Область изобретения
Настоящее изобретение относится к усовершенствованным конъюгатам тетрааминных хелатообразующих агентов с обеспечивающими направленную доставку биологическими молекулами, пригодным для образования металлических комплексов с радиоактивным металлом 99mTc. Эти радиоактивные металлические комплексы полезны в качестве 99mTc-радиофармацевтических средств. Предложены также наборы и предшественники.
Предшествующий уровень техники
В US 5489425 (Dow Chemical) раскрыт целый ряд функционализированных тетрааминных хелатообразующих агентов с открытой цепью и макроциклических, пригодных для образования комплексов металлов, в частности комплексов радиоактивного и нерадиоактивного родия, конкретно комплексов радиоактивного 105Rh или 101mRh. Конкретные раскрытые тетраамины включают:

Бифункциональные хелатообразующие агенты описаны как пригодные для конъюгирования с моноклональными антителами или их фрагментами в терапевтических или диагностических целях. В US 5489425 указано (Примеры 21, 22а и 23), что хелатный конъюгат антитело-радиометаллический комплекс получают сначала образованием комплекса металла 105Rh, затем взаимодействием с антителом с последующей очисткой. В US 5489425 не упоминаются конъюгаты антитело-хелатообразующий агент, которые находятся не в форме комплекса, то есть не имеют координированного радиоактивного металла. В US 5489425 нет сведений о том, как разграничивать боковую аминогруппу и четыре аминогруппы хелатообразующего агента в таких реакциях конъюгирования с антителом. В US 5489425 утверждается, что бифункциональные хелатообразующие агенты "могли бы быть использованы также в образовании комплексов технеция и рения", но не раскрыто, как этого можно достичь, не раскрыты также какие-либо реальные комплексы технеция.
В US 5650134 раскрыт конъюгат соматостатиновый пептид-хелатообразующий агент из ряда хелатообразующих агентов. В Примере 1 описано конъюгирование 6-(п-изотиоцианатобензил)-1,4,8,11-тетраазаундекана с октреотидным пептидом.
В ЕР 1181936 А1 раскрыты конъюгаты бомбезина (то есть тетрадекапептида) с тетрааминными хелатообразующими агентами, полученные с использованием бифункциональных хелатообразующих агентов BBN-1 и BBN-2, и их 99mTc-комплексы:

где Boc - трет-бутоксикарбонильная защитная группа.
Указано, что комплексы 99mTc демонстрируют быстрый клиренс из мышиного организма через почки и мочевую систему. В ЕР 1181936 А1 не дано, однако, никакого описания или ссылки на синтез BBN-1 или BBN-2, кроме ссылки на стадию, где их конъюгируют с N-концом бомбезина. Конъюгирование BBN-2 с бомбезином и 99mTc-мечение с образованием потенциального радиофармацевтического средства для визуализации опухолей было описано Nock et al. [Eur. J. Nucl. Med., 30(2), 247-258
(2003)]. Сообщается, что комплекс 99mTc придает улучшенную гидрофильность по сравнению с бомбезин-хелатными конъюгатами предшествующего уровня техники и поэтому ожидается, что он благоприятствует экскреции через почки и мочевую систему.
Конъюгирование BBN-1 с октреотидом и 99mTc-мечение с образованием потенциального радиофармацевтического средства для визуализации опухолей у пациентов-людей было описано Maina et al. [Eur. J. Nucl. Med., 30(9), 1211-1219 (2003)]. Ни в одной из вышеуказанных публикаций, касающихся BBN-1 или BBN-2, не приведен синтез BBN-1 или BBN-2.
Сущность изобретения
Согласно настоящему изобретению предложены конъюгаты тетрааминных хелатообразующих агентов с обеспечивающими направленную доставку биологическими группировками, которые связаны посредством линкерной группы, и их комплексы с технецием в качестве радиофармацевтических средств. Линкерная группа такова, что хелатообразующий агент монофункционализирован в положении головы мостика и обеспечивает как гибкость, так и отсутствие арильных групп для минимизации липофильности и стерической объемистости. Предложены соответствующим образом защищенные варианты хелатообразующих агентов, которые позволяют осуществлять конъюгирование с самыми разнообразными, обеспечивающими направленную доставку молекулами без взаимодействий с аминными атомами азота тетрааминного хелатообразующего агента. Описаны синтезы функционализированных хелатообразующих агентов вместе с бифункциональными хелатными предшественниками.
Описаны радиофармацевтические композиции, содержащие комплексы металла технеция по изобретению, и нерадиоактивные наборы для получения таких радиофармацевтических средств.
Подробное описание изобретения
В первом воплощении настоящего изобретения предложен катионный комплекс технеция 99mTc формулы (I)
 ,
,
где Х представляет собой -NR-, -CO2-, -CO-, -NR(C=S)-, -NR(C=O)-, -CONR- или группу Q;
каждый Y независимо представляет собой D- или L-аминокислоту, -СН2-, -CH2OCH2- или -OCH2CH2O-, или группу X;
Z представляет собой синтетическую обеспечивающую направленную доставку биологическую группировку;
n означает целое число от 1 до 8;
m означает целое число от 0 до 30;
R представляет собой Н, С1-4алкил, С2-4алкоксиалкил, С1-4гидроксиалкил или С1-4фторалкил;
Q представляет собой

А представляет собой противоион;
при условии, что цепь атомов X1-(Y)m не имеет связей, где один гетероатом непосредственно связан с другим.
Радиоактивный изотоп технеция может быть γ-излучателем, таким как 99mTc, или позитронным излучателем, подходящим для визуализации методом позитронно-эмиссионной томографии (PET), таким как 94mTc. Предпочтительно радиоактивный изотоп технеция представляет собой 99mTc или 94mTc, наиболее предпочтительно 99mTc.
Х предпочтительно представляет собой -CONR-, -NR(C=O)- или группу Q. Наиболее предпочтительно Х представляет собой -CONR- или -NR(C=O)-, причем -CONH- и -NH(C=O)- особенно предпочтительны.
Линкерная группа -(CH2)n-X-(Y)m- в соединении формулы (I) выбрана таким образом, что цепь атомов Х1-(Y)m не имеет связей, в которых один гетероатом непосредственно связан с другим, где термин "гетероатом" означает атом, не являющийся атомом углерода, такой как атом азота, кислорода или серы. Это означает, что эта цепь не имеет таких связей, как O-O, N-N или O-N.
Представляется, что роль линкерной группы -(CH2)n-X-(Y)m- в соединении формулы (I) заключается в том, чтобы дистанцировать комплекс технеция от активного сайта связывания обеспечивающей направленную доставку биологической группировки (Z) in vivo. Это дает возможность гарантировать, что относительно объемный комплекс технеция стерически не будет ингибировать связывание с активными сайтами in vivo. Алкиленовая группа -(СН2)n- имеет преимущество в том, что отсутствуют значительные взаимодействия с конъюгированной обеспечивающей направленную доставку биологической группировкой (Z) с образованием водородных связей, так что линкер не наматывается на Z. В предпочтительных алкиленовых группах n равно числу от 1 до 6, наиболее предпочтительно от 2 до 4 и особенно предпочтительно 2.
Линкерные группы по настоящему изобретению не имеют арильных колец. Это способствует минимизации липофильности технециевого комплекса с линкерной группой, которая присоединена к обеспечивающей направленную доставку биологической группировке (Z) конъюгата. Также минимизируются стерическая объемистость и молекулярная масса линкерной группы (и, следовательно, технециевого комплекса), при этом гибкость связи сохраняется.
Природа линкерной группы может быть также использована для модификации биораспределения визуализирующего агента. Так, например, введение простых эфирных групп в -(Y)m- будет способствовать минимизации связывания белков плазмы. В тех случаях, когда -(Y)m- содержит полиэтиленгликолевый (PEG) структурный элемент или пептидную цепь из 1-10 аминокислотных остатков, линкерная группа может выполнять функцию модификатора фармакокинетики и почечного клиренса визуализирующего агента in vivo. Такие линкерные группы-"биомодификаторы" могут ускорять выведение технециевого визуализирующего агента из фоновой ткани, такой как мышца или печень, и/или из крови, что позволяет получить более качественное диагностическое изображение благодаря меньшим фоновым помехам. Линкерная группа-биомодификатор может быть также использована для способствования конкретному пути экскреции, например через почки, а не через печень. Альтернативно такие линкерные группы могут пролонгировать срок пребывания в крови, обеспечивая аккумулирование большего количества агента в сайте-мишени in vivo.
В тех случаях, когда -(Y)m- содержит пептидную цепь из аминокислотных остатков, эти аминокислотные остатки предпочтительно выбраны из глицина, лизина, аспарагиновой кислоты, глутаминовой кислоты или серина. Количество аминокислот в пептидной цепи составляет предпочтительно от 1 до 10, наиболее предпочтительно от 1 до 3.
В тех случаях, когда -(Y)m- содержит группировку PEG, она предпочтительно содержит группу формулы (-OCH2CH2O)w-, где w означает целое число, имеющее значение от 3 до 25. Целым числом w предпочтительно является число от 6 до 22. Особенно предпочтительной PEG-содержащей группой -(Y)m- является группировка, образующаяся в результате полимеризации монодисперсной PEG-подобной структуры 17-амино-5-оксо-6-аза-3,9,12,15-тетраоксагептадекановой кислоты формулы (IV)
 ,
,
где р означает целое число от 1 до 10.
Под термином "фторалкил" подразумевается алкильная группа с по меньшей мере одним заместителем фтором, то есть этот термин охватывает группы от монофторалкильной (например, -CH2F) до перфторалкильной (например, CF3).
Группа -(Y)m- предпочтительно содержит группировку дигликолевой кислоты, малеимидную группировку, группировку глутаровой кислоты, группировку янтарной кислоты, группировку на основе полиэтиленгликоля или PEG-подобную группировку формулы IV.
Термин "синтетический" имеет общепринятое значение этого термина, то есть созданный человеком в отличие от выделенного из природных источников, например из организма млекопитающего. Такие соединения имеют преимущество в том, что их производство и показатели чистоты можно полностью контролировать. Таким образом, моноклональные антитела и их фрагменты не входят в объем настоящих притязаний.
Под термином "обеспечивающая направленную доставку биологическая группировка" подразумеваются 3-100-мерные пептиды или пептидные аналоги, которые могут представлять собой линейные пептиды или циклические пептиды, или их комбинации; или субстраты, антагонисты или ингибиторы ферментов; синтетические рецептор-связывающие соединения; олигонуклеотиды или олиго-ДНК- или олиго-РНК-фрагменты.
Термин "циклический пептид" означает последовательность из 5-15 аминокислот, в которой две концевые аминокислоты связаны вместе ковалентной связью, которая может представлять собой пептидную или дисульфидную связь, или синтетическую непептидную связь, такую как тиоэфирная, фосфодиэфирная, дисилоксановая или уретановая связь.
Под термином "аминокислота" подразумевается L- или D-аминокислота, аналог аминокислоты или миметик аминокислоты, которые могут быть оптически чистыми, то есть могут представлять собой единственный энантиомер и, следовательно, хиральный, или смесь энантиомеров. Предпочтительно аминокислоты по настоящему изобретению являются оптически чистыми.
Термин "миметик аминокислоты" относится к синтетическим аналогам встречающихся в природе аминокислот, которые являются изостерами, то есть имитируют стерическую и электронную структуру природного соединения. Такие изостеры общеизвестны в данной области и включают депсипептиды, ретроинверсопептиды, тиоамиды, циклоалканы или 1,5-дизамещенные тетразолы [смотри М.Goodman, Biopolymers, 24, 137, (1985)], но не ограничиваются ими.
Подходящие пептиды для использования в настоящем изобретении включают:
- соматостатин, октреотид и аналоги;
- пептиды, которые связываются с ST-рецептором, где ST относится к термостабильному токсину, продуцируемому E.coli и другими микроорганизмами;
- фрагменты ламинина, например YIGSR, PDSGR, IKVAV, LRE и KCQAGTFALRGDPQG;
- N-формильные пептиды для направленной доставки к сайтам аккумуляции лейкоцитов;
- тромбоцитарный фактор 4 (PF4) и его фрагменты;
- RGD (Arg-Cly-Asp)-содержащие пептиды, которые могут, например, направлено воздействовать на ангиогенез [R.Pasqualian et al., Nat Biotechnol. 1997 Jun; 15(6): 542-6; E.Ruoslahti, Kidney Int. 1997 May, 51(5); 1413-7];
- пептидные фрагменты α2-антиплазмина, фибронектина или бета-казеина, фибриногена или тромбоспондина; аминокислотные последовательности α2-антиплазмина, фибронектина, бета-казеина, фибриногена и тромбоспондина можно найти в следующих публикациях: предшественник α2-антиплазмина [М.Tone et al., J. Biochem, 102, 1033, (1987)], бета-казеин [L.Hansson et al., Gene, 139. 193 (1994)], фибронектин [A.Gutman et al., FEBS Lett, 207. 145 (1996)], предшественник тромбоспондина-1 [V.Dixit et al., Proc. Natl. Acad. Sci., USA, 83, 5449 (1986)], R.F.Doolittle, Ann. Rev. Biochem., 53, 195 (1984);
- пептиды, которые являются субстратами или ингибиторами ангиотензина, такие как ангиотензин II Asp-Arg-Val-Tyr-lle-His-Pro-Phe (E.C.Jorgensen et al., J. Med. Chem., 1979, Vol.22, 9, 1038-1044),
[Sar, lie] ангиотензин II: Sar-Arg-Val-Tyr-lle-His-Pro-lle (R.K.Turker et al., Science, 1972, 177, 1203),
- ангиотензин I: Asp-Arg-Val-Tyr-lle-His-Pro-Phe-His-Leu.
Предпочтительно пептиды по настоящему изобретению содержат пептиды антиплазмина или ангиотензина II. Пептиды антиплазмина содержат аминокислотную последовательность, взятую с N-конца:
1) α2-антиплазмина,
то есть NH2-Asn-Gln-Glu-Gln-Val-Ser-Pro-Leu-Thr-Leu-Thr-Leu-Leu-Lys-OH,
или варианты этой последовательности, в которых одна или более аминокислот заменены, добавлены или удалены, такие как:
NH2-Asn-Gln-Glu-Gln-Val-Ser-Pro-Leu-Thr-Leu-Thr-Leu-Leu-Lys-Gly-OH,
NH2-Asn-Gln-Glu-Ala-Val-Ser-Pro-Leu-Thr-Leu-Thr-Leu-Leu-Lys-Gly-OH,
NH2-Asn-Gln-Glu-Gln-Val-Gly-OH, или
2) казеина,
то есть Ac-Leu-Gly-Pro-Gly-Gln-Ser-Lys-Val-lle-GIy.
Синтетические пептиды по настоящему изобретению могут быть получены стандартным твердофазным синтезом, как описано в Р.Lloyd-Williams, F.Albericio and E.Girald; Chemical Approaches to the Synthesis of Peptides and Proteins, CRC Press, 1997.
Подходящие субстраты, антагонисты или ингибиторы ферментов включают глюкозу и аналоги глюкозы, такие как фтордезоксиглюкоза; жирные кислоты, или ингибиторы эластазы, ангиотензина II или металлопротеиназы. Предпочтительным непептидным антагонистом ангиотензина II является лозартан.
Подходящие синтетические рецептор-связывающие соединения включают эстрадиол, эстроген, прогестин, прогестерон и другие стероидные гормоны; лиганды для дофаминового D-1 или D-2 рецептора или транспортеры дофамина, например тропаны, и лиганды для серотонинового рецептора.
Обеспечивающая направленную доставку биологическая группировка (Z) предпочтительно имеет молекулярную массу менее 5000, наиболее предпочтительно менее 4000, идеально менее 3000. Это дает преимущество в том, что улучшенные биологические характеристики тетрааминных комплексов технеция по изобретению могут оказывать влияние на общее биораспределение, в частности клиренс, технециевого комплекса конъюгата формулы I. Когда n равно 3 и Х содержит атом азота, непосредственно связанный с группой (СН2)n, тогда выбранный Z является синтетическим и имеющим молекулярную массу менее 4000. Предпочтительными обеспечивающими направленную доставку биологическими группировками являются 3-20-мерные пептиды или субстраты ферментов, антагонисты ферментов или ингибиторы ферментов.
Противоион (А-) представляет собой анион, который присутствует в эквимолярном количестве и, таким образом, уравновешивает положительный заряд на Tc(V) диоксотехнециевом комплексе формулы I. Анион (А) соответственно является одно- или многозарядным при условии, что присутствует в уравновешивающем заряд количестве. Подходящий анион образует неорганическая или органическая кислота. Примеры подходящих анионов включают галогенидные ионы, такие как хлорид или бромид, сульфатный, нитратный, цитратный, ацетатный, фосфатный и боратный ионы. Предпочтительными анионами являются хлоридные.
Комплексы технеция формулы I имеют преимущество в том, что они стабильны после комплексообразования и содержат авидный хелатообразующий агент (cheland), который связывает технеций предпочтительно с обеспечивающей направленную доставку биологической группировкой. Поэтому маловероятно, что комплекс технеция будет подвергаться реакциям трансхелатирования с биологическими макромолекулами или конкурирующими лигандами in vivo. Комплексы технеция небольшие и компактные, что полезно, поскольку они оказывают минимальное стерическое влияние на конъюгированную обеспечивающую направленную доставку биологическую группировку (Z). Постоянный катионный обмен и Tc(V) диоксоядро означают, что комплексы также являются гидрофильными и поэтому вряд ли будут распределяться внутриклеточно в другие компартменты, и, следовательно, будут более быстро выводиться из фоновых органов и тканей in vivo, например из кровотока.
Комплексы технеция формулы I могут быть получены путем взаимодействия подходящего источника технеция с конъюгатом хелатообразующего агента, имеющим формулу II, как описано во втором воплощении ниже.
Во втором воплощении настоящего изобретения предложен конъюгат хелатообразующего агента формулы II

где X, Y, Z, n и m такие, как определено выше;
Q1-Q6 независимо представляют собой группы Q, где Q представляет собой Н или защитную группу для аминогрупп.
Эти конъюгаты хелатообразующего агента используют в получении комплексов технеция формулы I первого воплощения.
Под термином "защитная группа" подразумевается группа, которая ингибирует или подавляет нежелательные химические реакции, но которая достаточно реакционноспособна, чтобы ее можно было отщепить от функциональной группы в достаточно мягких условиях, в которых остальная часть молекулы не подвергается модификации. После удаления защитных групп получают целевой продукт. Защитные группы для аминогрупп общеизвестны специалистам в данной области и соответственно выбраны из Boc (Boc означает трет-бутилоксикарбонил), Fmoc (Fmoc означает флуоренилметоксикарбонил), трифторацетила, аллилоксикарбонила, Dde [то есть 1-(4,4-диметил-2,6-диоксоциклогексилиден)этила] или Npys (то есть 3-нитро-2-пиридинсульфенила). В некоторых случаях характер защиты может быть таким, что защитными группами являются как группы Q1/Q2, так и Q5/Q6, то есть на ассоциированном атоме азота амина связь NH отсутствует. Использование других защитных групп описано в "Protective Groups in Organic Synthesis", Theorodora W.Greene and Peter G.M.Wuts (John Wiley & Sons, 1991). Предпочтительными защитными группами для аминогрупп являются Вое и Fmoc, наиболее предпочтительно Вос. Если используют Вос, то обе группы Q1 и Q6 представляют собой Н, а группы Q2, Q3, Q4 и
Q5, каждая, представляют собой трет-бутоксикарбонил.
В конъюгатах формулы II защитные группы для аминогрупп служат в основном для защиты функциональных аминогрупп тетрааминного хелатообразующего агента в ходе химического синтеза до реакции образования комплекса с технецием. Однако, если обеспечивающая направленную доставку биологическая группировка (Z) способна вступать в реакцию с первичными и/или вторичными аминами, то эти защитные группы могут быть также полезны для предотвращения нежелательных химических реакций между хелатообразующими аминами и Z перед образованием комплекса с технецием.
Предпочтительные конъюгаты формулы II имеют по меньшей мере один незащищенный аминный атом азота (то есть одна из групп Q3 или Q4 представляет собой Н или обе группы Q1/Q2 или Q5/Q6 представляют собой Н). Одна или более свободных аминогрупп означает, что конъюгат более быстро растворяется в водной среде, которая является предпочтительным растворителем для получения комплекса технеция формулы I. Свободная аминогруппа также означает, что образование комплекса с технецием происходит более быстро, поскольку комплексообразование не зависит от предварительного удаления защитной группы, что также препятствовало бы образованию металлического комплекса. Если конъюгированная обеспечивающая направленную доставку биологическая группировка (Z) не восприимчива к дальнейшей реакции с аминами, то удобно использовать конъюгат формулы II в полностью лишенной защиты форме (то есть каждая из групп Q1-Q6 представляет собой Н), и такой конъюгат хелатообразующего агента формулы II является особенно предпочтительным. Полностью лишенная защиты форма предпочтительна для реакции комплексообразования с получением комплекса технеция формулы I.
Комплексы технеция формулы I по настоящему изобретению могут быть получены путем взаимодействия раствора радиоактивного металла в подходящем состоянии окисления с конъюгатом хелатообразующего агента формулы II при подходящем рН. Раствор возможно может содержать лиганд, который образует слабый комплекс с технецием (такой как глюконат или цитрат), то есть комплекс технеция получают путем обмена лигандов или трансхелатированием. Такие условия часто используют для подавления нежелательных побочных реакций, таких как гидролиз иона технеция, но они менее важны при использовании хелатообразующих агентов по настоящему изобретению, поскольку они быстро образуют комплексы с технецием. Если радиоактивный изотоп представляет собой 99mTc, то обычно исходным веществом является пертехнетат технеция из 99Мо генератора. Технеций присутствует в 99mTc-пертехнетате в состоянии окисления Tc(VII), в котором он относительно нереакционноспособен. Поэтому получение комплексов технеция в более низком состоянии окисления от Тс(I) до Tc(V) обычно требует добавления подходящего фармацевтически приемлемого восстановителя, такого как дитионат натрия, бисульфит натрия, аскорбиновая кислота, формамидин-сульфиновая кислота, ион олова, Fe(II) или Cu(I), чтобы способствовать комплексообразованию. Фармацевтически приемлемый восстановитель предпочтительно представляет собой соль двухвалентного олова, наиболее предпочтительно хлорид двухвалентного олова, фторид двухвалентного олова или тартрат двухвалентного олова.
Конъюгаты хелатообразующего агента формулы II могут быть получены конъюгированием обеспечивающей направленную доставку биологической молекулы (Z) с бифункциональным хелатообразующим агентом формулы III, как описано в пятом воплощении ниже.
В третьем воплощении настоящего изобретения предложено радиофармацевтическое средство, которое содержит комплекс технеция первого воплощения, где А представляет собой фармацевтически приемлемый противоион, вместе биосовместимым носителем в форме, подходящей для введения человеку.
Фраза "в форме, подходящей для введения человеку" означает, что композиция стерильна, апирогенна, не содержит соединений, которые вызывают токсические и вредные эффекты, и приготовлена в виде препарата с биосовместимым рН (приблизительно рН 4,0-10,5). В таких композициях нет включений, которые могли бы стать причиной эмболии in vivo, и они приготовлены так, что при контакте с биологическими жидкостями (например, кровью) преципитация не происходит. Кроме того, такие композиции содержат только биологически совместимые эксципиенты и предпочтительно являются изотоническими.
"Биосовместимый носитель" представляет собой текучую среду, в частности жидкость, в которой радиофармацевтическое средство суспендировано или предпочтительно растворено, такую, чтобы композиция была физиологически приемлемой, то есть чтобы ее можно было вводить в организм млекопитающего без токсического воздействия или чрезмерного дискомфорта. Подходящим биосовместимым носителем является инъекционный носитель-жидкость, такой как стерильная, апирогенная вода для инъекций; водный раствор, такой как физиологический раствор (который предпочтительно может быть сбалансирован так, чтобы конечный продукт для инъецирования был или изотоническим, или не гипотоническим); водный раствор одного или более веществ, регулирующих тоничность (например, солей катионов плазмы с биосовместимыми противоионами), сахаров (например, глюкозы или сахарозы), сахарных спиртов (например, сорбита или маннита), гликолей (например, глицерина) или других неионных полиолов (например, полиэтиленгликолей, пропиленгликолей и тому подобного).
Под термином "фармацевтически приемлемый противоион" подразумевается анион (то есть отрицательный ион), который не вызывает токсических или вредных эффектов при введении в организм млекопитающего in vivo и совместим химически и/или токсикологически с другими ингредиентами фармацевтической композиции. Химическая совместимость для радиофармацевтических средств на основе технеция по настоящему изобретению означает, что анион не конкурирует эффективно с тетрааминным хелатообразующим агентом за технеций. Подходящие такие анионы включают галогениды (например, хлорид, йодид и бромид); С1-2алкилсульфонаты (например, мезилат или этилсульфонат); арилсульфонаты (например, фенилсульфонат или тозилат); С1-2алкилфосфонаты; ди(С1-2)алкилфосфаты (например, диметилфосфат, диэтилфосфат или диглицеролфосфат); арилфосфонаты; арилфосфаты; алкиларилфосфонаты; алкиларилфосфаты; С1-2алкилкарбоксилаты (например, ацетаты, пропионаты, глутаматы или глицераты); арилкарбоксилаты (например, бензоаты) и тому подобное, но не ограничиваются ими. Предпочтительными фармацевтически приемлемыми противоионами являются хлорид, фторид, ацетат, тартрат, гидроксид и фосфат.
Такие радиофармацевтические средства соответственно поставляются в контейнере, который снабжен уплотняющей прокладкой, подходящей для однократного или многократного прокалывания иглой для подкожных инъекций (например, обжимной уплотняющей прокладкой-крышкой) и в то же время сохраняющей стерильность. Такие контейнеры могут содержать однократные или многократные дозы для пациентов. Предпочтительные многодозовые контейнеры представляют собой отдельный объемистый флакон (например, емкостью от 10 до 100 см3), который содержит многократные дозы для пациента, поэтому однократные дозы для пациента можно отбирать в клинические шприцы через различные промежутки времени в течение срока годности препарата в соответствии с клинической ситуацией. Предварительно заполненные шприцы спроектированы таким образом, чтобы содержать однократную дозу для человека и, следовательно, предпочтительно представляют собой одноразовые или другие шприцы, подходящие для клинического применения. Предварительно заполненные шприцы могут быть снабжены защитным экраном для защиты оператора от радиоактивной дозы. Подходящие защитные экраны для шприца с радиофармацевтическим средством известны в данной области и предпочтительно содержат либо свинец, либо вольфрам.
Радиофармацевтические средства по настоящему изобретению содержат радиоактивные изотопы технеция 99mTc или 94mTc, наиболее предпочтительно 99mTc. Если изотоп технеция представляет собой 99mTc, то величина радиоактивности, подходящая для радиофармацевтического средства для диагностической визуализации, находится в пределах от 180 до 1500 МБк 99mTc в зависимости от участка, предназначенного для визуализации in vivo, поглощения и соотношения мишень/фон.
Радиофармацевтические средства на основе технеция по настоящему изобретению могут быть получены различными способами:
1) асептические производственные технологии, когда образование технециевого комплекса, описанное выше для второго воплощения, осуществляют в помещении с чистой окружающей средой;
2) конечная стерилизация, когда образование технециевого комплекса проводят не в условиях асептического производства и затем стерилизуют на последней стадии (например, гамма-излучением или автоклавированием);
3) методология набора, когда стерильный, лиофилизированный нерадиоактивный препарат-набор, содержащий конъюгат хелатообразующего агента формулы II и фармацевтически приемлемый восстановитель плюс другие возможные эксципиенты, подвергают взаимодействию с аликвотой стерильного 99mTc-пертехнетата из генератора 99mTc.
Способ (3) является предпочтительным, а наборы для использования в этом способе описаны в четвертом воплощении (ниже).
В четвертом воплощении настоящего изобретения предложен нерадиоактивный набор для приготовления радиофармацевтической композиции, описанной выше, который включает в себя конъюгат формулы (II) вместе с биосовместимым восстановителем. Такие наборы предназначены для приготовления стерильных радиофармацевтических продуктов, подходящих для введения человеку, например, инъецированием прямо в кровоток. Конъюгаты лиганда и их предпочтительные аспекты описаны во втором воплощении выше.
Для 99mTc набор предпочтительно лиофилизирован и предназначен для разведения стерильным 99mTc-пертехнетатом (TCO4 -) из генератора радиоизотопа 99mTc для получения раствора, подходящего для введения человеку без дополнительных манипуляций. Подходящие наборы включают в себя контейнер (например, флакон с уплотняющей прокладкой), содержащий конъюгат хелатообразующего агента в форме либо свободного основания, либо соли с кислотой, вместе с биосовместимым восстановителем, таким как дитионит натрия, бисульфит натрия, аскорбиновая кислота, формамидинсульфиновая кислота, ион олова, Fe(II) или Cu(I). Биосовместимый восстановитель предпочтительно представляет собой соль двухвалентного олова, такую как хлорид олова или тартрат олова. Альтернативно набор возможно может содержать комплекс нерадиоактивного металла, который при добавлении технеция подвергается трансметаллированию (то есть металлообмену) с получением целевого продукта.
Нерадиоактивные наборы возможно могут также включать в себя дополнительные компоненты, такие как трансхелатообразующий агент, радиопротектор, противомикробный консервант, рН-регулирующий агент или наполнитель. "Трансхелатообразующий агент" представляет собой соединение, которое быстро взаимодействует с образованием слабого комплекса с технецием, а затем вытесняется хелатообразующим агентом. Это минимизирует риск образования восстановленного гидролизованного технеция (RHT) вследствие быстрого восстановления пертехнетата, конкурирующего с процессом образования комплекса с технецием. Подходящими такими трансхелатообразующими агентами являются соли слабой органической кислоты, то есть органической кислоты, имеющей рКа в пределах от 3 до 7, с биосовместимым катионом. Под термином "биосовместимый катион" подразумевается положительно заряженный противоион, который образует соль с ионизированной, отрицательно заряженной анионной группой, где указанный положительно заряженный противоион также является нетоксичным и, следовательно, подходящим для введения в организм млекопитающего, в частности в организм человека. Примеры подходящих биосовместимых катионов включают щелочные металлы, натрий или калий; щелочно-земельные металлы, кальций и магний, и ион аммония. Предпочтительными биосовместимыми катионами являются натрий и калий, наиболее предпочтительно натрий. Подходящими такими слабыми органическими кислотами являются уксусная кислота, лимонная кислота, винная кислота, глюконовая кислота, глюкогептоновая кислота, бензойная кислота, фенолы или фосфоновые кислоты. Соответственно, подходящими солями являются ацетаты, цитраты, тартраты, глюконаты, глюкогептонаты, бензоаты, феноляты или фосфонаты. Предпочтительными такими солями являются тартраты, глюконаты, глюкогептонаты, бензоаты или фосфонаты, более предпочтительно фосфонаты, наиболее предпочтительно дифосфонаты. Предпочтительным таким трансхелатообразующим агентом является соль MDP, то есть метилендифосфоновой кислоты, с биосовместимым катионом.
Под термином "радиопротектор" подразумевается соединение, которое ингибирует реакции деградации, такие как окислительно-восстановительные процессы, путем захвата высокореакционноспособных свободных радикалов, таких как кислородсодержащие свободные радикалы, образующиеся в результате радиолиза воды. Радиопротекторы по настоящему изобретению соответственно выбраны из аскорбиновой кислоты, пара-аминобензойной кислоты (то есть 4-аминобензойной кислоты или РАВА), гентизиновой кислоты (то есть 2,5-дигидроксибензойной кислоты) и их солей с биосовместимым катионом, как он определен выше.
Термин "противомикробный консервант" означает агент, который подавляет рост потенциально опасных микроорганизмов, таких как бактерии, дрожжи или плесени. Противомикробный консервант может также проявлять некоторые бактерицидные свойства в зависимости от дозы. Главная роль противомикробного(ых) консерванта(ов) по настоящему изобретению заключается в подавлении роста любого такого микроорганизма в радиофармацевтической композиции после ее разведения, то есть в самом радиоактивном диагностическом продукте. Однако противомикробный консервант возможно может быть использован также для подавления роста потенциально опасных микроорганизмов в одном или более чем одном компоненте нерадиоактивного набора по настоящему изобретению перед разведением. Подходящие противомикробные консерванты включают парабены, то есть метил-, этил-, пропил- или бутилпарабен или их смеси, бензиловый спирт; фенол; крезол; цетримид и тиомерсал. Предпочтительным противомикробным(и) консервантом(ами) являются парабены.
Термин "рН-регулирующий агент" означает соединение или смесь соединений, используемое(ая) для обеспечения рН разведенного набора в приемлемых пределах (приблизительно рН от 4,0 до 10,5) для введения человеку или млекопитающему. Подходящие рН-регулирующие агенты включают фармацевтически приемлемые буферы, такие как трицин, фосфат или TRIS (то есть трис(гидроксиметил)аминометан), и фармацевтически приемлемые основания, такие как карбонат натрия, бикарбонат натрия или их смеси. При использовании конъюгата в форме соли с кислотой рН-регулирующий агент возможно может находиться в отдельном флаконе или контейнере, чтобы пользователь набора мог регулировать рН, выполняя это как часть многостадийной процедуры.
Под термином "наполнитель" подразумевается фармацевтически приемлемый, увеличивающий объем агент, который может облегчить обращение с материалом в процессе производства и лиофилизации. Подходящие такие наполнители включают неорганические соли, например хлорид натрия, и водорастворимые сахара или сахарные спирты, такие как сахароза, мальтоза, маннит или трегалоза.
В пятом воплощении настоящего изобретения предложено соединение формулы III
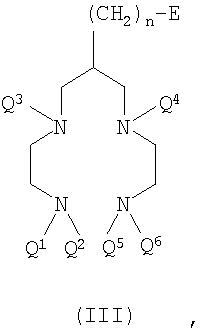
где Q1-Q6 и n такие, как определено для формул I и II выше;
Е представляет собой функциональную группу, подходящую для конъюгирования с обеспечивающей направленную доставку группировкой (Z) первого воплощения,
с условиями, что
1) когда n равно 3, тогда по меньшей мере один из Q1-Q6 представляет собой защитную группу для аминогрупп;
2) когда n равно 3 или 5, тогда Е не является ОН.
Соединение формулы III представляет собой "бифункциональный хелатообразующий агент", то есть хелатообразующий агент с одной или более присоединенными функциональными группами (Е). Функциональная группа (Е) является группой, подходящей для конъюгирования с биологической обеспечивающей направленную доставку группировкой (Z). Подходящие такие функциональные группы (Е) включают амин, тиоцианат, малеимид и активные сложные эфиры. Предпочтительно Е не содержит неактивированную гидроксильную (-ОН) группу. Термин "активный сложный эфир" означает эфирное производное карбоновой кислоты, которое является лучшей уходящей группой и, следовательно, обеспечивает более легкое взаимодействие с нуклеофилами, присутствующими на биологической обеспечивающей направленную доставку группировке, например аминами. Примерами подходящих активных сложных эфиров являются N-гидроксисукцинимид (NHS), пентафторфенол, пентафтортиофенол, пара-нитрофенол, гидроксибензотриазол и РуВОР (то есть гексафторфосфат бензотриазол-1-илокситрипирролидинофосфония). Предпочтительными активными сложными эфирами являются N-гидроксисукцинимидные или пентафторфенольные сложные эфиры.
Е предпочтительно представляет собой первичный амин (-NH2), -CO2M, -NCS, -NCO, малеимид или акриламид, где М представляет собой Н, катион, защитную группу или активный сложный эфир. Наиболее предпочтительно Е представляет собой -NH2, -CO2М или малеимид, идеально -NH2 или -CO2М.
Соединения формулы III подвергают взаимодействию с соответствующими эквивалентными функциональными группами на биологической обеспечивающей направленную доставку группировке (Z) до образования целевого конъюгата формулы II. Такие соответствующие функциональные группы на биологической обеспечивающей направленную доставку группировке включают:
карбоксилы (для образования амидной связи с амин-функционализированным бифункциональным хелатообразующим агентом);
амины (для образования амидной связи с карбоксил- или активный сложный эфир-функционализированным бифункциональным хелатообразующим агентом);
галогены, мезилаты и тозилаты (для N-алкилирования амин-функционализированого бифункциональным хелатообразующим агентом) и
тиолы (для взаимодействия с малеимид-функционализированным бифункциональным хелатообразующим агентом).
Когда Е представляет собой группу (например, активный сложный эфир), которая предназначена для взаимодействия с аминогруппой биологической обеспечивающей направленную доставку группировки (Z), ясно, что есть потенциальная возможность для нежелательных побочных реакций с аминогруппами хелатообразующего агента. Для таких групп Е Q1-Q6 в формуле III предпочтительно представляют собой защитные группы для азота, такие что каждый из четырех атомов азота амина тетрааминного хелатообразующего агента защищен. Когда Е представляет собой аминогруппу, ясно, что важно, чтобы реакция с биологической обеспечивающей направленную доставку молекулой (Z) происходила только по амину Е, а не по аминным атомам азота тетрааминного хелатообразующего агента. Поэтому в этой ситуации Q1-Q6 в формуле III предпочтительно представляют собой защитные группы для азота. Защитные группы для азота и их предпочтительные примеры описаны во втором воплощении (выше).
Соединения формулы III могут быть получены по Схемам 1 и 2. На Схеме 1 изображен гибкий путь синтеза карбокси-функционализированных, N-защищенных тетрааминных хелатообразующих агентов, который может быть адаптирован к множеству значений n в формуле III. Синтез Вос-защищенного тетрааминного аналога с заместителем -(CH2)5OH в голове мостика описан Turpin et al. [J.Lab. Comp. Radiopharm., 45, 379-393 (2002)]. На Схеме 2 представлен гибкий путь синтеза амин-функционализированных, N-защищенных тетрааминных хелатообразующих агентов, который может быть адаптирован к множеству значений n. Конъюгирование биологических обеспечивающих направленную доставку пептидов может быть осуществлено способом, аналогичным способам, описанным Nock et al. [Eur. J. Nucl. Med., 30(2), 247-258 (2003)] и Maina et al. [Ew. J. Nucl. Med., 30(9), 1211-1219 (2003)].
Схема 1: синтез соединения 1.

где ВОС - трет-бутоксикарбонильная защитная группа
IBX - 1-гидрокси-1,2-бензиодоксол-3(1Н)-он-1-оксид
NHS - N-гидроксисукцинимид
Bz-бензил.
Схема 2: синтез соединения 2.

где Ts - п-толуолсульфонил.
Изобретение иллюстрируют приведенные ниже не ограничивающие примеры. В Примере 1 описан синтез Соединения 1, представляющего собой карбокси-функционализированный N-защищенный тетрааминный хелатообразующий агент по настоящему изобретению. В Примере 2 описан синтез Соединения 2, представляющего собой амин-функционализированный N-защищенный тетрааминный хелатообразующий агент по настоящему изобретению. В Примере 3 представлен синтез Соединения 3, соединения, демонстрирующего конъюгирование Соединения 1 с амином (бензиламином). В Примере 4 описан синтез Соединения 6, иллюстрирующий конъюгирование Соединения 2 с активным сложным эфиром карбоновой кислоты. В Примере 5 описан синтез Соединения 4, представляющего собой конъюгат хелатообразующего агента по изобретению с производным лозартана. В Примере 6 представлен синтез конъюгата хелатообразующего агента с лозартаном, содержащего линкерную группу PEG. В Примере 7 описан синтез Соединения 8, представляющего собой конъюгат хелатообразующего агента по изобретению с ангиотензиновым пептидом. В Примере 8 описано 99mTc-радиомечение нескольких хелатообразующих агентов по изобретению. В Примере 9 представлены результаты измерений значений липофильности (logP) для различных 99mTc-комплексов по изобретению и показано, что эти комплексы относительно гидрофильны. В Примере 10 представлены результаты по биораспределению для нескольких 99mTc-комплексов по изобретению, демонстрирующие умеренный фон печени и высокий мочевой клиренс. В Примере 11 представлен синтез Соединения 1 с более высоким выходом. В Примере 12 представлен синтез защищенного тетрааминного хелатообразующего агента по настоящему изобретению с активированным сложным эфиром, конъюгированным с ним (Соединение 9).
Пример 1: синтез Соединения 1.
Стадия (а): диэтил[2-(бензилокси)этил]малонат
Это соединение получали путем модификации способа Ramalingam et al. [Ramalingam et al., Tetrahedron, 51, 2875-2894 (1995)]. Так, натрий (1,20 г) растворяли в абсолютном этаноле (25 мл) в атмосфере аргона. Добавляли диэтилмалонат (14,00 г) и эту смесь кипятили с обратным холодильником в течение 30 мин. Добавляли бензилбромэтиловый эфир (10 г) и эту смесь перемешивали при температуре дефлегмации в течение 16 часов. Роторным выпариванием удаляли этанол и остаток распределяли между эфиром (100 мл) и водой (50 мл). Эфирный слой промывали водой (3×50 мл) и сушили над сульфатом натрия. Эфир удаляли роторным выпариванием и остаток подвергали перегонке в вакууме. Фракцию, отгоняющуюся при 40-55°С, отбрасывали (непрореагировавший диэтилмалонат). Продукт перегоняли при 140-150°С (1 мм рт.ст.) [лит. т.кип. 138-140°С (1 мм рт.ст.)]. Выход составил 12,60 г бесцветного масла.
1H ЯМР (270 МГц, CDCl3, 25°C, TMS (тетраметилсилан)) δ=7.28 (m, 5H С6Н5), 4.47 (s, 2Н, СН2-Ph), 4.16 (m, 4Н, СООСН2), 3.58 (t, 1H, CH), 3.50 (t, 2H, O-СН2-СН2), 2.21 (t, 2H, O-СН2-СН2), 1.20 (t, 6H, СООСН2-СН2). 13С ЯМР (67,5 МГц, CDCl3, 25°C, TMS) δ=169.20 (CO), 138.10, 128.60, 127.80 (ароматический), 73.00 (CH2Ph), 67.30 (O-CH2-CH2), 61.70 (СООСН2), 49.10 (CH), 28.90 (O-CH2-CH2), 14.10 (СООСН2СН3).
Стадия (б): N,N'-бис(2-аминоэтил-2-(2-бензилоксиэтил)малонамид
Диэтил[2-(бензилокси)этил]малонат (4,00 г) добавляли к этилендиамину (30 мл) и раствор перемешивали при комнатной температуре в течение двух суток. Избыток этилендиамина удаляли роторным выпариванием и остаток сушили под высоким вакуумом в течение 2 суток до образования желтого масла (4,28 г), которое кристаллизовалось при стоянии. ЯМР-спектр показал, что продукт все еще содержит следы этилендиамина.
1H ЯМР (270 МГц, CDCl3, 25°С, TMS) δ=7.74 (br t, 2H, CO-NH), 7.32 (m, 5H, С6Н5), 4.46 (s, 2H, CH2-Ph), 3.50 (t, 2H, ОСН2-СН2-), 3.33 (t, 1H, CH), 3.23 (m, 4Н, CO-NH-СН2), 2.74 (t, 4Н, CH2-NH2), 2.18 (q, 2H, О-CH2-СН2-), 1.55 (br s, 4Н, NH2). 13C ЯМР (67,5 МГц, CDCl3, 25°C, TMS) δ=171.10 (CO), 138.20, 128.30, 127.70 (ароматический), 73.00 (CH2-Ph), 67.80 (O-CH2-CH2), 51.40 (CH), 42.40 (CO-NH-CH2), 41.20 (CH2-NH2), 31.90 (O-CH2-CH2-).
Стадия (в): N,N'-бис(2-аминоэтил)-2-(2-бензилоксиэтил)-1,3-диаминопропан
N,N'-бис(2-аминоэтил)-2-(2-бензилоксиэтил)малонамид (3,80 г) растворяли в ТГФ (20 мл) и колбу погружали в ледяную баню. Колбу продували струей аргона и через шприц добавляли ТГФ-борановый комплекс (80 мл, 1 М в ТГФ). Реакционной смеси давали возможность нагреться до комнатной температуры, а затем перемешивали ее при 40°С в течение 2 суток и кипятили с обратным холодильником в течение 1 часа. По каплям добавляли метанол (50 мл) и раствор перемешивали при 40°С в течение ночи. Растворители удаляли на роторном испарителе и остаток растворяли в метаноле (20 мл). Добавляли гидроксид натрия (10 г в 15 мл воды), и метанол выкипал. Выделялось бесцветное масло, которое экстрагировали в СН2СН2 (3×50 мл). Этот раствор сушили над Na2SO4. После удаления растворителя получили 3,40 г бесцветного масла.
1H ЯМР (270 МГц, CDCl3, 25°C, TMS) δ=7.34 (m, 5H, C6H5), 4.49 (s, 2H, CH2-Ph), 3.55 (t, 2H, OCH2-CH2-), 2.76 (t, 4H, N-CH2), 2.63 (m, 8H, N-CH2), 1.84 (m, 1H, CH), 1.58 (m, 2H, СН-СН2-СН2-O), 1.41 (br s, 6H, NH). 13C ЯМР (67,5 МГц, CDCl3, 25°C, TMS) δ=138.60, 128.30, 127.60 (ароматический), 72.80 (CH2-Ph), 68.70 (O-СН2-СН2), 53.50 (N-CH2), 52.80 (N-CH2), 41.60 (N-CH2), 36.40 (CH), 31.30 (СН-СН2-СН2-O). МС-ЭУ (масс-спектрометрия с ионизацией электронным ударом): 295 [M+H]+, (вычисл. 295,2).
Стадия (г): N,N'-бис(2-трет-бутоксикарбониламиноэтил)-2-(2-бензилоксиэтил)-1,3-ди(трет-бутоксикарбониламино)пропан
N,N'-бис(2-аминоэтил-2-(2-бензилоксиэтил)-1,3-диаминопропан (3,30 г) растворяли в СН2Cl2 (100 мл) и добавляли триэтиламин (5,40 г) и трет-бутилдикарбонат (10,30 г). Реакционную смесь перемешивали при комнатной температуре в течение 2 суток. Смесь промывали водой (100 мл), раствором лимонной кислоты (100 мл, 10% в воде) и водой (2×100 мл). Органический слой сушили над Na2SO4 и растворитель удаляли роторным выпариванием с получением желтого масла, которое сушили до постоянной массы под высоким вакуумом. Неочищенный продукт (7,70 г) очищали на силикагелевой колонке (250 г, 230-400 меш, CH2Cl2, CH2Cl2-Et2O, 1:1) с получением 6,10 г (78,3%) прозрачного масла.
1H ЯМР (270 МГц, CDCl3, 25°С, TMS) δ=7.32 (m, 5H, C6H5), 5.12 (br d, 2H, NH), 4.47 (s, 2H, CH2-Ph), 3.49 (t, 2H, OCH2-CH2-), 3.24 (br, 12H, N-CH2), 2.14 (br, 1H, CH), 1.59 (m, 2H, CH-CH2-CH2-O), 1.45 (s, 18H, t-Bu), 1.42 (s, 18H, t-Bu). 13C ЯМР (67,5 МГц, CDCl3, 25°C, TMS) δ=155.90 (NH-CO), 138.20, 128.30, 127.60, 127.50 (ароматический), 79.90, 78.90 (СМе3), 72.80 (CH2-Ph), 68.00 (O-СН2-СН2), 50.00 (br, N-CH2), 46.90 (br, N-CH2), 39.20 (N-CH2), 34.40 (br, CH), 29.80 (СН-СН2-СН2-O), 28.30 (t-Bu). МС-ЭУ: 695 [M+H]+, (вычисл. 695,5).
Стадия (д): N,N'-бис(2-трет-бутоксикарбониламиноэтил)-2-(2-гидроксиэтил)-1,3-ди(трет-бутоксикарбониламино)пропан
N,N'-бис(2-трет-бутоксикарбониламиноэтил)-2-(2-бензилоксиэтил)-1,3-ди(трет-бутоксикарбониламино)пропан (3,16 г) растворяли в абсолютном этаноле (100 мл) и добавляли Pd на активированном угле (1,00 г, сухой, 10%).
Смесь гидрировали в аппарате Парра при давлении 35 фунт/кв.дюйм (241,5 кПа) в течение двух суток. Катализатор отфильтровывали, промывали этанолом (3×20 мл). Этанол удаляли роторным выпариванием с получением бесцветного масла, которое сушили до постоянной массы (2,67 г, 97,1%) под высоким вакуумом.
1H ЯМР (270 МГц, CDCl3, 25°С, TMS) δ=5.25 (br d, 2H, NH), 3.69 (t, 2H, OCH2-СН2-), 3.28 (br, 12H, N-CH2), 2.71 (br, ОН), 2.23 (br, 1H, СН), 1.56 (уступ, m, 2H, СН-СН2-СН2-O), 1.48 (s, 18H, t-Bu), 1.44 (s, 18H, t-Bu). 13C ЯМР (67,5 МГц, CDCl3, 25°C, TMS) δ=156.10 (NHCO), 80.00, 79.20 (СМе3), 59.60 (O-СН2-СН2), 49.90 (br, N-CH2), 47.00 (br, N-CH2), 39.34 (N-CH2), 33.80 (СН), 32.30 (СН-СН2-СН2-O), 28.30 (t-Bu). МС-ЭУ: 605 [M+H]+, (вычисл. 605,4).
Стадия (е): N,N'-бис(2-трет-бутоксикарбониламиноэтил)-2-(2-карбоксиметил)-1,3-ди(трет-бутоксикарбониламино)пропан (Соединение 1)
Использовали способ Mazitschek et al. [Ang. Chem. Int. Ed., 41, 4059-4061 (2002)]. Так, N,N'-бис(2-трет-бутоксикарбониламиноэтил)-2-(2-гидроксиэтил)-1,3-ди(трет-бутоксикарбониламино)пропан (2,60 г) растворяли в DMSO (диметилсульфоксид) (15 мл) и 1-гидрокси-1,2-бензиодоксол-3(1Н)-он-1-оксиде (IBX, 3,50 г). Смесь перемешивали при комнатной температуре в течение 1 часа, затем добавляли N-гидроксисукцинимид (2,50 г). Эту реакционную смесь перемешивали при комнатной температуре в течение 2 суток. Добавляли раствор гидроксида натрия (2 М, 40 мл) и эту смесь перемешивали при комнатной температуре в течение 4 часов. Раствор погружали в ледяную баню и подкисляли 2 М соляной кислотой до рН 2. Водный слой экстрагировали эфиром (4×100 мл) и объединенные эфирные экстракты промывали водой (3×50 мл). Эфирный слой сушили над Na2SO4 и растворитель удаляли роторным выпариванием с получением желтого твердого остатка, который содержал продукт и 2-иодозобензойную кислоту. Большую часть иодозобензойной кислоты (2,1 г) удаляли кристаллизацией из смеси хлороформ-гексаны (1:3) (80 мл). Выпаривание хлороформ-гексановой маточной жидкости дало желтое масло (3 г), которое наносили на силикагелевую колонку (300 г, CH2Cl2-Et2O, 1:1). Оставшуюся иодозобензойную кислоту элюировали эфиром. Продукт элюировали смесью эфир-метанол (9:1). Фракции, содержащие продукт, объединяли и после удаления растворителя получали 1,5 г бледно-желтого масла. Его подвергали хроматографии на силикагелевой колонке (50 г, Et2O). Продукт элюировали смесью эфир-уксусная кислота (95:5). Фракции, содержащие продукт, объединяли и растворитель удаляли роторным выпариванием с получением масла, которое сушили под высоким вакуумом. Выход составил 1,10 г (41,3%).
1H ЯМР (270 МГц, CDCl3, 25°C, TMS) δ=7.61 (br s, 1H, COOH), 5.19 (br d, 2H, NH), 3.22 (br, 12H, N-CH2), 2.47 (br m, 1H, CH), 2.26 (br, 2H, CH-CH2-COOH), 1.41 (s, 18H, t-Bu), 1.37 (s, 18H, t-Bu). 13C ЯМР (67,5 МГц, CDCl3, 25°C, TMS) δ=175.90 (COOH), 156.10 (NHCO), 80.40, 79.10 (СМе3), 49.50 (N-CH2), 46.80 (N-CH2), 39.00 (N-CH2), 34.70 (СН-СН2-СООН), 34.20 (СН-СН2-СООН), 28.30, 28.20 (t-Bu). МС-ЭУ: 619 [М+Н]+, (вычисл. 619,4).
Пример 2: Синтез трет-бутилового эфира (8-амино-2-{[трет-бутоксикарбонил-(2-трет-карбониламиноэтил)амино]метил}октил-(2-трет-бутилоксикарбониламиноэтил)карбаминовой кислоты (Соединения 2)
Стадия (а): 2-(6-хлоргексилокси)тетрагидропиран
6-Хлоргексанол (6,85 г, 10 ммоль) и п-толуолсульфоновую кислоту (500 мг) растворяли в сухом эфире (75 мл) и охлаждали до 0-5°С в ледяной бане. По каплям добавляли дигидропиран (4,3 г, 10 ммоль) в сухом эфире (25 мл) при постоянном перемешивании за период времени 30 минут. После окончания добавления охлаждающую баню убирали и перемешивание продолжали в течение 16 часов. Раствор экстрагировали водой (50 мл × 2), сушили (MgSO4), фильтровали и растворитель выпаривали при пониженном давлении с получением бледно-желтого масла. 13С ЯМР спектроскопия показала, что это масло достаточно чистое для использования без очистки в последующих реакциях. Выход 10,1 г (91%).
13С ЯМР (CDCl3) δ=19.7 (СН2), 25.5 (СН2), 25.6 (СН2), 26.7 (СН2), 29.6 (СН2), 30.8 (СН2), 32.6 (СН2), 45.0 (CH2Cl), 62.3 (ОСН2), 67.4 (ОСН2), 98.8 (ОСНО).
1H ЯМР (CDCl3): δ 1.30-1.71 (14Н, m, СН2 × 7), 3.24-3.32 (1H), 3.41-3.48 (3Н, m, CH и СН2), 3.60-3.67 (1H, m, CH), 3.72-3.82 (1H, bm, CH), 4.44-4.49 (1H, bm, ОСНО).
Стадия (б): диэтиловый эфир 2-[6-(тетрагидропиран-2-илокси)гексил]малоновой кислоты
Натрий (1,13 г, 49 ммоль) небольшими количествами растворяли в сухом этаноле (100 мл) при постоянном перемешивании под подушкой сухого азота. Одной порцией добавляли диэтилмалонат (8,0 г, 50 ммоль) и раствор нагревали при 60°С в течение 1 часа. Одной порцией добавляли 2-(6-хлоргексилокси)тетрагидропиран (9,3 г, 42,2 ммоль), температуру поднимали до 75-80°С и выдерживали при этой температуре в течение 24 часов. После охлаждения неорганическое твердое вещество удаляли фильтрованием и растворитель выпаривали из фильтрата. Остаток растворяли в CH2Cl2 (50 мл), экстрагировали водой (30 мл × 2), сушили (MgSO4), фильтровали и удаляли летучие вещества с получением светло-желтого масла. Это масло подвергали хроматографии на силикагеле с использованием смеси петролейный эфир (40:60)/эфир (200:40) в качестве элюента. Целевой продукт элюировался с rf=0,15 и был выделен в виде бесцветного масла. Выход 8,7 г (60%).
13С ЯМР (CDCl3): δ 14.0 (СН3 × 2), 19.6 (СН2), 25.5 (СН2), 27.2 (CH2), 28.6 (CH2), 29.0 (CH2), 29.6 (CH2), 30.0 (CH2), 30.8 (CH2), 52.0 (CH), 61.2 (ОСН2 × 2), 62.2 (ОСН2), 76.4 (ОСН2), 98.8 (OCHO), 169.4 (С=O × 2).
1H ЯМР (CDCl3): δ 1.10-1.25 (14H, m, СН2 × 2, CH2 × 4), 1.36-1.50 (6Н, bm, СН2 × 3), 1.70-1.81 (2Н, bm, СН2), 3.17-3.28 (2Н, m, СН2), 3.56-3.66 (1Н, m, CH), 3.70-3.80 (1Н, m, ОСН), 4.04-4.16 (4Н, m, OCH2 × 2), 4.03-4.08 (1Н, m, OCHO).
Стадия (в): N,N'-бис-(2-аминоэтил)-2-[6-(тетрагидропиран-2-илокси)гексил]малонамид
Диэтиловый эфир 2-[6-(тетрагидропиран-2-илокси)гексил]малоновой кислоты (5,1 г, 14,8 ммоль) растворяли в 1,2-диаминоэтане (10 г, 167 ммоль) и перемешивали при комнатной температуре в течение 16 часов. Летучие вещества удаляли в вакууме (40-50°С при 0,01 мм рт.ст.) с получением светло-зеленого вязкого остатка, который подвергали колоночной хроматографии, элюируя смесью CH2Cl2/MeOH/NH4OH
(50:50:5). Указанное в заголовке соединение элюировалось с rf=0,2 и было собрано в виде светло-зеленого вязкого масла, которое затвердевало при стоянии (выход 3,9 г, 71%).
13С ЯМР (CDCl3): δ 19.8 (СН2), 25.5 (СН2), 26.0 (СН2), 27.5 (СН2), 29.2 (СН2), 29.7 (СН2), 30.8 (СН2), 31.9 (СН2), 41.0 (NCH2 × 2), 41.9 (NCH2 × 2), 54.6 (CH), 62.5 (ОСН2), 67.5 (ОСН2), 98.9 (OCHO), 171.6 (С=O × 2).
1H ЯМР (CDCl3): δ 1.15-1.28 (6Н, bs, CH2 × 3), 1.39-1.44 (6Н, bm, CH2 × 3), 1.69-1.74 (4Н, bm, CH2 × 2), 2.64 (4Н, bs, NH2 × 2), 2.73 (4Н, t, J=6 Гц, CH2 × 2), 3.08-3.29 (6Н, m,
CH2 × 3), 3.35-3.41 (1Н, m, CH), 3.55-3.63 (1Н, m, CH), 3.70-3.78 (1Н, m, CH), 4.43 (1Н, bt, J=4 Гц, OCHO), 7.78 (2Н, bt, J=5 Гц, OCNH × 2).
ИК (тонкая пленка), см-1: 3417, 3082, 2936, 2862, 1663, 1558, 1439, 1354, 1323, 1261, 1200, 1189, 1076, 1026, 956, 907, 867, 810.
Стадия (г): N,N'-бис(2-аминоэтил)-2-(6-гидроксигексил]малонамид
N,N'-Бис(2-аминоэтил)-2-[6-(тетрагидропиран-2-илокси)гексил]малонамид (3,9 г, 10,6 ммоль), моногидрат п-толуолсульфоновой кислоты (8,5 г, 3 ммоль) и этанол (50 мл) нагревали с обратным холодильником при 70-75°С в течение 16 часов. После охлаждения по каплям добавляли концентрированный гидроксид аммония (.880) до достижения постоянного значения рН 9. Выпавшее в осадок белое твердое вещество удаляли фильтрованием через целит и осадок на фильтре промывали этанолом (30 мл). Этанол удаляли при пониженном давлении (15 мм рт.ст., 40°С) с получением полутвердого воска. Этот остаток подвергали хроматографии на силикагеле, элюируя смесью CH2Cl2/MeOH/NH4OH (100:50:10), было обнаружено, что указанное в заголовке соединение имеет rf=0,2. Этот продукт собирали и выпаривали совместно с этанолом (100 мл × 3) для удаления остаточной воды. Был получен светло-зеленый вязкий остаток, который затвердевает при стоянии (выход 2,1 г, 69%).
13С ЯМР (CD3OD): δ 25.4 (CH2), 27.3 (CH2), 28.9 (CH2), 30.4 (CH2), 32.2 (CH2), 40.6 (NCH2 × 2), 41.7 (NCH2 × 2), 54.1 (CH), 61.6 (CH2OH), 171.7 (С=O × 2).
1H ЯМР (CD3OD): δ 1.28-1.38 (6Н, bs, CH2 × 3), 1.46-1.55 (2Н, bm, CH2), 1.79-1.87 (2Н, bm, CH2), 2.73 (4Н, t, J=6 Гц, H2NCH2 × 2), 3.13 (1Н, t, J=7 Гц, CH), 3.27 (4Н, dt, J=6 и 2 Гц, HNCH2 × 2), 3.53 (2Н, t, J=7 Гц, ОСН2).
ИК (тонкая пленка), см-1: 3364, 2932, 2862, 2527, 1663, 1558, 1462, 1327, 1223, 1192, 1034.
Масс-спектр (ББА (бомбардировка быстрыми атомами)) m/e. Вычислено для С13Н29N4O3 (М+Н) 289. Найдено 289.
Стадия (д): трет-бутиловый эфир (2-трет-бутоксикарбониламиноэтил-2-{[(трет-бутоксикарбонил-(2-трет-бутоксикарбониламиноэтил)амино]метил}-8-гидроксиоктил)карбоновой кислоты
Под подушкой сухого азота чистый аддукт боран-диметилсульфид (15 мл, 150 ммоль) добавляли по каплям через шприц к перемешиваемой смеси N,N'-бис(2-аминоэтил)-2-(6-гидроксигексил)малонамида (2,1 г, 7,3 ммоль) в диоксане (50 мл). После окончания добавления смесь осторожно нагревали с обратным холодильником при 110°С в течение 5 суток. За это время осталось некоторое количество белого твердого вещества. После охлаждения летучие вещества удаляли при пониженном давлении с получением белого твердого вещества, к которому по каплям добавляли метанол (50 мл) с получением бесцветного раствора. Этот раствор нагревали с обратным холодильником в течение 3 часов, охлаждали, добавляли концентрированную HCl (5 мл) и нагревание с обратным холодильником продолжали при 70-75°С в течение 48 часов. Растворитель удаляли с получением вязкого зеленого остатка, который подвергали совместному выпариванию с метанолом (100 мл × 3) с получением светло-зеленого твердого вещества. Это твердое вещество снова растворяли в сухом метаноле и добавляли безводный карбонат калия (4,0 г, 30 ммоль), а затем ди-трет-бутилдикарбонат (7,0 г, 32 ммоль). Смесь перемешивали при комнатной температуре в течение 48 часов. Неорганическое твердое вещество удаляли фильтрованием через целит и растворитель выпаривали из фильтрата с получением вязкого остатка. Этот остаток смешивали с водой (50 мл) и экстрагировали CH2Cl2 (50 мл × 3). Органические фракции объединяли, сушили (MgsO4), фильтровали и растворитель выпаривали с получением светло-желтого остатка.
Примечание: в этой точке удобно проконтролировать реакцию по данным 13С ЯМР.
Остаток подвергали хроматографии на силикагеле с использованием смеси CH2Cl2/MeOH (95:5) в качестве элюента. Указанное в заголовке соединение элюировалось с rf=0,41 и его выделяли в виде бесцветного вязкого масла (выход 2,5 г, 52%).
13С ЯМР (CDCl3): δ 25.6 (CH2), 26.4 (СН2), 28.4 (СН2 × 12), 29.8 (CH2 × 2), 32.6 (СН2), 36.5 (очень широкий, СН), 39.2 (NCH2 × 2, соседний СН), 46.9 (широкий синглет, HNCH2 × 2), 50.0 (широкий синглет, NCH2 × 2), 62.4 (HOCH2), 79.0 (ОС × 2), 79.9 (ОС × 2), 156.4 (широкий синглет, С=O × 4).
1H ЯМР (CDCl3): δ 1.05-1.18 (8Н, bs, CH2 × 4), 1.27 (18H, s, СН2 × 6, трет-бутил), 1.31 (18H, s, СН2 × 6, трет-бутил), 1.41 (2H, m, СН2), 1.81 (1Н, bs, СН), 2.63 (1Н, bs, ОН), 2.98 (4Н, bs, NCH2 × 2), 3.11 (8H, bs, NCH2 × 4), 3.44 (2Н, t, J=8 Гц, СН2O), 5.2 (2Н, bs, NH × 2).
ИК (тонкая пленка), см-1: 3350, 2976, 2931, 2859, 1674, 1516, 1455, 1418, 1393, 1260, 1250, 1165, 1069, 965, 871, 775.
Масс-спектр (ББА) m/e. Вычислено для C33H65N4O9 (M+H) 661. Найдено 661.
Стадия (е): 8-[трет-бутоксикарбонил-(2-трет-бутоксикарбониламиноэтиламино]-7-{[трет-бутоксикарбонил-(2-трет-бутоксикарбониламиноэтил)амино]метил}октиловый эфир толуол-4-сульфоновой кислоты
трет-Бутиловый эфир (2-трет-бутоксикарбониламиноэтил-2-{[(трет-бутоксикарбонил-(2-трет-бутоксикарбониламиноэтил)амино]метил}-8-гидроксиоктил)карбоновой кислоты (2,52 г, 3,82 ммоль), п-толуолсульфонилхлорид (1,0 г, 5,2 ммоль), триэтиламин (1,3 г, 12,8 ммоль) и CH2Cl2 (30 мл) перемешивали при комнатной температуре с медленным выпариванием растворителя. Реакцию контролировали 13С ЯМР, и через 3 суток осталось небольшое количество исходных веществ. Объем реакционной смеси доводили до 30 мл добавлением CH2Cl2, экстрагировали водой (50 мл × 3), сушили (MgsO4), фильтровали и растворитель выпаривали с получением коричневого остатка. Этот остаток подвергали хроматографии на силикагеле с использованием смеси CH2Cl2/MeOH (100:5) в качестве элюента. Первым элюировался непрореагировавший тозилхлорид с rf=0,95. Указанное в заголовке соединение элюировалось с rf=0,2, и его выделяли в виде светло-желтого вязкого масла (выход 1,20 г, 39%).
13С ЯМР (CDCl3): δ 21.7 (СН3, тозил), 25.3 (СН2), 26.3 (СН2), 28.5 (СН2 × 12), 28.8 (СН2), 29.5 (СН2), 36.5 (СН очень широкий), 39.4 (NCH2 × 2), 47.0 (широкий NCH2 × 2), 50.5 (широкий, NCH2 × 2), 70.6 (TsOCH2), 79.1 (ОС × 2), 80.0 (ОС × 2), 127.9 (СН × 2), 129.9 (СН × 2), 133.2 (С), 144.7 (C-S Ts), 156.1 (широкий, С=O × 4).
1H ЯМР (CDCl3): δ 1.16 (8H, bs, СН2 × 4), 1.35 (18Н, s, СН2 × 6), 1.39 (18H, s, СН3 × 6), 1.88 (1Н, bs, СН), 2.38 (3Н, s, СН2, тозил), 3.10-3.12 (4Н, bs, NCH2 × 2), 3.19 (8H, bs, NCH2 × 4), 3.93 (2Н, t, J=7 Гц, CH2OTs), 5.0 (1Н, bs, NH), 5.08 (1H, bs, NH), 7.29 (2Н, d, J=8 Гц, СН × 2, Ar), 7.72 (2Н, d, J=8 Гц, СН × 2, Ar).
ИК (тонкая пленка), см-1: 3360, 2974, 2932, 2862, 1693, 1516, 1479, 1418, 1391, 1366, 1250, 1177, 1069, 959, 816, 775.
Масс-спектр (ББА) m/e. Вычислено для C40H71N4O11S (M+H) 815. Найдено 815.
Стадия (ж): трет-бутиловый эфир (8-азидо-2-{[трет-бутоксикарбонил-(2-трет-карбониламиноэтил)амино]метил}октил)-(2-трет-бутоксикарбониламиноэтил)карбаминовой кислоты
8-[трет-Бутоксикарбонил-(2-трет-бутоксикарбониламиноэтиламино]-7-{[трет-бутоксикарбонил-(2-трет-бутоксикарбониламиноэтил)амино]метил}октиловый эфир толуол-4-сульфоновой кислоты (1,105 г, 1,36 ммоль), азид натрия (350 мг, 5,4 ммоль) и метанол (10 мл) нагревали с обратным холодильником при 70-75°С в течение 16 часов. После охлаждения метанол удаляли при комнатной температуре при пониженном давлении до тех пор, пока не осталось примерно 1-2 мл. Этот остаток разбавляли водой (25 мл) и экстрагировали CH2Cl2 (25 мл × 4). Органические экстракты объединяли, сушили (MgSO4), фильтровали и летучие вещества выпаривали при комнатной температуре (примечание: азиды потенциально взрывоопасны, и эту стадию следует проводить за защитным экраном) с получением светло-желтого вязкого остатка, который представлял собой целевое соединение в чистом состоянии (выход 820 мг, 88%).
13С ЯМР (CDCl3): δ 26.3 (СН2), 26.5 (CH2), 28.3 (СН2 × 12), 28.7 (CH2), 29.6 (CH2), 29.8 (СН2), 36.8 (широкий, СН), 39.3 (NCH2 × 2), 46.9 (широкий, NCH2 × 2), 50.0 (широкий, NCH2 × 2), 51.3 (CH2N3), 79.0 (ОС × 2), 79.8 (ОС × 2), 156.0 (С=O × 4).
1H ЯМР (CDCl3): δ 1.16 (8Н, bs, CH2 × 4), 1.29 (18H, s, СН2 × 6), 1.33 (18Н, s, СН2 × 6), 1.47 (2Н, bt, J=6.5 Гц, СН2, соседний СН), 1.86 (1Н, bs, СН), 2.95-3.05 (4Н, bs, NCH2 × 2), 3.05-3.20 (10Н, bs, NCH2 × 4 и CH2N3), 5.09 (2Н, bs, NH × 2).
ИК (тонкая пленка), см-1: 3350, 2974, 2932, 2860, 2097 (сильная полоса N3), 1694, 1520, 1470, 1418, 1391, 1366, 1250, 1167, 1069, 870, 777.
Стадия (з): трет-бутиловый эфир (8-амино-2-{[трет-бутоксикарбонил-(2-трет-карбониламиноэтил)амино]метил}октил)-(2-трет-бутилоксикарбониламиноэтил)карбаминовой кислоты (Соединение 2)
трет-Бутиловый эфир (8-азидо-2-{[трет-бутоксикарбонил-(2-трет-карбониламиноэтил)амино]метил}октил)-(2-трет-бутоксикарбониламиноэтил)карбаминовой кислоты (820 мг, 1,20 ммоль), 10% палладий на древесном угле (100 мг) и метанол (10 мл) обрабатывали газообразным водородом при давлении 30 атмосфер (примерно 3000 кПа) при комнатной температуре в течение 16 часов. Твердые вещества удаляли фильтрованием через целит и осадок на фильтре промывали метанолом (50 мл). Летучие вещества удаляли из фильтрата с получением вязкого масла, которое представляло собой целевое соединение в чистом состоянии (выход 700 мг, 89%).
13С ЯМР (CDCl3): δ 26.4 (CH2), 26.6 (CH2), 28.4 (СН2 × 12), 32.9 (CH2 × 2), 36.8 (широкий, СН), 39.2 (NCH2 × 2), 41.8 (H2NCH2), 46.9 (широкий, NCH2 × 2), 49.8 (широкий, NCH2 × 2), 78.9 (ОС × 2), 79.7 (ОС × 2), 156.0 (С=O × 4).
1H ЯМР (CDCl3): δ 1.08 (8H, bs, CH2 × 4), 1.23 (18Н, s, СН2 × 6), 1.27 (20Н, bs, СН2 × 6 и CH2), 1.77 (1Н, bs, СН), 2.40 (2Н, bs, NH2), 2.50 (2H, t, J=7 Гц, CH2NH2), 2.97 (4Н, bm, NCH2 × 2), 3.00-3.16 (8H, bm, NCH2 × 4), 5.21 (1H, bs, NH), 5.30 (1H, bs, NH).
ИК (тонкая пленка), см-1: 3360, 1693, 1520, 1459, 1418, 1392, 1367, 1250, 1170, 1068, 964, 922, 871, 775, 733.
Масс-спектр (ББА) m/e. Вычислено для C33H66N5O8 (M+H) 660. Найдено 660.
Пример 3: синтез Соединения 3.
Стадия (а): сочетание Соединения 1 с бензиламином
Соединение 1 (61,8 мг, 0,1 ммоль) в СН2Cl2 (5 мл) обрабатывали бензиламином (10,7 ммоль), хлорангидридом дифенилфосфиновой кислоты (25,9 мг) и диизопропиламидом (29 мг, 0,22 ммоль) в круглодонной колбе емкостью 50 мл при 20°С в течение 18 часов. Затем реакционную смесь разбавляли CH2Cl2 (20 мл) и промывали 1 н. соляной кислотой (5 мл) и насыщенным водным карбонатом калия (5 мл). СН2Cl2-слой отделяли, сушили (Na2SO4) и концентрировали в вакууме до смолы (приблизительно 50 мг). Это неочищенное вещество затем подвергали хроматографии на силикагеле с градиентом этилацетата в бензине (по 100 мл каждого 50%, 70% и 90%). Собирали небольшое количество быстрее текущей примеси, а затем непосредственно основную фракцию.
1H и 13С ЯМР спектры снимали в CDCl3. Эти спектры показали, что основная фракция представляла собой чистое целевое соединение.
Стадия (б): удаление Вос-защитных групп
Продукт со стадии (а) (27,8 мг, 0,039 ммоль) в СН2Cl2 (0,5 мл) обрабатывали трифторуксусной кислотой (0,5 мл) и реакционную смесь оставляли стоять при комнатной температуре в течение 3 часов. Затем реакционную смесь концентрировали в вакууме до смолы для удаления избыточной кислоты и взвешивали (53 мг). 1H и 13С ЯМР (CDCl3) спектры показали, что Boc-группы были удалены полностью. Взвешенный образец Соединения 2 растворяли в воде с получением 10 ммолярного раствора соли трифторуксусной кислоты (TFA), которую использовали для экспериментов по радиомечению.
Пример 4: синтез Соединения 6.
Стадия (а): трет-Бутиловый эфир (8-бензоиламино-2-{[трет-бутоксикарбонил-(2-трет-карбониламиноэтил)амино]метил}октил)-(2-трет-бутилоксикарбониламиноэтил)карбаминовой кислоты
2,5-Диоксопирролидин-1-иловый эфир бензойной кислоты (20 мг, 0,091 ммоль) в сухом CH2Cl2 добавляли одной порцией к раствору Соединения 2 (50 мг, 008 ммоль) в CH2Cl2 (1 мл) и этот раствор перемешивали при комнатной температуре в течение 16 часов. Эту реакционную смесь разбавляли CH2Cl2 (10 мл), экстрагировали водой (15 мл × 2), сушили (MgSO4), фильтровали и растворитель удаляли роторным выпариванием. Оставшийся остаток очищали хроматографией на силикагеле с использованием смеси СН2Cl2/метанол (94:6, rf=0,23) в качестве элюента с получением бесцветного вязкого масла (выход 25 мг, 41%).
13С ЯМР (CDCl3): δ 26.4 (СН2), 26.8 (CH2), 28.5 (СН2 × 12), 29.6 (СН2 × 2), 29.7 (СН2), 29.9 (СН2), 36.6 (широкий, СН), 39.4 (NCH2 × 2), 40.0 (O=CNCH2 × 2), 47.0 (широкий, NCH2 × 2), 49.8 (NCH2 × 2), 79.7 (ОС × 2), 80.0 (ОС × 2), 127.0 (Ar СН × 2), 128.5 (Ar СН × 2), 131.3 (Ar СН), 134.9 (Ar C), 156.1 (С=O × 4), 167.6 (ArC=O).
1H ЯМР (CDCl3): δ 1.28 (8H, bs, СН2 × 4), 1.38 (18Н, s, СН2 × 6), 1.42 (20Н, bs, СН2 × 6 и СН2), 1.95 (1Н, bs, СН), 3.1 (4Н, bs, NCH2 × 2), 3.22 (8H, bs, NCH2 × 4), 3.42 (2Н, bq, J=6 Гц, СН2N-бензоил), 5.08 (2Н, bs, NH × 2), 6.18 (1Н, bs, NH-бензоил), 7.38-7.45 (3Н, m, Ar СН × 3), 7.74 (2Н, bd, J=7 Гц, Ar СН × 2).
ИК (тонкая пленка), см-1: 3350, 2976, 2932, 2859, 1693 (широкий), 1652, 1520, 1419, 1391, 1367, 1251, 1166, 732.
Масс-спектр (ББА) m/e. Вычислено для C40H70N5O9 (M+H) 764. Найдено 764.
Стадия (б): удаление Вос-защитных групп
Вос-тетрааминбензамид со стадии (а) (42 мг, 0,056 ммоль) в CH2Cl2 (0,5 мл) обрабатывали трифторуксусной кислотой (0,5 мл) и реакционную смесь оставляли стоять при комнатной температуре в течение 3 часов. Затем реакционную смесь концентрировали в вакууме для удаления избыточной кислоты с получением смолы. Ожидаемая масса (45 мг), найденная масса (45,7 мг). 1H и 13С ЯМР (CD3OD) спектры показали, что Вос-группы были удалены полностью и что она содержала целевое соединение. Взвешенный образец этого соединения растворяли в воде с получением 10 ммолярного раствора соли трифторуксусной кислоты (TFA) (56 мкмоль в 5,6 мл), которую использовали для экспериментов по радиомечению.
Пример 5. Синтез Соединения 4.
Все реакции проводили в ручном аппарате-барботере азота.
Стадия (а): присоединение лозартана к тритил-дериватизированной твердой подложке

Лозартан (MSD, 0,236 г, 0,558) и триэтиламин (Fluka, 0,233 мл, 1,67 ммоль) добавляли к суспензии тритилхлоридной смолы (Novabiochem, замещение 1,24 ммоль/г, 0,300 г) в DMF (диметилформамид) (5 мл). Через 4 суток эту смолу фильтровали и промывали. Аликвоту этой смолы расщепляли (дихлорметан/TFA/триизопропилсилан, 92,5:5,0:2,5, 15 минут). ВЭЖХ-анализ (колонка Phenomenex Luna C18(2) 3 мкм, 4,6×50 мм, растворители: А=вода/0,1% TFA и Б = ацетонитрил/0,1% TFA; градиент 10-40% Б за 10 минут; скорость потока 2,0 мл/мин, УФ-детектирование при 214 и 254 нм) дал пик с tR 6,7 минут, что соответствует лозартану. Смолу обрабатывали раствором дихлорметан/метанол/диизопропилэтиламин (17:2:1, 20 мл, 1 ч), промывали дихлорметаном и сушили.
Стадия (б): замещение гидроксильной группы азидом

Дифенилфосфорилазид (Aldrich, 0,481 мл, 2,23 ммоль) и DBU (1,8-диазабицикло[5.4.0]ундец-7-ен) (0,611 мл, 4,09 ммоль) добавляли к суспензии связанного со смолой лозартана со стадии (а) (0,372 ммоль) в THF (тетрагидрофуран) (10 мл). Реакционную смесь оставляли стоять в течение ночи. Аликвоту смолы расщепляли, как описано для стадии (а). Анализ методом ЖХ-МС (жидкостная хроматография-масс-спектрометрия) (колонка Phenomenex Luna C18(2) 3 мкм, 50×4,60 мм, растворители: А = вода/0,1% TFA и Б = ацетонитрил/0,1% TFA; градиент 20-80% Б за 10 минут; скорость потока 1,0 мл/мин, УФ-детектирование при 214 нм, МС-ЭРИ) дал пик, tR 7,3 минут с m/z 448,1 (МН+), соответствующим структуре.
Стадия (в): восстановление азидной группы до амина

К суспензии смолы со стадии (б) в THF (4 мл) добавляли хлорид олова(II) (Acros, 0,141 г, 0,744 ммоль), тиофенол (Fluka, 0,304 мл, 2,976 ммоль) и триэтиламин (Fluka, 0,311 мл, 2,23 ммоль). Через 1,5 часа аликвоту смолы расщепляли, как описано в (а). ЖХ-МС анализ (колонка Phenomenex Luna C18(2) 3 мкм, 50×4,60 мм, растворители: А = вода/0,1% TFA и Б = ацетонитрил/0,1% TFA; градиент 20-80% Б за 10 минут; скорость потока 1,0 мл/мин, УФ-детектирование при 214 нм, МС-ЭРИ) дал пик при 1,9 минутах с m/z 422,2 (MH+), как ожидалось для амина.
Стадия (г): хелатообразующий агент лозартан-Leu-тетраамин (Соединение 4)
Fmoc-Leu-OH (Novabiochem, 0,030 г, 0,084 ммоль) подвергали реакции сочетания с аликвотой связанного со смолой амино-лозартана со стадии (в) (0,042 ммоль) в DMF с использованием стандартных реагентов сочетания (HATU (гексафторфосфат O-(7-азабензотриазол-1-ил)-N,N,N',N'-тетраметилурония) и DIEA (диизопропилэтиламин)) и стандартного протокола Fmoc-отщепления (20% пиперидина в DMF). Окончание реакции сочетания проверяли стандартным тестом Кайзера. Затем с этой смолой связывали Соединение 1 (0,026 г, 0,042 ммоль), используя те же реагенты сочетания (HATU и DIEA) в DMF. Через четыре часа продукт отщепляли от смолы и на той же стадии удаляли Вос-группы (дихлорметан/TFA/триизопропилсилан, 47,5:50:2,5 раствор в течение одного часа). Раствор фильтровали, концентрировали и очищали препаративной ВЭЖХ (колонка Phenomenex Luna С18(2) 5 мкм, 21,2×250 мм, растворители: А = вода/0,1% TFA и Б = ацетонитрил/0,1% TFA; градиент 20-40% Б за 60 минут; скорость потока 10,0 мл/мин, УФ-детектирование при 214 нм) с получением 5 мг продукта после лиофилизации. ЖХ-МС анализ (колонка Phenomenex Luna C18(2) 3 мкм, 50×4,60 мм, растворители: А = вода/0,1% TFA и Б = ацетонитрил/0,1% TFA; градиент 10-80% Б за 10 минут; скорость потока 0,3 мл/мин, УФ-детектирование при 214 и 254 нм, МС-ЭРИ (электрораспылительная ионизация)) tR 5,1 минут, m/z 735,4 (МН+)) подтвердил структуру.
Пример 6: синтез Соединения 5
Это соединение синтезировали на твердой подложке, как описано в Примере 4. Fmoc-Leu-OH (Novabiochem, 0,033 г, 0,092 ммоль) и Fmoc-амино PEG дигликолевую кислоту (Polypure, 0,049 мг, 0,092 ммоль) последовательно сочетали с аликвотой связанного со смолой амино-лозартана из Примера 4(в) (0,046 ммоль) в DMF с использованием стандартных реагентов сочетания (HATU и DIEA) и стандартного протокола Fmoc-отщепления (20% пиперидина в DMF). Окончание сочетания проверяли стандартным тестом Кайзера. С этой смолой затем связывали Соединение 1 (0,029 г, 0,046 ммоль), используя те же реагенты сочетания (HATU и DIEA) в DMF. Реакционную смесь оставляли стоять в течение ночи, затем продукт отщепляли от смолы и на той же стадии удаляли Вос-группы (дихлорметан/TFA/триизопропилсилан, 47,5:50:2,5 раствор в течение одного часа). Раствор фильтровали, концентрировали и очищали препаративной ВЭЖХ (колонка Phenomenex Luna C18(2) 5 мкм, 21,2×250 мм, растворители: А = вода/0,1% НСООН и Б = ацетонитрил/0,1% НСООН; градиент 10-40% Б за 60 минут; скорость потока 10,0 мл/мин, УФ-детектирование при 214 нм) с получением 3,5 мг продукта после лиофилизации. ЖХ-МС анализ (колонка Phenomenex Luna С18(2) 3 мкм 50×4,60 мм, растворители: А = вода/0,1% НСООН и Б = ацетонитрил/0,1% НСООН; градиент 10-40% Б за 10 минут; скорость потока 0,3 мл/мин, УФ-детектирование при 214 и 254 нм, МС-ЭРИ) tR 4,7 минут, m/z 1025,4 (МН+)) подтвердил структуру.
Пример 7: синтез Соединения 8
Стадия (а): синтез N-Boc-N-[FmocNH-CH2CH2]-Gly-OH
1 г N-[FmocNH-CH2CH2]-Gly-OtBu·HCl (Fluka 09660) обрабатывали 20 мл 50% трифторуксусной кислоты (TFA) в дихлорметане, содержащем 0,5 мл триизопропилсилана в течение 60 минут. Эту смесь упаривали в вакууме и остаток перерастворяли в 20 мл 50% тетрагидрофурана в воде. Добавляли 2,6 г трет-бутилоксикарбонил-ангидрида и 1,2 мл N-метилморфолина и эту смесь перемешивали в течение четырех суток. Тетрагидрофуран затем выпаривали в вакууме и остаток перерастворяли в дихлорметане. Органический слой промывали водой и сушили над MgsO4. Дихлорметан выпаривали в вакууме и остаток перерастворяли в 5 мл диметилформамида. Диметилформамидный раствор разбавляли 400 мл 60% ацетонитрила в воде и закачивали на колонку для препаративной ОФ-ВЭЖХ (ВЭЖХ с обращенной фазой) для очистки (30-80% Б за 40 минут, где А = H2O/0,1% TFA и Б = CH3CN/0,1% TFA, при скорости потока 50 мл/мин на колонке Phenomenex Luna 10 мк С 18(2) 250×50 мм) с получением 450 мг чистого продукта. Продукт анализировали аналитической ВЭЖХ (градиент, 20-70% Б за 10 минут, где А = Н2O/0,1% TFA и Б = СН3CN/0,1% TFA; скорость потока 0,3 мл/мин; колонка Phenomenex Luna 3 мк С18(2) 50×2 мм; УФ-детектирование при 214 нм; время удерживания продукта 8,66 минут). После этого проводили определение характеристик продукта масс-спектрометрией с электрораспылительной ионизацией (MH+ вычисленный 441,2; МН+ найденный 440,8).
Стадия (б): синтез N-((CH2)2-NHCOCH2-тетраамин)-Glv-Arg-Val-Tvr-lle-His-Pro-lle-OH (Соединение 8)
Пептидный аналог ангиотензина II синтезировали на синтезаторе пептидов Applied Biosystems 433A, начиная с 0,1 ммоль смолы Fmoc-lle-Wang. Избыток 1 ммоль предварительно активированных аминокислот [с использованием гексафторфосфата O-бензотриазол-1-ил-N,N,N',N'-тетраметилурония (HBTU)] применяли на стадиях сочетания с аргинином. 123 мг N-Boc-N-{FmocNH-CH2CH2]-Gly-OH, 114 мг N-[(диметиламино)-1Н-1,2,3-триазоло[4,5-b]пиридин-1-илметилен]-N-метилметанаминия
гексафторфосфата N-оксид (HATU) и 60 мкл N-метилморфолина растворяли в диметилформамиде и перемешивали в течение 5 минут, затем добавляли к смоле в аппарате-барботере азота. Реагенты удаляли через 2 часа и смолу промывали диметилформамидом и дихлорметаном. Смолу обрабатывали 20% пиперидином в диметилформамиде (3×10 мл) и промывали диметилформамидом. 23 мг Соединения 1, 14 мг HATU и 7,5 мкл N-метилморфолина растворяли в диметилформамиде в течение 10 минут и добавляли к смоле. Реагенты удаляли через 4 часа и смолу промывали диметилформамидом и дихлорметаном. Одновременное удаление защитных групп боковых цепей и отщепление пептида от смолы проводили в 10 мл трифторуксусной кислоты, содержащей 2,5% триизопропилсилана и 2,5% воды, в течение 90 минут. Трифторуксусную кислоту удаляли в вакууме, диэтиловый эфир добавляли к остатку, выпавший в осадок продукт промывали диэтиловым эфиром и сушили на воздухе. Очистка продукта препаративной ОФ-ВЭЖХ (0-30% Б за 40 минут, где А = Н2O/0,1% TFA и Б = СН3CN/0,1% TFA при скорости потока 10 мл/мин на колонке Phenomenex Luna 10 мк С18(2) 250×21,20 мм) дала 32 мг чистого конъюгата хелат-пептид. Продукт анализировали аналитической ВЭЖХ (градиент, 5-50% Б за 20 минут, где А = Н2O/0,1% TFA и Б = СН3CN/0,1% TFA; скорость потока 1 мл/мин; колонка Phenomenex Luna 3 мк С18(2) 50×2 мм; УФ-детектирование при 214 нм; время удерживания продукта 5,22 минут). После этого проводили определение характеристик продукта масс-спектрометрией с электрораспылительной ионизацией (MH+ вычисленный 1197,8; MH+ найденный 1197,8).
Пример 8: 99mTc радиоактивное мочение Соединений 3-7
Был приготовлен лиофилизированный набор ("Chelakit A plus"), содержащий следующие ингредиенты:
25-50 мкг соединения, предназначенного для мечения (растворенного в 25-50 мкл растворителя), добавляли к CHELAKIT-A plus, после чего добавляли элюат генератора (99mTc4 - в физиологическом растворе, 1,0 мл). Раствор смешивали и оставляли стоять при комнатной температуре в течение 20-30 минут. Соединения 3 и 6 метили технецием при комнатной температуре при рН 9 с получением соответствующих катионных комплексов 99mTc с высоким выходом (RCP (радиохимическая чистота) > 90%). Тетрааминные комплексы очищали ВЭЖХ (подвижная фаза: 0,1% TFA в воде, 0,1% TFA в ацетонитриле; колонка XTERRA RP18 3,5 мкм, 4,6×150 мм), и они стабильны в 50 мМ фосфатном буфере при 37°С в течение 2 часов (RCP > 95% по результатам ВЭЖХ после 2 часов).
Пример 9: измерение липофильности (LogP) 99mTc комплексов
Коэффициенты распределения октанол-вода (LogP) 99mTc комплексов Примера 8 определяли следующим образом.
10 мкл ВЭЖХ-очищенного 99mTc комплекса из Примера 8 смешивали с 1-октанолом (2 мл) и 50 мМ фосфатным буфером (рН 7,4, 2,0 мл) в центрифужной пробирке. Пробирку встряхивали на вортексе при комнатной температуре в течение 1 минуты и затем центрифугировали при высокой скорости в течение 60 минут. По 0,1 мл образцов обеих фаз закапывали пипеткой в другие тест-пробирки с соответствующими предосторожностями для избежания перекрестного загрязнения между фазами и считывали в гамма-счетчике Wallac Wizzard. Измерение повторяли три раза.
Коэффициент распределения Р рассчитывали следующим образом:
Р = (имп/мин в октаноле - имп/мин фона)/(имп/мин в воде - имп/мин фона).
Обычно окончательное значение коэффициента распределения выражали как log P.
Результаты приведены в таблице 1.
Значения Log Р технециевых комплексов тетрааминных соединений
Пример 10: биораспределение 99mTc комплексов
99mTc комплексы Соединений 4, 5 и 7 получали, как описано в Примере 8. Эксперименты осуществляли в двух предопределенных точках времени (22 и 120 минут) после инъекции (p.i.) тестируемого продукта нормальным самцам крыс Wistar (180-220 г). Животным делали анестезию галотаном (Halothane) (6% в кислороде), делали им инъекцию 0,1 мл (500 МБк/мл) тестируемого продукта, умерщвляли их, препарировали, и образцы анализировали на радиоактивность. Результаты приведены в Таблице 2.
Биораспределение 99mTc комплексов
Пример 11: альтернативное получение Соединения 1
N,N'-Бис(2-трет-бутоксикарбониламиноэтил)-2-(2-гидроксиэтил)-1,3-ди(трет-бутоксикарбониламино)пропан из Примера 1, стадия (д), растворяли в четыреххлористом углероде (14 мл) и ацетонитриле (14 мл). Добавляли воду (21 мл) с получением двухфазной смеси, после чего добавляли перйодат натрия (45 г, 21 ммоль) и гидрат хлорида рутения (35 мг, 0,026 ммоль). Полученный темно-коричневый раствор перемешивали при комнатной температуре в течение 1 часа, а затем разбавляли CH2Cl2 (40 мл). Органический слой отделяли и водную смесь экстрагировали дополнительным количеством CH2Cl2 (40 мл × 3). Все органические экстракты объединяли, сушили (MgSO4), фильтровали и летучие вещества выпаривали при пониженном давлении с получением натриевой соли Соединения 1 в виде темного вязкого остатка, который использовали без дополнительной очистки (4,15 г, 96%).
13С ЯМР (CDCl3): δс 28.2 (х12)(СН3), 34.1 (СН2), 34.4 (СН), 38.6 (x2)(NCH2), 46.8 (x2)(NCH2), 49.3 (x2)(NCH2), 79.0 (x2)(OC), 80.2 (x2)(OC), 155.9 (x4)(C=O), 175.4 (COOH).
1H ЯМР (CDCl3): δн 1.29 (18Н, s, СН2 × 6), 1.35 (18Н, s, СН2 × 6), 2.19 (1H, br, СН), 2.40 (2Н, br, CH2), 3.05-3.23 (12H, br, NCH2 × 6), 5.10-5.24 (2H, br, NH × 2).
Масс-спектр (ЭРИ) m/e. Вычислено для (M+Na) C29H54N4Na 641,3738. Найдено 641,3787.
Пример 12: получение Соединения 9
1,3-Дициклогексилкарбодиимид (DCC; 2,16 г, 10,5 ммоль) добавляли одной порцией к перемешиваемому раствору Соединения 1 (4,15 г, 6,90 ммоль) и N-гидроксисукцинимида (1,81 г, 15,7 ммоль) в сухом THF (30 мл). Эту смесь перемешивали при комнатной температуре в течение 16 часов и затем выпавшую в осадок DCU (1,3-дициклогексилмочевина) удаляли фильтрованием. Летучие вещества выпаривали из фильтрата с получением воскообразного остатка, к которому добавляли сухой эфир (50 мл), при этом в осадок выпадало дополнительное количество DCU, которое удаляли фильтрованием. Эфирный раствор промывали водой (25 мл × 2), сушили (MgSO4), фильтровали и растворитель выпаривали при пониженном давлении с получением воскообразного твердого вещества. Это твердое вещество очищали хроматографией на силикагеле, элюируя смесью СН2Cl2/эфир (1:1) до тех пор, пока DCC не был удален. Элюент заменяли эфиром и целевой продукт (rf=0,4, DCM (дихлорметан)/Et2O 1:1) выделяли в виде бесцветного твердого вещества (2,7 г, 57%), т.пл. 66-68°С.
13С ЯМР (CDCl3): δс 25.6 (x2)(CH2), 28.4 (x12)(СН2), 31.8 (СН2, 35.2 (СН), 39.3 (x2)(NCH2), 47.1 (x2)(NCH2), 49.1 (x2)(NCH2), 79.9 (x2)(OC), 80.5 (x2)(OC), 156.1 (x4)(C=O), 167.7 (C=0), 169.1 (x2)(C=O).
1H ЯМР (CDCl3): δн 1.35 (18Н, s, СН2 × 6), 1.41 (18Н, s, СН2 × 6), 2.52 (3Н, brs, СН & СН2), 2.77 (4Н, s, СН2 × 2), 3.10-3.35 (12H, brs, NCH2 × 6), 5.08 (2H, brs, NH×2).
Масс-спектр (ЭРИ) m/e. Вычислено для (M+Na) С33Н54N6O12Na 738,3896. Найдено 738,3893.
| название | год | авторы | номер документа |
|---|---|---|---|
| УЛУЧШЕННЫЕ ХЕЛАТНЫЕ КОНЪЮГАТЫ | 2002 |
|
RU2298012C2 |
| Субстрат для производства радиофармпрепарата на основе Tc | 2015 |
|
RU2614695C2 |
| СОЕДИНЕНИЕ ДЛЯ ДИАГНОСТИКИ ОПУХОЛЕЙ, ЭКСПРЕССИРУЮЩИХ ПСМА, И КОМПОЗИЦИЯ НА ЕГО ОСНОВЕ | 2019 |
|
RU2730507C1 |
| МЕЧЕННЫЕ РАДИОАКТИВНЫМ ИЗОТОПОМ КОНЪЮГАТЫ RGD-СОДЕРЖАЩИХ ПЕПТИДОВ И СПОСОБЫ ИХ ПОЛУЧЕНИЯ С ПОМОЩЬЮ CLICK-ХИМИИ | 2005 |
|
RU2419627C2 |
| СПОСОБ ВИЗУАЛИЗАЦИИ | 2008 |
|
RU2505316C2 |
| КОМПЛЕКСЫ МЕТАЛЛОВ С БИЦИКЛОПОЛИАЗАМАКРОЦИКЛОМ, СПОСОБ ИХ ПОЛУЧЕНИЯ И ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ ДЛЯ ЛЕЧЕНИЯ РАКА | 1992 |
|
RU2118325C1 |
| ЦИКЛИЧЕСКИЕ ПЕПТИДЫ, РАДИОФАРМАЦЕВТИЧЕСКИЙ ПРЕПАРАТ, ЦИКЛИЧЕСКИЕ ПЕПТИДЫ, ИМЕЮЩИЕ РАДИОАКТИВНУЮ МЕТКУ | 1994 |
|
RU2145608C1 |
| КОМПЛЕКСЫ ТЕХНЕЦИЯ И РЕНИЯ С БИС(ГЕТЕРОАРИЛАМИ) И СПОСОБЫ ИХ ПРИМЕНЕНИЯ | 2009 |
|
RU2539584C2 |
| ЛИГАНДЫ, ТРОПНЫЕ К ПРОСТАТИЧЕСКОМУ СПЕЦИФИЧЕСКОМУ МЕМБРАННОМУ АНТИГЕНУ И ИХ ПРИМЕНЕНИЕ ДЛЯ ПОЛУЧЕНИЯ ДВОЙНЫХ КОНЪЮГАТОВ С ТЕРАПЕВТИЧЕСКИМИ АГЕНТАМИ НА ИХ ОСНОВЕ ДЛЯ КОМБИНИРОВАННОЙ ТЕРАПИИ ПСМА ЭКСПРЕССИРУЮЩИХ ОПУХОЛЕЙ | 2023 |
|
RU2841078C1 |
| СОЕДИНЕНИЕ ПЕПТИДНОЙ ПРИРОДЫ, ОБЛАДАЮЩЕЕ СПОСОБНОСТЬЮ СВЯЗЫВАТЬСЯ С ПСМА, СПОСОБ ЕГО ПОЛУЧЕНИЯ И ПРИМЕНЕНИЯ | 2021 |
|
RU2823164C2 |
Настоящее изобретение относится к хелатообразующим агентам и их комплексам с технецием, которые могут быть использованы в качестве радиофармацевтических средств и охарактеризованы формулой I

где X является -NR-, -CO2-, -СО-,
-NR(C=S)-, -NR(C=O)-, -CONR- или Q; Y представляет собой аминокислоту, -СН2-, -СН2OCH2-, -ОСН2СН2O- или X; Z - группировка из пептидов, их аналогов, субстратов, антагонистов или ингибиторов ферментов, рецептор-связывающих соединений, олигонуклеотидов, олиго-ДНК- или олиго-РНК-фрагментов; n - число от 1 до 8; m - число от 0 до 30; R представляет собой Н, С1-4алкил, С2-4алкоксиалкил, С1-4гидроксиалкил или С1-4фторалкил; Q представляет собой остаток сукцинимида, А - фармацевтически приемлемый анион. Технический результат - получение новых хелатообразующих агентов, пригодных для получения комплексов технеция. 5 н. и 17 з.п. ф-лы, 3 ил., 2 табл.
1. Комплекс технеция формулы (I)

где X представляет собой -NR-, -CO2-, -СО-, -NR(C=S)-, -NR(C=O)-, -CONR- или группу Q;
каждый Y независимо представляет собой D- или L-аминокислоту, -СН2-, -СН2OCH2- или -ОСН2СН2O-, или группу X;
Z представляет собой синтетическую обеспечивающую направленную доставку биологическую группировку, выбранную из 3-100 мерных пептидов или пептидных аналогов, которые могут представлять собой линейные пептиды или циклические пептиды или их комбинации; или субстратов, антагонистов или ингибиторов ферментов; синтетических рецептор-связывающих соединений; олигонуклеотидов или олиго-ДНК- или олиго-РНК-фрагментов, причем Z не является моноклональным антителом или его фрагментом и имеет молекулярную массу менее 5000;
n означает целое число от 1 до 8;
m означает целое число от 0 до 30;
R представляет собой Н, С1-4алкил, С2-4алкоксиалкил, С1-4гидроксиалкил или С1-4фторалкил;
Q представляет собой
А представляет собой фармацевтически приемлемый анион, выбранный из гидроксида и анионов органических или неорганических кислот;
при условии, что цепь атомов X1-(Y)m не имеет связей, где один гетероатом непосредственно связан с другим.
2. Комплекс технеция по п.1, где радиоактивный изотоп технеция представляет собой 99mTc или 94mТс.
3. Комплекс технеция по п.1, где n означает число от 1 до 6, и Х представляет собой -CONR- или -NR(C=O)-.
4. Комплекс технеция по п.3, где Х представляет собой -CONH- или -NH(C=O)-.
5. Комплекс технеция по п.1, где -(Y)m- содержит PEG группу формулы
(-OCH2CH2O-)w, где w означает целое число от 3 до 25.
6. Комплекс технеция по п.5, где w означает число от 6 до 22.
7. Комплекс технеция по п.1, где -(Y)m- содержит от 1 до 10 аминокислот.
8. Комплекс технеция по п.7, где аминокислоты независимо выбраны из глицина, лизина, аспарагиновой кислоты, глутаминовой кислоты или серина.
9. Комплекс технеция по пп.1-8, где Z выбран из:
1) 3-30-мерного пептида;
2) субстрата фермента, антагониста фермента или ингибитора фермента.
10. Конъюгат хелатообразующего агента, пригодный для получения комплексов технеция по пп.1-9, причем указанный конъюгат представляет собой конъюгат формулы II
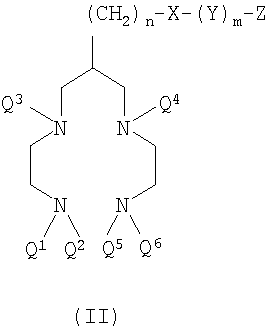
где X, Y, Z, n и m такие, как определено в п.1;
Q1-Q6 независимо представляют собой группы Q, где Q представляет собой Н или защитную группу для аминогрупп.
11. Конъюгат хелатообразующего агента по п.10, где каждый из Q1-Q6 представляет собой Н.
12. Конъюгат хелатообразующего агента по п.10, где Q1 и Q6 оба представляют собой Н, а Q2, Q3, Q4 и Q5 представляют собой трет-бутоксикарбонил.
13. Радиофармацевтическое средство, которое содержит комплекс технеция формулы I по любому из пп.1-9 вместе с жидким биосовместимым носителем, выбранным из воды и водных растворов.
14. Радиофармацевтическое средство по п.13, где радиоактивный изотоп технеция представляет собой 99mTc или 94mТс.
15. Набор для приготовления радиофармацевтического средства по пп.13 и 14, который включает в себя:
1) конъюгат хелатообразующего агента по пп.10-12;
2) биосовместимый восстановитель, выбранный из дитионита натрия, бисульфита натрия, аскорбиновой кислоты, формамидин-сульфиновой кислоты, иона двухвалентного олова, Fe(II) или Cu(II).
16. Набор по п.15, где биосовместимый восстановитель содержит ион олова.
17. Набор по п.15, где каждый из Q1-Q6 представляет собой Н.
18. Набор по п.15, где Z такой, как определено в п.9.
19. Соединение формулы III
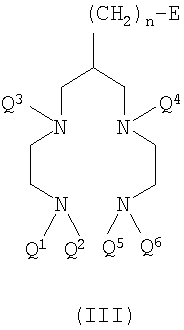
где Q1-Q6 и n такие, как определено в п.10;
Е представляет собой функциональную группу, подходящую для конъюгирования с обеспечивающей направленную доставку биологической группировкой (Z), как она определена в п.1, для получения коньюгата хелатообразующего агента по п.10, где Е выбрана из групп СО2Н, NH2 и CO2(NHS), где (NHS) представляет собой N-гидроксисукцинимидный остаток при условии, что а) когда n=3, тогда по меньшей мере один из Q1-Q6 представляет собой защитную группу для аминогрупп.
20. Соединение по п.19, где Е представляет собой -NH.
21. Соединение по п.19, где Е представляет собой -СО2Н.
22. Соединение по п.19, где n означает число от 1 до 6.
| THEODOSIA MAINA ET ALL, European Journal of Nuclear Medicine, 2002, Vol.29, №6, 742-753 | |||
| A.HEPPELER ET ALL, Current medicinal chemistry, 2000, 7, 971-994 | |||
| УСТАНОВКА ДЛЯ ОПРЕДЕЛЕНИЯ БИОЛОГИЧЕСКОЙ ФИКСАЦИИ АТМОСФЕРНОГО АЗОТА | 0 |
|
SU296522A1 |
| US 5650134, 22.07.1997 | |||
| Устройство для прокладки и подъема подводного кабеля | 1983 |
|
SU1181936A1 |
| US 5489425, 06.02.1996 | |||
| WO 03006070, 23.01.2003 | |||
| WO 03051859, 26.06.2003 | |||
| WO 03006491, 23.01.2003 | |||
| МЕЧЕНЫЕ РАДИОАКТИВНЫМ ИЗОТОПОМ ПЕПТИДЫ ДЛЯ ДИАГНОСТИКИ И ТЕРАПИИ | 1996 |
|
RU2171117C2 |

