Настоящее изобретение относится к новым производным циклогексана, к их получению и их применению в качестве лекарственных средств. Настоящее изобретение прежде всего относится к соединениям формулы (I)
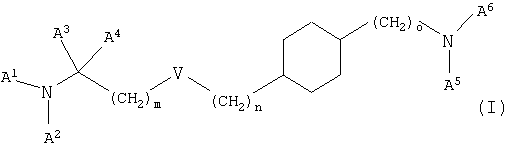
где V означает простую связь, О, S, -CH=СН-CH2-O-, -CH=СН- или -C≡С-, m и n независимо друг от друга равны 0-7 и m+n равно 0-7 при условии, что m не равно 0, если V означает О или S,
о равно 0-2,
А1 означает водород, (низш.)алкил, гидрокси(низш.)алкил или (низш.)алкенил,
А2 означает (низш.)алкил, циклоалкил, циклоалкил(низш.)алкил или (низш.)алкенил, необязательно замещенный группой R1, или
А1 и А2 связаны друг с другом с образованием цикла и -А1-А2- означает (низш.)алкилен или (низш.)алкенилен, необязательно замещенный группой R1, в которой одна из -СН2-групп в составе -А1-А2- необязательно замещена группой NR2, S или О,
А3 и А4 независимо друг от друга означают водород или (низш.)алкил или А3 и А4 связаны друг с другом и вместе с атомом углерода, к которому они присоединены, образуют цикл, а -А3-А4- означает -(СН2)2-5-,
А5 означает водород, (низш.)алкил или (низш.)алкенил,
А6 означает пиридинил, пиридазинил, пиримидинил или пиразинил, необязательно замещенный одним или двумя заместителями, независимо выбранными из группы, включающей (низш.)алкил, (низш.)алкилциклоалкил, тио(низш.)алкокси, циклоалкил, карбамоил, карбокси, карбокси(низш.)алкил, циано, амино, моно- и диалкиламино, (низш.)алкокси, (низш.)алкокси(низш.)алкил, (низш.)алкоксикарбонил, (низш.)алкоксикарбонил(низш.)алкил, (низш.)алкенил, (низш.)алкинил, арил, арил(низш.)алкил, арилокси, галоген, гетероарил, гетероциклил, гетероциклил(низш.)алкил и трифторметил,
R1 означает гидрокси, гидрокси(низш.)алкил, (низш.)алкокси, (низш.)алкоксикарбонил, галоген, CN, N(R3, R4) или тио(низш.)алкокси,
R2, R3 и R4 независимо друг от друга означают водород или (низш.)алкил,
и к их фармацевтически приемлемым солям, при условии, что соединение формулы (I) не означает транс-[4-(2-дипропиламиноэтил)циклогексил]пиримидин-2-иламин.
Соединения по настоящему изобретению ингибируют 2,3-оксидосквален-ланостеринциклазу (ЕС 5.4.99), которая необходима для биосинтеза холестерина, эргостерина и других стеринов. Факторы риска, которые непосредственно вызывают развитие коронарного или периферического атеросклероза, включают повышенный уровень липопротеинов низкой плотности (LDL-C), содержащих холестерин, липопротеинов высокой плотности (HDL-C), гипертензию, дым от сигарет и сахарный диабет. Прочие синергические факторы риска включают повышенную концентрацию липопротеинов, обогащенных триглицеридами (TG), небольшие плотные частицы липопротеина низкой плотности, липопротеин (a) (Lp(a)) и гомоцистеин. Предрасполагающие факторы риска модифицируют причинные и условные факторы риска и, следовательно, вызывают атерогенез опосредованным образом. Предрасполагающими факторами являются ожирение, физическая пассивность (инертность), преждевременный семейный анамнез CVD и принадлежность к мужскому полу. Тесная взаимосвязь между коронарной болезнью (CHD) и высоким уровнем LDL-C в плазме (крови) и вместе с тем терапевтическая эффективность от снижения повышенного уровня LDL-C являются известными фактами (Gotto и др., Circulation, 81, 1721-1733 (1990), Stein и др., Nutr. Metab. Cardiovasc. Dis., 2, 113-156 (1992), Illingworth, Med. Clin. North. Am., 84, 23-42 (2000)). Обогащенные холестрином, в некоторых случаях нестабильные, атеросклеротические бляшки вызывают закупорку сосудов, что приводит к ишемии или инфаркту. По результатам ранней профилактики установлено, что снижение уровня LDL-C в плазме (крови) снижает частоту несмертельных случаев CHD, в то время как общий уровень заболеваемости остается неизменным. Снижение уровня LDL-C в плазме (крови) пациентов с предварительно установленной CHD (повторное вмешательство) снижает смертность от CHD и общий уровень заболеваемости, причем метаанализ различных обследований свидетельствует о том, что такое снижение пропорционально уменьшению уровня LDL-C (Ross и др., Arch. Intern. Med., 159, 1793-1802 (1999)).
Клиническая эффективность благодаря снижению уровня холестерина выше у пациентов с предварительно установленной CHD, чем у лиц с бессимптомной гипохолестеринемией. По современным рекомендациям снижение уровня холестерина следует проводить у пациентов, перенесших инфаркт миокарда, или у пациентов со стенокардией или другим атеросклеротическим заболеванием, у которых установленный уровень LDL-C составляет 100 мг/дл.
Для проведения обычной стандартной терапии используются такие препараты, как секвестранты желчных кислот, фибраты, никотиновая кислота, пробукол, а также статины, т.е. ингибиторы HMG-Co-A-редуктазы, такие как симвастатин и аторвастатин. Наиболее эффективные статины снижают уровень LDL-C в плазме (крови) с эффективностью по меньшей мере 40%, а кроме того, снижают уровень триглицеридов, синергический фактор риска, но с меньшей эффективностью. Напротив, фибраты эффективно снижают уровень триглицеридов в плазме (крови), но не LDL-C. Оказалось, что комбинация статина и фибрата очень эффективно снижает уровень как LDL-C, так и триглицеридов (Ellen и Mc Pherson, J.Cardiol., 81, 60В-63В (1998)), однако безопасность такой комбинации еще подлежит изучению (Shepherd, Eur. Heart J., 16, 5-13 (1995)). Единое лекарственное средство со смешанным профилем, объединяющее эффективность в отношении снижения уровня как LDL-C, так и триглицеридов, обладает дополнительным клиническим преимуществом для асимптоматических и симптоматических пациентов.
В организме человека статины хорошо переносятся в стандартной дозе, однако снижение нестероидных промежуточных соединений при биосинтезе холестерина, таких как изопреноиды и кофермент Q, может ассоциироваться с отрицательными побочными действиями при высоких дозах (Davignon и др., Can. J.Cardiol., 8, 843-864 (1992), Pederson и Tobert, Drug Safety, 14, 11-24 (1996)).
Указанные выше факторы стимулировали исследование и разработку соединений, которые ингибируют биосинтез холестерина, но при этом косвенным образом (по периферическуму механизму) влияют на синтез этих важных нестероидных промежуточных соединений. Микросомальный фермент, 2,3-оксидосквален-ланостеринциклаза (OSC), является уникальной мишенью для лекарственного средства, снижающего уровень холестерина (Morand и др., J.Lipid Res., 38, 373-390 (1997), Mark и др., J.lipid Res., 37, 148-158 (1996)). Наряду с участием в синтезе изопреноидов и кофермента Q OSC потребляет фарнезилпирофосфат. У хомячков фармакологически активные дозы ингибитора OSC не проявляют неблагоприятных побочных действий, в отличие от статина, который снижает потребление пищи и массу тела и увеличивает уровень билирубина в плазме (крови), массу печени и содержание в печени триглицеридов (Morand и др., J.Lipid Res., 38, 373-390 (1997). К таким соединеням, которые ингибируют OSC и снижают уровень общего холестерина в плазме (крови), относятся соединения, описанные в ЕР 636367.
Ингибирование OSC не индуцирует сверхэкспрессию HMGR, поскольку непрямой регуляторный механизм отрицательной обратной связи включает продуцирование 24(S),25-эпоксихолестерина (Peffley и др., Biochem. Pharmacol., 56, 439-449 (1998), Nelson и др., J.Biol. Chem., 256, 1067-1068 (1981), Spencer и др., J.Biol. Chem., 260, 13391-13394 (1985), Panini и др., J.Lipid Res., 27, 1190-1204 (1986), Ness и др. Arch. Biochem. Biophys., 308, 420-425 (1994). Этот регуляторный механизм отрицательной обратной связи является фундаментальным в концепции ингибирования OSC, поскольку (I) он синергически усиливает первичное ингибирущее действие при непрямой понижающей регуляции HMGR и (II) предупреждает массивное накопление предшественника монооксидсквалена в печени. Кроме того, установлено, что 24(S),25-эпоксихолестерин является одним из наиболее активных агонистов ядерного рецептора LXR (Janowski и др., Proc. Nat. Acad. Sci. USA, 96, 266-271 (1999)). Что касается того, что 24(S),25-эпоксихолестерин является побочным продуктом ингибирования OSC, то предполагается, что ингибиторы OSC по настоящему изобретению также могут косвенным образом активировать LXR-зависимые пути, такие как (I) увеличение потребления холестерина ферментом холестерин-7α-гидроксилаза через путь желчных кислот, (II) экспрессия белков АВС, обладающих способностью стимулировать обратный транспорт холестерина и увеличивать уровень в плазме (крови) HDL-C (Venkateswaran и др., J.Biol. Chem., 275, 14700-14707 (2000), Costet и др., J.Biol. Chem. (2000) в печати, Ordovas, Nutr. Rev., 58, 76-79 (2000), Schmitz и Kaminsky, Front Biosci., 6, D505-D514 (2001)) и/или ингибировать кишечное всасывание холестерина (Mangelsdorf, XIIth International Symposium on Atherosclerosis, Stockholm, июнь 2000). Кроме того, предполагается, что возможная взаимосвязь между метаболизмом жирной кислоты и холестерином опосредуется LXR печени (Tobin и др., Mol. Endocrinol., 14, 741-752 (2000)).
Настоящие соединения формулы I ингибируют OSC и, следовательно, также ингибируют биосинтез холестерина, эргостерина и других стеринов, и снижают уровень холестерина в плазме (крови). Следовательно, в общем случае они могут использоваться для лечения и профилактики гиперхолестеринемии, гиперлипемии, артериосклероза и сосудистых заболеваний. Более того, они могут использоваться для лечения и/или профилактики микозов, паразитарных инфекций, желчнокаменной болезни, холестатических нарушений в печени, опухолей и гиперпролиферативных нарушений, например гиперпролиферативных заболеваний кожи, и сосудистых нарушений. Кроме того, неожиданно было установлено, что соединения по настоящему изобретению могут найти терапевтическое применение для улучшения толерантности к глюкозе с целью лечения и/или профилактики ассоциированных заболеваний, таких как диабет. Кроме того, соединения по настоящему изобретению проявляют улучшенные фармакологические свойства по сравнению с известными соединениями.
Если не указано иное, различные термины, используемые в описании заявки для иллюстрации и определения сущности и объема изобретения, имеют следующие значения.
Термин "низш.", используемый в описании, означает группу, содержащую от одного до семи, предпочтительно от одного до четырех, атомов углерода.
Термин "свободная пара" означает несвязанную электронную пару, прежде всего несвязанную электронную пару атома азота, например, в аминогруппе.
Термин "галоген" означает фтор, хлор, бром и иод, предпочтительное фтор, хлор и бром.
Термин "алкил", используемый отдельно или в комбинации с другими группами, означает разветвленный или прямой одновалентный насыщенный алифатический углеводородный радикал, содержащий от 1 до 20, предпочтительно от 1 до 16, атомов углерода, более предпочтительно от 1 до 10 атомов углерода. Описанные ниже (низш.)алкильные группы также означают предпочтительные алкильные группы.
Термин "(низш.)алкил", используемый отдельно или в комбинации с другими группами, означает разветвленный или прямой одновалентный алкильный радикал, содержащий от 1 до 7 атомов углерода, предпочтительно от 1 до 4 атомов углерода. Примеры радикала (низш.)алкил включают метил, этил, н-пропил, изопропил, н-бутил, втор-бутил, трет-бутил и т.п.
Термин "циклоалкил" означает одновалентный карбоциклический радикал, содержащий от 3 до 10 атомов углерода, предпочтительно от 3 до 6 атомов углерода, такой как циклопропил, циклобутил, циклопентил или циклогексил.
Термин "алкокси" означает группу R'-O-, где R' означает алкил. Термин "(низш.)алкокси" означает группу R'-O-, где R' означает (низш.)алкил. Термин "тиоалкокси" означает группу R'-S-, где R' означает алкил. Термин "тио(низш.)алкокси" означает группу R'-S-, где R' означает (низш.)алкил.
Термин "алкенил", используемый отдельно или в комбинации с другими группами, означает прямой или разветвленный углеводородный остаток, содержащий олефиновую связь и до 20, предпочтительно до 16, атомов углерода, более предпочтительно до 10 атомов углерода. Описанные ниже (низш.)алкениловые группы также означают предпочтительные алкениловые группы. Термин (низш.)алкенил означает прямой или разветвленный углеводородный остаток, содержащий олефиновую связь и до 7, предпочтительно до 4, атомов углерода, такой, например, как 2-пропенил.
Термин "алкинил", используемый отдельно или в комбинации с другими группами, означает прямой или разветвленный углеводородный остаток, содержащий тройную связь и до 20, предпочтительно до 16, атомов углерода, более предпочтительно до 10 атомов углерода. Описанные ниже (низш.)алкиниловые группы также означают предпочтительные алкиниловые группы. Термин (низш.)алкинил означает прямой или разветвленный углеводородный остаток, содержащий тройную связь и до 7, предпочтительно до 4, атомов углерода, такой, например, как 2-пропинил.
Термин "алкилен" означает двухвалентную насыщенную алифатическую углеводородную группу с прямой или разветвленной цепью, содержащую от 1 до 20 атомов углерода, предпочтительно от 1 до 16 атомов углерода, более предпочтительно до 10 атомов углерода. Описанные ниже (низш.)алкиленовые группы также означают предпочтительные алкиленовые группы. Термин "(низш.)алкилен", означает двухвалентную насыщенную алифатическую углеводородную группу с прямой или разветвленной цепью, содержащую от 1 до 7, предпочтительно от 1 до 6 или от 3 до 6, атомов углерода. Предпочтительны алкиленовые и (низш.)алкиленовые группы с прямой цепью.
Термин "алкенилен" означает двухвалентную углеводородную группу с прямой или разветвленной цепью, содержащую олефиновую связь и до 20 атомов углерода, предпочтительно до 16 атомов углерода, более предпочтительно до 10 атомов углерода. Описанные ниже (низш.)алкениленовые группы также означают предпочтительные алкениленовые группы. Термин "(низш.)алкенилен" означает двухвалентную углеводородную группу с прямой или разветвленной цепью, содержащую олефиновую связь и до 7, предпочтительно до 5, атомов углерода. Предпочтительны алкениленовые и (низш.)алкениленовые группы с прямой цепью.
Термин "арил" означает фенил или нафтил, предпочтительно фенил, который необязательно содержит от 1 до 3 заместителей, независимо выбранных из группы, включающей (низш.)алкил, (низш.)алкенил, (низш.)алкинил, диокси(низш.)алкилен (образующий, например, бензодиоксилгруппу), галоген, гидрокси, CN, CF3, NH2, N(H, (низш.)алкил), N((низш.)алкил)2, аминокарбонил, карбокси, NO2, (низш.)алкокси, тио(низш.)алкокси, (низш.)алкилкарбонил, (низш.)алкилкарбонилокси, (низш.)алкоксикарбонил. Предпочтительными заместителями являются галоген, CF3, CN, (низш.)алкил и/или (низш.)алкокси.
Термин "гетероарил" означает ароматический 5- или 6-членный цикл, содержащий 1, 2 или 3 атома, выбранных из ряда азот, кислород и/или сера, такой как фурил, пиридил, пиридазинил, пиримидинил, пиразинил, тиенил, изоксазолил, оксазолил, имидазолил или пирролил. Гетероарильная группа может содержать заместители, указанные для группы "арил".
Термин "гетероциклил", используемый в описании заявки, означает неароматические моноциклические гетероциклы, содержащие в цикле 5 или 6 атомов и включающие 1, 2 или 3 атома, выбранных из ряда азот, кислород и сера. Примеры пригодных гетероциклов включают пирролидинил, пирролинил, имидазолидинил, имидазилинил, пиразолидинил, пиразолинил, пиперидил, пиперазинил, морфолинил, пиранил, 4,5-дигидрооксазолил, 4,4-дигидротиазолил. Гетероциклильная группа может содержать заместители, указанные для группы "арил".
Термин "фармацевтически приемлемые соли" включает соли соединений формулы (I) с неорганическими или органическими кислотами, такими, как хлористоводородная кислота, бромистоводородная кислота, азотная кислота, серная кислота, фосфорная кислота, лимонная кислота, муравьиная кислота, малеиновая кислота, уксусная кислота, фумаровая кислота, янтарная кислота, винная кислота, метансульфоновая кислота, пара-толуолсульфоновая кислота и т.п., которые являются нетоксичными в живом организме. Предпочтительными солями являются фосфаты, цитраты, фумараты, формиаты, гидрохлориды, гидробромиды и метансульфонаты.
Более подробно изобретение относится к соединениям формулы (I)
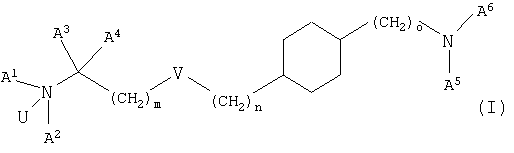
где U означает О или свободную пару электронов,
V означает простую связь, О, S, -СН=СН-СН2-О-, -СН=СН- или -С≡С-,
m и n независимо друг от друга равны 0-7, a m+n равно 0-7 при условии, что m не равно 0, если V означает О или S,
о равно 0-2,
А1 означает водород, (низш.)алкил, гидрокси(низш.)алкил или (низш.)алкенил,
А2 означает (низш.)алкил, циклоалкил, циклоалкил(низш.)алкил или (низш.)алкенил, необязательно замещенный группой R1, или
А1 и А2 связаны друг с другом с образованием цикла и -А1-А2- означает (низш.)алкилен или (низш.)алкенилен, необязательно замещенный группой R1, в которой одна из групп -СН2- в составе -А1-А2- необязательно заменена на NR2, S или О,
А3 и А4 независимо друг от друга означают водород или (низш.)алкил или
А3 и А4 вместе с атомом углерода, к которому они присоединены, образуют цикл и -А3-А4- означает -(СН2)2-5-,
А5 означает водород, (низш.)алкил или (низш.)алкенил,
А6 означает пиридинил, пиридазинил, пиримидинил или пиразинил, необязательно замещенные одним или двумя заместителями, независимо выбранными из группы, включающей (низш.)алкил, (низш.)алкилциклоалкил, тио(низш.)алкокси, циклоалкил, карбамоил, карбокси, карбокси(низш.)алкил, циано, амино, моно- и диалкиламино, (низш.)алкокси, (низш.)алкокси(низш.)-алкил, (низш.)алкоксикарбонил, (низш.)алкоксикарбонил(низш.)алкил, (низш.)алкенил, (низш.)алкинил, арил, арил(низш.)алкил, арилокси, галоген, гетероарил, гетероциклил, гетероциклил(низш.)алкил и трифторметил,
R1 означает гидрокси, гидрокси(низш.)алкил, (низш.)алкокси, (низш.)алкоксикарбонил, галоген, CN, N(R3, R4) или тио(низш.)алкокси,
R2, R3 и R4 независимо друг от друга означают водород или (низш.)алкил,
и к их фармацевтически приемлемым солям при условии, что соединение формулы (I) не означает транс-[4-(2-дипропиламиноэтил)циклогексил]пиримидин-2-иламин.
Предпочтительными соединениями являются соединения формулы (I) и/или их фармацевтически приемлемые соли. Другие предпочтительные варианты настоящего изобретения относятся к соединениям формулы (I), где U означает свободную пару электронов, или к соединениям формулы (I), где U означает О.
Предпочтительным вариантом настоящего изобретения являются соединения формулы (I), описанные выше, где V означает простую связь. О, -СН=СН-СН2-O- или -С≡С-. Наиболее предпочтительными соединениями, как описано выше, являются соединения, в которых V означает -С≡С-.
В другом предпочтительном варианте настоящего изобретения m равно 0-3, более предпочтительно m равно 0. Предпочтительны также соединения формулы (I), где n равно 0 или 1, причем более предпочтительны соединения, в которых n равно 0. Предпочтительны также соединения, описанные выше, в которых суммарное число атомов углерода в составе групп (СН2)m, V и (СН2)n равно 7 или менее. Другими предпочтительными соединениями формулы (I), как описано выше, являются соединения, в которых о равно 0 или 1.
Предпочтительными соединениями по настоящему изобретению являются также соединения, в которых А1 означает (низш.)алкил, предпочтительно метил или этил. Другая группа предпочтительных соединений по настоящему изобретению включает соединения, в которых А2 означает(низш.)алкенил или (низш.)алкил, необязательно замещенный группой R2, причем R2 означает гидрокси или (низш.)алкокси, и прежде всего предпочтительны соединения, в которых А2 означает метил, пропил или 2-гидроксиэтил.
Предпочтительны также соединения формулы (I), где А1 и А2 связаны друг с другом с образованием цикла, а -А1-А2- означает (низш.)алкилен, и прежде всего предпочтительны соединения, в которых -А1-А2- означает -(СН2)5-.
Предпочтительными соединениями, в которых А1 и А2 вместе образуют цикл, являются соединения, в которых указанный цикл означает 4-, 5- или 6-членное кольцо, например такое как пиперидинил или пирролидинил.
Еще одним предпочтительным вариантом настоящего изобретения являются соединения формулы (I), где А3 и А4 означают водород.
Еще одним вариантом настоящего изобретения являются также соединения формулы (I), где А5 означает водород или (низш.)алкил, прежде всего предпочтительны соединения, в которых А5 означает метил. Другими предпочтительными соединениями являются соединения, в которых А6 означает пиридинил, пиридазинил, пиримидинил или пиразинил, необязательно замещенные одним или двумя заместителями, независимо выбранными из группы, включающей (низш.)алкил, (низш.)алкокси, галоген, пиридил и тиенил. Наиболее предпочтительные соединения формулы (I) включают соединения, в которых А6 означает пиридазинил или пиримидинил, необязательно замещенные одним или двумя заместителями, независимо выбранными из группы, включающей бром, хлор, этил и пиридил, причем прежде всего предпочтительны соединения, в которых А6 означает 5-бромпиримидин-2-ил, 6-хлорпиридазин-3-ил, 5-хлорпиримидин-2-ил, 5-пиридин-4-илпиримидин-2-ил, 5-этилпиримидин-2-ил.
Предпочтительные соединения общей формулы (I) выбирают из группы, включающей
транс-(4-[3-(аллилметиламино)проп-1-инил]циклогексил}(5-бромпиримидин-2-ил)метиламин,
транс-(5-бромпиримидин-2-ил)метил{4-[3-(метилпропиламино)проп-1-инил]циклогексил}амин,
транс-(5-бромпиримидин-2-ил)[4-(3-диметиламинопроп-1-инил)циклогексил]метиламин,
транс-(5-бромпиримидин-2-ил)(4-{3-[этил(2-метоксиэтил)амино]проп-1-инил}циклогексил)метиламин,
транс-{4-[3-(аллилметиламино)пропил]циклогексил}(5-бромпиримидин-2-ил)метиламин,
транс-(5-бромпиримидин-2-ил)метил{4-[3-(метилпропиламино)пропил]циклогексил}амин,
транс-(5-бромпиримидин-2-ил)[4-(3-диметиламинопропил)циклогексил]метиламин,
транс-(5-бромпиримидин-2-ил)(4-{3-[этил(2-метоксиэтил)амино]пропил}циклогексил)метиламин,
транс-[4-(3-диметиламинопроп-1-инил)циклогексил]метилпиримидин-2-иламин,
транс-(6-хлорпиридазин-3-ил)[4-(3-диметиламинопроп-1-инил)циклогексил]метиламин,
транс-(5-хлорпиримидин-2-ил)[4-(3-диметиламинопроп-1-инил)циклогексил]метиламин,
транс-(5-бромпиридин-2-ил)[4-(3-диметиламинопроп-1-инил)циклогексил]метиламин,
транс-[4-(3-диметиламинопроп-1-инил)циклогексил]метилпиридин-2-иламин,
транс-[4-(3-диметиламинопроп-1-инил)циклогексил]метилпиразин-2-иламин,
транс-[2-[(3-{4-[(5-бромпиримидин-2-ил)метиламино]циклогексил}проп-2-инил)этиламино]этанол],
транс-[(5-бромпиримидин-2-ил)метил[4-(3-пиперидин-1-илпроп-1-инил)циклогексил]амин],
транс-[(5-бромпиримидин-2-ил)[4-(3-диэтиламинопроп-1-инил)циклогексил]метиламин],
транс-2-[(3-{4-[(6-хлорпиридазин-3-ил)метиламино]циклогексил}проп-2-инил)этиламино]этанол,
транс-2-[(3-{4-[(5-хлорпиримидин-2-ил)метиламино]циклогексил}проп-2-инил)этиламино]этанол,
транс-2-[(3-{4-[(5-бромпиридин-2-ил)метиламино]циклогексил}проп-2-инил)этиламино]этанол,
транс-[4-(3-диметиламинопроп-1-инил)циклогексил]метил(5-пиридин-4-илпиримидин-2-ил)амин,
транс-[4-(3-диметиламинопроп-1-инил)циклогексил]метил(5-тиофен-3-илпиримидин-2-ил)амин,
транс-6-(метил{4-[3-(метилпропиламино)проп-1-инил]циклогексил}амино)-никотинонитрил,
транс-6-{метил[4-(3-пиперидин-1-илпроп-1-инил)циклогексил]амино}никотинонитрил,
транс-6-{[4-(3-диметиламинопроп-1-инил)циклогексил]метиламино}никотинонитрил,
транс-(5-этилпиримидин-2-ил)метил[4-(3-пиперидин-1-илпроп-1-инил)циклогексил]амин,
транс-[4-(3-диметиламинопроп-1-инил)циклогексил](5-этилпиримидин-2-ил)метиламин,
транс-(5-бромпиримидин-2-ил)[4-(4-диметиламинобут-1-инил)циклогексил]метиламин,
транс-(6-хлорпиридазин-3-ил)метил[4-(3-пиперидин-1-илпроп-1-инил)циклогексил]амин,
транс-(5-хлорпиримидин-2-ил)метил[4-(3-пиперидин-1-илпроп-1-инил)циклогексил]амин,
транс-(5-бромпиримидин-2-ил)[4-(4-диметиламинобутил)циклогексил]метиламин,
транс-(5-бромпиримидин-2-ил)[2-(4-диметиламинометилциклогексил)этил]амин,
транс-(5-бромпиримидин-2-ил)метил[4-(4-пиперидин-1-илбут-1-инил)циклогексил]амин,
транс-(2Е)-(5-бромпиримидин-2-ил)[4-(4-диметиламинобут-2-енилокси)циклогексил]метиламин,
транс-(2Е)-(5-бромпиримидин-2-ил)метил[4-(4-пиперидин-1-илбут-2-енилокси)циклогексил]амин,
транс-(6-хлорпиридазин-3-ил)метил[4-(3-пирролидин-1-илпроп-1-инил)циклогексил]амин,
транс-(5-бромпиримидин-2-ил)метил[4-(4-пиперидин-1-илбутил)циклогексил]амин,
транс-(5-бромпиримидин-2-ил)[2-(4-пиперидин-1-илметилциклогексил)этил]амин,
транс-(6-хлорпиридазин-3-ил)метил{4-[3-(метилпропиламино)проп-1-инил]циклогексил}амин,
транс-(6-хлорпиридазин-3-ил)[4-(3-диэтиламинопроп-1-инил)циклогексил]метиламин,
транс-(6-хлорпиридазин-3-ил)[4-(4-диметиламинобут-2-инил)циклогексил]метиламин,
транс-(6-хлорпиридазин-3-ил)метил[4-(4-пиперидин-1-илбут-2-инил)циклогексил]амин,
транс-(5-бромпиримидин-2-ил)[4-(4-диметиламинобут-2-инил)циклогексил]метиламин,
транс-(5-бромпиримидин-2-ил)метил[4-(4-пиперидин-1-илбут-2-инил)циклогексил]амин,
транс-(5-бромпиримидин-2-ил)метил[4-(2-пирролидин-1-илэтокси)циклогексил]амин,
транс-(5-бромпиримидин-2-ил)[2-(4-диметиламинометилциклогексил)этил]метиламин,
транс-(5-бромпиримидин-2-ил)метил[2-(4-пиперидин-1-илметилциклогексил)этил]амин,
транс-[4-(3-диметиламинопроп-1-инил)циклогексил]метил(6-метилпиридазин-3-ил)амин,
транс-2-[этил(3-{4-[метил(6-метилпиридазин-3-ил)амино]циклогексил}проп-2-инил)амино]этанол,
транс-[4-(3-диметиламинопроп-1-инил)циклогексил](6-метоксипиридазин-3-ил)метиламин,
транс-(5-бромпиримидин-2-ил)[4-(3-диметиламинопроп-1-инил)циклогексилметил]метиламин,
транс-2-{[3-(4-{[(5-бромпиримидин-2-ил)метиламино]метил}циклогексил)проп-2-инил]этиламино}этанол,
транс-(5-бромпиримидин-2-ил)метил[4-(3-пиперидин-1-илпроп-1-инил)циклогексилметил]амин,
транс-[4-(3-диметиламинопроп-1-инил)циклогексилметил](5-этилпиримидин-2-ил)метиламин,
транс-2-{этил[3-(4-{[(5-этилпиримидин-2-ил)метиламино]метил}циклогексил)проп-2-инил]амино}этанол,
транс-(5-этилпиримидин-2-ил)метил[4-(3-пиперидин-1-илпроп-1-инил)циклогексилметил]амин,
транс-(6-хлорпиридазин-3-ил)[4-(3-диметиламинопроп-1-инил)циклогексилметил]метиламин,
транс-2-{[3-(4-{[(6-хлорпиридазин-3-ил)метиламино]метил}циклогексил)проп-2-инил]этиламино}этанол,
транс-(6-хлорпиридазин-3-ил)метил[4-(3-пиперидин-1-илпроп-1-инил)циклогексилметил]амин,
транс-2-[(4-{2-[(5-бромпиримидин-2-ил)метиламино]этил}циклогексилметил)этиламино]этанол,
транс-[4-(3-диметиламинопроп-1-инил)циклогексилметил]метил(5-пропилпиримидин-2-ил)амин,
транс-2-{этил[3-(4-{[метил(5-пропилпиримидин-2-ил)амино]метил}циклогексил)проп-2-инил]амино}этанол,
транс-(5-хлорпиримидин-2-ил)[4-(3-диметиламинопроп-1-инил)циклогексилметил]метиламин,
транс-2-{[3-(4-{[(5-хлорпиримидин-2-ил)метиламино]метил}циклогексил)проп-2-инил]этиламино}этанол,
транс-3-[(4-{2-[(5-бромпиримидин-2-ил)метиламино]этил}циклогексилметил)амино]пропан-1-ол и
транс-3-[(4-{2-[(5-бромпиримидин-2-ил)метиламино]этил}циклогексилметил)метиламино]пропан-1-ол
и их фармацевтически приемлемые соли.
Прежде всего предпочтительными соединениями общей формулы (I) являются соединения, выбранные из группы, включающей
транс-(5-бромпиримидин-2-ил)метил(4-[3-(метилпропиламино)проп-1-инил]циклогексил}амин,
транс-(5-бромпиримидин-2-ил)[4-(3-диметиламинопроп-1-инил)циклогексил]метиламин,
транс-(6-хлорпиридазин-3-ил)[4-(3-диметиламинопроп-1-инил)циклогексил]метиламин,
транс-(5-хлорпиримидин-2-ил)[4-(3-диметиламинопроп-1-инил)циклогексил]метиламин,
транс-[(5-бромпиримидин-2-ил)метил[4-(3-пиперидин-1-илпроп-1-инил)циклогексил]амин],
транс-[4-(3-диметиламинопроп-1-инил)циклогексил]метил(5-пиридин-4-илпиримидин-2-ил)амин,
транс-[4-(3-диметиламинопроп-1-инил)циклогексил](5-этилпиримидин-2-ил)метиламин,
транс-(5-бромпиримидин-2-ил)[4-(3-диметиламинопроп-1-инил)циклогексилметил]метиламин,
транс-2-{[3-(4-{[(5-бромпиримидин-2-ил)метиламино]метил}циклогексил)проп-2-инил]этиламино}этанол,
транс-2-{этил[3-(4-{[(5-этилпиримидин-2-ил)метиламино]метил}циклогексил)проп-2-инил]амино}этанол,
транс-(5-этилпиримидин-2-ил)метил[4-(3-пиперидин-1-илпроп-1-инил)циклогексилметил]амин и
транс-2-{[3-(4-{[(6-хлорпиридазин-3-ил)метиламино]метил}циклогексил)проп-2-инил]этиламино}этанол,
и их фармацевтически приемлемые соли.
Соединения формулы (I) могут содержать один или более асимметрических атомов углерода и могут существовать в форме оптически чистых энантиомеров или рацематов. Они могут существовать в форме цис- или транс-изомеров. В объем изобретения включены все указанные формы. Предпочтительны соединения формулы (I) в форме транс-изомеров (в отношении циклогексильного цикла).
Следует отметить, что соединения общей формулы (I) по настоящему изобретению можно модифицировать по функциональной группе с образованием производных, которые способны снова превращаться в исходное соединение in vivo.
Настоящее изобретение относится также к способу получения соединений формулы (I), описанных выше, который включает
а) взаимодействие соединения формулы (II)
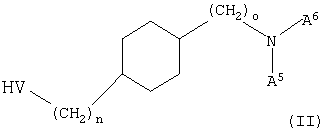
с соединением (А1,А2,U)N-C(A3,А4)-(СН2)m-М, где V означает О или S, М означает мезилат, тозилат, трифлат, Cl, Br или I и U, А1, А2, А3, А4, А5, А6, m, n и о имеют значения, указанные выше, или где HV означает мезилат, тозилат, трифлат, Cl, Br или I, а М означает ОН, SH, или
б) взаимодействие соединения формулы (III)
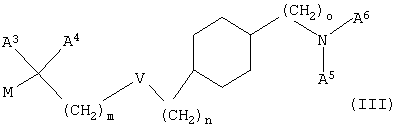
с соединением NHA1А2, где М означает мезилат, тозилат, трифлат, Cl, Br или I, и А1, А2, А3, А4, А5, А6, V, m, n и о имеют значения, указанные выше, и необязательно превращение соединения формулы (I), указанного выше, в фармацевтически приемлемую соль, и
необязательно превращение соединения формулы (I), указанного выше, где U означает свободную пару электронов, в соответствующее соединение, где U означает О.
Взаимодействие соединения формулы (II) с соединением (А1,А2,U)N-С(А3,А4)-(CH2)m-M проводят по известным методикам, показанным на схеме 5, в растворителе, таком как N,N-диметилформамид, N,N-диметилацетамид или нитрометан, в присутствии основания, такого как гидрид натрия или 2,6-ди-трет-бутилпиридин, при температуре, например, от 0 до 80°С. Взаимодействие соединения формулы (III) с соединением NHA1А2 проводят по известным методикам, описаннным в примерах, предпочтительно в растворителях, таких как N,N-диметилацетамид, N,N-диметилформамид или метанол, предпочтительно от комнатной температуры до 80°С. Соединение, указанное выше, можно превратить в фармацевтически приемлемую соль по известной методике, такой как обработка соответствующей кислотой в растворителе, таком как этанол, метанол или дихлорметан, при температуре, например, от -20 до +40°С. Соединение, описанное выше, в котором U означает свободную пару электронов, можно превратить в соединение, в котором U означает О, по известной методике, такой как взаимодействие в смеси аддукта перекись водорода/мочевина и фталевого ангидрида в дихлорметане при комнатной температуре.
Кроме того, изобретение относится к соединениям формулы (I), указанным выше, полученным по способу, описанному выше.
Соединения формулы (I) по настоящему изобретению, указанные выше, можно использовать для лечения и/или профилактики заболеваний, ассоциированных с OSC, таких как гиперхолестеринемия, гиперлипемия, артериосклероз, сосудистые заболевания, микозы, паразитарные инфекции и желчнокаменная болезнь, и/или для лечения и/или профилактики нарушенной толерантности к глюкозе, диабета, опухолей и/или гиперпролиферативных заболеваний, предпочтительно для лечения и/или профилактики гиперхолестеринемии и/или гиперлипемии. Гиперпролиферативные заболевания прежде всего означают гиперпролиферативные заболевания кожи и сосудистые нарушения.
Кроме того, настоящее изобретение относится к фармацевтическим композициям, включающим соединение, указанное выше, и фармацевтически приемлемый носитель и/или адьювант.
Кроме того, изобретение относится к соединениям, указанным выше, для применения в качестве терапевтически активных соединений, прежде всего терапевтически активных соединений для лечения и/или профилактики заболеваний, ассоциировнных с OSC, таких как гиперхолестеринемия, гиперлипемия, артериосклероз, сосудистые заболевания, микозы, паразитарные инфекции, желчнокаменная болезнь, опухоли и/или гиперпролиферативные заболевания и/или для лечения и/или профилактики нарушенной толерантности к глюкозе и диабета, предпочтительно для лечения и/или профилактики гиперхолестеринемии и/или гиперлипемии.
В другом варианте изобретение относится к способу лечения и/или профилактики заболеваний, ассоциированных с OSC, таких как гиперхолестеринемия, гиперлипемия, артериосклероз, сосудистые заболевания, микозы, паразитарные инфекции, желчнокаменная болезнь, опухоли и/или гиперпролиферативные заболевания и/или для лечения и/или профилактики нарушенной толерантности к глюкозе и диабета, предпочтительно для лечения и/или профилактики гиперхолестеринемии и/или гиперлипемии, причем способ включает введение соединения, указанного выше, человеку или животному.
Кроме того, изобретение относится к применению соединений, указанных выше, для лечения и/или профилактики заболеваний, ассоциированных с OSC, таких как гиперхолестеринемия, гиперлипемия, артериосклероз, сосудистые заболевания, микозы, паразитарные инфекции, желчнокаменная болезнь, опухоли и/или гиперпролиферативные заболевания и/или для лечения и/или профилактики нарушенной толерантности к глюкозе и диабета, предпочтительно для лечения и/или профилактики гиперхолестеринемии и/или гиперлипемии.
Изобретение относится также к применению соединений, указанных выше, для получения лекарственных средств, предназначенных для лечения и/или профилактики заболеваний, ассоциированных с OSC, таких как гиперхолестеринемия, гиперлипемия, артериосклероз, сосудистые заболевания, микозы, паразитарные инфекции, желчнокаменная болезнь, опухоли и/или гиперпролиферативные заболевания и/или для лечения и/или профилактики нарушенной толерантности к глюкозе и диабета, предпочтительно для лечения и/или профилактики гиперхолестеринемии и/или гиперлипемии. Такие лекарственные средства включают соединение, указанное выше.
Соединения формулы (I) получают по методам, указанным ниже, по методам, описанным в примерах, или аналогичными методами. Соответствующие условия реакций для каждой конкретной стадии синтеза известны для специалистов в данной области техники. Исходные материалы являются коммерческими препаратами или их можно получить по методам, указанным ниже, по методам, описанным в примерах, или известными методами.
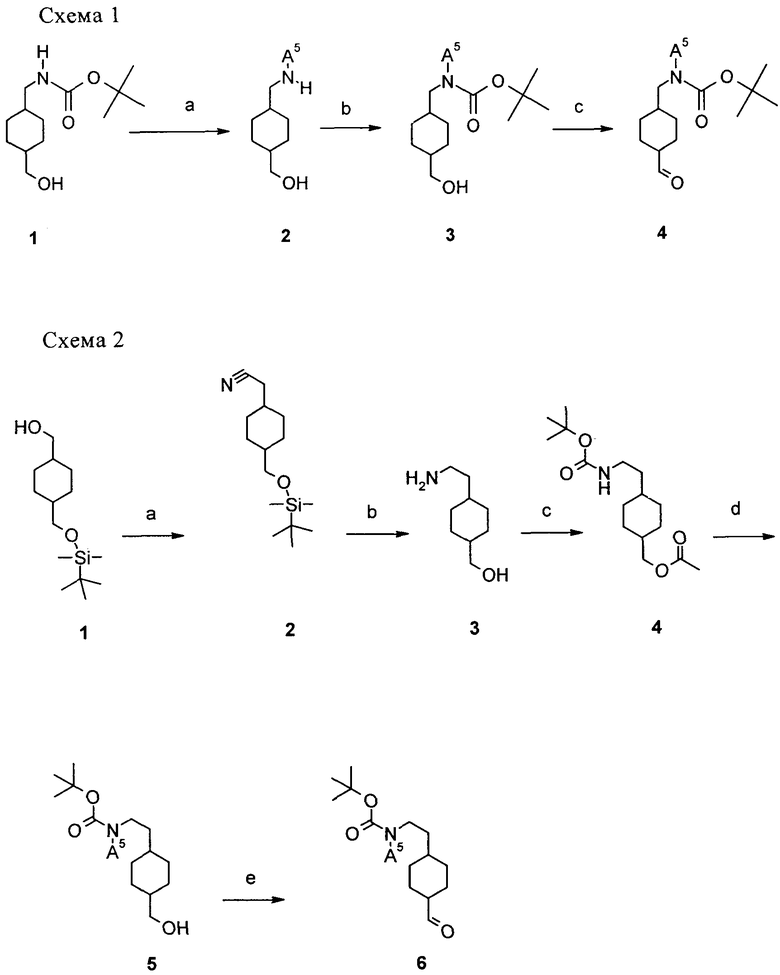
На схемах 1-4 показан синтез промежуточных соединений. цис- или транс-(4-Метиламинометилциклогексил)метанол 2 (А5 означает Me) получаютт из трет-бутилового эфира цис- или транс-(4-гидроксиметилциклогексилметил)карбаминовой кислоты 1 (US 5843973 (1998) или US 6022969 А (2000)) при обработке литийалюминийгидридом в тетрагидрофуране при температуре от комнатной температуры до температуры кипения тетрагидрофурана (стадия а). Затем при взаимодействии с ди-трет-бутилдикарбонатом в метаноле/триэтиламине при температуре от -10°С до комнатной температуры вводят трет-бутоксикарбонильную защитную группу, при этом получают соединение 3 (А5 означает Me) (стадия б). В соединение 1 можно сначала ввести O-защитную группу, а затем ввести заместители А5 по реакции N-алкилирования аминогруппы, защищенной трет-бутоксикарбонильной группой, с использованием алкил- или алкенилгалогенида в присутствии основания, такого как гидрид натрия в растворителе, таком как N,N-диметилформамид или ацетонитрил, при температуре от комнатной температуры до 80°С. Затем можно удалить O-защитную группу и получить соединение 3. Соединение 3 затем окисляют в соответствующий альдегид 4 в условиях Сверна: оксалилхлорид/диметилсульфоксид/триэтиламин в дихлорметане при температуре от -78°С до комнатной температуры (стадия в).
Схема 2
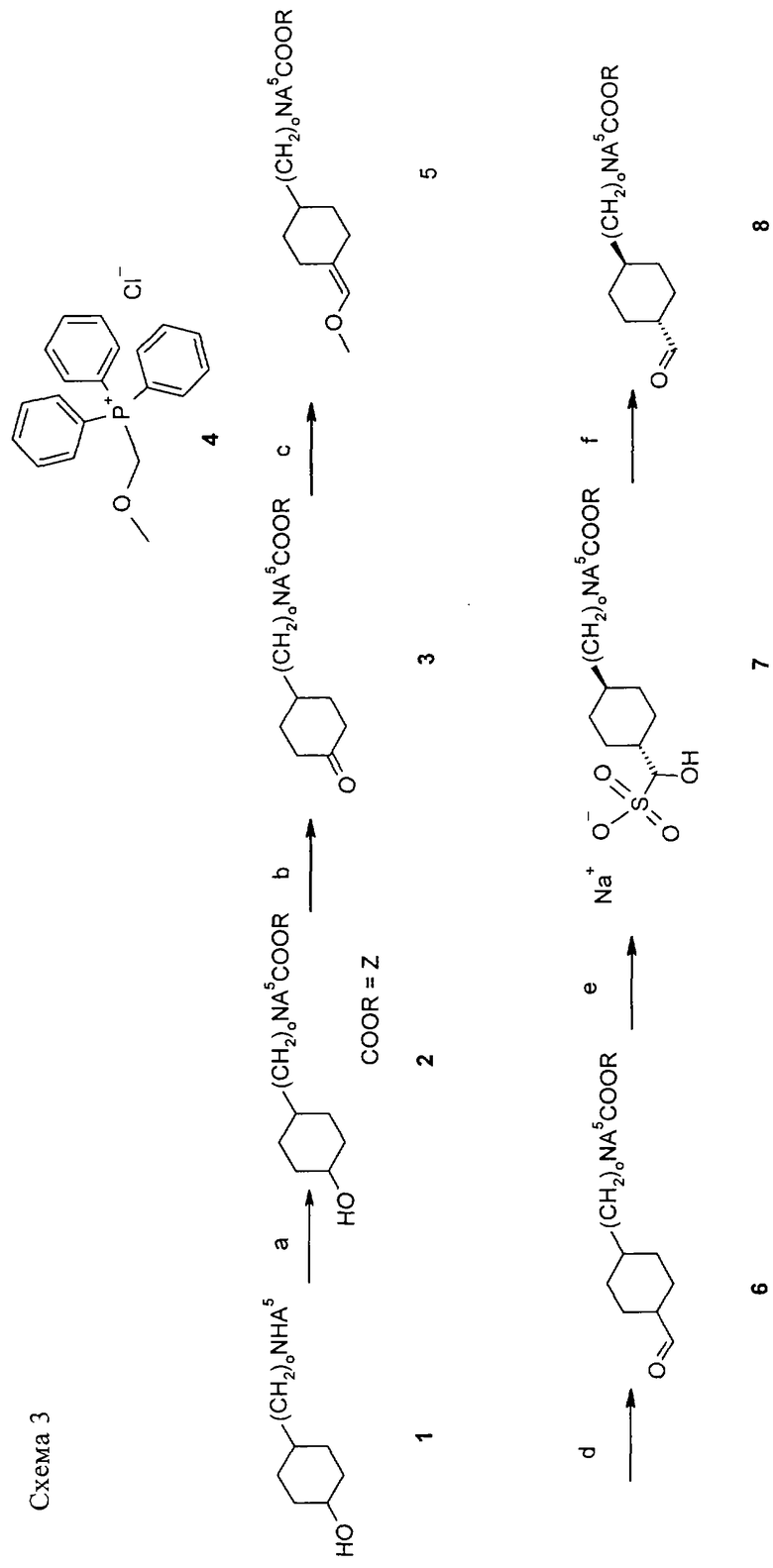
цис- или транс-[4-(трет-Бутилдиметилсиланилоксиметил)циклогексил]метанол 1 получают из соответствующих производных бис-гидроксиметилциклогексана при взаимодействии с одним эквивалентом н-бутиллития в тетрагидрофуране при -78°С с последующей обработкой одним эквивалентом трет-бутилдиметилхлорсилана при температуре от -65°С до комнатной температуры. При мезилировании [4-(трет-бутилдиметилсиланилоксиметил)циклогексил]метанола 1 (метансульфонилхлорид в дихлорметане и триэтиламине при 0-10°С) получают соответствующий метансульфонат, который обрабатывают цианидом натрия в N,N-диметилформамиде при 80°С, при этом получают цианопроизводное 2 (стадия а). Прямое восстановление цианопроизводного 2, например, гидрированием в присутствии платинового катализатора в кислом метаноле, приводит к образованию первичного O-деблокированного амина 3 (стадия б). При взаимодействии аминоспирта 3 сначала с ди-трет-бутилдикарбонатом в дихлорметане в присутствии триэтиламина, а затем с уксусным ангидридом и пиридином в дихлорметане получают дизащищенное соединение 4 (стадия в). Для введения заместителей А5 соединение 4 можно N-алкилировать по первичной аминогруппе, защищенной трет-бутоксикарбонильной группой, по реакции с алкилгалогенидом в присутствии основания, такого, как гидрид натрия, в растворителе, таком, как N,N-диметилформамид или ацетонитрил, при температуре от комнатной температуры до 80°С. При этом после отщепления ацетатной группы в присутствии основания получают гидроксисоединение 5 (стадия г). Затем первичное гидроксисоединение 5 можно окислить до соответствующего альдегида 6 в условиях Сверна: оксалилхлорид/диметилсульфоксид/триэтиламин в дихлорметане при температуре от -78°С до комнатной температуры (стадия д).
Схема 3
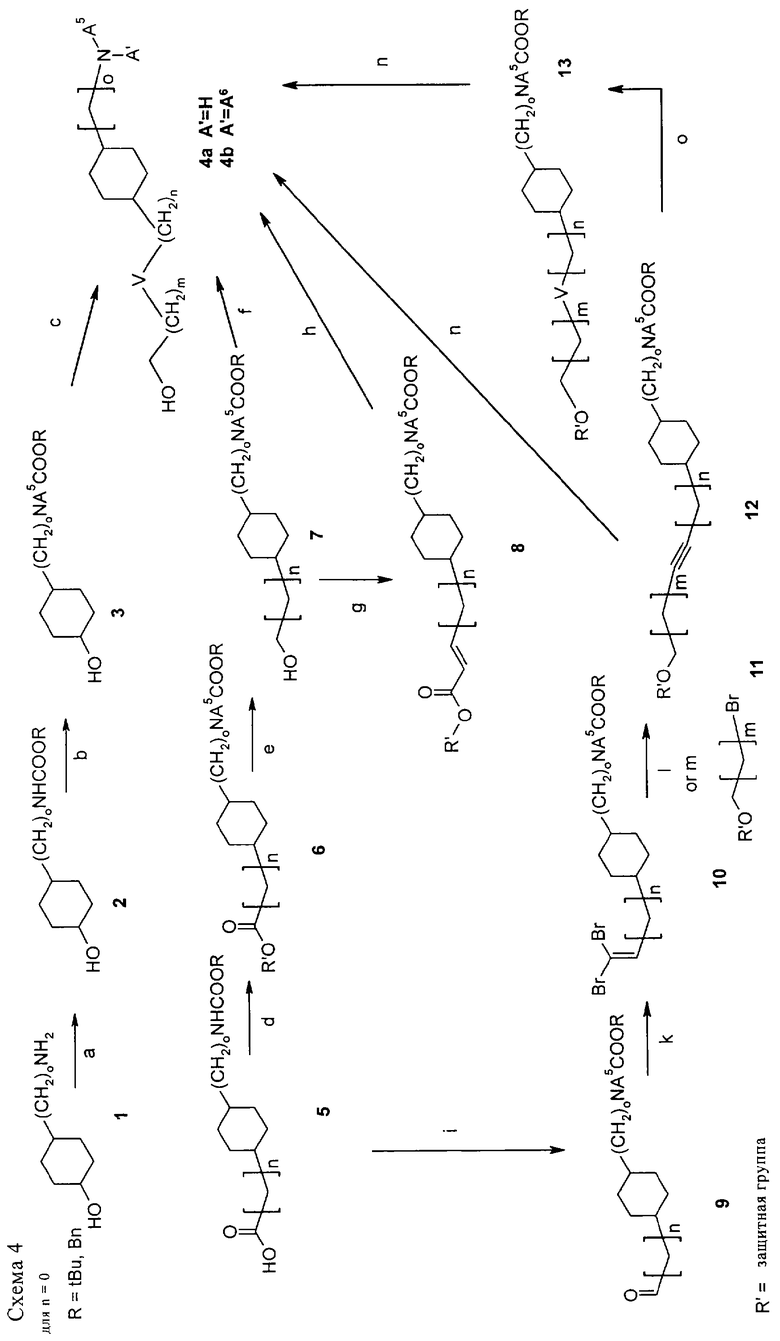
На схеме 3 показан синтез чистого транс-альдегида (структурного фрагмента 8). Необязательно замещенный группой А5 циклогексанол 1 синтезируют гидрированием соответствующих 4-аминофенола, 4-гидроксибензиламина или тирамина. Из амина 1 получают N-защищенное производное 2 (например, ZCl, Na2CO3/ТГФ/H2O) (стадия а). После окисления с использованием TEMPO (2,2,6,6-тетраметилпиперидин-1-оксил, радикал) и гипохлорита натрия получают кетон 3 (стадия б). При взаимодействии по реакции Виттига с хлоридом (метоксиметил)трифенилфосфония 4 в ТГФ и трет-бутоксидом калия, использованном в качестве основания, получают енолэфир 5 (стадия в). На этой стадии можно ввести заместители А5 (с использованием А5-галогенида/NaH в ДМФА или ДМА). После гидролиза енолэфира 5 в 1 н. HCl в ТГФ при кипячении с обратным холодильником (стадия г) получают альдегид 6. Неочищенный альдегид 6 (в виде смеси цис/транс-изомеров) подвергают изомеризации с использованием аддукта с бисульфитом 7 (с использованием динатрийпиросульфита в воде/ТВМЕ, стадия д). Аддукт с бисульфитом 7 можно превратить в чистый транс-альдегид 8 в присутствии водного Na2СО3 в воде/ТВМЕ (стадия е).
Схема 4
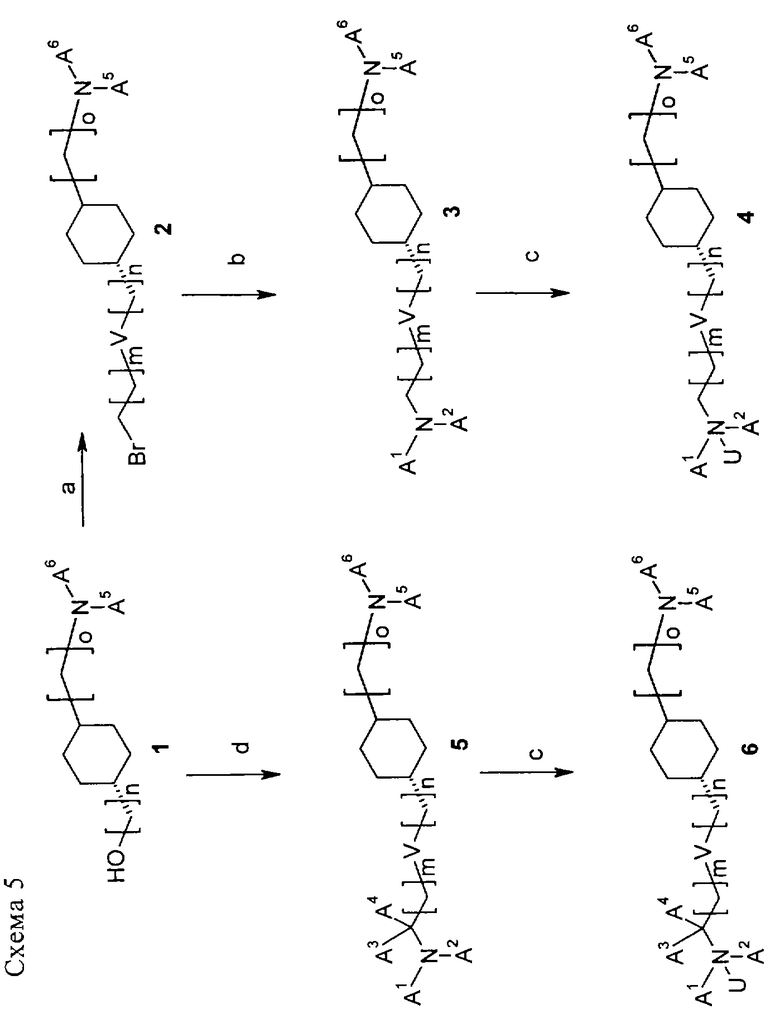
На схеме 4 показано получение исходных соединений для синтеза циклогексилпроизводных формулы (I), где V означает простую связь. О, S, -СН=СН-СН2-O-, -СН=СН- или -С≡С-. Если n равно 0, в качестве исходного соединения используют циклогексанол 1, который превращают в Z-производное или ВОС-производное 2, например, ZCl, Na2СО3, ТГФ, Н2O или (BOC)2O, iPrOH, CH2Cl2 соответственно (стадия а). Необязательно необходимую группу А5 можно ввести двумя способами. При восстановлении литийалюминийгидридом получают метиламинопроизводное, которое, например, защищают ВОС-группой и получают соединение 3. В соединение 2 можно сначала ввести O-защитную группу, а затем для введения заместителей А5 провести N-алкилирование по аминогруппе, защищенной трет-бутоксикарбонильной группой, по реакции с А5-галогенидом в присутствии основания, такого, как гидрид натрия, в растворителе, таком, как N,N-диметилформамид или ацетонитрил, при температуре от комнатной температуры до 80°С. После удаления O-защитной группы получают соединение 3 (стадия б), которое затем превращают в требуемое А6-производное 4b (стадия в).
Реакцию на стадии в можно проводить двумя способами.
Первый способ
При необходимости после введения мостиковой группы НОСН2(СН2)mV (V означает О или СН=СНСН2O) в условиях фазового переноса (например, α,ω-дигалоидалканы или α,ω-дигалоидалкены, NaOH, nBu4NHSO4) получают соответствующий галогенид, который гидролизуют с образованием спирта (например, в водном растворе NaOH в ТГФ или ДМА). В другом варианте защищенную группой R" мостиковую группу R"OCH2(CH2)mV вводят при образовании in situ R"ОСН2(СН2)mО-трифтата (из соответствующего R"O-алканола в присутствии трифторметансульфонового ангидрида/2,6-ди-трет-бутилпиридина в СН2Cl2 при 0°С). Затем указанный трифлат вводят в реакцию со спиртом 3 в присутствии 2,6-ди-трет-бутилпиридина, использованного в качестве основания, в нитрометане при температуре от КТ до 60°С, при этом получают продукт 3 с удлиненной цепью R"OCH2(CH2)mV (по методике, описанной Belostotskii Anatoly M., Hassner Alfred, Synthetic methods, 41, Etherification of hydroxysteroids via Inflates, Tetrahedron Lett. 35 (28), 5075-6 (1994)). В указанном продукте 3 с удлиненной цепью R"ОСН2(СН2)mV полностью удаляют защитные О- и N-группы (например, если R" означает Bzl, в присутствии Pd/C и Н2 в EtOH или МеОН/АсОН и в случае NA5COOtBu в присутствии ТФУ в CH2Cl2, при этом получают продукт 4а).
Второй способ
Введение гетероарильной группы А6 с образованием продукта 4b можно проводить в различных условиях. Метод А: взаимодействие соединения 4а с 2-галогенгетероарилом/N-этилдиизопропиламином в течение от 1 ч до 5 сут при температуре от 80 до 120°С в ДМА или в отсутствии растворителя. Метод Б (для менее реакционноспособных соединений): взаимодействие соединения 4а с 2-галогенгетероарилом/N-этилдиизопропиламином/CuI или NaI в течение 1-10 ч при 120°С или нагревание в микроволновой печи в течение от 0,5 до 6 ч при 120-150°С в ДМА.
Если n равно 0, в качестве исходного материала используют циклогексанкарбоновую кислоту 5, которая является коммерческим препаратом или ее можно синтезировать, например, при окислении альдегида 6 по схеме 3. Кислоту 5 превращают в производное 6 при образовании сложного эфира (например, карбонилдиимидазол в метаноле/ТФУ) или необязательно при А5-алкилировании с использованием гидрида натрия и реакционноспособного алкил- или алкенилпроизводного (стадия г). При восстановлении литийалюминийгидридом получают N-защищенный спирт 7, который можно превратить в соединение 4b (стадия е), как описано для соединений 3-4b.
Если n равно 1, в качестве исходного материала используют циклогексилуксусную кислоту 5 (которую можно получить из 4-нитрофенилуксусной кислоты по методике, описанной Karpavichyus K.I., Palaima A.I., Knunyants I.L., BACCAT, Bull. Acad. Sci. USSR Div. Chem. Sci. (Engl. TransL), EN, 29, 1689-1694 (1980); IASKA6, Izv. Akad. Nauk SSSR Ser. Khim, RU, 10, 2374-2379 (1980), или T.P.Johnston и др., Journal of Medicinal Chemistry, т. №2, 279-290 (1977)), которую можно затем превратить с соответствующий спирт по методике, описанной для соединений 5-4b. В другом варианте циклогексилуксусную кислоту 5 можно синтезировать (например, из кетона 3, как показано на схеме 3, методом С2-удлинения цепи по реакции Хорнера-Эммонса в присутствии триэтилфосфоноацетата и алкоголята натрия) и введения защитных групп, как описано ранее.
Если n равно 2 или более, в качестве исходного материала используют циклогексанкарбоновую кислоту 5. Удлинение цепи кислоты (n>1) проводят по известным методикам или как описано ниже.
С2-удлинение цепи проводят с использованием окисления спирта 7 по Сверну до соответствующего альдегида с последующим взаимодействием по реакции Хорнера-Эммонса с триэтилфосфоноацетатом и алкоголятом натрия в спирте, при этом получают ненасыщенный сложный эфир 8 (стадия ж). Полученный эфир подвергают гидрированию в присутствии 10% палладия на угле в метаноле и восстановлению литийалюминийгидридом в ТГФ, при этом получают спирт с удлиненной цепью, который можно превратить в соединение 4b (стадия з), как описано для соединений 3-4b. При необходимости дальнейшего С2-удлинения цепи в полученных соединениях трансформацию 7→8 можно повторить.
Для С(m)-удлинения цепи можно использовать методику Корей-Фухса. Из кислоты 5 получают производное Вейнреба при обработке гидрохлоридом N,O-диметилгидроксиламина в присутствии EDCI и НОВТ в СН2Cl2 при комнатной температуре с последующим А5-алкилированием (А5-галогенид в присутствии NaH в ДМФ или ДМА при температуре от 0°С до КТ) и восстановлением литийалюминийгидридом до соответствующего альдегида 9 (стадия и). Указанный альдегид 9 можно обработать трифенилфосфином, тетрабромметаном и триэтиламином в CH2Cl2 при температуре от 0°С до КТ, при этом получают 2,2-дибромвинилпроизводное 10. Перегруппировка в присутствии n-BuLi (приблизительно 1,6 М в гексане) в ТГФ при -78°С с последующим взаимодействием с формальдегидом (при температуре от -78°С до КТ) позволяет получить пропаргиловый спирт 12 (стадия 1, в условиях, описанных в книге Marshall James A., Bartley Gary S., Wallace Eli M., Total Synthesis of the Pseudopterane (-)-Kallolide B, the Enantiomer of Natural (+)-Kallolide B, J.Org. Chem., 61 (17), 5729-5735 (1996), и Baker Raymond, Boyes Alastair L., Swain Christopher J., Synthesis of talaromycins А, В, С and E, J.Chem. Soc., Perkin Trans. 1 (5), 1415-21 (1990)). Для получения соединений с более длинной цепью перегруппировку проводят в присутствии n-BuLi (приблизительно 1,6 М в гексане) в ТГФ при -78°С, как описано выше, с последующим добавлением сорастворителей, таких как DMPU, и взаимодействием с O-защищенными 1-бромспиртами 11 (стадия н), при этом получают O-защищенные соединения 12.
После удаления O-защитных групп (при необходимости) и N-защитных групп в соединении 12 и последующего взаимодействия с 2-галогенгетероарилом, как описано выше (стадия о) получают соединение 4b (V означает -С≡С-). Если V означает -СН=СН- или простую связь, после гидрирования соединения 12, например, в присутствии никеля Ренея, 10% Pd/C или PtO2·Н2О/Н2 (стадия п), и взаимодействия соединения 13 с 2-галогенгетероарилом, как описано выше (стадия о), получают производные 4b.
И наконец, введение заместителя А6 в продукт 4b можно осуществлять, например, по реакции Сузуки, если А6 означает галогенгетероарил, или нуклеофильным замещением, например, если А6 означает 6-хлорпиридазин, и после взаимодействия с алгоголятом натрия в ДМА при 80°С получить алкоксизамещенное соединение.
Схема 5
На схеме 5 показан синтез производных простых эфиров формулы (I) (V означает О или S). Для получения производных, в которых n равно 0, производное циклогексанола 1 (синтез см. на схемах 1-4) вводят в реакцию в условиях фазового переноса, например, с α,ω-дигалоидалканами или α,ω-дигалоидалкенами, NaOH, nBu4NHSO4, при этом получают бромид 2. Если n>0, производное спирта 1 можно ввести в реакцию с α,ω-дигалоидалканом (алкан содержит 4 или более атомов углерода) в присутствии NaH в ДМФА при температуре от 0°С до КТ, при этом получают бромид 2. При использовании более коротких алканов выбирают метод образования in situ галоидалкантрифлата (из соответствующего галоидалканола в присутствии трифторметансульфонового ангидрида/2,6-ди-трет-бутилпиридина в CH2Cl2 при 0°С). Затем полученный галоидалкантрифлат взаимодействует со спиртом 1 в присутствии 2,6-ди-трет-бутилпиридина, использованного в качестве основания, в нитрометане при температуре от КТ до 60°С, при этом получают бромид 2 (по методике, описанной Belostotskii Anatoly M., Hassner Alfred, Synthetic methods, 41, Etherification of hydroxysteroids via triflates, Tetrahedron Lett. 35 (28), 5075-6 (1994)).
После аминирования бромида 2 амином A1A2NH в ДМА или ДМФА при КТ или в МеОН при температуре от КТ до температуры кипения растворителя получают конечный амин 3, при этом необязательно можно добавить DBU и NaI. В случае, если А1 или А2 означает Н, второй заместитель можно вводить на второй стадии, например, при N-метилировании в присутствии NaH2РО3/формальдегида. Амин 3 можно превратить в соль или в N-оксид 4 с использованием смеси аддукта пероксид водорода/мочевина и фталевого ангидрида в CH2Cl2 при КТ.
В другом варианте спирт 1 можно превратить в амин 5 присоединением предварительно полученного фрагмента A1A2NC(A3A4)(CH2)m-VH (V означает О и S), который можно синтезировать известными методами, к мезилату/галогениду производного 1 в условиях алкилирования (стадия г). В другом варианте можно также получить мезилат/галогенат фрагмента A1A2NC(A3A4)(СН2)m-ОН и провести реакцию с производным 1 в условиях алкилирования (стадия г). Амин 5 можно превратить в соль или в N-оксид 6, как описано выше (стадия в).
И наконец, заместители А6 в продукте 5 можно модифицировать, например, с помощью гидролиза N-ацетилгруппы с образованием NH2-группы или по реакции Сузуки, если А6 означает галогенгетероарил, или с использованием нуклеофильного замещения, например, если А6 означает 6-хлорпиридазин, по реакции с алкоголятом натрия в ДМА при 80°С, при этом получают алкоксизамещенное соединение.
Кроме того, положение заместителей А1 или А2 можно модифицировать, например, при обработке гидроксиэтиламина реактивом DAST.
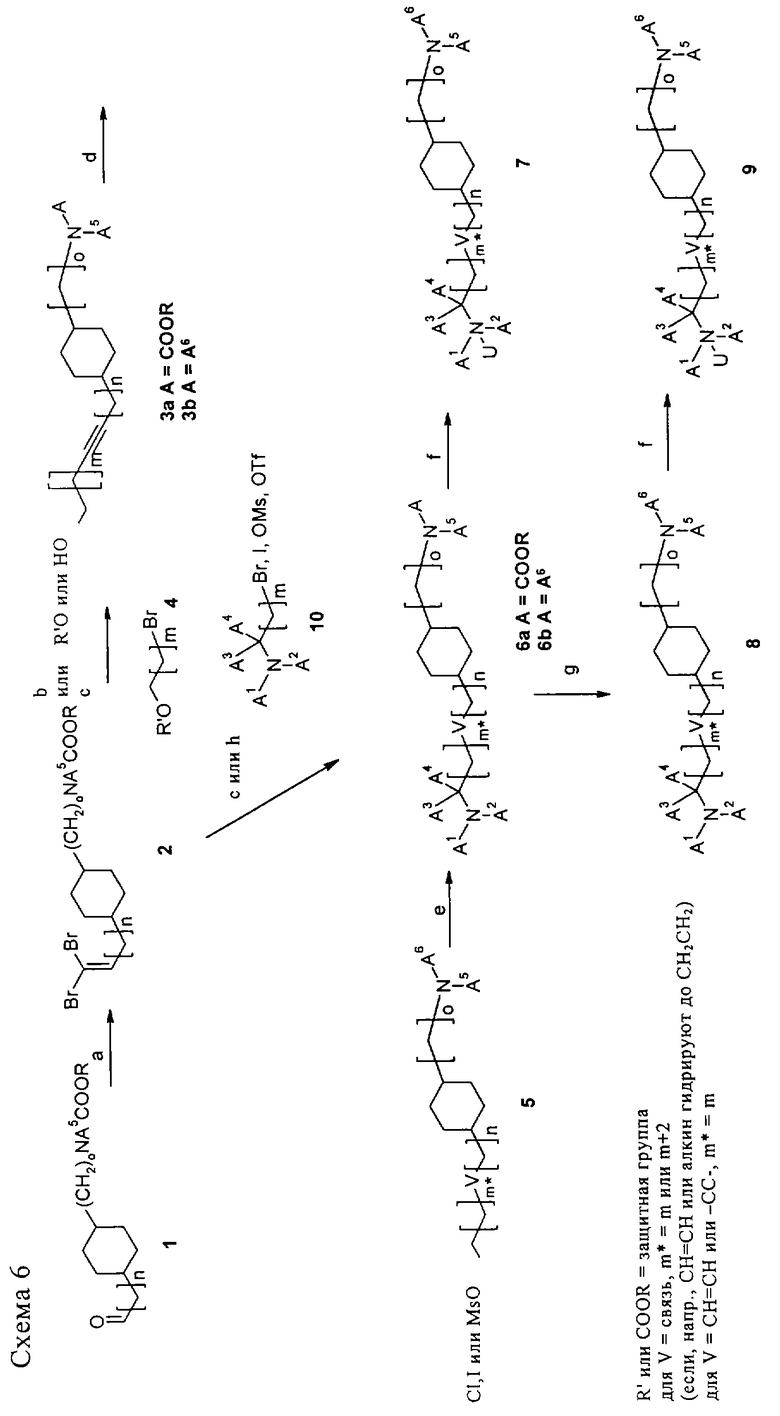
Схема 6
На схеме 6 показан синтез С-аналогов циклогексанов общей формулы I, где V означает простую связь, -СН=СН- или -С≡С-. В качестве исходного соединения для синтеза используют альдегид 1, показанный на схемах 1-4. Наращивание боковой цепи осуществляют с использованием метода Корея-Фухса. Альдегид 1 обрабатывают трифенилфосфином, тетрабромметаном и триэтиламином в CH2Cl2 при температуре от 0°С до КТ, при этом получают 2,2-дибромвинилпроизводное 2. После перегруппировки в присутствии n-BuLi (приблизительно 1,6 М в гексане) в ТГФ при -78°С с последующим взаимодействием с формальдегидом (от -78°С до КТ, стадия б) получают пропаргиловый спирт 3а (условия реакции см. в книге Marshall James A., Bartley Gary S., Wallace Eli M., Total Synthesis of the Pseudopterane (-)-Kallolide B, the Enantiomer of Natural (+)-Kallolide B, J.Org. Chem., 61 (17), 5729-5735 (1996), и Baker Raymond, Boyes Alastair L., Swain Christopher J., Synthesis of talaromycins А, В, С and E, J.Chem. Soc., Perkin Trans, 1 (5), 1415-21 (1990)). После удаления ВОС-защитных групп (ТФУ, CH2Cl2) с последующей обработкой А6-гетероарилом, как описано выше (схема 4) получают соединения формулы 3b.
Для получения соединений с более длинной боковой цепью проводят перегруппировку дибромалкена 2 в присутствии n-BuLi (приблизительно 1,6 М в гексане) в ТГФ при -78°С, как описано выше, а затем добавляют сорастворители, такие как DMPU, и проводят реакцию с O-защищенными 1-бромспиртами 4, при этом получают O-защищенные соединения 3а, в которых можно удалить защитные группы в МеОН при 50-60°С в присутствии каталитического количества толуол-4-сульфоната пиридиния с образованием соответствующего производного алкинола 3а. После удаления ВОС-защитных групп (ТФУ, CH2Cl2) с последующей обработкой А6-гетероарилом, как описано выше (схема 4), получают соединения формулы 3b (стадия в).
После мезилирования спирта 3b метансульфонилхлоридом, пиридином или лутидином в присутствии или отсутствие DMAP в CH2Cl2 при температуре от 0°С до КТ получают производное мезилата/хлорида или пиридиния 5, которое можно превратить в амин 6b в следующих условиях: в ДМА или МеОН при КТ или при 50-70°С в присутствии избытка соответствующего амина NHA1А2 (стадия д). В случае, если А1 или А2 означает Н, второй заместитель можно вводить на второй стадии, например, с использованием N-метилирования в присутствии NaH2PO3/формальдегида.
Соединения 6b, в которых А3 и/или А4 не означают Н, а М>0, можно получить при взаимодействии соединений 2 с соединениями 10 в условиях, описанных на стадии в. Структурный фрагмент 10 можно получить известными методами.
Для введения группы (А1,А2)N-C(A3,А4)-, где А3 и/или А4 не означают Н, а m равно 0, используют двустадийный метод: сначала проводят перегруппировку дибромида 2 в присутствии n-BuLi (приблизительно 1,6 М в гексане) в ТГФ при -78°С с последующим взаимодействием с соответствующим альдегидом (А3 или А4-СОН) или кетоном (А3СОА4, при температуре от -78°С до КТ), при этом получают А3,А4-замещенный пропаргиловый спирт, который можно превратить в фосфоэфир (см. Bartlett Paul A., McQuaid Loretta A., Total synthesis of (±)-methyl shikimate and (±)-3-phosphoshikimic acid, J.Am. Chem. Soc. 106 (25), 7854-60 (1984)). Затем полученный спирт вводят в реакцию с требуемым (А1,А2)-амином в присутствии тетракис(трифенилфосфин)палладия в ТГФ, при этом получают требуемое А3,А4-замещенное соединение 6а (стадия з). После удаления ВОС-защитных групп (ТФУ, СН2Cl2) с последующей обработкой А6-гетероарилом, как описано выше (схема 4) получают соединения формулы 6b. Соединения, в которых V означает простую связь или -СН=СН-, можно получить гидрированием соединения 6b в присутствии PtO2·Н2О/Н2 (получают насыщенный аналог 8) или гидрированием другими известными методами (например, в присутствии никеля Ренея получают аналог 8 с двойной связью). В другом варианте алкиленовая группа может быть уже восстановлена на предыдущей стадии, например, спирт 3а (например, при восстановлении с использованием LAH, если m равно 0, получают V, который означает транс-СН=СН-, или при гидрировании в присутствии Pt/C или PtO2·Н2О получают V, который означает CH2CH2- (простая связь соответственно), затем полученное соединение можно превратить в конечные соединения 8 и/или 9.
И наконец, заместители А6 в продукте 6b или 8 можно модифицировать, например, с использованием гидролиза N-ацетилгруппы с образованием NH2-группы, или по реакции Сузуки, если А6 означает галогенгетероарил, или с использованием нуклеофильного замещения, например, если А6 означает 6-хлорпиридазин, по реакции с алкоголятом натрия в ДМА при 80°С, при этом получают алкоксизамещенное соединение.
Кроме того, заместители А1 или А2 можно модифицировать, например, при обработке гидроксиэтиламина реактивом DAST. В случае, если А1 или А2 означает Н, второй заместитель можно вводить на второй стадии, например, с использованием N-метилирования в присутствии NaH2РО3/формальдегида.
Амины 6b и 8 можно превратить в соль или, как описано на стадии е, в N-оксид 7 и 9 соответственно с использованием смеси аддукта пероксид водорода/мочевина и фталевого альдегида в CH2Cl2 при КТ.
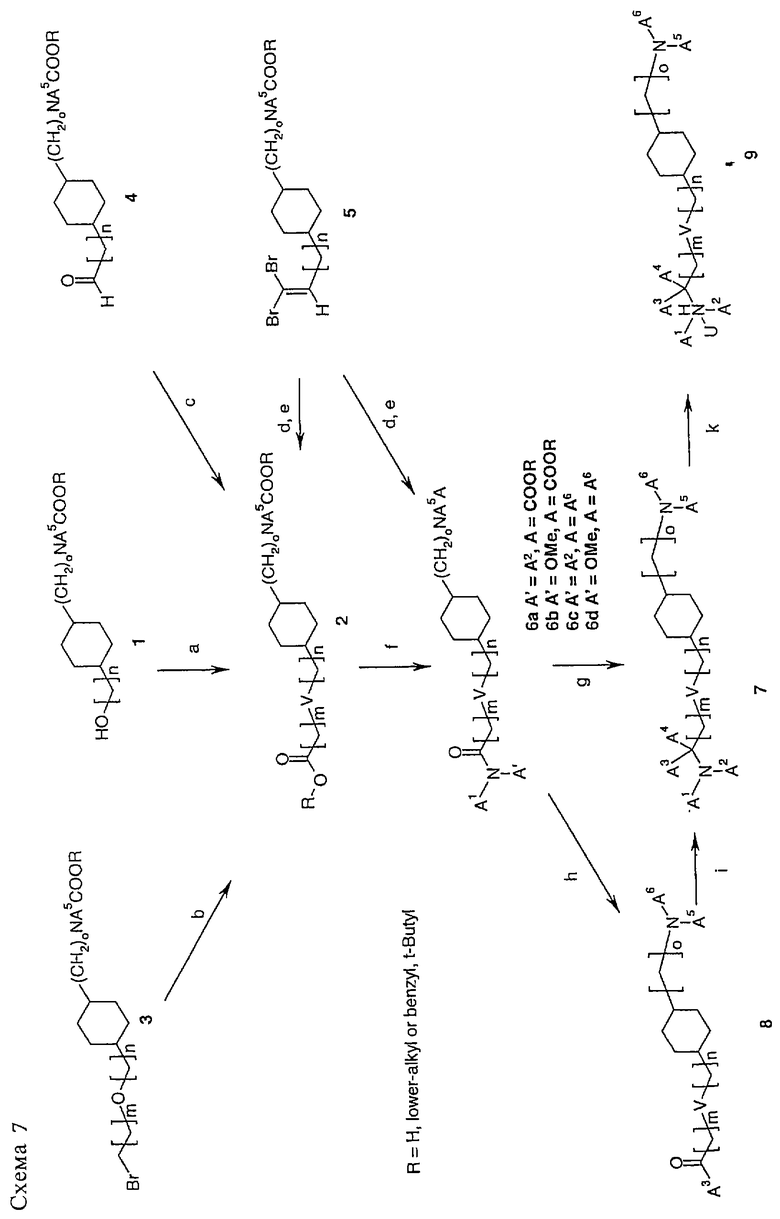
Схема 7
Другой возможный подход к введению замещенной боковой цепи показан на схеме 7. Для синтеза основного промежуточного производного 2 в качестве исходной стадии используют присоединение эфира ω-гидроксиалкилкарбоновой кислоты к спирту 1 через образование in situ трифлата аналогично тому, как описано Belostotskii Anatoly M., Hassner Alfred, Synthetic methods. 41, Etherification of hydroxysteroids via triflates, Tetrahedron Lett. 35 (28), 5075-6 (1994) (стадия а). В другом варианте сложный эфир 2 можно получить из бромида 3 (синтез показан на схеме 5) при обработке, например, ацетоциангидрином в ацетонитриле с последующим взаимодействием по реакции Пиннера и гидролизом имидата с образованием соответствующего сложного эфира (стадия б).
Если V означает СН=СН, сложный эфир 2 или соответствующую кислоту можно получить из альдегида 4 (синтез показан на схемах 1-4) при обработке соответствующим реагентом Виттига Ph3Р(СН2)m+1CO2R/Н. Если V означает связь, после гидрирования продукта Виттига в стандартных условиях получают насыщенный продукт 2.
Если V означает -С≡С-, сложный эфир 2 или амид 6а можно получить из дибромпроизводного 5 (синтез показан на схеме 4) с помощью перегруппировки в присутствии n-BuLi (приблизительно 1,6 М в гексане) в ТГФ при -78°С с последующим взаимодействием с хлорформиатом (с образованием эфира 2) или диалкилкарбамоилхлоридом (с образованием амида 6а) (при температуре от -78°С до КТ, стадия г). Для введения более длинной боковой цепи перегруппировку дибромалкена 5 проводят в присутствии n-BuLi (приблизительно 1,6 М в гексане) в ТГФ при -78°С, как описано выше, а затем добавляют сорастворители, такие как DMPU, и проводят реакцию с пригодным защищенным 1-бромалкилспиртом Br-(CH2)mCH2OH с последующим окислением, при этом получают соединение 2 в виде кислоты (стадия д).
Амид 6а или 6b получают после омыления эфира 2 в стандартных условиях, например, LiOH в EtOH, МеОН или ТГФ с последующей обработкой NHA1А2 или NHA1A', EDCI, НОВТ или основанием, таким как основание Хюнига, NEt3, NMM в CH2Cl2, ДМФА, ДМА или диоксане. После удаления N-защитных групп в соединениях 6а или 6b и последующего взаимодействия с 2-галогенгетероарилом, как показано выше на схеме 4, получают производные 6с и 6d.
Амид 6с можно превратить в амин 7 (А3, А4 означают Me) при взаимодействии с бромидом метилмагния, ZrCl4 в ТГФ при низкой температуре (см. Stephen M. Denton, Anthony Wood, A Modified Bouveault Reaction for the Preparation of α,α-dimethylammes from Amides, Synlett 1, 55-56 (1999)) или при обработке другими реагентами Гриньяра в присутствии ZrCl4 или Ti(OiPr)4 (см. V.Chalinski, A. de Meijere, A versatile New Preparation of Cyclopropylamines from acid dialkylamides, Angew. Chem. Int. Ed. Engl. 35, №4, 413-4 (1996)).
В случае, если А1 означает Me, А' означает ОМе, амид 6d можно обработать реагентом Гриньяра A3MgX с образованием соответствующего кетона 8. Амин 7 получают восстановительным алкилированием кетона 8 при обработке NHA1А2 в присутствии тетраизопропилортотитаната с последующим восстановлением реагентом NaCNBH3 в этаноле (см. R.J.Mattson, K.M.Pham, D.J.Leuck, K.A.Cowen, J.О.С., 55, 2552-4 (1990)).
И наконец, заместители А6 в продукте 7 можно модифицировать, например, с использованием гидролиза N-ацетилгруппы с образованием NH2-группы, или по реакции Сузуки, если А6 означает галогенгетероарил, или с использованием нуклеофильного замещения, например, если А6 означает 6-хлорпиридазин, при взаимодействии с алкоголятом натрия в ДМА при 80°С, при этом получают алкоксизамещенное соединение.
Кроме того, заместители А1 и А2 можно модифицировать, например, при обработке гидроксиэтиламина реактивом DAST. В случае, если А1 или А2 означает Н, второй заместитель можно вводить на второй стадии, например, с использованием N-метилирования в присутствии NaH2PO3/формальдегида.
Амины 7 можно превратить в соль или в N-оксид 9 с использованием смеси аддукта пероксид водорода/мочевина и фталевого альдегида в СН2Cl2 при КТ.
Чистые производные цис- или транс-аминоциклогексана можно получить разделением смесей методом ЖХВР или при использовании стереохимически чистых исходных соединений.
Активность соединений формулы I и их солей определяли по следующим методикам.
Ингибирование микросомальной 2,3-оксидоскваленланостеринциклазы (OSC) из печени человека
Микросомальную фракцию из печени здоровых добровольцев получали в натрий-фосфатном буферном растворе (рН 7,4). Активность OSC определяли в аналогичном буферном растворе, содержащем, кроме того, 1 мМ ЭДТА и 1 мМ дитиотреит. Микросомальную фракцию разбавляли холодным фосфатным буферным раствором до концентрации 0,8 мг/мл белка. Сухой [14C]R,S-монооксидосквален (MOS, 12,8 мКи/ммоль) растворяли в этаноле (20 нКи/мкл) и смешивали с фосфатным буферным раствором, содержащим 1% БСА (бычий сывороточный альбумин). Исходный 1 мМ раствор исследуемого соединения в ДМСО разбавляли до требуемой концентрации фосфатным буферным раствором, содержащим 1% БСА. Затем 40 мкл микросомальной фракции смешивали с 20 мкл раствора исследуемого соединения и реакцию инициировали добавлением 20 мкл раствора [14C]R,S-MOS, при этом получали реакционную смесь следующего состава: 0,4 мг/мл микросомального белка и 30 мкл [14C]R,S-MOS в фосфатном буферном растворе, рН 7,4, содержащем 0,5% альбумина, <0,1% ДМСО и <2% этанола в конечном объеме 80 мкл.
После инкубации смеси при 37°С в течение 1 ч реакцию останавливали добавлением раствора следующего состава: 0,6 мл 10% раствора КОН в метаноле, 0,7 мл воды и 0,1 мл гексана/эфира (1:1, об./об.), содержащего в качестве носителей 25 мкг нерадиоактивного MOS и 25 мкг ланостерина. После встряхивания в каждую пробирку добавляли по 1 мл гексана/эфира (1:1, об./об.), снова встряхивали и центрифугировали. Верхнюю фазу переносили в стеклянную пробирку, нижнюю фазу снова экстрагировали гексаном/эфиром и экстракт объединяли с первым экстрактом. Объединенные экстракты упаривали досуха в атмосфере азота, остаток суспендировали в 50 мкл гексана/эфира и наносили на пластинку с силикагелем. Хроматографическое разделение проводили в системе гексан/эфир (1:1, об./об.). Величины Rf субстрата MOS и ланостеринового продукта составляли 0,91 и 0,54 соответственно. После высушивания на пластине с силикагелем регистрировали полосы, соответствующие радиоактивным MOS и ланостерину. Выход реакции и степень ингибирования OSC определяли по соотношению MOS к ланостерину, которое рассчитывали по радиоактивности в соответствующих полосах.
Во-первых, испытание проводили при постоянной концентрации исследуемого соединения, равной 100 нМ, и процент ингибирования OSC рассчитывали по сравнению с контрольными образцами. Ингибирующая активность наиболее предпочтительных соединений по настоящему изобретению составляла более 50%. Во-вторых, испытания проводили при различных концентрациях исследуемых соединений и определяли величину IC50, то есть концентрацию, при которой наблюдается снижение превращения MOS в ланостерин на 50% по сравнению с контролем. Предпочтительные соединения по настоящему изобретению характеризуются величинами IC50 от 1 нМ до 10 мкМ, предпочтительно от 1 до 100 нМ.
Соединения формулы I и/или их фармацевтически приемлемые соли можно использовать в качестве лекарственных средств, например, в форме фармацевтических препаратов для энтерального, парентерального или местного введения. Препараты можно вводить, например, пероральным способом, например, в форме таблеток, таблеток с покрытием, драже, твердых и мягких желатиновых капсул, растворов, эмульсий или суспензий; ректальным способом, например, в форме суппозиториев; парентеральным способом, например, в форме растворов для инъекции или вливания, или местным способом, например, в форме мазей, кремов или масел. Предпочтительным способом является пероральное введение.
Фармацевтические препараты можно получать методами, известными специалистам в данной области, при переработке описанных соединений формулы I и/или их фармацевтически приемлемых солей, наобязательно в комбинации с другими терапевтически ценными соединениями, в галеновую готовую форму в смеси с пригодными, нетоксичными, инертными, терапевтически совместимыми твердыми или жидкими материалами-носителями и при необходимости со стандартными фармацевтическими адъювантами.
Пригодными материалами-носителями являются не только неорганические материалы, но и органические материалы. Таким образом, для получения таблеток, таблеток с покрытием, драже и твердых желатиновых капсул в качестве материалов-носителей можно использовать, например, лактозу, кукурузный крахмал или его производные, тальк, стеариновую кислоту или ее соли. Пригодными материалами-носителями для мягких желатиновых капсул являются, например, растительные масла, воски, жиры и полутвердые и жидкие полиолы (однако, в случае мягких желатиновых капсул, в зависимости от природы активного ингредиента использование носителя может не потребоваться). Пригодными материалами-носителями для получения растворов и сиропов являются, например, вода, полиолы, сахароза, инвертированный сахар и т.п. Пригодными носителями для растворов для инъекции являются, например, вода, спирты, полиолы, глицерин и растительные масла. Пригодными носителями для суппозиториев являются, например, природные или отвержденные масла, воски, жиры и полужидкие или жидкие полиолы. Пригодными носителями препаратов для местного введения являются глицериды, полусинтетические и синтетические глицериды, гидрированные масла, жидкие воски, жидкое вазелиновое масло, жидкие жирные спирты, стерины, полиэтиленгликоли и производные целлюлозы.
В качестве фармацевтических адъювантов можно использовать стандартные стабилизаторы, консерванты, смачивающие и эмульгирующие агенты, агенты для улучшения консистенции, ароматизаторы, соли для изменения осмотического давления, буферные вещества, солюбилизаторы, красители и маскирующие агенты и антиоксиданты.
Дозы соединений формулы I могут изменяться в широком диапазоне в зависимости от типа излечиваемого заболевания, возраста и индивидуального состояния пациента, а также от способа введения, причем дозу можно изменять в зависимости от индивидуальных требований в каждом конкретном случае. Суточная доза для взрослых пациентов составляет приблизительно от 1 до 1000 мг, прежде всего от 1 до 100 мг. В зависимости от тяжести заболевания и конкретных фармакокинетических свойств соединение можно вводить в одной или нескольких стандартных суточных дозах, например, от 1 до 3 доз.
Фармацевтические препараты обычно содержат приблизительно 1-500 мг, предпочтительно 1-100 мг, соединения формулы I.
Изобретение иллюстрируется следующими примерами, не ограничивающими его объема.
Сокращения
АсОН уксусная кислота, ВОС трет-бутилоксикарбонил, BuLi бутиллитий, CH2Cl2 дихлорметан, DAST трифторид диэтиламиносеры, DEAD диэтиловый эфир азодикарбоновой кислоты, DBU 1,8-диазабицикло[5.4.0]ундец-7-ен(1,5-5), DIBALH гидрид ди-изо-бутилалюминия, ДМА N,N-диметилацетамид, DMAP 4-диметиламинопиридин, ДМФА N,N-диметилформамид, DMPU 1,3-диметил-3,4,5,6-тетрагидро-2(1Н)-пиримидинон, EDCI гидрохлорид N-(3-диметиламинопропил)-N'-этилкарбодиимида, EtOAc этилацетат, EtOH этанол, Et2O диэтиловый эфир, Et3N триэтиламин, НОВТ 1-гидроксибензотриазол, основание Хюнига iPr2NEt-N-этилдиизопропиламин, LAH литийалюминийгидрид, LDA диизопропиламид лития, LiBH4 борогидрид лития, МеОН метанол, NaI иодид натрия, PdCl2(dppf) (1,1'-бис(дифенилфосфино)ферроцен)дихлорпалладия (II)/CH2Cl2 (1:1), Pd(Ph3Р)4 тетракис(трифенилфосфин)палладий, Red-Al натрий-бис(2-метоксиэтокси)алюминийгидрид, TEMPO 2,2,6,6-тетраметилпиперидин-1-оксил, радикал, TBDMSCl трет-бутилдиметилсилилхлорид, ТВМЕ трет-бутилметиловый эфир, ТФУ трифторуксусная кислота, ТГФ тетрагидрофуран.
Общие условия проведения реакций
Все реакции проводили в атмосфере аргона.
Пример 1
1.1. Раствор 20 г (82,2 ммоля) транс-4-трет-бутоксикарбониламиноциклогексанкарбоновой кислоты в 1,2 л CH2Cl2 обрабатывали 12,83 г (131,5 ммоля) гидрохлорида N,O-диметилгидроксиламина, 10,85 мл (98,6 ммоля) N-метилморфолина и при 0°C добавляли 18,91 г (98,64 ммоля) EDCI и 12,62 г (82,2 ммоля) НОВТ. Реакционную смесь перемешивали при комнатной температуре в течение 2 ч и трижды экстрагировали 10% раствором KHSO4/Et2O. Органические фазы промывали насыщенным раствором NaHCO3, 10% раствором NaCl и сушили над Na2SO4, при этом получали 24,25 г (выход количественный) трет-бутилового эфира транс-[4-(метоксиметилкарбамоил)циклогексил]карбаминовой кислоты, tпл. 130-140°С (медленное разл.). МС: 287 (МН+).
1.2. Раствор 24,18 г (82 ммоля) трет-бутилового эфира транс-[4-(метоксиметилкарбамоил)циклогексил]карбаминовой кислоты в 80 мл ДМФА при 0°С небольшими порциями обрабатывали 5,37 г (123 ммоля) NaH (55% в масле). Реакционную смесь перемешивали при 0°С в течение 1 ч, а затем медленно обрабатывали (20 мин) 40,9 мл (656 ммолей) иодметана и нагревали при КТ в течение ночи. Реакционную смесь охлаждали, нейтрализовали 10% раствором KHSO4 и выливали в воду/Et2О (3х). Органические фазы промывали 10% раствором NaCl, сушили над Na2SO4, упаривали и очищали экспресс-хроматографией на колонке с силикагелем (элюент: градиент CH2Cl2/EtOAc, от 9:1 до 1:1), при этом получали 20,69 г (84%) трет-бутилового эфира транс-[4-(метоксиметилкарбамоил)циклогексил]метилкарбаминовой кислоты. МС: 301 (МН+).
1.3. Раствор 2,09 г (55 ммолей) LAH в 250 мл ТГФ охлаждали (-50°С) и в течение 25 мин обрабатывали раствором 15,02 г (50 ммолей) трет-бутилового эфира транс-[4-(метоксиметилкарбамоил)циклогексил]метилкарбаминовой кислоты в 250 мл ТГФ. Реакционную смесь нагревали при 15°С в течение 3,5 ч, охлаждали (-78°С) и гидролизовали суспензией 15 г MgSO4·7H2O, 15 г силикагеля в 50 мл 10% раствора KHSO4. Охлаждающую баню удаляли, добавляли ТГФ, смесь перемешивали в течение 30 мин и фильтровали. После упаривания фильтрата остаток растворяли в СН2Cl2, раствор сушили над Na2SO4 и упаривали, при этом получали 12,83 г (выход количественный) трет-бутилового эфира транс-(4-формилциклогексил)метилкарбаминовой кислоты. МС: 241 (М).
1.4. Раствор 52,45 г (200 ммолей) трифенилфосфина в 200 мл CH2Cl2 обрабатывали 33,16 г (100 ммолей) тетрабромметана (при этом реакционная смесь разогревалась до температуры кипения растворителя) и через 50 мин обрабатывали 32,06 мл (230 ммолей) триэтиламина (при этом реакционная смесь разогревалась до температуры кипения растворителя и цвет смеси изменялся на темно-фиолетовый). После охлаждения (0°С) к полученной смеси в течение 10 мин добавляли 12,83 г (50 ммолей) трет-бутилового эфира транс-(4-формилциклогексил)метилкарбаминовой кислоты в 125 мл CH2Cl2. Раствор перемешивали при КТ в течение 16 ч, упаривали и фильтровали через силикагель (инактивированный гексаном/0,5% Et3N) (элюент: градиент гексан/Et3N, от 4:1 до 1:1), при этом получали 13,28 г (67%) трет-бутилового эфира транс-[4-(2,2-дибромвинил)циклогексил]метилкарбаминовой кислоты, tпл. 93-99°С (разл.). МС: 396 (MH+, 2Br).
1.5. Следующие реакции проводили аналогично тому, как описано в книге Marshall James A., Bartley Gary S., Wallace Eli M., Total Synthesis of Pseudopterane (-)-Kallolide B, the Enantiomer of Natural (+)-Kallolide B, J.org. Chem. 61 (17), 5729-5735 (1996), и в статье Baker Raymond, Boyes Alastair L., Swain Christopher J., Synthesis of talaromycins А, В, С и E, J, Chem. Soc., Perkin Trans., 1 (5), 1415-21, (1990). Раствор 993 мг (2,5 ммоля) трет-бутилового эфира транс-[4-(2,2-дибромвинил)циклогексил]метилкарбаминовой кислоты в 20 мл ТГФ при -78°С обрабатывали 3,28 мл (5,25 ммоля) BuLi (приблизительно 1,6 М раствор в гексане). Через 2 ч при той же температуре добавляли 790 мг (25 ммолей) параформальдегида. Реакционную смесь нагревали до КТ в течение 3 ч и через 1 ч при указанной температуре трижды экстрагировали водой/Et2О. Органические фазы промывали 10% раствором NaCl, сушили над Na2SO4 и упаривали. После очистки экспресс-хроматографией на силикагеле (элюент: гексан/EtOAc, 4:1) получали 530 мг (79%) трет-бутилового эфира транс-[4-(3-гидроксипроп-1-инил)циклогексил]метилкарбаминовой кислоты. МС: 268 (МН+).
1.6. Раствор 9,0 г (33,66 ммоля) трет-бутилового эфира транс-[4-(3-гидроксипроп-1-инил)циклогексил]метилкарбаминовой кислоты в 185 мл CH2Cl2 при 0°С в течение 30 мин обрабатывали 136 мл ТФУ. После выдерживания при указанной температуре в течение 15 мин реакционную смесь упаривали, обрабатывали холодным (0°С) 1 н. NaOH (насыщенным NaCl) и трижды экстрагировали CH2Cl2/MeOH, 9:1. Органические фазы сушили над Na2SO4 и упаривали, при этом получали 5,84 г (выход количественный) транс-3-(4-метиламиноциклогексил)проп-2-ин-1-ола. МС: 167 (М).
1.7. Смесь 0,51 г (3,05 ммоля) транс-3-(4-метиламиноциклогексил)проп-2-ин-1-ола, 0,87 г (3,66 ммоля) 2,5 дибромпиримидина (Brown Desmond J., Arantz B.W., Pyrimidine reactions. XXII. Relative reactivities of corresponding chloro-, bromo- and iodopyrimidines in aminolysis, J.Chem. Soc. C, Issue 10, 1889-91 (1971)), и 1,78 мл (10,34 ммоля) N-этилдиизопропиламина нагревали при 80°С в течение 2,5 ч. Реакционную смесь охлаждали, упаривали и распределяли между насыщенным раствором NaHCO3/Et2O (3х). Органические фазы промывали 10% раствором NaCl, сушили (Na2SO4) и упаривали. После очистки экспресс-хроматографией на силикагеле (элюент: гексан/EtOAc, 95:5) получали 0,72 г (73%) транс-3-{4-[(5-бромпиримидин-2-ил)метиламино]циклогексил}проп-2-ин-1-ола. tпл. 156-157°С. МС: 324 (МН+, 1Br).
1.8. Транс-3-[4-(метилпиримидин-2-иламино)циклогексил]проп-2-ин-1-ол, tпл. 138-140°С, МС: 245 (М), получали при взаимодействии транс-3-(4-метиламиноциклогексил)проп-2-ин-1-ола с 3,2 экв. 2-хлорпиримидина при 80°С в течение 3 ч аналогично тому, как описано в примере 1.7.
1.9. Транс-6-{[4-(3-гидрокспроп-1-инил)циклогексил]метиламино}никотинонитрил, tпл. 126,1-127,4°C, МС: 270 (МН+), получали при взаимодействии транс-3-(4-метиламиноциклогексил)проп-2-ин-1-ола с 1,2 экв. 2-хлорпиридин-5-карбонитрила при 80°С в течение 29 ч, аналогично тому, как описано в примере 1.7.
1.10. транс-3-{4-[(5-Хлорпиримидин-2-ил)метиламино]циклогексил}проп-2-ин-1-ол, tпл. 148-150°С (разл.), МС: 280 (МН+, 1Cl), получали при взаимодействии транс-3-(4-метиламиноциклогексил)проп-2-ин-1-ол и 2-бром-5-хлорпиримидина, аналогично тому, как описано в примере 1.7.
Исходное соединение, 2-бром-5-хлорпиримидин получали из 5-хлор-2-гидроксипиримидина аналогично тому, как описано Brown Desmond J., Arantz B.W., Pyrimidine reactions. XXII. Relative reactivities of corresponding chloro-, bromo- and iodopyrimidines in aminolysis, J.Chem. Soc. C, Issue 10, 1889-91 (1971). транс-3-(4-Метиламиноциклогексил)проп-2-ин-1-ол и 1,5 экв. 2-бром-5-хлорпиримидина нагревали при 80°С в течение 0,5 ч, при 120°С в течение 1 ч, а затем добавляли 0,5 экв. 2-бром-5-хлорпиримидина и нагревали при 120°С в течение 1 ч. Указанное соединение получали после соответствующей обработки реакционной смеси.
1.11. Смесь 0,67 г (4 ммоля) транс-3-(4-метиламиноциклогексил)проп-2-ин-1-ола, 2,76 г (18 ммолей) 3,6-дихлорпиридазина и 1,76 мл (13,6 ммоля) N-этилдиизопропиламина нагревали при 80°С в течение 3,5 ч, разбавляли 1 мл ДМФА и нагревали при 80°С в течение 4 сут, а затем при 120°С в течение 1 сут. Реакционную смесь охлаждали, упаривали и распределяли между насыщенным водным NaHCO3/Et2O (3х). Органические фазы промывали 10% раствором NaCl, сушили (Na2SO4) и упаривали. После очистки экспресс-хроматографией на силикагеле (элюент: градиент MeCl2/Et2O, от 95:5 до 9:1) получали 0,61 г (54%) транс-3-{4-[(6-хлорпиридазин-3-ил)метиламино]циклогексил}проп-2-ин-1-ола. МС: 280 (МН+, 1Cl).
1.12. Смесь 0,67 г (4 ммоля) транс-3-(4-метиламиноциклогексил)проп-2-ин-1-ола, 1,24 мл (12 ммолей) 5-бром-2-фторпиридина и 1,76 мл (13,6 ммоля) N-этилдиизопропиламина нагревали при 80°С в течение 3 ч и при 120°С в течение 24 ч. Смесь разбавляли 1 мл ДМФА, обрабатывали каталитическим количеством NaI и нагревали при 120°С в течение 2 сут. Реакционную смесь охлаждали, упаривали и распределяли между насыщенным водным NaHCO3/Et2O (3х). Органические фазы промывали 10% раствором NaCl, сушили (Na2SO4) и упаривали. После очистки экспресс-хроматографией на силикагеле (элюент: градиент MeCl2/Et2O, от 97,5:2,5 до 92,5:7,5) получали 0,57 г (44%) транс-3-{4-[(5-бромпиридин-2-ил)метиламино]циклогексил}проп-2-ин-1-ола. МС: 323 (МН+, 1Br).
1.13. транс-3-[4-(Метилпиридин-2-иламино)циклогексил]проп-2-ин-1-ол, МС: 245 (МН+), получали при взаимодействии транс-3-(4-метиламиноциклогексил)проп-2-ин-1-ола с 2-фторпиридином при 120°С в течение 5 сут, аналогично тому, как описано в примере 1.12.
1.14. транс-3-[4-(Метилпиразин-2-иламино)циклогексил]проп-2-ин-1-ол, tпл. 147-149°С (разл.), МС: 246 (МН+), получали из транс-3-(4-метиламиноциклогексил)проп-2-ин-1-ола и 2-хлорпиразина аналогично тому, как описано в примере 1.12.
1.15. Раствор 0,24 г (1,44 ммоля) транс-3-(4-метиламиноциклогексил)проп-2-ин-1-ола, 0,7 мл (5,74 ммоля) 2-хлор-5-этилпиримидина, 0,83 мл (4,88 ммоля) N-этилдиизопропиламина и каталитического количества NaI в 1,5 мл ДМА нагревали в микроволновой печи при 120°С в течение 3,75 ч. Реакционную смесь охлаждали и распределяли между насыщенным водным NaHCO3/Et2О (3х). Органические фазы промывали 10% раствором NaCl, сушили (Na2SO4) и упаривали. После очистки экспресс-хроматографией на силикагеле (элюент: градиент гексан/EtOAc, от 9:1 до 1:1) получали 0,24 г (61%) транс-3-{4-[(5-этилпиримидин-2-ил)метиламино]циклогексил}проп-2-ин-1-ола. МС: 274 (МН+).
1.16. транс-3-{4-[Метил-(6-метилпиридазин-3-ил)амино]циклогексил}проп-2-ин-1-ол, МС: 260 (МН+), получали при взаимодействии транс-3-(4-метиламиноциклогексил)-проп-2-ин-1-ола и 3-хлор-6-метилпиридазина при 150°С в течение 4 ч и при 120°С в течение 3/4 ч в микроволновой печи аналогично тому, как описано в примере 1.15, однако при отутствии NaI.
1.17. Раствор 420 мг (1,3 ммоля) транс-3-{4-[(5-бромпиримидин-2-ил)метиламино]циклогексил}проп-2-ин-1-ола в 10 мл CH2Cl2 при 0°C обрабатывали 0,11 мл (1,43 ммоля) метансульфонилхлорида, 0,16 мл (1,95 ммоля) пиридина и 159 мг (1,3 ммоля) DMAP. Реакционную смесь перемешивали при комнатной температуре в течение 3,5 ч, добавляли воду (2 мл) и перемешивали в течение 5 мин. После экстракции насыщенным раствором NaHCO3/Et2O (3х) органические фазы промывали 10% раствором NaCl, сушили над Na2SO4 и упаривали, при этом получали 540 мг (выход количественный) 3-{4-[(5-бромпиримидин-2-ил)метиламино]циклогексил}проп-2-инилового эфира транс-метансульфоновой кислоты. МС: 402 (МН+, 1Br).
1.18. 3-[4-(Метилпиримидин-2-иламино)циклогексил]проп-2-иниловый эфир транс-метансульфоновой кислоты, МС: 402 (МН+), получали из транс-3-[4-(метилпиримидин-2-иламино)циклогексил]проп-2-ин-1-ол аналогично тому, как описано в примере 1.17.
1.19. 3-{4-[(5-Цианопиридин-2-ил)метиламино]циклогексил}проп-2-иниловый эфир транс-метансульфоновой кислоты, МС: 348 (МН+), получали из транс-6-{[4-(3-гидроксипроп-1-инил)циклогексил]метиламино}никотинонитрила аналогично тому, как описано в примере 1.17.
1.20. 3-{4-[(5-Хлорпиримидин-2-ил)метиламино]циклогексил}проп-2-иниловый эфир транс-метансульфоновой кислоты, МС: 357 (МН+, 1Cl), получали из транс-3-{4-[(5-хлорпиримидин-2-ил)метиламино]циклогексил}проп-2-ин-1-ола аналогично тому, как описано в примере 1.17.
1.21. 3-{4-[(6-Хлорпиридазин-3-ил)метиламино]циклогексил}проп-2-иниловый эфир транс-метансульфоновой кислоты, МС: 358 (МН+, 1Cl), получали из транс-3-{4-[(6-хлорпиридазин-3-ил)метиламино]циклогексил}проп-2-ин-1-ола, аналогично тому, как описано в примере 1.17.
1.22. 3-{4-[(5-Бромпиридин-2-ил)метиламино]циклогексил}проп-2-иниловый эфир транс-метансульфоновой кислоты, МС: 401 (МН+, 1Br), получали из транс-3-{4-[(5-бромпиридин-2-ил)метиламино]циклогексил}проп-2-ин-1-ола аналогично тому, как описано в примере 1.17.
1.23. 3-[4-(Метилпиридин-2-иламино)циклогексил]проп-2-иниловый эфир транс-метансульфоновой кислоты, МС: 323 (МН+), получали из транс-3-[4-(метилпиридин-2-иламино)циклогексил]проп-2-ин-1-ола аналогично тому, как описано в примере 1.17.
1.24. 3-[4-(Метилпиразин-2-иламино)циклогексил]проп-2-иниловый эфир транс-метансульфоновой кислоты, МС: 324 (МН+), получали из транс-3-[4-(метилпиразин-2-иламино)циклогексил]проп-2-ин-1-ола аналогично тому, как описано в примере 1.17.
1.25. Метансульфонат транс-1-(3-{4-[(5-этилпиримидин-2-ил)метиламино]циклогексил}проп-2-инил)пиридиния, МС: 335 (МН+), получали из транс-3-{4-[(5-этилпиримидин-2-ил)метиламино]циклогексил}проп-2-ин-1-ола аналогично тому, как описано в примере 1.17.
1.26. Раствор 0,246 г (0,95 ммоля) транс-3-{4-[метил(6-метилпиридазин-3-ил)амино]циклогексил}проп-2-ин-1-ола в 7 мл CH2Cl2 при 0°С обрабатывали 0,081 мл (1,04 ммоля) метансульфонилхлорида и 0,17 мл (1,42 ммоля) 2,6-лутидина. Реакционную смесь перемешивали при комнатной температуре в течение 22 ч, добавляли воду (1 мл) и перемешивали в течение еще 5 мин. После экстракции насыщенным раствором NaHCO3/Et2O (3х) органические фазы промывали 10% раствором NaCl, сушили над Na2SO4 и упаривали, при этом получали 0,285 г неочищенного транс-[4-(3-хлорпроп-1-инил)циклогексил]метил(6-метилпиридазин-3-ил)амина. МС: 278 (МН+, 1Cl).
Пример 2
Раствор 125 мг (соответствуют 0,30 ммоля) неочищенного 3-{4-[(5-бромпиримидин-2-ил)метиламино]циклогексил}проп-2-инилового эфира транс-метансульфоновой кислоты в 3 мл метанола охлаждали (0°С), обрабатывали 0,54 мл (3 ммоля) диметиламина (33% в EtOH, 5,6 М) и перемешивали при КТ в течение 20 ч. Растворитель упаривали и остаток экстрагировали насыщенным раствором NaHCO3/Et2O (3х), органические фазы сушили над Na2SO4, фильтровали и упаривали. После очистки экспресс-хроматографией на колонке с силикагелем (элюент: градиент CH2Cl2/MeOH, от 99:1 до 97,5:2,5) получали 65 мг (62%) чистого транс-(5-бромпиримидин-2-ил)[4-(3-диметиламинопроп-1-инил)циклогексил]метиламина, tпл. 83-84°С (разл.). МС: 351 (МН+, 1Br).
Следующие соединения получали из соответствующих мезилатов, хлоридов или производных пиридиния и вторичных аминов. В случае, если реакция не завершена в течение 20 ч, добавляли дополнительное количество амина (5 экв.), а в случаях, обозначенных знаком *, добавляли также каталитическое количество NaI и реакционную смесь перемешивали в течение 24 ч.
Таблица 1
Пример 3
3.1. Суспензию 3,4 г (12,72 ммоля) трет-бутилового эфира транс-[4-(3-гидроксипроп-1-инил)циклогексил]метилкарбаминовой кислоты в 125 мл этанола и 810 мг PtO2·H2O гидрировали (1 атм) в течение 7 ч. Реакционную смесь фильтровали (целит) и фильтрат упаривали, при этом получали 3,5 г (выход количественный) трет-бутилового эфира транс-[4-(3-гидроксипропил)циклогексил]метилкарбаминовой кислоты. МС: 271 (М).
3.2. 3-(4-Метиламиноциклогексил)пропан-1-ол, МС: 172 (МН+), получали из трет-бутилового эфира транс-[4-(3-гидроксипропил)циклогексил]метилкарбаминовой кислоты аналогично тому, как описано в примере 1.6.
3.3. транс-3-{4-[(5-Бромпиримидин-2-ил)метиламино]циклогексил}пропан-1-ол, МС: 328 (МН+, 1Br), получали из 3-(4-метиламиноциклогексил)пропан-1-ола аналогично тому, как описано в примере 1.7.
3.4. 3-{4-[(5-Бромпиримидин-2-ил)метиламино]циклогексил}пропиловый эфир транс-метансульфоновой кислоты, МС: 406 (МН+, 1Br), получали из транс-3-{4-[(5-бромпиримидин-2-ил)метиламино]циклогексил}пропан-1-ола при обработке в течение 5 ч аналогично тому, как описано в примере 1.17.
Пример 4
Раствор 209 мг (соответствуют 0,50 ммоля) неочищенного 3-{4-[(5-бромпиримидин-2-ил)метиламино]циклогексил}пропилового эфира транс-метансульфоновой кислоты в 5 мл метанола обрабатывали 0,89 мл (5 ммолей) диметиламина (33% в EtOH, 5,6 М) и перемешивали при КТ в течение ночи. После добавления дополнительного количества 0,45 мл (2,5 ммоля) диметиламина (33% в EtOH, 5,6 М) реакционную смесь перемешивали в течение 66 ч, затем нагревали при 70°С в течение 2 ч, охлаждали, упаривали и остаток экстрагировали насыщенным раствором NaHCO3/Et2O (3х). Органические фазы сушили над Na2SO4, фильтровали и упаривали. После очистки экспресс-хроматографией на колонке с силикагелем (элюент: градиент СН2Cl2/МеОН, от 97:3 до 94:6) получали 157 мг (88%) транс-(5-бромпиримидин-2-ил)[4-(3-диметиламинопропил)циклогексил]метиламина. МС: 355 (МН+, 1Br).
Следующие соединения получали из соответствующих мезилатов и вторичных аминов. Если реакция была не завершена, реакционную смесь кипятили с обратным холодильником до завершения реакции.
Таблица 2
Пример 5
5.1. Раствор 10,0 г (25,2 ммоля) трет-бутилового эфира транс-[4-(2,2-дибромвинил)циклогексил]метилкарбаминовой кислоты в 400 мл ТГФ при -78°С обрабатывали 33,0 мл (68,3 ммоля) BuLi (приблизительно 1,6 М в гексане) и перемешивали в течение 2 ч, затем добавляли 27,8 мл (230,4 ммоля) DMPU и через 10 мин по каплям в течение 20 мин добавляли 19,0 мл (125,9 ммоля) 2-(2-бромэтокси)тетрагидро-2Н-пирана, растворенного в 20 мл. Реакционную смесь нагревали до комнатной температуры и перемешивали в течение ночи (приблизительно 16 ч). Затем к смеси добавляли насыщенный раствор NH4Cl и смесь экстрагировали Et2O (3х). Органическую фазу промывали Н2О (2х), 10% раствором NaCl, сушили над Na2SO4, фильтровали и упаривали. Остаток очищали экспресс-хроматографией на колонке с силикагелем (элюент: гексан/EtOAc, от 19:1 до 3:1), при этом получали 3.5 г (38%) трет-бутилового эфира транс-метил{4-[4-(тетрагидропиран-2-илокси)бут-1-инил]циклогексил}карбаминовой кислоты. МС: 366 (МН+).
5.2. Раствор 3,45 г (9,44 ммоля) трет-бутилового эфира транс-метил {4-[4-(тетрагидропиран-2-илокси)бут-1-инил]циклогексил}карбаминовой кислоты и 0,7 г (2,83 ммоля) толуол-4-сульфоната пиримидиния в 25 мл МеОН перемешивали при 55°С в течение 1,5 ч. Реакционную смесь распределяли между раствором 10% KHSO4/Et2O (3х). Органические фазы промывали насыщенным раствором NaHCO3, 10% раствором NaCl, сушили над Na2SO4 и упаривали, при этом получали 2,85 г (количественный выход) трет-бутилового эфира транс-[4-(4-гидроксибут-1-инил)циклогексил]метилкарбаминовой кислоты. МС: 281 (М).
5.3. транс-4-(4-Метиламиноциклогексил)бут-3-ин-1-ол, МС: 182 (МН+), получали из трет-бутилового эфира транс-[4-(4-гидроксибут-1-инил)циклогексил]метилкарбаминовой кислоты при обработке ТФУ аналогично тому, как описано в примере 1.6.
5.4. Смесь 1,06 г (5,85 ммоля) транс-4-(4-метиламиноциклогексил)бут-3-ин-1-ола, 1,67 г (7,02 ммоля) 2,5-дибромпиримидина (Brown Desmond J., Arantz В. W., Pyrimidine reactions. XXII. Relative reactivities of corresponding chloro-, bromo- and iodopyrimidines in aminolysis, J.Chem. Soc. C, Issue 10, 1889-91 (1971)) и 3,38 мл (19,88 ммоля) N-этилдиизопропиламина нагревали при 85°С в течение 2 ч, разбавляли 1 мл ДМА и нагревали при 85°С в течение 3,5 ч. Реакционную смесь охлаждали, упаривали и распределяли между насыщенным раствором NaHCO3/Et2O (3х). Органические фазы промывали 10% раствором NaCl, сушили (Na2SO4) и упаривали. После очистки экспресс-хроматографией на силикагеле (элюент: гексан/EtOAc, от 9:1 до 1:1) получали 1,37 г (69%) транс-4-{4-[(5-бромпиримидин-2-ил)метиламино]циклогексил}бут-3-ин-1-ола. МС: 338 (MH+, 1Br).
5.5. 4-{4-[(5-Бромпиримидин-2-ил)метиламино]циклогексил}бут-3-иниловый эфир транс-метансульфоновой кислоты, МС: 416 (МН+, 1Br), получали из транс-4-{4-[(5-бромпиримидин-2-ил)метиламино]циклогексил}бут-3-ин-1-ола аналогично тому, как описано в примере 1.17.
Пример 6
6.1. Раствор 211 мг (0,51 ммоля) неочищенного 4-{4-[(5-бромпиримидин-2-ил)метиламино]циклогексил}бут-3-инилового эфира транс-метансульфоновой кислоты в 5 мл метанола обрабатывали 0,91 мл (5,1 ммоля) диметиламина (33% в EtOH, 5,6 М) и нагревали при 65°С в течение 4 ч. После охлаждения и упаривания остаток экстрагировали насыщенным водным раствором NaHCO3/Et2O (3 раза). Органическую фазу сушили (Na2SO4), фильтровали и упаривали. После очистки экспресс-хроматографией на колонке с силикагелем (элюент: CH2Cl2/MeOH, от 99:1 до 95:5) получали 141 мг (76%) транс-(5-бромпиримидин-2-ил)[4-(4-диметиламинобут-1-инил)циклогексил]метиламина. МС: 365 (МН+, 1Br).
6.2. транс-(5-Бромпиримидин-2-ил)метил[4-(4-пиперидин-1-илбут-1-инил)циклогексил]амин, МС: 405 (МН+, 1Br), получали из 4-{4-[(5-бромпиримидин-2-ил)метиламино]циклогексил}бут-3-инилового эфира транс-метансульфоновой кислоты и пиперидина после нагревания при 65°С в течение 4,5 ч аналогично тому, как описано в примере 6.1.
Пример 7
7.1. трет-Бутиловый эфир транс-[4-(4-гидроксибутил)циклогексил]метилкарбаминовой кислоты, МС: 286 (МН+), получали из трет-бутилового эфира транс-[4-(4-гидроксибут-1-инил)циклогексил]метилкарбаминовой кислоты аналогично тому, как описано в примере 3.1.
7.2. транс-4-(4-Метиламиноциклогексил)бутан-1-ол, МС: 186 (МН+), получали из трет-бутилового эфира транс-[4-(4-гидроксибутил)циклогексил]метилкарбаминовой кислоты аналогично тому, как описано в примере 1.6.
7.3. транс-4-{4-[(5-Бромпиримидин-2-ил)метиламино]циклогексил}бутан-1-ол, МС: 342 (МН+, 1Br), получали из транс-4-(4-метиламиноциклогексил)бутан-1-ола и 2,5-дибромпиримидина аналогично тому, как описано в примере 1.7.
7.4. Транс-4-{4-[(5-бромпиримидин-2-ил)метиламино]циклогексил}бутан-1-ол, МС: 420 (МН+, 1Br), получали из 4-{4-[(5-бромпиримидин-2-ил)метиламино]циклогексил}бутилового эфира транс-метансульфоновой кислоты после инкубации в течение 2,5 ч аналогично тому, как описано в примере 1.17.
Пример 8
8.1. транс-(5-Бромпиримидин-2-ил)[4-(4-диметиламинобутил)циклогексил]метиламин, МС: 369 (МН+, 1Br), получали после инкубации 4-{4-[(5-бромпиримидин-2-ил)метиламино]циклогексил}бутилового эфира транс-метансульфоновой кислоты с диметиламином (33% в EtOH, 5,6 М) при 65°С в течение 4 ч аналогично тому, как описано в примере 6.1.
8.2. транс-(5-Бромпиримидин-2-ил)метил[4-(4-пиперидин-1-илбутил)циклогексил]амин, МС: 409 (МН+, 1Br), получали после инкубации 4-{4-[(5-бромпиримидин-2-ил)метиламино]циклогексил}бутилового эфира транс-метансульфоновой кислоты и пиперидина при 65°С в течение 7 ч аналогично тому, как описано в примере 6.1.
Пример 9
9.1. Интенсивно перемешиваемый раствор 100 г (774 ммоля) цис-4-метиламиноциклогексанола (Schut Robert N., Analgetic 3-(methylamino)-1,2,3,4-tetra-hydrocarbazole from 4-(methylamino)cyclohexanone, Fr. (1968), 3 pp. FR 1515629 19680301) в 775 мл EtOAc обрабатывали 1,55 л 1 М раствора NaHCO3 и 110 мл (774 ммоля) бензилхлорформиата (30 мин, t макс 30°С). После выдерживания смеси при КТ в течение 2 ч фазы разделяли. Водную фазу экстрагировали (EtOAc), органические фазы сушили (Na2SO4), фильтровали и упаривали. После очистки хроматографией на колонке с силикагелем (элюент: гексан/EtOAc, 2:1) получали 139 г (68%) бензилового эфира цис-(4-гидроксициклогексил)метилкарбаминовой кислоты. МС: 263 (М).
9.2. Раствор 2,63 г (10 ммолей) бензилового эфира цис-(4-гидроксицикло-гексил)метилкарбаминовой кислоты в 16 мл CH2Cl2 обрабатывали раствором 0,24 г (2 ммоля) KBr и 0,28 г (3,33 ммоля) NaHCO3 в 5 мл воды. Суспензию охлаждали (0-5°С) и в течение 20 мин добавляли 8 мг (0,05 ммоля) TEMPO и затем 5,7 мл (12,5 ммоля) NaOCl (13%, 2,18 М раствор в воде). Через 1 ч добавляли еще 8 мг (0,05 ммоля) TEMPO и затем 2,85 мл (6,25 ммоля) NaOCl (13%, 2,18 М раствор в воде). Через 1 ч добавляли 5 мл 1 М раствора тиосульфата натрия. Водную фазу экстрагировали CH2Cl2 (2x), органическую фазу сушили (Na2SO4), фильтровали и упаривали, при этом получали 2,57 г (99%) бензилового эфира метил(4-оксоциклогексил)карбаминовой кислоты. МС: 261 (М).
9.3. Суспензию 749,88 г (2187,5 ммоля) хлорида (метоксиметил)трифенилфосфония в 2,5 л ТГФ охлаждали (-10°С) и депротонировали в присутствии 245,5 г (2187,5 ммоля) трет-бутоксида калия. Раствор темно-красного цвета перемешивали при 0-5°С в течение 0,5 ч, охлаждали (-20°С) и к полученному раствору в течение 1,25 ч по каплям добавляли 457,32 г (261,33 ммоля) бензилового эфира метил(4-оксоциклогексил)карбаминовой кислоты в 1,25 л ТГФ. После выдерживания при КТ в течение 1,3 ч реакционную смесь обрабатывали 1,75 л 1 М раствора NaHCO3 и перемешивали в течение 45 мин. Фазы разделяли, водную фазу экстрагировали ТВМЕ (700 мл), органическую фазу сушили (Na2SO4), фильтровали и упаривали. Остаток суспендировали в гексане (5 л), охлаждали (0°С), фильтровали, фильтрат упаривали, при этом получали 495, 2 г (98%) бензилового эфира (4-метоксиметиленциклогексил)метилкарбаминовой кислоты. МС: 289 (М).
9.4. Раствор 495 г (1710,6 ммоля) бензилового эфира (4-метоксиметиленциклогексил)метилкарбаминовой кислоты в 1,7 л ТГФ при КТ обрабатывали 3,42 л 1 н. HCl и кипятили с обратным холодильником в течение 2 ч. Реакционную смесь охлаждали до КТ и экстрагировали ТВМЕ (1,7 и 0,9 л). Органическую фазу промывали 1 М раствором NaHCO3, сушили (Na2SO4), фильтровали и упаривали, при этом получали 457,4 (97%) неочищенного бензилового эфира (4-формилциклогексил)метилкарбаминовой кислоты (отношение транс/цис составляет приблизительно 70:30).
Раствор 327 г (1188 ммолей) неочищеного бензилового эфира (4-формилциклогексил)метилкарбаминовой кислоты в 1,64 л ТВМЕ при КТ добавляли к раствору 451,5 г (2375 ммолей) динатриевой соли пиросульфита в 1,64 л воды. Реакционную смесь перемешивали в течение 15 ч, фильтровали и промывали (1,1 л ТВМЕ), при этом получали 191,7 г (45%) натриевой соли [4-(бензилоксикарбонилметиламино)циклогексил]гидроксиметансульфоновой кислоты (отношение транс/цис 95:5). Полученное соединение суспендировали в 0,5 л ТВМЕ и 1,01 л 1 М раствора Na2СО3 и перемешивали при КТ в течение 1 ч. Фазы разделяли, водную фазу экстрагировали ТВМЕ (1 л), органическую фазу сушили (Na2SO4), фильтровали и упаривали, при этом получали 128 г (36% после 2 стадий) бензилового эфира транс-(4-формилциклогексил)метилкарбаминовой кислоты (отношение транс/цис 99:1). МС: 275 (М).
9.5. Суспензию 128,6 г (375 ммоля) хлорида (метоксиметил)трифенилфосфония в 540 мл ТГФ при -8°С обрабатывали 43,1 г (375 ммолей) трет-бутоксида калия. Раствор красного цвета перемешивали при 0°С в течение 30 мин, охлаждали (-20°С) и к полученному раствору по каплям в течение 60 мин добавляли 82,6 г (300 ммолей) бензилового эфира транс-(4-формилциклогексил)метилкарбаминовой кислоты в 240 мл ТГФ. Реакционную смесь нагревали до КТ, перемешивали в течение 2 ч, а затем промывали насыщенным раствором NaHCO3 (540 мл). Водную фазу экстрагировали 0,5 л ТВМЕ, объединенные органические фазы окисляли 16 мл раствора пероксида водорода (35%) и смешивали с водой (150 мл). Органический растворитель упаривали, остаток экстрагировали МеОН (350 мл)/гексаном (2×2000 мл). Гексан дважды промывали 500 мл МеОН/водой (7:3), сушили (Na2SO4) и упаривали, при этом получали 81,7 г (90%) бензилового эфира транс-(2Е/Z)-[4-(2-метоксивинил)циклогексил]метилкарбаминовой кислоты. МС: 303 (М).
9.6. Бензиловый эфир транс-метил-[4-(2-оксоэтил)циклогексил]карбаминовой кислоты, МС: 290 (МН+), получали из бензилового эфира транс-(2Е/Z)[4-(2-метоксивинил)циклогексил]метилкарбаминовой кислоты после кипячения с обратным холодильником в течение 1 ч аналогично тому, как описано в примере 9.4.
9.7. Суспензию 77,4 г (267,4 ммоля) бензилового эфира транс-метил[4-(2-оксоэтил)циклогексил]карбаминовой кислоты, 63,86 г (292,6 ммоля) ди-трет-бутилового эфира дикарбоновой кислоты и 7,7 г 10% Pd/C в 775 мл EtOAc гидрировали (1 атм) при 38°С в течение 48 ч (в течение дня каждый час подавали необходимое количество водорода). Реакционную смесь фильтровали (целит) и упаривали, после очистки экспресс-хроматографией на колонке с силикагелем (элюент: гексан/EtOAc, 4:1) получали 38,4 г (56%) трет-бутилового эфира транс-метил[4-(2-оксоэтил)циклогексил]карбаминовой кислоты. МС: 255 (М).
9.8. трет-Бутиловый эфир транс-[4-(3,3-дибромаллил)циклогексил]метилкарбаминовой кислоты, МС: 352 (М-бутен, 2Br), получали из трет-бутилового эфира транс-метил[4-(2-оксоэтил)циклогексил]карбаминовой кислоты аналогично тому, как описано в примере 1.4.
9.9. трет-Бутиловый эфир транс-[4-(4-гидроксибут-2-инил)циклогексил]метилкарбаминовой кислоты, МС: 281 (М), получали из трет-бутилового эфира транс-[4-(3,3-дибромаллил)циклогексил]метилкарбаминовой кислоты аналогично тому, как описано в примере 1.5.
9.10. транс-4-(4-Метиламиноциклогексил)бут-2-ин-1-ол, МС: 182 (МН+), получали из трет-бутилового эфира транс-[4-(4-гидроксибут-2-инил)циклогексил]метилкарбаминовой кислоты аналогично тому, как описано в примере 1.6.
9.11. транс-4-{4-[(6-Хлорпиридазин-3-ил)метиламино]циклогексил}бут-2-ин-1-ол, МС: 294 (МН+, 1Cl), получали из транс-4-(4-метиламиноциклогексил)бут-2-ин-1-ола и 3,6-дихлорпиридазина при нагревании при 80°С в течение 6 ч (при в отсутствии NaI) и после нагревания в микроволновой печи при 120°С в течение 3/4 ч аналогично тому, как описано в примере 1.15.
9.12. транс-4-{4-[(5-Бромпиримидин-2-ил)метиламино]циклогексил}бут-2-ин-1-ол, МС: 338 (МН+, 1Br), получали из транс-4-(4-метиламиноциклогексил)бут-2-ин-1-ола и 1,2 экв. 2,5-дибромпиримидина (Brown Desmond J., Arantz B.W., Pyrimidine reactions. XXII. Relative reactivities of corresponding chloro-, bromo- and iodopyrimidines in aminolysis. J.Chem. Soc, C, Issue 10, 1889-91 (1971)) при нагреваниии при 80°С в течение 6 ч (в отсутствие NaI) и после нагревания в микроволновой печи при 120°С в течение 1/4 ч аналогично тому, как описано в примере 1.15.
9.13. Раствор 1,08 г (3,68 ммоля) транс-4-{4-[(6-хлорпиридазин-3-ил)метиламино]циклогексил}бут-2-ин-1-ола в 30 мл CH2Cl2 при 0°С обрабатывали 0,31 мл (4,04 ммоля) метансульфонилхлорида и 0,64 мл (5,51 ммоля) 2,6-лутидина. Реакционную смесь перемешивали при комнатной температуре в течение 46 ч, добавляли воду (4 мл) и перемешивали в течение 5 мин. После экстракции насыщенным раствором NaHCO3/Et2O (3х) органическую фазу промывали 10% раствором NaCl, сушили над Na2SO4 и упаривали, при этом получали 1,45 г неочищенного 4-{4-[(6-хлорпиридазин-3-ил)метиламино]циклогексил}бут-2-инилового эфира транс-метансульфоновой кислоты. МС: 372 (МН+, 1Cl).
9.14. 4-{4-[(5-Бромпиримидин-2-ил)метиламино]циклогексил}бут-2-иниловый эфир транс-метансульфоновой кислоты, МС: 416 (МН+, 1Br), получали из транс-4-{4-[(5-бромпиримидин-2-ил)метиламино]циклогексил}бут-2-ин-1-ола аналогично тому, как описано в примере 9.13.
Пример 10
Следующие соединения получали аналогично тому, как описано в примере 2, из соответствующих мезилатов и вторичных аминов.
Таблица 3
Пример 11
11.1. К охлажденному сухим льдом раствору 30,0 г (208 ммолей) транс-(4-гидроксиметилциклогексил)метанола в 450 мл тетрагидрофурана при температуре от -60° до -67°С в течение 30 мин по каплям добавляли 130 мл (208 ммолей) 1,6 М раствора бутиллития (1,6 М раствор в гексане). После перемешивания в течение 30 мин при -78°С в течение 10 мин добавляли 32,3 г (208 ммолей) трет-бутилдиметилхлорсилана. Реакционную смесь выдерживали при -65°С в течение 15 мин, перемешивали при комнатной температуре в течение ночи, а затем распределяли между Et2O, 1 н. соляной кислотой и водой. Органический слой сушили над сульфатом магния и концентрировали при пониженном давлении, остаток очищали хроматографией на силикагеле (элюент: гексан/этилацетат, 3:1 (об./об.)), при этом получали 27,7 г (51%) чистого транс-[4-(трет-бутилдиметилсиланилоксиметил)циклогексил]метанола в виде бесцветного вязкого масла. МС: 259 (МН+).
11.2. К охлажденному сухим льдом раствору 27,6 г (107 ммолей) транс-[4-(трет-бутилдиметилсиланилоксиметил)циклогексил]метанола и 9,99 мл (128 ммолей) метансульфонилхлорида в 350 мл дихлорметана в течение 20 мин при перемешивании при 0-10°С добавляли 29,6 мл (213 ммолей) триэтиламина и реакционную смесь перемешивали при комнатной температуре в течение 1 ч. Затем смесь распределяли между дихлорметаном, 1 н. HCl и водой. Фазу дихлорметана сушили над сульфатом магния и концентрировали, при этом получали 38,2 г неочищенного 4-(трет-бутилдиметилсиланилоксиметил)циклогексилметилового эфира транс-метансульфоновой кислоты в виде бесцветного вязкого масла, МС: 354 (M+NH4 +).
11.3. 38,2 г неочищенного 4-(трет-бутилдиметилсиланилоксиметил)циклогексилметилового эфира транс-метансульфоновой кислоты и 16,7 г (340 ммолей) цианида цинка, растворенных в 380 мл N,N-диметилформамида, перемешивали при 80°С в течение 2 ч. После охлаждения до комнатной температуры реакционную смесь распределяли между Et2O и водой. Органический слой сушили над сульфатом магния и концентрировали при пониженном давлении, полученный остаток очищали хроматографией на силикагеле (элюент: гексан/этилацетат, 9:1 (об./об.)), при этом получали 24,2 г (78% после двух стадий) чистого транс-[4-(трет-бутилдиметилсиланилоксиметил)циклогексил]ацетонитрила в виде бесцветного вязкого масла. МС: 290 (MNa+).
11.4. Раствор 24,2 г (90,5 ммоля) транс-[4-(трет-бутилдиметилсиланилоксиметил)циклогексил]ацетонитрила, 22 мл (270 ммолей) хлороформа и 2,4 г PtO2·H2O (Degussa 223) в 250 мл этанола перемешивали в атмосфере водорода при комнатной температуре в течение 20 ч. Катализатор отделяли фильтрованием, растворитель упаривали при пониженном давлении, при этом получали 17,1 г (97%) чистого гидрохлорида транс-[4-(2-аминоэтил)циклогексил]метанола в виде бесцветного твердого вещества. МС: 158 (МН+).
11.5. транс-{4-[2-(5-Бромпиримидин-2-иламино)этил]циклогексил}метанол, tпл. 151,7-153,4°С, МС 314 (МН+, 1Br), получали из гидрохлорида транс-[4-(2-аминоэтил)циклогексил]метанола, 5,4 экв. N-этилдиизопропиламина и 1,2 экв. 2,5-дибромпиримидина в ДМА при нагревании при 85°С в течение 7,5 ч аналогично тому, как описано в примере 5.4.
11.6. Раствор 481 мг (1,53 ммоля) транс-{4-[2-(5-бромпиримидин-2-иламино)этил]циклогексил} метанола в 14 мл CH2Cl2 при 0°С обрабатывали 0,13 мл (1,68 ммолей) метансульфонилхлорида и 0,27 мл (2,30 ммоля) 2,6-лутидина. Реакционную смесь перемешивали при комнатной температуре в течение 20 ч, добавляли воду (2 мл) и перемешивали в течение 5 мин. После экстракции насыщенным раствором NaHCO3/Et2O (3х) органическую фазу промывали 10% раствором NaCl, сушили над Na2SO4 и упаривали, при этом получали 970 мг неочищеного 4-[2-(5-бромпиримидин-2-иламино)этил]циклогексилметилового эфира транс-метансульфоновой кислоты. МС: 392 (МН+, 1Br).
11.7. К раствору 17,6 г (90,9 ммоля) гидрохлорида транс-[4-(2-аминоэтил)циклогексил] метанола и 13,9 мл (100 ммолей) триэтиламина в 120 мл дихлорметана при перемешивании при комнатной температуре в течение 10 мин добавляли раствор 21,8 г (100 ммолей) ди-трет-бутилдикарбоната в 70 мл дихлорметана. После перемешивания при комнатной температуре в течение 3 ч реакционную смесь распределяли между дихлорметаном, 1 н. соляной кислотой и водой. Фазу дихлорметана сушили над сульфатом магния и концентрировали, при этом получали 27,9 г неочищенного трет-бутилового эфира транс-[2-(4-гидроксиметилциклогексил)этил]карбаминовой кислоты в виде бесцветного вязкого масла. МС: 275 (MNH4 +).
11.8. Раствор 27,9 г (86,7 ммоля) трет-бутилового эфира транс-[2-(4-гидроксиметилциклогексил)этил]карбаминовой кислоты, 41 мл (434 ммоля) уксусного ангидрида и 35 мл (434 ммоля) пиридина в 140 мл дихлорметана перемешивали при комнатной температуре в течение 16 ч. Реакционную смесь переносили в Et2O и промывали 1 н. соляной кислотой, раствором гидрокарбоната натрия и водой. Затем фазу Et2O сушили над сульфатом магния и концентрировали, при этом получали 26,0 г неочищенного 4-(2-трет-бутоксикарбониламиноэтил)циклогексилметилового эфира транс-уксусной кислоты в виде бесцветного вязкого масла, МС: 200 [(М-(трет-бутоксикарбонил))H+].
11.9. К охлажденному сухим льдом и перемешиваемому раствору 26,0 г неочищенного 4-(2-трет-бутоксикарбониламиноэтил)циклогексилметилового эфира транс-уксусной кислоты и 5,77 мл (92,6 ммоля) метилиодида в 300 мл N,N-диметилформамида в течение 15 мин добавляли 4,04 г (92,58 ммоля) гидрида натрия (55% в масле). После перемешивания в течение ночи при комнатной температуре добавляли еще 1,65 мл (26,5 ммоля) метилиодида и 1,16 г (26,5 ммоля) гидрида натрия и реакционную смесь перемешивали при комнатной температуре в течение 1 ч. Смесь распределяли между Et2O, 1 н. соляной кислотой и водой. Органический слой сушили над сульфатом магния и концентрировали при пониженном давлении, остаток очищали хроматографией на силикагеле (элюент: гексан/этилацетат, 4:1 (об./об.)), при этом получали 18,7 г (68% после 3 стадий) чистого 4-[2-(трет-бутоксикарбонилметиламино)этил]циклогексилметилового эфира транс-уксусной кислоты в виде бесцветного вязкого масла. МС: 214 [(М-(трет-бутоксикарбонил))Н+].
11.10. К раствору 18,7 г (59,7 ммоля) 4-[2-(трет-бутоксикарбонилметиламино)этил]циклогексилметилового эфира транс-уксусной кислоты в 110 мл метанола добавляли 41,25 г (298,5 ммоля) карбоната калия и реакционную смесь перемешивали при комнатной температуре в течение 2 ч. Избыток карбоната калия удаляли фильтрованием, метанол удаляли упариванием при пониженном давлении. Неочищенный остаток распределяли между Et2O, 1 н. соляной кислотой и водой. Органический слой сушили над сульфатом магния и концентрировали при пониженном давлении, остаток очищали хроматографией на силикагеле (элюент: гексан/этилацетат, 2:1 (об./об.)), при этом получали 13,9 г (86%) чистого трет-бутилового эфира транс-[2-(4-гидроксиметилциклогексил)этил]метилкарбаминовой кислоты в виде бесцветного вязкого масла. МС: 272 (МН+).
11.11. Раствор 1,45 г (5,34 ммоля) трет-бутилового эфира транс-[2-(4-гидроксиметилциклогексил)этил]метилкарбаминовой кислоты в 10 мл диоксана обрабатывали при 10°С 13,4 мл (53,4 ммоля) 4 М раствора HCl в диоксане. После выдерживания при КТ в течение 3,5 ч реакционную смесь упаривали, при этом получали 1,6 г (выход количественный) гидрохлорида транс-[4-(2-метиламиноэтил)циклогексил]метанола. МС: 172 (МН+).
11.12. транс-4-{2-[(5-Бромпиримидин-2-ил)метиламино]этил}циклогексилметанол, tпл. 61-63°С, МС: 328 (МН+, 1Br), получали из гидрохлорида транс-[4-(2-метиламиноэтил)циклогексил]метанола, 5,4 экв. N-этилдиизопропиламина и 1,2 экв. 2,5-дибромпиримидина (Brown Desmond J., Arantz B.W., Pyrimidine reactions. XXII. Relative reactivities of corresponding chloro-, bromo- and iodopyrimidines in aminolysis. J.Chem. Soc, C, Issue 10, 1889-91 (1971)) при нагревании в отсутствие растворителя при 85°С в течение 1/2 ч и при 85°С в ДМА в течение 6 ч аналогично тому, как описано в примере 5.4.
11.13. 4-{2-[(5-Бромпиримидин-2-ил)метиламино]этил}циклогексилметиловый эфир транс-метансульфоновой кислоты, МС: 406 (МН+, 1Br), получали из транс-4-{2-[(5-бромпиримидин-2-ил)метиламино]этил}циклогексилметанола аналогично тому, как описано в примере 11.6.
Пример 12
Раствор 258 мг (соответствуют 0,41 ммоля) неочищенного 4-[2-(5-бромпиримидин-2-иламино)этил]циклогексилметилового эфира транс-метансульфоновой кислоты в 5 мл метанола обрабатывали 0,73 мл (4,1 ммоля) диметиламина (33% в EtOH, 5,6 М), нагревали при 65°С в течение 4 ч, добавляли каталитическое количество NaI и нагревали в течение 16 ч. После охлаждения и упаривания остаток экстрагировали насыщенным раствором NaHCO3/Et2O (3х). Органическую фазу сушили над Na2SO4, фильтровали и упаривали. После очистки экспресс-хроматографией на колонке с силикагелем (элюент: CH2Cl2/MeOH, от 99:1 до 9:1) получали 105 мг (76%) транс-(5-бромпиримидин-2-ил)[2-(4-диметиламинометилциклогексил)этил]амина, tпл. 108,3-109,5°C. MC: 341 (МН+, 1Br).
Следующие соединения получали из соответствующих мезилатов и вторичных аминов.
Таблица 4
Пример 13
13.1. Для проведения реакции растворы дегазировали аргоном в течение 10 мин. Суспензию 7,3 мг PdCl2(dppf), 29,4 мг (0,24 ммоля) 4-пиридилбороновой кислоты и 70 мг (0,2 ммоля) транс-(5-бромпиримидин-2-ил)[4-(3-диметиламинопроп-1-инил)циклогексил]метиламина в 3,5 мл диоксана обрабатывали 1 мл 2 М раствора Na2СО3. После выдерживания смеси при 85°С в течение 17 ч добавляли 7 мг PdCl2(dppf) и реакционную смесь нагревали при 85°С в течение еще 24 ч. Смесь распределяли между насыщенным раствором NaHCO3/Et2O (3х) и объединенные органические фазы экстрагировали 0,1 М HCl. Водную фазу подщелачивали (1 н. NaOH) до рН 14 и экстрагировали Et2O (3х). Органическую фазу промывали 10% раствором NaCl и сушили над Na2SO4, после очистки экспресс-хроматографией на силикагеле (элюент: CH2Cl2/MeOH, от 99:1 до 9:1) получали 9 мг (13%) транс-[4-(3-диметиламинопроп-1-инил)циклогексил]метил(5-пиридин-4-илпиримидин-2-ил)амина. МС: 350 (МН+).
13.2. транс-[4-(3-Диметиламинопроп-1-инил)циклогексил]метил(5-тиофен-3-илпиримидин-2-ил)амин, МС: 355 (МН+), получали из транс-(5-бромпиримидин-2-ил)[4-(3-диметиламинопроп-1-инил)циклогексил]метиламина и тиофен-3-бороновой кислоты аналогично тому, как описано в примере 13.1.
Пример 14
Раствор 92,1 мг (0,3 ммоля) транс-(6-хлорпиридазин-3-ил)[4-(3-диметиламинопроп-1-инил)циклогексил]метиламина в 0,6 мл ДМА обрабатывали 0,56 мл (3 ммоля) метилата натрия (5,4 М в МеОН) и нагревали при 80°С в течение 54 ч. Реакционную смесь экстрагировали насыщенным раствором NaHCO3/Et2O (3х). Органическую фазу сушили (Na2SO4), фильтровали и упаривали. После очистки экспресс-хроматографией на колонке с силикагелем (элюент: CH2Cl2/MeOH, от 99:1 до 97:3) получали 67 мг (74%) транс-[4-(3-диметиламинопроп-1-инил)циклогексил](6-метоксипиридазин-3-ил)метиламина. МС: 303 (МН+).
Пример 15
15.1. Раствор 81 г (314,77 ммоля) транс-4-[(трет-бутоксиформамидо)метил]циклогексанкарбоновой кислоты в 4 л СН2Cl2 обрабатывали 50,13 г (503,63 ммоля) гидрохлорида N,O-диметилгидроксиламина, 55,37 мл (503,63 ммоля) N-метилморфолина и при 0°С добавляли 78,45 г (409, 2 ммоля) EDCI и 9,67 г (62,95 ммоля) НОВТ. Реакционную смесь перемешивали при комнатной температуре в течение 16 ч, упаривали и экстрагировали 10% раствором KHSO4/Et2O (3х). Органические фазы промывали насыщенным раствором NaHCO3, 10% раствором NaCl, сушили над Na2SO4 и упаривали, при этом получали 100,03 г (выход количественный) трет-бутилового эфира транс-[4-(метоксиметилкарбамоил)циклогексилметил]карбаминовой кислоты. МС: 301 (МН+).
15.2. К раствору 95 г (соответствуют 301,26 ммоля) неочищенного трет-бутилового эфира транс-[4-(метоксиметилкарбамоил)циклогексилметил]карбаминовой кислоты в 300 мл ДМА при 0°С небольшими порциями добавляли 19,72 г (451,9 ммоля) NaH (55% в масле). Реакционную смесь перемешивали при 0°С в течение 1 ч, а затем медленно (в течение 1,5 ч) добавляли 150 мл (2,41 моля) иодметана. После добавления 60 мл иодметана (в течение 1 ч) начиналась реакция и добавление реагента прекращали, после охлаждения реакционной смеси снова добавляли иодметан. После нагревания до КТ в течение ночи реакционную смесь охлаждали, нейтрализовали добавлением 10% раствора KHSO4 и выливали в воду/Et2О (3х). Органическую фазу промывали 10% раствором NaCl, сушили над Na2SO4, упаривали и очищали экспресс-хроматографией на колонке с силикагелем (элюент: CH2Cl2/EtOAc, от 9:1 до 1:1), при этом получали 99 г (выход количественный) трет-бутилового эфира транс-[4-(метоксиметилкарбамоил)циклогексилметил]метилкарбаминовой кислоты. МС: 315 (МН+).
15.3. Раствор 12,25 г (313,11 ммоля) LAH в 1,3 л ТГФ охлаждали (-50°С) и в течение 30 мин обрабатывали раствором 89,5 г (284,64 ммоля) трет-бутилового эфира транс-[4-(метоксиметилкарбамоил)циклогексилметил]метилкарбаминовой кислоты в 1,3 л ТГФ. После выдерживания при указанной температуре в течение 20 мин реакционную смесь нагревали до 0°С, охлаждали (-78°С) и гидролизовали добавлением суспензии 90 г MgSO4·7H2O, 90 г силикагеля в 292 мл 10% раствора KHSO4. Охлаждающую баню удаляли, добавляли ТГФ, смесь перемешвали в течение 30 мин и фильтровали. После упаривания остаток растворяли в CH2Cl2, раствор сушили над Na2SO4 и упаривали, при этом получали 90,4 г (выход количественный) трет-бутилового эфира транс-(4-формилциклогексилметил)метилкарбаминовой кислоты. МС: 255 (М).
15.4. Раствор 257,6 г (982 ммоля) трифенилфосфина в 1 л CH2Cl2 обрабатывали 162,8 г (491 ммоль) тетрабромметана (реакционная смесь нагревалась до кипения растворителя и ее охлаждали на ледяной бане), затем после выдерживания смеси при КТ в течение 40 мин добавляли 157, 4 мл (1129 ммолей) триэтиламина (реакционная смесь разогревалась до температуры кипения растворителя и смеси приобретали темно-фиолетовый цвет). После охлаждения до 0°С к смеси в течение 20 мин добавляли 77,96 г (соответствуют 245,5 ммоля) неочищенного трет-бутилового эфира транс-(4-формилциклогексилметил)метилкарбаминовой кислоты в 600 мл CH2Cl2. Раствор перемешивали при КТ в течение 20 ч, концентрировали и фильтровали через силикагель (инактивированный гексаном/0,5% Et3N, элюент: гексан/Et2О, от 99:1 до 4:1), при этом получали 61,5 г (61%) трет-бутилового эфира транс-[4-(2,2-дибромвинил)циклогексилметил]метилкарбаминовой кислоты. МС: 409 (М, 2Br).
15.5. Следующую реакцию проводили аналогично тому, как описано в книге Marshall James A., Bartley Gary S., Wallace Eli M., Total Synthesis of the Pseudopteran (-)-Kallolode B, the Enantiomer of Natural (+)-Kallolide В., J.Org. Chem. 61(17), 5729-5735 (1996), и в статье Baker Raymond, Boyes Alastair L., Swain Christopher J., Synthesis of talaromycins А, В, С and E., J.Chem. Soc., Perkin Trans. 1(5), 1415-21 (1990). Раствор 32,9 г (80 ммолей) трет-бутилового эфира транс-[4-(2,2-дибромвинил)циклогексилметил]метилкарбаминовой кислоты в 640 мл ТГФ обрабатывали при -78°С 105 мл (168 ммолей) BuLi (приблизительно 1,6 М в гексане). После выдерживания смеси при указанной температуре в течение 2 ч добавляли 24 г (800 ммолей) параформальдегида. Реакционную смесь нагревали до КТ в течение 3 ч и после выдерживания при указанной температуре в течение 0,5 ч смесь экстрагировали водой/Et2О (3х). Органические фазы промывали 10% раствором NaCl, сушили над Na2SO4, упаривали, остаток очищали экспресс-хроматографией на силикагеле (элюент: гексан/EtOAc, от 9:1 до 2:1), при этом получали 12,1 г (54%) трет-бутилового эфира транс-[4-(3-гидроксипроп-1-инил)циклогексилметил]метилкарбаминовой кислоты. МС: 282 (МН+).
15.6. транс-3-(4-Метиламинометилциклогексил)проп-2-ин-1-ол, tпл. 97-99°С, МС: 182 (МН+), получали из трет-бутилового эфира транс-[4-(3-гидроксипроп-1-инил)циклогексилметил]метилкарбаминовой кислоты аналогично тому, как указано в примере 1.6.
15.7. транс-3-(4-{[(5-Бромпиримидин-2-ил)метиламино]метил }циклогексил)проп-2-ин-1-ол, tпл. 121-122°C, МС: 338 (МН+, 1Br), получали при взаимодействии транс-3-(4-метиламинометилциклогексил)проп-2-ин-1-ола с 5,4 экв. N-этилдиизопропиламина и 1,2 экв. 2,5-дибромпиримидина (Brown Desmond J., Arantz B.W., Pyrimidine reactions. XXII. Relative reactivities of corresponding chloro-, bromo-, iodo-pyrimidines in aminolysis, J.Chem. Soc. C, Issue 10, 1889-91 (1971)) в в отсутствие NaI при нагревании при 120°С в течение 3 ч в микроволновой печи аналогично тому, как описано в примере 1.15.
15.8. 3-(4-{[(5-Бромпиримидин-2-ил)метиламино]метил}циклогексил)проп-2-иниловый эфир транс-метансульфоновой кислоты, МС: 416 (МН+, 1Br), получали из транс-3-(4-{[(5-бромпиримидин-2-ил)метиламино]метил}циклогексил)проп-2-ин-1-ола аналогично тому, как описано в примере 1.26.
15.9. транс-3-(4-{[(5-Этилпиримидин-2-ил)метиламино]метил}циклогексил)проп-2-ин-1-ол, tпл. 69-71°С, МС: 228 (МН+), получали при взаимодествии транс-3-(4-метиламинометилциклогексил)проп-2-ин-1-ол с 5 экв. N-этилдиизопропиламина и 4 экв. 2-хлор-5-этилпиримидина в отсутствие NaI при нагревании при 120°С в течение 3,75 ч в микроволновой печи аналогично тому, как описано в примере 1.15.
15.10. 3-(4-{[(5-Этилпиримидин-2-ил)метиламино]метил}циклогексил)проп-2-иниловый эфир транс-метансульфоновой кислоты, МС: 366 (МН+), получали из транс-3-(4-{[(5-этилпиримидин-2-ил)метиламино]метил}циклогексил)проп-2-ин-1-ола аналогично тому, как описано в примере 1.26.
15.11. транс-3-(4-{[(6-Хлорпиридазин-3-ил)метиламино]метил}циклогексил)проп-2-ин-1-ол, МС: 294 (МН+, 1Cl), получали при взаимодействии транс-3-(4-метиламинометилциклогексил)проп-2-ин-1-ола с 3,4 экв. N-этилдиизопропиламина и 4 экв. 3,6-дихлорпиридазина в отсутствие NaI при нагревании при 120-140°С в течение 30 мин в микроволновой печи аналогично тому, как описано в примере 1.15.
15.12. 3-(4-{[(6-Хлорпиридазин-3-ил)метиламино]метил}циклогексил)проп-2-иниловый эфир транс-метансульфоновой кислоты, МС: 372 (МН+, Cl), получали из транс-3-(4-{[(6-хлорпиридазин-3-ил)метиламино]метил}циклогексил)проп-2-ин-1-ола аналогично тому, как описано в примере 1.26.
Пример 16
Раствор 323 мг (соответствуют 0,49 ммоля) 3-(4-{[(5-бромпиримидин-2-ил)метиламино]метил}циклогексил)проп-2-инилового эфира транс-метансульфоновой кислоты в 5 мл метанола охлаждали (0°С), обрабатывали каталитическим количеством NaI, 0,88 мл (4,94 ммоля) диметиламина (33% в EtOH, 5,6 М) и перемешивали при КТ в течение 16 ч. Растворитель упаривали, остаток экстрагировали насыщенным раствором NaHCO3/Et2O (3х). Органические фазы сушили над Na2SO4, фильтровали и упаривали. После очистки экспресс-хроматографией на колонке с силикагелем (элюент: СН2Cl2/МеОН, 40:1) получали 137 мг (76%) чистого транс-(5-бромпиримидин-2-ил)[4-(3-диметиламинопроп-1-инил)циклогексилметил]метиламина. tпл. 71-72°С. МС: 365 (МН+, 1Br).
Следующие соединения получали из соответствующих мезилатов и вторичных аминов.
Таблица 5
Пример 17
17.1. К суспензии 50 г (0,33 моля) гидрохлорида транс-4-аминоциклогексанола и 77 г (0,726 моля, 2,2 экв.) Na2СО3 в 650 мл ТГФ и 150 мл воды при 5°С в течение 20 мин добавляли 51,2 мл (0,363 моля, 1,1 экв.) бензилхлорформиата. Реакционную смесь перемешивали при КТ в течение 2 ч, разбавляли EtOAc и фазы разделяли. Органический слой промывали солевым раствором, сушили над Na2SO4, фильтровали и упаривали. После растирания в гексане получали 162,4 г (98%) бензилового эфира транс-4-гидроксициклогексилкарбаминовой кислоты в виде кристаллов белого цвета. МС: 249 (М) (аналогично тому, как описано Venuti Michael С., Jones Gordon H., Alvarez Robert, Bruno John J., J.Med. Chem., 30, 2, 303-318 (1987)).
17.2. К суспензии 37,9 г (0,94 моля, 2,0 экв.) LAH в 1,3 л ТГФ через трубку при температуре 5-10°С в течение 6 ч добавляли суспензию 117 г (0,47 моля) бензилового эфира транс-4-гидроксициклогексилкарбаминовой кислоты в 1 л ТГФ. Реакционную смесь кипятили с обратным холодильником в течение ночи, добавляли смесь Na2SO4, силикагеля и воды (160 г, 50 г, 80 мл), перемешивали в течение еще 30 мин, фильтровали и концентрировали. Неочищенный продукт растирали в гексане, при этом получали 27,9 г (46%) транс-4-метиламиноциклогексанола. После хроматографии маточного раствора на колонке с силикагелем получали дополнительное количество (17,1 г, 28%) транс-4-метиламиноциклогексанола в виде твердого вещества белого цвета. МС: 129 (МН+) (аналогично тому, как описано Venuti Michael С., Jones Gordon H., Alvarez Robert, Bruno John J., J. Med. Chem., 30, 2, 303-318 (1987)).
17.3. транс-4-[(5-Бромпиримидин-2-ил)метиламино]циклогексанол, tпл. 140-142, МС: 286 (МН+, 1Br), получали из транс-4-метиламиноциклогексанол и 2,5-дибромпиримидина аналогично тому, как описано в примере 5.4.
17.4. Раствор 2,47 г (8,62 ммоля) транс-4-[(5-бромпиримидин-2-ил)метиламино]циклогексанола, 5,53 г (25,86 ммоля) транс-1,4-дибром-2-бутена и 0,87 г (2,57 ммоля, 0,3 экв.) гидросульфата тетрабутиламмония в 55 мл CH2Cl2 обрабатывали 55 мл 50% раствора NaOH. Смесь перемешивали при КТ в течение 40 ч, добавляли 2,76 г (12,93 ммоля) транс-1,4-дибром-2-бутена и перемешивали в течение еще 60 ч. Затем добавляли CH2Cl2 и слои разделяли. Водный слой и слой экстрагировали CH2Cl2 (3х), объединенные органические слои промывали солевым раствором и сушили над Na2SO4. Остаток очищали хроматографией на колонке с силикагелем (элюент: гексан/EtOAc, от 9:1 до 2:1), при этом получали 0,8 г (22%) транс-(2Е)-[4-(4-бромбут-2-енилокси)циклогексил](5-бромпиримидин-2-ил)метиламина в виде твердого вещества светло-желтого цвета. МС: 418 (МН+, 2Br).
Пример 18
Следующие соединения получали из соответствующих бромида и вторичных аминов аналогично тому, как описано в примере 2.
Таблица 6
Пример 19
Раствор 0,2 г (0,7 ммоля) транс-4-[(5-бромпиримидин-2-ил)метиламино]циклогексанола и 0,24 г (1,4 ммоля) гидрохлорида 1-(2-хлорэтил)пирролидина в 3,5 мл ДМА при 0°С обрабатывали небольшими порциями 0,24 г (5,59 ммоля) NaH (55% в масле) и реакционную смесь перемешивали при 0°С в течение 30 мин. После нагревания до КТ к смеси добавляли каталитическое количество NaI и перемешивали при 80°С в течение 1 ч. Реакционную смесь охлаждали и выливали в воду/Et2О (3х). Органическую фазу сушили над Na2SO4 и упаривали, остаток очищали хроматографией на колонке с силикагелем (элюент: СН2Cl2/МеОН, от 99:1 до 9:1), при этом получали 13 г (5%) транс-(5-бромпиримидин-2-ил)метил[4-(2-пирролидин-1-илэтокси)циклогексил]амина. МС: 383 (МН+, 1Br).
Пример 20
20.1. транс-2-[(4-{(5-Бромпиримидин-2-ил)метиламино]этил}циклогексилметил)этиламино]этанол, МС: 399 (МН+, 1Br), получали при взаимодействии 4-{2-[(5-бромпиримидин-2-ил)метиламино]этил}циклогексилметилового эфира транс-метансульфоновой кислоты с этил(2-гидроксиэтил)амином в присутствии 1 экв. NaI в ДМА при 60°С в течение 22 ч, аналогично тому, как описано в примере 12.
20.2. транс-3-[(4-{2-[(5-Бромпиримидин-2-ил)метиламино]этил}циклогексилметил)амино]пропан-1-ол, МС: 385 (МН+, 1Br), получали при взаимодействии 4-{2-[(5-бромпиримидин-2-ил)метиламино]этил}циклогексилметилового эфира транс-метансульфоновой кислоты с 3-амино-1-пропанолом в присутствии 1 экв. NaI в ДМА при 60°С в течение 46 ч аналогично тому, как описано в примере 12.
Пример 21
Раствор 0,21 г (0,6 ммоля) транс-3-[(4-{2-[(5-бромпиримидин-2-ил)метиламино]этил}циклогексилметил)амино]пропан-1-ола в 3 мл диоксана обрабатывали 3 мл 1 н. раствора NaH2PO3 и 3 мл 36% раствора формальдегида (Loibner H., A. Pruckner и др.. Reductive methylation of primary and secondary amines with formaldehyde and phosphorous acid salts, Tetrahedron Lett., 25 (24), 2535-6 (1984)). Смесь нагревали при 60°С в течение 30 мин, затем охлаждали и экстрагировали 2 н. NaOH/эфиром (3х). Органическую фазу промывали 10% раствором NaCl, сушили над Na2SO4 и упаривали, остаток очищали экспресс-хроматографией на колонке с силикагелем (элюент: СН2Cl2/МеОН, от 98:2 до 9:1), при этом получали 0,17 г (76%) транс-3-[(4-{2-[(5-бромпиримидин-2-ил)метиламино]этил}циклогексилметил)метиламино]пропан-1-ола. МС: 399 (МН+, 1Br).
Пример 22
22.1. транс-3-(4-{[Метил(5-пропилпиримидин-2-ил)амино]метил}циклогексил)проп-2-ин-1-ол, tпл. 78-79°С, МС: 302 (МН+), получали при взаимодействии транс-3-(4-метиламинометилциклогексил)проп-2-ин-1-ола с 5,4 экв. N-этилдиизопропиламина, 1,2 экв. 2-хлор-5-н-пропилпиримидина в отсутствие NaI при нагревании в микроволновой печи при 120°С в течение 4 ч аналогично тому, как описано в примере 1.15.
22.2. 3-(4-{[Метил(5-пропилпиримидин-2-ил)амино]метил}циклогексил)проп-2-иниловый эфир транс-метансульфоновой кислоты, МС: 380 (МН+), получали из транс-3-(4-{[метил(5-пропилпиримидин-2-ил)амино]метил}циклогексил)проп-2-ин-1-ола аналогично тому, как описано в примере 1.26.
22.3. транс-3-(4-{[(5-Хлорпиримидин-2-ил)метиламино]метил }циклогексил)проп-2-ин-1-ол, tпл. 108-110°C, МС: 294 (МН+, 1Cl), получали при взаимодействии транс-3-(4-метиламинометилциклогексил)проп-2-ин-1-ола с 5,4 экв. N-этилдиизопропиламина и 1,2 экв. 2-бром-5-хлорпиримидина (полученного из 5-хлор-2-гидроксипиримидина аналогично тому, как описано Brown Desmond J., Arantz B.W., Pyrimidine reactions. XXII. Relative reactivities of corresponding chloro-, bromo-, iodo-pyrimidines in aminolysis, J. Chem. Soc. C, Issue 10, 1889-91 (1971)) в отсутствие NaI при нагревании при 120°С в течение 2 ч в микроволновой печи аналогично тому, как описано в примере 1.15.
22.4. 3-(4-{[(5-Хлорпиримидин-2-ил)метиламино]метил}циклогексил)проп-2-иниловый эфир транс-метансульфоновой кислоты, МС: 372 (МН+, 1Cl), получали из транс-3-(4-{[(5-хлорпиримидин-2-ил)метиламино]метил}циклогексил)проп-2-ин-1-ола аналогично тому, как описано в примере 1.26.
Пример 23
23.1. транс-[4-(3-Диметиламинопроп-1-инил)циклогексилметил]метил(5-пропилпиримидин-2-ил)амин, tпл. 49-50°С, МС: 329 (МН+), получали из 3-(4-{[метил(5-пропилпиримидин-2-ил)амино]метил}циклогексил)проп-2-инилового эфира транс-метансульфоновой кислоты и диметиламина (33% в EtOH, 5,6 М) в ДМА, аналогично тому, как описано в примере 16.
Следующие соединения пролучали из соответствующих мезилатов и вторичных аминов.
Таблица 7
Примеры
Пример А
Таблетки с пленочным покрытием следующего состава получают обычным способом:
Активный ингредиент просеивают, смешивают с микрокристаллической целлюлозой и смесь гранулируют при добавлении раствора поливинилпирролидона в воде. Гранулят смешивают с натрийгликолятом крахмалом и стеаратом магния и прессуют в ядра массой 120 мг или 350 мг соответственно. Ядра покрывают водным раствором/суспензией пленочного покрытия указанного выше состава.
Пример Б
Капсулы следующего состава получают обычным способом.
Компоненты просеивают, смешивают и смесью заполняют капсулы размера 2.
Пример В
Растворы для инъекции получают следующего состава.
Активный ингредиент растворяют в смеси полиэтиленгликоля 400 и воды для инъекции (часть объема). Добавляют уксусную кислоту до 5,0, добавляют воду до 1 мл. Раствор фильтруют, заполняют флаконы с использованием соответствующего избытка раствора, затем стерилизуют.
Пример Г
Мягкие желатиновые капсулы следующего состава получают обычным способом.
Активный ингредиент растворяют в теплом расплаве остальных ингредиентов и смесью наполняют мягкие желатиновые капсулы соответствующего размера. Заполненные мягкие желатиновые капсулы обрабатывают стандартными методами.
Пример Д
Пакетики следующего состава получают стандартным способом.
Активный ингредиент смешивают с лактозой, микрокристаллической целлюлозой и натрийкарбоксиметилцеллюлозой, затем гранулируют в смеси с поливинилпирролидоном в воде. Гранулят смешивают со стеаратом магния и ароматизирующими добавками, смесью заполняют пакетики.
| название | год | авторы | номер документа |
|---|---|---|---|
| ЗАМЕЩЕННЫЕ ПРОИЗВОДНЫЕ ЦИКЛОГЕКСАНА | 2002 |
|
RU2288221C2 |
| Новые производные аминоциклогексана | 2001 |
|
RU2225393C1 |
| ПРОИЗВОДНЫЕ ПИРРОЛИДИНА | 2010 |
|
RU2562605C2 |
| ПРОИЗВОДНЫЕ 1,2-ДИАМИНОПЕНТАНА КАК АНТАГОНИСТЫ РЕЦЕПТОРА CCR-3 И ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ НА ИХ ОСНОВЕ | 2002 |
|
RU2286339C2 |
| ПРОИЗВОДНЫЕ НИТРИЛОВ β-АМИНОКИСЛОТ | 2001 |
|
RU2245871C2 |
| ПРОИЗВОДНЫЕ ДОЛАСТАТИНА 10 | 2002 |
|
RU2296750C2 |
| Способ получения соединений 7H-пирроло[2,3-d]пиримидина | 2017 |
|
RU2699034C1 |
| ПРОИЗВОДНЫЕ 2-АМИНОНИКОТИНАМИДА И ИХ ПРИМЕНЕНИЕ В КАЧЕСТВЕ ИНГИБИТОРОВ VEGF-РЕЦЕПТОРА ТИРОЗИНКИНАЗЫ | 2001 |
|
RU2296124C2 |
| АЗОЛЬНЫЕ И ТИАЗОЛЬНЫЕ ПРОИЗВОДНЫЕ И ИХ ПРИМЕНЕНИЯ | 2006 |
|
RU2436779C2 |
| ПРОИЗВОДНЫЕ ЦИКЛОАЛКИЛАМИНА | 2005 |
|
RU2402526C2 |
Изобретение относится к новым производным гетероарилзамещенного аминоциклогексана формулы (I) и их фармацевтически приемлемым солям, обладающим ингибирующим действием в отношении 2,3-оксидосквален-ланостеринциклазы (OSC). Соединения могут найти применение для лечения таких заболеваний, как гиперхолестеринемия, гиперлипемия, артериосклероз, сосудистые заболевания, микозы, паразитарные инфекции, желчнокаменная болезнь, опухоли и/или гиперпролиферативные нарушения, и/или нарушенной толерантности к глюкозе и диабета. В формуле (I)
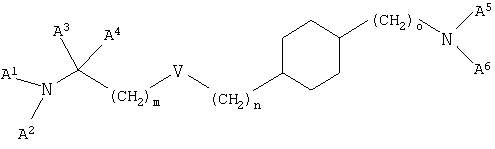
V означает простую связь, О, -CH=СН-CH2-O-, или -C≡С-, m и n независимо друг от друга равны 0-7 и m+n равно 0-7 при условии, что m не равно 0, если V означает O, o равно 0-2, А1 означает водород, низший алкил, гидрокси-низший алкил или низший алкенил, А2 означает низший алкил или А1 и А2 связаны друг с другом с образованием 5-6-членного цикла и -А1-А2- означает С4-C5алкилен, А3 и А4 независимо друг от друга означают водород; А5 означает водород, низший алкил; А6 означает пиридинил, пиридазинил, пиримидинил или пиразинил, необязательно замещенный одним заместителем, независимо выбранными из группы, включающей галоген, низший алкил, низший алкокси и 5-6-членный гетероарил, содержащий в качестве гетероатома азот или серу. Изобретение также относится к фармацевтической композиции и применению соединений для получения лекарственных средств. 4 н. и 20 з.п. ф-лы, 7 табл.
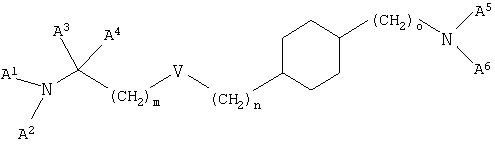
V означает простую связь, -О, -CH=СН-CH2-O- или -C≡С-, m и n независимо друг от друга равны 0-7 и m+n равно 0-7 при условии, что m не равно 0, если V означает О,
о равно 0-2;
А1 означает водород, низший алкил, гидроксинизший алкил или низший алкенил;
А2 означает низший алкил, или
А1 и А2 связаны друг с другом с образованием 5-6-членного цикла и -А1-А2- означает С4-C5алкилен;
А3 и А4 независимо друг от друга означают водород;
А5 означает водород, низший алкил;
А6 означает пиридинил, пиридазинил, пиримидинил или пиразинил, необязательно замещенный одним заместителем, независимо выбранными из группы, включающей галоген, низший алкил, низший алкокси и 5-6-членный гетероарил, содержащий в качестве гетероатома азот или серу,
и их фармацевтически приемлемые соли, при условии, что соединение формулы (I) не означает транс-[4-(2-дипропиламиноэтил)циклогексил]пиримидин-2-иламин.
транс-(5-бромпиримидин-2-ил)метил-{4-[3-(метилпропиламино)проп-1-инил]циклогексил}амин,
транс-(5-бромпиримидин-2-ил)[4-(3-диметиламинопроп-1-инил)циклогексил] метиламин,
транс-(6-хлорпиридазин-3-ил)[4-(3-диметиламинопроп-1-инил)циклогексил]метиламин,
транс-(5-хлорпиримидин-2-ил)[4-(3-диметиламинопроп-1-инил)циклогексил]метиламин,
транс-[(5-бромпиримидин-2-ил)метил[4-(3-пиперидин-1-илпроп-1-инил)циклогексил]амин],
транс-[4-(3-диметиламинопроп-1-инил)циклогексил]метил-(5-пиридин-4-илпиримидин-2-ил)амин,
транс-[4-(3-диметиламинопроп-1-инил)циклогексил](5-этилпиримидин-2-ил)метиламин,
транс-(5-бромпиримидин-2-ил)[4-(3-диметиламинопроп-1-инил)циклогексилметил]метиламин,
транс-2-{[3-(4-{[(5-бромпиримидин-2-ил)метиламино]метил}циклогексил)проп-2-инил]этиламино}этанол,
транс-2-{этил[3-(4-{[(5-этилпиримидин-2-ил)метиламино]метил}циклогексил)проп-2-инил]амино}этанол,
транс-(5-этилпиримидин-2-ил)метил[4-(3-пиперидин-1-илпроп-1-инил)циклогексилметил]амин и
транс-2-{[3-(4-{[(6-хлорпиридазин-3-ил)метиламино]метил}циклогексил)проп-2-инил]этиламино}этанол,
и их фармацевтически приемлемые соли.
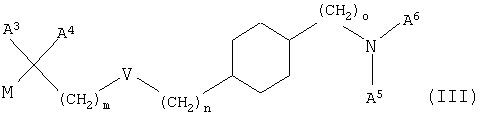
с соединением NHA1A2, где М означает мезилат, тозилат, трифлат, Cl, Br или I, a А1, А2, А3, А4, А5, А6, V, m, n и о имеют значения, указанные в п.1, и необязательно превращение полученного соединения в его фармацевтически приемлемую соль.
| ЕР 778264 А1, 11.06.1997 | |||
| WERUTH et al | |||
| The Practise of Medicinal Chemistry | |||
| Practice Medicinal Chemistry | |||
| Предохранительное устройство для паровых котлов, работающих на нефти | 1922 |
|
SU1996A1 |
| АМИДЫ, ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ, СПОСОБЫ СНИЖЕНИЯ СЕКРЕЦИИ АПОЛИПОПРОТЕИНА В, СОЕДИНЕНИЕ | 1996 |
|
RU2141478C1 |

