ОБЛАСТЬ ТЕХНИКИ
Настоящее изобретение относится к новому производному 2-циано-4-фторпирролидина или его соли, которое может быть использовано в качестве лекарственного препарата, в частности в качестве ингибитора дипептидилпептидазы-IV (обозначаемой далее "DPP-IV"), и к фармацевтической композиции, содержащей данное соединение в качестве активного ингредиента.
ПРЕДПОСЫЛКИ ИЗОБРЕТЕНИЯ
Дипептидилпептидаза-IV (DPP-IV) является сериновой протеазой, которая распознает и вырезает последовательность с пролином, гидроксипролином или аланином во второй позиции с ее N-конца (H-Xaa-Pro, H-Xaa-Hyp или H-Xaa-Ala, где Хаа ообзначает аминокислоту). Известно, что DPP-IV широко представлена в организме человека, не только в тканях почек, печени и слюнных желез, но также и в жидкостях организма, таких как сыворотка, моча и слюна. Хотя ее физиологическая роль к настоящему времени окончательно не прояснена, DPP-IV может участвовать в регулировании биологических функций, поскольку она отщепляет различные физиологически активные пептиды (непатентная ссылка 1). В частности, в настоящее время особо отмечено, что DPP-IV может контролировать активность гормона инкретина, который участвует в ингибировании увеличения глюкозы в крови.
Инкретин является гормоном, который секретируется в кишечнике человека после приема пищи, и он воздействует на β-клетки поджелудочной железы, усиливая секрецию инсулина, регулируя таким образом уровень глюкозы в крови. Известно, что активность инкретина ослаблена у больных диабетом 2 типа (непатентная ссылка 2), и это рассматривается как то, что ослабление активности может являться одной из причин проявления диабета. Соответственно, это предполагает, что гипергликемия после приема пищи у диабетиков может повышаться.
Пептид, сходный с глюкагоном (в дальнейшем именуемый "GLP-1"), в настоящее время известен как соединение, которое обладает наиболее эффективной инкретиновой активностью в организме человека. GLP-1 после секреции в кровь немедленно инактивируется, и известно, что данная инактивация в основном происходит вследствие действия DPP-IV, которая его расщепляет (непатентная ссылка 3). Далее, инактивированный GLP-1, расщепленный DPP-IV, связывается с рецептором GLP-1 и препятствует активной GLP-1 связываться с рецептором. Следовательно, предполагают, что инкретиновая активность GLP-1 поэтому ослабевает (непатентная ссылка 4).
В силу данных причин предполагают, что ингибитор DPP-IV может препятствовать инактивации GLP-1, таким образом повышая воздействие инкретина на активный GLP-1, и в результате может быть предотвращена гипергликемия после приема пищи у диабетиков. Кроме того, поскольку инкретин усиливает зависимую от глюкозы секрецию инсулина в организме человека, предполагается, что ингибитор DPP-IV может быть безопасным лекарственным средством, не обладающим побочным эффектом, таким как гипогликемия, которая часто наблюдается при использовании существующих лекарственных средств, вызывающих секрецию инсулина.
С другой стороны, известны некоторые производные 2-цианопирролидина, обладающие ингибирующей DPP-IV активностью (патентные ссылки 1-7).
Из них в международной публикации WO 02/30890 (патентная ссылка 5) конкретно описаны соединения общей формулы (А), и сказано, что эти соединения будут эффективны для профилактики и лечения диабета и для профилактики и лечения других заболеваний, вызванных или обострившихся в результате нарушенной толерантности к глюкозе, гиперинсулинемии и осложнений при диабете.
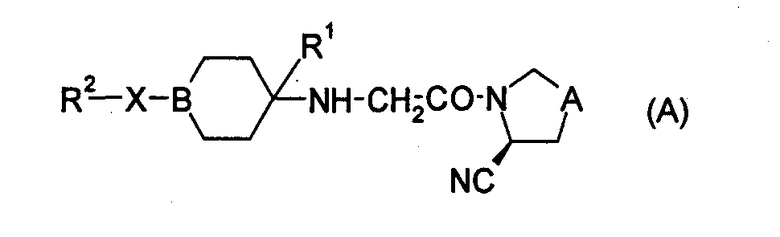
(Использованные в данной формуле символы имеют значения, указанные в патентном документе).
С другой стороны, в международной публикации WO 02/38541 (патентная ссылка 6) описаны соединения общей формулы (В), и сказано, что эти соединения значительно ингибируют повышение уровня глюкозы в крови в тесте на пероральную толерантность к глюкозе на крысах линии ожирения Цуккера (крысы Zucker Fatty).

(Использованные в данной формуле символы имеют значения, указанные в патентном документе).
В международной публикации WO 03/002553 (патентная ссылка 7) описаны соединения общих формул (C), (D), (E), (F), (G) и (H) и сказано, что эти соединения могут быть использованы для лечения заболеваний, таких как диабет, ожирение.
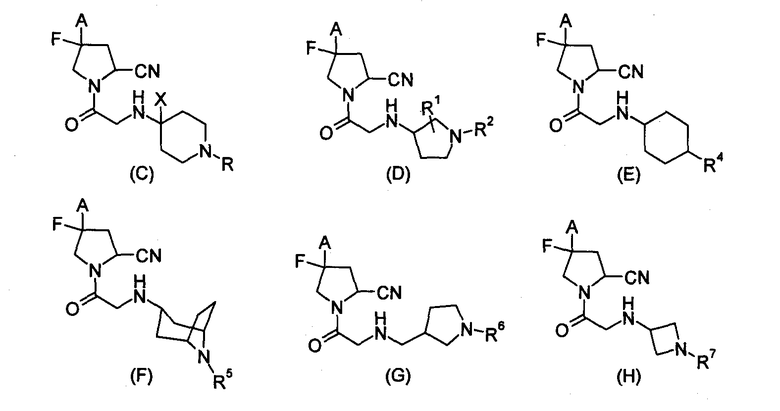
(Использованные в данной формуле символы имеют значения, указанные в патентном документе).
С учетом данной ситуации весьма желательна разработка лекарственных препаратов, которые обладают более превосходной ингибирующей DPP-IV активностью.
Непатентная ссылка 1: Mentlein R., Regulatory Peptide, 1999, т. 85, стр.9-24.
Непатентная ссылка 2: Nauck M.A. Diabetologia, 1986, т. 29, стр.46-52.
Непатентная ссылка 3: Drucker D.J. Diabets, 1998, т. 47, стр.159-169.
Непатентная ссылка 4: Knudsen L.B. European Journal of Pharmacology, 1996, т.318, стр.429-435.
Патентная ссылка 1: сборник международной публикации WO 98/19998.
Патентная ссылка 2: сборник международной публикации WO 01/96295.
Патентная ссылка 3: сборник международной публикации WO 00/34241.
Патентная ссылка 4: сборник международной публикации WO 01/55105.
Патентная ссылка 5: сборник международной публикации WO 02/30890.
Патентная ссылка 6: сборник международной публикации WO 02/38541.
Патентная ссылка 7: сборник международной публикации WO 03/002553.
ОПИСАНИЕ ИЗОБРЕТЕНИЯ
Авторы настоящего изобретения провели широкие и тщательные исследования в отношении соединений, обладающих ингибирующей DPP-IV активностью, которые, как ожидалось, являются эффективными для лечения инсулинзависимого диабета (диабет 1 типа), инсулиннезависимого диабета (диабет 2 типа), заболеваниях, резистентных к инсулину, и ожирения. В результате было установлено, что новое производное 2-циано-4-фторпирролидина или его соль по изобретению проявляет превосходную ингибирующую DPP-IV активность, что и привело к созданию данного изобретения.
Таким образом, данное изобретение относится к производному 2-циано-4-фторпирролидина общей формулы (I) или его фармацевтически приемлемой соли, которое может использоваться в качестве ингибитора DPP-IV.
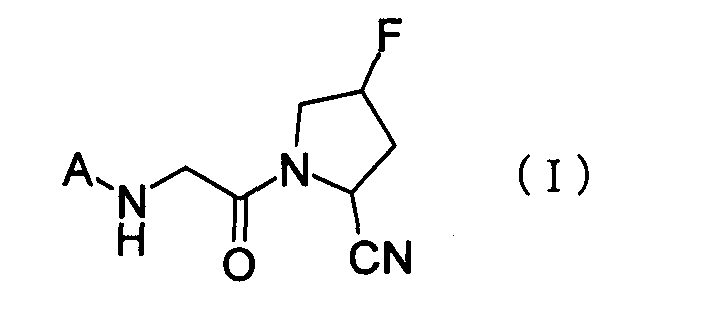
где А представляет собой пиперидин-4-ил, оксетан-3-ил, пирролидин-3-ил, тетрагидро-2Н-пиран-4-ил, пиразолидин-4-ил, 1,3-диоксан-5-ил, 8-азабицикло[3.2.1]окт-3-ил или тетрагидро-2Н-тиопиран-4-ил, каждый из которых может быть замещен, за исключением пиперидин-4-ила, который замещен группой, выбранной из группы, включающей пропан-2-сульфонил, 2,4,6-триметилбензенсульфонил, фенилметансульфонил, 2-нафталин-1-илэтансульфонил, 7,7-диметил-6-оксонорборнан-1-илметансульфонил, 4-фторфенил, 3,5-дифторфенил, 4-нитрофенил, 4-трифторметилфенил, 4-цианофенил, 4-циано-3-фторфенил, 4-циано-3,5-дифторфенил, 3-циано-5-фторфенил, бензоксазол-2-ил и бензил; пирролидин-3-ил, который замещен группой, выбранной из группы, включающей пропан-2-сульфонил, 3-цианопиридин-6-ил, 4-трифторметилфенил, 4-фторфенил и 4-фторбензил; и 8-азабицикло[3.2.1]окт-3-ил, который замещен этоксикарбонилом.
В формуле (I) А, предпочтительно, представляет собой пиперидин-4-ил или 8-азабицикло[3.2.1]окт-3-ил, каждый из которых может быть замещен, более предпочтительно, группу общей формулы (II):
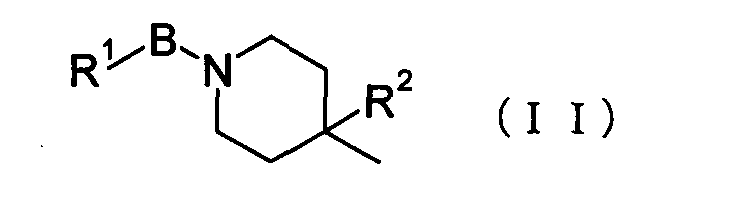
где В представляет собой карбонил, сульфонил или одинарную связь;
R1 представляет собой низший алкил, арил или ароматический гетероцикл, каждый из которых может быть замещен;
R2 представляет собой низший алкил, который необязательно замещен группой, выбранной из группы, включающей -ОН и О-низший алкил, или представляет собой -Н.
В формуле (II) В, предпочтительно, представляет собой карбонил или сульфонил.
В формуле (II) R1,предпочтительно, представляет собой низший алкил, который необязательно замещен, более предпочтительно, низший алкил, который необязательно замещен группой, выбираемой из группы, включающей -ОН или фтор, еще более предпочтительно, метил или этил, каждый из которых необязательно замещен группой, выбранной из группы, включающей -О или фтор, более предпочтительно, метил или этил, каждый из которых необязательно замещен -ОН.
В формуле (II) R2,предпочтительно, представляет собой низший алкил, который необязательно замещен -ОН, более предпочтительно, метил или этил, каждый из которых необязательно замещен -ОН, еще более предпочтительно, метил, этил или гидроксиметил, наиболее предпочтительно, метил.
Химическая структура производных 2-циано-4-фторпирролидина по данному изобретению характеризуется тем, что в них необязательно замещенный неароматический гетероцикл присоединен к 1-положению скелета 2-циано-4-фторпирролидина через аминометиленкарбонил, и его фармацевтической характеристикой является то, что данные производные обладают ингибирующей DPP-IV активностью.
Среди соединений формулы (I) предпочтительными являются такие, где А представляет собой пиперидин-4-ил или 8-азабицикло[3.2.1]окт-3-ил, каждый из которых необязательно замещен; более предпочтительными являются такие, где А является группой формулы (II); еще более предпочтительными являются такие, где А представляет собой группу формулы (II), В представляет собой карбонил или сульфонил, R1 необязательно замещен низшим алкилом, R2 представляет собой метил, этил или гидроксиметил; и наиболее предпочтительными являются такие, где А представляет собой группу формулы (II), В представляет собой карбонил или сульфонил, R1 представляет собой метил или этил, каждый из которых необязательно замещен группой, выбранной из группы, включающей в себя -ОН или фтор, R2 представляет собой метил.
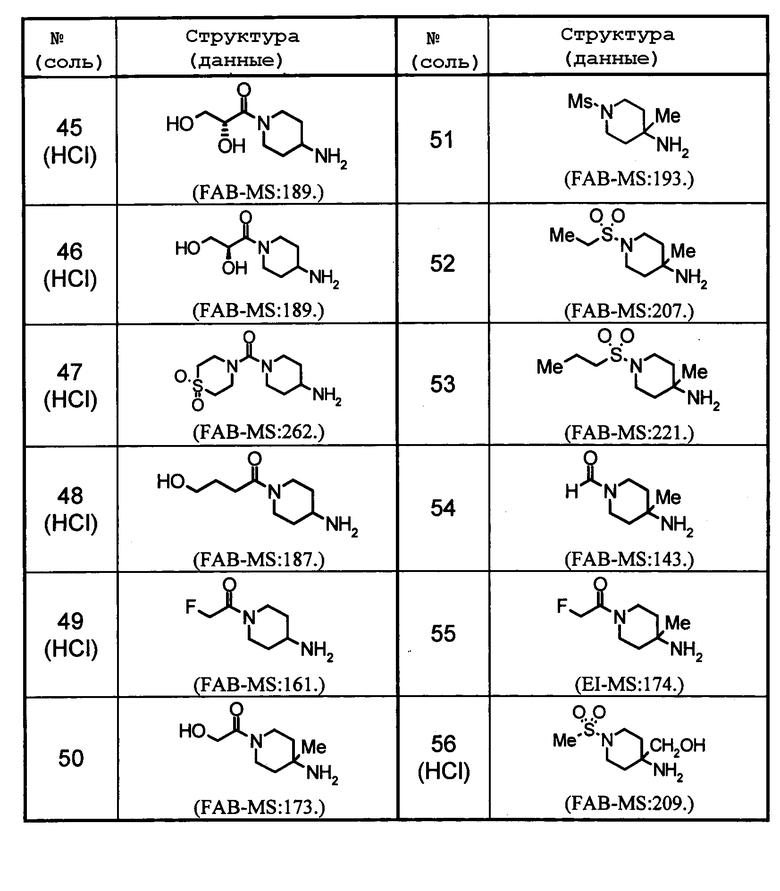
Из данных соединений особенно предпочтительными являются следующие:
4-фтор-1-({[1-(метансульфонил)пиперидин-4-ил]амино}аетил)пирролидин-2-карбонитрил,
4-фтор-1-({[4-метил-1-(метансульфонил)пиперидин-4-ил]амино}аетил)пирролидин-2-карбонитрил,
4-фтор-1-{[(1-гликолоилпиперидин-4-ил)амино]ацетил}прролидин-2-карбонитрил,
4-фтор-1-{[(1-гликолоил-4-метилпиперидин-4-ил)амино]ацетил}прролидин-2-карбонитрил,
4-фтор-1-{[(1-фторацетил-4-метилпиперидин-4-ил)амино]ацетил}прролидин-2-карбонитрил,
4-фтор-1-{[(1-формилпиперидин-4-ил)амино]ацетил}прролидин-2-карбонитрил,
4-фтор-1-{[(1-формил-4-метилпиперидин-4-ил)амино]ацетил}прролидин-2-карбонитрил или
4-фтор-1-({[1-(морфолин-4-илкарбонил)пиперидин-4-ил]амино}аетил)пирролидин-2-карбонитрил или их фармацевтически приемлемом соль;
более предпочтительными являются следующие:
4-фтор-1-({[4-метил-1-(метансульфонил)пиперидин-4-ил]амино}аетил)пирролидин-2-карбонитрил,
4-фтор-1-{[(1-гликолоил-4-метилпиперидин-4-ил)амино]ацетил}прролидин-2-карбонитрил или их фармацевтически приемлемая соль.
В другом предпочтительном примере соединений формулы (I) А представляет собой группу формулы (II) и В представляет собой карбонил.
Изобретение далее относится к фармацевтической композиции, содержащей в качестве активного ингредиента соединение, указанное выше, в частности, фармацевтической композиции, содержащей в качестве активного ингредиента соединение, указанное выше для лечения и/или профилактики инсулинзависимого диабета (диабета типа 1), инсулиннезависимого диабета (диабета типа 2), заболеваний, связанных с резистантностью к инсулину или ожирения; и к фармацевтической композиции, содержащей в качестве активного ингредиента соединение, указанное выше, в качестве ингибитора дипептидилпептидазы IV.
Соединения по данном изобретению описаны здесь и далее.
В настоящем описании «низший алкил» означает С1-6 линейный или разветвленный алкил, например, более конкретно, включающий метил, этил, пропил, изопропил, бутил, трет-бутил, пентил, неопентил и гексил, и т.д. Предпочтительно, это метил, этил, пропил или изопропил, более предпочтительно, метил или этил.
«Арил» означает С6-14 моноциклическую-трициклическую ароматическую моновалентную группу, состоящую из атомов углерода, например, более конкретно, включающую фенил, нафтил и т.д. Предпочтительно, это фенил.
«Ароматический гетероцикл» означает моноциклическую-трициклическую ароматическую моновалентную группу, содержащую по крайней мере один гетероатом, выбранный из группы, содержащей атомы азота, кислорода и серы, например, более конкретно, включающую фуранил, тиенил, пирролил, пиридил, оксазолил, тиазолил, изотиазолил, имидазолил, пиразолил, тетразолил, пиразинил, пиримидинил, пиридазинил, триазинил, изоксазолил, триазолил, бензофуранил, бензотиенил, бензотиадиазолил, бензотиазолил, бензоксазолил, бензимидазолил, бензотриазолил, индолил, изоиндолил, квиназолил, изоквинолил, квиноксаил, имидазопиридинил и имидазопиримидинил и т.д. Предпочтительно это пиридил.
Приемлемым заместителем для необязательно замещенных «пиперидин-4-ила, оксетан-3-ила, пирролидин-3-ила, тетрагидро-2Н-пиран-4-ила, пиразолидин-4-ила, 1,3-диоксан-5-ила, 8-азабицикло[3.2.1]окт-3-ила или тетрагидро-2Н-тиопиран-4-ила» для А может быть любой и каждый из обычно используемых для этих групп, и группа А может содержать один или несколько, предпочтительно, от 1 до 4 заместителей. Включая атом углерода в А, который напрямую присоединен к группе NH в формуле (I), атом углерода или атом азота кольца могут быть замещены. Кроме того, атом серы кольца может быть окислен.
Заместитель, присоединенный к атому углерода, включает заместитель Х, -ОН, -О-Х, галоген, -СО-Х, -СОО-Х, -SO2-X и -CONRR'. Предпочтительно, он представляет собой низший алкил или арил, каждый из которых необязательно замещен одной или несколькими группами, выбранными из группы, включающей -ОН, -О-низший алкил, -О-арил, галоген, циано и нитро, более предпочтительно низший алкил, необязательно замещенный заместителем, выбранным из -ОН и фтора.
R и R' могут быть одинаковыми или различными и представляют собой низший алкил, необязательно замещенный -ОН, или -Н (то же справедливо и в данном случае). «Группа заместителей Х» обозначает низший алкил, низший алкенил, низший алкинил, циклоалкил, циклоалкенил, арил, неароматический гетероцикл и ароматический гетероцикл, каждый из которых необязательно замещен одной или несколькими группами, выбранными из группы, включающей -ОН, -О-низший алкил, -О-арил, галоген, циано и нитро (то же справедливо и в данном случае).
С другой стороны, заместитель, присоединенный к атому азота, содержит заместитель Х, -СО-Х, -СОО-Х, -SO2-X и -CONRR'. Предпочтительно, он представляет собой низший алкил, циклоалкил, арил или ароматический гетероцикл, каждый из которых необязательно замещен одной или несколькими группами, выбранными из группы, включающей -ОН, -О-низший алкил, -О-арил, галоген, циано и нитро; или -SO2-низший алкил (необязательно замещенный одной или несколькими группами, выбранными из группы, включающей -ОН, -О-низший алкил, -О-арил, галоген, циано и нитро); или -SO2-арил (необязательно замещенный одной или несколькими группами, выбранными из группы, включающей -ОН, -О-низший алкил, -О-арил, галоген, циано и нитро).
Термин «низший алкенил» означает С2-6 алкенил, например, более конкретно, включающий винил, аллил, 1-пропенил, 1-бутенил, 2-бутенил и 3-бутенил.
Термин «низший алкинил» означает С2-6 алкинил, например, более конкретно, включающий этинил, 1-пропинил, 1-бутинил, 2-бутинил и 3-бутинил.
Термин «циклоалкил» означает С3-14 моновалентный остаток углеродного цикла, который может быть конденсирован с образованием мостиковой связи. Конкретно, например, он включает циклопропил, циклопентил, циклогексил, циклооктил, адамантил, борнил, нонборнил, бицикло[2.2.1]гептил, бицикло[3.1.1]гептил, бицикло[2.2.2]октил, бицикло[3.2.1]октил и бицикло[3.3.1]нонил.
Термин «циклоалкенил» означает остаток С3-14 карбонового кольца, который соответствует «циклоалкилу», но является частично ненасыщенным, например, более конкретно, включающий циклопентенил, циклогексенил и норборненил.
Термин «галоген» включает фтор, хлор, бром и йод. Предпочтительно, это фтор, хлор или бром, более предпочтительно, фтор.
Соединения по данному изобретению включают в себя смеси различных стереоизомеров, таких как таутомерные изомеры и оптические изомеры, и одни отделены от других.
Соединения по данному изобретению могут образовывать соли добавления кислот. В зависимости от типа заместителя они могут образовывать соли с основаниями. Более конкретно, соли включают соли добавления кислот, образованные минеральными кислотами, такими как хлористоводородная кислота, бромистоводородная кислота, йодистоводородная кислота, серная кислота, азотная кислота, фосфорная кислота, или органическими кислотами, такими как муравьиная кислота, уксусная кислота, пропионовая кислота, оксалиновая кислота, малоновая кислота, янтарная кислота, фумаровая кислота, малеиновая кислота, молочная кислота, яблочная кислота, винная кислота, лимонная кислота, метансульфоновая кислота, этансульфоновая кислота, п-толуолсульфоновая кислота; соли с кислотными аминокислотами, такими как аспарагиновая кислота, глутаминовая кислота, или с неорганическими основаниями, такими как натрий, калий, магний, кальций, алюминий, или с органическими основаниями, такими как метиламин, этиламин, этаноламин; или с основными аминокислотами, такими как лизин, орнитин; и соли аммония.
Соединения по данному изобретению далее включают в себя гидраты, различные фармацевтически приемлемые сольваты и полиморфные кристаллы. Естественно, данное изобретение не следует ограничивать соединениями, описанными в приведенных здесь примерах, и оно включает все производные формулы (I) и их фармацевтически приемлемые соли.
В этой связи, соединение по данному изобретению включает все так называемые пролекарства, т.е. соединения, которые будут подвергнуты метаболизму и преобразованы в соединение приведенной выше общей формулы (I) или в его соли в организме человека. В качестве групп, подходящих для образования пролекарства, можно указать группы, описанные в Prog. Med., 5, 2157-2161 (1985) и Iyakuhin No Kaihatsu (Development of Drugs), т.7, «Molecular Desighn», 163-198 (1990), Hirokawa Publishing Co.
(Способ получения)
Соединение по изобретению и его фармацевтически приемлемая соль могут быть получены различными способами синтеза с учетом характерных свойств его основного скелета или вида заместителей. В этой связи, в некоторых случаях является эффективным в методе получения в зависимости от вида функциональной группы, на стадии исходного или промежуточного материала заменять функциональную группу защитной группы, т.е. группой, которая может быть легко преобразована в функциональную группу. В дальнейшем, если это желательно, защитная группа удаляется, таким образом позволяя получить желаемое соединение. Примеры такой функциональной группы включают гидроксильную группу, карбоксильную группу и аминогруппу. Примеры защитных групп включают защитные группы, описанные в Green and Wuts, Protective Groups in Organic Synthesis (third edition), и они могут использоваться соответствующим способом в зависимости от условий реакции.
Один типичный способ получения описан ниже.
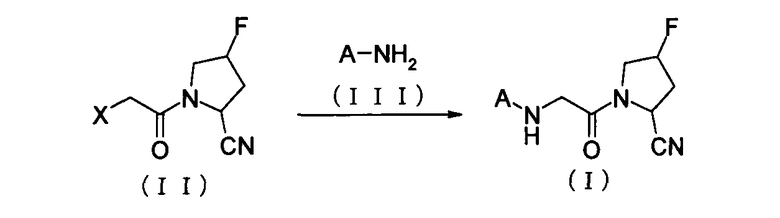
где А имеет значения, указанные выше; и Х представляет собой удаляемую группу, такую как галоген или сульфонилоксигруппу.
Данный способ включает алкилирование соединения (II) амином (III) общей формулы A-NH2 с образованием соединения (I) по изобретению. Взаимодействие может быть осуществлено в отсутствие или в присутствии растворителя. Растворитель может быть любым из ароматических углеводородов, таких как толуол, ксилол; кетонов, таких как метилэтилкетон, ацетон; простых эфиров, таких как диоксан, тетрагидрофуран, диглим; спиртов, таких как метанол, этанол, изопропанол; хлороформа, метиленхлорида, ацетонитрила, диметилформамида, диметилсульфоксида, воды; и их смеси. Для реакции может быть выбран подходящий растворитель в зависимости от типа реакционного вещества и условий реакции.
Добавление в реакцию основания является предпочтительным для мягкого протекания реакции. Конкретными примерами такого основания являются карбонаты щелочных металлов, такие как карбонат натрия, карбонат калия; гидрокарбонаты щелочных металлов, такие как гидрокарбонат натрия, гидрокарбонат калия; и органические амины, такие как триэтиламин, диизопропилэтиламин, пиридин.
Некоторые соединения по изобретению могут быть получены из соединений (I), полученных, как описано выше, путем объединения, любым желанным образом, некоторых известных стадий, которые обычно используются специалистами в данной области, таких как алкилирование, ацилирование, окисление, восстановление, гидролиз и т.д.
Полученные таким образом соединения по изобретению могут быть обычным методом выделены и очищены в виде свободных соединений или после преобразования их в соли в виде их солей. Выделение и очистку можно осуществить обычным химическим методом, таким как экстракция, концентрирование, дистилляция, кристаллизация, фильтрация, перекристаллизация, различные методы хроматографии и т.д.
В случае, когда соединения по изобретению имеют асимметрический углерод, они включают оптические изомеры. Оптические изомеры могут быть разделены обычным способом, например, путем фракционной кристаллизации с перекристаллизацией в виде соответствующей соли или путем колоночной хроматографии. Оптически активные соединения могут быть получены исходя из соответствующего оптически активного соединения.
ПРОМЫШЛЕННАЯ ПРИМЕНИМОСТЬ
Соединения по изобретению обладают ингибирующей DPP-IV активностью. В частности, они обладают активностью по ингибированию разложения GLP-1, гормона, который воздействует на β-клетки поджелудочной железы, усиливая секрецию инсулина и регулируя тем самым содержание глюкозы в крови.
Основываясь на их действии, соединения по изобретению могут использоваться для лечения и/или профилактики инсулинзависимого диабета (диабета 1 типа), инсулиннезависимого диабета (диабет 2 типа), заболеваний, связанных с резистентностью к инсулину, и ожирения.
Превосходная ингибирующая DPP-IV активность соединений по изобретению подтверждена методами исследований, указанных далее.
(1) Исследование по определению DPP-IV-ингибирующей активности
Реакцию проводили на плоскодонном 96-луночном планшете для микротитрования. Меняющаяся концентрация исследуемого соединения была добавлена в водный раствор, содержащий 25 мМ Трис-HCl, 140 мМ хлорида натрия, 10 мМ хлорида калия, 1% бычьего сывороточного альбумина для радиоиммунного анализа и 0,01 мМ Gly-Pro-AMC (Bachem). К этому реакционному раствору (95 мкл/лунка) добавляли 5 мкл плазмы, собранной у взрослых здоровых добровольцев, и инкубировали при комнатной температуре в течение 20 мин. После завершения реакции измеряли интенсивность флуоресценции (излучение 355 нм/испускание 460 нм) в каждой лунке (ARVO, Perkin Elmer). Результаты измерений для 3 лунок в одинаковых условиях усредняли.
Рассчитывали ингибирование исследуемой группы относительно добавленных с раствором групп и значение IC50 получали путем логического анализа. Результаты представлены в таблице 1.
Вышеуказанное подтверждает DPP-IV-ингибирующую активность соединений по данному изобретению.
(2) Исследование по определению продолжительности DPP-IV-ингибирующей активности у мышей
Самцов мышей ICR (Nippon SLC) разделяли на исследуемую группу и контрольную группу по 5 особей в каждой. Исследуемое соединение (10 мг/кг) растворяли в очищенной воде и вводили перорально. Мышам контрольной группы перорально вводили только очищенную воду. Спустя полчаса, 6 и 12 ч после введения собирали кровь каждой мыши из глазничного венозного сплетения. Собранную кровь сразу центрифугировали для отделения плазмы и измеряли активность DPP-IV плазмы.
Способ определения активности DPP-IV плазмы следующий: Реакцию проводят на 96-луночном планшете. 5 мкл собранной плазмы добавляли в водный раствор (95 мкл/лунка), содержащий 25 мМ Трис-HCl, 140 мМ хлорида натрия, 10 мМ хлорида калия, 1% бычьего сывороточного альбумина и 0,01 мМ Gly-Pro-AMC (Bachem) и инкубировали при комнатной температуре в течение 20 мин. Измеряли интенсивность флуоресценции (излучение 355 нм/испускание 460 нм) в каждой лунке (ARVO, Perkin Elmer).
Интенсивность флуоресценции лунки, в которую была добавлена плазма, собранная из контрольной группы, принималась за 100%. Основываясь на этом, вычисляли активность DPP-IV плазмы, собранной у мышей с введенным исследуемым соединением, и получали разницу в активности различие между контрольной группой и исследуемой группой. Данные показали эффект ингибирования в исследуемой группе. Результаты приведены в таблице 2.
(3) Исследование по продолжительности DPP-IV-ингибирующей активности у крыс
Самцы крыс SD (Clea Japan) были разделены на исследуемую группу и контрольную группу по 5 особей в каждой. Исследуемое соединение (10 мг/кг) растворяли в очищенной воде и вводили перорально. Крысам контрольной группы вводили только очищенную воду. Спустя полчаса, 6 и 12 ч после введения собирали кровь каждой крысы из хвостовой вены. Собранную кровь сразу центрифугировали для отделения плазмы, измеряли активность DPP-IV плазмы в соответствии с тем же способом по определению продолжительности ингибирующей активности дипептидилпептидазы-IV (DPP-IV), описанным в (2).
Интенсивность флуоресценции лунки, в которую была помещена плазма, собранная из контрольной группы, принималась за 100%. Основываясь на этом, рассчитывали активность DPP-IV плазмы, собранной у крыс с введенным исследуемым соединением, и было определено различие в активности между контрольной группой и исследуемой группой. Данные свидетельствуют об ингибировании в исследуемой группе. Результаты приведены в таблице 3.
через 6 ч/%
через 12 ч/%
через 6 ч/%
через 12 ч/%
В таблице 3 сравниваемое соединение 1 представляет собой соединение по примеру 4-9, описанное в патентной ссылке 5, сравниваемое соединение 2 представляет собой соединение по примеру 4-17, описанное в патентной ссылке 5, сравниваемое соединение 3 представляет собой соединение по примеру 33, описанное в патентной ссылке 7; каждая патентная ссылка указана выше. Структуры сравниваемых соединений 1-3 представлены далее.
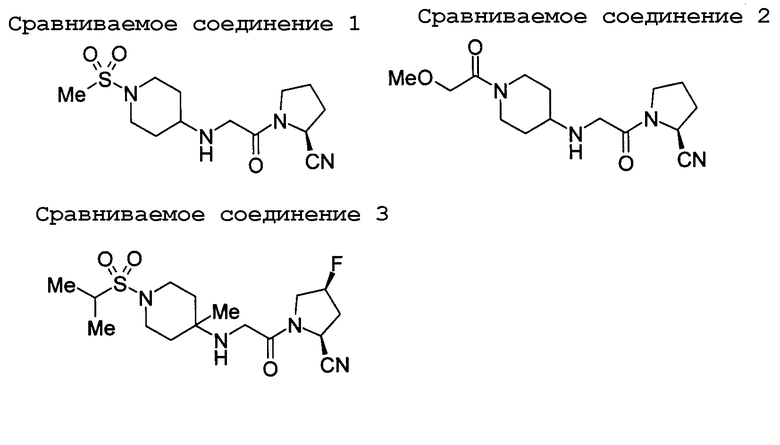
В таблице 2 и таблице 3, приведенных выше, показано, что соединения по настоящему изобретению обладают хорошей активностью при пероральном введении, и их активность сохраняется даже спустя 6 ч и 12 ч после введения. Эти результаты подтверждают, что соединения по настоящему изобретению проявляют улучшенные активность при пероральном введении и продолжительность in vivo в сравнении со сравниваемыми соединениями 1-3.
Фармацевтическая композиция, которая содержит в качестве активного ингредиента одно или несколько соединений и их фармацевтически приемлемые соли по изобретению, может быть получена с использованием носителя, наполнителя и других добавок, используемых обычно при составлении фармацевтических композиций. Она может вводиться перорально или парентерально в виде таблеток, порошков, мелких гранул, гранул, капсул, пилюль, жидкостей, инъекций, суппозиториев, мазей или припарок.
Клиническая доза соединения по изобретению может быть соответствующим образом определена в зависимости от состояния, массы тела, возраста и пола пациентов, которым его вводят, но благоприятной является обычно от 0,1 до 500 мг/день для взрослого человека при пероральном введении и от 0,01 до 100 мг/день для взрослого человека при парентеральном введении. Доза может вводиться пациентам вся за один прием или может быть разделена на несколько порций для введения в несколько приемов. Поскольку дозировка изменяется в зависимости от различных условий, ее можно уменьшить в ряде случаев, указанных выше.
Может быть использована твердая композиция для перорального введения соединений по изобретению, таблетки, порошки, гранулы и т.д. Твердая композиция этого вида содержит одно или несколько активных веществ, смешанных, по крайней мере, с одним инертным разбавителем, таким как лактоза, маннит, глюкоза, гидроксипропилцеллюлоза, микрокристаллическая целлюлоза, крахмал, поливинилпирролидон, метасиликаталюминат магния. При обычном способе композиция может включать в себя некоторые другие добавки, кроме инертных разбавителей, указанных выше, например, связывающее вещество, такое как стеарат магния, разрыхлитель, такой как целлюлозогликолат кальция, стабилизатор, такой как лактоза, солюбилизатор или стимулятор растворения, такой как глутаминовая кислота или аспарагиновая кислота. Если желательно, таблетки и пилюли могут быть покрыты пленкой из сахара или стабильным в желудке или кишечнике соединением, таким как сахароза, желатин, гидроксипропилцеллюлоза, фталат гидроксипропилметилцеллюлозы.
Жидкая композиция для перорального введения включает, например, фармацевтически приемлемые эмульсии, растворы, суспензии, сиропы, эликсиры и тому подобное, которые содержат обычные неактивные растворители, такие как чистая вода или этиловый спирт. В добавление к инертным растворителям такие композиции могут содержать также фармацевтические вспомогательные средства, такие как солюбилизаторы, стимуляторы растворения, промоторы смачивания, промоторы суспензий, а также подсластители, вкусовые добавки, ароматизаторы и консерванты.
Инъекции для парентерально введения включают, например, стерильные водные или неводные растворы, суспензии и эмульсии. Растворитель для водных растворов и суспензий включает, например, дистиллированную воду и физиологический раствор для инъекций. Разбавители для неводных растворов и суспензий включают, например, пропиленгликоль, полиэтиленглколь, растительные масла, такие как оливковое масло, спирты, такие как этиловый спирт, Polysolvate 80 (торговая марка).
Эти композиции могут также содержать добавки, такие как регуляторы изотоничности, консерванты, промоторы смачивания, эмульгаторы, разрыхлители, стабилизаторы (например, лактоза), солюбилизаторы, стимуляторы растворения. Они стерилизуются фильтрованием через бактериальные фильтры или добавлением соединений, убивающих микроорганизмы, или облучением их радиацией. Стерильные твердые композиции могут быть получены заранее, и они могут быть растворены в стерильной воде или стерильных растворителях для инъекций перед их использованием.
НАИЛУЧШИЕ СПОСОБЫ ПРИМЕНЕНИЯ ИЗОБРЕТЕНИЯ
Настоящее изобретение более конкретно проиллюстрировано следующими примерами, которые, однако, не предполагают ограничение объема изобретения. Некоторые исходные соединения, используемые в примерах, являются новыми, и способы их получения из известных соединений описаны в ссылочных примерах.
Ссылочный пример 1
Суспензию 1,4 г моногидрохлорида (2S,4S)-4-фторпирролидин-2-карбоксамида, полученную методом, подобным описанному в патентной ссылке (международная публикация WO02/38451), и 3,0 мл N,N-диизопропилэтиламина в 10 мл хлороформа добавляли по каплям к раствору 0,73 мл хлорацетилхлорида в 14 мл хлороформа при охлаждении на водно-ледяной бане, и реакционную смесь перемешивали в течение 30 мин при охлаждении на водно-ледяной бане. Затем реакционную смесь концентрировали при пониженном давлении. К раствору полученного остатка в 14 мл хлороформа добавляли по каплям 2,4 мл ангидрида трифторуксусной кислоты при охлаждении на водно-ледяной бане. Затем реакционной смеси давали нагреться до комнатной температуры и перемешивали в течение 1 часа. Смесь концентрировали при пониженном далении, к образовавшемуся остатку добавляли 0,1 М соляную кислоту и экстрагировали этилацетатом. Органический слой сушили над безводным сульфатом магния. Осушитель удаляли и растворитель удаляли при пониженном давлении. Полученный остаток очищали хроматографией на колонке с силикагелем (элюент: от хлороформа до хлороформа/метанола =30/1) с получением 0,84 г (2S,4S)-1-(хлорацетил)-4-фторпирролидин-2-карбонитрила.
ЯМР: 2,33-2,67 (2H, м), 3,60-4,05 (2H, м), 4,35-4,55 (2H, м), 4,95-5,05, 5,30-5,60(2H, м).
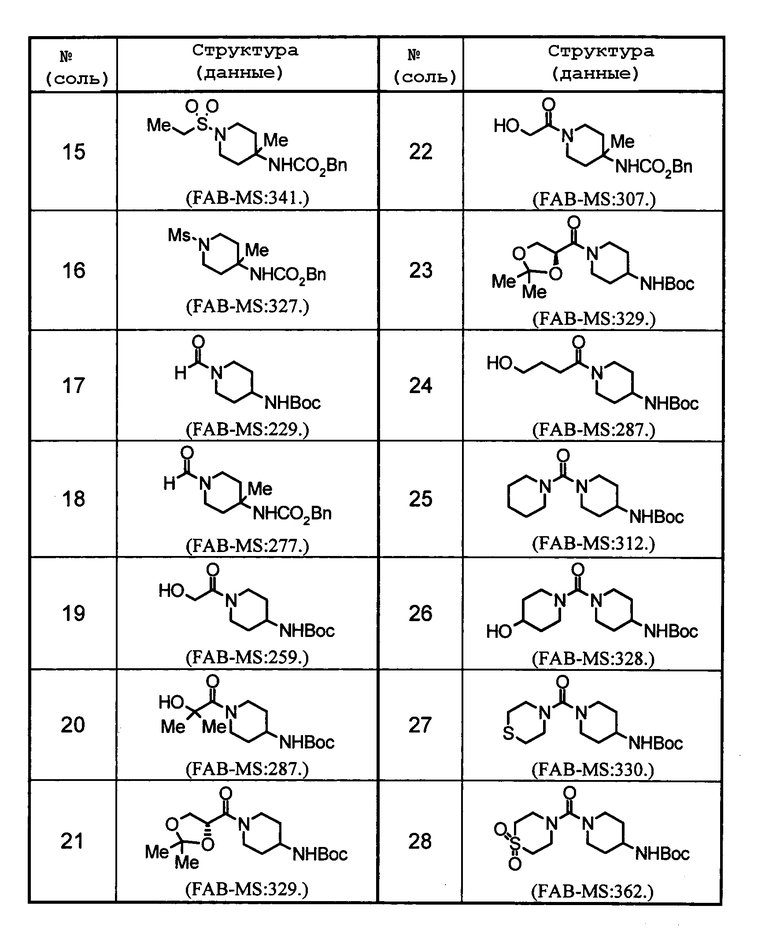
Тем же способом, что в ссылочном примере 1, из соответствующих исходных соединений получали соединения ссылочных примеров 2-4, показанных в таблице 4.
Ссылочный пример 5
К суспензии 790 мг гидрохлорида трет-бутил экзо-8-азабицикло[3.2.1]окт-3-илкарбамата, полученного по способу, описанному в J. Med. Chem. (1991), 34, 656-663 или в J.Heterocycl. Chem. (1892), 19, 485-488, в 10 мл метиленхлорида и 5 мл N,N-диметилформамида добавляли 1,3 г триэтиламина и 1,05 г метансульфонилхлорида. Реакционную смесь перемешивали при комнатной температуре в течение 1 дня и концентрировали при пониженном давлении. К полученному остатку добавляли воду и экстрагировали этилацетатом. Органический слой промывали насыщенным раствором соли и затем сушили над безводным сульфатом магния, растворитель удаляли при пониженном давлении. Полученный остаток очищали хроматографией на колонке с силикагелем (элюент: гексан/этилацетат = 7/3) и кристаллизовали из диэтилового эфира - гексана с получением 600 мг трет-бутил экзо-8-(метансульфонил)-8-азабицикло[3.2.1]окт-3-илкарбамата в виде бесцветного твердого продукта.
Тем же способом, что и в ссылочном примере 5, из соответствующих исходных веществ получали соединения ссылочных примеров 6-16, показанных в таблице 4.
Ссылочный пример 17
Раствор 2,0 г трет-бутил пиперидин-4-илкарбамата в хлороформе добавляли к смешанному раствору 4,7 мл ангидрида уксусной кислоты и 1,9 мл муравьиной кислоты и реакционную смесь перемешивали при комнатной температуре в течение 15 ч. К реакционной смеси добавляли воду и экстрагировали EtOAc. Органический слой промывали 1 М соляной кислотой, насыщенным водным раствором гидрокарбоната натрия и насыщенным раствором соли. Сушили над безводным сульфатом магния и растворитель удаляли при пониженном давлении. Полученный осадок очищали хроматографией на колонке с силикагелем (элюент: хлороформ/метанол (MeOH) = 50/1) с получением 1,7 г трет-бутил (1-формилпиперидин-4-ил)карбамата.
Тем же способом, что и в ссылочном примере 17, из соответствующего исходного соединения получали соединение по ссылочному примеру 18, показанное в таблице 4.
Ссылочный пример 19
К раствору 3,0 г трет-бутил пиперидин-4-илкарбамата в 60 мл метиленхлорида добавляли 2,5 мл триэтиламина, 2,4 г бутанола (HOBt), 1,3 г гидроксиуксусной кислоты и 3,5 г гидрохлорида WSCD и реакционную смесь перемешивали при комнатной температуре в течение 18 ч. К реакционной смеси добавляли 1М соляную кислоту и экстрагировали EtOAc. Органический слой промывали насыщенным водным раствором гидрокарбоната натрия, водой и насыщенным раствором соли. Сушили над безводным сульфатом магния и растворитель удаляли при пониженном давлении. Полученный осадок очищали хроматографией на колонке с силикагелем (элюент: хлороформ/метанол (MeOH) = 50/1) с получением 2,8 г трет-бутил [1-(гидроксиацетил)пиперидин-4-ил)]карбамата.
Тем же способом, что и в ссылочном примере 19, из соответствующего исходного соединения были получены соединения ссылочных примеров 20-24, показанные в таблице 4.
Ссылочный пример 25
Суспензию 1,0 г гидрохлорида трет-бутил пиперидин-4-илкарбамата и 0,6 мл триэтиламина в 15 мл метиленхлорида добавляли к раствору 418 мг трифосгена в 10 мл метиленхлорида при охлаждении на водно-ледяной бане. Реакционную смесь перемешивали 2 часа при охлаждении льдом и затем добавляли раствор 358 мг пиперидина и 0,6 мл триэтиламина в 5 мл метиленхлорида и перемешивали при комнатной температуре в течение 15 ч. К реакционной смеси добавляли 10% водный раствор лимонной кислоты и экстрагировали EtOAc. Органический слой промывали насыщенным раствором соли и затем сушили над безводным сульфатом магния и растворитель удаляли при пониженном давлении. Полученный твердый продукт промыли эфиром с получением 440 мг трет-бутил [1-(пиперидин-1-карбонил)пиперидин-4-ил]карбамата.
Тем же способом, что и в ссылочном примере 25, из соответствующего исходного вещества получали соединения ссылочных примеров 26 и 27, показанные в таблице 4.
Ссылочный пример 28
2,0 г м-хлорнадбензойной кислоты добавляли к 1,0 г трет-бутил [1-(тиоморфолин-4-карбонил)пиперидин-4-ил]карбамата в 10 мл метиленхлорида при охлаждении на водно-ледяной бане. Реакционной смеси давали достичь комнатной температуры и перемешивали в течение 18 ч. К реакционной смеси добавляли насыщенный водный раствор тиосульфата натрия и экстрагировали EtOAc. Органический слой промывали насыщенным водным раствором гидрокарбоната натрия и насыщенным раствором соли, сушили над безводным сульфатом магния и растворитель удаляли при пониженном давлении. Полученный твердый продукт промывали диизопропиловым эфиром с получением 607 мг трет-бутил [1-(1,1-диоксотиоморфолин-4-карбонил)пиперидин-4-ил]карбамата.
Ссылочный пример 29
Раствор 500 мг трет-бутил пиперидин-4-илкарбамата и 345 мг этилфторацетата в 1 мл трифторэтанола нагревали с обратным холодильником в течение 8 часов. Реакционную смесь концентрировали при пониженном давлении, к полученному остатку добавляли 1 М соляную кислоту. Затем экстрагировали EtOAc и органический слой промывали насыщенным водным раствором гидрокарбоната натрия и насыщенным раствором соли. Сушили над безводным сульфатом магния и растворитель удаляли при пониженном давлении. Полученный осадок очищали хроматографией на колонке с силикагелем (элюент: от хлороформа до хлороформа/метанола = 30/1) с получением 345 мг трет-бутил [1-(фторацетил)пиперидин-4-ил]карбамата.
Тем же способом, что и в ссылочном примере 29, из соответствующего исходного соединения получали соединение ссылочного примера 30, показанное в таблице 4.
Ссылочный пример 31
15 мл 4 М хлористого водорода в EtOAc добавляли к раствору 500 мг соединения ссылочного примера 5 в 15 мл EtOAc и реакционную смесь перемешивали при комнатной температуре в течение 9 часов. Полученный твердый продукт собирали фильтрацией с получением 400 мг бесцветного твердого продукта гидрохлорида экзо-8-(метансульфонил)-8-азобицикло[3.2.1]окт-3-иламина.
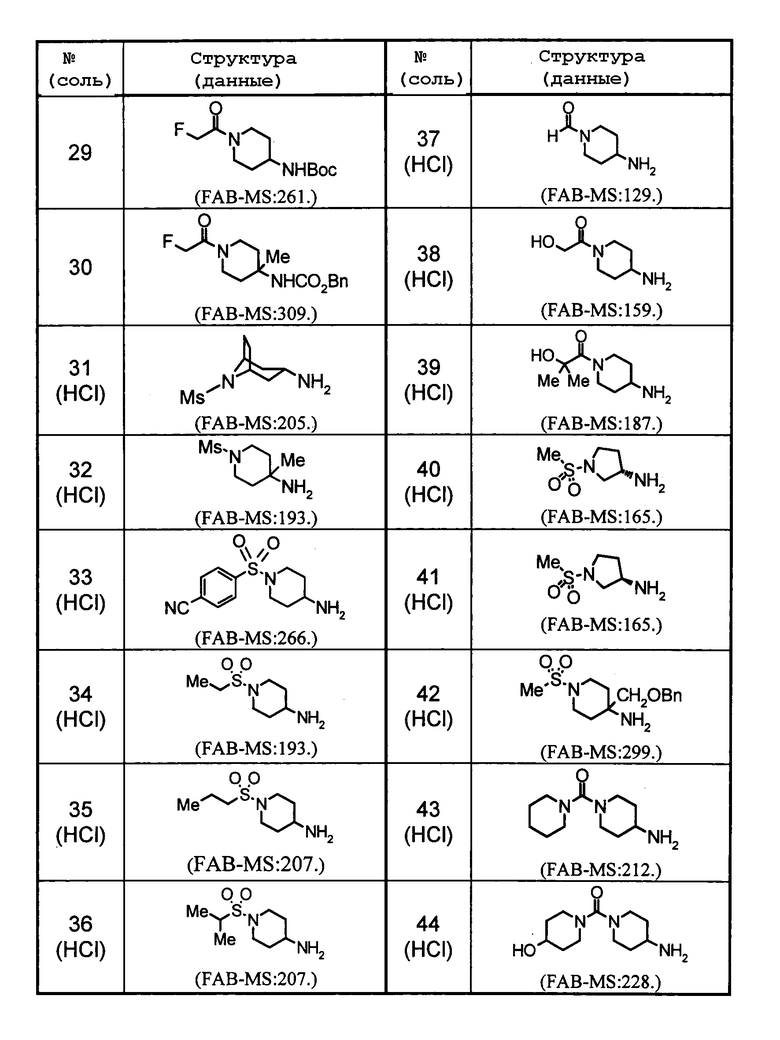
Тем же способом, что в ссылочном примере 31, из соответствующих исходных соединений получали соединения ссылочных примеров 32-49, показанные в таблице 4.
Ссылочный пример 50
300 мг 10%-го палладия-на-угле добавляли к раствору 1,4 г соединения по ссылочному примеру 22 в 30 мл MeOH и реакционную смесь перемешивали в атмосфере водорода при комнатной температуре. Нерастворимые продукты удаляли фильтрацией и фильтрат концентрировали при пониженном давлении с получением 400 мг бесцветного твердого продукта 2-(4-амино-4-метилпиперидин-1-ил)-2-оксоэтанола.
Тем же способом, что и в ссылочном примере 50, из соответствующих исходных соединений получали соединения ссылочных примеров 51-55, показанные в таблице 4.
Ссылочный пример 56
20 мл 6 М соляной кислоты добавляли к 2,2 г соединения ссылочного примера 42 и реакционную смесь кипятили с обратным холодильником в течение 24 часов. Реакционную смесь концентрировали, к полученному остатку добавляли толуол и MeOH и вновь концентрировали. Полученный остаток кристаллизовали из MeOH-диэтилового эфира и собирали с получением 1,22 г [4-амино-1-(метансульфонил)пиперидин-4-ил]метанола.
Значения сокращений в таблице указаны далее (то же справедливо и здесь).
№: номер ссылочного примера;
Соль: соль (HCl: гидрохлорид, фум: фумарат, не описано: свободное вещество);
Структура: структурная формула;
Данные: физико-химические данные (ЯМР: пик δ (м.д.) в 1Н-ЯМР, в котором внутренний стандарт представлен (CH3)4Si, и растворитель для измерений представляет собой диметилсульфоксид (ДМСО-d6), FAB-MS: масс-спектрометрия с бомбардировкой ускоренными атомами, т.пл. - температура плавления);
Ме: метил, Boc: трет-бутилоксикарбонил, Ms: метансульфонил, Bn:бензил.

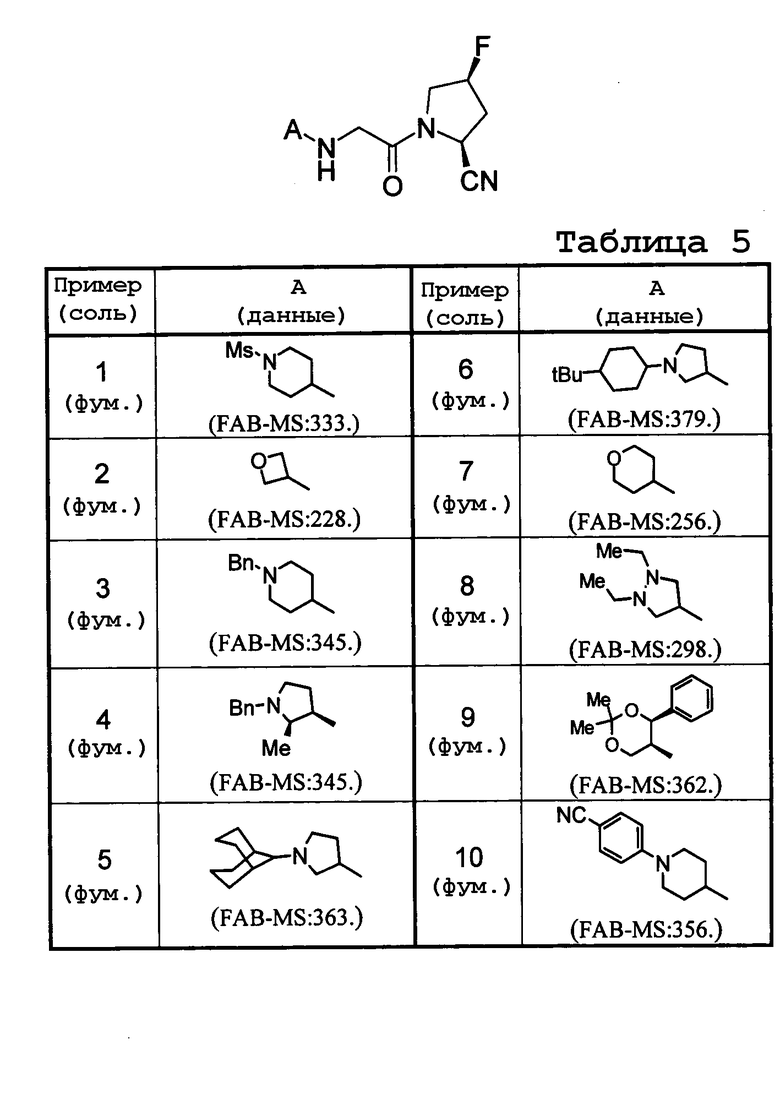
Пример 1
К суспензии 451 мг моногидрохлорида 1-(метансульфонил)пиперидин-4-амина, полученного по способу, описанному в международной публикации WO0218380, и 435 мг карбоната калия в 8 мл ацетонитрила добавляли 200 мг (2S,4S)-1-(хлорацетил)-4-фторпирролидин-2-карбонитрила, и реакционную смесь перемешивали при комнатной температуре в течение 4 дней. Нерастворимые продукты отделяли фильтрацией и к фильтрату добавляли 1,20 г силикагеля, реакционную смесь концентрировали при пониженном давлении. Полученный осадок очищали хроматографией на колонке с силикагелем (элюент: хлороформ/метанол/28% водный аммиак = от 100/1/0,1 до 20/1/0,1) с получением 487 мг бесцветного аморфного продукта. К полученному аморфному бесцветному продукту добавляли 10 мл этанола и реакционную смесь перемешивали при комнатной температуре в течение 30 мин. Нерастворимые продукты собирали фильтрацией, промывали этанолом и сушили при пониженном давлении. 230 мг полученного бесцветного твердого продукта растворяли в 2 мл тетрагидрофурана и 4 мл метанола, к которому добавляли 80 мг фумаровой кислоты. Полученный раствор концентрировали при пониженном давлении до тех пор, пока количество растворителя не становилось приблизительно 2 мл, и затем добавляли 5 мл этанола, и смесь перемешивали при комнатной температуре в течение 30 минут. Осадок собирали фильтрацией, промывали этанолом и сушили при пониженном давлении с получением 217 мг бесцветных кристаллов монофумарата (2S,4S)-4-фтор-1-({[1-(метансульфонил)пиперидин-4-ил]амино}аетил)пирролидин-2-карбонитрила.
Тем же способом, что и в примере 1, из соответствующих исходных веществ получали соединения по примерам 2-47, приведенные в таблице 5 и таблице 6.
Значения сокращений в таблицах указаны далее (то же справедливо и далее).
Пример: номер примера
А: заместитель в общей формуле
tBu: трет-бутил, Et: этил, n-Pr: н-пропил, i-Pr: изопропил, Ac: ацетил (таблица 5).



Здесь и далее данные ЯМР для соединений некоторых примеров показаны в таблице 7.
(соль)
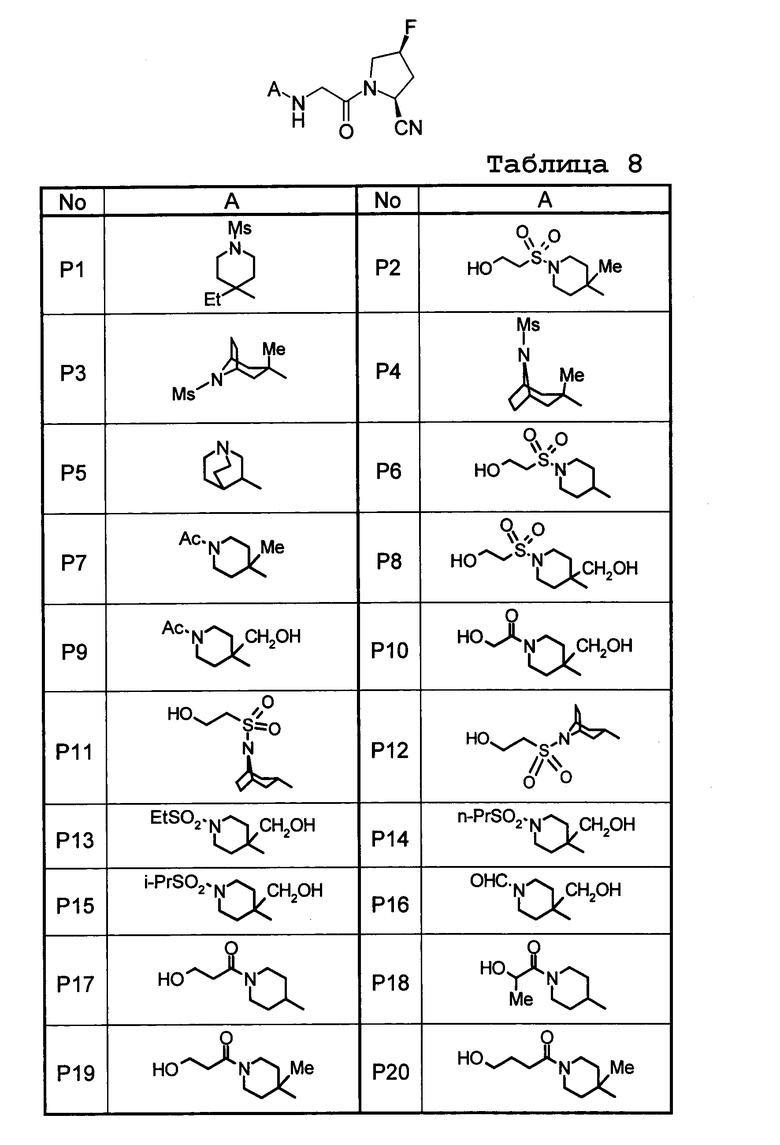
Структурные формулы других соединений по изобретению приведены в таблице 8. Их можно легко получить в соответствии со способами получения, указанными выше, в соответствии со способами, описанными в примерах, в соответствии с любыми другими методами, очевидными для специалистов в данной области, или в соответствии с модификациями данных методов.
Значение сокращения в таблице указано далее.
№: номер соединения

| название | год | авторы | номер документа |
|---|---|---|---|
| БИЦИКЛИЧЕСКИЕ ПРОИЗВОДНЫЕ ПИРРОЛА | 2005 |
|
RU2382786C2 |
| ИНГИБИТОРЫ ДИПЕПТИДИЛПЕПТИДАЗЫ IV | 2010 |
|
RU2574410C2 |
| ПРОИЗВОДНЫЕ ДИЦИКЛОАЗААЛКАНА, СПОСОБЫ ИХ ПОЛУЧЕНИЯ И ИХ ПРИМЕНЕНИЕ В МЕДИЦИНЕ | 2008 |
|
RU2487866C2 |
| ПРОИЗВОДНЫЕ ПИРРОЛИДИНА | 2009 |
|
RU2494094C2 |
| ПРОИЗВОДНЫЕ АЗАБИЦИКЛООКТАНА, СПОСОБ ИХ ПОЛУЧЕНИЯ И ИХ ПРИМЕНЕНИЯ В КАЧЕСТВЕ ИНГИБИТОРОВ ДИПЕПТИДИЛПЕПТИДАЗЫ IV | 2007 |
|
RU2447063C2 |
| АМИНОТЕТРАГИДРОПИРАНЫ В КАЧЕСТВЕ ИНГИБИТОРОВ ДИПЕПТИДИЛПЕПТИДАЗЫ-IV ДЛЯ ЛЕЧЕНИЯ ИЛИ ПРЕДУПРЕЖДЕНИЯ ДИАБЕТА | 2010 |
|
RU2550508C2 |
| ПРОИЗВОДНЫЕ ПИРИДИНА И ХИНОЛИНА, СПОСОБ ИХ ПОЛУЧЕНИЯ, ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ, ПРИМЕНЕНИЕ СОЕДИНЕНИЙ ДЛЯ ЛЕЧЕНИЯ ЗАБОЛЕВАНИЙ, АССОЦИИРОВАННЫХ С DPP-IV | 2003 |
|
RU2285693C2 |
| ПРОИЗВОДНЫЕ ПИРИДИНА И ПИРИМИДИНА, СПОСОБЫ ИХ ПОЛУЧЕНИЯ (ВАРИАНТЫ), ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ И ПРИМЕНЕНИЕ | 2003 |
|
RU2293731C2 |
| ПРОИЗВОДНЫЕ 4-АМИНОПИПЕРИДИНА | 2005 |
|
RU2396257C2 |
| НОВЫЕ ПИРИМИДИНИМИДАЗОЛАМИНЫ В КАЧЕСТВЕ МОДУЛЯТОРОВ АКТИВНОСТИ КИНАЗЫ | 2015 |
|
RU2690679C2 |
Изобретение относится к новым производным 2-циано-4-фторпирролидина формулы (I) или его фармацевтически приемлемой соли:
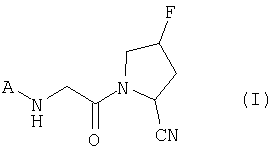
где А представляет собой группу общей формулы (II):

где В представляет собой карбонил или сульфонил;
R1 представляет собой C1-6алкил, который может необязательно замещен группой, выбранной из группы, включающей -ОН или фтор, хлор, бром или йод; фенил, необязательно замещенный CN; или морфолинил или когда В представляет собой карбонил, R1 может быть водородом. R2 представляет собой C1-6алкил, необязательно замещенный гидрокси, или водород. Соединение 1 представляет собой ингибитор дипептидилпептидазы IV, что позволяет использовать его в фармацевтической композиции, которая предназначена для лечения инсулинзависимого диабета (диабет 1 типа), инсулиннезависимого диабета (диабет 2 типа), заболеваний, связанных с резистентностью к инсулину, или ожирения. 2 н. и 6 з.п. ф-лы, 8 табл.
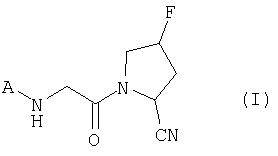
где А представляет собой группу следующей общей формулы (II):
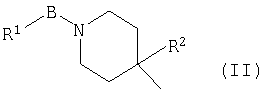
где В представляет собой карбонил или сульфонил;
R1 представляет собой C1-6алкил, который может необязательно замещен группой, выбранной из группы, включающей -ОН или фтор, хлор, бром или йод; фенил, необязательно замещенный CN; или морфолинил, или когда В представляет собой карбонил, R1 может быть водородом.
R2 представляет собой С1-6алкил, необязательно замещенный гидрокси, или водород;
4-фтор-1-({[1-(метансульфонил)пиперидин-4-ил]амино}ацетил)пирролидин-2-карбонитрил,
4-фтор-1-({[4-метил-1-(метансульфонил)пиперидин-4-ил]амино}ацетил)пирролидин-2-карбонитрил,
4-фтор-1-{[(1-гликолоилпиперидин-4-ил)амино]ацетил}пирролидин-2-карбонитрил,
4-фтор-1-{[(1-гликолоил-4-метилпиперидин-4-ил)амино]ацетил}пирролидин-2-карбонитрил,
4-фтор-1-{[(1-фторацетил-4-метилпиперидин-4-ил)амино] ацетил} пирролидин-2-карбонитрил,
4-фтор-1-{[(1-формилпиперидин-4-ил)амино]ацетил}пирролидин-2-карбонитрил,
4-фтор-1-{[(1-формил-4-метилпиперидин-4-ил)амино]ацетил}пирролидин-2-карбонитрил, или
4-фтор-1-({[1-(морфолин-4-илкарбонил)пиперидин-4-ил]амино}ацетил)пирролидин-2-карбонитрил,
или его фармацевтически приемлемая соль.
| N-ЗАМЕЩЕННЫЕ 2-ЦИАНОПИРРОЛИДИНЫ | 1997 |
|
RU2180901C2 |
| WO 00/34241 A1, 15.06.2000 | |||
| Аппарат для очищения воды при помощи химических реактивов | 1917 |
|
SU2A1 |

