Изобретение относится к парентеральным фармацевтическим композициям, включающим аналог соматостатина и новые аналоги соматостатина.
Соматостатин является тетрадекапептидом, имеющим структуру

После выделения и описания соматостатина продолжаются интенсивные поиски более мощных и более стабильных его аналогов.
Аналоги соматостатина описаны, например, в WO 97/01579. Указанные аналоги соматостатина включают последовательность аминокислот формулы I

в которой X1 обозначает радикал формулы (а) или (б)
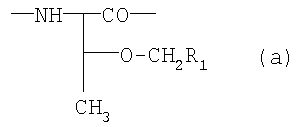

где R1 обозначает необязательно замещенный фенил,
R2 обозначает -Z1-CH2-R1, -CH2-CO-О-CH2-R1,
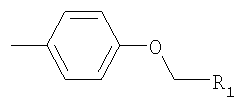 или
или 
где Z1 обозначает О или S, и
Х2 обозначает α-аминокислоту, имеющую ароматический остаток в боковой цепи Сα, или аминокислотную единицу, выбранную из Dab, Dpr, Dpm, His, (Bzl)HyPro, тиенил-Ala, циклогексил-Ala и тетра-бутл-Ala, остаток Lys в указанной последовательности соответствует остатку Lys9 в нативном соматостатине-14.
В контексте настоящего изобретения под аналогом соматостатина подразумевается пептид с прямой цепью или циклический, являющийся производным природного пептида соматостатина-14 и включающий последовательность формулы I, в котором дополнительно один или несколько аминокислотных единиц были изъяты и/или замещены одним или несколькими другими аминокислотными радикалами, и/или в котором одна или несколько функциональных групп были замещены одной или несколькими другими функциональными группами, и/или одна или несколько групп были замещены одной или несколькими другими изостерическими группами. В целом данное понятие относится ко всем модифицированным производным нативного соматостатина-14, включающим указанную выше последовательность формулы I, которые обладают связывающим сродством в наномолярном диапазоне, по меньшей мере, к одному подтипу рецептора соматостатина, описанному ниже.
Предпочтительными являются аналоги соматостатина, у которых остатки в положениях с 8 по 11 соматостатина-14 представлены последовательностью формулы I, приведенной выше.
Более предпочтительными являются приведенные выше аналоги соматостатина, включающие гексапептидную единицу, остатки в положениях с 3 по 6 указанной гексапептидной единицы, включающие последовательность формулы I. Еще более предпочтительно, если остатки в положениях 1 и 2 гексапептидной единицы гексапептида соматостатина могут быть любыми известными в данной области, например, описанными A.S.Dutta в кн.: «Small Peptides», изд-во Elsevier, 1993, т. 19, сс.292-354, или заместителями для Phe6 и/или Phe7 соматостатина-14.
Еще более предпочтительными являются циклические гексапептиды соматостатина, например, циклические гексапептиды соматостатина, включающие гексапептидную единицу под номерами с 1 по 6, остатки в положениях с 3 по 6 указанной гексапептидной единицы, имеющие аминокислотную последовательность формулы I, приведенной выше, например, соединение формулы Ia

в которой обозначения X1 и Х2 соответствуют приведенным выше обозначениям, А обозначает двухвалентный остаток, выбранный из Pro,

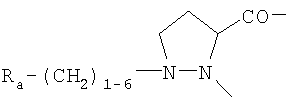


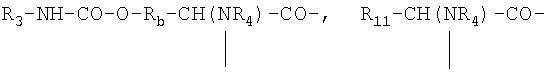 и
и 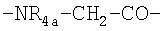
где R3 обозначает NR8R9-С2-6алкилен, гуанидино-С2-6алкилен или
С2-6алкилен-СООН, R3а обозначает Н, С1-4алкил или независимо имеет одно из значений, данных для R3, R3b обозначает Н или С1-4алкил, Ra обозначает ОН или
NR5R6, Rb обозначает -(CH2)1-3- или -СН(СН3)-, R4 обозначает Н или СН3, R4a обозначает необязательно замещенный в кольце бензил, каждый из R5 и R6 независимо обозначает Н, С1-4алкил, ω-амино-С1-4алкилен, ω-гидрокси-С1-4алкилен или ацил, R7 обозначает простую связь или С1-6алкилен, каждый из R8 и R9 независимо обозначает Н, С1-4алкил, ω-гидрокси-С2-4алкилен, ацил или СН2OH-(СНОН)с-СН2-, где с обозначает 0, 1, 2, 3 или 4, или R8 и R9 образуют вместе с атомом азота, к которому они присоединены, гетероциклическую группу, которая может содержать дополнительный гетероатом, и R11 обозначает необязательно замещенный в кольце бензил, -(CH2)1-3-OH, СН3-СН(ОН)- или -(CH2)1-5-NR5R6, и ZZa обозначает природную или искусственную α-аминокислотную единицу.
Особенно предпочтительными являются соединения формулы II

в которой конфигурация в положении С-2 является (R) или (S) или их смесью, и где R обозначает NR1R2-C2-6алкилен или гуанидин-С2-6алкилен, и каждый из R1 и R2 независимо обозначает Н или С1-4алкил, в свободной форме, в форме соли или в защищенной форме.
Предпочтительно R обозначает NR1R2-C2-6алкилен. Предпочтительные соединения формулы II обозначают соединения, в которых R обозначает 2-аминоэтил, а именно цикло[{4-(NH2-C2H4-NH-CO-О-)Pro}-Phg-DTrp-Lys-Tyr(4-Bzl)-Phe] (упоминается в настоящей заявке как соединение А) и цикло[{4-(NH2-C2H4-NH-CO-О-)Pro}-DPhg-DTrp-Lys-Tyr(4-Bzl)-Phe], в свободной форме, в форме соли или в защищенной форме. Phg обозначает -HN-СН(С6Н5)-СО- и Bzl обозначает бензил.
Эти соединения в свободной форме, в форме соли или в защищенной форме упоминаются ниже как «соединения настоящего изобретения».
Из-за протеолитического распада аналогов соматостатина настоящего изобретения весьма желательна системная доставка, например, парентеральное введение. Однако парентеральное введение может быть очень болезненным в месте введения, особенно при повторном введении.
В настоящее время обнаружено, что парентеральные композиции, включающие соединение настоящего изобретения и винную кислоту, показывают особенно интересные свойства, например, хорошую переносимость и высокую стабильность.
Соединение настоящего изобретения в защищенной форме соответствует аналогу соматостатина, в котором, по меньшей мере, одна из аминогрупп защищена и которое после снятия защиты приводит к образованию соединения формулы II, предпочтительно физиологически элиминируемого. Приемлемые аминозащитные группы описаны, например, Т.W.Greene в кн.: «Protective Groups in Organic Synthesis», изд-во J.Wiley & Sons, Нью-Йорк, 1981, cc. 219-287, содержание которой приводится в настоящей заявке в виде ссылки. Примером такой аминозащитной группы является ацетил.
Соединение настоящего изобретения может существовать, например, в свободной форме или в форме соли. К солям относятся кислотные аддитивные соли, например, неорганических кислот, полимерных кислот или органических кислот, например, соляной кислоты, уксусной кислоты, молочной кислоты, аспарагиновой кислоты, бензойной кислоты, янтарной кислоты или памоиновой кислоты. Кислотные аддитивные соли могут существовать в качестве моно- и дивалентных солей, например, в зависимости от того, внесен 1 или 2 эквивалента кислоты. Предпочтительными солями являются лактат, аспартат, бензоат, сукцинат и памоат, в том числе моно- и ди- соли, более предпочтительны диаспартат и монопамоат.
Соединения настоящего изобретения могут приготовляться с помощью традиционных способов.
Одним из объектов настоящего изобретения является парентеральная композиция, включающая соединение настоящего изобретения и винную кислоту.
Согласно настоящему изобретению обычно концентрация соединения настоящего изобретения в композиции настоящего изобретения составляет примерно от 0,05 до примерно 1 мг/мл композиции, особенно от 0,1 до 1 мг/мл.
Удобно, когда отношение массы соединения настоящего изобретения (в количестве, соответствующем свободной форме) к массе винной кислоты составляет примерно от 0,001 до примерно 2, предпочтительно примерно от 0,05 до примерно 0,6.
Количество соединения настоящего изобретения в композиции настоящего изобретения составляет примерно от 0,005 мас.% до примерно 0,1 мас.% от общей массы состава.
Предпочтительно используется винная кислота в мелкоизмельченной кристаллической форме. Более предпочтительным является использование кристаллической D(-) или L(+) винной кислоты. Количество винной кислоты в составе предпочтительно составляет примерно от 0,01 мас. % до примерно 1,5 мас.%, предпочтительно примерно от 0,01 мас.% до примерно 0,3 мас.%, более предпочтительно примерно 0,15 мас.%. Предпочтительно молярность винной кислоты в итоговой композиции составляет примерно 10 мМ.
В соответствии с настоящим изобретением дополнительно к винной кислоте и соединению настоящего изобретения фармацевтическая композиция предпочтительно содержит также основной компонент, выбранный и добавленный в композицию таким образом, что величина рН фармацевтической композиции, определяемая винной кислотой, доводится до значения рН, составляющей примерно от 4 до примерно 4,5, предпочтительно примерно 4,2.
Предпочтительно основным компонентом является основание, например, гидроксид натрия или гидроксид калия, или основная соль, например, гидрокарбонат натрия, карбонат натрия, гидрокарбонат калия, карбонат калия. Предпочтительно основной компонент вносят в таком количестве, что образующаяся в результате этого фармацевтическая композиция имеет доведенное буфером значение рН, согласно указанному выше.
Предпочтительно фармацевтическая композиция настоящего изобретения приготавливается на водной основе.
Композиции настоящего изобретения могут дополнительно включать тонический агент, например, маннит, хлорид натрия, глюкозу, декстрозу, сахарозу или глицерины. Предпочтительно тоническим агентом является маннит.
Количество тонического агента выбирают таким образом, чтобы скорректировать изотоничность композиции настоящего изобретения, например, предпочтительно маннита может быть примерно от 1 мас.% до примерно 5 мас.%, предпочтительно примерно 4,95 мас.%. Удобно, когда маннит присутствует в соотношении маннита к винной кислоте в пропорции примерно от 20 до примерно 40, предпочтительно примерно 33.
Композиции настоящего изобретения могут содержать дополнительные эксципиенты, обычно применяемые в парентеральных композициях с целью обеспечения требуемой стабильности и терапевтической эффективности. К эксципиентам могут относиться, например, антиоксидант или консервант.
Антиоксиданты могут применяться для защиты действующего агента от окислительного распада, в частности, в условиях повышенной температуры при стерилизации. В качестве антиоксидантов может быть выбрано любое из соединений, известных в данной области. Сходным образом количество применяемого антиоксиданта может быть определено с помощью обычных экспериментов. Предпочтительно композиции настоящего изобретения не содержат антиоксиданта.
Консервант, например, фенол, может предпочтительно добавляться в композицию, когда она перерабатывается для многодозовых флаконов, картриджей или шприцов. Предпочтительно композиции настоящего изобретения не содержат консерванта.
Ссылки приводятся на большое количество литературы по данному вопросу для этих и других упоминаемых в настоящем изобретении эксципиентов и методик, в частности, в кн.: «Handbook of Pharmaceutical Excipients» под ред. Ainley Wade, Paul J.Weller, изд-во Американской фармацевтической ассоциации, Вашингтон, США и изд-во «Pharmaceutical Press», Лондон, 2-е изд.; в кн.: «Lexikon der Hilfsstoffe für Pharmazie, Kosmetik und angrenzende Gebiete», под ред. H.P.Fiedler, изд-во Cantor, Aulendorf, 4-е изд., а также в предыдущих изданиях, упоминаемых в настоящей заявке в виде ссылки.
Предпочтительно композиция настоящего изобретения содержит в качестве действующего ингредиента только соединение настоящего изобретения, например, соединение формулы II, например, соединение А.
Действия, которые могут применяться для приготовления композиций настоящего изобретения, могут быть обычными, или известными в данной области, или основывающимися на таких методиках, которые, описаны, например, в книге L. Lachman и др. «The Theory and Practice of Industrial Pharmacy», 1986, 3-е изд.; в книге Н. Sucker и др. «Pharmazeutische Technologie», Thieme, 1991; в кн.: «Hager′s Handbuch der pharmazeutischen Praxis», изд-во Springer Verlag, 1971, 4-е изд.; в кн.: «Remington′s Pharmaceutical Sciences», изд-во Mack Publ., Co., 1970, 13-е изд.; или в последующих изданиях.
Обычно соединение настоящего изобретения, винная кислота и необязательно другие ингредиенты, как упоминалось, в требуемом количестве, растворяются в водном растворителе, предпочтительно в воде для инъекций, и значение рН корректируется основанием. Получаемый раствор затем может разбавляться водой для получения итогового желаемого объема. Получаемый раствор может фильтроваться через стерильный фильтр, например, Millipak® фильтр. Предпочтительно в ходе описанного выше приготовления не допускается контакт кислорода (воздуха) с раствором соединения настоящего изобретения. Обычно это достигается продувкой, например, азота в содержащем раствор контейнере. Фармацевтическая композиция может быть упакована в атмосфере двуокиси углерода или другого инертного газа для предупреждения распада, предпочтительно в атмосфере двуокиси углерода, например, наполненного во флаконы, например стеклянные флаконы, ампулы, например стеклянные ампулы, или шприцы, например заполненные шприцы, и стерилизована паром или прогревом.
Раствор может быть высушен заморозкой обычным способом в асептических условиях для получения порошка для инъекций, который может применяться для восстановления требуемого раствора для парентерального введения незадолго перед введением путем смешивания порошка с необходимым количеством растворителя, например с водой для инъекций.
В еще одном варианте осуществления настоящего изобретении предусматривается другой вариант композиции для парентерального введения, забуференный при значении рН примерно от 4 до примерно 4,5, предпочтительно примерно 4,2, и включающий в качестве действующего ингредиента соединение А или его фармацевтически приемлемую соль, например, лактат, моно- или диаспартат, сукцинат, предпочтительно диаспартат.
Эти композиции могут включать те же компоненты, которые описаны выше для композиций, включающих винную кислоту, в которой винная кислота/тартрат замещены другим буфером, например, ацетатом/уксусной кислотой, лактатом/молочной кислотой и глицином/HCl.
Композиции настоящего изобретения применяются:
а) для предупреждения или лечения расстройств с этиологией, включающей или ассоциированной с избыточной GH-секрецией и/или с избытком IGF-1, например, при лечении акромегалии, а также при лечении сахарного диабета I или II типа, особенно его осложнений, например, ангиопатии, диабетической пролиферативной ретинопатии, диабетического макулярного отека, нефропатии, нейропатии и феномена «утренней зари», а также других метаболических расстройств, связанных с высвобождением инсулина или глюкагона, например, при ожирении, например, патологическом ожирении или гипоталамическом, или гиперинсулинемическом ожирении,
б) для лечения кишечно-кожных и панкреатико-кожных свищей, синдрома раздраженного кишечника, воспалительных заболеваний, например, базедовой болезни, воспалительного заболевания кишечника, псориаза или ревматоидного артрита, поликистозной болезни почек, демпинг-синдрома, синдрома водянистого стула, СПИД-ассоциированной диареи, индуцированной химиотерапией диареи, острого или хронического панкреатита и опухолей, секретирующих желудочно-кишечные гормоны (например, GEP-опухоли, например, випомас, глюкагономас, инсулиномас, карциноиды и т.п.), злокачественных лимфоцитов, например, лимфом или лейкемий, гепатоцеллюлярной карциномы, а также желудочно-кишечного кровотечения, например, варикозного кровотечения пищевода,
в) для предупреждения или лечения ангиогенеза, воспалительных расстройств, описанных выше, включая воспаления глаз, макулярного отека, например, кистоидного макулярного отека, идиопатического кистоидного макулярного отека, экссудативного возрастного макулярного перерождения, расстройств, связанных с хориоидальной реваскуляризацией, и пролиферативной ретинопатии,
г) для предупреждения или лечения заболеваний сосудистых трансплантатов, например, васкулопатии алло- или ксенотрансплантатов, например, атеросклероза трансплантированных сосудов, например, в трансплантатах органов, например, сердца, легких, сочетанных сердца - легких, печени, почек или поджелудочной железы, или для предупреждения или лечения стеноза венозного трансплантата, рестеноза и/или закупорки сосудов с последующим повреждением сосудов, например, вызванной процедурами катетеризации или процедурами васкулярного соскабливания, например, чрескожной транслюминальной ангиопластикой, обработкой лазером или другими инвазивными процедурами, которые нарушают целостность внутренней оболочки стенки сосудов или эндотелия,
д) для лечения опухолей, экспрессирующих или аккумулирующих рецепторы соматостатина, например, опухолей гипофиза, например, болезни Кушинга, гастроэнтеропанкреатита, карциноидов, центральной нервной системы, груди, простаты (включая прогрессирующий, не поддающийся лечению рак простаты), опухолей яичника или толстой кишки, мелкоклеточного рака легких, злокачественной обструкции кишечника, параганглиомы, рака почек, рака кожи, нейробластомы, феохромоцитомы, медуллярных карцином щитовидной железы, миелом, лимфом, лимфомы Ходкина и неходкинской лимфомы, опухолей костей и их метастаз, а также аутоиммунных и воспалительных расстройств, например, ревматоидного артрита, базедовой болезни или других воспалительных заболеваний глаз.
Предпочтительно композиции настоящего изобретения полезны для лечения акромегалии и рака, например, болезни Кушинга.
Активность и свойства композиций настоящего изобретения могут быть показаны с помощью стандартных клинических тестов или тестов на животных.
Подходящие дозы композиции настоящего изобретения, конечно, могут варьировать, например, в зависимости от состояния, требующего лечения (например, типа заболевания или природы устойчивости), применяемого лекарственного средства, ожидаемого эффекта и способа введения.
При постоянном применении эффективное количество лекарственного средства может вводиться в виде двух или трех доз, распределенных по времени, например, путем парентерального введения, например, внутривенозным капельным вливанием, внутримышечной или подкожной инъекцией (инъекциями), или подкожным вливанием, например, постоянным подкожным вливанием, предпочтительно подкожной инъекцией или вливанием, с полной суточной дозой, поделенной на порции, или за единый период введения. При введении подкожной инъекцией наиболее предпочтительно введение от 3 раз в неделю до 3 раз в сутки, предпочтительно от двух раз в неделю до двух раз в сутки. Соединение настоящего изобретения также может вводиться в форме, например, подкожной болюсной инъекции.
Предпочтительно композиция настоящего изобретения подходит для подкожного введения.
После инъекции композиция настоящего изобретения хорошо переносится местно. В частности, парентеральное введение композиции настоящего изобретения, например, подкожной инъекцией, в месте инъекции переносится спокойно или приводит к чувству жжения.
Помимо хорошей местной переносимости после инъекции, композиция настоящего изобретения проявляет свойства хорошей стабильности. Например, менее 2,5% продуктов распада было обнаружено после 4 недель хранения при 60°С. Например, если хранить, защищая от света при температуре от 2°С до 8°С, композиции настоящего изобретения стабильны на протяжении 24 месяцев. Особенно хорошая стабильность может наблюдаться у диаспартата соединения А.
Как правило, удовлетворительные результаты получают при введении, например, подкожном введении, доз в количестве примерно от 0,01 до примерно 1,2 мг, предпочтительно примерно от 0,1 до примерно 0,6 мг соединения настоящего изобретения на инъекцию, или примерно от 0,001 до примерно 0,009 мг/кг массы тела животного в сутки, введенных однократно или поделенных на дозы для приема до 4 раз в сутки. Приемлемые суточные дозы для пациентов, таким образом, составляют примерно от 0,1 до примерно 0,6 мг соединения настоящего изобретения, например, соединения формулы II, например, соединения А.
Приводящиеся ниже примеры иллюстрируют композиции настоящего изобретения.
Примеры 1-7.
Винную кислоту и маннит растворяют в воде для инъекций, при этом через раствор пропускают азот. Затем вносят диаспартат соединения А, раствор корректируют гидроксидом натрия до величины рН 4,20 и добавляют воду для инъекций до 1,0 мл. В асептических условиях раствор фильтруют через стерильный фильтр Millipak-200® с размером пор ≤0,22 мкм, после чего его вносят в ампулы и стерилизуют автоклавированием.
В качестве еще одного объекта настоящего изобретения предусматриваются новые соединения формулы III

в которой R обозначает NR1R2-С2-6алкилен или гуанидин-С2-6алкилен, и каждый из R1 и R2 независимо обозначает Н или С1-4алкил, в свободной форме, в форме соли или в форме комплекса, или в защищенной форме.
Предпочтительно R обозначает NR1R2-С2-6алкилен. Предпочтительным соединением формулы III является соединение, в котором R обозначает 2-амино-этил, также называемое цикло[{4-(NH2-C2H4-NH-CO-О-)Pro}-DPhg-DTrp-Lys-Tyr(4-Bzl)-Phe] и упоминаемое в настоящем описании как соединение Б, в свободной форме, в форме соли, или в форме комплекса, или в защищенной форме. Обозначения Phg и Bzl указаны выше. Эти соединения в свободной форме, в форме соли, или в форме комплекса, или в защищенной форме упоминаются ниже в качестве «новых соединений настоящего изобретения».
Соединение формулы III, например, соединение Б, в защищенной форме соответствует указанной выше молекуле, в которой, по меньшей мере, одна из аминогрупп является защищенной и которая в результате снятия защитной группы приводит к образованию соединения формулы III, предпочтительно физиологически элиминируемого. Приемлемые аминозащитные группы описаны, например, Т.W. Greene в кн.: «Protective Groups in Organic Synthesis», изд-во J.Wiley & Sons, Нью-Йорк, 1981, сс.219-287, содержание которых упоминается в настоящем изобретении в виде ссылок. Примером такой аминозащитной группы является ацетил.
Когда соединение формулы III, например, соединение Б, представлено в форме комплекса, оно может быть соединением формулы III, содержащим хелатирующую группу в боковой цепи аминогруппы Pro и образующим комплекс с выявляемым или радиотерапевтическим элементом. Соединение Б, содержащее хелатирующую группу, упоминается в настоящем описании в качестве конъюгирующего соединения Б.
Примерами хелатирующих групп являются, например, производные поли-аминополикарбоновых кислот или ангидридов, например, производных нециклических лигандов, например, диэтилентриаминпентауксусная кислота (DTPA), этиленгликоль- -0,0′- бис(2-аминоэтил)-N,N,N′,N′-тетрауксусная кислота (EGTA), N,N′-бис(гидроксибензил)этилендиамин-N,N′-диуксусная кислота (HBED) и триэтилентетраамингексауксусная кислота (ТТНА), являющаяся производным замещенной DTPA, например, пара-изотиоцианатобензил -DTPA, являющийся производным макроциклических лигандов, например, 1,4,7,10-тетра-азациклододекан-N,N′,N′′,N′′′-тетрауксусная кислота (DOTA), 1,4,8,11-тетра-азациклотетрадекан-N,N′,N′′,N′′′-тетрауксусная кислота (ТЕТА), или 1,4,7,10-тетра-азациклотридекан-N,N′,N′′,N′′′-тетрауксусная кислота (TITRA).
Хелатирующая группа присоединена непосредственно или через спейсер к боковой цепи аминогруппы Pro. К приемлемым спейсерам относятся известные в данной области, например, описанные в GB-A-2225579, например, двухвалентный остаток аминокарбоновой кислоты, например, β-Ala или двухвалентный остаток, полученный из 6-аминокапроновой кислоты.
Предпочтительными являются хелатирующие группы, производные DTPA, DOTA или ТЕТА. Хелатирующие группы, производные DTPA или DOTA, наиболее предпочтительны.
Понятие «выявляемый элемент» означает любой элемент, предпочтительно ион металла, который проявляет свойство, обнаруживаемое с помощью диагностической техники in vivo, например, ион металла, который испускает фиксируемую радиацию, или ион металла, который может влиять на релаксацию ЯМР-свойств. Понятие «радиотерапевтический элемент» означает любой элемент, который испускает радиацию, оказывающую благотворный эффект на состояние, подвергаемое лечению.
К приемлемым элементам относятся, например, тяжелые элементы или редкоземельные ионы, например, такие, которые используются в САТ-сканировании (компьютерная осевая томография), парамагнитные ионы, например, Gd3+, Fe3+, Mn2+ и Cr2+, флуоресцирующие ионы металла, например, Eu3+, и радионуклиды, например, радиолантаниды, в частности, γ-эмитирующий радионуклид, β-эмитирующий радионуклид, α-эмитирующий радионуклид, Оже-е--эмитирующий радионуклид или позитрон-эмитирующий радионуклид, например, 68Ga, 18F или 86Y.
К подходящим γ-эмитирующим радионуклидам относятся те, которые пригодны для методов диагностики, γ-эмитирующие радионуклиды преимущественно имеют период полураспада от 1 ч до 40 суток, предпочтительно от 5 ч до 4 суток, более предпочтительно от 12 ч до 3 суток. Примерами являются радиоизотопы галлия, индия, технеция, иттербия, рения, тербия, лютеция, таллия и самария, например, 67Ga, 111In, 99mTc, 161Tb, 169Yb, 186Re или 177Lu.
К приемлемым β-эмитирующим радионуклидам относятся те, которые подходят для радиотерапевтических целей, например, 90Y, 67Cu,186Re, 188Re, 169Er, 121Sn, 127Te, 177Lu, 143Pr, 198Au, 109Pd, 165Dy, 142Pr или l53Sm.
К приемлемым α-эмитирующим радионуклидам относятся те, которые используются в терапевтической практике, например. 211At, 212Bi или 201Tl.
Соединения формулы III, например, соединение Б, могут существовать, например, в свободной форме или в форме соли. К солям относятся кислотные аддитивные соли, например, соли неорганических кислот, полимерных кислот или органических кислот, например, соляной кислоты, уксусной кислоты, молочной кислоты, аспарагиновой кислоты, бензойной кислоты, янтарной кислоты или памоиновой кислоты. Кислотные аддитивные соли могут существовать в качестве моно- и дивалентных солей, например, в зависимости от того, добавлен 1 или 2 кислотных эквивалента к соединению Б в свободной основной форме. Предпочтительными солями являются лактаты, аспартаты, бензоаты, сукцинаты и памоаты, в том числе моно- и дисоли, более предпочтительны диаспартат и монопамоат.
Конъюгированные соединения формулы III, например, соединение Б, могут дополнительно существовать в форме солей, образуемых с группами карбоновых кислот, когда присутствуют в хелатирующих группах, например, соли щелочных металлов, например, натрия или калия, или замещенные или незамещенные соли аммония.
Настоящее изобретение также предусматривает способ получения соединения формулы III, например, соединение Б. Оно может быть получено по аналогии с известными способами, например:
а) циклизацией линейного пептида в защищенной, полимер-связанной или незащищенной форме таким путем, что получают соединение формулы III, например, соединение Б, и затем необязательно удаляют защитную группу(группы),
б) для получения конъюгированного соединения формулы III, например, конъюгированного соединения Б, связывают вместе хелатирующую группу и соединение формулы III, например, соединение Б, в защищенной или незащищенной форме и затем необязательно удаляют защитную группу и восстанавливают соединение формулы III, например, соединение Б, или конъюгированное соединение формулы III, например, конъюгированное соединение Б, получаемое таким образом в свободной форме, в форме соли или необязательного комплекса с обнаруживаемым радиотерапевтическим элементом.
Обычно не является принципиальным, какая аминокислота выбирается в положении на С-конце для начала образования пептидной цепи, поскольку линейный пептид будет циклизован, важно только, чтобы последовательность аминокислот в линейном пептиде соответствовала требуемому соединению формулы III, например, соединению Б. Однако могут быть другие факторы, которые могут отдавать предпочтение одной стартовой аминокислоте над другой. Когда соединение формулы III, например, соединение Б, получают твердофазным синтезом, первая аминокислота предпочтительно присоединяется к смоле, например, коммерчески доступной смоле на основе полистирола, через подходящий линкер, например, линкер, который может расщепляться в мягких условиях для сохранения в интактном состоянии защиты боковой цепи, например, SASRIN или необязательно замещенный линкер на основе тритила, например, 4-(гидроксидифенилметил)бензойная кислота, в которой одна из фенильных групп может необязательно быть замещенной, например, С1. Сборка определенной пептидной цепи может быть эффективным при использовании обычного метода, например, с использованием аминокислотных единиц, в которых концевые аминогруппы являются Fmoc-защищенными, имеющиеся аминогруппы боковых цепей защищены различными аминозащитными группами, например, Вос или СВО. Предпочтительно линейный пептид циклизуется таким образом, что образуется связь между Tyr(4-Bzl)-OH и Phe, например, Phe-{4-(NHR1-C2H4-NH-CO-O-)Pro}-DPhg-DTrp(R2)-Lys(ε-NHR3)-Tyr(4-Bzl)-OH или его функциональное производное, где каждый из R1, R2 и R3 является аминозащитной группой. Стадия циклизации а) может быть легко выполнена с помощью известных методов, например, через азид, активный сложный эфир, смешанный ангидрид или карбодиимид. Затем защитная группа снимается, например, расщеплением, например, с помощью трифторуксусной кислоты или путем гидрогенизации.
Циклизация пептида также может выполняться непосредственно на твердом носителе, первая аминокислота на Nα- и С-концах защищена от присоединения по боковой цепи, например, ε-амино-функция Lys или путем заякоривания остова молекулы. Линейная последовательность затем синтезируется методами стандартного твердофазного синтеза (SPPS). После снятия С-концевой защиты пептид циклизуется, например, согласно описанному выше. После этого циклический пептид отщепляется от смолы и теряет защиту.
При желании боковую цепь в Pro могут внедрять в аминокислоту до или после стадии циклизации пептида а). Так, Pro в качестве стартовой аминокислоты или стартовый линейный или циклический пептид, в котором в каждом случае Pro замещен в кольце ОН, может быть преобразован с целью получения соединения формулы III, например, соединения Б, или желаемой единицы Pro или соответствующего линейного пептида, соответственно, в котором Pro замещен NHR1-C2H4-NH-CO-O-.
Комплексообразование конъюгированного соединения формулы III, например, конъюгированного соединения Б, может быть произведено путем взаимодействия конъюгированного соединения формулы III, например, конъюгированного соединения Б, с соответствующим выявляемым или радиотерапевтическим элемент-несущим соединением, например, солью металла, предпочтительно водорастворимой солью. Реакция может проводиться по аналогии с известными методами, например, описанными Perrin в кн.: «Organic Ligand, Chemical Data», изд-во Pergamon Press, Нью-Йорк, 1982, серия 22; Krejcant, Tucker в Biophys. Biochem. Res. Com. 77. 1977, с.581; Wagner, Welch в J.Nucl. Med. 20, 1979, с.428.
Последующие примеры иллюстрируют новые соединения формулы III. Все значения температуры приводятся в °С.
Аббревиатуры:
Пример 8. цикло[{4-(NH2-C2H4-NH-CO-O-)Pro}-DPhg-DTrp-Lvs-Tyr(4-Bzl)-Phe]
А. Синтез Fmoc-Pro(4-OCO-NH-CH2-CH2-NH-Boc)-OH
Гидрохлорид метилового эфира L-гидроксипролина взаимодействует с Fmoc-Osu в водном 1,0 н. углекислом натрии/THF при комнатной температуре. После завершения реакции Fmoc-Pro(4-OH)-Ome выделяют осаждением. Затем Fmoc-Pro(4-OH)-Ome добавляют по каплям в раствор трифосгена (0,6 эквивалента) в THF для получения хлоркарбоната промежуточного соединения. Через 1 ч вносят диметиламинопиридин (1,0 эквивалент) и N-Boc-диаминоэтан (6,0 эквивалентов) и реакцию проводят при перемешивании при комнатной температуре. После завершения реакции растворитель удаляют in vacuo и образуемый Fmoc-Pro(4-OCO-NH-CH2-CH2-NH-Boc)-Ome экстрагируют из двухфазной системы этилацетат/0,1 М НС1 для получения сырца продукта (МН+=554), который очищают кристаллизацией из этилацетата. Затем метиловый эфир удаляют с образованием свободной кислоты путем обработки 1 н. NaOH в диоксане/воде и продукт Fmoc-Pro(4-OCO-NH-CH2-CH2-NH-Boc)-OH очищают на силикагеле, [(M+Na)]+=562.
Б. H-Phe-Pro(4-OCO-NH-CH2-CH2-NH-Boc)-DPhg-DTrp(Boc)-Lvs(Boc)-Tyr(Bzl)-OH
Коммерчески доступную Fmoc-Tyr(Bzl)-O-CH2-Ph(3-OCH3)-O-CH2-полистирольную смолу (SASRIN-смола, 2,4 ммоля) применяют в качестве исходного материала и выполняют стандартные действия, состоящие из повторяющихся циклов по снятию защиты с Nα (пиперидин/DMF, 2:8), повторяющихся промываний DMF и присоединений (DIPCI: 4,8 ммолей/НОВТ: 6 ммолей, DMF). Следующие аминокислотные производные последовательно соединяются: Fmoc-Lys(Boc)-OH, Fmoc-DTrp(Boc)-OH, Fmoc-DPhg-OH, Fmoc-Pro(4-OCO-NH-CH2-CH2-NH-Boc)-OH, Fmoc-Phe-OH. Присоединения (2 эквивалента аминокислот) продолжаются или повторяются до завершения реакции, т.е. до полного исчезновения остаточных аминокислотных групп, мониторинг которых осуществляют по отрицательному нингидриновому Кайзер-тесту. Перед снятием полностью синтезированного защищенного линейного пептида со смолы снимают Nα-Fmoc защиту с последнего остатка.
B. H-Phe-Pro(4-OCO-NH-CH2-CH2-NH-Boc)-DPhg-DTrp(Boc)-Lys(Boc)-Tyr(Bzl)-ОН
После промывания СН2Cl2 соединенные пептид-смолу переносят на колонку или вакуум-фильтр с мешалкой и фрагмент пептида удаляют и элюируют с помощью краткой обработки 2% TFA в СН2Cl2 в течение 1 ч. Элюат немедленно нейтрализуют насыщенным раствором NaHCO3. Органический растворитель отделяют и упаривают, а предшественник с защищенной боковой цепью (МН+=1366) циклизируют без последующей очистки.
Г. Трифторацетат цикло[-Pro(4-ОСО-NH-СН2-СН2-NH2)-DPhg-DTrp-Lys-Tyr(Bzl)-Phe-]
Указанный выше линейный фрагмент растворяют в DMF (4 ммолях), охлаждают до минус 5°С и обрабатывают 2 эквивалентами DIPEA, затем 1,5 эквивалентами DPPA и перемешивают до завершения реакции (примерно 20 ч) при 0-4°С. Растворитель почти полностью удаляют in vacua; концентрат разводят этилацетатом, промывают NaHCO3, водой, сушат и упаривают in vacua.
Для снятия защиты остаток растворяют при 0°С в TFA/H2O 95:5 (примерно 50 ммолей) и перемешивают на холоде в течение 30 мин. Затем продукт осаждают эфиром, содержащим примерно 10 эквивалентов HCl, фильтруют, промывают эфиром и сушат. Для того чтобы полностью разрушить остающуюся индол-N-карбаминовую кислоту, продукт растворяют в 5% АсОН и лиофилизируют после 15 ч примерно при 5°С. Препаративную RP-ВЭЖХ проводят на колонке С-18 10 мкм STAGROMA (5-25 см), используя градиент от 0,5% TFA до 0,5% TFA в 70% ацетонитриле. Фракции, содержащие чистое основное вещество, объединяют, разводят водой и лиофилизируют. Лиофилизат растворяют в воде, после чего осаждают 10% Na2СО3 в воде. Твердое свободное основание отфильтровывают, промывают водой и высушивают в вакууме при комнатной температуре. Образовавшийся белый порошок непосредственно используют для получения различных солей.
Пример 9. цикло[{4-(NH2-C2H4-NH-CO-O-)Pro}-DPhg-DTrp-Lys-Tyr(4-Bzl)-Phe] в форме соли
А. Ацетат
Преобразование в форму соли уксусной кислоты производят с использованием ионообменной смолы (например, AG 3-X4). MS (ESI):m/z 524,5 [М+2Н]2+
[α]D 20=-41,6°; с=0,56; АсОН 95%; Т=20°С; 589,3 нм
Б. Аспартат
Преобразование в моно- или диаспартат проводят путем реакции 1 эквивалента соединения из примера 8 с 1 или 2 эквивалентами аспарагиновой кислоты в смеси ацетонитрил/вода 1:3. Образующуюся смесь замораживают и лиофилизируют.
Диаспартат также могут получать путем растворения соединения из примера 8 в воде/ацетонитриле 4:1, фильтрации, нагрузки на ионообменную смолу, например, в колонку BioRad AG4X4, и элюции водой/ацетонитрилом 4:1. Элюат концентрируют, замораживают и лиофилизируют.
В. Бензоат
Преобразование в бензоат могут производить путем растворения соединения из примера 8 в 2 эквивалентах бензойной кислоты в смеси ацетонитрил/вода 1:2. Образующуюся смесь замораживают и лиофилизируют.
Г. Памоат
1 эквивалент соединения из примера 8 растворяют вместе с 1 эквивалентом эмбоновой кислоты в смеси ацетонитрил/THF/вода 2:2:1. Образующуюся смесь замораживают и лиофилизируют.
Пример 10. Цикло[{4-(DOTA-NH-C2H4-NH-CO-O-)Pro}-DPhg-DTrp-Lys-Tyr(4-Bzl)-Phe
а) Трифторацетат цикло[-Pro(4-OCO-NH-CH2-CH2-NH2)-DPhg-DTrp-Lys(Cbo)-Tyr(Bzl)-Phe-]
Соединение синтезируют тем же способом, что и трифторацетат цикло[-Pro(4-OCO-NH-CH2-CH2-NH2)-DPhg-DTrp-Lys(Cbo)-Tyr(Bzl)-Phe-], используя Fmoc-Lys(Cbo)-OH вместо Fmoc-Lys(Boc)-OH.
б) 400 мг коммерчески доступного DOTA×2H2O (фирма SYMAFEX, Франция) растворяют в 20 мл воды. После добавления 20 мл DMF вносят 170 мг цикло[-Pro(4-OCO-NH-CH2-CH2-NH2)-DPhg-DTrp-Lys(CBO)-Tyr(Bzl)-Phe-] вместе со 190 мг DCCI и 60 мг N-гидроксисукцинимида. Образующуюся суспензию хранят при комнатной температуре в течение 72 ч. После фильтрации растворитель удаляют при пониженном давлении и остающийся сырец очищают на силикагеле (DCM/MeOH/HOAc50% 8/2/0,25→7/3/1 в качестве подвижной фазы).
в) Для снятия защиты указанный выше DOTA-конъюгат обрабатывают 5 мл трифторуксусной кислоты/тиоанизола (9/1) в течение 2 ч при комнатной температуре. После этого раствор вливают в смесь 100 мл диэтилового эфира +5 мл 3 н. HCl/диэтилэфира и образующийся осадок отделяют фильтрацией. Очистку проводят на силикагеле, используя DCM/MeOH/HOAc50% 7/4/2→7/5/4 в качестве подвижной фазы. Чистый для анализа конечный продукт получают после стадии обессоливания, используя градиент от 0,1% TFA до 0,1% TFA в 90 % CH3CN на RP18-HPLC колонке (Spherisorb 250×4,6 мм). МН+: 1434,7.
Соединения формулы III, например, соединение Б, в свободной форме или в форме фармацевтически приемлемых солей и комплексов, проявляют полезные фармакологические свойства, что было показано в тестах in vitro и in vivo, и поэтому показаны для терапии.
Более предпочтительные соединения формулы III, например, соединение Б, проявляют интересный профиль связывания по отношению к рецепторам соматостатина человека (hsst). 5 подтипов рецепторов соматостатина, sst1, sst2, sst3, sst4 и sst5, клонировали и описали. Рецепторы hsst1, hsst2 и hsst3, а также их последовательности, описаны Y. Yamada и др. в Proc. Nat. Acad. Sci., 89, 1992, cc.251-255. Рецептор hsst4 и его последовательность описаны L. Rohrer и др. в Proc. Acad. Sci., 90, 1993, cc.4196-4200. Рецептор hsst5 и его последовательность описаны R. Panetta и др. в Mol. Pharmacol. 45, 1993, cc.417-427.
Исследования связывания могут проводить согласно описанному в настоящем изобретении способу с применением мембран клеток клеточных линий, которые экспрессируют селективно и стабильно hsst1, hsst2, hsst3, hsst4 или hsst5, например, клеток СНО или COS.
Мембраны приготавливают известными методами, например, описанными С.Bruns и др. в Biochem. J. 65, 1990, cc.39-44. Мембраны приготавливают из hsst-селективных клеточных линий, например, клеток СНО или COS, стабильно экспрессирующих hsst1, или hsst2, или hsst3, или hsst4, или hsst5, которые инкубируют в трех повторностях в общем объеме 300 мкл при 22°С в течение 30 мин при возрастающих концентрациях [125I-Tyr11]-SRIF-14 в количестве 10 ммолей/л Hepes буфера (рН 7,6), содержащего 0,5% БСА. Инкубирование заканчивают быстрой фильтрацией и фильтры обсчитывают в счетчике. Специфическое связывание составляет общее связывание минус неспецифическое связывание в присутствии 1 мкмоля/л соматостатина-14. Эксперименты проводят в трех повторностях. Константу связывания (КD) и число сайтов связывания подсчитывают с применением соответствующих статистических и графических программ.
Соединения формулы III, например, соединение Б, не обладают существенной способностью к связыванию в описанных выше экспериментах по связыванию по отношению к hsst1, hsst2 и hsst4, низкой способностью к связыванию по отношению к hsst3 и хорошей способностью к связыванию по отношению к hsst5, проявляемой в виде значения IC50 в наномолярном диапазоне (IC50=концентрация, соответствующая половине максимального ингибирования при исследовании конкурентного связывания с применением [125I-Tyr11]-SRIF-14 в качестве специфического радиолиганда).
IC50
Соединения формулы III, например, соединение Б, демонстрируют активность по ингибированию GH-высвобождения, которая проявляется в виде ингибирования высвобождения GH in vitro из культивируемых клеток гипофиза. Например, передние доли гипофиза взрослых самцов крыс нарезают на мелкие кусочки и диспергируют, используя 0,1 % трипсин в 20 мМ буфере HEPES. Диспергированные клетки культивируют в течение четырех суток в среде MEM (фирма Gibco), дополнительно содержащей 5 % сыворотки теленка, 5% сыворотки лошади, 1 ммоль NaHCO3, 2,5 нмоля дексаметазона, 2,5 мг/мл инсулина и 20 ЕД/мл пенициллина/стрептомицина. В день эксперимента прикрепившиеся клетки промывают два раза средой Крэбса-Рингера, забуференной 20 мМ HEPES и содержащей добавки 5 ммолей глюкозы и 0,2 % БСА. Затем клетки инкубируют в течение 3 ч с соединением Б в присутствии 3×10-10 молей фактора высвобождения гормона роста. Количество гормона роста, поступающее в среду, измеряют с помощью RIA.
Соединения формулы III, например, соединение Б, ингибируют высвобождение гормона роста (GH) у крыс. Соединение Б вводят подкожно анестезированным крысам. Через 1 ч после введения соединения животных обезглавливают и собирают кровь. Длительность действия оценивают на основании ингибирования базальной GH-секреции через 6 ч после обработки лекарственным средством. Уровни гормонов определяют с помощью RIA через 1 и 6 ч после лечения. Значения ID50 для подавления секреции гормона определяют графически (log-пробит) для каждого эксперимента и получаемые значения усредняют логарифмически. В данной модели in vivo соединение Б ингибирует высвобождение гормона роста.
Соединения формулы III, например, соединение Б, также применимы для лечения hsst3- и/или hsst5-рецептор-позитивных опухолей, что показано в тестах на пролиферацию с различными линиями опухолевых клеток, несущих hsst3 и/или hsst5.
Соединения формулы III, например, соединение Б, соответственно применимы для предупреждения или лечения расстройств с этиологией, включающей или ассоциированной с присутствием или активацией hsst3 и/или hsst5, например, расстройств или заболеваний, связанных с повышенной GH-секрецией, например, при лечении акромегалии или для лечения заболеваний злокачественной пролиферации клеток, например, раковых опухолей, несущих hsst3 и/или hsst5, например, описанных ниже в связи с комплексным конъюгированным соединением Б.
Для всех указанных выше показаний требуемая доза варьирует в зависимости, например, от хозяина, способа введения и тяжести состояния, подвергаемого лечению. Однако обычно удовлетворительные результаты получают путем введения примерно от 1 мкг до 0,7 мг/кг/сутки соединения формулы III, например, соединения Б. Выявленная суточная доза для пациентов находится в диапазоне примерно от 2 мкг до примерно 50 мг, предпочтительно примерно от 0,01 до примерно 40 мг, например, примерно от 0,01 до примерно 3 мг при подкожном введении соединения, которое удобно вводить при разделении суточной дозы до трех введений в сутки в единой дозовой форме, содержащей, например, примерно от 0,5 мкг до примерно 25 мг, например, примерно от 2 мкг до 20 мг, например, примерно от 2 мкг до 1,5 мг соединения формулы III, например, соединения Б.
Соединения формулы III, например, соединение Б, может вводить в свободной форме или в форме фармацевтически приемлемой соли или комплексов. Такие соли и комплексы могут приготовлять обычным образом, они проявляют ту же активность, что и соединение в свободной форме. Настоящее изобретение также предусматривает фармацевтическую композицию, включающую соединение формулы III, например, соединение Б, в свободной форме или в форме фармацевтически приемлемой соли или комплексной форме, вместе с одним или несколькими фармацевтически приемлемыми растворителями или носителями. Такие композиции могут перерабатываться традиционным способом. Соединения формулы III, например, соединение Б, также могут вводиться в форме замедленного высвобождения, например, в форме имплантатов, микрокапсул, микросфер или наносфер, включающих, например, биодеградируемый полимер или сополимер, в форме липосомального состава, или в форме аутогеля, например, твердой или полутвердой композиции, способной образовывать гель после взаимодействия с тканевыми жидкостями организма пациента.
Соединения формулы III, например, соединение Б, или его фармацевтически приемлемая соль или комплекс, могут вводить любым обычным способом, например, парентерально, например, в форме растворов или суспензий для инъекций (включая, например, форму замедленного высвобождения, описанную выше), орально, используя обычный усилитель абсорбции, в назальной или суппозитарной форме, или местно, например, в форме глазной жидкости, геля, мази, или суспензионного препарата, например, липосомального, в виде состава микросфер или наносфер, например, для закапывания или субконъюнктивальных, или внутриглазных, или окологлазных инъекций.
В связи со сказанным выше настоящее изобретение также предусматривает:
1) соединение формулы III, например, соединение Б, или его фармацевтически приемлемую соль или комплекс для применения в качестве фармацевтического средства;
2) способ предупреждения или лечения заболеваний или расстройств, приводимых в настоящем изобретении, у субъекта, нуждающегося в таком лечении, который включает введение указанному субъекту эффективного количества соединения формулы III, например, соединения Б, или его фармацевтически приемлемой соли или комплекса; или
3) соединение формулы III, например, соединение Б, или его фармацевтически приемлемую соль или комплекс, для применения в приготовлении фармацевтической композиции для использования в любом способе, согласно описанному выше в п.2.
Конъюгированное соединение формулы III, например, соединение Б, или его фармацевтически приемлемая соль применимо в качестве визуализирующего агента, например, для визуализации тканей и клеток, позитивных в отношении рецепторов hsst3 и/или hsst5, например, опухолей и метастаз, позитивных в отношении рецепторов hsst3 и/или hsst5, воспалительных или аутоиммунных расстройств, проявляющих рецепторы соматостатина, туберкулеза или отторжения органов после трансплантации, когда образуется комплекс с выявляемым элементом, например, γ- или позитрон-эмитирующим нуклидом, флуоресцирующим ионом металла или парамагнитным ионом, например. 111In, 161Tb, 177Lu, 86Y, 68Ga, Eu3+, Gd3+, Fe3+, Mn2+ или Cr2+, или в качестве радиофармацевтического средства для лечения in vivo hsst3- и/или hsst5-рецептор-позитивных опухолей и метастаз, ревматоидного артрита и тяжелых воспалительных состояний, когда формируется комплекс с α- или β-эмитирующим нуклидом или нуклидом с Оже-е--каскадами, например, 90Y, 161Tb, 177Lu, 211At, 213Bi или 201Tl, что было показано с помощью стандартных тестов.
В частности было показано, что конъюгированное соединение Б связывается с рецепторами соматостатина со значениями pKi примерно от 8 до 10. Соединение примера 10 образует комплекс, например, с 111In, 88Y, 90Y или 177Lu, который связывается в наномолярном диапазоне с соответствующими подтипами sst в соответствии с профилем связывания соединения Б.
Связывание конъюгированного соединения формулы III, например, конъюгированного соединения Б и его комплексов с рецепторами hsst3 и/или hsst5 также может быть показано в опытах in vivo, соответствующих стандартным тест-методам, например, описанным в GB-A-2225579. Например, соединение из примера 10 в комплексе, например, с 111In, 88Y, 90Y или 177Lu, существенно аккумулируется в опухоли через 4 ч после инъекции мыши или крысе, у которых имеются экзокринные панкратические опухоли, экспрессирующие hsst5-рецепторы.
После введения конъюгированного соединения формулы III, например, конъюгированного соединения Б, в форме комплекса, например, радиомеченого 111In, 177Lu, 86Y или 161Tb в дозе от 1 до 5 мкг/кг, меченного в количестве от 0,1 до 5 мкюри радионуклидом, предпочтительно от 0,1 до 2 мкюри, место расположения опухоли становится выявляемым.
Конъюгированное соединение формулы III, например, конъюгированное соединение Б, при внесении радиометки в виде α- или β-радионуклида или нуклида с Оже-е--каскадами оказывает антипролиферативный и/или цитотоксический эффект на клетки опухоли, несущие hsst3- и/или hsst5-рецепторы, например, согласно показанному в опытах на голых мышах.
Голых мышей инокулируют опухолевыми клетками с рецепторами hsst5. Когда опухоли достигают размера 1-2 см3, животных рандомизированно разделяют на контрольную группу и группу лечения. Конъюгированное соединение формулы III, например, конъюгированное соединение Б в форме комплекса, вводят внутрибрюшинными или внутривенными инъекциями. Мышам вводят дозы, составляющие до 40 мкюри/кг. Размер опухолей определяют с помощью циркуля согласно описанному выше. Для статистических подсчетов применяют t-тест Стьюдента. В этом тесте кратковременное уменьшение опухоли, которое наблюдается через неделю роста опухоли, замедляется на две недели в результате одной аппликации соединения из примера 10 в комплексе с 90Y или 177Lu. Напротив, в контрольных группах наблюдается непрерывный рост опухолей, объем которых удваивается примерно каждые семь суток.
Таким образом, в сериях специфических или взаимоисключающих вариантов осуществления настоящего изобретения также предусматривается:
4. Применение конъюгированного соединения формулы III, например, конъюгированного соединения Б, в комплексе с выявляемым элементом для детекции in vivo hsst3- и/или hsst5-позитивных клеток и тканей, например, hsst3-или hsst5-позитивных опухолей и метастаз, у субъекта и распознавания локализации рецепторов, являющихся мишенями указанного комплекса.
5. Способ обнаружения in vivo hsst3- и/или hsst5-позитивных тканей и клеток, например, hsst3- или hsst5-позитивных опухолей и метастаз, у субъекта, включающий введение указанному субъекту конъюгированного соединения формулы III, например, конъюгированного соединения Б, в комплексе с выявляемым элементом, или его фармацевтически приемлемой солевой формы, и записи локализации рецепторов, на которые нацелен указанный комплекс.
Конъюгированное соединение формулы III, например, конъюгированное соединение Б, в форме комплекса для использования в качестве визуализирующего агента может быть введено, например, внутривенно, например, в форме инъекционных растворов или суспензий, предпочтительно разовой инъекции. Установление радиометки предпочтительно может быть незадолго до введения субъекту.
У животных показанные дозы могут варьировать от 0,01 до 1 мкг/кг конъюгированного соединения формулы III, например, конъюгированного соединения Б, в комплексе с 0,02-0,5 мкюри γ-эмитирующим радионуклидом. У более крупных млекопитающих, например, у человека, указанный диапазон доз может быть от 1 до 100 мкг/м2 конъюгированного соединения Б в комплексе, например, с 1-100 мкюри/м2 выявляемым элементом, например, 111In, 86Y или 177Lu.
6. Применение конъюгированного соединения формулы III, например, конъюгированного соединения Б, в комплексе с α- или β-эмитирующим нуклидом или с нуклидом с Оже-е--каскадами, для лечения in vivo hsst3- и/или hsst5-позитивных опухолей и метастаз.
7. Способ для лечения in vivo hsst3- и/или hsst5-позитивных опухолей и метастазов, например, для лечения инвазивности таких опухолей или симптомов, ассоциированных с таким опухолевым ростом, у субъекта, нуждающегося в таком лечении, которое включает введение указанному субъекту терапевтически эффективного количества конъюгированного соединения формулы III, например, конъюгированного соединения Б, в комплексе с α- или β-эмитирующим нуклидом или нуклидом с Оже-е--каскадами.
8. Применение конъюгированного соединения формулы III, например, конъюгированного соединения Б, или его фармацевтически приемлемой соли для получения визуализирующего агента или радиофармацевтической композиции.
Дозировки, применяемые в радиотерапевтической практике согласно настоящему изобретению, безусловно, варьируют в зависимости, например, от конкретного состояния, которое предстоит лечить, например, известную радиотоксичность по отношению к здоровым органам, экспрессирующим hsst5, объему опухоли и необходимого лечения. Обычно доза рассчитывается исходя из фармакокинетики и данных по распределению радиоактивности, полученных на здоровых органах и основывающихся на наблюдавшемся целевом потреблении, β-эмитирующий комплекс конъюгированного соединения формулы III, например, конъюгированного соединения Б, могут вводить многократно, например, на протяжении периода от 1 до 3 месяцев.
У животных указанный диапазон доз может составлять от 20 до 100 мкг/кг конъюгированного соединения формулы III, например, конъюгированного соединения Б, в комплексе с 15-70 мкюри α- или β-эмитирующего нуклида или нуклида с Оже-е--каскадами, например, 90Y, 177Lu или 161Tb. У более крупных млекопитающих, например, у людей, указанный диапазон доз может составлять от 1 до 100 мкг/м2 конъюгированного соединения формулы III, например, конъюгированного соединения Б, в комплексе, например, с 1-100 мкюри α- или β-эмитирующего нуклида или нуклида с Оже-е--каскадами, например, 90Y, 177Lu или 161Tb.
Конъюгированное соединение формулы III, например, конъюгированное соединение Б, в форме комплекса для применения в качестве радиотерапевтического агента может вводиться любым традиционным способом, например, внутривенно, например, в форме инъекционных растворов. Оно также может вводиться преимущественно путем инфузии, например, инфузии на протяжении от 15 до 60 мин. В зависимости от места расположения опухоли оно может вводиться как можно ближе к опухоли, например, с помощью катетера. Настоящее изобретение также предусматривает фармацевтическую композицию, включающую конъюгированное соединение формулы III, например, конъюгированное соединение Б, в свободной основной форме, или в форме фармацевтически приемлемой соли или комплекса с выявляемым или радиотерапевтическим агентом, вместе с одним или несколькими фармацевтически приемлемыми растворителями или носителями.
Соединение формулы III или конъюгированное соединение формулы III, например, соединение Б или конъюгированное соединение Б, в форме комплекса может быть пригодно для визуализации или лечения hsst3 и/или hsst5 экспрессирующих или аккумулирующих опухолей слизистых, например, аденом или пролактином, опухолей желудка и поджелудочной железы, карциноидов, центральной нервной системы, груди, простаты (включая прогрессирующий не поддающийся лечению рак простаты), опухолей яичника или толстой кишки, мелкоклеточного рака легких, злокачественной обструкции кишечника, параганглиомы, рака почки, рака кожи, нейробластомы, феохромоцитомы, медуллярных тироидных карцином, миелом, лимфом, лимфомы Ходкина и неходкинской лимфомы, опухолей костей и их метастаз, а также аутоиммунных и воспалительных расстройств, например, ревматоидного артрита, базедовой болезни или других воспалительных заболеваний глаз.
Согласно другому объекту настоящего изобретения предусмотрена фармацевтическая композиция, включающая конъюгированное соединение формулы III, например, конъюгированное соединение Б, или его комплекс с одним или несколькими его фармацевтически приемлемыми носителями или растворителями. Такие композиции могут быть получены обычным способом и могут быть представлены, например, для визуализации, в форме набора, включающего две раздельные дозы, одна из них радионуклид, а вторая - конъюгированное соединение формулы III, например, конъюгированное соединение Б, а также инструкции по их смешиванию. Для радиотерапии конъюгированное соединение формулы III, например, конъюгированное соединение Б, в форме комплекса может предпочтительно быть в форме радиоактивного жидкого состава.
Соединение формулы III, необязательно конъюгированное, например, соединение Б или конъюгированное соединение Б, в форме комплекса может вводиться в качестве единственного действующего ингредиента или в соединении, например, в качестве вспомогательного средства, с другими лекарственными средствами. Например, соединение формулы III, например, соединение Б, может применяться в комбинации с иммуносупрессирующим агентом, например, ингибитором кальциневрина, например, циклоспорином А, Isa Т×247 или FK 506; ингибитором mTOR, например, рапамицином, CCI779, АВТ578 или 40-O-(2-гидроксиэтил)-рапамицином; аскомицином, обладающим иммуносупрессирующими свойствами, например, АВТ-281, ASM981, etc.; кортикостероидами; циклофосфамидом; азатиопреном; метотрексатом; лефлуномидом; мизорибином; микофеноловой кислотой или ее солью, например MyforticR; микофенолятом мофетила; 15-дезоксиспергуалином или его иммуносупрессирующим гомологом, аналогом или производным; агонистом рецептора S1P, например, FTY720; иммуносупрессирующими моноклональными антителами, например, моноклональными антителами к рецепторам лейкоцитов, например, МНС, CD2, CD3, CD4, CD7, CD8, CD25, CD28, CD40, CD45, CD58, CD80, CD86 или к их лигандам; другими иммуномодулирующими соединениями, например, рекомбинантными связывающими молекулами, имеющими, по меньшей мере, часть внеклеточного домена CTLA4 или его мутанта, например, по меньшей мере, внеклеточной части CTLA4 или его мутанта, присоединенного к non-CTLA4 белковой последовательности, например, CTLA4Ig (например, коллекционный АТСС 68629) или его мутант, например, LEA29Y; ингибиторами адгезивных молекул, например, антагонистами LFA-1, антагонистами ICAM-1 или -3, антагонистами VCAM-4 или антагонистами VLA-4. Соединение формулы III, например, соединение Б, также может применяться в комбинации с противовоспалительным агентом, агентом моделирования рецептора стимулятора секреции GH, например, грелином или гексарелином, антагонистом рецептора GH, например, пегвисомантом.
Соединение формулы III, необязательно конъюгированное, например, соединение Б или конъюгированное соединение Б, в форме комплекса также может применяться с антипролиферативным агентом, например, химиотерапевтическим лекарственным средством, например, паклитакселем, гемцитабином, цисплатином, доксорубицином, 5-фторурацилом или таксолом, гормональным агентом или антагонистом, например, антиандрогеном или митоксантроном (особенно в случае рака простаты), антиэстрогеном, таким как летрозол (особенно в случае рака груди), антиметаболитом, растительным алкалоидом, модификатором биологического ответа, предпочтительно лимфокином или интерферонами, ингибитором протеинтирозинкиназы и/или серин/треонинкиназы, а также с агентом с иным или неизвестным механизмом действия, например, любым эпотилоном или производным эпотилона, или ингибитором mTOR, например, указанным выше.
Если соединение формулы III, необязательно конъюгированное, например, соединение Б или конъюгированное соединение Б, в форме комплекса вводят в конъюгации с другим лекарственным средством, дозы одновременно вводимых лекарственных средств, конечно, могут варьировать в зависимости от типа применяемого сопутствующего лекарственного средства, от специфики применяемого лекарственного средства, от состояния, требующего лечения, и др. Понятия «совместное введение» или «комбинированное введение» или близкие им понятия, применяемые в настоящем изобретении, обозначают осуществление введения выбранных терапевтических агентов одному пациенту, распространяются на режимы лечения, при которых агенты необязательно вводятся одинаковым способом и в одно и то же время.
В соответствии со сказанным выше в настоящем изобретении предусматривается еще один объект:
9. Фармацевтическая комбинация, включающая а) первый агент, являющийся необязательно конъюгированным соединением формулы III, например, соединением Б или конъюгированным соединением Б, в форме комплекса и б) сопутствующий агент, например, подобный описанному выше.
10. Способ, подобный описанному выше, включающий совместное введение, например, одновременное или последовательное, терапевтически эффективного количества необязательно конъюгированного соединения формулы III, например, соединения Б или конъюгированного соединения Б в форме комплекса, и второго лекарственного вещества, указанное второе лекарственное вещество является, например, таким, как описано выше.
| название | год | авторы | номер документа |
|---|---|---|---|
| АНАЛОГИ СОМАТОСТАТИНА | 2001 |
|
RU2287533C2 |
| ДЕПО-ФОРМА АНАЛОГА СОМАТОСТАТИНА И СПОСОБ ЕЕ ПОЛУЧЕНИЯ | 2005 |
|
RU2411031C2 |
| СОМАТОСТАТИНОВЫЕ ПЕПТИДЫ | 1996 |
|
RU2160741C2 |
| КОМПОЗИЦИЯ С ЗАМЕДЛЕННЫМ ВЫСВОБОЖДЕНИЕМ, ВКЛЮЧАЮЩАЯ БИСФОСФОНАТ | 2005 |
|
RU2395274C2 |
| ПОЛУЧЕНИЕ ПЕПТИДОВ СОМАТОСТАТИНА | 2004 |
|
RU2360921C2 |
| МИКРОЧАСТИЦЫ, СОДЕРЖАЩИЕ АНАЛОГИ СОМАТОСТАТИНА | 2004 |
|
RU2404749C2 |
| ФАРМАЦЕВТИЧЕСКИЕ КОМПОЗИЦИИ | 2009 |
|
RU2504360C2 |
| ХИМЕРНЫЕ АНАЛОГИ СОМАТОСТАТИНА-ДОФАМИНА | 2002 |
|
RU2277539C2 |
| ХИМЕРНЫЕ АНАЛОГИ ЛИГАНДОВ СОМАТОСТАТИНОВЫХ И ДОПАМИНОВЫХ РЕЦЕПТОРОВ, ФАРМАЦЕВТИЧЕСКИЕ КОМПОЗИЦИИ НА ИХ ОСНОВЕ И СПОСОБЫ ВОЗДЕЙСТВИЯ НА РЕЦЕПТОРЫ СОМАТОСТАТИНА И/ИЛИ ДОПАМИНА | 2004 |
|
RU2329273C2 |
| ПРИМЕНЕНИЕ АНАЛОГОВ СОМАТОСТАТИНА ПРИ МЕНИНГИОМЕ | 2008 |
|
RU2523416C2 |
Изобретение относится к медицине и биохимии и описывает парентеральные фармацевтические композиции, включающие аналог соматостатина и винную кислоту. Изобретение обеспечивает высокую стабильность и одновременно хорошую местную применимость. 3 н. и 5 з.п. ф-лы.
1. Фармацевтическая композиция для парентерального введения, включающая соединение формулы II

в которой конфигурация в положении С-2 является (R), или (S), или их смесью и в которой R обозначает NR1R2-С2-6алкилен или гуанидин-С2-6алкилен и каждый из R1 и R2 независимо обозначает Н или С1-4алкил,
в свободной форме, в форме соли или в защищенной форме, и винную кислоту.
2. Композиция по п.1, соединение формулы II представляет собой цикло[{4-(NH2-C2H4-NH-CO-O-)Pro}-Phg-DTrp-Lys-Tyr(4-Bzl)-Phe].
3. Композиция по п.1, в которой соединение формулы II присутствует в форме диаспартата.
4. Композиция по одному из предшествующих пунктов, в которой рН доводится до значения примерно от 4 до примерно 4,5.
5. Композиция для парентерального введения по п.1, забуференная при рН примерно от 4 до примерно 4,5 и включающая действующее вещество
цикло[{4-(NH2-С2Н4-NH-СО-O-)Pro}-Phg-DTrp-Lys-Tyr(4-Bzl)-PheJ или его фармацевтически приемлемую соль.
6. Применение фармацевтической композиции по одному из пп.1-5 для приготовления средства для лечения акромегалии или рака.
7. Применение по п.6 для приготовления средства для лечения болезни Кушинга.
8. Способ лечения акромегалии или рака у субъекта, нуждающегося в этом, включающий введение субъекту фармацевтической композиции по одному из пп.1 -7.
| WO9701579, 16.01.1997 | |||
| УСТРОЙСТВО ДЛЯ РЕЗАНИЯ ЛЬДА | 0 |
|
SU210192A1 |
| Приспособление для выжигания травы на железнодорожном полотне | 1923 |
|
SU1632A1 |
| Способ регулирования турбоустановки с отбором пара | 1985 |
|
SU1276839A1 |

