Изобретение относится к новым ретиноидным соединениям и способам их синтеза. Изобретение также относится к применению таких соединений при приготовлении лекарственных средств для лечения или профилактики различных болезней, к способам применения этих новых ретиноидных соединений и к фармацевтическим композициям на их основе.
Ретиноиды являются структурными аналогами витамина А и включают как природные, так и синтетические соединения. Ретиноидные соединения такие, как все транс-ретиноивые кислоты ("ATRA"), 9-цис-ретиноивая кислота, транс-3-4-дидегидроретиноивая кислота, 4-оксоретиноивая кислота, 13-цис-ретиноивая кислота и ретинол, являются политрофическими регуляторными соединениями, которые влияют на большое количество воспалительных, иммунных и структурных клеток.
Например, ретиноиды модулируют быстрое увеличение эпителиальной клетки, морфогенез в легком и дифференцирование через ряд гормональных ядерных рецепторов, которые принадлежат суперсемейству рецепторов стероидов щитовидной железы. Ретиноидные рецепторы классифицируют на рецепторы ретиноивой кислоты (RAR) и ретиноидные Х рецепторы (RXR), каждый из которых состоит из трех различных подтипов (α-, β- и γ-).
ATRA являются естественным лигандом для рецепторов ретиноивых кислот и проявляют аналогичное сродство с α-, β- и γ-подтипами. Было установлено количественное отношение структуры/активности для ряда синтетических α-, β- и γ-ретиноидных агонистов RAR, которое объясняет основные электронные и структурные характеристики, обеспечивающие избирательное сродство к каждому подтипу (Douget и др.. Quant. Struct.Akt.Relat., 18, 107, 1999).
ATRA не связываются с RXR, для которого 9-цис-ретиноивая кислота является естественным лигандом. Ряд синтетических α-, β- и γ-ретиноидных агонистов RXR также был описан в литературе, посвященной данной области техники (см., например, Billoni и др., US № 5962508; Klaus и др., US № 5986131).
В других тканях, кроме легочной ткани, ретиноиды как правило проявляют противовоспалительные эффекты, могут изменить прогрессию эпителиального дифференцирования клетки и могут ингибировать стромальное продуцирование матрицы клетки. Эти биологические эффекты ретиноидов ведут к развитию многих топических агентов дерматологических нарушений, таких как псориаз, прыщи и гипертрофические кожные шрамы. Ретиноиды также применяют в лечении свето- и возрастных повреждений кожи, заживлении вызванных ран, например операционных и ожоговых (Mustoe и др.. Science 237, 1333 1987; Sprugel и др., J.Pathol., 129, 601, 1987; Boyd, Am.J.Med., 86, 568, 1989), и как противовоспалительные агенты при лечении артрита. Другие лекарственные применения ретиноидов включают препятствование развитию острой промиелоцитарной лейкемии, адено- и скамозоклеточной карциномы и цирроза печени. Ретиноиды также широко используют в лечении предзлокачественных эпителиальных повреждений и злокачественных опухолей (карцином) эпителиального происхождения (Bollag и др., US № 5248071; Sporn и др., Fed.Proc. 1976, 1332; Hong и др., "Retinoids and Human Cancer" in The Retinoids: Biology, Chemistry and Medicine, M.B. Sporn, A.B. Roberts и D.S. Goodman (eds.) Raven Press, New York, 1994, 597-630). Однако многим ранее изученным ретиноидам часто недостает селективности, вследствие чего они проявляют опасные эффекты политрофии и, когда их использует в терапевтически эффективных дозах, могут вызвать смерть пациента. Таким образом, терапевтическое применение ретиноидов при других заболеваниях, кроме рака, ограничено токсическими побочными эффектами. Общий обзор ретиноидов можно найти в Goodman & Gilman's "The Pharmacological Basis of Therapeutics", издание 9-е (1996, McGrawHill), главы 63-64.
Хроническая обструктивная легочная болезнь (ХОЛБ) относится к большой группе болезней легкого, которые препятствуют нормальному дыханию. Приблизительно 11% населения Соединенных Штатов страдают ХОЛБ и, основываясь на доступных данных, полагают, что распространенность ХОЛБ увеличивается. В настоящее время ХОЛБ-четвертая ведущая причина смертности в Соединенных Штатах.
ХОЛБ-болезнь, при которой дыхание затруднено из-за присутствия по крайней мере одной болезни, такой как астма, эмфизема и хронический бронхит. Термин ХОЛБ был введен потому, что такие состояния часто сосуществуют, и в отдельных случаях бывает трудно установить, какая болезнь является ответственной за причину обструкции в легком (1987 Merck Manual). Клинически ХОЛБ проявляется уменьшением выдыхаемого из легких потока воздуха, которое оказывается постоянным в течение более чем нескольких месяцев, а в случае хронического бронхита сохраняется в течение двух или больше последовательных лет. Самые серьезные проявления ХОЛБ как правило включают характерные признаки эмфиземы.
Эмфизема-болезнь, при которой газообменные структуры (например, альвеолы) легкого разрушаются, что приводит к неадекватному кислородонасыщению, которое может привести к нетрудоспособности и смерти. Анатомически эмфизему определяет постоянное расширение воздушного пространства отдаленных, периферийных бронхиол (например, дыхательных трубок), которое характеризуется уменьшением эластичности легкого, уменьшением альвеолярной поверхности и газообменными и альвеолярными нарушениями, которые заканчиваются уменьшением дыхания. Таким образом, характерные физиологические отклонения при эмфиземе уменьшают газовый обмен и выдыхаемый газовый поток.
Курение сигарет - самая частая причина эмфиземы, хотя разрушению альвеол могут также способствовать и другие токсины окружающей среды. Вредные соединения, встречающиеся среди таких опасных для здоровья веществ, могут активизировать разрушительные процессы, включающие, например, выделение чрезмерных количеств протеаз, которые разрушают нормальные защитные механизмы, в частности присутствующие в легком ингибиторы протеаз. Дисбаланс между протеазами и присутствующими в легком ингибиторами протеаз может привести к матричному разрушению эластина, потере упругости отдачи, повреждению ткани и непрерывному снижению функции легкого. Скорость повреждения легкого можно уменьшить уменьшением количеств токсинов в легком (т.е. прекращением курения). Однако поврежденные альвеолярные структуры не восстанавливаются, и функция легкого не восстанавливается. Были описаны по крайней мере четыре различных типа эмфиземы согласно их местоположениям во вторичной доле: пандолевая эмфизема, центролобулярная эмфизема, эмфизема отдаленной от центра доли и парацикатрикальная эмфизема.
Главным признаком эмфиземы служит хроническая одышка. Другие важные признаки эмфиземы включают, хотя ими их список не ограничен, хронический кашель, окраску кожи, вызванную недостатком кислорода, одышку при минимальной физической нагрузке и хрипы. Дополнительные признаки, которые могут быть связаны с эмфиземой, включают, хотя ими их список не ограничен, нарушения зрения, головокружение, временное прекращение дыхания, беспокойство, одутловатость, усталость, бессонницу и потерю памяти. Эмфизему как правило диагностируют при физикальном обследовании, которое показывает ослабленные и ненормальные звуки дыхания, хрипы и длительный выдох. Для подтверждения диагноза эмфиземы можно воспользоваться результатами функциональных легочных тестов, пониженным содержанием кислорода в крови и рентгеновским обследованием грудной клетки.
В настоящее время в данной области техники отсутствуют какие-либо эффективные методы, позволяющие полностью излечить клинические признаки эмфиземы. В некоторых случаях лечение такими средствами, как бронхолитики, β-агонисты, теофиллин, антихолинергисты, мочегонные средства и кортикостероиды, вводимые в легкое ингалятором или распылителем, могут улучшить дыхание, которое нарушено при эмфиземе. В ситуациях, когда функции легкого нарушены настолько серьезно, что поглощение из воздуха достаточного количества кислорода невозможно, часто прибегают к лечению кислородом. Для лечения пациентов с серьезной эмфиземой можно использовать оперативные сокращения объема легкого. При этом поврежденные части легкого удаляют, что позволяет нормальным частям легкого более полно расшириться и приносит пользу благодаря увеличенной вентиляции. Наконец, трансплантация легкого - другая хирургическая альтернатива, доступная индивидуумам с эмфиземой, которая может улучшить качество жизни, но продолжительность жизни существенно не увеличит.
Альвеолы формируются в период развития посредством деления саккулесов, которые составляют газово-обменные элементы незрелого легкого. Точные механизмы, управляющие формированием септ и пространства между ними у приматов, остаются в настоящее время неизвестными. Ретиноиды, такие как ATRA, которые являются многофункциональным модулятором клеточного поведения, способные изменить как внеклеточный матричный метаболизм, так и нормальное эпителиальное дифференцирование, играют решающую регулирующую роль у млекопитающих, таких как крыса. Так, например, ATRA модулируют основные аспекты дифференцирования легкого, которые селективно экспрессируются во времени и пространстве, через связывание с конкретными рецепторами ретиноивых кислот. Скоординированная активация различных подтипов рецепторов ретиноивых кислот связана с разветвлением в легком, переходом альвеола/перегородка в легком и активацией гена тропоэластина у новорожденных крыс.
В процессе разделения альвеол перегородками гранулы хранения ретиноивых кислот в мезенхиме фибробластов, окружающих альвеолярные стенки, увеличиваются (Liu и др., Am.J.Physiol. 1993, 265, L430; McGowan и др., Am.J.Physiol., 1995, 269, L463), и в верхушках легкого экспрессируются рецепторы ретиноивой кислоты (Ong и др., Proc.Natl.Acad. of Sci., 1976, 73, 3976; Grummer и др., Pediatr.Pulm. 1994, 17, 234). Депозиция новой эластиновой матрицы и разделительные параллели истощают эти гранулы хранения ретиноивых кислот. Было показано, что введение ретиноивой кислоты в послеродовой период увеличивает у крыс число альвеол; это поддерживает концепцию того, что ATRA и другие ретиноиды могут вызывать формирование альвеол (Massaro и др., Am.J.Physiol., 270, L305, 1996). Лечение новорожденных детенышей крысы дексаметазоном, глюкокортикоидом, предотвращает разделение и уменьшает экспрессию некоторых подтипов рецептора ретиноивой кислоты. Было показано, что дополнительные количества ATRA предотвращают блокировку формирования альвеол дексаметазоном. Далее, ATRA препятствуют вызываемому дексаметазоном уменьшению экспрессии рецептора ретиноевой кислоты и последующему разделению альвеол перегородками в развивающемся легком крысы.
Согласно сообщениям, в моделях эмфиземы на животных ATRA вызывают формирование новых альвеол и возвращают упругую отдачу в легком к приблизительно нормальным значениям (Massaro и др.. Nature Med., 1997, 3, 675; "Strategies to Augment Alveolization", National Heart, Lung, and Blood Institute, RFA: HL-98-011, 1998; Massaro и др., US № 5998486). Однако механизм действия ATRA в этих исследованиях остается невыясненным, хотя Массаро сообщает, что ATRA генерируют новые альвеолы. Важнее то, что применение ATRA сопряжено с некоторой токсичностью или озабоченностью из-за отрицательных эффектов. Таким образом, существует настоятельная потребность в применении новых ретиноидных агонистов, которые могут быть использованы при лечении дерматологических нарушений, эмфиземы и рака без проблем токсичности ATRA или других ретиноидов.
По настоящему изобретению предлагаются новые ретиноидные агонисты, способ синтеза, применение таких соединений при приготовлении лекарственных средств для лечения или профилактики эмфиземы, рака и дерматологических нарушений, способы лечения или профилактики таких заболеваний, фармацевтические композиции, которые приемлемы для лечения или профилактики таких заболеваний, и способы введения композиций на основе новых ретиноидов в легкое млекопитающего, страдающего эмфиземой, раковым заболеванием и дерматологическими нарушениями.
По одному варианту выполнения изобретения предлагаются соединения, отвечающие структурной формуле (I):
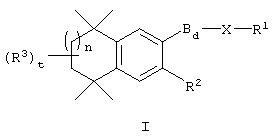
или их фармацевтически приемлемые соли, где
n обозначает 1;
d обозначает 0 или 1;
В обозначает -CR7=CR8-, -СН2О-;
R7 и R8 каждый независимо обозначает водородный атом;
Х обозначает фенил, необязательно замещенный галогеном, или 5-ти членный гетероарил, содержащий S в качестве гетероатома;
R1 обозначает -C(=O)-R9;
R обозначает алкил, гидроксил, амино-, гетероалкилоксигруппу, содержащую О, или 6-ти членный гетероциклил, содержащий N в качестве гетероатома; а
R2 обозначает:
(a)-(CR10R11)m-Yp-R12;
m обозначает целое число от 1 до 10;
р обозначает 0 или 1;
R10 и R11 обозначают водородный атом;
Y обозначает -О-, -S- или -NR13-; и
R13 обозначает водородный атом;
R12 обозначает водородный атом, алкил, циклоалкил, фенил, 5-ти или 6-ти членный гетероарил, содержащий N, S, О в качестве гетероатома, 5-ти или 6-ти членный гетероарилалкил, содержащий N, S, О в качестве гетероатома, гетероалкил, содержащий N, S, О в качестве гетероатома, 5-ти или 6-ти членный гетероциклил, содержащий N, S, О в качестве гетероатома, или 5-ти или 6-ти членный гетероциклилалкил, содержащий N, S, О в качестве гетероатома;
при условии, что когда р обозначает 0, тогда R12 не обозначает водородный атом или алкил;
(б) 5-ти или 6-ти членный гетероарил, содержащий N, S, О в качестве гетероатома;
(в) -Z-L;
Z обозначает -CR14=CR15-, 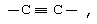 -С(=O) или -S-;
-С(=O) или -S-;
R14, R15 обозначают водородный атом; а
L обозначает 5-ти или 6-ти членный гетероарил, содержащий N, S, О в качестве гетероатома,
(г) -CR14=CR15-L1, где l1 обозначает S(O)2R17 или SO2NR18R19, где R17 обозначает алкил, а R18 и R19 обозначают водородный атом; каждый R3 независимо обозначает водородный атом, гидроксил или оксогруппу, a t обозначает 1 или 2.
Понятием "ацил" обозначают радикал -C(O)R, где R3 обозначает водородный атом, алкил, циклоалкил, циклоалкилалкил, арил или арилалкил, у которого алкил, циклоалкил, циклоалкилалкил, арил и арилалкил имеют значения, указанные в настоящем описании. Типичные примеры включают, без ограничения перечисленных, формил, ацетил, циклогексилкарбонил, циклогексилметилкарбонил, бензоил, бензилкарбонил и т.п.
Понятием "ациламино" обозначают радикал -NR'C(O)R, где R' обозначает водородный атом или алкил; а R обозначает водородный атом, алкил, циклоалкил, циклоалкилалкил, арил или арилалкил, у которого алкил, циклоалкил, циклоалкилалкил, арил и арилалкил имеют значения, указанные в настоящем описании. Типичные примеры включают, хотя ими их список не ограничен, формиламино-, ацетиламино-, циклогексилкарбониламино-, циклогексилметилкарбониламино-, бензоиламино", бензилкарбониламиногруппу и т.п.
Понятием "алкокси" обозначают радикал -OR, где R обозначает алкильную группу, как она представлена в настоящем описании, например метокси, этокси, пропокси, бутокси и т.п.
Понятием "алкоксикарбонил" обозначают радикал -C(O)-R, где R обозначает алкоксирадикал, как он представлен в настоящем описании.
Понятием "алкил" обозначают линейный насыщенный одновалентный углеводородный радикал, содержащий от одного до шести углеродных атомов, или разветвленный насыщенный одновалентный углеводородный радикал, содержащий от трех до шести углеродных атомов, например метил, этил, пропил, 2-пропил, н-бутил, изобутил, трет-бутил, пентил и т.п.
Понятием "алкиламиногруппа" обозначают радикал -NHR, где R обозначает алкильную, циклоалкильную или циклоалкилалкильную группу, как она представлена в настоящем описании. Типичные примеры включают, хотя ими их список не ограничен, метиламино-, этиламино-, 1-метилэтиламино-, циклогексиламиногруппу и т.п.
Понятием "алкилен" обозначают линейный насыщенный двухвалентный углеводородный радикал, содержащий от одного до десяти углеродных атомов, или разветвленный насыщенный двухвалентный углеводородный радикал, содержащий от трех до десяти углеродных атомов, например метилен, этилен, 2,2-диметилэтилен, пропилен, 2-метилпропилен, бутилен, пентилен и т.п.
Понятием "алкилсульфонил" обозначают радикал -S(O)2R, где R обозначает алкильную, циклоалкильную или циклоалкилалкильную группу, как она представлена в настоящем описании, например метилсульфонил, этилсульфонил, пропилсульфонил, бутилсульфонил, циклогексилсульфонил и т.п.
Понятием " алкилсульфинил" обозначают радикал -S(O)R, где R обозначает алкильную, циклоалкильную или циклоалкилалкильную группу, как она представлена в настоящем описании, например метилсульфинил, этилсульфинил, пропилсульфинил, бутилсульфинил, циклогексилсульфинил и т.п.
Понятием "алкилтио" обозначают радикал -SR, где R обозначает алкильную, циклоалкильную или циклоалкилалкильную группу, как она представлена в настоящем описании, например метилтио, этилтио, пропилтио, бутилтио, циклогексилтио и т.п.
Понятием "арил" обозначают моноциклический или бициклический ароматический углеводородный радикал, который необязательно замещен одним или несколькими заместителями, предпочтительнее одним, двумя или тремя заместителями, предпочтительно выбранными из группы, включающей алкил, ацил, ациламино-, алкоксикарбонил, алкиламино-, алкилсульфинил, алкилсульфонил, -SO2NR'R" (где каждый из R' и R" независимо обозначает водородный атом или алкил), алкилтио, алкокси, амино-, арилокси, карбамоил, пиано-, диалкиламино-, гало, галоалкил, гетероалкил, гетероциклил, гидрокси, гидроксиалкил, метилендиокси, этилендиокси, нитрогруппу и тио. Более конкретно понятие "арил" включает, хотя ими их список не ограничен, фенил, хлорфенил, фторфенил, метоксифенил, 1-нафтил, 2-нафтил и их производные.
Понятие "арилалкил" относится к алкильному радикалу, как он представлен в настоящем описании, у которого один из водородных атомов (алкильной группы) замещен арильной группой. Типичные арилалкильные группы включают, хотя ими их список не ограничен, бензил, 2-фенилэтан-1-ил, нафтилметил, 2-нафтилэтан-1-ил, нафтобензил, 2-нафтофенилэтан-1-ил и т.п.
Понятием "арилокси" обозначают радикал -O-R, где R обозначает арильную группу, как она представлена в настоящем описании.
Понятием "арилалкилокси" обозначают радикал -O-R, где R обозначает арилалкил, как он представлен в настоящем описании.
Понятием "карбамоил" обозначают радикал -C(O)N(R)2, где каждая группа R независимо представляет собой водородный атом, алкил или арил, как он представлен в настоящем описании.
Понятием "карбокси" обозначают радикал -С(O)ОН.
Понятием "цианогруппа" обозначают радикал -CN.
Понятие "циклоалкил" относится к насыщенному одновалентному циклическому углеводородному радикалу, содержащему от трех до семи кольцевых углеродных атомов, например к циклопропилу, циклобутилу, циклогексилу, 4-метилциклогексилу и т.п.
Понятием "циклоалкилалкил" обозначают радикал -RaRb, где Ra обозначает алкиленовую группу, a Rb обозначает циклоалкильную группу, как она представлена в настоящем описании, например циклогексилметил и т.п.
Понятием "замещенный циклоалкил" обозначают циклоалкильный радикал, как он представлен в настоящем описании, содержащий один, два или три (предпочтительно один) водородных атома, замещенных группой -Y-C(O)R (где Y отсутствует или обозначает алкиленовую группу, а R обозначает водородный атом, ацил, ациламино-, алкил, алкоксикарбонил, алкиламино-, алкилсульфинил, алкилсульфонил, алкилтио, алкокси, амино-, арилокси, арилалкилокси, карбамоил, циано-, диалкиламино-, гало, галоалкил, гетероалкил, гидроксил, гидроксиалкил, нитро- или тиогруппу).
Понятием "диалкиламино" обозначают радикал -NRR', где каждый из R и R' независимо обозначает алкильную, циклоалкильную или циклоалкилалкильную группу, как она представлена в настоящем описании. Типичные примеры включают, хотя ими их список не ограничен, диметиламино-, метилэтиламино-, ди(1-метилэтил)амино-, (циклогексил)(метил)амино-, (циклогексил)(этил)амино-, (циклогексил)(пропил)амино-, (циклогексилметил)(метил)амино-, (циклогексилметил)(этил)аминогруппы и т.п.
Понятием "гало" обозначают атом фтора, хлора, брома или иода, предпочтительно фтора или хлора.
Понятием "галоалкил" обозначают алкильную группу, замещенную одним или несколькими одинаковыми или разными атомами галогена, например, -СН2Cl, -CF3, -СН2CF3, -CH2CCl3 и т.п.
Понятием "гетероарил" обозначают моноциклический или бициклический радикал, содержащий от 5 до 12 кольцевых атомов, включающий по меньшей мере одно ароматическое кольцо, содержащее один, два или три кольцевых гетероатома, выбранных из N, О или S, а остальные кольцевые атомы приходятся на долю С, причем при этом подразумевается, что точка присоединения гетероарильного радикала находится на ароматическом кольце. Такое гетероарильное кольцо необязательно независимо замещено одним или несколькими заместителями, предпочтительно одним или двумя заместителями, выбранными из ацила, ациламино-, алкила, алкоксикарбонила, алкиламино-, алкилсульфинила, алкилсульфонила, -SO2NR'R" (где каждый из R' и R" независимо обозначает водородный атом или алкил), алкилтио, алкокси, амино-, арилокси, карбамоила, циано-, диалкиламино-, этилендиокси, гало, галоалкила, гетероалкила, гетероциклила, гидрокси, гидроксиалкила, метилендиокси, нитро- и тиогрупп. Более конкретно понятие "гетероарил" включает, хотя ими их список не ограничен, пиридил, фуранил, тиенил, тиазолил, изотиазолил, триазолил, имидазолил, изоксазолил, пирролил, пиразолил, пиримидинил, бензофуранил, тетрагидробензофуранил, изобензофуранил, бензотиазолил, бензизотиазолил, бензотриазолил, индолил, изоиндолил, бензоксазолил, хинолил, тетрагидрохинолинил, изохинолил, бензимидазолил, бензизоксазолил, бензотиенил и их производные.
Понятием "гетероарилалкил" обозначают алкильный радикал, как он представлен в настоящем описании, в котором один из водородных атомов алкильной группы замещен гетероарильной группой.
Понятием "гетероалкил" обозначают алкильный радикал, как он представлен в настоящем описании, в котором один или несколько водородных атомов замещены заместителем, независимо выбранным из группы, включающей -ORa, -NRbRc и -S(O)nRd (где n обозначает целое число от 0 до 2), причем при этом подразумевается, что гетероалкильный радикал присоединяется посредством углеродного атома, где Ra обозначает водородный атом, ацил, алкил, циклоалкил или циклоалкилалкил; каждый из Rb и Rc независимо друг от друга обозначает водородный атом, ацил, алкил, циклоалкил или циклоалкилалкил; когда n обозначает 0, Rd обозначает водородный атом, алкил, циклоалкил или циклоалкилалкил, а когда n обозначает 1 или 2, Rd обозначает алкил, циклоалкил, циклоалкилалкил, амино-, ациламино-, моноалкиламино- или диалкиламиногруппу. Типичные примеры включают, хотя ими их список не ограничен, 2-гидроксиэтил, 3-гидроксипропил, 2-гидрокси-1-гидроксиметилэтил, 2,3-дигидроксипропил, 1-гидроксиметилэтил, 3-гидроксибутил, 2,3-дигидроксибутил, 2-гидрокси-1-метилпропил, 2-аминоэтил, 3-амино пропил, 2-метилсульфонилэтил, аминосульфонилметил, аминосульфонилэтил, аминосульфонилпропил, метиламиносульфонилметил, метиламиносульфонилэтил, метиламиносульфонилпропил и т.п.
Понятием "гетероалкиламиногруппа" обозначают радикал -NHR, где R обозначает гетероалкильную группу, как она представлена в настоящем описании.
Понятием "гетероалкилокси" обозначают радикал -O-R, где R обозначает гетероалкильную группу, как она представлена в настоящем описании.
Понятием "гетероалкилзамещенный циклоалкил" обозначают циклоалкильный радикал, как он представлен в настоящем описании, причем один, два или три водородных атома в циклоалкильном радикале независимо замещены гетероалкильной группой и при этом подразумевается, что такой гетероалкильный радикал присоединен к циклоалкильному радикалу посредством углерод-углеродной связи. Типичные примеры включают, хотя ими их список не ограничен, 1-гидроксиметилциклопентил, 2-гидроксиметилциклогексил и т.п.
Понятием "гетерозамещенный циклоалкил" обозначают циклоалкильный радикал, как он представлен в настоящем описании, причем один, два или три водородных атома в этом циклоалкильном радикале замещены заместителем, независимо выбранным из группы, включающей гидрокси, алкокси, амино-, ациламино-, моноалкиламино-, диалкиламино-, оксо (С=O), имино-, гидроксиминогруппу (=NOH), NR'SO2Rd (где R' обозначает водородный атом или алкил, а Rd обозначает алкил, циклоалкил, амино-, моноалкиламино или диалкиламиногруппу), -X-C(O)R (где Х обозначает О или NR', R обозначает водородный атом, алкил, галоалкил, гидрокси, алкокси, амино-, моноалкиламино-, диалкиламиногруппу или необязательно замещенный фенил, а R' обозначает Н или алкил) или -S(O)nRd (где n обозначает целое число от 0 до 2), таким образом, что когда n обозначает 0, R обозначает водородный атом, алкил, циклоалкил или циклоалкилалкил, а когда n обозначает 1 или 2, R обозначает алкил, циклоалкил, циклоалкилалкил, амино-, ациламино-, моноалкиламино или диалкиламиногруппу. Типичные примеры включают, хотя ими их список не ограничен, 2-,3- и 4-гидроксициклогексилы, 2-, 3- и 4-аминоциклогексилы, 2-, 3- и 4-сульфонамидоциклогексилы и т.п., предпочтительно 4-гидроксициклогексил, 2-аминоциклогексил, 4-сульфонамидоциклогексил.
Понятием "гетерозамещенный циклоалкилалкил" обозначают радикал RaRb -, где Ra обозначает гетерозамещенный циклоалкильный радикал, а Rb обозначает алкиленовый радикал.
Понятием "гетероциклил" обозначают насыщенный или ненасыщенный неароматический циклический радикал, содержащий от 3 до 8 кольцевых атомов, в котором один или два кольцевых атома представляют собой гетероатомы, выбранные из N, О или S(O)n (где n обозначает целое число от 0 до 2), а остальные кольцевые атомы приходятся на долю С, где один или два атома С могут, но необязательно, быть замененными карбонильной группой. Такое гетероциклильное кольцо может быть необязательно независимо замещенным одним, двумя или тремя заместителями, выбранными из алкила, галоалкила, гетероалкила, гало, нитро-, цианоалкила, гидроксила, алкокси, амино-, моноалкиламино-, диалкиламиногрупп, арилалкила, -(X)n-C(O)R (где Х обозначает О или NR', n обозначает 0 или 1, R обозначает водородный атом, алкил, галоалкил, гидрокси, алкокси, амино-, моноалкиламино-, диалкиламиногруппу или необязательно замещенный фенил, а R' обозначает Н или алкил), -алкилен-С(O)R (где R обозначает водородный атом, алкил, галоалкил, гидроксил, алкокси, амино-, моноалкиламино-, диалкиламиногруппу или необязательно замещенный фенил) и -S(O)nRd [где n обозначает целое число от 0 до 2, а Rd обозначает водородный атом (при условии, что n обозначает 0), алкил, галоалкил, циклоалкил, циклоалкилалкил, амино-, моноалкиламино-, диалкиламино- или гидроксиалкил]. Более конкретно понятие гетероциклил включает, хотя ими их список не ограничен, тетрагидропиранил, пиперидино, N-метилпиперидин-3-ил, пиперазино-, N-метилпирролидин-3-ил, 3-пирролидино-, морфолино, тиоморфолино, тиоморфолино-1-оксид, тиоморфолино-1,1-диоксид, пирролинил, имидазолинил и их производные.
Понятием "гетероциклилалкил" обозначают радикал -RaRb, где Ra обозначает алкиленовую группу, а Rb обозначает гетероциклильную группу, как они представлены выше, например тетрагидропиран-2-илметил, 1, 2- и 3-пиперидинилметил, 1-пиперазинилметил, 4-метилпиперазин-1-илметил и т.п.
Понятием "гидроксиалкил" обозначают алкильный радикал, как он представлен в настоящем описании, замещенный одной или несколькими гидроксильными группами, при условии, что один и тот же углеродный атом не несет больше одной гидроксильной группы. Типичные примеры включают, хотя ими их список не ограничен, 2-гидроксиэтил, 2-гидроксипропил, 3-гидроксипропил, 1-(гидроксиметил)-2-метилпропил, 2-гидроксибутил, 3-гидроксибутил, 4-гидроксибутил, 2,3-дигидроксипропил, 2-гидрокси-1-гидроксиметилэтил, 2,3-дигидроксибутил, 3,4-дигидроксибутил и 2-(гидроксиметил)-3-гидроксипропил, предпочтительно 2-гидроксиэтил, 2,3-дигидроксипропил и 1-(гидроксиметил)-2-гидроксиэтил. Таким образом, в настоящем описании понятие "гидроксиалкил" используют для определения совокупности гетероалкильных групп.
Понятие "уходящая группа" используют в значении, которое обычно связано с ним в химии органического синтеза, т.е. для обозначения атома или группы, способной замещаться нуклеофилом, и которое охватывает гало (в частности атомы хлора, брома и иода), алкансульфонилокси, аренсульфонилокси, алкилкарбонилокси (например, ацетокси), арилкарбонилокси, мезилокси, тозилокси, трифторметансульфонилокси, арилокси (например, 2,4-динитрофенокси), метокси, N,O-диметилгидроксиламино группу и т.п.
Понятием "оксо" обозначают двухвалентный радикал (С=0). Понятием "фармацевтически приемлемый наполнитель" обозначают наполнитель, который может быть использован в приготовлении фармацевтической композиции, которая в общем безвредна, нетоксична и ни с биологической, ни с какой-либо другой точек зрения не является нежелательной и включает наполнитель, который приемлем для применения в ветеринарии, а также при применении с фармацевтическими целями в отношении человека. Понятие "фармацевтически приемлемый наполнитель" в том смысле, в котором оно использовано в описании и формуле изобретения, охватывает как один, так и больше одного такого наполнителя.
Понятием "фармацевтически приемлемая соль" соединения обозначают соль, которая фармацевтически приемлема и которая обладает целевым фармакологическим действием исходного соединения. Такие соли включают: (1) кислотно-аддитивные соли, полученные с использованием неорганических кислот, таких как соляная кислота, бромистоводородная кислота, серная кислота, азотная кислота, фосфорная кислота и т.п., или полученные с применением органических кислот, таких как уксусная кислота, пропионовая кислота, гексановая кислота, циклопентанпропионовая кислота, гликолевая кислота, пировиноградная кислота, молочная кислота, малоновая кислота, янтарная кислота, яблочная кислота, малеиновая кислота, фумаровая кислота, винная кислота, лимонная кислота, бензойная кислота, 3-(4-гидроксибензоил)бензойная кислота, коричная кислота, миндальная кислота, метансульфоновая кислота, этансульфоновая кислота, 1,2-этандисульфоновая кислота, 2-гидроксиэтансульфоновая кислота, бензолсульфоновая кислота, 4-хлорбензолсульфоновая кислота, 2-нафталинсульфоновая кислота, 4-толуолсульфоновая кислота, камфорсульфоновая кислота, 4-метилбицикло[2.2.2]окт-2-ен-1-карбоновая кислота, глюкогептоновая кислота, 3-фенилпропионовая кислота, триметилуксусная кислота, третичная бутилуксусная кислота, лаурилсерная кислота, глюконовая кислота, глутаминовая кислота, гидроксинафтойная кислота, салициловая кислота, стеариновая кислота, муконовая кислота и т.п.; и (2) соли, полученные, когда протонкислоты, содержащийся в исходном соединении, либо замещают ионом металла, например ионом щелочного металла, ионом щелочноземельного металла или ионом алюминия, либо он образует координационную связь с органическим основанием, таким, как этаноламин, диэтаноламин, триэтаноламин, трометамин, N-метилглюкамин и т.п.
Понятия "пролекарство" и "пролекарственное средство" в настоящем описании использованы как взаимозаменяющие и относящиеся к любому соединению, которое, когда такое пролекарство вводят в организм млекопитающего, in vivo выделяет активное исходное лекарственное средство, соответствующее одной из структурных формул (I-VII). Пролекарства соединения одной из структурных формул (I-VII) готовят модификацией одной или нескольких функциональных групп, имеющихся у соединения одной из структурных формул (I-VII), таким образом, чтобы модифицирующие группы были способны in vivo отщепляться с выделением исходного соединения. Пролекарства включают соединения структурных формул (I-VII), у которых гидроксильная, амино- или сульфгидрильная группа [в соединениях структурных формул (I-VII)] связана с любой группой, которая in vivo способна отщепляться с восстановлением соответственно свободной гидроксильной, амино- или сульфгидрильной группы. Примеры пролекарств включают, хотя ими их список не ограничен, сложные эфиры (например, ацетатные, формиатные, бензоатные производные), карбаматы (например, N,N-диметиламинокарбонил) гидроксильных функциональных групп в соединениях структурных формул (I-VIII), N-ацильные производные (например, N-ацетил), N-основания Манниха, шиффовы основания и енаминоны аминовых функциональных групп, оксимы, ацетали, кетали и енольные сложные эфиры кетоновых и альдегидных функциональных групп в соединениях формул I-VII и т.п.[см. Bundegaard, Н. "Design of prodrugs", с. 1-92, Elesevier, New York-Oxford (1985)].
Понятие "защитная группа" относится к группировке атомов, которая, когда ее присоединяют к реакционноспособной группе молекулы, маскирует, уменьшает или предотвращает проявление этой группой реакционной способности. Примеры защитных групп можно найти в работах T.W. Green и P.G.Futs, "Protective Groups in Organic Chemistry" (Wiley, 2nd ed. 1991) и Harrison и др., "Compendium of Synthetic Organic Methods", Vols. 1-8 (John Wiley and Sons, 1971-1996). Типичные защитные группы для аминогруппы включают, хотя ими их список не ограничен, формильную, ацетильную, трифторацетильную, бензильную, бензилоксикарбонильную (БЗК), трет-бутоксикарбонильную (БОК), триметилсилильную (ТМС), 2-триметилсилилэтансульфонильную (СЭС), тритильную и замещенную тритильную группы, аллилоксикарбонил, 9-флуоренилметилоксикарбонил (ФМОК), нитровератрилоксикарбонил (НВОК) и т.п. Типичные примеры защитной группы для гидроксила включают, хотя ими их список не ограничен, те, которые образуются когда гидроксильную группу либо ацилируют, либо алкилируют, в частности бензил, остатки тритиловых эфиров, а также алифатических эфиров, тетрагидропираниловых эфиров, триалкилсилиловых эфиров и аллилиловых эфиров.
В настоящем описании понятие "млекопитающее" охватывает человека. Понятия "человек" и "пациент" использованы в настоящем описании как взаимозаменяемые.
"Лечение" или "излечение" эмфиземы, рака или дерматологических нарушений включают профилактику болезни (т.е. подавление развития по крайней мере одного из клинических признаков у млекопитающего, которое болеет или предрасположено к болезни, но еще не испытывает или не проявляет признаков болезни), ингибирование болезни (т.е. прекращение или уменьшение развития болезни или по крайней мере одного из клинических признаков) или ослабление болезни (т.е. регрессию болезни или по крайней мере одного из клинических признаков). Профилактика или предотвращение охватывает введенние до проявления болезни или нарушения.
Понятием "терапевтически эффективное количество" называют то количество соединения, которое, когда его вводят млекопитающему для лечения болезни, является достаточным для осуществления такого лечения болезни. Терапевтически эффективное количество обычно варьируют в зависимости от соединения, заболевания, его серьезности и возраста, массы тела и т.д. млекопитающего, которому требуется лечение.
Используемым в настоящем описании понятием "соединения по изобретению" обозначают соединения общих формул (I-VII), включающие, хотя ими их список не ограничен, конкретные соединения, охватываемые этими формулами, представленными в настоящем описании. Соединения по изобретению в настоящем описании идентифицируют их химическим строением и/или химическим наименованием. Когда соединение обозначено как химическим строением, так и химическим наименованием, и химическое строение и химическое наименование друг другу противоречат, в идентификации этого соединения химическое строение является определяющим. Соединения по изобретению могут содержать по одному или несколько хиральных центров и/или двойных связей и, следовательно, могут существовать в виде стереоизомеров, таких, как изомеры по двойным связям (т.е. геометрические изомеры), энантиомеры или диастереоизомеры. В соответствии с изобретением химические структуры, приведенные в настоящем описании и, следовательно, соединения по изобретению, охватывают все соответствующие энантиомеры и стереоизомеры соединений, т.е. стереоизомерно чистую форму (например, геометрически чистую, энантиомерно чистую или диастереоизомерно чистую) и энантиомерные и стереоизомерные смеси. Энантиомерные и стереоизомерные смеси могут быть разделены на являющиеся их компонентами энантиомеры с применением либо технологии разделения, либо технологии хирального синтеза, которые известны в данной области техники.
Предпочтительные соединения по изобретению являются агонистами RAR, в частности селективными агонистами гамма-RAR, и связываются с рецептором гамма-RAR по меньшей мере в пять раз лучше, чем они связываются рецептором альфа-RAR. Сродство к связыванию агонистов RAR как правило составляет меньше 10 мкМ, предпочтительно меньше 1 мкМ.
В одном варианте n обозначает 1.
В предпочтительном варианте В обозначает -CR7=CR8- или -СН2О-, наиболее предпочтительно -CR7=CR8-, а в частности каждый из R7 и R8 обозначает водородный атом, когда В обозначает транс-СН=СН-, т.е. алкеновый остаток характеризуется Е-стереохимическим строением.
В одном варианте Х обозначает фенил. В другом варианте Х обозначает тиенил. В одном варианте R3 обозначает водородный атом. В другом варианте R3 обозначает гидроксил или оксогруппу. В одном варианте R9 обозначает гетероалкилокси.
В другом варианте R9 обозначает гидроксил. В одном предпочтительном варианте по изобретению предлагаются соединения, соответствующие структурной формуле (II):
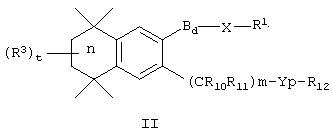
или их фармацевтически приемлемые соли, сольваты или гидраты, где В, d, X, R1, R3, n, R10, R11, m, Y, р и R12 имеют вышеуказанные значения. В предпочтительном варианте m обозначает число от 1 до 4. В одном из вариантов р обозначает 0. В другом варианте р обозначает 1.
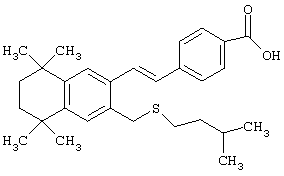
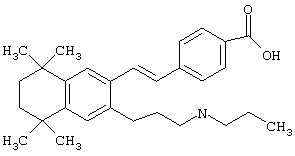
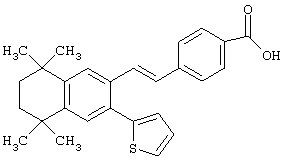
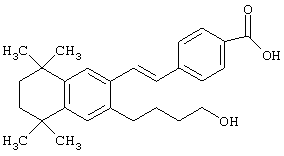
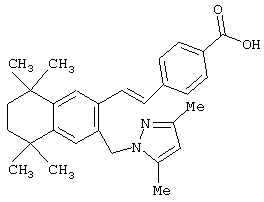
В предпочтительном варианте соединений, отвечающих структурной формуле (II), m обозначает 1, р обозначает 1, а Y обозначает -О-. В предпочтительном варианте R12 обозначает водородный атом, ацил, алкил, карбамоил, циклоалкил, арил, гетероарил или гетероалкил. Примерами этого варианта служат соединения 1, 5 и 15 в таблице 10.
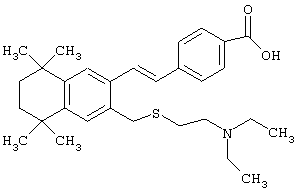
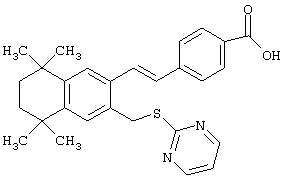
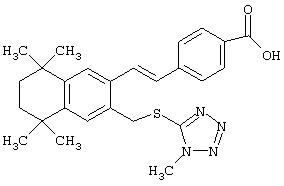
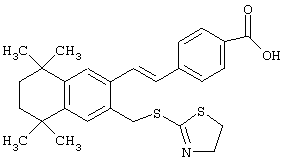
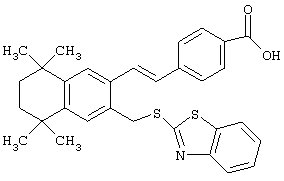
В другом предпочтительном варианте соединений, отвечающих структурной формуле (II), m обозначает 1, р обозначает 1, а Y обозначает -S. В одном варианте R12 обозначает алкил, циклоалкил или гетероалкил. Примерами этого варианта служат соединения 2, 3, 4, 9, 17 и 18 в таблице 1. В другом варианте R12 обозначает гетероарил, гетероарилалкил, гетероциклил или гетероциклилалкил. Примерами этого варианта являются соединения 8, 19, 22, 23, 25, 32, 34 и 35 в таблице 1.
Тем не менее, в другом предпочтительном варианте соединений, отвечающих структурной формуле (II), m обозначает 3, р обозначает 1, а Y обозначает -О-. В предпочтительном варианте R12 обозначает водородный атом, ацил, алкил, карбамоил, циклоалкил, арил, гетероарил или гетероалкил. Примерами этого варианта служат соединения 10, 11 и 12 в таблице 1.
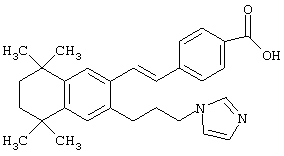
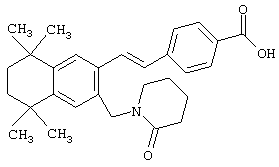
Однако в другом предпочтительном варианте соединений, отвечающих структурной формуле (II), m обозначает 3, р обозначает 1, a Y обозначает -NR13-. В предпочтительном варианте R обозначает ацил, алкил, циклоалкил, арил, гетероарил или гетероциклил. Примером этого варианта является соединение 33 в таблице 1.
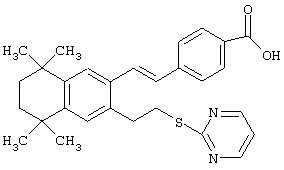
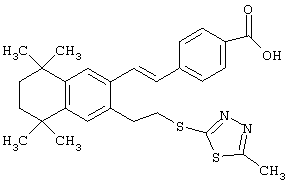
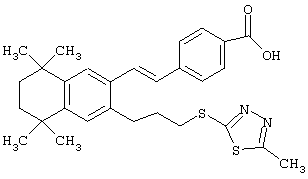
Тем не менее, в другом предпочтительном варианте соединений, отвечающих структурной формуле (II), m обозначает 3, р обозначает 1, а Y обозначает -S-. В предпочтительном варианте R12 обозначает арил, арилалкил, гетероарил, гетероалкил, гетероциклил или гетероциклилалкил. Примерами этого варианта служат соединения 24 и 28 в таблице 1.
Тем не менее, в другом предпочтительном варианте соединений, отвечающих структурной формуле (II), m обозначает 2, р обозначает 1, а Y обозначает -О-. В предпочтительном варианте R12 обозначает водородный атом, ацил, алкил, карбамоил, циклоалкил, арил, гетероарил или гетероалкил. Примером этого варианта является соединение 31 в таблице 1.
Тем не менее, в другом предпочтительном варианте соединений, отвечающих структурной формуле (II), m обозначает 2, р обозначает 1, а Y обозначает -S -. В предпочтительном варианте R12 обозначает арил, арилалкил, гетероарил, гетероалкил, гетероциклил или гетероциклилалкил. Примерами этого варианта служат соединения 26 и 27 в таблице 1.
Тем не менее, в другом предпочтительном варианте соединений, отвечающих структурной формуле (II), m обозначает 4, р обозначает 1, а Y обозначает -(О) -. В предпочтительном варианте R12 обозначает водородный атом, ацил, алкил, карбамоил, циклоалкил, арил, гетероарил или гетероалкил. Примером этого варианта является соединение 51 в таблице 1.
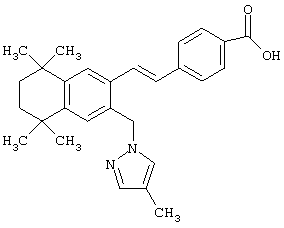
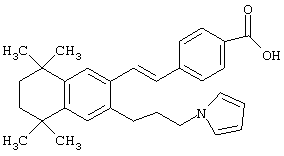
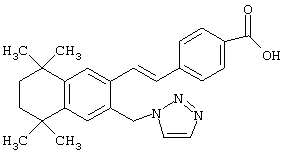
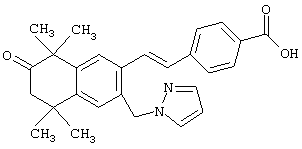
Тем не менее, в еще одном предпочтительном варианте соединений, отвечающих структурной формуле (II), m обозначает 1, а р обозначает 0. В одном варианте R12 обозначает гетероарил, гетероарилалкил, гетероциклил или гетероциклилалкил. Примерами этого варианта служат соединения 6, 7, 44, 45, 47, 50, 53, 54, 55, 138, 139, 143, 146, 149 и 150 в таблице 1. Соединение 6 является особенно предпочтительным представителем вышеупомянутой группы соединений.
В другом варианте R12 обозначает арил, арилалкил, циклоалкил или замещенный циклоалкил. Примерами этого варианта служат соединения 42 и 54 в таблице 1.
Тем не менее, в другом предпочтительном варианте соединений, отвечающих структурной формуле (II), m обозначает 2, а р обозначает 0. В предпочтительном варианте R12 обозначает арил, арилалкил, гетероарил, гетероарилалкил, гетероалкил, гетероциклил или гетероциклилалкил. Примерами этого варианта служат соединения 29, 37, 38, 40, 41, 132, 134, 140, 147 и 152 в таблице 1.
Однако в еще одном предпочтительном варианте соединений, отвечающих структурной формуле (II), m обозначает 3, а р обозначает 0. В предпочтительном варианте R12 обозначает арил, арилалкил, гетероарил, гетероарилалкил, гетероциклил или гетероциклилалкил. Примерами этого варианта являются соединения 30, 36, 46, 52, 130, 131, 135, 141 и 142 в таблице 1.
В другом предпочтительном варианте по изобретению предлагаются соединения, соответствующие структурной формуле (III):
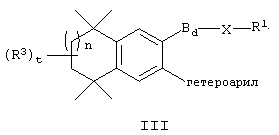
или их фармацевтически приемлемые соли, сольваты или гидраты, где В, d, X, R1,
R3 и n имеют вышеуказанные значения. Примерами предшествующего варианта являются соединения 48, 49, 156 и 157 в таблице 1.
В другом варианте по изобретению предлагаются соединения, отвечающие структурной формуле (IV):
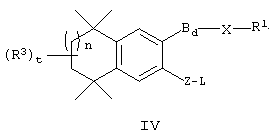
или их фармацевтически приемлемые соли, сольваты или гидраты, где В, d, X, R1, R3, n, Z и L имеют вышеуказанные значения. В одном варианте L обозначает гетероарил или гетероарилалкил. В другом варианте Z обозначает -О- или -S-. Примерами этого варианта служат соединения 154, 155, 159 и 160 в таблице 1.
В другом варианте общей формулы (I) d обозначает 1, В обозначает -CR7=CR8-, a n, R1, R2 R3 и Х имеют вышеуказанные значения. В предпочтительном варианте каждый из R7 и R8 обозначает водородный атом. В одном варианте Х обозначает фенил, необязательно замещенный галогеном. В более конкретном варианте по изобретению предлагаются соединения, соответствующие структурной формуле (V):
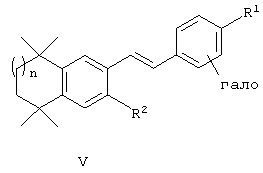
или их фармацевтически приемлемые соли, сольваты или гидраты, где n, R1, R2 и R3 имеют вышеуказанные значения. В другом варианте по изобретению предлагаются соединения, соответствующие структурной формуле (VI):
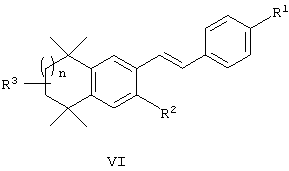
или их фармацевтически приемлемые соли, сольваты или гидраты, где n, R1, R2 и R3 имеют вышеуказанные значения.
В другом варианте Х обозначает гетероарил. В этом варианте по изобретению предлагаются соединения, соответствующие структурной формуле (VII):
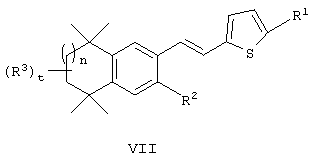
или их фармацевтически приемлемые соли, сольваты или гидраты, где n, R1, R2 и
R3 имеют вышеуказанные значения.
Другой вариант выполнения настоящего изобретения представлен соединениями структурной формулы VIII
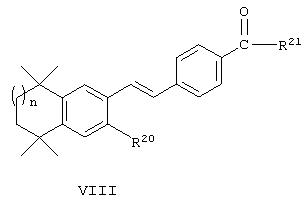
в которой R20 обозначает алкил;
R21 обозначает: (а) гетероалкилоксигруппу, содержащую N, S, О, гетероалкиламиногруппу, содержащую О, или гетероалкилтиогруппу, содержащую О; или
(б) Q-R22, где Q обозначает -NR23 - или -S- (где R23 обозначает водородный атом),
R22 обозначает карбоксиалкил,
а n обозначает 1.
Эти соединения являются пролекарствами соединений формулы VIII, где R21 обозначает гидроксил, и in vivo превращаются в соединения, у которых R21 обозначает гидроксил. Примерами этого варианта служат соединения 56, 57, 58 и 59.
Однако в другом варианте общей формулы (I) d обозначает 1, В обозначает -CR7=CR8-, a n, R1, R2 R3, R7, R8 и Х имеют вышеуказанные значения. В предпочтительном варианте каждый из R7 и R8 обозначает водородный атом.
В более конкретном варианте Х обозначает фенил, необязательно замещенный галогеном.
Во всех вышеописанных вариантах также предпочтительны те соединения, у которых R1 обозначает -CO2H или -СО2-алкил, в частности -СО2Н. Более того, также предпочтительны те варианты, где R3 обозначает водородный атом, а каждый из n и t обозначает 1.
Предпочтительные соединения по изобретению включают те, которые представлены в таблице 1.
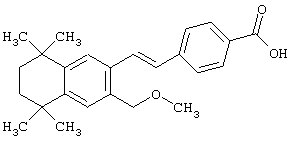
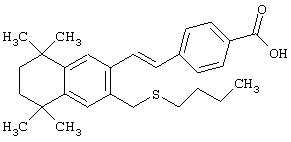
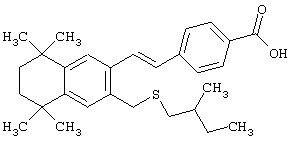
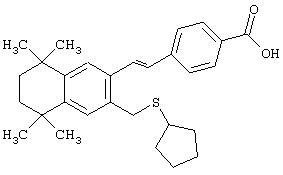
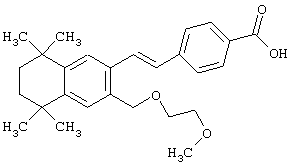
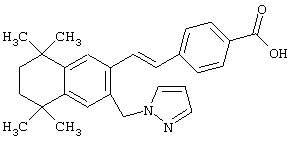
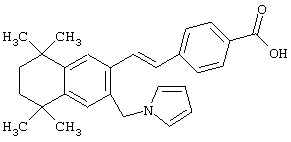
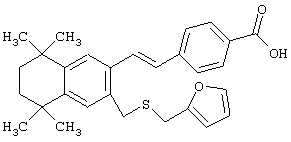
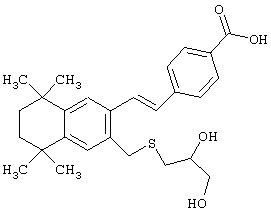
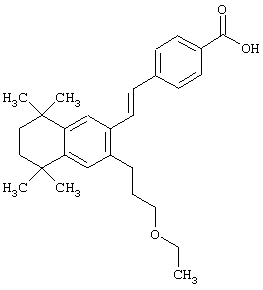
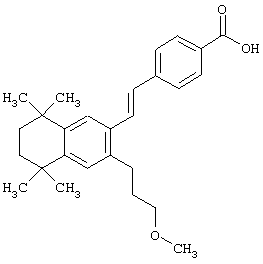
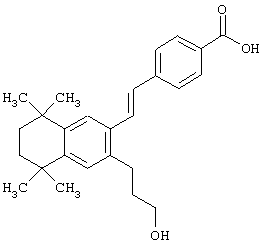
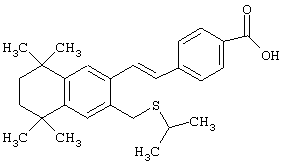
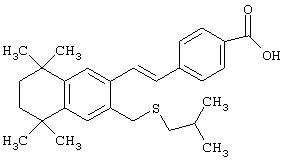
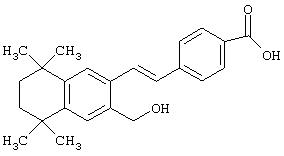

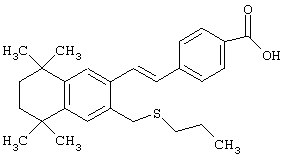
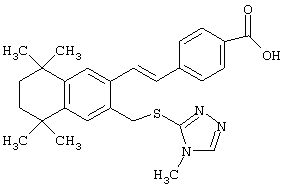
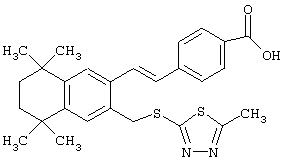
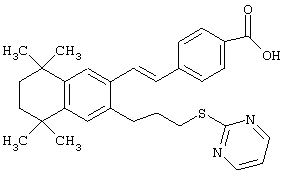
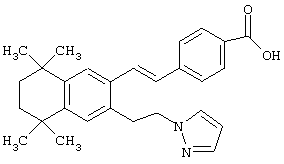
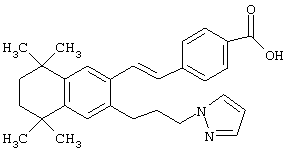
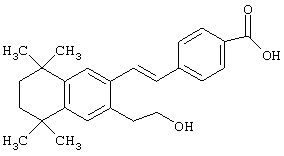
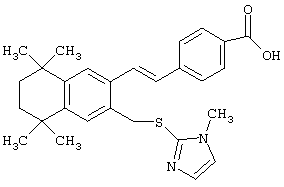
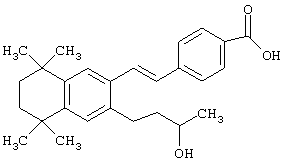
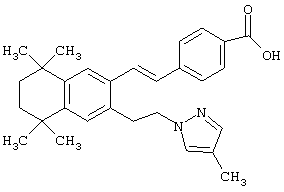
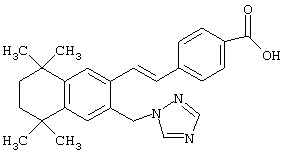
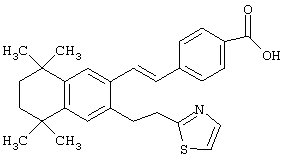
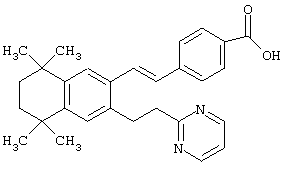
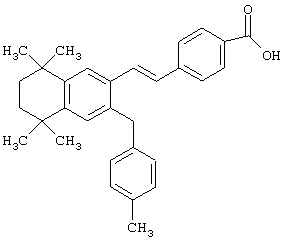
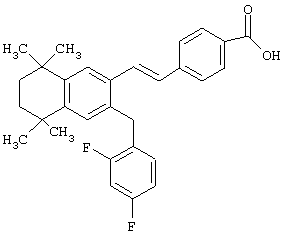
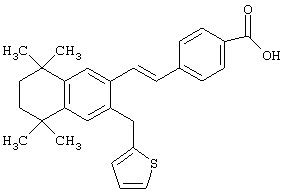
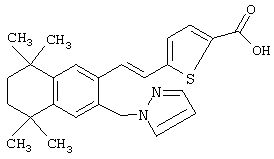
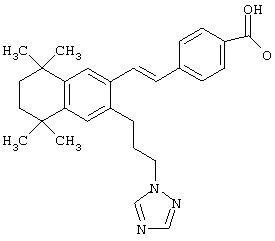
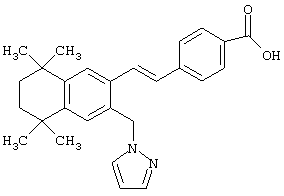
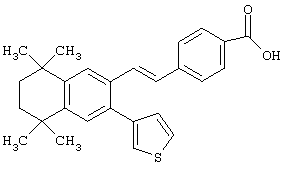
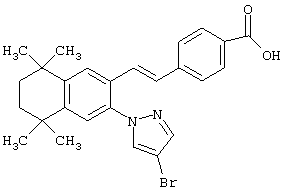
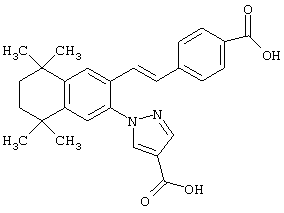
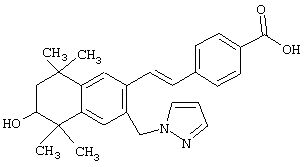
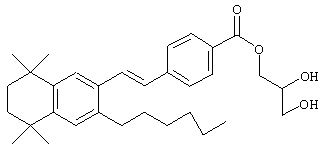
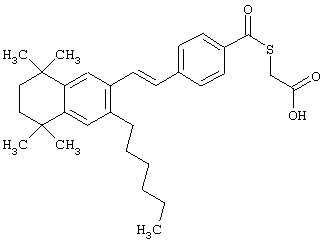
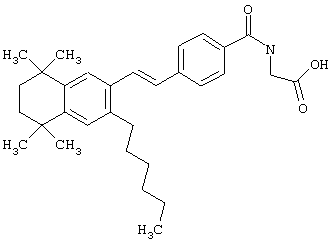
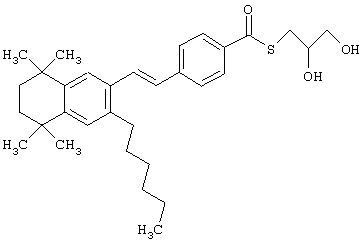
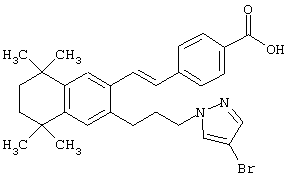
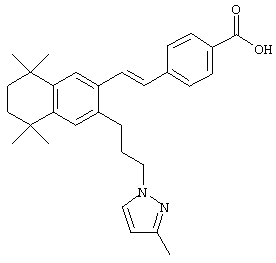
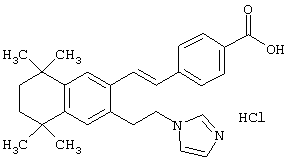
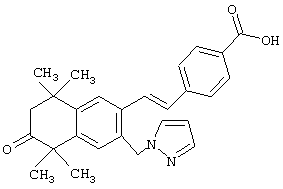
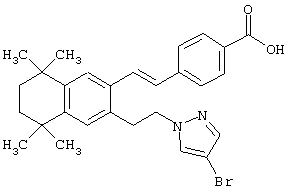
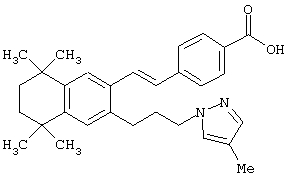
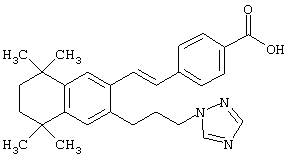
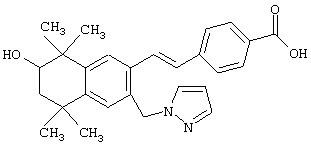
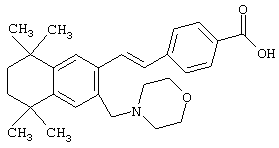
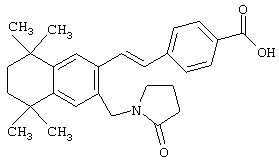
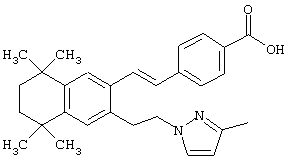
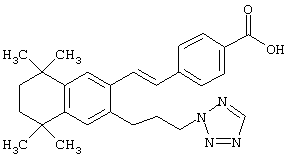
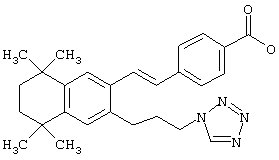
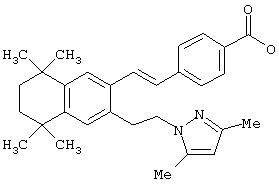
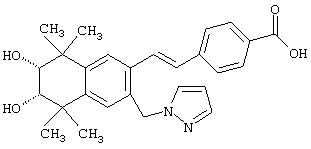
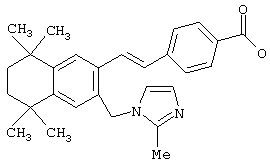
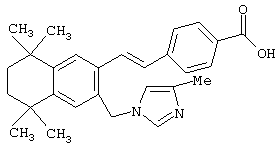
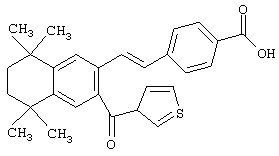
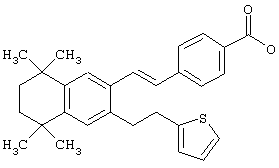
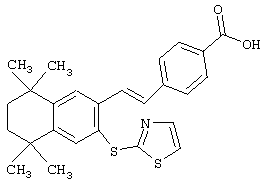
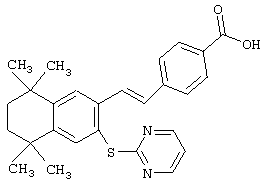
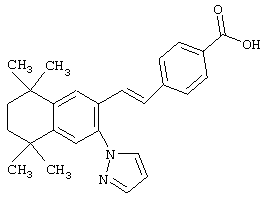
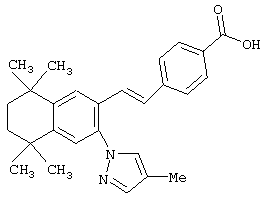
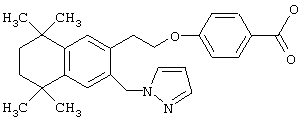
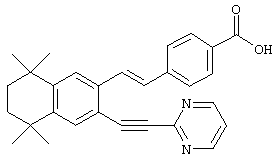
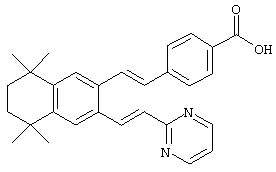
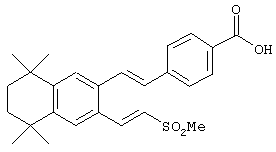
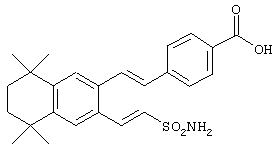
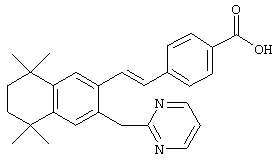
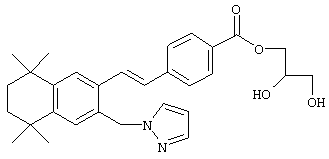
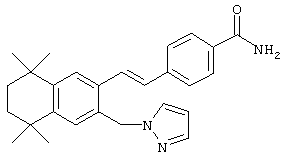
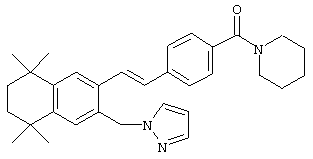
Объем изобретения включает лечение эмфиземы и связанных с ней нарушений, рака и дерматологических нарушений, предпочтительно при одновременном ослаблении или устранении неблагоприятных эффектов, связанных с естественными и синтетическими ретиноидами, когда их используют в терапевтических дозах. Неблагоприятные эффекты, связанные с ретиноидами в терапевтических дозах, включают, хотя ими их список не ограничен, токсические эффекты гипервитаминоза А, такие как головная боль, лихорадка, сухость кожи и слизистых, боли в костях, тошнота и рвота, психические расстройства и желудочно-кишечные расстройства.
Объем настоящего изобретения охватывает также применение соединений по изобретению для лечения или профилактики некоторых хронических обструктивных нарушений дыхательных путей, особенно хронической обструктивной легочной болезни, включая хронический бронхит, эмфизему и астму у млекопитающих, преимущественно у людей, которые курят или курили сигареты. В предпочтительном варианте объем изобретения включает лечение или профилактику панлобулярной эмфиземы, центральнодолевой эмфиземы или эмфиземы отдаленной от центра доли у млекопитающих с использованием терапевтически эффективных доз соединений по изобретению.
В одном из вариантов выполнения настоящего изобретения его объем охватывает применение соединений по изобретению для лечения или профилактики эмфиземы. Далее, объем настоящего изобретения включает применение фармацевтических композиций соединений изобретения для лечения или профилактики эмфиземы. Кроме того, объем изобретения охватывает применение электрогидродинамических аэрозольных устройств, обычных аэрозольных устройств и распылителей для введения композиций соединений по изобретению в легкое млекопитающего, страдающего эмфиземой или подвергающегося опасности ее возникновения.
Объем изобретения включает системное применение, а также местное применение соединений по изобретению или сочетание того и другого. Любое из них или и то, и другое можно осуществлять введением перорально, через слизистые оболочки или парентерально. Как упомянуто выше, объем изобретения охватывает средство доставки соединений по изобретению непосредственно в легкое распылителем, ингалятором или другими известными устройствами.
Объем изобретения охватывает также способ лечения эмфиземы сочетанием соединений по изобретению с одним или несколькими дополнительными терапевтическими средствами, такими как прекращение курения (когда это приемлемо), бронхолитики, антибиотики, терапия кислородом и т.п.
По другому варианту объектом настоящего изобретения являются способы профилактики эмфиземы у человека в случае опасности эмфиземы путем введения в организм некоторого количества соединения по изобретению или его пролекарства, которого достаточно для профилактики эмфиземы. По еще одному варианту объем настоящего изобретения охватывает фармацевтические композиции для профилактики эмфиземы у человека в случае опасности эмфиземы путем введения некоторого количества соединения по изобретению или его пролекарства, в фармацевтически приемлемом носителе, которого достаточно для профилактики эмфиземы.
В другом варианте объем настоящего изобретения охватывает применение соединений по изобретению для лечения или профилактики рака. Далее, объем настоящего изобретения включает применение фармацевтических композиций соединений по изобретению для лечения или профилактики рака. Кроме того, объем настоящего изобретения охватывает применение электрогидродинамических аэрозольных устройств, обычных аэрозольных устройств и распылителей для доставки композиций на основе соединений по изобретению в легкое млекопитающего, страдающего раковым заболеванием или в случае его опасности. Раковые образования включают твердые опухоли, такие как опухоли молочной железы, легкого, простаты и печени, промиелоцитарные лейкемии, предзлокачественные изменения слизистой оболочки рта, языка, гортани, пищевода, мочевого пузыря, шейки матки и толстой кишки.
Объемом изобретения охватывается также способ лечения рака сочетанием соединений по изобретению с одним или несколькими дополнительными терапевтическими средствами. Дополнительные терапевтические средства лечения включают интеркалирующие DNA средства, такие как цис-платина и иммунотерапевтические агенты, такие как гамма-интерферон и другие цитокины.
В другом варианте объем настоящего изобретения охватывает способы профилактики рака у человека в случае опасности рака введением некоторого количества соединения по изобретению или его пролекарства, которого достаточно для профилактики рака. В еще одном варианте объем настоящего изобретения охватывает фармацевтические композиции для профилактики рака у человека в случае опасности рака введением некоторого количества соединения по изобретению или его пролекарства в фармацевтически приемлемом носителе, которого достаточно для профилактики рака.
В еще одном варианте объем настоящего изобретения охватывает применение соединений по изобретению для лечения или профилактики дерматологических нарушений. Далее, объем настоящего изобретения охватывает применение фармацевтических композиций на основе соединений по изобретению для лечения или профилактики дерматологических нарушений. Дерматологические нарушения включают прыщи, псориаз, фотоповреждения кожи и другие дерматозы, сопровождаемые ороговением. Охватываются также заживление ран, например, порезов, ожогов, послеоперационных ран и других ран, связанных с кожной травмой.
Объем изобретения охватывает также способ лечения дерматологических нарушений сочетанием соединений по изобретению с одним или несколькими дополнительными терапевтическими средствами и т.п.
По другому варианту объем настоящего изобретения охватывает способы профилактики дерматологических нарушений у человека в случае опасности дерматологических нарушений введением некоторого количества соединения по изобретению или его пролекарства, которого достаточно для профилактики дерматологических нарушений. Объем настоящего изобретения охватывает, наконец, фармацевтические композиции для профилактики эмфиземы у человека в случае опасности дерматологических нарушений введением некоторого количества соединения по изобретению или его пролекарства в фармацевтически приемлемом носителе, которого достаточно для профилактики дерматологических нарушений.
Объектом настоящего изобретения является также применение соединений формулы I при приготовлении лекарственных средств для лечения или профилактики эмфиземы, рака и дерматологических нарушений. Всякий раз, когда в описании настоящего изобретения упоминается способ профилактики или лечения вышеупомянутых болезней, объектом настоящего изобретения является также применение соединений формулы I при приготовлении лекарственных средств для лечения или профилактики вышеупомянутых болезней.
Другой объект изобретения охватывает способ лечения эмфиземы у млекопитающего, который включает введение млекопитающему, нуждающемуся в таком лечении, терапевтически эффективного количества соединения по изобретению или его пролекарства. В одном варианте эмфизема является панлобулярной эмфиземой, центродолевой эмфиземой или эмфиземой отдаленной от центра доли.
В предпочтительном варианте терапевтически эффективное количество соединения по изобретению или его пролекарства при лечении эмфиземы находится в пределах от примерно 0,1 мкг/день до примерно 30,0 мг/день, более предпочтительно в пределах от примерно 1,0 мкг/день до примерно 1,0 мг/день. В одном варианте, преимущественно при пероральном введении, терапевтически эффективное количество соединения по изобретению или его пролекарства находится в пределах от примерно 10,0 мкг/день до примерно 30 мг/день, предпочтительно от 30,0 до примерно 300,0 мкг/день. По другому варианту, преимущественно при введении ингаляцией, терапевтически эффективное количество соединения по изобретению или его пролекарства находится в пределах от примерно 0,1 до примерно 100 мкг/день, более предпочтительно в пределах от примерно 10,0 до примерно 100 мкг/день, наиболее предпочтительно в пределах от примерно 1,0 до примерно 30,0 мкг/день.
Данный объект изобретения охватывает способ лечения эмфиземы у млекопитающего посредством восстановления альвеол у млекопитающего. В предпочтительном варианте млекопитающим является человек. В предпочтительном варианте такой человек в прошлом или в настоящее время - курильщик сигарет. В другом предпочтительном варианте для введения терапевтически эффективного количества соединения по изобретению или его пролекарства применяют электрогидродинамическое аэрозольное устройство или распылительное устройство, или обычное аэрозольное устройство.
С другой стороны, объем изобретения охватывает фармацевтическую композицию для лечения млекопитающего, страдающего эмфиземой, включающую определенное количество соединения по изобретению или его пролекарства в фармацевтически приемлемом носителе, причем количества этого соединения достаточно для ослабления одного симптома эмфиземы. В одном из вариантов эмфизема представляет собой панлобулярную эмфизему, центродолевую эмфизему или эмфизему отдаленной от центра доли. В предпочтительном варианте, млекопитающим является человек. В предпочтительном варианте такой человек в прошлом или в настоящее время - курильщик сигарет.
Главные признаки эмфиземы включают, хотя ими их список не ограничен, хроническую одышку, хронический кашель, окраску кожи, вызванную недостатком кислорода, одышку при минимальном физическом напряжении и хрипы. Дополнительные признаки, которые могут быть связаны с эмфиземой, включают, хотя ими их список не ограничен, нарушения зрения, головокружение, временную остановку дыхания, беспокойство, одутловатость, усталость, бессонницу и потерю памяти.
В предпочтительном варианте количество соединения по изобретению или его пролекарства в фармацевтической композиции находится в пределах от примерно 0,1 мкг до примерно 30,0 мг, более предпочтительно в пределах от примерно 1,0 мкг до примерно 1,0 мг, наиболее предпочтительно в пределах от примерно 100,0 до примерно 300,0 мкг.
В одном варианте фармацевтически приемлемый носитель пригоден для электрогидродинамического аэрозольного устройства, распылительного устройства или аэрозольного устройства. В одном предпочтительном варианте этот фармацевтически приемлемый носитель представляет собой жидкость, такую как вода, спирт, полиэтиленгликоль и перфторуглерод. Количество соединения по изобретению или его пролекарства в фармацевтической композиции по этому предпочтительному варианту находится в пределах от примерно 0,1 мкг до примерно 1,0 мг, более предпочтительно в пределах от примерно 1,0 до примерно 100,0 мкг, наиболее предпочтительно в пределах от примерно 50,0 до примерно 150,0 мкг.
Другой объект изобретения охватывает способ лечения эмфиземы и связанных с ней нарушений введением композиции на основе соединения по изобретению или его пролекарства в легкие млекопитающего. Предпочтительным млекопитающим является человек, в более предпочтительном варианте такой человек в прошлом или в настоящее время - курильщик сигарет. В одном варианте эту композицию вводят в легкие млекопитающего с помощью распылительного устройства. Во втором варианте композицию вводят в легкие млекопитающего с помощью аэрозольного устройства. В третьем варианте композицию вводят в легкие млекопитающего с помощью электрогидродинамического аэрозольного устройства.
В служащем примером варианте композиция представляет собой фармацевтическую композицию на основе соединения по изобретению. В предпочтительном варианте количество соединения по изобретению или его фармацевтически приемлемой соли, гидрата, сольвата или пролекарства в такой фармацевтической композиции находится в пределах от примерно 1,0 мкг до примерно 10,0 мг, более предпочтительно в пределах от примерно 10,0 мкг до примерно 1,0 мг, наиболее предпочтительно в пределах от примерно 50,0 до примерно 150,0 мкг. В одном предпочтительном варианте фармацевтически приемлемый носитель представляет собой жидкость, такую как вода, спирт, полиэтиленгликоль и перфторуглерод. В другом предпочтительном варианте в композицию добавляют материал, который изменяет аэрозольные свойства такой композиции. В предпочтительном варианте этот материал представляет собой спирт, гликоль, полигликоль или жирную кислоту.
В еще одном варианте объем изобретения включает способ лечения эмфиземы, в котором соединение по изобретению применяют в сочетании с одним или несколькими дополнительными терапевтическими средствами. Такие дополнительные терапевтические средства включают, хотя ими их список не ограничен, прекращение курения, антибиотики, бронхолитики и кислородную терапию. В предпочтительном варианте фармацевтическую композицию на основе соединения по изобретению используют в сочетании с другими терапевтическими средствами.
В еще одном варианте по настоящему изобретению предлагается способ профилактики эмфиземы у человека в случае опасности эмфиземы введением некоторого количества соединения по изобретению или его пролекарства, достаточного для профилактики эмфиземы. В предпочтительном варианте такой человек в прошлом или в настоящее время - курильщик сигарет.
В другом варианте по настоящему изобретению предлагается фармацевтическая композиция, которая предотвращает эмфизему у человека в случае опасности ее возникновения. Эта композиция включает некоторое количество соединения по изобретению или его пролекарства и фармацевтически приемлемый носитель, достаточное для профилактики эмфиземы.
Другой объект изобретения охватывает способ лечения рака у млекопитающего, который включает введение млекопитающему, нуждающемуся в таком лечении, терапевтически эффективного количества соединения по изобретению или его пролекарства. В предпочтительном варианте рак является заболеванием эпителиального происхождения; такие заболевания включают, хотя ими их список не ограничен, рак молочной железы, рак кожи, рак толстой кишки, опухоли живота, рак гортани и рак легкого.
В предпочтительном варианте терапевтически эффективное количество соединения по изобретению или его пролекарства при лечении рака находится в пределах от примерно 50 мкг/день до примерно 500 мг/день, более предпочтительно в пределах от примерно 300 мкг/день до примерно 30 мг/день. В одном варианте, преимущественно при пероральном введении, терапевтически эффективное количество соединения по изобретению или его пролекарства находится в пределах от примерно 3 до примерно 120 мг/день. По другому варианту, преимущественно при введении ингаляцией, терапевтически эффективное количество соединения по изобретению или его пролекарства находится в пределах от примерно 50 до примерно 500 мкг/день, более предпочтительно в пределах от примерно 50 до примерно 150 мкг/день.
В предпочтительном варианте млекопитающим является человек. В другом предпочтительном варианте для введения в организм терапевтически эффективного количества соединения по изобретению или его пролекарства применяют электрогидродинамическое аэрозольное устройство или распылительное устройство, или обычное аэрозольное устройство.
С другой стороны, объем изобретения охватывает фармацевтическую композицию для лечения млекопитающего, страдающего раковым заболеванием, включающую определенное количество соединения по изобретению или его пролекарства в фармацевтически приемлемом носителе, причем количества этого соединения достаточно для ослабления одного симптома рака. В предпочтительном варианте рак представляет собой заболевание эпителиальной природы, включая, хотя ими их список не ограничен, рак молочной железы, кожный рак, рак толстой кишки, желудочные опухоли, рак гортани и рак легкого. В предпочтительном варианте млекопитающим является человек.
В предпочтительном варианте количество соединения по изобретению или его пролекарства в фармацевтической композиции находится в пределах от примерно 250 мкг до примерно 500 мг, более предпочтительно в пределах от примерно 2,5 до примерно 100 мкг, наиболее предпочтительно в пределах от примерно 10 до примерно 50 мкг.
В одном варианте фармацевтически приемлемый носитель пригоден для электрогидродинамического аэрозольного устройства, распылительного устройства или обычного аэрозольного устройства. В одном предпочтительном варианте этот фармацевтически приемлемый носитель представляет собой жидкость, такую, как вода, спирт, полиэтиленгликоль или перфторуглерод. Количество соединения по изобретению или его пролекарства в фармацевтической композиции по этому предпочтительному варианту находится в пределах от примерно 50 мкг до примерно 1,5 мг, более предпочтительно в пределах от примерно 150 мкг до примерно 1,5 мг, наиболее предпочтительно в пределах от примерно 150 до примерно 300 мкг.
С другой стороны, объем изобретения охватывает способ лечения рака введением композиции на основе соединения по изобретению или его пролекарства в легкие млекопитающего. В предпочтительном варианте млекопитающим является человек, более предпочтительно человек, страдающий раком легких. В одном варианте композицию вводят в легкие млекопитающего с помощью распылительного устройства. Во втором варианте композицию вводят в легкие млекопитающего с помощью обычного аэрозольного устройства. В третьем варианте композицию вводят в легкие млекопитающего с помощью электрогидродинамического аэрозольного устройства.
В служащем примером варианте композиция представляет собой фармацевтическую композицию соединения по изобретению. В предпочтительном варианте количество соединения по изобретению или его пролекарства в такой фармацевтической композиции находится в пределах от примерно 50 мкг до примерно 1,5 мг, более предпочтительно в пределах от примерно 50 до примерно 1,5 мкг, наиболее предпочтительно в пределах от примерно 100 до примерно 300 мкг. В одном предпочтительном варианте фармацевтически приемлемый носитель представляет собой жидкость, такую, как вода, спирт, полиэтиленгликоль и перфторуглерод. В другом предпочтительном варианте в композицию добавляют материал, который изменяет аэрозольные свойства такой композиции. В предпочтительном варианте этот материал представляет собой спирт, гликоль, полигликоль или жирную кислоту.
По еще одному варианту объем настоящего изобретения охватывает способ лечения рака, в котором соединение по изобретению применяют в сочетании с одним или несколькими дополнительными терапевтическими средствами. Такие дополнительные терапевтические средства включают, хотя ими их список не ограничен, химиотерапию, лучевую терапию или хирургию. В предпочтительном варианте фармацевтическую композицию на основе соединения по изобретению используют в сочетании с другими терапевтическими средствами.
Тем не менее в другом варианте по настоящему изобретению предлагается способ профилактики рака у человека в случае опасности рака (например, у курильщиков, рабочих асбестовых и урановых предприятий) введением некоторого количества соединения по изобретению или его пролекарства, достаточного для профилактики рака. Примеры предзлокачественных и предраковых патологических изменений или опухолей, которые могут быть предотвращены соединениями по изобретению, включают, хотя ими их список не ограничен, актинический и мышьяковый кератозы, нарушения роста и папилломы слизистых оболочек и предраковые изменения мочевого пузыря.
В другом варианте по настоящему изобретению предлагается фармацевтическая композиция, которая предотвращает рак у человека в случае опасности рака. Эта композиция включает некоторое количество соединения по изобретению или его пролекарства и фармацевтически приемлемый носитель, достаточное для профилактики рака.
В другом варианте объект изобретения охватывает способ лечения дерматологических нарушений у млекопитающего, который включает введение млекопитающему, нуждающемуся в таком лечении, терапевтически эффективного количества соединения по изобретению или его пролекарства. В предпочтительном варианте такие дерматологические нарушения включают, хотя ими их список не ограничен, повреждение кожи, вызванное светом и возрастом, хирургические раны, ожоги, раны, вызванные кожной травмой, прыщи и псориаз.
В предпочтительном варианте терапевтически эффективное количество соединения по изобретению или его пролекарства при лечении дерматологических нарушений находится в пределах от примерно 5 мкг/день до примерно 50 мг/день, более предпочтительно в пределах от примерно 50 мкг/день до примерно 5 мг/день. Мягчительные средства местного применения (для кожи) как правило представляют собой кремы, лосьоны или мази концентрацией примерно от 1 до 0,005%, предпочтительно от 0,5 до 0,01%, наиболее предпочтительно 0,05 до 0,01%.
Соединения по изобретению, отвечающие формулам (I-VII), могут быть получены осуществлением методов синтеза, проиллюстрированных на схемах с 1 по 7, и методов, описанных в литературе, посвященной данной области техники [Douget и др.. Quant. Struct. Act.Relat., 18, 107 (1999)], и в литературных источниках, которые приведены в настоящем описании и которые включены в настоящее описание в качестве ссылок. Исходные материалы, которые могут быть использованы для получения соединений по изобретению, и промежуточные продукты для их получения технически доступны или могут быть получены по хорошо известным методам синтеза.
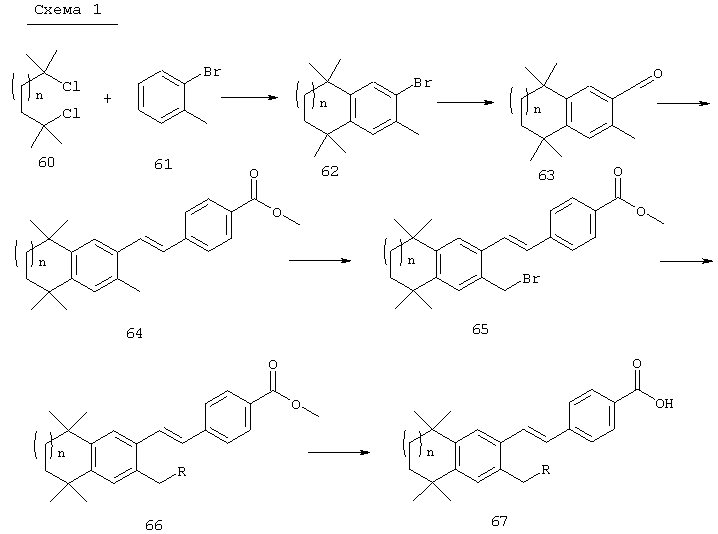
Соединения 67 формулы (I), где n обозначает 0, 1 или 2, m обозначает 1, а R обозначает алкокси, алкилтио, гетероарил, гетероциклил, амино-, алкиламино и т.д., могут быть получены так, как представлено на схеме 1. Бромзамещенные 5,5,8,8-тетраметил-5,6,7,8-тетрагидронафталины 62 и соответствующие пяти- и семичленные циклические аналоги могут быть синтезированы осуществлением ряда методов, известных специалисту в данной области техники. В предпочтительном варианте алкилированием по Фриделю-Крафтсу 2-бромтолуола 61 2,4-дихлор-2,4-диметилпентаном, 2,5-дихлор-2,5-диметилгексаном или 2,6-дихлор-2,6-диметилгептаном 60 получают соединения 62. Арилбромиды 62 могут быть гомологизированы до альдегидов 64 заменой атома галогена атомом металла (т.е. с помощью н-бутиллития) с получением промежуточного литийорганического соединения, которое затем "гасят" N-формилпиперидином. По другому варианту альдегиды 63 могут быть получены гомологизацией бромидов 62 (т.е. Cu(I)CN) до цианового соединения, которое можно восстанавливать (т.е. диизобутилалюминийгидрид). Другие методы синтеза с целью превращения бромидов 62 в альдегиды 63 для специалиста в данной области техники очевидны.
Для получения Е олефинов 64 можно прибегнуть к олефинированию альдегидов 63 по Хорнеру-Эммонсу соответствующим фосфонатным эфиром. Соответствующие Z олефины могут быть получены обычными реакциями Виттига с последующим, если необходимо, разделением. Бромированием соединений 64 (т.е. N-бромсукцинимид, бензоилпероксид и свет) получают бензилбромиды 65. Эти бромиды могут быть замещены азот-, серу- или кислородсодержащими нуклеофилами с получением соответствующих замещенных сложных эфиров 66, которые могут быть гидролизованы (с помощью кислоты или основания) с получением кислот 67. Кислоты 67 могут быть этерифицированы осуществлением хорошо известных методов с получением большого числа сложных эфиров.
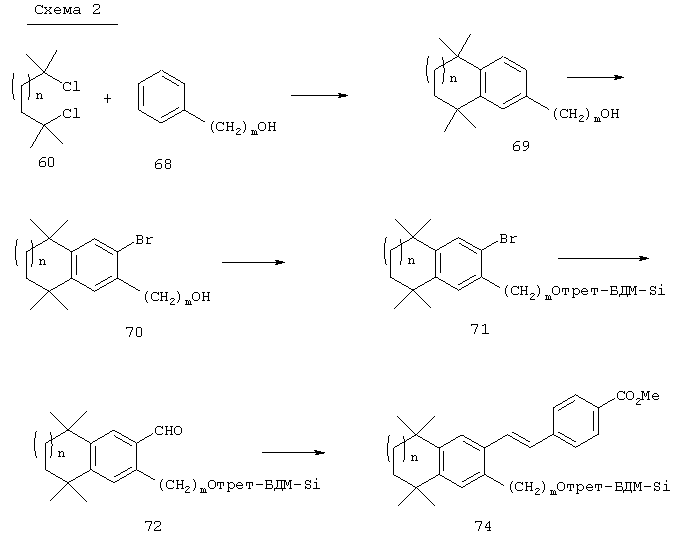
Соединения 78 формулы (I), где n обозначает 0, 1 или 2, m обозначает число от 2 до 10, a R12 обозначает алкокси, алкилтио, гетероарил, гетероциклил, амино-, алкиламиногруппу и т.д., могут быть получены так, как представлено на схеме 2. Гидроксиалкилзамещенные 5,5,8,8-тетраалкил-5,6,7,8-тетрагидронафталины 69 могут быть легко получены реакцией по Фриделю-Крафтсу 2,4-дихлор-2,4-диметилпентана, 2,5-дихлор-2,5-диметилгексана или 2,6-дихлор-2,6-диметилгептана 60 с гидроксиалкилбензолами 68. Бромированием гидроксиалкил-5,5,8,8-тетраалкил-5,6,7,8-тетрагидронафталинов 69 получают арилбромиды 70. Гидроксильную группу соединения 70 можно защитить (т.е. трет-бутилдиметилсилилхлорид и имидазол) с получением соединений 71. Бромиды 71 можно превращать в альдегиды 72 в одну стадию (т.е. заменой атома галогена атомом металла с использованием н-бутиллития с последующей обработкой N-формилпиперидином). По другому варианту альдегиды 72 могут быть получены из бромидов 70 по двухстадийному методу (т.е. Cu(I)CN с получением нитрила и восстановлением диизобутилалюминийгидридом). Другие методы превращения бромидов 70 в альдегиды 72 находятся в компетенции специалистов в данной области техники.
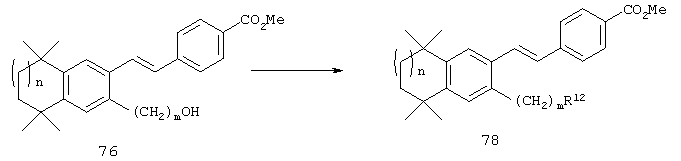
Для получения Е олефинов 74 можно прибегнуть к олефинированию альдегидов 72 по Хорнеру-Эммонсу соответствующим фосфонатным эфиром. Защитная группа у соединения 74 может быть удалена (т.е. водный тетрабутиламмонийфторид) с получением спиртов 76. В предпочтительном варианте спирты 76 можно подвергать превращению реакцией Мицонобу (т.е. алкилтиолы, трифенилфосфин и диизопропилазодикарбоксилат) в тиоловые аналоги 78 (R обозначает алкилтио). По другому варианту гидроксильная функциональная группа соединений 76 может быть активирована превращением в мезилат (MsCl, Et3N) с последующими реакциями замещения с азот- или кислородсодержащими нуклеофилами с получением соединений 78 (R обозначает алкокси, амино-, алкиламино-, диалкиламиногруппу и т.д.). Другие методы превращения спиртов 76 в соединения по изобретению специалистам в данной области техники известны. Для получения свободных кислот соединений 78 можно прибегнуть к гидролизу сложных эфиров.
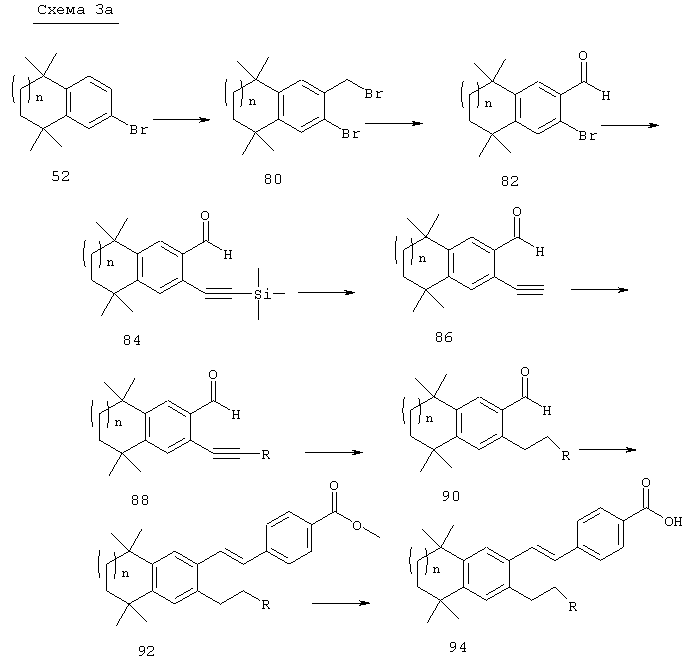
Для соединений формулы I, у которых m обозначает 2, альтернативный метод проиллюстрирован на схеме 3. Бромзамещенные 5,5,8,8-тетраметил-5,6,7,8-тетрагидронафталины 62, представленные на схеме 1, можно превращать в бромальдегиды 82 бензилбромированием N-бромсукцинимидом и бензоилпероксидом, получая соединение 80, с последующей обработкой 2-нитропропаном и гидридом натрия. Обработкой бромальдегида 82 триметилсилилацетиленом, дихлорбис(трифенилфосфин)палладием(II), иодидом одновалентной меди и триэтиламином получают силилированные ацетиленовые соединения 84. Удалением триметилсилильной группы основанием получают соединение 86, после чего проводят реакцию с галоидированными гетероароматическими соединениями, дихлорбис(трифенилфосфин)палладием(II), одновалентным иодидом меди и триэтиламином с получением ацетиленовых гетероароматических промежуточных продуктов 88. В результате каталитической гидрогенизации ацетиленовых соединений 88 получают насыщенные гетероароматические промежуточные продукты 90. Олефинированием соединения 90 по Хорнеру-Эммонсу соответствующим фосфонатным эфиром получают Е олефины 92. Затем этот сложный эфир может быть гидролизован с получением ретиноидных аналогов 94.
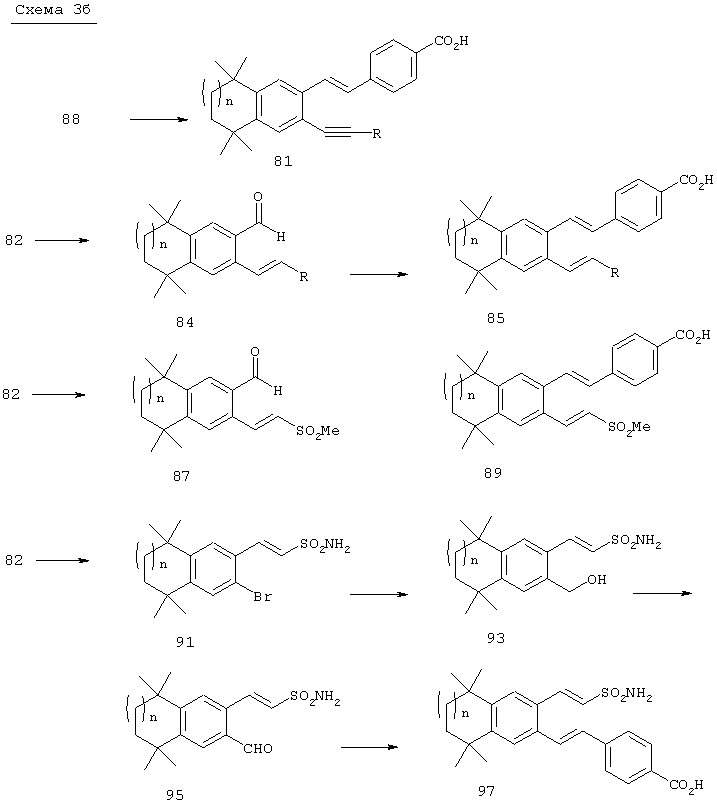
Что касается соединений формулы I, где Z обозначает ацетилен, a L обозначает гетероарил, то, как показано на схеме 3б, промежуточный продукт 88 может быть обработан в условиях олефинирования по Хорнеру-Эммонсу соответствующим фосфонатом с получением Е олефинов и в дальнейшем гидролизован до образования ретиноидных аналогов 81.
Что касается соединений формулы I, где Z обозначает олефин, a L обозначает гетероарил, то промежуточный продукт 82 может быть обработан транс-1,2-бис(три-н-бутилстаннил)этиленом и тетракис(трифенилфосфин)палладием в толуоле при кипячении с обратным холодильником с последующими добавлением галоидированных гетероароматических соединений и получением олефина 83. В результате олефинирования по Хорнеру-Эммонсу продукта 83 соответствующим фосфонатным эфиром с последующим гидролизом получают ретиноидные аналоги 85. По другому варианту, когда R2 обозначает винилсульфон, обработкой промежуточного продукта 82 метилвинилсульфоном, тетракис(трифенилфосфин)палладием и ТЭА в ДМФ получают винилсульфоновый промежуточный продукт 87. Олефинированием с последующим гидролизом получают ретиноидные аналоги 89. По другому варианту, когда R2 обозначает винилсульфонамид, в результате обработки промежуточного продукта 82 трет-бутил[(дифенилфосфорил)метил]сульфонилкарбаматом и NaH в ДМФ образуется винилсульфонамидный промежуточный продукт 91. Обработкой продукта 91 трибутилстаннилметаном и тетракис(трифенилфосфин)палладием в диоксане получают гидроксиметиловый промежуточный продукт 93. Окислением продукта 93 1,1,1-триацетокси-1,1,1-1,1-дигидро-1,2-бензииодоксол-З (1Н)-оном получают альдегид 95, а олефинированием с последующим гидролизом получают ретиноидный аналог 97.
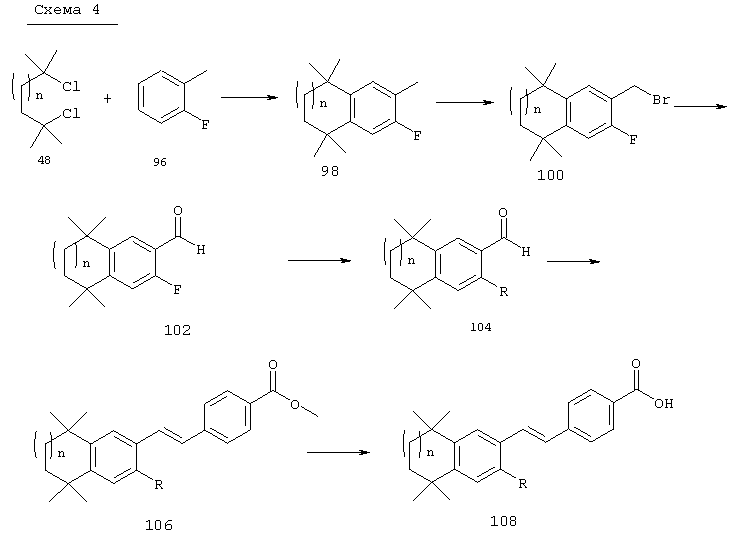
Соединения 108, у которых R обозначает азотсодержащий гетероароматический радикал с атомом азота, который непосредственно присоединен к ароматическому кольцу, или тиогетероароматический радикал с атомом серы, который непосредственно присоединен к ароматическому кольцу, могут быть получены по методу, представленному на схеме 4. Фторзамещенные 5,5,8,8-тетраметил-5,6,7,8-тетрагидронафталины 98 могут быть получены реакцией по Фриделю-Крафтсу 2,3-дихлор-2,5-диметилгексана с 2-фтортолуолом 96. Фторсодержащие альдегиды 102 могут быть получены бензилбромированием продукта 98 N-бромсукцинимидом и бензоилпероксидом с получением продукта 100 и последующей обработкой бромида 100 анионом 2-нитропропана. В результате прямого замещения фторной группы продукта 102 азотсодержащим гетероароматическим соединением в основных условиях (карбонат калия) в апротонном растворителе с нагреванием получают промежуточные продукты 104. Олефинированием продукта 104 по Хорнеру-Эммонсу получают сложноэфирные промежуточные продукты 106, а вследствие гидролиза сложного эфира образуются продукты 108 с ароматическим кольцом, замещенным азотсодержащим гетероароматическим радикалом.
По другому варианту обработкой тиогетероароматических соединений гидридом натрия в полярном апротонном растворителе с последующим добавлением фторированного альдегида 102 получают промежуточные продукты 104 с тиогетероароматической группой, которая непосредственно присоединена к ароматическому кольцу. Как и в предыдущем случае, в результате олефинирования продукта 104 по Хорнеру-Эммонсу получают сложноэфирный промежуточный продукт 106, после чего следует гидролиз сложного эфира с образованием продуктов 108 с ароматическим кольцом, замещенным серусодержащим гетероароматическим радикалом.
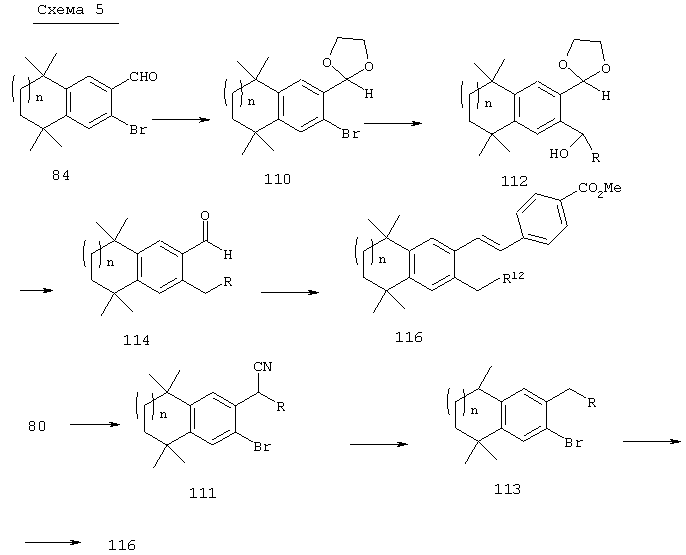
Соединения 116, у которых m обозначает 1, а R12 обозначает гетероарильную группу, связанную через углеродный атом, или арильную группу, могут быть получены в соответствии с методом, представленным на схеме 5. Бромальдегидные промежуточные продукты 84, описанные выше, могут быть защищены в форме ацеталей 110. Обработкой продукта 110 металлорганическим реагентом, таким, как н-BuLi, с последующим добавлением гетероарильных альдегидов получают спирты 112. Каталитическим гидрогенолизом с катализаторами на основе благородных металлов в присутствии водорода удаляют как гидроксильную группу, так и ацетальную защитную группу с получением альдегида 114. Олефинированием по Хорнеру-Эммонсу соответствующим фосфонатным эфиром с последующим гидролизом сложного эфира получают соединение 116.
По другому варианту обработкой продукта 84 арилцинковым реагентом в условиях катализа палладием после удаления в кислых условиях ацетальной группы получают альдегид 114, у которого R обозначает замещенный арил. В результате олефинирования продукта 114 по Хорнеру-Эммонсу соответствующим фосфонатным эфиром с последующим гидролизом сложного эфира получают соединение 116.
По другому варианту дибромированный промежуточный продукт 80 может быть обработан NaCN, после чего проводят реакцию с металлорганическим гетероароматическим реагентом с получением продукта 111. Гидролизом продукта 111 до соответствующей кислоты с последующим декарбоксилированием получают промежуточный продукт 113. В результате двойной реакции Хека (Heck), вначале с триметоксивинилсиланом, а затем с метил-4-бромбензоатом, с последующим гидролизом получают ретиноидные аналоги 116.
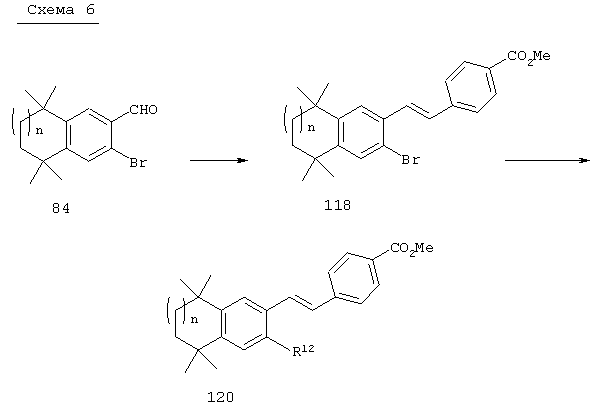
Соединения 120, у которых R12 обозначает гетероарильную группу или арильную группу, непосредственно присоединенную к ароматическому кольцу тетрагидронафталина, могут быть получены в соответствии с методом, представленным на схеме 6. Олефинированием по Хорнеру-Эммонсу продукта 84 соответствующим фосфонатным эфиром получают бромид 118. Обработкой продукта 118 гетероарилборонатами или арилборонатами в присутствии палладиевого катализатора получают соответствующие гетероарилзамещенные аналоги или арилзамещенные аналоги, из которых при гидролизе сложного эфира получают соединения 120 с гетероарильной группой или арильной группой, непосредственно присоединенной к ароматическому кольцу.
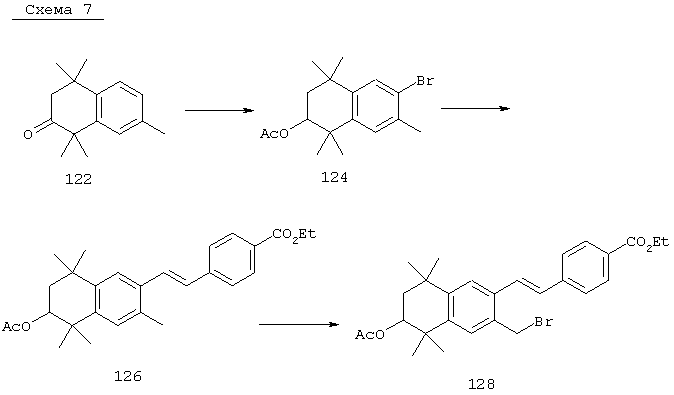
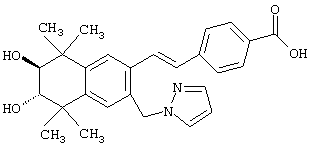
Соединения формулы I, у которых R3 обозначает гидроксил, могут быть получены так, как в качестве примера представлено на схемах 7 и 8. Тетралон 122 может быть получен реакцией конденсации дигидро-2,2,5,5-тетраметил-3(2Н)-фуранона с толуолом. В результате восстановления и защиты с использованием стандартных реагентов получают ацетат 124. Биспалладиевой реакцией перекрестного сочетания с использованием 4-бромэтилбензоата и триметоксивинилсилана получают соединение 126, которое можно превращать в бромид 128 бромированием по свободным радикалам. Бромид 128 может быть непосредственно замещен соответствующим нуклеофилом с получением соединений, у которых m обозначает 1, или может быть гомологизирован соответствующими углеродными нуклеофилами с получением соединений, у которых значение m превышает 1.
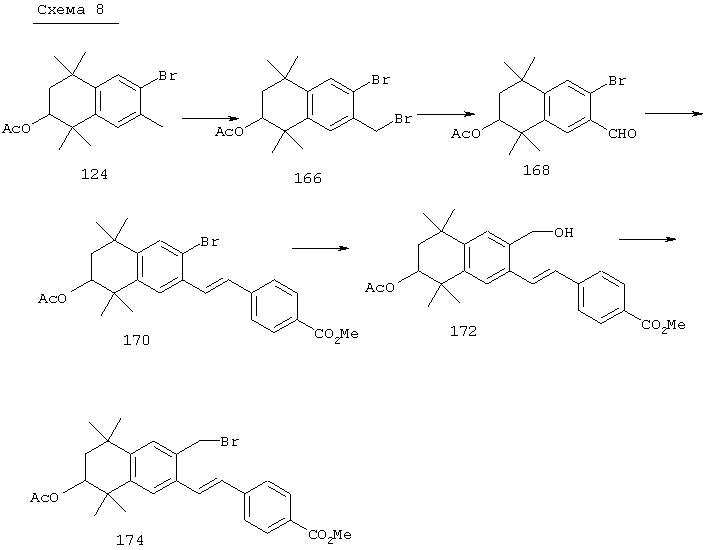
По другому варианту соединения формулы I, у которых R3 обозначает гидроксил, могут быть получены так, как в качестве примера представлено на схеме 8. Промежуточный ацетат 124 может быть бромирован с образованием продукта 166, после чего проводят обработку анионом, который образуется из 2-нитропропана, с получением альдегида 168. Олефинированием по Хорнеру-Эммонсу получают соединение 170, а реакцией сочетания по Стиллу (Stille) с гидроксиметилтрибутилоловом получают продукт 172. В результате бромирования продукта 172 с использованием НБС получают бромид 174. Бромид 174 может быть непосредственно замещен с помощью соответствующего нуклеофила с получением соединений, у которых m обозначает 1, или может быть гомологизирован соответствующими углеродными нуклеофилами с получением соединений, у которых значение m превышает 1.
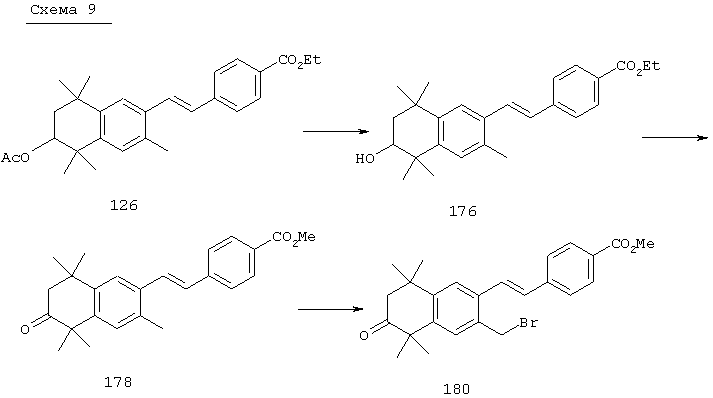
Соединения формулы I, у которых R3 обозначает оксогруппу, могут быть получены так, как в качестве примера представлено на схемах 9 и 10. В основных условиях отщепляют остаток ацетатного эфира 126, а затем с использованием триметилсилилдиазометана проводят переэтерификацию с получением продукта 176. Окислением продукта 176 реактивом Десса-Мартина (Dess-Martin) получают кетон 178, который бромированием по свободным радикалам можно превратить в бромид 180. Бромид 180 можно непосредственно замещать с использованием соответствующего нуклеофила, получая соединения, у которых m обозначает 1, или можно гомологизировать соответствующими углеродными нуклеофилами, когда значение m превышает 1.
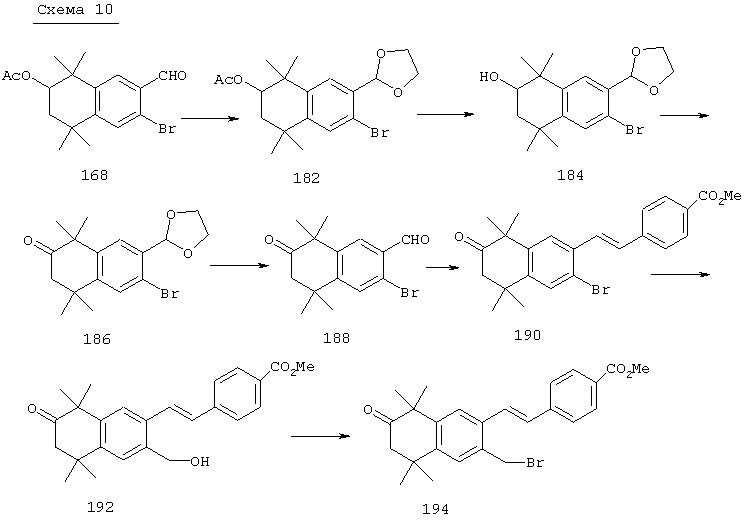
По другому варианту соединения формулы I, у которых R3 обозначает оксогруппу, могут быть получены так, как в качестве примера представлено на схеме 10. Альдегид 168 может быть защищен в форме ацеталя 182 обработкой этиленгликолем с кислотным катализом, и в основных условиях можно удалить ацетатную группу с получением спирта 184. Окислением реактивом Десса-Мартина получают кетон 186 и отщеплением ацетальной группы в кислых условиях получают альдегид 188. Олефинированием по Хорнеру-Эммонсу с использованием соответствующего фосфоната получают продукт 190 и реакцией сочетания по Стиллу с гидроксиметилтрибутилоловом получают продукт 192. Бромированием НБС получают бромид 194, который может быть непосредственно замещен с использованием соответствующего нуклеофила с получением соединений, у которых m обозначает 1, или может быть гомологизирован соответствующими углеродными нуклеофилами с получением соединений, у которых значение m превышает 1.
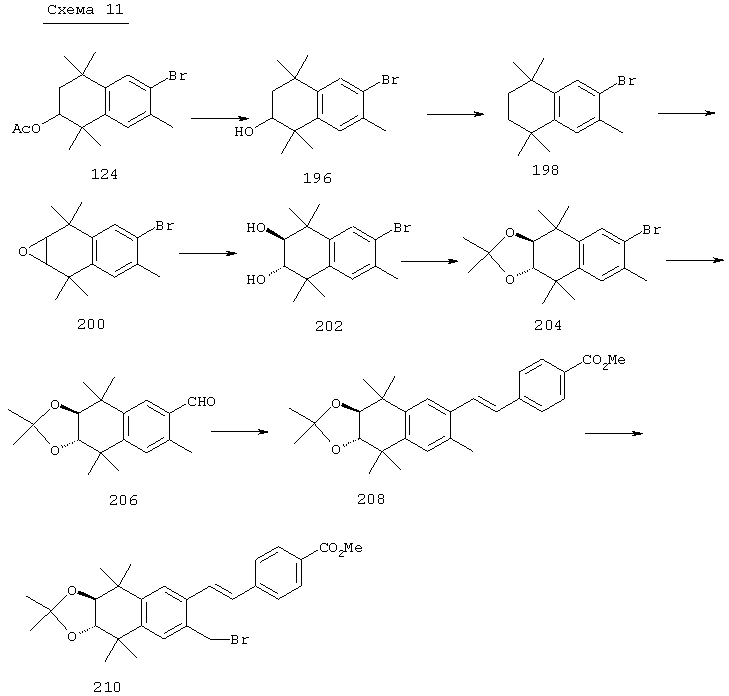
Соединения формулы I, у которых R3 обозначает диоловый остаток, могут быть получены так, как в качестве примера представлено на схемах 11 и 12. Омылением ацетатного промежуточного продукта 124 в основных условиях получают спирт 196 и дегидратацией при обработке POCl3 и пиридином получают олефин 198. Эпоксидированием продукта 198 м-ХПБК получают продукт 200. Этот эпоксид можно раскрыть в кислых условиях с получением транс-ацетатдиолов, которые затем можно гидролизовать в основных условиях с получением транс-диола 202. Защиту этого диола в форме диметилкеталя 204 с использованием в кислых условиях 2,2-диметоксипропана сопровождают превращением в альдегид 206 обработкой н-бутиллитием и N-формилпиперидином. Олефинированием по Хорнеру-Эммонсу альдегида 206 с использованием соответствующего фосфоната получают продукт 208, который бромированием по свободным радикалам можно превращать в бромид 210. Бромид 210 может быть непосредственно замещен с использованием соответствующего нуклеофила с получением соединений, у которых m обозначает 1, или может быть гомологизирован соответствующими углеродными нуклеофилами с получением соединений, у которых значение m превышает 1.
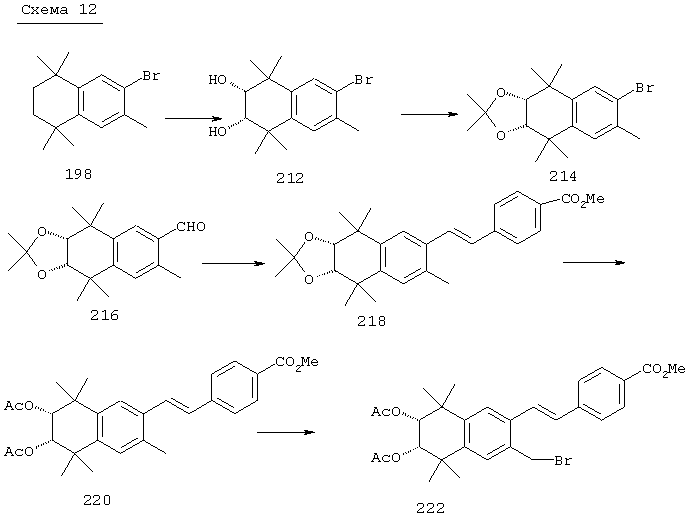
По другому варианту соединения формулы I, у которых R3 обозначает диоловый остаток, могут быть получены так, как в качестве примера представлено на схеме 12. Олефин 198 может быть обработан тетроксидом осмия с получением цис-диола 212. Защиту продукта 212 в форме кеталя 214 сопровождают превращением в альдегид 216 последовательной обработкой н-бутиллитием и N-формилпиперидином. Олефинированием по Хорнеру-Эммонсу с использованием соответствующего фосфоната получают продукт 218, а за удалением защитной группы следуют повторная защита с использованием уксусного ангидрида в пиридине и образование бисацетата 220. Бромированием продукта 220 по свободным радикалам получают бромид 222, который может быть непосредственно замещен с использованием соответствующего нуклеофила с получением соединений, у которых m обозначает 1, или может быть гомологизирован с помощью соответствующих углеродных нуклеофилов с получением соединений, у которых значение m превышает 1.
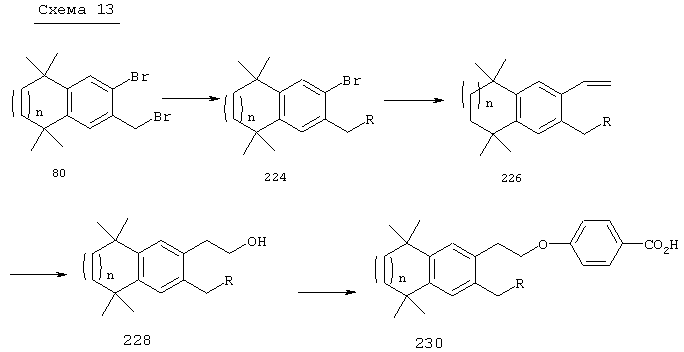
Соединения формулы I, в которой А обозначает СН2, а В обозначает СН2О, могут быть получены так, как представлено на схеме 13. Обработкой промежуточного продукта 80 соответствующим гетероароматическим нуклеофилом в основных условиях (например, пиразол и трет-бутоксид калия в ТГФ) получают продукт 224. Обработкой продукта 224 триметоксивинилсиланом, ацетатом палладия, три-о-толуилфосфином в N-МП получают виниловые промежуточные продукты 226. Гидроборированием/окислением продукта 226 с использованием 9BBN в ТГФ с последующим окислением 30%-ным пероксидом водорода получают гидроксиэтиловый промежуточный продукт 228. Реакцией сочетания по Мицонобу продукта 228 с метил-4-гидроксибензоатом совместно с трифенилфосфином и диэтилазодикарбоксилатом в ТГФ, после чего проводят омыление сложного эфира, получают ретиноидные аналоги 230.
Предлагается также способ получения соединения формулы VI, в которой каждый из n и t обозначает 1, R1 обозначает СО2Н или СО2-алкил, R2 обозначает -(CR10R11)m-R12, R3 обозначает Н, а R12 обозначает гетероарил,
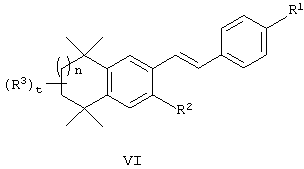 включающий обработку соединения формулы VII
включающий обработку соединения формулы VII
где G обозначает уходящую группу, нуклеофилом R12-Н, а когда R обозначает СО2-алкил, гидролизом с использованием основания.
Соединения по изобретению, представленные в настоящем описании, могут быть использованы для ускорения восстановления поврежденных альвеол и разделения альвеол. Таким образом, способы по изобретению можно применять для лечения легочных заболеваний, таких как эмфизема. Способы лечения с использованием представленного в настоящем описании соединения по изобретению можно также применять для лечения рака и дерматологических нарушений.
Селективность соединения по изобретению как агониста рецепторов ретиноивой кислоты можно определить по анализу связывания лиганда, как это известно специалисту в данной области техники (Apfel и др., Proc.Natl.Acad.Sci., (1992), 89, 7129; Teng и др., J.Med. Chem., (1997), 40, 2445; Вгусе и др., US № 5807900, которые включены в настоящее описание в качестве ссылок). Лечение RAR агонистами, в частности агонистами γ-RAR, может ускорить восстановление альвеолярной матрицы и перегородок, что важно в лечении эмфиземы. Предпочтительные соединения по изобретению являются γ-селективными агонистами, которые связываются с γ-рецепторами со сродством в пределах от примерно 25 до примерно 1000 нМ и проявляют в пять-десять раз более высокую селективность, чем при связывании с α-RAR рецептором. Следует отметить, что при лечении эмфиземы могут оказаться эффективными RAR агонисты, которым не свойственна γ-селективность. Трансактивацию, которая является способностью ретиноида активировать транскрипцию гена, когда транскрипцию гена вызывают связыванием лиганда с конкретным тестируемым рецептором ретиноивой кислоты, можно определить осуществлением методов, описанных в литературе, посвященной данной области техники (Apfel и др., Proc.Natl.Acad.Sci. (1992), 89, 7129; Bernard и др., Biochem. and Biophys. Res.Comm. (1992), 186, 977), которая включена в настоящее описание в качестве ссылки.
Пригодность соединений изобретения для лечения дерматологических нарушений, вызванных светом или возрастом, и заживления ран можно определить по методам, описанными в литературе, посвящанной данной области техники (Mustoe и др., Science 237, 1333 (1987); Sprugel и др., J.Pathol., 129. 601 (1987), которая включена в настоящее описание в качестве ссылки). Методы, описанные в литературе, посвящанной данной области техники, можно использовать для определения полезности соединений по изобретению при лечении дерматологических нарушений, таких как прыщи или псориаз (Boyd, Am.J.Med., 86, 568 (1989) и приведенные в этой работе ссылки; Doran и др., Methods in Enzymology, 190, 34 (1990), которые включены в настоящее описание в качестве ссылок). Наконец, способность соединений по изобретению лечить рак также можно определить по методам, описанным в литературе (Sporn и др., Fed.Proc. (1976) 1332; Hong и др., "Retinoids and Human Cancer" in The Retinoids: Biology, Chemistry and Medicine, M.B. Sporn, A.B. Roberts and D.S. Goodman (eds.). Raven Press, New York, 1994, 597-630, которые включены в настоящее описание в качестве ссылок).
Когда их используют при лечении или профилактике эмфиземы или связанных с ней заболеваний, рака или дерматологических расстройств, соединения по изобретению можно вводить в организм или применять самостоятельно или в сочетании с другими лекарственными средствами. Соединения по изобретению можно также вводить или применять самостоятельно или в сочетании с другими фармацевтически активными агентами, включая другие соединения по изобретению. Соединение по изобретению можно вводить в организм или применять самостоятельно или в форме фармацевтических композиций. Характеристики конкретной фармацевтической композиции обычно зависят от целевого пути введения, и для специалистов в данной области техники очевидны. Многочисленные композиции для местного применения или системного введения ретиноидных агонистов в данной области техники известны. С использованием соединения по изобретению можно готовить любые из таких композиций.
Фармацевтические композиции, включающие соединение по изобретению, могут быть приготовлены с помощью обычных методов смешения, растворения, гранулирования, изготовления драже, растирания материала в жидкой среде, эмульгирования, инкапсулирования, улавливания или лиофилизации. Фармацевтические композиции могут быть приготовлены обычным путем с использованием одного или нескольких физиологически приемлемых носителей, разбавителей, наполнителей или вспомогательных веществ, которые упрощают переработку соединений по изобретению в препараты, которые могут быть использованы с фармацевтическими целями. Состав подходящей композиции зависит от выбранного пути ее введения.
Для местного введения в организм на основе соединения по изобретению можно готовить композиции в виде растворов, гелей, мазей, кремов, суспензий и т.д., как это хорошо известно в данной области техники.
Системные композиции включают те, которые предусмотрены для введения инъекцией, например подкожной, внутривенной, внутримышечной, интратекальной или внутрибрюшинной инъекцией, а так же те, которые предназначены для чрезкожного, трансмукозного, перорального или внутрилегочного введения. Системные композиции можно готовить в сочетании с другим действующим веществом, которое улучшает мукоцилиарный клиренс слизи дыхательных путей или снижает вязкость слизи. Эти действующие вещества включают, хотя ими их список не ограничен, блокаторы натриевых канальцев, антибиотики, N-ацетилцистеин, гомоцистеин и фосфолипиды.
Для инъекции соединение по изобретению можно включать в композиции в виде водных растворов, предпочтительно в физиологически совместимых буферах, таких как раствор Ханкса, раствор Рингера или физиологический буферный раствор. Такой раствор может содержать такие компоненты композиций, как суспендирующие, стабилизирующие и/или диспергаторные вещества.
В другом варианте соединения по изобретению могут находиться в порошкообразной форме для разведения перед применением в подходящем носителе, например в стерильной апирогенной воде.
Для трансмукозного введения в композиции используют смачивающие вещества, приемлемые для проникновения через барьеры. Такие смачивающие вещества в данной области техники общеизвестны.
Для перорального введения композиции на основе соединения по изобретению могут быть легко приготовлены совмещением с фармацевтически приемлемыми носителями, хорошо известными в данной области техники.
Наличие таких носителей дает возможность на основе соединений по изобретению готовить композиции в форме таблеток, пилюль, драже, капсул, жидкостей, гелей, сиропов, шламов, суспензий и т.п. для глотания пациентом, которому назначено лечение. При приготовлении твердых композиций для перорального применения, таких как, например порошки, капсулы и таблетки, приемлемые наполнители включают обычные наполнители, такие как сахара, в частности лактозу, сахарозу, маннит и сорбит; целлюлозные материалы, такие как кукурузный крахмал, пшеничный крахмал, рисовый крахмал, картофельный крахмал, желатина, трагакантная камедь, метилцеллюлоза, гидроксипропилметилцеллюлоза, натрийкарбоксиметилцеллюлоза и/или поливинилпирролидон (ПВП); гранулирующие агенты и связующие вещества. При необходимости можно добавлять разрыхлители, такие как сшитый поливинилпирролидон, агар и альгиновая кислота или ее соль, такая как альгинат натрия. При необходимости твердые дозированные препаративные формы с применением стандартных технологий могут быть покрыты сахаром или снабжены покрытием для усвоения в кишечнике. Методы приготовления композиций на основе ретиноидных агонистов для перорального введения описаны в литратуре, посвящанной данной области техники (см., например, приготовление препарата Accutane®, Physicians' Desk Reference 54th Ed., сс.2610, 2000).
Используемые при приготовлении жидких препаратов для перорального введения, таких как, например, суспензии, эликсиры и растворы, приемлемые носители, наполнители или разбавители включают воду, солевой раствор, алкиленгликоли (например, пропиленгликоль), полиалкиленгликоли (например, полиэтиленгликоль), масла, спирты, слабокислые буферы с рН в пределах от 4 до 6 (например, ацетат, цитрат, аскорбат при концентрации в пределах от примерно 5,0 до примерно 50,0 мМ) и т.д. Дополнительно можно добавлять корригенты, консерванты, красители, соли желчных кислот, ацилкарнитины и т.п.
Для трансбуккального введения композициям можно придавать форму таблеток, лепешек и т.д. с приготовлением препарата обычным путем.
При лечении эмфиземы соединения по изобретению можно также вводить непосредственно в легкое ингаляцией (см., например, Tong и др., заявка РСТ WO 97/39745; Clark и др., заявка РСТ WO 99/47196, которые включены в настоящее описание в качестве ссылок). При введении в организм ингаляцией может оказаться целесообразной доставка соединения по изобретению в легкое с помощью ряда различных устройств. Так, например, для доставки соединений по изобретению непосредственно в легкое можно применять ингалятор с дозатором ("ИсД"), который снабжен емкостями, в которых содержится подходящий низкокипящий пропеллент, например дихлордифторметан, трихлорфторметан, дихлортетрафторэтан, диоксид углерода или другой приемлемый газ. Устройства типа ИсД можно приобрести у ряда поставщиков, таких, как 3М Corporation, Aventis, Boehringer Ingleheim, Forest Laboratories, Glaxo-Wellcome, Sobering Plough и Vectura.
По другому варианту для доставки соединения по изобретению в легкое можно применять такое устройство, как ингалятор для сухого порошка (ИСП) (см., например, Raleigh и др., материалы Proc.Amer.Assoc.Cancer Research Annual Meeting (1999), 40, 397, которые включены в настоящее описание в качестве ссылки). В устройствах типа ИСП как правило применяют такой механизм, как импульсный дозатор газа, который внутри контейнера создает облако сухого порошка, которое затем может быть ингалировано пациентом. Устройства типа ИСП также хорошо известны в данной области техники и могут быть приобретены у ряда продавцов, который включают, например, фирмы Fisons, Glaxo-Wellcome, Inhale Therapeutic Systems, ML Laboratories, Qdose и Vectura. Популярным вариантом является система ИСП на множество доз ("ИСПМД"), наличие которой позволяет вводить больше одной терапевтической дозы. Устройства типа ИСПМД доступны на таких фирмах, как AstraZeneca, GlaxoWellcome, IVAX, Scbering Plough, SkyePharma и Vectura. Так, например, желатиновые капсулы и картриджи для применения в ингаляторе или аппарате для вдувания можно изготовить таким образом, чтобы они содержали порошкообразную смесь соединения по изобретению с приемлемой порошкообразной основой, такой как лактоза и крахмал, для этих систем.
Для введения соединения по изобретению в легкое можно применять устройство другого типа, которое представляет собой устройство для распыления жидкости, поставляемое, например, фирмой Aradigm Corporation. В системах для распыления жидкости применяют распылительные головки с исключительно малыми отверстиями с целью перевода в аэрозольное состояние жидких лекарственных препаратов, которые при этом можно ингалировать непосредственно в легкое.
В одном предпочтительном варианте для введения соединения по изобретению в легкое применяют распылительное устройство. Распылительные устройства с использованием, например, энергии ультразвука из жидких лекарственных композиций создают аэрозоли с получением тонкодисперсных частиц, которые можно легко ингалировать (см., например, работу Verschoyle и др., British J.Cancer (1999), u, Suppl. 2, 96, которая включена в настоящее описание в качестве ссылки). Примеры распылительных устройств включают устройства, поставляемые фирмами Sheffield/Systemic Pulmonary Delivery Ltd. (см., Armer и др., US № 5954047; van der Linden и др., US № 5950619; van der Linden и др., US № 5970974, которые включены в настоящее описание в качестве ссылок), Aventis и Batelle Pulmonary Therapeutics.
В другом предпочтительном варианте для введения соединения по изобретению в легкое применяют электрогидродинамическое ("ЭГД") аэрозольное устройство. В ЭГД аэрозольных устройствах для перевода жидких лекарственных растворов или суспензий в аэрозольное состояние используют электрическую энергию (см., например, Noakes и др., US № 4765539; Coffee, US № 4962885; Coffee, заявка РСТ WO 94/12285; Coffee, заявка РСТ WO 94/14543; Coffee, заявка РСТ WO 95/26234, Coffee, заявка РСТ WO 95/26235, Coffee, заявка РСТ WO 95/32807, которые включены в настоящее описание в качестве ссылок). Для оптимизации процесса важными параметрами могут служить электрохимические свойства соединения по изобретению, входящего в состав композиции, когда это соединение вводят в легкое с помощью ЭГД аэрозольного устройства, причем такую оптимизацию обычно осуществляет специалист в данной области техники. ЭГД аэрозольные устройства способны вводить в легкое лекарственные средства более эффективно, чем существующие средства технологии введения в легкие. Другие методы введения соединения по изобретению внутрь легких специалистам в данной области техники известны и охватываются объемом изобретения.
Жидкие лекарственные композиции, приемлемые для применения с распылителями и устройствами для распыления жидкости и ЭГД аэрозольными устройствами, как правило включают соединение по изобретению совместно с фармацевтически приемлемым носителем. В предпочтительном варианте фармацевтически приемлемый носитель представляет собой жидкость, такую как спирт, вода, полиэтиленгликоль или перфторуглерод. Для изменения аэрозольных свойств раствора или суспензии соединений по изобретению можно добавлять, что необязательно, другой материал. В предпочтительном варианте этим материалом является жидкость, такая как спирт, гликоль, полигликоль или жирная кислота. Специалистам в данной области техники известны и другие методы приготовления жидких лекарственных растворов или суспензий, приемлемых для применения в аэрозольных устройствах (см., например. Biesalski, US № 5112598; Biesalski, US № 5556611, которые включены в настоящее описание в качестве ссылок).
Соединение по изобретению может также быть использовано для приготовления композиций для ректального или вагинального введения, таких как суппозитории или retention enemas, например содержащие обычную основу для суппозиториев, такую, как масло какао или другие глицериды.
Помимо композиций, которые описаны выше, соединение по изобретению можно также использовать при приготовлении композиций длительного действия. Такие композиции длительного действия можно вводить имплантацией (например, подкожно или внутримышечно) или внутримышечной инъекцией. Так, например, соединение по изобретению можно совмещать в композиции с приемлемыми полимерными или гидрофобными материалами (например, в форме эмульсии в приемлемом масле) или ионообменными смолами, или в виде слаборастворимых производных, например в виде слаборастворимой соли.
По другому варианту можно применять другие системы доставки фармацевтических препаратов. Хорошо известными примерами носителей для доставки, которые могут быть использованы при доставке соединения по изобретению, являются липосомы и эмульсии. Можно также применять некоторые органические растворители, такие как диметилсульфоксид, хотя обычно за счет более высокой токсичности. Соединение по изобретению можно также вводить в составе системы с регулируемым высвобождением. По одному из вариантов можно применять насос (Sefton, CRC Crit.Ref.Biomed.Eng. (1987), u, 201; Buchwald и др.. Surgery (1980), u, 507; Saudek и др., N.Engl.J.Med. (1989), 321, 574). В другом варианте могут быть использованы полимерные материалы (см. Medical Applications of Controlled Release, Langer и Wise (eds.), CRC Pres., Boca Raton, Florida (1974); Controlled Drug Bioavailability, Drug Product Design and Performance, Smolen and Ball (eds.), Wiley, New York (1984); Ranger and Peppas, J.Macromol.Sci.Rev.Macromol.Chem. (1983), 23, 61; см. также Levy и др., Science (1985), 228, 190; During и др., Ann.Neurol. (1989), 25, 351; Howard и др. (1989), J.Neurosurg. 71, 105). Тем не менее по еще одному варианту систему с регулируемым высвобождением можно размещать вблизи от цели применения соединения по изобретению, например вблизи легкого, вследствие чего требуется только доля системной дозы (см., например, Goodson, in Medical Applications of Controlled Release, упомянут выше, том 2, с. 115 (1984)). Может быть использована и другая система с регулируемым высвобождением (см., например, Langer, Science (1990), 249. 1527).
Когда соединение по изобретению проявляет кислую природу, его можно включать в любую из вышеописанных композиций в виде свободной кислоты, фармацевтически приемлемой соли, пролекарства, сольвата или гидрата. Фармацевтически приемлемые соли по существу сохраняют активность свободной кислоты и могут быть получены реакцией с основанием. Фармацевтически приемлемые соли включают любые известные приемлемые соли ретиноевых кислот, которые известны в данной области техники как вводимые млекопитающим. Фармацевтическим солям свойственна более высокая растворимость в водных и других протонных растворителях, чем у соответствующей свободной кислотной формы. Подобным же образом соединение по изобретению можно включать в любую из вышеописанных композиций в виде сольвата, гидрата или пролекарства. Предпочтительные пролекарства включают гидролизуемые эфирные производные, такие, как ароматические сложные эфиры, бензиловые эфиры и низшие алифатические эфиры, такие, как этиловый, циклопентиловый и т.д. Специалистам в области фармации известны и другие пролекарства.
Соединение по изобретению или композиции на его основе обычно используют в количестве, эффективном для достижения поставленной цели. Необходимо осознавать, разумеется, что используемое количество обычно зависит от метода введения в организм.
В случае использования при лечении или профилактике эмфиземы, рака или дерматологических нарушений соединения по изобретению или композиции на их основе вводят в организм или применяют в терапевтически эффективном количестве. Терапевтически эффективные количества соединений по изобретению для системного введения можно установить, изучив то, что подробно изложено в настоящем описании.
Фармакокинетический профиль соединений по изобретению предсказуем и может быть описан с использованием линейной фармакокинетической теории. Важно то, что фармакокинетика соединений по изобретению у людей может быть легко определена специалистом в данной области техники. Такой специалист с помощью методов, описанных в литературе, посвященной данной области техники (см., например, Khoo и др., J. Clin.Pharm. (1982), 22, 395; Colburn и др., J. Clin.Pharm. (1983) 23, 534; Colburn и др., Eur.J.Clin.Pharm. (19), 23. 689), после применения одной пероральной дозы соединения по изобретению способен определить диапазон фармакокинетических параметров. Такой специалист в соответствии с методами, описанными в литературе, посвященной данной области техники (Brazzel и др., Eur.J.Clin.Pharm. (1983), 24, 695; Lucek и др., Clin.Pharmacokinetics (1985), 10, 38), способен также определить значения этих фармакокинетических параметров после применения нескольких доз для того, чтобы сделать вывод о том, происходит ли в этих обстоятельствах индукция под действием соединения по изобретению или его аккумуляция. С помощью фармакокинетических параметров, которые определены по вышеупомянутым методам, в сочетании с полученными в тестах на животных-моделях результатами, касающимися дозировок, специалисты в данной области техники способны рассчитать соответствующие уровни системных доз соединений по изобретению, необходимых для лечения эмфиземы, рака или дерматологических нарушений у млекопитающих (предпочтительно, у людей).
Дозируемые количества и интервалы можно регулировать индивидуально для достижения в плазме такого уровня соединения по изобретению, которого достаточно для сохранения терапевтического эффекта. Обычные дозы для введения пациенту инъекцией находятся в интервале от 0,1 мкг до примерно 10,0 мг, предпочтительно в пределах от примерно 1,0 мкг до примерно 1,0 мг, более предпочтительно в пределах от примерно 100,0 до примерно 300,0 мкг. Терапевтически эффективное содержание в сыворотке может быть достигнуто введением по одной дозе или по несколько доз каждый день.
Количество вводимого соединения по изобретению обычно находится, разумеется, в зависимости, помимо прочих факторов, от проходящего курс лечения суб"екта, массы тела суб"екта, серьезности заболевания, метода введения и мнения лечащего врача. Так, например дозу можно вводить в составе фармацевтической композиции в один прием, в несколько приемов или посредством регулируемого высвобождения. Дозирование можно скачкообразно повторять, лекарственное средство можно вводить самостоятельно или в сочетании с другими лекарственными средствами и продолжать так долго, как это окажется необходимым для эффективного лечения эмфиземы.
В предпочтительном варианте терапевтически эффективная доза соединения по изобретению, представленного в настоящем описании, обычно оказывает терапевтически благоприятное воздействие без существенного токсикологического действия. Токсичность соединений по изобретению можно определить с использованием стандартных фармацевтических методов, она может быть легко установлена специалистом. Соотношение доз, вызывающих токсическое и терапевтическое действие, является терапевтическим индексом. В предпочтительном варианте в сравнении с другими ретиноидными агонистами соединение по изобретению обычно проявляет особенно высокие терапевтические индексы при лечении эмфиземы, рака или дерматологических расстройств. В предпочтительном варианте доза соединения по изобретению, представленного в настоящем описании, обычно находится в диапазоне of circulating concentrations, которые включают эффективную дозу со слабым токсическим действием или без него. Дозу можно варьировать в этом диапазоне в зависимости от применяемой дозированной формы и используемого пути введения. Конкретные композиции, путь введения и доза могут быть выбраны индивидуальным врачем с учетом состояния пациента (см., например, Fingl и др., 1975, In: The Pharmacological Basis of Therapeutics, Ch.1, c.1). Так, например, терапевтически эффективную дозу соединения по изобретению можно вводить либо перорально, либо непосредственно в легкое.
Далее изобретение представлено со ссылкой на следующие примеры, в которых подробно описаны получение соединения и композиций по изобретению. Для специалистов в данной области техники вполне очевидно, что как в материалы, так и способы можно вносить множество модификаций, не выходя при этом из объема изобретения.
Пример 1: пероральная композиция с соединением по изобретению
В таблице 2 приведены компоненты дозированной формы соединения по изобретению в таблетках.
Действующий компонент (т.е. соединение по изобретению) смешивают с лактозой до тех пор, пока не образуется однородная смесь. С лактозной смесью гомогенно смешивают остальные компоненты, а затем прессованием формуют таблетки, снабженные одним надрезом.
Пример 2: пероральная композиция с соединением по изобретению
Капсулы с соединением по изобретению, приемлемые для лечения эмфиземы, могут быть изготовлены с использованием компонентов, приведенных в таблице 3.
Вышеуказанные компоненты гомогенно смешивают и загружают в полутвердые желатиновые капсулы.
Пример 3: суспензия композиции с соединением по изобретению
Вышеуказанные компоненты, перечисленные в таблице 4, смешивают с получением суспензии для перорального введения.
Пример 4: приемлемая для инъекции композиция с соединением по изобретению
Вышеуказанные компоненты, перечисленные в таблице 5, смешивают с получением приемлемой для инъекции композиции.
Пример 5: приемлемая для инъекции композиция с соединением по изобретению
Вышеуказанные компоненты смешивают с получением приемлемой для инъекции композиции.
Пример 6: композиция с соединением по изобретению для интраназального введения
Вышеуказанные компоненты смешивают с получением суспензии для интраназального введения.
Пример 7: композиция с соединением по изобретению для ингаляции

Соединение по изобретению осторожно растворяют в 1,1,2-трихлор-1,2,2-трифторэтане без выпаривания какого-либо растворителя и полученный раствор фильтруют и хранят в герметизированном контейнере. Образовавшийся раствор и газообразный пропеллент можно вводить в аэрозольные баллоны (для подачи) в процентных количествах, указанных в таблице 8, с применением известных специалисту методов. С целью точного дозирования соединения по изобретению можно применять дозирующий клапан, который сконструирован для выпуска содержимого за один распылительный импульс в пределах от 100 до 300 мкг.
Пример 8: композиция с соединением по изобретению для ингаляции

Продукт Cremophor pH 40 может быть приобретен на фирме BASF Corporation. Другие эмульгаторы или ускорители растворения специалистам в данной области техники известны, их можно добавлять в водный растворитель вместо продукта Cremaphor рH 40. Соединение по изобретению, эмульгатор, 1,2-пропиленгликоль и воду смешивают между собой с получением раствора. Вышеуказанную жидкую композицию можно использовать, например, в аэрозольном препарате со сжатым газом в качестве соответствующего газообразного носителя (например, с азотом или диоксидом углерода).
Пример 9: композиция с соединением по изобретению для ЭГД
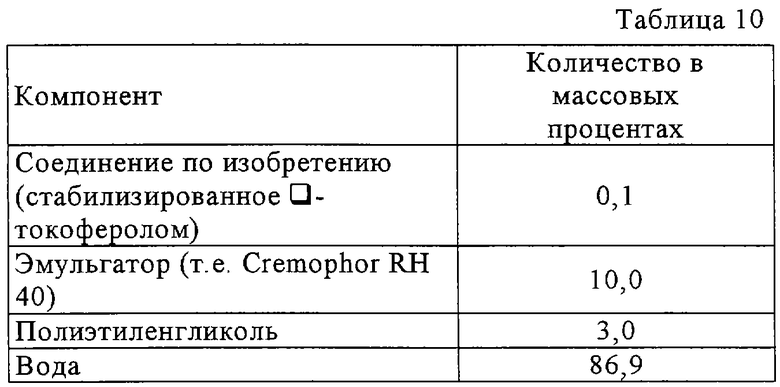
Соединение по изобретению, эмульгатор, полиэтиленгликоль и воду смешивают между собой с получением раствора. Вышеуказанную жидкую композицию можно использовать в типичных ЭГД устройствах, известных в данной области техники.
Пример 10: определение альвеолярной репорации в легком крысы с помощью соединения по изобретению
Эффективность соединения по изобретению можно оценивать по альвеолярной репорации на крысиной модели эластаз-вызванной эмфиземы (Massaro и др.. Nature, 1997, том 3, № 6: 675; Massaro и др., US № 5998486). В предпочтительном варианте животных разделяют на группы лечения приблизительно по восемь особей. Воспаление легкого и повреждение альвеол можно вызвать у самцов крыс Sprague Dawley отдельной инстиляцией приблизительно 2 ед/г массы тела панкреатической эластазы (полученной от свиньи, Calbiochem).
Животных можно лечить соединением по изобретению, полученным в Capmul, в подходящих пероральных диапазонах доз (предпочтительно от примерно 10,0 до 0,0001 мг/кг), которое обычно вводят перорально один раз в день, начиная через 21 день после повреждения. Животных контрольных групп провоцируют эластазой и 21 день спустя лечат носителем (раствор Capmul) в течение 14 дней. По прошествии 24 ч после введения последней дозы животных умерщвляют обескровливанием под глубокой анастезией. Во время обескровливания для анализа собирают кровь.
Легкие раздувают внутритрахеальным введением 10%-ного формалина с нейтральным буфером по постоянной схеме (1 мл/г массы тела/мин). Легкое изымают и погружают до обработки в фиксирующий раствор на 24 ч. Альвеолярные измерения проводят в четырех участках легкого крысы. Среднее значение для лечебной группы можно определить сложением средней площади у животного для всех восьми особей относительно животных лечебной группы, обработанных эластазой + носитель. В некоторых случаях разница между особями в пределах лечебной группы оказывается слишком большой относительно среднего значения для группы, чтобы быть статистически показательной. Для подготовки 5-микрометрических парафиновых блоков можно прибегнуть к стандартным методам. Блоки окрашивают гематоксилином и эозином. Для определения среднего альвеолярного размера и альвеолярного числа осуществляют компьютеризованный морфометрический анализ.
Количество триглицеридов, содержавшихся в плазме крысы, можно определить с помощью общепризнанных методов. Вкратце, последовательной обработкой плазмы липазой и глицерокиназой согласно указаниям, составленным изготовителем комплекта триглицеридов/GPO (#1488872, фирма Boehringer Mannheim), триглицериды плазмы можно превращать в дигидррксиацетон и пероксид водорода. Пероксид водорода можно определять колориметрическим путем в химическом анализаторе Hitachi 911. У крыс нормальные уровни триглицерида находятся в пределах от примерно 75 до примерно 175 мг/дл. Уровень триглицерида является удобной мерой токсичности.
В следующих примерах описан синтез конкретных соединений по изобретению, включая многие соединения, представленные в таблице 1.
Пример 11: получение (Е)-метил-4-[2-(3-бромметил-5.5.8.8-тетраметил-5,6.7,8-тетрагидронафталин-2-ил)винил]бензоата
Стадия А: получение 2,5-дихлор-2,5-диметилгексана
В раствор 100 г (684 ммоля) 2,5-диметил-2,5-гександиола в 300 мл этанола посредством диспергаторной трубки барботированием вводили газообразный HCl. Реакционную смесь медленно, в течение 3 ч, нагревали от комнатной температуры до 60°С. Реакционную смесь охлаждали на бане из мокрого льда и отфильтровывали белое твердое вещество. Это твердое вещество промывали водой и холодным этанолом, затем сушили с получением 65,2 г (65%-ный выход) 2,5-дихлор-2,5-диметилгексана (М+=181).
Стадия Б: получение 2-бром-3-метил-5,5,8,8-тетраметил-5,6,7,8-тетрагидронафталина
В раствор 20 г (117 ммолей) 2-бромтолуола и 14,4 г (97,4 ммоля) 2,5-дихлор-2,5-диметилгексана в 100 мл дихлорметана вводили 1,56 г (16,9 ммоля) хлорида алюминия и смесь выдерживали при температуре кипения с обратным холодильником. По прошествии 16 ч смесь охлаждали до комнатной температуры, разбавляли 150 мл гексана и добавляли 100 мл 1н. HCl. Органический слой отделяли, а водный слой экстрагировали гексаном. Объединенные органические слои промывали насыщенным раствором NaCl, сушили над сульфатом натрия и концентрировали под пониженным давлением. Продукт очищали фильтрованием через слой силикагеля с элюированием гексаном и получали в виде твердого вещества 23,9 г (87%-ный выход) 2-бром-3-метил-5,5,8,8-тетраметил-5,6,7,8-тетрагидронафталина (tпл: 81,1-85°С).
Стадия В: получение 2-формил-3-метил-5,5,8,8-тетраметил-5,6,7,8-тетрагидронафталина
Добавляли 22,2 мл (35,2 ммоля) н-бутиллития (1,6 М в гексанах). По прошествии 1 ч в раствор 5 г (17,8 ммоля) 2-бром-3-метил-5,5,8,8-тетраметил-5,6,7,8-тетрагидронафталина в 50 мл тетрагидрофурана, охлажденный на бане из сухого льда с ацетоном, добавляли раствор 3,95 мл (35,5 ммоля) N-формилпиперидина. Спустя 30 мин в реакционную смесь добавляли 30 мл насыщенного водного аммонийхлорида. Реакционную смесь нагревали до комнатной температуры и экстрагировали этилацетатом. Органическую фазу отделяли, промывали рассолом, сушили над сульфатом натрия и концентрировали под пониженным давлением. Остаток очищали экспресс-хроматографией на силикагеле с градиентным элюированием (гексан-10% этилацетата/гексан) с получением 3,5 г (85%-ный выход) 2-формил-3-метил-5,5,8,8-тетраметил-5,6,7,8-тетрагидронафталина (tпл: 82,4-84,1°С).
Получение 2-формил-3-метил-5,5,8,8-тетраметил-5,6,7,8-тетрагидронафталина по другому варианту
К 22,7 г (80,7 ммоля) 2-бром-3-метил-5,5,8,8-тетраметил-5,6,7,8-тетрагидронафталина в 270 мл N-метилпирролидина добавляли 28,9 г (322 ммоля) цианида меди(I). Реакционную смесь выдерживали при 175°С. По прошествии 16 ч смесь охлаждали до комнатной температуры и обрабатывали 400 мл 10%-ного водного гидроксида аммония. Реакционную смесь фильтровали для удаления соли и твердые частицы экстрагировали горячим этилацетатом. Объединенные органические фракции промывали рассолом, сушили над сульфатом натрия и концентрировали под пониженным давлением. Продукт очищали градиентным элюированием через слой силикагеля (гексан-5% этилацетата/гексан) с получением 18 г (95%-ный выход) 2-циано-3-метил-5,5,8,8-тетраметил-5,6,7,8-тетрагидронафталина (М = 227).
В раствор 18,7 г (82,3 ммоля) 2-циано-3-метил-5,5,8,8-тетраметил-5,6,7,8-тетрагидронафталина в 280 мл дихлорметана, охлажденный до -78°С, добавляли 123 мл (123 ммоля) диизобутилалюминийгидрида (1,0 М в толуоле). Реакционную смесь перемешивали и давали постепенно нагреться до комнатной температуры. По прошествии 16 ч реакционную смесь обрабатывали 30 мл добавляемой по каплям уксусной кислоты, а затем 150 мл воды. Органический слой отделяли, разбавляли 200 мл гексана, промывали рассолом, сушили над сульфатом натрия и концентрировали под пониженным давлением. Остаток очищали экспресс-хроматографией (градиентное элюирование 5-10% этилацетата/гексаном) на силикагеле с получением 11,8 г (63%-ный выход) 2-формил-3-метил-5,5,8,8-тетраметил-5,6,7,8-тетрагидронафталина.
Стадия Г: получение метил-4-[(Е)-2-(3,5,5,8,8-пентаметил-5,6,7,8-тетрагидронафталин-2-ил)винил]бензоата
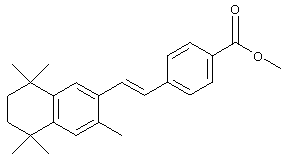
В раствор 3,5 г (13,5 ммоля) диметил-4-метилкарбоксилбензилфосфоната в 80 мл толуола при 0° добавляли 7,6 мл (23 ммоля) трет-пентилата калия (фирма Fluka Chemical Co.). По прошествии 15 мин добавляли раствор 2,3 г (10 ммоля) 2-формил-3-метил-5,5,8,8-тетраметил-5,6,7,8-тетрагидронафталина в 20 мл толуола и реакционную смесь перемешивали и нагревали до комнатной температуры. По прошествии 16 ч реакционную смесь выливали в 50 мл 2н. HCl, экстрагировали этилацетатом, промывали рассолом, сушили над сульфатом натрия и концентрировали под пониженным давлением. Остаток перемешивали со 100 мл гексана, фильтровали и фильтрат концентрировали под пониженным давлением. Остаток перемешивали со 100 мл метилового спирта и продукт отфильтровывали с получением 2,32 г (64%-ный выход) метил-4-[(Е)-2-(3,5,5,8,8-пентаметил-5,6,7,8-тетрагидронафталин-2-ил)винил]бензоата (М+=362).
Стадия Д: получение метил-4-[(Е)-2-(3-бромметил-5,5,8,8-тетраметил-5,6,7,8-тетрагидронафталин-2-ил)винил]бензоата.
Смесь 1,0 г (2,76 ммоля) (Е)-метил-4-[2-(3-метил-5,5,8,8-тетраметил-5,6,7,8-тетрагидронафталин-2-ил)винил]бензоата, 0,64 г (3,6 ммоля) N-бромсукцинимида и 0,033 г (0,13 ммоля) бензоилпероксида в 20 мл тетрахлорида углерода выдерживали при кипячении с обратным холодильником под лампой высокой интенсивности излучения. По прошествии 2 ч реакционную смесь охлаждали до комнатной температуры и выливали в 10%-ный водный раствор бисульфита натрия. Органический слой отделяли, промывали рассолом, сушили над сульфатом натрия и концентрировали под пониженным давлением. Остаток перемешивали с метиловым спиртом с получением 0,88 г метил-4-[2-(3-бромметил-5,5,8,8-тетраметил-5,6,7,8-тетрагидронафталин-2-ил)винил]бензоата (72%-ный выход) (М+ = 440).
Пример 12: получение 4-[(Е)-2-(5,5,8,8-тетраметил-3-пиразол-1-илметил-5,6,7,8-тетрагидронафталин-2-ил)винил]бензойной кислоты (6)
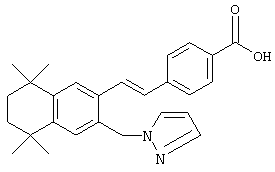
Смесь 2,0 г (4,5 ммоля) (Е)-метил-4-[2-(3-бромметил-5,5,8,8-тетраметил-5,6,7,8-тетрагидронафталин-2-ил)винил]бензоата и 0,65 г (9,5 ммоля) пиразола в 15 мл N-метилпирролидина выдерживали при 100°С. По прошествии 2 ч реакционную смесь охлаждали до комнатной температуры, выливали в рассол и экстрагировали этилацетатом. Органические экстракты промывали рассолом, сушили над сульфатом натрия и концентрировали под пониженным давлением. Остаток перемешивали с гексаном и продукт отфильтровывали, промывали гексаном и сушили с получением 1,6 г (83%-ный выход) метил-4-[2-(5,5,8,8-тетраметил-3-пиразол-1-илметил-5,6,7,8-тетрагидронафталин-2-ил)винил]бензоата (М+=429).
Смесь 27,6 г (64,4ммоля) метил-4-[2-(5,5,8,8-тетраметил-3-пиразол-1-илметил-5,6,7,8-тетрагидронафталин-2-ил)винил]бензоата и 97 мл (193 ммоля) 2 н. гидроксида натрия в 300 мл этилового спирта выдерживали при температуре кипения с обратным холодильником. По истечении 1 ч реакционную смесь охлаждали до комнатной температуры и разбавляли 900 мл воды. Реакционную смесь подкисляли 2 н. HCl и продукт выделяли фильтрованием, промывали водой и пентаном и сушили с получением 25,9 г (97%-ный выход) 4-[(Е)-2-(5,5,8,8-тетраметил-3-пиразол-1-илметил-5,6,7,8-тетрагидронафталин-2-ил)винил]бензойной кислоты (tпл = 246,5-248°С) 6.
Проведением процесса так, как изложено в вышеприведенном примере, но с заменой пиразола пирролом, 4-метилпиразолом, 1,2,4-триазолом, морфолином, 2-пирролидоном, 3,5-диметилпиразолом, δ-валеролактоном, 2-метилимидазолом и 4-метилимидазолом получали соответственно 4-[(Е)-2-(5,5,8,8-тетраметил-3-пиррол-1-илметил-5,6,7,8-тетрагидронафталин-2-ил)винил]бензойную кислоту 7, 4-{(Е)-2-[5,5,8,8-тетраметил-3-(4-метилпиразол-1-илметил-5,6,7,8-тетрагидронафталин-2-ил)винил]бензойную кислоту 20, 4-{[(Е)-2-(5,5,8,8-тетраметил-3-[1,2,4]триазол-1-илметил-5,6,7,8-тетрагидронафталин-2-ил]винил}бензойную кислоту 39, 4-[(Е)-2-(5,5,8,8-тетраметил-3-морфолин-4-илметил-5,6,7,8-тетрагидронафталин-2-ил)винил]бензойную кислоту 138, 4-[(Е)-2-(5,5,8,8-тетраметил-3-(2-оксопирролидин-1-илметил-5,6,7,8-тетрагидронафталин-2-ил)винил]бензойную кислоту 139, 4-{(Е)-2-[5,5,8,8-тетраметил-3-(3,5-диметилпиразол-1-илметил-5,6,7,8-тетрагидронафталин-2-ил)винил]бензойную кислоту 143, 4-[(Е)-2-(5,5,8,8-тетраметил-3-(2-оксопиперидин-1-илметил-5,6,7,8-тетрагидронафталин-2-ил)винил]бензойную кислоту 146, 4-{(Е)-2-[5,5,8,8-тетраметил-3-(2-метилимидазол-1-илметил-5,6,7,8-тетрагидронафталин-2-ил)винил]бензойную кислоту 149 и 4-{(Е)-2-[5,5,8,8-тетраметил-3-(4-метилимидазол-1-илметил-5,6,7,8-тетрагидронафталин-2-ил)винил]бензойную кислоту 150.
Пример 13: получение 4-[(Е)-2-(3-бутилтиометил-5,5,8,8-тетраметил-5,6,7,8-тетрагидронафталин-2-ил)винил]бензойной кислоты
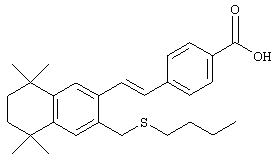
Раствор 445 мг (1 ммоль) метил-4-[(Е)-2-(3-бромметил-5,5,8,8-тетраметил-5,6,7,8-тетрагидронафталин-2-ил)винил]бензоата, 418 мг (3 ммоля) карбоната калия и 180 мг (2 ммоля) 1-бутантиола в 10 мл диметилформамида выдерживали при температуре кипения с обратным холодильником. По прошествии 40 мин реакционную смесь охлаждали до комнатной температуры, затем разбавляли водой. Смесь экстрагировали этилацетатом, органические экстракты промывали рассолом, сушили над сульфатом натрия, затем концентрировали под пониженным давлением. Остаток очищали экспресс-хроматографией на силикагеле (5% этилацетата/гексан) с получением 263 мг (58%-ный выход) метил-4-[(Е)-2-(3-бутилтиометил-5,5,8,8-тетраметил-5,6,7,8-тетрагидронафталин-2-ил)винил]бензоата.
Это, вышеуказанное соединение растворяли в 10 мл метилового спирта и 5 мл 1 н. LiOH и выдерживали при температуре кипения с обратным холодильником. По прошествии 2 ч реакционную смесь охлаждали до комнатной температуры, разбавляли рассолом и подкисляли концентрированной HCl. Смесь экстрагировали этиловым эфиром и органические фракции сушили над сульфатом натрия. Раствор концентрировали под пониженным давлением с получением 200 мг (78%-ный выход) 4-[(Е)-2-(3-бутилтиометил-5,5,8,8-тетраметил-5,6,7,8-тетрагидронафталин-2-ил)винил]бензойной кислоты (М+= 456).
Проведением процесса так, как изложено в вышеприведенном примере, но с заменой 1-бутантиола фурфурилмеркаптаном, 1-меркапто-2,3-пропандиолом, 1-меркапто-2-метилбутаном, циклопентилмеркаптаном, изопропилмеркаптаном, изобутилмеркаптаном, 3-метилбутилмеркаптаном, 2-диэтиламиноэтилмеркаптаном, 2-циклогексиламиноэтилмеркаптаном, 2-меркаптопиримидином, пропилмеркаптаном, 4-метил-4Н-1,2,4-триазол-3-тиолом, 5-метил-1,3,4-тиадиазол-2-тиолом, 5-меркапто-1-метилтетразолом, 2-меркапто-1-метилимидазолом, 2-меркаптотиазолином и 2-меркаптобензотиазолом получали соответственно соединения 8, 9, 3, 4, 13, 14, 16, 17, 18, 19, 21, 22, 23, 25, 32, 34 и 35.
Пример 14: получение 2-фтор-4-[(Е)-2-(5,5,8,8-тетраметил-3-пиразол-1-илметил-5,6,7,8-тетрагидронафталин-2-ил]винил)бензойной кислоты
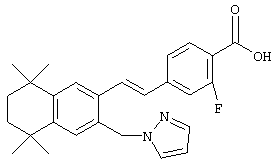
При 0°С 3 М тетрагидрофурановый раствор 350 мг (1,62 ммоля) 2-формил-3,5,5,8,8-пентаметил-5,6,7,8-тетрагидронафталина и 541 мг (1,78 ммоля) диэтил-2-фтор-4-карбометоксибензилфосфоната обрабатывали добавлением 10-миллиграммовыми порциями в течение 10 мин 84 мг (2,10 ммоля, 60 мас.% в минеральном масле) NaH. Реакционную смесь перемешивали при комнатной температуре в течение 2 ч с последующими разбавлением этилацетатом и промывкой рассолом. Органические экстракты сушили над сульфатом магния, концентрировали под вакуумом и растирали в порошок в метаноле с получением 330 мг (54%-ный выход) метил-2-фтор-4-[(Е)-2-(3,5,5,8,8-пентаметил-5,6,7,8-тетрагидронафталин-2-ил)винил]бензоата.
Суспензию 315 мг (0,828 ммоля) метил-2-фтор-4-[(Е)-2-(3,5,5,8,8-пентаметил-5,6,7,8-тетрагидронафталин-2-ил)винил]бензоата, 147 мг N-бромсукцинимида и 10 мг бензоилпероксида в 6 мл тетрахлорида углерода нагревали до кипения и выдерживали под облучением солнечной лампой с вольфрамовой нитью в течение 1,5 ч. Далее реакционную смесь фильтровали и концентрировали под вакуумом. Полученный остаток очищали экспресс-хроматографией на силикагеле (2% этилацетата в гексане) с получением 247 мг (65%-ный выход) метил-2-фтор-4-[(Е)-2-(3-бромметил-5,5,8,8-тетраметил-5,6,7,8-тетрагидронафтилен-2-ил)винил]бензоата.
Смесь 231 мг (0,503 ммоля) метил-2-фтор-4-[(Е)-2-(3-бромметил-5,5,8,8-тетраметил-5,6,7,8-тетрагидронафтилен-2-ил)винил]бензоата, 120 мг (1,76 ммоля) пиразола и 1,5 мл N-метилпирролидина выдерживали при 125°С в течение 6 ч. Образовавшуюся суспензию разделяли между этилацетатом и водой. Органический слой промывали рассолом, сушили над сульфатом магния и концентрировали под пониженным давлением, получая 207 мг (92%-ный выход) метил-2-фтор-4-{(Е)-2-[5,5,8,8-тетраметил-3-пиразол-1-илметил-5,6,7,8-тетрагидронафталин-2-ил] винил} бензоата.
Суспензию 199 мг (0,446 ммоля) метил-2-фтор-4-{[(Е)-2-[5,5,8,8-тетраметил-3-пиразол-1-илметил-5,6,7,8-тетрагидронафталин-2-ил]винил}бензоата, 1,5 мл этанола и 1 мл 2 М водного гидроксида натрия перемешивали в течение 16 ч. Смесь нейтрализовали водным аммонийхлоридом и экстрагировали этилацетатом. Далее органический слой промывали рассолом, сушили над сульфатом магния и концентрировали под вакуумом. Полученные 145 мг остатка растирали в порошок в метаноле с получением 85 мг (44%-ный выход) 2-фтор-4-[(Е)-2-[5,5,8,8-тетраметил-3-пиразол-1-илметил-5,6,7,8-тетрагидронафталин-2-ил]винил)бензойной кислоты 47.
Пример 15: получение 5-[(Е)-2-(5,5,8,8-тетраметил-3-пиразол-1-илметил-5,6,7,8-тетрагидронафталин-2-ил)винил]тиофен-2-карбоновой кислоты
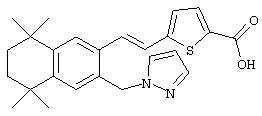
3 М тетрагидрофурановый раствор 434 мг (1,46 ммоля) 2-формил-5,5,8,8-тетраметил-3-пиразол-1-илметил-5,6,7,8-тетрагидронафталина и 494 мг (1,61 ммоля) диэтил(5-карбоэтокситиофен-2-ил)метилфосфоната при 0°С в течение 10 мин обрабатывали 5-миллиграммовыми порциями 76 мг NaH (1,91 ммоля, 60 мас.% в минеральном масле). Далее суспензию перемешивали при комнатной температуре в течение 2 ч. Затем реакционную смесь разбавляли этилацетатом и промывали водой и рассолом. Далее органический слой сушили над сульфатом магния и концентрировали под вакуумом. Полученный остаток растирали в порошок в метаноле, получая продукт 45.
Пример 16: получение 4-{(Е)-2-[3-(2-метоксиэтоксиметил)-5,5,8,8-тетраметил-5,6,7,8-тетрагидронафталин-2-ил]винил}бензойной кислоты
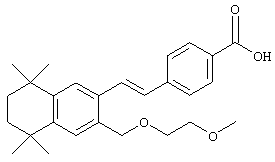
В смесь 63 мг (1,57 ммоля) гидрида натрия в 5 мл тетрагидрофурана добавляли 110 мг (1,47 ммоля) 2-метоксиэтанола, после чего 16 мг (0,098 ммоля) иодида калия. Реакционную смесь охлаждали до -30°С и добавляли раствор 434 мг (0,98 ммоля) метил-4-[(Е)-2-(3-бромметил-5,5,8,8-тетраметил-5,6,7,8-тетрагидронафталин-2-ил)винил]бензоата в 5 мл тетрагидрофурана. Реакционной смеси давали медленно нагреться до комнатной температуры. По прошествии 5 ч реакционную смесь разбавляли водой и экстрагировали этилацетатом. Органические фракции промывали рассолом, сушили над сульфатом натрия и концентрировали под пониженным давлением. Остаток очищали экспресс-хроматографией на силикагеле (13% этилацетата/гексан).
Вышеупомянутый продукт растворяли в 10 мл метилового спирта и 5 мл 1 н. LiOH. Реакционную смесь нагревали до кипения. По истечении 4 ч реакционную смесь охлаждали до комнатной температуры, разбавляли водой и этиловым эфиром, а затем подкисляли концентрированной HCl. Смесь экстрагировали этилацетатом, органические экстракты промывали рассолом, сушили над сульфатом натрия и концентрировали под пониженным давлением. Остаток перекристаллизовывали (этилацетат/гексан) с получением 50 мг (12%-ный выход) 4-{(Е)-2-[3-(2-метоксиэтоксиметил-5,5,8,8-тетраметил-5,6,7,8-тетрагидронафталин-2-ил]винил}бензойной кислоты (tпл: 55,9-58,2°С) 5.
Пример 17: получение 4-{(Е)-2-[3-(3-гидроксипропил)-5,5,8,8-тетраметил-5,6,7,8-тетрагидронафталин-2-ил]винил)бензойной кислоты
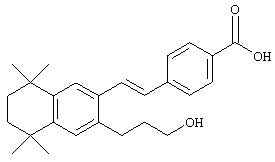
Стадия А: получение 2-(3-гидроксипропил)-5,5,8,8-тетраметил-5,6,7,8-тетрагидронафталина
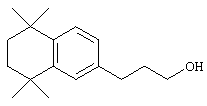
В раствор 14 г (103 ммоля) 3-фенил-1-пропанола и 18,2 г (123 ммоля) 2,5-дихлор-2,5-диметилгексана в 100 мл дихлорметана добавляли 15 г (113 ммолей) алюминийхлорида. После завершения добавления алюминийхлорида реакционную смесь кипятили с обратным холодильником. По прошествии 16 ч реакционную смесь охлаждали до комнатной температуры и добавляли 100 мл воды, после чего 100 мл 1 н. HCl. Реакционную смесь перемешивали в течение 2 ч, фильтровали через слой броунмиллерита и слои разделяли. Водный слой экстрагировали диэтиловым эфиром и объединенные органические фракции промывали рассолом, сушили над сульфатом натрия и концентрировали под пониженным давлением. Остаток очищали экспресс-хроматографией на силикагеле (30% этилацетата/гексан) с получением 13,45 г (53%-ный выход) 2-(3-гидроксипропил)-5,5,8,8-тетраметил-5,6,7,8-тетрагидронафталина.
Стадия Б: получение 2-бром-3-(3-гидроксипропил)-5,5,8,8-тетраметил-5,6,7,8-тетрагидронафталина
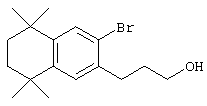
В раствор 12,3 г (49,8 ммоля) 2-(3-гидроксипропил)-5,5,8,8-тетраметил-5,6,7,8-тетрагидронафталина в 100 мл тетрахлорида углерода при 0°С добавляли следовое количество порошкообразного железа, после чего раствор 8,76 г (54,8 ммоля) брома в 80 мл тетрахлорида углерода. По прошествии 4,5 ч выдержки при 0°С реакционную смесь нагревали до комнатной температуры. Спустя 3 ч выдержки при комнатной температуре реакционную смесь выливали в смесь воды со льдом и реакционную смесь экстрагировали дихлорметаном. Органические фракции промывали рассолом, сушили над сульфатом натрия и концентрировали под пониженным давлением. Остаток очищали экспресс-хроматографией на силикагеле (30% этилацетата/гексан) с получением 10,2 г (63%-ный выход) 2-бром-3-(3-гидроксипропил)-5,5,8,8-тетраметил-5,6,7,8-тетрагидронафталина. (М+ = 324).
Стадия В: получение 2-бром-3-(3-трет-бутилдиметилсилоксипропил)-5,5,8,8-тетраметил-5,6,7,8-тетрагидронафталина
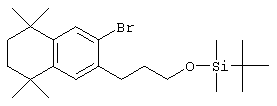
В раствор 10,2 г (31,55 ммоля) 2-бром-3-(3-гидроксипропил)-5,5,8,8-тетраметил-5,6,7,8-тетрагидронафталина в 80 мл диметилформамида добавляли 9,46 г (138,8 ммоля) имидазола и 10,46 г (69,4 ммоля) трет-бутилдиметилсилилхлорида. По прошествии 4 ч реакционную смесь разбавляли диэтиловым эфиром и промывали 1 н. водным аммонийхлоридом и рассолом. Органические фракции сушили над сульфатом натрия, а затем концентрировали под пониженным давлением. Остаток очищали экспресс-хроматографией на силикагеле (градиентное элюирование: от 1 до 2% этилацетата/гексан) с получением 9,17 г (66%-ный выход) 2-бром-3-(3-трет-бутилдиметилсилоксипропил)-5,5,8,8-тетраметил-5,6,7,8-тетрагидронафталина.
Стадия Г: получение 2-циано-3-(3-трет-бутилдиметилсилоксипропил)-5,5,8,8-тетраметил-5,6,7,8-тетрагидронафталина
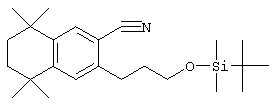
В раствор 9,0 г (20,5 ммоля) 2-бром-3-(3-трет-бутилдиметилсилоксипропил)-5,5,8,8-тетраметил-5,6,7,8-тетрагидронафталина в 70 мл N-метилпирролидина добавляли 7,36 г (82 ммоля) цианида меди(I) и реакционную смесь выдерживали при 175°С. По истечении 16 ч реакционную смесь охлаждали до комнатной температуры и разбавляли 10%-ным водным гидроксидом аммония. Полученные соли удаляли фильтрованием и промывали горячим этилацетатом. Фильтрат экстрагировали этилацетатом и объединенные органические фракции промывали рассолом, сушили над сульфатом натрия и концентрировали под пониженным давлением. Остаток очищали экспресс-хроматографией на силикагеле (градиентное элюирование: 3-25% этилацетата/гексан) с получением 4,8 г (61%-ный выход) 2-циано-3-(3-трет-бутилдиметилсилоксипропил)-5,5,8,8-тетраметил-5,6,7,8-тетрагидронафталина.
Стадия Д: получение 2-формил-3-(3-трет-бутилдиметилсилоксипропил)-5,5,8,8-тетраметил-5,6,7,8-тетрагидронафталина
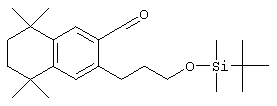
В раствор 4,6 г (11,9 ммоля) 2-циано-3-(3-трет-бутилдиметилсилоксипропил)-5,5,8,8-тетраметил-5,6,7,8-тетрагидронафталина в 40 мл дихлорметана при -78°С добавляли 17,9 мл (17,9 ммоля) диизобутилалюминийгидрида (1,0 М в толуоле). Реакционную смесь перемешивали и ей давали медленно нагреться до комнатной температуры. По прошествии 16 ч по каплям добавляли уксусной кислоты с последующим добавлением воды и дихлорметана. Органические фракции отделяли, промывали рассолом, сушили над сульфатом натрия и концентрировали под пониженным давлением. Остаток очищали экспресс-хроматографией на силикагеле (градиентное элюирование: 4-25% этилацетата/гексан) с получением 2,48 г (53%-ный выход) 2-формил-3-(3-трет-бутилдиметилсилоксипропил)-5,5,8,8-тетраметил-5,6,7,8-тетрагидронафталина (М = 389).
Другой вариант получения 2-формил-3-(3-трет-бутилдиметилсилилоксипропил)-5,5,8,8-тетраметил-5,6,7,8-тетрагидронафталина
В раствор 14 г (32 ммоля) 2-бром-3-(3-трет-бутилдиметилсилилоксипропил)-5,5,8,8-тетраметил-5,6,7,8-тетрагидронафталина в 200 мл сухого тетрагидрофурана при -78°С посредством шприца в атмосфере N2 добавляли 24,9 мл (64 ммоля) н-BuLi (2,5 М в гексане). В этих условиях реакционную смесь перемешивали в течение 1 ч. Затем реакцию в ней гасили раствором 7 мл (64 ммоля) N-формилпиперидина в 10 мл сухого тетрагидрофурана. Образовавшийся раствор перемешивали в течение еще 30 мин, после чего реакцию в нем гасили 100 мл раствора NH4Cl. Реакционную смесь экстрагировали 3 порциями по 100 мл этилацетата. Органические слои объединяли, сушили над MgSO4 и под пониженным давлением удаляли растворитель. Остаток очищали экспресс-хроматографией на силикагеле (градиентное элюирование: 30% этилацетата/гексаны) с получением 11,1 г (90%-ный выход) 2-формил-3-(3-трет-бутилдиметилсилилоксипропил)-5,5,8,8-тетраметил-5,6,7,8-тетрагидронафталина (M+= 386).
Стадия Е: получение метил-4-{(Е)-2-[3-[3-трет-бутилдиметилсилоксипропил)-5,5,8,8-тетраметил-5,6,7,8-тетрагидронафталин-2-ил] винил} бензоата
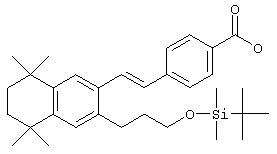
В суспензию 0,85 г (21,3 ммоля) 2-формил-3-(3-трет-бутилдиметилсилоксипропил)-5,5,8,8-тетраметил-5,6,7,8-тетрагидронафталина в 20 мл диметилсульфоксида при 0°С добавляли раствор 6,25 г (21,8 ммоля) диметил-4-метилкарбоксибензилфосфоната в 20 мл диметилсульфоксида. По прошествии 2 ч добавляли раствор 4 г (10,4 ммоля) диизопропиламида лития в 10 мл диметилсульфоксида. Спустя 3,5 ч реакционную смесь выливали в лед. Водный раствор подкисляли 1 н. HCl и экстрагировали этилацетатом. Органические фракции промывали рассолом, сушили над сульфатом натрия и концентрировали под пониженным давлением. Остаток очищали хроматографией посредством нетолстого слоя силикагеля (4% этилацетата/гексан) с получением 4,22 г (78%-ный выход) метил-4-{(Е)-2-[3-(3-трет-бутилдиметилсилоксипропил)-5,5,8,8-тетраметил-5,6,7,8-тетрагидронафталин-2-ил]винил}бензоата (tпл: 73,2-76,5°С).
Стадия Ж: получение метил-4-{(Е)-2-[3-(3-гидроксипропил)-5,5,8,8-тетраметил-5,6,7,8-тетрагидронафталин-2-ил]винил}бензоата
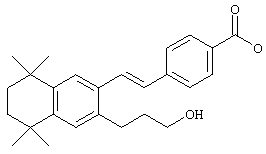
В раствор 4,0 г (7,7 ммоля) метил-4-{(Е)-2-[3-(3-трет-бутилдиметилсилоксипропил)-5,5,8,8-тетраметил-5,6,7,8-тетрагидронафталин-2-ил]винил}бензоата в 20 мл тетрагидрофурана добавляли раствор 8 мл (8,08 ммоля) тетрабутиламмонийфторида (1,0 М в тетрагидрофуране). Спустя 30 мин реакционную смесь разбавляли водой и экстрагировали этилацетатом. Органические фракции промывали рассолом, сушили над сульфатом натрия и концентрировали под пониженным давлением. Остаток очищали экспресс-хроматографией на силикагеле (этилацетат) с получением 2,67 г (85%-ный выход) метил-4-{(Е)-2-[3-(3-гидроксипролил)-5,5,8,8-тетраметил-5,6,7,8-тетрагидронафталин-2-ил]винил}бензоата (tпл: 109-115,5°С).
Стадия 3: получение 4-{(Е)-2-[3-(3-гидроксипропил)-5,5,8,8-тетраметил-5,6,7,8-тетрагидронафталин-2-ил] винил} бензойной кислоты
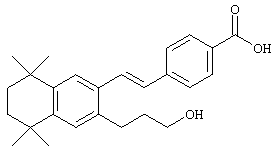
В раствор 27,0 г (0,66 ммоля) метил-4-{(Е)-2-[3-(3-гидроксипропил)-5,5,8,8-тетраметил-5,6,7,8-тетрагидронафталин-2-ил]винил}бензоата в 10 мл метилового спирта добавляли 5 мл 1 н. LiOH. Реакционную смесь нагревали до кипения. По прошествии 4 ч реакционную смесь охлаждали до комнатной температуры, разбавляли диэтиловым эфиром и подкисляли концентрированной HCl. Органический слой отделяли, промывали рассолом, сушили над сульфатом натрия и концентрировали под пониженным давлением. Остаток очищали кристаллизацией (гексан/этилацетат) с получением 200 мг (76%-ный выход) 4-{(Е)-2-[3-(3-гидроксипропил)-5,5,8,8-тетраметил-5,6,7,8-тетрагидронафталин-2-ил]винил} бензойной кислоты (tпл: 212,8-213,2°С) 12.
Проведением процесса так, как изложено в вышеприведенном примере, но с заменой 3-гидроксипропилбензола 2-гидроксиэтилбензолом получали 4-{(Е)-2-[3-(2-гидроксиэтил)-5,5,8,8-тетраметил-5,6,7,8-тетрагидронафталин-2-ил]винил} бензойной кислоты 31.
Пример 18: получение 4-{(Е)-2-[3-(3-метоксипропил)-5,5,8,8-тетраметил-5,6,7,8-тетрагидронафталин-2-ил]винил}бензойной кислоты
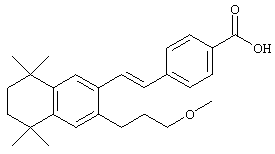
К 390 мг (0,96 ммоля) метил-4-[(Е)-2-[3-(3-гидроксипропил)-5,5,8,8-тетраметил-5,6,7,8-тетрагидронафталин-2-ил]винил}бензоата в 5 мл диметилформамида добавляли 480 мг (4,78 ммоля) метилиодида и при 0°С добавляли 38 мг (0,96 ммоля) гидрида натрия (60%-ная дисперсия в масле). По прошествии 4 ч реакционную смесь разбавляли насыщенным водным аммонийхлоридом и экстрагировали дихлорметаном. Органические фракции промывали рассолом, сушили над сульфатом натрия и концентрировали под пониженным давлением. Остаток очищали экспресс-хроматографией на силикагеле (6% этилацетата/гексан) с получением 200 мг продукта. Этот материал растворяли в 10 мл метилового спирта и 5 мл 1 н. LiOH и выдерживали при температуре кипения с обратным холодильником. По прошествии 2 ч реакционную смесь охлаждали до комнатной температуры, разбавляли водой и подкисляли концентрированной HCl. Реакционную смесь экстрагировали этилацетатом, органические фракции промывали рассолом, сушили над сульфатом натрия и концентрировали под пониженным давлением. Остаток очищали экспресс-хроматографией на силикагеле (40% этилацетата/гексан/0,5% уксусной кислоты) с получением 100 мг 4-{(Е)-2-[3-(3-метоксипропил}-5,8,8-тетраметил-5,6,7,8-тетрагидронафталин-2-ил]винил}бензойной кислоты 11.
Проведением процесса аналогично изложенному в вышеприведенном примере, но с заменой метилиодида этилиодидом получали 4-{(Е)-2-[3-(3-этоксипропил)-5,5,8,8-тетраметил-5,6,7,8-тетрагидронафталин-2-ил]винил}бензойной кислоты 10.
Пример 19: получение 4-{(Е)-2-[5,5,8,8-тетраметил-3-(пиримидин-2-илтиопропил)-5,6,7,8-тетрагидронафталин-2-ил]винил}бензойной кислоты
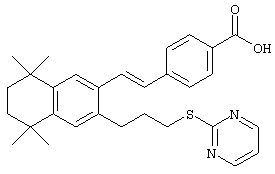
Смесь 250 мг (0,62 ммоля) метил-4-{(Е)-2-[3-(3-гидроксипропил)-5,5,8,8-тетраметил-5,6,7,8-тетрагидронафталин-2-ил]винил}бензоата, 1 мл (4,9 ммоля) диизопропилазодикарбоксилата, 1,3 г (4,9 ммоля) трифенилфосфина и 550 мг (4,9 ммоля) 2-меркаптопиримидина в 10 мл тетрагидрофурана перемешивали при комнатной температуре. По прошествии 48 ч реакционную смесь выливали в рассол и экстрагировали этилацетатом, органические экстракты промывали рассолом, сушили над сульфатом натрия и концентрировали под пониженным давлением. Остаток очищали экспресс-хроматографией на силикагеле (10% этилацетата/гексан).
Очищенный материал растворяли в 20 мл метилового спирта и 10 мл 1 н. LiOH и кипятили с обратным холодильником. По истечении 1 ч реакционную смесь охлаждали и под пониженным давлением удаляли метанол. Раствор подкисляли уксусной кислотой, экстрагировали этилацетатом, промывали рассолом, сушили над сульфатом натрия и концентрировали под пониженным давлением. Остаток очищали экспресс-хроматографией на силикагеле (градиентное элюирование: 10-20% этилацетата/гексан) и продукт перекристаллизовывали (этилацетат/гексан) с получением 160 мг (55%-ный выход) 4-{(Е)-2-[5,5,8,8-тетраметил-3-(пиримидин-1-илтиопропил)-5,6,7,8-тетрагидронафталин-2-ил]винил}бензойной кислоты 24 (tпл: 177-177,5°С).
Проведением процесса, аналогично изложенному выше, но с заменой 2-меркаптопиримидина 5-метил-1,3,4-тиадиазол-2-тиолом получали 4-((Е)-2-{5,5,8,8-тетраметил-3-[3-(5-метил[1,3,4]тиадиазол-2-илсульфанил)пропил]-5,6,7,8-тетрагидронафталин-2-ил}винил)бензойной кислоты 28.
Проведением процесса, аналогично изложенному выше, но с заменой (Е)-метил-4-[2-(3-{3-гидроксипропил}-5,5,8,8-тетраметил-5,6,7,8-тетрагидронафталин-2-ил)винил]бензоата (Е)-метил-4-[2-(3-{2-гидроксиэтил}-5,5,8,8-тетраметил-5,6,7,8-тетрагидронафталин-2-ил)винил]бензоатом получали 4-{(Е)-2-[5,5,8,8-тетраметил-3-(пиримидин-2-илтиометил)-5,6,7,8-тетрагидронафталин-2-ил]винил}бензойную кислоту 26 и 4-((Е)-2-{5,5,8,8-тетраметил-3-[3-(5-метил[1,3,4]тиадиазол-2-илсульфанил)этил]-5,6,7,8-тетрагидронафталин-2-ил}винил)бензойную кислоту 27.
Пример 20: получение 4-{(Е)-2-[5,5,8,8-тетраметил-3-(3-пиразол-1-илпропил)-5,6,7,8-тетрагидронафталин-2-ил]винил}бензойной кислоты
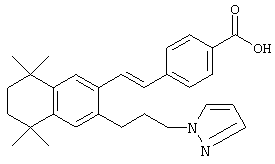
В раствор 210 мг (0,54 ммоля) метил-4-{(Е)-2-[3-(3-гидроксипропил)-5,5,8,8-тетраметил-5,6,7,8-тетрагидронафталин-2-ил]винил}бензоата в 20 мл дихлорметана при 0°С добавляли 0,22 мл (1,62 ммоля) триэтиламина и 0,083 мл (1,1 ммоля) метансульфонилхлорида. Спустя 3 ч выдержки при 0°С реакционную смесь разбавляли водой и экстрагировали дихлорметаном. Органические фракции отделяли, промывали рассолом, сушили над сульфатом натрия и концентрировали под пониженным давлением. Остаток очищали хроматографией посредством нетолстого слоя силикагеля. В смесь этого продукта в 5 мл тетрагидрофурана при 0°С добавляли смесь 65 мг (0,25 ммоля) 18-краун-6 и 28 мг (0,27 ммоля) калийтрет-бутоксида в 5 мл тетрагидрофурана. Реакционную смесь перемешивали и в течение ночи давали нагреться до комнатной температуры. По прошествии 16 ч реакционную смесь разбавляли водой и экстрагировали этилацетатом. Органические фракции отделяли, промывали рассолом и концентрировали под пониженным давлением. Остаток очищали экспресс-хроматографией на силикагеле (25% этилацетата/гексан). Очищенный продукт растворяли в 10 мл метилового спирта и 5 мл 1 н. LiOH. Реакционную смесь выдерживали при температуре кипения с обратным холодильником. По истечении 1,5 ч реакционную смесь охлаждали до комнатной температуры, разбавляли водой и подкисляли 1 н. HCl. Реакционную смесь экстрагировали этилацетатом, промывали рассолом, сушили над сульфатом натрия и концентрировали под пониженным давлением. Остаток очищали экспресс-хроматографией на силикагеле (10% метилового спирта/дихлорметан) с получением 84 мг (24%-ный выход) 4-{(Е)-2-[5,5,8,8-тетраметил-3-(3-пиразол-1-илпропил)-5,6,7,8-тетрагидронафталин-2-ил]винил}бензойной кислоты 30 (tпл:181,5-182,5°С).
Проведением процесса так, как изложено в вышеприведенном примере, но с заменой пиразола пирролом, 3-аминопропаном, 4-бромпиразолом, 3-метилпиразолом, 4-метилпиразолом и тетразолом получали соответственно 4-{(Е)-2-[5,5,8,8-тетраметил-3-(3-пиррол-1-илпропил)-5,6,7,8-тетрагидронафталин-2-ил]винил}бензойную кислоту 36, 4-{(Е)-2-[5,5,8,8-тетраметил-3-(3-пропиламинопропил)-5,6,7,8-тетрагидронафталин-2-ил)винил]бензойную кислоту 33, 4-{(Е)-2-[5,5,8,8-тетраметил-3-[3-(4-бромпиразол-1-ил)пропил]-5,6,7,8-тетрагидронафталин-2-ил]винил}бензойную кислоту 130, 4-{(Е)-2-[5,5,8,8-тетраметил-3-[3-(3-метилпиразол-1-ил)пропил]-5,6,7,8-тетрагидронафталин-2-ил]винил}бензойную кислоту 131, 4-{(Е)-2-[5,5,8,8-тетраметил-3-[3-(4-метилпиразол-1-ил)пропил]-5,6,7,8-тетрагидронафталин-2-ил]винил}бензойную кислоту 135, 4-{(Е)-2-[5,5,8,8-тетраметил-3-(3-тетразол-3-ил)пропил)-5,6,7,8-тетрагидронафталин-2-ил]винил}бензойную кислоту 141 и 4-{(Е)-2-[5,5,8,8-тетраметил-3-(3-тетразол-1-ил)пропил)-5,6,7,8-тетрагидронафталин-2-ил]винил}бензойную кислоту 142.
Проведением процесса так, как изложено в вышеприведенном примере, но с заменой метил-4-{(Е)-2-[3-(3-гидроксипропил)-5,5,8,8-тетраметил-5,6,7,8-тетрагидронафталин-2-ил]винил} бензоата (метил-4- {(Е)-2-[3-(2-гидроксиэтил)-5,5,8,8-тетраметил-5,6,7,8-тетрагидронафталин-2-ил]винил}бензоатом с использованием пиразола, 4-метилпиразола, 4-бромпиразола, имидазола, триазола, 3-метилпиразола и 3,5-диметилпиразола получали соответственно 4-{(Е)-2-[5,5,8,8-тетраметил-3-(2-пиразол-1-илэтил)-5,6,7,8-тетрагидронафталин-2-ил]винил}бензойную кислоту 29, 4-{(Е)-2-[5,5,8,8-тетраметил-3-[2-(4-метилпиразол-1-ил)этил]-5,6,7,8-тетрагидронафталин-2-ил]винил) бензойную кислоту 38, 4-{(Е)-2-[5,5,8,8-тетраметил-3-[2-(4-бромпиразол-1-ил)этил]-5,6,7,8-тетрагидронафталин-2-ил]винил}бензойную кислоту 134, 4-{(Е)-2-[5,5,8,8-тетраметил-3-[2-(имидазол-1-ил)этил]-5,6,7,8-тетрагидронафталин-2-ил]винил}бензойную кислоту 132, 4-{(Е)-2-[5,5,8,8-тетраметил-3-(2-[1,2,4]триазол-1-илэтил)-5,6,7,8-тетрагидронафталин-2-ил]винил}бензойную кислоту 136, 4-{(Е)-2-[5,5,8,8-тетраметил-3-[2-(3-метилпиразол-1-ил)этил]-5,6,7,8-тетрагидронафталин-2-ил]винил}бензойную кислоту 140 и 4-{(Е)-2-[5,5,8,8-тетраметил-3-[2-(3,5-диметилпиразол-1-ил)этил]-5,6,7,8-тетрагидронафталин-2-ил]винил}бензойную кислоту 147.
Пример 21: получение 3-бром-5,5,8,8-тетраметил-5,6,7,8-тетрагидро-2-нафтальдегида
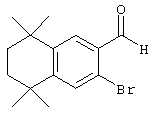
Стадия А
В раствор 12,0 г (281 ммоль) 2-бром-3-метил-5,5,8,8-тетраметил-5,6,7,8-тетрагидронафталина в 84 мл тетрахлорида углерода добавляли 7,59 г (42,7 ммоля) N-бромсукцинимида и 0,310 г (1,28 ммоля) бензоилпероксида. Смесь кипятили с обратным холодильником в течение 40 мин, а затем охлаждали до комнатной температуры. В охлажденный раствор добавляли 170 мл петролейного эфира и раствор фильтровали и концентрировали под вакуумом с получением 17,3 г 2-бром-3-бромметил-5,5,8,8-тетраметил-5,6,7,8-тетрагидронафталина, который использовали без дополнительной очистки.
Стадия Б
0,981 г (42,7 ммоля) натрия растворяли в 50 мл этанола. В этот раствор добавляли 4,94 г (55,5 ммоля) 2-нитропропана, после чего 17,3 г (42,7 ммоля) сырого 2-бром-3-бромметил-5,5,8,8-тетраметил-5,6,7,8-тетрагидронафталина в 75 мл этанола. По истечении 8 ч эту смесь концентрировали под вакуумом, а затем разделяли между этилацетатом и водой. Органический слой последовательно промывали 1 М водным гидроксидом натрия, водой, рассолом, сушили над безводным сульфатом натрия и концентрировали под вакуумом. Продукт очищали экспресс-хроматографией на силикагеле с градиентным элюированием (1-2% этилацетата/гексан) и получением 7,63 г (61%-ный выход) 3-бром-5,5,8,8-тетраметил-5,6,7,8-тетрагидро-2-нафтальдегида (tпл: 113,9-114,3°С).
Пример 22: получение 3-(2-триметилсилилэтинил)-5,5,8,8-тетраметил-5,6,7,8-тетрагидро-2-нафтальдегида
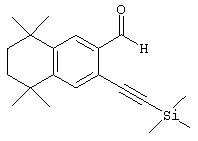
Раствор 6,97 г (23,6 ммоля) 3-бром-5,5,8,8-тетраметил-5,6,7,8-тетрагидро-2-нафтальдегида, 4,66 г (47,4 ммоля) триметилсилилацетилена, 700 мг (0,997 ммоля) дихлорбис(трифенилфосфин)палладия(II), 350 мг (1,84 ммоля) иодида одновалентной меди и 3,60 г (35,5 ммоля) триэтиламина в 95 мл диметилформамида выдерживали при 45°С в течение 2,5 ч, охлаждали и разделяли между этилацетатом и водой. Органическую фазу промывали рассолом, сушили над безводным сульфатом натрия и концентрировали под вакуумом. Продукт очищали фильтрованием через слой силикагеля с элюированием 10% этилацетата/гексаном и получали 7,23 г (98%-ный выход) 3-(2-триметилсилилэтинил)-5,5,8,8-тетраметил-5,6,7,8-тетрагидро-2-нафтальдегида (tпл: 78,4-82,0°С).
Пример 23: получение 3-этинил-5,5,8,8-тетраметил-5,6,7,8-тетрагидро-2-нафтальдегида
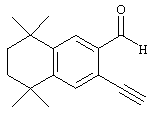
В раствор 7,21 г (23,1 ммоля) 3-(2-триметилсилилэтинил)-5,5,8,8-тетраметил-5,6,7,8-тетрагидро-2-нафтальдегида в 150 мл метанола добавляли 6,38 г (46,2 ммоля) карбоната калия. По истечении 1 ч реакционную смесь выливали в воду и экстрагировали этилацетатом. Органическую фазу промывали рассолом, сушили над безводным сульфатом натрия и концентрировали под вакуумом. Продукт очищали экспресс-хроматографией на силикагеле (3% этилацетата/гексан) с получением 4,04 г (73%-ный выход) 3-этинил-5,5,8,8-тетраметил-5,6,7,8-тетрагидро-2-нафтальдегида (tпл: 94,0-94,6°С).
Пример 24: 4-[(Е)-2-(5,5,8,8-тетраметил-3-((пиримидин-2-ил)этинил)-5,6,7,8-тетрагидро-2-нафталин-2-ил)винил]бензойной кислоты
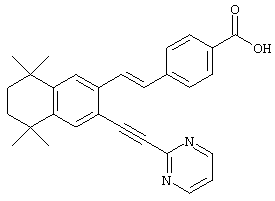
Стадия А: получение 5,5,8,8-тетраметил-3-пиримидин-2-илэтинил-5,6,7,8-тетрагидро-2-нафтальдегида
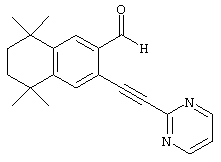
Раствор 0,400 г (1,66 ммоля) 3-этинил-5,5,8,8-тетраметил-5,6,7,8-тетрагидро-2-нафтальдегида, 0,278 г (1,75 ммоля) 2-бромпиримидина, 0,053 г (0,075 ммоля) дихлорбис(трифенилфосфин)палладия(II), 0,026 г (0,14 ммоля) иодида одновалентной меди и 0,253 г (2,50 ммоля) триэтиламина в 12 мл диметилформамида выдерживали при 45°С в течение 3 ч, охлаждали до комнатной температуры и разделяли между этилацетатом и водой. Органический слой промывали рассолом, сушили над безводным сульфатом натрия и концентрировали под вакуумом. Продукт очищали экспресс-хроматографией на силикагеле (25% этилацетата/гексан) с получением 0,372 г (70%-ный выход) 5,5,8,8-тетраметил-3-пиримидин-2-илэтинил-5,6,7,8-тетрагидро-2-нафтальдегида (М+=318).
Стадия Б: получение метил-4-[(Е)-2-(5,5,8,8-тетраметил-3-((пиримидин-2-ил)этинил)-5,6,7,8-тетрагидро-2-нафталин-2-ил)винил]бензоата
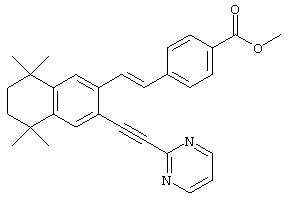
В суспензию 0,057 г (1,43 ммоля) 60%-ного NaH в 1,5 мл тетрагидрофурана добавляли 0,180 г (0,70 ммоля) метилового эфира 4-(диметоксифосфорилметил)бензоиной кислоты в 2,5 мл ТГФ и реакционную смесь перемешивали при комнатной температуре. По прошествии 20 мин медленно добавляли 0,182 г (0,57 ммоля) 3-((пиримидин-2-ил)этинил)-5,5,8,8-тетраметил-5,6,7,8-тетрагидро-2-нафтальдегида в 3 мл ТГФ и реакционную смесь перемешивали при комнатной температуре. По истечении 1 ч 20 мин реакцию гасили 6 мл 1 М соляной кислоты и экстрагировали этиловым эфиром. Органический слой промывали водой, рассолом, сушили над безводным сульфатом натрия и концентрировали под вакуумом. Сырой материал очищали экспресс-хроматографией (элюирование 40% этилацетата/гексаном) с получением 0,055 г (21%-ный выход) метил-4-[(Е)-2-(5,5,8,8-тетраметил-3-((пиримидин-2-ил)этинил)-5,6,7,8-тетрагидро-2-нафталин-2-ил)винил]бензоата (М+1 =451).
Стадия В: получение 4-[(Е)-2-(5,5,8,8-тетраметил-3-((пиримидин-2-ил)этинил)-5,6,7,8-тетрагидро-2-нафталин-2-ил)винил]бензойной кислоты
Раствор 0,055 г (0,123 ммоля) метил-4-[(Е)-2-(5,5,8,8-тетраметил-3-((пиримидин-2-ил)этинил)-5,6,7,8-тетрагидро-2-нафталин-2-ил)винил]бензоата в 2,5 мл 1 М LiOH и 5 мл этилового спирта кипятили с обратным холодильником. По прошествии 55 мин реакционную смесь охлаждали до комнатной температуры и подкисляли 4 мл 1 М соляной кислоты. Водную смесь экстрагировали этиловым эфиром, промывали водой, рассолом, сушили над безводным сульфатом магния и выпаривали под вакуумом с получением 0,054 г (99%-ный выход) 4-[(Е)-2-(5,5,8,8-тетраметил-3-((пиримидин-2-ил)этинил)-5,6,7,8-тетрагидро-2-нафталин-2-ил)винил]бензойной кислоты 159 (М+=436).
Пример 25: получение 5,5,8,8-тетраметил-3-(2-пиримидин-2-илэтил)-5,6,7,8-тетрагидро-2-нафтальдегида
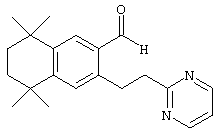
В раствор 0,362 г (1,14 ммоля) 3-(2-(2-пиримидил)этинил)-5,5,8,8-тетраметил-5,6,7,8-тетрагидро-2-нафтальдегида в 20 мл этанола добавляли 0,060 г 10%-ного палладия на угле и суспензию встряхивали в течение 3 ч под давлением газообразного водорода 40 фунтов/кв.дюйм. Образовавшийся раствор фильтровали и концентрировали под вакуумом. Продукт очищали экспресс-хроматографией на силикагеле (30% этилацетата/гексан) с получением 0,251 г (68%-ный выход) 3-(2-(2-пиримидил)этил)-5,5,8,8-тетраметил-5,6,7,8-тетрагидро-2-нафтальдегида (tпл: 78,0-84,5°C).
В результате замены 2-бромпиридина 2-бромтиазолом получали 2-формил-3-[2-(тиазол-2-ил)этил]-5,5,8,8-тетраметил-5,6,7,8-тетрагидронафталина.
Пример 26: получение 4-{(Е)-2-[5,5,8,8-тетраметил-3-(2-тиазол-2-илэтил)-5,6,7,8-тетрагидронафталин-2-ил]винил}бензойной кислоты
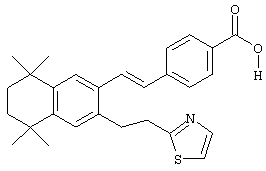
Стадия А: получение этил-4-{(Е)-2-[5,5,8,8-тетраметил-3-(2-тиазол-2-илэтил)-5,6,7,8-тетрагидронафталин-2-ил]винил} бензилового эфира
При 0°С 0,3 М тетрагидрофурановый раствор 229 мг (0,699 ммоля) 2-формил-3-[2-(тиазол-2-ил)этил]-5,5,8,8-тетраметил-5,6,7,8-тетрагидронафталина и 202 мг (0,706 ммоля) диэтил-4-карбоэтоксибензилфосфоната в течение десяти минут обрабатывали 5-миллиграммовыми порциями 30 мг (0,768 ммоля, 60 мас.% в минеральном масле) NaH. Раствору давали нагреться до комнатной температуры и перемешивали в течение 2 ч. Реакционную смесь разбавляли этилацетатом и промывали водой и рассолом. Далее органический слой сушили над сульфатом магния, концентрировали под пониженным давлением и полученный остаток растирали в порошок в метаноле с получением 278 мг (86%-ный выход) этил-4-{(Е)-2-[5,5,8,8-тетраметил-3-(2-тиазол-2-илэтил)-5,6,7,8-тетрагидронафталин-2-ил]винил}бензоата.
Стадия Б: получение 4-{(Е)-2-[5,5,8,8-тетраметил-3-(2-тиазол-2-илэтил)-5,6,7,8-тетрагидронафталин-2-ил]винил}бензойной кислоты
Суспензию, содержавшую 278 мг (0,604 ммоля) этил-4-{(Е)-2-[5,5,8,8-тетраметил-3-(2-тиазол-2-илэтил)-5,6,7,8-тетрагидронафталин-2-ил]винил}бензоата в 2,3 мл этанола и 1,7 мл 2 М водного гидроксида натрия, перемешивали в течение 7 ч. Реакционную смесь нейтрализовали и промывали насыщенным водным раствором аммонийхлорида. Образовавшийся осадок несколько раз экстрагировали этилацетатом. Органические слои объединяли, промывали рассолом и сушили над сульфатом магния с последующим концентрированием под вакуумом. Далее полученный остаток (95 мг) растворяли в дихлорметане и осаждали добавлением избытка гексана, в результате чего выделяли 25 мг (10%-ный выход) 4-{(Е)-2-[5,5,8,8-тетраметил-3-((2-тиазол-2-илэтил)-5,6,7,8-тетрагидронафталин-2-ил)винил]бензойной кислоты 40 (tпл: 215,8-217,5°С, М+ = 446).
Пример 27: получение 2-фтор-3-метил-5,5,8,8-тетраметил-5,6,7,8-тетрагидронафталина
В раствор 15 г (136 ммоля) 2-фтортолуола и 24,9 г (136 ммоля) 2,5-дихлор-2,5-диметилгексана в 120 мл дихлорметана добавляли 1,82 г (13,6 ммоля) алюминийхлорида и раствор выдерживали при температуре кипения с обратным холодильником. По истечении 18 ч реакционную смесь охлаждали до комнатной температуры и добавляли 5%-ную водную HCl. Смесь разделяли между гексаном и водой. Водную фазу один раз промывали гексаном. Объединенные органические слои промывали рассолом, сушили над безводным сульфатом натрия и концентрировали под вакуумом с получением 25,8 г (86%-ный выход) 2-фтор-3-метил-5,5,8,8-тетраметил-5,6,7,8-тетрагидронафталина (tпл: 90,0-91,8°С).
Пример 28: получение 3-фтор-5,5,8,8-тетраметил-5,6,7,8-тетрагидро-2-нафтальдегида
Стадия А
В раствор 10,0 г (45,4 ммоля) 2-фтор-3-метил-5,5,8,8-тетраметил-5,6,7,8-тетрагидронафталина в 91 мл тетрахлорида углерода вводили 8,48 г (47,7 ммоля) N-бромсукцинимида и 0,330 г (1,36 ммоля) бензоилпероксида. Раствор выдерживали при температуре кипения с обратным холодильником в течение 35 мин, а затем охлаждали до комнатной температуры. Добавляли 200 мл петролейного эфира и раствор фильтровали и концентрировали под вакуумом с получением 16,9 г 2-фтор-3-бромметил-5,5,8,8-тетраметил-5,6,7,8-тетрагидронафталина, который использовали без дополнительной очистки.
Стадия Б
5,26 г (59,0 ммоля) 2-нитропропана вводили в раствор, приготовленный растворением 1,04 г (45,4 ммоля) натрия в 60 мл этанола. Образовавшийся раствор добавляли в раствор 16,9 г (45,4 ммоля) сырого 2-фтор-3-бромметил-5,5,8,8-тетраметил-5,6,7,8-тетрагидронафталина в 60 мл этанола. По прошествии 5 ч реакционную смесь разделяли между этилацетатом и водой. Органический слой промывали 1 М водным гидроксидом натрия, водой, рассолом, сушили над безводным сульфатом натрия и концентрировали под вакуумом. Продукт очищали экспресс-хроматографией на силикагеле (1% этилацетата/гексан) с получением 6,17 г (58%-ный выход) 3-фтор-5,5,8,8-тетраметил-5,6,7,8-тетрагидро-2-нафтальдегида (tпл: 119,4-120,0°C).
Пример 29: получение 5,5,8,8-тетраметил-3-пиразол-1-ил-5,6,7,8-тетрагидро-2-нафтальдегида
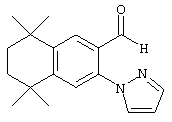
Раствор 0,200 г (0,845 ммоля) 3-фтор-5,5,8,8-тетраметил-5,6,7,8-тетрагидро-2-нафтальдегида, 0,058 г (0,854 ммоля) пиразола и 0,130 г (0,939 ммоля) карбоната калия в 2 мл диметилсульфоксида выдерживали при 95°С в течение 18 ч. Реакционную смесь охлаждали до комнатной температуры и разделяли между этилацетатом и водой. Органический слой промывали рассолом, сушили над безводным сульфатом натрия и концентрировали под вакуумом. Продукт очищали экспресс-хроматографией на силикагеле (5% этилацетата/гексан) с получением 0,101 г (42%-ный выход) 5,5,8,8-тетраметил-3-пиразол-1-ил-5,6,7,8-тетрагидро-2-нафтальдегида (tпл: 95,7-97,6).
Олефинированием по Хорнеру-Эммонсу с последующим омылением сложного эфира получали 4-{(Е)-2-[5,5,8,8-тетраметил-3-(пиразол-1-ил)-5,6,7,8-тетрагидронафталин-2-ил)винил]бензойной кислоты 156 (МН+ = 401).
Заменой пиразола 3-метилпиразолом получали 4-{(Е)-2-[5,5,8,8-тетраметил-3-(3-метилпиразол-1-ил)-5,6,7,8-тетрагидронафталин-2-ил)винил]бензойную кислоту 157 (МН+ = 415).
Пример 30: получение 5,5,8,8-тетраметил-3-(пиримидин-2-илтио)-5,6,7,8-тетрагидро-2-нафтальдегида
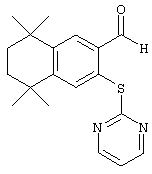
В раствор 0,479 г (4,27 ммоля) 2-меркаптопиримидина в 11 мл диметилформамида вводили 0,108 г (4,27 ммоля) 95%-ного гидрида натрия. По прошествии 20 мин добавляли 1,00 г (4,27 ммоля) 3-фтор-5,5,8,8-тетраметил-5,6,7,8-тетрагидро-2-нафтальдегида и полученный раствор выдерживали при температуре кипения с обратным холодильником. По истечении 18 ч реакционную смесь охлаждали до комнатной температуры и разделяли между этилацетатом и водой. Органическую фазу промывали водой, рассолом, сушили над безводным сульфатом натрия и концентрировали под вакуумом. Продукт очищали экспресс-хроматографией на диоксиде кремния (15% этилацетата/гексан) с получением 0,219 г (16%-ный выход) 5,5,8,8-тетраметил-3-(пиримидин-2-илтио)-5,6,7,8-тетрагидро-2-нафтальдегида (tпл: 143,2-145,8). Олефинированием по Хорнеру-Эммонсу с последующим омылением сложного эфира получали 4-{(Е)-2-[5,5,8,8-тетраметил-3-(меркаптопиримидин-2-ил)-5,6,7,8-тетрагидронафталин-2-ил)винил]бензойную кислоту 155 (tпл: 283-283,5). Заменой 2-меркаптопиримидина 2-меркаптотиазолом получали 4-{(Е)-2-[5,5,8,8-тетраметил-3-(меркаптотиазол-2-ил)-5,6,7,8-тетрагидронафталин-2-ил)винил]бензойную кислоту 154 (МН+= 450).
Пример 31: получение 2-(1,3-диоксолан-2-ил)-3-бром-5,5,8,8-тетраметил-5,6,7,8-тетрагидронафталина
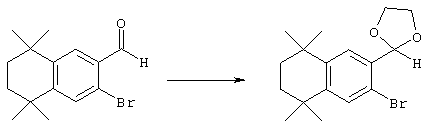
В раствор 1,8 г (6,3 ммоля) 2-формил-3-бром-5,5,8,8-тетраметил-5,6,7,8-тетрагидронафталина в 70 мл бензола вводили 3,5 мл этиленгликоля (63 ммоля), после чего моногидрат п-толуолсульфоновой кислоты (180 мг, 0,9 ммоля). Реакционную смесь выдерживали при температуре кипения с обратным холодильником в течение примерно 3 ч, а затем концентрировали под пониженным давлением. Остаток разделяли между диэтиловым эфиром и насыщенным раствором бикарбоната натрия. Эфирный слой промывали рассолом, сушили над MgSO4 и концентрировали под пониженным давлением. Остаток очищали экспресс-хроматографией (1,4% этилацетата/гексан) с помощью сменного элемента колонки Biotage 40M SiO2 с получением в виде кристаллического твердого вещества 1,5 г (72%-ный выход) 2-(1,3-диоксолан-2-ил)-3-бром-5,5,8,8-тетраметил-5,6,7,8-тетрагидронафталина.
Пример 32: получение 2-формил-3-[(тиофен-2-ил)метил]-5,5,8,8-тетраметил-5,6,7,8-тетрагидронафталина
В охлажденный до -78°С тетрагидрофурановый (3,0 мл) раствор 2-(1,3-диоксолан-2-ил)-3-бром-5,5,8,8-тетраметил-5,6,7,8-тетрагидронафталина (253 мг, 0,75 ммоля) вводили н-BuLi (1,6 М в гексане, 0,49 мл). Реакционная смесь постепенно загустевала в течение 45 мин. По каплям в вышеуказанный шлам добавляли раствор тиофен-2-карбоксальдегида (0,09 мл, 0,94 ммоля) в тетрагидрофуране (0,75 мл). Реакционная смесь быстро становилась гомогенной и ее перемешивали при -78°С в течение 45 мин. Добавляли насыщенного раствора аммонийхлорида и этилацетата и слои разделяли. Органический слой промывали рассолом, сушили над сульфатом магния и концентрировали с получением в виде желтого масла вещества, которое очищали препаративной тонкослойной хроматографией (15% этилацетата/гексан), выделяя в виде воскоподобного твердого вещества 2-(1,3-диоксолан-2-ил)-3-[1-гидрокси-1-(тиофен-2-ил)метил]-5,5,8,8-тетраметил-5,6,7,8-тетрагидронафталин (190 мг, 67%-ный выход).
Спирт, полученный по вышеизложенному, растворяли в 7 мл этилацетата, добавляли 75 мг 10% Pd/C и реакционную смесь перемешивали в атмосфере водорода в течение примерно 4 ч. Реакционную смесь фильтровали через броунмиллерит и концентрировали под вакуумом. Полученный остаток очищали препаративной тонкослойной хроматографией (10% этилацетата/гексан) с получением непосредственно 2-формил-3-[(тиофен-2-ил)метил]-5,5,8,8-тетраметил-5,6,7,8-тетрагидронафталина (94 мг, 62%-ный выход).
Пример 33: получение 4-[(Е)-2-(5,5,8,8-тетраметил-3-тиофен-2-илметил-5,6,7,8-тетрагидронафталин-2-ил)винил]бензойной кислоты
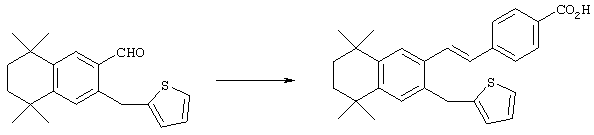
Олефинированием по Хорнеру-Эммонсу (после описанного в примере 26, стадия А, процесса) 2-формил-3-[(тиофен-2-ил)метил]-5,5,8,8-тетраметил-5,6,7,8-тетрагидронафталина (94 мг, 0,3 ммоля) с последующим гидролизом сложного эфира (после описанного в примере 26, стадия Б процесса), этил(Е)-4-{2-[3-((тиофен-2-ил)метил)-5,5,8,8-тетраметил-5,6,7,8-тетрагидронафталин-2-ил] винил }бензоата (53 мг, 0,11 ммоля) в виде белого кристаллического твердого вещества получали 4-[(Е)-2-(5,5,8,8-тетраметил-3-тиофен-2-илметил-5,6,7,8-тетрагидронафталин-2-ил)винил]бензойную кислоту (30 мг, 63%-ный выход) 44 (tпл 229,1-229,6°С).
Пример 34: получение 4-{(Е)-2-[5,5,8.8-тетраметил-3-(4-метилбензил)-5,6,7,8-тетрагидронафталин-2-ил]винил}бензойной кислоты
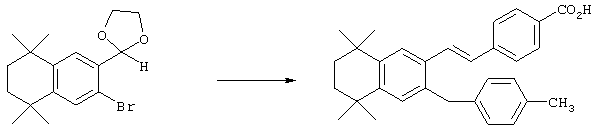
В смесь 2-(1,3-диоксолан-2-ил)-3-бром-5,5,8,8-тетраметил-5,6,7,8-тетрагидронафталина (250 мг, 0,74 ммоля) и тетракис(трифенилфосфин)палладия(0) (21 мг, 0,018 ммоля) в аргоновой атмосфере вводили 0,5 М тетрагидрофурановый раствор 4-метилбензилцинкхлорида (7,3 мл, 3,68 ммоля). Реакционную смесь перемешивали при температуре кипения с обратным холодильником в течение ночи и разделяли между насыщенным раствором хлорида аммония и этилацетатом. Органический слой промывали рассолом, сушили над MgSO4 и концентрировали под вакуумом. Остаток очищали препаративной тонкослойной хроматографией на силикагеле (5% этилацетата/гексан) с получением в виде белого кристаллического твердого вещества 2-(1,3-диоксолан-2-ил)-3-(4-метилбензил)-5,5,8,8-тетраметил-5,6,7,8-тетрагидронафталина (240 мг, 89%-ный выход).
Раствор ацеталя, полученного по изложенному выше (240 мг, 0,66 ммоля), в 3 мл тетрагидрофурана обрабатывали 2 мл 1 М HCl. Реакционную смесь перемешивали при комнатной температуре в течение 1,5 ч, а затем разделяли между насыщенным раствором хлорида аммония и этилацетатом. Органический слой промывали рассолом, сушили над MgSO4 и концентрировали с получением в виде прозрачного масла 2-формил-3-(4-метилбензил)-5,5,8,8-тетраметил-5,6,7,8-тетрагидронафталина, который при стоянии медленно кристаллизовался (210 мг, 99%-ный выход).
Олефинированием по Хорнеру-Эммонсу (после завершения описанного в примере процесса) 2-формил-3-(4-метилбензил)-5,5,8,8-тетраметил-5,6,7,8-тетрагидронафталина (210 мг, 0,65 ммоля) с последующим гидролизом сложного эфира (после завершения описанного в примере процесса), этил-4-{(Е)-2-[5,5,8,8-тетраметил-3-(4-метилбензил)-5,6,7,8-тетрагидронафталин-2-ил]винил}бензоата в виде белого кристаллического твердого вещества получали 4-{(Е)-2-[5,5,8,8-тетраметил-3-(4-метилбензил)-5,6,7,8-тетрагидронафталин-2-ил]винил}бензойную кислоту (185 мг, 64%-ный выход) (tпл: 216,3-217,3°C) (СН2Cl2/гексан) 42.
Пример 35: получение 4-{(Е)-2-[5,5,8,8-тетраметил-3-(2,4-дифторбензил)-5,6,7,8-тетрагидронафталин-2-ил]винил}бензойной кислоты
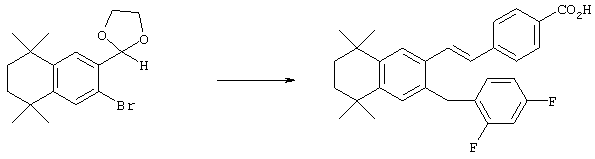
В смесь 2-(1,3-диоксолан-2-ил)-3-бром-5,5,8,8-тетраметил-5,6,7,8-тетрагидронафталина (250 мг, 0,74 ммоля) и тетракис(трифенилфосфин)палладия(0) (21 мг, 0,018 ммоля) в аргоновой атмосфере вводили 0,5 М тетрагидрофурановый раствор 2,4-дифторбензилцинкхлорида (7,3 мл, 3,68 ммоля). Реакционную смесь перемешивали при температуре кипения с обратным холодильником в течение ночи и разделяли между насыщенным раствором хлорида аммония и этилацетатом. Органический слой промывали рассолом, сушили над MgSO4 и концентрировали. Остаток очищали препаративной тонкослойной хроматографией на силикагеле (5% этилацетата/гексан) с получением в виде белого кристаллического твердого вещества 2-(1,3-диоксолан-2-ил)-3-(2,4-дифторбензил)-5,5,8,8-тетраметил-5,6,7,8-тетрагидронафталина (245 мг, 96%-ный выход).
Раствор вышеописанного ацеталя (245 мг, 0,71 ммоля) в 3 мл тетрагидрофурана обрабатывали 2 мл 1 М HCl. Реакционную смесь перемешивали при комнатной температуре в течение 1,5 ч, а затем разделяли между насыщенным раствором хлорида аммония и этилацетатом. Органический слой промывали рассолом, сушили над сульфатом магния и концентрировали с получением в виде прозрачного масла 2-формил-3-(2,4-дифторбензил)-5,5,8,8-тетраметил-5,6,7,8-тетрагидронафталина (239 мг, 97%-ный выход).
Олефинированием по Хорнеру-Эммонсу (после описанного в примере 26, стадия А процесса) 2-формил-3-(4-метилбензил)-5,5,8,8-тетраметил-5,6,7,8-тетрагидронафталина (239 мг, 0,69 ммоля) с последующим гидролизом сложного эфира (после описанного в примере 26, стадия Б процесса), этил-4-{(Е)-2-[5,5,8,8-тетраметил-3-(2,4-дифторбензил)-5,6,7,8-тетрагидронафталин-2-ил]винил}бензоата в виде белого кристаллического твердого вещества получали 4-{(Е)-2-[5,5,8,8-тетраметил-3-(2,4-дифторбензил)-5,6,7,8-тетрагидронафталин-2-ил] винил }бензойную кислоту (230 мг, 72%-ный выход) (tпл: 204,3-205,7°С; перекристаллизовывали из СН2Cl2/гексана) 43.
Пример 36: получение 4-{(Е)-2-[5,5,8,8-тетраметил-3-(тиофен-3-ил)-5,6,7,8-тетрагидронафталин-2-ил]винил}бензойной кислоты
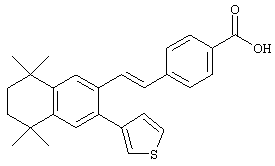
При 0°С 0,3 М тетрагидрофурановый раствор 1,832 г (6,21 ммоля) 2-формил-3-бром-5,5,8,8-тетраметил-5,6,7,8-тетрагидронафтилена и диэтил-4-карбоэтоксибензилфосфоната обрабатывали в течение десяти минут 301 мг NaH (7,51 ммоля, концентрацией 60 мас.% в минеральном масле, в виде 30-миллиграммовых порций). Далее смесь перемешивали при комнатной температуре в течение 2 ч с последующими разбавлением этилацетатом и промывкой водой. Органический слой промывали рассолом, сушили над сульфатом магния и концентрировали под вакуумом. Полученный остаток растирали в порошок в метаноле, получая 1,334 г (50%-ный выход) этил-4-[(Е)-2-(3-бром-5,5,8,8-тетраметил-5,6,7,8-тетрагидронафтилен-2-ил)винил]бензоата.
В раствор 375 мг (0,825 ммоля) этил-4-[(Е)-2-(3-бром-5,5,8,8-тетраметил-5,6,7,8-тетрагидронафтилен-2-ил)винил]бензоата и 48 мг (0,0413 ммоля) тетракис(трифенилфосфин)палладия(0) в 17 мл толуола вводили 158 мг 3-тиофенбороновой кислоты (фирмы Aldrich) в 5 мл этанола, а затем 8,5 мл насыщенного водного бикарбоната натрия. Смесь кипятили с обратным холодильником в течение 16 ч с последующими разбавлением этилацетатом и промывкой водой. Органический слой промывали рассолом, сушили над сульфатом магния и концентрировали под вакуумом, получая 504 мг сырого остатка. Сырую смесь очищали препаративной тонкослойной хроматографией на силикагеле (2% этилацетата в гексане), получая 160 мг (44%-ный выход) этил-4-{(Е)-2-[5,5,8,8-тетраметил-3-(тиофен-3-ил)-5,6,7,8-тетрагидронафтилен-2-ил]винил}бензоата.
Суспензию 160 мг (0,359 ммоля) этил-4-{(Е)-2-[5,5,8,8-тетраметил-3-(тиофен-3-ил)-5,6,7,8-тетрагидронафтилен-2-ил]винил}бензоата, 1,5 мл этанола и 1 мл 2 М водного NaOH перемешивали в течение 16 ч с последующими нейтрализацией хлоридом аммония и экстракцией этилацетатом. Органический слой промывали рассолом, сушили над сульфатом магния и концентрировали под вакуумом, получая 115 мг сырого остатка. Этот остаток очищали препаративной тонкослойной хроматографией на силикагеле (10% МеОН/дихлорметан) с получением 42 мг 4-{(Е)-2-[5,5,8,8-тетраметил-3-(тиофен-3-ил)-5,6,7,8-тетрагидронафталин-2-ил]винил}бензойной кислоты 48.
Пример 37: получение 4-[(Е)-2-(5,5,8,8-тетраметил-3-пиразол-1-илметил-6-гидрокси-5,6,7,8-тетрагидронафталин-2-ил)винил]бензойной кислоты
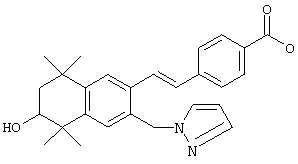
Стадия А: получение 1,1,4,4,7-пентаметил-2-тетралона
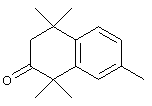
В раствор 20 г (140,6 ммоля) дигидро-2,2,5,5-тетраметил-3(2Н)-фуранона в 240 мл толуола, охлажденный на бане из мокрого льда, отдельными порциями в течение 15 мин вводили 38,4 г (288 ммоля) алюминийхлорида. После завершения добавления реакционной смеси давали нагреться до комнатной температуры. По истечении 16 ч реакционную смесь осторожно выливали на лед и образовавшийся водный раствор экстрагировали этилацетатом. Органические фракции промывали рассолом, сушили над сульфатом натрия и концентрировали под пониженным давлением. Остаток очищали экспресс-хроматографией (элюирование гексаном) с получением 26 г (86%-ный выход) 1,1,4,4,7-пентаментил-2-тетралона (М+ = 216).
Стадия Б: получение 3-метил-6-гидрокси-5,5,8,8-тетраметил-5,6,7,8-тетрагидронафталина
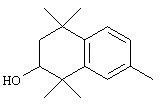
В раствор 10 г (46,2 ммоля) 1,1,4,4,7-пентаментил-2-тетралона в 100 мл этилового спирта отдельными порциями в течение 15 мин вводили 7 г (185 ммолей) боргидрида натрия. После завершения добавления реакционную смесь перемешивали при комнатной температуре. По истечении 16 ч реакционную смесь концентрировали под пониженным давлением и остаток разделяли между водой и этилацетатом. Органические экстракты промывали рассолом, сушили над сульфатом натрия и концентрировали под пониженным давлением. Остаток очищали экспресс-хроматографией (градиентное элюирование 5-10% этилацетата/гексаном) с последующей кристаллизацией из гексана и получением 4 г (40%-ный выход) 3-метил-6-гидрокси-5,5,8,8-тетраметил-5,6,7,8-тетрагидронафталина (М+ = 218).
Стадия В: получение 2-бром-3-метил-6-ацетокси-5,5,8,8-тетраметил-5,6,7,8-тетрагидронафталина
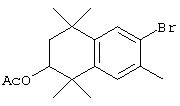
В раствор 4 г (18,3 ммоля) З-метил-6-гидрокси-5,5,8,8-тетраметил-5,6,7,8-тетрагидронафталина в 20 мл уксусной кислоты по каплям вводили 3,25 г (20,15 ммоля) брома и реакционную смесь перемешивали при комнатной температуре. По прошествии 16 ч реакционную смесь выливали в рассол и экстрагировали этилацетатом. Органические экстракты промывали рассолом, сушили над сульфатом натрия и концентрировали под пониженным давлением. Остаток очищали экспресс-хроматографией (градиентное элюирование гексаном - 15% этилацетата/гексаном) с получением 6 г (97%-ный выход) 2-бром-3-метил-6-ацетокси-5,5,8,8-тетраметил-5,6,7,8-тетрагидронафталина (М+= 338).
Стадия Г: получение этил-4-[(Е)-2-(5,5,8,8-тетраметил-3-метил-6-ацетокси-5,6,7,8-тетрагидронафталин-2-ил)винил]бензоата
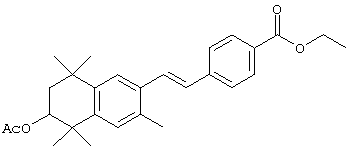
Смесь 1 г (3 ммоля) 2-бром-3-метил-6-ацетокси-5,5,8,8-тетраметил-5,6,7,8-тетрагидронафталина, 0,68 мл (4,42 ммоля) триметоксивинилсилана, 0,12 г (0,53 ммоля) ацетата палладия, 0,36 г три-о-толилфосфина (1,2 ммоля) и 0,82 мл триэтиламина (5,9 ммоля) в 10 мл N-метилпирролидина выдерживали при 90°С. Спустя 3 ч реакционную смесь охлаждали до комнатной температуры и добавляли 0,57 мл (3,5 ммоля) этил-4-бромбензоата, 0,82 мл триэтиламина (5,9 ммоля) и 5 мл (5 ммоля) тетрабутиламмонийфторида. Реакционную смесь вновь нагревали до 90°С. По прошествии 2,5 ч реакционную смесь охлаждали до комнатной температуры, выливали в рассол и экстрагировали этилацетатом. Органические экстракты промывали рассолом, сушили над сульфатом натрия и концентрировали под пониженным давлением. Полученный остаток очищали экспресс-хроматографией (градиентное элюирование 2-5% этилацетата/гексаном) с получением 0,2 г (20%-ный выход) этил-4-[(Е)-2-(5,5,8,8-тетраметил-3-метил-6-ацетокси-5,6,7,8-тетрагидронафталин-2-ил)винил]бензоата (М+ = 434).
Стадия Д: получение этил-4-[(Е)-2-(5,5,8,8-тетраметил-3-бромметил-6-ацетокси-5,6,7,8-тетрагидронафталин-2-ил)винил]бензоата
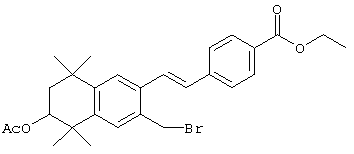
Раствор 0,68 г (1,57 ммоля) этил-4-[(Е)-2-(5,5,8,8-тетраметил-3-метил-6-ацетокси-5,6,7,8-тетрагидронафталин-2-ил)винил]бензоата, 0,36 г (2,03 ммоля) N-бромсукцинимида и 0,019 г (0,078 ммоля) бензоилпероксида в 15 мл тетрахлорида углерода выдерживали при температуре кипения с обратным холодильником. Спустя 3 ч реакционную смесь охлаждали до комнатной температуры, промывали 10%-ным водным бисульфитом натрия и рассолом, сушили над сульфатом натрия и концентрировали под пониженным давлением. Остаток очищали экспресс-хроматографией (градиентное элюирование гексаном, затем 5% этилацетата/гексаном) с получением 0,45 г (56%-ный выход) этил-4-[(Е)-2-(5,5,8,8-тетраметил-3-бромметил-6-ацетокси-5,6,7,8-тетрагидронафталин-2-ил)винил]бензоата (М+=512).
Стадия Е: получение этил-4-[(Е)-2-(5,5,8,8-тетраметил-3-пиразол-1-илметил-6-ацетокси-5,6,7,8-тетрагидронафталин-2-ил)винил]бензоата
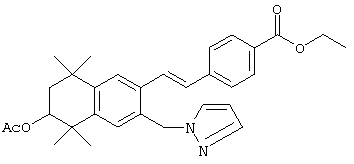
Раствор 0,45 г (0,88 ммоля) этил-4-[(Е)-2-(5,5,8,8-тетраметил-3-бромметил-6-ацетокси-5,6,7,8-тетрагидронафталин-2-ил)винил]бензоата и 0,24 г (3,5 ммоля) пиразола в 15 мл N-метилпирролидина выдерживали при 100°С. По прошествии 2 ч реакционную смесь охлаждали до комнатной температуры, выливали в рассол и экстрагировали этилацетатом. Органические экстракты промывали рассолом, сушили над сульфатом натрия и концентрировали под пониженным давлением. Остаток очищали экспресс-хроматографией (градиентное элюирование гексаном - 16% этилацетата/гексаном) с получением 32 г (73%-ный выход) этил-4-[(Е)-2-(5,5,8,8-тетраметил-3-пиразол-1-илметил-6-ацетокси-5,6,7,8-тетрагидронафталин-2-ил)винил]бензоата (М+=500).
Стадия Ж: получение 4-[(Е)-2-(5,5,8,8-тетраметил-3-пиразол-1-илметил-6-гидрокси-5,6,7,8-тетрагидронафталин-2-ил)винил]бензойной кислоты
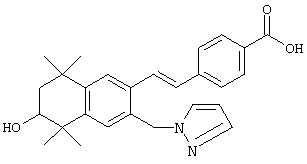
Смесь 0,32 г (0,64 ммоля) этил-4-[(Е)-2-(5,5,8,8-тетраметил-3-пиразол-1-илметил-6-ацетокси-5,6,7,8-тетрагидронафталин-2-ил)винил]бензоата в 10 мл 1 н. раствора LiOH и 20 мл этилового спирта кипятили с обратным холодильником. По истечении 1 ч реакционную смесь охлаждали до комнатной температуры, концентрировали под пониженным давлением и подкисляли 2 н. HCl. Водный раствор экстрагировали этилацетатом, промывали рассолом, сушили над сульфатом натрия и концентрировали под пониженным давлением. Остаток перекристаллизовывали из этилацетата/гексана с получением 0,22 г (82%-ный выход) 4-[(Е)-2-(5,5,8,8-тетраметил-3-пиразол-1-илметил-6-гидрокси-5,6,7,8-тетрагидронафталин-2-ил)винил]бензойной кислоты 55 (tпл: 241,6-242,0°).
Пример 38: получение 4-[(Е)-2-(5,5,8,8-тетраметил-3-пиразол-1-илметил-7-гидрокси-5,6,7,8-тетрагидронафталин-2-ил)винил]бензойной кислоты
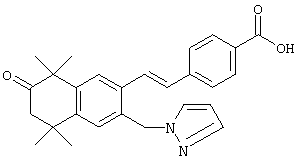
Стадия А: получение 7-ацетокси-3-бром-2-бромметил-5,5,8,8-тетраметил-5,6,7,8-тетрагидронафталина
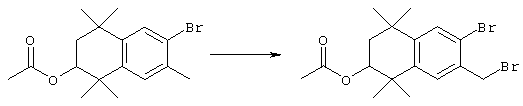
Смесь 7-ацетокси-3-бром-2,5,5,8,8-пентаметил-5,6,7,8-тетрагидронафталина, включавшего небольшое количество 2-дебромированного производного (10 г, 29,5 ммоля), растворяли в тетрахлориде углерода. Добавляли N-бромсукцинимида (5,25 г, 29,5 ммоля, перекристаллизованный из воды) и раствор нагревали до 50°С. Добавляли бензоилпероксида (0,36 г, 1,47 ммоля) и раствор доводили до кипения с обратным холодильником при одновременном облучении лампой с вольфрамовой нитью. За ходом реакции следили с помощью ТСХ (SiO2, 5% этилацетата/гексаны) и в совокупности в течение 3 ч дополнительно добавляли 0,15 г N-бромсукцинимида. Реакционную смесь охлаждали до комнатной температуры, фильтровали и выпаривали с получением полутвердого остатка. Хроматографией (SiO2, 5% этилацетата/гексаны) в виде масла получали продукт (9,85 г), который включал неразделявшуюся смесь 7-ацетокси-3-бром-2-бромметил-5,5,8,8-тетраметил-5,6,7,8-тетрагидронафталина и соответствовавшего 3-дебромированного производного, которую использовали непосредственно в последующей реакции.
Стадия Б: получение 7-ацетокси-3-бром-2-формил-5,5,8,8-тетраметил-5,6,7,8-тетрагидронафталина
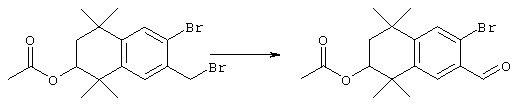
В раствор метоксида натрия (7 мл, 30,6 ммоля, раствор в метаноле концентрацией 25 мас.%), разбавленный 7 мл дополнительного количества метанола, добавляли раствор 2-нитропропана (3,63 мл, 35,33 ммоля) в 3 мл метанола. Реакционную смесь перемешивали в течение 10 мин и медленно с перемешиванием добавляли раствор смешанного продукта (9,85 г) предыдущей реакции в 84 мл метанола. В дальнейшем за ходом реакции следили с помощью ТСХ (SiO2, 5% этилацетата/гексаны) и после 5 ч выдержки при КТ убеждались в ее завершении. Реакционную смесь концентрировали и разделяли между этилацетатом и насыщенным раствором NaHCO3. Органический слой промывали рассолом, сушили над сульфатом натрия и выпаривали с получением в виде густого масла продукта, который очищали хроматографией (SiO2, 2-5 % этилацетата/гексан) с выделением в виде прозрачного масла 7-ацетокси-З-бром-2-формил-5,5,8,8-тетраметил-5,6,7,8-тетрагидронафталина (3 г, 36%-ный выход), который при стоянии медленно затвердевал.
Стадия В: получение 7-ацетокси-3-бром-2-(1,3-диоксолан-2-ил)-5,5,8,8-тетраметил-5,6,7,8-тетрагидронафталина
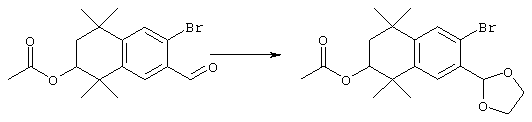
7-ацетокси-3-бром-2-формил-5,5,8,8-тетраметил-5,6,7,8-тетрагидронафталин (0,3 г, 0,849 ммоля) из предыдущей реакции растворяли в 2 мл бензола. Добавляли этиленгликоля (0,104 мл, 1,87 ммоля) и моногидрата п-толуолсульфоновой кислоты (0,024 г, 0,127 ммоля). На реакционной колбе устанавливали аппарат Дина-Старка и реакционную смесь кипятили с обратным холодильником в течение нескольких часов. Когда с помощью ТСХ (SiO2, 20% этилацетата/гексаны) убеждались в завершении реакции, добавляли дихлорметана (25 мл) и раствора бикарбоната натрия (50 мл) и слои разделяли.
Водный слой еще раз экстрагировали дихлорметаном. Объединенные органические слои промывали рассолом, сушили над сульфатом натрия и концентрировали с получением в виде маслянистого остатка 7-ацетокси-3-бром-2-(1,3-диоксолан-2-ил)-5,5,8,8-тетраметил-5,6,7,8-тетрагидронафталина, который использовали непосредственно на следующей стадии.
Стадия Г: получение 3-бром-2-(1,3-диоксолан-2-ил)-7-гидрокси-5,5,8,8-тетраметил-5,6,7,8-тетрагидронафталина
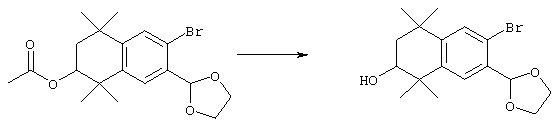
Сырой ацеталь с предыдущей стадии растворяли в 8 мл тетрагидрофурана/метанола в соотношении 1:1 и медленно добавляли раствор LiOH (0,17 г, 4 ммоля) в 2 мл воды. Реакционную смесь перемешивали в течение 2 ч, а затем разделяли между этилацетатом и водой. 1 М HCl значение рН водной фазы доводили до 6. Органический слой промывали рассолом, сушили над сульфатом натрия и концентрировали с получением в виде пенистого остатка 3-бром-2-(1,3-диоксолан-2-ил)-7-гидрокси-5,5,8,8-тетраметил-5,6,7,8-тетрагидронафталина (0,29 г, 96%-ный выход).
Стадия Д: получение 3-бром-2-(1,3-диоксолан-2-ил)-7-оксо-5,5,8,8-тетраметил- 5,6,7,8-тетрагидронафталина
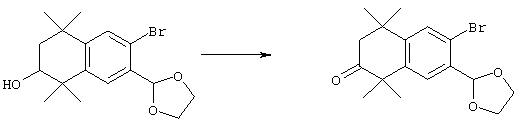
Вышеупомянутый спирт (0,29 г, 0,816 ммоля) растворяли в 16 мл дихлорметана. Добавляли периодинана Десс-Мартина (0,38 г, 0,9 ммоля, фирма Lancaster). Реакционную смесь перемешивали в течение 1 ч и обрабатывали выливанием в насыщенный раствор бикарбоната натрия и 1 М тиосульфата натрия. Эту смесь дважды экстрагировали дихлорметаном и объединенные органические слои промывали водой, раствором бикарбоната натрия и сушили над сульфатом натрия. Растворитель выпаривали и остаток хроматографировали (SiO2, 20% этилацетата/гексаны) с получением в виде прозрачного масла 3-бром-2-(1,3-диоксолан-2-ил)-7-оксо-5,5,8,8-тетраметил-5,6,7,8-тетрагидронафталина (200 мг, 69%-ный выход).
Стадия Е: получение 3-бром-2-формил-7-оксо-5,5,8,8-тетраметил-5,6,7,8-тетрагидронафталина
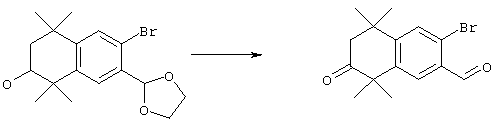
Вышеупомянутый кетон растворяли в приблизительно 5 мл тетрагидрофурана и добавляли 0,75 мл 3 М HCl. Смесь перемешивали в течение 3 ч при комнатной температуре, после чего в течение 1 ч при 40°С. Реакционную смесь разделяли между этилацетатом и насыщенным раствором бикарбоната натрия. Органический слой промывали рассолом, сушили над сульфатом натрия и концентрировали с получением в виде твердого вещества 3-бром-2-формил-7-оксо-5,5,8,8-тетраметил-5,6,7,8-тетрагидронафталина, который направляли непосредственно в последующую реакцию.
Стадия Ж: получение этил-(Е)-4-[2-(3-бром-7-оксо-5,5,8,8-тетраметил-5,6,7,8-тетрагидронафталин-2-ил)винил]бензоата
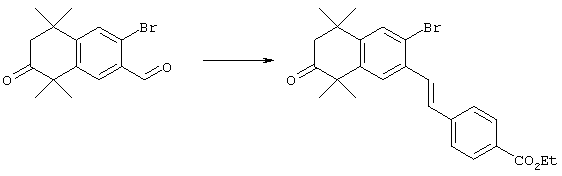
К вышеупомянутому альдегиду (0,19 г, 0,612 ммоля) в 3 мл тетрагидрофурана добавляли этил-4-(диэтоксифосфорилметил)бензоата (0,275 г, 0,919 ммоля). Образовавшийся раствор охлаждали в смеси воды со льдом и добавляли NaH (0,029 г, 0,73 ммоля, 60%-ная дисперсия в масле). Реакционную смесь перемешивали, одновременно давая ей нагреться в течение 2,5 ч до КТ. С помощью ТСХ (SiO2, 20% этилацетата/гексаны) убеждались в завершении реакции и реакционную смесь разделяли между 1 М HCl и этилацетатом. Органический слой сушили над сульфатом натрия, выпаривали до получения вещества в виде пены и очищали хроматографией на силикагеле (20% этилацетата/гексаны), выделяя этил-(Е)-4-[2-(3-бром-7-оксо-5,5,8,8-тетраметил-5,6,7,8-тетрагидронафталин-2-ил)винил]бензоат (230 мг, 82%-ный выход).
Стадия 3: получение этил-(Е)-4-[2-(3-гидроксиметил-7-оксо-5,5,8,8-тетраметил- 5,6,7,8-тетрагидронафталин-2-ил)винил] бензоата
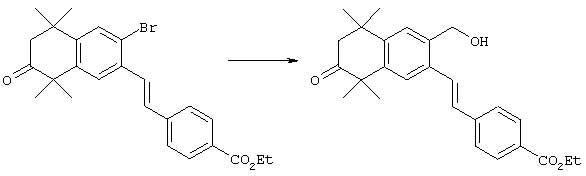
Вышеупомянутый бромированный сложный эфир (0,23 г, 0,5 ммоля) растворяли в 2 мл безводного 1,4-диоксана и добавляли гидроксиметилтрибутилолова (241 мг, 0,75 ммоля, см.: Seitz, D.E. и др., Synth.Comm. 1983, 13(2), 129; Kosugi, M. и др., Chem.Lett. 1985, 997) при одновременном дегазировании реакционного раствора аргоном. Добавляли тетракис(трифенилфосфин)палладия(0) (0,043 г, 0,038 ммоля) и реакционную смесь перемешивали при температуре кипения с обратным холодильником в течение ночи. Реакционную смесь охлаждали до комнатной температуры и разделяли между этилацетатом и насыщенным раствором бикарбоната натрия. Слои разделяли и водный слой еще раз экстрагировали этилацетатом. Объединенные органические слои промывали рассолом, сушили над сульфатом натрия и концентрировали. Полученный остаток очищали хроматографией на силикагеле (25% этилацетата/гексан) с получением этил-(Е)-4-[2-(3-гидроксиметил-7-оксо-5,5,8,8-тетраметил-5,6,7,8-тетрагидронафталин-2-ил)винил]бензоата (150 мг, 73%-ный выход).
Стадия И: получение этил-4-[2-(3-бромметил-7-оксо-5,5,8,8-тетраметил-5,6,7,8-тетрагидронафталин-2-ил)винил]бензоата
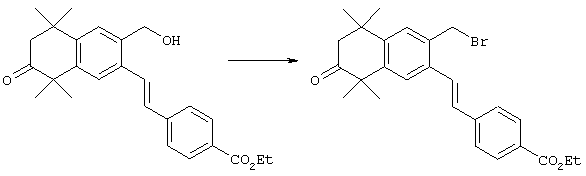
Вышеупомянутый гидроксиметильный материал (0,15 г, 0,368 ммоля) растворяли в 4 мл дихлорметана и обрабатывали трифенилфосфином (0,11 г, 0,42 ммоля) и N-бромсукцинимидом (0,076 г, 0,42 ммоля). Реакционную смесь перемешивали при КТ в течение 30 мин, а затем концентрировали с получением пенистого остатка, который хроматографировали (SiO2, 25% этилацетата/гексаны) с получением в виде масла этил-4-[2-(3-бромметил-7-оксо-5,5,8,8-тетраметил-5,6,7,8-тетрагидронафталин-2-ил)винил]бензоата (130 мг, 75%-ный выход).
Стадия К: получение этил (Е)-4-[2-(3-(пиразол-1-илметил)-7-оксо-5,5,8,8-тетраметил-5,6,7,8-тетрагидронафталин-2-ил)винил]бензоата
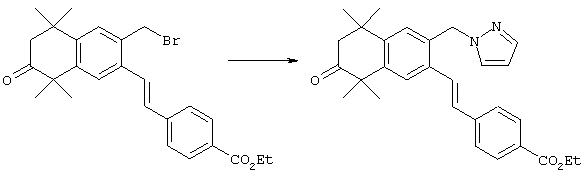
В раствор 18-краун-6 (0,092 г, 0,35 ммоля) и трет-бутоксида калия (0,04 г, 0,36 ммоля) в 2 мл тетрагидрофурана добавляли пиразола (0,024 г, 0,346 ммоля). Реакционную смесь перемешивали в течение 10 мин и по каплям в течение 5 мин добавляли раствор вышеупомянутого бромметильного материала (0,13 г, 0,277 ммоля) в 1 мл тетрагидрофурана. Реакционную смесь перемешивали в течение 45 мин при КТ, а затем разделяли между этилацетатом и раствором хлорида аммония. Этилацетатный слой сушили над сульфатом натрия, концентрировали и остаток хроматографировали (SiO2, 30% этилацетата/гексаны) с получением этил (Е)-4-[2-(3-(пиразол-1-илметил)-7-оксо-5,5,8,8-тетраметил-5,6,7,8-тетрагидронафталин-2-ил)винил]бензоата (120 мг, 94%-ный выход).
Стадия Л: получение (Е)-4-[2-(3-(пиразол-1-илметил)-7-оксо-5,5,8,8-тетраметил-5,6,7,8-тетрагидронафталин-2-ил)винил]бензойной кислоты
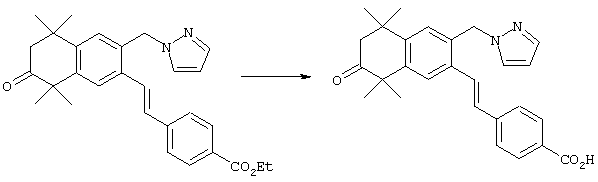
В раствор вышеупомянутого сложного эфира (0,12 г, 0,262 ммоля) в 4 мл этанола медленно добавляли раствор LiOH (0,055 г, 1,3 ммоля) в 0,65 мл воды. Мутную реакционную смесь нагревали до температуры 50°С, по достижении которой реакционная смесь становилась гомогенной. Реакционную смесь перемешивали при 50°С в течение 2 ч, а затем охлаждали до комнатной температуры и обрабатывали добавлением 1,1 мл 1 М раствора HCl. Белое твердое вещество отделяли и перемешивание продолжали в течение 30 мин. Это твердое вещество отфильтровывали, промывали 20% этанола/водой и сушили под вакуумом с получением в виде белого твердого вещества (Е)-4-[2-(3-(пиразол-1-илметил)-7-оксо-5,5,8,8-тетраметил-5,6,7,8-тетрагидронафталин-2-ил)винил]бензойной кислоты (71 мг, 63%-ный выход) 144; М-Н = 427; tпл: 264,8-2б5,9°С.
Пример 39: получение (Е)-4-[2-(3-(пиразол-1-илметил)-7-гидрокси-5,5,8,8-тетраметил-5,6,7,8-тетрагидронафталин-2-ил)винил]бензойной кислоты
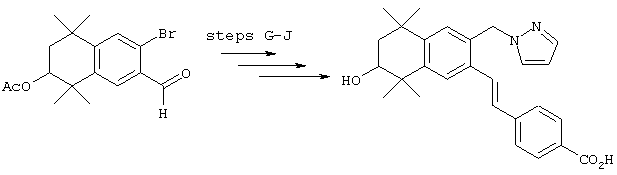
Проведение процессов так, как указано в описании приведенных выше стадий Ж-Л Примера 38, за исключением замены 3-бром-2-формил-7-оксо-5,5,8,8-тетраметил-5,6,7,8-тетрагидронафталина в начале стадии Ж 7-ацетокси-3-бром-2-формил-5,5,8,8-тетраметил-5,6,7,8-тетрагидронафталином, приводило к образованию в виде белого твердого вещества (Е)-4-[2-(3 -(пиразол-1 -илметил)-7-гидрокси-5,5,8,8-тетраметил-5,6,7,8-тетрагидронафталин-2-ил)винил]бензойной кислоты (158 мг, 81%-ный выход) 137: М-Н = 429; tпл: 247,6-248,4°C.
Пример 40: получение 4-[(Е)-2-(5,5,8,8-тетраметил-3-пиразол-1-илметил-6-кето-5,6,7,8-тетрагидронафталин-2-ил)винил]бензойной кислоты
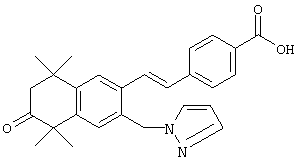
Стадия А: получение 4-[(Е)-2-(5,5,8,8-тетраметил-3-метил-6-гидрокси-5,6,7,8-тетрагидронафталин-2-ил)винил]бензойной кислоты
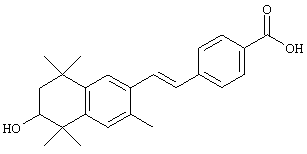
Суспензию 6 г (13,8 ммоля) этил-4-[(Е)-2-(5,5,8,8-тетраметил-3-метил-6-ацетокси-5,6,7,8-тетрагидронафталин-2-ил)винил]бензоата, полученного на стадии Г в примере 37, при температуре кипения с обратным холодильником перемешивали с 30 мл 1 н. LiOH и 30 мл МеОН. По истечении 1,5 ч реакционную смесь охлаждали до комнатной температуры, подкисляли 2 н. HCl и экстрагировали этилацетатом. Органические фракции промывали рассолом, сушили над сульфатом натрия и концентрировали под пониженным давлением с получением 3,5 г (70%-ный выход) 4-[(Е)-2-(5,5,8,8-тетраметил-3-метил-6-гидрокси-5,6,7,8-тетрагидронафталин-2-ил)винил]бензойной кислоты.
Стадия Б: получение метил-4-[(Е)-2-(5,5,8,8-тетраметил-3-метил-6-гидрокси-5,6,7,8-тетрагидронафталин-2-ил)винил]бензойной кислоты
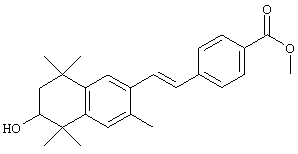
В смесь из 3,4 г (9,3 ммоля) 4-[(Е)-2-(5,5,8,8-тетраметил-3-метил-6-гидрокси-5,6,7,8-тетрагидронафталин-2-ил)винил]бензойной кислоты в 100 мл MeOH/CH2Cl2 в соотношении 1:1 при комнатной температуре добавляли 5,4 мл 2 М (11,2 ммоля) триметилсилилдиазометана. По прошествии 0,5 ч дополнительно добавляли 5,4 мл триметилсилилдиазометана. По прошествии 16 ч добавляли 0,5 мл уксусной кислоты и реакционную смесь концентрировали под пониженным давлением. Полученный остаток разделяли между водой и этилацетатом и органические фракции отделяли, промывали водным бикарбонатом натрия и рассолом, сушили над сульфатом натрия и адсорбировали на силикагеле.
Очисткой экспресс-хроматографией (градиентное элюирование гексаном - 15% этилацетата/гексаном) получали 1,8 г (51%-ный выход) метил-4-[(Е)-2-(5,5,8,8-тетраметил-3-метил-6-гидрокси-5,6,7,8-тетрагидронафталин-2-ил)винил]бензойную кислоту (М+ = 378).
Стадия В: получение метил-4-[(Е)-2-(5,5,8,8-тетраметил-3-метил-6-оксо-5,6,7,8-тетрагидронафталин-2-ил)винил]бензойной кислоты
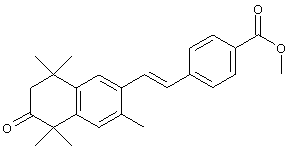
В раствор 1,6 г (4,2 ммоля) метил-4-[(Е)-2-(5,5,8,8-тетраметил-3-метил-6-гидрокси-5,6,7,8-тетрагидронафталин-2-ил)винил]бензойной кислоты в 50 мл метиленхлорида при комнатной температуре добавляли 1,97 г (4,65 ммоля) периодинана Десса-Мартина. По истечении 1 ч реакционную смесь выливали в рассол и экстрагировали метиленхлоридом. Органические фракции сушили над сульфатом натрия, адсорбировали на силикагеле и очищали экспресс-хроматографией (градиентное элюирование гексаном - 8% этилацетата/гексаном) с получением 1,45 г (92%-ный выход) метил-4-[(Е)-2-(5,5,8,8-тетраметил-3-метил-6-оксо-5,6,7,8-тетрагидронафталин-2-ил)винил]бензойной кислоты (М+ = 376).
Стадия Г: получение метил-4-[(Е)-2-(5,5,8,8-тетраметил-3-бромметил-6-оксо-5,6,7,8-тетрагидронафталин-2-ил)винил]бензойной кислоты
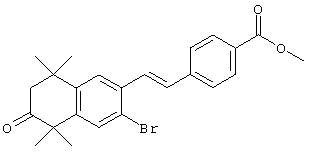
В раствор 1,4 г (3,7 ммоля) метил-4-[(Е)-2-(5,5,8,8-тетраметил-3-метил-6-оксо-5,6,7,8-тетрагидронафталин-2-ил)винил]бензойной кислоты и 0,86 г (4,8 ммоля) N-бромсукцинимида в 50 мл тетрахлорида углерода добавляли 45 мг (0,19 ммоля) бензоилпероксида и реакционную смесь выдерживали при температуре кипения с обратным холодильником. По истечении 1 ч дополнительно добавляли 45 мг бензоилпероксида и 25 мл тетрахлорида углерода. Спустя в общем 3 ч добавляли 0,2 г N-бромсукцинимида. По прошествии в совокупности 6 ч смесь охлаждали до комнатной температуры, промывали 10%-ным водным бисульфитом натрия и рассолом, сушили над сульфатом натрия и адсорбировали на силикагеле. Очищали экспресс-хроматографией (гексаном - 8% этилацетата/гексаном) с получением 1,0 г (59%-ный выход) метил-4-[(Е)-2-(5,5,8,8-тетраметил-3-бромметил-6-оксо-5,6,7,8-тетрагидронафталин-2-ил)винил]бензойной кислоты.
Стадия Д: получение 4-[(Е)-2-(5,5,8,8-тетраметил-3-пиразол-1-илметил-6-оксо-5,6,7,8-тетрагидронафталин-2-ил)винил]бензойной кислоты
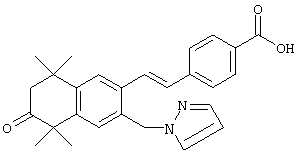
Раствор 1,0 г (2,2 ммоля) метил-4-[(Е)-2-(5,5,8,8-тетраметил-3-бромметил-6-оксо-5,6,7,8-тетрагидронафталин-2-ил)винил]бензойной кислоты и 0,6 г (8,8 ммоля) пиразола в 15 мл N-метилпирролидина выдерживали при 100°С. По прошествии 2 ч реакционную смесь охлаждали до комнатной температуры, выливали в рассол, экстрагировали этилацетатом, промывали рассолом и сушили над сульфатом натрия. Органический раствор адсорбировали на силикагеле и очищали экспресс-хроматографией (градиентное элюирование гексаном - 18% этилацетата/гексаном) с получением 0,71 г (73%-ный выход) метил-4-[(Е)-2-(5,5,8,8-тетраметил-3-пиразол-1-илметил-6-оксо-5,6,7,8-тетрагидронафталин-2-ил)винил]бензойной кислоты (МН+ = 443).
Смесь из 0,7 г (1,58 ммоля) метил-4-[(Е)-2-(5,5,8,8-тетраметил-3-пиразол-1-илметил-6-оксо-5,6,7,8-тетрагидронафталин-2-ил)винил]бензойной кислоты в 40 мл МеОН и 20 мл 1 н. LiOH выдерживали при температуре кипения с обратным холодильником. По истечении 1 ч реакционную смесь охлаждали до комнатной температуры и под пониженным давлением удаляли МеОН. Водный раствор подкисляли 2 н. HCl, экстрагировали этилацетатом, промывали рассолом, сушили над сульфатом натрия и адсорбировали на силикагеле. Продукт очищали экспресс-хроматографией (градиентное элюирование: 10-30% этилацетата/гексан с 0,2% уксусной кислоты) и перекристаллизацией (этилацетат/гексан) получали 100 мг (15%-ный выход) 4-[(Е)-2-(5,5,8,8-тетраметил-3-пиразол-1-илметил-6-оксо-5,6,7,8-тетрагидронафталин-2-ил)винил]бензойной кислоты (tпл: 233-233,5) 133.
Пример 41: получение 4-[(Е)-2-(5,5,8,8-тетраметил-3-пиразол-1-илметил-6,7-транс-дигидрокси-5,6,7,8-тетрагидронафталин-2-ил)винил]бензойной кислоты
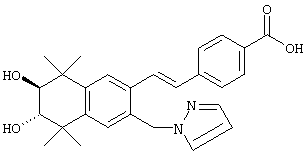
Стадия А: получение 2-бром-3-метил-6-гидрокси-5,5,8,8-тетраметил-5,6,7,8-тетрагидронафталина
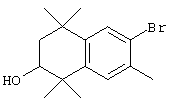
Смесь 20 г (59 ммоля) 2-бром-3-метил-6-ацетокси-5,5,8,8-тетраметил-5,6,7,8-тетрагидронафталина, 177 мл 1 н. LiOH и 350 мл метилового спирта выдерживали при температуре кипения с обратным холодильником. По истечении 1 ч реакционную смесь охлаждали до комнатной температуры, концентрировали под пониженным давлением, подкисляли 2 н. HCl, экстрагировали этилацетатом, сушили над сульфатом натрия и адсорбировали на силикагеле. Продукт очищали экспресс-хроматографией (градиентное элюирование 5-20% этилацетата/гексаном) с получением 15,3 г (87%-ный выход) 2-бром-3-метил-6-гидрокси-5,5,8,8-тетраметил-5,6,7,8-тетрагидронафталина.
Стадия Б: получение 2-бром-3,5,5,8,8-пентаметил-5,8-дигидронафталина
В раствор 15,3 г (51,5 ммоля) 2-бром-3-метил-6-гидрокси-5,5,8,8-тетраметил-5,6,7,8-тетрагидронафталина в 120 мл пиридина вводили 17,9 мл (192 ммоля) оксихлорида трехвалентного фосфора. Реакционную смесь выдерживали при 100°С. По прошествии 6 ч ее охлаждали до комнатной температуры и осторожно с перемешиванием выливали на лед. По истечении 1 ч экстрагировали этилацетатом, промывали 2 н. HCl и рассолом, сушили над сульфатом натрия и концентрировали под пониженным давлением. Полученный остаток очищали гравитационной хроматографией (градиентное элюирование гексаном - 10% этилацетата/гексаном) с получением 12,5 г (87%-ный выход) 2-бром-3,5,5,8,8-пентаметил-5,8-дигидронафталина.
Стадия В: получение 4-бром-2,2,5,7,7-пентаметил-1а,2,7,7а-тетрагидро-1-оксациклопропа[b]нафталина
В раствор 10 г (35,8 ммоля) 2-бром-3,5,5,8,8-пентаметил-5,8-дигидронафталина в 300 мл дихлорметана при 0° отдельными порциями в течение 20 мин вводили 12,4 г (35,8 ммоля) метахлорпербензойной кислоты. Спустя один час после завершения добавления реакционную смесь промывали 10%-ным водным бисульфитом натрия и рассолом, сушили над сульфатом натрия и адсорбировали на силикагеле. Очищали экспресс-хроматографией (градиентное элюирование гексаном - 10% этилацетата/гексаном) с получением 9 г (85%-ный выход) 4-бром-2,2,5,7,7-пентаметил-1а,2,7,7а-тетрагидро-1-оксациклопропа[b]нафталина.
Стадия Г: получение 2-бром-3-метил-6,7-дигидрокси-5,5,8,8-тетраметил-5,6,7,8-тетрагидронафталина
Раствор 2,0 г (6,8 ммоля) 4-бром-2,2,5,7,7-пентаметил-1а,2,7,7а-тетрагидро-1 -оксациклопропа[b]нафталина в 20 мл уксусной кислоты с 0,2 мл H2SO4 выдерживали при температуре кипения с обратным холодильником. По прошествии 0,5 ч реакционную смесь охлаждали до комнатной температуры и концентрировали под пониженным давлением. Полученный остаток растворяли в толуоле и повторно концентрировали под пониженным давлением. Продукт растворяли в 40 мл метилового спирта и добавляли 20 мл 1 н. LiOH, Реакционный раствор выдерживали при температуре кипения с обратным холодильником. По истечении 1 ч реакционную смесь охлаждали до комнатной температуры и концентрировали под пониженным давлением. Реакционную смесь подкисляли 2 н. HCl, экстрагировали этилацетатом, промывали рассолом, сушили над сульфатом натрия, а затем адсорбировали на силикагеле. Продукт очищали экспресс-хроматографией (градиентное элюирование гексаном - 20% этилацетата/гексаном) с получением 1,2 г (56%-ный выход) 2-бром-3-метил-6,7-дигидрокси-5,5,8,8-тетраметил-5,6,7,8-тетрагидронафталина.
Стадия Д: получение 6-бром-2,2,4,4,7,9,9-гептаметил-3а,4,9,9а-тетрагидронафтао[2,3-d][1,3]-транс-диоксола
В суспензию 1,0 г (3,2 ммоля) 2-бром-3-метил-6,7-дигидрокси-5,5,8,8-тетраметил-5,6,7,8-тетрагидронафталина в 30 мл 2,2-диметоксипропана вводили 100 мг п-толуолсульфоновой кислоты. Реакционную смесь перемешивали при комнатной температуре. По истечении 1 ч реакционную смесь выливали в водный бикарбонат натрия и экстрагировали этилацетатом. Органические экстракты промывали рассолом, сушили над сульфатом натрия, концентрировали под пониженным давлением и очищали экспресс-хроматографией (градиентное элюирование гексаном - 5% этилацетата) с получением 1,2 г (97%-ный выход) 6-бром-2,2,4,4,7,9,9-гептаметил-3а,4,9,9а-тетрагидронафтао[2,3-d][1,3]-транс-диоксола.
Стадия Е: получение 2,2,4,4,7,9,9-гептаметил-3а,4,9,9а-тетрагидронафто[2,3-d][1,3]-трансдиоксол-6-карбальдегида
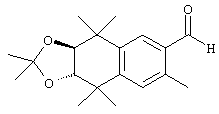
В раствор 3,8 г (10,8 ммоля) 6-бром-2,2,4,4,7,9,9-гептаметил-3а,4,9,9а-тетрагидронафтао[2,3-d][1,3]-транс-диоксола в 50 мл ТГФ при -78°С вводили 13,5 мл 1,6 М (21,5 ммоля) н-BuLi. По прошествии 1 ч добавляли раствор 2,4 мл (21,5 ммоля) N-формилпиперидина в 10 мл ТГФ. По истечении 1,5 ч добавляли насыщенного водного раствора хлорида аммония, нагревали до комнатной температуры, экстрагировали этилацетатом, промывали рассолом, сушили над сульфатом натрия и адсорбировали на силикагеле. Очищали экспресс-хроматографией (градиентное элюирование гексаном - 3% этилацетата/гексаном). Выделяли 2,1 г (64%-ный выход) 2,2,4,4,7,9,9-гептаметил-3а,4,9,9а-тетрагидронафто[2,3-d][1,3]-трансдиоксол-6-карбальдегида (М+= 302).
Стадия Ж: получение метилового эфира 4-[(Е)-2-(2,2,4,4,7,9,9)гептаметил-3а,4,9,9а-тетрагидронафто[2,3d][1,3]-транс-диоксол-6-ил)винил]бензойной кислоты
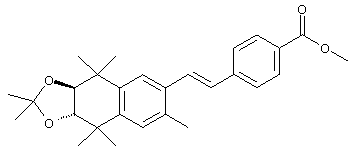
В раствор 2,0 г (6,6 ммоля) 2,2,4,4,7,9,9-гептаметил-3а,4,9,9а-тетрагидронафто[2,3-d][1,3]-транс-диоксол-6-карбальдегида и 2,2 г (8,6 ммоля) метилобензилового эфира 4-(диметоксифосфорилметил)бензойной кислоты в 50 мл толуола при 0° добавляли 5,6 мл 1,7 М (8,6 ммоля) трет-пентилата калия в толуоле. По истечении 1,5 ч смесь выливали в рассол, экстрагировали этилацетатом, сушили над сульфатом натрия и адсорбировали на силикагеле. Очищали экспресс-хроматографией (градиентное элюирование гексаном - 2% этилацетата/гексаном) с получением 2,5 г (87%-ный выход) метилового эфира 4-[(Е)-2-(2,2,4,4,7,9,9)гептаметил-3а,4,9,9а-тетрагидронафто[2,3d][1,3]-транс-диоксол-6-ил)винил]бензойной кислоты.
Стадия 3: получение метилового эфира 4-[(Е)-2-(7-бромметил-2,2,4,4,7,9,9)гептаметил-3а,4,9,9а-тетрагидронафто[2,3d][1,3]-транс-диоксол-6-ил)винил]бензойной кислоты
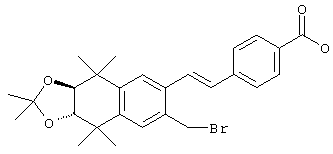
Раствор 2,4 г (5,5 ммоля) метилового эфира 4-[2-(2,2,4,4,7,9,9-гептаметил-3а,4,9,9а-тетрагидронафто[2,3d][1,3]-транс-диоксол-6-ил)винил]бензойной кислоты, 1,47 г (8,28 ммоля) N-бромсукцинимида и 67 мг (0,28 ммоля) бензоилпероксида в 50 мл CCl4 выдерживали при температуре кипения с обратным холодильником. По прошествии 2 ч дополнительно добавляли 34 мг (0,14 ммоля) бензоилпероксида. Спустя в общей сложности 3 ч реакционную смесь фильтровали и фильтрат промывали водным бисульфитом натрия, сушили над сульфатом натрия и адсорбировали на силикагеле. Продукт очищали экспресс-хроматографией (градиентное элюирование гексаном - 3% этилацетата/гексаном) с получением 1,7 г метилового эфира 4-[(Е)-2-(7-бромметил-2,2,4,4,9,9)гексаметил-3а,4,9,9а-тетрагидронафто[2,3d][1,3]-транс-диоксол-6-ил)винил]бензойной кислоты.
Стадия И: получение метилового эфира 4-[(Е)-2-(2,2,4,4,9,9)гексаметил-7-пиразол-1-илметил-3а,4,9,9а-тетрагидронафто[2,3d][1,3]-транс-диоксол-6-ил)винил]бензойной кислоты
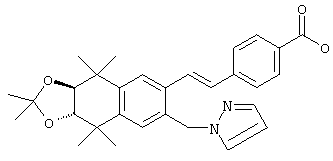
Раствор 170 мг (0,33 ммоля) метилового эфира 4-[(Е)-2-(7-бромметил-2,2,4,4,9,9)гексаметил-3а,4,9,9а-тетрагидронафто[2,3d][1,3]-транс-диоксол-6-ил)винил]бензойной кислоты и 47 мг (0,7 ммоля) пиразола в 10 мл N-МП выдерживали при 100°С. По прошествии 2 ч реакционную смесь охлаждали до комнатной температуры, выливали в рассол, экстрагировали этилацетатом, промывали рассолом, сушили над сульфатом натрия и адсорбировали на силикагеле. Продукт очищали экспресс-хроматографией (градиентное элюирование гексаном - 15% этилацетата/гексаном) с получением 75 мг (45%-ный выход) метилового эфира 4-[(Е)-2-(2,2,4,4,9,9-гексаметил-7-пиразол-1-илметил-3а,4,9,9а-тетрагидронафто[2,3d][1,3]-транс-диоксол-6-ил)винил]бензойной кислоты.
Стадия К: получение 4-[(Е)-2-(5,5,8,8-тетраметил-3-пиразол-1-илметил-6,7-транс-дигидрокси-5,6,7,8-тетрагидронафталин-2-ил)винил] бензойной кислоты
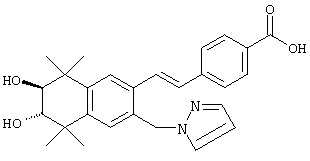
Смесь из 75 мг (0,15 ммоля) метилового эфира 4-[(Е)-2-(2,2,4,4,9,9-гексаметил)-7-пиразол-1-илметил-3а,4,9,9а-тетрагидронафто[2,3d][1,3]-транс-диоксол-6-ил)винил]бензойной кислоты в 10 мл 1 н. HCl и 10 мл ТГФ перемешивали при комнатной температуре. По истечении 1 ч реакционную смесь концентрировали под пониженным давлением, экстрагировали этилацетатом, концентрировали под пониженным давлением и растворяли в 20 мл метилового спирта и 10 мл LiOH. Реакционную смесь кипятили с обратным холодильником. По истечении 1 ч реакционную смесь охлаждали до комнатной температуры и концентрировали под пониженным давлением. Реакционную смесь подкисляли 2 н. HCl, экстрагировали этилацетатом, промывали рассолом, сушили над сульфатом натрия и концентрировали под пониженным давлением с получением 60 мг (90%-ный выход) 4-[(Е)-2-(5,5,8,8-тетраметил-3-пиразол-1-илметил-6,7-транс-дигидрокси-5,6,7,8-тетрагидронафталин-2-ил)винил]бензойной кислоты (МН+ = 447) 145.
Пример 42: получение 4-[(Е)-2-(5,5,8,8-тетраметил-3-пиразол-1-илметил-6,7-цис-дигидрокси-5,6,7,8-тетрагидронафталин-2-ил)винил]бензойной кислоты
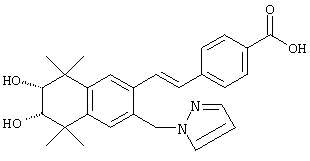
Стадия А: получение 2-бром-цис-6,7-дигидрокси-3,5,5,8,8-пентаметил-5,6,7,8-тетрагидронафталина
В раствор 9,41 г (33,7 ммоля) 2-бром-3,5,5,8,8-пентаметил-5,8-дигидронафталина в 110 мл пиридина в азотной атмосфере вводили 8,65 г (34,0 ммоля) тетроксида осмия. После перемешивания при комнатной температуре в течение 18 ч добавляли 17,3 г (166 ммолей) бисульфита натрия в 110 мл воды. По прошествии 2 ч образовавшийся раствор разделяли между этилацетатом и водной соляной кислотой. Органический слой промывали водой, промывали рассолом, сушили над безводным сульфатом натрия и концентрировали под вакуумом. Сырой материал очищали экспресс-хроматографией с элюированием 30% этилацетата/гексаном и получением в виде белого твердого вещества 7,97 г (75%-ный выход) 2-бром-цис-6,7-дигидрокси-3,5,5,8,8-пентаметил-5,6,7,8-тетрагидронафталина (М+= 312).
Стадия Б: получение 2-бром-цис-6,7-дигидрокси-3,5,5,8,8-пентаметил-5,6,7,8-тетрагидронафталинацетонида
Раствор 7,73 г (24,7 ммоля) 2-бром-цис-6,7-дигидрокси-3,5,5,8,8-пентаметил-5,6,7,8-тетрагидронафталина и 445 мг (2,39 ммоля) моногидрата п-толуолсульфоновой кислоты в 100 мл 2,2-диметоксипропана кипятили с обратным холодильником в течение 90 мин. После охлаждения образовавшийся раствор разделяли между этилацетатом и разбавленным водным бикарбонатом натрия. Органический слой промывали рассолом, сушили над безводным бикарбонатом натрия и концентрировали под вакуумом с получением 9,10 г 2-бром-цис-6,7-дигидрокси-3,5,5,8,8-пентаметил-5,6,7,8-тетрагидронафталинацетонида (М+=352), который использовали без дополнительной очистки.
Стадия В: получение 3-метил-цис-6,7-дигидрокси-5,5,8,8-тетраметил-5,6,7,8-тетрагидро-2-нафтальдегидацетонида
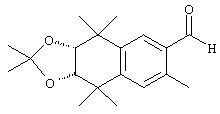
В охлажденный до -78°С раствор 9,04 г (25,6 ммоля) 2-бром-цис-6,7-дигидрокси-3,5,5,8,8-пентаметил-5,6,7,8-тетрагидронафталинацетонида в 110 мл тетрагидрофурана вводили 32,0 мл (51,1 ммоля) 1,6 М раствора н-бутиллития в гексанах. По истечении 1 ч выдержки при -78°С добавляли 5,79 г (51,1 ммоля) 1-формилпиперидина. По прошествии 20 мин выдержки при -78°С реакцию в образовавшейся смеси гасили водой. После нагревания до комнатной температуры смесь разделяли между этилацетатом и водой. Органический слой промывали рассолом, сушили над безводным сульфатом натрия и концентрировали под вакуумом. Сырой материал очищали экспресс-хроматографией с элюированием 5% этилацетата/гексаном и получением 5,65 г (73%-ный выход) 3-бром-цис-6,7-дигидрокси-5,5,8,8-тетраметил-5,6,7,8-тетрагидро-2-нафтальдегидацетонида (МН+ = 303).
Стадия Г: получение метилового эфира 4-[(Е)-2-(2,2,4,4,7,9,9)гептаметил-3а,4,9,9а-тетрагидронафто[2,3d][1,3]-цис-диоксол-6-ил)винил]бензойной кислоты
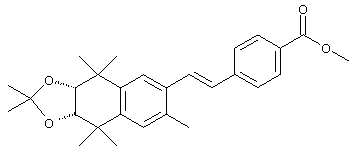
В раствор 5,3 г (17,5 ммоля) 2,2,4,4,7,9,9-гептаметил-3а,4,9,9а-тетрагидронафто[2,3-d][1,3]-цис-диоксол-6-карбальдегида и 5,9 г (22,8 ммоля) метилового эфира 4-(диметоксифосфорилметил)бензойной кислоты в 50 мл толуола при 0° вводили 13,4 мл 1,7 М (22,2 ммоля) трет-пентилата калия. По истечении 1 ч реакционную смесь выливали в рассол, экстрагировали этилацетатом, промывали рассолом, сушили над сульфатом натрия и адсорбировали на силикагеле. Продукт очищали экспресс-хроматографией (градиентное элюирование гексаном - 15% этилацетата/гексаном) с получением 6,0 г (79%-ный выход) метилового эфира 4-[(Е)-2-(2,2,4,4,7,9,9)гептаметил-3а,4,9,9а-тетрагидронафто[2,3d][1,3]-цис-диоксол-6-ил)винил]бензойной кислоты.
Стадия Д: получение метилового эфира 4-[(Е)-2-(5,5,8,8-тетраметил-3-метил-6,7-цис-диацетокси-5,6,7,8-тетрагидронафталин-2-ил)винил]бензойной кислоты
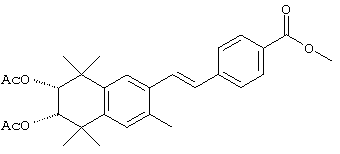
Смесь 4,7 г (10,8 ммоля) метилового эфира 4-[(Е)-2-(2,2,4,4,7,9,9)гептаметил-3а,4,9,9а-тетрагидронафто[2,3d][1,3]-цис-диоксол-6-ил)винил]бензойной кислоты в 50 мл ТГФ и 50 мл 1 н. HCl перемешивали при комнатной температуре. Спустя 3 ч смесь концентрировали под пониженным давлением, экстрагировали этилацетатом, промывали рассолом, сушили над сульфатом натрия и концентрировали под пониженным давлением. Полученный остаток растворяли в 50 мл пиридина и добавляли 5 мл (53 ммоля) уксусного ангидрида. Реакционную смесь выдерживали при 60°С. По истечении 2 ч реакционную смесь охлаждали до комнатной температуры, разбавляли водой и экстрагировали этилацетатом. Органические фракции промывали 2 н. HCl и рассолом, сушили над сульфатом натрия и очищали экспресс-хроматографией (градиентное элюирование гексаном - 10% этилацетата/гексаном) с получением 5,2 г (100%-ный выход) метилового эфира 4-[(Е)-2-(5,5,8,8-тетраметил-3-метил-6,7-цис-диацетокси-5,6,7,8-тетрагидронафталин-2-ил)винил]бензойной кислоты.
Стадия Е: получение метилового эфира 4-[(Е)-2-(5,5,8,8-тетраметил-3-бромметил-6,7-цис-диацетокси-5,6,7,8-тетрагидронафталин-2-ил)винил]бензойной кислоты
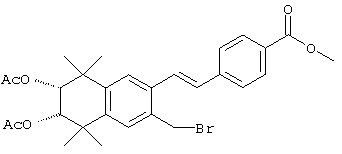
Раствор 5,2 г (10,8 ммоля) метилового эфира 4-[(Е)-2-(5,5,8,8-тетраметил-3-метил-6,7-цис-диацетокси-5,6,7,8-тетрагидронафталин-2-ил)винил]бензойной кислоты и 2,4 г (13,3 ммоля) N-бромсукцинимида со 124 мг (0,5 ммоля) бензоилпероксида в 50 мл тетрахлорида углерода выдерживали при температуре кипения с обратным холодильником. По прошествии 5 ч смесь охлаждали до комнатной температуры, фильтровали и фильтрат промывали 10%-ным водным бисульфитом и рассолом, сушили над сульфатом натрия и очищали экспресс-хроматографией (градиентное элюирование гексаном - 10% этилацетата/гексаном) с получением 4,0 г (66%-ный выход) метилового эфира 4-[(Е)-2-(5,5,8,8-тетраметил-3-бромметил-6,7-цис-диацетокси-5,6,7,8-тетрагидронафталин-2-ил)винил]бензойной кислоты.
Стадия Ж: получение метилового эфира 4-[(Е)-2-(5,5,8,8-тетраметил-3-пиразол-1-илметил-6,7-цис-диацетокси-5,6,7,8-тетрагидронафталин-2-ил)винил]бензойной кислоты
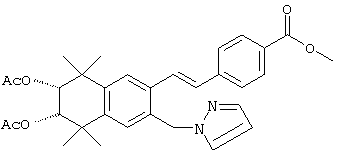
В раствор 730 мг (2,76 ммоля) 18-краун-6 и 338 мг (3 ммоля) трет-бутоксида калия в 30 мл ТГФ вводили 205 мг (3 ммоля) пиразола. По прошествии 20 мин реакционную смесь охлаждали до 0 и по каплям добавляли раствор 1,4 г (2,5 ммоля) метилового эфира 4-[(Е)-2-(5,5,8,8-тетраметил-3-бромметил-6,7-цис-диацетокси-5,6,7,8-тетрагидронафталин-2-ил)винил]бензойной кислоты в 20 мл ТГФ. Спустя 3 ч реакционную смесь выливали в рассол, экстрагировали этилацетатом, промывали рассолом, сушили над сульфатом натрия и адсорбировали на силикагеле. Продукт очищали экспресс-хроматографией (градиентное элюирование гексаном - 30% этилацетата/гексаном) с получением 1,05 г (77%-ный выход) метилового эфира 4-[(Е)-2-(5,5,8,8-тетраметил-3-пиразол-1-илметил-6,7-цис-диацетокси-5,6,7,8-тетрагидронафталин-2-ил)винил]бензойной кислоты.
Стадия 3: получение 4-[(Е)-2-(5,5,8,8-тетраметил-3-пиразол-1-илметил-6,7-цис-дигидрокси-5,6,7,8-тетрагидронафталин-2-ил)винил]бензойной кислоты
Смесь из 1,0 г (2,2 ммоля) метилового эфира 4-[(Е)-2-(5,5,8,8-тетраметил-3-пиразол-1-илметил-6,7-цис-диацетокси-5,6,7,8-тетрагидронафталин-2-ил)винил]бензойной кислоты в 20 мл метилового спирта и 10 мл 1 н. LiOH кипятили с обратным холодильником. По истечении 1 ч смесь охлаждали до комнатной температуры, концентрировали под пониженным давлением, подкисляли 2 н. HCl, экстрагировали этилацетатом, промывали рассолом, сушили над сульфатом натрия и адсорбировали на силикагеле. Продукт очищали экспресс-хроматографией (градиентное элюирование 20-60% этилацетата/гексаном с 0,2% уксусной кислоты) с получением 560 мг 4-[(Е)-2-(5,5,8,8-тетраметил-3-пиразол-1-илметил-6,7-цис-дигидрокси-5,6,7,8-тетрагидронафталин-2-ил)винил]бензойной кислоты (tпл: 238,3-241,5) 148.
Пример 43: получение 4-[(Е)-2-(5,5,8,8-тетраметил-3-((Е)-2-(пиримидин-2-ил)винил)-5,6,7,8-тетрагидро-2-нафталин-2-ил)винил]бензойной кислоты
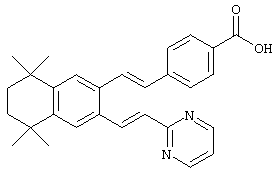
Стадия А: получение 3-((Е)-2-(пиримидин-2-ил)винил)-5,5,8,8-тетраметил-5,6,7,8-тетрагидро-2-нафтальдегида
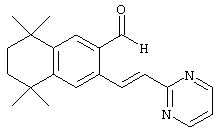
Раствор 0,874 г (2,96 ммоля) 3-бром-5,5,8,8-тетраметил-5,6,7,8-тетрагидро-2-нафтальдегида, 1,886 г (3,11 ммоля) транс-1,2-бис(три-н-бутилстаннил)этилена и 0,068 г (0,059 ммоля) тетракис(трифенилфосфин)палладия в 20 мл толуола кипятили с обратным холодильником в аргоновой атмосфере в течение 1,75 ч. Реакционную смесь слегка охлаждали и добавляли 0,518 г (3,26 ммоля) 2-бромпиримидина и 0,068 г (0,059 ммоля) тетракис(трифенилфосфин)палладия в 3,5 мл толуола. Реакционную смесь выдерживали при температуре кипения с обратным холодильником в течение 3 ч. Реакционную смесь охлаждали до комнатной температуры, реакцию в ней гасили 5% фторида калия и разбавляли этилацетатом. Обе фазы интенсивно перемешивали в течение 16 ч. Смесь фильтровали через броунмиллерит и органический слой промывали рассолом, сушили над безводным сульфатом натрия и концентрировали под вакуумом. Сырой материал очищали экспресс-хроматографией (градиентное элюирование 20% этилацетата/гексаном - 25% этилацетата/гексаном) с получением 0,254 г (27%-ный выход) 3-((Е)-2-(пиримидин-2-ил)винил)-5,5,8,8,-тетраметил-5,6,7,8-тетрагидро-2-нафтальдегида (М+1 = 321).
Стадия Б: получение метил-4-[(Е)-2-(5,5,8,8-тетраметил-3-((Е)-2-(пиримидин-2-ил)винил)-5,6,7,8-тетрагидро-2-нафталин-2-ил)винил]бензоата
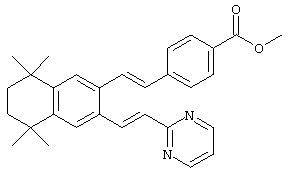
В суспензию 0,079 г (1,98 ммоля) 60%-ного NaH в 1,5 мл тетрагидрофурана вводили 0,248 г (0,96 ммоля) метилового эфира 4-(диметоксифосфорилметил)бензойной кислоты в 2,5 мл ТГФ и реакционную смесь перемешивали при комнатной температуре в течение 1 ч 20 мин. Добавляли 0,254 г (0,79 ммоля) 3-((Е)-2-(пиримидин-2-ил)винил)-5,5,8,8-тетраметил-5,6,7,8-тетрагидро-2-нафтальдегида в 3 мл ТГФ и реакционную смесь перемешивали при комнатной температуре. По истечении 17 ч реакцию гасили 5 мл 1 М соляной кислоты и экстрагировали этиловым эфиром. Органический слой промывали водой, рассолом, сушили над безводным сульфатом магния и концентрировали под вакуумом. Сырой материал очищали экспресс-хроматографией (элюирование 30% этилацетата/гексаном) с получением 0,180 г (50%-ный выход) метил-4-[(Е)-2-(5,5,8,8-тетраметил-3-((Е)-2-(пиримидин-2-ил)винил)-5,6,7,8-тетрагидро-2-нафталин-2-ил)винил]бензоата (М+1 = 453).
Стадия В: получение 4-[(Е)-2-(5,5,8,8-тетраметил-3-((Е)-2-(пиримидин-2-ил)винил)-5,6,7,8-тетрагидро-2-нафталин-2-ил)винил]бензойной кислоты
Раствор 0,180 г (0,40 ммоля) метил-4-[(Е)-2-(5,5,8,8-тетраметил-3-((Е)-2-(пиримидин-2-ил)винил)-5,6,7,8-тетрагидро-2-нафталин-2-ил)винил]бензоата в 5 мл 1 М LiOH и 10 мл этилового спирта кипятили с обратным холодильником. По прошествии 50 мин реакционную смесь охлаждали до комнатной температуры и подкисляли 1 М соляной кислотой. Водный слой экстрагировали этиловым эфиром, промывали водой, рассолом, сушили над безводным сульфатом магния и выпаривали под вакуумом с получением 0,160 г (91%-ный выход) 4-[(Е)-2-(5,5,8,8-тетраметил-3-((Е)-2-(пиримидин-2-ил)винил)-5,6,7,8-тетрагидро-2-нафталин-2-ил)винил]бензойной кислоты 160 (М-1 = 437).
Пример 44: получение 5,5,8,8-тетраметил-3-((Е)-2-тиазол-2-ил-винил)-5,6,7,8-тетрагидро-2-нафтальдегида
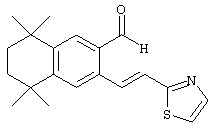
Раствор 0,147 г (0,498 ммоля) 3-бром-5,5,8,8-тетраметил-5,6,7,8-тетрагидро-2-нафтальдегида, 0,302 г (0,498 ммоля) транс-1,2-бис(три-н-бутилстаннил)этилена и 0,012 г (0,00996 ммоля) тетракис(трифенилфосфин)палладия(0) в 5 мл толуола кипятили с обратным холодильником в аргоновой атмосфере в течение 1 ч. Смесь охлаждали и добавляли 0,082 г (0,498 ммоля) 2-бромтиазола и 0,012 г (0,00996 ммоля) тетракис(трифенилфосфин)палладия(0). Смесь перемешивали при температуре кипения с обратным холодильником в течение 2 ч и при комнатной температуре в течение 16 ч. Добавляли 20 мл 5%-ного водного фторида калия и 15 мл этилацетата. Образовавшуюся смесь интенсивно перемешивали в течение 2 ч и фильтровали через броунмиллерит. Органический слой промывали рассолом, сушили над безводным сульфатом натрия и концентрировали под вакуумом. Продукт очищали экспресс-хроматографией с элюированием 15% этилацетата/гексаном и получением 0,065 г (40%-ный выход) 5,5,8,8-тетраметил-3-((Е)-2-тиазол-2-илвинил)-5,6,7,8-тетрагидро-2-нафтальдегида (М+Н = 326).
Пример 45: получение (Е)-4-{2-[3-((тиофен-3-ил)оксометил)-5,5,8,8-тетраметил-5,6,7,8-тетрагидронафталин-2-ил]винил}бензойной кислоты
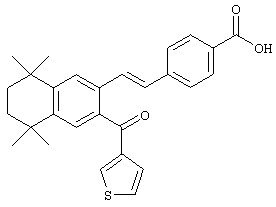
Раствор 2-(1,3-диоксолан-2-ил)-3-[(тиофен-3-ил)гидроксиметил]-5,5,8,8-тетраметил-5,6,7,8-тетрагидронафталина (1,31 г, 3,86 ммоля) [получен из 3-бром-2-(1,3-диоксолан-2-ил)-5,5,8,8-тетраметил-5,6,7,8-тетрагидронафталина и 3-тиофенкарбоксальдегида так, как изложено выше для получения 2-(1,3-диоксолан-2-ил)-3-[(тиофен-2-ил)гидроксиметил)-5,5,8,8-тетраметил-5,6,7,8-тетрагидронафталина] в 13 мл безводного дихлорметана охлаждали до 0°С и в течение 3 мин добавляли периодинан Десса-Мартина (1,80 г, 4,25 ммоля). Реакционную смесь нагревали до КТ и перемешивали в течение 4 ч. Мутный раствор разбавляли 200 мл дихлорметана и промывали раствором бикарбоната натрия. Органический слой промывали рассолом, сушили над сульфатом магния и концентрировали под вакуумом. Далее полученный остаток растворяли в 10 мл тетрагидрофурана и интенсивно перемешивали при одновременном добавлении 10 мл водного 1 н. раствора HCl. Смесь перемешивали в течение двух часов и разделяли между этилацетатом и водой. Полученный органический слой промывали рассолом, сушили над сульфатом магния и сушили под вакуумом. Хроматографическое разделение осуществляли с применением колонки с силикагелем, элюируя с градиентом от 2% этилацетата/гексанов вначале до 8% этилацетата/гексанов, с получением 2-формил[3-((тиофен-3-ил)оксометил)-5,5,8,8-тетраметил-5,6,7,8-тетрагидронафталина (386 мг).
В соответствии со стандартными методами гидролиза по Хорнеру-Эммансу 2-формил[3-((тиофен-3-ил)оксометил)-5,5,8,8-тетраметил-5,6,7,8-тетрагидронафталин превращали в (Е)-4-{2-[3-((тиофен-3-ил)оксометил)-5,5,8,8-тетраметил-5,6,7,8-тетрагидронафталин-2-ил]винил}бензойную кислоту в виде белого твердого вещества (М+= 444) 151.
Пример 46: получение 4-[(Е)-2-(5,5,8,8-тетраметил-3-((Е)-2-метилсульфонилвинил)-5,6,7,8-тетрагидро-2-нафталин-2-ил)винил]бензойной кислоты
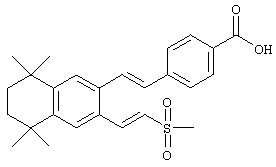
Стадия А: получение 3-((Е)-2-метилсульфонилвинил)-5,5,8,8-тетраметил-5,6,7,8-тетрагидро-2-нафтальдегида
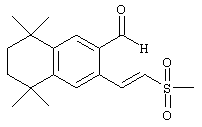
Раствор 0,537 г (1,82 ммоля) 3-бром-5,5,8,8-тетраметил-5,6,7,8-тетрагидро-2-нафтальдегида, 0,579 г (5,46 ммоля) метилвинилсульфона, 0,191 г (0,27 ммоля) тетракис(трифенилфосфин)палладия и 5,23 г (51,7 ммоля) триэтиламина в 12 мл диметилформамида выдерживали при 100°С в аргоновой атмосфере в течение 5 ч. Реакционную смесь выливали в воду и экстрагировали этилацетатом. Органический слой промывали рассолом, сушили над безводным сульфатом натрия и концентрировали под вакуумом. Сырой материал очищали экспресс-хроматографией (элюирование 25% этилацетата/гексаном) с получением 0,359 г (62%-ный выход) 3-(2-метилсульфонилвинил)-5,5,8,8-тетраметил-5,6,7,8-тетрагидро-2-нафтальдегида(М+1 = 321).
Стадия Б: получение метил-4-[(Е)-2-(5,5,8,8-тетраметил-3-((Е)-2-метилсульфонилвинил)-5,6,7,8-тетрагидро-2-нафталин-2-ил)винил]бензоата
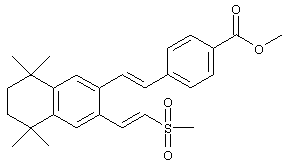
В суспензию 0,065 г (1,63 ммоля) 60%-ного NaH в 1,5 мл тетрагидрофурана вводили 0,205 г (0,79 ммоля) метилового эфира 4-(диметоксифосфорилметил)бензойной кислоты в 2,5 мл ТГФ и при комнатной температуре реакционную смесь перемешивали в течение 35 мин. Добавляли 0,209 г (0,65 ммоля) 3-(2-метилсульфонилвинил)-5,5,8,8-тетраметил-5,6,7,8-тетрагидро-2-нафтальдегида в 2 мл ТГФ и реакционную смесь перемешивали при комнатной температуре. По истечении 16 ч реакцию гасили 4 мл 1 М соляной кислоты и смесь экстрагировали этиловым эфиром. Органический слой промывали водой, рассолом, сушили над безводным сульфатом натрия и концентрировали под вакуумом. Сырой материал очищали экспресс-хроматографией (элюирование 30% этилацетата/гексаном) с получением 0,085 г (29%-ный выход) метил-4-[(Е)-2-(5,5,8,8-тетраметил-3-((Е)-2-метилсульфонилвинил)-5,6,7,8-тетрагидро-2-нафталин-2-ил)винил]бензоата (М+1 = 453).
Стадия В: получение 4-[(Е)-2-(5,5,8,8-тетраметил-3-((Е)-2-метилсульфонилвинил)-5,6,7,8-тетрагидро-2-нафталин-2-ил)винил] бензойной кислоты
Раствор 0,085 г (0,18 ммоля) метил-4-[(Е)-2-(5,5,8,8-тетраметил-3-((Е)-2-метилсульфонилвинил)-5,6,7,8-тетрагидро-2-нафталин-2-ил)винил]бензоата в 5 мл 1 М LiOH и 10 мл этилового спирта кипятили с обратным холодильником. Спустя 30 мин реакционную смесь охлаждали до комнатной температуры и подкисляли 1 М соляной кислотой. Водный слой экстрагировали этиловым эфиром, промывали водой, рассолом, сушили над безводным сульфатом натрия и концентрировали под вакуумом. Остаток очищали экспресс-хроматографией (элюирование 50% этилацетата/гексаном с 0,5% уксусной кислоты) с получением 0,013 г (15%-ный выход) 4-[(Е)-2-(5,5,8,8-тетраметил-3-((Е)-2-метилсульфонилвинил)-5,6,7,8-тетрагидро-2-нафталин-2-ил)винил]бензойной кислоты (М-1 = 437) 161.
Пример 47: получение 4-[(Е)-2-(5,5,8,8-тетраметил-3-((Е)-2-сульфонамидилвинил)-5,6,7,8-тетрагидро-2-нафталин-2-ил)винил]бензойной кислоты
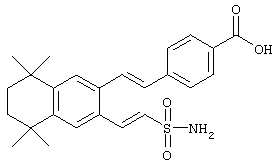
Стадия А: получение 2-бром-3-((Е)-2-сульфонамидилвинил)-5,5,8,8-тетраметил-5,6,7,8-тетрагидронафталина
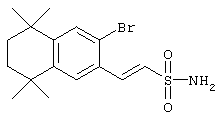
В раствор 0,943 г (2,39 ммоля) трет-бутил[(дифенилфосфорил)метил]-сульфонилкарбамата в 6 мл диметилформамида при 0°С вводили 0,133 г (5,25 ммоля) 95%-ного гидрида натрия. Смесь нагревали до комнатной температуры. По истечении 15 мин добавляли 0,704 г (2,39 ммоля) 3-бром-5,5,8,8-тетраметил-5,6,7,8-тетрагидро-2-нафтальдегида в 5 мл диметилформамида. После перемешивания в течение 18 ч образовавшийся раствор разделяли между 5%-ной водной соляной кислотой и этилацетатом. Органический слой промывали водой, рассолом, сушили над безводным сульфатом натрия и концентрировали под вакуумом. Полученный остаток растворяли в 10 мл дихлорметана и 5 мл трифторуксусной кислоты и перемешивали при комнатной температуре в течение 3 ч. Реакционную смесь концентрировали под вакуумом и один раз выпаривали совместно с толуолом. Остаток очищали экспресс-хроматографией (элюирование 25% этилацетата/гексаном) с получением 0,417 г (47%-ный выход) 2-бром-3-((Е)-2-сульфонамидилвинил)-5,5,8,8-тетраметил-5,6,7,8-тетрагидронафталина (M+Na = 396).
Стадия Б: получение 2-гидроксиметил-3-((Е)-2-сульфонамидилвинил)-5,5,8,8-тетраметил-5,6,7,8-тетрагидронафталина
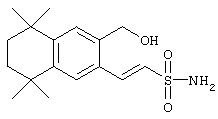
Раствор 0,412 г (1,11 ммоля) 2-бром-3-((Е)-2-сульфонамидилвинил)-5,5,8,8-тетраметил-5,6,7,8-тетрагидронафталина, 0,533 г (1,66 ммоля) трибутилстаннилметанола и 0,060 г тетракис(трифенилфосфин)палладия в 10 мл 1,4-диоксана кипятили с обратным холодильником в аргоновой атмосфере в течение 3,5 ч. Реакционную смесь охлаждали и перемешивали в течение 18 ч при комнатной температуре. Образовавшийся раствор разделяли между этилацетатом и водой. Органический слой промывали рассолом, сушили над безводным сульфатом натрия и концентрировали под вакуумом. Сырой материал очищали экспресс-хроматографией (элюирование 45% этилацетата/гексаном) с получением 0,162 г (45%-ный выход) 2-гидроксиметил-3-((Е)-2-сульфонамидилвинил)-5,5,8,8-тетраметил-5,6,7,8-тетрагидронафталина (М-Н = 322).
Стадия В: получение 3-((Е)-2-сульфонамидилвинил)-5,5,8,8-тетраметил-5,6,7,8-тетрагидро-2-нафтальдегида
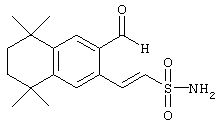
В суспензию 0,155 г (0,479 ммоля) 2-гидроксиметил-3-((Е)-2-сульфонамидилвинил)-5,5,8,8-тетраметил-5,6,7,8-тетрагидронафталина в 4 мл дихлорметана вводили 0,225 г (0,527 ммоля) 1,1,1-триацетокси-1,1-дигидро-1,2-бензиодоксол-3(1Н)-она. Реакционную смесь перемешивали при комнатной температуре. Спустя 3,5 ч раствор разделяли между этилацетатом и насыщенным водным аммонийхлоридом. Органический слой промывали рассолом, сушили над безводным сульфатом натрия и концентрировали под вакуумом. Полученный остаток очищали экспресс-хроматографией (элюирование 30% этилацетата/гексаном) с получением 0,117т (76%-ный выход) 3-((Е)-2-сульфонамидилвинил)-5,5,8,8-тетраметил-5,6,7,8-тетрагидро-2-нафтальдегида (М+Н = 322).
Стадия Г: получение метил-4-[(Е)-2-(5,5,8,8-тетраметил-3-((Е)-2-сульфонамидилвинил)-5,6,7,8-тетрагидро-2-нафталин-2-ил)винил]бензоата
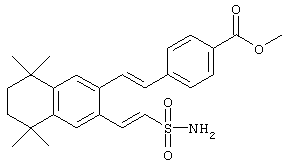
В суспензию 0,036 г (0,90 ммоля) 60%-ного NaH в 1,5 мл тетрагидрофурана вводили 0,114 г (0,44 ммоля) метилового эфира 4-(диметоксифосфорилметил)бензойной кислоты в 2,5 мл ТГФ и реакционную смесь перемешивали при комнатной температуре в течение 35 мин. Добавляли 0,117 г (0,36 ммоля) 3-((Е)-2-сульфонамидилвинил)-5,5,8,8-тетраметил-5,6,7,8-тетрагидро-2-нафтальдегида в 2 мл ТГФ и реакционную смесь перемешивали при комнатной температуре. По истечении 17 ч реакцию гасили 2 мл 1 М соляной кислоты, насыщенным раствором бикарбоната натрия значение рН доводили до 7 и смесь экстрагировали этиловым эфиром. Органический слой промывали водой, рассолом, сушили над безводным сульфатом натрия и концентрировали под вакуумом. Сырой материал очищали экспресс-хроматографией (элюирование 35% этилацетата/гексаном) с получением 0,015 г (9%-ный выход) метил-4-[(Е)-2-(5,5,8,8-тетраметил-3-((Е)-2-сульфонамидилвинил)-5,6,7,8-тетрагидро-2-нафталин-2-ил)винил]бензоата (М-1=452).
Стадия Д: получение 4-[(Е)-2-(5,5,8,8-тетраметил-3-((Е)-2-сульфонамидилвинил)-5,6,7,8-тетрагидро-2-нафталин-2-ил)винил]бензойной кислоты
Раствор 0,015 г (0,034 ммоля) метил-4-[(Е)-2-(5,5,8,8-тетраметил-3-((Е)-2-сульфонамидилвинил)-5,6,7,8-тетрагидро-2-нафталин-2-ил)винил]бензоата в 0,5 мл 1 М LiOH и 1 мл этилового спирта выдерживали при 60°С. По истечении 15 мин реакционную смесь охлаждали до комнатной температуры и подкисляли 1 М соляной кислотой. Водный слой экстрагировали этиловым эфиром, промывали рассолом, сушили над безводным сульфатом натрия и концентрировали под вакуумом. Остаток растворяли в этилацетате и пропускали через 4-микрометрический фильтр. Фильтрат выпаривали под вакуумом с получением 0,012 г (82%-ный выход) 4-[(Е)-2-(5,5,8,8-тетраметил-3-((Е)-2-сульфонамидилвинил)-5,6,7,8-тетрагидро-2-нафталин-2-ил)винил]бензойной кислоты (М-1 = 438) 162.
Пример 48: получение 4-[2-(5,5,8,8-тетраметил-3-((пиразол-1-ил)метил)-5,6,7,8-тетрагидронафталин-2-ил)этил]феноксибензойной кислоты
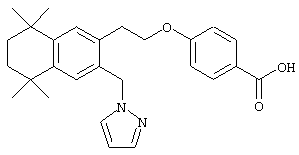
Стадия А: получение 2-бром-5,5,8,8-тетраметил-3-((пиразол-1-ил)метил)-5,6,7,8-тетрагидронафталина
В суспензию 0,955 г (8,52 ммоля) трет-бутоксида калия и 1,877 г (7,10 ммоля) 18-краун-6 эфира в 35 мл тетрагидрофурана вводили 0,580 г (8,52 ммоля) пиразола. По истечении 10 мин добавляли 2,557 г (7,10 ммоля) 2-бром-5,5,8,8-тетраметил-3-бромметил-5,6,7,8-тетрагидронафталина в 15 мл ТГФ. По прошествии 17 ч реакционную смесь концентрировали под пониженным давлением и подкисляли 1 М соляной кислотой. Водный раствор экстрагировали этиловым эфиром, промывали насыщенным раствором бикарбоната натрия, водой, рассолом, сушили над безводным сульфатом натрия и концентрировали под вакуумом. Сырой материал очищали экспресс-хроматографией (градиентное элюирование гексаном - 25% этилацетата/гексаном) с получением 1,185 г (48%-ный выход) 2-бром-5,5,8,8-тетраметил-3-((пиразол-1-ил)метил)-5,6,7,8-тетрагидронафталина (М+1 = 348).
Стадия Б: получение 2-винил-3-((пиразол-1-ил)метил)-5,5,8,8-тетраметил-5,6,7,8-тетрагидронафталина
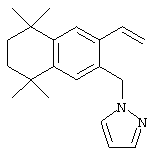
В суспензию 2,076 г (3,99 ммоля) 2-бром-5,5,8,8-тетраметил-3-((пиразол-1-ил)метил)-5,6,7,8-тетрагидронафталин-п-толуолсульфоната, 0,045 г (0,20 ммоля) ацетата палладия, 0,122 г (0,40 ммоля) три-о-толуилфосфина и 1,22 мл (7,97 ммоля) триметоксивинилсилана в 8 мл N-метилпирролидинона вводили 1,80 мл (12,90 ммоля) триэтиламина. В реакционном сосуде создавали вакуум и его три раза заполняли азотом, а затем выдерживали при 90°С в течение 1,5 ч. Реакционную смесь охлаждали и перемешивали в течение 17 ч при комнатной температуре. Реакцию в образовавшейся суспензии гасили 1 М соляной кислотой и смесь экстрагировали этиловым эфиром. Органический слой промывали 1 М HCl, водой, рассолом, сушили над безводным сульфатом натрия и концентрировали под вакуумом. Остаток очищали экспресс-хроматографией (элюирование 20% этилацетата/гексаном) с получением 0,249 г (21%-ный выход) 2-винил-5,5,8,8-тетраметил-3-((пиразол-1-ил)метил)-5,6,7,8-тетрагидронафталина (М+1 = 295).
Стадия В: получение 2-(2-гидрокси)этил-3-(пиразол-1-илметил)-5,5,8,8-тетраметил-5,6,7,8-тетрагидронафталина
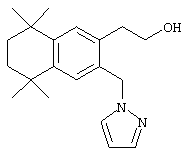
В раствор 0,249 г (0,85 ммоля) 2-винил-5,5,8,8-тетраметил-3-(пиразол-1-илметил)-5,6,7,8-тетрагидронафталина в 3 мл тетрагидрофурана вводили 1,86 мл (0,93 ммоля) 9-борабицикло[3,3,1]нонана в виде 0,5 М раствора в ТГФ. Реакционную смесь перемешивали при комнатной температуре. По прошествии 7 ч реакцию в растворе гасили 2 мл воды и 4 мл 1 М гидроксида натрия. По истечении 15 мин добавляли 10 мл 30%-ного пероксида водорода и реакционную смесь перемешивали при комнатной температуре. Спустя 30 мин раствор экстрагировали этилацетатом. Органическую фазу промывали 10%-ным сульфитом натрия, водой и рассолом, сушили над безводным сульфатом натрия и концентрировали под вакуумом. Полученный остаток очищали экспресс-хроматографией (элюирование 50% этилацетата/гексаном) с получением 0,106 г (40%-ный выход) 2-(2-гидрокси)этил-3-((пиразол-1-ил)метил)-5,5,8,8-тетраметил-5,6,7,8-тетрагидронафталина (М+1 =313).
Стадия Г: получение метил-4-[2-(5,5,8,8-тетраметил-3-(пиразол-1-илметил)-5,6,7,8-тетрагидронафталин-2-ил)этил]феноксибензоата
В раствор 0,106 г (0,34 ммоля) 2-(2-гидрокси)этил-3-(пиразол-1-илметил)-5,5,8,8-тетраметил-5,6,7,8-тетрагидронафталина, 0,057 г (0,37 ммоля) метил-4-гидроксибензоата и 0,097 г (0,37 ммоля) трифенилфосфина в 5 мл тетрагидрофурана добавляли 0,066 г (0,38 ммоля) диэтилазодикарбоксилата и реакционную смесь выдерживали при 70°С. По прошествии 2 ч реакционную смесь охлаждали до комнатной температуры и реакцию гасили водой. Водный раствор экстрагировали этиловым эфиром, промывали водой, рассолом, сушили над безводным сульфатом натрия и концентрировали под вакуумом. Сырой материал очищали экспресс-хроматографией (элюирование 30% этилацетата/гексаном) с получением 0,131 г (86%-ный выход) метил-4-[2-(5,5,8,8-тетраметил-3-(пиразол-1-илметил)-5,6,7,8-тетрагидронафталин-2-ил)этил]феноксибензоата (М+1 = 447).
Стадия Д: получение 4-[2-(5,5,8,8-тетраметил-3-(пиразол-1-илметил)-5,6,7,8-тетрагидронафталин-2-ил)этил]феноксибензойной кислоты
Раствор 0,131 г (0,293 ммоля) метил-4-[2-(5,5,8,8-тетраметил-3-(пиразол-1-илметил)-5,6,7,8-тетрагидронафталин-2-ил)этил]феноксибензоата в 3 мл 1 М LiOH и 10 мл этилового спирта кипятили с обратным холодильником. По прошествии 2 ч реакционную смесь охлаждали до комнатной температуры и подкисляли 1 М соляной кислотой. Водный слой экстрагировали этиловым эфиром, промывали рассолом, сушили над безводным сульфатом натрия и концентрировали под вакуумом. Сырой материал очищали экспресс-хроматографией (градиентное элюирование 10% метанола/дихлорметаном - 10% метанола/дихлорметаном с 5% уксусной кислоты) с получением 0,101 г (79%-ный выход) 4-[2-(5,5,8,8-тетраметил-3-((пиразол-1-ил)метил)-5,6,7,8-тетрагидронафталин-2-ил)этил]феноксибензойной кислоты (М+1 = 433) 158.
Пример 49: получение метил-4-[2-5,5,8,8-тетраметил-3-пиримидин-2-илметил-5,6,7,8 -тетрагидронафталин-2-ил)винил] бензоата
Стадия А: получение 2-бром-3-цианометил-5,5,8,8-тетраметил-5,6,7,8-тетрагидронафталина
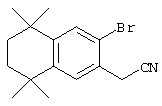
Смесь 14,2 г (39,4 ммоля) 2-бром-3-бромметил-5,5,8,8-тетраметил-5,6,7,8-тетрагидронафталина (процесс получения см. в примере 5.21, стадия А) и 6,15 г (39,4 ммоля) тетраэтиламмонийцианида в 50 мл ДМФ перемешивали при комнатной температуре. По прошествии 48 ч реакционную смесь выливали в рассол и добавляли 50 мл 2 н. HCl. Смесь экстрагировали этилацетатом, сушили (MgSO4), концентрировали досуха и очищали экспресс-хроматографией (10% этилацетата/гексан) с получением 9,2 г (75%-ный выход) 2-бром-3-цианометилметил-5,5,8,8-тетраметил-5,6,7,8-тетрагидронафталина.
Стадия Б: получение 2-бром-3-[1,1-(2-пиримидинил)циано]метил-5,5,8,8-тетраметил-5,6,7,8-тетрагидронафталина
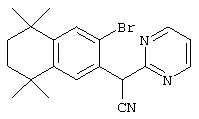
В суспензию 1,5 г (65,6 ммоля) гидрида натрия в 80 мл ДМФ на бане из мокрого льда вводили раствор 9,2 г (29,1 ммоля) 2-бром-3-цианометилметил-5,5,8,8-тетраметил-5,6,7,8-тетрагидронафталина в 20 мл ДМФ. Реакционную смесь выдерживали при комнатной температуре. По истечении 1 ч добавляли раствор 10,4 г (65,6 ммоля) 2-бромпиримидина в 20 мл ДМФ. По прошествии 12 ч реакционную смесь выливали в смесь воды со льдом, нейтрализовали 2 н. HCl и экстрагировали этилацетатом. Органическую фазу сушили (MgSO4), концентрировали под вакуумом и продукт очищали экспресс-хроматографией (15% этилацетата/гексан) с получением 5,5 г (47%-ный выход) 2-бром-3-[1,1(2-пиримидинил)циано]метил-5,5,8,8-тетраметил-5,6,7,8-тетрагидронафталина.
Стадия В: получение 2-бром-3-(2-пиримидинил)метил-5,5,8,8-тетраметил-5,6,7,8-тетрагидронафталина
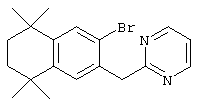
Смесь 5,5 г (14,3 ммоля) 2-бром-3-[1,1-(2-пиримидинил)циано]метил-5,5,8,8-тетраметил-5,6,7,8-тетрагидронафталина, 40 мл концентрированной HCl, 20 мл уксусной кислоты и 20 мл воды кипятили с обратным холодильником. По истечении 15 ч реакционную смесь выливали на лед, добавляли рассола, затем экстрагировали этилацетатом. Органическую фазу сушили (MgSO4), концентрировали под вакуумом и продукт очищали экспресс-хроматографией (20% этилацетата/гексан) с получением 2,8 г (54%-ный выход) 2-бром-3-(2-пиримидинил)метил-5,5,8,8-тетраметил-5,6,7,8-тетрагидронафталина.
Стадия Г: получение метил-4-[2-(5,5,8,8-тетраметил-3-(пиримидин-2-илметил)-5,6,7,8-тетрагидронафталин-2-ил)винил]бензоата
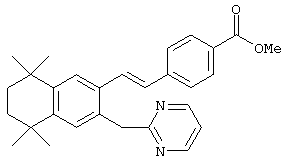
Смесь 200 мг (0,55 ммоля) 2-бром-3-(2-пиримидинил)метил-5,5,8,8-тетраметил-5,6,7,8-тетрагидронафталина, 106 мг (0,72 ммоля) винилтриметилсилана и 150 мкл (1,1 ммоля) триэтиламина в 30 мл N-МП помещали в аргоновую атмосферу и добавляли 64 мг (0,22 ммоля) три-о-толилфосфина и 24 мг (0,11 ммоля) ацетата палладия. Реакционную смесь выдерживали при 90°С. По прошествии 2 ч реакционную смесь охлаждали до комнатной температуры и добавляли 64 мг (0,22 ммоля) три-о-толилфосфина, 24 мг ацетата палладия, 106 мкл (0,66 ммоля) этил-4-бромбензоата и 900 мкл (0,93 ммоля) тетрабутиламмонийфторида. Реакционную смесь выдерживали при 100°С. По прошествии 6 ч реакционную смесь охлаждали до комнатной температуры, выливали в рассол, экстрагировали этилацетатом, сушили (MgSO4), концентрировали досуха и очищали экспресс-хроматографией (30% этилацетата/гексан) с получением 38 мг (15%-ный выход) метил-4-[2-(5,5,8,8-тетраметил-3-пиримидин-2-илметил)-5,6,7,8-тетрагидронафталин-2-ил)винил]бензоата. Омылением сложного эфира получали 4-[2-(5,5,8,8-тетраметил-3-пиримидин-2-илметил-5,6,7,8-тетрагидронафталин-2-ил)винил]бензойную кислоту (MH+= 427).
Пример 50: получение глицерин-4-[(Е)-2-(5,5,8,8-тетраметил-3-пиразол-1-илметил-5,6,7,8-тетрагидронафталин-2-ил)винил]бензоата
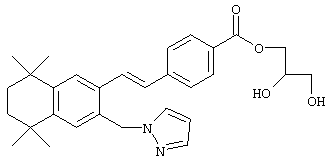
К 200 мг (0,48 ммоля) 4-[(Е)-2-(5,5,8,8-тетраметил-3-пиразол-1-илметил-5,6,7,8-тетрагидронафталин-2-ил)винил]бензойной кислоты в 30 мл бензола добавляли 0,17 мл (1,9 ммоля) оксалилхлорида и одну каплю диизопропиламина. Спустя 30 мин реакционную смесь концентрировали под пониженным давлением, дополнительно добавляли бензола и смесь вновь концентрировали. Полученный хлорангидрид кислоты растворяли в бензоле и добавляли в смесь 0,3 мл (2,4 ммоля) золькеталя, 293 мг (2,4 ммоля) ДМАП и 0,14 мл (1 ммоля) ТЭА в бензоле. Реакционную смесь перемешивали при комнатной температуре. По истечении 8 ч реакцию гасили 1 н. HCl, смесь разбавляли рассолом, органический слой отделяли, сушили (MgSO4), концентрировали под пониженным давлением и продукт очищали экспресс-хроматографией (25% этилацетата/гексан). Очищенный продукт растворяли в метиленхлориде и ТГФ в соотношении 1:1 и при комнатной температуре обрабатывали избытком п-ТоСК (п-толуолсульфокислота). По прошествии 2 ч реакционную смесь концентрировали под пониженным давлением, разбавляли водой и экстрагировали этилацетатом. Экстракты сушили (MgSO4), концентрировали под пониженным давлением и очищали экспресс-хроматографией (5% MeOH/CH2Cl2) с получением 62 мг (27%-ный выход) глицерин-4-[(Е)-2-(5,5,8,8-тетраметил-3-пиразол-1-илметил-5,6,7,8-тетрагидронафталин-2-ил)винил]бензоата (МН+ = 489) 164.
Пример 51: получение 4-[(Е)-2-(5,5,8,8-тетраметил-3-пиразол-1-илметил-5,6,7,8-тетрагидронафталин-2-ил)винил]бензамида
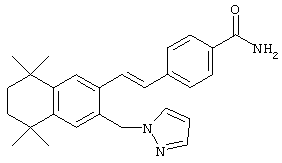
Хлорангидрид кислоты, полученный так, как изложено в вышеприведенном примере для случая со сложным эфиром глицерина, в таком же количестве в ТГФ вводили в 15 мл концентрированного гидроксида аммония. По истечении 1 ч реакционную смесь разбавляли рассолом, экстрагировали EtOAc, сушили (MgSO4), концентрировали под пониженным давлением и очищали экспресс-хроматографией (5% метилового спирта/дихлорметан) с получением 156 мг (78%-ный выход) 4-[(Е)-2-(5,5,8,8-тетраметил-3-пиразол-1-илметил-5,6,7,8-тетрагидронафталин-2-ил)винил]бензамида (tпл: 248-249) 165.
Пример 52: получение пиперидинамида 4-[(Е)-2-(5,5,8,8-тетраметил-3-пиразол-1-илметил- 5,6,7,8-тетрагидронафталин-2-ил)винил] бензойной кислоты
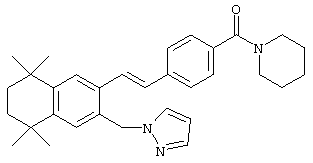
Хлорангидрид кислоты, полученный так, как изложено в вышеприведенном примере для случая со сложным эфиром глицерина, в таком же количестве в ТГФ вводили в раствор 0,12 мл (1,2 ммоля) пиперидина в ТГФ. По прошествии 0,5 ч реакционную смесь разбавляли рассолом, экстрагировали EtOAc, сушили (MgSO4), концентрировали под пониженным давлением и очищали экспресс-хроматографией (60% этилацетата/гексан) с получением 125 мг (54%-ный выход) пиперидинамида 4-[(Е)-2-(5,5,8,8-тетраметил-3-пиразол-1-илметил-5,6,7,8-тетрагидронафталин-2-ил)винил]бензойной кислоты (tпл: 171,6-172,5) 166.
Пример 53: получение 2.3-дигидроксипропилбензилового эфира 4-[(Е)-2-(3-гексил-5,5,8,8-тетраметил-5,6,7,8-тетрагидронафталин-2-ил)винил]бензойной кислоты
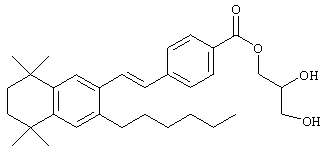
4-[(Е)-2-(3-гексил-5,6,7,8-тетрагидро-5,5,8,8-тетраметил-2-нафтил)винил]бензойную кислоту (100 мг) растворяли в бензоле (2,5 мл). В этот раствор добавляли оксалилхлорида (44 мкл) и диметилформамида (5 мкл) и наблюдали интенсивное выделение газа. В дальнейшем реакционную смесь перемешивали при комнатной температуре в течение 1 ч, добавляли триэтиламина (70 мкл), после чего раствор N,N-диметиламинопиридина (147 мг) и золькеталя (149 мкл) в бензоле (3 мл). Реакционную смесь перемешивали при комнатной температуре в течение ночи. Реакционную смесь подкисляли 1 М соляной кислотой и экстрагировали этиловым эфиром (2 порции по 20 мл). Объединенные экстракты промывали водой, рассолом, сушили и выпаривали. Остаток очищали экспресс-хроматографией (силикагель, гексан/10% этилацетата) с получением в виде прозрачного стеклоподобного вещества 97 мг 2,2,4-триметил-1,3-диоксалан-4-илового эфира 4-[(Е)-2-(3-гексил-5,6,7,8-тетрагидро-5,5,8,8-тетраметил-2-нафтил)винил]бензойной кислоты. Этот сложный эфир растворяли в дихлорметане (4 мл) и добавляли моногидрата п-толуолсульфоновой кислоты (60 мг). Реакционную смесь перемешивали при комнатной температуре в течение ночи, разбавляли водой и экстрагировали этиловым эфиром. Органический слой промывали водой, 10%-ным бикарбонатом натрия и рассолом, сушили и концентрировали под вакуумом. Сырой продукт в виде желтого масла очищали экспресс-хроматографией (силикагель, СН2Cl2/6% МеОН) с получением в виде прозрачного стеклоподобного вещества с 42%-ным выходом 49 мг 2,3-дигидроксипропилового эфира [(Е)-2-(3-гексил-5,6,7,8-тетрагидро-5,5,8,8-тетраметил-2-нафтил)винил]бензойной кислоты 56 (М+ 492) 56.
Пример 54: сродство к связыванию ретиноидных рецепторов
Антагонистическую селективность соединений по изобретению в отношении α-RAR определяли по анализу связывания лиганда, описанному в работе С. Apfel и др. Proc.Nat.Sci.Acad. (USA), 89:7129-7133 (1992). Ниже представлены данные для соединений, выбранных из таблицы 1.
Описанные выше варианты выполнения изобретения представлены просто в качестве примеров, и для специалистов в данной области техники вполне очевидны или могут стать вполне очевидными, если воспользоваться не более чем обычными экспериментами, многочисленные эквиваленты приведенных в настоящем описании конкретных технических приемов. Все такие эквиваленты рассматриваются как входящие в объем изобретения и охватываемые следующей формулой изобретения.
Таким образом, все патенты, заявки на патенты и публикации, упоминаемые в данной заявке, включены в настоящее описание во всей полноте для всех целей в той же самой степени, как если бы было приведено самостоятельное указание на каждый патент, заявку на патенты и публикацию по отдельности.
Изобретение относится к новым ретиноидным соединениям структурной формулы (I) или их фармацевтически приемлемым солям, и к фармацевтическим композициям, обладающим агонистической активностью в отношении ретиноидных рецепторов и включающим указанные соединения 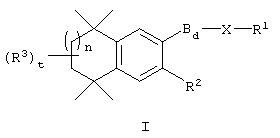 где n обозначает 1; d обозначает 0 или 1; В обозначает -CR7 =CR8-, -CH2O-; R7 и R8 каждый независимо обозначает водородный атом; Х обозначает фенил, необязательно замещенный галогеном, или 5-ти членный гетероарил, содержащий S в качестве гетероатома; R1 обозначает -C(=O)-R9; R9 обозначает алкил, гидроксил, амино-, гетероалкилоксигруппу, содержащую О, или 6-ти членный гетероциклил, содержащий N в качестве гетероатома; а R2 обозначает: (a) -(CR10R11)m-Yp-R12; m обозначает целое число от 1 до 10; р обозначает 0 или 1; R10 и R11 обозначают водородный атом; Y обозначает -О-, -S- или -NR13-; и R13 обозначает водородный атом; R12 обозначает водородный атом, алкил, циклоалкил, фенил, 5-ти или 6-ти членный гетероарил, содержащий N, S, О в качестве гетероатома, 5-ти или 6-ти членный гетероарилалкил, содержащий N, S, О в качестве гетероатома, гетероалкил, содержащий N, S, О в качестве гетероатома, 5-ти или 6-ти членный гетероциклил, содержащий N, S, О в качестве гетероатома, или 5-ти или 6-ти членный гетероциклилалкил, содержащий N, S, О в качестве гетероатома; при условии, что когда р обозначает 0, тогда R12 не обозначает водородный атом или алкил; (б) 5-ти или 6-ти членный гетероарил, содержащий N, S, О в качестве гетероатома; (в) -Z-L; Z обозначает -CR14=CR15-, -С=С-, -С(=O) или -S-; R14, R15 обозначают водородный атом; а L обозначает 5-ти или 6-ти членный гетероарил, содержащий N, S, О в качестве гетероатома, (г) -CR14=CR15-L1, где L1 обозначает S(O)2R17 или SO2NR18R19, где R17 обозначает алкил, а R18 и R19 обозначают водородный атом; каждый R3 независимо обозначает водородный атом, гидроксил или оксогруппу; a t обозначает 1 или 2. 3 н. и 56 з.п. ф-лы, 10 табл.
где n обозначает 1; d обозначает 0 или 1; В обозначает -CR7 =CR8-, -CH2O-; R7 и R8 каждый независимо обозначает водородный атом; Х обозначает фенил, необязательно замещенный галогеном, или 5-ти членный гетероарил, содержащий S в качестве гетероатома; R1 обозначает -C(=O)-R9; R9 обозначает алкил, гидроксил, амино-, гетероалкилоксигруппу, содержащую О, или 6-ти членный гетероциклил, содержащий N в качестве гетероатома; а R2 обозначает: (a) -(CR10R11)m-Yp-R12; m обозначает целое число от 1 до 10; р обозначает 0 или 1; R10 и R11 обозначают водородный атом; Y обозначает -О-, -S- или -NR13-; и R13 обозначает водородный атом; R12 обозначает водородный атом, алкил, циклоалкил, фенил, 5-ти или 6-ти членный гетероарил, содержащий N, S, О в качестве гетероатома, 5-ти или 6-ти членный гетероарилалкил, содержащий N, S, О в качестве гетероатома, гетероалкил, содержащий N, S, О в качестве гетероатома, 5-ти или 6-ти членный гетероциклил, содержащий N, S, О в качестве гетероатома, или 5-ти или 6-ти членный гетероциклилалкил, содержащий N, S, О в качестве гетероатома; при условии, что когда р обозначает 0, тогда R12 не обозначает водородный атом или алкил; (б) 5-ти или 6-ти членный гетероарил, содержащий N, S, О в качестве гетероатома; (в) -Z-L; Z обозначает -CR14=CR15-, -С=С-, -С(=O) или -S-; R14, R15 обозначают водородный атом; а L обозначает 5-ти или 6-ти членный гетероарил, содержащий N, S, О в качестве гетероатома, (г) -CR14=CR15-L1, где L1 обозначает S(O)2R17 или SO2NR18R19, где R17 обозначает алкил, а R18 и R19 обозначают водородный атом; каждый R3 независимо обозначает водородный атом, гидроксил или оксогруппу; a t обозначает 1 или 2. 3 н. и 56 з.п. ф-лы, 10 табл.
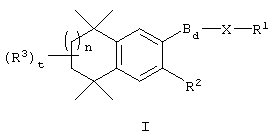
или его фармацевтически приемлемая соль,
где n обозначает 1;
d обозначает 0 или 1;
В обозначает -CR7=CR8-, -СН2О-;
R7 и R8 каждый независимо обозначает водородный атом;
Х обозначает фенил, необязательно замещенный галогеном или 5-членный гетероарил, содержащий S в качестве гетероатома;
R1 обозначает -C(=O)-R9;
R9 обозначает алкил, гидроксил, амино-, гетероалкилоксигруппу, содержащую О, или 6-членный гетероциклил, содержащий N в качестве гетероатома; а
R2 обозначает (a)-(CR10R11)m-Yp-R12;
m обозначает целое число от 1 до 10;
р обозначает 0 или 1;
R10 и R11 обозначают водородный атом;
Y обозначает -О-, -S- или -NR13- и
R13 обозначает водородный атом;
R12 обозначает водородный атом, алкил, циклоалкил, фенил, 5- или 6-членный гетероарил, содержащий N, S, О в качестве гетероатома, 5- или 6-членный гетероарилалкил, содержащий N, S, О в качестве гетероатома, гетероалкил, содержащий N, S, О в качестве гетероатома, 5- или 6-членный гетероциклил, содержащий N, S, О в качестве гетероатома, или 5- или 6-членный гетероциклилалкил, содержащий N, S, О в качестве гетероатома;
при условии, что когда р обозначает 0, тогда R не обозначает водородный атом или алкил;
(б) 5- или 6-членный гетероарил, содержащий N, S, О в качестве гетероатома;
(в) -Z-L;
Z обозначает -CR14=CR15-, -C≡C-, -C(=O) или -S-;
R14, R15 обозначают водородный атом; а
L обозначает 5- или 6-членный гетероарил, содержащий N, S, О в качестве гетероатома,
(г) -CR14=CR15-L1, где L1 обозначает S(O)2R17 или SO2NR18R19, где R17
обозначает алкил, а R18 и R19 обозначают водородный атом; каждый R независимо обозначает водородный атом, гидроксил или оксогруппу; а t обозначает 1 или 2.
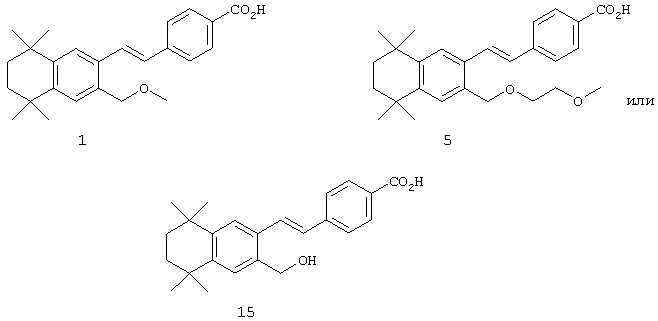
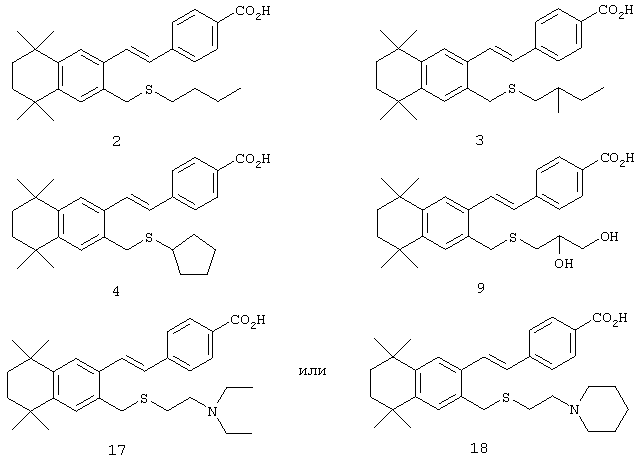
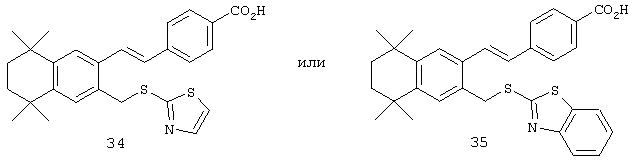
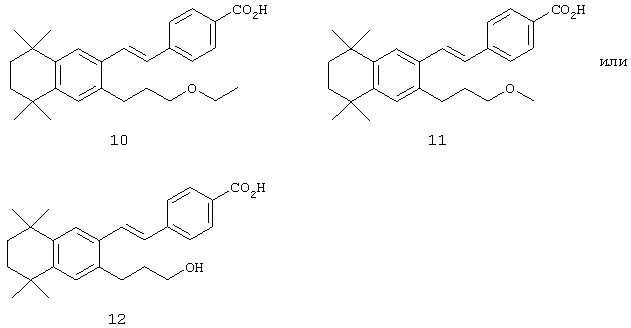
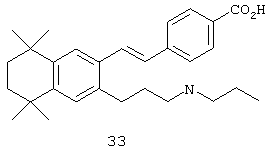
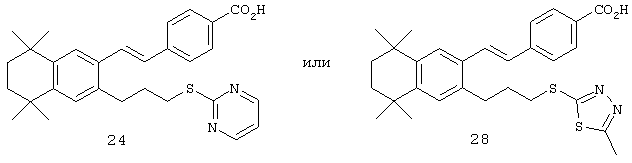
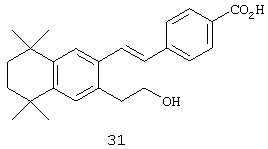
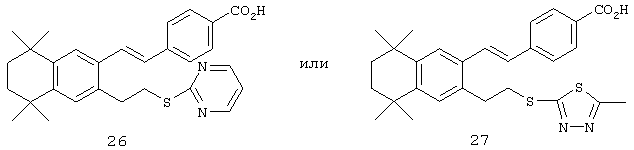
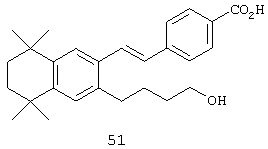
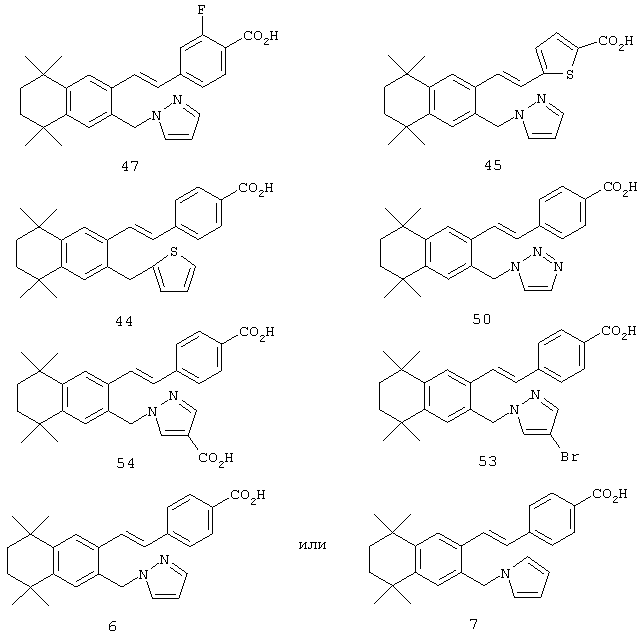
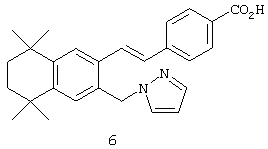
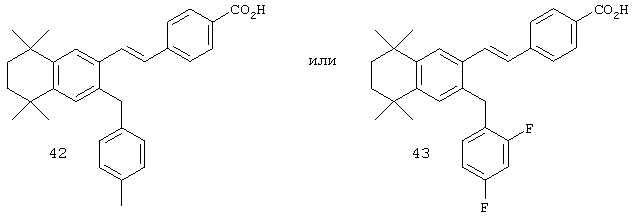
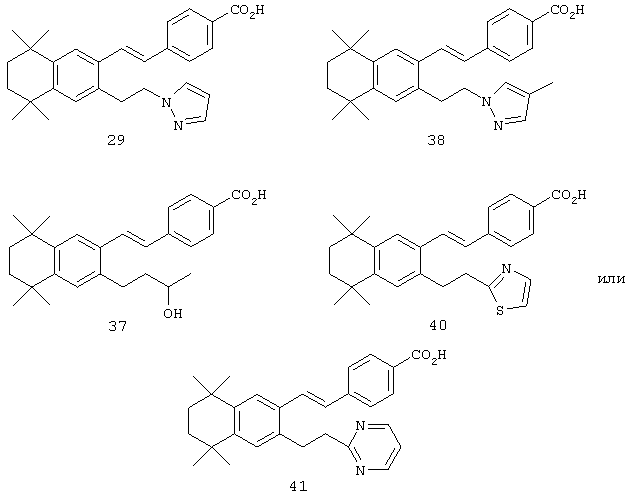
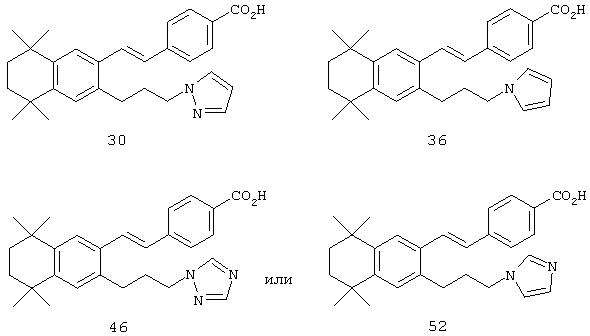
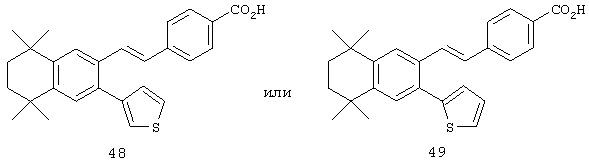
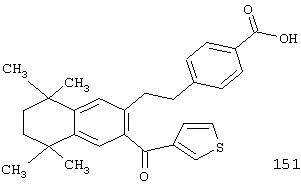
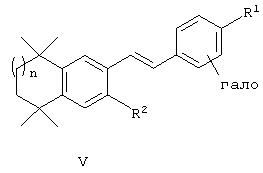
где R обозначает -СО2Н, а R2 и n имеют значения, указанные в п.1.
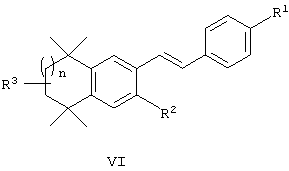
где R1 обозначает -СО2Н, R3 обозначает водородный атом, а R2 и n имеют значения, указанные в п.1.
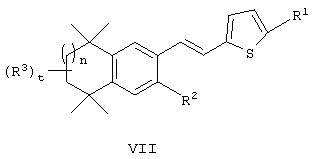
в которой R1 обозначает -СО2Н, R3 обозначает водородный атом, а R2, t и n имеют значения, указанные в п.1.
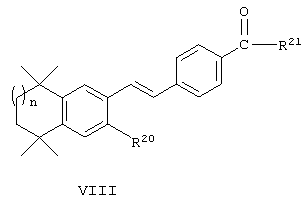
в которой R20 обозначает алкил,
R21 обозначает (а) гетероалкилоксигруппу, содержащую N, S, О, гетероалкиламиногруппу, содержащую О, или гетероалкилтиогруппу, содержащую О; или
(б) Q-R22, где Q обозначает -NR23- или -S- (где R23 обозначает водородный атом),
R22 обозначает карбоксиалкил,
a n обозначает 1.
| WO 9724116 А2, 10.07.1997 | |||
| ЕР 0641759 А, 08.03.1995 | |||
| US 5998486 А, 07.12.1999 | |||
| WO 9415902 А, 21.07.1994 | |||
| ЛИГАНДЫ Х-РЕЦЕПТОРА РЕТИНОЕВОЙ КИСЛОТЫ | 1995 |
|
RU2146241C1 |
| ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ | 1995 |
|
RU2120304C1 |
| СОЕДИНЕНИЯ, ОБЛАДАЮЩИЕ ИЗБИРАТЕЛЬНОСТЬЮ В ОТНОШЕНИИ РЕЦЕПТОРОВ РЕТИНОИДА, ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ (ВАРИАНТЫ), СПОСОБ МОДУЛИРОВАНИЯ ПРОЦЕССОВ (ВАРИАНТЫ), СПОСОБ ПОДАВЛЕНИЯ ПАТОЛОГИЧЕСКИХ СОСТОЯНИЙ | 1993 |
|
RU2144913C1 |

