Область изобретения
Настоящее изобретение относится к улучшенным хелатным конъюгатам с биологическими молекулами для направленной доставки, пригодными для образования комплексов с радиоактивными металлами. Комплексы с радиоактивными металлами пригодны в качестве радиоактивных фармацевтических препаратов, в особенности с 99mТс.
Предшествующий уровень техники
Диаминдиоксимы представляют собой известный класс хелатообразователей, которые, как было показано, образуют
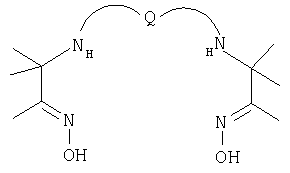
Q=-(СН2)3 -, т.е. пропиленаминоксим или PnAO;
Q=-(СН2)4 -, т.е. бутиленаминоксим или BnAO;
Q=-(СН2)5 -, т.е. пентиленаминоксим или PentAO;
комплексы с радиоактивным металлом 99mТс.
Лиганд PentAO был впервые описан S.Jurisson et al [Inorg. Chem., 26, 3576-82 (1987)], который показал, что его комплекс с долгоживущим радиоактивным металлом 99Tc является нейтральным, имеющим ядро диоксо Tc(V) (т.е. TcO2 +). J-M Lo et al [Appl. Rad. Inst, 44, 1139-46 (1993)] описал синтез PentAO и образование его комплекса с 99mTc.
US 5688487 описывает хелатные конъюгаты диаминдиоксимов, имеющие С2-5 алкиленовый мостик с нитроимидазольными биологическими молекулами для направленной доставки для визуализации гипоксии. Описана конъюгация нитроимидазола в положении С1 (метил оксима).
WO 95/04552 описывает конъюгаты нитроимидазола с BnAO и PentAO. Пример демонстрирует конъюгацию в положении С1 (метил оксима).
WO 95/19187 описывает конъюгаты линейных или циклических 3-50-членных синтетических пептидов с полидентатными хелатообразователями, присоединяемыми на карбоксильном конце пептида, для применения в качестве радиоактивных фармацевтических препаратов. Диаминдиоксимы, такие как PnAO, BnAO и PentAO, описаны в качестве пригодных хелатообразователей.
WO 99/60018 описывает диаминдиоксимовые хелатные конъюгаты диаминдиоксимовых лигандов с пептидами для визуализации тромба. Как утверждалось, таким предпочтительным хелатообразователем является диаминдиоксим с Q=-(CH2)2NR(CH2)2-.
Настоящее изобретение
Диаминдиоксим-пептидные хелатные конъюгаты по WO 99/60018 формулы I
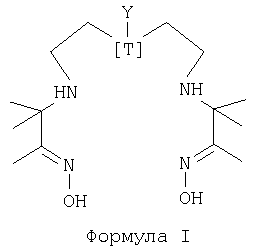
где Т=N и Y=-СН2СН2NH-[пептид],
тем не менее обладают существенными недостатками. Таким образом, при хелатировании с 99mТс этот аза-диаминдиоксим образует несколько частиц с технецием, которые могут быть разделены и обнаружены с помощью хроматографии. При температуре окружающей среды исходные радиоактивные частицы (промежуточные соединения) превращаются в течение времени (2-3 часа) в устойчивый продукт. Этому превращению промежуточное соединение - продукт может способствовать применение более высоких значений рН (рН>8) и нагревание. Эти условия не являются идеальными для госпитальной радиофармации, поэтому желателен хелатообразователь с меньшим количеством промежуточных соединений и/или более быстрым превращением промежуточное соединение - продукт. Очевидно, что необходимость в нагревании и возможно сравнительно высоком рН для достижения достаточной радиохимической чистоты (РХЧ) желаемых частиц 99mTc является нежелательной, поскольку такое нагревание может разрушить присоединенную биологическую молекулу для направленной доставки или пептид. Еще одна проблема с азадиаминдиоксимовыми хелатообразователями формулы I заключается в том, что атом азота третичного амина в основании мостика является сравнительно основным. Это означает, что при образовании соответствующего комплекса 99mТс в водном растворе третичный амин по меньшей мере частично протонируется, что приводит в результате к тому, что конъюгат является заряженным. Этот заряд может ограничивать применения меченой биологической группировки для направленной доставки, поскольку заряд может усложнять радиоактивно меченному конъюгату переход через клеточные мембраны.
Настоящее изобретение предлагает альтернативную хелатную систему (формула I, где Т=С), которая позволяет преодолеть эти проблемы предшествующего уровня техники, и предлагает конъюгаты, которые могут быть радиоактивно мечены с получением хорошей РХЧ при комнатной температуре в водной среде при почти нейтральном значении рН. Комплексы с радиоактивным металлом имеют хорошую стабильность. N2S2 и N3S тиолсодержащие бифункциональные хелатообразователи по предшествующему уровню техники имеют недостатки в том, что тиолы чувствительны к воздуху, быстро окисляются на воздухе до соответствующих дисульфидов при условиях от нейтральных до основных. Поэтому их следует держать в инертной атмосфере перед применением или в защитной матрице. В качестве альтернативы они могут применяться в виде защищенных частиц, таких как тиоацетат или тетрагидропиранила гемитиокеталь, но это требует обязательного удаления защитных групп перед применением с использованием кислоты или основания и нагревания. Все эти свойства уменьшают удобство этих хелатообразователей по сравнению с хелатообразователями по настоящему изобретению. Следовательно, хелатообразователи по настоящему изобретению полезны для конъюгации и радиоактивного мечения широкого диапазона биологических группировок для направленной доставки.
Подробное описание изобретения
В первом аспекте настоящего изобретения предложен хелатный конъюгат диаминдиоксимового лиганда с биологической группировкой для направленной доставки. Термин "хелатный конъюгат" означает соединение, где агент, образующий с металлом хелат, ковалентно связан ("конъюгирован") с биологической группировкой для направленной доставки. Хелатный конъюгат имеет формулу II
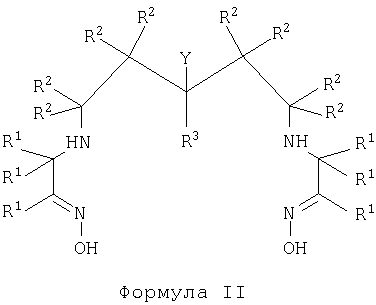
где каждый R1, R2 и R3 независимо представляет собой группу R;
Y представляет собой -(A)n-X-Z,
где Х представляет собой -NH-, -CO2-, -N(С=O)-;
Z представляет собой биологическую группировку для направленной доставки, которая выбрана из 3-20-членного пептида, субстрата фермента, ингибитора фермента или синтетического связывающегося с рецептором соединения;
-(A)n- представляет собой линкер, где каждый А независимо представляет собой
-CR2-, -NRCO-, -CONR-, -CR2OCR2- или группировку полиалкиленгликоля;
n представляет собой целое число, имеющее значение от 2 до 10;
каждая группа R независимо представляет собой Н или C1-10алкил, или 2 группы R вместе с атомами, к которым они присоединены, образуют С3-6карбоциклическое насыщенное кольцо.
Под термином "биологическая группировка для направленной доставки" понимают 3-100-членные пептиды или пептидные аналоги, которые могут представлять собой линейные пептиды или циклические пептиды или их комбинации; моноклональные антитела или их фрагменты; субстраты или ингибиторы ферментов; синтетические связывающиеся с рецептором соединения; олигонуклеотиды или фрагменты олиго-ДНК или олиго-РНК. Биологическая группировка для направленной доставки может иметь синтетическое или природное происхождение, но предпочтительно синтетическое. Предпочтительные биологические группировки для направленной доставки представляют собой 3-20-членные пептиды, которые могут иметь синтетическое или природное происхождение, но предпочтительно синтетическое. Под термином "циклический пептид" понимают последовательность, состоящую из 5-15 аминокислот, где две концевые аминокислоты связаны вместе ковалентной связью, которая может представлять собой пептидную или дисульфидную связь, или синтетическую непептидную связь, такую как тиоэфирная, фосфодиэфирная, дисилоксановая или уретановая связь.
Под термином "аминокислота" понимают L- или D-аминокислоту, аминокислотный аналог или аминокислотный миметик, который может встречаться в природе или иметь чисто синтетическое происхождение, и может быть оптически чистым, т.е. единственный энантиомер, и, следовательно, хиральный, или смесь энантиомеров. Предпочтительно аминокислоты по настоящему изобретению являются оптически чистыми. Под термином "аминокислотный миметик" понимают синтетические аналоги встречающихся в природе аминокислот, которые представляют собой изостеры, т.е. разработанны для того, чтобы имитировать стерическую и электронную структуру природного соединения. Такие изостеры хорошо известны специалистам в данной области техники и включают депсипептиды, ретро-инверзопептиды, тиоамиды, циклоалканы или 1,5-дизамещенные тетразолы, но не ограничиваются ими [описано в М.Goodman, Biopolymers, 24, 137, (1985)].
Подходящие пептиды для применения по настоящему изобретению включают в себя следующие:
- соматостатин, октреотид и аналоги,
- пептиды, которые связываются с рецептором ST, где ST относится к теплостабильному токсину, продуцируемому E.coli и другими микроорганизмами;
- фрагменты ламинина, например YIGSR, PDSGR, IKVAV, LRE и KCQAGTFALRGDPQG,
- N-формилпептиды, имеющие в качестве мишеней сайты аккумулирования лейкоцитов,
- тромбоцитарный фактор 4 (PF4) и его фрагменты,
- пептиды, содержащие RGD,
- фрагменты пептида α2-антиплазмина, фибронектина или бета-казеина, фибриногена или тромбоспондина. Последовательности аминокислот α2-антиплазмина, фибронектина, бета-казеина, фибриногена или тромбоспондина можно найти в следующих ссылках: предшественник α2-антиплазмина [М.Tone et al., J. Biochem, 102, 1033 (1987)]; бета-казеин [L.Hansson et al., Gene, 139, 193 (1994)]; фибронектин [A.Gutman et al., FEBS Lett., 207, 145, (1996)]; предшественник тромбоспондина-1 [V.Dixit et al., Proc. Natl. Acad. Sci., USA, 83, 5449 (1986)]; R.F.Doolittle, Ann. Rev. Biochem., 53, 195 (1984).
Предпочтительно пептиды по настоящему изобретению включают в себя последовательность аминокислот, взятую из N-конца:
(1) α2-антиплазмина,
т.е. NH2-Asn-Gln-Glu-Gln-Val-Ser-Pro-Leu-Thr-Leu-Thr-Leu-Leu-Lys-OH
или ее варианты, где замещена, добавлена или удалена одна или более чем одна аминокислота, такие как
NH2-Asn-Gln-Glu-Gln-Val-Ser-Pro-Leu-Thr-Leu-Thr-Leu-Leu-Lys-Gly-OH,
NH2-Asn-Gln-Glu-Ala-Val-Ser-Pro-Leu-Thr-Leu-Thr-Leu-Leu-Lys-Gly-OH,
NH2-Asn-Gln-Glu-Gln-Val-Gly-OH, или
(2) казеина,
т.е. Ac-Leu-Gly-Pro-Gly-Gln-Ser-Lys-Val-Ile-Gly.
Синтетические пептиды по настоящему изобретению могут быть получены путем обычного твердофазного синтеза, в соответствии с тем, как описано в Р.Lloyd-Williams, F.Albericio and E.Girald; Chemical Approaches to the Synthesis of Peptides and Proteins, CRC Press, 1997.
Подходящие моноклональные антитела или их фрагменты для применения по настоящему изобретению включают антитела к антигену CD-20, экспрессируемому на поверхности В-клеток; антилейкоцитарные или антигранулоцитарные антитела; антимиозиновые антитела или антитела в отношении карциноэмбрионального антигена (КЭА).
Подходящие субстраты или ингибиторы для ферментов включают в себя глюкозу и аналоги глюкозы, такие как фтордезоксиглюкоза; жирные кислоты или ингибиторы эластазы.
Подходящие синтетические связывающиеся с рецептором соединения включают в себя эстрадиол, эстроген, прогестин, прогестерон и другие стероидные гормоны; лиганды дофаминового рецептора D-1 или D-2, или переносчик дофамина, такой как тропаны; лиганды рецептора серотонина.
Под термином "фторалкил" понимают алкильную группу с по меньшей мере одним заместителем фтором, т.е. этот термин охватывает группы от монофторалкила (например, -CH2F) до перфторалкила (например, CF3).
В диаминдиоксимовых хелатообразователях по настоящему изобретению R3 предпочтительно представляет собой Н. Также предпочтительно, чтобы по меньшей мере одна группа R2 представляла собой Н, более предпочтительно, все группы R2 представляли собой Н. Каждый R1 предпочтительно представляет собой C1-3алкил. Особенно предпочтительно, чтобы все группы R1 представляли собой CH3.
Предпочтительные хелатные конъюгаты формулы II, где 2 группы R, которые вместе с атомами, к которым они присоединены, образуют карбоциклическое насыщенное кольцо, включают в себя такие кольца, которые имеют от 3 до 6 членов, в частности 5 или 6 членов. Предпочтительные карбоциклические кольца представляют собой кольца, где 2 группы R1, присоединенные либо к одному и тому же атому углерода, либо к соседним атомам углерода, соединены с образованием 3-6-членных, в частности 5 или 6-членных насыщенных колец.
Предусмотрено, что роль связывающей группы -(A)n- заключается в том, чтобы дистанционно отдалить относительно громоздкий комплекс с радиоактивным металлом, который получается в результате координации металла, от активного сайта биологической группировки для направленной доставки, таким образом, что, например, связывание с рецептором не нарушается. Этого можно достигнуть путем комбинации гибкости (например, обычные алкильные цепи), так что громоздкая группа обладает свободой располагаться дальше от активного сайта, и/или жесткости, так как при циклоалкильном или арильном разделителе, который ориентирует комплекс металла на расстоянии от активного сайта. Природа линкерной группы может также быть использована для модификации биологического распределения полученного комплекса конъюгата с радиоактивным металлом. Таким образом, например, введение эфирных групп в линкер, будет способствовать минимизации связывания с белками плазмы крови. Предпочтительные связывающие группы -(A)n- имеют основную цепь молекулы, состоящую из связанных атомов, которые образуют группировку -(A)n-, содержащую от 2 до 10 атомов, наиболее предпочтительно от 2 до 5 атомов, причем 2 или 3 атома являются особенно предпочтительными. Минимальная основная цепь молекулы линкерной группы, состоящая из 2 атомов, предоставляет преимущество, заключающееся в том, что хелатообразователь хорошо отделен от биологической группировки для направленной доставки, так что любое взаимодействие минимизировано. Еще одно преимущество заключается в том, что размер потенциального хелатного кольца групп Х и Z так велик (по меньшей мере 8 для 2-атомноой основной цепи), что эти группы вряд ли эффективно конкурируют с координацией хелатообразователя с радиоактивным металлом. Таким образом, в конъюгатах этого типа сохраняются как характеристики биологического нацеливания биологической группировки для направленной доставки, так и способность диаминдиоксимового хелатообразователя образовывать комплекс с металлом.
Непептидные линкерные группы, такие как алкиленовые группы, обладают тем преимуществом, что отсутствуют значительные взаимодействия между углеводородной связью и конъюгированной биологической группировкой для направленной доставки, так что линкер не обвивается вокруг биологической группировки для направленной доставки. Предпочтительные алкиленовые разделяющие группы представляют собой -(CH2)n-, где n представляет собой от 2 до 5.
Предпочтительная группа Y представляет собой, таким образом, -CH2CH2-X-Z, наиболее предпочтительно -CH2CH2-NR4-Z, причем особенно предпочтительно Y=-CH2CH2-NH-Z. Эти группы обладают дополнительным преимуществом, заключающемся в том, что происходят от промежуточного соединения R3C(CH2CH2NH2)3, предпочтительно от промежуточного соединения НС(СН2СН2NH2)3, которое, поскольку оно является симметричным, гораздо проще синтезировать, так как для триаминов с различной длиной цепи необходимо применение стратегий синтеза, чтобы химически различать различные амины (например, за счет использования защитных групп).
Группа Х представляет собой функциональную группу, которая обеспечивает легкую конъюгацию хелатирующего агента с биологической группировкой для направленной доставки Z. Так как большинство пептидов и белков имеют доступные карбоксильные или аминосайты для образования функциональной группы, то предпочтительными группами X, когда Z представляет собой пептид или белок, являются -NR4- и -CO2-, так как это обеспечивает легкую конъюгацию посредством амидных связей. Содержащие цистеин пептиды и белки могут обладать свободными тиоловыми группами, предпочтительными группами X, когда Z представляет собой содержащий цистеин пептид или белок, являются тиолфильные группы, такие как малеимид и акриламид, так как они обеспечивают легкую конъюгацию посредством тиоэфирных связей.
Предпочтительные диаминдиоксимовые хелатообразователи по настоящему изобретению являются симметричными, т.е. два заместителя -CR2 2R2 2NHCR1 2C(=N-OH)R1 на группировке -CY(R3)- выбраны таким образом, чтобы они были одинаковыми. Это обеспечивает преимущество, заключающееся в том, что хелатообразователь не содержит хирального центра, поскольку такие центры могут образовывать диастереоизомерные комплексы с радиоактивным металлом, и возможно для них требуется очистка конкретных изомеров.
Хелатные конъюгаты формулы II возможно могут применяться в форме соли кислоты, т.е. когда один или более чем один амин либо из набора диаминдиоксимовых доноров, либо из группы Y протонирован биосовместимой кислотой. Такие соли могут быть получены непосредственно, например, путем очистки с помощью высокоэффективной жидкостной хроматографии (ВЭЖХ), использующей такие кислоты в подвижной фазе (например, уксусная или трифторуксусная кислота), или путем добавления биосовместимой кислоты к раствору хелатного конъюгата. Форма соли может быть пригодной для облегчения очистки (например, путем осаждения или перекристаллизации), или может облегчать растворение в водных средах (после чего рН при необходимости можно легко отрегулировать).
Хелатные конъюгаты по настоящему изобретению могут быть получены путем взаимодействия бифункционального хелатообразователя формулы III с биологической группировкой для направленной доставки

где каждый R1, R2 и R3 независимо представляет собой группу R;
Е представляет собой -(A)n-J,
где J представляет собой -NR5R6 или -CO2M, где R5 и R6 независимо представляют собой группу R или РG, и М представляет собой Н, катион, РG или активный сложный эфир;
-(A)n- представляет собой линкер, где А независимо представляет собой -CR2-, -NRCO-, -CONR-, -CR2OCR2- или группировку полиалкиленгликоля;
n представляет собой целое число, имеющее значение от 2 до 10;
каждая группа R независимо представляет собой Н или C1-10алкил, или 2 группы R вместе с атомами, к которым они присоединены, образуют C3-6 карбоциклическое насыщенное кольцо;
PG представляет собой защитную группу.
В соединениях формулы III J представляет собой функциональную группу, пригодную для конъюгации. Под термином "функциональная группа, пригодная для конъюгации" понимают функциональную группу, которая вступает в реакцию с соответствующей функциональной группой Z (как правило, аминогруппой, карбоксильной группой или тиоловой группой) для химического связывания диаминдиоксимового хелатообразователя с Z. Предпочтительные такие функциональные группы, пригодные для конъюгации, т.е. группы J, представляют собой -NR5R6 или -CO2M, где R5 и R6 независимо представляют собой группу R или PG; М представляет собой H, катион, PG или активный сложный эфир; РG представляет собой защитную группу. Катион в подходящем случае представляет собой положительно заряженный противоион, такой как ион металла, ион аммония (NH4 +) или четвертичный аммониевый или фосфониевый ион. Предпочтительно катион представляет собой биосовместимый катион. Термины "биосовместимый катион", "активный сложный эфир" и "защитная группа" определены ниже. Когда функциональная группа представляет собой -NR5R6, тогда по меньшей мере один и предпочтительно оба R5, так и R6 представляют собой Н.
Под термином "защитная группа" понимают группу, которая ингибирует или подавляет нежелательные химические реакции, но которая разработана таким образом, что является достаточно реакционноспособной для того, чтобы она могла отщепляться от рассматриваемой функциональной группы при достаточно мягких условиях, которые изменяют оставшуюся часть молекулы. После удаления защиты рассматриваемая группа может использоваться для конъюгации бифункционального хелата формулы III с биологической группировкой для направленной доставки.
Защитные группы хорошо известны специалистам в данной области техники и, когда J представляет собой -NR5R6, подходящим образом выбраны из Вос (где Вос представляет собой трет-бутилоксикарбонил), Fmoc (где Fmoc представляет собой флуоренилметоксикарбонил), трифторацетила, аллилоксикарбонила, Dde (т.е. 1-(4,4-диметил-2,6-диоксоциклогексилиден)этила) или Npys (т.е. 3-нитро-2-пиридинсульфенила), и когда J представляет собой -CO2PG: метилового эфира, трет-бутилового эфира, бензилового эфира. Применение дополнительных защитных групп описано в "Protective Groups in Organic Synthesis", Theorodora W.Greene and Peter G.M.Wuts (John Wiley & Sons, 1991).
Под термином "биосовместимый катион" понимают положительно заряженный противоион, который образует соль с ионизированной отрицательно заряженной группой, где указанный положительно заряженный противоион также не является токсичным и, следовательно, пригоден для введения в организм млекопитающего, в частности в организм человека. Примеры пригодных биосовместимых ионов включают щелочные металлы (например, натрий или калий), щелочноземельные металлы (например, кальций или магний) и ион аммония. Предпочтительный биосовместимый катион представляет собой ион натрия (Na+).
Под термином "активный сложный эфир" понимают эфирное производное карбоновой кислоты, которое разработано для того, чтобы служить в качестве более хорошей уходящей группы и, следовательно, обеспечивать более легкую реакцию с нуклеофилами, присутствующими на биологической группировке для направленной доставки, например аминами. Примеры пригодных активных сложных эфиров представляют собой N-гидроксисукцинимид (NHS), пентафторфенол, пентафтортиофенол, пара-нитрофенол и гидроксибензотриазол.
Функционализированные амином хелатообразователи формулы III (т.е. J=-NR5R6), таким образом, могут быть конъюгированы с карбоксильной группой(ми) биологической группировки для направленной доставки посредством амидных связей. Это связывание может осуществляться непосредственно (например, путем применения твердофазного пептидного синтеза) или в присутствии пригодного активирующего агента, такого как ВОР (т.е. бензотриазол-1-илокситрис(диметиламино)фосфоний) или N,N'-дициклогексилкарбодиимид (DCCI). Связывание может также осуществляться через образование подходящих промежуточных соединений, как известно из уровня техники, таких как активированные сложные эфиры карбоксильной группы биологической группировки для направленной доставки. В качестве альтернативы дополнительная аминогруппа бифункционального хелатообразователя сначала может быть преобразована в изотиоцианатную (-NCS) или изоцианатную группы (-NCO), которые допускают конъюгацию с содержащими амин биологическими группировками для направленной доставки посредством образования связывающих групп тиомочевины и мочевины соответственно. В качестве альтернативы дополнительная аминогруппа бифункционального хелатообразователя может вступать в реакцию с двухосновной кислотой для введения концевой карбоксильной группы посредством связывающей группы. Бифункциональный хелатообразователь, несущий карбоксильную функциональную группу (т.е. J=-CO2M), может быть использован подобным образом для связывания непосредственно с содержащими амин биологическими группировками для направленной доставки посредством амидной связи. Бифункциональный хелат может также нести группу, разработанную для того, чтобы вступать в реакцию с тиоловыми группами на биологической группировке для направленной доставки с образованием устойчивых тиоэфирных связывающих групп. Примеры таких групп представляют собой малеимиды (которые могут быть получены путем реакции малеинового ангидрида с соответствующим амином с последующим нагреванием с уксусным ангидридом) и акриламиды (которые могут быть получены путем реакции акрилилхлорида с амином).
Во втором аспекте настоящего изобретения предложены комплексы хелатного конъюгата, описанного выше, с радиоактивным металлом. Пригодные радиоактивные металлы могут представлять собой либо источники излучения позитронов, такие как 64Cu, 48V, 52Fe, 55Co, 94mТс или 68Ga, либо гамма-излучатели, такие как 94mТс, 111In, 113mIn или 67Ga. Наиболее предпочтительные радиоактивные металлы для диагностической визуализации представляют собой гамма-излучатели, в частности 94mТс. Металлические комплексы с некоторыми радионуклидами могут быть полезны в качестве радиоактивных фармацевтических препаратов для радиоактивного лечения различных заболеваний, таких как рак, или лечения тромбоза или рестеноза. Пригодные радиоизотопы для таких радиотерапевтических применений включают 90Y, 89Sr, 67Cu, 103Pd, 186Re, 188Re, 169Er, 153Sm и 198Au. Весьма предпочтительно, чтобы биологическая группировка для направленной доставки Z была связана с хелатообразователем таким образом, чтобы связывающая группа не претерпевала легкого метаболизма в крови, который приводил бы в результате к расщеплению комплекса металла перед тем, как меченая биологическая группировка для направленной доставки достигала желаемого сайта-мишени in vivo. Поэтому биологическая группировка для направленной доставки предпочтительно ковалентно связана с комплексами металлов по настоящему изобретению посредством связывающих групп, которые не подвергаются легкому метаболизму (как, например, эфирные связывающие группы).
Предпочтительные комплексы с радиоактивным металлом по настоящему изобретению являются симметричными, т.е. два заместителя -CR2 2R2 2NHCR1 2C(=N-OH)R1 на группировке -CY(R3)- выбирают таким образом, чтобы они были одинаковыми. Это обеспечивает преимущество, заключающееся в том, что хелатообразователь не содержит хирального центра, поскольку такие центры могут образовывать диастереоизомерные комплексы с радиоактивным металлом и возможно для них требуется очистка конкретных изомеров. Также предпочтительно, чтобы комплексы хелатного конъюгата были электрически нейтральными.
Полагают, что комплексы хелатообразователей с 99mTc по настоящему изобретению представляют собой
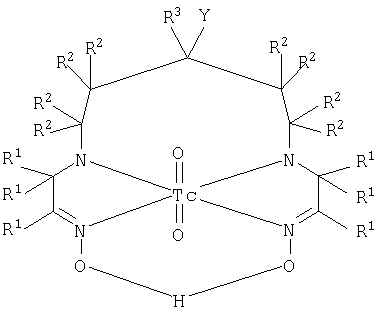
нейтральные диоксокомплексы с Tc(V) в соответствии с тем, как изображено выше.
В 99mTc-диаминдиоксимовых комплексах по настоящему изобретению R3 предпочтительно представляет собой Н. Также предпочтительно, чтобы по меньшей мере одна группа R2 представляла собой Н, более предпочтительно все группы R2 представляли собой Н. Каждая R1 предпочтительно представляет собой C1-3алкил. Особенно предпочтительно, чтобы все группы R1 представляли собой CH3. Предпочтительные группы Y для комплекса 99mTc являются такими, как было описано выше для хелатного конъюгата.
Предпочтительные комплексы с радиоактивным металлом по настоящему изобретению, где 2 группы R, которые вместе с атомами, к которым они присоединены, образуют карбоциклическое насыщенное кольцо, включают в себя такие кольца, которые имеют от 3 до 6 членов, в частности 5 или 6 членов. Предпочтительные карбоциклические кольца представляют собой кольца, где 2 группы R1, присоединенные либо к одному и тому же атому углерода, либо к соседним атомам углерода, соединены с образованием 3-6-членных, в частности 5 или 6-членных насыщенных колец.
Комплексы с радиактивным металлом по настоящему изобретению могут быть получены путем взаимодействия раствора радиоактивного металла в подходящем состоянии окисления с хелатным конъюгатом при подходящем значении рН. Раствор может предпочтительно содержать лиганд, который слабо вступает в комплекс с металлом (таким как глюконат или цитрат), т.е. получают комплекс с радиоактивным металлом путем обмена лигандами или трансхелатообразования. Такие условия пригодны для подавления нежелательных побочных реакций, таких как гидролиз иона металла. Когда ион радиоактивного металла представляет собой 99mTc, обычный исходный материал представляет собой пертехнетат натрия из генератора 99Mo. Технеций представлен в 99mTc-пертехнетате в состоянии окисления Tc(VII), которое является относительно нереакционноспособным. При получении комплексов технеция с более низким состоянием окисления от Tc(I) до Tc(V), таким образом, обычно для облегчения комплексообразования требуется добавление пригодного фармацевтически приемлемого восстановителя, такого как дитионит натрия, бисульфит натрия, аскорбиновая кислота, формамидинсульфиновая кислота, ион двухвалентного олова, Fe(II) или Cu(I). Фармацевтически приемлемый восстановитель предпочтительно представляет собой соль двухвалентного олова, наиболее предпочтительно хлорид двухвалентного олова, фторид двухвалентного олова или тартрат двухвалентного олова.
В третьем аспекте настоящего изобретения предложены радиоактивные фармацевтические препараты, которые включают в себя вышеупомянутые комплексы хелатных конъюгатов с радиоактивным металлом в стерильной форме, пригодной для введения человеку. Такие радиоактивные фармацевтические препараты удобным образом поставляются в контейнере, который оборудован крышкой, которая пригодна для разового или многократного укола шприцем для подкожных инъекций (например, изолирующая крышка с обжатой мембраной) при сохранении стерильности. Такие контейнеры могут содержать однократные или иногократные дозы для пациентов. Предпочтительные контейнеры для многократных доз включают одиночный объемный флакон (например, объемом от 10 до 30 см3), который содержит многократные дозы для пациентов, из которого в соответствии с клинической ситуацией в медицинские шприцы с различными интервалами времени в течение срока годности препарата могут быть отобраны однократные дозы для пациентов. Предварительно заполненные шприцы предназначены для содержания однократной дозы для человека и поэтому предпочтительно представляют собой одноразовые или другие шприцы, пригодные для клинического использования. Предварительно заполненный шприц возможно может быть снабжен защитным кожухом для защиты оператора от радиоактивной дозы. Такие пригодные защитные кожухи для шприца с радиоактивным фармацевтическим препаратом известны из уровня техники и предпочтительно включают в себя или свинец, или вольфрам.
Радиоактивность содержимого 99mТс, пригодная для диагностического радиоактивного фармацевтического препарата для визуализации, находится в диапазоне от 180 до 1500 мБк в зависимости от сайта визуализации in vivo, поглощения и соотношения мишень/фон. Для визуализации сердца с применением радиоактивного фармацевтического препарата, содержащего 99mTc, может применяться приблизительно 1110 мБк (30 мКи) для исследования в состоянии стресса и приблизительно 250 мБк (10 мКи) для исследования в состоянии покоя.
В четвертом аспекте настоящее изобретение предлагает нерадиоактивные наборы для приготовления радиоактивной фармацевтической композиции, содержащей 99mТс. Такие наборы предназначены для получения стерильных радиоактивных фармацевтических продуктов, пригодных для введения человеку, например, посредством прямой инъекции в кровоток. Для 99mТс набор предпочтительно лиофилизирован и предназначен для разведения стерильным 99mТс-пертехнетатом (TcO4 -) из генератора радиоизотопа 99mTc с получением раствора, пригодного для введения человеку без какой-либо дополнительной обработки. Пригодные наборы включают в себя контейнер (например, закрытый диафрагмой флакон), содержащий хелатный конъюгат формулы II или в форме свободного основания или соли кислоты, вместе с фармацевтически приемлемым восстановителем, таким как дитионит натрия, бисульфит натрия, аскорбиновая кислота, формамидинсульфиновая кислота, ион двухвалентного олова, Fe(II) или Cu(I). Фармацевтически приемлемый восстановитель предпочтительно представляет собой соль двухвалентного олова, такую как хлорид двухвалентного олова или тартрат двухвалентного олова. Альтернативно набор может возможно содержать комплекс металла, который при добавлении радиоактивного металла претерпевает переметаллирование (т.е. обмен металлом) с получением желаемого продукта.
Нерадиоактивные наборы могут возможно дополнительно включать в себя дополнительные компоненты, такие как трансхелатообразователь, радиопротектор, антимикробный консервант, агент, регулирующий рН, или наполнитель. "Трансхелатообразователь" представляет собой соединение, которое быстро вступает в реакцию с образованием слабого комплекса с технецием, затем замещается диаминдиоксимом. Это минимизирует риск образования восстановленного гидролизованного технеция (ВГТ) вследствие быстрого восстановления пертехнетата, конкурирующего с образованием комплекса с технецием. Такие пригодные трансхелатообразователи представляют собой соли слабой органической кислоты, т.е. органической кислоты, обладающие значением рКа, находящимся в диапазоне от 3 до 7, с биосовместимым катионом. Такие пригодные слабые органические кислоты представляют собой уксусную кислоту, лимонную кислоту, винную кислоту, глюконовую кислоту, глюкогептоновую кислоту, бензойную кислоту, фенолы или фосфоновые кислоты. Следовательно, пригодные соли представляют собой ацетаты, цитраты, тартраты, глюконаты, глюкогептонаты, бензоаты, феноляты или фосфонаты. Такие предпочтительные соли представляют собой тартраты, глюконаты, глюкогептонаты, бензоаты или фосфонаты, наиболее предпочтительно фосфонаты, в особенности дифосфонаты. Такой предпочтительный трансхелатообразователь представляет собой соль МДФ, т.е. метилендифосфоновой кислоты, с биосовместимым катионом.
Под термином "радиопротектор" понимают соединение, которое ингибирует реакции разрушения, такие как процессы окисления, путем захвата высокореакционных свободных радикалов, таких как кислородсодержащие свободные радикалы, возникающие в результате радиолиза воды. Радиопротекторы по настоящему изобретению удобным образом выбраны из аскорбиновой кислоты, пара-аминобензойной кислоты (т.е. 4-аминобензойной кислоты), гентизиновой кислоты (т.е. 2,5-дигидроксибензойной кислоты) и их солей с биосовместимым катионом в соответствии с тем, как было описано выше.
Под термином "антимикробный консервант" понимают агент, который ингибирует рост потенциально вредных микроорганизмов, таких как бактерии, дрожжи или плесневые грибы. Антимикробный консервант может также проявлять некоторые бактерицидные свойства в зависимости от дозы. Основная роль антимикробного консерванта(ов) по настоящему изобретению заключается в том, чтобы ингибировать рост любого микроорганизма в радиоактивной фармацевтической композиции после ее разведения, т.е. в самом радиоактивном диагностическом продукте. Тем не менее антимикробный консервант также возможно может использоваться для ингибирования роста потенциально вредных микроорганизмов в одном или более чем одном компоненте нерадиоактивного набора по настоящему изобретению перед разведением. Приемлемый(е) антимикробный(е) консервант(ы) включает(ют) в себя парабены, т.е. метил-, этил-, пропил- или бутилпарабен или их смеси; бензиловый спирт; фенол; крезол; цетавлон и тиомерсал. Предпочтительный(е) антимикробный(е) консервант(ы) представляет(ют) собой парабены.
Термин "агент, регулирующий рН" обозначает соединение или смесь соединений, пригодную для обеспечения того, что рН разведенного набора окажется в допустимых пределах (приблизительно рН от 4,0 до 10,5) для введения человеку и млекопитающим. Такие пригодные агенты, регулирующие рН, включают в себя фармацевтически приемлемые буферы, такие как трицин, фосфат или трис (т.е. трис(гидроксиметил)аминометан), и фармацевтически приемлемые основания, такие как карбонат натрия, бикарбонат натрия или их смеси. Когда конъюгат формулы II применяют в форме соли, агент, регулирующий рН, может поставляться в отдельном флаконе или контейнере, так что пользователь набора может довести рН как этап многостадийного способа.
Под термином "наполнитель" понимают фармацевтически приемлемый заполняющий агент, который может облегчать обработку материала в процессе производства и лиофилизации. Пригодные наполнители включают в себя неорганические соли, такие как хлорид натрия, и водорастворимые сахара, такие как сахароза, мальтоза или трегалоза.
В пятом аспекте настоящего изобретения предложены бифункциональные диаминдиоксимовые хелатообразователи формулы III, пригодные для получения конъюгатов хелатообразователь-биологическая группировка для направленной доставки
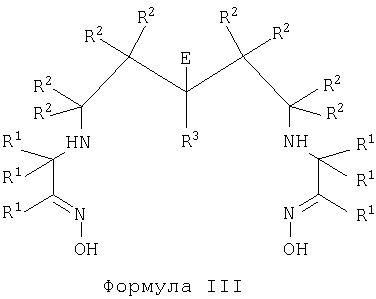
где каждый R1, R2 и R3 независимо представляет собой группу R;
Е представляет собой -(A)n-J,
где J представляет собой -NR5R6 или -CO2M, где R5 и R6 независимо представляют собой группу R или PG, и М представляет собой Н, катион, PG или активный сложный эфир;
-(A)n- представляет собой линкер, где А независимо представляет собой -CR2-, -NRCO-, -CONR-, -CR2OCR2- или группировку полиалкиленгликоля;
n представляет собой целое число, имеющее значение от 2 до 10;
каждая группа R независимо представляет собой Н или C1-10алкил, или 2 группы R вместе с атомами, к которым они присоединены, образуют C3-6карбоциклическое насыщенное кольцо;
PG представляет собой защитную группу.
В соединениях формулы III J представляет собой функциональную группу, пригодную для конъюгации. Под термином "функциональная группа, пригодная для конъюгации" понимают функциональную группу, которая вступает в реакцию с соответствующей функциональной группой Z (как правило, аминогруппой, карбоксильной группой или тиоловой группой) для химического связывания диаминдиоксимового хелатообразователя с Z. Предпочтительные такие функциональные группы, пригодные для конъюгации, т.е. группы J, представляют собой -NR5R6 или -CO2M, где R5 и R6 независимо представляют собой группу R или РG; М представляет собой Н, катион, PG или активный сложный эфир; РG представляет собой защитную группу. Катион в подходящем случае представляет собой положительно заряженный противоион, такой как ион металла, ион аммония (NH4 +) или четвертичный аммониевый или фосфониевый ион. Предпочтительно катион представляет собой биосовместимый катион. Термины "биосовместимый катион", "активный сложный эфир" и "защитная группа" определены ниже. Когда функциональная группа представляет собой -NR5R6, тогда по меньшей мере один и предпочтительно оба как R5, так и R6 представляют собой Н.
В бифункциональных хелатообразователях формулы III по настоящему изобретению R3 предпочтительно представляет собой Н. Также предпочтительно, чтобы по меньшей мере одна группа R2 представляла собой Н, более предпочтительно все группы R2 представляли собой Н. Каждый R1 предпочтительно представляет собой C1-3алкил. Особенно предпочтительно, чтобы все группы R1 представляли собой CH3.
Предпочтительные бифункциональные хелатообразователи, где 2 группы R, которые вместе с атомами, к которым они присоединены, образуют карбоциклическое насыщенное кольцо, включают в себя такие кольца, которые имеют от 3 до 6 членов, в частности 5 или 6 членов. Предпочтительные карбоциклические кольца представляют собой кольца, где 2 группы R1, присоединенные либо к одному и тому же, либо к соседним атомам углерода, соединены с образованием 3-6-членных, в частности 5 или 6-членных, насыщенных колец.
Хелатные конъюгаты формулы III возможно могут применяться в форме соли кислоты, т.е. когда один или более чем один амин либо из набора диаминдиоксимовых доноров, либо из группы Y протонирован биосовместимой кислотой. Такие соли могут быть получены непосредственно, например, путем очистки с помощью высокоэффективной жидкостной хроматографии (ВЭЖХ), использующей такие кислоты в подвижной фазе (например, уксусная или трифторуксусная кислота), или путем добавления биосовместимой кислоты к раствору хелатного конъюгата. Форма соли может быть пригодной для облегчения очистки (например, путем осаждения или перекристаллизации), или может облегчать растворение в водных средах (после чего рН при необходимости можно легко отрегулировать).
Предпочтительные линкерные группы -(A)n бифункционального хелатообразователя имеют основную цепь молекулы, состоящую из связанных атомов, которые образуют группировку -(A)n-, содержащую от 2 до 10 атомов, наиболее предпочтительно от 2 до 5 атомов, причем 2 или 3 атома являются особенно предпочтительными. Минимальная основная цепь молекулы линкерной группы, состоящая из 2 атомов, предоставляет преимущество, заключающееся в том, что хелатообразователь хорошо отделен от биологической группировки для направленной доставки, так что любое взаимодействие минимизировано. Еще одно преимущество заключается в том, что размер потенциального хелатного кольца групп Х и Z так велик (по меньшей мере 8 для 2-атомноой основной цепи), что эти группы вряд ли эффективно конкурируют с координацией хелатообразователя с радиоактивным металлом.
Непептидные линкерные группы, такие как алкиленовые группы, обладают тем преимуществом, что отсутствуют значительные взаимодействия между углеводородной связью и конъюгированной биологической группировкой для направленной доставки, так что линкер не обвивается вокруг биологической группировки для направленной доставки. Предпочтительные алкиленовые разделяющие группы представляют собой -(CH2)n-, где n представляет собой от 2 до 5.
Предпочтительная группа Е представляет собой, таким образом, -CH2CH2-J, наиболее предпочтительно -CH2CH2-NHR5 или -CH2CH2-CO2H или их активные сложные спирты, причем особенно предпочтительно Е=-CH2CH2-NH2. Кислота может также быть превращена в смешанный ангидрид, например, путем реакции с изобутилхлорформиатом и основанием. Смешанный ангидрид также вступает в реакцию с нуклеофилами, такими как амины. Группировка Е=-CH2CH2-NH2 обладает дополнительным преимуществом, заключающимся в том, что происходит из промежуточного соединения R3C(CH2CH2NH2)3, предпочтительно промежуточного соединения НС(CH2CH2NH2)3, которое, поскольку оно является симметричным, гораздо проще синтезировать, так как триаминам, обладающим различной длиной цепи, требуется применение стратегий синтеза, химически различающих различные амины (например, путем применения защитных групп).
Предпочтительные бифункциональные диаминдиоксимовые хелатообразователи формулы III по настоящему изобретению являются симметричными, т.е. два заместителя -C(R2)2(R2)2NHCR1 2C(=N-OH)R1 на группировке -CY(R3)- выбраны таким образом, чтобы они были одинаковыми. Это обеспечивает преимущество, заключающееся в том, что хелатообразователь не содержит хирального центра, поскольку такие центры могут образовывать диастереоизомерные комплексы с радиоактивным металлом и возможно для них требуется очистка конкретных изомеров.
В еще одном аспекте настоящего изобретения предложен особенно предпочтительный бифункциональный диаминдиоксимовый хелатообразователь, который имеет формулу
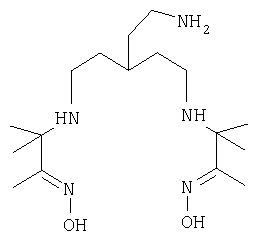
Кислые соли этого соединения также находятся в объеме настоящего изобретения.
Бифункциональные диаминдиоксимовые хелатообразователи по настоящему соединению могут быть подходящим образом получены путем алкилирования соединения формулы IV

где A, J, R2, R3 и n являются такими, как было определено для формулы III выше,
или (1) подходящим хлоронитрозопроизводным Cl-C(R1)2-CH(NO)R1,
или (2) альфа-хлороксимом формулы Cl-C(R1)2-C(=NOH)R1,
или (3) альфа-бромкетоном формулы Br-C(R1)2-C(=O)R1 с последующим превращением диаминдикетонового продукта в диаминдиоксим с использованием гидроксиламина.
Путь (1) описан S.Jurisson et al. [Inorg. Chem., 26, 3576-82 (1987)]. Хлоронитрозосоединения могут быть получены путем обработки подходящего алкена нитрозилхлоридом (NOCI) в соответствии с тем, как описано в Примере 3. Дополнительные подробности синтеза хлоронитрозосоединений приведены: Ramalingam, K. et al. Synth. Commun. (1995) 25(5) 743-52; Glaser et al. J. Org. Chem. (1996), 61(3), 1047-48; Clapp, Leallyn В. et al. J. Org Chem. (1971), 36(8) 1169-70; Saito, Giulichi et al. Shizen Kagaku (1995), 47, 41-9, и Schulz, Manfred Z. Chem. (1981), 21(11), 404-5. Путь (2) описан в общих чертах Nowotnik et al. [Tetrahedron, 50(29), р.8617-8632 (1994)]. Альфа-хлороксимы могут быть получены путем оксимирования соответствующего альфа-хлоркетона или альдегида, имеющихся в продаже. Альфа-бромкетоны имеются в продаже.
Когда J представляет собой -NH2, триамин формулы IV может возможно быть монозащищен первым, так что оказывается защищен первичный амин - группа J. Затем получают диаминдиоксим в соответствии с путями (1), (2) или (3), приведенными выше, затем защитную группу удаляют. Пригодные защитные группы известны из уровня техники и включают в себя Boc (т.е. трет-бутилоксикарбонил) или Fmoc в соответствии с описанным выше.
Соединения формулы IV подходящим образом получают из НС(CH2CH2OAc)3 путем гидролиза одного или более чем одного ацетатного эфира до первичного спирта(ов) и превращения до уходящей группы, такой как метансульфонатный эфир, с использованием метансульфонилхлорида и пиридина. Эта уходящая группа может затем быть замещена подходящим нуклеофилом, который может быть превращен в желательную функциональную группу. Для образования карбоновой кислоты (т.е. J=-CO2H) может быть использован цианид-анион. Кислотный гидролиз цианида может привести к образованию желаемой карбоновой кислоты. Для образования амина можно использовать азидный нуклеофил для образования алкилазида. Гидрирование алкилазида может привести к образованию амина.
В еще одном аспекте настоящего изобретения предложено соединение формулы V

где R7 и R8 независимо представляют собой Н или PG, или R7 и R8 вместе образуют PG, где PG представляет собой защитную группу, или его соль.
PG представляет собой защитную группу, как было определено выше. Соединения формулы V являются пригодными предшественниками для ряда бифункциональных хелатообразователей по настоящему изобретению. Предпочтительное соединение формулы V представляет собой HC(CH2CH2NH2)2(CH2CH2NR7R8), т.е. монозащищенный триамин в соответствии с тем, как было описано выше. Наиболее предпочтительно все группы R7 и R8 представляют собой Н, т.е. соединение HC(CH2CH2NH2)3 или его кислотная соль представляют собой предпочтительные соединения по настоящему изобретению.
Таким образом, в еще одном аспекте настоящего изобретения предложено соединение HC(CH2CH2NH2)3.
В еще одном аспекте настоящего изобретения предложен способ получения соединения

при котором алкилируют НС(CH2CH2NH2)3 хлоронитрозопроизводным формулы Cl-C(CH3)2-CH(NO)CH3.
На фиг.1 показаны химические структуры Соединений 1-6. На фиг.2 показана химическая структура Соединения 5 полностью. На фиг.3 показана реакционная схема для азидного синтеза 1,1,1-трис(2-аминоэтил)амина Примера 1. На фиг.4 показана реакционная схема для альтернативного синтеза 1,1,1-трис(2-аминоэтил)амина Примера 2.
Данное изобретение проиллюстрировано не ограничивающими его объема примерами, подробно описанными ниже. Пример 1 описывает синтез нового соединения - 1,1,1-трис(2-аминоэтил)метана. Пример 2 предлагает альтернативный синтез 1,1,1-трис(2-аминоэтил)метана, который позволяет избежать применения потенциально опасных азидных промежуточных соединений. Пример 3 описывает синтез различных хлорнитрозоалкановых предшественников. Пример 4 описывает синтез предпочтительного аминзамещенного бифункционального диаминдиоксима по настоящему изобретению (Соединение 1). Пример 5 описывает синтез бензамидного конъюгата (Соединение 2) Соединения 1. Пример 6 демонстрирует, каким образом может быть введена разделяющая группа, которая эффективно превращает концевую функциональную аминогруппу бифункционального хелатообразователя в концевую карбоксильную функциональную группу. Пример 7 описывает твердофазный синтез пептида, нацеленного на тромб. Пример 8 предлагает синтез Соединения 5, т.е. конъюгат Соединения 1 с нацеленным пептидом. Пример 9 описывает синтез Соединений 7 и 8, которые представляют собой диаминдиоксимовые аналоги, включающие в себя кольцевые структуры.
Пример 10 сравнивает введение радиоактивной метки 99mTc в Соединение 2 с введением радиоактивной метки 99mТс в аза-аналог хелатообразователя по предшествующему уровню техники (Соединение 3), и демонстрирует, что хелатообразователи по настоящему изобретению обеспечивают гораздо большую эффективность и быстрое мечение при более мягких условиях, т.е. комнатной температуре и менее щелочном рН. Таким образом, хелатообразователь по предшествующему уровню техники требует рН 10 и промежуток времени по меньшей мере 120 минут при комнатной температуре с получением радиохимической чистоты (РХЧ) приблизительно 80%, тогда как Соединение 3 дает мечение около 95% в течение 15 минут. Пример 11 демонстрирует, что Соединение 5 метят 99mТс с получением препарата высокой радиохимической чистоты. Пример 12 демонстрирует, что Соединение 5, меченное 99mТс, захватывается сгустком крови in vitro на уровне, сопоставимом с 99mTc-Соединением 6 по предшествующему уровню техники, что следовательно свойства биологического нацеливания пептида сохраняются при его конъюгации с диаминдиоксимовыми хелатообразователями по настоящему изобретению. Пример 13 демонстрирует радиоактивное мечение с помощью 99mТс в мягких условиях Соединения 9, представляющего собой конъюгат Соединения 1 с циклическим пептидом, обладающим относительно чувствительными дисульфидными связями.
Пример 1: синтез 1,1,1-трис(2-аминоэтил)метана
Стадия 1(а): диметиловый эфир 3-(метоксикарбонилметилен)глутаровой кислоты
Карбометоксиметилентрифенилфосфоран (167 г, 0,5 моль) в толуоле (600 мл) обрабатывали диметил-3-оксоглутаратом (87 г, 0,5 моль) и реакционную смесь нагревали до 100°С на масляной бане при 120°С в атмосфере азота в течение 36 ч. Реакционную смесь затем концентрировали в вакууме и масляный остаток растирали со смесью 40/60 петролейный эфир/диэтиловый эфир, 600 мл. Трифенилфосфина оксид выпадал в осадок, а жидкий супернатант сливали/отфильтровывали. Остаток после выпаривания в вакууме подвергали перегонке через трубку с шаровым расширением (Kugelrohr) в высоком вакууме (температура печи 180-200°С при давлении 26,6644 Па (0,2 торр)) с получением диметилового эфира 3-(метоксикарбонилметилен)глутаровой кислоты (89,08 г, 53%).
ЯМР 1H (CDCl3): δ 3,31 (2Н, s, CH2), 3,7 (9Н, s, 3×ОСН3), 3,87 (2Н, s, CH2), 5,79 (1H, s, =CH,) млн-1.
ЯМР 13C (CDCl3): δ 36,56, CH3, 48,7; 2×CH3, 52,09 и 52,5 (2×CH2); 122,3 и 146,16 С=СН; 165,9, 170,0 и 170,5 3×COO млн-1.
Стадия 1(б): гидрирование диметилового эфира 3-(метоксикарбонилметилен)глутаровой кислоты
Диметиловый эфир 3-(метоксикарбонилметилен)глутаровой кислоты (89 г, 267 ммоль) в метаноле (200 мл) перемешивали с 10% палладия на угле : 50 % воды (9 г) в атмосфере газообразного водорода (3,5×105 Па (3,5 бар) в течение 30 ч. Раствор фильтровали через кизельгур и концентрировали в вакууме с получением диметилового эфира 3-(метоксикарбонилметил)глутаровой кислоты в виде масла, выход 84,9 г (94%).
ЯМР 1H (CDCl3): δ 2,48 (6Н, d, J=8 Гц, 3×CH2), 2,78 (1Н, секстет, J=8 Гц CH,) 3,7 (9H, s, 3×CH3).
ЯМР 13C (CDCl3): δ 28,6, СН; 37,50, 3×CH3; 51,6, 3×CH2; 172,28, 3×COO.
Стадия 1 (в): восстановление и этерификация триметилового эфира в триацетат
В атмосфере азота в трехгорлой круглодонной колбе на 2 л алюмогидрид лития (20 г, 588 моль) в тетрагидрофуране (400 мл) осторожно обрабатывали трис(метилоксикарбонилметил)метаном (40 г, 212 ммоль) в тетрагидрофуране (200 мл) в течение 1 ч. Возникала сильно экзотермическая реакция, заставляющая растворитель сильно кипеть при температуре дефлегмации. Реакционную смесь нагревали на масляной бане при 90°С при дефлегмации в течение 3 суток. Реакционную смесь гасили путем осторожного добавления по каплям уксусной кислоты (100 мл) до прекращения выделения водорода. Перемешиваемую реакционную смесь осторожно обрабатывали раствором уксусного ангидрида (500 мл) с такой скоростью, чтобы вызвать слабую дефлегмацию. Колбу оборудовали для перегонки и перемешивали, а затем нагревали при 90°С (температура масляной бани) для отгонки тетрагидрофурана. Добавляли еще одну порцию уксусного ангидрида (300 мл), реакционную смесь возвращали к состоянию дефлегмации, перемешивали и нагревали на масляной бане при 140°С в течение 5 ч. Реакционной смеси давали возможность охладиться и фильтровали. Осадок оксида алюминия промывали этилацетатом и объединенные фильтраты концентрировали на роторном испарителе при температуре водяной бани 50°С в вакууме (666,61 Па (5 мм рт.ст.)) с получением масла. Масло собирали этилацетатом (500 мл) и промывали насыщенным водным раствором карбоната калия. Этилацетатный раствор разделяли, сушили над сульфатом натрия и концентрировали в вакууме с получением масла. Масло подвергали перегонке через трубку с шаровым расширением в высоком вакууме с получением трис(2-ацетоксиэтил)метана (45,3 г, 95,9%) в виде масла. Температура плавления (ТП) 220°С при 13,3322 Па (0,1 мм рт.ст.).
ЯМР 1H (CDCl3): δ 1,66 (7Н, m, 3×CH2, СН), 2,08 (1Н, s, 3×CH3); 4,1 (6Н, t, 3×CH2O).
ЯМР 13C (CDCl3): δ 20,9, CH3; 29,34, СН; 32,17, CH2; 62,15, CH2O; 171, СО.
Стадия 1(г): удаление ацетатных групп из триацетата
Трис(2-ацетоксиэтил)метан (45,3 г, 165 мМ) в метаноле (200 мл) и аммиаке 880 (100 мл) нагревали на масляной бане при 80°С в течение 2 суток. Реакционную смесь обрабатывали еще одной порцией аммиака 880 (50 мл) и нагревали при 80°С на масляной бане в течение 24 ч. Добавляли еще одну порцию аммиака 880 (50 мл) и реакционную смесь нагревали при 80°С в течение 24 ч. Реакционную смесь затем концентрировали в вакууме для удаления всех растворителей с получением масла. Его растворяли в аммиаке 880 (150 мл) и нагревали при 80°С в течение 24 ч. Реакционную смесь затем концентрировали в вакууме для удаления всех растворителей с получением масла. Перегонка через трубку с шаровым расширением позволила получить ацетамид с ТП 170-180°С при 26,6644 Па (0,2 мм рт.ст.). Сосуды, содержащие ацетамид, промывали начисто и перегонку продолжали. Трис(2-гидроксиэтил)метан (22,53 г, 92%) перегоняли при ТП 220°С при 26,6644 Па (0,2 мм рт.ст.).
ЯМР 1H (CDCl3): δ 1,45 (6Н, q, 3×CH2), 2,2 (1Н, квинтет, СН); 3,7 (6Н, t, 3×CH2OH); 5,5 (3Н, brs, 3×OH).
ЯМР 13C (CDCl3): δ 22,13, СН; 33,95, 3×CH2; 57,8, 3×CH2OH.
Стадия 1(д): превращение триола в трис(метансульфонат)
К перемешиваемому охлажденному на льду раствору трис(2-гидроксиэтил)метана (10 г, 0,0676 моль) в дихлорметане (50 мл) медленно добавляли по каплям раствор метансульфонилхлорида (40 г, 0,349 моль) в дихлорметане (50 мл) в атмосфере азота с такой скоростью, что температура не поднималась выше 15°С. Затем по каплям добавляли пиридин (21,4 г, 0,27 моль, 4 экв.), растворенный в дихлорметане (50 мл), с такой скоростью, что температура не поднималась выше 15°С (экзотермическая реакция). Реакционную смесь оставляли перемешиваться при комнатной температуре в течение 24 ч, затем обрабатывали 5 н. раствором (80 мл) и слои разделяли. Водный слой экстрагировали дополнительным количеством дихлорметана (50 мл) и органические экстракты объединяли, сушили над сульфатом натрия, фильтровали и концентрировали в вакууме с получением трис[2-(метилсульфонилокси)этил]метана, загрязненного избытком метансульфонилхлорида. Теоретический выход составляет 25,8 г.
ЯМР 1H (CDCl3): δ 4,3 (6Н, t, 2×CH2), 3,0 (9H, s, 3×CH3), 2 (1Н, секстет, СН), 1,85 (6H, q, 3×CH2).
Стадия 1(е): получение 1,1,1-трис(азидоэтил)метана
Перемешиваемый раствор трис[2-(метилсульфонилокси)этил]метана (со стадии 1(д), загрязненного избытком метансульфонилхлорида) (25,8 г, 67 ммоль, теоретический) в безводном диметилформамиде (ДМФ) (250 мл) в атмосфере азота обрабатывали азидом натрия (30,7 г, 0,47 моль) порциями в течение 15 минут. Наблюдали экзотермическую реакцию и реакционную смесь охлаждали на ледяной бане. Через 30 минут реакционную смесь нагревали на масляной бане при 50°С в течение 24 ч. Реакционная смесь становилась коричневая. Реакционной смеси давали возможность охладиться, обрабатывали разбавленным раствором карбоната калия (200 мл) и трижды экстрагировали смесью 40/60 петролейный эфир/диэтиловый эфир 10:1 (3×150 мл). Органические экстракты промывали водой (2×150 мл), сушили над сульфатом натрия и фильтровали. К раствору петролейного эфира добавляли этанол (200 мл) для удержания триазида в растворе и объем уменьшали в вакууме, но не больше чем до 200 мл. Добавляли этанол (200 мл) и повторно концентрировали в вакууме для удаления последних следов петролейного эфира, оставляя не менее 200 мл этанолового раствора. Этаноловый раствор триазида используют непосредственно на Стадии 1(ж).
Осторожно: НЕ УДАЛЯЙТЕ ВЕСЬ РАСТВОРИТЕЛЬ, ПОСКОЛЬКУ АЗИД ПОТЕНЦИАЛЬНО ВЗРЫВООПАСЕН И ДОЛЖЕН ХРАНИТЬСЯ В РАСТВОРЕННОМ СОСТОЯНИИ ПОСТОЯННО.
Менее чем 0,2 мл раствора выпаривали в вакууме для удаления этанола и ЯМР осуществляли на этом небольшом образце.
ЯМР 1H (CDCl3): δ 3,35 (6Н, t, 3×CH2), 1,8 (1Н, септет, СН,), 1,6 (6Н, q, 3×CH2).
Стадия 1(ж): получение 1,1,1-трис(аминоэтил)метана
Трис(2-азидоэтил)метан (15,06 г, 0,0676 моль) (полагая что выход предыдущей реакции 100%) в этаноле (200 мл) обрабатывали 10% палладием на угле (2 г, 50% воды) и гидрировали в течение 12 ч. Реакционный сосуд убирали каждые 2 часа для удаления азота, выделившегося в результате реакции, и повторно заполняли водородом. Образец подвергали анализу путем ЯМР для того, чтобы убедиться в полном превращении триазида в триамин.
Осторожно: окисленный азид может взорваться при перегонке. Реакционную смесь фильтровали через слой целита для удаления катализатора и концентрировали в вакууме с получением трис(2-аминоэтил)метана в виде масла. Его дополнительно очищали путем перегонки через трубку с шаровым расширением ТП 180-200°С при 53,3288 Па (0,4 мм рт.ст.) с получением бесцветного масла (8,1 г, 82,7% общий выход из триола).
ЯМР 1H (CDCl3): 2,72 (6Н, t, 3×CH2N), 1,41 (H, septet, CH), 1,39 (6Н, q, 3×CH2).
ЯМР 13C (CDCl3): δ 39,8 (CH2NH2), 38,2 (CH2), 31,0 (CH).
Пример 2: альтернативный способ получения 1,1,1-трис(2-аминоэтил)метана
Стадия 2 (а): амидирование триметилового эфира пара-метоксибензиламином
Трис(метилоксикарбонилметил)метан [2 г, 8,4 ммоль; полученный, как на Стадии 1(б) выше] растворяли в пара-метоксибензиламине (25 г, 178,6 ммоль). Аппарат настраивали для перегонки и нагревали до 120°С в течение 24 часов при потоке азота. За развитием реакции следили по количеству собираемого метанола. Реакционную смесь охлаждали до температуры окружающей среды и добавляли 30 мл этилацетата, затем осажденный продукт - триамид - перемешивали в течение 30 минут. Триамид выделяли путем фильтрации и фильтр промывали несколько раз достаточным количеством этилацетата для удаления избытка пара-метоксибензиламина. После сушки получали 4,6 г, 100%, белого порошка. Нерастворимый продукт использовали непосредственно на следующей стадии без дополнительной очистки или определения характеристик.
Стадия 2(б): получение 1,1,1-трис[2-(пара-метоксибензиламино)этил]метана
Триамид со стадии 2(а) (10 г, 17,89 ммоль) осторожно добавляли в трехгорлую круглодонную охлажденную на ледяной бане колбу на 1000 мл к 250 мл 1 М раствора борана (3,5 г, 244,3 ммоль). После завершения добавления ледяную баню удаляли и реакционную смесь медленно нагревали до 60°С. Реакционную смесь перемешивали при 60°С в течение 20 часов. Отбирали образец реакционной смеси (1 мл) и смешивали с 0,5 мл 5 н. HCl и оставляли на 30 мин. К образцу добавляли 0,5 мл 50 NaOH, далее 2 мл воды и раствор перемешивали до полного растворения белого осадка. Раствор экстрагировали простым эфиром (5 мл) и выпаривали. Остаток растворяли в ацетонитриле в концентрации 1 мг/мл и анализировали путем масс-спектрометрии (МС). Если на масс-спектре наблюдали моно- и диамид (M+H/z=520 и 534), то реакция не завершилась. Для завершения реакции добавляли дополнительно 100 мл 1 М тетрагидрофуранового (ТГФ) раствора борана и реакционную смесь перемешивали в течение еще 6 часов при 60°С, новый образец отбирали в соответствии с первой процедурой отбора. Добавление 1 М борана в ТГФ растворе продолжали по необходимости до наступления полного превращения в триамин.
Реакционную смесь охлаждали до температуры окружающей среды и медленно добавляли 5 н. HCl (осторожно: имеет место интенсивное образование пены!). Добавляли HCl до тех пор, пока не наблюдалось никакого выделения газа. Смесь перемешивали в течение 30 минут и затем упаривали. Остаток суспендировали в водном растворе NaOH (20-40%; 1:2 масса/объем) и перемешивали в течение 30 минут. Смесь затем разбавляли водой (3 объема). Смесь затем экстрагировали диэтиловым эфиром (2×150 мл) (осторожно: не использовать галогенированные растворители). Объединенные органические фазы затем промывали водой (1×200 мл), рассолом (150 мл) и сушили над сульфатом магния. Выход после выпаривания 7,6 г, 84% в виде масла.
ЯМР 1H (CDCl3): δ 1,45 (6Н, т, 3×CH2); 1,54 (1Н, septet, СН); 2,66 (6Н, t, 1, 3×CH2N); 3,68 (6Н, s, ArCH2); 3,78 (9Н, s, 3×CH3O); 6,94 (6Н, d, 6×Ar), 7,20 (6Н, d, 6×Ar).
ЯМР 13C (CDCl3): δ 32,17, CH; 34,44, CH2; 47,00, CH2; 53,56, ArCH2; 55,25, CH3O; 113,78, Ar; 129,29, Ar; 132,61, Ar; 158,60, Ar.
Стадия 2(в): получение 1,1,1-трис(2-аминоэтил)метана
1,1,1-Трис[2-(пара-метоксибензиламино)этил]метан (20,0 грамм, 0,036 ммоль) растворяли в метаноле (100 мл) и добавляли Pd(OH)2. Смесь гидрировали (3×105 Па (3 бар), 100°С, в автоклаве) и перемешивали в течение 5 часов. Добавляли еще две порции Pd(OH)2 (2×5 грамм) через 10 и 15 часов соответственно. Реакционную смесь фильтровали и фильтрат промывали метанолом. Объединенную органическую фазу упаривали и остаток перегоняли в вакууме (1×10-2, 110°С) с получением 2,60 грамм (50%) 1,1,1-трис(2-аминоэтил)метана, идентичного описанному в Примере 1.
Пример 3: получение 3-хлор-3-метил-2-нитрозобутана
Смесь 2-метилбут-2-ена (147 мл, 1,4 моль) и изоамилнитрита (156 мл, 1,16 моль) охлаждали до -30°С в бане с cardice и метанолом и интенсивно перемешивали при помощи мешалки с верхней подачей воздуха и по каплям обрабатывали концентрированной соляной кислотой (140 мл, 1,68 моль) с такой скоростью, что температура поддерживалась ниже -20°С. Для этого необходимо около 1 часа, поскольку имеет место значительная экзотермичность, и следует соблюдать осторожность для предотвращения перегревания. Добавляли этанол (100 мл) для уменьшения вязкости суспензии, которая образовалась к концу добавления, и реакционную смесь перемешивали при температуре от -20 до -10°С в течение еще 2 ч для завершения реакции. Осадок собирали путем фильтрации в вакууме, промывали 4×30 мл охлажденного (-20°С) этанола и 100 мл охлажденной на льду воды и сушили в вакууме с получением 3-хлор-3-метил-2-нитрозобутана в виде белого твердого вещества. Этаноловый фильтрат и промывки объединяли и разбавляли водой (200 мл), охлаждали и давали отстояться в течение 1 ч при -10°С, в течение которого выкристаллизовывалась еще одна порция 3-хлор-3-метил-2-нитрозобутана. Осадок собирали путем фильтрации, промывали минимальным количеством воды и сушили в вакууме с получением общего выхода 3-хлор-3-метил-2-нитрозобутана (115 г, 0,85 ммоль, 73%), чистота в соответствии с ЯМР>85%.
ЯМР 1H (CDCl3), как смесь изомеров: (изомер 1, 90%) 1,5 d, (2H, CH3), 1,65 d, (4H, 2×CH3), 5,85, q, и 5,95, q, вместе с 1Н; (изомер 2, 10%), 1,76 s, (6H, 2×CH3), 2,07 (3Н, CH3).
1-Хлор-1-(1-нитрозоэтил)циклопентан получали аналогичным образом из этилиденциклопентана (выход 55%) [J. Org. Chem., 36(8) p.1169-70].
1-Хлор-1-(1-нитрозоэтил)циклогексан получали аналогичным образом из этилиденциклогексана (выход 63%) [J. Org. Chem., 36(8) р.1169-70].
δH (CDCl3; 270 МГц): 1,52 (3Н, d JHH 7 Гц), 1,48-2,20 (10Н, m, CH2×5), 5,96 (1H, q, JHH 7 Гц, CH).
1-Хлор-1-метил-2-нитрозоциклогексан получали аналогичным образом из 1-метил-1-циклогексана (выход 57%) [Ind J. Chem Sect В (1978) 16B (10) 917-20, Z. Chem. (1981), 21 (11) 404-5, J. Pract. Chem. (1978) 320 (3) 433-51].
δH (CDCl3; 270 Мгц): 1,41-2,28 (11Н, m, CH3, CH2×4), 5,72-5,79 (1Н, m, СН).
Пример 4: синтез бис[N-(1,1-диметил-2-N-гидроксииминпропил)2-аминоэтил]-(2-аминоэтил)метана (Соединение 1)
К раствору трис(2-аминоэтил)метана (4,047 г, 27,9 ммоль) в безводном этаноле (30 мл) добавляли безводный карбонат калия (7,7 г, 55,8 ммоль, 2 экв.) при комнатной температуре при интенсивном перемешивании в атмосфере азота. Раствор 3-хлор-3-метил-2-нитрозобутана (7,56 г, 55,8 моль, 2 экв.) разводили в безводном этаноле (100 мл) и 75 мл этого раствора медленно вводили по каплям в реакционную смесь. После завершения реакции проводили тонкослойную хроматографию (ТСХ) на диоксиде кремния [хроматографию на пластинках осуществляли в дихлорметане, метаноле, концентрированном (0,88 относит. плотность) аммиаке, 100/30/5, и пластинки ТСХ проявляли путем разбрызгивания нингидрина и нагревания]. Моно-, ди- и триалкилированные продукты визуализировали по увеличению RF в этой последовательности. Проводили аналитическую ВЭЖХ, используя обращенно-фазовую колонку RPR, в градиенте 7,5-75% ацетонитрил в 3% водном аммиаке. Реакционную смесь концентрировали в вакууме для удаления этанола и ресуспендировали в воде (110 мл). Водную суспензию экстрагировали простым эфиром (100 мл) для удаления некоторых триалкилированных соединений и липофильных загрязнений, оставляя моно- и желаемые диалкилированные продукты в водном слое. Водный раствор забуферивали ацетатом аммония (2 экв., 4,3 г, 55,8 ммоль) для обеспечения хорошей хроматографии. Водный слой хранили при 4°С в течение ночи перед очисткой с использованием автоматической препаративной ВЭЖХ.
Выход 2,2 г (6,4 ммоль, 23%).
Масс-спектр; положительный ион 10 В напряжение. Обнаружено 344; рассчитано М+Н=344.
ЯМР 1H (CDCl3): δ 1,24 (6Н, s, 2×CH3), 1,3 (6Н, s, 2×CH3), 1,25-1,75 (7Н, т, 3×CH2CH), (3Н, s, 2×CH2), 2,58 (4Н, m, CH2N), 2,88 (2Н, t, CH2N2), 5,0 (6Н, s, NH2, 2×NH, 2×OH).
ЯМР 1H ((CD3)2SO): δ 1,1 4×CH; 1,29, 3×CH2; 2,1 (4Н, t, 2×CH2).
ЯМР 13C ((CD3)2SO): δ 9,0 (4×CH3), 25,8 (2×CH3), 31,0 2×CH2, 34,6 CH2, 56,8 2×CH2N; 160,3, C=N.
Условия ВЭЖХ: скорость потока 8 мл/мин с использованием колонки PRP 25 мм.
А=3% раствор аммиака (относит. плотн. 0,88) в воде.
Б=ацетонитрил.
Для проведения хроматографии вводят 3 мл водного раствора и собирают в диапазоне времени 12,5-13,5 мин.
Пример 5: получение Соединения 2 - бензамидного конъюгата Соединения 1
Соединение 1 (0,5 г, 1,45 ммоль) в безводном ацетонитриле (50 мл) и триэтиламине (150 мг, 1,45 ммоль) в атмосфере азота охлаждали на ледяной бане до 0°С. Добавляли бензангидрид (330 мг, 1,45 ммоль) к перемешиваемой реакционной смеси, давали нагреться до комнатной температуры и оставляли перемешиваться в течение ночи. Ацетонитрил удаляли в вакууме и остаток снова растворяли в 50 мл дихлорметана, промывали водным карбонатом калия (2×50 мл), разделяли и сушили над сульфатом натрия. Водный раствор карбоната калия экстрагировали дихлорметаном (2×50 мл), сушили над сульфатом натрия и объединенные дихлорметановые экстракты концентрировали в вакууме до смолы. Аналитическая ВЭЖХ указывала на то, что продукт не являлся чистым, как требуется, и материал поэтому очищали с использованием автоматической препаративной ВЭЖХ с получением Соединения 2. Продукт аналитически выявлялся в виде одного пятна на ТСХ и аналитической ВЭЖХ.
Условия ВЭЖХ: скорость потока 8 мл/мин с использованием колонки PRP 150 мм×25 мм.
Образец для одного анализа вводится в объеме 2 мл 30% этанола в воде.
А=3% раствор аммиака (относит. плотн. 0,88) в воде.
Б=ацетонитрил.
Требуемый продукт элюировали на 15,25-16,5 мин. Раствор продуктавыпаривали в вакууме с получением (304 мг, 0,68 ммоль, 47%) бесцветной стекловидной пены, т. пл. 55°С.
ЯМР 1H (CDCl3): 1,26 (12Н, s, 4×CH3), 1,43 (2Н, m, CH2), 1,57 (4Н, m, CH2), 1,75 (1H, m, СН), 1,823 (6Н, s, 2×CH3), 2,58 (4Н, m, 2×CH2N), 3,56 (2Н, m, CH2NHCO), 6,95 (1H, m, NHCO), 7,42 (3H, m, 3×ArH) 7,79 (2Н, d, ArH).
ЯМР 13C (CDCl3): 10,09, 25,7, 26,1, 28,5, 32,8, 33,3, 37,93, 57,57, 127,0, 128,4, 131,4, 158,98, 168,15.
МС (C24H41N5O3) M+H=448, обнаружено 448.
RF 0,8 в 100:30:5 / CH2Cl2:MeOH:880 аммиак, визуализировано с использованием нингидрина.
Пример 6: синтез бис[(1,1-диметил-2-N-гидроксииминпропил)-2-аминоэтил]-(2-(глутариламид)этил)метана [Соединение 4; глутариламидное производное Соединения 1]
Соединение 1 (0,5 г, 1,45 ммоль) в безводном ацетонитриле (50 мл) и триэтиламине (150 мг, 1,45 ммоль) в атмосфере азота охлаждали на ледяной бане до 0°С. Добавляли глутаровый ангидрид (165 мг, 1,45 ммоль) к перемешиваемой реакционной смеси, давали нагреться до комнатной температуры и оставляли перемешиваться в течение ночи. Образовавшийся за ночь осадок собирали путем фильтрации и сушили в вакууме с получением неочищенного образца указанного в заголовке соединения (267 мг, 0,583 ммоль, 40%). Фильтрат концентрировали в вакууме с получением бесцветного стекла, которое вместе с осадком, который был собран, вновь растворяли в 5% аммиаке, 0,880 относит. плотн., воде (50 мл) и очищали с использованием автоматической препаративной ВЭЖХ.
Условия ВЭЖХ: скорость потока 8 мл/мин с использованием колонки PRP 150 мм×25 мм.
Образец для одного анализа вводили в объеме 2 мл раствора.
А=3% раствор аммиака (относит. плотн. 0,88) в воде.
Б=ацетонитрил.
Требуемый продукт элюировали на 15,25-16,5 мин. Раствор продукта выпаривали в вакууме с получением (304 мг, 0,68 ммоль, 47%) бесцветной стекловидной пены, т. пл. 54,8°С. Продукт аналитически выявляли в виде одного пятна на ТСХ и аналитической ВЭЖХ.
ЯМР 1H (диметилсульфоксид (ДМСО)): 0,7 (12Н, s, 4×CH3), 0,85 (4Н, m, 2×CH2), 1,0 (1Н, m, СН), 1,3 (6Н, s, 2×CH3), 1,3 (4Н, m, 2×CH2), 1,6 (2Н, m, CH2), 1,75 (6Н, m, 3×CH3), 2,6 (2, m, 2×OH), 3,2 (2Н, t, NH), 7,3 (1Н, t, NH).
ЯМР 13C (CD3SO): 8,97, 20,51, 20,91, 25,09, 25,60, 31,06, 33,41, 33,86, 56,89, 66,99, 160,07, 1712,34, 174,35, 174,56.
МС (C22H43N5O5) М+Н=457, обнаружено 457,6.
Пример 7: синтез защищенного пептида Ац-NQEQVSP(3-I)YTLLKG
Защищенный пептид AC-Asn(Trt)-Gln(Trt)-Glu(OtBu)-Gln(Trt)-Val-Ser(tBu)-Pro-Tyr(3l)-Thr(tBu)-Leu-Leu-Lys(Boc)-Gly-OH закрепляли на 2-хлортритиловой твердофазной смоле путем закрепления Fmoc-Gly- на смоле и затем осуществляли последовательные циклы удаления защиты/сочетания с использованием подходящих защищенных аминокислот и агентов сочетания DCCl и 1-гидроксибензотриазол (HOBt). Концевой аспарагин ацетилировали, отщепляли от смолы с использованием 0,5% трифторуксуной кислоты (TFA) и пептид использовали без дополнительной очистки.
Пример 8: синтез Соединения 5 - пептидного конъюгата Соединения 1
Защищенный пептид Ac-NQEQVSPY(3I)TLLKG по Примеру 7 отщепляли от твердофазной смолы и затем связывали с Соединением 1 в растворе, используя гексафторфосфат бензотриазол-1-илокситриспирролидинофосфония и 1-гидроксибензотриазол в качестве агентов сочетания. Соединение 5 получали путем удаления защиты в реактиве К (реактив К представляет собой 82,5% TFA, 5% фенола, 5% обработанной воды, 5% тиоанизола, 2,5% этандитиола). Неочищенный пептид сначала очищали путем ВЭЖХ с обращенной фазой с использованием TFA с последующей второй очисткой и солевым обменом с уксусной кислотой, лиофилизацией, фильтрованием через фильтр 0,22 мкм и окончательной лиофилизацией с получением Соединения 5.
Аза-диаминдиоксимовый хелатный конъюгат того же самого пептида по предшествующему уровню техники (Соединение 6, показано на фиг.1), т.е. Ас-NQEQVSPY(3I)TLLKG был для сравнения получен тем же самым образом.
Пример 9: получение диоксима 1-(1-{3-(2-аминоэтил)-5-[1-(1-гидроксилиминоэтил)циклогексиламино]пентиламино}циклогексил)этанона [Соединение 7]
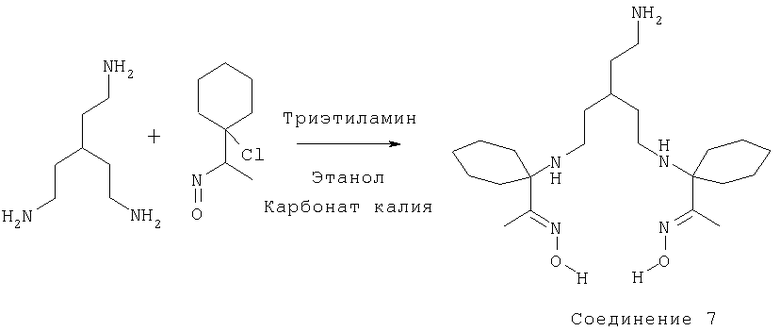
К раствору 1,1,1-трис(2-аминоэтил)метана (0,96 г, 6,6 ммоль) в безводном этаноле (7,5 мл) добавляли карбонат калия (безводный) (1,8 г, 13 ммоль) и триэтиламин (1,33 г, 13 ммоль) при комнатной температуре и интенсивном перемешивании в атмосфере азота. В течение 1 ч по каплям добавляли раствор 1-хлор-1-(1-нитрозоэтил)циклогексана (2,3 г, 13 ммоль) в дихлорметане (30 мл). Смесь затем оставляли перемешиваться при комнатной температуре в течение 18 ч. Растворитель затем удаляли при пониженном давлении. Затем в реакционный остаток добавляли воду (30 мл) и простой эфир (25 мл). Водную фазу и органическую фазу затем разделяли.
ВЭЖХ:
изократический: 90% Б (МеОН), 10% (NH3 3%). Эфирный экстракт: в соответствии с ВЭЖХ наблюдали две основные зоны - первая зона: диоксим, вторая зона: триоксим. Диоксим: (0,55 г, 20%), бомбардировка быстрыми атомами (FAB) m/z 424 (М+Н), масс-спектрометрия высокого разрешения (HRMS): обнаружено 424,3642, рассчитано 424,3652 (C23H45N5O2).
ЯМР δH (CDCl3; 270 МГц): 1,34-1,72 (33Н, m, CH, CH2×13, CH3×2), 2,18-2,33 (4Н, m, NCH2×2), 2,56-2,69 (2Н, m, NCH2).
Соединение 1-(1-{3-(2-аминоэтил)-5-[1-(1-гидроксилиминоэтил)циклогексиламино]пентиламино}циклогексил)этанон диоксим [Соединение 8] получали аналогичным способом.
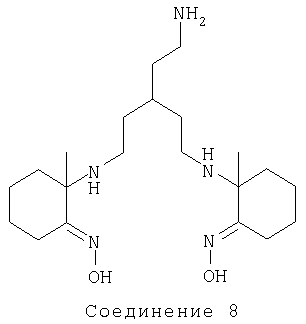
FAB, m/z 396 (M+H), MCBP, обнаружено 396,3322, рассчитано 396,3339 (C21H42N5O3).
Пример 10: введение радиоактивной метки 99mTc в Соединение 2 по сравнению с соответствующим аза-аналогом (Соединение 3, предшествующий уровень техники)
Сублимационно высушенный препарат, содержащий
23 мкг Соединения 2 (бензамидное производное Соединения 1 - описано в Примере 3),
36 мкг дигидрата хлорида двухвалентного олова,
90 мкг тринатриевого медроната,
4,0 мг ацетата натрия,
помещали в стеклянный сосуд на 10 мл, укупоривали в атмосфере азота (фармакопея США/национальный формуляр).
Его восстанавливали 99mTc-пертехнетатом в рассоле, полученным из генератора 99mTc, при комнатной температуре, и радиохимическую чистоту (РХЧ) изучали с использованием ВЭЖХ и мгновенной тонкослойной хроматографии (МТСХ). Результаты сравнивали с результатами, полученным для Соединения 3, и они приведены в таблицах 1 и 2.
Результаты определения радиохимической чистоты в соответствии с МТСХ (%)
Результаты определения радиохимической чистоты для Соединения 2 в соответствии с МТСХ и ВЭЖХ (%)
Пример 11: введение радиоактивной метки 99mTc в Соединение 5 - пептидный конъюгат Соединения 1
Сублимационно высушенный препарат, содержащий
50 мкг РАВА (пара-аминобензойной кислоты),
36 мкг SnCl2,
90 мкг MDP (метилендифосфоновой кислоты),
1,32 мг NaHCO3,
98 мкг Na2CO3,
4,0 мг NaOAc,
укупорили в атмосфере азота в стеклянном сосуде на 10 мл. Сосуд извлекали из холодильника и оставляли при комнатной температуре на 15 минут, а затем растворяли в 100 мкл раствора, содержащего Соединение 5, т.е. пептид-хелатный конъюгат Ac-Asn-Gln-Glu-Gln-Val-Ser-Pro-(I-Tyr)-Thr-Leu-Leu-Lys-Gly-[Соединение 1] (2 мг в 2 мл воды) и Х мл 99mТс-пертехнетата в рассоле с радиоактивной концентрацией 0,5 ГБк/мл, полученного из генератора 99mTc Amertec II, при комнатной температуре. Активность измеряли с использованием ионной камеры. РХЧ измеряют с использованием МТСХ и ВЭЖХ. Результаты для различных значений Х представлены в таблице 3.
Результаты определения радиохимической чистоты для Соединения 5 (%) с использованием МТСХ и ВЭЖХ
Пример 12: захват 99mTc-меченного Соединения 5 сгустком in vitro по сравнению с захватом 99mТс-меченного Соединения 6 (предшествующий уровень техники)
Введение радиоактивной метки 99mTc осуществляли в соответствии с Примером 10.
Плазму (5 мл на тестируемый образец) и тромбин (100 единиц мл-1) извлекали из морозильника (-20°С) и давали им разморозиться до комнатной температуры. Плазму проверяли перед использованием, чтобы убедиться, что в ней не наблюдается образования сгустков или разрушения образца. В один флакон на 5 мл с плазмой (крысы, кролика, собаки и человека) добавляли 10 мкл 99mTc-меченного Соединения 5 или 99mТс-меченного Соединения 6. Во второй флакон, содержащий 5 мл плазмы, параллельно в качестве отрицательного контроля добавляли 10 мкл 99mТс-меченной диэтилентриаминпентауксусной кислоты (DTPA). Образующие сгустки инкубационные смеси получали путем добавления 800 мкл кальциевого трис-буфера и 40 мкл раствора бычьего тромбина в четыре флакона (образующий сгустки буфер, обогащенный кальцием/тромбином). При не образующей сгустки инкубации фоновые смеси для анализа связывания получали путем добавления 800 мкл забуференного трис раствора рассола к 40 мкл воды AnalaR (не образующий сгустки буфер, обедненный кальцием/тромбином).
В четырех параллелях как в обогащенные кальцием/тромбином, так и обеденные кальцием/тромбином инкубационные смеси добавляли 400 мкл плазмы человека с введенным в нее тестируемым образцом (99mТс-меченное Соединение 5 или 99mТс-меченное Соединение 6) или радиоактивно меченный отрицательный контроль (99mTc-DTPA). В каждый флакон добавляли дефибринирующую палочку для облегчения сворачивания плазмы крови. Анализируемые флаконы инкубировали при температуре окружающей среды в течение 1 часа. Реакцию останавливали путем добавления в каждый флакон Р7 1 мл 0,4 М раствора этилендиаминтетрауксусной кислоты (EDTA).
Общую радиоактивность определяли (в четырех параллелях) путем добавления 400 мкл образцов плазмы крови с предварительно введенным тестируемым образцом и отрицательных контролей в отдельные стеклянные сцинтилляционные флаконы. Радиоактивность, связанную с этими стандартами, определяли с помощью натриево-иодидной гамма-топографии. Содержимое каждого флакона Р7 выливали на отдельные блокированные бычьим сывороточным альбумином (БСА) нитроцеллюлозные фильтры над вакуумным коллектором. Каждый флакон Р7 ополаскивали 2 мл раствора TBST (рассол, забуференный трис, 0,05% Tween 20). Каждый фильтр затем ополаскивали четыре раза 5 мл раствора TBST. Сгустки сушили в течение 1 часа над вакуумным коллектором. Фильтровальную бумагу затем переносили в отдельные сцинтилляционные флаконы и определяли присутствующую радиоактивность.
Неспецифическое связывание тестируемого образца с нитроцеллюлозным фильтром учитывали путем вычитания общей радиоактивности, присутствующей в смеси, образующей сгустки, из общей радиоактивности, присутствующей в смеси, не образующей сгустки. Захват сгустком (специфический и неспецифический) выражали в виде процентного захвата тестируемого образца в плазме крови путем деления присутствующей в самом сгустке радиоактивности на усредненную радиоактивность, присутствующую в стандартах плазмы крови, и умножения на 100:
% захвата = % захвата сгустком на фильтре - % захвата на фильтре × 100
(фон скорректирован).
Процентное специфическое связывание определяли как захват радиоактивной метки, происходящий благодаря исключительно Фактору XIIIa, образующему изопептидные ковалентные связи между фибрином и тестируемым образцом. Специфическое связывание рассчитывали путем вычитания фонового (нитроцеллюлозный фильтр) скорректированного процентного захвата радиоактивно меченного отрицательного контроля (99mТс-DPTA), который не обладал сродством в отношении Фактора XIIIa, из фонового (нитроцеллюлозный фильтр) скорректированного процентного захвата радиоактивно меченного тестируемого образца:

Влияния на эффективность in vitro
Данные сравнивали захват 99mТс-меченного Соединения 5 и 99mТс-меченного Соединения 6 формирующимся сгустком плазмы in vitro. He наблюдали существенного различия (p>0,05) в отношении эффективности этих двух молекул (30,66±5,01 по сравнению с 29,69±6,33) в этих моделях сворачивания.
Пример 13: 99mTc-мечение Соединения 9
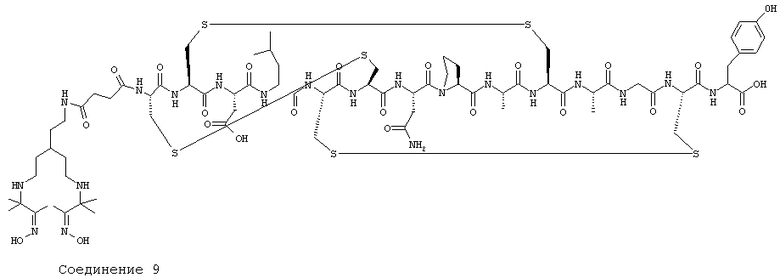
Соединение 9 представляет собой конъюгат Соединения 1 с изображенным циклическим петидом, т.е. [Соединение 1]-Cys-Cys-Glu-Leu-Cys-Cys-Asn-Por-Ala-Cys-Ala-Cys-Tyr-OH.
Соединение 9 получали аналогично Примерам 7 и 8 и метили 99mТс в растворе (Получение 1) или с помощью сублимационно высушенного набора в соответствии с Примером 10 (Получение 2).
Для Получения 1 100 мкг Соединения 9 растворяли в 1 мл боратного буфера, рН 8,5. Его переносили во флакон Р6 и закрывали. При комнатной температуре добавляли 1 мл 99mTc-пертехнетата в рассоле (1,0 ГБк/мл, из генератора Amertec II) вместе с 0,1 мл раствора SnCl2 (10 мг SnCl2 в 100 мл продуваемого N2 рассола). Активность измеряли с использованием ионной камеры. РХЧ измеряют с использованием МТСХ и ВЭЖХ.
Получение 1 позволяло получить РХЧ 96% в соответствии с МТСХ и Получение 2 позволяло получить РХЧ 82% в соответствии с ВЭЖХ.
Авторами изобретения были также получены непептидные конюъгаты Соединения 1, примеры получения которых были приведены в других патентных заявках, и эти примеры приведены ниже.
Так, приведенные ниже Примеры 14 и 15 соответствуют Примерам 13 и 14 из WO 04/032936, где описано получение конъюгатов с барбитуратными ингибиторами металлопротеиназ (MMPi).
Примеры 16-18 соответствуют Примерам 2-4 из WO 04/052357, где описано получение конъюгатов с антагонистами фагоцитарного рецептора макрофагов A (Macrophage Scavenger Receptor A (MSRA)).
Примеры 19, 20, 21, 22 и 23 соответствуют Примерам 1, 3, 5, 8 и 9 из WO 04/062568, где описано получение конъюгатов с непептидными соединениями, имеющими сродство к рецептору ангиотензина II.
Примеры 24, 25, 26 и 27 соответствуют Примерам 7, 8, 9 и 10 из WO 04/069365, где описано получение конъюгатов с MMPi.
Примеры 28 и 29 соответствуют примерам 9 и 11 из WO 05/049005, где описано получение конъюгатов с MMPi.
В Примере 30 описано получение конъюгата с соединением, которое связывается с лизилоксидазными ферментами (LOX).
В приведенных Примерах 14-30 Хелатообразователь 1 (имеющий также обозначение cPn216) представляет собой Соединение 1, получение которого описано в Примере 4; PEG - полиэтиленгликоль.
Примеры 14 и 15: получение конъюгатов с барбитуратными ингибиторами матриксных металлопротеиназ (MMPi) общей формулы
Пример 14: 3-(4-{5-[4-(4-бромфенокси)Фенил]-2,4,6-триоксогексагидропиримидин-5-ил}пиперазин-1-ил)-N-{5-(2-гидроксилимино-1,1-диметилпропиламино)-3-[2-(2-гидроксиламино-1,1-диметилпропиламино)этил]пентил}пропионамид (Соединение 10)
Стадия (а): 5-[4-(4-бромфенокси)фенил]-5-[4-(2-карбоксиэтил)пиперазин-1-ил]пиримидин-2,4,6-трион
200 мг (440 мкмоль) 5-бром-5-[4-(4-бром-фенокси)фенил]пиримидин-2,4,6-триона растворяли в 2 мл абсолютного метанола и обрабатывали 83,5 мг (5,28 ммоль, 1,2 экв.) 3-(пиперазин-1-ил)пропионовой кислоты. Эту реакционную смесь нагревали до дефлегмации в течение 6 ч и затем концентрировали. Желтый твердый остаток перекристаллизовывали из воды с получением 170 мг (320 мкмоль, 72%) бесцветного аморфного твердого вещества. Т. пл. 208-210°С (разложение).
1H-ЯМР (300 МГц, ДМСО-D6): δ [м.д.]: 7.64 (широкий, d, 3J=8.6 Гц, 2Н, HAryl), 7.50 (широкий, d, 3J=8.6 Гц, 2Н, HAryl), 7.14 (широкий, d, 3J=8.6 Гц, 2Н, HAryl), 7.10 (широкий, d, 3J=8.6 Гц, 2Н, HAryl), 2.75-2.39 (m, 12H, CH2).
Стадия (б): 3-(4-{5-[4-(4-бромфенокси)фенил]-2,4,6-триоксогексагидропиримидин-5-ил}пиперазин-1-ил)-N-{5-(2-гидроксилимино-1,1-диметилпропиламино)-3-[2-(2-гидроксиламино-1,1-диметилпропиламино)этил]пентил}пропионамид (Соединение 10)
К раствору соединения со стадии (а) (53 мг) в N,N-диметилформамиде (10 мл) в атмосфере азота последовательно добавляли O-(бензотриазол-1-ил)-N,N,N',N'-тетраметилурония тетрафторборат (TBTU) (85 мг) и N-метилморфолин (0,01 мл). Через десять минут добавляли Хелатообразователь 1 (35 мг) и эту реакционную смесь перемешивали при комнатной температуре в течение 24 часов. Растворитель удаляли при пониженном давлении и смесь растворяли в метаноле (5 мл). Неочищенную смесь разделяли ВЭЖХ. Продукт элюировался примерно через 10 минут (выход 75%). Структура была подтверждена масс-спектральным [электрораспылительная ионизация с регистрацией положительных ионов (ЭРИ(+)), 858,1] и 1Н ЯМР анализом.
Пример 15: синтез Соединения 11
Стадия (а): 5-[4-(2-аминоэтил)пиперазин-1-ил]-5-[4-(4-бромфенокси)фенил]пиримидин-2,4,6-трион
200 мг (440 мкмоль) 5-бром-5-[4-(4-бромфенокси)фенил]пиримидин-2,4,6-трион растворяли в 2 мл абсолютного метанола и обрабатывали 125 мг (127 мкл, 9,67 мкмоль) N-(2-аминоэтил)пиперазина. Эту реакционную смесь перемешивали при комнатной температуре и примерно через 30 минут образовался бесцветный осадок. Перемешивание продолжали в течение 16 ч, затем осадок собирали отсасыванием и сушили в вакууме с получением 100 мг (200 мкмоль, 45%) бесцветного твердого вещества, т. пл. 220-223°С (разложение).
1H-ЯМР (300 МГц, ДМСО-D6): δ [м.д.]: 7.67 (широкий, d, 3J=9.0 Гц, 2Н, HAryl, орто к C-Br), 7.55 (широкий, d, 3J=9.0 Гц, 2Н, HAryl, орто к Cquart., присоединен к Pyr.-С 5), 7.15 (широкий, d, 3J=9.0 Гц, 2Н, HAryl, мета к Cquart, присоединен к Pyr.-C 5), 7.12 (широкий, d, 3J=9.0 Гц, 2Н, HAryl, мета к C-Br), 2.89-2.79 (m, 2H, CH2-NH2), 2.77-2.65 (m, 4H, N 1-CH2), 2.39-2.58 (m, 6H, N 4-CH2).
Стадия (б): 4-[2-(4-{5-[4-(4-бромфенокси)фенил]-2,4,6-триоксогексагидропиримидин-5-ил}пиперазин-1-ил)этилкарбамоил]-1-масляная кислота
К раствору соединения со стадии (а) в N,N-диметилформамиде (30 мл) в атмосфере азота последовательно добавляли глутаровый альдегид (11 мг) и триэтиламин (0,01 мл). Через 24 часа растворитель удаляли при пониженном давлении. Неочищенную смесь растворяли в метаноле (5 мл) и разделяли ВЭЖХ. Продукт элюировался через 12 минут (выход 50%). Структура была подтверждена масс-спектральным [ЭРИ(+) 617,9] и 1Н ЯМР-анализом.
Стадия (в): конъюгирование 4-[2-(4-{5-[4-(4-бромфенокси)фенил]-2,4,6-триоксогексагидропиримидин-5-ил}пиперазин-1-ил)этилкарбамоил]масляной кислоты с Хелатообразователем 1.
К раствору соединения со стадии (б) (11 мг) в дихлорметане (5 мл) добавляли TBTU (8 мг) и N-метилморфолин (0,1 мл) в атмосфере азота. Через 5 минут добавляли Хелатообразователь 1 (6 мг) и эту смесь перемешивали в течение 24 часов. Растворитель удаляли при пониженном давлении и смесь растворяли в метаноле (5 мл). Смесь разделяли ВЭЖХ и продукт элюировался примерно через 10 минут (выход 58%). Структура была подтверждена масс-спектральным [ЭРИ(+) 943,2] и 1H ЯМР-анализом.
В приведенных ниже Примерах 16-18 описано получение конъюгатов с антагонистами фагоцитарного рецептора макрофагов A (Macrophage Scavenger Receptor A (MSRA)) (см. WO 04/052357).
Пример 16: присоединение хелатообразователя 1 к 4-карбокси-N-(4-бромфенил)-2-(4-хлорфенилсульфониламидо)бензамиду с образованием Соединения 12
К раствору 4-карбокси-N-(4-бромфенил)-2-(4-хлорфенилсульфониламидо)бензамида (1 мг) в дихлорметане (2 мл) при комнатной температуре добавляли 4 эквивалента TBTU и 1,1 эквивалент хелатообразователя 1 и 3 эквивалента N,N-диизопропилэтиламина (DIEA) в атмосфере азота в течение 24 часов. Неочищенную смесь очищали ВЭЖХ.
Масс-спектрометрический анализ: ЭРИ [М+Н] m/z 836.
Пример 17: присоединение хелатообразователя 1 к 4-бром-N-(4-бромфенил)-2-(4-карбоксифенилсульфониламидо)бензамиду с образованием соединения 13
Для присоединения хелатообразователя 1 к 4-бром-N-(4-бромфенил)-2-(4-карбоксифенилсульфониламидо)бензамиду с образованием соединения 13 использовали способ, описанный в Примере 16.
Пример 18: присоединение хелатообразователя 1 к 4-бром-N-(4-карбоксифенил)-2-(4-хлорфенилсульфониламидо)бензамиду с образованием соединения 14
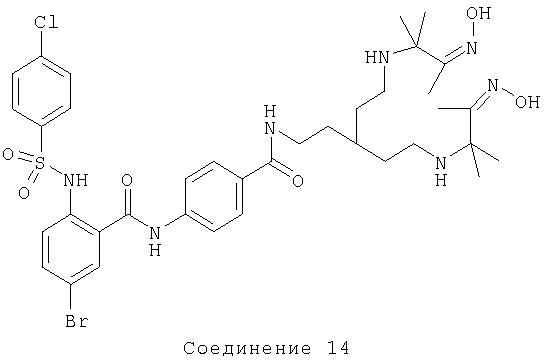
Для присоединения хелатообразователя 1 к 4-бром-N-(4-карбоксифенил)-2-(4-хлорфенилсульфониламидо)бензамиду с образованием предшественника 3 использовали способ, описанный в Примере 16.
В приведенных ниже Примерах 19-23 описано получение конъюгатов с антагонистами рецептора ангиотензина II, в частности с лозартаном (см. WO 04/062568).
Пример 19: аминофункционализированный лозартан
а) Замещение гидроксильной группы лозартана азидогруппой
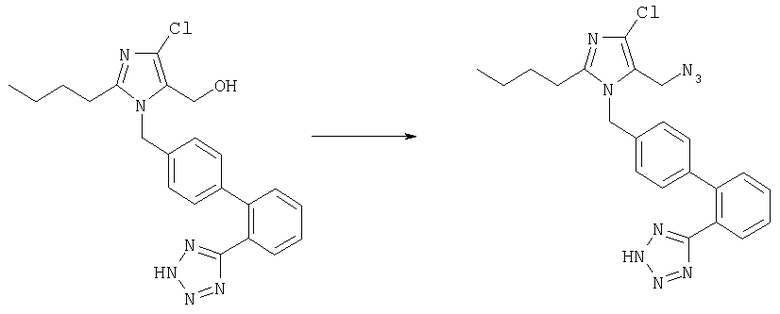
К перемешиваемой суспензии лозартана (MSD, 0,423 г, 1,00 ммоль) и дифенилфосфорилазида (Aldrich, 0,259 мл, 1,20 ммоль) в тетрагидрофуране (8 мл) добавляли 1,8-диазабицикло[5,4,0]ундец-7-ен (DBU) (0,329 мл, 2,20 ммоль). После перемешивания в течение ночи добавляли воду/ацетонитрил (1:1, 4,8 мл) и эту смесь фильтровали. После добавления неразбавленной TFA (трифторуксусная кислота) (до рН 2) смесь очищали препаративной ВЭЖХ (колонка Phenomenex Luna C18(2) 5 мкм 21,2×250 мм, растворители: А=вода/0,1% TFA и В=ацетонитрил/0,1% TFA; градиент 35-45% В за 60 минут; скорость потока 10,0 мл/мин, УФ детектирование при 214 нм) в несколько приемов с получением после лиофилизации 99 мг (22%) продукта в виде белых кристаллов. Анализ жидкостной хроматографией-масс-спектрометрией (ЖХ-МС) (колонка Phenomenex Luna C18(2) 3 мкм 50×4,60 мм, растворители: А=вода/0,1% TFA и В=ацетонитрил/0,1% TFA; градиент 20-80% В за 10 мин; скорость потока 1 мл/мин, УФ детектирование при 214 нм, ЭРИ-МС (масс-спектрометрия с электрораспылительной ионизацией) дал пик при 7,3 минутах с m/z 448,1 (МН+), соответствующий структуре.
б) Восстановление азидогруппы до аминогруппы
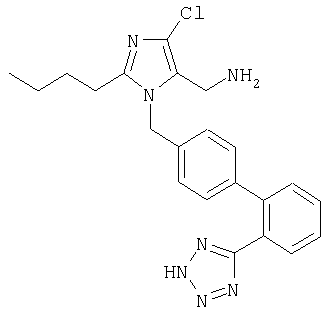
К раствору соединения со стадии (а) (5,0 мг, 0,011 ммоль) в метаноле (3 мл) добавляли Pd/C (Koch-Light, приблизительно 10 мг). Эту смесь перемешивали под водородом (1 атм (101,3 кПа)) в течение 10 мин, фильтровали и концентрировали. Остаток использовали на следующей стадии без дополнительной обработки. ЖХ-МС анализ (колонка Phenomenex Luna С18(2) 3 мкм 50 х 4,60 мм, растворители: А=вода/0,1% TFA и В=ацетонитрил/0,1% TFA; градиент 20-80% В за 10 мин; скорость потока 1 мл/мин, УФ детектирование при 214 нм, ЭРИ-МС) дал пик при 1,9 минутах с m/z 422,2 (МН+), соответствующий амину.
Пример 20: лозартан, связанный с глутаровой кислотой, модифицированной cPn216, для Тс-мечения
б) Синтез тетрафтортиофенилового эфира cPn216-глутаровой кислоты
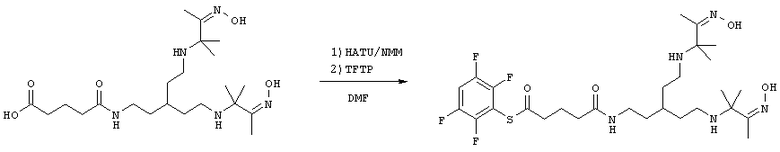
К Соединению 4 из Примера 6 (300 мг, 0,66 ммоль) в диметилформамиде (DMF) (2 мл) добавляли 2-(7-аза-1Н-бензотриазол-1-ил)-1,1,3,3-тетраметилурония гексафторфосфат (HATU) (249 мг, 0,66 ммоль) и N-метилморфолин (NMM) (132 мкл, 1,32 ммоль). Эту смесь перемешивали в течение 5 минут, затем добавляли тетрафтортиофенол (TFTP) (0,66 ммоль, 119 мг). Этот раствор перемешивали в течение 10 минут, затем реакционную смесь разбавляли смесью 20% ацетонитрила/вода (8 мл) и продукт очищали ОФ-ВЭЖХ (высокоэффективной жидкостной хроматографией с обращенной фазой) с получением после сублимационной сушки 110 мг целевого продукта.
в) Связывание активного эфира cPn216-глутаровой кислоты с аминопроизводным лозартана
К раствору аминопроизводного лозартана из Примера 19(б) (10 мкмоль) в DMF (1 мл) добавляли N-метилморфолин (3,3 мкл, 30 мкмоль), тетрафтортиофениловый эфир cPn216-глутаровой кислоты из (б) (6,8 мг, 11 мкмоль, получен из cPn216 и ангидрида глутаровой кислоты стандартными способами). Через 45 мин реакционную смесь концентрировали. Остаток переносили в ацетонитрил/воду и очищали препаративной ВЭЖХ (колонка Vydac 218ТР1022 С18 10 мкм, 22×250 мм, растворители: А=вода/0,1% TFA и В=ацетонитрил/0,1% TFA; градиент 10-40% В за 60 мин; скорость потока 10,0 мл/мин, УФ детектирование при 214 нм) с получением после лиофилизации 4,2 мг (49%) продукта. ЖХ-МС анализ (колонка Phenomenex Luna C18(2) 3 мкм 2,0×50 мм, растворители: А=вода/0,1% НСООН и В=ацетонитрил/0,1% НСООН; градиент 10-40% В за 10 мин; скорость потока 0,3 мл/мин, УФ детектирование при 214 и 254 нм, ЭРИ-МС) дал пик при 6,2 мин с m/z 861,6 (МН+), как и ожидалось. Дополнительно были определены параметры ЯМР-спектроскопии, подтверждающие структуру.
Пример 21: лозартан, связанный с глутаровой кислотой, модифицированной cPn216, для Тс-мечения
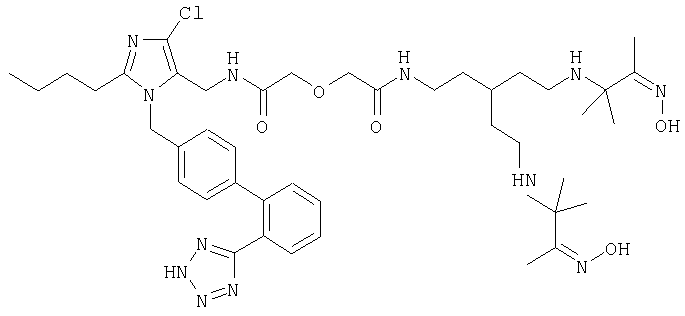
К раствору ацилированного дигликолевым ангидридом лозартана (WO 04/062568, Пример 4(а)) (5,4 мг, 0,010 ммоль) и HATU (3,8 мг, 0,010 ммоль) в DMF (1 мл) добавляли 2 М DIEA в NMP (15 мл, 0,030 ммоль). Эта реакционная смесь делалась желтой, и ее перемешивали в течение 15 мин. К активированной карбоновой кислоте добавляли раствор cPn216 (3,4 мг, 0,010 ммоль) в DMF (0,25 мл). Протекание реакции контролировали аналитической ВЭЖХ (колонка Phenomenex Luna С 18(2) 3 мкм 4,6×50 мм, растворители: А=вода/0,1% TFA и В=ацетонитрил/0,1% TFA; градиент 10-40% В за 10 мин; скорость потока 2,0 мл/мин, УФ детектирование при 214 и 254 нм). После добавления еще двух аликвот HATU была достигнута полная конверсия исходного вещества. Смесь очищали препаративной ВЭЖХ (колонка Phenomenex Luna C18(2) 5 мкм 21,2×250 мм, растворители: А=вода/0,1% TFA и В=ацетонитрил/0,1% TFA; градиент 10-40% В за 60 мин; скорость потока 10,0 мл/мин, УФ детектирование при 214 нм) с получением после лиофилизации 3,7 мг (43%) продукта. Анализ ЖХ-МС (колонка Phenomenex Luna C18(2) 3 мкм 2,0×50 мм, растворители: А=вода/0,1% НСООН и В=ацетонитрил/0,1% НСООН; градиент 10-40% В за 10 мин; скорость потока 0,3 мл/мин, УФ детектирование при 214 и 254 нм, ЭРИ-МС) дал пик при 6,1 мин с m/z 863,2 (МН+) в соответствии со структурой. Дополнительно структура была определена ЯМР-спектроскопией.
Пример 22: лозартан, связанный с PEG-глутаровая кислота-cPn216, для Тс-мечения
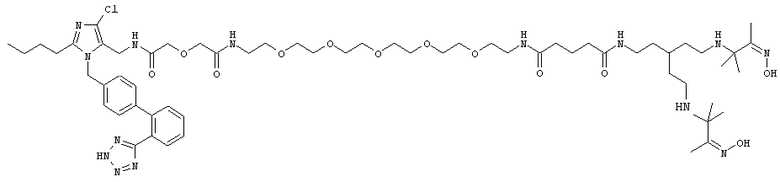
DIEA (2М в NMP, 8 мкл, 16 мкмоль) и раствор тетрафтортиофенилового эфира cPn216-янтарной кислоты из Примера 20 (б) (5 мг, 8 мкмоль) в DMF (0,5 мл) добавляли к раствору соединения

(Пример (6б) из WO 04/062568) (4 мкмоль) в DMF (1,5 мл). После перемешивания в течение ночи реакционную смесь разбавляли водой (3 мл) и рН доводили до 2 добавлением TFA. Продукт очищали препаративной ВЭЖХ (колонка Phenomenex Luna C18(2) 5 мкм 21,2×250 мм, растворители: А=вода/0,1% TFA и В=ацетонитрил/0,1% TFA; градиент 20-60% В за 60 мин; скорость потока 10,0 мл/мин, УФ детектирование при 214 нм) с получением после лиофилизации 2,0 мг (40%). МС анализ с прямым впрыском (растворители: А=вода/0,1% TFA и В=ацетонитрил/0,1% TFA; 50% В за 2 мин; скорость потока 0,3 мл/мин, ЭРИ-МС) дал m/z 1239,8 (МН+), соответствующий структуре.
Пример 23: лозартан, связанный с PEG-дигликолевая кислота-cPn216, для Тс-мечения
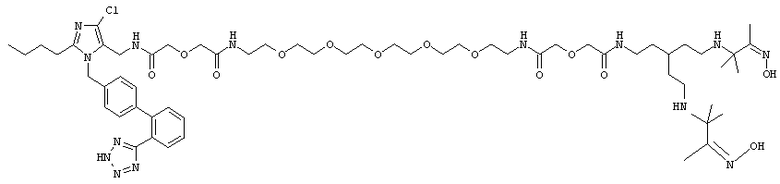
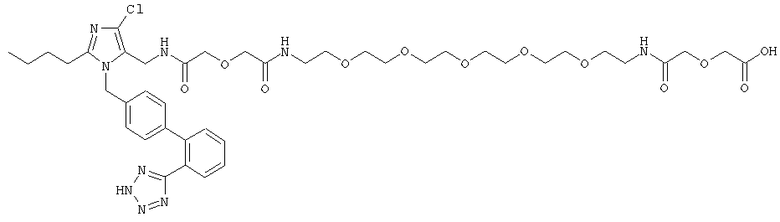
Указанное в Примере 22 соединение Примера (6б) из WO 04/062568 (5,0 мкмоль) растворяли в метаноле (2,5 мл) и рН доводили до 9 добавлением DIEA. К этому раствору добавляли дигликолевый ангидрид (Acros, 1,2 мг, 10 мкмоль). Через 50 минут реакционную смесь концентрировали и остаток очищали препаративной ВЭЖХ (колонка Phenomenex Luna C18(2) 5 мкм 21,2×250 мм, растворители: А=вода/0,1% TFA и В=ацетонитрил/0,1% TFA; градиент 20-60% В за 60 мин; скорость потока 10,0 мл/мин, УФ детектирование при 214 нм) с получением 4,5 мг (98%) продукта. МС-анализ с прямым впрыском (растворители: А=вода/0,1% TFA и В=ацетонитрил/0,1% TFA; 50% В за 2 мин; скорость потока 0,3 мл/мин, ЭРИ-МС; m/z 916,9 (МН+)) показал соответствие со структурой.
б) Конъюгирование cPn216
Соединение (а) (5 мкмоль) растворяли в DMF (2 мл) и активировали HATU (2 мг, 5 мкмоль) и DIEA (2M в NMP, 5 мкл, 10 мкмоль) в течение 5 минут. Добавляли cPn216 (3,4 мг, 10 мкмоль) и реакционную смесь перемешивали в течение ночи. Продукт очищали препаративной ВЭЖХ (колонка Phenomenex Luna C18(2) 5 мкм 21,2×250 мм, растворители: А=вода/0,1% TFA и В=ацетонитрил/0,1% TFA; градиент 20-60% В за 60 мин; скорость потока 10,0 мл/мин, УФ детектирование при 214 нм) с получением после лиофилизации 3,7 мг (57%). ЖХ-МС анализ (колонка Phenomenex Luna C18(2) 3 мкм 2,0×50 мм, растворители: А=вода/0,1% муравьиная кислота и В=ацетонитрил/0,1% муравьиная кислота; градиент 10-40% В за 10 мин; скорость потока 1 мл/мин, УФ детектирование при 214 нм, ЭРИ-МС) дал пик при 6,8 минутах с m/z 1241,2 (МН+), соответствующий структуре.
Примеры 24-27. Получение конъюгатов с соединениями (MMPi) общей формулы
Таблица 4
Пример 24: получение Соединений 15-18
24(а) 4-(1-трет-Бутоксикарбонил-2-метилпропилсульфамоилбензойной кислоты метиловый эфир
К перемешиваемому раствору 2-амино-3-метилмасляной кислоты трет-бутилового эфира (H-D-Val-OtBu·HCl) (500 мг, 2,38 ммоль) в ацетонитриле (20 мл) добавляли пиридин (767 мкл, 9,52 ммоль) при температуре окружающей среды. Быстро получался прозрачный и бесцветный раствор. Затем по каплям добавляли раствор 4-хлорсульфонилбензойной кислоты метилового эфира (670 мг, 2,86 ммоль) в ацетонитриле (6 мл), и эта смесь становилась светло-желтой, ее перемешивали при температуре окружающей среды в течение 4 часов. Реакцию контролировали тонкослойной хроматографией (ТСХ) (EtOAc/гексан, 1:1). Растворители выпаривали и добавляли этилацетат (50 мл) и насыщенный раствор бикарбоната натрия (10 мл). Смесь переносили в разделительную воронку и энергично встряхивали. Затем фазы разделяли и этилацетатную фазу экстрагировали один раз рассолом (10 мл), сушили (MgSO4), фильтровали и упаривали с получением неочищенного продукта. Использование флэш-хроматографии (этилацетат/гексан, 1:1) дало чистый продукт в виде белого твердого вещества. Выход 880 мг (99,55%).
24(б) 4-[(1-трет-Бутоксикарбонил-2-метилпропил)пиридин-3-илметилсульфамоил]бензойной кислоты метиловый эфир
К перемешиваемому раствору 4-(1-трет-бутоксикарбонил-2-метилпропилсульфамоил)бензойной кислоты метилового эфира со стадии (а) (884 мг, 2,38 ммоль) в диметилформамиде (30 мл) при температуре окружающей среды добавляли карбонат цезия (10,86 г, 33,34 ммоль). Затем к этой суспензии добавляли 3-пиколилхлорида гидрохлорид (546 мг, 3,33 ммоль) и реакционную смесь перемешивали при комнатной температуре в течение 24 часов, по истечении которых ТСХ (EtOAc/гексан, 1:1) показала, что реакция завершилась. Смесь упаривали до сухости и остаток перемешивали в этилацетате (50 мл). Этилацетатную фазу экстрагировали водой (1×50 мл), сушили (MgSO4), фильтровали и упаривали с получением неочищенного продукта в виде коричневого масла. Это масло очищали флэш-хроматографией с получением чистого продукта в виде бесцветного масла. Выход 800 мг (79,21%).
24(в) 4-[(1-Карбокси-2-метилпропил)пиридин-3-илметилсульфамоил]бензойной кислоты метиловый эфир
Эфир со стадии (б) (321 мг, 0,75 ммоль) растворяли в метиленхлориде (15 мл) и охлаждали до -10°С. Газ хлороводород барботировали в раствор в течение 10 минут. Реакционную смесь герметизировали, подогревали до комнатной температуры и перемешивали в течение 16 часов. Растворитель выпаривали и остаток выпаривали совместно с метиленхлоридом (2×10 мл) с получением продукта в виде белой пены (выход 278 мг, 83,73%).
24(г) 4-[(1-Гидроксикарбамоил-2-метилпропил)пиридин-3-илметилсульфамоил]бензойной кислоты метиловый эфир
Кислоту со стадии (в) (112 мг, 0,28 ммоль), 1-гидроксибензотриазол (39 мг, 0,29 ммоль), 4-метилморфолин (297 мкл, 1,4 ммоль) и O-трет-бутилдиметилсилил)гидроксиламин (124 мг, 0,84 ммоль) растворяли в метиленхлориде (8 мл). Добавляли N-[(диметиламино)пропил]-N'-этилкарбодиимида гидрохлорид (73 мг, 0,38 ммоль) и реакционную смесь перемешивали в течение 24 часов. Реакционную смесь разбавляли водой (10 мл) и экстрагировали метиленхлоридом (2×10 мл). Объединенные метиленхлоридные фазы сушили (Na2SO4) и фильтровали. К фильтрату добавляли несколько капель соляной кислоты в диоксане с получением прямо свободной гидроксамовой кислоты (108 мг).
24(д) 4-[(1-Гидроксикарбамоил-2-метилпропил)пиридин-3-илметилсульфамоилбензойная кислота
2 н. гидроксид натрия (200 мкл) добавляли к раствору эфира со стадии (г) (32 мг, 0,072 ммоль) в метаноле (1 мл) и эту смесь перемешивали при температуре окружающей среды. Через 30 минут смесь упаривали до сухости и остаток растворяли в воде (2 мл). Раствор делали кислотным, используя 2 н. HCl. Чистый продукт был получен после препаративной ВЭЖХ в виде белого порошка (выход 28 мг, 95,01%).
24(е) 4-[(1-Гидроксикарбамоил-2-метилпропил)пиридин-3-илметилсульФамоил]-N-{5-(2-гидроксиимино-1,1-диметилпропиламино)-3-[2-(2-гидроксиимино-1,1-диметилпропиламино)этил]пентил}бензамид (Соединение 18)
Кислоту со стадии (д) (12,5 мг, 0,031 ммоль), 1-гидроксибензотриазол (3,68 мг, 0,027 ммоль), 4-метилморфолин (17,4 мкл, 0,124 ммоль) и N-[(диметиламино)пропил]-N'-этилкарбодиимида гидрохлорид (8,06 мг, 0,042 ммоль) растворяли в диметилформамиде (2 мл). Добавляли хелатообразователь 1 (13 мг, 0,037 ммоль) и эту реакционную смесь перемешивали в течение 24 часов. После выпаривания растворителей неочищенный продукт прямо направляли на очистку ВЭЖХ (градиент 00-30-60). Продукт представлял собой светло-коричневую смолу (выход 2 мг, 9%).
Пример 25: получение Соединения 19
25(а) 2-(4-Метоксибензолсульфониламино)-3-метилмасляной кислоты трет-бутиловый эфир
К перемешиваемой суспензии 2-амино-3-метилмасляной кислоты трет-бутилового эфира (H-D-Val-OtBu·HCl) (500 мг, 2,38 ммоль) в ацетонитриле (20 мл) добавляли пиридин (767 мкл, 9,52 ммоль) при температуре окружающей среды. Быстро образовывался прозрачный и бесцветный раствор. Затем по каплям добавляли раствор 4-метоксибензолсульфонилхлорида (541 мг, 2,62 ммоль) в ацетонитриле (10 мл), смесь становилась светло-желтой, и ее перемешивали при температуре окружающей среды. ТСХ (EtOAc/гексан, 1:1) показала, что реакция закончилась через 3 часа. После выпаривания ацетонитрила остаток переносили в дихлорметан (30 мл) и экстрагировали один раз 10% раствором бикарбоната натрия (30 мл) и один раз водой (30 мл). Затем фазы разделяли и дихлорметановую фазу сушили (Na2SO4), фильтровали и упаривали с получением неочищенного продукта. Флэш-хроматография (этилацетат/гексан, 1:1) дала чистый продукт в виде белого твердого вещества. Выход 801 мг (93,90%).
25(б) 4-{[(1-трет-Бутоксикарбонил-2-метилпропил)-(4-метоксибензолсульфонил)амино]метил}бензойной кислоты метиловый эфир
К перемешиваемому раствору 2-(4-метоксибензолсульфониламино)-3-метилмасляной кислоты трет-бутилового эфира (140 мг, 0,41 ммоль) в ацетонитриле (5 мл) при температуре окружающей среды добавляли карбонат цезия (1,33 г, 4,10 ммоль). Затем к этой суспензии добавляли метил-4-(бромметил)бензоат (115 мг, 0,50 ммоль) и эту реакционную смесь перемешивали при 70°С в течение 1 часа, по истечении которого ТСХ (EtOAc/гексан, 1:1) показала, что реакция прошла до конца. После охлаждения до температуры окружающей среды смесь фильтровали для удаления избытка карбоната цезия и упаривали до сухости. Остаток очищали флэш-хроматографией (EtOAc/гексан, 1:1) с получением чистого продукта в виде светло-желтого масла. Выход 153 мг (76%).
25(в) 4-{[(1-трет-Бутоксикарбонил-2-метилпропил)-(4-метоксибензолсульфонил)амино]метил}бензойная кислота
Диэфир со стадии (б) (151 мг, 0,31 ммоль) растворяли в тетрагидрофуране (2 мл) и добавляли 4 н. LiOH (250 мкл) при температуре окружающей среды. Эту смесь нагревали до 60°С в течение 5 часов, когда ВЭЖХ-мониторинг показал, что гидролиз закончился. Смесь охлаждали до температуры окружающей среды и растворитель выпаривали. Остаток растворяли в воде и прозрачный раствор экстрагировали один раз диэтиловым эфиром. Затем водную фазу охлаждали до 5°С (лед/вода) и нейтрализовали 1 н. HCl и после этого ее экстрагировали этилацетатом (3×5 мл). Объединенные этилацетатные фазы экстрагировали водой (5 мл) и рассолом (5 мл), сушили (Na2SO4), фильтровали и выпаривали с получением продукта в виде белой пены. Выход 133 мг (90%).
25(г) 2-[(4-{5-(2-Гидроксиимино-1,1 -диметилпропиламино)-3-[2-(2-гидроксиимино-1,1-диметилпропиламино)этил]пентилкарбамоил}бензил)-4-метоксибензолсульфонил)амино]-3-метилмасляной кислоты трет-бутиловый эфир
К кислоте со стадии (в) (61 мг, 0,13 ммоль) в диметилформамиде (4 мл) добавляли N,N-диизопропилэтиламин (46 мкл, 0,26 ммоль), N-[(диметиламино)-1Н-1,2,3-триазоло[4,5-b]пиридин-1-илметилен]-N-метилметанаминия гексафторфосфата N-оксид, HATU (49 мг, 0,15 ммоль) и cPn216 (51 мг, 0,15 ммоль). Эту реакционную смесь перемешивали при температуре окружающей среды и через 1 час ВЭЖХ показала полную конверсию в новый продукт. Смесь упаривали до сухости и затем чистый продукт выделяли после флэш-хроматографии (хлороформ:метанол, 8/2) в виде белых кристаллов. Выход 66 мг.
25(д) 2-[(4-{5-(2-Гидроксиимино-1,1-диметилпропиламино)-3-[2-(2-гидроксиимино-1,1-диметилпропиламино)этил]пентилкарбамоил}бензил)-(4-метоксибензолсульфонил)амино]-3-метилмасляная кислота
Дихлорметан (4 мл) добавляли к трет-бутиловому эфиру со стадии (г) (64 мг, 0,08 ммоль) и в раствор молочного цвета, который был получен, при температуре окружающей среды барботировали газ хлороводород в течение 10 минут. Смесь упаривали до сухости и остаток выпаривали совместно с дихлорметаном (5×5 мл) с получением продукта в виде не совсем белого твердого вещества. Выход 58 мг (97%). М+1=747.
25(е) 4-{[(1-Гидроксикарбамоил-2-метилпропил)-(4-метоксибензолсульфонил)амино]метил}-N-{5-(2-гидроксиимино-1,1-диметилпропиламино)-3-[2-(2-гидроксиимино-1,1-диметилпропиламино)этил]пентил}бензамид (Соединение 19)
Гидроксамовую кислоту присоединяли посредством трет-бутилдиметилсилилзащищенного промежуточного соединения. Так, смесь кислоты со стадии (д) (57 мг, 0,76 ммоль), 4-метилморфолина (34 мкл, 0,30 ммоль), PyAOP [7-азабензотриазол-1-илокситрис(пирролидино)фосфонийгексафторфосфата] (40 мг, 0,076 ммоль) и O-(трет-бутилдиметилсилил)гидроксиламина (12 мг, 0,08 ммоль) в диметилформамиде (4 мл) перемешивали при температуре окружающей среды и реакцию контролировали ВЭЖХ. Реакцию останавливали через 3 часа и растворители выпаривали. Остаток снова растворяли в дихлорметане и при температуре окружающей среды газ хророводород барботировали через эту смесь в течение 10 минут. Смесь упаривали до сухости и остаток выпаривали совместно с дихлорметаном (5×5 мл). После ВЭЖХ был получен продукт в виде белого порошка, М+Н, 762. Выход 15 мг.
Пример 26: получение Соединения 23
26(а) 2-[[4-(2-{2-[2-(2-Азидоэтокси)этокси]этокси}этилкарбамоил)бензил]-(4-метоксибензолсульфонил)амино]-3-метилмасляной кислоты трет-бутиловый эфир
К кислоте со стадии (в) Примера 25 (140 мг, 0,30 ммоль) в диметилформамиде (6 мл) добавляли N,N'-диизопропилэтиламин (104,51 мкл, 0,60 ммоль), N-[(диметиламино)-1Н-1,2,3-триазоло[4,5-b]пиридин-1-илметилен]-N-метилметанаминия гексафторфосфата N-оксид, HATU (114 мг, 0,30 ммоль) и 2-{2-[2-(2-азидоэтокси)этокси]этокси}этиламин (65,50 мг, 0,30 ммоль). Эту реакционную смесь перемешивали при температуре окружающей среды и через 2 часа ВЭЖХ показала полную конверсию в новый продукт. Смесь выпаривали до сухости и затем после флэш-хроматографии (этилацетат) был выделен чистый продукт в виде бесцветного масла. Выход 142 мг (70%).
26(б) 2-[[4-(2-{2-[2-(2-Аминоэтокси)этокси]этокси}этилкарбамоил)бензил]-(4-метоксибензолсульФонил)амино]-3-метилмасляной кислоты трет-бутиловый эфир
К перемешиваемому раствору азида со стадии (а) (200 мг, 0,29 ммоль) в тетрагидрофуране (5 мл), который охлаждали до 0°С (лед/H2O), добавляли трифенилфосфин (84 мг, 0,32 ммоль). После перемешивания при этой температуре в течение 5 минут охлаждающую баню удаляли и перемешивание продолжали при температуре окружающей среды в течение 19 часов. Затем добавляли воду (200 мкл) и анализ (ВЭЖХ) после 15 часов показал, что гидролиз прошел до конца. Растворители выпаривали и маслянистый остаток очищали флэш-хроматографией с использованием сначала CHCl3/метанола (8:2), а затем CHCl3/MeOH/H2O с образованием данного соединения в виде бесцветного масла. Выход 134 мг (71%). М+652.
26(в) 2-[{4-[2-{2-(2-[2-(4-Карбоксибутириламино)этокси]этокси}этокси)этилкарбамоил]бензил}-(4-метоксибензолсульфонил)амино]-3-метилмасляной кислоты трет-бутиловый эфир-СА1
Раствор амина со стадии (б) (73 мг, 0,11 ммоль), N,N-диизопропилэтиламина (39 мкл, 0,22 ммоль) и активного эфира 4-{5-(2-гидроксиимино-1,1-диметилпропиламино)-3-[2-(2-гидроксиимино-1,1-диметилпропиламино)этил]пентилкарбамоил}тиомасляной кислоты 2,3,5,6-тетрафторфенилового эфира (б; 85 мг, 0,11 ммоль) в диметилформамиде (5 мл) перемешивали при температуре окружающей среды в течение 2 часов, когда ВЭЖХ показала, что реакция прошла до конца. Смесь выпаривали до сухости и остаток очищали флэш-хроматографией (CHCl3/МеОН, 8:2) с получением продукта в виде смолы. Выход 54 мг (45%).
26(г) 2-[{4-[2-(2-{2-[2-(4-Карбоксибутириламино)этокси]этокси}этокси)этилкарбамоил]бензил}-(4-метоксибензолсульфонил)амино]-3-метилмасляная кислота-cPn216
Дихлорметан (5 мл) добавляли к mpem-бутиловому эфиру со стадии (в) (50 мг, 0,046 ммоль) и в полученный молочного цвета раствор при температуре окружающей среды барботировали газ хлороводород в течение 10 минут. Смесь упаривали до сухости и остаток выпаривали совместно с дихлорметаном (5×5 мл) с получением продукта в виде белого твердого вещества. Выход 47 мг (99%). М+1=1035.
26(д) 4-[2-(2-{2-[2-(4-{[(1-Гидроксикарбамоил-2-метилпропил)-(4-метоксибензолсульфонил)амино]метил}бензоиламино)этокси]этокси}этокси)этилкарбамоил]масляная кислота-cPn216 (Соединение 23)
Функциональную группу гидроксамовой кислоты присоединяли через трет-бутилдиметилсилилзащищенное промежуточное соединение. Так, смесь кислоты со стадии (г) (47 мг, 0,045 ммоль), 4-метилморфолина (20 мкл, 0,18 ммоль), [7-азабензотриазол-1-илокситрис(пирролидино)фосфонийгексафторфосфата] PyAOP (а; 23,5 мг, 0,045 ммоль) и O-(трет-бутилдиметилсилил)гидроксиламина (10 мг, 0,07 ммоль) в диметилформамиде (3 мл) перемешивали при температуре окружающей среды и реакцию контролировали ВЭЖХ. Реакцию останавливали через 1 час и растворители выпаривали. Остаток снова растворяли в дихлорметане и при температуре окружающей среды газ хлороводород барботировали через эту смесь в течение 10 минут. Смесь упаривали до сухости и остаток выпаривали совместно с дихлорметаном (5×5 мл). Продукт был получен в виде белого порошка после ВЭЖХ, М+Н, 1050, выход 7 мг (15%).
Пример 27: получение Соединения 24
10(а) 2-[{4-[2-(2-{2-[2-(2-Карбоксиметоксиацетиламино)-этокси]этокси}этокси)этилкарбамоил]бензил}-(4-метоксибензолсульфонил)амино]-3-метилмасляной кислоты трет-бутиловый эфир
Смесь амина из Примера 26(б) (60 мг, 0,092 ммоль), N,N'-диизопропилэтиламина (96 мкл, 0,55 ммоль) и дигликолевого ангидрида (66 мг, 0,55 ммоль) в диметилформамиде (6 мл) перемешивали при температуре окружающей среды в течение 3 часов, когда ВЭЖХ показала завершение реакции. Растворитель удаляли при пониженном давлении и остаток растворяли в ацетонитриле, содержащем 0,1% TFA. После перемешивания в течение 5 минут смесь упаривали до сухости и неочищенный продукт очищали флэш-хроматографией с использованием CHCl3/МеОН/H2O, 65:25:4. Этот продукт был получен в виде белой пены. Выход 70,50 мг (99,80%).
27(б) Конъюгат 2-[{4-[2-(2-{2-[2-(2-карбоксиметоксиацетиламино)этокси]этокси}этокси)этилкарбамоил]бензил}-(4-метоксибензолсульфонил)амино]-3-метилмасляной кислоты трет-бутиловый эфир-СА1
К кислоте со стадии (а) (70,50 мг, 0,092 ммоль) в диметилформамиде (5 мл) добавляли N,N-диизопропилэтиламин (32 мкл, 0,184 ммоль), N-[(диэтиламино)-1Н-1,2,3-триазоло[4,5-b]пиридин-1-илметилен]-N-метилметанаминия гексафторфосфата N-оксид, HATU (38 мг, 0,10 ммоль) и хелатообразователь 1 (СА1; 34 мг, 0,10 ммоль). Эту реакционную смесь перемешивали при температуре окружающей среды и через 6 часов ВЭЖХ показала значительную конверсию в новый продукт. Смесь упаривали до сухости и затем после препаративной ВЭЖХ с использованием смеси ацетонитрил:вода:0,1% трифторуксусная кислота (10:80:60) выделяли чистый продукт в виде белых кристаллов. Выход 21 мг (21%).
27(в) Конъюгат 2-[{4-[2-(2-{2-[2-(2-Карбоксиметоксиацетиламино)этокси]этокси}этокси)этилкарбамоил]бензил}-(4-метоксибензолсульфонил)амино]-3-метилмасляная кислота-CA1
Дихлорметан (3 мл) добавляли к трет-бутиловому эфиру со стадии (б) (20 мг, 0,018 ммоль) и в полученный раствор молочного цвета при температуре окружающей среды барботировали газ хлороводород в течение 60 минут. Смесь упаривали до сухости и остаток выпаривали совместно с дихлорметаном (5×5 мл) с получением продукта в виде не совсем белого твердого вещества. Выход 18 мг (95%). М+1=1037.
27(г) Конъюгат {[2-(2-{2-[2-(4-{[(1-Гидроксикарбамоил-2-метилпропил)-(4-метоксибензолсульфонил)амино]метил}бензоиламино)этокси]этокси}этокси)этилкарбамоил]метокси}уксусная кислота-СА1 (Соединение 24)
Гидроксамовую кислоту присоединяли через трет-бутилдиметилсилилзащищенное промежуточное соединение. Так, смесь кислоты со стадии (в) (18 мг, 0,017 ммоль), 4-метилморфолина (8 мкл, 0,70 ммоль), [7-азабензтриазол-1-илокситрис(пирродидино)фосфонийгексафторфосфата] PyAOP (9,4 мг, 0,017 ммоль) и O-(трет-бутилдиметилсилил)гидроксиламина (4 мг, 0,026 ммоль) в диметилформамиде (2 мл) перемешивали при температуре окружающей среды и реакцию контролировали ВЭЖХ. Реакцию останавливали через 2 часа и растворители выпаривали. Остаток (Соединение 20*) снова растворяли в дихлорметане и при температуре окружающей среды газ хлороводород барботировали через эту смесь в течение 10 минут. Смесь упаривали до сухости и остаток выпаривали совместно с дихлорметаном (5×3 мл). После ВЭЖХ был получен продукт в виде белого порошка, М+Н, 1052. Выход 4 мг (22,35%).
Примеры 28-29: конъюгаты с MMPi формулы
Хелатообразователь 1 представляет собой
Пример 28: синтез конъюгата Хелатообразователь-MMPi (Соединение 26)
Соединение 25 (5,1 мг), PyAOP (6,0 мг) и N-метилморфолин (2 мкл) растворяли в диметилформамиде (0,5 мл) и эту смесь перемешивали в течение 2 минут. Добавляли Хелатообразователь 1 (3,4 мг) и реакционную смесь перемешивали в течение ночи. Добавляли смесь 20% ацетонитрила/вода (8 мл) и продукт очищали препаративной ВЭЖХ (колонка: Phenomenex Luna 10 мк С18(2) 250×10 мм, детектирование: 230 нм, растворитель А: H2O/0,1% TFA, растворитель В: CH3CN/0,1% TFA, скорость потока 5 мл/мин, градиент 20-60% В за 30 мин, tR 17 мин). После лиофилизации получили 1 мг чистого вещества и проводили ЖХ-МС (колонка: Phenomenex Luna 5 мк С18(2) 250×4,6 мм, детектирование 214 нм, растворитель А: H2O/0,1% TFA, растворитель В: CH3CN/0,1% TFA, скорость потока 1 мл/мин, градиент 20-60% В за 20 мин, tR 14,32 мин, обнаружено m/z: 792,5, ожидалось МН+ 792,4).
Пример 29: синтез хелатного конъюгата с PEG(3)-дигликолильным спейсером (Соединение 27)
Стадия (а) синтез (Соединение 25)-PEG(3)-дигликолевой кислоты
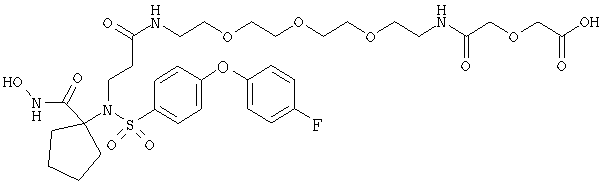
К раствору (Соединение 25)-PEG(3)-NH2 (25 мг, 39 мкмоль) в DMF (4 мл) добавляли дигликолевый ангидрид (Acros, 9 мг, 78 мкмоль). После перемешивания в течение 1,5 ч реакционную смесь концентрировали и остаток очищали препаративной ВЭЖХ (колонка Phenomenex Luna C18(2) 5 мкм 21,2×250 мм, растворители: А=вода/0,1% TFA и В=ацетонитрил/0,1% TFA; градиент 20-80% В за 60 мин; скорость потока 10,0 мл/мин, УФ детектирование при 214 нм) с получением 14,9 мг (51%) лиофилизированного материала. Этот продукт анализировали ЖХ-МС (колонка Phenomenex Luna С18(2) 3 мкм 50×4,60 мм, растворители: А=вода/0,1% TFA и В=ацетонитрил/0,1% TFA; градиент 20-100% В за 10 мин; скорость потока 1 мл/мин, УФ детектирование при 214 нм, ЭРИ-МС) и получили пик при 4,15 мин с m/z 757,3 (МН+), соответствующий продукту. Дополнительно проводили ЯМР-спектроскопию.
Стадия (б): синтез Соединения 27

Соединение 27
К раствору (Соединение 25)-PEG(3)-дигликолевой кислоты (6,6 мг, 9 мкмоль) в DMF (3 мл) добавляли Хелатообразователь 1 (3,1 мг, 9 мкмоль), HATU (Applied Biosystems, 3,4 мг, 9 мкмоль) и DIEA (Fluka, 3,1 мкл, 18 мкмоль). Через 20 мин реакционного времени смесь концентрировали и остаток очищали препаративной ВЭЖХ (колонка Phenomenex Luna C18(2) 5 мкм 21,2×250 мм, растворители: А=вода/0,1% TFA и В=ацетонитрил/0,1% TFA; градиент 10-80% В за 60 мин; скорость потока 10,0 мл/мин, УФ детектирование при 214 нм) с получением 4,2 мг (43%) лиофилизированного продукта. ЖХ-МС анализ (колонка Phenomenex Luna C18(2) 3 мкм 50×4,60 мм, растворители: А=вода/0,1% TFA и В=ацетонитрил/0,1% TFA; градиент 20-100% В за 10 мин; скорость потока 1 мл/мин, УФ детектирование при 214 нм, ЭРИ-МС; tR=4,17 мин, m/z 1082,5 (МН+)) и ЯМР-спектроскопия подтвердили структуру.
Пример 30: синтез 5-{4-[1-(4-хлорфенил)-5-(4'-фторбифенил-4-илокси)-6-оксо-1,6-дигидропиридазин-4-ил]пиперазин-1-ил}-5-оксопентановой кислоты {5-(2-гидроксиимино-1,1-диметилпропиламино)-3-[2-(2-гидроксиимино-1,1-диметилпропиламино)этил]пентил}амида
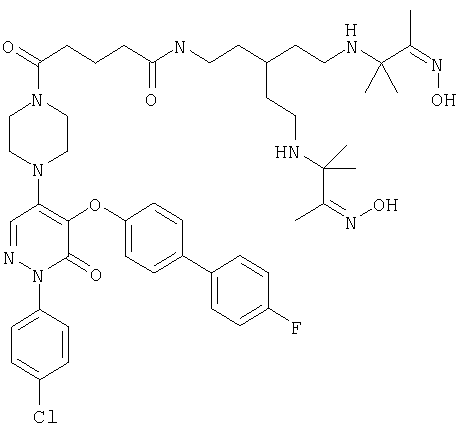
К глутариламидному производному хелата Х (300 мг, 0,66 ммоль) в DMF (2 мл) добавляли HATU (249 мг, 0,66 ммоль) и NMM (132 мкл, 1,32 ммоль). Эту смесь перемешивали в течение 5 минут и добавляли тетрафтортиофенол (0,66 ммоль, 119 мг). После перемешивания в течение 10 минут реакционную смесь разбавляли смесью 20% ацетонитрила/вода (8 мл) и продукт очищали ОФ-ВЭЖХ с получением после сублимационной сушки 110 мг целевого продукта.
К раствору 2-(4-хлорфенил)-4-(4'-фторбифенил-4-илокси)-5-пиперазин-1-ил-2Н-пиридазин-3-она (0,5 ммоль, синтезирован способами, описанными в WO 03/097612) в DMF (5 мл) добавляли N-метилморфолин (1 ммоль) и пентафторфениловый эфир, описанный выше (0,55 ммоль). Реакционную смесь перемешивали в течение 1 ч и концентрировали в вакууме. Остаток переносили в смесь воды и ацетонитрила, содержащую 0,1% TFA, и очищали хроматографией с обращенной фазой с использованием подходящего градиента вода/ацетонитрил (0,1% TFA).
| название | год | авторы | номер документа |
|---|---|---|---|
| УСОВЕРШЕНСТВОВАННЫЕ КОНЪЮГАТЫ N4 ХЕЛАТООБРАЗУЮЩИХ АГЕНТОВ | 2005 |
|
RU2360701C2 |
| СПОСОБ ВИЗУАЛИЗАЦИИ | 2008 |
|
RU2505316C2 |
| СОЕДИНЕНИЯ НА ОСНОВЕ ПЕПТИДОВ ДЛЯ НАПРАВЛЕННОЙ ДОСТАВКИ К РЕЦЕПТОРАМ ИНТЕГРИНОВ | 2002 |
|
RU2303042C2 |
| РАДИОАКТИВНО МЕЧЕНЫЕ ПЕПТИДЫ, СВЯЗЫВАЮЩИЕСЯ С HER2 | 2011 |
|
RU2592685C2 |
| МЕЧЕННЫЕ РАДИОАКТИВНЫМ ИЗОТОПОМ КОНЪЮГАТЫ RGD-СОДЕРЖАЩИХ ПЕПТИДОВ И СПОСОБЫ ИХ ПОЛУЧЕНИЯ С ПОМОЩЬЮ CLICK-ХИМИИ | 2005 |
|
RU2419627C2 |
| ДИАГНОСТИЧЕСКИЕ СОЕДИНЕНИЯ | 2005 |
|
RU2396272C9 |
| ЦИКЛИЧЕСКИЕ ПЕПТИДЫ, РАДИОФАРМАЦЕВТИЧЕСКИЙ ПРЕПАРАТ, ЦИКЛИЧЕСКИЕ ПЕПТИДЫ, ИМЕЮЩИЕ РАДИОАКТИВНУЮ МЕТКУ | 1994 |
|
RU2145608C1 |
| КОНЪЮГИРОВАННЫЕ БИСФОСФОНАТЫ ДЛЯ ДИАГНОСТИКИ И ТЕРАПИИ ЗАБОЛЕВАНИЙ КОСТЕЙ | 2015 |
|
RU2742660C2 |
| Способ получения производного мочевины с хелатным центром, тропного к простат-специфичному мембранному антигену для связывания технеция-99м/рения для диагностики/лечения рака предстательной железы | 2018 |
|
RU2692126C1 |
| КОНТРАСТНЫЙ АГЕНТ, НАЦЕЛЕННЫЙ НА РЕЦЕПТОР УРОКИНАЗНОГО АКТИВАТОРА ПЛАЗМИНОГЕНА | 2005 |
|
RU2394837C2 |
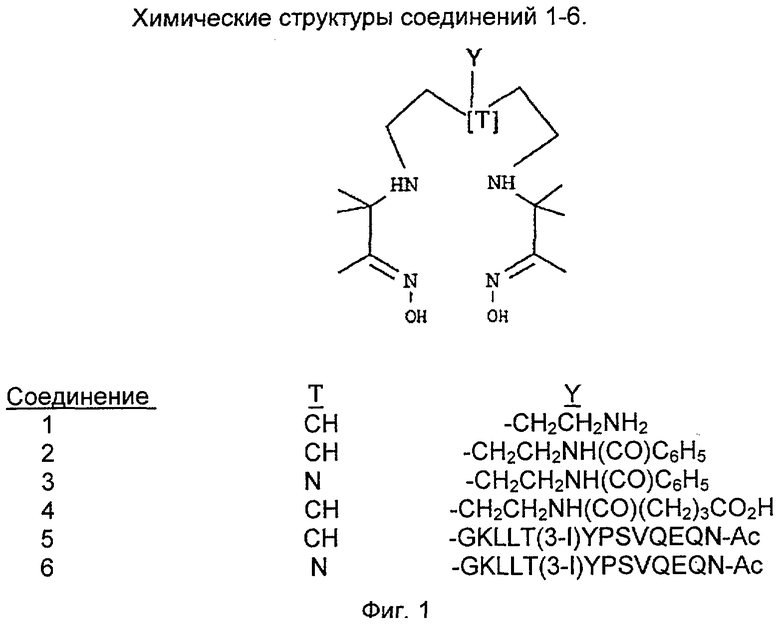
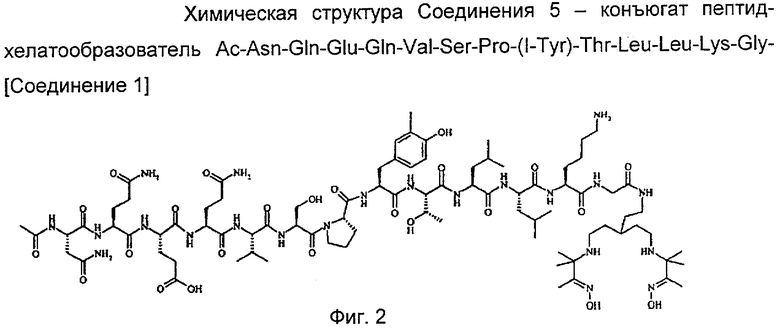
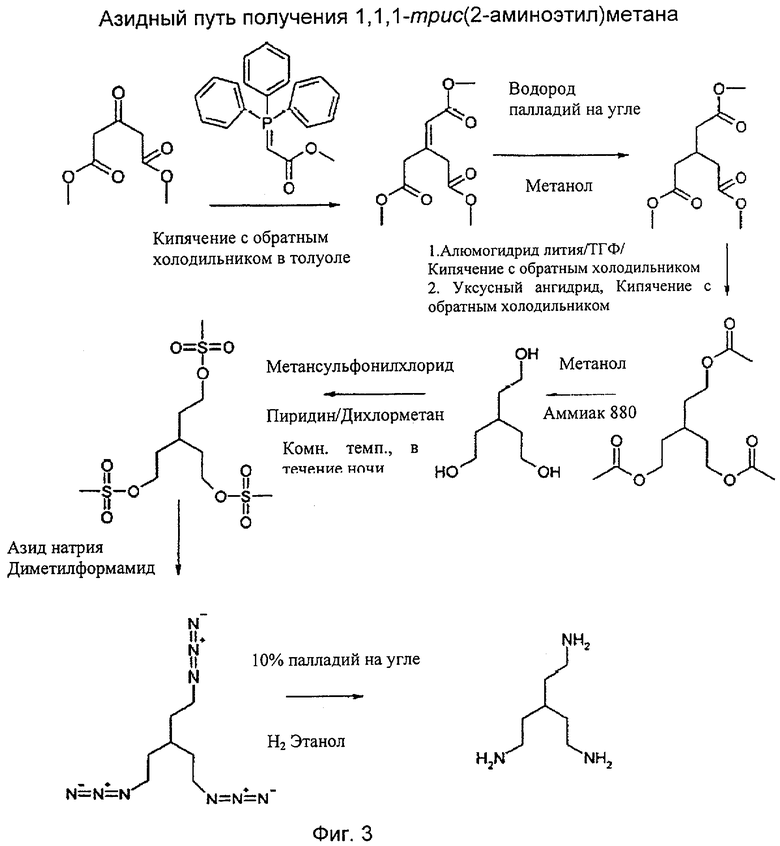
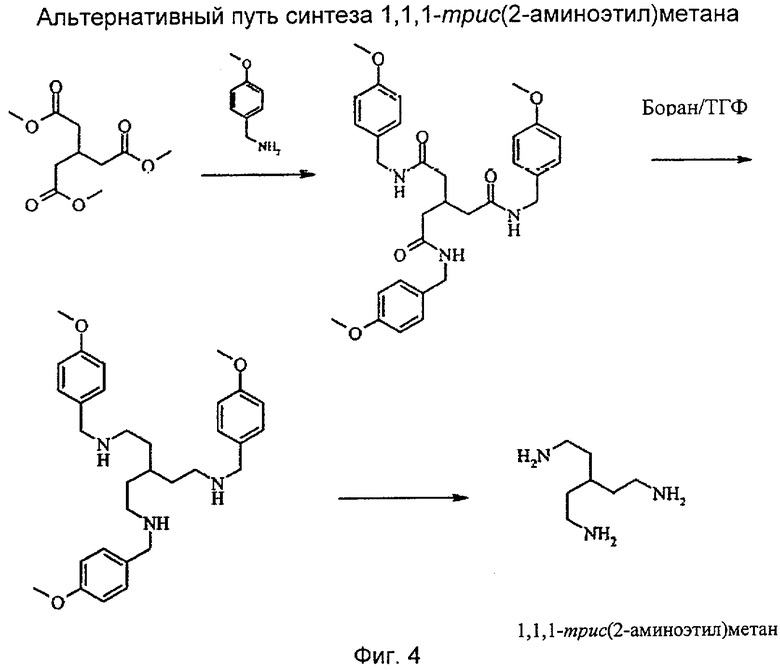
Настоящее изобретение относится к улучшенным хелатным конъюгатам формулы

где каждый R1, R2 и R3 независимо представляет собой группу R; Y представляет собой -(A)n-X-Z, где Х представляет собой -NH-, -CO2-, -N(C=O)-; Z представляет собой биологическую группировку для направленной доставки, которая выбрана из 3-20-членного пептида, субстрата фермента, ингибитора фермента или синтетического связывающегося с рецептором соединения; -(А)n- представляет собой линкер, где каждый А независимо представляет собой -CR2-, -NRCO-, -CONR-, -CR2OCR2- или группировку полиалкиленгликоля; n представляет собой целое число, имеющее значение от 2 до 10; каждая группа R независимо представляет собой Н или С1-10алкил, или 2 группы R вместе с атомами, к которым они присоединены, образуют C3-6карбоциклическое насыщенное кольцо. Комплексы с радиоактивными металлами пригодны в качестве радиоактивных фармацевтических препаратов, в особенности с 99mTc. 7 н. и 21 з.п. ф-лы, 4 табл., 4 ил.
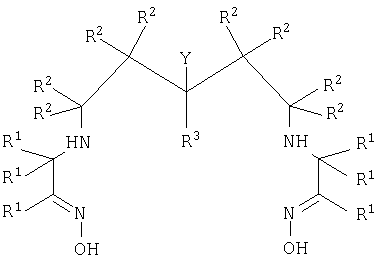
где каждый R1, R2 и R3 независимо представляет собой группу R;
Y представляет собой -(A)n-X-Z,
где Х представляет собой -NH-, -CO2-, -N(C=O)-;
Z представляет собой биологическую группировку для направленной доставки, которая выбрана из 3-20-членного пептида, субстрата фермента, ингибитора фермента или синтетического связывающегося с рецептором соединения;
-(A)n- представляет собой линкер, где каждый А независимо представляет собой
-CR2-, -NRCO-, -CONR-, -CR2OCR2- или группировку полиалкиленгликоля;
n представляет собой целое число от 2 до 10;
каждая группа R независимо представляет собой Н или С1-10алкил или две группы R вместе с атомами, к которым они присоединены, образуют С3-6карбоциклическое насыщенное кольцо.

где каждый R1 независимо представляет собой C1-3алкил.
(1) хелатный конъюгат по пп.1-10;
(2) биосовместимый восстанавливающий агент.
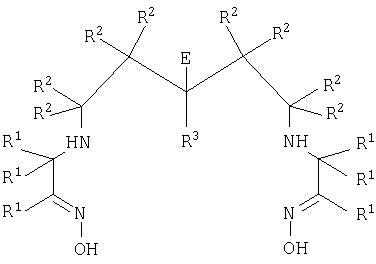
где каждый R1, R2 и R3 независимо представляет собой группу R;
Е представляет собой -(A)n-J,
где J представляет собой -NR5R6 или -CO2M, где R5 и R6 независимо представляют собой группу R или РG и М представляет собой Н, катион, РG или активный сложный эфир;
-(A)n- представляет собой линкер, где А независимо представляет собой -CR2-, -NRCO-, -CONR-, -CR2OCR2- или группировку полиалкиленгликоля;
n представляет собой целое число от 2 до 10;
каждая группа R независимо представляет собой Н или С1-10алкил или две группы R вместе с атомами, к которым они присоединены, образуют C3-6карбоциклическое насыщенное кольцо;
PG представляет собой защитную группу.
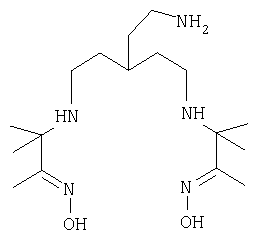
HC(CH2CH2NR7R8)3,
где R7 и R8 независимо представляют собой Н или PG или R7 и R8 вместе образуют РG, где РG представляет собой защитную группу,
или его соль.
| RU 97118152 А, 10.08.1999 | |||
| WO 9960018 A1, 25.11.1999 | |||
| US 5997843 А, 07.12.1999. |

