Область техники, к которой относится изобретение
Данное изобретение касается производных триазола как ингибиторов фермента 11-бета-гидроксистероиддегидрогеназы I-го типа (11β-ГСД-1 или ГСД-1) и способов лечения определенных состояний с их использованием. Соединения данного изобретения применяются для лечения диабета, например инсулиннезависимого диабета 2-го типа (ИНЗСД), инсулинорезистентности, ожирения, нарушений жирового обмена, гипертензии и других заболеваний и состояний.
Уровень техники данного изобретения
Причиной диабета является множество факторов и наиболее упрощенно диабет характеризуется повышенным содержанием глюкозы в плазме крови натощак (гипергликемия). Существуют две наиболее изученные формы диабета: диабет 1-го типа, или инсулинзависимый сахарный диабет (ИЗСД), при котором у пациентов продуцируется мало или не продуцируется инсулин, гормон, регулирующий усвоение глюкозы, и диабет 2-го типа, или инсулиннезависимый диабет 2-го типа (ИНЗСД), при котором у пациентов продуцируется инсулин и даже проявляется гиперинсулинемия (уровни инсулина в плазме крови такие же, или даже повышены, по сравнению с людьми, не страдающими диабетом), при этом у них проявляется гипергликемия. Диабет 1-го типа обычно лечат инъекционным введением инсулина. Однако при диабете 2-го типа часто развивается "инсулинорезистентность", так что стимулирующее воздействие инсулина на глюкозу и липидный метаболизм в основных инсулин-чувствительных тканях, а именно мышцах, печени и жировых тканях, снижено. Инсулинорезистентные пациенты, не являющиеся диабетиками, имеют повышенные уровни инсулина, компенсирующие их инсулинорезистентность, так что уровни глюкозы в сыворотке крови не повышены. У пациентов с ИНЗСД уровни инсулина в плазме крови, даже если они повышены, недостаточны для преодоления выраженной инсулинорезистентности, приводя к гипергликемии.
Инсулинорезистентность, главным образом, возникает из-за дефекта связывания с рецептором, что пока еще не совсем ясно. Резистентность к инсулину приводит к недостаточной активации усвоения глюкозы, сниженному окислению глюкозы и отложению гликогена в мышцах, неадекватному подавлению инсулином липолиза в жировой ткани и неадекватной продукции глюкозы и секреции ее печенью.
Хроническая или неконтролируемая гипергликемия, наблюдающаяся у диабетиков, связана с повышенной заболеваемостью и преждевременной смертностью. Ненормальный гомеостаз глюкозы так же как напрямую, так и опосредованно связан с ожирением, гипертензией и изменениями в липидном, липопротеиновом и аполипопротеиновом метаболизме. Диабет 2-го типа сопровождается повышенным риском развития сердечно-сосудистых осложнений, например атеросклероза, заболевания коронарных артерий сердца, инфаркта, заболевания периферических сосудов, гипертензии, нефропатии, нейропатии и ретинопатии. Следовательно, терапевтический контроль над гомеостазом глюкозы, липидным метаболизмом, ожирением и гипертензией является принципиально важным в клиническом управлении и лечении сахарного диабета.
Многие пациенты, имеющие инсулинорезистентность, но не имеющие выраженный диабет 2-го типа, также подвержены риску появления симптомов, называемых "синдромом Х" или "метаболическим синдромом". Синдром Х и метаболический синдром характеризуются инсулинорезистентностью, наряду с абдоминальным ожирением, гиперинсулинемией, высоким кровяным давлением, низким уровнем ЛВП (липопротеинов высокой плотности) и высоким уровнем ЛОНП (липопротеинов очень низкой плотности). Такие пациенты, в независимости от того, имеют ли они выраженный сахарный диабет или нет, подвержены высокому риску развития сердечно-сосудистых осложнений, приведенных выше.
Лечение диабета 2-го типа обычно включает физические упражнения и диету. Повышение уровня инсулина в плазме введением сульфонилмочевин (например, толбутамида и глипизида) или меглитинида, который стимулирует β-клетки поджелудочной железы секретировать больше инсулина, и/или инъекцией инсулина, когда сульфонилмочевины или меглитинид становятся неэффективными, может привести к созданию достаточно высокой концентрации инсулина для стимуляции инсулинрезистентных тканей. Однако в результате может возникнуть опасно низкий уровень глюкозы и в конечном итоге повышенный уровень инсулинорезистентности.
Бигуаниды повышают чувствительность к инсулину, приводя к некоторой коррекции гипергликемии. Однако многие бигуаниды, например фенформин и метформин, вызывают лактоацидоз, тошноту и диарею.
Глитазоны (а именно 5-бензилтиазолидин-2,4-дионы) образуют новый класс соединений со способностью улучшения гипергликемии и других симптомов диабета 2-го типа. Данные агенты существенно повышают чувствительность к инсулину мышц, печени и жировой ткани, приводя к частичной или полной коррекции повышенных уровней глюкозы в плазме крови, в основном, не вызывая гипогликемии. Глитазоны, недавно выпущенные на рынок, являются агонистами рецепторов, активируемых пероксисомным пролифератором подтипа гамма (РАПП). Полагают, что агонизм РАПП-гамма, в основном, ответственен за повышенную чувствительность к инсулину, которая наблюдается в отношении глитазонов. Новые РАПП-агонисты, созданные для лечения диабета 2-го типа и/или дислипидемии, являются агонистами одного или более РАПП-подтипов альфа, гамма и дельта. Обзор по инсулин-сенсибилизирующим агентам и другим механизмам лечения диабета 2-го типа, см. M. Tadayyon и S.A. Smith, "Insulin sensitisation in the treatment of Type 2 diabetes", Expert Opin. Investig. Drugs, 12: 307-324 (2003).
Существует постоянная потребность в новых способах лечения диабета и связанных с ним состояний, таких как метаболический синдром. Данное изобретение решает эту и другие задачи.
Сущность изобретения
Данное изобретение касается бицикло[2.2.2]-окт-1-ил-1,2,4-триазолов структурной формулы I:
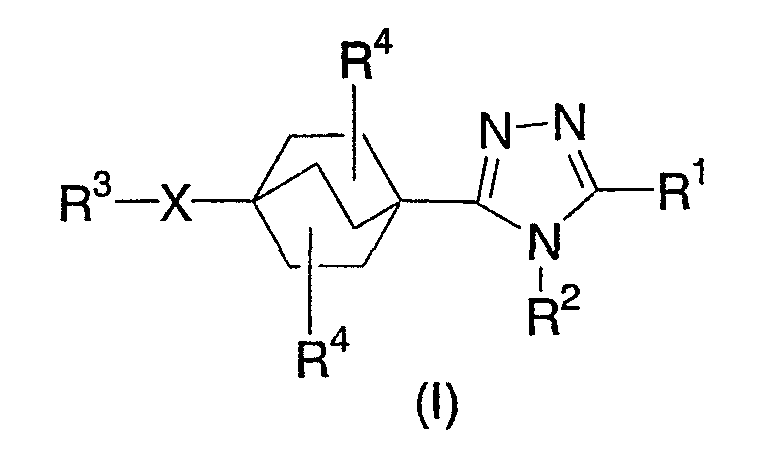
Данные производные бицикло[2.2.2]-октилтриазола эффективны в качестве ингибиторов 11β-гидроксистероиддегидрогеназы 1-го типа (11β-ГСД1). Таким образом, они эффективны в лечении, контроле и профилактике нарушений, реагирующих на ингибирование 11β-ГСД1, таких как гипергликемия, инсулинорезистентность, диабет 2-го типа, липидные нарушения, ожирение, атеросклероз и метаболический синдром.
Данное изобретение также касается фармацевтических композиций, содержащих соединения данного изобретения и фармацевтически приемлемый носитель.
Данное изобретение также касается способов лечения или контролирования гипергликемии, инсулинорезистентности, диабета 2-го типа, ожирения, липидных нарушений, атеросклероза и метаболического синдрома введением соединений и фармацевтических композиций данного изобретения.
Подробное описание изобретения
Данное изобретение касается производных бицикло[2.2.2]-окт-1-ил-1,2,4-триазола, применяемых в качестве ингибиторов 11β-ГСД1. Соединения данного изобретения описываются структурной формулой I:
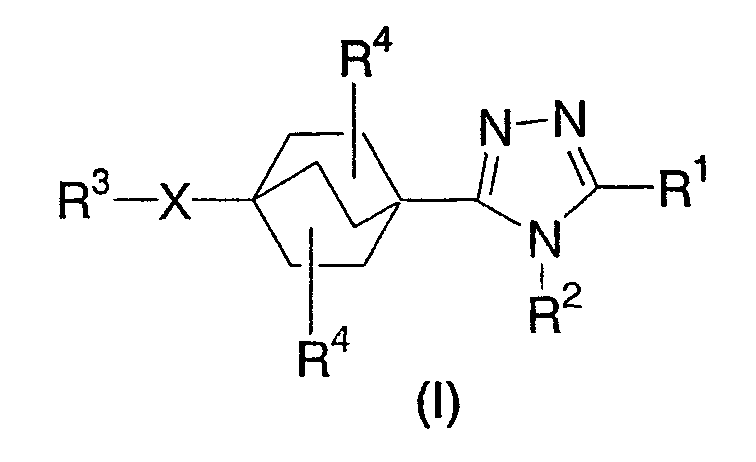
или их фармацевтически приемлемой солью; где
каждый p независимо представляет собой 0, 1, или 2;
каждый n независимо представляет собой 0, 1, или 2;
X выбирают из группы, состоящей из одинарной связи, O, S(O)p, NR6,
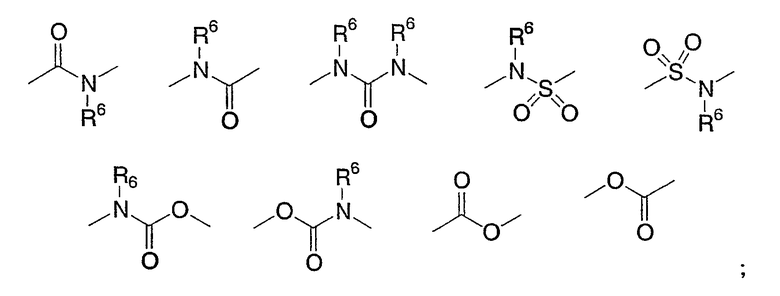
R1 выбирают из группы, состоящей из
арилкарбонила,
(CH2)n-арила и
(CH2)n-гетероарила;
где арил и гетероарил являются незамещенными или замещенными от одного до трех заместителями, независимо выбранными из R5;
R2 выбирают из группы, состоящей из
водорода,
C1-8алкила,
C2-6алкенила и
(CH2)n-C3-6циклоалкила,
в котором алкил, алкенил и циклоалкил являются незамещенными или замещенными от одного до трех заместителями, независимо выбранными из R8 и оксо;
каждый R4 независимо выбирают из группы, состоящей из
водорода,
галогена,
гидрокси,
оксо,
C1-3алкила и
C1-3алкокси;
R3 выбирают из группы, состоящей из
водорода,
C1-10алкила,
C2-10алкенила,
(CH2)n-C3-6циклоалкила,
(CH2)n-арила,
(CH2)n-гетероарила и
(CH2)n-гетероциклила;
в котором арил, гетероарил и гетероциклил являются незамещенными или замещенными от одного до трех заместителями, независимо выбранными из R5; и алкил, алкенил и циклоалкил являются незамещенными или замещенными от одной до пяти группами, независимо выбранными из R8 и оксо;
R5 и R8, каждый, независимо выбирают из группы, состоящей из
водорода,
формила,
C1-6алкила,
(CH2)n-арила,
(CH2)n-гетероарила,
(CH2)n-гетероциклила,
(CH2)n-C3-7циклоалкила,
галогена,
OR7,
(CH2)nN(R7)2,
циано,
(CH2)nCO2R7,
NO2,
(CH2)nNR7SO2R6,
(CH2)nSO2N(R7)2,
(CH2)nS(O)pR6,
(CH2)nSO2OR7,
(CH2)nNR7C(O)N(R7)2,
(CH2)nC(O)N(R7)2,
(CH2)nNR6C(O)R6,
(CH2)nNR6CO2R7,
O(CH2)nC(O)N(R7)2,
CF3,
CH2CF3,
OCF3,
OCHCF2 и
OCH2CF3;
где арил, гетероарил, циклоалкил и гетероциклил являются незамещенными или замещенными от одного до трех заместителями, независимо выбранными из галогена, гидрокси, C1-4алкила, трифторметила, трифторметокси и C1-4алкокси; и где любой углеродный атом метиленовой группы (СН2) в R5 и R8 является незамещенным или замещенным одной-двумя группами, независимо выбранными из галогена, гидрокси и C1-4алкила; или два заместителя, находящихся на одном и том же углеродном атоме метиленовой группы (СН2), взятые вместе с атомом углерода, к которому они присоединены, образуют циклопропильную группу;
каждый R6 независимо выбирают из группы, состоящей из
C1-8алкила,
(CH2)n-арила,
(CH2)n-гетероарила и
(CH2)n-C3-7циклоалкила;
где алкил и циклоалкил являются незамещенными или замещенными от одного до пяти заместителями, независимо выбранными из галогена, оксо, C1-4алкокси, C1-4алкилтио, гидрокси, амино; и арил и гетероарил являются незамещенными или замещенными от одного до трех заместителями, независимо выбранными из циано, галогена, гидрокси, амино, карбокси, трифторметила, трифторметокси, C1-4алкила и C1-4алкокси;
или двух R6 групп вместе с атомом, к которому они присоединены с образованием 5-8-членной моно- или бицикличной кольцевой системы, необязательно содержащей дополнительный гетероатом, выбранный из O, S и NC1-4алкила; и
каждый R7 является водородом или R6.
В одном воплощении соединений данного изобретения R2 является циклопропилом, C1-3алкилом или C2-3алкенилом и R1 является фенилом или нафтилом, в котором фенил или нафтил являются незамещенными или замещенными от одного до трех заместителями, независимо выбранными из R5. В классе этого воплощения R5 выбирают из группы, состоящей из галогена, гидрокси, трифторметила, трифторметокси, C1-3алкила, C1-3алкокси, C1-3алкилтио и C1-3алкилсульфонила. В подклассе этого класса R2 является метилом и R4 является водородом.
Во втором воплощении соединений данного изобретения
Х является одинарной связью;
R1 является фенилом или нафтилом, в котором фенил и нафтил являются незамещенными или замещенными от одного до трех заместителями, независимо выбранными из R5;
R2 является циклопропилом, C1-3алкилом или C2-3алкенилом; и
R3 является C1-6алкилом, незамещенным или замещенным одним или тремя заместителями, независимо выбранными из R8 и оксо.
В классе этого второго воплощения R5 выбирают из группы, состоящей из галогена, гидрокси, трифторметила, трифторметокси, C1-3алкила, C1-3алкокси, C1-3алкилтио и C1-3алкилсульфонила. В подклассе этого класса R2 является метилом и R4 является водородом.
В другом классе этого воплощения R8 выбирают из группы, состоящей из галогена, гидрокси, оксо, C1-4алкокси, C1-4алкилтио, С1-4алкилсульфинила, C1-4алкилсульфонила, и фенил незамещен или замещен от одной до трех групп, независимо выбранных из галогена и трифторметила. В подклассе этого класса R2 является метилом и R4 является водородом.
В третьем классе этого воплощения R5 выбирают из группы, состоящей из галогена, гидрокси, трифторметила, трифторметокси, C1-3алкила, C1-3алкокси, C1-3алкилтио и C1-3алкилсульфонила; и R8 выбирают из группы, состоящей из галогена, гидрокси, оксо, C1-4алкокси, C1-4алкилтио, C1-4алкилсульфонила, и фенил незамещен или замещен от одной до трех групп, независимо выбранных из галогена и трифторметила. В подклассе этого класса R2 является метилом и R4 является водородом.
В третьем воплощении соединений данного изобретения
Х является одинарной связью;
R1 является фенилом или нафтилом, в котором фенил и нафтил являются незамещенными или замещенными от одного до трех заместителями, независимо выбранными из R5;
R2 является циклопропилом, C1-3алкилом или C2-3алкенилом; и
R3 является фенилом или гетероарилом, в котором фенил и гетероарил являются незамещенными или замещенными от одного до трех заместителями, независимо выбранными из R5.
В классе этого воплощения R2 является метилом и R4 является водородом.
В другом классе данного воплощения R3 является фенилом, незамещенным или замещенным от одного до трех заместителями, независимо выбранными из R5. В подклассе этого класса R5 выбирают из группы, состоящей из галогена, гидрокси, трифторметила, трифторметокси, C1-3алкила, C1-3алкокси, C1-3алкилтио и C1-3алкилсульфонила. В подклассе этого класса R2 является метилом и R4 является водородом.
В третьем классе этого воплощения R3 является оксадиазолилом, незамещенным или замещенным от одного до двух заместителей, независимо выбранных из R5.
В подклассе этого класса R5 является фенилом, незамещенным или замещенным от одного до трех заместителей, независимо выбранных из галогена, гидрокси, C1-4алкила, трифторметила, трифторметокси и C1-4алкокси. В подклассе этого класса R2 является метилом и R4 является водородом.
В данном описании применяются следующие определения.
"Алкил", также как и другие группы, имеющие приставку "алк", такие как алкокси и алканоил, подразумевают углеродные цепи, которые могут быть линейными или разветвленными, и их комбинации, если углеродная цепь не определена иначе. Примеры алкильных групп включают метил, этил, пропил, изопропил, бутил, втор- и трет-бутил, пентил, гексил, гептил, октил, нонил и тому подобные. В случае, когда определенное число углеродных атомов позволяет, например от C3-10, термин "алкил" также включает циклоалкильные группы и комбинации линейных или разветвленных алкильных цепей, конденсированных с циклоалкильными структурами; когда число углеродных атомов не определено, подразумевается C1-6.
"Алкенил" подразумевает углеродные цепи, которые содержат, по меньшей мере, одну углерод-углеродную двойную связь, которые могут быть линейными или разветвленными или их комбинации, если углеродная цепь не определена иначе. Примеры алкенила включают винил, аллил, изопропенил, пентенил, гексенил, гептенил, 1-пропенил, 2-бутенил, 2-метил-2-бутенил и тому подобные. В случае, когда определенное число углеродных атомов позволяет, например от C5-10, термин "алкенил" также включает циклоалкенильные группы и комбинации линейных, разветвленных и циклических структур. Когда число углеродных атомов не определено, подразумевается C2-6.
"Алкинил" подразумевает углеродные цепи, которые содержат, по меньшей мере, одну углерод-углеродную тройную связь и которые могут быть линейными или разветвленными, или их комбинации. Примеры алкинила включают этинил, пропаргил, 3-метил-1-пентинил, 2-гептинил и тому подобные.
"Циклоалкил" является подтипом алкила и подразумевает насыщенное карбоциклическое кольцо, имеющее определенное число углеродных атомов. Примеры циклоалкила включают циклопропил, циклобутил, циклопентил, циклогексил, циклогептил, циклооктил и тому подобные. Циклоалкильная группа является, в основном, моноцикличной, если не определено иначе. Циклоалкильные группы являются насыщенными, если не определено иначе.
Термин "алкокси" касается прямой или разветвленной цепи алкоксидов с определенным числом атомов углерода (например, Cl-6алкокси) или с любым числом атомов углерода в пределах данного интервала [то есть метокси (MeO-), этокси, изопропокси и т.д.].
Термин "алкилтио" касается прямой или разветвленной цепи алкилсульфидов с определенным числом атомов углерода (например, Cl-6алкилтио) или с любым числом атомов углерода в пределах данного интервала [то есть метилтио (MeS-), этилтио, изопропилтио и т.д.].
Термин "алкиламино" касается прямой или разветвленной цепи алкиламинов с определенным числом атомов углерода (например, Cl-6алкиламино) или с любым числом атомов углерода в пределах данного интервала [то есть метиламино, этиламино, изопропиламино, т-бутиламино и т.д.].
Термин "алкилсульфонил" касается прямой или разветвленной цепи алкилсульфонов с определенным числом атомов углерода (например, Cl-6алкилсульфонил) или с любым числом атомов углерода в пределах данного интервала [то есть метилсульфонил (MeSO2-), этилсульфонил, изопропилсульфонил и т.д.].
Термин "алкилсульфинил" касается прямой или разветвленной цепи алкилсульфоксидов с определенным числом атомов углерода (например, Cl-6алкилсульфинил) или с любым числом атомов углерода в пределах данного интервала [то есть метилсульфинил (MeSO-), этилсульфинил, изопропилсульфинил и т.д.].
Термин "алкилоксикарбонил" касается прямых или разветвленных эфиров производных карбоновых кислот данного изобретения с определенным числом атомов углерода (например, Cl-6алкилоксикарбонил) или с любым числом атомов углерода в пределах данного интервала [то есть метилоксикарбонил (MeOCO-), этилоксикарбонил или бутилоксикарбонил].
"Арил" подразумевает моно- или полициклическую ароматическую кольцевую систему, содержащую атомы углеродного кольца. Предпочтительными арилами являются моноцикличные или бицикличные 6-10-членные кольцевые ароматические системы. Предпочтительными арилами являются фенил и нафтил. Наиболее предпочтительным арилом является фенил.
"Гетероцикл" и "гетероциклил" касаются насыщенных или ненасыщенных неароматических колец или кольцевых систем, содержащих, по меньшей мере, один гетероатом, выбранный из O, S и N, дополнительно содержащих окисленные формы серы, а именно SO и SO2. Примеры гетероциклов включают тетрагидрофуран (ТГФ), дигидрофуран, 1,4-диоксан, морфолин, 1,4-дитиан, пиперазин, пиперидин, 1,3-диоксолан, имидазолидин, имидазолин, пирролин, пирролидин, тетрагидропиран, дигидропиран, оксатиолан, дитиолан, 1,3-диоксан, 1,3-дитиан, оксатиан, тиоморфолин и тому подобные.
"Гетероарил" подразумевает ароматический или частично ароматический гетероцикл, который содержит, по меньшей мере, один кольцевой гетероатом, выбранный из O, S и N. Гетероарилы, следовательно, включают гетероарилы, конденсированные с другими типами колец, такими как арилы, циклоалкилы и гетероциклы, которые не являются ароматическими.
Примеры гетероарильных групп включают пирролил, изоксазолил, изотиазолил, пиразолил, пиридил, оксазолил, оксадиазолил, тиадиазолил, тиазолил, имидазолил, триазолил, тетразолил, фурил, триазинил, тиенил, пиримидил, бензизоксазолил, бензоксазолил, бензотиазолил, бензотиадиазолил, дигидробензофуранил, индолинил, пиридазинил, индазолил, изоиндолил, дигидробензотиенил, индолизинил, циннолинил, фталазинил, хиназолинил, нафтиридинил, карбазолил, бензодиоксолил, хиноксалинил, пуринил, фуразанил, изобензилфуранил, бензимидазолил, бензофуранил, бензотиенил, хинолил, индолил, изохинолил, дибензофуранил и тому подобные. В понятие гетероциклильной и гетероарильной групп включены кольца и кольцевые системы, содержащие 3-15 атомов, образуя 1-3 кольца.
"Галоген" касается фтора, хлора, брома и иода. В основном, предпочтительны хлор и фтор. Фтор наиболее предпочтителен, когда галогены замещены алкильной или алкоксигруппой (например, CF3О и CF3CH2О).
Термин "композиция", например фармацевтическая композиция, охватывает продукт, содержащий активный ингредиент(ы) и инертный ингредиент(ы), образующий носитель, так же как любой продукт, который получается напрямую или опосредованно, комбинацией, комплексообразованием, или агрегацией любых двух или более ингредиентов, или диссоциацией одного или более ингредиентов, или другими типами реакций или взаимодействий одного или более ингредиентов. Соответственно, фармацевтические композиции данного изобретения включают любую композицию, образованную смешиванием соединения данного изобретения и фармацевтически приемлемого носителя.
Следует понимать, что термины "введение" и "назначение" соединения подразумевают применение соединения данного изобретения или пролекарства соединения данного изобретения для индивидуального потребления. Соединения структурной формулы I могут содержать один или более асимметрических центров и, следовательно, могут существовать в виде рацематов и рацемических смесей, отдельных энантиомеров, диастереомерных смесей и индивидуальных диастереомеров. Подразумевается, что данное изобретение охватывает все данные изомерные формы соединений структурной формулы I.
Некоторые из описанных здесь соединений содержат олефиновые двойные связи и, если иначе не определено, подразумевается, что они содержат геометрические изомеры E и Z.
Некоторые из описанных здесь соединений могут существовать в виде таутомеров, таких как кетоенольные таутомеры. Индивидуальные таутомеры, так же как и их смеси, охвачены соединениями структурной формулы I.
Соединения структурной формулы I могут быть разделены на индивидуальные диастереоизомеры, например, фракционной кристаллизацией из подходящего растворителя, например метанола или этилацетата или их смеси, или посредством хиральной хроматографии с использованием оптически активной стационарной (неподвижной) фазы. Абсолютная стереохимия может быть определена рентгенокристаллографией кристаллических продуктов или кристаллических интермедиатов, которые дериватизируют, если необходимо, реактивом, содержащим асимметрический центр известной абсолютной конфигурации.
Альтернативно, любой стереоизомер общей структурной формулы I может быть получен стереоспецифическим синтезом, используя оптически чистые исходные материалы или реагенты известной абсолютной конфигурации.
В другом аспекте данного изобретения фармацевтическая композиция целенаправленно содержит соединение в соответствии со структурной формулой I или фармацевтически приемлемой солью, или их сольватами, в комбинации с фармацевтически приемлемым носителем. Под термином "сольват" подразумевается гидрат, алкоголят или другой кристаллизационный сольват.
Другой аспект данного изобретения направлен на способ лечения гипергликемии, диабета или инсулинорезистентности у млекопитающих, нуждающихся в таком лечении, который включает введение указанному пациенту эффективного количества соединения, соответствующего структурной формуле I или фармацевтически приемлемой соли или их сольватов.
В другом аспекте данного изобретения описан способ лечения инсулиннезависимого сахарного диабета (2-го типа) у млекопитающих, нуждающихся в таком лечении, включающий введение пациенту эффективного количества антидиабетического соединения, соответствующего структурной формуле I.
В следующем аспекте данного изобретения описан способ лечения ожирения у млекопитающих, нуждающихся в таком лечении, включающий введение указанному пациенту соединения, соответствующего структурной формуле I, в количестве, эффективном для лечения ожирения.
В ином аспекте данного изобретения описан способ лечения метаболического синдрома у млекопитающих, нуждающихся в таком лечении, включающий введение указанному пациенту соединения, соответствующего структурной формуле I в количестве, эффективном для лечения метаболического синдрома.
В следующем аспекте данного изобретения описан способ лечения нарушения жирового обмена, выбранного из группы, состоящей из дислипидемии, гиперлипидемии, гипертриглицеридемии, гиперхолестеринемии, низкого содержания ЛВП и высокого содержания ЛНП у млекопитающего-пациента, нуждающегося в таком лечении, включающий введение указанному пациенту соединения, соответствующего структурной формуле I, в количестве, эффективном для лечения указанного нарушения жирового обмена.
В другом аспекте данного изобретения описан способ лечения атеросклероза у млекопитающего-пациента, нуждающегося в таком лечении, включающий введение указанному пациенту соединения, соответствующего структурной формуле I, в количестве, эффективном для лечения атеросклероза.
Другой аспект данного изобретения касается использования соединений структурной формулы I для лечения гипергликемии, инсулинорезистентности, диабета 2-го типа, липидных нарушений, ожирения, атеросклероза и метаболического синдрома.
Кроме того, дополнительный аспект данного изобретения обеспечивает использование соединений структурной формулы I в производстве лекарственных средств для применения в лечении состояния, выбранного из группы, состоящей из гипергликемии, инсулинорезистентности, диабета 2-го типа, нарушений жирового обмена, ожирения, атеросклероза и метаболического синдрома.
Соединения данного изобретения могут быть введены в форме фармацевтически приемлемой соли. Термин "фармацевтически приемлемая соль" относится к солям, приготовленным из фармацевтически приемлемых нетоксичных оснований или кислот, включая неорганические или органические основания и неорганические или органические кислоты. Соли основных соединений, охваченные термином "фармацевтически приемлемая соль", относятся к нетоксичным солям соединений данного изобретения, которые, в основном, получают взаимодействием свободного основания с подходящей органической или неорганической кислотой. Представители солей основных соединений данного изобретения включают, не ограничиваясь ими, ацетат, бензолсульфонат, бензоат, бикарбонат, бисульфат, битартрат, борат, бромид, камсилат, карбонат, хлорид, клавуланат, цитрат, дигидрохлорид, эдетат, эдисилат, эстолат, эсилат, фумарат, глуцептат, глюконат, глютамат, гликоллиларсанилат, гексилрезорцинат, гидрабамин, гидробромид, гидрохлорид, гидроксинафтоат, иодид, изотионат, лактат, лактобионат, лаурат, малат, малеат, манделат, мезилат, метилбромид, метилнитрат, метилсульфат, мукат, напсилат, нитрат, аммониевую соль N-метилглюкамина, олеат, оксалат, памоат (эмбонат), пальмитат, пантотенат, фосфат/дифосфат, полигалактуронат, салицилат, стеарат, сульфат, субацетат, сукцинат, таннат, тартрат, теоклат, тозилат, триэтилиодид и валерат. Кроме того, когда соединения данного изобретения несут кислотный участок, их фармацевтически приемлемые соли включают, не ограничиваясь ими, соли, получаемые из неорганических оснований, содержащих алюминий, аммоний, кальций, медь, железо, двухвалентное железо, литий, магний, трехвалентный марганец, двухвалентный марганец, калий, натрий, цинк и тому подобные. Особенно предпочтительными являются аммониевые, кальциевые, магниевые, калиевые и натриевые соли. Соли, получаемые из органических нетоксичных оснований, включают соли первичных, вторичных и третичных аминов, циклических аминов и основные ионообменные смолы, такие как аргинин, бетаин, кофеин, холин, N,N-дибензилэтилендиамин, диэтиламин, 2-диэтиламиноэтанол, 2-диметиламиноэтанол, этаноламин, этилендиамин, N-этилморфолин, N-этилпиперидин, глюкамин, глюкозамин, гистидин, гидрабамин, изопропиламин, лизин, метилглюкамин, морфолин, пиперазин, пиперидин, полиаминовые смолы, прокаин, пурины, теобромин, триэтиламин, триметиламин, трипропиламин, трометамин и тому подобные.
Также в случае карбоновой кислоты (-COOH) или спиртовой группы, представленных в соединениях данного изобретения, могут быть применены фармацевтически приемлемые эфиры производных карбоновой кислоты, такие как метильные, этильные, или пивалоилоксиметильные, или ацильные производные спиртов, таких как ацетат или малеат. Включены те эфиры и ацильные группы, которые известны в уровне техники модифицирующими растворимость или гидролиз характеристиками при применении в качестве композиций с замедленным высвобождением или пролекарств.
Следует понимать, что используемые здесь ссылки на соединения структурной формулы I, как подразумевается, также включают фармацевтически приемлемые соли, а также соли, которые не являются фармацевтически приемлемыми при использовании их в качестве предшественников свободных соединений или их фармацевтически приемлемых солей или в других процессах синтеза.
Сольваты и, в частности, гидраты соединений структурной формулы I также включены в данное изобретение.
Соединения, описываемые здесь, являются селективными ингибиторами фермента 11β-ГСД1. Таким образом, данное изобретение касается применения ингибиторов 11β-ГСД1 для ингибирования редуктазной активности 11β-гидроксистероиддегидрогеназы, ответственной за превращение кортизона в кортизол. Избыток кортизола связан с множеством нарушений, включая ИНЗСД, ожирение, дислипидемию, инсулинорезистентность и гипертензию. Введение соединений данного изобретения снижает уровень кортизола и других 11β-гидроксистероидов в тканях-мишенях, таким образом, снижая эффекты избыточных количеств кортизола и других 11β-гидроксистероидов. Ингибирование 11β-ГСД1 может быть использовано в лечении и контролировании заболеваний, обусловленных ненормально высокими уровнями кортизола и других 11β-гидроксистероидов, таких как ИНЗСД, ожирения, гипертензии и дислипидемии. Ингибирование активности 11β-ГСД в мозге, например снижение уровня кортизола, может быть также полезно в лечении или снижении тревоги, депрессии и когнитивного ухудшения.
Данное изобретение также включает применение ингибитора 11β-ГСД1 для лечения, контроля, улучшения, предотвращения, отсрочки начала или снижения риска развития заболеваний и состояний, которые описаны здесь, обусловленных избытком или неконтролируемыми количествами кортизола и/или других кортикостероидов у млекопитающего-пациента, особенно у человека, введением эффективного количества соединения структурной формулы I или его фармацевтически приемлемой соли или сольвата. Ингибирование фермента 11β-ГСД1 ограничивает превращение кортизона, который в норме инертен, в кортизол, который может обусловливать или давать вклад в симптомы таких заболеваний и состояний, если присутствует в избыточных количествах.
ИНЗСД и гипертензия:
Соединения данного изобретения являются селективными ингибиторами в отношении 11β-ГСД1 по сравнению с 11β-ГСД2. В то время как ингибирование 11β-ГСД1 применяется для снижения уровня кортизола и лечения состояний, связанных с ним, ингибирование 11β-ГСД2 связано с серьезными побочными эффектами, такими как гипертензия.
Кортизол является важным и хорошо изученным противовоспалительным гормоном, который также действует как антагонист действия инсулина в печени, таким образом, что чувствительность к инсулину снижается, приводя к сниженному глюконеогенезу и повышенному уровню глюкозы в печени. Пациенты, уже имеющие развитую толерантность к глюкозе, имеют большую вероятность развития диабета 2-го типа при наличии ненормально высоких уровней кортизола.
Высокий уровень кортизола в тканях, где присутствует рецептор к минералокортикоиду, часто ведет к гипертензии. Ингибирование 11β-ГСД1 сдвигает соотношение кортизола и кортизона в определенных тканях в пользу кортизона.
Введение терапевтически эффективного количества ингибитора 11β-ГСД1 эффективно в лечении, контролировании и улучшении симптомов ИНЗСД, и регулярное введение терапевтически эффективного количества ингибитора 11β-ГСД1 отдаляет или предотвращает возникновение ИНЗСД, в частности, у людей.
Ожирение, метаболический синдром, дислипидемия:
Избыточные уровни кортизола связаны с ожирением, вероятно, благодаря повышенному печеночному глюконеогенезу. Абдоминальное ожирение тесно связано с нетолерантностью к глюкозе, гиперинсулинемией, гипертриглицеридемией и другими факторами метаболического синдрома, такими как высокое кровяное давление, повышенный уровень ЛОНП и сниженный уровень ЛВП. Montague и др., Diabetes, 2000, 49: 883-888. Таким образом, введение эффективного количества ингибитора 11β-ГСД1 применяется в лечении или контролировании ожирения. Длительное лечение ингибитором 11β-ГСД1 также применяется в оттягивании или предотвращении начала ожирения, особенно, если пациент применяет ингибитор 11β-ГСД1 в комбинации с контролируемой диетой и упражнениями.
Соединения данного изобретения могут найти применение в лечении и профилактике состояний, сопровождающих диабет 2-го типа и инсулинорезистентность, включая метаболический синдром или синдром Х, ожирение, реактивную гипогликемию и диабетическую дислипидемию, за счет снижения инсулинорезистентности и сохранения уровня глюкозы в плазме в нормальных концентрациях.
Атеросклероз:
Как описано выше, ингибирование активности 11β-ГСД1 и снижение количества кортизола полезны в лечении или контролировании гипертензии. Ввиду того что гипертензия и дислипидемия делают вклад в развитие атеросклероза, введение терапевтически эффективного количества ингибитора 11β-ГСД1 данного изобретения может также быть особенно полезно в лечении, контролировании, оттягивании начала или профилактике атеросклероза.
Другие варианты применения:
Следующие заболевания, нарушения и состояния можно лечить, контролировать, предотвращать или отдалять их возникновение лечением соединениями данного изобретения: (1) гипергликемия, (2) низкая толерантность к глюкозе, (3) инсулинорезистентность, (4) ожирение, (5) липидные нарушения, (6) дислипидемия, (7) гиперлипидемия, (8) гипертриглицеридемия, (9) гиперхолестеринемия, (10) низкий уровень ЛВП, (11) высокий уровень ЛНП, (12) атеросклероз и его последствия, (13) сосудистый рестеноз, (14) панкреатит, (15) абдоминальное ожирение, (16) нейродегенеративное заболевание, (17) ретинопатия, (18) нефропатия, (19) нейропатия, (20) метаболический синдром, (21) гипертензия и другие нарушения, в которых компонентом является инсулинорезистентность.
Вышеперечисленные заболевания и состояния можно лечить, используя соединения структурной формулы I, или можно вводить соединение для предотвращения или снижения риска развития заболеваний и состояний, описанных здесь. Ввиду того что конкурентное ингибирование 11β-ГСД2 может иметь опасные побочные эффекты или может фактически повысить количество кортизола в ткани-мишени, где снижение содержания кортизола желательно, желательны селективные ингибиторы 11β-ГСД1 с незначительным ингибированием 11β-ГСД2 или его отсутствием.
Ингибиторы 11β-ГСД1 структурной формулы I, в основном, имеют константу ингибирования IC50 менее чем примерно 500 нМ и предпочтительно менее чем примерно 100 нМ. В основном, значение IC50 для соединения 11β-ГСД2 в сравнении с 11β-ГСД1 равно, по меньшей мере, двум или более и предпочтительно примерно десяти или больше. Еще более предпочтительны соединения со значением IC50 для 11β-ГСД2 по сравнению с 11β-ГСД1 примерно 100 или больше. Например, соединения данного изобретения идеально демонстрируют константу ингибирования IC50 при действии против 11β-ГСД2 более чем примерно 1000 нМ и предпочтительно более чем 5000 нМ.
Любой подходящий путь введения может быть применен для обеспечения млекопитающего, особенно человека, эффективной дозой соединения данного изобретения. Например, пероральный, ректальный, местный, парентеральный, окулярный, ингаляционный, интраназальный и тому подобные. Дозированные формы включают таблетки, пастилки, дисперсии, суспензии, растворы, капсулы, кремы, мази, аэрозоли и тому подобные. Предпочтительно соединение структурной формулы I вводить перорально.
Эффективная дозировка активного ингредиента варьирует в зависимости от частного применяемого соединения, режима введения, излечиваемого состояния и выраженности состояния. Такие дозировки могут быть легко установлены квалифицированным специалистом в данной области.
При лечении или профилактике описываемых здесь заболеваний и состояний, для чего предназначены соединения структурной формулы I, получены удовлетворительные результаты при введении соединений данного изобретения в дневной дозе от примерно 0,1 до примерно 100 миллиграмм на килограмм (мнк) массы тела, предпочтительно вводимой в виде разовой дневной дозы или в виде отдельных доз примерно от двух до шести раз в день. Общая дневная доза, таким образом, находится в пределах от примерно 0,1 мг до примерно 1000 мг, предпочтительно от примерно 1 мг до примерно 50 мг. В случае стандартного взрослого человека массой 70 кг общая дневная доза будет варьировать от примерно 7 мг до примерно 350 мг. Данная дозировка может быть изменена для обеспечения оптимального терапевтического эффекта.
Другой аспект данного изобретения касается фармацевтической композиции, которая содержит соединение структурной формулы I или фармацевтически приемлемую соль или его сольват в комбинации с фармацевтически приемлемым носителем.
Данные композиции включают подходящие для перорального, ректального, местного, парентерального (включая подкожное, внутримышечное и внутривенное), окулярного (офтальмологического), трансдермального, ингаляционного (назальной или буккальной ингаляции) или интраназального введения, хотя наиболее подходящий путь введения в любом имеющемся случае будет зависеть от природы и выраженности излечиваемого состояния и активного ингредиента. Они могут быть удобно представлены в виде стандартной дозированной формы и приготовлены с помощью любых хорошо известных в фармации методов.
Соединение структурной формулы I может быть объединено с фармацевтически приемлемым носителем в соответствии с традиционными методиками приготовления фармацевтических препаратов. Носители могут иметь широкое разнообразие форм. Например, носители для пероральных жидких композиций включают, например, воду, гликоли, масла, спирты, ароматизирующие агенты, консерванты, красители и другие компоненты, используемые в производстве пероральных жидких суспензий, эликсиры и растворы. Носители, такие как крахмалы, сахара и микрокристаллическая целлюлоза, разбавители, гранулирующие агенты, смазывающие вещества, связующие вещества, дезинтегрирующие агенты и тому подобные используют в приготовлении твердых дозированных форм, например порошков, твердых и мягких капсул и таблеток. Твердые препараты для перорального применения предпочтительны по сравнению с жидкими.
Пероральные твердые дозированные формы могут также содержать связывающее вещество, такое как камедь трагаканта, акации, кукурузный крахмал или желатин; эксципиенты, такие как дикальций фосфат; дезинтегрирующий агент, такой как кукурузный крахмал, картофельный крахмал, альгиновая кислота; смазывающее вещество, такое как стеарат магния; и подсластитель, такой как сахароза, лактоза или сахарин. Капсулы могут также содержать жидкий носитель, такой как жирное масло.
Различные другие материалы могут присутствовать и действовать в качестве покрытий или модифицировать физическую форму стандартной дозы. Например, таблетки могут быть покрыты шеллаком, сахаром или ими обоими.
Таблетки могут быть покрыты с помощью стандартных водных или безводных методик. Обычное процентное содержание активного соединения в данных композициях может, конечно, варьировать от примерно 2 процентов до примерно 60 мас/мас процентов на основе. Таким образом, таблетки содержат соединение структурной формулы I или его соль или гидрат в количестве, варьирующем уже от примерно 0,1 мг до примерно 1,5 г, предпочтительно от примерно 1,0 мг до примерно 500 мг и более предпочтительно от примерно 10 мг до примерно 100 мг.
Жидкие формы для перорального применения, такие как сиропы или эликсиры, могут содержать, в дополнение к активному ингредиенту, сахарозу в качестве подсластителя, метил или пропилпарабены в качестве консервантов, краситель и ароматизатор, например вишневый или апельсиновый аромат.
Парентеральные формы обычно существуют в виде раствора или суспензии, обычно приготовлены на воде и необязательно содержат поверхностно-активное вещество, такое как гидроксипропилцеллюлоза. Дисперсии могут быть приготовлены на глицерине, жидких полиэтиленгликолях и их смесях в маслах. Обычно препараты в разбавленных формах также содержат консерванты.
Фармацевтические дозированные инъекционные формы, включая водные растворы и дисперсии и порошки для экстемпорального приготовления инъекционных растворов или дисперсий, также стерильны и должны быть жидкими в такой степени, чтобы их можно было легко набрать шприцом; они должны быть стабильны в условиях производства и хранения и традиционно содержать консерванты. Носитель, таким образом, включает растворитель или дисперсионную среду, содержащую, например, воду, этанол, полиол (например, глицерин, пропиленгликоль и жидкий полиэтиленгликоль), их подходящие смеси и растительные масла.
Анализы: измерение констант ингибирования:
С помощью Scintillation Proximity Assay (SPA) оценивали ферментную активность для тест-соединений. Вкратце, меченный тритием субстрат кортизона, кофактор НАДФН и титруемое соединение структурной формулы I инкубировали с 11β-ГСД1 при 37ºС для превращения в кортизол. После инкубации препаратом белка А покрывали SPA гранулы, предварительно смешанные с антикортизол-моноклональными антителами и неспецифическим ингибитором 11β-ГСД, например 18β-глицерритиновой кислотой, добавляли в каждую лунку. Смесь встряхивали при 15ºС и затем анализировали с помощью жидкостного сцинтилляционного счетчика, подходящего для 96 лунок. Процент ингибирования подсчитывали относительно неингибированной контрольной лунки и строили кривые IC50.
Данный анализ применяли сходным образом в отношении 11β-ГСД2, при этом меченный тритием кортизол и НАД использовали в качестве субстрата и кофактора соответственно. Для начала анализа добавляли 40 мкл субстрата (25 нМ 3H-кортизона + 1,25 мМ НАДФН в 50 мМ буфере HEPES, pH 7,4) в определенные лунки в 96-луночном планшете. Соединение растворяли в 10 мМ ДМСО с последующим 50-кратным разбавлением ДМСО. Разбавленный материал затем 4-кратно титровали, семь раз. 1 мкл каждого титрованного соединения затем добавляли в двойном количестве к субстрату. Для инициирования взаимодействия в каждую лунку добавляли 10 мкл 11β-ГСД1 микросомы из CHO-трансфектантов в соответствующей концентрации с получением приблизительно 10%-ного превращения исходного материала. Для окончательного подсчета процента ингибирования добавляли серии лунок, представлявшие минимум и максимум в анализе: один набор, содержащий субстрат без соединения или фермент (фон), и другой набор, содержащий субстрат и фермент без какого-либо соединения (максимальный сигнал). Планшеты кратковременно центрифугировали при низкой скорости в центрифуге для объединения реагентов, закрыли адгезивной полоской, аккуратно смешали и инкубировали при 37°С в течение 2 ч. После инкубации в каждую лунку добавляли 45 мкл, SPA гранулы, предварительно суспендированные с антикортизол-моноклональными антителами и соединением формулы I. Планшеты повторно закрывали и аккуратно встряхивали в течение более чем 1,5 ч при 15°C. Данные собирали с жидкостного сцинтилляционного счетчика для планшета, такого как Topcount. Для контроля над ингибированием связывания антикортизол-антител с кортизолом субстрат, скрепленный с 1,25 нМ 3H-кортизолом, добавляли в определенные стандартные лунки. 1 мкл 200 мкМ соединения добавляли в каждую из этих лунок вместе с 10 мкл буфера, вместо фермента. Любое вычисленное ингибирование наблюдалось благодаря тому, что соединение мешало связыванию кортизола с антителами на SPA гранулы.
Анализы: измерение ингибирования in vivo:
В общих терминах, тест-соединение перорально вводили млекопитающему и ожидали окончания описываемого интервала времени, обычно между 1 и 24 ч. Меченный тритием кортизон вводили внутривенно, с последующим, после нескольких минут, сбором крови. Стероиды экстрагировали из сепарированной сыворотки и анализировали с помощью ВЭЖХ. Относительные уровни 3H-кортизона и продукта его восстановления, 3H-кортизола, определяли для соединения и контрольных групп, принимавших носитель. Исходя из данных значений, рассчитывали абсолютное превращение и процент ингибирования. Более конкретно, соединения готовили для перорального введения растворением их в носителе (5% гидроксипропил-бета-циклодекстрин об./об. H2O, или эквивалент) в требуемой концентрации для получения дозировки обычно 10 мг на кг. После ночного голодания растворы вводили ICR мышам (получены от Charles River) пероральным зондом, 0,5 мл на дозу на животного, с тремя животными в тест-группе.
После окончания требуемого времени, обычно либо через 4 или 16 ч, вводили в хвостовую вену 0,2 мл 3 мкМ 3H-кортизона в dPBS. Животное отсаживали и через две минуты умерщвляли в CO2-камере. После смерти мышь удаляли и собирали кровь пункцией сердца. Кровь оставляли в пробирке для отделения сыворотки не менее чем на 30 мин при комнатной температуре для достижения адекватной коагуляции. После инкубационного периода кровь сепарировали с получением сыворотки центрифугированием при 3000Xg, 4ºС, в течение 10 мин.
Для анализа стероидов в сыворотке их сначала экстрагировали органическим растворителем. 0,2 мл сыворотки перенесли в чистую микроцентрифужную пробирку. К этому количеству добавляли 1,0 мл этилацетата, с последующим энергичным встряхиванием в течение 1 мин. Быстрое центрифугирование в микроцентрифуге приводит к образованию осадка водных сывороточных белков и просветленного органического супернатанта. 0,85 мл верхней органической фазы перенесли в чистую микроцентрифужную пробирку и высушили. Высушенный образец ресуспендировали в 0,250 мл ДМСО, содержащем высокую концентрацию кортизона и кортизола для анализа с помощью ВЭЖХ.
Образец объемом 0,200 мл вводили в хроматографическую колонку C-18 Metachem Inertsil, уравновешенную 30% метанолом. Медленный линейный градиент до 50% метанола разделил целевые стероиды; одновременный мониторинг с помощью УФ при 254 нм охлажденных стандартов в ресуспендированном растворе действовал как внутренний стандарт. Тритиевый сигнал фиксировали радиохроматографическим детектором и анализировали с помощью соответствующего программного обеспечения. Процент превращения 3H-кортизона в 3H-кортизол вычисляли как отношение AUC для кортизола к общей AUC для кортизона и кортизола.
Приготовление соединений данного изобретения:
Соединения данного изобретения структурной формулы I могут быть получены в соответствии с методиками следующих схем и примеров, используя соответствующие материалы, и далее представлены следующими частными примерами. Соединения, иллюстрированные примерами, не являются, однако, единственным классом соединений, который рассматривается как соединения данного изобретения. Примеры далее иллюстрируют в деталях получение соединений настоящего изобретения. Квалифицированные в этой области специалисты легко поймут, что известные вариации условий и процессов следующих препаративных методик могут быть использованы в получении данных соединений. Представленные соединения выделяют, в основном, в нейтральной форме, но фрагмент триазола может быть далее превращен в фармацевтически приемлемую соль растворением в органическом растворителе с последующим добавлением соответствующей кислоты и последующего выпаривания, осаждения или кристаллизацией. Все температуры выражены в градусах Цельсия, если другого не отмечено. Масс-спектры (МС) были измерены с помощью масс-спектроскопии с ионным электрораспылением (МСИЭР).
Фраза "условия стандартного пептидного синтеза" означает связывание карбоновой кислоты с амином с использованием агента, активирующего кислоту, такого как EDC, DCC и BOP в инертном растворителе, таком как дихлорметан, в присутствии катализатора, такого как HOBT. Использование защитных групп для амина и карбоновой кислоты предназначено для облегчения требуемого взаимодействия и минимизации нежелательных реакций, что хорошо известно. Условия, необходимые для удаления защитных групп, можно узнать из стандартных книг, таких как Greene, T., и Wuts, P. G. M., Protective Groups in Organic Synthesis, John Wiley & Sons, Inc., New York, NY, 1991. Cbz и BOC являются обычно используемыми защитными группами в органическом синтезе, и условия их удаления известны специалисту в данной области.
Аббревиатуры, используемые в описании получения соединений настоящего изобретения:
Схемы реакций 1-5 иллюстрируют способы, примененные в синтезе соединений настоящего изобретения структурной формулы I. Все заместители являются такими, как определено выше, если не указано иначе.
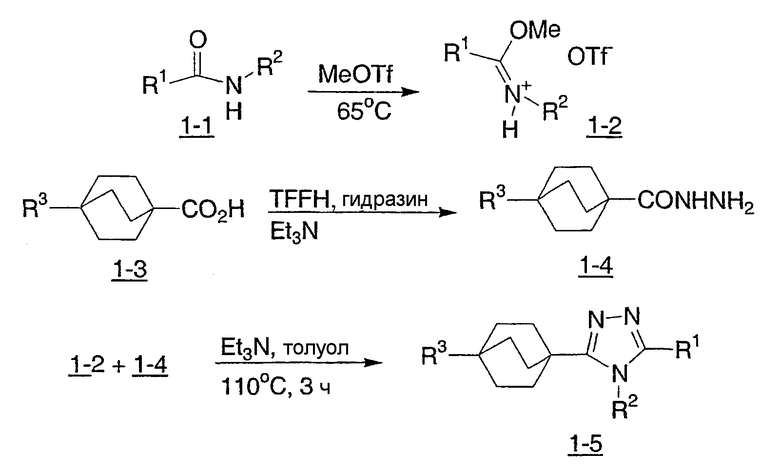
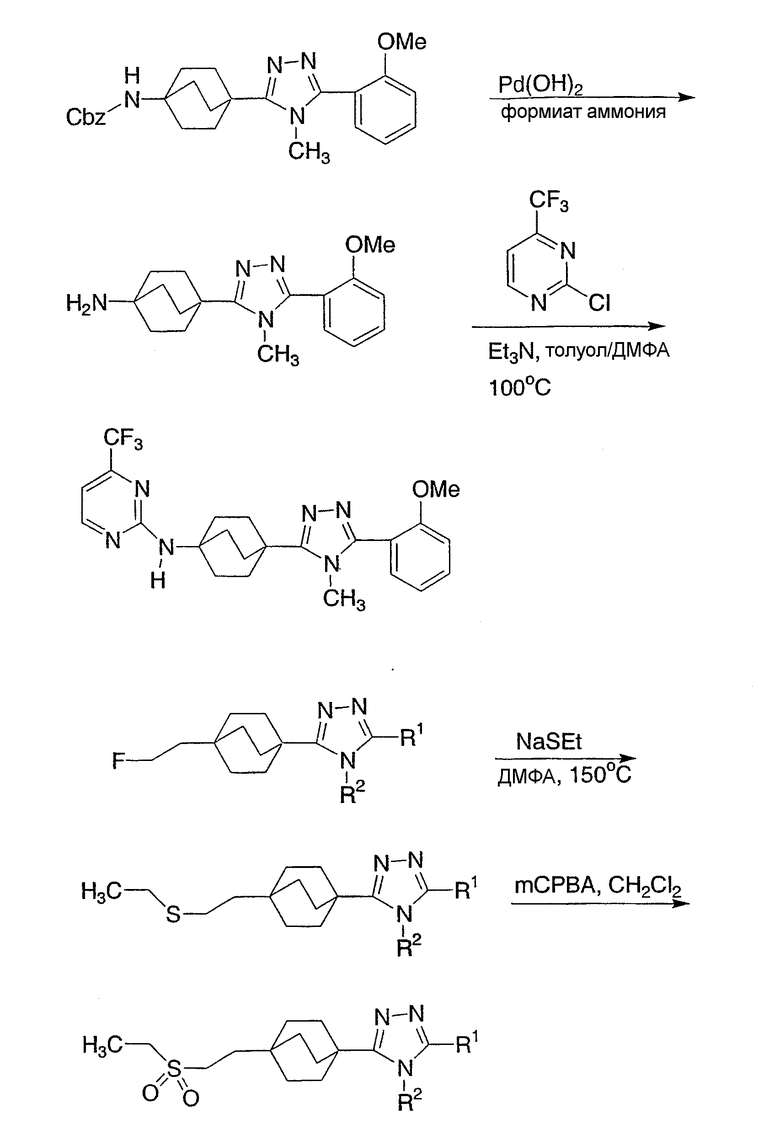
Схема реакции 1 иллюстрирует ключевую стадию в синтезе новых соединений структурной формулы I настоящего изобретения. Как показано на схеме реакции 1, вторичный амид (1-1) (N-Me или N-Et предпочтительны) может быть метилирован нагревом с чистым метилтрифлатом для получения иминоэфира (1-2). Альтернативно, могут быть использованы другие метилирующие реагенты, такие как метилиодид или метилсульфат без примесей или в ненуклеофильном органическом растворителе. Как показано на схеме 1, бицикло[2.2.2]октан-1-карбоновую кислоту (1-3) переводят в ацилгидразид (1-4), используя связывающий реактив TFFH и гидразин, в присутствии основания третичного амина, такого как триэтиламин. Альтернативно, для данной трансформации, наряду с гидразином, могут быть использованы другие связывающие реагенты, обычно используемые для получения амидов. Альтернативно, бицикло[2.2.2]октан-1-карбоновый эфир может быть нагрет с гидразином для получения ацилгидразидов (1-4). Ацилгидразид (1-4) и иминоэфир (1-2), получаемые таким образом, могут быть нагреты вместе в инертном высококипящем органическом растворителе, таком как толуол, в присутствии основания третичного амина, такого как триэтиламин, с получением бицикло[2.2.2]октилтриазолов (1-5) структурной формулы I.
Схема 1
Альтернативно, реакцию можно провести в обратном направлении, как это отображено на реакционной схеме 2. В этой методике вторичный амид (2-1) получают из бицикло[2.2.2]октан-1-карбоновой кислоты, используя реакцию стандартного пептидного синтеза. Это соединение метилируют с образованием иминоэфира (2-2) и подвергают взаимодействию с ацилгидразидом, как отображено на схеме 1, с образованием бицикло[2.2.2]октилтриазолов (2-3) структурной формулы I.
Схема 2
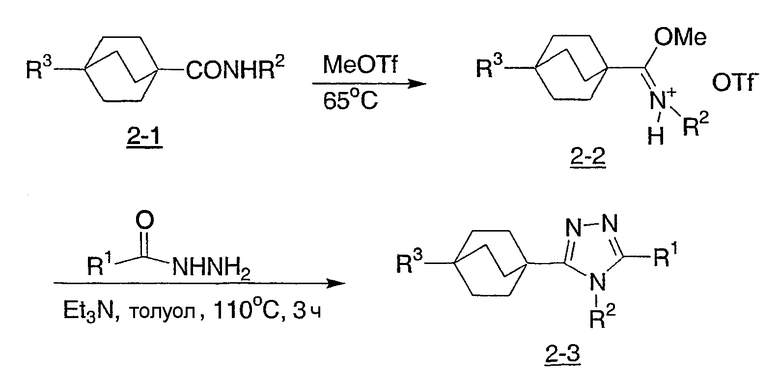
Реакционная схема 3 демонстрирует альтернативный подход к соединениям настоящего изобретения структурной формулы I, в котором ключевым этапом является реакция синтеза Сузуки, катализируемая палладием, между бицикло[2.2.2]октилбромтриазолом (3-1) и арилбороновой кислотой с получением триазолов (3-2) структурной формулы I. В предпочтительных условиях используется тетракис(трифенилфосфин)палладий(0) в качестве катализатора в растворителе ДМФА с карбонатом цезия, но могут быть применены другие катализаторы и условия, что известно специалистам в этой области.
Схема 3
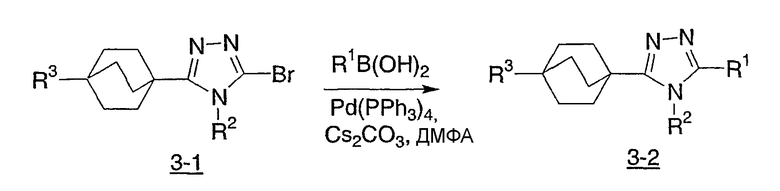
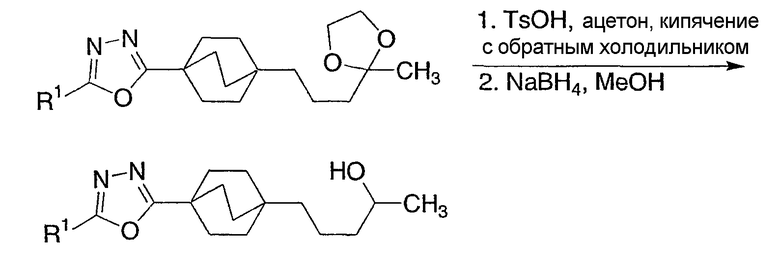
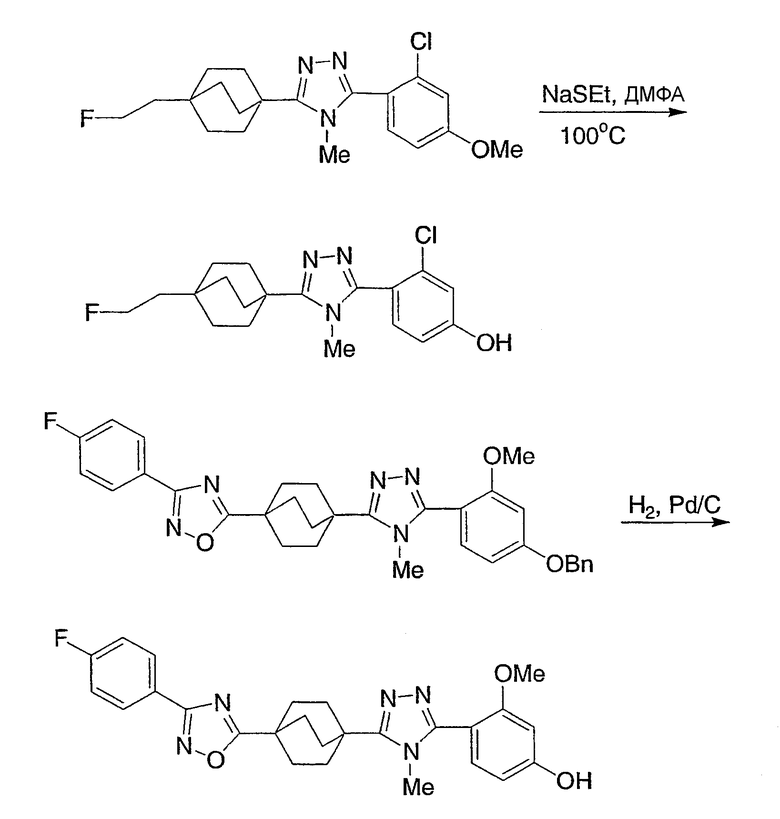
Реакционная схема 4 демонстрирует еще другой синтетический подход к получению соединений структурной формулы I. Используя эту методику, 4-(бицикло[2.2.2]октил)оксадиазолы (4-1) при дегидратации конденсируют с метиламином либо в чистом виде в расплаве с метиламмонийтрифторацетатом, либо в буферированном растворе MeOH. Данные реакции лучше проводить при повышенных температурах в реакторе под высоким давлением для предотвращения потери метиламина.
Схема 4
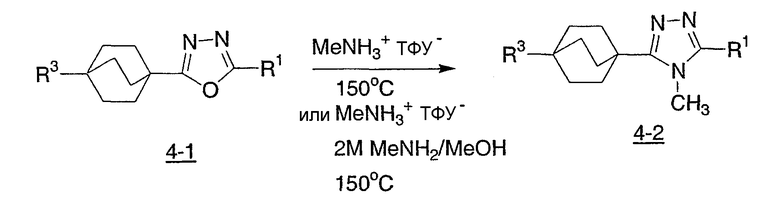
Реакционная схема 5 демонстрирует еще другой синтетический подход к получению соединений структурной формулы I. Используя эту методику, бицикло[2.2.2]октилкарбоксамиды (5-1) превращают в иминохлориды (5-2), с применением реагента, такого как оксалилхлорид, тионилхлорид, фосфороксихлорид или фосфорпентахлорид, необязательно в присутствии ДМФА. Иминохлорид (5-2) конденсируют с арилтетразолом в высококипящем инертном органическом растворителе, таком как толуол, с получением триазола (5-3).
Схема 5
Получение [2.2.2]бициклооктил интермедиатов:
Методики, применяемые в получении [2.2.2]бициклооктил интермедиатов для использования в получении соединений настоящего изобретения, представлены ниже.
Схемы получения промежуточных соединений 1-4 описывают получение оксадиазолов, которые являются важными промежуточными соединениями в синтезе соединений структурной формулы I. Они могут быть превращены в соединения структурной формулы I, используя, например, реакции, описанные в реакционной схеме 4.
Схема получения промежуточных соединений 1 демонстрирует предпочтительный способ получения оксадиазолов посредством дегидратации диацилгидразидов, используя дегидратирующий реагент, такой как тионилхлорид. Альтернативно, могут быть применены другие дегидратирующие реагенты, такие как фосфороксихлорид, фосфорпентахлорид или оксалилхлорид. Диацилгидразиды могут быть получены предпочтительно из гидразида и активированной кислоты, такой как хлорангидрид кислоты, в присутствии основания третичного амина. Альтернативно, могут быть применены реакции стандартного пептидного синтеза для получения диацилгидразида из гидразида и карбоновой кислоты.
Схема получения промежуточных соединений 2 демонстрирует подходящий реагент для дегидратации диацилгидразидов до оксадиазолов, а именно 2-хлор-1,3-диметил-4,5-дигидро-lH-имидазол-3-иний хлорид. Этот реагент в неполярном растворителе (предпочтителен метиленхорид) вместе с основанием третичного амина (предпочтителен триэтиламин) дает целевые интермедиаты оксадиазола наиболее эффективным образом.
Схема получения промежуточных соединений 3 демонстрирует предпочтительный реагент для однореакторного превращения (из гидразида и карбоновой кислоты) и дегидратацию диацилгидразидов до оксадиазолов: 2-хлор-1,3-диметил-4,5-дигидро-lH-имидазол-3-иний хлорид. Этот реагент в неполярном растворителе (предпочтителен метиленхлорид) вместе с основанием третичного амина (предпочтителен триэтиламин) дает требуемые интермедиаты оксадиазола наиболее эффективным образом.
Схема получения промежуточных соединений 4 демонстрирует эффективный способ получения оксадиазолов из вторичных амидов и гидразидов. Вторичный амид (предпочтительны N-Me или N-Et) может быть метилирован нагревом с чистым метилтрифлатом для получения иминоэфира. Альтернативно, могут быть использованы другие метилирующие реагенты, такие как метилиодид или метилсульфат без примесей или в ненуклеофильном органическом растворителе. Нагрев иминоэфира, образованный таким образом в высококипящем инертном органическом растворителе в присутствии гидразида, позволяет получить оксадиазолы, как показано на схеме.
Схема получения промежуточных соединений 1
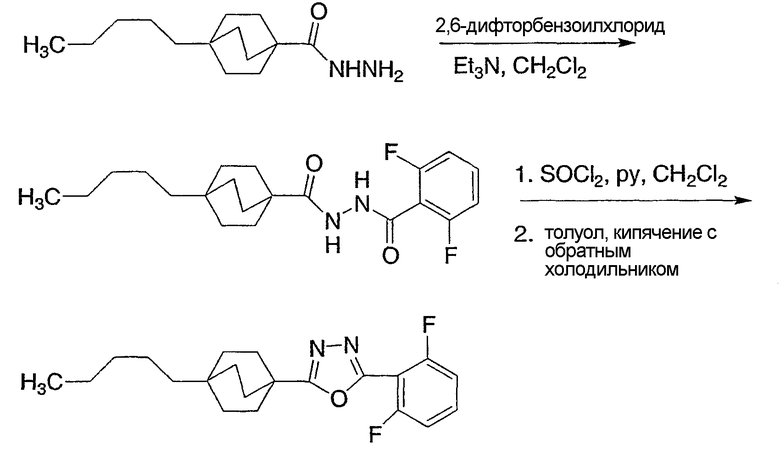
Схема получения промежуточных соединений 2
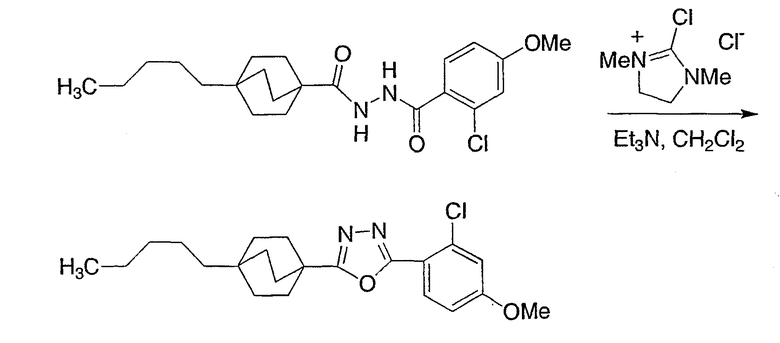
Схема получения промежуточных соединений 3
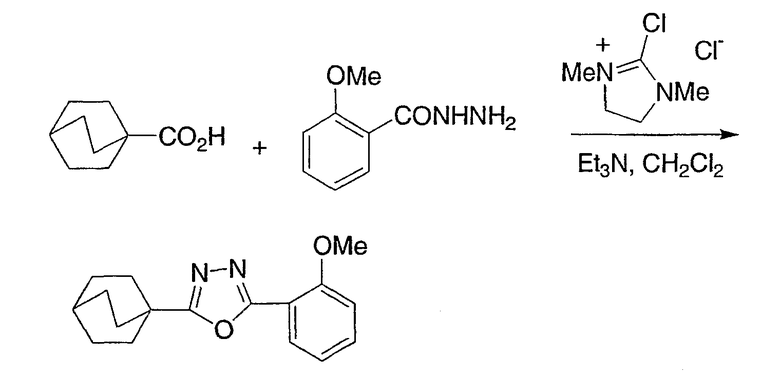
Схема получения промежуточных соединений 4
Промежуточная схема 5 демонстрирует предпочтительный метод синтеза бицикло[2.2.2]октан-1-карбоновой кислоты.
Схема получения промежуточных соединений 5
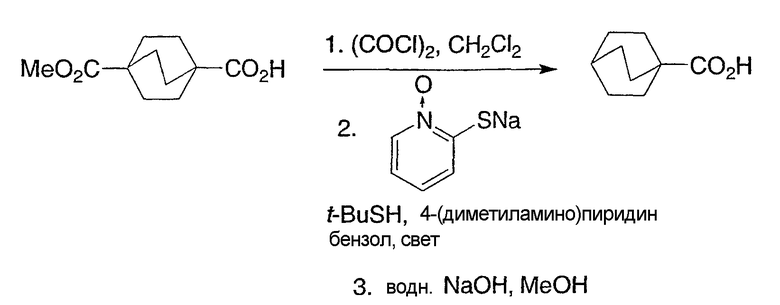
Схемы получения промежуточных соединений 6 и 7 демонстрируют предпочтительные способы получения бицикло[2.2.2]октан-1-карбоновых кислот с гетероарильной группой в положении R3, как представлено структурной формулой I. Оксадиазолы в положении R3 могут быть получены конденсацией бицикло[2.2.2]октан-1-карбоновой кислоты с амидоксимом, как показано в промежуточной схеме 6, подходящим реагентом для синтеза CDI. Альтернативно, могут быть использованы другие подходящие для дегидратации или реакций пептидного синтеза реагенты. Промежуточная схема 7 иллюстрирует предпочтительный способ синтеза интермедиатов соединений структурной формулы I, несущих тиазольную группу в R3-позиции.
Схема получения промежуточных соединений 6
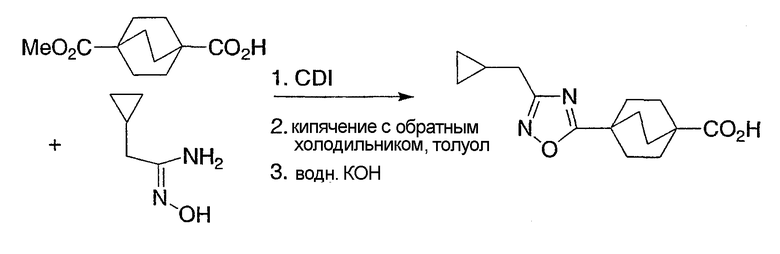
Схема получения промежуточных соединений 7
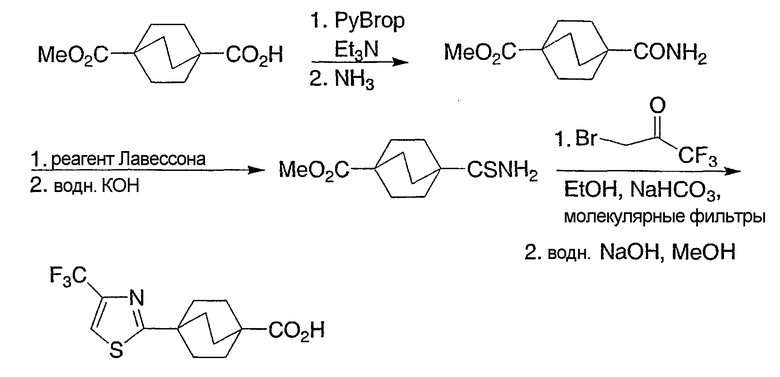
Схемы получения промежуточных соединений 8-14 показывают предпочтительные способы получения интермедиатов бицикло[2.2.2]октан-1-карбоновых кислот в синтезе соединений структурной формулы I с различными алкильными или алкенильными или замещенными алкильными группами в положении R3. Ключевой реакцией является реакция Виттига, осуществляемая с бицикло[2.2.2]октан-1-карбоксальдегидом, как показано на промежуточной схеме 8. Двойная связь в продукте этой реакции может быть гидрирована с получением алкильной группы различной длины и характера (которая станет R3-заместителем в структурной формуле I), в зависимости от реагента Виттига, как показано на промежуточной схеме 9. Альтернативно, двойная связь может быть использована для введения других функциональных групп, таких как гидрокси или фторогрупп, как показано на промежуточной схеме 10. Сам по себе альдегид может быть использован для получения дифторметильной группы в положении R3, как показано на промежуточной схеме 11. Алкен, продукт реакции Виттига, может претерпеть множество других трансформаций, например циклопропанирование, как показано на промежуточной схеме 12. Альтернативно, реагент Виттига может содержать отдаленную функциональную группу, например кеталь, как показано на промежуточной схеме 13. Эта функциональная группа может претерпеть характеристические трансформации функциональной группы после последовательного проведения реакции и Виттига и восстановления, например гидролиз кеталя до кетона, как показано на промежуточной схеме 13, или восстановление кеталя до спирта, как показано на промежуточной схеме 14. Таким образом, могут быть получены соединения структурной формулы I с многообразием различных R3-заместителей. Частные примеры представлены для отображения основных принципов и не стремятся ограничить объем R3-заместителей.
Схема получения промежуточных соединений 8
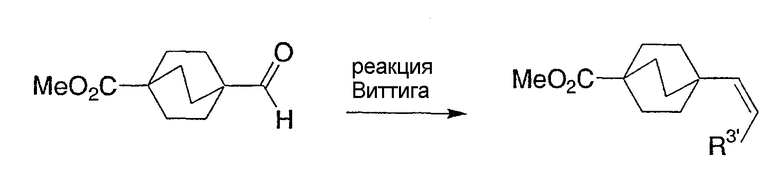
Схема получения промежуточных соединений 9
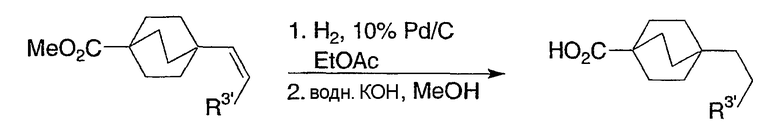
Схема получения промежуточных соединений 10
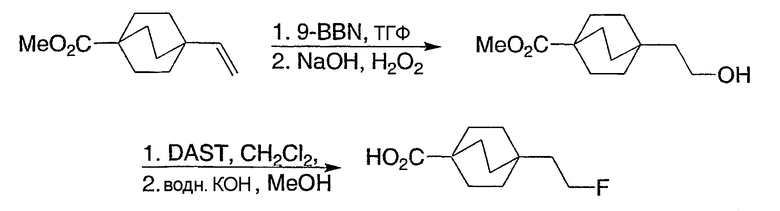
Схема получения промежуточных соединений 11
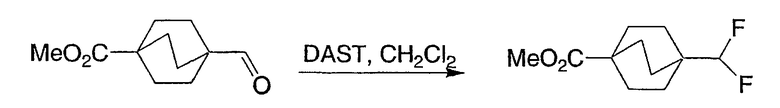
Схема получения промежуточных соединений 12
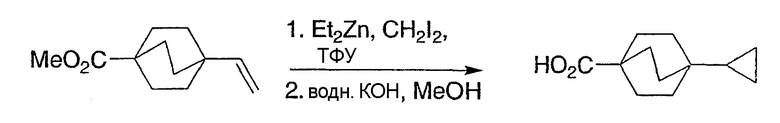
Схема получения промежуточных соединений 13
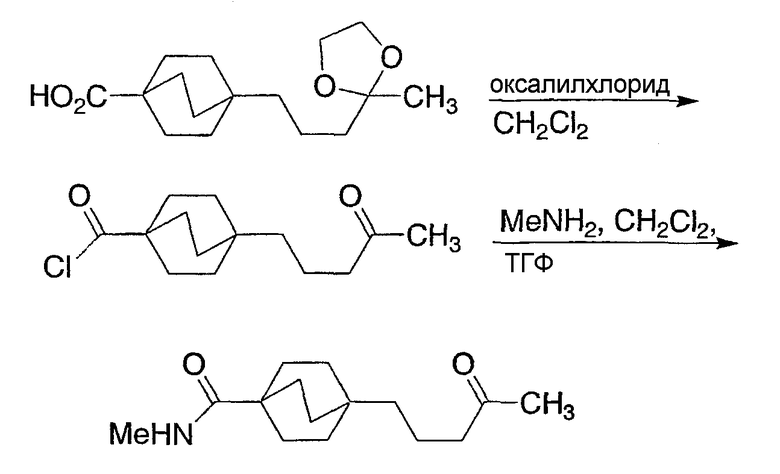
Схема получения промежуточных соединений 14
Основные химические трансформации функциональной группы, используемые в получении соединений настоящего изобретения, представлены ниже в получении частных соединений настоящего изобретения.
Эти трансформации функциональной группы в своем многообразии, в основном, хорошо понимаемы специалистами в данной области.
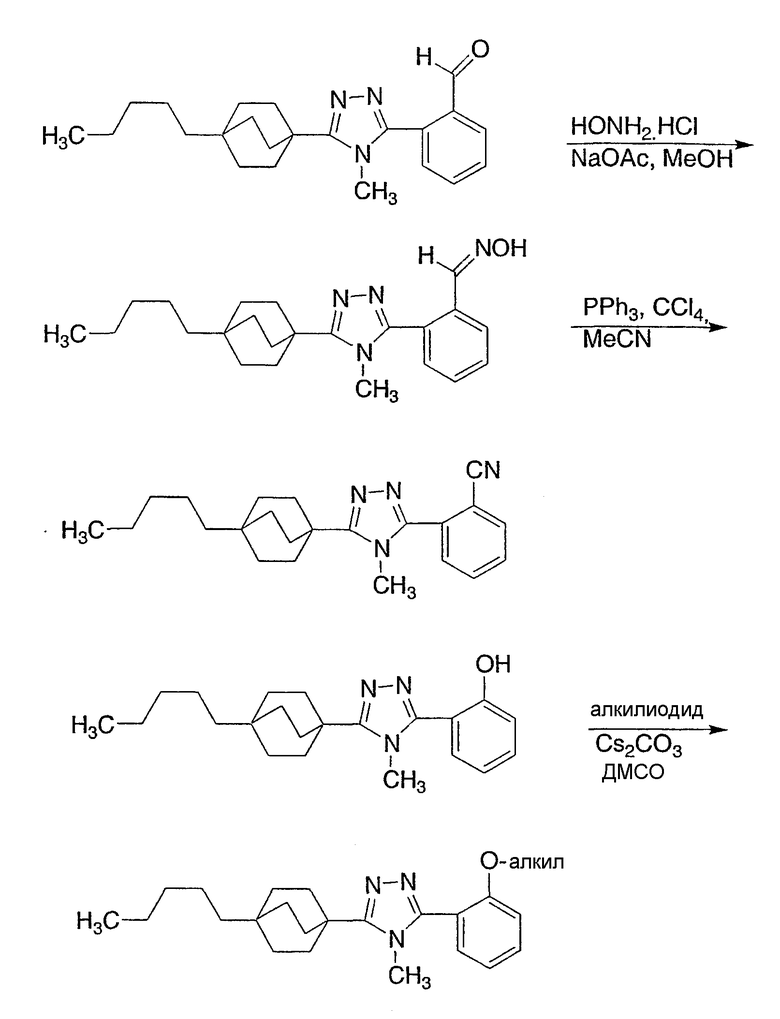
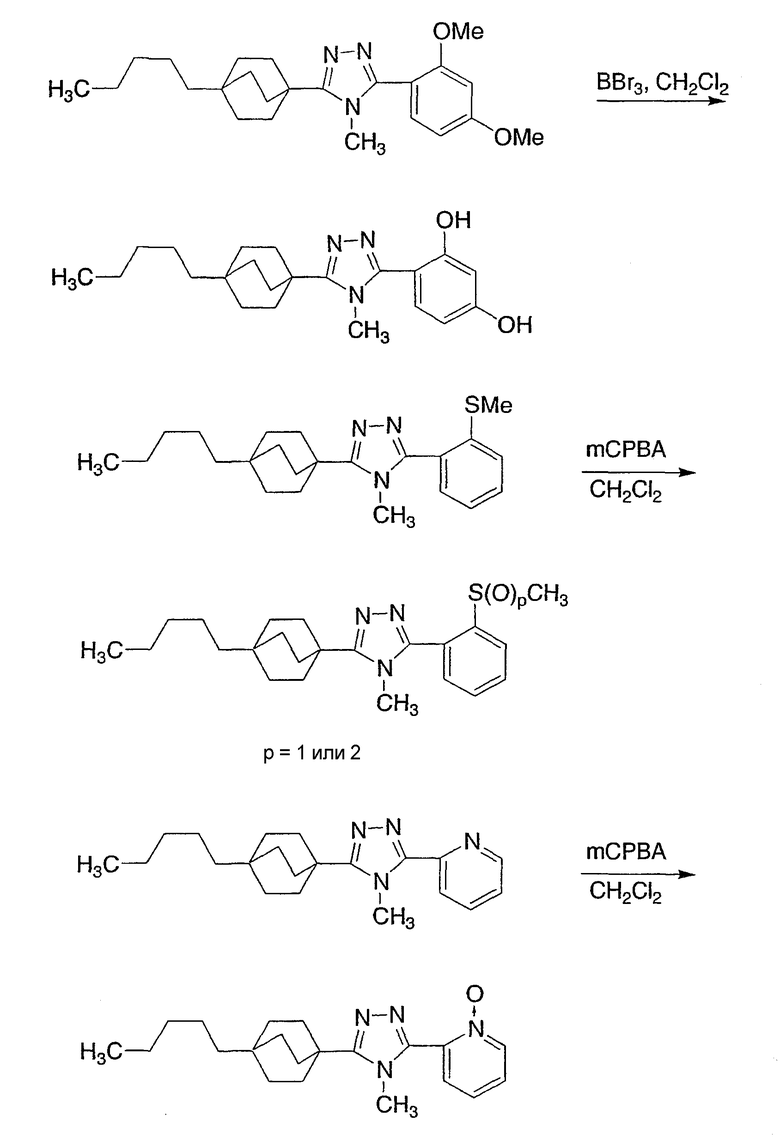
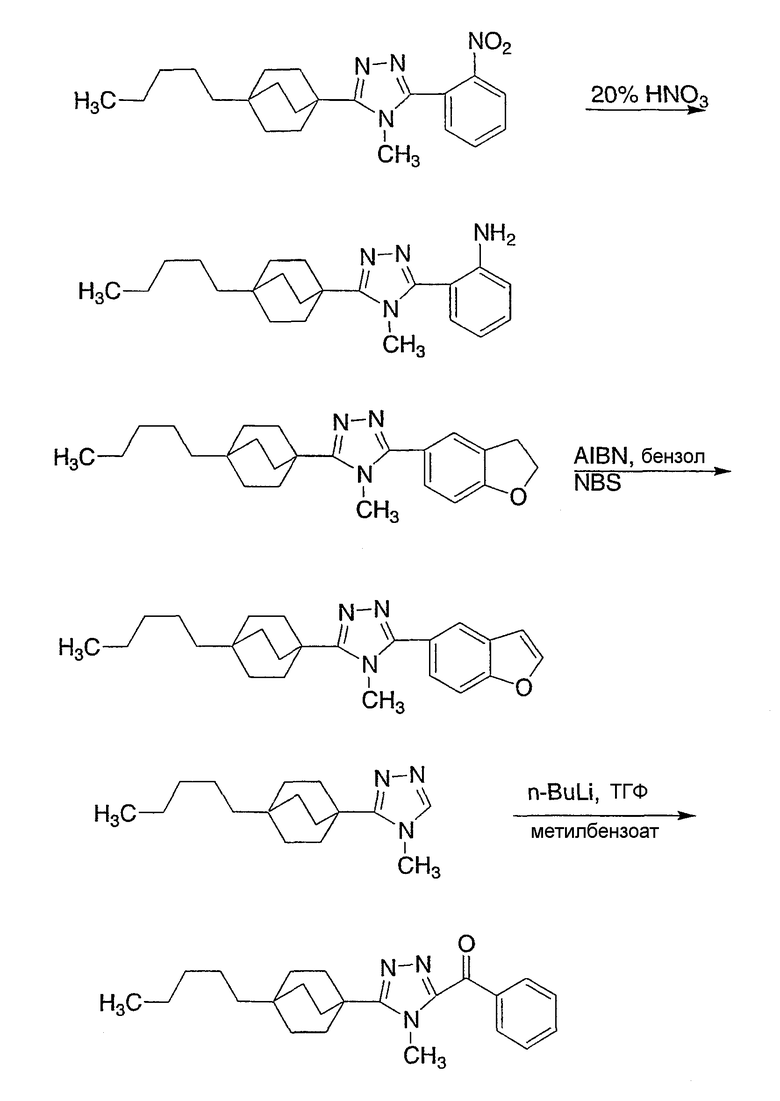
Следующие примеры обеспечивают иллюстрацию изобретения и не призваны каким-либо образом ограничить объем данного изобретения.
Пример 1
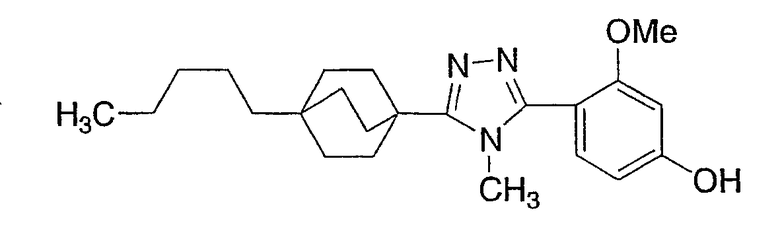
3-Метокси-4-[4-метил-5-(4-пентилбицикло[2.2.2]окт-1-ил)-4H-1,2,4-триазол-3-ил]фенол (1-F)
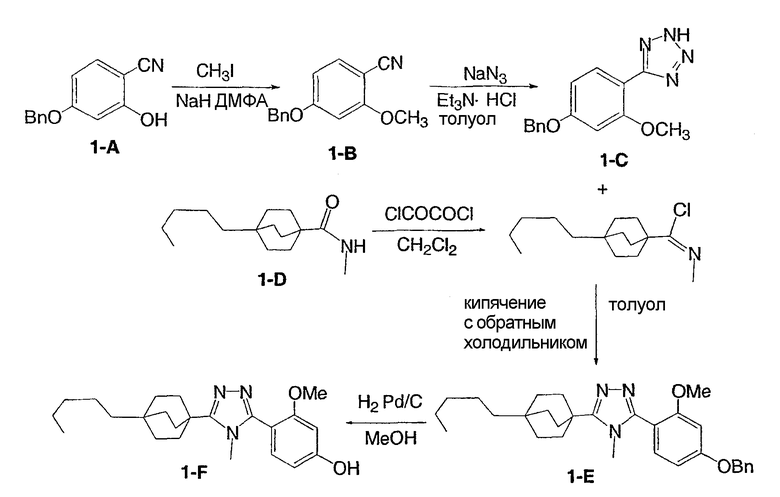
Стадия A:
К перемешиваемому магнитной мешалкой раствору 4-бензилокси-2-гидроксибензонитрила (1-A, WO 00/69841) (7,95 г, 35,3 ммоль) и иодометана (5,43 мл, 87,2 ммоль) в ДМФА (90 мл), охлажденному до -5°С, добавляли сразу все количество гидрида натрия (60% дисперсия, 2,17 г, 54,2 ммоль). Смесь перемешивали в течение 30 мин, нагревали до комнатной температуры и дополнительно перемешивали в течение 2 ч.
Большую часть ДМФА отгоняли в вакууме и остаток распределяли между водой и этилацетатом. Водную фазу экстрагировали три раза этилацетатом. Объединенные органические фазы промывали водой и насыщенным раствором соли и высушивали (MgSO4). Остаток после удаления растворителя в вакууме растирали с гексаном и хроматографировали на силикагеле с гексаном-CH2Cl2 (2:3) с получением 4-бензилокси-2-метоксибензонитрила (1-B). МС: m/z 240 (M+1); 1H ЯМР (500 МГц, CDCl3): δ 7,47 (д, 1H, J=8,4 Гц), 7,36-7,45 (м, 5H), 6,58 (дд, 1Н, J=2,3, 8,4 Гц), 6,57 (д, 1H, J=2,3 Гц), 5,10 (с, 2H), 3,88 (с, 3H) м.д.
Стадия В:
Энергично перемешиваемую суспензию 4-бензилокси-2-метоксибензонитрила (1-B) (1,20 г, 5,0 ммоль), азида натрия (732 мг, 11,3 ммоль) и триэтиламингидрохлорида (1,54 г, 11,3 ммоль) в толуоле (6 мл) нагревали при 110°С в течение 48 ч. Коричневую суспензию охлаждали, добавляли воду (15 мл) и смесь перемешивали в течение 30 мин. Органический слой отделяли и экстрагировали водой (5 мл). pH объединенных водных экстрактов доводили до примерно значения 1 с помощью концентрированной HCl. Первоначально осаждающаяся смола затвердевала при перемешивании в течение 30 мин.
Твердую часть промывали водой и высушивали с получением 5-[4-(бензилокси)-2-метоксифенил]-2H-тетразола (1-C). 1H ЯМР (500 МГц, CDCl3): δ 12,9 (ушир.с, 1H), 7,37 (д, 1Н, J=8,7 Гц), 7,34-7,48 (м, 5H), 6,78 (дд, 1H, J= 2,3, 8,7 Гц), 6,70 (д, 1H, J=2,3 Гц), 5,15 (с, 2H), 4,05 (с, 3H) м.д.
Стадия С:
Оксалилхлорид (3,49 мл, 40 ммоль) добавляли по каплям к раствору N-метил-4-пентилбицикло[2.2.2]октан-1-карбоксамида (1-D) (952 мг, 4,0 ммоль) в сухом CH2Cl2 при комнатной температуре. После снижения интенсивного выделения газа раствор перемешивали при комнатной температуре в течение 2 ч. CH2Cl2 осторожно удаляли в вакууме при комнатной температуре и затем при 50°С. Чистый остаток в виде сиропа растворяли в толуоле (8 мл) и добавляли 5-[4-(бензилокси)-2-метоксифенил]-2H-тетразол (1-C) (1,13 г, 4,0 ммоль). Смесь нагревали при 120°С в течение 9 ч. Смесь охлаждали и осадившееся твердое вещество фильтровали, промывали толуолом и высушивали с получением соли триазолгидрохлорида. Соль распределяли между CH2Cl2 и 10% водным K2CO3. Водную фазу экстрагировали дважды CH2Cl2. Объединенные экстракты CH2Cl2 высушивали (MgSO4) и упаривали в вакууме. Остаток хроматографировали на силикагеле с 5% MeOH/CH2Cl2 с получением 3-[4-(бензилокси)-2-метоксифенил]-4-метил-5-(4-пентилбицикло[2.2.2]окт-1-ил)-4H-1,2,4-триазола (1-E). МС: m/z 474 (М+1); 1H ЯМР (500 МГц, CDCl3): δ 7,33-7,47 (м, 6H), 6,65 (дд, 1H, J=2,3, 8,5 Гц), 6,60 (д, 1H, J=2,3 Гц), 5,10 (с, 2H), 3,75 (с, 3H), 3,48 (с, 3H), 2,08 (м, 6H), 1,51 (м, 6H), 1,00-1,35 (м, 8H), 0,89 (т, 3H, J=7,2) м.д.
Стадия D:
Раствор 3-[4-(бензилокси)-2-метоксифенил]-4-метил-5-(4-пентилбицикло[2.2.2]окт-1-ил)-4H-1,2,4-триазола (1-E) (272 мг, 0,572 ммоль) в MeOH (8 мл) гидрогенизировали в течение 19 ч с 10% катализатором Pd/C (27 мг) при комнатной температуре и атмосферном давлении. Катализатор фильтровали и промывали MeOH. MeOH удаляли в вакууме с получением 3-метокси-4-[4-метил-5-(4-пентилбицикло[2.2.2]окт-1-ил)-4H-1,2,4-триазол-3-ил]фенола (1-F). МС: m/z 384 (М+1); 1Н ЯМР (500 МГц, ДМСО-d6): δ 9,94 (с, 1H), 7,09 (д, 1H, J=8,3), 6,53 (д, 1H, J=1,6 Гц), 6,46 (дд, 1H, J=2,2, 8,2 Гц), 3,72 (с, 3H), 3,40 (с, 3H), 1,95 (м, 6H), 1,44 (м, 6H), 1,07-1,33 (м, 8H), 0,86 (т, 3H, J=7,2).
Пример 2
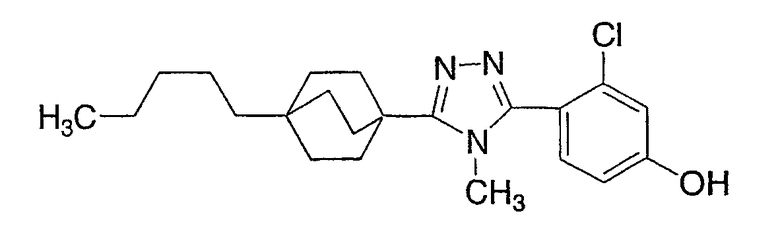
3-Хлор-4-[4-метил-5-(4-пентилбицикло[2.2.2]окт-1-ил)-4H-1,2,4-триазол-3-ил]фенол (2-G)
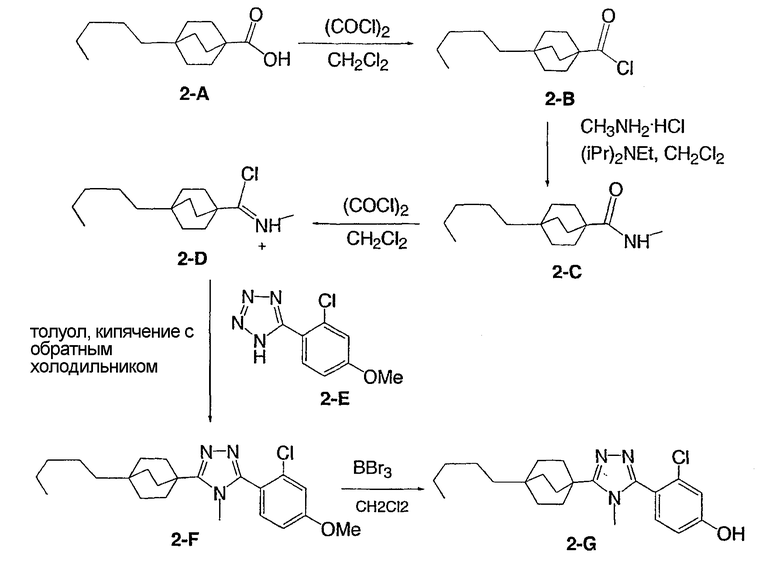
Стадия А:
Оксалилхлорид (505 мкл, 5,79 ммоль) добавляли по каплям к смеси 4-пентилбицикло[2.2.2]октан-1-карбоновой кислоты (2-A) в метиленхлориде (10 мл). Раствор перемешивали при комнатной температуре в течение 3 ч и затем концентрировали в вакууме с получением 4-пентилбицикло[2.2.2]октан-1-карбонилхлорида (2-B). 1H ЯМР (500 МГц, CDCl3): δ 0,90 (т, 3H); 1,21 (м, 8H); 1,45 (м, 6H); 1,88 (м, 6H) м.д.
Стадия В:
N,N-диизопропилэтиламин (1,44 мл, 11,1 ммоль) добавляли к смеси 4-пентилбицикло[2.2.2]октан-1-карбоновой кислоты (2-A) (1,09 г, 4,45 ммоль) и метиламингидрохлорида (1,5 г, 22,3 ммоль) в метиленхлориде (10 мл) и смесь перемешивали при комнатной температуре в течение 18 ч. После разбавления метиленхлоридом смесь промывали водой, насыщенным раствором соли, высушивали (MgSO4) и концентрировали в вакууме с получением N-метил-4-пентилбицикло[2.2.2]октан-1-карбоксамида (2-C). 1H ЯМР (500 МГц, CDCl3): δ 0,91 (т, 3H); 1,22 (м, 8H); 1,43 (м, 6H); 1,77 (м, 6H); 2,82 (д, 3H) м.д.
Стадия С:
Оксалилхлорид (846 мкл, 9,7 ммоль) добавляли по каплям к раствору N-метил-4-пентилбицикло[2.2.2]октан-1-карбоксамида (2-C) (230 мг, 0,97 ммоль) в метиленхлориде (2,0 мл) и смесь перемешивали при комнатной температуре в течение 4 ч. Растворитель и избыток реагента удаляли в вакууме с получением N-метил-4-пентилбицикло[2.2.2]октан-1-карбоксимидоилхлорида (2-D). Добавляли толуол (1,5 мл) с последующим добавлением 5-(2-хлор-4-метоксифенил)-lH-тетразола (2-E) (204 мг, 0,97 ммоль) и смесь нагревали с обратным холодильником в течение 18 ч. Реакционную смесь охлаждали до комнатной температуры и осадок фильтровали, промывали холодным толуолом, гексаном, растворяли в метиленхлориде, высушивали (MgSO4) и концентрировали в вакууме с получением 3-(2-хлор-4-метоксифенил)-4-метил-5-(4-пентилбицикло[2.2.2]окт-1-ил)-4H-1,2,4-триазола (2-F). Масс-спектр: 402 (М+1); 1H ЯМР (500 МГц, CDCl3): δ 0,94 (т, 3H), 1,27 (м, 8H), 1,56 (м, 6H), 2,13 (м, 6H), 3,56 (с, 3H), 3,89 (с, 3H), 6,95 (дд, 1H), 7,07 (д, 1H), 7,43 (д, 1H).
Стадия D:
Трибромид бора (135 мкл, 1,43 ммоль) добавляли по каплям к раствору 3-(2-хлор-4-метоксифенил)-4-метил-5-(4-пентилбицикло[2.2.2]окт-1-ил)-4H-1,2,4-триазола (2-F) (287 мг, 0,714 ммоль) в метиленхлориде (5 мл) при 0°C. Смесь перемешивали при комнатной температуре в течение 2,5 ч. Раствор промывали водой, 10% NaHCO3, высушивали (MgSO4) и концентрировали в вакууме и остаток очищали колоночной хроматографией (силикагель, 5% MeOH/метиленхлорид) с получением 3-хлор-4-[4-метил-5-(4-пентилбицикло[2.2.2]окт-1-ил)-4H-1,2,4-триазол-3-ил]фенола (2-G). Масс-спектр: 388 (М+1); 1H ЯМР (500 МГц, CDCl3): δ 0,93 (т, 3H), 1,26 (м, 8H), 1,56 (м, 6H), 2,13 (м, 6H), 3,58 (с, 3H), 6,69 (дд, 1H), 6,92 (д, 1H), 7,09 (д, 1H) м.д.
Пример 3
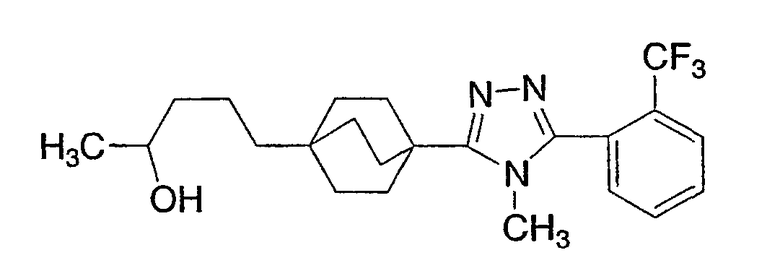
5-(4-{1-Метил-5-[2-(трифторметил)фенил]-1-H-1,2,4-триазол-3-ил}бицикло[2.2.2]окт-1-ил)пентан-2-ол (3-J)
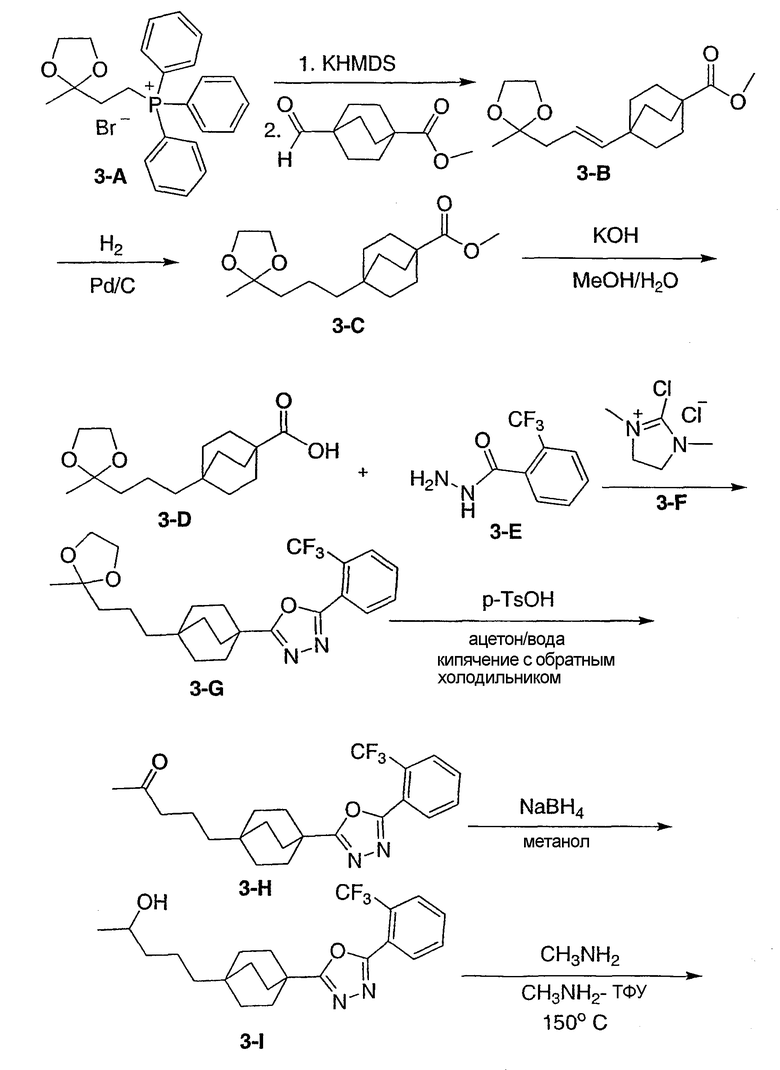
Стадия А:
[2-(2-Метил-1,3-диоксолан-2-ил)этил](трифенил)фосфонийбромид (3-A, Synthesis: 532 (1986)) (5,99 г, 12,7 ммоль) перемешивали в сухом ТГФ (200 мл). Добавляли бис(триметилсилил)амид калия (20,4 мл, 2M раствор в толуоле, 10,2 ммоль). Реакционную смесь оставляли для перемешивания в течение 30 мин. Затем реакционную смесь охлаждали до -78°C. С помощью канюли добавляли метил-4-формилбицикло[2.2.2]октан-1-карбоксилат при -78°C. Реакционную смесь оставляли для нагрева до комнатной температуры на ночь. Объем уменьшали выпариванием ТГФ в вакууме. Добавляли 100 мл воды. На смесь наслаивали 100 мл диэтилэфира. Эфир экстрагировали и высушивали (MgSO4). Продукт (метил-4-[(lE)-3-(2-метил-1,3-диоксолан-2-ил)проп-1-енил]бицикло[2.2.2]октан-1-карбоксилат (3-B)) очищали флэш-хроматографией на силикагеле со смесью 10/90 этилацетат-гексан.
Стадия В:
(Метил-4-[(lE)-3-(2-метил-1,3-диоксолан-2-ил)проп-1-енил]бицикло[2.2.2]октан-1-карбоксилат (3-B) (1,1 г) перемешивали в этаноле (75 мл). Добавляли на кончике шпателя 10% Pd на угле (150 мг). Вводили водород из баллона и смесь перемешивали в атмосфере водорода в течение 3 ч. Палладий на угле фильтровали и этанол удаляли в вакууме с получением 4-[3- (2-метил-1,3-диоксолан-2-ил)пропил]бицикло[2.2.2]октан-1-карбоксилата (3-C).
Стадия С:
Метил-4-[3-(2-метил-1,3-диоксолан-2-ил)пропил]бицикло[2.2.2]октан-1-карбоксилат (3-C) (1,0 г, 3,38 ммоль) перемешивали в растворе 90% метанола и 10% воды (50 мл). Добавляли избыток гидроксида калия (2,0 г). Смесь кипятили с обратным холодильником в течение ночи. В охлажденную смесь добавляли 1н. хлороводородную кислоту (100 мл) и затем промывали дважды этилацетатом (100 мл). Объединенные органические слои высушивали (MgSO4). Этилацетат удаляли в вакууме с получением чистой 4-[3-(2-метил-1,3-диоксолан-2-ил)пропил]бицикло[2.2.2]октан-1-карбоновой кислоты (3-D).
Стадия D:
4-[3-(2-Метил-1,3-диоксолан-2-ил)пропил]бицикло[2.2.2]октан-1-карбоновой кислоты (3-D) (0,200 г, 0,708 ммоль) объединяли с гидразидом 2-(трифторметил)бензойной кислоты (3-E) (0,173 г, 0,847 ммоль) и дважды азеотропно перегоняли из толуола. Затем смесь перемешивали в сухом метиленхлориде (10 мл). Добавляли 2-хлор-1,3-диметилимидазолинийхлорид (3-F) (0,718 г, 4,25 ммоль) с последующим добавлением 1,184 мл триэтиламина. Реакционную смесь оставляли для перемешивания в течение 2 ч. Реакционную смесь разбавляли метиленхлоридом и промывали водой. Полученный оксадиазол, 2-{4-[3-(2-метил-1,3-диоксолан-2-ил)пропил]бицикло[2.2.2]окт-1-ил}-5-[2-(трифторметил)фенил]-1,3,4-оксадиазол (3-G) очищали флэш-хроматографией на силикагеле со смесью 50/50 этилацетат-гексан.
Стадия Е:
2-{4-[3-(2-Метил-1,3-диоксолан-2-ил)пропил]бицикло[2.2.2]окт-1-ил}-5-[2-(трифторметил)фенил]-1,3,4-оксадиазол (3-G) (0,158 г) перемешивали в смеси 90% ацетон/10% вода (20 мл). Добавляли к раствору п-толуолсульфоновую кислоту (10 мг). Реакционную смесь кипятили с обратным холодильником в течение 1 ч. Объем уменьшали выпариванием в вакууме ацетона. На смесь затем наслаивали этилацетат (25 мл) и насыщенный раствор бикарбоната натрия (25 мл). Слой этилацетата экстрагировали и высушивали (MgSO4). Растворитель удаляли в вакууме с получением чистого 5-(4-{5-[2-(трифторметил)фенил]-1,3,4-оксадиазол-2-ил}бицикло[2.2.2]окт-1-ил)пентан-2-она (3-H).
Стадия F:
5-(4-{5-[2-(Трифторметил)фенил]-1,3,4-оксадиазол-2-ил}бицикло[2.2.2]окт-1-ил)пентан-2-он (3-H) (0,072 г) перемешивали в метаноле (2 мл) при 0°C. Добавляли боргидрид натрия (20 мг). Реакционную смесь оставляли для перемешивания при комнатной температуре. На смесь затем наслаивали этилацетат (15 мл) и воду (15 мл). Слой этилацетата экстрагировали и высушивали (MgSO4). Растворитель удаляли в вакууме с получением чистого 5-(4-{5-[2-(трифторметил)фенил]-1,3,4-оксадиазол-2-ил}бицикло[2.2.2]окт-1-ил)пентан-2-ола (3-I).
Стадия G:
5-(4-{5-[2-(Трифторметил)фенил]-1,3,4-оксадиазол-2-ил}бицикло[2.2.2]окт-1-ил)пентан-2-ол (3-I) (50 мг) помещали в герметичную колбу в раствор 2M метиламина (2,5 мл). Добавляли небольшой мерной ложечкой соль ТФУ и колбу герметично закрывали. Герметичную колбу нагревали до 150°C в течение 3 дней. Реакционную смесь разбавляли этилацетатом (15 мл), промывали водой (15 мл) и высушивали (MgSO4). Этилацетат удаляли в вакууме. Продукт, 5-(4-{1-метил-5-[2-(трифторметил)фенил]-1-Н-1,2,4-триазол-3-ил}бицикло[2.2.2]окт-1-ил)пентан-2-ол (3-J), очищали препаративной обращеннофазовой ВЭЖХ на колонке C-18 с силикагелем, используя градиент ацетонитрил-вода с буффером 0,1% трифторуксусной кислотой. Эффлюэнт, содержащий чистый триазол, делали более основным с помощью 10% NaHCO3, выпаривали в вакууме для удаления большей части ацетонитрила и экстрагировали метиленхлоридом. Органический экстракт высушивали (MgSO4) и упаривали и остаток высушивали в вакууме для получения желаемого соединения. МС (ЭСИ+) = 422,5 (М+1); 1H ЯМР (500 МГц, CDCl3): δ 1,21 (2H, м), 1,23 (3H, д, J=6,5 Гц), 1,29 (2H, м), 1,57 (6H, м), 2,13 (6H, м), 3,47 (3H, с), 3,85 (1H, м), 7,51 (1H, м), 7,70 (2H, м), 7,85 (1H, м) м.д.
Пример 4
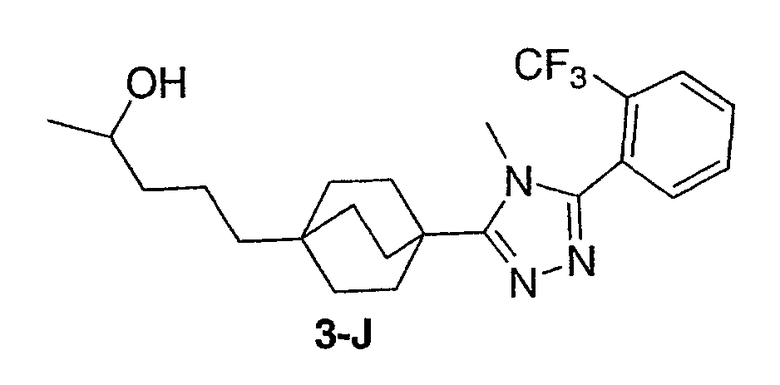
Стадия А:
К суспензии 4-(метоксикарбонил)бицикло[2.2.2]октан-1-карбоновой кислоты (4-A) (0,906 г, 4,27 ммоль) в дихлорметане (20 мл) добавляли 1,1'-карбонилдиимидазол (1,04 г, 6,41 ммоль). Реакционная смесь перешла в состояние прозрачного раствора с постоянным выделением газа. После перемешивания смеси при комнатной температуре в течение 1 ч, добавляли 4-фторбензамидоксим (1,98 г, 12,8 ммоль). Перемешивание продолжали в течение ночи. Затем смесь концентрировали и остаток кипятили с обратным холодильником в толуоле в течение 16 ч. Смесь концентрировали и остаток очищали колоночной хроматографией, используя в качестве элюэнта гексан/этилацетат (7/1) с получением метил-4-[3-(4-фторфенил)-1,2,4-оксадиазол-5-ил]бицикло[2.2.2]октан-1-карбоксилата (4-B) в виде белого твердого вещества. 1H ЯМР (500 МГц, CDCl3): δ 1,96-1,99 (м, 6H), 2,08-2,14 (м, 6H), 3,71 (с, 3H), 7,16-7,20 (м, 2H), 8,08-8,10 (м, 2H) м.д., ЭСИ-МС: m/z (M+H) 349,2.
Стадия B:
Эфир (4-B) (1,01 г, 3,06 ммоль) обрабатывали KOH (0,52 г, 9,18 ммоль) в метанол/вода (95/5, 20 мл). После нагрева его при 60°C в течение 12 ч реакционную смесь концентрировали, разбавляли водой, дважды экстрагировали этилацетатом. В водный слой добавляли водный раствор 1н. HCl, и выпадал белый твердый осадок. Осадок 4-[3-(4-фторфенил)-1,2,4-оксадиазол-5-ил]бицикло[2.2.2]октан-1-карбоновой кислоты (4-C) собирали и дополнительно высушивали совместной перегонкой с толуолом. ЭСИ-МС: m/z (M+H) 317,2.
Стадия C:
Смесь кислоты (4-C) (138,9 мг, 0,439 ммоль) и гидразида 2-(трифторметил)бензойной кислоты (4-D) (89,7 мг, 0,439 ммоль) сначала три раза перегоняли с толуолом. Дихлорметан (7 мл) добавляли к смеси в качестве растворителя. К полученной суспензии добавляли 2-хлор-1,3-диметилимидазолинийхлорид (743 мг, 4,39 ммоль) с последующим добавлением триэтиламина (1,2 мл, 8,78 ммоль). Смесь оставляли для перемешивания при комнатной температуре в атмосфере азота в течение 48 ч для подтверждения завершения взаимодействия. Затем реакционную смесь разбавляли дихлорметаном, промывали водой, 1н. HCl, насыщенным водным раствором бикарбоната натрия и в конце насыщенным раствором соли. Органическую фазу высушивали над безводным сульфатом натрия, фильтровали и концентрировали. Остаток очищали колоночной хроматографией, используя в качестве элюэнта гексан/этилацетат (3/1) с получением 3-(4-фторфенил)-5-(4-{5-[2-(трифторметил)фенил]-1,3,4-оксадиазол-2-ил}бицикло[2.2.2]окт-1-ил)-1,2,4-оксадиазола (4-E) в виде белого твердого вещества. 1H ЯМР (500 МГц, CDCl3): δ 2,25 (с, 12H), 7,21 (т, J=8,7 Гц, 2H), 7,74-7,76 (м, 2Н), 7,91 (м, 1H), 8,11-8,15 (м, 3H) м.д., ЭСИ-МС: m/z (M+H) 485,2.
Стадия D:
Смесь указанного выше 1,2,4-оксадиазола (4-E) (115,2 мг, 0,238 ммоль) и соли метиламина и трифторуксусной кислоты (1,73 г, 11,9 ммоль) в 2M растворе метиламина в метаноле (4 мл) нагревали при 150°C в герметичной пробирке в течение 48 ч. Затем смесь концентрировали и остаток переносили в дихлорметан, промывали насыщенным раствором бикарбоната натрия. Органическую фазу концентрировали и остаток очищали, используя в качестве элюэнта обращеннофазовую ВЭЖХ с ТФУ-буферированной смесью ацетонитрил/вода (40-80%). Фракции, содержащие продукт, объединяли, нейтрализовали насыщенным водным раствором бикарбоната натрия и лиофилизовали из смеси ацетонитрил/вода с получением 3-(4-фторфенил)-5-(4-{4-метил-5-[2-(трифторметил)фенил]-4Н-1,2,4-триазол-3-ил}бицикло[2.2.2]окт-1-ил)-1,2,4-оксадиазола (4-F). 1H ЯМР (CDCl3): δ 2,25-2,35 (м, 12H), 3,53 (с, 3H), 7,21 (т, J=8,7 Гц, 2H), 7,54 (м, 1H), 7,73 (м, 2H), 7,88 (м, 1H), 8,13 (м, 2H), ЭСИ-МС: m/z (M+H) 498,2.
Пример 5
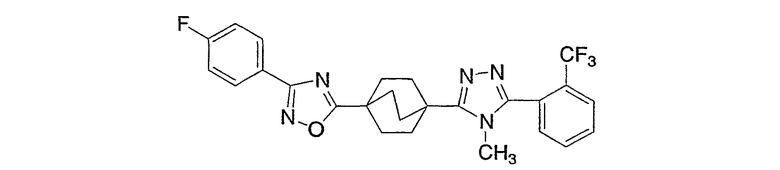
4-[4-Метил-5-(4-фенилбицикло[2.2.2]окт-1-ил)-4H-1,2,4-триазол-3-ил]-3-(трифторметил)фенол (5-F)
Получение 4-фенилбицикло[2.2.2]октан-l-карбоновой кислоты (5-A). Литературный источник: Chapman, N. B., Sotheeswaran, S., и Toyne, K. J., J. Org. Chem, 35: 917-923 (1970).
Стадия A:
К перемешиваемому на магнитной мешалке раствору 4-фенилбицикло[2.2.2]октан-l-карбоновой кислоты (5-A) (70 мг, 0,30 ммоль) в метиленхлориде (1 мл) при комнатной температуре добавляли 2M оксалилхлорид в метиленхлориде (0,61 мл, 1,22 ммоль). Добавляли две капли каталитического ДМФА для катализа взаимодействия. Реакционную смесь перемешивали в течение 30 мин и растворитель и реагент удаляли в вакууме. Метиленхлорид (1 мл) добавляли к остатку с последующим добавлением гидразида 4-(бензилокси)-2-(трифторметил)бензойной кислоты (5-B) (141 мг, 0,46 ммоль) и триэтиламина (0,07 мл, 0,46 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение ночи с получением интермедиата 5-C, N'-[4-(бензилокси)-2-(трифторметил)бензоил]-4-фенилбицикло[2.2.2]октан-l-карбогидразида, который не выделяли. К неочищенному продукту затем добавляли (5-C) 2-хлор-1,3-диметилимидазолинийхлорид (257 мг, 1,52 ммоль), еще триэтиламин (0,42 мл, 3,04 ммоль) и метиленхлорид (2 мл). Реакционную смесь перемешивали при комнатной температуре в течение 4 ч. Затем реакционную смесь разбавляли метиленхлоридом (30 мл) и промывали водой (30 мл) два раза и насыщенным раствором соли (30 мл) один раз. Объединенные водные слои экстрагировали метиленхлоридом (25 мл) один раз. Объединенные органические слои высушивали (MgSO4) и растворитель удаляли в вакууме. Остаток хроматографировали на силикагеле с 10% этилацетатом в гексане в качестве элюэнта с получением 2-[4-(бензилокси)-2-(трифторметил)фенил]-5-(4-фенилбицикло[2.2.2]окт-1-ил)-1,3,4-оксадиазола (5-D). МС: m/z 505 (М+1).
Стадия B:
Трифторацетатную соль метиламина (380 мг, 2,61 ммоль) и 2-[4-(бензилокси)-2-(трифторметил)фенил]-5-(4-фенилбицикло[2.2.2]окт-1-ил)-1,3,4-оксадиазол (5-D) суспендировали в 2M растворе метиламина в метаноле (1,3 мл, 2,61 ммоль) и нагревали при 150°C в течение ночи. После охлаждения до комнатной температуры реакционную смесь распределяли между этилацетатом (25 мл) и насыщенным водным раствором бикарбоната натрия (30 мл). Слои разделяли и водный слой экстрагировали дважды этилацетатом (25 мл). Объединенные органические слои промывали насыщенным раствором соли, высушивали (MgSO4) и растворитель удаляли в вакууме. Остаток растворяли в метаноле (8 мл) и очищали обращеннофазовой хроматографией, используя градиентную элюцию с 10% ацетонитрил (0,1% ТФУ)/вода (0,1% ТФУ) до 100% ацетонитрила (0,1% ТФУ) в течение 10 мин (20 мл/мин). Фракции, содержащие продукт, распределяли между насыщенным водным раствором бикарбоната натрия (25 мл) и метиленхлоридом (15 мл). Слои сепарировали и водный слой экстрагировали метиленхлоридом (15 мл) три раза, высушивали (MgSO4) и растворитель удаляли в вакууме с получением 3-[4-(бензилокси)-2-(трифторметил)фенил]-4-метил-5-(4-фенилбицикло[2.2.2]окт-1-ил)-4Н-1,2,4-триазола (5-E). МС: m/z 518 (М+1).
Стадия C:
3-[4-(Бензилокси)-2-(трифторметил)фенил]-4-метил-5-(4-фенилбицикло[2.2.2]окт-1-ил)-4Н-1,2,4-триазол (5-E) (27 мг, 0,05 ммоль) растворяли в этилацетат/метанол (1:1, 4 мл), к которому добавляли 10% палладий на угле (4 мг). Затем реакционную смесь помещали в атмосферу водорода и перемешивали в течение 3 ч при комнатной температуре и давлении. После соответствующего удаления атмосферы водорода палладий отфильтровывали через фильтр и промывали метанолом (40 мл). Фильтрат собирали и растворитель удаляли в вакууме с получением 4-[4-метил-5-(4-фенилбицикло[2.2.2]окт-1-ил)-4H-1,2,4-триазол-3-ил]-3-(трифторметил)фенола (5-F). МС: m/z 428 (М+1); 1H ЯМР (500 МГц, CDCl3): δ 1,92 (6H, м), 2,11 (6H, м), 3,41 (3H, с), 7,17 (2H, м), 7,24 (1H, м), 7,31 (2H, м), 7,38 (3H, м) м.д.
Пример 6
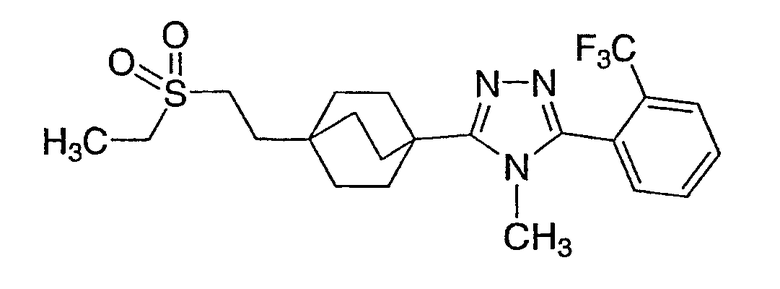
3-{4-[2-(Этилсульфонил)этил]бицикло[2.2.2]окт-1-ил}-4-метил-5-[2-(трифторметил)фенил]-4H-1,2,4-триазол (6-6)
Стадия А:
Диэтил(этилсульфонметан)фосфонат (1,12 г, 4,6 ммоль) (Popoff, I. C. и др., J. Org. Chem., 34: 1128-30 (1969)) и 4-карбометоксибицикло[2.2.2]октан-1-карбоксальдегид (6-1) (0,82 г, 4,2 ммоль) (Adcock, W., Kok, G.B., J. Org. Chem., 50: 1079-1087 (1985)) растворяли в 8 мл абсолютного метанола. Смесь помещали в атмосферу азота, охлаждали на ледяной бане и обрабатывали 0,5M раствором метоксида натрия в метаноле (8,8 мл, 4,4 ммоль). Реакционную смесь нагревали с обратным холодильником в течение 4 ч, затем охлаждали до комнатной температуры, концентрировали при пониженном давлении, затем обрабатывали 2 мл воды и позволяли осесть в холодильнике в течение ночи. Смесь фильтровали и твердое вещество промывали небольшим количеством холодного 1:1 MeOH/вода. Полученный белый осадок собирали и высушивали в вакууме с получением ненасыщенного сульфона 6-2. МС (ЭСИ+): m/z 287 (М+1).
Стадия B:
Сульфон 6-2 (880 мг, 3,08 ммоль) растворяли в смеси этилацетат/метанол 1:2 (30 мл), помещали в атмосферу азота, затем обрабатывали 10% Pd/C (800 мг). Реакционную смесь помещали в атмосферу водорода и энергично перемешивали в течение 90 мин. Полученный раствор фильтровали через целит, промывали метанолом и этилацетатом и упаривали с получением 4-[2-(этилсульфонил)этил]бицикло[2.2.2]октан-1-карбоксилата (6-3) в виде белого твердого вещества.
Стадия C:
Эфир 6-3 (880 мг, 3 ммоль) растворяли в растворе 10% вода/метанол (100 мл) и обрабатывали 1 г гидроксида калия. Реакционную смесь нагревали при 60°C в течение 1 ч, затем при 45°C в течение ночи. Смесь концентрировали в вакууме, затем доводили pH до значения 2 с помощью 1M HCl и экстрагировали тремя порциями метиленхлорида. Органические слои объединяли, высушивали над безводным сульфатом натрия и упаривали с получением 4-[2-(этилсульфонил)этил]бицикло[2.2.2]октан-1-карбоновой кислоты (6-4).
Стадия D:
Карбоновую кислоту 6-4 (810 мг, 2,96 ммоль) растворяли в 12 мл безводного метиленхлорида в атмосфере азота, обрабатывали оксалилхлоридом (2M в метиленхлориде, 4,4 мл, 8,8 ммоль) и последовательно 5 каплями ДМФА. Реакционную смесь перемешивали при комнатной температуре в атмосфере азота в течение 90 мин, затем упаривали и помещали в вакуум на 20 мин. Хлорангидрид кислоты растворяли в безводном метиленхлориде (12 мл), охлаждали на ледяной бане и затем капельно обрабатывали раствором метиламина (2M в ТГФ, 8,9 мл, 17,8 ммоль). После завершения добавления амина охлаждающую баню удаляли и реакционную смесь перемешивали при температуре окружающей среды в течение 30 мин. Смесь разбавляли 200 мл метиленхлорида и промывали водной 1н. HCl, насыщенным водным раствором бикарбоната натрия и насыщенным раствором соли. Органический слой высушивали над безводным сульфатом натрия и выпаривали. Остаток подвергали хроматографии на силикагеле, элюируя градиентом от 0 до 3,5% метанола в метиленхлориде с получением
4-[2-(этилсульфонил)этил]-N-метилбицикло[2.2.2]октан-1-карбоксамида 6-5 в виде белого порошка. МС (ЭСИ+): m/z 288 (М+1).
Стадия E:
Метиламид 6-5 (220 мг, 0,77 ммоль) растворяли в безводном метиленхлориде (2 мл) и обрабатывали оксалилхлоридом (2M в метиленхлориде, 0,77 мл, 1,54 ммоль) и ДМФА (2 капли). Реакционную смесь перемешивали при комнатной температуре в течение 1 ч, затем растворитель удаляли выпариванием при пониженном давлении. Остаток повторно растворяли в безводном толуоле (2 мл) и обрабатывали 5-[2-(трифторметил)фенил]-1H-тетразолом (214 мг, 1 ммоль). Смесь нагревали с обратным холодильником 18 ч. Реакционную смесь охлаждали до комнатной температуры и осадок кремового цвета отфильтровывали и промывали для получения 300 мг неочищенного продукта в виде соли HCl. Смесь поглощали метиленхлоридом/1н. HCl и водный слой промывали двумя дополнительными порциями метиленхлорида. Органические слои объединяли и выпаривали и остаток хроматографировали флэш-хроматографией на силикагеле. Элюцию проводили в градиенте в интервале от 0 до 5% метанол/метиленхлорид. Соответствующие фракции объединяли и выпаривали с получением 3-{4-[2-(этилсульфонил)этил]бицикло[2.2.2]окт-1-ил}-4-метил-5-[2-(трифторметил)фенил]-4H-1,2,4-триазола (6-6) в виде белого порошка. МС (ЭСИ+): 456,2 (М+1); 1H ЯМР (500 МГц, CDCl3): δ 1,46 (3H, т, J=7,3 Гц), 1,63 (6H, м), 1,78 (2H, м), 2,19 (6H, м), 2,96 (2H, м), 3,05 (2H, кв., J=7,2 Гц), 3,50 (3H, с), 7,56 (1Н, м), 7,72 (2H, м), 7,87 (1H, м) м.д.
Пример 7
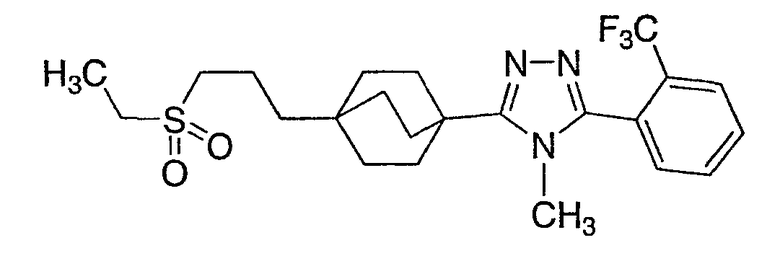
3-{4-[3-(Этилсульфонил)пропил]бицикло[2.2.2]окт-1-ил}-4-метил-5-[2-(трифторметил)фенил]-4H-1,2,4-триазол (7-J)
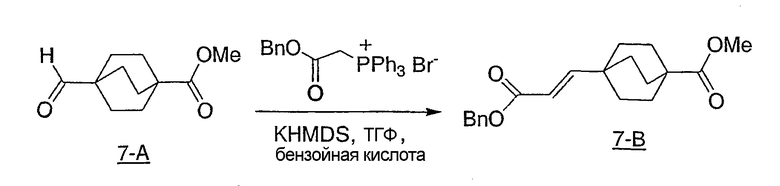
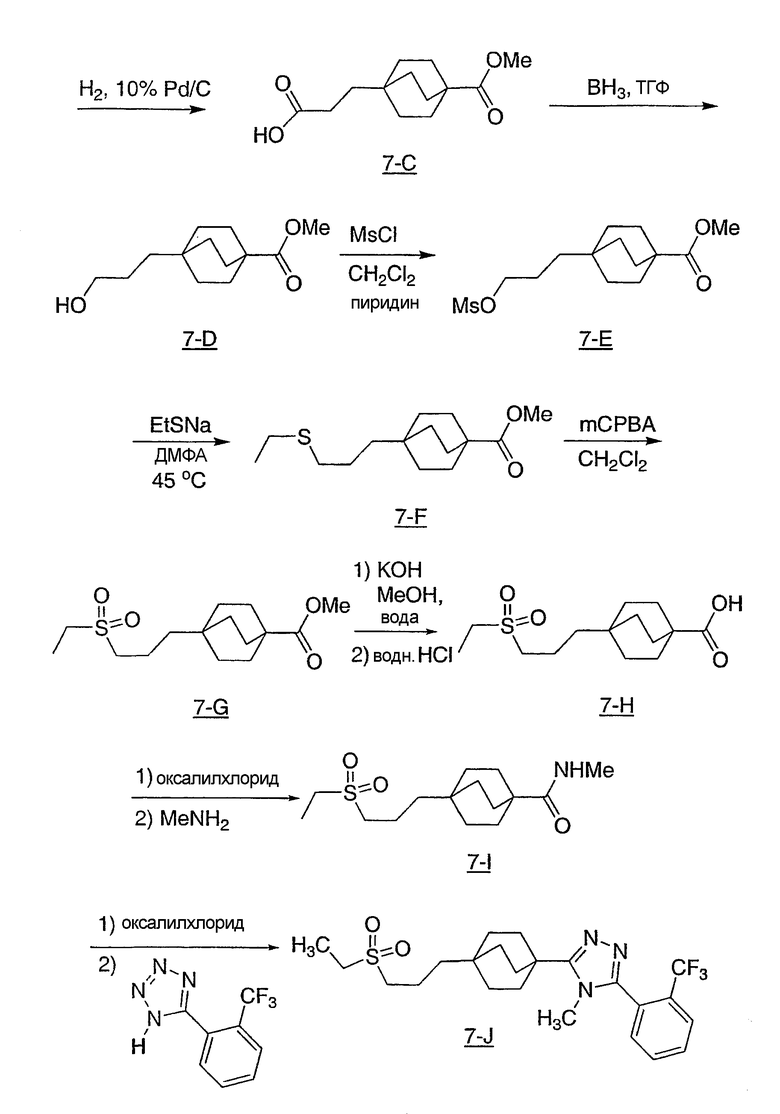
Стадия A:
(Бензилоксикарбонилметил)трифенилфосфонийбромид (4,6 г, 9,4 ммоль) дважды азеотропно перегоняли из толуола и затем суспендировали в 30 мл сухого ТГФ. По каплям добавляли гексаметилдисилазид калия (0,5 M в толуоле, 16,8 мл, 8,4 ммоль) при комнатной температуре и желтый раствор оставляли для перемешивания в течение 1 ч, после этого времени он становился молочно-белым. Готовили раствор 4-карбометокси-бицикло[2.2.2]октан-1-карбоксальдегида (7-A) (0,50 г, 2,55 ммоль) (Adcock, W., Kok, G. B., J. Org. Chem., 50: 1079-1087 (1985)) и бензойной кислоты (0,015 г, 0,13 ммоль) в 2 мл сухого ТГФ и добавляли по каплям шприцем при комнатной температуре. Смесь нагревали до 90°C и оставляли для перемешивания при температуре образования флегмы, после этого смесь разбавляли 200 мл этилацетата и последовательно промывали порциями по 50 мл 1н. HCl (дважды), насыщенным водным раствором бикарбоната натрия и насыщенным раствором соли. Органический слой высушивали, используя сульфат магния, и растворитель удаляли при пониженном давлении. Остаток хроматографировали на силикагеле, элюируя градиентом от 5% до 10% этилацетатом в гексане с получением метил-4-[(lE)-3-(бензилокси)-3-оксопроп-1-ен-1-ил]бицикло[2.2.2]октан-1-карбоксилата (7-B) в виде бесцветного масла. 1H ЯМР (500 МГц, CDCl3): δ 7,4 (5H, м), 6,94 (1H, д, J=17 Гц), 5,77 (1Н, д, J=17 Гц), 5,21 (2H, с), 3,69 (3H, с), 1,86 (6H, м), 1,63 (6H, м) м.д.
Стадия B:
Диэфир 7-B (0,625 г, 1,90 ммоль) растворяли в смеси 1:1 этилацетат/метанол (30 мл), помещали в атмосферу азота, затем обрабатывали 10% Pd/C (500 мг) и 0,1 мл уксусной кислоты. Реакционную смесь помещали в атмосферу водорода и энергично перемешивали в течение 2 ч. Полученный раствор фильтровали через целит и растворитель удаляли при пониженном давлении. Остаток распределяли между 200 мл этилацетата и 200 мл раствора 1н. NaOH. Водный слой сепарировали и нейтрализовали, затем три раза экстрагировали 50 мл метиленхлорида. Объединенные органические слои высушивали над сульфатом магния и растворитель удаляли при пониженном давлении с получением 3-[4-(метоксикарбонил)бицикло[2.2.2]окт-1-ил]пропановой кислоты (7-С). 1H ЯМР (500 МГц, CDCl3): δ 3,62 (3H, с), 2,20 (2H, ушир.т, J=9 Гц), 1,75 (6H, м), 1,47 (2H, ушир.т, J=9 Гц), 1,38 (6H, м) м.д.
Стадия C:
Карбоновую кислоту 7-C (400 мг, 1,67 ммоль) растворяли в тетрагидрофуране (5 мл) и боран (1M раствор в ТГФ, 2,17 мл, 1,3 экв.) по каплям добавляли при комнатной температуре. После 2 ч реакционную смесь добавляли к 50 мл 1н. HC1 и затем три раза экстрагировали 50 мл метиленхлорида. Объединенные органические слои высушивали над сульфатом магния и растворитель удаляли при пониженном давлении с получением неочищенного метил-4-(3-гидроксипропил)бицикло[2.2.2]октан-l-карбоксилата (7-D), который использовали без очистки на следующей стадии. 1Н ЯМР (500 МГц, CD3OD): δ 3,66 (3H, с), 3,62 (2H, т, J=6,5 Гц), 1,78 (6H, м), 1,50 (2H, м), 1,41 (2H, м), 1,17 (2H, м) м.д.
Стадия D:
Гидроксиэфир 7-D (430 мг, 1,9 ммоль) растворяли в 2,5 мл безводного метиленхлорида в атмосфере азота, обрабатывали пиридином (0,5 мл) и метансульфонилхлоридом (0,368 мл, 4,8 ммоль) и перемешивали в течение 4 ч при комнатной температуре. Смесь разбавляли 100 мл этилацетата и промывали водной 1н. HCl, насыщенным водным раствором бикарбоната натрия и насыщенным раствором соли. Органический слой высушивали над безводным сульфатом натрия и выпаривали. Неочищенный метил-
4-{3-[(метилсульфонил)окси]пропил}бицикло[2.2.2]октан-l-карбоксилат (7-E), полученный, таким образом, использовали без очистки в следующей реакции. 1H ЯМР (500 МГц, CDCl3): δ 4,22 (2H, т, J=7,5 Гц), 3,68 (3H, с), 3,04 (3H, с), 1,82 (6H, м), 1,70 (2H, м), 1,44 (6H, м), 1,24 (2H, м) м.д.
Стадия E:
Мезилат 7-E (3,30 г, 10,9 ммоль) растворяли в ДМФА (20 мл) и обрабатывали этантиолатом натрия (1,82 г, 21,7 ммоль). Раствор перемешивали при 45°C в течение 3 ч, затем смесь разбавляли 100 мл этилацетата и промывали дважды водной 1н. HCl, затем насыщенным водным раствором бикарбоната натрия и насыщенным раствором соли. Органический слой высушивали над безводным сульфатом натрия и выпаривали с получением метил-4-[3-(этилтио)пропил]бицикло[2.2.2]октан-l-карбоксилата (7-F) в виде неочищенного масла, которое использовали в следующей стадии без очистки. 1H ЯМР (500 МГц, CDCl3): δ 3,68 м.д. (3H, с), 2,56 (2H, кв., J=7 Гц), 2,51 (2H, т, J=7,5 Гц), 1,80 (6H, м), 1,52 (2H, м), 1,42 (6H, м), 1,28 (2H, т, J=7 Гц), 1,02 (2H, м).
Стадия F:
Сульфид 7-F (3,0 г, 11 ммоль) растворяли в метиленхлориде (50 мл) и обрабатывали м-хлорнадбензойной кислотой (75%, 6,2 г). Раствор перемешивали при комнатной температуре в течение 2 ч, затем смесь разбавляли 100 мл метиленхлорида и промывали насыщенным водным раствором бикарбоната натрия, затем дважды насыщенным водным раствором бисульфита натрия, затем дважды насыщенным водным раствором бикарбоната натрия и насыщенным раствором соли. Органический слой высушивали над безводным сульфатом натрия и выпаривали с получением метил-4-[3-(этилсульфонил)пропил]бицикло[2.2.2]октан-l-карбоксилата (7-G) в виде неочищенного масла, которое использовали в следующей стадии без очистки. 1H ЯМР (500 МГц, CDCl3): δ 3,68 м.д. (3H, с), 2,56 (2H, кв., J=7 Гц), 2,51 (2H, т, J=7,5 Гц), 1,80 (6H, м), 1,52 (2H, м), 1,42 (6H, м), 1,28 (2H, т, J=7 Гц), 1,02 (2H, м) м.д.
Стадия G:
Сульфон 7-G (3,1 г, 10 ммоль) растворяли в 9:1 MeOH/вода (50 мл) и обрабатывали гидроксидом калия (3 г). Раствор перемешивали при комнатной температуре в течение ночи, затем в смесь добавляли 1н. HC1 и экстрагировали четыре раза 50 мл метиленхлорида. Органический слой высушивали над безводным сульфатом натрия и выпаривали с получением 4-[3-(этилсульфонил)пропил]бицикло[2.2.2]октан-l-карбоновой кислоты (7-H), которую использовали в следующей стадии без очистки. 1H ЯМР (500 МГц, CDCl3): δ 3,03 (2H, кв., J=7 Гц), 2,94 (2H, дд, J=7,5 Гц), 1,84 (8H, м), 1,45 (8H, м), 1,30 (2H, м) м.д.
Стадия H:
Карбоновую кислоту 7-H (3,0 г, 11 ммоль) растворяли в 50 мл безводного метиленхлорида в атмосфере азота, обрабатывали оксалилхлоридом (2M в метиленхлориде, 16,2 мл, 32,4 ммоль) и далее 5 каплями ДМФА. Раствор перемешивали при комнатной температуре в атмосфере азота в течение 90 мин, затем упаривали и помещали в вакуум на 20 мин. Хлорангидрид кислоты растворяли в безводном метиленхлориде (12 мл), охлаждали на ледяной бане и затем капельно обрабатывали раствором метиламина (2M в ТГФ, 27 мл, 54 ммоль). После добавления метиламина охлаждающую баню убирали и реакционную смесь перемешивали при температуре окружающей среды в течение 30 мин. Смесь разбавляли 200 мл метиленхлорида и промывали водной 1н. HCl, насыщенным водным раствором бикарбоната натрия и насыщенным раствором соли. Органический слой высушивали над безводным сульфатом натрия и выпаривали. Остаток подвергали хроматографии на силикагеле, элюируя градиентом от 0 до 3% метанола в этилацетате с получением 4-[3-(этилсульфонил)пропил]-N-метилбицикло[2.2.2]октан-1-карбоксамида 7-I в виде белого порошка. МС (ЭСИ+) = 302 (М+1); 1H ЯМР (500 МГц, CDCl3): δ 5,56 (1H, ушир.с), 3,02 (2H, кв., J=7 Гц), 2,94 (2H, дд, J=7,5 Гц), 2,82 (3H, д, J=4 Гц), 1,80 (8H, м), 1,45 (9H, м), 1,28 (2H, м) м.д.
Стадия I:
Метиламид 7-I (0,470 г, 1,56 ммоль) растворяли в безводном метиленхлориде (5 мл) и обрабатывали оксалилхлоридом (2M в метиленхлориде, 1,56 мл, 3,12 ммоль) и ДМФА (2 капли). Раствор перемешивали при комнатной температуре в течение 1 ч, затем растворитель удаляли выпариванием при пониженном давлении. Остаток повторно растворяли в безводном толуоле (7 мл) и обрабатывали 5-[2-(трифторметил)фенил]-lH-тетразолом (368 мг, 1,72 ммоль). Смесь кипятили с обратным холодильником в течение 18 ч. Реакционную смесь охлаждали до комнатной температуры и осадок отфильтровывали и промывали с получением 300 мг неочищенного продукта в виде соли HCl. Соль поглощали метиленхлоридом/lн. HCl и водный слой промывали двумя дополнительными порциями метиленхлорида. Органические слои объединяли и выпаривали и остаток подвергали флэш-хроматографии на силикагеле. Элюцию проводили с градиентом в интервале от 0 до 5% метанол/метиленхлорид. Соответствующие фракции объединяли и выпаривали с получением 3-{4-[3-(этилсульфонил)пропил]бицикло[2.2.2]окт-1-ил}-4-метил-5-[2-(трифторметил)фенил]-4H-1,2,4-триазола (7-J) в виде белого порошка. МС (ЭСИ+) = 470,4 (М+1), 1H ЯМР (500 МГц, CDCl3): δ 7,87 (1H, м), 7,72 (2H, м), 7,56 (1H, м), 3,49 (3H, с), 3,05 (2H, кв., J=7,2 Гц), 2,96 (2H, м), 2,18 (6H, м), 1,86 (2H, м), 1,62 (6H, м), 1,46 (3H, т, J=7,3 Гц), 1,36 (2H, м) м.д.
Пример 8
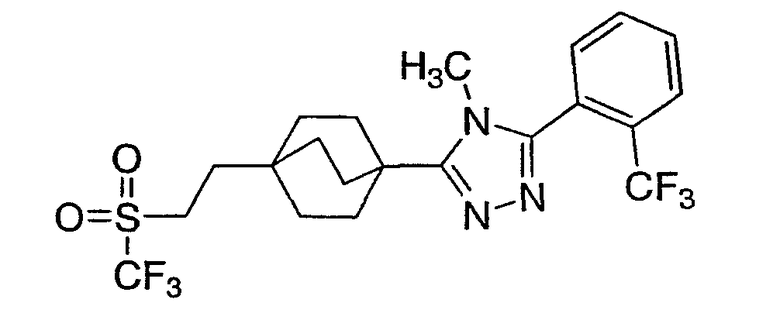
4-Метил-3-[2-(трифторметил)фенил]-5-(4-{2-[(трифторметил)сульфонил]этил}бицикло[2.2.2]окт-1-ил)-4H-1,2,4-триазол (8-G)
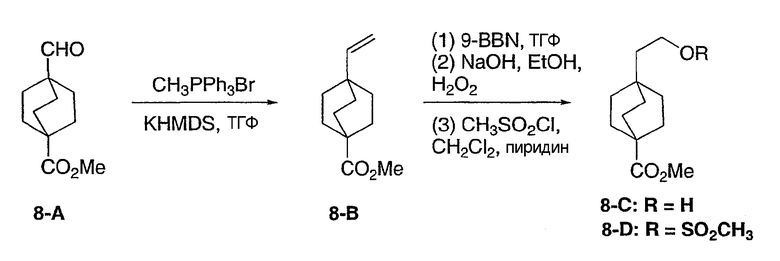
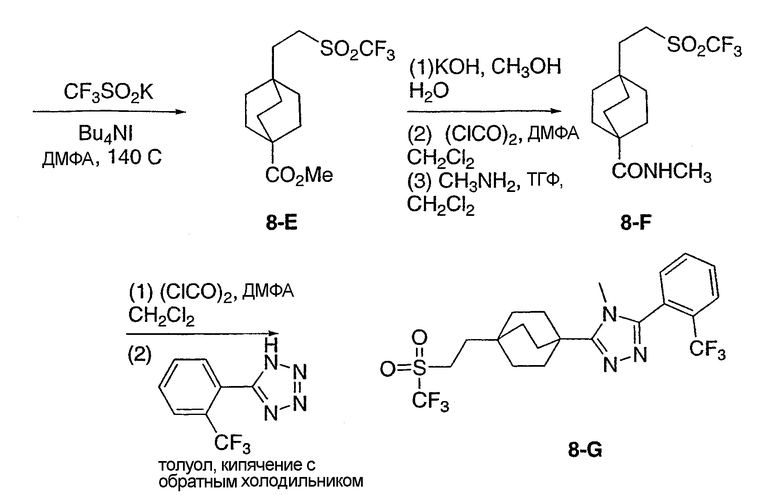
Стадия A:
К перемешиваемому раствору метилтрифенилфосфонийбромида (9,1 г, 12,8 ммоль) в ТГФ (50 мл) при 0°C добавляли по каплям гексаметилдисилазид калия (0,5M в толуоле, 48,6 мл) в течение 5 мин. Полученную смесь оставляли для нагрева до комнатной температуры в течение 1 ч, затем снова охлаждали до 0°C и обрабатывали метил-4-формилбицикло[2.2.2]октан-l-карбоксилатом 8-A (Chapman, N. B. и др., J. Org. Chem., 1970, 35, 917) (2,5 г, 12,8 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение 18 ч, затем разбавляли EtOAc (350 мл). Органическую фазу промывали водной HCl (1н.), насыщенным водным раствором бикарбоната натрия и насыщенным раствором соли, затем высушивали (Na2SO4) и концентрировали в вакууме. Полученное твердое вещество очищали флэш-хроматографией на силикагеле, элюируя градиентом 0-4% EtOAc/гексан. Полученный метил-4-винилбицикло[2.2.2]октан-l-карбоксилат (8-B) был выделен в виде прозрачного, бесцветного масла.
Стадия B:
К перемешиваемому раствору олефина 8-B (1,6 г, 8,3 ммоль) в ТГФ (20 мл) добавляли по каплям 9-BBN (0,5M в ТГФ, 49 мл). Раствор оставляли для перемешивания при комнатной температуре в течение 18 ч, затем последовательно обрабатывали этанолом (14,5 мл), водным NaOH (5н., 5 мл) и пероксидом водорода (30% водный раствор, 9,7 мл). pH реакционной смеси доводили до значения 2 водным раствором HCl (1н.) и три раза экстрагировали CH2C12. Органические слои объединяли, промывали насыщенным раствором соли, высушивали (Na2SO4) и соскабливали. Полученный спирт 8-C очищали хроматографией на силикагеле, элюируя градиентом 30-50% EtOAc/гексан, и выделяли в виде прозрачного, бесцветного масла.
Стадия C:
Раствор спирта 8-C (1,5 г, 7,1 ммоль) в CH2C12 (7,5 мл), пиридин (1,5 мл) охлаждали до 0°C и обрабатывали метансульфонилхлоридом (1,65 мл, 21,3 ммоль), капельно, в течение 5 мин. Реакционную смесь оставляли для нагрева до комнатной температуры, затем перемешивали в течение 3 ч. Добавляли EtOAc (300 мл) и органическую фазу промывали водным раствором HCl (1н.) три раза, насыщенным водным раствором бикарбоната натрия два раза и насыщенным раствором соли. Органический слой высушивали (Na2SO4) и соскабливали для получения метил-4-{2-[(метилсульфонил)окси]этил}бицикло[2.2.2]октан-l-карбоксилата 8-D в виде белого твердого вещества. 1H ЯМР (500 МГц, CDCl3): δ 1,52 (6H, м), 1,66 (2H, т, J=7,1 Гц), 1,84 (6H, м), 3,04 (3H, с), 3,69 (3H, с), 4,29 (2H, т, J=7,2 Гц) м.д.
Стадия D:
Раствор 8-D (0,25 г, 0,86 ммоль), трифторметансульфинат калия (0,3 г, 1,72 ммоль) и тетрабутиламмонийиодид (0,15 г, 0,4 ммоль) в ДМФА (5 мл) нагревали при 140ºC в течение 5 ч в атмосфере азота. Раствор затем охлаждали до комнатной температуры и разбавляли EtOAc (100 мл) и промывали два раза водным раствором HCl (1н.) и насыщенным раствором соли. Органический слой высушивали (Na2SO4), соскабливали и подвергали флэш-хроматографии на силикагеле, элюируя градиентом 5-20% EtOAc/гексан. Полученный трифторметилсульфон 8-E выделяли в виде белого твердого вещества. 1H ЯМР (500 МГц, CDCl3): δ 1,50 (6H, м), 1,78 (2H, м), 1,82 (6H, м), 3,17 (2H, м), 3,67 (3H, с) м.д.
Стадия E:
Метиловый эфир 8-E (0,035 г, 0,11 ммоль) переводили в метиламид 8-F, используя способы, описанные в примере 6, на стадиях C и D.
N-метил-4-{2-[(трифторметил)сульфонил]этил}бицикло[2.2.2]октан-l-карбоксамид выделяли в виде белого твердого вещества; МС (ЭСИ+) = 328,2 (М+1).
Стадия F:
Метиламид 8-F (0,030 г, 0,092 ммоль) превращали в триазол 8-G по методике примера 6, стадии E. 4-Метил-3-[2-(трифторметил)фенил]-5-(4-{2-[(трифторметил)сульфонил]этил}бицикло[2.2.2]окт-1-ил)-4H-1,2,4-триазол (8-G) выделяли в виде белого порошка; МС (ЭСИ+) = 496,4 (М+1).
Примеры 9-102
Следуя методикам, сходным с теми, что описаны выше, также были получены следующие соединения формулы II:
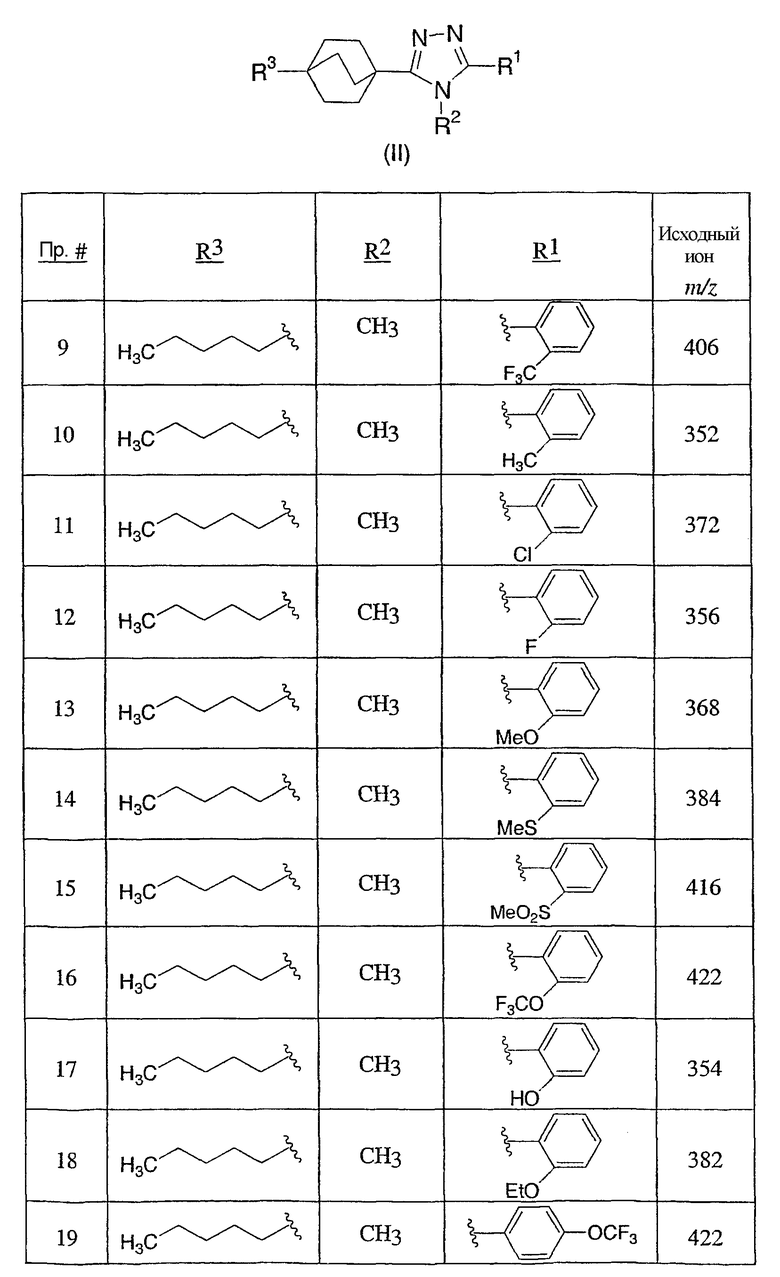
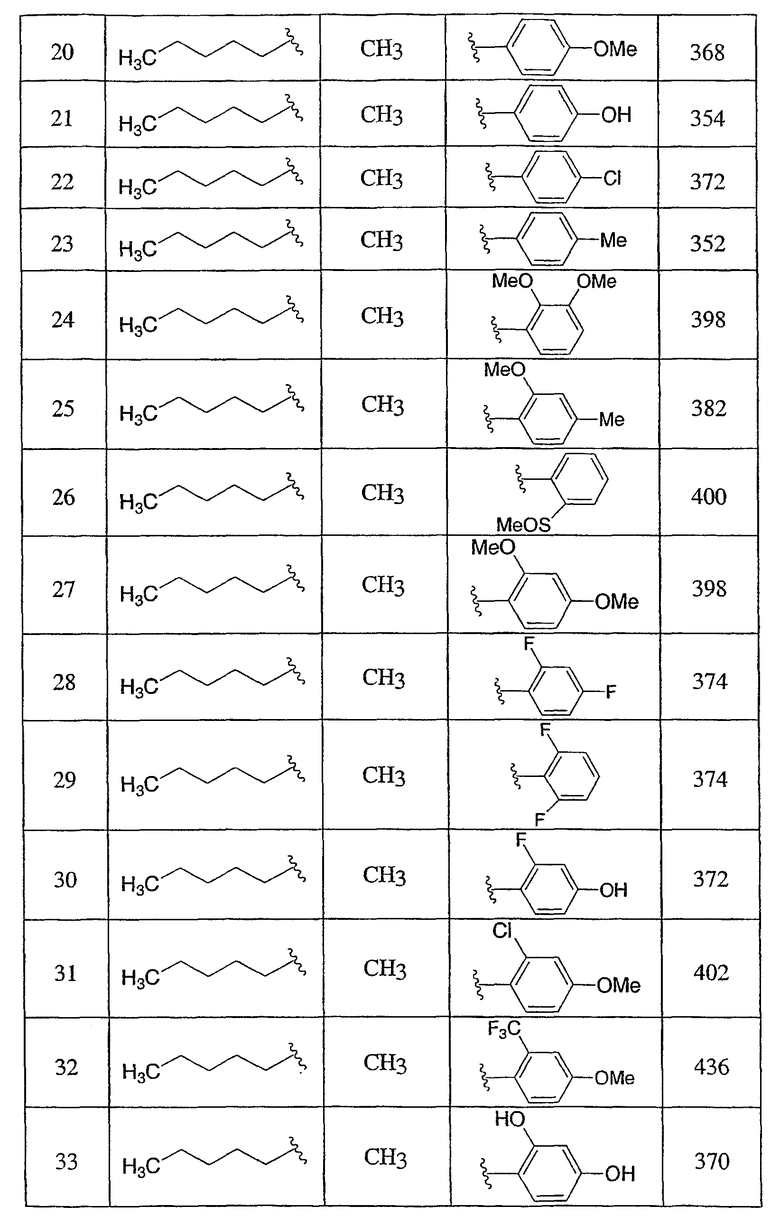
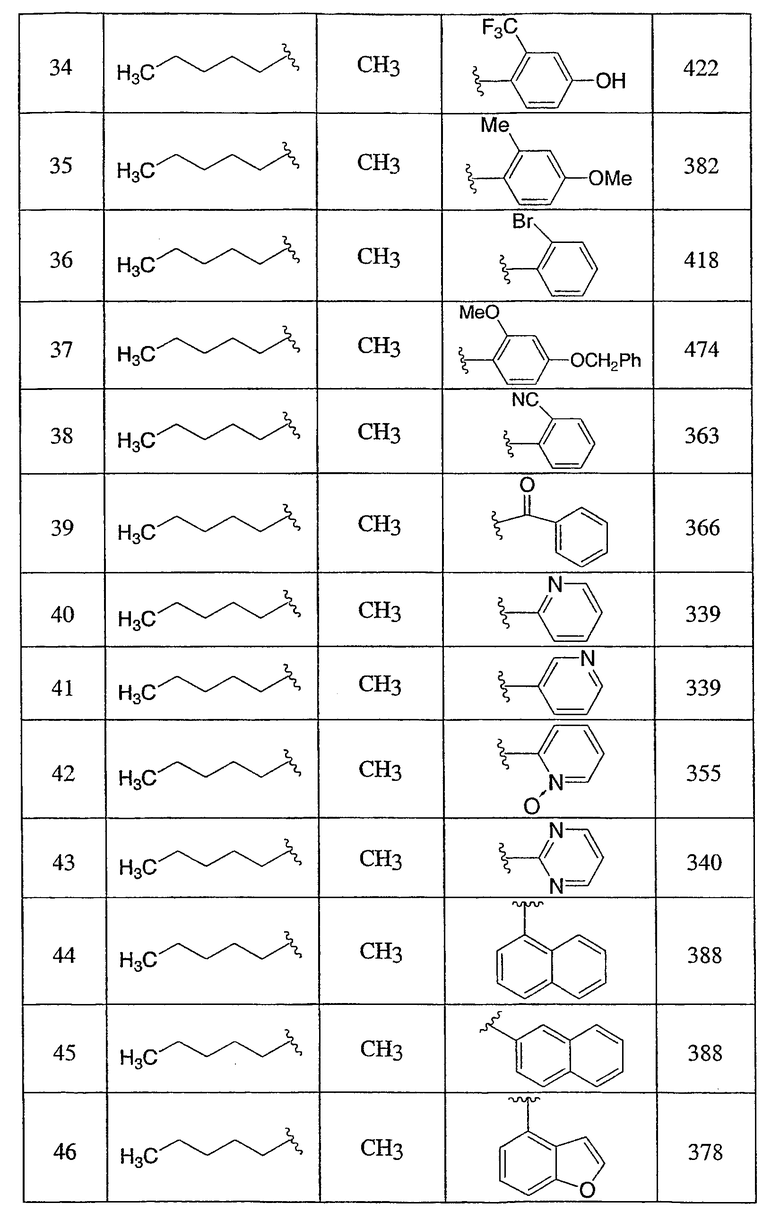
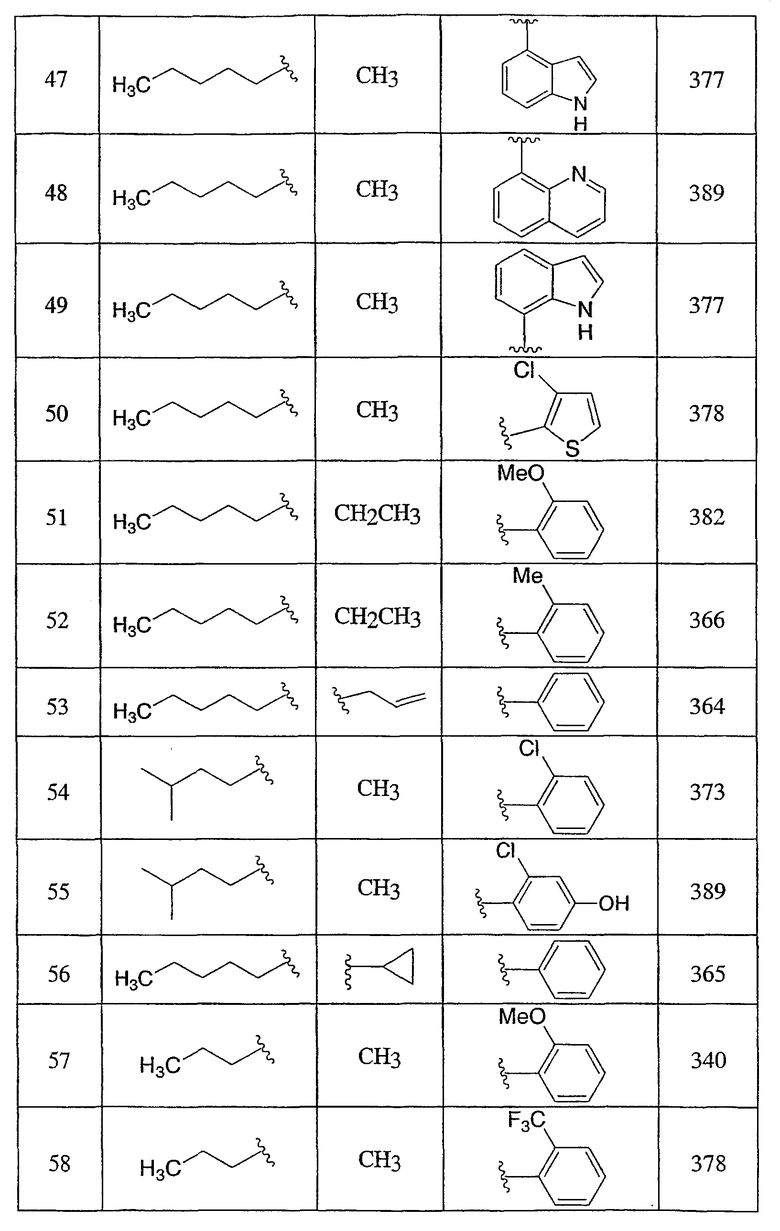
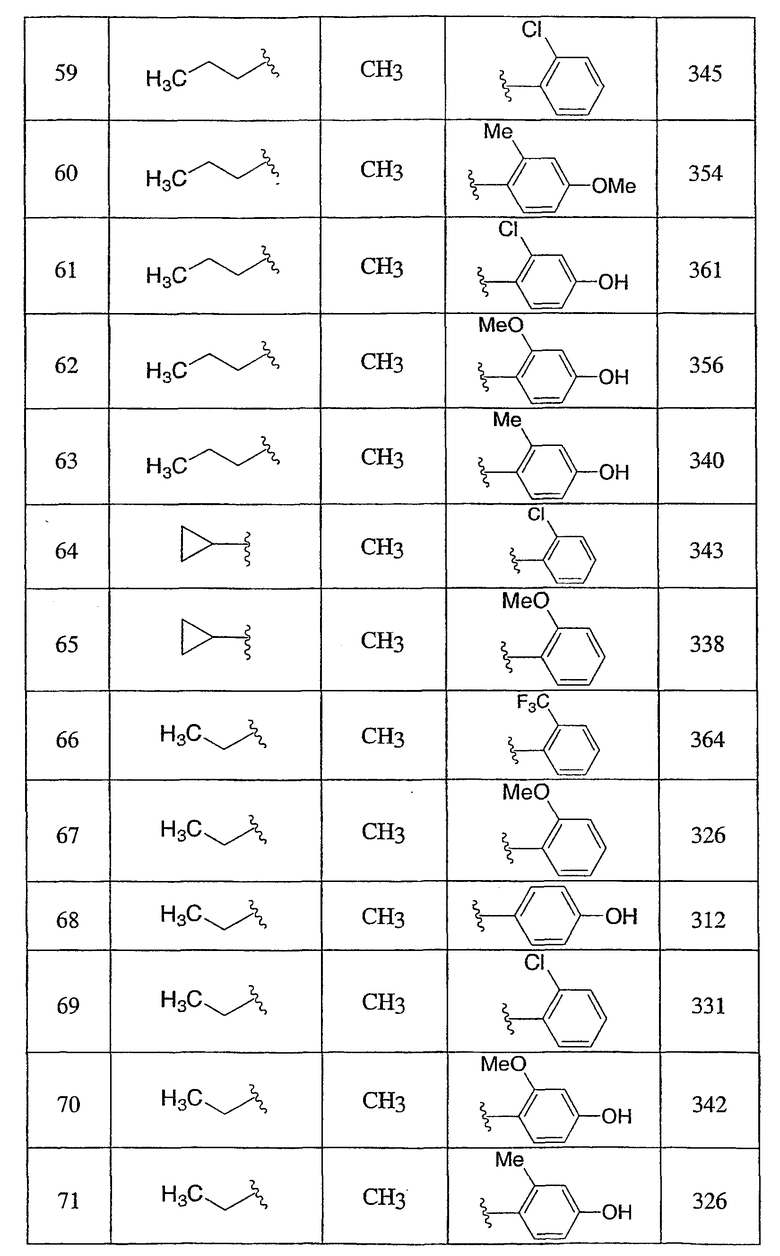
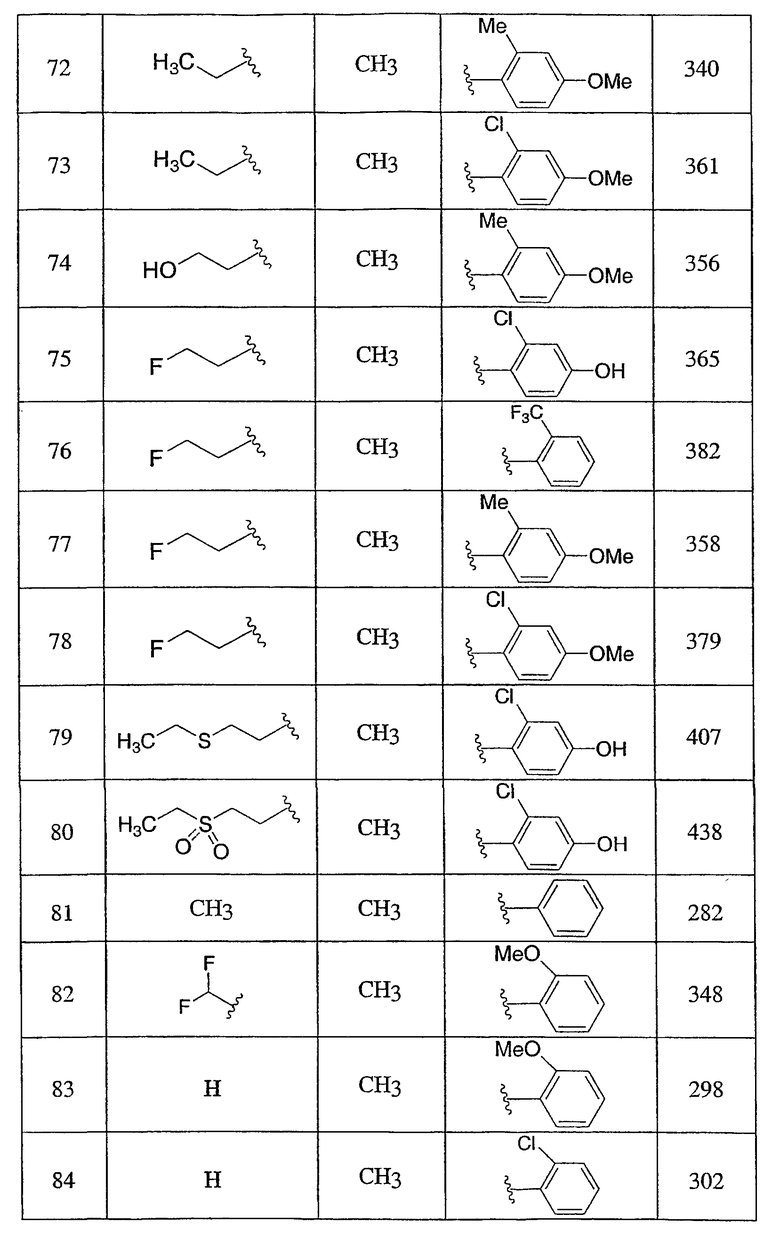
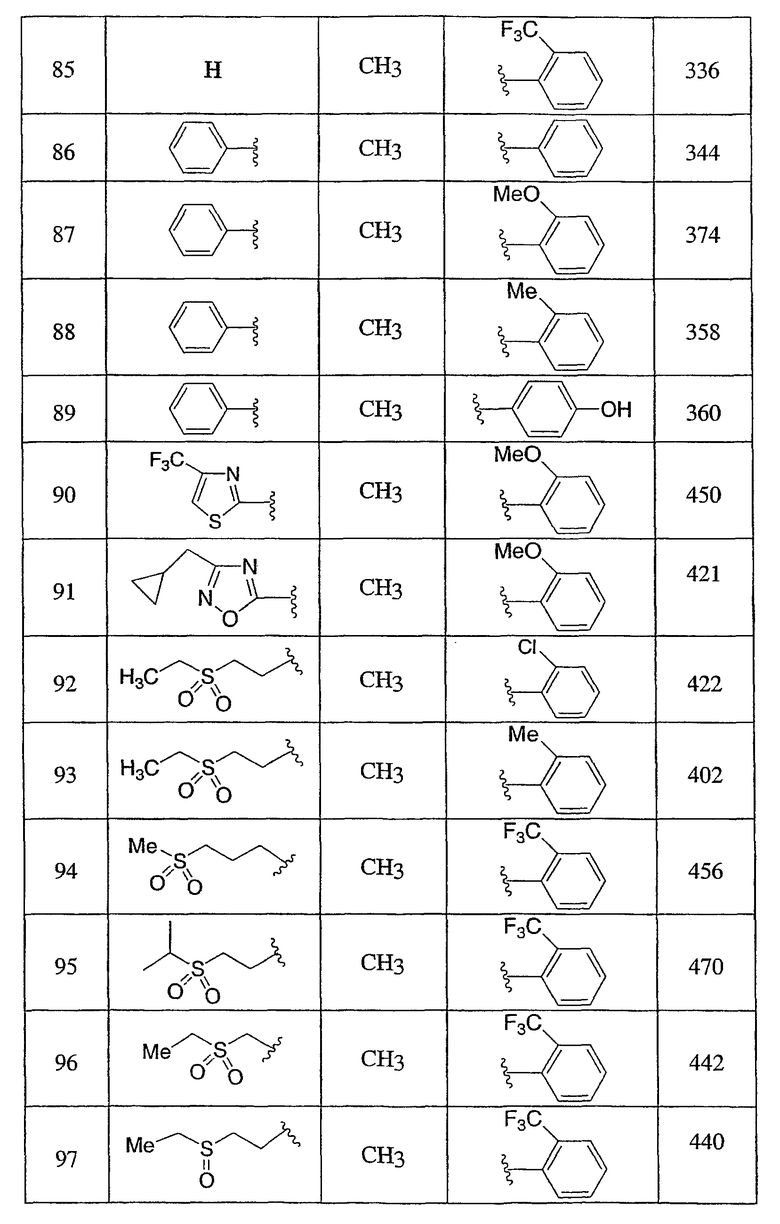
Пример фармацевтической композиции
В качестве частного воплощения пероральной композиции.
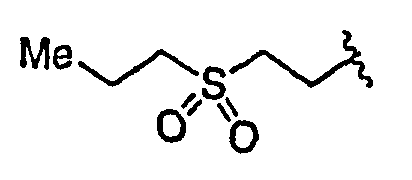
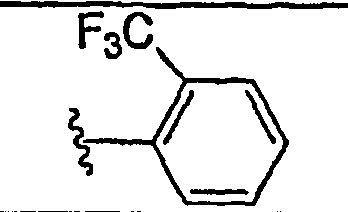
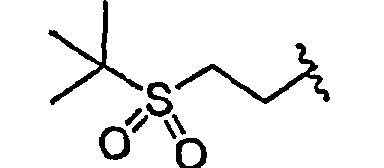
Следуя методикам, сходным с теми, что описаны выше, были получены соединения 103-654 формулы I данного изобретения и исследованы в соответствии с методикой приведенной выше на страницах 29-33 активности. В частности, для всех соединений представлена ингибирующая концентрация (IC50), выраженная в наномолях; для некоторых приведен процент превращения 3H-кортизона в 3H-кортизол.
В нижеприведенных таблицах представлены конкретные соединения с различными значениями групп, полностью подтверждающие наличие у всех изученных соединений данного изобретения свойств ингибиторов фермента 11-бета-гидроксистероиддегидрогеназы 1-го типа, ГСД-1 (hHSDl).
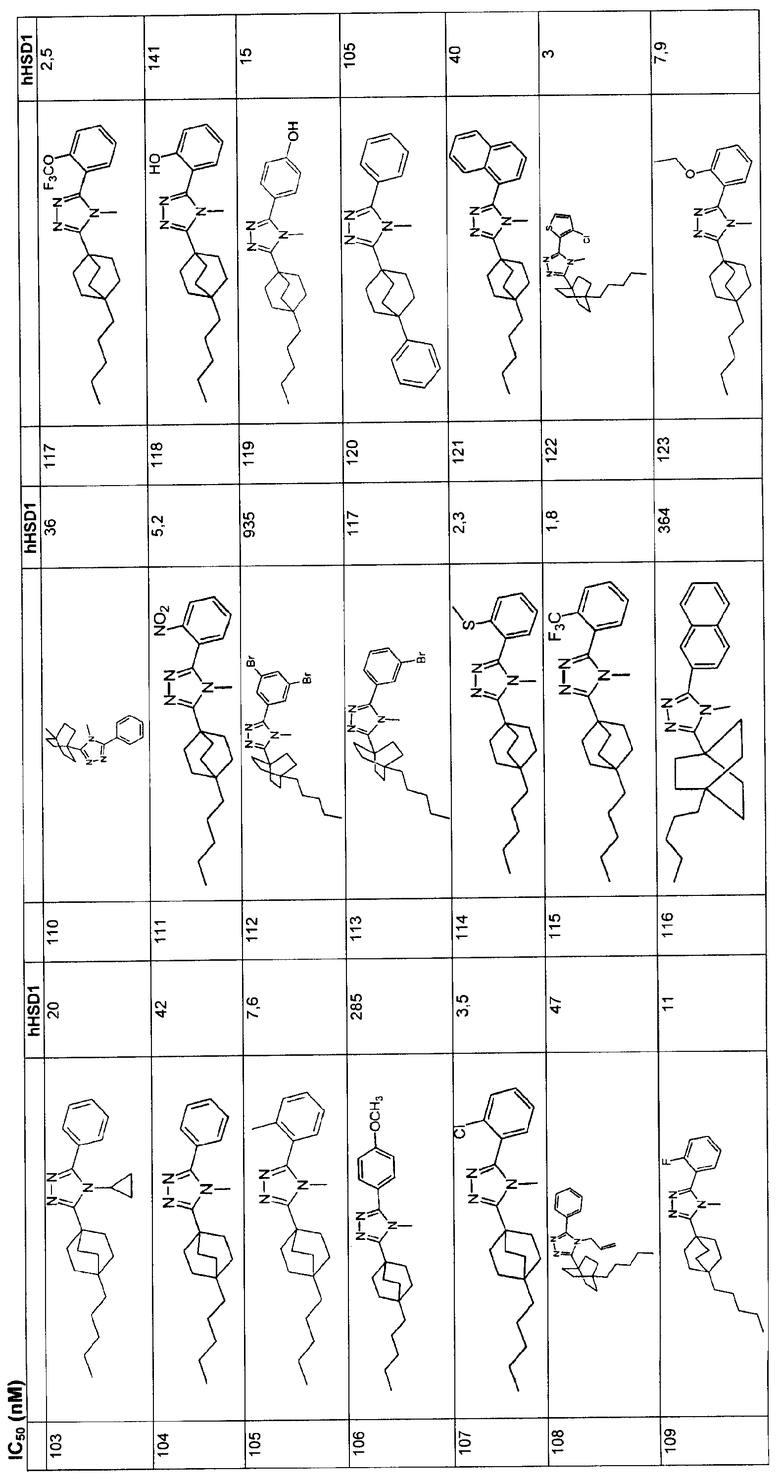
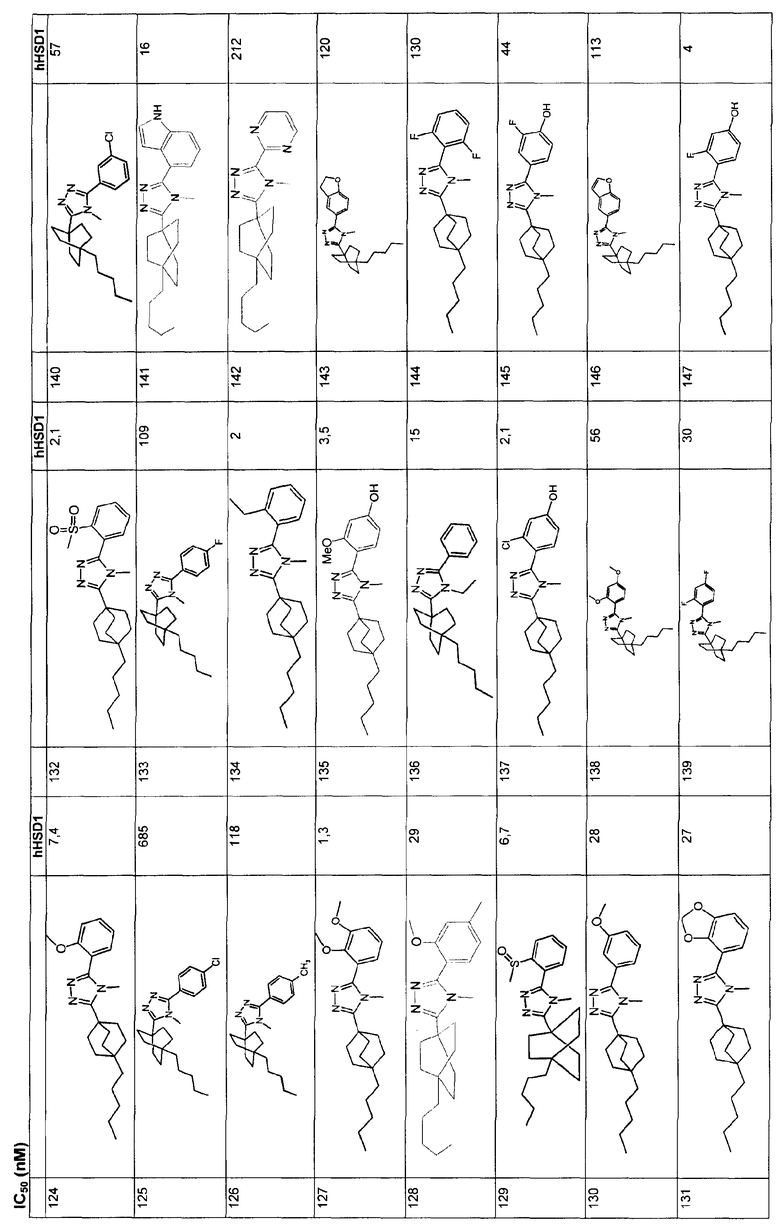
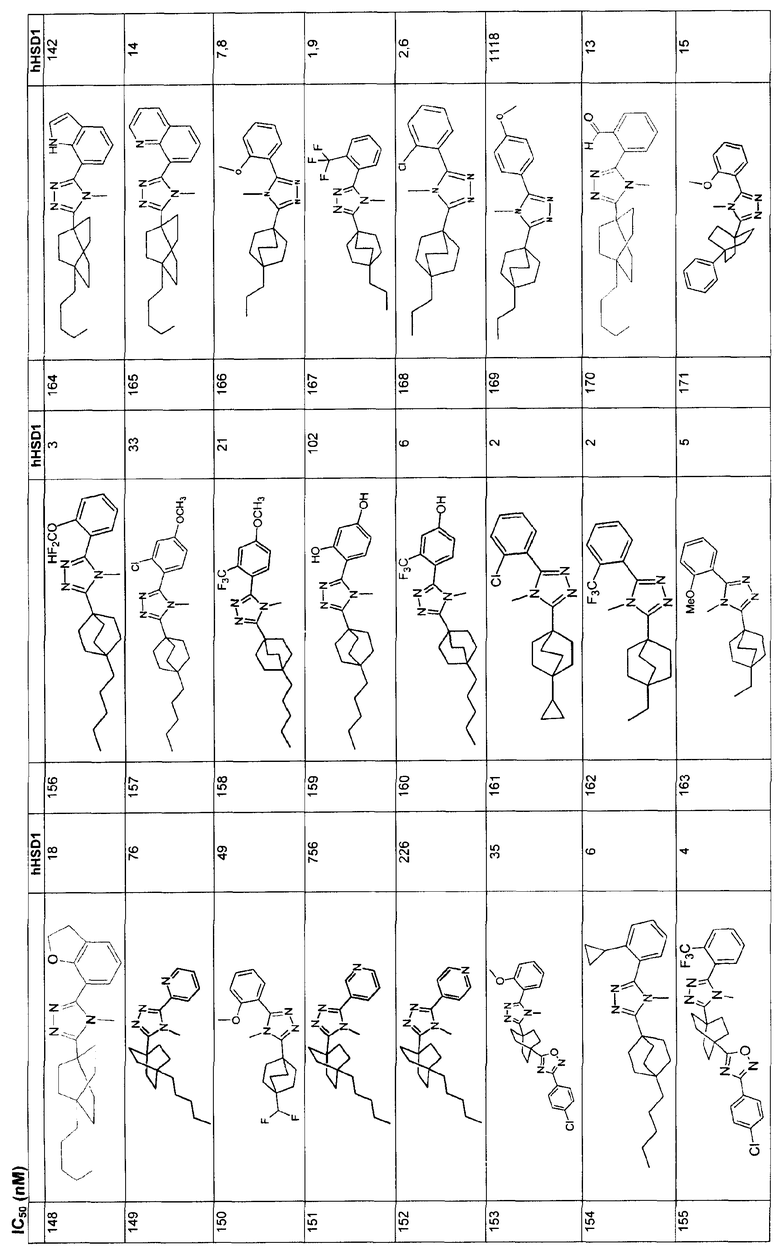
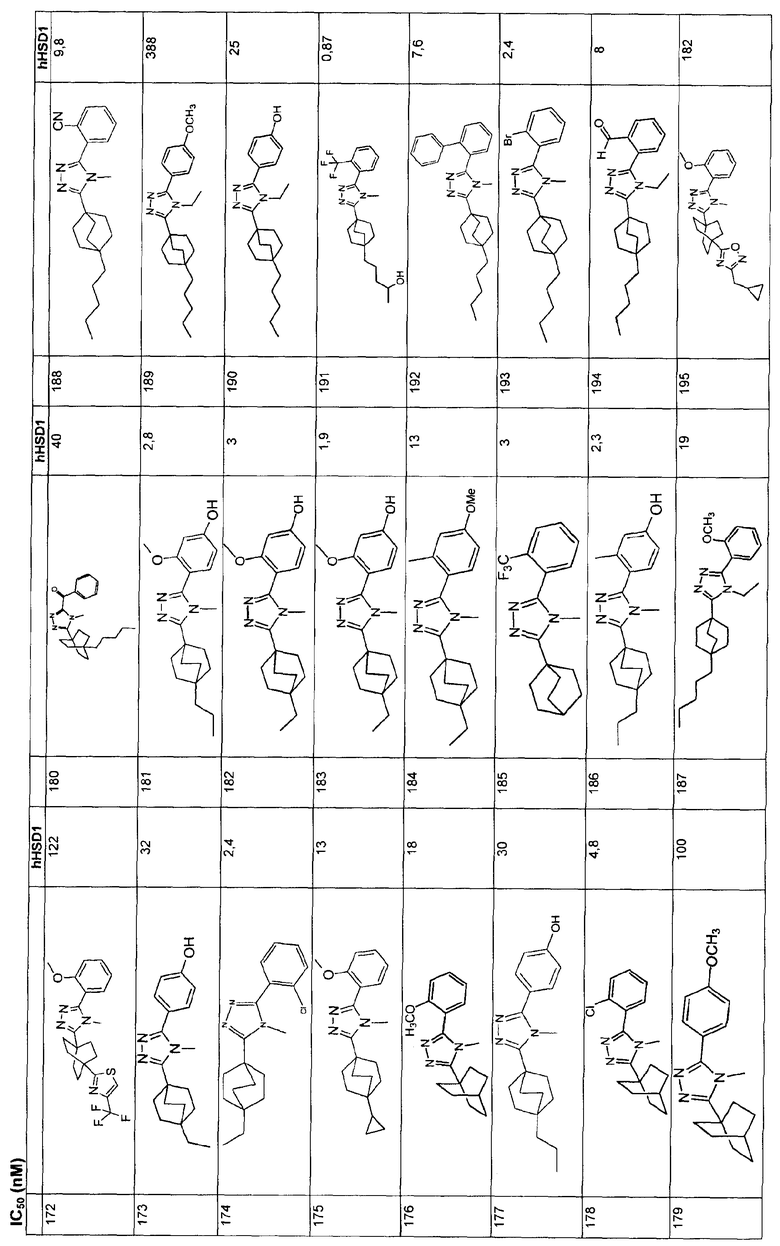
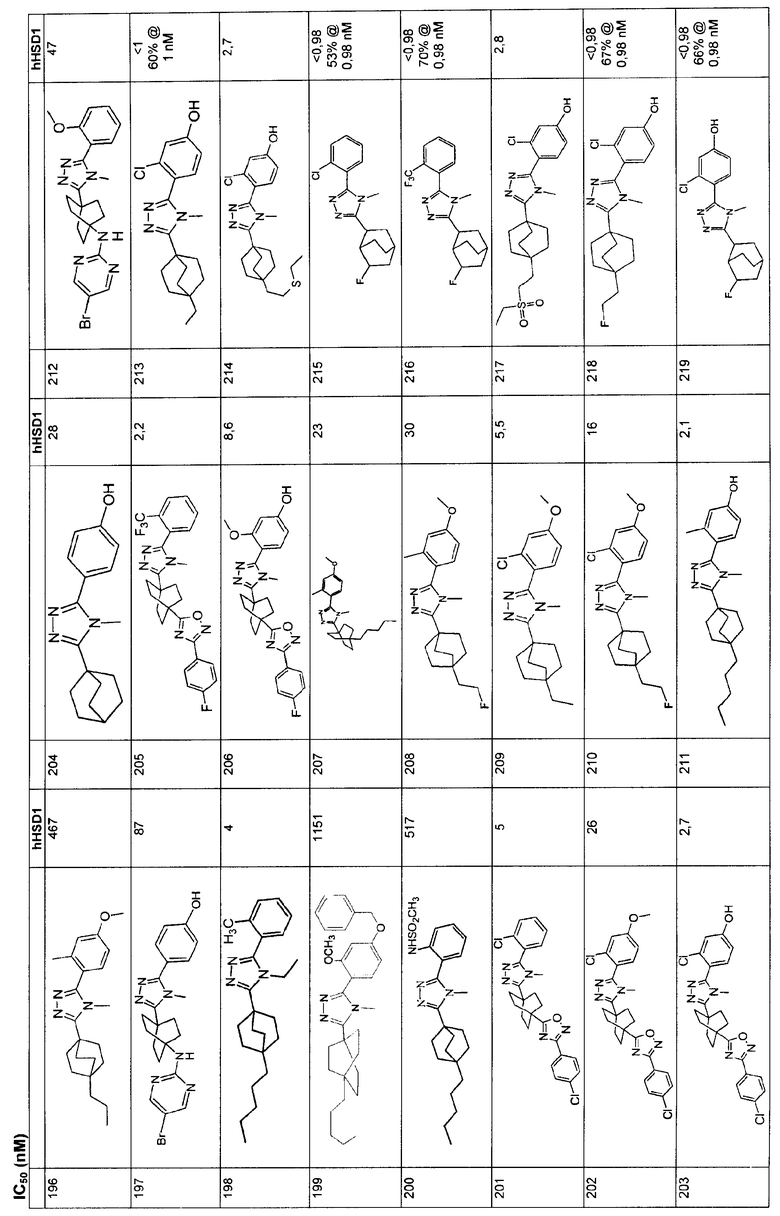
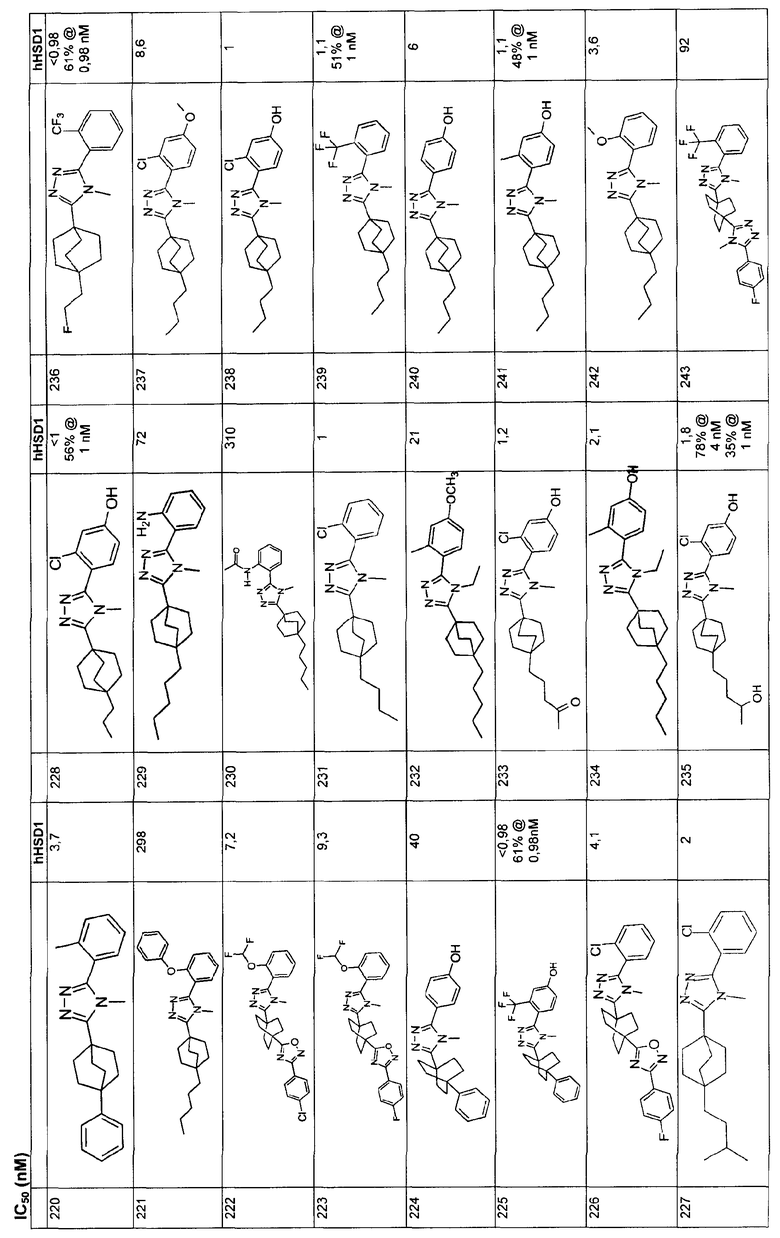
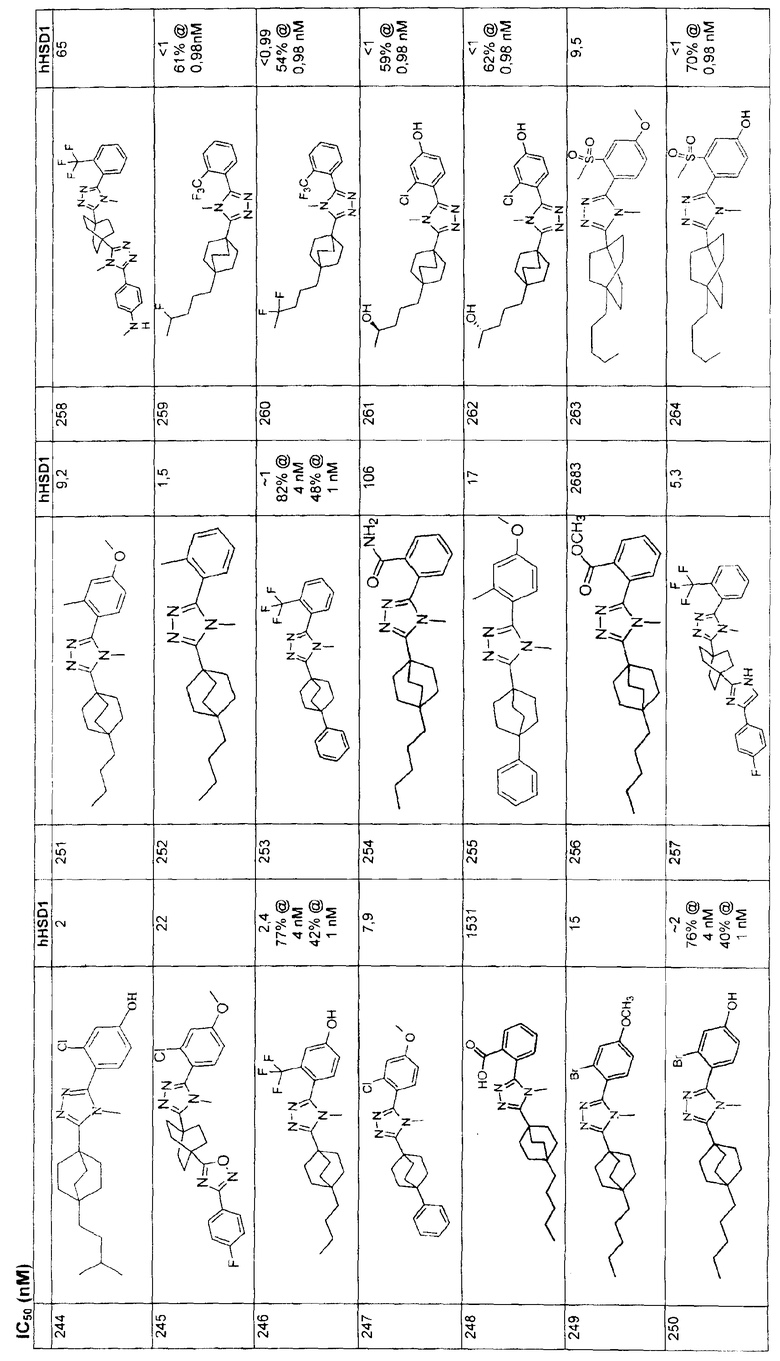
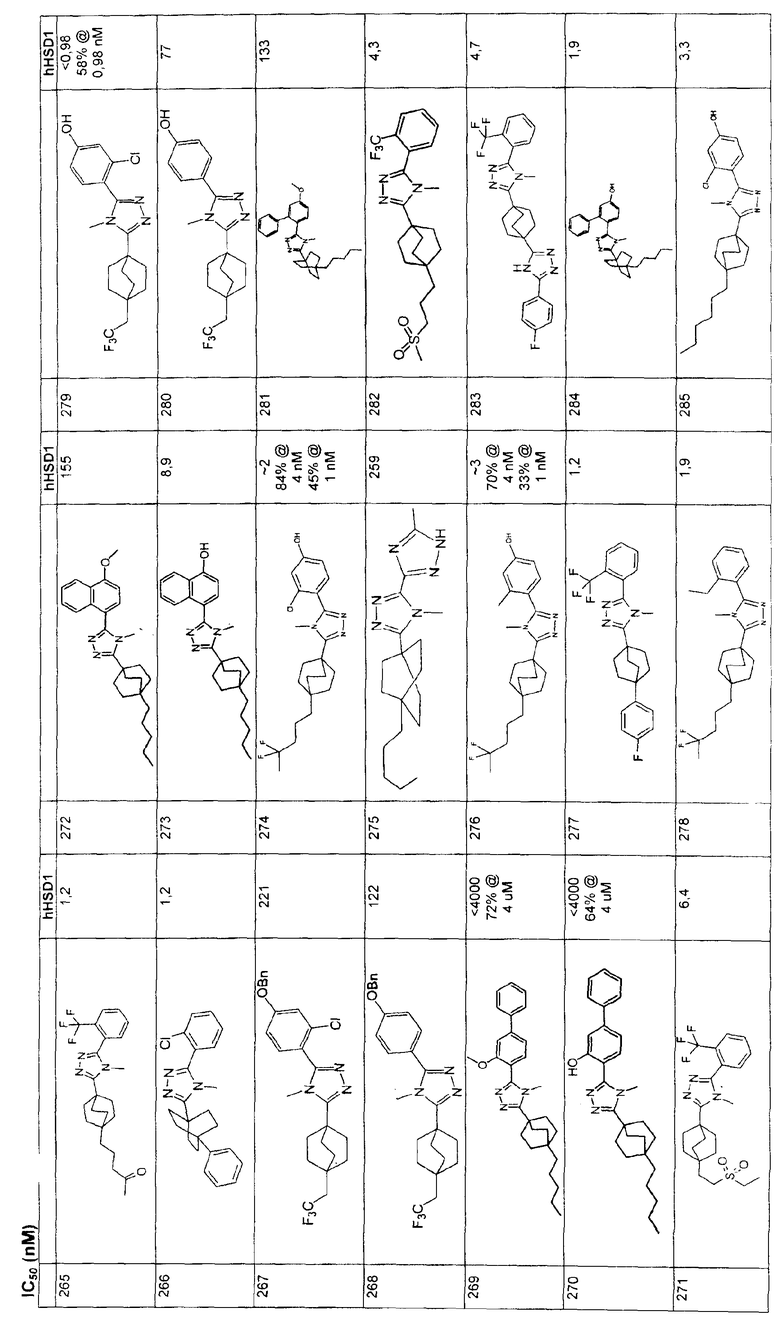
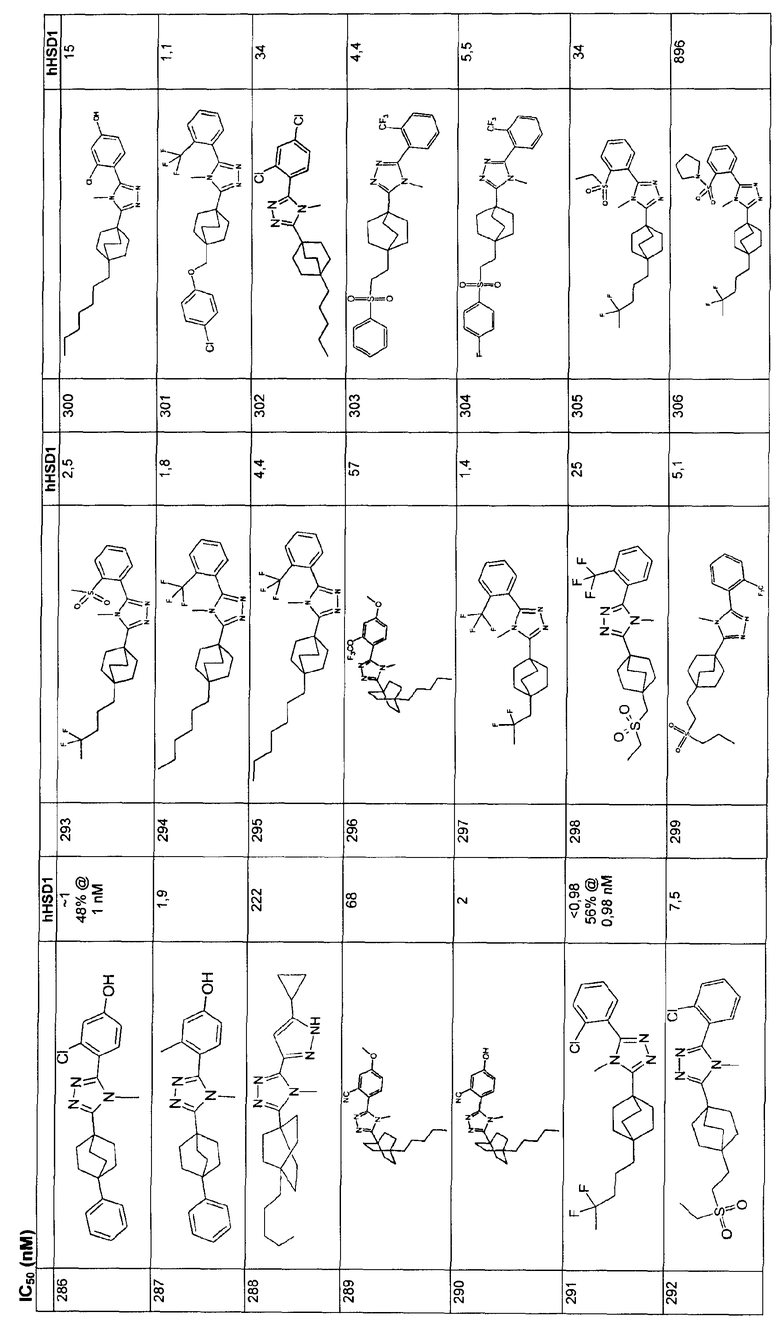
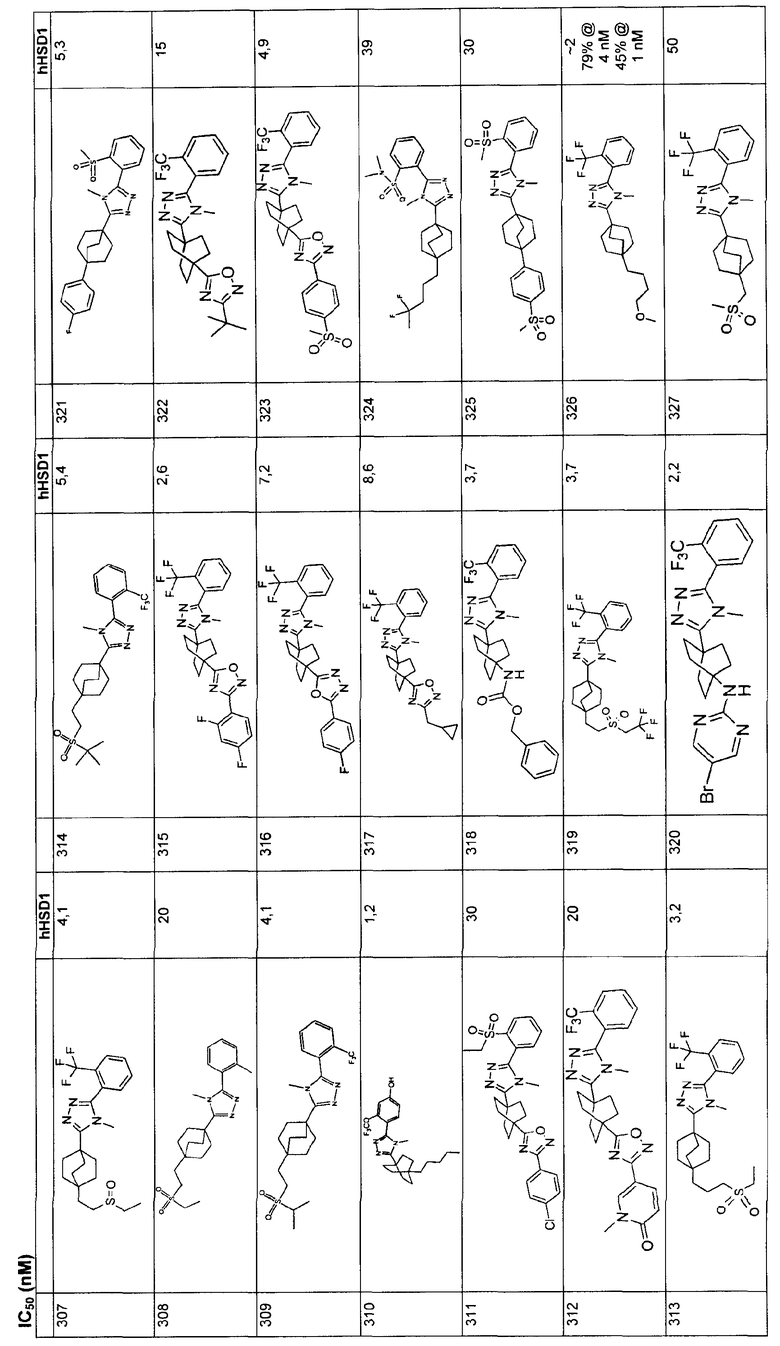
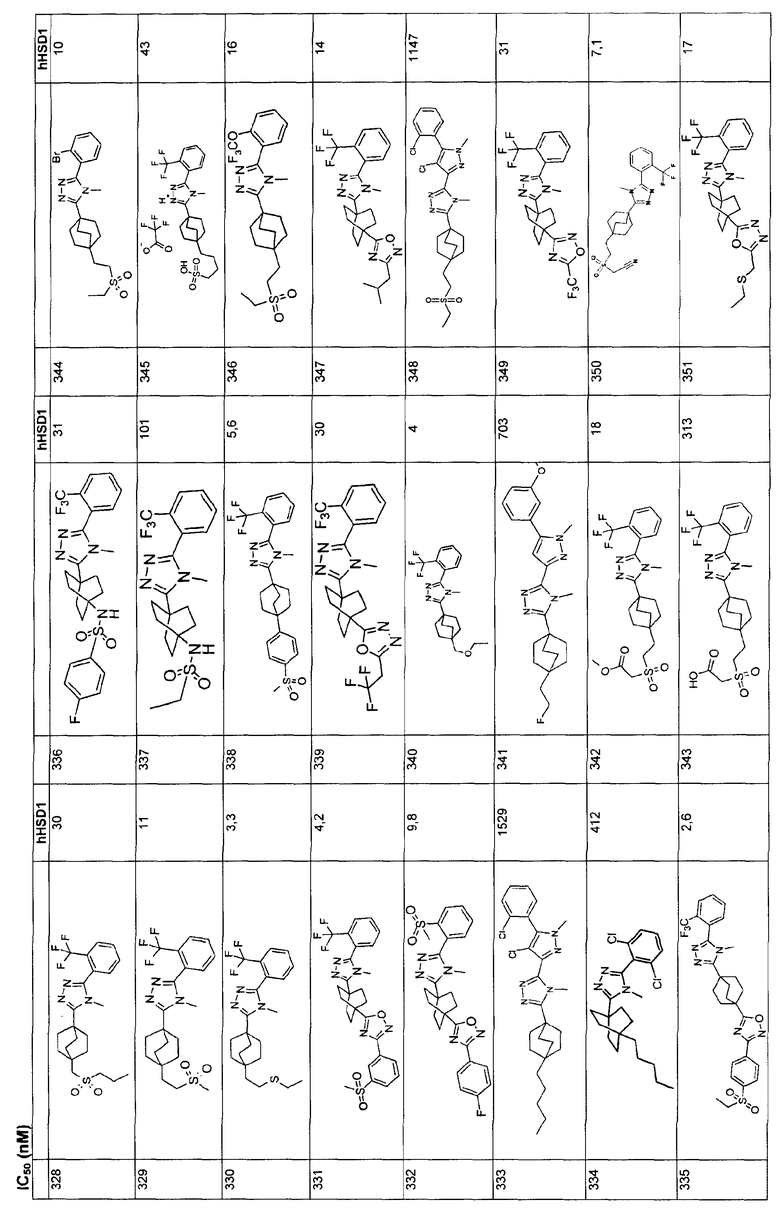
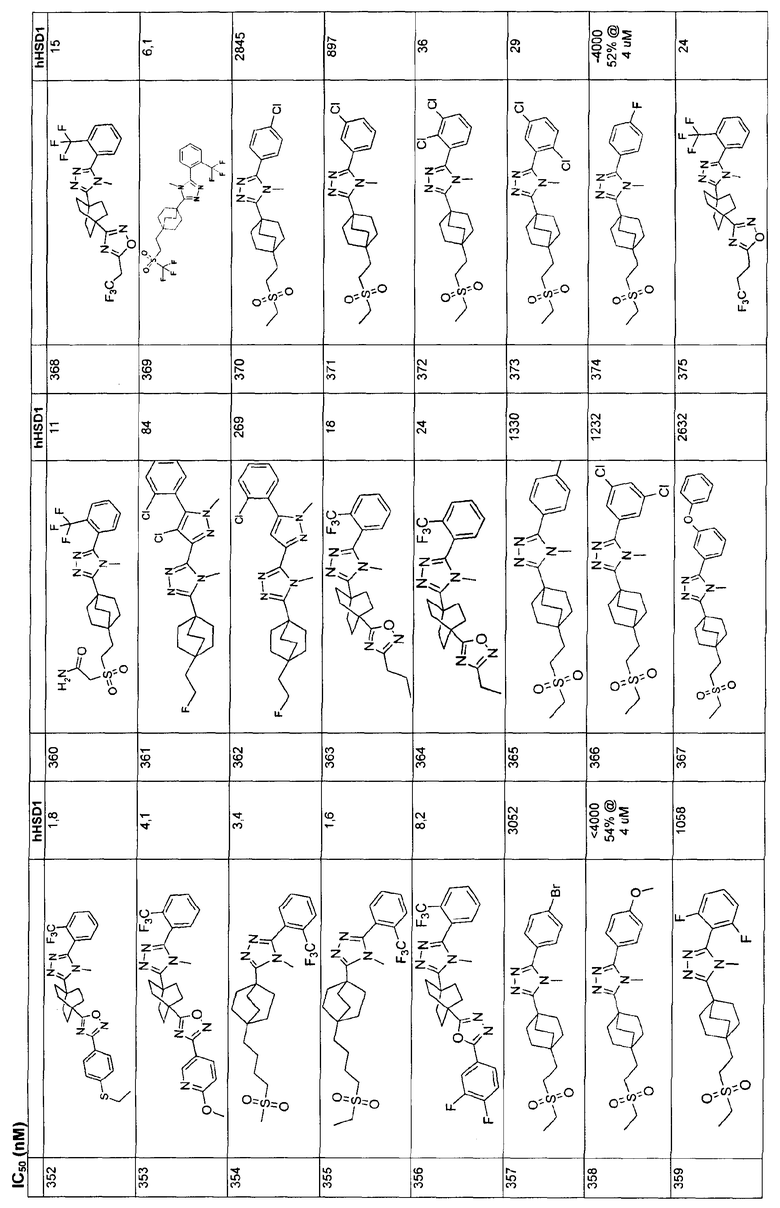
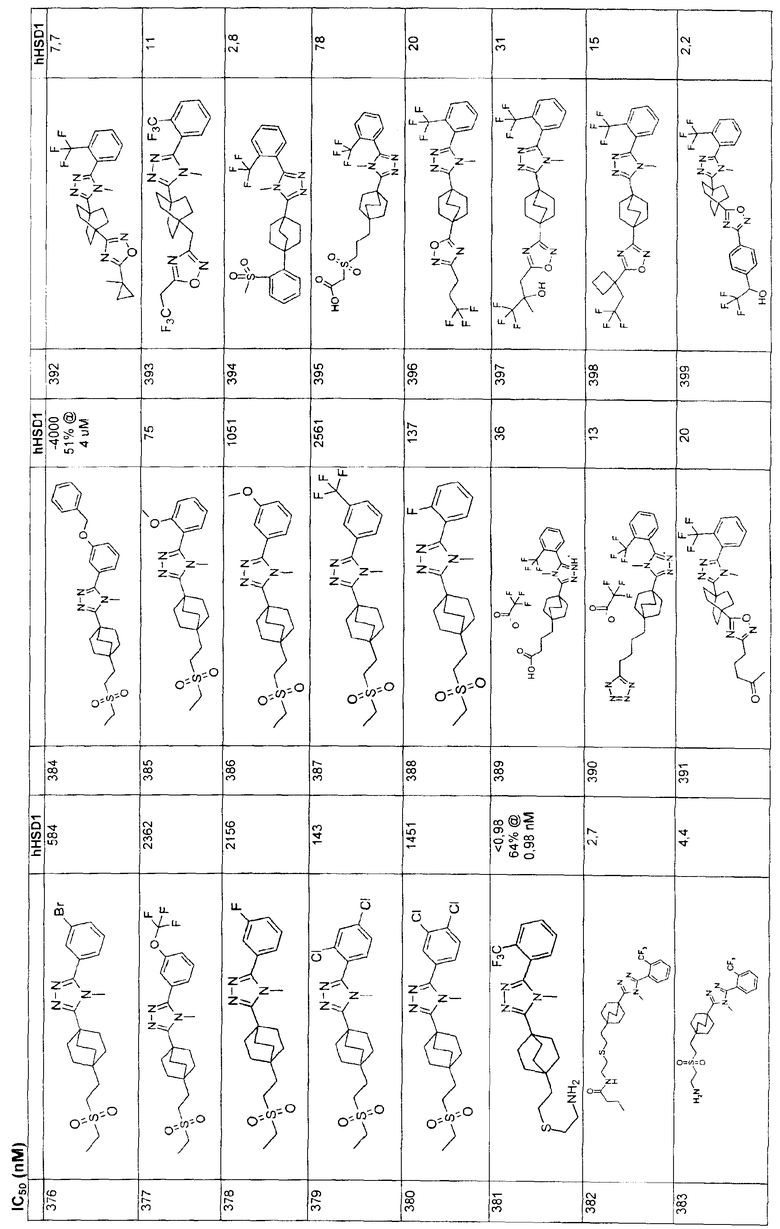
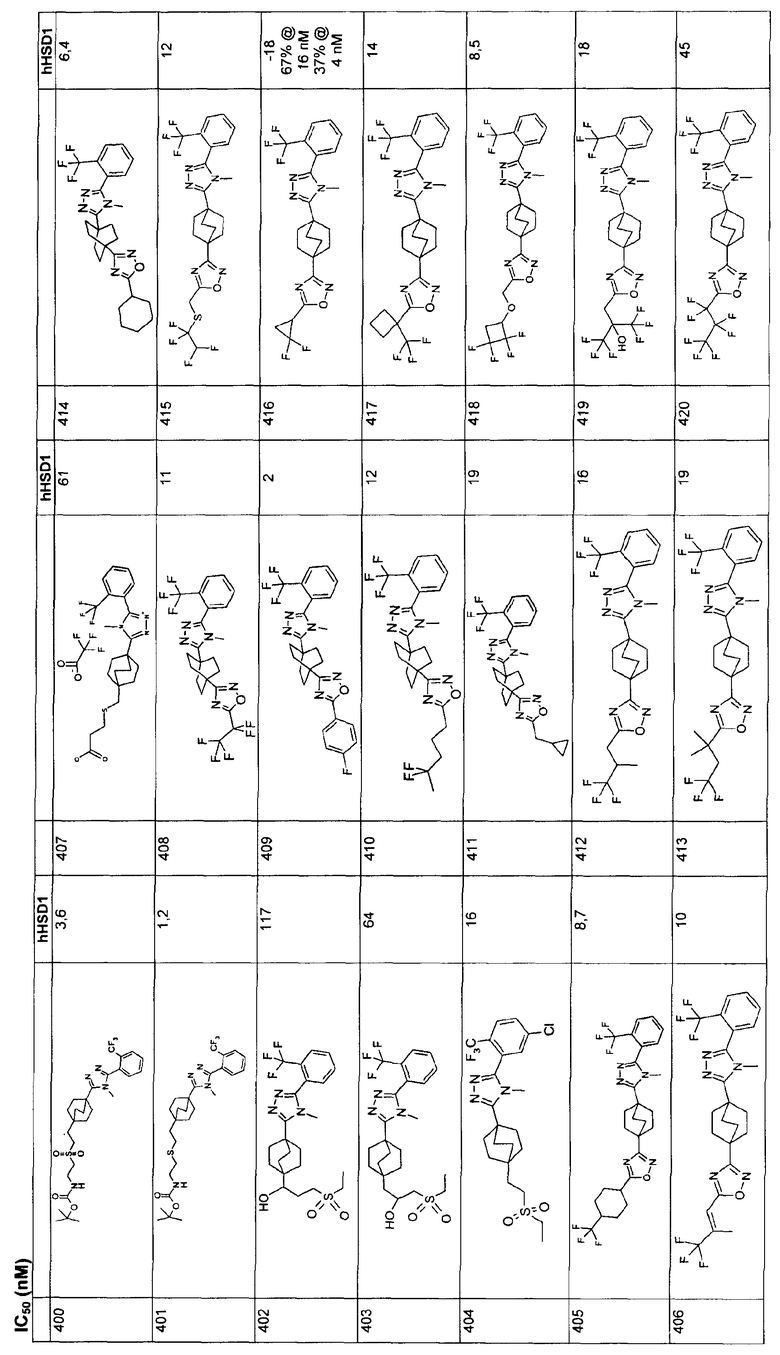
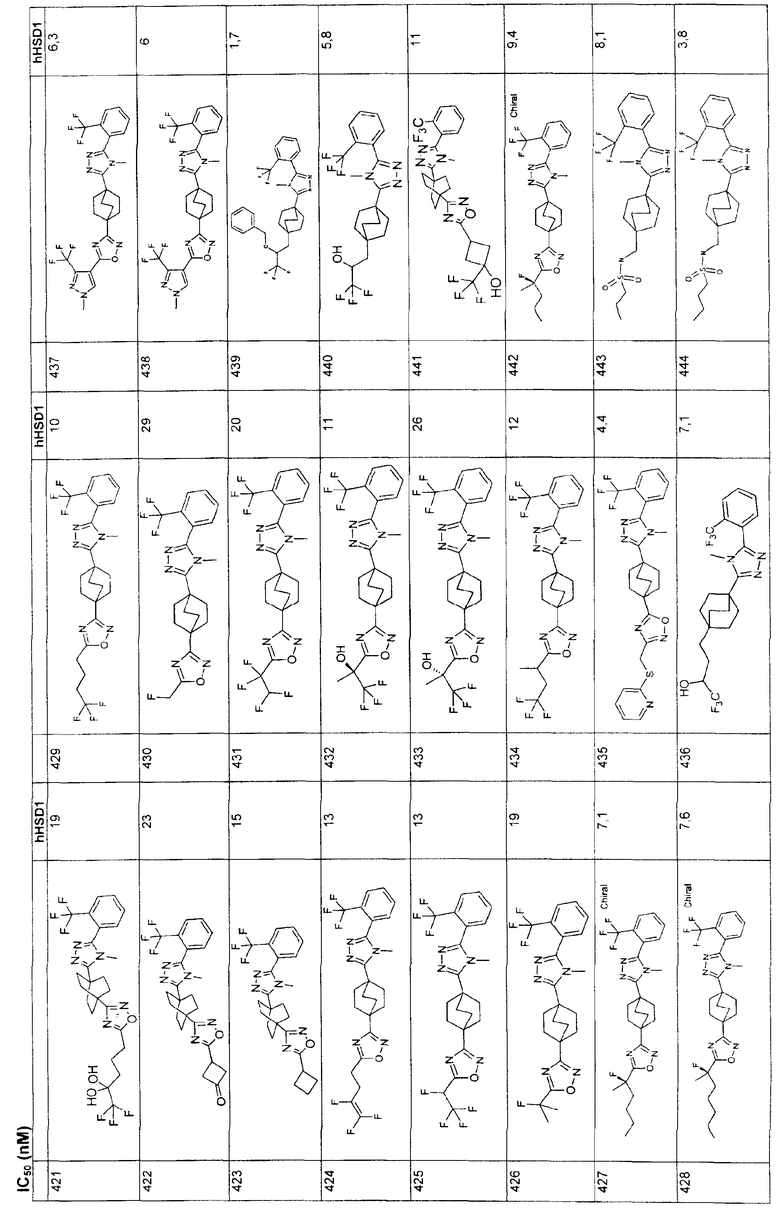
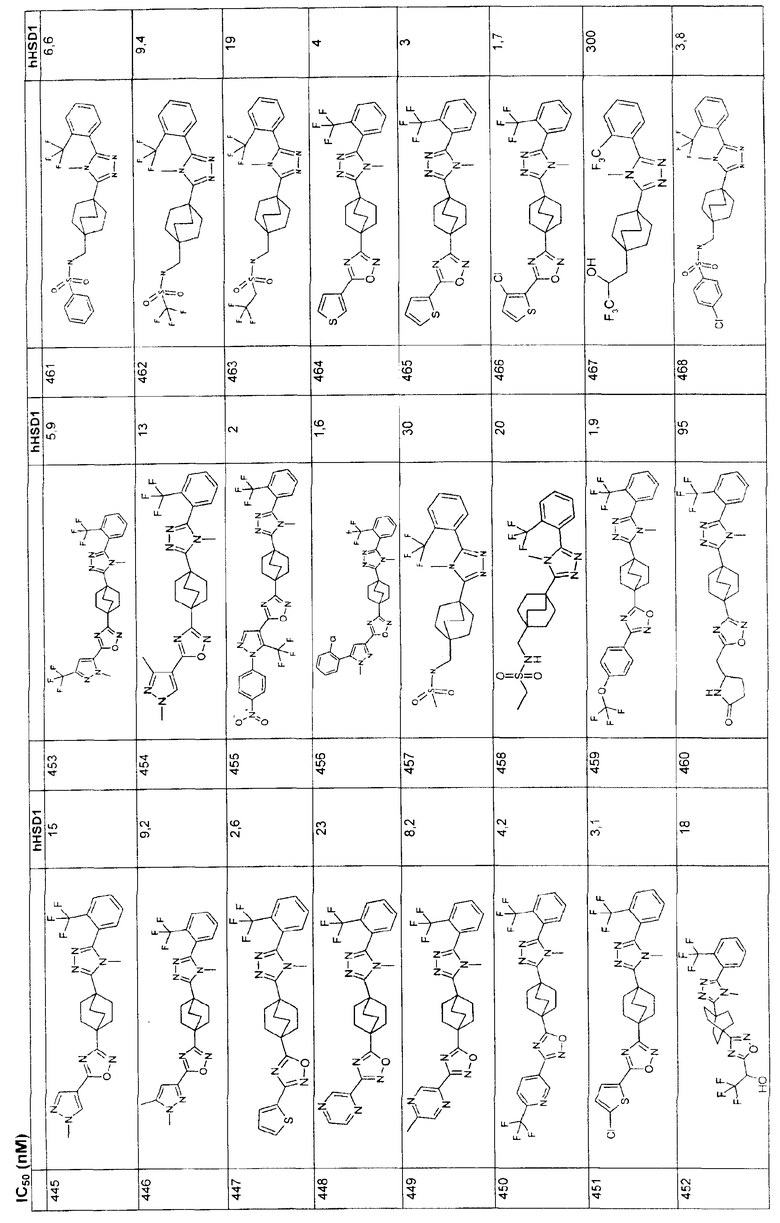
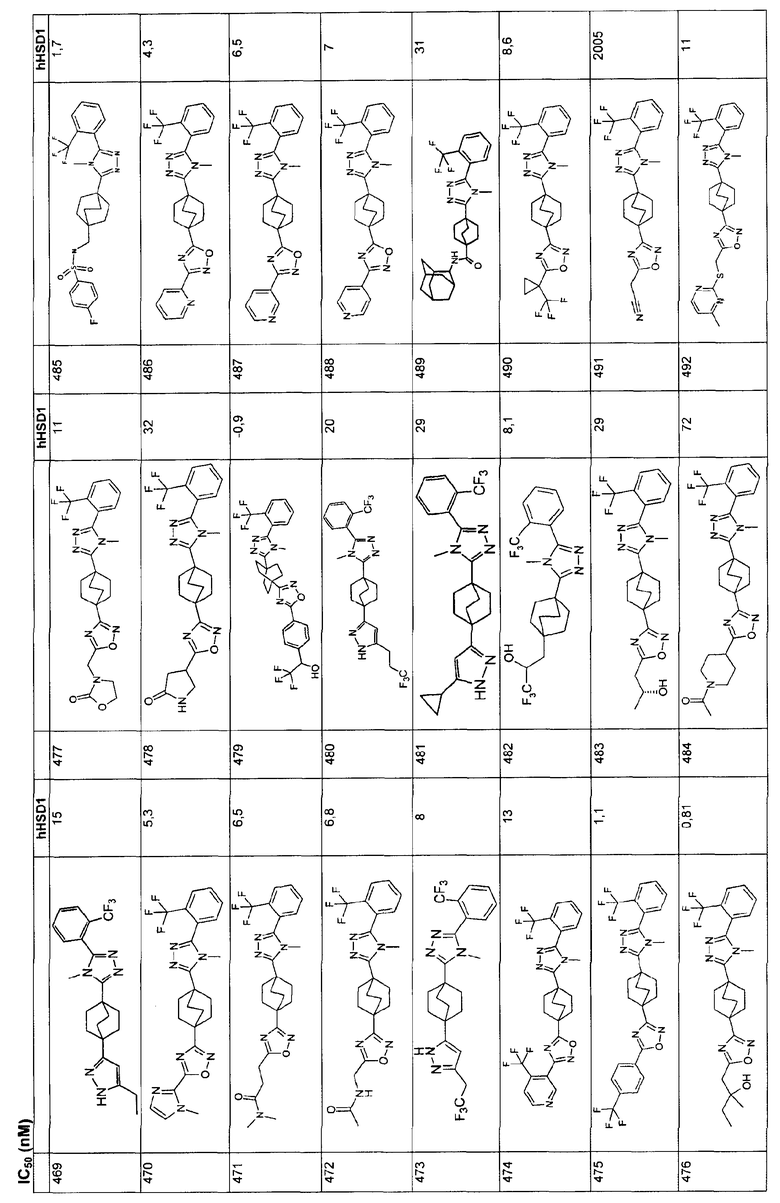
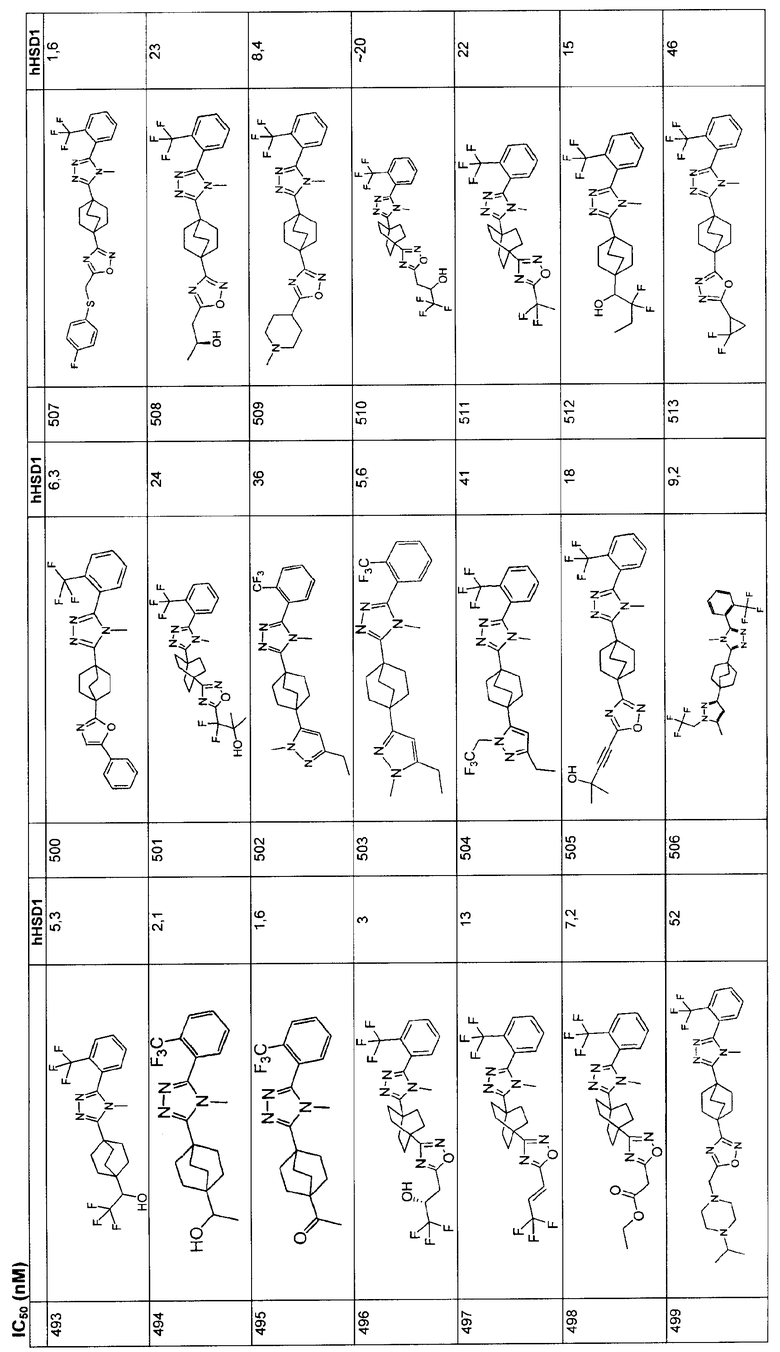
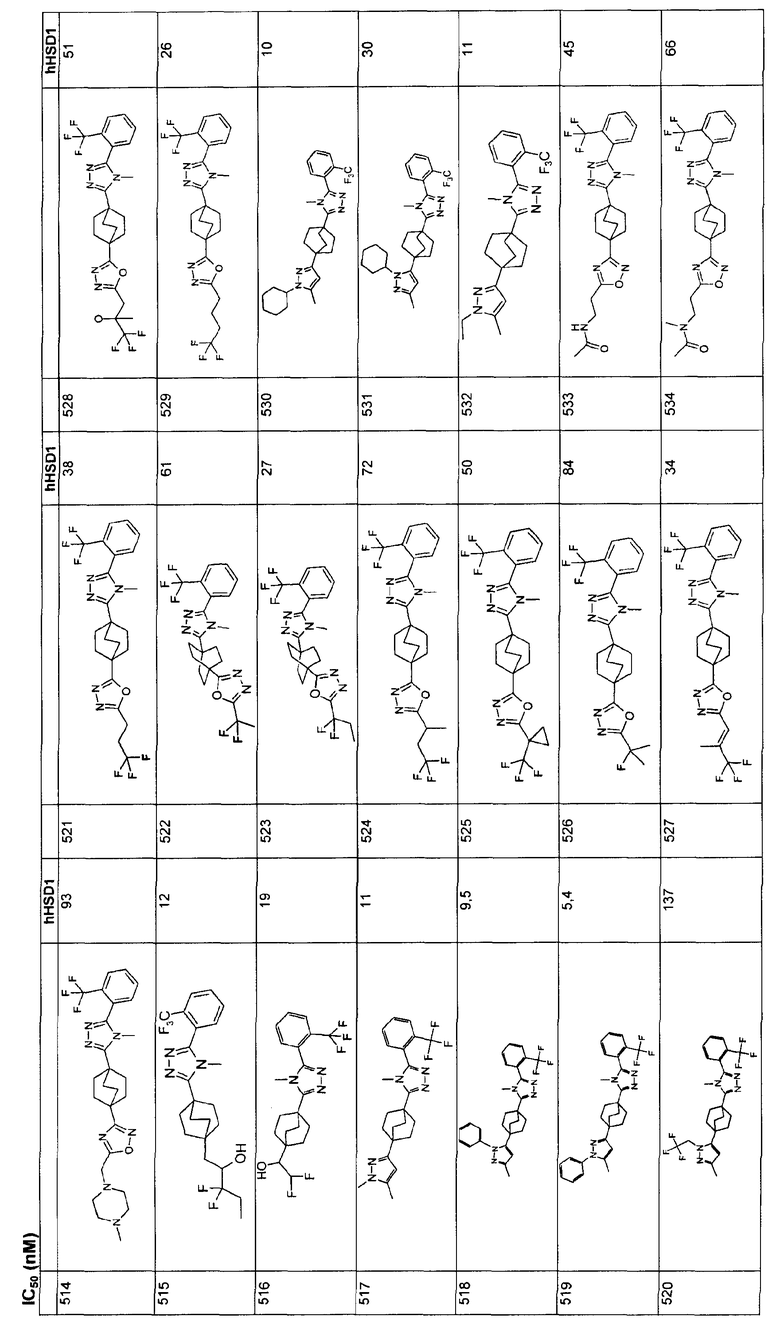
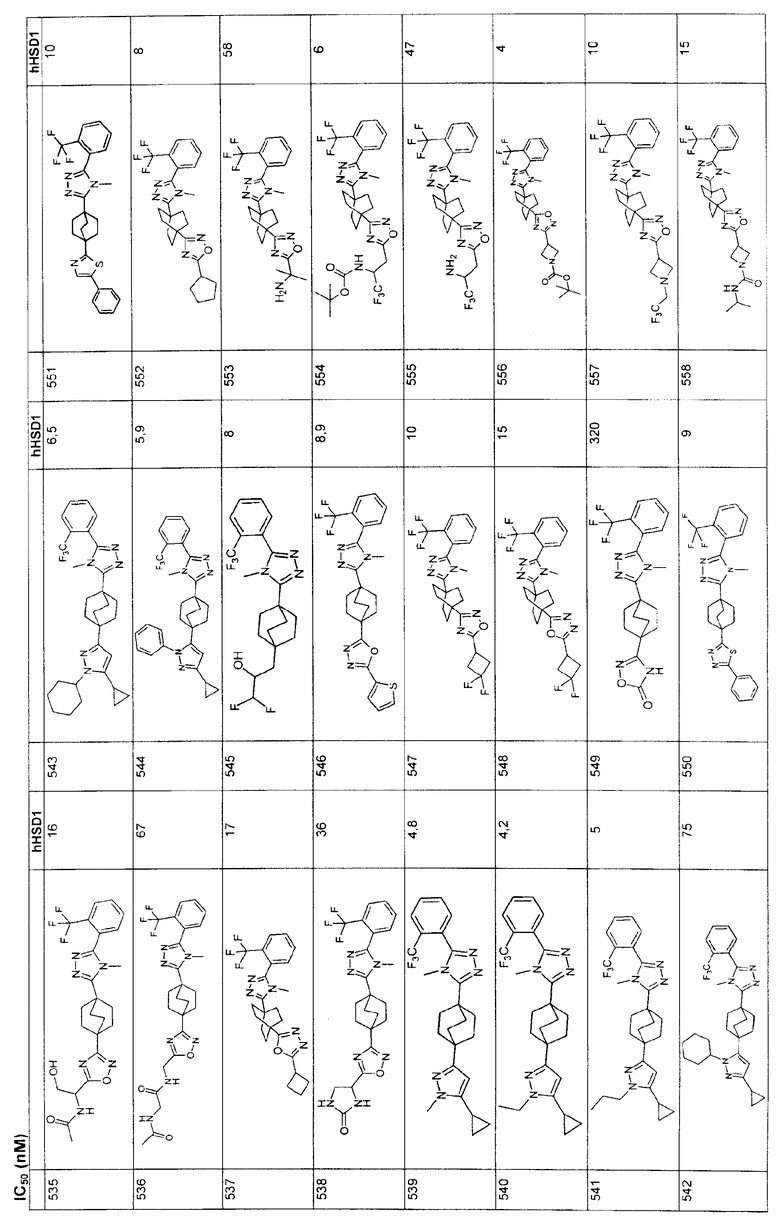
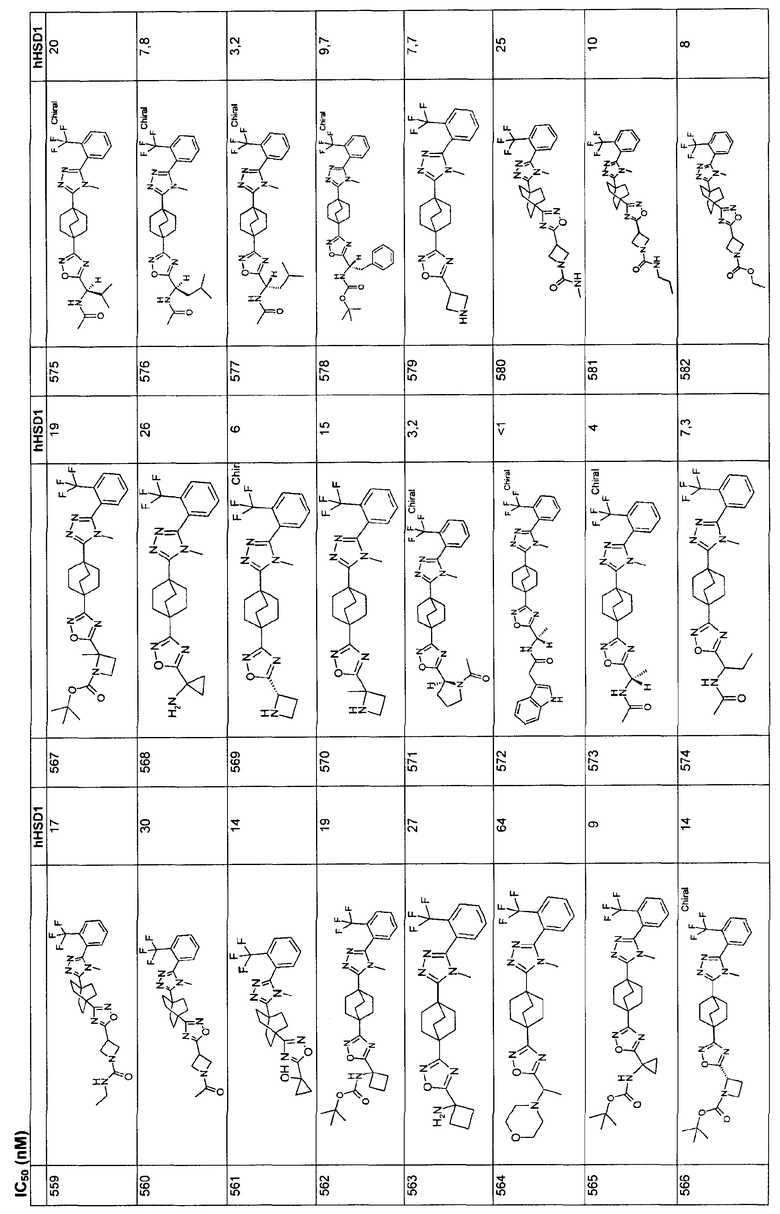
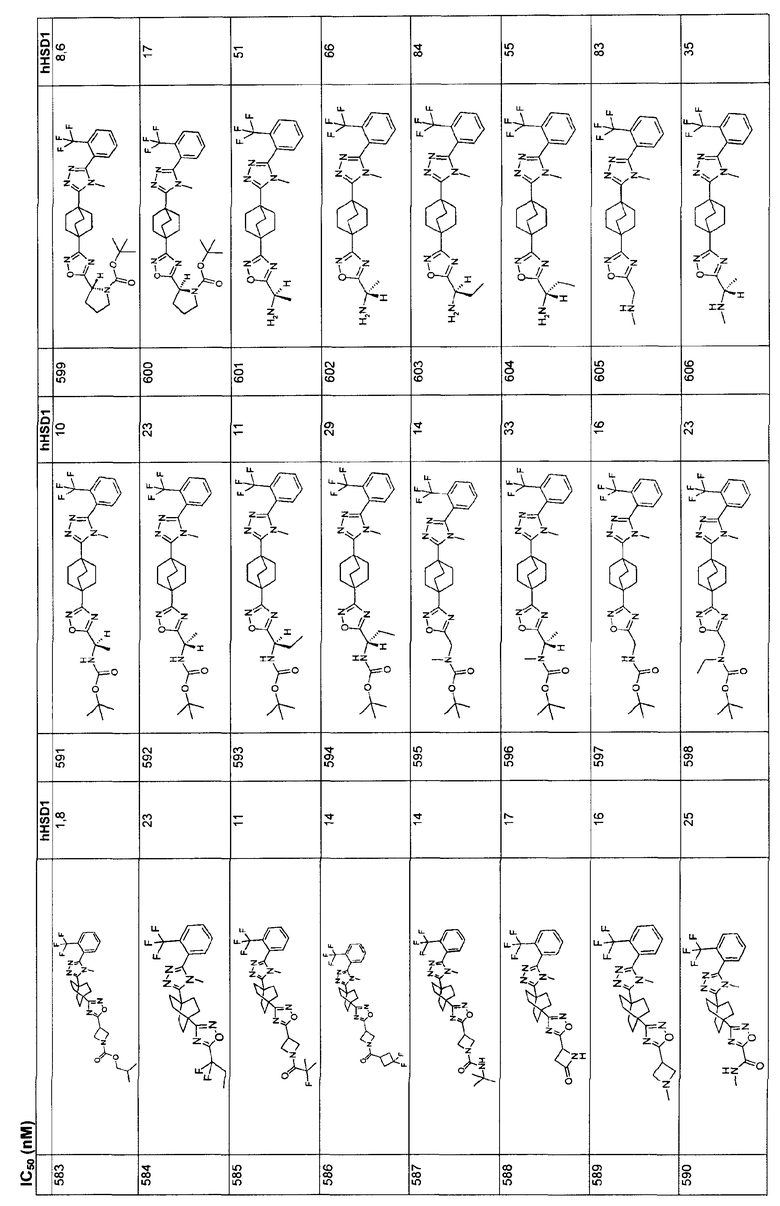
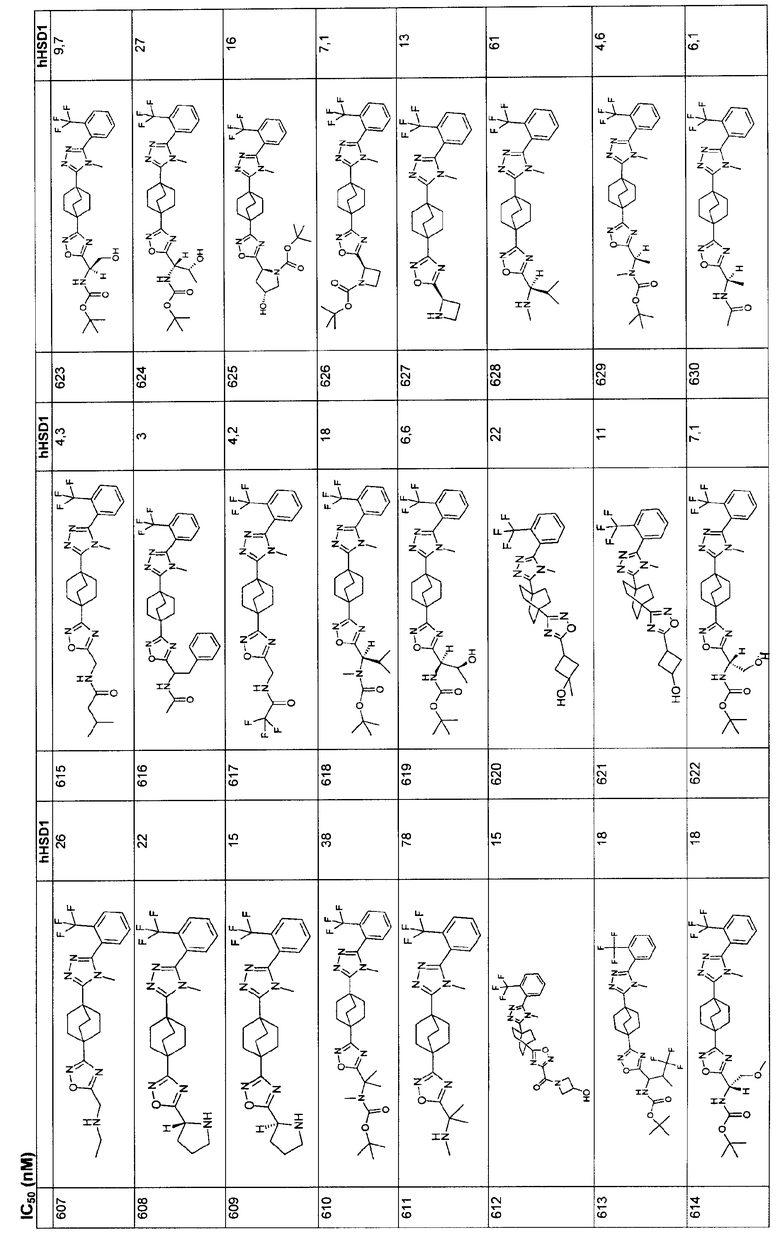
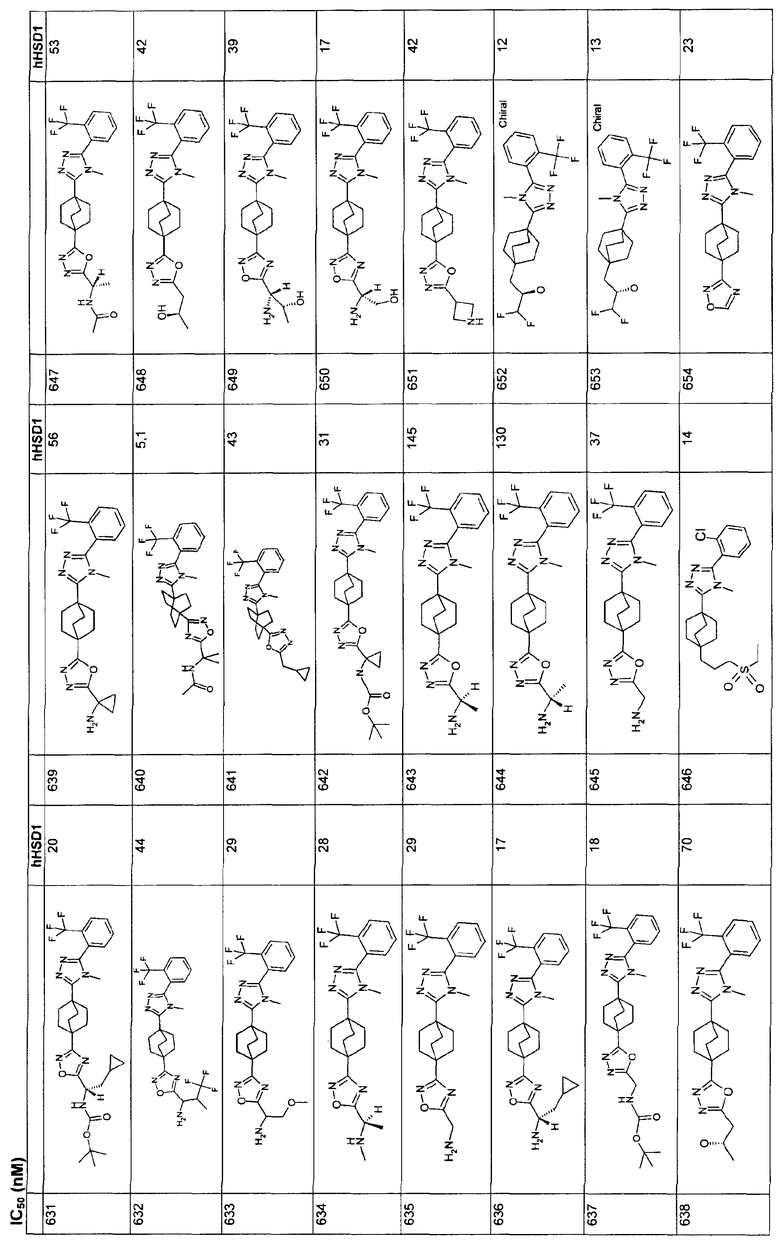
Пример фармацевтической композиции:
В качестве частного воплощения пероральной композиции соединения данного изобретения 50 мг любого из соединений примеров 1-8 формуют с достаточным количеством тонко измельченной лактозы с получением общего количества, равного от 580 до 590 мг, для заполнения твердой желатиновой капсулы 0-размера.
Несмотря на то, что данное изобретение описано и проиллюстрировано со ссылкой на его частные воплощения, специалисты в данной области поймут, что, не отходя от духа и объема данного изобретения, в нем могут быть сделаны изменения, модификации и замены. Например, эффективные дозировки, отличные от установленных выше, могут быть применены вследствие вариаций в реакции человека, которого лечат от определенного состояния. Более того, наблюдаемый фармакологический ответ может варьировать в соответствии с и зависит от выбранного активного соединения или от, если они присутствуют, фармацевтических носителей, так же как от типа композиции и режима введения, и такие ожидаемые вариации или различия в результатах ожидаются в соответствии с объектами и практическим воплощением настоящего изобретения. Следует, таким образом, ограничить данное изобретение только объемом следующей формулы изобретения и интерпретировать такую формулу изобретения настолько широко, насколько возможно.
| название | год | авторы | номер документа |
|---|---|---|---|
| СОЕДИНЕНИЯ И КОМПОЗИЦИИ В КАЧЕСТВЕ ИНГИБИТОРОВ ЭНДОСОМАЛЬНЫХ TOLL-ПОДОБНЫХ РЕЦЕПТОРОВ | 2017 |
|
RU2759678C2 |
| ТРИЦИКЛИЧЕСКИЕ СОЕДИНЕНИЯ, СОДЕРЖАЩАЯ ИХ ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ И ИХ ПРИМЕНЕНИЕ ДЛЯ ЛЕЧЕНИЯ ИММУНОЛОГИЧЕСКИХ И ОНКОЛОГИЧЕСКИХ СОСТОЯНИЙ | 2009 |
|
RU2545023C9 |
| СОЕДИНЕНИЕ, ОБЛАДАЮЩЕЕ ЦИКЛИЧЕСКОЙ СТРУКТУРОЙ | 2018 |
|
RU2795119C2 |
| МОДУЛЯТОРЫ ИНТЕГРИРОВАННОГО СИГНАЛЬНОГО ПУТИ СТРЕССА | 2017 |
|
RU2769327C2 |
| ЦИКЛИЧЕСКИЕ ИНГИБИТОРЫ 11БЕТА-ГИДРОКСИСТЕРОИД-ДЕГИДРОГЕНАЗЫ 1 | 2009 |
|
RU2539979C2 |
| СПОСОБ ПОЛУЧЕНИЯ НЕКОТОРЫХ 2-(ПИРИДИН-3-ИЛ)ТИАЗОЛОВ | 2013 |
|
RU2649000C2 |
| АЛКАЛОИДНЫЙ ЭФИР И КАРБАМАТНЫЕ ПРОИЗВОДНЫЕ И ИХ МЕДИЦИНСКИЕ КОМПОЗИЦИИ | 2012 |
|
RU2611627C2 |
| СПОСОБ ПОЛУЧЕНИЯ ЦИКЛИЧЕСКИХ ДИКЕТОНОВ | 2003 |
|
RU2316544C2 |
| СПОСОБЫ ПОЛУЧЕНИЯ НЕКОТОРЫХ 2-(ПИРИДИН-3-ИЛ)ТИАЗОЛОВ | 2013 |
|
RU2647851C2 |
| ИНГИБИТОРЫ ФЕРМЕНТА ДИАЦИЛГЛИЦЕРИН О-АЦИЛТРАНСФЕРАЗЫ ТИПА 1 | 2008 |
|
RU2497816C2 |
Описываются новые производные триазола общей формулы I
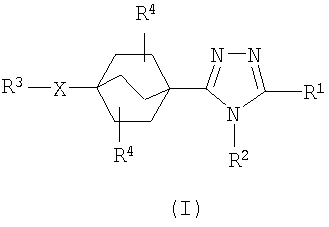
или их фармацевтически приемлемые соли, где R4 - водород;
Х выбирают из группы, состоящей из одинарной связи, NH- и групп:
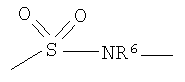 ,
,  и
и  ,
,
значения радикалов R1-R3 приведены в описании, фармацевтическая композиция, их содержащая, и применение новых соединений для получения лекарственного средства для лечения гипергликемии, инсулинорезистентности, диабета 2-го типа, нарушений жирового обмена, ожирения, атеросклероза и метаболического синдрома. 4 н. и 22 з.п. ф-лы, 2 табл.
1. Соединение структурной формулы I
или его фармацевтически приемлемая соль, где R4 - водород;
Х выбирают из группы, состоящей из одинарной связи, NH- и групп:
, и ,
в которых R6 означает водород;
R1 выбирают из группы, состоящей из фенилкарбонила или арила, где указанный в определении R1 арил представляет собой:
нафтил, возможно замещенный ОН или С1-3 алкоксигруппой, фенил, возможно замещенный от одного до трех заместителями, независимо выбранными из R5,
где R5 независимо выбирают из группы, состоящей из
формила,
С1-6алкила,
C1-3алкокси,
NO2,
NH2,
галогена,
циано,
CF3-
СН2CF3,
OCF2H,
OR7, где R7 означает водород,
OCF3,
фенила,
фенилокси,
бензилокси,
С3-7 циклоалкила,
CO2R7, где R7 означает водород или С1-6алкил,
S(O)pR6, где p равно 0, 1 или 2, а R6 означает С1-8алкил, диС1-6алкиламиногруппу, или пиразолил,
C(O)N(R7)2, где R7 - одинаковые и означают водород,
NR6C(O)R6, где R6 означает водород, R6' означает водород, C1-6алкил,
NR7SO2R6, где R7 означает водород, а R6 - C1-6 алкил,
SO2N(R7)2, где R7 означает водород или C1-6алкил,;
либо R1 выбирают из гетероарила, где указанный гетероарил представляет собой бензофуранил, бензопиррол, индолил, хинолинил, пиримидинил, пиридинил, 1,3-бензодиоксоланил или тиофенил, пиразолил и триазолил, возможно замещенный заместителями от одного до трех, независимо выбранными из водорода, галогена, С1-3алкила, С3-7 циклоалкила, фенила, возможно замещенного галогеном или С1-3 алкокси;
R2 выбирают из группы, состоящей из
водорода,
С1-8алкила,
С2-6алкенила и
С3-6циклоалкила;
R3 выбирают из группы, состоящей из
водорода,
С1-10алкила,
С2-10алкенила,
С3-6циклоалкила и
где указанные для R3 алкил, алкенил и циклоалкил незамещены или замещены от одной до пяти групп, независимо выбранных из R8:
оксо,
водорода,
галогена,
OR7, где R7=H или алкил;
CF3;
тетразолил;
(CH2)nCO2R7, где n=0-2, R7 означает водород или C1-6алкил;
(CH2)nNR7SO2R6, где n=0-2, R6 - возможно замещенный атомами галогена С1-6алкил, фенил, возможно замещенный атомами галогена; R7 означает водород или C1-6алкил;
(CH2)nSO2OR7, где R7-H;
(CH2)nS(O)pR6, где n=0-2, p=0, 1 или 2, и R6 представляет собой:
фенил,
фенил, возможно замещенный галогеном,
С1-6алкил;
С1-6алкил, возможно замещенный NH2,
С1-6алкил, возможно замещенный атомами галогена
либо
R3 выбирают из группы, состоящей из
фенила,
гетероарила и гетероциклила;
где указанные для R3 арил, гетероарил и гетероциклил незамещены или замещены от одного до трех заместителями, независимо выбранными из R9, где R9 выбран из:
водорода;
С1-6алкила;
CF3СН2;
С3-7циклоалкила;
арила;
гетероарила, или гетероциклила,
галогена;
OR7, где R7=H или R7=R6;
N(R7)2, где R7 означает С1-6алкил;
(CH2)nNR7SO2R6, где n=0-2, R6 - возможно замещенный C1-6алкил, фенил, R7 означает водород или С1-6алкил;
(CH2)nS(O)pR6, где n=0-2, р=0, 1 или 2, R6 - возможно замещенный галогеном или карбокси; C1-6алкил, возможно замещенный галогеном фенил или пиримидинил;
(CH2)nC(O)N(R7)2,;
(CH2)nNR6C(O)R6 где R6 - С1-6алкил и R6 (СН2)n-гетероарил;
(CH2)nCO2R7, где R7 означает водород или С1-6алкил;
(CH2)nN(R7)2;
(CH2)nNR6CO2R7;
и где указанные в определении R8 и R9:
арил (фенил), гетероарил, циклоалкил и гетероциклил незамещены или замещены от одного до трех заместителями, независимо выбранными из R10:
галогена;
гидрокси;
С1-4алкила;
С1-4алкокси;
трифторметила;
трифторметокси и
и где в определении R5, R8 и R9 любой метиленовый атом углерода (CH2) незамещен или замещен от одной-двумя группами, независимо выбранными из R11:
галогена;
гидрокси и
С1-4алкила;
или в определении R5, R8 и R9 два заместителя, находящихся на одном и том же углеродном атоме метиленовой группы (СН2), взятые вместе с атомом углерода, к которому они присоединены, образуют циклопропильную группу;
каждый R7 является водородом или R6;
а каждый R6 независимо выбирают из группы, состоящей из
водорода,
С1-8алкила,
С3-7циклоалкила;
и где указанные в определении R6 алкил и циклоалкил незамещены или замещены от одного до пяти заместителями, независимо выбранными из R12
галогена,
оксо,
С1-4алкокси,
С1-4алкилтио,
гидрокси,
амино;
либо R6 выбирают из группы, состоящей из
арила,
гетероарила;
где указанные в определении R6 арил и гетероарил незамещены или замещены и имеют от одного до трех заместителей, независимо выбранных из R13
галогена,
С1-4алкила,
гидрокси,
амино,
карбокси,
трифторметила,
трифторметокси,
пиано, и
С1-4алкокси;
либо две R6-группы вместе с атомом, с которым они связаны, образуют 5-8-членную моно- или бициклическую систему, необязательно содержащую дополнительный гетероатом, выбранный из О, S и NC1-4алкила, причем
каждый p представляет собой независимо 0, 1 или 2, если не указано иное значение;
каждый n представляет собой независимо 0, 1 или 2 если не указано иное значение.
2. Соединение по п.1, где R2 представляет собой циклопропил, C1-3алкил или С2-3алкенил и R1 представляет собой фенил или нафтил, в котором фенил незамещен или замещен и имеет от одного до трех заместителей, независимо выбранных из R5, и нафтил, возможно замещенный ОН-группой или С1-3алкокси группой.
3. Соединение по п.2, где R5 выбирают из группы, состоящей из галогена, гидрокси, трифторметила, трифторметокси, C1-3алкила, C1-3алкокси, C1-3алкилтио и С1-3алкилсульфонила.
4. Соединение по п.3, где R2 представляет собой метил.
5. Соединение по п.1, где
Х представляет собой одинарную связь;
R1 представляет собой фенил или нафтил, в котором фенил незамещен или замещен и имеет от одного до трех заместителей, независимо выбранных из R5, и нафтил, возможно замещенный ОН-группой или C1-3алкокси группой.
R2 представляет собой циклопропил, C1-3алкил или С2-3алкенил; и
R3 представляет собой незамещенный или замещенный C1-6алкил и имеет от одного до трех заместителей, независимо выбранных из R8.
6. Соединение по п.5, где R5 выбирают из группы, состоящей из галогена, гидрокси, трифторметила, трифторметокси, С1-3алкила, C1-3алкокси, C1-3алкилтио и C1-3алкилсульфонила.
7. Соединение по п.6, где R2 представляет собой метил.
8. Соединение по п.5, где R8 выбирают из группы, состоящей из галогена, гидрокси, оксо, С1-4алкокси, С1-4алкилтио, С1-4алкилсульфинила, C1-4алкилсульфонила, и R1 - фенил, незамещен или замещен от одной до трех групп, независимо выбранных из галогена и трифторметила.
9. Соединение по п.8, где R2 представляет собой водород.
10. Соединение по п.5, где R5 выбирают из группы, состоящей из галогена, гидрокси, трифторметила, трифторметокси, C1-3алкила, C1-3алкокси, C1-3алкилтио и С1-3алкилсульфонила; и R8 выбирают из группы, состоящей из галогена, гидрокси, оксо, С1-4алкокси, С1-4алкилтио, С1-4алкилсульфонила, и R1-фенил незамещен или замещен от одной до трех групп, независимо выбранных из галогена и трифторметила.
11. Соединение по п.10, где R2 представляет собой метил и R4 представляет собой водород.
12. Соединение по п.1, где
Х представляет собой одинарную связь;
R1 представляет собой фенил или нафтил, в котором фенил незамещен или замещен и имеют от одного до трех заместителей, независимо выбранных из R5, и нафтил, возможно замещенный ОН-группой или C1-3алкоксигруппой,
R2 представляет собой циклопропил, C1-3алкил или С2-3алкенил; и
R3 представляет собой фенил или гетероарил, где фенил и гетероарил незамещены или замещены и имеют от одного до трех заместителей, независимо выбранных из R9.
13. Соединение по п.12, где R2 представляет собой метил.
14. Соединение по п.12, где R3 представляет собой фенил, который возможно замещен галогеном или S(O)2R6, где R6 представляет собой С1-С6алкил.
15. Соединение по п.14, где R9 выбирают из группы, состоящей из галогена и C1-3алкилсульфонила.
16. Соединение по п.14, где R2 представляет собой метил.
17. Соединение по п.12, где R3 представляет собой оксадиазолил, который незамещен.
18. Соединение по п.17, где R9 представляет собой фенил, который незамещен или замещен галогеном.
19. Соединение по п.18, где R2 представляет собой метил.
20. Соединение, выбранное из группы, состоящей из
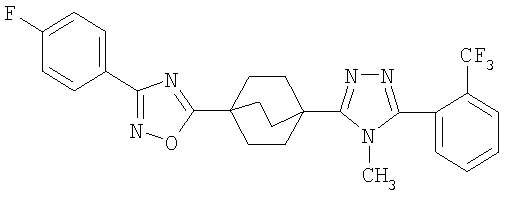
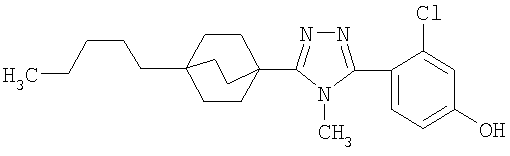
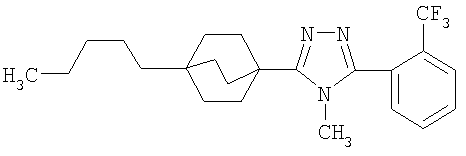
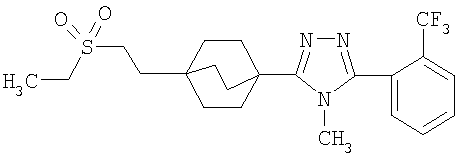
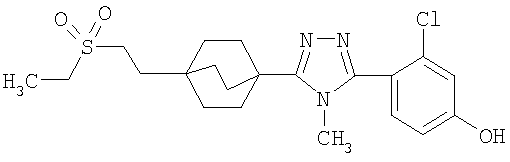
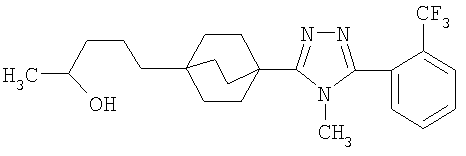
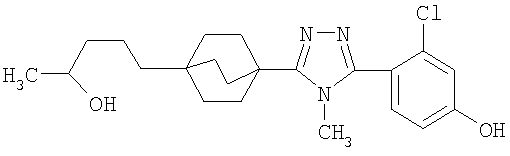
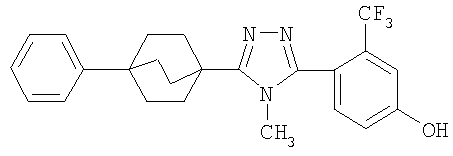
 и
и
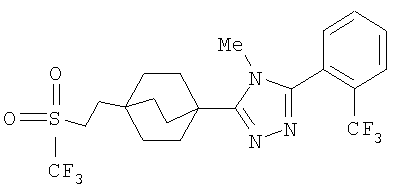
или его фармацевтически приемлемую соль.
21. Соединение по п.20, которое представляет собой
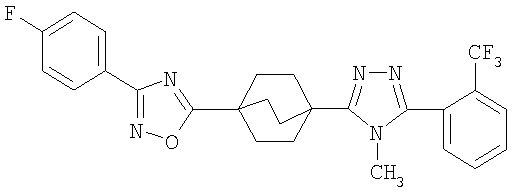
или его фармацевтически приемлемую соль.
22. Соединение по п.20, которое представляет собой
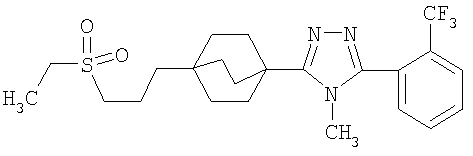
или его фармацевтически приемлемую соль.
23. Соединение по п.20, которое представляет собой
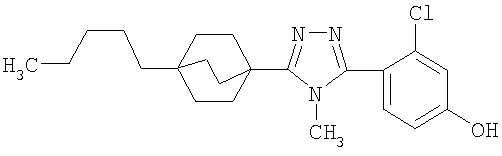
или его фармацевтически приемлемую соль.
24. Соединение по п.20, которое представляет собой
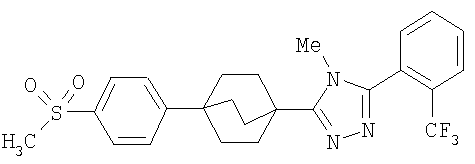
или его фармацевтически приемлемую соль.
25. Фармацевтическая композиция, проявляющая свойства ингибиторов фермента 11-бета-гидроксистероиддегидрогеназы 1-го типа (ГСД-1), содержащая соединение по п.1, в комбинации с фармацевтически приемлемым носителем.
26. Применение соединения по п.1 для получения лекарственного средства, используемого в лечении состояния, выбранного из группы, состоящей из гипергликемии, инсулинорезистентности, диабета 2-го типа, нарушений жирового обмена, ожирения, атеросклероза и метаболического синдрома.
| Печь для непрерывного получения сернистого натрия | 1921 |
|
SU1A1 |
| Переносная печь для варки пищи и отопления в окопах, походных помещениях и т.п. | 1921 |
|
SU3A1 |
| Переносная печь для варки пищи и отопления в окопах, походных помещениях и т.п. | 1921 |
|
SU3A1 |
| СОЕДИНЕНИЯ ТРИАЗОЛА И ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ НА ИХ ОСНОВЕ | 1995 |
|
RU2167869C2 |

