Область изобретения
Изобретение относится к синтетическим процианидиновым олигомерам и способам получения и применения олигомеров.
Предпосылки изобретения
Полифенолы являются очень разнообразной группой соединений (Ferreira D., Steynberg J.P., Roux D.G. и Brandt E.V., Tetrahedron, 48 (10), 1743-1803 (1992)), которые широко распространены в различных растениях, некоторые из которых входят в пищевые продукты. В некоторых случаях они представляют важную группу соединений для питания человека. Несмотря на то, что считается, что некоторые полифенолы не имеют пищевого значения, интерес к данным соединениям возрос в результате их возможных полезных эффектов на здоровье.
Например, было показано, что кверцетин обладает противоопухолевой активностью в опытах на экспериментальных животных (Decschner E.E., Ruperto J., Wong G. и Newmark H.L., Carcinogenesis, 7, 1193-1196 (1991) и Kato R., Nakadate Т., Yamamoto S. и Sugimura Т., Carcinogenesis, 4, 1301-1305 (1983)). Было показано, что (+)-катехин и (-)-эпикатехин ингибируют активность обратной транскриптазы вируса лейкемии (Chu S.-C., Hsieh Y.-S. и Lim J.-Y., J. of Natural Products, 55 (2), 179-183 (1992)). Также было показано, что нобатанин (олигомерный гидролизуемый таннин) обладает противоопухолевой активностью (Okuda Т., Yoshida Т. и Hatano Т., Molecular Structures and Pharmacological Activities of Polyphenols - Oligomeric Hydrolyzable Tannins and Others - Presented at the XVIth International Conference of the Group Polyphenols, Лиссабон, Португалия, 13-16 июля 1992). Статистические отчеты также показывали, что смертность в результате рака желудка значительно ниже в районах Японии, где производится чай. Сообщалось, что эпигаллокатехингаллат является фармакологически активным соединением в зеленом чае, которое ингибирует развитие рака кожи у мышей (Okuda et al., Ibid.). Было показано, что эллаговая кислота обладает противоопухолевой активностью на различных опухолевых моделях у животных (Boukharta M., Jalbert G. и Castonguay A., Efficacy of Ellagitannins and Ellagic Acid as Cancer Chemopreventic Agents - Presented at the XVIth International Conference of the Group Polyphenols, Лиссабон, Португалия, 13-16 июля 1992). Проантоцианидиновые олигомеры запатентованы Kikkoman Corporation для применения в качестве антимутагенов. Применение фенольных соединений в продуктах питания и модуляция под их воздействием развития опухолей на моделях экспериментальных животных недавно было представлено на 202-м Национальном собрании Американского химического общества (Phenolic Compounds in Foods and Their Effects on Health I, Analysis, Occurrence & Chemistry, Ho, C.-T., Lee C.Y. и Huang M.-T. editors, ACS Symposium Series 506, American Chemical Society, Вашингтон, D.C. (1992); Phenolic Compounds in Foods and Their Effects on Health II. Antioxidants & Cancer prevention, Huang M.-T., Ho С.-Т. и Lee C.Y. editors, ACS Symposium Series 506, American Chemical Society, Вашингтон, D.C. (1992)).
Однако данные ссылки не относятся к экстрактам какао или соединениям, входящим в их состав, или к каким-нибудь способам получения таких экстрактов или соединений из них, или к каким-нибудь из применений, описанных в патенте США 5554645 от 10 сентября 1996 года, Romanczyk et al., патенте США 5712305 от 27 января 1998 года, Romanczyk et al. и патенте США 5650432 от 22 июля 1997 года, Walker et al.
Были разработаны способы выделения, разделения, очистки и идентификации для извлечения ряда процианидиновых олигомеров в целях сравнительной оценки биологической активности в условиях in vitro и in vivo. Например, показана противоопухолевая активность для пентамерных-декамерных процианидинов, но не для мономерных-тетрамерных соединений. В настоящее время быстрыми способами получают граммовые количества чистого пентамера (>95%). Данные способы не подходят для получения значительных количеств пентамера для широкомасштабных фармакологических исследований и исследований биодоступности. Еще большие усилия необходимы для получения многограммовых количеств высших олигомеров (гексамеров-декамеров) для подобных исследований, поскольку их концентрация в натуральном продукте значительно ниже, чем для пентамера. Кроме того, увеличение размера олигомера увеличивает структурную сложность. Такие факторы, как хиральность мономерных единиц, составляющих олигомер, в различных местах межфлавановых связей, динамическая поворотная изомеризация межфлавановых связей, конформационные состояния пиранового кольца и многочисленные точки связывания в нуклеофильных центрах создают затруднения в отношении эффективности современных аналитических способов разделения и очистки для последующей идентификации.
Например, предыдущие попытки сочетать мономерные единицы в свободной фенольной форме с использованием минеральной кислоты в качестве катализатора в водной среде имели ограниченный успех. Выходы были низкими, реакции протекали с низкой селективностью, и олигомеры было трудно выделить (Stynberg P.J., Nel R.J. и Ferreira D., Tetrahedron, 54, 8153-8158 (1998); Botha J.J., Young D.A., Ferreira F. и Roux D.J.J., J.Chem.Soc., Perkins Trans. I, 1213-1219 (1981)).
Бензилированные мономеры получили способами, описанными Kawamoto H., Nakatsubo F. и Murkami К., Mokuzai Gakkashi, 37, 741-747 (1991), где использовали бензилбромид в сочетании с карбонатом калия (К2СО3) и диметилформамид (ДМФА). Однако выход составил только примерно 40%. Кроме того, конкурентное С-бензилирование приводит к образованию смеси продуктов, что затрудняет выделение целевого мономера. Кроме того, наблюдали частичную рацемизацию (+)-катехина в С-2- и С-3-положениях (Pierre M.-C. et al., Tetrahedron Letters, 38: 32, 5639-5642 (1997)).
В литературе описаны два основных способа окислительной функционализации (Betts M.J., Brown B.R. и Shaw M.R., J.Chem.Soc., C. 1178 (1969); Steenkamp J.A., Ferreira D. и Roux D.J., Tetrahedron Lett., 26, 3045-3048 (1985)). В более старом способе защищенный (+)-катехин обрабатывали тетраацетатом свинца (LTA) в бензоле с получением 4β-ацетокси-производного, которое затем успешно гидролизовали до 3,4-диола. Флаван-3,4-диолы представляют начальные электрофилы в биомимметическом синтезе процианидинов. Однако флаван-3,4-диолы, которые имеют кислородную функциональную группу в С-5-положении, недоступны из естественных источников, и их следует синтезировать. Таким образом окислительная функционализация прохирального бензильного положения с образованием 3,4-диолов предлагает значительную потенциальную возможность для синтеза процианидинов. Наибольшим недостатком данной реакции был низкий выход (30-36%) ацетата во время окисления с помощью LTA. Более поздний способ окислительной функционализации С-4-положения основан на использовании 2,3-дихлор-5,6-дициано-1,4-бензохинона (DDQ). В данном способе защищенный мономер обрабатывали DDQ в метаноле. Это позволяет ввести метоксильную группу в С-4-положение стереоспецифичным образом. Выход составляет примерно 40-50%.
Имеется ряд сообщений по реакции сочетания мономеров и их 3,4-диолов в водной кислоте. Данные способы являются неудовлетворительными за счет низких выходов, отсутствия специфичности и трудностей при выделении из водной среды. Kawamoto H., Nakatsubo F. и Murakami К., J. of Wood Chem. Tech., 9, 35-52 (1989) сообщили, что тетрахлорид титана (TiCl4) опосредует сочетание между 4-гидроксил тетра-O-бензил(+)-катехином и 5 эквивалентами (экв) тетра-O-бензил(+)-катехина с получением смеси 4α→8 и 4β→8 димеров 3:2.
Следовательно, имеется потребность в способах синтеза, которые обеспечивают большие количества структурно определенных олигомеров для оценки in vitro и in vivo. Подобные способы синтеза могут привести к получению олигомеров сложной конфигурации, некоторые из которых идентичны таковым, найденным в природе, а также редких или «неестественных» типов. Следовательно, будет полезным разработать универсальный способ синтеза, способный обеспечить большие количества любого желаемого процианидинового олигомера.
Краткое описание изобретения
Описан способ получения частично защищенного процианидинового димера. Он включает стадии:
(a) защиты каждой фенольной гидроксильной группы процианидинового мономера с помощью удаляемой защитной группы, которая не дезактивирует кольцо А мономера, где стадия защиты проводится в апротонном растворителе;
(b) активирования для сочетания С-4-положения соединения со стадии (а) путем введения ацилоксильной группы с использованием соли свинца (IV) органической кислоты с получением активированного соединения; и
(c) сочетания активированного соединения со стадии (b) с незащищенным процианидиновым мономером в присутствии катализатора сочетания с получением димера.
Также описан способ получения линейного процианидинового олигомера. Он включает стадии:
(a) получения частично защищенного процианидинового димера, в котором фенольные гидроксильные группы верхнего мера защищены удаляемой защитной группой, которая не дезактивирует кольцо А защищенного мера;
(b) маскирования незащищенных фенольных гидроксильных групп нижнего мера со стадии (а) и гидроксильных групп в С-3-положениях мера с помощью удаляемой маскирующей группы, которая дезактивирует нижний мер маскированного, защищенного димера;
(с) снятия защиты с верхнего мера димера со стадии (b) с получением маскированного димера со снятой защитой;
(d) сочетания димера со стадии (с) с неблокированным или блокированным, защищенным, активированным мономером с образованием тримера, в котором верхний мер тримера является защищенным, блокированным мером или защищенным, неблокированным мером, и где сочетание имеет место в С-8-положении;
(e) маскирования неблокированного или блокированного тримера со стадии (d);
(f) снятия защиты с неблокированного или блокированного, защищенного, маскированного тримера со стадии (е) с образованием неблокированного или блокированного маскированного тримера;
и
(g) необязательно повторяют или чередуют стадии (a)-(f) с получением высших олигомеров, в которых число мер составляет от 4 до 18.
В следующих иллюстративных соединениях Р представляет защитную группу, В представляет блокирующую группу, и М представляет маскирующую группу. Следующее соединение является иллюстрацией частично защищенного процианидинового димера, такого, как получено на стадии (а) выше:
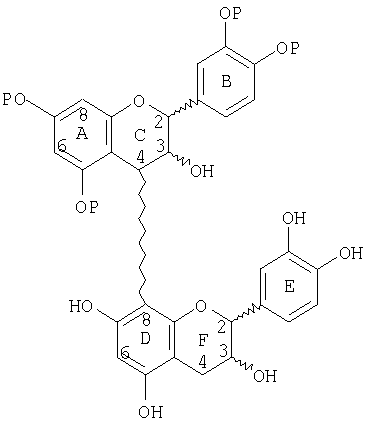
Следующее соединение является иллюстрацией защищенного, маскированного димера такого, как получено на стадии (b) выше:
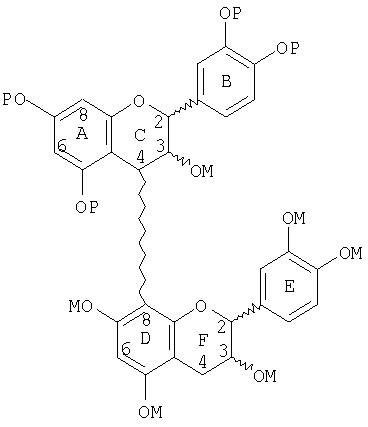
Следующее соединение является иллюстрацией маскированного димера со снятой защитой такого, как получено на стадии (с) выше:
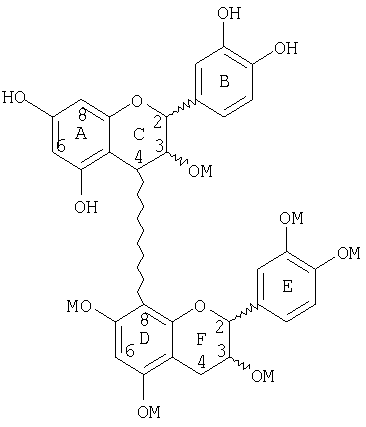
Следующие соединения являются иллюстрацией защищенного, маскированного, блокированного линейного триммера:
или
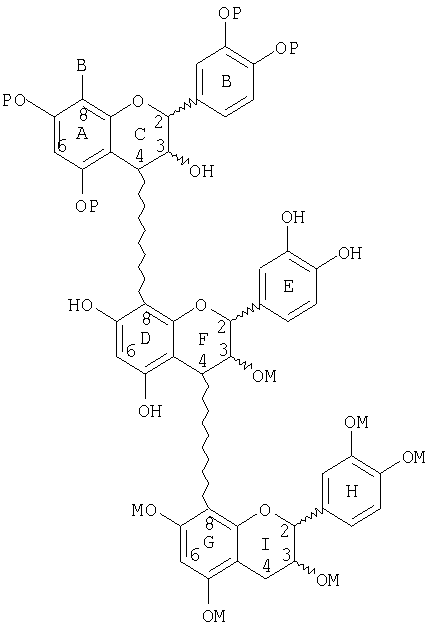
Следующие соединения являются иллюстрацией неблокированного и блокированного, защищенного, маскированного линейного тримера со стадии (d):
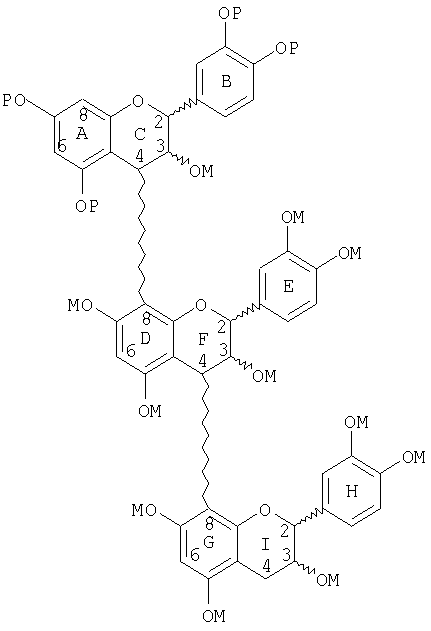
или
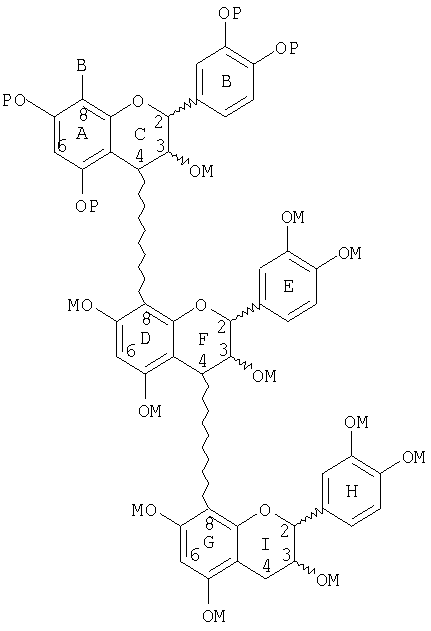
Следующие соединения являются иллюстрацией неблокированного и/или блокированного, маскированного линейного тримера со снятой защитой со стадии (е):
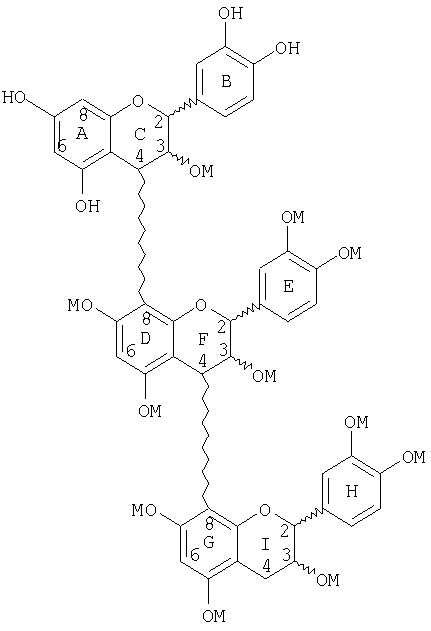
или
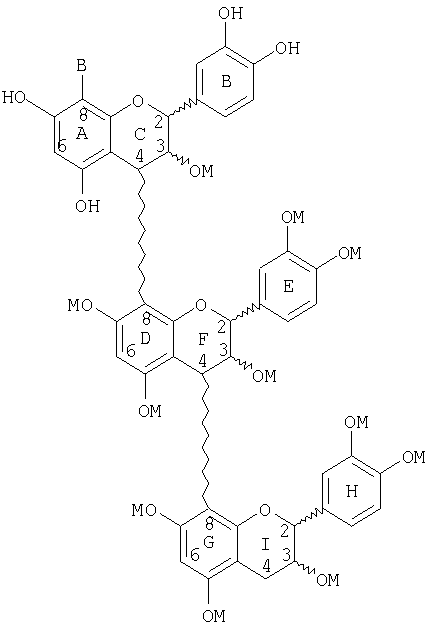
Следующие соединения являются иллюстрацией соединений, которые получают в результате необязательного повторения или чередования стадий (a)-(f) с получением высших олигомеров, в которых число меров равно 4:
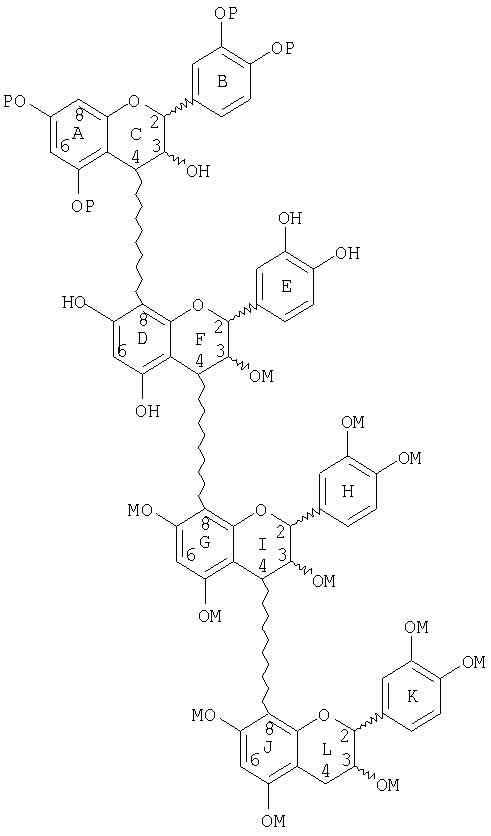
или
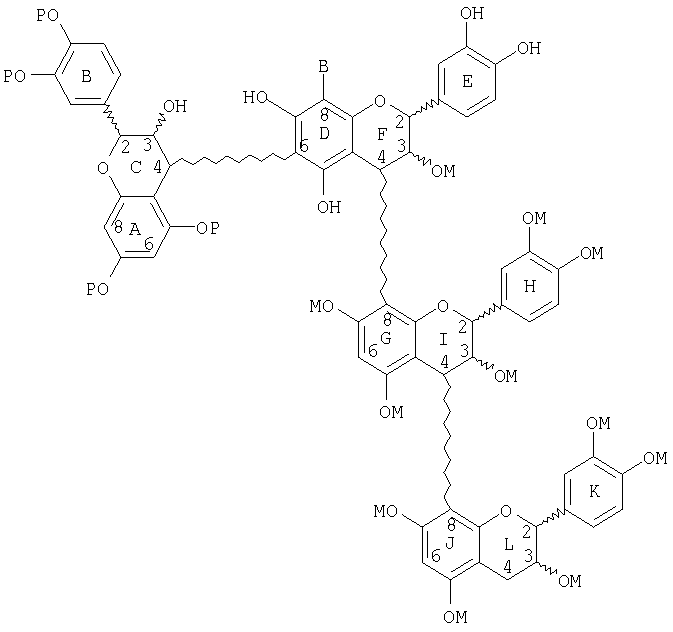
или
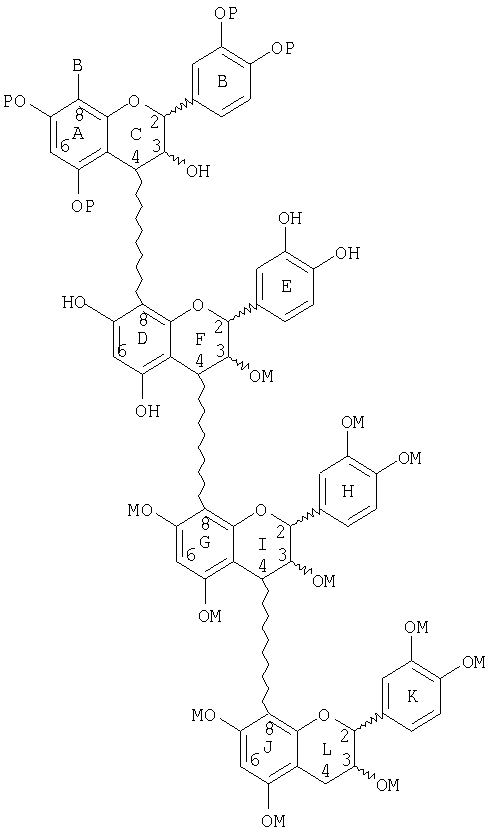
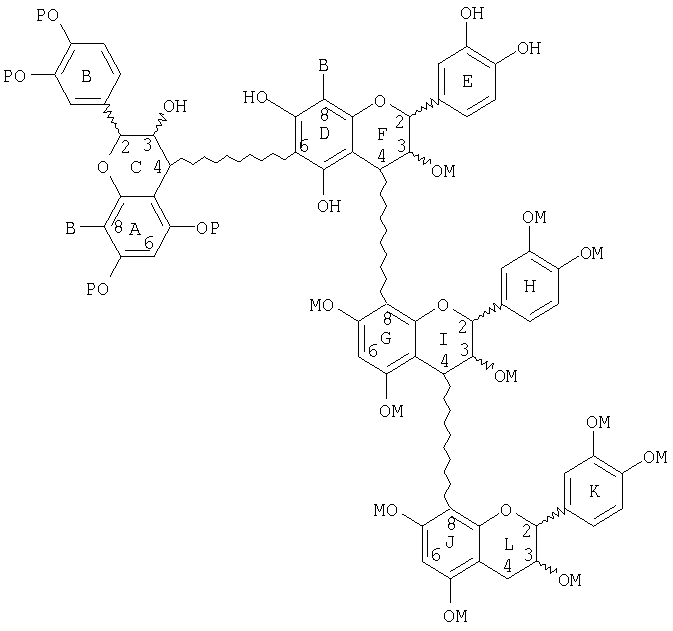
Также описан способ получения разветвленных процианидиновых олигомеров. Он включает стадии:
(a) получения неблокированного или блокированного, частично защищенного процианидинового димера, в котором фенольные гидроксильные группы верхнего мера защищены удаляемой защитной группой, которая не дезактивирует кольцо А защищенного мера, в то время как нижний мер имеет свободные фенольные гидроксильные группы;
(b) сочетания димера со стадии (а) с неблокированным или блокированным, защищенным, активированным процианидиновым мономером с образованием разветвленного тримера;
(c) снятия защиты с тримера со стадии (b); и
(d) необязательно проведения одной из следующих стадий в последовательном, чередующемся или комбинационном порядке с получением процианидиновых олигомеров, имеющих 4-18 меров,
включающих связи (4→8), (4→6), (6→4) и/или (8→4);
(i) сочетания олигомера со стадии (с) с неблокированным или блокированным, защищенным процианидиновым мономером;
(ii) маскирования олигомера со стадии (с), снятия защиты с маскированного олигомера и сочетания маскированного олигомера с незащищенным или защищенным, блокированным, активированным процианидиновым мономером.
Свободные фенольные формы процианидинового димера, линейного процианидинового олигомера или разветвленного процианидинового олигомера получают снятием защиты с димера или олигомера и, если необходимо, демаскированием и/или деблокированием димера или олигомера. Димеры или олигомеры могут включать одинаковые или различные эпикатехиновые или катехиновые меры. Предпочтительно n равно 5-12, более предпочтительно n равно 5. В линейных олигомерах связи представляют (4→6), (4→8) и/или (4→6). В разветвленных олигомерах связи представляют (4→6), (4→8), (6→4) и/или (8→4).
Защитные группы могут быть бензилом, п-метоксибензилом, трет-бутилом или тритилом; бензил является предпочтительным. На стадии защиты используется апротонный растворитель, например диметилформамид, диметилацетамид или диметилсульфоксид; диметилацетамид является предпочтительным. Ацилоксильные активирующие группы обычно представляют ацетокси, формилокси или пропионилокси; ацетокси является предпочтительным. Активирование проводят с использованием соли свинца (IV), например тетраацетата свинца, тетраформиата свинца или тетрапропионата свинца. Предпочтительно стадию архивирования проводят также с использованием органической кислоты, которая является такой же, как при получении соли свинца. Пригодные органические кислоты включают муравьиную кислоту, уксусную кислоту и пропионовую кислоту. Предпочтительным растворителем для стадии активирования является бензол. Блокирующая группа представляет галоген, предпочтительно бром или йод. Стадию деблокирования проводят с алкиллитием, например трет-бутиллитием или н-бутиллитием. Стадию демаскирования проводят основным гидролизом. Стадию снятия защиты проводят гидрогенолизом.
Дважды разветвленный олигомер, имеющий структуру:
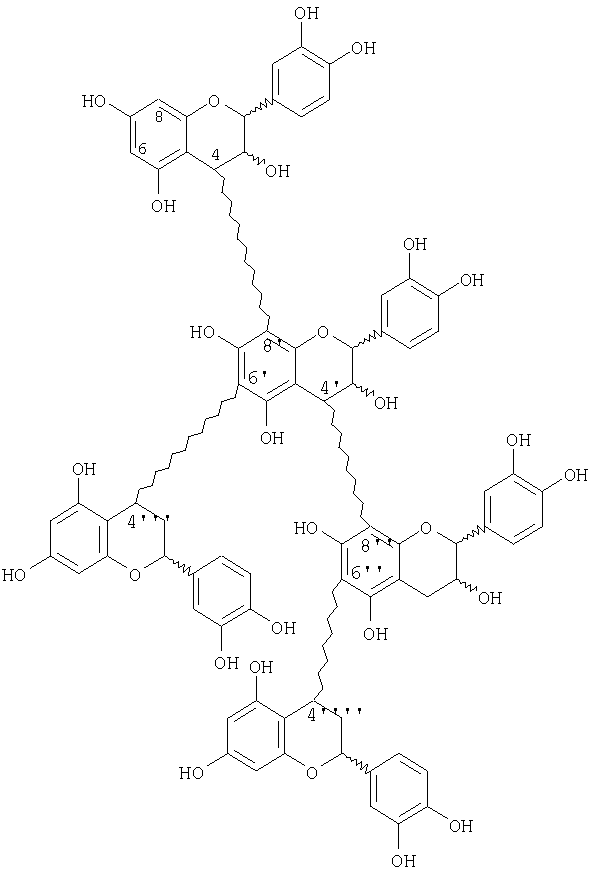
можно получить способом, который включает стадии:
(a) защиты каждой фенольной гидроксильной группы первого процианидинового мономера с помощью первой удаляемой защитной группы, которая не дезактивирует кольцо А мономера, где стадию защиты проводят в апротонном растворителе с получением защищенного мономера;
(b) активирования для сочетания С-4-положения соединения со стадии (а) введением ацилоксильной группы с использованием соли свинца органической кислоты с получением активированного, защищенного мономера;
(c) сочетания соединения со стадии (b) с незащищенным процианидиновым мономером в присутствии катализатора сочетания с получением частично защищенного димера;
(d) маскирования димера со стадии (с) с получением маскированного, частично защищенного димера;
(e) снятия защиты с маскированного, частично защищенного димера со стадии (d) с получением маскированного димера;
(f) сочетания маскированного димера со стадии (е) с 4β-ацетокси-защищенным процианидиновым мономером с получением тримера;
(g) сочетания тримера со стадии (f) с 4β-ацетокси-процианидиновым мономером с получением процианидинового тетрамера;
(h) демаскирования тетрамера со стадии (g); и
(i) сочетания тетрамера со стадии (h) с 4β-ацетокси-процианидиновым мономером с получением процианидинового пентамера.
Стадии до (i) можно повторить с получением множественно разветвленного процианидинового олигомера, включающего n меров, где n равно целому числу от 6 до 18.
Описан способ получения процианидинового димера, имеющего связь (8↔8). Он включает стадии:
(a) взаимодействия первого 8-бром-защищенного мономера с гексаалкилдистаннаном в присутствии палладия(0) с получением защищенного мономер-8-триалкилстаннана;
(b) сочетания соединения со стадии (а) со вторым 8-бром-защищенным мономером в присутствии тетракис(трифенилфосфин)-палладия(0) в бензоле с получением (8↔8) связанного димера; и
(с) снятия защиты с соединения со стадии (b) с получением (8↔8) димера.
Также описан способ получения процианидинового димера, имеющего связь (6↔6). Способ включает стадии:
(a) взаимодействия первого 6-бром-защищенного мономера с гексаалкилдистаннаном в присутствии палладия(0) с получением мономер-6-триалкилстаннана;
(b) сочетания соединения со стадии (а) со вторым 6-бром-защищенным мономером в присутствии тетракис(трифенилфосфин)-палладия(0) в бензоле с получением (6↔6) связанного димера; и
(c) снятия защиты с соединения со стадии (b) с получением (6↔6) димера.
Также описан способ получения процианидинового димера, имеющего связь (6↔8). Способ включает стадии:
(a) взаимодействия первого 6-бром-защищенного мономера с гексаалкилдистаннаном в присутствии палладия(0) с получением защищенного мономер-6-триалкилстаннана;
(b) сочетания соединения со стадии (а) со вторым 8-бром-защищенным мономером в присутствии тетракис(трифенилфосфин)-палладия(0) в бензоле с получением (6↔8) связанного димера; и
(c) снятия защиты с соединения со стадии (b) с получением (6↔8) димера.
Также описан способ получения процианидинового димера, имеющего связь (8↔6). Способ включает стадии:
(a) взаимодействия первого 8-бром-защищенного мономера с гексаалкилдистаннаном в присутствии палладия(0) с получением защищенного мономер-8-триалкилстаннана;
(b) сочетания соединения со стадии (а) со вторым 6-бром-защищенным мономером в присутствии тетракис(трифенилфосфин)-палладия(0) в бензоле с получением (8↔6) связанного димера; и
(c) снятия защиты с соединения со стадии (b) с получением (8↔6) димера.
Настоящий способ имеет важные преимущества и эффективность по сравнению с более ранними способами получения процианидиновых олигомеров, они включают лучший выход, лучшую селективность и более легкое выделение продукта. При проведении стадии защиты в диметилацетамиде вместо диметилформамида легче контролировать частичную и полную защиту фенольных гидроксильных групп.
Настоящее изобретение также относится к кристаллическому 8-бром-тетра-О-бензил(-)-эпикатехину, когда используется диметилацетамид в качестве растворителя на стадии защиты.
Подробное описание изобретения
Мономеры, входящие в состав процианидинов, имеют структуру
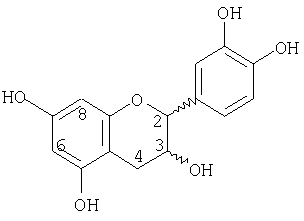
Процианидины включают таковые, обнаруженные в какао-бобах, полученных из Theobroma cacao и различных родственных видов какао, а также рода Herrania и их внутри- и межродовых гибридов.
Мономеры, входящие в состав процианидинов, включают (+)катехин, (-)-эпикатехин и их соответствующие эпимеры (например, (-)-катехин и (+)-эпикатехин).
Синтетические линейные и/или разветвленные олигомеры, имеющие следующие структуры, показаны в качестве таковых, которые можно получить вышеуказанным способом.
Линейные олигомеры, где n равно целому числу от 0 до 16
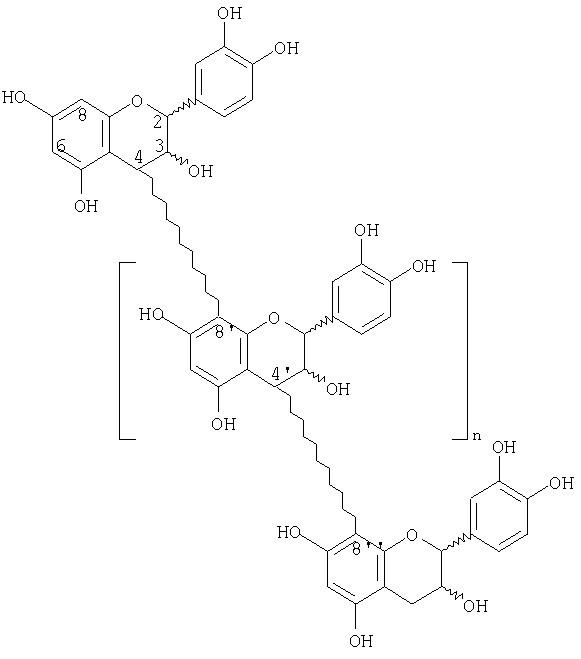
разветвленные олигомеры, где А и В независимо представляют олигомеры от 1 до 15, в целом 3-18 в конечном олигомере.
В олигомерах n равно целому числу от 2 до 18, предпочтительно от 3 до 12, более предпочтительно от 5 до 12 и наиболее предпочтительно равно 5. Олигомеры имеют межфлавановые связи (4→6) и/или (4→8). Олигомеры, полученные способом по изобретению, могут быть представлены вышеуказанными структурами. В отношении линейного олигомера, то, когда n равно 0, олигомер называют «димером»; когда n равно 1, олигомер называют «тримером»; когда n равно 2, олигомер называют «тетрамером»; когда п равно 3, олигомер называют «пентамером»; и аналогичными перечислениями можно обозначить олигомеры, имеющие n до и включая 16 и выше, так, например, когда n равно 16, олигомер называют «октадекамером». В отношении разветвленного олигомера, то, когда А или В равно 1, олигомер называют «тримером»; с аналогичными перечислениями, описанными для линейных олигомеров.
Стереоизомеры олигомеров включены в объем изобретения. Стереохимию мономеров, входящих в состав олигомера, можно описать терминами их относительной стереохимии, т.е. «альфа/бета» или «цис/транс» или терминами их абсолютной стереохимии, т.е. «R/S». Термин «альфа» (α) указывает, что заместитель ориентирован ниже плоскости флаванового кольца, в то время как термин «бета» (β) означает, что заместитель ориентирован выше плоскости кольца. Термин «цис» указывает, что два заместителя ориентированы на одной стороне кольца, в то время как «транс» указывает, что два заместителя ориентированы с противоположных сторон кольца. Термины «R» и «S» используются для обозначения расположения заместителей около стереогенного центра, основываясь на расположении групп соответственно атомному номеру атомов, непосредственно связанных со стереогенным центром (система КИП).
Имеются многочисленные стереохимические связи между С-4-положением флаван-3-ол-мономера и С-6- и С-8-положениями смежного мономера. Стереохимические связи между мономерными единицами обозначены здесь как (4α→6), или (4β→6), или (4α→8), или (4β→8) для линейных олигомеров. Для связей с разветвленным или соединительным мономером стереохимическими связями являются (6→4α), или (6→4β), или (8→4α), или (8→4β). Когда (+)-катехин, обозначенный здесь как С, связан с другим С или (-)-эпикатехином, обозначенным здесь как ЕС, связи предпочтительно представляют (4α→6) или (4α→8). Когда ЕС связан с С или другим ЕС, связи предпочтительно представляют (4β→6) или (4β→8).
Линейные или разветвленные олигомеры можно получить способами по настоящему изобретению с использованием стадий защиты, активирования, сочетания, маскирования, блокирования, снятия защиты, демаскирования и деблокирования. В каждой последовательности реакций мономеры можно использовать для получения линейных или разветвленных олигомеров, включающих одинаковые или различные мономеры. Высшие олигомеры можно получить повторением сочетания димера, тримера и т.п. с дополнительным мономером, используя вышеуказанные стадии.
Примеры соединений, которые можно синтезировать способом по изобретению, включают димеры;
ЕС-(4β→8)-ЕС и ЕС-(4β→6)-ЕС, где ЕС-(4β→8) -ЕС является предпочтительным; тримеры [ЕС-(4β→8)]2-ЕС, [ЕС-(4β→8)]2-С и [ЕС-(4β→6)]2-EC, где [ЕС-(4β→8)]2-EC является предпочтительным; тетрамеры [ЕС- (4β→8)]3-ЕС, [ЕС-(4β→8)]3-С и [ЕС-(4β→8)]2-ЕС-(4β→6)-С, где [ЕС-(4β→8)]3-ЕС является предпочтительным; и пентамеры [ЕС-(4β→8)]4-ЕС, [ЕС-(4β→8)]3-ЕС-(4β→6)-ЕС, [ЕС-(4β→8)]3-ЕС-(4β→8)-С и [ЕС-(4β→8)]3-ЕС-(4β→6)-С, где [ЕС-(4β→8)]4-ЕС является предпочтительным. Примером разветвленного тримера является
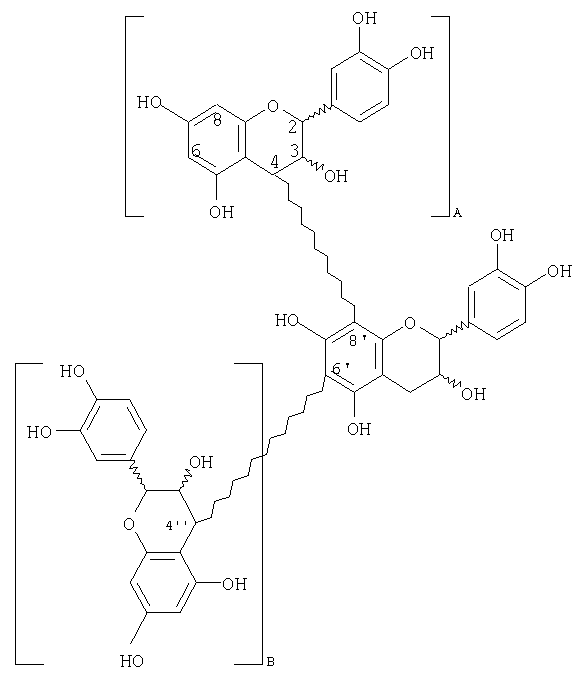
примером разветвленного тетрамера является
примером разветвленного пентамера является
Дополнительные соединения, которые можно синтезировать, включают следующие:
(i) гексамер, в котором один мономер (С или ЕС) связан с пентамером соединения, входящим в перечень выше, например [ЕС-(4β→8)]5-EC, [ЕС-(4β→8)]4-ЕС-(4β→6)-ЕС, [ЕС-(4β→8)]4-ЕС-(4β→6)-С и [ЕС-(4β→8)]4-ЕС-(4β→6)-С, где [ЕС-(4β→8)]5-EC является предпочтительным, с примером разветвленного гексамера
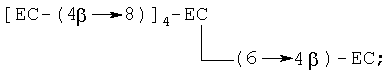
(ii) гептамер, в котором любая комбинация двух мономеров (С и/или ЕС) связана с одним из вышеуказанных пентамеров, например [ЕС-(4β→8)]6-ЕС, [ЕС-(4β→8)]5-ЕС-(4β→6)-ЕС, [ЕС-(4β→8)]5-ЕС-(4β→8)-С и [ЕС-(4β→8)]5-ЕС-(4β→6)-С, где [ЕС-(4β→8)]6-ЕС является предпочтительным, с примером разветвленного гептамера
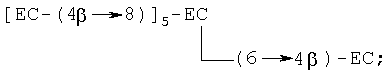
(iii) октамер, в котором любая комбинация трех мономеров (С и/или ЕС) связана с одним из вышеуказанных пентамеров, например [ЕС-(4β→8)]7-ЕС, [ЕС-(4β→8)]6-EC-(4β→6)-EC, [ЕС-(4β→8)]6-ЕС-(4β→8)-С, [ЕС-(4β→8)]6-ЕС-(4β→6)-С, где [ЕС-(4β→8)]7-ЕС является предпочтительным, с примером разветвленного октамера
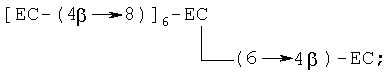
(iv) нонамер, в котором любая комбинация четырех мономеров (С и/или ЕС) связана с одним из вышеуказанных пентамеров, например [ЕС-(4β→8)]8-ЕС, [ЕС-(4β→8)]7-ЕС-(4β→6)-ЕС, [ЕС-(4β→8)]7-ЕС-(4β→8)-С, [ЕС-(4β→8)]7-ЕС-(4β→6)-С, где [ЕС-(4β→8)]8-ЕС является предпочтительным, с примером разветвленного нонамера
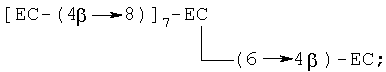
(v) декамер, в котором любая комбинация пяти мономеров (С и/или ЕС) связана с одним из вышеуказанных пентамеров, например [ЕС-(4β→8)]9-ЕС, [ЕС-(4β→8)]8-ЕС-(4β→6)-ЕС, [ЕС-(4β→8)]8-ЕС-(4β→8)-С, [ЕС-(4β→8)]8-ЕС-(4β→6)-С, где [ЕС-(4β→8)]9-ЕС является предпочтительным, с примером разветвленного декамера
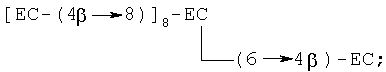
(vi) ундекамер, в котором любая комбинация шести мономеров (С и/или ЕС) связана с одним из вышеуказанных пентамеров, например [EC-(4β→8)]10-EC, [EC-(4β→8)]9-ЕС-(4β→6)-ЕС, [ЕС-(4β→8)]9-ЕС-(4β→8)-С, [ЕС-(4β→8)]9-ЕС-(4β→6)-С, где [ЕС-(4β→8)]10-ЕС является предпочтительным, с примером разветвленного ундекамера
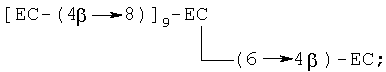
(vii) додекамер, в котором любая комбинация семи мономеров (С и/или ЕС) связана с одним из вышеуказанных пентамеров, например [ЕС-(4β→8)]11-ЕС, [ЕС-(4β→8)]10-ЕС-(4β→6)-ЕС, [ЕС-(4β→8)]10-ЕС-(4β→8)-С, [ЕС-(4β→8)]10-ЕС-(4β→6)-С, где [ЕС-(4β→8)]11-ЕС является предпочтительным, с примером разветвленного додекамера
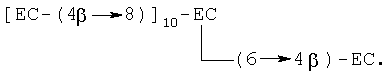
Вышеуказанный перечень олигомеров является иллюстративным и приводится для демонстрации типов соединений, которые можно получить способами по изобретению, и не представляет исчерпывающий перечень соединений, входящих в изобретение. Олигомеры можно разделить и очистить способами, раскрытыми в патенте США 5554645 от 10 сентября 1976, Romanczyk et al. и патенте США 5712305 от 27 января 1998, Romanczyk et al.
Специалистам в данной области очевидно понятно, что вращение ряда связей внутри олигомера может быть ограниченным за счет стерического препятствия особенно, если олигомер является замещенным, например, бензильными группами. Следовательно, все возможные региоизомеры и стереоизмеры соединений, полученных по изобретению, включены в объем изобретения.
Определения
Как здесь использовано, «защитная группа» является удаляемой группой, которая замещает водород фенольных гидроксильных групп в процианидиновых мономерах или олигомерах. Группу следует удалить в условиях, которые не влияют отрицательно на процианидиновые олигомеры.
Как здесь использовано, «блокирующая группа» является удаляемой группой, которая направляет сочетание блокированием С-8-положения кольца А процианидинового олигомера, направляя таким образом сочетание с другим процианидиновым мономером по С-6-положению кольца А. Группу следует удалить в условиях, которые не влияют отрицательно на процианидиновый олигомер.
Как здесь использовано, «маскирующая группа» является удаляемой группой в С-4-положении, которая маскирует незащищенную фенольную гидроксильную и С-3-гидроксильную группу(ы) процианидинового мономера или высшего олигомера во время сочетания димера высшего олигомера с другим процианидиновым мономером. Группу следует удалить в условиях, которые не влияют отрицательно на процианидиновый олигомер.
Как здесь использовано, «активирующая группа» является ацилоксильной группой, которая активирует С-4-положение С-кольца процианидинового димера или олигомера и приводит к сочетанию с другим процианидиновым мономером или олигомером в этом положении.
Термин «комбинационный», используемый здесь, относится к вероятным возможностям региохимических связей для получения любого данного процианидинового олигомера. Например, линейный процианидиновый тетрамер может включать связи (4→8) и (4→6) между мономерами, входящими в состав тетрамера. Для целей синтеза данные связи приводят к получению отдельных соединений, которые могут обладать различной биологической активностью. Для исследования зависимости структура-активность будет полезным обеспечить ряд таких олигомеров с целью определения важности региохимических связей в отношении биологической активности. Для линейного тетрамера возможные связи являются следующими:
что делает необходимым потребность в способе синтеза для получения 8 различных тетрамеров, для каждого из которых требуются различные стадии получения.
Защитные группы
Защитные группы, пригодные для данного изобретения, представляют электронодонорные группы, функция которых заключается в активировании процианидиновых мономеров в реакциях С-4-активирования и сочетания, описанных ниже. В реакции С-4-активирования электронодонорные фенольные защитные группы функционируют для стабилизации и таким образом они способствуют образованию промежуточного С-4-бензильного катиона, образующегося при окислении защищенного мономера солью свинца (IV). В реакции сочетания, реакции электрофильного ароматического замещения электронодонорные фенольные защитные группы также функционируют для стабилизации и таким образом способствуют образованию С-4-бензильного катиона, образующегося при обработке С-4-ацилокси-замещенного процианидинового мономера (активированного мономера) кислотой Льюиса в качестве катализатора. В реакции сочетания электронодонорные фенольные защитные группы также функционируют для того, чтобы увеличить различия в реакционной способности у различных арильных групп, которые могут находиться в реакции. Как описано ниже, незащищенные процианидиновые мономеры или выбранные незащищенные (со снятой защитой) мономерные единицы процианидинового олигомера используются в качестве нуклеофилов в реакции сочетания; С-4-ацилокси-замещенный, защищенный процианидиновый мономер при обработке кислотой Льюиса функционирует в качестве электрофила. Незащищенные процианидины функционируют в качестве нуклеофилов, поскольку они обладают более высокой электронной плотностью, которая выше нуклеофильности, чем у защищенных процианидиновых мономеров. Любое самосочетание между защищенными процианидиновыми мономерами ограничивается за счет сравнительно более высокой нуклеофильности у незащищенных процианидинов.
Среди различных защитных групп бензильные группы являются предпочтительными, поскольку они легче удаляются в мягких условиях, таких как гидрогенолиз. Другим преимуществом бензилирования (за исключением, например, п-нитробензилирования) является то, что оно не дезактивирует ароматическое кольцо перед сочетанием, когда процианидиновые мономеры или олигомеры действуют в качестве нуклеофилов. Является удивительным и совершенно неожиданным, что замена апротонного органического растворителя, используемого на стадии защиты с диметилформамида (ДМФА) на диметилацетамид (ДМА), приводила к повышенному выходу защищенного олигомера, возможно за счет того факта, что незначительно более высокая диэлектрическая константа ДМА может способствовать О-алкилированию. Выход составил по меньшей мере примерно 50%, обычно примерно от 60% до примерно 70%. Кроме того, при этом не требовалось дополнительной очистки, и продукты легко кристаллизовались. В примерах 1 и 2 описываются конкретные условия получения тетра-O-бензил-(+)-катехина и тетра-O-бензил-(-)-эпикатехина. Дальнейшее исследование системы растворителей показало, что карбонат калия (К2СО3) был предпочтителен по сравнению с карбонатом натрия (Na2СО3) в результате его растворимости в предпочтительной системе растворителей. Было установлено, что йодид калия можно применять в каталитических количествах в сочетании с бензилбромидом.
Другой пригодной защитной группой для (-)-эпикатехина является п-метоксибензильная группа (РМВ). Если выбрана РМВ в качестве защитной группы при получении частично защищенного процианидинового димера, тогда стадия защиты дополнительно включает ацетилирование процианидинового мономера с последующей обработкой гидридом натрия, РМВ-хлоридом и ДМФА в воде для удаления фенольных ацетатных групп, приводя к алкилированию феноксидных ионов с помощью РМВ. При использовании ДМА в качестве растворителя на стадии защиты РМВ-группы использовать не следует. Тетра-O-РМВ-(-)-эпикатехин можно получить с использованием способа Kawamoto H., Nakatsubo F. и Murakami К., Synth. Commun., 26, 531-534 (1996). Вначале получают пентаацетил (-)-эпикатехин (как описано в примере 3 ниже) обработкой гидридом натрия (NaH), п-метоксибензилхлоридом (PMBCl), диметилформамидом (ДМФА) и водой в количестве, достаточном (4 экв) для получения эквивалентного количества основания для последовательного удаления четырех фенольных ацетатных групп. Полученные феноксидные ионы подвергаются быстрому алкилированию с помощью PMBCl. В примерах 4 и 5 описаны конкретные условия, при которых получали пентаацетил(+)-катехин и тетра-О-п-метоксибензил-3-ацетил-(-)-эпикатехин.
Очевидно специалистам в данной области понятно, что можно использовать другие защитные группы, такие как трет-бутил, тритил и 2,4-диметоксибензил.
С-4-активирование
Изменение условий реакции при участии LTA на смесь бензол: уксусная кислота 1:1 в конечном итоге давало наиболее высокий выход (60-70%) и стереоспецифичный 4β-продукт. Другие пригодные смеси растворителей включают бензол, толуол, хлорбензол, циклогексан, гептан, четыреххлористый углерод или их смеси, смешанные с органической кислотой, которая является такой же, какая была использована для получения соли свинца (IV), примененного в реакции активирования.
На стадии активирования применяют соли свинца органических кислот, например тетраформиат свинца, тетрапропионат свинца и тому подобное. Предпочтительно использовать для улучшения выхода соответствующие органические кислоты, например муравьиную и пропионовую кислоты в сочетании с солью свинца. Предпочтительной солью является ацетат свинца и предпочтительную комбинацию представляют тетраацетат свинца и уксусная кислота.
В примерах 6-9 описаны конкретные условия получения 4β-ацетокси тетра-O-бензил-(-)-эпикатехина, 4β-гидроксил тетра-O-бензил-(+)-катехина, 4β-гидроксил тетра-O-бензил-(-)-эпикатехина и 4β-ацетокси тетра-O-бензил-(+)-катехина.
Маскирующие группы
Маскирующие группы, пригодные для данного изобретения, представляют электроноакцепторные группы, функция которых заключается в дезактивировании выбранных мономерных единиц процианидиновых олигомеров в реакции электрофильного ароматического заместительного сочетания, описанной ниже. Когда в реакции сочетания используются процианидиновые олигомеры, требуется, чтобы активированный мономер не взаимодействовал произвольно с различными мономерными единицами олигомера. Маскирующие группы используются для повышения различий в реакционной способности у различных мономерных единиц олигомера. Применение электроноакцепторных групп в качестве маскирующих групп дезактивирует мономерные единицы олигомера, имеющего маскирующие группы, перед сочетанием с активированным, защищенным мономером. Следовательно, в способе по данному изобретению активированный мономер селективно взаимодействует с незащищенной мономерной единицей частично маскированного олигомера за счет высокой реакционной способности (нуклеофильности) незащищенной мономерной единицы и пониженной реакционной способности маскированной мономерной единицы олигомера.
Маскирующие группы, пригодные для способа по изобретению, включают ацильные группы, такие как ацетил и пропионил, ароильные группы, такие как бензоил, карбаматные группы, такие как N-фенилкарбамат, карбонатные группы, такие как метилкарбонат, и арилсульфонильные группы, такие как 2,4-динитробензол-сульфонил. Предпочтительно маскирующей группой является ацетил. Маскирование незащищенных фенольных гидроксильных и С-3-гидроксильных групп процианидиновых олигомеров можно проводить с использованием обычного способа замещения водорода гидроксилов подходящими маскирующими группами, такими, как указаны выше. Реагенты, которые будут использоваться, зависят от вводимых маскирующих групп и хорошо известны в данной области.
Реакция сочетания
В способе по изобретению является предпочтительным применение катализатора сочетания, такого как кислота Льюиса (например, бромида лития). Также предпочтительно использование 4β-ацетокси-производного в качестве электрофила. Тем самым значительно повышается селективность реакции сочетания. Использование Li+ в качестве противоиона способствует протеканию С-алкилирования по сравнению с O-алкилированием.
Когда в нагреваемый с обратным холодильником метиленхлоридный раствор 4β-ацетокситетра-O-бензил-(+)-катехина и LiBr добавляют метанол, 4β-метокси тетра-O-бензил-(+)-катехин образуется со значительно более высоким выходом (смотри пример 10). β-стереохимия определяется на основе константы сочетания 3,5 Гц между Н-3 и Н-4, что указывает на цис-связь. Данная реакция не протекает, когда не используется галид, такой как LiBr, в качестве кислоты Льюиса. В данной реакции ацетокси замещается галидом, который затем сразу же взаимодействует с метанолом, действуя в качестве нуклеофила, приводя тем самым реакцию к равновесию.
Неожиданно было найдено, что применение LiBr в качестве кислоты Льюиса будет направлять реакцию сочетания между 4β-ацетокси тетра-O-бензил-мономером и другим мономером, действующим в качестве нуклеофила, тем самым исключая стадию получения 3,4-диола мономера.
Для проверки данной неожиданной находки и понимания сущности потенциального применения данной реакции 4β-ацетокси тетра-O-бензил-(+)-катехин взаимодействовал с (-)-эпикатехином в присутствии LiBr, как описано в примере 11. Полученный димер был 90% чистоты, и выход составлял 62%. 1H ЯМР-спектр указывал на наличие одного синглета при δ 5,85 для 1Н и пары дублетов при δ 6,19 и 6,16, каждый объединенный с 1Н типичным m-сочетанием 1,5 Гц, что указывало на образование только одного изомера. Обработка димера смесью уксусный ангидрид/пиридин давала гексаацетат (пример 13), и, как ожидалось, синглет С-6'-водорода смещался в сторону слабого поля к δ 6,52. Дублет С-4-водорода имел константу сочетания 9,6 Гц, что указывает на α-конфигурацию. С частично бензилированного димера снимали защиту с помощью водорода/палладия(0) (Н2/Pd) с получением димера (+)-катехин-(4α→8)-(-)-эпикатехина (смотри пример 12). Анализ ВЭЖХ (фиг.1В) показывал, что помимо вышеуказанного димера присутствовал другой неизвестный димер (13,5%), а также следовые количества тримера и тетрамера. Окончательное подтверждение структуры димера было сделано при получении октаацетата и сравнении 1Н ЯМР-спектра с литературными данными (Kawamoto Н., Nakatsubo F. и Murakami К., J.Wood Chem. Tech., 9, 35-92 (1989)).
Для исследования неожиданных результатов с использованием реакции сочетания с LiBr проводили сочетание между 4β-ацетокси тетра-O-бензил-(-)-эпикатехином и (-)-эпикатехином, как описано в примере 15 (смотри пример 17 для катехинового димера). Димер (-)-эпикатехин-(4β→8)-(-)-эпикатехин получали после гидрогенолиза (пример 16). Как показано в последующей таблице, в данной реакции присутствовали оцениваемые количества тримеров и тетрамеров.
Реакция сочетания с LiBr
13,5
9,3
**Выход по данным хроматографии на силикагеле.
В данной таблице показано, что в результате этой реакции получают только один основной димер. При изменении времени реакции и количеств реагирующих мономеров можно снизить присутствие высших олигомеров. Кроме того, наблюдали, что тетрабензил-мономеры не действуют в качестве нуклеофилов в реакции сочетания с участием бромида лития. Свободные фенольные гидроксильные группы необходимы для увеличения активности ароматического кольца перед сочетанием. Это важно, поскольку это предлагает средство дифференциации колец, способных к участию в сочетании, и колец, неспособных к участию в сочетании.
Выходы в реакции сочетания также были улучшены при использовании йодида лития (LiI) в качестве кислоты Льюиса (смотри пример 20). Кроме того, реакция между 4β-ацетокси тетра-O-бензил-(+)-катехином и (-)-эпикатехином завершалась только в течение 18 ч с выходом 85% после хроматографии. В примере 11, где использовали бромид лития, выход составил только 62% после 24 ч.
Данный способ сочетания можно использовать для получения высших олигомеров, иных, чем димер, как показано на следующей реакционной схеме:
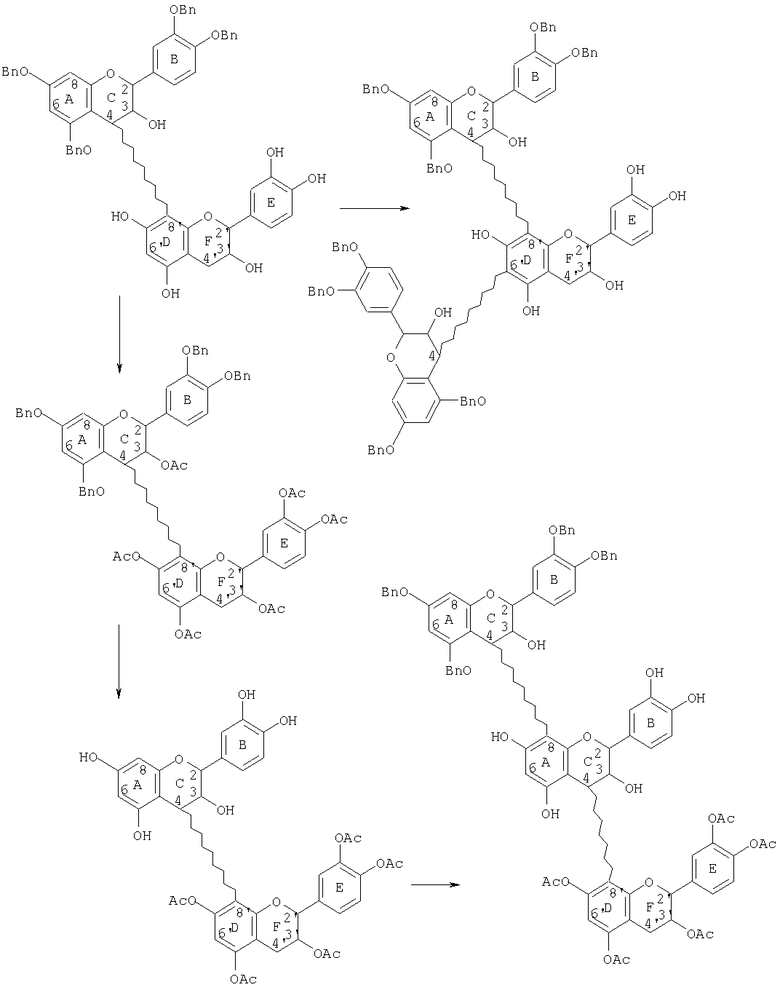
Поскольку фенольные гидроксильные группы в кольце А бензилированы, сочетание протекает только по С-6'-положению кольца D и приводит к образованию разветвленного тримера. Это было с успехом проверено, при взаимодействии тетра-O-бензил-(+)-катехин-(4α→8)-(-)-эпикатехина (смотри пример 11) с 4β-ацетокси тетра-O-бензил-(+)-катехином (смотри пример 6) с LiBr в смеси ТГФ-метиленхлорид. Тонкослойной хроматографией (ТСХ) было выделено пятно, 1H ЯМР-спектр которого сложно интепретировать. Однако масс-спектральный анализ показал, что молекулярный ионный пик при m/z 1861 соответствовал желаемой структуре, в которой одна из гидроксильных групп не была ацетилирована. Реакцию повторяли и выделяли тот же продукт. Масс-спектр четко показывал образование этого разветвленного тримера, где наблюдали (M+Na)+ при m/z 1610 и (М+Н)+ при m/z 1588 при обычной фрагментации расщеплением retro Diels Alder. Третью параллельную реакцию проводили в течение более длительного периода времени, и вновь масс-спектр соответствовал разветвленному тримеру, чья экспериментальная структура была определена, как тетра-O-бензил-(+)-катехин-(4α→8)-(-)-эпикатехин-(6→4α)-тетра-O-бензил-(+)-катехин (смотри пример 24).
Для синтеза линейных олигомеров была разработана стратегия селективного активирования-дезактивирования колец, способных участвовать в реакции сочетания. В данном случае частично бензилированный димер ацетилировали и затем гидрогенолизировали, получив димер со свободными фенольными гидроксильными группами (ОН-группами) в кольцах А/В и с ацетатными группами в кольцах D/E. Электроноакцепторные ацетильные группы дезактивировали кольцо D и таким образом позволяли протекать сочетанию с 4β-ацетокси-мономерами региоселективно по С-8-положению кольца А. Полученный тример можно подвергать такому же способу ацетилирования и дебензилирования с последующим сочетанием с другим 4β-ацетокси-мономером с получением тетрамера. Повторение этих стадий приводит к получению олигомеров возрастающего размера.
Данный способ был подтвержден, когда частично защищенный димер из примера 11 ацетилировали (смотри пример 13) и затем гидрогенолизировали (смотри пример 14). Наблюдали дублирование ЯМР-пиков, что указывает на присутствие ротамеров. ЯМР-спектры, полученные при более высокой температуре (39,8°С), упрощали спектр, подтверждая наличие ротамеров. Интересно, что проведение гидрогенолиза в этилацетате облегчало выделение моно-O-бензил-3-ацетил-(+)-катехин-(4α→8)-пентаацетил-(-)-эпикатехина, чей ЯМР-спектр представлен на фиг.7. Когда данный олигомер гидрогенолизировали, продукт был таким же, как получали ранее. Взаимодействие 3-ацетил-(+)-катехин-(4α→8)-пентаацетил-(-)-эпикатехина (смотри пример 14) с 4β-ацетокси тетра-O-бензил-(-)-катехином (смотри пример 6) приводило к образованию желаемого продукта, т.е. тетра-O-бензил(+)-катехин-(4α→8)-3-ацетил(+)-катехин(4α→8)-пентаацетил(-)-эпикатехина (смотри пример 20). Масс-спектр (APCI (Химическая ионизация при атмосферном давлении.), режим отрицательных ионов) указывал на высокий молекулярный ионный пик при m/z 1479, 6, который был идентичен вычисленной массе для С85Н74О24 (1479,5). Масс-фрагменты при m/z 1437, 1389 и 1347 соответствовали потере ацетильной, бензильной и ацетильной/бензильной групп в исходном соединении. FAB (Бомбардировки быстрыми атомами.) MC показывал молекулярный ионный пик при m/z 1482 (М+H)+ и характер фрагментации, соответствующие тетра-O-бензил-3-ацетил (+)-катехин-(4α→8)-пентаацетил(-)-эпикатехину.
Аналогичным образом получали линейный тетрамер приготовлением тетра-O-бензил-(+)-катехин-(4α→8)-3-ацетил-(+)-катехин-(4α→8)-пентаацетил(-)-эпикатехина (смотри пример 20), который ацетилировали до тетра-O-бензил-3-ацетил-(+)-катехин-(4α→8)-пентаацетил-(+)-катехин-(4α→8)-пентаацетил(-)-эпикатехина (смотри пример 21). Гидрогенолиз вышеуказанного соединения (смотри пример 22) давал 3-ацетил-(+)-катехин-(4α→8)-пентаацетил-(+)-катехин-(4α→8)-пентаацетил(-)-эпикатехин. Сочетание 3-ацетил-(+)-катехин-(4α→8)-пентаацетил-(+)-катехин- (4α→8)-пентаацетил-(-)-эпикатехина с 4β-ацетокси тетра-O-бензил-(+)-катехином (смотри пример 6) приводило к образованию тетрамера тетра-O-бензил-(+)-катехин-(4α→8)-3-ацетил-пентаацетил-(+)-катехин-(4α→8)-пентаацетил-(+)-катехин-(4α→)-пентаацетил (-)-эпикатехина (смотри пример 23). FAB масс-спектр показывал наличие молекулярного ионного пика при m/z 1978, который соответствовал структуре.
Разработано также замещение in situ фенольных ацетатных групп бензильными группами. Замещение in situ ацетильных групп на бензильные проводили с 3-ацетил-тетра-О-бензил-(+)-катехин-(4α→8)-пентаацетил-(-)-эпикатехином (смотри пример 13) в условиях, указанных в примере 18, с получением 3-ацетил-тетра-О-бензил-(+)-катехин-(4α→8)-3-ацетил-тетра-О-бензил-(-)-эпикатехина. Гидрогенолиз (смотри пример 19) обеспечивал получение тетра-O-бензил-(+)-катехин-(4α→8)-тетра-O-бензил-(-)-эпикатехина, который затем гидрогенолизировали до свободного димера, что доказывает пригодность данного способа.
Блокирующие группы
На основе вышеуказанных стадий были разработаны способы синтеза межфлавановых связей (4→6) между мономерами. Например, мономеры можно бензилировать с высокими выходами с использованием системы растворителей с ДМА, описанной выше. Бензилированные мономеры можно бромировать в С-8-положении с получением 8-бром-тетра-О-бензил-мономеров, как показано в примере 25, и нескольких вариантов, представленных в примерах 26 и 27, и в примере 28 показана абсолютная стереохимия 8-бром-тетра-О-(-)-эпикатехина. Снятие защиты с этих производных приводит к получению 8-бром-мономеров. Полученное бромсодержащее производное эффективно блокирует сочетание в С-8-положении, направляя таким образом сочетание по С-6-положению. Сочетание 8-бром-мономеров с 4β-ацетокси тетра-O-бензил-мономером в условиях способа с бромидом лития приводит к образованию (4→6) димера. Типичная реакционная схема показана ниже
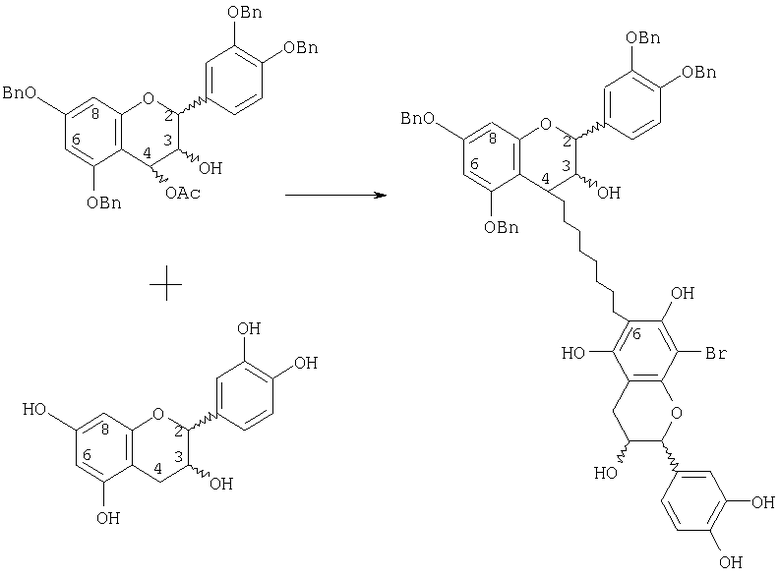
Дебромирование (т. е. деблокирование) проводят при низкой температуре (-78°С) в растворе с подходящим производным алкил-лития, таким как н-бутиллитий или трет-бутиллитий, с последующим протонированием полученного димера слабой протонной кислотой (например, водой). При использовании дополнительных стадий, воплощенных в изобретении, (4→6) димер можно увеличить до размера высших олигомеров, включая вариации регио- и стереохимии, описанных выше.
Снятие защиты и демаскирование
Реагенты, используемые на стадии снятия защиты, будут зависеть от группы, которая удаляется. Например, когда удаляют бензильные защитные группы, гидрогенолиз проводят в условиях, описанных в примерах 12, 16 и 22. Когда удаляют маскирующие группы, проводят щелочной гидролиз в условиях, описанных в примерах 5 и 18.
Альтернативно можно использовать промышленно доступную липазу для энзиматического деацетилирования защищенного олигомера. Удаление защитных или маскирующих групп можно проводить с использованием любых обычных способов при условии, что способы не влияют отрицательно на процианидиновый олигомер.
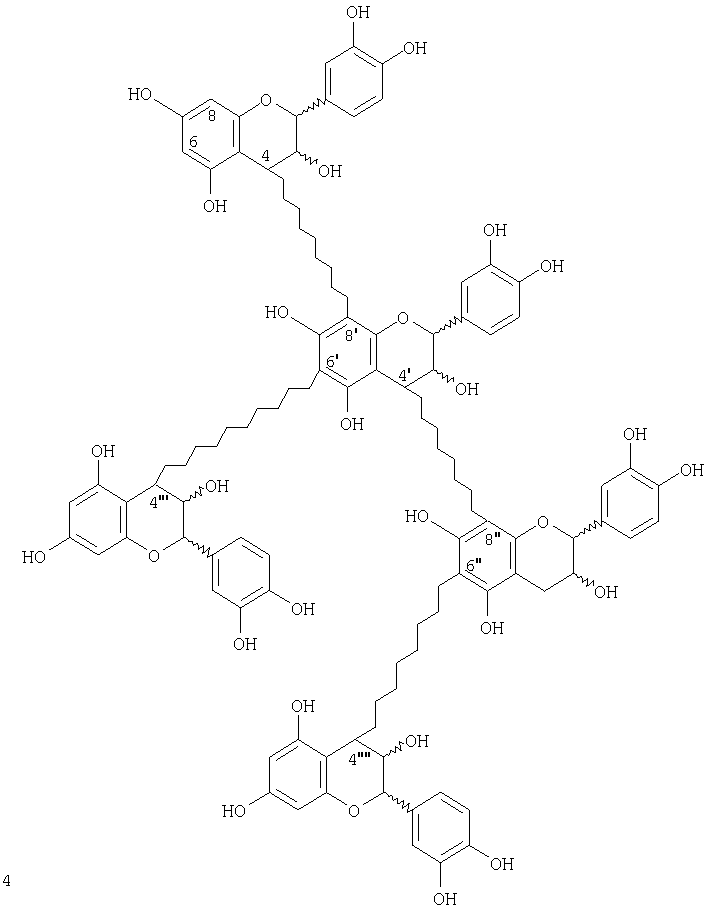
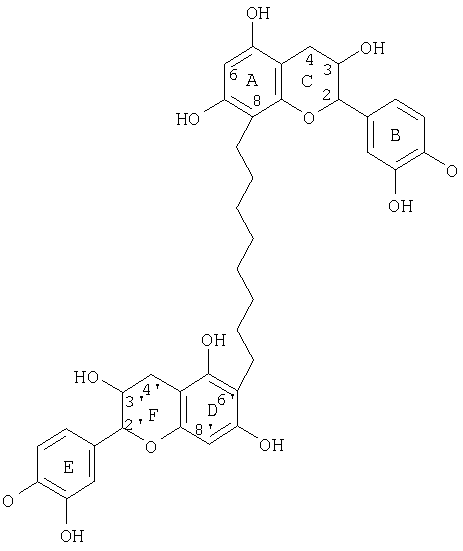
Соединения по изобретению
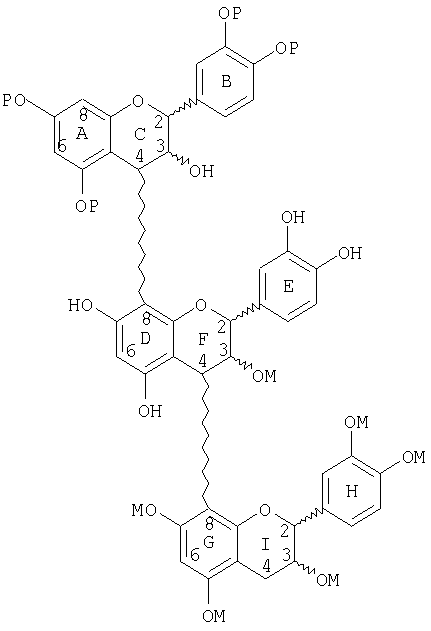
Новые соединения, которые можно получить способом по изобретению, включают новые множественно разветвленные, предпочтительно дважды разветвленные процианидиновые олигомеры, представленные следующей структурой:
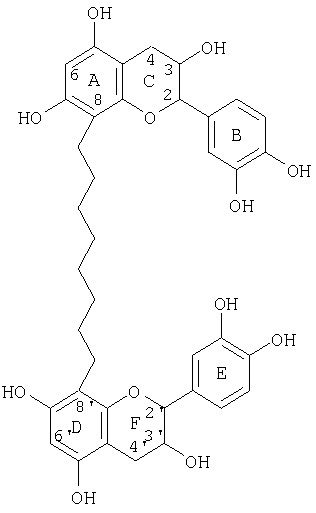
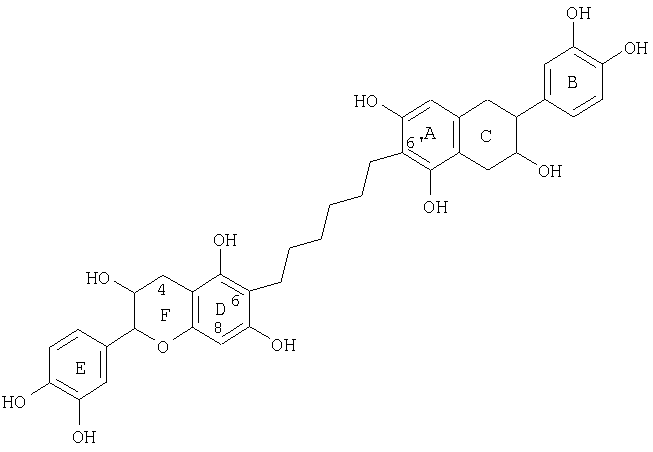
Другие соединения, которые можно получить, включают процианидиновые олигомеры, включающие связи (8↔8), (6↔8) и(6↔6), где типичные структуры показаны ниже.
Процианидиновый олигомер со связью (8↔8)
Процианидиновый олигомер со связью (6↔6)
Процианидиновый олигомер со связью (8↔6)
Стадии, схематично представленные в разделе «Блокирующие группы», можно увеличить для получения процианидиновых олигомеров, включающих межфлавановые связи (6↔6), (6↔8), (8↔8). Данные соединения могут быть получены из промежуточных 8-бром- или 6-бром-мономеров. Сочетание этих соединений с арилбороновой кислотой или арилстаннанами, полученными из этих же галогенированных промежуточных соединений реакциями сочетания Сузуки или Стилле, приводит к желаемым олигомерным связям (Suzuki A., Pure Appl. Chem. 57, 11749-11758 (1985), Stille J.K., Agnew, Chem. Internal. Ed. Engl., 25, 508-524 (1986)).
Применение процианидиновых олигомеров
Олигомеры имеют такое же применения и могут быть включены в состав лекарственных форм, очищены и вводиться таким же образом, как описано в патенте США 5554645 от 10 сентября 1996 г., Romanczyk et al. и патенте США 5712305 от 27 января 1998 г., Romanczyk et al. Данные применения включают применение в качестве антибластомных агентов, противораковых агентов, противоопухолевых агентов, антиоксидантов, ингибиторов ДНК-топоизомеразы, модуляторов циклооксигеназы и липоксигеназы, модуляторов окиси азота или синтазы окиси азота, нестероидных противовоспалительных агентов, модуляторов апоптоза, модуляторов агрегации тромбоцитов, модуляторов крови или глюкозы in vivo, антимикробных препаратов и ингибиторов повреждения ДНК в результате окисления.
ПРИМЕРЫ
В следующих примерах (+)-катехин и (-)-эпикатехин представляют примеры процианидиновых мономеров, использованных для демонстрации способов по изобретению, и при этом не подразумевается ограничение изобретения. Данные мономеры могут быть получены из промышленных источников или выделены и очищены из естественных источников, из таких как семена Theobroma cacao, родственные виды, род Herrania и их меж- и внутриродовые гибриды. Если не указано иначе, чистота соединений, полученных в примерах, была ˜85% или выше.
Пример 1
Получение тетра-O-бензил-(+)-катехина
К раствору (+)-катехина (580 мг, 2 ммоль) в ДМА (15 мл) добавляли бензилбромид (960 мкл, 4 экв) и К2СО3 (1,7 г, 6 экв) и смесь перемешивали при комнатной температуре в атмосфере аргона в течение 48 ч. Затем смесь распределяли между этилацетатом и водой (по 50 мл каждого). Органический слой промывали водой (3 х 50 мл), затем 50 мл насыщенного NaCl. Удаление растворителя давало вязкий остаток, из которого выделяли указанное в заголовке соединение кристаллизацией из 50 мл смеси метиленхлорид: метанол (9:1, об./об.) с получением 880 мг не совсем белых кристаллов с выходом 68%. 1H ЯМР (CDCl3) δн 7,44-7,24 (20Н, м, Ar-Н), 7,0 (1Н, с, Н-2'), 6,94 (2Н, с, Н-5', Н-6'), 6,25, 6,19 (2х1Н, 2×д, J=2,0 Гц, Н-6, Н-8), 5,16 (4Н, с, CH2Ph), 5,0, 4,97 (2×2Н, 2×с, CH2Ph), 4,61 (1Н, д, J=8,2 Гц, Н-2), 3,98 (1Н, м, Н-3), 3,10 (1Н, дд, J=16,5 Гц, Н-4α), 2,63 (1Н, дд, J=8,9, 16,5 Гц, Н-4β).
Пример 2
Получение тетра-O-бензил-(-)-эпикатехина
Указанное в заголовке соединение получали таким же способом, как в примере 1, за исключением того, что использовали (-)-эпикатехин вместо (+)-катехина, с получением 893 мг не совсем белых кристаллов с выходом 69%. 1H ЯМР (CDCl3) δн 7,43-7,30 (20Н, м, Ar-Н) 7,13 (1Н, с, Н-2'), 6,96 (2Н, с, Н-5', Н-6'), 6,26 (2Н, м, Н-6, Н-8), 5,18, 5,16 (2×2Н, с, СН2Ph), 5,01, 4,99 (2×2Н, 2×с. CH2Ph), 4,90 (1Н, с, Н-2), 4,19 (1Н, шир.м, Н-3), 2,45 (2Н, м, Н-4), 1,64 (1Н, д, J=3,8 Гц, ОН).
Пример 3
Получение пентаацетил (-)-эпикатехина
500 мг (-)-эпикатехина (1,6 ммоль) растворяли в 5 мл охлажденного (0°С) сухого пиридина. Добавляли 2 мл уксусного ангидрида, раствор перемешивали в течение 18 ч в атмосфере аргона. Затем раствор распределяли между 50 мл этилацетата: 50 мл 1Н HCl и органический слой промывали 3×50 мл 1Н HCl, затем 50 мл воды и 50 мл насыщенного NaCl. Промытый органический слой высушивали над MgSO4, фильтровали и высушивали с получением вязкого масла, которое затвердевало при добавлении 100 мл гексана с получением 720 мг продукта с выходом 90%. 1H ЯМР (CDCl3) δн 7,36 (1Н, д, J=2 Гц, Н-2'), 7,27 (1Н, дд, J=2,0, 8,4 Гц, Н-6'), 7,20 (1Н, д, J=8,4 Гц, Н-5'), 6,67 (1Н, д, J=2,3 Гц, Н-6, или Н-8), 6,56 (1Н, д, J=2,3 Гц, Н-8 или Н-6), 5,38 (1Н, м, Н-3), 5,11 (1Н, шир.с, Н-2), 2,98 (1Н, дд, J=4,4, 17,8 Гц, Н-4), 2,87 (1Н, дд, J=2, 17,8 Гц, Н-4), 2,299, 2,297, 2,295, 2,282, 1,920 (5×3Н, 5×с, 5×СОСН3).
Пример 4
Получение пентаацетил (+)-катехина
Указанное в заголовке соединение получали таким же способом, как в примере 3, за исключением того, что использовали (+)-катехин вместо (-)-эпикатехина, с получением 720 мг с выходом 90%. 1H ЯМР (CDCl3) δн 7,16 (1Н, д, J=2 Гц, Н-2'), 7,26 (1Н, дд, J=2,0, 8,4 Гц, Н-6'), 7,19 (1Н, д, J=8,4 Гц, Н-5'), 6,66 (1Н, д, J=2,3 Hz, Н-6, или Н-8), 6,59 (1Н, д, J=2,3 Гц, Н-8 или Н-6), 5,25 (1Н, м, Н-3), 5,15 (1Н, д, J=6, 1 Гц, Н-2), 2,87 (1Н, дд, J=5,1, 16,8 Гц, Н-4), 2,63 (1Н, дд, J=6,4, 16,8 Гц, Н-4), 2,290, 2,286, 2,279, 2,051, 2,006 (5×3H, 5×c, 5×СОСН3).
Пример 5
Получение тетра-O-п-метоксибензил-3-ацетил-(-)-эпикатехина
К смеси пентаацетил (-)-эпикатехина (50 мг, 0,2 ммоль), п-метоксибензилхлорида (69 мг, 4,4 экв), 60% NaH в минеральном масле (10 мг, 4 экв) и ДМФА (5 мл) добавляли по каплям воду (20 мкл, 4 экв) при 0°С в течение 5 мин. После перемешивания в атмосфере аргона в течение 12 ч при комнатной температуре реакционную смесь разбавляли этилацетатом (30 мл) и промывали водой (50 мл) и 30 мл насыщенного NaCl. Органический слой высушивали над MgSO4 и растворитель отгоняли с получением бледно-желтого масла, из которого выделяли указанное в заголовке соединение хроматографией на силикагеле в виде белого твердого вещества, кристаллизовали из смеси гексан: метиленхлорид (1:1, об./об.); выход (50 мг, 70%). 1H ЯМР (CDCl3) δн 7,35-7,29 (9Н, м, Ar-Н), 6,92-6,85 (10Н, м, Ar-Н), 6,26 (2Н, шир.с, Н-6, Н-8)), 5,38 (1Н, м, Н-3), 5,06-6,85 (8Н, м, 4×СН2), 4,92 (1Н, с, Н-2), 3,80 (12Н, перекрывающиеся синглеты, 4×ОСН3), 2,93 (2Н, м, Н-4), 1,85 (3Н, с, ОСОСН3).
Пример 6
Получение 4β-ацетокси тетра-O-бензил-(+)-катехина
Тетра-O-бензил-(+)-катехин (300 мг, 0,46 ммоль) и тетра-ацетат свинца (304 мг, 1,5 экв) объединяли в круглодонной колбе и высушивали в вакууме в течение 30 мин. Вводили аргон с последующим добавлением бензола и ледяной уксусной кислоты (по 5 мл каждого). Первоначальный желтый цвет исчезал при добавлении уксусной кислоты. Раствор перемешивали в течение 24 ч при комнатной температуре и переносили в делительную воронку. Смесь промывали охлажденным 1Н NaOH (4×50 мл), затем водой (50 мл) и наконец насыщенным NaCl (50 мл). Органический слой высушивали над Na2SO4 с последующим удалением растворителя с получением коричневатого остатка, из которого выделяли указанное в заголовке соединение хроматографией на силикагеле элюированием смесью гексан: этилацетат (7:3, об./об.). Элюат упаривали с получением 210 мг, 66% указанного в заголовке соединения. 1Н ЯМР (CDCl3) δн 7,44-7,28 (20Н, м, Ar-Н), 7,08 (1Н, с, Н-2'), 7,01, 6,95 (2Н, ABq, J=8,3 Гц, Н-5', Н-6'), 6,41 (1Н, д, J=3,6 Гц, Н-4), 6,23, 6,15 (2×1Н, 2×д, J=2,1 Гц, Н-6, Н-8), 5,16 (4Н, с, СН2Ph), 5,05, 4,97 (2×2Н, 2×с, CH2Ph), 4,83 (1Н, д, J=10,3 Гц, Н-2), 4,13 (1Н, дд, J=3,6, 10,3 Гц, Н-3), 2,23 (1Н, шир.с, ОН), 2,07 (3Н, с, СОСН3).
Пример 7
Получение 4β-ацетокси тетра-O-бензил-(-)-эпикатехина
Указанное в заголовке соединение получали таким же способом, как в примере 6, за исключением того, что использовали 1,01 г тетра-O-бензил-(-)-эпикатехина (1,55 ммоль) вместо тетра-O-бензил-(+)-катехина, с получением 62 мг, 59% продукта. 1H ЯМР (CDCl3) δн 7,44-7,24 (20Н, м, Ar-Н), 7,12 (1Н, с, Н-2'), 6,98, 6,95 (2Н, ABq, J=8,3 Гц, Н-5', Н-6'), 6,25 (2Н, с, Н-6, Н-8), 5,16 (4Н, с, CH2Ph), 6,10 (1Н, д, J=2,5 Гц, Н-4), 5,17, 5,16, 5,03 (4×2Н, 4×с CH2Ph), 4,97 (1Н, с, Н-2), 3,95 (1Н, м, Н-3), 2,0 (3Н, с, СОСН3).
Пример 8
Получение 4β-гидроксил тетра-O-бензил-(+)-катехина
К раствору 4-ацетокси тетра-O-бензил-(+)-катехина (692 мг, 1 ммоль) в ТГФ (9 мл) и метаноле (1 мл) добавляли порошкообразный КОН (168 мг, 3 экв) и раствор перемешивали при комнатной температуре в течение 2 ч. Добавляли насыщенный NH4Cl (25 мл) и раствор экстрагировали 2×25 мл этилацетата. Органический слой высушивали над Na2SO4 и растворитель отгоняли с получением не совсем белого твердого вещества (650 мг, количественно). 1H ЯМР (CDCl,) δн 7,45-7,28 (20Н, м, Ar-Н), 7,08 (1Н, с, Н-2'), 6,99, 6,95 (2Н, ABq, J=8,3 Гц, Н-5', Н-6'), 6,26, 6,15 (2×1Н, 2×д, J=2,1 Гц, Н-6, Н-8), 5,16 (4Н, с, CH2Ph), 5,06 (3Н, м, Н-4, CH2Ph), 4,97 (2Н, с, СН2Ph), 4,85 (1Н, д, J=9,9 Гц, Н-2), 3,95 (1Н, м, H-3), 2,75 (1Н, шир.с, ОН), 2,55 (1Н, шир.с, ОН).
Пример 9
Получение 4β-гидроксил тетра-O-бензил-(-)-эпикатехина
Указанное в заголовке соединение получали таким же способом, как в примере 8, за исключением того, что использовали 4β-ацетокси тетра-O-бензил-(-)-эпикатехин вместо 4β-ацетокси тетра-O-бензил-(+)-катехина, с получением 287 мг, 86% продукта. 1H ЯМР (CDCl3) δн 7,45-7,31 (20Н, м, Ar-Н), 7,16 (1Н, с, Н-2'), 6,99, 6,95 (2Н, ABq, J=8,3 Гц, Н-5', Н-6'), 6,29, 6,27 (2×1Н, 2×д, J=2,1 Гц, Н-6, Н-8), 5,18 (4Н, с, CH2Ph), 5,18 (4Н, с, CH2Ph), 5,07 (3Н, м, Н-4, CH2Ph), 5,01 (2Н, с, CH2Ph), 4,92 (1Н, с, Н-2), 3,98 (1Н, дд, J=2,5, 5,7 Гц, H-3), 2,43 (1Н, д, J=2,4 Гц, ОН), 1,58 (1Н, д, J=5,7 Гц, ОН).
Пример 10
Получение 4β-метокси тетра-O-бензил-(+)-катехина
К раствору 4-ацетокси тетра-O-бензил-(+)-катехина (70 мг, 0,1 ммоль) в метиленхлориде (5 мл) и метаноле (1 мл) добавляли LiBr (87 мг, 10 экв) и раствор кипятили с обратным холодильником в течение 4 ч. Затем смесь распределяли между метиленхлоридом и водой (по 25 мл каждого). Органический слой высушивали над Na2SO4, фильтровали и растворитель отгоняли. Остаток подвергали хроматографии на силикагеле с получением 54 мг, 80% указанного в заголовке соединения в виде бледно-желтого масла. 1H ЯМР (CDCl3) δн 7,42-7,27 (20Н, м, Ar-Н), 7,07 (1Н, д, J=1,6 Гц, Н-2'), 6,96 (2Н, м, Н-5', Н-6'), 6,26 (1Н, д, J=2,2 Гц, Н-6 или Н-8), 6,15 (1Н, д, J=2,2 Гц, Н-8, Н-6), 5,15 (4Н, с, CH2Ph), 5,02 (2Н, ABq, J=7,8 Гц, CH2Ph), 4,97 (2H, с, CH2Ph), 4,92 (1Н, д, J=10,4 Гц, Н-2), 4,72 (1Н, д, J=3,5 Гц, Н-4), 3,85 (1Н, дт, J=3,5, 9,2 Гц, Н-3), 3,47 (3Н, с, ОСН3).
Пример 11
Получение тетра-O-бензил-(+)-катехин-(4α→8)-(-)-эпикатехина 4β-ацетокси тетра-О-бензил-(+)-катехин (пример 6) (140 мг, 0,2 ммоль), (-)-эпикатехин (290 мг, 5 экв) и LiBr (87 мг, 5 экв) растворяли в смеси ТГФ и метиленхлорида (по 5 мл каждого) и раствор кипятили с обратным холодильником в течение 24 ч, после чего раствор распределяли между этилацетатом и водой (по 40 мл каждого). Органический слой высушивали над Na2SO4 с последующей отгонкой растворителя. Остаток ресуспендировали в этилацетате и фильтровали для удаления большей части (-)-эпикатехина. Фильтрат упаривали и подвергали хроматографии на силикагеле, где элюат из смеси метиленхлорид: этилацетат (1:1, об./об.) давал 116 мг, 62% димера в виде не совсем белого порошка. В ЯМР-спектре Hs, входящие в состав верхнего мономера димера, обозначены А, и Hs, входящие в состав нижнего мономера димера, обозначены В. 1Н ЯМR (CDCl3:d4 - метанол, 9:1) δн 7,36-7,23 (20Н, м, Ar-Н), 7,02-6,74 (5Н, м, А-5', А-6', А-2', В-2', В-5'), 6,35 (1Н, дд, J=1,7, 8,2 Гц, В-6'), 6,18-6,16 (2H, ABq, J=2,2 Гц, А-6, А-8), 5,86 (1Н, с, В-6), 5,12 (5Н, м, CH2Ph, В-2), 4,90 (2H, с, CH2Ph), 4,71 (1Н, д, J=8,2 Гц, А-2), 4,59 (1Н, д, J=10 Гц, СН2Ph), 4,47 (1Н, д, J=10 Гц, CH2Ph), 4,29 (1Н, дд, J=8,3, 8,3 Гц, А-4), 3,80 (1Н, м, Н-3), 2,71 (1Н, д, J=16,6 Гц, В-4), 2,53 (1Н, дд, J=4,4, 16,6 Гц, В-4); 13С ЯМР δ 156,5, 156,0, 154,6, 154,0, 152,5, 151,6, 151,4, 151,2, 150,6, 147,0, 146,7, 141,7, 141,6, 141,5, 139,8, 134,8, 134,4, 134,2, 133,9, 129,3, 128,7, 126,2, 126,1, 126,0, 125,8, 125,5, 125,4, 125,2, 125,1, 125,0, 124,9, 119,8, 115,0, 112,6, 111,2, 106,1, 104,5, 96,5, 94,9, 93,0, 92,8, 79,9, 70,5, 69,1, 69,0, 67,8, 67,7, 63,9, 35,0, 25,5; ИК-спектр (KBr, см-1) 3418, 3057, 3034, 2918, 1609, 1510, 1446, 1371, 1260, 1202, 1097, 806, 731, 696; Масс-спектр (бомбардировка быстрыми атомами, m/z) 939,6 (M+H)+ 649,1, 607,0, 559,0, 459,8.
Пример 12
Получение (+)-катехин (4α→8)-(-)-эпикатехина
Тетра-O-бензил-(+)-катехин-(4α→8)-(-)-эпикатехин, полученный в примере 11 (50 мг), растворяли в метаноле (10 мл) и дегазировали пропусканием аргона в течение 10 мин. Добавляли палладий на угле (30 мг) и проводили гидрогенолиз под давлением 45 фунтов/кв. дюйм в течение 3 ч. Раствор фильтровали через целит, который промывали метанолом. Объединенные фильтрат и промывные фракции упаривали и остаток растворяли в воде, затем лиофилизировали с получением количественного выхода димера в виде не совсем белого твердого вещества. В ЯМР-спектре Hs, входящие в состав верхнего мономера димера, обозначены А, и Hs, входящие в состав нижнего мономера димера, обозначены В. 1H ЯМР (CDCl3:d4-метанол, 9:1) δн 7,21 (1Н, шир.с, А-2'), 7,04 (1Н, шир.с, В-2'), 6,95-6,75 (2Н, м, А-5', В-5'), 6,62 (1Н, м, А-6'), 6,45 (1Н, м, В-6'), 6,20 (1Н, м, В-6), 6,05 (1Н, м, В-6), 5,89 (2Н, м, А-6, А-8), 4,98 (1Н, м, В-2, ), 4,85 (1Н, м, В-2), 4,42-4,25 (3Н, м, А-4, А-3, А-2), 3,05-2,62 (2Н, м, В-4).
Пример 13
Получение 3-ацетил-тетра-O-бензил-(+)-катехин-(4α→8)-пентаацетил-(-)эпикатехина
Тетра-O-бензил-(+)-катехин-(4α→8)-эпикатехин, полученный в примере 11, ацетилировали уксусным ангидридом в пиридине. 120 мг тетра-O-бензил-(+)-катехин (4α→8)-(-)-эпикатехина растворяли в 2 мл сухого пиридина и добавляли 500 мкл уксусного ангидрида. Реакционную смесь перемешивали в атмосфере аргона в течение 18 ч при комнатной температуре. Смесь распределяли между этилацетатом и 1Н HCl (по 25 мл каждого). Органический слой промывали 25 мл воды, затем 25 мл 5% бикарбоната натрия, затем 25 мл насыщенного раствора NaCl и органическую фазу высушивали над Na2SO4. Продукт очищали хроматографией с получением 140 мг указанного в заголовке соединения с выходом 91%. В ЯМР-спектре Hs, входящие в состав верхнего мономера димера, обозначены А, и Hs, входящие в состав нижнего мономера димера, обозначены В. 1Н ЯМР (CDCl3:d4-метанол, 9:1) δн 7,40-7,29 (20Н, м, Ar-Н), 7,19-7,13 (5Н, м, А-2', А-6', В-2', В-5', В-6'), 6,92 (1Н, д, J=8,3 Гц, А-5'), 6,52 (1Н, с, В-6), 6,21, 6,18 (2×1Н, 2×д, J=2,3 Гц, А-6, А-8), 5,67 (1Н, т, J=9,6, Гц, Н-3), 5,14 (3Н, с, CH2Ph, В-3), 5,0 (2Н, с, CH2Ph), 4,98 (2Н, с, CH2Ph,) 4,84 (1H, д, J=9,1 Гц, А-3), 4,75 (1Н, Д, J=10,1 Гц, А-2), 4,58, 4,41 (2Н, ABq, J=9,2 Гц, CH2Ph), 2,64 (2H, м, В-4), 2,29 (6Н, с, СОСН3), 2,26 (3Н, с, СОСН3), 1,76 (3Н, с, СОСН3), 1,74 (3Н, С, СОСН3), 1,64 (3Н, с, СОСН3); 13С ЯМR 169,6, 168,2, 168,0, 167,5, 158,0, 156,2, 153,2, 149,2, 148,7, 147,1, 146,2, 142,5, 142,0, 137,0, 136,8, 136,5, 136,2, 130,0, 129,8, 128,3, 128,2, 127,7, 127,6, 127,3, 127,1, 122,8, 121,6, 121,4, 114,9, 114,8, 110,2, 108,4, 106,0, 95,0, 94,6, 80,0, 75,5, 73,4, 71,4, 71,0, 70,5, 69,7, 66,5, 35,0, 26,2, 20,6, 20,5, 20,4, 20,3, 20,0; Масс-спектр (бомбардировка быстрыми атомами, m/z) 1192 (M+H)*, 1131, 1039, 949, 841, 691.
Пример 14
Получение 3-ацетил-(+)-катехин- (4α→8)-пентаацетил--(-)-эпикатехина
3-Ацетил-тетра-О-бензил-(+)-катехин (4α→8)-пентаацетил-(-)-эпикатехин, полученный в примере 13, растворяли в дегазированной смеси этилацетат-метанол (по 3 мл каждого) и гидрировали в присутствии 30% палладия на угле под давлением 45 фунтов/кв. дюйм в течение 4 ч. Раствор фильтровали через целит и растворитель отгоняли с получением количественного выхода димера в виде бледно-желтого твердого вещества. В ЯМР-спектре Hs, входящие в состав верхнего мономера димера, обозначены А, и Hs, входящие в состав нижнего мономера димера, обозначены В. 1Н ЯМР (CDCl3:d4-метанол, 9:1) δн 7,47, 6,98 (2×1Н, шир.с, В-2'), 7,36, 6,98 (2×1Н, 2×д, J=8,4, B-6'), 7,24, 7,16 (2×1Н, 2×д, J=8,4 Гц, В-5'), 6,89, 6,60 (2×1Н, 2×шир.с, А-2' ), 6,83, 6,79 (2×1H, ABq, J=8,4 Гц, A-5', А-6'), 6,66 (1H, д, J=8,4 Гц, A-5'), 6,47 (1H, д, J=8,4 Гц, А-6'), 6,51, 6,45 (2×1H, 2×c, B-6), 5,97, 5,84 (2×1H, 2×д, J=2 Гц, А-6, А-8), 5,96 (2Н, с, А-6, А-8), 5,71 (2Н, м, А-3), 5,50, 5,17 (2×1H, 2×шир.с, В-3), 5,28 (2×1H, 2×шир.с, В-2), 5,0, 4,54 (2×1H, 2×д, J=8,9, 9,4 Гц, А-2), 4,74, 4,61 (2×1H, 2×д, J=9, 9 Гц, А-4), 3,2, 2,74 (2×2Н, 2×м, В-4), 2,32, 2,29, 2,28, 2,26, 2,23, 2,01, 1,80, 1,77, 1,63 (с, СОСН3); 13С ЯМР 172,5, 172,0, 171,8, 171,5, 170,3, 170,2, 170,1, 170,0, 169,8, 169,2, 157,2, 157,1, 156,8, 154,8, 154,0, 149,2, 147,5, 146,0, 145,9, 145,6, 145,0, 143,0, 142,8, 137,5, 137,0, 129,8, 125,8, 124,9, 124,0, 123,0, 122,6, 121,8, 120,8, 120,2, 119,8, 115,9, 115,8, 115,5, 115,1, 110,8, 110,9, 109,5, 109,4, 105,0, 104,2, 98,0, 97,5, 96,8, 96,0, 81,0, 80,2, 79,0, 78,8, 78,6, 78,0, 75,0, 72,2, 68,1, 68,0, 37,5, 36,0, 26,5, 26,0, 20,7.
Пример 15
Получение тетра-O-бензил-(-)-эпикатехин-(4β→8)--(-)-эпикатехина
4β-Ацетокси тетра-O-бензил-(-)-эпикатехин, полученный в примере 7 (70 мг, 0,1 ммоль), (-)-эпикатехин (145 мг, 5 экв) и LiBr (44 мг, 5 экв) растворяли в смеси ТГФ и метиленхлорида (по 3 мл каждого) и раствор кипятили с обратным холодильником в течение 24 ч в атмосфере аргона. Раствор распределяли между этилацетатом и водой (по 25 мл каждого) и органический слой высушивали над Na2SO4. Растворитель отгоняли и остаток ресуспендировали в 25 мл этилацетата с последующим фильтрованием для удаления большей части непрореагировавшего (-)-эпикатехина. Фильтрат упаривали до остатка и указанное в заголовке соединение выделяли хроматографией на силикагеле элюированием смесью метиленхлорид: этилацетат (1:1, об./об.). Упаривание элюата давало 56 мг (60%) не совсем белого порошка. 1H ЯМР (CDCl3: d4-метанол, 9:1) δн [No assignment] 7,35-7,14 (20H, м), 6,92 (1H, шир.с), 6,82 (1Н, с), 6,29 (1Н, с) 6,18 (1Н, с), 5,85 (1Н, с), 5,01 (4Н, с), 4,94 (2Н, с), 4,93 (2Н, с), 4,38 (1Н, с), 3,93 (1Н, с), 2,85 (2Н, с).
Пример 16
Получение (-)-эпикатехин-(4β→8)-(-)эпикатехина
Тетра-O-(-)-бензил-эпикатехин(4β→8)-(-)-эпикатехин, полученный в примере 15 (40 мг, 0,043 ммоль), растворяли в 8 мл метанола и дегазировали пропусканием аргона в течение 10 мин. К раствору добавляли 25 мг 30% палладия на угле и смесь гидрогенолизовали под давлением 45 фунтов/кв. дюйм в течение 3 ч. Раствор фильтровали через целит с последующим промыванием 25 мл метанола. Объединенные фильтрат и промывную фракцию упаривали и остаток растворяли в воде. Лиофилизация давала 23 мг не совсем белого порошка. Анализ ВЭЖХ (фиг.1А) показывал наличие 18% мономера, 45% димера, 25% тримера и 8% тетрамера. 1H ЯМР-спектр представлен на фиг.2.
Пример 17
Получение тетра-O-бензил-(+)-катехин-(4α→8)-катехина
4β-Ацетокси тетра-O-бензил-(+)-катехин, полученный в примере 6 (70 мг, 0,1 ммоль), (+)-катехин (145 мг, 5 экв) и LiBr (44 мг, 5 экв) растворяли в смеси ТГФ и метиленхлорида (по 3 мл каждого) и раствор кипятили в течение 24 ч в атмосфере аргона. Раствор распределяли между этилацетатом и водой (по 25 мл каждого) и органическую фазу высушивали над Na2SO4. После выпаривания остаток ресуспендировали в этилацетате (25 мл) и фильтровали для удаления большей части непрореагировавшего (+)-катехина. После выпаривания остаток подвергали хроматографии на силикагеле, где элюирование смесью метиленхлорид: этилацетат (1:1, об./об.) давало не совсем белый порошок (81 мг, 68%) после выпаривания. 1H ЯМР (CDCl3:d4-метанол, 9:1) δн 7,39-7,06 (20Н, м), 6,84-6,68 (5Н, м), 6,47 (1Н, д, J=7,9 Гц), 6,32-5,98 (4Н, м), 5,00-4,33 (11Н, м), 3,58 (1Н, м), 2,98 (1Н, м), 2,35 (1Н, м); ИК-спектр (KBr, см-1) 3441, 3057, 3034, 2918, 1609, 1542, 1510, 1371, 1266, 1097, 812, 737, 696; Масс-спектр (химическая ионизация при атмосферном давлении, m/z) 938 (М-Н), 920, 848, 816, 696, 607, 558.
Пример 18
Получение 3-ацетил-тетра-O-бензил-(+)-катехин-(4α→8)--3-ацетил тетра-O-бензил-(-)-эпикатехина
3-Ацетил тетра-O-бензил-(+)-катехин-(4α→8)-пентаацетил-(-)-эпикатехин, полученный в примере 13 (119 мг, 0,1 ммоль), добавляли к сухому ДМФА (4 мл) при 0°С, затем добавляли NaH (29 мг, 4,2 экв), затем бензилбромид (54 мкл, 4,5 экв). Медленно в течение 2 мин добавляли воду (8 мкл, 4 экв) и смесь перемешивали в течение 24 ч при комнатной температуре. Раствор распределяли между этилацетатом и водой (по 25 мл каждого) и органическую фазу высушивали над MgSO4. После выпаривания остаток подвергали хроматографии на силикагеле, где элюирование смесью 20% этилацетат/гексан давало 105 мг (90%) указанного в заголовке соединения после выпаривания. В ЯМР-спектре Hs, входящие в состав верхнего мономера димера, обозначены А, и Hs, входящие в состав нижнего мономера димера, обозначены В. 1H ЯМР (CDCl3: d4метанол, 9:1) δн 7,45-7,24 (40Н, м, Ar-Н), 6,90-6,78 (6Н, м, А-2', А-5', А-6', В-2', В-5', В-6'), 6,22 (2Н, м, А-6, А-8), 6,21 (1Н, с, В-6), 5,92 (1Н, м, А-3), 5,21-4,40 (20Н, комплекс, CH2Ph, А-2, А-4, В-2, В-3), 2,70 (2Н, м, В-4), 1,67 (6Н, с, CH2Ph).
Пример 19
Получение тетра-O-бензил-(+)-катехин-(4α→8)-(+)-тетра-O-бензил-(-)-эпикатехина
К раствору 3-ацетил тетра-O-бензил-(+)-катехин-(4α→8)-3-ацетил тетра-O-бензил-(-)-эпикатехина, полученного в примере 18 (40 мг, 0,03 ммоль), в ТГФ (2 мл) и метаноле (200 мкл) добавляли порошкообразный КОН (5 мг, 3 экв) и раствор перемешивали при комнатной температуре в течение 18 ч в атмосфере аргона. Реакционную смесь распределяли между этилацетатом и насыщенным NH4Cl (по 25 мл каждого). Органический слой высушивали над MgSO4 и растворитель отгоняли. Остаток подвергали хроматографии на силикагеле, где элюирование смесью 20% этилацетат/метиленхлорид давало указанное в заголовке соединение (31 мг, 82/5%) в виде бесцветного масла после отгонки растворителя. В ЯМР-спектре Hs, входящие в состав верхнего мономера димера, обозначены А, и Hs, входящие в состав нижнего мономера димера, обозначены В. 1H ЯМР (CDCl3:d4-метанол, 9:1) δн 7,41-7,13 (40Н, м, Ar-Н), 6,97-6,79 (6Н, комплекс, А-2', А-5', А-6', В-2', В-5', В-6'), 6,22 (1Н, с, В-6), 6,20, 6,12 (2×1Н, 2×д, J=2,4 Гц, А-6, А-8), 5,18-4,51 (19Н, СН2Pb, А-2, А-4, В-2), 4,28 (1Н, м, А-3), 3,85 (1Н, м, В-3), 2,95 (1Н, д, J=16 Гц, В-4), 2,60 (1Н, дд, J=5, 16 Гц, В-4).
Пример 20
Получение тетра-O-бензил-(+)-катехин-(4α→8)-3-ацетил-(+)-катехин-(4α→8)-пентаацетил(-)-эпикатехина
К раствору 3-ацетил-(+)-катехин-(4α→8)-пентаацетил-(-)-эпикатехина, полученного в примере 14 (100 мг, 0,068 ммоль), и 4β-ацетокси тетра-O-бензил-(+)-катехина, полученного в примере 6 (334 мг, 2 экв) в ТГФ (7 мл) и метиленхлориде (7 мл) добавляли 161 мг LiI. Раствор кипятили с обратным холодильником в течение 24 ч с последующим распределением между этилацетатом и водой (по 25 мл каждого). Органическую фазу высушивали над Na2SO4, фильтровали и растворитель отгоняли. Остаток подвергали хроматографии на силикагеле, где элюирование смесью этилацетат:метиленхлорид (1:1, об./об.) давало коричневато-белое твердое вещество (100 мг, 28%) после отгонки растворителя. Полученный 1Н ЯМР-спектр представлен на фиг.3. Масс-спектр (бомбардировка быстрыми атомами, m/z) 1482 (М+Н)+, 1148, 1042, 962, 920, 650.
Пример 21
Получение тетра-O-бензил-(+)-катехин-(4α→8)-пентаацетил-(+)-катехин-(4α→8)-пентаацетил(-)-эпикатехина
Тетра-O-бензил-(+)-катехин-(4α→8)-3-ацетил-(+)-катехин-(4α→8)-пентаацетил(-)-эпикатехин, полученный в примере 20 (100 мг, 0,068 ммоль), перемешивали в сухом пиридине (2 мл) и уксусном ангидриде (1 мл) в атмосфере аргона в течение 24 ч. Затем раствор распределяли между 1Н HCl и этилацетатом (по 25 мл каждого), органический слой промывали 5% NaHCO3, насыщенным NaCl и высушивали над MgSO4. Отгонка растворителя давала маслянистый остаток, который подвергали хроматографии на силикагеле, где элюирование смесью 10% этилацетат/метиленхлорид давало белый порошок (70 мг, 61%) после отгонки растворителя. Полученный 1Н ЯМР-спектр представлен на фиг.4.
Пример 22
Получение 3-ацетил-(+)-катехин-(4α→8)-пентаацетил-(+)-катехин-(4α→8)-пентаацетил(-)-эпикатехина
Тетра-O-бензил-(+)-катехин-(4α→8)-пентаацетил-(+)-катехин-(4α→8)-пентаацетил(-)-эпикатехин, полученный в примере 21 (50 мг), растворяли в дегазированной смеси этилацетат-метанол (по 3 мл каждого) и гидрогенолизировали в течение 4 ч в присутствии 30% палладия на угле (30 мг) под давлением 45 фунтов/кв. дюйм. Удаление катализатора фильтрованием через целит и выпаривание давали указанное в заголовке соединение в виде бледно-коричневого порошка (35 мг, 91%). Полученный 1H ЯМР-спектр представлен на фиг.5.
Пример 23
Получение тетра-O-бензил-(+)-катехин-(4α→8)-3-ацетил-(+)-катехин-(4α→8)-пентаацетил(+)-катехин-(4α→8)-пентаацетил(-)-эпикатехина
К раствору 3-ацетил-(+)-катехин-(4α→8)-пентаацетил-(+)-катехин-(4α→8)-пентаацетил(-)-эпикатехина, полученного в примере 22 (30 мг, 0,0226 ммоль), и 4β-ацетокси тетра-O-бензил-(+)-катехина, полученного в примере 6 (31 мг, 2 экв), в ТГФ и метиленхлориде (по 2 мл каждого) добавляли LiI (16 мг, 5 экв) и раствор кипятили с обратным холодильником в течение 24 ч. Раствор распределяли между этилацетатом и водой (по 25 мл каждого) и органический слой высушивали над MgSO4, фильтровали и растворитель отгоняли. Остаток подвергали хроматографии на силикагеле, где элюирование смесью 10% метанол/метиленхлорид давало коричневато-белое твердое вещество (20 мг, 45%) после отгонки растворителя. Масс-спектр (бомбардировка быстрыми атомами, m/z) 1978 (М+Н)+, 1934 (M+-СОСН3), 1571 (М+-СОСН3, -3×СН2Ph), 1646, 1430, 1373, 1330, 1269.
Пример 24
Получение тетра-O-бензил-(+)-катехин-(4α→8)-(-)эпикатехин-(6→4α)-тетра-O-бензил-(+)-катехина
К раствору тетра-O-бензил-(+)-катехин-(4α→8)-(-)-эпикатехина, полученного в примере 11 (69 мг, 0,074 ммоль), и 4β-ацетокси тетра-O-бензил-(+)-катехина, полученного в примере 6 (51 мг, 0,074 ммоль), в смеси метиленхлорида и ТГФ (по 5 мл каждого) добавляли LiBr (65 мг, 10 экв) и смесь кипятили с обратным холодильником в течение 24 ч. Раствор распределяли между этилацетатом и водой (по 25 мл каждого) и органический слой высушивали над MgSO4. Растворитель отгоняли и остаток подвергали хроматографии на силикагеле, где элюирование смесью этил-ацетат: метиленхлорид (1:1, об./об.) давало белый порошок (35 мг, 30%) после отгонки растворителя. Полученный 1H ЯМР-спектр представлен на фиг.6. Масс-спектр (бомбардировка быстрыми атомами, m/z) 1588 (М + Н)+, 1255, 772, 648, 607, 560.
Пример 25
Получение 8-бром тетра-O-бензил-(-)-эпикатехина
К раствору тетра-O-бензил-(-)-эпикатехина (пример 2) (65 мг, 0,1 ммоль) в метиленхлориде (2 мл) добавляли N-бромсукцинимид (18 мг, 0,1 ммоль) и раствор перемешивали в атмосфере аргона в течение 10 мин. Смесь фильтровали через силикагель с последующим элюированием 20 мл смеси этилацетат: метиленхлорид (1:1, об./об.). Объединенный фильтрат и элюат упаривали. Остаток подвергали хроматографии на силикагеле, где элюирование метиленхлоридом давало указанное в заголовке соединение в виде блестящих розовато-белых кристаллов (66 мг, 90%) после отгонки растворителя. 1H ЯМР (CDCl3) δн, 7,45-7,21 (21 Н, м, Ar-Н), 7,01 (1Н, дд, J=1,4 8,3 Гц, Н-6'), 6,96 (1Н, д, J=8,3 Гц, Н-5'), 6,23 (2Н, с, Н-6), 5,38 (1Н, м, Н-3), 5,21, 5,18, 5,10, 4,97 (4×2H, 4×c, 4×CH2), 5,01 (1Н, с, 20 Н-2), 4,3 (1Н, м, Н-3), 3,03 (1Н, дд, J=1,9, 17,4 Гц, Н-4), 2,89 (1Н, дд, J=4, 17,4 Гц, Н-4), 1,55 (1Н, д, J=4,8 Гц, ОН).
Пример 26
Получение 8-бром пента-O-бензил-(-)-эпикатехина
К раствору пента-O-бензил-(-)-эпикатехина (55 мг, 0,074 ммоль) в метиленхлориде (2 мл) при 0°С добавляли N-бромсукцинимид (14 мг, 1 экв) и раствор перемешивали при комнатной температуре в течение 30 мин. Раствор пропускали через колонку диаметром 25 мм с силикагелем (7 г), которую элюировали метиленхлоридом (30 мл). Объединенные фильтрат и элюат упаривали с получением указанного в заголовке соединения в виде белой пены (50 мг, 82,5%) после отгонки растворителя. 1H ЯМР (CDCl3) δн, 7,43-6,90 (28Н, м, Ar-Н), 6,21 (1Н, с, Н-6), 5,17 (2Н, с, CH2), 5,09 (5Н, с, 2×СН2, Н-2), 4,96 (2Н, с, CH2), 4,37, 4,27 (2Н, АВ, J=12,6 Гц, 3-ОСН2), 3,95 (1Н, м, Н-3), 2,94 (1Н, дд, J=3,6, 17,1 Гц, Н-4), 2,78 (1Н, дд, J=4,4, 17,1 Гц, Н-4).
Пример 27
Получение 8-бром 4β-ацетокси пентабензил-(-)-эпикатехина
К смеси 8-бром пентабензил-(-)-эпикатехина (пример 26) (59 мг, 0,072 ммоль) и тетраацетата свинца (48 мг, 1,5 экв) в атмосфере аргона добавляли бензол (2 мл), затем 2 мл уксусной кислоты и смесь перемешивали в течение 60 ч при комнатной температуре. Раствор распределяли между этилацетатом и водой (по 50 мл каждого). Органический слой промывали 1Н NaOH (2×50 мл), затем водой (50 мл), насыщенным NaCl (50 мл) и высушивали над Na2SO4. Раствор фильтровали и упаривали с получением маслянистого остатка, который подвергали хроматографии на силикагеле, где элюирование смесью 20% этилацетат/гексан давало указанное в заголовке соединение в виде белой пены (38 мг, 60%) после выпаривания. 1Н ЯМР (CDCl3) δн 7,46-6,85 (28Н, м, Ar-Н), 6,25 (1Н, с, Н-6), 6,18 (1Н, д, J=2,3 Гц, Н-4), 5,20, 5,13, 5,02 (4×2Н, 4×с, 4×СН2), 4,99 (1Н, с, Н-2), 4,51, 4,33 (2Н, АВ, J=12,3 Гц, 3 -ОСН3), 3,65 (1Н, м, Н-3), 2,0 (2Н, с, ОСОСН3).
Пример 28
Определение абсолютной конфигурации 8-бром тетра-O-бензил-(-)-эпикатехина
Кристаллы 8-бром тетра-O-бензил(-)-эпикатехина помещали на стекловолокно и располагали в атмосфере охлажденного N2 при -44°С в рентгеновском дифрактометре Siemens SMART CCD. В основном кристаллы дифрагировали слабо с отражениями с несколькими или без больших углов и со слабой общей интенсивностью. Первые три кристалла не дифрагировали в достаточной мере, чтобы определить элементарную ячейку даже при времени экспозиции, превышающем обычные. Четвертый дифрагировал в достаточной степени, чтобы точно определить элементарную ячейку с использованием пятнадцати отражений. Были получены данные по данному кристаллу с использованием пятидесятисекундных экспозиций для более чем двух тысяч каркасов для того, чтобы примерно покрыть область дифракции с использованием МО-облучения.
Объем элементарной ячейки указывает на наличие двух молекул в элементарной ячейке. Несмотря на то что один угол ячейки был четко равен 90°, и другой четко отличался от 90° (92,6°), третий угол отличался от 90° на 0,1°, что является большей ошибкой, чем обычно для моноклинной ячейки. Однако исследование возможных систематических пропусков показывало на явную ось 21, соответствующую моноклинной пространственной группе P21, подходящей для хирального соединения. Последующее успешное разрешение структуры и уточнение P21 поддерживали этот выбор. Структуру разрешали прямыми методами с использованием упаковки SHELX (Sheldrick, G.M. SHELXTL Structure Determination Software Programs: Siemens Analytical X-ray Instruments Inc. Madison, WI, 1990) программ. Атомы водорода помещали в фиксированные вычисленные положения. Фенильные кольца в бензильных группах уточняли изотропически в качестве жестких групп. Не делали поправок на абсорбцию или экстинкцию. В следующей таблице перечислены данные по кристаллам.
Данные по кристаллам и уточнение структуры
Определение правильной абсолютной конфигурации проверяли вычислением параметра Флэка 'х'. Этот параметр не отличался от нуля, что указывает на определение правильной конфигурации. Тест уточнения инвертированной конфигурации приводил к значению параметра Флэка 'х', равному 0,95 (5), и значительному увеличению R-факторов, что в обоих случаях указывает на правильное определение. Конечная модель 8-бром тетра-O-бензил-(-)-эпикатехина представлена на фиг.7.
Пример 29
Получение (8↔8), (8↔6) и (6↔6) связанных процианидиновых олигомеров
Стадии, описанные в данном изобретении, могут быть увеличены с получением процианидиновых олигомеров, включающих межфлавановые связи (8↔8), (8↔6), (6↔6). Данные соединения получены из 6-бром- и/или 8-бром-(мономер)промежуточных соединений. Сочетание этих бромированных мономеров с производными органотина реакцией Стилле в присутствии катализатора палладия(0) приводит к желаемой олигомерной связи. (Stille J.K., Agnew, Chem. Internal. Ed. Engl., 25, 508-524 (1986)).
Например, 8-бром пентабензил-(-)-эпикатехин, полученный в примере 26, взаимодействует с гексабутилдистаннаном с получением алкилстаннана пентабензил(-)-эпикатехина. Сочетание данного станнана с другим 8-бром пентабензил(-)-эпикатехином в присутствии тетракис(трифенилфосфин)палладия(0) в бензоле обеспечивает декабензил (-)-эпикатехиновый димер со связью (8↔8). Снятие защиты с помощью H2/Pd обеспечивает (-)-эпикатехин-(8↔8)-(-)-эпикатехин в свободной фенольной форме.
Аналогичным образом можно синтезировать процианидиновые олигомеры, включающие (8↔6) или (6↔6) связи с использованием 6-бром- или 8-бром-(мономер)производных. Кроме того, сочетание 8-бром- или 6-бром-димеров, тримеров и высших олигомеров может обеспечить «одинаковые» по размеру процианидиновые олигомеры, включающие связи (8↔8), (8↔6) и (6↔6).
Кроме того, сочетание блокированных мономеров, использованных для получения (4→6) связанных олигомеров, описанных в изобретении, можно использовать в реакции Стилле с получением новых процианидиновых олигомеров, включающих комбинации (4→6) и (4→8) связанных олигомеров с (8↔8), (8↔6) и (6↔6) связями. В качестве примера следующая структура показывает (8↔8) и (4↔8) связанный процианидиновый триммер:
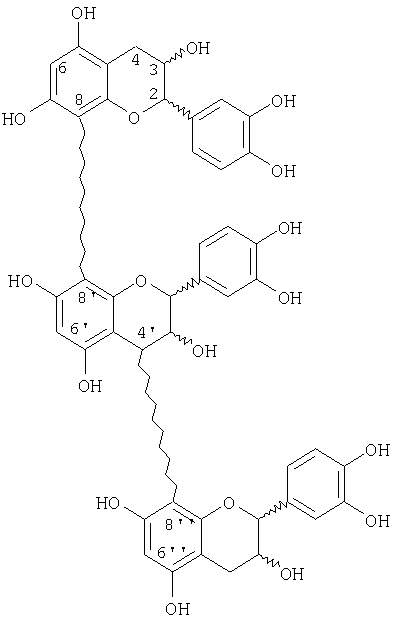
Краткое описание чертежей
Фиг.1А представляет разделение с помощью ВЭЖХ (-)-эпикатехин-(4β→8)-(-)-эпикатехина, полученного в примере 16.
Фиг.1В представляет разделение с помощью ВЭЖХ (+)-катехин-(4α→8)-(-)-эпикатехина, полученного в примере 12.
Фиг.2 представляет 1Н ЯМР-спектр (-)-эпикатехин-(4β→8)-(-)-эпикатехина, полученного в примере 16.
Фиг.3 представляет 1Н ЯМР-спектр тетра-O-бензил-(-)-катехин-(4α→8)-пентаацетил(-)-эпикатехина, полученного в примере 20.
Фиг.4 представляет 1H ЯМР-спектр тетра-O-бензил(+)-катехин-(4α→8)-петаацетил-(+)-катехин-(4α→8)-(-)-эпикатехина, полученного в примере 21.
Фиг.5 представляет 1H ЯМР-спектр тетра-O-бензил(+)-катехин-(4а→8)-пентаацетил(+)-катехин-(4α→8)-пентаацетил(-)-эпикатехина, полученного в примере 22.
Фиг.6 представляет 1Н ЯМР-спектр тетра-O-бензил(+)-катехин-(4α→8)-(-)-эпикатехин-(6→4α)тетра-O-бензил(+)-катехина, полученного в примере 24.
Фиг.7 представляет рентгеновскую модель 8-бром тетра-O-бензил(-)-эпикатехина.
| название | год | авторы | номер документа |
|---|---|---|---|
| СПОСОБЫ ПОЛУЧЕНИЯ (8↔8) ДИМЕРА КАТЕХИНА, И/ИЛИ ЭПИКАТЕХИНА, И/ИЛИ ЭПИКАТЕХИНГАЛЛАТА | 2000 |
|
RU2293081C2 |
| СИНТЕЗ 4АЛЬФА-АРИЛЭПИКАТЕХИНОВ И ПРОМЕЖУТОЧНЫХ СОЕДИНЕНИЙ | 2001 |
|
RU2281942C2 |
| СОЕДИНЕНИЕ ЭКСТРАКТА КАКАО И СПОСОБЫ ИХ ПОЛУЧЕНИЯ И ПРИМЕНЕНИЯ | 2004 |
|
RU2394562C2 |
| СПОСОБ ПОЛУЧЕНИЯ ПРОАНТОЦИАНИДИНОВОГО ОЛИГОМЕРА | 2006 |
|
RU2435579C2 |
| СНИЖЕНИЕ УРОВНЕЙ/АКТИВНОСТИ АРГИНАЗЫ | 2007 |
|
RU2469742C2 |
| УСОВЕРШЕНСТВОВАННЫЙ СПОСОБ ЭКСТРАГИРОВАНИЯ ПРОЦИАНИДИНОВ КАКАО (ВАРИАНТЫ) И ЭКСТРАКТ КАКАО (ВАРИАНТЫ) | 2001 |
|
RU2281653C2 |
| НЕПРЕРЫВНЫЙ СПОСОБ ЭКСТРАГИРОВАНИЯ ПРОЦИАНИДИНОВ КАКАО И ЭКСТРАКТ КАКАО, ПОЛУЧЕННЫЙ ЭТИМ СПОСОБОМ | 2001 |
|
RU2311041C1 |
| ЛЕЧЕНИЕ ЗАБОЛЕВАНИЙ, СВЯЗАННЫХ С ДЕФЕКТНОЙ КОММУНИКАЦИЕЙ ПОСРЕДСТВОМ ЩЕЛЕВЫХ КОНТАКТОВ | 2003 |
|
RU2351325C2 |
| КОНДЕНСИРОВАННЫЕ ГЕТЕРОЦИКЛИЧЕСКИЕ СУКЦИНИМИДНЫЕ СОЕДИНЕНИЯ | 2001 |
|
RU2298554C2 |
| ПРОДУКТЫ, СОДЕРЖАЩИЕ ПОЛИФЕНОЛЫ | 2006 |
|
RU2417711C2 |
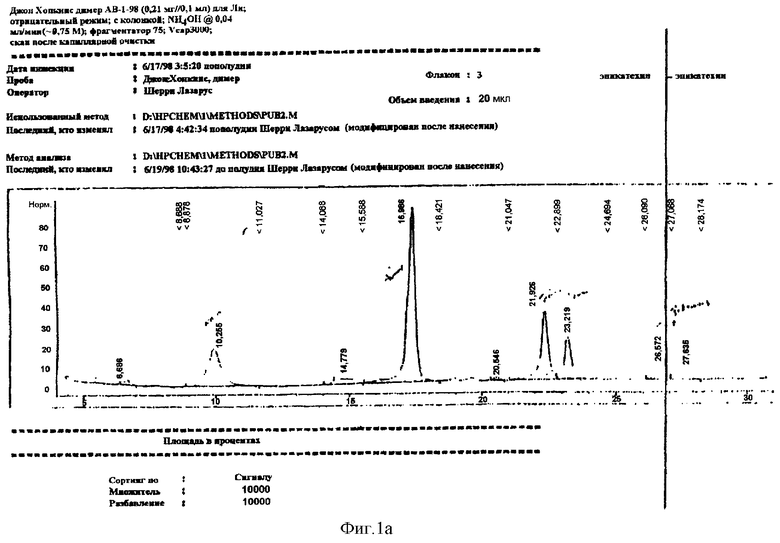
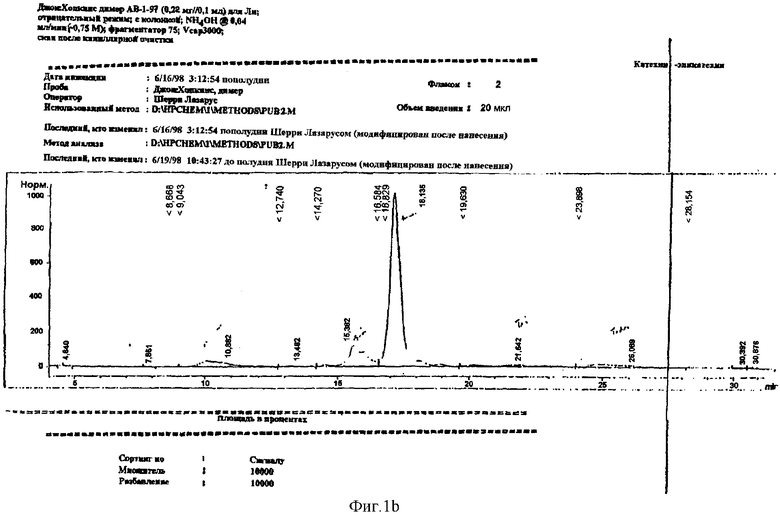
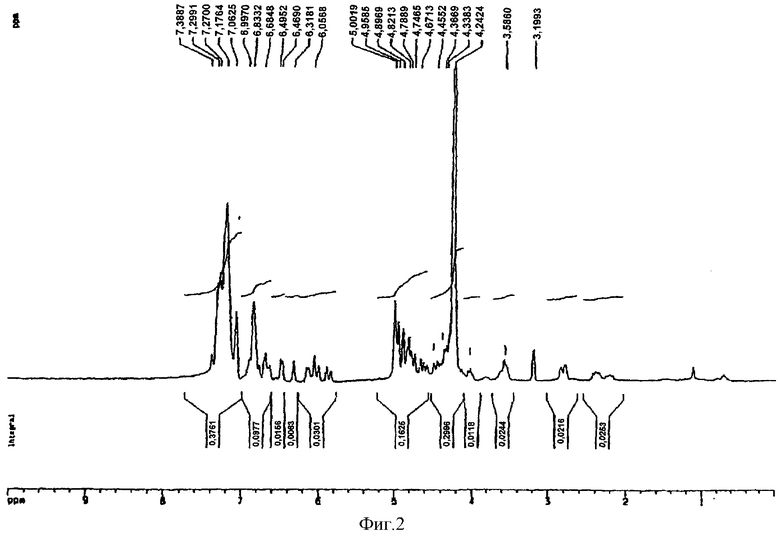
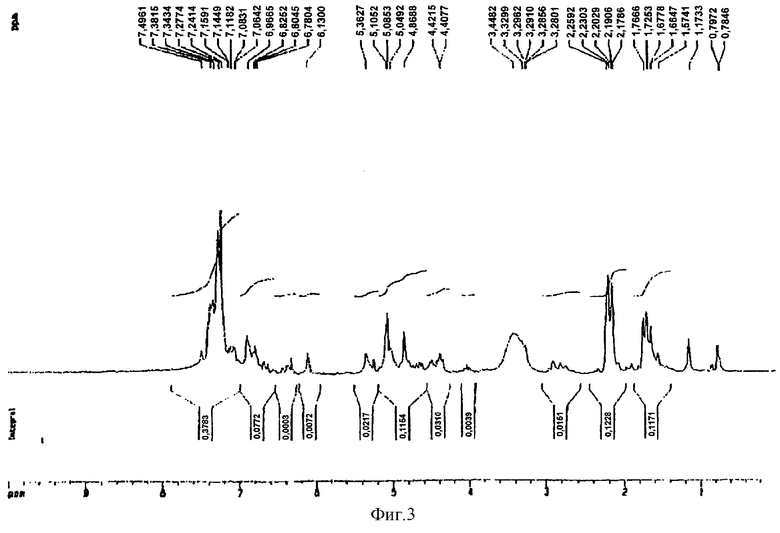
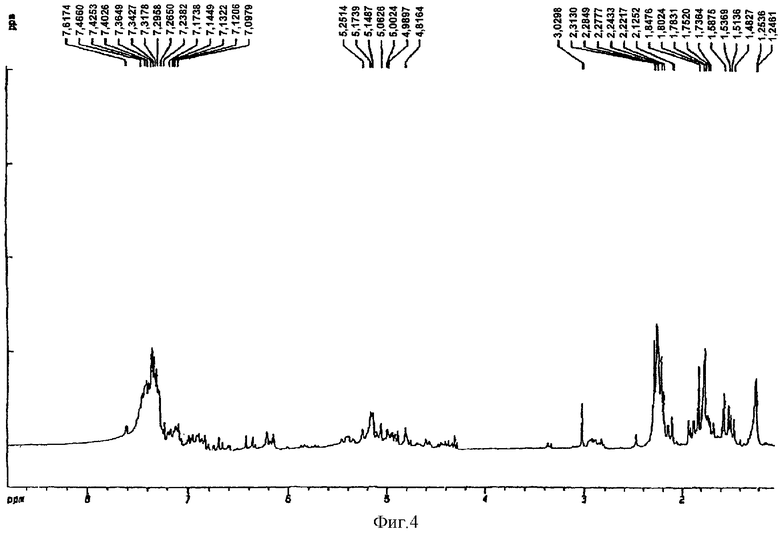
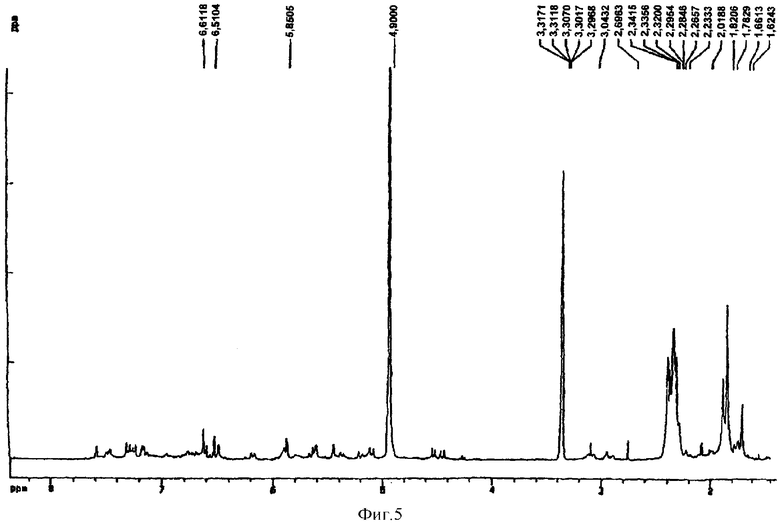
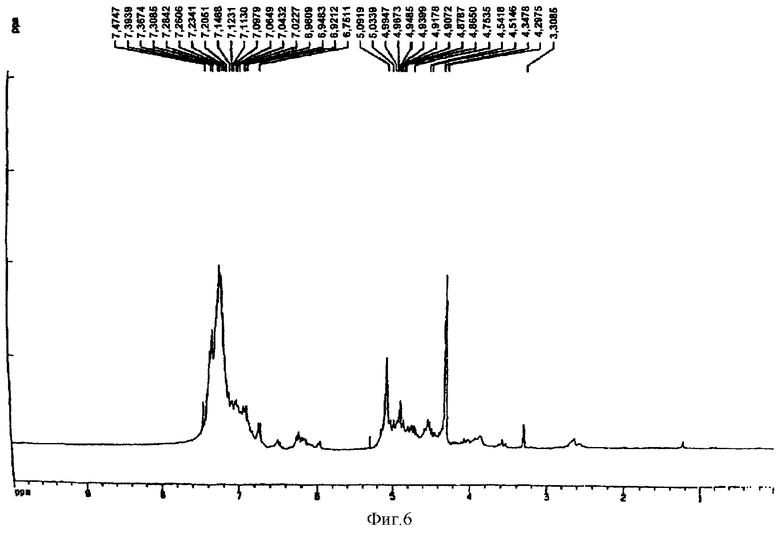
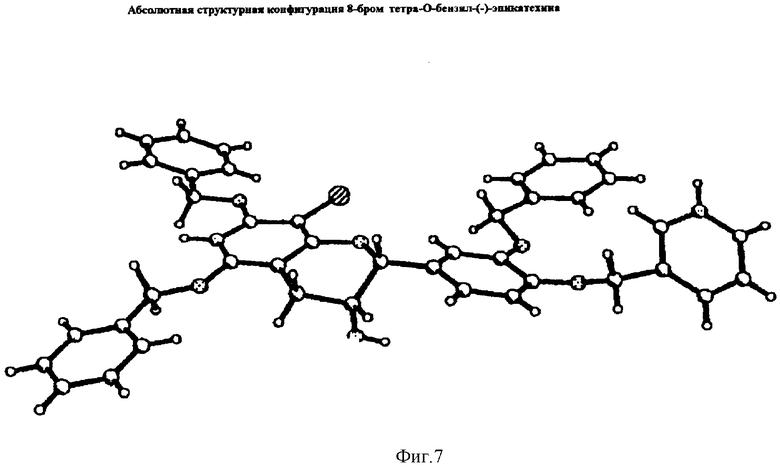
Изобретение относится к способу получения синтетических процианидиновых олигомеров, в частности к способу получения процианидинового димера, который включает стадии: (а) защиты каждой фенольной гидроксильной группы процианидинового мономера общей формулы 1: 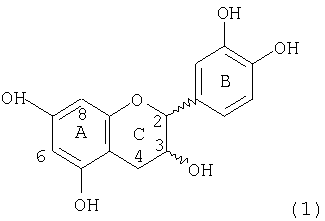 с помощью удаляемой защитной группы, которая не дезактивирует кольцо А мономера, причем стадия защиты проводится в апротонном растворителе; (b) необязательно блокирования С-8 положения мономера со стадии (а) галоидной группой; (c) активирования для сочетания С-4-положения соединения со стадии (а) или стадии (b) введением ацилоксильной группы с использованием соли свинца (IV) органической кислоты с получением активированного соединения; и (d) сочетания активированного соединения со стадии (с) с незащищенным процианидиновым мономером в присутствии катализатора сочетания с получением димера. В определенных условиях на стадии (b) получат кристаллический 8-бром-тетра-О-бензил(-)-эпикатехин в качестве промежуточного продукта. Технический результат - разработан универсальный способ получения олигомеров сложной конфигурации. 2 н. и 13 з.п. ф-лы., 1 табл., 8 ил.
с помощью удаляемой защитной группы, которая не дезактивирует кольцо А мономера, причем стадия защиты проводится в апротонном растворителе; (b) необязательно блокирования С-8 положения мономера со стадии (а) галоидной группой; (c) активирования для сочетания С-4-положения соединения со стадии (а) или стадии (b) введением ацилоксильной группы с использованием соли свинца (IV) органической кислоты с получением активированного соединения; и (d) сочетания активированного соединения со стадии (с) с незащищенным процианидиновым мономером в присутствии катализатора сочетания с получением димера. В определенных условиях на стадии (b) получат кристаллический 8-бром-тетра-О-бензил(-)-эпикатехин в качестве промежуточного продукта. Технический результат - разработан универсальный способ получения олигомеров сложной конфигурации. 2 н. и 13 з.п. ф-лы., 1 табл., 8 ил.
(а) защиты каждой фенольной гидроксильной группы процианидинового мономера общей формулы 1
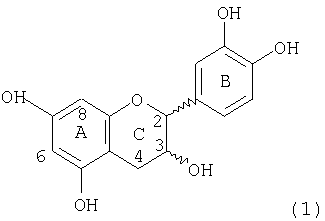
с помощью удаляемой защитной группы, которая не дезактивирует кольцо А мономера, причем стадия защиты проводится в апротонном растворителе;
(b) необязательно блокирования С-8 положения мономера со стадии (а) галоидной группой;
(c) активирования для сочетания С-4-положения соединения со стадии (а) или стадии (b) введением ацилоксильной группы с использованием соли свинца (IV) органической кислоты с получением активированного соединения; и
(d) сочетания активированного соединения со стадии (с) с незащищенным процианидиновым мономером в присутствии катализатора сочетания с получением димера.
| US 5554645 А, 10.09.1996 | |||
| US 5172305 A, 27.01.1998 | |||
| СПОСОБ ПОЛУЧЕНИЯ ПРОИЗВОДНЫХ КАТЕХИНА | 1987 |
|
RU2014319C1 |
| Способ получения растительного полимера-эпигалохина | 1981 |
|
SU1155601A1 |
| Delcour et al, J.Inst | |||
| of Brewing, 1986, v.92, p.244-249 | |||
| Betts M.J., Brown B.R | |||
| et al, J.Chem.Soc., 1969, p.1178 | |||
| Steen Kamp J.A | |||
| et al, Tetrahedron Letters, 1985, v.26, p.3045-3048. | |||

