Изобретение относится к соматостатиновым пептидам, способу их получения и фармацевтическим препаратам, содержащим их.
Соматостатин - это тетрадекапептид, имеющий структуру:
С момента выделения и характеристики соматостатина проводился интенсивный поиск более сильнодействующих и более стабильных аналогов.
Более конкретно в настоящем изобретении предлагается аналог соматостатина, содержащий аминокислотную последовательность формулы (I)
-(D/L)Trp-Lys-X1-X2- (I)
в которой X1 представляет собой радикал формулы (а) или (б)
или
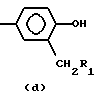
где R1 представляет собой возможно замещенный фенил,
R2 представляет собой -Z1-CH2-R1, -CH2-CO-O-CH2-R1,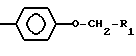
или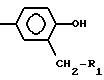
где Z1 представляет собой О или S, и
X2 представляет собой α-аминокислоту, имеющую ароматический остаток на боковой цепи Cg, или аминокислотную единицу, выбранную из Dab, Dpr, Dpm, His, (Bzl)HyPro, тиенил-Ala, циклогексил-Ala и трет-бутил-Ala, причем остаток Lys указанной последовательности соответствует остатку Lys9 природного соматостатина - 14.
Эти соединения именуются здесь и далее как соединения по изобретению.
В применяемом здесь значении аналог соматостатина представляет собой прямоцепочечный или циклический пептид, производный от существующего в природе соматостатина-14, содержащий последовательность формулы I, и в котором дополнительно одна или более чем одна аминокислотная единица удалена и/или замещена одним или более чем одним другим аминокислотным радикалом(ами), и/или в котором одна или более чем одна функциональная группа замещена одной или более чем одной другой функциональной группой, и/или одна или более чем одна группа замещена одной или несколькими другими изостерическими группами. В общем понимании этот термин охватывает все модифицированные производные природного соматостатина-14, содержащие вышеуказанную последовательность формулы I, которая обладает сродством связывания в nM диапазоне с по меньшей мере одним субтипом рецепторов соматостатина, как определено здесь далее.
Согласно предпочтительному воплощению изобретения предусматривается аналог соматостатина, в котором остатки в положениях 8-11 соматостатина-14 представлены последовательностью формулы I, как определено выше.
Более предпочтительно предусматривается аналог соматостатина, как определено выше, содержащий гексапептидную единицу, причем остатки в положениях 3-6 указанной гексапептидной единицы содержат последовательность формулы I. Наиболее предпочтительным является соматостатиновый гексапептид, в котором остатки в положениях 1 и 2 гексапептидной единицы могут быть любыми из известных из уровня техники (A.S.Dutta, Small Peptides, Vol.19, 292-354, Elsevier, 1993) или, в качестве заместителей, остатками Phe6 и/или Phe7 соматостатина-14.
Более конкретно, предусматривается аналог соматостатина, в котором гексапептидная единица является циклической, например, имеющей прямую пептидную связь между α-карбонильной группой остатка в положении 6 и α-аминогруппой остатка в положении 1.
Тогда как Lys, X1 и X2 в последовательности формулы I имеют L-конфигурацию, Trp может иметь D- или L-конфигурацию. Предпочтительно Trp имеет D-конфигурацию.
X1 предпочтительно представляет собой остаток формулы (а) или (б), причем R2 предпочтительно является
-Z1-CH2-R1
или
Когда X2 содержит ароматический остаток на боковой цепи Cg, он соответственно может являться природной или неприродной α-аминокислотой, например Phe, Tyr, Trp, Nal, Pal, бензотиенил-Ala, Tic и тиронином, предпочтительно Phe или Nal, более предпочтительно Phe. X2 предпочтительно представляет собой α-аминокислоту, несущую ароматический остаток на боковой цепи Cg.
Когда R1 представляет собой замещенный фенил, он соответственно может быть замещен, например, в орто и/или пара положении, галогеном, метилом, этилом, метоксилом или этоксилом. Более предпочтительно R1 представляет собой незамещенный фенил.
Z1 предпочтительно представляет собой О.
Представителем соединений по изобретению является, например, соединение формулы (II)
в которой X1 и X2 такие, как определено выше,
A представляет собой двухвалентный остаток, выбранный из Pro,
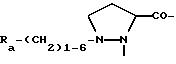
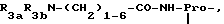


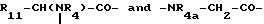
где R3 представляет собой NR8R9-C2-6-алкилен, гуанидино-C2-6-алкилен или C2-6-алкилен-COOH, R3a представляет собой H, C1-4алкил или независимо имеет одно из значений, данных для R3, R3b представляет собой H или C1-4алкил, Ra представляет собой OH или NR5R6, Rb представляет собой -(CH2)1-3- или -CH(CH3)-, R4 представляет собой H или CH3, R4a представляет собой возможно замещенный в кольце бензил, каждый из R5 и R6 независимо представляет собой H, C1-4 алкил, ω-амино-C1-4алкилен, ω-гидрокси-C1-4алкилен или ацил, R7 представляет собой прямую связь или C1-6алкилен, каждый из R8 и R9 независимо представляет собой H, C1-4 алкил ω-гидpoкcи-C2-4алкилен, ацил или CH2OH-(CHOH)c-CH2-, где c является 0, 1, 2, 3 или 4, или R8 и R9 образуют вместе с атомом азота, к которому они присоединены, гетероциклическую группу, которая может содержать дополнительный гетероатом, и R11 представляет собой возможно замещенный в кольце бензил, -(CH2)1-3-OH, CH3-CH(OH)- или (CH2)1-5-NR5R6, и
ZZa представляет собой единицу природной или неприродной α-аминокислоты.
ZZa может иметь D- или L-конфигурацию. Когда ZZa представляет собой единицу природной или неприродной α-аминокислоты, он соответственно может являться, например, Thr, Ser, Ala, Val, Ile, Leu, Nle, His, Arg, Lys, Nal, Pal, Tyr, Trp, возможно замещенным в кольце Phe или Ng-бензил-Cly. Когда ZZa представляет собой Phe, его бензольное кольцо может быть замещено, например, NH2, NO2, CH3, OCH3 или галогеном, предпочтительно в пара-положении. Когда ZZa представляет собой Phe, его бензольное кольцо предпочтительно незамещенно.
Если A содержит Pro аминокислотный остаток, то любой заместитель, присутствующий в пролиновом кольце, например R3-NH-CO-О- и так далее, находится предпочтительно в положении 4. Такой замещенный пролиновый остаток может существовать в цис-форме, например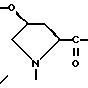
а также в транс-форме. Настоящее изобретение охватывает каждый геометрический изомер индивидуально, а также их смеси.
Если A представляет собой  в котором NR8R9 образует гетероциклическую группу, то такая группа может быть ароматической или насыщенной и может содержать один гетероатом азота или один гетероатом азота и второй гетероатом, выбранный из азота и кислорода. Предпочтительно гетероциклическая группа представляет собой, например, пиридил или морфолино. C2-6-алкилен в этом остатке представляет собой предпочтительно -CH2-CH2-.
в котором NR8R9 образует гетероциклическую группу, то такая группа может быть ароматической или насыщенной и может содержать один гетероатом азота или один гетероатом азота и второй гетероатом, выбранный из азота и кислорода. Предпочтительно гетероциклическая группа представляет собой, например, пиридил или морфолино. C2-6-алкилен в этом остатке представляет собой предпочтительно -CH2-CH2-.
Любой ацил, такой как R5, R6, R8 и R9 в A, может являться, например, R12CO-, где R12 представляет собой H, C1-4алкил, C1-4алкенил, C3-6циклоалкил или бензил, предпочтительно метил или этил. Когда R4a или R11 в A представляет собой замещенный в кольце бензил, бензольное кольцо может быть замещено, как указано выше для ZZa.
Предпочтительной группой соединений по изобретению являются, например, соединения формулы (II), в которой A является свободной боковой группировкой -NH-CO-O-. Дополнительной группой предпочтительных соединений по изобретению являются, например, соединения формулы (II), в которой A содержит основной боковой радикал, например группировку R3-NH-CO-O- или 
Еще одной дополнительной группой предпочтительных соединений по изобретению является группа соединений, в которых N-терминальная аминокислота содержит замещенный Pro, в частности 4-замещенный Pro, например соединения формулы II, в которых A представляет собой 4-замещенный Pro.
Предпочтительно A представляет собой 4-(R3-NH-CO-O)Pro.
Дополнительными представителями соединений по изобретению являются соединения, содержащие аминогруппу, несущую хелатирующую группу, в частности соединение формулы (II), где A содержит аминогруппу боковой цепи, которая несет хелатирующую группу, в свободной форме, в форме соли или комплексного соединения с обнаруживаемым элементом. Эти соединения наименованы здесь и далее как хелатные соединения по изобретению.
Подходящими хелатирующими группами являются физиологически приемлемые хелатирующие группы, способные к комплексообразованию с обнаруживаемым элементом. Предпочтительно хелатирующая группа имеет в значительной степени гидрофильный характер. Примеры хелатирующих групп включают в себя, например, группы, полученные из полиаминополикарбоновых кислот или ангидридов, например группы, полученные из нециклических лигандов, например, этилендиаминтетрауксусной кислоты (EDTA), диэтилентриаминпентауксусной кислоты (DTPA), этиленгликоль- O,O'-бис(2-аминоэтил)-N,N,N',N'-тетрауксусной кислоты (EGTA), N, N'-бис (гидроксибензил) этилендиамин-N, N'-диуксусной кислоты (HBED) и триэтилентетрамингексауксусной кислоты (TTHA), группы, полученные из замещенных EDTA или DTPA, например параизотиоцианато-бензил-EDTA или -DTPA, группы, полученные из макроциклических лигандов, например 1,4,7,10-тетраазациклододекан-N,N',N'',N'''-тетрауксусной кислоты (DOTA) и 1,4,8,11-тетраазациклотетрадекан-N, N',N'',N'''-тетрауксусной кислоты (TETA) или 1,4,7,10-тетраазациклотридекан-N, N', N'', N'''- тетрауксусной кислоты (TITRA).
Хелатирующая группа может быть присоединена либо непосредственно, либо через спейсерную группу к аминогруппе соединения по изобретению. Подходящие спейсерные группы включают в себя известные из уровня техники (GB-A-2225579) группы, например двухвалентный остаток амино-карбоновой кислоты, например β-Ala или двухвалентный остаток, полученный из 6-аминокапроновой кислоты.
Предпочтительными хелатирующими группами являются группы, полученные из DTPA, DOTA, TETA или замещенных EDTA или DTPA. Хелатирующие группы, полученные из DTPA или DOTA, наиболее предпочтительны.
Под обнаруживаемым элементом подразумевается любой элемент, предпочтительно ион металла, который проявляет свойство, обнаруживаемое в терапевтических или in vivo диагностических методах, например ион металла, который испускает обнаруживаемое излучение, или ион металла, который способен оказывать влияние на ЯМР релаксационные свойства.
Подходящие обнаруживаемые ионы металлов включают в себя, например, ионы тяжелых элементов или редкоземельных металлов, в частности, используемые при CAT сканировании (компьютерная осевая томография), парамагнитные ионы, например Gd3+, Fe3+, Mn2+ и Cr2+, ионы флуоресцентных металлов, например Eu3+, и радионуклиды, например радиолантаниды, в частности γ-испускающие радионуклиды, β-испускающие радионуклиды, α-испускающие радионуклиды, Auger-e--испускающие радионуклиды, позитрон-испускающие радионуклиды, например 68Ga.
Подходящие γ-испускающие радионуклиды включают в себя те из них, которые полезны в диагностических методах. γ-испускающие радионуклиды преимущественно имеют период полураспада от 1 часа до 40 дней, предпочтительно от 5 часов до 4 дней, более предпочтительно от 12 часов до 3 дней. Примерами являются радионуклиды, полученные из галлия, индия, технеция, иттербия, рения, тербия, таллия и самария, например 67Ga, 111In, 99mTc, 161Tb, 169Yb и 186Re.
Подходящие β-испускающие радионуклиды включают в себя те из них, которые полезны в терапевтических применениях, например 90Y, 67Cu, 186Re, 188Re, 169Er, 121Sn, 127Te, 143Pr, 198Au, 109Pd, 165Dy, 32P, 142Pr и 156Sm.
Подходящими α-испускающими радионуклидами являются те из них, которые применяют в терапевтическом лечении, например 211At, 212Bi или 201Tl.
Соединения по изобретению могут существовать, например, в свободной форме или в форме соли. Соли включают в себя соли, полученные присоединением кислот, например соли органических кислот, полимерных кислот или неорганических кислот, например гидрохлориды и ацетаты, и солевые формы, получаемые с группами карбоновых кислот при их наличии, например, в хелатирующей группе, например соли щелочных металлов, таких как натрий или калий, или замещенные или незамещенные соли аммония.
Настоящее изобретение также включает в себя способ получения соединений по изобретению. Они могут быть получены по аналогии с известными способами.
Соединения по изобретению могут быть получены, например, следующим образом:
а) удаляют, по меньшей мере, одну защитную группу, которая присутствует в соматостатиновом пептиде, содержащем остаток формулы 1, причем соматостатиновый пептид находится в защищенной форме, или
б) сшивают амидной связью две пептидные единицы, причем каждая из них содержит, по меньшей мере, одну аминокислоту в защищенной или незащищенной форме, при таком расположении амидной связи, при котором получают желаемую аминокислотную последовательность, и, если требуется, выполняют стадию, а) способа, или
в) удаляют функциональную группу незащищенного или защищенного соматостатинового пептида или превращают ее в другую функциональную группу таким образом, чтобы получить другой незащищенный или защищенный пептид и, в последнем случае, выполняют стадию а) способа, или
г) для получения хелатного соединения по изобретению сшивают хелатирующий агент и нехелатное соединение по изобретению в защищенной или незащищенной форме, содержащее свободную аминогруппу, таким образом, чтобы зафиксировать хелатирующую группу на желаемой аминогруппе соединения, и затем возможно выполняют стадию а) способа,
и выделяют полученное таким образом соединение по изобретению в свободной форме, в форме соли или возможно в форме комплексного соединения с обнаруживаемым элементом.
Стадия б) способа приводит к получению линейного пептида, но включает в себя также циклизацию посредством амидной связи линейного пептида с образованием циклического пептида, имеющего желаемую аминокислотную последовательность. При желании боковую цепь, которая присутствует в A, можно ввести в аминокислоту до стадии пептидного сочетания б) или в конечный линейный или циклический пептид согласно стадии в). Таким образом, в последнем случае соединение формулы II, в котором A представляет собой гидрокси-Pro, можно превратить в соединение формулы II, в котором A представляет собой R3-NH-CO-O-Pro.
Циклизацию также можно осуществить традиционным способом с помощью гидразида. Когда линейный пептид получают на смоле, обычно не имеет значения, какая аминокислота выбрана, для ее размещения в C-терминальном положении, при условии, что последовательность аминокислот в линейном пептиде соответствует последовательности в желаемом аналоге соматостатина. Как только циклизация линейного пептида проведена, можно уже не определять, какая аминокислота была на C-конце линейного пептида. Несмотря на то, что в общем случае выбор первой аминокислоты для начала цепи не играет роли, поскольку линейный пептид будет циклизован, могут иметь место другие факторы, которые могут предпочесть одну исходную аминокислоту другой. Предпочтительно линейный пептид циклизуют таким образом, чтобы получить связь от Tpr в положении 3 до ZZa в положении 2 или от X2 в положении 6 до X1 в положении 5.
Комплексообразование соединения по изобретению, содержащего аминогруппу, замещенную хелатирующей группой, может быть осуществлено взаимодействием хелатного соединения с соединением, дающим соответствующий обнаруживаемый элемент, например с металлической солью, предпочтительно водорастворимой солью. Взаимодействие можно проводить по аналогии с известными (Perrin, Organic Ligand, Cemical Data Series 22. NY Pergamon Press (1982); Krejcarit and Tucker, Biophys. Biochem. Res. Com. 77: 581 (1977); Wagner and Welch, J. Nucl. Med. 20: 428 (1979)) способами.
Поскольку получение исходных материалов детально не описано, соединения известны или могут быть получены по аналогии с известными и практикуемыми в технике способами.
Изобретение иллюстрируется следующими примерами.
Все температуры выражены в oC.
Используются следующие аббревиатуры:
Bzl = бензил(Bzl)= -CH2-фенил, присоединенный к кислороду или сере согласно (а) или (б)
DFM (ДМФ) = диметилформамид
BOC = трет-бутилоксикарбонил
Fmoc = 9-флуоренилметоксикарбонил
TFA = трифтороуксусная кислота
DIPCI = диизопропилкарбодиимид
DCCI = дициклогексилкарбодиимид
HOBt = гидроксибензотриазол
Dab = 2,4-диаминомасляная кислота
Dpr = 2,3-диаминопропановая кислота
Dpm = 2,6-диаминогептандиовая кислота
Dde = 4,4-диметил-2,6-диоксоциклогекс-1-илиденэтил
RT (KT) = комнатная температура
HyPro = 4-гидрокси-Pro (транс-, если не установлено иначе)
Tic = тетрагидроизохинолинкарбоновая кислота
FAB = бомбардировка быстрыми атомами
M.S. = масс-спектрометрия
E.S. = эмиссионная спектроскопия
ПРИМЕР 1
цикло [HyPro-Phe-DTrp-Lys-Tyr(Bzl)-Phe]:
Смолу Fmoc-Phe-SASRIN ® (1,00 г, 0,65 ммоль) подвергают процедурам твердофазного синтеза Fmoc, пока не соберут в структуру пептидную смолу Fmoc-(D)Tpr-Lys-(Boc)-Tyr(Bzl)-Phe-Pro(y-t-OH)-Phe SASRIN ® Fmoc лишают защиты, применяя пиперидин. Расщепление пептидной смолы проводят с применением гидразинолиза. К 1,00 г пептидной смолы добавляют 8,3 мл ДМФ и 1,24 мл гидрата гидразина (прибл.15%-ный гидрат гидразина в ДМФ). Смесь перемешивают в течение 15 ч при КТ. После реакции смолу фильтруют и тщательно промывают ДМФ. Фильтрат собирают и упаривают в глубоком вакууме до получения маслянистого остатка гидразида. Остаток растворяют в воде и лиофилизируют до получения 480 мг линейного гидразидного продукта H-(D)Trp-Lys(Boc)-Tyr(Bzl)-Phe-HyPro-Phe-NH-NH2. Этот гидразид растворяют в 16 мл ДМФ, охлаждают до -20o и обрабатывают 4N раствором HCl в эфире (2,4 мл, 11,6 ммоль), а затем трет-бутилнитритом (41,3 мкл, 0,348 ммоль). Реакционную смесь перемешивают в течение 20 мин. Добавляют диизопропилэтиламин (11,6 ммоль, 2 мл) и реакционную смесь перемешивают в течение 72 ч при комнатной температуре. После того как реакция закончится, ДМФ удаляют под глубоким вакуумом. К маслянистому остатку добавляют воду до выпадения осадка. Экстракцию проводят между этилацетатом и водой. Органические фазы сушат над сульфатом натрия, и продукт выделяют. Защиту удаляют с применением смеси TFA/вода (95/5), и продукт выделяют, применяя высокоэффективную жидкостную хроматографию (HPLC) с обращенной фазой. Осуществляют ионный обмен продукта, содержащего фракции, и лиофилизируют. В результате получают соединение, указанное в заголовке, в виде белого порошка, MH+ (FAB) 975, F = 1,24 [α]
ПРИМЕР 2
цикло [{4-(NH2-C2H4-NH-CO-O-)Pro}-Phe-DTrp-Lys- Tyr(Bzl)-Phe]
Fmoc-HyPro-OMe по каплям добавляют в раствор трисфосгена (0,6 экв.) в ТГФ. По истечении 1ч добавляют диметиламинопиридин (1,0 экв.) и N-BOC-диаминоэтан (6,0 экв. ), и реакционную смесь перемешивают при КТ. После того, как проведут операции тонкослойной хроматографии (TLC), растворитель удаляют в вакууме, и Fmoc-4-(N-BOC-аминоэтиламинокарбонилокси)Pro-OMe экстрагируют из двухфазной системы этилацетат/0,1М HCl с получением неочищенного продукта (MH+ = 554). Неочищенный метиловый эфир, выделенный ранее, затем расщепляют до свободной кислоты путем обработки 1N NaOH в системе диоксан/вода, и продукт, Fmoc-4- (аминоэтиламинокарбонилокси)-Pro-OH, очищают на силикагеле, (MNa+ = 562).
Смолу Fmoc-Phe-SASRIN ® (1,0 г, 0,65 ммоль) подвергают процедурам твердофазного синтеза Fmoc, пока не будет собрана в структуру пептидная смола Fmoc-(D)Trp(BOC)-Lys(ВOC)-Tyr(Bzl)- Phe-Pro(y-t-N-BOC-диаминоэтанкарбамоил)-Phe-SASRIN. Fmoc лишают защиты, применяя пиперидин. Расщепление пептидной смолы проводят путем ее обработки в стеклянной колонке 2%-ным раствором TFA в CH2Cl2. Нейтрализацию проводят 1М раствором NaHCO3. Растворитель испаряют в вакууме, и защищенный линейный пептид лиофилизируют (MH+ = 1379,8). Защищенный линейный пептид подвергают циклизации путем обработки DCCI (6,0 экв.) и HOBt (6,0 экв.) за период в 5 дней.
Затем удаляют защиту путем обработки смесью TFA:H2O (95:5), и циклический пептид очищают препаративной высокоэффективной жидкостной хроматографией (HPLC) и подвергают ионному обмену с получением ацетатной соли с помощью ионообменной смолы AG4-X4 с получением соединения, указанного в заголовке.
(FAB-MH+= 1061,7).
ПРИМЕР 3
цикло [{ 4-(морфолино-этил-аминокарбонилокси)-Pro} -Phe-DTrp-Lys- Tyr(Bzl)-Phe]
Синтез гидроксипролиновых производных осуществляют следующим образом.
Fmoc-HyPro-OMe по каплям добавляют в раствор трисфосгена (0,6 экв.) в ТГФ. По истечении 1ч добавляют диметиламинопиридин (1,0 экв.) и N-этиламиноморфолин (6,0 экв.) и перемешивают смесь при КТ. После операций тонкослойной хроматографии (TLC) растворитель удаляют в вакууме и Fmoc-4- (морфолиноэтиламинокарбонилокси)Pro-OMe очищают на силикагеле, (MH+ 524). Затем метиловый эфир расщепляют путем обработки 1N NaOH в системе диоксан/вода, и продукт, Fmoc-4-(морфолиноэтиламинокарбонилокси)Pro-OH, очищают на силикагеле, (MH+ 510).
Fmoc-Phe-SASRIN подвергают процедурам твердофазного синтеза Fmoc как в предыдущем примере, пока не будет собрана в структуру смола Fmoc-(D)Trp(BOC)-Lys(BOC)-Tyr(Bzl)- Phe(морфолиноэтиламинокарбамат)HyPro-Phe-SASRIN. Удаляют защиту Fmoc, применяя пиперидин. Расщепление пептидной смолы проводят путем ее обработки в стеклянной колонке 2%-ной TFA в CH2Cl2. Нейтрализацию проводят 1М раствором NaHCO3. Растворитель удаляют в вакууме, и защищенный линейный пептид лиофилизируют. Защищенный линейный пептид подвергают циклизации путем обработки DCCI (6,0 экв.) и HOBt (6,0 экв.) за период в 5 дней. Затем удаляют защиту путем обработки смесью TFA:H2O (95:5), и циклический пептид очищают препаративной высокоэффективной жидкостной хроматографией (HPLC) и подвергают ионному обмену с получением ацетатной соли с помощью ионообменной смолы AG4-X4 до получения соединения, указанного в заголовке.
MH+ (FAB): 1131.
[α]
Путем повторения операций, как показано выше, но с использованием соответствующих исходных материалов, могут быть получены соединения формулы:
цикло[X-Y-DTrp-Lys-Z-Phe]
в которой X, Y и Z определены ниже в Таблице 1.
Пептид из примера 29 может быть получен следующим образом.
Защищенный пептид, цикло[(NH2-C(=NH)-NH-C2 H4-NH-CO-O-)]-Pro-Tyr-DTrp-Lys(Dde)-Tyr(Bzl)-Phe, собирают на смоле, применяя процедуру твердофазного синтеза Fmoc, как описано в Примере 2. Вместо Ng-Boc-Lys используют Ng-Dde-Lys, чтобы предпочтительно ввести функциональность гуанидинила в основную боковую цепь остатка HyPro. После того, как сборку пептида в структуру заканчивают, концевую Fmoc группу удаляют, пептид подвергают циклизации и в конце удаляют защиту, как в Примере 2. Этот пептид растворяют в ДМФ, добавляют диизопропилэтиламин (3 экв.) и HOBt (4 экв.), затем добавляют 3,5-диметилпиразолилформамидиния нитрат (4 экв.) и раствор перемешивают в течение 72 ч при комнатной температуре. Реакционную смесь упаривают в вакууме, а затем подвергают обработке безводным гидразином (2% в ДМФ) в течение 30 мин для удаления группы Dde на Lys. Неочищенный пептид, указанный в заголовке (Пример 29), затем очищают высокоэффективной жидкостной хроматографией (HPLC) в системе ацетонитрила и водного фосфата триэтиламмония.
ПРИМЕР 41
цикло[4-(NH2-C2H4-NH-CO-O-)Pro-Ala-DTrp- Lys-Tyr(3-Bzl)-Phe]
MH+ (E.S.): 984,5
ПРИМЕР 42
цикло[{4-(NH2-C2H4-NH-CO-O-)Pro}- (p-NH2)-Phe-DTrp-Lys-Tyr(3-Bzl)-Phe]
MH+ (E.S.): 1076,6
ПРИМЕР 43
цикло[4-HyPro-Phe-DTrp-Lys-Tyr(Bzl)- β Nal]
MH+ (E.S.): 1025,5
ПРИМЕР 44
цикло[4-HyPro-Phe-DTrp-Lys-Tyr(Bzl)-Tyr]
MH+ (E.S.): 991,6
ПРИМЕР 45
цикло[MePhe-His-DTrp-Lys-Tyr(Bzl)-Dab]
MH+ (E.S.): 1005
ПРИМЕР 46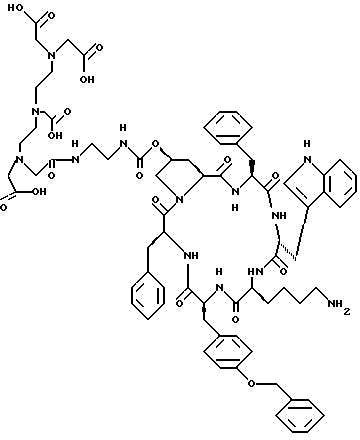
a) цикло[4-(NH2-C2H4-NH-CO-O-) Pro-Phe-DTrp-Lys( ε -Boc)-Tyr(Bzl)-Phe]
60 мг цикло [4-(NH2-C2H4-NH-CO-O)Pro-Phe- DTrp-Lys-Tyr(Bzl)-Phe], 12 мг NaHCO3 и 12 мг (BOC)2O растворяют в 10 мл смеси ДМФ/вода (7/3) и выдерживают при комнатной температуре при перемешивании в течение ночи. После удаления растворителя указанный в заголовке продукт выделяют хроматографией на силикагеле с применением системы метиленхлорид/метанол/уксусная кислота50% (8/2/0,25) в качестве подвижной фазы.
б) цикло[4-(DTPA-NH-C2H4-NH-CO-O-)Pro-Phe-DTrp- Lys) ε -Boc)-Tyr(Bzl)-Phe]
120 мг DTPA-гидразида растворяют в 5 мл ДМФ и доводят до pH 3 добавлением по каплям смеси диэтиловый эфир/3N HCl. После охлаждения до -15o в реакционную смесь добавляют 4 мкл трет- бутилнитрита и раствор из 15 мг соединения, полученного выше в а), в 3 мл ДМФ, содержащего 15 мкл основания Хюнига (Hunig). По истечении 4 часов растворитель удаляют выпариванием, и удаляют защиту полученного остатка без какой-либо дополнительной очистки.
в) цикло[4-(DTPA-NH-C2H4-NH-CO-O-)Pro-Phe-DTrp-Lys- yr(Bzl)-Phe]
Неочищенный продукт со стадии б) обрабатывают 5 мл смеси TFA/вода (95/5) при 0o в течение 10 минут. После разбавления 50 мл воды раствор непосредственно переносят на колонку RP18-HPLC и элюируют градиентом вода/ацетонитрил/TEA0,1%. Чистые фракции объединяют и лиофилизируют.
FAB-MS: 1436,6
ПРИМЕР 47
Соединение из Примера 46 в), меченое 111In
1 мг соединения из Примера 46 в) растворяют в 5 мл 0,01М раствора уксусной кислоты. Полученный раствор пропускают через 0,22-микронный фильтр Millex ® -GV (товарный знак зарегистрирован), делят на порции по 0,1 мл и хранят при -20o. 111InCl3 (Amersham, 1 мкюри/100 мкл) предварительно разбавляют равным объемом 0,5М раствора ацетата натрия и проводят мечение путем смешивания лиганда с раствором InCl3 и мягкой гомогенизации при комнатной температуре.
Затем добавляют буфер HEPES (N-2-гидроксиэтилпиперазин-N-2- этансульфоновая кислота) с pH 7,4 и получают раствор концентрации 10-6 М.
ПРИМЕР 48
цикло[4-(DOTA-NH-C2H4-NH-CO-O)Pro-Phe-DTrp-Lys-Ser (Bzl)-Phe]
M.S.: 1371,57
Это соединение метят изотопом 90Y следующим образом: 20 мкл 90Y (1,2 мкюри, 0,04М HCl) добавляют к 20 мкл из 50 мкМ указанного выше соединения (0,15 М NH4= Ac, 0,3% BSA (бычий сывороточный альбумин), pH 4,5). Этот раствор инкубируют при 100o в течение 15 минут. Отбирают аликвоту и разбавляют ее 4 мМ DTPA (pH 4,5) перед тем, как анализировать с помощью колонки C18 HPLC с обращенной фазой для определения количества свободного нехелатного 90Y в реакционной смеси (по присутствию [90YDTPA]2-).
ПРИМЕР 49
116 мг соединения из Примера 46 а), 12 мг NaCNBH3 и 2 эквивалента соответствующего альдегида растворяют в 25 мл смеси DMF/HOAc1% и выдерживают при 60o до тех пор, пока исходный материал можно будет определить тонкослойной хроматографией (TLC). После удаления растворителя остаток очищают хроматографией на силикагеле (метиленхлорид/метанол/HOAc50% 9/1/0,125 ---> 8/2/0,25) для того, чтобы разделить моно- и диалкилированный конечный продукт.
1) Альдегид: (D)-глюкоза
X1 = HOCH2-(CHOH)4-CH2- X2=H
E.S. -MH+ = 1225,4
2) Альдегид: (D)-глюкоза
X1 = X2= HOCH2-(CHOH)4-CH2-
E.S. -MH+ = 1389,6
3) Альдегид: 2,3-O-изопропилилен-(D)-глицеральдегид
X1 = HOCH2-CHOH-CH2- X2 = H
4) Альдегид: 2,3-O-изопропилиден-(D)-глицеральдегид
X1 = X2 = HOCH2-CHOH-CH2-
E.S. -MH+ = 1209,4
5) Альдегид: гидроксиацетальдегид
X1 = X2 = HOCH2-CH2-
E.S. -MH+ = 1149,4
Как в свободной форме, так и в форме фармацевтически приемлемых солей и комплексов соединения по данному изобретению демонстрируют ценные фармакологические свойства, о которых свидетельствуют испытания как in vivo, так и in vitro, и являются, таким образом, показанными для лечения.
В частности, соединения по данному изобретению связываются с по меньшей мере одним подтипом рецепторов к соматостатину. Клонированы и описаны 5 подтипов рецепторов к соматостатину: SST-1, SST-2, SST-3, SST-4 и SST-5.
hSST-1, hSST-2, hSST-3 и их последовательности известны (Y. Yamada et al. in Proc. Nat. Acad. Sci., 89, 251-255 (1992)). hSST-4 и его последовательность известны (L. Rohrer et al. in Proc. Acad. Sci., 90, 4196-4200 (1993)). hSST-5 и его последовательность известны (R. Panetta et al. in Mol. Pharmacol. 45, 417-427, 1993).
Анализ связывания может быть проведен, как описано ниже, используя мембраны, приготовленные из селективных по отношению к hSST-1, hSST-2, hSST-3, hSST-4 или hSST-5 линий клеток, например клеток CHO, стабильно экспрессирующих hSST-1, hSST-2, hSST-3, hSST-4 или hSST-5.
Используют ткань мозга или гипофиза, в которой визуализируют hSST посредством, например, гибридизации in situ и/или ауторадиографии рецепторов. Мембраны приготавливают в соответствии с известными способами (JC. Reubi et al. in J. Clin. Endocrinol. Metab. 1987, 65, 1127-1137). Мембраны, приготовленные из селективных по отношению к hSST линий клеток, например клеток CHO, стабильно экспрессирующих hSST-1, hSST-2, hSST-3, hSST-4 или hSST-5, трижды инкубируют в общем объеме 300 мкл при температуре 22oC в течение 30 минут с возрастающими концентрациями: [125I-Tyr3]-октреотида в буфере Hepes (pH 7,6) с концентрацией 10 ммоль/л, содержащем 0,5% BSA. Инкубацию прерывают посредством быстрой фильтрации через стекловолоконные фильтры Whatman GF/B, каждый из которых затем промывают четыре раза 5 мл охлажденного на льду раствора 10 ммоль/л Трис/150 ммоль/л NaCl. Фильтры обсчитывают в счетчике LKB с эффективностью подсчета 78%. Специфическое связывание представляет собой общее связывание минус неспецифическое связывание в присутствии 1 мкмоль/л соматостатина-14. Эксперименты проводят трижды. Константу сродства (Kd) и количество сайтов связывания рассчитывают, исходя из графиков данных по Statchard.
ИК50 (концентрация, при которой ингибирование составляет половину от максимального в конкурентном анализе связывания с использованием [125I-Tyr3] -октреотида, т. е. того же радиолиганда, что указан выше) соединений по данному изобретению, указанных, например, выше, в вышеописанных анализах связывания с hSST-1, hSST-2, hSST-3, hSST-4 и/или hSST-5 находится, соответственно, в диапазоне нМолей, а предпочтительно составляет от 0,1 до 10 нМ (см. табл. 2).
Более того, соединения по данному изобретению обладают способностью ингибировать высвобождение гормона роста (GH), о чем свидетельствует ингибирование высвобождения GH из культуры клеток гипофиза in vitro. Передние доли гипофизов взрослых крыс-самцов нарезают на маленькие кусочки и диспергируют, используя 0,1%-ный трипсин в 20 мМ буфере Hepes. Диспергированные клетки выращивают в течение четырех дней в MEM(Gibco), дополненной 5% фетальной телячьей сыворотки, 5% лошадиной сыворотки, 1 мМ NaHCO3, 2,5 нМ дексаметазона, 2,5 мг/мл инсулина и 20 Ед/мл пенициллина/стрептомицина. В день эксперимента присоединившиеся клетки дважды промывают средой Кребса-Рингера (Krebs-Ringer), забуференной 2 мМ Hepes и дополненной 5 мМ глюкозы и 0,2% BSA. Затем клетки инкубируют с исследуемым соединением в присутствии фактора высвобождения гормона роста в концентрации 3•10-10 М в течение от 2-4 часов. Количество высвобождаемого в среду гормона роста измеряют посредством радиоиммунного анализа (RIA). Соединения по изобретению ингибируют высвобождение GH зависимым от концентрации образом от 10-11 до 10-6 М. ИК50 соединения по Примеру 2 составляет 0,4 нМ.
Соединения по изобретению также ингибируют высвобождение инсулина и/или глюкагона, о чем свидетельствуют стандартные опыты на крысах-самцах. Исследуемое вещество вводят в различных, изменяющихся логарифмически, дозах, применяя каждую дозу у по меньшей мере 5 крыс. Через 1 час после подкожного введения исследуемого вещества производят забор крови. Определение уровня инсулина и глюкагона в крови производят посредством радиоиммунного анализа. В данном опыте соединения по данному изобретению активны при введении их в дозах от 0,02 до 1000, например 10 мкг/кг подкожно. ЕК50 соединения по Примеру 9 в отношении секреции инсулина составляет 1,8 мкг/кг подкожно.
Соединения по изобретению, соответственно, являются полезными в лечении заболеваний, этиология которых состоит в избыточной секреции гормона роста или связана с ней, например в лечении акромегалии, а также в лечении сахарного диабета (в особенности его осложнений, таких как ангиопатия, пролиферативная ретинопатия, феномен "утренней зари" и нефропатия) и прочих метаболических расстройств, связанных с высвобождением инсулина или глюкагона.
Соединения по изобретению также ингибируют секрецию кислого желудочного содержимого, эндокринную и экзокринную секрецию поджелудочной железы и секрецию различных пептидов в желудочно-кишечном тракте, о чем свидетельствуют стандартные опыты, например на крысах с желудочными или панкреатическими фистулами, в которых соединения по данному изобретению активны в дозах от 0,01 до 10 мг/кг.
Таким образом, соединения по изобретению также являются полезными в лечении желудочно-кишечных заболеваний, например в лечении пептических язв, кишечных и панкреатических свищей, синдрома и болезни "раздраженной" кишки, демпинг-синдрома, синдрома водянистой диарреи, диарреи, связанной со СПИДом или обусловленной химиотерапией, острого или хронического панкреатита и опухолей, секретирующих гастроинтестинальные гормоны (например, випом, глюкагоном, инсулином, карциноидов и т.п.), а также желудочно-кишечных кровотечений.
Соединения по изобретению также эффективны при лечении опухолей, содержащих рецепторы к соматостатину, в особенности опухолей, содержащих hSST-1, hSST-2, hSST-3, hSST-4 и/или hSST-5, на что указывают результаты исследования пролиферации различных линий раковых клеток, содержащих подобные рецепторы соматостатина.
Линию клеток опухоли поджелудочной железы крысы AR42J получают из вызванной азасерином экзокринной опухоли поджелудочной железы (Jessop и Hay, 1980). Отсутствие микоплазмы регулярно подтверждают, используя окрашивание бизбензимидом и анализ гибридизации GenProbe (San Diego, CA). Культуры распространяют в среде DMEM, дополненной 10% фетальной телячьей сыворотки, при 5% CO2. Клетки выращивают без антибиотиков или противогрибковых агентов. Сливающиеся клетки AR42J трипсинизируют, растворяют в DMEM + 2,5% фетальной овечьей сыворотки и высевают в непокрытые 96-луночные планшеты. После 48-часового инкубационного периода (День 0) определяют количество клеток в отдельном контрольном планшете, как подсчетом в счетчике Coulter, так и колориметрическим анализом SRB. Затем клетки подвергают воздействию исследуемого соединения в различных концентрациях в течение 2-5 дней, после чего подсчитывают. В подобных условиях соединения по данному изобретению ингибируют пролиферацию опухолевых клеток в концентрации от 10-12 до 10-6 М. ИК50 соединения по Примеру 2 составляет 0,7 ± 0,3 (CO) нМ.
Исследования роста опухолей in vivo
Самок бесшерстных мышей (nu/nu Balbc-A, IFFA Credo, Lyon, France) массой 19-22 г содержат в группах по 5 животных в макролоновых клетках (тип III, 16х22х11см). Клетки помещают в вентилируемые шкафы (IFFA Credo), температура в которых поддерживается на уровне 24±1oC. Животные получают беспрепятственный доступ к питьевой воде и не содержащую патогенов диету для грызунов (Диета A, Kliba, Basel, Switzerland). Чтобы инициировать опухоли из культивируемых клеток, клетки AR42J трипсинизируют, после чего 5-10•106 опухолевых клеток (в 0,2 мл) вводят подкожно в оба бока бесшерстных мышей. Лечение начинают через 2-4 дня после инокуляции опухолевых клеток, причем предпочтительно введение исследуемого соединения в виде постоянной инфузии, например со скоростью 10-50 мкг/кг в час. Размер опухолей определяют с помощью циркуля. Для расчета объема опухолей используют следующее уравнение: "объем (эллипсоида) = длина х ширина х высота х 0,52". Для статистических расчетов применяют t-тест Стьюдента. В этой серии опытов соединение по Примеру 2 ингибирует рост опухоли в день 11 на 51% по сравнению с контрольным введением физиологического раствора.
Таким образом, соединения по данному изобретению являются полезными в лечении злокачественных пролиферативных заболеваний, например раковых опухолей, в особенности опухолей, содержащих те типы рецепторов к соматостатину, на которые направлено действие данных соединений, так, например, как изложено ниже для хелатных соединений.
Соединения по изобретению также оказывают ингибирующее действие на ангиогенез, на что указывают результаты стандартных исследований, например на бесшерстных мышах, как описано ниже.
Мышей анестезируют посредством внутрибрюшинного введения 400 мг/кг хлоралгидрата (Sigma). Опухолевые клетки в количестве 0,1-10•106 в 0,1 мл (клетки SiHa и клетки MDA-MB-231, приготовленные, как описано (Angiogenesis, Ed. by R. Steiner, P.В. Weisz and R. Langer, 1992, Switzerland)), инокулируют внутрикожно. Обычно инъекцию у каждой мыши производят в две удаленные от основных сосудов кожи живота точки в мезогастрии, так, чтобы исходное количество сосудов было невелико. Контрольным группам животных вводят 0,1 мл 0,02% трипановой сини в PBS. Через 10 дней после инъекции анестезированных мышей убивают посредством ингаляции CO2. Кожу натягивают на пластиковое кольцо диаметром 40 мм для исследования посредством инвертированного микроскопа (Zeiss IM) с 12,5- и 25-кратным увеличением. Для измерения ангиогенеза сосуды фотографируют и подсчитывают те из них, что непосредственно связаны с опухолью. У контрольных животных подсчитывают сосуды, связанные с определенной областью вокруг места инъекции. Эта область соответствует средней площади опухолей кожи. Последнюю определяют с помощью циркуля, в соответствии с уравнением 3,14 х r2. Исследуемые соединения вводят подкожно либо в день инокуляции опухоли, либо тремя днями позже. Контрольным животным вводят носитель. В этой серии опытов соединения по изобретению ингибируют формирование кровеносных сосудов при введении в дозах от, например, 0,01 до 1000 мкг/кг подкожно.
Таким образом, соединения по изобретению являются полезными для профилактики или лечения ангиогенеза, воспалительных заболеваний и ретинопатии.
Соединения по изобретению также оказывают ингибирующее действие на пролиферацию и миграцию гладкомышечных клеток, на что указывают результаты нижеследующих исследований.
Хроническое отторжение аллотрансплантата.
Соединения по данному изобретению ингибируют хроническое отторжение аллотрансплантата почки у крыс. Почку самца крысы DA (RT1a) ортотопически пересаживают самцу-реципиенту Lewis (RT11). В целом пересадку производят у 24 крыс. В течение 14 дней, начиная со дня трансплантации, все животные получают циклоспорин A в дозе 7,5 мг/кг в день внутрь для предотвращения острого отторжения клеток. Нефрэктомию с противоположной стороны не выполняют. Каждая группа экспериментальных животных, получающих различные дозы соединения по данному изобретению или плацебо, состоит из шести животных.
Начиная с 53-64-го дня после трансплантации животные-реципиенты получают соединение по данному изобретению в виде инфузии или плацебо в течение последующих 69-72-х дней. На 14-й день после трансплантации перфузию органа измеряют с помощью MRI. Эту процедуру повторяют на 53-64-й день после трансплантации и в конце эксперимента. После этого производят вскрытие животных. Применение к данной модели аллотрансплантата почки крысы соединения по данному изобретению, например соединения по Примеру 31, в дозе от 1 до 10 мкг/кг в час вызывает улучшение перфузии органов. Также отмечено резкое снижение уровня IGF-1.
Ангиопластика
Исследования ангиопластики проводят на модели повреждения баллонным катетером. Баллонную катетеризацию проводят в день О, практически так, как описано (Powell et al. (1989)). Под анестезией изофлюраном катетер Фогерти (Fogarty) 2F вводят в левую общую сонную артерию через наружную сонную артерию, после чего раздувают баллон (растяжение - около 10 мкл H2O). Раздутый баллон извлекают вдоль общей сонной артерии трижды, причем два последних раза слегка вращают, чтобы добиться равномерной деэндотелизации. Затем катетер удаляют, на наружную сонную артерию накладывают лигатуру во избежание кровотечения и животным дают восстановиться.
В данном исследовании используют две группы по 12 крыс RoRo (400 г, приблизительный возраст - 24 недели): одну - контрольную и одну - получающую соединение по данному изобретению. Весь уход, экспериментальные процедуры и анализ проводят на основе полной рандомизации.
Исследуемое соединение вводят в виде постоянной инфузии с использованием минидозаторов со скоростью 10-50 мкг/кг в час, начиная за 3 дня до повреждения баллоном (день -3) и до окончания исследования через 14 дней после повреждения (день +14). Крыс содержат в отдельных клетках и предоставляют им пищу и воду ad libitum.
Затем крыс анестезируют изофлюраном, через левый желудочек вводят перфузионный катетер, который фиксируют в дуге аорты, а аспирационную канюлю вводят в правый желудочек. Выполняют перфузию под перфузионным давлением 150 мм Hg, вначале в течение одной минуты 0,1 М забуференным фосфатом физиологическим раствором (PBS, pH 7,4), а затем в течение 15 минут 2,5%-ным глютаральдегидом в фосфатном буфере (pH 7,4). Затем сонные артерии иссекают, отделяют от окружающих тканей, погружают в 0,1 М какодилатный буфер (pH 7,4), содержащий 7% сахарозы, и инкубируют в течение ночи при температуре 4oC. На следующий лень сонные артерии погружают в 0,05%-ный KMnO4 в 0,1 М какодилате и встряхивают в течение 1 часа при комнатной температуре. Затем ткани дегидратируют последовательным воздействием различных концентраций этанола: 2х10 минут в 75%-ном, 2х10 минут в 85%-ном, 3х10 минут в 95%-ном и 3х10 минут в 100%-ном этаноле. Затем дегидратированные сонные артерии укладывают в Technovit 7100 в соответствии с рекомендациями производителя. Окружающую среду оставляют полимеризоваться в течение ночи в эксикаторе под аргоном. Из середины каждой сонной артерии твердым металлическим ножом на дисковом микротоме вырезают срезы толщиной 4 мкм, которые окрашивают по Гимзе (Giemse) в течение 2 минут. Таким образом приготавливают около 5 срезов каждой сонной артерии, после чего поперечное сечение среднего слоя, неоинтимы и просвета оценивают морфометрически с помощью системы анализа изображений (MCID, Toronto, Canada).
Согласно результатам данного исследования, соединения по изобретению ингибируют пролиферацию миоинтимы при их введении путем постоянной инфузии в суточной дозе от 0,2 до 10 мг/кг, а предпочтительно - от 0,05 до 5 мг/кг.
Таким образом, соединения по изобретению также являются полезными для профилактики заболеваний сосудов в трансплантатах или борьбы с подобными заболеваниями, например васкулопатиями алло- или ксенотрансплантатов, такими как атеросклероз сосудов в трансплантатах, например в пересаженном органе: сердце, легком, сочетанием трансплантате сердце-легкие, печени, почке или поджелудочной железе, или для профилактики или лечения рестеноза и/или окклюзии сосудов после повреждения сосудов, например ангиопластики.
Необходимые для применения по всем этим показаниям дозы будут, естественно, варьироваться в зависимости, например, от применяемого соединения по изобретению, реципиента, способа введения и тяжести подлежащего лечению состояния. Тем не менее, в целом удовлетворительных результатов удается достичь при введении порядка от 1 мкг до 0,5 мг/кг в день соединения по данному изобретению. Показанная пациентам суточная доза составляет от 2 мкг до примерно 20 мг, предпочтительно от 0,01 до примерно 20 мг, в частности от примерно 10 до примерно 5000 мкг соединения подкожно, удобным образом вводимого в разделенных дозах до трех раз в сутки в форме стандартной дозы, содержащей, например, от 0,5 мкг до примерно 10 мг, в частности от примерно 2 мкг до 10 мг соединения в форме замедленного высвобождения.
Соединения по изобретению могут вводиться как в свободной форме, так и в форме фармацевтически приемлемых солей или комплексов. Подобные соли и комплексы могут быть приготовлены обычным способом и обладать такой же выраженной активностью, как и свободные соединения. В данном изобретении также предложена фармацевтическая композиция, содержащая соединение по изобретению, например соединение формулы II, в форме свободного основания или в форме фармацевтически приемлемых солей или комплексов, в сочетании с фармацевтически приемлемым разбавителем или носителем. Подобные композиции могут быть изготовлены в виде препаратов обычным образом. Соединения по изобретению или их фармацевтически приемлемые соли и комплексы могут вводиться обычным путем, например парентерально, в форме растворов или суспензий для инъекций, или в назальной форме, или в форме свечей.
В соответствии с вышеизложенным в данном изобретении дополнительно предложены:
а) соединение по данному изобретению, например соединение формулы II, или его фармацевтически приемлемая соль, или комплекс для использования в качестве фармацевтического средства.
б) способ профилактики или лечения нарушений, как указано выше, у субъекта, нуждающегося в подобном лечении, заключающийся во введении указанному субъекту эффективного количества соединения по изобретению, например соединения формулы II, или его фармацевтически приемлемой соли, или комплекса, или
в) соединения по изобретению, или его фармацевтически приемлемая соль, или комплекс для использования в приготовлении фармацевтической композиции для использования по любому определенному выше в п. б) способу.
Как указывают результаты стандартных исследований, хелатные соединения по данному изобретению или их фармацевтически приемлемые соли пригодны либо для применения в комплексе с γ - или позитрон-испускающим нуклидом, например 111In, 161Tb или 86Y, в качестве визуализирующего агента, например для визуализации содержащих рецепторы к соматостатину тканей и клеток, таких, как содержащие рецепторы к соматостатину опухоли и метастазы, воспалительные и аутоиммунные заболевания, характеризующиеся наличием рецепторов к соматостатину, туберкулез или отторжение органов после трансплантации, либо для применения в комплексе с α - или β -испускающим нуклидом или нуклидом с Auger-e-каскадом, таким, например, как 90Y, 61Tb, 211At, 213Bi или 201Tl, в качестве радиофармацевтического средства для лечения in vivo содержащих рецепторы к соматостатину опухолей и метастазов, ревматоидного артрита и тяжелых воспалительных состояний.
В частности, отмечено, что хелатные соединения по данному изобретению, например 111In, 88Y, 90Y, 153Sm, 186Re или 161Tb хелатные соединения по изобретению, связываются с хорошим сродством с рецепторами к соматостатину со значением pKi от примерно 8 до 10. ИК50 соединения по Примеру 47 составляет 1,2 нМ для hSST-2, 0,65 нМ для hSST-3, и 0,30 нМ для hSST-5.
Сродство хелатных соединений по данному изобретению и их комплексов к рецепторам к соматостатину может также быть продемонстрировано посредством исследований in vivo, в соответствии со стандартными способами исследований, описанными (GB-A-2,225,579). Соединение по Примеру 47, например, демонстрирует значительное накопление в опухоли через 4 часа после инъекции его мышам или крысам, имеющим экзокринные опухоли поджелудочной железы, содержащие рецепторы SST-2.
После введения хелатного соединения по изобретению в форме комплекса, например 111In, 88Y или 161Tb хелатного соединения по изобретению, маркированного 0,1-5 мКи, а предпочтительно - 0,1-2 мКи радионуклида, в дозе 1-5 мкг/кг место расположения опухоли становится обнаруживаемым, как и органы, посредством которых происходит выделение.
Как указывают результаты исследований на бесшерстных мышах, хелатные соединения по изобретению, меченные радиоактивной меткой в виде α - или β - испускающего нуклида или нуклида с Auger-e-каскадом, обладают антипролиферативным и/или цитотоксическим действием на опухолевые клетки, содержащие рецепторы к соматостатину.
Клетки опухоли поджелудочной железы крысы AR42J или клетки мелкоклеточного рака легких человека NCI-H69 инокулируют бесшерстным мышам описанным выше способом. Когда опухоли достигают объема 1-2 см3, животных рандомизируют в опытную и контрольную группы. Контрольные животные получают либо немеченое соединение, либо хелатное соединение в форме комплекса в виде внутрибрюшинных или внутривенных инъекций в дозах, соответствующих максимальной дозе, примененной в опытной группе. Каждая мышь получает дозы до 40 мКи/кг. Размер опухолей определяют с помощью циркуля, как описано выше. Для статистических расчетов применяют t-тест Стьюдента. В этой серии опытов отмечены временное уменьшение размеров опухолей на 50% от исходного и задержка роста опухолей на две недели после однократного введения соединения по Примеру 48. В контрольной группе, напротив, отмечен постоянный рост опухолей с удвоением объема примерно в течение семи дней.
Соответственно, в сериях особых или альтернативных воплощений данного изобретения также предложены:
1. Применение хелатного соединения по данному изобретению, например хелатного соединения формулы II в форме комплекса для обнаружения у субъекта in vivo клеток и тканей, содержащих рецепторы к соматостатину, и регистрации расположения рецепторов, с которыми связывается данный комплекс.
Меченные радиоактивной меткой соединения по изобретению могут вводиться внутрибрюшинно, а предпочтительно внутривенно, например в форме инъекционных растворов или суспензий, предпочтительно в виде однократной инъекции. Предпочтительно, чтобы введение радиоактивной метки производилось незадолго до введения субъекту.
Удобным является введение хелатного соединения по изобретению в дозе, содержащей 0,2-20 мКи, а предпочтительно 1-10 мКи, нуклида в виде стабильного комплекса.
У животных показанный диапазон доз может составлять 0,01-1 мкг/кг хелатного соединения по данному изобретению в комплексе с 0,02-0,5 мКи γ - испускающего нуклида. У более крупных млекопитающих, например у человека, показанный диапазон доз может составлять 1-20 мкг хелатного соединения в комплексе, например с 1-10 мКи 111In или 86Y.
2. Применение хелатного соединения по данному изобретению, например хелатного соединения формулы II в форме комплекса для лечения in vivo опухолей и метастазов, содержащих рецепторы к соматостатину.
Дозы, применяемые в практике радиотерапевтического использования данного изобретения, будут, конечно, варьироваться в зависимости от конкретного подлежащего лечению состояния, учитывая, например известную радиотоксичность по отношению к нормальным органам, содержащим рецепторы к соматостатину, объем опухоли и желаемую терапию. В целом, дозы рассчитывают, исходя из фармакокинетики, распределения радиоактивности к здоровым органам и наблюдаемого поглощения мишенями, β - испускающий комплекс хелатного соединения может вводиться неоднократно, например в течение периода, составляющего 1-3 месяца.
У животных показанный диапазон доз может составлять 20-100 мкг/кг хелатного соединения в комплексе с 15-70 мКи 90Y или 161Tb.
Хелатные соединения по данному изобретению в форме комплексов могут вводиться любым общепринятым способом, в частности внутрибрюшинно или внутривенно, например в форме инъекционных растворов или суспензий. Они также могут удобным образом применяться в виде инфузии, например, в течение 30-60 минут. В зависимости от локализации опухоли, они могут быть введены как можно ближе к опухоли, например, посредством катетера.
Хелатные соединения по данному изобретению в форме комплексов могут быть пригодны для визуализации или лечения таких опухолей, как гипофизарные и гастроэнтеропанкреатические опухоли, карциноиды, опухоли центральной нервной системы, молочной железы, предстательной железы, яичников или толстой кишки, мелкоклеточный рак легких, параганглиомы, рак почки, кожи, нейробластомы, феохромоцитомы, медуллярные карциномы щитовидной железы, миеломы, Ходжкинские и не-Ходжкинские лимфомы, опухоли костей и их метастазы, а также ревматоидного артрита.
В соответствии с дальнейшим аспектом данного изобретения предложена фармацевтическая композиция, содержащая хелатное соединение по данному изобретению в свободной форме или в форме комплекса вместе с одним или несколькими фармацевтически приемлемыми носителями или растворителями. Подобные композиции могут быть изготовлены общепринятыми способами и представлены, например, для визуализирующих исследований, в виде набора, содержащего две отдельных дозировки с инструкциями по их смешиванию: одну - представляющую собой радионуклид, а другую - свободное хелатное соединение. Для, радиотерапии предпочтительно, чтобы хелатное соединение по данному изобретению в свободной форме или в форме комплекса было представлено в виде горячего жидкого препарата.
| название | год | авторы | номер документа |
|---|---|---|---|
| ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ | 2004 |
|
RU2355418C2 |
| АНАЛОГИ СОМАТОСТАТИНА | 2001 |
|
RU2287533C2 |
| ПОЛУЧЕНИЕ ПЕПТИДОВ СОМАТОСТАТИНА | 2004 |
|
RU2360921C2 |
| КОМПОЗИЦИЯ С ЗАМЕДЛЕННЫМ ВЫСВОБОЖДЕНИЕМ, ВКЛЮЧАЮЩАЯ БИСФОСФОНАТ | 2005 |
|
RU2395274C2 |
| ДЕПО-ФОРМА АНАЛОГА СОМАТОСТАТИНА И СПОСОБ ЕЕ ПОЛУЧЕНИЯ | 2005 |
|
RU2411031C2 |
| КОМБИНАЦИЯ АНАЛОГОВ СОМАТОСТАТИНА С РАЗЛИЧНОЙ СЕЛЕКТИВНОСТЬЮ В ОТНОШЕНИИ ПОДТИПОВ РЕЦЕПТОРОВ СОМАТОСТАТИНА ЧЕЛОВЕКА | 2007 |
|
RU2451520C2 |
| МИКРОЧАСТИЦЫ, СОДЕРЖАЩИЕ АНАЛОГИ СОМАТОСТАТИНА | 2004 |
|
RU2404749C2 |
| ХИМЕРНЫЕ АНАЛОГИ ЛИГАНДОВ СОМАТОСТАТИНОВЫХ И ДОПАМИНОВЫХ РЕЦЕПТОРОВ, ФАРМАЦЕВТИЧЕСКИЕ КОМПОЗИЦИИ НА ИХ ОСНОВЕ И СПОСОБЫ ВОЗДЕЙСТВИЯ НА РЕЦЕПТОРЫ СОМАТОСТАТИНА И/ИЛИ ДОПАМИНА | 2004 |
|
RU2329273C2 |
| ФАРМАЦЕВТИЧЕСКИЕ КОМПОЗИЦИИ | 2009 |
|
RU2504360C2 |
| ПЕПТИДНЫЕ ВЕКТОРЫ | 2004 |
|
RU2361876C2 |



Описывается новый циклический гексапептид формулы (II) цикло [А - Zzа - (D/L)Trp - Lys - Х1 - Х2] (II), где Х1 означает радикал формулы (а) или (б), где R1 означает необязательно замещенный фенил; R2 означает -Z1-СН2-R1, -СН2-СО-О-СН2-R1; или (с) или (d), где Z1 означает О или S; Х2 представляет собой α-аминокислоту, имеющую ароматический остаток на боковой цепи Cα, или является аминокислотным звеном Dаb; А представляет собой двухвалентный остаток, выбранный из Pro,  где R3 представляет собой NR8R9-(С2-С6)алкилен или гуанидино (С2-С6)алкилен; R4 представляет собой водород или СН3; R11 представляет собой необязательно замещенный в кольце бензил, -(СН2)1-3 - ОН или -(СН2)1-5 -NН2; Rb представляет собой -(СН2)1-3; каждый из R8 и R9 независимо друг от друга представляют собой водород, (С1-С4)алкил, ацил или СН2ОН -(СНОН)с-СН2-, в котором с = 0, 1, 2, 3 или 4, или R8 и R9 образуют вместе с атомом азота, к которому они присоединены, гетероциклическую группу, которая может содержать дополнительный гетероатом, например пиридил или морфолино; Zzа представляет собой звено природной или неприродной α-аминокислоты, выбранной из группы, включающей Ala, Val, Thr, Ser, Leu, Nle, Jle, His, Trp, Arg, Tyr, Phe и NН2-Phe, в свободной форме или в форме соли или комплексного соединения. Соединения обладают сродством по отношению к рецепторам соматостатина. 2 с. и 5 з.п.ф-лы, 2 табл.
где R3 представляет собой NR8R9-(С2-С6)алкилен или гуанидино (С2-С6)алкилен; R4 представляет собой водород или СН3; R11 представляет собой необязательно замещенный в кольце бензил, -(СН2)1-3 - ОН или -(СН2)1-5 -NН2; Rb представляет собой -(СН2)1-3; каждый из R8 и R9 независимо друг от друга представляют собой водород, (С1-С4)алкил, ацил или СН2ОН -(СНОН)с-СН2-, в котором с = 0, 1, 2, 3 или 4, или R8 и R9 образуют вместе с атомом азота, к которому они присоединены, гетероциклическую группу, которая может содержать дополнительный гетероатом, например пиридил или морфолино; Zzа представляет собой звено природной или неприродной α-аминокислоты, выбранной из группы, включающей Ala, Val, Thr, Ser, Leu, Nle, Jle, His, Trp, Arg, Tyr, Phe и NН2-Phe, в свободной форме или в форме соли или комплексного соединения. Соединения обладают сродством по отношению к рецепторам соматостатина. 2 с. и 5 з.п.ф-лы, 2 табл.

или


где X1 (означает радикал формулы (а) или (б),
или
где R1 означает необязательно замещенный фенил;
R2 означает -Z1-CH2-R1, -CH2-CO-O-CH2-R1;

где Z1 означает O или S;
X2 представляет собой α-аминокислоту, имеющую ароматический остаток на боковой цепи Cα, или является аминокислотным звеном Dab;
A представляет собой двухвалентный остаток, выбранный из Pro,  где R3 представляет собой NR8R9-(C2-C6)алкилен или гуанидино(C2-C6)алкилен;
где R3 представляет собой NR8R9-(C2-C6)алкилен или гуанидино(C2-C6)алкилен;
R4 представляет собой водород или CH3;
R11 представляет собой необязательно замещенный в кольце бензил, -(CH2)1-3-OH или -(CH2)1-5-NH2;
Rb представляет собой -(CH2)1-3;
каждый из R8 и R9 независимо друг от друга представляют собой водород, (C1-C4)алкил, ацил или CH2OH-(CHOH)c-CH2-, в котором c = 0, 1, 2, 3 или 4, или R8 и R9 образуют вместе с атомом азота, к которому они присоединены, гетероциклическую группу, которая может содержать дополнительный гетероатом, например, пиридил или морфолино;
ZZa представляет собой звено природной или неприродной α-аминокислоты, выбранной из группы, включающей Ala, Val, Thr, Ser, Leu, Nle, Jle, His, Trp, Arg, Tyr, Phe и NH2-Phe,
в свободной форме или в форме соли или комплексного соединения.
| "Способ получения альбумина,меченного технецием тс-99 4 | 1975 |
|
SU581945A1 |
| Способ получения производных соматостатина | 1974 |
|
SU586837A3 |
| ПНЕВМАТИЧЕСКОЕ ВЫЧИСЛИТЕЛЬНОЕ УСТРОЙСТВО | 0 |
|
SU203031A1 |
| Устройство для автоматической стабилизации уровней полного сигнала цветного телевидения системы секам | 1974 |
|
SU515313A1 |
| Гидроимпульсатор | 1976 |
|
SU657174A1 |
| Прибор для очистки паром от сажи дымогарных трубок в паровозных котлах | 1913 |
|
SU95A1 |
