Область изобретения
Данное изобретение относится к применению новых антибактериальных соединений N-формил-N-гидроксиламина и фармацевтических композиций, содержащих эти соединения, в качестве ингибиторов пептиддеформилазы.
Предпосылки изобретения
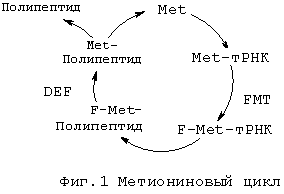
Бактериальная инициаторная метионил-тРНК модифицируется метионил-тРНК-формилтрансферазой (FMT) с образованием формилметионил-тРНК. Затем формилметионин (f-Met) присоединяется к N-концу синтезированных полипептидов. Затем полипептиддеформилаза (PDF или Def) деформилирует первичные продукты трансляции с образованием N-метионилполипептидов. Большинство внутриклеточных белков дополнительно процессируются метионинаминопептидазой (MAP) с образованием зрелого пептида и свободного метионина, который рециркулируется. Ферменты PDF и MAP, оба, являются существенными для бактериального роста, и PDF является необходимым для активности MAP. Эту серию реакций называют метиониновым циклом (фиг.1).
К настоящему времени гомологичные гены полипептиддеформилазы были обнаружены в бактериях, в содержащих хлоропласты растениях, у мышей и у человека. Растительные белки являются ядерно кодируемыми, но, по-видимому, несут сигнал локализации в хлоропластах. Это согласуется с наблюдением, что хлоропластная РНК и процессы синтеза белка являются высоко подобными РНК и синтезу белка эубактерий. Хотя имеется ограниченная информация об экспрессии белка гомологов гена PDF млекопитающих (Bayer Aktiengesellschaft, Pat. WO 2001/42431), функциональная роль таких белков до настоящего времени не была обнаружена (Meinnel T. 2000, Parasitology Today, 16(4), 165-168).
Полипептиддеформилаза обнаружена во всех эубактериях, для которых информация высокого охвата геномной последовательности является доступной. Разнообразие последовательности среди гомологов PDF является высоким с всего лишь 20% идентичностью между отдаленно родственными последовательностями. Однако консервативность около активного сайта является очень высокой, с несколькими полностью консервативными остатками, включающими один цистеин и два гистидина, которые являются необходимыми для координации металла активного сайта (Meinnel, T. et al., 1997, Journal of Molecular Biology, 267, 749-761).
Считают, что фермент PDF является привлекательной антибактериальной мишенью, так как было продемонстрировано, что этот фермент является важным для бактериального роста in vitro (Mazel, D. et al., EMBO J. 13(4), 914-923, 1994), не считают, что он участвует в синтезе эукариотических белков (Rajagopalan et al., J. Am. Chem. Soc. 119, 12418-12419, 1997), и он является универсально консервативным в прокариотах (Kozak, M. Microbiol. Rev. 47, 1-45, 1983). Таким образом, ингибиторы PDF потенциально могут служить в качестве антибактериальных агентов широкого спектра.
Сущность изобретения
Данное изобретение относится к новым антибактериальным соединениям формулы (I) и их применению в качестве ингибиторов PDF.
Подробное описание изобретения
Соединения, применимые в способах данного изобретения, выбраны из соединений формулы (I), ниже:
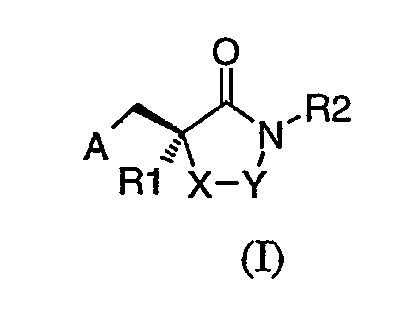
где
R1 выбран из группы, состоящей из С1-9алкила, С1-2алкилAr и Ar;
R2 выбран из группы, состоящей из водорода, С1-9алкила, С1-4алкилAr', NR4, NC(O)R4, С2-4алкилNR3R4, С1-3алкилС(О)NR3R4, С1-3алкилС(О)Ar', С2-3алкилNHC(O)NR3R4, С2-3алкилNHC(O)Ar' и С1-2алкилSO2R4;
R3 выбран из группы, состоящей из С1-9алкила, С1-2алкилAr и Ar;
R4 обозначает R3, Ar' или R4 может быть взят вместе с R3 и атомом азота, к которому они присоединены, для образования гетероциклического кольца, которое является необязательно замещенным одним, двумя или тремя заместителями, выбранными из группы, состоящей из С1-3алкила, арила, С1-3алкокси (необязательно замещенного от одного до трех F), арилокси, карбокси, оксо, гидрокси, амино, нитро и циано, или которое может быть необязательно конденсировано с арилом, гетероарилом или вторым гетероциклическим кольцом;
Ar выбран из группы, состоящей из фенила, фурила и тиенила, которые все могут быть необязательно замещены одним, двумя или тремя заместителями, выбранными из группы, состоящей из С1-3алкила, CN, F, Cl, Br и I;
Ar' выбран из группы, состоящей из фенила, нафтила, фурила, пиридила, тиенила, тиазолила, изотиазолила, пиразолила, триазолила, тетразолила, имидазолила, имидазолидинила, бензофуранила, индолила, тиазолидинила, изоксазолила, оксадиазолила, тиадиазолила, пирролила и пиримидила, которые все могут быть необязательно замещены одним, двумя или тремя заместителями из группы, состоящей из С1-6алкила, С1-6алкокси, (СН2)0-5СО2R1, C(O)N(R1)2, CN, (CH2)0-5OH, NO2, F, Cl, Br, I, CF3, N(R1)2 и NHC(O)R1;
А выбран из группы C(O)NHOH или N(CHO)OH;
Х обозначает NH, когда Y обозначает С(О), или Х обозначает СН2, когда Y обозначает С(О) или СН2.
"Алкил" обозначает необязательно замещенную углеводородную группу, соединенную вместе связями углерод-углерод. Алкильная углеводородная группа может быть линейной, разветвленной или циклической.
"Арил" обозначает необязательно замещенную ароматическую группу с, по меньшей мере, одним кольцом, имеющим конъюгированную пи-электронную систему, содержащую до двух конъюгированных или конденсированных кольцевых систем. "Арил" включает карбоциклическую арильную, гетероциклическую арильную и биарильную группы, которые все могут быть необязательно замещенными.
"Гетероциклический" означает трех-семичленное кольцо, содержащее один или несколько гетероатомных фрагментов, выбранных из S, SO, SO2, O или N. Такое кольцо может быть насыщенным или иметь одну или несколько степеней ненасыщенности. Примеры "гетероциклических" частей молекулы включают, но не ограничиваются ими, морфолинил, пиперидинил и пиперазинил.
Предпочтительные соединения, применимые в данном изобретении, выбраны из группы, включающей:
N-[(S)-1-бензил-4-пентил-2,5-диоксоимидазолидин-4-илметил]-N-гидроксиформамид;
N-[(S)-1,4-дибензил-2,5-диоксоимидазолидин-4-илметил]-N-гидроксиформамид;
N-[(S)-1-бензил-4-бутил-2,5-диоксоимидазолидин-4-илметил]-N-гидроксиформамид;
N-[(S)-2,5-диоксо-4-пентил-1-фенилимидазолидин-4-илметил]-N-гидроксиформамид;
N-[(S)-4-бутил-(3,4-дихлорбензил)-2,5-диоксоимидазолидин-4-илметил]-N-гидроксиформамид;
N-[(S)-4-бутил-2,5-диоксо-1-(2-оксо-2-фенилэтил)имидазолидин-4-илметил]-N-гидроксиформамид;
N-[(S)-1-бифенил-4-илметил-4-бутил-2,5-диоксоимидазолидин-4-илметил]-N-гидроксиформамид;
N-[(S)-1-бензил-4-циклогексилметил-2,5-диоксоимидазолидин-4-илметил]-N-гидроксиформамид;
N-{(S)-4-бутил-1-[2-(5-хлор-3-метил-1-бензо[b]тиофен-2-ил)-2-оксоэтил]-2,5-диоксоимидазолидин-4-илметил}-N-гидроксиформамид;
2-{(S)-4-бутил-4-[(формилгидроксиамино)метил]-2,5-диоксоимидазолидин-1-ил}-N-(3,5-дихлорфенил)ацетамид;
Метиловый эфир 2-{(S)-4-бутил-4-[(формилгидроксиамино)метил]-2,5-диоксоимидазолидин-1-илметил}бензойной кислоты;
N-[(S)-4-бутил-1-(2-морфолин-4-илэтил)-2,5-диоксоимидазолидин-4-илметил]-N-гидроксиформамид;
N-[(S)-1-(2-бензофуран-2-ил-2-оксоэтил)-4-бутил-2,5-диоксоимидазолидин-4-илметил]-N-гидроксиформамид и
2-{(S)-4-бутил-4-[(формилгидроксиамино)метил]-2,5-диоксоимидазолидин-1-илметил}бензойной кислоты.
В данное изобретение включены также фармацевтически приемлемые соли и комплексы, такие как хлористоводородные, бромистоводородные, трифторацетатные соли, соли натрия, калия и магния. Соединения данного изобретения могут содержать один или более асимметричных атомов углерода и могут существовать в рацемической и оптически активных формах. Предполагается, что все эти соединения и диастереомеры находятся в рамках данного изобретения.
Соединения формулы (I) могут быть получены в соответствии со следующими репрезентативными схемами, которые являются иллюстрацией используемых способов и не предназначены для ограничения объема данного изобретения, определяемого прилагаемой формулой изобретения. Соединения формулы (I) могут быть получены способом, аналогичным схеме 1.
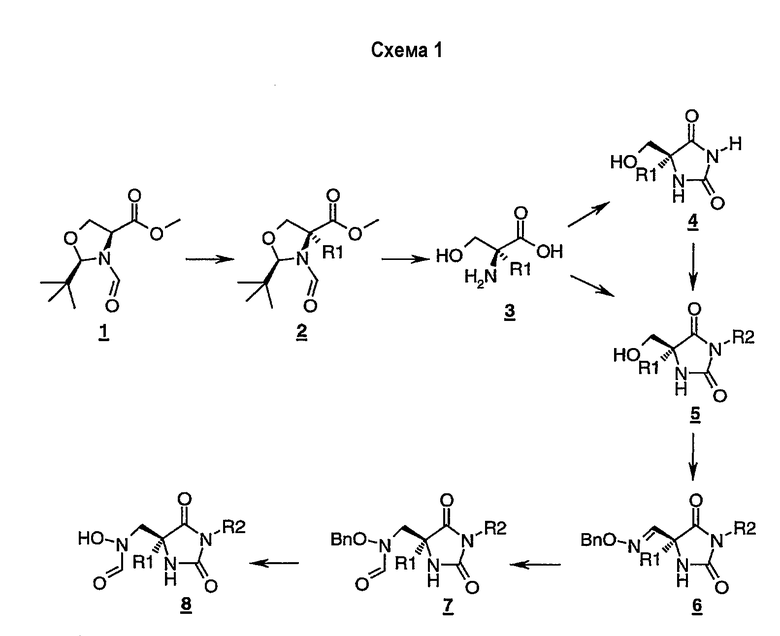
Производное оксазолидина 1-схема-1 может быть получено из метилового эфира L-серина, как описано в литературе [D. Seebach, J.D.Aebi, M. Gander-Coquoz and R. Naef, Helv. Chim. Acta, 70, 1194 (1987)]. Обработка 1-схема-1 подходящим основанием, таким как бис(триметилсилил)амид натрия, в присутствии реакционноспособного галогенида R1Х в смешанном растворителе тетрагидрофуран/гексаметилфосфорамид (10:1) дает сложный эфир 2-схема-1, который может быть гидролизован в кислотных условиях с получением замещенного серина 3-схема-1. Производное N-замещенного гидантоина 5-схема-1 может быть получено прямой обработкой 3-схема-1 изоцианатом R2NCO, или, альтернативно, при обработке 3-схема-1 цианатом калия получают гидантоин 4-схема-1, который может быть затем алкилирован галогенидом R2Х. Спирт 5-схема-1 может быть окислен подходящим реагентом, таким как периодинан Десса-Мартина, с получением альдегида, который может быть обработан О-бензилгидрокисламином с получением оксима 6-схема-1. Восстановление оксима 6-схема-1 цианоборгидридом натрия с последующей обработкой смешанным ангидридом, образованным из муравьиной кислоты и уксусного ангидрида, дает формамид 7-схема-1. Наконец, N-формил-N-гидроксиламин 8-схема-1 получают гидрогенолизом 7-схема-1 в спиртовом растворителе в присутствии катализатора, такого как палладий на активированном угле.
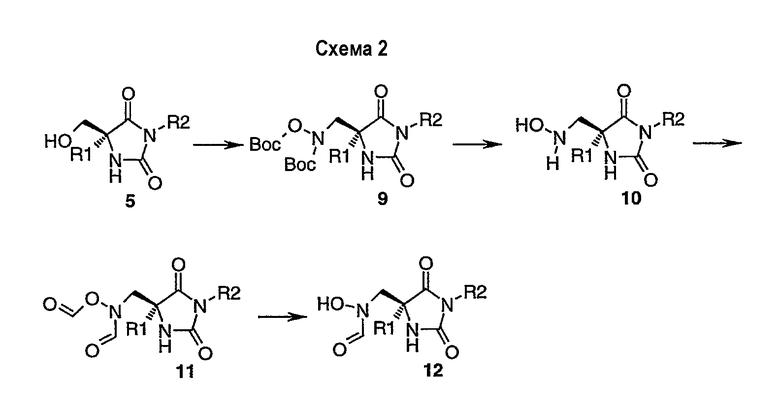
Альтернативно, соединения формулы (I) могут быть получены из спирта 5-схема-1, как показано в схеме 2. Реакция Митцунобу 5-схема-1 дает соединение 9-схема-2. Удаление защитных групп в кислотных условиях обеспечивает производное гидроксиламина 10-схема-2. Обработка 10-схема-2 смешанным ангидридом, образованным из муравьиной кислоты и уксусного ангидрида, приводит к N,O-формилированному соединению 11-схема-2, и удаление О-формильной группы щелочным гидролизом дает N-формил-N-гидроксиламин 12-схема-2.
Вышеописанные способы могут быть более понятными со ссылкой на следующие примеры, которые иллюстрируют способы, при помощи которых могут быть получены соединения данного изобретения, и не предназначены для ограничения объема данного изобретения, определяемого прилагаемой формулой изобретения.
Пример 1
Получение N-[(S)-1-бензил-4-бутил-2,5-диоксоимидазолидин-4-илметил]-N-гидроксиформамида, SB-725291
1а. Метиловый эфир (2R,4S)-4-бутил-2-трет-бутил-3-формилоксазолидин-4-карбоновой кислоты
К раствору метилового эфира (2R,4S)-2-трет-бутил-3-формилоксазолидин-4-карбоновой кислоты (4,6 г, 21,4 ммоль) [см. D. Seebach, J.D.Aebi, M. Gander-Coquoz and R. Naef, Helv. Chim. Acta, 70, 1194 (1987)] в сухом ТГФ (120 мл) в атмосфере N2 добавляли 1-иодбутан (12,2 мл, 106,8 ммоль) и НМРА (12 мл). Смесь охлаждали до -78°С и добавляли по каплям раствор бис(триметилсилил)амида натрия в ТГФ (1 М, 32 мл, 32 ммоль) на протяжении 15 минут. Спустя 2 часа реакционную смесь нагревали до 0°С и гасили насыщенным водным NH4Cl (200 мл). Погашенную реакционную смесь разбавляли эфиром (400 мл) и промывали водой (3·200 мл) и солевым раствором (200 мл), затем сушили (Na2SO4) и фильтровали. Концентрирование фильтрата и флэш-хроматография остатка (20% этилацетат/гексаны) давали указанное в заголовке соединение в виде бледно-коричневого твердого вещества (4,0 г, 69%). MC (ES) m/e 272 [M+H]+.
1b. (S)-5-бутил-5-гидроксиметилимидазолидин-2,4-дион
Раствор соединения примера 1а (4,0 г, 14,7 ммоль) в 40 мл смеси концентрированная водная HCl/диоксан (1:1) нагревали с обратным холодильником в течение 2 часов. После охлаждения до комнатной температуры реакционную смесь концентрировали в вакууме и повторно растворяли в воде (15 мл). К этому раствору добавляли гидроксид калия (1 г, 17,8 ммоль) и цианат калия (2,39 г, 29,4 ммоль) и эту смесь нагревали до 115°С в течение 1 часа. Реакционную смесь охлаждали до комнатной температуры, медленно обрабатывали концентрированной водной HCl (5 мл) и затем нагревали с обратным холодильником в течение 2 часов. Растворитель удаляли в вакууме и полученное твердое вещество экстрагировали CH2Cl2/Н2О (2:1) (3·20 мл). Объединенные органические слои концентрировали с получением белого твердого вещества (4,8 г), которое очищали автоматизированной ВЭЖХ Гилсона с получением указанного в заголовке соединения в виде белого твердого вещества (0,85 г, 31%). MC (ES) m/e 187 [M+H]+.
1с.(S)-3-бензил-5-бутил-5-гидроксиметилимидазолидин-2,4-дион
К раствору соединения примера 1b (0,13 г, 0,70 ммоль) в ДМФ (3 мл) добавляли K2CO3 (0,1 г, 0,74 ммоль) и бензилбромид (0,087 мл, 0,74 ммоль). Реакционную смесь перемешивали в течение ночи. Твердое вещество отфильтровывали и органический раствор очищали автоматизированной ВЭЖХ Гилсона с получением указанного в заголовке соединения в виде белого твердого вещества (0,14 г, 73%). MC (ES) m/e 277 [M+H]+.
1d.О-бензилоксим (S)-1-бензил-4-бутил-2,5-диоксоимидазолидин-4-карбальдегида
К перемешиваемому раствору соединения примера 1с (0,14 г, 0,51 ммоль) в 10 мл смеси ацетонитрил/дихлорметан (1:1) при 0°С добавляли периодинан Десс-Мартина (0,32 г, 0,77 ммоль). Реакционную смесь перемешивали при 0°С в течение 1 часа, затем нагревали до комнатной температуры. После перемешивания при комнатной температуре в течение ночи органические растворители удаляли в вакууме. Белое твердое вещество растворяли в пиридине (10 мл) и обрабатывали гидрохлоридом О-бензилгидроксиламина (0,123 г, 0,77 ммоль). После 1 часа при комнатной температуре реакционную смесь концентрировали досуха. Остаток растворяли в дихлорметане (25 мл) и промывали насыщенным водным NaHCO3 (20 мл), водой (20 мл), сушили (Na2SO4) и концентрировали. Твердое вещество очищали автоматизированной ВЭЖХ Гилсона с получением указанного в заголовке соединения в виде коричневатого твердого вещества (0,09 г, 47%). MC (ES) m/e 380 [M+H]+.
1е.N-[(S)-1-бензил-4-бутил-2,5-диоксоимидазолидин-4-илметил]-N-бензилоксиформамид
При перемешивании цианоборгидрид натрия (45 мг, 0,72 ммоль) добавляли медленно к раствору соединения примера 1d (0,09 г, 0,24 ммоль) в уксусной кислоте (5 мл). После перемешивания при комнатной температуре в течение 2 часов реакционную смесь концентрировали в вакууме. Остаток помещали в насыщенный водный NaHCO3 (15 мл) и экстрагировали дихлорметаном (3·10 мл). Объединенные органические экстракты сушили (Na2SO4), фильтровали и концентрировали с получением неочищенного (S)-3-бензил-5-(бензилоксиаминометил)-5-бутилимидазолидин-2,4-диона. MC (ES) m/e 382 [M+H]+.
Вышеуказанное неочищенное промежуточное соединение растворяли в дихлорметане (5 мл). К этому раствору добавляли триэтиламин (0,035 мл) с последующим добавлением свежеприготовленного смешанного ангидрида (полученного нагреванием смеси 0,019 мл муравьиной кислоты и 0,045 мл уксусного ангидрида при 50°С в течение 1 часа и охлаждения до комнатной температуры). Реакционную смесь перемешивали при комнатной температуре в течение 1 часа и затем концентрировали досуха. Остаток очищали автоматизированной ВЭЖХ Гилсона с получением указанного в заголовке соединения в виде белого твердого вещества (0,06 г, 62%). MC (ES) m/e 410 [M+H]+.
1f.N-[(S)-1-бензил-4-бутил-2,5-диоксоимидазолидин-4-илметил]-N-гидроксиформамид
Соединение примера 1е (0,06 г, 0,15 ммоль) растворяли в метаноле (5 мл) и перемешивали под давлением подаваемого из баллона водорода в присутствии палладия на активированном угле (0,02 г) в течение 4 часов. Реакционную смесь фильтровали и концентрировали и остаток очищали автоматизированной ВЭЖХ Гилсона с получением указанного в заголовке соединения в виде белого твердого вещества (0,04 г, 84%). MC (ES) m/e 320 [M+H]+.
Подобным образом, но с заменой описанных выше промежуточных продуктов подходящими промежуточными продуктами получали следующие соединения:
N-[(S)-1-бензил-4-циклогексилметил-2,5-диоксоимидазолидин-4-илметил]-N-гидроксиформамид. MC (ES) m/e 360 [M+H]+.
2-{(S)-4-бутил-4-[(формилгидроксиамино)метил]-2,5-диоксоимидазолидин-1-ил}-N-(3,5-дихлорфенил)ацетамид. MC(ES) m/e 431 [M+H]+.
Метиловый эфир 2-{(S)-4-бутил-4-[(формилгидроксиамино)метил]-2,5-диоксоимидазолидин-1-илметил}бензойной кислоты. MC (ES) m/e 378 [M+H]+.
N-[(S)-4-бутил-(2-морфолин-4-илэтил)-2,5-диоксоимидазолидин-4-илметил]-N-гидроксиформамид. MC (ES) m/e 343 [M+H]+.
2-{(S)-4-бутил-4-[(формилгидроксиамино)метил]-2,5-диоксоимидазолидин-1-илметил}бензойной кислоты. MC (ES) m/e 364 [M+H]+.
Пример 2
Получение N-[(S)-2,5-диоксо-4-пентил-1-фенилимидазолидин-4-илметил]-N-гидроксиформамида, SB-728794
2а. (S)-5-пентил-5-гидроксиметил-3-фенилимидазолидин-2,4-дион
Согласно процедуре примера 1а, за исключением применения пентилиодида вместо иодбутана, и затем согласно процедуре примера 1b, с заменой цианата калия фенилизоцианатом, указанное в заголовке соединение получали в виде белого твердого вещества (75%). MC (ES) m/e 277 [M+H]+.
2b. N-[(S)-2,5-диоксо-4-пентил-1-фенилимидазолидин-4-илметил]-N-гидроксиформамид
Согласно процедуре примеров 1d-f, за исключением замены соединения примера 1с соединением примера 2а, указанное в заголовке соединение получали в виде белого твердого вещества. MC (ES) m/e 320 [M+H]+.
Подобным образом, но с заменой описанных выше промежуточных продуктов подходящими промежуточными продуктами получали следующие соединения:
N-[(S)-1-бензил-4-пентил-2,5-диоксоимидазолидин-4-илметил]-N-гидроксиформамид. MC (ES) m/e 334 [M+H]+.
N-[(S)-1,4-дибензил-2,5-диоксоимидазолидин-4-илметил]-N-гидроксиформамид. MC (ES) m/e 354 [M+H]+.
Пример 3
Получение N-[(S)-4-бутил-2,5-диоксо-1-(2-оксо-2-фенилэтил)имидазолидин-4-илметил]-N-гидроксиформамида, SB-736063
3а. (S)-5-бутил-5-гидроксиметил-3-(2-оксо-2-фенилэтил)имидазолидин-2,4-дион
Согласно процедуре примера 1с, за исключением замены бензилбромида 2-бром-1-фенилэтаноном, указанное в заголовке соединение получали в виде белого твердого вещества (93%). MC (ES) m/e 305 [M+H]+.
3b. Трет-бутил-N-[(S)-4-бутил-2,5-диоксо-1-(2-оксо-2-фенилэтил)имидазолидин-4-илметил]-N-(трет-бутоксикарбоксикарбонилокси)карбамат
Соединение примера 3а (0,15 г, 0,49 ммоль) и трет-бутил-N-(трет-бутоксикарбоксикарбонилокси)карбамат (0,18 г, 0,74 ммоль) растворяли в ТГФ (3 мл) в атмосфере N2 при 0°С. К этому раствору добавляли предварительно смешанный раствор трибутилфосфина (0,19 мл, 0,74 ммоль) и ди-трет-бутилазодикарбоксилата (0,176 г, 0,74 ммоль) в ТГФ (2 мл) в атмосфере N2 при 0°С. Реакционную смесь выдерживали при 0°С в течение 1 часа, нагревали до комнатной температуры и перемешивали в течение ночи. Реакцию гасили насыщенным водным NaHCO3 (15 мл) и экстрагировали дихлорметаном (3·10 мл). Объединенные органические слои сушили (Na2SO4), фильтровали и концентрировали. Остаток очищали автоматизированной ВЭЖХ Гилсона с получением указанного в заголовке соединения в виде белого твердого вещества (0,11 г, 42%). MC (ES) m/e 520 [M+H]+.
3с.N-[(S)-4-бутил-2,5-диоксо-1-(2-оксо-2-фенилэтил)имидазолидин-4-илметил]-N-гидроксиформамид
Соединение примера 3b (0,11 г, 0,21 ммоль) растворяли в смеси 15% ТФУ/1,2-дихлорэтан (5 мл). После перемешивания при комнатной температуре в течение 1 часа реакционную смесь концентрировали в вакууме. Остаток растворяли в дихлорметане (5 мл) и обрабатывали триэтиламином (0,3 мл) с последующим добавлением свежеприготовленного смешанного ангидрида, (0,031 мл муравьиной кислоты и 0,069 мл уксусного ангидрида, 50°С, 1 час). После перемешивания при комнатной температуре в течение 1 часа органический растворитель удаляли в вакууме и добавляли метанол (10 мл) с последующим добавлением насыщенного водного Na2CO3 (3 мл) с интенсивным перемешиванием. Спустя 3 часа реакционную смесь концентрировали досуха и твердое вещество экстрагировали смесью 30% метанол/дихлорметан (3·5 мл). Объединенные экстракты фильтровали и концентрировали и полученный осадок очищали автоматизированной ВЭЖХ Гилсона с получением указанного в заголовке соединения в виде белого твердого вещества (0,03 г, 41%). MC (ES) m/e 348 [M+H]+.
Подобным образом, но с заменой описанных выше промежуточных продуктов подходящими промежуточными продуктами получали следующие соединения:
N-[(S)-4-бутил-(3,4-дихлорбензил)-2,5-диоксоимидазолидин-4-илметил]-N-гидроксиформамид. MC (ES) m/e 388 [M+H]+.
N-[(S)-1-бифенил-4-илметил-4-бутил-2,5-диоксоимидазолидин-4-илметил]-N-гидроксиформамид. MC (ES) m/e 396 [M+H]+.
N-{(S)-4-бутил-1-[2-(5-хлор-3-метил-1-бензо[b]тиофен-2-ил)-2-оксоэтил]-2,5-диоксоимидазолидин-4-илметил}-N-гидроксиформамид. MC (ES) m/e 452 [M+H]+.
N-{(S)-1-(2-бензофуран-2-ил-2-оксоэтил)-4-бутил-2,5-диоксоимидазолидин-4-илметил]-N-гидроксиформамид. MC (ES) m/e 388 [M+H]+.
С использованием подходящей манипуляции и защиты любой химической функциональной группы синтез остальных соединений формулы (I) выполняют способами, аналогичными описанным способам.
Соединения данного изобретения применимы для лечения бактериальных инфекций. Для использования соединения формулы (I) или его фармацевтически приемлемой соли их обычно готовят в соответствии со стандартной фармацевтической практикой в виде фармацевтической композиции.
Соединения формулы (I) и их фармацевтически приемлемые соли могут вводиться стандартным для антибиотиков способом, например, перорально, парентерально, сублингвально, дермально, трансдермально, ректально, ингаляцией или посредством буккального введения.
Композиции формулы (I) и их фармацевтически приемлемые соли, которые являются активными при пероральном введении, могут быть приготовлены в виде сиропов, таблеток, капсул, кремов и пастилок. Формы сиропов обычно состоят из суспензии или раствора соединения или его соли в жидком носителе, таком как, например, этанол, арахисовое масло, оливковое масло, глицерин или вода, с ароматизирующим агентом или красителем. Если эта композиция находится в форме таблетки, может быть использован любой фармацевтический носитель, традиционно используемый для приготовления твердых форм. Примеры таких носителей включают стеарат магния, гипс, тальк, желатин, аравийскую камедь, стеариновую кислоту, крахмал, лактозу и сахарозу. Если эта композиция находится в форме капсулы, пригодным является любое инкапсулирование, например, использование вышеупомянутых носителей в оболочке твердой желатиновой капсулы. Если эта композиция находится в форме желатиновой капсулы с мягкой оболочкой, может рассматриваться любой фармацевтический носитель, используемый для приготовления дисперсий или суспензий, например, водные камеди, целлюлозы, силикаты или масла, и они могут быть включены в оболочку мягкой желатиновой капсулы.
Типичные парентеральные композиции состоят из раствора или суспензии соединения или соли в стерильном водном или неводном носителе, необязательно содержащем парентерально приемлемое масло, например, полиэтиленгликоле, поливинилпирролидоне, лецитине, арахисовом масле или кунжутном масле.
Типичные композиции для ингаляции находятся в форме раствора, суспензии или эмульсии, которые могут вводиться в виде сухого порошка или в форме аэрозоля с использованием общепринятого пропеллента, такого как дихлордифторметан или трихлорфторметан.
Типичная форма суппозитория содержит соединение формулы (I) или его фармацевтически приемлемую соль, которая является активной при введении таким образом, со связывающим и/или смазывающим агентом, таким как полимерные гликоли, желатины, какао-масло или другие низкоплавящиеся растительные воски или жиры или их синтетические аналоги.
Типичные дермальные и трансдермальные формы содержат общепринятый водный или неводный носитель, например, крем, мазь, лосьон или пасту, или они находятся в форме медицинского пластыря, подушечки или мембраны.
Предпочтительно, композиция находится в стандартной лекарственной форме, например, в виде таблетки, капсулы или отмеренной аэрозольной дозе, так что пациент может вводить дозу для одного приема.
Каждая дозированная единица для перорального введения содержит 0,1 мг - 500 мг/кг и предпочтительно 1 мг - 100 мг/кг, а каждая дозированная единица для парентерального введения содержит 0,1 мг - 100 мг/кг соединения формулы (I) или его фармацевтически приемлемой соли в расчете на свободную кислоту. Каждая дозированная единица для интраназального введения содержит 1-400 мг, предпочтительно 10-200 мг на одно лицо. Форма для местного введения содержит предпочтительно 0,01-5,0% соединения формулы (I).
Схема введения суточной дозы для перорального введения предусматривает предпочтительно приблизительно 0,01 мг/кг - 40 мг/кг соединения формулы (I) или его фармацевтически приемлемой соли в расчете на свободную кислоту. Схема введения суточной дозы для парентерального введения предусматривает приблизительно 0,001 мг/кг - 40 мг/кг соединения формулы (I) или его фармацевтически приемлемой соли в расчете на свободную кислоту.
Схема введения суточной дозы для интраназального введения и ингаляции ротовой полости предусматривает приблизительно 10 - приблизительно 500 мг на одно лицо. Активный ингредиент может вводиться 1-6 раз в день, что является достаточным для проявления желаемой активности.
Не ожидаются неприемлемые токсикологические эффекты при введении соединений данного изобретения в соответствии с данным изобретением.
Биологическая активность соединений формулы (I) демонстрируется следующим тестом.
Биологический тест
Активность PDF S. aureus или E. coli измеряют при 25°С с использованием непрерывного ферментсвязывающего анализа, разработанного Lazennec & Meinnel (1997) "Formate dehydeogenase-coupled spectrophotometric assay of peptide deformolase" Anal. Biochem. 244, pp.180-182, с небольшими модификациями. Реакционная смесь содержится в 50 мкл с 50 мМ калий-фосфатным буфером (рН 7,6), 15 мМ NAD, 0,25 Е формиатдегидрогеназы. Субстратный пептид, f-Met-Ala-Ser, включают при концентрации КМ. Реакцию запускают добавлением 10 нМ фермента Def1 и оптическую плотность подвергают мониторингу в течение 20 минут при 340 нм.
Тест антимикробной активности
Антимикробную активность против целых клеток определяли микроразведением бульона с использованием процедуры, рекомендованной Национальным Комитетом для Клинических Лабораторных Стандартов (NCCLS), Документ М7-А4, "Methods for Dilution Susceptibility Tests for Bacteria that Grow Aerobically" (включенный здесь в качестве ссылки). Соединение испытывали в серийных двукратных разведениях в диапазоне от 0,06 до 64 мкг/мл. В данном испытании оценивали панель из 12 штаммов. Эта панель состояла из следующих лабораторных штаммов: Staphylococcus aureus Oxford, Staphylococcus aureus WCUH29, Enterococcus faecalis I, Enterococcus faecalis 7, Haemophilus influenzae Q1, Haemophilus influenzae NEMC1, Moraxella catarrhalis 1502, Streptococcus pneumoniae 1629, Streptococcus pneumoniae N1387, Streptococcus pneumoniae N1387, E. coli 7623 (AcrABEFD+) и E. coli 120 (AcrAB-). Минимальную ингибиторную концентрацию (MIC) определяли как самую низкую концентрацию соединения, которая ингибировала видимый рост. Использовали зеркальный планшет-ридер в определении конечной точки MIC.
Все публикации, включающие, но не ограничивающиеся ими, патенты и заявки на патенты, цитируемые в этом описании, включены в качестве ссылки, как если бы каждая отдельная публикация была специально и индивидуально указана как включенная в качестве ссылки в ее полном объеме.
Приведенное выше описание полностью раскрывает данное изобретение, в том числе предпочтительные варианты. Модификации и усовершенствования вариантов, особо раскрытых здесь, находятся в объеме нижеследующей формулы изобретения. Предполагается, что без особого труда специалист с квалификацией в данной области сможет, с использованием данного описания, использовать данное изобретение в его полном объеме. Таким образом, приведенные примеры должны рассматриваться лишь как иллюстрирующие, а не как ограничивающие каким-либо образом данное изобретение. Варианты данного изобретения, в которых заявляется исключительное право собственности или преимущественное право, определяются следующей формулой изобретения.
| название | год | авторы | номер документа |
|---|---|---|---|
| КОМПОЗИЦИЯ ДЛЯ ПОДДЕРЖАНИЯ ФУНКЦИИ ТРОМБОЦИТОВ | 2011 |
|
RU2578607C2 |
| ПРОИЗВОДНОЕ N-ГИДРОКСИФОРМАМИДА И СОДЕРЖАЩЕЕ ЕГО ЛЕКАРСТВЕННОЕ СРЕДСТВО | 2011 |
|
RU2569851C2 |
| ИНГИБИТОРЫ МЕТАЛЛОПРОТЕИНАЗ, ИХ ПРИМЕНЕНИЕ И ФАРМАЦЕВТИЧЕСКИЕ КОМПОЗИЦИИ НА ИХ ОСНОВЕ | 2002 |
|
RU2293729C2 |
| СПИРОИМИДАЗОЛИДИНОВЫЕ ПРОИЗВОДНЫЕ, СПОСОБ ИХ ПОЛУЧЕНИЯ И ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ НА ИХ ОСНОВЕ | 2000 |
|
RU2245879C2 |
| АНТИБАКТЕРИАЛЬНЫЕ АГЕНТЫ | 2000 |
|
RU2269525C2 |
| ПИРАЗОЛОПИРИМИДИНЫ И РОДСТВЕННЫЕ ГЕТЕРОЦИКЛЫ КАК СК2 ИНГИБИТОРЫ | 2010 |
|
RU2607453C2 |
| ПИПЕРИДИН- И ПИПЕРАЗИНЗАМЕЩЕННЫЕ N-ГИДРОКСИФОРМАМИДЫ В КАЧЕСТВЕ ИНГИБИТОРОВ МЕТАЛЛОПРОТЕИНАЗ | 2001 |
|
RU2283306C2 |
| АМИДОЗАМЕЩЕННЫЕ ПРОИЗВОДНЫЕ КСАНТИНА, ОБЛАДАЮЩИЕ ИНГИБИРУЮЩИМ ДЕЙСТВИЕМ ФОСФОЕНОЛПИРУВАТКАРБОКСИКИНАЗЫ (ФЕПКК), СПОСОБ ИХ ПОЛУЧЕНИЯ, ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ И ПРИМЕНЕНИЕ | 2003 |
|
RU2295525C2 |
| МОДУЛЯТОРЫ АДЕНОЗИНОВЫХ РЕЦЕПТОРОВ | 2001 |
|
RU2277911C2 |
| ГЕТЕРОЦИКЛИЧЕСКИЕ СОЕДИНЕНИЯ В КАЧЕСТВЕ ПОЛОЖИТЕЛЬНЫХ МОДУЛЯТОРОВ МЕТАБОТРОПНОГО ГЛУТАМАТНОГО РЕЦЕПТОРА 2 (РЕЦЕПТОРА MGLU2) | 2008 |
|
RU2479577C2 |
Изобретение относится к новым производным имидазолидина. Описывается соединение формулы (I):

где R1 выбран из группы, состоящей из С1-9алкила, С1-2алкилAr; R2 выбран из группы, состоящей из фенила, С1-4алкилAr', NC(O)R4, C2-4алкилNR3R4, С1-3алкилС(O)NR3R4, С1-3алкилС(O)Ar'; R3 выбран из группы, состоящей из Н, С1-2алкилAr и Ar; R4 является R3 или R4 может быть взят вместе с R3 и атомом азота, к которому они присоединены, для образования морфолинила; Ar выбран из группы, состоящей из фенила, который может быть необязательно замещены одним, двумя или тремя заместителями, выбранными из группы, состоящей из F, Cl, Br и I; Ar' выбран из группы, состоящей из фенила, бифенила, бензофуранила и бензо[b]тиофена, которые все могут быть необязательно замещены одним, двумя или тремя заместителями из группы, состоящей из С1-6алкила, (CH2)0-5CO2R1, F, Cl, Br, I, и СООН; А выбран из группы C(O)NHOH или N(CHO)OH; Х обозначает NH, когда Y обозначает С(O), или его фармацевтически приемлемая соль. Также описывается способ лечения бактериальной инфекции на основе соединений формулы (I). Технический результат - получены новые соединения, обладающие полезными биологическими свойствами 2 н. и 2 з.п. ф-лы.
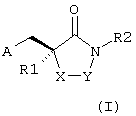
где R1 выбран из группы, состоящей из С1-9алкила, С1-2алкилAr;
R2 выбран из группы, состоящей из фенила, С1-4алкилAr', NC(O)R4, С2-4алилNR3R4, C1-3алкилC(O)NR3R4, С1-3алкилС(O)Ar';
R3 выбран из группы, состоящей из Н, С1-2алкилAr и Ar;
R4 является R3, или R4 может быть взят вместе с R3 и атомом азота, к которому они присоединены, для образования морфолинила;
Ar выбран из группы, состоящей из фенила, который может быть необязательно замещен одним, двумя или тремя заместителями, выбранными из группы, состоящей из F, Cl, Br и I;
Ar' выбран из группы, состоящей из фенила, бифенила, бензофуранила и бензо[b]тиофена, которые все могут быть необязательно замещены одним, двумя или тремя заместителями из группы, состоящей из С1-6алкила, (СН2)0-5CO2R1, F, Cl, Br, I, и СООН;
А выбран из группы C(O)NHOH или N(CHO)OH;
Х обозначает NH, когда Y обозначает С(O),
или его фармацевтически приемлемая соль.
N-[(S)-1-бензил-4-пентил-2,5-диоксоимидазолидин-4-илметил]-N-гидроксиформамид;
N-[(S)-1,4-дибензил-2,5-диоксоимидазолидин-4-илметил]-N-гидроксиформамид;
N-[(S)-1-бензил-4-бутил-2,5-диоксоимидазолидин-4-илметил]-N-гидроксиформамид;
N-[(S)-2,5-диоксо-4-пентил-1-фенилимидазолидин-4-илметил]-N-гидроксиформамид;
N-[(S)-4-бутил-1-(3,4-дихлорбензил)-2,5-диоксоимидазолидин-4-илметил]-N-гидроксиформамид;
N-[(S)-4-бутил-2,5-диоксо-1-(2-оксо-2-фенилэтил)имидазолидин-4-илметил]-N-гидроксиформамид;
N-[(S)-1-бифенил-4-илметил-4-бутил-2,5-диоксоимидазолидин-4-илметил]-N-гидроксиформамид;
N-[(S)-1-бензил-4-циклогексилметил-2,5-диоксоимидазолидин-4-илметил]-N-гидроксиформамид;
N-{(S)-4-бутил-1-[2-(5-хлор-3-метил-1-бензо[b]тиофен-2-ил)-2-оксоэтил]-2,5-диоксоимидазолидин-4-илметил}-N-гидроксиформамид;
2-{(S)-4-бутил-4-[(формилгидроксиамино)метил]-2,5-диоксоимидазолидин-1-ил}-N-(3,5-дихлорфенил)ацетамид;
Метиловый эфир 2-{(S)-4-бутил-4-[(формилгидроксиамино)метил]-2,5-диоксоимидазолидин-1-илметил}бензойной кислоты;
N-[(S)-4-бутил-1-(2-морфолин-4-илэтил)-2,5-диоксоимидазолидин-4-илметил]-N-гидроксиформамид;
N-[(S)-1-(2-бензофуран-2-ил-2-оксоэтил)-4-бутил-2,5-диоксоимидазолидин-4-илметил]-N-гидроксиформамид и
2-{(S)-4-бутил-4-[(формилгидроксиамино)метил]-2,5-диоксоимидазолидин-1-илметил}бензойной кислоты
или его фармацевтически приемлемая соль.
| НОВЫЕ ПРОИЗВОДНЫЕ ГИДРОКСАМОВОЙ КИСЛОТЫ, ПОЛЕЗНЫЕ ДЛЯ ЛЕЧЕНИЯ ЗАБОЛЕВАНИЙ, СВЯЗАННЫХ С ДЕГЕНЕРАЦИЕЙ СОЕДИНИТЕЛЬНОЙ ТКАНИ | 1997 |
|
RU2168497C2 |
| Кипятильник для воды | 1921 |
|
SU5A1 |
| Способ получения производных пирролидинона | 1985 |
|
SU1360583A3 |

