Настоящее изобретение относится к полифункциональным агентам сочетания, пригодным, в частности, для связки усиливающих неорганических наполнителей с диеновыми эластомерами в резиновых смесях, предназначенных, например, для производства пневмошин.
Известно, что обычно для получения оптимальных усиливающих свойств, сообщаемых наполнителем, конечная форма, в которой наполнитель будет присутствовать в эластомерной матрице, должна быть наиболее диспергированной и одновременно наиболее равномерно распределенной. Однако такие условия могут быть выполнены только в том случае, когда наполнитель обладает очень хорошей способностью, с одной стороны, инкорпорироваться при смешении с эластомером в матрицу и при этом дезагломерироваться и, с другой стороны, равномерно диспергироваться в матрице.
Очень хорошо известно, что такими свойствами обладает углеродная сажа, что, как правило, не свойственно неорганическим наполнителям. Действительно, по причине взаимного сродства частицы неорганического наполнителя обладают неприятной тенденцией агломерировать между собой в эластомерной матрице. Вредным последствием этих взаимодействий является то, что они ограничивают диспергирование наполнителя и, следовательно, усиливающие свойства значительно более низким уровнем по сравнению с уровнем, который мог бы быть теоретически достигнут, если бы были эффективно реализованы все связи (в системе неорганический наполнитель/эластомер), которые могли бы возникнуть при операции смешения. С другой стороны, названные взаимодействия имеют тенденцию повышать консистенцию резиновых смесей в сыром состоянии и, следовательно, затруднять их переработку (ухудшать технологичность) по сравнению с переработкой в присутствии углеродной сажи.
С тех пор как экономия горючего и необходимость защиты окружающей среды стали приоритетом, пришли к необходимости производить пневмошины с пониженным сопротивлением качению без ущерба для их износостойкости. Это стало, в частности, возможным благодаря открытию новых резиновых смесей, усиленных специальными неорганическими наполнителями, квалифицируемыми как "усиливающие", которые способны конкурировать в качестве усилителей с традиционной углеродной сажей для пневмошин, сообщая при этом резиновым смесям более низкий гистерезис, что является синонимом более низкого сопротивления качению содержащих эти смеси пневмошин.
Такие резиновые смеси, содержащие усиливающие неорганические наполнители кремнеземного или глиноземного типа, были, в частности, описаны в патентах и патентных заявках ЕР-А-0501227 (или US-А-5227425), ЕР-А-0735088 (или US-А-5852099), ЕР-А-0810258 (или US-А-5900449), ЕР-А-0881252, WO99/02590, WO99/02601, WO99/02602, WO99/28376, WO00/05300, WO00/05301.
Следует, в частности, упомянуть документы ЕР-A-0501227, A-0735088 или ЕР-A-0881252, в которых раскрываются диеновые резиновые смеси, усиленные высокодисперсными осажденными кремнеземами, которые позволяют изготовлять протекторы покрышек, обладающие значительно улучшенным сопротивлением качению, не влияя при этом на другие свойства, в частности на сцепление с поверхностью, усталостную прочность и износостойкость. Такого рода смеси, обладающие указанным выше балансом взаимоисключающих свойств, описаны также в заявках ЕР-А-0810258 и WO99/28376, где в качестве усиливающих неорганических наполнителей использованы специальные глиноземные наполнители (оксиды алюминия и оксидо-гидроксиды алюминия) с повышенной диспергируемостью, или же в заявках WO00/73372 и WO00/73373, в которых описываются специальные оксиды титана с усиливающим действием.
Использование этих обладающих высокой диспергируемостью неорганических наполнителей в качестве преобладающего или не преобладающего усиливающего наполнителя, безусловно, уменьшило трудности при применении содержащих эти наполнители резиновых смесей, однако применение этих смесей все же продолжает оставаться более трудным по сравнению с применением традиционных резиновых смесей с наполнителем из углеродной сажи.
В частности, необходимо использовать связывающий агент, называемый также соединительным агентом, задачей которого является обеспечить связи между поверхностью частиц неорганического наполнителя и эластомером, облегчая при этом диспергирование этого неорганического наполнителя внутри эластомерной матрицы.
Напомним здесь, что под "связывающим агентом" (для системы неорганический наполнитель/эластомер) следует, как обычно, понимать агент, способный обеспечивать достаточно хорошую связь химической и/или физической природы между неорганическим наполнителем и диеновым эластомером. Такой связывающий агент, по крайней мере бифункциональный, имеет, например, общую формулу Y-W-X, в которой:
- Y означает функциональную группу (функцию Y), которая способна физически и/или химически связываться с неорганическим наполнителем, причем такого рода связь может быть установлена, например, между атомом кремния связывающего агента и гидроксильными группами (ОН) поверхности неорганического наполнителя (например, силанольными поверхностными группами, если речь идет о кремнеземе);
- X означает функциональную группу (функцию X), способную физически и/или химически связываться с диеновым эластомером, например через атом серы;
- W означает двухвалентную группу, соединяющую Y и X.
Связывающие агенты не следует, в частности, путать с обычными покровными агентами для неорганического наполнителя, которые, как это известно, могут содержать функцию Y, обладающую активностью в отношении неорганического наполнителя, но не имеют функции X, обладающей активностью в отношении диенового эластомера.
Связывающие агенты, в частности для системы кремнезем/диеновый эластомер, были описаны во множестве документов, причем наиболее известными из этих агентов являются бифункциональные органосиланы, имеющие в качестве функции Y, по меньшей мере, одну алкоксильную функцию, а в качестве функции X, по меньшей мере, одну функцию, способную взаимодействовать с диеновым эластомером, такую, например, как серусодержащая функция.
Далее, в патентных заявках FR-A-2094859 или GB-A-1310379 было предложено использовать для изготовления протекторов пневмошин меркаптоалкоксисилановый связывающий агент. При этом скоро выяснилось, и сегодня об этом хорошо известно, что хотя меркаптоалкоксисиланы и способны обеспечить прекрасную связь кремнезем/эластомер, однако промышленное применение этих связывающих агентов невозможно по причине очень высокой реакционной способности серусодержащих групп типа тиоловых -SH (функции X), что в процессе приготовления резиновых смесей в закрытом резиносмесителе быстро приводит к преждевременной вулканизации, называемой также "подгоранием" ("scorching"), к повышенной вязкости в сыром состоянии и, в конечном итоге, к резиновым смесям, с которыми почти невозможно работать и применять в промышленном масштабе. Для иллюстрации этой проблемы можно сослаться, например, на документы FR-A-2206330, US-A-3873489, US-A-4002594.
Для преодоления названного выше недостатка было предложено заменить меркаптоалкоксисиланы полисульфидными алкоксисиланами, в частности полисульфидами бис(алкоксисилилпропила), такими как те, которые описаны в очень большом количестве документов (см., например, FR-A-2149339, FR-A-2206330, US-A-3842111, US-A-3873489, US-A-3997581, EP-A-680997 или US-A-5650457, EP-A-791622 или US-A-5733963, DE-A-19951281 или EP-A-1043357, WO00/53671). Из этих полисульфидов следует, в частности, назвать тетрасульфид бис(3-триэтоксисилилпропила) (сокращенно TESPT) и дисульфид бис(3-триэтоксисилилпропила) (сокращенно TESPD).
Названные полисульфиды алкоксисиланов, в частности TESPT, обычно рассматривают как продукты, которые придают вулканизатам, содержащим усиливающий неорганический наполнитель, в частности кремнезем, наилучший компромисс между предупреждением подгорания, легкости при применении и усиливающей способности. По этой причине названные продукты являются связывающими агентами, наиболее широко используемыми сегодня в резиновых смесях для пневмошин, даже несмотря на то, что они являются относительно дорогостоящими и к тому же чаще всего должны использоваться в относительно больших количествах.
Однако у этих полисульфидов алкоксисиланов имеется недостаток, состоящий в том, что они значительно замедляют кинетику вулканизации содержащих их резиновых смесей по сравнению с кинетикой вулканизации традиционных смесей, усиленных углеродной сажей. Обусловленная этим увеличенная продолжительность вулканизации может в некоторых случаях затруднить промышленное применение такого рода резиновых смесей, усиленных неорганическими наполнителями, а также использование содержащих их резиновых изделий.
В то же время заявители при проведении своих исследований неожиданным образом обнаружили новые особые органосиланы, которые неожиданным образом позволяют преодолеть недостаток, связанный с кинетикой вулканизации, не ухудшая при этом связывающую и, следовательно, усиливающую способность и придавая, таким образом, резиновым смесям улучшенный баланс свойств по сравнению с балансом свойств, достигнутых к настоящему времени с использованием полисульфидных алкоксисиланов, в частности с TESPT.
Эти особые органосиланы не создают, кроме того, названных выше проблем преждевременного подгорания и проблем, обусловленных слишком высокой вязкостью резиновых смесей в сыром состоянии, - недостатков, свойственных меркаптоалкоксисиланам.
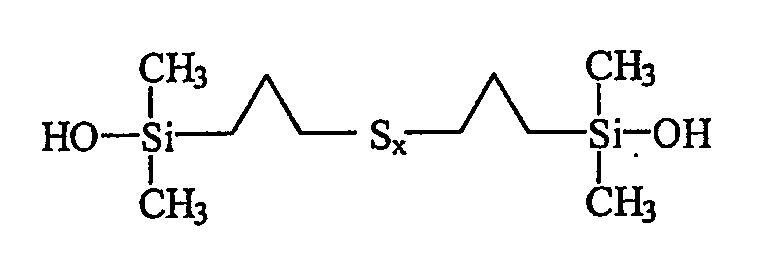
Таким образом, первым объектом изобретения является полисульфид моногидроксисилана формулы (I):
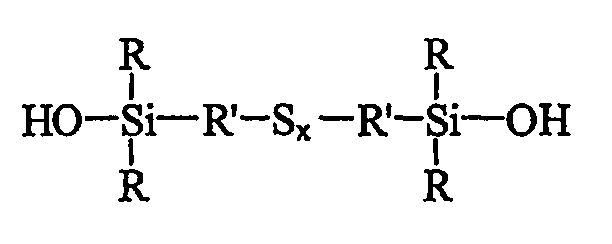
в которой радикалы R, одинаковые или разные, означают углеводородные группы; радикалы R', одинаковые или разные, означают двухвалентные связывающие группы; x больше или равен 2.
Насколько известно заявителям, такой полисульфидный моногидроксисилан никогда ранее не был синтезирован, в частности с целью его использования в качестве связывающего агента по причине сильных предубеждений относительно известной неустойчивости органосиланов, имеющих гидроксильные функции. Уместно здесь напомнить, что эта неустойчивость обусловлена высокой склонностью гидроксисилильных групп (Si-OH), в частности в присутствии сильных кислот (образующихся при синтезе органосиланов из галогеносиланов), к самоконденсации с образованием связей Si-O-Si, называемых силоксановыми связями. С другой стороны, этот механизм используют для синтеза полисилоксанов (см., например, следующие работы: "The Chemistry of organic silicon compounds", S. Patai и Z. Rappoport, John Wiley & Sons, 1989, Part I, 722-725; "Siloxane polymers", S.I. Clarson, S.J.Semlyen, Ellis Horwood Pretice-Hall, Elgelwood Cliffs N.J., 1993, 577-578; 597).
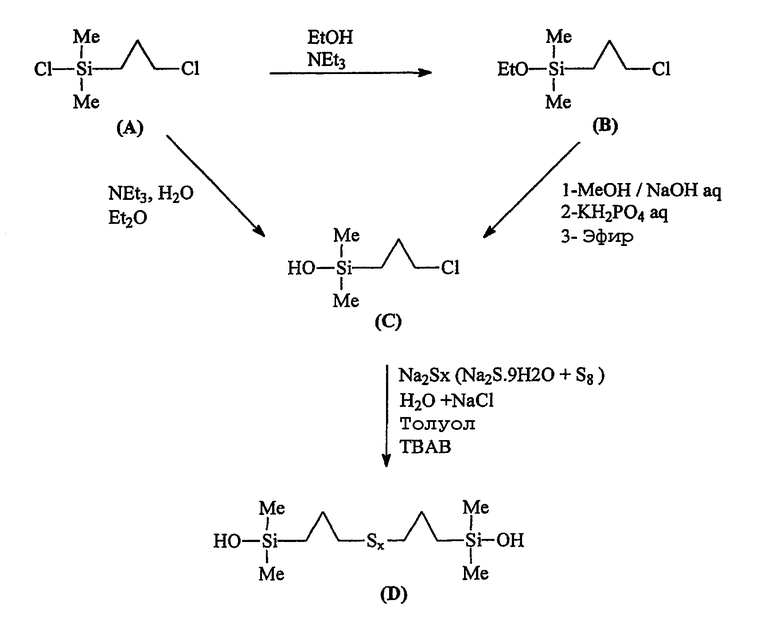
Другим объектом изобретения является способ получения моногидроксисилана по изобретению, включающий следующие стадии (R и R' имеют указанные выше значения):
• исходят из галогенированного органосилана (далее именуемого продуктом А) формулы А (Hal означает галоген):
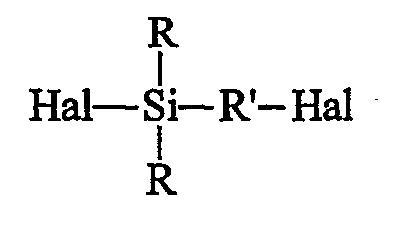
• этот галогенированный органосилан может быть при желании подвергнут алкоголизу с помощью спирта (R"-OH) в присутствии органического основания, предназначенного для связывания образующейся галогеноводородной кислоты, с получением моноалкоксисилана формулы B (далее именуемого продуктом В):
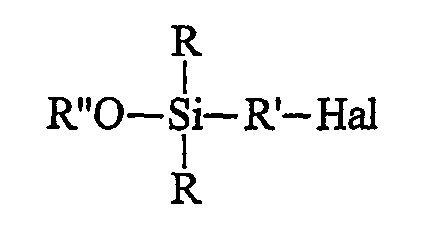
• осуществляют в инертном органическом растворителе с использованием донора гидроксилов гидролиз:
- либо продукта А в присутствии (в этом случае) органического основания для связывания образующейся галогеноводородной кислоты, используя в качестве донора гидроксилов воду;
- либо (при желании) продукта В, используя в качестве донора гидроксилов минеральное основание и в качестве органического растворителя полярный растворитель, получая моногидроксисилан (продукт С) формулы С:
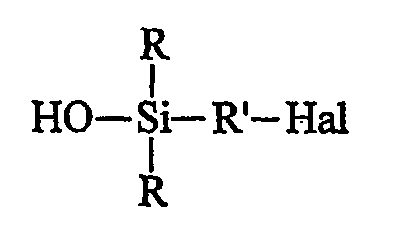
• в заключение осуществляют стадию сульфидирования продукта С с помощью полисульфида, получая целевой продукт формулы I.
Объектом изобретения является также применение в качестве связывающего агента моногидроксисилана по изобретению, в частности в качестве связывающего агента (для системы неорганический наполнитель/диеновый эластомер) в резиновой смеси.
Изобретение, а также его преимущества станут яснее из приведенных ниже описания и примеров осуществления изобретения, а также из соответствующих этим примерам фигур, которые представляют:
- фиг.1: реограммы (кривые вулканизации), зарегистрированные для резиновых смесей, содержащих в качестве связывающего агента полифункциональные органосиланы, соответствующие или не соответствующие изобретению;
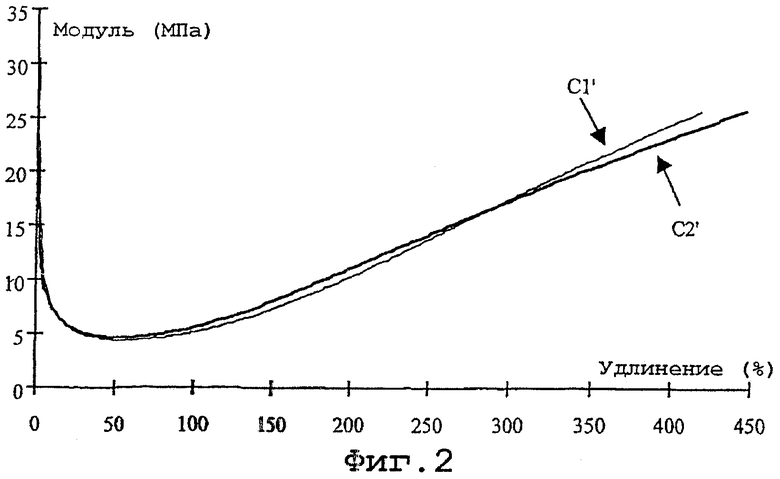
- фиг.2: кривые зависимости модуля от удлинения для этих резиновых смесей.
I. Использованные измерения и тесты
Резиновые смеси, в которых протестированы органосилановые связывающие агенты, охарактеризованы до и после вулканизации, как указано ниже.
I-1. Пластичность по Муни
Использован осциллирующий консистометр, описанный во французском стандарте NF T 43-005 (1991). Измерение пластичности по Муни производится в соответствии со следующей методикой: смесь в сыром состоянии (т.е. до вулканизации) формуют в цилиндрическом сосуде, нагретом до 100оС. После предварительного нагрева в течение 1 мин вводят в действие находящийся внутри образца ротор со скоростью 2 об/мин и измеряют крутящий момент, необходимый для поддержания работы ротора после 4 мин его вращения. Пластичность по Муни (ML 1+4) выражена в "единицах Муни" (UM, где UM=0,83 Н·м).
I-2. Время пригорания
Измерения проводят при 130°С в соответствии с французским стандартом NF T 43-005 (1991). Временная зависимость консистометрического показателя позволяет найти время пригорания резиновых смесей, измеряемое в соответствии с указанным выше стандартом с помощью параметра Т5 (в случае большого ротора), выраженное в минутах и определяемое как время, необходимое для получения увеличения консистометрического показателя (выраженного в UM) до 5 единиц выше минимального значения, измеренного для этого показателя.
I-3. Испытания на растяжение
Эти испытания позволяют определить напряжения упругости и свойства при разрыве. Если не оговорено особо, эти испытания проводятся в соответствии с французским стандартом NF T 46-002 от сентября 1988 г. Измеряют при втором растяжении (т.е. после аккомодационного цикла со степенью удлинения, предусмотренной для самого измерения) номинальные секущие модули (или кажущиеся напряжения, в МПа) при удлинении 10% (обозначаемом М10), при удлинении 100% (обозначаемом М100) и удлинении 300% (обозначаемом М300). Измеряют также напряжения при разрыве (в МПа) и удлинения при разрыве (в %). Все измерения растяжения проводят в условиях нормальных температуры и влажности в соответствии с французским стандартом NF T 40-101 (декабрь 1979).
Обработка данных по растяжению позволяет также построить кривую зависимости модуля от удлинения (см. фиг.2), где использованным модулем является истинный секущий модуль, измеренный при первом растяжении, рассчитанный не для начального отрезка, как это делалось выше для номинальных модулей, а для реального отрезка образца.
I-4. Динамические свойства
Динамические свойства измеряют на анализаторе вязкости (Metravib VA4000) в соответствии со стандартом ASTM D5992-96. Регистрируют реакцию образца вулканизированной смеси (цилиндрический образец толщиной 4 мм и 400 мм2 в сечении), подвергнутого синусоидальной нагрузке при простом знакопеременном напряжении сдвига с частотой 10 Гц в условиях нормальной температуры (23°С) в соответствии со стандартом ASTM D 1349-99. Производят развертку амплитуды деформации от 0,1 до 50% (ход вперед) и затем от 50 до 1% (обратный ход), регистрируя при обратном ходе максимальное значение, наблюдаемое для тангенса угла потерь tg(δ), обозначаемое tg(δ)max.
I-5. Реологические измерения
Измерения производят при 150°С на осциллирующем камерном реометре в соответствии со стандартом DIN 53529, часть 3 (июнь 1983). Временная зависимость реометрического крутящего момента характеризует увеличение жесткости смеси в результате реакции вулканизации (см. фиг.1). Измеренные данные обрабатывают в соответствии со стандартом DIN 53529, часть 2 (март 1983). Минимальный и максимальный крутящие моменты, измеренные в дециньютонах·метр (дН·м), обозначают, соответственно, Cmin и Cmax; ti означает время индукции, т.е. время, необходимое для начала вулканизации; tα (например, t99) означает время, необходимое для достижения конверсии α%, т.е. α% (например, 99%), разницы между минимальным и максимальным крутящими моментами. Измеряют также разницу, обозначаемую Δ крутящий момент (в дН·м), между минимальным и максимальным крутящими моментами, а также константу скорости конверсии К (в мин-1), которая позволяет оценить кинетику вулканизации.
II. Условия осуществления изобретения
II-1. Полисульфидный гидроксисилан по изобретению
Первым объектом изобретения является полисульфид (с х 2, т.е. включая дисульфид) моногидроксисилана, отвечающий приведенной выше формуле I, в которой радикалы R, одинаковые или разные, являются углеводородными группами, радикалы R', одинаковые или разные, являются двухвалентными связывающими группами, и x больше или равен 2.
Такой органосилан хорошо подходит под определение связывающего агента в том случае, когда он содержит в своей молекуле:
- с одной стороны, по меньшей мере, одну функциональную группу (полисульфидную функцию Sx в качестве функции X), способную образовать устойчивую связь с полимером, таким как диеновый эластомер;
- с другой стороны, гидроксильную группу (ОН) у каждого атома кремния с образованием функции, называемой гидроксисилильной (Si-OH), в качестве функции Y, позволяющей органосилану быть привитым к различным подложкам (например металлическим или минеральным), таким как поверхность неорганического усиливающего наполнителя.
Радикалы R, линейные или разветвленные, содержащие преимущественно от 1 до 15 атомов углерода, выбираются преимущественно из алкилов, циклоалкилов и арилов, в частности из С1-С6-алкилов, С5-С8-циклоалкилов и радикала фенила. Из этих радикалов можно в качестве примеров назвать радикалы, выбираемые из группы, в которую входят метил, этил, н-пропил, изопропил, н-бутил, втор-бутил, трет-бутил, н-пентил, неопентил, н-гексил, 2-этилгексил, н-октил, изооктил, циклопентил, циклогексил, 2-метилциклогексил, фенил, толуил и бензил.
Еще более предпочтительны С1-С3-алкилы (а именно метил, этил, н-пропил и изопропил) и наиболее предпочтительны метил и этил.
Радикалы R', замещенные или незамещенные, являются преимущественно насыщенными или ненасыщенными углеводородными радикалами, содержащими от 1 до 18 атомов углерода, причем в эти углеводородные радикалы R' может быть встроен, по меньшей мере, один гетероатом, такой как O, S или N. Подходящими являются, в частности, алкиленовые группы С1-С18 или С6-С12-ариленовые группы, более конкретно С1-С10-алкилены, в частности С1-С4, в особенности выбранные из метилена, этилена и пропилена.
Иными словами, полисульфидный моногидроксисилан по изобретению является преимущественно полисульфидом (симметричным или несимметричным в отношении природы радикалов R и R') бис(моногидрокси-С1-С18-алкилсилил-С1-С15-алкила), отвечающим приведенной выше общей формуле I, в частности полисульфидом бис(моногидрокси-С1-С3-алкилсилил(метила, этила или пропила)).
Особо предпочтительными гидроксисиланами формулы I являются гидроксисиланы, у которых радикалы R являются С1-С3-алкильными группами, радикалы R' являются С1-С4-алкиленами, а x больше или равен 2.
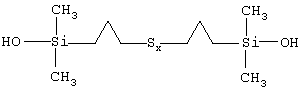
Из последних можно, в частности, назвать полисульфиды бис(С1-С4-алкил-диметилсиланола), т.е. полисульфиды, у которых R есть Ме (метил), отвечающие приведенной ниже формуле II:
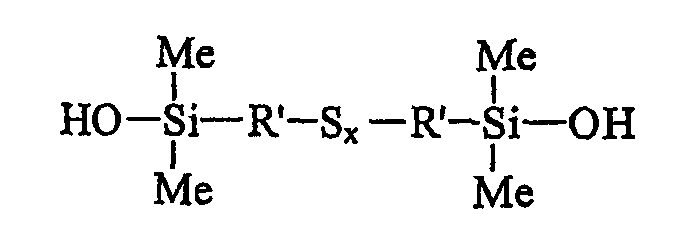
в которой x больше или равен 2, а радикалы R' являются С1-С4-алкиленами, в частности метиленом, этиленом или пропиленом, предпочтительно пропиленом.
В качестве примера можно назвать полисульфид бис(пропилдиметилсиланола) приведенной ниже индивидуальной формулы III:
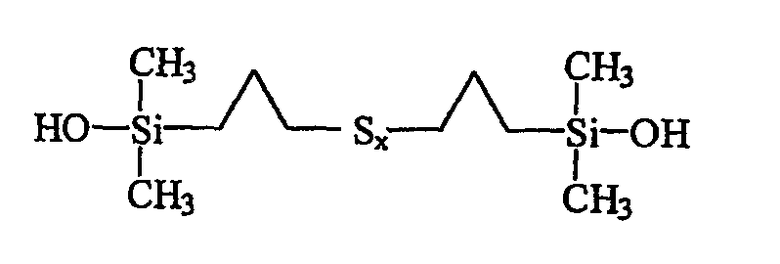
В формулах I и III число атомов серы х может варьировать в широких пределах, например от 2 до 9, в зависимости от конкретных условий синтеза моногидроксисилана, однако значения х выбирают преимущественно в пределах от 2 (дисульфиды) до 6 (гексасульфиды), включая соответствующие трисульфиды (х=3), тетрасульфиды (х=4) и пентасульфиды (х=5). Еще более предпочтительно х выбирают в пределах от 2 до 4.
Приведенные выше полисульфидные моногидроксисиланы получают с помощью нового метода синтеза, который составляет другой объект настоящего изобретения.
II-2. Метод синтеза
Способ по изобретению для получения моногидроксисилана приведенной выше формулы I включает следующие стадии:
• исходят из галогенированного органосилана (далее именуемого продуктом А) формулы А (Hal означает галоген):
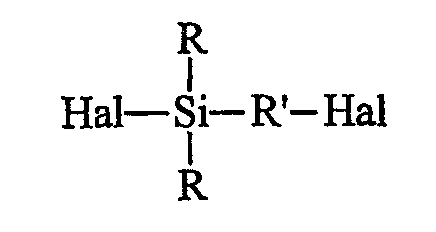
• этот галогенированный органосилан может быть, при желании, подвергнут алкоголизу с помощью спирта (R"-OH) в присутствии органического основания, предназначенного для связывания образующейся галогеноводородной кислоты, с получением моноалкоксисилана (далее именуемого продуктом В) формулы B:
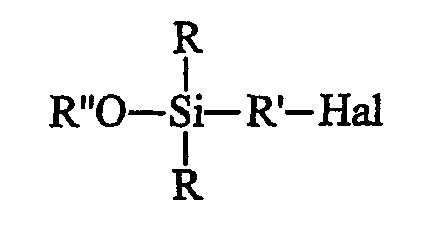
• осуществляют в инертном органическом растворителе с использованием донора гидроксилов гидролиз:
- либо продукта А в присутствии (в этом случае) органического основания для связывания образующейся галогеноводородной кислоты, используя в качестве донора гидроксилов воду;
- либо (при желании) продукта В, используя в качестве донора гидроксилов минеральное основание и в качестве органического растворителя полярный растворитель, получая моногидроксисилан (продукт С) формулы С:
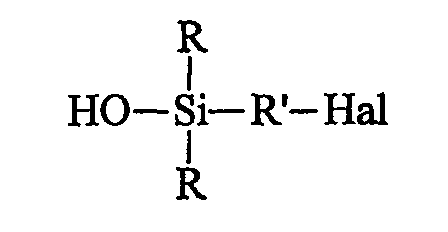
• в заключение осуществляют стадию сульфидирования продукта С с помощью полисульфида, получая целевой продукт формулы I.
Некоторые стадии (алкоголиз, сульфидирование) этого способа по изобретению, по крайней мере, в их общих чертах, уже применялись для синтеза серусодержащих алкоксисиланов, таких как меркаптоалкоксисиланы или полисульфидные алкоксисиланы (см., например, FR-A-2149339 или US-A-4076550, FR-A-2206330, EP-A-0694552 или US-A-5405985). Однако, насколько известно заявителям, приведенные выше стадии никогда не были описаны в комбинации для получения полисульфидных моногидроксисиланов.
Галогены (Hal) исходного силана (продукта А) могут быть одинаковыми или разными, и их преимущественно выбирают из брома и хлора, предпочтительно используя хлор. В общем случае исходные галогенсиланы (продукты А) и их промежуточные производные (продукты В или С) являются жидкими продуктами и, следовательно, могут быть использованы на различных стадиях способа по изобретению как таковые или будучи разбавленными подходящим растворителем.
Первая, необязательная, стадия алкоголиза состоит, таким образом, в замене галогена (Hal) у атома кремния в продукте А алкоксильной группой (OR") спирта в присутствии органического основания, предназначенного для связывания образующейся в процессе реакции галогеноводородной кислоты. Углеводородный радикал R" спирта (R"OH) содержит преимущественно от 1 до 8 атомов углерода. Его преимущественно выбирают из С1-С6-алкилов, предпочтительнее из С1-С3-алкилов и, главным образом, из метила или этила. В качестве органического основания, предназначенного для связывания образующейся галогеноводородной кислоты, можно использовать амин, преимущественно третичный амин, такой как триэтиламин. Для лучшего связывания галогеноводородной кислоты алкоголиз проводят при температуре предпочтительно ниже 15°С, более предпочтительно ниже 10°С.
Стадия гидролиза может быть осуществлена непосредственно с исходным галогенсиланом (продуктом А) при действии воды в инертном органическом растворителе, например в эфире, в присутствии органического основания, предназначенного, как и выше, для связывания образующейся галогеноводородной кислоты.
Однако исходный галогенсилан предпочитают перед гидролизом подвергать алкоголизу. Такой гидролиз продукта В проводят в полярном органическом растворителе, преимущественно в спирте, действием неорганического основания в водном растворе, причем неорганическим основанием преимущественно является гидроксид щелочного или щелочноземельного металла, в частности гидроксид натрия (NaOH). Основание используют преимущественно в небольшом избытке, который в конце реакции нейтрализуют слабой кислотой, такой как дигидрофосфат калия. Полярным органическим растворителем преимущественно является спирт С1-С6, предпочтительнее С1-С3 и, главным образом, метанол.
Для конечной стадии сульфидирования может быть использован полисульфид (x) аммония или металла формулы M2Sx или M'Sx (M означает щелочной металл или NH4; M' означает Zn или щелочноземельный металл). Примерами таких соединений являются полисульфиды Na, K, Cs, Rb, Ca, Mg, Zn и NH4, а x находится преимущественно в пределах от 2 до 6, предпочтительнее от 2 до 4. Используется преимущественно полисульфид натрия Na2Sx, в частности Na2S2, Na2S3, Na2S4, Na2S5, Na2S6, причем этот полисульфид преимущественно образуется при действии серы (S8) на Na2S. Получение полисульфидов осуществляют известным способом в органическом или неорганическом растворителе, таком, например, как вода, спирты, кетоны и эфиры, - растворителе, в котором реагирующие вещества частично или полностью растворимы.
Однако для того, чтобы исключить всякий риск образования побочных продуктов типа полисульфидов алкоксисиланов в результате побочной реакции алкоголиза силанольных функций продукта С, предпочитают осуществлять стадию сульфидирования в отсутствие любого спирта. В этом случае работают преимущественно в водной фазе, предпочтительнее в двухфазной среде вода/органический растворитель (например, толуол, ксилол, бензол, гептан или им подобные), как это описано в упомянутых выше документах ЕР-А-0694552 или US-А-5405985 в отношении синтеза полисульфидных алкоксисиланов. Реакцию сульфидирования в этом случае проводят известным способом в присутствии катализатора фазового перехода и соли формулы M"Hal или M"SO4 (M" выбирают из Li, Na, K; Hal выбирают из F, Cl и Br). Используемую соль преимущественно выбирают из NaCl, NaBr и Na2SO4, отдавая предпочтение NaCl. Количество соли может варьироваться, например, от 10% от массы водного раствора до полного насыщения раствора. Катализатором фазового перехода является, например, тетрабутиламмонийбромид (TBAB).
Стадию сульфидирования осуществляют преимущественно в атмосфере инертного газа, такого как аргон. Температура реакционной среды не является определяющей: можно, например, работать при комнатной температуре, однако для увеличения скорости реакции предпочитают все же работать при нагревании, например, от 60 до 100°С и в некоторых случаях до температуры кипения растворителя. Мольное отношение гидроксисилана (продукта С) к полисульфиду (аммония или металла) подбирают преимущественно таким образом, чтобы иметь небольшой избыток полисульфида по отношению к его стехиометрическому количеству.
Если сульфидирование проводится в органической фазе, продукт С сам предпочтительно заранее разбавляют инертным органическим растворителем, таким как спирт, кетон или эфир. Когда реакция закончена, удаляют фильтрацией образовавшуюся соль (галогенид металла) и освобождают фильтрат от растворителя с помощью перегонки в вакууме. В случае сульфидирования в водной фазе или в двухфазной системе (вода/органический растворитель) органическая фаза, содержащая продукт D, может быть, при желании, выделена, после чего избыточный растворитель отгоняют в вакууме.
Полисульфидные моногидроксисиланы, синтезированные в соответствии с описанным выше способом, фактически являются смесями полисульфидов (например, с х, равным от 2 до 9), в результате чего среднее значение х не является целой величиной. Желаемое среднее значение х лежит преимущественно в пределах от 2 до 6 и предпочтительнее от 2 до 4.
III-3. Применение в качестве связывающего агента
Как было указано выше, соединение по изобретению, благодаря его двойной функциональности, находит предпочтительное промышленное применение в качестве связывающего агента, предназначенного, например, обеспечивать связь или адгезию между реакционно-способной полимерной матрицей (в частности, каучуковой матрицей) и любым материалом с гидроксилированной поверхностью, в частности минеральной (например, стекловолокна) или металлической (например, углеродистой или нержавеющей стали).
В качестве не имеющего ограничительного характера примера, соединение по изобретению может быть, в частности, использовано для связывания белой сажи или неорганических усиливающих наполнителей с диеновыми эластомерами, например в резиновых смесях, предназначенных для производства пневмошин. Под "неорганическим усиливающим наполнителем" подразумевают, как это принято, неорганический или минеральный наполнитель вне зависимости от его цвета и происхождения (природного или синтетического), называемый также белой (или иногда светлой) сажей, в отличие от углеродной сажи, причем такой неорганический наполнитель способен один, без какого-либо другого агента, кроме выполняющего функции посредника связывающего агента, усиливать резиновую смесь, предназначенную для производства пневмошин, иными словами, способного заменить в качестве усилителя традиционный усилитель - углеродную сажу категории "для пневмошин".
Для такого рода применения диеновый эластомер выбирают преимущественно из группы высоконенасыщенных диеновых эластомеров, в которую входят полибутадиены (BR), синтетические полиизопрены (IR), натуральный каучук (NR), бутадиен-стирольные сополимеры (SBR), бутадиен-изопреновые сополимеры (BIR), бутадиен-акрилонитрильные сополимеры (NBR), изопрен-стирольные сополимеры (SIR), бутадиен-стирол-изопреновые сополимеры (SBIR) и смеси этих эластомеров.
В том случае, когда моногидроксисилан по изобретению предназначен для связывания (для системы неорганический наполнитель/диеновый эластомер) в резиновой смеси, образующей, например, весь или часть протектора для пневмошины легкового транспорта, диеновым эластомером преимущественно является SBR или смесь SBR с каким-либо другим диеновым эластомером, таким как BR, NR или IR. В случае эластомера SBR используется, более конкретно, SBR с содержанием стирола в пределах от 20 до 30 мас.%, содержанием виниловых связей в бутадиеновой части в пределах от 15 до 65%, содержанием связей транс-1,4 в пределах от 15 до 75% и температурой стеклования (Tg - измеренной в соответствии со стандартом ASTM D3418-82) в пределах от -20 до -55°С, причем этот сополимер SBR, приготовляемый преимущественно в растворе (SSBR), может также применяться в смеси с полибутадиеном (BR), имеющим преимущественно более 90% связей цис-1,4.
В том случае, когда протектор предназначен для пневмошины транспортных средств утилитарного назначения, таких как большегрузные транспортные средства, диеновым эластомером преимущественно является изопреновый эластомер, т.е. диеновый эластомер, выбранный из группы, в которую входят натуральный каучук (NR), синтетические полиизопрены (IR), различные сополимеры изопрена и смеси этих эластомеров. В этом случае преимущественно используется натуральный каучук или синтетический полиизопрен типа цис-1,4 с содержанием связей цис-1,4 выше 90 мол.% и более предпочтительно выше 98 мол.%.
Полисульфидные моногидроксисиланы по изобретению оказались сами по себе достаточно эффективными для связывания диенового эластомера с усиливающим неорганическим наполнителем, таким как кремнезем, при их применении в количестве более 1 чсэ (весовой части на 100 весовых частей эластомера), предпочтительно от 2 до 20 чсэ. Преимущественно, они могут являться единственным связывающим агентом, присутствующим в резиновых смесях, усиленных неорганическим наполнителем и предназначенных для производства пневмошин.
В качестве усиливающих неорганических наполнителей можно назвать минеральные наполнители кремнеземного типа, в частности оксид кремния (SiO2), или глиноземного типа, в частности оксид алюминия (Al2O3), или оксидо-гидроксиды алюминия, или также усиливающие оксиды титана, такие как те, которые описаны в упомянутых выше патентах и патентных заявках.
Гидроксисиланы по изобретению могли бы быть также использованы после предварительной прививки к усиливающему неорганическому наполнителю (через гидроксисилильную функцию): "предварительно связанный" таким образом неорганический наполнитель может после этого быть связан с диеновым эластомером через свободную полисульфидную функцию.
III. Примеры осуществления изобретения
В приведенных ниже примерах осуществления изобретение осуществляется с полисульфидом бис(пропилдиметилсиланолом) индивидуальной формулы III:
III-1. Синтез полисульфидного моногидроксисилана
В этом примере прежде всего описывается сначала синтез соединения приведенной выше формулы III (называемого далее продуктом D) способом по изобретению, осуществляемым в две или три стадии, исходя из хлорпропилдиметилхлорсилана (называемого далее продуктом А) через хлорпропилдиметилэтоксисилан (называемый далее продуктом В; факультативный путь) и хлорпропилдиметилсиланол (называемый далее продуктом С).
Применена следующая схема синтеза:
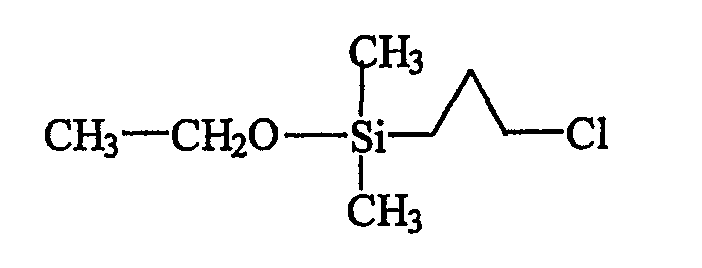
а) Получение хлорпропилдиметилэтоксисилана (продукта В)
Первая стадия состоит в алкоголизе, позволяющем заменить хлор у атома кремния продукта А на этоксильную группу этанола, путем проведения реакции в присутствии триэтиламина, предназначенного для связывания выделяющейся в процессе реакции хлористоводородной кислоты.
В 2-литровую трехгорлую колбу (предварительно высушиваемую в сушильном шкафу в течение 24 часов), на которой установлен холодильник и которая снабжена магнитной мешалкой, вводят с помощью шприца в токе аргона 950 мл этанола (марки Normapur) и затем 288 мл триэтиламина (2,07 моль, или 209 г). Смесь после этого охлаждают до температуры приблизительно 5°С и затем с помощью перистальтического насоса прибавляют продукт А (237,7 г или 1,38 моль - продукт фирмы ABCR, поставляемый в продажу под названием SIC2336.0), в процессе чего выделяющаяся хлористоводородная кислота сразу же связывается триэтиламином с образованием гидрохлорида триэтиламина.
Как только заливка прекращается (спустя приблизительно 8 ч), ледяную баню убирают и продолжают перемешивание при комнатной температуре в токе аргона в течение ночи. Спустя восемь часов анализ с помощью ГФХ (газофазной хроматографии) указывает на исчезновение пика, соответствующего исходному продукту А, и на образование хлорпропилдиметилэтоксисилана (продукта В). Реакционную среду после этого фильтруют на воронке Аллина, отделяя продукт В в растворе этанола от гидрохлорида триэтиламина.
Фильтрат, содержащий продукт В, упаривают и затем перегоняют в вакууме (2 мм рт.ст., температура масляной бани 70°С, температура верха колонки 45°С) с целью удаления избытка свободного триэтиламина и выделения продукта В в чистом состоянии.
В итоге получают 160 г бесцветной жидкости, ЯМР-анализ и масс-спектрометрия которой подтверждают, что речь идет о желаемом продукте В, отвечающем следующей формуле:
b) Получение хлорпропилдиметилсиланола (продукта С)
Эта стадия (вторая) состоит в гидролизе полученного выше этоксисилана (в метанольном растворе) с целью получения гидроксисилана. Реакцию проводят, действуя водным раствором NaOH. После реакции введенный вначале избыток основания нейтрализуют дигидрофосфатом калия.
В снабженную магнитной мешалкой 100-мл трехгорлую колбу с установленным на ней холодильником вводят 2,62 г гидроксида натрия (65 ммоль, или 2,4 эквивалента по отношению к продукту В), который растворяют в 15 мл деминерализованной воды. После полного растворения и возвращения температуры раствора к комнатной добавляют 20 мл метанола и затем, с помощью капельной воронки, полученный выше продукт В (5 г, или 27,7 ммоль), разбавленный 35 мл метанола. После этого реакционную среду перемешивают в течение 90 мин и вливают в водный раствор дигидрофосфата калия (16 г KH2PO4 в 200 мл воды). Полученный раствор, имеющий рН 7, перемешивают несколько минут и затем смешивают с 200 мл эфира с целью экстракции образовавшегося продукта С. Двухфазную среду после этого перемешивают приблизительно 30-45 мин и помещают в делительную воронку. Отделенную органическую фазу промывают один раз водой и сушат на MgSO4, после чего фильтруют и упаривают в вакууме.
Хроматограмма ЖФХ собранного таким образом неочищенного продукта реакции имеет три пика, которые можно приписать, соответственно, (i) хлорфенилдиметилметоксисилану, образовавшемуся, по-видимому, в результате реакции метанола с продуктом В (порядка 2% по структурным элементам на основании 1Н ЯМР), (ii) целевому продукту С, который существенно преобладает (85% по структурным элементам на основании 1Н ЯМР), а также (iii) бис(хлорпропил)тетраметилдисилоксану (присутствующему в количестве 13% по структурным элементам).
Перегонка в вакууме, выполненная на колонке с шаровыми расширениями (Kügelrohr), позволяет выделить продукт С. В процессе перегонки поддерживают температуру в колонке преимущественно ниже 45°С для снижения риска конденсации продукта С в соответствующий дисилоксан. После удаления хлорпропилдиметилметоксисилана при вакууме 1,3 мбар и температуре 35°С выделяют продукт С при вакууме 1 мбар и нагревании до 40°С, в то время как более высоко кипящий бис(хлорпропил)тетраметилдисилоксан остается в перегонной колбе.
В результате выделяют 2,48 г чистой бесцветной жидкости, анализ ЯМР и масс-спектрометрия которой подтверждают получение продукта С, отвечающего следующей формуле:
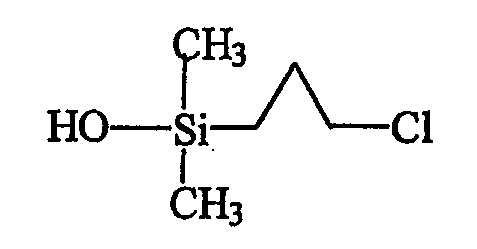
Как было указано выше, приведенный выше продукт С может быть также получен непосредственно гидролизом исходного продукта А в инертном органическом растворителе (эфире) в присутствии воды в качестве донора гидроксилов и триэтиламина, предназначенного для связывания выделяющейся хлористоводородной кислоты. Воду предпочтительно вводят в избытке, создавая тем самым более благоприятные условия для желаемой реакции и позволяя избежать реакцию конденсации силанола, образующегося из вводимого хлорсилана. Применение небольшого избытка триэтиламина обеспечивает полное связывание хлористоводородной кислоты, а оставшийся триэтиламин отгоняют после завершения реакции.
Более конкретно, реакцию проводят следующим образом: в снабженную магнитной мешалкой 500-мл трехгорлую колбу с установленным на ней холодильником вводят 9,78 мл триэтиламина (70,1 ммоль, или 1,5 эквивалента по отношению к продукту А), 3,36 г воды (187 ммоль, или 4 эквивалента по отношению к продукту А) и затем 150 мл эфира. Полученный раствор охлаждают на ледяной бане (температура <10°С) и медленно прибавляют раствор продукта А (8,0 г, или 46,7 ммоль в 80 мл эфира). Сразу же наблюдается появление белого осадка, соответствующего гидрохлориду триэтиламина. По завершении прибавления продукта А реакционную среду продолжают перемешивать в течение еще 30 мин, сохраняя температуру ниже 10°С. Образовавшийся осадок удаляют после этого фильтрацией, фильтрат собирают и высушивают над сульфатом магния, фильтруют и упаривают в вакууме. Оставшийся триэтиламин удаляют перегонкой. В результате получают 6,1 г ярко-желтой жидкости, которая по данным анализа ЯМР и масс-спектрометрии соответствует целевому продукту С (чистота продукта выше 95%).
с) получение полисульфидного гидроксисилана (продукта D)
На этой последней стадии полисульфид натрия, образующийся путем введения серы в сульфид натрия Na2S в водной среде, замещает атомы хлора двух молекул продукта С в толуольном растворе. Реакцию проводят в присутствии катализатора фазового перехода (ТБАБ) и хлорида натрия NaCl.
В снабженную магнитной мешалкой 250-мл трехгорлую колбу с установленным на ней холодильником вводят 3,50 г (14,5 ммоль) Na2S·9H2O, а также 1,40 г (43,7 ммоль) серы, которые растворяют в 40 мл водного раствора NaCl (5,0 г, или 85,8 ммоль) и 8 мл толуола. Эту смесь нагревают до 85°С, наблюдая во время подъема температуры изменение цвета реакционной среды от желтого до темно-красного.
После достижения желаемой температуры вводят за один прием 0,25 г ТВАВ (0,77 ммоль) и начинают прибавлять по каплям продукт С (4,60 г, или 28,6 ммоль), растворенный в 30 мл толуола. В процессе прибавления толуольная фаза имеет ярко-красную окраску, которая постепенно переходит в оранжевую, в то время как водная фаза, вначале ярко-красная, бледнеет, становясь после прекращения прибавления продукта С бесцветной и прозрачной. Реакцию продолжают в течение 75 мин при температуре 85°С, после чего реакционную среду охлаждают в атмосфере аргона.
Реакционную среду переливают затем в делительную воронку для отделения толуольной фазы, которую промывают водой и высушивают над сульфатом магния. Органическую фазу после этого фильтруют, поглощают эфиром и перегоняют на колонке с шаровыми расширениями (40°С) с целью удаления остаточного хлорфенилдиметилсиланола (продукта С).
В итого получают 3,82 г вязкой красно-оранжевой жидкости, ЯМР-анализ которой и масс-спектрометрия подтверждают, что она содержит (в мол.%):
• приблизительно 95% полисульфида бис(пропилдиметилсиланола) формулы III:
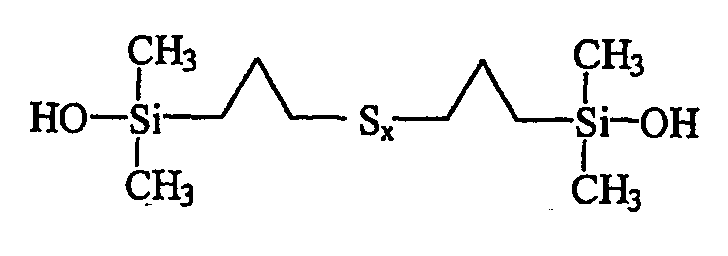
• приблизительно 5% циклического силоксанового соединения приведенной ниже формулы IV:
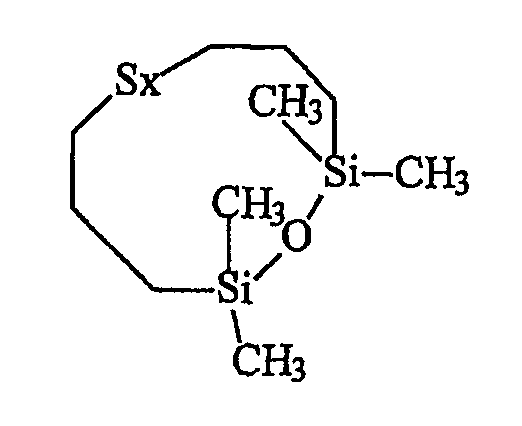
Синтезированный таким образом продукт D представляет собой в действительности распределение полисульфидов от дисульфида (х=2) до гексасульфидов (х=6) с усредненным значением х=3,7. Содержание дисульфида S2, определенное с помощью ЯМР, равно приблизительно 18% от полисульфидных звеньев.
Таким образом, этот продукт представляет собой, как это имеет место, в частности, в случае полисульфидов алкоксисиланов, таких как TESPT, распределение полисульфидов, среднее значение х которых близко к 4. Понятно, что изменение условий синтеза позволило бы получить другие распределения полисульфидов с разными средними значениями х, но преимущественно в пределах от 2 до 6 и главным образом от 2 до 4.
III-2. Применение в качестве связывающего агента
Целью этого испытания является подтверждение улучшенных характеристик гидроксисилана по изобретению в качестве связывающего агента в резиновой смеси для пневмошин по сравнению с традиционным связывающим агентом (полисульфидом алкоксисилана TESPT), который в данном случае является агентом сравнения.
С этой целью приготовляют две усиленные кремнеземом резиновые смеси на основе эластомеров SBR и BR, обозначаемые С-1 и С-2, которые предназначены для протекторов пневмошин легкового автомобиля. Эти две смеси одинаковы, за исключением используемого связывающего агента:
- смесь С-1: TESPT
- смесь С-2: синтезированный выше продукт D.
Эти смеси приготовляют следующим известным способом: в заполняемый на 70% закрытый резиносмеситель с исходной температурой в емкости приблизительно 60°С вводят диеновый эластомер (или, при желании, смесь диеновых эластомеров), усиливающий наполнитель, связывающий агент и затем различные другие ингредиенты, за исключением вулканизационной системы. После этого осуществляют первую термомеханическую операцию (так называемая непродуктивная фаза) в одну или две стадии (общая продолжительность перемешивания составляет примерно 7 мин) до достижения максимальной температуры "усадки" около 165°С. Полученную таким образом смесь выгружают, охлаждают и добавляют вулканизационную систему (серу и сульфенамидный ускоритель) во внешнем резиносмесителе (конечном гомогенизаторе) при 30°С, перемешивая содержимое смесителя (так называемая производительная фаза) в течение 3-4 мин. Смеси после этого прокатывают в форме пластин (толщиной 2-3 мм) или тонких листов каучука для измерения их физических или механических свойств или экструдируют в виде прокатных профилей, пригодных для непосредственного применения (после разрезки и/или сборки до желаемых размеров), в частности в качестве протекторов пневмошин для легкового транспорта.
Напомним, что TESPT (смесь С-1) представляет собой тетрасульфид бис(3-триэтоксисилилпропила) формулы [(C2H5O)3Si(CH2)3S2]2. Его поставляет в продажу, например, фирма Degussa под названием "Si69" (или "X50S" в том случае, когда он нанесен в количестве 50 мас.% на углеродную сажу) или фирмой Witco под названием "Silquest A1289" (в обоих случаях коммерческая смесь полисульфидов Sx со средним значением х, которое близко к 4).
Ниже приведена развернутая формула TESPT (Et означает этил):
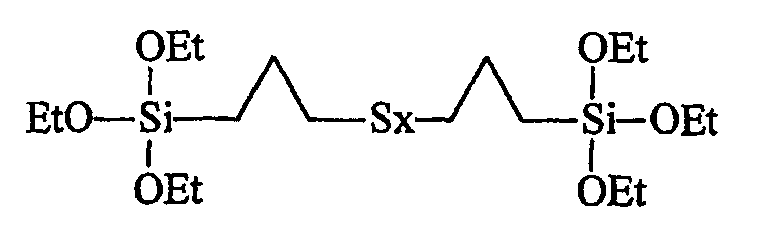
Отметим, что приведенная структура очень близка к структуре тестируемого здесь моногидроксисилана по изобретению (формула III), который отличается только наличием гидроксильной группы и двух метилов на месте трех традиционных алкоксильных групп.
В таблицах 1 и 2 приведен состав двух смесей (таблица 1: содержание различных продуктов, выраженное в чсе - вес. частях на 100 вес. частей эластомера), их свойства до и после вулканизации (приблизительно 30 мин при 150°С); вулканизационная система состоит из серы и сульфенамидного ускорителя. Две протестированные смеси содержат кроме того (не указано в таблице 1) небольшое количество углеродной сажи N330 (6 чсе), используемой в качестве агента черной пигментации для защиты от ультрафиолетового излучения.
Фиг. 1 и 2 воспроизводят, соответственно, реограммы (зависимость крутящего момента в дН·м от продолжительности вулканизации в мин) и кривые зависимости модуля (в МПа) от удлинения (в %); эти кривые на фиг.1 обозначены С1 и С2, а на фиг.2 C1' и C2', соответствуя в обоих случаях смесям С-1 и С-2 соответственно.
Изучение различных результатов таблицы 2 приводит к следующим соображениям:
- смесь С-2, содержащая продукт D, характеризуется временем подгорания более коротким, чем смесь сравнения С-1, но это время, Т5, остается достаточно большим для того, чтобы обеспечить удовлетворительный запас безопасности в отношении проблемы подгорания;
- значения пластичности по Муни остаются низкими (85-90 UM) во всех случаях, в то же время самое низкое значение зарегистрировано для смеси С-2, что является показателем очень хорошей пригодности резиновых смесей для использования в сыром состоянии, по крайней мере столь же хорошей, как и у традиционных смесей, в которых использованы полисульфиды алкоксисиланов;
- после вулканизации смесь С-2, при сравнении со смесью сравнения, обладает очень близкими значениями модуля при сильной деформации (М100 и М300) и отношением М300/М100, которые являются достаточными для специалиста показателями качества связывания, придаваемого продуктом D;
- гистерезисные свойства оказываются несколько улучшенными для смеси С-2, о чем свидетельствует более низкое значение tg(δ)max (синоним низкого сопротивления качению);
- наконец, и это особенно важно, отмечается, что смесь С-2 неожиданным образом отличается константой скорости конверсии K, которая более чем вдвое превосходит константу скорости конверсии смеси сравнения или, иными словами, вулканизация смеси может быть осуществлена за значительно более короткое время.
Реограммы на фиг.1 хорошо подтверждают преимущество смеси С-2, содержащей гидроксисилан по изобретению: время индукции близко к времени индукции смеси сравнения; максимальный крутящий момент идентичен максимальному крутящему моменту смеси сравнения, но достигается за значительно более короткое время; константа скорости конверсии K более высокая.
Фиг.2 также подтверждает предыдущие соображения: кривая C2' (смесь C-2) и кривая C1' (контрольная смесь C-1) почти совпадают, в особенности при самых больших удлинениях, которые являются показательными для усиления и, следовательно, для износостойкости резиновых смесей.
В заключение следует сказать, что в целом поведение смеси, содержащей моногидроксисилан по изобретению, иллюстрирует не только высокое качество связывания (соединения) между неорганическим усиливающим наполнителем и диеновым эластомером, по крайней мере такое же, какое достигается с традиционными полисульфидами алкоксисиланов, такими как TESPT, но также, неожиданным образом, существенно улучшенную способность вулканизироваться.
Замена полисульфидного алкоксисилана, такого как TESPT, полисульфидным гидроксисиланом по изобретению обеспечивает также значительную выгоду с точки зрения экологии и проблемы, связанной с выделением VOC (volatile organic compounds). В частности, известно, что алкоксильные группы алкоксисилана (такие как этоксильные группы TESPT) действительно являются причиной выделения спирта (этанола) как при приготовлении самих резиновых смесей, так и в процессе вулканизации изделий из каучука, включающих эти смеси.
Таким образом, моногидроксисилан по изобретению особенно выгодно применять в качестве связывающего агента в резиновых смесях, пригодных для использования в производстве пневмошин, в частности протекторов пневмошин, обладающих низким сопротивлением качению и повышенной износостойкостью, особенно в тех случаях, когда протекторы предназначены для пневмошин легковых автомашин или для промышленного транспорта типа большегрузных транспортных средств.
BR (2)
кремнезем (3)
алкоксисилан (4)
продукт D (5)
DPG (6)
ZnO
стеариновая кислота
антиоксидант (7)
25
80
6,4
-
1,5
2,5
2
1,9
25
80
-
4,6
1,5
2,5
2
1,9
Ускоритель (8)
2
2
(1) SBR с 59,5% звеньев полибутадиена 1-2; 26,5% стирола; дополненный ароматическим маслом (37,5 чсэ масла на 100 чсэ сухого SBR); Tg = -29°С; в расчете на сухой SBR;
(2) BR с 4,3% 1-2; 2,7% транс; 93% цис 1-4 (Tg = -106°С);
(3) кремнезем типа "HD" - "Zeosil 1165MP" фирмы Rhodia в форме микробусин (BET и CTAB: примерно 150-160 м2/г);
(4) TESPT ("Si69" фирмы DEGUSSA-HÜLS);
(5) синтезированный продукт D (полисульфид бис(пропилдиметилсиланола), 95 мол.%);
(6) дифенилгуанидин ("Vulcacit D" фирмы Bayer);
(7) N-1,3-диметилбутил-N-фенил-п-фенилендиамин ("Santoflex 6-PPD" фирмы Flexsys);
(8) N-циклогексил-2-бензотиазил-сульфенамид ("Santocure CBS" фирмы Flexsys).
T5 (мин)
ti (мин)
t99 (мин)
K (мин-1)
t99-ti (мин)
Δ крутящий момент
20
9
41
0,14
32
17,8
13
7
21
0,32
14
19,6
M100 (МПа)
M300 (МПа)
M300/М100
tg(δ)max
Усилие при разрыве (МПа)
Удлинение при разрыве (%)
2,6
4,4
1,7
0,310
20,6
416
2,8
4,3
1,6
0,299
20,8
443
Описывается полисульфид моногидроксисилана формулы
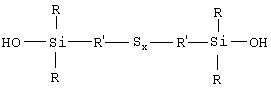
в которой радикалы R, одинаковые или разные, означают углеводородные группы, содержащие от 1 до 15 атомов; радикалы R', одинаковые или разные, означают двухвалентные связывающие группы, содержащие от 1 до 15 атомов углерода; и х больше или равен 2, способ его получения и применение этого гидроксисилана в качестве связывающего агента в резиновых смесях. 3 н. и 18 з.п. ф-лы, 2 табл., 2 ил.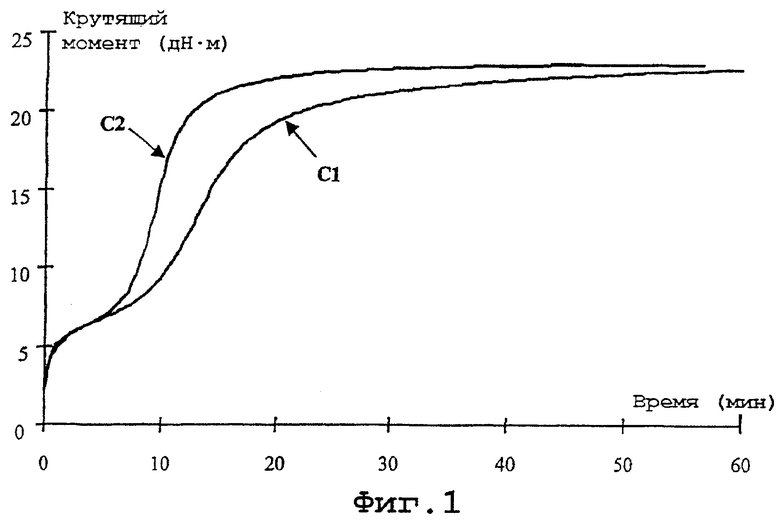
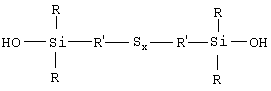
в которой радикалы R, одинаковые или разные, означают углеводородные группы, содержащие от 1 до 15 атомов углерода; радикалы R', одинаковые или разные, содержащие от 1 до 18 атомов углерода, означают двухвалентные связывающие группы; х обозначает число от 2 до 9.
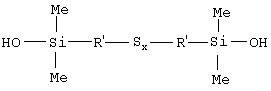
в которой радикалы R' являются С1-С4-алкиленами.
исходят из галогенированного органосилана (далее именуемого продуктом А) формулы (A) (Hal означает галоген)
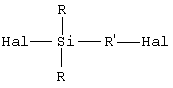
этот галогенированный органосилан может быть подвергнут алкоголизу с использованием спирта (R''-OH) в присутствии органического основания, предназначенного для связывания образующейся галогеноводородной кислоты, с получением моноалкоксисилана (далее именуемого продуктом В) формулы В
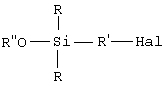
осуществляют в инертном органическом растворителе с использованием донора гидроксилов гидролиз:
- либо продукта А в присутствии (в этом случае) органического основания для связывания образующейся галогеноводородной кислоты, используя в качестве донора гидроксилов воду;
- либо (в отдельных случаях) продукта В, используя в качестве донора гидроксилов минеральное основание и в качестве органического растворителя полярный растворитель, получая моногидроксисилан (продукт С) формулы (С)
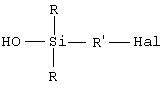
при этом радикалы R, одинаковые или разные, означают углеводородные группы, содержащие от 1 до 15 атомов углерода; радикалы R', одинаковые или разные, содержащие от 1 до 18 атомов углерода, означают двухвалентные связывающие группы, радикал R'' спирта (R''ОН) содержит от 1 до 8 атомов углерода и является алкилом;
в заключение осуществляют стадию сульфидирования продукта С с помощью полисульфида, получая целевой продукт формулы I.
| WO 00/05301 А, 03.02.2000 | |||
| DE 19734295 С, 25.02.1999 | |||
| 0 |
|
SU211863A1 | |

