), 6,52 (с, 1H), 4,04 (дд, 2H), 3,44 (т, 2H), 3,19 (д, J=4 Гц, 2H), 1,97 (м, 1H), 1,79 (д, J-12 Гц, 2H), 1,41 (м, 2H).
Пример 140: Получение [3-бром-(5-морфолин-4-ил)метил-2-фенил-1H-индол-7-ил]-(тетрагидропиран-4-ил)амина
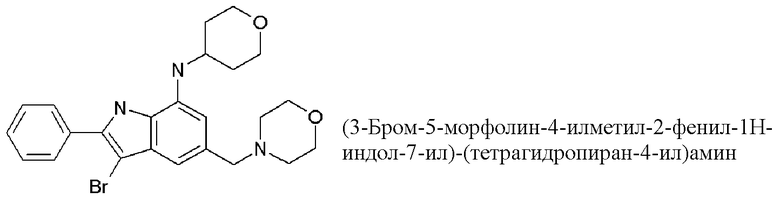
Соединение, полученное в синтезе 64, и тетрагидропиран-4-он подвергали взаимодействию в соответствии с такими же методами, как на стадии 3 синтеза 35 и в синтезе 36, с получением названного соединения.
1H ЯМР (500 МГц, DMSO-d6): δ 8,13 (ушир.с, 1H), 7,81 (м, 2H), 7,49 (м, 2H), 7,40 (м, 1H), 7,02 (с, 1H), 6,45 (с, 1H), 4,40 (м, 2H), 3,72 (м, 4H), 3,67 (м, 1H), 3,6-3,53 (м, 4H), 2,48 (м, 4H), 2,10 (м, 2H), 1,56 (м, 4H)
Пример 141: Получение [3-бром-5-(1,1-диоксотиоморфолин-4-илметил)-2-фенил-1H-индол-7-ил]циклопентиламина
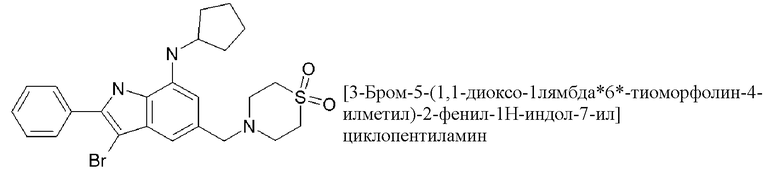
Соединение, полученное в синтезе 63, и циклопентанон подвергали взаимодействию в соответствии с такими же методами, как на стадии 3 синтеза 35 и в синтезе 36, с получением названного соединения.
1H-ЯМР (500 МГц, CDCl3); δ 8,29 (ушир.с, 1H), 7,82 (д, J=7,3 Гц, 1H), 7,49 (т, J=7,3 Гц, 2H), 7,39 (т, J=7,3 Гц, 1H), 6,93 (с, 1H), 6,53 (д, J=1,85 Гц, 1H), 3,95 (м, 1H), 3,70 (с, 2H), 3,02 (м, 8H), 2,10 (м, 2H), 1,77 (м, 2H), 1,68 (м, 2H), 1,56 (м, 2H).
Пример 142: Получение [3-бром-5-(1,1-диоксотиоморфолин-4-илметил)-2-фенил-1H-индол-7-ил]-(тетрагидропиран-4-илметил)амина
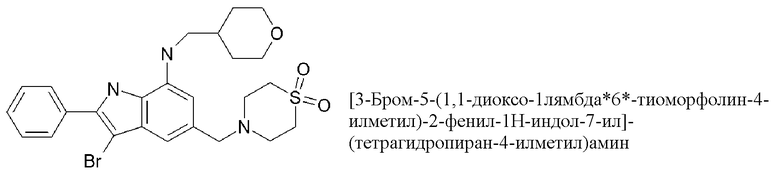
Соединение, полученное в синтезе 63, и тетрагидропиран-4-карбоксиальдегид подвергали взаимодействию в соответствии с такими же методами, как на стадии 3 синтеза 35 и в синтезе 36, с получением названного соединения.
1H-ЯМР (500 МГц, CDCl3); δ 8,20 (ушир.с, 1H), 7,80 (д, J=7,3 Гц, 1H), 7,49 (т, J=7,3 Гц, 2H), 7,40 (т, J=7,3 Гц, 1H), 6,97 (с, 1H), 6,50 (д, J=1,85 Гц, 1H), 4,03 (м, 2H), 3,71 (с, 2H), 3,42 (м, 2H), 3,18 (м, 2H), 3,03 (м, 8H), 1,94 (м, 1H), 1,78 (м, 2H), 1,47 (м, 2H).
Синтез 65: Получение 5-хлор-3-фенил-7-нитро-1H-индола
(Стадия 1)
Производимый в промышленности 4-хлор-2-нитрофениламин (17,4 г, 131,5 ммоль) растворяли в этаноле (300 мл), и добавляли к нему нитрат серебра (27 г, 157,7 ммоль) и йод (40 г, 157,7 ммоль). Смесь перемешивали в течение 8 часов при комнатной температуре. В конце реакции смесь фильтровали через целит и промывали с помощью 100 мл этилацетата и концентрировали. К концентрату добавляли воду и экстрагировали с помощью этилацетата. Органический слой промывали с помощью насыщенного раствора хлорида натрия и сушили над безводным сульфатом магния с получением 2-амино-5-хлор-3-нитрофенилйодида (27,4 г, выход 69%).
1H-ЯМР (500 МГц, CDCl3); δ 7,94 (с, 1H), 7,75 (с, 1H), 6,48 (ушир.с, 2H), 2,23 (с, 3H).
(Стадия 2)
2-Амино-5-хлор-3-нитрофенилйодид (1,5 г, 4,90 ммоль), полученный на стадии 1, и 1-фенил-2-триметилсилилацетилен (4,3 г, 24,50 ммоль) растворяли в DMF (50 мл), и добавляли к ним ацетат палладия (0,11 г, 0,5 ммоль), хлорид лития (0,21 г, 4,90 ммоль) и триэтиламин (2,48 г, 24,50 ммоль). Смесь нагревали при перемешивании в течение 3 часов при 100°C. В конце реакции добавляли воду и экстрагировали с помощью этилацетата. Экстракт промывали с помощью насыщенного раствора хлорида натрия, сушили над безводным сульфатом магния и фильтровали. Растворитель удаляли при пониженном давлении, и остаток отделяли с помощью колоночной хроматографии с получением 5-хлор-7-нитро-3-фенил-2-триметилсилил-1H-индола (1,05 г, выход 87%).
1H-ЯМР (400 МГц, CDCl3); δ 9,78 (ушир.с, 1H), 8,15 (с, 1H), 7,78 (с, 1H), 7,38-7,48 (м, 5H), 0,26 (с, 9H).
(Стадия 3)
5-Хлор-7-нитро-3-фенил-2-триметилсилил-1H-индол (1,5 г, 4,35 ммоль), полученный на стадии 2, растворяли в тетрагидрофуране (30 мл), и добавляли по каплям 1M раствор фторида тетрабутиламмония (5,2 мл, 5,2 ммоль) при 0°C. В конце реакции полученную смесь разбавляли с помощью воды и экстрагировали с помощью этилацетата. Экстракт промывали с помощью насыщенного раствора хлорида натрия, сушили над безводным сульфатом магния и фильтровали. Растворитель удаляли при пониженном давлении, и остаток отделяли при помощи колоночной хроматографии с получением 5-хлор-3-фенил-7-нитро-1H-индола (1,2 г, выход 100%).
1H-ЯМР (400 МГц, CDCl3); δ 10,03 (ушир.с, 1H), 8,24 (д, J=8 Гц, 2H), 7,62 (м, 3H), 7,55 (м, 2H), 7,43 (м, 1H).
Пример 143: Получение (5-хлор-3-фенил-1H-индол-7-ил)-(тетрагидропиран-4-ил)амина

5-Хлор-3-фенил-7-нитро-1H-индол, полученный в синтезе 65, и тетрагидропиран-4-он подвергали взаимодействию в соответствии с такими же методами, как на стадии 3 синтеза 35 и в синтезе 36, с получением названного соединения.
1H-ЯМР (400 МГц, CDCl3); δ 8,10 (ушир.с, 1H), 7,61 (д, J=8 Гц, 2H), 7,43 (т, 2H), 7,38 (с, 1H), 7,31 (м, 2H), 6,56 (с, 1H), 4,05 (м, 2H), 3,65 (м, 1H), 3,62 (т, 2H), 2,15 (д, J=12 Гц, 2H), 1,62 (м, 5H).
Пример 144: Получение (5-хлор-3-фенил-1H-индол-7-ил)-(циклопентил)амина
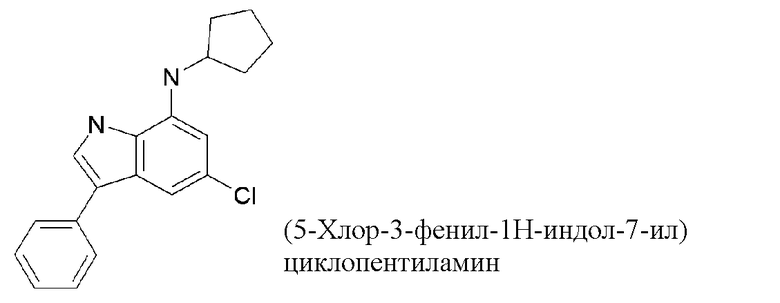
5-Хлор-3-фенил-7-нитро-1H-индол, полученный в синтезе 65, и циклопентанон подвергали взаимодействию в соответствии с такими же методами, как на стадии 3 синтеза 35 и в синтезе 36 с получением названного соединения.
1H-ЯМР (400 МГц, CDCl3); δ 8,06 (ушир.с, 1H), 7,58 (д, J=8 Гц, 2H), 7,39 (т, 2H), 7,33 (с, 1H), 7,26 (м, 1H), 7,17 (с, 1H), 6,51 (с, 1H), 3,91 (м, 1H), 2,08 (м, 2H), 1,74 (м, 2H), 1,66 (м, 2H), ,54 (м, 2H).
Пример 145: Получение (5-хлор-3-фенил-1H-индол-7-ил)-(тетрагидропиран-4-илметил)амина

5-Хлор-3-фенил-7-нитро-1H-индол, полученный в синтезе 65, и тетрагидропиран-4-карбоксилальдегид подвергали взаимодействию в соответствии с такими же методами, как на стадии 3 синтеза 35 и в синтезе 36, с получением названного соединения.
1H-ЯМР (400 МГц, CDCl3); δ 8,08 (ушир.с, 1H), 7,61 (д, J=8 Гц, 2H), 7,44 (т, 2H), 7,37 (с, 1H), 7,29 (м, 2H), 6,52 (с, 1H), 4,04 (дд, 2H), 3,44 (т, 2H), 3,19 (д, J=4 Гц, 2H), 1,97 (м, 1H), 1,79 (д, J=12 Гц, 2H), 1,41 (м, 2H).
Пример 146: Получение [5-(1,1-диоксотиоморфолин-4-илметил)-3-фенил-2-триметилсиланил-1H-индол-7-ил]-(тетрагидропиран-4-ил)-амина

(Стадия 1)
Этиловый эфир 4-амино-3-нитробензойной кислоты и триметил-фенилэтинилсилан подвергали взаимодействию в соответствии с такими же методами, как в синтезе 19, с получением этилового эфира 7-нитро-3-фенил-2-триметилсиланил-1H-индол-5-карбоновой кислоты.
(Стадия 2)
Соединение, полученное на стадии 1, тетрагидропиран-4-он и 1,1-диоксотиоморфолин подвергали взаимодействию в соответствии с синтезом 62 и 36 с получением названного соединения.
1H-ЯМР (400 МГц, CDCl3); δ 8,00 (ушир.с, 1H), 7,43 (м, 4H), 7,36 (м, 1H), 6,92 (с, 1H), 6,55 (с, 1H), 4,08 (д, J=12 Гц, 2H), 3,67 (м, 1H), 3,66 (с, 2H), 3,60 (т, 2H), 3,01 (д, 8H), 2,13 (д, J=12 Гц, 2H), 1,64 (м, 2H).
Синтез 66: Получение 5-хлор-7-нитро-3-(2-оксопиперазин-4-ил)метил-2-фенил-1H-индола
(Стадия 1)
7-нитро-5-хлор-2-фенил-1H-индол (1,0 г, 3,67 ммоль), полученный в примере 59, растворяли в дихлорметане (20 мл), и добавляли к нему по каплям при 0°C фосфорилоксихлорид (0,84 г, 5,50 ммоль) и DMF (0,80 г, 11,01 ммоль). Смесь перемешивали при комнатной температуре в течение 6 часов. В конце реакции добавляли насыщенный водный раствор гидрокарбоната натрия для прерывания реакции и экстрагировали с помощью этилацетата. Экстракт промывали с помощью насыщенного раствора хлорида натрия, сушили над безводным сульфатом магния и фильтровали. Растворитель удаляли при пониженном давлении, и остаток отделяли с помощью колоночной хроматографии с получением 5-хлор-3-формил-7-нитро-2-фенил -1H-индола (0,5 г, выход 45%).
1H-ЯМР (400 МГц, DMSO-d6); δ 9,89 (с, 1H), 8,46 (с,1H), 7,93 (с, 1H), 7,81 (д, J=9,2 Гц, 2H), 7,53 (м, 3H).
(Стадия 2)
5-Хлор-3-формил-7-нитро-2-фенил-1H-индол (0,5 г, 1,66 ммоль), полученный на стадии 1, растворяли в дихлорметане (20 мл), и добавляли к нему по каплям уксусную кислоту (0,10 г, 1,66 ммоль), 2-оксопиперазин (0,3 г, 3,32 ммоль) и триацетоксиборгидрид натрия (0,71 г, 3,32 ммоль). Смесь перемешивали при комнатной температуре в течение 4 часов. В конце реакции смесь разбавляли с помощью воды, и экстрагировали с помощью дихлорметана. Экстракт промывали с помощью насыщенного раствора хлорида натрия, сушили над безводным сульфатом магния и фильтровали. Растворитель удаляли при пониженном давлении, и остаток отделяли с помощью колоночной хроматографии с получением 5-хлор-7-нитро-3-(2-оксопиперазин-4-ил)метил-2-фенил-1H-индола (0,55 г, выход 86%).
1H-ЯМР (400 МГц, DMSO-d6); δ 9,89 (с, 1H), 8,46 (с,1H), 7,93 (с, 1H), 7,81 (д, J=9,2 Гц, 2H), 7,53 (м, 3H).
Пример 147: Получение 4-(5-хлор-7-циклопентиламино-2-фенил-1H-индол-3-илметил)пиперазин-2-она

Соединение, полученное в синтезе 66, и циклопентанон подвергали взаимодействию в соответствии с такими же методами, как на стадии 3 синтеза 35 и в синтезе 36, с получением названных соединений.
1H-ЯМР (400 МГц, DMSO-d6); δ 11,07 (с, 1H), 7,82 (д, J=8 Гц, 2H), 7,71 (с, 1H), 7,53 (т, 2H), 7,40 (т, 1H), 6,94 (с, 1H), 6,20 (с, 1H), 5,70 (д, J=8 Гц, 1H), 3,88 (м, 1H), 3,61 (с, 2H), 3,31 (с, 2H), 2,96 (с, 2H), 2,59 (м, 2H), 2,03 (м, 2H), 1,74 (м, 2H), 1,64 (м, 2H), 1,54 (м, 2H).
Пример 148: Получение 4-[5-хлор-2-фенил-7-(тетрагидропиран-4-иламино)-1H-индол-3-илметил]пиперазин-2-она
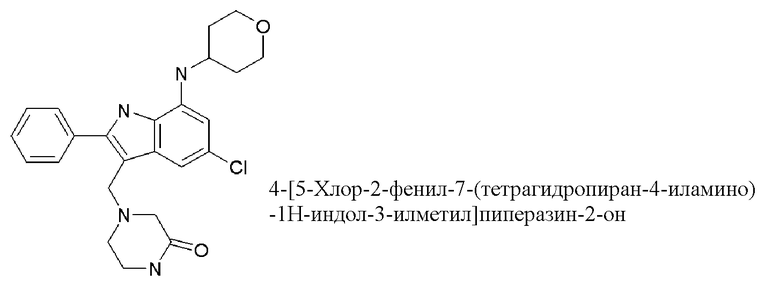
Соединение, полученное в синтезе 66, и тетрагидропиран-4-он подвергали взаимодействию в соответствии с такими же методами, как на стадии 3 синтеза 35 и в синтезе 36, с получением названных соединений.
1H-ЯМР (400 МГц, DMSO-d6); δ 11,09 (с, 1H), 7,83 (д, J=8 Гц, 2H), 7,70 (с, 1H), 7,54 (т, 2H), 7,40 (т, 1H), 6,96 (с, 1H), 6,31 (с, 1H), 5,64 (д, J=8 Гц, 1H), 3,90 (д, J= 12 Гц, 2H), 3,62 (м, 1H), 3,61 (с, 3H), 3,50 (м, 2H), 3,31 (с, 2H), 2,96 (с, 2H), 2,59 (с, 2H), 2,00 (д, J=12 Гц, 2H), 1,46 (м, 2H).
Пример 149: Получение 4-{5-хлор-2-фенил-7-[(тетрагидропиран-4-илметил)амино]-1H-индол-3-илметил}пиперазин-2-она

Соединение, полученное в синтезе 66, и тетрагидропиран-4-карбоксиальдегид подвергали взаимодействию в соответствии с такими же методами, как на стадии 3 синтеза 35 и в синтезе 36, с получением названных соединений.
1H-ЯМР (400 МГц, DMSO-d6); δ 11,12 (с, 1H), 7,83 (д, J=8 Гц, 2H), 7,71 (с, 1H), 7,54 (т, 2H), 7,40 (т, 1H), 6,96 (с, 1H), 6,21 (с, 1H), 5,73 (м, 1H), 3,91 (д, J=12 Гц, 2H), 3,89 (с, 2H), 3,36 (м, 2H), 3,31 (с, 2H), 3,10 (м, 2H), 2,96 (с, 2H), 2,59 (м, 2H), 1,90 (м, 1H), 1,74 (д, J=12 Гц, 2H), 1,34 (м, 2H).
Синтез 67: Получение 3-(4-метоксифенил-1-ил)-7-нитро-1H-индазола
(Стадия 1)
7-Нитроиндазол (1,60 г, 9,80 ммоль) растворяли в DMF (100 мл), и добавляли к нему по каплям гидроксид калия (2,20 г, 39,20 ммоль) и йод (4,98 г, 19,61 ммоль). Смесь перемешивали в течение 2 часов и разбавляли с помощью 10% раствора бисульфита натрия. Полученное твердое вещество собирали и сушили с получением 3-йод-7-нитро-1H-индазола (2,50 г, выход 88%).
1H-ЯМР (400 МГц, CDCl3); δ 11,49 (ушир.с, 1H), 8,45 (д, J=8 Гц, 1H), 7,94 (д, J=8 Гц, 1H), 7,41 (т, 1H).
(Стадия 2)
3-Йод-7-нитро-1H-индазол (2,50 г, 8,65 ммоль), полученный на стадии 1, растворяли в ацетоне (50 мл), и добавляли к нему по каплям при 0°C гидроксид калия (0,73 г, 12,97 ммоль) и 4-метоксибензилхлорид (1,63 г, 10,38 ммоль). Смесь перемешивали в течение 2 часов. В конце реакции смесь разбавляли с помощью воды, и экстрагировали с помощью этилацетата. Экстракт промывали с помощью насыщенного раствора хлорида натрия, сушили над безводным сульфатом магния и фильтровали. Растворитель удаляли при пониженном давлении, и остаток отделяли с помощью колоночной хроматографии с получением 3-йод-7-нитро-1-(4-метоксибензил)-1H-индазола (3,2 г, выход 91%).
1H-ЯМР (400 МГц, CDCl3); δ 8,08 (д, J=8 Гц, 1H), 7,83 (д, J=8 Гц, 1H), 7,28 (м, 1H), 6,97 (д, J=8 Гц, 1H), 6,75 (д, J=8 Гц, 1H), 5,83 (с, 2H), 3,72 (с, 3H).
(Стадия 3)
3-Йод-7-нитро-1-(4-метоксибензил)-1H-индазол (1,50 г, 3,67 ммоль), полученный на стадии 2, растворяли в диметоксиэтане (20 мл), и добавляли к нему по каплям карбонат натрия (1,17 г, 11,01 ммоль), 4-метоксифенилбороновую кислоту (0,84 г, 5,50 ммоль) и тетракис(трифенилфосфин)палладий(0) (0,43 г, 0,37 ммоль). Смесь кипятили с обратным холодильником при перемешивании в течение 2 часов. После охлаждения раствора реакционную смесь разбавляли с помощью воды, и экстрагировали с помощью этилацетата. Экстракт промывали с помощью насыщенного раствора хлорида натрия, сушили над безводным сульфатом магния и фильтровали. Растворитель удаляли при пониженном давлении, и остаток растворяли в трифторуксусной кислоте (20 мл), и полученный раствор кипятили с обратным холодильником при перемешивании в течение 5 часов, и затем растворитель удаляли при пониженном давлении. Полученный остаток разбавляли с помощью воды и экстрагировали с помощью этилацетата. Экстракт промывали с помощью насыщенного раствора хлорида натрия, сушили над безводным сульфатом магния и фильтровали. Растворитель удаляли при пониженном давлении, и остаток отделяли с помощью колоночной хроматографии с получением 3-(4-метоксифенил-1-ил)-7-нитро-1H-индазола (0,85 г, выход 86%).
1H-ЯМР (400 МГц, CDCl3); δ 11,33 (ушир.с, 1H), 8,39 (т, 2H), 7,90 (д, J=8 Гц, 2H), 7,36 (т, 1H), 7,08 (д, J=8 Гц, 2H), 3,91 (с, 3H).
Пример 150: Получение циклопентил-[3-(4-метоксифенил)-1H-индазол-7-ил]амина

Соединение, полученное в синтезе 67, и циклопентанон подвергали взаимодействию в соответствии с такими же методами, как на стадии 3 синтеза 35 и в синтезе 36, с получением названного соединения.
1H-ЯМР (400 МГц, CDCl3); δ 7,83 (д, J=12 Гц, 2H), 7,31 (д, J=8 Гц, 1H), 7,10 (т, 1H), 7,00 (дд, 2H), 6,53 (д, J=8 Гц, 1H), 3,91 (м, 1H), 3,86 (с, 1H), 2,03 (м, 2H), 1,69 (м, 2H), 1,59 (м, 2H), 1,25 (м, 2H)
Пример 151: Получение [3-(4-метоксифенил)-1H-индазол-7-ил]-(тетрагидропиран-4-ил)амина

Соединение, полученное в синтезе 67, и тетрагидропиран-4-он подвергали взаимодействию в соответствии с такими же методами, как на стадии 3 синтеза 35 и в синтезе 36, с получением названного соединения.
1H-ЯМР (400 МГц, CDCl3); δ 7,78 (д, J=12 Гц, 2H), 7,24 (д, J=8 Гц, 1H), 7,06 (т, 1H), 6,98 (д, J=8 Гц, 2H), 6,45 (д, J=8 Гц, 1H), 3,93 (м, 2H), 3,86 (с, 1H), 3,46 (т, 2H), 3,44 (м, 1H), 1,92 (д, J=12 Гц, 2H), 1,39 (м, 2H).
Пример 152: Получение [3-(4-метоксифенил)-1H-индазол-7-ил]-(тетрагидропиран-4-илметил)амина
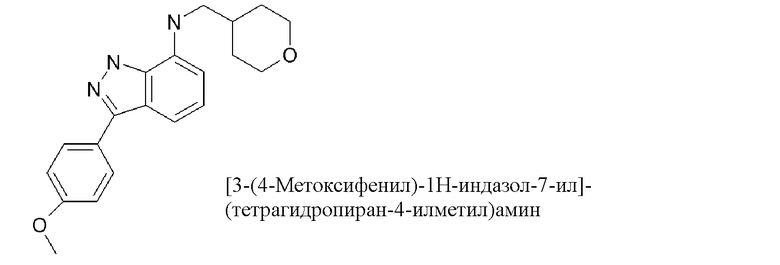
Соединение, полученное в синтезе 67, и тетрагидропиран-4-карбоксиальдегид подвергали взаимодействию в соответствии с такими же методами, как на стадии 3 синтеза 35 и в синтезе 36, с получением названных соединений.
1H-ЯМР (400 МГц, CDCl3); δ 7,78 (д, J=8 Гц, 2H), 7,29 (д, J=8 Гц, 1H), 7,07 (т, 1H), 6,97 (д, J=8 Гц, 2H), 6,45 (д, J=8 Гц, 1H), 3,89 (м, 2H), 3,86 (с, 1H), 3,24 (т, 2H), 3,04 (д, J=8 Гц, 2H), 1,73 (м, 1H), 1,57 (д, J=12 Гц, 2H), 1,30 (м, 2H).
Примеры испытаний
Далее эффекты, обеспечиваемые настоящим изобретением, далее будут разъяснены при помощи следующих примеров испытаний. Однако эффекты настоящего изобретения не ограничиваются теми эффектами, которые иллюстрируются следующими примерами испытаний.
Пример испытания 1: Защитный эффект по отношению к гепатоцитам
Если клетки выделяют in vivo из окружающей среды и начинают их культивирование или консервирование in vitro, то это будет воздействовать на клетки в качестве стресса, и начнет работать механизм апоптоза. При консервации органов или тканей необходимо защищать их от такого первичного стресса.
Для изучения того, насколько эффективно соединения могут защищать выделенные из крыс первичные гепатоциты от первичного стресса, эти гепатоциты подвергали обработке соединениями демонстрационных примеров. Первичные гепатоциты выделяли при помощи метода Seglen PO (Experimental Cell Research 74 (1972) pp. 450-454). Короче говоря, гепатоциты извлекали в соответствии с двухстадийным методом перфузии коллагеназой и затем центрифугировали с использованием градиента перколла (Kreamer BL и etc, In Vitro Cellular & Developmental Biology 22 (1986) pp. 201-211) при низкой скорости вращения (500 об/мин) в течение 10 минут для удаления мертвых клеток. Жизнеспособность гепатоцитов поддерживалась на уровне 90% или более. Клетки суспендировали в среде HepatoZYME media (Gibco BRL) и подсчитывали. 1,5×104 клеток в 100 мкл помещали в 96-луночный планшет (BD biocoat), покрытый прикрепленным к нижней части коллагеном, в течение 3-4 часов. Затем, как показано на фигуре 1, обрабатывали ингибитором каспазы, IDN6556 (Liver Transplantation (2003) 9: pp. 278-284), и соединением примера 21 при концентрации 1 или 10 мкМ. Для оценки защитного эффекта по отношению к гепатоцитам измеряли жизнеспособность клеток через 24, 72 или 144 часов.
Жизнеспособность клеток измеряли по оптической плотности при 440 нм в соответствии с методом WST-1 (MK-400, Takeda). Как показано на фигуре 1, жизнеспособность быстро падала в контрольной группе, которую обрабатывали только с помощью DMSO. В отличие от этого, IDN6556 поддерживал жизнеспособность через 72 часа до примерно 50%, по сравнению с жизнеспособностью через 24 часа, и соединение примера 21 поддерживало жизнеспособность через 144 часа до 80% или более, по сравнению с жизнеспособностью через 24 часа.
Пример испытания 2: Эффект по защите клеток от холодового шока
Для того чтобы воспроизвести условия для консервирования перед трансплантацией органа, приготавливали первично культивированные гепатоциты в соответствии с примером испытания 1, и обрабатывали их соединениями демонстрационных примеров при 9 различных концентрациях, которые готовили путем трехкратного последовательного разбавления при использовании начальной концентрации 25 мкМ. Смесь культивировали при 37°C в течение 1 часа, хранили при 4°C в течение 22 часов, и затем переносили в инкубатор при 37°C и подвергали культивированию. Жизнеспособность клеток измеряли по оптической плотности при 440 нм в соответствии с методом WST-1 (MK-400, Takeda), и определяли значение IC50 (единица измерения: мкМ) при помощи программного обеспечения Prism Software (смотрите таблицу 1).
Результаты, аналогичные приведенным выше результатам, были также получены и для других типов клеток, таких как линия клеток легкого эмбриона человека (LB-HEL), а также гепатоцитов (смотрите фигуру 2 и таблицу 2). Более конкретно, LB-HEL клетки высевали при 1,5×106 клеток/лунка с использованием среды DMEM (Gibco BRL) и обрабатывали с помощью DMSO, nec-1, IM-54 или соединениями примеров 126, 127, 137 и 138 при указанных концентрациях. После хранения при 4°C в течение 24 часов смесь переносили в инкубатор при 37°C и проводили культивирование. Для проверки способности к восстановлению при 37°C, измеряли жизнеспособность клеток через 24 часа после их переноса в инкубатор по оптической плотности при 440 нм в соответствии с методом WST-1 (MK-400, Takeda) и сравнивали с жизнеспособностью клеток до холодового шока (фигура 2). Соединения примеров 126, 127, 137 и 138 проявляли защитный эффект против холодового шока. Однако, ни nec-1, который ингибирует некропоптоз (Nat Chem Biol. (2005) 1:112-119), ни IDN6556, который ингибирует апоптоз (ингибитор pan-caspase), не проявляли существенного защитного эффекта.
Эти результаты подтверждают тот факт, что соединения настоящего изобретения проявляют значительный эффект по предотвращению повреждения и защите от повреждения, вызванного холодовым шоком и повторным подогревом, которые неизбежно происходят при трансплантации органа или консервации клеток.
Пример испытания 3: Защитный эффект от повреждения при консервировании при низких температурах на модели перфузии печени, удаленной у крыс
Для того чтобы проверить эффект консервирования органа, использовали модель перфузии печени, удаленной у крыс, для оценки защитного эффекта соединений демонстрационных примеров от повреждения при консервировании при низких температурах. Белую крысу подвергали анестезированию путем интраперитонеальной инъекции пентобарбитала натрия (50 мг/кг) и вводили гепарин (500 Е/кг) путем внутривенной инъекции для предотвращения коагуляции крови. После разреза по абдоминальной средней линии, для измерения образование желчи в желчный проток вставляли полиэтиленовую трубку-10, и перевязывали коронарную вену. Полиэтиленовую трубку-190 вставляли в воротную вену печени, и пропускали перфузируемую жидкость, бикарбонатный буфер Кребса-Хенселейта (KHBB) (при pH 7,4 при 37°C), при скорости 4 мл/минут/грамм. Для того чтобы предотвратить расширение печени, вызываемое перфузируемой жидкостью, открывали полую нижнюю вену, и затем печень извлекали из окружающих ее тканей. Во время перфузии, непрерывно вводили в KHBB смесь газов (O2 95% и CO2 5%) для обеспечения кислородом. Извлеченную печень подвергали перфузии с помощью KHBB в течение 5 минут для стабилизации, и затем промывали струей 10 мл смеси гистидин-триптофан-кетоглутарат (HTK) (при 4°C) для удаления KHBB. Контрольную группу и испытуемые группы консервировали при 4°C: первую в 60 мл раствора HTK, а последние в 60 мл раствора HTK, к которому добавляли соединение примера 126 с концентрацией 30 мкМ.
Для оценки эффекта консервирования испытуемыми соединениями в зависимости от периода консервирования при низких температурах, извлеченную печень консервировали путем хранения в условиях низких температур (4°C) в течение 6, 24, 48 и 72 часов, и промывали путем введения 10 мл HTK (4°C) в воротную вену печени. Выделяемую в этот момент перфузируемую жидкость собирали, и проводили измерения маркеров повреждения печени, активность лактатдегидрогеназы (LDH), аспартатаминотрансферазы (AST) и аланинаминотрансферазы (ALT) (фигура 3). LDH, ALT и AST активность в перфузируемой жидкости измеряли в соответствии со стандартным методом адсорбционного анализа, используемым в биохимических автоматических анализаторах Hitachi 7150.
Пример испытания 4: Защитный эффект от повреждения при холодовой ишемии/подогретой реперфузии на модели перфузии печени, удаленной у крыс
Для того чтобы проверить защитный эффект от повреждения в результате ишемии и реперфузии, которые обязательно происходят во время трансплантации органа, использовали модель перфузии печени, удаленной у крыс, из примера испытания 3 для оценки защитного эффекта соединений демонстрационных примеров. Также как в примере испытания 3, извлеченную печень подвергали перфузии с помощью KHBB в течение 5 минут для стабилизации, и затем промывали струей 10 мл смеси гистидин-триптофан кетоглутарат (HTK) (при 4°C) для удаления KHBB. Контрольную группу и группу, подвергнутую обработке, консервировали при 4°C: первую в 60 мл раствора HTK, а вторую в 60 мл раствора HTK, к которому добавляли соединение примера 126 с концентрацией 30 мкМ. Через 24 часа после консервирования при низких температурах извлеченную печень подсоединяли к установке для перфузии печени, которая поддерживает температуру 37°C, и затем повторно перфузировали с помощью KHBB (при pH 7,4 при 37°C) через воротную вену печени при скорости 4 мл/минут/грамм в течение 80 минут. В случае IPRL группы без консервирования при низких температурах, извлеченную печень не хранили в HTK, а немедленно перфузировали с помощью KHBB в течение 2 часов.
Для того чтобы наблюдать защитный эффект от повреждения в результате ишемии и реперфузии, измеряли образование желчи и активность LDH в перфузируемой жидкости. Собирали перфузируемую жидкость KHBB, которая высвобождалась из печени через 5, 10, 20, 30, 40, 50, 60, 70 и 80 минут после реперфузии, и измеряли активность LDH в перфузируемой жидкости при помощи биохимического автоматического анализатора Hitachi 7150 (фигура 4). Собирали желчь, выделяемую через полиэтиленовую трубку, которая была вставлена в желчный проток, в период 0-20, 20-40, 40-60 и 60-80 минут после реперфузии, и измеряли образование желчи (фигура 5).
Результатами измерения активности LDH, которую выделяли с помощью перфузируемой жидкости путем реперфузии после 24-часового консервирования извлеченной печени при низких температурах, являются следующими. Активность LDH в перфузируемой жидкости IPRL группы без консервирования при низких температурах составляла около 20-110 Е/л в течение 80 минут реперфузии, которая является самой низкой активностью LDH во время реперфузии. Однако активность LDH в перфузируемой жидкости группы HTK при консервировании при низких температурах в растворе HTK в течение 24 часов составляла около 250-680 U/л в течение 80 минут реперфузии, что означает, что активность LDH была исключительно повышена по сравнению с IPRL группой во время реперфузии. В отличие от этого группа, подвергнутая обработке при помощи 30 мкМ испытуемого лекарственного средства, соединения примера 126, ингибировала активность LDH в перфузируемой жидкости на 50-70%, до тех пор, пока не проходили 50 минут реперфузии, по сравнению с HTK группой, и значительно ингибировала повреждение клеток печени, вызываемое консервированием при низких температурах и реперфузией (фигура 4).
В IPRL группе без консервирования при низких температурах, желчь выделялась со скоростью 1,61-2,40 мкл/минута/грамм при 0-20, 20-40, 40-60 и 60-80 минут перфузии, и, таким образом, суммарное количество образовавшейся желчи составляло 0,154±0,017 мл/грамм печени в течение 80 минут реперфузии. Однако суммарное количество образовавшейся желчи для HTK группы при консервировании при низких температурах в течение 24 часов составляло 0,044±0,006 мл/грамм печени, что означает, что образование желчи существенно повышалось на 70% или более, по сравнению с IPRL группой. Суммарное количество образовавшейся желчи для группы, подвергнутой обработке при помощи 30 мкМ соединения примера 126, составляло 0,070 ± 0,006 мл/грамм печени, что означает, что соединение значительно ингибировало снижение образования желчи, вызываемое консервированием при низких температурах и реперфузией (фигура 5).
ПРОМЫШЛЕННАЯ ПРИМЕНИМОСТЬ
При сопоставлении полученных результатов становится очевидным, что соединения настоящего изобретения могут предотвращать некроцитоз органов, вызываемый реперфузией после хранения при низких температурах, и ингибируют снижение их функции. Поэтому, считают, что соединения настоящего изобретения являются исключительно эффективными в качестве консервирующего средства при трансплантации органа.
| название | год | авторы | номер документа |
|---|---|---|---|
| СОЕДИНЕНИЯ ИНДОЛА И ИНДАЗОЛА В КАЧЕСТВЕ ИНГИБИТОРА НЕКРОЗА КЛЕТКИ | 2008 |
|
RU2437883C1 |
| СОЕДИНЕНИЯ ИНДОЛА В КАЧЕСТВЕ ИНГИБИТОРОВ КЛЕТОЧНОГО НЕКРОЗА | 2008 |
|
RU2477282C2 |
| АКТИВАТОРЫ ГЛЮКОКИНАЗЫ И ФАРМАЦЕВТИЧЕСКИЕ КОМПОЗИЦИИ, СОДЕРЖАЩИЕ ИХ В КАЧЕСТВЕ АКТИВНОГО ИНГРЕДИЕНТА | 2008 |
|
RU2450001C2 |
| ГЕТЕРОЦИКЛИЧЕСКИЕ ПРОИЗВОДНЫЕ И ИХ ПРИМЕНЕНИЕ | 2014 |
|
RU2681849C2 |
| НОВЫЕ СОЕДИНЕНИЯ, ИХ ИЗОМЕР ИЛИ ИХ ФАРМАЦЕВТИЧЕСКИ ПРИЕМЛЕМЫЕ СОЛИ В КАЧЕСТВЕ АНТАГОНИСТА ВАНИЛОИДНОГО РЕЦЕПТОРА И СОДЕРЖАЩАЯ ИХ ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ | 2007 |
|
RU2448108C2 |
| 2-ПИРИДИЛЗАМЕЩЕННЫЕ ИМИДАЗОЛЫ В КАЧЕСИВЕ ТЕРАПЕВТИЧЕСКИХ ИНГИБИТОРОВ ALK5 И/ИЛИ ALK4 | 2011 |
|
RU2518069C1 |
| ГЕТЕРОЦИКЛИЧЕСКИЕ ПРОИЗВОДНЫЕ И ИХ ПРИМЕНЕНИЕ | 2015 |
|
RU2711502C2 |
| ПРОИЗВОДНЫЕ ТИЕНО[3,2-d]ПИРИМИДИНА, ОБЛАДАЮЩИЕ ИНГИБИРУЮЩЕЙ АКТИВНОСТЬЮ В ОТНОШЕНИИ ПРОТЕИНКИНАЗЫ | 2011 |
|
RU2524210C2 |
| ПРОИЗВОДНЫЕ ХИНАЗОЛИНА ДЛЯ ТОРМОЖЕНИЯ РОСТА РАКОВЫХ КЛЕТОК И СПОСОБ ИХ ПОЛУЧЕНИЯ | 2005 |
|
RU2362773C2 |
| НОВЫЕ ПРОИЗВОДНЫЕ ОКСАЗОЛИДИНОНА С ЦИКЛИЧЕСКИМ АМИДОКСИМОМ ИЛИ ЦИКЛИЧЕСКИМ АМИДРАЗОНОМ И СОДЕРЖАЩИЕ ИХ ФАРМАЦЕВТИЧЕСКИЕ КОМПОЗИЦИИ | 2009 |
|
RU2468023C1 |
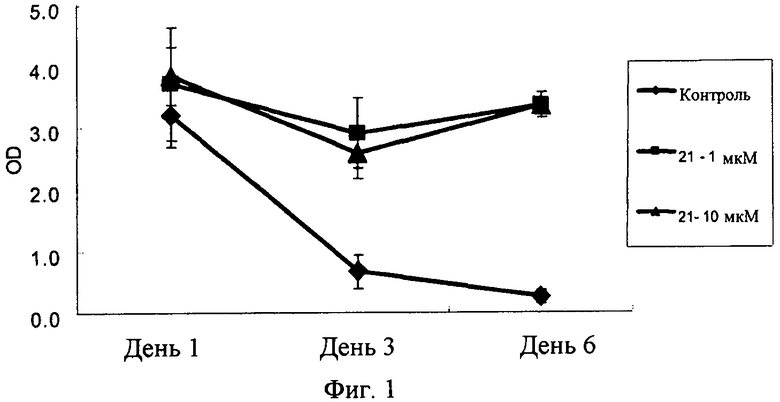
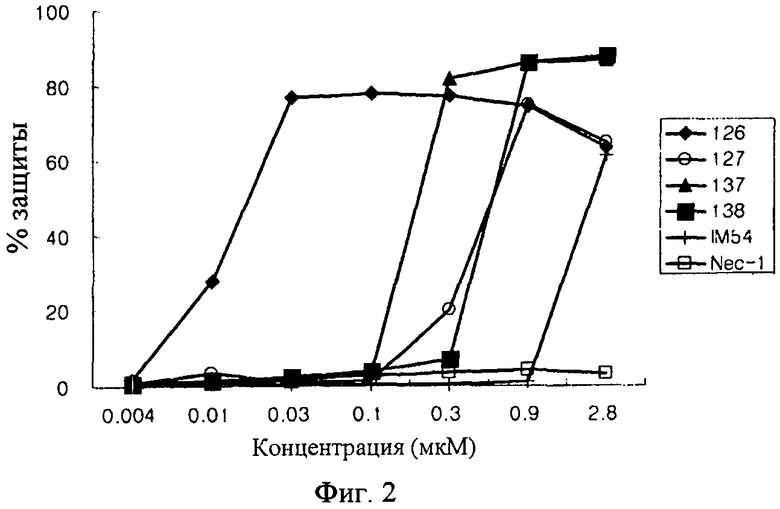
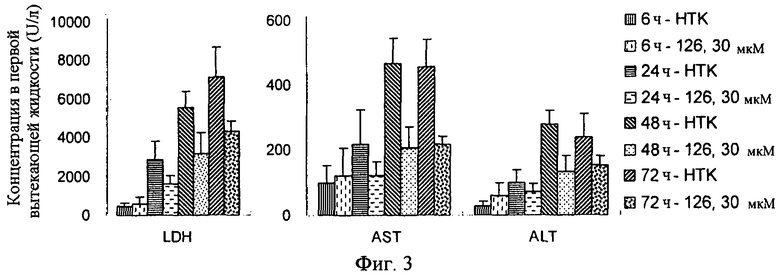

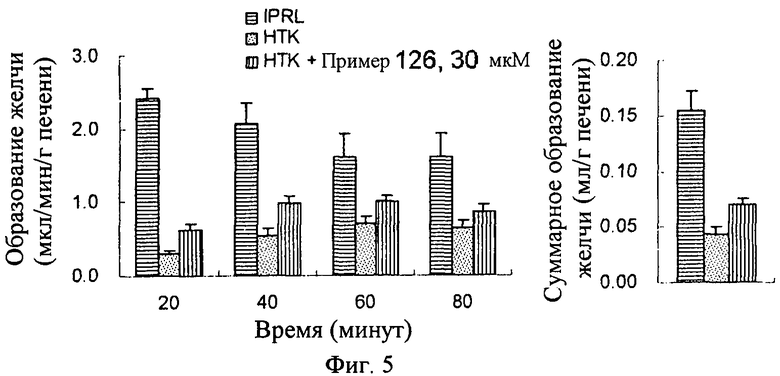
Изобретение относится к композиции для консервирования клеток, тканей и органов, включающей в качестве активного ингредиента соединения индола и индазола формулы (1), или их фармацевтически приемлемые соли или изомеры, которые являются эффективными для предотвращения повреждения органов, выделенных клеточных систем или тканей, вызванного хранением при низких температурах, операцией по трансплантации или посттрансплантационной реперфузией. Изобретение также относится к способу получения указанной композиции. 3 н. и 18 з.п. ф-лы, 5 ил., 6 табл, 156 пр.
1. Композиция для консервирования клеток, тканей и органов, включающая в качестве эффективного ингредиента соединение следующей формулы (1) или его фармацевтически приемлемую соль или изомер:
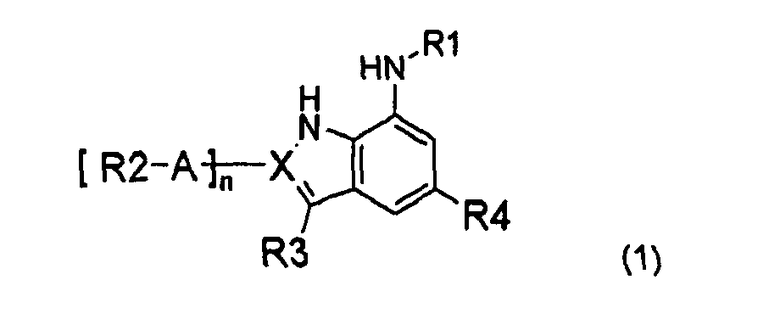
где Х представляет С или N,
n является 0 или 1, и n является 1, когда Х является С, и n является 0, когда Х является N,
А представляет непосредственную химическую связь, С3-С8-циклоалкил, фенил, или 5-6-членный гетероарил или гетероцикл, каждый из которых включает 1-3 гетероатома, выбранных из атомов N, О и S,
R1 представляет водород, -С(O)-В-Х'-R7 или -(CR5R6)m-B-X'-R7,
m является целым числом от 0 до 4,
каждый из R5 и R6 независимо представляет водород или С1-С5-алкил,
В представляет непосредственную химическую связь, С3-С8-циклоалкил, необязательно содержащий оксо, или 3-10-членный гетероцикл или гетероарил, каждый из которых включает 1-3 гетероатома, выбранных из атомов О, S и N,
X' представляет непосредственную химическую связь, -С(O)-, -SO2-, -СО2- или -C(O)NR5-,
R7 представляет водород, C1-С6-алкил, галоген-С1-С6-алкил, галоген, (CR5R6)m-фенил, (CR5R6)m-гидрокси или (CR5R6)m-гетероцикл, где гетероцикл необязательно содержит оксо и является 3-10-членным кольцом, включающим 1-3 гетероатома, выбранных из атомов N, О и S,
R2 представляет - (CR5R6)m-D-X''-R8,
D представляет непосредственную химическую связь или 3-10-членный гетероцикл или гетероарил, каждый из которых необязательно содержит оксо и необязательно конденсирован, и включает 1-4 гетероатома, выбранных из атомов N, О и S,
X'' представляет непосредственную химическую связь, -С(O)-, -С(O)O-, -NR5C(O)-, -C(O)NR5- или -O-,
R8 представляет водород, галоген, C1-С6-алкил, галоген-С1-С6-алкил, три(C1-С6-алкил)силан или гидрокси-С1-С6-алкил,
R3 представляет водород, галоген, циано, нитро, арил-R9 или (CR5R6)m-D-R9,
R9 представляет водород, галоген, C1-С6-алкил, циано, нитро или C1-С6-алкокси,
R4 представляет -(CR5R6)m-Y-D-R10,
Y представляет непосредственную химическую связь, -С(O)O- или -О-, и
R10 представляет водород, нитро, галоген, C1-С6-алкил, карбокси-С1-С6-алкил, арил или -C(O)O-R5,
где каждый из указанных алкил, алкокси, арил, циклоалкил, гетероцикл и гетероарил необязательно замещен при помощи одного или более заместителей, выбранных из группы, состоящей из гидрокси, галогена, нитрила, амино, C1-С6-алкиламино, ди(C1-С6-алкил)амино, C1-С6-алкила, галоген-C1-С6-алкила, C1-С6-алкилсульфонила, арил-C1-С6-алкокси и оксо.
2. Композиция по п.1, где
Х представляет С или N,
n является 0 или 1, и n является 1, когда Х является С, и n является 0, когда Х является N,
А представляет непосредственную химическую связь, фенил, или 5-6-членный гетероарил или гетероцикл, каждый из которых включает 1-3 гетероатома, выбранных из атомов N, О и S,
R1 представляет водород, -С(O)-В-Х'-R7 или -(CR5R6)m-B-X'-R7,
m является целым числом от 0 до 2,
каждый из R5 и R6 независимо представляет водород или C1-С5-алкил,
В представляет непосредственную химическую связь, С4-С7-циклоалкил, необязательно содержащий оксо, или 4-8-членный гетероцикл или гетероарил, каждый из которых включает 1-3 гетероатома, выбранных из атомов О, S и N,
Х' представляет непосредственную химическую связь, -С(O)-, -SO2-, -СО2- или -C(O)NH-,
R7 представляет водород, C1-С6-алкил, галоген-C1-С6-алкил, галоген, (CR5R6)m-фенил, (CR5R6)m-гидрокси или (CR5R6)m-гетероцикл, где гетероцикл необязательно содержит оксо и является 4-8-членным кольцом, включающим 1-3 гетероатома, выбранных из атомов N, О и S,
R2 представляет -(CR5R6)m-D-X''-R8,
D представляет непосредственную химическую связь или 4-8-членный гетероцикл или гетероарил, каждый из которых необязательно содержит оксо и необязательно конденсирован, и включает 1-4 гетероатома, выбранных из атомов N, О и S,
X' представляет -С(O)-, -С(O)O-, -NR5C(O)-, -C(O)NR5- или -O-,
R8 представляет водород, галоген, C1-С6-алкил, галоген-C1-С6-алкил, три(C1-С6-алкил)силан или гидрокси-C1-С6-алкил,
R3 представляет водород, галоген, циано, нитро, арил-R9 или (CR5R6)m-D-R9,
R9 представляет водород, галоген, C1-С6-алкил, циано, нитро или C1-С6-алкокси,
R4 представляет -(CR5R6)m-Y-D-R10,
Y представляет непосредственную химическую связь, -С(O)O- или -O-, и
R10 представляет водород, нитро, галоген, C1-С6-алкил, карбокси-C1-С6-алкил, арил или -C(O)O-R5.
3. Композиция по п.2, где соединение формулы (1) имеет структуру индола следующей формулы (1а) или структуру индазола следующей формулы (1b):
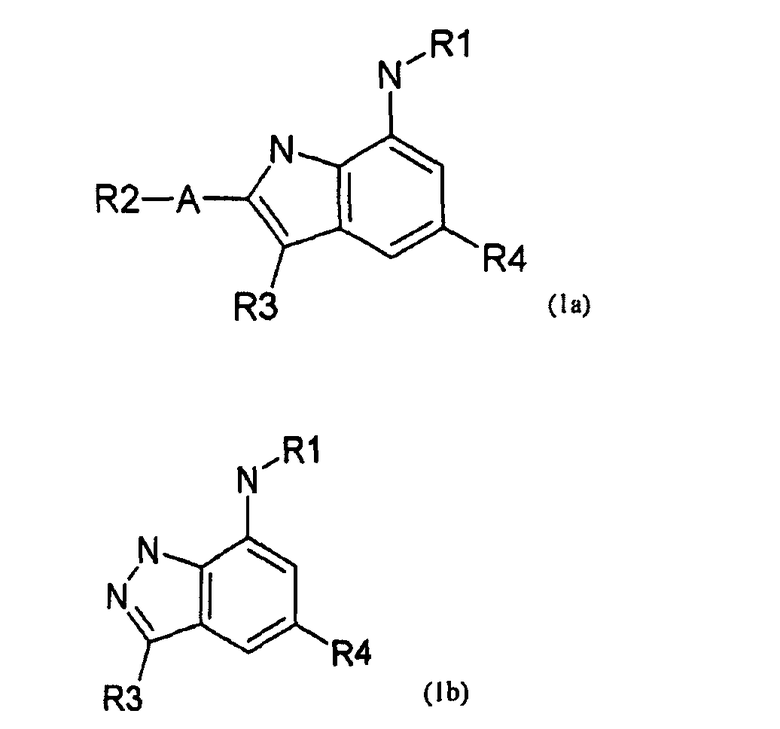
4. Композиция по п.2, где А выбирают из группы, состоящей из фенила, пиридина, 1,4-пиразина, 4, 5-дигидротиазола, тиазола, 4,5-дигидрооксазола, [1,2,4]оксадиазола и [1,3,4]оксадиазола.
5. Композиция по п.2, где R1 представляет -С(O)-В-Х'-R7 или - (CHR5)m-В-Х'-R7, где m является целым числом от 0 до 2; R5 представляет C1-С3-алкил; В представляет непосредственную химическую связь, C5-С6-циклоалкил, необязательно содержащий оксо, или 5-6-членный гетероцикл или гетероарил, каждый из которых включает 1-3 гетероатома, выбранных из атомов О, S и N;
Х' представляет непосредственную химическую связь, -С(O)-, -SO2-, -CO2- или -C(O)NH-; и R7 представляет водород, C1-С3-алкил, галоген-C1-С3-алкил, галоген, (СН2)m-фенил, (СН2)m-гидрокси или (CH2)m-гетероцикл, где гетероцикл необязательно содержит оксо и является 5-6-членным кольцом, включающим 1-3 гетероатома, выбранных из атомов N, О и S.
6. Композиция по п.5, где В выбирают из группы, состоящей из циклопентила, циклогексила, пиперидина, тетрагидропирана, оксоциклогексила, пирролидина, дифторциклогексила и тетрагидрофурана.
7. Композиция по п.5, где R7 выбирают из группы, состоящей из водорода, метила, этила, изопропила, бензила, гидроксиметила, (морфолин-4-ил)этила, тетрагидрофурана, 2,2,2-трифторэтила, гидроксиэтила, 1,1-диоксотиоморфолина, тетрагидропирана, (тетрагидропиран-4-ил)метила и трифторметила.
8. Композиция по п.2, где D представляет непосредственную химическую связь, или его выбирают из группы, состоящей из пиперазина, пирролидина, морфолина, 1,1-диоксотиоморфолина и оксопиперазина.
9. Композиция по п.2, где R8 выбирают из группы, состоящей из водорода, этила, гидроксиметила, метила и фтора.
10. Композиция по п.2, где R3 представляет водород; галоген; фенил, необязательно замещенный при помощи алкокси; или 6-членный гетероциклилметил, включающий 1-3 гетероатома, выбранных из атомов N, S и О в качестве членов кольца, и необязательно содержащий оксо.
11. Композиция по п.10, где R3 выбирают из группы, состоящей из водорода, брома, фенила, метоксифенила, морфолин-4-илметила, оксопиперазин-4-илметила и 1,1-диоксотиоморфолин-4-ил-метила.
12. Композиция по п.2, где R4 представляет -(CH2)m-Y-D-R10, где m является целым числом от 0 до 2; Y представляет непосредственную химическую связь, -С(O)O- или -O-; D представляет пиридин или 5-6-членный гетероцикл, включающий 1-3 гетероатома, выбранных из атомов N, S и О, и необязательно содержащий оксо; и R10 представляет водород, галоген, C1-С3-алкил, -(СН3)-CO2H, арил или -C(O)O-R5.
13. Композиция по п.12, где D выбирают из группы, состоящей из 1,1-диоксотиоморфолина, оксопиперазина, пиридина, морфолина и 4,5-дигидротиазола.
14. Композиция по п.12, где R10 выбирают из группы, состоящей из водорода, фтора, хлора, брома, метила, этила и -(СН2)-CO2H.
15. Композиция по п.2, где соединение выбирают из группы, состоящей из следующих соединений:
циклопентил-[2-(4,5-дигидро-1,3-тиазол-2-ил)-1Н-индол-7-ил]амин;
[2-(4,5-дигидротиазол-2-ил)-1Н-индол-7-ил]-(4-метил-циклогексил)амин;
[2-(4,5-дигидротиазол-2-ил)-1H-индол-7-ил]пиперидин-4-ил-амин;
2-{5-[7-(тетрагидропиран-4-иламино)-1Н-индол-2-ил]-[1,2,4]оксадиазол-3-ил}этанол;
[(R)-2-(7-циклопентиламино-1Н-индол-2-ил)-4,5-дигидро-1,3-тиазол-4-ил]метанол;
циклопентил[2-((R)-4-пирролидин-1-илметил-4,5-дигидротиазол-2-ил)-1Н-индол-7-ил]амин;
{(R)-2-[7-(тетрагидропиран-4-иламино)-1Н-индол-2-ил]-4,5-дигидротиазол-4-ил}метанол;
[(R)-2-(7-циклопентиламино-5-фтор-1Н-индол-2-ил)-4,5-дигидротиазол-4-ил]метанол;
{(R)-2-[5-фтор-7-(тетрагидропиран-4-иламино)-1Н-индол-2-ил]-4,5-дигидротиазол-4-ил}метанол;
{(R)-2-[5-(пиридин-3-илокси)-7-(тетрагидропиран-4-иламино)-1Н-индол-2-ил]-4,5-дигидротиазол-4-ил}метанол;
[(R)-2-(5-хлор-7-циклопентиламино-1Н-индол-2-ил)-4,5-дигидротиазол-4-ил]уксусная кислота;
этиловый эфир [(R)-2-(5-хлор-7-циклопентиламино-1Н-индол-2-ил)-4,5-дигидротиазол-4-ил]уксусной кислоты;
2-{(R)-2-[5-хлор-7-(тетрагидропиран-4-иламино)-1Н-индол-2-ил]-4,5-дигидротиазол-4-ил}этанол;
1-[4-(2-{(R)-2-[5-хлор-7-(тетрагидропиран-4-иламино)-1Н-индол-2-ил]-4,5-дигидротиазол-4-ил}этил)пиперазин-1-ил]-2-гидроксиэтанон;
1-(2-{(R)-2-[5-хлор-7-(тетрагидропиран-4-иламино)-1Н-индол-2-ил]-4,5-дигидротиазол-4-ил}этил)пирролидин-3-ол;
[(R)-2-(5-бром-7-циклопентиламино-1Н-индол-2-ил)-4,5-дигидротиазол-4-ил]уксусная кислота;
[(R)-2-(7-циклопентиламино-5-этокси-1Н-индол-2-ил)-4,5-дигидротиазол-4-ил]уксусная кислота;
[2-(7-циклопентиламино-5-фенокси-1Н-индол-2-ил)-4,5-дигидротиазол-4-ил]уксусная кислота;
[(R)-2-(7-циклопентиламино-5-фенокси-1Н-индол-2-ил)-4,5-дигидротиазол-4-ил]уксусная кислота;
[(S)-2-(7-циклопентиламино-5-фенокси-1Н-индол-2-ил)-4,5-дигидротиазол-4-ил]уксусная кислота;
этиловый эфир 3-[(R)-2-(5-хлор-7-циклопентиламино-1Н-индол-2-ил)-4,5-дигидротиазол-4-ил]пропионовой кислоты;
3-[(R)-2-(5-хлор-7-циклопентиламино-1Н-индол-2-ил)-4,5-дигидротиазол-4-ил]пропионовая кислота;
циклопентил(2-пиридин-2-ил-1Н-индол-7-ил)амин;
циклопентил(2-пиразин-2-ил-1Н-индол-7-ил)амин;
(2-пиразин-2-ил-1Н-индол-7-ил)-(тетрагидропиран-4-ил)амин;
циклопентил(2-тиазол-2-ил-1Н-индол-7-ил)амин;
этиловый эфир 2-(7-циклопентиламино-5-метил-1Н-индол-2-ил)тиазол-4-карбоновой кислоты;
2-(7-циклопентиламино-5-метил-1Н-индол-2-ил)-тиазол-4-карбоновая кислота;
[2-(7-циклопентиламино-5-метил-1Н-индол-2-ил)-тиазол-4-ил]-метанол;
[2-(7-циклопентиламино-5-метил-1Н-индол-2-ил)-тиазол-5-ил]-метанол;
циклопентил(5-метил-2-[1,3,4]оксадиазол-2-ил-1Н-индол-7-ил)амин;
циклопентил(5-метил-2-пиридин-2-ил-1Н-индол-7-ил)амин;
(5-метил-2-пиридин-2-ил-1Н-индол-7-ил)-(тетрагидропиран-4-ил)амин;
циклогексил(5-метил-2-пиридин-2-ил-1Н-индол-7-ил)амин;
1-[4-(5-метил-2-пиридин-2-ил-1Н-индол-7-иламино)пиперидин-1-ил]этанон;
(1-метилпиперидин-4-ил)-(5-метил-2-пиридин-2-ил-1Н-индол-7-ил)амин;
4-(5-метил-2-пиридин-2-ил-1Н-индол-7-иламино)-циклогексанон;
(1-бензилпирролидин-3-ил)-(5-метил-2-пиридин-2-ил-1Н-индол-7-ил)амин;
циклопентилметил-(5-метил-2-пиридин-2-ил-1Н-индол-7-ил)-амин;
N-(5-метил-2-пиридин-2-ил-1Н-индол-7-ил)бензамид;
циклопентил(5-метил-2-пиразин-2-ил-1Н-индол-7-ил)амин;
циклопентил(5-этокси-2-пиридин-2-ил-1Н-индол-7-ил)амин;
циклопентил(5-фенокси-2-пиридин-2-ил-1Н-индол-7-ил)амин;
циклопентил(3,5-диметил-2-фенил-1Н-индол-7-ил)амин;
циклопентил(5-метил-2-фенил-1Н-индол-7-ил)амин;
(2-циклогексил-5-метил-1Н-индол-7-ил)циклопентиламин;
циклопентил[5-метил-2-(6-метилпиридин-2-ил)-1Н-индол-7-ил]-амин;
(5-метил-2-фенил-1Н-индол-7-ил)-(тетрагидропиран-4-ил)-амин;
(5-метил-2-фенил-1Н-индол-7-ил)-(1-метилпиперидин-4-ил)-амин;
1-[4-(5-метил-2-фенил-1Н-индол-7-иламино)пиперидин-1-ил]-этанон;
гидрохлорид(5-метил-2-фенил-1Н-индол-7-ил)пиперидин-4-ил-амина;
2-гидрокси-1-[4-(5-метил-2-фенил-1Н-индол-7-иламино)-пиперидин-1-ил]этанон;
(1-метансульфонилпиперидин-4-ил)-(5-метил-2-фенил-1Н-индол-7-ил)амин;
4-(5-метил-2-фенил-1Н-индол-7-иламино)циклогексан-карбоновая кислота;
(2-морфолин-4-илэтил)амид 4-(5-метил-2-фенил-1Н-индол-7-иламино)циклогексан-карбоновая кислота;
циклопентилметил(5-метил-2-фенил-1Н-индол-7-ил)амин;
(5-метил-2-фенил-1Н-индол-7-ил)-(тетрагидропиран-4-илметил)амин;
(5-хлор-2-фенил-1Н-индол-7-ил)циклопентиламин;
(5-хлор-2-фенил-1Н-индол-7-ил)-(тетрагидропиран-4-ил)амин;
(5-хлор-2-фенил-1Н-индол-7-ил)-(1-метилпиперидин-4-ил)-амин;
(5-хлор-2-фенил-1Н-индол-7-ил)циклогексиламин;
(1-бензилпирролидин-3-ил)-(5-хлор-2-фенил-1Н-индол-7-ил)-амин;
метиловый эфир 4-(5-хлор-7-циклопентиламино-1Н-индол-2-ил)бензойной кислоты;
4-(5-хлор-7-циклопентиламино-1Н-индол-2-ил)бензойная кислота;
[4-(5-хлор-7-циклопентиламино-1Н-индол-2-ил)фенил]метанол;
метиловый эфир 4-(7-циклопентиламино-5-метил-1Н-индол-2-ил)бензойной кислоты;
метиловый эфир 2-(5-хлор-7-циклопентиламино-1Н-индол-2-ил)бензойной кислоты;
2-(5-хлор-7-циклопентиламино-1Н-индол-2-ил)бензойная кислота;
[2-(5-хлор-7-циклопентиламино-1Н-индол-2-ил)фенил]метанол;
этиловый эфир 7-циклопентиламино-2-фенил-1Н-индол-5-карбоновой кислоты;
7-циклопентиламино-2-фенил-1Н-индол-5-карбоновая кислота;
(7-циклопентиламино-2-фенил-1Н-индол-5-ил)метанол;
этиловый эфир (7-циклопентиламино-2-фенил-1Н-индол-5-ил)уксусной кислоты;
(7-циклопентиламино-2-фенил-1Н-индол-5-ил)уксусная кислота;
2-[(4S)-2-[5-метил-7-(оксан-4-илметиламино)-1Н-индол-2-ил]-4,5-дигидро-1,3-тиазол-4-ил]уксусная кислота;
2-[(4S)-2-[5-хлор-7-(оксан-4-илметиламино)-1Н-индол-2-ил]-4,5-дигидро-1,3-тиазол-4-ил]уксусная кислота;
2-[(4S)-2-[7-[(4,4-Дифторциклогексил)амино]-5-метил-1Н-индол-2-ил]-4,5-дигидро-1,3-тиазол-4-ил]уксусная кислота;
2-[(4S)-2-[7-(оксан-4-иламино)-5-фенокси-1Н-индол-2-ил]-4,5-дигидро-1,3-тиазол-4-ил]уксусная кислота;
2-[(4R)-2-[7-(оксан-4-иламино)-5-фенокси-1Н-индол-2-ил]-4,5-дигидро-1,3-тиазол-4-ил]уксусная кислота;
2-[(4R)-2-[7-(оксан-4-илметиламино)-5-фенокси-1Н-индол-2-ил]-4,5-дигидро-1,3-тиазоле-4-ил]уксусная кислота;
2-[(4S)-2-[7-(циклопентиламино)-1Н-индол-2-ил]-4,5-дигидро-1,3-тиазол-4-ил]уксусная кислота;
2-[(4S)-2-[7-[(1-ацетилпирролидин-3-ил)амино]-5-метил-1Н-индол-2-ил]-4,5-дигидро-1,3-тиазол-4-ил]уксусная кислота;
2-[(4S)-2-[7-(оксан-4-илметиламино)-1Н-индол-2-ил]-4,5-дигидро-1,3-тиазол-4-ил]уксусная кислота;
2-[(4S)-2-[7-(оксан-4-иламино)-1Н-индол-2-ил]-4,5-дигидро-1,3-тиазол-4-ил]уксусная кислота;
2-[(4S)-2-[7-(оксан-2-илметиламино)-1Н-индол-2-ил]-4,5-дигидро-1,3-тиазол-4-ил]уксусная кислота;
2-[(4S)-2-[5-метил-7-[[1-(3,3,3-трифторпропаноил)-пиперидин-4-ил]амино]-1Н-индол-2-ил]-4,5-дигидро-1,3-тиазол-4-ил]уксусная кислота;
2-[(4R)-2-[7-(циклопентиламино)-5-метил-1Н-индол-2-ил]-4,5-дигидро-1,3-тиазол-4-ил]уксусная кислота;
2-[(4R)-2-[5-метил-7-(оксан-4-илметиламино)-1Н-индол-2-ил]-4,5-дигидро-1,3-тиазол-4-ил]уксусная кислота;
4-[2-[(4S)-2-[5-метил-7-(оксан-4-илметиламино)-1Н-индол-2-ил]-4,5-дигидро-1,3-тиазол-4-ил]этил]пиперазин-2-он;
2-[(4S)-4-[2-(1,1-диоксо-1,4-тиазинан-4-ил)этил]-4,5-дигидро-1,3-тиазол-2-ил]-5-метил-N-(оксан-4-илметил)-1Н-индол-7-иламин;
N-(4,4-дифторциклогексил)-5-метил-2-[(4S)-4-(2-морфолин-4-илэтил)-4,5-дигидро-1,3-тиазол-2-ил]-1Н-индол-7-иламин;
4-[2-[(4S)-2-[7-[(4,4-дифторциклогексил)амино]-5-метил-1Н-индол-2-ил]-4,5-дигидро-1,3-тиазол-4-ил]этил]пиперазин-2-он;
4-[2-[(4S)-2-[7-(оксан-4-илметиламино)-5-фенокси-1Н-индол-2-ил]-4,5-дигидро-1,3-тиазоле-4-ил]этил]пиперазин-2-он;
2-[(4S)-4-(2-морфолин-4-илэтил)-4,5-дигидро-1,3-тиазол-2-ил]-N-(оксан-4-илметил)-5-фенокси-1Н-индол-7-амин;
5-метил-2-[(4S)-4-(2-морфолин-4-илэтил)-4,5-дигидро-1,3-тиазол-2-ил]-N-(оксан-4-илметил)-1Н-индол-7-амин;
1-[2-[(4S)-2-[5-метил-7-(оксан-4-илметиламино)-1Н-индол-2-ил]-4,5-дигидро-1,3-тиазол-4-ил]этил]пиперидин-4-карбоксиамид;
[(2R)-1-[2-[(4S)-2-[5-метил-7-(оксан-4-илметиламино)-1Н-индол-2-ил]-4,5-дигидро-1,3-тиазол-4-ил]этил]пирролидин-2-ил]-метанол;
(2S)-1-[2-[(4S)-2-[5-метил-7-(оксан-4-илметиламино)-1Н-индол-2-ил]-4,5-дигидро-1,3-тиазол-4-ил]этил]пирролидин-2-карбоксиамид;
4-[2-[(4R)-2-[7-(циклопентиламино)-5-метил-1Н-индол-2-ил]-4,5-дигидро-1,3-тиазол-4-ил]этил]пиперазин-2-он;
2-[(4S)-2-[7-(циклопентиламино)-5-метил-1Н-индол-2-ил]-4,5-дигидро-1,3-оксазол-4-ил]уксусная кислота;
{(S)-2-[5-метил-7-(тетрагидропиран-4-иламино)-1Н-индол-2-ил]-4,5-дигидро-оксазол-4-ил}уксусная кислота;
2-[(4S)-2-[5-метил-7-(тетрагидропиран-4-иламино)-1Н-индол-2-ил]-4,5-дигидро-1,3-оксазол-4-ил]этанол;
{5-метил-2-[(S)-4-(2-морфолин-4-илэтил)-4,5-дигидро-1,3-оксазол-2-ил]-1Н-индол-7-ил}-(тетрагидропиран-4-ил)амин;
4-[(5-хлор-2-фенил-1Н-индол-7-ил)амино]-N-этилпиперидин-1-карбоксиамид;
[4-[(5-хлор-2-фенил-1Н-индол-7-ил)амино]пиперидин-1-ил]-(оксолан-3-ил)метанон;
2-[7-(оксан-4-иламино)-2-фенил-1Н-индол-5-ил]уксусная кислота;
2-[7-(циклопентилметиламино)-2-фенил-1Н-индол-5-ил]-уксусная кислота;
5-фтор-N-(1-метилпиперидин-4-ил)-2-фенил-1Н-индол-7-амин;
2-[4-[(5-фтор-2-фенил-1Н-индол-7-ил)амино]пиперидин-1-ил]этанон;
5-фтор-N-[1-(оксан-4-ил)пиперидин-4-ил]-2-фенил-1Н-индол-7-амин;
N-[1-(1,1-диоксан-4-ил)пиперидин-4-ил]-5-фтор-2-фенил-1Н-индол-7-амин;
N-(оксан-4-ил)-5-фенокси-2-фенил-1Н-индол-7-амин;
метил 2-[(5-фтор-2-фенил-1Н-индол-7-ил)амино]ацетат;
2-[(5-фтор-2-фенил-1Н-индол-7-ил)амино]уксусная кислота;
метил 2-[(5-хлор-2-фенил-1Н-индол-7-ил)амино]пропаноат;
2-[(5-хлор-2-фенил-1Н-индол-7-ил)амино]пропановая кислота;
2-[(5-фенокси-2-фенил-1Н-индол-7-ил)амино]уксусная кислота;
2-[(5-фенокси-2-фенил-1Н-индол-7-ил)амино]пропановая кислота;
2-[(4S)-2-[7-(оксан-4-илметиламино)-2-фенил-1Н-индол-5-ил]-4,5-дигидро-1,3-тиазол-4-ил]уксусная кислота;
2-[(4S)-2-[7-(циклопентиламино)-2-фенил-1Н-индол-5-ил]-4,5-дигидро-1,3-тиазол-4-ил]уксусная кислота;
метил 2-[4-[5-хлор-7-(оксан-4-иламино)-1Н-индол-2-ил]-фенил]ацетат;
метил 2-[4-[5-хлор-7-(оксан-4-илметиламино)-1H-индол-2-ил]фенил] ацетат;
2-[4-[5-хлор-7-(оксан-4-иламино)-1Н-индол-2-ил]фенил]-уксусная кислота;
5-[(1,1-диоксо-1,4-тиазинан-4-ил)метил]-N-(оксан-4-ил)-2-фенил-1Н-индол-7-амин;
5-[(1,1-диоксо-1,4-тиазинан-4-ил)метил]-N-(оксан-4-ил-метил)-2-фенил-1Н-индол-7-амин;
4-[[7-(оксан-4-иламино)-2-фенил-1Н-индол-5-ил]метил]-пиперазин-2-он;
5-[(1,1-диоксо-1,4-тиазинан-4-ил)метил]-2-фенил-N-пиперидин-4-ил-1Н-индол-7-амин;
[4-[[5-[(1,1-диоксо-1,4-тиазинан-4-ил)метил]-2-фенил-1Н-индол-7-ил]амино]пиперидин-1-ил]-(оксолан-3-ил)метанон;
N-[4-[5-[(1,1-диоксо-1,4-тиазинан-4-ил)метил]-7-(оксан-4-иламино)-1Н-индол-2-ил]фенил]ацетамид;
N-[4-[7-(дициклопентиламино)-5-[(1,1-диоксо-1,4-тиазинан-4-ил)метил]-1Н-индол-2-ил]фенил]ацетамид;
N-[4-[5-[(1,1-диоксо-1,4-тиазинан-4-ил)метил]-7-(оксан-4-илметиламино)-1Н-индол-2-ил]фенил]ацетамид;
N-циклопентил-5-[(1,1-диоксо-1,4-тиазинан-4-ил)метил]-2-(4-метоксифенил)-1Н-индол-7-амин;
5-[(1,1-диоксо-1,4-тиазинан-4-ил)метил]-2-(4-метокси-фенил)-N-(оксан-4-ил)-1Н-индол-7-амин;
5-[(1,1-диоксо-1,4-тиазинан-4-ил)метил]-N-(3-метокси-бутил)-2-фенил-1Н-индол-7-амин;
5-[(1,1-диоксо-1,4-тиазинан-4-ил)метил]-2-(3-фторфенил)-N-(оксан-4-ил)-1Н-индол-7-амин;
N-циклопентил-5-[(1,1-диоксо-1,4-тиазинан-4-ил)метил]-2-(3-фторфенил)-1Н-индол-7-амин;
3-бром-5-[(1,1-диоксо-1,4-тиазинан-4-ил)метил]-N-(оксан-4-ил)-2-фенил-1Н-индол-7-амин;
3-бром-5-(морфолин-4-илметил)-N-(оксан-4-ил)-2-фенил-1Н-индол-7-амин;
3-бром-(циклопентил-5-[(1,1-диоксо-1,4-тиазинан-4-ил)-метил]-2-фенил-1Н-индол-7-амин;
3-бром-5-[(1,1-диоксо-1,4-тиазинан-4-ил)метил]-N-(оксан-4-ил)-2-фенил-1Н-индол-7-амин;
5-хлор-N-(оксан-4-ил)-3-фенил-1Н-индол-7-амин;
5-хлор-N-циклопентил-3-фенил-1Н-индол-7-амин;
5-хлор-N-(оксан-4-илметил)-3-фенил-1Н-индол-7-амин;
5-[(1,1-диоксо-1,4-тиазинан-4-ил)метил]-N-(оксан-4-ил)-3-фенил-2-триметилсилил-1Н-индол-7-амин;
4-[[5-хлор-7-(циклопентиламино)-2-фенил-1Н-индол-3-ил]метил]пиперазин-2-он;
4-[[5-хлор-7-(оксан-4-иламино)-2-фенил-1Н-индол-3-ил]метил]пиперазин-2-он;
4-[[5-хлор-7-(оксан-4-илметиламино)-2-фенил-1Н-индол-3-ил]метил]пиперазин-2-он;
N-циклопентил-3-(4-метоксифенил)-1Н-индазол-7-амин;
3-(4-метоксифенил)-N-(оксан-4-ил)-1Н-индазол-7-амин;
3-(4-метоксифенил)-N-(оксан-4-илметил)-1Н-индазол-7-амин и 2-(7-циклопентиламино-2-фенил-1Н-индол-5-ил)этанол.
16. Композиция по п.1, где клеткой является клетка животного, выделенная из тканей или органов человека или не относящихся к человеку животных, и выбранная из группы, состоящей из клетки печени, клетки кожи, клетки слизистой оболочки, клетки островка Лангерганса, клетки нерва, клетки хряща, клетки эндотелия, клетки эпителия, клетки кости и клетки мышцы; или мужской половой клетки, яйцеклетки или оплодотворенной яйцеклетки животного или рыбы.
17. Композиция по п.1, где орган выбирают из группы, состоящей из кожи, роговой оболочки глаза, почки, сердца, печени, поджелудочной железы, кишечника, нерва, легкого, плаценты, пуповины и кровеносного сосуда.
18. Композиция по п.1, где ткань выбирают из группы, состоящей из кожи, роговой оболочки глаза, почки, сердца, печени, поджелудочной железы, кишечника, нерва, легкого, плаценты, пуповины и кровеносного сосуда.
19. Композиция по п.1 для предотвращения повреждения органов, выделенных клеточных систем или тканей, вызванного хранением при низких температурах, операцией по трансплантации или посттрансплантационной реперфузией.
20. Применение композиции по п.1 для консервирования клеток, тканей или органов животных для трансплантации.
21. Способ получения композиции для консервирования клеток, тканей или органов животных, включающий стадию смешения соединения формулы (1) по п.1 или его фармацевтически приемлемой соли или изомера в качестве эффективного ингредиента, вместе с фармацевтически приемлемым носителем.
| WO 2006112549 A1, 26.10.2006 | |||
| WO 2006062982 A2, 15.06.2006 | |||
| Способ разделения углеводородов @ или @ разной степени насыщенности | 1982 |
|
SU1057484A1 |
| RU 2060254 C1, 20.05.1996. | |||

