Область техники, к которой относится изобретение
Настоящее изобретение относится к новым замещенным аминотетрагидропиранам, которые являются ингибиторами фермента дипептидилпептидазы-IV (“ингибиторами DPP-4”) и которые являются применимыми при лечении или предупреждении заболеваний, в которых принимает участие фермент дипептидилпептидаза-IV, таких как диабет и, конкретно, диабет типа 2. Изобретение относится также к фармацевтическим композициям, содержащим указанные соединения, и применению указанных соединений и композиций при предупреждении или лечении таких заболеваний, в которых участвует фермент дипептидилпептидаза-IV.
Уровень техники изобретения
Диабет относится к заболеванию, вызываемому множественными этиологическими факторами и характеризующемуся повышенными уровнями глюкозы плазмы или гипергликемией при состоянии голода или после введения глюкозы во время тестирования на пероральную толерантность к глюкозе. Устойчивая или нерегулируемая гипергликемия связана с повышенной и преждевременной заболеваемостью и летальностью. Часто аномальный гомеостаз глюкозы связан, как непосредственно, так и опосредованно, с изменениями метаболизма липидов, липопротеинов и аполипопротеинов и другим метаболическим и гемодинамическим заболеванием. Поэтому у пациентов с сахарным диабетом типа 2 имеется особенно повышенный риск макровоскулярных и микроваскулярных осложнений, включающих ишемическую болезнь сердца, удар, периферическое васкулярное заболевание, гипертензию, нефропатию, невропатию и ретинопатию. Следовательно, терапевтические устранения гомеостаза глюкозы, метаболизма липидов и гипертензии являются критически важными при клиническом устранении и лечении сахарного диабета.
Имеется две обычно различаемые формы диабета. При диабете типа 1, или инсулинзависимом сахарном диабете (IDDM), пациенты продуцируют мало или не продуцируют инсулин, гормон, который регулирует утилизацию глюкозы. При диабете типа 2, или инсулиннезависимом сахарном диабете (NIDDM), пациенты часто имеют уровни инсулина плазмы, которые являются такими же или даже повышенными по сравнению с недиабетическими субъектами; однако, у этих пациентов развивается резистентность к стимулирующему действию инсулина на глюкозу и метаболизм липидов в основных чувствительных к инсулину тканях, которые являются мышечными, печеночными и жировыми тканями, и уровни инсулина плазмы, хотя и повышенные, являются недостаточными для преодоления значительной инсулинорезистентности.
Инсулинорезистентность является в основном не результатом уменьшенного числа рецепторов инсулина, а дефектом после связывания с рецепторами инсулина, который еще не является понятным. Эта резистентность к инсулиновому ответу приводит к недостаточной активации инсулина в отношении потребления, окисления и сохранения глюкозы в мышце и неадекватной репрессии инсулином липолиза в жировой ткани и продуцирования и секреции глюкозы в печени.
Имеющиеся способы лечения диабета типа 2, которые не изменялись по существу много лет, обнаруживают ограничения. Хотя физическое упражнение и уменьшение калорий в режиме питания могут резко улучшить диабетическое состояние, комплаентность с таким лечением является очень недостаточной вследствие широко распространенного сидячего образа жизни и потребления избыточных пищевых продуктов, особенно пищевых продуктов, содержащих большие количества жира с высоким содержанием насыщенных жирных кислот. Увеличение уровня инсулина плазмы введением сульфонилмочевин (например, толбутамида и глипизида) или меглитинида, которые стимулируют панкреатические β-клетки для секреции большего количества инсулина, и/или инъекцией инсулина, когда сульфонилмочевины или меглитинид становятся неэффективным, может привести к концентрациям инсулина, достаточно высоким для стимуляции очень инсулинорезистентных тканей. Однако, опасно низкие уровни глюкозы в плазме могут быть результатом введения инсулина или средств, усиливающих секрецию инсулина (сульфонилмочевин или меглитинида), и может иметь место повышенный уровень инсулинорезистентности вследствие даже более высоких уровней инсулина плазмы. Бигуаниды повышают чувствительность к инсулину, приводя к некоторой коррекции гипергликемии. Однако, два бигуанида, фенформин и метформин, могут индуцировать молочный ацидоз и тошноту/диарею. Метформин имеет меньше побочных действий, чем фенформин, и часто прописывается для лечения диабета типа 2.
Глитазоны (т.е. 5-бензилтиазолидин-2,4-дионы) составляют дополнительный класс соединений с потенциалом для уменьшения интенсивности многих симптомов диабета типа 2. Эти агенты значительно повышают чувствительность к инсулину в мышечной, печеночной и жировой ткани на некоторых животных моделях диабета типа 2, приводя к частичной или полной коррекции повышенных уровней глюкозы плазмы без появления гипогликемии. Глитазоны, которые в настоящее время имеются в продаже, являются агонистами активированного пролифератором рецептора пероксисомы (PPAR), главным образом подтипа PPAR-гамма. Обычно считается, что агонизм PPAR-гамма является восприимчивым к повышенной сенсибилизации инсулина, которую наблюдают с глитазонами. Более новые агонисты PPAR, которые испытывают для лечения диабета типа 2, являются агонистами альфа-, гамма- или дельта-подтипа или их комбинациями, и во многих случаях являются химически отличными от глитазонов (т.е. они не являются по структуре тиазолидиндионами). У некоторых глитазонов, таких как троглитазон, проявляются серьезные побочные действия (например, печеночная токсичность).
До сих пор исследуются дополнительные способы лечения такого заболевания. Новые биохимические подходы, которые за последнее время были введены или до сих пор разрабатываются, включают применение ингибиторов альфа-глюкозидазы (например, акарбозы), миметики GLP-1 (например, эксенатид и лираглутид), антагонисты рецептора глюкагона, активаторы глюкокиназы и агонисты GPR-119.
Обнаружено, что соединения, которые являются ингибиторами фермента дипептидилпептидазы-IV (“DPP-4”), являются применимыми для лечения диабета, конкретно диабета типа 2 [см. WO 97/40832; WO 98/19998; патент США № 5939560; патент США № 6303661; патент США № 6699871; патент США № 6166063; Bioorg. Med. Chem. Lett., 6: 1163-1166 (1996); Bioorg. Med. Chem. Lett., 6: 2745-2748 (1996); D.J. Drucker in Exp. Opin. Invest. Drugs, 12: 87-100 (2003); K. Augustyns, et al., Exp. Opin. Ther. Patents. 13: 499-510 (2003); Ann E. Weber, J. Med. Chem., 47: 4135-4141 (2004); J.J. Holst, Exp. Opin. Emerg. Drugs, 9: 155-166 (2004); D. Kim, et al., J. Med. Chem., 48: 141-151 (2005); K. Augustyns, Exp. Opin. Ther. Patents, 15: 1387-1407 (2005); H.-U. Demuth in Biochim. Biophys. Acta, 1751: 33-44 (2005) и R. Mentlein, Exp. Opin. Invest. Drugs. 14: 57-64 (2005).
Дополнительными патентными публикациями, в которых описываются ингибиторы DPP-4, применимые для лечения диабета, являются следующие: WO 2006/009886 (26 января 2006); WO 2006/039325 (13 апреля 2006); WO 2006/058064 (1 июня 2006); WO 2006/127530 (30 ноября 2006); WO 2007/024993 (1 марта 2007); WO 2007/070434 (21 июня 2007); WO 2007/087231 (2 августа 2007); WO 07/097931 (30 августа 2007); WO 07/126745 (8 ноября 2007); WO 07/136603 (29 ноября 2007) и WO 08/060488 (22 мая 2008).
Применимость ингибиторов DDP-4 при лечении диабета типа 2 основана на том факте, что DPP-4 in vivo легко инактивирует подобный глюкагону пептид-1 (GLP-1) и пептид, ингибирующий секрецию желудочного сока (GIP). GLP-1 и GIP являются инкретинами и продуцируются, когда потребляется пища. Инкретины стимулируют продуцирование инсулина. Ингибирование DPP-4 приводит к пониженной инактивации инкретинов, и это, в свою очередь, приводит к повышенной эффективности инкретинов при стимуляции продуцирования инсулина поджелудочной железой. Поэтому ингибирование DPP-4 приводит к повышенному уровню инсулина сыворотки. Поскольку инкретины продуцируются преимущественно организмом только тогда, когда потребляется пища, предполагается, что ингибирование DPP-4 не повышает уровень инсулина в неподходящее время, такое как время между приемами пищи, которое может привести к чрезмерно низкому уровню сахара в крови (гипогликемии). Поэтому предполагается, что ингибирование DDP-4 повышает уровень инсулина без повышения риска гипогликемии, которая является опасным побочным действием, связанные с применением средств, усиливающих секрецию инсулина.
Ингибиторы DPP-4 имеют также другие терапевтические эффективности, описываемые в контексте. Требуются новые соединения, так чтобы можно было найти улучшенные ингибиторы DPP-4 для лечения диабета и потенциально других заболеваний и состояний. В частности, имеется потребность в ингибиторах DPP-4, которые являются селективными по сравнению с другими членами семейства серинпептидазы, которое включает пролиндипептидазу (QPP), DPP8 и DPP покоящихся клеток [см. G. Lankas, et al., "Dipeptidyl Peptidase-IV Inhibition for the Treatment of Type 2 Diabetes: Potential Importance of Selectivity Over Dipeptidyl Peptidases 8 and 9", Diabetes, 54: 2988-2994 (2005); N.S. Kang, et al., "Docking-based 3D-QSAR study for selectivity of DPP4, DPP8, and DPP9 inhibitors", Bioorg. Med. Chem. Lett., 17: 3716-3721 (2007)].
Терапевтический потенциал ингибиторов DPP-4 для лечения диабета типа 2 обсуждается (i) D.J. Drucker, Exp. Opin. Invest. Drugs, 12: 87-100 (2003); (ii) K. Augustyns, et al., Exp. Opin. Ther. Patents, 13: 499-510 (2003); (iii) J.J. Holst, Exp. Opin. Emerg. Drugs, 9: 155-166 (2004); (iv) H.-U. Demuth, et al., Biochim. Biophvs. Acta, 1751: 33-44 (2005); (v) R. Mentlein, Exp. Opin. Invest. Drugs. 14: 57-64 (2005); (vi) K. Augustyns, "Inhibitors of proline-specific dipeptidyl peptidases: DPP IV inhibitors as a novel approach for the treatment of Type 2 diabetes", Exp. Opin. Ther. Patents. 15: 1387-1407 (2005); (vii) D.J. Drucker and M.A. Nauck, "The incretin system: GLP-1 receptor agonists and dipeptidyl peptidase-4 inhibitors in Type 2 diabetes", The Lancet. 368: 1696-1705 (2006); (viii) T.W. von Geldern and J.M. Trevillyan, ""The Next Big Thing" in Diabetes: Clinical Progress on DPP-IV Inhibitors", Drug Dev. Res., 67: 627-642 (2006); (ix) B.D. Green et al., "Inhibition of dipeptidyl peptidase IV activity as a therapy of Type 2 diabetes", Exp. Opin. Emerging Drugs. 11: 525-539 (2006); (x) J. J. Holst and C.F. Deacon, "New Horizons in Diabetes Therapy", Immun., Endoc. & Metab. Agents in Med. Chem., 7: 49-55 (2007); (xi) R.K. Campbell, "Rationale for Dipeptidyl Peptidase 4 Inhibitors: a New Class of Oral Agents for the Treatment of Type 2 Diabetes Mellitus", Ann. Pharmacother., 41: 51-60 (2007); (xii) Z. Pei, "From the bench to the bedside: Dipeptidyl peptidase IV inhibitors, a new class of oral antihyperglycemic agents", Curr. Opin. Drug Discovery Development, 11: 512-532 (2008); and (xiii) J.J. Holst, et al., "Glucagon-like peptide-1, glucose homeostasis, and diabetes, Trends in Molecular Medicine, 14: 161-168 (2008). Конкретные ингибиторы DPP-4, либо уже одобренные, либо подвергающиеся клиническому исследованию для лечения диабета типа 2, включают ситаглиптин, вилдаглиптин, саксаглиптин, алоглиптин, кармеглиптин, мелоглиптин и дутоглиптин.
Сущность изобретения
Настоящее изобретение относится к новым замещенным 3-аминотетрагидропиранам, которые являются ингибиторами фермента дипептидилпептидаза-IV (“ингибиторами DPP-4”) и которые являются применимыми при лечении или предотвращении заболеваний, в которых принимает участие фермент дипептидилпептидаза-IV, таких как диабет и особенно диабет типа 2. Изобретение относится также к фармацевтическим композициям, содержащим указанные соединения, и применению указанных соединений и композиций при предупреждении или лечении таких заболеваний, в которых принимает участие фермент дипептидилпептидаза-IV.
Подробное описание изобретения
Настоящее изобретение относится к новым замещенным 3-аминотетрагидропиранам, которые являются применимыми в качестве ингибиторов дипептидилпептидазы-IV. Соединения, описанные в контексте, имеют преимущества по сравнению с существующими терапиями, поскольку они действуют посредством глюкозазависимого механизма, тем самым снижая риск гипогликемии. Кроме того, описанные в контексте соединения, имеют более благоприятные фармакокинетические свойства, включающие проникновение в головной мозг и/или более длительную продолжительность действия по сравнению с другими ингибиторами DPP-4. Соединения настоящего изобретения описаны структурной формулой I
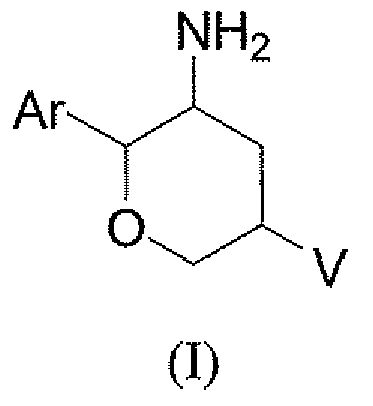
и их фармацевтически приемлемыми солями, в которой
Ar представляет собой фенил, незамещенный или замещенный одним-пятью атомами галогена;
V выбран из группы, состоящей из
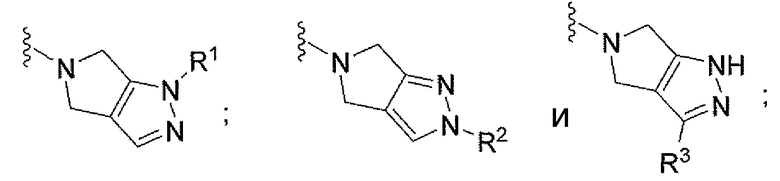
каждый из R1 и R2 независимо выбран из группы, состоящей из
С1-С6алкила;
циклоалкила;
гетероциклила и
гетероарила;
R3 выбран из группы, состоящей из
С1-С6алкила;
циклоалкила;
гетероциклила;
гетероарила;
циано;
-С(О)ОС1-С6алкила и
-С(О)NH2,
где С1-С6алкил, циклоалкил, гетероциклил и гетероарил являются незамещенными или замещенными 1-4 заместителями, независимо выбранными из группы, состоящей из
циано;
-ОН;
-С(О)NH2;
-CO2H;
-C(O)OC1-6алкила;
галогена;
оксо и
-С(О)гетероциклила.
В некоторых вариантах осуществления соединений, описанных в контексте, Ar необязательно замещен одним-тремя заместителями, независимо выбранными из группы, состоящей из атома фтора, хлора, брома. Например, Ar замещен одним-тремя заместителями, независимо выбранными из группы, состоящей из атома фтора, хлора, брома. В одном варианте осуществления Ar замещен двумя-тремя атомами фтора. В других вариантах осуществления Ar замещен двумя атомами фтора. В других вариантах осуществления Ar замещен тремя атомами фтора. В классе этих вариантов осуществления Ar представляет собой 2,5-дифторфенил или 2,4,5-трифторфенил.
В некоторых вариантах осуществления описанных в контексте соединений V выбран из группы, состоящей из
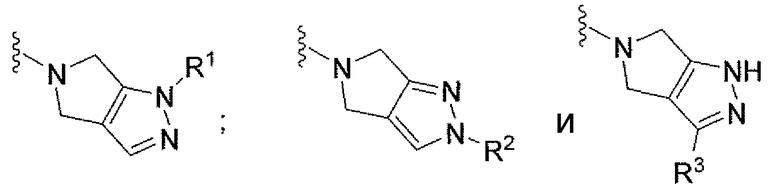
где каждый из R1 и R2 независимо выбран из группы, состоящей из
С1-С6алкила;
циклоалкила и
гетероциклила;
R3 выбран из группы, состоящей из
С1-С6алкила;
циклоалкила;
циано;
гетероциклила и
-С(О)NH2;
где С1-С6алкил, циклоалкил и гетероциклил являются незамещенными или замещенными 1-4 заместителями, независимо выбранными из группы, состоящей из
циано;
-ОН;
-С(О)NH2;
-CO2H;
-C(O)OC1-6алкила;
галогена;
оксо и
-С(О)гетероциклила.
В другом варианте осуществления каждый из R1 и R2 независимо выбран из группы, состоящей из С1-С6алкила и гетероциклила. В другом варианте осуществления каждый из R1 и R2 независимо выбран из группы, состоящей из С1-С6алкила и гетероарила. В другом варианте осуществления каждый из R1 и R2 представляет собой С1-С6алкил. Еще в одном варианте осуществления каждый из R1 и R2 независимо выбран из группы, состоящей из С1-С6алкила и циклоалкила. Во всех описанных в контексте вариантах осуществления С1-С6алкил может быть неразветвленным или разветвленным. В некоторых вариантах осуществления С1-С6алкил является неразветвленным. В некоторых вариантах осуществления С1-С6алкил является разветвленным.
В другом варианте осуществления R3 выбран из группы, состоящей из С1-С6алкила, гетероарила, гетероциклила и -С(О)NH2. В другом варианте осуществления R3 выбран из группы, состоящей из С1-С6алкила, гетероарила и -С(О)NH2. В другом варианте осуществления каждый R3 выбран из группы, состоящей из С1-С6алкила и -С(О)NH2. В другом варианте осуществления каждый R3 представляет собой циано. Еще в другом варианте осуществления R3 представляет собой С1-С6алкил. Еще в другом варианте осуществления R3 выбран из группы, состоящей из С1-С6алкила и циклоалкила. Еще в другом варианте осуществления каждый из R1, R2 и R3 представляет собой С1-С6алкил. Во всех вариантах осуществления, описанных в контексте, С1-С6алкил может быть неразветвленным или разветвленным. В некоторых вариантах осуществления С1-С6алкил является неразветвленным. В некоторых вариантах осуществления С1-С6алкил является разветвленным.
В некоторых вариантах осуществления, описанных в контексте, циклоалкил включает, но не ограничивается перечисленным, циклопропил, циклопентил и циклогексил. Кроме того, в вариантах осуществления, описанных в контексте, гетероциклил включает, но не ограничивается перечисленным, морфолин. В вариантах осуществления, описанных в контексте, гетероарил включает, но не ограничивается перечисленным, тетразол.
В некоторых вариантах осуществления соединений, описанных в контексте, V выбран из группы, состоящей из
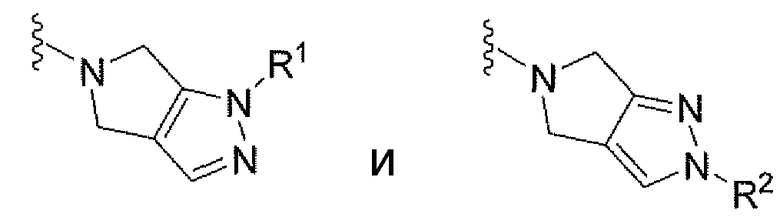
где R1 и R2 имеют значения, указанные выше. В другом варианте осуществления каждый из R1 и R2 независимо выбран из группы, состоящей из
С1-С6алкила;
циклоалкила;
гетероциклила и
гетероарила;
где С1-С6алкил, циклоалкил, гетероциклил и гетероарил являются незамещенными или замещенными 1-4 заместителями, независимо выбранными из группы, состоящей из
-ОН;
-С(О)NH2;
-CO2H;
-C(O)OC1-6алкила;
галогена и
оксо.
Еще в одном варианте осуществления каждый из R1 и R2 представляет собой C1-C6алкил, где C1-C6алкил замещен 1-4 заместителями, независимо выбранными из группы, состоящей из
циано;
-ОН;
-С(О)NH2;
-CO2H;
-C(O)OC1-6алкила;
галогена;
оксо и
-С(О)гетероциклила.
Еще в одном варианте осуществления каждый из R1 и R2 представляет собой C1-C6алкил, где C1-C6алкил замещен -С(О)NH2.
В другом варианте осуществления каждый из R1, R2 и R3 независимо выбран из группы, состоящей из С1-С6алкила, циклоалкила, гетероциклила и гетероарила, где С1-С6алкил, циклоалкил, гетероциклил и гетероарил замещен 1-4 заместителями, независимо выбранными из группы, состоящей из
циано;
-ОН;
-С(О)NH2;
-CO2H;
-C(O)OC1-6алкила и
галогена.
В другом варианте осуществления каждый из R1, R2 и R3 независимо выбран из группы, состоящей из С1-С6алкила, циклоалкила, гетероциклила и гетероарила, где С1-С6алкил, циклоалкил, гетероциклил и гетероарил замещены 1-4 заместителями, независимо выбранными из группы, состоящей из -ОН; циано; -С(О)NH2; -CO2H и -С(О)С1-6алкила. В другом варианте осуществления каждый из R1, R2 и R3 независимо выбран из группы, состоящей из С1-С6алкила, циклоалкила, гетероциклила и гетероарила, где С1-С6алкил, циклоалкил, гетероциклил и гетероарил замещены 1-4 заместителями, независимо выбранными из группы, состоящей из -ОН; -С(О)NH2 и -СО2Н. В другом варианте осуществления каждый из R1, R2 и R3 независимо выбран из группы, состоящей из С1-С6алкила, циклоалкила, гетероциклила и гетероарила, где С1-С6алкил, циклоалкил, гетероциклил и гетероарил замещены 1-4 заместителями, независимо выбранными из группы, состоящей из -ОН; циано; -С(О)NH2 и -СО2Н. В другом варианте осуществления каждый из R1, R2 и R3 независимо выбран из группы, состоящей из С1-С6алкила, циклоалкила, гетероциклила и гетероарила, где С1-С6алкил, циклоалкил, гетероциклил и гетероарил замещены 1-4 заместителями, независимо выбранными из группы, состоящей из -ОН; -С(О)NH2; галогена и -СО2Н. В другом варианте осуществления каждый из R1 и R2 независимо выбран из группы, состоящей из С1-С6алкила, циклоалкила, гетероциклила и гетероарила, где С1-С6алкил, циклоалкил, гетероциклил и гетероарил замещены 1-4 заместителями, независимо выбранными из группы, состоящей из -ОН; -С(О)NH2 и -СО2Н.
Например, вариант осуществления, описанный в контексте, включает соединение формулы 1 или его фармацевтически приемлемую соль, где V представляет собой
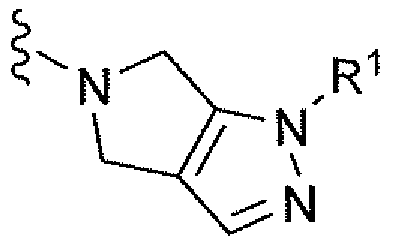
Ar представляет собой фенил, замещенный двумя-тремя атомами фтора;
R1 представляет собой С1-С6алкил;
где С1-С6алкил замещен 1-4 заместителями, независимо выбранными из группы, состоящей из
-ОН;
-С(О)NH2;
-CO2H;
-C(O)OC1-6алкила;
галогена;
оксо и
-С(О)гетероциклила.
Например, вариант осуществления, описанный в контексте, включает соединение формулы 1 или его фармацевтически приемлемую соль, где V представляет собой
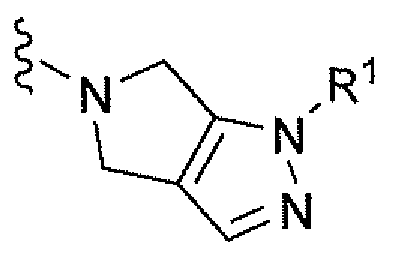
Ar представляет собой фенил, замещенный двумя-тремя атомами фтора;
R1 представляет собой С1-С6алкил;
где С1-С6алкил замещен 1-4 заместителями, независимо выбранными из группы, состоящей из -ОН и -С(О)NH2.
Например, вариант осуществления, описанный в контексте, включает соединение формулы 1 или его фармацевтически приемлемую соль, где V представляет собой
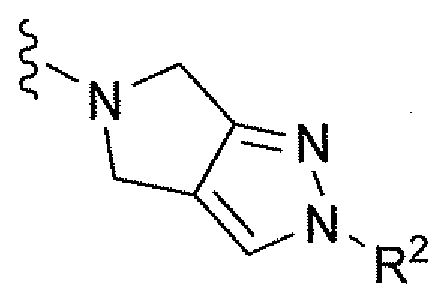
Ar представляет собой фенил, замещенный двумя-тремя атомами фтора;
R2 представляет собой С1-С6алкил;
где С1-С6алкил замещен 1-4 заместителями, независимо выбранными из группы, состоящей из
-ОН;
-С(О)NH2;
-CO2H;
-C(O)OC1-6алкила;
галогена;
оксо и
-С(О)гетероциклила.
Например, вариант осуществления, описанный в контексте, включает соединение формулы 1 или его фармацевтически приемлемую соль, где V представляет собой
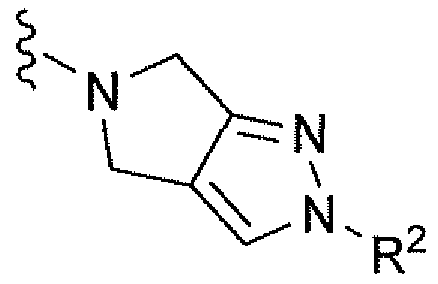
Ar представляет собой фенил, замещенный двумя-тремя атомами фтора;
R2 представляет собой С1-С6алкил;
где С1-С6алкил замещен 1-4 заместителями, независимо выбранными из группы, состоящей из -ОН и -С(О)NH2.
Например, вариант осуществления, описанный в контексте, включает соединение формулы 1 или его фармацевтически приемлемую соль, где V представляет собой

Ar представляет собой фенил, замещенный двумя-тремя атомами фтора;
R2 представляет собой С1-С6алкил;
где С1-С6алкил замещен -ОН.
Например, другой вариант осуществления, описанный в контексте, включает соединение формулы 1 или его фармацевтически приемлемую соль, где V представляет собой
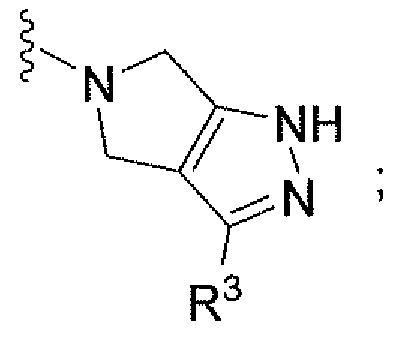
Ar представляет собой фенил, замещенный одним-пятью атомами галогена;
R3 независимо выбран из группы, состоящей из
С1-С6алкила;
циклоалкила;
гетероциклила,
гетероарила;
циано;
-С(О)ОС1-С6алкила и
-С(О)NH2;
где С1-С6алкил, циклоалкил, гетероциклил и гетероарил могут быть замещены 1-4 заместителями, независимо выбранными из группы, состоящей из
циано;
-ОН;
-С(О)NH2;
-СО2Н;
-С(О)ОС1-6алкила;
галогена;
оксо и
-С(О)гетероциклила.
Например, другой вариант осуществления, описанный в контексте, включает соединение формулы 1 или его фармацевтически приемлемую соль, где V представляет собой
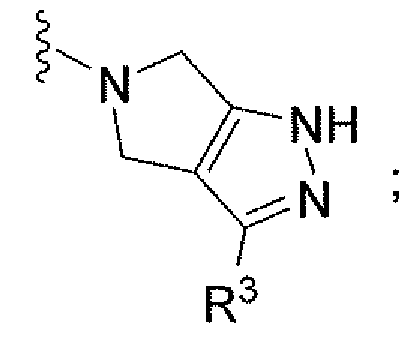
Ar представляет собой фенил, замещенный двумя-тремя атомами галогена;
R3 независимо выбран из группы, состоящей из
С1-С6алкила;
гетероарила;
циано;
-С(О)ОС1-6алкила и
-С(О)NH2;
где С1-С6алкил и гетероарил могут быть замещены 1-4 заместителями, независимо выбранными из группы, состоящей из
-ОН;
-С(О)NH2;
-СО2Н и
-С(О)ОС1-6алкила.
Например, другой вариант осуществления, описанный в контексте, включает соединение формулы 1 или его фармацевтически приемлемую соль, где V представляет собой

Ar представляет собой фенил, замещенный двумя-тремя атомами галогена;
R3 независимо выбран из группы, состоящей из
С1-С6алкила;
циано и
-С(О)NH2;
где С1-С6алкил и гетероарил могут быть замещены 1-4 заместителями, независимо выбранными из группы, состоящей из
-ОН;
-С(О)NH2;
галогена;
-СО2Н и
-С(О)ОС1-6алкила.
Например, другой вариант осуществления, описанный в контексте, включает соединение формулы 1 или его фармацевтически приемлемую соль, где V представляет собой
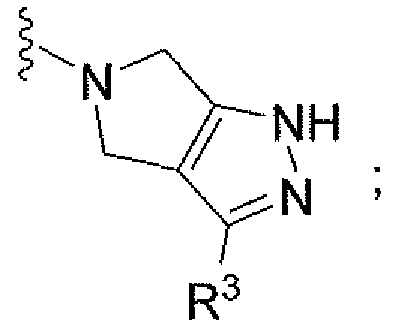
Ar представляет собой фенил, замещенный двумя-тремя атомами галогена;
R3 независимо выбран из группы, состоящей из
циано;
-С(О)NH2;
-СО2Н;
-С(О)ОС1-6алкила и
гетероциклила,
где гетероциклил не замещен или замещен 1-4 заместителями, независимо выбранными из группы, состоящей из
циано;
-ОН;
-С(О)NH2;
-СО2Н;
-С(О)ОС1-6алкила;
галогена;
оксо и
-С(О)гетероциклила.
В другом варианте осуществления соединений настоящего изобретения предложены соединения структурных формул Ia и Ib указанной стереохимической конфигурации, имеющие транс-ориентацию заместителей Ar и NH2 у двух стереогенных атомов углерода тетрагидропирана, отмеченных знаком *
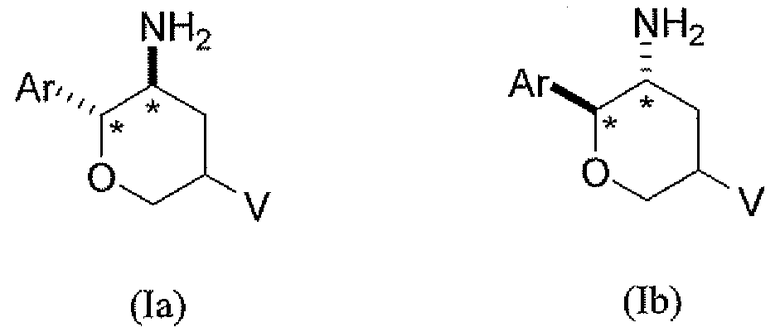
или их фармацевтически приемлемые соли, где Ar и V имеют значения, указанные выше.
В классе данного варианта осуществления предложены соединения структурной формулы Ia указанной абсолютной стереохимической конфигурации, имеющие транс-ориентацию заместителей Ar и NH2 у двух стереогенных атомов углерода тетрагидропирана, отмеченных знаком *
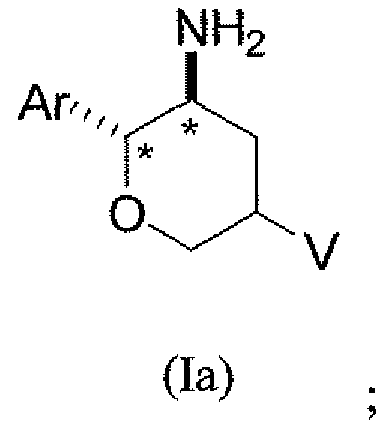
и их фармацевтически приемлемые соли.
Во втором классе данного варианта осуществления предложены соединения структурных формул Ic и Id указанной стереохимической конфигурации, имеющие транс-ориентацию заместителей Ar и NH2, транс-ориентацию заместителей Ar и V и цис-ориентацию заместителей NH2 и V у трех стереогенных атомов углерода тетрагидропирана, отмеченных знаком *
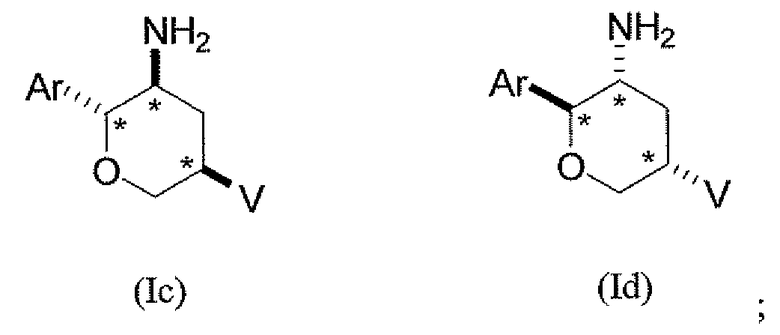
или их фармацевтически приемлемые соли.
В подклассе данного класса предложены соединения структурной формулы Ic указанной стереохимической конфигурации, имеющие транс-ориентацию заместителей Ar и NH2, транс-ориентацию заместителей Ar и V и цис-ориентацию заместителей NH2 и V у трех стереогенных атомов углерода тетрагидропирана, отмеченных знаком *
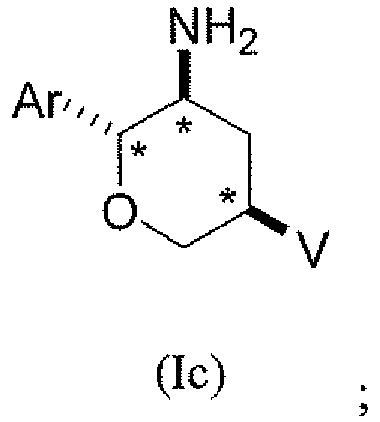
или их фармацевтически приемлемые соли.
В подклассе этого подкласса V выбран из группы, состоящей из
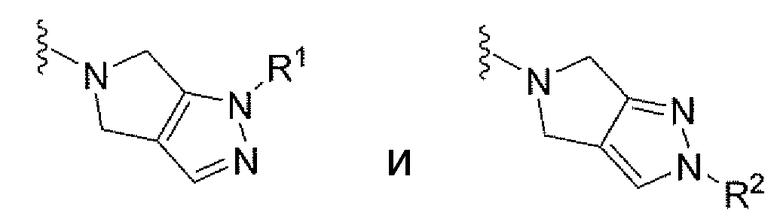
или их фармацевтически приемлемых солей, где R1 и R2 имеют значения, указанные выше. В подклассе этого подкласса каждый из R1 и R2 независимо выбран из группы, состоящей из С1-С6алкила, циклоалкила, гетероциклила и гетероарила, где С1-С6алкил, циклоалкил, гетероциклил и гетероарил может быть замещен 1-4 заместителями, независимо выбранными из группы, состоящей из -ОН; -С(О)NH2 и -СО2Н.
В третьем классе данного варианта осуществления предложены соединения структурных формул Ie и If указанной стереохимической конфигурации, имеющие транс-ориентацию заместителей Ar и NH2, цис-ориентацию заместителей Ar и V и транс-ориентацию заместителей NH2 и V трех стереогенных атомов углерода тетрагидропирана, отмеченных знаком *
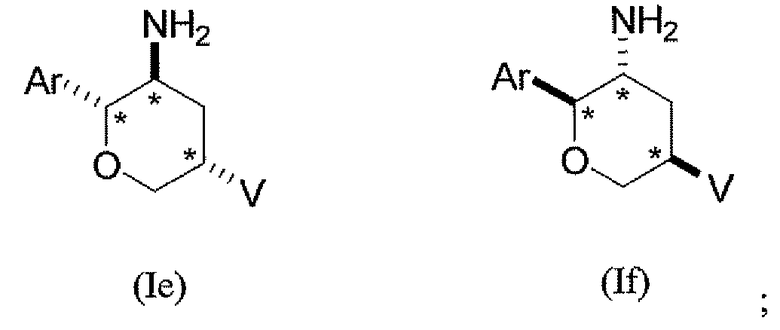
или их фармацевтически приемлемые соли.
В подклассе данного класса предложены соединения структурной формулы Ie указанной абсолютной стереохимической конфигурации, имеющие транс-ориентацию заместителей Ar и NH2, цис-ориентацию заместителей Ar и V и транс-ориентацию заместителей NH2 и V трех стереогенных атомов углерода тетрагидропирана, отмеченных знаком *
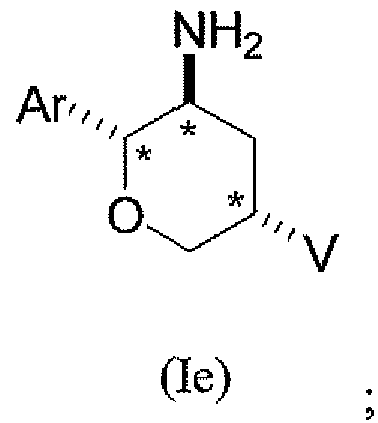
или их фармацевтически приемлемые соли.
В подклассе этого подкласса V выбран из группы, состоящей из
или их фармацевтически приемлемых солей, где R1 и R2 имеют значения, указанные выше. В подклассе этих подклассов каждый из R1 и R2 независимо выбран из группы, состоящей из С1-С6алкила, циклоалкила, гетероциклила и гетероарила, где С1-С6алкил, циклоалкил, гетероциклил и гетероарил может быть замещен 1-4 заместителями, независимо выбранными из группы, состоящей из -ОН; -С(О)NH2; -СО2Н.
Неограничивающимися примерами соединений настоящего изобретения, которые являются применимыми в качестве ингибиторов дипептидилпептидазы-IV, являются следующие структуры, имеющие указанные абсолютные стереохимические конфигурации у трех стереогенных атомов углерода тетрагидропирана.
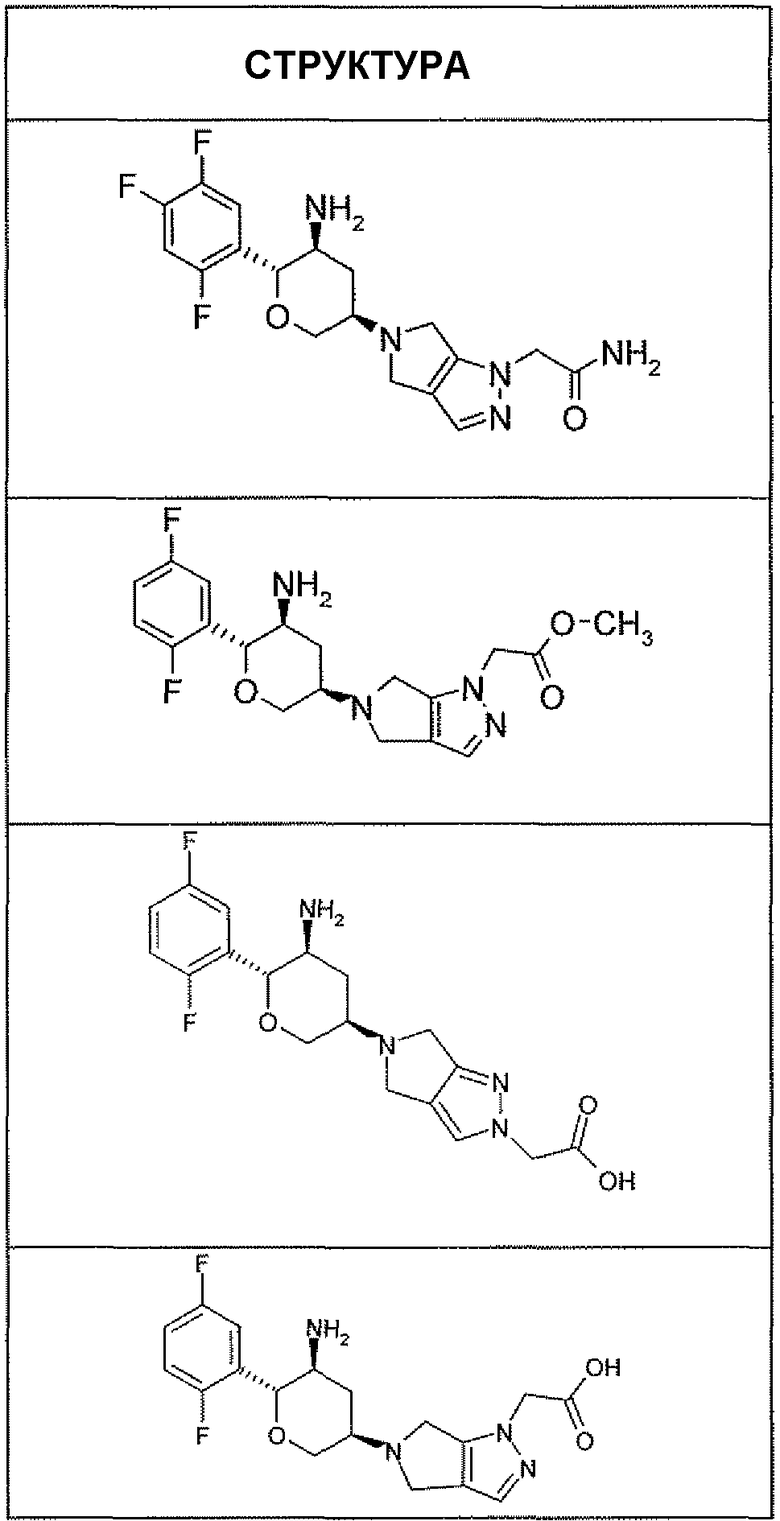

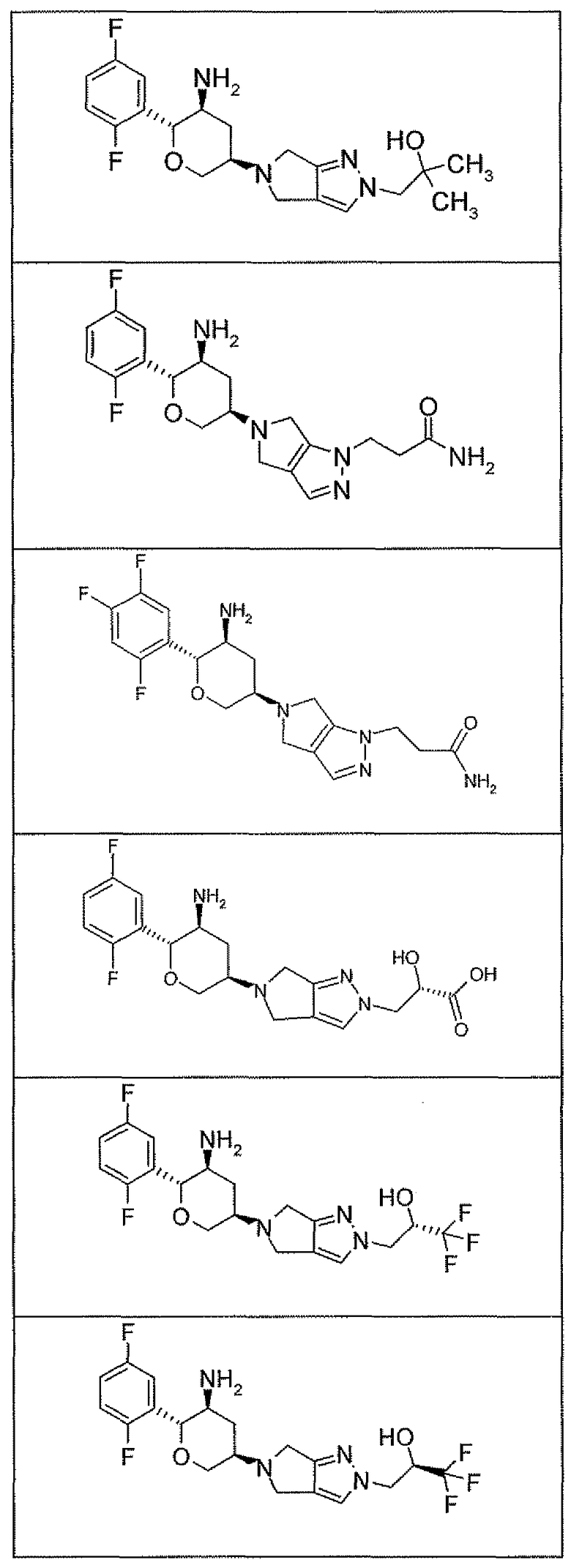
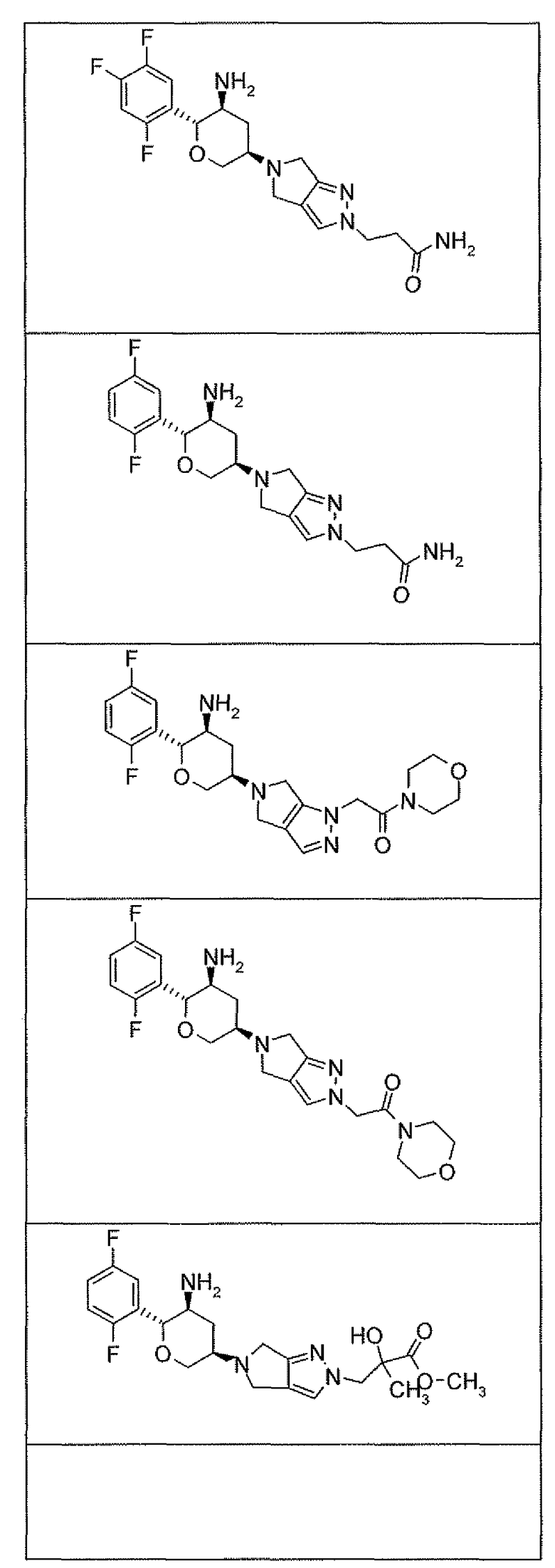
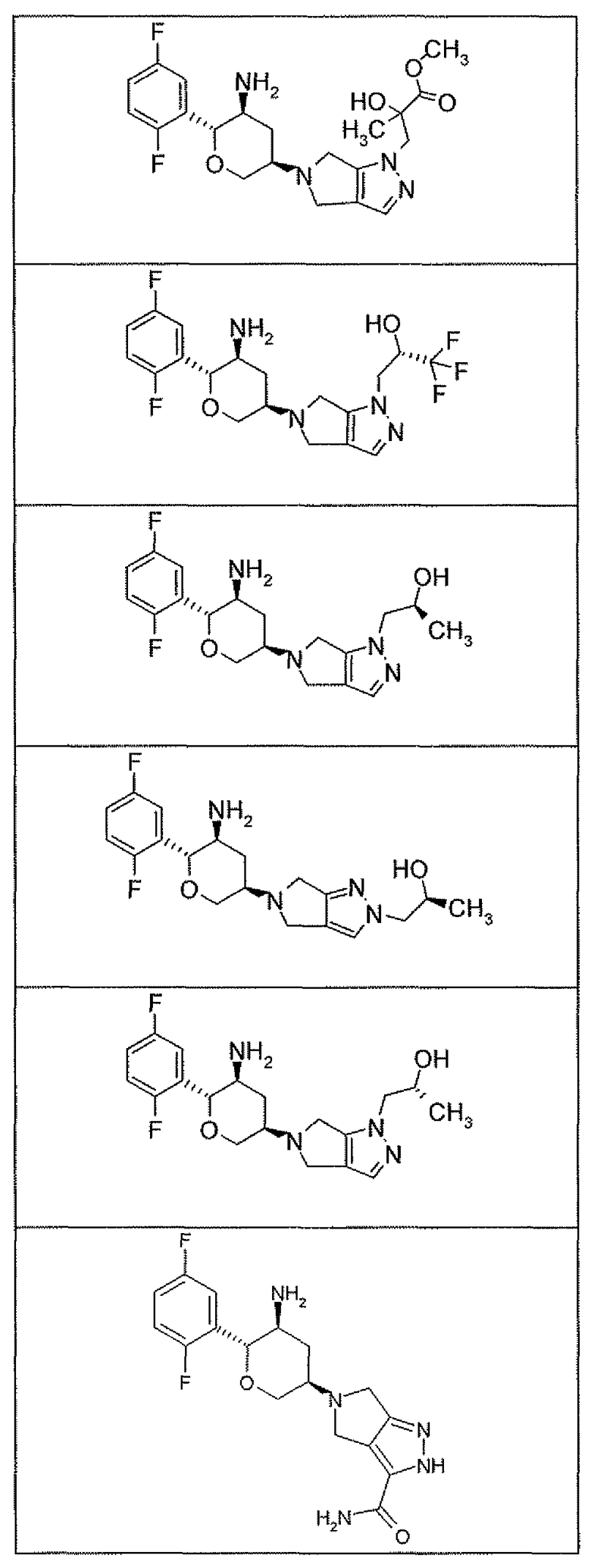
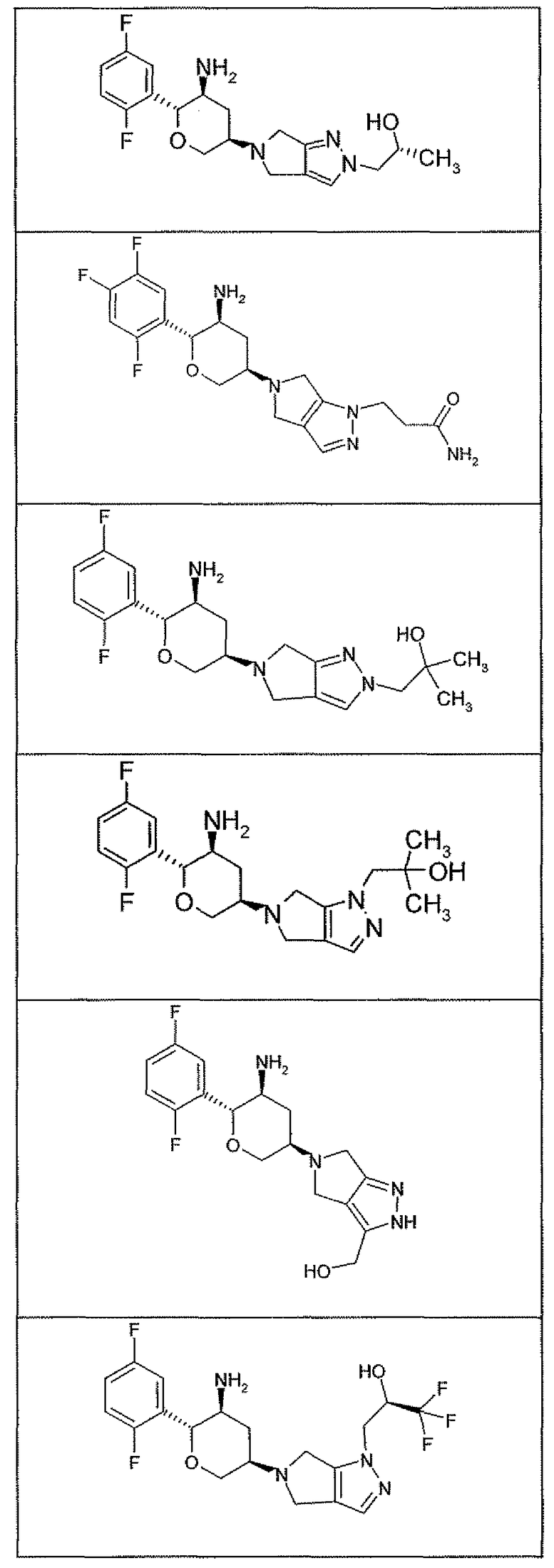
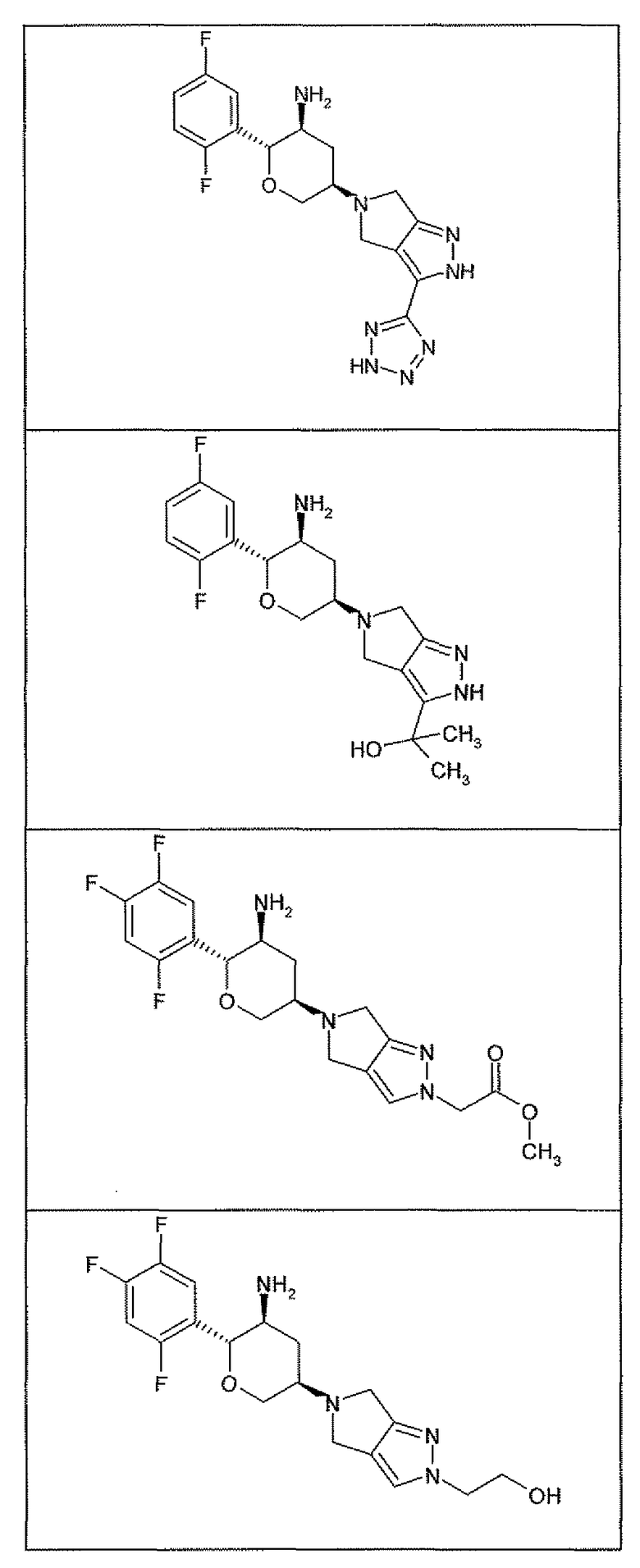
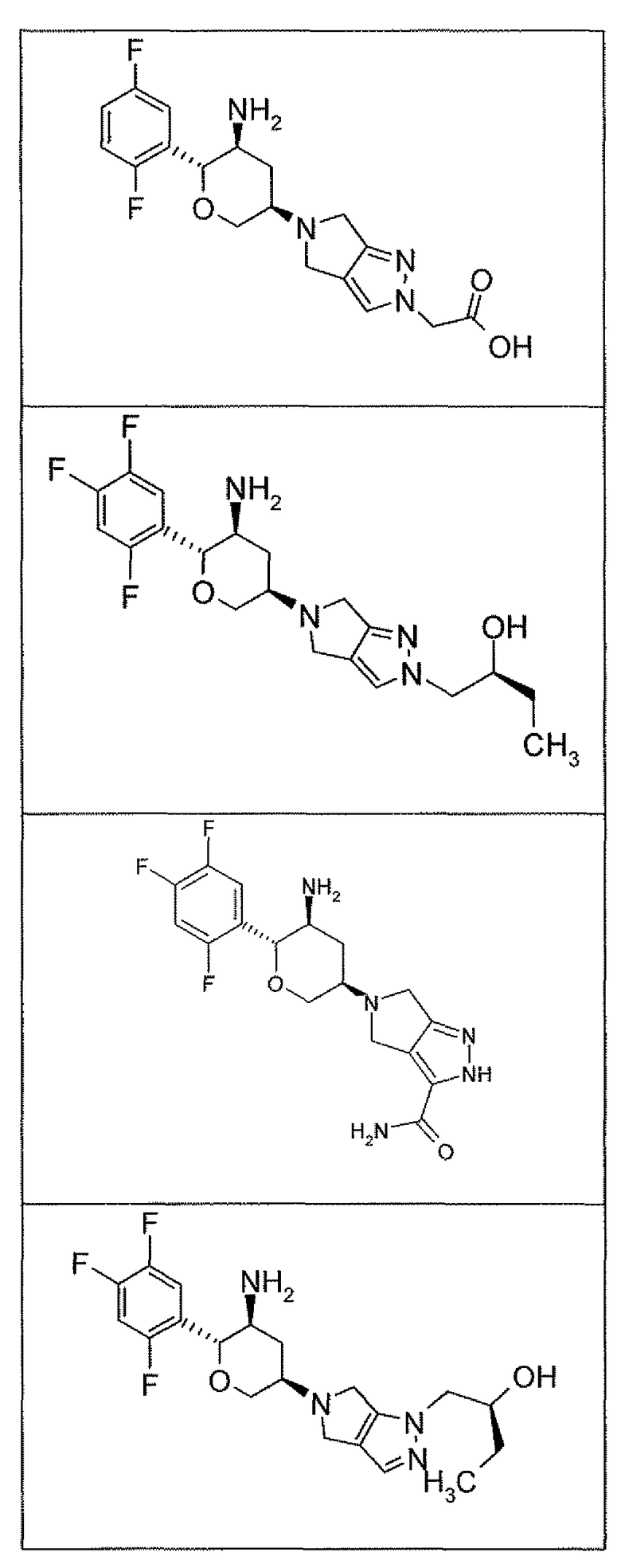
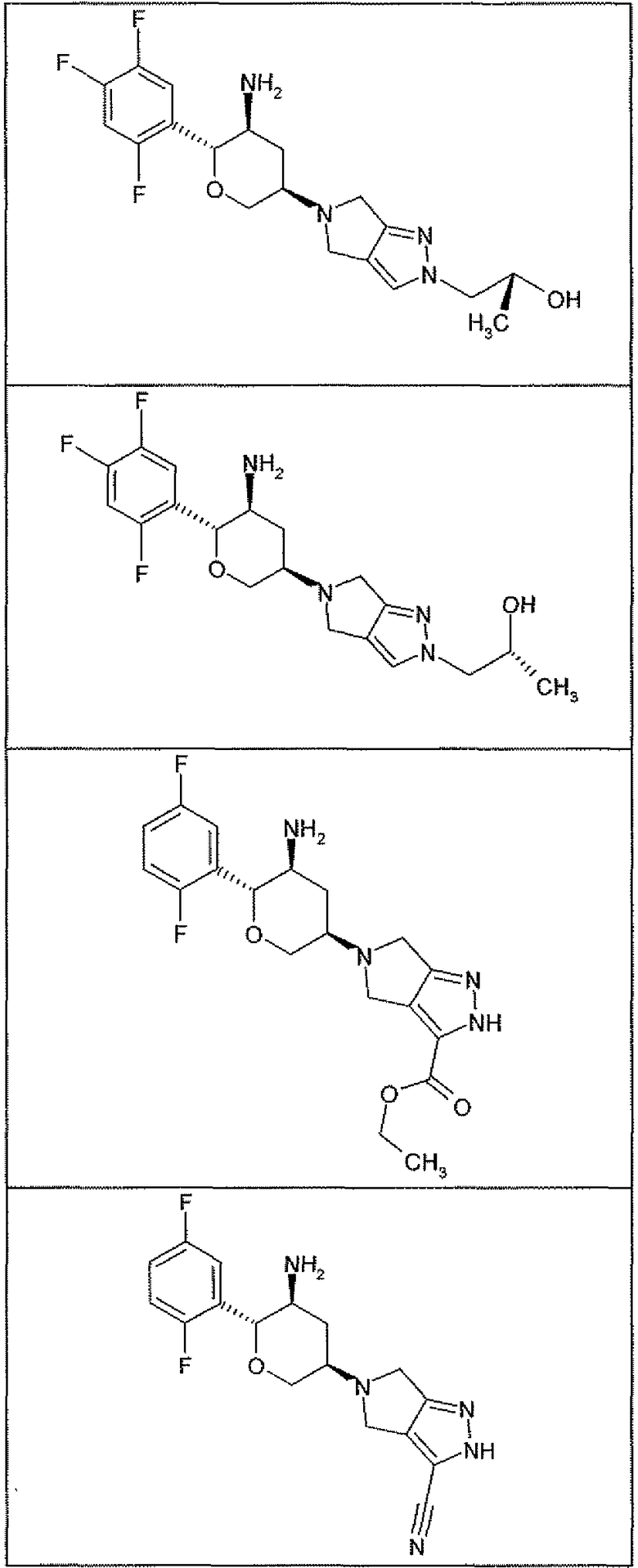
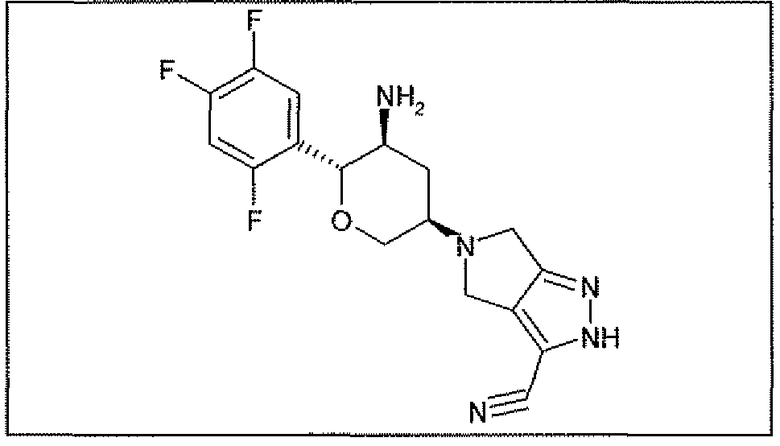
или их фармацевтически приемлемые соли.
Определения
Применяемые в контексте следующие определения являются общепринятыми.
“Алкил”, а также другие группы, имеющие префикс “алк”, такие как алкокси и алканоил, означают углеродные цепи, которые могут быть неразветвленными или разветвленными и их сочетаниями, если углеродная цепь не указана иначе. Примеры алкильных групп включают метил, этил, пропил, изопропил, бутил, втор- и трет-бутил, пентил, гексил, гептил, октил, нонил и тому подобное. Когда допускается определенное число атомов углерода, например, С3-10, термин "алкил” включает также циклоалкильные группы и сочетания неразветвленных или разветвленных алкильных цепей, комбинированных с циклоалкильными структурами. Когда число атомов углерода не указывается, подразумевается С1-6.
“Циклоалкил” означает насыщенное карбоциклическое кольцо, имеющее указанное число атомов углерода. Примеры циклоалкила включают циклопропил, циклобутил, циклопентил, циклогексил, циклогептил, циклооктил и тому подобное. Циклоалкильная группа обычно является моноциклической, если не указано иначе. Циклоалкильные группы являются насыщенными, если не указано иначе.
Термин “алкиламино” относится к неразветвленным или разветвленным алкиламинам с указанным числом атомов углерода (например, С1-6алкиламино) или любым числом в этом диапазоне [т.е. метиламино, этиламино, изопропиламино, трет-бутиламино и т.д.].
Термин “алкилоксикарбонил” относится к имеющим неразветвленную или разветвленную цепь эфирам производного карбоновой кислоты настоящего изобретения с указанным числом атомов углерода (например, С1-6алкилоксикарбонил) или любым числом в этом диапазоне [т.е. метилоксикарбонил (МеОСО-), этилоксикарбонил или бутилоксикарбонил].
“Арил” означает систему моно- или полициклических ароматических колец, содержащих в кольце атомы углерода. Предпочтительными арилами являются системы моноциклических или бициклических 6-10-членных ароматических колец. Фенил и нафтил являются предпочтительными арилами. Наиболее предпочтительным арилом является фенил.
Термин “гетероциклил” относится к насыщенным или ненасыщенным неароматическим кольцам или системам колец, содержащим по меньшей мере один гетероатом, выбранный из О, S и N, дополнительно включая окисленные формы серы, а именно SO и SO2. Примеры гетероциклов включают тетрагидрофуран (ТГФ), дигидрофуран, 1,4-диоксан, морфолин, 1,4-дитиан, пиперазин, пиперидин, 1,3-диоксолан, имидазолидин, имидазолин, пирролин, пирролидин, тетрагидропиран, дигидропиран, оксатиолан, дитиолан, 1,3-диоксан, 1,3-дитиан, оксатиан, тиоморфолин, пирролидинон, оксазолидин-2-он, имидазолидин-2-он, пиридон и тому подобное.
“Гетероарил” означает ароматический или частично ароматический гетероцикл, который содержит по меньшей мере один гетероатом кольца, выбранный из О, S и N. Гетероарилы включают также гетероарилы, конденсированные с другими типами колец, такими как арилы, циклоалкилы и гетероциклилы, которые не являются ароматическими. Примеры гетероарильных групп включают пирролил, изоксазолил, изотиазолил, пиразолил, пиридинил, 2-оксо-(1H)-пиридинил, (2-гидроксипиридинил), оксазолил, 1,2,4-оксадиазолил, 1,3,4-оксадиазолил, тиадиазолил, тиазолил, имидазолил, триазолил, тетразолил, фурил, триазинил, тиенил, пиримидинил, пиразинил, бензизоксазолил, бензоксазолил, бензотиазолил, бензотиадиазолил, дигидробензофуранил, индолинил, пиридазинил, индазолил, изоиндолил, дигидробензотиенил, индолизинил, циннолинил, фталазинил, хиназолинил, нафтиридинил, карбазолил, бензодиоксолил, хиноксалинил, пуринил, фуразанил, изобензилфуранил, бензимидазолил, бензофуранил, бензотиенил, хинолил, индолил, изохинолил, дибензофуранил, имидазо[1,2-α]пиридинил, [1,2,4-триазоло][4,3-α]пиридинил, пиразоло[1,5-α]пиридинил, [1,2,4-триазоло][1,5-α]пиридинил, 2-oxo-1,3-бензоксазолил, 4-оксо-3H-хиназолинил, 3-оксо-[1,2,4]-триазоло[4,3-α]-2H-пиридинил, 5-oxo-[1,2,4]-4H-оксадиазолил, 2-oxo-[1,3,4]-3H-оксадиазолил, 2-oxo-1,3-дигидро-2H-имидазолил, 3-оксо-2,4-дигидро-3H-1,2,4-триазолил и тому подобное. В гетероциклильные и гетероарильные группы включены кольца и системы колец, содержащие 3-15 атомов и образующие 1-3 кольца.
"Галоген" относится к атому фтора, хлора, брома и йода. Предпочтительными являются обычно атомы хлора и фтора. Атом фтора является наиболее предпочтительным, когда галогены являются заместителями у алкила или алкоксигруппы (например, CF3O и CF3CH2O).
Соединения настоящего изобретения содержат один или несколько асимметричных центров и поэтому могут существовать в виде рацематов, рацемических смесей, индивидуальных энантиомеров, диастереомерных смесей и индивидуальных диастереомеров. В частности, соединения настоящего изобретения имеют асимметричный центр у стереогенных атомов углерода, отмеченных знаком * в формулах Ia, Ib, Ic, Id, Ie и If. В зависимости от природы различных заместителей у молекулы могут присутствовать дополнительные асимметричные центры. Каждый такой асимметричный центр может независимо образовывать два оптических изомера, и предполагается, что все из возможных оптических изомеров и диастереомеров в смесях и в виде чистых и частично очищенных соединений включены в объем данного изобретения. Имеется в виду, что настоящее изобретение включает все такие изомерные формы этих соединений.
Имеется в виду, что некоторые из описанных в контексте соединений содержат олефиновые двойные связи и, если не оговорено иначе, включают как Е-, так и Z-геометрические изомеры.
Некоторые из описанных в контексте соединений могут существовать в виде таутомеров, которые имеют разные точки присоединения водорода, причем образование таутомеров сопровождается перемещениями одной или нескольких двойных связей. Например, кетон и его енольная форма являются кето-енольными таутомерами. Индивидуальные таутомеры, а также их смеси включены в соединения настоящего изобретения. Пример таутомеров, которые предполагается включить в соединения настоящего изобретения, иллюстрируется ниже:
Формула I показывает структуру класса соединений без предпочтительной стереохимии. Формулы Ia и Ib показывают предпочтительную стереохимию у стереогенных атомов углерода, к которым присоединены группы NH2 и Ar в кольце тетрагидропирана. Формулы Ic и Id показывают предпочтительную стереохимию у стереогенных атомов углерода, к которым присоединены группы NH2, Ar и V в кольце тетрагидропирана.
Независимого синтеза этих диастереомеров или их хроматографических разделений можно достичь, как известно в данной области, подходящей модификацией описанных в контексте методологии. Их абсолютную стереохимию можно определить рентгеновской кристаллографией кристаллических продуктов или кристаллических промежуточных продуктов, которые превращают в производные, если необходимо, реагентом, содержащим асимметричный центр известной абсолютной конфигурации.
Если необходимо, рацемические смеси соединений можно разделить так, чтобы выделить индивидуальные энантиомеры. Разделение можно проводить способами, хорошо известными в данной области, такими как сочетание рацемической смеси соединений с энантиомерно чистым соединением с образованием диастереомерной смеси с последующим разделением индивидуальных диастереомеров стандартными способами, такими как фракционная кристаллизация или хроматография. Реакцией сочетания часто является образование солей с применением энантиомерно чистой кислоты или основания. Диастереомерные производные можно затем превратить в чистые энантиомеры отщеплением присоединенного хирального остатка. Рацемическую смесь соединений можно также разделить непосредственно хроматографическими способами с применением хиральных неподвижных фаз, причем эти способы хорошо известны в данной области.
Альтернативно, любой энантиомер соединения можно получить стереоселективным синтезом с применением оптически чистых исходных соединений или реагентов известной конфигурации способами, хорошо известными в данной области.
Имеется в виду, как должно быть понятно, что применяемые в контексте ссылки на соединения структурной формулы I включают также фармацевтически приемлемые соли, а также соли, которые не являются фармацевтически приемлемыми, когда их применяют в качестве предшественников свободных соединений или их фармацевтически приемлемых солей или в других синтетических операциях.
Соединения настоящего изобретения можно вводить в форме фармацевтически приемлемой соли. Термин “фармацевтически приемлемая соль” относится к солям, полученным из фармацевтически приемлемых нетоксичных оснований или кислот, включающих неорганические или органические основания и неорганические или органические кислоты. Соли основных соединений, включенных в термин “фармацевтически приемлемая соль”, относятся к нетоксичным солям соединений данного изобретения, которые обычно получают реакцией свободного основания с подходящей органической или неорганической кислотой. Репрезентативные соли основных соединений настоящего изобретения включают, но не ограничиваются перечисленным, следующие соли: ацетат, бензолсульфонат, бензоат, бикарбонат, бисульфат, битартрат, борат, бромид, камсилат, карбонат, хлорид, клавуланат, цитрат, дигидрохлорид, эдетат, эдисилат, эстолат, эзилат, фумарат, глюцептат, глюконат, глутамат, гликоллиларсанилат, гексилрезорцинат, гидрабамин, гидробромид, гидрохлорид, гидроксинафтоат, иодид, изотионат, лактат, лактобионат, лаурат, малат, малеат, манделат, мезилат, метилбромид, метилнитрат, метилсульфат, мукат, напсилат, нитрат, N-метилглюкаминаммониевая соль, олеат, оксалат, памоат (эмбонат), пальмитат, пантотенат, фосфат/дифосфат, полигалактуронат, салицилат, стеарат, сульфат, себацетат, сукцинат, таннат, тартрат, теоклат, тозилат, триэтиодид и валерат. Кроме того, когда соединения изобретения имеют кислотную часть, их подходящие фармацевтически приемлемые соли включают, но не ограничиваются перечисленным, соли, полученные из неорганических оснований, включающих соли алюминия, аммония, кальция, меди, железа(III), железа(II), лития, магния, марганца(III), марганца(II), калия, натрия, цинка и тому подобное. Особенно предпочтительными являются соли аммония, кальция, магния, калия и натрия. Соли, полученные из фармацевтически приемлемых органических нетоксичных оснований, включают соли первичных, вторичных и третичных аминов, циклических аминов и основных ионообменных смол, таких как аргинин, бетаин, кофеин, холин, N,N-дибензилэтилендиамин, диэтиламин, 2-диэтиламиноэтанол, 2-диметиламиноэтанол, этаноламин, этилендиамин, N-этилморфолин, N-этилпиперидин, глюкамин, глюкозамин, гистидин, гидрабамин, изопропиламин, лизин, метилглюкамин, морфолин, пиперазин, пиперидин, полиаминовые смолы, прокаин, пурины, теобромин, триэтиламин, триметиламин, трипропиламин, трометамин и тому подобное.
Кроме того, в случае группы карбоновой кислоты (-СООН) или спирта, присутствующей в соединениях настоящего изобретения, можно применять фармацевтически приемлемые эфиры производных карбоновой кислоты, такие как метиловый, этиловый или пивалоилоксиметиловый эфир, или ацилпроизводные спиртов, такие как O-ацетил-, О-пивалоил-, О-бензоил- и О-аминоацилпроизводные. Указанные группы сложных эфиров и ацильные группы, известные в данной области, включают для модификации характеристик растворимости или гидролиза для применения в препаратах со стойко поддерживаемым высвобождением или препаратах с пролекарством.
Сольваты, и особенно гидраты, соединений структурной формулы I также включены в настоящее изобретение.
Кроме того, в соединениях структурной формулы I атомы могут быть в виде их распространенных в природе изотопов или один или несколько атомов могут быть искусственно обогащены конкретным изотопом, имеющим такой же атомный номер, но атомную массу или массовое число, отличное от атомной массы или массового числа изотопа, преимущественно находимого в природе. Имеется в виду, что настоящее изобретение включает все подходящие изотопные варианты соединений общей формулы I. Например, различные изотопные формы водорода (Н) включают протий (1Н) и дейтерий (2Н). Протий является преобладающим изотопом водорода, найденным в природе. Обогащение дейтерием может обеспечить определенные терапевтические преимущества, такие как увеличение in vivo полупериода существования или уменьшение требуемых доз, или может обеспечить получение соединения, применимого в качестве стандарта для характеризации биологических образцов. Изотопно обогащенные соединения в пределах родовой формулы I можно получить без излишнего экспериментирования общепринятыми методиками, хорошо известными специалисту в данной области, или способами, аналогичными способам, описанным на схемах и примерах данного контекста с применением подходящих, обогащенных изотопами реагентов и/или промежуточных продуктов.
Иллюстрацией изобретения является применение соединений, описанных в примерах и контексте.
Способы лечения
Рассматриваемые соединения являются применимыми в способе ингибирования фермента дипептидилпептидазы-IV у пациента, такого как млекопитающее, нуждающееся в таком ингибировании, содержащем введение эффективного количества соединения. Настоящее изобретение относится к применению описанных в контексте соединений в качестве ингибиторов активности фермента дипептидилпептидазы-IV.
Согласно способу настоящего изобретения, помимо приматов, таких как люди, можно лечить различных других млекопитающих. Например, можно лечить млекопитающих, включающих, но не ограничивающихся перечисленным, коров, овец, коз, лошадей, собак, кошек, морских свинок, крыс или других бычьих, овечьих, лошадиных, собачьих, кошачьих, грызуновых или мышиных видов. Однако способ можно также применять на практике для других видов, таких как птичьи виды (например, куры).
Настоящее изобретение далее относится к способу получения лекарственного средства для ингибирования активности фермента дипептидилпептидазы-IV у людей и животных, содержащему смешивание соединения настоящего изобретения с фармацевтически приемлемым носителем или разбавителем. Более конкретно, настоящее изобретение относится к применению соединения структурной формулы I при изготовлении лекарственного средства для применения при лечении состояния, выбранного из группы, состоящей из гипергликемии, диабета типа 2, ожирения и липидного нарушения у млекопитающего, где липидное нарушение выбрано из группы, состоящей из дислипидемии, гиперлипидемии, гипертриглицеридемии, гиперхолестеринемии, низкого HDL и высокого LDL.
Субъектом, которого лечат настоящими способами, обычно является млекопитающее, предпочтительно человек, мужчина или женщина, для которого требуется ингибирование фермента дипептидилпептидазы-IV. Термин “терапевтически эффективное количество” означает количество рассматриваемого соединения, которое может вызвать биологическую или лечебную реакцию ткани, системы, животного или человека, которой добивается исследователь, ветеринар, врач или другой клиницист.
Предполагается, что термин “композиция”, применяемый в контексте, включает продукт, содержащий указанные ингредиенты в указанных количествах, а также любой продукт, который является результатом непосредственного или опосредованного сочетания определенных ингредиентов в определенных количествах. Предполагается, что такой термин в отношении фармацевтической композиции включает продукт, содержащий активный ингредиент(ы) и инертный ингредиент(ы), которые образуют носитель, а также любой продукт, который является результатом, непосредственно или опосредованно, сочетания, комплексообразования или агрегации любых двух или более ингредиентов или диссоциации одного или нескольких ингредиентов, или других типов реакций или взаимодействий одного или нескольких ингредиентов. Согласно этому, фармацевтические композиции настоящего изобретения включают любую композицию, изготовленную смешиванием соединения настоящего изобретения и фармацевтически приемлемого носителя. Термин “фармацевтически приемлемый” означает, что носитель, разбавитель или эксципиент должен быть совместимым с другими ингредиентами препарата и не должен быть вредным для его реципиента.
Должно быть понятно, что термины “введение” и/или “вводимое” соединение означают обеспечение организма индивидуума, нуждающегося в лечении, соединением изобретения или пролекарством соединения изобретения.
Эффективность соединений согласно настоящему изобретению в качестве фермента дипептидилпептидазы-IV можно демонстрировать методикой, известной в данной области. Константы ингибирования определяют следующим образом. Для непрерывного флуорометрического анализа применяют субстрат Gly-Pro-AMC, который расщепляется DPP-4 с высвобождением флуоресцентной уходящей группы АМС. Кинетическими параметрами, которые описывают данную реакцию, являются следующие параметры: Km = 50 мкМ; κcat=75 сек-1; kcat/Km=1,5×106 M-1сек-1. Типичная реакция содержит приблизительно 50 пМ фермента, 50 мкМ Gly-Pro-AMC и буфер (100 мМ HEPES, pH 7,5, 0,1 мг/мл BSA) в общем реакционном объеме 100 мкл. Мониторинг выделения AMC проводят непрерывно в 96-луночном планшете-флуорометре с применением длины волны возбуждения 360 нм и длины волны эмиссии 460 нм. В этих условиях приблизительно 0,8 мкМ AMC продуцируется за 30 мин при 25 градусах C. Фермент, применяемый в этих исследованиях, был растворимым (исключая трансмембранный домен и цитоплазмический инсерционный материал) белком человека, продуцированным в системе экспрессии бакуловирусов (Bac-To-Bac, Gibco BRL). Обнаружено, что кинетические константы гидролиза Gly-Pro-AMC и GLP-1 согласуются с литературными величинами для нативного фермента. Для измерения констант диссоциации соединений растворы ингибитора в ДМСО добавляли к реакционным смесям, содержащим фермент и субстрат (конечная концентрация ДМСО 1%). Все эксперименты проводили при комнатной температуре с применением стандартных условий реакции, описанных выше. Для определения констант диссоциации (Ki), скорости реакции подбирали нелинейной регрессией для уравнения Михаэлис-Ментона для конкурентного ингибирования. Ошибки в воспроизведении констант диссоциации обычно меньше, чем двукратные.
Соединения структурной формулы (I), конкретно указанные ниже соединения примеров 1-14, имели активность при ингибировании фермента дипептидилпептидазы-IV в указанных выше анализах обычно с IC50 меньше, чем приблизительно 1 мкМ и более обычно меньше, чем 0,1 мкМ. Такие результаты являются показателем присущей активности соединений настоящего изобретения для применения в качестве ингибиторов активности фермента дипептидилпептидазы-IV.
Фермент дипептидилпептидаза-IV (DPP-4) является белком клеточной поверхности, который участвует в широком диапазоне биологических функций. Он имеет широкое распространение в тканях (кишечнике, почках, печени, поджелудочной железе, плаценте, тимусе, селезенке, эпителиальных клетках, васкулярном эндотелии, лимфоидных и миелоидных клетках, сыворотке) и имеет определенные уровни экспрессии в тканях и типах клеток. DPP-4 является идентичным с маркером CD26 активации Т-клеток и может расщеплять ряд иммунорегуляторных, эндокринных и неврологических пептидов in vitro. Это предполагало потенциальную роль данной пептидазы в различных патологических процессах у людей или у других видов животных.
Согласно этому, рассматриваемые соединения являются применимыми в способе предотвращения или лечения следующих заболеваний, нарушений и состояний.
Диабет типа II и родственные нарушения. Хорошо установлено, что инкреатины GLP-1 и GIP быстро инактивируются in vivo DPP-4. Исследования на мышах с DPP-4(-/-)-дефицитом и предварительные клинические испытания показали, что ингибирование DPP-4 повышает концентрации GLP-1 и GIP в стационарном состоянии, приводя к повышенной толерантности к глюкозе. Вероятно, что по аналогии с GLP-1 и GIP другие пептиды семейства глюкагона принимают участие в регуляции уровня глюкозы и также инактивируются DPP-4 (например, РАСАР). Инактивация этих пептидов DDP может также играть роль в гомеостазе глюкозы. Поэтому ингибиторы DPP-4 настоящего изобретения являются применимыми при лечении диабета типа II и лечении и предотвращении многочисленных состояний, которые часто сопровождаются диабетом типа II, включая синдром Х (известный также как метаболический синдром), реактивную гипогликемию и диабетическую дислипидемию. Ожирение, обсуждаемое ниже, является другим состоянием, которое часто обнаруживают с диабетом типа II, который может реагировать на лечение соединениями данного изобретения.
Нижеследующие заболевания, нарушения и состояния связаны с диабетом типа 2, и поэтому их можно лечить, устранять или в некоторых случаях предотвращать лечением соединениями данного изобретения: (1) гипергликемия, (2) низкая толерантность к глюкозе, (3) инсулинорезистентность, (4) ожирение, (5) липидные нарушения, (6) дислипидемия, (7) гиперлипидемия, (8) гипертриглицеридемия, (9) гиперхолестеринемия, (10) низкие уровни HDL, (11) высокие уровни LDL, (12) атеросклероз и его осложнения, (13) васкулярный рестеноз, (14) синдром раздраженной толстой кишки, (15) воспалительное заболевание кишечника, включающее болезнь Крона и неспецифический язвенный колит, (16) другие воспалительные состояния, (17) панкреатит, (18) ожирение брюшной полости, (19) нейродегенеративное заболевание, (20) ретинопатия, (21) нефропатия, (22) невропатия, (23) синдром Х, (24) овариальный гиперандрогенизм (синдром поликистоза яичников) и другие нарушения, у которых компонентом является инсулинорезистентность. Считается, что при синдроме Х, известном также как метаболический синдром, ожирение способствует инсулинорезистентности, диабету, дислипидемии, гипертензии и повышенному сердечно-сосудистому риску. Следовательно, ингибиторы DPP-4 могут быть также применимыми для лечения гипертензии, связанной с указанным состоянием.
Ожирение. Ингибиторы DPP-4 могут быть применимы для лечения ожирения. Это основано на наблюдаемых ингибирующих действиях на потребление пищи и эвакуацию GLP-1 и GLP-2 из желудка. Экзогенное введение GLP-1 людям значительно уменьшает потребление пищи и замедляет опорожнение желудка (Am. J. Physiol., 277: R910-R916 (1999)). ICV-введение GLP-1 крысам и мышам оказывает также сильное влияние на потребление корма (Nature Medicine, 2: 1254-1258 (1996)). Такое подавление потребления корма не наблюдают у GLP-1R(-/-)-мышей, это указывает на то, что эти действия опосредуются посредством рецепторов GLP-1 головного мозга. Вероятно, что по аналогии с GLP-1 GLP-2 также регулируется DPP-4. ICV-введение GLP-2 также подавляет потребления корма, аналогично действиям, наблюдаемым с GLP-1 (Nature Medicine, 6: 802-807 (2000)). Кроме этого, исследования на мышах с DPP-4-дефицитом позволяют предположить, что эти животные являются резистентными к индуцированному кормом ожирению и связанной с этим патологией (например, гиперинсулинонемией).
Сердечно-сосудистое заболевание. Показано, что GLP-1 является благоприятным при введении пациентам после острого инфаркта миокарда, приводя к улучшенной функции левого желудочка, и пониженной смертности после первой пластической операции на сосудах (Circulation, 109: 962-965 (2004)). Введение GLP-1 является также применимым для лечения систолической дисфункции левого желудочка у собак с дилатированной кардиомиопатией и индуцированной ишемией дисфункции левого желудочка и, таким образом, может оказаться применимым для лечения пациентов с сердечной недостаточностью (US 2004/0097411). Предполагается, что ингибиторы DPP-4 проявляют аналогичные действия посредством их способности стабилизировать эндогенный GLP-1.
Дефицит гормона роста. Ингибирование DPP-1 может быть применимым для лечения дефицита гормона роста, основанного на гипотезе, что фактор, высвобождающий гормон роста (GRF), пептид, который стимулирует высвобождение гормона роста из передней доли гипофиза, расщепляется ферментом DPP-4 in vivo (WO 00/56297). Следующие данные обеспечивают доказательство того, что GRF является эндогенным субстратом: (1) GRF эффективно расщепляется in vitro с образованием неактивного продукта GRF[3-44] (BBA 1122: 147-153 (1992)); (2) GRF быстро разрушается в плазме с образованием GRF[3-44]; это предотвращается дипротином А, ингибитором DPP-4; и (3) GRF[3-44] обнаружен в плазме трансгенной свиньи с GRF человека (J. Clin. Invest., 83: 1533-1540 (1989)). Таким образом, ингибиторы DPP-4 могут быть применимыми для такого же спектра показаний, которые рассматривали для средств, усиливающих секрецию гормона роста.
Кишечные повреждения. Возможность применения ингибиторов DPP-4 для лечения кишечного повреждения позволяют предположить результаты исследований, показывающие, что подобный глюкагону пептид-2 (GLP-2), возможный эндогенный субстрат для DPP-4, может оказывать трофические действия на кишечный эпителий (Regulatory Peptides, 90: 27-32 (2000)). Введение GLP-2 приводит к повышенной массе тонкой кишки у грызунов и ослабляет кишечное повреждение у грызунов-моделей колита и энтерита.
Иммуносупрессия. Ингибирование DPP-4 может быть применимо для модуляции иммунной реакции на основе исследований, включающих активацию фермента DPP-4 в Т-клетках и процессинг хемокина и эффективности ингибиторов DPP-4 на моделях заболевания in vivo. Показано, что DPP-4 является идентичным CD26, маркеру клеточной поверхности для активированных иммунных клеток. экспрессия CD26 регулируется статусом дифференциации и активации иммунных клеток. В общем признано, что CD26 функционирует в качестве состимулирующей молекулы в моделях активации Т-клеток in vitro. Ряд хемокинов содержат пролин в предпоследнем положении, вероятно для защиты их от деградации неспецифическими аминопептидазами. Показано, что многие из них процессируются in vitro посредством DPP-4. В некоторых случаях (RANTES, LD78-бета, MDC, эотаксин, SDF-1-alpha) расщепление приводит к измененной активности в анализах хемотаксиса и передачи сигнала. Оказывается, что селективность рецептора в некоторых случаях также модифицируется (RANTES). Множественные усеченные в N-концевых положениях формы ряда хемокинов были идентифицированы in vitro в системах культур клеток, в том числе предсказанные продукты гидролиза DPP-4.
Показано, что ингибиторы DPP-4 являются эффективными иммуносупрессантами на животных моделях трансплантации и артрита. Показано, что продипин (Pro-Pro-дифенилфосфонат), необратимый ингибитор DPP-4, удваивает выживаемость сердечного аллотрансплантата у крыс от 7 дней до 14 дней (Transplantation, 63: 1495-1500 (1997)). Ингибиторы DPP-4 испытывали при коллаген- и алкилдиамин-индуцированном артрите у крыс и показали статистически значимое ослабление опухания задней лапы на этой модели [Int. J. Immunopharmacology, 19:15-24 (1997) and Immunopharmacology, 40: 21-26 (1998)]. DPP-4 позитивно регулируется при ряде аутоиммунных заболеваний, включающих ревматоидный артрит, рассеянный склероз, болезнь Граве и тироидит Хашимото (Immunology Today, 20: 367-375 (1999)).
Инфекция ВИЧ. Ингибирование DPP-4 может быть применимым для лечения или предотвращения инфекции ВИЧ или СПИД, потому что ряд хемокинов, которые ингибируют проникновение клеток ВИЧ, являются потенциальными субстратами для DPP-4 (Immunology Today 20: 367-375 (1999)). В случае SDF-1альфа расщепление снижает противовирусную активность (PNAS, 95: 6331-6 (1998)). Таким образом, можно предположить, что стабилизация SDF-1альфа посредством ингибирования DPP-4 уменьшает инфективность ВИЧ.
Гемопоэз. Ингибирование DPP-4 может быть применимым для лечения или предотвращения гемопоэза, потому что DPP-4 может быть вовлечен в гемопоэз. Ингибитор DPP-4, Val-Boro-Pro, стимулировал гемопоэз в мышиной модели индуцированной циклофосфамидом нейтропении (WO 99/56753).
Нейронные нарушения. Ингибирование DPP-4 может быть применимо для лечения или предотвращения различных нейронных или психиатрических нарушений, потому что различные пептиды, участвующие в различных нейронных процессах, расщепляются DPP-4 in vitro. Таким образом, ингибитор DPP-4 может иметь терапевтическую пользу при лечении нейронных нарушений. Показано, что каждый из эндоморфина-2, бета-казоморфина и вещества Р является in vitro субстратом для DPP-4. Во всех случаях расщепление in vitro является очень эффективным с отношением kcat/Km приблизительно 106 M-1сек-1 или больше. На модели испытания аналгезии у крыс на основе вздрагивании из-за электрошока ингибитор DPP-4 показал значительное действие, которое не зависело от присутствия экзогенного эндоморфина-2 (Brain Research, 815: 278-286 (1999)). Нейрозащитные и нейрорегенеративные действия ингибиторов DPP-4 доказывали также посредством способности ингибиторов защищать двигательные нейроны от гибели эксайтотоксичных клеток, защищать стриарную иннервацию дофаминергических нейронов при введении одновременно с МРТР и стимулировать восстановление плотности стриарной иннервации при введении терапевтическим способом после лечения МРТР [см. Yong-Q. Wu, et al., "Neuroprotective Effects of Inhibitors of Dipeptidyl peptidase-IV In Vitro and In Vivo", Int. Conf. On Dipeptidyl Aminopeptidases: Basic Science and Clinical Applications, September 26-29, 2002 (Berlin, Germany)].
Тревога. Крысы с природным дефицитом DPP-4 имеют анксиолитический фенотип (WO 02/34243; Karl et al., Physiol. Behav. 2003). Мыши с дефицитом DPP-4 имеют также анксиолитический фенотип при применении порсольта и моделей свет/темнота. Таким образом, можно доказать, что DPP-4 являются применимыми для лечения тревоги и родственных нарушений.
Память и познавательная способность. Агонисты GLP-1 являются активными на моделях обучаемости (пассивное уклонение, водный лабиринт Морриса) и нейронного повреждения (индуцированный каинатом нейронный апоптоз), как показали During et al. (Nature Med, 9: 1173-1179 (2003)). Результаты предполагают физиологическую роль GLP-1 в обучаемости и нейрозащите. Предполагается, что стабилизация GLP-1 ингибиторами DPP-4 проявляет аналогичные действия.
Инфаркт миокарда. Показано, что GLP-1 является благоприятным при введении пациентам после острого инфаркта миокарда (Circulation, 109: 962-965 (2004)). Предполагается, что ингибиторы DPP-4 проявляют аналогичные действия вследствие их способности стабилизировать эндогенный GLP-1.
Прорастание опухоли и метастаз. Ингибирование DPP-4 может быть применимым для лечения или предотвращения прорастание опухоли и метастаза, потому что увеличение или уменьшение экспрессии некоторых эктопептидаз, включающих DPP-4, наблюдали во время трансформации нормальных клеток в клетки злокачественного фенотипа (J. Exp. Med., 190: 301-305 (1999)). Оказывается, что позитивная или негативная регуляция этих белков является специфичной для типа ткани и клеток. Например, повышенную экспрессию CD26/DPP-4 наблюдали при Т-клеточной лимфоме, T-клеточном остром лимфофластном лейкозе, карциномах, происходящих из клеток щитовидной железы, карциномах базальных клеток и карциномах молочной железы. Таким образом, ингибиторы DPP-4 могут быть пригодными при лечении таких карцином.
Доброкачественная гипертрофия простаты. Ингибирование DPP-4 может быть применимым для лечения доброкачественной гипертрофии простаты, потому что повышенная активность DPP-4 не отмечалась в ткани простаты у пациентом с ВРН (Eur. J. Clin. Chem. Clin. Biochem., 30: 333-338 (1992)).
Подвижность сперматозоидов/мужская контрацепция. Ингибирование DPP-4 может быть применимым для изменения подвижности сперматозоидов и для мужской контрацепции, потому что семенная жидкость, простатосомы, происходящие из простаты органеллы, важные для подвижности сперматозоидов, обладают очень высокими уровнями активности DPP-4 (Eur. J. Clin. Chem. Clin. Biochem., 30: 333-338 (1992)).
Гингивит. Ингибирование DPP-4 может быть применимым для лечения гингивита, потому что активность DPP-4 была обнаружена в жидкости десневой борозды и в некоторых исследованиях коррелировала с тяжестью периодонтального заболевания (Arch. Oral Biol., 37: 167-173 (1992)).
Остеопороз. Ингибирование DPP-4 может быть применимым для лечения или предотвращения остеопороза, потому что рецепторы GIP присутствуют в остеобластах.
Трансплантация стволовых клеток. Показано, что ингибирование DPP-4 на стволовых клетках донора приводит к повышению их эффективности хоминга в костный мозг и приживления и увеличению выживаемости мышей (Christopherson, et al., Science, 305:1000-1003 (2004)). Таким образом, ингибиторы DPP-4 могут быть применимыми при трансплантации костного мозга.
Соединения настоящего изобретения являются применимыми при лечении или предотвращении одного или нескольких из следующих состояний или заболеваний: (1) гипергликемии, (2) низкой толерантности к глюкозе, (3) инсулинорезистентности, (4) ожирения, (5) липидных нарушений, (6) дислипидемии, (7) гиперлипидемии, (8) гипертриглицеридемии, (9) гиперхолестеринемии, (10) низких уровней HDL, (11) высоких уровней LDL, (12) атеросклероза и его осложнений, (13) васкулярного рестеноза, (14) синдрома раздраженной толстой кишки, (15) воспалительного заболевания кишечника, включающего болезнь Крона и неспецифический язвенный колит, (16) других воспалительных состояний, (17) панкреатита, (18) ожирения брюшной полости, (19) нейродегенеративного заболевания, (20) ретинопатии, (21) нефропатии, (22) невропатии, (23) синдрома Х, (24) овариального гиперандрогенизма (синдрома поликистоза яичников), (25) диабета типа 2, (26) дефицита гормона роста, (27) нейтропении, (28) нейронных нарушений, (29) метастаза опухоли, (30) доброкачественной гипертрофии простаты, (32) гингивита, (33) гипертензии, (34) остеопороза, (35) состояния страха, (36) дефицита памяти, (37) дефицита познавательной способности, (38) удара, (39) болезни Альцгеймера и других состояний, которые можно лечить или предотвратить ингибированием DPP-4.
КОМБИНАЦИИ
Соединения настоящего изобретения можно применять в комбинации с одним или несколькими другими лекарственными средствами при лечении, предотвращении, подавлении или уменьшении интенсивности симптомов заболевания или состояний, для которых можно могут быть применимыми соединения формулы I или другие лекарственные средства, причем комбинация таких лекарственных средств является безопасной или более эффективной, чем любое одно лекарственное средство. Такое другое лекарственное средство(а) можно вводить путем и в количестве, обычно применяемом для этого, одновременно или последовательно с соединением формулы I. Когда соединение формулы I применяют одновременно с одним или несколькими другими лекарственными средствами, предпочтительной является фармацевтическая композиция в виде дозированной лекарственной формы, содержащей такие другие лекарственные средства и соединение формулы I, особенно в комбинации с фармацевтически приемлемым носителем. Однако комбинированная терапия может включать в себя также терапии, в которых соединение формулы I и один или несколько других лекарственных средств вводят по различным перекрываемым схемам применения. Предполагается также, что при применении в комбинации с одним или несколькими другими активными ингредиентами соединения настоящего изобретения и другие активные ингредиенты можно применять в более меньших дозах, чем при применении каждого отдельно. Соответственно этому, фармацевтические композиции настоящего изобретения включают композиции, которые содержат один или несколько других активных ингредиентов помимо соединения формулы I.
Когда соединение настоящего изобретения применяют одновременно с одним или несколькими другими лекарственными средствами, предпочтительной является фармацевтическая композиция, содержащая такие другие лекарственные средства помимо соединения настоящего изобретения. Согласно этому фармацевтические композиции настоящего изобретения включают композиции, которые содержат также один или несколько других активных ингредиентов помимо соединения настоящего изобретения.
Массовое соотношение соединения настоящего изобретения и второго активного ингредиента может изменяться и будет зависеть от эффективной дозы каждого ингредиента. Обычно применяют эффективную дозу каждого ингредиента. Так, например, когда соединение настоящего изобретения сочетают с другим агентом, массовое соотношение соединения настоящего изобретения к другому агенту обычно бывает в диапазоне от приблизительно 1000:1 до приблизительно 1:1000, предпочтительно от приблизительно 200:1 до приблизительно 1:200. Комбинации соединения настоящего изобретения и других активных ингредиентов обычно также бывают в указанном выше диапазоне, но в каждом случае следует применять эффективную дозу каждого активного ингредиента.
В таких комбинациях соединение настоящего изобретения и другие активные ингредиенты можно вводить по отдельности или в сочетании. Кроме того, введение одного элемента может быть проведено до, одновременно или после введения другого агента(ов).
Примеры других активных ингредиентов, которые можно вводить в комбинации с соединением формулы I и либо вводить по отдельности, либо в одной и той же фармацевтической композиции, включают, но не ограничиваются перечисленным
(1) сенсибилизирующие инсулин вещества, включающие (i) агонисты PPARγ, такие как глитазоны (например, пиоглитазон, розиглитазон, нетоглитазон, ривоглитазон и балаглитазон), и другие лиганды PPAR, включающие (1) двойные агонисты PPARα/γ, такие как мураглитазар, алеглитазар, соделглитазар и навеглитазар, (2) агонисты PPARα, такие как производные фенофибриновой кислоты (гемфиброзил, клофибрат, ципрофибрат, фенофибрат и безафибрат), (3) селективные модуляторы PPARγ (SPPARγM), такие как модуляторы, описанные в WO 02/060388, WO 02/08188, WO 2004/019869, WO 2004/020409, WO 2004/020408 и WO 2004/066963 и (4) частичные агонисты PPARγ; (ii) бигуаниды, такие как метформин и его фармацевтически приемлемые соли, в частности, гидрохлорид метформина, и их препараты с пролонгированным высвобождением, такие как Glumetza®, Fortamet® и GlucophageXP®, (iii) ингибиторы протеинтирозинфосфатазы-1В (РТР-1В);
(2) инсулин и аналоги или производные инсулина, такие как инсулин лиспро, инсулин детемир, инсулин гларгин, инсулин глулизин и вдыхаемые препараты каждого из них;
(3) лептин и производные, агонисты и аналоги лептина, такие как метрелептин;
(4) амилин; аналоги амилина, такие как давалинтид; и агонисты амилина, такие как прамлинтид;
(5) сульфонилмочевиновые и несульфонилмочевиновые средства, усиливающие секрецию инсулина, такие как толбутамид, глибурид, глипизид, глимепирид, митиглинид и меглитиниды, такие как натеглинид и репаглинид;
(6) ингибиторы α-глюкозидазы (такие как акарбоза, воглибоза и миглитол);
(7) антагонисты рецептора глюкагона, такие как антагонисты, описанные в WO 98/04528, WO 99/01423, WO 00/39088 и WO 00/69810;
(8) миметики инкретина, такие как GLP-1, аналоги, производные и миметики GLP-1 (см., например, WO 2008/011446, US 5545618, US 6191102 и US 56583111); и агонисты рецептора GLP-1, такие как оксинтомодулин и его аналоги и производные (см., например, WO 2003/022304, WO 2006/134340, WO 2007/100535), глюкагон и его аналоги и производные (см., например, WO 2008/101017), эксенатид, лираглутид, таспоглутид, албиглутид, AVE0010, CJC-1134-PC, NN9535, LY2189265, LY2428757 и BIM-51077, включающие интраназальные, трансдермальные и их вводимые один раз в неделю препараты, такие как эксенатид QW;
(9) агенты, снижающие уровень холестерина LDL, такие как (i) ингибиторы HMG-CoA-редуктазы (ловастатин, симвастатин, правастатин, церивастатин, флувастатин, аторвастатин, питавастатин и розувастатин), (ii) агенты, усиливающие секрецию желчных кислот (такие как холестирамин, колестимид, гидрохлорид колесевелама, колестипол и диалкиламиноалкилпроизводные сшитого декстрана, (iii) ингибиторы абсорбции холестерина, такие как азетимиб, и (iv) ингибиторы ацил-CoA:холестеринацилтрансферазы, такие как авазимиб;
(10) HDL-увеличивающие лекарственные средства, такие как ниацин или его соль и их варианты с пролонгированным высвобождением; MK-524A, который является комбинацией ниацина с пролонгированным высвобождением и DP-1-антагониста MK-524; и агонисты рецептора никотиновой кислоты;
(11) соединения, действующие против ожирения;
(12) агенты, предназначенные для применения при воспалительных состояниях, такие как аспирин, нестероидные противовоспалительные лекарственные средства (NSAID), глюкокортикоиды и селективные ингибиторы циклооксигеназы-2 (COX-2);
(13) гипотензивные агенты, такие как ингибиторы ACE (такие как эналаприл, лизиноприл, рамиприл, каптоприл, хинаприл и тандолаприл), блокаторы рецептора A-II (такие как лозартан, кандесартан, ирбесартан, олмесартан медоксомил, валсартан, телмисартан и эпросартан), ингибиторы ренина (такие как алискирен), бета-блокаторы (такие как блокаторы кальциевых каналов);
(14) активаторы глюкокиназы (GKA), такие как LY2599506;
(15) ингибиторы 11β-гидроксистероиддегидрогеназы типа 1, такие как ингибиторы, описанные в патенте США № 6730690; WO 03/104207 и WO 04/058741 ;
(16) ингибиторы белка-переносчика холестерилового сложного эфира (CETP), такие как торцетрапиб и MK-0859;
(17) ингибиторы фруктоза-1,6-бисфосфатазы, такие как ингибиторы, описанные в патентах США № 6054587; 6110903; 6284748; 6399782 и 6489476;
(18) ингибиторы ацетил-CoA-карбоксилазы-1 или -2 (ACC1 или ACC2);
(19) активаторы AMP-активированной протеинкиназы (AMPK);
(20) агонисты G-белок-связанных рецепторов: GPR-109, GPR-116, GPR-119 и GPR-40;
(21) антагонисты SSTR3, такие как антагонисты, описанные в WO 2009/011836;
(22) агонисты рецептора 1 нейромедина U (NMUR1) и/или рецептора 2 нейромедина U (NMUR2), такие как агонисты, описанные в WO2007/109135 и WO2009/042053, включающие, но не ограничивающиеся перечисленным, нейромедин U (NMU) и нейромидин S (NMS) и их аналоги и производные;
(23) ингибиторы стеароилкофермент A-дельта-9-десатуразы (SCD);
(24) антагонисты GPR-105 (P2YR14), такие как антагонисты, описанные в WO 2009/000087;
(25) ингибиторы поглощения глюкозы, такие как ингибиторы переносчика натрия-глюкозы (SGLT) и его различных изоформ, такие как SGLT-1; SGLT-2, такие как дапаглифлозин и ремоглифлозин; и SGLT-3;
(26) ингибиторы ацилкофермент A:диацилглицеринацилтрансферазы 1 и 2 (DGAT-1 и DGAT-2);
(27) ингибиторы синтазы жирных кислот;
(28) ингибиторы ацилкофермент A:моноацилглицеринацилтрансферазы 1 и 2 (MGAT-1 и MGAT-2);
(29) агонисты рецептора TGR5 (известные также как GPBAR1, BG37, GPCR19, GPR131 и M-BAR);
(30) бромкриптина мезилат и его препараты с быстрым высвобождением;
(31) агонисты рецептора гистамина Н3 и
(32) агонисты α2-адренергического или β3-адренергического рецептора.
Соединения, применяемые против ожирения, которые можно комбинировать с соединениями формулы I, включают топирамат; зонисамид; налтрексон; фентермин; бупропион; комбинацию бупропиона и налтрексона, комбинацию бупропиона и зонисамида; комбинацию топирамата и фентермина; фенфлурамин; дексфенфлурамин; сибутрамин; ингибиторы липазы, такие как орлистат и цетилистат; агонисты рецептора меланокортина, в частности, агонисты рецептора меланокортина-4; агонисты CCK-1; антагонисты рецептора гормона, концентрирующего меланин (MCH); антагонисты нейропептида Y1 или Y5 (такие как MK-0557); инверсные агонисты и антагонисты рецептора CB1 (такие как римонабант и таранабант); агонисты β3-адренергического рецептора; антагонисты грелина; агонисты рецептора бомбезина (такие как агонисты рецептора подтипа-3 бомбезина); инверсные агонисты рецептора гистамина Н3; агонисты 5-гидрокситриптамина-2c (5-HT2c), такие как лоркасерин; и ингибиторы синтазы жирных кислот (FAS). Для ознакомления с обзором соединений с действием против ожирения, которые можно комбинировать с соединениями настоящего изобретения, см. S. Chaki et al., "Recent advances in feeding suppressing agents: potential therapeutic strategy for the treatment of obesity", Expert Opin. Ther. Patents, 11: 1677-1692 (2001); D. Spanswick and K. Lee, "Emerging antiobesity drugs", Expert Opin. Emerging Drugs, 8: 217-237 (2003); J.A. Fernandez-Lopez, et al., "Pharmacological Approaches for the Treatment of Obesity", Drugs, 62: 915-944 (2002); and K.M. Gadde, et al., "Combination pharmaceutical therapies for obesity", Exp. Opin. Pharmacother., 10: 921-925 (2009).
Антагонисты рецептора глюкагона, которые можно применять в комбинации с соединениями формулы I, включают, но без ограничения
N-[4-((1S)-1-{3-(3,5-дихлорфенил)-5-[6-(трифторметокси)-2-нафтил]-1H-пиразол-1-ил}этил)бензоил]-β-аланин;
N-[4-((1R)-1-{3-(3,5-дихлорфенил)-5-[6-(трифторметокси)-2-нафтил]-1H-пиразол-1-ил}этил)бензоил]-β-аланин;
N-(4-{1-[3-(2,5-дихлорфенил)-5-(6-метокси-2-нафтил)-1Н-пиразол-1-ил]этил}бензоил)-β-аланин;
N-(4-{(1S)-1-[3-(3,5-дихлорфенил)-5-(6-метокси-2-нафтил)-1H-пиразол-1-ил]этил}бензоил)-β-аланин;
N-(4-{(1S)-1-[(R)-(4-хлорфенил)(7-фтор-5-метил-1H-индол-3-ил)метил]бутил}бензоил)-β-аланин и
N-(4-{(1S)-1-[(4-хлорфенил)(6-хлор-8-метилхинолин-4-ил)метил]бутил}бензоил)-β-аланин и
их фармацевтически приемлемые соли.
Ингибиторы стеароилкофермент A дельта-9-десатуразы (SCD), которые можно применять в комбинации с соединениями формулы I, включают, но без ограничения
[5-(5-{4-[2-(трифторметил)фенокси]пиперидин-1-ил}-1,3,4-тиадиазол-2-ил)-2Н-тетразол-2-ил]уксусную кислоту;
(2'-{4-[2-(трифторметил)фенокси]пиперидин-1-ил}-2,5'-би-1,3-тиазол-4-ил)уксусную кислоту;
(5-{3-[4-(2-бром-5-фторфенокси)пиперидин-1-ил]изоксазол-5-ил}-2H-тетразол-2-ил)уксусную кислоту;
(3-{3-[4-(2-бром-5-фторфенокси)пиперидин-1-ил]-1,2,4-оксадиазол-5-ил}-1H-пиррол-1-ил)уксусную кислоту;
(5-{5-[4-(2-бром-5-фторфенокси)пиперидин-1-ил]пиразин-2-ил}-2H-тетразол-2-ил)уксусную кислоту и
(5-{2-[4-(5-бром-2-хлорфенокси)пиперидин-1-ил]пиримидин-5-ил}-2H-тетразол-2-ил)уксусную кислоту и
их фармацевтически приемлемые соли.
Активаторы глюкокиназы, которые можно применять в комбинации с соединениями формулы I, включают, но без ограничения
3-(6-этансульфонилпиридин-3-илокси)-5-(2-гидрокси-1-метилэтокси)-N-(1-метил-1H-пиразол-3-ил)бензамид;
5-(2-гидрокси-1-метилэтокси)-3-(6-метансульфонилпиридин-3-илокси)-N-(1-метил-1H-пиразол-3-ил)бензамид;
5-(1-гидроксиметилпропокси)-3-(6-метансульфонилпиридин-3-илокси)-N-(1-метил-1H-пиразол-3-ил)бензамид;
3-(6-метансульфонилпиридин-3-илокси)-5-(1-метоксиметилпропокси)-N-(1-метил-1H-пиразол-3-ил)бензамид;
5-изопропокси-3-(6-метансульфонилпиридин-3-илокси)-N-(1-метил-1H-пиразол-3-ил)бензамид;
5-(2-фтор-1-фторметилэтокси)-3-(6-метансульфонилпиридин-3-илокси)-N-(1-метил-1Н-пиразол-3-ил)бензамид;
3-({4-[2-(диметиламино)этокси]фенил}тио)-N-(3-метил-1,2,4-тиадиазол-5-ил)-6-[(4-метил-4H-1,2,4-триазол-3-ил)тио]пиридин-2-карбоксамид;
3-({4-[(1-метилазетидин-3-ил)окси]фенил}тио)-N-(3-метил-1,2,4-тиадиазол-5-ил)-6-[(4-метил-4Н-1,2,4-триазол-3-ил)тио]пиридин-2-карбоксамид;
N-(3-метил-1,2,4-тиадиазол-5-ил)-6-[(4-метил-4Н-1,2,4-триазол-3-ил)тио]-3-{[4-(2-пирролидин-1-илэтокси)фенил]тио}пиридин-2-карбоксамид и
3-[(4-{2-[(2R)-2-метилпирролидин-1-ил]этокси}фенил)тио-N-(3-метил-1,2,4-тиадиазол-5-ил)-6-[(4-метил-4H-1,2,4-триазол-3-ил)тио]пиридин-2-карбоксамид и их фармацевтически приемлемые соли.
Агонисты рецептора GPR-119, которые можно применять в комбинации с соединениями формулы I, включают, но без ограничения
рац-цис-5-хлор-2-{4-[2-(2-{[5-(метилсульфонил)пиридин-2-ил]окси}этил)циклопропил]пиперидин-1-ил}пиримидин;
5-хлор-2-{4-[(1R,2S)-2-(2-{[5-(метилсульфонил)пиридин-2-ил]окси}этил)циклопропил]пиперидин-1-ил}пиримидин;
рац-цис-5-хлор-2-[4-(2-{2-[4-(метилсульфонил)фенокси]этил}циклопропил)пиперидин-1-ил]пиримидин;
5-хлор-2-[4-((1S,2R)-2-{2-[4-(метилсульфонил)фенокси]этил}циклопропил)пиперидин-1-ил]пиримидин;
5-хлор-2-[4-((1R,2S)-2-{2-[4-(метилсульфонил)фенокси]этил}циклопропил)пиперидин-1-ил]пиримидин;
рац-цис-5-хлор-2-[4-(2-{2-[3-(метилсульфонил)фенокси]этил}циклопропил)пиперидин-1-ил]пиримидин и
рац-цис-5-хлор-2-[4-(2-{2-[3-(5-метил-1,3,4-оксадиазол-2-ил)фенокси]этил}циклопропил)пиперидин-1-ил]пиримидин и
их фармацевтически приемлемые соли.
Селективные модуляторы PPARγ (SPPARγM's), которые можно применять в комбинации с соединениями формулы I, включают, но без ограничения
(2S)-2-({6-хлор-3-[6-(4-хлорфенокси)-2-пропилпиридин-3-ил]-1,2-бензизоксазол-5-ил}окси)пропановую кислоту;
(2S)-2-({6-хлор-3-[6-(4-фторфенокси)-2-пропилпиридин-3-ил]-1,2-бензизоксазол-5-ил}окси)пропановую кислоту;
(2S)-2-{[6-хлор-3-(6-фенокси-2-пропилпиридин-3-ил)-1,2-бензизоксазол-5-ил]окси}пропановую кислоту;
(2R)-2-({6-хлор-3-[6-(4-хлорфенокси)-2-пропилпиридин-3-ил]-1,2-бензизоксазол-5-ил}окси)пропановую кислоту;
(2R)-2-{3-[3-(4-метокси)бензоил-2-метил-6-(трифторметокси)-1H-индол-1-ил]фенокси}бутановую кислоту;
(2S)-2-{3-[3-(4-метокси)бензоил-2-метил-6-(трифторметокси)-1H-индол-1-ил]фенокси}бутановую кислоту;
2-{3-[3-(4-метокси)бензоил-2-метил-6-(трифторметокси)-1H-индол-1-ил]фенокси}-2-метилпропановую кислоту и
(2R)-2-{3-[3-(4-хлор)бензоил-2-метил-6-(трифторметокси)-1H-индол-1-ил]фенокси}пропановую кислоту и
их фармацевтически приемлемые соли.
Ингибиторы 11β-гидроксистероиддегидрогеназы типа 1, которые можно применять в комбинации с соединениями формулы I, включают, но без ограничения
3-[1-(4-хлорфенил)-транс-3-фторциклобутил]-4,5-дициклопропил-r-4H-1,2,4-триазол;
3-[1-(4-хлорфенил)-транс-3-фторциклобутил]-4-циклопропил-5-(1-метилциклопропил)-r-4H-1,2,4-триазол;
3-[1-(4-хлорфенил)-транс-3-фторциклобутил]-4-метил-5-[2-(трифторметокси)фенил]-r-4H-1,2,4-триазол;
3-[1-(4-хлорфенил)циклобутил]-4-метил-5-[2-(трифторметил)фенил]-4Н-1,2,4-триазол;
3-{4-[3-(этилсульфонил)пропил]бицикло[2.2.2]окт-1-ил}-4-метил-5-[2-(трифторметил)фенил]-4Н-1,2,4-триазол;
4-метил-3-{4-[4-(метилсульфонил)фенил]бицикло[2.2.2]окт-1-ил}-5-[2-(трифторметил)фенил]-4H-1,2,4-триазол;
3-(4-{4-метил-5-[2-(трифторметил)фенил]-4Н-1,2,4-триазол-3-ил}бицикло[2.2.2]окт-1-ил)-5-(3,3,3-трифторпропил)-1,2,4-оксадиазол;
3-(4-{4-метил-5-[2-(трифторметил)фенил]-4H-1,2,4-триазол-3-ил}бицикло[2.2.2]окт-1-ил)-5-(3,3,3-трифторэтил)-1,2,4-оксадиазол;
5-(3,3-дифторциклобутил)-3-(4-{4-метил-5-[2-(трифторметил)фенил]-4Н-1,2,4-триазол-3-ил}бицикло[2.2.2]окт-1-ил)-1,2,4-оксадиазол;
5-(1-фтор-1-метилэтил)-3-(4-{4-метил-5-[2-(трифторметил)фенил]-4H-1,2,4-триазол-3-ил}бицикло[2.2.2]окт-1-ил)-1,2,4-оксадиазол;
2-(1,1-дифторэтил)-5-(4-{4-метил-5-[2-(трифторметил)фенил]-4H-1,2,4-триазол-3-ил}бицикло[2.2.2]окт-1-ил)-1,3,4-оксадиазол;
2-(3,3-дифторциклобутил)-5-(4-{4-метил-5-[2-(трифторметил)фенил]-4H-1,2,4-триазол-3-ил}бицикло[2.2.2]окт-1-ил)-1,3,4-оксадиазол и
5-(1,1-дифторэтил)-3-(4-{4-метил-5-[2-(трифторметил)фенил]-4H-1,2,4-триазол-3-ил}бицикло[2.2.2]окт-1-ил)-1,2,4-оксадиазол и
их фармацевтически приемлемые соли.
Антагонисты подтипа рецептора 3 соматостатина (SSTR3), которые можно применять в комбинации с соединениями формулы I, включают, но без ограничения
и их фармацевтически приемлемые соли.
Активаторы AMP-активированной протеинкиназы (AMPK), которые можно применять в комбинации с соединениями формулы I, включают, но без ограничения
и их фармацевтически приемлемые соли.
Ингибиторы ацетил-СоА-карбоксилазы-1 и 2 (АСС-1 и АСС-2), которые можно применять в комбинации с соединениями формулы I, включают, но без ограничения
3-{1'-[(1-циклопропил-4-метокси-1H-индол-6-ил)карбонил]-4-оксоспиро[хроман-2,4'-пиперидин]-6-ил}бензойную кислоту;
5-{1'-[(1-циклопропил-4-метокси-1H-индол-6-ил)карбонил]-4-оксоспиро[хроман-2,4'-пиперидин]-6-ил}никотиновую кислоту;
1'-[(1-циклопропил-4-метокси-1H-индол-6-ил)карбонил]-6-(1H-тетразол-5-ил)спиро[хроман-2,4'-пиперидин]-4-он;
1'-[(1-циклопропил-4-этокси-3-метил-1H-индол-6-ил)карбонил]-6-(1H-тетразол-5-ил)спиро[хроман-2,4'-пиперидин]-4-он;
5-{1'-[(1-циклопропил-4-метокси-3-метил-1H-индол-6-ил)карбонил]-4-оксоспиро[хроман-2,4'-пиперидин]-6-ил}никотиновую кислоту;
4'-({6-(5-карбамоилпиридин-2-ил)-4-оксоспиро[хроман-2,4'-пиперидин]-1'-ил}карбонил)-2',6'-диэтоксибифенил-4-карбоновую кислоту;
2',6'-диэтокси-4'-{[6-(1-метил-1H-пиразол-4-ил)-4-оксоспиро[хроман-2,4'-пиперидин]-1'-ил]карбонил}бифенил-4-карбоновую кислоту;
2',6'-диэтокси-3-фтор-4'-{[6-(1-метил-1H-пиразол-4-ил)-4-оксоспиро[хроман-2,4'-пиперидин]-1'-ил]карбонил}бифенил-4-карбоновую кислоту;
5-[4-({6-(3-карбамоилфенил)-4-оксоспиро[хроман-2,4'-пиперидин]-1'-ил}карбонил)-2,6-диэтоксифенил]никотиновую кислоту;
4'-({6-(5-карбамоилпиридин-2-ил)-4-оксоспиро[хроман-2,4'-пиперидин]-1'-ил}карбонил)-2',6'-диэтоксибифенил-4-карбоксилат натрия;
метил-4'-({6-(5-карбамоилпиридин-2-ил)-4-оксоспиро[хроман-2,4'-пиперидин]-1'-ил}карбонил)-2',6'-диэтоксибифенил-4-карбоксилат;
1'-[(4,8-диметоксихинолин-2-ил)карбонил]-6-(1H-тетразол-5-ил)спиро[хроман-2,4'-пиперидин]-4-он;
(5-{1'-[(4,8-диметоксихинолин-2-ил)карбонил]-4-оксоспиро[хроман-2,4'-пиперидин]-6-ил}-2H-тетразол-2-ил)метилпивалат;
5-{1'-[(8-циклопропил-4-метоксихинолин-2-ил)карбонил]-4-оксоспиро[хроман-2,4'-пиперидин]-6-ил}никотиновую кислоту;
1'-(8-метокси-4-морфолин-4-ил-2-нафтоил)-6-(1H-тетразол-5-ил)спиро[хроман-2,4'-пиперидин]-4-он и
1'-[(4-этокси-8-этилхинолин-2-ил)карбонил]-6-(1H-тетразол-5-ил)спиро[хроман-2,4'-пиперидин]-4-он и
их фармацевтически приемлемые соли и сложные эфиры.
Соединения настоящего изобретения можно вводить пероральным, парентеральным (например, внутримышечной, внутрибрюшинной, внутривенной, ICV, внутриполостной инъекцией или инфузией, подкожной инъекцией или имплантатом) путем или ингаляционным спреем, назальным, вагинальным, ректальным, сублингвальным или местным путем введения и их можно применять отдельно или вместе в составе препаратов с подходящими унифицированными дозами, содержащих общепринятые нетоксичные фармацевтически приемлемые носители, адъюванты и наполнители, подходящие для каждого пути введения. Помимо лечения теплокровных животных, таких как мыши, крысы, лошади, крупный рогатый скот, овцы, собаки, обезьяны и т.д., соединения изобретения являются эффективными для применения при лечении людей.
Фармацевтические композиции для введения соединений настоящего изобретения можно преимущественно представить в виде дозированной лекарственной формы и можно изготовить любым из способов, хорошо известных в области фармации. Все способы включают стадию получения смеси активного ингредиента с носителем, который состоит из одного или нескольких дополнительных ингредиентов. Фармацевтические композиции обычно получают однородным и тесным смешиванием активного ингредиента с жидким носителем, или тонкоизмельченным твердым носителем, или обоими и затем, если необходимо, формованием продукта для получения требуемого препарата. В фармацевтическую композицию активное рассматриваемое соединение включают в количестве, достаточном для обеспечения требуемого действия при развитии заболеваний или состояния заболеваний. Имеется в виду, что применяемый в контексте термин “композиция” включает продукт, содержащий определенные ингредиенты в определенных количествах, а также любой продукт, который является результатом непосредственного или опосредованного комбинирования определенных ингредиентов в определенных количествах.
ВВЕДЕНИЕ
Фармацевтические композиции, содержащие активный ингредиент, могут быть в форме, подходящей для перорального применения, например, таблеток, пастилок, лепешек, водных или масляных суспензий, диспергируемых порошков или гранул, эмульсий, твердых или мягких капсул, или сиропов, или эликсиров. Композиции, предназначенные для перорального введения, можно получать согласно любому способу, известному в области изготовления фармацевтических композиций, и такие композиции могут содержать один или несколько агентов, выбранных из группы, состоящей из подслащивающих агентов, корригентов, окрашивающих агентов и консервантов, для получения фармацевтически изящных и приятных на вкус препаратов. Таблетки содержат активный ингредиент в смеси с нетоксичными фармацевтически приемлемыми эксципиентами, которые являются подходящими для изготовления таблеток. Этими эксципиентами могут быть, например, инертные разбавители, такие как карбонат кальция, карбонат натрия, лактоза, фосфат кальция или фосфат натрия; гранулирующие и дезинтегрирующие агенты, например, кукурузный крахмал или альгиновая кислота; связывающие агенты, например, крахмал, желатин или аравийская камедь, и смазывающие агенты, например, стеарат магния, стеариновая кислота или тальк. Таблетки могут быть непокрытыми или их можно покрыть известными методиками для замедления дезинтеграции и абсорбции в желудочно-кишечном тракте и тем самым обеспечить стойко поддерживаемое действие на протяжении более длительного периода. Например, можно применять вещество, замедляющее время высвобождения (активного ингредиента), такое как глицерилмоностеарат или глицерилдистеарат. Таблетки можно также покрыть способами, описанными в патентах США 4256108; 4166452 и 4265874, для получения осмотических терапевтических таблеток для регуляции высвобождения.
Препараты для перорального применения можно также представить в виде твердых желатиновых капсул, в которых активный ингредиент смешивают с инертным твердым разбавителем, например, карбонатом кальция, фосфатом кальция или каолином, или мягких желатиновых капсул, в которых активный ингредиент смешивают с водой или масляной средой, например, арахисовым маслом, жидким парафином или оливковым маслом.
Водные суспензии содержат активные вещества в смеси с эксципиентами, подходящими для изготовления водных суспензий. Такими эксципиентами являются суспендирующие агенты, например, натрий-карбоксиметилцеллюлоза, метилцеллюлоза, гидроксипропилметилцеллюлоза, альгинат натрия, поливинилпирролидон, трагакантовая камедь и аравийская камедь; диспергирующими или смачивающими агентами могут быть существующий в природе фосфатид, например лецитин, или продукты конденсации алкиленоксида с жирными кислотами, например полиоксиэтиленстеарат, или продукты конденсации этиленоксида с алифатическими спиртами с длинной цепью, например гептадекаэтиленоксицетанол, или продукты конденсации этиленоксида с неполными эфирами, полученными из жирных кислот и гексита, такие как моноолеат полиоксиэтиленсорбита, или продукты конденсации этиленоксида с неполными эфирами, полученными из жирных кислот и ангидридов гексита, например, моноолеат полиэтиленсорбитана. Водные суспензии могут содержать также один или несколько консервантов, например этил- или н-пропил-п-гидроксибензоат, один или несколько окрашивающих агентов, один или несколько корригентов и один или несколько подслащивающих агентов, таких как сахароза или сахарин.
Масляные суспензии можно изготовить суспендированием активного ингредиента в растительном масле, например, арахисовом масле, оливковом масле, кунжутном масле или кокосовом масле, или в минеральном масле, таком как жидкий парафин. Масляные суспензии могут содержать загущающий агент, например, пчелиный воск, твердый парафин или цетиловый спирт. Для получения перорального препарата с приятным вкусом можно добавлять подслащивающие агенты, такие как агенты, указанные выше, и корригенты. Эти композиции можно предохранять от порчи добавлением антиоксиданта, такого как аскорбиновая кислота.
Диспергируемые порошки и гранулы, подходящие для получения водной суспензии добавлением воды, содержат активный ингредиент в смеси с диспергирующим или смачивающим агентом, суспендирующим агентом и одним или несколькими консервантами. Примерами подходящих диспергирующих или смачивающих агентов и суспендирующих агентов являются агенты, уже указанные выше. Могут также присутствовать дополнительные эксципиенты, например, подслащивающие агенты, корригенты и окрашивающие агенты.
Фармацевтические композиции изобретения могут быть также в форме эмульсий типа масло-в-воде. Масляной фазой может быть растительное масло, например, оливковое масло или арахисовое масло, или минеральное масло, например, жидкий парафин, или их смеси. Подходящими эмульгирующими агентами могут быть существующие в природе камеди, например, аравийская камедь или трагакантовая камедь, существующие в природе фосфатиды, например, фосфатиды сои культурной, лецитин и эфиры или неполные эфиры, полученные из жирных кислот и ангидридов гексита, например, моноолеат сорбитана, и продукты конденсации указанных неполных сложных эфиров с этиленоксидом, например, моноолеат полиоксиэтиленсорбитана. Эмульсии могут содержать также подслащивающие агенты и корригенты.
Сиропы и эликсиры можно изготовлять с подслащивающими агентами, например, глицерином, пропиленгликолем, сорбитом или сахарозой. Такие препараты могут содержать также средство, уменьшающее раздражение, консервант, корригент и окрашивающий агент.
Фармацевтические композиции могут быть в форме стерильной инъецируемой водной или маслянистой суспензии. Суспензию можно изготовить согласно известному способу с применением таких подходящих диспергирующих или смачивающих агентов и суспендирующих агентов, которые были указаны выше. Стерильным инъецируемым препаратом может быть также стерильный инъецируемый раствор или суспензия в нетоксичном, парантерально приемлемом разбавителе или растворителе, например, в виде раствора в 1,3-бутандиоле. Среди приемлемых наполнителей и растворителей, которые можно применять, имеются вода, раствор Рингера и изотонический раствор хлорида натрия. Кроме того, в качестве растворителя или суспендирующей среды преимущественно применяют стерильные нелетучие масла. Для этой цели можно применять любое легкое нелетучее масло, в том числе синтетические моно- или диглицериды. Кроме того, при получении инъецируемых препаратов находят применение жирные кислоты, такие как олеиновая кислота.
Соединения настоящего изобретения можно также вводить в форме суппозиториев для ректального введения лекарственного средства. Эти композиции можно получить смешиванием лекарственного средства с подходящим нераздражающим эксципиентом, который является твердым при обычных температурах, но становится жидким при ректальной температуре и поэтому будет плавиться в прямой кишке с высвобождением лекарственного средства. Такими веществами-эксципиентами являются какао-масло и полиэтиленгликоли.
Для местного введения применяют кремы, мази, желе, растворы или суспензии и т.д., содержащие соединение настоящего изобретения. (Для целей такого применения местный препарат может включать жидкости для полоскания рта и растворы и жидкости для полоскания рта и горла).
Фармацевтическая композиция и способ настоящего изобретения могут дополнительно содержать другие терапевтически активные соединения, указываемые в контексте, которые обычно применяют при лечении указанных выше патологических состояний.
При лечении или предотвращении состояний, которые требуют ингибирования активности фермента дипептидилпептидазы-IV, подходящий уровень дозы обычно бывает приблизительно 0,01-500 мг на кг массы тела пациента в день, такую дозу можно вводить в виде целой или дробных доз. Уровень доз предпочтительно будет составлять от приблизительно 0,1 до приблизительно 250 мг/кг в день; более предпочтительно от приблизительно 0,5 до приблизительно 100 мг/кг в день. Подходящий уровень доз может быть от приблизительно 0,01 до 250 мг/кг в день, от приблизительно 0,05 до 100 мг/кг в день или от приблизительно 0,1 до 50 мг/кг в день. В этом диапазоне дозой может быть 0,05-0,5, 0,5-5 или 5-50 мг/кг в день. Для перорального введения композиции предпочтительно представляют в форме таблеток, содержащих 1,0-1000 мг активного ингредиента, конкретно 1,0, 5,0, 10,0, 15,0, 20,0, 25,0, 50,0, 75,0, 100,0, 150,0, 200,0, 250,0, 300,0, 400,0, 500,0, 600,0, 750,0, 800,0, 900,0, и 1000,0 мг активного ингредиента, для симптоматического подбора дозы для подвергаемого лечению пациента. Соединения можно вводить при схеме введения 1-4 раза в день, предпочтительно один или два раза в день.
При лечении или предотвращении сахарного диабета и/или гипергликемии, или гипертриглицеридемии или других заболеваний, для которых показаны соединения настоящего изобретения, обычно удовлетворительные результаты получают, когда соединения настоящего изобретения вводят при суточной дозе от приблизительно 0,1 мг до приблизительно 100 мг на килограмм массы тела животного, предпочтительно введенной в виде одной суточной дозы или в виде дробных доз от двух до шести раз в день, или в виде формы со стойко поддерживаемым высвобождением. Для большинства крупных животных общая суточная доза составляет от приблизительно 1,0 мг до приблизительно 1000 мг, предпочтительно от приблизительно 1 мг до приблизительно 50 мг. В случае 70 кг взрослого человека общая суточная доза обычно составляет от приблизительно 7 мг до приблизительно 350 мг. Эту схему приема лекарственного средства можно регулировать для обеспечения оптимальной терапевтической реакции.
Однако должно быть понятно, что определенный уровень доз и частота дозирования для любого конкретного пациента может изменяться и будет зависеть от различных факторов, включающих активность конкретного применяемого соединения, метаболической стабильности и продолжительности действия данного соединения, возраста, массы тела, общего состояния здоровья, пола, питания, способа и времени введения, скорости экскреции, комбинации лекарственных средств, тяжести конкретного состояния и хозяина, подвергаемого терапии.
ПРИМЕРЫ
Синтетические способы получения соединений настоящего изобретения иллюстрируются в следующих схемах и примерах. Исходные вещества являются коммерчески доступными или их можно получить согласно процедурам, известным в данной области или иллюстрируемым в контексте.
Соединения настоящего изобретения можно получить из промежуточных продуктов, таких как промежуточные продукты формул II и III, с применением стандартных условий восстановительного аминирования с последующим снятием защиты.
где Ar и V имеют значения, указанные выше, и Р представляет собой подходящую защитную группу атома азота, такую как трет-бутоксикарбонил (ВОС), бензилоксикарбонил (Cbz) или 9-флуоренилметоксикарбонил (Fmoc). Получение указанных промежуточных продуктов описано на следующих схемах.
СХЕМА 1
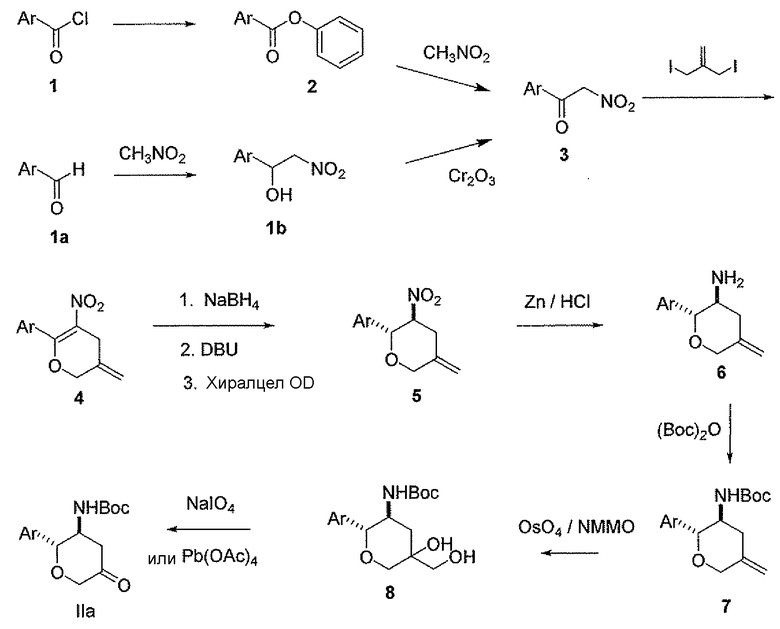
Промежуточные продукты формулы II являются известными в литературе, или их можно подходящим образом получить различными способами, известными специалисту в данной области. Один общий путь иллюстрируется на схеме 1. Замещенный бензоилгалогенид 1 обрабатывают фенолом в присутствии основания, такого как N,N-диизопропилэтиламин, получая при этом сложный эфир 2. Обработка 2 анионом, образованным из нитрометана с применением гидрида натрия, дает нитрокетон 3. Альтернативно, нитрокетон 3 можно получить реакцией альдегида 1а с нитрометаном в присутствии основания и окислением образовавшегося нитроспирта 1b окисляющим агентом, таким как реагент Джона. Нагревание нитрокетона 3 с 3-йод-2-(йодметил)проп-1-еном дает пиран 4, который при восстановлении боргидридом натрия и изомеризации основанием, таким как 1,8-диазабицикло[5.4.0]ундец-7-ен (DBU), дает транс-пиран 5. Энантиомеры 5 можно разделить на этой стадии различными способами, известными специалисту в данной области. Рацемат можно преимущественно разделить ВЭЖХ с применением хиральной колонки. Нитрозамещенный пиран 5 затем восстанавливают, например, с применением цинка и кислоты, такой как хлористоводородная кислота, и образовавшийся амин 6 защищают, например, в виде его ВОС-производного, обработкой ди-трет-бутилдикарбонатом с получением 7. Обработка 7 тетраоксидом осмия и N-оксидом N-метилморфолина дает диол 8, который при обработке периодатом натрия образует промежуточный пиранон IIa.
СХЕМА 2
Промежуточные продукты формул IIIa и IIIb являются известными в литературе, или их можно подходящим образом получить различными способами, знакомыми специалисту в данной области. Один общий путь получения тетрагидропирролопиразолов IIIa и IIIb иллюстрируется на схеме 2. Тритил- или Вос-защищенный пирролидинол 9 можно окислить различными способами, такими как процедура Сверна, обычно известная специалисту в данной области, для получения кетона 10, который при обработке с нагреванием диметилацеталем N,N-диметилформамида (ДМФА-DMA) дает 11. Требуемые промежуточные продукты IIIa и IIIb можно затем легко получить нагреванием раствора 11 с гидразином (R = H) или производным гидразина 12 в подходящем растворителе, таком как этанол, необязательно в присутствии основания, такого как этоксид натрия, с последующим удалением защитной группы кислотой и хроматографическим разделением двух региоизомеров либо до, либо после удаления защитной группы Р.
СХЕМА 3
Как показано на схеме 3, соединения настоящего изобретения структурной формулы (I) можно получить восстановительным аминированием промежуточного продукта II в присутствии промежуточного продукта III с применением таких реагентов, как цианоборгидрид натрия, декаборан или триацетоксиборгидрид натрия, в растворителях, таких как дихлорметан, тетрагидрофуран или метанол, с образованием промежуточного продукта IV. Реакцию необязательно проводят в присутствии кислоты Льюиса, такой как тетрахлорид титана или тетраизопропоксид титана. Реакцию можно также облегчить добавлением кислоты, такой как уксусная кислота. В некоторых случаях промежуточный продукт III может быть в форме соли, такой как соль хлористоводородной кислоты или трифторуксусной кислоты, и в этих случаях к реакционной смеси удобно добавлять основание, обычно N,N-диизопропилэтиламин. Защитную группу затем удаляют, например, трифторуксусной кислотой или раствором хлорида водорода в метаноле в случае Вос, или палладием-на-углероде и газообразным водородом в случае Cbz, получая при этом требуемый амин формулы I. Продукт очищают, если необходимо, перекристаллизацией, растиранием, препаративной тонкослойной хроматографией, флэш-хроматографией на силикагеле, например, с применением аппаратуры Biotage®, или ВЭЖХ. Соединения, которые очищают ВЭЖХ, можно выделить в виде соответствующей соли.
В некоторых случаях продукт I или синтетические промежуточные продукты, иллюстрированные в указанных выше схемах, можно далее модифицировать, например, превращением заместителей у Ar или V. Эти превращения могут включать, но не ограничиваются перечисленным, реакции восстановления, окисления, алкилирования, ацилирования и гидролиза, которые обычно известны специалисту в данной области.
В некоторых случаях порядок проведения реакций вышеупомянутых схем можно изменять, чтобы облегчить реакцию или избежать образования нежелательных продуктов реакции.
Соединения структурной формулы I настоящего изобретения далее иллюстрируются нижеследующими конкретными примерами. Соединения, иллюстрированные в примерах, однако, не следует истолковывать как образующие единственный вид, который рассматривают как изобретение. В примерах дополнительно иллюстрируются подробности получения соединений настоящего изобретения. Специалист в данной области может легко понять, что известные варианты условий и способов нижеследующих препаративных процедур можно применять для получения этих соединений. Настоящие соединения обычно выделяют в форме их фармацевтически приемлемых солей, таких как соли, предварительно описанные в контексте выше. Основания в виде свободных аминов, соответствующих выделенным солям, можно получить нейтрализацией солей подходящим основанием, таким как водный гидрокарбонат натрия, карбонат натрия, гидроксид натрия и гидроксид калия, и экстракцией образованного основания в виде свободного амина органическим растворителем с последующим упариванием. Выделенное таким способом основание в виде свободного амина можно далее превратить в другую фармацевтически приемлемую соль растворением в органическом растворителе с последующим добавлением подходящей кислоты и затем упариванием, осаждением или кристаллизацией. Все температуры являются градусами Цельсия, если не указывается иначе. Масс-спектры (МС) измеряли масс-спектроскопией с электрораспылением.
Нижеследующее перечисление является списком аббревиатур, применяемых в описании синтеза промежуточных продуктов и примеров, показанных ниже.
Список аббревиатур:
ПРОМЕЖУТОЧНЫЙ ПРОДУКТ 1
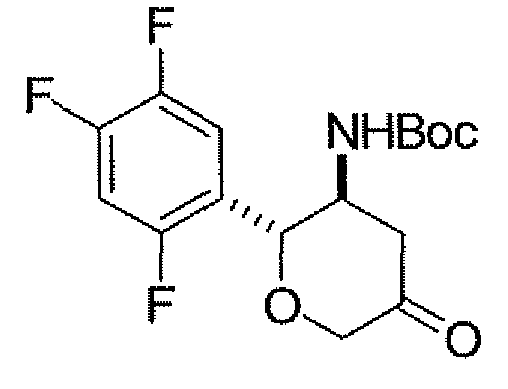
трет-Бутил-[(2R,3S)-5-оксо-2-(2,4,5-трифторфенил)тетрагидро-2Н-пиран-3-ил]карбамат
Стадия А: Фенил-2,4,5-трифторбензоат
Раствор фенола (13,3 г, 141 ммоль) в сухом дихлорметане (370 мл) охлаждали на ледяной бане и обрабатывали N,N-диизопропилэтиламином (34 мл, 193 ммоль) с последующим добавлением по каплям 2,4,5-трифторбензоилхлорида (25 г, 129 ммоль) в течение 15 мин. Ледяную баню удаляли, перемешивание продолжали в течение двух часов при комнатной температуре и раствор затем переносили в делительную воронку и органический слой промывали последовательно раствором хлористоводородной кислоты (2 н, 150 мл), насыщенным водным раствором бикарбоната натрия (150 мл) и насыщенным раствором соли (150 мл), сушили над безводным сульфатом натрия, фильтровали, упаривали и образовавшийся твердый продукт порциями очищали на силикагеле с элюированием последовательно гексаном и затем градиентным способом 0-5% раствором простого эфира в гексане, получая при этом фенил-2,4,5-трифторбензоат в виде белого твердого вещества.
Стадия B: 2-Нитро-1-(2,4,5-трифторфенил)этанон
Гидрид натрия (12 г, 60% в масле, 297 ммоль) промывали гексаном (4×100 мл), промывали струей сухого азота, суспендировали в N,N-диметилформамиде (350 мл) и затем обрабатывали нитрометаном (44 мл, 81 ммоль). Образовавшуюся смесь перемешивали при комнатной температуре в течение 2,5 час, охлаждали до 0°С и затем обрабатывали раствором фенил-2,4,5-трифторбензоатом (22,8 г, 90,0 ммоль) в N,N-диметилформамиде (180 мл) в течение двух часов. Реакционную смесь выдерживали при такой же температуре на протяжении ночи и перемешивали непрерывно в течение дополнительного часа при комнатной температуре. Смесь выливали на лед (400 г) и концентрированную хлористоводородную кислоту (48 мл). Водную смесь экстрагировали этилацетатом (3×250 мл). Объединенные органические слои промывали насыщенным раствором соли (40 мл), сушили над безводным сульфатом натрия, фильтровали и упаривали при пониженном давлении. Сырой продукт растворяли в смеси простой эфир-гексан (1:1, 240 мл) и воде (200 мл). Органический слой отделяли и кристаллы, которые образовались при стоянии и охлаждении в холодильнике, выделяли фильтрованием и сушили, получая при этом 2-нитро-1-(2,4,5-трифторфенил)этанон в виде не совсем белого твердого вещества.
Стадия C: 3-Метилен-5-нитро-6-(2,4,5-трифторфенил)-3,4-дигидро-2H-пиран
Смесь 3-хлор-2-(хлорметил)проп-1-ена (1,0 г, 8 ммоль) и иодида натрия (6,6 г, 44 ммоль) в ацетоне (60 мл) перемешивали при комнатной температуре в течение 20 час, упаривали при пониженном давлении и растворяли в дихлорметане (150 мл) и воде (50 мл). Органический слой сушили над сульфатом натрия, фильтровали и упаривали, получая при этом 3-йод-2-(йодметил)проп-1-ен в виде красноватого масла. К раствору 2-нитро-1-(2,4,5-трифторфенил)этанона (110 мг, 0,5 ммоль) в N,N-диметилформамиде (3 мл) и 3-йод-2-(йодметил)проп-1-ену (170 мг, 0,55 ммоль) добавляли N,N-диизопропилэтиламин (0,20 мл) и смесь нагревали при 60°С в течение 2,5 час, упаривали и очищали хроматографией на системе Biotage Horizon® (силикагель, градиент 0-30% дихлорметана в гексане), получая при этом 3-метилен-5-нитро-6-(2,4,5-трифторфенил)-3,4-дигидро-2H-пиран.
Стадия D: (2R,3S)-5-Метилен-3-нитро-2-(2,4,5-трифторфенил)тетрагидро-2H-пиран
К раствору 3-метилен-5-нитро-6-(2,4,5-трифторфенил)-3,4-дигидро-2H-пирана (798 мг, 2,94 ммоль) в хлороформе (42 мл) и изопропиловом спирте (7,8 мл) добавляли силикагель (5,1 г) и боргидрид натрия (420 мг, 11,1 ммоль) и реакционную смесь перемешивали в течение 30 мин при комнатной температуре. Реакционную смесь затем гасили добавлением по каплям хлористоводородной кислоты (6 мл, 2 н) и фильтровали. Образовавшийся твердый остаток промывали этилацетатом (100 мл). Объединенный фильтрат промывали последовательно насыщенным водным раствором бикарбоната натрия и насыщенным раствором соли, сушили над безводным сульфатом натрия и упаривали. Образовавшееся янтарное масло растворяли в тетрагидрофуране (15 мл) и добавляли 1,8-диазабицикло[5.4.0]ундец-7-ен (DBU, 40 мкл). Раствор перемешивали в течение 105 мин и затем переносили в делительную воронку, содержащую этилацетат (100 мл) и 1 н хлористоводородную кислоту. Органический слой промывали насыщенным раствором соли и водный слой экстрагировали этилацетатом. Объединенный органический слой сушили над безводным сульфатом натрия, фильтровали и упаривали, получая при этом сырой продукт, который очищали флэш-хроматографией (силикагель, 8-10% простого эфира в гексане), получая при этом транс-5-метилен-3-нитро-2-(2,4,5-трифторфенил)тетрагидро-2H-пиран. Часть этого продукта разделяли ВЭЖХ (хиралцел OD (ChiralCel OD), 1,5% изопропиловый спирт в гептане), получая при этом более медленно движущийся энантиомер, (2R,3S)-5-метилен-3-нитро-2-(2,4,5-трифторфенил)тетрагидро-2H-пиран.
Стадия Е: (2R,3S)-5-Метилен-2-(2,4,5-трифторфенил)тетрагидро-2H-пиран-3-амин
К энергично перемешиваемой суспензии (2R,3S)-5-метилен-3-нитро-2-(2,4,5-трифторфенил)тетрагидро-2H-пирана (200 мг, 0,73 ммоль) и порошка цинка (561 мг, 8,59 ммоль) в этаноле (7 мл) добавляли 6 н хлористоводородную кислоту (2,3 мл, 14 ммоль). Спустя один час смесь обрабатывали простым эфиром (100 мл) и водным раствором гидроксида натрия (2,5 н, 40 мл). Органический слой промывали насыщенным раствором соли, сушили над безводным сульфатом натрия и упаривали, получая при этом (2R,3S)-5-метилен-2-(2,4,5-трифторфенил)тетрагидро-2H-пиран-3-амин, который применяли в следующей стадии без дополнительной очистки.
Стадия F: трет-Бутил-[(2R,3S)-5-метилен-2-(2,4,5-трифторфенил)тетрагидро-2H-пиран-3-ил]карбамат
К раствору (2R,3S)-5-метилен-2-(2,4,5-трифторфенил)тетрагидро-2H-пиран-3-амина (177 мг, 0,73 ммоль) в дихлорметане (5 мл) добавляли ди-трет-бутилдикарбонат (239 мг, 1,1 ммоль) и смесь перемешивали в течение 2,5 час при комнатной температуре. Раствор упаривали при пониженном давлении, получая при этом трет-бутил-[(2R,3S)-5-метилен-2-(2,4,5-трифторфенил)тетрагидро-2H-пиран-3-ил]карбамат в виде белого твердого вещества. Его применяли в следующей стадии без дополнительной очистки.
Стадия G: трет-Бутил-[(2R,3S)-5-гидрокси-5-(гидроксиметил)-2-(2,4,5-трифторфенил)тетрагидро-2H-пиран-3-ил]карбамат
К раствору трет-бутил-[(2R,3S)-5-метилен-2-(2,4,5-трифторфенил)тетрагидро-2H-пиран-3-ил]карбамата (203 мг, 0,59 ммоль) в трет-бутиловом спирте (6 мл), ацетоне (3 мл) и воде (1,5 мл) добавляли тетраоксид осмия (0,113 мл 2,5% раствора в трет-бутиловом спирте, 0,009 ммоль). Образовавшуюся смесь перемешивали при комнатной температуре в течение 10 мин и затем обрабатывали N-оксидом N-метилморфолина (92 мг, 0,79 ммоль) и перемешивали в течение двух дней. Реакционную смесь затем обрабатывали водным раствором бисульфита натрия (5 мл, 2,0 н) с последующей обработкой этилацетатом спустя 10 мин. Органический слой промывали последовательно 2 н хлористоводородной кислотой и насыщенным водным раствором бикарбоната натрия, сушили над безводным сульфатом натрия, фильтровали и упаривали, получая при этом трет-бутил-[(2R,3S)-5-гидрокси-5-(гидроксиметил)-2-(2,4,5-трифторфенил)тетрагидро-2H-пиран-3-ил]карбамат, который применяли в следующей стадии без дополнительной очистки.
Стадия H: трет-Бутил-[(2R,3S)-5-okco-2-(2,4,5-трифторфенил)тетрагидро-2H-пиран-3-ил]карбамат
К раствору трет-бутил-[(2R,3S)-5-гидрокси-5-(гидроксиметил)-2-(2,4,5-трифторфенил)тетрагидро-2H-пиран-3-ил]карбамата (223 мг, 0,59 ммоль) в тетрагидрофуране (4 мл) добавляли раствор периодата натрия (143 мг, 0,67 ммоль) в воде (1,3 мл) и смесь перемешивали в течение 3 час. Смесь концентрировали и очищали флэш-хроматографией (силикагель, градиент 5-20% этилацетата в хлороформе), получая при этом трет-бутил-[(2R,3S)-5-оксо-2-(2,4,5-трифторфенил)тетрагидро-2H-пиран-3-ил]карбамат в виде белого твердого вещества.
ПРОМЕЖУТОЧНЫЙ ПРОДУКТ 2
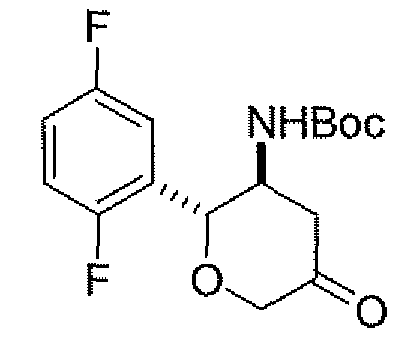
трет-Бутил-[(2R,3S)-5-оксо-2-(2,5-дифторфенил)тетрагидро-2H-пиран-3-ил]карбамат
Стадия A: 1-(2,5-Дифторфенил)-2-нитроэтанол
К гидроксиду натрия (1 н, 3 л) и метанолу (1500 мл) при 5°С добавляли по каплям раствор 2,5-дифторбензальдегида (350 г, 2,46 моль) и нитрометана (157 мл, 2,9 моль) в метаноле (350 мл) на протяжении периода 1 час. Реакционную смесь затем нейтрализовали ледяной уксусной кислотой (165 мл). Водная обработка давала требуемый нитроспирт.
Стадия B: 2-Нитро-1-(2,5-дифторфенил)этанон
Раствор периодинана Dess-Martin (125 г) в дихлорметане (600 мл) добавляли к раствору нитроспирта, полученного на стадии A (46,3 г), при 10°С на протяжении периода 30 мин. Перемешивание продолжали в течение 2 час и реакционную смесь затем выливали на смесь бикарбоната натрия (300 г) и тиосульфата натрия (333 г) в воде (3 л). Требуемый продукт экстрагировали метил-трет-бутиловым простым эфиром (MTBE) (2 л). Водный слой нейтрализовали HCl (2 н, 1,5 л) и экстрагировали MTBE (3 л). Объединенные органические слои сушили над безводным сульфатом магния, фильтровали, упаривали и остаток очищали хроматографией (силикагель, элюирование дихлорметаном), получая при этом требуемый нитрокетон.
Стадия C: 3-Йод-2-(йодметил)проп-1-ен
Смесь 3-хлор-2-(хлорметил)проп-1-ена (1,0 г, 8 ммоль) и иодида натрия (6,6 г, 44 ммоль) в ацетоне (60 мл) перемешивали при комнатной температуре в течение 20 час, упаривали при пониженном давлении и распределяли между дихлорметаном (150 мл) и водой (50 мл). Органический слой сушили над сульфатом натрия, фильтровали и упаривали, получая при этом 3-йод-2-(йодметил)проп-1-ен в виде красноватого масла.
Стадия D: 3-Метилен-5-нитро-6-(2,5-дифторфенил)-3,4-дигидро-2H-пиран
N,N-Диизопропилэтиламин (184 мл) добавляли к раствору 2-нитро-1-(2,5-дифторфенил)этанона (92,7 г, 461 ммоль) в N,N-диметилформамиде (1000 мл) и 3-йод-2-(йодметил)проп-1-ену (156 г, 507 ммоль). Смесь нагревали при 60°С в течение 2 час, упаривали и очищали хроматографией (силикагель, градиент 0-30% дихлорметана в гексане), получая при этом 3-метилен-5-нитро-6-(2,5-дифторфенил)-3,4-дигидро-2H-пиран.
Стадия E: (2R,3S)-5-Метилен-3-нитро-2-(2,5-дифторфенил)тетрагидро-2H-пиран
Данное соединение получали таким же способом, какой описан при получении промежуточного продукта 1, стадия D.
Стадия F: (2R,3S)-5-Метилен-2-(2,5-дифторфенил)тетрагидро-2H-пиран-3-амин
Данное соединение получали таким же способом, какой описан при получении промежуточного продукта 1, стадия Е.
Стадия G: трет-Бутил-(2R,3S)-5-метилен-2-(2,5-дифторфенил)тетрагидро-2H-пиран-3-ил]карбамат
Данное соединение получали таким же способом, какой описан при получении промежуточного продукта 1, стадия F.
Стадия H: трет-Бутил-[(2R,3S)-5-гидрокси-5-(гидроксиметил)-2-(2,5-дифторфенил)тетрагидро-2H-пиран-3-ил]карбамат
Данное соединение получали таким же способом, какой описан при получении промежуточного продукта 1, стадия G.
Стадия I: трет-Бутил-[(2R,3S)-5-оксо-2-(2,5-дифторфенил)тетрагидро-2H-пиран-3-ил]карбамат
К раствору трет-бутил-[(2R,3S)-5-гидрокси-5-(гидроксиметил)-2-(2,5-трифторфенил)тетрагидро-2H-пиран-3-ил]карбамат (10,5 г) в метаноле (100 мл) при 0°С добавляли пиридин (7,8 мл) и тетраацетат свинца (21,7 г). Реакционную смесь перемешивали в течение 20 мин. Водная обработка этилацетатом давала сырой продукт, который очищали хроматографией (силикагель, 0-50% раствор этилацетат/гептан), получая при этом трет-бутил-[(2R,3S)-5-оксо-2-(2,5-дифторфенил)тетрагидро-2H-пиран-3-ил]карбамат в виде белого твердого вещества.
ПРОМЕЖУТОЧНЫЙ ПРОДУКТ 3
Стадия A: трет-Бутил-(3Z)-3-[(диметиламино)метилен]-4-оксопирролидин-1-карбоксилат
Раствор трет-бутил 3-оксопирролидин-1-карбоксилата (40 г, 216 ммоль) обрабатывали смесью ДМФА-DMA (267 г, 2241 ммоль) и нагревали при 105°С в течение 40 мин. Раствор охлаждали и упаривали при пониженном давлении и образовавшееся оранжевое твердое вещество обрабатывали гексаном (200 мл) и охлаждали в холодильнике на протяжении времени с субботы до понедельника. Образовавшееся коричневато-желтое твердое вещество собирали фильтрованием, сушили и применяли на следующей стадии без дополнительной очистки.
Стадия B: 1,4,5,6-Тетрагидропирроло[3,4-c]пиразол
Раствор гидразина (3 мл) и трет-бутил-(3Z)-3-[(диметиламино)метилен]-4-оксопирролидин-1-карбоксилата (19,22 г) в этаноле (40 мл) нагревали при 85°С в герметизированной трубке в течение 4 час. Растворитель удаляли при пониженном давлении и остаток растирали с дихлорметаном (160 мл) и этилацетатом (15 мл). Образовавшееся твердое вещество отделяли фильтрованием. Фильтрат концентрировали и образовавшееся твердое вещество снова растирали и отделяли фильтрованием. Объединенное твердое вещество обрабатывали 4 н хлористоводородной кислотой (250 мл) в метаноле и перемешивали в течение 6 час. Реакционную смесь концентрировали и сушили. Образовавшееся твердое вещество снова обрабатывали в течение 6 час 4 н хлористоводородной кислотой (250 мл) в метаноле. После концентрирования и сушки образовавшуюся гидрохлоридную соль обрабатывали аммиаком в метаноле (2 н, 300 мл) и раствором гидроксида аммония в воде (28%, 30 мл) и концентрировали досуха. Полученное твердое вещество обрабатывали метанолом (70 мл) и водой (5 мл) и очищали в виде трех порций на системе Biotage Horizon® [диоксид кремния, градиент 5-17% метанола (содержащего 10% концентрированного гидроксида аммония) в этилацетате], получая при этом 1,4,5,6-тетрагидропирроло[3,4-c]пиразол. 1ЯМР (500 МГц, CD3OD): δ 4,04 (д, 4Н); 7,39 (с, 1H).
ПРОМЕЖУТОЧНЫЙ ПРОДУКТ 4
трет-Бутил-4,6-дигидропирроло[3,4-c]пиразол-5(1H)-карбоксилат
К суспензии 1,4,5,6-тетрагидропирроло[3,4-c]пиразола (промежуточный продукт 3) (207 г) в CH2Cl2 (1,0 л) добавляли триэтиламин (147 мл, 1058 ммоль) с последующим добавлением жидкого ди-трет-бутилдикарбоната (95 мл, 445 ммоль) при помощи капельной воронки на протяжении 45 мин. Смесь перемешивали при комнатной температуре на протяжении ночи. Смесь затем переносили в делительную воронку и промывали последовательно 3×500 мл 2,0 н HC1, 2×500 мл 10% NaOH и 400 мл насыщенного раствора соли. Смесь затем сушили над Na2SO4 и упаривали, получая при этом промежуточный продукт 4 в виде коричневого твердого вещества. ЖХ-МС=210,1 [M+1].
ПРОМЕЖУТОЧНЫЕ ПРОДУКТЫ 5 И 6
трет-Бутил-2-(2-амино-2-оксоэтил)-2,6-дигидропирроло[3,4-c]пиразол-5(4H)-карбоксилат (5)
трет-Бутил-1-(2-амино-2-оксоэтил)-4,6-дигидропирроло[3,4-c]пиразол-5(1Н)-карбоксилат (6)
В 2-л трехгорлую колбу, снабженную термометром, механической мешалкой и капельной воронкой, загружали суспензию промежуточного продукта 4 (18,01 г, 86,5 ммоль) в безводном ацетонитриле (1,0 л). К суспензии в виде одной порции добавляли гидрид натрия (60% дисперсия в масле, 4,15 г, 104 ммоль) в атмосфере азота. Реакционную смесь перемешивали при комнатной температуре в течение 2 час. Образовавшуюся белую суспензию затем охлаждали на ледяной бане и добавляли йодацетамид (31,95 г, 173 ммоль). Ледяную баню затем удаляли и смесь перемешивали 18 час при комнатной температуре. Смесь гасили водой (50 мл) и растворитель удаляли при пониженном давлении. Остаток распределяли между разбавленным NaCl (50 мл насыщенного раствора соли и 100 мл воды) и 1,0 л EtOAc. Водный слой экстрагировали 2×1,0 л EtOAc. Органические слои объединяли и сушили над Na2SO4 и растворитель упаривали при пониженном давлении. Сырое вещество очищали на силикагеле, элюируя 20-50% раствором EtOAc/CH2Cl2 для вымывания избыточного йодацетамида и затем 2-10% раствором MeOH/CH2Cl2, получая при этом смесь двух продуктов, которые разделяли на колонке хирального AD с элюированием 30% раствором MeOH/CO2, получая при этом промежуточный продукт 5 (менее подвижная фракция) и промежуточный продукт 6 (более подвижная фракция). ЖХ-МС=267,32 [M+1].
ПРОМЕЖУТОЧНЫЙ ПРОДУКТ 7
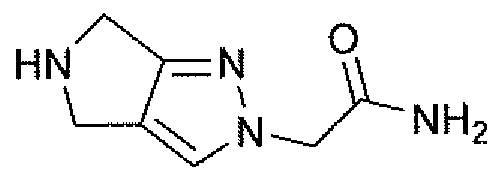
2-(5,6-Дигидропирроло[3,4-c]пиразол-2(4H-ил)ацетамид
К раствору промежуточного продукта 5 (25,04 г, 94 ммоль) в CH2Cl2 (200 мл) добавляли трифторуксусную кислоту (100 мл) при 0°С. Смесь перемешивали при комнатной температуре в течение 3 час. Смесь концентрировали и нейтрализовали раствором 25% MeOH (содержащей 10% NH4OH)/CH2Cl2. Остаток затем очищали на двух колонках 65i Biotage®, элюируя раствором 12,5-25% MeOH (содержащий 10% NH4OH)/CH2Cl2, получая при этом промежуточный продукт 7 в форме свободного основания. ЖХ-МС=167,10 [M+1].
ПРОМЕЖУТОЧНЫЙ ПРОДУКТ 8
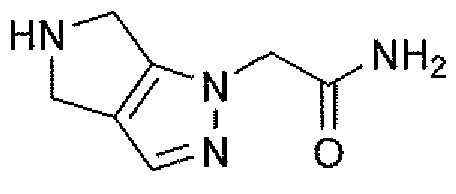
2-(5,6-Дигидропирроло[3,4-с]пиразол-1(4Н)-ил)ацетамид
Промежуточный продукт 6 (2,4 г, 9,01 ммоль) в CH2Cl2 (60 мл) обрабатывали TFA (30 мл) в течение 1,5 час и сырой продукт очищали, как описано для промежуточного продукта 7, получая при этом требуемый промежуточный продукт 8. ЖХ-МС=167,09 [M+1].
ПРОМЕЖУТОЧНЫЙ ПРОДУКТ 9
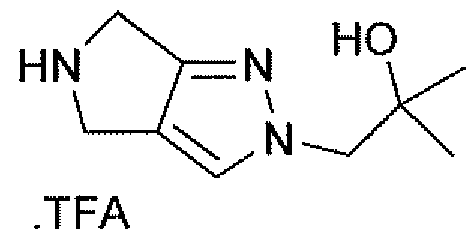
Трифторацетат 2-(2-гидрокси-2-метилпропил)-2,4,5,6-тетрагидропирроло[3,4-c]пиразол-5-ия
К перемешиваемому раствору промежуточного продукта 4 (35 г, 167 ммоль) в ДМФА (500 мл) при 0°С в атмосфере N2 добавляли гексаметилдисилазид натрия в ТГФ (351 мл, 351 ммоль) и смесь перемешивали при 0°С в течение 30 мин. Затем медленно добавляли оксид изобутилена (74,3 мл, 836 ммоль). Раствор перемешивали при 0°С в течение 0,5 час и затем перемешивали при комнатной температуре в течение 1 час. Раствор нагревали до 80°С в течение 100 мин в микроволновой печи. Остаток очищали колоночной хроматографией на силикагеле, элюируя с градиентом 0-6% раствора CH2Cl2/МеОН (содержащего 10% NH4OH), получая при этом смесь двух региоизомеров. Смесь двух региоизомеров А и В разделяли хроматографией на колонке ChiralPak AD-H, элюируя 4-40% раствором MeOH/CO2, получая при этом изомер А как быстрее элюируемый изомер и изомер В как медленнее элюируемый изомер. 1H ЯМР (500 МГц, CD3OD): для изомера В: δ 7,42 (д, 1H); 4,42 (с, 2H); 4,41 (с, 2H); 4,07 (с, 2H); 1,51 (д, 9H); 1,16 (с, 6H). ЖХ-МС: 226,27 (M+1-56).
Требуемый изомер B обрабатывали смесью 1:1 TFA/CH2Cl2 в течение 1 час, получая при этом указанное в заголовке соединение. 1H ЯМР (500 МГц, CD3OD): δ 7,55 (с, 1H); 4,43 (с, 2H); 4,39 (с, 2H); 4,10 (с, 2H); 1,17 (с, 6H). ЖХ-МС: 182,31 (M+1).
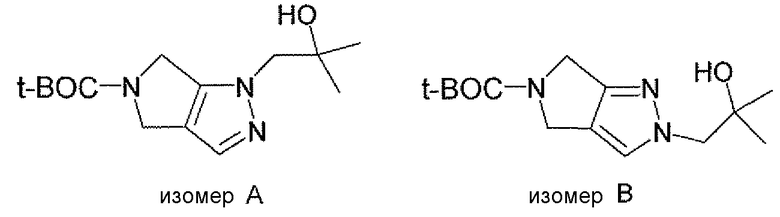
ПРОМЕЖУТОЧНЫЙ ПРОДУКТ 10
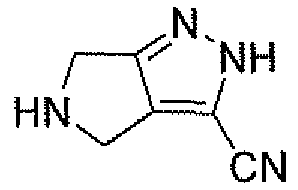
Стадия А: трет-Бутил-2-{[2-(триметилсилил)этокси]метил}-2,6-дигидропирроло[3,4-c]пиразол-5(4H)-карбоксилат
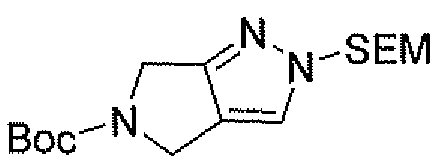
К перемешиваемому раствору промежуточного продукта 4 (20 г, 96 ммоль) в ДМФА (200 мл) при 0°С добавляли гидрид натрия (4,21 г, 105 ммоль). После перемешивания в течение 1 час при комнатной температуре добавляли 2-триметилсилилэтоксиметилхлорид (SEM-Cl) (4,65 мл, 26,3 ммоль). Образовавшуюся смесь перемешивали при комнатной температуре на протяжении ночи. Смесь гасили насыщенным NH4OH и растворители удаляли. Остаток разбавляли этилацетатом (500 мл), промывали водой, насыщенным раствором соли, сушили над безводным сульфатом натрия и концентрировали. Остаток очищали колоночной хроматографией на колонке силикагеля Biotage 65i®, элюируя раствором 0-20% этилацетата в гексанах, получая при этом указанное в заголовке соединение в виде бесцветной смолы. ЖХ-МС: 340,1 (M+H).
Стадия B: 5-трет-Бутил-3-этил-2-{[2-(триметилсилил)этокси]метил}-2,6-дигидропирроло[3,4-c]пиразол-3,5(4H)-дикарбоксилат

К перемешиваемому раствору трет-бутил-2-{[2-(триметилсилил)этокси]метил}-2,6-дигидропирроло[3,4-c]пиразол-5(4H)-карбоксилата (26 г, 77 ммоль) в ТГФ (400 мл) при -78°С в атмосфере азота добавляли раствор н-бутиллития (1,6 M, 57,4 мл, 92 ммоль). Смесь перемешивали при -78°С в течение 40 мин, добавляли этилхлорформиат (9,19 мл, 96 ммоль) и смесь перемешивали при -78°С в течение дополнительных 4 час. Смесь гасили насыщенным хлоридом аммония (200 мл) и водой (50 мл), экстрагировали этилацетатом (500 мл), промывали насыщенным раствором соли (200 мл), сушили (Na2SO4), фильтровали и концентрировали. Остаток очищали колоночной хроматографией на колонке силикагеля Biotage 65i®, элюируя 0-25% раствором EtOAc/гексаны, получая при этом указанное в заголовке соединение. ЖХ-МС: 412,2 (M+H).
Стадия C: 5-(трет-Бутоксикарбонил)-2-{[2-(триметилсилил)этокси]метил}-2,4,5,6-тетрагидропирроло[3,4-c]пиразол-3-карбоновая кислота
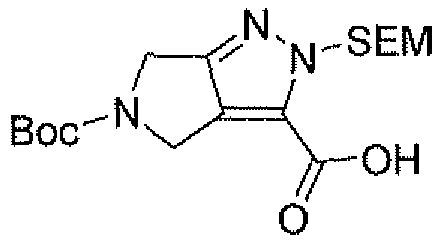
К перемешиваемой смеси продукта, полученного на стадии B (2,2 г, 5,35 ммоль), в 1,4-диоксане (75 мл) добавляли гидроксид лития (1 M, 26,7 мл). Реакционную смесь перемешивали при 35°С в течение 24 час. Растворители удаляли и остаток промывали гексанами (75 мл). Водный слой разбавляли водой, подкисляли 2 н HCl до рН приблизительно 3, экстрагировали EtOAc (2×200 мл). Органические слои промывали водой, насыщенным раствором соли, сушили над Na2SO4 и концентрировали, получая при этом сырой продукт. ЖХ-МС: 384,2 (M+H).
Стадия D: трет-Бутил-3-карбамоил-2-{[2-(триметилсилил)этокси]метил}-2,6-дигидропирроло[3,4-c]пиразол-5(4Н)-карбоксилат
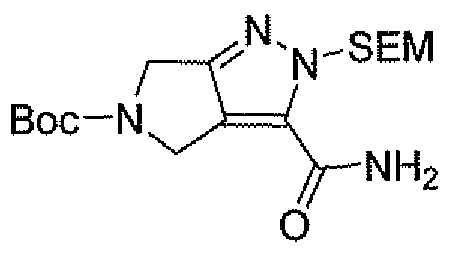
К перемешиваемому раствору продукта, полученного на стадии С (600 мг, 1,564 ммоль), в ДМФА (15 мл) добавляли DIPEA (1,366 мл, 7,82 ммоль) и хлорид аммония (2,5 г, 46,9 ммоль). После перемешивания в течение 30 мин добавляли HATU (1190 мг, 3,13 ммоль). Образовавшуюся смесь перемешивали при комнатной температуре в атмосфере азота в течение 22 час, разбавляли этилацетатом (200 мл), промывали 1 н хлористоводородной кислотой (2×100 мл), сушили над Na2SO4, фильтровали и концентрировали, получая при этом сырое указанное в заголовке соединение. ЖХ-МС: 383,2 (M+H).
Стадия Е трет-Бутил-3-циано-2-{[2-(триметилсилил)этокси]метил}-2,6-дигидропирроло[3,4-c]пиразол-5(4H)-карбоксилат
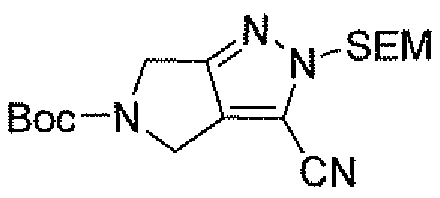
К перемешиваемому раствору продукта, полученного на стадии D (500 мг, 1,307 ммоль), в ДМФА (10 мл) добавляли хлорангидрид циануровой кислоты (482 мг, 2,61 ммоль). Образовавшуюся смесь перемешивали при комнатной температуре в атмосфере N2 на протяжении ночи, гасили насыщенным NaHCO3 и экстрагировали EtOAc (2×100 мл). Органические слои промывали насыщенным раствором соли, сушили над Na2SO4 и концентрировали. Остаток очищали колоночной хроматографией на колонке силикагеля Biotage 40S®, элюируя 0-20% этилацетатом в гексанах, получая при этом указанное в заголовке соединение в виде бесцветной смолы. ЖХ-МС: 265,1 (M-Boc).
Стадия F: 2,4,5,6-Тетрагидропирроло[3,4-c]пиразол-3-карбонитрил
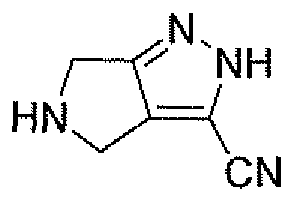
К перемешиваемому раствору продукта, полученного на стадии Е (250 мг, 0,686 ммоль), в EtOH (4 мл) добавляли 1 н HCl (8 мл). Образовавшуюся смесь нагревали при 90°С в атмосфере азота в течение 3 час. Растворители удаляли и сырой продукт подвергали колоночной хроматографии на силикагеле Biotage 40S®, элюируя 5-14% раствором MeOH (содержащим 10% концентрированной NH4OH) в CH2Cl2, получая при этом указанное в заголовке соединение. ЖХ-МС: 135,1 (M+H).
ПРОМЕЖУТОЧНЫЙ ПРОДУКТ 11
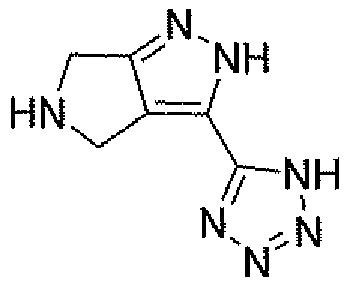
Стадия A: трет-Бутил-3-(1H-тетразол-5-ил)-2-{[2-(триметилсилил)этокси]метил)-2,6-дигидропирроло[3,4-c]пиразол-5-(4H)-карбоксилат
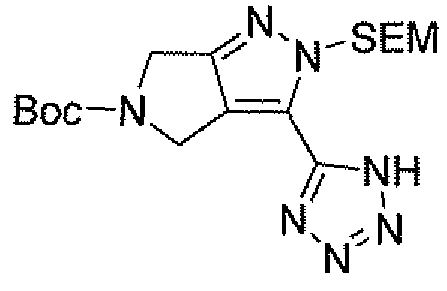
К перемешиваемому раствору продукта, полученного в разделе промежуточного продукта 10, стадия D (330 мг, 0,905 ммоль), в толуоле (10 мл) добавляли азид триметилолова (1,86 г, 9,05 ммоль). Образовавшуюся смесь кипятили с обратным холодильником в атмосфере азота на протяжении ночи, гасили насыщенным NaHCO3 и экстрагировали EtOAc (2×150 мл) и CH2Cl2 (2×150 мл). Органические слои промывали насыщенным раствором соли, сушили над Na2SO4 и концентрировали. Остаток очищали колоночной хроматографией на колонке силикагеля Biotage 40S®, элюируя 0-4% раствором EtOH (содержащего 10% HCO2H) в CH2Cl2, получая при этом указанное в заголовке соединение. ЖХ-МС: 408,1 (M+H).
Стадия В: 3-(1H-Тетразол-5-ил)-2,4,5,6-тетрагидропирроло[3,4-c]пиразол
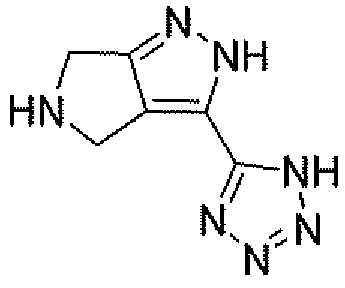
Посредством применения трет-бутил-3-(1H-тетразол-5-ил)-2-{[2-(триметилсилил)этокси]метил}-2,6-дигидропирроло[3,4-c]пиразол-5(4H)-карбоксилата (320 мг, 0,785 ммоль) указанное в заголовке соединение получали, как описано для промежуточного продукта 10, стадия F. ЖХ-MC: 178,1 (M+1).
ПРИМЕР 1

Стадия A: трет-Бутил-[(2R,3S,5R)-5-[2-(2-амино-2-оксоэтил)-2,6-дигидропирроло[3,4-c]пиразол-5(4Н)-ил]-2-(2,5-дифторфенил)тетрагидро-2Н-пиран-3-ил]карбамат
Смесь промежуточного продукта 2 (28,8 г, 88 ммоль) и промежуточного продукта 7 (14,6 г, 88 ммоль) в MeOH (1,5 л) перемешивали при комнатной температуре в течение 1 час. Затем добавляли декаборан (3,22 г, 25,4 ммоль). После перемешивания при комнатной температуре в течение 18 час реакционную смесь концентрировали и остаток очищали на двух отдельных колонках 65i Biotage®, элюируя раствором 1,25% метанола (содержащего 10% NH4OH) и 2,5% метанола (содержащего 10% NH4OH) в CH2Cl2, получая при этом требуемый продукт. ЖХ/МС=478,20 [M+1].
Стадия B: 2-{5-[(3R,5S,6R)-5-амино-6-(2,5-дифторфенил)тетрагидро-2H-пиран-3-ил]-5,6-дигидропирроло[3,4-c]пиразол-2(4Н)-ил}ацетамид
Продукт из стадии A (433 мг, 0,907 ммоль) в CH2Cl2 (60 мл) обрабатывали TFA (60 мл) при комнатной температуре в течение 2 час. После удаления смеси TFA/CH2Cl2 остаток очищали на колонке силикагеля и элюировали смесью 2,5-5% MeOH (содержащий 10% NH4OH)/CH2Cl2, получая при этом указанное в заголовке соединение. ЖХ-МС=378,04 [M+1].
ПРИМЕР 2
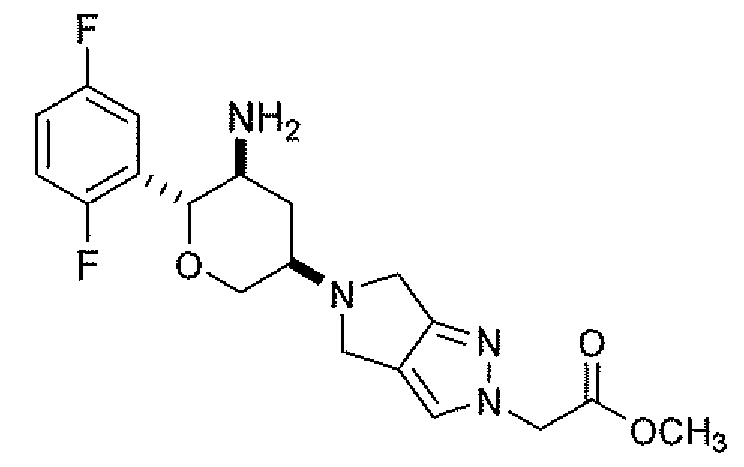
Метил-{5-[(3R,5S,6R)-5-амино-6-(2,5-дифторфенил)тетрагидро-2H-пиран-3-ил]-5,6-дигидропирроло[3,4-c]пиразол-2(4H)-ил}ацетат
Продукт, полученный в примере 1, стадии В (6,2 г, 16,44 ммоль), в MeOH (400 мл) обрабатывали 12 M HCl в течение 5 мин. Растворитель удаляли и сырое вещество очищали на силикагеле, элюируя смесью 2,5% МеОН (содержащий 10% NH4OH)/CH2Cl2, получая при этом указанное в заголовке соединение. МС 392,96 (M+1).
ПРИМЕР 3
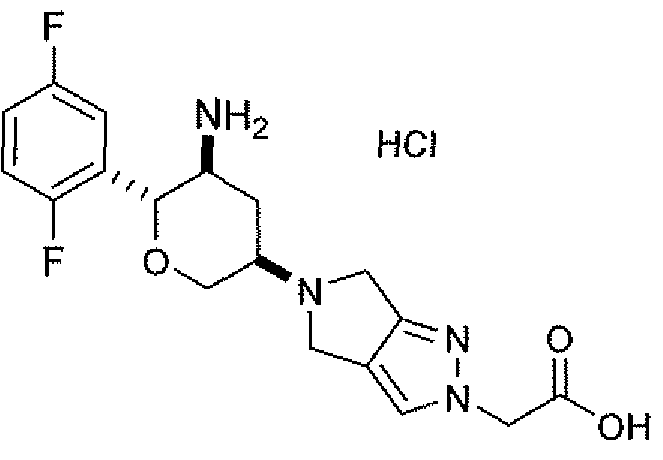
Гидрохлоридная соль {5-[(3R,5S,6R)-5-амино-6-(2,5-дифторфенил)тетрагидро-2H-пиран-3-ил]-5,6-дигидропирроло[3,4-c]пиразол-2(4H)-ил}уксусной кислоты
К суспензии продукта, полученного в примере 2 (200 мг, 0,406 ммоль), в Н2О (2 мл) добавляли концентрированную HCl (1,0 мл) при комнатной температуре. Смесь затем нагревали при 100°С в микроволновом реакторе в течение 1 час. Хлористоводородную кислоту и воду удаляли, получая при этом указанное в заголовке соединение в виде соли HCl. ЖХ/МС=378,97 [M+1].
ПРИМЕР 4
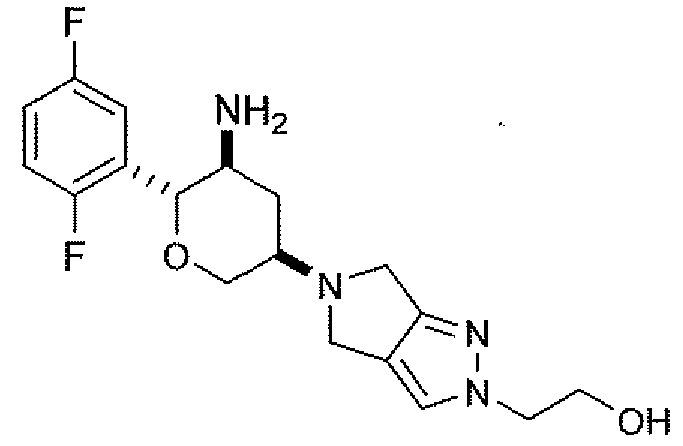
2-{5-[(3R,5S,6R)-5-амино-6-(2,5-дифторфенил)тетрагидро-2H-пиран-3-ил]-5,6-дигидропирроло[3,4-c]пиразол-2(4H)-ил}этанол
К перемешиваемому раствору продукта, полученного в примере 3 (400 мг, 1,019 ммоль), в МеОН (10 мл) добавляли боргидрид натрия (154 мг, 4,18 ммоль). Смесь перемешивали при комнатной температуре в течение 2 час и затем концентрировали. Образовавшееся вещество очищали на силикагеле, элюируя смесью 0-5% МеОН (содержащий 10% NH4OH)/CH2Cl2, получая при этом указанное в заголовке соединение в виде белого твердого вещества. ЖХ/МС=365,25 [M+1].
ПРИМЕР 5
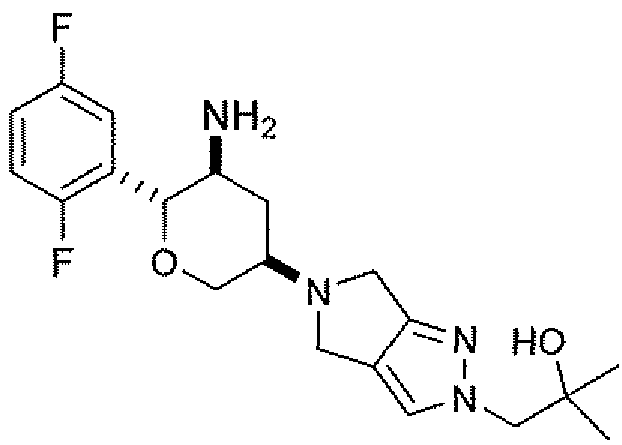
1-{5-[(3R,5S,6R)-5-Амино-6-(2,5-дифторфенил)тетрагидро-2H-пиран-3-ил]-5,6-дигидропирроло[3,4-c]пиразол-2(4H)-ил}-2-метилпропан-2-ол
К перемешиваемому раствору промежуточного продукта 9, изомер B (40,17 г, 105 ммоль), в MeOH (1184 мл) при комнатной температуре в атмосфере N2 медленно добавляли триэтиламин (26,3 мл, 189 ммоль). После перемешивания в течение 45 мин при комнатной температуре добавляли промежуточный продукт 2 (29,5 г, 90 ммоль) и смесь перемешивали при комнатной температуре в течение 20 мин. Затем добавляли декаборан (3,30 г, 27,0 ммоль). Смесь перемешивали при комнатной температуре в течение 4 час. Остаток очищали колоночной хроматографией на силикагеле, элюируя с градиентом 2-7% CH2Cl2/MeOH (содержащий 10% NH4OH), получая при этом BOC-защищенный продукт. ЖХ-МС=493,31 (M+1). BOC-защищенный продукт (30 г, 60,9 ммоль) растворяли в 4 M HCl в MeOH (914 мл, 3654 ммоль). Смесь перемешивали при комнатной температуре в течение 3 час, упаривали при пониженном давлении и остаток очищали колоночной хроматографией на колонке силикагеля Biotage 65i®, элюируя с градиентом от 2,5% до 11,5% CH2Cl2/EtOH (содержащий 10% NH4OH), получая при этом указанный в заголовке продукт в виде бесцветного твердого вещества. 1Н ЯМР (500 МГц, CD3OD): δ 7,37 (с, 1Н); 7,18-7,23 (м, 1H); 7,05-7,16 (м, 2H); 4,24-4,29 (м, 2H); 4,05 (с, 2H); 3,87 (с, 4H); 3,40 (т, J=11 Гц, 1H); 3,01-3,08 (м, 1Н); 2,87-2,92 (м, 1Н); 2,46-2,49 (м, 1Н); 1,50 (квт, J=12 Гц, 1H); 1,15 (с, 6H). ЖХ-МС=393,35 (M+1).
ПРИМЕР 6
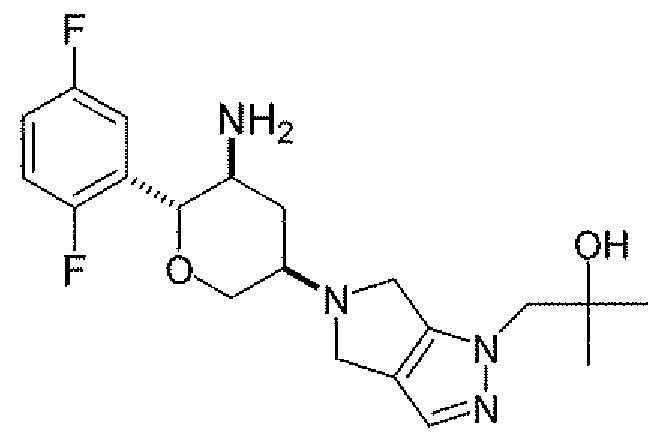
1-{5-[(3R,5S,6R)-5-Амино-6-(2,5-дифторфенил)тетрагидро-2H-пиран-3-ил]-5,6-дигидропирроло[3,4-c]пиразол-1(4H)-ил}-2-метилпропан-2-ол
Указанное в заголовке соединение получают по способу, описанному для примера 5, но с применением промежуточного продукта 9 с Вос-удаленной группой, изомер А, для реакции восстановительного аминирования с декабораном. Удаление Вос-защитной группы выполняют обработкой продукта 4 М HCl в МеОН.
1H ЯМР (500 МГц, CD3OD): δ 7,23 (с, 1H); 7,18-7,22 (м, 1H); 7,06-7,17 (м, 2H); 4,27-4,30 (м, 1H); 4,21-4,25 (м, 1Н); 3,99 (с, 4H); 3,84-3,90 (м, 2H); 3,38 (т, J=11 Гц, 1H); 3,02-3,09 (м, 1H); 2,89-2,94 (м, 1Н); 2,43-2,47 (м, 1H); 1,48 (квт, J=11,5 Гц, 1Н); 1,17 (с, 6H). ЖХ-МС=393,09 (M+1).
Нижеследующие соединения примеров получали с применением способов, описанных в примерах 1-6.
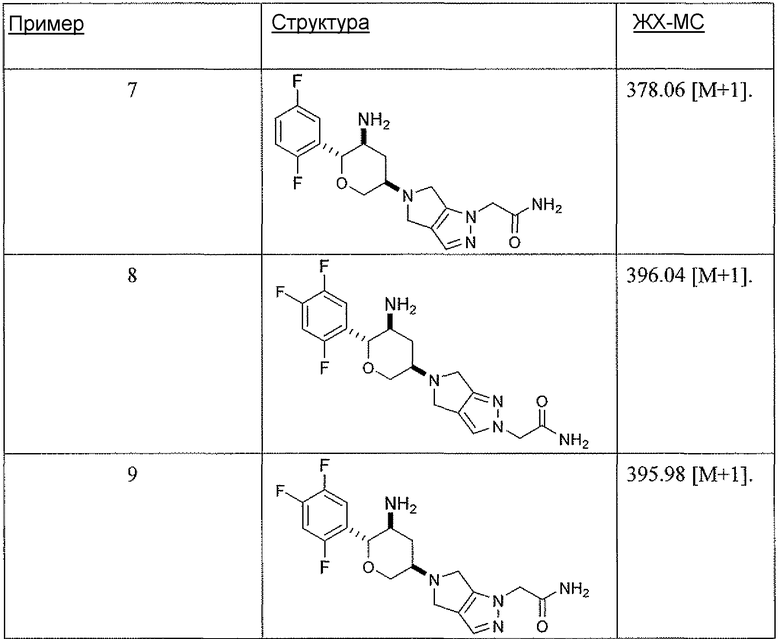
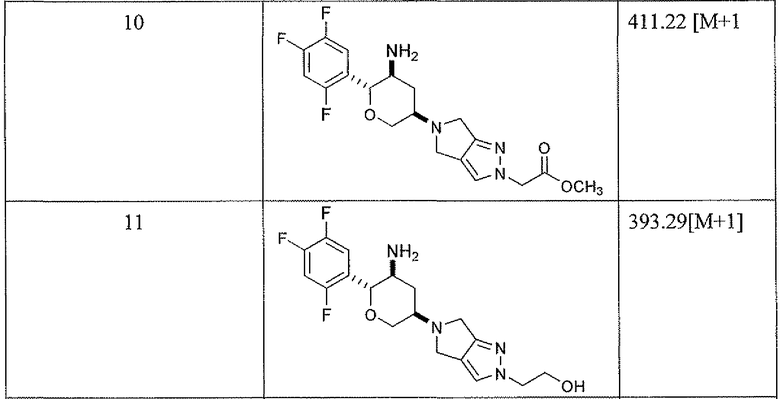
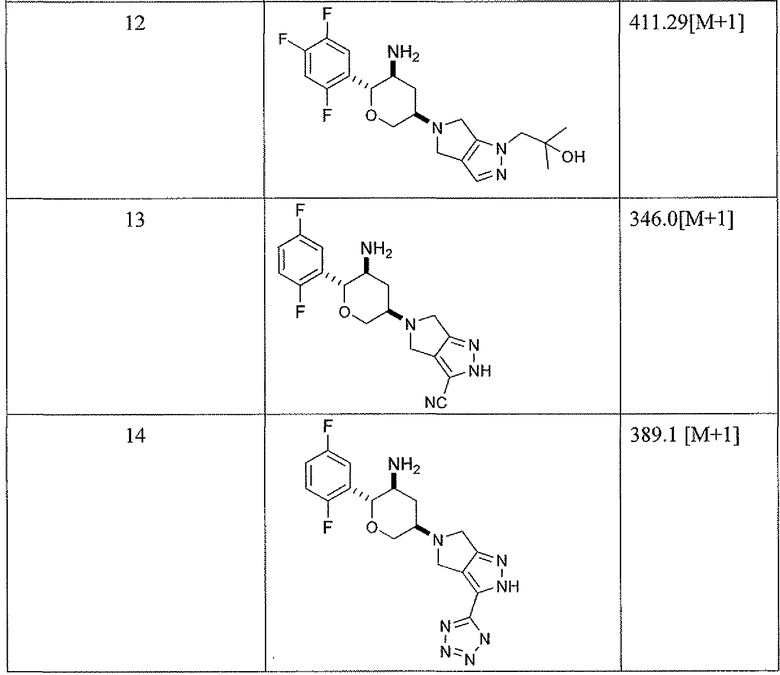
Ингибирование DPP-4 (IC50, нМ)
Пример фармацевтического препарата
В качестве конкретного варианта осуществления пероральной фармацевтической композиции таблетка со 100 мг активного лекарственного вещества состоит из 100 мг соединения любого из примеров, 268 мг микрокристаллической целлюлозы, 20 мг натрий-кроскармеллозы и 4 мг стеарата магния. Сначала смешивают активный ингредиент, микрокристаллическую целлюлозу и кроскармеллозу. Смесь затем смазывают стеаратом магния и прессуют в таблетки.
Хотя изобретение было описано и иллюстрировано обращением к некоторым его конкретным вариантам осуществления, специалисту в данной области должно быть понятно, что могут быть произведены различные адаптации, изменения, модификации, замены, исключения или добавления процедур и протоколов, не выходящие за пределы существа и объема изобретения. Например, эффективные дозы, другие, чем конкретные дозы, указываемые в контексте выше, могут быть применимыми в результате различий в восприимчивости подвергаемого лечению млекопитающего для любого из показаний с соединениями изобретения, указанными выше. Конкретные наблюдаемые фармакологические реакции могут варьироваться в соответствии и в зависимости от конкретных выбранных активных соединений или от того, присутствуют ли фармацевтические носители, а также типа препарата и применяемого способа введения, и такие предполагаемые варианты или различия в результатах рассматриваются в соответствии с задачами и практикой настоящего изобретения. Таким образом, предполагается, что изобретение должно определяться объемом формулы изобретения, которая приводится ниже, и что формула изобретения должна интерпретироваться в разумных широких пределах.
| название | год | авторы | номер документа |
|---|---|---|---|
| ЗАМЕЩЕННЫЕ АМИНО ШЕСТИЧЛЕННЫЕ НАСЫЩЕННЫЕ ГЕТЕРОАЛИЦИКЛЫ В КАЧЕСТВЕ ИНГИБИТОРОВ DPP-IV ДЛИТЕЛЬНОГО ДЕЙСТВИЯ | 2016 |
|
RU2720488C2 |
| ПИРАЗОЛО[3,4-b]ПИРИДИНОВЫЕ СОЕДИНЕНИЯ И ИХ ПРИМЕНЕНИЕ В КАЧЕСТВЕ ИНГИБИТОРОВ ФОСФОДИЭСТЕРАЗ | 2003 |
|
RU2348633C2 |
| ПРОИЗВОДНОЕ 2-ЦИАНО-4-ФТОРПИРРОЛИДИНА ИЛИ ЕГО СОЛЬ | 2003 |
|
RU2288222C2 |
| ХИМИЧЕСКИЕ СОЕДИНЕНИЯ В КАЧЕСТВЕ ИНГИБИТОРОВ АКТИВНОСТИ ИНТЕРЛЕЙКИНА-1 | 2018 |
|
RU2792143C2 |
| СОЕДИНЕНИЯ СУЛЬФОНИМИДАМИДА В КАЧЕСТВЕ ИНГИБИТОРОВ АКТИВНОСТИ ИНТЕРЛЕЙКИНА-1 | 2019 |
|
RU2820289C2 |
| ПРОИЗВОДНЫЕ 2-ПИРАЗИНОНА ДЛЯ ЛЕЧЕНИЯ ЗАБОЛЕВАНИЯ ИЛИ СОСТОЯНИЯ, ПРИ КОТОРЫХ ПОЛЕЗНО ИНГИБИРОВАНИЕ АКТИВНОСТИ НЕЙТРОФИЛЬНОЙ ЭЛАСТАЗЫ | 2007 |
|
RU2448098C2 |
| ЗАМЕЩЕННЫЕ ПИРИДОПИРАЗИНЫ КАК НОВЫЕ ИНГИБИТОРЫ Syk | 2012 |
|
RU2569635C9 |
| СОЕДИНЕНИЯ ДЛЯ ЛЕЧЕНИЯ ОПОСРЕДОВАННЫХ КОМПЛЕМЕНТОМ НАРУШЕНИЙ | 2015 |
|
RU2703995C2 |
| АРИЛ- И ГЕТЕРОАРИЛЗАМЕЩЕННЫЕ ГЕТЕРОЦИКЛИЧЕСКИЕ ПРОИЗВОДНЫЕ МОЧЕВИНЫ, СПОСОБ ИНГИБИРОВАНИЯ КИНАЗЫ RAF И ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ | 1998 |
|
RU2265597C2 |
| НОВЫЕ СОЕДИНЕНИЯ, КОТОРЫЕ ЯВЛЯЮТСЯ ИНГИБИТОРАМИ ERK | 2013 |
|
RU2660429C2 |
Настоящее изобретение относится к новым замещенным аминотетрагидропиранам структурной формулы I или к их фармацевтически приемлемым солям, в которой V выбран из групп, имеющих приведенные ниже формулы, Ar представляет собой фенил, незамещенный или замещенный одним-пятью атомами галогена, каждый из R1 и R2 независимо выбран из С1-С6алкила; R3 выбран из группы, состоящей из С1-С6алкила; циано; тетразолила; -С(О)ОС1-С6алкила и -C(O)NH2; где С1-С6алкил замещен 1-4 заместителями, независимо выбранными из группы, состоящей из ОН; -C(O)NH2 и -CO2H. Заявленные соединения являются ингибиторами фермента дипептидилпептидазы-IV и могут быть применимыми при лечении или предупреждении заболеваний, в которых участвует фермент дипептидилпептидаза-IV, таких как диабет и, особенно, диабет типа 2. Изобретение относится также к фармацевтической композиции, содержащей указанные соединения, и применению указанных соединений и композиций при предупреждении или лечении таких заболеваний, в которых участвует фермент дипептидилпептидаза-IV. 4 н. и 8 з.п. ф-лы, 14 пр.

1. Соединение структурной формулы I

или его фармацевтически приемлемая соль, где
V выбран из группы, состоящей из
Ar представляет собой фенил, незамещенный или замещенный одним-пятью атомами галогена,
каждый из R1 и R2 независимо выбран из С1-С6алкила;
R3 выбран из группы, состоящей из
С1-С6алкила;
циано;
тетразолила;
-С(О)ОС1-С6алкила и
-C(O)NH2;
где С1-С6алкил, замещен 1-4 заместителями, независимо выбранными из группы, состоящей из ОН; -C(O)NH2 и -CO2H.
2. Соединение по п. 1 или его фармацевтически приемлемая соль, где Ar замещен одним-тремя заместителями, независимо выбранными из группы, состоящей из атомов фтора, хлора и брома.
3. Соединение по п. 1 или его фармацевтически приемлемая соль, где V выбран из группы, состоящей из
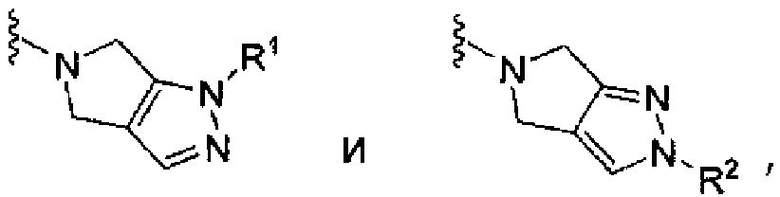
где каждый из R1 и R2 представляет собой С1-С6алкил, где алкил замещен -ОН.
4. Соединение по п. 1 или его фармацевтически приемлемая соль, где V выбран из группы, состоящей из
Ar представляет собой фенил, замещенный двумя-тремя атомами фтора;
R1 представляет собой С1-С6алкил;
где С1-С6алкил замещен 1-4 заместителями, независимо выбранными из группы, состоящей из
-ОН;
-C(O)NH2;
-CO2H;
R2 представляет собой С1-С6алкил;
где С1-С6алкил замещен 1-4 заместителями, независимо выбранными из группы, состоящей из
-ОН;
-С(O)NH2 и
-CO2H.
5. Соединение по п. 1 или его фармацевтически приемлемая соль, где V представляет собой
Ar представляет собой фенил, замещенный двумя-тремя атомами фтора;
R2 представляет собой С1-С6алкил;
где С1-С6алкил замещен 1-4 заместителями, независимо выбранными из группы, состоящей из
-ОН;
-C(O)NH2;
-CO2H.
6. Соединение по п. 1 структурной формулы Ia, имеющей указанную абсолютную стереохимическую конфигурацию у двух стереогенных атомов углерода, отмеченных знаком *
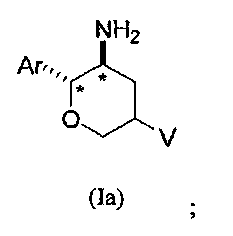
или его фармацевтически приемлемая соль.
7. Соединение по п. 6 структурной формулы Ic, имеющей указанную абсолютную стереохимическую конфигурацию у трех стереогенных атомов углерода, отмеченных знаком *
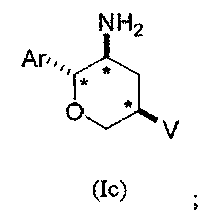
или его фармацевтически приемлемая соль.
8. Соединение по п. 7, где V выбран из группы, состоящей из
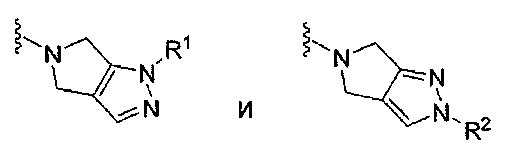
или его фармацевтически приемлемая соль,
где каждый из R1 и R2 независимо представляет собой С1-С6алкила;
где С1-С6алкил замещены 1-4 заместителями, независимо выбранными из группы, состоящей из
-ОН;
-С(O)NH2;
-CO2H.
9. Соединение по п. 1 или его фармацевтически приемлемая соль, где R3 выбран из группы, состоящей из циано, С1-С6алкила и -C(O)NH2, где С1-С6алкил замещен 1-4 заместителями, независимо выбранными из группы, состоящей из -ОН, -C(O)NH2 и -CO2H.
10. Соединение, которое выбрано из группы, состоящей из
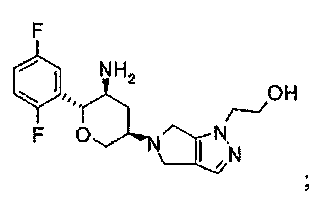
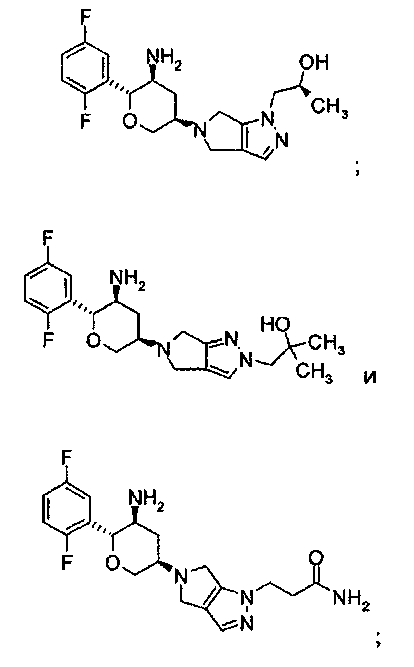
или его фармацевтически приемлемая соль.
11. Фармацевтическая композиция для лечения состояния, выбранного из группы, состоящей из инсулинорезистентности, гипергликемии, диабета типа 2, которая содержит соединение по п. 1 и фармацевтически приемлемый носитель.
12. Применение соединения по п. 1 при изготовлении лекарственного средства для лечения состояния, выбранного из группы, состоящей из инсулинорезистентности, гипергликемии, диабета типа 2.
| EA 200870380 A1 27.02.2009 & US 20070232676 A1, 04.10.2007 | |||
| Колосоуборка | 1923 |
|
SU2009A1 |
| Колосоуборка | 1923 |
|
SU2009A1 |

