Настоящее изобретение относится к новым производным имидазолидина формулы I

где A, E, Z, R1, R2, R3, R4 и R5 имеют указанные ниже значения. Соединения формулы I являются ценными фармацевтически активными соединениями, пригодными для лечения воспалительных заболеваний, например ревматоидного артрита, или аллергических заболеваний. Соединения формулы I являются ингибиторами адгезии и миграции лейкоцитов и/или антагонистами адгезионного рецептора VLA-4, относящегося к группе интегринов. Данные вещества пригодны для лечения заболеваний, вызываемых нежелательной степенью адгезии и/или миграции лейкоцитов или связанных с указанными процессами, а также заболеваний, в которых важное значение имеет межклеточное взаимодействие или взаимодействие клеток с матриксом, обусловленное взаимодействием рецепторов VLA-4 с их лигандами. Настоящее изобретение далее относится к способам получения соединений формулы I, их применению и к фармацевтическим препаратам, содержащим соединения формулы I.
Интегрины представляют группу адгезионных рецепторов, которые играют важную роль в процессах межклеточного связывания и связывания клеток с внеклеточным матриксом. Указанные вещества имеют αβ-гетеродимерную структуру, широко распространены в клетках и характеризуются высокой степенью эволюционной стабильности. Интегрины включают, например, рецептор фибриногена тромбоцитов, который предпочтительно взаимодействует с последовательностью RGD фибриногена, или рецептор витронектина остеокластов, который предпочтительно взаимодействует с последовательность RGD витронектина или остеопонтина. Интегрины можно разделить на три основные группы, включающие подсемейство β2, типичными представителями которого являются LFA-1, Mac-1 и р150/95, ответственные, в частности, за межклеточное взаимодействие иммунной системы, и подклассы β1 и β3, типичные представители которых опосредуют адгезию клеток к компонентам внеклеточного матрикса (Ruoslahti, Annu. Ran. Biochem. 1988, 57, 375). Интегрины подсемейства β1, именуемые также белками VLA (очень поздний (активационный) антиген), включают по крайней мере шесть рецепторов, которые специфически взаимодействуют с фибронектином, коллагеном и/или ламинином, представляющими лиганды. В семействе VLA интегрин VLA-4 (α4β1) является атипичным, так как он в основном ограничен лимфоидными и миелоидными клетками и отвечает за межклеточное взаимодействие указанных клеток с большим числом других клеток. VLA-4 опосредует, например, взаимодействие Т- и В-лимфоцитов с гепарин II-связывающим фрагментом фибронектина плазмы человека (FN). Связывание VLA-4 с гепарин II-связывающим фрагментом фибронектина плазмы основано главным образом на взаимодействии с последовательностью LDVP. В отличие от рецептора фибриногена или витронектина VLA-4 не является типичным RGD-связывающим интегрином (Kilger and Holzmann, J. Mol. Meth. 1995, 73, 347).
Лейкоциты, циркулирующие в крови, обычно обладают очень низким сродством к эндотелиальным клеткам, образующих выстилку кровеносных сосудов. Цитокины, высвобождаемые из воспаленных тканей, активируют эндотелиальные клетки и таким образом стимулируют экспрессию большого числа антигенов на поверхности клеток. Указанные антигены включают, например, адгезионные молекулы ELAM-1 (фактор адгезии эндотелиальных клеток 1, именуемый также Е-селектином), которые наряду с прочим связывают нейтрофилы, ICAM-1 (фактор межклеточной адгезии 1), взаимодействующие с LFA-1 (функционально-связанный антиген лейкоцитов 1) лейкоцитов, и VCAM-1 (фактор адгезии клеток сосудов 1), которые связывают разные лейкоциты, включая лимфоциты (Osborn et al., Cell 1989, 59, 1203). VCAM-1 подобно ICAM-1 является членом суперсемейства иммуноглобулиновых генов. VCAM-1 (ранее известный как INCAM-110) представляет адгезионную молекулу, которая индуцируется на эндотелиальных клетках воспалительными цитокинами, такими как TNF и IL-1, и липополисахаридами (LPS). Элисез и др. (Elices et al., Cell 1990, 60, 577) показали, что VLA-4 и VCAM-1 образуют пару рецептор-лиганд, которая опосредует связывание лимфоцитов с активированным эндотелием. VCAM-1 не связывается с VLA-4 из-за отсутствия в нем последовательности RGD, с которой обычно взаимодействует VLA-4 (Bergelson et al., Current Biology 1995, 5, 615). VLA-4 имеется также в других лейкоцитах, и механизм адгезии VCAM-1/VLA-4 опосредует адгезию лейкоцитов, не являющихся лимфоцитами. Таким образом, VLA-4 является конкретным примером рецептора β1-интегрина, который благодаря лигандам VCAM-1 и фибронектину играет важную роль в межклеточном взаимодействии и взаимодействии клеток с внеклеточным матриксом.
Индуцируемые цитокином адгезионные молекулы играют важную роль в рекрутинге лейкоцитов во внесосудистую ткань. Лейкоциты направляются в очаги воспаления адгезионными молекулами клеток, которые экспрессируются на поверхности эндотелиальных клеток и являются лигандами для поверхностных белков или белковых комплексов (рецепторов) лейкоцитов (термины "лиганд" и "рецептор" являются взаимозаменяемыми). Прежде чем мигрировать в синовиальную оболочку, лейкоциты крови должны связаться с эндотелиальными клетками. Так как VCAM-1 связывается с клетками, несущими интегрин VLA-4 (α4β1), такими как эозинофилы, Т- и В-лимфоциты, моноциты или нейтрофилы, указанная молекула и механизм действия VCAM-1/VLA-4 выполняют функцию рекрутинга клеток подобного типа из кровотока в очаги появления инфекции и воспаления (Elices et al., Cell 1990, 60, 577; Osborn, Cell 1990, 62, 3; Issekutz et al., J. Exp. Med. 1996, 183, 2175).
Механизм адгезии VCAM-1/VLA-4 связан с целым рядом физиологических и патологических процессов. Помимо индуцированного цитокином эндотелия, VCAM-1 экспрессируется также такими клетками, как миобласты, лимфоидные дендритные клетки и тканевые макрофаги, ревматоидная синовиальная оболочка, стимулированные цитокином нервные клетки, париетальные эпителиальные клетки капсулы Боумена, цилиндрический эпителий почек, воспаленные ткани при отторжении трансплантатов сердца и почки, а также тканями кишечника при реакции "трансплантат против хозяина". VCAM-1 экспрессируется также в тех областях ткани артериального эндотелия, которые соответствуют расположению первичных атеросклеротических бляшек в животной модели с использованием кроликов. Кроме того, VCAM-1 экспрессируется на фолликулярных дендритных клетках лимфатических узлов человека и обнаружен на стромальных клетках костного мозга мышей. Последнее открытие указывает на роль VCAM-1 в развитии В-клеток. Помимо клеток гемопоэтического происхождения VLA-4 обнаружен также в линиях клеток меланомы, и механизм адгезии VCAM-1/VLA-4 связан с образованием метастазов таких опухолей (Rice et al., Science 1989, 246, 1303).
Основная форма, в которой VCAM-1 существует in vivo на эндотелиальных клетках и которая является доминирующей формой in vivo, представляет VCAM-7D и имеет семь доменов иммуноглобулина. Аминокислотные последовательности доменов 4, 5 и 6 аналогичны доменам 1, 2 и 3. В другой форме, состоящей из шести доменов, определяемой в данном описании изобретения как VCAM-6D, четвертый домен удален альтернативным сплайсингом. VCAM-6D может также связывать VLA-4-экспрессирующие клетки.
Более подробная информация, относящаяся к VLA-4, VCAM-1, интегринам и адгезионным белкам, приведена, например, в статьях Kilger and Holzmann, J. Mol. Meth. 1995, 73, 347; Elices, Cell Adhesion in Human Disease, Wiley, Chichester 1995, p. 79; Kuijpers, Springer Semin. Immunopathol. 1995, 16, 379.
Что касается роли механизма VCAM-1/VLA-4 в процессах адгезии клеток, которые имеют важное значение в случае инфекций, например воспалений или атеросклероза, то предпринимаются попытки воздействовать на указанные процессы адгезии с целью лечения некоторых болезней, в частности воспалений (Osborn et al., Cell 1989, 59, 1203). Для этого используют моноклональные антитела против VLA-4. Известны моноклональные антитела (mABs) указанного типа, которые, будучи антагонистами VLA-4, блокируют взаимодействие между VCAM-1 и VLA-4. Так, например, антитела против VLA-4 mABs HP2/1 и HP1/3 ингибируют присоединение VLA-4-экспрессирующих клеток Рамоса (В-клеткоподобные клетки) к эндотелиальным клеткам пуповины человека и к VCAM-1-трансфецированным клеткам COS. Аналогичным образом антитела против VCAM-1 mAB 4B9 ингибируют адгезию клеток Рамоса, клеток Джурката (Т-клеткоподобные клетки) и клеток HL60 (гранулоцитоподобные клетки) к клеткам COS, трансфецированным генетическими конструкциями, вызывающими экспрессию VCAM-6D и VCAM-7D. Данные, полученные in vitro с использованием антител против субъединицы α4 VLA-4, показывают, что указанные антитела блокируют адгезию лимфоцитов к эндотелиальным клеткам синовиальной оболочки, которая играет важную роль в возникновении ревматоидного артрита (van Dinther-Janssen et al., J. Immunol. 1991, 147, 4207).
Выполненные in vivo эксперименты показывают, что искусственно (экспериментально) вызванный аутоиммунный энцефаломиелит можно ингибировать при помощи антител против α4 mAB. Миграцию лейкоцитов в очаг воспаления можно аналогичным образом блокировать моноклональным антителом против цепи α4 VLA-4. Воздействие на VLA-4-зависимый механизм адгезии с использованием антител было также исследовано в модели астмы с целью изучения роли VLA-4 в рекрутинге лейкоцитов в воспаленные ткани легкого (WO-A-93/13798). Введение антител против VLA-4 подавляет реакцию на поздней стадии заболевания и аллергическую реакцию воздушных путей у страдающих аллергией овец. Значение VLA-4 в качестве мишени для лечения астмы подробно описано в статье Metzger, Springer Semin. Immunopathol. 1995, 16, 467.
VLA-4-зависимый механизм адгезии клеток исследован также при помощи модели воспаления кишечника (IBD) у приматов. В данной модели, которая соответствует неспецифическому язвенному колиту у человека, введение антител против α4 значительное уменьшает острое воспаление.
Кроме того, можно показать, что VLA-4-зависимая адгезия клеток играет важную роль в нижеследующих клинических заболеваниях, включающих хронические воспалительные процессы, таких как ревматоидный артрит (Cronstein and Weismann, Arthritis Rheum. 1993, 36, 147; Elices et al., J. Clin. Invest. 1994, 93, 405), сахарный диабет (Yang et al., Proc. Natl. Acad. Sci. USA 1993, 90, 10494), системная красная волчанка (Takeuchi et al., J. Clin. Invest. 1993, 92, 3008), аллергические реакции замедленного типа (аллергия типа IV) (Elices et al., Clin. Exp. Rheumatol. 1993, 11, S77), рассеянный склероз (Yednock et al., Nature 1992, 356, 63), малярия (Ockenhouse et al., J. Exp. Med. 1992, 176, 1183), атеросклероз (O'Brien et al., J. Clin. Invest. 1993, 92, 945; Shih et al., Circ. Res. 1999, 84, 345), трансплантация (Isobe et al., Transplantation Proceedings 1994, 26, 867), разные злокачественные опухоли, например меланома (Renkonen et al., Am. J. Pathol. 1992, 140, 763), лимфома (Freedman et al., Blood 1992, 79, 206) и другие болезни (Albelda et al., J. Cell Biol. 1991, 114, 1059).
Взаимодействие VLA-4 с VCAM-1 и фибронектином связано с некоторыми патофизиологическими процессами в случае сердечно-сосудистых заболеваний. В исследуемой in vitro клеточной системе инфильтрованные нейтрофилы ингибируют сокращение клеток (негативная инотропия) кардиомиоцитов на 35%. Негативное инотропное действие нейтрофилов можно ингибировать антителом против α4 и нельзя ингибировать антителом против CD18 (Poon et al., Circ. Res. 1999, 84, 1245). Значение VLA-4 в патогенезе атеросклероза продемонстрировано на модели атеросклероза у мышей. Так, пептид CS-1, воздействующий на сайт связывания VLA-4 фибронектина, ингибирует рекрутинг лейкоцитов и жировые отложения в аорте, препятствуя таким образом образованию атеросклеротических бляшек у мышей с отсутствием рецептора LDL, получающих корм, способствующий развитию атеросклероза (Shih et al., Circ. Res. 1999, 84, 345). Используя пептид CS-1, можно далее продемонстрировать при помощи модели гетеротопической трансплантации сердца у кролика возможность значительного уменьшения васкулита трансплантата в результате подавления взаимодействия VLA-4 и фибронектина (Molossi et al., J. Clin. Invest. 1995, 95, 2601).
Блокирование VLA-4 приемлемыми антагонистами открывает широкие возможности для лечения разных воспалительных заболеваний, включая астму и IBD. Как указывалось выше, возможность использования антагонистов VLA-4 для лечения ревматоидного артрита заключается в том, что, прежде чем мигрировать в синовиальную оболочку, лейкоциты крови должны связаться с эндотелиальными клетками и что рецептор VLA-4 играет важную роль в такой адгезии. Выше было описано, что VCAM-1 индуцируется в эндотелиальных клетках воспалительными веществами (Osborn, Cell 1990, 62, 3; Stoolman, Cell 1989, 56, 907), и происходит рекрутинг разных лейкоцитов в очаги появления инфекции и воспаления. Т-клетки связываются с активированным эндотелием главным образом под действием механизмов адгезии LFA-1/ICAM-1 и VLA-4/VCAM-1 (Springer, Cell 1994, 76, 301). В большинстве синовиальных Т-клеток способность связывания VLA-4 с VCAM-1 увеличивается в случае ревматоидного артрита (Postigo et al., J. Clin. Invest. 1992, 89, 1445). Кроме того, обнаружена повышенная адгезия синовиальных Т-клеток к фибронектину (Laffon et al., J. Clin. Invest. 1991, 88, 546; Morales-Ducret et al., J. Immunol. 1992, 149, 1424). Таким образом, увеличивается экспрессия VLA-4 и возрастает его воздействие на Т-лимфоциты ревматоидной синовиальной мембраны. Ингибирование связывания VLA-4 с физиологическими лигандами VCAM-1 и фибронектином позволяет эффективно предотвращать или облегчать определенные воспалительные процессы. Данное положение также подтверждено экспериментами по воздействию антитела НР2/1 на крыс Льюиса, страдающих адъювантным артритом, при выполнении которых наблюдалось эффективное излечивание болезни (Barbadillo et al., Springer Semin. Immunopathol. 1995, 16, 427). Вещества, имитирующие пептид CS-1, которые содержат в молекуле звено аспарагиновой кислоты или ее производного и ингибируют связывание VLA-4 с последовательностью CS-1 матриксного белка фибронектина, описаны в заявке WO-A-00/02903. Таким образом, VLA-4 является важной молекулой-мишенью для лечения.
Вышеуказанные антитела против VLA-4 и их применение в качестве антагонистов VLA-4 описаны в заявках на патент WO-A-93/13798, WO-A-93/15764, WO-A-94/16094, WO-A-94/17828 и WO-A-95/19790. В заявках на патент WO-A-94/15958, WO-A-95/15973, WO-A-96/00581, WO-A-96/06108 и WO-A-96/20216 описаны соединения пептидов, являющиеся антагонистами VLA-4. Однако при использовании антител и соединений пептидов в качестве фармацевтических препаратов возникают трудности, связанные, например, с биологической недоступностью при пероральном введении, быстрым разрушением или иммуногенным действием при длительном введении, поэтому существует потребность в антагонистах VLA-4, обладающих необходимыми свойствами для лечения и профилактики разных заболеваний.
В заявках на патент WO-A-95/14008, WO-A-93/18057, US-A-5658935, US-A-5686421, US-A-5389614, US-A-5397796, US-A-5424293 и US-A-5554594 описаны замещенные гетероциклы с 5-членным кольцом, имеющие функциональную амино-, амидино- или гуанидиногруппу у N-конца молекулы и обладающие способностью ингибировать агрегацию тромбоцитов. В заявке на патент ЕР-А-796855 описаны гетероциклы, являющиеся ингибиторами резорбции костной ткани. В заявках на патент ЕР-А-842943, ЕР-А-842945 и ЕР-А-842944 указано, что соединения данного ряда и другие соединения ингибируют также адгезию лейкоцитов и являются антагонистами VLA-4.
В заявках на патент ЕР-А-903353, ЕР-А-905139, ЕР-А-918059, WO-99/23063, WO-A-99/24398, WO-A-99/54321 и WO-A-99/60015 описаны другие соединения, ингибирующе адгезию лейкоцитов и являющиеся антагонистами VLA-4. Однако свойства указанных соединений являются неудовлетворительными в разных отношениях, поэтому существует потребность в соединениях, обладающих улучшенными свойствами. В заявке на патент ЕР-А-918059 наряду с другими соединениями описаны производные имидазолидина, в которых имидазолидиновое кольцо связано в 1-положении с атомом углерода в 2-положении 2-(циклоалкилалкил)ацетиламиногруппы или 2-изобутилацетиламиногруппы. Однако в данной заявке не рассмотрены производные имидазолидина формулы I по настоящему изобретению, которые отличаются гораздо лучшими свойствами и, в частности, более сильным действием.
Настоящее изобретение относится к соединениям формулы I
где А означает циклопропилметил- или изобутил;
Е означает -СО-R6, -CO-H или -СН2-О-R7;
Z означает кислород или серу;
R1 означает водород или метил;
R2 означает фенил, пиридил или (С1-С4)алкил, причем алкильный остаток может быть замещен одним или несколькими атомами фтора и фенильный остаток может быть замещен одним или несколькими одинаковыми или разными заместителями, выбираемыми из группы, включающей (С1-С4)алкил, (С1-С4)алкокси, метилендиокси, этилендиокси, галоген, трифторметил и трифторметокси;
R3 и R4 означают метил или трифторметил;
R5 означает водород или (С1-С4)алкил, причем алкильный остаток может быть замещен одним или несколькими атомами фтора;
R6 означает гидроксил, (С1-С10)алкокси, фенил(С1-С8)алкокси, фенилокси, (С1-С8)алкилкарбонилокси(С1-С6)алкокси, фенилкарбонилокси(С1-С6)алкокси, фенил(С1-С6)алкилкарбонилокси(С1-С6)алкокси, (С1-С8)алкоксикарбонилокси(С1-С6)алкокси, фенилоксикарбонилокси(С1-С6)алкокси, фенил(С1-С6)алкоксикарбонилокси(С1-С6)алкокси, амино, моно((С1-С10)алкил)амино или ди((С1-С10)алкил)амино;
R7 означает водород или (С1-С4)алкил;
во всех стереоизомерных формах и их смесях во всех соотношениях и к физиологически толерантным солям указанных соединений.
Алкильные остатки могут иметь прямую или разветвленную цепь. Данное определение относится также к алкильным остаткам, имеющим заместители или используемым в качестве заместителей других остатков, например к фторалкильным остаткам, алкоксильным остаткам или алкоксикарбонильным остаткам. Примеры алкильных остатков включают метил, этил, н-пропил, изопропил (= 1-метилэтил = iC3H7), н-бутил, изобутил (= 2-метилпропил), втор-бутил (= 1-метилпропил), трет-бутил (= 1,1-диметилэтил), н-пентил, изопентил, трет-пентил, неопентил, н-гексил, 3-метилпентил, изогексил, неогексил, н-гептил, 2,3,5-триметилгексил, н-октил, н-нонил, н-децил. Предпочтительными алкильными остатками являются метил, этил, н-пропил, изопропил, н-бутил, изобутил, втор-бутил, трет-бутил. Один или несколько, например 1, 2, 3, 4 или 5, атомов водорода в алкильных остатках могут быть замещены атомами фтора. Примеры таких фторалкильных остатков включают трифторметил, 2,2,2-трифторэтил, пентафторэтил, гептафторизопропил. Замещенные алкильные остатки, например фенилалкильные остатки или фторалкильные остатки, могут быть замещены в любых требуемых положениях.
Фенильные остатки могут быть не замещены или моно-, или полизамещены, например, моно-, ди-, три-, тетра- или пентазамещены одинаковыми или разными заместителями. Замещенный фенильный остаток предпочтительно имеет один или два одинаковых или разных заместителя. Данное определение относится также к замещенным фенильным остаткам в группах, таких как фенилалкил, фенилкарбонил и т.д. Фенилалкильные остатки представляют, например, бензил, 1-фенилэтил или 2-фенилэтил, в частности бензил, которые также могут быть замещены.
В монозамещенных фенильных остатках заместитель может находиться в 2-положении, 3-положении или 4-положении. Дизамещенный фенил может быть замещен в 2,3-положении, 2,4-положении, 2,5-положении, 2,6-положении, 3,4-положении или 3,5-положении. В тризамещенных фенильных остатках заместители могут находиться в 2,3,4-положении, 2,3,5-положении, 2,4,5-положении, 2,4,6-положении, 2,3,6-положении или 3,4,5-положении. Если фенильный остаток имеет заместители, выбираемые из группы, включающей метилендиокси (-О-СН2-О-) и этилендиокси (-О-СН2-СН2-О-), то такой остаток предпочтительно имеет только один заместитель из указанной группы (при желании дополнительно к другим заместителям).
Примеры замещенных фенильных остатков, которые могут представлять R2, включают 2-метилфенил, 3-метилфенил, 4-метилфенил, 2,3-диметилфенил, 2,4-диметилфенил, 2,5-диметилфенил, 2,6-диметилфенил, 3,4-диметилфенил, 3,5-диметилфенил, 2,4,5-триметилфенил, 2,4,6-триметилфенил, 3,4,5-триметилфенил, 2-(н-бутил)фенил, 3-(н-бутил)фенил, 4-(н-бутил)фенил, 2-изобутилфенил, 3-изобутилфенил, 4-изобутилфенил, 3-трет-бутилфенил, 4-трет-бутилфенил, 2-метоксифенил, 3-метоксифенил, 4-метоксифенил, 2,3-диметоксифенил, 2,4-диметоксифенил, 2,5-диметоксифенил, 2,6-диметоксифенил, 3,4-диметоксифенил, 3,5-диметоксифенил, 2,4,5-триметоксифенил, 2,4,6-триметоксифенил, 3,4,5-триметоксифенил, 2-(н-бутокси)фенил, 3-(н-бутокси)фенил, 4-(н-бутокси)фенил, 2-изобутоксифенил, 3-изобутоксифенил, 4-изобутоксифенил, 2-трет-бутоксифенил, 3-трет-бутоксифенил, 4-трет-бутоксифенил, 2,3-метилендиоксифенил, 3,4-метилендиоксифенил, 2,3-этилендиоксифенил, 3,4-этилендиоксифенил, 2-фторфенил, 3-фторфенил, 4-фторфенил, 2,3-дифторфенил, 2,4-дифторфенил, 2,5-дифторфенил, 2,6-дифторфенил, 3,4-дифторфенил, 3,5-дифторфенил, 2,4,5-трифторфенил, 2,4,6-трифторфенил, 3,4,5-трифторфенил, 2,3,5,6-тетрафторфенил, 2,3,4,5,6-пентафторфенил, 2-хлорфенил, 3-хлорфенил, 4-хлорфенил, 2,3-дихлорфенил, 2,4-дихлорфенил, 2,5-дихлорфенил, 2,6-дихлорфенил, 3,4-дихлорфенил, 3,5-дихлорфенил, 2-бромфенил, 3-бромфенил, 4-бромфенил, 3-иодфенил, 4-иодфенил, 2-трифторметилфенил, 3-трифторметилфенил, 4-трифторметилфенил, 3,4-бис(трифторметил)фенил, 3,5-бис(трифторметил)фенил, 2-трифторметоксифенил, 3-трифторметоксифенил, 4-трифторметоксифенил и т.д. Однако в замещенных фенильных остатках разные заместители могут находиться в любом требуемом сочетании, таком как, например, в остатках 3-метокси-4-метилфенил, 4-фтор-3-метоксифенил, 3-фтор-4-метоксифенил, 3,5-дифтор-4-метоксифенил, 3-фтор-4,5-метилендиоксифенил, 3-фтор-4,5-этилендиоксифенил, 2-хлор-3-метилфенил, 3-хлор-4-метилфенил, 3-хлор-4-фторфенил и т.д.
Галоген представляет фтор, хлор, бром или иод, предпочтительно фтор или хлор.
Пиридил представляет 2-пиридил, 3-пиридил или 4-пиридил. В пиридильных остатках атом азота может быть также окислен, и соответствующее соединение формулы I может находиться в форме N-оксида пиридина, который также входит в объем настоящего изобретения.
Физиологически толерантные соли соединений формулы I являются нетоксичными или фармацевтически приемлемыми солями. Соединения формулы I, содержащие кислотные группы, например группу карбоновой кислоты, представляющую группу Е, могут находиться в форме солей щелочных или щелочно-земельных металлов, таких как соли натрия, соли калия, соли магния и соли кальция, или солей аммония, таких как, например, соли с физиологически толерантными ионами четвертичного аммония, кислотно-аддитивные соли с аммиаком и соли с физиологически толерантными органическими аминами, такими как метиламин, этиламин, триэтиламин, 2-гидроксиэтиламин, трис(2-гидроксиэтил)амин, α,α,α-трис(гидроксиметил)метиламин (трометамин), или с аминокислотами, в частности основными аминокислотами. Соли кислотного соединения формулы I с органическим амином могут содержать два компонента в соотношении 1:1, около 1:1 или в другом соотношении, например в соотношении от около 1:0,5 до около 1:4 (1 молекула соединения формулы I на 0,5-4 молекулы амина), в частности в соотношении от около 1:0,5 до около 1:2 (1 молекула соединения формулы I на 0,5-2 молекулы амина).
Соединения формулы I, содержащие основные группы, например пиридильную группу, могут находиться в форме кислотно-аддитивных солей, в частности солей с неорганическими кислотами, такими как хлористоводородная кислота, серная кислота или фосфорная кислота, или в форме солей с органическими карбоновыми кислотами или сульфоновыми кислотами, такими как уксусная кислота, лимонная кислота, бензойная кислота, малеиновая кислота, фумаровая кислота, винная кислота, метансульфоновая кислота или п-толуолсульфоновая кислота. Соединения, содержащие как кислотные, так и основные группы, могут также находиться в форме внутренних солей, цвиттерионов или бетаинов, которые также входят в объем настоящего изобретения.
Соли могут быть получены из соединений формулы I обычными способами, известными специалистам в данной области, например, в результате объединения с органической или неорганической кислотой или основанием в растворителе или разбавителе, либо из других солей в результате анионного или катионного обмена.
Соединения формулы I могут находиться в стереоизомерных формах. Благодаря наличию в соединениях формулы I асимметричных центров, не зависящих от других асимметричных центров, можно получить S-конфигурацию, R-конфигурацию или смеси R/S-конфигураций. Так, асимметричный атом углерода, к которому присоединен остаток R2, может иметь R-конфигурацию или S-конфигурацию, либо соединение формулы I может представлять смесь R/S-конфигураций по отношению к данному атому углерода. Аналогичным образом асмметричный атом углерода, к которому присоединены группа А и имидазолидиновое кольцо, может иметь R-конфигурацию или S-конфигурацию, либо соединение формулы I может представлять смесь R/S-конфигураций по отношению к данному атому углерода. Все другие асимметричные атомы углерода могут аналогичным образом иметь R-конфигурацию или S-конфигурацию, либо соединение формулы I может представлять смесь R/S-конфигураций по отношению к указанным атомам углерода. В смесях R/S-конфигураций отдельные стереоизомеры могут находиться в любых соотношениях, включая соотношение 1:1.
В объем данного изобретения входят все возможные стереоизомеры соединений формулы I, например чистые или в основном чистые энантиомеры и чистые или в основном чистые диастереомеры, а также смеси двух или более стереоизомерных форм, например смеси энантиомеров и/или диастереомеров во всех соотношениях. Таким образом, данное изобретение относится к энантиомерам в энантиомерно чистой форме в виде левовращающих и правовращающих изомеров, в форме рацематов или смесей двух энантиомеров во всех соотношениях. Данное изобретение относится также к диастереомерам в диастереомерно чистой форме или в форме смесей во всех соотношениях. Примеры отдельных стереоизомеров, входящих в объем данного изобретения, включают соединения формул Ia, Ib, Ic и Id.

Отдельные стереоизомеры при желании могут быть получены при использовании стереохимически однородных исходных веществ в процессе синтеза, при помощи стереоизбирательного синтеза или в результате разделения смеси обычными методами, такими как хроматография или кристаллизация, например в случае энантиомеров при помощи хроматографии с хиральными фазами. При необходимости до разделения стереоизомеров могут быть получены производные соединения. Смесь стереоизомеров можно разделить на стадии получения соединений формулы I, на стадии введения исходного вещества или получения промежуточного продукта в процессе синтеза.
Соединения формулы I по данному изобретению могут содержать подвижные атомы водорода, то есть могут находиться в разных таутомерных формах. Настоящее изобретение относится ко всем таутомерам соединений формулы I. В объем настоящего изобретения входят также сольваты и продукты присоединения или аддукты соединений формулы I, например аддукты с водой, то есть гидраты, или аддукты со спиртами или аминами. Данное изобретение далее относится к производным соединений формулы I, таким как, например, сложные эфиры, амиды, пролекарства и другие физиологически толерантные производные, и к активным метаболитам соединений формулы I. Данное изобретение, в частности, относится к пролекарствам соединений формулы I, которые необязательно являются фармацевтически активными in vitro, но в физиологических условиях in vivo превращаются в активные соединения формулы I. Специалистам в данном области известны приемлемые пролекарства для соединений формулы I, то есть химически модифицированные производные соединений формулы I, обладающие должным образом улучшенными свойствами. Более подробно пролекарства описаны, например, в статьях Fleisher et al., Advanced Drug Delivery Reviews 19 (1996) 115; Design of Prodrugs, H. Bundgaard, Ed., Elsevier, 1985; H. Bundgaard, Drugs of the Future 16 (1991) 443. Приемлемыми пролекарствами для соединений формулы I являются, в частности, сложноэфирные пролекарства, амидные пролекарства, альдегидные пролекарства и спиртовые пролекарства на основе групп карбоновой кислоты, например группы карбоновой кислоты, представляющей группу Е. Так, соединения формулы I, в которых группа Е означает гидроксиметил, алкоксиметил или формил и которые являются антагонистами VLA-4 in vivo, являются пролекарствами соединений формулы I, в которых группа Е означает гидроксикарбонил. В качестве примеров сложноэфирных пролекарств и амидных пролекарств можно привести сложные (С1-С4)алкиловые эфиры, такие как метиловые эфиры, этиловые эфиры, изопропиловые эфиры, изобутиловые эфиры, замещенные алкиловые эфиры, такие как гидроксиалкиловые эфиры, ацилоксиалкиловые эфиры, аминоалкиловые эфиры, ациламиноалкиловые эфиры, диалкиламиноалкиловые эфиры, незамещенные амиды или N-(C1-C4)алкиламиды, такие как метиламиды или этиламиды.
В качестве примеров соединений формулы I по данному изобретению можно привести нижеследующие соединения формул Ie и If, которые имеют S-конфигурацию в положении атома углерода, несущего группу А, и S-конфигурацию в положении атома углерода, несущего группу R2, если R2 означает фенил или пиридил, и имеют R-концигурацию в положении атома углерода, несущего группу R2, если R2 означает метил. Настоящее изобретение относится также к физиологически толерантным солям соединений формул Ie и If, таким как соли металлов или соли с катионами органического аммония соединений формул Ie и If, которые содержат группу карбоновой кислоты, или к кислотно-аддитивным солям соединений формул Ie и If, содержащим пиридильные остатки, таким как гидрохлориды.

Соединения формул Ie и If:
Отдельные структурные элементы в соединениях формулы I по данному изобретению предпочтительно имеют нижеследующие значения, не зависящие друг от друга.
R2 предпочтительно означает (С1-С4)алкил, который может быть замещен одним или несколькими атомами фтора, пиридил, незамещенный фенил или фенил, замещенный метилендиокси-остатком или этилендиокси-остатком, и фенил, замещенный одной или двумя (С1-С4)алкоксильными группами. Алкильная группа, представляющая R2, которая может быть необязательно замещена фтором, является, в частности, одной из групп, включающих метил, этил, изопропил, трифторметил и 2,2,2-трифторэтил. Алкоксильные заместители в фенильной группе, представляющие R2, являются, в частности, метоксильными группами. Более предпочтительно R2 означает метил, пиридил, незамещенный фенил или фенил, замещенный метилендиокси-остатком или этилендиокси-остатком, и фенил, замещенный одной или двумя метоксильными группами. Особенно предпочтительно R2 означает метил, незамещенный фенил или пиридил.
R3 и R4 могут иметь одинаковые или разные значения. Обе группы R3 и R4 предпочтительно имеют одинаковые значения. В одном варианте осуществления настоящего изобретения R3 и R4 оба означают метил. В другом варианте осуществления настоящего изобретения R3 и R4 оба означают трифторметил.
Алкильная группа, представляющая R5, которая может быть замещена одним или несколькими атомами фтора, предпочтительно является метильной группой, этильной группой или трифторметильной группой. R5 предпочтительно означает (С1-С4)алкил, который может быть замещен одним или несколькими атомами фтора. Более предпочтительно R5 означает метил или трифторметил и особенно предпочтительно метил.
R6 предпочтительно означает гидроксил, (С1-С6)алкокси, фенил(С1-С4)алкокси, фенилокси или амино (NH2), более предпочтительно гидроксил, (С1-С6)алкокси или амино, особенно предпочтительно гидроксил или (С1-С6)алкокси, наиболее предпочтительно гидроксил или (С1-С4)алкокси, в частности гидроксил.
R7 предпочтительно означает водород или (С1-С3)алкил, более предпочтительно водород или метил, особенно предпочтительно водород.
Е предпочтительно означает -CO-R6, -CO-H, -CH2-OH или -CH2-OCH3, более предпочтительно -CO-R6, -CH2-OH или -СН2-ОСН3, особенно предпочтительно -CO-R6 или -СН2-ОН, наиболее предпочтительно -СООН, -СООС2Н5, -COOiC3Н7 или -СН2-ОН, в частности -СООН.
В одном варианте осуществления настоящего изобретения Z означает серу, в другом варианте осуществления изобретения Z означает кислород.
В одном варианте осуществления настоящего изобретения А означает изобутильный остаток (2-метилпропильный остаток; (СН3)2СН-СН2-), в другом варианте осуществления изобретения А означает циклопропилметильный остаток (циклопропил-СН2-). Кроме того, в одном варианте осуществления настоящего изобретения R1 означает водород и в другом варианте осуществления изобретения R1 означает метил.
Предпочтительными соединениями формулы I являются соединения, которые имеют однородную конфигурацию в положении одного или нескольких хиральных центров, например в положении атома углерода, несущего остаток R2, и/или в положении атома углерода, несущего остаток А и имидазолидиновый остаток, то есть предпочтительны соединения, имеющие однородную или по существу однородную конфигурацию в положении одного или нескольких хиральных центров, в частности R-конфигурацию или S-конфигурацию, и не представляющие смесь R/S-конфигураций. Однако, как отмечалось, отдельные хиральные центры в соединениях формулы I могут независимо друг от друга иметь R-конфигурацию или S-конфигурацию и одинаковые или разные конфигурации. Особенно предпочтительными соединениями формулы I являются соединения, в которых атом углерода, несущий остаток А и имидазолидиновый остаток, имеет S-конфигурацию, то есть конфигурацию стереоцентра, представленную в формулах Ia и Ib. Особенно предпочтительными соединениями формулы I являются также соединения, в которых атом углерода, несущий группу R2, имеет конфигурацию, представленную в формулах Ia и Ic. Если R2 означает фенил, замещенный фенил или пиридил, в указанных предпочтительных соединениях атом углерода, несущий группу R2, имеет S-конфигурацию, если R2 означает метил, этил или изобутил, указанный атом имеет R-конфигурацию. Наиболее предпочтительными соединениями формулы I являются соединения, в которых два вышеуказанных стереоцентра имеют конфигурации, представленные в формуле Ia.
Предпочтительными соединениями формулы I являются соединения, в которых один или несколько остатков имеют предпочтительные или специальные значения, выбираемые из приведенных значений, причем настоящее изобретение относится ко всем комбинациям предпочтительных и/или специальных значений остатков. Примеры предпочтительных соединений включают, например, соединения, в которых R1, R3, R4 и R5 одновременно означают метил и А означает изобутил; соединения, в которых R1, R3, R4 и R5 одновременно означают метил и А означает циклопропилметил; соединения, в которых R1 означает метил, R3 и R4 означают трифторметил, R5 означает метил и А означает изобутил; соединения, в которых R1 означает метил, R3 и R4 означают трифторметил, R5 означает метил и А означает циклопропилметил; соединения, в которых R1 означает водород, R3, R4 и R5 означают метил и А означает изобутил; соединения, в которых R1 означает водород, R3, R4 и R5 означают метил и А означает циклопропилметил; соединения, в которых R1 означает водород, R3 и R4 означают трифторметил, R5 означает метил и А означает изобутил; соединения, в которых R1 означает водород, R3 и R4 означают трифторметил, R5 означает метил и А означает циклопропилметил, и другие группы имеют значения, указанные выше в формуле I, а также предпочтительные или специальные значения, выбираемые из соответствующих определений.
Более предпочтительными соединениями являются, например, соединения формулы I, в которой:
А означает циклопропилметил- или изобутил;
Е означает -CO-R6 или -СН2-ОН;
Z означает кислород;
R1 означает водород или метил;
R2 означает пиридил, незамещенный фенил, фенил, замещенный метилендиокси-остатком или этилендиокси-остатком, фенил, замещенный одной или двумя (С1-С4)алкоксильными группами, или (С1-С4)алкил, который может быть замещен одним или несколькими атомами фтора;
R3 и R4 означают метил;
R5 означает метил;
R6 означает гидрокси, (С1-С6)алкокси, фенил(С1-С4)алкокси, фенилокси или амино;
во всех стереоизомерных формах и их смесях во всех соотношениях и физиологически толерантные соли указанных соединений.
Особенно предпочтительными соединениями являются, например, соединения формулы I, в которой
А означает циклопропилметил- или изобутил;
Е означает -СООН, -СООС2Н5, -COOiC3Н7 или -СН2-ОН;
Z означает кислород;
R1 означает метил;
R2 означает пиридил, незамещенный фенил, фенил, замещенный метилендиокси-остатком или этилендиокси-остатком, фенил, замещенный одной или двумя метоксильными группами, или (С1-С4)алкил, который может быть замещен одним или несколькими атома фтора;
R3 и R4 означают метил;
R5 означает метил;
во всех стереоизомерных формах и их смесях во всех соотношениях и физиологически толерантные соли указанных соединений.
Наиболее предпочтительными соединениями являются, например, соединения формулы I, в которой
А означает циклопропилметил- или изобутил;
Е означает -СООН, -СООС2Н5, -COOiC3Н7 или -СН2-ОН;
Z означает кислород;
R1 означает метил;
R2 означает незамещенный фенил, пиридил, метил или 2,2,2-трифторэтил;
R3 и R4 означают метил;
R5 означает метил;
во всех стереоизомерных формах и их смесях во всех соотношениях и физиологически толерантные соли указанных соединений.
Еще более предпочтительными соединениями являются, например, соединения формулы I, в которой
А означает циклопропилметил- или изобутил;
Е означает -СООН, -СООС2Н5, -COOiC3Н7 или -СН2-ОН;
Z означает кислород;
R1 означает метил;
R2 означает незамещенный фенил, пиридил или метил;
R3 и R4 означают метил;
R5 означает метил;
во всех стереоизомерных формах и их смесях во всех соотношениях и физиологически толерантные соли указанных соединений.
Все вышеуказанные определения подгрупп в соединениях формулы I аналогичным образом относятся к соединениям формулы I, в которой R3 и R4 оба означают трифторметил вместо метила. Так, например, особенно предпочтительными соединениями являются также соединения формулы I, в которой
А означает циклопропилметил- или изобутил;
Е означает -СООН, -СООС2Н5, -COOiC3Н7 или -СН2-ОН;
Z означает кислород;
R1 означает метил;
R2 означает незамещенный фенил, пиридил или метил;
R3 и R4 означают трифторметил;
R5 означает метил;
во всех стереоизомерных формах и их смесях во всех соотношениях и физиологически толерантные соли указанных соединений.
Соединения формулы I можно получить конденсацией соединения формулы II

с соединением формулы III
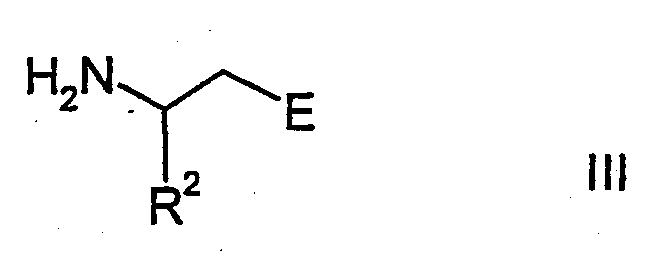
где группы А, E, Z, R1, R2, R3, R4 и R5 в формулах II и III имеют указанные выше значения, альтернативно функциональные группы могут быть защищены или могут находиться в форме предшественников, и G означает гидроксикарбонил, (С1-С6)алкоксикарбонил или производные активированной карбоновой кислоты, такие как, например, хлорангидриды кислот или активные сложные эфиры.
В процессе конденсации соединений формул II и III группу карбоновой кислоты, присутствующую в соединении, но не участвующую в реакции конденсации, как правило, необходимо защитить обратимой защитной группой, в результате чего она приобретает форму приемлемого (С1-С6)алкилового сложного эфира, такого как трет-бутиловый эфир или бензиловый эфир. При необходимости получения соединений формулы I, в которой группа Е представляет, например, гидроксикарбонильную группу или группу, получаемую из гидроксикарбонильной группы, остаток Е в соединениях формулы III сначала может быть защищенной гидроксикарбонильной группой, с которой снимают защиту после конденсации соединений формул II и III, и/или требуемую конечную группу Е можно синтезировать в результате выполнения еще одной или нескольких стадий.
Предшественниками функциональных групп являются группы, которые можно превратить в требуемую функциональную группу обычными способами синтеза, известными специалистам в данной области. Например, предшественником может быть цианогруппа, которую можно превратить в группу карбоновой кислоты гидролизом. Предшественником может быть также спиртовая группа, которую можно окислить в альдегидную группу. Примеры защитных групп, которые можно ввести в соединение до выполнения отдельной реакции или последовательности реакций и затем удалить, приведены выше.
Для конденсации соединений формул II и III можно успешно использовать способы связывания, применяемые в химии пептидов, которые хорошо известны специалистам в данной области (см., например, Houben-Weyl, Methoden der Organischen Chemie [Methods of Organic Chemistry], Volume 15/1 and 15/2, Georg Thieme Verlag, Stuttgart, 1974). Приемлемые конденсирующими агентами или связующими реагентами являются, например, карбонилдиимидазол, карбодиимиды, такие как дициклогексилкарбодиимид (DCC) или диизопропилкарбодиимид, тетрафторборат О-((циано(этоксикарбонил)метилен)амино)-N,N,N',N'-тетраметилурония (TOTU) или пропилфосфоновый ангидрид (РРА). Конденсацию выполняют в стандартных условиях, хорошо известных специалистам в данной области. Указанные реакции обычно осуществляют в инертном растворителе или разбавителе, например в апротонном растворителе, таком как N,N-диметилформамид (ДМФ), диметилсульфоксид (ДМСО), N-метил-2-пирролидон (NMP), тетрагидрофуран (ТГФ) или диметоксиэтан (DME). В зависимости от типа конденсации, выполняемой в каждом отдельном случае, хорошие результаты могут быть получены при использовании основания, такого как третичный амин, или вспомогательных реагентов, например N-гидроксильного соединения, такого как 1-гидроксибензотриазол (HOBT). Обработку реакционной смеси и очистку продукта можно производить обычными способами. После выполнения конденсации имеющиеся защитные группы удаляют приемлемым способом. Например, бензильные группы в бензиловых эфирах можно удалить каталитическим гидрированием и защитные трет-бутильные группы можно удалить, обрабатывая продукт приемлемой кислотой. Соединения формулы I можно также получить в результате поэтапного синтеза в твердой фазе известными способами, благодаря чему можно ввести отдельные структурные элементы молекулы в разные последовательности.
Аминосоединения формулы III можно приобрести коммерческим путем или синтезировать хорошо известными стандартными способами из исходных соединений, которые можно приобрести коммерческим путем или получить способами, описанными в научной литературе. Например, оптически активные 3-замещенные 3-аминопропионовые кислоты формулы III или их сложные эфиры, в частности сложные эфиры 3-фенил-3-аминопропионовой кислоты, можно получить из соответствующих 3-замещенных акриловых кислот, которые, в свою очередь, можно получить из соответствующих альдегидов. 3-Замещенные акриловые кислоты превращают при помощи оксалилхлорида в хлорангидриды кислот, которые при помощи спирта превращают в сложные эфиры, например, используя трет-бутанол, осуществляют превращение в сложные трет-бутиловые эфиры. Чтобы ввести аминогруппу, сложные эфиры подвергают взаимодействию с литиевой солью оптически активного амина, например с литиевой солью (R)-(+)-N-бензил-N-(1-фенилэтил)амина, после чего бензильную группу и фенилэтильную группу в полученном 3-замещенном трет-бутил-3-(N-бензил-N-(1-фенилэтил)амино)пропионате удаляют каталитическим гидрированием. Для получения соединений формулы III, в которой Е представляет гидроксиметильную группу СН2ОН или этерифицированную гидроксиметильную группу, можно использовать полученные в результате реакции конденсации 3-замещенные 3-аминопропанолы или их простых эфиры, которые получают из 3-замещенных 3-аминопропионовых кислот или их сложных эфиров восстановлением кислотной или сложноэфирной группы, например, из сложного этилового эфира или трет-бутилового эфира, используя алюмогидрид лития или алюмогидрид лития/трихлорид алюминия.
Соединения формулы II можно получить, осуществляя взаимодействие соединений формулы IV
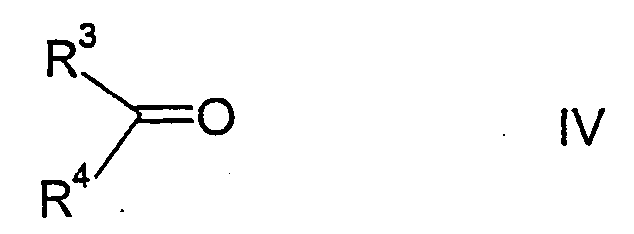
с карбонатом аммония и цианидом калия при помощи реакции Бухерера, в результате чего образуются соединения формулы V
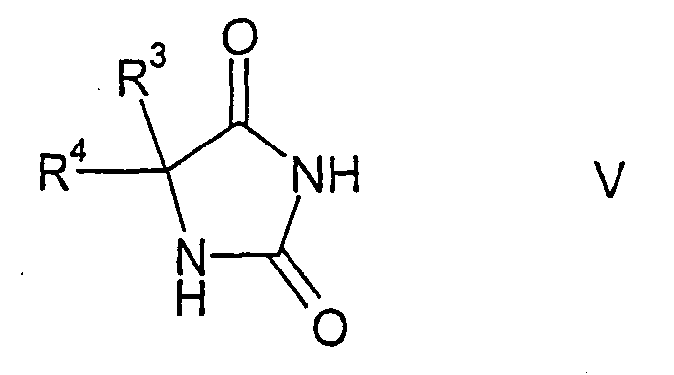
которые затем подвергают взаимодействию с алкилирующим реагентом формулы LG-CHA-G, позволяющим ввести в молекулу остаток формулы -CHA-G, в результате чего образуются соединения формулы VI
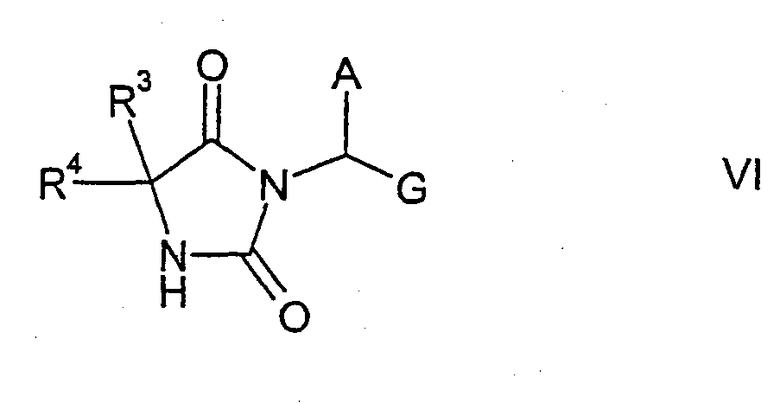
где А, R3, R4 и G имеют указанные выше значения. Соединения формулы VI подвергают взаимодействию со вторым алкилирующим реагентом формулы VII
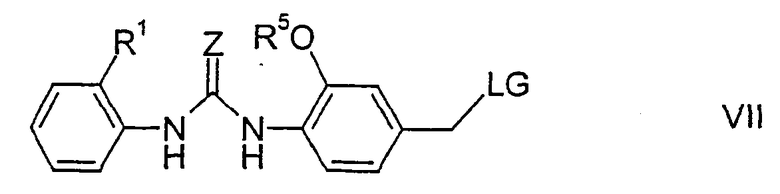
где Z, R1 и R5 имеют указанные выше значения, что позволяет получить соответствующие соединения формулы II. Группа LG представляет нуклеофильно замещаемую удаляемую группу, например галоген, в частности хлор или бром, или сульфонилокси, такой как тозилокси, метилсульфонилокси или трифторметилсульфонилокси.
Соединения формулы II можно также получить, осуществляя взаимодействие соединения формулы VI с реагентом формулы 4-(PG-NH)-C6H3-(OR5)-CH2-LG, где LG означает вышеуказанную нуклеофильно замещаемую удаляемую группу, с образованием соединения формулы VIII,

где А, G, R3, R4 и R5 имеют указанные выше значения и PG представляет аминозащитную группу, например трет-бутоксикарбонил или бензилоксикарбонил. После удаления защитной группы PG соединения формулы II получают, осуществляя взаимодействие аминогруппы H2N с фенилизоцианатом, фенилизотиоцианатом, 2-метилфенилизоцианатом или 2-метилфенилизотиоцианатом. Аналогично соединениям формулы VIII указанные соединения можно получить и использовать в процессе синтеза, в котором группу PG-NH- соединения формулы VIII заменяют группой, являющейся предшественником аминогруппы, которую превращают в аминогруппу на последующей стадии реакции. Например, соединение формулы VI можно сначала подвергнуть взаимодействию с нитросоединением формулы 4-O2N-C6H3(OR5)-CH2-LG с образованием соединения, соответствующего соединению формулы VIII, после чего нитрогруппу можно превратить в аминогруппу, например, каталитическим гидрированием, после чего аминогруппу можно превратить в требуемое соединение формулы II, используя фенилизоцианат, фенилизотиоцианат, 2-метилфенилизоцианат или 2-метилфенилизотиоцианат.
Отдельные стадии синтеза соединений формулы I обычно осуществляют обычными способами, известными специалистам в данной области. Как указывалось выше, в некоторых случаях на любых стадиях синтеза соединений формулы I можно временно блокировать функциональные группы, которые могут вызвать побочные реакции или нежелательные взаимодействия, при помощи защитной группы, пригодной для данного способа синтеза, способом, известным специалистам в данной области.
Соединения формулы I можно также получить описанными ниже способами. N-замещенные аминокислоты или предпочтительно их сложные эфиры, например метиловые эфиры, этиловые эфиры, трет-бутиловые эфиры или бензиловые эфиры, получаемые стандартными способами, в частности соединения формулы IX,
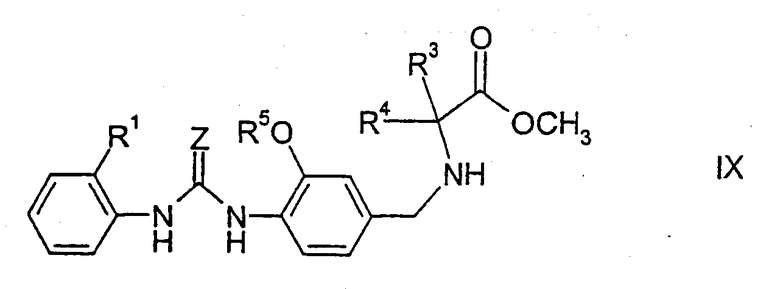
где Z, R1, R3, R4 и R5 имеют указанные выше значения, подвергают взаимодействию с изоцианатом формулы Х

где А, Е и R2 имеют указанные выше значения, который получают стандартными способами из соответствующего соединения, содержащего группу H2N вместо изоцианатной группы, с образованием производных мочевины, например соединений формулы XI,
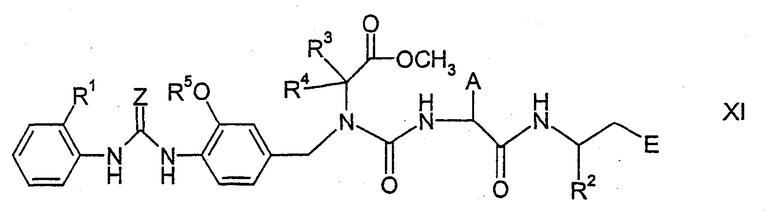
где все символы имеют указанные выше значения. Соединения формулы XI затем можно подвергнуть циклизации, нагревая с кислотой, в результате чего образуются соединения формулы I. Циклизацию соединений формулы XI в соединения формулы I можно выполнить путем обработки основаниями в инертных растворителях, например гидридом натрия в апротонном растворителе, таком как диметилформамид. Как указывалось выше, во время выполнения реакций функциональные группы могут быть защищены.
Соединения формулы I можно также получить, осуществляя взаимодействие соединения формулы IX с изоцианатом формулы XII
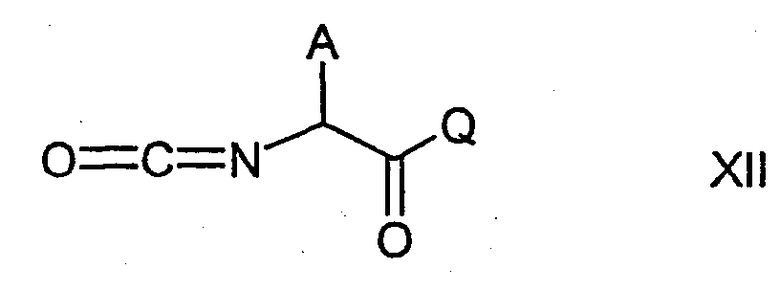
где А имеет указанные выше значения и Q означает, например, алкоксильную группу, например (С1-С4)алкоксильную группу, такую как метокси, этокси или трет-бутокси, или (С6-С14)арил(С1-С4)алкоксильную группу, такую как бензилокси. В результате выполнения данной реакции получают соединение формулы XIII
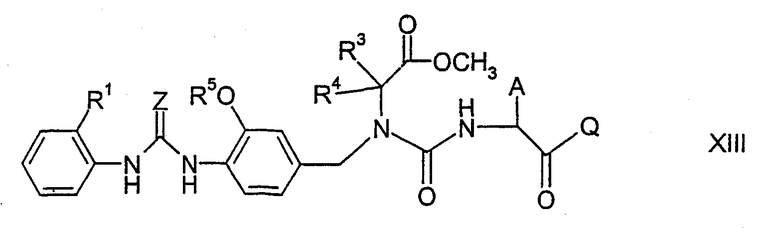
где А, Q, Z, R1, R3, R4 и R5 имеют указанные выше значения, которое затем подвергают циклизации в присутствии кислоты или основания, как было описано выше для циклизации соединений формулы XI, с образованием соединения формулы XIV
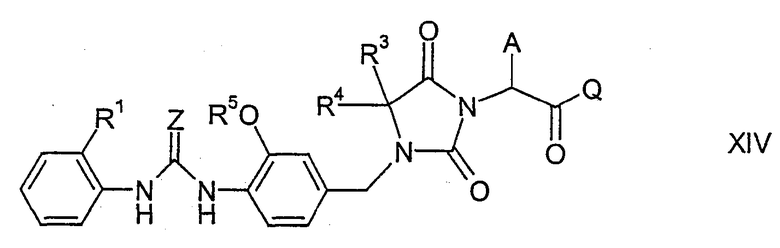
где А, Q, Z, R1, R3, R4 и R5 имеют указанные выше значения. Например, гидролизуя соединение формулы XIV, группу CO-Q можно превратить в группу карбоновой кислоты СООН. Если циклизация соединения формулы XIII в соединение формулы XIV протекает с использованием кислоты, то группу СО-Q можно превратить в группу СООН одновременно с циклизацией. Соединение формулы I получают, осуществляя последующее связывание с соединением формулы III, как было описано выше для связывания соединений формулы II и III. В процессе синтеза функциональные группы могут быть защищены или могут находиться в форме предшественников.
Еще один способ получения соединений формулы I заключается в том, что соединения формулы XV
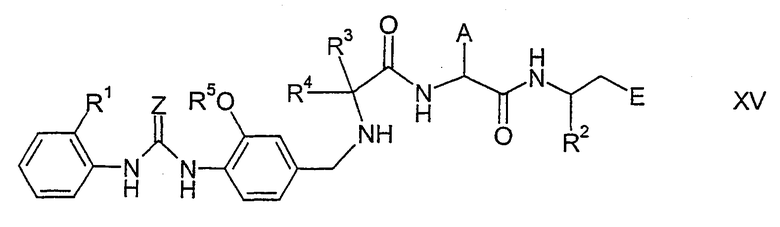
где символы имеют указанные выше значения, подвергают взаимодействию с фосгеном или их эквивалентами (аналогично способу, описанному S. Goldschmidt and M. Wick, Liebigs, Ann. Chem. 575 (1952), 217 and C. Tropp. Chem. Ber. 61 (1928), 1431).
Соединения формулы I можно также получить, связывая соединение формулы XVI

где R3 и R4 имеют указанные выше значения и PG представляет аминозащитную группу, такую как, например, бензилоксикарбонильная группа, с соединением формулы XVII

где А имеет указанные выше значения и Q' представляет защищенную гидроксильную группу карбоновой кислоты, например алкоксильную группу, такую как трет-бутокси, с образованием соединения формулы XVIII,
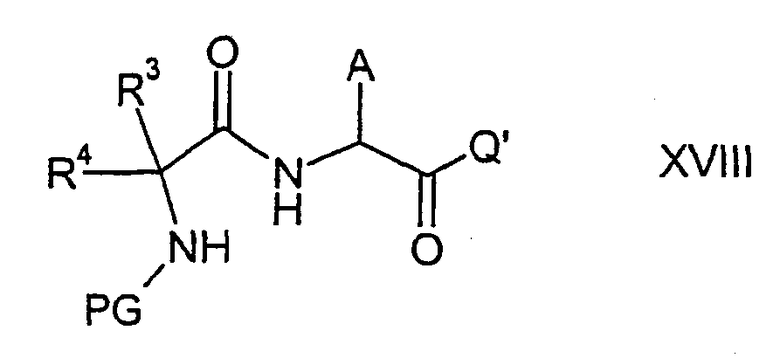
где A, R3, R4, PG и Q' имеют указанные выше значения. В соединении формулы XVIII защитную группу PG можно избирательно удалить от аминогруппы, например гидрированием в случае бензилоксикарбонильной группы, и ввести группу СО в место замыкания кольца с образованием соединения формулы XIX

где А, R3, R4 и Q' имеют указанные выше значения. Для введения карбонильной группы можно использовать, например, фосген или эквивалент фосгена, такой как дифосген (аналогично вышеописанному взаимодействию соединений формулы XV). При превращении соединения формулы XVIII в соединение формулы XIX в качестве промежуточного продукта, например, может образоваться или быть специально получен изоцианат. Превращение соединения формулы XVIII в соединение формулы XIX можно осуществлять в одну или нескольких стадий. Например, сначала можно ввести карбонильную группу и затем на отдельной стадии можно осуществить циклизацию в присутствии основания, такого как гидрид натрия, как описано для вышеуказанных способов циклизации. Соединения формулы XVIII, в которых PG представляет бензилоксикарбонильную группу, можно сразу же превратить в соединения формулы XIX без использования синтетического структурного элемента, такого как фосген, для введения карбонильной группы. Если соединения формулы XVIII, в которой PG представляет бензилоксикарбонил, обработать основанием, таким как гидрид натрия, можно сразу же получить соединения формулы XIX.
Как описано выше для соединений формулы VI, соединения формулы XIX затем можно алкилировать в положении группы NH, используя реагент формулы VII, и после превращения защищенной группы карбоновой кислоты CO-Q' в группу карбоновой кислоты СООН требуемые соединения формулы I можно синтезировать способом, описанным выше для соединений формул VI и II. В процессе данного синтеза функциональные группы также могут быть защищены или могут находиться в форме предшественников.
Соединения формулы I можно далее получить, осуществляя взаимодействие соединения формулы ХХ
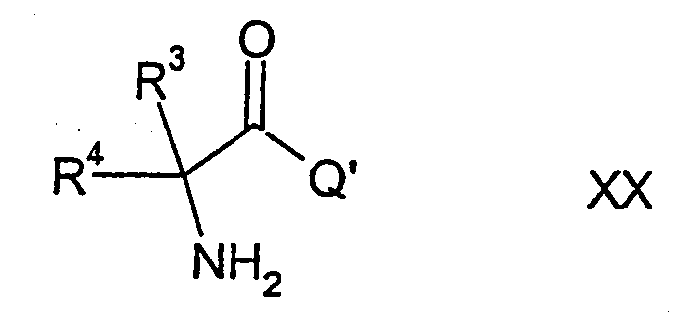
где R3, R4 и Q' имеют указанные выше значения, с изоцианатом формулы XII, в результате чего образуется соединение формулы XXI,

где А, R3, R4, Q ии Q' имеют указанные выше значения. Соединение формулы XXI затем циклизуют, обрабатывая сильной кислотой, например полуконцентрированной хлористоводородной кислотой, с образованием соединения формулы XXII.

Соединения формулы XXII можно также получить, синтезировав сначала соединение формулы XVIII, где А, R3, R4 и Q' имеют указанные выше значения и PG представляет алкоксикарбонильную группу, такую как (С1-С4)алкоксикарбонил, (С6-С14)арил(С1-С4)алкоксикарбонильную группу, такую как фенил(С1-С4)алкоксикарбонил, или (С6-С14)арилоксикарбонильную группу, такую как фенилоксикарбонил, которое после удаления защитной группы карбоновой кислоты СО-Q' превращают в соединение формулы XVIII, где СО-Q' представляет свободную группу карбоновой кислоты СО-ОН, PG представляет (С1-С4)алкоксикарбонил, (С6-С14)арил(С1-С4)алкоксикарбонил или (С6-С14)арилоксикарбонил и А, R3 и R4 имеют указанные выше значения, и циклизуют основанием, таким как, например, карбонат натрия, с образованием соединения формулы XXII.
Соединения формулы IIa

где А, Z, R1, R3, R4 и R5 имеют указанные выше значения, можно получить, осуществляя взаимодействие соединений формулы XXII с алкилирующим реагентом формулы VII в присутствии избытка основания, например в присутствии избытка н-бутиллития, и последующее окисление. 4-(3-Арилуреидо)бензильную группу или 4-(3-арилтиоуреидо)бензильную группу можно вводить поэтапно аналогично получению соединений формулы VIII, после чего полученные соединения формулы II можно превратить в соединения формулы XXII.
Соединения формулы I, в которой остатки R3 и R4 представляют трифторметил, можно успешно получить, осуществляя взаимодействие изонитрила формулы XXIII с 2-трет-бутокси-4,4-бис(трифторметил)-1,3-оксазабута-1,3-диеном формулы XXIV, с образованием соединения формулы XXV,
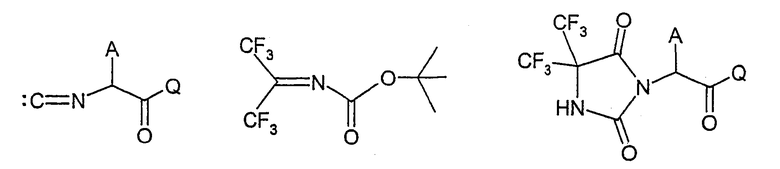
где А и Q имеют указанные выше значения. Группа С(=О)-Q представляет, например, сложноэфирную группу и Q представляет, например, алкоксильную группу, такую как (С1-С4)алкокси, в том числе метокси, этокси и трет-бутокси, или (С6-С14)арил(С1-С4)алкокси, включая бензилокси. Соединения формул XXIII и XXIV можно успешно подвергнуть взаимодействию в растворителе, например в углеводороде или простом эфире, таком как бензол или толуол, при нагревании, например, до температуры от около 40°С до около 80°С, например около 60°С, с образованием соединений формулы XXV.
Изонитрилы (изоцианиды) формулы XXIII можно получить обычными способами, известными специалистам в данной области, из соответствующих сложных эфиров аминокарбоновой кислоты формулы H2N-CHA-C(=O)-Q, где А и Q имеют указанные выше значения. Сложный эфир аминокарбоновой кислоты формулы H2N-CHA-C(=O)-Q сначала превращают в результате взаимодействия с реакционноспособным сложным эфиром муравьиной кислоты, таким как цианометилформиат, в сложный эфир N-формиламинокислоты формулы НС(=О)-NH-CHA-C(=O)-Q, который затем превращают, например, в результате взаимодействия с фосгеном или эквивалентом фосгена, таким как дифосген или трифосген, в присутствии третичного амина, такого как триэтиламин, в изоцианид формулы XXIII. 2-Трет-бутокси-4,4-бис(трифторметил)-1,3-оксазабута-1,3-диен формулы XXIV получают способом, описанным Steglich et al., Chemische Berichte 107 (1974), 1488, из трет-бутилкарбамата ((СН3)3С-О-СО-NH2) и безводного гексафторацетона с последующей обработкой 2-трет-бутоксикарбониламино-2-гидрокси-1,1,1,3,3,3-гексафторпропаном, первоначально полученным при помощи трифторуксусного ангидрида в присутствии основания, такого как хинолин.
Соединения формулы XXV можно затем алкилировать соединениями формулы VII в положении группы NH с образованием соединений формулы XIV, которые при желании после превращения сложноэфирной группы CO-Q в группу карбоновой кислоты СО-ОН подвергают взаимодействию с соединениями формулы III, получая при этом вышеописанные соединения формулы I. В соединениях формулы XXV сложноэфирную группу СО-Q можно сначала превратить в группу карбоновой кислоты СО-ОН обычными способами, после чего соединение формулы XXII, полученное вышеописанным способом с использованием алкилирующего реагента формулы VII в присутствии избытка основания, можно превратить в соединение формулы IIa, которое далее позволяет получить соединение формулы I в результате взаимодействия с соединением формулы III. Аналогично вышеописанным способам получения соединений формулы VIII и способам получения из них соединений формулы II или IIa в соединения формулы XXV можно также поэтапно ввести 4-(3-арилуреидо)бензильную группу или 4-(3-арилтиоуреидо)бензильную группу. При выполнении указанных реакций функциональные группы могут быть защищены или могут находиться в форме предшественников.
Соединения формулы I, в которой Е означает, например, гидроксикарбонил или гидроксиметил, можно стандартными способами превратить в соединения формулы I, где Е имеет другие значения, в другие пролекарства или производные соединений формулы I. Так, для получения сложных эфиров соединения формулы I, где Е означает гидроксикарбонил, можно этерифицировать приемлемыми спиртами, например, в присутствии конденсирующего реагента, такого как DCC, или соединения формулы I, где Е означает гидроксикарбонил, можно алкилировать алкилгалогенидами, такими как алкилхлориды или алкилбромиды, с использованием ацилоксиалкилгалогенидов, получая при этом соединения формулы I, где Е означает ацилоксиалкокси-СО-. Соединения формулы I, где Е означает гидроксикарбонил, можно превратить в амиды, используя аммиак или органические амины в присутствии конденсирующего реагента. Соединения формулы I, где Е означает СО-NH2, можно также успешно получить в твердой фазе, связывая соединение, в котором Е означает СООН, с амидной смолой Ринка в присутствии конденсирующего агента, такого как TOTU, с последующим удалением его смолы при помощи трифторуксусной кислоты. Соединения формулы I, где Е означает гидроксиметильную группу СН2ОН, можно этерифицировать в положении гидроксиметильной группы обычными способами. Обычными способами, применяемыми для избирательного окисления спиртов в альдегиды, например, с использованием гидрохлорита натрия в присутствии 4-ацетамидо-2,2,6,6-тетраметилпиперидин-1-оксила (4-ацетамидо-ТЕМРО), соединения формулы I, в которой Е означает СН2ОН, можно превратить в соединения формулы I, где Е представляет альдегидную группу -СО-Н.
Соединения формулы I, где R5 означает водород, можно также получить, отщепляя группу простого эфира у соединений формулы I, где R5 означает метил. Например, метоксильную группу, представляющую R5O, можно превратить в гидроксильную группу путем обработки трибромидом бора.
Соединения формулы I являются ценными фармацевтически активными соединениями, пригодными, например, для лечения воспалительных заболеваний, аллергических заболеваний или астмы. Соединения формулы I, их физиологически толерантные соли и производные можно вводить в соответствии с данным изобретением животным, предпочтительно млекопитающим, в частности человеку, в виде фармацевтических препаратов для лечения различных болезней, причем термин "лечение" обычно предполагает как лечение симптомов острых или хронических заболеваний, так и профилактику или предотвращение возникновения симптомов заболевания, то есть, например, предотвращение появления симптомов острого аллергического или астматического заболевания либо предотвращение инфаркта миокарда или повторного инфаркта миокарда у нуждающихся в лечении субъектов. Соединения формулы I, их соли и производные можно вводить отдельно, в смеси друг с другом или в форме фармацевтических препаратов, которые пригодны для введения в тонкую кишку или парентерального введения и в качестве активного компонента содержат эффективную дозу, по крайней мере, одного соединения формулы I и/или его физиологически толерантных солей и/или производных и фармацевтически толерантный носитель.
Таким образом, настоящее изобретение относится к соединениям формулы I и/или их физиологически толерантным солям и/или производным, предназначенным для использования в качестве фармацевтических препаратов (или лекарственных средств), к применению соединений формулы I и/или их физиологически толерантных солей и/или производных для получения фармацевтических препаратов для лечения выше- и нижеуказанных заболеваний, например для лечения воспалительных заболеваний, и к применению соединений формулы I и/или их физиологически толерантных солей и/или производных для лечения указанных заболеваний. Настоящее изобретение далее относится к фармацевтическим препаратам (или фармацевтическим композициям), содержащим эффективную дозу по крайней мере одного соединения формулы I и/или его физиологически толерантных солей и/или производных и фармацевтически толерантный носитель, который представляет один или несколько фармацевтически безвредных наполнителей и/или добавок.
Фармацевтические препараты можно вводить системно или местно. Указанные препараты можно вводить, например, перорально в форме пилюль, таблеток, таблеток с пленочной оболочкой, таблеток с сахарным покрытием, гранул, твердых и мягких желатиновых капсул, порошков, растворов, сиропов, эмульсий, суспензий или в других фармацевтических формах. Однако указанные препараты можно также вводить вагинально или ректально, например в форме суппозиториев, парентерально или в виде имплантатов, например в форме растворов для инъекций или вливаний, микрокапсул или палочек, местно или подкожно, например в форме кремов, мазей, порошков, растворов, эмульсий или настоек, или другими способами, например в форме распыляемых в нос растворов или аэрозольных смесей. Парентеральное введение растворов включает, например, внутривенное, внутримышечное, подкожное, внутрисуставное или другие способы введения.
Фармацевтические препараты по данному изобретению получают известными способами, в соответствии с которыми одно или несколько соединений формулы I и/или их физиологически толерантные соли и/или производные смешивают с фармацевтически инертными неорганическими и/или органическими наполнителями и/или добавками и изготавливают приемлемую дозированную лекарственную форму или форму для введения. Для получения пилюль, таблеток, таблеток с сахарным покрытием и твердых желатиновых капсул можно использовать, например, лактозу, кукурузный крахмал или его производные, тальк, стеариновую кислоту или ее соли, полиэтиленгликоли и т.д., для получения мягких желатиновых капсул и суппозиториев можно использовать, например, жиры, воски, полутвердые и жидкие полиолы, полиэтиленгликоли, натуральные или гидрированные масла и т.д. Приемлемыми наполнителями для получения растворов, например инъекционных растворов, эмульсий или сиропов, являются, например, вода, спирты, глицерин, диолы, полиолы, сахароза, инвертный сахар, глюкоза, растительные масла и т.д. Приемлемыми наполнителями для микрокапсул, имплантатов или палочек являются, например, сополимеры гликолевой кислоты и молочной кислоты. Фармацевтические препараты обычно содержат от около 0,5 до около 90 мас.% соединений формулы I и/или их физиологически толерантных солей и/или производных. Количество активного соединения формулы I и/или его физиологически толерантных солей и/или производных в фармацевтических препаратах обычно составляет от около 0,2 до около 1000 мг, предпочтительно от около 1 до около 500 мг. Однако в зависимости от формы фармацевтического препарата количество активного соединения может быть больше.
Помимо активных соединений и носителей фармацевтические препараты могут также содержать наполнители или добавки, например дезинтеграторы, связывающие вещества, смазывающие вещества, смачивающие вещества, стабилизаторы, эмульгаторы, консерванты, подсластители, красители, отдушки, ароматизаторы, загустители, разбавители, буферные вещества, растворители, солюбилизаторы, вещества для достижения эффекта депо, соли для изменения осмотического давления, вещества для нанесения покрытий или антиоксиданты. Фармацевтические препараты могут также содержать два или более соединений формулы I и/или их физиологически толерантные соли и/или производные. Кроме того, помимо по крайней мере одного соединения формулы I и/или его физиологически толерантных солей и/или производных, указанные препараты могут также содержать одно или несколько других фармацевтически активных соединений, например веществ, обладающих противовоспалительным действием.
Если соединения формулы I или содержащие их фармацевтические препараты предназначены для введения в виде аэрозолей, например распыляемых в нос аэрозолей, или путем ингаляции, такие препараты можно вводить, например, при помощи распылителя, насосного распылителя, ингалятора, дозирующего ингалятора или ингалятора сухого порошка. Лекарственные формы для введения соединений формулы I в виде аэрозоля можно получить способами, хорошо известными специалистам в данной области. Для их получения можно использовать, например, растворы или дисперсии соединений формулы I в воде, смесях воды/спирта или приемлемых физиологических растворах, включающие обычные добавки, например бензиловый спирт или другие приемлемые консерванты, усилители поглощения для увеличения биологической доступности, солюбилизаторы, диспергаторы и другие вещества, и при необходимости обычные пропелленты, например хлорфторуглероды и/или фторуглероды.
Другие фармацевтически активные соединения, которые могут присутствовать в фармацевтических препаратах по данному изобретению помимо соединений формулы I и с которыми соединения формулы I могут быть объединены другими способами в режиме комбинированной терапии, представляют активные соединения, пригодные для лечения или профилактики тех же выше- или нижеуказанных заболеваний, что и соединения формулы I. В качестве примеров отдельных классов активных соединений такого типа можно привести стероиды, нестероидные противовоспалительные средства, нестероидные противовоспалительные производные уксусной кислоты, нестероидные противовоспалительные производные пропионовой кислоты, нестероидные противоастматические средства, производные салициловой кислоты, пиразолоны, оксикамы, антагонисты лейкотриена, ингибиторы биосинтеза лейкотриена, ингибиторы циклооксигеназы, ингибиторы циклооксигеназы-2 (ингибиторы СОХ-2), антигистаминные средства, антагонисты Н1-гистамина, неседативные антигистаминные средства, соединения золота, β2-агонисты, антихолинергические средства, антагонисты мускарина, средства, снижающие содержание липидов, средства, снижающие содержание холестерина, ингибиторы редуктазы HMG-CoA, статины, производные никотиновой кислоты, иммуносупрессанты, циклоспорины, β-интерфероны, противоопухолевые средства, цитостатические средства, ингибиторы образования метастазов, антиметаболиты, производные 5-аминосалициловой кислоты, антидиабетические средства, инсулины, сульфонилмочевины, бигуаниды, глитазоны, ингибиторы α-глюкозидазы и другие. Примерами приемлемых активных соединений являются ацетилсалициловая кислота, бенорилат, сульфазалазин, фенилбутазон, оксифенбутазон, метамизол, мофебутазон, фепразон, селекоксиб, рофекоксиб, диклофенак, фентиазак, сулиндак, зомепирак, толметин, индометацин, ацеметацин, ибупрофен, напроксен, карпрофен, фенбуфен, индопрофен, кетопрофен, пирпрофен, тиапрофеновая кислота, дифлунизал, флуфенамовая кис-лота, меклофенамовая кислота, мефенамовая кислота, нифлумовая кислота, толфенамовая кислота, пироксикам, изоксикам, теноксикам, никотиновая кислота, преднизон, дексаметазон, гидрокортизон, метилпреднизолон, бетаметазон, беклометазон, будезонид, монтелукаст, пранлукаст, зафирлукаст, зилеутон, циклоспорин, циклоспорин А, рапамицин, такролимус, метотрексат, 6-меркаптопурин, азатиоприн, интерферон-бета-1а, интерферон-бета-1b, 5-аминосалициловая кислота, лефлуномид, D-пеницилламин, хлорохин, глибенкламид, глимепирид, троглитазон, метформин, акарбоза, аторвастатин, флувастатин, ловастатин, симвастатин, правастатин, колестипол, колестирамин, пробукол, клофибрат, фенофибрат, безафибрат, гемфиброзил, бромид ипатропия, кленбутерол, фенотерол, метапротеренол, пирбутерол, тулобутерол, салбутамол, салметерол, тербуталин, изоэтарин, кетотифен, эфедрин, бромид окситропия, атропин, кромоглициновая кислота, теофиллин, фексофенадин, терфенадин, цетиризин, диметинден, дифенгидрамин, дифенилпиралин, фенирамин, бромфенирамин, хлорфенирамин, дексхлорфенирамин, алимезаин, антазолин, астемизол, азатадин, клемастин, ципрогептадин, гидроксизин, лоратидин, мепирамин, прометазин, трипеленнамин, трипролидин и другие.
Если соединения формулы I и/или их физиологически толерантные соли и/или производные предназначены для использования в комбинированной терапии вместе с одним или несколькими другими активными соединениями, все активные соединения можно вводить вместе в одном фармацевтическом препарате, например таблетке или капсуле. Настоящее изобретение в равной степени относится к фармацевтическим препаратам такого типа, для которых верны все вышеуказанные положения. Количество активных соединений в таких фармацевтических препаратах обычно выбирают с учетом наличия эффективного количества каждого активного соединения. Однако используемые в комбинированной терапии активные соединения могут входить в состав двух или более отдельных фармацевтических препаратов, находящихся в одной упаковке или в двух или большем количестве отдельных упаковок. Соединения формулы I и/или их физиологически толерантные соли и/или производные и другие активные соединения можно вводить вместе или раздельно и одновременно или последовательно в любом порядке. Можно использовать разные способы введения, например одно активное соединение можно вводить перорально, а другое - в виде инъекции, ингаляции или местно. Все такие способы лечения входят в объем настоящего изобретения.
Соединения формулы I обладают, например, способностью ингибировать процессы межклеточного взаимодействия и взаимодействия клеток с матриксом, обусловленные взаимодействием между VLA-4 и его лигандами. Активность соединений формулы I можно продемонстрировать при помощи анализа, позволяющего измерить связывание клеток, содержащих рецептор VLA-4, например лейкоцитов, с лигандами данного рецептора, такими как VCAM-1, которые можно получить методами рекомбинантных ДНК. Такой анализ подробно описан ниже. Соединения формулы I, в частности, обладают способностью ингибировать адгезию и миграцию лейкоцитов, например адгезию лейкоцитов к эндотелиальным клеткам, которая, как указывалось выше, регулируется механизмом адгезии VCAM-1/VLA-4. Поэтому помимо противовоспалительных средств соединения формулы I и их физиологически толерантные соли и производные можно использовать для лечения и профилактики заболеваний, обусловленных взаимодействием между рецептором VLA-4 и его лигандами и поддающихся лечению в результате подавления такого взаимодействия, в частности, указанные соединения пригодны для лечения заболеваний, по крайней мере частично вызываемых нежелательной степенью адгезии и/или миграции лейкоцитов или связанных с такими процессами, и в случае которых необходимо предотвратить, уменьшить или устранить адгезию и/или миграцию лейкоцитов.
Настоящее изобретение относится также к соединениям формулы I и/или их физиологически толерантным солям и/или производным, предназначенным для ингибирования адгезии и/или миграции лейкоцитов или подавления рецептора VLA-4, и к применению соединений формулы I и/или их физиологически толерантных солей и/или производных для получения фармацевтических препаратов для лечения заболеваний, обусловленных нежелательной степенью адгезии и/или миграции лейкоцитов, или для лечения заболеваний, в которых важную роль играют процессы VLA-4-зависимой адгезии, и к применению соединений формулы I и/или их физиологически толерантных солей и/или производных для лечения заболеваний указанного типа.
Соединения формулы I можно использовать в качестве противовоспалительных средств при появлении симптомов воспалительного характера, вызываемых разными причинами, для предотвращения, уменьшения или подавления нежелательных или вредных последствий воспаления. Указанные соединения используют, например, для лечения или профилактики артрита, ревматоидного артрита, полиартрита, воспаления кишечника (неспецифический язвенный колит, болезнь Крона), системной красной волчанки, воспалительных заболеваний центральной нервной системы, таких как, например, рассеянный склероз, астмы или аллергических реакций, таких как, например, аллергические реакции замедленного типа (аллергия типа IV). Кроме того, указанные соединения можно использовать для профилактики сердечных заболеваний и инсульта, для вторичной профилактики инсульта, для лечения и профилактики сердечно-сосудистых заболеваний, атеросклероза, инфаркта миокарда, повторного инфаркта миокарда, острого коронарного синдрома, инсульта, рестеноза, диабета, поражения трансплантатов органов, иммунных заболеваний, аутоиммунных заболеваний, роста опухоли или метастазов опухоли при разных злокачественных заболеваниях, малярии и других заболеваний, которые можно предотвратить, облегчить или вылечить, блокируя интегрин VLA-4 и/или подавляя активность лейкоцитов. Предпочтительным применением является профилактика инфаркта миокарда или повторного инфаркта миокарда.
Доза соединений формулы I может изменяться в широких пределах, и, как известно лечащему врачу, должна быть подобрана в каждом отдельном случае с учетом состояния нуждающегося в лечении субъекта. Например, величина дозы зависит от характера и тяжести заболевания, состояния пациента, используемого соединения, острой или хронической стадии заболевания и использования других активных соединений дополнительно к соединениям формулы I. В случае перорального введения суточная доза от около 0,01 до около 100 мг/кг, предпочтительно от около 0,1 до около 10 мг/кг (во всех случаях мг/кг массы тела), обычно является достаточной для достижения эффективных результатов при введении взрослому человеку с массой тела около 75 кг. В случае внутривенного введения суточная доза обычно составляет от около 0,01 до около 50 мг/кг, предпочтительно от около 0,01 до около 10 мг/кг (во всех случаях мг/кг массы тела). При введении относительно больших количеств лекарственного средства суточная доза может быть разделена на 2, 3 или 4 части. В зависимости от индивидуальной реакции указанная суточная доза может быть увеличена или уменьшена.
Помимо применения в качестве фармацевтически активных соединений в медицине и ветеринарии соответствующие соединения формулы I, их соли и производные можно использовать в диагностических целях, например для диагностики in vitro по образцам клеток или тканей, и в качестве вспомогательных или научных инструментов при выполнении биохимических исследований, в которых необходимо блокировать VLA-4 или подавить межклеточное взаимодействие или взаимодействие клеток с матриксом. Кроме того, соединения формулы I и их соли можно использовать в качестве промежуточных продуктов для получения других соединений, в частности других фармацевтически активных соединений, синтезируемых из соединений формулы I, например, в результате модификации или введения остатков или функциональных групп, например, при помощи этерификации, восстановления, окисления или других способов преобразования функциональных групп.
Примеры
Пример 1
(R)-3-((S)-2-(4,4-Диметил-3-(4-(3-(2-метилфенил)уреидо)-3-метоксибензил)-2,5-диоксоимидазолидин-1-ил)-2-(циклопропилметил)ацетиламино)-3-метилпропионовая кислота

1а) 4-(3-(2-Метилфенил)уреидо)-3-метоксибензиловый спирт
15 г (81,8 ммоль) 3-метокси-4-нитробензилового спирта гидрируют на 1,3 г палладия на угле (10% крепость, 50% воды) в 500 мл метил-трет-бутилового эфира (метилового эфира трет-бутилового спирта) при охлаждении льдом. После прекращения поглощения водорода катализатор отфильтровывают и к фильтрату добавляют 10,14 мл (81,8 ммоль) 2-метилфенилизоцианата, перемешивая смесь в течение 30 минут. Реакционную смесь оставляют выстаиваться в течение ночи, осажденное твердое вещество отфильтровывают через вакуум-фильтр и промывают метил-трет-бутиловым эфиром. Выход: 20,5 г (88%).
1b) 4-(3-(2-Метилфенил)уреидо)-3-метоксибензилхлорид
7,65 мл (104,8 ммоль) Тионилхлорида добавляют по каплям при охлаждении льдом к суспензии 15 г (52,4 ммоль) соединения по примеру 1а в 300 мл дихлорметана. Реакционную смесь перемешивают при комнатной температуре в течение 3 часов, оставляют выстаиваться в течение ночи и выливают в 1000 мл гептана. Гептан декантируют с отделенного масла, остаток снова суспендируют в гептане и гептан декантируют. Указанную процедуру повторяют еще два раза. Остаток растворяют в дихлорметане и выливают в 800 мл охлаждаемого льдом диизопропилового эфира. Смесь перемешивают в течение 2 часов при охлаждении льдом, продукт отфильтровывают через вакуум-фильтр, промывают диизопропиловым эфиром и сушат над пентоксидом фосфора. Выход: 12 г (75%).
1с) Бензил-(S)-2-амино-3-циклопропилпропионат
1 н. Раствор гидроксида натрия добавляют при 0°С к суспензии 10 г (77,5 ммоль) (S)-2-амино-3-циклопропилпропионовой кислоты в 160 мл диоксана до достижения рН 8-9. Затем добавляют 16,9 г (77,5 ммоль) ди-трет-бутилдикарбоната, баню со льдом удаляют и рН поддерживают равным 8-9, добавляя 1 н. раствор гидроксида натрия. Реакционную смесь оставляют выстаиваться в течение ночи, диоксан удаляют в вакууме, к водной фазе добавляют этилацетат и фазы разделяют. Водную фазу доводят до рН 4,5, используя 1 н. раствор хлористоводородной кислоты, и экстрагируют этилацетатом. Полученную этилацетатную фазу сушат над сульфатом натрия, затем сульфат натрия отфильтровывают и фильтрат концентрируют в вакууме. Остаток растворяют в 1000 мл дихлорметана и обрабатывают 53,4 мл бензилового спирта, 8,37 г 4-диметиламинопиридина и 18,8 г DCC. Смесь перемешивают в течение 6 часов, оставляют выстаиваться в течение ночи и фильтруют, фильтрат концентрируют и остаток обрабатывают 300 мл трифторуксусной кислоты крепостью 90%. Смесь перемешивают при комнатной температуре в течение 10 минут, трифторуксусную кислоту удаляют в вакууме и остаток дважды хроматографируют на силикагеле (дихлорметан/метанол, 95/5). Выход: 11,48 г (68%).
1d) (S)-2-(4,4-Диметил-2,5-диоксоимидазолидин-1-ил)-2-(циклопропилметил)уксусная кислота
321 мг НОВТ и 4,75 г (23,7 ммоль) DCC добавляют к раствору 3,82 г (23,7 ммоль) 2-метоксикарбониламино-2-метилпропионовой кислоты (полученной из 2-амино-2-метилпропионовой кислоты и метилхлорформиата) и 5,2 г (23,7 ммоль) соединения по примеру 1с в 100 мл ТГФ и смесь перемешивают при комнатной температуре в течение 4 часов. Реакционную смесь оставляют выстаиваться в течение ночи и фильтруют, ТГФ удаляют в вакууме, остаток поглощают метил-трет-бутиловым эфиром и раствор дважды промывают насыщенным раствором NaHCO3 и водным раствором KHSO4/K2SO4. Органическую фазу сушат над сульфатом натрия, фильтруют и растворитель удаляют в вакууме. Остаток растворяют в этилацетате и гидрируют в присутствии палладия на угле (10% крепость, 50% воды). Катализатор отфильтровывают и к органической фазе добавляют 500 мл воды и 10,1 г карбоната натрия. Смесь экстрагируют встряхиванием, фазы разделяют и водную фазу перемешивают при 100°С в течение 24 часов. Реакционную смесь оставляют выстаиваться в течение ночи, добавляют 500 мл 6 н. раствора хлористоводородной кислоты и водную фазу трижды экстрагируют метил-трет-бутиловым эфиром. Объединенные органические фазы сушат над сульфатом натрия, фильтруют и концентрируют в вакууме. Остаток кристаллизуют, используя диизопропиловый эфир, и продукт отфильтровывают. Выход: 2,88 г (51%).
1е) (S)-2-(4,4-Диметил-3-(4-(3-(2-метилфенил)уреидо)-3-метоксибензил)-2,5-диоксоимидазолидин-1-ил)-2-(циклопропилметил)уксусная кислота

9,44 мл Раствора н-бутиллития (2,5 М раствор в гексане) добавляют при -40°С в атмосфере аргона к раствору 2,85 г (11,8 ммоль) соединения по примеру 1d в 60 мл абсолютного ТГФ. Реакционную смесь перемешивают при -40°С в течение 30 минут, оставляют нагреваться до 0°С и добавляют раствор 3,6 г (11,8 ммоль) соединения по примеру 1b в 20 мл N-метил-2-пирролидона. Реакционную смесь оставляют нагреваться до 0°С и перемешивают в течение 2 часов при 0°С. Добавляют 15 мл 1 н. раствора хлористоводородной кислоты и ТГФ удаляют в вакууме. Остаток выливают в 300 мл метил-трет-бутилового эфира. Фазы разделяют, органическую фазу промывают водой, сушат над сульфатом натрия, фильтруют и концентрируют в вакууме. Остаток очищают препаративной ВЭЖХ. Фракции продукта концентрируют и сушат вымораживанием, получая при этом 1,33 г (22%) указанного в заголовке соединения.
1f) Трет-бутил-(R)-3-((S)-2-(4,4-диметил-3-(4-(3-(2-метилфенил)уреидо)-3-метоксибензил)-2,5-диоксоимидазолидин-1-ил)-2-(циклопропилметил)ацетиламино)-3-метилпропионат
626 мг (1,91 ммоль) TOTU и 308 мкл (1,81 ммоль) N,N-диизопропилэтиламина последовательно добавляют при охлаждении льдом к раствору 974 мг (1,91 ммоль) соединения по примеру 1е и 305 мг (1,91 ммоль) трет-бутил-(R)-3-аминобутаноата в 10 мл абсолютного ДМФ. Реакционную смесь перемешивают при комнатной температуре в течение 2 часов, растворитель удаляют в вакууме, остаток растворяют в этилацетате и этилацетатный раствор дважды последовательно промывают водным раствором KHSO4/K2SO4, насыщенным раствором NaHCO3 и водой. Органическую фазу сушат над сульфатом натрия и фильтруют, растворитель удаляют в вакууме и остаток хроматографируют на силикагеле, используя этилацетат/гептан (1/1). Фракции продукта концентрируют и получают 880 мг (71%) указанного в заголовке соединения.
1g) (R)-3-((S)-2-(4,4-Диметил-3-(4-(3-(2-метилфенил)уреидо)-3-метоксибензил)-2,5-диоксоимидазолидин-1-ил)-2-(циклопропилметил)ацетиламино)-3-метилпропионовая кислота
880 мг (1,35 ммоль) Соединения по примеру 1f обрабатывают 10 мл трифторуксусной кислоты крепостью 90%. Реакционную смесь выдерживают в течение 15 минут при комнатной температуре и концентрируют в вакууме. Остаток поглощают дихлорметаном и концентрируют в вакууме. Указанную процедуру повторяют еще раз. Полученный остаток поглощают дихлорметаном, дихлорметановую фазу трижды промывают водой и сушат над сульфатом натрия. Продукт фильтруют и концентрируют в вакууме, остаток поглощают ацетонитрилом/водой и сушат вымораживанием. Выход: 730 мг (91%).
ES(+)-МС: 594,2 (М+Н)+
Пример 2
Натриевая соль (R)-3-((S)-2-(4,4-диметил-3-(4-(3-(2-метилфенил)уреидо)-3-метоксибензил)-2,5-диоксоимидазолидин-1-ил)-2-(циклопропилметил)ацетиламино)-3-метилпропионовой кислоты
1,64 мл 0,1 н. Раствора гидроксида натрия добавляют порциями при перемешивании к суспензии 100 мг (0,168 ммоль) соединения по примеру 1 в 10 мл воды и смесь перемешивают при комнатной температуре в течение 1 часа. Реакционную смесь фильтруют и фильтрат сушат вымораживанием, получая при этом 104 мг (100%) указанного в заголовке соединения.
ES(+)-MC: 594,3 ((R)-3-((S)-2-(4,4-диметил-3-(4-(3-(2-метилфенил)уреидо)-3-метоксибензил)-2,5-диоксоимидазолидин-1-ил)-2-(циклопропилметил)ацетиламино)-3-метилпропионовая кислота + Н)+, 616,2 (М+)
Пример 3
(R)-3-((S)-2-(4,4-Диметил-3-(4-(3-(2-метилфенил)уреидо)-3-метоксибензил)-2,5-диоксоимидазолидин-1-ил)-2-(циклопропилметил)ацетиламино)-3-метилпропанол
535 мг (1,05 ммоль) Соединения по примеру 1е в 15 мл абсолютного ДМФ обрабатывают при охлаждении льдом 140 мг (1,05 ммоль) НОВТ и 260 мг (1,26 ммоль) DCC. Смесь перемешивают в течение 45 минут при охлаждении льдом, добавляют 112 мг (1,26 ммоль) (R)-3-амино-3-метилпропанола и продолжают перемешивать при комнатной температуре еще 2 часа. Смесь оставляют выстаиваться в течение ночи и фильтруют, фильтрат концентрируют, остаток растворяют в этилацетате и этилацетатную фазу дважды промывают водным раствором KHSO4/K2SO4. Реакционную смесь сушат над сульфатом натрия, фильтруют, концентрируют и остаток хроматографируют на силикагеле, используя этилацетат. Фракции продукта концентрируют и получают 423 мг (70%) указанного в заголовке соединения.
ES(+)-MC: 580,3 (М+Н)+
Получение (R)-3-амино-3-метилпропанола
5,68 г (149 ммоль) Алюмогидрида лития добавляют порциями в атмосфере аргона к раствору 19,9 г (149 ммоль) трихлорида алюминия в 250 мл абсолютного диэтилового эфира и смесь нагревают с обратным холодильником в течение 30 минут. К реакционной смеси медленно по каплям добавляют 6 г (37,7 ммоль) трет-бутил-(R)-3-аминобутаноата в 50 мл абсолютного диэтилового эфира и нагревают с обратным холодильником в течение 2 часов. При охлаждении льдом осторожно добавляют по каплям 10,8 мл воды и 25,3 г гидроксида калия, растворенного в 43 мл воды. Смесь оставляют выстаиваться в течение ночи при комнатной температуре, эфирную фазу декантируют и остаток трижды кипятят с дихлорметаном. Объединенные органические фазы сушат над сульфатом натрия. Продукт фильтруют и удаляют растворитель в вакууме, получая при этом 2,5 г (75%) указанного в заголовке соединения.
Пример 4
(R)-3-((S)-2-(4,4-Диметил-3-(4-(3-(2-метилфенил)уреидо)-3-метоксибензил)-2,5-диоксоимидазолидин-1-ил)-2-(циклопропилметил)ацетиламино)-3-метилпропионамид

131 мг (0,636 ммоль) DCC добавляют к раствору 330 мг (0,555 ммоль) соединения по примеру 1 и 125 мг (0,926 ммоль) НОВТ в 5 мл абсолютного ДМФ, смесь перемешивают при комнатной температуре в течение 1 часа и добавляют 47 мкл (0,555 ммоль) водного раствора аммиака крепостью 25%. Смесь оставляют выстаиваться в течение ночи при комнатной температуре, добавляют еще 16 мкл водного раствора аммиака крепостью 25% и смесь перемешивают в течение 4 часов. Реакционную смесь фильтруют, фильтрат концентрируют в вакууме, остаток растворяют в этилацетате и этилацетатную фазу дважды промывают водным раствором KHSO4/K2SO4, насыщенным раствором NaHCO3 и водой. Органическую фазу сушат над сульфатом натрия и фильтруют, растворитель удаляют в вакууме и остаток хроматографируют на силикагеле, используя этилацетат. Фракции продукта концентрируют и сушат вымораживанием, получая при этом 272 мг (82%) указанного в заголовке соединения.
ES(+)-MC: 593,3
Пример 5
(R)-3-((S)-2-(4,4-Диметил-3-(4-(3-(2-метилфенил)уреидо)-3-гидроксибензил)-2,5-диоксоимидазолидин-1-ил)-2-(циклопропилметил)ацетиламино)-3-метилпропионовая кислота

211 мкл Трибромида бора добавляют в атмосфере аргона к раствору 100 мг (0,169 ммоль) соединения по примеру 1 в 20 мл абсолютного дихлорметана при -78°С и реакционную смесь оставляют нагреваться до 0°С при охлаждении льдом. Смесь выдерживают при 0°С в течение 30 минут и осторожно добавляют воду. Фазы разделяют и органическую фазу сушат над сульфатом натрия. Реакционную смесь фильтруют, растворитель удаляют в вакууме, фракции продукта очищают препаративной ВЭЖХ и сушат вымораживанием, получая при этом 35 мг (36%) указанного в заголовке соединения.
ES(+)-MC: 580,2 (М+Н)+
Пример 6
(R)-3-((S)-2-(4,4-Диметил-3-(4-(3-(2-метилфенил)уреидо)-3-гидроксибензил)-2,5-диоксоимидазолидин-1-ил)-2-(циклопропилметил)ацетиламино)-3-метилпропанол

488 мкл Трибромида бора добавляют в атмосфере аргона к раствору 220 мг (0,39 ммоль) соединения по примеру 3 в 40 мл абсолютного дихлорметана при -78°С и реакционную смесь оставляют нагреваться до 0°С при охлаждении льдом. Смесь выдерживают при 0°С в течение 30 минут и осторожно добавляют воду. Фазы разделяют, органическую фазу четыре раза промывают водой и сушат над сульфатом натрия. Реакционную смесь фильтруют, растворитель удаляют в вакууме и фракции продукта очищают препаративной ВЭЖХ и сушат вымораживанием, получая при этом 81 мг (37%) указанного в заголовке соединения.
ES(+)-MC: 566,3 (М+Н)+
Пример 7
(R)-3-((S)-2-(4,4-Диметил-3-(4-(3-фенилуреидо)-3-метоксибензил)-2,5-диоксоимидазолидин-1-ил)-2-(циклопропилметил)ацетиламино)-3-метилпропионовая кислота
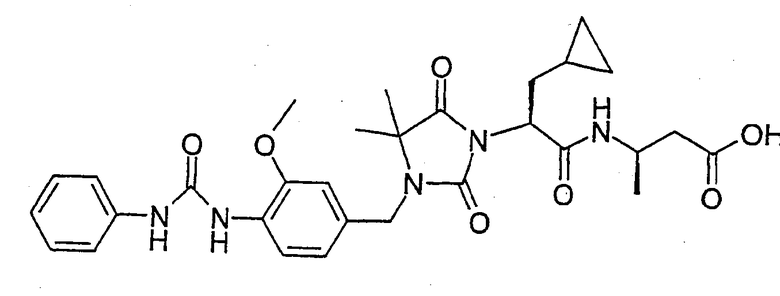
7а) 4-(3-Фенилуреидо)-3-метоксибензилхлорид
7,55 мл (103,4 ммоль) Тионилхлорида добавляют по каплям к суспензии 14,07 г (51,7 ммоль) 4-(3-фенилуреидо)-3-метоксибензилового спирта (полученного способом по примеру 1 с использованием фенилизоцианата вместо 2-метилфенилизоцианата) в 200 мл дихлорметана. Смесь перемешивают при комнатной температуре в течение 2 часов, оставляют выстаиваться в течение ночи и выливают в 800 мл гептана. Гептан декантируют с отделенного масла, остаток несколько раз суспендируют в гептане и каждый раз гептан декантируют. Остаток растворяют в 100 мл дихлорметана и по каплям добавляют к 800 мл диизопропилового эфира. Смесь перемешивают в течение 1 часа при охлаждении льдом, продукт отфильтровывают через вакуум-фильтр, промывают диизопропиловым эфиром и сушат в вакууме.
7b) (S)-2-(4,4-Диметил-3-(4-(3-фенилуреидо)-3-метоксибензил)-2,5-диоксоимидазолидин-1-ил)-2-(циклопропилметил)уксусная кислота
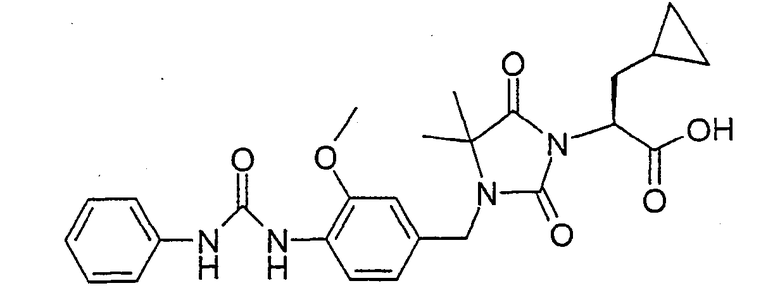
9,32 мл раствора н-бутиллития (2,5 М раствор в гексане) добавляют при -40°С в атмосфере аргона к раствору 2,8 г (11,6 ммоль) соединения по примеру 1d в 60 мл абсолютного ТГФ. Реакционную смесь перемешивают при -40°С в течение 30 минут, оставляют нагреваться до 0°С и добавляют раствор 5,07 г (17,4 ммоль) соединения по примеру 7а в 20 мл N-метил-2-пирролидона. Реакционную смесь оставляют нагреваться до 0°С и перемешивают в течение 2 часов при 0°С. Затем добавляют 15 мл 1 н. раствора хлористоводородной кислоты, ТГФ удаляют в вакууме и остаток выливают в 300 мл метил-трет-бутилового эфира. Фазы разделяют, органическую фазу промывают водой, сушат над сульфатом натрия, фильтруют и концентрируют в вакууме. Остаток очищают препаративной ВЭЖХ. Фракции продукта концентрируют и сушат вымораживанием, получая при этом 484 мг (8%) указанного в заголовке соединения.
7с) (R)-3-((S)-2-(4,4-Диметил-3-(4-(3-фенилуреидо)-3-метоксибензил)-2,5-диоксоимидазолидин-1-ил)-2-(циклопропилметил)ацетиламино)-3-метилпропионовая кислота
Указанное в заголовке соединение получают способом по примеру 1 из 120 мг (0,242 ммоль) соединения по примеру 7b и 38 мг (0,242 ммоль) трет-бутил-(R)-3-аминобутаноата, для чего реакционную смесь подвергают реакции сочетания, очищают хроматографией, отщепляют трет-бутиловый эфир и сушат вымораживанием. Выход: 113 мг (81%).
ES(+)-MC: 580,2 (М+Н)+
Пример 8
(R)-3-((S)-2-(4,4-Диметил-3-(4-(3-фенилуреидо)-3-метоксибензил)-2,5-диоксоимидазолидин-1-ил)-2-(циклопропилметил)ацетиламино)-3-метилпропанол
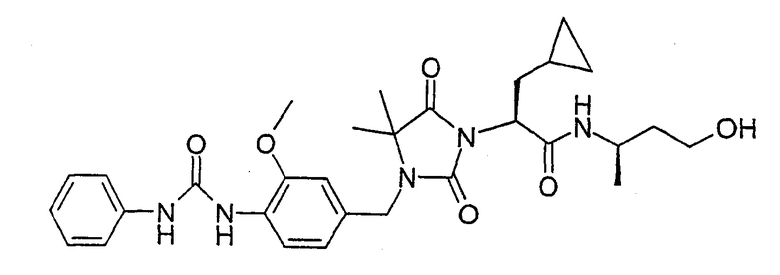
Указанное в заголовке соединение получают способом по примеру 3 из 172 мг (0,348 ммоль) соединения по примеру 7b и 31 мг (0,417 ммоль) (R)-3-амино-3-метилпропанола (см. пример 3), для чего реакционную смесь подвергают реакции сочетания и очищают хроматографией (этилацетат/гептан, 9/1), фракции продукта концентрируют и сушат вымораживанием. Выход: 117 мг (59%).
ES(+)-MC: 566,3 (М+Н)+
Пример 9
Этил-3-((S)-2-(4,4-диметил-3-(4-(3-(2-метилфенил)уреидо)-3-метоксибензил)-2,5-диоксоимидазолидин-1-ил)-2-(циклопропилметил)ацетиламино)-3-(3-пиридил)пропионат

129 мг (0,393 ммоль) TOTU и 64 мкл (0,374 ммоль) N,N-диизопропилэтиламина добавляют при охлаждении льдом к раствору 200 мг (0,393 ммоль) соединения по примеру 1d и 76,4 мг (0,393 ммоль) этил-3-амино-3-(3-пиридил)пропионата (для ознакомления со способом получения см. J.G. Rico et al., J. Org. Chem. 58 (1993) 7948) в 5 мл абсолютного ДМФ. Реакционную смесь перемешивают при комнатной температуре в течение 30 минут, растворитель удаляют в вакууме и остаток поглощают этилацетатом. Этилацетатный раствор дважды последовательно промывают насыщенным раствором NaHCO3 и водой. Органическую фазу сушат над сульфатом натрия и фильтруют, растворитель удаляют в вакууме и остаток хроматографируют на силикагеле, используя этилацетат. Фракции продукта концентрируют и получают 195 мг (72%) указанного в заголовке соединения.
ES(+)-MC: 685,4 (М+Н)+
Пример 10
Гидрохлорид 3-((S)-2-(4,4-диметил-3-(4-(3-(2-метилфенил)уреидо)-3-метоксибензил)-2,5-диоксоимидазолидин-1-ил)-2-(циклопропилметил)ацетиламино)-3-(3-пиридил)пропионовой кислоты
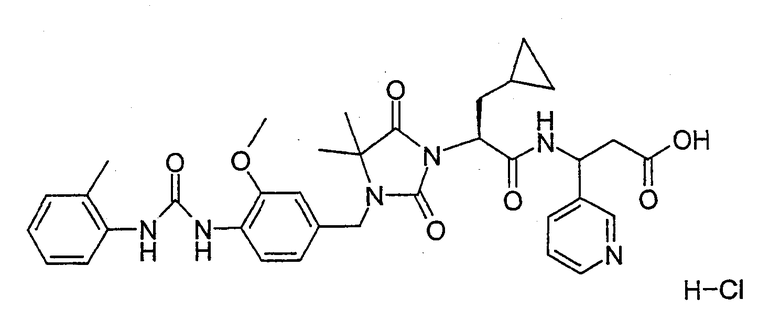
0,82 мл (0,82 ммоль) 1 М водного раствора гидроксида лития добавляют к раствору 141 мг (0,206 ммоль) соединения по примеру 9 в 7,25 мл метанола и реакционную смесь оставляют выстаиваться в течение ночи при комнатной температуре. Метанол удаляют в вакууме, остаток доводят до рН 2, используя 1 н. раствор хлористоводородной кислоты, и смесь концентрируют в вакууме. Остаток хроматографируют на силикагеле, используя дихлорметан/метанол/ледяную уксусную кислоту/воду (95/5/0,5/0,5). Фракции продукта концентрируют, остаток обрабатывают 1,1 эквивалента 1 н. раствора хлористоводородной кислоты и сушат вымораживанием. Выход: 120 мг (89%).
ES(+)-MC: 657,4 (М+Н)+
Пример 11
Изопропил-3-((S)-2-(4,4-диметил-3-(4-(3-(2-метилфенил)уреидо)-3-метоксибензил)-2,5-диоксоимидазолидин-1-ил)-2-(циклопропилметил)ацетиламино)-3-(3-пиридил)пропионат
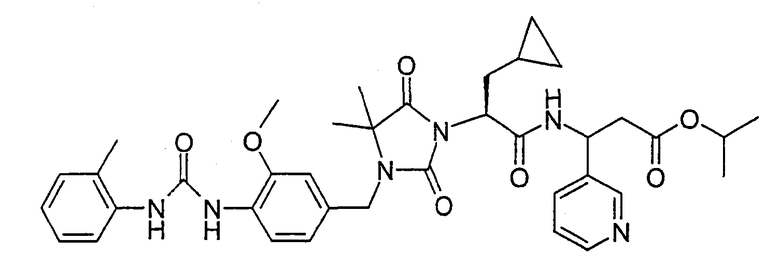
56 мкл (0,731 ммоль) изопропанола и 23,6 мг (0,193 ммоль) 4-диметиламинопиридина добавляют к суспензии 80 мг (0,122 ммоль) соединения по примеру 11 в 3 мл дихлорметана. 38 мг (0,183 ммоль) DCC растворяют в 1 мл дихлорметана и добавляют к ставшему прозрачным раствору. Смесь перемешивают при комнатной температуре в течение 2 часов и оставляют выстаиваться в течение ночи при комнатной температуре. Затем реакционную смесь фильтруют, фильтрат концентрируют в вакууме и остаток хроматографируют на силикагеле, используя гептан/этилацетат (3/1) и этилацетат/гептан (20/1). Фракции продукта концентрируют и получают 70 мг (82%) указанного в заголовке соединения.
ES(+)-MC: 699,4 (М+Н)+
Пример 12
Этил-3-((S)-2-(4,4-диметил-3-(4-(3-(2-метилфенил)уреидо)-3-метоксибензил)-2,5-диоксоимидазолидин-1-ил)-2-(циклопропилметил)ацетиламино)-3-(4-пиридил)пропионат
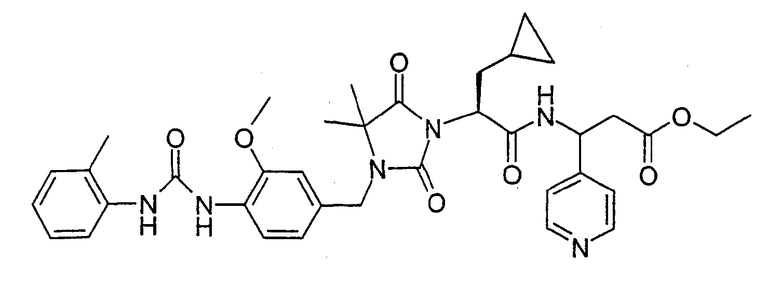
Указанное в заголовке соединение получают способом по примеру 9 из 200 мг (0,393 ммоль) соединения по примеру 1d и 76,4 мг (0,393 ммоль) этил-3-амино-3-(4-пиридил)пропионата (для ознакомления со способом получения см. J.G. Rico et al., J. Org. Chem. 58 (1993) 7948). Выход: 199 мг (74%).
ES(+)-MC: 685,4 (М+Н)+
Пример 13
Гидрохлорид 3-((S)-2-(4,4-диметил-3-(4-(3-(2-метилфенил)уреидо)-3-метоксибензил)-2,5-диоксоимидазолидин-1-ил)-2-(циклопропилметил)ацетиламино)-3-(4-пиридил)пропионовой кислоты

Указанное в заголовке соединение получают способом по примеру 10 из 143 мг (0,209 ммоль) соединения по примеру 12. Выход: 126 мг (87%).
ES(+)-MC: 657,2 (М+Н)+
Пример 14
Изопропил-3-((S)-2-(4,4-диметил-3-(4-(3-(2-метилфенил)уреидо)-3-метоксибензил)-2,5-диоксоимидазолидин-1-ил)-2-(циклопропилметил)ацетиламино)-3-(4-пиридил)пропионат
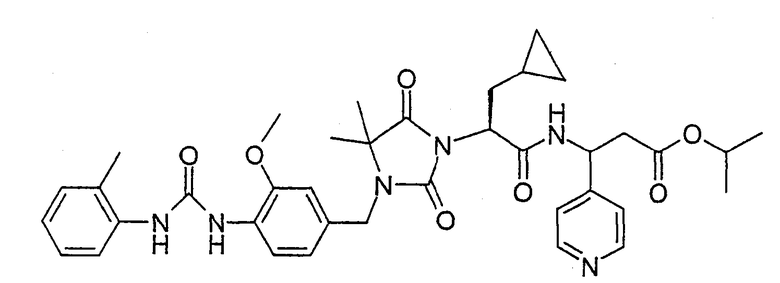
Указанное в заголовке соединение получают способом по примеру 11 из 83 мг (0,126 ммоль) соединения по примеру 13. Выход: 34,6 мг (39%).
ES(+)-MC: 699,4 (М+Н)+
Пример 15
Этил-3-((S)-2-(4,4-диметил-3-(4-(3-(2-метилфенил)уреидо)-3-метоксибензил)-2,5-диоксоимидазолидин-1-ил)-2-(циклопропилметил)ацетиламино)-3-(2-пиридил)пропионат
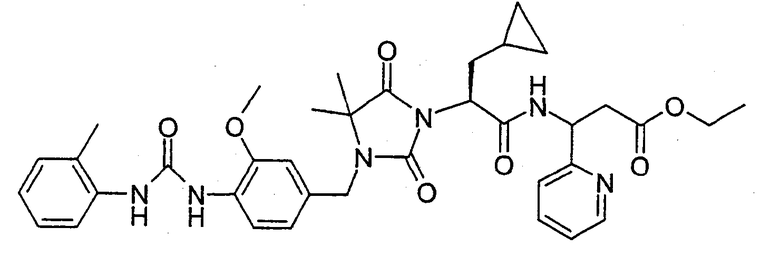
Указанное в заголовке соединение получают способом по примеру 9 из 200 мг (0,393 ммоль) соединения по примеру 1d и 76,4 мг (0,393 ммоль) этил-3-амино-3-(2-пиридил)пропионата (для ознакомления со способом получения см. J.G. Rico et al., J. Org. Chem. 58 (1993) 7948). Выход: 226 мг (84%).
ES(+)-MC: 685,4 (М+Н)+
Пример 16
3-((S)-2-(4,4-Диметил-3-(4-(3-(2-метилфенил)уреидо)-3-метоксибензил)-2,5-диоксоимидазолидин-1-ил)-2-(циклопропилметил)ацетиламино)-3-(2-пиридил)пропионовая кислота
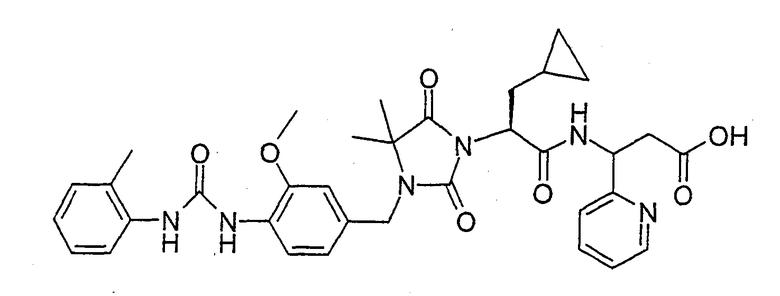
Указанное в заголовке соединение получают способом по примеру 10 из 170 мг (0,248 ммоль) соединения по примеру 15, но полученный продукт не превращают в гидрохлорид, добавляя хлористоводородную кислоту. Выход: 160 мг (98%).
ES(+)-MC: 657,4 (М+Н)+
Пример 17
Изопропил-3-((S)-2-(4,4-диметил-3-(4-(3-(2-метилфенил)уреидо)-3-метоксибензил)-2,5-диоксоимидазолидин-1-ил)-2-(циклопропилметил)ацетиламино)-3-(2-пиридил)пропионат
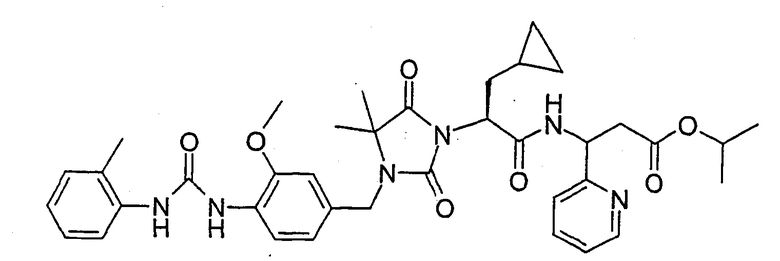
Указанное в заголовке соединение получают способом по примеру 11 из 90 мг (0,137 ммоль) соединения по примеру 16. Выход: 39 мг (41%).
ES(+)-MC: 699,4 (М+Н)+
Пример 18
(R)-3-((S)-2-(4,4-Бис(трифторметил)-3-(4-(3-(2-метилфенил)-уреидо)-3-метоксибензил)-2,5-диоксоимидазолидин-1-ил)-2-(циклопропилметил)ацетиламино)-3-метилпропионовая кислота
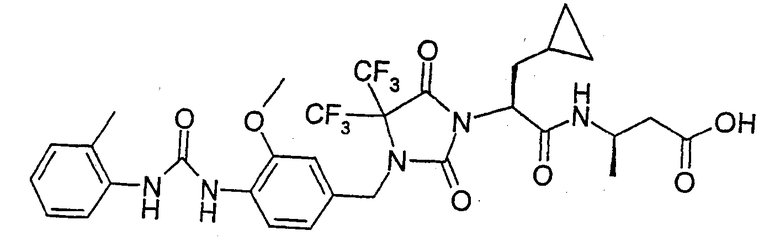
18а) 2-Трет-бутокси-4,4-бис(трифторметил)-1,3-оксазабута-1,3-диен
Указанное в заголовке соединение получают способом, описанным W. Steglich et al., Chem. Ber. 107 (1974), 1488-1498. Для получения безводного гексафторацетона (HFA) тригидрат HFA добавляют по каплям к концентрированной серной кислоте, нагретой до 80°С. Образовавшийся газ еще раз промывают концентрированной серной кислотой и подают в газовое пространство реакционной колбы. Обратный холодильник, заполненный смесью ацетона и сухого льда, присоединяют к отверстию колбы для выпуска газа.
Как указано выше, раствор 20 г (170 ммоль) трет-бутилкарбамата в 150 мл дихлорметана подвергают взаимодействию с безводным газообразным HFA до насыщения реакционного раствора. Растворитель удаляют в вакууме и образовавшийся сырой 2-трет-бутоксикарбониламино-2-гидрокси-1,1,1,3,3,3-гексафторпропан (выход 48,3 г, 100%) используют на следующей стадии реакции.
13,6 г трифторуксусного ангидрида и 5 капель хинолина добавляют по каплям при 0°С к раствору 50,05 г (176 ммоль) 2-трет-бутоксикарбониламино-2-гидрокси-1,1,1,3,3,3-гексафторпропана в 300 мл диэтилового эфира. Смесь перемешивают при 0°С в течение 10 минут и добавляют по каплям еще 27,2 г трифторуксусного ангидрида. Реакционную смесь перемешивают при 0°С (внешняя температура) в течение 30 минут, при этом внутренняя температура смеси повышается до 8-10°С. Смесь охлаждают до 0°С и добавляют 50,01 г (388 ммоль) хинолина, в результате чего соль трифторуксусной кислоты хинолина начинает кристаллизоваться. Затем смесь перемешивают при 0°С в течение 2 часов и фильтруют. Остаточную соль удаляют из фильтрата, перегоняя ее в вакууме в колбу-приемник, охлаждаемую смесью ацетона с сухим льдом. Дистиллят перегоняют через колонку Вигрекса. Получают 36,2 г (77%) указанного в заголовке соединения. Температура кипения: 126-130°С.
18b) Трет-бутиловый эфир (S)-β-циклопропилаланина
3,5 г (27,1 ммоль) (S)-β-циклопропилаланина добавляют при комнатной температуре к смеси 50 мл диоксана и 5 мл концентрированной серной кислоты (которую получают, осторожно добавляя по каплям кислоту к диоксану при 5°С). Раствор переносят в запаиваемую трубку, в которой конденсируют 40 мл изобутилена при -78°С. Запаянную трубку встряхивают в шейкере при комнатной температуре в течение 24 часов. Запаянную трубку открывают (при охлаждении), реакционную смесь осторожно вводят в охлажденную до 0°С перемешиваемую смесь 30 мл триэтиламина и 50 мл воды. Избыток изобутилена удаляют и продукт экстрагируют простым эфиром (2×50 мл). Эфирные фазы сушат над сульфатом магния и фильтруют, растворитель удаляют в вакууме, получая при этом сырой продукт (бледно-желтое масло), который используют при выполнении следующей реакции без дальнейшей очистки. Выход: 4,2 г (84%).
18с) Трет-бутиловый эфир (S)-N-формил-β-циклопропилаланина
Смесь 10 г (54 ммоль) трет-бутилового эфира (S)-β-циклопропилаланина и 4,7 г (55,2 ммоль) цианометилформиата в 100 мл дихлорметана перемешивают в течение ночи при комнатной температуре. Растворитель удаляют в вакууме и образовавшийся остаток перегоняют в вакууме. Выход: 8,8 г (76%). Температура кипения: 120°С/40 Па (0,3 тор).
18d) Трет-бутил-(S)-2-(4,4-бис(трифторметил)-2,5-диоксоимидазолидин-1-ил)-2-(циклопропилметил)ацетат
2,4 г (12,1 ммоль) дифосгена добавляют при -30°С к раствору 2,5 г (11,7 ммоль) трет-бутилового эфира (S)-N-формил-β-циклопропилаланина и 2,5 г (24,7 ммоль) триэтиламина в 100 мл сухого дихлорметана. Реакционный раствор оставляют нагреваться до -15°С в течение 1 часа и перемешивают при указанной температуре до окончания реакции. Затем реакционный раствор дважды промывают при комнатной температуре раствором гидрокарбоната натрия крепостью 7% и органическую фазу сушат над сульфатом магния. Смесь фильтруют, растворитель удаляют в вакууме и остаток поглощают 70 мл бензола. 3,05 г (11,5 ммоль) 2-трет-бутокси-4,4-бис(трифторметил)-1,3-оксазабута-1,3-диена в 10 мл бензола добавляют по каплям к полученному раствору при комнатной температуре. Реакционный раствор нагревают в течение ночи при 60°С и удаляют бензол в вакууме. Остаток хроматографируют на силикагеле (элюент: петролейный эфир/этилацетат = 8/1). Выход: 3,7 г (78%). Температура плавления: 76-77°С. [α]20 = -28° (с = 1, CHCl3).
18е) (S)-2-(4,4-Бис(трифторметил)-2,5-диоксоимидазолидин-1-ил)-2-(циклопропилметил)уксусная кислота
Раствор 7 г (17,3 ммоль) соединения по примеру 18d в 20 мл дихлорметана добавляют при 10°С к смеси 30 мл трифторуксусной кислоты и 50 мл дихлорметана и перемешивают смесь при комнатной температуре в течение 16 часов. Трифторуксусную кислоту и дихлорметан удаляют в вакууме, получая при этом 5,9 г (98%) указанного в заголовке соединения. Температура плавления: 123-125°С. [α]22 = -26° (с = 2, метанол).
18f) (S)-2-(4,4-Бис(трифторметил)-3-(4-(3-(2-метилфенил)уреидо)-3-метоксибензил)-2,5-диоксоимидазолидин-1-ил)-2-(циклопропилметил)уксусная кислота

3,2 мл раствора н-бутиллития (2,5 М раствор в гексане) добавляют при -40°С в атмосфере аргона к раствору 1,39 г (4 ммоль) соединения по примеру 18е в 40 мл абсолютного ТГФ. Реакционную смесь оставляют нагреваться до 0°С при перемешивании, добавляют раствор 2,43 г (8 ммоль) 4-(3-(2-метилфенил)уреидо)-3-метоксибензилхлорида в 20 мл абсолютного ТГФ и перемешивают при комнатной температуре в течение 3 часов. Добавляют 20 мл 1 н. раствора хлористоводородной кислоты и ТГФ удаляют в вакууме. Водную фазу дважды экстрагируют метил-трет-бутиловым эфиром. Объединенные органические фазы сушат над сульфатом натрия, фильтруют и концентрируют в вакууме. Остаток очищают препаративной ВЭЖХ. Фракции продукта концентрируют и сушат вымораживанием, получая при этом 1,41 г (57%) указанного в заголовке соединения.
18g) (R)-3-((S)-2-(4,4-Бис(трифторметил)-3-(4-(3-(2-метилфенил)уреидо)-3-метоксибензил)-2,5-диоксоимидазолидин-1-ил)-2-(циклопропилметил)ацетиламино)-3-метилпропионовая кислота
Указанное в заголовке соединение можно получить способом по примерам 1f и 1g из соединения по приеру 18f и трет-бутил-(R)-3-аминобутаноата, выполняя реакцию сочетания с последующим отщеплением трет-бутилового эфира.
Пример 19
(S)-3-((S)-2-(4,4-Бис(трифторметил)-3-(4-(3-(2-метилфенил)уреидо)-3-метоксибензил)-2,5-диоксоимидазолидин-1-ил)-2-(циклопропилметил)ацетиламино)-3-фенилпропионовая кислота
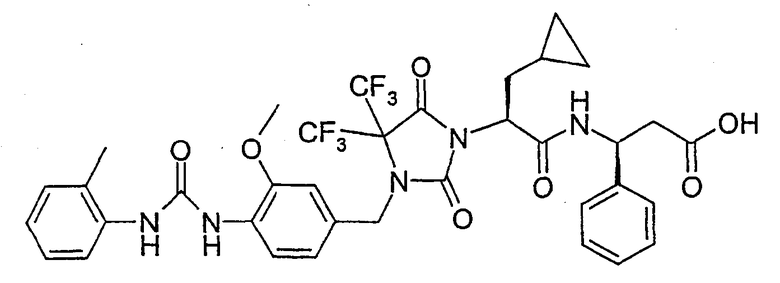
19а) Этил-(S)-3-((S)-2-(4,4-бис(трифторметил)-3-(4-(3-(2-метилфенил)уреидо)-3-(метоксибензил)-2,5-доксоимидазолидин-1-ил)-2-(циклопропилметил)ацетиламино)-3-фенилпропионат
748 мг (2,28 ммоль) TOTU (тетрафторборат О-((циано(этоксикарбонил)метилен)амино)-N,N,N',N'-тетраметилурония) и 368 мкл N,N-диизопропилэтиламина добавляют при 0°С к раствору 1,41 г (2,28 ммоль) соединения по примеру 18f и 442 мг (2,28 ммоль) этил-(S)-3-амино-3-фенилпропионата в 20 мл абсолютного диметилформамида (ДМФ). Реакционную смесь перемешивают при комнатной температуре в течение 1 часа, ДМФ удаляют в вакууме, остаток поглощают этилацетатом и этилацетатный раствор последовательно промывают водным раствором KHSO4/K2SO4, насыщенным раствором NaHCO3 и водой. Органическую фазу сушат над сульфатом натрия и фильтруют, растворитель удаляют в вакууме и остаток хроматографируют на силикагеле, используя гептан/этилацетат (3/2). Фракции продукта концентрируют и получают 1,48 г (82%) указанного в заголовке соединения.
19b) (S)-3-((S)-2-(4,4-Бис(трифторметил)-3-(4-(3-(2-метилфенил)уреидо)-3-метоксибензил)-2,5-диоксоимидазолидин-1-ил)-2-(циклопропилметил)ацетиламино)-3-фенилпропионовая кислота
Раствор 1,46 г (1,84 ммоль) соединения по примеру 19а в 40 мл N-метил-2-пирролидона и 20 мл 6 н. раствора хлористоводородной кислоты нагревают до 60°С в течение 6 часов. Реакционную смесь охлаждают до комнатной температуры и выливают в 300 мл воды, осадок отфильтровывают через вакуум-фильтр, промывают водой и сушат над пентоксидом фосфора. Сырой продукт дважды хроматографируют на силикагеле (элюент: дихлорметан/метанол/уксусная кислота/вода = 95/5/0,5/0,5). Фракции продукта концентрируют, остаток поглощают дихлорметаном и органическую фазу промывают водой и сушат над сульфатом натрия. Продукт фильтруют, растворитель удаляют в вакууме и сушат вымораживанием, получая при этом 1,19 г (85%) указанного в заголовке соединения.
ES(+)-MC: 764,2 (М+Н)+
Пример 20
(R)-3-((S)-2-(4,4-Бис(трифторметил)-3-(4-(3-(2-метилфенил)уреидо)-3-метоксибензил)-2,5-диоксоимидазолидин-1-ил)-2-(2-метилпропил)ацетиламино)-3-метилпропионовая кислота
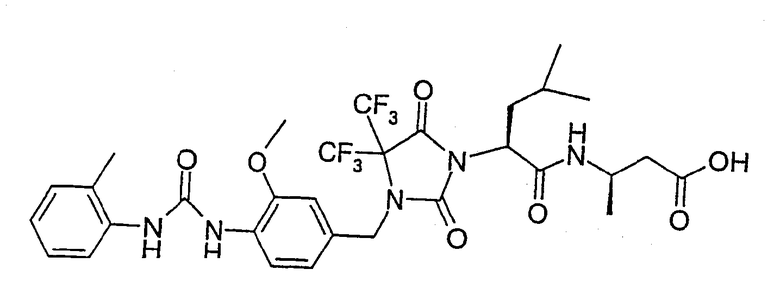
20а) Трет-бутиловый эфир N-формил-L-лейцина
Данный способ получения аналогичен способу, описанному W. Duczek et al., Synthesis 1996, 37-38. Раствор 4,04 г (40 ммоль) триэтиламина в 10 мл дихлорметана добавляют при 0°С к раствору 8,94 г (40 ммоль) гидрохлорида трет-бутилового эфира L-лейцина и 3,4 г (40 ммоль) цианометилформиата в 60 мл дихлорметана. Реакционный раствор оставляют нагреваться до комнатной температуры, перемешивают в течение ночи при комнатной температуре и дважды промывают насыщенным раствором NaCl. Фазы разделяют и органическую фазу сушат над сульфатом магния. Реакционную смесь фильтруют, растворитель удаляют в вакууме и образовавшийся остаток перегоняют в вакууме. Выход: 7,5 г (87%). Температура кипения: 118°С/2,7 Па (0,02 тор).
20b) Трет-бутил-(S)-2-(4,4-бис(трифторметил)-2,5-диоксоимидазолидин-1-ил)-2-(2-метилпропил)ацетат
2,4 г (12,1 ммоль) дифосгена добавляют при -30°С к раствору 2,5 г (11,6 ммоль) трет-бутилового эфира N-формил-L-лейцина и 2,5 г (24,7 ммоль) триэтиламина в 100 мл сухого дихлорметана. Реакционный раствор оставляют нагреваться до -10°С в течение 1 часа и перемешивают при указанной температуре до окончания реакции. Затем реакционный раствор дважды промывают при комнатной температуре раствором гидрокарбоната натрия крепостью 7%. Фазы разделяют и органическую фазу сушат над сульфатом магния. Реакционную смесь фильтруют, растворитель удаляют в вакууме и остаток поглощают 70 мл бензола. К полученному раствору по каплям добавляют при комнатной температуре 3 г (11,3 ммоль) 2-трет-бутокси-4,4-бис(трифторметил)-1,3-оксазабута-1,3-диена в 10 мл бензола. Реакционный раствор нагревают до 60°С в течение ночи и бензол удаляют в вакууме. Остаток хроматографируют на силикагеле (элюент: петролейный эфир/этлацетат = 10/1) и получают 3,7 г (80%) указанного в заголовке соединения. Температура плавления: 105-106°С. [α]20 = -24° (с = 1, CHCl3).
20с) (S)-2-(4,4-Бис(трифторметил)-2,5-диоксоимидазолидин-1-ил)-2-(2-метилпропил)уксусная кислота
Раствор 7 г (17,2 ммоль) соединения по примеру 20b в 20 мл дихлорметана добавляют при 10°С к смеси 30 мл трифторуксусной кислоты и 50 мл дихлорметана и реакционную смесь перемешивают при комнатной температуре в течение 16 часов. Трифторуксусную кислоту и дихлорметан удаляют в вакууме и получают 6,0 г (99%) указанного в заголовке соединения. Температура плавления: 154-156°С. [α]22 = -23° (с = 2, метанол).
20d) (R)-3-((S)-2-(4,4-Бис(трифторметил)-3-(4-(3-(2-метилфенил)уреидо)-3-метоксибензил)-2,5-диоксоимидазолидин-1-ил)-2-(2-метилпропил)ацетиламино)-3-метилпропионовая кислота
Указанное в заголовке соединение получают способами по примерам 1f и 1g из 500 мг (0,809 ммоль) (S)-2-(4,4-бис(трифторметил)-3-(4-(3-(2-метилфенил)уреидо)-3-метоксибензил)-2,5-диоксоимидазолидин-1-ил)-2-(2-метилпропил)уксусной кислоты формулы
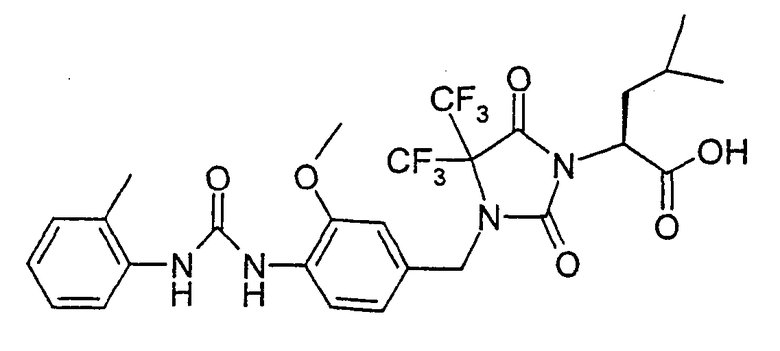
полученной из (S)-2-(4,4-бис(трифторметил)-2,5-диоксоимидазолидин-1-ил)-2-(2-метилпропил)уксусной кислоты и 4-(3-(2-метилфенил)уреидо)-3-метоксибензилхлорида способом по примеру 18f, и 128 мг (0,809 ммоль) трет-бутил-(R)-3-аминобутаноата. Реакционную смесь подвергают реакции сочетания, очищают хроматографией на силикагеле (элюент: гептан/этилацетат = 3/2) и отщепляют трет-бутиловый эфир, получая при этом 299 мг (53%) указанного в заголовке соединения.
ES(+)-MC: 704,5 (М+Н)+
Пример 21
(S)-3-((S)-2-(4,4-Бис(трифторметил)-3-(4-(3-(2-метилфенил)уреидо)-3-метоксибензил)-2,5-диоксоимидазолидин-1-ил)-2-(2-метилпропил)ацетиламино)-3-фенилпропионовая кислота
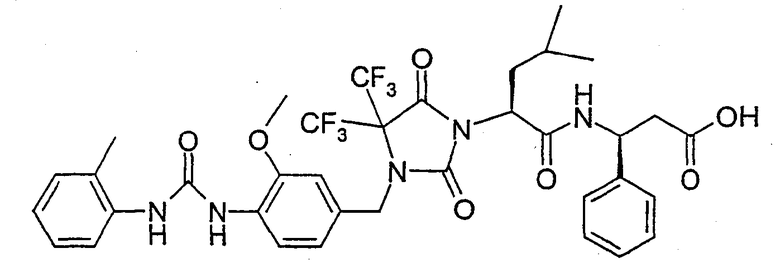
21а) Этил-(S)-3-((S)-2-(4,4-бис(трифторметил)-3-(4-(3-(2-метилфенил)уреидо)-3-метоксибензил)-2,5-диоксоимидазолидин-1-ил)-2-(2-метилпропил)ацетиламино)-3-фенилпропионат
1,89 г (5,77 ммоль) TOTU и 932 мкл N,N-диизопропилэтиламина добавляют при 0°С к раствору 3,57 г (5,77 ммоль) (S)-2-(4,4-бис(трифторметил)-3-(4-(3-(2-метилфенил)уреидо)-3-метоксибензил)-2,5-диоксоимидазолидин-1-ил)-2-(2-метилпропил)уксусной кислоты (полученной из (S)-2-(4,4-бис(трифторметил)-2,5-диоксоимидазолидин-1-ил)-2-(2-метилпропил)уксусной кислоты и 4-(3-(2-метилфенил)уреидо)-3-метоксибензилхлорида способом по примеру 18f) и 1,11 г (5,77 ммоль) этил-(S)-3-амино-3-фенилпропионата в 30 мл абсолютного ДМФ. Реакционную смесь перемешивают при комнатной температуре в течение 1 часа, ДМФ удаляют в вакууме, остаток поглощают этилацетатом и этилацетатный раствор последовательно промывают водным раствором KHSO4/K2SO4, насыщенным раствором NaHCO3 и водой. Органическую фазу сушат над сульфатом натрия и фильтруют, растворитель удаляют в вакууме и остаток хроматографируют на силикагеле, используя этилацетат/гептан (2/3). Фракции продукта концентрируют и получают 3,26 г (71%) указанного в заголовке соединения.
21b) (S)-3-((S)-2-(4,4-Бис(трифторметил)-3-(4-(3-(2-метилфенил)уреидо)-3-метоксибензил)-2,5-диоксоимидазолидин-1-ил)-2-(2-метилпропил)ацетиламино)-3-фенилпропионовая кислота
45 мл 6 н. раствора хлористоводородной кислоты добавляют к раствору 3,25 г (4,09 ммоль) соединения по примеру 21а в 90 мл N-метил-2-пирролидона и смесь нагревают до 60°С в течение 6 часов. Реакционную смесь охлаждают до комнатной температуры и выливают в 600 мл воды. Осадок отфильтровывают через вакуум-фильтр, промывают водой и сушат над пентоксидом фосфора. Сырой продукт дважды очищают хроматографией на силикагеле (элюент: дихлорметан/метанол/уксусная кислота/вода = 95/5/0,5/0,5), фракции продукта концентрируют и остаток поглощают дихлорметаном. Органическую фазу дважды промывают водой и сушат над сульфатом магния. Продукт фильтруют, концентрируют и сушат вымораживанием, получая при этом 2,7 г (86%) указанного в заголовке соединения.
ES(+)-MC: 766,2 (М+Н)+
Пример 22
Натриевая соль (S)-3-((S)-2-(4,4-бис(трифторметил)-3-(4-(3-(2-метилфенил)уреидо)-3-метоксибензил)-2,5-диоксоимидазолидин-1-ил)-2-(2-метилпропил)ацетиламино)-3-фенилпропионовой кислоты
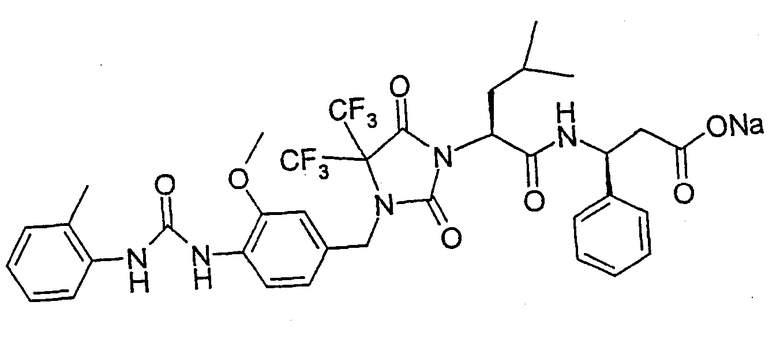
1,24 мл 1 н. раствора гидроксида натрия (разбавленного 20 мл воды) добавляют при перемешивании к суспензии 1 г (1,3 ммоль) соединения по примеру 21 в 100 мл ацетонитрила и 200 мл воды. Раствор сушат вымораживанием и получают 1,01 г (79%) указанного в заголовке соединения.
ES(+)-MC: 766,2 (3-((S)-2-(4,4-бис(трифторметил)-3-(4-(3-(2-метилфенил)уреидо)-3-метоксибензил)-2,5-диоксоимидазолидин-1-ил)-2-(2-метилпропил)ацетиламино)-3-фенилпропионовая кислота + Н)+
Пример 23
Натриевая соль (S)-3-((S)-2-(4,4-бис(трифторметил)-3-(4-(3-(2-метилфенил)уреидо)-3-метоксибензил)-2,5-диоксоимидазолидин-1-ил)-2-(циклопропилметил)ацетиламино)-3-фенилпропионовой кислоты
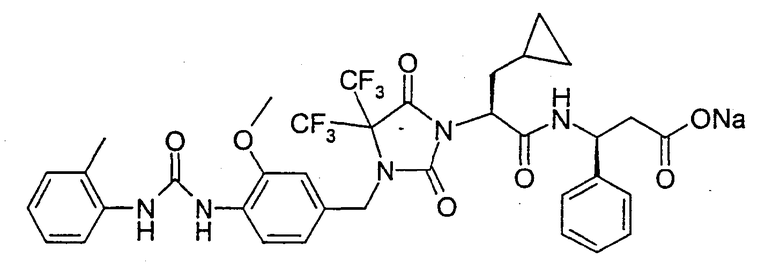
720 мг (99%) указанного в заголовке соединения получают из 720 мг соединения по примеру 19b способом, описанным в примере 22.
ES(+)-MC: 764,3 ((S)-3-((S)-2-(4,4-бис(трифторметил)-3-(4-(3-(2-метилфенил)уреидо)-3-метоксибензил)-2,5-диоксоимидазолидин-1-ил)-2-(циклопропилметил)ацетиламино)-3-фенилпропионовая кислота + Н)+
Исследование биологической активности
А) Испытание адгезии клеток U937/VCAM-1
Описанный ниже анализ представляет метод испытания активности соединений формулы I в отношении подавления взаимодействия между VCAM-1 и VLA-4, который является специфичным для данного взаимодействия. Связывающие компоненты клетки, то есть интегрины VLA-4, получены в природной форме в виде поверхностных молекул на клетках U937 человека (АТСС CRL 1593), относящихся к группе лейкоцитов. В качестве специфических связывающих компонентов использованы рекомбинантные растворимые слитые белки, полученные методами рекомбинантных ДНК, которые включают экстрацитоплазматический домен VCAM-1 человека и константную область иммуноглобулина подкласса IgG1 человека.
Метод измерения адгезии клеток U937 (ATCC CRL 1593) к hVCAM-1(1-3)-IgG
1. Получение VCAM-1(1-3)-IgG и CD4-IgG человека
В данном анализе использована генетическая конструкция для экспрессии внеклеточного домена VCAM-1 человека в сочетании с генетической последовательностью тяжелой цепи иммуноглобулина IgG1 человека (шарнирная область, области СН2 и СН3) (предоставлена Dr. Brian Seed, Massachusetts General Hospital, Boston, USA; cf. Damle and Aruffo, Proc. Natl. Acad. Sci. USA 1991, 88, 6403). Растворимый слитый белок hVCAM-1-(1-3)-IgG содержит три аминоконцевых внеклеточных иммуноглобулиноподобных домена VCAM-1 человека (Damle and Aruffo, Proc. Natl. Acad. Sci. USA 1991, 88, 6403). CD4-IgG (Zettlmeissl et al., DNA and Cell Biology 1990, 9, 347) служит в качестве слитого белка для отрицательных контрольных образцов. Рекомбинантные белки экспрессированы в виде растворимых белков после трансфекции DEAE/декстранопосредованной ДНК в клетках яичника китайского хомячка (COS) (ATCC CRL 1651) стандартными методами (Ausubel et al., Current protocols in molecular biology, John Willey & Sons, Inc., 1994).
2. Метод измерения адгезии клеток U937 к hVCAM-1-(1-3)-IgG
2.1. 96-Луночные титрационные планшеты для микроанализа (Nunc Maxisorb) инкубируют при комнатной температуре в течение 1 часа с 100 мкл/лунку раствором антитела козы против IgG человека (10 мкг/мл в 50 мМ трис, рН 9,5). Затем раствор антитела удаляют и лунки один раз промывают физиологическим раствором с фосфатным буфером (PBS).
2.2. 150 мкл/лунку блокирующего буфера (1% BSA в PBS) инкубируют на планшетах при комнатной температуре в течение 0,5 часа. Затем блокирующий буфер удаляют и лунки один раз промывают PBS.
2.3. 100 мкл/лунку супернатанта культуры трансфецированных клеток COS инкубируют на планшетах при комнатной температуре в течение 1,5 часов. Клетки COS трансфецируют плазмидой, кодирующей три N-концевых иммуноглобулиноподобных домена VCAM-1, связанных с Fc-фрагментом IgG1 человека (hVCAM-1(1-3)-IgG). Количество hVCAM-1(1-3)-IgG составляет примерно 0,5-1 мкг/мл. Супернатант культуры удаляют и лунки один раз промывают PBS.
2.4. Планшеты инкубируют при комнатной температуре в течение 20 минут с 100 мкл/лунку блокирующего Fc-рецептор буфера (1 мг/мл γ-глобулина, 100 мМ NaCl, 100 мкМ MgCl2, 100 мкМ MnCl2, 100 мкМ CaCl2, 1 мг/мл BSA в 50 мМ HEPES, рН 7,5). Затем блокирующий Fc-рецептор буфер удаляют и лунки один раз промывают PBS.
2.5. Вводят 20 мкл связывающего буфера (100 мМ NaCl, 100 мкМ MgCl2, 100 мкМ MnCl2, 100 мкМ CaCl2, 1 мг/мл BSA в 50 мМ HEPES, рН 7,5), добавляют испытуемые вещества в 10 мкл связывающего буфера и инкубируют в течение 20 минут. Антитела против VCAM-1 (BBT, No. BBA6) и против VLA-4 (Immunotech, No. 0764) служат в качестве контрольных образцов.
2.6. Клетки U937 инкубируют в течение 20 минут в блокирующем Fc-рецептор буфере и добавляют пипеткой в концентрации 1×106/мл и в количестве 100 мкл/лунку (конечный объем 125 мкл/лунку).
2.7. Планшеты медленно погружают под углом 45° в стоп-буфер (100 мМ NaCl, 100 мкМ MgCl2, 100 мкМ MnCl2, 100 мкМ CaCl2 в 25 мМ трис, рН 7,5) и встряхивают. Данную операцию повторяют.
2.8. 50 мкл/лунку окрашивающего раствора (16,7 мкг/мл красителя Хехста 33258, 4% формальдегида, 0,5% тритона Х-100 в PBS) инкубируют на планшетах в течение 15 минут.
2.9. Планшеты встряхивают и медленно погружают под углом 45° в стоп-буфер (100 мМ NaCl, 100 мкМ MgCl2, 100 мкМ MnCl2, 100 мкМ CaCl2 в 25 мМ трис, рН 7,5). Данную операцию повторяют. Затем вместе с оставшейся жидкостью (стоп-буфер) планшеты измеряют в цитофлуориметре (Millipore) (чувствительность: 5, фильтр: длина волны возбуждения: 360 нм, длина волны излучения: 460 нм).
Интенсивность света, излучаемого окрашенными клетками U937, соответствует количеству клеток U937, связанных с hVCAM-1(1-3)-IgG и оставшихся на планшете, и, таким образом, является показателем способности добавленного испытуемого вещества ингибировать данную адгезию. С учетом ингибирования адгезии при разных концентрациях испытуемого вещества можно вычислить концентрацию IC50, вызывающую 50% ингибирование адгезии.
3. Результаты
При испытании адгезии клеток U937/VCAM-1 (значения IC50 указаны в нМ (наномоль/литр)) получены нижеследующие результаты.
Фармакологические свойства соединений формулы I можно также исследовать с использованием нижеследующих моделей.
В) Адгезия лейкоцитов у крыс
Воздействие соединений формулы I на адгезию лейкоцитов в венулах крыс исследуют при помощи модели адгезии лейкоцитов у крыс. Адгезия лейкоцитов к эндотелию посткапиллярных венул считается важной стадией развития воспалительных реакций (J. M. Harlan, Blood 1985, 65, 513). В процессе рекрутинга лейкоцитов из крови в очаги воспаления происходит хорошо скоординированная динамическая последовательность событий, в которой активную роль играют хемотактические цитокины и адгезионные молекулы клеток. Установлено, что взаимодействие VCAM-1/VLA-4 имеет важное значение в процессе адгезии и миграции лейкоцитов и увеличении проницаемости сосудов для макромолекул, индуцируемых разными веществами-посредниками и цитокинами (D. Seiffge, Int. J. Microcirc. 1995, 15, 301). В настоящей модели генерализованное воспаление или ревматоидный артрит, вызывающий адгезию лейкоцитов и их миграцию в очаги воспаления органа, провоцируют местной или системной инъекцией эндотоксинов, например зимосана, бактериальных токсинов, таких как липополисахариды (LPS), или адъюванта Фрейнда. Затем определяют увеличение адгезии к эндотелию венул, вызываемое эндотоксином.
Для определения адгезии лейкоцитов используют инвертированный микроскоп с камерой (Zeiss), оснащенный видеосистемой. Зимосан или бактериальный эндотоксин инъецируют самцам крыс Sprague-Dawley (с массой тела около 250 г), производя легкую премедикацию галотаном. Контрольным животным вводят такой же объем физиологического раствора крепостью 0,9%. Затем животным подкожно или перорально вводят испытуемое вещество в виде одной или нескольких доз. Для выполнения измерений крыс анестезируют при помощи внутримышечной инъекции 1,25 г/кг уретана. У животных сохраняют самопроизвольное дыхание через трахеотомическую трубку. Температуру тела поддерживают равной 37°С при помощи термостата с регулируемым нагревом. В нагреваемом термостатом (37°С) окне предметного столика микроскопа осторожно обнажают мезентерий, делая надрез в подчревной области, и покрывают жидким парафином при 37°С. Илеоцекальную область мезентерия фиксируют при помощи трех тупоконечных игл и формовочной глины. После 30-минутного уравновешивания, в течение которого ткань стабилизируется, определяют адгезию лейкоцитов в посткапиллярных венулах диаметром 20-30 мкм и длиной около 100 мкм, производя подсчет в 2-3 сегментах венул с 10-минутными интервалами в течение 1 часа. Лейкоцит считается связанным с эндотелием, если он остается неподвижным в течение 30 секунд. После выполнения данного эксперимента производят системный подсчет лейкоцитов и содержания фибриногена в крови. Ингибирование адгезии лейкоцитов испытуемым веществом определяют с учетом уменьшения (в %) количества связанных лейкоцитов у подопытных животных по сравнению с количеством таких лейкоцитов у контрольных животных.
С) Аллергическая реакция замедленного типа у мышей
Противоаллергическое и противовоспалительное действие соединений формулы I исследуют при помощи модели аллергической реакции замедленного типа (DTH). DTH представляет воспалительную реакцию кожи, вызываемую повышенной чувствительностью к антигенным веществам. Для определения соответствующей воспалительной реакции и рекрутинга лейкоцитов в очаги воспаления in vivo вещества испытывают при помощи нижеследующей модели DTH у мышей (см. также T.B. Issekutz, J. Immunol. 1991, 147, 4178).
У самок мышей BALB/c (с массой тела около 20 г) в нескольких группах сенсибилизируют эпителиальный слой на бритом участке кожи 150 мкл раствора оксазалона крепостью 3%, который вызывает сильную воспалительную реакцию DTH. Через 6 дней снова вызывают указанную реакцию, вводя в правое ухо животных 20 мкл раствора оксазалона крепостью 1%. Испытуемые вещества вводят подкожно или перорально за 44 часа до провоцирования реакции, за 20 часов до провоцирования реакции и через 4 часа после провоцирования реакции. Непосредственно перед провоцированием реакции и через 24 часа после провоцирования реакции при помощи микрометра фирмы Mitutoyo Engineering измеряют толщину правого уха, изменившуюся вследствие возникновения воспалительной припухлости. Для каждого животного в группе определяют разность между двумя измерениями. Затем сравнивают средние значения разности в группе животных, которым вводили испытуемое вещество, и в контрольной группе животных, которым не вводили испытуемое вещество. Мерой эффективности вещества является процентное уменьшение припухлости уха.
D) Противоастматическое воздействие на морских свинок
Воздействие соединений формулы I на функцию легких и противоастматическое действие указанных веществ определяют при помощи модели с использованием морских свинок методом, описанным G. Moacevic, Arch. Toxicol. 1975, 34, 1. В техническом отношении данное исследование выполняют в соответствии с описанием, приведенным Moacevic. Используют самцов морских свинок-альбиносов с массой тела 300-500 г. Животных помещают в плетизмограф (фирмы FMI) и регистрируют три исходных значения параметров частоты и амплитуды дыхания. В данной модели астматическое дыхание характеризуется уменьшением амплитуды дыхания (= уменьшение дыхательного объема вследствие бронхостеноза) и увеличением частоты дыхания (= рефлекторная реакция). Такое состояние известно у больных астмой как одышка.
Морских свинок-альбиносов сенсибилизируют за 22 дня до начала исследования 1 мл/животное раствора овальбумина крепостью 0,1% на протяжении двух последовательных дней. Экспериментальный приступ астмы вызывают ингаляцией раствора овальбумина крепостью 0,3% в течение 1 минуты. После восстановительной фазы продолжительностью 40-60 минут животные вдыхают испытуемое вещество в виде водного раствора. Сразу же вслед за этим животным вводят раствор овальбумина крепостью 0,3% в течение 1 минуты. В следующую восстановительную фазу продолжительностью 30 минут животные вдыхают обычный воздух. Указанную процедуру повторяют дважды. Если приступ астмы угрожает жизни, животным вводят кислород.
Противоастматическое действие можно определить методом, описанным Abraham et al., J. Clin. Invest. 1994, 93, 776.
Е) Исследование противоатеросклеротического действия при помощи нижеследующих животных моделей
Модель исследования образования неоинтимы с использованием манжеты
Мыши дикого типа линии С57ВL/6J предоставлены службой разведения Charles River Wiga GmbH (Sulzfeld, FRG), и гомозиготные мыши КО линии C57BL/6J-ApoE tm1Unc (ApoE KO) предоставлены фирмой Jackson Laboratory (Maine, USA). В начале эксперимента всех мышей в возрасте 10-12 недель содержат в полностью кондиционируемых помещениях при температуре 22°С. Цикл чередования дня и ночи, устанавливаемый программой регулируемого светового дня, равен 12 часам. Мышей анестезируют внутрибрюшинной инъекцией 60 мг/кг массы тела пентобарбитала натрия. Всем животным дополнительно внутримышечно вводят 0,01 мг ксилазина/10 г массы тела.
Мышей фиксируют в положении супинации, бреют и дезинфицируют внутренние поверхности обеих задних лап. На коже с внутренней стороны левого бедра делают продольный разрез длиной примерно 1 см, бедренную артерию отделяют от окружающих тканей, от бедренной вены и седалищного нерва. Отрезают кусок полиэтиленовой трубки длиной примерно 2 мм (внутренний диаметр 0,58 мм, наружный диаметр 0,965 мм, Becton Dickinson, Sparks, MD, USA), накладывают на бедренную артерию и фиксируют пролиновыми нитями (7/0, метрический размер 0,5, фирмы Ethicon, Norderstedt, FRG). Кожу ушивают непрерывным швом. Правую заднюю лапу оперируют аналогичным образом, но на бедренную артерию манжету не накладывают. Затем животное возвращают в клетку. После операции животным ежедневно вводят испытуемое вещество.
В конце эксперимента мышей снова анестезируют внутрибрюшинной инъекцией 60 мг/кг массы тела пентобарбитала натрия и внутримышечно вводят 0,01 мг ксилазина/10 г массы тела. Для фиксации сосудов in situ каждой мыши инъецируют в брюшную аорту раствор формалина крепостью 4%. Затем удаляют правую и левую бедренные артерии. С левой стороны удаляют часть артерии с прилегающим к манжете участком около 1 мм, указанная часть включает манжету и дистальный участок сосуда длиной 1 мм. С правой стороны указанная часть соответствует участку, выделенному во время операции без наложения манжеты.
Части левой и правой бедренных артерий, фиксированных раствором формалина крепостью 4%, погружают в парафин. В части левой артерии с наложенной манжетой и в соответствующей части контрольной правой артерии делают несколько поперечных срезов ткани, которые окрашивают гематоксилином и эозином для выполнения морфометрического анализа с использованием программного обеспечения (LeicaQWin фирмы Leica Imaging Systems, Cambridge, GB).
Для каждой мыши оценивают три поперечных среза ткани с участка левой бедренной артерии с наложенной манжетой и три поперечных среза ткани с соответствующего участка контрольной правой артерии. Окрашивают наружный эластический слой, внутренний эластический слой и граничный слой между полостью и эндотелием и при помощи анализирующей программы вычисляют размеры следующих областей: полости, неоинтимы и срединной части. Размеры указанных областей выражают в мкм2. Воздействие соединения определяют по уменьшению соотношения неоинтима/срединная часть по сравнению с контрольной группой.
Трансплантация сердца
В модели аллогенной трансплантации сердца выполняют трансплантацию между двумя генетически несовместимыми линиями крыс. Для указанной цели используют крыс Wistar-Furth в качестве животных-доноров и крыс Lewis в качестве животных-реципиентов. Животные предоставлены службой разведения Charles River Wiga GmbH (Sulzfeld, FRG). Самцов крыс Lewis с массой тела 270-330 г в возрасте 2,5-3 месяцев и самцов крыс Wistar-Furth с массой тела 200-250 г в возрасте 1,5-2 месяцев содержат в постоянных контролируемых условиях (температура 19-22°С; относительная влажность 50-55%; цикл чередования дня и ночи, устанавливаемый программой регулируемого светового дня, равен 12 часам).
Перед операцией крысам вводят 3,3 мг/кг массы тела ксилазина и 115 мг/кг массы тела кетамина. После начала действия анестезии делают срединный разрез брюшной полости реципиента. Брюшную аорту и нижнюю полую вену отделяют друг от друга между почечной артерией, веной и сосудами подвздошно-поясничной области. Затем аорту пережимают при помощи зажима для сосуда. Оба сосуда обвивают шелковой нитью и затягивают. Вторую шелковую нить свободно накладывают на конец нижней полой вены. После вскрытия брюшной полости животное-донора умерщвляют, перерезая крупные брюшные сосуды. В данный момент начинается ишемический период донорского органа. Затем вскрывают диафрагму и обнажают сердце. Верхнюю и нижнюю полую вену лигируют и перерезают со стороны лигатуры, удаленной от сердца. Легочные вены перевязывают шелковой нитью. Аорту и легочную артерию поднимают пинцетом и перерезают. Трансплантат освобождают от крови, оставшейся в сосудистой системе. Затем сердце поднимают, удаляют вместе с лигатурой из легкого и помещают в холодный физиологический раствор NaCl на одну-две минуты. Аорту и легочную артерию донорского органа соединяют "конец в конец" соответственно с брюшной артерией и нижней полой веной животного-реципиента. После выполнения анастомоза сосудов успешно восстанавливают венозное кровообращение и затем артериальное кровообращение. Брюшную полость закрывают, накладывая швы на брюшину/мышцы и кожу. После возобновления кровообращения и короткого восстановительного периода трансплантированное сердце начинает биться с синусным ритмом около 100-120 ударов/минуту. Для подавления иммунитета животным вводят циклоспорин А (CSA) подкожно или перорально с питьевой водой. После прохождения периода острого отторжения дозу можно уменьшить с 25 мг/кг массы тела до 5 мг/кг массы тела, начиная с 15-го дня после операции. Животным делают инъекции в шейную область один раз в сутки утром.
Подкожное введение CSA заменяют пероральным введением CSA на 22-ой день после операции для безопасного преодоления периода острого отторжения. Исследуемое вещество вводят в течение 100 дней после операции. По истечении периода наблюдения (100 дней) животных анестезируют и вскрывают брюшную полость. Сердце удаляют вместе с концами брюшных сосудов, делают срезы и хранят в 4% растворе формалина. Фиксированные срезы сердца погружают в парафин и окрашивают эластический слой стандартным гистологическим методом ван Гисона. Пролиферацию неоинтимы и обусловленное этим процессом сужение полости сосуда определяют методом Адамса и др. (Adams et al., Transplantation 1993, 56, 794). Определяют адгезию между внутренним эластическим слоем и эндотелием. Данное определение облегчает специальный краситель, используемый в методе ван Гисона, который избирательно окрашивает волокна эластического слоя. Действие соединения определяют по уменьшению пролиферации неоинтимы и, следовательно, атеросклероза трансплантата по сравнению с контрольной группой.
Модель атеросклероза у мышей с отсутствием АроЕ (КО)
Гомозиготные мыши КО линии C57BL/6J-ApoE tm1Unc (ApoE KO) предоставлены фирмой Jackson Laboratory (Maine, USA). В начале эксперимента всех животных в возрасте 10-12 недель содержат на стандартной подстилке для лабораторных животных (Altromin, Lage, FRG) в полностью кондиционируемых помещениях при температуре 22°С. Цикл чередования дня и ночи, устанавливаемый программой регулируемого светового дня, равен 12 часам. Животным вводят испытуемое вещество в течение 4 месяцев.
В конце эксперимента мышей анестезируют внутрибрюшинной инъекцией 60 мг/кг массы тела пентобарбитала натрия и внутримышечно вводят 0,01 мг ксилазина/10 г массы тела. Сердце, дугу аорты и нисходящую грудную аорту удаляют и фиксируют в растворе формалина крепостью 4%. Нисходящую аорту обрабатывают масляным красным О для окрашивания жировых отложений. Морфометрический анализ жировых отложений выполняют при помощи микроскопа (Leitz DM RBE фирмы Leica, Bensheim), присоединенной к нему камеры с устройством управления (CF 15 MCC фирмы Kappa Meβtechnik, Gleichen) и компьютера (Leica, Bensheim). Измерения выполняют при помощи компьютерной программы для анализа изображений (LeicaQWin фирмы Leica Imaging Systems, Cambridge, GB). Сердце и дугу аорты разрезают в продольном направлении и окрашивают гематоксилином и эозином для выполнения морфометрического анализа. В каждом случае анализируют 15-20 срезов. Другие срезы подвергают иммуногистохимическому анализу для определения макрофагов и Т-лимфоцитов. Действие соединения определяют по уменьшению образования бляшек в аорте по сравнению с контрольной группой.
F) Исследование кардиозащитного действия при помощи нижеследующей животной модели
Величина инфаркта сердца у крыс
Самцы крыс Wistar в возрасте 2,5-3 месяцев с массой тела 270-330 г предоставлены службой разведения Charles River Wiga GmbH (Sulzfeld, FRG). Животных содержит в постоянных контролируемых условиях (температура 19-22°С; относительная влажность 50-55%; цикл чередования дня и ночи, устанавливаемый программой регулируемого светового дня, равен 12 часам). Перед операцией крысам вводят 3,3 мг/кг массы тела ксилазина и 115 мг/кг массы тела кетамина. Затем животных интубируют и вентилируют легкие 30% кислородом. Грудную клетку бреют, дезинфицируют и вскрывают при помощи левосторонней боковой торакотомии. Левую коронарную артерию лигируют на 2-3 см ниже ушка предсердия на 48 часов или 4 недели, либо указанную артерию лигируют на 30 минут и перфузируют в течение 47,5 часа или 4 недель.
После операции грудную клетку закрывают и животных экстубируют после восстановления самопроизвольного дыхания. Испытуемое вещество вводят через 30 минут после наложения лигатуры или непосредственно перед реперфузией. Затем животным ежедневно вводят испытуемое вещество. В конце эксперимента животных снова анестезируют 3,3 мг/кг массы тела ксилазина и 115 мг/кг массы тела кетамина. Для анализа подвижности стенок животных с реперфузией сердца исследуют методом "ядерно-магнитно-резонансной томографии". Животным без реперфузии сердца вводят катетер с наконечником через правую сонную артерию для измерения вентрикулярного давления и сократимости левой полости сердца. Затем у всех животных удаляют сердце, которое перфузируют через аорту в обратном направлении в аппарате Лангендорфа теплым раствором голубого Эванса крепостью 1% при 37°С для определения анатомической зоны риска и не пораженной ишемией области. Делают 5-6 тонких срезов сердца и инкубируют в растворе хлорида 2,3,5-трифенилтетразолия в течение 15 минут для определения жизнеспособной и мертвой ткани сердца. Планиметрический анализ зоны риска и области инфаркта выполняют при помощи камеры (Leica, Bensheim), присоединенной к компьютеру с программой анализа (Leitz, Bensheim). Величину зоны риска выражают в процентах к площади левого желудочка сердца вместе с перегородкой и величину области инфаркта выражают в процентах к площади зоны риска. Действие соединения определяют по уменьшению области инфаркта относительно зоны риска по сравнению с контрольной группой.
| название | год | авторы | номер документа |
|---|---|---|---|
| СПИРОИМИДАЗОЛИДИНОВЫЕ ПРОИЗВОДНЫЕ, СПОСОБ ИХ ПОЛУЧЕНИЯ И ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ НА ИХ ОСНОВЕ | 2000 |
|
RU2245879C2 |
| НОВЫЕ ПРОИЗВОДНЫЕ ИМИДАЗОЛИДИНА, ИХ ПОЛУЧЕНИЕ И ИХ ПРИМЕНЕНИЕ В КАЧЕСТВЕ АНТАГОНИСТОВ VLA-4 | 2002 |
|
RU2318815C2 |
| ЗАМЕЩЕННЫЕ ПРОИЗВОДНЫЕ ИМИДАЗОЛИДИНА, СПОСОБ ПОЛУЧЕНИЯ И ФАРМАЦЕВТИЧЕСКИЙ ПРЕПАРАТ | 1998 |
|
RU2239641C2 |
| ПЯТИЧЛЕННЫЕ ГЕТЕРОЦИКЛЫ В КАЧЕСТВЕ ИНГИБИТОРОВ АДГЕЗИИ ЛЕЙКОЦИТОВ И VLA-4-АНТАГОНИСТОВ | 1997 |
|
RU2229296C2 |
| НОВЫЕ ПЯТИЧЛЕННЫЕ ГЕТЕРОЦИКЛЫ, ИХ ПОЛУЧЕНИЕ, ИХ ПРИМЕНЕНИЕ И СОДЕРЖАЩИЕ ИХ ФАРМАЦЕВТИЧЕСКИЕ ПРЕПАРАТЫ | 1998 |
|
RU2240326C2 |
| 4-ПИРИДИНИЛ-N-АЦИЛ-L-ФЕНИЛАЛАНИНЫ | 2000 |
|
RU2270193C2 |
| ЗАМЕЩЕННЫЕ АНИЛИДЫ, ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ И СПОСОБ ЛЕЧЕНИЯ | 1998 |
|
RU2233269C2 |
| ПРОИЗВОДНЫЕ ТИОАМИДА И ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ | 2000 |
|
RU2245874C2 |
| ПРОИЗВОДНЫЕ КАРБОНОВЫХ КИСЛОТ (ВАРИАНТЫ), ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ И СПОСОБ СЕЛЕКТИВНОГО ИНГИБИРОВАНИЯ СВЯЗЫВАНИЯ αβ ИНТЕГРИНА У МЛЕКОПИТАЮЩЕГО | 2000 |
|
RU2263109C2 |
| 4-ПИРИМИДИНИЛ-N-АЦИЛ-L-ФЕНИЛАЛАНИНЫ И ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ | 2000 |
|
RU2266901C2 |
Настоящее изобретение относится к новым производным имидазолидина формулы (I),
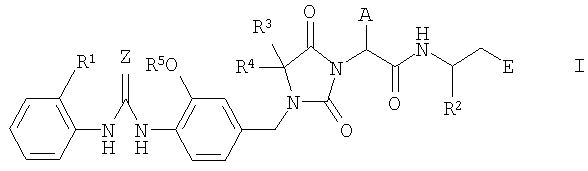
в которой А означает циклопропилметил- или изобутил; Е означает -CO-R6 или -СН2-O-R7; Z означает кислород; R1 означает водород или метил; R2 означает фенил, пиридил или (С1-С4)алкил; R3 и R4 означают метил или трифторметил; R5 означает водород или (C1-С4)алкил; R6 означает гидроксил, (С1-С10)алкокси или амино; R7 означает водород; или его стереоизомерам R-конфигурации или S-конфигурации, или смеси стереоизомеров R/S-конфигурации, или физиологически толерантным солям указанного соединения. Соединения формулы (I) являются ценными фармацевтически активными соединениями, пригодными для лечения воспалительных заболеваний, например ревматоидного артрита, или аллергических заболеваний. Соединения формулы (I) являются ингибиторами адгезии и миграции лейкоцитов и/или антагонистами адгезионного рецептора VLA-4, относящегося к группе интегринов. Указанные соединения пригодны для лечения заболеваний, вызываемых нежелательной степенью адгезии и/или миграции лейкоцитов или связанных с указанными процессами, а также заболеваний, в которых важное значение имеет межклеточное взаимодействие или взаимодействие клеток с матриксом, обусловленное взаимодействием рецепторов VLA-4 с их лигандами. Настоящее изобретение далее относится к способу получения соединений формулы (I) и фармацевтическому препарату на их основе. 3. н. и 11 з.п. ф-лы.
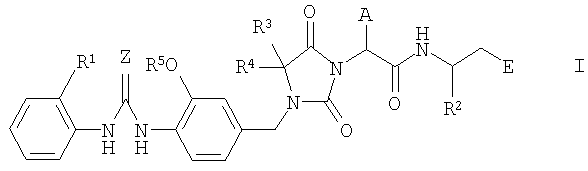
где А означает циклопропилметил- или изобутил;
Е означает -CO-R6 или -CH2-O-R7;
Z означает кислород;
R1 означает водород или метил;
R2 означает фенил, пиридил или (С1-С4)алкил;
R3 и R4 означают метил или трифторметил;
R5 означает водород или (С1-С4)алкил;
R6 означает гидроксил, (С1-С10)алкокси или амино;
R7 означает водород; или
его стереоизомеры R-конфигурации или S-конфигурации или смесь стереоизомеров R/S-конфигурации или физиологически толерантные соли указанного соединения.
А означает циклопропилметил- или изобутил;
Е означает -СООН, -СООС2Н5, -COOiC3H7 или -СН2-ОН;
Z означает кислород;
R1 означает метил;
R2 означает незамещенный фенил, пиридил или метил;
R3 и R4 означают метил;
R5 означает метил; или
его стереоизомеры R-конфигурации или S-конфигурации или смесь стереоизомеров R/S-конфигурации или физиологически толерантные соли указанного соединения.
А означает циклопропилметил- или изобутил;
Е означает -СООН, -СООС2Н5, -COOiC3Н7 или -CH2-ОН;
Z означает кислород;
R1 означает метил;
R2 означает незамещенный фенил, пиридил или метил;
R3 и R4 означают трифторметил;
R5 означает метил; или
его стереоизомеры R-конфигурации или S-конфигурации или смесь стереоизомеров R/S-конфигурации или физиологически толерантные соли указанного соединения.
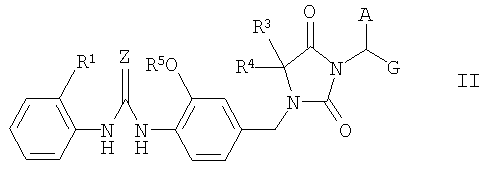
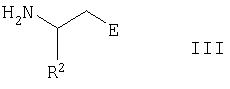
где А, Е, Z, R1, R2, R3, R4 и R5 имеют значения, указанные в пп.1-6 и G означает гидроксикарбонил, (С1-С6)алкоксикарбонил или производные активированной карбоновой кислоты.
| Устройство для доводки конического отверстия | 1979 |
|
SU918059A1 |
| WO 00/69831 A1, 23.11.2000 | |||
| ЗАМЕЩЕННЫЕ 5-ЧЛЕННЫЕ ГЕТЕРОЦИКЛЫ, СПОСОБ ИХ ПОЛУЧЕНИЯ, ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ | 1994 |
|
RU2151143C1 |
| RU 97119628 A, 10.09.1999. | |||

