[0001] В настоящей заявке заявляется приоритет по предварительной заявке на патент США с серийным №61/550782, поданной 24 октября 2011 года, полное содержание которой включено в настоящий документ посредством ссылки.
УРОВЕНЬ ТЕХНИКИ
[0002] Рецептор N-метил-d-аспартата (НМДА) представляет собой постсинаптический, ионотропный рецептор, который отвечает, среди прочего, за глутамат возбуждающей аминокислоты и глицин, а также за синтетическое соединение НМДА. Рецептор НМДА контролирует поток двухвалентных и одновалентных ионов в постсинаптическую нервную клетку через рецептор-связанный канал (Foster et al., Nature 1987, 329:395-396; Mayer et al., Trends in Pharmacol. Sci. 1990, 11:254-260). Рецептор НМДА участвует в создании определенной нейронной структуры и конфигурации синаптических связей, и может участвовать в синаптических модификациях, зависящих от жизненного опыта. Кроме того, рецепторы НМДА предположительно участвуют также в долговременной потенциации и расстройствах центральной нервной системы.
[0003] Рецептор НМДА играет важную роль в синаптической пластичности, которая лежит в основе многих высших когнитивных функций, таких как запоминание, сохранение в памяти и обучение, а также в некоторых когнитивных путях и в восприятии боли (Collingridge et al., The NMDA Receptor, Oxford University Press, 1994). Кроме того, некоторые свойства рецепторов НМДА позволяют предположить, что они могут участвовать в обработке информации в головном мозге, что является основной самого сознания.
[0004] Рецептор НМДА привлекает особый интерес, поскольку он участвует в широком ряде расстройств ЦНС. Например, при ишемии головного мозга, вызванной инсультом или травмой, из поврежденных нейронов или нейронов, испытывающих кислородную недостаточность, выделяются избыточные количества глутамата возбуждающей аминокислоты. Этот избыточный глутамат связывается с рецепторами НМДА, что открывает их управляемые лигандами ионные каналы; в свою очередь, приток кальция вызывает высокую концентрацию внутриклеточного кальция, что активирует биохимический каскад, приводящий к разрушению белка и гибели клетки. Это явление, известное как эксайтотоксичность, предположительно отвечает также за неврологическое повреждение, связанное с другими расстройствами, от гипогликемии и остановки сердца до эпилепсии. Кроме того, существуют предварительные отчеты, указывающие на такое же участие в хронической нейродегенерации при болезни Хантингтона, Паркинсона и Альцгеймера. Было показано, что активация рецептора НМДА отвечает за судороги после инсульта и, в некоторых моделях эпилепсии, было показано, что рецептор НМДА необходим для возникновения пароксизмов. Было также установлено нейропсихиатрическое участие рецептора НМДА, поскольку блокирование Са++ канала рецептора НМДА с помощью анестезирующего РСР (фенциклидина) животного происхождения вызывает у людей психотическое состояние, схожее с шизофренией (рассмотрено в публикации Johnson, K. and Jones, S., 1990). Более того, рецепторы НМДА также участвуют в некоторых типах пространственного обучения.
[0005] Рецептор НМДА предположительно состоит из нескольких белковых цепей, встроенных в постсинаптическую мембрану. Первые два типа субъединиц, открытых до настоящего времени, образуют крупную внеклеточную область, которая вероятно содержит большинство аллостерических связывающих сайтов, несколько трансмембранных областей, образующих петли и сложенных с образованием поры или канала, который является проницаемым для Са++, а также карбоксиконцевую область. Открывание и закрывание этого канала регулируется связыванием различных лигандов с доменами (аллостерическими сайтами) белка, находящегося на внеклеточной поверхности. Связывание этих лигандов предположительно влияет на конформационное изменение общей структуры белка, что в конечном итоге отражается на открывании, частичном открывании, частичном закрывании или закрывании канала.
[0006] Соединения рецепторов НМДА могут проявлять двойное (агонистическое/антагонистическое) действие на рецептор НМДА через аллостерические сайты. Эти соединения обычно называют «частичными агонистами». В присутствии лиганда основного сайта частичный агонист заменяет некоторые из лигандов и за счет этого уменьшает поток Са++ через рецептор. В отсутствие лиганда основного сайта или при его пониженной концентрации, частичный агонист действует для увеличения потока Са++ через рецепторный канал.
[0007] В данной области сохраняется необходимость в новых и более специфичных/эффективных соединениях, которые способны связывать глицин-связывающий сайт рецепторов НМДА и обеспечивать фармацевтическое преимущество. Кроме того, в области медицины сохраняется необходимость в перорально доставляемых формах таких соединений.
КРАТКОЕ ОПИСАНИЕ
[0008] В настоящем документе, по меньшей мере в его части, представлены соединения, которые являются модуляторами НМДА, например, частичными агонистами НМДА. Например, в настоящем документе описаны соединения, представленные формулой:

где
R11, R12 и R13, каждый независимо, выбраны из группы, состоящей из Н, галогена, С1-3алкокси и С1-3алкила (необязательно замещенного одним, двумя или тремя галогенами);
X1 представляет собой ОН или NH2,
X2 представляет собой Η или ОН;
и их фармацевтически приемлемые соли, стереоизомеры и гидраты.
[0009] В некоторых вариантах реализации галоген рассматриваемого соединения может быть независимо выбран из группы, состоящей из Cl, Br, и F. В некоторых случаях X2 в представленной выше структуре может быть ОН, а X1 в представленной выше структуре может быть ΝΗ2.
[0010] В некоторых случаях каждый из R11, R12 и R13 в представленной выше структуре может быть Н. Альтернативно, R11 и R13 в представленной выше структуре может быть Н. R12 в представленной выше структуре может быть выбран из группы, состоящей из Н, галогена, С1-3алкокси и С1-3алкила (необязательно замещенного одним, двумя или тремя галогенами). Например, R11 и R13 в представленной выше структуре может быть Η, a R12 в представленной выше структуре может быть выбран из группы, состоящей из F, Br, Cl, СН3, CF3 и -ОСН3.
[0011] В некоторых вариантах реализации рассматриваемое соединение может быть значительно более эффективным при пероральном введении, по сравнению с
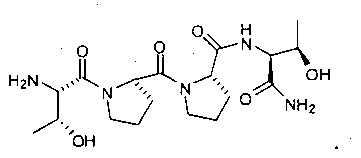
[0012] В другом аспекте представлено соединение, представленное структурой:

где:
R1, R2, R3 и R4, каждый независимо, выбраны из группы, состоящей из водорода; галогена, C1-С6алкила или -ОН;
R5 представляет собой Η или -СН2-фенил (где указанный фенил замещен одним, двумя или тремя заместителями, выбранными из группы, состоящей из галогена, С1-3алкокси и С1-3алкила (необязательно замещенного одним, двумя или тремя галогенами)), при условии, что R5 представляет собой -СН2-фенил, если R1 и R3 представляют собой -ОН, a R2 и R4 представляют собой метил; и
X выбран из группы, состоящей из ORx и NRxRx, где RX в каждом случае независимо выбран из группы, состоящей из водорода и С1-С6алкила; его фармацевтически приемлемые соли, стереоизомеры и гидраты.
[0013] Например, рассматриваемое соединение может быть представлено структурами:
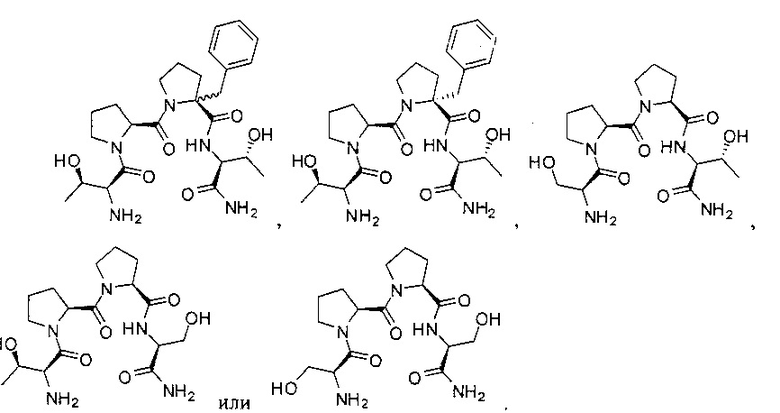
[0014] В других примерах рассматриваемое соединение может быть представлено структурами:


[0015] В настоящем документе представлены также фармацевтически приемлемые композиции, содержащие описанное соединение и фармацевтически приемлемое вспомогательное вещество. Например, такие композиции могут быть пригодными для перорального введения пациенту. В других вариантах реализации такие композиции могут быть пригодными для инъекции.
[0016] В другом аспекте описан способ лечения депрессии, болезни Альцгеймера, расстройства дефицита внимания, синдрома дефицита внимания с гиперактивностью (СДВГ), шизофрении или беспокойства у пациента, нуждающегося в этом, включающий введение, например, перорально, внутривенно или подкожно, указанному пациенту фармацевтически эффективного количества рассматриваемого соединения.
КРАТКОЕ ОПИСАНИЕ ГРАФИЧЕСКИХ МАТЕРИАЛОВ
[0017] ФИГУРА 1 иллюстрирует график концентрации СМ-4А в плазме в зависимости от времени после введения дозы.
[0018] ФИГУРА 2 иллюстрирует дозозависимые графики анализа связывания [3Н] МК-801 для оценки агонистических свойств рассматриваемых соединений.
[0019] ФИГУРА 3 иллюстрирует дозозависимые графики анализа связывания [3Н] МК-801 для оценки агонистических свойств рассматриваемых соединений.
[0020] ФИГУРА 4 иллюстрирует дозозависимый график анализа связывания [3Н] МК-801 для оценки агонистических свойств рассматриваемого соединения.
[0021] ФИГУРА 5 иллюстрирует график, изображающий динамику влияния СМ-4А на одиночные спровоцированные коллатералями Шаффера фармакологически выделенные возбуждающие постсинаптические токи (ВПСТ) НМДА, записанные в пирамидальный нейронах СА1.
[0022] ФИГУРА 6 иллюстрирует график, изображающий влияние концентрации СМ-4А на долговременное потенцирование после высокочастотной стимуляции спровоцированных коллатералями Шаффера ВПСТ НМДА, записанных в пирамидальных нейронах СА1.
[0023] ФИГУРА 7 иллюстрирует график, изображающий дозозависимое влияние СМ-4А на долговременную депрессию (ДВД).
[0024] ФИГУРА 8 иллюстрирует диаграмму процентного долговременного потенцирования угла наклона полевого возбуждающего постсинаптического потенциала (пВПСТ) для рассматриваемых соединений.
[0025] ФИГУРА 9 иллюстрирует график, изображающий среднее время плавания в испытании по Порсолту в зависимости от концентрации СМ-4А.
[0026] ФИГУРА 10 иллюстрирует диаграмму среднего времени плавания в испытании по Порсолту для СМ-4А и СМ-4А в комбинации с антагонистом рецептора АМРА, 2,3-дигидрокси-6-нитро-7-сульфамоил-бензо[f]хиноксалин-2,3-дионом (NBQX).
[0027] ФИГУРЫ 11А-11С иллюстрируют диаграммы, изображающие результаты испытания по Порсолту для рассматриваемых соединений.
[0028] ФИГУРА 12 иллюстрирует диаграмму, изображающую результаты испытания по Порсолту для рассматриваемых соединений через 1 час и через 24 часа.
[0029] ФИГУРА 13 иллюстрирует диаграмму, изображающую результаты испытания гипофагии в непривычных условиях (ГНУ) для СМ-4А.
[0030] ФИГУРЫ 14А и 14В иллюстрируют диаграммы, изображающие результаты испытания ультразвуковой вокализации (УЗВ) крыс для СМ-4А. ФИГУРА 14А представляет количественную оценку положительного эмоционального изучения, а ФИГУРА 14В представляет количественную оценку положительной и отрицательной УЗВ и удовлетворенности.
[0031] ФИГУРА 15 иллюстрирует диаграмму, изображающую результаты теста открытого поля на крысах для СМ-4А.
[0032] ФИГУРА 16 иллюстрирует диаграмму, изображающую результаты теста ускорения вращающегося стержня на крысах для СМ-4А.
ПОДРОБНОЕ ОПИСАНИЕ
[0033] Настоящее описание относится, в основном, к соединениям, которые могут модулировать НМДА, например, антагонистам или частичным агонистам НМДА, а также к композициям и/или способам применения описанных соединений.
Определения
[0034] В некоторых вариантах реализации соединения, описанные в настоящем документе, могут быть замещены любым количеством заместителей или функциональных фрагментов. Как правило, термин «замещенный», используемый после термина «необязательно» или без него, а также заместители, содержащиеся в формулах, относятся к замене водородных радикалов в данной структуре на радикал указанного заместителя.
[0035] В некоторых случаях, если в данной структуре может быть замещено более одного положения более чем одним заместителем, выбранным из определенной группы, то заместитель может быть одинаковым или различным в каждом положении.
[0036] При использовании в настоящем документе, термин «замещенный» включает все допустимые заместители органических соединений. В широком аспекте, допустимые заместители включают ациклические и циклические, разветвленные и неразветвленные, карбоциклические и гетероциклические, ароматические и неароматические заместители органических соединений. В некоторых вариантах реализации гетероатомы, такие как азот, могут иметь водородные заместители и/или любые допустимые заместители органических соединений, описанные в настоящем документе, которые заполняют валентности этих гетероатомов. Не ограничивающие примеры заместителей включают ацил; алифатический; гетероалифатический; арил; гетероарил; арилалкил; гетероарилалкил; алкокси; циклоалкокси; гетероциклилалкокси; гетероциклилокси; гетероциклилоксиалкил; алкенилокси; алкинилокси; арилокси; гетероалкокси; гетероарилокси; алкилтио; арилтио; гетероарилтио; оксо; -F; -Cl; -Br; -I; -OH; -NO2; -N3; -CN; -SCN; -SRX; -CF3; -CH2CF3; -CHCl2; -CH2OH; -CH2CH2OH; -CH2NH2; -CH2SO2CH3; -ORx, -C(O)Rx; -CO2(Rx); -C(O)N(Rx)2; -C(NRx)N(Rx)2; -OC(O)Rx; -OCO2Rx; -OC(O)N(Rx)2; -N(Rx)2; -SORx; -S(O)2Rx; -NRxC(O)Rx; -NRxC(O)N(Rx)2; -NRxC(O)ORx; -NRxC(NRx)N(Rx)2; и -C(Rx)3; при этом Rx в каждом случае независимо включает, но не ограничиваясь этим, водород, галоген, ацил, алифатический, гетероалифатический, арил, гетероарил, арилалкил или гетероарилалкил, при этом любой из алифатических, гетероалифатических, арилалкильных или гетероарилалкильных заместителей, описанных выше и в настоящем документе, может быть замещенным или незамещенным, разветвленным или неразветвленным, циклическим или ациклическим, и при этом любой из арильных или гетероарильных заместителей, описанных выше и в настоящем документе, может быть замещенным или незамещенным. Более того, соединения, описанные в настоящем документе, не предполагаются ограниченными каким-либо образом допустимыми заместителями органических соединений. В некоторых вариантах реализации комбинации заместителей и переменных, описанных в настоящем документе, могут быть предпочтительно такими, которые приводят к образованию устойчивых соединений. Термин «устойчивые», при использовании в настоящем документе, относится к соединениям, которые обладают достаточной устойчивостью для обеспечения возможности производства, и которые сохраняют целостность соединения в течение достаточного периода времени, чтобы быть обнаруженными и, предпочтительно, в течение достаточного периода времени, чтобы быть полезными для целей, детализированных в настоящем документе.
[0037] Термин «алифатический», при использовании в настоящем документе, включает насыщенные и ненасыщенные прямые (то есть неразветвленные), разветвленные, ациклические, циклические или полициклические алифатические углеводороды, которые необязательно замещены одной или более функциональными группами. Специалистам в данной области понятно, что «алифатический» в настоящем документе включает, но не ограничиваясь этим, алкильные, алкенильные, алкинильные, циклоалкильные, циклоалкенильные и циклоалкинильные фрагменты.
[0038] Термины «арил» и «гетероарил», используемые в настоящем документе, относятся к моно- или полициклическим ненасыщенным фрагментам, имеющим, предпочтительно, 3-14 углеродных атомов, каждый из которых может быть замещенным или незамещенным. В некоторых вариантах реализации «арил» относится к моно- или бициклической карбоциклической кольцевой системе, имеющей одно или два ароматических кольца, включая, но не ограничиваясь этим, фенил, нафтил, тетрагидронафтил, инданил, инденил и тому подобные. В некоторых вариантах реализации «гетероарил» относится к моно- или бициклической гетероциклической кольцевой системе, имеющей одно или два ароматических кольца, в которых один, два или три кольцевых атома представляют собой гетероатомы, независимо выбранные из группы, состоящей из S, О и N, а остальные кольцевые атомы представляют собой углерод. Не ограничивающие примеры гетероарильных групп включают пиридил, пиразинил, пиримидинил, пирролил, пиразолил, имидазолил, тиазолил, оксазолил, изооксазолил, тиадиазолил, оксадиазолил, тиофенил, фуранил, хинолинил, изохинолинил и тому подобные.
[0039] Термин «алкенил», используемый в настоящем документе, относится к ненасыщенному прямому или разветвленному углеводороду, имеющему по меньшей мере одну двойную углерод-углеродную связь, такому как прямая или разветвленная группа из 2-12, 2-10 или 2-6 углеродных атомов, упоминаемая в настоящем документе как С2-С12алкенил, С2-С10алкенил и С2-С6алкенил, соответственно. Иллюстративные алкенильные группы включают, но не ограничиваясь этим, винил, аллил, бутенил, пентенил, гексенил, бутадиенил, пентадиенил, гексадиенил, 2-этилгексенил, 2-пропил-2-бутенил, 4-(2-метил-3-бутен)-пентенил и так далее.
[0040] Термин «алкокси», используемый в настоящем документе, относится к алкильной группе, присоединенной к кислороду (-О-алкил). Иллюстративные алкокси-группы включают, но не ограничиваясь этим, группы с алкильной группой из 1-12, 1-8 или 1-6 углеродных атомов, упоминаемые в настоящем документе как С1-12алкокси и С1-8алкокси и С1-6алкокси, соответственно. Иллюстративные алкокси-группы включают, но не ограничиваясь этим, метокси, этокси и так далее. Точно так же, иллюстративные «алкенокси» группы включают, но не ограничиваясь этим, винилокси, аллилокси, бутенокси и так далее.
[0041] Термин «алкоксикарбонил», используемый в настоящем документе, относится к прямой или разветвленной алкильной группе, присоединенной к кислороду, присоединенному к карбонильной группе (алкил-О-С(О)-). Иллюстративные алкоксикарбонильные группы включают, но не ограничиваясь этим, алкоксикарбонильные группы из 1-6 углеродных атомов, упоминаемые в настоящем документе как С1-6алкоксикарбонил. Иллюстративные алкоксикарбонильные группы включают, но не ограничиваясь этим, метоксикарбонил, этоксикарбонил, трет-бутоксикарбонил и так далее.
[0042] Термин «алкинилокси», используемый в настоящем документе, относится к прямой или разветвленной алкинильной группе, присоединенной к кислороду (алкинил-О)). Иллюстративные алкинилокси-группы включают, но не ограничиваясь этим, пропинилокси.
[0043] Термин «алкил», используемый в настоящем документе, относится к насыщенному прямому или разветвленному углеводороду, например, такому как прямая или разветвленная группа из 1-6, 1-4 или 1-3 углеродных атомов, упоминаемая в настоящем документе как C1-С6алкил, С1-С4алкил и C1-С3алкил, соответственно. Иллюстративные алкильные группы включают, но не ограничиваясь этим, метил, этил, пропил, изопропил, 2-метил-1-пропил, 2-метил-2-пропил, 2-метил-1-бутил, 3-метил-1-бутил, 2-метил-3-бутил, 2,2-диметил-1-пропил, 2-метил-1-пентил, 3-метил-1-пентил, 4-метил-1-пентил, 2-метил-2-пентил, 3-метил-2-пентил, 4-метил-2-пентил, 2,2-диметил-1-бутил, 3,3-диметил-1-бутил, 2-этил-1-бутил, бутил, изобутил, трет-бутил, пентил, изопентил, неопентил, гексил, гептил, октил и так далее.
[0044] Термин «алкилкарбонил», используемый в настоящем документе, относится к прямой или разветвленной алкильной группе, присоединенной к карбонильной группе (алкил-С(О)-). Иллюстративные алкил карбонильные группы включают, но не ограничиваясь этим, алкилкарбонильные группы из 1-6 атомов, упоминаемые в настоящем документе как С1-С6алкилкарбонильные группы. Иллюстративные алкилкарбонильные группы включают, но не ограничиваясь этим, ацетил, пропаноил, изопропаноил, бутаноил и так далее.
[0045] Термин «алкинил», используемый в настоящем документе, относится к ненасыщенному прямому или разветвленному углеводороду, имеющему по меньшей мере одну тройную углерод-углеродную связь, такому как прямая или разветвленная группа из 2-6 или 3-6 углеродных атомов, упоминаемая в настоящем документе как С2-6алкинил и С3-6алкинил, соответственно. Иллюстративные алкиниловые группы включают, но не ограничиваясь этим, этинил, пропинил, бутинил, пентинил, гексинил, метилпропинил и так далее.
[0046] Алкильные, алкенильные и алкинильные группы могут быть необязательно замещены, если не указано иное, одной или более группами, выбранными из алкокси, алкила, циклоалкила, амино, галогена и -С(O)алкила. В некоторых вариантах реализации алкильные, алкенильные и алкинильные группы не замещены, то есть они являются незамещенными.
[0047] Термин «амид» или «амидо», используемый в настоящем документе, относится к радикалу формы -RaC(O)N(Rb)-, -RaC(O)N(Rb)Rc- или -C(O)NRbRc, где Ra, Rb и Rc, каждый независимо, выбраны из алкокси, алкила, алкенила, алкинила, амида, амино, арила, арилалкила, карбамата, циклоалкила, сложного эфира, простого эфира, формила, галогена, галоалкила, гетероарила, гетероциклила, водорода, гидроксила, кетона и нитро. Амид может быть присоединен к другой группе через углерод, азот, Rb, Rc или Ra. Амид также может быть циклическим, например, Rb и Rc, Ra и Rb или Ra и Rc могут быть соединены с образованием 3-12-членного кольца, такого как 3-10-членное кольцо или 5-6-членное кольцо. Термин «карбоксамидо» относится к структуре -С(O)NRbRc.
[0048] Термин «амин» или «амино», используемый в настоящем документе, относится к радикалу формы -NRdRe, где Rd и Re независимо выбраны из водорода, алкила, алкенила, алкинила, арила, арилалкила, циклоалкила, галоалкила, гетероарила и гетероциклила. «Амино» также может быть циклическим, например, Rd и Re соединены вместе с N с образованием 3-12-членного кольца, например, морфолино или пиперидинила. Термин «амино» включает также соответствующую четвертичную аммониевую соль любой аминогруппы, например, -[N(Rd)(Re)(Rf)]+. Иллюстративные аминогруппы включают аминоалкильные группы, в которых по меньшей мере один из Rd, Re или Rf представляет собой алкильную группу. В некоторых вариантах реализации Rd и Re представляют собой водород или алкил.
[0049] Термин «циклоалкил», используемый в настоящем документе, относится к моноциклической насыщенной или частично ненасыщенной углеводородной группе, например, из 3-6 или 4-6 углеродных атомов, упоминаемой в настоящем документе, например, как С3-6циклоалкил или С4-6циклоалкил, и полученной из циклоалкана. Иллюстративные циклоалкильные группы включают, но не ограничиваясь этим, циклогексил, циклогексенил, циклопентил, циклобутил или циклопропил.
[0050] Термины «гало» или «галоген», или «Hal», используемые в настоящем документе, относятся к F,Cl, Br или I. Термин «галоалкил», используемый в настоящем документе, относится к алкильной группе, замещенной одним или более атомами галогена.
[0051] Термины «гетероциклил» или «гетероциклическая группа» известны в данной области и относятся к насыщенным или частично ненасыщенным 3-10-членным кольцевым структурам, альтернативно, - к 3-7-членным кольцам, кольцевые структуры которых содержат от одного до четырех гетероатомов, таких как азот, кислород и сера. Гетероциклы также могут быть моно-, би- или иными полициклическими кольцевыми системами. Гетероцикл может быть конденсирован с одним или более арильными, частично ненасыщенными или насыщенными кольцами. Гетероциклильные группы включают, например, биотинил, хроменил, дигидрофурил, дигидроиндолил, дигидропиранил, дигидротиенил, дитиазолил, гомопиперидинил, имидазолидинил, изохинолил, изотиазолидинил, изоксазолидинил, морфолинил, оксоланил, оксазолидинил, феноксатенил, пиперазинил, пиперидинил, пиразинил, пиразолидинил, пиразолинил, пиридил, пиримидинил, пирролидинил, пирролидин-2-онил, пирролинил, тетрагидрофурил, тетрагидроизохинолил, тетрагидропиранил, тетрагидрохинолил, тиазолидинил, тиоланил, тиоморфолинил, тиопиранил, ксантенил, лактоны, лактамы, такие как азетидиноны и пирролидиноны, сультамы, сультоны и тому подобные. Гетероциклическое кольцо может быть замещено в одном или более положениях заместителями, такими как алканоил, алкокси, алкил, алкенил, алкинил, амидо, амидино, амино, арил, арилалкил, азидо, карбамат, карбонат, карбокси, циано, циклоалкил, сложный эфир, простой эфир, формил, галоген, галоалкил, гетероарил, гетероциклил, гидроксил, имино, кетон, нитро, фосфат, фосфонато, фосфинато, сульфат, сульфид, сульфонамидо, сульфонил и тиокарбонил. В некоторых вариантах реализации гетероциклическая группа не замещена, то есть гетероциклическая группа является незамещенной.
[0052] Термин «гетероциклилалкокси», используемый в настоящем документе, относится к группе гетероциклил-алкил-О-.
[0053] Термин «гетероциклилокси» относится к группе гетероциклил-О-.
[0054] Термин «гетероциклилоксиалкил» относится к группе гетероциклил-О-алкил.
[0055] Термины «гидрокси» и «гидроксил», используемые в настоящем документе, относятся к радикалу -ОН.
[0056] Термин «оксо», используемый в настоящем документе, относится к радикалу =O.
[0057] «Фармацевтически или фармакологически приемлемый» включает молекулярные вещества и композиции, которые не вызывают побочных, аллергических или других неблагоприятных реакций при введении животному или человеку, в зависимости от конкретного случая. Для введения человеку, препараты должны соответствовать стандартам стерильности, пирогенности, общей безопасности и чистоты, обязательным по нормативам Отдела биологических веществ Управления США по надзору за качеством пищевых продуктов и лекарственных средств (FDA).
[0058] При использовании в настоящем описании, термин «частичный агонист рецептора НМДА» определяется как соединение, которое способно связываться с глицин-связывающим сайтом рецептора НМДА; при низких концентрациях агонист рецептора НМДА действует, в основном, как агонист, а при высоких концентрациях он действует, в основном, как антагонист. Эти концентрации определяют экспериментально для всех и каждого «частичного агониста».
[0059] При использовании в настоящем документе, «фармацевтически приемлемый носитель» или «вспомогательное вещество» включает любой и все растворители, дисперсионные среды, покрытия, антибактериальные и противогрибковые агенты, изотонические средства и агенты замедления абсорбции, и тому подобные, которые являются физиологически совместимыми. В одном из вариантов реализации носитель пригоден для парентерального введения. Альтернативно, носитель может быть пригодным для внутривенного, внутрибрюшинного, внутримышечного, сублингвального или перорального введения. Фармацевтически приемлемые носители включают стерильные водные растворы или дисперсии, а также стерильные порошки для приготовления стерильных растворов или дисперсий для инъекций непосредственно перед применением. Использование такой среды и агентов для фармацевтически активных веществ хорошо известно в данной области. За исключением случаев несовместимости какой-либо стандартной среды или агента с активным компонентом, их использование предусмотрено в фармацевтических композициях настоящего изобретения. В состав композиций также могут быть введены дополнительные активные соединения.
[0060] Термин «фармацевтически приемлемая соль(-и)», используемый в настоящем документе, относится к солям кислотных или основных групп, которые могут присутствовать в соединениях, используемых в композициях настоящего изобретения. Соединения, входящие в состав композиций настоящего изобретения, которые являются основными по своей природе, способны образовывать широкий ряд солей с различными неорганическими и органическими кислотами. Кислоты, которые могут быть использованы для получения фармацевтически приемлемых солей присоединения кислот таких основных соединений, представляют собой те, которые образуют нетоксичные соли присоединения кислот, то есть соли, содержащие фармакологически приемлемые анионы, включая, но не ограничиваясь этим, малатные, оксалатные, хлоридные, бромидные, йодидные, нитратные, сульфатные, бисульфатные, фосфатные, гидрофосфатные, изоникотинатные, ацетатные, лактатные, салицилатные, цитратные, тартратные, олеатные, таннатные, пантотенатные, битартратные, аскорбатные, сукцинатные, малеатные, гентизинатные, фумаратные, глюконатные, глюкуронатные, сахаратные, формиатные, бензоатные, глутаматные, метансульфонатные, этансульфонатные, бензолсульфонатные, п-толуолсульфонатные и памоатные (то есть 1,1'-метилен-бис-(2-гидрокси-3-нафтоатные)) соли. Соединения, входящие в композиции настоящего изобретения, которые содержат амино-фрагмент, могут образовывать фармацевтически приемлемые соли с различными аминокислотами, помимо кислот, упомянутых выше. Соединения, входящие в композиции настоящего изобретения, которые являются кислотными по своей природе, способны образовывать основные соли с различными фармакологически приемлемыми катионами. Примеры таких солей включают соли щелочных металлов или щелочноземельных металлов и, в частности, соли кальция, магния, натрия, лития, цинка, калия и железа.
[0061] Соединения настоящего описания могут содержать один или более хиральных центров и/или двойных связей и поэтому могут существовать в виде стереоизомеров, таких как геометрические изомеры, энантиомеры или диастереомеры. Термин «стереоизомеры», используемый в настоящем документе, состоит из всех геометрических изомеров, энантиомеров или диастереомеров. Эти соединения могут быть обозначены символами «R» или «S», в зависимости от конфигурации заместителей вокруг стереогенного атома углерода. Настоящее изобретение охватывает различные стереоизомеры этих соединений и их смеси. Стереоизомеры включают энантиомеры и диастереомеры. Смеси энантиомеров или диастереомеров могут быть обозначены в номенклатуре «(±)», но специалистам в данной области понятно, что структура может неявно обозначать хиральный центр.
[0062] Отдельные стереоизомеры соединений настоящего изобретения могут быть получены синтетически из имеющихся в продаже исходных материалов, которые содержат асимметричные или стереогенные центры, или получением рацемических смесей с последующими способами разделения, хорошо известными специалистам в данной области. Эти способы разделения представлены на примере (1) присоединения смеси энантиомеров к хиральному вспомогательному веществу, разделения полученной смеси диастереомеров перекристаллизацией или хроматографией, и отделения оптически чистого продукта от вспомогательного вещества, (2) образования соли с использованием оптически активного разделительного агента, или (3) прямого разделения смеси оптических энантиомеров на хиральных хроматографических колонках. Стереоизомерные смеси также могут быть разделены на их составляющие стереоизомеры с помощью хорошо известных способов, таких как хирально-фазовая газовая хроматография, хирально-фазовая высокоэффективная жидкостная хроматография, кристаллизация соединения в виде комплекса хиральной соли или кристаллизация соединения в хиральном растворителе. Стереоизомеры также могут быть получены из стереомерно чистых промежуточных соединений, реагентов и катализаторов по общеизвестным способам асимметричного синтеза.
[0063] В соединениях настоящего изобретения также могут существовать геометрические изомеры. Символ  обозначает связь, которая может быть одинарной, двойной или тройной связью, как описано в настоящем документе. Настоящее изобретение охватывает различные геометрические изомеры и их смеси, образующиеся в результате расположения заместителей вокруг двойной углерод-углеродной связи или расположения заместителей относительно карбоциклического кольца. Заместители вокруг двойной углерод-углеродной связи обозначают как существующие в «Ζ» или «E» конфигурации, при этом термины «Ζ» и «E» используют в соответствии с правилами ИЮПАК. Если не указано иное, то структуры, изображающие двойную связь, охватывают оба изомера «Ζ» и «E».
обозначает связь, которая может быть одинарной, двойной или тройной связью, как описано в настоящем документе. Настоящее изобретение охватывает различные геометрические изомеры и их смеси, образующиеся в результате расположения заместителей вокруг двойной углерод-углеродной связи или расположения заместителей относительно карбоциклического кольца. Заместители вокруг двойной углерод-углеродной связи обозначают как существующие в «Ζ» или «E» конфигурации, при этом термины «Ζ» и «E» используют в соответствии с правилами ИЮПАК. Если не указано иное, то структуры, изображающие двойную связь, охватывают оба изомера «Ζ» и «E».
[0064] Альтернативно, заместители вокруг двойной углерод-углеродной связи могут быть упомянуты как «цис» или «транс», при этом «цис» представляет собой заместители с одной стороны двойной связи, а «транс» представляет собой заместители с противоположных сторон двойной связи. Расположение заместителей относительно карбоциклического кольца обозначают как «цис» или «транс». Термин «цис» представляет заместители с одной и той же стороны плоскости кольца, а термин «транс» представляет заместители с противоположных сторон плоскости кольца. Смеси соединений, в которых заместители расположены с одной или с противоположных сторон плоскости кольца, обозначают как «цис/транс».
[0065] Соединения, описанные в настоящем документе, могут существовать в сольватированных, а также в не сольватированных формах с фармацевтически приемлемыми растворителями, такими как вода, этанол и тому подобные, и подразумевается, что настоящее изобретение включает и сольватированные, и не сольватированные формы. В одном из вариантов реализации соединение является аморфным. В одном из вариантов реализации, соединение представляет собой полиморф. В другом варианте реализации соединение находится в кристаллической форме.
[0066] Настоящее изобретение охватывает также меченные изотопами соединения настоящего изобретения, которые идентичны соединениям, упомянутым в настоящем документе, за исключением того, что один или более атомов замещены атомами, имеющими атомную массу или массовое число, отличное от атомной массы или массового числа, обычно встречающегося в природе. Примеры изотопов, которые могут быть внедрены в соединения настоящего изобретения, включают изотопы водорода, углерода, азота, кислорода, фосфора, фтора и хлора, такие как 2Н, 3Н, 13С, 14С, 15N, 18O, 17O, 31Р, 32Р, 35S, 18F и 36Cl, соответственно.
[0067] Некоторые описанные соединения с изотопной меткой (например, соединения, меченные 3Н и 14С) применимы в анализах распределения соединения и/или субстрата в ткани. Изотопы трития (то есть 3Н) и углерода-14 (то есть 14С), являются наиболее предпочтительными, благодаря простоте получения и обнаружения. Кроме того, замена на более тяжелые изотопы, такие как дейтерий (то есть Н), может давать определенное терапевтическое преимущество из-за более высокой метаболической устойчивости (например, увеличенного периода полувыведения in vivo или снижения необходимых дозировок), и поэтому может быть предпочтительна в некоторых условиях. Соединения настоящего изобретения с изотопной меткой могут быть получены, в основном, по таким же способам, как описаны, например, в Примерах, представленных в настоящем документе, путем замены реагента без изотопной метки на реагент с изотопной меткой.
[0068] При использовании в настоящем описании, «НМДА» определяют как N-метил-d-аспартат.
[0069] В настоящем описании термин «терапевтически эффективное количество» означает количество рассматриваемого соединения, которое вызывает биологический или медицинский ответ ткани, системы, животного или человека, видимый для исследователя, ветеринара, врача или другого клинициста. Соединения настоящего изобретения вводят в терапевтически эффективных количествах для лечения заболевания. Альтернативно, терапевтически эффективное количество соединения представляет собой количество, необходимое для достижения заданного терапевтического и/или профилактического эффекта, такое как количество, которое приводит к определенному результату, или количество, которое необходимо для достижения максимального улучшения поведения (например, обучаемости), физиологического ответа (например, индукции ДВП) или подавления невропатической боли.
Соединения
[0070] Описанные соединения включают соединения, представленные формулой:
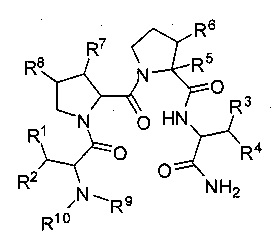
и их фармацевтически приемлемые соли, стереоизомеры, метаболиты и гидраты, где:
R1, R2, R3 и R4 могут быть независимо выбраны из группы, состоящей из водорода; галогена; циклического или ациклического, замещенного или незамещенного, разветвленного или неразветвленного алифатического; циклического или ациклического, замещенного или незамещенного, разветвленного или неразветвленного гетероалифатического; замещенного или незамещенного арила; замещенного или незамещенного гетероарила; -ORx; -NO2; -N3; -CN; -SCN; -SRx; -C(O)Rx; -CO2(Rx); -C(O)N(Rx)2; -C(NRx)N(Rx)2; -OC(O)Rx; -OCO2Rx; -OC(O)N(Rx)2; -N(Rx)2; -SORx; -S(O)2Rx; -NRxC(O)Rx; -NRxC(O)N(Rx)2; -NRxC(O)ORx; -NRxC(NRx)N(Rx)2; и -C(Rx)3; где Rx в каждом случае незаисимо выбран из группы, состоящей из водорода; галогена; ацила; необязательно замещенного алифатического; необязательно замещенного гетероалифатического; необязательно замещенного арила; и необязательно замещенного гетероарила;
R5 и R6 могут быть независимо выбраны из группы, состоящей из -Q-Ar и водорода, при условии, что по меньшей мере один из R5 и R6 представляет собой -Q-Ar; при этом Q независимо выбран из группы, состоящей из циклического или ациклического, замещенного или незамещенного, разветвленного или неразветвленного алифатического; циклического или ациклического, замещенного или незамещенного, разветвленного или неразветвленного гетероалифатического; и связи; и при этом Ar выбран из группы, состоящей из замещенного или незамещенного арила и замещенного или незамещенного гетероарила; или R5 и R6, вместе с атомами, к которым они присоединены, образуют замещенное или незамещенное 4-6-членное гетероциклическое или циклоалкильное кольцо;
R7 и R8 могут быть независимо выбраны из группы, состоящей из водорода; галогена; гидроксила; замещенного или незамещенного С1-С6 алкила; замещенного или незамещенного C1-С6 алкокси; и замещенного или незамещенного арила; или R7 и R8, вместе с атомами, к которым они присоединены, образуют замещенное или незамещенное 4-6-членное гетероциклическое или циклоалкильное кольцо;
R9 и R10 могут быть независимо выбраны из группы, состоящей из водорода; C1-С6 алкила, необязательно замещенного одним или более заместителями, каждый независимо выбран из группы, состоящей из галогена, оксо и гидроксила; С2-6алкенила, необязательно замещенного одним или более заместителями, каждый независимо выбран из группы, состоящей из галогена, оксо и гидроксила; С2-6алкинила, необязательно замещенного одним или более заместителями, каждый независимо выбран из группы, состоящей из галогена, оксо и гдроксила; С3-6циклоалкила, необязательно замещенного одним или более заместителями, каждый независимо выбран из группы, состоящей из С1-6алкила, галогена, оксо и гидроксила; фенила, необязательно замещенного одним или более заместителями, каждый независимо выбран из группы, состоящей из C1-6алкила; С1-6алкокси; галогена; гидроксила; -C(O)Rx; -CO2(Rx); -C(O)N(Rx)2; -C(NRx)N(Rx)2; и -C(Rx)3; где Rx в каждом случае независимо выбран из группы, состоящей из водорода; галогена; С1-6алкила; С2-6алкенила; С2-6алкинила; С3-6циклоалкила; и фенила; или R9 и R10, вместе с N, образуют 4-6-членное гетероциклическое кольцо, необязательно замещенное одним или более заместителями, каждый независимо выбран из группы, состоящей из С1-6алкила, галогена, оксо и гидроксила.
[0071] В некоторых вариантах реализации R1, R2, R3 и R4 могут быть независимо выбраны из группы, состоящей из водорода; галогена; C1-6алкила; С2-6алкенила; С2-6алкинила; С3-6циклоалкила; фенила; нафтола; гетероарила; гетероциклила; С3-6циклоалкил-С1-6алкил-; фенил-С1-6алкил-; нафтил-С1-6алкил-; гетероарил-С1-6алкил-; и гетероциклил-С1-6алкил-; -ORx; -NO2; -N3; -CN; -SCN; -SRx; -C(O)Rx; -CO2(Rx); -C(O)N(Rx)2; -C(NRx)N(Rx)2; -OC(O)Rx; -OCO2Rx; -OC(O)N(Rx)2; -N(Rx)2; -SORx; -S(O)2Rx; -NRxC(O)Rx; -NRxC(O)N(Rx)2; -NRxC(O)ORx; -NRxC(NRx)N(Rx)2; и -C(Rx)3; где гетероарил представляет собой 5-6-членное кольцо, имеющее один, два или три гетероатома, каждый независимо выбран из N, О или S; при этом гетероарил необязательно замещен одним или более заместителями, каждый независимо выбран из Rb; при этом гетероциклил представляет собой 4-7-членное кольцо, необязательно замещенное одним или более заместителями, каждый независимо выбран из Rc; при этом если гетероциклил содержит фрагмент -NH-, то фрагмент -NH- необязательно замещен Rd; при этом С2-6алкенил и С2-6алкинил, каждый независимо, необязательно замещены одним или более заместителями, каждый независимо выбран из Re; при этом С1-6алкил необязательно замещен одним или более заместителями, каждый независимо выбран из Rf; при этом С3-6циклоалкил независимо необязательно замещен одним или более заместителями, каждый независимо выбран из Rg;
Rb в каждом случае независимо может быть выбран из группы, состоящей из галогена; гидроксила; -NO2; -N3; -CN; -SCN; С1-6алкила; С2-6алкенила; С2-6алкинила; С3-6циклоалкила; C1-6алкокси; С3-6алкенилокси; С3-6алкинилокси; С3-6циклоалкокси; С1-6алкил-S(O)w-, где w равен 0, 1 или 2; С1-6алкилС3-6циклоалкил-; С3-6циклоалкил-С1-6алкил-; С1-6алкоксикарбонил-N(Ra)-; C1-6алкил-N(Ra)-; С1-6алкил-N(Ra)карбонил-; RaRa'N-; RaRa'N-карбонил-; RaRa'N-карбонил-N(Ra)-; RaRa'N-SO2-; и С1-6алкил-карбонил-N(Ra)-;
Ra и Ra'' в каждом случае независимо могут быть выбраны из группы, состоящей из водорода и С1-6алкила, или Ra и Ra'', взятые вместе с атомом азота, к которому они присоединены, образуют 4-6-членное гетероциклическое кольцо, при этом С1-6алкил необязательно замещен одним или более заместителями, каждый независимо выбран из группы, состоящей из галогена, оксо и гидроксила, и при этом указанное гетероциклическое кольцо необязательно замещено одним или более заместителями, каждый независимо выбран из группы, состоящей из галогена, алкила, оксо или гидроксила;
Rc в каждом случае независимо может быть выбран из группы, состоящей из галогена; гидроксила; -NO2; -N3; -CN; -SCN; оксо; С1-6алкила; С2-6алкенила; С2-6алкинила; С3-6циклоалкила; C1-6алкокси; С3-6алкенилокси; С3-6алкинилокси; С3-6циклоалкокси; C1-6алкил-S(O)w-, где w равен 0, 1 или 2; C1-6алкилС3-6циклоалкил-; С3-6циклоалкил-С1-6алкил-; С1-6алкоксикарбонил-N(Ra)-; С1-6алкил-N(Ra)-; С1-6алкил-N(Ra)карбонил-; RaRa'N-; RaRa'N-карбонил-; RaRa'N-карбонил-N(Ra)-; RaRa'N-SO2-; и С1-6алкил-карбонил-N(Ra)-;
Rd в каждом случае независимо может быть выбран из группы, состоящей из С1-6алкила, С1-6алкилкарбонила и С1-6алкилсульфонила, при этому C1-6алкил необязательно замещен одним или более заместителями, каждый независимо выбран из галогена, гидроксила и RaRa'N-;
Re в каждом случае независимо может быть выбран из группы, состоящей из галогена; гидроксила; -NO2; -N3; -CN; -SCN; С1-4алкокси; С1-4алкоксикарбонила; RaRa'N-; RaRa'N-карбонила; RaRa'N-SO2-; и С1-4алкилS(O)w-, где w равен 0, 1 или 2;
Rf в каждом случае независимо может быть выбран из группы, состоящей из галогена; гидроксила; -NO2; -N3; -CN; -SCN; С1-4алкокси; С1-4алкоксикарбонила; RaRa'N-; RaRa'N-карбонила; RaRa'N-SO2-; и C1-4алкил(O)w-, где w равен 0, 1 или 2;
Rg в каждом случае независимо может быть выбран из группы, состоящей из галогена, гидроксила, -NO2; -N3; -CN; -SCN; C1-6алкила; С1-4алкокси; C1-4алкоксикарбонила; RaRa'N-; RaRa'N-карбонила; RaRa'N-SO2-; и С1-4алкилS(O)w-, где w равен 0, 1 или 2;
Rx может быть независимо выбран из группы, состоящей из водорода; галогена; C1-6алкила; С2-6алкенила; С2-6алкинила; С3-6циклоалкила; фенила; нафтила; гетероарила; гетероциклила; С3-6циклоалкил-С1-6алкил-; фенил-С1-6алкил-; нафтил-С1-6алкил-; гетероарил-С1-6алкил-; и гетероциклил-С1-6алкил-; при этом гетероарил представляет собой 5-6-членное кольцо, имеющее один, два или три гетероатома, каждый независимо выбран из N, О или S; при этом гетероарил необязательно замещен одним или более заместителями, каждый независимо выбран из Rb; при этом гетероциклил представляет собой 4-7-членное кольцо, необязательно замещенное одним или более заместителями, каждый независимо выбран из Rc; при этом если гетероциклил содержит фрагмент -NH-, то фрагмент -NH- необязательно замещен Rd; при этом С2-6алкенил и С2-6алкинил, каждый независимо, необязательно замещены одним или более заместителями, каждый независимо выбран из Re; при этом С1-6алкил необязательно замещен одним или более заместителями, каждый независимо выбран из Rf; при этом С3-6циклоалкил независимо необязательно замещено одним или более заместителями, каждый независимо выбран из Rg.
[0072] В некоторых вариантах реализации по меньшей мере один из R1, R2, R3 и R4 может быть гидроксилом.
[0073] В некоторых случаях по меньшей мере один из R1, R2, R3 и R4 может быть C1-С6 алкилом, необязательно замещенным одним, двумя или тремя заместителями, независимо выбранными из группы, состоящей из галогена, гидроксила, -NH2 и циано.
[0074] В некоторых вариантах реализации по меньшей мере один из R5 и R6 может быть -(C1-С6 алкилен)-Ar. По меньшей мере один из R5 и R6 также может быть -СН2-Ar. В некоторых случаях по меньшей мере один из R5 и R6 представляет собой -Q-фенил. В некоторых примерах один из R5 и R6 может быть водородом.
[0075] В некоторых случаях R7 и R8 могут быть независимо выбраны из группы, состоящей из водорода; галогена; гидроксила; C1-С6 алкила; фенила; и нафтола; или R7 и R8, вместе с атомами, к которым они присоединены, образуют 4-6-членное гетероциклическое или циклоалкильное кольцо; при этом каждый С1-С6 алкил, фенил, нафтол, циклоалкильное кольцо или гетероциклическое кольцо могут быть независимо замещены одним или более заместителями, выбранными из группы, состоящей из галогена; гидроксила; -NO2; -N3; -CN; -SCN; С1-4алкокси; С1-4алкоксикарбонила; RaRa'N-; RaRa'N-карбонила; RaRa'N-SO2-; и С1-4алкилS(O)w-, где w равен 0, 1 или 2; при этом Ra и Ra' могут быть каждом случае независимо выбраны из группы, состоящей из водорода и C1-6алкила, или Ra и Ra', взятые вместе с атомом азота, к которому они присоединены, образуют 4-6-членное гетероциклическое кольцо, при этом С1-6алкил необязательно замещен одним или более заместителями, каждый независимо выбран из группы, состоящей из галогена, оксо и гидроксила, и при этом указанное гетероциклическое кольцо необязательно замещено одним или более заместителями, каждый независимо выбран из группы, состоящей из галогена, алкила, оксо или гидроксила.
[0076] В некоторых случаях R7 и R8 могут быть водородом.
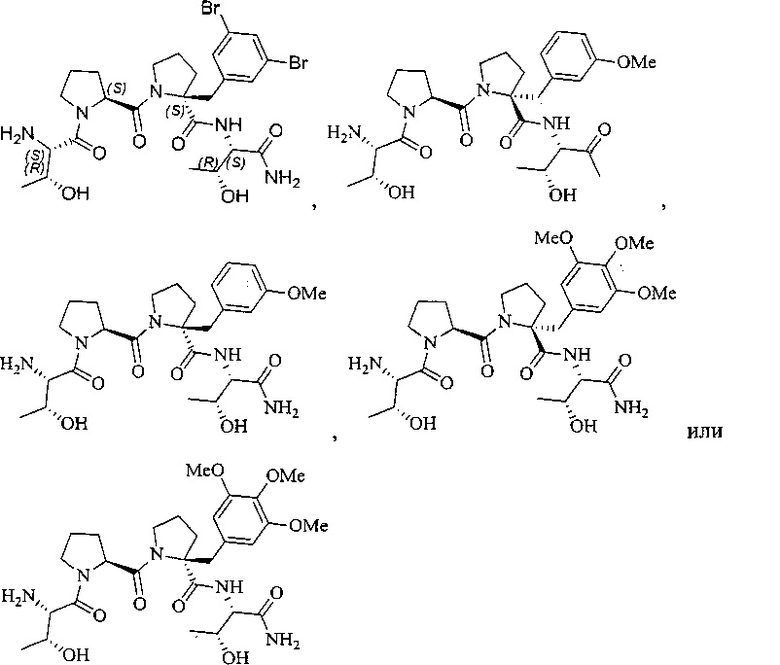
[0077] В иллюстративном варианте реализации соединение может быть представлено структурой:

[0078] В другом иллюстративном варианте реализации соединение может быть представлено структурой:

[0079] В другом иллюстративном варианте реализации соединение может быть представлено структурой:

[0080] В другом иллюстративном варианте реализации соединение может быть представлено структурами:
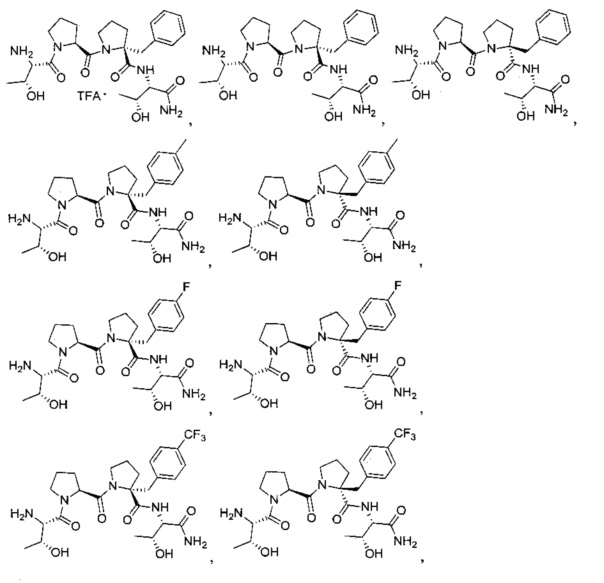

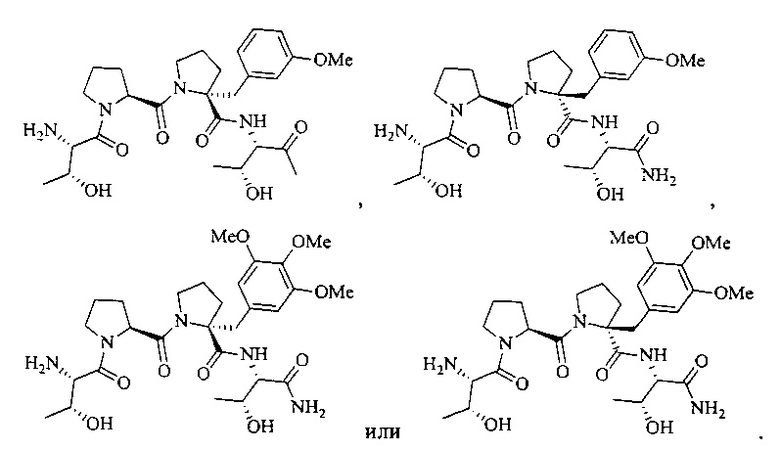
[0081] Описанные соединения включают также соединения, представленные формулой:

и их фармацевтически приемлемые соли, стереоизомеры, метаболиты и гидраты, где:
R1 и R3 могут быть независимо выбраны из группы, состоящей из водорода; галогена; циклического или ациклического, замещенного или незамещенного, разветвленного или неразветвленного алифатического; циклического или ациклического, замещенного или незамещенного, разветвленного или неразветвленного гетероалифатического; замещенного или незамещенного арила; замещенного или незамещенного гетероарила; -ORx; -NO2; -N3; -CN; -SCN; -SRx; -C(O)Rx; -CO2(Rx); -C(O)N(Rx)2; -C(NRx)N(Rx)2; -OC(O)Rx; -OCO2Rx; -OC(O)N(Rx)2; -N(Rx)2; -SORx; -S(O)2Rx; -NRxC(O)Rx; -NRxC(O)N(Rx)2; -NRxC(O)ORx; -NRxC(NRx)N(Rx)2; и -C(Rx)3; где Rx в каждом случае независимо выбран из группы, состоящей из водорода; галогена; ацила; необязательно замещенного алифатического; необязательно замещенного гетероалифатического; необязательно замещенного арила; и необязательно замещенного гетероарила;
R2 и R4 могут быть независимо выбраны из группы, состоящей из водорода и -ORx, при условии, что по меньшей мере один из R2 и R4 представляет собой водород, при этом Rx выбран из группы, состоящей из водорода; галогена; ацила; необязательно замещенного алифатического; необязательно замещенного гетероалифатического; необязательно замещенного арила; и необязательно замещенного гетероарила;
R5 и R6 могут быть независимо выбраны из группы, состоящей из -Q-Ar и водорода; при этом Q независимо выбран из группы, состоящей из циклического или ациклического, замещенного или незамещенного, разветвленного или неразветвленного алифатического; циклического или ациклического, замещенного или незамещенного, разветвленного или неразветвленного гетероалифатического; и связи; и при этом Ar выбран из группы, состоящей из замещенного или незамещенного арила, и замещенного или незамещенного гетероарила; или R5 и R6, вместе с атомами, к которым они присоединены, образуют замещенное или незамещенное 4-6-членное гетероциклическое или циклоалкильное кольцо;
R7 и R8 независимо выбраны из группы, состоящей из водорода; галогена; гидроксила; замещенного или незамещенного С1-С6 алкила; замещенного или незамещенного С1-С6 алкокси; и замещенного или незамещенного арила; или R7 и R8, вместе с атомами, к которым они присоединены, образуют замещенное или незамещенное 4-6-членное гетероциклическое или циклоалкильное кольцо;
R9 и R10 могут быть независимо выбраны из группы, состоящей из водорода; С1-С6 алкила, необязательно замещенного одним или более заместителями, каждый независимо выбран из группы, состоящей из галогена, оксо и гидроксила; С2-6алкенила, необязательно замещенного одним или более заместителями, каждый независимо выбран из группы, состоящей из галогена, оксо и гидроксила; С2-6алкинила, необязательно замещенного одним или более заместителями, каждый независимо выбран из группы, состоящей из галогена, оксо и гдроксила; С3-6циклоалкила, необязательно замещенного одним или более заместителями, каждый независимо выбран из группы, состоящей из С1-6алкила, галогена, оксо и гидроксила; фенила, необязательно замещенного одним или более заместителями, каждый независимо выбран из группы, состоящей из C1-6алкила; C1-6алкокси; галогена; гидроксила; -C(O)Rx; -CO2(Rx); -C(O)N(Rx)2; -C(NRx)N(Rx)2; и -C(Rx)3; где Rx в каждом случае независимо выбран из группы, состоящей из водорода; галогена; С1-6алкила; С2-6алкенила; С2-6алкинила; С3-6циклоалкила; и фенила; или R9 и R10, вместе с N, образуют 4-6-членное гетероциклическое кольцо, необязательно замещенное одним или более заместителями, каждый независимо выбран из группы, состоящей из C1-6алкила, галогена, оксо и гидроксила.
[0082] В некоторых вариантах реализации R1 и R3 могут быть независимо выбраны из группы, состоящей из водорода; галогена; C1-6алкила; С2-6алкенила; С2-6алкинила; С3-6циклоалкила; фенила; нафтила; гетероарила; гетероциклила; С3-6циклоалкил-С1-6алкил-; фенил-С1-6алкил-; нафтил-С1-6алкил-; гетероарил-С1-6алкил-; и гетероциклил-С1-6алкил-; -ORx; -NO2; -N3; -CN; -SCN; -SRx; -C(O)Rx; -CO2(Rx); -C(O)N(Rx)2; -C(NRx)N(Rx)2; -OC(O)Rx; -OCO2Rx; -OC(O)N(Rx)2; -N(Rx)2; -SORx; -S(O)2Rx; -NRxC(O)Rx; -NRxC(O)N(Rx)2; -NRxC(O)ORx; -NRxC(NRx)N(Rx)2; и -C(Rx)3; где гетероарил представляет собой 5-6-членное кольцо, имеющее один, два или три гетероатома, каждый независимо выбран из N, О или S; при этом гетероарил необязательно замещен одним или более заместителями, каждый независимо выбран из Rb; при этом гетероциклил представляет собой 4-7-членное кольцо, необязательно замещенное одним или более заместителями, каждый независимо выбран из Rc; при этом если гетероциклил содержит фрагмент -NH-, то фрагмент -NH- необязательно замещен Rd; при этом С2-6алкенил и С2-6алкинил, каждый независимо, необязательно замещены одним или более заместителями, каждый независимо выбран из Re; при этом С1-6алкил необязательно замещен одним или более заместителями, каждый независимо выбран из Rf; при этом С3-6циклоалкил независимо необязательно замещен одним или более заместителями, каждый независимо выбран из Rg;
Rb в каждом случае независимо может быть выбран из группы, состоящей из галогена; гидроксила; -NO2; -N3; -CN; -SCN; С1-6алкила; С2-6алкенила; С2-6алкинила; С3-6циклоалкила; C1-6алкокси; С3-6алкенилокси; С3-6алкинилокси; С3-6циклоалкокси; С1-6алкил-S(O)w-, где w равен 0, 1 или 2; C1-6алкилС3-6циклоалкил-; С3-6циклоалкил-С1-6алкил-; C1-6aлкoкcикapбoнил-N(Ra)-; C1-6aлκилN(Ra)-; С1-6алкил-N(Ra)карбонил-; RaRa'N-; RaRa'N-карбонил-; RaRa'N-карбонил-N(Ra)-; RaRa'N-SO2-; и С1-6алкил-карбонил-N(Ra)-;
Ra и Ra'' в каждом случае независимо могут быть выбраны из группы, состоящей из водорода и C1-6алкила, или Ra и Ra', взятые вместе с атомом азота, к которому они присоединены, образуют 4-6-членное гетероциклическое кольцо, при этом C1-6алкил необязательно замещен одним или более заместителями, каждый независимо выбран из группы, состоящей из галогена, оксо и гидроксила, и при этом указанное гетероциклическое кольцо необязательно замещено одним или более заместителями, каждый независимо выбран из группы, состоящей из галогена, алкила, оксо или гидроксила;
Rc в каждом случае независимо может быть выбран из группы, состоящей из галогена; гидроксила; -NO2; -N3; -CN; -SCN; оксо; С1-6алкила; С2-6алкенила; С2-6алкинила; С3-6циклоалкила; С1-6алкокси; С3-6алкенилокси; С3-6алкинилокси; С3-6диклоалкокси; C1-6алкил-S(O)w-, где w равен 0, 1 или 2; C1-6алкилС3-6циклоалкил-; С3-6циклоалкил-С1-6алкил-; С1-6алкоксикарбонил-N(Ra)-; С1-6алкилN(Ra)-; С1-6алкил-N(Ra)карбонил-; RaRa'N-; RaRa'N-карбонил-; RaRa'N-карбонил-N(Ra)-; RaRa'N-SO2-; и С1-6алкил-карбонил-N(Ra)-;
Rd в каждом случае независимо может быть выбран из группы, состоящей из C1-6алкила, C1-6алкилкарбонила и С1-6алкилсульфонила, при этому C1-6алкил необязательно замещен одним или более заместителями, каждый независимо выбран из галогена, гидроксила и RaRa'N-;
Re в каждом случае независимо может быть выбран из группы, состоящей из галогена; гидроксила; -NO2; -N3; -CN; -SCN; С1-4алкокси; С1-4алкоксикарбонила; RaRa'N-; RaRa'N-карбонила; RaRa'N-SO2-; и С1-4алкилS(O)w-, где w равен 0, 1 или 2;
Rf в каждом случае независимо может быть выбран из группы, состоящей из галогена; гидроксила; -NO2; -N3; -CN; -SCN; С1-4алкокси; С1-4алкоксикарбонила; RaRa'N-; RaRa'N-карбонила; RaRa'N-SO2-; и С1-4алкилS(O)w-, где w равен 0, 1 или 2;
Rg в каждом случае независимо может быть выбран из группы, состоящей из галогена, гидроксила, -NO2; -N3; -CN; -SCN; C1-6алкила; С1-4алкокси; С1-4алкоксикарбонила; RaRa'N-; RaRa'N-карбонила; RaRa'N-SO2-; и C1-4алкилS(O)w-, где w равен 0, 1 или 2;
Rx может быть независимо выбран из группы, состоящей из водорода; галогена; C1-6алкила; С2-6алкенила; С2-6алкинила; С3-6циклоалкила; фенила; нафтила; гетероарила; гетероциклила; С3-6циклоалкил-С1-6алкил-; фенил-С1-6алкил-; нафтил-С1-6алкил-; гетероарил-С1-6алкил-; и гетероциклил-С1-6алкил-; при этом гетероарил представляет собой 5-6-членное кольцо, имеющее один, два или три гетероатома, каждый независимо выбран из N, О или S; при этом гетероарил необязательно замещен одним или более заместителями, каждый независимо выбран из Rb; при этом гетероциклил представляет собой 4-7-членное кольцо, необязательно замещенное одним или более заместителями, каждый независимо выбран из Rc; при этом если гетероциклил содержит фрагмент -NH-, то фрагмент -NH- необязательно замещен Rd; при этом С2-6алкенил и С2-6алкинил, каждый независимо, необязательно замещены одним или более заместителями, каждый независимо выбран из Re; при этом С1-6алкил необязательно замещен одним или более заместителями, каждый независимо выбран из Rf; при этом С3-6циклоалкил независимо необязательно замещено одним или более заместителями, каждый независимо выбран из Rg.
[0083] В некоторых случаях R2 и R4 могут быть независимо выбраны из труппы, состоящей из водорода и -ORx, при условии, что по меньшей мере один из R2 и R4 представляет собой водород, при этом Rx может быть выбран из группы, состоящей из водорода; галогена; C1-6алкила; С1-6алкенила; С1-6алкинила; С3-6циклоалкила; фенила; нафтила; гетероциклила; С3-6циклоалкил-С1-6алкил-; фенил-С1-6алкил-; нафтил-С1-6алкил-; гетероарил-С1-6алкил; и гетероциклил-С1-6алкил-; при этом гетероарил представляет собой 5-6-членное кольцо, имеющее один, два или три гетероатома, каждый независимо выбран из N, О или S; при этом гетероарил необязательно замещен одним или более заместителями, каждый независимо выбран из Rb; при этом гетероциклил представляет собой 4-7-членное кольцо, необязательно замещенное одним или более заместителями, каждый независимо выбран из Rc; при этом если гетероциклил содержит фрагмент -NH-, то фрагмент -NH- необязательно замещен Rd; при этом С2-6алкенил и С2-6алкинил, каждый независимо, необязательно замещены одним или более заместителями, каждый независимо выбран из Re; при этом С1-6алкил необязательно замещен одним или более заместителями, каждый независимо выбран из Rf; при этом С3-6циклоалкил независимо необязательно замещен одним или более заместителями, каждый независимо выбран из Rg;
Rb в каждом случае независимо может быть выбран из группы, состоящей из галогена; гидроксила; -NO2; -N3; -CN; -SCN; С1-6алкила; С2-6алкенила; С2-6алкинила; С3-6циклоалкила; C1-6алкокси; С3-6алкенилокси; С3-6алкинилокси; С3-6циклоалкокси; С1-6алкил-S(O)w-, где w равен 0, 1 или 2; C1-6алкилС3-6циклоалкил-; С3-6циклоалкил-C1-6алкил-; С1-6алкоксикарбонил-N(Ra)-; C1-6aлκилN(Ra)-; C1-6aлкил-N(Ra)карбонил-; RaRa'N-; RaRa'N-карбонил-; RaRa'N-карбонил-N(Ra)-; RaRa'N-SO2-; и C1-6алкил-карбонил-N(Ra)-;
Ra и Ra' в каждом случае независимо могут быть выбраны из группы, состоящей из водорода и C1-6алкила, или Ra и Ra', взятые вместе с атомом азота, к которому они присоединены, образуют 4-6-членное гетероциклическое кольцо, при этом С1-6алкил необязательно замещен одним или более заместителями, каждый независимо выбран из группы, состоящей из галогена, оксо и гидроксила, и при этом указанное гетероциклическое кольцо необязательно замещено одним или более заместителями, каждый независимо выбран из группы, состоящей из галогена, алкила, оксо или гидроксила;
Rc в каждом случае независимо может быть выбран из группы, состоящей из галогена; гидроксила; -NO2; -N3; -CN; -SCN; оксо; C1-6алкила; С2-6алкенила; С2-6алкинила; С3-6циклоалкила; C1-6алкокси; С3-6алкенилокси; С3-6алкинилокси; С3-6циклоалкокси; C1-6алкли-S(O)w-, где w равен 0, 1 или 2; С1-6алкилС3-6циклоалкил-; С3-6циклоалкил-С1-6алкил-; C1-6алкоксикарбонил-N(Ra)-; С1-6алкилN(Ra)-; C1-6алкил-N(Ra)карбонил-; RaRa'N-; RaRa'N-карбонил-; RaRa'N-карбонил-N(Ra)-; RaRa'N-SO2-; и С1-6aлκил-карбонил-N(Ra)-;
Rd в каждом случае независимо может быть выбран из группы, состоящей из С1-6алкила, C1-6алкилкарбонила и С1-6алкилсульфонила, при этому С1-6алкил необязательно замещен одним или более заместителями, каждый независимо выбран из галогена, гидроксила и RaRa'N-;
Re в каждом случае независимо может быть выбран из группы, состоящей из галогена; гидроксила; -NO2; -N3; -CN; -SCN; С1-4алкокси; C1-6алкоксикарбонила; RaRa'N-; RaRa'N-карбонила; RaRa'N-SO2-; и С1-4алкилS(O)w-, где w равен 0, 1 или 2;
Rf в каждом случае независимо может быть выбран из группы, состоящей из галогена; гидроксила; -NO2; -N3; -CN; -SCN; С1-4алкокси; C1-6алкоксикарбонила; RaRa'N-; RaRa'N-карбонила; RaRa'N-SO2-; и C1-4алкилS(O)w-, где w равен 0, 1 или 2;
Rg в каждом случае независимо может быть выбран из группы, состоящей из галогена, гидроксила, -NO2; -N3; -CN; -SCN; C1-6алкила; С1-4алкокси; С1-4алкоксикарбонила; RaRa'N-; RaRa'N-карбонила; RaRa'N-SO2-; и С1-4алкилS(O)w-, где w равен 0, 1 или 2.
[0084] В некоторых вариантах реализации R5 и R6 могут быть независимо выбраны из группы, состоящей из -Q-Ar и водорода; при этом Q независимо выбран из группы, состоящей из C1-6алкила; С2-6алкенила; С2-6алкинила; С3-6циклоалкила; гетероциклила; С3-6циклоалкил-С1-6алкил-; гетероциклил-С1-6алкил-; и связи; и при этом Ar выбран из группы, состоящей из замещенного или незамещенного фенила, нафтила или гетероарила; или R5 и R6, вместе с атомами, к которым они присоединены, образуют 4-6-членное гетероциклическое или циклоалкильное кольцо, необязательно замещенное одним или более заместителями, каждый независимо выбран из галогена, гидроксила, -NO2; -N3; -CN; -SCN; C1-6алкила; С1-4алкокси; С1-4алкоксикарбонила; RaRa'N-; RaRa'N-карбонила; RaRa'N-SO2-; и С1-4алкилS(O)w-, где w равен 0, 1 или 2; и
где Ra и Ra' в каждом случае независимо могут быть выбраны из группы, состоящей из водорода и C1-6алкила, или Ra и Ra', взятые вместе с атомом азота, к которому они присоединены, образуют 4-6-членное гетероциклическое кольцо, при этом С1-6алкил необязательно замещен одним или более заместителями, каждый независимо выбран из группы, состоящей из галогена, оксо и гидроксила, и при этом указанное гетероциклическое кольцо необязательно замещено одним или более заместителями, каждый независимо выбран из группы, состоящей из галогена, алкила, оксо или гидроксила.
[0085] В некоторых вариантах реализации по меньшей мере один из R1, R2, R3 и R4 может быть гидроксилом.
[0086] В некоторых случаях по меньшей мере один из R1, R2, R3 и R4 может быть C1-С6 алкилом, необязательно замещенным одним, двумя или тремя заместителями, независимо выбранными из группы, состоящей из галогена, гидроксила, -NH2 и циано.
[0087] В некоторых вариантах реализации по меньшей мере один из R5 и R6 может быть -(C1-С6 алкилен)-Ar, например, один из R5 и R6 также может быть -СН2-Ar. В некоторых случаях по меньшей мере один из R5 и R6 представляет собой -Q-фенил. В некоторых примерах один из R5 и R6 может быть водородом.
[0088] В некоторых случаях R7 и R8 могут быть независимо выбраны из группы, состоящей из водорода; галогена; гидроксила; C1-С6 алкила; фенила; и нафтила; или R7 и R8, вместе с атомами, к которым они присоединены, образуют 4-6-членное гетероциклическое или циклоалкильное кольцо; при этом каждый C1-С6 алкил, фенил, нафтил, циклоалкильное кольцо или гетероциклическое кольцо могут быть независимо замещены одним или более заместителями, выбранными из группы, состоящей из галогена; гидроксила; -NO2; -N3; -CN; -SCN; С1-4алкокси; С1-4алкоксикарбонила; RaRa' N-; RaRa' N-карбонила; RaRa'N-SO2-; и С1-4алкилS(O)w, где w равен 0, 1 или 2; при этом Ra и Ra' могут быть каждом случае независимо выбраны из группы, состоящей из водорода и C1-6алкила, или Ra и Ra', взятые вместе с атомом азота, к которому они присоединены, образуют 4-6-членное гетероциклическое кольцо, при этом С1-6алкил необязательно замещен одним или более заместителями, каждый независимо выбран из группы, состоящей из галогена, оксо и гидроксила, и при этом указанное гетероциклическое кольцо необязательно замещено одним или более заместителями, каждый независимо выбран из группы, состоящей из галогена, алкила, оксо или гидроксила.
[0089] В некоторых случаях R7 и R8 могут быть водородом.
[0090] В иллюстративном варианте реализации соединение может быть представлено структурой:

[0091] В другом иллюстративном варианте реализации соединение может быть представлено структурой:

[0092] В другом иллюстративном варианте реализации соединение может быть представлено структурой:
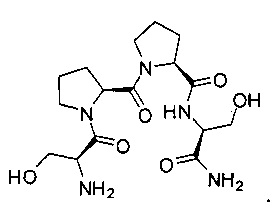
[0093] Соединения настоящего описания и их композиции могут иметь множество хиральных центров. Каждый хиральный центр может быть независимо R, S или смесью R и S. Например, в некоторых вариантах реализации хиральный центр может иметь соотношение R:S от около 100:0 до около 50:50, от около 100:0 до около 75:25, от около 100:0 до около 85:15, от около 100:0 до около 90:10, от около 100:0 до около 95:5, от около 100:0 до около 98:2, от около 100:0 до около 99:1, от около 0:100 до 50:50, от около 0:100 до около 25:75, от около 0:100 до около 15:85, от около 0:100 до около 10:90, от около 0:100 до около 5:95, от около 0:100 до около 2:98, от около 0:100 до около 1:99, от около 75:25 до 25:75, и около 50:50. Композиции описанных соединений, содержащие более высокие соотношения одного или более изомеров (то есть R и/или S) могут обладать улучшенными терапевтическими характеристиками, по сравнению с рацемическими композициями описанных соединений или смесей соединений.
[0094] Описанные соединения могут обеспечивать эффективное открывание катионного канала у рецептора НМДА, например, могут связываться или ассоциироваться с глутаматным сайтом рецептора НМДА, способствуя открыванию катионного канала. Описанные соединения могут быть использованы для регулирования (включения или выключения) рецептора НМДА за счет их агонистического действия.
[0095] Соединения, описанные в настоящем документе, могут быть частичными агонистами глицинового сайта рецептора НМДА. Частичный агонист, при использовании в данном контексте, следует понимать как обозначение того, что при низких концентрациях указанный аналог действует в качестве агониста, а при высоких концентрациях указанный аналог действует в качестве антагониста. Связывание глицина не ингибируется глутаматом или конкурентными ингибиторами глутамата, а также он не связывается с тем же сайтом, что и глутамат на рецепторе НМДА. На рецепторе НМДА находится второй и отдельный сайт связывания глицина. Таким образом, управляемый лигандом ионный канал рецептора НМДА управляется по меньшей мере двумя этими отдельными аллостерическими сайтами. Описанные соединения могут быть способны связываться или ассоциироваться с глицин-связывающим сайтом рецептора НМДА. В некоторых вариантах реализации описанные соединения могут обладать эффективностью, которая в 10 раз или более превышает активность существующих частичных агонистов глицинового сайта рецептора НМДА. Например, описанные соединения могут обладать в 10-20 раз более высокой эффективностью, по сравнению с GLYX-13. GLYX-13 представлен структурой:
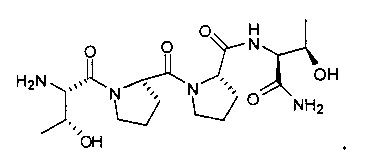
[0096] Например, в настоящем документе представлены соединения, которые могут быть по меньшей мере примерно в 20 раз более эффективными, по сравнению с GLYX-13, по результатам измерения активируемой импульсом проводимости рецептор-управляемого единичного нейрона НМДА (INMDA) в культуре гиппокампальных пирамидальных нейронов СА1 в концентрации 50 нМ. В другом варианте реализации представленное соединение может быть способно создавать увеличенную проводимость рецептор-управляемого единичного нейрона НМДА, спровоцированную однократным импульсом (INMDA), в гиппокампальных пирамидальных нейронах СА1 в концентрации от 100 нМ до 1 мкМ. Описанные соединения могут обладать улучшенной эффективностью по сравнению с GLYX-13, по результатам измерения амплитуды долговременного потенцирования (ДВП) в синапсах коллатерали Шаффера-СА-1 в гиппокампальных срезах in vitro.
[0097] Описанные соединения могут демонстрировать высокий терапевтический индекс. Терапевтический индекс, при использовании в настоящем документе, относится к соотношению дозы, которая вызывает токсичность в 50% популяции (то есть TD50), к минимальной эффективной дозе для 50% указанной популяции (то есть ED50). Следовательно, терапевтический индекс =(TD50):(ED50). В некоторых вариантах реализации описанное соединение может иметь терапевтический индекс, составляющий по меньшей мере около 10:1, по меньшей мере около 50:1, по меньшей мере около 100:1, по меньшей мере около 200:1, по меньшей мере около 500:1 или по меньшей мере около 1000:1.
Композиции
[0098] В других аспектах представлены препараты и композиции, содержащие описанные соединения и необязательно фармацевтически приемлемое вспомогательное вещество. В некоторых вариантах реализации рассматриваемая композиция содержит рацемическую смесь одного или более из описанных соединений.
[0099] Рассматриваемые композиции могут быть приготовлены в любой из многочисленных форм применения. В качестве примера, а не ограничения, указанные соединения могут быть приготовлены в композиции, пригодной для перорального введения, подкожной инъекции или для других способов введения активного агента животному, известных в области фармацевтики.
[00100] Количество описанного соединения в композиции, описанной в настоящем документе, может варьироваться в зависимости от таких факторов как состояние заболевания, возраст, пол и вес индивидуума. Режимы дозирования могут быть подобраны для обеспечения оптимального терапевтического ответа. Например, может быть введен однократный болюс, могут быть введены несколько дробных доз в течение определенного времени, или доза может пропорционально уменьшаться или увеличиваться, в соответствии с требованиями терапевтической ситуации. Особенно преимущественно составлять парентеральные композиции в единичной дозированной форме для простоты введения и равномерности дозирования. Единичная дозированная форма, при использовании в настоящем документе, относится к физически дискретным единицам, пригодным в качестве однократных доз для млекопитающих субъектов, подлежащих лечению; каждая единица содержит определенное количество активного соединения, рассчитанное для обеспечения заданного терапевтического эффекта, в сочетании с заданным фармацевтическим носителем.
[00101] Характеристики единичных лекарственных форм настоящего изобретения обусловлены и напрямую зависят от (а) уникальных характеристик выбранного соединения и конкретного ожидаемого терапевтического эффекта, а также от (b) ограничений, существующих в области составления композиций, таких как активный агент для лечения сенситивности у пациентов.
[00102] Терапевтические композиции, как правило, должны быть стерильными и стабильными в условиях производства и хранения. Композиция может быть составлена в виде раствора, микроэмульсии, липосомы или другой упорядоченной структуры, пригодной для высокой концентрации лекарства. Носителем может быть растворитель или дисперсионная среда, содержащая, например, воду, этанол, полиол (например, глицерин, пропиленгликоль и жидкий полиэтиленгликоль, и так далее) и их подходящие смеси. Необходимая текучесть может поддерживаться, например, за счет использования покрытий, таких как лецитин, путем сохранения нужного размера частиц в случае дисперсий, а также при помощи поверхностно-активных веществ. Во многих случаях предпочтительно включать в композицию изотонические агенты, например, сахара, многоатомные спирты, такие как маннит или сорбит, или как хлорид натрия. Пролонгированное поглощение инъецируемых композиций может быть осуществлено за счет включения в композицию агента, замедляющего абсорбцию, например, моностеаратных солей и желатина.
[00103] Соединения могут быть введены в форме композиции с временным высвобождением, например, в композиции, которая содержит полимер с замедленным высвобождением. Соединения могут быть приготовлены с носителями, которые защищают соединение от быстрого высвобождения, как в композиции с контролируемым высвобождением, включая имплантаты и микроинкапсулированные системы доставки. Могут быть использованы биоразлагаемые, биосовместимые полимеры, такие как этиленвинилацетат, полиангидриды, полигликолевая кислота, коллаген, полиортоэфиры, полимолочная кислота и сополимеры полимолочной и полигликолевой кислот (PLG). Многие способы получения таких композиций хорошо известны специалистам в данной области.
[00104] Стерильные инъецируемые растворы могут быть получены введением необходимого количества соединения в подходящий растворитель, при необходимости, с одним компонентом или комбинацией компонентов, перечисленных выше, с последующей стерилизацией фильтрованием. Как правило, дисперсии получают введением активного соединения в стерильный жидкий носитель, который содержит основную дисперсионную среду и другие необходимые ингредиенты из перечисленных выше. В случае стерильных порошков для приготовления стерильных растворов для инъекций, предпочтительные способ приготовления представляют собой вакуумную сушку и вымораживание, в результате которых из предварительно стерильно отфильтрованного раствора получают порошок активного компонента плюс любой дополнительный заданный компонент.
[00105] В некоторых вариантах реализации, определенные описанные соединения могут доставлять эффективное количество соединения при пероральном введении пациенту. Например, в некоторых вариантах реализации определенные описанные соединения являются более эффективными при пероральном введении пациенту, по сравнению с пероральным введением пациенту пептидилового соединения, представленного структурой:

[00106] В соответствии с альтернативным аспектом настоящего изобретения, соединение может быть составлено в композицию с одним или более дополнительными соединениями, которые улучшают растворимость указанного соединения.
Способы
[00107] Представлены способы лечения когнитивных расстройств и улучшения обучаемости. Такие способы включают введение фармацевтически приемлемой композиции одного или более из описанных соединений пациенту, нуждающемуся в этом. Также рассмотрены способы лечения пациентов, страдающих от дефицита внимания, связанного со старением, шизофрении, нестандартных расстройств обучаемости, пароксизма, судорог после инсульта, ишемии головного мозга, гипогликемии, остановки сердца, эпилепсии, мигрени, а также болезни Хантингтона, Паркинсона и Альцгеймера.
[00108] Другие рассмотренные способы включают лечение церебральной ишемии, инсульта, травмы головного мозга, опухолей головного мозга, острой невропатической боли, хронической невропатической боли, расстройств сна, наркотической зависимости, депрессии, некоторых расстройств зрения, синдрома отмены алкоголя, боязни, нарушений памяти и обучаемости, аутизма, эпилепсии, СПИД-деменции, множественной системной атрофии, прогрессирующего надъядерного паралича, атаксии Фридриха, синдрома Дауна, синдрома ломкой Х-хромосомы, туберозного склероза, оливо-понто-церебеллярной атрофии, церебрального паралича, медикаментозного неврита зрительного нерва, периферической невропатии, миелопатии, ишемической ретинопатии, диабетической ретинопатии, глаукомы, остановки сердца, поведенческих нарушений, расстройств импульсного контроля, болезни Альцгеймера, потери памяти, которая сопутствует болезни Альцгеймера на ранней стадии, расстройства дефицита внимания, СДВГ, шизофрении, облегчения опиатной, никотиновой зависимости, алкогольной зависимости, травматического повреждения головного мозга, повреждения спинного мозга, пост-травматического стрессового синдрома и хореи Хантингтона.
[00109] Например, в настоящем документе представлен способ лечения депрессии у пациента, нуждающегося в это, включающий введение описанного соединения, например, острое введение описанного соединения. В некоторых вариантах реализации пациента, резистентного к лечению, идентифицируют как пациента, который проходил лечение по меньшей мере двумя типами антидепрессантных лекарств до введения описанного соединения. В других вариантах реализации пациент, резистентный к лечению, представляет собой пациента, которого определяют как не желающего или не способного переносить побочные эффекты по меньшей мере одного типа антидепрессантного лечения.
[00110] Наиболее распространенные состояния депрессии включают большое депрессивное расстройство и дистимическое расстройство. Другие состояния депрессии развиваются при уникальных обстоятельствах. Такие состояния депрессии включают, но не ограничиваясь этим, психотическую депрессию, послеродовую депрессию, сезонное аффективное расстройство (САР), расстройство настроения, депрессии, обусловленные хроническими медицинскими состояниями, такими как рак или хроническая боль, химиотерапия, хронический стресс, пост-травматические стрессовые расстройства и биполярное расстройство (или маниакально-депрессивное расстройство).
[00111] Рефрактерная депрессия возникает у пациентов, страдающих от депрессии, которые являются резистентными к стандартным фармакологическим способам лечения, включая трициклические антидепрессанты, ингибиторы моноаминоксидазы (ИМАО), селективные ингибиторы обратного захвата серотонина (СИОЗС) и двойные и тройные ингибиторы захвата и/или анксиолитические лекарства, а также к нефармакологическим способам лечения, таким как психотерапия, электросудорожная терапия, стимуляция блуждающего нерва и/или транскраниальная магнитная стимуляция. Резистентный к лечению пациент может быть определен как пациент с неудачной попыткой облегчения одного или более симптомов депрессии (например, постоянное тревожное или мрачное чувство, ощущение беспомощности, безнадежности, пессимизма), несмотря на то, что он прошел одно или более стандартных фармакологических или нефармакологических лечений. В некоторых вариантах реализации пациент, резистентный к лечению, представляет собой пациента с неудачной попыткой облечения одного или более симптомов депрессии, несмотря на то, что он прошел лечение двумя различными антидепрессантными лекарствами. В других вариантах реализации пациент, резистентный к лечению, представляет собой пациента с неудачной попыткой облегчения одного или более симптомов депрессии, несмотря на то, что Он прошел лечение четырьмя различными антидепрессантными лекарствами. Пациент, резистентный к лечению, также может быть определен как пациент, который не желает или не в состоянии переносить побочные эффекты одного или более стандартных фармакологических или нефармакологических лечений.
[00112] В другом аспекте представлен способ усиления облегчения боли и обеспечения обезболивания у животного.
[00113] В некоторых вариантах реализации представлены способы лечения шизофрении. Например, с помощью способов и композиций, рассмотренных в настоящем документе, можно лечить шизофрению параноидального типа, шизофрению гебефренического типа (то есть гебефреническую шизофрению), шизофрению кататонического типа, шизофрению недифференцированного типа, шизофрению остаточного типа, пост-шизофреническую депрессию и простую шизофрению. С помощью композиций, рассмотренных в настоящем документе, можно лечить также психотические расстройство, такие как шизоаффективные расстройства, бредовые расстройства, кратковременные психотические расстройства, индуцированные психотические расстройства и психотические расстройствами с бредом или галлюцинациями.
[00114] Параноидальная шизофрения может характеризоваться наличием бреда или слуховых галлюцинаций, хотя отсутствует психическое расстройство, дезорганизованное поведение или аффективная тупость. Бред может быть бредом преследования и/или претенциозным бредом, но помимо этого могут также присутствовать другие темы, такие как ревнивость, религиозность или соматизация.
[00115] Шизофрения гебефренического типа может характеризоваться при одновременном наличии психического расстройства и уплощенного аффекта.
[00116] Шизофрения кататонического типа может характеризоваться в случае если субъект может быть почти неподвижным или проявлять возбужденное, бесцельное движение. Симптомы могут включать кататонический ступор и восковую гибкость.
[00117] Шизофрения недифференцированного типа может характеризоваться при наличии психотических симптомов, но при несоответствии критериям параноидального, гебефренического или кататонического типа.
[00118] Шизофрения остаточного типа может характеризоваться при наличии лишь положительных симптомов низкой интенсивности.
[00119] Пост-шизофреническая депрессия может характеризоваться при возникновении депрессивного эпизода вследствие шизофренической болезни, при этом могут присутствовать некоторые слабые шизофренические симптомы.
[00120] Простая шизофрения может характеризоваться постепенным и прогрессирующим развитием явных негативных симптомов в отсутствие истории психотических эпизодов.
[00121] В некоторых вариантах реализации представлены способы лечения психотических симптомов, которые могут присутствовать при других психических расстройствах, включая, но не ограничиваясь этим, биполярное расстройство, пограничное расстройство личности, наркотическая интоксикация и наркотический психоз.
[00122] В другом варианте реализации представлены способы лечения бреда (например, «не причудливого»), который может иметь место, например, при бредовом расстройстве.
[00123] Также представлены способы лечения социального отчуждения в состояниях, включающих, но не ограничиваясь этим, социальное тревожное расстройство, личностное расстройство избегания и шизотипическое расстройство личности.
[00124] Дополнительно представлены способы лечения обсессивно-компульсивного расстройства (ОКР).
ПРИМЕРЫ
[00125] Следующие примеры представлены лишь для иллюстративных целей, и не предназначены для ограничения рамок настоящего описания.
Пример 1 - Синтез (S)-N-((2S,3R)-1-амино-3-гидрокси-1-оксобутан-2-ил)-1-((S)-1-((S)-2-амино-3-гидроксипропаноил)пирролидин-2-карбонил)пирролидин-2-карбоксамида (CM-1).
[00126] Для синтеза (S)-N-((2S,3R)-1-амино-3-гидрокси-1-оксобутан-2-ил)-1-((S)-1-((S)-2-амино-3-гидроксипропаноил)пирролидин-2-карбонил)пирролидин-2-карбоксамида (CM-1) использовали следующую последовательность реакций (Схема А):
Схема А. Синтез CM-1.

[00127] Синтез (S)-трет-бутил 1-((S)-3-ацетокси-2-(бензилоксикарбониламино)-пропаноил)-пирролидин-2-карбоксилата (2):
(S)-3-Ацетокси-2-(бензилоксикарбониламино)-пропановую кислоту (1,5 г, 5,33 ммоль) растворили в CH2Cl2 (15 мл). Добавили N-метилфорфолин (NMM) (0,64 мл, 5,87 ммоль) и изобутилхлорформиат (IBCF) (0,72 мл, 6,12 ммоль) при -15°С и перемешивали в течение 30 минут под инертной атмосферой. К реакционной смеси по каплям добавили смесь (S)-трет-бутил пирролидин-2-карбоксилата (1) (998 мг, 5,87 ммоль) и NMM (0,64 мл, 5,87 ммоль) в ДМФ (5 мл) и продолжали перемешивание еще 3 часа при комнатной температуре. Реакционную смесь разбавили ДХМ (200 мл), промыли водой (50 мл), раствором лимонной кислоты (10 мл) и насыщенным солевым раствором (10 мл). Отделенный органический слой высушили над безводным Na2SO4 и концентрировали под пониженным давлением. Полученный неочищенный остаток очистили силикагелевой колоночной хроматографией, элюируя 30% EtOAc в гексане, с получением соединения 2 (1,6 г, 69,5%).
1H-ЯМР: (200 МГц, ДМСО-d6): δ 7.81-7.76 (d, J=20.5 Гц, 1H), 7.35-7.30 (m, 5Н), 5.03-4.97 (m, 2Н), 4.61-4.55 (m, 1Н), 4.32-4.16 (m, 2H), 4.08-3.87 (m, 2H), 3.65-3.59 (m, 1H), 2.21-2.11 (m, 2H), 1.98 (s, 3H), 1.91-1.75 (m, 2H), 1.37 (s, 9H).
Масса m/z: 435,0 [М++1].
[00128] Синтез (S)-1-((S)-3-ацетокси-2-(бензилоксикарбониламино)-пропаноил)-пирролидин-2-карбоновой кислоты (3):
К раствору соединения 2 (1 г, 2,30 ммоль) в CH2Cl2 (5 мл) добавили 20% ТФК в ДХМ (10 мл) и перемешивали при комнатной температуре в течение 2 часов. Реакционную смесь разбавили водой (10 мл) и экстрагировали EtOAc (2×15 мл). Органический слой высушили над безводным Na2SO4 и концентрировали под пониженным давлением с получением соединения 3 (800 мг, 92%).
1Н-ЯМР: (200 МГц, ДМСО-d6): δ 12.58 (br s, 1Н), 7.81-7.77 (d, J=8.0 Гц, 1H), 7.35-7.27 (m, 5H), 5.04-4.96 (m, 2H), 4.66-4.60 (m, 1H), 4.32-4.24 (m, 2H), 4.04-3.86 (m, 1H), 3.66-3.59 (t, J=12.6 Гц, 2H), 2.17-2.07 (m, 3H), 1.98-1.80 (m, 4H).
Масса m/z: 379,0 [М++1].
[00129] Синтез (2S,3R)-метил 2-((S)-1-((S)-1-((R)-3-ацетокси-2-(бензилоксикарбониламино)-пропаноил)-пирролидин-2-карбонил)пирролидин-2-карбоксамидо)-3-гидроксибутаноата (S):
Соединение 3 (1,0 г, 2.64 ммоль) растворили в CH2Cl2 (10 мл), к реакционной смеси добавили NMM (0,32 г, 3,17 ммоль) и IBCF (0,41 г, 3,04 ммоль) при -15°С и перемешивали в течение 30 минут под инертной атмосферой. К реакционной смеси по каплям добавили смесь (2S,3R)-метил 3-гидрокси-2-((S)-пирролидин-2-карбоксамидо)-бутаноата (4) (0,73 г, 3,17 ммоль) и NMM (0,35 мл) в ДМФ (3 мл) при -15°С и продолжали перемешивание еще 3 часа при комнатной температуре. Реакционную смесь разбавили ДХМ (200 мл), промыли водой (20 мл), раствором лимонной кислоты (2×20 мл) и насыщенным солевым раствором (2×50 мл). Отделенный органический слой высушили над безводным Na2SO4 и концентрировали под пониженным давлением. Полученный неочищенный остаток очистили силикагелевой колоночной хроматографией, элюируя 5% СН3ОН в EtOAc, с получением соединения (S) (0,29 г, 19%).
1H-ЯМР: (500 МГц, ДМСО-d6): δ 7.83-7.81 (m, 1Н), 7.72-7.70 (m, 1H), 7.36-7.35 (m, 5H), 5.07-5.01 (m, 2H), 4.99-4.93 (m, 1H), 4.58 (s, 1H), 4.50-4.48 (m, 1H), 4.26-4.22 (m, 2H), 4.07-4.00 (m, 2H), 3.89-3.86 (m, 1H), 3.61-3.55 (m, 5H), 3.53 (s, 1H), 3.39 (s, 1H), 2.12 (s, 1H), 1.98 (s, 3H), 1.94-1.83 (m, 4H), 1.81-1.80 (m, 3H), 1.05 (d, J=6.5 Гц, 3H).
Масса m/z: 591,0 [M++1].
[00130] Синтез бензил-(R)-1-((S)-2-((S)-2-((2S,3R)-1-(аминоокси)-3-гидрокси-1-оксобутан-2-илкарбамоил)-пирролидин-1-карбонил)-пирролидин-1-ил)-3-гидрокси-1-оксопропан-2-илкарбамата (6):
Метанольный раствор аммиака (3 мл) добавили к соединению 5 (0,28 г, 0,47 ммоль) и перемешивали при комнатной температуре в течение 18 часов. Летучие вещества выпарили под пониженным давлением с получением соединения 6 (0,21 г, 82,3%).
1H-ЯМР: (500 МГц, ДМСО-d6): δ 7.38-7.31 (m, 5Η), 7.26 (s, 1H), 7.10-7.03 (m, 2H), 6.65 (br s, 1H), 5.04-5.01 (m, 2H), 4.98-4.84 (m, 1H), 4.76-4.75 (m, 1H), 4.61 (s, 1H), 4.38-4.31 (m, 2H), 4.02-4.00 (m, 2H), 3.77-3.74 (m, 1H), 3.67-3.56 (m, 3H), 3.44-3.37 (m, 2H), 2.14-1.86 (m, 8H), 1.01-1.00 (m, 3H).
Масса m/z: 550 [M++1].
[00131] Синтез (S)-N-((2S,3R)-1-амино-3-гидрокси-1-оксобутан-2-ил)-1-((S)1-((S)-2-амино-3-гидроксипропаноил)-пирролидин-2-карбонил)-пирролидин-2-карбоксамида (CM-1).
К раствору соединения 6 (0,21 г, 0,39 ммоль) в метаноле (5 мл) добавили 10% Pd/C (30 мг) и перемешивали реакционную смесь под атмосферой водорода в течение 2 часов. Реакционную смесь отфильтровали через целит, растворитель выпарили in vacuo, а полученный неочищенный остаток растерли с диэтиловым эфиром с получением CM-1 (130 мг, 83,3%).
1Н-ЯМР: (500 МГц, ДМСО-d6) (ротамеры): δ 7.39 (d, J=8.0 Гц, 1Н), 7.08-7.03 (m, 2H), 6.65 (br s, 1H), 4.89-4.85 (m, 1H), 1.61-1.59 (m, 1H), 4.39-4.38 (m, 1H), 4.02-4.00 (m, 2H), 3.68-3.52 (m, 4H), 3.43-3.36 (m, 2H), 3.22-3.10 (m, 2H), 2.19-2.13 (m, 1H), 2.07-1.98 (m, 1H), 1.93-1.81 (m, 5H), 1.75 (s, 2H), 1.01-1.00 (m, 3H).
ЖХМС m/z: 400,2 [M++1].
Чистота по ВЭЖХ: 99,27%.
[00132] Синтез (S)-1-(бензилоксикарбонил)пирролидин-2-карбоновой кислоты (8):
К перемешанному раствору (S)-пирролидин-2-карбоновой кислоты (7) (2,0 г, 17,39 ммоль) в ТГФ: H2O (20 мл, 1:1) добавили Na2CO3 (2,76 г, 26,08 ммоль) и Cbz-Cl (3,54 г, 20,80 ммоль) и перемешивали при комнатной температуре в течение 18 часов. Реакционную смесь промыли EtOAc (10 мл), а водный слой подкислили с помощью 3 н. HCl и экстрагировали EtOAc (2×20 мл). Объединенный органический слой промыли насыщенным солевым раствором, высушили над безводным Na2SO4 и концентрировали под пониженным давлением с получением соединения 8 (3,0 г, 69,7%).
1H-ЯМР: (500 МГц, ДМСО-d6): δ 12.62 (br s, 1Н), 7.36-7.22 (m, 5H), 5.12-5.00 (m, 2H), 4.24-4.15 (dd, J=5.0, 36.0 Гц, 1H), 3.46-3.31 (m, 2H), 2.25-2.15 (m, 1H), 1.94-1.79 (m, 3H).
Масса m/z: 250,0 [М++1].
[00133] Синтез (S)-бензил 2-((2S, 3R)-3-гидрокси-1-метокси-1-оксобутан-2-илкарбамоил)пирролидин-1-карбосилата (9):
Соединение 8 (5,0 г, 20,08 ммоль) растворили в CH2Cl2 (50 мл), добавили NMM (2,43 мл, 22,08 ммоль) и D3CF (2,74 мл, 23,09 ммоль) и перемешивали при -15°С в течение 30 минут под инертной атмосферой. По каплям добавили смесь (2S,3R)-метил 2-амино-3-гидроксибутаноата (2,93 г, 22,08 ммоль) и NMM (2,43 мл, 22,08 ммоль) в ДМФ (15 мл) при -15°С. Полученную реакционную смесь перемешивали при комнатной температуре в течение 3 часов. Смесь разбавили ДХМ (200 мл), а органический слой промыли водой (50 мл), насыщенным солевым раствором (50 мл), высушили над безводным Na2SO4 и концентрировали под пониженным давлением. Полученное неочищенное вещество очистили силикагелевой колоночной хроматографией, элюируя 30% EtOAc в гексане, с получением соединения 9 (3,1 г, 42%).
1H-ЯМР: (500 МГц, ДМСО-d6) (ротамеры): δ 7.98-7.94 (m, 1H), 7.35-7.27 (m, 5Н), 5.09-4.94 (m, 3Н), 4.44 (dd, J=5.5, 8.5 Гц, 1Н), 4.29-4.27 (m, 1H), 4.12 (s, 1H), 3.62 (s, 3H), 3.44-3.30 (m, 2H), 2.20-2.08 (m, 1H), 1.87-1.78 (m, 3H), 1.08-0.94 (2d, 3H).
Масса m/z: 365,0 [М++1].
Пример 2 - Синтез (S)-N-((S)-1-амино-3-гидрокси-1-оксопропан-2-ил)-1-((S)-1-((2S,3R)-2-амино-3-гидроксибутаноил)пирролидин-2-карбонил)пирролидин-2-карбоксамида (CM-2):
[00134] Для синтеза (S)-N-((S)-1-амино-3-гидрокси-1-оксопропан-2-ил)-1-((S)-1-((2S,3R)-2-амино-3-гидроксибутаноил)пирролидин-2-карбонил)пирролидин-2-карбоксамида (CM-2) использовали следующую последовательность реакций (Схема В):
Схема В. Синтез CM-2.

[00135] Синтез (S)-1-(трет-бутоксикарбонил)-пирролидин-2-карбоновой кислоты (2):
К ледяному перемешанному раствору (S)-пирролидин-2-карбоновой кислоты (1) (3,0 г, 26,08 ммоль) в ТТФ:H2O (60 мл, 1:1) добавили Na2CO3 (5,52 г, 52,16 ммоль), Boc2O (6,25 г, 26,69 ммоль) и перемешивали при комнатной температуре в течение 16 часов. Реакционную смесь разбавили водой и промыли EtOAc (50 мл). Водный слой подкислили с помощью 2 н. HCl и экстрагировали EtOAc (2×100 мл). Объединенный органический слой высушили над безводным Na2SO4 и концентрировали под пониженным давлением с получением (S)-1-(трет-бутоксикарбонил)-пирролидин-2-карбоновой кислоты (2) (4,8 г, 86%).
1H-ЯМР: (500 МГц, ДМСО-d6): δ 12.49 (br s, 1Н), 4.08-4.03 (m, 1H), 3.36-3.24 (m, 2H), 2.22-2.11 (m, 1H), 1.87-1.76 (m, 3H), 1.39 (s, 9H).
Масса m/z: 216,0 [M++1].
[00136] Синтез (S)-трет-бутил 2-((S)-3-гидрокси-1-метокси-1-оксопропан-2-илкарбамоил)-пирролидин-1-карбоксилата (3):
Соединение 2 (2,0 г, 9,00 ммоль) растворили в CH2Cl2 (10 мл), охлажденном до -15°С, добавили NMM (1,12 мл, 10,2 ммоль) и IBCF (1,26 мл, 1,15 ммоль) и перемешивали при 0°С в течение 20 минут. По каплям добавили смесь (S)-метил 2-амино-3-гидроксипропаноата (1,59 г, 10,2 ммоль) и NMM (1,12 мл) в ДМФ (3 мл) при -15°С и перемешивали полученную реакционную смесь при комнатной температуре в течение 1 часа. Смесь разбавили ДХМ (200 мл), водой (50 мл) и промыли 2 н. HCl (20 мл) и насыщенным солевым раствором (2×50 мл). Отделенный органический слой высушили над безводным Na2SO4 и концентрировали под пониженным давлением. Полученный неочищенный остаток очистили силикагелевой колоночной хроматографией, элюируя 20% EtOAc в гексане, с получением соединения 3 (2,3 г) в виде сиропообразного вещества.
Масса m/z: 317,0 [М++1].
[00137] Синтез (S)-метил 3-гидрокси-2-((S)-пирролидин-2-карбоксамидо)пропионата (4):
(S)-трет-Бутил-2-((S)-3-гидрокси-1-метокси-1-оксопропан-2-илкарбамоил)-пирролидин-1-карбоксилат (3) (500 мг, 1,58 ммоль) растворили в 1,4-диоксане (3 мл) и добавили раствор HCl в диоксане (3,16 мл, 3,16 ммоль), перемешивали при комнатной температуре в течение 4 часов. Летучие вещества выпарили под пониженным давлением с получением соединения 4 (280 мг) в виде твердого вещества.
1Н-ЯМР: (200 МГц, ДМСО-d6): δ 9.99 (br s, 1Н), 9.12-9.08 (m, 1H), 8.53 (br s, 1H), 5.48 (br s, 2H), 4.43-4.22 (m, 2H), 3.82-3.67 (m, 4H), 3.56 (s, 3H), 2.36-2.27 (m, 1H), 1.93-1.86 (m, 3H).
Масса m/z: 217,0 [М++1].
[00138] Синтез (S)-метил 2-((S)-1-((S)-1-((2R,3S)-3-ацетокси-2-(бензилоксикарбониламино)-бутаноил)-пирролидин-2-карбонил)-пирролидин-2-карбоксамидо)-3-гидроксипропаноата (6):
(2S)-1-((2R)-3-ацетокси-2-(бензилоксикарбониламино)-бутаноил)-пирролидин-2-карбоновую кислоту (S) (1,3 г, 2,62 ммоль) растворили в CH2Cl2 (15 мл), добавили NMM (0,43 мл) и IBCF (0,51 мл) при -10°С и перемешивали в течение 30 минут под инертной атмосферой. К реакционной смеси по каплям добавили смесь (S)-метил-3-гидрокси-2-((S)-пирролидин-2-карбоксамидо)-пропионата (4) (992 мг, 3,93 ммоль) и NMM (0,43 мл) в ДМФ (5 мл) и продолжали перемешивание еще 3 часа при комнатной температуре. Реакционную смесь разбавили ДХМ (200 мл), промыли водой (20 мл), раствором лимонной кислоты (2×20 мл) и насыщенным солевым раствором (2×50 мл). Отделенный органический слой высушили над безводным Na2SO4 и концентрировали под пониженным давлением. Полученный неочищенный материал очистили силикагелевой колоночной хроматографией, элюируя 5% СН3ОН в CH2Cl2, с получением соединения 6 (270 мг, 17,5%).
1H-ЯМР: (500 МГц, ДМСО-d6): δ 8.13 (d, J=8.0 Гц, 1H), 7.74 (d, J=7.5 Гц, 1Н), 7.38-7.31 (m, 5H), 5.08-4.96 (m, 3Н), 4.85-4.82 (m, 1H), 4.56 (d, J=8.0 Гц, 1H), 4.44-4.42 (m, 2H), 4.27 (d, J=7.0 Гц, 1H), 4.10 (d, J=10.5 Гц, 2H), 3.81-3.78 (m, 1H), 3.72-3.70 (m, 1H), 3.61-3.59 (m, 3H), 3.54-3.50 (m, 2H), 2.16-2.14 (m, 1H), 2.05-2.01 (m, 1H), 1.90 (s, 3H), 1.87-1.86 (m, 3H), 1.85-1.84 (m, 3H), 1.21-1.20 (d, J=6.0 Гц, 3Н).
Масса m/z: 591,0 [М++1].
[00139] Синтез бензил-(2S,3S)-1-((S)-2-((S)-2-((S)-1-(аминоокси)-3-гидрокси-1-оксопропан-2-илкарбамоил)пирролидин-1-карбонил)пирролидин-1-ил)-3-гидрокси-1-оксобутан-2-илкарбамата (7):
К раствору соединения 6 (250 г, 0,42 ммоль) в СН3ОН (2 мл) добавили MeOH-NH3 (10 мл) и перемешивали при комнатной температуре в течение 16 часов. Летучие вещества выпарили под пониженным давлением с получением соединения 7 (190 мг, 84%).
1H-ЯМР: (500 МГц, ДМСО-d6): δ 7.60 (d, J=7.5 Гц, 1Н), 7.35-7.30 (m, 5H), 7.18 (d, J=7.0 Гц, 1H), 7.11-7.06 (m, 2H), 5.05-4.97 (m, 2H), 4.82-4.81 (m, 1H), 4.60-4.59 (m, 2H), 4.33-4.31 (m, 1H), 4.15-4.08 (m, 2H), 3.81-3.79 (m, 1H), 3.72-3.64 (m, 2H), 3.59-3.53 (m, 4H), 2.14 (s, 1H), 2.03 (d, 7=9.0 Гц, 1H), 1.95-1.85 (m, 5H), 1.75 (s, 1H), 1.10 (d,J=6.5 Гц, 3Н).
Масса m/z: 550,0 [М++1].
[00140] Синтез (S)-N-((S)-1-амино-3-гидрокси-1-оксопропан-2-ил)-1-((S)-1-((2S,3R)-2-амино-3-гидроксибутаноил)пирролидин-2-карбонил)-пирролидин-2-карбоксамида (CM-2):
К раствору соединения 7 (190 мг, 0,35 ммоль) в метаноле (5 мл) добавили 10% Pd/C (50 мг) и перемешивали реакционную смесь под атмосферой водорода в течение 2 часов. Реакционную смесь отфильтровали через слой целита, растворитель выпарили in vacuo, а неочищенное вещество очистили колоночной хроматографии на щелочном оксиде алюминия, используя 0-5% СН3ОН в CH2Cl2 в качестве элюента, с получением CM-2 (130 мг, 73%).
1Н-ЯМР: (500 МГц, ДМСО-d6): δ 7.65-7.60 (m, 1Н), 7.12-7.03 (m, 2H), 4.81 (br s, 1H), 4.58-4.57 (m, 1H), 4.49 (m, 1H), 4.38-4.19 (m, 1H), 4.10-4.06 (m, 1H), 3.69-3.62 (m, 2H), 3.59-3.56 (m, 4H), 3.49-3.45 (m, 2H), 3.37-3.26 (m, 2H), 2.19-2.15 (m, 1H), 2.09-1.99 (m, 1H), 1.95-1.84 (m, 5H), 1.75 (s, 1H), 1.06 (d, J=13.0 Гц, 3Н).
ЖХМС m/z: 400,8 [М++1].
Чистота по ВЭЖХ: 97,71%.
Пример 3 - Синтез (S)-N-((S)-1-амино-3-гидрокси-1-оксопропан-2-ил)-1-((S)-1-((S)-2-амино-3-гидрокси-пропаноил)-пирролидин-2-карбонил)-пирролидин-2-карбоксамида (CM-3):
[00141] Для синтеза (S)-N-((S)-1-амино-3-гидрокси-1-оксопропан-2-ил)-1-((S)-1-((S)-2-амино-3-гидрокси-пропаноил)-пирролидин-2-карбонил)-пирролидин-2-карбоксамида (CM-3) использовали следующую последовательность реакций (Схема С):
Схема С. Синтез CM-3.

[00142] Синтез (S)-1-(трет-бутоксикарбонил)-пирролидин-2-карбоновой кислоты (2):
К перемешанному раствору (S)-пирролидин-2-карбоновой кислоты (3,0 г, 26,08 ммоль) в ТГФ:H2O (60 мл, 1:1) при 0°С добавили Na2CO3 (5,52 г, 52,16 ммоль) и Вос2О (6,25 г, 26,69 ммоль) и перемешивали при комнатной температуре в течение 16 часов. Реакционную смесь разбавили водой и промыли EtOAc (50 мл). Водный слой подкислили с помощью 2 н. HCl и экстрагировали EtOAc (2×50 мл). Объединенный органический слой высушили над безводным Na2SO4 и концентрировали под пониженным давлением с получением (S)-1-(трет-бутоксикарбонил)-пирролидин-2-карбоновой кислоты 2 (4,8 г, 85,7%).
1Н-ЯМР: (500 МГц, ДМСО-d6): δ 12.49 (br s, 1Н), 4.08-4.03 (m, 1H), 3.36-3.24 (m, 2H), 2.22-2.11 (m, 1H), 1.87-1.76 (m, 3H), 1.39 (s, 9H).
Масса m/z: 216,0 [М++1].
[00143] Синтез (S)-трет-бутил 2-((S)-3-гидрокси-1-метокси-1-оксопропан-2-илкарбамоил)-пирролидин-1-карбоксилата (3):
Соединение 2 (2,0 г, 9,00 ммоль) растворили в CH2Cl2 (10 мл), охлажденном до -15°С, добавили NMM (1,12 мл, 10,2 ммоль) и IBCF (1,26 мл, 1,15 ммоль) и перемешивали при 0°С в течение 20 минут. По каплям добавили смесь (S)-метил-2-амино-3-гидроксипропаноата (1,59 г, 10,2 ммоль) и NMM (1,12 мл) в ДМФ (3 мл) при -15°С. Полученную Смесь перемешивали при комнатной температуре в течение 1 часа. Реакционную смесь разбавили ДХМ (200 мл) и водой (25 мл) и промыли 2 н. HCl (20 мл) и насыщенным солевым раствором (10 мл). Отделенный органический слой высушили над безводным Na2SO4 и концентрировали под пониженным давлением. Полученный неочищенный материал очистили силикагелевой колоночной хроматографией, элюируя 20% EtOAc в гексане, с получением соединения 3 (2,3 г) в виде твердого вещества.
Маеса m/z: 317,0 [М++1].
[00144] Синтез (S)-метил 3-гидрокси-2-((S)-пирролидин-2-карбоксамидо)пропаноата (4):
К раствору (S)-трет-бутил-2-((S)-3-гидрокси-1-метокси-1-оксопропан-2-илкарбамоил)пирролидин-1-карбоксилата 3 (500 мг, 1,58 ммоль) в 1,4-диоксане (3 мл) добавили раствор HCl в диоксане (3,16 мл, 3,16 ммоль) и перемешивали при комнатной температуре в течение 4 часов. Летучие вещества выпарили под пониженным давлением с получением соединения 4 (280 мг) в виде твердого вещества.
1Н-ЯМР: (200 МГц, ДМСО-d6): δ 9.99 (br s, 1Н), 9.12-9.08 (m, 1H), 8.53 (br s, 1H), 5.48 (br s, 2H), 4.43-4.22 (m, 2H), 3.82-3.67 (m, 4H), 3.56 (s, 3H), 2.36-2.27 (m, 1H), 1.93-1.86 (m, 3H).
Масса m/z: 217,0 [М++1].
[00145] Синтез (S)-метил 2-((S)-1-((S)-1-((S)-2-(бензилоксикарбониламино)-3-гидроксипропаноил)-пирролидин-2-карбонил)-пирролидин-2-карбоксамидо)-3-гидроксипропаноата (6):
(S)-1-((S)-3-Ацетокси-2-(бензилоксикарбониламино)-пропаноил)-пирролидин-2-карбоновую кислоту (S) (400 мг, 1,05 ммоль) растворили в CH2Cl2 (2 мл), добавили NMM (0,13 мл) и IBCF (0,14 мл) при -15°С и перемешивали в течение 30 минут под инертной атмосферой. К реакционной смеси по каплям добавили смесь (S)-метил-3-гидрокси-2-((S)-пирролидин-2-карбоксамидо)-пропаноата гидрохлорида (4) (293 мг, 1,16 ммоль) и NMM (0,13 мл) в ДМФ (2 мл) и продолжали перемешивание еще 3 часа при комнатной температуре. Реакционную смесь разбавили ДХМ (200 мл), промыли водой (20 мл) и насыщенным солевым раствором (10 мл). Отделенный органический слой высушили над безводным Na2SO4 и концентрировали под пониженным давлением. Полученный неочищенный материал очистили силикагелевой колоночной хроматографией, элюируя 5% СН3ОН в CH2Cl2, с получением соединения 6 (80 мг, 13%).
1Н-ЯМР: (500 МГц, ДМСО-d6): δ 8.09 (d, J=7.5 Гц, 1Н), 7.71 (d, J=8.0 Гц, 1H), 7.36-7.31 (m, 6H), 5.07-4.99 (m, 3H), 4.59-4.58 (m, 2H), 4.41-4.40 (m, 1H), 4.29-4.24 (m, 3H), 3.86 (t, J=9.5 Гц, 1H), 3.72-3.68 (m, 1H), 3.64-3.57 (m, 3H), 3.40-3.38 (m, 3H), 2.14-2.01 (m, 2H), 1.98 (s, 3H), 1.90-1.80 (m, 6H).
Масса m/z: 535,0 [M++1].
[00146] Синтез бензил-(S)-1-((S)-2-((S)-2-((S)-1-амино-3-гидрокси-1-оксопропан-2-илкарбамоил)пирролидин-1-карбонил)пирролидин-1-ил)-3-гидрокси-1-оксопропан-2-илкарбамата (7):
К раствору (S)-метил-2-((S)-1-((S)-1-((S)-2-(бензилоксикарбониламино)-3-гидроксипропаноил)-пирролидин-2-карбонил)-пирролидин-2-карбоксамидо)-3-гидроксипропаноата (6) (60 мг, 1,04 ммоль) в МеОН добавили MeOH-NH3 (3 мл) и перемешивали при комнатной температуре в течение 16 часов. Летучие вещества выпарили под пониженным давлением с получением соединения 7 (30 мг, 55%).
1Н-ЯМР: (500 МГц, ДМСО-d6): δ 7.60 (d, J=7.5 Гц, 1Н), 7.36-7.31 (m, 6H), 7.11-7.06 (m, 2H), 5.04-4.98 (m, 2H), 4.82-4.74 (m, 2H), 4.61-4.59 (m, 1H), 4.36-4.30 (m, 2H), 4.10-4.07 (m, 1H), 3.67-3.65 (m, 2H), 3.59-3.55 (m, 6H), 3.44-3.40 (m, 2H), 1.95-1.92 (m, 6H).
Масса m/z: 520,0 [M++1].
[00147] Синтез (S)-N-((S)-1-амино-3-гидрокси-1-оксопропан-2-ил)-1-((S)-1-((S)-2-амино-3-гидрокси-пропаноил)-пирролидин-2-карбонил)-пирролидин-2-карбоксамида (CM-3):
Бензил-(S)-1-((S)-2-((S)-2-((S)-1-амино-3-гидрокси-1-оксопропан-2-илкарбамоил)пирролидин-1-карбонил)пирролидин-1-ил)-3-гидрокси-1-оксопропан-2-илкарбамат 7 (300 мг, 0,57 ммоль) растворили в метаноле (8 мл), добавили 10% Pd/C (50 мг) и перемешивали реакционную смесь под атмосферой водорода в течение 2 часов. Реакционную смесь отфильтровали, а фильтрат концентрировали под пониженным давлением с получением СМ-3 (150 мг, 68%).
1Н-ЯМР: (500 МГц, ДМСО-d6) (ротамеры): δ 7.62 (d, J=8.0 Гц, 1Н), 7.24 (br s, 1H), 7.14-7.07 (m, 2H), 4.87-4.82 (m, 2H), 4.59-4.57 (m, 1H), 4.37-4.31 (m, 2H), 4.11-4.07 (m, 2H), 3.70-3.39 (m, 8H), 2.17-2.01 (m, 2H), 1.95-1.79 (m, 6H).
ЖХМС m/z: 386,4 [M++1].
Чистота по ВЭЖХ: 98.45%.
Пример 4 - Синтез (R)-N-((2S,3R)-1-амино-3-гидрокси-1-оксобутан-2-ил)-1-((S)-1-(2S,3R)-2-амино-3-гидроксибутаноил)пирролидин-2-карбонил)-2-бензилпирролидин-2-карбоксамида (CM-4А) и (S)-N-((2S,3R)-1-амино-3-гидрокси-1-оксобутан-2-ил)-1-((S)-1-(2S,3R)-2-амино-3-гидроксибутаноил)пирролидин-2-карбонил)-2-бензилпирролидин-2-карбоксамида (CM-4В):
[00148] Для синтеза (R)-N-((2S,3R)-1-амино-3-гидрокси-1-оксобутан-2-ил)-1-((S)-1-((2S,3R)-2-амино-3-гидроксибутаноил)пирролидин-2-карбонил)-2-бензилпирролидин-2-карбоксамида (CM-4А) и (S)-N-((2S,3R)-1-амино-3-гидрокси-1-оксобутан-2-ил)-1-((S)-1-((2S,3R)-2-амино-3-гидроксибутаноил)-пирролидин-2-карбонил)-2-бензилпирролидин-2-карбоксамида (CM-4В) использовали следующую последовательность реакций (Схема D):
Схема D. Синтез CM-4А и CM-4В.

[00149] Синтез 1-бензил 2-этил 2-бензилпирролидин-1,2-дикарбоксилата (2):
К раствору (S)-1-бензил 2-этил-пирролидин-1,2-дикарбоксилата 1 (10 г, 36,10 ммоль) в ТПГФ (150 мл) под инертной атмосферой добавили LiHMDS (1M в ТГФ) (43,3 мл, 43,3 ммоль) при -25°С и перемешивали в течение 2 часов. К реакционной смеси по каплям добавили бензилбромид (5,17 мл, 43,26 ммоль) при -25°С. Смесь оставили нагреваться до комнатной температуры и перемешивали в течение 2 часов. Реакционную смесь охладили до 5°С, погасили насыщенным раствором NH4Cl, а водный слой экстрагировали EtOAc (2×200 мл). Объединенные органические экстракты высушили над безводным Na2SO4 и концентрировали под пониженным давлением. Полученный неочищенный остаток очистили силикагелевой колоночной хроматографией, элюируя 5% EtOAc в гексане, с получением соединения 2 (13 г, 75%) в виде жидкого вещества.
1H-ЯМР: (200 МГц, ДМСО-d6): δ 7.47-7.32 (m, 5Н), 7.27-7.16 (m, 3Н), 7.07-7.04 (m, 2Н), 5.29-5.06 (m, 2Н), 4.16-3.89 (m, 2Н), 3.57-3.33 (m, 2Н), 3.02-2.78 (m, 2Н), 2.13-1.89 (m, 2Н), 1.56-1.51 (m, 1Н), 1.21-1.04 (m, 3Н), 0.93-0.79 (m, 1H).
Масса m/z: 368,2 [М++1].
[00150] Синтез 1-бензил 2-этил-2-бензилпирролидин-1,2-дикарбоксилата (3):
К перемешанному раствору соединения 2 (8,0 г, 21,79 ммоль) в СН3ОН (20 мл) добавили 2 н. водный раствор КОН (20 мл) и нагревали до 100°С, и перемешивали в течение 16 часов. Летучие вещества выпарили под пониженным давлением. Полученный остаток разбавили ледяной водой (50 мл) и промыли эфиром (50 мл). Водный слой подкислили до рН~2 с помощью раствора HCl и экстрагировали EtOAc (2×100 мл). Объединенный органический слой высушили над безводным Na2SO4 и концентрировали под пониженным давлением с получением соединения 3 (6 г, 81%) в виде грязновато-белого твердого вещества.
1Н-ЯМР: (200 МГц, ДМСО-d6): δ 12.71 (br s, 1Н), 7.40-7.30 (m, 5H), 7.25-7.19 (m, 3H), 7.07-7.00 (m, 2H), 5.27-5.02 (m, 2H), 3.59-3.32 (m, 2H), 3.02-2.83 (m, 2H), 2.13-1.91 (m, 2H), 1.58-1.49 (m, 1H), 0.90-0.77 (m, 1H).
Масса m/z: 340,1 [M++1].
[00151] Синтез бензил 2-бензил-2-((2S,3R)-3-гидрокси-1-метокси-1-оксобутан-2-илкарбамоил)пирролидин-1-карбоксилата (4):
К суспензии 2-бензил-1-(бензилоксикарбонил)пирролидин-2-карбоновой кислоты (1,0 г, 2,94 ммоль), метилового эфира L-треонина (471 мг, 3,53 ммоль) в ДМФ (20 мл) добавили HATU (1,12 г, 2,94 ммоль) и DIPEA (1,58 мл, 8,84 ммоль) при 5°С. Реакционную смесь перемешивали при комнатной температуре в течение 16 часов. Смесь разбавили EtOAc (150 мл) и промыли водой (2×30 мл). Органический слой промыли насыщенным солевым раствором, высушили над Na2SO4, концентрировали и очистили силикагелевой колоночной хроматографией, используя 50% EtOAc в гексане в качестве элюента, с получением соединения 4 (1,0 г, 74%).
1Н-ЯМР: (200 МГц, ДМСО-d6): δ 7.62-7.59 (m, 1Н), 7.44-7.31 (m, 5H), 7.21-7.18 (m, 3Н), 7.06-6.99 (m, 2H), 5.25-5.24 (m, 1H), 5.12-4.94 (m, 2H), 4.30 (s, 1H), 4.15-4.08 (m, 1H), 3.66-3.64 (m, 3H), 3.63-3.49 (m, 2H), 3.14 (s, 1H), 2.89 (s, 1H), 2.09-2.02 (m, 2H), 1.56-1.51 (m, 1H), 1.09-0.98 (m, 4H).
Масса m/z: 455,1 [M++1], 477,3 [M+Na].
[00152] Синтез бензил 2-((2S,3R)-3-ацетокси-1-метокси-1-оксобутан-2-илкарбамоил)-2-бензилпирролидин-1-карбоксилата (5)
2-Бензил-2-((2S,3R)-3-гидрокси-1-метокси-1-оксобутан-2-илкарбамоил)пирролидин-1-карбоксилат (3 г, 6,60 ммоль) растворили в ТУФ (30 мл), добавили Et3N (1,11 мл, 7,92 ммоль) и Ac2O (742 мг, 7,26 ммоль) при комнатной температуре. Реакционную смесь перемешивали при комнатной температуре в течение 2 часов. Летучие вещества выпарили под пониженным давлением, а полученный остаток разбавили CH2Cl2 и промыли разбавленной HCl. Объединенные органические экстракты высушили над Na2SO4 и концентрировали под пониженным давлением. Неочищенный остаток очистили колоночной хроматографией, используя 30% EtOAc в гексане в качестве элюента, с получением соединения 5 (2,5 г, 76%).
1H-ЯМР: (500 МГц, ДМСО-d6) (ротамеры): δ 8.15-7.71 (m, 1H), 7.42-7.04 (m, 10Н), 5.30-5.19 (m, 2Н), 5.11-5.09 (m, 1Н), 4.99-4.93 (m, 1H), 4.67-4.62 (m, 1H), 3.66-3.64 (m, 3H), 3.55-3.46 (m, 2H), 3.38-3.35 (m, 1H), 2.88-2.69 (m, 1H), 2.17-2.00 (m, 2H), 1.98-1.92 (m, 3H), 1.56-1.46 (m, 1H), 1.23-1.17 (m, 3H), 1.02-0.86 (m, 1H).
ЖХМС m/z: 497,4 [М++1].
[00153] Синтез (2S, 3R)-метил 3-ацетокси-2-(2-бензилпирролидин-2-карбоксамидо)бутаноата (6):
К перемешанному раствору соединения 5 (4 г, 8,06 ммоль) в этаноле (50 мл) добавили 10% Pd/C (1,2 г) и перемешивали реакционную смесь под атмосферой Н2 (баллонное давление) в течение 4 часов. Смесь отфильтровали через слой целита, а фильтрат концентрировали под пониженным давлением с получением соединения 6 (2,2 г, 75%).
1Н-ЯМР: (500 МГц, ДМСО-d6) (ротамеры): δ 8.22-8.17 (m, 1H), 7.24-7.16 (m, 5Н), 5.17 (t, J=11.5 Гц, 1Н), 4.48-4.42 (m, 1H), 3.60-3.54 (s, 3H), 3.20 (t, J=13.5 Гц, 1H), 3.06-2.97 (m, 1H), 2.82-2.68 (m, 3H), 2.08-2.02 (m, 1H), 1.89 (s, 3H), 1.72-1.51 (m, 3H), 1.10 (2d, 3H).
ЖХМС m/z: 363 [M++1], 385 [M+Na].
[00154] Синтез (S)-бензил 2-(2-((2S,3R)-3-ацетокси-1-метокси-1-оксобутан-2-илкарбамоил)-2-бензилпирролидин-1-карбонил)пирролидин-1-карбоксилата (7):
К перемешанному раствору соединения 6 (1 г, 2,76 ммоль) и Na2CO3 (732 мг, 6,90 ммоль) в CH2Cl2:H2O (20 мл, 1:1) добавили раствор хлорангидрида кислоты [К раствору (S)-1-(бензилоксикарбонил)пирролидин-2-карбоновой кислоты (825 мг, 3,31 ммоль) в CH2Cl2 (20 мл) по каплям добавили SOCl2 (0,60 мл) при 0°С и нагревали с дефлегматором в течение 2 часов. Летучие вещества удалили под пониженным давлением с получением (S)-бензил 2-(хлоркарбонил)пирролидин-1-карбоксилата] в CH2Cl2, и перемешивали реакционную смесь при комнатной температуре в течение 2 часов. Летучие вещества выпарили под пониженным давлением. Остаток разбавили CH2Cl2 (100 мл), отфильтровали, а фильтрат концентрировали под вакуумом. Неочищенный остаток очистили колоночной хроматографией, используя 60% EtOAc в гексане в качестве элюента, с получением соединения 7 (750 мг, 45%).
1H-ЯМР: (500 МГц, ДМСО-d6) (ротамеры): δ 7.36-7.23 (m, 8Н), 7.15-7.12 (m, 3Н), 5.21-5.15 (m, 2Н), 5.04-4.92 (m, 1Н), 4.57-4.50 (m, 2H), 3.88 (d, J=14.5 Гц, 1H), 3.65 (s, 3H), 3.54-3.46 (m, 3H), 3.21-3.13 (m, 1H), 3.02-2.90 (m, 2H), 2.19-2.02 (m, 4H), 1.97 (s, 3H), 1.89 (s, 1H), 1.77-1.65 (m, 1H), 1.17 (s, 2H), 1.06 (s, 2H).
Масса m/z: 594,1 [М++1].
[00155] Синтез (2S,3R)-метил 3-ацетокси-2-(2-бензил-1-((S)-пирролидин-2-карбонил)пирролидин-2-карбоксамидо)бутаноата (8):
К раствору соединения 7 (200 мг, 0,336 ммоль) в EtOAc (15 мл) добавили 10% Pd/C (40 мг) под инертной атмосферой и перемешивали в течение 12 часов под атмосферой Н2 (баллонное давление). Реакционную смесь отфильтровали через слой целита и концентрировали под пониженным давлением. Полученный остаток растерли с эфиром (10 мл) с получением соединения 8 (125 мг, 81%) в виде твердого вещества.
1Н-ЯМР: (500 МГц, CDCl3) (ротамеры): δ 7.88-7.87 (d, 1Н, J=8.5), 7.30-7.26 (m, 2H), 7.24-7.21 (m, 1H), 7.13-7.12 (d, 2H, J=7), 5.44-5.43 (m, 1H), 4.76-4.74 (m, 1H), 3.94-3.92 (m, 1H), 3.84-3.81 (m, 1H), 3.75 (s, 3H), 3.50 (m, 1H), 3.26-3.12 (m, 3H), 2.90-2.88 (m, 1H), 2.23-2.15 (m, 4H), 2.04 (s, 3H), 1.87-1.77 (m, 5H), 1.27-1.24 (m, 3H).
Масса m/z: 460 (M+1).
[00156] Синтез бензил 2-(трет-бутоксикарбониламино)-3-гидроксибутаноата (10):
К раствору 2-(трет-бутоксикарбониламино)-3-гидроксибутановой кислоты (3,0 г, 13,69 ммоль) в ДМФ (50 мл) добавили K2СО3 (3,73 г, 27,39 ммоль) и перемешивали при комнатной температуре в течение 15 минут. Добавили (бромметил)бензол (2,81 г, 16,43 ммоль) и перемешивали при комнатной температуре в течение 6 часов. Реакционную смесь разбавили водой (50 мл) и экстрагировали EtOAc (2×50 мл). Объединенный органический слой промыли насыщенным солевым раствором (50 мл), высушили над безводным Na2SO4 и концентрировали под пониженным давлением. Неочищенный материал очистили силикагелевой колоночной хроматографией, используя 20% EtOAc в гексане в качестве элюента, с получением бензил 2-(трет-бутоксикарбониламино)-3-гидроксибутаноата 10 (2,8 г, 66%).
1H-ЯМР: (500 МГц, ДМСО-d6): δ 7.37-7.30 (m, 5Н), 6.60 (d, J=8.5 Гц, 1H), 5.18-5.08 (m, 2H), 4.76 (d, J=7 Гц, 1H), 4.08-4.00 (m, 2H), 1.38 (s, 9H), 1.09 (d, J=6.0 Гц, 3Н).
Масса m/z: 310,0 [M++1], 210 [М+-De Вос].
[00157], Синтез бензил 3-ацетокси-2-(трет-бутоксикарбониламино)бутаноата (11):
К перемешанному раствору бензил 2-(трет-бутоксикарбониламино)-3-гидроксибутаноата (2,8 г, 9,06 ммоль) в ТГФ (80 мл) добавили Ac2O (1,1 г, 10,87 ммоль), Εt3Ν (1,51 мл, 10,87 ммоль) и DMAP (280 мг) и перемешивали при комнатной температуре в течение 15 минут. Летучие вещества удалили под пониженным давлением. Полученный остаток разбавили EtOAc (150 мл) и промыли холодным 0,5 н. раствором HCl (2×20 мл). Органический слой промыли насыщенным солевым раствором, высушили над безводным Na2SO4 и концентрировали под пониженным давлением с получением 3-ацетокси-2-(трет-бутоксикарбониламино)бутаноата 11 (2,8 г, 88%).
1H-ЯМР: (500 МГц, ДМСО-d6): δ 7.35-7.34 (m, 5Н), 7.27-7.25 (d, J=8.5 Гц, 1H), 5.18-5.06 (m, 3Н), 4.34-4.32 (m, 1H), 1.90 (s, 3H), 1.39 (s, 9H), 1.16 (d, J=3 Гц, 3Н).
Масса m/z: 252 [M++1-De Boc].
[00158] Синтез (2S,3R)-3-ацетокси-2-(трет-бутоксикарбониламино)бутановой кислоты (12):
Бензил-3-ацетокси-2-(трет-бутоксикарбониламино)бутаноат 11 (1,4 г, 3,98 ммоль) растворили в EtOAc (40 мл), добавили 10% Pd/C (600 мг) и перемешивали смесь под атмосферой водорода в течение 16 часов. Реакционную смесь отфильтровали через целит, растворитель выпарили in vacuo, а неочищенный остаток растерли с гексаном с получением (2S, 3R)-3-ацетокси-2-(трет-бутоксикарбониламино)бутановой кислоты 12 (0,7rg, 70%).
1Н-ЯМР: (500 МГц, ДМСО-d6): δ 12.78 (br s, 1Н), 6.94 (d, J=9.5 Гц, 1H), 5.16-5.14 (m, 1H), 4.17-4.15 (m, 1H), 1.95 (s, 3H), 1.39 (s, 9H), 1.10 (d, J=6.0 Гц, 3Н).
Масса m/z: 260,0 [М-1].
[00159] Синтез (2S,3R)-метил 3-ацетокси-2-(1-((S)-1-((2S,3R)-3-ацетокси-2-((трет-бутоксикарбонил)-амино)бутаноил)пирролидин-2-карбонил)-2-бензилпирролидин-2-карбоксамидо)бутаноата (13):
К раствору соединения (2S,3R)-3-ацетокси-2-(трет-бутоксикарбониламино)бутановой кислоты 12 (199 мг, 0,76 ммоль) в CH2Cl2 (6 мл) под инертной атмосферой добавили IBCF (125 мг, 0,91 ммоль) и NMM (154 мг, 1,52 ммоль) при -15°С и перемешивали в течение 1 часа. К реакционной смеси добавили раствор (2S,3R)-метил 3-ацетокси-2-(2-бензил-1-((S)-пирролидин-2-карбонил)пирролидин-2-карбоксамидо)бутаноата 8 (350 мг, 0,76 ммоль) в ДМФ (2 мл) и перемешивали в течение 1 часа при -15°С. Полученную реакционную Смесь оставили нагреваться до комнатной температуры и перемешивали в течение 19 часов. Реакционную смесь экстрагировали EtOAc, а отделенный органический слой промыли водой (20 мл), затем насыщенным солевым раствором (20 мл), высушили над Na2SO4 и концентрировали под пониженным давлением. Неочищенный материал очистили препаративной ВЭЖХ с получением соединения 13 (100 мг, 20%).
1H-ЯМР: (500 МГц, CD3OD) (ротамеры): δ 7.30-7.24 (m, 3Н), 7.15-7.13 (m, 2Н), 4.62-4.55 (m, 2Н), 4.29-3.97 (m, 1Н), 3.98-3.79 (m, 4H), 3.75 (s, 3Н), 3.62-3.22 (m, 2H), 3.23 (d, J=13.5 Гц, 1H), 3.00-2.95 (q, 1H), 2.37-2.31 (m, 1H), 2.23-2.10 (m, 2H), 2.02-1.88 (m, 3H), 1.46-1.28 (m, 2H), 0.97 (d, J=7.0 Гц, 6H).
[00160] Синтез трет-бутил ((2S,3R)-1-((S)-2-(2-(((2S,3R)-1-амино-3-гидрокси-1-оксобутан-2-ил)карбамоил)-2-бензилпирролидин-1-карбонил)пирролидин-1-ил)-3-гидрокси-1-оксобутан-2-ил)карбамата (NT-13-10).
Раствор соединения 13 (100 мг, 0,153 ммоль) в метанольном NH3 (10 мл) перемешивали в закрытой пробирке при комнатной температуре в течение 72 часов. Реакционную смесь концентрировали под пониженным давлением. Полученный неочищенный остаток промыли эфиром (2×2 мл) с получением NT-13-10 (85 мг).
[00161] Синтез (R)-N-((2S,3R)-1-амино-3-гидрокси-1-оксобутан-2-ил)-1-((S)-1-((2S,3R)-2-амино-3-гидроксибутаноил)пирролидин-2-карбонил)-2-бензилпирролидин-2-карбоксамида (CM-4А) и (S)-N-((2S,3R)-1-амино-3-гидрокси-1-оксобутан-2-ил)-1-((S-)-1-((2S,3R)-2-амино-3-гидроксибутаноил)пирролидин-2-карбонил)-2-бензилпирролидин-2-карбоксамида (CM-4В):
[00162] NT-13-10 (85 мг) дополнительно очистили хиральной препаративной ВЭЖХ с получением двух изомеров, в результате чего после очистки с HCl в диоксане и выпаривания получили по 15 мг CM-4А и CM-4В.
1Н-ЯМР: (500 МГц, CD3OD) (ротамеры): δ 7.33-7.26 (m, 3Н), 7.16 (s, 2Н), 4.55-4.54 (m, 1H), 4.39 (s, 1H), 4.14 (s, 1H), 4.01-3.98 (m, 1H), 3.91-3.71 (m, 3H), 3.59 (s, 2H), 3.25-3.16 (m, 1H), 3.04-3.00 (m, 1H), 2.33-2.10 (m, 3H), 2.01-1.91 (m, 2H), 1.86-1.80 (m, 1H), 1.46-1.44 (m, 1H), 1.34-1.29 (m, 1H), 1.25-1.19 (m, 3H), 0.99-0.97 (d, J=14.0 Гц, 3Н).
Масса m/z: 503 [M+]
Чистота по ВЭЖХ: 98.1%.
[00163] Синтез (R)-N-((2S,3R)-1-амино-3-гидрокси-1-оксобутан-2-ил)-1-((S)-1-((2S,3R)-2-амино-3-гидроксибутаноил)-пирролидин-2-карбонил)-2-бензилпирролидин-2-карбоксамида трифторацетатной соли (CM-4А-ТФК):
К раствору CM-4А в метаноле добавили избыток трифторуксусной кислоты. Полученный осадок собрали фильтрацией, промыли метанолом и высушили in vacuo с получением CM-4А-ТФК.
Пример 5 - Синтез (R)-N-((2S,3R)-1-амино-3-гидрокси-1-оксобутан-2-ил)-1-((S)-1-((2S,3R)-2-амино-3-гидроксибутаноил)пирролидин-2-карбонил)-2-(4-метилбензил)пирролидин-2-карбоксамида (CM-5А) и (S)-N-((2S,3R)-1-амино-3-гидрокси-1-оксобутан-2-ил)-1-((S)-1-((2S,3R)-2-амино-3-гидроксибутаноил)пирролидин-2-карбонил)-2-(4-метилбензил)пирролидин-2-карбоксамида (CM-5В):
[00164] Для синтеза (R)-N-((2S,3R)-1-амино-3-гидрокси-1-оксобутан-2-ил)-1-((S)-1-((2S,3R)-2-амино-3-гидроксибутаноил)пирролидин-2-карбонил)-2-(4-метилбензил)пирролидин-2-карбоксамида (CM-5А) и (S)-N-((2S,3R)-1-амино-3-гидроокси-1-оксобутан-2-ил)-1-((S)-1-((2S,3R)-2-амино-3-гидроксибутаноил)пирролидин-2-карбонил)-2-(4-метилбензил)пирролидин-2-карбоксамида (CM-5В) использовали следующую последовательность реакций (Схема Е):
[00165] Схема Е. Синтез CM-5А и CM-5В:
[00166] Синтез (S)-этил пирролидин-2-карбоксилата гидрохлорида (1):
К перемешанному раствору L-пролина (исходный материал) (110 г, 956,5 ммоль) в этаноле добавили тионилхлорид (141 мл, 1911,3 ммоль) и нагревали с дефлегматором в течение 16 часов. Реакционную смесь довели до комнатной температуры и концентрировали под вакуумом с получением соединения 1 в виде гидрохлоридной соли (170 г, 99%).
[00167] Синтез (S)-1-бензил 2-этил пирролидин-1,2-дикарбоксилата (2):
К перемешанному раствору соединения 1 (170 г, 947 ммоль) в CH2Cl2 добавили Et3N (398 мл, 2,83 моль). После перемешивания в течение 30 минут к реакционной смеси добавили Cbz-Cl (1,136 ммоль) и продолжали перемешивание еще 12 часов при комнатной температуре. Реакционную смесь промыли водой и экстрагировали CH2Cl2. Органический слой высушили над безводным Na2SO4 и концентрировали под вакуумом. Неочищенное вещество очистили колоночной хроматографией с получением соединения 2 (230 г, 88%).
1H-ЯМР: (200 МГц, ДМСО-d6): δ 7.42-7.31 (m, 5Н), 5.09 (m, 2Н), 4.32-4.23(m, 1Н), 4.08-3.98(m, 2H), 3.50-3.38 (m, 2H), 2.30-2.18 (m, 1H), 1.90-1.81(m, 3H), 1.10-1.04 (t, 3H).
Масса m/z: 278 [M++1].
[00168] Синтез 1-бензил 2-этил 2-(4-метилбензил)пирролидин-1,2-дикарбоксилата (3):
К перемешанному раствору соединения 2 (5 г, 0,018 моль) в ТТФ (40 мл) под инертной атмосферой добавили LiHMDS (1M в ТТФ) (36 мл, 0,036 моль) при -78°С и перемешивали в течение 30 минут. К смеси по каплям добавили 4-метил-бензилбромид (5 г, 0,027 моль) при -30°С и оставили нагреваться до комнатной температуры и перемешивали в течение 2 часов. Реакционную смесь охладили до 5°С, погасили насыщенным раствором NH4Cl, а водный слой экстрагировали EtOAc (2×100 мл). Объединенные органические экстракты высушили над безводным Na2SO4 и концентрировали под пониженным давлением. Неочищенный остаток очистили силикагелевой колоночной хроматографией, элюируя 20% EtOAc в гексане, с получением соединения 3 (5,2 г, 76%) в виде жидкого вещества.
1Н-ЯМР: (500 МГц, ДМСО-d6): δ 7.47-7.32 (m, 5Н), 7.17-7.12 (m, 1H), 7.05-7.01 (m, 1H), 6.95-6.90 (m, 2H), 5.25-5.00 (m, 2H), 4.16-4.12 (m, 1H), 4.00-3.89 (m, 1H), 3.52 (d, 1H), 3.38-3.33 (m, 1H), 2.99-2.89 (m, 2H), 2.23 (s, 3H), 2.12-1.90 (m, 1H), 1.56-1.51 (m, 1H), 1.05-1.01 (m, 3H), 1.00-0.97 (m, 1H), 0.92-0.89 (m, 1H).
[00169] Синтез 1-((бензилокси)карбонил)-2-(4-метилбензил)пирролидин-2-карбоновой кислоты (4):
К перемешанному раствору соединения 3 (5,2 г, 0,0136 моль) в метаноле (25 мл) и воде (15 мл) добавили 2 н. водный раствор NaOH (2 г, 0,052 моль) и нагревали до 85°С в течение 4 часов. Летучие вещества выпарили под пониженным давлением, а полученный остаток разбавили ледяной водой (50 мл) и промыли эфиром (50 мл). Водный слой подкислили до рН~2, используя 2 н. HCl, и экстрагировали CH2Cl2 (2×100 мл). Объединенные органические слои высушили над безводным Na2SO4 и концентрировали под пониженным давлением с получением соединения 4 (4,3 г, 89%) в виде грязновато-белого твердого вещества.
1H-ЯМР: (400 МГц, CDCl3): δ 7.45-7.38 (m, 5Н), 7.09-6.89 (m, 4Н), 5.35-5.26 (m, 2Н), 3.67-3.47 (m, 2Н), 3.14-2.99 (m, 2Н), 2.42-2.39 (m, 1Н), 2.29 (s, 3Н), 2.22-1.99 (m, 1H), 1.74-1.61 (m, 1H), 1.31-1.22 (m, 1H).
ЖХМС (ESI): 354 [М++1].
[00170] Синтез бензил 2-(((2R,3S)-3-гидрокси-1-метокси-1-оксобутан-2-ил)карбамоил))-2-(4-метилбензил)пирролидин-1-карбоксилата (S):
К перемешанному раствору соединения 4 (4,3 г, 12,1 ммоль) в CH2Cl2 (40 мл) добавили EDCI.HCl (2,5 г, 13,4 ммоль), затем HOBt (1,82 г, 13,4 ммоль) и DIPEA (6,4 мл, 36,5 ммоль) при 0°С. После перемешивания в течение 10 минут к реакционной смеси добавили метиловый эфир L-треонина (2,2 г, 13,4 ммоль) и перемешивали еще 4 часа при комнатной температуре. После расходования исходного материала (по ТСХ) реакционную смесь разбавили CH2Cl2 (150 мл) и промыли водой (2×30 мл). Органический слой промыли насыщенным солевым раствором, высушили над Na2SO4, концентрировали и очистили силикагелевой колоночной хроматографией, элюируя 2% МеОН/CH2Cl2, с получением соединения 5 (4,1 г, 72%).
1Н-ЯМР: (500 МГц, CDCl3): δ 7.44-7.31 (m, 5Н), 7.09-6.85 (m, 5Н), 5.32-5.25 (m, 1Н), 5.05-4.94 (m, 2H), 4.25-4.20 (m, 1H), 4.15-4.08 (m, 1H), 3.66-3.64 (m, 2H), 3.45-3.41 (m, 2H), 3.14-3.09 (m, 1H), 2.89-2.84 (m, 1H), 2.20 (s, 3H), 2.05-2.02 (m, 2H), 1.55-1.51 (m, 1H), 1.09-0.98 (m, 4H).
ЖХМС (ESI): 469 [M++1].
[00171] Синтез бензил 2-(((2S,3R)-3-ацетокси-1-метокси-1-оксобутан-2-ил)карбамоил)-2-(4-метилбензил)пирролидин-1-карбоксилата (6):
К перемешанному раствору соединения 5 (4,1 г, 8,75 ммоль) в CH2Cl2 (40 мл) добавили Et3N (3,0 мл, 21,9 ммоль), затем Ас2О (1,0 г, 10,5 ммоль) при 0°С и перемешивали в течение 1 часа. К смеси добавили DMAP (25 мг, каталитическое количество) и перемешивали при комнатной температуре в течение 2 часов. После расходования исходного материала (по ТСХ) реакционную смесь разбавили водой и экстрагировали EtOAc (2×100 мл). Отделенный органический слой высушили над безводным Na2SO4, отфильтровали и концентрировали под вакуумом с получением соединения 6 (4,2 г, неочищенный). Это вещество напрямую использовали на следующей стадии без дополнительной очистки.
1Н-ЯМР: (500 МГц, CDCl3): δ 7.42-7.30 (m, 5Н), 7.09-6.85 (m, 5Н), 5.25-5.21 (m, 2Н), 5.11-5.09 (m, 1Н), 4.67-4.62 (m, 1H), 3.66-3.64 (m, 2H), 3.55-3.46 (m, 3H), 3.15-3.10 (m, 1H), 2.92-2.85 (m, 1H), 2.20 (s, 3H), 2.05-1.95 (m, 5H), 1.56-1.46 (m, 1H), 1.15-1.11 (m, 4H).
ЖХМС (ESI): 511 [M++1].
[00172] Синтез (2S,3R)-метил 3-ацетокси-2-(2-(4-метилбензил)пирролидин-2-карбоксамидо)бутаноата (7):
К перемешанному раствору соединения 6 (4,2 г, 8,22 ммоль) в метаноле (35 мл) добавили 10% Pd/C (1,0 г) под атмосферой N2. Реакционную смесь перемешивали под атмосферой Н2 (баллонное давление) в течение 3 часов. После расходования исходного материала (по ТСХ) реакционную смесь отфильтровали через слой целита, а фильтрат концентрировали под пониженным давлением с получением соединения 7 (3,0 г, 100%) в виде сиропообразного вещества.
1Н-ЯМР: (500 МГц, CDCl3): δ 8.22 (br s, 1H), 7.09-7.06 (m, 4H), 5.23-5.19 (m, 1H), 4.50-4.42 (m, 1H), 3.60 (d, 3H), 3.20 (m, 1H), 3.00-2.97 (m, 1H), 2.75-2.68 (m, 2H), 2.28 (s, 3H), 2.05-2.02 (m, 1H), 1.90 (d, 3H), 1.65-1.51 (m, 3H), 1.10 (dd, 3H).
ЖХМС m/z: 377 [M1+1].
[00173] Синтез (2S)-бензил 2-(2-(((2S,3R)-3-ацетокси-1-метокси-1-оксобутан-2-ил)карбамоил)-2-(4-метилбензил)пирролидин-1-карбонил)пирролидин-1-карбоксилата (8):
К перемешанному раствору соединения 7 (3,0 г, 7,97 ммоль) в CH2Cl2 (30 мл) и воде (20 мл) добавили Na2CO3 (2,1 г, 19,9 ммоль) и перемешивали при 0°С в течение 5 минут. К смеси добавили хлорангидрид кислоты [К раствору (S)-1-(бензилоксикарбонил)пирролидин-2-карбоновой кислоты (2,3 г, 9,56 ммоль) в CH2Cl2 (10 мл) по каплям добавили SOCl2 (1,4 мл) при 0°С и нагревали с дефлегматором в течение 2 часов. Летучие вещества удалили под пониженным давлением] и перемешивали реакционную смесь при комнатной температуре в течение 2 часов. Отделенный органический слой концентрировали под вакуумом, а полученные изомеры разделили промыванием диэтиловым эфиром (2×20 мл) с получением соединения 8-F1 (1,6 г) и соединения 8-F2 (2,6 г).
Аналитические данные для Соединения 8-F1:
1H-ЯМР: (400 МГц, CDCl3): δ 7.39-7.28 (m, 5Н), 7.15-6.85 (m, 5Н), 5.21-5.05 (m, 2Н), 5.04-4.92 (m, 1Н), 4.65-4.50 (m, 1H), 4.53-4.45 (m, 1H), 3.65 (s, 3H), 3.54-3.46 (m, 4H), 3.21-3.13 (m, 2H), 2.25-2.16 (m, 4H), 2.05-2.00 (m, 2H), 1.95-1.85 (m, 4H), 1.56-1.51 (m, 2H), 1.15 (dd, 3H).
ЖХМС m/z: 608 [M++1].
СЭЖХ (чистота): 97%.
Аналитические данные для Соединения 8-F2:
1H-ЯМР: (400 МГц, CDCl3): δ 8.50 (br s, 1H), 7.36-7.23 (m, 5H), 7.15-6.85 (m, 5H), 5.21-5.05 (m, 2H), 5.04-4.92 (га, 1H), 4.65-4.50 (m, 1H), 4.53-4.45 (га, 1H), 3.65 (s, 3H), 3.54-3.46 (га, 4H), 3.21-3.13 (m, 2H), 2.25-2.16 (m, 4H), 2.05-2.00 (m, 2H), 1.95-1.85 (m, 4H), 1.56-1.51 (m,2H), 1.15 (dd, 3H).
ЖХМС m/z: 608 [M++1]
СЭЖХ (чистота): 96%.
[00174] Синтез (2S,3R)-метил 3-ацетокси-2-((R)-2-(4-метилбензил)-1-((S)-пирролидин-2-карбонил)пирролидин-2-карбоксамидо)бутаноата (9-F1):
К перемешанному раствору соединения 8-F1 (1,6 г, 2,63 ммоль) в МеОН (20 мл) и EtOAc (20 мл) добавили 10% Pd/C (0,5 г) под инертной атмосферой и перемешивали в течение 4 часов под атмосферой Н2 (баллонное давление). Реакционную смесь отфильтровали через слой целита и концентрировали под пониженным давлением с получением соединения 9-F1 (1.1 г, 92%).
1Н-ЯМР: (400 МГц, CDCl3): δ 8.23 (dd, 1H), 7.20-6.85 (m, 5Н), 5.20-5.13 (m, 1H), 4.63-4.59 (m, 1Н), 3.85-3.81 (m, 1H), 3.65-3.61 (m, 5H), 3.32-3.25 (m, 2H), 3.12-3.05 (m, 3H), 2.75-2.71 (m, 1H), 2.25 (s, 3H), 2.15-2.13 (m, 2H), 2.00 (d, 3H), 1.77 (m, 3H), 1.27 (dd, 4H).
ЖХМС m/z: 474 [М++1].
[00175] Синтез (2S,3R)-метил 3-ацетокси-2-((S)-2-(4-метилбензил)-1-((S)-пирролидин-2-карбонил)пирролидин-2-карбоксамидо)бутаноата (9-F2):
К перемешанному раствору соединения 8-F2 (2,6 г, 4,20 ммоль) в МеОН (25 мл) добавили 10% Pd/C (1,0 г) под инертной атмосферой и перемешивали в течение 3 часов под атмосферой Н2 (баллонное давление). Реакционную смесь отфильтровали через слой целита и концентрировали под пониженным давлением с получением соединения 9-F2 (1,1 г, 55%).
1H-ЯМР: (400 МГц, CDCl3): δ 8.23 (dd, 1Н), 7.20-6.85 (m, 5H), 5.20-5.13 (m, 1H), 4.63-4.59 (m, 1H), 3.85-3.81 (m, 1H), 3.65-3.61 (m, 5H), 3.32-3.25 (m, 2H), 3.12-3.05 (m, 3H), 2.75-2.71 (m, 1H), 2.25 (s, 3H), 2.15-2.13 (m, 2H), 2.00 (d, 3H), 1.77 (m, 3H), 1.27 (dd, 4H).
ЖХМС m/z: 474 [M++1].
[00176] Синтез бензил 2-(трет-бутоксикарбониламино)-3-гидроксибутаноата (А):
К раствору 2-(трет-бутоксикарбониламино)-3-гидроксибутановой кислоты (50 г, 228,3 ммоль) в ДМФ (500 мл) добавили K2CO3 (63 г, 456,6 ммоль) и перемешивали при комнатной температуре в течение 15 минут. К смеси добавили бензилбромид (46,83 г, 273,9 ммоль) и перемешивали при комнатной температуре в течение 6 часов. Реакционную смесь разбавили водой (500 мл) и экстрагировали EtOAc (2×750 мл). Объединенные органические слои промыли насыщенным солевым раствором (50 мл), высушили над безводным Na2SO4 и концентрировали под пониженным давлением. Неочищенный материал очистили силикагелевой колоночной хроматографией, элюируя 20% EtOAc в гексане, с получением бензил 2-(трет-бутоксикарбониламино)-3-гидроксибутаноата А (52 г, 73%).
1Н-ЯМР: (500 МГц, ДМСО-d6): δ 7.37-7.30 (m, 5Н), 6.60 (d, 1Н), 5.18-5.08 (m, 2H), 4.76 (d, 1H), 4.08-4.00 (m, 2H), 1.38 (s, 9H), 1.09 (d, 3H).
Масса m/z: 310,0 [M++1], 210 [M+-De Boc].
[00177] Синтез бензил 3-ацетокси-2-(трет-бутоксикарбониламино)бутаноата (В):
К перемешанному раствору соединения А (52 г, 168,2 ммоль) в CH2Cl2 (500 мл) добавили Ac2O (20,5 г, 201,9 ммоль), Et3N (25,4 г, 252,4 ммоль) и DMAP (3,5 г) и перемешивали при комнатной температуре в течение 2 часов. Летучие вещества удалили под пониженным давлением. Полученный остаток разбавили EtOAc (750 мл) и промыли холодным 0,5 н. раствором HCl (2×200 мл). Органический слой промыли насыщенным солевым раствором, высушили над безводным Na2SO4 и концентрировали под пониженным давлением с получением 3-ацетокси-2-(трет-бутоксикарбониламино)бутаноата В (52 г, 88%).
1H-ЯМР: (500 МГц, ДМСО-d6): δ 7.35-7.34 (m, 5Н), 7.27-7.25 (d, 1Н), 5.18-5.06 (m, 3Н), 4.34-4.32 (m, 1H), 1.90 (s, 3H), 1.39 (s, 9H), 1.16 (d, 3H).
Масса m/z: 252 [M+-De Boc]
[00178] Синтез (2S,3R)-3-ацетокси-2-(трет-бутоксикарбониламино)бутановой кислоты (С):
К перемешанному раствору соединения В (52 г, 148,1 ммоль) в МеОН (1 л) добавили 10% Pd/C под атмосферой N2 и перемешивали реакционную смесь под атмосферой водорода в течение 16 часов. Реакционную смесь отфильтровали через слой целита, полученный фильтрат выпарили под пониженным давлением, а неочищенный остаток растерли с гексаном с получением (2S,3R)-3-ацетокси-2-(трет-бутоксикарбониламино)бутановой кислоты С (35 г, 90%).
1Н-ЯМР: (500 МГц, ДМСО-d6): δ 12.78 (br s, 1Н), 6.94 (d, 1H), 5.16-5.14 (m, 1H), 4.17-4.15 (m, 1H), 1.95 (s, 3H), 1.39 (s, 9H), 1.10 (d, 3H).
Масса m/z: 260,0 [М-1].
[00179] Синтез (2S,3R)-метил 3-ацетокси-2-((R)-1-((S)-1-((2S,3R)-3-ацетокси-2-((трет-бутоксикарбонил)амино)бутаноил)пирролидин-2-карбонил)-2-(4-метилбензил)пирролидин-2-карбоксамидо)бутаноата (10-F1):
К перемешанному раствору соединения С (0,66 г, 2,55 ммоль) в CH2Cl2 (15 мл) добавили EDCI (0,49 г, 2,55 ммоль), HOBt (0,345 г, 2,55 ммоль), затем DIPEA (1,2 мл, 6,96 ммоль) при 0°С и перемешивали в течение 10 минут. К смеси добавили соединение 9-F1 (1,1 г, 2,32 ммоль) и перемешивали в течение 15 часов. Реакционную Смесь экстрагировали EtOAc (2×75 мл), а отделенный органический слой промыли водой (200 мл), затем насыщенным солевым раствором (200 мл), высушили над Na2SO4 и концентрировали под пониженным давлением. Неочищенный материал очистили колоночной хроматографией, элюируя 70% EtOAc в гексане, с получением соединения 10-F1 (1,6 г, 100%) в виде сиропообразного вещества.
1H-ЯМР: (500 МГц, CDCl3): δ 7.24-6.91 (m, 5Н), 5.42-5.01 (m, 2Н), 4.73-4.68 (m, 1Н), 4.54-4.41 (m, 1H), 3.90-3.85 (m, 1H), 3.67 (s, 3H), 3.65-3.60 (m, 2H), 3.59-3.55 (m, 1H), 3.59-3.55 (m, 1H), 3.30 (s, 3H), 3.12-3.05 (m, 1H), 2.33 (s, 3H), 2.05 (s, 3H), 2.00-1.96 (m, 4H), 1.94-1.85 (m, 4H), 1.43 (s, 9H), 1.25 (d, 3H), 1.15 (d, 3H).
ЖХМС (ESI): m/z 717 [M++1].
[00180] Синтез (2S,3R)-метил 3-ацетокси-2-((S)-1-((S)-1-((2S,3R)-3-ацетокси-2-((трет-бутоксикарбонил)амино)бутаноил)пирролидин-2-карбонил)-2-(4-метилбензил)пирролидин-2-карбоксамидо)бутаноата (10-F2):
К перемешанному раствору соединения С (0,66 г, 2,55 ммоль) в CH2Cl2 (10 мл) добавили EDCI.HCl (0,48 г, 2,55 ммоль), HOBt (0,345 г, 2,55 ммоль), затем DIPEA (1,2 мл, 6,96 ммоль) при 0°С и перемешивали в течение 10 минут. К смеси добавили соединение 9-F2 (1,1 г, 2,32 ммоль) и перемешивали в течение 15 часов. Реакционную смесь экстрагировали EtOAc (2×75 мл), а отделенный органический слой промыли водой (200 мл), затем насыщенным солевым раствором (200 мл), высушили над Na2SO4 и концентрировали под пониженным давлением. Неочищенный материал очистили колоночной хроматографией, элюируя 70% EtOAc в гексане, с получением соединения 10-F2 (0,8 г, 50%) в виде сиропообразного вещества.
1H-ЯМР: (400 МГц, ДМСО-d6): δ 7.24-6.91 (m, 5Н), 5.42-5.01 (m, 2Н), 4.73-4.68 (m, 1Н), 4.54-4.41 (m, 1H), 3.90-3.85 (m, 1H), 3.67 (s, 3H), 3.65-3.60 (m, 2H), 3.59-3.55 (m, 1H), 3.59-3.55 (m, 1H), 3.30 (s, 3H), 3.12-3.05 (m, 1H), 2.33 (s, 3H), 2.05 (s, 3H), 2.00-1.96 (m, 4H), 1.94-1.85 (m, 4H), 1.43 (s, 9H), 1.25 (d, 3H), 1.15 (d, 3H).
ЖХМС (ESI): m/z 717 [M++1].
[00181] Синтез трет-бутил ((2S,3R)-1-((S)-2-((R)-2-(((2S,3R)-1-амино-3-гидрокси-1-оксобутан-2-ил)карбамоил)-2-(4-метилбензил)пирролидин-1-карбонил)пирролидин-1-ил)-3-гидрокси-1-оксобутан-2-ил)карбамата (11-F1):
Смесь соединения 10-F1 (1,6 г, 2,20 ммоль) и метанольного раствора ΝΗ3 (20 мл) поместили в автоклав и нагревали до 70°С в течение 16 часов. После расходования исходного материала (по ТСХ) реакционную смесь концентрировали под пониженным давлением с получением соединения 11-F1 (0,6 г, 46%).
1Н-ЯМР: (400 МГц, ДМСО-d6): δ 7.32 (br s, 1Н), 7.15-7.10 (m, 2H), 7.05-6.91 (m, 3H), 6.65 (br s, 1H), 4.75-4.59 (m, 2H), 4.24-3.45 (m, 8H), 3.35 (s, 1H), 3.05-3.00 (m, 1H), 2.32 (s, 3H), 2.26-1.85 (m, 6H), 1.75 (s, 6H), 1.60-1.55 (m, 1H), 1.47 (s, 9H), 1.15 (d, 3H).
ЖХМС (ESI): m/z 618,5 [М++1].
[00182] Синтез трет-бутил ((2S,3R)-1-((S)-2-((S)-2-(((2S,3R)-1-амино-3-гидрокси-1-оксобутан-2-ил)карбамоил)-2-(4-метилбензил)пирролидин-1-карбонил)пирролидин-1-ил)-3-гидрокси-1-оксобутан-2-ил)карбамата (11-F2):
Смесь соединения 10-F2 (0,8 г, 1,11 ммоль) и метанольного раствора NH3 (20 мл) перемешивали при комнатной температуре в течение 16 часов. После расходования исходного материала (по ТСХ и ЖХМС) реакционную смесь концентрировали под пониженным давлением с получением соединения 11-F2 (0,4 г, 58%).
1H-ЯМР: (400 МГц, ДМСО-d6): δ 7.32 (br s, 1Н), 7.15-7.10 (m, 2H), 7.05-6.91 (m, 3H), 6.65 (br s, 1H), 4.75-4.59 (m, 2H), 4.24-3.45 (m, 8H), 3.35 (s, 1H), 3.05-3.00 (m, 1H), 2.32 (s, 3H), 2.26-1.85 (m, 6H), 1.75 (s, 6H), 1.60-1.55 (m, 1H), 1.47 (s, 9H), 1.15 (d, 3H).
ЖХМС (ESI): m/z 618,5 [M++1].
[00183] Синтез (R)-N-((2S,3R)-1-амино-3-гидрокси-1-оксобутан-2-ил)-1-((S)-1-((2S,3R)-2-амино-3-гидроксибутаноил)пирролидин-2-карбонил)-2-(4-метилбензил)пирролидин-2-карбоксамида (CM-5А):
Перемешивали раствор соединения 11-F1 (0,25 г, 0,40 ммоль) в 2 н. HCl в 1,4-диоксане (7 мл) при комнатной температуре в течение 2 часов. Реакционную смесь концентрировали под пониженным давлением и очистили препаративной ВЭЖХ с получением СМ-5А (0,1 г, 44%) в виде гидрохлоридной соли.
1Н-ЯМР: (400 МГц, D2O): δ 7.35 (t, 2Н), 7.25 (d, 2Н), 4.95-4.90 (m, 1Н), 4.43-4.35 (m, 4H), 3.94-3.89 (m, 1H), 3.87-3.78 (m, 2H), 3.63 (d, 1H), 3.49-3.40 (m, 1H), 3.25 (d, 1H), 2.56-2.34 (m, 5H), 2.28-2.20 (m, 4H), 1.75-1.71 (m, 1H), 1.56 (d, 3H), 1.32 (d, 3H), 1.15-1.11 (m, 1H).
ЖХМС (ESI) m/z: 518 [M++1].
СЭЖХ чистота: 99%.
Оптическое вращение [α25D]:+30,51 (С=1% в воде).
Препаративная ВЭЖХ: Rt=7,56 мин. (Chiralpak ΙΑ, 250×4.6 мм, 5 мкм; подвижная фаза (А) 0,1% ТФК в н-гексане (В) EtOH (4/1): А: В (80:20); скорость потока: 1,00 мл/мин).
[00184] Синтез (S)-N-((2S,3R)-1-амино-3-гидрокси-1-оксобутан-2-ил)-1-((S)-1-((2S,3R)-2-амино-3-гидроксибутаноил)пирролидин-2-карбонил)-2-(4-метилбензил)пирролидин-2-карбоксамида (CM-5В):
Перемешивали раствор соединения 11-F2 (0,4 г, 0,64 ммоль) в 2 н. HCl в 1,4-диоксане (5 мл) при комнатной температуре в течение 2 часов. Реакционную смесь концентрировали под пониженным давлением с получением неочищенного вещества, которое промыли EtOAc и н-пентаном с получением CM-5В (0,15 г, 42%) в виде гидрохлоридной соли.
1Н-ЯМР: (400 МГц, D2O): δ 7.35 (t, 2Н), 7.25 (d, 2Н), 4.95-4.90 (m, 1Н), 4.43-4.35 (m, 4H), 3.94-3.89 (m, 1H), 3.87-3.78 (m, 2H), 3.63 (d, 1H), 3.49-3.40 (m, 1H), 3.25 (d, 1H), 2.56-2.34 (m, 5H), 2.28-2.20 (m, 4H), 1.75-1.71 (m, 1H), 1.56 (d, 3H), 1.32 (d, 3H), 1.15-1.11 (m, 1H).
ЖХМС (ESI) m/z: 518 [M++1].
СЭЖХ чистота: 96%.
Оптическое вращение [α25D]: -124,6 (С=1% в воде).
Препаративная ВЭЖХ: Rt=9,68 мин. (Chiralpak ΙΑ, 250×4,6 мм, 5 мкм; подвижная фаза (А) 0,1% ТФК в н-гексане (В) EtOH (4/1): А: В (80:20); скорость потока: 1,00 мл/мин).
Пример 6 - Синтез (R)-N-((2S,3R)-1-амино-3-гидрокси-1-оксобутан-2-ил)-1-((S)-1-((2S,3R)-2-амино-3-гидроксибутаноил)пирролидин-2-карбонил)-2-(4-фторбензил)пирролидин-2-карбоксамида (CM-6А) и (S)-N-((2S,3R)-1-амино-3-гидрокси-1-оксобутан-2-ил)-1-((S)-1-((2S,3R)-2-амино-3-гидроксибутаноил)пирролидин-2-карбонил)-2-(4-фторбензил)пирролидин-2-карбоксамида (CM-6В):
[00185] Для синтеза (R)-N-((2S,3R)-1-амино-3-гидрокси-1-оксобутан-2-ил)-1-((S)-1-((2S,3R)-2-амино-3-гидроксибутаноил)пирролидин-2-карбонил)-2-(4-фторбензил)пирролидин-2-карбоксамида (CM-6А) и (S)-N-((2S,3R)-1-амино-3-гидрокси-1-оксобутан-2-ил)-1-((S)-1-((2S,3R)-2-амино-3-гидроксибутаноил)пирролидин-2-карбонил)-2-(4-фторбензил)пирролидин-2-карбоксамида (CM-6В) использовали следующую последовательность реакций (Схема F):
[00186] Схема F. Синтез CM-6А и CM6B:

[00187] Синтез (S)-этил пирролидин-2-карбоксилата гидрохлорида (1):
К перемешанному раствору L-пролина (исходный материал) (110 г, 956,5 ммоль) в этаноле добавили тионилхлорид (141 мл, 1911,3 ммоль) и нагревали с дефлегматором в течение 16 часов. Реакционную смесь довели до комнатной температуры и концентрировали под вакуумом с получением соединения 1 в виде гидрохлоридной соли (170 г, 99%).
[00188] Синтез (S)-1-бензил 2-метил пирролидин-1,2-дикарбоксилата (2):
К перемешанному раствору соединения 1 (170 г, 947 ммоль) в CH2Cl2 добавили Et3N (398 мл, 2,83 моль). После перемешивания в течение 30 минут к реакционной смеси добавили Cbz-Cl (1,136 моль) и продолжали перемешивание еще 12 часов при комнатной температуре. Реакционную смесь промыли водой и экстрагировали CH2Cl2. Органический слой высушили над безводным Na2SO4 и концентрировали под вакуумом. Неочищенное вещество очистили колоночной хроматографией с получением соединения 2 (230 г, 88%).
1Н-ЯМР: (200 МГц, ДМСО-d6): δ 7.42-7.3l(m, 5Н), 5.09 (m, 2Н), 4.32-4.23(m, 1Н), 4.08-3.98(m, 2Н), 3.50-3.38 (m, 2H), 2.30-2.18 (m, 1H), 1.90-1.81(m, 3H), 1.10-1.04 (t, 3H).
Масса m/z: 278 [М++1].
[00189] Синтез 1-бензил 2-этил 2-(4-фторбензил)пирролидин-1,2-дикарбоксилата (3):
К перемешанному раствору соединения 2 (10 г, 36,0 ммоль) в сухом ТГФ (100 мл) под инертной атмосферой добавили LiHMDS (1M в ΤГΦ) (72,1 мл, 72,1 ммоль) при -20°С и перемешивали в течение 1 часа. К смеси по каплям добавили 4-фторбензилбромид (5,34 мл, 43,2 ммоль) при -20°С и оставили нагреваться до комнатной температуры, и перемешивали в течение 1 часа. Реакционную смесь охладили до 5°С, погасли водным раствором NH4Cl, а водный слой экстрагировали EtOAc (2×100 мл). Объединенные органические экстракты высушили над безводным Na2SO4 и концентрировали под пониженным давлением. Полученный неочищенный остаток очистили силикагелевой колоночной хроматографией, элюируя 30% EtOAc в гексане, с получением соединения 3 (10 г, 72%).
1Н-ЯМР: (400 МГц, ДМСО-d6) (ротамеры): δ 7.46-7.32 (m, 5Н), 7.15-7.01 (m, 4Н), 5.18-5.09 (m, 2Н), 4.16-3.91 (m, 2Н), 3.53 (d, 1Н), 3.62 (d, 1H), 3.38-3.32 (m, 1H), 2.97-2.81 (m, 1H), 2.14-1.91 (m, 2H), 1.65-1.51 (m, 1H), 1.25 (t, 2H), 1.09 (t, 1H), 0.99-0.85 (m, 1H).
ЖХМС (ESI): 386 [M++1].
[00190] Синтез 1-((бензилокси)карбонил)-2-(4-фторбензил)пирролидин-2-карбоновой кислоты (4):
К перемешанному раствору соединения 3 (10 г, 25,9 ммоль) в метаноле (50 мл) и воде (50 мл) добавили 2 н. водный раствор NaOH (3,11 г, 77,8 ммоль) и нагревали до 70°С в течение 16 часов. Летучие вещества выпарили под пониженным давлением, а полученный остаток разбавили ледяной водой (50 мл) и промыли эфиром (50 мл). Водный слой подкислили до рН~2, используя водный раствор HCl, и экстрагировали EtOAc (2×100 мл). Объединенные органические слои промыли насыщенным солевым раствором, высушили над безводным Na2SO4 и концентрировали под пониженным давлением с получением неочищенного вещества, которое очистили силикагелевой колоночной хроматографией, элюируя 2% МеОН в CH2Cl2, с получением соединения 4 (9 г, 97%).
[00191] Синтез бензил 2-(4-фторбензил)-2-(((2S,3R)-3-гидрокси-1-метокси-1-оксобутан-2-ил)карбамоил)пирролидин-1-карбоксилата (S):
К перемешанному раствору соединения 4 (9,5 г, 26,6 ммоль) в CH2Cl2 (95 мл) добавили EDCI.HCl (7,61 г, 39,87 ммоль), затем HOBt (5,42 г, 39,87 ммоль) и DIPEA (14,6 мл, 79,7 ммоль) при 0°С под атмосферой N2. После перемешивания в течение 10 минут к реакционной смеси добавили гидрохлоридную соль метилового эфира L-треонина (5,4 г, 31,89 ммоль) и перемешивали еще 16 часов при комнатной температуре. После расходования исходного материала (по ТСХ) реакционную смесь разбавили водой (100 мл) и экстрагировали CH2Cl2 (2×100 мл). Органический слой высушили над Na2SO4 и концентрировали с получением неочищенного вещества, которое очистили силикагелевой колоночной хроматографией, элюируя 2% МеОН в CH2Cl2, с получением соединения 5 (13 г, неочищенное).
[00192] Синтез бензил 2-(((2S,3R)-3-ацетокси-1-метокси-1-оксобутан-2-ил)карбамоил)-2-(4-фторбензил)пирролидин-1-карбоксилата (6):
К перемешанному раствору соединения 5 (13 г, 27,5 ммоль) в CH2Cl2 (130 мл) добавили Εt3Ν (9,58 мл, 68,7 ммоль), затем Ас2О (4,13 мл, 41,26 ммоль) при 0°С и перемешивали в течение 1 часа. К смеси добавили DMAP (0,13 г) и перемешивали при комнатной температуре еще 16 часов. После расходования исходного материала (по ТСХ) реакционную смесь разбавили водой (100 мл) и экстрагировали CH2Cl2 (2×100 мл). Органический слой высушили над Na2SO4 и концентрировали с получением неочищенного вещества, которое очистили силикагелевой колоночной хроматографией, элюируя 30% EtOAc в гексане, с получением соединения 6 (10 г, 71%).
1Н-ЯМР: (500 МГц, ДМСО-d6) (ротамеры): δ 7.22-7.14 (m, 5Н), 7.11-6.98 (m, 4Н), 5.26-5.21 (m, 2Н), 5.09 (t, 1Н), 4.67-4.63 (m, 1H), 3.66-3.64 (m, 3H), 3.69-3.65 (m, 2H), 3.19-3.16 (m, 1H), 2.92-2.85 (m, 1H), 2.10-2.02 (m, 1H), 2.98 (s, 4H), 1.56-1.46 (m, 1H), 1.20-1.14 (m, 3H), 1.12 (t, 1H).
ЖХМС (ESI): 515 [М++1].
Масса m/z: 515 [M++1].
[00193] Синтез (2S,3R)-метил 3-ацетокси-2-(2-(4-фторбензил)пирролидин-2-карбоксамидо)бутаноата (7):
К перемешанному раствору соединения 6 (10 г, 19,4 ммоль) в метаноле (100 мл) добавили влажный 10% Pd/C (2 г) под атмосферой N2. Реакционную смесь перемешивали под атмосферой Н2 (баллонное давление) в течение 4 часов. После расходования исходного материала (по ТСХ) реакционную смесь отфильтровали через слой целита, а фильтрат концентрировали под пониженным давлением с получением соединения 7 (6,7 г, 91%).
Синтез (2S)-бензил 2-(2-(((2S,3R)-3-ацетокси-1-метокси-1-оксобутан-2-ил)карбамоил)-2-(4-фторбензил)пирролидин-1-карбонил)пирролидин-1-карбоксилата (8):
К перемешанному раствору соединения 7 (6,7 г, 17,6 ммоль) в CH2Cl2 (67 мл) и воде (67 мл) добавили Na2CO3 (4,67 г, 44,0 ммоль) и перемешивали при 0°С в течение 5 минут. К смеси добавили хлорангидрид кислоты [К раствору (S)-1-(бензилоксикарбонил)пирролидин-2-карбоновой кислоты (20,6 г, 82,8 ммоль) в CH2Cl2 (20 мл) по каплям добавили SOCl2 (3,19 мл, 44,0 ммоль) при 0°С и нагревали с дефлегматором в течение 2 часов. Летучие вещества удалили под пониженным давлением с получением (S)-бензил 2-(хлоркарбонил)пирролидин-1-карбоксилата] (5,26 г, 21,15 ммоль) и перемешивали реакционную смесь при комнатной температуре в течение 2 часов. Отделенный органический слой концентрировали под вакуумом, а полученный неочищенный материал очистили колоночной хроматографией, элюируя 2% МеОН в CH2Cl2, с получением соединения 8 (8,9 г, 83%).
ЖХМС m/z: 612 [М++1].
Масса m/z: 612 [М++1].
[00194] Синтез (2S,3R)-метил 3-ацетокси-2-(2-(4-фторбензил)-1-((S)-пирролидин-2-карбонил)пирролидин-2-карбоксамидо)бутаноата (9):
К перемешанному раствору соединения 8 (8,9 г, 14,5 ммоль) в МеОН (180 мл) добавили влажный 10% Pd/C (1,8 г) под инертной атмосферой и перемешивали в течение 4 часов под атмосферой Н2 (баллонное давление). Реакционную смесь отфильтровали через слой целита и концентрировали под пониженным давлением с получением соединения 9 (6,9 г, неочищенное).
1Н-ЯМР: (400 МГц, CD3OD) (ротамеры): δ 7.22-7.14 (m, 2Н), 7.03 (t, 2Н), 5.39-5.34 (m, 1H), 4.52 (t, 1H), 3.78-3.72 (m, 2H), 3.74 (d, 3H), 3.35 (s, 2H), 3.29-3.24 (m, 1H), 3.18-3.12 (m, 1H), 2.85-2.83 (m, 1H), 2.31-2.25 (m, 2H), 2.18-2.15 (m, 1H), 2.06 (d, 3H), 1.89-1.75 (m, 3H), 1.71-1.65 (m, 1H), 1.52-1.40 (m, 1H), 1.29 (dd, 3H).
Масса m/z: 478 [M++1].
[00195] Синтез бензил 2-(трет-бутоксикарбониламино)-3-гидроксибутаноата (А):
К раствору 2-(трет-бутоксикарбониламино)-3-гидроксибутановой кислоты (Вос-Thr) (50 г, 228,3 ммоль) в ДМФ (500 мл) добавили K2CO3 (63 г, 456,6 ммоль) и перемешивали при комнатной температуре в течение 15 минут. К смеси добавили бензилбромид (46,83 г, 273,9 ммоль) и перемешивали при комнатной температуре в течение 6 часов. Реакционную смесь разбавили водой (500 мл) и экстрагировали EtOAc (2×750 мл). Объединенные органические слои промыли насыщенным солевым раствором (50 мл), высушили над безводным Na2SO4 и концентрировали под пониженным давлением. Неочищенный материал очистили силикагелевой колоночной хроматографией, элюируя 20% EtOAc в гексане, с получением бензил 2-(трет-бутоксикарбониламино)-3-гидроксибутаноата А (52 г, 73%).
1H-ЯМР: (500 МГц, ДМСО-d6): δ 7.37-7.30 (m, 5Н), 6.60 (d, 1H), 5.18-5.08 (m, 2H), 4.76 (d, 1H), 4.08-4.00 (m, 2H), 1.38 (s, 9H), 1.09 (d, 3H).
Масса m/z: 310,0 [M++1], 210 [M+-De Boc].
[00196] Синтез бензил 3-ацетокси-2-(трет-бутоксикарбониламино)бутаноата (В):
К перемешанному раствору соединения А (52 г, 168,2 ммоль) в CH2Cl2 (500 мл) добавили Ac2O (20,5 г, 201,9 ммоль), Et3N (25,4 г, 252,4 ммоль) и DMAP (3,5 г) и перемешивали при комнатной температуре в течение 2 часов. Летучие вещества удалили под пониженным давлением. Полученный остаток разбавили EtOAc (750 мл) и промыли холодным 0,5 н. раствором HCl (2×200 мл). Органический слой промыли насыщенным солевым раствором, высушили над безводным Na2SO4 и концентрировали под пониженным давлением с получением 3-ацетокси-2-(трет-бутоксикарбониламино)бутаноата В (52 г, 88%).
1Н-ЯМР: (500 МГц, ДМСО-d6): δ 7.35-7.34 (m, 5Н), 7.27-7.25 (d, 1Н), 5.18-5.06 (m, 3Н), 4.34-4.32 (m, 1H), 1.90 (s, 3H), 1.39 (s, 9H), 1.16 (d, 3H).
Масса m/z: 252 [M++1-De Boc].
[00197] Синтез (2S,3R)-3-ацетокси-2-(трет-бутоксикарбониламино)бутановой кислоты (С):
К перемешанному раствору соединения В (52 г, 148,1 ммоль) в МеОН (1 л) добавили 10% Pd/C под атмосферой N2 и перемешивали реакционную смесь под атмосферой водорода в течение 16 часов. Реакционную смесь отфильтровали через слой целита, полученный фильтрат выпарили под вакуумом, а неочищенный остаток растерли с гексаном с получением (2S,3R)-3-ацетокси-2-(трет-бутоксикарбониламино)бутановой кислоты С (35 г, 90%).
1H-ЯМР: (500 МГц, ДМСО-d6): δ 12.78 (br s, 1Н), 6.94 (d, 1H), 5.16-5.14 (m, 1H), 4.17-4.15 (m, 1H), 1.95 (s, 3H), 1.39 (s, 9H), 1.10 (d, 3H).
Масса m/z: 260,0 [M+-1].
[00198] Синтез (2S;3R)-метил 3-ацетокси-2-(1-((S)-1-((2S,3R)-3-ацетокси-2-((трет-бутоксикарбонил)амино)бутаноил)пирролидин-2-карбонил)-2-(4-фторбензил)пирролидин-2-карбоксамидо)бутаноата (10):
К перемешанному раствору соединения С (4,53 г, 17,35 ммоль) в CH2Cl2 (69 мл) добавили EDCI.HCl (4,14 г, 21,69 ммоль), HOBt (2,95 г, 21,7 ммоль), затем DIPEA (7,99 мл, 43,4 ммоль) при 0°С и перемешивали в течение 10 минут. К смеси добавили соединение 9 (6,9 г, 14,46 ммоль) и перемешивали в течение 16 часов. Реакционную Смесь разбавили водой (75 мл) и экстрагировали EtOAc (2×75 мл). Отделенный органический слой высушили над Na2SO4 и концентрировали под пониженным давлением. Неочищенный материал очистили колоночной хроматографией, элюируя 2% МеОН в CH2Cl2, с получением соединения 10 (8,0 г, 77%).
1Н-ЯМР: (400 МГц, ДМСО-d6) (ротамеры): δ 7.35-7.29 (m, 1Н), 7.23 (m, 1H), 7.13 (d, 2H), 7.08-7.05 (m, 1H), 5.30-5.12 (m, 2H), 4.67-4.34 (m, 3H), 3.98-3.70 (m, 3H), 3.65 (s, 1H), 3.49-3.45 (m, 1H), 3.25-3.10 (m, 1H), 2.32-1.96 (m, 8H), 1.75-1.62 (m, 2H), 1.49 (d, 9H), 1.32-1.29 (m, 3H), 1.25-1.20 (m, 3H).
ЖХМС m/z: 621 [M++1].
[00199] Синтез трет-бутил ((2S,3R)-1-((2S)-2-(2-(((2S,3R)-1-амино-3-гидрокси-1-оксобутан-2-ил)карбамоил))-2-(4-фторбензил)пирролидин-1-карбонил)пирролидин-1-ил)-3-гидрокси-1-оксобутан-2-ил)карбамата (11):
Перемешанный раствор соединения 10 (4 г, 5,55 ммоль) и метанольного NH3 (20 мл) поместили в закрытую пробирку и перемешивали в течение 48 часов. Реакционную смесь концентрировали под вакуумом с получением неочищенного вещества, которое очистили силикагелевой колоночной хроматографией, элюируя 2% МеОН в CH2Cl2, с получением соединения 11 (1,6 г) в виде смеси. Этот неочищенный материал подвергли хиральной препаративной очистке.
Хиральная препаративная ВЭЖХ изомеров:
Изомеры соединения 11 (1,6 г, неочищенное) разделили хиральной препаративной ВЭЖХ с получением Соединения 11-F1 (0,6 г) и Соединения 11-F2 (0,25 г).
Аналитические данные для Соединения 11-F1:
1H-ЯМР: (400 МГц, ДМСО-d6): δ 8.15 (br s, 1Н), 7.39-7.35 (m, 2H), 7.24 (s, 1H), 7.11 (t, 2H), 7.04-7.00 (m, 1H), 6.79-6.74 (m, 2H), 4.75 (d, 1H), 4.21 (t, 2H), 4.02-3.95 (m, 1H), 3.81-3.75 (m, 2H), 3.69-3.56 (m, 2H), 3.53-3.49 (m, 1H), 3.27-3.09 (m, 2H), 2.29-2.10 (m, 1H), 2.05-1.91 (m, 5H), 1.69-1.51 (m, 1H), 1.40 (s, 9H), 1.19-1.11 (m, 6H), 0.92-0.84 (m, 1H).
ВЭЖХ: 85%.
ЖХМС (ESI): m/z 622 [M++1].
Хиральная ВЭЖХ: Rt=10,69 мин. (Chiralpak ΙΑ, 250×4,6 мм, 5 мкм; подвижная фаза (А) 0,1% ТФК в н-гексане (В) EtOH (4/1): А: В (80:20); скорость потока: 1,00 мл/мин).
Аналитические данные для Соединения 11-F2:
1Н-ЯМР: (400 МГц, ДМСО-d6): δ 8.15 (br s, 1Н), 7.39-7.35 (m, 2H), 7.24 (s, 1H), 7.11 (t, 2H), 7.04-7.00 (m, 1H), 6.79-6.74 (m, 2H), 4.75 (d, 1H), 4.21 (t, 2H), 4.02-3.95 (m, 1H), 3.81-3.75 (m, 2Н), 3.69-3.56 (m, 2Н), 3.53-3.49 (m, 1Н), 3.27-3.09 (m, 2H), 2.29-2.10 (m, 1H), 2.05-1.91 (m, 5H), 1.69-1.51 (m, 1H), 1.40 (s, 9H), 1.19-1.11 (m, 6H), 0.92-0.84 (m, 1H).
ВЭЖХ: 84%.
ЖХМС (ESI): m/z 622 [M++1].
Харальная ВЭЖХ: Rt=18,55 мин. (Chiralpak ΙΑ, 250×4,6 мм, 5 мкм; подвижная фаза (А) 0,1% ТФК в н-гексане (В) EtOH (4/1): А: В (80:20); скорость потока: 1,00 мл/мин).
[00200] Синтез (R)-N-((2S,3R)-1-амино-3-гидрокси-1-оксобутан-2-ил))-1-((S)-1-((2S,3R)-2-амино-3-гидроксибутаноил)пирролидин-2-карбонил)-2-(4-фторбензил)пирролидин-2-карбоксамида (CM-6А):
К перемешанному раствору соединения 11-F1 (0,6 г, 0,96 ммоль) в CH2Cl2 (12 мл) добавил 4 Μ HCl в 1,4-диоксане (6 мл) при 0°С и перемешивали при комнатной температуре в течение 1 часа. Реакционную смесь концентрировали под вакуумом с получением неочищенного вещества, которое дополнительно промыли EtOAc и высушили под вакуумом с получением СМ-6А (0,28 г, 52%) в виде гидрохлоридной соли.
1Н-ЯМР: (400 МГц, D2O): δ 7.41-7.32 (m, 2Н), 7.21 (t, 2Н), 4.95-4.90 (m, 1H), 4.43-4.35 (m, 4H), 3.94-3.62 (m, 5H), 3.27 (d, 1H), 2.56-2.50 (m, 1H), 2.35-2.29 (m, 1H), 2.14-2.05 (m, 4H), 1,81-1.75 (m, 1H), 1.35 (d, 3H), 1.32 (d, 3H), 1.19-1.11 (m, 1H).
ЖХМС (ESI) m/z: 522 [M++1].
СЭЖХ чистота: 96%.
Хиральная ВЭЖХ: Rt=7,73 мин. (Chiralpak ΙΑ, 250×4,6 мм, 5 мкм; подвижная фаза (А) 0,1% ТФК в н-гексане (В) EtOH (4/1): А: В (80:20); скорость потока: 1,00 мл/мин).
Оптическое вращение [α20D]:+7,15 (С=1% в воде).
[00201] Синтез (S)-N-((2S,3R)-1-амино-3-гидрокси-1-оксобутан-2-ил)-1-((S)-1-((2S,3R)-2-амино-3-гидроксибутаноил)пирролидин-2-карбонил)-2-(4-фторбензил)пирролидин-2-карбоксамида (CM-6В):
К перемешанному раствору соединения 11-F2 (0,25 г, 0,40 ммоль) в CH2Cl2 (5 мл) добавили 4 Μ HCl в 1,4-диоксане (2,5 мл) при 0°С и перемешивали при комнатной температуре в течение 3 часов. Реакционную смесь концентрировали под вакуумом с получением неочищенного вещества, которое дополнительно промыли EtOAc и высушили под вакуумом с получением СМ-6В (0,1 г, 45%) в виде гидрохлоридной соли.
1Н-ЯМР: (400 МГц, D2O): δ 7.41-7.32 (m, 2H), 7.21 (t, 2H), 4.95-4.90 (m, 1H), 4.43-4.35 (m, 4H), 3.94-3.62 (m, 5H), 3.27 (d, 1H), 2.56-2.50 (m, 1H), 2.35-2.29 (m, 1H), 2.14-2.05 (m, 4H), 1.81-1.75 (m, 1H), 1.35 (d, 3H), 1.32 (d, 3H), 1.19-1.11 (m, 1H).
ЖХМС (ESI) m/z: 522 [M1+1].
СЭЖХ чистота: 98,38%.
Хиральная ВЭЖХ: Rt=11,28 мин. (Chiralpak ΙΑ, 250×4,6 мм, 5 мкм; подвижная фаза (А) 0,1% ТФК в н-гексане (В) EtOH (4/1): А: В (80:20); скорость потока: 1,00 мл/мин).
Оптическое вращение [α20D]: -102,77 (С=1% в воде).
Пример 7 - Синтез (R)-N-((2S,3R)-1-амино-3-гидрокси-1-оксобутан-2-ил)-1-((S)-1-((2S,3R)-2-амино-3-гидроксибутаноил)пирролидин-2-карбонил)-2-(4-(трифторметил)бензил)пирролидин-2-карбоксамида (CM-7А) и (S)-N-((2S,3R)-1-амино-3-гидрокси-1-оксобутан-2-ил)-1-((S)-1-((2S,3R)-2-амино-3-гидроксибутаноил)пирролидин-2-карбонил)-2-(4-(трифторметил)бензил)пирролидин-2-карбоксамида (CM-7В):
[00202] Для синтеза (R)-N-((2S,3R)-1-амино-3-гидрокси-1-оксобутан-2-ил)-1-((S)-1-((2S,3R)-2-амино-3-гидроксибутаноил)пирролидин-2-карбонил)-2-(4-(трифторметил)бензил)пирролидин-2-карбоксамида (CM-7А) и (S)-N-((2S,3R)-1-амино-3-гидрокси-1-оксобутан-2-ил)-1-((S)-1-((2S,3R)-2-амино-3-гидроксибутаноил)пирролидин-2-карбонил)-2-(4-(трифторметил)бензил)пирролидин-2-карбоксамида (CM-7В) использовали следующую последовательность реакций (Схема G):
[00203] Схема G. Синтез CM-7А и CM-7В:

[00204] Синтез (S)-этил пирролидин-2-карбоксилата гидрохлорида (1):
К перемешанному раствору L-пролина (исходный материал) (110 г, 956,5 ммоль) в этаноле добавили тионилхлорид (141 мл, 1911,3 ммоль) и нагревали с дефлегматором в течение 16 часов. Реакционную смесь довели до комнатной температуры и концентрировали под вакуумом с получением соединения 1 в виде гидрохлоридной соли (170 г, 99%).
[00205] Синтез (S)-1-бензил 2-метил пирролидин-1,2-дикарбоксилата (2):
К перемешанному раствору соединения 1 (170 г, 947 ммоль) в CH2Cl2 добавили Et3N (398 мл, 2,83 моль). После перемешивания в течение 30 минут к реакционной смеси добавили Cbz-Cl (1,136 моль) и продолжали перемешивание еще 12 часов при комнатной температуре. Реакционную смесь промыли водой и экстрагировали CH2Cl2. Органический слой высушили над безводным Na2SO4 и концентрировали под вакуумом. Неочищенное вещество очистили колоночной хроматографией с получением соединения 2 (230 г, 88%).
1H-ЯМР: (200 МГц, ДМСО-d6): δ 7.42-7.31(m, 5Н), 5.09 (m, 2Н), 4.32-4.23(m, 1Н), 4.08-3.98(m, 2H), 3.50-3.38 (m, 2H), 2.30-2.18 (m, 1H), 1.90-1.81(m, 3H), 1.10-1.04 (t, 3H).
Масса m/z: 278 [M++1].
[00206] Синтез 1-бензил 2-этил 2-(4-(трифторметил)бензил)пирролидин-1,2-дикарбоксилата (3):
К перемешанному раствору соединения 2 (5 г, 0,018 моль) в ТТФ (50 мл) под инертной атмосферой добавили LiHMDS (1Mb ТТФ) (27 мл, 0,026 моль) при -78°С и перемешивали в течение 30 минут. К смеси по каплям добавили 1-(бромметил)-4-(трифторметил)бензол (3,3 мл, 0,021 моль) при -40°С и оставили нагреваться до комнатной температуры, и перемешивали в течение 3 часов. Реакционную смесь охладили до 5°С, погасили водным раствором NH4Cl, а водный слой экстрагировали EtOAc (2×100 мл). Объединенные органические экстракты высушили над безводным Na2SO4 и концентрировали под пониженным давлением. Полученный неочищенный остаток очистили силикагелевой колоночной хроматографией, элюируя 10% EtOAc в гексане, с получением соединения 3 (6 г, 76%) в виде жидкого вещества.
1Н-ЯМР: (500 МГц, ДМСО-d6): δ 7.64-7.58 (m, 2Н), 7.44-7.25 (m, 7Н), 5.22-5.12 (m, 2Н), 4.16-4.12 (m, 1H), 4.05-3.89 (m, 1Н), 3.62 (d, 1H), 3.48-3.39 (m, 1H), 3.16-3.10 (m, 1H), 2.99-2.89 (m, 1H), 2.19-1.99 (m, 2H), 1.63-1.58 (m, 1H), 1.19 (t, 2H), 1.05 (t, 1H), 0.92-0.89 (m, 1H).
[00207] Синтез 1-((бензилокси)карбонил)-2-(4-(трифторметил)бензил)пирролидин-2-карбоновой кислоты (4):
К перемешанному раствору соединения 3 (6 г, 0,014 моль) в метаноле (50 мл) и воде (15 мл) добавили 2 н. водный раствор NaOH (1,1 г, 0,027 моль) и нагревали до 70°С в течение 3 часов. Летучие вещества выпарили под пониженным давлением, а полученный остаток разбавили ледяной водой (50 мл) и промыли эфиром (50 мл). Водный слой подкислили до рН~2 с помощью 2 н. HCl и экстрагировали EtOAc (2×100 мл). Объединенные органические слои высушили над безводным Na2SO4 и концентрировали под пониженным давлением с получением соединения 4 (4 г, 71%) в виде грязновато-белого твердого вещества.
1H-ЯМР: (400 МГц, ДМСО-d6): δ 7.59-7.22 (m, 10Н), 5.24-5.19 (m, 1Н), 5.05-5.00 (m, 1H), 3.65 (d, 1H), 3.35-3.20 (m, 1H), 3.19 (t, 1H), 3.01-2.89 (m, 1H), 2.00-1.97 (m, 2H), 1.65-1.61 (m, 1H), 0.85-0.77 (m, 1H).
ЖХМС (ESI): 406 [M+-1].
Масса m/z: 406 [М+-1].
[00208] Синтез бензил 2-(((2R,3S)-3-гидрокси-1-метокси-1-оксобутан-2-ил)карбамоил)-2-(4-(трифторметил)бензил)пирролидин-1-карбоксилата (5):
К перемешанному раствору соединения 4 (4 г, 9,80 ммоль) в CH2Cl2 (40) добавили EDCl (2,8 г, 14,7 ммоль), затем HOBt (2,6 г, 19,6 ммоль) и DIPEA (3,5 мл, 19,6 ммоль) при 0°С. После перемешивания в течение 10 минут к реакционной смеси добавили гидрохлоридную соль метилового эфира L-треонина (2 г, 11,8 ммоль) и перемешивали еще 16 часов при комнатной температуре. После расходования исходного материала (по ТСХ) реакционную смесь разбавили CH2Cl2 (150 мл) и промыли водным раствором NaHCO3 (2×30 мл), затем лимонной кислотой (2×30 мл). Органический слой высушили над Na2SO4 и концентрировали с получением соединения 5 (5 г, неочищенное).
[00209] Синтез бензил 2-(((2S,3R)-3-ацетокси-1-метокси-1-оксобутан-2-ил)карбамоил)-2-(4-(трифторметил)бензил)пирролидин-1-карбоксилата (6):
К перемешанному раствору соединения 5 (5 г, 9,57 ммоль) в CH2Cl2 (50 мл) добавили Et3N (4 мл, 28,7 ммоль), затем Ас2О (2,7 мл, 28,7 ммоль) при 0°С и перемешивали в течение 1 часа. К смеси добавили DMAP (0,1 г) и перемешивали при комнатной температуре в течение 16 часов. Летучие вещества выпарили под пониженным давлением, а полученный неочищенный материал очистили колоночной хроматографией, эюируя 20% EtOAc в гексане, с получением соединения 6 (3,4 г, 63%) в виде светло-желтого жидкого вещества.
1Н-ЯМР: (500 МГц, ДМСО-d6) (ротамеры): δ 7.62-7.50 (m, 2Н), 7.43-7.24 (m, 7Н), 5.27-5.21 (m, 2Н), 5.11-5.09 (m, 1Н), 4.67-4.62 (m, 1H), 3.66-3.64 (m, 4H), 3.55-3.46 (m, 2H), 2.92-2.85 (m, 2H), 2.05-1.95 (m, 1H), 1.99 (s, 3H), 1.56-1.46 (m, 1H), 1.15-1.11 (m, 4H).
ЖХМС (ESI): 565 [М++1].
[00210] Синтез (2S,3R)-метил 3-ацетокси-2-(2-(4-(трифторметил)бензил)пирролидин-2-карбоксамидо)бутаноата (7):
К перемешанному раствору соединения 6 (3,4 г, 6,02 ммоль) в метаноле (20 мл) добавили влажный 10% Pd/C (0,34 г) под атмосферой N2. Реакционную смесь перемешивали под атмосферой Н2 (баллонное давление) в течение 6 часов. После расходования исходного материала (по ТСХ) реакционную смесь отфильтровали через слой целита, а фильтрат концентрировали под пониженным давлением с получением соединения 7 (2,1 г, 77%) в виде густого сиропообразного вещества.
1Н-ЯМР: (400 МГц, ДМСО-d6) (ротамеры): δ 8.22 (br s, 1H), 7.62-7.56 (m, 2H), 7.45-7.39 (m, 2H), 5.23-5.19 (m, 1H), 4.50-4.42 (m, 1H), 3.60 (d, 3H), 3.05-3.00 (m, 1H), 2.97-2.85 (m, 3H), 2.18-2.09 (m, 1H), 1.95 (d, 3H), 1.65-1.51 (m, 3H), 1.10 (d, 2H), 0.75 (d, 1H).
ЖХМС m/z: 431 [M++1].
[00211] Синтез (2S)-бензил 2-(2-(((2S,3R)-3-ацетокси-1-метокси-1-оксобутан-2-ил)карбамоил)-2-(4-(трифторметил)бензил)пирролидин-1-карбонил)пирролидин-1-карбоксилата (8):
К перемешанному раствору соединения 7 (4,5 г, 10,4 ммоль) в CH2Cl2 (60 мл) и воде (40 мл) добавили Na2CO3 (2,2 г, 20,9 ммоль) и перемешивали при 0°С в течение 5 минут. Добавили (S)-бензил 2-(хлоркарбонил)пирролидин-1-карбоксилат (3,3 г, 12,5 ммоль) и перемешивали реакционную смесь при комнатной температуре в течение 2 часов. Отделенный органический слой концентрировали под вакуумом, а полученный неочищенный материал очистили колоночной хроматографией, элюируя 50% EtOAc в гексане, с получением соединения 8 (4,5 г, 65%) в виде желтого жидкого вещества. (S)-бензил 2-(хлоркарбонил)пирролидин-1-карбоксилат синтезировали следующим образом. К раствору (S)-1-(бензилоксикарбонил)пирролидин-2-карбоновой кислоты (20,6 г, 82,8 ммоль) в CH2Cl2 (20 мл) по каплям добавили SOCl2 (20,5 г, 172,6 ммоль) при 0°С и нагревали с дефлегматором в течение 2 часов. Летучие вещества удалили под пониженным давлением с получением (S)-бензил 2-(хлоркарбонил)пирролидин-1-карбоксилата.
1H-ЯМР: (400 МГц, ДМСО-d6) (ротамеры): δ 8.50 (br s, 1Н), 7.79-7.65 (m, 2H), 7.52-7.37 (m, 7H), 5.42-5.05 (m, 3H), 4.79-4.59 (m, 1H), 4.15 (q, 1H), 3.78 (s, 3H), 3.69-3.50 (m, 3H), 3.21-3.13 (m, 1H), 2.45-2.16 (m, 2H), 2.10 (s, 6H), 2.05-2.00 (m, 2H), 1.95-1.85 (m, 1H), 1.44-1.19 (m, 6H).
ЖХМС m/z: 662 [M++1].
[00212] Синтез (2S,3R)-метил 3-ацетокси-2-(1-((S)-пирролидин-2-карбонил)-2-(4-(трифторметил)бензил)пирролидин-2-карбоксамидо)бутаноата (9):
К перемешанному раствору соединения 8 (4,5 г, 6,8 ммоль) в МеОН (45 мл) добавили влажный 10% Pd/C (0,45 г) под инертной атмосферой и перемешивали в течение 16 часов под атмосферой Н2 (баллонное давление). Реакционную смесь отфильтровали через слой целита и концентрировали под пониженным давлением с получением соединения 9 (3,5 г, неочищенное) в виде полутвердого вещества.
[00213] Синтез бензил 2-(трет-бутоксикарбониламино)-3-гидроксибутаноата (А):
К раствору 2-(трет-бутоксикарбониламино)-3-гидроксибутановой кислоты (Вос-Thr) (50 г, 228,3 ммоль) в ДМФ (500 мл) добавили K2CO3 (63 г, 456,6 ммоль) и перемешивали при комнатной температуре в течение 15 минут. К смеси добавили бензилбромид (46,83 г, 273,9 ммоль) и перемешивали при комнатной температуре в течение 6 часов. Реакционную смесь разбавили водой (500 мл) и экстрагировали EtOAc (2×750 мл). Объединенные органические слои промыли насыщенным солевым раствором (50 мл), высушили над безводным Na2SO4 и концентрировали под пониженным давлением. Неочищенный материал очистили силикагелевой колоночной хроматографией, элюируя 20% EtOAc в гексане, с получением бензил 2-(трет-бутоксикарбониламино)-3-гидроксибутаноата А (52 г, 73%).
1Н-ЯМР: (500 МГц, ДМСО-d6): δ 7.37-7.30 (m, 5Н), 6.60 (d, 1Н), 5.18-5.08 (m, 2H), 4.76 (d, 1H), 4.08-4.00 (m, 2H), 1.38 (s, 9H), 1.09 (d, 3H).
Масса m/z: 310,0 [M++1], 210 [M+-De Boc].
[00214] Синтез бензил 3-ацетокси-2-(трет-бутоксикарбониламино)бутаноата (В):
К перемешанному раствору соединения А (52 г, 168,2 ммоль) в CH2Cl2 (500 мл) добавили Ac2O (20,5 г, 201,9 ммоль), Et3N (25,4 г, 252,4 ммоль) и DMAP (3,5 г) и перемешивали при комнатной температуре в течение 2 часов. Летучие вещества удалили под пониженным давлением. Полученный остаток разбавили EtOAc (750 мл) и промыли холодным 0,5 н. раствором HCl (2×200 мл). Органический слой промыли насыщенным солевым раствором, высушили над безводным Na2SO4 и концентрировали под пониженным давлением с получением 3-ацетокси-2-(трет-бутоксикарбониламино)бутаноата В (52 г, 88%).
1H-ЯМР: (500 МГц, ДМСО-d6): δ 7.35-7.34 (m, 5Н), 7.27-7.25 (d, 1Н), 5.18-5.06 (m, 3Н), 4.34-4.32 (m, 1H), 1.90 (s, 3H), 1.39 (s, 9H), 1.16 (d, 3H).
Масса m/z: 252 [M++1-De Boc].
[00215] Синтез (2S,3R)-3-ацетокси-2-(трет-бутоксикарбониламино)бутановой кислоты (С):
К перемешанному раствору соединения В (52 г, 148,1 ммоль) в МеОН (1 л) добавили 10% Pd/C под атмосферой N2 и перемешивали реакционную смесь под атмосферой водорода в течение 16 часов. Реакционную смесь отфильтровали через слой целита, полученный фильтрат выпарили под вакуумом, а неочищенный остаток растерли с гексаном с получением (2S,3R)-3-ацетокси-2-(трет-бутоксикарбониламино)бутановой кислоты С (35 г, 90%).
1Н-ЯМР: (500 МГц, ДМСО-d6): δ 12.78 (br s, 1H), 6.94 (d, 1H), 5.16-5.14 (m, 1H), 4.17-4.15 (m, 1H), 1.95 (s, 3H), 1.39 (s, 9H), 1.10 (d, 3H).
Масса m/z: 260,0 [М-1].
[00216] Синтез (2S,3R)-метил 3-ацетокси-2-(1-((S)-1-((2S,3R)-3-ацетокси-2-((трет-бутоксикарбонил)амино)бутаноил)пирролидин-2-карбонил)-2-(4-(трифторметил)бензил)пирролидин-2-карбоксамидо)бутаноата (10):
К перемешанному раствору соединения С (1,78 г, 6,83 ммоль) в CH2Cl2 (30 мл) добавили EDCI (1,63 г, 8,53 ммоль), HOBt (1,53 г, 11,38 ммоль), затем DIPEA (2 мл, 14,2 ммоль) при 0°С и перемешивали в течение 10 минут. К смеси добавили соединение 9 (3 г, 5,69 ммоль) и перемешивали в течение 16 часов. Реакционную смесь экстрагировали EtOAc (2×75 мл), а отделенный органический слой промыли водным раствором NaHCO3 (100 мл), водным раствором лимонной кислоты (100 мл), затем насыщенным солевым раствором (100 мл), высушили над Na2SO4 и концентрировали под пониженным давлением. Неочищенный материал очистили колоночной хроматографией, элюируя 70% EtOAc в гексане, с получением соединения 10 (2,1 г, 51%) в виде грязновато-белого твердого вещества.
1H-ЯМР: (400 МГц, ДМСО-d6) (ротамеры): δ 7.71-7.55 (m, 3Н), 7.34-7.05 (m, 2Н), 5.35-5.20 (m, 1Н), 5.09-5.01 (m, 1H), 4.78-4.35 (m, 3H), 3.90-3.85 (m, 2H), 3.67 (s, 4H), 3.39-3.25 (m, 1H), 3.25-3.05 (m, 1H), 2.21-1.96 (m, 12H), 1.39 (d, 9H), 1.35-1.25 (m, 8H), 0.85 (d, 1H).
[00217] Синтез трет-бутил ((2S,3R)-1-((2S)-2-(2-(((2S,3R)-1-амино-3-гидрокси-1-оксобутан-2-ил)карбамоил)-2-(4-(трифторметил)бензил)пирролидин-1-карбонил)пирролидин-1-ил)-3-гидрокси-1-оксобутан-2-ил)карбамата (11):
К перемешанному раствору соединения 10 (1 г, 1,29 ммоль) в МеОН (10 мл) добавили метанольный NH3 (20 мл) при 0°С и перемешивали при комнатной температуре в течение 24 часов. Реакционную смесь концентрировали под вакуумом с получением соединения 11 (0,87 г, неочищенное) в виде грязновато-белого твердого вещества.
Хиральная препаративная ВЭЖХ изомеров:
Изомеры соединения 11 (0,3 г, неочищенное) разделили хиральной препаративной ВЭЖХ с получением Соединения 11 (0,3 г) и Соединения 11-F2 (0,15 г).
Аналитические данные для Соединения 11:
1H-ЯМР: (400 МГц, ДМСО-d6): δ 7.59-7.51 (m, 4Н), 7.24 (s, 1Н), 7.02 (s, 1H), 6.79-6.60 (m, 2H), 5.09 (d, 1H), 4.77-4.74 (m, 1H), 4.64-4.61 (m, 1H), 4.21 (t, 2H), 3.98 (dd, 1H), 3.85-3.80 (m, 2H), 3.71-3.60 (m, 3H), 3.25-3.19 (m, 2H), 2.21-1.85 (m, 5H), 1.75-1.70 (m, 1H), 1.44 (s, 10H), 1.21 (d, 3H), 1.09 (d, 3H), 0.91-0.85 (m, 1H).
ВЭЖХ: 93%.
ЖХМС (ESI): m/z 671,6 [M++1].
Хиральная ВЭЖХ: Rt=11,70 мин. (Chiralpak ΙΑ, 250×4,6 мм, 5 мкм; подвижная фаза (А) н-гексан (В) EtOH (4/1): А: В (80:20); скорость потока: 1,00 мл/мин).
Аналитические данные для Соединения 11-F2:
1Н-ЯМР: (400 МГц, ДМСО-d6): δ 7.59-7.51 (m, 4Н), 7.24 (s, 1Н), 7.02 (s, 1H), 6.79-6.60 (m, 2H), 5.09 (d, 1H), 4.77-4.74 (m, 1H), 4.64-4.61 (m, 1H), 4.21 (t, 2H), 3.98 (dd, 1H), 3.85-3.80 (m, 2H), 3.71-3.60 (m, 3H), 3.25-3.19 (m, 2H), 2.21-1.85 (m, 5H), 1.75-1.70 (m, 1H), 1.44 (s, 10H), 1.21 (d, 3H), 1.09 (d, 3H), 0.91-0.85 (m, 1H).
ВЭЖХ: 91%.
ЖХМС (ESI): m/z 671,6 [M++1].
Хиральная ВЭЖХ: Rt=19,68 мин. (Chiralpak ΙΑ, 250×4,6 мм, 5 мкм; подвижная фаза (А) н-гексан (В) EtOH (4/1): А: В (80:20); скорость потока: 1,00 мл/мин).
[00218] Синтез (R)-N-((2S,3R)-1-амино-3-гидрокси-1-оксобутан-2-ил)-1-((S)-1-((2S,3R)-2-амино-3-гидроксибутаноил)пирролидин-2-карбонил)-2-(4-(трифторметил)бензил)пирролидин-2-карбоксамида (СМ-7А):
К перемешанному раствору соединения 11-F1 (0,3 г, 0,45 ммоль) в CH2Cl2 (5 мл) добавили 4 н. HCl в 1,4-диоксане (1 мл) при 0°С и перемешивали при комнатной температуре в течение 3 часов. Реакционную смесь концентрировали под вакуумом с получением CM-7А (0,15 г, 59%) в виде гидрохлоридной соли.
1Н-ЯМР: (400 МГц, D2O): δ 7.89 (d, 2Н), 7.57 (d, 2Н), 4.95-4.90 (m, 1Н), 4.43-4.35 (m, 4H), 3.94-3.79 (m, 5H), 3.35 (q, 2H), 2.56-2.50 (m, 1H), 2.35-2.29 (m, 2H), 2.14-2.05 (m, 3H), 1.81-1.75 (m, 1H), 1.35 (d, 3H), 1.32 (d, 3H), 1.19-1.11 (m, 1H).
ЖХМС (ESI) m/z: 572,6 [M++1].
Масса m/z: 572,4 [М++1].
СЭЖХ чистота: 97%.
Оптическое вращение [α20D]:+14,86 (С=1% в воде).
[00219] Синтез (S)-N-((2S,3R)-1-амино-3-гидрокси-1-оксобутан-2-ил)-1-((S)-1-((2S,3R)-2-амино-3-гидроксибутаноил)пирролидин-2-карбонил)-2-(4-(трифторметил)бензил)пирролидин-2-карбоксамида (CM-7 В):
К перемешанному раствору соединения 11-F2 (0,15 г, 0,22 ммоль) в CH2Cl2 (5 мл) добавили 4 н. HCl в 1,4-диоксане (0,5 мл) при 0°С и перемешивали при комнатной температуре в течение 3 часов. Реакционную смесь концентрировали под вакуумом с получением CM-7В (140 мг, 59%) в виде гидрохлоридной соли.
1H-ЯМР: (400 МГц, D2O): δ 7.89 (d, 2Н), 7.57 (d, 2Н), 4.95-4.90 (m, 1H), 4.43-4.35 (m, 4H), 3.94-3.79 (m, 5H), 3.35 (q, 2H), 2.56-2.50 (m, 1H), 2.35-2.29 (m, 2H), 2.14-2.05 (m, 3H), 1.81-1.75 (m, 1H), 1.35 (d, 3H), 1.32 (d, 3H), 1.19-1.11 (m, 1H).
ЖХМС (ESI) m/z: 572,8 [M++1].
Масса m/z: 572,4 [М++1].
СЭЖХ чистота: 97,67%.
Оптическое вращение [α20D]: -105,51 (С=1% в воде).
Пример 8 - Синтез (R)-N-((2S,3R)-1-амино-3-гидрокси-1-оксобутан-2-ил)-1-((S)-1-((2S,3R)-2-амино-3-гидроксибутаноил)пирролидин-2-карбонил)-2-(3-метилбензил)пирролиди-2-карбоксамида (CM-8А) и (S)-N-(2S,3R)-1-амино-3-гидрокси-1-оксобутан-2-ил)-1-((S)-1-((2S,3R)-2-амино-3-гидроксибутаноил)пирролидин-2-карбонил)-2-(3-метилбензил)пирролидин-2-карбоксамида (CM-8В):
[00220] Для синтеза (R)-N-((2S,3R)-1-амино-3-гидрокси-1-оксобутан-2-ил)-1-((S)-1-((2S,3R)-2-амино-3-гидроксибутаноил)пирролидин-2-карбонил)-2-(3-метилбензил)пирролидин-2-карбоксамида (CM-8А) и (S)-N-{(2S,3R)-1-амино-3-гидрокси-1-оксобутан-2-ил)-1-((S)-1-((2S,3R)-2-амино-3-гидроксибутаноил)пирролидин-2-карбонил)-2-(3-метилбензил)пирролидин-2-карбоксамида (CM-8В) использовали следующую последовательность реакций (Схема Н):
[00221] Схема Н. Синтез СМ-8А и СМ-8В:

[00222] Синтез (S)-этил пирролидин-2-карбоксилата гидрохлорида (1):
К перемешанному раствору L-пролина (исходный материал) (110 г, 956,5 ммоль) в этаноле добавили тионилхлорид (141 мл, 1911,3 ммоль) и нагревали с дефлегматором в течение 16 часов. Реакционную смесь довели до комнатной температуры и концентрировали под вакуумом с получением соединения 1 в виде гидрохлоридной соли (170 г, 99%).
[00223] Синтез (S)-1-бензил 2-метил пирролидин-1,2-дикарбоксилата (2):
К перемешанному раствору соединения 1 (170 г, 947 ммоль) в CH2Cl2 добавили Et3N (398 мл, 2,83 моль). После перемешивания в течение 30 минут к реакционной смеси добавили Cbz-Cl (1,136 моль) и продолжали перемешивание еще 12 часов при комнатной температуре. Реакционную смесь промыли водой и экстрагировали CH2Cl2. Органический слой высушили над безводным Na2SO4 и концентрировали под вакуумом. Неочищенное вещество очистили колоночной хроматографией с получением соединения 2 (230 г, 88%).
1H-ЯМР: (200 МГц, ДМСО-d6): δ 7.42-7.3l(m, 5Н), 5.09 (m, 2Н), 4.32-4.23(m, 1Н), 4.08-3.98(m, 2H), 3.50-3.38 (m, 2H), 2.30-2.18 (m, 1H), 1.90-1.81(m, 3Н), 1.10-1.04 (t, 3Н).
Масса m/z: 278 [М++1].
[00224] Синтез 1-бензил 2-этил 2-(3-метилбензил)пирролидин-1,2-дикарбоксилата (3):
К перемешанному раствору соединения 2 (5 г, 0,018 моль) в ТТФ (50 мл) под инертной атмосферой добавили LiHMDS (1Mb ТТФ) (37 мл, 0,036 моль) при -78°С и перемешивали в течение 30 минут. К смеси по каплям добавили 3-метил-бензилбромид (4 г, 0,021 моль) при -40°С и оставили нагреваться до комнатной температуры, и перемешивали в течение 2 часов. Реакционную смесь охладили до 5°С, погасили насыщенным раствором NH4Cl, а водный слой экстрагировали EtOAc (2×100 мл). Объединенные органические экстракты высушили над безводным Na2SO4 и концентрировали под пониженным давлением. Полученный неочищенный остаток очистили силикагелевой колоночной хроматографией, элюируя 20% EtOAc в гексане, с получением соединения 3 (6 г, 87%) в виде жидкого вещества.
1H-ЯМР: (500 МГц, ДМСО-d6): δ 7.47-7.32 (m, 5Н), 7.17-7.12 (m, 1H), 7.05-7.01 (m, 1H), 6.95-6.90 (m, 2H), 5.25-5.00 (m, 2H), 4.16-4.12 (m, 1H), 4.00-3.89 (m, 1H), 3.52 (d, 1H), 3.38-3.33 (m, 1H), 2.99-2.89 (m, 2H), 2.23 (s, 3H), 2.12-1.90 (m, 1H), 1.56-1.51 (m, 1H), 1.05-1.01 (m, 3H), 1.00-0.97 (m, 1H), 0.92-0.89 (m, 1H).
ЖХМС (ESI): 382 [М++1].
ВЭЖХ (чистота): 99%.
[00225] Синтез 1-((бензилокси)карбонил)-2-(3-метилбензил)пирролидин-2-карбоновой кислоты (4):
К перемешанному раствору соединения 3 (6 г, 0,015 моль) в метаноле (30 мл) и воде (15 мл) добавили 2 н. водный раствор NaOH (2 г, 0,052 моль) и нагревали до 85°С в течение 6 часов. Летучие вещества выпарили под пониженным давлением, а полученный остаток разбавили ледяной водой (50 мл) и промыли эфиром (50 мл). Водный слой подкислили до рН~2, используя 2 н. HCl, и экстрагировали EtOAc (2×100 мл). Объединенные органические слои высушили над безводным Na2SO4 и концентрировали под пониженным давлением с получением соединения 4 (3,3 г, 78%) в виде грязновато-белого твердого вещества.
1Н-ЯМР: (400 МГц, ДМСО-d6): δ 12.71 (br s, 1H), 7.40-7.30 (m, 5Н), 7.15-7.11 (m, 1Н), 7.07-7.00 (m, 1H), 6.97-6.85 (m, 2H), 5.27-5.20 (m, 1H), 5.05-5.00 (m, 1H), 3.65 (d, 1H), 3.35-3.20 (m, 1H), 3.00-2.85 (m, 2H), 2.26 (s, 3H), 2.00-1.97 (m, 2H), 1.65-1.61 (m, 1H), 0.85-0.77 (m, 1H).
ВЭЖХ (чистота): 99,78%.
[00226] Синтез бензил 2-(((2R,3S)-3-гидрокси-1-метокси-1-оксобутан-2-ил)карбамоил)-2-(3-метилбензил)пирролидин-1-карбоксилата (S):
К перемешанному раствору соединения 4 (3,3 г, 9,0 ммоль) в CH2Cl2 (30) добавили EDCI.HCl (2,2 г, 11,4 ммоль), затем HOBt (1,82 г, 13,5 ммоль) и DIPEA (6,1 мл, 27,0 ммоль) при 0°С. После перемешивания в течение 10 минут к реакционной смеси добавили гидрохлоридную соль метилового эфира L-треонина (1,95 г, 11,2 ммоль) и перемешивали еще 16 часов при комнатной температуре. После расходования исходного материала (по ТСХ) реакционную Смесь разбавили EtOAc (150 мл) и промыли водой (2×30 мл). Органический слой промыли насыщенным солевым раствором, высушили над Na2SO4, концентрировали и очистили силикагелевой колоночной хроматографией, элюируя 2% МеОН в CH2Cl2, с получением соединения 5 (3,15 г, 73%).
1Н-ЯМР: (500 МГц, ДМСО-d6): δ 7.64 (br s, 1Н), 7.44-7.31 (m, 5H), 7.12-7.00 (m, 2H), 6.95-6.85 (m, 2H), 5.32-5.25 (m, 1H), 5.05-4.94 (m, 2H), 4.25-4.20 (m, 1H), 4.15-4.08 (m, 1H), 3.66-3.64 (m, 2H), 3.45-3.41 (m, 2H), 3.14-3.09 (m, 1H), 2.89-2.84 (m, 1H), 2.20 (s, 3H), 2.05-2.02 (m, 2H), 1.55-1.51 (m, 1H), 1.09-0.98 (m, 4H).
ЖХМС (ESI): 469 [M++1].
[00227] Синтез бензил 2-(((2S,3R)-3-ацетокси-1-метокси-1-оксобутан-2-ил)карбамоил)-2-(3-метилбензил)пирролидин-1-карбоксилата (6):
К перемешанному раствору соединения 5 (3,1 г, 6,4 ммоль) в CH2Cl2 (30 мл) добавили Et3N (1,4 мл, 5,6 ммоль), затем Ас2О (0,8 мл, 8,4 ммоль) при 0°С и перемешивали в течение 1 часа. К смеси добавили DMAP (0,1 г) и перемешивали при комнатной температуре в течение 6 часов. Летучие вещества выпарили под пониженным давлением, а полученный неочищенный материал очистили колоночной хроматографией, элюируя 20% EtOAc в гексане, с получением соединения 6 (3,15 г, 92,5%).
1Н-ЯМР: (400 МГц, ДМСО-d6) (ротамеры): δ 8.25 (br s, 1Н), 7.42-7.30 (m, 5H), 7.12-7.00 (m, 2H), 6.98-6.89 (m, 2H), 5.25-5.21 (m, 2H), 5.11-5.09 (m, 1H), 4.67-4.62 (m, 1H), 3.66-3.64 (m, 2H), 3.55-3.46 (m, 3H), 3.15-3.10 (m, 1H), 2.92-2.85 (m, 1H), 2.20 (s, 3H), 2.05-1.95 (m, 5H), 1.56-1.46 (m, 1H), 1.15-1.11 (m, 4H).
ЖХМС (ESI): 511 [М++1].
ВЭЖХ (чистота): 98%.
[00228] Синтез (2S,3R)-метил 3-ацетокси-2-(2-(3-метилбензил)пирролидин-2-карбоксамидо)бутаноата (7):
К перемешанному раствору соединения 6 (3,15 г, 5,97 ммоль) в метаноле (30 мл) добавили влажный 10% Pd/C (1,0 г) под атмосферой N2. Реакционную смесь перемешивали под атмосферой Н2 (баллонное давление) в течение 4 часов. После расходования исходного материала (по ТСХ) реакционную смесь отфильтровали через слой целита, а фильтрат концентрировали под пониженным давлением с получением соединения 7 (2,35 г, 99,5%) в виде сиропообразного вещества.
1H-ЯМР: (400 МГц, ДМСО-d6) (ротамеры): δ 8.22 (br s, 1Н), 7.19-7.16 (m, 1H), 7.00-6.95 (m, 3H), 5.23-5.19 (m, 1H), 4.50-4.42 (m, 1H), 3.60 (d, 3H), 3.20 (m, 1H), 3.00-2.97 (m, 1H), 2.75-2.68 (m, 2H), 2.28 (s, 3H), 2.05-2.02 (m, 1H), 1.90 (d, 3H), 1.65-1.51 (m, 3H), 1.10(dd,3H).
ЖХМС m/z: 377 [M++1].
ВЭЖХ (чистота): 98,5%.
[00229] Синтез (2S)-бензил 2-(2-(((2S,3R)-3-ацетокси-1-метокси-1-оксобутан-2-ил)карбамоил)-2-(3-метилбензил)пирролидин-1-карбонил)пирролидин-1-карбоксилата (8):
К перемешанному раствору соединения 7 (2,35 г, 6,30 ммоль) в CH2Cl2 (60 мл) и воде (40 мл) добавили Na2CO3 (2,0 г, 18,8 ммоль) и перемешивали при 0°С в течение 5 минут. Добавили (S)-бензил 2-(хлоркарбонил)пирролидин-1-карбоксилат (2,12 г, 7,6 ммоль) и перемешивали реакционную смесь при комнатной температуре в течение 2 часов. Отделенный органический слой концентрировали под вакуумом, а полученный неочищенный материал очистили колоночной хроматографией, элюируя 3% МеОН в CH2Cl2, с получением соединения 8 (2,5 г, 63%). (S)-бензил 2-(хлоркарбонил)пирролидин-1-карбоксилат синтезировали следующим образом. К раствору (S)-1-(бензилоксикарбонил)пирролидин-2-карбоновой кислоты (20,6 г, 82,8 ммоль) в CH2Cl2 (20 мл) по каплям добавили SOCl2 (20,5 г, 172,6 ммоль) при 0°С и нагревали с дефлегматором в течение 2 часов. Летучие вещества удалили под пониженным давлением с получением (S)-бензил 2-(хлоркарбонил)пирролидин-1-карбоксилата.
1Н-ЯМР: (400 МГц, ДМСО-d6) (ротамеры): δ 8.50 (br s, 1Н), 7.36-7.23 (m, 5H), 7.15-6.85 (m, 5H), 5.21-5.05 (m, 2H), 5.04-4.92 (m, 1H), 4.65-4.50 (m, 1H), 4.53-4.45 (m, 1H), 3.65 (s, 3H), 3.54-3.46 (m, 4H), 3.21-3.13 (m, 2H), 2.25-2.16 (m, 4H), 2.05-2.00 (m, 2H), 1.95-1.85 (m, 4H), 1.56-1.51 (m, 2H), 1.15 (dd, 3H).
ЖХМС m/z: 608 [М++1].
ЖХМС (чистота): 91,3%.
[00230] Синтез (2S,3R)-метил 3-ацетокси-2-(2-(3-метилбензил)-1-((S)-пирролидин-2-карбонил)пирролидин-2-карбоксами до)бутаноата (9):
К перемешанному раствору соединения 8 (2,5 г, 4,0 ммоль) в МеОН (30 мл) добавили влажный 10% Pd/C (0,5 г) под инертной атмосферой и перемешивали в течение 4 часов под атмосферой Н2 (баллонное давление). Реакционную смесь отфильтровали через слой целита и концентрировали под пониженным давлением с получением соединения 9 (1,56 г, 79,5%).
1H-ЯМР: (400 МГц, ДМСО-d6) (ротамеры): δ 8.23 (dd, 1Н), 7.20-6.85 (m, 5H), 5.20-5.13 (m, 1H), 4.63-4.59 (m, 1H), 3.85-3.81 (m, 1H), 3.65-3.61 (m, 5H), 3.32-3.25 (m, 2H), 3.12-3.05 (m, 3H), 2.75-2.71 (m, 1H), 2.25 (s, 3H), 2.15-2.13 (m, 2H), 2.00 (d, 3H), 1.77 (m, 3H), 1.27(dd,4H).
ЖХМС m/z: 474 [M++1].
ЖХМС (чистота): 87,3%.
[00231] Синтез бензил 2-(трет-бутоксикарбониламино)-3-гидроксибутаноата (А):
К раствору 2-(трет-бутоксикарбониламино)-3-гидроксибутановой кислоты (Вос-Thr) (50 г, 228,3 ммоль) в ДМФ (500 мл) добавили K2CO3 (63 г, 456,6 ммоль) и перемешивали при комнатной температуре в течение 15 минут. К смеси добавили бензилбромид (46,83 г, 273,9 ммоль) и перемешивали при комнатной температуре в течение 6 часов. Реакционную смесь разбавили водой (500 мл) и экстрагировали EtOAc (2×750 мл). Объединенные органические слои промыли насыщенным солевым раствором (50 мл), высушили над безводным Na2SO4 и концентрировали под пониженным давлением. Неочищенным материал очистили силикагелевой колоночной хроматографией, элюируя 20% EtOAc в гексане, с получением бензил 2-(трет-бутоксикарбониламино)-3-гидроксибутаноата А (52 г, 73%).
1H-ЯМР: (500 МГц, ДМСО-d6): δ 7.37-7.30 (m, 5Н), 6.60 (d, 1Н), 5.18-5.08 (m, 2H), 4.76 (d, 1H), 4.08-4.00 (m, 2H), 1.38 (s, 9H), 1.09 (d, 3H).
Масса m/z: 310,0 [M++1], 210 [M+De Boc].
[00232] Синтез бензил 3-ацетокси-2-(трет-бутоксикарбониламино)бутаноата (В):
К перемешанному раствору соединения А (52 г, 168,2 ммоль) в CH2Cl2 (500 мл) добавили Ac2O (20,5 г, 201,9 ммоль), Et3N (25,4 г, 252,4 ммоль) и DMAP (3,5 г) и перемешивали при комнатной температуре в течение 2 часов. Летучие вещества удалили под пониженным давлением. Полученный остаток разбавили EtOAc (750 мл) и промыли холодным 0,5 н. раствором HCl (2×200 мл). Органический слой промыли насыщенным солевым раствором, высушили над безводным Na2SO4 и концентрировали под пониженным давлением с получением 3-ацетокси-2-(трет-бутоксикарбониламино)бутаноата В (52 г, 88%).
1H-ЯМР: (500 МГц, ДМСО-d6): δ 7.35-7.34 (m, 5Н), 7.27-7.25 (d, 1Н), 5.18-5.06 (m, 3Н), 4.34-4.32 (m, 1H), 1.90 (s, 3H), 1.39 (s, 9H), 1.16 (d, 3H).
Масса m/z: 252 [M++1-De Boc].
[00233] Синтез (2S,3R)-3-ацетокси-2-(трет-бутоксикарбониламино)бутановой кислоты (С):
К перемешанному раствору соединения В (52 г, 148,1 ммоль) в МеОН (1 л) добавили 10% Pd/C под атмосферой N2 и перемешивали реакционную смесь под атмосферой водорода в течение 16 часов. Реакционную смесь отфильтровали через слой целита, полученный фильтрат выпарили под вакуумом, а неочищенный остаток растерли с гексаном с получением (2S,3R)-3-ацетокси-2-(трет-бутоксикарбониламино)бутановой кислоты С (35 г, 90%).
1Н-ЯМР: (500 МГц, ДМСО-d6): δ 12.78 (br s, 1H), 6.94 (d, 1H), 5.16-5.14 (m, 1H), 4.17-4.15 (m, 1H), 1.95 (s, 3H), 1.39 (s, 9H), 1.10 (d, 3H).
Масса m/z: 260,0 [М-1].
[00234] Синтез (2S,3R)-метил-3-ацетокси-2-(1-((S)-1-((2S,3R)-2-((трет-бутоксикарбонил)амино)-3-гидроксибутаноил)пирролидин-2-карбонил)-2-(3-метилбензил)пирролидин-2-карбоксамидо)бутаноата (10):
К перемешанному раствору соединения С (10 г, 0,02 моль) в CH2Cl2 (100 мл) добавили EDCI (5 г, 0,02 моль), HOBt (5 г, 0,04 моль), затем DBPEA (10,6 мл, 0,06 моль) при 0°С и перемешивали в течение 10 минут. К смеси добавили соединение 9 (7 г, 0,025 моль) и перемешивали в течение 15 часов. Реакционную смесь экстрагировали EtOAc (2×75 мл), а отделенный органический слой промыли водой (200 мл), затем насыщенным солевым раствором (200 мл), высушили над Na2SO4 и концентрировали под пониженным давлением. Неочищенный материал очистили колоночной хроматографией, элюируя 4% МеОН в CH2Cl2, с получением соединения 10 (10 г, 67%) в виде сиропообразного вещества.
1H-ЯМР: (400 МГц, ДМСО-d6) (ротамеры): δ 7.24-6.91 (m, 5Н), 5.42-5.01 (m, 2Н), 4.73-4.68 (m, 1H), 4.54-4.41 (m, 1H), 3.90-3.85 (m, 1H), 3.67 (s, 3H), 3.65-3.60 (m, 2H), 3.59-3.55 (m, 1H), 3.59-3.55 (m, 1H), 3.30 (s, 3H), 3.12-3.05 (m, 1H), 2.33 (s, 3H), 2.05 (s, 3H), 2.00-1.96 (m, 4H), 1.94-1.85 (m, 4H), 1.43 (s, 9H), 1.25 (d, 3H), 1.15 (d, 3H).
ЖХМС (ESI): m/z 675 [M++1].
ВЭЖХ: 71%.
[00235] Синтез трет-бутил ((2S,3R)-1-((2S)-2-(2-(((2S,3R)-1-амино-3-гидрокси-1-оксобутан-2-ил)карбамоил)-2-(3-метилбензил)пирролидин-1-карбонил)пирролидин-1-ил)-3-гидрокси-1-оксобутан-2-ил)карбамата (11):
К перемешанному раствору соединения 10 (10 г, 0,014 моль) в МеОН (20 мл) добавили метанольный NH3 (100 мл) и поместили в закрытую пробирку. Реакционную смесь перемешивали при комнатной температуре в течение 20 часов. Реакционную смесь концентрировали под пониженным давлением. Полученный неочищенный материал очистили силикагелевой колоночной хроматографией, элюируя 4% МеОН в CH2Cl2, с получением соединения 11 (8,0 г, выход 99%).
1H-ЯМР: (400 МГц, ДМСО-d6) (ротамеры): δ 7.32 (br s, 2Н), 7.15-7.10 (m, 4Н), 7.05-6.91 (m, 4Н), 6.65 (br s, 2H), 4.75-4.59 (m, 2H), 4.24-3.45 (m, 8H), 3.35 (s, 1H), 3.05-3.00 (m, 1H), 2.32 (s, 3H), 2.26-1.85 (m, 6H), 1.75 (s, 6H), 1.60-1.55 (m, 1H), 1.47 (s, 9H), 1.15 (d, 3H), 1.05 (d, 3H).
ЖХМС (ESI): m/z 618 [M++1].
Хиральная препаративная ВЭЖХ изомеров:
Изомеры соединения 11 (8,0 г, 12,9 ммоль) разделили хиральной препаративной ВЭЖХ с получением Соединения 11-F1 (0,4 г) и Соединения 11-F2 (0,25 г).
Аналитические данные для Соединения 11-F1: ВЭЖХ: 91,1%.
ЖХМС (ESI): m/z 618 [М++1].
Хиральная ВЭЖХ: Rt=9,20 мин. (Chiralpak ΙΑ, 250×4,6 мм, 5 мкм; подвижная фаза (А) 0,1% ТФК в н-гексане (В) EtOH (4/1): А: В (80:20); скорость потока: 1,00 мл/мин).
Аналитические данные для Соединения 11-F2: ВЭЖХ: 84%.
ЖХМС (ESI): m/z 618 [М++1].
Хиральная ВЭЖХ: Rt=13,39 мин. (Chiralpak LA, 250×4,6 мм, 5 мкм, подвижная фаза (А) 0,1% ТФК в н-гексане (В) EtOH (4/1): А: В (80:20); скорость потока: 1,00 мл/мин).
[00236] Синтез (R)-N-((2S,3R)-1-амино-3-гидрокси-1-оксобутан-2-ил)-1-((S)-1-((2S,3R)-2-амино-3-гидроксибутаноил)пирролидин-2-карбонил)-2-(3-метилбензил)пирролидин-2-карбоксамида (CM-8А):
К перемешанному раствору соединения 12-F1 (0.4 г, 0,60 ммоль) в CH2Cl2 (20 мл) добавили 2 н. HCl в 1,4-диоксане (2 мл) при 0°С и перемешивали при комнатной температуре в течение 3 часов. Реакционную смесь концентрировали под пониженным давлением, а полученный материал растерли с EtOAc и высушили под вакуумом с получением СМ-8А (0,22 г, 66%) в виде гидрохлоридной соли.
1Н-ЯМР: (400 МГц, D2O): δ 7.35 (t, 1Н), 7.25 (s, 1H), 7.21 (d, 1H), 7.15 (d, 1H), 4.95-4.90 (m, 1H), 4.43-4.35 (m, 4H), 3.94-3.89 (m, 1H), 3.87-3.78 (m, 2H), 3.63 (d, 1H), 3.49-3.40 (m, 1H), 3.25 (d, 1H), 2.56-2.34 (m, 5H), 2.28-2.20 (m, 4H), 1.75-1.71 (m, 1H), 1.56 (d, 3H), 1.32 (d, 3H), 1.15-1.11 (m, 1H).
ЖХМС (ESI) m/z: 518 [М++1].
Чистота по ВЭЖХ: 97%.
Оптическое вращение [α25D]:+18,8 (С=1% в воде).
[00237] Синтез (S)-N-((2S,3R)-1-амино-3-гидрокси-1-оксобутан-2-ил)-1-((S)-1-((2S,3R)-2-амино-3-гидроксибутаноил)пирролидин-2-карбонил)-2-(3-метилбензил)пирролидин-2-карбоксамида (CM-8В):
К перемешанному раствору соединения 12-F2 (0,25 г, 0,40 ммоль) в CH2Cl2 (20 мл) добавили 2 н. HCl в 1,4-диоксане (2 мл) при 0°С и перемешивали при комнатной температуре в течение 3 часов. Реакционную смесь концентрировали под пониженным давлением, а полученный материал растерли с EtOAc и высушили под вакуумом с получение СМ-8В (130 мг, 62%) в виде гидрохлоридной соли.
1H-ЯМР: (400 МГц, D2O): δ 7.35-7.23 (m, 2Η), 7.12-7.00 (m, 2Н), 4.45-4.40 (m, 1Н), 4.32-4.10 (m, 3Н), 3.94-3.75 (m, 3Н), 3.60 (d, 1H), 3.25 (d, 1H), 3.14-3.11 (m, 1H), 2.56-2.05 (m, 10H), 1.85-1.79 (m, 2H), 1.45 (d, 3H), 1.25 (d, 3H).
ЖХМС (ESI) m/z: 518 [M++1].
Чистота по ВЭЖХ: 92%.
Оптическое вращение [α25D]: -104,9 (C=1% в воде).
Пример 8 - Синтез (R)-N-((2S,3R)1-амино-3-гидрокси-1-оксобутан-2-ил)-1-((S)-1-((2S,3R)-2-амино-3-гидроксибутаноил)пирролидин-2-карбонил)-2-(3-фторбензил)пирролидин-2-карбоксамида (CM-9А) и (S)-N-((2S,3R)-1-амино-3-гидрокси-1-оксобутан-2-ил)-1-((S)-1-((2S,3R)-2-амино-3-гидроксибутаноил)пирролидин-2-карбонил)-2-(3-фторбензил)пирролидин-2-карбоксамида (CM-9В):
[00238] Для синтеза (R)-N-((2S,3R)-1-амино-3-гидрокси-1-оксобутан-2-ил)-1-((S)-1-((2S,3R)-2-амино-3-гидроксибутаноил)пирролидин-2-карбонил)-2-(3-фторбензил)пирролидин-2-карбоксамида (CM-9А) и (S)-R-((2S,3R)-1-амино-3-гидрокси-1-оксобутан-2-ил)-1-((S)-1-((2S,3R)-2-амино-3-гидроксибутаноил)пирролидин-2-карбонил)-2-(3-фторбензил)пирролидин-2-карбоксамида (CM-9В) использовали следующую последовательность реакций (Схема I):
[00239] Схема I. Синтез CM-9А и CM-9В:
[00240] Синтез (S)-этил пирролидин-2-карбоксилата гидрохлорида (1):
К перемешанному раствору L-пролина (исходный материал) (110 г, 956,5 ммоль) в этаноле добавили тионилхлорид (141 мл, 1911,3 ммоль) и нагревали с дефлегматором в течение 16 часов. Реакционную смесь довели до комнатной температуры и концентрировали под вакуумом с получением соединения 1 в виде гидрохлоридной соли (170 г, 99%).
[00241] Синтез (S)-1-бензил 2-метил пирролидин-1,2-дикарбоксилата (2):
К перемешанному раствору соединения 1 (170 г, 947 ммоль в CH2Cl2 добавили Et3N (398 мл, 2,83 моль). После перемешивания в течение 30 минут, к реакционной смеси добавили Cbz-Cl (1,136 моль) и продолжали перемешивание еще 12 часов при комнатной температуре. Реакционную смесь промыли водой и экстрагировали CH2Cl2. Органический слой высушили над безводным Na2SO4 и концентрировали под вакуумом. Неочищенное вещество очистили колоночной хроматографией с получением соединения 2 (230 г, 88%).
1Н-ЯМР: (200 МГц, ДМСО-d6): δ 7.42-7.31 (m, 5Н), 5.09 (m, 2Н), 4.32-4.23(m, 1Н), 4.08-3.98(m, 2H), 3.50-3.38 (m, 2H), 2.30-2.18 (m, 1H), 1.90-1.81(m, 3H), 1.10-1.04 (t, 3H).
Масса m/z: 278 [M++1].
[00242] Синтез 1-бензил 2-этил-2-(3-фторбензил)пирролидин-1,2-дикарбоксилата (3):
К перемешанному раствору соединения 2 (10 г, 0,018 моль) в ТТФ (100 мл) под инертной атмосферой добавили LiHMDS (1Mb ТТФ) (76 мл, 0,076 моль) при -78°С и перемешивали в течение 1 часа. К этой смеси по каплям добавили 3-фторбензилбромид (5,6 мл, 0,045 моль) при -30°С и перемешивали в течение 3 часов. Реакционную смесь охладили до 5°С, погасили насыщенным раствором NH4Cl, а водный слой экстрагировали EtOAc (2×100 мл). Объединенные органические экстракты высушили над безводным Na2SO4 и концентрировали под пониженным давлением. Полученный неочищенный остаток очистили силикагелевой колоночной хроматографией, элюируя 12% EtOAc в гексане, с получением соединения 3 (11 г, 78%) в виде жидкого вещества.
1H-ЯМР: (400 МГц, ДМСО-d6): δ 7.64-7.58 (m, 2Н), 7.44-7.25 (m, 7Н), 5.22-5.12 (m, 2Н), 4.16-4.12 (m, 1Н), 4.05-3.89 (m, 1H), 3.62 (d, 1H), 3.48-3.39 (m, 1Н), 3.16-3.10 (m, 1Н), 2.99-2.89 (m, 1Н), 2.19-1.99 (m, 2Н), 1.63-1.58 (m, 1H), 1.19 (t, 2Н), 1.05 (t, 1Н), 0.92-0.89 (m, 1Н).
ЖХМС (ESI): 372 [М++1].
[00243] Синтез 1-((бензилокси)карбонил)-2-(3-фторбензил)пирролидин-2-карбоновой кислоты (4):
К перемешанному раствору соединения 3(11 г, 0,029 моль) в метаноле (50 мл) и воде (50 мл) добавили 2 н. водный раствор NaOH (3,5 г, 0,089 моль) и нагревали до 60°С в течение 3 часов. Летучие вещества выпарили под пониженным давлением, а полученный остаток разбавили ледяной водой (50 мл) и промыли эфиром (50 мл). Водный слой подкислили до рН~2 с помощью 2 н. HCl и экстрагировали EtOAc (2×100 мл). Объединенные органические слои высушили над безводным Na2SO4 и концентрировали под пониженным давлением с получением соединения 4 (9,5 г, 90%) в виде жидкого вещества.
1Н-ЯМР: (400 МГц, ДМСО-d6): δ 7.59-7.22 (m, 10Н), 5.24-5.19 (m, 1H), 5.05-5.00 (m, 1H), 3.65 (d, 1H), 3.35-3.20 (m, 1H), 3.19 (t, 1H), 3.01-2.89 (m, 1H), 2.00-1.97 (m, 2H), 1.65-1.61 (m, 1H), 0.85-0.77 (m, 1H).
ЖХМС (ESI): 358 [M++1].
[00244] Синтез бензил 2-(3-фторбензил)-2-(((2R,3S)-3-гидрокси-1-метокси-1-оксобутан-2-ил)карбамоил)пирролидин-1-карбоксилата (S):
К перемешанному раствору соединения 4 (9,5 г, 26,6 ммоль) в CH2Cl2 (100 мл) добавили EDCI.HCl (6,1 г, 31,9 ммоль), затем HOBt (5,4 г, 39,9 ммоль) и DLPEA (14 мл, 79,8 ммоль) при 0°С. После перемешивания в течение 10 минут, к реакционной смеси добавили гидрохлоридную соль метилового эфира L-треонина (4,97 г, 29,2 ммоль) и перемешивали еще 16 часов при комнатной температуре. После расходования исходного материала (по ТСХ), реакционную смесь разбавили CH2Cl2 (150 мл), промыли насыщенным солевым раствором, высушили над Na2SO4 и концентрировали с получением соединения 5 (13 г, неочищенное).
ЖХМС (ESI): 473,7 [М++1].
[00245] Синтез бензил 2-(((2S,3R)-3-ацетокси-1-метокси-1-оксобутан-2-ил)карбамоил)-2-(3-фторбензил)пирролидин-1-карбоксилата (6):
К перемешанному раствору соединения 5(13 г, 27,5 ммоль) в CH2Cl2 (100 мл) добавили Et3N (5,8 мл, 41,3 ммоль), затем Ас2О (3,23 мл, 33,0 ммоль) при 0°С и перемешивали в течение 10 минут. К этой смеси добавили DMAP (1,2 г) и перемешивали при комнатной температуре в течение 2 часов. Реакционную смесь промыли лимонной кислотой и экстрагировали CH2Cl2 (2×150 мл). Отделенный органический слой высушили над безводным Na2SO4, отфильтровали и концентрировали под пониженным давлением с получением неочищенного вещества, которое очистили колоночной хроматографией, элюируя 20% EtOAc в гексане, с получением соединения 6 (9,5 г, 67%) в виде светло-желтого жидкого вещества.
1Н-ЯМР: (400 МГц, ДМСО-d6) (ротамеры): δ 7.39-7.23 (m, 6Н), 7.05 (t, 1Н), 6.95-6.78 (m, 2H), 5.27-5.21 (m, 2H), 5.11-5.09 (m, 1H), 4.67-4.62 (m, 1H), 3.66-3.64 (m, 4H), 3.55-3.46 (m, 2H), 2.92-2.85 (m, 2H), 2.05-1.95 (m, 1H), 1.99 (s, 3H), 1.56-1.46 (m, 1H), 1.15-1.11 (m,4H).
ЖХМС (ESI): 515 [M++1].
[00246] Синтез (2S,3R)-метил 3-ацетокси-2-(2-(3-фторбензил)пирролидин-2-карбоксамидо)бутаноата (7):
К перемешанному раствору соединения 6 (9,5 г, 18,4 ммоль) в метаноле (100 мл) добавили влажный 10% Pd/C (1,2 г) под атмосферой N2. Реакционную смесь перемешивали под атмосферой Н2 (баллонное давление) в течение 3 часов. После расходования исходного материала (по ТСХ), реакционную смесь отфильтровали через слой целита, а фильтрат концентрировали под пониженным давлением с получением соединения 7 (6,3 г, 90%) в виде жидкого вещества.
1H-ЯМР: (400 МГц, ДМСО-d6) (ротамеры): δ 8.22 (br s, 1Н), 7.62-7.56 (m, 2H), 7.45-7.39 (m, 2H), 5.23-5.19 (m, 1H), 4.50-4.42 (m, 1H), 3.60 (d, 3H), 3.05-3.00 (m, 1H), 2.97-2.85 (m, 3H), 2.18-2.09 (m, 1H), 1.95 (d, 3H), 1.65-1.51 (m, 3H), 1.10 (d, 2H), 0.75 (d, 1H).
ЖХМС m/z: 381 [M++1].
[00247] Синтез (2S)-бензил 2-(2-(((2S,3R)-3-ацетокси-1-метокси-1-оксобутан-2-ил)карбамоил)-2-(3-фторбензил)пирролидин-1-карбонил)пирролидин-1-карбоксилата (8):
К перемешанному раствору соединения 7 (6,2 г, 16,3 ммоль) в CH2Cl2 (50 мл) и воде (50 мл) добавили Na2CO3 (4,3 г, 40,7 ммоль) и перемешивали при 0°С в течение 5 минут. Добавили (S)-бензил 2-(хлоркарбонил)пирролидин-1-карбоксилат (17,93 ммоль) и перемешивали реакционную смесь при комнатной температуре в течение 2 часов. Реакционную смесь промыли водой и экстрагировали CH2Cl2 (2×100 мл). Отделенный органический слой концентрировали под вакуумом, а полученный неочищенный материал очистили колоночной хроматографией, элюируя 30% EtOAc в гексане, с получением соединения 8 (7,3 г, 73%) в виде жидкого вещества. (S)-бензил 2-(хлоркарбонил)пирролидин)-1-карбоксилат синтезировали следующим образом. К раствору (S)-1-(бензилоксикарбонил)пирролидин-2-карбоновой кислоты (4,87 г, 19,5 ммоль) в CH2Cl2 (20 мл) по каплям добавили SOCl2 (3,0 мл, 40,75 ммоль) при 0°С и нагревали с дефлегматором в течение 2 часов. Летучие вещества удалили под пониженным давлением с получением (S)-бензил 2-(хлоркарбонил)пирролидин-1-карбоксилата.
1H-ЯМР: (400 МГц, ДМСО-d6) (Ротамеры): δ 8.50 (br s, 1Н), 7.79-7.65 (m, 2H), 7.52-7.37 (m, 7H), 5.42-5.05 (m, 3H), 4.79-4.59 (m, 1H), 4.15 (q, 1H), 3.78 (s, 3H), 3.69-3.50 (m, 2H), 3.21-3.13 (m, 1H), 2.45-2.16 (m, 2H), 2.10 (s, 6H), 2.05-2.00 (m, 2H), 1.95-1.85 (m, 1H), 1.44-1.19 (m,6H).
ЖХМС m/z: 612 [M++1], 613 [M++2].
[00248] Синтез (2S,3R)-метил 3-ацетокси-2-(2-(3-фторбензил)-1-((S)-пирролидин-2-карбонил)пирролидин-2-карбоксамидо)бутаноата (9):
К перемешанному раствору соединения 8 (7,2 г, 11,78 ммоль) в МеОН (80 мл) добавили влажный 10% Pd/C (1,2 г) под инертной атмосферой и перемешивали в течение 16 часов под атмосферой Н2 (баллонное давление). Реакционную смесь отфильтровали через слой целита и концентрировали под пониженным давлением с получением соединения 9 (5,2 г, 92%) в виде жидкого вещества.
[00249] Синтез бензил 2-(трет-бутоксикарбониламино)-3-гидроксибутаноата (А):
К раствору 2-(трет-бутоксикарбониламино)-3-гидроксибутановой кислоты (Вос-Thr) (50 г, 228,3 ммоль) в ДМФ (500 мл) добавили К2С03 (63 г, 456,6 ммоль) и перемешивали при комнатной температуре в течение 15 минут. К этой смеси добавили бензилбромид (46,83 г, 273,9 ммоль) и перемешивали при комнатной температуре в течение 6 часов. Реакционную смесь разбавили водой (500 мл) и экстрагировали EtOAc (2×750 мл). Объединенные органические слои промыли насыщенным солевым раствором (50 мл), высушили над безводным Na2SO4 и концентрировали под пониженным давлением. Неочищенным материал очистили силикагелевой колоночной хроматографией, элюируя 20% EtOAc в гексане, с получением бензил 2-(трет-бутоксикарбониламино)-3-гидроксибутаноата А (52 г, 73%).
1H-ЯМР: (500 МГц, ДМСО-d6): δ 7.37-7.30 (m, 5Н), 6.60 (d, 1Н), 5.18-5.08 (m, 2H), 4.76 (d, 1H), 4.08-4.00 (m, 2H), 1.38 (s, 9H), 1.09 (d, 3H).
Масса m/z: 310,0 [M++1], 210 [M+-De Boc].
[00250] Синтез бензил 3-ацетокси-2-(третя-бутоксикарбониламино)бутаноата (В):
К перемешанному раствору соединения А (52 г, 168,2 ммоль) в CH2Cl2 (500 мл) добавили Ac2O (20,5 г, 201,9 ммоль), Et3N (25,4 г, 252,4 ммоль) и DMAP (3,5 г) и перемешивали при комнатной температуре в течение 2 часов. Летучие вещества удалили под пониженным давлением. Полученный остаток разбавили EtOAc (750 мл) и промыли холодным 0,5 н. раствором HCl (2×200 мл). Органический слой промыли насыщенным солевым раствором, высушили над безводным Na2SO4 и концентрировали под пониженным давлением с получением 3-ацетокси-2-(трет-бутоксикарбониламино)бутаноата В (52 г, 88%).
1H-ЯМР: (500 МГц, ДМСО-d6): δ 7.35-7.34 (m, 5Н), 7.27-7.25 (d, 1Н), 5.18-5.06 (m, 3Н), 4.34-4.32 (m, 1H), 1.90 (s, 3H), 1.39 (s, 9H), 1.16 (d, 3H).
Масса m/z: 252 [M++1-De Boc].
[00251] Синтез (2S,3R)-3-ацетокси-2-(трет-бутоксикарбониламино)бутановой кислоты (С):
К перемешанному раствору соединения В (52 г, 148,1 ммоль) в МеОН (1 л) добавили 10% Pd/C под атмосферой N2 и перемешивали реакционную смесь под атмосферой водорода в течение 16 часов. Реакционную смесь отфильтровали через слой целита, полученный фильтрат выпарили под вакуумом, а неочищенный остаток растерли с гексаном с получением (2S,3R)-3-ацетокси-2-(трет-бутоксикарбониламино)бутановой кислоты С (35 г, 90%).
1H-ЯМР: (500 МГц, ДМСО-d6): δ 12.78 (br s, 1Н), 6.94 (d, 1H), 5.16-5.14 (m, 1H), 4.17-4.15 (m, 1H), 1.95 (s, 3H), 1.39 (s, 9H), 1.10 (d, 3H).
Масса m/z: 260,0 [М-1].
[00252] Синтез (2S,3R)-метил 3-ацетокси-2-(1-((S)-1-((2S,3R)-2-((трет-бутоксикарбонил)Амино)-3-гидроксибутаноил)пирролидин-2-карбонил)-2-(3-фторбензил)пирролидин-2-карбоксамидо)бутаноата (10):
К перемешанному раствору соединения С (3,41 г, 13,0 ммоль) в CH2Cl2 (50 мл) добавили EDCI (3,12 г, 16,3 ммоль), HOBt (2,2 г, 16,3 ммоль), затем DIPEA (5,7 мл, 32,7 ммоль) при 0°С и перемешивали в течение 10 минут. К этой смеси добавили соединение 9 (5,2 г, 10,9 ммоль) и перемешивали в течение 16 часов. Реакционную смесь экстрагировали CH2Cl2 (2×75 мл), а отделенный органический слой промыли водным раствором NaHCO3 (100 мл), водным раствором лимонной кислоты (100 мл), затем насыщенным солевым раствором (100 мл), высушили над Na2SO4 и концентрировали под пониженным давлением. Неочищенный материал очистили колоночной хроматографией, элюируя 2% МеОН в CH2Cl2, с получением соединения 10 (6,0 г, 77%) в виде жидкого вещества.
1Η-ΗΜΡ: (400 МГц, ДМСО-d6) (ротамеры): δ 7.71-7.55 (m, 3Н), 7.34-7.05 (m, 2Н), 5.35-5.20 (m, 1Н), 5.09-5.01 (m, 1H), 4.78-4.35 (m, 3H), 3.90-3.85 (m, 2H), 3.67 (s, 3H), 3.39-3.25 (m, 1H), 3.25-3.05 (m, 1H), 2.21-1.96 (m, 12H), 1.39 (d, 9H), 1.35-1.25 (m, 8H), 0.85 (d, 1H).
ЖХМС (ESI): m/z 478,4 [M++1], 479,4 [М++2].
[00253] Синтез трет-бутил ((2S,3R)-1-((2S)-2-(2-(((2S,3R)-1-амино-3-гидрокси-1-оксобутан-2-ил)карбамоил)-2-(3-фторбензил)пирролидин-1-карбонил)пирролидин-1-ил)-3-гидрокси-1-оксобутан-2-ил)карбамата (11):
Раствор соединения 10 (3 г, 4,1 ммоль) в метанольном растворе аммиака (50 мл) перемешивали при комнатной температуре в течение 48 часов. Реакционную Смесь концентрировали под вакуумом с получением неочищенного вещества, которое очистили силикагелевой колоночной хроматографией, элюируя 4% МеОН в CH2Cl2, с получением соединения 11 (1,7 г, неочищенное) в виде белого твердого вещества.
1H-ЯМР: (400 МГц, ДМСО-d6): δ 7.59-7.51 (m, 4Н), 7.24 (s, 1Н), 7.02 (s, 1H), 6.79-6.60 (m, 2H), 5.09 (d, 1H), 4.77-4.74 (m, 1H), 4.64-4.61 (m, 1H), 4.21 (t, 2H), 3.98 (dd, 1H), 3.85-3.80 (m, 2H), 3.71-3.60 (m, 3H), 3.25-3.19 (m, 2H), 2.21-1.85 (m, 5H), 1.75-1.70 (m, 1H), 1.44 (s, 10H), 1.21 (d, 3H), 1.09 (d, 3H), 0.91-0.85 (m, 1H).
ЖХМС (ESI): m/z 622 [M++1].
Хиральная препаративная ВЭЖХ изомеров:
Изомеры соединения 11 (1,7 г) разделили хиральной препаративной ВЭЖХ с получением Соединения 11 (0,4 г) и Соединения 11-F2 (0,3 г).
Аналитические данные для Соединения 11:
1H-ЯМР: (400 МГц, ДМСО-d6): δ 7.59-7.51 (m, 4Н), 7.24 (s, 1Н), 7.02 (s, 1Н), 6.79-6.60 (m, 2Н), 5.09 (d, 1Н), 4.77-4.74 (m, 1H), 4.64-4.61 (m, 1Н), 4.21 (t, 2Н), 3.98 (dd, 1Н), 3.85-3.80 (m, 2Н), 3.71-3.60 (m, 3Н), 3.25-3.19 (m, 2Н), 2.21-1.85 (m, 5Н), 1.75-1.70 (m, 1H), 1.44 (s, 10Н), 1.21 (d, 3Н), 1.09 (d, 3Н), 0.91-0.85 (m, 1H).
СЭЖХ: 87%.
ЖХМС (ESI): m/z 622 [М++1].
Хиральная ВЭЖХ: Rt=10,32 мин. (Chiralpak ΙΑ, 250×4,6 мм, 5 мкм; подвижная фаза (А) 0,1% ТФК в н-гексане (В) EtOH (4/1): А: В (80:20); скорость потока: 1,00 мл/мин).
Аналитические данные для Соединения 11-F2:
1Н-ЯМР: (400 МГц, ДМСО-d6): δ 7.59-7.51 (m, 4Н), 7.24 (s, 1Н), 7.02 (s, 1H), 6.79-6.60 (m, 2H), 5.09 (d, 1H), 4.77-4.74 (m, 1H), 4.64-4.61 (m, 1H), 4.21 (t, 2H), 3.98 (dd, 1H), 3.85-3.80 (m, 2H), 3.71-3.60 (m, 3H), 3.25-3.19 (m, 2H), 2.21-1.85 (m, 5H), 1.75-1.70 (m, 1H), 1.44 (s, 10Н), 1.21 (d, 3H), 1.09 (d, 3H), 0.91-0.85 (m, 1H).
СЭЖХ: 91%.
ЖХМС (ESI): m/z 622 [M++1].
Хиральная ВЭЖХ: Rt=15,37 мин. (Chiralpak ΙΑ, 250×4,6 мм, 5 мкм; подвижная фаза (А) 0,1% ТФК в н-гексане (В) EtOH (4/1): А: В (80:20); скорость потока: 1,00 мл/мин).
[00254] Синтез (R)-Ν-((2S,3R)-1-амино-3-гидрокси-1-оксобутан-2-ил)-1-((S)-1-((2S,3R)-2-амино-3-гидроксибутаноил)пирролидин-2-карбонил)-2-(3-фторбензил)пирролидин-2-карбоксамида (CM-9А):
К перемешанному раствору соединения 11-F1 (0,4 г, 0,64 ммоль) в CH2Cl2 (5 мл) добавили 4 н. HCl в 1,4-диоксане (2 мл) при 0°С и перемешивали при комнатной температуре в течение 2 часов. Реакционную смесь концентрировали под вакуумом. Полученный неочищенный материал промыли EtOAc (20 мл), затем н-пентаном (20 мл) и высушили под вакуумом с получением СМ-9А (0,2 г, 59%) в виде гидрохлоридной соли.
1Н-ЯМР: (400 МГц, D2O): δ 7.89 (d, 2Н), 7.57 (d, 2Н), 4.95-4.90 (m, 1Н), 4.43-4.35 (m, 4H), 3.94-3.79 (m, 5H), 3.35 (q, 2H), 2.56-2.50 (m, 1H), 2.35-2.29 (m, 2H), 2.14-2.05 (m, 3H), 1.81-1.75 (m, 1H), 1.35 (d, 3H), 1.32 (d, 3H), 1.19-1.11 (m, 1H).
ЖХМС (ESI) m/z: 522 [M++1], 523 [M++2].
Чистота по СЭЖХ: 97%.
Чистота по хиральной ВЭЖХ: 99%.
Оптическое вращение [α20D]:+15,6 (С=1% в воде).
[00255] Синтез (S)-N-((2S,3R)-1-амино-3-гидрокси-1-оксобутан-2-ил)-1-((S)-1-((2S,3R)-2-амино-3-гидроксибутаноил)пирролидин-2-карбонил)-2-(3-фторбензил)пирролидин-2-карбоксамида (CM-9В):
К перемешанному раствору соединения 11-F2 (0,2 г, 0,32 ммоль) в CH2Cl2 (5 мл) добавили 4 н. HCl в 1,4-диоксане (2 мл) при 0°С и перемешивали при комнатной температуре в течение 2 часов. Реакционную смесь концентрировали под вакуумом. Полученный неочищенный материал промыли EtOAc (20 мл), затем н-пентаном (20 мл) и высушили под вакуумом с получением СМ-9В (0,1 г, 59%) в виде гидрохлоридной соли.
1H-ЯМР: (400 МГц, D2O): δ 7.89 (d, 2Н), 7.57 (d, 2Н), 4.95-4.90 (m, Ш), 4.43-4.35 (m, 4H), 3.94-3.79 (m, 5H), 3.35 (q, 2H), 2.56-2.50 (m, 1H), 2.35-2.29 (m, 2H), 2.14-2.05 (m, 3H), 1.81-1.75 (m, 1H), 1.35 (d, 3H), 1.32 (d, 3H), 1.19-1.11 (m, 1H).
ЖХМС (ESI) m/z: 522 [M++1], 523 [M++2].
Чистота по СЭЖХ: 98%.
Чистота по хиральной ВЭЖХ: 98%.
Оптическое вращение [α20D]: -101,46 (С=1% в воде).
Пример 10 - Синтез N-((2S,3R)-1-амино-3-гидрокси-1-оксобутан-2-ил)-1-((S)-1-((S)-2-амино-3-гидроксипропаноил)пирролидин-2-карбонил)-2-бензилпирролидин-2-карбоксамида (CM-10):
[00256] Для синтеза N-((2S,3R)-1-амино-3-гидрокси-1-оксобутан-2-ил)-1-((S)-1-((S)-2-амино-3-гидроксипропаноил)пирролидин-2-карбонил)-2-бензилпирролидин-2-карбоксамида (CM-10) использовали следующую последовательность реакций (Схема J):
Схема J. Синтез CM-10.

[00257] Синтез (S)-этил пирролидин-2-карбоксилата гидрохлорида (1):
К перемешанному раствору L-пролина (исходный материал) (110 г, 956,5 ммоль) в этаноле добавили тионилхлорид (141 мл, 1911,3 ммоль) и нагревали с дефлегматором в течение 16 часов. Реакционную смесь довели до комнатной температуры и концентрировали под вакуумом с получением соединения 1 в виде гидрохлоридной соли (170 г, 99%).
[00258] Синтез (S)-1-бензил 2-метил-пирролидин-1,2-дикарбоксилата (2):
К перемешанному раствору соединения 1 (170 г, 947 ммоль) в CH2Cl2 добавили ТЭА (398 мл, 2,83 моль) и через 30 минут добавили Cbz-Cl (1,136 моль). Реакционную смесь перемешивали при комнатной температуре в течение 12 часов. Реакционную смесь промыли водой и экстрагировали CH2Cl2. Органический слой высушили над безводным Na2SO4 и концентрировали под вакуумом. Полученный неочищенный материал очистили колоночной хроматографией с получением соединения 2 (230 г, 88%).
1H-ЯМР: (200 МГц, ДМСО-d6): δ 7.42-7.31 (m, 5Н), 5.09 (m, 2Н), 4.32-4.23(m, 1Н), 4.08-3.98(m, 2Н), 3.50-3.38 (m, 2Н), 2.30-2.18 (m, 1H), 1.90-1.81(m, 3Н), 1.10-1.04 (t, 3Н).
Масса m/z: 278 [М++1].
[00259] Синтез 1-бензил 2-этил-2-бензилпирролидин-1,2-дикарбоксилата (3):
К раствору (S)-1-бензил 2-этил-пирролидин-1,2-дикарбоксилата 2 (87 г, 314 ммоль) в ТТФ (800 мл) под инертной атмосферой добавили LiHMDS (1Mb ТТФ) (351 мл, 351 ммоль) при -25°С и перемешивали в течение 2 часов. К реакционной смеси по каплям добавили бензилбромид (45 мл, 376 ммоль) при -25°С. Смесь оставили нагреваться до комнатной температуры и перемешивали в течение 2 часов. Реакционную смесь охладили до 5°С, погасили насыщенным раствором NH4Cl, а водный слой экстрагировали EtOAc (2×200 мл). Объединенные органические экстракты высушили над безводным Na2SO4 и концентрировали под пониженным давлением. Полученный неочищенный остаток очистили силикагелевой колоночной хроматографией, элюируя 5% EtOAc в гексане, с получением соединения 3 (80 г, 69%) в виде жидкого вещества.
1Н-ЯМР: (200 МГц, ДМСО-d6): δ 7.47-7.32 (m, 5Н), 7.27-7.16 (m, 3Н), 7.07-7.04 (m, 2Н), 5.29-5.06 (m, 2Н), 4.16-3.89 (m, 2Н), 3.57-3.33 (m, 2Н), 3.02-2.78 (m, 2Н), 2.13-1.89 (m, 2Н), 1.56-1.51 (m, 1Н), 1.21-1.04 (m, 3Н), 0.93-0.79 (m, 1H).
Масса m/z: 368,2 [М++1].
[00260] Синтез 1-бензил 2-этил-2-бензилпирролидин-1,2-дикарбоксилата (4):
К перемешанному раствору соединения 3 (125 г, 340 ммоль) в СН3ОН (700 мл) добавили 2 н. водный раствор NaOH (680 мл) и нагревали до 100°С в течение 16 часов. Летучие вещества выпарили под пониженным давлением. Полученный остаток разбавили ледяной водой (50 мл) и промыли эфиром (50 мл). Водный слой подкислили до рН~2 с помощью 2 н. раствора HCl и экстрагировали EtOAc (2×100 мл). Объединенные органические слои высушили над безводным Na2SO4 и концентрировали под пониженным давлением с получением соединения 4 (90 г, 78%) в виде грязновато-белого твердого вещества.
1Η-ΗΜΡ: (200 МГц, ДМСО-d6): δ 12.71 (br s, 1Н), 7.40-7.30 (m, 5H), 7.25-7.19 (m, 3H), 7.07-7.00 (m, 2H), 5.27-5.02 (m, 2H), 3.59-3.32 (m, 2H), 3.02-2.83 (m, 2H), 2.13-1.91 (m, 2H), 1.58-1.49 (m, 1H), 0.90-0.77 (m, 1H).
Масса m/z: 340,1 [М++1].
[00261] Синтез бензил 2-бензил-2-((2S,3R)-3-гидрокси-1-метокси-1-оксобутан-2-илкарбамоил)пирролидин-1-карбоксилата (5):
К перемешанной суспензии соединения 4 (50 г, 147,9 ммоль), метилового эфира L-треонина (25 г, 147,9 ммоль) в ДХМ (500 мл) добавили HATU (56,2 г, 147,9 ммоль) и DIPEA (64 мл, 36,98 ммоль) при 5°С. Реакционную смесь перемешивали при комнатной температуре в течение 3 часов. Смесь разбавили EtOAc (150 мл) и промыли водой (2×30 мл). Органический слой промыли насыщенным солевым раствором, высушили над Na2SO4, концентрировали и очистили силикагелевой колоночной хроматографией, используя 50% EtOAc в гексане в качестве элюента, с получением соединения 5 (49,6 г, 74%).
1H-ЯМР: (200 МГц, ДМСО-d6): δ 7.62-7.59 (m, 1Н), 7.44-7.31 (m, 5H), 7.21-7.18 (m, 3Н), 7.06-6.99 (m, 2H), 5.25-5.24 (m, 1H), 5.12-4.94 (m, 2H), 4.30 (s, 1H), 4.15-4.08 (m, 1H), 3.66-3.64 (m, 3H), 3.63-3.49 (m, 2H), 3.14 (s, 1H), 2.89 (s, 1H), 2.09-2.02 (m, 2H), 1.56-1.51 (m, 1H), 1.09-0.98 (m, 4H).
Масса m/z: 455,1 [M++1], 477,3 [M+Na].
[00262] Синтез бензил 2-((2S,3R)-3-ацетокси-1-метокси-1-оксобутан-2-илкарбамоил)-2-бензилпирролидин-1-карбоксилата (6):
К перемешанному раствору соединения 5 (49 г, 107,9 ммоль) в ТТФ (30 мл) добавили Et3N (22,7 мл, 161,8 ммоль) и Ac2O (13,2 г, 129,5 ммоль) при комнатной температуре. Реакционную смесь перемешивали при комнатной температуре в течение 2 часов. Летучие вещества выпарили под пониженным давлением, а полученный остаток разбавили CH2Cl2 и промыли разбавленным раствором HCl. Объединенные органические экстракты высушили над Na2SO4 и концентрировали под пониженным давлением. Неочищенный остаток очистили колоночной хроматографией, используя 30% EtOAc в гексане в качестве элюента, с получением соединения 6 (42 г, 80%).
1H-ЯМР: (500 МГц, ДМСО-d6) (ротамеры): δ 8.15-7.71 (m, 1Н), 7.42-7.04 (m, 10Н), 5.30-5.19 (m, 2H), 5.11-5.09 (m, 1H), 4.99-4.93 (m, 1H), 4.67-4.62 (m, 1H), 3.66-3.64 (m, 3H), 3.55-3.46 (m, 2H), 3.38-3.35 (m, 1H), 2.88-2.69 (m, 1H), 2.17-2.00 (m, 2H), 1.98-1.92 (m, 3H), 1.56-1.46 (m, 1H), 1.23-1.17 (m, 3H), 1.02-0.86 (m, 1H).
ЖХМС m/z: 497,4 [M++1].
[00263] Синтез (2S,3R)-метил-3-ацетокси-2-(2-бензилпирролидин-2-карбоксамидо)-бутаноата (7):
К перемешанному раствору соединения 6 (50 г, 100,9 ммоль) в метаноле (1,5 л) добавили 10% Pd/C под атмосферой N2. Реакционную смесь перемешивали под атмосферой Н2 (баллонное давление) в течение 4 часов. После расходования исходного материала (по ТСХ), реакционную смесь отфильтровали через слой целита, а фильтрат концентрировали под пониженным давлением с получением соединения 7 (28 г, 77%).
1H-ЯМР: (500 МГц, ДМСО-d6) (ротамеры): δ 8.22-8.17 (m, 1H), 7.24-7.16 (m, 5Н), 5.17 (t, 1Н), 4.48-4.42 (m, 1H), 3.60-3.54 (s, 3H), 3.20 (t, 1H), 3.06-2.97 (m, 1H), 2.82-2.68 (m, 3H), 2.08-2.02 (m, 1H), 1.89 (s, 3H), 1.72-1.51 (m, 3H), 1.10 (2d, 3H).
ЖХМС m/z: 363 [M++1], 385 [M+Na].
[00264] Синтез (S)-бензил 2-(2-((2S,3R)-3-ацетокси-1-метокси-1-оксобутан-2-илкарбамоил)-2-бензилпирролидин-1-карбонил)-пирролидин-1-карбоксилата (8):
К перемешанному раствору соединения 7 (25 г, 69,1 ммоль) и Na2CO3 (18,3 г, 172,6 ммоль) в CH2Cl2:H2O (200 мл, 1:1) добавили раствор (S)-бензил 2-(хлоркарбонил)пирролидин-1-карбоксилата в CH2Cl2 и перемешивали реакционную смесь при комнатной температуре в течение 2 часов. Летучие вещества выпарили под пониженным давлением. Остаток разбавили CH2Cl2 (100 мл), отфильтровали, а фильтрат концентрировали под вакуумом. Неочищенный остаток очистили колоночной хроматографией, используя 60% EtOAc в гексане в качестве элюента, с получением соединения 8 (30 г, 73%). (S)-бензил 2-(хлоркарбонил)пирролидин-1-карбоксилат получили следующим образом. К раствору (S)-1-(бензилоксикарбонил)пирролидин-2-карбоновой кислоты (20,6 г, 82,8 ммоль) в CH2Cl2 (20 мл) по каплям добавили SOCl2 (20,5 г, 172,6 ммоль) при 0°С и нагревали полученный раствор с дефлегматором в течение 2 часов. Летучие вещества удалили под пониженным давлением с получением (S)-бензил 2-(хлоркарбонил)пирролидин-1-карбоксилата.
1H-ЯМР: (500 МГц, ДМСО-d6) (ротамеры): δ 7.36-7.23 (m, 8Н), 7.15-7.12 (m, 3Н), 5.21-5.15 (m, 2Н), 5.04-4.92 (m, 1Н), 4.57-4.50 (m, 2H), 3.88 (d, 1H), 3.65 (s, 3H), 3.54-3.46 (m, 3H), 3.21-3.13 (m, 1H), 3.02-2.90 (m, 2H), 2.19-2.02 (m, 4H), 1.97 (s, 3H), 1.89 (s, 1H), 1.77-1.65 (m, 1H), 1.17 (s, 2H), 1.06 (s, 2H).
Масса m/z: 594,1 [М++1].
[00265] Синтез (2S,3R)-метил 3-ацетокси-2-(2-бензил-1-((S)-пирролидин-2-карбонил)пирролидин-2-карбоксамидо)бутаноата (9):
К перемешанному раствору соединения 8 (30 г, 50,05 ммоль) в МеОН (300 мл) добавили 10% Pd/C под инертной атмосферой и перемешивали в течение 12 часов под атмосферой Н2 (баллонное давление). Реакционную смесь отфильтровали через слой целита и концентрировали под пониженным давлением. Полученный остаток растерли с эфиром (10 мл) с получением соединения 9 (21 г, 90%) в виде твердого вещества.
1Н-ЯМР: (500 МГц, CDCl3) (ротамеры): δ 7.88-7.87 (m, 1Н), 7.30-7.26 (m, 2H), 7.24-7.21 (m, 1H), 7.13-7.12 (d, 2H), 5.44-5.43 (m, 1H), 4.76-4.74 (m, 1H), 3.94-3.92 (m, 1H), 3.84-3.81 (m, 1H), 3.75 (s, 3H), 3.50 (m, lH), 3.26-3.12 (m, 3H), 2.90-2.88 (m, 1H), 2.23-2.15 (m, 4H), 2.04 (s, 3H), 1.87-1.77 (m, 5H), 1.27-1.24 (m, 3H).
Масса m/z: 460 (M+1).
[00266] Синтез 2-((трет-бутоксикарбонил)амино)бутановой кислоты (В):
К перемешанному раствору 2-аминобутановой кислоты А (50 г, 0,47 моль) в ТТФ: H2O (1 л, 1: 1) добавили NaHCO3 (80 г, 0,95 моль) и охладили реакционную смесь до 0°С. К этой смеси по каплям добавили Вос-ангидрид (124 г, 0,57 моль) и оставили при комнатной температуре на 16 часов. После расходования исходного материала (по ТСХ), реакционную смесь промыли эфиром, а водный слой нейтрализовали водным раствором HCl (рН~4-5) и экстрагировали EtOAc (2×250 мл). Объединенные органические экстракты высушили над безводным Na2SO4, отфильтровали и концентрировали под вакуумом с получением соединения В (40 г, 41%) в виде бесцветного густого сиропообразного вещества.
[00267] Синтез (2S,3R)-метил 3-ацетокси-2-(2-бензил-1-((S)-1-((S)-2-((трет-бутоксикарбонил)амино)-3-гидроксипропаноил)пирролидин-2-карбонил)пирролидин-2-карбоксамидо)бутаноата (10):
К перемешанному раствору соединения 9 (0,6 г, 1,30 ммоль) и соединения В (267 мг, 1,30 ммоль) в CH2Cl2 (10 мл) добавили EDCI (248 мг, 1,30 ммоль), HOBt (351 мг, 2,60 ммоль), затем DLPEA (167 мг, 1,30 ммоль) под атмосферой аргона. Полученную реакционную смесь перемешивали в течение 3 часов и дополнительно перемешивали при комнатной температуре в течение 16 часов. После расходования исходного материала (по ТСХ), реакционную смесь разбавили водой и экстрагировали CH2Cl2 (2×150 мл). Отделенный органический слой промыли насыщенным солевым раствором, высушили над безводным Na2SO4 и концентрировали под пониженным давлением. Неочищенный материал очистили силикагелевой колоночной хроматографией, элюируя 50% EtOAc в гексане, с получением соединения 10 (0,5 г, 59%) в виде грязновато-белого твердого вещества.
1H-ЯМР: (400 МГц, ДМСО-d6) (ротамеры): δ 7.30-7.24 (m, 6Н), 7.12-7.10 (m, 1H), 5.29-5.20 (m, 1Н), 4.79-4.60 (m, 2H), 4.55 (t, 1H), 4.38-4.30 (m, 2H), 3.79-3.60 (m, 11H), 2.21-1.87 (m, 11H), 1.36 (s, 9H), 1.25-1.15 (m, 4H).
ЖХМС (ESI): 647 (M+1).
[00268] Синтез трет-бутил ((2S)-1-((2S)-2-(2-(((2S,3R)-1-амино-3-гидрокси-1-оксобутан-2-ил)карбамоил)-2-бензилпирролидин-1-карбонил)пирролидин-1-ил)-3-гидрокси-1-оксопропан-2-ил)карбамата (11):
Раствор соединения 10 (0,5 г, 0,77 ммоль) в метанольном растворе NH3 (5 мл) поместили в закрытую пробирку и перемешивали в течение 24 часов. После расходования исходного материала (по ТСХ), реакционную смесь концентрировали под пониженным давлением с получением неочищенного вещества. Полученный материал очистили силикагелевой колоночной хроматографией, элюируя 10% МеОН в CH2Cl2, с получением соединения 11 (275 мг, 60%) в виде грязновато-белого твердого вещества.
1Н-ЯМР: (400 МГц, ДМСО-d6) (ротамеры): δ 7.35-7.20 (m, 6Н), 7.15 (d, 1Н), 7.01-6.97 (m, 2H), 6.77 (d, 1H), 5.09 (d, 1H), 4.75 (t, 2H), 4.39-4.35 (m, 1H), 4.24-4.20 (m, 1H), 3.92 (dd, 1H), 3.74-3.49 (m, 6H), 3.21-3.11 (m, 2H), 2.23-1.91 (m, 6H), 1.80-1.56 (m, 2H), 1.41 (s, 9H), 1.09 (d, 3H), 0.89-0.80 (m, 1H).
ЖХМС (ESI): 590(M+1).
Масса (m/z): 590 (M+1).
[00269] Синтез N-((2S,2R)-1-амино-3-гидрокси-1-оксобутан-2-ил)-1-((S)-1-((S)-2-амино-3-гидроксипропаноил)пирролидин-2-карбонил)-2-бензилпирролидин-2-карбоксамида (CM-10):
К перемешанному раствору соединения 11 (175 мг, 0,29 ммоль) в диоксане (3 мл) добавили 4 н. раствор HCl в 1,4-диоксане (2 мл) под атмосферой N2. Реакционную смесь перемешивали при комнатной температуре в течение 2 часов. Реакционную смесь концентрировали под пониженным давлением, разбавили диэтиловым эфиром и дополнительно перемешивали еще 15 минут. Эфирный слой декантировали и высушили под вакуумом с получением СМ-10 (108 мг, 74%) в виде грязновато-белого твердого вещества.
1H-ЯМР: (400 МГц, ДМСО-d6) (ротамеры): δ 8.29 (br s, 3Н), 7.39-7.20 (m, 7Н), 7.07-7.01 (m, 1Н), 5.45-5.40 (m, 1H), 5.06 (d, 1H), 4.79-4.76 (m, 1H), 4.27-4.20 (m, 2H), 4.01 (d, 1H), 3.91-3.85 (m, 4H), 3.69-3.55 (m, 5H), 3.29-3.21 (m, 2H), 2.10-1.87 (m, 6H), 1.08 (s, 3H).
ЖХМС (ESI): 490(M+2).
Пример 11 - Синтез (R)-N-((2S,3R)-1-амино-3-гидрокси-1-оксобутан-2-ил)-1-((S)-1-((2S,3R)-2-амино-3-гидроксибутаноил)пирролидин-2-карбонил)-2-(4-метоксибензил)пирролидин-2-карбоксамида (CM-11А) и (S)-N-(2S,3R)-1-амино-3-гидрокси-1-оксобутан-2-ил)-1-((S)-1-((2S,3R)-2-амино-3-гидроксибутаноил)пирролидин-2-карбонил)-2-(4-метоксибензил)пирролидин-2-карбоксамида (CM-11В):
[00270] Для синтеза (R)-N-((2S,5R)-1-амино-3-гидрокси-1-оксобутан-2-ил)-1-((S)-1-((2S,5R)-2-амино-3-гидроксибутаноил)пирролидин-2-карбонил)-2-(4-метоксибензил)пирролидин-2-карбоксамида (CM-11А) и (S)-N-((2S,3R)-1-амино-3-гидрокси-1-оксобутан-2-ил)-1-((S)-1-((2S,3R)-2-амино-3-гидроксибутаноил)пирролидин-2-карбонил)-2-(4-метоксибензил)пирролидин-2-карбоксамида (CM-11В) использовали следующую последовательность реакций (Схема К): Схема К.
Синтез CM-11А и CM-11В.

[00271] Синтез (S)-этил пирролидин-2-карбоксилата гидрохлорида (1):
К раствору L-пролина (исходный материал) (110 г, 956,5 ммоль) в этаноле добавили тионилхлорид (141 мл, 1911,3 ммоль) и нагревали с дефлегматором в течение 16 часов. Реакционную смесь довели до комнатной температуры и концентрировали под пониженным давлением с получением соединения 1 в виде гидрохлоридной соли (170 г, 99%). Этот материал напрямую использовали на следующей стадии без дополнительной очистки.
[00272] Синтез (S)-1-бензил 2-этил-пирролидин-1,2-дикарбоксилата (2):
К раствору соединения 1 (170 г, 947 ммоль) в ДХМ добавили ТЭА (398 мл, 2,83 ммоль), и через 30 минут добавили Cbz-Cl (1,136 моль). Реакционную смесь перемешивали при комнатной температуре в течение 12 часов. Реакционную смесь промыли водой и экстрагировали ДХМ. Органический слой высушили над безводным Na2SO4 и концентрировали под пониженным давлением. Неочищенное вещество очистили колоночной хроматографией с получением соединения 2 (230 г, 88%).
1H-ЯМР: (200 МГц, ДМСО-d6): δ 7.42-7.31 (m, 5Н), 5.09 (m, 2Н), 4.32-4.23 (m, 1Н), 4.08-3.98 (m, 2H), 3.50-3.38 (m, 2H), 2.30-2.18 (m, 1H), 1.90-1.81(m, 3H), 1.10-1.04 (t, 3H).
Масса m/z: 278 [M++1].
[00273] Синтез 1-бензил 2-этил-2-(4-метоксибензил)пирролидин-1,2-дикарбоксилата (3):
К раствору соединения 2 (10 г, 0,038 моль) в ТТФ (100 мл) под инертной атмосферой добавили LiHMDS (1Mb ТТФ) (76 мл, 0,076 моль) при -25°С и перемешивали в течение 2 часов. По каплям добавили 4-метоксибензилбромид (11,5 г, 0,057 моль) при -25°С, оставили нагреваться до комнатной температуры и перемешивали в течение 2 часов. Реакционную смесь охладили до 5°С, погасили насыщенным раствором NH4Cl, а водный слой экстрагировали EtOAc (2×200 мл). Объединенные органические экстракты высушили над безводным Na2SO4 и концентрировали под пониженным давлением. Полученный неочищенный остаток очистили силикагелевой колоночной хроматографией, элюируя 10% EtOAc в гексане, с получением соединения 3(11 г, 75%).
1H-ЯМР: (400 МГц, CDCl3): δ 7.44-7.29 (m, 6Н), 7.01 (t, 2Н), 6.81-6.79 (m, 2Н), 5.32 (t, 1Н), 5.15 (d, 1H), 3.82 (d, 5H), 3.55 (d, 1H), 3.50 (s, 2H), 3.02 (d, 2H), 2.14-1.99 (m, 2H), 1.65-1.62 (m, 1H), 1.09-0.95 (m, 1H).
ЖХМС (m/z): 384,3 [M++1].
[00274] Синтез 1-((бензилокси)карбонил)-2-(4-метоксибензил)пирролидин-2-карбоновой кислоты (4):
К перемешанному раствору соединения 3 (1 г, 0,026 моль) в СН3ОН (50 мл) добавили 2 н. водный раствор NaOH (25 мл) и нагревали до 100°С в течение 16 часов. Летучие вещества выпарили под пониженным давлением. Полученный остаток разбавили ледяной водой (50 мл) и промыли эфиром (50 мл). Водный слой подкислили до рН~2 с помощью 2 н. HCl и экстрагировали EtOAc (2×100 мл). Объединенные органические слои высушили над безводным Na2SO4 и концентрировали под пониженным давлением с получением соединения 4 (10 г, 94%).
1H-ЯМР: (400 МГц, ДМСО-d6): δ 12.55 (br s, 1Н), 7.49-7.65 (m, 5H), 6.99 (t, 2H), 6.78-6.75 (m, 2H), 5.25 (d, 1H), 5.11 (t, 1H), 3.72 (s, 3H), 3.51-3.42 (m, 3H), 2.99-2.95 (m, 2H), 2.15-2.09 (m, 2H), 1.62-1.57 (m, 1H), 0.98-0.94 (m, 1H).
ЖХМС (m/z): 368,3 [M+-1]
ВЭЖХ (чистота): 95%.
[00275] Синтез бензил 2-(((2R,3S)-3-гидрокси-1-метокси-1-оксобутан-2-ил)карбамоил)-2-(4-метоксибензил)пирролидин-1-карбоксилата (5):
К суспензии соединения 4 (10 г, 0,027 моль), метилового эфира L-треонина (4,5 г, 0,027 моль) в ДХМ (100 мл) добавили HATU (10,2 г, 0,027 моль) и DIPEA (11,8 мл, 0,067 моль) при 5°С. Реакционную смесь перемешивали при комнатной температуре в течение 2 часов. Реакционную смесь разбавили ДХМ (150 мл), промыли водой (2×30 мл), насыщенным солевым раствором и высушили над Na2SO4, концентрировали и очистили силикагелевой колоночной хроматографией, используя 50% EtOAc в гексане в качестве элюента, с получением соединения 5 (7,8 г, 59%).
1H-ЯМР: (400 МГц, ДМСО-d6): δ 7.47-7.42 (m, 6Н), 6.99-6.95 (m, 2Н), 6.84-6.77 (m, 2Н), 5.35-5.24 (m, 1Н), 5.12-4.95 (m, 2H), 4.35-4.32 (m, 1H), 4.19-4.15 (m, 1H), 3.74 (s, 3H), 3.65 (d, 2H), 3.59-3.50 (m, 2H), 3.14-3.05 (m, 1H), 3.92-3.85 (m, 1H), 2.71 (s, 5H), 2.19-2.05 (m, 2H), 1.68-1.60 (m, 1H), 1.14-1.05 (m, 3H).
ЖХМС (m/z): 4858,3 [M++1].
[00276] Синтез бензил 2-(((2S,3R)-3-ацетокси-1-метокси-1-оксобутан-2-ил)-карбамоил)-2-(4-метоксибензил)пирролидин-1-карбоксилата (6):
К перемешанному раствору соединения 5 (7,8 г, 0,016 моль) в ДХМ (100 мл) добавили Et3N (3,36 мл, 0,24 моль) и Ac2O (1,9 г, 0,019 моль) при комнатной температуре и перемешивали в течение 2 часов. Летучие вещества выпарили под пониженным давлением, а полученный остаток разбавили CH2Cl2 и промыли разбавленной HCl. Объединенные органические экстракты высушили над Na2SO4 и концентрировали под пониженным давлением. Неочищенный остаток очистили колоночной хроматографией, используя 30% EtOAc в н-гексане в качестве элюента, с получением соединения 6 (7,6 г, 90%).
1Н-ЯМР: (400 МГц, ДМСО-d6): δ 7.45-7.40 (m, 5Н), 6.99-6.95 (m, 2Н), 6.82-6.75 (m, 2Н), 5.31-5.25 (m, 2Н), 4.62-4.55 (m, 1Н), 3.74 (s, 3Н), 3.65 (t, 2H), 3.09-3.01 (m, 2H), 3.19-3.10 (m, 1H), 2.92-2.87 (m, 1H), 2.21-1.99 (m, 6H), 1.59-1.45 (m, 1H), 1.25-1.15 (m, 4H).
ЖХМС (m/z): 527,4 [M++1].
[00277] Синтез (2S,3R)-метил 3-ацетокси-2-(2-бензилпирролидин-2-карбоксамидо)бутаноата (7):
К перемешанному раствору соединения 6 (7,6 г, 0,014 моль) в метаноле (100 мл) добавили 10% Pd/C (1,5 г) и перемешивали реакционную смесь под атмосферой Н2 (баллонное давление) в течение 12 часов. Реакционную смесь отфильтровали через слой целита, а фильтрат концентрировали под пониженным давлением с получением соединения 7 (4,5 г, 80%).
1H-ЯМР: (400 МГц, ДМСО-d6): δ 8.25 (br s, 1Н), 6.81 (t, 2H), 5.2-5.17 (m, 1H), 4.52 (dd, 1H), 3.73 (d, 3H), 3.64 (d, 3H), 3.05 (br s, 1H), 2.86-2.80 (m, 2H), 2.12 (br s, 1H), 1.95 (d, 3H), 1.71-1.62 (br m, 3H), 1.12 (d, 1H), 0.82 (d, 1H).
ЖХМС (m/z): 393,3 [M++1].
[00278] Синтез (2S)-бензил 2-(2-(((2S,3R)-2-ацетокси-1-метокси-1-окосбутан-2-ил)карбамоил)-2-(4-метоксибензил)пирролидин-1-карбонил)пирролидин-1-карбоксилата (8):
К перемешанному раствору соединения 7 (4,5 г, 11,4 ммоль) и Na2CO3 (3,0 г, 28,0 ммоль) в CH2Cl2 (54 мл) и H2O (36 мл) добавили раствор хлорангидрида кислоты (3,4 г, 13,0 ммоль) в CH2Cl2 и перемешивали реакционную смесь при комнатной температуре в течение 2 часов. Летучие вещества выпарили под пониженным давлением. Остаток разбавили CH2Cl2 (100 мл), отфильтровали, а фильтрат концентрировали под пониженным давлением. Неочищенный остаток очистили колоночной хроматографией, используя 30% EtOAc в гексане в качестве элюента, с получением соединения 8 (7,0 г, 96%).
1H-ЯМР: (400 МГц, ДМСО-d6): δ 7.45-7.36 (m, 4Н), 7.22-6.69 (m, 3Н), 5.27-5.05 (m, 2Н), 4.69-4.55 (m, 1H), 3.75-3.62 (m, 4H), 3.55-3.47 (m, 3Н), 3.25-3.01 (m, 1H), 2.31-1.85 (m, 6H), 1.75-1.45 (m, 1H), 1.22-1.09 (m, 3H).
[00279] Синтез (2S,3R)-метил 3-ацетокси-2-(2-(4-метоксибензил)-1-((S)-пирролидин-2-карбонил)пирролидин-2-карбоксамидо)бутаноата (9):
К раствору соединения 8 (7,0 г, 11,2 ммоль) в МеОН (100 мл) добавили 10% Pd/C (2,0 г) и перемешивали под атмосферой Н2 (баллонное давление) в течение 12 часов. Реакционную смесь отфильтровали через слой целита и концентрировали под пониженным давлением с получением соединения 9 (5,4 г, 99%).
1H-ЯМР: (400 МГц, ДМСО-d6): δ 9.25 (br m, 1Н), 8.01-7.85 (m, 1H), 7.11-6.99 (m, 2H), 6.85 (d, 2H), 5.29-5.22 (m, 1H), 4.65-4.60 (m, 1H), 4.44-4.22 (m, 1H), 3.72 (s, 3H), 3.55 (s, 3H), 3.49 (d, 3H), 3.39-3.30 (m, 5H), 3.15-3.09 (m, 3H), 2.25-2.20 (m, 1H), 2.02 (s, 9H), 1.65-1.60 (m, 2H), 1.21 (t, 3H).
ЖХМС (m/z): 490,4 [M++1].
[00280] Синтез (2S,3R)-метил 3-ацетокси-2-(1-((S)-1-((2S,3R)-3-ацетокси-2-((трет-бутоксикарбонил)амино)бутаноил)пирролидин-2-карбонил)-2-(4-метоксибензил)пирролидин-2-карбоксамидо)бутаноата (10):
К раствору соединения 9 (5,4 г, 81,1 ммоль) в CH2Cl2 (100 мл) под инертной атмосферой добавили EDCI.HCl (3,1 г, 16,0 ммоль), HOBt (2,2 г, 16,0 ммоль), затем DIPEA (4,2 г, 33,0 ммоль) и соединение С (3,4 г, 13,0 ммоль) при 0°С. Реакционную смесь перемешивали при комнатной температуре в течение 12 часов. Реакционную смесь экстрагировали EtOAc (2×75 мл), а отделенный органический слой промыли водой (200 мл), затем насыщенным солевым раствором (200 мл), высушили над Na2SO4 и концентрировали под пониженным давлением. Неочищенный материал очистили колоночной хроматографией с получением соединения 10 (5,8 г, 72%).
1Н-ЯМР: (400 МГц, ДМСО-d6): δ 7.22-7.01 (m, 2Н), 6.85-6.82 (m, 1Н), 5.25-5.05 (m, 1H), 4.65-4.20 (m, 2H), 3.85-3.42 (m, 7H), 3.22-3.12 (m, 1H), 2.22-1.85 (m, 8H), 1.52 (d, 6H), 1.29-1.10 (m, 5H).
[00281] Синтез трет-бутил ((2S,3R)-1-((2S)-2-(2-(((2S,3R)-1-амино-3-гидрокси-1-оксобутан-2-ил)карбамоил)-2-(4-метоксибензил)-пирролидин-1-карбонил)пирролидин-1-ил)-3-гидрокси-1-оксобутан-2-ил)карбамата (11):
Раствор соединения 10 (4 г, 5,45 ммоль) в метанольном растворе NH3 (40 мл) перемешивали при комнатной температуре в течение 36 часов. Реакционную смесь концентрировали под пониженным давлением. Полученный неочищенный материал очистили силикагелевой колоночной хроматографией, затем препаративной очисткой с получением соединения 11-Fr-1 (200 мг) и соединения 11-Fr-2 (150 мг).
[00282] Синтез (R)-N-((2S,3R)-1-амино-3-гидрокси-1-оксобутан-2-ил)-1-((S)-1-((2S,3R)-2-амино-3-гидроксибутаноил)пирролидин-2-карбонил)-2-(4-метоксибензил)пирролидин-2-карбоксамида (СН-11 А):
К раствору соединения 11-Fr-1 (0,2 г, 0,3 ммоль) в ДХМ (3 мл) добавили 4 н. раствор HCl в 1,4-диоксане (2 мл) и перемешивали при комнатной температуре в течение 2 часов. Реакционную смесь концентрировали под пониженным давлением с получением соединения СН-11А (0,12 г, 66%) в виде гидрохлоридной соли.
1H-ЯМР: (400 МГц, D2O): δ 7.32 (d, 2Н), 7.05 (d, 2Н), 4.99-4.95 (m, 2Н), 4.42 (d, 1Н), 4.37 (s, 3Н), 3.81 (s, 3Н), 3.74 (d, 1H), 3.72-3.65 (m, 1H), 3.62 (d, 1H), 3.35-3.30 (m, 1H), 3.29-3.25 (m, 1H), 3.22 (d, 1H), 2.51-2.50 (m, 1H), 2.35-2.30 (m, 1H), 2.19-2.05 (m, 4H), 1.75-1.72 (m, 1H), 1.55 (d, 3H), 1.25 (d, 3H), 1.21-1.15 (m, 1H).
ЖХМС (m/z): 534,5 [M++1].
Чистота по ВЭЖХ: 97%.
Оптическое вращение [α20D]: 34,33 (С=1% в воде).
[00283] Синтез (S)-N-((2S,3R)-1-амино-3-гидрокси-1-оксобутан-2-ил)-1-((S)-1-((2S,5R)-2-амино-3-гидроксибутаноил)пирролидин-2-карбонил)-2-(4-метоксибензил)пирролидин-2-карбоксамида (СН-11В):
К раствору соединения 11-Fr-2 (0,15 г, 0,2 ммоль) в ДХМ (3 мл) добавили 4 н. раствор HCl в 1,4-диоксане (2 мл) и перемешивали при комнатной температуре в течение 2 часов. Реакционную смесь концентрировали под пониженным давлением с получением соединения СМ-11В (90 мг, 66%) в виде гидрохлоридной соли.
1H-ЯМР: (400 МГц, D2O): δ 7.32 (d, 2Н), 7.05 (d, 2Н), 4.99-4.95 (m, 2Н), 4.42 (d, 1Н), 4.37 (s, 3Н), 3.81 (s, 3Н), 3.74 (d, 1H), 3.72-3.65 (m, 1H), 3.62 (d, 1H), 3.35-3.30 (m, 1H), 3.29-3.25 (m, 1H), 3.22 (d, 1H), 2.51-2.50 (m, 1H), 2.35-2.30 (m, 1H), 2.19-2.05 (m, 4H), 1.75-1.72 (m, 1H), 1.55 (d, 3H), 1.25 (d, 3H), 1.21-1.15 (m, 1H).
ЖХМС (m/z): 534,5 [M++1].
Чистота по ВЭЖХ: 92%.
Оптическое вращение [α20D]:-108,63 (С=1% в воде).
[00284] Синтез бензил 2-(трет-бутоксикарбониламино)-3-гидроксибутаноата (А):
К раствору 2-(трет-бутоксикарбониламино)-3-гидроксибутановой кислоты (Вос-Thr) (50 г, 228,3 ммоль) в ДМФ (500 мл) добавили K2CO3 (63 г, 456,6 ммоль) и перемешивали при комнатной температуре в течение 15 минут. Добавили бензилбромид (46,83 г, 273,9 ммоль) и перемешивали при комнатной температуре в течение 6 часов. Реакционную смесь разбавили водой (500 мл) и экстрагировали EtOAc (2×750 мл). Объединенные органические слои промыли насыщенным солевым раствором (50 мл), высушили над безводным Na2SO4H концентрировали под пониженным давлением. Неочищенный материал очистили силикагелевой колоночной хроматографией, используя 20% EtOAc в гексане в качестве элюента, с получением бензил 2-(трет-бутоксикарбониламино)-3-гидроксибутаноата А (52 г, 73%).
1H-ЯМР: (500 МГц, ДМСО-d6): δ 7.37-7.30 (m, 5Н), 6.60 (d, J=8.5 Гц, 1Н), 5.18-5.08 (m, 2H), 4.76 (d, J=7 Гц, 1H), 4.08-4.00 (m, 2H), 1.38 (s, 9H), 1.09 (d, J=6.0 Гц, 3Н).
Масса m/z: 310,0 [M++1], 210 [M+-De Boc].
[00285] Синтез бензил 3-ацетокси-2-(трет-бутоксикарбониламино)бутаноата (В):
К перемешанному раствору бензил 2-(трет-бутоксикарбониламино)-3-гидроксибутаноата А (52 г, 168,2 ммоль) в ДХМ (500 мл) добавили Ac2O (20,5 г, 201,9 ммоль), Et3N (25,4 г, 252,4 ммоль) и DMAP (3,5 г) и перемешивали при комнатной температуре в течение 2 часов. Летучие вещества удалили под пониженным давлением. Полученный остаток разбавили EtOAc (750 мл) и промыли холодным 0,5 н. раствором HCl (2×200 мл). Органический слой промыли насыщенным солевым раствором, высушили над безводным Na2SO4 и концентрировали под пониженным давлением с получением 3-ацетокси-2-(трет-бутоксикарбониламино)бутаноата В (52 г, 88%).
1H-ЯМР: (500 МГц, ДМСО-d6): δ 7.35-7.34 (m, 5Н), 7.27-7.25 (d, J=8.5 Гц, 1H), 5.18-5.06 (m, 3Н), 4.34-4.32 (m, 1H), 1.90 (s, 3H), 1.39 (s, 9H), 1.16 (d, J=3 Гц, 3Н).
Масса m/z: 252 [M++1-De Boc].
[00286] Синтез (2S,3R)-3-ацетокси-2-(трет-бутоксикарбониламино)бутановой кислоты (С):
Бензил-3-ацетокси-2-(трет-бутоксикарбониламино)бутаноат (52 г, 148,1 ммоль) растворили в МеОН (1 л), добавили 10% Pd/C и перемешивали реакционную Смесь под атмосферой водорода в течение 16 часов. Реакционную смесь отфильтровали через целит, растворитель выпарили под пониженным давлением, а неочищенный остаток растерли с гексаном с получением (2S,3R)-3-ацетокси-2-(отрет-бутоксикарбониламино)бутановой кислоты С (35 г, 90%).
1H-ЯМР: (500 МГц, ДМСО-d6): δ 12.78 (br s, 1Н), 6.94 (d, J=9.5 Гц, 1H), 5.16-5.14 (m, 1H), 4.17-4.15 (m, 1H), 1.95 (s, 3H), 1.39 (s, 9H), 1.10 (d, J=6.0 Гц, 3Н).
Масса m/z: 260,0 [М-1].
Пример 12 - Синтез (R)-N-((2S,3R)-1-амино-3-гидрокси-1-оксобутан-2-ил)-1-(S)-1-((2S,3R)-2-амино-3-гидроксибуганоил)пирролидин-2-карбонил)-2-(3,5-дибромбензил)пирролидин-2-карбоксамида (CM-12А) и (S)-N-((2S,3R)-1-амино-3-гидрокси-1-оксобутан-2-ил)-1-((S)-1-((2S,3R)-2-амино-3-гидроксибутаноил)пирролидин-2-карбонил)-2-(3,5-дибромбензил)пирролидин-2-карбоксамида (CM-12В):
[00287] Для синтеза (R)-N-((2S,3R)-1-амино-3-гидрокси-1-оксобутан-2-ил)-1-((S)-1-((2S,3R)-2-амино-3-гидроксибутаноил)пирролидин-2-карбонил)-2-(3,5-дибромбензил)пирролидин-2-карбоксамида (CM-12А) и (S)-N-((2S,3R)-1-амино-3-гидрокси-1-оксобутан-2-ил)-1-((S)-1-((2S,3R)-2-амино-3-гидроксибутаноил)пирролидин-2-карбонил)-2-(3,5-дибромбензил)пирролидин-2-карбоксамида (CM-12В) использовали следующую последовательность реакций (Схема L): Схема L.
Синтез CM-12А и CM-12В:

[00288] Синтез (S)-этил пирролидин-2-карбоксилата (1):
К перемешанному раствору пирролидин-2-карбоновой кислоты (исходный материал) (100 г, 0,87 моль) в метаноле (800 мл) медленно добавили тионилхлорид (76,9 мл, 1,04 моль) при 0°С. Реакционную смесь нагревали с дефлегматором в течение 12 часов. После расходования исходного материала (по ТСХ), реакционную смесь концентрировали под пониженным давлением с получением соединения 1 (143,9 г, HCl соль).
1Η-ΗΜΡ: (400 МГц, CDCl3) (ротамеры): δ 3.89 (s, 3Н), 3.68-3.62 (m, 2Н), 3.59-3.47 (m, 2Н), 2.49-2.37 (m, 1Н), 2.27-2.05 (m, 3Н).
ЖХМС (m/z): 166 [М++1].
[00289] Синтез 1-трет-бутил 2-метил-пирролидин-1,2-дикарбоксилата (2):
К перемешанному раствору соединения 1 (35 г, 0,22 моль) в CH2Cl2 (175 мл) добавили Et3N (90 мл, 0,65 моль), затем Вос-ангидрид (56,9 мл, 0,26 моль) при 0°С. Реакционную смесь перемешивали при комнатной температуре в течение 16 часов. После расходования исходного материала (по ТСХ), реакционную смесь разбавили водой (100 мл) и экстрагировали CH2Cl2 (2×100 мл). Органический слой промыли водой, насыщенным солевым раствором, высушили над Na2SO4 и концентрировали. Полученный неочищенный материал очистили силикагелевой колоночной хроматографией, элюируя 30% EtOAc в н-гексане, с получением соединения 2 (41 г, 95%).
1Н-ЯМР: (400 МГц, CDCl3) (ротамеры): δ 4.25-4.21 (m, 1Н), 3.75 (s, 3Н), 3.57-3.26 (m, 2H), 2.29-2.10 (m, 1H), 1.99-1.75 (m, 3H), 1.45 (s, 9H).
ЖХМС (m/z): 130 [(M++1)-Boc].
[00290] Синтез 1-трет-бутил 2-метил-2-(3,5-дибромбензил)пирролидин-1,2-дикарбоксилата (3):
К перемешанному раствору соединения 2 (10 г, 0,043 моль) в NUA (50 vk) добавили LiHMDS (65,5 мл, 0,065 моль) при -25°С и перемешивали в течение 2 часов. К этой смеси по каплям добавили 3,5-дибромбензилбромид (17,1 г, 0,052 моль) при -25°С и перемешивали в течение 2 часов при комнатной температуре. После расходования исходного материала (по ТСХ), реакцию погасили с помощью NH4Cl при 0°С. Отделенный органический слой промыли водой и экстрагировали EtOAc. Отделенный органический слой высушили над Na2SO4 и концентрировали под пониженным давлением с получением неочищенного продукта, который очистили силикагелевой колоночной хроматографией с получением соединения 3 (10 г, 50%).
1H-ЯМР: (500 МГц, ДМСО-d6): δ 7.72 (s, 1H), 7.41 (d, 2H), 3.69 (d, 3Н), 3.37-3.35 (m, 2H), 3.07-3.01 (m, 1H), 2.89-2.81 (m, 1H), 2.08-2.01 (m, 2H), 1.68-1.62 (m, 1H), 1.45 (s, 9H), 1.19-1.12 (m, 1H).
ЖХМС (m/z): 419,1 [(M++1)-Boc].
[00291] Синтез метил 2-(3,5-дибромбензил)пирролидин-2-карбоксилата (4):
К перемешанному раствору соединения 3 (10 г, 0,016 моль) в метаноле (20 мл) добавили МеОН. HCl (80 мл) под атмосферой N2 и перемешивали в течение 8 часов при комнатной температуре. Летучие вещества выпарили под пониженным давлением с получением соединения 4 (8 г, 93%) в виде твердого вещества белого цвета.
1H-ЯМР: (400 МГц, ДМСО-d6): δ 10.95 (br s, 1Н), 9.45 (br s, 1H), 7.81 (s, 1H), 7.65 (s, 2H), 3.75 (s, 3H), 3.55 (d, 1H), 3.39 (d, 1H), 3.22 (br s, 1H), 2.43 (t, 1H), 2.15-2.10 (m, 2H), 1.85-1.81 (m, 1H).
ЖХМС (m/z): 378 [(М+-HCl].
[00292] Синтез (S)-1-((аллилокси)карбонил)пирролидин-2-карбоновой кислоты (A):
К перемешанному раствору (S)-пирролидин-2-карбоновой кислоты (исходный материал) (5 г, 0,043 моль) в 1,4-диоксане (50 мл) и воде (100 мл) добавили Na2CO3 (11,5 г, 0,11 моль), затем аллилхлорформиат (6,2 г, 0,052 моль) при 0°С и перемешивали в течение 12 часов. Реакционную смесь экстрагировали EtOAc (2×75 мл). Отделенный органический слой высушили над безводным Na2SO4, отфильтровали и концентрировали под пониженным давлением с получением неочищенного вещества, которое очистили силикагелевой колоночной хроматографией с получением соединения А (3,28 г, 37%).
[00293] Синтез (S)-аллил 2-((R)-2-(3,5-дибромбензил)-2-(метоксикарбонил)пирролидин-1-карбонил)пирролидин 1-карбоксилата (S):
К перемешанному раствору соединения 4 (8 г, 0,019 моль) и Na2CO3 (5,1 г, 0,048 моль) в ДХМ:H2O (150 мл, 3:2) добавили раствор хлорангидрида кислоты В и перемешивали в течение 2 часов при комнатной температуре. Водный слой экстрагировали CH2Cl2 (2×50 мл). Отделенный органический слой промыли водой (50 мл), высушили над безводным Na2SO4, отфильтровали и концентрировали под пониженным давлением. Полученный неочищенный материал очистили силикагелевой колоночной хроматографией с получением соединения 5 (8 г, 74%). Хлорангидрид кислоты В получили следующим образом. К раствору соединения А (4,6 г, 0,023 моль) в ДХМ по каплям добавили SOCl2 (5,7 г, 0,048 моль) при 0°С, и полученный раствор нагревали с дефлегматором в течение 2 часов; растворитель удалили из реакционной смеси под пониженным давлением.
1Н-ЯМР: (400 МГц, ДМСО-d6): δ 7.75-7.42 (m, 2Н), 7.37 (s, 1Н), 5.95 (br s, 1H), 9.45 (br s, 1H), 7.81 (s, 1H), 7.65 (s, 2H), 3.75 (s, 3H), 3.55 (d, 1H), 3.39 (d, 1H), 3.22 (br s, 1H), 2.43 (t, 1H), 2.15-2.10 (m, 2H), 1.85-1.81 (m, 1H).
ЖХМС (m/z): 559,3 [(M++1].
[00294] Синтез (R)-1-((S)-1-((аллилокси)карбонил)прролидин-2-карбонил)-2-(3,5-дибромбензил)пирролидин-2-карбоновой кислоты (6):
К перемешанному раствору соединения 5 (8 г, 0.014 моль) в МеОН (50 мл) добавили 2 н. раствор NaOH (10 мл) и нагревали с дефлегматором в течение 5 часов. Летучие вещества выпарили под пониженным давлением, а полученный остаток разбавили водой и промыли эфиром. Водный слой подкислили до рН~2 с помощью 2 н. раствора HCl и экстрагировали EtOAc. Отделенный органический слой высушили над безводным Na2SO4, отфильтровали и концентрировали под пониженным давлением с получением соединения 6 (7 г, 90%). Этот материал напрямую использовали на следующей стадии без дополнительной очистки.
ЖХМС (m/z): 543,3 [(М+-1].
[00295] Синтез (2S,3R)-метил 2-амино-3-гидроксибутаноата (С):
К перемешанному раствору (2S,3R)-2-амино-3-гидроксибутановой кислоты (200 г, 1,68 моль) в метаноле (1,2 л) по каплям добавили SOCl2 (244 мл, 3,36 моль) при 0°С и перемешивали в течение 1 часа. Полученную реакционную смесь нагревали с дефлегматором в течение 24 часов. После расходования исходного материала (по ТСХ), реакционную смесь нагрели до комнатной температуры и концентрировали под пониженным давлением, и промыли н-гексаном (2×50 мл). Остаток растворили в EtOH (1 л) и нейтрализовали с помощью Et3N (471 мл, 3,36 моль) и снова перемешивали в течение 2 часов. Выпавшее в осадок твердое вещество отфильтровали; полученный фильтрат концентрировали под пониженным давлением с получением соединения С (195 г, 80%).
1H-ЯМР: (400 МГц, ДМСО-d6): δ 8.51 (br s, 3Н), 4.13-4.10 (m, 1Н), 3.91 (br s, 1H), 1.20 (d, 3H).
ЖХМС (m/z): 134,1 [M++1].
[00296] Синтез (2S,3R)-2-амино-3-гидроксибутанамида (D):
Раствор соединения С (190 г, 1,35 моль) в LPA (2 л) поместили в автоклав и продували газообразный NH3 (7-8 кг) и перемешивали при 35°С в течение 24 часов. Затем удалили газообразный NH3, а реакционную смесь концентрировали под пониженным давлением и добавили CH2Cl2, и отфильтровали. Полученное твердое вещество нагревали с дефлегматором в EtOH в течение 1 часа при 78°С. Реакционную массу отфильтровали при нагревании, а к фильтрату добавили н-гексан и снова перемешивали еще 4 часа. Полученное осажденное твердое вещество отфильтровали и высушили под пониженным давлением с получением соединения D (160 г, 47%).
1H-ЯМР: (500 МГц, ДМСО-d6): δ 7.38 (br s, 1Н), 7.02 (br s, 1H), 4.66 (br s, 1H), 3.77-3.70 (m, 1H), 2.93 (d, 1H), 2.72 (br m, 1H), 1.05 (d, 3H).
ЖХМС (m/z): 119,1 [М++1].
СЭЖХ (чистота по ИДСР): 99,9%.
[00297] Синтез (S)-аллил 2-((R)-2-(((2S,3R)-1-амино-3-гидрокси-1-оксобутан-2-ил)карбамоил)-2-(3,5-дибромбензил)пирролидин-1-карбонил)пирролидин-1-карбоксилата(7):
К перемешанному раствору соединения 6 (5 г, 9,19 ммоль) в CH2Cl2 (100 мл) добавили HOBt (1,8 г, 13,7 ммоль), EDCI.HCl (2,6 г, 13,7 ммоль), затем DLPEA (3,5 г, 27,0 ммоль) и соединение D (1,3 г, 11,0 ммоль) при 0°С. Реакционную смесь перемешивали при комнатной температуре в течение 12 часов. После расходования исходного материала (по ТСХ), реакцию погасили водой и экстрагировали CH2Cl2 (2×100 мл). Органический слой высушили над безводным Na2SO4 и концентрировали под пониженным давлением. Неочищенное вещество очистили колоночной хроматографией с получением соединения 7 (5 г, 84%).
[00298] Синтез (R)-N-((2S,3R)-1-амино-3-гидрокси-1-оксобутан-2-ил)-2-(3,5-дибромбензил)-1-((S)-пирролидин-2-карбонил)Пирролидин-2-карбоксамида (8):
К перемешанному раствору соединения 7 (1 г, 1,35 ммоль) в ТГФ (50 мл) добавили DABCO (1.3 г, 12,4 ммоль), затем Pd(PPh3)4 (358 мг, 0,3 ммоль) при комнатной температуре и перемешивали в течение 30 минут. Реакционную смесь разбавили EtOAc (75 мл) и насыщенным раствором NaHCO3 (75 мл). Отделенный органический слой высушили над безводным Na2SO4 и концентрировали под пониженным давлением с получением соединения 8 (560 мг, 64%). Этот материал напрямую использовали на следующей стадии без дополнительной очистки.
[00299] Синтез трет-бутил ((2S,3R)-1-((S)-2-((R)-2-(((2S,3R)-1-амино-3-гидрокси-1-оксобутан-2-ил)карбамоил)-2-(3,5-дибромбензил)пирролидин-1-карбонил)пирролидин-1-ил-3-гидрокси-1-оксобутан-2-ил)карбамата (9):
К перемешанному раствору соединения 8 (1,8 г, 3,20 ммоль) в CH2Cl2 (100 мл) добавили EDCI.HCl (0,916 г, 4,80 ммоль), НОВ: (0,65 г, 4,80 ммоль), затем DLPEA (1,7 мл, 9,60 ммоль) и (2S,3R)-2-((трет-бутоксикарбонил)амино)-3-гидроксибутановую кислоту (0,84 г, 3,80 ммоль) при 0°С. Реакционную смесь перемешивали при комнатной температуре в течение 12 часов. Реакционную смесь погасили водой (100 мл) и экстрагировали CH2Cl2 (2×150 мл). Отделенные органические слои промыли насыщенным солевым раствором, высушили над безводным Na2SO4, отфильтровали и концентрировали под пониженным давлением с получением неочищенного продукта, который очистили силикагелевой колоночной хроматографией, затем препаративной ВЭЖХ очисткой с получением соединения 9-Fr-1 (140 мг) и соединения 9-Fr-2 (150 мг).
Данные хиральной ВЭЖХ для соединения 9-Fr-1:
1Н-ЯМР: (400 МГц, ДМСО-d6): δ 7.71 (d, 3Н), 7.21 (s, 1Н), 6.99-6.75 (m, 2H), 6.59 (d, 1H), 5.01 (d, 1H), 4.65-4.60 (m, 2H), 4.19 (t, 2H), 3.99 (d, 1H), 3.89-3.85 (m, 2H), 3.65-3.61 (m, 2H), 3.44-3.39 (m, 3H), 2.25-2.15 (m, 1H), 2.02-1.95 (m, 5H), 1.75-1.72 (m, 1H), 1.35 (s, 9H), 1.25 (d, 3H), 1.19 (d, 3H), 0.99-0.95 (m, 1H).
ВЭЖХ: 99,83%.
ЖХМС (m/z): 762,3 [М++1].
Данные хиральной ВЭЖХ для соединения 9-Fr-2:
1H-ЯМР: (400 МГц, ДМСО-d6): δ 7.71 (d, 3Н), 7.21 (s, 1H), 6.99-6.75 (m, 2H), 6.59 (d, 1H), 5.01 (d, 1H), 4.65-4.60 (m, 2H), 4.19 (t, 2H), 3.99 (d, 1H), 3.89-3.85 (m, 2H), 3.65-3.61 (m, 2H), 3.44-3.39 (m, 3H), 2.25-2.15 (m, 1H), 2.02-1.95 (m, 5H), 1.75-1.72 (m, 1H), 1.35 (s, 9H), 1.25 (d, 3H), 1.19 (d, 3H), 0.99-0.95 (m, 1H).
ВЭЖХ: 92%.
ЖХМС (m/z):762,2 [M++1].
[00300] Синтез (R)-N-((2S,3R)-1-амино-3-гидрокси-1-оксобутан-2-ил)-1-((S)-1-((2S,3R)-2-амино-3-гидроксибутаноил)пирролидин-2-карбонил)-2-(3,5-дибромбензил)пирролидин-2-карбоксамида (CM-12A):
К перемешанному раствору соединения 9-Fr-l (140 мг, 0,183 ммоль) в CH2Cl2 (3 мл) добавили диоксан. HCl (2 мл) при 0°С под атмосферой N2. Реакционную смесь перемешивали при комнатной температуре в течение 2 часов. Реакционную смесь концентрировали под пониженным давлением с получением CM-12А (120 мг, 93%).
1Н-ЯМР: (400 МГц, D2O): δ 7.84 (s, 1Н), 7.61 (s, 2H), 4.39 (d, 4H), 3.92-3.85 (m, 4H), 3.65-3.62 (m, 2H), 3.45 (d, 1H), 2.59-2.55 (m, 1H), 2.48-2.44 (m, 2H), 2.15-2.19 (m, 3H), 1.91-1.87 (m, 1Ή), 1.60 (s, 3H), 1.25 (s, 3H).
ЖХМС (m/z):662 (M+1).
ВЭЖХ: 97,55%.
Оптическое вращение [α20D]:+29,21 (С=0,5% в воде).
[00301] Синтез (S)-N-((2S,3R)-1-амино-3-гидрокси-1-оксобутан-2-ил)-1-((S)-1-((2S,3R)-2-амино-3-гидроксибутаноил)пирролидин-2-карбонил)-2-(3,5-дибромбензил)пирролидин-2-карбоксамида (CM-12В):
К перемешанному раствору соединения 9-Fr-2 (150 мг, 0,197 ммоль) в CH2Cl2 (3 мл) добавили диоксан. HCl (2 мл) при 0°С под атмосферой N2. Реакционную смесь перемешивали при комнатной температуре в течение 2 часов. Реакционную смесь концентрировали под пониженным давлением с получением CM-12 В (130 мг, 94%).
1Н-ЯМР: (400 МГц, D2O): δ 7.84 (s, 1H), 7.61 (s, 2H), 4.39 (d, 4H), 3.92-3.85 (m, 4H), 3.65-3.62 (m, 2H), 3.45 (d, 1H), 2.59-2.55 (m, 1H), 2.48-2.44 (m, 2H), 2.15-2.19 (m, 3H), 1.91-1.87 (m, 1H), 1.60 (s, 3H), 1.25 (s, 3H).
ЖХМС (m/z): 662 (M+1).
ВЭЖХ: 92,11%.
Оптическое вращение [α20D]: -78,70 (С=0,5% в воде).
Пример 13 - Синтез (R)-N-((2S,3R)-1-амино-3-гидрокси-1-оксобутан-2-ил)-1-((S)-1-((2S,3R)-2-амино-3-гидроксибутаноил)пирролидин-2-карбонил)-2-(3-метоксибензил)пирролидин-2-карбоксамида (СМ-13А) и N-((2S,3R)-1-амино-3-гидрокси-1-оксобутан-2-ил)-1-((S)-1-((2S,3R)-2-амино-3-гидросибутаноил)пирролидин-2-карбонил)-2-(3-метоксибензил)пирролидин-2-карбоксамида (CM-13В):
[00302] Для синтеза (R)-N-((2S,3R)-1-амино-3-гидрокси-1-оксобутан-2-ил)-1-((S)-1-((2S,3R)-2-амино-3-гидроксибутаноил)пирролидин-2-карбонил)-2-(3-метоксибензил)пирролидин-2-карбоксамида (CM-13А) и N-((2S,3R)-2-амино-3-гидрокси-1-оксобутан-2-ил)-1-((S)-1-((2S,3R)-2-амино-3-гидроксибутаноил)пирролидин-2-карбонил)-2-(3-метоксибензил)пирролидин-2-карбоксамида (CM-13В) использовали следующую последовательность реакций (Схема М):
Схема Μ. Синтез CM-13А и CM-13В:
[00303] Синтез (S)-1-((бензилокси)карбонил)пирролидин-2-карбоновой кислоты (А):
К перемешанному раствору (S)-пирролидин-2-карбоновой кислоты (250 г, 2,17 моль) в воде (1 л) добавили Na2CO3 (576 г, 5,43 моль) и перемешивали в течение 1 часа. После охлаждения до 0°С, к реакционной смеси по каплям добавили бензилхлорформиат (444 г, 2,61 моль) и снова перемешивали в течение 1 часа. Полученную смесь нагрели до комнатной температуры и дополнительно перемешивали в течение 24 часов. После расходования исходного материала (по ТСХ), реакционную смесь разбавили водой (1 л) и эфиром (1,5 л). Отделенный водный слой обработали РрСН3 (1,5 л) и подкислили с помощью 6 н. HCl. Водный слой экстрагировали EtOAc (3×1,5 л); объединенные органические экстракты промыли насыщенным солевым раствором, высушили над безводным Na2SO4, отфильтровали и концентрировали под пониженным давлением с получением соединения А (450 г, 84%) в виде светло-желтого сиропообразного вещества.
1H-ЯМР: (400 МГц, ДМСО-d6): δ 12.71 (br s, 1Н), 7.40-7.30 (m, 5H), 5.19-5.01 (m, 2H), 4.25 (dd, 1H), 3.51-3.50 (m, 2H), 2.29-2.15 (m, 1H), 1.89-1.80 (m, 3H).
ЖХМС (m/z): 250 [M++1].
[00304] Синтез (S)-бензил 2-(хлоркарбонил)пирролидин-1-карбоксилата (В):
К перемешанному раствору соединения А (2,5 г, 0,01 моль) в CH2Cl2 (50 мл) добавили SOCl2 (2,7 г, 0,02 моль) при 0°С и перемешивали в течение 2 часов. Реакционную смесь концентрировали под пониженным давлением с получением неочищенного соединения В. Этот материал напрямую использовали на следующей стадии без дополнительной очистки.
[00305] Синтез (2S,5R)-2-(((бензилокси)карбонил)амино-3-гидроксибутановой кислоты (Е):
К перемешанному раствору NaHCO3 (529 г, 6,30 моль) в воде (1 л) добавили (2S,3R)-2-амино-3-гидроксибутановую кислоту (250 г, 2,10 моль) при комнатной температуре и перемешивали в течение 30 минут. Реакционную смесь охладили до 0°С, к реакционной смеси по каплям добавили Cbz-Cl (850 мл, 2,52 моль, 50% на PhCH3) и перемешивали в течение 1 часа. Реакционную смесь нагрели до комнатной температуры и снова перемешивали в течение 28 часов. К этой смеси добавили МТБЭ (1 л) и перемешивали в течение 20 минут. Отделенный водный слой С1-4ешали с толуолом и перемешивали в течение 20 минут. Водный слой подкислили с помощью 1 н. HCl (рН~1-2) и экстрагировали EtOAc (3×1,5 л). Органический слой промыли насыщенным солевым раствором, высушили над безводным Na2SO4 и концентрировали под пониженным давлением. Неочищенный материал перемешивали с дициклогексиламином (819 мл, 4,20 моль) в течение 4 часов с получением белого твердого вещества, которое отфильтровали и высушили. Полученное твердое вещество нагревали с дефлегматором с EtOAc (1,5 л) в течение 1 часа, а затем отфильтровали. Твердый материал растворили в воде (1 л) и подкислили разбавленной H2SO4, и снова перемешивали в течение 30 минут. Водный слой экстрагировали EtOAc (3×1 L). Отделенный органический слой промыли насыщенным солевым раствором, высушили над безводным Na2SO4, отфильтровали и концентрировали под пониженным давлением. Полученный неочищенный материал растирали с н-гексаном в течение 2 часов с получением белого твердого вещества. Полученный твердый материал отфильтровали и высушили под пониженным давлением с получением соединения Ε (230 г, 43%).
1H-ЯМР: (400 МГц, ДМСО-d6): δ 7.35-7.32 (m, 5Н), 6.91 (d, 1Н), 5.06 (s, 2H), 4.13-4.09 (m, 1H), 3.99-3.95 (m, 1H), 1.02 (d, 3H).
ЖХМС (m/z): 254,1(M+1).
СЭЖХ (чистота по ИДСР): 99,82%.
[00306] Синтез (2S,3R)-метил 2-амино-3-гидроксибутаноата (С):
К перемешанному раствору (2S,3R)-2-амино-3-гидроксибутановой кислоты (200 г, 1,68 моль) в метаноле (1,2 л) по каплям добавили SOCl2 (244 мл, 3,36 моль) при 0°С и перемешивали в течение 1 часа. Полученную реакционную смесь нагревали с дефлегматором в течение 24 часов. После расходования исходного материала (по ТСХ), реакционную смесь нагрели до комнатной температуры и концентрировали под пониженным давлением, и промыли н-гексаном (2×50 мл). Остаток растворили в EtOH (1 л) и нейтрализовали с помощью Et3N (471 мл, 3,36 моль), и снова перемешивали в течение 2 часов. Выпавшее в осадок твердое вещество отфильтровали; полученный фильтрат концентрировали под пониженным давлением с получением соединения С (195 г, 80%).
1Н-ЯМР: (400 МГц, ДМСО-d6): δ 8.51 (br s, 3Н), 4.13-4.10 (m, 1H), 3.91 (br s, 1H), 1.20 (d, 3H).
ЖХМС (m/z): 134.1 [M++1].
[00307] Синтез (2S,3R)-2-амино-3-гидроксибутанамида (D):
Раствор соединения F (190 г, 1,35 моль) в IPA (2 л) поместили в автоклав и продували газообразный NH3 (7-8 мг), и перемешивали при 35°С в течение 24 часов. Затем удалили газообразный NH3 и концентрировали реакционную смесь под пониженным давлением, и добавили CH2Cl2, и отфильтровали. Полученное твердое вещество нагревали с дефлегматором в EtOH в течение 1 часа при 78°С. Реакционную массу отфильтровали при нагревании и добавили к фильтрату н-гексан, и снова перемешивали еще 4 часа. Полученное осажденное твердое вещество отфильтровали и высушили под пониженным давлением с получением соединения D (160 г, 47%).
1Н-ЯМР: (500 МГц, ДМСО-d6): δ 7.38 (br s, 1Н), 7.02 (br s, 1H), 4.66 (br s, 1H), 3.77-3.70 (m, 1H), 2.93 (d, 1H), 2.72 (br m, 1H), 1.05 (d, 3H).
ЖХМС (m/z): 119,1 [M++1].
СЭЖХ (чистота по ИДСР): 99,9%.
[00308] Синтез (S)-этил пирролидин-2-карбоксилата гидрохлорида (1):
К раствору L-пролина (исходный материал) (110 г, 956,5 ммоль) в этаноле добавили тионилхлорид (141 мл, 1911,3 ммоль) и нагревали с дефлегматором в течение 16 часов. Реакционную смесь довели до комнатной температуры и концентрировали под пониженным давлением с получением соединения 1 в виде гидрохлоридндой соли (170 г, 99%).
[00309] Синтез (S)-1-бензил 2-этил-пирролидин-1,2-дикарбоксилата (2):
К раствору соединения 1 (170 г, 947 ммоль) в ДХМ добавили ТЭА (398 мл, 2,83 моль), а через 30 минут добавили Cbz-Cl (1,136 моль). Реакционную смесь перемешивали при комнатной температуре в течение 12 часов. Реакционную смесь промыли водой и экстрагировали ДХМ. Органический слой высушили над безводным Na2SO4 и концентрировали под пониженным давлением. Неочищенное вещество очистили колоночной хроматографией с получением соединения 2 (230 г, 88%).
1H-ЯМР: (200 МГц, ДМСО-d6): δ 7.42-7.3l(m, 5Н), 5.09 (m, 2Н), 4.32-4.23(m, 1H), 4.08-3.98(m, 2Н), 3.50-3.38 (m, 2Н), 2.30-2.18 (m, 1Н), 1.90-1.81(m, 3Н), 1.10-1.04 (t, 3Н).
Масса m/z: 278 [М+ +1].
[00310] Синтез 1-бензил 2-этил-2-(3-метоксибензил)пирролидин-1,2-дикарбоксилата (3):
К раствору соединения 2 (10 г, 37,9 ммоль) в сухом ТТФ (100 мл) под инертной атмосферой добавили LiHMDS (1Mb ТГФ) (76 мл, 0,076 моль) при -25°С и перемешивали в течение 2 часов. К реакционной смеси по каплям добавили 3-метоксибензилбромид (9,21 г, 45,59 ммоль) при -25°С, оставили нагреваться до комнатной температуры и перемешивали в течение 30 минут. Реакционную смесь погасили насыщенным раствором NH4Cl, а водный слой экстрагировали EtOAc (2×200 мл). Объединенные органические экстракты высушили над безводным Na2SO4 и концентрировали под пониженным давлением. Полученный неочищенный остаток очистили силикагелевой колоночной хроматографией, элюируя 10% EtOAc в гексане, с получением соединения 3 (10 г, 69%).
1H-ЯМР: (400 МГц, CDCl3): δ 7.41-7.32 (m, 5Н), 7.19-7.09 (m, 1Н), 6.80 (t, 1H), 6.65 (d, 2H), 5.30 (t, 1H), 5.19 (t, 1H), 3.79-3.70 (m, 5H), 3.55-3.52 (m, 2H), 3.10-3.07 (m, 2H), 2.15-2.08 (m, 1H), 1.79-1.72 (m, 1H), 1.10-0.99 (m, 1H).
[00311] Синтез метил 2-(3-метоксибензил)пирролидин-2-карбоксилата (4):
К перемешанному раствору соединения 3 (10 г, 26,17 ммоль) в СН3ОН (100 мл) добавили Pd/C (3 г) и перемешивали под атмосферой Н2 при комнатной температуре в течение 10 часов. После расходования исходного материала (по ТСХ), реакционную смесь отфильтровали через слой целита и промыли МеОН. Полученный фильтрат концентрировали под пониженным давлением с получением соединения 4 (5,3 г, 82%). Этот материал использовали напрямую на следующей стадии без дополнительной очистки.
1H-ЯМР: (400 МГц, ДМСО-d6): δ 7.15 (t, 1H), 6.59-6.55 (m, 3Н), 3.71 (s, 3Н), 3.55 (s, 3Н), 2.99 (d, 1H), 2.85-2.81 (m, 3Н), 2.09-2.05 (m, 1H), 1.75-1.67 (m, 3Н).
ЖХМС (m/z): 250,2 [М++1].
[00312] Синтез (2S)-бензил 2-(2-(3-метоксибензил)-2-(метоксикарбонил)пирролидин-1-карбонил)пирролидин-1-карбоксилата (S):
К суспензии соединения 4 (4,3 г, 17,47 ммоль) в ДХМ: H2O (700 мл, 2:5) добавили Na2CO3 (4,6 г, 43,69 ммоль), затем раствор Промежуточного соединения В (5,2 г, 20,97 ммоль) в ДХМ при комнатной температуре и перемешивали в течение 2 часов. Органический слой промыли насыщенным солевым раствором, высушили над Na2SO4, концентрировали под пониженным давлением с получением неочищенного соединения 5 (7 г). Этот материал напрямую использовали на следующей стадии без дополнительной очистки.
[00313] Синтез (S)-1-(1-((бензилокси)карбонил)пирролидин-2-карбонил)-2-(3-метоксибензил)пирролидин-2-карбоновой кислоты (6):
К перемешанному раствору соединения 5 (7 г, 14,58 ммоль) в МеОН (100 мл) добавили 1 н. раствор NaOH (100 мл) и перемешивали при температуре дефлегмации в течение 5 часов. Летучие вещества выпарили под пониженным давлением, а полученный остаток разбавили водой и довели до рН~2 с помощью насыщенного раствора лимонной кислоты. Водный слой экстрагировали EtOAc. Объединенные органические экстракты высушили над Na2SO4 и концентрировали под пониженным давлением с получением неочищенного соединения 6 (7,6 г).
1Н-ЯМР: (400 МГц, ДМСО-d6): δ 12.45 (br s, 1Н), 7.85-7.75 (m, 4H), 6.99-6.82 (m, 2H), 5.34-5.29 (m, 1H), 3.74 (d, 1H), 3.65-3.41 (m, 2H), 3.39-3.34 (m, 1H), 3.01 (t, 1H), 2.25-2.21 (m, 1H), 2.01-1.92 (m, 3H), 1.12 (t, 1H).
ЖХМС (m/z): 467 [M++1].
[00314] Синтез (S)-бензил 2-(2-(((2S,3R)-1-амино-3-гидрокси-1-оксобутин-2-ил)карбамоил)-2-(3-метоксибензил)пирролидин-1-карбонил)пирролидин-1-карбоксилата (7):
К перемешанному раствору соединения 6 (2 г, 4,29 ммоль) в CH2Cl2 (30 мл) добавили EDCI.HCl (1,23 г, 6,43 ммоль), HOBt (869 мг, 6,43 ммоль), затем DIPEA (1,66 г, 12,87 ммоль) и соединение D (506 мг, 4,29 ммоль) при 0°С. Реакционную смесь перемешивали при комнатной температуре в течение 12 часов. После расходования исходного материала (по ТСХ), реакцию погасили водой и экстрагировали CH2Cl2 (2×100 мл). Органический слой высушили над безводным Na2SO4 и концентрировали под пониженным давлением. Неочищенное вещество очистили колоночной хроматографией с получением соединения 7 (1,5 г, 61%).
1Н-ЯМР: (400 МГц, ДМСО-d6): δ 7.49-7.37 (m, 6Н), 7.11-6.92 (m, 2Н), 6.88-6.82 (m, 2Н), 5.35-5.01 (m, 2Н), 4.89-4.52 (m, 1Н), 4.22 (d, 1H), 4.01-3.84 (m, 1H), 3.72 (d, 2H), 3.62-3.55 (m, 4H), 3.22-3.18 (m, 1H), 2.22-1.75 (m, 5H), 1.09 (br s, 3H).
[00315] Синтез N-((2S, 3R)-1-амино-3-гидрокси-1-оксобутан-2-ил)-2-(3-метоксибензил)-1-((S)-пирролидин-2-карбонил)пирролидин-2-карбоксамида (8):
К перемешанному раствору соединения 7 (1,5 г, 2,65 ммоль) в МеОН (20 мл) добавили 10% Pd/C (0,4 г) и перемешивали под атмосферой Н2 при комнатной температуре в течение 6 часов. После расходования исходного материала (по ТСХ), реакционную смесь отфильтровали через слой целита, а фильтрат концентрировали под пониженным давлением с получением соединения 8 (1 г, 87%) в виде белого твердого вещества.
1H-ЯМР: (400 МГц, ДМСО-d6): δ 7.22-7.18 (m, 2Н), 7.08-7.01 (m, 1H), 6.98 (br s, 1H), 6.84 (t, 1H), 6.75-6.71 (m, 2H), 5.05 (d, 1H), 4.23 (t, 1H), 5.09 (br s, 1H), 3.95 (d, 1H), 3.74 (s, 4H), 3.61 (t, 2H), 3.09-3.01 (m, 2H), 2.72-2.68 (m, 1H), 2.17-2.01 (m, 3H), 1.85-1.80 (m, 5H), 1.08 (d, 3H).
ЖХМС (m/z): 433 [M++1].
[00316] Синтез бензил ((2S,3R)-1-((S)-2-(2-(((2S,3R)-1-амино-3-гидрокси-1-оксобутан-2-ил)карбамоил)-2-(3-метоксибензил)пирролидин-1-карбонил)пирролидин-1-ил)-3-гидрокси-1-оксобутан-2-ил)карбамата (9):
К перемешанному раствору соединения 8 (2 г, 4,62 ммоль) в CH2Cl2 (30 мл) добавили HOBt (937 мг, 6,94 ммоль), EDCI.HCl (1,32 г, 6,93 ммоль), затем DIPEA (1,79 г, 13,82 ммоль) и соединение Ε (1,17 г, 4,62 ммоль) при 0°С. Реакционную смесь перемешивали при комнатной температуре в течение 12 часов. После расходования исходного материала (по ТСХ), реакцию погасили водой и экстрагировали CH2Cl2 (2×100 мл). Органический слой высушили над безводным Na2SO4 и концентрировали под пониженным давлением. Неочищенное вещество очистили колоночной хроматографией, затем препаративной ВЭЖХ очисткой с получением соединения 9-Fr-l (0,5 г) и соединения 9-Fr-2 (0,1 г) в виде белого твердого вещества.
Данные хиральной ВЭЖХ для соединения 9-Fr-l:
1Н-ЯМР: (400 МГц, ДМСО-d6): δ 7.39 (s, 4Н), 7.19-7.15 (m, 2Н), 7.09-7.02 (m, 2Н), 6.75-6.71 (m, 2Н), 5.10-4.99 (m, 2Н), 4.76-4.71 (m, 1Н), 4.29-4.21 (m, 1H), 4.07-3.87 (m, 2H), 3.71 (d, 2H), 3.65-3.62 (m, 1H), 3.45 (d, 1H), 3.21-3.14 (m, 1H), 2.15-2.01 (m, 5H), 1.72 (br m, 1H), 1.22-1.15 (m, 2H), 1.05 (t, 2H).
ВЭЖХ: 41,56%.
ЖХМС (m/z): 668 [М++1].
Данные хиральной ВЭЖХ для соединения 9-Fr-2:
1H-ЯМР: (400 МГц, ДМСО-d6): δ 7.391 (s, 4Н), 7.19-7.15 (m, 2Н), 7.09-7.02 (m, 2Н), 6.75-6.71 (m, 2Н), 5.10-4.99 (m, 2Н), 4.76-4.71 (m, 1H), 4.29-4.21 (m, 1H), 4.07-3.87 (m, 2H), 3.71 (d, 2H), 3.65-3.62 (m, 1H), 3.45 (d, 1H), 3.21-3.14 (m, 1H), 2.15-2.01 (m, 5H), 1.72 (br m, 1H), 1.22-1.15 (m, 2H), 1.05 (t, 2H).
ВЭЖХ: 58,43%.
ЖХМС (m/z): 762,2 [М++1].
[00317] Синтез (R)-N-((2S,3R)-1-амино-3-гидрокси-1-оксобутан-2-ил)-1-((S)-1-((2S,3R)-2-амино-3-гидроксибутаноил)пирролидин-2-карбонил)-2-(3-метоксибензил)пирролидин-2-карбоксамида (CM-13A):
К раствору соединения 9-Fr-l (0,5 г, 0,0,09 моль) в ДХМ (2 мл) добавили эфирный раствор HCl (2 мл) и перемешивали при комнатной температуре в течение 1 часа. Реакционную смесь концентрировали под пониженным давлением с получением СМ-13А (45 мг, 84%) в виде белого твердого вещества.
1Н-ЯМР: (400 МГц, ДМСО-d6): δ 8.01 (br s, 3Н), 7.35 (d, 1H), 7.21 (t, 1H), 7.09 (s, 1H), 7.01 (s, 1H), 6.89 (d, 1H), 6.69 (s, 2H), 5.49 (br s, 1H), 4.69 (t, 1H), 4.35-4.31 (m, 1H), 4.15 (d, 1H), 4.09 (br m, 1H), 3.91-3.85 (m, 4H), 3.75-3.71 (m, 1H), 3.49 (d, 2H), 3.19 (s, 2H), 3.12 (d, 1H), 2.21-2.15 (m, 1H), 2.12-2.05 (m, 3H), 1.97-1.92 (m, 3H), 1.75-1.71 (m, 1H), 1.41 (d, 3H), 1.09 (d, 3H).
ЖХМС (m/z): 534 [M++1].
Чистота по ВЭЖХ: 94%.
[00318] Синтез N-((2S,3R)-1-амино-3-гидрокси-1-оксобутан-2-ил)-1-((S)-1-((2S,3R)-2-амино-3-гидроксибутаноил)пирролидин-2-карбонил)-2-(3-метоксибензил)пирролидин-2-карбоксамида (CM-13В):
К раствору соединения 9-Fr-2 (0,1 г, 0,187 ммоль) в ДХМ (3 мл) добавили эфирный раствор HCl (2 мл) и перемешивали при комнатной температуре в течение 1 часа. Реакционную смесь концентрировали под пониженным давлением с получением CM-13В (80 мг, 75%) в виде белого твердого вещества.
1H-ЯМР: (400 МГц, ДМСО-d6): δ 8.01 (br s, 3Н), 7.35 (d, 1Н), 7.21 (t, 1H), 7.09 (s, 1H), 7.01 (s, 1H), 6.89 (d, 1H), 6.69 (s, 2H), 5.49 (br s, 1H), 4.69 (t, 1H), 4.35-4.31 (m, 1H), 4.15 (d, 1H), 4.09 (br m, 1H), 3.91-3.85 (m, 4H), 3.75-3.71 (m, 1H), 3.49 (d, 2H), 3.19 (s, 2H), 3.12 (d, 1H), 2.21-2.15 (m, 1H), 2.12-2.05 (m, 3H), 1.97-1.92 (m, 3H), 1.75-1.71 (m, 1H), 1.41 (d, 3H), 1.09 (d, 3H).
ЖХМС (m/z): 534 [М++1].
Чистота по ВЭЖХ: 92%.
Пример 14 - Синтез (R)-N-((2S,3R)-1-амино-3-гидрокси-1-оксобутан-2-ил)-1-((S)-1-(2S,3R)-2-амино-3-гидроксибутаноил)пирролидин-2-карбонил)-2-(3,4,5-триметоксибензил)пирролидин-2-карбоксамида (CM-14А) и (S)-N-(2S,3R)-1-амино-3-гидрокси-1-оксобутан-2-ил)-1-((S)-1-((2S,3R)-2-амино-3-гидроксибутаноил)приролидин-2-карбонил)-2-(3,4,5-триметоксибензил)пирролидин-2-карбоксамида (CM-14В):
[00319] Для синтеза (R)-N-((2S,3R)-1-амино-3-гидрокси-1-оксобутан-2-ил)-1-((S)-1-((2S,3R)-2-амино-3-гидроксибутаноил)пирролидин-2-карбонил)-2-(3,4,5-триметоксибензил)пирролидин-2-карбоксамида (CM-14А) и (S)-N-((2S,3R)-1-амино-3-гидрокси-1-оксобутан-2-ил)-1-((S)-1-((2S,3R)-2-амино-3-гидроксибутаноил)пирролидин-2-карбонил)-2-(3,4,5-триметоксибензил)пирролидин-2-карбоксамида (CM-14В) использовали следующую последовательность реакций (Схема N): Схема N.
Синтез С1-4-14А и С1-4-14 В:

[00320] Синтез (S)-1-((бензилокси)карбонил)пирролидин-2-карбоновой кислоты (А):
К перемешанному раствору (S)-пирролидин-2-карбоновой кислоты (250 г, 2,17 моль) в воде (1 л) добавили Na2CO3 (576 г, 5,43 моль) и перемешивали в течение 1 часа. После охлаждения до 0°С, к реакционной смеси по каплям добавили бензилхлорформиат (444 г, 2,61 моль) и снова перемешивали в течение 1 часа. Полученную реакционную смесь нагрели до комнатной температуры и дополнительно перемешивали в течение 24 часов. После расходования исходного материала (по ТСХ), реакционную смесь разбавили водой (1 л) и эфиром (1,5 л). Отделенный водный слой обработали PhCH3 (1,5 л) и подкислили с помощью 6 н. HCl. Водный слой экстрагировали EtOAc (3×1,5 л). Объединенные органические экстракты промыли насыщенным солевым раствором, высушили над безводным Na2SO4, отфильтровали и концентрировали под пониженным давлением с получением соединения А (450 г, 84%) в виде светло-желтого сиропообразного вещества.
1H-ЯМР: (400 МГц, ДМСО-d6): δ 12.71 (br s, 1Н), 7.40-7.30 (m, 5H), 5.19-5.01 (m, 2H), 4.25 (dd, 1H), 3.51-3.50 (m, 2H), 2.29-2.15 (m, 1H), 1.89-1.80 (m, 3H).
ЖХМС (m/z): 250 [M++1].
[00321] Синтез (S)-бензил 2-(хлоркарбонил)пирролидин-1-карбоксилата (В):
К перемешанному раствору соединения А (2,5 г, 0,01 моль) в CH2Cl2 (50 мл) добавили SOCl2 (2,7 г, 0,02 моль) при 0°С и перемешивали в течение 2 часов. Реакционную смесь концентрировали под пониженным давлением с получением неочищенного соединения В. Этот материал напрямую использовали на следующей стадии без дополнительной очистки.
[00322] Синтез (2S,3R)-метил 2-амино-3-гидроксибутаноата (С):
К перемешанному раствору (2S,3R)-2-амино-3-гидроксибутановой кислоты (200 г, 1,68 моль) в метаноле (1,2 л) по каплям добавили SOCl2 (244 мл, 3,36 моль) при 0°С и перемешивали в течение 1 часа. Полученную реакционную смесь нагревали с дефлегматором в течение 24 часов. После расходования исходного материала (по ТСХ), реакционную смесь нагрели до комнатной температуры и концентрировали под пониженным давлением, и декантировали с н-гексаном (2×50 мл). Остаток растворили EtOH (1 л) и нейтрализовали с помощью Et3N (471 мл, 3,36 моль), и снова перемешивали в течение 2 часов. Выпавшее в осадок твердое вещество отфильтровали; полученный фильтрат концентрировали под пониженным давлением с получением соединения С (195 г, 80%).
1H-ЯМР: (400 МГц, ДМСО-d6): δ 8.51 (br s, 3Н), 4.13-4.10 (m, 1H), 3.91 (br s, 1H), 1.20 (d, 3H).
ЖХМС (m/z): 134,1 [M++1].
[00323] Синтез (2S,3R)-2-амино-3-гидроксибутанамида (D):
Раствор соединения F (190 г, 1,35 моль) в IPA (2 л) поместили в автоклав и продували газообразный NH3 (7-8 мг), и перемешивали при 35°С в течение 24 часов. Затем удалили газообразный NH3 и концентрировали реакционную смесь под пониженным давлением, и добавили CH2Cl2, и отфильтровали. Полученное твердое вещество нагревали с дефлегматором в EtOH в течение 1 часа при 78°С. Реакционную массу отфильтровали при нагревании, а к фильтрату добавили н-гексан и снова перемешивали еще 4 часа. Полученное осажденное твердое вещество отфильтровали и высушили под пониженным давлением с получением соединения D (160 г, 47%).
1H-ЯМР: (500 МГц, ДМСО-d6): δ 7.38 (br s, 1Н), 7.02 (br s, 1H), 4.66 (br s, 1H), 3.77-3.70 (m, 1H), 2.93 (d, 1H), 2.72 (br m, 1H), 1.05 (d, 3H).
ЖХМС (m/z): 119,1 [М++1].
СЭЖХ (чистота по ИДСР): 99,9%.
[00324] Синтез бензил 2-(трет-бутоксикарбониламино)-3-гидроксибутаноата (Е):
К раствору 2-(трет-бутоксикарбониламино)-3-гидроксибутановой кислоты (50 г, 228,3 ммоль) в ДМФ (500 мл) добавили К2СО3 (63 г, 456,6 ммоль) и перемешивали при комнатной температуре в течение 15 минут. К этой смеси добавили бензилбромид (46,83 г, 273,9 ммоль) и перемешивали при комнатной температуре в течение 6 часов. Реакционную смесь разбавили водой (500 мл) и экстрагировали EtOAc (2×750 мл). Объединенные органические слои промыли насыщенным солевым раствором (50 мл), высушили над безводным Na2SO4 и концентрировали под пониженным давлением. Неочищенный материал очистили силикагелевой колоночной хроматографией, элюируя 20% EtOAc в гексане, с получением бензил 2-(трет-бутоксикарбониламино)-3-гидроксибутаноата Ε (52 г, 73%).
1Η-ΗΜΡ: (500 МГц, ДМСО-d6): δ 7.37-7.30 (m, 5Н), 6.60 (d, 1Н), 5.18-5.08 (m, 2H), 4.76 (d, 1H), 4.08-4.00 (m, 2H), 1.38 (s, 9H), 1.09 (d, 3H).
Масса m/z: 310,0 [М++1], 210 [M+-De Boc].
[00325] Синтез бензил 3-ацетокси-2-(трет-бутоксикарбониламино)бутаноата (F):
К перемешанному раствору соединения Ε (52 г, 168,2 ммоль) в CH2Cl2 (500 мл) добавили Ac2O (20,5 г, 201,9 ммоль), Et3N (25,4 г, 252,4 ммоль) и DMAP (3,5 г) и перемешивали при комнатной температуре в течение 2 часов. Летучие вещества удалили под пониженным давлением. Полученный остаток разбавили EtOAc (750 мл) и промыли холодным 0,5 н. раствором HCl (2×200 мл). Органический слой промыли насыщенным солевым раствором, высушили над безводным Na2SO4 и концентрировали под пониженным давлением с получением 3-ацетокси-2-(трет-бутоксикарбониламино)бутаноата F (52 г, 88%).
1H-ЯМР: (500 МГц, ДМСО-d6): δ 7.35-7.34 (m, 5Н), 7.27-7.25 (d, 1Н), 5.18-5.06 (m, 3Н), 4.34-4.32 (m, 1H), 1.90 (s, 3H), 1.39 (s, 9H), 1.16 (d, 3H).
Масса m/z: 252 [M++1-De Boc].
[00326] Синтез (2S,3R)-3-ацетокси-2-(трет-бутоксикарбониламино)бутановой кислоты (G):
К перемешанному раствору соединения F (52 г, 148,1 ммоль) в МеОН (1 л) добавили 10% Pd/C под атмосферой N2 и перемешивали реакционную смесь под атмосферой водорода в течение 16 часов. Реакционную смесь отфильтровали через слой целита, полученный фильтрат выпарили под пониженным давлением, а неочищенный остаток растерли с гексаном с получением (2S,3R)-3-ацетокси-2-(трет-бутоксикарбониламино)бутановой кислоты G (35 г, 90%).
1H-ЯМР: (500 МГц, ДМСО-d6): δ 12.78 (br s, 1Н), 6.94 (d, 1H), 5.16-5.14 (m, 1H), 4.17-4.15 (m, 1H), 1.95 (s, 3H), 1.39 (s, 9H), 1.10 (d, 3H).
Масса m/z: 260,0 [М-1].
[00327] Синтез (S)-метил пирролидин-2-карбоксилата гидрохлорида (1):
К раствору L-пролина (исходный материал) (110 г, 956,5 ммоль) в метаноле добавили тионилхлорид (141 мл, 1911,3 ммоль) и нагревали с дефлегматором в течение 16 часов. Реакционную смесь довели до комнатной температуры и концентрировали под пониженным давлением с получением соединения 1 в виде гидрохлоридной соли (170 г, 99%).
[00328] Синтез (S)-1-бензил 2-метил-пирролидин-1,2-дикарбоксилата (2):
К раствору соединения 1 (170 г, 947 ммоль) в ДХМ добавили ТЭА (398 мл, 2,83 моль), а через 30 минут добавили Cbz-Cl (1,136 моль). Реакционную смесь перемешивали при комнатной температуре в течение 12 часов. Реакционную смесь промыли водой и экстрагировали ДХМ. Органический слой высушили над безводным Na2SO4 и концентрировали под пониженным давлением. Неочищенное вещество очистили колоночной хроматографией с получением соединения 2 (230 г, 88%).
1H-ЯМР: (200 МГц, ДМСО-d6): δ 7.42-7.31(m, 5Н), 5.09 (m, 2Н), 4.32-4.23(m, 1Н), 4.08-3.98(m, 2H), 3.50-3.38 (m, 2H), 2.30-2.18 (m, 1H), 1.90-1.81(m, 3H), 1.10-1.04 (t, 3H).
Масса m/z: 278 [Μ++1].
[00329] Синтез 1-бензил 2-метил-2-(3,4,5-триметоксибензил)пирролидин-1,2-дикарбоксилата (3):
К раствору соединения 2 (3,4 г, 12,9 ммоль) в сухом ТГФ (60 мл) под инертной атмосферой добавили LiHMDS (1Mb ТГФ) (25 мл, 25,0 ммоль) при -25°С и перемешивали в течение 2 часов. К реакционной смеси по каплям добавили 5-(бромметил)-1,2,3-триметоксибензол (4 г, 15,5 ммоль) при -25°С. Смесь оставили нагреваться до комнатной температуры и перемешивали в течение 2 часов. Реакционную смесь погасили насыщенным раствором NH4Cl, а водный слой экстрагировали EtOAc (2×200 мл). Объединенные органические экстракты высушили над безводным Na2SO4 и концентрировали под пониженным давлением. Полученный неочищенный остаток очистили силикагелевой колоночной хроматографией, элюируя 15% EtOAc в гексане, с получением соединения 3 (4,8 г, 84%).
ЖХМС (m/z): 444,4 [М++1].
ВЭЖХ: 92%.
[00330] Синтез метил 2-(3,4,5-триметоксибензил)пирролидин-2-карбоксилата (4):
К перемешанному раствору соединения 3 (4,8 г, 10,8 ммоль) в СН3ОН (40 мл) добавили Pd/C (1,5 г) и перемешивали под атмосферой Н2 при комнатной температуре в течение 16 часов. После расходования исходного материала (по ТСХ), реакционную смесь отфильтровали через слой целита и промыли МеОН. Полученный фильтрат концентрировали под пониженным давлением с получением соединения 4 (3,0 г, 90%).
1H-ЯМР: (400 МГц, ДМСО-d6): δ 6.49 (s, 2Н), 3.72 (s, 6Н), 3.61 (s, 6Н), 2.99 (d, 1Н), 2.85-2.81 (m, 2H), 2.78 (d, 1H), 2.12-2.07 (m, 1H), 1.79-1.71 (m, 3H).
ЖХМС (m/z): 310,2 [M++1].
[00331] Синтез (2S)-бензил 2-(2-(метоксикарбонил)-2-(3,4,5-триметоксибензил)-пирролидин-1-карбонил)пирролидин-1-карбоксилата (5):
К суспензии соединения 5 (3 г, 9,70 ммоль) в ДХМ: H2O (45 мл, 5:4) добавили Na2CO3 (2,5 г, 24,20 ммоль), затем раствор Промежуточного соединения В (3,2 г, 11,60 ммоль) в ДХМ при комнатной температуре и перемешивали в течение 2 часов. Органический слой промыли насыщенным солевым раствором, высушили над Na2SO4, концентрировали под пониженным давлением с получением неочищенного соединения 5 (4,6 г). Этот материал напрямую использовали на следующей стадии без дополнительной очистки.
ЖХМС (m/z): 541,3 [М++1].
[00332] Синтез (S)-1-(1-((бензилокси)карбонил)пирролидин-2-карбонил)-2-(3,4,5-триметоксибензил)пирролидин-2-карбоновой кислоты (6):
К перемешанному раствору соединения 5 (4,6 г, 8,80 ммоль) в МеОН (40 мл) добавили 2 н. раствор NaOH (10 мл) и перемешивали при температуре дефлегмации в течение 16 часов. Летучие вещества выпарили под пониженным давлением, а полученный остаток разбавили водой и довели до рН~2 с помощью 1 н. раствора HCl. Водный слой экстрагировали EtOAc. Объединенные органические экстракты высушили над Na2SO4 и концентрировали под пониженным давлением с получением неочищенного соединения 6 (4 г).
[00333] Синтез (S)-бензил 2-(2-(((2S,3R)-1-амино-3-гидрокси-1-оксобутан-2-ил) карбамоил)-2-(3,4,5-триметоксибензил)пирролидин-1-карбонил)пирролидин-1-карбоксилата(7):
К перемешанному раствору соединения 6 (4 г, 7,60 ммоль) в CH2Cl2 (50 мл) добавили HOBt (1,5 г, 11,40 ммоль), EDCI.HCl (2,10 г, 11,40 ммоль), затем DIPEA (4,1 мл, 22,8 ммоль) и соединение D (987 мг, 8,00 ммоль) при 0°С. Реакционную смесь перемешивали при комнатной температуре в течение 16 часов. После расходования исходного материала (по ТСХ), реакцию погасили водой и экстрагировали CH2Cl2 (2×100 мл). Органический слой высушили над безводным Na2SO4 и концентрировали под пониженным давлением с получением неочищенного продукта, который очистили колоночной хроматографией с получением соединения 7 (3 г, 63%).
ЖХМС (m/z): 625,6 [М+-1].
[00334] Синтез N-((2S, 3R)-1-амино-3-гидрокси-1-оксобутан-2-ил)-1-((S)-пирролидин-2-карбонил)-2-(3,4,5-триметоксибензил)пирролидин-2-карбоксамида (8):
К перемешанному раствору соединения 7 (3 г, 4,79 ммоль) в МеОН (30 мл) добавили 10% Pd/C (0,7 г) под атмосферой N2. Реакционную смесь перемешивали при комнатной температуре в течение 16 часов под атмосферой Н2. После расходования исходного материала (по ТСХ), реакционную смесь отфильтровали через слой целита, а фильтрат концентрировали под пониженным давлением с получением соединения 8 (2,1 г, 91%) в виде белого твердого вещества.
[00335] Синтез трет-бутил ((2S,3R)-1-((S)-2-(2-(((2S,3R)-1-амино-3-гидрокси-1-оксобутан-2-ил)карбамоил)-2-(3,4,5-триметоксибензил)пирролидин-1-карбонил)пирролидин-1-ил)-3-гидрокси-1-оксобутан-2-ил)карбамата (9):
К перемешанному раствору соединения 8 (2 г, 4,00 ммоль) в CH2Cl2 (30 мл) добавили HOBt (823 мг, 6,00 ммоль), EDCI.HCl (1,16 г, 6,00 ммоль), затем DIPEA (2,2 мл, 11,6 ммоль) и соединение G (1,068 г, 4,80 ммоль) при 0°С. Реакционную смесь перемешивали при комнатной температуре в течение 16 часов. После расходования исходного материала (по ТСХ), реакцию погасили водой и экстрагировали CH2Cl2 (2×100 мл). Органический слой высушили над безводным Na2SO4 и концентрировали под пониженным давлением. Неочищенный материал очистили колоночной хроматографией, затем препаративной ВЭЖХ очисткой с получением соединения 9-Fr-1 (0,12 г) и соединения 9-Fr-2 (0,2 г) в виде белого твердого вещества.
Данные хиральной ВЭЖХ для соединения 9-Fr-1:
ЖХМС (m/z): 594,5 [(М++1)-Вос].
ВЭЖХ: 94%.
Данные хиральной ВЭЖХ для соединения 9-Fr-2: ЖХМС (m/z): 594,5 [(М++1)-Вос].
ВЭЖХ: 95%.
[00336] Синтез (R)-N-((2S,3R)-1-амино-3-гидрокси-1-оксобутан-2-ил)-1-((S)-1-{(2S,3R)-2-амино-3-гидроксибутаноил)пирролидйн-2-карбонил)-2-(3,4,5-триметоксибензил)пирролидин-2-карбоксамида (CM-14А):
К раствору соединения 9-Fr-1 (0,12 г, 0.17 ммоль) в ДХМ (2,5 мл) добавили HCl в 1,4-диоксане (2,5 мл) и перемешивали при комнатной температуре в течение 2 часов. Реакционную смесь концентрировали под пониженным давлением с получением CM-14А (80 мг) в виде HCl соли.
1H-ЯМР: (400 МГц, D2O): δ 6.89 (s, 2Н), 4.99 (t, 1H), 4.45-4.40 (m, 4H), 3.98 (s, 6H), 3.85 (s, 4H), 3.71-3.65 (m, 4H), 3.51 (d, 1H), 3.39 (d, 1H), 2.62-2.55 (m, 1H), 2.44-2.37 (m, 1H), 2.24-2.20 (m, 4H), 1.84-1.82 (m, 1H), 1.51 (d, 3H), 1.27 (d, 3H), 1.15-1.09 (m, 1H). ЖХМС (m/z): 594,5 [M++1]
Чистота по ВЭЖХ: 93%.
Оптическое вращение [α19,9D]:+21,91 (С=0,5% в воде).
[00337] Синтез (S)-N-((2S,3R)-1-амино-3-гидрокси-1-оксобутан-2-ил)-1-((S)-1-((2S,3R)-2-амино-3-гидроксибутаноил)пирролидин-2-карбонил)-2-(3,4,5-триметоксибензил)пирролидин-2-карбоксамида (CM-14 В):
К раствору соединения 9-Fr-2 (0,2 г, 0,20 ммоль) в ДХМ (2,5 мл) добавили HCl в 1,4-диоксане (2,5 мл) и перемешивали при комнатной температуре в течение 2 часов. Реакционную смесь концентрировали под пониженным давлением с получением CM-14В (170 мг) в виде HCl соли.
1H-ЯМР: (400 МГц, D2O): δ 6.89 (s, 2Η), 4.99 (t, 1H), 4.45-4.40 (m, 4H), 3.98 (s, 6H), 3.85 (s, 4H), 3.71-3.65 (m, 4H), 3.51 (d, 1H), 3.39 (d, 1H), 2.62-2.55 (m, 1H), 2.44-2.37 (m, 1H), 2.24-2.20 (m, 4H), 1.84-1.82 (m, 1H), 1.51 (d, 3H), 1.27 (d, 3H), 1.15-1.09 (m, 1H). ЖХМС (m/z): 594,5 [M++1]
Чистота по ВЭЖХ: 95%.
Оптическое вращение [α19,9D]: -92,57(C=0,5% в воде).
Пример 15 - Метаболизм и фармакокинетика лекарства
[00338] Этот пример демонстрирует лекарственный метаболизм и фармакокинетические свойства исследуемых соединений.
[00339] Соединения CM-5А, CM-6А, CM-7А, CM-8А и CM-9А испытали в in vitro анализе растворимости в воде, проницаемости в клетки MDCK и метаболической стабильности. Соединения CM-5А, CM-6А и CM-7А испытали также на фармакокинетику у крыс. Краткое описание способа, результаты и выводы представлены ниже.
Растворимость в воде:
[00340] Способ. Оценивали кинетическую растворимость в воде (способ pION) с использованием представленных ниже параметров:
[00341] Результаты. Ниже представлены результаты анализа растворимости:


[00342] Выводы. Результаты позволяют предположить, что исследуемые соединения обладают хорошей растворимостью в воде при рН 7,4.
Метаболическая стабильность:
[00343] Метаболическую стабильность оценивали в микросомах печени, а также в гепатоцитах с некоторыми соединениями.
[00344] Способ для микросомальной стабильности:
соединению, ЖХ-МС/МС (отношения площади пиков).
[00345] Способ для гепатоцитарной стабильности:
[00346] Результаты:
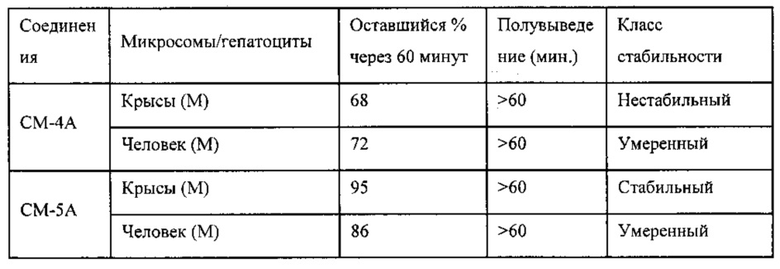

(Г): Гепатоциты; (М): Микросомы
[00347] Классификация метаболической стабильности основана на представленном ниже критерии.

[00348] Выводы. Результаты позволяют предположить, что исследуемые соединения обладают от умеренной до высокой стабильностью в микросомах/гепатоцитах.
Проницаемость в клетки MDCK:
[00349] Способ. Проницаемость исследуемых соединений оценили с помощью клеток MDCK (клетки почек собак Мадин-Дарби), используя представленные ниже параметры.
- Исследуемая концентрация: 5 мкМ (n=3)
- Буфер:HBSScpH7,4
- Временные точки: 0, 15, 30, 60 и 90 минут
- Анализ: Контролирование по исходному соединению с помощью ЖХ-МС/МС
[00350] Результаты. Ниже представлены результаты анализа проницаемости в клетки MDCK:

[00351] Выводы. Результаты позволяют предположить, что исследуемые соединения обладают слабой/низкой проницаемостью через клетки MDCK. Классификация MDCK проницаемости основана на представленном ниже критерии.
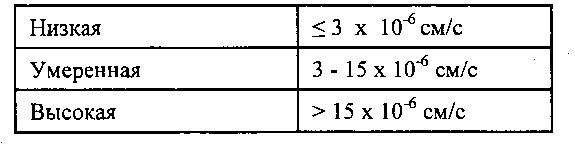
Биодоступность CM-4А
[00352] Способ. Самцам в возрасте 2-3 месяца (1 мг/кг внутривенно и 10 мг/кг перорально), а также самцам и самкам (0,1 и 1 мг/кг перорально) крыс Спрага-Доули вводили дозы CM-4А (1 мг/кг внутривенно [хвостовая вена] или 0, 1, 1 или 10 мг/кг перорально [желудочный зонд]; n=5-6 на группу) в стерильном солевом носителе, а повторные образцы крови брали в рзаличные временные точки из боковой хвостой вены или с помощью сердечных игл, используя шприцы, обработанные К2-ЭДТА. Данные для самцов и самок объединяли для пероральной дозы 1 мг/кг, при условии, что не наблюдали разницы между полами. Плазму разделяли центрифугированием (1000×G в течение 10 минут при 4°С) и хранили образцы плазмы при -80°С до выполнения анализа. Образцы хранили при -80°С до выполнения анализа. В день анализа образцы и маркированные стандарты плазмы оттаивали при 4°С, экстрагировали в 3 объемах ледяного ацетонитрила, центрифугировали при 25000×G в течение 10 минут при 4°С, а надосадочную жидкость загружали в пробирки автоматического пробоотборника (4°С). Образцы анализировали с помощью прибора ЖХ/МС/МС Agilent QQQ в режиме контролирования нескольких реакций (504,3 ->171,1, 199,1, 386,1 m/z), а время удерживания С1-4-4А составило 9,3 минуты с симметричным пиком -10 секунд.
[00353] Как показано на Фигуре 1, пероральная биодоступность CM-4А является, основном, линейной от 0,1 до 10 мг/кг.
Пример 16 - Анализ связывания [3Н] МК-801
[00354] Этот пример демонстрирует анализ связывания [3Н] МК-801, который может быть использован для оценки агонистических и/или антагонистических свойств потенциальных модуляторов рецептора НМДА.
[00355] Неочищенные синаптические мембраны приготовили из переднего мозга крыс, как описано в публикации Moskal et al. (2001 г.), "The use of antibody engineering to create novel drugs that target N-methyl-D-aspartate receptors," Curr. Drug Targets, 2:331-45. Самцов крыс в возрасте 2-3 месяцев обезглавливали без анестезии с помощью гильотины, головной мозг быстро вынимали (~90 секунд), а цельную кору и гиппокамп препарировали на ледяной платформе, замораживали на сухом льду и хранили при -80°С. Образцы гомогенизировали в 20 объемах ледяного 5 мМ Tris-HCl с рН 7,4 с помощью прибора Brinkman Polytron и осаждали центрифугированием при 48000×g в течение 20 минут при 4°С, и промывали еще 3 раза, как описано выше. Затем мембраны повторно суспендировали в 5 мМ ЭДТА и 15 мМ Tris-HCl с рН 7,4 и инкубировали в течение 1 часа при 37°С, мембраны осаждали центрифугированием при 48000×g в течение 20 минут при 4°С, мгновенно замораживали в жидком азоте и хранили при -80°С. В день эксперимента мембраны оттаивали при комнатной температуре и еще 7 раз промывали в ледяном 5 мМ Tris-HCl (рН 7,4), как описано выше. После последнего промывания мембраны повторно суспендировали в аналитическом буфере (5 мМ Tris-ацетата с рН 7,4), а содержание белка определяли анализом ВСА.
[00356] Анализ связывания [3Н] МК-801 выполняли так, как описано в публикации Urwyler et al. (2009 г.), "Drug design, in vitro pharmacology, and structure-activity relationships of 3-acylamino-2-aminopropionic acid derivatives, a novel class of partial agonists at the glycine site on the N-methyl-D-aspartate (NMDA) receptor complex," J. Med. Chem., 52:5093-10. Мембранный белок (200 мкг) инкубировали с различными концентрациями исследуемых соединений (10-3-10-17 М) с 50 мкМ глутамата в течение 15 минут при 23°С. Затем аналитические пробирки инкубировали в неравновесных условиях с [3Н]МК-801 (5 нМ; 22,5 Ки/ммоль) в течение 15 минут при 23°С, затем фильтровали чирез фильтры Whatman GF7B с помощью харвестера клеток Brandel М-24R. Затем пробирки три раза промывали аналитическим буфером (5 мМ Tris-ацетата с рН 7,4), а фильтры анализировали жидкостной сцинтилляцией для подсчета количества распадов в минуту (DPM). Нулевой уровень определили в отсутствие какого-либо глицинового лиганда и в присутствии 30 мкМ 5,7-дихлоркинуреновой кислоты (5,7-DCKA). Максимальную стимуляцию измеряли в присутствии 1 мМ глицина. Во всех образцах присутствовал 50 мкМ глутамат.
[00357] Для каждой точки данных (то есть одной концентрации исследуемого соединения) рассчитали % от максимального связывания [3Н] МК-801 по следующей формуле:
[00358] % от максимального связывания [3Н] МК-801=((DPM(исследуемое соединение)-DPM5,7-DCKA)/(DPM1 мМ глицин DPM5,7-DCKA))×100%
[00359] Эффективность каждого соединения, выраженную в % увеличения связывания [3Н] МК-801, рассчитывали подбором данных к уравнению «log(агониста) в зависимости от ответа (три параметра)» с помощью программы Graph Pad Prism, а действенность (ЕС50, выраженное в пМ) и максимальную активность (% от максимального стимулирования) рассчитывали для каждого исследуемого соединения, где эффективность для исследуемого соединения представляет собой наиболее соответствующее максимальное значение (Фигуры 2-4).

Пример 17 - Токи рецептора НМДА (НМДАР)
[00360] Этот пример демонстрирует анализ для определения влияния исследуемых соединений на токи НМДАР.
[00361] Эксперименты выполнили на гиппокампальных срезах из крыс Спрага-Доули возрастом 14-18 дней, как описано в публикации Zhang et al. (2008 г.) "A NMDA receptor glycine site partial agonist, GLYX-13 ("GLYX"), simultaneously enhances LTP and reduces LTD at Schaffer collateral-CAl synapses in hippocampus," Neuropharmacology, 55:1238-50. Значения для цельных клеток получили из пирамидальных нейронов СА1 с фиксацией потенциала при -60 мВ, в срезах, обрызганных ACSF (искусственной цереброспинальной жидкостью), содержащей 0 мМ [Mg2+] и 3 мМ [Са2+], плюс 10 мкМ бикукуллина и 20 мкМ CNQX для фармакологического выделения НМДАР-зависимых возбуждающих постсинаптических токов (ВПСТ). Изменение концентрации исследуемого соединения (от 10 нМ до 1 мкМ) применяли ко всей кювете, а коллатеральные волокна Шаффера стимулировали однократными электрическими импульсами (продолжительностью 80 мкс), один раз каждые 30 секунд. ВПСТ НМДАР характеризовались длительным временем нарастания и спада, и полностью блокировались в конце каждого эксперимента введением в кювету НМД АР-специфического антагониста 0-2-амино-5-фосфонопентановой кислоты (D-AP5; 50 мкМ). Эффективность исследуемого соединения рассчитали как % увеличение тока НМДАР от исходного значения. Исходное значение измерили как ток НМДАР до внесения исследуемого соединения.

[00362] Фигура 5 иллюстрирует динамику влияния CM-4А (1 мкМ) на одиночные спровоцированные коллатералями Шаффера фармакологически выделенные ВПСТ НМДА, записанные в пирамидальный нейронах СА1 (n=5). (Каждая точка представляет собой среднее значение±стандартная погрешность среднего значения для амплитуды пика ВПСТ или полевого возбуждающего постсинаптического потенциала (пВПСТ) для n пирамидальных нейронов).
Пример 18 - Анализ долговременного потенцирования (ДВГГ), способ 1
[00363] Этот пример демонстрирует анализ для определения влияния исследуемых соединений на ДВП.
[00364] Гиппокампальные срезы от крыс Спрага-Доули возрастом 14-18 дней перенесли в интерфейсную записывающую камеру и непрерывно обрызгивали при 3 мл/мин. оксигенированной ACSF при 32±0,5°С. Измерительные электроды с низким сопротивлением были выполнены из тонкостенного боросиликатного стекла (1-2 МОм после наполнения ACSF), их вставили в апикальную дендритную область конечного поля коллатералей Шаффера в stratum radiatum региона СА1 для записи полевых возбуждающих постсинаптических потенциалов (пВПСТ). Биполярный стимулирующий электрод из нержавеющей стали (FHC Со.) поместили в коллатерально-комиссуральные волокна Шаффера в stratum radiatum СА3, а интенсивность стимула постоянным током отрегулировали для инициации приблизительно полумаксимальных пВПСТ, каждый по 30 секунд (50-100 пА; продолжительность 100 мс). Угол наклона пВПСТ измерили линейной интерполяцией из 20%-80% от максимально отрицательного отклонения, и подтвердили, что углы наклона стабильны в пределах ±10% по меньшей мере в течение 10 минут до начала эксперимента. Долговременное потенцирование (ДВП) инициировали высокочастотным стимулированием (3×100 Гц/500 мс; стрелка) в синапсах коллатерали Шаффера-CAl в контрольном образце (носитель), необработанных срезах или срезах, предварительно обработанных исследуемым соединением (от 10 нМ до 100 мкМ). Сигналы долговременного потенцирования записывали с помощью амплификатора Multiclamp 700 В и оцифровывали на приборе Digidata 1322 (Axon Instruments, Фостер Сити, штат Калифорния). Данные анализировали с помощью программы pClamp (версия 9, Axon Instruments) на IBM-совместимом персональном компьютере. Эффективность рассчитали как % увеличения долгосрочного потенцирования, измеренное для срезов, предварительно обработанных исследуемым соединением, по сравнению с носителем.
Таблица 3. Данные анализа ДВП.

[00365] Фигура 6 иллюстрирует дозозависимую взаимосвязь влияния на ДВП для CM-2, CM-1 и CM-4А.
[00366] Фигура 7 иллюстрирует дозозависимую взаимосвязь влияния GLYX-13 ("GLYX") на долговременную депрессию (ДВД), по сравнению с CM-4А. 1 мкМ GLYX-13 и CM-4А существенно уменьшают амплитуду ДВП, тогда как более низкие концентрации приводят к меньшим изменениям амплитуды. (Следует отметить, что 0,001 мкМ представляет собой отсутствие добавления лекарства; каждая точка представляет собой среднее значение±стандартную ошибку среднего значения для 8-10 срезов).
Пример 19 - Анализ долговременного потенцирования (ДВП), способ 2
[00367] Этот пример демонстрирует анализ для определения влияния исследуемых соединений на ДВП.
[00368] Анализ выполнили так, как описано в публикации Zhang et al. (2008 г.). Крыс Спрага-Доули (возрастом 12-18 дней; Taconic Farms) глубоко анестезировали изофлураном и обезглавили. Быстро вынули головной мозг, погрузили в ледяную искусственную цереброспинальную жидкость (ACSF, 2-4°С), которая содержала (в мМ): 124 NaCl, 4 KCl, 2 MgSO4, 2 CaCl2, 1,25 NaH2PO4, 26 NaHCO3, 10 глюкозы; с рН 7,4, непрерывно насыщали газом, 95% 02/5% CO2). Выполнили гемисекцию головного мозга, отрезали лобные доли, а отдельные полусферы наклеили с помощью цианоакрилатного клея на пластину, погруженную в ледяную ACSF, непрерывно насыщаемую газом, 95% 02/5% CO2 при разрезании. Коронарные срезы толщиной 400 мкм отрезали с помощью Vibratome (Leica VT1200S), и перенесли в интерфейсную портативную камеру для инкубации при комнатной температуре в течение не менее одного часа, затем перенесли в интерфейсную записывающую камеру Haas, непрерывно обрызгиваемую при 3 мл/мин. оксигенированной ACSF при 32±0,5°С. Измерительные электроды с низким сопротивлением были выполнены из тонкостенного боросиликатного стекла (1-2 МОм после наполнения ACSF). Их вставили в апикальную дендритную область конечного поля коллатералей Шаффера в stratum radiatum региона СА1 для записи полевых возбуждающих постсинаптических потенциалов (пВПСП). Биполярный стимулирующий электрод из нержавеющей втали (FHC Со.) поместили в коллатерально-комиссуральные волокна Шаффера в stratum radiatum СА3, а интенсивность стимула постоянным током отрегулировали для инициации приблизительно полумаксимальных пВПСТ, каждый по 30 секунд (50-100 пА; продолжительность 100 мкс). Угол наклона пВПСТ измерили до и после инициации ДВП линейной интерполяцией из 20-80% от максимально отрицательного отклонения, и подтвердили, что углы наклона стабильны в пределах ±10% по меньшей мере в течение 15 минут до начала эксперимента. Введение исследуемых соединений (1 мкМ) в кювету выполняли за 30 минут до применения испытания стимулом коллатерали Шаффера для достижения ДВП. ДВП индуцировали стимуляцией аксонов коллатералей Шаффера четырьмя испытаниями высокочастотными тета-импульсными стимулами по 10×100 Гц/5 пакетов импульсов в каждом, которые выполняли с интервалами между импульсами 200 мс. Продолжительность каждого испытания составляла 2 с, и испытания выполняли с интервалом 15 секунд. Сигналы записывали с помощью амплификатора Multiclamp 700 В и оцифровывали на приборе Digidata 1322 (Axon Instruments, США). Данные анализировали с помощью программы pClamp (версия 9, Axon Instruments) на IBM-совместимом персональном компьютере.
[00369] Как показано на Фигуре 8, CM-6А, CM-7А, CM-8А и CM-9А (1 мкМ) увеличивают долговременное потенцирование после высокочастотной стимуляции спровоцированных коллатералями Шаффера ВПСТ НМДА, записанное в пирамидальных нейронах СА1. *Р<0,05.
Пример 20 - Испытание Порсолта, способ 1
[00370] Этот пример демонстрирует испытание Порсолта для оценки антидепрессивной активности исследуемых соединений.
[00371] Эксперименты выполнили так, как описано в публикации Burgdorf et al. (2009 г.) "The effect of selective breeding for differential rates of 50-кГц ultrasonic vocalizations on emotional behavior in rats," Devel. Psychobiol., 51:34-46. Самцам крыс Спрага-Доули (возрастом 2-3 месяца) вводили дозу исследуемого соединения (от 0,3 до 30 мг/кг; внутривенно путем инъекции в хвостовую вену или перорально через желудочный зонд) или носителя (1 мл/кг стерильного солевого раствора, или 1 мл/кг ДМСО для 2,5-диазаспиро[3.4]октан-1-она) слепым образом, за 1 час до испытания. В день испытания животных поместили в прозрачный цилиндр высотой 46 см × диаметром 20 см, наполненный до 30 см водопроводной водой при комнатной температуре (23°С±0,5°С) на 5 минут. Всех животных экспериментаторы вытерли полотенцем после окончания сеанса плавания. Воду меняли после каждого животного. Поведение животных записывали на видео, а общую продолжительность (с) плавания (по определению минимального движения, необходимого для сохранения головы животного над поверхностью воды) выполнил «слепой» экспериментатор.

[00372] На Фигуре 9 показано среднее время плавания±стандартная погрешность среднего значения в испытании крыс по Порсолту, у самцов крыс SD возрастом 2-3 месяца, которым вводили СМ-4А (от 0,1 до 10 мг/кг, перорально через желудочный зонд) или стерильный солевой носитель (1 мл/кг, перорально) за 1 час до первого сеанса испытаний. За день до первого 5-минутного испытания Порсолта животных адаптировали однократным 15-минутным сеансом Порсолта. N=9-10 в группе. Результаты демонстрируют дозо- и времязависимую антидепрессивную активность.
[00373] Фигура 10 иллюстрирует среднее время плавания±стандартная погрешность среднего значения в испытании крыс по Порсолту, у самцов крыс SD возрастом 2-3 месяца, которым вводили антагонист рецептора АМРА 2,3-дигидрокси-6-нитро-7-сульфамоил-бензо[f]хиноксалин-2,3-дион (NBQX) (10 мг/кг, внутрибрюшинно) или дистиллированную воду (1 мг/кг) за 10 минут до введения дозы С1-4-4А (1 мг/кг, перорально) или стерильный солевой носитель (1 мл/кг, перорально через желудочный зонд) за 1 час до испытания. За день до первого 5-минутного испытания Порсолта животных адаптировали однократным 15-минутным сеансом Порсолта. N=6 в группе. *Ρ<0,05, тест-критерий PLSD Фишера полученных результатов, по сравнению с другими группами. Результаты демонстрируют, что антагонист АМРА NBQX блокирует антидепрессант-подобное действие CM-4А.
Пример 21 - Испытание Порсолта. Способ 2
[00374] Этот пример демонстрирует испытание Порсолта для оценки антидепрессивной активности исследуемых соединений.
[00375] Антидепрессант-подобное действие исследуемых соединений проверили с помощью теста Порсолта на крысах, как описано в публикациях Page et al. (1999 г.) и Burgdorf et al. (2009 г.). В этом исследовании использовали самцов крыс Спрага-Доули (возрастом 2-3 месяца). Животным вводили исследуемые соединения (0,3 мг/кг, внутривенно) или стерильный солевой носитель (1 мл/кг, внутривенно в хвостовую вену) за 1 час до одного сеанса испытания. Второй группе самцов крыс Спрага-Доули (возрастом 2-3 месяца) вводили дозу исследуемые соединения (0,1 мг/кг, перорально) стерильного солевого носителя (1 мл/кг, перорально через желудочный зонд) за 1 час до первого сеанса испытания, и повторно испытывали через 24 часа после введения дозы. За 1 день до введения дозы животных поместили в прозрачный пластиковый цилиндр высотой 46 см × диаметром 20 см, наполненный до 30 см водопроводной водой при комнатной температуре (22-23°С) на 15 минут (адаптация) и на 5 минут - на следующий день (дни) испытания. Воду меняли после каждого животного. Поведение животных записывали на видео, а общую продолжительность (с) плавания количественно определил «слепой» экспериментатор. В тесте Порсолта испытывали СМ-6А, СМ-7А, СМ-8А и СМ-9А. СМ-5А в тесте Порсолта не испытывали.
[00376] Как показано на Фигуре 11А, СМ-6А, СМ-7А, СМ-8А и СМ-9А вызывали антидепрессант-подобный эффект в испытании Порсолта, что демонстрируется снижением времени плавания после однократной дозы (0,3 мг/кг, внутривенно), по сравнению с носителем, через 1 час после введения дозы, что демонстрируется значительным действием лекарства по дисперсионному анализу (F(2, 24)=17,5, Ρ<0,0001), затем значительным тест-критерием PLSD Фишера полученных результатов для каждого испытанного соединения, по сравнению с группой носителя (* все Ρ<0,0001). Ν=5-6 крыс в группе.
[00377] Как показано на Фигурах 11В и 11С, СМ-6А, СМ-7А, СМ-8А и СМ-9А вызывали антидепрессант-подобный эффект в испытании Порсолта, что демонстрируется снижением времени плавания после однократной дозы (0,3 мг/кг, внутривенно), по сравнению с носителем через 1 час (Фигура 11 В) и через 24 часа (Фигура 11С) после введения дозы, что демонстрируется значительным действием лекарства в повторяющихся измерениях дисперсионным анализом (F(4, 25)=58,2, Ρ<0,0001), затем значительным тест-критерием PLSD Фишера полученных результатов для каждого испытанного соединения в каждой временной точке, по сравнению с соответствующей группой носителя (* все Ρ<0,0001). n=6 крыс в группе.
[00378] Как показано на Фигуре 12, устойчивые антидепрессант-подобные эффекты наблюдали по среднему значению времени плавания в испытании крыс по Порсолту±стандартная погрешность среднего значения, у самцов крыс SD возрастом 2-3 месяца, испытанных по Порсолту, которым вводили дозу положительного контроля CM-4А (1 мг/кг, перорально), исследуемые соединения (0,1 мг/кг, перорально) или стерильный солевой носитель (1 мл/кг, перорально через желудочный зонд) за 1 час до испытания и через 24 часа после введения дозы. За день до первого 5-минутного испытания Порсолта животных адаптировали однократным 15-минутным сеансом Порсолта. N=6 в группе. *Ρ<0,05, тест-критерий PLSD Фишера полученных результатов, по сравнению с носителем.
Пример 22 - Тест гипофагии, вызванной новизной (ΝΙΗ), у крыс
[00379] Тест ΝΙΗ на крысах выполнили следующим образом. Животных оставили без еды в течение ночи перед испытанием, а лабораторную пищу поместили в центр камеры открытого поля. После испытания ΝΙΗ задержку приема пищи в домашних клетках животных определяли как контрольное значение. Антидепрессанты экстренного действия, а также обработка хроническими, но не экстренными СИОЗС вызывали задержку приема пищи в новой клетке, но не в домашних клетках животных. На Фигуре 13 представлена средняя линейная задержка приема пищи±стандартная погрешность среднего значения в испытании ΝΙΗ самцов крыс SD возрастом 2-3 месяца, обработанных CN-4А (1 мг/кг, перорально) или стерильным солевым носителем (1 мл/кг, перорально) за 1 час до однократного 10-минутного сеанса испытания. N=12 в группе. *Ρ<0,05. CM-4А (1 мг/кг, перорально) вызывает антидепрессивный/анксиолитический эффект у крыс в испытании ΝIΗ.
Пример 23 - Испытание ультразвуковой вокализации крыс (УЗВ)
[00380] На Фигурах 14А и 14В представлены результаты испытания УЗВ на крысах. Положительное эмоциональное познавание измеряли во время испытаний условно-рефлекторными раздражителями (УРР), затем выполняли испытания безусловным раздражителем щекотки (БУР) (Фигура 14А). На животных выполняли 15-секундные испытания, состоящие из 6 УРР и 6 БУР испытаний (в общем 3 минуты). Измеряли также скорость движения животных к месту почесывания в конце 3-минутного сеанса. На Фигуре 14 В представлено среднее значение±стандартная погрешность среднего значения для гедонического и отвращающего БУР в испытании БУР на самцах крыс SD возрастом 2-3 месяца, обработанных CM-4А (1 мг/кг, перорально) или стерильным солевым носителем (1 мл/кг, перорально) за 1 час до испытания. N=7-8. * Ρ<0,05. Результаты демонстрируют, что CM-4А увеличивает положительное эмоциональное познание и снижает отвращающие 20-кГц БУР в испытании БУР на крысах, указывая, таким образом, на антидепрессивный эффект.
Пример 24 - Тест открытого поля на крысах
[00381] На Фигуре 15 представлены средние значения±стандартные погрешности среднего значения для пересечений линий, пересечений центральной части и времени, проведенного в центральной части, для самцов крыс SD возрастом 2-3 месяца, обработанных CM-4А (1 мг/кг, перорально) или стерильным солевым носителем (1 мл/кг, перорально) за 1 час до однократного 10-минутного сеанса испытания в открытом поле. N=7-8. * Ρ<0,05. Результаты демонстрируют, что CM-4А вызывает анксиолитический эффект в тесте открытого поля на крысах, без ухудшения двигательного поведения.
Пример 24 - Тест ускорения вращающегося стержня на крысах
[00382] Фигура 16 иллюстрирует среднее значение±стандартная погрешность среднего значения для задержки падения (с) в тесте ускорения вращающегося стержня (4-40 об/мин за 300 с) на самцах крыс SD возрастом 2-3 месяца, предварительно обработанных солевым носителем (1 мл/кг, перорально), CM-4А (1, 10 или 100 мг/кг, перорально), или положительным контролем кетамином (30 мг/кг, подкожно). Животных испытывали непосредственно до введения дозы (0 мин), через 30 минут после введения дозы и через 60 минут после введения дозы, с помощью внутригруппового дизайна эксперимента. За один день до испытания животные прошли 3 сеанса адаптации с вращающимся стержнем, с интервалом между сеансами по меньшей мере 30 минут, и характеристики с вращающимся стержнем в последнем сеансе адаптации существенно не отличались от испытания во временной точке 0 минут. N=5 для носителя и доз CM-4А, N=3 для кетамина. * Ρ<0,05. Результаты демонстрируют, что CM-4А (10-100 мг/кг, перорально) не вызывает седативного/атаксия-подобного эффекта у крыс в испытании с ускорением вращающегося стержня.
ЭКВИВАЛЕНТЫ
[00383] Специалисты в данной области понимают или могут установить с помощью не более чем стандартных экспериментов множество эквивалентов конкретных вариантов реализации настоящего изобретения, описанных в настоящем документе. Подразумевается, что такие эквиваленты входят в рамки следующей формулы изобретения.
ВКЛЮЧЕНИЕ ПОСРЕДСТВОМ ССЫЛКИ
[00384] Полное содержание всех патентов, опубликованных патентных заявок, веб-сайтов и других ссылок, цитируемых в настоящем документе, явным образом включено в настоящий документ в полном объеме посредством ссылки.
| название | год | авторы | номер документа |
|---|---|---|---|
| ФАРМАЦЕВТИЧЕСКАЯ КОМБИНАЦИЯ АТИПИЧНОГО АНТИПСИХОТИКА И МОДУЛЯТОРА NMDA ДЛЯ ЛЕЧЕНИЯ ШИЗОФРЕНИИ, БИПОЛЯРНОГО РАССТРОЙСТВА, КОГНИТИВНОГО НАРУШЕНИЯ И КЛИНИЧЕСКОЙ ДЕПРЕССИИ | 2016 |
|
RU2802972C2 |
| ПОЛИЦИКЛИЧЕСКИЕ СОЕДИНЕНИЯ И СПОСОБЫ ЦЕЛЕНАПРАВЛЕННОЙ ДЕГРАДАЦИИ ПОЛИПЕПТИДОВ БЫСТРО УСКОРЕННОЙ ФИБРОСАРКОМЫ | 2019 |
|
RU2830173C2 |
| ПРОТИВОВИРУСНЫЕ СОЕДИНЕНИЯ | 2010 |
|
RU2541571C2 |
| ТРИТЕРПЕНОВЫЕ ПРОИЗВОДНЫЕ ЛУПЕОЛЬНОГО ТИПА КАК ПРОТИВОВИРУСНЫЕ ПРЕПАРАТЫ | 2010 |
|
RU2561604C2 |
| ПРОИЗВОДНЫЕ 3-АМИНОПИРРОЛИДИНА В КАЧЕСТВЕ МОДУЛЯТОРОВ РЕЦЕПТОРОВ ХЕМОКИНОВ | 2003 |
|
RU2355679C2 |
| ЦИТОТОКСИЧЕСКИЕ ПЕПТИДЫ И ИХ КОНЪЮГАТЫ АНТИТЕЛО-ЛЕКАРСТВЕННОЕ СРЕДСТВО | 2012 |
|
RU2586885C2 |
| СОЕДИНЕНИЯ И КОМПОЗИЦИИ, ПРИМЕНЯЕМЫЕ ДЛЯ ЛЕЧЕНИЯ РАССТРОЙСТВ, СВЯЗАННЫХ С NTRK | 2016 |
|
RU2744974C2 |
| ПИРАЗОЛ-4-ИЛ-ГЕТЕРОЦИКЛИЛ-КАРБОКСАМИДНЫЕ СОЕДИНЕНИЯ И СПОСОБЫ ПРИМЕНЕНИЯ | 2012 |
|
RU2638552C2 |
| АГОНИСТЫ РЕЦЕПТОРА МЕЛАНОКОРТИНА | 2007 |
|
RU2411240C2 |
| НОВОЕ СОЕДИНЕНИЕ БИФЕНИЛА ИЛИ ЕГО СОЛЬ | 2016 |
|
RU2726622C2 |








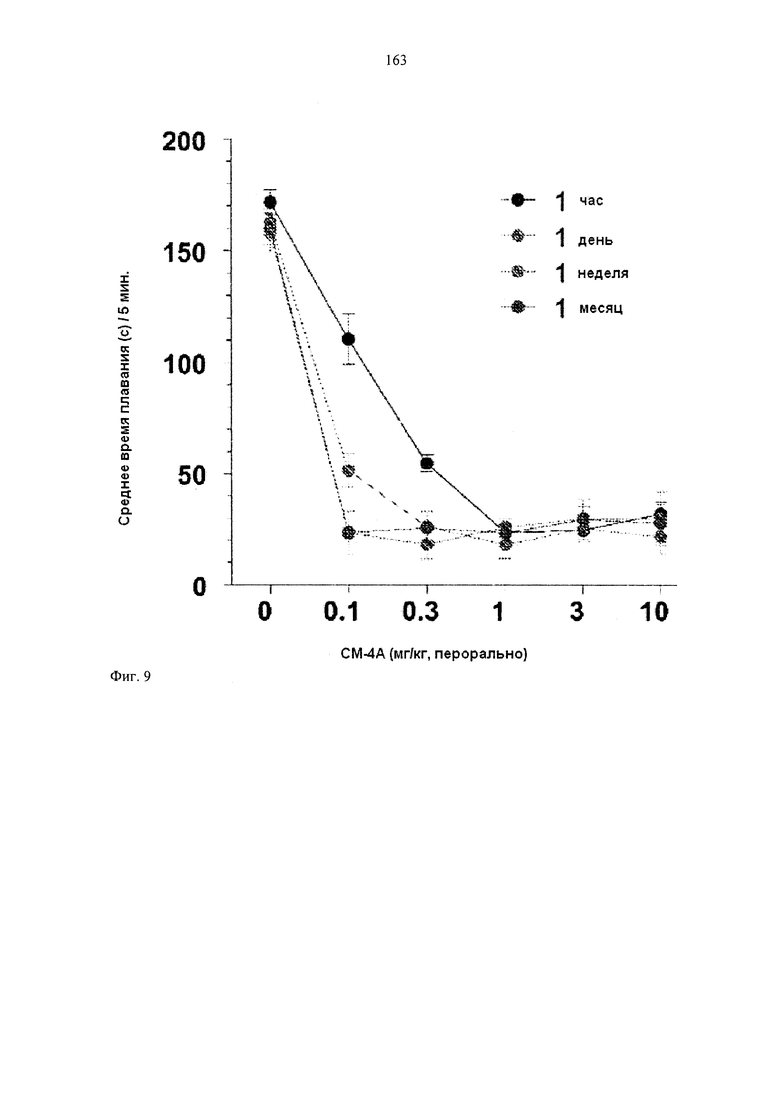
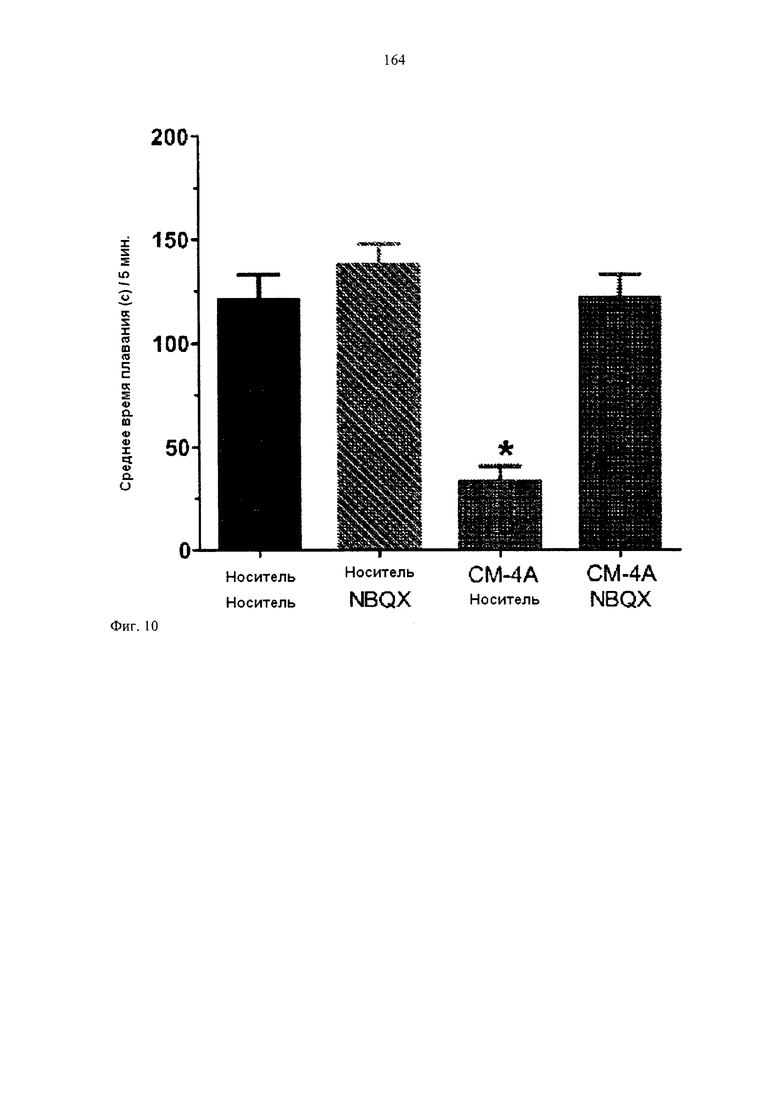






Изобретение относится к соединениям, обладающим улучшенной эффективностью модулирования активности рецептора НМДА, и их фармацевтически приемлемым солям. Такие соединения предназначены для применения при лечении заболеваний и расстройств, таких как заболевания и расстройства познавательной способности, когнитивной деятельности, и аналгезии, в частности для облегчения и/или уменьшения невропатической боли. 6 н. и 6 з.п. ф-лы, 16 ил., 4 табл., 24 пр.

1. Соединение, представленное структурой:

где
R11, R12 и R13, каждый независимо, выбраны из группы, состоящей из Н, галогена, С1-3алкокси и C1-3алкила (необязательно замещенного одним, двумя или тремя галогенами); при этом R11, R12 и R13 одновременно не являются Н,
X1 представляет собой ОН или NH2,
X2 представляет собой Н или ОН;
и его фармацевтически приемлемые соли и стереоизомеры.
2. Соединение по п. 1, отличающееся тем, что галоген в каждом случае независимо выбран из группы, состоящей из Cl, Br и F.
3. Соединение по п. 1, отличающееся тем, что указанное соединение значительно более эффективно при пероральном введении пациенту, по сравнению с пероральным введением пациенту пептидилового соединения, представленного структурой:

4. Соединение по п. 1, отличающееся тем, что X2 представляет собой ОН, а X1 представляет собой NH2.
5. Соединение по п. 1, отличающееся тем, что R11 и R13 представляют собой Н, a R12 выбран из группы, состоящей из F, Br, Cl, СН3, CF3 и -ОСН3.
6. Соединение, представленное структурой:
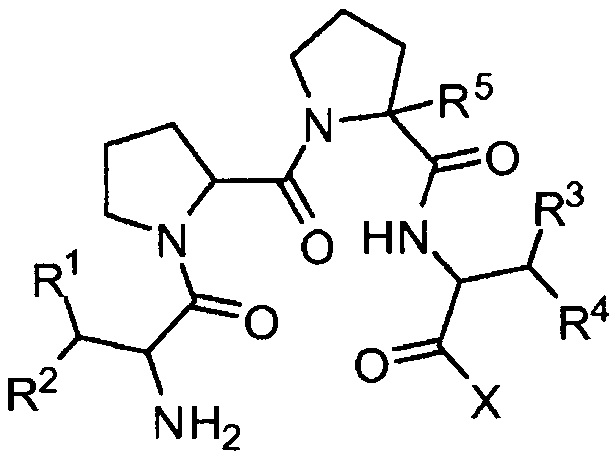
где:
R1 и R3 представляют собой -ОН, a R2 и R4 представляют собой метил;
R5 представляет собой -СН2-фенил (где указанный фенил замещен одним, двумя или тремя заместителями, выбранными из группы, состоящей из галогена, C1-3алкокси и C1-3алкила (необязательно замещенного одним, двумя или тремя галогенами)); и
X выбран из группы, состоящей из ORx и NRxRx, где RX в каждом случае независимо выбран из группы, состоящей из водорода и С1-С6алкила; его фармацевтически приемлемые соли и стереоизомеры.
7. Соединение по п. 6, представленное структурой:

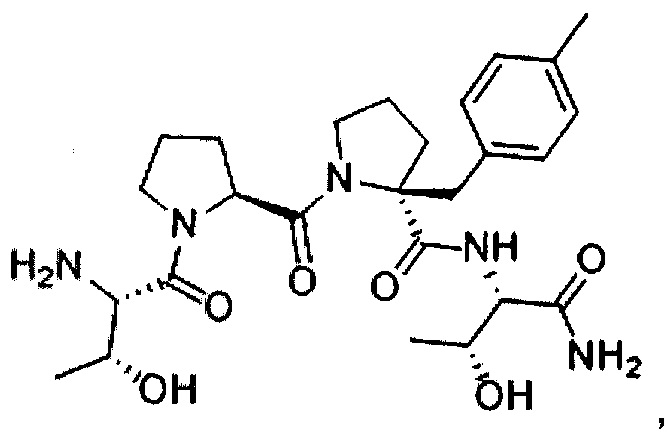

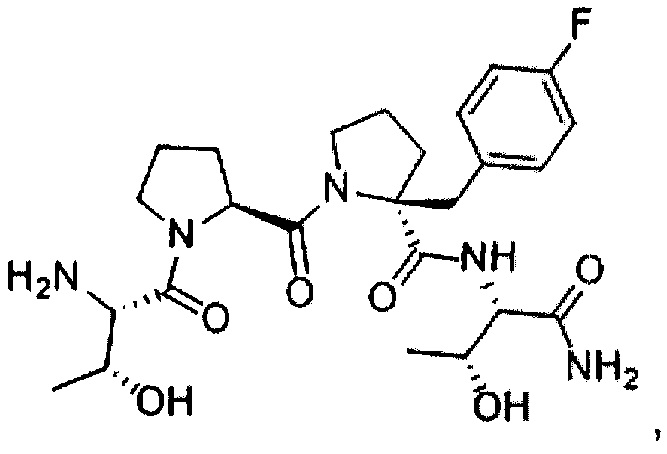

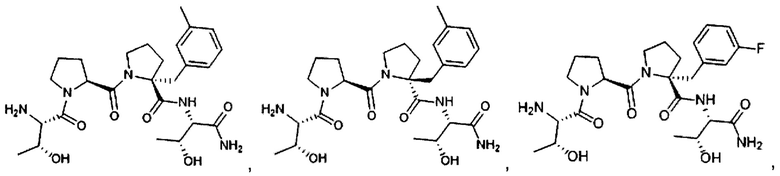
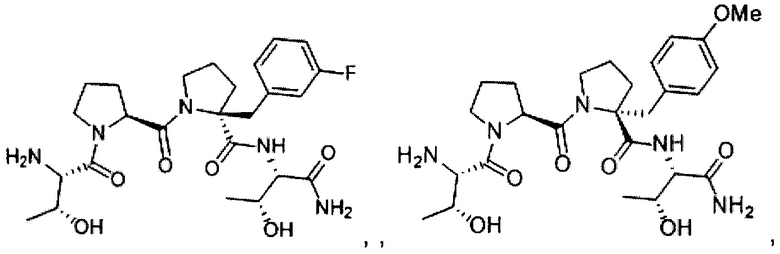



 или
или

8. Фармацевтическая композиция для модулирования рецептора НМДА, содержащая терапевтически эффективное количество соединения по любому из пп. 1-7 и фармацевтически приемлемый носитель.
9. Фармацевтическая композиция по п. 8, пригодная для инъекций.
10. Способ лечения депрессии, болезни Альцгеймера, расстройства дефицита внимания, СДВГ, шизофрении или тревоги у пациента, нуждающегося в этом, включающий введение указанному пациенту фармацевтически эффективного количества соединения по любому из пп. 1-7.
11. Способ лечения депрессии, болезни Альцгеймера, расстройства дефицита внимания, СДВГ, шизофрении или тревоги у пациента, нуждающегося в этом, включающий пероральное введение указанному пациенту фармацевтически эффективного количества соединения по любому из пп. 1-5.
12. Способ лечения депрессии, болезни Альцгеймера, расстройства дефицита внимания, СДВГ, шизофрении или тревоги у пациента, нуждающегося в этом, включающий подкожное или внутривенное введение указанному пациенту фармацевтически эффективного количества соединения по п. 7.
| РАЗЛИВНОЙ АВТОМАТ ДЛЯ НАПИТКА | 2005 |
|
RU2371375C2 |
| US 6902886 B1, 07.06.2005 | |||
| US 6897028 B1, 24.05.2005 | |||
| US 4683221 A, 28.07.1987 | |||
| US 20050176649 A1, 11.08.2005 | |||
| JP 3318622 B2, 26.08.2002 | |||
| WO 2007027559 A2, 08.03.2007 | |||
| WO 2011044089 A2, 14.04.2011 | |||
| RU 2011114982 A, 27.10.2012 | |||
| OHMORI T ET AL, "Isolation of Prolylendopeptidase-Inhibiting Peptides from Bovine Brain", BIOCHEMICAL AND BIOPHYSICAL RESEARCH COMMUNICATIONS, 1994, vol | |||
| Приспособление к тростильной машине для прекращения намотки шпули | 1923 |
|
SU202A1 |
| Аппарат для очищения воды при помощи химических реактивов | 1917 |
|
SU2A1 |

