Изобретение относится к применению соединений, обладающих специфичной или селективной активностью по отношению к ΠΠΚα (рецепторам ретиновой кислоты), для лечения болезней и состояний, подвергающихся воздействию таких ретиноидов. Более конкретно, настоящее изобретение направлено на применение специфичных или селективных агентов для PPKα для лечения опухолей.
Соединения, обладающие ретиноидподобной активностью, хорошо известны и описаны в многочисленных американских и других патентах и в научных публикациях. Общеизвестно и общепризнано, что такая ретиноидная активность полезна для лечения млекопитающих, включая людей, для устранения и облегчения симптомов и состояний при многочисленных болезнях. Другими словами, общепризнано, что фармацевтические композиции, включающие в качестве активного вещества ретиноидное соединение или соединения, используются в качестве регуляторов пролиферации и дифференциации клеток и, в частности, как агенты для лечения кожных заболеваний, включая лучевые кератозы, арсениазные кератозы, воспалительные и невоспалительные угри, псориаз, ихтиозы, и другой кератинизации и гиперпролиферативных нарушений кожи, экземы, атопического дерматита, болезни Дарье, плоского лихена, предотвращения и реверсирования глюкокортикоидных повреждений (стероидной атрофии), как антимикробные агенты для местного применения, как антипигментационные агенты для кожи, и для лечения и полного устранения возрастных и световых повреждений кожи. Ретиноиды также пригодны для профилактики и лечения раковых и предраковых состояний, включая предраковые и раковые гиперпролиферативные заболевания, такие как рак груди, кожи, простаты, шейки матки, прямой кишки, мочевого пузыря, пищевода, желудка, легких, гортани, пространства рта, кровяной и лимфатической системы, метаплазии, дисплазии, неоплазии, лейкоплакии, и папиллом слизистой оболочки и для лечения саркомы Капоши. Кроме того, ретиноидные соединения могут быть использованы как активные вещества для лечения глазных болезней, включая (но не ограничивая) пролиферативную витреоретинопатию (ПВР), отслоение сетчатки, сухость глаз и другие корнеопатии, а также при лечении и профилактике различных сердечно-сосудистых болезней, включая, без ограничения, болезни, связанные с метаболизмом липидов, такие как дислипидемии, при профилактике рестеноза после ангиопластики и в качестве активного вещества для повышения концентрации активатора плазминогена тканей (АТП). Другие применения ретиноидов включают профилактику и лечение состояний и болезней, связанных с вирусом папилломы человека (ВПЧ), включая бородавки и остроконечные бородавки, различных воспалительных заболеваний, таких как легочный фиброз, илеит, колит и болезнь Крона, нервнодегенеративных болезней, таких как болезнь Альцгеймера, болезнь Паркинсона, удар, нарушение функции гипофиза, включая недостаточное образование гормона роста, модуляцию аппоптоза, включая как индукцию аппоптоза, так и ингибирование активированного Т-клетками аппоптоза, восстановление роста волос, включая комбинированную терапию с применением этих соединений и других активных веществ, таких как MinoxidilR, болезней, связанных с иммунной системой, включая применение этих соединений в качестве иммуностимуляторов, модуляции отторжения пересаженных органов, облегчения заживления ран, включая модуляцию хелоза.
Патенты США 4740519 (Shroot и др.), 4826969 (Maignan и др.), 4326055 (Lоеligеr и др. ), 5130335 (Chandraratna и др.), 5037825 (Klaus и др.), 5231113 (Chandraratna и др.), 5324840 (Chandraratna), 5344959 (Chandraratna), 5130335 (Chandraratna и др.), Европейские заявки 0170105 (Shudo), 0 176 034 A (Wuest и дp.), 0350846 A (Klaus и др.), 0 176 032 A (Frickel и др.), 0 176 033 A (Frickel и др.), 0 253 302 A (Klaus и др.), 0 303 915 A (Bryce и др.), Заявка GB 2190378 А (Klaus и др.), Заявки DE N.N. DE 3715955 А1 (Klaus и др.), DE 3602473 Al (Wuest и дp.), и статьи в J. Amer. Acad. Derm. 15: 756-764 (1986) (Sporn и др.), Chem. Pharm. Bull. 33: 404-407 (1985) (Shudo и др.), J. Med. Chem. 1988 31, 2182-2192 (Каgесhiка и др. ). Chemistry and Biology of Synthetic Retinoids CRC Press Inc. 1990 p. 334-335, 354 (Dawson и др.), раскрывают и относятся к соединениям, которые включают тетрагидронафтильные группы и обладают ретиноидной или связанной с ней биологической активностью.
В патентах США 4980369, 5006550, 5015658, 5045551, 5089509, 5134159, 5162546, 5234926, 5248777, 5264578, 5272156, 5278318, 5324744, 5346895, 5346915, 5348972, 5348975, 5380877, 5399561, 5407937 (переуступленных тому же правопреемнику, что и данная заявка) и патентах и публикациях, приведенных выше, описаны производные хромана, тиохромана и 1,2,3,4-тетрагидрохинолина, которые обладают биологической активностью ретиноидного типа.
В патентах США 4723028 (Shudo), Европейской заявке DE 0170105 (Shuda), заявке Германии DE 3524199 Al (Shudo), PCT WO 85/04652 (Polus) и J. Med. Chem. 1988, 31, 2182-2192 (Kagechika и др.), описаны арил- и гетероарил- или диарилзамещенные олефины или амиды, которые имеют ретиноидную или связанную с ней биологическую активность.
В патентах США 4992468, 5013744, 5068252, 5175185, 5202471, 5264456, 5324840, 5326898, 5349105, 5391753, 5414007 и 5434173 (принадлежащих тому же правопреемнику, что и данная заявка) и патентах и публикациях, указанных выше, описаны соединения, имеющие биологическую активность ретиноидного типа и характеризующиеся структурой, где фенил и гетероарил или фенил и второй фенил соединены олефиновой или ацетиленовой связью. Кроме того, несколько находящихся на рассмотрении заявок и недавно выданных патентов, принадлежащих этому же правопреемнику, что и данная заявка, направлены на получение соединений с активностью ретиноидного типа.
Общеизвестно, что в организмах млекопитающих (и в других организмах) существуют два основных типа ретиноидных рецепторов. Эти два основных типа или семейства рецепторов обозначаются соответственно РРК и РХР. Внутри каждого типа есть подтипы: в семействе РРК подтипы обозначаются PPKα,PPKβ и РРКr, в семействе РХР существуют подтипы PXPα,PXPβ и РХРr. Было также установлено, что распределение двух основных типов ретиноидных рецепторов и нескольких подтипов в различных тканях и органах организмов млекопитающих не является однородным.
Известно также, что применение ретиноидов для лечения различных болезней и состояний не обходится без возникновения различных проблем и побочных эффектов. Побочные эффекты при использовании терапевтических доз включают головную боль, тератогенез, кожно-слизистую токсичность, мускульную токсичность, дислипидемию, воспаление кожи, токсичность для печени и т.д. Эти побочные эффекты ограничивают применимость и полезность ретиноидов для лечения болезней. Продолжаются исследования по определению того, какое из семейств РРК и РХР и, внутри каждого семейства, какой подтип или подтипы ответственны за один или несколько нежелательных побочных эффектов. Соответственно, для соединений, способных связываться с ретиноидными рецепторами, специфичность или селективность одного из основных типов или семейств и даже специфичность или селективность одного или нескольких подтипов внутри семейства рецепторов рассматриваются как желательное фармакологическое свойство.
Такая селективность или специфичность полезна как инструмент для исследований при установлении ролей нескольких типов рецепторов и подтипов в вызывании различных эффектов ретиноидов в биологических системах, а также как средство для создания ретиноидных лекарственных средств со специфичными терапевтическими эффектами и/или с уменьшенными побочными эффектами и токсичностью. В этом аспекте в патенте США N 5324840 описывается класс соединений, у которых активность ретиноидного типа сопровождается пониженной токсичностью для кожи и пониженными тератогенными эффектами. В патенте США N 5455265 описаны способы лечения млекопитающих с применением соединений, обладающих агонистической активностью по отношению к рецепторам РХР. Опубликованная Заявка WO 93/11755 также посвящена применению соединений, являющихся селективными агонистами рецепторов РХР.
Данное изобретение предусматривает способы лечения опухолей при помощи соединений, которые являются специфичными или селективными по отношению к PPKα.
Согласно данному изобретению было обнаружено, что соединения ретиноидного типа, которые действуют селективно или, предпочтительно даже специфично, на подтипы рецептора PPKα скорее, чем на подтипы рецепторов PPKβ и РРКr, обладают желательными фармацевтическими свойствами, связанными с ретиноидами, и особенно пригодны для лечения новообразований, таких как острый моноцитарный лейкоз, цервикальная карцинома, миеломы, овариальные карциномы, карциномы головы и шеи, без одного или нескольких побочных эффектов, вызываемых ретиноидами, таких как потеря веса, токсичность для кожно-слизистой оболочки, кожные воспаления и тератогенность.
Соответственно настоящее изобретение относится к применению специфичных или селективных по отношению к PPKα ретиноидов для лечения болезней и состояний, которые поддаются такому лечению, и особенно для лечения новообразований, прежде всего острого моноцитарного лейкоза, цервикальной карциномы, миеломы, овариальной карциномы, карциномы шеи и головы при помощи ретиноидов, специфичных или селективных по отношению к PPKα. В соответствии с данным изобретением селективные по отношению к PPKα соединения также используют для лечения пролиферативной витреоретинопатии (ПРР) и возрастной пятнистой дегенерации (ВДП).
Для целей данного описания соединение рассматривается как специфичное или селективное по отношению к PPKα, если в процессе трансактивационного анализа (описанного ниже) соединение трансактивирует рецепторы PPKα при значительно более низких концентрациях, чем PPKβ и РРКr. Вместо измерения трансактивации можно также определить связывание соединения с тремя подтипами РРК рецептора соответственно. Данные, выраженные в величинах Kd, полученные при реакции связывания (описано ниже), также свидетельствуют о способности соединения действовать специфично или селективно на рецепторы PPKα в большей степени, чем на PPKβ и РРКr рецепторы. Соединение считается специфичным или селективным в отношении PPKα для целей данного изобретения, если величина Kd его связывания с рецепторами PPKα приблизительно в 500 раз меньше, чем Kd его сродства к рецепторам PPKβ и РРКr.
На фиг.1 показан график, показывающий результаты анализа клеточных культур клеток RPMI 8226, осуществленного со всеми селективными соединениями - транс-ретиновой кислотой (АТРК) и двумя PPKα в соответствии с данным изобретением.
На фиг. 2 представлен другой график, показывающий результаты анализа клеточных культур AML 193, осуществленного с двумя селективными соединениями к PPKα в соответствии с данным изобретением и с двумя соединениями, которые не являются селективными к PPKα.
На фиг. 3 представлен еще один график, показывающий результаты анализа клеточных культур AML 193, осуществленного с тремя селективными к PPKα соединениями в соответствии с настоящим изобретением и со всеми транс-ретиновыми кислотами (АТРК).
На фиг. 4 приведен график, показывающий пролиферацию клеток овариальной опухоли при анализе клеточной культуры (метод EDR) в присутствии различных концентраций Соединения 2 согласно данному изобретению.
На фиг. 5 показан график, показывающий пролиферацию клеток RPE в присутствии всех транс-форм ретиновой кислоты или Соединения 42 в культуральной среде.
На фиг. 6 показан график, показывающий величины веса группы подопытных крыс, которым вводили в течение 3 дней различные дозы селективного к PPKα соединения согласно данному изобретению.
На фиг.7 показана диаграмма, демонстрирующая вес группы подопытных крыс в конце 4-дневного периода, когда в течение 3 дней крысам вводили различные дозы Соединения 18 согласно данному изобретению.
На фиг. 8 представлен график, показывающий вес морских свинок, которых лечили различными дозами Соединения 42 в течение 15 дней.
Определения, касающиеся химических соединений, используемых согласно данному изобретению
Термин алкил относится к любой и всем группам, известным как нормальный алкнл, разветвленный алкил и циклоалкил. Термин алкенил охватывает нормальный алкенил, разветвленный алкенил и циклоалкенил, имеющие одну или несколько ненасыщенных связей. Точно так же термин алкинил относится к нормальному алкинилу и разветвленному алкинилу, содержащим одну или несколько тройных связей.
Низший алкил означает алкильные группы, содержащие 1-6 атомов углерода в случае нормального низшего алкила и 3-6 атомов углерода в случае разветвленного и циклоалкильного радикалов. Низший алкенил означает группу, содержащую 2-6 атомов углерода в случае нормального низшего алкенила и 3-6 атомов углерода в случае разветвленного и циклоалкенила. Низший алкинил означает также группу, содержащую 2-6 атомов углерода для нормальных низших алкинилов и 4-6 атомов углерода для разветвленных низших алкинилов.
Термин сложный эфир охватывает любое соединение, охватываемое значением этого термина, используемым в классической органической химии. Он относится к органическим и неорганическим сложным эфирам. Когда В в общей формуле предпочтительных соединений по изобретению обозначает -СООН, этот термин охватывает продукты, полученные при обработке этой функциональной группы спиртами или тиоспиртами, предпочтительно алифатическими, содержащими 1-6 атомов углерода. Если сложный эфир является производным соединений, в которых В обозначает -СН2ОН, этот термин охватывает соединения, полученные из органических кислот, способных образовывать сложные эфиры, включая фосфор- и серусодержащие кислоты, или соединения формулы -CH2OCOR11, где R11 обозначает любую замещенную или незамещенную алифатическую, ароматическую, гетероароматическую или аралифатическую группу, содержащую предпочтительно 1-6 атомов углерода в алифатических фрагментах.
Если не указано иное, предпочтительные эфиры получают из насыщенных алифатических спиртов или кислот, содержащих 10 или менее атомов углерода или циклических или насыщенных алифатических циклических спиртов и кислот, содержащих 5-10 атомов углерода. Особенно предпочтительны алифатические сложные эфиры, полученные из кислот и спиртов, содержащих низшие алкилы. Также предпочтительными являются фениловые или фениловые, содержащие низшие алкилы, сложные эфиры.
Амиды означают соединения, охватываемые классическим в органической химии определением. В этом случае этот термин включает незамещенные амиды и все алифатические и ароматические моно- или дизамещенные амиды. Если не указано иное, предпочтительны моно- и дизамещенные амиды, содержащие насыщенные алифатические радикалы, с 10 или менее атомами углерода или циклические или насыщенные алифатические циклические радикалы с 5-10 атомами углерода. Особенно предпочтительными амидами являются амиды, полученные из замещенных и незамещенных низших алкиламинов. Также предпочтительны моно- и дизамещенные фениламиды, полученные из замещенных и незамещенных фенил- или низших алкилфениламинов. Предпочтительны также незамещенные амиды.
Ацетали и кетали включают радикалы формулы -СК, где К означает (-OR)2. В этом случае R обозначает низший алкил. Кроме того К может обозначать -OR7O-, где R7 является низшим С2-5-алкилом, с прямой или разветвленной цепью.
Фармацевтически приемлемая соль может быть получена из любого соединения, используемого согласно данному изобретению и имеющего функциональную группу, способную образовать такую соль, например, кислотную группу. Фармацевтически приемлемая соль является любой солью, которая сохраняет активность родительского соединения и не оказывает вредного или нецеленаправленного воздействия на субъект, которому она вводится и в условиях, в которых она вводится. Фармацевтически приемлемые соли могут быть получены при помощи органических или неорганических оснований. Соль может содержать одно- или поливалентный ион. Особенный интерес представляют собой неорганические ионы натрия, калия, кальция и магния. Органические соли можно получить из аминов, особенно аммониевые соли из моно-, ди- и триалкиламинов или этаноламинов. Соли можно также получить на основе кофеина, трометамина и подобных соединений. Если в молекуле есть азот с достаточной для образования солей с кислотой основностью, такие соли могут быть получены с неорганическими или органическими кислотами или алкилирующим агентом, таким как метилиодид. Предпочтительны соли, полученные из неорганических кислот, таких как соляная, серная или фосфорная. Можно использовать любую из простых органических кислот с одной, двумя или тремя карбоксильными группами.
Некоторые из соединений согласно данному изобретению могут иметь транс- и цис-(Е и Z) изомеры. Кроме того, соединения по изобретению могут содержать один или несколько хиральных центров и, следовательно, могут существовать в энантиомерной и диастереомерной формах. Изобретение охватывает применение всех таких изомеров per se, а также смесей цис- и транс-изомеров, смесей диастереомеров и рацемических смесей энантиомеров (оптические изомеры).
Описание соединений, предпочтительно используемых в способах по изобретению
Ретиноидные соединения, применяемые в способах лечения согласно данному изобретению, являются специфичными или селективными по отношению к PPKα. Тот факт, что соединение является специфичным или селективным по отношению к PPKα, может быть установлен методом трансактивационного анализа, описанным ниже, когда специфичное или селективное по отношению к PPKα соединение трансактивирует PPKα при значительно меньшей концентрации, чем PPKβ или РРКr. При проведении связывающего анализа, когда оценивается способность соединения связывать эти подтипы рецепторов, соединение считается специфичным или селективным по отношению к PPKα, если оно связывает PPKα примерно в 500 раз сильнее, чем PPKβ или РРКr. Кроме того, соединение считается специфичным или селективным по отношению к PPKα, если в реакции связывания его величина Kd находится примерно в наномолярном интервале от 10-1 до 5•102 и величина Kd для PPKβ или РРrК больше 1000 нмоль. Последнее показано как 0,00 в нижеприведенных таблицах, где приведены величины связывания (величины Kd) для некоторых соединений согласно изобретению.
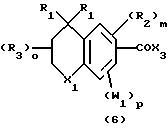
Примерами соединений, которые являются селективными по отношению к PPKα, предпочтительно используемых согласно настоящему изобретению, являются соединения формулы 1 и формулы 2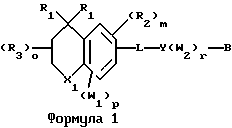
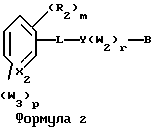
где X1 обозначает О или X1 обозначает [C(R1)2]n, где n - целое число от 0 до 2;
R1 независимо обозначает Н или алкил, содержащий 1-6 атомов углерода;
R2 независимо обозначает водород или низший С1-С6, алкил;
R3 обозначает водород, низший С1-С6 алкил или F;
m - целое число 0-5;
о - целое число 0-4;
р - целое число 0-2;
r - целое число 0-2;
Х2 обозначает N или СН;
Y обозначает фенил или нафтил, или гетероарил, выбранный из группы, состоящей из пиридила, тиенила, фурила, пиридазинила, пиримидинила, пиразинила, тиазолила, оксазолила, имидазолила и пиразолила, причем указанные фенил, нафтил и гетероарил могут быть замещены одним или двумя R2;
W1 обозначает заместитель, независимо выбранный из группы, состоящей из F, Br, Cl, I, фторзамещенного С1-6 алкила, NO2, и ОН, при условии, что:
(i) когда соединение соответствует формуле 1 и Z означает О, сумма р и r равна по меньшей мере 1 и W1 не является F в положении 3 тетрагидронафталинового кольца;
(ii) когда соединение соответствует формуле 1 и r равно 0 и р равно 1 и W1 обозначает ОН, ОН находится в α-положении по отношению к группе L;
W2 обозначает заместитель, независимо выбранный из группы, состоящей из F, Br, Cl, I, фторзамещенного C1-6 алкила, NO2, и ОН;
W3 обозначает заместитель, независимо выбранный из группы, состоящей из F, Br, Cl, I, фторзамещенного C1-6 алкила, NO2, и ОН, при условии, что когда соединение соответствует формуле 2 и X2 обозначает СН и r равен 0, р не равен 0 и, по меньшей мере, один W3 не является алкилом;
L обозначает -(C=Z)-NH- или -NH-(C=Z)-
Z обозначает О или S, и
В обозначает СООН или фармацевтически приемлемую соль, COOR8, CONR9R10, -CH2OH, CH2OR11, CH2OCOR11, CHO, CH(OR12)2, СНОR13О, -COR7, CR7(ОR12)2, CR7ОR13О, где R7 обозначает алкил, циклоалкил или алкенил, содержащие 1-5 атомов углерода, R8 обозначает алкил, содержащий 1-10 атомов углерода или триметилсилилалкил, где алкильная группа содержит 1-10 атомов углерода или циклоалкил с 5-10 атомами углерода, или R8 обозначает фенил или низший алкилфенил, R9 и R10 независимо обозначают водород, С1-10-алкил, C5-10-циклоалкил, или фенил или низший алкилфенил, R11 обозначает низший алкил, фенил или низший алкилфенил, R12 обозначает низший алкил и R13 обозначает двухвалентный С2-5-алкил.
В отношении X1 в формуле 1 следует указать, что в способах согласно изобретению предпочтительно применять соединения, в которых X1 обозначает [C(R1)2] n и n равен 1 (производные тетрагидронафталина). В отношении Х2 в формуле 2 предпочтительны соединения, в которых Х2 обозначает СН или N. Если Х2 обозначает СН, бензольное кольцо предпочтительно является 1,3,5-замещенным, L находится в положении 1 и W3 и/или R2 находятся в положениях 3 и 5. Если Х2 обозначает N, пиридиновое кольцо является предпочтительно 2,4,6-замещенным, L находится в положении 4 и W3 и/или R2 находятся в положениях 2 и 6.
R1 в формуле 1 предпочтительно обозначает Н или СН3•R3 в формуле 1 предпочтительно обозначает Н. В в предпочтительных соединениях обозначает СООН или фармацевтически приемлемую соль, COOR8 или CONR9R10, где R8, R9 и R10 указаны выше.
Что касается W1 и W2 в формуле 1, эти группы вообще оттягивают электроны, которые находятся в соединениях по изобретению или в ароматической части конденсированных ядер, или в виде заместителя арила или гетероарила Y. Предпочтительно W2 находится в группе Y и W1 также содержится в ароматической части конденсированной системы. Если Z обозначает S (тиоамиды), W1 или W2 необязательно должны содержаться в соединениях по изобретению в соответствии с формулой 1, хотя предпочтительно, по меньшей мере, один из W1 или W2 тем не менее содержится. В ариле или гетероариле Y в соединениях формулы 1 и формулы 2 W2 предпочтительно расположен в положении, смежном с группой В; предпочтительно В находится в пара-положении в фенильном кольце относительно "амидных" фрагментов и, следовательно, W2 предпочтительно находится в мета-положении относительно амидных фрагментов. Когда в ароматической части конденсированной системы соединений формулы 1 содержится W1, предпочтительно, чтобы W1 находился в положении 8 хроманового ядра и группа Z=C=NH- занимала положение 6. В тетрагидронафталинах формулы 1 группа Z=C=NH- занимает предпочтительно положение 2 и W1 находится предпочтительно в положении 4. Однако, если W1 обозначает ОН в соединениях формулы 1, ОН предпочтительно находится в положении 3 в тетрагидронафталиновом кольце.
Предпочтительными W1 и W2 группами являются F, NO2, Br, I, СF3, СlN3 и ОН. Особенно предпочтительным является наличие одного или двух фтор-заместителей в Y (W2). Если Y обозначает фенил, фторзаместители предпочтительно находятся в орто- и орто'-положениях по отношению к В, которая предпочтительно обозначает СООН или COOR8.
Что касается группы W3 в формуле 2, эта группа также является группой, оттягивающей электроны, или алкильной группой, наиболее предпочтительными W3 группами являются F, NO2, Br, I, СF3, N3 и ОН. Кроме того, в фенильном или пиридиновом кольце (показанном в формуле 2 в виде заместителя "(W3)р") W3 является алкилом, предпочтительно разветвленным алкилом, таким как трет. -бутил, и р предпочтительно равен 2.
Что касается символа Y в формуле 1, а также в формуле 2 предпочтительные соединения, используемые в способах согласно данному изобретению, представляют собой такие соединения, в которых Y обозначает фенил, пиридил, 2-тиазолил, тиенил или фурил, более предпочтительно фенил. Что касается заместителей у Y (фенил) и Y (пиридил), то предпочтительны соединения, у которых фенильная группа является 1,4(пара)-замещенной L и В группами и пиридиновое кольцо является 2,5-замещенным L и В группами. (Замещение в 2,5-положениях по номенклатуре для "пиридинов" соответствует замещению в 6-положение по номенклатуре для "никотиновой" кислоты). В предпочтительных соединениях по изобретению нет возможного заместителя R1 (кроме Н) у группы Y.
L в формуле 1 и в формуле 2 предпочтительно обозначает -(C=Z)-NH- и Z предпочтительно обозначает О. Другими словами, согласно изобретению предпочтительны те карбамоильные или амидные соединения, у которых -NH- присоединена к Y.
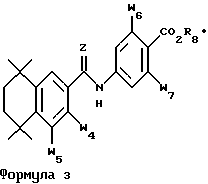
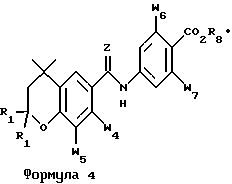
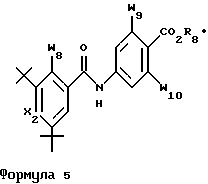
Соединения, которые наиболее предпочтительно применять в способах лечения по изобретению, указаны ниже в таблице 1 со ссылкой на формулы 3 и 4 и в таблице 2 со ссылкой на формулу 5.
Способы введения
Соединения, специфичные или селективные по отношению к PPKα, применяемые согласно изобретению, можно вводить системно или локально, в зависимости от таких факторов, как состояние пациента, необходимость специфичного к определенному участку лечения, количество лекарства, которое нужно ввести, и многочисленные другие факторы.
При лечении дерматозов предпочтительно вводить лекарство локально, хотя в некоторых случаях, например, при лечении острых кистозных угрей или псориаза, можно применять пероральное введение. Можно использовать любой обычный препарат для локального введения, например, раствор, суспензию, гель, мазь или т.п. Приготовление таких локальных препаратов широко известно в области лекарственных средств, как описано, например, в Remington, Pharmaceutical Science, Edition 17, Mack Publishing Company, Easton, Pennsylvania. Локально эти соединения могут быть также введены в виде порошка или лака, в частности, аэрозоля. Если лекарство надо вводить системно, оно может быть в виде порошка, пилюль, таблеток и т.п. или в виде сиропа или эликсира, пригодного для введения перорально. Для внутривенного или внутрибрюшинного введения соединение вводят в виде раствора или суспензии, которые можно инъецировать. В некоторых случаях полезно приготавливать на основе этих соединений суппозитории или препарат с замедленным выделением лекарства для введения под кожу или путем внутримышечной инъекции.
К таким препаратам для местного введения могут быть добавлены другие медикаменты для вторичных целей, например, для лечения сухости кожи; защиты от действия света; другие лекарства для лечения дерматозов; лекарства для предотвращения инфекции, уменьшения раздражения, воспаления и т.п.
Лечение дерматозов или других симптомов, поддающихся лечению соединениями типа ретиновой кислоты, будет эффективным при введении терапевтически эффективной дозы одного или нескольких соединений по изобретению. Терапевтической концентрацией считается такая концентрация, которая вызывает улучшение конкретного состояния или замедляет его распространение. В некоторых случаях соединение можно применять профилактически для предотвращения появления болезненного состояния.
Терапевтическая или профилактическая концентрация меняется в зависимости от состояния и в некоторых случаях будет меняться с ухудшением состояния и восприимчивости пациента к лечению. Соответственно, не является установленной какая-то определенная концентрация, но она требует изменения в зависимости от особенностей болезни. Такие концентрации определяют при рутинном эксперименте. Однако, установлено, что при лечении, например, угрей или похожих дерматозов терапевтически эффективная концентрация составляет 0,01-1,0 мг на мл препарата. Если вводить лекарство системно, то ожидается, что для получения терапевтического действия при лечении многих болезней надо применять 0,01-5,0 мг на кг веса тела в день.
Анализ селективной биологической активности в отношении PPKα и ее значение, приводящее к снижению побочных эффектов и токсичности
Во вводной части данного описания отмечалось, что у млекопитающих (и в других организмах) существуют два основных типа рецепторов ретиновой кислоты (РРК и РХР). Внутри каждого типа существуют подтипы (PPKα,PPKβ, РРКr, PXPα,PXPβ и РХРr), распределение которых является неоднородным в различных тканях и органах организма млекопитающих. Селективное связывание только одного или двух подтипов ретиноидных рецепторов внутри одного семейства этих рецепторов может привести к выгодным фармакологическим свойствам вследствие меняющегося распределения подтипов в нескольких тканях или органах млекопитающего. По вышеприведенным причинам связывание любого или всех ретиноидных рецепторов, а также специфичная или селективная активность к любому подтипу рецепторов являются желательными фармакологическими свойствами.
В свете сказанного были разработаны методы анализа для испытания агонистической активности соединений в отношении подтипов PPKα,PPKβ, РРКr, PXPα,PXPβ и РХРr. Например, метод анализа трансактивации химерного рецептора, который позволяет определить агонистическую активность в отношении PPKα,PPKβ, РРКr и PXPα подтипов и который основан на работе Feigner P.L. and Holm M. (1989) Focus, 112, описан подробно в патенте США N 5 455 265, включенном в данное описание в качестве ссылки.
Способ анализа трансактивации голорецептора и метод связывания лигандов, которые основаны на измерении способности соединений связывать некоторые подтипы ретиноидных рецепторов, описаны соответственно в WO 93/11755 (в частности, на стр. 30-33 и 37-41), опубликованной 24 июня 1993, включенной в данное описание в качестве ссылки. Описание метода связывания приведено также ниже.
ИССЛЕДОВАНИЕ СВЯЗЫВАНИЯ
Все исследования связывания осуществляют одинаково. Все шесть типов рецепторов были получены из экспрессионного типа рецептора (РАР α,β, Г и РХР α,β, Г), экспрессию осуществляли в Baculovirus. Исходные растворы всех соединений были приготовлены в виде растворов в этаноле с концентрацией 10 мМ и была приготовлена серия разбавленных растворов 1:1 ДМСО:этанолом. Буферные растворы для анализа всех шести рецепторов имели следующей состав: 8% глицерина, 120 мМ КСl, 8 мМ Трис, 5 мМ CHAPS, 4 мМ DTT и 0,24 мМ ФМСФ, рН 7,4 при комнатной температуре.
Все исследования связывания рецепторов осуществляли одинаково. Конечный объем был равен 250 мкл и раствор содержал 20-40 мкг белкового экстракта в зависимости от исследуемого рецептора вместе с 5 нМ [3Н]-полностью транс-ретиновой кислоты или 10 нМ [3Н]-9-цис-ретиновой кислоты и изменяющихся концентраций конкурентного лиганда, находящихся в интервале 0-10-5 М. Для анализа использовали систему с 96 лунками. Инкубирование осуществляли при 4oС до достижения равновесия. Неспецифичные связывания определяли как остаточное связывание в присутствии 1000 нМ соответствующего изомера ретиновой кислоты, не содержащего метки. В конце инкубационного периода добавляли в соответствующем отмывающем буфере 50 мкл 6,25%-ного гидроксиапатита. Отмывающий буфер состоял из 100 мМ КСl, 10 мМ Трис и/или 5 мМ CHAPS (PXP α,β, r), или 0.5% Triton X-100 (PAP α,β, r). Смесь перемешивали и осуществляли инкубирование в течение 10 минут при 4oС, центрифугировали и удаляли надосадочную жидкость. Гидроксиапатит промывали еще три раза подходящим буфером. Комплекс рецептор-лиганд адсорбировался гидроксиапатитом. Количество комплекса рецептор-лиганд определялось при помощи жидкостного сцинтилляционного счетчика на гранулах гидроксиапатита.
После корректировки неспецифичного связывания определяли величины IC50. Величина IC50 определяется как концентрация конкурентного лиганда, необходимая для уменьшения специфичного связывания на 50%. Величина IC50 была определена графически по логарифмическому графику. Величины Kd определяли с применением уравнения Ченга-Прусоффа для IC50, концентрации меченого лиганда и Кd меченого лиганда. Результаты исследования связывания лиганда выражены в величинах Кd. (См. Cheng и др. Biochemical Pharmacology, Vol. 22, рр. 3099-3108, включенную в данное описание в качестве ссылки).
В таблице 3 приведены результаты исследования связывания лиганда для некоторых заявленных соединений.
Как можно видеть из вышеприведенных данных, соединения по изобретению специфично или селективно связываются с PPKα-рецепторами. Было установлено согласно настоящему изобретению, что этот уникальный вид селективности позволяет соединениям сохранять ценные свойства ретиноидов при уменьшении ряда побочных эффектов и токсичности. Более конкретно некоторые исследования клеточных культур in vitro описаны ниже, причем показана способность соединений, специфичных или селективных к PPKα, значительно ингибировать рост раковых клеток.
ИСПЫТАНИЯ РАКОВЫХ КЛЕТОЧНЫХ ЛИНИЙ
МАТЕРИАЛЫ И МЕТОДЫ
Гормоны
Полностью транс-форму ретиновой кислоты (т-РК) (Sigma Chemicals Co., St. Louis, МО) хранили при -70oС. Перед каждым опытом соединение растворяли в 100%-ном этаноле с концентрацией 1 мМ и разбавляли в культуральной среде непосредственно перед использованием. Все опыты проводили при слабом освещении. Контрольные образцы анализировали при той же концентрации этанола и эта концентрация растворителя не оказывала влияния на опыты.
Клеточные линии RPMI 8226, ME-180 и AML-193 получали из коллекции American Type Culture Collection (ATCC, Rockville, MD). RPMI 8226 представляет собой линию гемапоэтических клеток человека, полученных из периферической крови пациента с множественной миеломой. Клетки походят на лимфобластозные клетки другой клеточной линии лимфоцитных клеток человека и выделяют легкие цепи иммуноглобулина α-тип. Клетки RPMI 8226 выращены в среде RPMI (Gibco), дополненной 10% плодной сыворотки коровы, глутамином и антибиотиками. Клетки выдерживали по мере роста культур в суспензии при 37oС во влажной атмосфере 5% СО2 в воздухе. Клетки разбавляли до концентрации, равной 1•105/мл дважды в неделю.
ME-180 представляет собой линию эпидермоидных клеток карциномы человека, полученных из шейки. Опухоль представляла собой высокоинвазивную сквамозную клеточную карциному с неравномерным скоплением клеток и незначительной кератинизацией. Клетки ME-180 были выращены и выдерживались в среде McCoy 5a (Gibco), дополненной 10% плодной сыворотки коровы, глутамином и антибиотиками. Клетки выдерживали в виде однослойных культур, выращенных при 37oС во влажной атмосфере 5% СО2 в воздухе. Клетки были разбавлены до концентрации 1•105/мл дважды в неделю.
AML-193 получали из бластных клеток, классифицируемых как клетки М5 острой моноцитарной лейкемии. Фактор роста, фактор стимуляции колонии гранулоцитов (GM-CSF) требовался для создания этой клеточной линии, а для ее непрерывного размножения в химически определенной среде были необходимы факторы роста. Клетки AML-193 выращивались и выдерживались в Iscove's модифицированной среде Dulbecco, дополненной 10% плодной сыворотки коровы, глутамином и антибиотиками с 5 мкг/мл инсулина (Sigma Chemical Co.) и 2 нг/мл агглютинин антирезуса GM-CSF (системы R и D). Клетки разбавляли до концентрации 3•105/мл дважды в неделю.
Внедрение 3Н-тимидина
Метод, используемый для определения внедрения радиомеченого тимидина, был адаптирован на основе методики, описанной Shrivastav и др. Клетки RPMI-8226 помещали в титрационный микропланшет с круглым дном, содержащий 96-лунок (Costar) с плотностью 1000 клеток/лунку. В соответствующие лунки добавляли испытуемые ретиноиды с конечными концентрациями, указанными для конечного объема 150 мкл/лунку. Планшеты инкубировали в течение 96 час при 37oС во влажной атмосфере 5% СО2 в воздухе. Затем в каждую лунку добавляли 1 мкКе [5'-3Н]-тимидина (Amersham, U.K., специфичная активность 43 К.е/ммол) в 25 мкл культуральной среды и культивировали клетки еще в течение 6 часов. Затем культуры обрабатывали, как описано ниже.
Лунки с МЕ-180, полученными трипсинизацией, помещали в титрационный микропланшет с плоским дном с 96-лунками с плотностью 2000 клеток/лунку. Культуры обрабатывали, как описано выше для RPMI 8226 со следующими исключениями. После инкубирования с тимидином надосадочную жидкость аккуратно удаляют и промывают клетки 0.5 мМ раствором тимидина в физиологическом растворе с фосфатным буфером. Клетки МЕ-180 кратковременно обрабатывали 50 мкл 2.5% трипсина для удаления клеток из планшета.
Клетки AML-193 помещали в титрационный микропланшет с круглым дном с 96-лунками (Costar) с плотностью 1000 клеток/лунку. В соответствующие лунки добавляли испытуемые ретиноидные соединения с конечной концентрацией, указанной для конечного объема 150 мкл/лунку. Планшеты инкубировали в течение 96 час при 37oС во влажной атмосфере 5% СО2 в воздухе. Затем в каждую лунку добавляли 1 мкКе [5'-3Н]-тимидина (Amersham, U.K., специфичная активность 43 Ке/ммол) в 25 мкл культуральной среды и инкубировали клетки еще в течение 6 часов.
Линии клеток затем обрабатывали следующим образом: клеточную ДНК осаждали 10% трихлоруксусной кислотой на стекловолокнистом фильтре с использованием Скатроновского харвестера клеток с множеством лунок (Skatron Instruments, Sterling VA). Радиоактивность, внедренная в ДНК, как непосредственная мера роста клеток, измерялась при помощи сцинтилляционного счетчика. Величины являются показателем среднего числа распадов в минуту внедренного тимидина из трипликатных лунок ± РЭМ.
График на фиг. 1 показывает, что при проведении вышеописанного анализа клеточной культуры RPMI 8226 (злокачественная миелома) Соединения 4 и 12 (два соединения по изобретению) ингибировали рост этих злокачественных клеток почти так же, как соединение для сравнения, полностью транс-форма ретиновой кислоты (ПТРК). График на фиг. 1 также показывает, что в то время как при низких концентрациях (10-12 до примерно 10-9) полностью транс-форма ретиновой кислоты (ПТРК) действительно ускоряет рост этих клеток, селективные по отношению к PPKα Соединения 4 и 12 по изобретению не стимулируют, а уже в таких низких концентрациях ингибируют рост этих злокачественных клеток.
График на фиг. 2 показывает, что при проведении анализа клеточной культуры AML-193 (острая моноцитарная лейкемия) Соединения 22 и 42 согласно настоящему изобретению ингибировали рост этих злокачественных клеток. Два других соединения, для которых также приведены данные на этом графике, обозначены AGN 193090 и AGN 193459. (Номер AGN - это произвольный номер, используемый правопреемником по данной заявке). Соединения AGN 193090 и AGN 193459 не являются селективными в отношении к PPKα. Эти соединения - это соответственно 4-[(8-циан-5,6-дигидро-5,5-диметилнафт-2-ил)этинил]бензойная кислота и 4-[(5,6-дигидро-5,5-диметилнафт-7(6Н)-8-(1-2,2-диметилпропилиден)нафт-2-ил)этинил]бензойная кислота, их Kd для PPKα, PPKβ и РРКr равны 109, 34, 77 и 6, 2, 7 соответственно. График на фиг. 2 показывает, что селективные и специфичные по отношению к PPKα соединения ингибируют рост злокачественных клеток при низких концентрациях, в то время как панагонисты AGN 193090 и AGN 193459 не ингибируют, а скорее даже стимулируют рост таких клеток при этих низких концентрациях.
на фиг. 3 показан другой график, показывающий результаты анализа клеток AML-193, где испытаны Соединения 4, 12 и 18 в соответствии с данным изобретением и полностью транс-ретиновая кислота (ПТРК). Данные показывают, что соединения, селективные по отношению к PPKα, уменьшают размножение клеток при низких концентрациях, тогда как ПТРК при тех же низких концентрациях промотирует размножение клеток.
Для другого случая испытывалось действие ретиноидных соединений на клетки, полученные из твердых опухолей пациентов. Этот метод анализа (ЭДР) описан ниже.
Свежие биопсии твердых опухолей были получены в течение 24 час хирургической операции. Образцы обрабатывались после заливки части опухоли парафином и гистопатологического подтверждения жизнеспособности образца и диагноза болезни. Остальные образцы были разделены на маленькие части стерильными ножницами. Маленькие фрагменты опухоли затем подвергались обработке коллагеназой и ДНКазой в течение 2 час при перемешивании в термостате с СО2 для того, чтобы выделить клетки опухоли из стромы соединительной ткани. Полученную суспензию клеток промывали и определяли количество клеток из цистоспинального препарата. Клетки опухоли снова суспендировали с концентрацией 40000 клеток/мл в 0,3% агарозе в RPMI 1640 с добавлением 15% плодной сыворотки коровы, глютамина и антибиотиков, 0,5 мл суспензии помещали в каждую лунку 24-луночного планшета над 0,5 мл слоем 0,5%-ной агарозы. Эти условия предотвращают адгезию клеток, тем самым способствуя размножению только трансформированных клеток. Клетки растут с получением трехмерных сферических частиц, рекапитулирующих их in vivo морфологию.
Ретиноиды добавляли через 24 час после посева для того, чтобы образцы восстановили равновесие в среде роста после передвижения и обработки. Клетки выращивали в течение четырех дней в присутствии лекарства, причем за 48 час до сбора клеток добавляли 3H-тимидин (5 к.е./мл) для обеспечения нужной метки быстро размножающихся клеток. После того как суспензия агароза-клетки была сжижена при 90oС, клетки собирали на стекловолокнистых фильтрах, количество клеток определяли с использованием 5 мл сцинтилляционной жидкости и жидкого сцинтилляционного счетчика Beckman 6500.
Результаты выражены в виде реакции быстроразмножающихся необработанных контрольных клеток. Обработка осуществлялась дважды или трижды, а контрольные опыты проводились четыре раза.
На графике на фиг.4 показано действие соединения 2 на яичниковые опухоли, полученные от 4 пациентов, график показывает, что это соединение ингибирует пролиферацию клеток этой опухоли, эффект зависит от концентрации.
Специалистам в данной области понятно, что способность соединений, селективных по отношению к PPKα ингибировать в значительной степени рост злокачественных клеток в вышеописанных способах анализа, является показателем того, что эти соединения можно вводить, получая положительный результат, млекопитающим (включая людей), у которых есть опухоли, для лечения этих опухолей, в особенности острого моноцитарного лейкоза, цервикальной карциномы, миеломы, овариальной карциномы и карцином на голове и шеи.
Согласно настоящему изобретению было также установлено, что пролиферация ретинальных пигментных эпителиальных клеток ингибируется соединениями, селективными по отношению к PPKα. Известно, что после ретинального отслоения ретинальный пигментный эпителий (РПЭ) становится дифференцированным, быстро размножается и мигрирует в подретинальное пространство (Campochiaro et al. Invest. Opthal and Vis. Sci. 32:65-72 (1991)). Следовательно такие процессы влияют на успех ретинального присоединения. РРК агонисты, такие как полностью транс-ретиновая кислота (ПТРК), оказывают антипролиферативное действие на скорость роста первичных РПЭ культур человека (Campochiaro et. al. , там же) и было показано, что они уменьшают вероятность ретинального отслоения после хирургических методов ретинального присоединения в опытах на людях (Fekrat et al., Opthtmology 102:412-418 (1994)).
График на фиг. 5 показывает влияние ингибирующего действия (в зависимости от концентрации) полностью транс-ретиновой кислоты (ПТРК) и соединения 42 на пролиферацию РПЭ в процессе анализа, методика которого описана ниже.
Анализ первичных РПЭ культур
Первичные культуры ретинального пигментного эпителия (РПЭ) человека были получены из глаза, как это было ранее описано (Campochiaro et. al., Invest. Opthal and Vis. Sci. 32:65-72 (1991)). 5•104 клеток помещали в 16-мм лунки многолуночного планшета с 24 лунками в Dulbecco's модифицированную среду Eagle (DMEM Gibco), содержащую 10% плодной сыворотки коровы (FBS). Клетки обрабатывали одним этанолом (контроль), ПТРК (10-10-10-6 M) в этаноле и соединением 42 (10-10-10-6 M) в этаноле. Клетки питали свежей средой, содержащей соответствующие концентрации этих соединений каждые два дня в течение шести дней. Клетки удаляли из планшета путем обработки трипсином и определяли число клеток при помощи электронного счетчика клеток. Как можно видеть на фиг. 5, обработка первичных РПЭ клеток ПТРК и соединением 42 приводит к зависящему от дозы уменьшению пролиферации РПЭ клеток.
Влияние локального введения экспериментальным безволосым мышам ретиноидов, селективных по отношению к PPKα согласно данному изобретению, изучалось также методом анализа при локальном раздражении кожи с использованием селективного по отношению к PPKα Соединения 18 по изобретению. Более подробно, раздражение кожи измерялось полуколичественно путем ежедневного субъективного осмотра шелушащихся и оцарапанных участков кожи. Отдельное число, показатель локального раздражения кожи, суммирует раздраженные участки кожи животного во время опыта. Показатель локального раздражения представляет собой алгебраическую сумму сложного показателя шелушения и сложного показателя царапин. Сложные показатели имеют значение 0-9 и 0-8 для шелушения и для царапин соответственно, они учитывают максимальную тяжесть, время появления и среднюю тяжесть шелушения и царапин.
Тяжесть шелушения оценивается по пятибалльной шкале и тяжесть царапин оценивается по 4-балльной шкале, более высокий балл означает более сильное поражение. Компонент максимальной тяжести сложного показателя является самым высоким дневным баллом, который дается конкретному животному в период наблюдения.
Компонент времени появления, входящий в сложный показатель, оценивается от 0 до 4 баллов следующим образом:
Время до появления шелушащихся участков или царапин с показателем тяжести 2 или более (дни) - Балл, оценивающий время появления
8 - 0
6-7 - 1
5 - 2
3-4 - 3
1-2 - 4
Компонент, оценивающий среднюю тяжесть, входящий в сложный показатель, представляет собой сумму показатель ежедневных шелушащихся участков или трещин (царапин), деленную на число дней наблюдения. Первый день лечения не учитывается, так как лекарственное соединение еще не имело возможности подействовать во время первой обработки.
Для подсчета сложных показателей шелушения и царапин, средние показатели тяжести и времени появления суммируются и делятся на 2. Результат прибавляется к показателю максимальной тяжести. Сложные показатели шелушения и царапин затем складываются с получением общего показателя локального раздражения. Каждое животное характеризуется показателем локального раздражения и величины выражены как среднее значение ±SD (ст. отклонение) индивидуальных показателей группы животных. Числа округляются до ближайшего целого числа.
Таким образом, самки безволосых мышей [Crl: SKH1-hrBR] (возраст 8-12 недель, n=4) обрабатывались локально в течение 5 дней соединением 18 дозами, выраженными в наномолях/25 г, указанными в таблице 4. Обработке подвергалась кожа на спине, общий объем составлял 4 мл/кг (-0,1 мл). За мышами наблюдали каждый день и оценивали шелушение и трещины каждый день и через 3 дня после последнего применения, то есть 8 дней.
Эти данные показывают, что соединение селективное по отношению к РРК, не вызывает раздражения кожи и потери веса до дозы 1000 нмол/25 г испытуемого животного. Для сравнения соединение ретиноидного типа 4-(Е)-2-[(5,6,7,8-тетрагидро-5,5,8,8-тетраметилнафталин-2-ил)пропен-1-ил] бензойная кислота (TTNPB), которое не является селективным по отношению к PPKα, вызывает гораздо более сильное раздражение кожи, как это видно из таблицы 4.
Другое важное преимущество введения млекопитающему ретиноидов селективных по отношению к PPKα состоит в значительно пониженной тератогенной активности таких селективных к PPKα соединений по сравнению со многими другими ретиноидами, как это показывает метод подавления хондрогенеза. Этот анализ проводят следующим образом:
Для сравнения способности испытуемых лекарственных средств подавлять хондрогенную дифференциацию при осуществлении биоанализа использовали культуры мезенхимных клеток почки конечности в виде пятен с высокой плотностью. Почки передней конечности эмбриона мыши на 12 день беременности (54±2 сомита) подвергают расщеплению в растворе трипсин-ЭДТК и полученную одноклеточную суспензию помещают в виде пятен объемом 20 мкл (200 000 клеток/пятно) на пластиковые чашки. Ретиноиды с концентрациями от 0,3 нг/мл до 3 мкг/мл (1 нМ - 10 мкМ) добавляли в культуральную среду (Eagle's MEM + 10% фетальной бычьей сыворотки, GIBCO) через 24 час после посева. Контрольные культуры получали только разбавитель (этанол, концентрация ≤ 1об.%). В качестве контрольного образца в другой серии опытов использовали ретиновую кислоту.
Рост культур останавливали через 96 час после посева, при этом среду удаляли и клетки фиксировали в течение 1 часа в 10% формалине, содержащем 0,5% цетилпиридинийхлорида. Культуры промывали уксусной кислотой и окрашивали в течение 1 часа в 0,5%-ном Alcian голубом растворе при рН 1,0, дифференцировали в 3% уксусной кислоты и затем дегидратировали в этаноле и оценивали хондрогенез под микроскопом. Отсутствие или уменьшение числа хрящевых узелков в окрашенных культурах по сравнению с контрольными культурами является показателем подавления хондрогенеза. Число хрящевых узелков, окрашенных в пятне; среднее число узелков и стандартные отклонения рассчитывались для четырех дублированных культур на одну обработку. Средняя величина концентрации, вызывающая 50%-ное ингибирование хондрогенеза в сравнении с контрольными образцами (IC50), рассчитывалась по логарифмической кривой, отражающей зависимость доза-ответ. Величины IC50 выражены в нанограммах на мл (нг/мл). Согласно данному методу анализа более высокое значение IC50 означает меньшую тератогенность. В таблице 5 показаны результаты, полученные для Соединений 10, 18 и 42 согласно данному изобретению, и для полностью транс-ретиновой кислоты (ПТРК) и 4-(Е)-2-[ (5,6,7,8-тетрагидро-5,5,8,8-тетраметилнафталин-2-ил)пропен-1-ил]бензойной кислоты (TTNPB).
Как видно из таблицы 5, соединения по изобретению являются менее тератогенными, чем полностью транс-ретиновая кислота и значительно (порядка 104) менее тератогенными, чем известное TTNPB.
Уменьшение или увеличение веса экспериментальных животных от введения ретиноидов представляет собой еще один опыт по определению токсичности, причем значительная потеря веса при сравнительно низких дозах свидетельствует о значительной токсичности ретиноида. В одном опыте группам из 5 крыс вводили различные дозы (в кукурузном масле) испытуемого соединения в течение 3 дней. Крыс умерщвляли через 24 часа после введения последней дозы. На графике на фиг. 6 показан средний вес каждой группы крыс, которым вводили дневные дозы 10, 30 и 90 мкмол/кг/день соединения 42, а также средний вес группы контрольных крыс, которым ретиноид не вводили. Как можно видеть, селективное по отношению к ΠΠΚα соединение 42 не вызывает потери веса по сравнению с контрольными животными, за исключением введения очень высокой дозы (90 мкмол/кг/день). На графике на фиг. 7 показан вес крыс на четвертый день (через 24 часа после последнего введения ретиноида) в таком же опыте при введении различных доз соединения 18, причем ноль показывает контрольных животных. Как видно, этот селективный по отношению к PPKα ретиноид не вызывает потери веса даже при дозе 90 мкмол/кг/день. Примечательно, что в опытах с TTNPB, который связывается со всеми тремя подтипами РРК (см. таблицу 3), это соединение вызывает значительную потерю веса. В этом опыте, когда крысам вводили соединение 42, не наблюдалось значительной токсичности для кожно-слизистой оболочки.
В другом опыте трехнедельным самцам морских свинок Hartley имплантировали внутрибрюшинно осмотические насосы, содержащие 20% ДМСО/80% полиэтиленгликоля (разбавитель) или соединение 42 с концентрацией в разбавителе 4,4, 13,3 или 40 мг/мл. Исходя из первоначального веса животных и известной скорости подачи определяли приблизительные дозы соединения 42 - 0, 2, 6 и 18 мг/кг/день. Вес животных и клинические наблюдения регистрировали по меньшей мере каждый второй день в течение 14 дней после имплантации. Свинки были умерщвлены через 14 дней, насосы проверяли на наличие повреждений. На графике на фиг. 8 показан вес животных в течение 15 дней эксперимента. Как видно из графика, низкие и средние дозы ретиноида, являющегося селективным по отношению к PPKα (соединение 42), не вызывают уменьшения веса или вызывают статистическое уменьшение веса животного по сравнению с контрольными животными. Значительное подавление увеличения веса наблюдалось только при высокой дозе (18 мг/кг/день) соединения 42. Важно, что при любой дозе соединения 42 в этом опыте это соединение не было токсичным для кожно-слизистой оболочки. Эта значительно сниженная токсичность, наблюдавшаяся при введении животным селективных по отношению к PPKα соединений согласно настоящему изобретению, является значительным преимуществом, так как токсичность для кожно-слизистой оболочки является основным и неприятным побочным эффектом ретиноидов, являющихся токсичными для людей.
Способы синтеза предпочтительных соединений по изобретению, являющихся селективными по отношению к PPKα
Общая структура соединений, предпочтительно используемых в способах лечения согласно данному изобретению, представлены выше формулой 1 и формулой 2. Эти соединения могут быть получены методами синтеза, описанными ниже. Химику-органику очевидно, что условия, описанные ниже, являются конкретными формами воплощения изобретения, которые могут быть использованы для получения любого или всех соединений, представленных указанными формулами.
В общем способ получения соединений, предпочтительно используемых в способах по изобретению, соответствующих формуле 1, включает образование амида путем реакции соединения общей формулы 6 с соединением общей формулы 7, или путем реакции соединения общей формулы 6а с соединением общей формулы 7а. Точно так же способ получения соединений формулы 2 включает образование амида реакцией соединения общей формулы 8 с соединением общей формулы 7, или реакцией соединения общей формулы 8а с соединением общей формулы 7а.
Соединение формулы 6 представляет собой кислоту или "активированную форму" карбоновой кислоты, присоединенной к ароматической части тетрагидронафталина, (X1=[C(R1)2]n и n=1), дигидроиндена ([C(R1)2]n где n=0) или хромана (X1 обозначает 0). Карбоновая кислота или ее "активированная форма" присоединена в 2- или 3-положении тетрагидронафталина и в 6- или 7-положении хромановых ядер. В соединениях, предпочтительно используемых по изобретению, присоединение происходит в 2-положении тетрагидронафталина и в 6-положении хромана.
Термин "активированная форма" карбоновой кислоты следует понимать как производное карбоновой кислоты, которое способно образовать амид при взаимодействии с первичным амином формулы 7. В случае "обратных амидов" активированная форма карбоновой кислоты представляет собой производное (формула 7а), которое способно образовать амид при реакции с первичным амином формулы 6а. Это в общем означает такие производные карбоновой кислоты, которые известны и используются для образования амидных связей с амином. Примерами подходящих форм или производных для этой цели служат хлорангидриды кислот, бромангидриды кислот и эфиры карбоновой кислоты, в частности активные эфиры, где спиртовой фрагмент эфира образует отщепляемую группу. Наиболее предпочтительными в качестве реагентов по изобретению, описываемых формулой 6 (или формулой 7а), являются хлорангидриды кислот (Х3 обозначает Сl). Хлорангидриды формулы 6 (или формулы 7а) можно получать традиционными методами из соответствующих эфиров (X означает, например, этил) гидролизом и обработкой хлористым тионилом (SOCl2). Хлорангидриды формулы 2 (или формулы 3а) могут быть получены традиционными способами из соответствующих сложных эфиров (Х3 обозначает, например, этил) путем гидролиза и обработки хлористым тионилом (SO2Cl). Хлорангидриды формулы 6 (или формулы 7а) можно также получить прямой обработкой карбоновых кислот тионилхлоридом, когда карбоновая кислота является более доступной, чем ее эфир, или известным методом синтеза. Хлорангидриды формулы 6 (или формулы 7а) обычно реагируют с амином формулы 7 (или амином формулы 6а) в инертном растворителе, например, метиленхлориде, в присутствии акцептора кислоты, например, пиридина.
Сами карбоновые кислоты формулы 6 (или формулы 7а) также пригодны для образования амида при реакции с амином, катализатор (4-диметиламинопиридин) в присутствии дегидратирующего агента, например, дициклогексилкарбодиимида (ДЦК) или, что более предпочтительно, гидрохлорида 1-(3-диметиламинопропил)-3-этилкарбодиимида (ЭКД).
Карбоновые кислоты, соответствующие эфирам формулы 6, получают обычно как описано в химической научной или патентной литературе, описанные способы могут быть изменены, если необходимо, с использованием таких химических реакций или методов, которые per se известны. Например, 2,2-, 4,4- и/или 2,2,4,4-замещенные хроман-6-карбоновые кислоты и хроман-7-карбоновые кислоты известны из патентов США NN 5006550; 5314159; 5324744 и 5348975, включенных в данное описание в качестве ссылок. 5,6,7,8-Тетрагидронафталин-2-карбоновые кислоты можно получить в соответствии с патентом США N. 5130335, также включенным в качестве ссылки.
Вышеописанный принцип реакций, ведущих к образованию амидов формулы 1, также применим для получения амидов формулы 2. Реагенты, которые используются для получения амидов формулы 2, следующие: активированные карбоновые кислоты формулы 8 и формулы 7а и амины формулы 7 и формулы 8а.
H2N-Y(W2)r-B (7)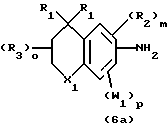
X3OC-Y(W2)r-B (7a)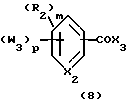
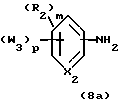
Карбоновые кислоты, соответствующие эфирам формулы 8, получают обычно, как описано в химической научной или патентной литературе, методики могут быть модифицированы, если необходимо, такими химическими методами или реакциями, которые известны per se.
Реакционные схемы 1 и 2 (см. в конце описания) представляют примеры синтеза производных 5,6,7,8-тетрагидро-5,5,8,8-тетраметилнафталин-2-карбоновой кислоты, которые подпадают под формулу 6 и которые реагируют с амином формулы 7 с получением 5,6,7,8-тетрагидро-5,5,8,8-тетраметилнафталин-2-карбамоильных производных, подпадающих под формулу 1. Так, как показано на реакционной схеме 1, Этил-5,6,7,8-тетрагидро-5,5,8,8-тетраметилнафталин-2-карбоксилат (соединение А) нитруют с получением соответствующего 3-нитросоединения (соединение В). Нитрогруппа соединения В восстанавливается с получением соответствующего 3-аминосоединения (соединение С), которое описано в работе Lehman et. al. Cancer Research, 1991, 51, 4804. Этил-5,6,7,8-тетрагидро-5,5,8,8-тетраметил-3-аминонафталин-2-карбоксилат (соединение С) бромируют с получением соответствующего 4-бромпроизводного (соединение D), которое при обработке изоамилнитритом и восстановлением Н3РO4 превращается в этил-5,6,7,8-тетрагидро-5,5,8,8-тетраметил-4-бромнафталин-2-карбоксилат (соединение Е). Соединение Е приводит к образованию 5,6,7,8-тетрагидро-5,5,8,8-тетраметил-4-бромнафталин-2-карбоновой кислоты (соединение F), которое используют как реагент в соответствии с Формулой 6. Этил-5,6,7,8-тетрагидро-5,5,8,8-тетраметил-3-аминонафталин-2-карбоксилат (соединение С) также диазотируют и подвергают реакции с HBF4 с получением этил-5,6,7,8-тетрагидро-5,5,8,8-тетраметил-3-фторнафталин-2-карбоксилата (соединение G), которое служит per se или после омыления реагентом в соответствии с Формулой 6.
5,6,7,8-Тетрагидро-5,5,8,8-тетраметил-2-гидроксинафталин (соединение Н, можно получить в соответствии с работой Krause Synthesis 1972, 140) является исходным веществом согласно реакционной схеме 2. Соединение Н бромируют с образованием соответствующего 3-бромсоединения (соединение I), в котором затем защищают гидроксильную группу обработкой метоксиметилхлоридом (МОМСl), с получением 5,6,7,8-тетрагидро-5,5,8,8-тетраметил-3-метоксиметокси-2-бромнафталина (соединение J). Соединение J реагирует с трет.-бутиллитием и двуокисью углерода с образованием соответствующей карбоновой кислоты (соединение К), из которой при помощи кислоты удаляют метоксиметилзащитную группу с получением 5,6,7,8-тетрагидро-5,5,8,8-тетраметил-2-гидроксинафталин-3-карбоновой кислоты (соединение L). Соединение L бромируют с образованием 5,6,7,8-тетрагидро-5,5,8,8-тетраметил-1-бром-2-гидроксинафталин-3-карбоновой кислоты (соединение М). Соединение L и соединение М служат реагентами в соответствии с Формулой 6. Гидроксильная группа соединения М замещается метоксиметилхлоридом (МОМСl) в присутствии основания с образованием 5,6,7,8-тетрагидро-5,5,8,8-тетраметил-1-бром-2-метоксиметоксинафталин-3-карбоновой кислоты (соединение N).
Реакционные схемы 3, 4 и 5 (см. в конце описания) являются примерами синтеза производных 2,2,4,4- и 4,4-замещенных хроман-6-карбоновых кислот, которые могут служить реагентами согласно Формуле 6 для синтеза карбамоил (амидных) соединений, входящих в объем данного изобретения. Так, что касается реакционной схемы 3, 2,2,4,4-тетраметилхроман-6-карбоновую кислоту (соединение О, см. патент США N 5006550) бромируют бромом в уксусной кислоте с получением соответствующего 8-бромпроизводного (соединение Р). Соединение Р превращают в хлорангидирид обработкой тионилхлоридом и полученный хлорангидрид пригоден для реакции с амином формулы 3 с образованием карбамоил- (амидных) соединений согласно изобретению. Хлорангидрид также реагирует со спиртом (метанолом) в присутствии основания с получением соответствующего эфира, метил-2,2,4,4-тетраметил-8-бромхроман-6-карбоксилата (соединение R). Бром в соединении R превращают в трифторметильную группу обработкой трифторацетатом натрия в присутствии катализатора йодистой меди и 1-метил-2-пирролидинона (NMP), и эфирную группу карбоксилата омыляют с получением 2,2,4,4-тетраметил-8-трифторметилхроман-6-карбоновой кислоты (соединение S). Соединение S подпадает под формулу 6 и пригодно per se или в виде хлорангидирида или в другой "активированной" форме для реакции с аминами формулы 7 с получением карбамоил- (амидных) соединений по изобретению. 2,2,4,4-Тетраметилхроман-6-карбоновую кислоту (соединение О) превращают также в метиловый эфир (соединение Т), которое затем нитруют с получением 2,2,4,4-тетраметил-8-нитрохроман-6-карбоновой кислоты (соединение V), еще один реагент, подпадающий под формулу 6. Далее, как показано на реакционной схеме 3, 2,2,4,4-тетраметилхроман-6-карбоновая кислота превращается в этиловый эфир и затем ее нитруют с получением этил-2,2,4,4-тетраметил-8-нитрохроман-6-карбоксилата (соединение W). Далее, соединение О взаимодействует с ICl с образованием 2,2,4,4-тетраметил-8-иодхроман-6-карбоновой кислоты (соединение X).
В соответствии с реакционной схемой 4 2-метилфенол подвергают ряду превращений в соответствии с патентом США N 5045551 (введен в качестве ссылки в данное описание) с получением 2,2,4,4,8-пентаметилхромана (соединение Y). Соединение Y бромируют бромом в уксусной кислоте с образованием 2,2,4,4,8-пентаметил-6-бромхромана (соединение Z), которое реагирует с трет.-бутиллитием и затем с двуокисью углерода с получением 2,2,4,4,8-пентаметилхроман-6-карбоновой кислоты (соединение А1).
На реакционной схеме 5 показан синтез 4,4-диметил-8-бромхроман-6-карбоновой кислоты (соединение В,) бромированием 4,4-диметилхроман-6-карбоновой кислоты, которую можно получить согласно патенту США 5059621, который включен в данное описание в качестве ссылки. 2,2,4,4,8-Пентаметилхроман-6-карбоновая кислота (соединение А, ) и 4,4-диметил-8-бромхроман-6-карбоновая кислота (соединение В1) служат реагентами per se, или в виде хлорангидридов кислот (или в других "активированных" формах) в соответствии с Формулой 6 для синтеза карбамоил (амидных) соединений согласно изобретению.
Возвращаясь к реакции между реагентом формулы 6 с амином формулы 7, следует отметить, что амины широко известны из научной и патентной литературы. Более конкретно амины формулы 7 могут быть получены, как описано в научной и патентной литературе или из известных соединений такими реакциями или превращениями, которые очевидны химику-органику. Реакционная схема 6 показывает примеры получения аминов формулы 7 (где Y обозначает фенил) из промышленного сырья (Aldrich Chemical Company или Research Plus, Inc.). Соединения формулы 7 используют для синтеза нескольких предпочтительных соединений по изобретению.
Так, согласно реакционной схеме 6 (см. в конце описания), 3-нитро-6-метилфторбензол (Aldrich) подвергают окислению, превращению полученной карбоновой кислоты в хлорангидрид и затем в ее этиловый эфир с последующим восстановлением нитрогруппы с получением этил-2-фтор-4-аминобензоата (соединение С1). 3-Нитро-6-метилбромбензол (Aldrich) и 3-нитро-6-метилхлорбензол (Aldrich) подвергают практически той же серии превращений с получением этил-2-бром-4-аминобензоата (соединение D1) и этил-2-хлор-4-аминобензоата (соединение Е1) соответственно. 2-Нитро-4-аминобензойную кислоту (Research Plus) превращают в ее метиловый эфир (соединение F1) через соответствующий хлорангидрид. 2,3,5,6-Тетрафтор-4-аминобензойную кислоту этерифицируют обработкой этанолом в присутствии 1-(3-диметиламинопропил)-3-этилкарбодиимида гидрохлорида (ЭДК) и 4-диметиламинопиридина в CH2Cl2 с получением этил-2,3,5,6-тетрафтор-4-аминобензоата (соединение G1). 2,4,6-Трифторбензойную кислоту (Aldrich) превращают в ее метиловый эфир через хлорангидрид и 4-фтор удаляют реакцией с азидом натрия с последующим гидрированием с получением метил-2,6-дифтор-4-аминобензоата (соединение Н1). Соединения С1, D1, Е1, F1 и G1 служат в качестве аминов-реагентов, подпадающих под формулу 7. Далее, примерами реагентов формулы 7 являются нитро-, фтор-, хлор, бром- и трифторметилпроизводные аминозамещенных гетероарилкарбоновых кислот или их низших алкильных эфиров, например, этил-2-амино-4-хлорпиридин-2-карбоксилат, этил-5-амино-3-хлорпиридин-5-карбоксилат и 3,4-дибром-5-аминотиофен-2-карбоновая кислота. Последние могут быть получены соответственно хлорированием или бромированием 2-аминопиридин-5-карбоновой кислоты или ее эфира, 3-аминопиридин-6-карбоновой кислоты или ее эфира (описано в WO 93/06086) и 2-аминотиофен-5-карбоновой кислоты (описано в PCT/US92/06585).
Реакции между соединениями формулы 6 и формулы 7 или соединениями формулы 6а и формулы 7а, описанные выше, представляют собой синтез карбамоил (амидных) соединений по изобретению. Многочисленные примеры этого синтеза описаны подробно ниже. Карбамоил-(амидные) соединения по изобретению могут быть превращены в тиокарбамоил (амидные) соединения по изобретению, в формуле 1 в этом случае Z обозначает S, взаимодействием карбамоил (амидных) соединений с 2,4-бис-(4-метоксифенил)-1,3-дитиа-2,4-дифосфетан-2,4-дисульфидом (реагент Лавессона). Эта реакция представлена на реакционной схеме 7 для двух конкретных примеров соединений, используемых в способах по изобретению.
На реакционной схеме 7 (см. в конце описания) одно исходное соединение этил 4-[5',6',7',8'-тетрагидро-5',5',8',8'-тетраметилнафталин-2-ил)карбамоил] бензоат (соединение I1) получают в соответствии с публикацией Kagechika et al. T. Med. Chem., 1988, 31, 2182-2192. Другое исходное соединение, этил-2-фтор-4-[5', 6', 7', 8'-тетрагидро-5',5',8',8'-тетраметилнафталин-2-ил)карбамоил] бензоат (соединение I) получено в соответствии с данным изобретением.
Реакционные схемы 8, 9 и 10 (см. в конце описания) отражают примеры получения карбамоил (амидных) соединений по изобретению путем реакции сочетания соединений формулы 6 с соединениями формулы 7 с последующей реакцией или реакциями карбамоил (амидного) соединения, которое получается сразу в ходе реакции сочетания. Так, как показано на реакционной схеме 8, 5,6,7,8-тетрагидро-5,5,8,8-тетраметил-3-метоксиметоксинафталин-2-карбоновая кислота (соединение К) вступает в реакцию сочетания с этил-4-амино-2-фторбензоатом (соединение С1) в CH2Cl2 в присутствии 1-(3-диметиламинопропил)-3-этилкарбодиимида гидрохлорида (ЭДК) и диметиламинопиридина (ДМАП) с образованием 2-фтор-4-[5', 6', 7',8'-тетрагидро-5',5',8',8'-тетраметил-2'-метоксиметоксинафталин-3'-ил)карбамоил]бензоата (соединение К1). Метоксиметильная защитная группа удаляется из соединения К1 обработкой тиофенолом и эфиратом трехфтористого бора с получением этил-2-фтор-4-[5',6',7',8'-тетрагидро-5',5',8', 8'-тетраметил-2'-гидроксинафталин-3'-ил)карбамоил] бензоата (соединение 5). Гидроксильная группа Соединения 5 превращается в н-гексилэфирную обработкой гексилиодидом в присутствии слабого основания.
Согласно реакционной схеме 9 5,6,7,8-тетрагидро-5,5,8,8-тетраметил-1-бром-2-метоксиметоксинафталин-3-карбоновая кислота (соединение N) вступает в реакцию сочетания с метил-4-амино-2,6-дифторбензоатом (соединение Н1) в СH2Cl2 в присутствии 1-(3-диметиламинопропил)-3-этилкарбодиимида гидрохлорида (ЭДК) и ДМАП с получением метил-2,6-дифтор-4-[5',6',7',8'-тетрагидро-5', 5', 8', 8'-тетраметил-1'-бром-2'-метоксиметоксинафталин-3'-ил)карбамоил] бензоата (соединение М1), из которого этерифицирующая метильная группа и метоксиметильная защитная группа удаляются обработкой соответственно основанием и кислотой с образованием 2,6-дифтор-4-[5',6',7',8'-тетрагидро-5',5', 8',8'-тетраметил-1'-бром-2'-гидроксинафталин-3'-ил)карбамоил]бензойной кислоты (соединение 32).
Реакционная схема 10 отражает пример превращения 2,2,4,4-тетраметил-8-нитрохроман-6-карбоновой кислоты (V) в соответствующий хлорангидрид обработкой тионилхлоридом с последующим сочетанием с этил-4-амино-2-фторбензоатом (соединение С1) и гидрированием с получением этил-2-фтор-4-[2',2',4', 4'-тетраметил-8'-амино-6'-хроманил)карбамоил] бензоата (соединение N1). Соединение N1 превращается в соответствующее 8-азидосоединение, этил-2-фтор-4-[2',2',4',4'-тетраметил-8'-азидо-6'-хроманил)карбамоил]бензоата (соединение 13), при обработке изоамилнитратом и NaN3.
Реакционная схема 11 (см. в конце описания) иллюстрирует процесс синтеза первичных аминов формулы 6а из хлорангидридов кислот (Х3=Сl) или другой формы активированных кислот формулы 6, когда получение первичного амина формулы 6а не описано в литературе. Так, в соответствии с перегруппировкой Курциуса (Curtius) хлорангидрид формулы 6 реагирует с азидом натрия в ацетоне с получением азидосоединения формулы 9. Азид формулы 9 нагревают в полярном высококипящем растворителе, например, трет.-бутаноле, с получением промежуточного изоцианата формулы 10, который гидрируют с получением соединения формулы 6а.
Реакционная схема 12 (см. в конце описания) отражает примеры получения соединений формулы 7а, когда получение таких соединений не описано или они не являются промышленными продуктами. Так, например, 2,5-дифтор-4-бромбензойную кислоту (описана в публикации Sugawara et al., Kogyo Kaguku Lasshi, 1970, 73, 972-979) вначале этерифицируют обработкой этиловым спиртом и кислотой с получением соответствующего эфира и затем подвергают взаимодействию с бутиллитием и затем с двуокисью углерода с получением моноэфира 2,5-дифтортерефталевой кислоты (соединение Т1). Похожая последовательность реакций с 2,3,5,6-тетрафтор-4-бромбензойной кислотой (получение описано в Reuman et al., J. Med. Chem. 1995, 38, 2531-2540) приводит к получению 2,3,5,6-тетрафтортерефталевой кислоты (соединение V1). Эта последовательность реакций, вообще говоря, может быть использована для синтеза всех соединений формулы 7а, при этом нужные изменения очевидны для специалиста в данной области, если такие соединения нельзя получить известными методами.
Реакционная схема 13 отражает пример получения 2,6-дитрет.-бутилизоникотиновой кислоты (соединение С3), которая является реагентом формулы 8 для получения нескольких предпочтительных соединений по изобретению. Так, 2,6-дитрет. -бутил-4-метилпиридин (производится Aldrich Chemical Со.) подвергают взаимодействию с N-бромсукцинимидом и перекисью бензоила с получением 4-бромметил-2,6-дитрет. -бутилпиридина (соединение А3). Соединение А3 реагирует с основанием (гидроокисью натрия) с образованием соответствующего оксиметильного соединения (соединение В3), которое затем окисляется по реакции Джонса с образованием 2,6-дитрет.-бутилизоникотиновой кислоты (соединение С3).
Еще один пример получения соединения, являющегося реагентом при синтезе карбамоильных (или амидных) соединений по изобретению, представлен на реакционной схеме 13 (см. в конце описания). 2,4-Дитрет.-бутилфенол (Aldrich) бромируют в среде ледяной уксусной кислоты с получением 2-бром-4,6-дитрет. -бутилфенола (соединение D3), которое затем реагирует с метоксиметилхлоридом (МОМСl) с образованием о-метоксиметил-2-бром-4,6-дитрет. -бутилфенола (соединение Е3). Соединение Е3 обрабатывают трет.-бутиллитием и затем двуокисью углерода с получением о-метоксиметил-3,5-дитрет.-бутилсалициловой кислоты (соединение F3). Соединение F3 является реагентом, который отличается от соединений, охватываемых формулой 8, только тем, что гидроксильная группа этого соединения защищена метоксиметильной (MOM) группой. Однако, метоксиметильная защитная группа удаляется после образования карбамоильной (амидной) связи, как показано на реакционной схеме 14 (см. в конце описания). Реакция ароматического бромсодержащего соединения (такого как Соединение D3) с трет.-бутиллитием и затем с двуокисью углерода является предпочтительным методом получения нескольких ароматических карбоновых кислот формулы 8 и формулы 7а, описанных в данной заявке.
Первичные амины формулы 8а, которые не производятся или не описаны в литературе, могут быть получены из хлорангидридов кислот (Х3=Сl) или другой формы активированных кислот формулы 8 практически в соответствии с перегруппировкой Курциуса, аналогично реакции, описанной выше и показанной на реакционной схеме 11.
Реакционная схема 14 отражает примеры получения карбамоил (амидных) соединений формулы 2 путем реакции реагента формулы 8 с реагентом формулы 7. Так, 2,6-дитрет.-бутилизоникотиновая кислота (соединение С3) реагирует с тионилхлоридом (SOCl2) с образованием промежуточного хлорангидрида кислоты, который затем реагирует с этил-2-фтор-4-аминобензоатом (соединение С1) в присутствии акцептора кислоты (пиридина) с образованием этил-2-фтор-4-[2', 6'-дитрет. -бутилпирид-4'-ил)карбамоил] бензоата (соединение 41). Другим примером является 3,5-дитрет.-бутилбензойная кислота (получаемая по известной методике Kagechika et al., J. Med. Chem. 1988, 31, 2182, эта публикация включена в качестве ссылки), которая реагирует с тионилхлоридом, затем с этил-2-фтор-4-аминобензоатом (соединение С1) с образованием этил-2-фтор-4-[3', 5'-дитрет. -бутилфенил)-карбамоил] бензоата (соединение 45). Далее, о-метоксиметил-3,5-дитрет.-бутилсалициловая кислота (соединение F3) реагирует с этил-2-фтор-4-аминобензоатом (соединение С1) в присутствии 4-диметиламинопиридина (ДМАП), являющегося катализатором и 1-(3-диметиламинопропил)-3-этилкарбодиимида гидрохлорида (ЭДК) с образованием этил-2-фтор-4-[2'-метоксиметил-3', 5'-дитрет. -бутилфенил)карбамоил] бензоата (соединение G3). Метоксиметильную защитную группу из соединения G3 удаляют обработкой эфиратом трехфтористого бора и тиофенолом с получением этил-2-фтор-4-[2'-гидрокси-3', 5'-дитрет.-бутилфенил)карбамоил]бензоата (соединение 47).
В еще одном примере, отраженном на реакционной схеме 14, 2,6-дитрет.-бутилизоникотиновая кислота (соединение С3) реагирует с тионилхлоридом (SOCl2), полученный промежуточный хлорангидрид кислоты реагирует с метил-2,6-дифтор-4-аминобензоатом (соединение Н1) с последующим омылением сложноэфирной группы с образованием 2,6-дифтор-4-[2',6'-дитрет.-бутилпирид-4'-ил)карбамоил1 бензойной кислоты (соединение 50). 3,5-Дитрет.-бутилбензойная кислота подвергается аналогичной последовательности реакций и приводит к 2,6-дифтор-4-[3', 5'-дитрет. -бутилфенил)карбамоил] бензойной кислоте (соединение 52).
Реакционная схема 14 отражает еще один пример, когда 2,6-дитрет.-бутилизоникотиновая кислота (соединение С3) реагирует с тионилхлоридом (SOCl2), затем с метил-2-нитро-4-аминобензоатом (соединение F1), затем проводят омыление сложноэфирной группы с получением 2-нитро-4-[2',6'-дитрет.-бутилпирид-4'-ил)карбамоил]-бензойной кислоты (соединение 54).
Многочисленные другие способы синтеза по изобретению и превращения соединений формулы 1 и/или формулы 2 в соединения, которые могут применяться в способах лечения согласно данному изобретению, а также способы получения реагентов формулы 6, формулы 7, формулы 8, формулы 6а, формулы 7а и формулы 8а станут очевидными для специалистов в свете данного описания. В этом отношении следует отметить следующую общую методику синтеза, пригодную для превращения соединений формулы 1 и/или формулы 2 в их гомологи и/или производные, а также для получения реагентов формулы 6, формулы 7 и формулы 8 (а также Формул 6а, 7а и 8а).
Обычно карбоновые кислоты этерифицируют путем нагревания с обратным холодильником раствора кислоты в подходящем спирте в присутствии кислого катализатора, такого как хлористый водород или тионилхлорид. Или же, карбоновая кислота подвергается конденсации с соответствующим спиртом в присутствии дициклогексилкарбодиимида и диметиламинопиридина. Эфир выделяют и очищают обычными методами. Ацетали и кетали легко получить методом, описанным в March, "Advabced Organic Chemistry", 2nd Edition, McGraw-Hill Book Company, p. 810). Спирты, альдегиды и кетоны могут быть защищены путем получения, соответственно, простых и сложных эфиров, ацеталей и кеталей известными методами, например, описанными в McOmie, Plenum Publishing Press, 1973 и Protecting Groups, Ed. Greene, John Wiley and Sons, 1981.
Кислоты и соли, полученные из соединений формулы 1 и формулы 2, легко получить из соответствующих сложных эфиров. Щелочное омыление, например, при помощи основания щелочного металла приведет к получению кислоты. Например, сложный эфир можно растворить в полярном растворителе, например, спирте, предпочтительно, в инертной атмосфере при комнатной температуре с примерно трехмольным избытком основания, например, гидроокиси калия или лития. Раствор перемешивают в течение длительного промежутка времени порядка 15-20 час, охлаждают, подкисляют и выделяют гидролизат обычными методами.
Амид (В обозначает CONR9R10 в формуле 1 или формуле 2) можно получить любым подходящим методом амидирования из соответствующих сложных эфиров карбоновых кислот, который известен из литературы. Один из методов получения таких соединений состоит в превращении кислоты в хлорангидрид и в последующей обработке последнего гидроокисью аммония или подходящим амином.
Спирты получают превращением соответствующих кислот в хлорангидрид при помощи тионилхлорида или других средств (J. March, "Advabced Organic Chemistry", 2 nd Edition, McGraw-Hill Book Company), затем восстанавливают хлорангидрид боргидридом натрия (March, там же, р. 1124), что приводит к получению спиртов. Или же можно восстановить сложные эфиры литийалюмогидридом при пониженных температурах. Алкилирование этих спиртов подходящими алкилгалогенидами в условиях реакции Вильямсона (Williamson) (March, там же, р. 357) приводит к получению простых эфиров. Эти спирты можно превратить в сложные эфиры путем взаимодействия с подходящими кислотами в присутствии кислых катализаторов или дициклогексилкарбодиимида и диметиламинопиридина.
Альдегиды можно получить из первичных спиртов с помощью слабых окислителей, таких как пиридиний дихромат в метиленхлориде (Corey, E.J., Schmidt, G. , Tet. Lett. , 399, 1979), или диметилсульфоксид/ оксалилхлорид в метиленхлориде (Omura, К., Swern, D., Tetrahedron, 1978, 34, 1651).
Кетоны можно получить из альдегидов путем обработки альдегида алкилирующим реактивом Гриньяра или похожим реагентом с последующим окислением.
Ацетали или кетали можно получить из соответствующего альдегида или кетона методом, описанным March, там же, р. 810.
Конкретные примеры
Этил 4-амино-2-фторбензоат (соединение С1)
К смеси 2-фтор-4-нитротолуола (1.0 г, 6,4 ммол, Aldrich) и Na2Cr2O7 (2.74 г, 8,4 ммол) в 13.7 мл НОАс медленно добавляют 6.83 мл Н2SО4. Эту смесь медленно нагревают до 90oС в течение 1 часа, при этом образуется зеленоватый гетерогенный раствор. Смесь охлаждают до комнатной температуры и разбавляют этилацетатом. рН раствора доводят до 4 с помощью NaOH (водн.). Смесь экстрагируют дополнительно этилацетатом. Органический слой промывают NaHCO3 (насыщен. ), затем рассолом и сушат над Na2SO4. После фильтрования раствор упаривают до сухого остатка, который затем растворяют в 6 мл SOCl2 и нагревают при 80oС в течение 1 часа. Избыток SOCl2 удаляют при пониженном давлении и остаток растворяют в 5 мл СН2Сl2, 2 мл EtOH и 2 мл пиридина. Смесь перемешивают при комнатной температуре в течение 2 часов и концентрируют досуха. Этиловый эфир 2-фтор-4-нитробензойной кислоты получают в виде белого твердого вещества после хроматографии на колонке с помощью этил ацетата/гексана (1/9). Это твердое вещество затем растворяют в 10 мл этилацетата и добавляют Pd/C (50 мг). Гидрированием с помощью балонного Н2 превращают этил 2-фтор-4-нитробензоат в обозначенное в заголовке соединение. 1Н ЯМР δ 7,77 (т, J= 8,4 Гц, 1Н), 6,41 (дд, J1=8,6, J2=2,2 Гц, 1Н), 6.33 (дд, J1=13.0, J2=2,2 Гц, 1Н), 4,33 (к, J=7,1 Гц, 2Н), 4,3 (ш.с, 2Н), 1,37 (т, J=7,1 Гц, 3Н).
Метил 4-амино-2,6-дифторбензоат (соединение Н1)
Раствор трифторбензойной кислоты (150 мг, 0.85 ммол, Aldrich) в 0,5 мл SOCl2 нагревают при кипячении в течение 2 часов. Реакционную смесь охлаждают до комнатной температуры и избыток SOCl2 удаляют при пониженном давлении. Остаток растворяют в 1 мл пиридина и 0.2 мл метанола. После перемешивания при комнатной температуре в течение 30 минут растворитель удаляют и остаток очищают с помощью хроматографии на колонке (этилацетат/гексан 1/10) с получением метил трифторбензоата в виде бесцветного масла. Это масло затем растворяют в 1 мл CH3CN и добавляют затем раствор NaN3 (100 мг, 1.54 ммол) в 0,5 мл воды. Реакционную смесь кипятят с обратным холодильником в течение двух дней. Соль удаляют фильтрованием и полученный раствор концентрируют до масла. Это масло затем растворяют в 1 мл метанола и добавляют каталитическое количество Pd/C (10 вес.%). Реакционную смесь гидрируют в течение 12 часов. Катализатор удаляют и раствор концентрируют до масла. После хроматографии на колонке (этилацетат/гексан 1/3) получают обозначенное в заголовке соединение в виде бесцветных кристаллов. 1Н ЯМР δ 6.17 (д, J=10,44 Гц, 2Н), 4.2 (ш.с, 2Н), 3.87 (с, 3Н).
8-Бром-2,2,4,4-тетраметилхроман-6-карбоновая кислота (соединение Р)
К раствору 2,2,4,4-тетраметилхроман-6-карбоновой кислоты (200 мг, 0.85 ммол) в 0,5 мл АсОН добавляют Вr2 (0,07 мл, 1,28 ммол). Полученный темно-оранжевый раствор перемешивают при комнатной температуре в течение ночи. Избыток брома удаляют при пониженном давлении. Затем раствор вносят в 5 мл воды и экстрагируют этилацетатом (343 мл). Объединенные этилацетатные слои далее промывают NaHCO3 (насыщен.), рассолом и сушат над MgSO4. После концентрирования остаток очищают с помощью хроматографии на колонке (силикагель, этилацетат/гексан 1/3) с получением нужного продукта (170 мг) в виде белого твердого вещества. 1Н ЯМР δ 8,11 (д, J=2,2 Гц, 1Н), 8,00 (д, J= 2,2 Гц, 1Н), 1,90 (с, 2Н), 1,43 (с, 6Н), 1,39 (с, 6Н).
8-Иод-2,2,4,4-тетраметилхроман-6-карбоновая кислота (соединение X)
К раствору 2,2,4,4-тетраметилхроман-6-карбоновой кислоты (66 мг, 0.28 ммол) в 0.8 мл АсОН добавляют ICl (0,07 мл, 1,4 ммол). Полученный окрашенный раствор перемешивают при комнатной температуре в течение ночи. Использование той же методики, что и для получения 8-бром-2,2,4,4-тетраметилхроман-6-карбоновой кислоты (соединение Р) приводит к получению обозначенного в заголовке соединения (107 мг) в виде белого твердого вещества. 1Н ЯМР δ 8,35 (д, J= 2,2 Гц, 1Н), 8,03 (д, J=2,2 Гц, 1Н), 1,87 (с, 2Н), 1,43 (с, 6Н), 1,38 (с, 6Н).
2,2,4,4-Тетраметил-8-трифторметилхроман-6-карбоновая кислота (соединение S)
Раствор 8-бром-2,2,4,4-тетраметилхроман-6-карбоновой кислоты (соединение R, 150 мг, 0,48 ммол) в 1 мл SOCl2 нагревают при кипячении в течение 2 часов. После охлаждения до комнатной температуры, избыток SOCl2 удаляют при пониженном давлении и остаток растворяют в 1 мл пиридина и 0.2 мл метанола. Смесь перемешивают при комнатной температуре в течение 30 минут. Растворитель удаляют и остаток пропускают через хроматографическую колонку (силикагель, этилацетат/гексан 1/10) с получением метилового эфира 8-бром-2,2,4,4-тетраметилхроман-6-карбоновой кислоты (158 мг) в виде бесцветного масла. К раствору этого метилового эфира в 3 мл N-метилпирролидона (NMP) добавляют NaCO2CF3 (502 мг, 3,7 ммол) и CuI (350 мг, 1,84 ммол). Полученную смесь нагревают до 175oС (температура бани) в течение 2 часов. Полученную смесь охлаждают до комнатной температуры и вносят в ледяную воду. Продукт экстрагируют этилацетатом (343 мл). Объединенные органические слои далее сушат и концентрируют досуха. Сырое вещество очищают с помощью хроматографии на колонке (силикагель, этилацетат/хлороформ 1/10) с получением обозначенного в заголовке соединения в виде бесцветного масла (120 мг). Его гидролизуют в стандартных условиях с получением обозначенного в заголовке соединения. 1Н ЯМР δ 8,21 (д, J=2,1 Гц, 1Н), 8,17 (д, J=2,1 Гц, 1Н), 1,92 (с, 2Н), 1,41 (с, 12Н).
Этил 8-нитро-2,2,4,4-тетраметил-6-хроманоат (соединение W)
Этил 2,2,4,4-тетраметил-6-хроманоат (150 мг, 0,57 ммол) медленно добавляют к 0,3 мл конц. Н2SО3 при 0oС. К этой смеси очень медленно добавляют 0,03 мл HNO3. Реакционную смесь перемешивают при 0oС в течение 30 мин и вносят в ледяную воду. Продукт экстрагируют 5 мл этилацетата, промывают NaHCO3 (насыщен. ), рассолом и сушат над MgSO4. После концентрирования продукт очищают с помощью хроматографии на колонке (этилацетат/гексан 1/10) с получением 74 мг светло-желтого масла. 1Н ЯМР δ 8,24 (д, J=2,1 Гц, 1Н), 8,17 (д, J=2,1 Гц, 1Н), 4,38 (к, J=7,1 Гц, 2Н), 1,95 (с, 2Н), 1,43 (с, 6Н), 1,42 (с, 6Н), 1,40 (т, J=7,1 Гц, 3Н).
2-Оксо-4,4,8-триметилхроман (соединение Р1)
В круглодонной колбе на 500 мл помещают NaH (1,66 г, 60% суспензия в масле, 0,046 мол) и промывают его сухим гексаном. Затем добавляют сухой ТГФ (22 мл) и потом о-крезол (5 г, 0,046 мол) в 10 мл сухого ТГФ. Реакционную смесь перемешивают при 0oС в течение 30 мин и затем добавляют 3,3-диметилакрилоилхлорид в 10 мл ТГФ. Полученную белую суспензию перемешивают при комнатной температуре в течение 12 ч, затем медленно разбавляют водой. Смесь затем экстрагируют этилацетатом. Органический слой промывают рассолом, водой и сушат над MgSO4. После фильтрования и удаления растворителя получают желтое масло (10,44 г). Это масло затем растворяют в 50 мл сухого CH2Cl2 и вводят шприцом в раствор АlСl3 (10,8 г, 0,069 мол) в 10 мл СH2Cl2. Реакционную смесь перемешивают при комнатной температуре в течение 12 ч. Затем осторожно добавляют ледяную воду, органический слой отделяют, промывают NaHCO3 (насыщен. ), рассолом, водой и сушат над MgSO4. После удаления осушительного агента и растворителя остаток очищают хроматографией на колонке (силикагель, этилацетат/гексан 1/9) с получением обозначенного в заголовке соединения (4,408 г) в виде масла. 1Н ЯМР δ 7,1 (м, 3Н), 2,62 (с, 2Н), 2,33 (с, 3Н), 1,36 (с, 6Н).
2.4-Диметил-4-(2'-гидрокси-3'-метилфенил)пентан-2-ол (соединение R1)
К раствору 2-оксо-4,4,8-триметилхромана (соединение Р1, 2,20 г, 11.5 ммол) в 40 мл сухого этилового эфира добавляют метилмагнийбромид (12,67 мл, 38 ммол, 3 М раствор в ТГФ). Реакционную смесь перемешивают при комнатной температуре в течение 12 ч, затем добавляют NH4Cl (насыщен.), пока весь осадок не растворится. Смесь экстрагируют диэтиловым эфиром и объединенные органические слои отделяют и промывают рассолом, водой и сушат над MgSO4. После фильтрования и удаления растворителя выделяют обозначенное в заголовке соединение в виде рыжевато-коричневого твердого вещества (2,215 г). 1Н ЯМР δ 7,16 (д, J= 7,88 Гц, 1Н), 7,00 (д, J=6.72 Гц, 1Н), 6,81 (т, J=7,6 Гц, 1Н), 5.89 (ш.с, 1Н), 2,21 (с, 3Н), 2,17 (с, 2Н), 1,48 (с, 6Н), 1,10 (с, 6Н).
2,2,4,4,8-Пентаметил-6-бромхроман (соединение Z)
Раствор 2,4-диметил-4-(2'-гидрокси-3'-метилфенил)пентан-2-ола (соединение R1, 2,215 г, 9,98 ммол) в 30 мл 15%-ной Н2SO4 нагревают до 110oC. После охлаждения до комнатной температуры реакционную смесь экстрагируют диэтиловым эфиром. Органический слой промывают NaHCO3 (насыщен.), рассолом и водой. После фильтрования и удаления растворителя остаток пропускают через колонку (силикагель, чистый гексан) с получением нужного соединения в виде прозрачного масла (1,636 г). Это масло затем растворяют в 1.5 мл НОАс, затем добавляют Вr2 (0,4113 мл, 7,98 ммол). Реакционную смесь перемешивают при комнатной температуре в течение 12 ч. Растворитель удаляют при пониженном давлении и к остатку добавляют этилацетат, полученную смесь промывают NaHCO3 (насыщен.), рассолом, водой и сушат над MgSO4. После фильтрования и удаления растворителя остаток пропускают через колонку (силикагель, чистый гексан) с получением обозначенного в заголовке соединения в виде белого твердого вещества (2,227 г). 1Н ЯМР δ 7,21 (с, 1Н), 7,06 (с, 1Н), 2,14 (с, 3Н), 1,79 (с, 2Н), 1,32 (с, 6Н), 1,31 (с, 6Н).
2,2,4,4,8-Пентаметил-хроман-6-карбоновая кислота (соединение А1)
К раствору 2,2,4,4,8-пентаметил-6-бромхромана (соединение Z, 1,2 г, 4.24 ммол) в 18 мл сухого ТГФ при -78oС в атмосфере аргона медленно добавляют 5.48 мл трет.-BuLi (1,7 М в гексане, 9,33 ммол). Реакционную смесь перемешивают при -78oС в течение 1 ч. Затем через раствор пропускают СО2 в течение 1 ч. После прекращения тока СО2 реакционную смесь перемешивают дополнительно в течение 1 часа при -78oС. Затем добавляют 10% НСl. После достижения комнатной температуры реакционную смесь экстрагируют этилацетатом. Органический слой затем промывают рассолом и сушат над MgSO4. После концентрирования остаток очищают с помощью хроматографии на колонке (этилацетат/гексан 5/95) с получением обозначенного в заголовке соединения в виде белого твердого вещества (774 г). 1Н ЯМР δ 7,96 (с, 3Н), 7,75 (с, 1Н), 2,23 (с, 3Н), 1,88 (с, 2Н), 1,39 (с, 6Н).
8-Бром-4,4-диметил-хроман-6-карбоновая кислота (соединение В1)
Применяя методику, аналогичную приведенной для синтеза 8-бром-2,2,4,4-тетраметилхроман-6-карбоновой кислоты (соединение Р), но с использованием 4,4-диметилхроманкарбоновой кислоты (100 мг, 0,49 ммол) получают обозначенное в заголовке соединение в виде белого твердого вещества. 1Н ЯМР δ 8,10 (д, J=2,1 Гц, 1Н), 7,98 (д, J=2,1 Гц, 1Н), 4,39 (т, J=5.44 Гц, 2Н), 1,89 (т, J=5.4 Гц, 1Н), 1,38 (с, 6Н).
Этил 2-амино-1-бром-5,5,8,8-тетрагидро-5,5,8,8-тетраметилнафталин-3-карбоксилат (соединение D)
К раствору этил 5,6,7,8-тетрагидро-5,5,8,8-тетраметил-3-аминонафталин-2-карбоксилата (соединение С, 58 мг, 0.21 ммол) в 2 мл НОАс добавляют Вr2 (0,02 мл, 0,42 ммол). Оранжевый раствор перемешивают при комнатной температуре в течение 2 дней. Избыток Вr2 и НОАс удаляют при пониженном давлении и остаток пропускают через колонку (силикагель, этилацетат/гексан 1/10) с получением обозначенного в заголовке соединения в виде светло-оранжевого масла (59 мг, 79,5%). 1Н ЯМР δ 7,90 (с, 1Н), 6,41 (ш.с, 2Н), 4,36 (к, J=7,2 Гц, 2Н), 1,70 (м, 4Н), 1.58 (с, 6Н), 1,40 (т, J=7,2 Гц, 3Н), 1,28 (с, 6Н).
Этил 5,6,7,8-тетрагидро-5.5.8,8-тетраметил-4-бромнафталин-2-карбоксилат (соединение Е)
Этил 2-амино-1-бром-5,5,8,8-тетрагидро-5,5,8,8-тетраметил-нафталин-3-карбоксилат (соединение D, 59 мг, 0,17 ммол) растворяют в 2 мл EtOH при 0oС. К этому раствору добавляют 1 мл трифторуксусной кислоты и 1 мл изоамилнитрита. Реакционную смесь перемешивают при 0oС в течение 30 мин, затем добавляют Н3РO4 (0,325 мл, 3.14 ммол). Реакционную смесь доводят до комнатной температуры и перемешивают в течение 12 ч. Добавляют NаНСО3 (насыщен.), реакционную смесь экстрагируют этилацетатом, сушат над MgSO4, фильтруют и концентрируют с получением масла. Продукт очищают с помощью хроматографии на колонке (силикагель, этилацетат/гексан 1/10) с получением обозначенного в заголовке соединения в виде бесцветного масла. 1Н ЯМР δ 8,02 (д, J=2,0 Гц, 1Н), 7,95 (д, J=2,0 Гц, 1Н), 4,35 (к, J=7,1 Гц, 2Н), 1,71 (м, 4Н), 1.56 (с, 6Н), 1,38 (т, J=7,1 Гц, 3Н), 1,31 (с, 6Н).
Этил 5,6,7,8-тетрагидро-5,5,8,8-тетраметил-3-фторнафталин-2-илкарбоксилат (соединение G)
К раствору этил 5,6,7,8-тетрагидро-5,5,8,8-тетраметил-3-аминонафталин-2-карбоксилата (соединение С, 150 мг, 0,55 ммол), охлажденному ледяной баней, добавляют 0.24 мл HBF4 (48% раствор в воде) и затем раствор NaNO3 (81 мг, 1,16 ммол) в 1 мл воды. Суспензию оставляют в холодильнике на 3 дня. Реакционную смесь последовательно промывают этилацетатом, пока ТСХ не покажет отсутствие УФ-чувствительного пятна на старте. Этилацетатный слой сушат над MgSO4 и раствор концентрируют с получением масла. Далее масло растворяют в 1 мл толуола и смесь кипятят с обратным холодильником в течение 2 ч. После охлаждения реакции до комнатной температуры растворитель упаривают и остаток пропускают через колонку (силикагель, этилацетат/гексан 1/10) с получением обозначенного в заголовке соединения в виде масла. 1H ЯМР δ 7,85 (д, J=7,8 Гц, 1Н), 7,04 (д, J=12.3 Гц, 1Н), 4,38 (к, J=7,1 Гц, 2Н), 1,71 (м, 4Н), 1,69 (с, 4Н), 1,38 (т, J=7,1 Гц, 3Н), 1,30 (с, 6Н), 1,28 (с, 6Н).
2-Бром-3-гидрокси-5,5,8,8-тетрагидро-5,5,8,8-тетраметилнафталин (соединение I)
Применяя методику, аналогичную приведенной для синтеза 8-бром-2,2,4,4-тетраметилхроман-6-карбоновой кислоты (соединение Р), но с использованием 2-гидрокси-5,5,8,8-тетрагидро-5,5,8,8-тетраметилнафталина (700 мг, 3.43 ммол) и Вr2 (0,177 мл, 3.43 ммол) в 1.5 мл НОАс, получают обозначенное в заголовке соединение в виде белого твердого вещества (747 мг). 1Н ЯМР δ 7,36 (с, 1Н), 6.96 (с, 2Н), 5.32 (ш.с, 1Н), 1,66 (с, 4Н), 1,25 (с, 12Н).
5,6,7,8-Тетрагидро-5,5,8,8-тетраметил-3-метоксиметокси-2-бромнафталин (соединение J)
К раствору 2-бром-3-гидрокси-5,5,8,8-тетрагидро-5,5,8,8-тетраметилнафталина (соединение I, 600 мг, 2,12 ммол) и каталитического количества Bu4NBr в 20 мл сухого СН2Сl2 при 0oС добавляют диизопропилэтиламин (1,138 мл, 12.75 ммол), и затем метоксиметилхлорид (0,484 мл, 6.39 ммол). Реакционную смесь нагревают при 45oС в течение 12 ч. Реакционную смесь промывают 10% лимонной кислотой, затем NaHCO3 (насыщен. ), рассолом и сушат над MgSO4. После фильтрования и удаления растворителя остаток очищают с помощью хроматографии на колонке (этилацетат/гексан 1/9) с получением обозначенного в заголовке соединения (772 мг) в виде белого твердого вещества. 1Н ЯМР δ 7,43 (с, 1Н), 7,06 (с, 1Н), 5.21 (с, 2Н), 3.54 (с, 3Н), 1,66 (с, 4Н), 1,26 (с, 6Н), 1,25 (с, 6Н).
3-Метоксиметокси-5,5,8,8-тетраметил-5,6,7,8-тетрагидронафталин-2-илкарбоновая кислота (соединение К)
Применяя методику, аналогичную приведенной для синтеза 2,2,4,4,8-пентаметилхроман-6-карбоновой кислоты (соединение А1), но с использованием 5,6,7,8-тетрагидро-5,5,8,8-тетраметил-3-метоксиметокси-2-бромнафталина (соединение J, 722 мг, 2,21 ммол) и 2.86 мл трет.-BuLi (4.87 ммол, 1,7 М раствор в гексане), получают обозначенное в заголовке соединение в виде белого твердого вещества (143 мг). 1Н ЯМР δ 8,12 (с, 1Н), 7,19 (с, 1Н), 5.40 (с, 2Н), 3.58 (с, 3Н), 1,70 (с, 4Н), 1,30 (с, 12Н).
Этил 2-фтор-4-[(5', 6', 7', 8'-тетрагидро-5',5',8',8'-тетраметил-нафталин-2'-ил)карбамоил]бензоат (соединение 1)
К 5,5,8,8-тетраметил-5,6,7,8-тетрагидро-2-нафталинкарбоновой кислоте (46 мг, 0.2 ммол) добавляют 1 мл тионилхлорида. Эту смесь нагревают при кипячении в течение 2 часов. Избыток тионилхлорида удаляют при пониженном давлении и остаток растворяют в 2 мл СН2Сl2. К этому раствору добавляют этил 4-амино-2-фтор-бензоат (соединение С1, 37 мг, 0.2 ммол) и затем 0,5 мл пиридина. Реакционную смесь перемешивают при комнатной температуре в течение 4 часов и концентрируют при пониженном давлении. Остаток очищают с помощью хроматографии на колонке (этилацетат/гексан (1/10) и получают обозначенное в заголовке соединение в виде белого твердого вещества. 1Н ЯМР δ 8,06 (ш.с, 1Н), 7,93 (т, J= 8,4 Гц, 1Н), 7,85 (д, J=2,0 Гц, 1Н), 7,78 (дд, J1=2,0, J2=12,9 Гц, 1Н), 7,55 (дд, J1=2,0, J2=8,2 Гц, 1Н), 7,40 (д, J=8,3 Гц, 1Н), 7,32 (дд, J1= 2,02, J2= 8,8 Гц, 1Н), 4,38 (к, J=7,2 Гц, 2Н), 1,71 (с, 4Н), 1,40 (т, J=7,2 Гц, 3Н), 1,32 (с, 6Н), 1,30 (с, 6Н).
Этил 2-фтop-4-[(5',6',7',8'-тeтpaгидpo-4'-6poм-5',5',8',8'-тeтpaмeтил-нaфтaлин-2'-ил)карбамоил]бензоат (соединение 3)
Применяя методику, аналогичную приведенной для синтеза этил 2-фтор-4-[(5',6',7',8'-тетрагидро-5',5',8',8'-тетраметилнафталин-2'-ил)карбамоил]бензоата (соединение 1), но с использованием 5,6,7,8-тетрагидро-5,5,8,8-тетраметил-4-бромнафталин-2-карбоновой кислоты (соединение F) получают обозначенное в заголовке соединение в виде белого твердого вещества. 1Н ЯМР δ 8,30 (ш.с, 1Н), 7,92 (т, J=8,4 Гц, 1Н), 7,84 (д, J=2,1 Гц, 1Н), 7,81 (д, J= 2,1 Гц, 1Н), 7,74 (дд, J1=2,1, J2=12,8 Гц, 1Н), 7,35 (дд. J1=2,0, J2=8,4 Гц, 1Н), 4,36 (к, J=7,2 Гц, 2Н), 1,67 (м, 4Н), 1,55 (с, 6Н), 1,39 (т, J=7,2 Гц, 3Н), 1,31 (с, 6Н).
Этил 2-фтop-4-[(3'-мeтокcимeтoкcи-5',6',7',8'-тeтpaгидpo-5',5',8',8'-тетраметилнафталин-2'-ил)карбамоил]бензоат (соединение К1)
Применяя методику, аналогичную приведенной для синтеза этил 2-фтор-4-[(3'-метоксиметокси-4'-бром-5', 6', 7',8'-тетрагидро-5',5',8',8'-тетраметилнафталин-2'-ил)карбамоил]бензоата (соединение S1), но с использованием 3-метоксиметокси-5,5,8,8-тетраметил-5,6,7,8-тетрагидронафталин-2-илкарбоновой кислоты (соединение К, 143 мг, 0,49 ммол) и этил 4-амино-2-фтор-бензоата (соединение С1, 98,5 мг, 0,54 ммол) получают обозначенное в заголовке соединение в виде белого твердого вещества. 1Н ЯМР δ 10,1 (ш.с, 1Н), 8,20 (с, 1Н), 7,93 (т, J= 8,8 Гц, 1Н), 7,83 (д, J=13.4 Гц, 1Н), 7,29 (д, J=8,0 Гц, 1Н), 5.41 (с, 2Н), 4,39 (к, J=7,1 Гц, 2Н), 3.59 (с, 3Н), 1,70 (с, 4Н), 1,31 (с, 12Н), 1,26 (т, J=7,1 Гц, 3Н).
Этил 2-фтop-4-[(3'-гидpoкcи-5',6',7',8'-тeтpaгидро-5',5',8',8'-тeтpaмeтил-2-нафталинил)карбамоил]бензоат (соединение 5)
К раствору этил 2-фтор-4-[(3'-метоксиметокси-5',6',7',8'-тетрагидро-5', 5', 8', 8'-тетраметилнафталин-2'-ил)карбамоил]-бензоата (соединение К1, 50.7 мг, 0,11 ммол) в 2 мл CH2Cl2 добавляют тиофенол (0,061 мл, 0,55 ммол). Реакционную смесь перемешивают при 0oС в течение 2 ч, затем добавляют NaHCO3 (насыщен. ). Органический слой отделяют и промывают рассолом, водой и сушат над MgSO4. После фильтрования и удаления растворителя остаток пропускают через колонку (силикагель, этилацетат/гексан 1/3) с получением обозначенного в заголовке соединения в виде белого твердого вещества (44.2 мг). 1Н ЯМР δ 8,61 (ш.с, 1Н), 7,94 (т, J=8,42 Гц, 1Н), 7,71 (дд, J=10,8, 2,0 Гц, 1Н), 7,53 (дд, J=6,4, 2,0 Гц, 1Н), 6.97 (с, 1Н), 4,39 (к, J=7,1 Гц, 2Н), 1,69 (с, 4Н), 1,40 (т, J=7,1 Гц, 3Н), 1,29 (с, 6Н), 1,27 (с, 6Н).
Этил 2-фтор-4-[(4', 4'-диметил-8'-бромхроман-6'-ил)-карбамоил] бензоат (соединение 7)
В круглодонную колбу на 10 мл помещают 4,4-диметил-8-бром-хроман-6-карбоновой кислоты (соединение В1, 139 мг, 0,485 ммол) и добавляют SOCl2 (1 мл, большой избыток). Полученный раствор нагревают при 90oС в течение 2 часов и охлаждают до комнатной температуры. Избыток SOCl2 удаляют при пониженном давлении. Остаток растворяют CH2Cl2 (3 мл). Добавляют этил 4-амино-2-фтор-бензоат (соединение С1, 90 мг, 0,49 ммол) и затем пиридин (0,5 мл, большой избыток). Реакционную смесь перемешивают при комнатной температуре в течение ночи и затем концентрируют досуха. Остаток очищают с помощью хроматографии на колонке (этилацетат/гексан (1/5) и получают обозначенное в заголовке соединение в виде белого твердого вещества (190 мг). 1Н ЯМР δ 7,95 (т, J= 8,31 Гц, 1Н), 7,88 (ш.с, 1Н), 7,83 (д, J=2,2 Гц, 1Н), 7,80 (д, J=2,2 Гц, 1Н), 7,75 (дд, J1=12.89, J2=2,0 Гц, 1Н), 7,30 (дд, J1=8,55, J2=2,0 Гц, 1Н), 4,37 (м, 5Н), 1,89 (т, J=5.49 Гц, 2Н), 1,40 (т, J=7,1 Гц, 3Н), 1,39 (с, 6Н).
Этил 2-фтор-4-[(2',2',4',4'-тетраметил-8'-бромхроман-6'-ил)карбамоил]бензоат (соединение 9)
Применяя методику, аналогичную приведенной для синтеза этил 2-фтор-4-[(4', 4'-диметил-8'-бромхроман-6'-ил)карбамоил]бензоата (соединение 7), но с использованием 2,2,4,4-диметил-8-бром-хроман-6-карбоновой кислоты (соединение Р, 70 мг, 0.22 ммол) и этил 4-амино-2-фтор-бензоата (соединение С1, 38 мг, 0.22 ммол) получают обозначенное в заголовке соединение в виде белого твердого вещества (80 мг, 76%). 1Н ЯМР δ 8,25 (ш.с, 1Н), 7,92 (т, J= 8,4 Гц, 1Н), 7,83 (с, 2Н), 7,74 (дд, J1=2,0, J2=13,0 Гц, 1Н), 7,34 (дд. J1= 2,0, J2= 8,7 Гц, 1Н), 4,37 (к, J=7,1 Гц, 2Н), 1,88 (с, 2Н), 1,41 (с, 6Н), 1,39 (т, J=7,1 Гц, 3Н), 1,37 (с, 6Н).
Этил 2-фтор-4-[(2', 2', 4', 4'-тетраметил-8'-трифторметил-хроман-6'-ил)карбамоил]бензоат (соединение 11)
Применяя методику, аналогичную приведенной для синтеза этил 2-фтор-4-[(4', 4'-диметил-8'-бромхроман-6'-ил)карбамоил]бензоата (соединение 7), но с использованием 2,2,4,4-диметил-8-трифторметилхроман-6-карбоновой кислоты (соединение S, 57 мг, 0,19 ммол) и этил 4-амино-2-фтор-бензоата (соединение С1, 35 мг, 0,19 ммол) получают обозначенное в заголовке соединение в виде белого твердого вещества. 1Н ЯМР δ 8,06 (д, J=2,2 Гц, 1Н), 7,99 (ш.с, 2Н), 7,95 (т, J=8,55 Гц, 1Н), 7,83 (с, 2Н), 7,81 (д, J=2,2 Гц, 1Н), 7,76 (дд, J1= 12.8, J2=2,1 Гц, 1Н), 7,33 (дд, J=8,55, 1,9 Гц, 1Н), 4,37 (к, J=7,1 Гц, 2Н), 1,93 (с, 2Н), 1,41 (с, 12Н), 1,40 (т, J=7,1 Гц, 3Н).
Этил 2-фтор-4-[(2', 2',4',4'-тетраметил-8'-аминохроман-6'-ил)карбамоил] бензоат (соединение N1)
С использованием 8-нитро-2,2,4,4-тетраметил-6-хроманкарбоновой кислоты (соединение V) и применяя методику, аналогичную приведенной для синтеза этил 2-фтор-4-1(4', 4'-диметил-8'-бромхроман-6'-ил)карбамоил]бензоата (соединение 7), получают этил 2-фтор-4-[(2', 2', 4',4'-тетраметил-8'-нитрохроман-6'-ил)карбамоил] бензоат в виде белого твердого вещества. Это соединение (50 мг, 0,12 ммол) растворяют в 2 мл метанола. К раствору добавляют каталитическое количество Pd/C и раствор выдерживают в атмосфере Н2 (водород из баллона) в течение ночи. Катализатор удаляют фильтрованием, растворитель упаривают и получают обозначенное в заголовке соединение в виде белого твердого. 1Н ЯМР δ 7,93 (т, J=8,43 Гц, 1Н), 7,90 (ш.с, 1Н), 7,73 (дд, J=12.9, 2,0 Гц, 1Н), 7,29 (дд, J1=8,43, 1,96 Гц, 1Н), 7,23 (д, J=2,14 Гц, 1Н), 7,01 (д, J= 2,2 Гц, 1Н), 4,35 (к, J=7,1 Гц, 2Н), 1,88 (с, 2Н), 1,39 (с, 6Н), 1,38 (т, J=7,1 Гц, 3Н), 1,37 (с, 6Н).
Этил 2-фтор-4-[(2', 2',4',4'-тетраметил-8'-азидохроман-6'-ил)карбамоил] бензоат (соединение 13)
К раствору этил 2-фтор-4-[(2',2',4',4'-тетраметил-8'-аминохроман-6'-ил)карбамоил]бензоата (соединение N1, 32 мг, 0,077 ммол) в 3 мл EtOH добавляют 0,5 мл трифторуксусной кислоты (TFA) и 0,5 мл изоамилнитрита при 0oС. Реакцию перемешивают в течение 2 ч, затем добавляют раствор NaN3 (5 мг) в 0.2 мл воды. Реакционную смесь доводят до комнатной температуры и перемешивают в течение ночи. Растворитель удаляют и остаток очищают с помощью хроматографии на колонке (силикагель, этилацетат/гексан (1/10) с получением обозначенного в заголовке соединения в виде бесцветного масла. 1Н ЯМР δ 8,0 (ш. с, 2Н), 7,94 (т, J=7,8 Гц, 1Н), 7,73 (д, J=12,1 Гц, 1Н), 7,64 (с, 1Н), 7,31 (дд, J= 8,5, 2,0 Гц, 1Н), 7,21 (д, J=2,0 Гц, 1Н), 4,37 (к, J=7,1 Гц, 2Н), 1,90 (с, 2Н), 1,39 (т, J=7,1 Гц, 3Н), 1,45 (с, 6Н), 1,40 (с, 6Н).
Метил 2,6-дифтор-4-[(2',2',4',4'-тетраметил-8'-трифтор-метилхроман-6'-ил)карбамоил]бензоат (соединение 15)
Применяя методику, аналогичную приведенной для синтеза этил 2-фтор-4-[(4', 4'-диметил-8'-бромхроман-6'-ил)карбамоил]бензоата (соединение 7), но с использованием 2,2,4,4-тетраметил-8-трифторметилхроманкарбоновой кислоты (соединение S, 11,2 мг, 0,037 ммол) и метил 4-амино-2,6-дифтор-бензоата (соединение Н1, 6.6 мг, 0,035 ммол) получают обозначенное в заголовке соединение в виде белых кристаллов. 1Н ЯМР δ 8,21 (ш.с, 1Н), 8,05 (с, 1Н), 7,82 (с, 1Н), 7,36 (д, J= 10.20 Гц, 1Н), 3.93 (с, 3Н), 1,92 (с, 2Н), 1,40 (с, 12Н).
Этил 2-фтop-4-[(2',2',4',4'-тeтpaмeтил-8'-иoдxpoмaн-6'-ил)кapбaмoил]бензоат (соединение 17)
Применяя методику, аналогичную приведенной для синтеза этил 2-фтор-4-[(4', 4'-диметил-8'-бромхроман-6'-ил)карбамоил]бензоата (соединение 7), но с использованием 2,2,4,4-тетраметил-8-иодхроманкарбоновой кислоты (соединение X, 81 мг, 0.25 ммол) и этил 4-амино-2-фтор-бензоата (соединение С1, 55 мг, 0,30 ммол) получают обозначенное в заголовке соединение в виде белого твердого вещества. 1Н ЯМР δ 8,05 (ш.с, 1Н), 8,01 (д, J=2,2 Гц, 1Н), 7,94 (т, J= 8,4 Гц, 1Н), 7,86 (д, J=2,2 Гц, 1Н), 7,75 (дд, J=12.8, 2,1 Гц, 1Н), 7,33 (дд, J= 8,8, 2,1 Гц, 1Н), 4,37 (к, J=7,1 Гц, 2Н), 1,89 (с, 2Н), 1,42 (с, 12Н), 1,38 (с, 6Н).
Этил 2-фтор-4-[(2',2',4',4',8'-пентаметилхроман-6'-ил)карбамоил]бензоат (соединение 19)
Применяя методику, аналогичную приведенной для синтеза этил 2-фтор-4-[(4', 4'-диметил-8'-бромхроман-6'-ил)карбамоил]бензоата (соединение 7), но с использованием 2,2,4,4,8-пенетаметилхроман-6-карбоновой кислоты (соединение А1, 92 мг, 0,37 ммол) и этил 4-амино-2-фтор-бензоата (соединение С1, 75 мг, 0,41 ммол) получают обозначенное в заголовке соединение в виде белого твердого вещества (100 мг). 1Н ЯМР δ 8,31 (ш.с, 1Н), 7,90 (т, J=8,24 Гц, 1Н), 7,76 (дд, J=14.29, 1,7 Гц, 1Н), 7,74 (с, 1Н), 7,43 (с, 1Н), 7,35 (дд, J= 8,67, 1,7 Гц, 1Н), 4,32 (к, J=7,1 Гц, 2Н), 2,18 (с, 3Н), 1,84 (с. 2Н), 1,38 (т, J=7,1 Гц, 3Н), 1,35 (с, 6Н), 1,34 (с, 6Н).
Этил 4-[(5',6',7',8'-тeтpaгидpo-5',5',8',8'-тeтpaмeтил-2'-нaфтaлинил)-тиокарбамоил]бензоат (соединение 21)
К раствору этил 4-[(5',6',7',8'-тетрагидро-5',5',8',8'-тетраметил-2'-нафталинил)карбамоил] бензоата (соединение I1, 61 мг, 0,16 ммол) в 2 мл безводного бензола добавляют реагент Лавессона (45 мг, 0,112 ммол). Полученный желтый раствор кипятят с обратным холодильником в атмосфере N2 в течение 2 ч. Растворитель удаляют и остаток очищают с помощью хроматографии на колонке (силикагель, этилацетат/гексан (1/5) с получением обозначенного в заголовке соединения в виде желтого твердого вещества (55 мг, 87%). 1Н ЯМР δ 9.04 (ш. с, 1Н), 8,11 (д, J=8,70 Гц, 2Н), 7,85 (ш.с, 2Н), 7,75 (ш.с, 2Н), 7,55 (дд, J=8,2, 1,9 Гц, 1Н), 7,36 (д, J=8,3 Гц, 1Н), 4,35 (к, J=7,1 Гц, 2Н), 1,71 (с, 4Н), 1,40 (т, J=7,1 Гц, 3Н), 1,30 (с, 12Н).
Этил 2-фтop-4-[(5', 6', 7',8'-тeтpaгидpo-5',5',8',8'-тeтpaмeтил-2'-нафталинил)тиокарбамоил]бензоат (соединение 23)
Применяя методику, аналогичную приведенной для синтеза этил 4-[(5',6', 7',8'-тетрагидро-5',5',8',8'-тетраметил-2'-нафталинил)-тиокарбамоил]бензоата (соединение 21), но с использованием этил 2-фтор-4-[(5',6',7',8'-тетрагидро-5', 5', 8',8'-тетраметил-2'-нафталинил)-карбамоил]бензоата (соединение 1, 167 мг, 0,42 ммол) в 8 мл бензола и реагента Лавессона (220 мг, 0,544 ммол), получают обозначенное в заголовке соединение в виде светло-желтого твердого вещества (127,5 мг). 1Н ЯМР δ 9,30 (ш.с, 1Н), 8,05 (ш.с, 1Н), 7,95 (т, J= 8,37 Гц, 1Н), 7,77 (д, J=1,89 Гц, 1Н), 7,53 (дд, J=8,24, 2,1 Гц, 1Н), 7,49 (ш. с, 1Н), 7,35 (д, J=8,24 Гц, 1Н), 4,33 (к, J=7,1 Гц, 2Н), 1,71 (с, 4Н), 1,32 (с, 6Н), 1,30 (с, 6Н).
3-Гидрокси-5,5,8,8-тетраметил-5,6,7,8-тетрагидронафталин-2-илкарбоновая кислота (соединение L)
К раствору 2-бром-3-метоксиметокси-5,5,8,8-тетрагидро-5,5,8,8-тетраметилнафталина (соединение J, 722 мг, 2,2 ммол) в 10 мл сухого ТГФ при -78oС в атмосфере аргона медленно добавляют 2.86 мл трет.-BuLi (1,7 М в гексане, 4.8 ммол). Реакционную смесь перемешивают при -78oС в течение 1 ч. Затем через раствор пропускают СО2 в течение 1 ч. После прекращения тока СО2 реакционную
смесь перемешивают дополнительно в течение 1 часа при -78oС. Затем добавляют 10% HCl. После достижения комнатной температуры реакционную смесь оставляют стоять в течение ночи и затем экстрагируют этилацетатом. Органический слой промывают рассолом и сушат над MgSO4. После концентрирования остаток очищают с помощью хроматографии на колонке (этилацетат/гексан 1/3) с получением обозначенного в заголовке соединения в виде белого твердого вещества. 1Н ЯМР δ 7,85 (с, 1Н), 6,93 (с, 1Н), 1,68 (с, 4Н), 1,28 (с, 12Н).
4-Бром-3-гидрокси-5,5,8,8-тетраметил-5,6,7,8-тетрагидронафталин-2-илкарбоновая кислота (соединение М)
3-Гидрокси-5,5,8,8-тетраметил-5,6,7,8-тетрагидронафталин-2-илкарбоновую кислоту (соединение L, 155 мг, 0,62 ммол) растворяют в 1 мл НОАс. К этому раствору добавляют Вr2 (0,033 мл, 0,62 ммол). Реакционную смесь оставляют при комнатной температуре в течение ночи. Для удаления непрореагировавшего Вr2 пропускают через реакционную смесь струю воздуха. Оставшийся твердый остаток растворяют в небольшом количестве ТГФ и очищают с помощью хроматографии на колонке (этилацетат/гексан 1/1) с получением нужного продукта в виде кремоватого твердого вещества. 1Н ЯМР δ 7,91 (с, 1Н), 1,75 (м, 2Н), 1,64 (м, 2Н), 1,62 (с, 6Н), 1,30 (с, 6Н).
4-Бром-3-метоксиметокси-5,5,8,8-тетраметил-5,6,7,8-тетрагидронафталин-2-ил карбоновая кислота (соединение N)
К раствору 4-бром-3-гидрокси-5,5,8,8-тетраметил-5,6,7,8-тетрагидронафталин-2-илкарбоновой кислоты (соединение М, 233 мг, 0,71 ммол) в 6 мл СН2Сl2 добавляют хлорметилметиловый эфир (0,162 мл, 2,1 ммол), диизопропилэтиламин (0,764 мл, 4,2 ммол) и каталитическое количество тетрабутиламмоний бромида. Реакционную смесь нагревают при 45oС в течение 2 ч. Реакционную смесь концентрируют и остаток очищают с помощью хроматографии на колонке (этилацетат/гексан 1/9) с получением нужного соединения в виде белого твердого вещества (200 г). Это белое твердое вещество растворяют в 20 мл EtOH. Добавляют водный раствор NaOH (0,5 мл, 1 М). Реакционную смесь перемешивают при комнатной температуре в течение ночи. EtOH удаляют и к остатку добавляют 2 мл этилацетата и 3 мл воды. Эту смесь очень медленно подкисляют 10% НСl до рН 7. Этилацетатный слой отделяют и промывают рассолом, сушат над Na2SO4. После отфильтровывания осушающего агента и удаления растворителя получают обозначенное в заголовке соединение в виде белого твердого вещества (155 г). 1Н ЯМР δ 7,99 (с, 1Н), 6,93 (с, 1Н), 5.20 (с, 2Н), 3,66 (с, 3Н), 1,74 (м, 2Н), 1,67 (м, 2Н), 1,60 (с, 6Н), 1,32 (с, 6Н).
Этил 2-фтор-4-[(3'-метоксиметокси-4'-бром-5',6',7',8'-тетрагидро-5',5', 8',8'-тетраметил-2'-нафталинил)карбамоил]бензоат (соединение S1)
К раствору 4-бром-3-метоксиметокси-5,5,8,8-тетраметил-5,6,7,8-тетрагидронафталин-2-илкарбоновой кислоты (соединение N, 80 мг, 0,22 ммол) в 4 мл СН2Сl2 добавляют DMAP (60 мг, 0,26 ммол), этил 2-фтор-4-аминобензоат (соединение С1, 43 мг, 0,24 ммол) и EDC (50 мг, 0,26 ммол). Реакционную смесь перемешивают при комнатной температуре в течение ночи и затем концентрируют досуха. Остаток очищают с помощью хроматографии на колонке (этилацетат/гексан 1/3) с получением обозначенного в заголовке соединения в виде бесцветного масла (45 г). 1Н ЯМР δ 9.92 (ш.с, 1Н), 8,10 (ш.с, 1Н), 7,94 (т, J= 8,4 Гц, 1Н), 7,81 (дд, J=12,9, 1,9 Гц, 1Н), 7,35 (дд, J=8,5, 1,8 Гц, 1Н), 5.20 (с, 2Н), 4,39 (к, J=7,1 Гц, 2Н), 3,61 (с, 3Н), 1,74 (м, 4Н), 1,64 (м, 2Н), 1,60 (с, 6Н), 1,40 (т, J=7,1 Гц, 3Н), 1,34 (с, 6Н).
Метил 2,6-дифтор-4-[(3'-метоксиметокси-4'-бром-5',6',7',8'-тетрагидро-5',5',8',8'-тетраметил-2'-нафталинил)карбамоил]бензоат (соединение М1)
Применяя методику, аналогичную приведенной для синтеза этил 2-фтор-4-[(3'-метоксиметокси-4'-бром-5', 6',7',8'-тетрагидро-5',5',8',8'-тетраметил-2'-нафталинил)карбамоил] бензоата (соединение S1), но с использованием 4-бром-3-метоксиметокси-5,5,8,8-тетраметил-5,6,7,8-тетрагидронафталин-2-ил карбоновой кислоты (соединение N, 80 мг, 0,22 ммол), DMAP (60 мг, 0,26 ммол), метил 2,5-дифтор-4-аминобензоата (соединение Н1, 52 мг, 0,24 ммол) и EDC (50 мг, 0,26 ммол), получают обозначенное в заголовке соединение в виде прозрачного масла. 1H ЯМР δ 10,01 (ш.с, 1Н), 8,11 (с, 1Н), 7,42 (д, J=10,0 Гц, 2Н), 5.2 (с, 2Н), 3,95 (с, 3Н), 3,63 (с, 3Н), 1,75 (м, 2Н), 1,65 (м, 2Н), 1,61 (с, 6Н), 1,35 (с, 6Н).
4-Бромметил-2,6-ди-трет.-бутилпиридин (соединение А3)
К смеси 2,6-ди-трет.-бутил-4-метилпиридина (Aldrich, 2,0 г, 9.73 ммол) в 25 мл сухого ССl4 добавляют перекись бензоила (24 мг, 0,097 ммол) и NBS (1,9 г, 10,7 ммол). Реакционную смесь кипятят в течение 24 часов. Затем ее охлаждают до комнатной температуры, растворитель удаляют в вакууме и остаток очищают с помощью хроматографии на колонке (силикагель, гексан) с получением масла (1,957 г), которое содержит 82% нужного продукта и 18% исходного вещества. 1Н ЯМР δ 7,09 (с, 2Н), 4,39 (с, 2Н), 1,35 (с, 18Н).
4-Гидроксиметил-2,6-ди-трет.-бутилпиридин (соединение В3)
Гетерогенный раствор 4-бромметил-2,6-ди-трет.-бутилпиридина (соединение А3, 1,743 г, 82%-ной чистоты) в 20 мл 12% NaOH в воде и 10 мл 1,4-диоксана кипятят в течение 12 часов. Раствор самопроизвольно разделяется на два слоя, когда его охлаждают до комнатной температуры. Верхний слой отделяют и добавляют этилацетат. Этот органический слой затем промывают рассолом, водой и сушат над MgSO4. Нужный продукт очищают с помощью хроматографии на колонке (этилацетат/гексан 1/9) с получением белого твердого вещества. 1Н ЯМР δ 7,09 (с, 2Н), 4,67 (д, J=4,4 Гц, 2Н), 2,3 (ш.с, 1Н), 1,36 (с, 18Н).
2,6-Ди-трет.-бутилизоникотиновая кислота (соединение С3)
Реагент Джонса добавляют по каплям к раствору 4-гидроксиметил-2,6-ди-трет. -бутилпиридина (соединение В3, 302 мг, 1,37 ммол) в 5 мл ацетона, пока раствор не изменит цвет от светло-желтого до оранжевого (необходимо 55 капель реагента Джонса). Спустя 5 минут к реакционной смеси добавляют 2 мл изопропанола, образуется зеленый осадок Сr+3-соли. Осадок отделяют фильтрованием и раствор разбавляют этилацетатом, затем промывают рассолом, водой и сушат над MgSO4. После фильтрования растворитель удаляют с получением нужного продукта в виде белого твердого вещества (227 мг). 1Н ЯМР δ 7,71 (с, 2Н), 1,34 (с, 18Н).
2-Бром-4,6-ди-трет.-бутилфенол (соединение D3)
К раствору 2,4-ди-трет. -бутилфенола (Aldrich, 2,0 г, 9.7 ммол) в 2 мл НОАс добавляют Вr2 (0,5 мл, 9.7 ммол). Реакционную смесь перемешивают при комнатной температуре в течение 12 часов. Растворитель удаляют при пониженном давлении и остаток очищают с помощью хроматографии на колонке (этилацетат/гексан 1/20) с получением нужного продукта (2,54 г) в виде белого твердого вещества. 1Н ЯМР δ 7,33 (д, J=2,3 Гц, 1Н), 7,24 (д, J=2,3 Гц, 1Н), 1,41 (с, 9Н), 1,29 (с, 9Н).
O-Метоксиметил-2-бром-4,6-ди-трет.-бутилфенол (соединение Е3)
К раствору 2-бром-4,6-ди-трет.-бутилфенола (соединение D3, 2,54 г, 8,88 ммол) и каталитического количества Bu4NI в 20 мл сухого СН2Сl2 при 0oС добавляют диизопропилэтиламин (9.51 мл, 53 ммол) и затем метоксиметилхлорид (2,02 мл, 26,6 ммол). Реакционную смесь нагревают при 45oС в течение 12 часов. Реакционную смесь затем промывают 10% лимонной кислотой, затем NaHCO3 (насыщенный раствор), рассолом и сушат над MgSO4. После фильтрования и упаривания растворителя при пониженном давлении остаток очищают с помощью хроматографии на колонке (чистый гексан) с получением вышеназванного соединения (2,79 г) в виде бесцветного масла. 1Н ЯМР δ 7,40 (д, J=2,44 Гц, 1Н), 7,30 (д, J=2,4 Гц, 1Н), 5.22 (с, 2Н), 3,70 (с, 3Н), 1,43 (с, 9Н), 1,29 (с, 9Н).
O-Метоксиметил-3',5'-ди-трет.-бутилсалициловая кислота (соединение F3)
К раствору O-метоксиметил-2-бром-4,6-ди-трет. -бутилфенола (соединение Е3, 2,79 г, 8,5 ммол) в 30 мл сухого ТГФ при -78oС в атмосфере аргона добавляют 11 мл трет.-BuLi (1,7 М в гексане, 18,7 ммол). Эту смесь перемешивают при -78oС в течение 1 часа. Затем пропускают через раствор СО2 (газ) при -78oС в течение 1 часа. После прекращения тока СО2 реакционную смесь перемешивают дополнительно один час при -78oС. Затем добавляют 10% НСl, дают возможность смеси нагреться до комнатной температуры и экстрагируют этилацетатом. Органический слой промывают рассолом и сушат над Na2SO4. После концентрирования остаток очищают с помощью хроматографии на колонке (этилацетат/гексан 1/1) с получением вышеназванного соединения в виде белого твердого вещества (429 мг). 1Н ЯМР δ 7,75 (д, J=2,81 Гц, 1Н), 7,60 (д, J=2,8 Гц, 1Н), 5,07 (с, 2Н), 3,62 (с, 3Н), 1,33 (с, 9Н), 1,26 (с, 9Н).
Этил 2-фтор-4-[(2', 6'-ди-трет. -бутилпирил-4'-ил)-карбамоил] бензоат (соединение 41).
Раствор 2,6-ди-трет. -бутилизоникотиновой кислоты (соединение С3, 47,3 мг, 0,20 ммол) в 2 мл SOCl2 нагревают при кипячении в течение 2 часов. Избыток SOCl2 удаляют в вакууме, остаток растворяют в 2 мл сухого CH2Cl2 и добавляют этил 2-фтор-4-аминобензоат (соединение С1, 40,4 мг, 0,22 ммол) и пиридин (0,0835 мл, 0,69 ммол). Реакционную смесь перемешивают при комнатной температуре в течение 12 часов. Растворитель удаляют, остаток очищают с помощью хроматографии на колонке (этилацетат/гексан (1/9) и получают обозначенное в заголовке соединение (71,2 мг) в виде белых кристаллов. 1Н ЯМР δ 8,56 (ш. с, 1Н), 7,91 (т, J=8,36 Гц, 1Н), 7,53 (дд, J=12,82, 2,0 Гц, 1Н), 7,39 (дд, J=8,7, 2,0 Гц, 1Н), 4,33 (к, J=7,1 Гц, 2Н), 1,37 (т, J=7,1 Гц, 3Н), 1,35 (с, 18Н).
Этил 4-[2', 6'-ди-трет.-бутилпирид-4'-ил)-карбамоил]бензоат (соединение 43).
Применяя методику, аналогичную приведенной для синтеза этил 2-фтор-4-[(2', 6'-ди-трет.-бутилпирид-4'-ил)-карбамоил]бензоата (соединение 41), но с использованием 2,6-ди-трет.-бутилизоникотиновой кислоты (соединение С3, 101 мг, 0,43 ммол) и этил 4-амино-бензоата (78 мг, 0,47 ммол) получают обозначенное в заголовке соединение в виде белого твердого вещества (135 мг). 1Н ЯМР δ 8,43 (ш.с, 1Н), 8,02 (д, J=8,7 Гц, 2Н), 7,75 (д, J=8,7, Гц, 2Н), 7,48 (с, 2Н), 4,33 (к, J=7,1 Гц, 2Н), 1,38 (т, J=7,1 Гц, 3Н), 1,35 (с, 18Н).
Этил 2-фтор-4-[(3',5'-ди-трет.-бутилфенил)-карбамоил]бензоат (соединение 45).
Применяя методику, аналогичную приведенной для синтеза этил 2-фтор-4-[(2', 6'-ди-трет.-бутилпирид-4'-ил)-карбамоил]бензоата (соединение 41), но с использованием 3,5-ди-трет. -бутилбензойной кислоты (60 мг, 0,26 ммол, полученной по литературной методике, см. Kagechicka et al., J. Med. Chem. 1988, 31, 2182-2192) и этил 2-фтор-4-аминобензоата (соединение С1, 51,5 мг, 0,28 ммол) получают обозначенное в заголовке соединение в виде белого твердого вещества (135 мг). 1Н ЯМР δ 8,21 (ш.с, 1Н), 7,93 (т, J=8,3 Гц, 1Н), 7,79 (дд, J= 12,8, 2,0 Гц, 1Н), 7,67 (д, J=1,8 Гц, 2Н), 7,65 (т, J=1,7 Гц, 1Н), 7,35 (дд, J=8,7, 2,1 Гц, 1Н), 4,36 (к, J=7,2 Гц, 2Н), 1,39 (т, J=7,2 Гц, 3Н), 1,36 (с, 18Н).
Этил 2-фтop-4-[(2'-мeтoкcимeтил-3', 5',-ди-тpeт.-бутилфeнил)-кapбaмoил] бeнзоaт (соединение G3).
К смеси O-метоксиметил-3',5'-ди-трет.-бутилсалициловой кислоты (соединение F3, 150 мг, 0,51 ммол), 4-диметиламинопиридина (142 мг, 0,61 ммол) и этил 2-фтор-4-аминобензоата (соединение С1, 102 мг, 0,56 ммол) в 5 мл сухого СН2Сl2 добавляют гидрохлорид 1-(3-диметиламинопропил)-3-этилкарбодиимида (117 мг, 0,61 ммол). Реакционную смесь перемешивают при комнатной температуре в течение 12 часов. Растворитель упаривают в вакууме и остаток растворяют в этилацетате, затем промывают рассолом, водой и сушат над MgSO4. Удаляют растворитель и остаток очищают хроматографией на колонке (этилацетат/гексан 1/3) с получением обозначенного в заголовке соединения (58 мг). 1Н ЯМР δ 8,97 (ш.с, 1Н), 7,94 (т, J=8,37 Гц, 1Н), 7,78 (д, J=2,7 Гц, 1Н), 7,61 (д, J=13,0 Гц, 1Н), 7,56 (д, J=2,6 Гц, 1Н), 7,35 (д, J=8,7 Гц, 1Н), 5,0 (с, 2Н), 3,53 (с, 3Н), 4,38 (к, J=7,1 Гц, 2Н), 1,47 (с, 9Н), 1,39 (т, J=7,2 Гц, 3Н), 1,33 (с, 9Н).
Этил 2-фтор-4-[(2'-гидрокси-3',5'-ди-трет.-бутилфенил)-карбамоил]-бензоат (соединение 47).
К раствору этил 2-фтор-4-[(2'-метоксиметил-3',5'-ди-трет.-бутилфенил)-карбамоил] бензоата (соединение G3, 34 мг, 0,07 ммол) в 1 мл ТГФ добавляют 10 капель НОАс. Реакционную смесь нагревают при кипячении в течение 12 часов. Растворитель удаляют и добавляют этилацетат. Раствор промывают NаНСО3 (насыщенный раствор), рассолом, водой и сушат над MgSO4. Растворитель удаляют в вакууме с получением масла. Масло выдерживают при обычной атмосфере 12 часов, в течение которых образуется кристаллическое вещество. Кристаллы собирают и промывают несколько раз гексаном, что приводит к обозначенному в заголовке соединению в виде белого твердого вещества (13,5 мг). 1Н ЯМР δ 10,73 (с, 1Н), 7,98 (д, J=2,56 Гц, 1Н), 7,88 (ш.с, 1Н), 7,75 (т, J=8,26 Гц, 1Н), 7,60 (д, J=2,44 Гц, 1Н), 7,32 (дд, J=12,3, 2,0 Гц, 1Н), 7,02 (дд, J= 8,6, 2,0 Гц, 1Н), 4,35 (к, J=7,2 Гц, 2Н), 1,39 (с, 9Н), 1,37 (т, J=7,2 Гц, 3Н), 1,5 (с, 9Н).
2,6-Дифтор-4-[(2',6'-ди-трет.-бутилпирид-4'-ил)-карбамоил]бензойная кислота (соединение 50).
К 2,6-ди-трет.-бутилизоникотиновой кислоте (соединение С3, 20 мг, 0,085 ммол) добавляют 1 мл SOCl2. Смесь нагревают при кипячении в течение 2 часов. После охлаждения до комнатной температуры избыток SOCl2 удаляют и остаток растворяют в 2 мл СН2Сl2. К этому раствору добавляют метил 2,6-дифтор-4-аминобензоат (соединение Н1, 16 мг, 0,085 ммол) и триэтиламин (0,015 мл, 0,1 ммол). Реакционную смесь выдерживают при комнатной температуре в течение 2 часов и затем концентрируют досуха. Остаток очищают с помощью хроматографии на колонке с использованием этилацетата/гексана (1/10) и получают метиловый эфир обозначенного в заголовке соединения. Его омыляют (гидролизуют) согласно общей методике (см. ниже) с получением обозначенного в заголовке соединения в виде бесцветного твердого вещества. 1Н ЯМР δ 7,44 (с, 2Н), 7,40 (д, J=11,8, 2Н), 1,37 (с, 18Н).
2,6-Дифтop-4-[(3', 5'-ди-тpeт. -бутилфeнид)-кapбaмoил] бeнзoйнaя кислота (соединение 52).
Применяя методику, аналогичную приведенной для синтеза 2,6-дифтор-4-[(2', 6'-ди-трет. -бутилпирид-4'-ил)-карбамоил]бензойной кислоты (соединение 50), но с использованием 3,5-ди-трет.-бутил-бензойной кислоты (37 мг, 0,16 ммол) и метил 2,6-дифтор-4-амино-бензоата (соединение Н1, 29 мг, 0,16 ммол) получают обозначенное в заголовке соединение в виде бесцветных кристаллов. 1Н ЯМР δ 7,92 (ш.с, 1Н), 7,60 (м, 3Н), 7,42 (д, J=10,0 Гц, 2Н), 1,38 (с, 18Н).
2-Нитро-4-[(2',6'-ди-трет.-бутилпирид-4'-ил)-карбамоил]бензойная кислота (соединение 54).
Применяя методику, аналогичную приведенной для синтеза 2,6-дифтор-4-[(2', 6'-ди-трет. -бутилпирид-4'-ил)-карбамоил]бензойной кислоты (соединение 50), но с использованием 2,6-ди-трет.-бутилизоникотиновой кислоты (40 мг, 0,17 ммол) и метил 2-нитро-4-аминобензоата (соединение F1, 33 мг, 0,17 ммол) получают обозначенное в заголовке соединение в виде светло-желтого масла. 1Н ЯМР δ (ацетон-d6) 10,25 (ш.с, 1Н), 8,32 (с, 1Н), 7,97 (д, J=8,1 Гц, 1Н), 7,93 (ш.с, 1Н), 7,70 (с, 2Н), 1,36 (с, 18Н).
Метил 2-нитро-4-[(4'-бром-5', 6', 7',8'-тетрагидро-5',5',8',8'-тетраметилнафталин-2'-ил)карбамоил]бензоат (соединение 25)
Применяя методику, аналогичную приведенной для синтеза соединения 1, но с использованием соединения F и соединения F1, получают нужный продукт в виде белого твердого вещества. 1Н ЯМР δ 9.24 (ш.с, 1Н), 9.23 (д, J=1,8 Гц, 1Н), 7,92 (дд, J=8,4, 2,4 Гц, 1Н), 7,87 (д, J=2,1 Гц, 1Н), 7,84 (д, J=2,1 Гц, 1Н), 7,80 (д, J=8,7 Гц, 1Н), 3,91 (с, 3Н), 1,75 (м, 2Н), 1,65 (м, 2Н), 1,58 (с, 6Н), 1,33 (с, 6Н).
Общая методика получения производных бензойной кислоты гидролизом соответствующих метиловых и этиловых эфиров.
К раствору эфира (3,0 ммол) в 20 мл EtOH добавляют 5 мл 1 N NaOH в воде. Реакционную смесь перемешивают при комнатной температуре в течение ночи и нейтрализуют 10% НСl до рН 5. Спирт удаляют упариванием и водный слой экстрагируют этилацетатом. Этилацетатный слой далее промывют NaHCO3 (насыщенный раствор), рассолом и сушат над MgSO4. После концентрирования желаемую карбоновую кислоту выделяют и при необходимости перекристаллизовывают из этилацетата или ацетонитрила.
2-Фтор-4-[(5', 6', 7', 8'-тетрагидоо-5',5',8',8'-тетраметилнафталин-2'-ил)карбамоил]бензойная кислота (соединение 2)
1Н ЯМР (ацетон-D6) δ 9.86 (ш.с, 1Н), 7,95 (м, 3Н), 7,75 (дд, J=7,9, 2,2 Гц, 1Н), 7,62 (дд, J=8,5, 1,6 Гц, 1Н), 7,50 (д, J=8,3 Гц, 1Н), 1,73 (с, 4Н), 1,32 (с, 6Н), 1,30 (с, 6Н).
2-Фтop-4-[(4'-бpoм-5', 6', 7',8'-тeтpaгидpo-5',5',8',8'-тeтpaмeтилнaфтaлин-2'-ил)карбамоил]бензойная кислота (соединение 4)
1Н ЯМР (ацетон-D6) δ 9.97 (ш.с, 1Н), 8,04 (д, J=1,89 Гц, 1Н), 8,01 (д, J= 1,90 Гц, 1Н), 7,95 (т, J=8,55 Гц, 1Н), 7,90 (дд, J=12,28, 2,0 Гц, 1Н), 7,59 (дд, J=8,67, 1,50 Гц, 1Н), 1,76 (м, 4Н), 1,58 (с, 6Н), 1,35 (с, 6Н).
2-Фтор-4-[(3'-гидрокси-5',6',7',8'-тетрагидро-5',5',8',8'-тетраметил-нафталин-2'-ил)карбамоил]бензойная кислота (соединение 6)
1Н ЯМР (ацетон-D6) δ 11,3 (ш.с, 1Н), 10,2 (ш.с, 1Н), 7,94 (м, 2Н), 7,85 (дд, J= 11,4, 1,95 Гц, 1Н), 7,53 (дд, J=6,59, 2,08 Гц, 1Н), 6,94 (с, 1Н), 2,85 (ш.с, 1Н), 1,70 (с, 4Н), 1,29 (с, 6Н), 1,28 (с, 12Н).
2-Фтop-4-[(8'-бpoм-4',4'-димeтилxpoмaн-6'-ил)кapбaмoил]бeнзoйнaя кислота (соединение 8)
1Н ЯМР (ацетон-d6) δ 9.87 (ш.с, 1Н), 8,04 (д, J=2,1 Гц, 1Н), 8,03 (д, J= 2,1 Гц, 1Н), 7,94 (т, J=8,66 Гц, 1Н), 7,91 (дд, J=13,8, 2,0 Гц, 1Н), 7,57 (дд, J= 8,6, 2,0 Гц, 1Н), 4,37 (т, J=5.44 Гц, 2Н), 1,92 (т, J=5.44 Гц, 2Н), 1,40 (с, 6Н).
2-Фтор-4-[(2', 2'-4', 4'-тетраметил-8'-бромхроман-6'-ил)карбамоил] бензойная кислота (соединение 10)
1Н ЯМР (ацетон-d6) δ 9.87 (ш.с, 1Н), 8,06 (д, J=2,2 Гц, 1Н), 8,04 (д, J= 2,1 Гц, 1Н), 7,94 (т, J=8,54 Гц, 1Н), 7,91 (дд, J=14,0, 2,0 Гц, 1Н), 7,59 (дд, J=8,5, 2,3 Гц, 1H), 1,96 (с, 2Н), 1,42 (с, 6Н), 1,41 (с, 6Н).
2-Фтop-4-[(2',2'-4',4'-тeтpaмeтил-8'-тpифтоpмeтилxpoмaн-6'-ил)кapбaмoил] бeнзoйнaя кислота (соединение 12)
1Н ЯМР (ацетон-d6) δ 10,02 (ш.с, 1H), 8,31 (с, 1H), 8,09 (с, 1H), 7,92 (м, 2Н), 7,56 (д, J=7,69 Гц, 1H), 2,00 (с, 2Н), 1,44 (с, 6Н), 1,41 (с, 6Н).
2-Фтор-4-[(2', 2'-4', 4'-тетраметил-8'-азидохроман-6'-ил)карбамоил] бензойная кислота (соединение 14)
1Н ЯМР δ 8,03 (т, J=8,4 Гц, 1H), 7,87 (ш.с, 1H), 7,79 (дд, J=13, 2,0 Гц, 1H), 7,64 (д, J=2,2 Гц, 1H), 7,32 (дд, J=8,66, 1,9 Гц, 1H), 7,22 (д, J=2,1 Гц, 1H), 1,91 (с, 2Н), 1,45 (с, 6Н), 1,41 (с, 6Н).
2,6-Дифтop-4-[(2', 2'-4', 4'-тeтpaмeтил-8'-тpифтopмeтилoxpoмaн-6'-ил)карбамоил]бензойная кислота (соединение 16)
1Н ЯМР (ацетон-d6) δ 8,30 (д, J=2,3 Гц, 1H), 8,06 (д, J=2,2 Гц, 1H), 7,59 (д, J=10,32 Гц, 1H), 1,954 (с, 2Н), 1,44 (с, 6Н), 1,41 (с, 6Н).
2-Фтop-4-[(2',2'-4',4'-тeтpaмeтил-8'-иoдxpoмaн-6'-ил)кapбaмoил]бeнзoйнaя кислота (соединение 18)
1Н ЯМР (ацетон-d6) δ 10,0 (ш.с, 1H), 8,24 (с, 1H), 8,07 (с, 1H), 7,94 (м, 1H), 7,57 (д, J=8,67 Гц, 1H), 1,95 (с, 2Н), 1,41 (с, 12Н).
2-Фтop-4-[(2', 2'-4', 4', 8'-пeнтaмeтилxpoмaн-6'-ил)кapбaмоил] бeнзoйнaя кислота (соединение 20)
1Н ЯМР (ацетон-d6) δ 9.77 (ш.с, 1H), 7,90 (м, 3Н), 7,65 (д, J=2,0 Гц, 1H), 7,56 (дд, J= 8,61, 2,0 Гц, 1H), 2,19 (с, 3Н), 1,90 (с, 2Н), 1,38 (с, 6Н), 1,37 (с, 6Н).
4-[(5',6',7',8'-Teтрaгидро-5',5',8',8'-тетрамeтилнaфтaлин-2'-ил)тиокарбамоил]бензойная кислота (соединение 22)
1Н ЯМР δ 9,08 (ш.с, 1H), 8,17 (д, J=8,61 Гц, 2Н), 7,95 (ш.с, 2Н), 7,77 (ш. с, 1H), 7,57 (дд, J=8,1, 2,1 Гц, 1H), 7,37 (д, J=8,2 Гц, 1H), 1,72 (с, 4Н), 1,32 (с, 6Н), 1,31 (с, 12Н).
2-Фтop-4-[(5', 6', 7', 8'-тeтpaгидpo-5',5',8',8'-тeтpaмeтилнaфтaлин-2'-ил)тиокарбамоил]бензойная кислота (соединение 24)
1Н ЯМР (ацетон-d6) δ 11,1 (ш.с, 1H), 8,27 (д, J=13,2 Гц, 1H), 8,02 (т, J= 8,3 Гц, 1H), 7,89 (с, 1H), 7,86 (д, J=10,0 Гц, 1H), 7,62 (д, J=8,3 Гц, 1H), 7,41 (д, J=8,37 Гц, 1H), 1,72 (с, 4Н), 1,30 (с, 12Н).
2-Фтор-4-[(3'-гидрокси-4'-бром-5',6',7',8'-тетрагидро-5',5',8',8'-тетраметилнафталин-2'-ил)карбамоил]бензойная кислота (соединение 30)
Раствор этил 2-фтор-4-[(3'-метоксиметокси-4'-бром-5', 6', 7',8'-тетрагидро-5', 5', 8',8'-тетраметил-2'-нафталинил)-карбамоил]бензоата (соединение S1, 45 мг, 0,084 ммол) в 1 мл EtOH добавляют 1 мл водного раствора NaOH (1M) в воде. Реакционную смесь перемешивают при комнатной температуре в течение ночи и подкисляют до рН 1 10% НСl. EtOH удаляют и к раствору добавляют этилацетат и много воды. Органический слой отделяют и промывают NaHCO3, рассолом и сушат над MgSO4. После фильтрования и концентрирования получают 2-фтор-4-[(3'-метоксиметокси-4'-бром-5', 6', 7',8'-тетрагидро-5',5',8',8'-тетраметилнафталин-2'-ил)карбамоил]бензойную кислоту в виде белого твердого вещества. Метоксиметильную группу удаляют путем растворения белого твердого вещества в 2 мл МеОН с 3 каплями НСl (конц.). После перемешивания в течение ночи реакционную смесь концентрируют досуха. Остаток распределяют между этилацетатом и водой. Органический слой отделяют и промывют NaHCO3, рассолом и сушат над MgSO4. После фильтрования и концентрирования твердый остаток очищают на миниколонке (пипетка) с помощью этилацетата/гексана (1/1) с получением обозначенного в заголовке соединения в виде белого твердого вещества (5,0 мг). 1Н ЯМР (ацетон-d6) δ 10,19 (ш.с, 1Н), 8,01 (с, 1Н), 7,96 (т, J= 8,6 Гц, 1Н), 7,76 (дд, J=11,2, 2,0 Гц, 1Н), 7,54 (дд, J=8,8, 2,0 Гц, 1Н), 1,75 (м, 2Н), 1,65 (м, 2Н), 1,61 (с, 6Н), 1,32 (с, 6Н).
2,6-Дифтop-4-[(3'-гидpoкcи-4'-бpoм-5', 6',7',8'-тeтpaгидpo-5',5',8',8'-тетраметилнафталин-2'-ил)карбамоил]бензойная кислота (соединение 32)
Применяя методику, аналогичную приведенной для синтеза 2-фтор-4-[(3'-гидрокси-4'-бром-5', 6', 7',8'-тетрагидро-5',5',8',8'-тетраметил-2'-нафталинил)карбамоил]бензойной кислоты (соединение 30) получают обозначенное в заголовке соединение в виде белого твердого вещества. 1Н ЯМР (ацетон-d6) δ 10,23 (ш.с, 1Н), 8,01 (с, 1Н), 7,52 (д, J=10,2 Гц, 2Н), 4,8 (ш.с, 1Н), 1,75 (м, 2Н), 1,65 (м, 2Н), 1,60 (с, 6Н), 1,31 (с, 6Н).
2,6-Дифтop-4-[(5', 6', 7',8'-тeтpaгидpo-5',5',8',8'-тeтpaмeтилнaфтaлин-2'-ил)карбамоил]бензойная кислота (соединение 34)
К 5,5,8,8-тетраметил-5,6,7,8-тетрагидро-2-нафталинкарбоновой кислоте (43 мг, 0,19 ммол) добавляют 1 мл тионилхлорида. Эту смесь кипятят с обратным холодильником в течение 2 часов. Избыток тионилхлорида удаляют при пониженном давлении и остаток растворяют в 2 мл СН2Сl2. К этому раствору добавляют метил 4-амино-2,6-дифторбензоат (соединение Н1, 7 мг, 0,2 ммол) и затем 0,5 мл пиридина. Реакционную смесь перемешивают при комнатной температуре в течение 4 ч и концентрируют при пониженном давлении. Остаток очищают с помощью хроматографии на колонке (этилацетат/гексан (1/5) и получают метиловый эфир нужного продукта в виде бесцветного масла. 1Н ЯМР δ 8,11 (д, J=1,9 Гц, 1Н), 8,05 (ш. с, 1H), 7,86 (дд, J=6,2, 2,2 Гц, 1H), 7,41 (м, 3Н), 3,93 1,69 (с, 4Н), 1,29 (с, 6Н), 1,28 (с, 6Н). Это бесцветное масло гидролизуют до нужного продукта с использованием NаОН/Н2О/ЕtOН согласно общей методике. 1Н ЯМР (ацетон-d6) δ 9.74 (ш.с, 1H), 7,95 (с, 1H), 7,70 (д, J=6,8 Гц, 1H), 7,43 (д, J=8,4 Гц, 3Н), 1,71 (с, 4Н), 1,29 (с, 6Н), 1,28 (с, 6Н).
2-Hитpo-4-[(4'-бpoм-5', 6', 7',8'-тeтpaгидpo-5',5',8',8'-тeтpaмeтил-нaфтaлин-2'-ил)карбамоил]бензойная кислота (соединение 26)
1Н ЯМР δ (ацетон-d6): 10,16 (ш.с, 1H), 8,42 (д, J=2,0 Гц, 1H), 8,09 (дд, J=8,6, 2,1 Гц, 1H), 8,06 (д, J=2,2 Гц, 1H), 8,04 (д, J=2,2 Гц, 1H), 7,93 (д, J=8,6 Гц, 1H), 1,75 (м, 2Н), 1,65 (м, 2Н), 1,57 (с, 6Н), 1,34 (с, 6Н).
2-Фтор-4-[(2', 6'-ди-трет.-бутилпирид-4'-ил)-карбамоил]бензойная кислота (соединение 42).
1Н ЯМР δ (СD3OD) 7,92 (т, J=8,36 Гц, 1H), 7,82 (дд, J=12,82, 2,0 Гц, 1H), 7,63 (с, 2Н), 7,55 (дд, J=8,7, 2,1 Гц, 1H), 1,39 (с, 18Н).
4-[(2', 6'-Ди-тpeт. -бутилпиpид-4'-ил)-кapбaмоил] бeнзoйнaя кислота (соединение 44).
1Н ЯМР δ (CD3OD) 8,02 (д, J=8,85 Гц, 2Н), 7,85 (д, J=8,85 Гц, 2Н), 7,63 (с, 2Н), 1,40 (с, 18Н).
2-Фтop-4-[(3', 5'-ди-тpeт. -бутил)фенилкapбaмoил] бeнзoйнaя кислота (соединение 46).
1Н ЯМР δ (СD3OD) 7,92 (т, J=8,3 Гц, 1Н), 7,80 (дд, J=12,8, 2,0 Гц, 1Н), 7,79 (д, J= 1,8 Гц, 2Н), 7,69 (т, J=1,7 Гц, 1Н), 7,57 (дд, J=8,7, 2,1 Гц, 1Н), 1,37 (с, 18Н).
2-Фтop-4-[(2'-гидpoкcи-3', 5'-ди-тpeт. -бутил)фeнилкapбaмоил] бeнзoйнaя кислота (соединение 48).
1Н ЯМР δ (ацетон-d6) 12,3 (с, 1Н), 10,07 (ш.с, 1Н), 7,98 (т, J=8,48 Гц, 1Н), 7,80 (м, 2Н), 7,58 (д, J=2,3 Гц, 1Н), 7,56 (дд, J=8,8, 2,0 Гц, 1Н), 7,02 (дд, J=8,6, 2,0 Гц, 1Н), 4,35 (к, J=7,2 Гц, 2Н), 1,44 (с, 9Н), 1,31 (с, 9Н).
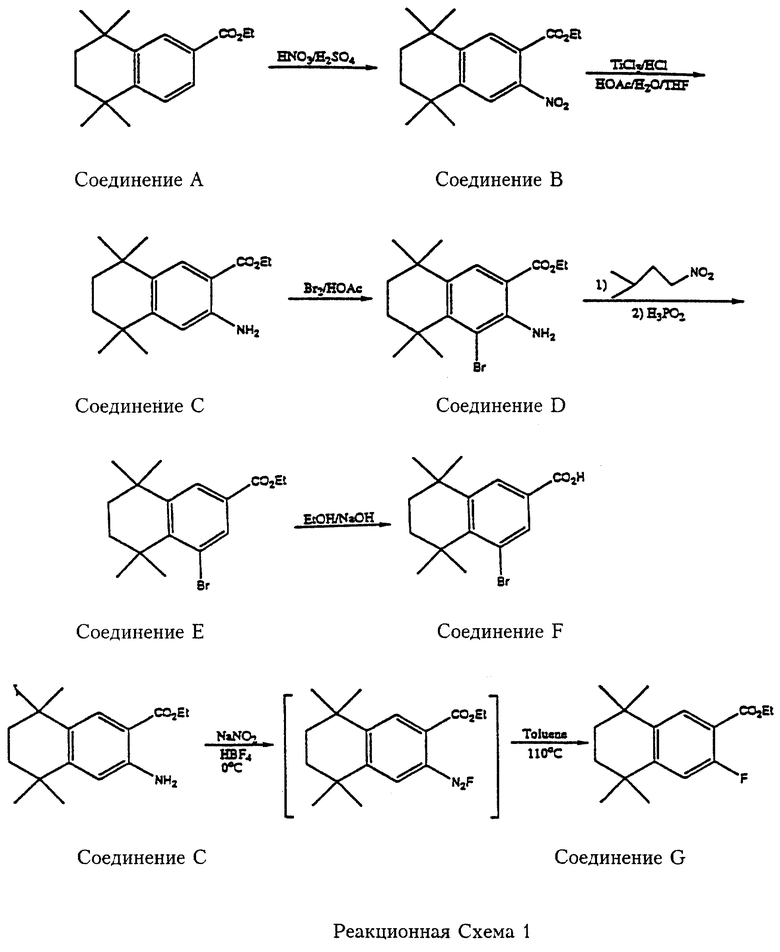
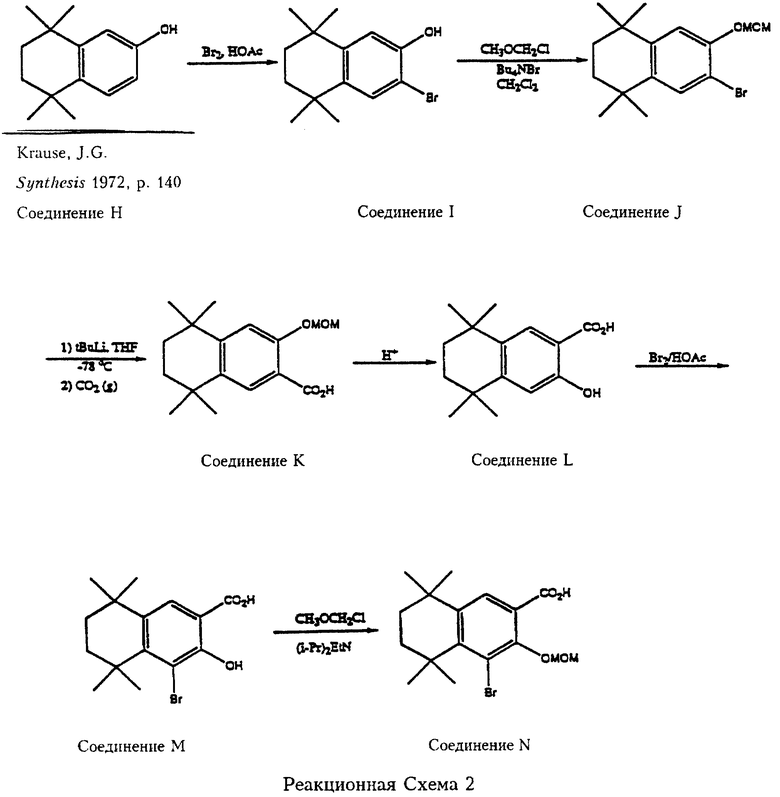
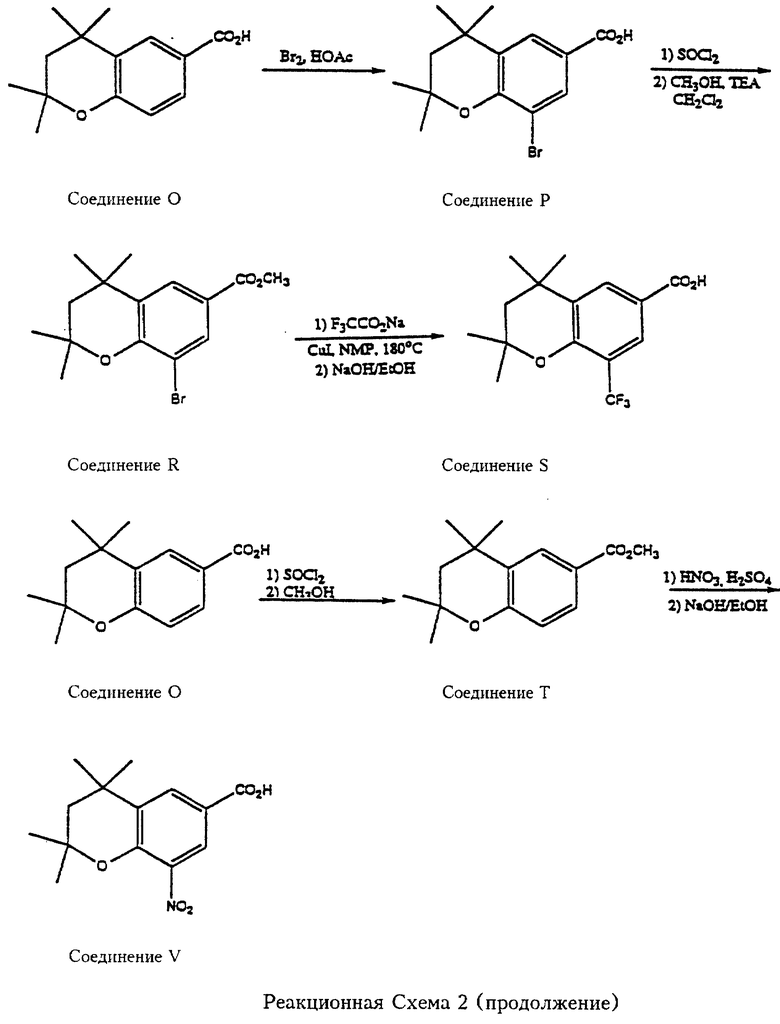
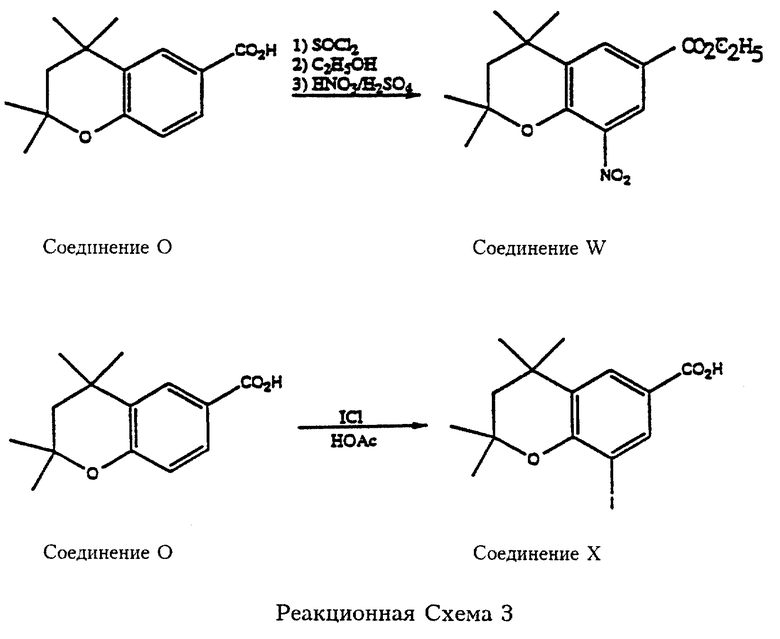
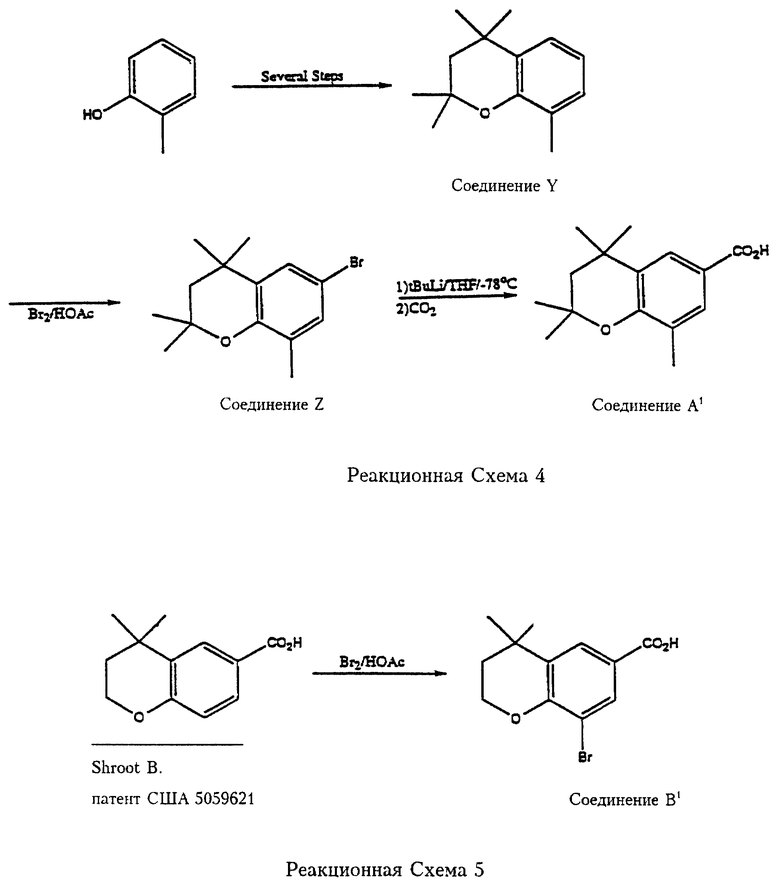
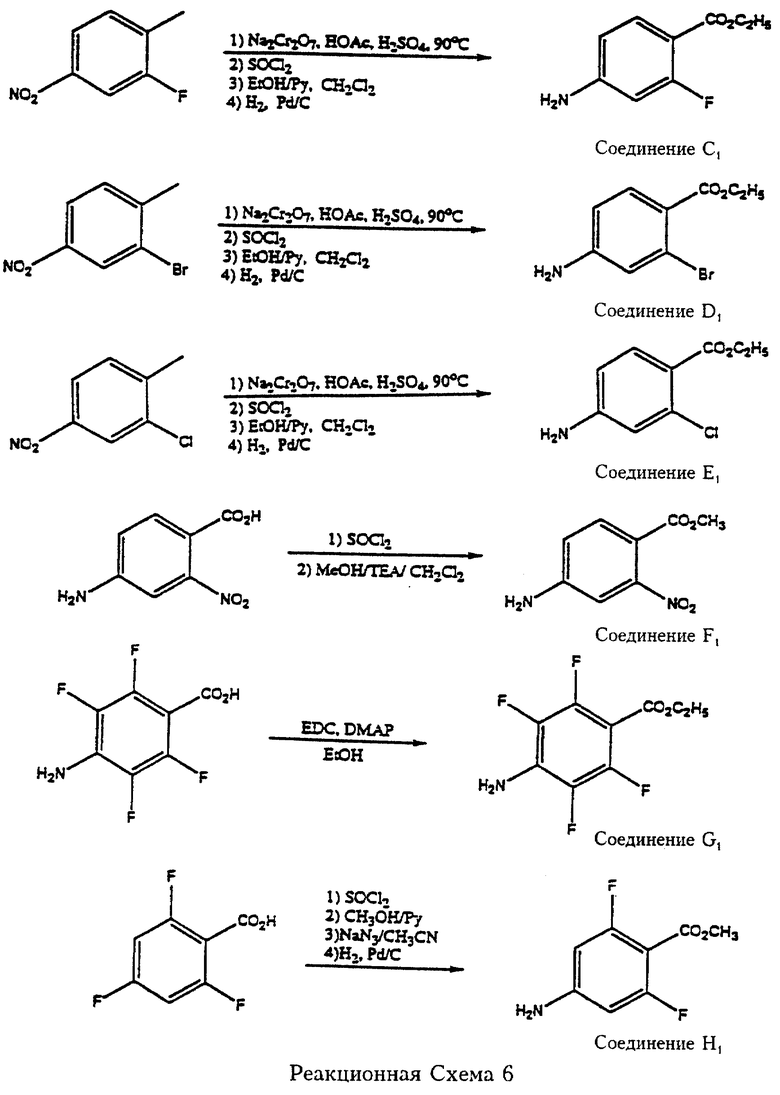
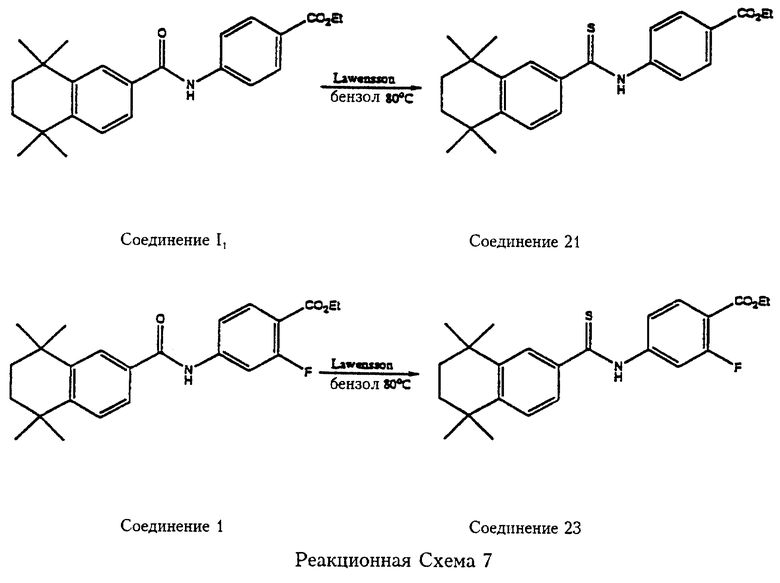
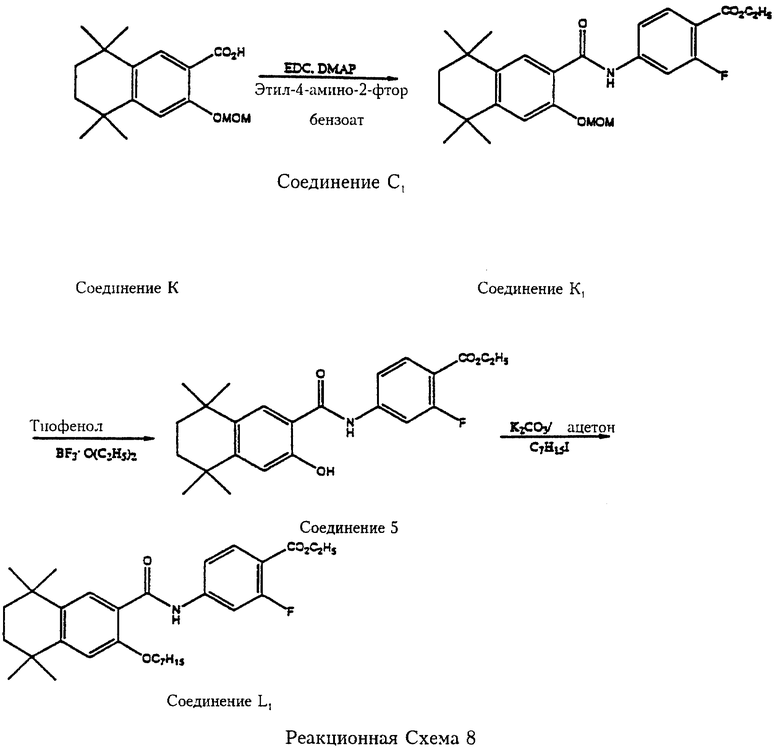
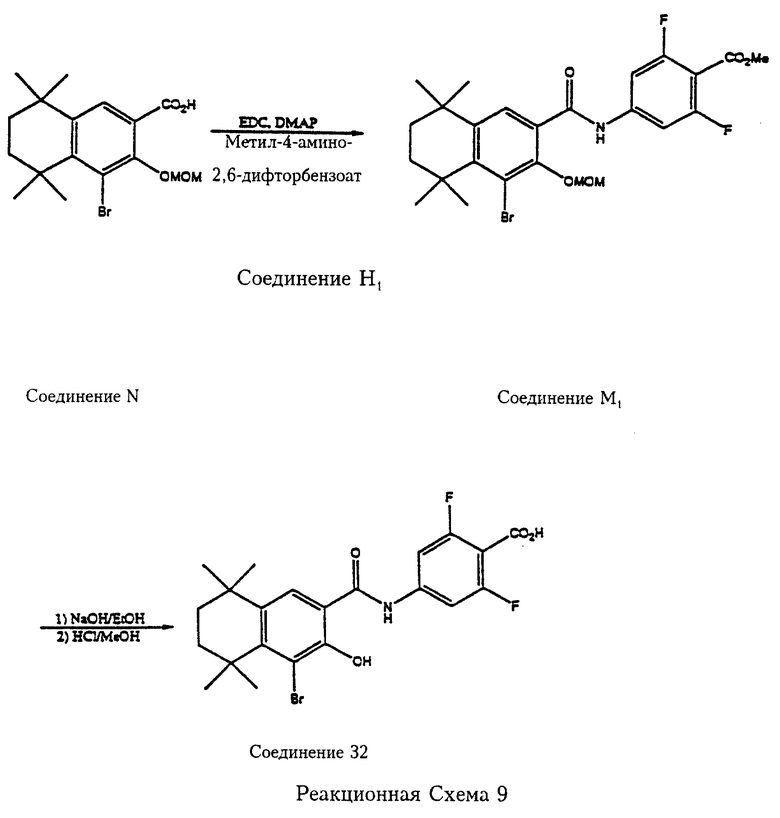
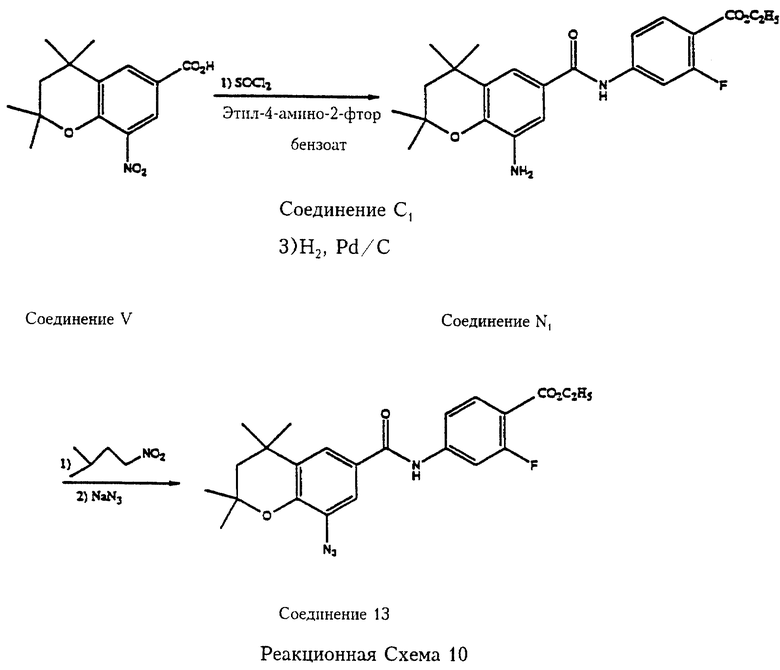
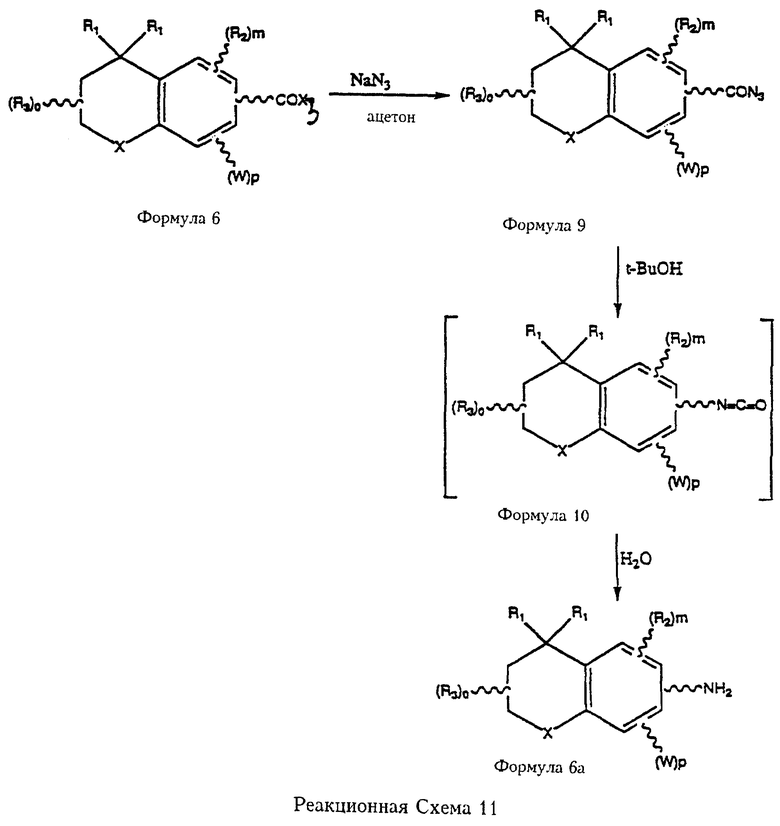
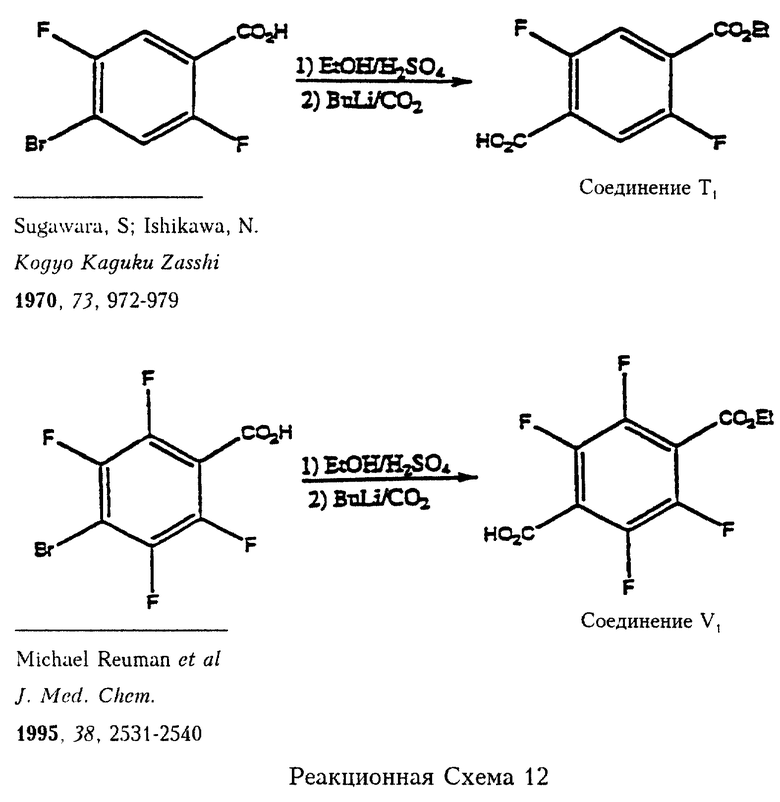
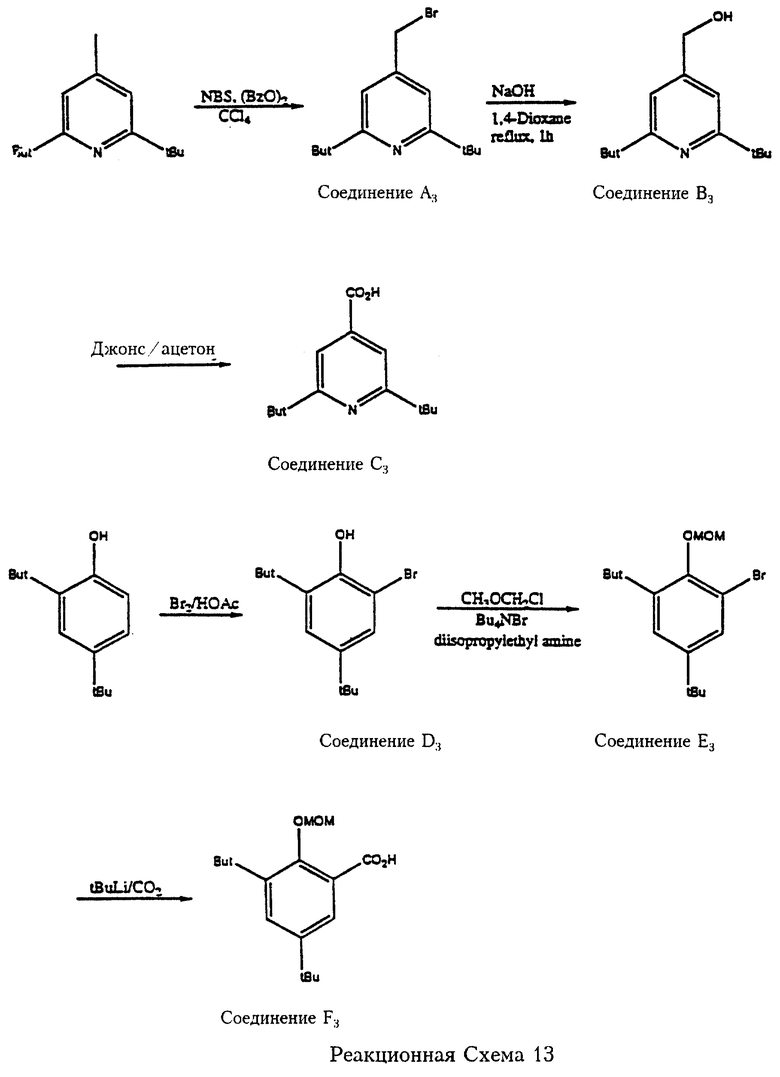
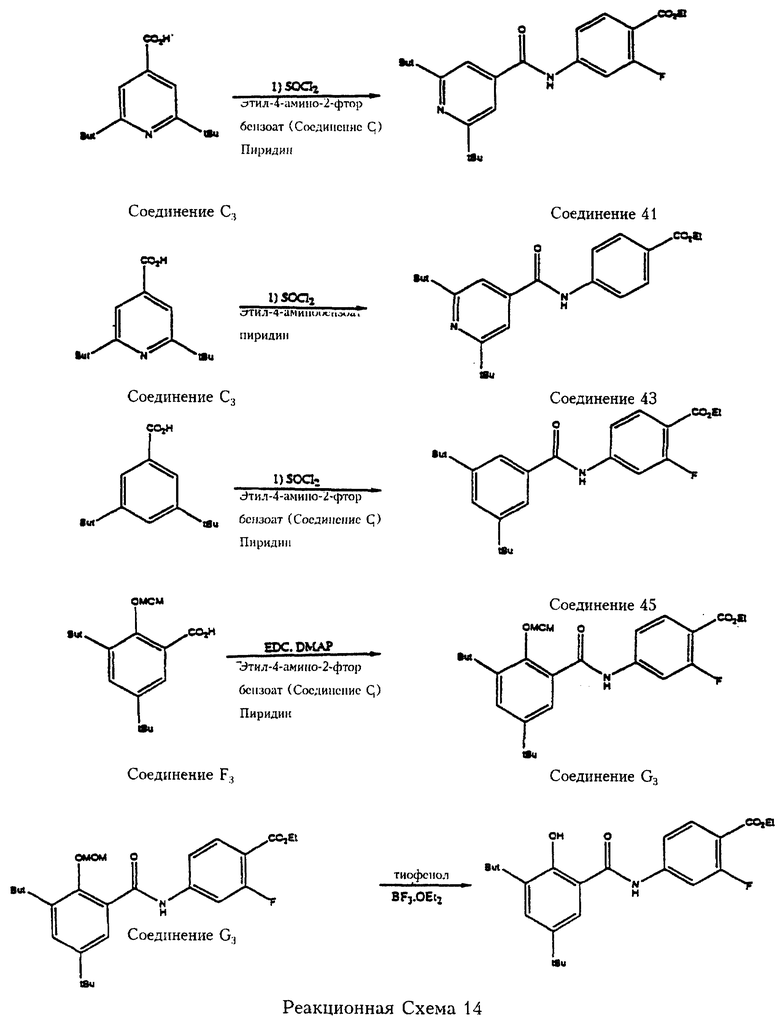
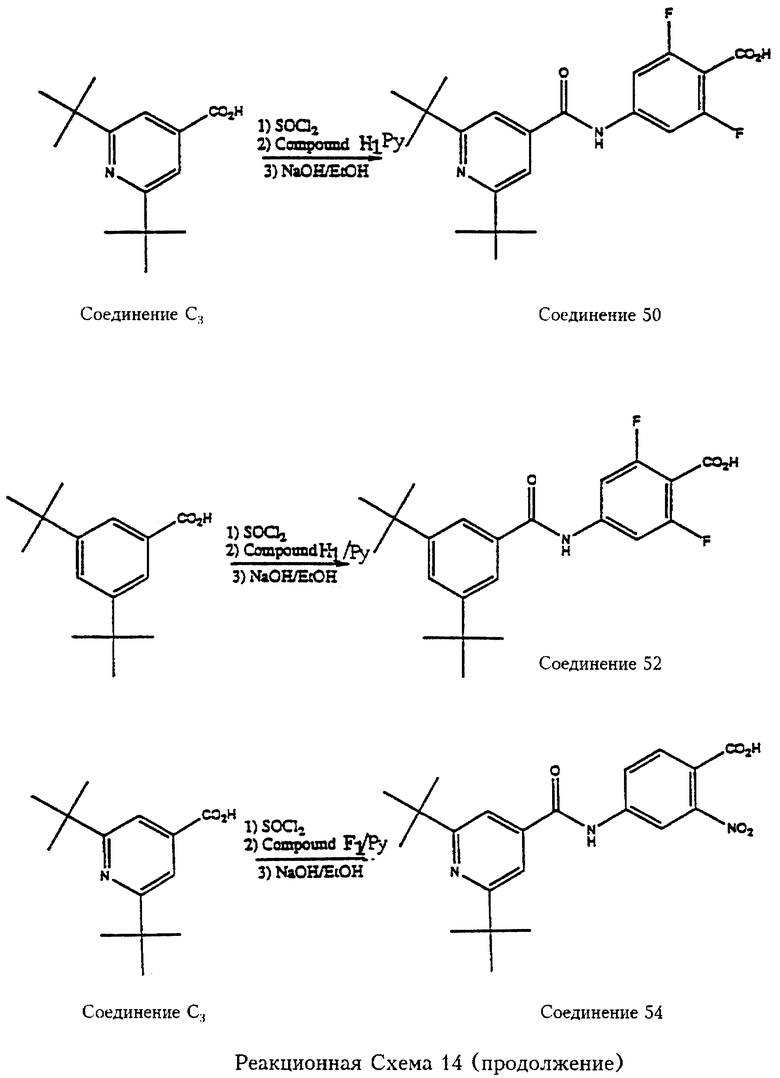
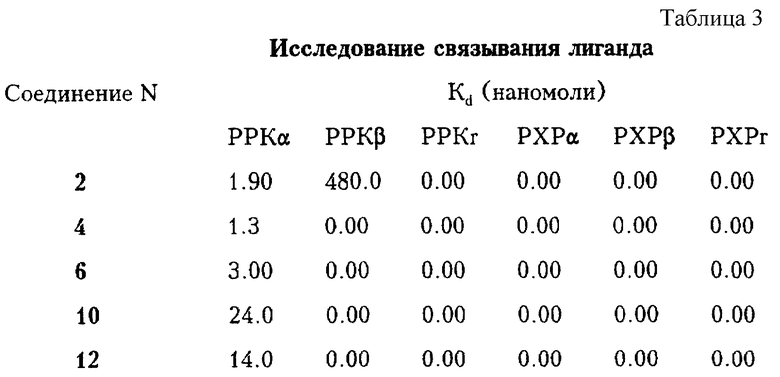
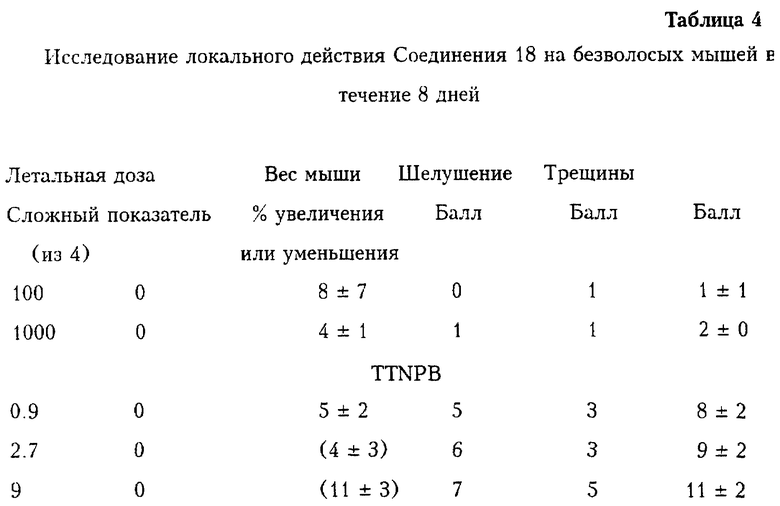

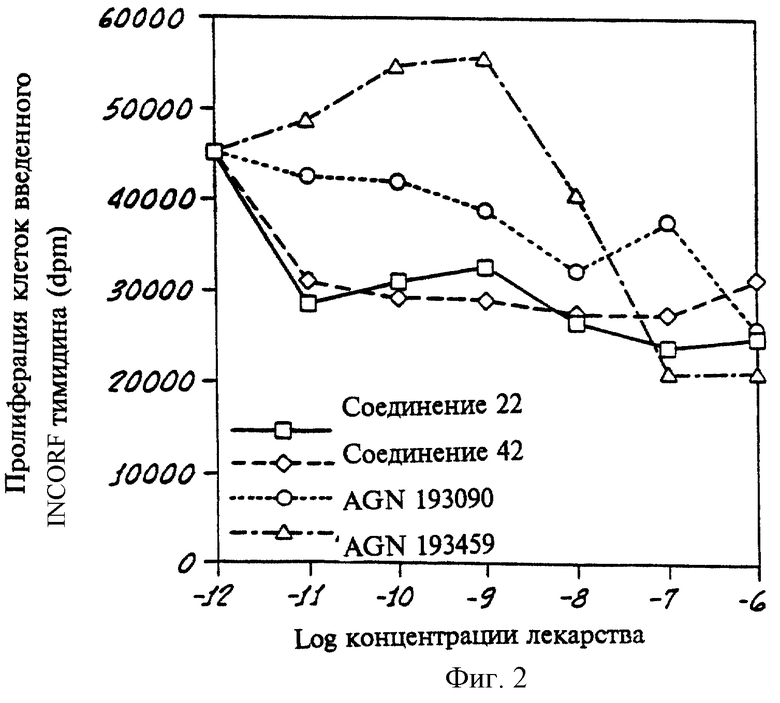
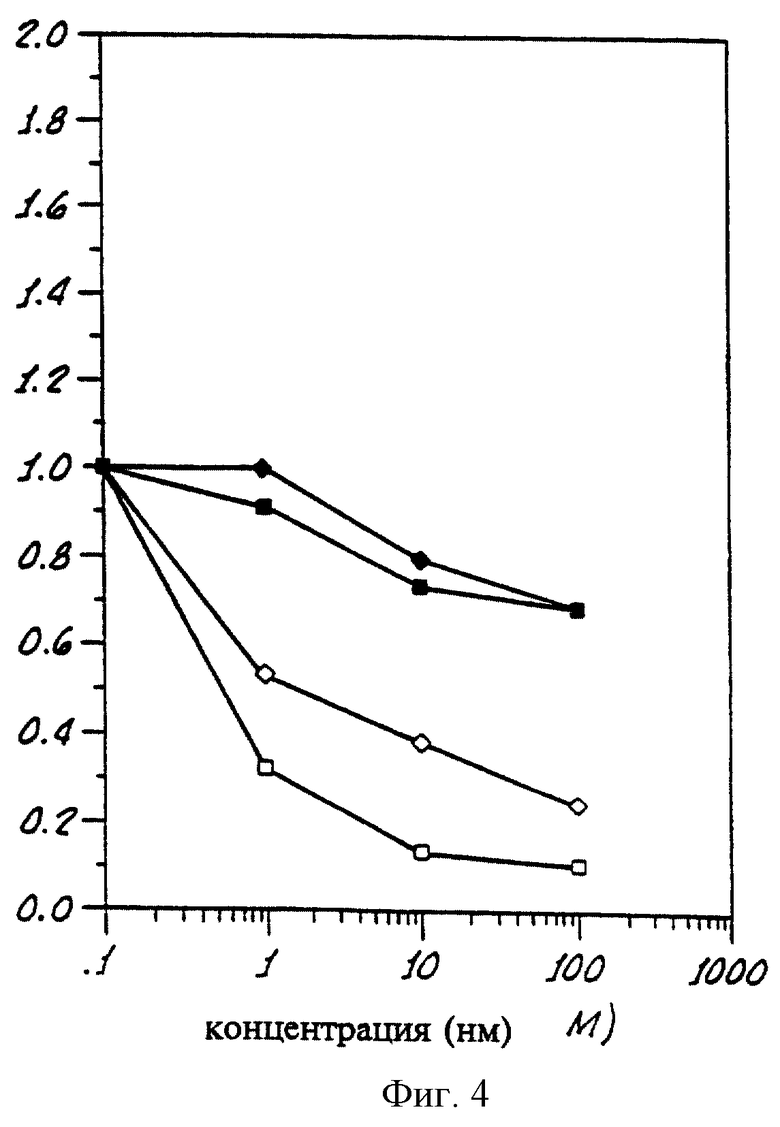
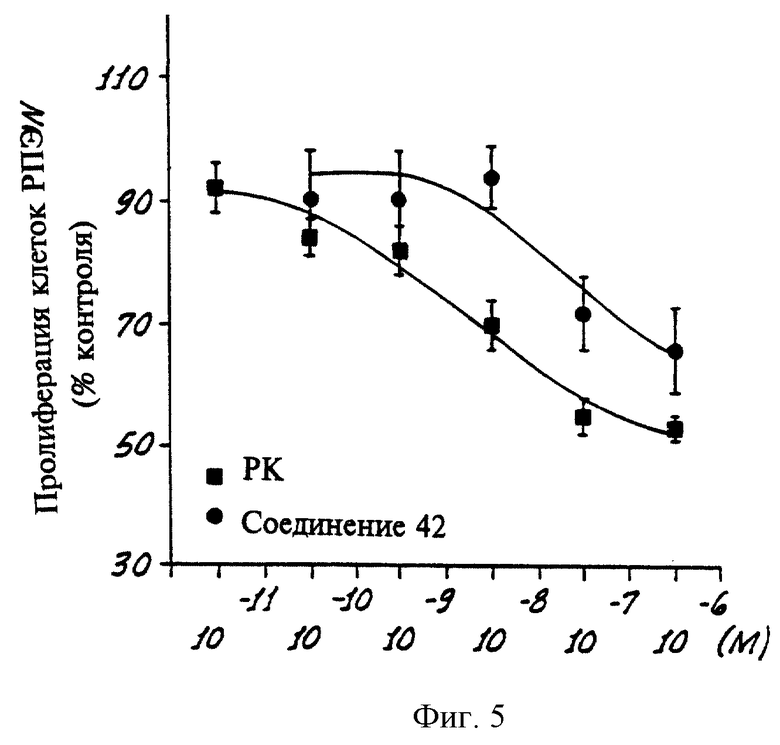
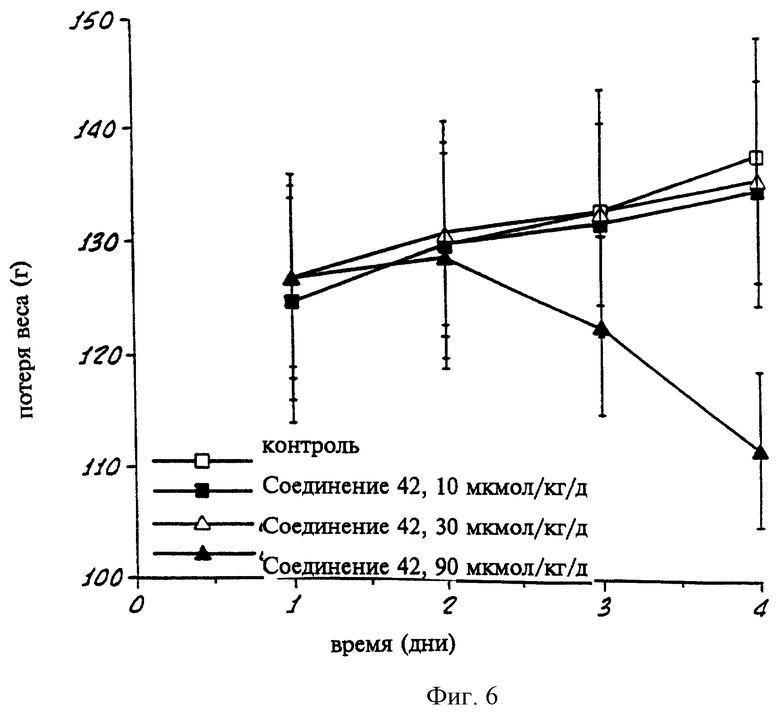
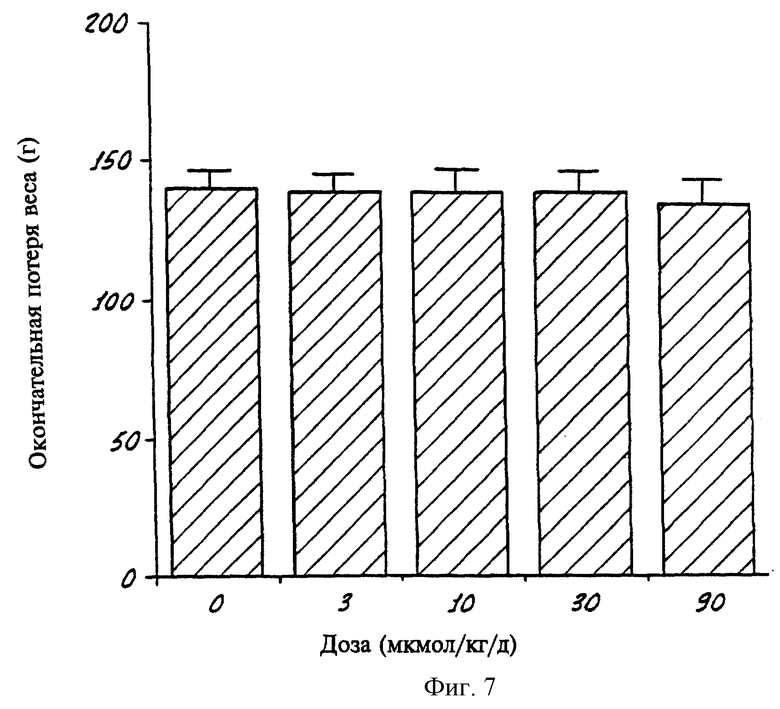
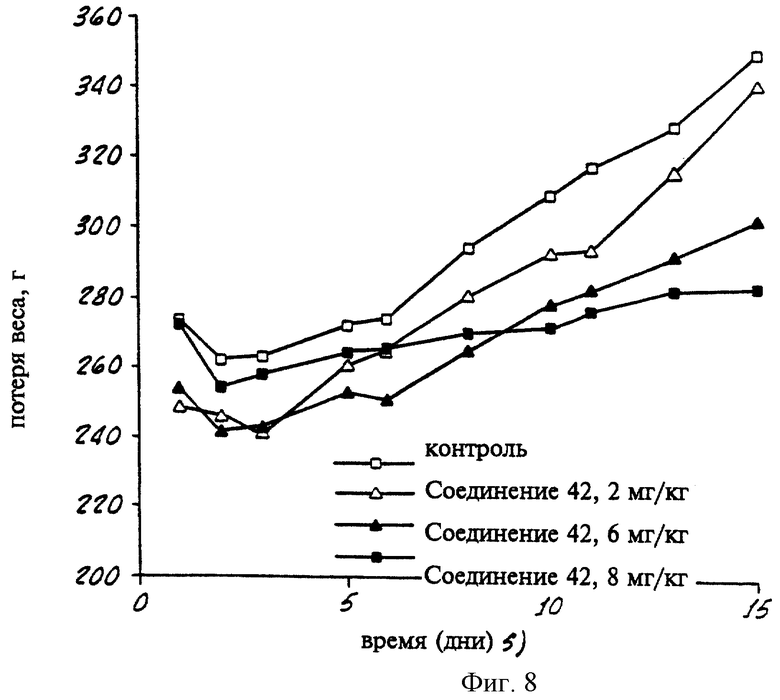
Изобретение относится к области медицины. Ретиноидные соединения, которые действуют специфично или селективно более предпочтительно на подтипы PPKα, чем на подтипы PPKβ и РРКr, обладают желательными фармацевтическими свойствами, характерными для ретиноидов, особенно вводят млекопитающему для лечения опухолей, например острого моноцитарного лейкоза, цервикальной карциномы, миеломы, овариальных карцином и карцином на шее и голове. Предложенный способ не имеет нежелательных побочных эффектов, свойственных ретиноидам: при введении, например, потеря веса, токсичность для кожно-слизистой оболочки, раздражение кожи и терратогенность. 5 с. и 21 з.п. ф-лы, 8 ил., 5 табл.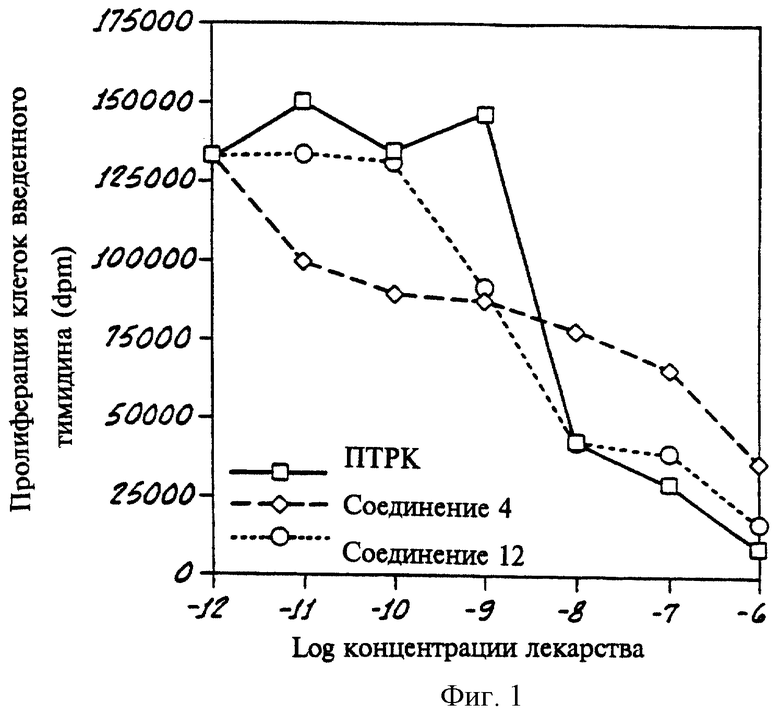
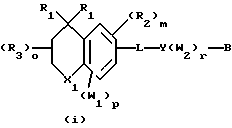
или (ii)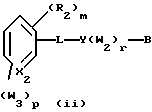
где Х1 - О или [C(R1)2]n, где n = 0, 1, 2;
R1 независимо - Н или С1-С6 алкил;
R2 независимо - водород или С1-С6 алкил;
R3 - водород, низший С1-С6 алкил или F;
m = 0 - 5, целое число;
о = 0 - 4, целое число;
р = 0, 1, 2;
r = 0, 1, 2;
Х2 - N или СН;
Y - фенил, или нафтил, или гетероарил, выбранный из группы, состоящей из пиридила, тиенила, фурила, пиридазинила, пиримидинила, пиразинила, тиазолила, оксазолила, имидазолила, пиразолила, причем указанные фенил, нафтил и гетероарил могут быть замещены одним или двумя R2;
W1 - заместитель, независимо выбранный из группы, состоящей из F, Br, C1, I, фторзамещенного С1-С6 алкила, NO2, и ОН, при условии, что (i) когда соединение соответствует Формуле (i) и Z обозначает О, сумма р и r равна по меньшей мере 1 и W1 не является фтором в 3-положении тетрагидронафталинового кольца; (ii) когда соединение соответствует Формуле (i), r = 0, р = 1 и W1 - ОН, группа ОН находится в α-положении к группе L;
W2 - заместитель, независимо выбранный из группы, состоящей из F, Br, C1, I, фторзамещенного С1-С6 алкила, NO2, и ОН;
W3 - заместитель, независимо выбранный из группы, состоящей из F, Br, C1, I, С1-С6 алкила, фторзамещенного С1-С6 алкила, NO2, OН при условии, что когда соединение соответствует Формуле 2 и Х2 - СН и r = 0, р ≠ 0 и, по меньшей мере одна группа W3 не является алкилом;
L - - (С=Z)-NH- или -NH-(C=Z)-;
Z - О или S;
В - СООН или фармацевтически приемлемая соль, СООR8, CONR9R10, -CH2OH, CH2OR11, CH2OCOR11, CHO, CH(OR12)2, CHOR13O, -COR7, CR7(OR12)2, CR7OR13O, где R7 - алкил, циклоалкил или алкенил, содержащий 1-5 атомов углерода; R8 - алкил, содержащий 1-10 атомов углерода или триметилсилилалкил, где алкильная группа содержит 1-10 атомов углерода, или циклоалкил с 5-10 атомами углерода, или R8 - фенил или низший алкилфенли; R9 и R10 независимо - водород, С1-С10 алкил, С5-С10 циклолалкил, или фенил, или низший алкилфенил; R11 - низший алкил, фенил или низший алкилфенил, R12 - низший алкил и R13 - двухвалентный С2-С5-алкил.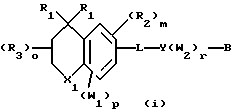
или (ii)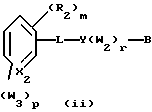
где Х1 - О или [C(R1)2]n, где n = 0, 1, 2;
R1 независимо - Н или С1-С6 алкил;
R2 независимо - водород или С1-С6 алкил;
R3 - водород, низший С1-С6 алкил или F;
m = 0-5, целое число;
о = 0-4, целое число;
р = 0, 1, 2;
r = 0, 1, 2;
Х2 - N или СН;
Y - фенил, или нафтил, или гетероарил, выбранный из группы, состоящей из пиридила, тиенила, фурила, пиридазинила, пиримидинила, пиразинила, тиазолила, оксазолила, имидазолила, пиразолила, причем указанные фенил, нафтил и гетероарил могут быть замещены одним или двумя R2;
W1 - заместитель, независимо выбранный из группы, состоящей из F, Br, C1, I, фторзамещенного С1-С6 алкила, NO2, и ОН, при условии, что (i) когда соединение соответствует Формуле (i) и Z - О, сумма р и r равна по меньшей мере 1 и W1 не является фтором в 3-положении тетрагидронафталинового кольца; (ii) когда соединение соответствует Формуле (i), r = 0, р = 1 и W1 - ОН, группа ОН находится в α- положении к группе L;
W2 - заместитель, независимо выбранный из группы, состоящей из F, Br, C1, I, фторзамещенного С1-С6 алкила, NO2, и ОН;
W3 - заместитель, независимо выбранный из группы, состоящей из F, Br, C1, I, С1-С6 алкила, фторзамещенного С1-С6 алкила, NO2, OН при условии, что когда соединение соответствует Формуле 2 и Х2 - СН и r = 0, р ≠ 0 и по меньшей мере одна группа W3 не является алкилом;
L обозначает - (С=Z)-NH- или -NH-(C=Z)-;
Z - О или S;
В - СООН или фармацевтически приемлемая соль, СООR8, CONR9R10, -CH2OH, CH2OR11, CH2OCOR11, CHO, CH(OR12)2, CHOR13O, -COR7, CR7(OR12)2, CR7OR13O, где R7 - алкил, циклоалкил или алкенил, содержащий 1-5 атомов углерода, R8 - алкил, содержащий 1-10 атомов углерода, или триметилсилилалкил, где алкильная группа содержит 1-10 атомов углерода, или циклоалкил с 5-10 атомами углерода, или R8 - фенил или низший алкилфенил; R9 и R10 независимо - водород, С1-С10 алкил, С5-С10 циклолалкил, или фенил, или низший алкилфенил; R11 - низший алкил, фенил или низший алкилфенил; R12 - низший алкил и R13 - двухвалентный С2-С5 - алкил.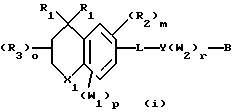
или (ii)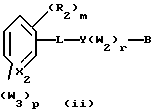
где Х1 - О или [C(R1)2]n, где n = 0, 1, 2;
R1 независимо - Н или С1-С6 алкил;
R2 независимо - водород или С1-С6 алкил;
R3 - водород, низший С1-С6 алкил или F;
m = 0-5, целое число;
o = 0-4, целое число;
р = 0, 1, 2;
r = 0, 1, 2;
Х2 - N или СН;
Y - фенил, или нафтил, или гетероарил, выбранный из группы, состоящей из пиридила, тиенила, фурила, пиридазинила, пиримидинила, пиразинила, тиазолила, оксазолила, имидазолила, пиразолила, причем указанные фенил, нафтил и гетероарил могут быть замещены одним или двумя R2;
W1 - заместитель, независимо выбранный из группы, состоящей из F, Br, C1, I, фторзамещенного С1-С6 алкила, NO2, и ОН, при условии, что (i) когда соединение соответствует Формуле (i) и Z - О, сумма р и r равна по меньшей мере 1 и W1 не является фтором в 3- положении тетрагидронафталинового кольца; (ii) когда соединение соответствует Формуле (i), r - 0, р = 1 и W1 - ОН, группа ОН находится в α- положении к группе L;
W2 - заместитель, независимо выбранный из группы, состоящей из F, Br, C1, I, фторзамещенного С1-С6 алкила, NO2, и ОН;
W3 - заместитель, независимо выбранный из группы, состоящей из F, Br, C1, I, С1-С6 алкила, фторзамещенного С1-С6 алкила, NO2, OН, при условии, что когда соединение соответствует Формуле 2, Х2 - СН и r = 0, р ≠ 0 и по меньшей мере одна группа W3 не является алкилом;
L - -(С=Z)-NH- или -NH-(C=Z)-;
Z - О или S;
В - СООН или фармацевтически приемлемая соль, СООR8, CONR9R10, -CH2OH, CH2OR11, CH2OCOR11, CHO, CH(OR12)2, CHOR13O, -COR7, CR7(OR12)2, CR7OR13O, где R7 - алкил, циклоалкил или алкенил, содержащий 1-5 атомов углерода; R8 - алкил, содержащий 1-10 атомов углерода, или триметилсилилалкил, где алкильная группа содержит 1-10 атомов углерода, или циклоалкил с 5-10 атомами углерода, или R8 - фенил или низший алкилфенил; R9 и R10 независимо - водород, С1-С10 алкил, С5-С10 циклолалкил, или фенил, или низший алкилфенил; R11 - низший алкил, фенил или низший алкилфенил; R12 - низший алкил и R13 - двухвалентный С2-С5-алкил.
этил 2-фтор-4-[(5', 6', 7',8'-тетрагидро-5',5',8',8'-тетраметилнафталин -2'- ил) карбамоил]бензоат;
2-фтор-4-[(5', 6', 7', 8'-тетрагидро-5', 5', 8',8'-тетраметилнафтил-2'-ил)карбамоил] бензойную кислоту;
этил-2-фтор-4-[(5', 6', 7',8'-тетрагидро-4'-бром-5',5',8',8'-тетраметилнафталин-2'-ил)карбамоил]бензоат;
2-фтор-4-[(4'-бром-5', 6', 7',8'-тетрагидро-5',5',8,'8'-тетраметилнафталин-2'-ил) карбамоил]бензойную кислоту;
этил 2-фтор-4-[(2,2', 4', 4'-тетраметил-8'-бромхромат-6'-ил)карбамоил] бензоат;
2-фтор-4-[(2',2',4',4'-тетраметил-8'-бромхромат-6'-ил)карбамоил]бензойную кислоту;
этил-2-фтор-4-[(2',2',4',4'-тетраметил-8'-трифторметилхромат-6'-ил) карбамоил]бензоат;
2-фтор-4-[(2',2',4',4'-тетраметил-8'-трифторметилхромат-6'-ил)карбамоил] бензойную кислоту);
этил 2-фтор-4[(2', 2', 4',4'-тетраметил-8'-азидохроман-6'-ил)карбамоил] бензоат;
2-фтор-4-[(2', 2', 4',4'-тетраметил-8'-азидохроман-6'-ил)карбамоил] бензойную кислоту;
этил 2-фтор-4-[(2', 2', 4', 4'-тетраметил-8'-иодхроман-6'-ил)карбамоил] бензоат;
2-фтор-4-[(2',2',4,'4-тетраметил-8'-иодхроман-6'-ил)карбамоил] бензойную кислоту;
этил 4-[(5', 6', 7', 8'-тетрагидро-5', 5', 8', 8'-тетраметил-2-нафталинил)тиокарбамоил]бензоат, и
4-[(5',6',7',8'-тетрагидро-5',5',8',8'-тетраметилнафталин-2'ил)тиокарбамоил] бензойную кислоту.
этил 2-фтор-4-[(2',6'-дитрет.бутилпирид-4'-ил)карбамоил] бензоат или
2-фтор-4-[(2',6'-дитрет.бутилпирид-4'-ил)карбамоил] бензойную кислоту.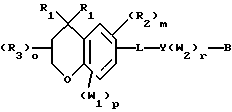
где R1 независимо - Н или С1-С6 алкил;
R2 независимо - водород или низший С1-С6 алкил;
R3 - водород, низший С1-С6 алкил или F;
m = 0-5, целое число;
о = 0-4, целое число;
р = 0, 1, 2;
r = 0, 1, 2;
Y - фенил, или нафтил, или гетероарил, выбранный из группы, состоящей из пиридила, тиенила, фурила, пиридазинила, пиримидинила, пиразинила, тиазолила, оксазолила, имидазолила, пиразолила, причем указанный фенил, нафтил и гетероарил могут быть замещены одним или двумя R2;
W1 - заместитель, независимо выбранный из группы, состоящей из F, Br, Cl, J, фторзамещенного C1-C6-алкила, NO2 и OH, при условии, что когда Z - О, сумма р и r равна по меньшей мере 1 и когда Z - О, р + r = 1 и Y - фенил, тогда W1 не является С1 в положении 8 хроманового кольца;
W2 - заместитель, независимо выбранный из группы, состоящей из F, Br, C1, I, фторзамещенного С1-С6 алкила, NO2, и ОН;
W3 - заместитель, независимо выбранный из группы, состоящей из F, Br, C1, I, фторзамещенного С1-С6 алкила, NO2, и OН;
L - -(С=Z)-NH- или -NH-(C=Z)-;
Z - О или S;
В - СООН или фармацевтически приемлемую соль, СООR8, где R8 - С1-С10 алкил или триметилсилилалкил, где алкил содержит 1-10 атомов углерода, или циклоалкил, содержащий 5-10 атомов углерода, или R8 - фенил или низший алкилфенил.
этил 2-фтор-4-[(2', 2', 4',4'-тетраметил-8'-бромхроман-6'-ил)карбамоил] бензоат;
2-фтор-4-[(2', 2', 4', 4'-тетраметил-8'-бромхроман-6'-ил)карбамоил] бензойную кислоту;
этил 2-фтор-4-[2', 2', 4', 4-тетраметил-8'-трифторметилхроман-6'-ил)карбамоил]бензоат;
2-фтор-4-[2', 2',4',4'-тетраметил-8'-трифторметилхроман-6'-ил)карбамоил] бензойную кислоту;
этил 2-фтор-4-[(2', 2',4',4'-тетраметил-8'-азидохроман-6'-ил)карбамоил] бензоат;
2-фтор-4-[(2',2',4',4'-тетраметил-8'-азидохроман-6'-ил)карбамоил] бензойную кислоту;
этил 2-фтор-4-[(2',2',4',4-тетраметил-8'-иодхроман-6'-ил)карбамоил]бензоат;
2-фтор-4-[2',2',4',4'-тетраметил-8'-иодхроман-6'-ил)карбамоил] бензойную кислоту.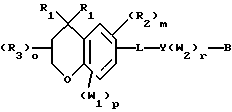
где R1 независимо - Н или С1-С6 алкил;
R2 независимо - водород или низший С1-С6 алкил;
R3 - водород, низший С1-С6 алкил или F;
m = 0-5, целое число;
о = 0-4, целое число;
р = 0, 1, 2;
r = 0, 1, 2;
Y - фенил, или нафтил, или гетероарил, выбранный из группы, состоящей из пиридила, тиенила, фурила, пиридазинила, пиримидинила, пиразинила, тиазолила, оксазолила, имидазолила, пиразолила, причем указанные фенил, нафтил и гетероарил могут быть замещены одним или двумя R2;
W1 - заместитель, независимо выбранный из группы, состоящей из F, Br, C1, I, фторзамещенного С1-С6 алкила, NO2, и ОН, при условии, что когда Z - О, сумма р и r равна по меньшей мере 1 и когда Z - О, р + r = 1 и Y - фенил, тогда W1 не является С1 в положении 8 хроманового кольца;
W2 - заместитель, независимо выбранный из группы, состоящей из F, Br, C1, I, фторзамещенного С1-С6 алкила, NO2, и ОН;
W3 - заместитель, независимо выбранный из группы, состоящей из F, Br, C1, I, фторзамещенного С1-С6 алкила, NO2, и OН;
L - -(С=Z)-NH- или -NH-(C=Z)-;
Z - О или S;
В - СООН или фармацевтически приемлемую соль, СООR8, где R8 - С1-С10 алкил или триметилсилилалкил, где алкил содержит 1-10 атомов углерода, или циклоалкил, содержащий 5-10 атомов углерода, или R8 - фенил или низший алкилфенил.
| WO 9303713 А1, 04.03.1993 | |||
| US 5399586 А, 21.03.1995 | |||
| ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ | 1995 |
|
RU2120304C1 |
| База данных Lexis-Nexis | |||
| Реферат, J | |||
| of Medicinal chemistry, vol | |||
| Способ очистки нефти и нефтяных продуктов и уничтожения их флюоресценции | 1921 |
|
SU31A1 |
| Механическая топочная решетка с наклонными частью подвижными, частью неподвижными колосниковыми элементами | 1917 |
|
SU1988A1 |
| ЧАСЫ С ПРИСПОСОБЛЕНИЕМ ДЛЯ ВКЛЮЧЕНИЯ И ВЫКЛЮЧЕНИЯ ТОКА В ОСВЕТИТЕЛЬНОЙ СЕТИ | 1924 |
|
SU2182A1 |

