Область изобретения
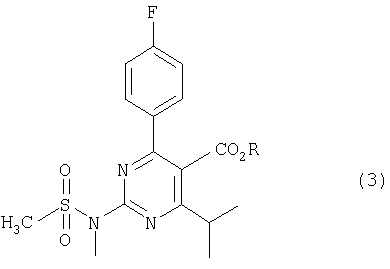
Настоящее изобретение относится к получению аминопиримидиновых соединений, имеющих следующую формулу (8):

[в формуле (8) R представляет собой низший алкил и каждый из R1 и R2 независимо представляет собой атом водорода, алкильную группу, алкилсульфонильную группу или арилсульфонильную группу], более конкретно, к получению 2-(N-метил-N-метансульфониламино)пиримидина, имеющего следующую формулу (3):

где R представляет собой низший алкил.
Предшествующий уровень техники
Bioorg. Med. Chem., 5, 437 (1997), раскрывает, что соединение, представляющее собой 2-(N-метил-N-метансульфониламино)пиримидин, применяют как промежуточное соединение для получения агента, снижающего уровень холестерина (ингибитора гидроксиметилглутарил-КоА-редуктазы: S-4522), имеющего следующую формулу:
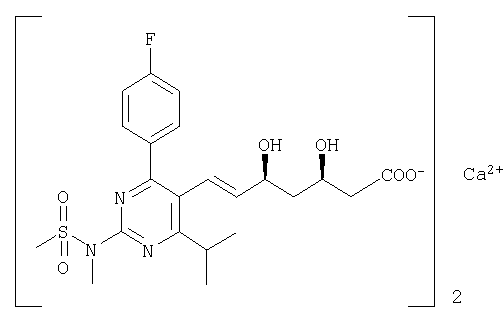
который сейчас общеизвестен как кальциевая соль розувастатина или розувастатина кальций.
WO 01/04100 раскрывает способ получения соединения, представляющего собой 2-(N-метил-N-метансульфониламино)пиримидин, причем этот способ включает в себя стадии, на которых осуществляют взаимодействие метилизобутирилацетата с 4-фторбензонитрилом с получением метил-2-[1-амино-1-(4-фторфенил)метилен]-4-метил-3-оксопентаната и осуществляют взаимодействие 2-[1-амино-1-(4-фторфенил)метилен-4-метил-3-оксопентаната с N-циано-N-метилметансульфонамидом, который получают реакцией между N-метилметансульфонамидом и цианхлоридом, с получением 4-(4-фторфенил)-6-изопропил-5-метоксикарбонил-2-(N-метансульфонил-N-метиламино)пиримидина.
Раскрыто, что суммарный выход (по количеству метилизобутирилацетата) составляет 45,5%.
Способ, раскрытый в WO 01/04100, оказывается невыгодным для промышленного получения из-за невысокого выхода и необходимости применять токсичный цианхлорид как одно из исходных соединений.
Соответственно, цель данного изобретения состоит в том, чтобы предложить новый способ получения 2-(N-метил-N-метансульфониламино)пиримидина или аналогичного ему аминопиримидинового соединения, более конкретно, предложить новый способ, который позволяет получить это соединение более удобным образом и/или без применения токсичного соединения и/или позволяет получить это соединение с высоким выходом и/или высокой чистотой.
Другая цель этого изобретения состоит в том, чтобы предложить новый способ получения соединения, представляющего собой 2-(N-метил-N-метансульфониламино)пиримидин, или аналогичного ему аминопиримидинового соединения, который выгодно применять в промышленном получении.
Краткое изложение сущности изобретения
Настоящее изобретение относится к способу получения 2-(N-метил-N-метансульфониламино)пиримидина, имеющего формулу (3)

[R представляет собой низший алкил],
включающему стадии, на которых
гидроксипиримидиновое соединение, имеющее формулу (1)
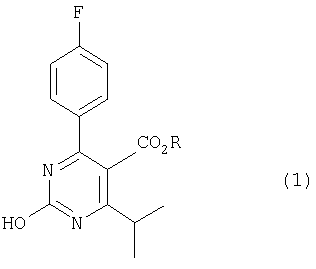
где R такой, как указано выше,
подвергают взаимодействию с органическим сульфонилгалогенидом, имеющим формулу (2)

где R' представляет собой низший алкил, возможно замещенный атомами галогена, фенил, возможно замещенный 1-3 группами, выбранными из нитро, атомов галогена, разветвленного или неразветвленного низшего алкила, низшей алкоксигруппы,
Х представляет собой атом галогена,
или с органическим сульфоновым ангидридом, имеющим формулу (2а)
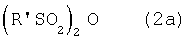
где R' такой, как указано выше, в присутствии основания,
и получившийся продукт реакции подвергают взаимодействию с N-метил-N-метансульфонамидом в присутствии основания.
Предпочтительно соединение формулы (1) подвергают взаимодействию с соединением формулы (2) и получившийся продукт реакции подвергают взаимодействию с N-метил-N-метансульфонамидом.
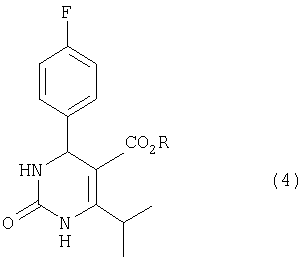
Предпочтительно используют гидроксипиримидиновое соединение, полученное путем окисления дигидропиримидинонового соединения, имеющего формулу (4)

где R такой, как определено выше.
Окисление дигидропиримидинонового соединения можно проводить с использованием азотной кислоты.
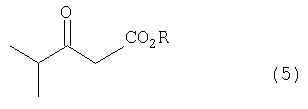
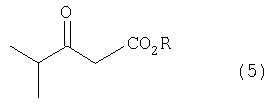
Предпочтительно используют дигидропиримидиноновое соединение, полученное путем взаимодействия изобутирилацетатного эфира, имеющего формулу (5)

где R такой, как определено выше,
с 4-фторбензальдегидом и мочевиной в присутствии протонного соединения и соли металла.
Протонное соединение может представлять собой протонную кислоту.
Предпочтительно протонная кислота представляет собой серную кислоту.
Предпочтительно соль металла представляет собой хлорид меди(I).
Это изобретение также относится к гидроксипиримидиновому соединению, имеющему вышеуказанную формулу (1).
Предпочтительно R в гидроксипиримидиновом соединении формулы (1) представляет собой метил.
Это изобретение далее относится к способу получения гидроксипиримидинового соединения формулы (1), при котором окисляют дигидропиримидиноновое соединение, имеющее формулу (4)
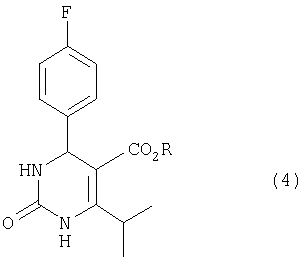
где R представляет собой низший алкил.
Окисление дигидропиримидинонового соединения можно проводить с использованием азотной кислоты.
Предпочтительно используют дигидропиримидиноновое соединение, полученное путем взаимодействия изобутирилацетатного эфира, имеющего формулу (5)
где R такой, как определено выше,
с 4-фторбензальдегидом и мочевиной в присутствии протонного соединения и соли металла.
Протонное соединение может представлять собой протонную кислоту.
Предпочтительно протонная кислота представляет собой серную кислоту.
Предпочтительно соль металла представляет собой хлорид меди(I).
Это изобретение далее относится к дигидропиримидиноновому соединению, имеющему формулу (4).
Предпочтительно R в дигидропиримидиноновом соединении формулы (4) представляет собой метил.
Это изобретение далее относится к способу получения дигидропиримидинонового соединения формулы (4), при котором изобутирилацетатный эфир, имеющий формулу (5)
где R представляет собой низший алкил,
подвергают взаимодействию с 4-фторбензальдегидом и мочевиной в присутствии протонного соединения и соли металла.
Протонное соединение может представлять собой протонную кислоту. Предпочтительно протонная кислота представляет собой серную кислоту.
Предпочтительно соль металла представляет собой хлорид меди(I).
Это изобретение далее относится к способу получения аминопиримидинового соединения, имеющего формулу (8)
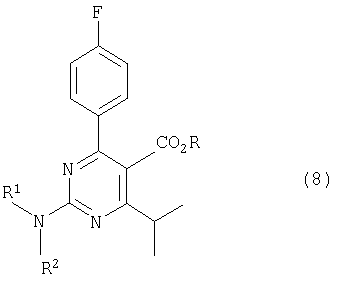
где R представляет собой низший алкил и каждый из R1 и R2 независимо представляет собой атом водорода, алкильную группу, алкилсульфонильную группу или арилсульфонильную группу, при котором 2-замещенное пиримидиновое соединение, имеющее формулу (6)

где R такой, как указано выше, и Х представляет собой атом галогена или R'-замещенную сульфонилоксигруппу, где R' является таким, как указано выше,
подвергают взаимодействию с аминным соединением, имеющим формулу (7)

где каждый из R1 и R2 такой, как указано выше.
Предпочтительно R1 представляет собой метил и R2 представляет собой метансульфонил.
Взаимодействие 2-замещенного пиримидинового соединения с аминным соединением можно проводить в присутствии основания.
Это изобретение далее относится к галогенопиримидиновому соединению, имеющему формулу (9)
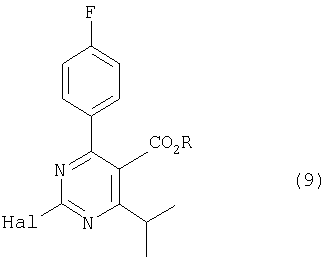
где R представляет собой низший алкил и Hal представляет собой атом галогена.
Предпочтительно R представляет собой метил и Hal представляет собой атом хлора.
Это изобретение далее относится к способу получения галогенопиримидинового соединения формулы (9), при котором осуществляют взаимодействие гидроксипиримидинового соединения вышеупомянутой формулы (1) с галогенирующим агентом.
Предпочтительно галогенирующий агент представляет собой оксихлорид фосфора или тионилхлорид.
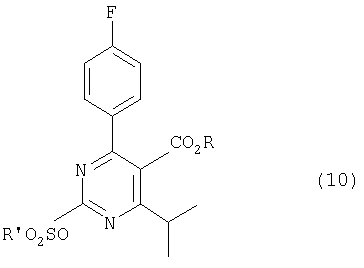
Это изобретение далее относится к органическому сульфонилоксипиримидиновому соединению, имеющему формулу (10)
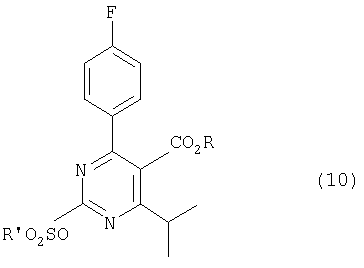
где R представляет собой низший алкил и R' представляет собой низший алкил, возможно замещенный атомами галогена, фенил, возможно замещенный 1-3 группами, выбранными из нитро, атомов галогена, разветвленного или неразветвленного низшего алкила, низшей алкоксгруппы.
Предпочтительно в органическом сульфонилоксипиримидиновом соединении каждый из R и R' независимо представляет собой метил.
Это изобретение далее относится к способу получения органического сульфонилоксипиримидинового соединения формулы (10), при котором осуществляют взаимодействие гидроксипиримидинового соединения вышеупомянутой формулы (1) с органическим сульфонилгалогенидом, имеющим формулу (2)
где R' представляет собой низший алкил, возможно замещенный атомами галогена, фенил, возможно замещенный 1-3 группами, выбранными из нитро, атомов галогена, разветвленного или неразветвленного низшего алкила, низшей алкоксигруппы, и Х представляет собой атом галогена,
или органическим сульфоновым ангидридом, имеющим формулу (2а):
где R' такой, как указано выше,
в присутствии основания.
Это изобретение далее относится к способу получения 2-(N-метил-N-метансульфониламино)пиримидина вышеуказанной формулы (3), причем этот способ включает в себя стадии, на которых
(I) изобутирилацетатный эфир вышеуказанной формулы (5) подвергают взаимодействию с 4-фторбензальдегидом и мочевиной в присутствии протонного соединения и соли металла;
(II) окисляют продукт реакции стадии (I);
(III) продукт окисления стадии (II) подвергают взаимодействию с органическим сульфонилгалогенидом вышеуказанной формулы (2) или органическим сульфоновым ангидридом вышеуказанной формулы (2а) в присутствии основания и
(IV) продукт реакции стадии (III) подвергают взаимодействию с N-метил-N-метансульфонамидом в присутствии основания.
Это изобретение также относится к способу получения розувастатина или его фармацевтически приемлемой соли, включающему получение соединения вышеуказанной формулы (3) указанными выше способами или соединения вышеуказанной формулы (8) вышеуказанным способом и последующее превращение полученного соединения в розувастатин или его фармацевтически приемлемую соль.
Подробное описание изобретения
Репрезентативный способ получения 2-(N-метил-N-метансульфониламино)пиримидина формулы (3) по настоящему изобретению схематически проиллюстрирован нижеследующим образом:

Каждая стадия в проиллюстрированной выше реакционной схеме раскрыта ниже более детально.
Стадия (I)
На стадии (I) изобутирилацетатный эфир следующей формулы (5):
[R представляет собой низший алкил]
подвергают взаимодействию с 4-фторбензальдегидом и мочевиной в присутствии протонного соединения и соли металла.
Группа R в формулах соединений, участвующих в реакциях по этому изобретению, может представлять собой алкильную группу, такую как метил, этил, пропил, бутил, пентил, гексил, гептил, октил, нонил или децил, более конкретно, алкильную группу, имеющую 1-6 атомов углерода, и особенно алкильную группу, имеющую 1-4 атома углерода. Группа R может принимать любые изомерные конфигурации, такие как нормальная, изо и третичная.
Протонным соединением может быть неорганическая кислота или ее соль, такая как соляная кислота, серная кислота, бисульфат калия, бисульфат натрия, азотная кислота или фосфорная кислота; органическая сульфоновая кислота, такая как метансульфоновая кислота, этансульфоновая кислота, бензосульфоновая кислота, пара-толуолсульфоновая кислота или пара-бромбензосульфоновая кислота; органическая карбоновая кислота, такая как уксусная кислота, пропионовая кислота, масляная кислота или бензойная кислота; спирт, такой как метанол, этанол или пропанол. Предпочтительны такие протонные кислоты, как соляная кислота, серная кислота, пара-толуолсульфоновая кислота и уксусная кислота. Наиболее предпочтительна серная кислота. Протонные соединения можно применять по отдельности или в комбинации.
Протонные соединения можно применять в количестве предпочтительно от 0,01 до 3 моль, более предпочтительно от 0,1 до 1 моль на один моль изобутирилацетатного эфира.
Солью металла, применяемой в реакции, может быть хлорид меди(I), хлорид меди(II), ацетат меди(II), хлорид железа(II), хлорид железа(III), хлорид алюминия, бромид никеля(II), хлорид олова(IV), тетрахлорид титана или бромид магния. Предпочтительны хлорид меди(I), хлорид меди(II), хлорид железа(III) и бромид никеля(II). Наиболее предпочтителен хлорид меди(I). Соли металлов могут содержать кристаллизационную воду. Соли металлов можно применять по отдельности или в комбинации.
Соль металла можно применять в количестве предпочтительно от 0,001 до 5 моль, более предпочтительно от 0,01 до 0,1 моль, на один моль изобутирилацетатого эфира.
4-Фторбензальдегид можно применять в количестве предпочтительно от 0,5 до 10 моль, более предпочтительно от 0,9 до 1,1 моль, на один моль изобутирилацетатого эфира.
Мочевину можно применять в количестве предпочтительно от 0,5 до 10 моль, более предпочтительно от 1,5 до 2 моль, на один моль изобутирилацетатного эфира.
Реакцию можно проводить в присутствии или в отсутствие растворителя. В отношении применяемых растворителей особых ограничений нет, поскольку растворитель не нарушает желаемую реакцию. Примеры применимых растворителей включают в себя спирты, такие как метанол, этанол, н-пропиловый спирт, изопропиловый спирт, н-бутиловый спирт, изобутиловый спирт, втор-бутиловый спирт и трет-бутиловый спирт; эфиры, такие как диэтиловый эфир, диизопропиловый эфир, тетрагидрофуран и диметоксиэтан; нитрилы, такие как ацетонитрил, пропионитрил, бутиронитрил и изобутиронитрил; галогенированные алифатические углеводороды, такие как дихлорметан, дихлорэтан, хлороформ и тетрахлорид углерода; ароматические углеводороды, такие как бензол, толуол и ксилол; галогенированные ароматические углеводороды, такие как хлорбензол, и нитрованные ароматические углеводороды, такие как нитробензол. Предпочтительны метанол, этанол, н-пропиловый спирт, изопропиловый спирт, н-бутиловый спирт, диизопропиловый эфир, тетрагидрофуран, диметоксиэтан, ацетонитрил, бутиронитрил, изобутилонитрил, дихлорметан, дихлорэтан, хлороформ, толуол, ксилол и хлорбензол. Особенно предпочтительны метанол, этанол и изопропиловый спирт. Растворители можно применять по отдельности или в комбинации.
Растворитель можно применять в количестве предпочтительно от 0,1 до 10 литров, более предпочтительно от 0,3 до 2 литров, на один моль изобутирилацетатного эфира. Это количество может изменяться в зависимости от гомогенности и диспергируемости реакционной смеси.
Реакцию можно осуществлять путем взаимодействия изобутирилацетатного эфира, 4-фторбензальдегида и мочевины в растворителе в присутствии протонного соединения и соли металла в атмосфере инертного газа. Реакцию можно проводить при температуре предпочтительно от -10 до 200°С, более предпочтительно от 30 до 100°С. В отношении окружающего давления особых ограничений нет.
Получившийся продукт реакции, а именно дигидропиримидиноновое соединение формулы (4), можно выделить и очистить в соответствии с общепринятыми процедурами, такими как дистилляция, кристаллизация, перекристаллизация и колоночная хроматография.
Стадия (II)
На стадии (II) дигидропиримидиноновое соединение формулы (4), то есть продукт реакции стадии (I), окисляют с образованием гидроксипиримидинового соединения формулы (1).
Окисление (или дегидрогенирующее окисление) можно проводить различными общепринятыми способами. Предпочтительно окисление с использованием азотной кислоты, потому что эту процедуру окисления легко проводить, и последующая обработка продукта реакции проста.
Азотную кислоту можно применять в количестве предпочтительно от 1 до 20 моль, более предпочтительно от 3 до 15 моль, на один моль дигидропиримидинонового соединения формулы (4). Предпочтительно применять азотную кислоту в концентрации от 40 до 80%, более предпочтительно от 50 до 70%.
Окисление можно осуществлять в присутствии или в отсутствие растворителя. Особые ограничения в отношении применяемого растворителя отсутствуют, поскольку этот растворитель не нарушает желаемую реакцию. Примеры предпочтительных растворителей включают в себя карбоновые кислоты, такие как уксусная кислота, пропионовая кислота и масляная кислота. Растворители можно применять по отдельности или в комбинации.
Растворитель можно применять в количестве предпочтительно 0,1 до 7 мл, более предпочтительно от 0,5 до 3 мл, на 1 г дигидропиримидинонового соединения. Это количество может изменяться в зависимости от гомогенности и диспергируемости реакционной смеси.
Окисление можно осуществлять путем взаимодействия дигидропиримидинонового соединения и азотной кислоты в растворителе в атмосфере инертного газа. Окисление можно осуществлять при температуре предпочтительно от -10 до 100°С, более предпочтительно от 0 до 50°С. Отсутствуют особые ограничения в отношении окружающего давления. Для ускорения окисления в реакционную систему можно включить инициатор реакции, такой как нитрит натрия.
Получившийся продукт реакции, а именно гироксипиримидиновое соединение формулы (1), можно выделить и очистить в соответствии с общепринятыми процедурами, такими как дистилляция, кристаллизация, перекристаллизация и колоночная хроматография.
Стадии (III) и (IV)
На стадиях (III) и (IV) гидроксипиримидиновое соединение формулы (1), то есть продукт реакции стадии (II), подвергают взаимодействию с органическим сульфонилгалогенидом формулы (2)
или органическим сульфоновым ангидридом формулы (2а)
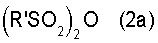
в присутствии основания и получившийся продукт реакции подвергают взаимодействию с N-метил-N-метансульфонамидом в присутствии основания.
В формулах (2) и (2a) R' представляет собой низший алкил, возможно замещенный атомами галогена, фенил, возможно замещенный 1-3 группами, выбранными из нитро, атомов галогена, разветвленного или неразветвленного низшего алкила, низшей алкоксигруппы. Примеры включают в себя алкильные группы, такие как метил, этил, пропил, бутил, пентил, гептил, октил, нонил и децил, более конкретно, алкильные группы, имеющие 1-6 атомов углерода, и особенно алкильные группы, имеющие 1-4 атома углерода; фторированные алкильные группы, такие как трифторметил, нонафторбутил, тридекафторгексил, гептадекафтороктил и ункозафтордецил; незамещенную или замещенную фенильную группу, такую как фенил, толил, ксилил, мезитил, триизопропилфенил, метоксифенил, хлорфенил и нитрофенил. Таким образом, группа R' может иметь один или более чем один заместитель при условии, что эти заместители не нарушают осуществляемую реакцию. Эта группа может принимать любые изомерные конфигурации, такие как нормальная, изо и третичная. Особенно подходящее значение R' представляет собой фенильную группу, которая является незамещенной или несет 1, 2 или 3 заместителя. Эти заместители можно независимо выбирать из, например, алкила, имеющего 1-4 атома углерода, алкокси, имеющей 1-4 атома углерода, галогено и нитро.
В формуле (2) Х представляет собой атом галогена, такого как фтор, хлор, бром или йод.
Примеры сульфонилгалогенидов включают в себя метансульфонилфторид, метансульфонилхлорид, этансульфонилхлорид, 1-пропансульфонилхлорид, 2-пропансульфонилхлорид, трифторметансульфонилфторид, трифторметансульфонилхлорид, нонафторбутансульфонилфторид, тридекафторгексансульфонилфторид, гептадекафтороктансульфонилфторид, ункозафтордекансульфонилфторид, бензосульфонилхлорид, пара-толуолсульфонилфторид, пара-толуолсульфонилхлорид, 2,4,6-триметилбензосульфонилхлорид, 2,4,6-триизопропилбензосульфонилхлорид, пара-метоксибензосульфонилхлорид, пара-хлорбензосульфонилхлорид и 2-нитробензосульфонилхлорид. Предпочтительны трифторметансульфонилфторид, бензосульфонилхлорид, пара-толуолсульфонилхлорид, 2,4,6-триметилбензосульфонилхлорид, 2,4,6-триизопропилбензосульфонилхлорид, пара-метоксибензосульфонилхлорид и пара-хлорбензосульфонилхлорид. Особенно предпочтительны пара-толуолсульфонилхлорид, 2,4,6-триметилбензосульфонилхлорид, 2,4,6-триизопропилбензосульфонилхлорид и пара-метоксибензосульфонилхлорид.
Примеры сульфоновых ангидридов включают в себя метансульфоновый ангидрид, трифторметансульфоновый ангидрид, бензолсульфоновый ангидрид, пара-толуолсульфоновый ангидрид. Предпочтительны трифторметансульфоновый ангидрид, бензолсульфоновый ангидрид и пара-толуолсульфоновый ангидрид. Особенно предпочтительны трифторметансульфоновый ангидрид и пара-толуолсульфоновый ангидрид.
Сульфонилгалогенид или сульфоновый ангидрид можно применять в количестве предпочтительно 0,1 до 20 моль, более предпочтительно от 0,5 до 5 моль, наиболее предпочтительно от 1 до 2 моль, на один моль гидроксипиримидинового соединения.
На следующей стадии N-метилметансульфонамид можно применять в количестве предпочтительно от 0,1 до 30 моль, более предпочтительно от 1 до 5 моль, на один моль гидроксипиримидинового соединения.
Реакции стадий (III) и (IV) можно предпочтительно проводить в присутствии основания. Примеры оснований включают в себя карбонаты щелочных металлов, такие как карбонат натрия и карбонат калия; бикарбонаты щелочных металлов, такие как бикарбонат натрия; гидроксиды щелочных металлов, такие как гидроксид лития, гидроксид натрия и гидроксид калия; алкоксиды щелочных металлов, такие как метоксид натрия, трет-бутоксид натрия, трет-бутоксид калия и трет-пентоксид натрия, и третичные амины, такие как триэтиламин, триизопропиламин, диизопропилэтиламин и пиридин. Предпочтительны карбонат натрия, карбонат калия, трет-бутоксид калия, трет-пентоксид натрия, триэтиламин и пиридин. Особенно предпочтительны карбонат калия, трет-пентоксид натрия и триэтиламин. Наиболее предпочтительны карбонат калия и трет-пентоксид натрия. Основания можно применять по отдельности или в комбинации.
Основание можно применять в количестве предпочтительно от 0,1 до 30 моль, более предпочтительно от 1 до 5 моль, на один моль гидроксипиримидинового соединения. Все количество основания можно включать в реакционную систему до начала реакции, или же основание можно добавлять в реакционную систему порциями после начала реакции.
Реакцию можно проводить в присутствии или в отсутствие растворителя. Особые ограничения в отношении растворителя отсутствуют, так как растворитель не влияет на реакцию. Примеры растворителей включают в себя воду; кетоны, такие как ацетон, метилэтилкетон и диэтилкетон; простые эфиры, такие как диэтиловый эфир и тетрагидрофуран, сложные эфиры, такие как этилацетат, пропилацетат и бутилацетат; нитрилы, такия как ацетонитрил и пропионитрил; амиды, такие как N,N-диметилформамид и N-метилпирролидон; сульфоксиды, такие как диметилсульфоксид; мочевины, такие как N,N'-диметилимидазолинон. Предпочтительны ацетон, тетрагидрофуран, этилацетат, бутилацетат, ацетонитрил, N,N-диметилформамид и диметилсульфоксид. Особенно предпочтительны этилацетат, бутилацетат и ацетонитрил. Наиболее предпочтительны бутилацетат и ацетонитрил. Растворители можно применять по отдельности или в комбинации.
Растворитель можно применять в количестве предпочтительно от 0,01 до 100 литров, более предпочтительно от 0,5 до 5 литров, на один моль гидроксипиримидинового соединения. Это количество может изменяться в зависимости от гомогенности и диспергируемости реакционной смеси.
Реакцию можно осуществлять путем взаимодействия гидроксипиримидинового соединения и органического сульфонилгалогенида или сульфонового ангидрида в растворителе в присутствии основания при перемешивании в атмосфере инертного газа. Основание можно добавлять порциями. Реакцию можно проводить при температуре предпочтительно от -30 до 250°С, более предпочтительно от 0 до 150°С. Отсутствуют особые ограничения в отношении окружающего давления.
Получившийся продукт реакции, а именно соединение формулы (3), представляющее собой 2-(N-метил-N-метанкарбосульфониламино)пиримидин, можно выделить и очистить в соответствии с общепринятыми процедурами, такими как дистилляция, кристаллизация, перекристаллизация и колоночная хроматография.
Соединение формулы (3), представляющее собой 2-(N-метил-N-метансульфониламино)-пиримидин, и другие пиримидиновые соединения формулы (8) можно получать из гидроксипиримидинового соединения формулы (1) через 2-замещенное пиримидиновое соединение формулы (6) на следующих стадиях (V) и (VI):
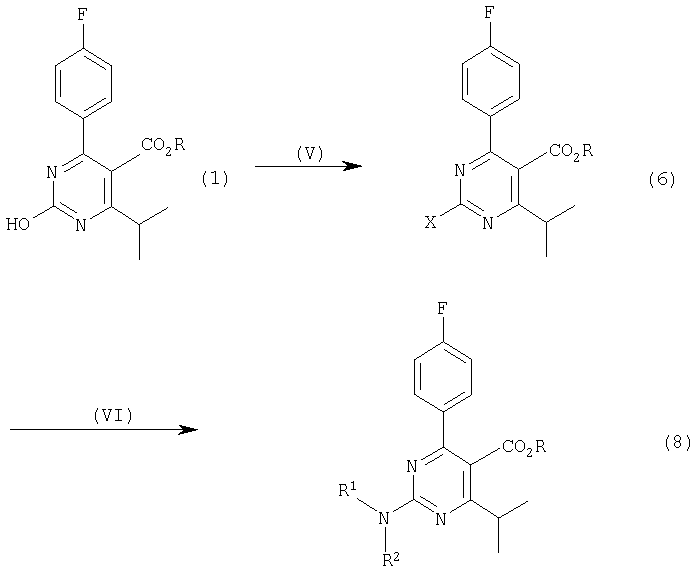
В формуле (8) R имеет значение, как указано выше, и каждый из R1 и R2 независимо представляет собой атом водорода, алкильную группу, алкилсульфонильную группу или арилсульфонильную группу.
Стадия (V)
На стадии (V) гидроксипиримидиновое соединение формулы (1) подвергают взаимодействию с галогенирующим агентом, таким как хлорирующий агент, органическим сульфонилгалогенидом формулы (2)
где R' имеет значение, как указано выше, и Х представляет собой атом галогена,
или органическим сульфоновым ангидридом формулы (2а)
где R' имеет значение, как указано выше.
Примеры галогенирующих агентов включают в себя оксихлорид фосфора и тионилхлорид. Галогенирующие агенты можно применять по отдельности или в комбинации.
Галогенирующий агент можно применять в количестве предпочтительно 0,1 до 50 моль, более предпочтительно от 1 до 20 моль, наиболее предпочтительно от 1,5 до 10 моль, на один моль гидроксипиримидинового соединения.
Примеры органических сульфонилгалогенидов и сульфоновых ангидридов те же, что описаны здесь ранее.
Органический сульфонилгалогенид или сульфоновый ангидрид можно применять в количестве предпочтительно 0,1 до 20 моль, более предпочтительно от 0,5 до 5 моль, наиболее предпочтительно от 1 до 2 моль, на один моль гидроксипиримидинового соединения.
Реакцию можно проводить в присутствии или в отсутствие растворителя. Особые ограничения в отношении растворителя отсутствуют, поскольку растворитель не нарушает реакцию. Примеры растворителей включают в себя ароматические углеводороды, такие как толуол; галогенированные ароматические углеводороды, такие как хлорбензол; нитрованные углеводороды, такие как нитробензол; галогенированные алифатические углеводороды, такие как метиленхлорид и 1,2-дихлорэтан; амиды, такие как N,N-диметилформамид; воду (не для галогенирующего агента); нитрилы, такие как ацетонитрил и пропионитрил; эфиры карбоновых кислот, такие как этилацетат, пропилацетат, бутилацетат; кетоны, такие как ацетон, метилэтилкетон, диэтилкетон, и простые эфиры, такие как диэтиловый эфир и тетрагидрофуран. Предпочтительны бутилцетат, толуол, метиленхлорид, ацетонитрил, хлорбензол, нитробензол и N,N-диметилформамид. Растворители можно применять по отдельности или в комбинации.
В реакции с использованием галогенирующего агента растворитель можно применять в количестве предпочтительно от 0,01 до 10 литров, более предпочтительно от 0,1 до 2 литров, на один моль гидроксипиримидинового соединения. Количество может изменяться в зависимости от гомогенности и диспергируемости реакционной смеси.
В реакции с использованием сульфонилхлорида или сульфонового ангидрида растворитель можно применять в количестве предпочтительно от 0,1 до 50 литров, более предпочтительно от 0,5 до 2 литров, на один моль гидроксипиримидинового соединения. Количество может изменяться в зависимости от гомогенности и диспергируемости реакционной смеси.
Реакцию можно осуществлять путем взаимодействия гидроксипиримидинового соединения и галогенирующего агента в растворителе при перемешивании в атмосфере инертного газа. Реакцию можно проводить при температуре предпочтительно от 0 до 200°С, более предпочтительно от 50 до 120°С. Отсутствуют особые ограничения в отношении окружающего давления.
Реакцию можно осуществлять путем взаимодействия гидроксипиримидинового соединения и сульфонилгалогенида или сульфонильного ангидрида в растворителе при перемешивании в атмосфере инертного газа. Реакцию можно проводить при температуре предпочтительно от -30 до 200°С, более предпочтительно от 0 до 50°С. Отсутствуют особые ограничения в отношении окружающего давления.
Получившийся продукт реакции, а именно 2-замещенное пиримидиновое соединение, такое как хлорпиримидиновое соединение или сульфонилоксипиримидиновое соединение, можно выделить и очистить в соответствии с общепринятыми процедурами, такими как дистилляция, кристаллизация, перекристаллизация и колоночная хроматография.
Стадия (VI)
На стадии (VI) 2-замещенное пиримидиновое соединение, такое как хлорпиримидиновое соединение или сульфонилоксипиримидиновое соединение, полученное на стадии (V), подвергают взаимодействию с аминным соединением, имеющим формулу (7)
где каждый из R1 и R2 такой, как указано выше.
Примеры групп R1 и R2 включают в себя атом водорода, алкильные группы, такие как метил, этил, пропил, бутил, пентил и гексил; алкилсульфонильные группы, такие как метансульфонил, и арилсульфонильные группы, такие как бензосульфонил и пара-толуолсульфонил.
Аминное соединение можно применять в количестве предпочтительно от 0,1 до 30 моль, более предпочтительно от 1 до 5 моль, на один моль 2-замещенного пиримидинового соединения.
Реакцию предпочтительно проводить в присутствии основания. Примеры оснований такие, какие указано ранее.
Основание можно предпочтительно применять в количестве предпочтительно от 0,1 до 30 моль, более предпочтительно от 1 до 5 моль, на один моль 2-замещенного пиримидинового соединения.
Реакцию можно проводить в присутствии или в отсутствие растворителя. Особые ограничения в отношении растворителя отсутствуют, поскольку растворитель не влияет на реакцию. Примеры растворителей включают в себя воду; кетоны, такие как ацетон, метилэтилкетон и диэтилкетон; простые эфиры, такие как диэтиловый эфир и тетрагидрофуран, сложные эфиры, такие как этилацетат, пропилацетат и бутилацетат; нитрилы, такие как ацетонитрил и пропионитрил; амиды, такие как N,N-диметилформамид и N-метилпирролидон; сульфоксиды, такие как диметилсульфоксид; мочевины, такие как N,N'-диметилимидазолидон. Предпочтительны ацетон, тетрагидрофуран, этилацетат, бутилацетат, ацетонитрил, N,N-диметилформамид и диметилсульфоксид. Особенно предпочтительны этилацетат, бутилацетат и ацетонитрил. Растворитель можно применять по отдельности или в комбинации.
Растворитель можно применять в количестве предпочтительно от 0,01 до 100 литров, более предпочтительно от 0,5 до 5 литров, на один моль 2-замещенного пиримидинового соединения. Это количество может изменяться в зависимости от гомогенности и диспергируемости реакционной смеси.
Реакцию можно осуществлять путем взаимодействия 2-замещенного пиримидинового соединения и аминного соединения в растворителе в присутствии основания при перемешивании в атмосфере инертного газа. Реакцию можно проводить при температуре предпочтительно от -20 до 250°С, более предпочтительно от 25 до 150°С. Отсутствуют особые ограничения в отношении окружающего давления.
Реакцию можно проводить в двух отдельных жидких фазах в присутствии катализатора фазового перехода. Примеры катализаторов фазового перехода включают в себя хлорид тетраметиламмония, бромид тетраметиламмония, фторид тетраэтиламмония, хлорид тетраэтиламмония, бромид тетраэтиламмония, бромид тетрапропиламмония, йодид тетрапропиламмония, фторид тетрабутиламмония, хлорид тетрабутиламмония, бромид тетрабутиламмония, йодид тетрабутиламмония, бромид тетрапентиламмония, бромид тетрагексиламмония, бромид тетрагептиламмония, бромид тетраоктиламмония, хлорид бензилдиметилтетрадециламмония, хлорид бензилтриэтиламмония, хлорид фенилтриметиламмония, йодид фенилтриметиламмония и хлорид гексадецилтриметиламмония. Предпочтительны хлорид тетрабутиламмония, бромид тетрабутиламмония, йодид тетрабутиламмония, хлорид бензилтриэтиламмония и хлорид гексадецилтриметиламмония. Наиболее предпочтительны бромид тетрабутиламмония, хлорид бензилтриэтиламмония и хлорид гексадецилтриметиламмония.
Катализатор фазового перехода можно применять в количестве от 0,01 до 0,5 моль, предпочтительно от 0,05 до 0,2 моль, на один моль 2-замещенного пиримидинового соединения.
Получившийся продукт реакции, а именно соединение формулы (3), представляющее собой 2-(N-метил-N-метансульфониламино)пиримидин, или другие аминопиримидиновые соединения формулы (8), можно выделить и очистить в соответствии с общепринятыми процедурами, такими как дистилляция, кристаллизация, перекристаллизация или колоночная хроматография.
Настоящее изобретение далее раскрывают следующие неограничивающие примеры.
Пример 1. Получение 4-(4-фторфенил)-6-изопропил-5-метоксикарбонил-3,4-2(1Н)-дигидропиримидинона.
В стеклянную колбу объемом 500 мл, снабженную мешалкой, термометром и дефлегматором, помещали 28,8 г (0,2 моль) метилизобутирилацетата, 24,8 г (0,2 моль) 4-фторбензальдегида, 21,0 г (0,35 моль) мочевины, 200 мг (2 ммоль) хлорида меди(I), 2 мл серной кислоты и 200 мл метанола. Содержимое колбы нагревали до 64-65°С в течение 24 часов при дефлегмации и перемешивании для проведения реакции. В результате получили осажденный кристаллический продукт. Этот кристаллический продукт собирали на фильтровальной бумаге и промывали метанолом с получением 49,7 г 4-(4-фторфенил)-6-изопропил-5-метоксикарбонил-3,4-2(1Н)-дигидропиримидинона в виде бесцветного кристаллического продукта, имеющего нижеуказанные характеристики. Выход составил 85% (на основании количества метилизобутирилацетата).
Точка плавления 223-225°С.
УФλмакс (СН3CN, нм): 194,3; 278,6.
ИК (KBr, см-1): 3296, 3229, 3137, 2963, 1685, 1629, 1504, 1225, 1097.
1H-ЯМР (ДМСО-d6, δ (м.д.)): 1.14 (6Н, dd, J=6.8, 6.9 Гц), 3.52 (3Н, s), 4.0-4.2 (1Н, m), 5.15 (1Н, d, J=3.4 Гц), 7.1-7.2 (2H, m), 7.2-7.3 (2Н, m), 7.76 (1Н, d, J=3.2 Гц), 8.91 (1H, s).
МСВР (масс-спектрометрия высокого разрешения): 292, 1247 (теоретическое значение (C15H17FN2O3 (M+)) 292, 1223).
Пример 2. Получение 4-(4-фторфенил)-6-изопропил-5-метоксикарбонил-3, 4-2(1Н)-дигидропиримидинона.
Повторяли процедуры Примера 1 за исключением того, что заменяли 200 мг (2 ммоль) хлорида меди(I) на 5,41 г (20 ммоль) гексагидрата хлорида железа(III). Получали 35,6 г 4-(4-фторфенил)-6-изопропил-5-метоксикарбонил-3,4-2(1Н)-дигидропиримидинона. Выход составил 61% (на основании количества метилизобутирилацетата).
Пример 3. Получение 4-(4-фторфенил)-2-гидрокси-6-изопропил-5-метоксикарбонилпиримидина.
В стеклянную колбу объемом 50 мл, снабженную мешалкой и термометром, помещали 11 мл (144 ммоль) азотной кислоты (60-61%, уд. вес 1,38). К азотной кислоте медленно добавляли при комнатной температуре 4,00 г (13,7 ммоль) 4-(4-фторфенил)-6-изопропил-5-метоксикарбонил-3,4-2(1Н)-дигидропиримидинона, полученного таким же образом, как в Примере 1, и эту смесь подвергали реакции в течение 30 минут при комнатной температуре. После завершения реакции реакционную смесь нейтрализовали путем помещения этой смеси в 140 мл насыщенного водного раствора бикарбоната натрия. Затем экстрагировали реакционную смесь этилацетатом. Органическую жидкую часть отделяли и концентрировали при пониженном давлении. Остаток кристаллизовали из толуола. Кристаллический продукт собирали на фильтре и промывали толуолом с получением 3,64 г 4-(4-фторфенил)-2-гидрокси-6-изопропил-5-метоксикарбонилпиримидина в виде бесцветного кристаллического продукта, имеющего нижеуказанные характеристики. Выход составил 92% (на основании количества 4-(4-фторфенил)-6-изопропил-5-метоксикарбонил-3,4-2(1Н)-дигидропиримидинона).
Точка плавления 193°С (с разложением).
УФλмакс (CH3CN, нм): 196,6; 243,2; 317.9.
ИК (KBr, см-1): 2991, 2887, 1717, 1653, 1589, 1433, 1280, 1223.
1H-ЯМР (ДМСО-d6, δ (м.д.)): 1.23 (6Н, d, J=6.8H4), 3.0-3.2 (1Н, m), 3.56 (3Н, s), 7.3-7.4 (2Н, m), 7.5-7.6 (2Н, m), 12.25 (1Н, brs).
MCBP: 290,1054 (теоретическое значение (C15H15FN2O3 (M+)) 290,1067).
Пример 4. Получение 4-(4-фторфенил)-2-гидрокси-6-изопропил-5-метоксикарбонилпиримидина.
В стеклянную колбу объемом 50 мл, снабженную мешалкой и термометром, помещали 2,92 г (10 ммоль) 4-(4-фторфенил)-6-изопропил-5-метоксикарбонил-3,4-2(1Н)-дигидропиримидинона, полученного таким же образом, как в Примере 1, и 5 мл уксусной кислоты. К этой смеси медленно добавляли 3,74 мл (50 ммоль) азотной кислоты (60-61%, уд. вес 1,38). К этой смеси далее добавляли 0,07 г (1 ммоль) нитрита натрия и проводили реакцию в течение одного часа при комнатной температуре. После завершения реакции реакционную смесь нейтрализовали путем помещения этой смеси в 50 мл насыщенного водного раствора бикарбоната натрия. Реакционную смесь затем экстрагировали этилацетатом. Органическую жидкую часть отделяли и концентрировали при пониженном давлении. Остаток кристаллизовали из толуола. Кристаллический продукт собирали на фильтре и промывали толуолом с получением 2,61 г 4-(4-фторфенил)-2-гидрокси-6-изопропил-5-метоксикарбонилпиримидина в виде бесцветного кристаллического продукта. Выход составил 90% (на основании количества 4-(4-фторфенил)-6-изопропил-5-метоксикарбонил-3,4-2(1Н)-дигидропиримидинона).
Пример 5. Получение 4-(4-фторфенил)-2-гидрокси-6-изопропил-5-метоксикарбонилпиримидина.
В стеклянную колбу объемом 200 мл, снабженную мешалкой и термометром, помещали 54,0 г (735 ммоль) азотной кислоты (60-61%, уд. вес 1,38). К азотной кислоте медленно добавляли при комнатной температуре 30,6 г (105 ммоль) 4-(4-фторфенил)-6-изопропил-5-метоксикарбонил-3,4-2(1Н)-дигидропиримидинона, полученного таким же образом, как в Примере 1, и эту смесь подвергали реакции в течение 30 минут при комнатной температуре. После завершения реакции реакционную смесь вливали в 162 мл воды. Водную смесь нейтрализовали добавлением 61 г водного раствора гидроксида натрия (48 мас.%), чтобы осадить кристаллический продукт. Кристаллический продукт собирали фильтрацией и сушили с получением 27,6 г 4-(4-фторфенил)-2-гидрокси-6-изопропил-5-метоксикарбонилпиримидина в виде бесцветного кристаллического продукта.
Выход составил 91% (на основании количества 4-(4-фторфенил)-6-изопропил-5-метоксикарбонил-3,4-2(1Н)-дигидропиримидинона).
Пример 6. Получение 4-(4-фторфенил)-2-гидрокси-6-изопропил-5-метоксикарбонилпиримидина.
В стеклянную колбу объемом 2 л, снабженную мешалкой и термометром, помещали 323,3 г (3,09 моль) азотной кислоты (60-61%, плотность 1,38). Затем концентрированную азотную кислоту охлаждали до 10°С. К азотной кислоте добавляли 2,36 г (34,2 ммоль) нитрита натрия и далее медленно добавляли 100 г (342 ммоль) 4-(4-фторфенил)-6-изопропил-5-метоксикарбонил-3,4-2(1Н)-дигидропиримидинона, полученного таким же образом, как в Примере 1. Эту смесь подвергали реакции в течение 2 часов при температуре 10-12°С. После завершения реакции в реакционную смесь вливали 970 мл воды. Водную смесь затем нейтрализовали добавлением 257 г водного раствора гидроксида натрия (48 мас.%), чтобы осадить кристаллический продукт. Кристаллический продукт собирали фильтрацией и сушили с получением 93,3 г 4-(4-фторфенил)-2-гидрокси-6-изопропил-5-метоксикарбонилпиримидина в виде бесцветного кристаллического продукта. Выход составил 94% (на основании количества 4-(4-фторфенил)-6-изопропил-5-метоксикарбонил-3,4-2(1Н)-дигидропиримидинона).
Пример 7. Получение 4-(4-фторфенил)-6-изопропил-5-метоксикарбонил-2-(N-метил-N-метансульфониламино)пиримидина.
В стеклянную колбу объемом 200 мл, снабженную мешалкой, термометром и дефлегматором, помещали 5,81 г (20 ммоль) 4-(4-фторфенил)-2-гидрокси-6-изопропил-5-метоксикарбонилпиримидина, 3,59 г (26 ммоль) карбоната калия (можно получать от Asahi Glass Works, Co., Ltd., Lot No. 1111632; распределение частиц по размерам: 75-250 мкм 14%; прохождение через 75 мкм 86%) и 40 мл бутилацетата. К этой смеси медленно добавляли 4,19 г (22 ммоль) п-толуолсульфонилхлорида при перемешивании и проводили реакцию при 40°С в течение 4 часов. После этого реакционную смесь охлаждали до комнатной температуры. К охлажденной реакционной смеси добавляли 2,84 г (26 ммоль) N-метилметансульфонамида и 4,15 г (30 ммоль) карбоната калия (такого же, как указано выше). Смесь нагревали до 110-125°С в течение 2 часов с обратным холодильником для проведения реакции. После завершения реакции смесь охлаждали до комнатной температуры. К охлажденной смеси добавляли 25 мл воды и 20 мл ацетона и отделяли органическую жидкую часть. Органическую жидкую часть промывали насыщенным водным раствором хлорида натрия и сушили над безводным сульфатом магния. Сухую органическую жидкую часть фильтровали и концентрировали под пониженным давлением. Остаток кристаллизовали из гептана с получением 6,58 г 4-(4-фторфенил)-6-изопропил-5-метоксикарбонил-2-(N-метил-N-метансульфониламино)пиримидина в виде бледно-желтого кристаллического продукта. Выход составил 86% (на основании количества 4-(4-фторфенил)-2-гидрокси-6-изопропил-5-метоксикарбонилпиримидина).
Пример 8. Получение 4-(4-фторфенил)-6-изопропил-5-метоксикарбонил-2-(N-метил-N-метансульфониламино)пиримидина.
В стеклянную колбу объемом 1000 мл, снабженную мешалкой, термометром и дефлегматором, помещали 50,0 г (172 ммоль) 4-(4-фторфенил)-2-гидрокси-6-изопропил-5-метоксикарбонилпиримидина, 20,8 г (189 ммоль) трет-пентоксида натрия и 344 мл ацетонитрила и перемешивали получившуюся смесь при 0-10°С в течение 30 минут. К этой смеси медленно добавляли 36,1 г (189 ммоль) пара-толуолсульфонилхлорида и проводили реакцию в течение 5 часов при комнатной температуре. После этого реакционную смесь охлаждали до температуры 0-10°С. К охлажденной реакционной смеси добавляли 28,2 г (258 ммоль) N-метилметансульфонамида и 26,5 г (241 ммоль) трет-пентоксида натрия. Смесь держали при 0-10°С в течение одного часа и затем нагревали до 75-82°С в течение 2 часов с обратным холодильником для проведения реакции. После завершения реакции смесь охлаждали до комнатной температуры. К охлажденной смеси добавляли 344 мл воды. Водную смесь охлаждали до 0-10°С и перемешивали в течение одного часа, при этом осаждался кристаллический продукт. Кристаллический продукт собирали фильтрацией и сушили с получением 45,3 г 4-(4-фторфенил)-6-изопропил-5-метоксикарбонил-2-(N-метил-N-метансульфониламино)пиримидина в виде бледно-желтого кристаллического продукта. Выход составил 68% (на основании количества 4-(4-фторфенил)-2-гидрокси-6-изопропил-5-метоксикарбонилпиримидина).
Пример 9. Получение 4-(4-фторфенил)-6-изопропил-5-метоксикарбонил-2-(N-метил-N-метансульфониламино)пиримидина из метилизобутирилацетата, 4-фторбензальдегида и мочевины.
1) В эмалированный реакционный сосуд объемом 200 л, снабженный мешалкой, термометром и дефлегматором, помещали 24,4 кг (169 моль) метилизобутирилацетата, 20,0 кг (161 моль) 4-фторбензальдегида, 16,9 кг (282 моль) мочевины, 0,2 кг (2 моль) хлорида меди(I), 3,0 кг серной кислоты и 80,4 кг метанола. Смесь нагревали до 64-66°С в течение 20 часов при дефлегмации для проведения реакции. После завершения реакции реакционную смесь охлаждали до комнатной температуры для осаждения кристаллического продукта. Кристаллический продукт собирали на фильтровальной бумаге и промывали метанолом с получением 43,4 кг 4-(4-фторфенил)-6-изопропил-5-метоксикарбонил-3,4-2(1Н)-дигидропиримидинона в виде бесцветного кристаллического продукта.
2) В эмалированный реакционный сосуд объемом 200, снабженный мешалкой и термометром, помещали 62,5 кг (615,6 моль) разбавленной азотной кислоты и 0,5 кг (6,8 моль) нитрита натрия. К этой смеси медленно добавляли при охлаждении 20,0 кг (68,4 ммоль) 4-(4-фторфенил)-6-изопропил-5-метоксикарбонил-3,4-2(1Н)-дигидропиримидинона, полученного, как указано выше. Получившуюся смесь подвергали реакции при низкой температуре (10°С). После завершения реакции реакционную смесь нейтрализовали добавлением раствора гидроксида натрия в водном метаноле. Затем к этой смеси добавляли водный раствор гидроксида натрия. Получившуюся смесь помещали под пониженное давление для отгонки метанола. К остатку добавляли 96,5 кг ацетона и 96,5 кг воды. Водный остаток затем нейтрализовали добавлением уксусной кислоты, чтобы осадить кристаллический продукт. Кристаллический продукт собирали на фильтровальной бумаге и промывали смесью ацетона и воды с получением 17,9 кг 4-(4-фторфенил)-2-гидрокси-6-изопропил-5-метоксикарбонилпиримидина.
3) В эмалированный реакционный сосуд объемом 200 л, снабженный мешалкой, термометром и дефлегматором, помещали 17,9 кг (62,0 моль) 4-(4-фторфенил)-2-гидрокси-6-изопропил-5-метоксикарбонилпиримидина, полученного, как указано выше, 107,7 кг бутилацетата, 11,1 кг (80,3 моль) карбоната калия (можно получать от Asahi Glass Works, Co., Ltd., Lot №1111632, распределение частиц по размерам: 75-250 мкм 14%; прохождение через 75 мкм 86%) и 12,9 кг (67,7 моль) пара-толуолсульфонилхлорида. Смесь нагревали при 60°С в течение 2 часов для проведения реакции. После этого реакционную смесь охлаждали до комнатной температуры. К охлажденной смеси добавляли 8,8 кг (80,6 моль) N-метилметансульфонамида и 12,9 кг (93,3 моль) карбоната калия и нагревали получившуюся смесь при 122-125°С в течение 3 часов для проведения реакции. После завершения реакции реакционную смесь охлаждали до комнатной температуры. К охлажденной смеси добавляли ацетон и воду и отделяли органическую жидкую часть. Органическую жидкую часть затем последовательно промывали водным раствором гидроксида натрия (3 мас.%) и насыщенным водным раствором хлорида натрия. Промытую органическую жидкую часть концентрировали под пониженным давлением. К остатку добавляли изопропиловый спирт и воду, что приводило к осаждению кристаллического продукта. Кристаллический продукт фильтровали на фильтровальной бумаге и промывали изопропиловым спиртом. Промытый кристаллический продукт и 85,7 кг ацетона помещали в эмалированный реакционный сосуд объемом 200 л, снабженный мешалкой, термометром и дефлегматором. Смесь перемешивали при 50-55°С для растворения кристаллического продукта в ацетоне. Нерастворимую часть удаляли линейным фильтром. После этого к раствору добавляли 58,3 кг воды, чтобы осадить кристаллический продукт. Кристаллический продукт собирали на фильтровальной бумаге и промывали смесью ацетона и воды с получением 19,5 кг 4-(4-фторфенил)-6-изопропил-5-метоксикарбонил-2-(N-метил-N-метансульфониламино)пиримидина.
Пример 10. Получение 2-хлор-4-(4-фторфенил)-6-изопропил-5-метоксикарбонилпиримидина.
В стеклянную колбу объемом 25 мл, снабженную мешалкой, термометром и дефлегматором, помещали 1,00 г (3,43 ммоль) 4-(4-фторфенил)-2-гидрокси-6-изопропил-5-метоксикарбонилпиримидина и 3,4 мл (3,7 ммоль) оксихлорида фосфора. Смесь нагревали до 100-106°С в течение полутора часов с обратным холодильником для проведения реакции. После завершения реакции реакционную смесь охлаждали до комнатной температуры и выливали в смесь воды и льда. Получившуюся водную смесь нейтрализовали насыщенным водным раствором бикарбоната натрия. Нейтрализованную водную смесь экстрагировали этилацетатом. Этилацетатную часть отделяли, промывали насыщенным водным раствором хлорида натрия и сушили над безводным сульфатом магния. Высушенную этилацетатную часть фильтровали и концентрировали при пониженном давлении с получением 1,03 г 2-хлор-4-(4-фторфенил)-6-изопропил-5-метоксикарбонилпиримидина в виде бесцветного кристаллического продукта, имеющего нижеуказанные характеристики. Выход составил 97% (на основании количества 4-(4-фторфенил)-2-гидрокси-6-изопропил-5-метоксикарбонилпиримидина).
Точка плавления 99-101°С.
УФλмакс (СН3CN, нм): 194,7; 276,5.
ИК (KBr, см-1): 2980, 1728, 1542, 1508, 1227, 1086.
1H-ЯМР (ДМСО-d6, δ (м.д.)): 1.33 (6Н, d, J=6.8 Гц), 3.1-3.2 (1Н, m), 3.76 (3Н, s), 7.15 (2Н, t, J=8.5 Гц), 7.6-7.7 (2Н, m).
МСВР: 308,0695 (теоретическое значение (C15H14CIFN2O2 (M+)) 308,0728).
Пример 11. Получение 2-хлор-4-(4-фторфенил)-6-изопропил-5-метоксикарбонилпиримидина.
В стеклянную колбу объемом 25 мл, снабженную мешалкой, термометром и дефлегматором, помещали 1,00 г (3,43 ммоль) 4-(4-фторфенил)-2-гидрокси-6-изопропил-5-метоксикарбонилпиримидина, 0,5 мл (3,9 ммоль) тионилхлорида, 3,44 мл толуола и 0,11 мл N,N-диметилформамида. Смесь нагревали до 80°С в течение 3 часов для проведения реакции. После завершения реакции реакционную смесь охлаждали до комнатной температуры и выливали в смесь воды и льда. Получившуюся водную смесь нейтрализовали насыщенным водным раствором бикарбоната натрия. Нейтрализованную водную смесь экстрагировали этилацетатом. Этилацетатную часть отделяли, промывали насыщенным водным раствором хлорида натрия и сушили над безводным сульфатом магния. Высушенную этилацетатную часть фильтровали и концентрировали под пониженным давлением с получением 0,80 г 2-хлор-4-(4-фторфенил)-6-изопропил-5-метоксикарбонилпиримидина в виде бесцветного кристаллического продукта. Выход составил 76% (на основании количества 4-(4-фторфенил)-2-гидрокси-6-изопропил-5-метоксикарбонилпиримидина).
Пример 12. Получение 4-(4-фторфенил)-6-изопропил-5-метоксикарбонил-2-(N-метил-N-метансульфониламино)пиримидина.
В стеклянную колбу объемом 25 мл, снабженную мешалкой, термометром и дефлегматором, помещали 546 мг (5 ммоль) N-метилметансульфонамида, 551 мг (5 ммоль) трет-пентоксида натрия, 10 мл ацетонитрила и 309 мг (1 ммоль) 2-хлор-4-(4-фторфенил)-6-изопропил-5-метоксикарбонилпиримидина. Смесь нагревали до 81-82°С в течение 3 часов с обратным холодильником для проведения реакции. После завершения реакции реакционную смесь охлаждали до комнатной температуры. К охлажденной смеси добавляли 10 мл воды и экстрагировали водную смесь этилацетатом. Этилацетатную часть отделяли и сушили над безводным сульфатом магния. Высушенную этилацетатную часть фильтровали и концентрировали при пониженном давлении. Остаток очищали колоночной хроматографией на силикагеле (колонка Wako Gel C-200, элюент: гексан/этилацетат, объемное соотношение 2:1). Получали 339 мг 4-(4-фторфенил)-6-изопропил-5-метоксикарбонил-2-(N-метил-N-метансульфониламино)пиримидина. Выход составил 89% (на основании количества 2-хлор-4-(4-фторфенил)-6-изопропил-5-метоксикарбонилпиримидина).
Пример 13. Получение 4-(4-фторфенил)-6-изопропил-5-метоксикарбонил-2-метансульфонилоксипиримидина.
В стеклянную колбу объемом 100 мл помещали 10,0 г (34,4 ммоль) 4-(4-фторфенил)-2-гидрокси-6-изопропил-5-метоксикарбонилпиримидина, 5,22 г (58,5 ммоль) триэтиламина и 34 мл ацетонитрила. Смесь в колбе охлаждали до 0-5°С в ледяной бане. К охлажденной смеси медленно добавляли 5,12 г (44,7 ммоль) метансульфонилхлорида и подвергали получившуюся смесь реакции при 20-25°С в течение 2 часов. После завершения реакции к реакционной смеси добавляли 60 мл воды. Водную реакционную смесь экстрагировали толуолом и толуольную часть отделяли. Толуольную часть промывали насыщенным водным раствором хлорида натрия и сушили над безводным сульфатом магния. Высушенную смесь фильтровали и концентрировали при пониженном давлении. Остаток кристаллизовали из метанола с получением 11,3 г 4-(4-фторфенил)-6-изопропил-5-метоксикарбонил-2-метансульфонилоксипиримидина в виде бесцветного кристаллического продукта, имеющего нижеуказанные характеристики. Выход составил 89% (на основании количества 4-(4-фторфенил)-2-гидрокси-6-изопропил-5-метоксикарбонилпиримидина).
Точка плавления 110-111°С.
УФ λмакс (CH3CN, нм): 193,7; 276,8.
ИК (KBr, см-1): 2980, 1724, 1562, 1391, 1250, 1175, 1079, 971.
1H-ЯМР (CDCI2, δ (м.д.)): 1.33 (6Н, d, J=6.6 Гц), 3.20 (1Н, m), 3.60 (3Н, s), 7.1-7.2 (2H, s), 7.6-7.8 (2H, m).
МСВР: 368,0842 (теоретическое значение (C15H17FN2O5S (M+)) 368,0892).
Пример 14. Получение 4-(4-фторфенил)-6-изопропил-5-метоксикарбонил-2-(пара-толуолсульфонилокси)пиримидина.
В стеклянную колбу объемом 200 мл помещали 27,6 г (95,1 ммоль) 4-(4-фторфенил)-2-гидрокси-6-изопропил-5-метоксикарбонилпиримидина, 12,5 г (123 ммоль) триэтиламина и 95 мл ацетонитрила. Смесь в колбе охлаждали до 0-5°С в ледяной бане. К охлажденной смеси медленно добавляли 20,0 г (105 ммоль) пара-толуолсульфонилхлорида и подвергали получившуюся смесь реакции при 20-25°С в течение одного часа. После завершения реакции к реакционной смеси добавляли 95 мл воды. Водную реакционную смесь экстрагировали толуолом и толуольную часть отделяли. Толуольную часть промывали насыщенным водным раствором хлорид натрия и сушили над безводным сульфатом магния. Высушенную смесь фильтровали и концентрировали под пониженным давлением. Остаток кристаллизовали из метанола с получением 35,9 г 4-(4-фторфенил)-6-изопропил-5-метоксикарбонил-2-(пара-толуолсульфонилокси)пиримидина в виде бесцветного кристаллического продукта, имеющего нижеуказанные характеристики. Выход составил 85% (на основании количества 4-(4-фторфенил)-2-гидрокси-6-изопропил-5-метоксикарбонилпиримидина).
Точка плавления 94-96°С.
УФλмакс (СН3CN, нм): 194,9; 275,2.
ИК (KBr, см-1): 2961, 1734, 1539, 1389, 1352, 1247, 1090, 980.
1H-ЯМР (CDCI3, δ (м.д.)): 1.23 (6Н, d, J=6.8 Гц), 2.45 (3Н, s), 3.0-3.2 (1Н, m), 3.74 J=8.5 Гц).
МСВР: 444,1155 (теоретическое значение (C32H21FN2O5S (M+)) 444,1194).
Пример 15. Получение 4-(4-фторфенил)-6-изопропил-5-метоксикарбонил-2-бензосульфонилоксипиримидина.
Повторяли процедуры Примера 13 за исключением того, что заменяли пара-толуолсульфонилхлорид на 18,5 г (105 ммоль) бензосульфонилхлорида.
Получали 39,3 г 4-(4-фторфенил)-6-изопропил-5-метоксикарбонил-2-бензосульфонилоксипиримидина в виде бледно-желтого кристаллического продукта, имеющего нижеуказанные характеристики. Выход составил 96% (на основании количества 4-(4-фторфенил)-2-гидрокси-6-изопропил-5-метоксикарбонилпиримидина).
1H-ЯМР (CDCI3, δ (м.д.)): 1.21 (6Н, d, J=6.4 Гц), 3.0-3.1 (1Н, m), 3.73 (3Н, s), 7.1-7.2 (2Н, m), 7.5-7.7 (5Н, m), 8.1-8.2 (2Н, m).
Пример 16. Получение 4-(4-фторфенил)-6-изопропил-5-метоксикарбонил-2-(2,4,6-триметилбензосульфонилокси)пиримидина.
Повторяли процедуры Примера 13 за тем исключением, что заменяли пара-толуолсульфонилхлорид на 23,0 г (105 ммоль) 2,4,6-триметилбензосульфонилхлорида.
Получали 37,7 г 4-(4-фторфенил)-6-изопропил-5-метоксикарбонил-2-(2,4,6-триметилбензосульфонилокси)пиримидина в виде бледно-желтого кристаллического продукта, имеющего нижеуказанные характеристики. Выход составил 84% (на основании количества 4-(4-фторфенил)-2-гидрокси-6-изопропил-5-метоксикарбонилпиримидина).
1H-ЯМР (CDCI3, δ (м.д.)): 1.17 (6Н, d, J=6.8 Гц), 2.34 (3Н, s), 2.67 (6Н, s), 3.0-3.1 (1Н, m), 3.73 (3Н, s), 7.00 (2Н, s), 7.0-7.2 (2Н, m), 7.4-7.5 (2Н, m).
Пример 17. Получение 4-(4-фторфенил)-6-изопропил-5-метоксикарбонил-2-(2,4,6-триизопропилбензосульфонилокси)пиримидина.
Повторяли процедуры Примера 13 за тем исключением, что заменяли пара-толуолсульфонилхлорид на 31,8 г (105 ммоль) 2,4,6-триизопропилбензосульфонилхлорида.
Получали 47,1 г 4-(4-фторфенил)-6-изопропил-5-метоксикарбонил-2-(2,4,6-триизопропилбензосульфонилокси)пиримидина в виде бледно-желтого кристаллического продукта, имеющего нижеуказанные характеристики. Выход составил 89% (на основании количества 4-(4-фторфенил)-2-гидрокси-6-изопропил-5-метоксикарбонилпиримидина).
1H-ЯМР (CDCl3, δ (м.д.)): 1.12 (6Н, d, J=6.6 Гц), 1.19 (12Н, d, J=6.8 Гц), 1.27 (6Н, d, J=7.1 Гц), 2.8-2.95 (1H, m), 2.95-3.1 (1Н, m), 3.73 (3Н, s), 4.1-4.3 (2Н, m), 7.0-7.1 (2Н, m), 7.20 (2Н, s), 7.4-7.5 (2Н, m).
Пример 18. Получение 4-(4-фторфенил)-6-изопропил-5-метоксикарбонил-2-(пара-метоксибензосульфонилокси)пиримидина.
Повторяли процедуры Примера 13 за тем исключением, что заменяли пара-толуолсульфонилхлорид на 21,7 г (105 ммоль) пара-метоксибензосульфонилхлорида.
Получали 39,9 г 4-(4-фторфенил)-6-изопропил-5-метоксикарбонил-2-(лара-метоксибензосульфонилокси)пиримидина в виде бесцветного кристаллического продукта, имеющего нижеуказанные характеристики. Выход составил 91% (на основании количества 4-(4-фторфенил)-2-гидрокси-6-изопропил-5-метоксикарбонилпиримидина).
1Н-ЯМР (CDCI3, δ (м.д.)): 1.25 (6Н, d, J=6.8 Гц), 3.0-3.2 (1H, m), 3.74 (3Н, s), 3.88 (3Н, s), 6.99 (2Н, dd, J=2.0, 9.0 Гц), 7.0-7.2 (2Н, m), 7.5-7.7 (2Н, m), 8.07 (2Н, dd, J=2.2, 9.0 Гц).
Пример 19. Получение 4-(4-фторфенил)-6-изопропил-5-метоксикарбонил-2-(пара-хлорбензосульфонилокси)пиримидина.
Повторяли процедуры Примера 13 за тем исключением, что заменяли пара-толуолсульфонилхлорид на 22,2 г (105 ммоль) пара-хлорбензосульфонилхлорида.
Получали 39,9 г 4-(4-фторфенил)-6-изопропил-5-метоксикарбонил-2-(пара-хлорбензосульфонилокси)пиримидина в виде бесцветного кристаллического продукта, имеющего нижеуказанные характеристики. Выход составил 89% (на основании количества 4-(4-фторфенил)-2-гидрокси-6-изопропил-5-метоксикарбонилпиримидина).
1H-ЯМР (CDCI3, δ (м.д.)): 1.23 (6Н, d, J=6.6 Гц), 3.0-3.2 (1Н, m), 3.74 (3Н, s), 7.1-7.2 (2Н, m), 7.5-7.7 (4Н, m), 8.0-8.1 (2Н, m).
Пример 20. Получение 4-(4-фторфенил)-6-изопропил-5-метоксикарбонил-2-(2-нитробензосульфонилокси)пиримидина.
Повторяли процедуры Примера 13 за тем исключением, что заменяли пара-толуолсульфонилхлорид на 23,3 г (105 ммоль) 2-нитробензосульфонилхлорида.
Получали 28,0 г 4-(4-фторфенил)-6-изопропил-5-метоксикарбонил-2-(2-нитробензосульфонилокси)пиримидина в виде непрозрачного кристаллического продукта, имеющего нижеуказанные характеристики. Выход составлял 62% (на основании количества 4-(4-фторфенил)-2-гидрокси-6-изопропил-5-метоксикарбонилпиримидина).
1H-ЯМР (CDCI3, δ (м.д.)): 1.17 (6Н, d, J=6.8 Гц), 3.0-3.2 (1Н, m), 3.75 (3Н, s), 7.1-7.2 (2Н, m), 7.5-7.6 (2Н, m), 7.7-8.0 (3Н, m), 8.33 (1Н, dd, J=1.7, 8.1 Гц).
Пример 21. Получение 4-(4-фторфенил)-6-изопропил-5-метоксикарбонил-2-(N-метил-N-метансульфониламино)пиримидина.
В стеклянную колбу объемом 25 мл, снабженную мешалкой, термометром и дефлегматором, помещали 196 мг (1,8 ммоль) N-метилметансульфонамида, 198 мг (1,8 ммоль) трет-пентоксида натрия, 7,5 мл ацетонитрила и 667 мг (1,5 ммоль) 4-(4-фторфенил)-6-изопропил-5-метоксикарбонил-2-(пара-толуолсульфокси)пиримидина. Смесь нагревали до 81-82°С в течение полутора часов с обратным холодильником для проведения реакции. После завершения реакции реакционную смесь охлаждали до комнатной температуры. К охлажденной смеси добавляли 10 мл воды и экстрагировали водную смесь этилацетатом. Этилацетатную часть отделяли и сушили над безводным сульфатом магния. Высушенную этилацетатную часть фильтровали и концентрировали под пониженным давлением. Остаток очищали колоночной хроматографией на силикагеле (колонка Wako Gel C-200; элюент: гексан/этилацетат (объемное соотоншение 2:1)). Получали 428 мг 4-(4-фторфенил)-6-изопропил-5-метоксикарбонил-2-(N-метил-N-метансульфониламино)пиримидина. Выход составил 75% (на основании количества 4-(4-фторфенил)-6-изопропил-5-метоксикарбонил-2-(п-толуолсульфонилокси)пиримидина).
Пример 22. Получение 2-амино-4-(4-фторфенил)-6-изопропил-5-метоксикарбонилпиримидина.
В стеклянную колбу объемом 25 мл, снабженную мешалкой, термометром и входом для газа, помещали при охлаждении на льду 1,00 г (2,71 ммоль) 4-(4-фторфенил)-6-изопропил-5-метоксикарбонил-2-метансульфонилоксипиримидина и 8,1 мл тетрагидрофурана. Смесь перемешивали при комнатной температуре в течение 12 часов в атмосфере газообразного аммиака для проведения реакции. После завершения реакции в реакционную смесь добавляли 10 мл воды. Затем водную смесь подвергали экстракции толуолом. Толуольную часть отделяли, промывали насыщенным водным раствором хлорида натрия и сушили над безводным сульфатом магния. Высушенную толуольную часть фильтровали и концентрировали под пониженным давлением. Остаток очищали колоночной хроматографией на силикагеле (колонка Wako Gel C-200; элюент: гексан/этилацетат (объемное соотношение 2:1)). Получали 0,63 г 2-амино-4-(4-фторфенил)-6-изопропил-5-метоксикарбонилпиримидина. Выход составил 80% (на основании количества 4-(4-фторфенил)-6-изопропил-5-метоксикарбонил-2-метансульфонилокси-пиримидина).
6-изопропил-5-метоксикарбонилпиримидин
Пример 23. Получение 4-(4-фторфенил)-6-изопропил-5-метоксикарбонил-2-N-метиламинопиримидина.
В стеклянную колбу объемом 50 мл, снабженную мешалкой, термометром и капельной воронкой, помещали 6,00 г (16,3 ммоль) 4-(4-фторфенил)-6-изопропил-5-метоксикарбонил-2-метансульфонилоксипиримидина. В колбу медленно добавляли по каплям при охлаждении льдом 5,06 г (65,2 ммоль) водного 40%-ного (мас.) раствора метиламина. Получившуюся смесь перемешивали в течение одного часа при той же температуре для проведения реакции. После завершения реакции в реакционную смесь добавляли 16 мл воды. Затем водную смесь подвергали экстракции толуолом. Толуольную часть отделяли, промывали насыщенным водным раствором хлорида натрия и сушили над безводным сульфатом магния. Высушенную толуольную часть фильтровали и концентрировали при пониженном давлении с получением 4,81 г 4-(4-фторфенил)-6-изопропил-5-метоксикарбонил-2-N-метиламинопиримидина. Выход составил 97% (на основании количества 4-(4-фторфенил)-6-изопропил-5-метоксикарбонил-2-метансульфонилоксипиримидина).
Пример 24. Получение 4-(4-фторфенил)-6-изопропил-5-метоксикарбонил-2-трифторметансульфонилоксипиримидина.
В стеклянную колбу объемом 300 мл, снабженную мешалкой, термометром и дефлегматором, помещали 8,7 г (30,0 ммоль) 4-(4-фторфенил)-2-гидрокси-6-изопропил-5-метоксикарбонилпиримидина, 3,0 г (30,0 ммоль) триэтиламина и 150 мл толуола. Смесь в колбе охлаждали до 0°С на ледяной бане. К охлажденной смеси медленно добавляли 8,46 г (30,0 ммоль) трифторметансульфонового ангидрида и подвергали получившуюся смесь реакции в течение 3 часов при той же температуре. После завершения реакции в реакционную смесь добавляли 90 мл воды. От водной реакционной смеси отделяли органическую жидкую часть. Органическую жидкую часть концентрировали под пониженным давлением. Остаток очищали колоночной хроматографией на силикагеле (колонка Wako Gel C-200; элюент: гексан/этилацетат (объемное соотношение 8:2)). Получали 8,46 г 4-(4-фторфенил)-6-изопропил-5-метоксикарбонил-2-трифторметансульфонилоксипиримидина, имеющего нижеуказанные характеристики, в виде бесцветного масла. Выход составил 74% (на основании количества 4-(4-фторфенил)-2-гидрокси-6-изопропил-5-метоксикарбонилпиримидина).
ИК (KBr, см-1): 3421, 2978, 1737, 1570, 1429, 1222, 1136, 973, 851.
1H-ЯМР (CDCI3, δ (м.д.)): 1.33 (6Н, d, J=6.6 Гц), 3.1-3.2 (1Н, m), 3.80 (3Н, s), 7.1-7.2 (2H, m), 7.7-7.8 (2H, m).
МСВР: 422,0585 (теоретическое значение (C16H14F4N2O5S (M+)) 422,0560).
Пример 25. Получение 4-(4-фторфенил)-6-изопропил-5-метоксикарбонил-2-трифторметансульфонилоксипиримидина.
В стеклянную колбу объемом 300 мл, снабженную мешалкой, термометром и дефлегматором, помещали 2,9 г (10,0 ммоль) 4-(4-фторфенил)-2-гидрокси-6-изопропил-5-метоксикарбонилпиримидина, 1,7 г (16,8 ммоль) триэтиламина и 50 мл толуола. Смесь в колбе охлаждали до 0°С на водяной бане. К охлажденной смеси медленно добавляли 2,4 г (14,1 ммоль) трифторметансульфонилхлорида и подвергали получившуюся смесь реакции в течение 3 часов при той же температуре. После завершения реакции к реакционной смеси добавляли 30 мл воды. От водной реакционной смеси отделяли органическую жидкую часть. Органическую жидкую часть концентрировали при пониженном давлении. Остаток очищали колоночной хроматографией на силикагеле (колонка Wako Gel C-200; элюент: гексан/этилацетат (объемное соотношение 8:2)). Получали 2,8 г 4-(4-фторфенил)-6-изопропил-5-метоксикарбонил-2-трифторметансульфонилоксипиримидина, имеющего нижеуказанные характеристики, в виде бесцветного масла. Выход составил 66% (на основании количества 4-(4-фторфенил)-2-гидрокси-6-изопропил-5-метоксикарбонилпиримидина).
Пример 26. Получение 4-(4-фторфенил)-6-изопропил-5-метоксикарбонил-2-(N-метил-N-метансульфониламино)пиримидина.
В стеклянную колбу объемом 50 мл, снабженную мешалкой, термометром и дефлегматором, помещали 3,0 г (7 ммоль) 4-(4-фторфенил)-6-изопропил-5-метоксикарбонил-2-трифторметансульфонилоксипиримидина, 0,99 г (9,1 ммоль) N-метил-N-метансульфонамида, 1,45 г (10,5 ммоль) карбоната калия (можно получать от Wako Junyaku Co., Ltd., особый сорт) и 14 мл бутилацетата. Смесь нагревали до 122-125°С в течение 3 часов при дефлегмации для проведения реакции. После завершения реакции реакционную смесь охлаждали до комнатной температуры. К реакционной смеси добавляли 10 мл воды и 7 мл ацетона и отделяли органическую жидкую часть. Органическую жидкую часть промывали насыщенным водным раствором хлорида натрия и концентрировали под пониженным давлением. Остаток очищали колоночной хроматографией на силикагеле (колонка Wako Gel C-200; элюент: гексан/этилацетат (объемное соотношение 5:1)). Получали 2,1 г 4-(4-фторфенил)-6-изопропил-5-метоксикарбонил-2-(N-метил-N-метансульфониламино)пиримидина в виде белого кристаллического продукта. Выход составил 78% (на основании количества 4-(4-фторфенил)-6-изопропил-5-метоксикарбонил-2-трифторметансульфонилоксипиримидина).
Пример 27. Получение 4-(4-фторфенил)-6-изопропил-5-метоксикарбонил-2-(N-метил-N-метансульфониламино)пиримидина.
В стеклянную колбу объемом 50 мл, снабженную мешалкой, термометром и дефлегматором, помещали 1,1 г (2,5 ммоль) 4-(4-фторфенил)-6-изопропил-5-метоксикарбонил-2-(пара-толуолсульфонилокси)пиримидина, 0,55 г (5,0 ммоль) N-метилметансульфонамида, 0,69 г (5,0 ммоль) карбоната калия (можно получать от Wako Junyaku Co., Ltd., особый сорт), 0,32 г (1,0 ммоль) бромида тетрабутиламмония, 20 мл толуола и 5 мл воды. Смесь нагревали до 85°С в течение 28 часов с обратным холодильником для проведения реакции. После завершения реакции реакционную смесь охлаждали до комнатной температуры. К реакционной смеси добавляли 10 мл воды и 7 мл ацетона и отделяли органическую жидкую часть. Органическую жидкую часть анализировали с помощью высокоэффективной жидкостной хроматографии. Подтвердили, что получили 0,6 г 4-(4-фторфенил)-6-изопропил-5-метоксикарбонил-2-(N-метил-N-метансульфониламино)пиримидина. Выход составил 63% (на основании количества 4-(4-фторфенил)-6-изопропил-5-метоксикарбонил-2-(пара-толуолсульфонилокси)пиримидина).
Промышленное применение
Пиримидиновое соединение, особенно соединение, представляющее собой 2-(N-метил-N-метансульфониламино)пиримидин, которое получают по этому изобретению, является полезным в качестве промежуточного соединения для получения агента, понижающего уровень холестерина (агента, представляющего собой гидроксиметилглутарил-КоА-редуктазу). Соединение формулы (3) можно превращать в ингибитор гидроксиметилглутарил-КоА-редуктазы способом, раскрытым в публикации заявки на Европейский патент №0521471, в Bioorg. Med. Chem., 5, 437 (1997), и в заявке на Международный патент № WO 0049014. Описания этих источников включены сюда посредством ссылки для демонстрации того, как соединение формулы (3) или формулы (8) можно превращать в ингибитор гидроксиметилглутарил-КоА-редуктазы, в частности розувастатин или его фармацевтически приемлемую соль, такую как розувастатина кальций.
Изобретение относится к новым способам получения (вариантам) 2-(N-метил-N-метансульфониламино)пиримидина, имеющего формулу (3), и аминопиримидинового соединения, имеющего формулу (8), которые могут найти применение для получения известного лекарственного соединения - розувастатина. В соединениях (3) и (8) 
R представляет собой низший алкил, каждый из R1 и R2 независимо представляет собой атом водорода, алкильную группу, алкилсульфонильную группу или арилсульфонильную группу. Способ получения соединения формулы (3) включает в себя стадии, на которых (I) изобутирилацетатный эфир формулы (5)
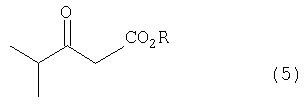
где R представляет собой низший алкил, подвергают взаимодействию с 4-фторбензальдегидом и мочевиной в присутствии протонного соединения и соли металла; (II) окисляют продукт реакции стадии (I); (III) продукт окисления стадии (II) подвергают взаимодействию с органическим сульфонилгалогенидом, имеющим формулу (2)

где R' представляет собой низший алкил, возможно замещенный атомами галогена, фенил, возможно замещенный 1-3 группами, выбранными из нитро, атомов галогена, разветвленного или неразветвленного низшего алкила, низшей алкоксигруппы, и Х представляет собой атом галогена, или органическим сульфоновым ангидридом, имеющим формулу (2а)

где R' имеет значения, указаные выше, в присутствии основания, и (IV) продукт реакции стадии (III) подвергают взаимодействию с N-метил-N-метансульфонамидом в присутствии основания. Способ получения соединения (8) включает стадию взаимодействия соответствующего 2-галоид- или 2-замещенного сульфонилпиримидина с соответствующим аминосоединением. Изобретение также относится к новым промежуточным продуктам и способам их получения. Предлагаемые способы позволяют избавиться от токсичных соединений и получать продукты с высоким выходом и чистотой. 11 н. и 24 з.п. ф-лы.

где R представляет собой низший алкил,
включающий стадии, на которых
гидроксипиримидиновое соединение, имеющее формулу (1)

где R такой, как указано выше,
подвергают взаимодействию с органическим сульфонилгалогенидом, имеющим формулу (2)

где R' представляет низший алкил, возможно замещенный атомами галогена, фенил, возможно замещенный 1-3 группами, выбранными из нитро, атомов галогена, разветвленного или неразветвленного низшего алкила, низшей алкоксигруппы, и Х представляет собой атом галогена, или с органическим сульфоновым ангидридом, имеющим формулу (2а)
где R' имеет значение, как указано выше, в присутствии основания, и получившийся продукт реакции подвергают взаимодействию с N-метил-N-метансульфонамидом в присутствии основания.

где R такой, как определено в п.1.
где R такой, как определено в п.1,
с 4-фторбензальдегидом и мочевиной в присутствии протонного соединения и соли металла.
где R представляет собой низший алкил.
при котором окисляют дигидропиримидиноновое соединение, имеющее формулу (4)

где R представляет собой низший алкил.
где R такой же, как определено в п.10,
с 4-фторбензальдегидом и мочевиной в присутствии протонного соединения и соли металла.
где R представляет низший алкил.
где R такой, как указано в п.17,
подвергают взаимодействию с 4-фторбензальдегидом и мочевиной в присутствии протонного соединения и соли металла.

где R представляет собой низший алкил, и каждый из R1 и R2 независимо представляет собой атом водорода, алкильную группу, алкилсульфонильную группу или арилсульфонильную группу,
при котором 2-замещенное пиримидиновое соединение, имеющее формулу (6)

где R такой, как указано выше, и Х представляет собой атом галогена или R'-замещенную сульфонилоксигруппу, где R' такой, как указано выше,
подвергают взаимодействию с аминосоединением, имеющим формулу (7)

где каждый из R1 и R2 такой, как указано выше.

где R представляет собой низший алкил и Hal представляет собой атом галогена.
где R представляет собой низший алкил,
подвергают взаимодействию с галогенирующим агентом.
где R представляет собой низший алкил и R1 - низший алкил, возможно замещенный атомами галогена, фенил, возможно замещенный 1-3 группами, выбранными из нитро, атомов галогена, разветвленного или неразветвленного низшего алкила, низшей алкоксигруппы.
где R представляет собой низший алкил,
подвергают взаимодействию с органическим сульфонилгалогенидом, имеющим формулу (2)

где R' представляет собой низший алкил, возможно замещенный атомами галогена, фенил, возможно замещенный 1-3 группами, выбранными из нитро, атомов галогена, разветвленного или неразветвленного низшего алкила, низшей алкоксигруппы, и Х представляет собой атом галогена, или органическим сульфоновым ангидридом, имеющим формулу (2а)

где R' имеет значение, как указано выше, в присутствии основания.
где R представляет собой низший алкил,
причем этот способ включает в себя стадии, на которых
(I) изобутирилацетатный эфир формулы (5)
где R представляет собой низший алкил, подвергают взаимодействию с 4-фторбензальдегидом и мочевиной в присутствии протонного соединения и соли металла;
(II) окисляют продукт реакции стадии (I);
(III) продукт окисления стадии (II) подвергают взаимодействию с органическим сульфонилгалогенидом, имеющим формулу (2)
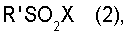
где R' представляет собой низший алкил, возможно замещенный атомами галогена, фенил, возможно замещенный 1-3 группами, выбранными из нитро, атомов галогена, разветвленного или неразветвленного низшего алкила, низшей алкоксигруппы, и Х представляет собой атом галогена, или органическим сульфоновым ангидридом, имеющим формулу (2a)

где R' имеет значение, как указано выше, в присутствии основания,
и (IV) продукт реакции стадии (III) подвергают взаимодействию с N-метил-N-метансульфонамидом в присутствии основания.
Приоритет по пунктам и признакам:
| Печь для непрерывного получения сернистого натрия | 1921 |
|
SU1A1 |
| Способ формовки трубной заготовки | 1974 |
|
SU555931A1 |
| Кипятильник для воды | 1921 |
|
SU5A1 |
| MA YUN et al | |||
| "Lantanide Triflate Catalized Biginelli Reaction | |||
| One - Pot synthesis of dihydropyrimidinones under Solvent - Pree Conditions" Joumal of Organic Chemystry, 2000, 65 (12), 3864-3868 | |||
| WO 00/49014 A1, 24.08.2000. | |||

