Настоящее изобретение относится к новым производным оксазола, полезным в качестве сенсибилизаторов инсулина, особенно в качестве активаторов PPAR.
Изобретение относится к соединениям формулы I
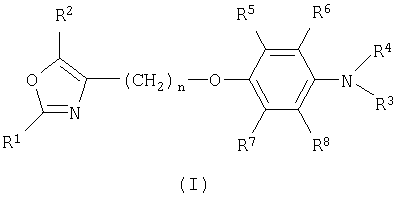
и их фармацевтически приемлемым солям и эфирам, где
R1 представляет собой арил;
R2 представляет собой водород, алкил или циклоалкил;
R3 представляет собой водород, алкил, аралкил, арил, алкилкарбонил, арилкарбонил, алкил-S(O)2- или арил-S(O)2-;
R4 представляет собой аралкил;
R5, R6, R7 и R независимо выбраны из водорода, алкила или циклоалкила; и n имеет значение 1, 2, 3, 4 или 5.
Соединения формулы I и их фармацевтически приемлемые соли и эфиры являются новыми и обладают ценными фармакологическими свойствами. Они являются сенсибилизаторами инсулина, особенно активаторами PPAR.
Рецепторы, активирующие пролиферацию пероксисом (PPAR) являются членами семейства рецептора ядерного гормона, которые являются лиганд-активируемыми факторами транскрипции, регулирующими экспрессию генов. Идентифицированы и клонированы различные их подтипы. Они включают PPARα, PPARβ (также известный как PPARδ) и PPARγ. Существует по крайней мере две основные изоформы PPARγ. Тогда как PPARγ1 экспрессируется во многих тканях, более длинная изоформа PPARγ2 обнаружена почти только в адипоцитах. Наоборот, PPARα в основном экспрессируется в печени, почках и сердце. PPAR модулируют различные реакции организма, включая глюкозный и липидный гомеостаз, клеточную дифференциацию, воспалительные реакции и сердечно-сосудистые реакции.
Диабет представляет собой заболевание, при котором нарушена способность пациента контролировать уровни глюкозы в крови из-за того, что он частично потерял способность должным образом реагировать на действие инсулина. При диабете II типа (T2D), часто называемом неинсулинзависимым сахарным диабетом (NIDDM), который составляет 80-90% всех пациентов, страдающих диабетом в развитых странах, при том, что панкреатические островки все еще производят инсулин в поджелудочной железе. Однако целевые органы, главным образом мышцы, печень и жировая ткань, проявляют сильную резистентность на стимулирование инсулином, и орган компенсирует это выработкой нефизиологически высоких уровней инсулина. На более поздней стадии заболевания, однако, секреция инсулина уменьшается из-за истощения поджелудочной железы. Кроме того, возникает T2D - метаболический сердечно-сосудистый синдром. Среди осложнений, связанных с T2D, можно указать, например, инсулиновую резистентность, дислипидемию, гипертонию, эндотелиальную дисфункцию и воспалительный атеросклероз.
В настоящее время первичное лечение диабета обычно включает диету с низким содержанием жиров и глюкозы и физические упражнения. Однако результаты могут быть средними и при развитии заболевания становится необходимым лечение гипоглицемическими лекарственными средствами, например, сульфонилмочевинами или метформином. Недавно был описан перспективный новый класс лекарственных средств, который обеспечивает возвращение чувствительности пациентов к их собственному инсулину (сенсибилизаторы инсулина), таким образом возвращая нормальные уровни глюкозы и триглицеридов в крови, и таким образом отменяя или по крайней мере сокращая потребность в экзогенном инсулине. Пиоглитазон (ActosTM) и росиглитазон (AvandiaTM) относятся к тиазолидиндионам (TZD), классу PPARγ - агонистов, и были первыми представителями, одобренными для NIDDM в нескольких странах. Эти соединения, однако, обладают побочными действиями, включая редкую, но сильную токсичность для печени (для троглитазона), и они увеличивают вес тела у людей. Поэтому срочно необходимы новые, улучшенные и более эффективные лекарственные средства для лечения NIDDM. Последние исследования подтверждают, что соагонизм PPARα и PPARγ привел бы к соединениям с увеличенной терапевтической активностью, то есть с улучшенным липидным профилем для нормализации уровней глюкозы и инсулина (Keller и Wahli: Trends Endocrin. Metab. 1993; 4:291-296, Macdonald and Lane: Current Biology, vol. 5, pp. 618-621 (1995)).
Новые соединения настоящего изобретения превосходят соединения, известные из уровня техники, поскольку они связываются и активизируют PPARα и PPARγ одновременно и очень эффективно. Поэтому эти соединения объединяют антиглицемическое действие на активацию PPARγ с антидислипидемическим действием на активацию PPARα. Следовательно, уровни глюкозы и инсулина в плазме уменьшаются (= сенсибилизация инсулина), уровни триглицеридов понижаются и уровни HDL холестерина увеличиваются (= улучшенный липидный профиль). Кроме того, такие соединения могут также понижать уровень LDL холестерина, уменьшать давление крови и противодействовать воспалительному атеросклерозу. Так как многочисленные аспекты T2D синдрома связаны с соагонистами PPARα и γ, ожидается, что они будут иметь увеличенную терапевтическую активность по сравнению с соединениями, уже известными в уровне техники.
Соответственно, соединения формулы I могут использоваться для профилактики и/или лечения диабетов, особенно неинсулинзависимых сахарных диабетов, повышенного давления крови, повышенных уровней липидов и холестерина, атеросклеротических заболеваний или метаболического синдрома.
Объектами настоящего изобретения являются соединения формулы I и их упомянутые выше фармацевтически приемлемые соли и эфиры и их применение в качестве терапевтически активных веществ, способ получения указанных соединений, промежуточные соединения, фармацевтические композиции, лекарственные средства, включающие указанные соединения, их фармацевтически приемлемые соли и эфиры, применение указанных соединений, эфиров и солей для профилактики и/или лечения заболеваний, особенно для лечения и/или профилактики диабетов, неинсулинзависимых сахарных диабетов, повышенного давления крови, повышенных уровней липидов и холестерина, атеросклеротических заболеваний или метаболического синдрома и особенно для профилактики и/или лечения неинсулинзависимых сахарных диабетов, и применение указанных соединений, солей и эфиров для изготовления лекарственных средств для лечения и/или профилактики заболеваний, особенно для лечения и/или профилактики диабетов, неинсулинзависимых сахарных диабетов, повышенного давления крови, повышенных уровней липидов и холестерина, атеросклеротических заболеваний или метаболического синдрома.
В настоящем описании термин "алкил", отдельно или в комбинации, обозначает алкильную группу с прямой цепью или разветвленной цепью с 1-8 атомами углерода, предпочтительно алкильную группу с прямой или разветвленной цепью с 1-6 атомами углерода и особенно предпочтительно алкильную группу с прямой или разветвленной цепью с 1-4 атомами углерода. Примерами C1-C8 алкильных групп с прямой и разветвленной цепью являются метил, этил, пропил, изопропил, бутил, изобутил, трет-бутил, изомерные пентилы, изомерные гексилы, изомерные гептилы и изомерные октилы, предпочтительно метил и этил, и наиболее предпочтительно метил.
Термин "циклоалкил", отдельно или в комбинации, обозначает циклоалкильное кольцо с 3-8 атомами углерода и предпочтительно циклоалкильное кольцо с 3-6 атомами углерода. Примерами С3-C8 циклоалкила являются циклопропил, метилциклопропил, диметилциклопропил, циклобутил, метилциклобутил, циклопентил, метилциклопентил, циклогексил, метилциклогексил, диметилциклогексил, циклогептил и циклооктил, предпочтительно циклопропил.
Термин "алкокси", отдельно или в комбинации, обозначает группу формулы алкил-O-, где термин "алкил" имеет указанное ранее значение, такую как метокси, этокси, н-пропокси, изопропокси, н-бутокси, изо-бутокси, втор-бутокси и трет-бутокси, 2-гидроксиэтокси, 2-метоксиэтокси, предпочтительно метокси и этокси, и наиболее предпочтительно метокси.
Термин "арилокси", отдельно или в комбинации, обозначает группу формулы арил-O-, где термин "арил" имеет указанное ранее значение, такую как фенилокси.
Термин "арил", отдельно или в комбинации, обозначает фенильную или нафтильную группу, предпочтительно фенильную группу, которая необязательно имеет один или несколько заместителей, каждый независимо выбранный из галогена, трифторметила, амино, алкила, алкокси, алкилкарбонила, циано, карбамоила, алкоксикарбамоила, метилендиокси, карбокси, алкоксикарбонила, аминокарбонила, алкиламинокарбонила, диалкиламинокарбонила, гидрокси, нитро и т.п., такую как фенил, хлорфенил, трифторметилфенил, хлорфторфенил, аминофенил, метилкарбонилфенил, метоксифенил, метилендиоксифенил, 1-нафтил и 2-нафтил. Предпочтительным является фенил, 3-хлорфенил, 3-трифторметилфенил, 3-аминофенил, 4-метилкарбонилфенил, 4-метоксифенил и особенно фенил.
Термин "аралкил", отдельно или в комбинации, обозначает определенную выше алкильную или циклоалкильную группу, в которой один или несколько, предпочтительно один атом водорода замещен арильной группой, определенной выше. Предпочтительными являются бензил, бензил, замещенный гидрокси, алкокси или галогеном, предпочтительно фтором. Особенно предпочтительным является бензил.
Термин "амино", отдельно или в комбинации, обозначает первичную, вторичную или третичную аминогруппу, связанную через атом азота, со вторичной аминогруппой, несущей алкильный или циклоалкильный заместитель и третичной аминогруппой, несущей два одинаковых или различных алкильных или циклоалкильных заместителя, или два азотных заместителя вместе образуют кольцо, такие как, например, -NH2, метиламино, этиламино, диметиламино, диэтиламино, метилэтиламино, пирролидинил-1-ил или пиперидино и т.д., предпочтительно амино, диметиламино и диэтиламино и особенно первичная аминогруппа.
Термин "галоген" обозначает фтор, хлор, бром или йод и предпочтительно фтор, хлор или бром.
Термин "карбонил", отдельно или в комбинации, обозначает группу -С(O)-.
Термин "циано", отдельно или в комбинации, обозначает группу -CN.
Термин "фармацевтически приемлемые соли" обозначает соли, которые сохраняют биологическую эффективность и свойства свободных оснований или свободных кислот, которые не являются биологически или иным образом нежелательными. Такие соли получают с неорганическими кислотами, такими как соляная кислота, бромоводородная кислота, серная кислота, азотная кислота, фосфорная кислота и др., предпочтительно соляная кислота, и органическими кислотами, такими как уксусная кислота, пропионовая кислота, гликолевая кислота, пирувиновая кислота, щавелевая кислота, малеиновая кислота, малоновая кислота, янтарная кислота, фумаровая кислота, винная кислота, лимонная кислота, бензойная кислота, коричная кислота, миндальная кислота, метансульфоновая кислота, этансульфоновая кислота, п-толуолсульфоновая кислота, салициловая кислота, N-ацетилцистеин и т.д. Кроме того, эти соли могут быть получены добавлением неорганического основания или органического основания со свободной кислотой. Соли, полученные из неорганического основания, включают, но не ограничиваются ими, соли натрия, калия, лития, аммония, кальция, магния и т.д. Соли, полученные из органических оснований, включают, но не ограничиваются ими, соли первичных, вторичных и третичных аминов, замещенных аминов, включая природные замещенные амины, циклические амины и основные ионообменные смолы, такие как изопропиламин, триметиламин, диэтиламин, триэтиламин, трипропиламин, этаноламин, лизин, аргинин, N-этилпиперидин, пиперидин, полиминовые смолы и т.д. Соединение формулы I может также присутствовать в форме цвиттерионов. Особенно предпочтительными фармацевтически приемлемыми солями соединений формулы I являются гидрохлориды.
Соединения формулы I могут быть также сольватированы, например, гидратированы. Сольват может быть получен в процессе производства или может быть получен, например, в результате гигроскопических свойств первоначально безводного соединения формулы I (гидратация). Термин фармацевтически приемлемые соли также включает физиологически приемлемые сольваты.
"Фармацевтически приемлемые эфиры" обозначает, что соединения общей формулы (I) могут быть дериватизированы по функциональным группам с получением производных, которые способны превращаться обратно в исходные соединения in vivo. Примеры таких соединений включают физиологически приемлемые и метаболически лабильные эфирные производные, такие как метоксиметиловые эфиры, метилтиометиловые эфиры и пивалоилоксиметиловые эфиры. Кроме того, любые физиологически приемлемые эквиваленты соединений общей формулы (I), подобные метаболически лабильным эфирам, которые способны преобразовываться в исходные соединения общей формулы (I) in vivo, входят в объем настоящего изобретения.
Термин "ингибитор липазы" относится к соединениям, которые способны ингибировать действия липаз, например, желудочных и панкреатических липаз. Например, орлистат и липстатин, как описано в US 4,598,089, являются сильными ингибиторами липаз. Липстатин является природным продуктом микробного происхождения, а орлистат является результатом гидрирования липстатина. Другой ингибитор липаз включает класс соединений, обычно упоминаемый как панклицины. Панклицины являются аналогами орлистата (Mutoh и др., 1994). Термин "ингибитор липазы" относится также к полимерным связывающим липазы ингибиторам, например, описанным в WO 99/34786 (Geltex Pharmaceuticals Inc). Эти полимеры характеризуются тем, что они были замещены одной или несколькими группами, которые ингибируют липазы. Термин "ингибитор липазы" также включает фармацевтически приемлемые соли этих соединений. Термин "ингибитор липазы" предпочтительно относится к орлистату.
Орлистат является известным соединением, полезным для контроля или профилактики ожирения и гиперлипидемии. См. US 4,598,089 от 01.07.1986, который также раскрывает способы получения орлистата, и US 6,004,996, который раскрывает соответствующие фармацевтические композиции. Далее подходящие фармацевтические композиции описаны, например, в WO 00/09122 и WO 00/09123. Дополнительные способы получения орлистата раскрыты в ЕР 185,359, 189,577, 443,449 и 524,495.
Предпочтительно орлистат вводится орально в дозе от 60 до 720 мг в день в раздельных дозах два-три раза в день. Предпочтительно, субъекту вводится доза от 180 до 360 мг, наиболее предпочтительно 360 мг в день ингибитора липазы, предпочтительно в раздельных дозах два или, особенно, три раза в день. Субъект - предпочтительно полный или толстый человек, то есть человек с показателем массы тела 25 или более. Вообще предпочтительно, чтобы ингибитор липазы вводился приблизительно через один или два часа после приема пищи, содержащей жир. Вообще, для введения определенного выше ингибитора липазы предпочтительно, чтобы лечение проводилось для человека с наследственным ожирением и индексом массы тела 25 или более.
Орлистат может вводиться людям в обычных оральных композициях, таких как, таблетки, покрытые таблетки, твердые и мягкие желатиновые капсулы, эмульсии или суспензии. Примерами носителей, которые могут использоваться для таблеток, покрытых таблеток, драже и твердых желатиновых капсул, являются лактоза, другие сахара и сахарные спирты, подобно сорбиту, манниту, мальтодекстрину или другие наполнители; поверхностно-активные вещества, подобно лаурилсульфату натрия, Brij 96 или Tween 80; дезинтегрирующие вещества, подобно натрийгликолированному крахмалу, кукурузному крахмалу или его производным; полимеры, подобно повидону, кросповидону; тальк; стеариновая кислота или ее соли и т.п. Подходящими носителями для мягких желатиновых капсул являются, например, растительные масла, воски, жиры, полутвердые и жидкие полиолы и т.п. Кроме того, фармацевтические составы могут содержать консервирующие агенты, солюбилизаторы, стабилизирующие агенты, увлажняющие агенты, эмульсифицирующие агенты, подслащивающие агенты, красители, отдушки, соли для изменения осмотического давления, буферы, покрывающие агенты и антиоксиданты. Они могут также содержать другие терапевтически полезные вещества. Составы могут быть удобно представлены в единичных дозированных формах и могут быть получены любыми методами, известными в фармацевтической области. Предпочтительно, орлистат вводится в соответствии с формулировкой, показанной в примерах и в US 6,004,996, соответственно.
Соединения формулы I могут содержать несколько асимметричных центров и могут присутствовать в форме оптически чистых энантиомеров, смесей энантиомеров, таких как, например, рацематы, оптически чистых диастереоизомеров, смесей диастереоизомеров, диастереоизомерных рацематов или смесей диастереоизомерных рацематов.
Предпочтительными являются соединения формулы I и их фармацевтически приемлемые соли, особенно соединения формулы I.
Также предпочтительными являются соединения формулы I, где R1 представляет собой фенил.
Предпочтительным вариантом осуществления настоящего изобретения являются соединения формулы I, где R2 представляет собой алкил, предпочтительно метил.
Также предпочтительными являются соединения формулы I, где R3 представляет собой алкил, аралкил, арил, алкилкарбонил, арилкарбонил, алкил-S(O)2- или арил-S(O)2-. Также предпочтительными являются соединения, где R3 представляет собой метил, пропил, бензил, метилкарбонил, фенилкарбонил, метил-S(O)2-, фенил-S(O)2-, фенил или фенил, замещенный одним или несколькими, предпочтительно одним или двумя заместителями, независимо выбранными из алкила, галогена, трифторметила и алкокси.
Другим предпочтительным вариантом осуществления настоящего изобретения являются соединения формулы I, где R3 представляет собой алкил или фенил, где фенил необязательно моно- или дизамещен галогеном. Особенно предпочтительными являются соединения формулы I, где R3 представляет собой пропил, фенил, фторфенил или дифторфенил.
Также предпочтительными соединениями формулы I являются соединения, где R4 представляет собой арилметил. Особенно предпочтительными являются соединения формулы I, где R4 представляет собой бензил, замещенный карбокси и необязательно также замещенный фтором, хлором, трифторметилом или алкокси.
Также предпочтительными являются соединения формулы I, где R5 представляет собой водород.
Другими предпочтительными соединениями являются соединения формулы I, где R6 представляет собой водород.
Другим предпочтительным вариантом осуществления настоящего изобретения являются соединения формулы I, где R7 представляет собой водород.
Также предпочтительными являются соединения формулы I, где R8 представляет собой водород.
Особенно предпочтительными соединениями формулы I являются соединения, где R5, R6, R7 и R8 представляют собой водород.
Другим предпочтительным вариантом осуществления настоящего изобретения являются соединения формулы I, где n имеет значение 1, 2 или 3. Особенно предпочтительными соединениями формулы I являются соединения, где n имеет значение 2.
Примерами предпочтительных соединений формулы (I) являются
1. 2-[(Ацетил-{4-[2-(5-метил-2-фенилоксазол-4-ил)этокси]фенил}амино)метил]бензойная кислота;
2. 2-[(Бензоил-{4-[2-(5-метил-2-фенилоксазол-4-ил)этокси]фенил}амино)метил]бензойная кислота;
3. 2-[(Метансульфонил-{4-[2-(5-метил-2-фенилоксазол-4-ил)этокси]фенил}амино)метил]бензойная кислота;
4. 2-[(Бензолсульфонил-{4-[2-(5-метил-2-фенилоксазол-4-ил)этокси]фенил}амино)метил]бензойная кислота;
5. 2-[(Метил-{4-[2-(5-метил-2-фенилоксазол-4-ил)этокси] фенил} амино)метил]бензойная кислота;
6. 2-[({4-[2-(5-Метил-2-фенилоксазол-4-ил)этокси]фенил}пропиламино)метил]бензойная кислота;
7. 2-[(Бензил-{4-[2-(5-метил-2-фенилоксазол-4-ил)этокси]фенил}амино)метил]бензойная кислота;
8. 2-[({4-[2-(5-Метил-2-фенилоксазол-4-ил)этокси]фенил}-о-толиламино)метил]бензойная кислота;
9. 2-[((3-Фторфенил)-{4-[2-(5-метил-2-фенилоксазол-4-ил)этокси]фенил}амино)метил]бензойная кислота;
10. 2-[({4-[2-(5-Метил-2-фенилоксазол-4-ил)этокси]фенил}-м-толиламино)метил]бензойная кислота;
11. 2-{[{4-[2-(5-Метил-2-фенилоксазол-4-ил)этокси]фенил}-(3-трифторметилфенил)амино]метил}бензойная кислота;
12. 2-[((4-Фторфенил)-{4-[2-(5-метил-2-фенилоксазол-4-ил)этокси]фенил}амино)метил]бензойная кислота;
13. 2-[({4-[2-(5-Метил-2-фенилоксазол-4-ил)этокси]фенил}-п-толиламино)метил]бензойная кислота;
14. 2-[((4-Метоксифенил)-{4-[2-(5-метил-2-фенилоксазол-4-ил)этокси]фенил}амино)метил]бензойная кислота;
15. 2-[((3,4-Диметилфенил)-{4-[2-(5-метил-2-фенилоксазол-4-ил)этокси]фенил}амино)метил]бензойная кислота;
16. 2-[((3,4-Дифторфенил)-{4-[2-(5-метил-2-фенилоксазол-4-ил)этокси] фенил} амино)метил]бензойная кислота;
17. 2-[((4-Фтор-3-метилфенил)-{4-[2-(5-метил-2-фенилоксазол-4-ил)этокси]фенил}амино)метил]бензойная кислота;
18. 3-Фтор-2-[({4-[2-(5-метил-2-фенилоксазол-4-ил)этокси] фенил} фениламино)метил]бензойная кислота;
19. 3-Хлор-2-[({4-[2-(5-метил-2-фенилоксазол-4-ил)этокси]фенил}фениламино)метил]бензойная кислота;
20. 2-[({4-[2-(5-Метил-2-фенилоксазол-4-ил)этокси]фенил}фениламино)метил]-3-трифторметилбензойная кислота;
21. 3-Метокси-2-[({4-[2-(5-метил-2-фенилоксазол-4-ил)этокси]фенил}фениламино)метил]бензойная кислота;
22. 4-Хлор-2-[({4-[2-(5-метил-2-фенилоксазол-4-ил)этокси]фенил}фениламино)метил]бензойная кислота;
23. 4-Метокси-2-[({4-[2-(5-метил-2-фенилоксазол-4-ил)этокси]фенил}фениламино)метил]бензойная кислота;
24. 5-Фтор-2-[({4-[2-(5-метил-2-фенилоксазол-4-ил)этокси]фенил}фениламино)метил]бензойная кислота;
25. 2-[({4-[2-(5-Метил-2-фенилоксазол-4-ил)этокси]фенил}фениламино)метил]бензойная кислота и
26. 2-Метокси-6-[({4-[2-(5-метил-2-фенилоксазол-4-ил)этокси]фенил}фениламино)метил]бензойная кислота.
Примерами особенно предпочтительных соединений формулы (I) являются
2-[({4-[2-(5-метил-2-фенилоксазол-4-ил)этокси] фенил}фениламино)метил]бензойная кислота;
2-[({4-[2-(5-метил-2-фенилоксазол-4-ил)этокси]фенил}пропиламино)метил]бензойная кислота;
2-[((3-фторфенил)-{4-[2-(5-метил-2-фенилоксазол-4-ил)этокси]фенил}амино)метил]бензойная кислота;
2-[((3,4-дифторфенил)-{4-[2-(5-метил-2-фенилоксазол-4-ил)этокси]фенил}амино)метил]бензойная кислота;
3-фтор-2-[({4-[2-(5-метил-2-фенилоксазол-4-ил)этокси]фенил}фениламино)метил]бензойная кислота и
5-фтор-2-[({4-[2-(5-метил-2-фенилоксазол-4-ил)этокси]фенил}фениламино)метил]бензойная кислота.
Способы получения соединений формулы I являются объектами настоящего изобретения.
Заместители и обозначения, используемые в следующем описании способов, имеют приведенные выше значения, если не указано иное.
Соединения общей формулы I, где R1, R2, R4-R8 и n являются такими, как определено выше, и где R3 представляет собой водород, алкил, аралкил, арил, алкилкарбонил, арилкарбонил, алкил-S(O)2- или арил-S(O)2-, могут быть получены в соответствии со схемами I и II:
Схема I
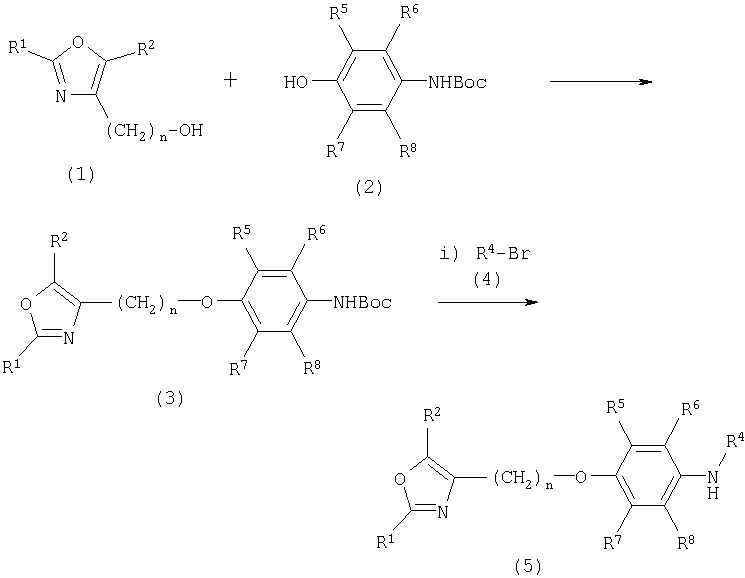
Boc обозначает трет-бутоксикарбонил
Оксазолэфир (3) может быть получен реакцией спирта (1) и монозамещенного анилина (2) в условиях Митсунобу в ТГФ (О.Mitsunobu, Synthesis, I, 1981). Алкилирование (3) с помощью (4) может осуществляться в КОН в ДМСО с последующим снятием защиты Вос-группы с помощью CF3COOH с получением соединения (5).
Схема II
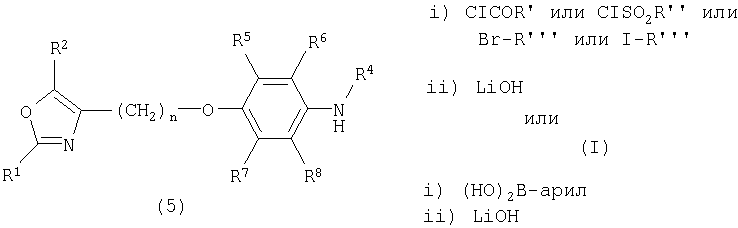
R' представляет собой алкил или арил,
R'' представляет собой алкил или арил,
R''' представляет собой алкил или аралкил.
Ацилирование и сульфонилирование амина (5) может осуществляться соответствующими хлоридами. Алкилирование (5) требует предпочтительно соответствующих алкилйодидов или бензилбромида. Все реакции могут осуществляться в присутствии основания, например, NEt3 в ТГФ, с получением предшественника (I) в виде его эфиров. На второй стадии эфиры могут быть гидролизованы с помощью LiOH в смеси растворителя ТГФ, МеОН и воды с получением конечных соединений (I). Арилирование амина (5) может осуществляться соответствующими борными кислотами в соответствии со способом, описанным Lam (P.Y.S.Lam, G.Vincent, C.G.Clark, S.Deudon, P.K.Jadhav, Tetrahedron Lett. 42, 3415, 2001) с получением предшественника (I) в виде эфиров, которые могут быть гидролизованы как описано выше.
В случае присутствия свободной карбоксигруппы, например, в заместителе R4, эта карбоксигруппа может быть замещена способами, известными из уровня техники, например, этиловым эфиром. Присутствующие группы ОН могут быть защищены с помощью подходящих защитных групп, таких как, например, этиловый эфир.
Альтернативно, соединения общей формулы I, где R1, R2, R4-R8 и n являются такими, как определено выше, и где R3 представляет собой водород, алкил, аралкил, арил, алкилкарбонил, арилкарбонил, алкил-S(O)2- или арил-S(O)2-, могут быть получены в соответствии со схемой III:
Схема III
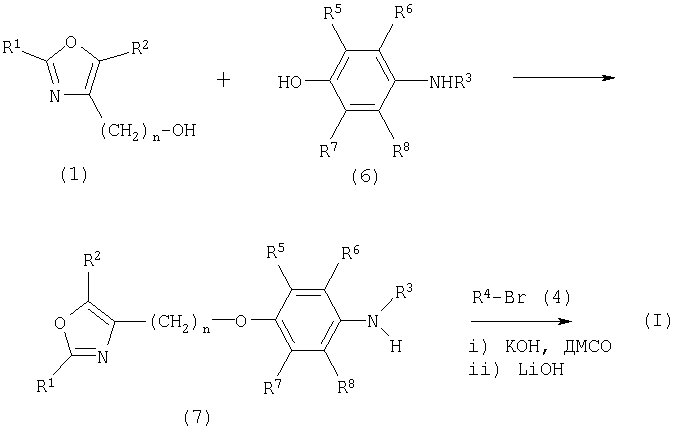
Альтернативное получение (I) в соответствии со схемой III предпочтительно используется, когда R3 постоянный и R4 изменяется в зависимости от типа реакции, как описано на схемах I и II.
В случае присутствия свободной карбоксигруппы, например, в заместителе R4, эта карбоксигруппа может быть замещена способами, известными в уровне техники, например, этиловым эфиром.
Исходное соединение (1) может быть получено, например, в соответствии со схемами IV или V:
Схема IV
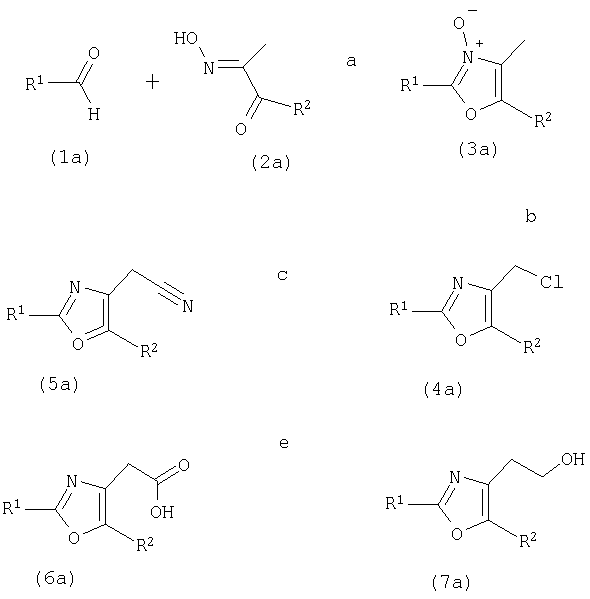
Альдегиды (1а) являются коммерчески доступными или известными. Они конденсируются с дикетомонооксимами (2а) известным способом (Goto, Y.; Yamazaki, M.; Hamana, M.; Chem Pharm Bull (1971), 19, 2050) в присутствии сильной кислоты, обычно HCl, в полярном растворителе, таком как АсОН, с получением оксазол-N-оксидов (3а) (стадия а). Последующая обработка POCl3 в дихлорметане при кипячении с обратным холодильником приводит к получению соответствующих первичных хлоридов (4а) (Goto, Y.; Yamazaki, M.; Hamana, M.; Chem Pharm Bull (1971), 19, 2050, стадия b). Эти промежуточные соединения используются в том же виде, превращаясь известными способами в соответствующие спирты или активированные спирты, такие как мезилаты или тозилаты, или в бромиды или йодиды, или далее преобразуются SN2-реакцией с NaCN с получением, через нитрилы 5 (стадия с), полный гидролиз (стадия d) и восстановление (стадия е), например, бором в тетрагидрофуране, соединений (7а).
4-Хлорметил-2-арил или 2-гетероарилоксазолы (4а) с R2, являющимся водородом, предпочтительно получают из соответствующих арил- или гетероарилкарбоксамидов и 1,3-дихлорацетона, как описано, например, в Bioorg. Med. Chem. Lett. (2000), 10(17), 2041-2044.
Соединения формулы (I), где n обозначает 1, могут быть получены реакцией соединения формулы 4а с соединением формулы 6 по реакциям, показанным на схеме III.
Схема V
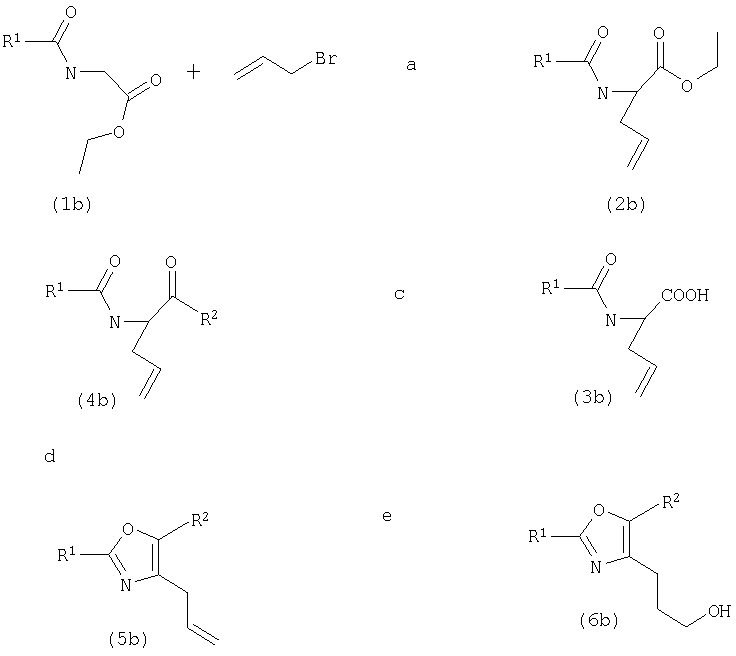
Эфиры N-ацилглицина (1b) являются коммерчески доступными, известными или могут быть получены стандартными реакциями N-ацилирования. Моноаллильные эфиры (2b) могут быть легко получены двойным депротонированием (1b) сильным ненуклеофильным основанием, таким как LiHMDS, в апротонном растворителе, таком как ТГФ, обычно при -78°С, с последующей обработкой аллилбромидом с получением селективно С-алкилированных продуктов (2b) (стадия а). Стандартный гидролиз приводит к получению промежуточных кислот (3b) (стадия b), которые затем преобразуют хорошо известными способами (J. Med. Chem. (1996), 39, 3897) в соединения (4b) (стадия с). Закрытие кольца в оксазол с помощью трифторуксусной кислоты и трифторуксусного ангидрида приводит к получению ключевых промежуточных соединений (5b) (стадия d), которые, наконец, подвергают гидроборированию с получением целевых спиртов (6b), например, с помощью 9-BBN в ТГФ и последующему окислению H2O2 и NaOH (стадия е).
Исходные соединения формулы (1), где n обозначает 4, могут быть получены следующим образом:
1) мезилирование соединения формулы (6b);
2) SN2-реакция с NaCN с получением соответствующего нитрила;
3) гидролиз нитрила;
4) восстановление, например, бором.
Исходные соединения формулы (1), где n обозначает 5, могут быть получены следующим образом:
1) мезилирование соединения формулы 1, где n обозначает 4;
2) SN2-реакция с NaCN с получением соответствующего нитрила;
3) гидролиз нитрила;
4) восстановление, например, бором.
Исходные соединения (2) и (6) являются известными или могут быть синтезированы способами, известными из уровня техники, например, нитрованием фенола с помощью серной и salpetric кислоты с последующим восстановлением соответствующего нитрофенола каталитическим гидрированием или железом в соляной кислоте. Вос-защита первичного амина может вводиться Вос-ангидридом в пиридине с получением (2) или, альтернативно, незащищенный анилин может быть превращен в соединение (6) арилированием, описанным Lam (P.Y.S.Lam, G.Vincent, C.G.Clark, S.Deudon, P.K.Jadhav, Tetrahedron Lett. 42, 3415, 2001).
Соединения формулы (4) и соответствующие защищенные аналоги могут быть получены следующим образом: замещенную толуолкарбоновую кислоту защищают как эфир известными способами, например, этерификацией спиртом и соляной кислотой. Бромирование метильной группы осуществляется также хорошо известными способами, например, с помощью N-бромсукцинимида и каталитического количества дибензоилпероксида в галогенированном растворителе, таком как CCl4.
Превращение соединения формулы I в фармацевтически приемлемую соль может осуществляться обработкой такого соединения неорганической кислотой, например, галогенводородной кислотой, такой как, например, соляная кислота или бромоводородная кислота, серная кислота, азотная кислота, фосфорная кислота и т.д., или органической кислотой, такой как, например, уксусная кислота, лимонная кислота, малеиновая кислота, фумаровая кислота, винная кислота, метансульфоновая кислота или п-толуолсульфоновая кислота. Соответствующие карбоксилатные соли также могут быть получены из соединений формулы I обработкой физиологически приемлемых оснований.
Превращение соединений формулы I в фармацевтически приемлемые эфиры или амиды может осуществляться, например, обработкой подходящих амино- или гидроксильных групп в молекулах карбоновой кислоты, такой как уксусная кислота, конденсирующим реагентом, таким как гексафторфосфат бензотриазол-1-илокситрис(диметиламино)фосфония (ВОР) или N,N-дициклогексилкарбодиимид (DCCI), с получением эфира карбоновой кислоты или амида карбоновой кислоты.
Далее предпочтительным является способ получения соединения формулы I, включающий одну из следующих реакций:
а) реакцию соединения формулы (5)
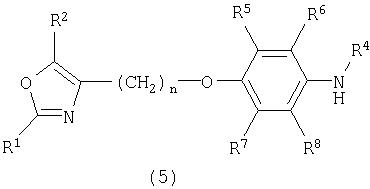
в присутствии R3-Hal и последующую реакцию в присутствии гидроксида, предпочтительно LiOH, с получением соединения формулы I, где Hal обозначает хлор, бром или йод и R1 to R8 и n определены выше. Особенно предпочтительной является указанная выше реакция, где R3 обозначает алкилкарбонил или арилкарбонил и Hal обозначает хлор. Далее предпочтительной является указанная выше реакция, где R3 обозначает алкил или аралкил и Hal обозначает бром или йод.
б) реакцию соединения формулы (5) в присутствии (НО)2В-арила и последующую реакцию в присутствии гидроксида, предпочтительно LiOH, с получением соединения формулы I, где R1, R2, R4-R8 и n определены выше;
в) реакцию соединения формулы (7)
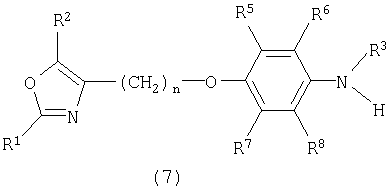
в присутствии R4-Hal и последующую реакцию в присутствии гидроксида, предпочтительно LiOH, с получением соединения формулы I, где Hal обозначает хлор, бром или йод и R1-R8 и n определены выше. Предпочтительной является указанная выше реакция, где Hal обозначает бром.
Предпочтительными промежуточными соединениями являются:
трет-бутиловый эфир {4-[2-(5-метил-2-фенилоксазол-4-ил)этокси] фенил} карбоновой кислоты,
этиловый эфир 2-[(трет-бутоксикарбонил-{4-[2-(5-метил-2-фенилоксазол-4-ил)этокси] фенил} амино)метил]бензойной кислоты,
этиловый эфир 2-({4-[2-(5-метил-2-фенилоксазол-4-ил)этокси]фениламино}метил)бензойной кислоты и
{4-[2-(5-метил-2-фенилоксазол-4-ил)этокси]фенил}фениламин.
Описанные выше соединения формулы I для применения в качестве терапевтически активных веществ являются следующим объектом данного изобретения. Предпочтительным является применение в качестве терапевтически активных веществ для профилактики и/или лечения диабетов, неинсулинзависимых сахарных диабетов, повышенного давления крови, повышенных уровней липидов и холестерина, атеросклеротических заболеваний или метаболического синдрома и особенно предпочтительно неинсулинзависимых сахарных диабетов.
Также объектом настоящего изобретения являются описанные выше соединения для получения лекарственных средств для профилактики и/или лечения заболеваний, которые модулируются агонистами PPARα и/или PPARγ, предпочтительно для изготовления лекарственных средств для профилактики и/или лечения диабетов, неинсулинзависимых сахарных диабетов, повышенного давления крови, повышенных уровней липидов и холестерина, атеросклеротических заболеваний или метаболического синдрома и особенно предпочтительно неинсулинзависимых сахарных диабетов.
Аналогично объектом изобретения являются фармацевтические композиции, включающие описанное выше соединение формулы I и терапевтически инертный носитель. Другим объектом настоящего изобретения является указанная выше фармацевтическая композиция, также включающая терапевтически эффективное количество ингибитора липазы, особенно, где ингибитором липазы является орлистат.
Объектом настоящего изобретения также является применение описанных выше соединений для изготовления лекарственных средств, особенно для лечения и/или профилактики заболеваний, которые модулируются агонистами PPARα и/или PPARγ, предпочтительно диабетов, неинсулинзависимых сахарных диабетов, повышенного давления крови, повышенных уровней липидов и холестерина, атеросклеротических заболеваний или метаболического синдрома и особенно предпочтительно неинсулинзависимых сахарных диабетов.
Другим объектом настоящего изобретения является применение соединения формулы I для получения лекарственного средства для лечения и/или профилактики заболеваний, которые модулируются агонистами PPARα и/или PPARγ у пациента, который также подвергается лечению ингибитором липазы. Предпочтительным является указанное выше применение, где ингибитором липазы является орлистат. Особенно предпочтительным является указанное выше применение для лечения и/или профилактики заболеваний, где заболевания выбраны из диабетов, неинсулинзависимых сахарных диабетов, повышенного давления крови, повышенных уровней липидов и холестерина, атеросклеротических заболеваний или метаболического синдрома и особенно предпочтительно неинсулинзависимых сахарных диабетов.
Следующий объект данного изобретения включает соединения, которые получены в соответствии с одним из описанных выше способов.
Следующим объектом изобретения является способ лечения и/или профилактики заболеваний, которые модулируются агонистами PPARα и/или PPARγ, предпочтительно диабетов, неинсулинзависимых сахарных диабетов, повышенного давления крови, повышенных уровней липидов и холестерина, атеросклеротических заболеваний или метаболического синдрома и особенно предпочтительно неинсулинзависимых сахарных диабетов, при котором вводят эффективное количество соединения формулы I. Другим объектом настоящего изобретения является указанный выше способ, который также включает введение человеку терапевтически эффективного количества ингибитора липазы, особенно где ингибитором липазы является орлистат. Указанный выше способ для одновременного, отдельного или последовательного введения также является объектом настоящего изобретения.
Биологические испытания
Следующие тесты могут использоваться для определения активности соединений формулы I.
Ранее приведенная информация об испытаниях может быть обнаружена в: Nichols JS и др. "Development of а scintillation proximity assay for peroxisome proliferator-activated receptor gamma ligand binding domain", (1998) Anal. Biochem. 257: 112-119.
Клоны кДНК полной длины PPARα человека и PPARγ мыши получали с помощью RT-PCR из кРНК жировой ткани человека и печени мыши, соответственно, клонированной в плазмидные векторы и измененные ДНК-секвенированием. Векторы экспрессии бактерий и млекопитающих конструировали для получения глутатион-s-трансферазы (GST) и белков Gal4 ДНК-связывающего домена, конденсированных с лиганд-связывающими доменами (LBD) PPARγ (aa 174-476) и PPARα (аа 167-469). Для этого часть клонированных последовательностей, кодирующих LBD, амплифицировали из клонов полной длины PCR и затем субклонировали в плазмидные векторы. Конечные клоны верифицировали анализом последовательности ДНК.
Индукцию, экспрессию и очистку GST-LBD слитых белков осуществляли в клетках штамма Е.coli BL21 (pLysS) стандартными методами (См. Current Protocols in Molecular Biology, Wiley Press, edited by Ausubel и др.).
Анализ на связывание радиолиганда
Связывание рецептора PPARα осуществляли в ТКЕ10 (10 мМ Tris-HCl, pH 8, 50 мМ KCl, 2 мМ EDTA, 0,1 мг/мл БСА без жирных кислот и 10 мМ DTT). В каждых 96 ячейках 2,4 мкг эквивалента GST-PPARα-LBD слитого белка и радиолиганда, например, 40000 распадов в минуту 2(S)-(2-бензоилфениламино)-3-{4-[1,1-дитритио-2-(5-метил-2-фенилоксазол-4-ил)этокси]фенил}пропановой кислоты, инкубировали в 100 мкл при комнатной температуре в течение 2 часов. Связавшийся лиганд удаляли из несвязавшегося лиганда твердофазным разделением с помощью плашек MultiScreen (Millipore), наполненных 80 мкл SG25 в соответствии с инструкциями производителя.
Связывание рецептора PPARγ осуществляли в ТКЕ50 (50 мМ Tris-HCl, pH 8, 50 мМ KCl, 2 мМ EDTA, 0,1 мг/мл БСА без жирных кислот и 10 мМ DTT). В каждых 96 реакционных ячейках 140 нг эквивалента GST-PPARγ-LBD слитого белка связывали с 10 мкг гранулами SPA (PharmaciaAmersham) в конечном объеме 50 мкл при встряхивании. Полученную суспензию инкубировали в течение 1 ч при комнатной температуре и центрифугировали в течение 2 мин при 1300 об/мин. Супернатант, содержащий несвязанный белок, удаляли и подсушенный остаток, содержащий покрытые рецептором гранула, растворяли в 50 мкл ТКЕ. Для связывания радиолиганда добавляли, например, 50 мкл 10000 распадов в минуту 2(S)-(2-бензоилфениламино)-3-{4-[1,1-дитрио-2-(5-метил-2-фенилоксазол-4-ил)этокси]фенил}пропановой кислоты, реакцию инкубировали при комнатной температуре в течение 1 ч и проводили стинцилляционные подсчеты. Все анализы на связывание осуществляли в 96-ячеечных подложках и количество связавшегося лиганда измеряли на Packard TopCount с помощью OptiPlates (Packard). Неспецифическое связывание определяли в присутствии 10-4 М немеченого соединения. Кривые зависимости от дозы строили трижды в области концентраций от 10-10 М до 10-4 М.
Испытания с транскрипционным репортерным геном люциферазы
Клетки почек молодых хомячков (ВНК21 АТСС CCL10) выращивали в DMEM среде, содержащей 10% FBS при 37°С в атмосфере 95%O2:5%CO2. Клекти высаживали в 6-ячеечные плашки плотностью 105 клеток/ячейка и затем трансфицировали порциями с помощью pFA-PPARγ-LBD или pFA-PPARα-LBD плазмид экспрессии плюс pFR-luc репортерная плазмида и плазмида экспрессии, кодирующая секретируемую форму щелочной фосфатазы (SEAP) в качестве нормализационного контроля. Трансфекцию осуществляли с помощью реагента Fugene 6 (Roche Molecular Biochemicals) в соответствии с предлагаемой методикой. Через шесть часов после трансфекции клетки собирали трипсинизацией и высаживали в 96-ячеечные плашки плотностью 104 клеток/ячейка. Через 24 часа связывания клеток среду удаляли и замещали 100 мкл среды без фенола красного, содержащей тестируемые вещества или контрольные лиганды (конечн. 0,1% ДМСО). После инкубирования клеток в течение 24 часов с веществами, отбирали 50 мкл супернатанта и анализировали на SEAP активность (Roche Molecular Biochemicals). Остаток супернатанта декантировали с осадка, добавляли 50 мкл PBS на ячейку и затем один объем реагента Luciferase Constant-Light (Roche Molecular Biochemicals) для лизиса клеток и инициирования реакции с люциферазой. Люминесценцию SEAP и люциферазы измеряли на Packard TopCount. Активность люциферазы нормализовали до SEAP контроля и транскрипционную активацию в присутствии тестируемого вещества проводили как активацию клеток, инкубированных без вещества. Значения ЕС50 рассчитывали с помощью программы XLfit (ID Business Solutions Ltd. UK).
Соединения настоящего изобретения имеют значения IC50 от 0,1 нМ до 50 мкМ, предпочтительно от 1 нМ до 10 мкМ, особенно 1-3500 нМ, более предпочтительно от 1 до 500 нМ, относительно PPARα и PPARγ. Соединения также имеют значения ЕС50 от 0,1 нМ до 50 мкМ, предпочтительно от 1 нМ до 10 мкМ, более предпочтительно от 1 до 3500 нМ, особенно от 1 до 500 нМ, относительно PPARα и PPARγ.
Соединения формулы I и их фармацевтически приемлемые соли и эфиры могут использоваться в качестве лекарственных средств, например, в форме фармацевтических составов для энтерального, парентерального или местного введения. Они могут вводиться, например, перорально, например, в форме таблеток, покрытых таблеток, драже, твердых и мягких желатиновых капсул, растворов, эмульсий или суспензий, ректально, например, в форме свечей, парентерально, например, в форме инъекционных растворов или вливаемых растворов, или местно, например, в форме мазей, кремов или масла.
Получение фармацевтических составов может осуществляться способом, известным специалисту в данной области техники, введением описанных соединений формулы I и их фармацевтически приемлемых солей в галеновую форму для введения, вместе с подходящим, нетоксичным, инертным, терапевтически совместимым твердым или жидким носителем и, если желательно, обычными фармацевтическими адъювантами.
Подходящие носители являются не только неорганическими носителями, но также органическими носителями. Таким образом, в качестве носителя для таблеток, покрытых таблеток, драже и твердых желатиновых капсул могут использоваться, например, лактоза, кукурузный крахмал или его производные, тальк, стеариновая кислота или ее соли. Подходящими носителями для мягких желатиновых капсул являются, например, растительные масла, воски, жиры и полутвердые и жидкие полиолы (в зависимости от природы активных компонентов, однако, никакого носителя не требуется в случае мягких желатиновых капсул). Подходящими носителями для изготовления растворов и сиропов являются, например, вода, полиолы, сахароза, инвертированный сахар и им подобные. Подходящими носителями для инъекционных растворов являются, например, вода, спирты, полиолы, глицерин и растительные масла. Подходящими носителями для свечей являются, например, природные или утяжеленные масла, воски, жиры и полужидкие или жидкие полиолы. Подходящими носителями для местных составов являются глицериды, полусинтетические и синтетические глицериды, гидрированные масла, жидкие воски, жидкие керосины, жидкие жирные спирты, стеролы, полиэтиленгликоли и производные целлюлозы.
В качестве фармацевтических адъювантов входят также обычные стабилизаторы, консерванты, увлажняющие и эмульсифицирующие агенты, агенты для улучшения консистенции, улучшающие аромат агенты, соли для изменения осмотического давления, буферные вещества, солюбилизаторы, красители и маскирующие агенты и антиоксиданты.
Дозировка соединений формулы I может изменяться в широких пределах в зависимости от излечиваемого заболевания, возраста и индивидуального состояния пациента и способа введения, и будет, конечно, зависеть от индивидуальных требований в каждом конкретном случае. Для взрослых пациентов ежедневная дозировка составляет приблизительно от 1 мг приблизительно до 1000 мг, особенно приблизительно от 1 мг приблизительно до 100 мг. В зависимости от дозировки ежедневную дозировку удобно вводить в нескольких единичных дозировках.
Фармацевтические составы обычно содержат приблизительно 0,5-500 мг, предпочтительно 0,5-100 мг соединения формулы I.
Следующие примеры служат для более подробного иллюстрирования настоящего изобретения. Они, однако, никаким образом не предназначены для ограничения его возможностей.
Примеры
Получение исходных реагентов примеров 1-17
а) трет-Бутиловый эфир {4-[2-(5-метил-2-фенилоксазол-4-ил)этокси] фенил} карбоновой кислоты
К раствору 5,00 г трет-бутилового эфира (4-гидроксифенил)карбоновой кислоты, 7,28 г 2-(5-метил-2-фенил-1,3-оксазол-4-ил)этан-1-ола и 9,40 г трифенилфосфина в 100 мл ТГФ добавляли при 0°С раствор 7,25 г диизопропилазодикарбоксилата в 50 мл ТГФ в течение 30 мин и перемешивание продолжали при 22°С в течение 16 ч. Добавляли другую порцию 1,88 г трифенилфосфина и 1,45 г диизопропилазодикарбоксилата в 10 мл ТГФ при 0°С и перемешивание продолжали при 22°С в течение 2 ч, после чего конверсия завершалась. Смесь упаривали и остаток очищали хроматографией (SiO2, н-гексан/AcOEt 4:1) с получением 7,5 г указанного в заголовке соединения в виде бесцветного твердого вещества. MS: (M+H)+ 395,4.
б) Этиловый эфир 2-[(трет-бутоксикарбонил-{4-[2-(5-метил-2-фенилоксазол-4-ил)этокси]фенил}амино)метил]бензойной кислоты
К суспензии 3,41 г порошка КОН в 110 мл ДМСО добавляли при 22°С 6,00 г трет-бутилового эфира {4-[2-(5-метил-2-фенилоксазол-4-ил)этокси] фенил }карбоновой кислоты и суспензию перемешивали в течение 25 мин. Медленно добавляли раствор 7,40 г этилового эфира 2-бромметилбензойной кислоты в 10 мл ДМСО, хранящийся при температуре 15-20°С, и перемешивание продолжали при 22°С в течение 2,5 ч. Темную смесь разделяли между 500 мл насыщенного водного NH4Cl и 200 мл AcOEt, органический слой промывали насыщенным водным NH4Cl и водой, высушивали и упаривали. Остаток очищали хроматографией (SiO2, н-гексан/AcOEt 4:1) с получением 7,96 г указанного в заголовке соединения в виде светло-желтого масла. MS: (M+H)+ 557,3.
в) Этиловый эфир 2-({4-[2-(5-метил-2-фенилоксазол-4-ил)этокси] фениламино} метил)бензойной кислоты
К раствору 7,00 г этилового эфира 2-[(трет-бутоксикарбонил-{4-[2-(5-метил-2-фенилоксазол-4-ил)этокси]фенил}амино)метил]бензойной кислоты в 40 мл дихлорметана добавляли 9,6 мл трифторуксусной кислоты и перемешивание продолжали при 22°С в течение 2,5 ч. Смесь разделяли между водным насыщенным Na2CO3 и дихлорметаном, органический слой высушивали и упаривали. Остаток очищали хроматографией (SiO2, н-гексан/AcOEt 4:1) с получением 3,14 г указанного в заголовке соединения в виде светло-желтого масла. MS: (M+H)+ 457,5.
Общее описание получения примеров 1-7
К раствору 0,2 ммоль этилового эфира 2-({4-[2-(5-метил-2-фенилоксазол-4-ил)этокси]фениламино}метил)бензойной кислоты, полученной как описано выше, и 0,4 ммоль триэтиламина в 2 мл ТГФ добавляли 0,22 ммоль соответствующего ацилхлорида или сульфохлорида или 0,22-2 ммоль алкилйодида или бензилбромида с последующим добавлением каталитического количества диметиламинопиридина в случае медленного превращения. Реакционные смеси перемешивали при 22-55°С до полного превращения. Суспензии отфильтровывали, фильтрат упаривали и остаток очищали препаративной ВЭЖХ хроматографией (RP-18, СН3CN/H2O градиент) с получением продукта в виде эфира.
Эфиры (0,1 ммоль) гидролизовали 0,3 ммоль LiOH·H2O в 1 мл ТГФ, 0,5 мл МеОН и 0,5 мл воды с последующей очисткой препаративной ВЭЖХ хроматографией (RP-18, СН3CN/H2O градиент).
Пример 1
Этиловый эфир 2-[(ацетил-{4-[2-(5-метил-2-фенилоксазол-4-ил)этокси]фенил}амино)метил]бензойной кислоты получали с 66% выходом в виде светло-желтого вязкого вещества. MS: (M+H)+ 499,3. После гидролиза получали 2-[(ацетил-{4-[2-(5-метил-2-фенилоксазол-4-ил)этокси]фенил}амино)метил]бензойную кислоту с 61% выходом в виде белого твердого вещества. MS: (M-H) 469,2.
Пример 2
Этиловый эфир 2-[(бензоил-{4-[2-(5-метил-2-фенилоксазол-4-ил)этокси]фенил}амино)метил]бензойной кислоты получали с 65% выходом в виде бесцветного вязкого вещества. MS: (M+H)+ 561,4. После гидролиза получали 2-[(бензоил-{4-[2-(5-метил-2-фенилоксазол-4-ил)этокси]фенил}амино)метил]бензойную кислоту с 71% выходом в виде белого твердого вещества. MS: (M-H) 531,1.
Пример 3
Этиловый эфир 2-[(метансульфонил-{4-[2-(5-метил-2-фенилоксазол-4-ил)этокси]фенил}амино)метил]бензойной кислоты получали с 57% выходом в виде желтого вязкого вещества. MS: (M+H)+ 535,3. После гидролиза получали 2-[(метансульфонил-{4-[2-(5-метил-2-фенилоксазол-4-ил)этокси]фенил}амино)метил]бензойную кислоту с 75% выходом в виде белого твердого вещества. MS: (M-H) 505,2.
Пример 4
Этиловый эфир 2-[(бензолсульфонил-{4-[2-(5-метил-2-фенилоксазол-4-ил)этокси]фенил}амино)метил]бензойной кислоты получали с 89% выходом в виде светло-желтого вязкого вещества. MS: (M+H)+ 597,1. После гидролиза получали 2-[(бензолсульфонил- {4-[2-(5-метил-2-фенилоксазол-4-ил)этокси]фенил}амино)метил]бензойную кислоту с 66% выходом в виде белого твердого вещества. MS: (M-H) 567,1.
Пример 5
Этиловый эфир 2-[(метил-{4-[2-(5-метил-2-фенилоксазол-4-ил)этокси]фенил}амино)метил]бензойной кислоты получали с 29% выходом в виде светло-коричневого вязкого вещества. MS: (M+H)+ 471,1. После гидролиза получали 2-[(метил-{4-[2-(5-метил-2-фенилоксазол-4-ил)этокси]фенил}амино)метил]бензойную кислоту с 64% выходом в виде светло-желтого вязкого вещества. MS: (M+H)+ 443,4.
Пример 6
Этиловый эфир 2-[({4-[2-(5-метил-2-фенилоксазол-4-ил)этокси]фенил}пропиламино)метил]бензойной кислоты получали с 59% выходом в виде желтого вязкого вещества. MS: (M+H)+ 499,3. После гидролиза получали 2-[({4-[2-(5-метил-2-фенилоксазол-4-ил)этокси]фенил}пропиламино)метил]бензойную кислоту с 83% выходом в виде светло-желтого вязкого вещества. MS: (M+H)+ 471,3.
Пример 7
Этиловый эфир 2-[(бензил-{4-[2-(5-метил-2-фенилоксазол-4-ил)этокси]фенил}амино)метил]бензойной кислоты получали с 55% выходом в виде светло-желтого вязкого вещества. MS: (M+H)+ 547,2. После гидролиза получали 2-[(бензил-{4-[2-(5-метил-2-фенилоксазол-4-ил)этокси]фенил}амино)метил]бензойную кислоту с 59% выходом в виде светло-желтого вязкого вещества. MS: (M+H)+ 519,3.
Общее описание получения примеров 8-17
В соответствии со способом, описанным P.Y.S. Lam и др., Tetrahedron Letters 42, 3415, 2001, суспензию 0,44 ммоль соответствующей борной кислоты и 0,25 г молекулярных сит в 3 мл дихлорметана обрабатывали последовательно 0,22 ммоль этилового эфира 2-({4-[2-(5-метил-2-фенилоксазол-4-ил)этокси]фениламино}метил)бензойной кислоты, полученной как описано выше, 0,22 ммоль ацетата меди(II), 0,24 ммоль 2,2,6,6-тетраметилпиперидин-1-оксила (TEMPO) и 0,44 ммоль NEt3 и смесь перемешивали при 22°С в открытой колбе (доступ кислорода) в течение 16 ч. Смесь отфильтровывали через небольшой слой силикагеля и фильтрат очищали препаративной ВЭЖХ хроматографией (RP-18, СН3CN/H2O градиент) с получением продукта в виде эфира. Эфир гидролизовали как описано выше.
Пример 8
Этиловый эфир 2-[({4-[2-(5-метил-2-фенилоксазол-4-ил)этокси]фенил}-о-толиламино)метил]бензойной кислоты получали с 22% выходом в виде коричневого вязкого вещества. MS: (M+H)+ 547,3. После гидролиза получали 2-[({4-[2-(5-метил-2-фенилоксазол-4-ил)этокси]фенил}-о-толиламино)метил]бензойную кислоту с 78% выходом в виде светло-желтого вязкого вещества. MS: (M+H)+ 519,3.
Пример 9
Этиловый эфир 2-[((3-фторфенил)-{4-[2-(5-метил-2-фенилоксазол-4-ил)этокси]фенил}амино)метил]бензойной кислоты получали с 22% выходом в виде светло-желтого вязкого вещества. MS: (M+H)+ 551,1. После гидролиза получали 2-[((3-фторфенил)-{4-[2-(5-метил-2-фенилоксазол-4-ил)этокси]фенил}амино)метил]бензойную кислоту с 29% выходом в виде светло-желтого вязкого вещества. MS: (M+H)+ 523,2.
Пример 10
Этиловый эфир 2-[({4-[2-(5-метил-2-фенилоксазол-4-ил)этокси]фенил}-м-толиламино)метил]бензойной кислоты получали с 33% выходом в виде светло-желтого вязкого вещества. MS: (M+H)+ 547,2. После гидролиза получали 2-[({4-[2-(5-метил-2-фенилоксазол-4-ил)этокси]фенил}-м-толиламино)метил]бензойную кислоту с 43% выходом в виде светло-желтого вязкого вещества. MS: (M+H)+ 519,3.
Пример 11
Этиловый эфир 2-{[{4-[2-(5-метил-2-фенилоксазол-4-ил)этокси]фенил}-(3-трифторметилфенил)амино]метил}бензойной кислоты получали с 19% выходом в виде светло-желтого вязкого вещества. MS: (M+H)+ 601,1. После гидролиза получали 2-{[{4-[2-(5-метил-2-фенилоксазол-4-ил)этокси]фенил}-(3-трифторметилфенил)амино]метил}бензойную кислоту с 31% выходом в виде светло-желтого вязкого вещества. MS: (M+H)+ 573,1.
Пример 12
Этиловый эфир 2-[((4-фторфенил)-{4-[2-(5-метил-2-фенилоксазол-4-ил)этокси]фенил}амино)метил]бензойной кислоты получали с 17% выходом в виде светло-желтого вязкого вещества. MS: (M+H)+ 551,3. После гидролиза получали 2-[((4-фторфенил)-{4-[2-(5-метил-2-фенилоксазол-4-ил)этокси]фенил}амино)метил]бензойную кислоту с 70% выходом в виде светло-желтого вязкого вещества. MS: (M+H)+ 523,2.
Пример 13
Этиловый эфир 2-[({4-[2-(5-метил-2-фенилоксазол-4-ил)этокси]фенил}-п-толиламино)метил]бензойной кислоты получали с 31% выходом в виде светло-желтого вязкого вещества. MS: (M+H)+ 547,2. После гидролиза получали 2-[({4-[2-(5-метил-2-фенилоксазол-4-ил)этокси]фенил}-п-толиламино)метил]бензойную кислоту с 81% выходом в виде светло-желтого вязкого вещества. MS: (M+H)+ 519,2.
Пример 14
Этиловый эфир 2-[((4-метоксифенил)-{4-[2-(5-метил-2-фенилоксазол-4-ил)этокси]фенил}амино)метил]бензойной кислоты получали с 37% выходом в виде светло-желтого вязкого вещества. MS: (M+H)+ 563,4. После гидролиза получали 2-[((4-метоксифенил)-{4-[2-(5-метил-2-фенилоксазол-4-ил)этокси]фенил}амино)метил]бензойную кислоту с 43% выходом в виде светло-желтого вязкого вещества. MS: (M+H)+ 535,3.
Пример 15
Этиловый эфир 2-[((3,4-диметилфенил)-{4-[2-(5-метил-2-фенилоксазол-4-ил)этокси]фенил}амино)метил]бензойной кислоты получали с 31% выходом в виде светло-желтого вязкого вещества. MS: (M+H)+ 561,4. После гидролиза получали 2-[((3,4-диметилфенил)-{4-[2-(5-метил-2-фенилоксазол-4-ил)этокси]фенил}амино)метил]бензойную кислоту с 12% выходом в виде светло-желтого вязкого вещества. MS: (M+H)+ 533,3.
Пример 16
Этиловый эфир 2-[((3,4-дифторфенил)-{4-[2-(5-метил-2-фенилоксазол-4-ил)этокси]фенил}амино)метил]бензойной кислоты получали с 9% выходом в виде светло-желтого вязкого вещества. MS: (M+H)+ 569,2. После гидролиза получали 2-[((3,4-дифторфенил)-{4-[2-(5-метил-2-фенилоксазол-4-ил)этокси]фенил}амино)метил]бензойную кислоту с 57% выходом в виде светло-желтого вязкого вещества. MS: (M+H)+ 541,2.
Пример 17
Этиловый эфир 2-[((4-фтор-3-метилфенил)-{4-[2-(5-метил-2-фенилоксазол-4-ил)этокси]фенил}амино)метил]бензойной кислоты получали с 10% выходом в виде желтого вязкого вещества. MS: (M+H)+ 565,4. После гидролиза получали 2-[((4-фтор-3-метилфенил)-{4-[2-(5-метил-2-фенилоксазол-4-ил)этокси]фенил}амино)метил]бензойную кислоту с 39% выходом в виде белого твердого вещества. MS: (M+H)+ 537,5.
Получение исходного реагента примеров 18-26
{4-[2-(5-Метил-2-фенилоксазол-4-ил)этокси]фенил}фениламин
К раствору 0,50 г 4-гидроксифениланилина, 0,83 г 2-(5-метил-2-фенил-1,3-оксазол-4-ил)этан-1-ола и 1,06 г трифенилфосфина в 20 мл ТГФ добавляли при 0°С раствор 0,82 г диизопропилазодикарбоксилата в 10 мл ТГФ в течение 30 мин и перемешивание продолжали при 22°С в течение 5 ч. Смесь упаривали и остаток очищали хроматографией (SiO2, н-гексан/AcOEt 6:1) с получением 0,62 г указанного в заголовке соединения в виде бесцветного твердого вещества. MS: (М+Н)+ 371,4.
Общее описание получения примеров 18-26
К суспензии 0,76 ммоль порошка КОН в 2,5 мл ДМСО добавляли при 22°С 0,19 ммоль амина и суспензию перемешивали в течение 5 мин. Медленно добавляли раствор 0,38 ммоль соответствующего бензилбромида в 0,5 мл ДМСО при температуре 15-20°С и перемешивание продолжали при 22°С до окончания превращения. рН реакционной смеси доводили до 2-3 муравьиной кислотой и продукт очищали препаративной ВЭЖХ хроматографией (RP-18, СН3CN/Н2О градиент) с получением продукта в виде эфира.
Эфиры гидролизовали, как описано в примерах 1-7.
Пример 18
Из амина и метилового эфира 2-бромметил-3-фторбензойной кислоты получали метиловый эфир 3-фтор-2-[({4-[2-(5-метил-2-фенилоксазол-4-ил)этокси]фенил}фениламино)метил]бензойной кислоты с 55% выходом в виде светло-желтого масла. MS: (M+H)+ 537,3. После гидролиза получали 3-фтор-2-[({4-[2-(5-метил-2-фенилоксазол-4-ил)этокси]фенил}фениламино)метил]бензойную кислоту с 90% выходом в виде светло-коричневого твердого вещества. MS: (M+H)+ 523,1.
Пример 19
Из амина и метилового эфира 2-бромметил-3-хлорбензойной кислоты получали метиловый эфир 3-хлор-2-[({4-[2-(5-метил-2-фенилоксазол-4-ил)этокси] фенил }фениламино)метил]бензойной кислоты с 43% выходом в виде светло-желтого масла. MS: (M+H)+ 553,1 и 555,3 (Cl изотопы). После гидролиза получали 3-хлор-2-[({4-[2-(5-метил-2-фенилоксазол-4-ил)этокси] фенил }фениламино)метил]бензойную кислоту с 74% выходом в виде бесцветного твердого вещества. MS: (М-Н) 537,1 и 539,3 (Cl изотопы).
Пример 20
Из амина и метилового эфира 2-бромметил-3-трифторметилбензойной кислоты получали метиловый эфир 2-[({4-[2-(5-метил-2-фенилоксазол-4-ил)этокси]фенил}фениламино)метил]-3-трифторметилбензойной кислоты с 59% выходом в виде бесцветного масла. MS: (M+H)+ 587,2. После гидролиза получали 2-[({4-[2-(5-метил-2-фенилоксазол-4-ил)этокси]фенил}фениламино)метил]-3-трифторметилбензойную кислоту с 87% выходом в виде бесцветного твердого вещества. MS: (М-Н) 571,0.
Пример 21
Из амина и метилового эфира 2-бромметил-3-метоксибензойной кислоты получали метиловый эфир 3-метокси-2-[({4-[2-(5-метил-2-фенилоксазол-4-ил)этокси]фенил}фениламино)метил]бензойной кислоты с 36% выходом в виде бесцветного масла. MS: (M+H)+ 549,2. После гидролиза получали 3-метокси-2-[({4-[2-(5-метил-2-фенилоксазол-4-ил)этокси]фенил}фениламино)метил]бензойную кислоту с 77% выходом в виде светло-желтого твердого вещества. MS: (М-Н) 533,2.
Пример 22
Из амина и метилового эфира 2-бромметил-4-хлорбензойной кислоты получали метиловый эфир 4-хлор-2-[({4-[2-(5-метил-2-фенилоксазол-4-ил)этокси]фенил}фениламино)метил]бензойной кислоты с 15% выходом в виде светло-желтого масла. MS: (M+H)+ 553,2 и 555,1 (Cl изотопы). После гидролиза получали 4-хлор-2-[({4-[2-(5-метил-2-фенилоксазол-4-ил)этокси]фенил}фениламино)метил]бензойную кислоту с 72% выходом в виде желтого твердого вещества. MS: (M-H) 537,1 и 539,2 (Cl изотопы).
Пример 23
Из амина и метилового эфира 2-бромметил-4-метоксибензойной кислоты получали метиловый эфир 4-метокси-2-[({4-[2-(5-метил-2-фенилоксазол-4-ил)этокси]фенил}фениламино)метил]бензойной кислоты с 14% выходом в виде бесцветного масла. MS: (M+H)+ 549,2. После гидролиза получали 4-метокси-2-[({4-[2-(5-метил-2-фенилоксазол-4-ил)этокси]фенил}фениламино)метил]бензойную кислоту с 93% выходом в виде светло-коричневого твердого вещества. MS: (M-H) 533,2.
Пример 24
Из амина и метилового эфира 2-бромметил-5-фторбензойной кислоты получали метиловый эфир 5-фтор-2-[({4-[2-(5-метил-2-фенилоксазол-4-ил)этокси]фенил}фениламино)метил]бензойной кислоты с 44% выходом в виде бесцветного масла. MS: (M+H)+ 537,3. После гидролиза получали 5-фтор-2-[({4-[2-(5-метил-2-фенилоксазол-4-ил)этокси]фенил}фениламино)метил]бензойную кислоту с 96% выходом в виде светло-желтой пены. MS: (M-H) 521,1.
Пример 25
Из амина и этилового эфира 2-бромметилбензойной кислоты получали этиловый эфир 2-[({4-[2-(5-метил-2-фенилоксазол-4-ил)этокси]фенил}фениламино)метил]бензойной кислоты с 48% выходом в виде светло-желтого масла. (M+H)+ 533,4. После гидролиза получали 2-[({4-[2-(5-метил-2-фенилоксазол-4-ил)этокси]фенил}фениламино)метил]бензойную кислоту с 29% выходом в виде бесцветного твердого вещества. (M-H) 503,2.
Пример 26
Из амина и этилового эфира 2-бромметил-6-метоксибензойной кислоты получали этиловый эфир 2-метокси-6-[({4-[2-(5-метил-2-фенилоксазол-4-ил)этокси]фенил}фениламино)метил]бензойной кислоты с 67% выходом в виде бесцветного масла. (M+H)+ 563,3. После гидролиза получали 2-метокси-6-[({4-[2-(5-метил-2-фенилоксазол-4-ил)этокси]фенил}фениламино)метил]бензойную кислоту с 29% выходом в виде бесцветного масла. (M+H)+ 535,3.
Пример А
Таблетки, включающие следующие ингредиенты, могут быть получены обычным способом:
Пример Б
Капсулы, включающие следующие ингредиенты, могут быть получены обычным способом:
Пример В
Инъекционные растворы могут иметь следующий состав:
| название | год | авторы | номер документа |
|---|---|---|---|
| N-ЗАМЕЩЕННЫЕ 1Н-ИНДОЛ-5-ПРОПИОНОВЫЕ КИСЛОТЫ, ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ, СОДЕРЖАЩАЯ ЭТИ СОЕДИНЕНИЯ, И ИХ ПРИМЕНЕНИЕ (ВАРИАНТЫ) | 2003 |
|
RU2296759C2 |
| ХИРАЛЬНЫЕ ПРОИЗВОДНЫЕ ОКСАЗОЛАРИЛПРОПИОНОВОЙ КИСЛОТЫ И ИХ ПРИМЕНЕНИЕ В КАЧЕСТВЕ АГОНИСТОВ РЕЦЕПТОРОВ, АКТИВИРОВАННЫХ ПРОЛИФЕРАТОРОМ ПАРОКСИСОМЫ (PPAR АГОНИСТОВ) | 2003 |
|
RU2303593C2 |
| ИНДОЛИЛПРОИЗВОДНЫЕ, СПОСОБ ИХ ПОЛУЧЕНИЯ, ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ, СПОСОБ ЛЕЧЕНИЯ И/ИЛИ ПРОФИЛАКТИКИ ЗАБОЛЕВАНИЙ | 2003 |
|
RU2315767C2 |
| ПРОИЗВОДНЫЕ ПИРАЗОЛО[1,5-a]ПИРИМИДИНА И ТИЕНО[3,2-b]ПИРИМИДИНА В КАЧЕСТВЕ МОДУЛЯТОРОВ IRAK-4 | 2011 |
|
RU2604062C2 |
| ЗАМЕЩЕННЫЕ БЕНЗАМИДНЫЕ ПРОИЗВОДНЫЕ | 2010 |
|
RU2595902C2 |
| ГЕТЕРОЦИКЛИЧЕСКИЕ СОЕДИНЕНИЯ И СПОСОБЫ ПРИМЕНЕНИЯ | 2002 |
|
RU2345983C2 |
| ДИАРИЛЦИКЛОАЛКИЛЬНЫЕ ПРОИЗВОДНЫЕ, СПОСОБ ИХ ПОЛУЧЕНИЯ И ИХ ПРИМЕНЕНИЕ В КАЧЕСТВЕ PPAR-АКТИВАТОРОВ | 2002 |
|
RU2330846C2 |
| ЗАМЕЩЕННЫЕ ПРОИЗВОДНЫЕ ФЕНИЛМЕТАНОНА | 2007 |
|
RU2437872C2 |
| ПРОИЗВОДНЫЕ ПИРАЗОЛПИРИМИДИНА | 2005 |
|
RU2378277C2 |
| НОВЫЕ СОЕДИНЕНИЯ 2-АРИЛТИАЗОЛА В КАЧЕСТВЕ АГОНИСТОВ PPAR α И PPAR γ | 2003 |
|
RU2296754C2 |
Настоящее изобретение относится к новым производным оксазола, полезным в качестве сенсибилизаторов инсулина, особенно в качестве активаторов PPAR. Изобретение относится к соединениям формулы I
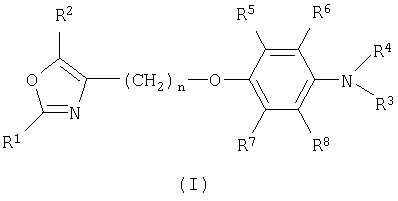
и его фармацевтически приемлемым солям и эфирам, где R1 представляет собой фенил; R2 представляет С1-С8алкил; R3 представляет собой С1-С8алкил, аралкил, фенил, С1-С8алкилкарбонил, фенилкарбонил, C1-C8алкил-S(O)2- или фенил-S(O)2-; R4 представляет собой аралкил; R5, R6, R7 и R8 представляют собой водород и n имеет значение 1, 2, 3, 4 или 5. Целью настоящего изобретения является получение лекарственных средств для профилактики и/или лечения заболеваний, которые модулируются агонистами PPARα и/или PPARγ. 3 н. и 13 з.п. ф-лы.
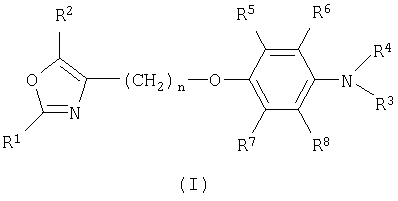
где R1 представляет собой фенил;
R2 представляет собой С1-С8алкил;
R3 представляет собой С1-С8алкил, аралкил, фенил, С1-С8алкилкарбонил, фенилкарбонил, С1-С8алкил-S(O)2- или фенил-S(O)2-, причем аралкил и фенил необязательно имеют один или несколько заместителей, каждый независимо выбранный из галогена, трифторметила, С1-С8алкила и алкоксигруппы;
R4 представляет собой аралкил, который необязательно имеет один или несколько заместителей, каждый независимо выбранный из карбоксигруппы, галогена, трифторметила, С1-С8алкила и алкоксигруппы;
R5, R6, R7 и R8 представляют собой водород;
n имеет значение 1, 2, 3, 4 или 5,
и их фармацевтически приемлемые соли и эфиры.
2-[({4-[2-(5-метил-2-фенилоксазол-4-ил)этокси]фенил}фениламино)метил]бензойная кислота;
2-[({4-[2-(5-метил-2-фенилоксазол-4-ил)этокси]фенил}пропиламино)метил]бензойная кислота;
2-[((3-фторфенил)-{4-[2-(5-метил-2-фенилоксазол-4-ил)этокси]фенил}амино)метил]бензойная кислота;
2-[((3,4-дифторфенил)-{4-[2-(5-метил-2-фенилоксазол-4-ил)этокси]фенил}амино)метил]бензойная кислота;
3-фтор-2-[({4-[2-(5-метил-2-фенилоксазол-4-ил)этокси]фенил}фениламино)метил]бензойная кислота и
5-фтор-2-[({4-[2-(5-метил-2-фенилоксазол-4-ил)этокси]фенил}фениламино)метил]бензойная кислота.
| WO 200100603 A1, 04.01.2001 | |||
| ПРОИЗВОДНЫЕ ОКСАЗОЛА ИЛИ ИХ ФАРМАЦЕВТИЧЕСКИ ПРИЕМЛЕМЫЕ КАТИОННЫЕ СОЛИ ИЛИ КИСЛОТНО-АДДИТИВНЫЕ СОЛИ | 1992 |
|
RU2079497C1 |

