Настоящее изобретение относится к пептидным миметикам соматостатина, способу их получения и к содержащим их фармацевтическим композициям.
Более конкретно настоящее изобретение относится к соединению формулы
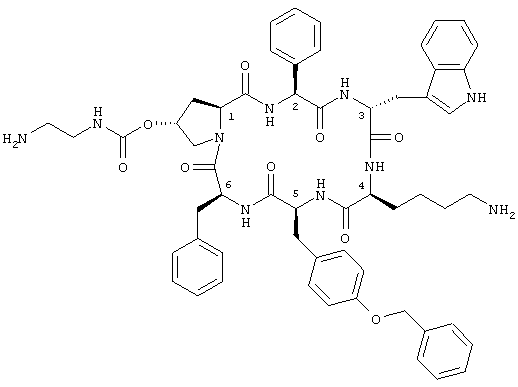
которое называют также цикло[{4-(NH2-C2H4-NH-CO-O-)Pro}-Phg-DTrp-Lys-Tyr(4-Bzl)-Phe], обозначенному в настоящем описании как соединение А, а также к его диастереоизомерам и их смесям, в свободной форме, в форме соли или комплекса или в защищенной форме. Phg обозначает -HN-СН(С6Н5)-СО-, а Bzl обозначает бензил.
Соединение А в защищенной форме соответствует указанной выше молекуле, в которой по меньшей мере одна из аминогрупп является защищенной и после удаления защитной группы, предпочтительно удаляемой в физиологических условиях, превращается в соединение А. Приемлемыми аминозащитными группами являются группы, описанные, например, в "Protective Groups in Organic Synthesis", Т. W. Greene, J. Wiley & Sons NY (1981), 219-287, содержание публикации включено в настоящее описание в качестве ссылки. Примером аминозащитной группы является ацетил.
Когда соединение А присутствует в форме комплекса, то оно может представлять собой соединение А, несущее хелатирующую группу на боковой цепи аминогруппы Pro и образующее комплекс с обнаруживаемым или меченным с помощью радиоизотопа терапевтическим элементом (радиотерапевтическим элементом). Соединение А, несущее хелатирующую группу, обозначено в настоящем описании как конъюгированное соединение А.
Примеры хелатирующих групп включают, например, группы, являющиеся производными полиаминополикарбоновых кислот или ангидридов, например, группы, являющиеся производными нециклических лигандов, например диэтилентриаминпентауксусной кислоты (ДТПК), этиленгликоль-0,0'-бис(2-аминоэтил)-N,N,N',N'-тетрауксусной кислоты (ЭГТК), N,N'-бис(гидроксибензил)этилендиамин-N,N'-диуксусной кислоты (ГБЭД) и триэтилентетрамингексауксусной кислоты (ТТГК), группы, являющиеся производными замещенной ДТПК, например пара-изотиоцианатбензил-ДТПК, группы, являющиеся производными макроциклических лигандов, например, 1,4,7,10-тетраазациклододекан-N,N',N'',N'''-тетрауксусной кислоты (ДОТК) и 1,4,8,11-тетраазациклотетрадекан-N,N',N'',N'''-тетрауксусной кислоты (ТЕТК) или 1,4,7,10-тетраазациклотридекан-N,N',N'',N'''-тетрауксусной кислоты (ТИТРК).
Хелатирующие группы можно присоединять к боковой цепи аминогруппы Pro либо непосредственно, либо через спейсер. Приемлемые спейсеры включают спенсеры, известные в данной области, например, описанные в GB-A-2225579, например двухвалентный остаток аминокарбоновой кислоты, например, β-Ala, или двухвалентный остаток 6-аминокапроновой кислоты.
Предпочтительные хелатирующие группы являются производными ДТПК, ДОТК или ТЕТК. Наиболее предпочтительными являются хелатирующие группы, являющиеся производными ДТПК или ДОТК.
Под обнаруживаемым элементом понимают любой элемент, предпочтительно ион металла, который можно обнаруживать in vivo с помощью диагностических методов, например ион металла, испускающий изучение, которое можно обнаруживать, или ион металла, который может оказывать воздействие на релаксацию ЯМР. Под радиотерапевтическим элементом понимают любой элемент, испускающий излечение, оказывающее положительное воздействие на подлежащее лечению состояние.
Приемлемые элементы включают, например, элементы тяжелых металлов или ионы редкоземельных металлов, например, применяемые для КАТ-сканирования (компьютерная трансаксиальная томография), парамагнитные ионы, например, Gd3-, Fe3+, Mn2+ и Cr2+, флуоресцентные ионы металлов, например Eu3+ и радионуклиды, например радиолантанид, в частности радионуклид, испускающий γ-лучи, радионуклид, испускающий β-лучи, радионуклид, испускающий α-лучи, радионуклид, испускающий Оже-е--излучение или испускающий позитроны радионуклид, например, 68Ga, 18F или 86Y.
Приемлемые испускающие γ-лучи радионуклиды включают радионуклиды, которые можно применять в методах диагностики. Испускающие γ-лучи радионуклиды предпочтительно имеют период полураспада от 1 ч до 40 дней, предпочтительно от 5 ч до 4 дней, более предпочтительно от 12 ч до 3 дней. Примерами радиоизотопов галлия, индия, технеция, иттербия, рения, тербия, лютеция, таллия и самария являются, например 67Ga, 111In, 99mTc, 161Tb, 169Yb, 186Re или 177Lu.
Приемлемые испускающие β-лучи радионуклиды включают радионуклиды, которые можно применять в радиотерапии, например, 90Y, 67Cu, 186Re, 188Re, 169Er, 121Sn, 127Те, 177Lu, 143Pr, 198Au, 109Pd, 165Dy, 142Pr или 153Sm.
Приемлемые испускающие α-лучи радионуклиды представляют собой радионуклиды, которые можно применять в терапевтических целях, например 211At, 212Bi или 201Tl.
Соединение А может находится, например, в свободной форме или в форме соли. Соли включают кислотно-аддитивные соли, например неорганических кислот, полимерных кислот или органических кислот, например, соляной кислоты, уксусной кислоты, молочной кислоты, аспарагиновой кислоты, бензойной кислоты, янтарной кислоты или памовой кислоты. Кислотно-аддитивные соли могут присутствовать в виде одно- или двухвалентных солей, например в зависимости от того, один или два эквивалента кислоты добавляют к соединению А в форме свободного основания. Предпочтительными солями являются лактат, аспартат, бензоат, сукцинат и памоат, включая их одно- и двухвалентные соли, более предпочтительно двукислый аспартат и монокислый памоат.
Конъюгированное соединение А может существовать также в формах солей, получаемых с помощью групп карбоновых кислот, присутствующих в хелатирующей группе, например, солей щелочных металлов, таких как натрий или калий, или замещенных или незамещенных аммонийных солей.
Настоящее изобретение относится также к способу получения соединения А. Его можно получать по аналогии с известными методами, например:
а) путем циклизации линейного пептида в защищенной, связанной с полимером или незащищенной форме, таким образом, чтобы получать соединение А, и затем необязательно удаления защитных(ой) групп(ы),
б) для получения конъюгированного соединения А путем связывания хелатирующей группы и соединения А в защищенной или незащищенной форме и затем необязательного удаления защитной группы и путем выделения полученного таким образом соединения А или конъюгированного соединения А в свободной форме, в форме соли или необязательно в форме комплекса с обнаруживаемым или радиотерапевтическим элементом.
Как правило, выбор аминокислоты, находящейся на С-конце относительно начала пептидной цепи, не имеет решающего значения, поскольку линейный пептид должен быть циклизован, единственным условием является то, что последовательность аминокислот в линейном пептиде должна соответствовать их последовательности в соединении А. Однако известны другие факторы, которые могут обусловливать предпочтение одной стартовой аминокислоты относительно другой. Когда соединение А получают с помощью твердофазного синтеза, то первую аминокислоту предпочтительно связывают со смолой, например, с поступающим в продажу полистиролом, через приемлемый линкер, например линкер, который отщепляется в мягких условиях, сохраняя при этом защиту боковой цепи, например, такой линкер, как SASRIN, или необязательно замещенный содержащий тритил линкер, например, 4-(гидроксидифенилметил)бензойная кислота, в которой одна из фенильных групп необязательно может быть замещена, например Cl. Создание требуемой пептидной цепи можно осуществлять общепринятым методом, например с помощью остатков аминокислот, в которых концевая аминогруппа защищена с помощью Fmoc, боковые цепи аминогрупп, если они присутствуют, должны быть защищены различными аминозащитными группами, например Вос или КБО. Предпочтительно линейный пептид циклизуют таким образом, чтобы получать связь между Tyr(4-Bzl)-OH и Phe, например, Phe-{4-(NHR1-C2H4-NH-CO-O-)Pro}-Phg-DTrp(R2)-Lys(ε-NHR3)-Tyr(4-Bzl)-OH или его функционально активное производное, где R1, R2 и R3 каждый обозначает аминозащитную группу. Стадию циклизации а) можно осуществлять с помощью известного метода, например с помощью азида, активного сложного эфира, смешанного ангидрида или карбодиимида. После этого защитные группы удаляют, например, путем расщепления, например с помощью трифторуксусной кислоты или гидрированием.
Циклизацию пептида можно также осуществлять непосредственно на твердой подложке, при этом первая аминокислота защищена на Nα-и С-конце и присоединена через боковую цепь, например, через ε-аминофункцию Lys или путем прикрепления к основе. Затем линейную последовательность синтезируют с помощью стандартного твердофазного синтеза (ТФС). После расщепления С-концевой защитной группы пептид циклизуют, например, согласно описанному выше методу. После чего циклический пептид отщепляют от смолы и удаляют защитные группы.
При необходимости боковую цепь, присутствующую на Pro, можно вводить в аминокислоту до или после циклизации пептида на стадии а). Так, Pro в качестве исходной аминокислоты или исходного линейного или циклического пептида, в которых в каждом случае Pro содержит в качестве заместителя в кольце ОН, можно подвергать взаимодействию с получением соединения А или требуемого содержащего фрагмент Pro пептида или соответствующего линейного пептида соответственно, где Pro замещен NHR1-C2H4-NH-CO-O-.
Комплексообразование конъюгированного соединения А можно осуществлять путем взаимодействия конъюгированного соединения А с соответствующим обнаруживаемым или радиотерапевтическим элементом, получая соединение, например соль металла, предпочтительно водорастворимую соль. Реакцию можно осуществлять аналогично известным методам, описанным, например, у Perrin, Organic Ligand, Chemical Data, серии 22, NY Pergamon Press (1982); у Krejcarit и Tucker, Biophys. Biochem. Res. Corn. 77: 581 (1977) и у Wagner и Welch, J. Nucl. Med. 20: 428 (1979).
Ниже изобретение проиллюстрировано на примерах. Все температуры даны в градусах Цельсия. Сокращения:
Пример 1: Цикло[{4-(NH2-C2H4-NH-CO-O-)Pro}-Phe-DTrp-Lys-Tyr(4-Bzl)-Phe]
а) Синтез Fmoc-Pro(4-OCO-NH-CH2-CH2-NH-Boc)-OH
Гидрохлорид метилового эфира L-гидроксипролина подвергают взаимодействию с Fmoc-OSu в водном 1,0н. растворе карбонат натри/ТГФ при комнатной температуре. После завершения реакции Fmoc-Pro(4-OH)-OMe выделяют осаждением. Затем Fmoc-Pro(4-OH)-OMe добавляют по каплям к раствору трисфосгена (0,6 экв.) в ТГФ, получая в качестве промежуточного продукта хлоркарбонат. Через 1 ч добавляют диметиламинопиридин (1,0 экв.) и N-Boc-диаминоэтан (6,0 экв.) и реакционную смесь перемешивают при комнатной температуре. После завершения реакции растворитель удаляют в вакууме и образовавшийся Fmoc-Pro(4-OCO-NH-CH2-CH2-NH-Boc)-OMe экстрагируют из двухфазной системы этилацетат/0,1 М HCl, получая неочищенный продукт (МН+= 554), который очищают кристаллизацией из этилацетата. Затем метиловый эфир расщепляют с получением свободной кислоты обработкой 1н./NaOH в смеси диоксан/вода и продукт, т.е. Fmoc-Pro(4-OCO-NH-CH2-CH2-NH-Boc)-OH, очищают на силикагеле, [(M+Na)]+=562).
б) H-Phe-Pro(4-OCO-NH-CH2-CH2-NH-Boc)-Phg-DTrp(Boc)-Lys(Boc)-Tyr(Bzl)-ОН
В качестве исходного продукта используют имеющуюся в продаже Fmoc-Tyr(Bzl)-O-СН2-Ph(3-ОСН3)-O-СН2-полистироловую смолу (SASRIN-смола, 2,4 мМ) и подвергают обработке согласно стандартному протоколу, включающему повторные циклы удаления защитных групп Nα-концов (пиперидин/ДМФ, 2:8), повторные промывки ДМФ и реакцию сочетания (ДИПКИ: 4,8 мМ/ГОБТ: 6 мМ, ДМФ). Для осуществления реакции сочетания используют следующие аминокислотные производные: Fmoc-Lys(Boc)-OH, Fmoc-DTrp(Boc)-OH, Fmoc-Phg-OH, Fmoc-Pro(4-OCO-NH-CH2-CH2-NH-Boc)-OH, Fmoc-Phe-OH. Реакции сочетания (2 экв. аминокислот) продолжают или повторяют до завершения, т.е. до полного исчезновения остаточных аминогрупп, которые можно обнаруживать с использованием отрицательного "Kaiser"-теста на основе нингидрина. Перед отщеплением полностью собранного защищенного линейного пептида от подложки, представляющей собой смолу, удаляют Nα-Fmoc-защиту последнего остатка.
B)H-Phe-Pro(4-OCO-NH-CH2-CH2-NH-Boc)-Phg-DTrp(Boc)-Lys(Boc)-Tyr(Bzl)-OH
После промывки с помощью CH2Cl2 комплекс пептид-смола переносят на колонку или при перемешивании на вакуум-фильтр и пептидный фрагмент отщепляют и элюируют кратковременной обработкой 2%-ной ТФК в CH2Cl2 в течение 1 ч. Элюат сразу нейтрализуют насыщенным раствором NaHCO3. Органический раствор отделяют и упаривают и соединение-предшественник с защищенной боковой цепью (MH+=1366) подвергают циклизации без дополнительной очистки.
г) Трифторацетат цикло[-Pro(4-OCO-NH-CH2-CH2-NH2)-Phg-DTrp-Lys-Tyr(Bzl)-Phe-]
Вышеуказанный линейный фрагмент растворяют в ДМФ (4 мМ), охлаждают до -5°С и обрабатывают 2 экв. ДИПЭА, а затем 1,5 экв. ДФФА и перемешивают до завершения реакции (примерно, 20 ч) при 0-4°С. Растворитель практически полностью удаляют в вакууме; концентрат разбавляют этилацетатом, промывают NaHCO3, водой, сушат и упаривают в вакууме.
Для удаления защитных групп остаток растворяют при 0°С в ТФК/Н2О, 95:5 (примерно, 50 мМ) и перемешивают на холоде в течение 30 мин. Затем продукт осаждают простым эфиром, содержащим примерно 10 экв. HCl, фильтруют, промывают простым эфиром и сушат. Для полного расщепления оставшейся индол-N-карбаминовой кислоты продукт растворяют в 5%-ной АсОН и лиофилизируют после выдерживания в течение 15 ч примерно при 5°С. Осуществляют препаративную ОФ-ЖХВР с применением колонки С-18, 10 мкм, типа STAGROMA (5-25 см), используя градиент от 0,5% ТФК до 0,5% ТФК в 70%-ном ацетонитриле. Фракции, содержащие чистое указанное в заголовке соединение, объединяют, разбавляют водой и лиофилизируют. Лиофилизат растворяют в воде, а затем осаждают 10%-ным Na2CO3 в воде. Твердое свободное основание фильтруют, промывают водой и сушат в вакууме при комнатной температуре. Образовавшийся порошок белого цвета используют непосредственно для получения различных солей.
Пример 2: Цикло[{4-NH2-C2H4-NH-CO-O-)Pro}-Phg-DTrp-Lys-Tyr(4-Bzl)-Phe] в форме соли
а. Ацетат
Превращение в ацетат осуществляют с помощью ионообменной смолы (например, типа AG 3-X4). МС (ESI): m/z 524,5 [М+2Н]2+ [α]D20=-42°, с=0,26 в 95% АсОН
б. Аспартат
Превращение в моно- или диаспартата осуществляют путем взаимодействия 1 эквивалента соединения из примера 1 с 1 или 2 эквивалентами аспарагиновой кислоты в смеси ацетонил/вода, 1:3. Образовавшуюся смесь замораживают и лиофилизируют.
Диаспартат можно получать также путем растворения соединения из примера 1 в смеси вода/ацетонитрил, 4:1, фильтрации, введения в ионобменную смолу, например, используя колонку типа BioRad AG4X4, и элюирования с помощью смеси вода/ацетонитрил, 4:1. Элюат концентрируют, замораживают и лиофилизируют. [α]D20=-47.5°, с=2,5 мг/мл в метаноле.
в. Бензоат
Превращение в бензоат можно осуществлять путем растворения соединения из примера 1 с 2 эквивалентами бензойной кислоты в смеси ацетонитрил/вода, 1:2. Образовавшуюся смесь замораживают и лиофилизируют.
г. Памоат
1 эквивалент соединения из примера 1 растворяют с 1 эквивалентом эмбоновой кислоты в смеси ацетонитрил/ТГФ/вода, 2:2:1. Образовавшуюся смесь замораживают и лиофилизируют.
Пример 3: Цикло-[{4-(ДОТК-NH-C2H4-NH-CO-O-)Pro}-Phg-DTrp-Lys-Tyr(4-Bzl)-Phe
а) Трифторацетат цикло[-Pro(4-OCO-NH-CH2-CH2-NH2)-Phg-DTrp-Lys(КБО)-Tyr(Bzl)-Phe-]
Соединение синтезируют аналогично трифторацетату цикло[-Pro(4-ОСО-NH-CH2-CH2-NH2)-Phg-DTrp-Lys(KEO)-Tyr(Bzl)-Phe], используя Fmoc-Lys(КБО)-OH вместо Fmoc-Lys(Boc)-OH.
б) 400 мг поступающего в продажу ДОТК × 2Н2O (фирма SYMAFEX, Франция) растворяют в 20 мл воды. После добавления 20 мл ДМФ добавляют 170 мг цикло[-Pro(4-OCO-NH-CH2-CH2-NH2)-Phg-DTrp-Lys(КБО)-Tyr(Bzl)-Phe-] вместе с 190 мг ДЦКИ и 60 мг N-гидроксисукцинимида. Образовавшуюся суспензию выдерживают при комнатной температуре в течение 72 ч. После фильтрации растворитель удаляют при пониженном давлении и оставшийся неочищенный продукт очищают на силикагеле (ДХМ/МеОН/НОАс50% 8/2/0,25 → 7/3/1 в качестве подвижной фазы).
в) Для удаления защитных групп описанный выше ДОТК - конъюгат обрабатывают 5 мл смеси трифторуксусная кислота/тиоанизол (9/1 )в течение 2 ч при комнатной температуре. Затем раствор сливают на смесь, содержащую 100 мл диэтилового эфира +5 мл 3н. HCl/диэтиловый эфир, и образовавшийся осадок осаждают фильтрацией. Очистку осуществляют на силикагеле, используя ДХМ/МеОН/НОАс50% 7/4/2 → 7/5/4 в качестве подвижной фазы. Аналитически чистый конечный продукт получают после стадии обессоливания, используя градиент от 0,1% ТФК до 0,1% ТФК в 90% CH3CN и колонку ОФ18-ЖХВР (типа Spherisorb, 250×4,6 мм). МН+: 1434,7
Соединение А в свободной форме или в форме фармацевтически приемлемых солей и комплексов обладает ценными фармакологическими свойствами, что продемонстрировано с помощью опытов in vitro и in vivo, благодаря чему их можно применять в терапии.
Более конкретно, для соединения А характерен интересный профиль связывания с человеческими рецепторами соматостатина (hsst), прежде всего с рецепторами hsst1, hsst2, hsst3 и hsst5. Ранее были клонированы и охарактеризованы пять подтипов рецептора соматостатина, а именно sst1, sst2, sst3, sst4 и sst5. Подтипы hssti, hsst2 и hsst3 и их последовательности описаны у Y.Yamada и др. в: Proc. Nat. Acad. Sci., 89, 251-255 (1992). hsst4 и его последовательность описаны у L. Rohrer и др. в: Proc. Acad. Sci., 90, 4196-4200 (1993). hsst5 и его последовательность описаны у R. Panetta и др. в: Mol. Pharmacol. 45, 417-427, (1993).
Анализы связывания можно осуществлять согласно описанному ниже методу с использованием мембран, полученных из клеточных линий, которые избирательно и стабильно экспрессируют hsst1, hsst2, hsst3, hsst4 или hsst5, например, СНО- или COS-клеток.
Мембраны получают согласно известным методам, например описанным у С. Bruns и др. в: Biochem. J., 65. 39-44 (1990). Мембраны, полученные из hsst-селективных линий клеток, например, СНО- или COS-клеток, которые стабильно экспрессируют hsst1 или hsst2, или hsst3, или hsst4, или hsst5, инкубируют в трех повторностях в общем объеме 300 мкл при 22°С в течение 30 мин с возрастающими концентрациями [125I-Tyr11]-SRIF-14 в Hepes-буфере с концентрацией 10 ммолей/л (рН 7,6), содержащем 0,5% БСА. Инкубацию прекращают путем быстрой фильтрации и подсчитывают радиоактивность фильтратов. Специфическое связывание рассчитывают как общее связывание минус неспецифическое связывание в присутствии 1 мкмоля/л соматостатина-14. Эксперименты осуществляют в трех повторностях. Аффинную константу (КD) и количество сайтов связывания рассчитывают с помощью соответствующих статистических и графических программ.
С помощью вышеописанных анализов связывания с hsst1, hsst2, hsst3 и/или hsst5 установлено, что значения IC50 для соединения А находятся в наномолярном диапазоне, предпочтительно значение IC50 составляет от 0.1 до 10 нМ (IC50 представляет собой концентрацию, при которой ингибирование при анализе конкурентного связывания с использованием [125I-Tyr11]-SRIF-14 в качестве специфического для hsst1-5 радиолиганда составляет половину от максимального).

Соединение А связывается также с рецепторами стимулятора секреции гормона роста. Такие рецепторы описаны у G. Muccioli и др., J. Endocrinol., 157, 99-106 (1998), H. Ong и др., в: Endocrinology, 139, 432-435 (19998) и R.G. Smith и др., Horm. Res. 3), 1-8 (1999). Анализ связывания с этими рецепторами можно осуществлять согласно методу, описанному в: J. Endocrinol. Invest. 24: RC1-RC3 (2001). При осуществлении указанного анализа соединение А замещает 125I-Tyr-Ala-гексарелин. Таким образом, соединение А можно применять для модуляции активности рецепторов стимулятора секреции гормона роста, например, с учетом их возможной роли в прибавлении веса тела и регуляции метаболизма.
Кроме того, соединение А проявляет ингибирующую активность в отношении секреции РГ (гормон роста, соматотропин), что установлено по ингибированию секреции РГ in vitro из культивируемых гипофизных клеток. Например, передние доли гипофиза взрослых самцов крыс нарезают на мелкие кусочки и диспергируют с помощью 0,1%-ного раствора трипсина в 20 мМ HEPES-буфере. Диспергированные клетки культивируют в течение 4 дней в среде MEM (фирма Gibco), дополненной 5% фетальной телячьей сыворотки, 5% лошадиной сыворотки, 1 мМ NaHCO3, 2,5 нМ дексаметазоном, 2,5 мг/мл инсулина и 20 ед./мл Pen/Strep. В день эксперимента прикрепившиеся клетки дважды промывают средой Кребса-Рингера, забуференной 20 мМ HEPES и дополненной 5 мМ глюкозой и 0,2 % БСА. Затем клетки инкубируют в течение 3 ч с соединением А в присутствии 3×10-10 М рилизинг-фактора гормона роста. Количество высвободившегося в среду гормона роста оценивают с помощью РИА. В этом опыте установлено, что значение IC50 соединения А составляет 0,4 нМ.
Соединение А ингибирует высвобождение гормона роста (РГ) у крыс. Соединение А вводят подкожно (s.c.) крысам после анестезии. Образцы крови собирают после декапитации, которую осуществляют через 1 ч после введения соединения. Продолжительность действия оценивают на основе данных о ингибировании основного уровня секреции РГ через 6 ч после введения лекарственного средства. Концентрации гормона оценивают с помощью РИА через 1 и 6 ч после обработки. Значение ID50 для ингибирования секреции гормона для каждого эксперимента оценивают графически (log-пробит) и усредняют логарифмы полученных значений. С помощью этой модели in vivo установлено, что соединение А в значительной степени ингибирует высвобождение гормона роста и обладает продолжительным действием (среднее основное значение ID50=5,5 мкг/кг, s.c., 6 ч). С помощью аналогичного опыта оценивают воздействие на высвобождение инсулина, установлено, что соединение А ингибирует секрецию инсулина.
Уровень и эффективность ингибирования РН также была подтверждена в опытах на обезьянах. Оценка метаболизма у страдающих диабетом обезьян позволила установить наличие у соединения А выраженного антидиабетического/повышающего чувствительность к инсулину действия.
Кроме того, соединение А ингибирует уровни в плазме IGF-1 in vivo, что установлено с помощью стандартных тестов с использованием самцов крыс. В целом, тест состоит в следующем: соединение А вводят с помощью осмотического насоса, имплантированного подкожно самцам крыс линии Lewis. Собирают образцы крови из расположенного позади глазного яблока (ретробульбарного) сплетения после непродолжительной анестезии, например изофлураном. С помощью этого анализа установлено, что соединение А в значительной степени понижает уровни IGF-1 в плазме и отличается длительным остаточным действием: например, уровень ингибирования, превышающий 60%, наблюдают через 14 дней после обработки 10 мкг/кг/ч соединения А. Еще более четко отсутствие клиренса было обнаружено в опытах по длительной обработке крыс-реципиентов с аллотрансплантатами аорты или почки, которым непрерывно в течение периода времени до 126 дней вводили путем инфузии соединение А в дозе 10 мкг/кг/ч, что индуцирует значительное и сохраняющееся в течение продолжительного времени понижение уровней IGF-1 в плазме.
Таким образом, соединение А можно применять для предупреждения или лечения патологий, этиология которых включает или связана с избыточной секрецией РГ и/или избыточным уровнем IGF-1, например, для лечения акромегалии, а также для лечения сахарного диабета типа I или типа II, прежде всего связанных с ними осложнений, например, ангиопатии, диабетической пролиферативной ретинопатии, диабетического отека желтого пятна, нефропатии, нейропатии и синдрома Дауна, и других метаболических патологий, связанных с секрецией инсулина или глюкагона, например ожирения, например, ожирения, препятствующего нормальному функционированию организма, или ожирения, связанного с гипоталамусом или гиперинсулинемией. Соединение А можно также применять для лечения проникающего через кожу и проникающего через оболочку поджелудочной железы свища, синдрома раздраженной толстой кишки, воспалительных заболеваний, например, болезни Грейвса, воспалительного заболевания кишечника, псориаза или ревматоидного артрита, поликистозного заболевания почки, демпинг-синдрома, синдрома водянистого стула, связанной со СПИДом диареи, вызванной химиотерапией диареи, острого и хронического панкреатита и опухолей, секретирующих желудочно-кишечный гормон (например, GEP-опухолей, например, випом, глюкагоном, инсулином, карциноидов и т.п.), злокачественных связанных с лимфоцитами заболеваний, например лимфом или лейкозов, печеночно-клеточного рака, а также желулочно-кишечного кровотечения, например, кровотечения из варикозно расширенных вен пищевода.
Соединение А можно также применять для лечения опухолей, которые являются позитивными в отношении рецепторов соматостатина, например, опухолей, несущих hsst1, hsst2, hsst3 и/или hsst5, что установлено с помощью экспериментов по оценке пролиферации с использованием различных линий раковых клеток, несущих указанные рецепторы соматостатина.
Линию опухолевых клеток поджелудочной железы крысы AR42J получают из вызванной азасерином экзокринной опухоли поджелудочной железы (Jessop и Hay, 1980). Не содержащие микоплазматических клеток культуры размножают в среде DMEM, дополненной 10% фетальной телячьей сыворотки (ФТС) в атмосфере 5% СО2. Клетки выращивают без антибиотиков или противогрибковых агентов. Находящиеся на субконфлюэнтной стадии клетки линии AR42J обрабатывают трипсином, разбавляют DMEM+2,5% ФТС и высевают в несенсибилизированные 96-луночные планшеты. После 48-часового периода инкубации (день 0) количество клеток в различных контрольных планшетах определяют как подсчетом количества клеток с помощью счетчика Коултера (Coulter), так и помощью SRB-колориметрического анализа. Далее клетки обрабатывают различными концентрациями соединения А в течение 2-5 дней и затем подсчитывают. В этих условиях установлено, что соединение А ингибирует пролиферацию опухолевых клеток в концентрациях от 10-12 до 10-6 М.
Оценка роста опухолей in vivo
Самок бестимусных мышей весом 19-22 г содержат группами по 5 животных в каждой в условиях, когда они имеют свободный доступ к воде для питья и не содержащему патогенов корму для грызунов. С помощью культивируемых клеток линии AR42J инициируют развитие подкожных опухолей. Обработку начинают через 2-4 дня после инокуляции опухолевых клеток, причем соединение А вводят путем непрерывной инфузии, например, с скоростью от 10 до 50 мкг/кг/ч. Размеры опухолей определяют с помощью циркуля. Для статистического анализа применяют t-критерий Стьюдента. С помощью указанного анализа установлено, что к 11 дню соединение А ингибирует рост опухолей на 51% по сравнению с контролем, в котором для обработки используют физиологический раствор.
Таким образом, соединение А можно применять для лечения пролиферативных болезней, связанных со злокачественным ростом клеток, например раковых опухолей, прежде всего опухолей, несущих рецепторы соматостатина таких типов, с которыми оно связывается с достаточной аффинностью, например, как это описано ниже для входящего в состав комплекса конъюгированного соединения А.
Соединение А также оказывает ингибирующее действие в отношении ангиогенеза, что доказано с помощью стандартных тестов, например, с использованием бестимусных мышей. В целом, тест состоит в следующем: опухолевые клетки (от 0,1 до 10×106 в 0,1 мл) (клетки линии SiHa и клетки линии MDA МВ-231, полученные согласно методам, описанным в: Angiogenesis, R. Steiner, Р.В. Weisz и R. Langer (ред.), 1992, Швейцария), инокулируют внутрикожно. Как правило, мышам вводят по две инъекции в середину брюшного отдела в области, локализованные далеко от основных брюшных кожных сосудов, для того чтобы количество исходных сосудов было низким. Мышам контрольной группы вводят по 0,1 мл 0,02%-ного трипанового синего в ЗФР. Через 10 дней после инъекции мышей после анестезии умерщвляют аспирацией СО2. Кожу монтируют на пластиковое кольцо (диаметром 40 мм) для оценки с помощью инвертированного микроскопа (типа Zeiss IM) при 12,5-и 25-кратном увеличении. Для оценки ангиогенеза сосуды фотографируют и подсчитывают количество сосудов, непосредственно связанных с опухолью. У контрольных животных подсчитывают количество сосудов, непосредственно связанных с определенной областью вокруг мест инъекции. Эта область соответствует средней площади кожных опухолей. Последнюю определяют позднее с помощью циркуля из уравнения 3,14×r2. Соединение А вводят s.c. либо в день инокуляции опухоли, либо через 3 дня. Контрольных животных обрабатывают носителем. В этом опыте установлено, что соединение А ингибирует образование кровеносных сосудов при введении в дозе, например, от 0,01 до 1000 мкг/кг s.c.
Таким образом, соединение А можно применять для предупреждения или лечения ангиогеназа, указанных выше воспалительных заболеваний, включая воспалительные заболевания глаз, отек желтого пятна, например, кистозный отек желтого пятна, идиопатический кистозный отек желтого пятна, эксудативную связанную с возрастом дегенерацию желтого пятна, болезни, связанные с неоваскуляризацией сосудистой оболочки глаза, и пролиферативную ретинопатию.
Соединение А может также оказывать ингибирующее действие в отношении пролиферации и миграции клеток гладких мышц, что установлено с помощью описанных ниже тестов.
Хроническое отторжение аллотрансплантата
Почку самца крыс линии DA (RT1a) ортотопически трансплантируют реципиенту - самцу крыс линии Lewis (RT11). Всего для трансплантации используют 24 животных. Всем животным вводят циклоспорин А в дозе 7,5 мг/кг/день перорально (per os) в течение 14 дней, начиная со дня трансплантации, предупреждая тем самым острое клеточное отторжение. Контралатеральную нефрэктомию не проводят. Каждая экспериментальная группа, которую обрабатывают конкретной дозой соединения А или плацебо, состоит из 6 животных. Начиная с 14 дня после трансплантации животных-реципиентов обрабатывают вплоть до 112 дня путем инфузии соединения А, или им вводят плацебо. Через 14 дней после трансплантации перфузию органа оценивают с помощью ЯМР-томографии. Эту процедуру повторяют через 53-64 дня после трансплантации и в конце эксперимента. Затем проводят аутопсию животных. Установлено, что введение соединения А в дозе 10 мкг/кг/ч при использовании указанной модели аллотрансплантата почки улучшает перфузию органа, а также снижает степень хронического отторжения, связанного с сосудистой реконструкцией и инфильтрацией трансплантата (клеточное отторжение). Выявлено также значительное и сохраняющееся в течение длительного времени снижение уровней IGF-1. Эти результаты подтверждены во второй серии экспериментов, основанных на использовании в качестве модели гетеротопической аллотрансплантации сердца мыши, в которых продемонстрированы полезные воздействия на сосудистую реконструкцию, а также на инфильтрацию трансплантата.
Соединение А также изучено при применении в качестве модели трансплантации петли сонной артерии с использованием мышей линии В 10. А(2R) (H-2h2) в качестве донора и мышей линии B10.BR (Н-2k) в качестве реципиента. В целом опыт состоит в следующем: сонную артерию донора трансплантируют паратопически в виде петли в сонную артерию реципиента путем анастомоза "конец в бок". Мини-насос вводят s.c. сразу же после трансплантации для введения соединения А в дозе 50 мкг/кг/ч. Трансплантаты сонной артерии удаляют через 30 дней после трансплантации для анализа сосудистой реконструкции, например, с помощью морфометрического анализа парафиновых срезов, окрашенных эластином Верхоффа (Verhoeff), с использованием компьютеризированной системы. С помощью этой модели установлено, что соединение А ингибирует образование новой интимы по сравнению с необработанными животными, у которых происходит образование массивной новой интимы.
Ангиопластика (реконструкция сосудов)
Изучение ангиопластики осуществляют с использованием в качестве модели сосудов крыс, поврежденных с помощью катетера-баллона. Катетеризацию с помощью баллона осуществляют в день 0, в целом, согласно методу, описанному у Powell и др. (1989). Анестизированным с помощью изофлуорана животным в левую общую сонную артерию вводят катетер-баллон Фогарти 2F, при этом происходит однородное удаление эндотелия. Затем катетер удаляют, накладывают лигатуру вокруг наружной сонной артерии для предупреждения кровотечения и животным дают отойти от наркоза. Для опыта используют 2 группы по 12 крыс линии RoRo (вес 400 г, возраст примерно 24 недели): одну контрольную группу и одну группу, которую обрабатывают соединением А, крыс по группам распределяют совершенно случайным образом. Соединение А вводят с помощью непрерывной инфузии с использованием мини-наносов со скоростью 10 мкг/кг/ч, начиная обработку за 2 дня до катетеризации баллоном (день - 3) и проводя ее вплоть до окончания эксперимента, т.е. через 14 дней после повреждения баллоном. Затем крыс анестезируют изофлуораном и подвергают перфузии с помощью 0,1 М забуференного фосфатом физиологического раствора (ЗФР, рН 7,4) и далее в течение 15 мин с помощью 2,5%-ного глутарового альдегида в фосфатном буфере (рН 7,4). Затем сонные артерии вырезают, отделяют от окружающей ткани и погружают в 0,1М какодилатный буфер (рН 7,4), содержащий 7% сахарозы, и инкубируют в течение ночи при 4°С. На следующий день сонные артерии погружают в Technovit 7100, следуя рекомендации производителя. Площадь поперечных сечений средней оболочки кровеносных сосудов (медия), новой интимы и полости оценивают морфометрически с помощью системы для анализа изображения (фирма MCID, Торонто, Канада). С помощью этого анализа установлено, что соединение А в значительной степени ингибирует толщину новой интимы.
Таким образом, соединение А можно применять также для предупреждения или лечения заболеваний сосудистого трансплантата, например, сосудистых патологий алло- или ксенотрансплантатов, например, атеросклероза сосудистого трансплантата, например при трансплантации органа, например, трансплантатов сердца, легкого, объединенного трансплантата сердце-легкое, печени, почки или поджелудочной железы, или для предупреждения или лечения стеноза трансплантатов вен, рестеноза и/или сосудистой окклюзии после повреждения сосудов, например, вызванного процедурами катетеризации или процедурами кюретажа сосудов, такими как чрескожная внутриполостная ангиопластика, лазерного лечения или другими инвазивными процедурами, при которых нарушается целостность сосудистой интимы или эндотелия.
Соединение А обладает достаточно продолжительным временем полужизни в плазме. Период его полувыведения составляет 15-30 ч.
Для всех указанных выше показаний требуемая доза, естественно, должна варьироваться в зависимости, например, от хозяина, пути введения и серьезности подлежащего лечению состояния. Однако, как правило, удовлетворительные результаты получают при введении примерно от 1 мкг до 0,7 мг/кг/день соединения А. Рекомендуемая суточная доза для пациентов составляет от примерно 2 мкг до примерно 50 мг, предпочтительно от примерно 0,01 до примерно 40 мг, например, от примерно 0,01 до примерно 3 мг s.c. соединения, которое целесообразно вводить в виде разделенных доз до 3 раз в день в виде стандартной дозируемой формы, содержащей, например, от примерно 0,5 мкг до примерно 25 мг, например, примерно от 2 мкг до 20 мг, например, от 2 мкг до 1,5 мг соединения А.
Соединение А можно вводить в свободной форме или форме фармацевтически приемлемой соли или комплексов. Такие соли и комплексы можно получать общепринятыми методами, и они обладают активностью, соответствующей активности свободного соединения. Настоящее изобретение относится также к фармацевтической композиции, которая содержит соединение А в форме свободного основания или в форме фармацевтически приемлемой соли или в форме комплекса в сочетании с одним или несколькими фармацевтически приемлемыми разбавителем или носителем. Такие композиции можно приготавливать общепринятыми методами. Соединение А можно также применять в виде формы с непрерывным высвобождением, например в виде имплантатов, микрокапсул, микросфер или наносфер, содержащих, например, способный к биологическому разложению полимер или сополимер, в форме липосом или в форме аутогеля, например в случае, когда твердая или полутвердая композиция может образовывать гель после взаимодействия с общей водой организма пациента.
Соединение А или его фармацевтически приемлемую соль или комплекс можно вводить с помощью любого обычного пути введения, например, парентерально, например, в форме инъецируемых растворов или суспензий (включая, например, указанную выше форму с непрерывным высвобождением), перорально с помощью общепринятого усилителя абсорбции, назально или в форме суппозитория или местно, например, в форме глазной жидкости, геля, мази или суспензионного препарата, например, композиции в виде липосомы, микросферы или наносферы, например, для инстилляции или введения под конъюнктиву или внутри- или периокулярных инъекций.
Согласно вышеизложенному под объем настоящего изобретения подпадает также:
1. Соединение А или его фармацевтически приемлемая соль или комплекс, предназначенные для применения в качестве фармацевтического агента,
2. Способ предупреждения или лечения указанных выше болезней или нарушений у пациента, который нуждается в таком лечении, предусматривающий введение пациенту эффективного количества соединения А или его фармацевтически приемлемой соли или комплекса, или
3. Соединение А или его фармацевтически приемлемая соль или комплекс для приготовления фармацевтической композиции, предназначенной для применения согласно любому способу, указанному выше в п. 2.
Конъюгированное соединение А или его фармацевтически приемлемая соль можно применять в качестве агента для визуализации, например, для визуализации тканей и клеток, позитивных в отношении рецептора соматостатина, например, позитивных в отношении рецептора соматостатина опухолей и метастазов, воспалительных или аутоиммунных заболеваний, связанных с рецепторами соматостатина, туберкулеза или отторжения органа после трансплантации, при объединении в комплекс с обнаруживаемым элементом, например нуклидом, испускающим γ-лучи или позитроны, флуоресцентным ионом металла или парамагнитным ионом, например, 111In, 161Tb, 177Lu, 86Y, 68Са Eu3+, Gd3+, Fe3+, Mn3+ или Cr3+, или в качестве радиофармацевтического агента для лечения in vivo позитивных в отношении рецептора соматостатина опухолей и метастазов, ревматоидного артрита и серьезных воспалительных состояний, при объединении в комплекс с испускающими α- или β-лучи нуклидами или нуклидом с каскадным Оже-е--излучением, например, 90Y, 161Tb, 177Lu, 211At, 213Bi или 201Tl, с использованием стандартных тестов.
В частности, установлено, что конъюгированное соединение А связывается в рецепторами соматостатина, при этом значения pKi составляют примерно от 8 до 10. Концентрации, в которых соединение из примера 3 в комплексе, например, с 111In, 88Y, 90Y или 177Lu связывается с соответствующими подтипами sst, находятся в наномолярном диапазоне согласно профилю связывания соединения А.
Аффинность конъюгированного соединения А и его комплексов к рецепторам соматостатина можно продемонстрировать также с помощью анализа in vivo с использованием стандартных методов анализа, например, описанных в GB-A-2225579. Например, обнаружено, что соединение из примера 3 в комплексе, например, с 111In, 88Y, 90Y или 177Lu, в значительной степени накапливается в опухоли через 4 ч после инъекции мышам или крысам, несущим экзокринную опухоль поджелудочной железы, клетки которой экспрессируют hsst2-рецепторы.
Введение конъюгированного соединения А в форме комплекса, например, соединения А в комплексе с 111In, 177Lu, 86Y или 161Tb, в дозе от 1 до 5 мкг/кг, меченного с помощью нуклида с радиоактивностью от 0,1 до 5 мКи, предпочтительно от 0,1 до 2 мКи, обеспечивает возможность определения месторасположения опухоли.
Конъюгированное соединение А, в которое с помощью испускающего α- или β-лучи радионуклида или нуклида с каскадным Оже-е--излучением включена радиоактивная метка, обладает антипролиферативным и/или цитотоксическим действием в отношении опухолевых клеток, несущих рецепторы соматостатина, что, например, установлено с помощью опытов на бестимусных мышах.
Бестимусных мышей инокулируют опухолевыми клетками поджелудочной железы крысы линии AR42J или клетками мелкоклеточного рака легкого человека линии NCI-H69 согласно описанному выше методу. Когда объем опухолей достигает 1-2 см3, животных случайным образом разделяют на контрольную и опытную (подвергающуюся обработке) группу. Конъюгированное соединение А в форме комплекса вводят с помощью внутрибрюшинной (i.p.) или внутривенной (i.v.) инъекций. Каждой мыши вводят дозу до 40 мКи/кг. Размер опухолей определяют с помощью циркуля, как описано выше. Для статистической обработки используют t-критерий Стьюдента. С помощью этого опыта установлено, что после однократного введения соединения из примера 3 в комплексе с 90Y, через одну неделю происходит временное уменьшение размера опухоли до 50% от первоначального объема, и рост опухоли замедляется в течение двух недель. В противоположность этому в контрольных группах обнаружен непрерывный рост опухоли, причем ее объем удваивался в течение примерно 7 дней.
Таким образом, под объем конкретных или альтернативных вариантов осуществления настоящего изобретения подпадает также:
4. Применение конъюгированного соединения А в комплексе с обнаруживаемым элементом для выявления in vivo у пациента позитивных в отношении рецептора соматостатина клеток и тканей, например, позитивных в отношении рецептора соматостатина опухолей и метастазов, и определение локализации рецепторов, являющихся мишенью для указанного комплекса.
5. Способ обнаружения in vivo у пациента позитивных в отношении рецептора соматостатина тканей и клеток, например, позитивных в отношении рецептора соматостатина опухолей и метастазов, предусматривающий введение пациенту конъюгированного соединения А в комплексе с обнаруживаемым элементом или в форме его фармацевтически приемлемой соли, и определение локализации рецепторов, являющихся мишенью для указанного комплекса.
Конъюгированное соединение А в форме комплекса, предназначенное для применения в качестве агента для визуализации, можно вводить, например, внутривенно, например, в форме инъецируемых растворов или суспензий, предпочтительно в форме однократной инъекции. Введение радиоактивной метки предпочтительно можно осуществлять непосредственно перед обработкой пациента.
Рекомендованные для применения на животных дозы могут составлять от 0,01 до 1 мкг/кг конъюгированного соединения А в комплексе с испускающим γ-лучи радионуклидом, имеющим радиоактивность от 0,02 до 0,5 мКи. Для более крупных млекопитающих, например, людей, рекомендованные дозы могут составлять от 1 до 100 мкг/м конъюгированного соединения А в комплексе, например, с обнаруживаемым элементом, имеющим радиоактивность от 1 до 100 мКи/м2, например, 111In, 86Y или 177Lu.
6. Применение конъюгированного соединения А в комплексе с испускающим α- или β-лучи нуклидом или нуклидом с каскадным Оже-е -излучением, для лечения in vivo позитивных в отношении рецептора соматостатина опухолей и метастазов.
7. Способ лечения in vivo позитивных в отношении рецептора соматостатина опухолей и метастазов, например, лечения инвазии таких опухолей или симптомов, связанных с ростом таких опухолей, у пациента, который нуждается в таком лечении, предусматривающий введение пациенту терапевтически эффективного количества конъюгированного соединения А в комплексе с испускающим α- или β-лучи нуклидом или нуклидом с каскадным Оже-е--излучением.
8. Применение конъюгированного соединения А или его фармацевтически приемлемой соли для приготовления агента для визуализации или радиофармацевтической композиции.
Дозы, применяемые для практического воплощения радиотерапевтического применения по настоящему изобретению, естественно, должны варьироваться в зависимости, например, от конкретного состояния подлежащего лечению, например, от известной радиотоксичности в отношении здоровых органов, экспрессирующих рецепторы соматостатина, объема опухоли и требуемой терапии. Как правило, дозу рассчитывают на основе фармакокинетических данных и данных о распределении радиоактивности, полученных для здоровых органов, и с учетом данных о поглощении мишенью. Испускающий β-лучи комплекс, включающий конъюгированное соединение А, можно вводить повторно, например, через 1-3 месяца.
Рекомендованные для применения на животных дозы могут составлять от 20 до 100 мкг/кг конъюгированного соединения А в комплексе с имеющим радиоактивность от 15 до 70 мКи испускающим α-или β-лучи нуклидом или нуклидом с каскадным Оже-е--излучением, например, 90Y, 177Lu или 161Tb. Для более крупных млекопитающих, например, людей рекомендованная доза конъюгированного соединения А в комплексе с имеющим радиоактивность от 1 до 70 мКи/м испускающим α-или β-лучи нуклидом или нуклидом с каскадным Оже-е--излучением, например, 90Y, 177Lu или 161Tb, может составлять от 1 до 100 мкг/м2.
Конъюгированное соединение А в форме комплекса, предназначенное для применения в качестве радиотерапевтического агента, можно вводить любым общепринятым путем, например, внутривенно, например, в фоме инъецируемых растворов. Его целесообразно вводить с помощью инфузии, например, инфузии продолжительностью 15-60 мин. В зависимости от локализации опухоли его можно вводить по возможности максимально близко к месторасположению опухоли, например, с помощью катетера. Настоящее изобретение относится также к фармацевтической композиции, содержащей конъюгированное соединение А в форме свободного основания или в форме фармацевтически приемлемой соли или комплекса с обнаруживаемым или радиотерапевтическим агентом, в сочетании с одним или несколькими фармацевтически приемлемыми разбавителем или носителем.
Соединение А или конъюгированное соединение А в форме комплекса можно применять для визуализации или лечения экспрессирующих или накапливающих рецепторы соматостатина опухолей, таких как опухоли гипофиза, гастро-энтеропанкреатической системы, карциноидные опухоли, опухоли центральной нервной системы, молочной железы, предстательной железы (включая запущенный невосприимчивый к действию гормонов рак предстательной железы), яичника или ободочной кишки, мелкоклеточный рак легкого, злокачественная обструкция кишечника, параганглиомы, рак почки, рак кожи, нейробластомы, феохромоцитомы, медуллярные карциномы щитовидной железы, миеломы, лимфомы, Ходжкинская и не-ходжкинская лимфомы, опухоли кости и их метастазы, а также аутоиммунные или воспалительные заболевания, например, ревматоидный артрит, болезнь Грейвса или другие вспалительные заболевания глаз.
Таким образом, еще одним объектом изобретения является фармацевтическая композиция, содержащая конъюгированное соединение А или его комплекс в сочетании с одним или несколькими фармацевтически приемлемыми носителями или разбавителями. Такие композиции можно приготавливать общепринятым методом, и их можно применять, например для визуализации, в форме набора, содержащего различные дозируемые формы, одна из которых представляет собой радионуклид, а другая - конъюгированное соединение А, с инструкцией по их смешению. Для радиотерапии конъюгированное соединение А в форме комплекса предпочтительно может иметь форму горячей жидкой композиции.
Соединение А или конъюгированное соединение А в форме комплекса можно применять в качестве единственного действующего вещества или в сочетании, например, с другими лекарственными средствами в качестве адъювантов. Например, соединение А можно применять в сочетании с иммунодепрессантом, например, ингибитором кальцинеурина, например, циклоспорином А или FK 506; макроциклическим лактоном, обладающим иммуносупрессорными свойствами, например рапамицином или 40-O-(2-гидроксиэтил)рапамицином (RAD); аскомицином, обладающим иммуносупрессорыми свойствами, например АВТ-281, ASM981, и т.д.; кортикостеридами; циклофосфамидом; азатиопреном; метотрексатом; лефлуномидом; мизорибином; микофенольной кислотой или ее солью, например, MyfbrticR; микофенолятом мофетила; 15-дезоксиспергуалином или его обладающим иммуносупрессорной активностью гомологом, аналогом или производным; агентом, ускоряющим хоминг лимфоцитов, например, FTY720; иммуносупрессорными мононоклональными антителами, например, моноклональными антителами к рецепторам лейкоцитов, например, МНС, CD2, CD3, CD4, CD7, CD8, CD25, CD28, CD40, CD45, CD58, CD80, CD86 или к их лигандам; с другими иммуномодуляторами, например, рекомбинантной связывающей молекулой, несущей последнюю область внеклеточного домена CTLA4 или ее мутантом, например, по меньшей мере с внеклеточной областью CTLA4 или ее мутантом, соединенным с не относящейся к CTLA4 последовательностью протеина, например, CTLA4Ig (например, который имеет регистрационный номер АТСС 68629) или ее мутантом, например, LEA29Y; с ингибиторами молекулы адгезии, например, антагонистами LFA-1, антагонистами ICAM-1 или -3, антагонистами VCAM-4 или антагонистами VLA-4. Соединение А можно также применять в сочетании с противовоспалительным агентом, модулятором стимулятора секреции РГ, например, грелином или гексарелином, антагонистом рецептора РГ, например, пегвисомантом, стимулятором секреции инсулина или усилителетем секреции инсулина, например, сульфонилмочевиной, например, толбутамидом, хлорпропамидом, толазамидом, ацетогексамидом, 4-хлор-N-[(1-пиролидиниламино)карбонил]бензолсульфонамидом (гликопирамид), глибенкламидом (глибурид), гликлазидом, 1-бутил-3-метанилилмочевиной, карбутамидом, глибонуридом, глипизидом, глихидоном, глисоксепидом, глибутиазолом, глибузолом, глихексамидом, глимидином, глипинамидом, фенбутамидом или толилцикламидом, не содержащими сульфонильную группу мочевинами непродолжительного действия, пероральным инсулинтропным производным, например усилителями инсулина непродолжительного действия, например меглитинидом, репаглинидом, производным фенилуксусной кислоты, например, натеглинидом, ингибитором DPP IV, например дигидрохлоридом 1-{2-[(5-цианпиридин-2-ил)амино]этиламино}ацетил-(2S)-цианпирролидина, LAF237, аналогом агониста GLP-1 или a GLP-1, сенсибилизатором инсулина, например, агонистом γ-рецептора, активируемого пероксисомным пролифератором (PPARγ), например, глитазоном, например (S)-((3,4-дигидро-2-(фенилметил)-2Н-1-бензопиран-6-ил)метилтиазолидин-2,4-дионом (энглитазон), 5-{[4-(3-(5-метил-2-фенил-4-оксазолил)-1-оксопропил)фенил]метил}тиазолидин-2,4-дионом (дарглитазон), 5-{[4-(1-метилциклогексил)метокси)фенил]метил}тиазолидин-2,4-дионом (циглитазон), 5-{[4-(2-(1-индолил)этокси)фенил]метил}тиазолидин-2,4-дионом (DRF2189), 5-{4-[2-(5-метил-2-фенил-4-оксазолил)этокси)]бензил}тиазолидин-2,4-дионом (ВМ-13.1246), 5-(2-нафтилсульфонил)тиазолидин-2,4-дионом (AY-31637), бис{4- [(2,4-диоксо-5-тиазолидинил)метил]фенил} метаном (YM268), 5-{4-[2-(5-метил-2-фенил-4-оксазолил)-2-гидроксиэтокси]бензил}тиазолидин-2,4-дионом (AD-5075), 5-[4-(1-фенил-1-циклопропанкарбониламино)бензил]тиазолидин-2,4-дионом (DN-108), 5-{[4-(2-(2,3-дигидроиндол-1-ил)этокси)фенил]метил}тиазолидин-2,4-дионом, 5-[3-(4-хлорфенил])-2-пропинил]-5-фенилсульфонил)тиазолидин-2,4-дионом, 5-[3-(4-хлорфенил])-2-пропинил]-5-(4-фторфенилсульфонил)тиазолидин-2,4-дионом, 5-{[4-(2-(метил-2-пиридиниламино)этокси)фенил]метил}тиазолидин-2,4-дионом (росиглитазон), 5-{[4-(2-(5-этил-2-пиридил)этокси)фенил]метил}тиазолидин-2,4-дионом (пиоглитазон), 5-{[4-((3,4-дигидро-6-гидрокси-2,5,7,8-тетраметил-2Н-1-бензопиран-2-ил)метокси)фенил]метил }тиазолидин-2,4-дионом (троглитазон), 5-[6-(2-фторбензилокси)нафталин-2-илметил]тиазолидин-2,4-дионом (МСС555), 5-{[2-(2-нафтил)бензоксазол-5-ил]метил}тиазолидин-2,4-дионом (Т-174) или 5-(2,4-диоксотиазолидин-5-илметил)-2-метокси-N-(4-трифторметилбензил)бензамидом (KRP297), агонистом неглитазонного типа, таким как аналог N-(2-бензоилфенил)-L-тирозина, например, GI-262570, или оксолидиндионом, например, JTT501, двойным агонистом PPARγ/PPARα, например DRF-554158, NC-2100 или NN-622, агонистом ретиноидного X-рецептора или рексиноидом, например, 2-[1-(3,5,5,8,8-пентаметил-5,6,7,8-тетрагидро-2-нафтил)циклопропил]пиридин-5-карбоновой кислотой, 4-[(3,5,5,8,8-пентаметил-5,6,7,8-тетрагидро-2-нафтил)-2-карбонил]бензойной кислотой, 9-цис-ретиноевой кислотой или ее аналогом, производным или фармацевтически приемлемой солью, протеин-тирозин-фосфатазо-киназой 1В, ингибитором глюкоген-синтазо-киназой-3, непептидильными небольшими миметиками инсулина, например, L-783,281 или CLX-901, или с низкой дозой инсулина, ингибитором глутамин:фруктозо-6-фосфат-амидотрансферазы, ингибитором глюкозо-6-фосфатазы, бигуанидом, например, метформином, ингибитором фруктозо-1,6-бифосфатазы, ингибитором гликогенфосфорилазы, например, СР-91149, антагонистом рецептора глюкагона, например, СР-99711, NNC 92-1687, L-168,049 или BAY27-9955, фосфоенолпируват-карбоксикиназой, ингибитором [пируватдегидрогена]-киназы, ингибитором α-глюкозидазы, например, 4",6"-дидезокси-4"-[(18)-(1,4,6/5)-4,5,6-тригидрокси-3-гидроксиметил-2-циклогексениламино}мальтотриозой или 0-4,6-дидезокси-4- {[1S,4R,5S,6S]-4,5,6-тригидрокси-3-(гидроксиметил)-2-циклогексен-1-ил]-амино}-α-D-глюкопиранозил-(1→4)-O-α-D-глюкопиранозил-(1→4)-D-глюкопиранозой (акарбоза), N-(1,3-дигидрокси-2-пропил)валиоламидом (воглибоза) или миглитом, или с ингибитором опорожнения желудка, например, GLP-1, ССК-8 и амилином (например, прамлинтидом), агентом, обладающим антиангиогенными действиями, например, бензопорфирином, например, вертепорфином, мидостаурином или 4-пиридилметилфталазином.
Соединение А или конъюгированное соединение А в форме комплекса можно применять также в сочетании с антипролиферативным агентом, например, химиотерапевтическим лекарственным средством, например, паклитакселом, гемцитабином, цисплатином, доксорубицином, 5-флуороурацилом или таксолом, гормональным агентом или антагонистом, например, антиандрогеном или митоксантроном (прежде всего, в случае рака предстательной железы), или антиэстрогеном, типа летрозола (прежде всего, в случае рака молочной железы), антиметаболитом, растительным алкалоидом, модификатором биологических реакций, предпочтительно лимфокином или интерферонами, ингибитором протеин-тирозин-киназы и/или серин/треонин-киназы, или с агентом иного или неизвестного механизма действия, например, с любым эпотилоном или производным эпотилона, или макроциклическим лактоном, например, рапамицином, RAD или CCI779.
Когда соединение А или конъюгированное соединение А в форме комплекса вводят в сочетании с другим лекарственным средством, дозы применяемого для совместной терапии лекарственного средства, естественно, должны варьироваться в зависимости от типа применяемого для совместной терапии лекарственного средства, конкретного применяемого лекарственного средства, подлежащего лечению состояния и т.д. Понятия "совместное введение" или "объединенное введение" или т.п. в контексте настоящего описания относятся к введению выбранных терапевтических агентов одному и тому же пациенту, и подразумевается, что они включают режимы лечения, согласно которым агенты не обязательно вводят с помощью одного и того же пути введения или в один и тот же момент времени.
Согласно вышеизложенному объектами настоящего изобретения являются также:
9. Фармацевтическая комбинация, содержащая а) первый агент, представляющий собой соединение А или конъюгированное соединение А в форме комплекса, и б) дополнительный агент, например, указанный выше.
10. Способ, как он определен выше, предусматривающий совместное введение, например, одновременное или последовательное, терапевтически эффективного количества соединения А или конъюгированного соединения А в форме комплекса, и второго лекарственного средства, причем второе лекарственное средства представляет собой, например, указанное выше соединение.
Конкретную комбинацию по изобретению можно выбирать в зависимости от болезней или состояний, подлежащих предупреждению или лечению; например, использовать комбинацию с иммунодепрессантом, например, для предупреждения или лечения хронического отторжения трансплантата, комбинацию со стимулятором секреции инсулина, усилителем секреции инсулина, сенсибилизатором инсулина или с низкой дозой инсулина - для лечения диабета или связанных с ним осложнений, комбинацию с противовоспалительным агентом - для предупреждения или лечения воспалительных заболеваний или нарушений, комбинацию с агентом, обладающим антиангиогенными действиями - для предупреждения или лечения, например, отека или дегенерации желтого пятна или рака, комбинацию с химиотерапевтическим агентом - при раке.
| название | год | авторы | номер документа |
|---|---|---|---|
| ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ | 2004 |
|
RU2355418C2 |
| СОМАТОСТАТИНОВЫЕ ПЕПТИДЫ | 1996 |
|
RU2160741C2 |
| ПЕПТИДНЫЕ ВЕКТОРЫ | 2004 |
|
RU2361876C2 |
| ДЕПО-ФОРМА АНАЛОГА СОМАТОСТАТИНА И СПОСОБ ЕЕ ПОЛУЧЕНИЯ | 2005 |
|
RU2411031C2 |
| ПОЛУЧЕНИЕ ПЕПТИДОВ СОМАТОСТАТИНА | 2004 |
|
RU2360921C2 |
| КОМПОЗИЦИЯ С ЗАМЕДЛЕННЫМ ВЫСВОБОЖДЕНИЕМ, ВКЛЮЧАЮЩАЯ БИСФОСФОНАТ | 2005 |
|
RU2395274C2 |
| ХИМЕРНЫЕ АНАЛОГИ ЛИГАНДОВ СОМАТОСТАТИНОВЫХ И ДОПАМИНОВЫХ РЕЦЕПТОРОВ, ФАРМАЦЕВТИЧЕСКИЕ КОМПОЗИЦИИ НА ИХ ОСНОВЕ И СПОСОБЫ ВОЗДЕЙСТВИЯ НА РЕЦЕПТОРЫ СОМАТОСТАТИНА И/ИЛИ ДОПАМИНА | 2004 |
|
RU2329273C2 |
| МИКРОЧАСТИЦЫ, СОДЕРЖАЩИЕ АНАЛОГИ СОМАТОСТАТИНА | 2004 |
|
RU2404749C2 |
| КОМБИНАЦИЯ АНАЛОГОВ СОМАТОСТАТИНА С РАЗЛИЧНОЙ СЕЛЕКТИВНОСТЬЮ В ОТНОШЕНИИ ПОДТИПОВ РЕЦЕПТОРОВ СОМАТОСТАТИНА ЧЕЛОВЕКА | 2007 |
|
RU2451520C2 |
| ПРИМЕНЕНИЕ АНАЛОГОВ СОМАТОСТАТИНА ПРИ МЕНИНГИОМЕ | 2008 |
|
RU2523416C2 |
В заявке описано соединение цикло[{4-(NH2-C2H4-NH-CO-O-)Pro}-Phg-DTrp-Lys-Tyr(4-Bzl)-Phe], необязательно в защищенной форме, или его фармацевтически приемлемая соль или комплекс. Соединение обладает ингибирующей активностью на высвобождение гормона роста и инсулина. 3 н. и 6 з.п. ф-лы.
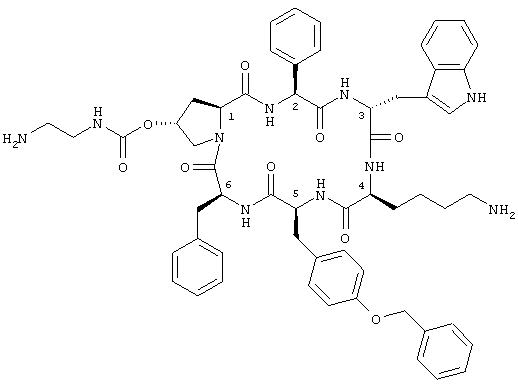
одна из аминогрупп которого необязательно находится в защищенной форме, или его соль или комплекс.
| RU 98101506 А, 20.12.1999 | |||
| Клетка для пчелиных маток | 1928 |
|
SU29310A1 |

