Настоящее изобретение относится к способу получения циклических аналогов соматостатина и к промежуточным продуктам, которые применяют в этом способе.
Более конкретно изобретение относится к способу получения циклического аналога соматостатина формулы I
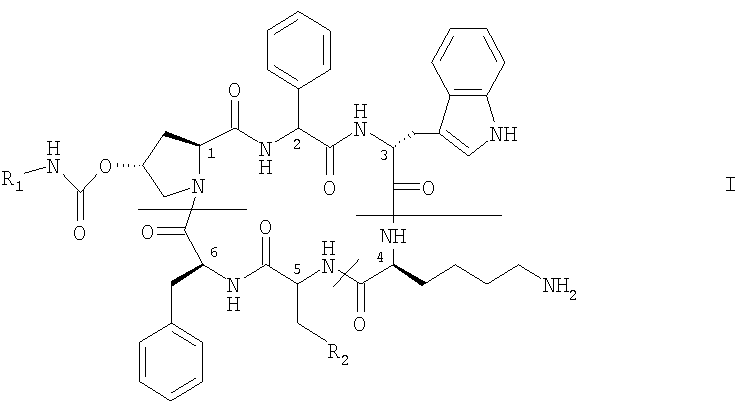
в которой
R1 обозначает -С2-C6алкилен-NR3R4, -С2-C6алкиленгуанидин или -С2-C6алкилен-СООН, где R3 и R4 каждый независимо друг от друга обозначает Н, С1-С4алкил, ω-гидрокси-С2-С4алкилен или ацил или R3 и R4 вместе с атомом азота, к которому они присоединены, образуют гетероциклическую группу, которая может содержать дополнительный гетероатом, и
R2 обозначает Z1-CH2-R5, -CH2-CO-O-CH2-R5
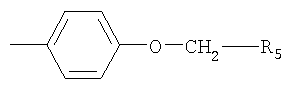 или
или 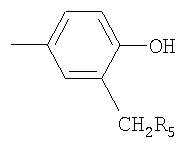
где Z1 обозначает О или S и R5 обозначает необязательно замещенный фенил, или его соли.
Ацил может представлять собой любой ацил, например RaCO-, где Ra обозначает Н, C1-C4алкил, С2-С4алкенил, С3-С6циклоалкил или бензил. Когда NR3R4 образует гетероциклическую группу, то такая группа может быть ароматической или насыщенной и может содержать один атом азота или один атом азота и второй гетероатом, выбранный из азота и кислорода. Предпочтительно гетероциклическая группа представляет собой, например, пиридил или морфолиногуппу. С2-С6алкилен предпочтительно обозначает -СН2-СН2-. Когда R5 обозначает замещенный фенил, то фенильное кольцо может быть замещено галогеном, метилом, этилом, метоксигруппой или этоксигруппой, например, в орто- и/или пара-положении. Предпочтительно R5 обозначает незамещенный фенил.
Аминокислоты в положении 2 и/или 5 могут иметь D- или L-конфигурацию. Предпочтительно каждая из них имеет L-конфигурацию.
Соединения формулы I могут находиться, например, в свободной форме или в форме соли. Соли представляют собой кислотно-аддитивные соли, образованные, например, с органическими кислотами, полимерными кислотами или неорганическими кислотами, например, с соляной кислотой, уксусной кислотой, молочной кислотой, аспарагиновой кислотой, бензойной кислотой, янтарной кислотой или памоновой кислотой, или, например, соли щелочных металлов, когда R1 несет СООН-группу. Кислотно-аддитивные соли могут представлять собой одно- или двухвалентные соли, например, в зависимости от того 1 или 2 кислотных эквивалента добавляют к соединению формулы I в форме свободного основания. Предпочтительными солями являются двухвалентный аспартат или одновалентный памоат.
Способ получения, предлагаемый в изобретении, включает стадию циклизации соответствующего линейного пептида в защищенной форме. При создании изобретения неожиданно было установлено, что стадия циклизации зависит прежде всего от выбора двух концевых аминокислот соответствующего линейного пептида. При создании изобретения неожиданно было установлено, что из 6 возможных сайтов циклизации циклизация между аминокислотами 3 и 4 или 4 и 5, или 6 и 1 приводит к очень важным результатам. Циклизация между аминокислотами 4 и 5 является наиболее предпочтительной. Циклизация, предлагаемая в изобретении, между аминокислотами 3 и 4 или 4 и 5, или 6 и 1 приводит к улучшенному выходу с пониженной изомеризацией в хиральных сайтах. Кроме того, стадии циклизации можно осуществлять в менее жестких условиях и с использованием менее сильных реактантов: например, можно исключать циклизацию с использованием азида.
Первым вариантом осуществления изобретения является способ получения указанного выше соединения формулы I или его соли, заключающийся в том, что осуществляют циклизацию линейного аналога соматостатина формулы II

или формулы III

или формулы IV

в которых R1 и R2 имеют указанные выше значения,
R11 и R12 каждый независимо друг от друга обозначает аминозащитную группу, причем, когда R1 содержит концевую NH2-группу, то эта концевая NH2-группа также защищена аминозащитной группой, и при необходимости удаляют защитную(ые) группу(ы) и выделяют соединение формулы I, полученное таким образом, в свободной форме или в форме соли.
Приемлемые аминозащитные группы описаны, например, в «Protective Groups in Organic Synthesis», Т.W.Greene и др., изд-во John Wiley & Sons Inc., 2-e изд., 1991. Примерами таких аминозащитных групп являются, например, ацетил или аминогруппы, которые используют в пептидном синтезе, например, трет-бутоксикарбонил, карбобензоксигруппа (КБО), флуоренилметоксикарбонил, аллоксикарбонил, 1-(4,4-диметил-2,6-диоксоциклогексилиден)этил, 1-(4,4-диметил-2,6-диоксоциклогексилиден)-3-метилбутил, 4-метилтритил (W.C.Chan и P.D.White, Fmoc solid Phase Peptide Synthesis, изд-во Oxford University Press, 2000).
Циклизацию удобно осуществлять в присутствии производного аминия или фосфония для активации карбоксигруппы in situ, например, гексафторфосфата O-(бензотриазол-1-ил)-N,N,N',N'-тетраметилурония, гексафторфосфата бензотриазол-1-илокситрис(пирролидино)фосфония, N-оксида тетрафторбората N-[(1Н-бензотриазол-1-ил)(диметиламино)метилен]-N-метилметанаминия, N-[(диметиламино)-1Н-1,2,3-триазоло[4,5-b]пиридин-1-илметилен]-N-метилметанаминия, гексафторфосфата 7-азабензотриазол-1-илокситрис(пирролидино)фосфония. Предпочтительно реакцию можно осуществлять в присутствии основания, например, органического амина, например, N-этилдиизопропиламина, N-метилморфолина, триэтиламина или трибензиламина, и в присутствии вспомогательного нуклеофила, например, 1-гидроксибензотриазола, N-гидроксисукцинимида, 3-гидрокси-3,4-дигидро-1,2,3-бензотриазин-4-она или 1-гидрокси-7-азабензотриазола.
Циклизация соединения формулы II, III или IV приводит к получению соединения формулы I в защищенной форме, т.е. соединения формулы I, в котором одна или несколько или все аминогруппы, присутствующие в молекуле, защищены аминозащитной группой. Примерами таких соединений являются, например, цикло[(2S,4R)-4-(Boc-NH-CH2-CH2-NH-CO-O)-Pro-Phg-D-Trp(Boc)-Lys(Boc)-Tyr(Bzl)-Phe-], цикло[(2S,4R)-4-(Boc-NH-CH2-CH2-NH-CO-O)-Pro-Phg-D-Trp-Lys(Boc)-Tyr(Bzl)-Phe-] и цикло[(2S,4R)-4-(Boc-NH-CH2-CH2-NH-CO-O)-Pro-DPhg-D-Trp-Lys(Boc)-Tyr(Bzl)-Phe-].
Аминозащитные группы можно удалять с помощью методов, хорошо известных в данной области, например, путем отщепления, например, с помощью трифторукусусной кислоты; 3М HCl, EtOAc; Ме3CCl, феонола, СН2Cl2; 10%-ной H2SO4, диоксана; бромкатехолборана.
Соединения формул II, III и IV или их соли являются новыми и являются частью изобретения. Эти соединения можно получать, связывая вместе с помощью амидной связи два пептидных звена, каждое из которых содержит по меньшей мере одну аминокислоту в защищенной или незащищенной форме, где амидная связь расположена так, чтобы получить требуемую аминокислотную последовательность, указанную в формулах II, III или IV.
Синтез можно осуществлять с помощью методов, известных в данной области, например, в растворе или путем твердофазного синтеза, начиная процесс с первой аминокислоты. При твердофазном синтезе первую аминокислоту связывают со смолой, например, с поступающей в продажу смолой на основе полистирола, необязательно с помощью приемлемого линкера, например, линкера, который может отщепляться в мягких условиях, оставляя при этом неповрежденной защиту боковой цепи, например, с использованием необязательно замещенного тритильного линкера, например, 4-(гидроксилдифенилметил)бензойной кислоты, при этом одна из фенильных групп необязательно может быть замещена, например, с помощью Cl. Построение требуемой пептидной цепи, либо в растворе, либо в помощью твердофазного синтеза, можно осуществлять общепринятым методом, например, используя аминокислоты, при этом концевые аминогруппы являются Fmoc-защищенными, боковые цепи аминогрупп, если они присутствуют, защищены различными аминозащитными группами, например, Вос или КБО.
Когда синтез осуществляют с помощью твердофазного синтеза, то синтезированный пептид затем отщепляют от смолы с помощью методов, известных в данной области, например, с помощью уксусной кислоты; трифторуксусной кислоты; смеси уксусная кислота-трифторэтанол-дихлорметан; гексафторизопропанола в дихлорметане. Предпочтительным методом отщепления синтезированного пептида от твердой фазы, например, при использовании нехлорированного тритильного линкера, является, например, обработка метанолом/дихлорметаном, предпочтительно при комнатной температуре, или обработка кетоном, например этилметилкетоном, предпочтительно при температуре примерно 50°С.
Примерами соединений формул II, III и IV являются, например,
H-Lys(Boc)-D-Tyr(Bzl)-Phe-(2S,4R)-4-(Boc-NH-CH2-CH2-NH-CO-O)-Pro-Phg-D-Tyr(Вос)-ОН,
H-Lys(Boc)-D-Tyr(Bzl)-Phe-(2S,4R)-4-(Boc-NH-CH2-CH2-NH-CO-O)-Pro-Phg-D-Tyr-ОН,
H-(2S,4R)-4-(Boc-NH-CH2-CH2-NH-CO-O)-Pro-Phg-D-Trp(Boc)-Lys(Boc)-Tyr(Bzl)-Phe-OH,
H-Tyr(Bzl)-Phe-(2S,4R)-4-(Boc-NH-CH2-CH2-NH-CO-O)-Pro-DPhg-DTrp(Boc)-Lys(Boc)-OH,
H-Tyr(Bzl)-Phe-(2S,4R)-4-(Boc-NH-CH2-CH2-NH-CO-O)-Pro-Phg-DTrp-Lys(Boc)-OH,
H-Tyr(Bzl)-Phe-(2S,4R)-4-(Boc-NH-CH2-CH2-NH-CO-O)-Pro-Phg-D-Trp(Boc)Lys(Boc)-OH.
Соединения формулы II, III или IV могут находиться в форме соли, как указано выше для соединений формулы I.
Соединения формулы I являются ценными агонистами соматостатина и обладают важными фармакологическими свойствами, которые описаны, например, в WO 97/01579 или WO 02/10192, содержание которых, касающееся фармакологических свойств, включено в настоящее описание в качестве ссылки. Предпочтительным соединением является цикло[{(4-NH2-С2Н4-NH-CO-O)-Pro}-Phg-DTrp-Lys-Tyr(4-бензил)-Phe] или его соль. Предпочтительными солями являются аспартат (моно- или диаспартат) или памоат.
При использовании способа, предлагаемого в изобретении, с помощью циклизации можно получать соответствующий линейный пептид формулы II, III или IV с выходом, превышающим 70%.
Ниже изобретение проиллюстрировано на примерах. Все температуры даны в °С.
Использовали следующие сокращения:
Пример 1: [H-Tyr(Bzl)-Phe-(2S,4R)-4-(Boc-NH-CH2-CH2-NH-CO-O)-Pro-Phg-D-Trp-Lys(Boc)-OH]
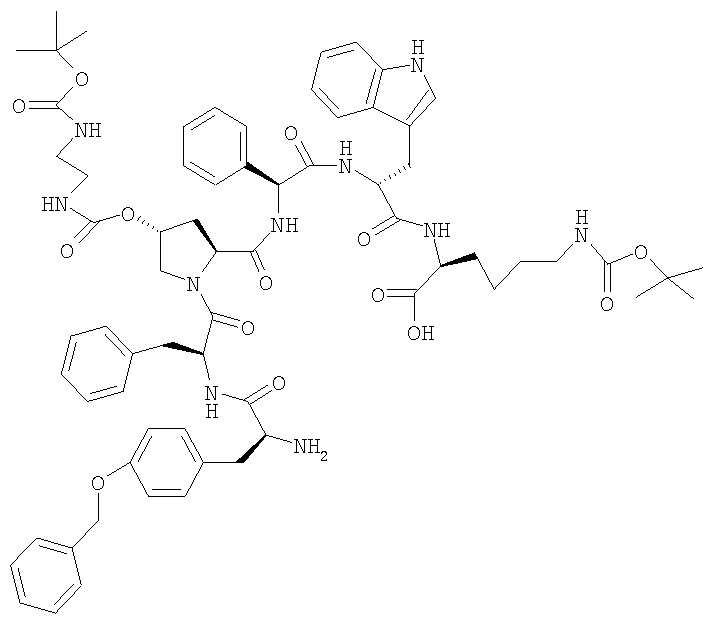
10,4 г (FMOC-Tyr(Bzl)-Phe-[(2S,4R)-4-(Boc-NH-CH2-CH2-NH-CO-O)]-Pro-Phg-D-Trp-Lys(BOC)-OH) растворяют в 100 мл ДМФ. При КТ добавляют 2,0 мл диэтиламина и перемешивание продолжают в течение 4 ч при КТ. Прозрачный раствор желтого цвета упаривают при 40°. К остатку добавляют 150 мл изопропилацетата. Вносят затравку и перемешивают в течение 18 ч при КТ, фильтруют и промывают 20 мл изопропилацетата. Сушат при 40°. ЖХВР 87,5% b.a. (17,6 мин). ESI-MC: 1287,5 (M+Na)+ 1Н-ЯМР (ДМСО) (∂, ДМСО): 1,14(2Н, m), 1,33(2Н, m), 1,37(18Н, m), 1,53(1Н, m), 1,64(1Н, m), 2,06(1Н, m), 2,22(1Н, m), 2,78-3,15(12Н, m), 3,38(1Н, m), 3,79(2Н, br), 4,12(1Н, m), 4,55-4,75(3Н, m), 5,06(2Н, s), 5,15(1H, d), 5,54(1H, d), 6,75(1H, br), 6,83(1H, br), 6,89(2H, d), 6,94(1Н, t), 6,99(1H, s), 7,43(2Н, d), 7,10-7,50(аром.Н), 7,59(1Н, d), 8,14(1Н, d), 8,45-8,55(2Н, m), 8,60(1Н, d), 10,71(1Н, br).
Применяемое в качестве исходного продукта соединение получают следующим образом:
a) Z-D-Trp-Lys(BOC)-OMe

К раствору, содержащему 23,7 г Z-D-Trp-ОН в 230 мл ТГФ, добавляют при КТ 9,5 г ГОБТ. Образуется прозрачный бесцветный раствор. Затем добавляют при КТ 20,8 г H-Lys(BOC)-OMe×HCl, а затем 7,7 мл N-метилморфолина.
Тонкую суспензию охлаждают до 0° и добавляют 11,4 мл диизопропилкарбодиимида. Перемешивают в течение 1 ч при 0°, затем 3 ч при КТ. К суспензии добавляют раствор, содержащий 7,0 г конц. серной кислоты в 120 мл воды. Экстрагируют 150 мл этилацетата, разделяют фазы и органическую фазу промывают последовательно 100 мл насыщенного соляного раствора, 100 мл 25%-ного соляного раствора, 5%-ным раствором бикарбоната натрия. Затем органическую фазу сушат над MgSO4 и упаривают. Остаток растворяют в 250 мл изопропилацетата и перемешивают 2 ч при КТ. После фильтрации и сушки при 40° получают 40,9 г Z-D-Trp-Lys(BOC)-OMe в виде вещества белого цвета. Условия ЖХВР: MN Nucleosil 100 А/С18; 5 мкм; 250×4 мм; фаза А: 0,24%-ная фосфорная кислота, фаза Б ацетонитрил; градиент: от 20 до 80% Б в течение 30 мин; длина волны 220 нм; скорость потока 1,3 мл/мин; температура 35°; чистота
97,8% b.a. (24,2 мин).
б) H-D-Trp-Lys(BOC)-OMe
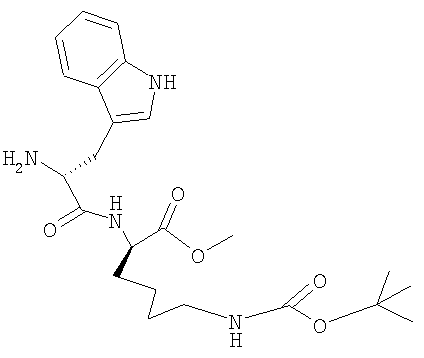
19,8 г (34,1 ммоля) Z-D-Trp-Lys(BOC)-OMe растворяют в 220 мл метанола, добавляют 2,2 г катализатора (10% PdC) и гидрируют при КТ. Гидрирование прекращают после выдерживания в течение 1 ч при КТ, катализатор отфильтровывают и фильтрат упаривают при 30°; получают твердое вещество белого цвета. ЖХВР: 98,1% b.a. (16,2 мин).
в) Z-Phg-D-Trp-Lys(BOC)-OMe
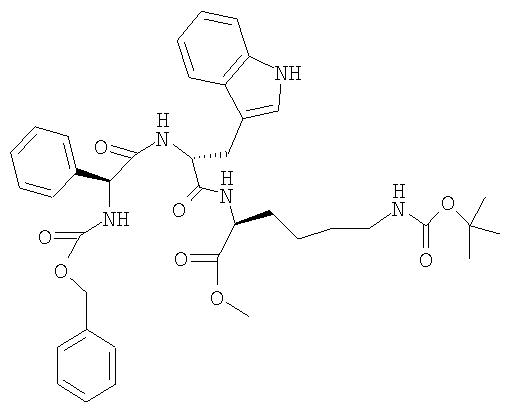
17,0 г H-D-Trp-Lys(BOC)-OMe смешивают с 80 мл ТГФ. К суспензии серого цвета добавляют при КТ 9,2 г Z-Phg-OH. Добавляют 4,4 г ГОБТ, промывают 20 мл ТГФ. Мутный раствор желтого цвета охлаждают до 0° и добавляют в течение 10 мин раствор, содержащий 5,3 мл ДИКИ в 15 мл ТГФ. Перемешивают в течение 2 ч при 0°, затем 2 ч при КТ. Затем добавляют раствор, содержащий 3,6 г серной кислоты в 57 мл воды, экстрагируют 70 мл этилацетата, промывают водой, насыщенным соляным раствором, 5%-ным бикарбонатом натрия, 25%-ным соляным раствором, сушат над MgSO4 и упаривают. Остаток белого цвета перемешивают с 125 мл изопропилацетата в течение 2 ч при 40° и суспензию фильтруют. Остаток сушат при 40°: получают субстанцию желтоватого цвета ЖХВР: 96,3% b.a. (25,3 мин).
г) H-Phg-D-Trp-Lys(BOC)-OMe
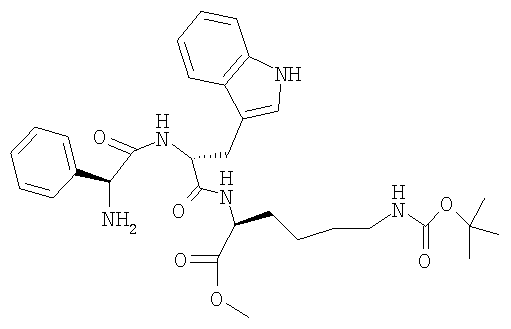
23,4 г (23,8 ммоля) (Z-Phg-D-Trp-Lys(BOC)-OMe) растворяют в 260 мл метанола, добавляют 2,6 г 10%-ного Pd/C и гидрируют при КТ и нормальном давлении. Через 3 ч катализатор отфильтровывают и фильтрат упаривают при 30°: получают твердое вещество белого цвета. ЖХВР 95,9% b.a. (17,6 мин).
д) Z-[(2S,4R)-4-(Boc-NH-CH2-CH2-NH-CO-O)]-Pro-Phg-D-Trp-Lys(BOC)-OMe
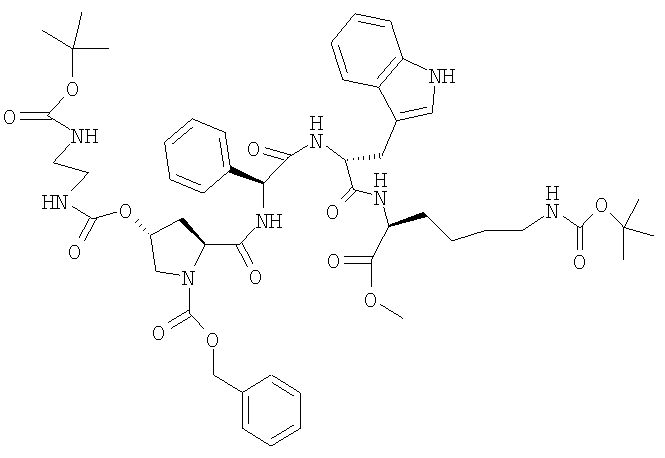
11,1 г бензилового эфира 2-карбоновой кислоты (2S,4R)-4-(2-трет-бутоксикарбониламиноэтилкарбамоилокси)пирролидин-1-карбоновой кислоты и 19,0 г H-Phg-D-Trp-Lys(BOC)-OMe растворяют в 290 мл ТГФ при КТ. Добавляют 3,32 г ГОБТ и мутный раствор охлаждают до 0°. Добавляют раствор, содержащий 4,56 мл ДИКИ в 90 мл ТГФ. Перемешивают в течение 24 ч при 0°. Затем при КТ добавляют раствор, содержащий 2,2 г серной кислоты в 22 мл воды, слегка опалесцирующий раствор перемешивают в течение 15 мин и вносят по каплям в 500 мл воды. Суспензию белого цвета упаривают при 50° до прекращения отгонки ТГФ. Суспензию фильтруют и остаток промывают 4 раза водой, используя каждый раз по 80 мл, затем 250 мл метанола и затем дважды метанолом, используя всего 80 мл. Сушат в течение ночи при 50°. ЖХВР 91,0% b.a. (19,5 мин).
e) H-[(2S,4R)-4-(Boc-NH-CH2-CH2-NH-CO-O)]-Pro-Phg-D-TrD-Lys(BOC)-Оме
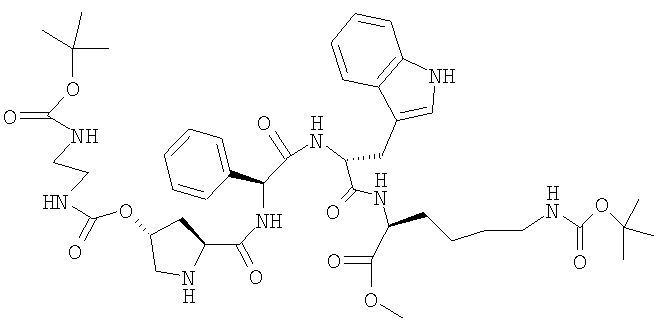
22,0 г (12,7 ммоля) Z-[(2S,4R)-4-(Boc-NH-CH2-CH2-NH-CO-O)]-Pro-Phg-D-Trp-Lys(BOC)-OMe растворяют в 220 мл ДМФ и добавляют 4,4 г 10%-ного Pd/C. Гидрируют в течение 4 ч при КТ. Затем катализатор отфильтровывают и фильтрат добавляют к смеси, содержащей 600 г льда и 400 мл воды. Осадившийся продукт отфильтровывают и промывают водой. Сушат при 30°, получая твердое вещество серого цвета. ЖХВР: 97,0% b.a. (12,3 мин).
ж) Z-Phe-[(2S,4R)-4-(Boc-NH-CH2-CH2-NH-CO-O)]-Pro-Phg-D-Trp-Lys(BOC)-Ome
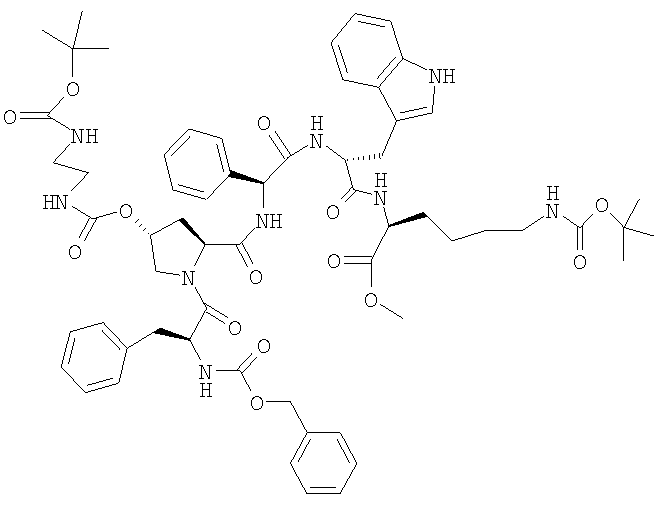
5,1 г Z-Phe-OH и 16,5 г H-[(2S,4R)-4-(Boc-NH-CH2-CH2-NH-CO-O)]-Pro-Phg-D-Trp-Lys(BOC)-Ome растворяют в 250 мл ТГФ, добавляют 2,29 г ГОБТ и темный раствор охлаждают до 0°. 3,1 мл ДИКИ растворяют в 80 мл ТГФ и добавляют к реакционной смеси при 0°. Перемешивают в течение 24 ч при 0°. Реакционную смесь добавляют к 200 мл 10%-ной серной кислоты, осадившиеся твердые частицы отфильтровывают и промывают водой. После фильтрации остаток смешивают с 200 мл метанола и суспензию фильтруют. Сушат при 40°. ЖХВР: 97,2% b.a. (21,1 мин).
з) (H-Phe-[(2S,4R)-4-(Boc-NH-CH2-CH2-NH-CO-O)]-Pro-Phg-D-Trp-Lys(BOC)-OMe)
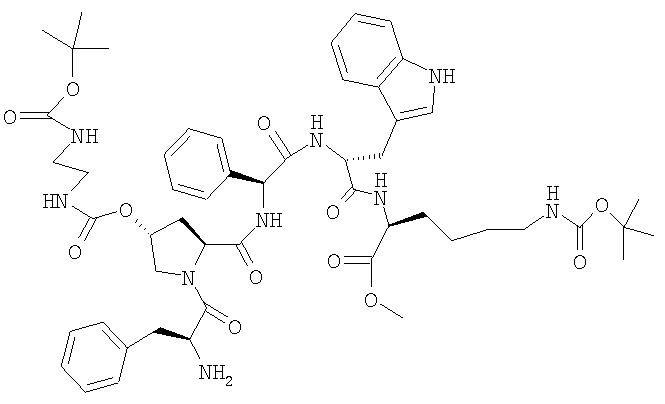
8,5 г Z-Phe-[(2S,4R)-4-(Boc-NH-CH2-CH2-NH-CO-O)]-Pro-Phg-D-Trp-Lys(BOC)-OMe растворяют в 180 мл ДМФ, добавляют 4,25 г 10%-ного Pd/C и смесь гидрируют при КТ в течение 6 ч. Затем катализатор отфильтровывают и фильтрат упаривают при 40°. К остатку (30 г) добавляют по каплям при КТ 600 мл метил-трет-бутилового эфира, суспензию фильтруют и остаток промывают 300 мл МТБЭ. Сушат при 40°. ЖХВР 93,1% b.a. (16,6 мин).
и) FMOC-Tyr(Bzl)-Phe-[(2S,4R)-4-(Boc-NH-CH2-CH2-NH-CO-O)]-Pro-Phg-D-Trp-Lys(BOC)-OMe
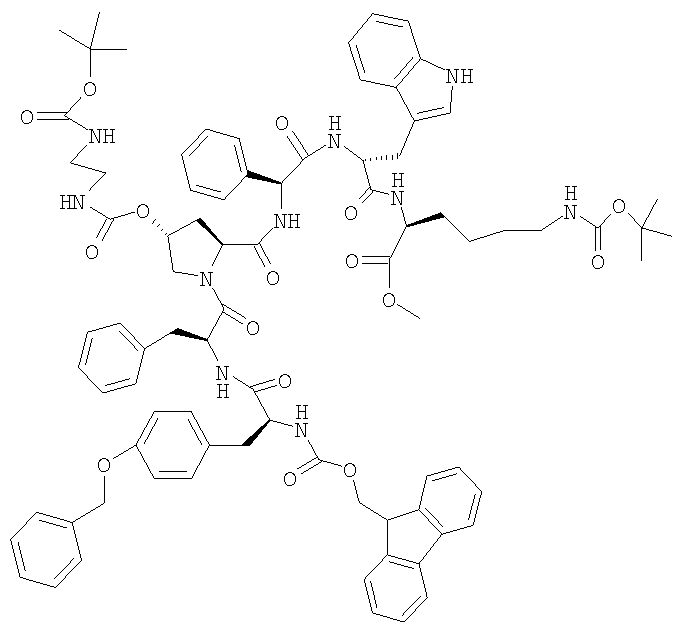
6,5 г H-Phe-[(2S,4R)-4-(Boc-NH-CH2-CH2-NH-CO-O)]-Pro-Phg-D-Trp-Lys(BOC)-OMe и 3,1 г FMOC-Tyr(Bzl)-OH и 0,86 г ГОБТ суспендируют в 180 мл ТГФ и охлаждают до 0°. Добавляют 5,5 г LiBr и затем 20 мл ТГФ. Затем добавляют 1,18 мл ДИКИ, растворенного в 50 мл ТГФ, и темную суспензию перемешивают при 0° в течение 2 ч. Охлаждающую баню удаляют и перемешивание продолжают в течение 24 ч. Затем добавляют раствор, содержащий 0,65 г серной кислоты и 6,5 мл воды, а затем добавляют 160 мл воды. Упаривают при 40°, фильтруют остаток, промывают водой до тех пор, пока значение рН последнего водного смыва не достигает 4,0. Осадок на фильтре перемешивают в 30 мл метанола, суспензию фильтруют и остаток промывают 10 мл метанола. Сушат при 35°, получая вещество серого цвета. ЖХВР 95,4% b.a. (26.0 мин).
к) FMOC-Tyr(Bzl)-Phe-[(2S,4R)-4-(Boc-NH-CH2-CH2-NH-CO-O)]-Pro-Phg-D-Trp-Lys(BOC)-OH
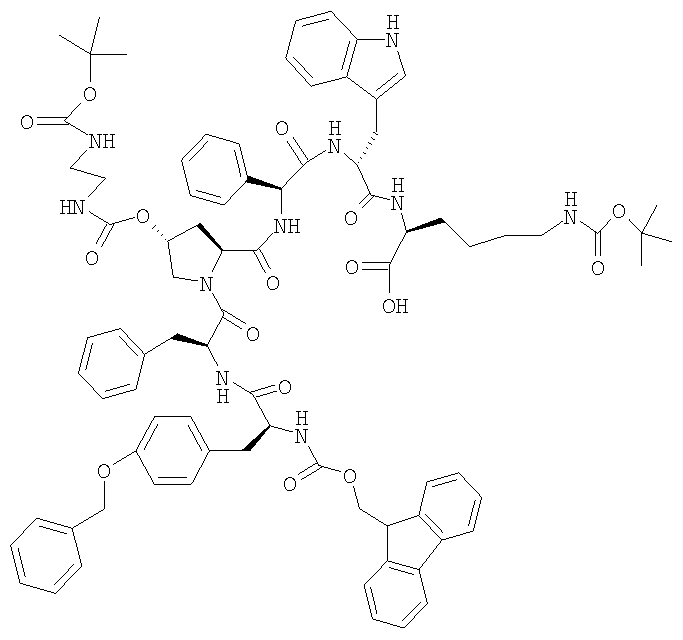
13,0 г FMOC-Tyr(Bzl)-Phe-[(2S,4R)-4-(Boc-NH-CH2-CH2-NH-CO-O)]-Pro-Phg-D-Trp-Lys(BOC)-OMe суспендируют в 250 мл ТГФ. Добавляют 7,3 г LiBr и 75 мл ТГФ. Затем медленно в течение 20 мин добавляют при КТ 8,7 мл 1М NaOH. Перемешивание при КТ продолжают в течение 3 ч, затем в течение последующих 15 ч добавляют еще 5 мл 1М NaOH. Конечный реакционный раствор добавляют к раствору, содержащему 1,7 г серной кислоты в 34 мл воды. Полученную двухфазную смесь добавляют в 50 мл этилацетата. После разделения фаз органическую фазу трижды промывают соляным раствором и затем упаривают. К остатку добавляют 25 мл DIF. Образовавшийся прозрачный DIF-раствор добавляют в 260 мл воды. Полученную суспензию фильтруют и осадок на фильтре промывают трижды водой и сушат в течение ночи при 40°. ЖХВР 71,9% b.a. (28,6 мин).
Пример 2:
Получение 4-(хлор(дифенил)метил)бензоиламинометилполистироловой смолы
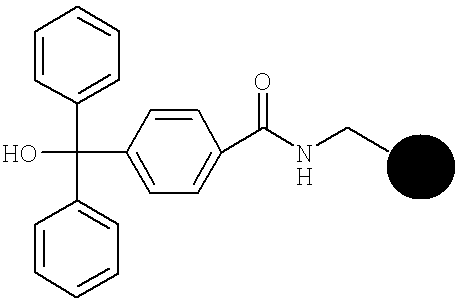
Синтез осуществляют в управляемом вручную 1-литровом реакторе с периодической загрузкой с мешалкой, снабженном фриттой из спекшегося стекла в атмосфере азота. Поступающую в продажу аминометилполистироловую смолу (30,4 г, 41,07 ммоля), предварительно обработанную ДМФ, подвергают взаимодействию при КТ в течение ночи с раствором, содержащем 4-(гидроксилдифенилметил) бензойную кислоту (15,0 г, 49,28 ммоля, 1,2 экв.), гидроксибензотриазолом (ГОБТ) (7,54 г, 49,28 ммоля, 1,2 экв.) и ДИКИ (12,43 г, 98,57 ммоля, 2,4 экв.) в ДМФ (140 мл). Растворитель удаляют фильтрацией через фритту при пониженном давлении и смолу последовательно промывают 5 раз ДМФ и 5 раз метанолом. После сушки в вакууме при 40° получают 44,69 г смолы. Эту смолу используют в качестве исходного продукта для следующего синтеза гексапептидов.
Процесс синтеза защищенного линейного пептида
Вручную осуществляют сборку связанных со смолой линейных гексапептидов в направлении от С- к N-концу с помощью повторяющихся реакций сочетания в реакторе с периодической загрузкой с мешалкой, снабженном фриттой из спекшегося стекла, в атмосфере азота. 4-(Хлор(дифенил)метил)бензоиламинометилполистироловую смолу используют в качестве исходного продукта и применяют стандартный протокол, который заключается в повторяющихся циклах удаления Nα-защитных групп (20 об.% диэтиламин (ДЭАМ) в ДМФ), повторяющихся последовательных отмывках с помощью ДМФ и IPK и осуществлении реакций сочетания (DIPCI/ГОБТ, ЭДИПА и ДМФ) при КТ. Небольшим изменением этой процедуры сочетания является то, что предпринимали специальные меры для минимизации рацемизации фенилглицина, для чего сочетание этой аминокислоты проводили при 0°. Перед отщеплением от смолы полностью собранного защищенного линейного пептида удаляли защиту Nα-Fmoc.
Fmoc-(2S,4R)-4-(Boc-NH-CH2-CH2-NH-CO-O)-Pro-OH синтезируют согласно методу, описанному в WO 02/101192. Все другие аминокислоты поступают в продажу.
Синтез H-Tyr(Bzl)-Phe-(2S,4R)-4-(Boc-NH-CH2-CH2-NH-CO-O)-Pro-Phg-D-Trp(Boc)Lys(Boc)-OH
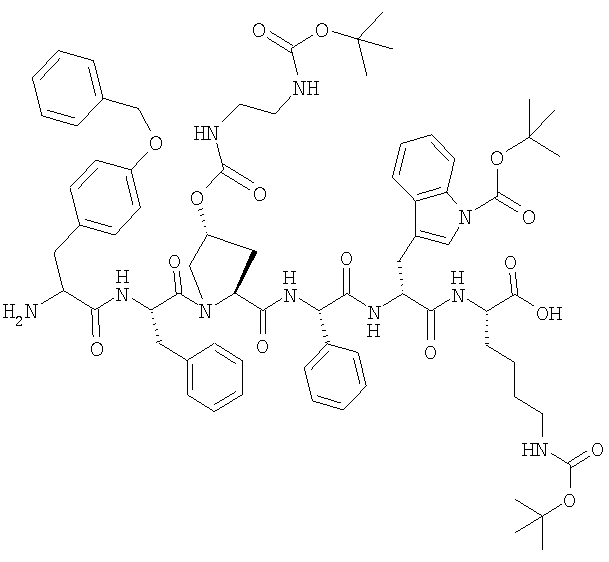
а) Синтез Fmoc-Lys(Boc)-О-смолы
4-(Хлор(дифенил)метил)бензоиламинометилполистироловую смолу (20 г, 19,4 ммоля), предварительно обработанную толуолом, обрабатывают в течение 4 ч ацетилхлоридом (7,6 т, 97 ммолей) в толуоле при КТ. После фильтрации процедуру повторяют в течение ночи, после чего смолу фильтруют и промывают толуолом и дихлорметаном. Сочетание осуществляют в смеси Fmoc-Lys(Boc)-ОН (18,2 г, 38,8 ммоля; 2 экв.) и N-метилморфолина (3,94 г, 38,8 ммоля, 2 экв.) в течение 4 ч при КТ. После фильтрации смолу последовательно трижды промывают ДМФ и ИПС и сушат в вакууме, получая 26,5 г Fmoc-Lys(Boc)-О-смолы в виде продукта желтоватого цвета, производительность 0,566 ммоля/г (определено с помощью Fmoc-метода).
(Литература: Fmoc-method: Meienhofer J.; Waki M; Heimer E.P.; Lambros T.J.; Makofske R.C.; Chang C.D. Int. J. Pep. Prot. Res.13, 1979, с.35)
б) Смола-О-Lys(Boc)-D-TrD(Boc)-Phg-(2S,4R)-4-(Boc-NH-CH2-CH2-NH-CO-O)-Pro-Phe-Tyr(Bzl)-H
Fmoc-Lys(Boc)-O-смолу (28,0 г, 19,96 ммоля) суспендируют в ДМФ и обрабатывают раствором ДЭАМ в ДМФ (20 об.%) в течение 10 мин при КТ. После фильтрации процедуру повторяют и затем последовательно трижды промывают ДМФ и ИПС, а затем трижды промывают ДМФ. Эту процедуру удаления защитной группы Nα-Fmoc и промывки повторяют после каждой стадии сочетания.
Реакцию сочетания осуществляют в смеси аминокислоты, ГОБТ и ДИКИ, которую перемешивают в течение 30 мин при КТ и затем в виде одной порции добавляют к смоле. Реакцию сочетания продолжают до завершения, т.е. до полного исчезновения остаточных аминогрупп, что определяют по отрицательному результату нингидринового теста «Kaiser». После осуществления реакции сочетания смолу 5 раз промывают ДМФ, после чего она готова для защиты с помощью Fmoc-группы.
Следующие производные аминокислот последовательно подвергают сочетанию:
Fmoc-D-Trp(Boc)-OH (16.81 г, 31,92 ммоля, 2 экв.), DIF (100 мл), ГОБТ (4,93 г, 32,24 ммоля, 2,02 экв.), ДИКИ (5,35 г, 42,45 ммоля, 2,66 экв.). Fmoc-Phe-OH (11,92 г, 31,92 ммоля, 2 экв.), ТГФ (70 мл), ГОБТ (4,93 г, 32,24 ммоля, 2,02 экв.), ДИКИ (5,35 г, 42,45 ммоля, 2,66 экв.). Предпринимают специальные меры для минимизации рацемизации фенилглицина, осуществляя сочетание этой аминокислоты при 0°С.
Fmoc-(2S,4R)4-(Boc-NH-CH2-CH2-NH-CO-O)-Pro-OH (17,26 г, 31,92 ммоля, 2 экв.), DIF (100 мл), ГОБИ (4,93 г, 32,24 ммоля, 2,02 экв.), ДИКИ (5,35 г, 42,45 ммоля, 2,66 экв.).
Fmoc-Phe-OH (12.36 г, 31,92 ммоля, 2 экв.), DIF (100 мл), ГОБТ (4,93 г, 32,24 ммоля, 2,02 экв.), ДИКИ (5,35 г, 42,45 ммоля, 2,66 экв.). Fmoc-Tyr(Bzl)-OH (15,75 г, 31,92 ммоля, 2 экв.), DIF (100 мл), ГОБТ (4,93 г, 32,24 ммоля, 2,02 экв.), ДИКИ (5,35 г, 42,45 ммоля, 2,66 экв.).
Перед отщеплением полностью собранного защищенного линейного пептида от подложки из смолы удаляют защиту Nα-Fmoc, обрабатывая смолу раствором ДЭАМ в ДМФ (20 об.%) в течение 10 мин при КТ. После фильтрации процедуру повторяют и последовательно трижды промывают ДМФ и ИПС, а затем трижды промывают ДМФ.
(После сушки смолы в вакууме при 40°С получают смолу желтоватого цвета).
в) Отщепление линейного пептида от подложки из смолы:
H-Tyr(Bzl)-Phe-(2S,4R)-4-(Boc-NH-CH2-CH2-NH-CO-O)-Pro-Phg-D-Trp(Boc)Lys(Boc)-OH
ва) Метод с использованием АсОН/СН2Cl2/Н2О 45/45/5 об./об./об.
Полностью собранный защищенный линейный пептид смола-O-Lys(Boc)-D-Trp(Boc)-Phg-(2S,4R)-4-(Boc-NH-CH2-CH2-NH-CO-O)-Pro-Phe-Tyr(Bzl)-H(24,5 г) суспендируют в смеси AcOH/CH2Cl2/H2O 45/45/5 об./об./об. (150 мл) и перемешивают в течение 1 ч при КТ, фильтруют и промывают СН2Cl2. Фильтрат упаривают досуха, остаток перемешивают в течение 1 ч в смеси МТБЭ и гептана 7/3 об./об., фильтруют и сушат в вакууме. Получают твердый продукт желтоватого цвета; содержание: 93,5% по данным ЖХВР г/г; чистота 91,6% по данным (F)-ЖХВР и содержание D-Phg-эпимера 2,5% по данным (F)-ЖХВР.
Смолу (4-(хлор(дифенил)метил)бензоиламинометилполистироловая смола) промывают трижды метанолом и сушат, после чего ее можно использовать повторно.
вб) Метод с использованием СН2Cl2 и МеОН
Полностью собранный защищенный линейный пептид смола-O-Lys(Boc)-D-Trp(Boc)-Phg-(2S,4R)-4-(Boc-NH-CH2-CH2-NH-CO-O)-Pro-Phe-Tyr(Bzl)-H(6,2r) суспендируют в смеси СН2Cl2/МеОН 1/1 об./об. (115 мл) и перемешивают в течение 3 дней при КТ, фильтруют и промывают CH2Cl2. Фильтрат упаривают досуха, остаток перемешивают в течение 1 ч со смесью МТБЭ и гептана 7/3 об./об. (60 мл), фильтруют и сушат в вакууме. Содержание: 93,5% по данным ЖХВР г/г; чистота 92,6% по данным (F)-ЖХВР и содержание D-Phg-эпимера 1,1% по данным (F)-ЖХВР.
Смолу (4-(хлор(дифенил)метил)бензоиламинометилполистироловая смола) промывают трижды метанолом и сушат, после чего ее можно использовать повторно.
вв) Метод с использованием этилметилкетона/МеОН
Полностью собранный защищенный линейный пептид смола-O-Lys(Boc)-D-Trp(Boc)-Phg-(2S,4R)-4-(Boc-NH-CH2-CH2-NH-CO-O)-Pro-Phe-Tyr(Bzl)-H(4,0 г) суспендируют в смеси с диэтилметилкетоном 1/1 об./об. (24 мл) и перемешивают в течение 15 ч при 50°С, фильтруют и промывают МеОН. Фильтрат упаривают досуха, остаток перемешивают в течение 1 ч в смеси МТБЭ и гептана 7/3 об./об. (60 мл), фильтруют и сушат в вакууме. Содержание: 88,1% по данным ЖХВР г/г; чистота 95,2% по данным (Р)-ЖХВР и содержание D-Phg-эпимера 1,8% по данным (F)-ЖХВР.
Смолу (4-(хлор(дифенил)метил)бензоиламинометилполистироловая смола) промывают трижды метанолом и сушат, после чего ее можно использовать повторно.
Очистка
Для аналитических целей линейные пептиды очищают с помощью хроматографии с обращенной фазой (ОФ).
Характеристики
Структуру аналитического образца, очищенного с помощью ОФ-хроматографии подтверждают с помощью FAB-MC, ЖХ-МС и ЯМР (ДМСО в част/млн, 1,16, 1,34, 1,55, 1,61 (3Н), 1,66, 2,05, 2,20, 2,51, 2,83 (2Н), 2,91, 2,96, 2,98, 3,02, 3,41, 3,78, 4,13, 4,61, 5,13, 5,51, 6,74, 6,83, 6,88 (2Н), 7,01 (2Н), 7,11 (2Н), 7,38, 7,42 (2Н), 7,49, 7,72, 8,01, 8,29, 8,48, 8,62, 8,75).
Конфигурацию аминокислот определяют с помощью анализа аминокислот: соединение гидролизуют в кислотных условиях, превращают в производные и конфигурацию каждой индивидуальной аминокислоты оценивают путем энантиоселективной газовой хроматографии/масс-спектрометрии с химической ионизацией.
Дополнительное подтверждение структуры осуществляют путем превращения различных линейных пептидов в циклический пептид с хорошо известными характеристиками.
С помощью описанного метода синтезируют следующие соединения.
H-Lys(Boc)-Tyr(Bzl)-Phe-(2S,4R)-4-(Boc-NH-CH2-CH2-NH-CO-O)-Pro-Phg-D-Tyr(Вос)-ОН,
H-Lys(Boc)-Tyr(Bzl)-Phe-(2S,4R)-4-(Boc-NH-CH2-CH2-NH-CO-O)-Pro-Phg-D-Trp-ОН,
H-(2S,4R)-4-(Boc-NH-CH2-CH2-NH-CO-O)-Pro-Phg-D-Trp(Boc)-Lys(Boc)-Tyr(Bzl)-Phe-OH,
H-Phe-(2S,4R)-4-(Boc-NH-CH2-CH2-NH-CO-O)-Phg-D-Trp(Boc)-Lys(Boc)-Try(Bzl)-OH,
H-Tyr(Bzl)-Phe-(2S,4R)-4-(Boc-NH-CH2-CH2-NH-CO-O)-Pro-DPhg-DTrp(Boc)-Lys(Boc)-OH.
Пример 3: Циклизация линейных защищенных пептидов для синтеза
Цикло[(2S,4R)-4-(Вос-NH-СН2-СН2-NH-CO-O)-Pro-Phg-D-Tyr(Вос)-Lys(Boc)-Tyr(Bzl)-Phe-]
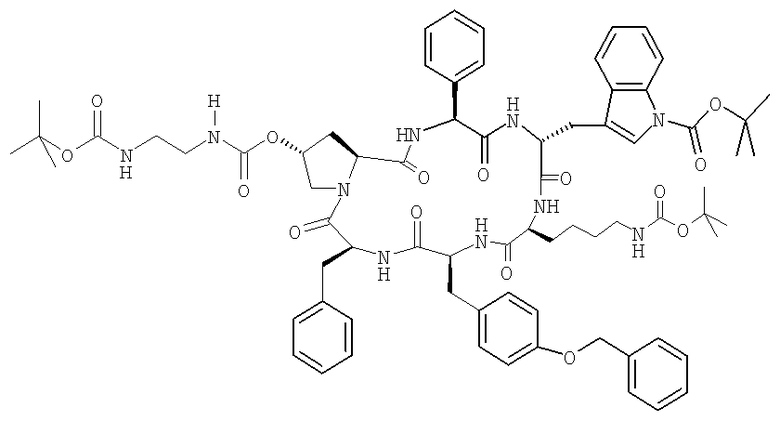
Циклизация H-Phe-(2S,4R)-4-(Boc-NH-CH2-CH2-NH-CO-O)-Pro-Phg-D-Trp(Boc)-Lys(Boc)-Tyr(Bzl)-OH
Азидный метод
Для циклизации линейный фрагмент (2,5 г, 1,83 ммоля) растворяют в ДМФ (391 мл), охлаждают до -5°, обрабатывают ЭДИПА (0,47 г, 3,66 ммоля, 2 экв.) и ДФФА (0,75 г, 2,75 ммоля, 1,5 экв.) и перемешивают при указанной температуре до завершения реакции (примерно 20 ч). К реакционной смеси по каплям добавляют воду (391 мл), осадок фильтруют и промывают водой до тех пор, пока содержание азида не снизится до уровня, ниже обнаруживаемого. Получают 4,9 г смоченного водой твердого вещества белого цвета (значение Rf при ЖХВР идентично стандарту), которое применяют в реакции удаления защитной группы без дополнительной очистки. Соединение характеризуют с помощью ЖХВР путем непосредственного сравнения с соединением-стандартом.
Циклизация H-Tyr(Bzl)-Phe-(2S,4R)-4-(Boc-NH-CH2-CH2-NH-CO-O)-Pro-Phg-D-Trp(Boc)Lys(Boc)-OH
а) ГБТУ-метод
Для циклизации линейный фрагмент (6,0 г, 3,5 ммоля) растворяют в ДМФ (780 мл), охлаждают до -5°, обрабатывают ЭДИПА (1,13 г, 8,75 ммоля, 2,5 экв.), ГОБТ (1,18 г, 8,75 ммоля, 2.5 экв.), ГБТУ (3,3 г, 8,78 ммоля, 2,5 экв.) и перемешивают при указанной температуре до завершения реакции (примерно 2 ч). К реакционной смеси по каплям добавляют воду (391 мл), осадок фильтруют и промывают водой и гептаном и сушат в вакууме в течение ночи. Получают 5,4 г твердого вещества желтовато-белого цвета. Соединение характеризуют с помощью ЖХВР путем непосредственного сравнения с соединением-стандартом. Содержание 55 мас.%. по данным ЖХВР, чистота 78 (А%)-ЖХВР.
б) ГБТУ-метод б
Для циклизация линейный фрагмент (6,0 г, 4,16 ммоля) растворяют в ДМФ (60 мл) и добавляют по каплям в смесь ГОБТ (4,15 г, 10,4 ммоля, 2,5 экв.), ГБТУ (4,15 г, 10,4 ммоля, 2,5 экв.) и ЭДИПА (1,41 г, 10,4 ммоля, 2,5 экв.) в ДМФ (135 мл) при -5° и перемешивают при указанной температуре до завершения реакции (примерно 2 ч). К реакционной смеси по каплям добавляют воду (559 мл) при КТ, осадок фильтруют и промывают водой и гептаном и сушат в вакууме в течение ночи. Получают твердое вещество желтовато-белого цвета. Соединение характеризуют с помощью ЖХВР путем непосредственного сравнения с соединением-стандартом. Содержание 77 мас.% по данным ЖХВР, чистота 84 (А%)-ЖХВР.
Следующие линейные пептиды циклизуют с помощью вышеописанной процедуры:
H-Lys(Boc)-Tyr(Bzl)-Phe-(2S,4R)-4-(Boc-NH-CH2-CH2-NH-CO-O)-Pro-Phg-D-Tyr(Вос)-ОН,
H-Lys(Boc)-Tyr(Bzl)-Phe-(2S,4R)-4-(Boc-NH-CH2-CH2-NH-CO-O)-Pro-Phg-D-Trp-ОН,
H-(2S,4R)-4-(Boc-NH-CH2-CH2-NH-CO-O)-Pro-Phg-D-Trp(Boc)-Lys(Boc)-Tyr(Bzl)-Phe-OH,
H-Phe-(2S,4R)-4-(Boc-NH-CH2-CH2-NH-CO-O)-Phg-D-Trp(Boc)-Lys(Boc)-Try(Bzl)-OH,
H-Tyr(Bzl)-Phe-(2S,4R)-4-(Boc-NH-CH2-CH2-NH-CO-O)-Pro-DPhg-DTrp(Boc)-Lys(Boc)-OH.
Пример 4: Синтез трифторацетата цикло[(2S,4R)-4-(Boc-NH-CH2-CH2-NH-CO-O)-Pro-Phg-D-Trp-Lys-Tyr(Bzl)-Phe-]
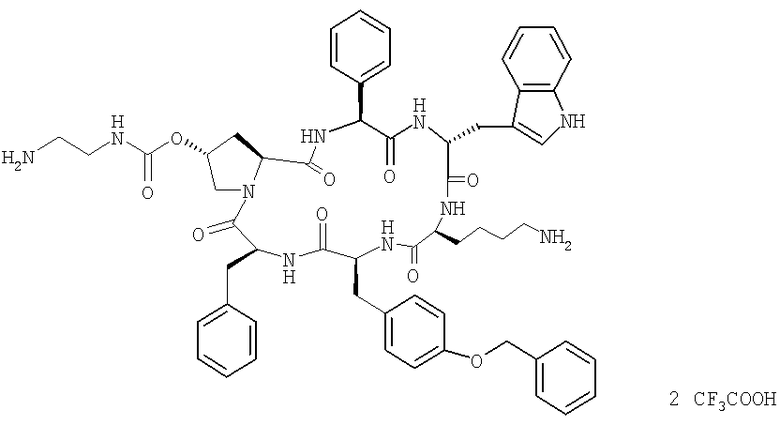
Для завершения удаления защитной группы остаток (1,5 г, 0,79 ммоля) растворяют при 0° в смеси ТФК/Н2О, 95:5 (8,3 мл) и смесь перемешивают при охлаждении в течение 30 мин. Холодную реакционную смесь вносят по каплям в смесь МТБЭ (29 мл) и гептана (13 мл) при КТ и перемешивают в течение 2 ч. Осадок фильтруют, промывают смесью МТБЭ /гептан, 1:1 (об./об.) и сушат в вакууме. Получают твердое вещество бежевого цвета, содержание 53 мас.% по данным ЖХВР, чистота: 79 (А%)-ЖХВР.
| название | год | авторы | номер документа |
|---|---|---|---|
| ПЕПТИДНЫЕ ВЕКТОРЫ | 2004 |
|
RU2361876C2 |
| АНАЛОГИ СОМАТОСТАТИНА | 2001 |
|
RU2287533C2 |
| СОМАТОСТАТИНОВЫЕ ПЕПТИДЫ | 1996 |
|
RU2160741C2 |
| СОЕДИНЕНИЯ, СПОСОБСТВУЮЩИЕ ВЫСВОБОЖДЕНИЮ ГОРМОНА РОСТА | 1994 |
|
RU2167881C2 |
| ИНГИБИТОРЫ ФАКТОРА ХА | 1995 |
|
RU2152954C1 |
| ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ | 2004 |
|
RU2355418C2 |
| ЦИКЛОПЕПТИДЫ, СПОСОБ ИХ ПОЛУЧЕНИЯ, СОДЕРЖАЩИЕ ИХ ФАРМАЦЕВТИЧЕСКИЕ КОМПОЗИЦИИ И СПОСОБ ИХ ПОЛУЧЕНИЯ | 1994 |
|
RU2130030C1 |
| СПОСОБ ПОЛУЧЕНИЯ ПРОИЗВОДНЫХ ПЕПТИДАМИДОВ ИЛИ ИХ ФИЗИОЛОГИЧЕСКИ СОВМЕСТИМЫХ АЦЕТАТОВ ИЛИ ГИДРОХЛОРИДОВ | 1991 |
|
RU2036200C1 |
| ПЕПТИДЫ ИЛИ ИХ ФИЗИОЛОГИЧЕСКИ ПРИЕМЛЕМЫЕ СОЛИ | 1992 |
|
RU2083586C1 |
| ПРОИЗВОДНЫЕ МЕТАСТИНА И ИХ ПРИМЕНЕНИЕ | 2007 |
|
RU2454425C2 |
Изобретение относится к способу получения циклических аналогов соматостатина формулы I и промежуточным продуктам, применяемым в способе. Способ проводят путем циклизации линейного аналога соматостатина формулы II, где R1 обозначает -С2-С6алкилен-NR3R4, R3 и R4 каждый независимо друг от друга обозначает Н или ацил, и R2 обозначает 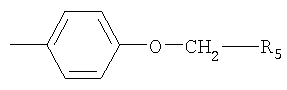 , где R5 обозначает фенил, R11 и R12 каждый независимо друг от друга обозначает аминозащитную группу, при этом, когда R1 содержит концевую NH2-группу, то концевая NH2-группа также защищена аминозащитной группой, и при необходимости удаляют защитную(ые) группу(ы), и восстанавливают полученное таким образом соединение формулы I в свободной форме или в форме соли. 3 н. и 3 з.п. ф-лы.
, где R5 обозначает фенил, R11 и R12 каждый независимо друг от друга обозначает аминозащитную группу, при этом, когда R1 содержит концевую NH2-группу, то концевая NH2-группа также защищена аминозащитной группой, и при необходимости удаляют защитную(ые) группу(ы), и восстанавливают полученное таким образом соединение формулы I в свободной форме или в форме соли. 3 н. и 3 з.п. ф-лы.
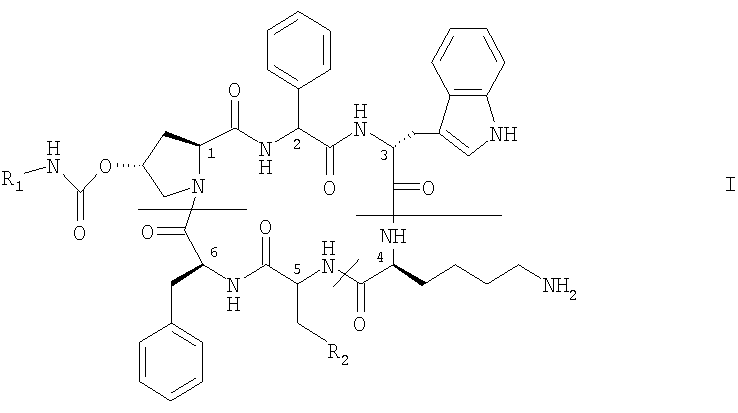

1. Способ получения соединения формулы I
в которой
R1 обозначает -С2-С6алкилен-NR3R4, где R3 и R4 каждый независимо друг от друга обозначает Н или ацил, и
R2 обозначает
где R5 обозначает фенил, или его соли,
заключающийся в том, что осуществляют циклизацию линейного аналога соматостатина формулы II

в которой R1 и R2 имеют указанные выше значения,
R11 и R12 каждый независимо друг от друга обозначает аминозащитную группу, при этом, когда R1 содержит концевую NH2-группу, то концевая NH2-группа также защищена аминозащитной группой, и при необходимости удаляют защитную(ые) группу(ы), и восстанавливают полученное таким образом соединение формулы I в свободной форме или в форме соли.
2. Способ по п.1, в котором осуществляют циклизацию линейного аналога и соматостатина формулы II

в которой R1 обозначает -СН2-СН2-NR3R4, R2 обозначает 4-бензилоксифенил и R3,
R4, R11 и R12 имеют значения, указанные в п.1,
при этом, когда R1 содержит концевую NH2-группу, то концевая NH2-группа также защищена аминозащитной группой,
и при необходимости удаляют защитную(ые) группу(ы),
и восстанавливают полученное таким образом соединение формулы I в свободной форме или в форме соли, где R1 обозначает -CH2-CH2-NR3R4 и R2 обозначает 4-бензилоксифенил.
3. Соединение формулы II

в которых R1 и R2 имеют значения, указанные в п.1,
R11 и R12 каждый независимо друг от друга обозначает аминозащитную группу, при этом, когда R1 содержит концевую NH2-группу, то концевая NH2-группа также защищена аминозащитной группой, или его соль.
4. Соединение формулы II по п.3, в которой R1 обозначает -CH2-CH2-NR3R4, R2 обозначает 4-бензилоксифенил и R11 и R12 каждый независимо друг от друга обозначает аминозащитную группу, при этом, когда R1 содержит концевую NH2-группу, то концевая NH2-группа также защищена аминозащитной группой, или его соль.
5. Соединение формулы II по п.3, выбранное из ряда, включающего Н-Tyr(Bzl)-Phe-(2S,4R)-4-(Boc-NH-CH2-CH2-NH-CO-O)-Pro-DPhg-DTrp(Boc)-Lys(Boc)-OH,
H-Tyr(Bzl)-Phe-(2S,4R)-4-(Boc-NH-CH2-CH2-NH-CO-O)-Pro-Phg-DTrp-Lys(Boc)-OH и
H-Tyr(Bzl)-Phe-(2S,4R)-4-(Boc-NH-CH2-CH2-NH-CO-O)-Pro-Phg-DTrp(Boc)Lys(Boc)-OH, или его соль.
6. Способ получения соединения формулы II,

в которой R1 обозначает -С2-С6алкилен-NR3R4, где R3 и R4 каждый независимо друг от друга обозначает Н или ацил,
R2 обозначает
где R5 обозначает фенил,
R11 и R12 каждый независимо друг от друга обозначает аминозащитную группу, при этом, когда R1 содержит концевую NH2-группу, то концевая NH2-группа также защищена аминозащитной группой, заключающийся в том, что связывают друг с другом с помощью амидной связи два пептидных звена, каждое из которых содержит по меньшей мере одну аминокислоту в защищенной или незащищенной форме, где амидная связь расположена так, чтобы получить требуемую аминокислотную последовательность, указанную в формуле II, и при необходимости удаляют по меньшей мере одну защитную группу, и выделяют полученное таким образом соединение формулы II в свободной форме или в форме соли.
| СОМАТОСТАТИНОВЫЕ ПЕПТИДЫ | 1996 |
|
RU2160741C2 |
| Аппарат для очищения воды при помощи химических реактивов | 1917 |
|
SU2A1 |
| HUANG ZIWEI ет al | |||
| Main chain and side chain chiral methylated somatostatin analogs: synthethes and conformational analyses | |||
| Journal of American Chemical Society | |||
| American Chemical Society | |||
| Пуговица для прикрепления ее к материи без пришивки | 1921 |
|
SU1992A1 |

