Настоящее изобретение относится к полиолефиновым композициям, отличающимся улучшенным сопротивлением разрыву, но одновременно сохраняющим мягкость и хорошие механические свойства, которые можно выгодно использовать в листовом материале, таком как однослойные мембранные кровельные покрытия для плоских кровель промышленного и гражданского назначения, и в других применениях для кровельных покрытий. Данные композиции можно получить при помощи способа последовательной полимеризации.
В строительной промышленности в качестве предпочтительного материала для изготовления кровли традиционно использовали обычное кровельное покрытие из асфальта или стекловолокна. Однако совсем недавно как предпочтительный материал обычно используемые материалы были заменены на материалы мембранных кровельных покрытий, что обуславливается их стойкостью к растрескиванию на холоде, легкостью укладки и общей улучшенной и повышенной защитой от протечек во времени. Кроме этого, мембранные системы легче и безопаснее для укладки, и поэтому они более желательны для подрядчика, а также и для заказчика.
В данной области используют два типа мембран: термопластичные и эластомерные.
Термопластичные мембраны, такие как те, что получают из поливинилхлорида (ПВХ), хлорированного полиэтилена (СРЕ), хлорсульфонированного полиэтилена и тому подобного, можно подвергать термосварке или сварке под действием растворителя с получением надежных сварных швов с высокой прочностью; однако данным пленкам также свойственны и серьезные недостатки. Например, термопластичные материалы необходимо пластифицировать для получения гибкости, требуемой для мембранного кровельного покрытия, а пластификаторы достаточно дорого стоят, и они вымываются из мембраны с течением времени, что, таким образом, приводит в результате к потере гибкости, появлению хрупкости и уменьшению стойкости мембраны к растрескиванию на холоде. Кроме этого, ограничена и способность термопластичных материалов по принятию наполнителей. Помимо этого, с учетом наличия тенденции к созданию окружения, не содержащего хлора, существует потребность в альтернативе для листового материала на основе ПВХ.
Вследствие наличия превосходных атмосферостойкости и гибкости быстро приобрели признание эластомерные мембраны, такие как вулканизованные каучуки EPDM на основе этилен/пропилен/диеновых терполимеров. Данный материал обычно получают в результате вулканизации композиции в присутствии серы или серосодержащих соединений. Однако данные мембраны трудно сваривать вследствие отсутствия адгезии у EPDM, в особенности у вулканизованного EPDM, к самому себе, и в данном случае для соединения мембраны требуется клей, для того чтобы получить непрерывное кровельное покрытие без протечек. Данные клеи вносят значительный вклад в стоимость материала, а операция по их нанесению трудна и требует много времени. Кроме этого, клеевые соединения зачастую ослабевают с течением времени, что приводит к отслаиванию мембран и к последующему возникновению протечек в кровельном покрытии. Для эластомерных мембран также требуется и дополнительная дорогостоящая стадия вулканизации.
Для того чтобы избежать стадии вулканизации, на современном уровне техники в случае применения для кровельных покрытий предложили использовать листовые материалы, содержащие EPDM в комбинации с высококристаллическими промоторами термопластичности, такими как полиэтилен высокой плотности (ПЭВП), полиэтилен низкой плотности (ПЭНП) и другие подобные полимеры олефинового типа. Например, патент США № 5256228 описывает термосвариваемый листовой материал для кровельного покрытия, полученный из невулканизованной полимерной композиции, которая содержит 100 массовых частей полимерной смеси, содержащей от 50 до 90 массовых частей полиолефинов со степенью кристалличности вплоть до 2 массовых процентов, и от 10 до 50 массовых частей высококристаллического промотора термопластичности, такого как ПЭВП или ПЭНП; 50-250 массовых частей наполнителя; и 20-150 массовых частей материала технологической добавки.
Тем не менее, присутствие кристаллических полиэтиленовых смол приводит к появлению многих недостатков. Например, более высокие уровни степени кристалличности в результате дают меньшую формоустойчивость при нагревании, приводят к расширению или сжатию в большом температурном диапазоне, наблюдаемом в данной сфере; это может в результате привести к короблению листа или возникновению в нем напряжений. Кроме этого, более низкие характеристики плавления полиэтилена в результате становятся причиной ограничений для использования в приложениях при повышенных температурах, таких как в листе с темной окраской. В конце концов, полиэтилен характеризуется узким диапазоном температур плавления, что в результате становится причиной узости технологического окна термосварки.
Полиолефиновые композиции, обладающие хорошими механическими свойствами, использовали во многих сферах приложений благодаря характеристикам, которые типичны для полиолефинов (таким как химическая инертность, механические свойства и нетоксичность); помимо этого, данным композициям свойственны экономически выгодные соотношения стоимость/эксплуатационные свойства. На современном уровне техники данные композиции получали в результате последовательной сополимеризации пропилена, необязательно содержащего незначительные количества олефиновых сомономеров, а после этого этилен/пропиленовых или этилен/альфа-олефиновых смесей. Для данной цели обычно использовали катализаторы на основе галогенированных соединений титана, нанесенных на хлорид магния.
Например, патент США № 5286564, выданный на имя тех же самых заявителей, описывает гибкие полиолефиновые композиции, содержащие, в массовых частях:
А) 10-50 частей изотактического пропиленового гомополимера или сополимера;
В) 5-20 частей этиленового сополимера, нерастворимого в ксилоле при комнатной температуре; и
С) 40-80 частей этилен/пропиленового сополимера, содержащего менее 40% (масс.) этилена и растворимого в ксилоле при комнатной температуре; характеристическая вязкость упомянутого сополимера предпочтительно находится в диапазоне от 1,7 до 3 дл/г.
Упомянутые композиции характеризуются значениями модуля упругости при изгибе, меньшими 150 МПа, и твердостью D по Шору в диапазоне от 20 до 35. Несмотря на то что данные механические свойства выгодны для определенных областей применения, композиции, полученные в примерах 1-5, демонстрируют значения предела прочности при растяжении в диапазоне от 13,8 до 17,3 МПа; для многих применений, таких как кровельные листы, данные полимеры не обеспечивают удовлетворительного сопротивления разрыву, где последнее представляет собой свойство, тесно связанное с характеристиками предела прочности при растяжении и относительного удлинения при разрыве. Сопротивление разрыву представляет собой фундаментальное свойство при применениях для кровельных покрытий, где для кровельного покрытия наблюдается тенденция к разрушению под действием ветра.
Как хорошо известно, на современном уровне техники, прочность и гибкость термопластичных полиолефиновых композиций можно улучшить в результате уменьшения содержания каучука; тем не менее, с увеличением прочности обычно ассоциируется увеличение жесткости, что становится причиной возникновения различных недостатков. Например, увеличение жесткости у гибкой мембраны накладывает ограничения на укладку мембраны в области угла, дымовых труб, вентиляционных отверстий и тому подобного, где пленка должна быть гибкой для сгибания вокруг препятствия.
Европейская патентная заявка № 1202876.7, выданная на имя тех же самых заявителей, описывает полиолефиновую композицию, содержащую:
(А) от 8 до 25% (масс.) кристаллического пропиленового гомополимера или сополимера; и
(В) от 75 до 92% (масс.) эластомерной фракции, содержащей первый эластомерный сополимер пропилена с 15-32% (масс.) этилена, для которого характеристическая вязкость фракции, растворимой в ксилоле, находится в диапазоне от 3,0 до 5,0 дл/г; и второй эластомерный сополимер пропилена с более чем 32% и вплоть до 45% (масс.) этилена, для которого характеристическая вязкость фракции, растворимой в ксилоле, находится в диапазоне от 4,0 до 6,5 дл/г.
Данные полиолефиновые композиции характеризуются значениями модуля упругости при изгибе, меньшими 60 МПа, и очень хорошими значениями твердости (твердость А по Шору меньше 90); тем не менее, они демонстрируют низкие значения сопротивления разрыву. Собственно говоря, полиолефиновые композиции, полученные в примерах 1-3 упомянутой выше заявки, обнаруживают значения предела прочности при растяжении, равные 5,5, 11,7 и 11,2 МПа соответственно. Как уже сообщалось выше, такие значения не вполне удовлетворительны для кровельного листа, потому что увеличение гибкости связано с усиленной жесткостью.
Поэтому существует настоятельная потребность в более мягких материалах, которые будет легче укладывать и которые в то же самое время будут обладать удовлетворительными сопротивлением разрыву, прочностью и сопротивлением раздиру и проколу. Другими словами, ощущается потребность в полиолефиновых композициях, которые бы демонстрировали хороший баланс гибкости в переменном температурном диапазоне и сопротивления разрыву, а также хорошие механические свойства; кроме этого, данные композиции должны быть пригодны для термосварки и должны обладать маслостойкостью, и они не должны подвергаться деструкции при воздействии атмосферных условий.
Настоящее изобретение относится к полиолефиновой композиции, содержащей:
(А) от приблизительно 15 до приблизительно 40% (масс.) кристаллического сополимера пропилена, по меньшей мере, с одним альфа-олефином, описываемым формулой Н2С=CHR1, где R1 представляет собой Н или линейный или разветвленный С2-8 алкил, содержащего, по меньшей мере, приблизительно 90% (масс.) пропилена и характеризующегося растворимостью в ксилоле при комнатной температуре, меньшей, чем приблизительно 15% (масс.);
(В) от приблизительно 60 до приблизительно 85% (масс.) эластомерной фракции, содержащей:
(1) сополимер пропилена с этиленом, необязательно содержащий приблизительно от 0,5 до 5% (масс.) диена, содержащий от приблизительно 20 до приблизительно 35% (масс.) этилена и характеризующийся растворимостью в ксилоле при комнатной температуре, большей, чем приблизительно 70% (масс.), характеристической вязкостью фракции, растворимой в ксилоле, большей, чем приблизительно 3,0 дл/г, и вплоть до приблизительно 6,0 дл/г; и
(2) сополимер этилена, по меньшей мере, с одним альфа-олефином, описываемым формулой Н2С=CHR2, где R2 представляет собой линейный или разветвленный С2-8 алкил, необязательно содержащий приблизительно от 0,5 до 5% (масс.) диена, содержащий приблизительно от 15% до 40% (масс.) альфа-олефина и характеризующийся растворимостью в ксилоле при комнатной температуре, большей, чем приблизительно 25% (масс.), характеристической вязкостью фракции, растворимой в ксилоле, в диапазоне от приблизительно 0,5 до приблизительно 5,0 дл/г;
при этом массовое отношение (1)/(2) находится в диапазоне от приблизительно 1:5 до приблизительно 5:1.
Настоящее изобретение, кроме этого, относится к способу получения полиолефиновой композиции, описанной выше, включающему, по меньшей мере, три стадии последовательной полимеризации, при этом каждую реакцию последовательной полимеризации проводят в присутствии полимерного материала, образованного в непосредственно предшествующей реакции полимеризации, где кристаллический сополимер (А) получают на первой стадии, а эластомерную фракцию (В) получают, по меньшей мере, на двух последующих стадиях. В соответствии с предпочтительным вариантом реализации все стадии полимеризации проводят в присутствии катализатора, включающего соединение триалкилалюминия, необязательно донор электронов, и твердый компонент катализатора, содержащий галогенид или галоген-алкоголят Ti и электронодонорное соединение, нанесенные на носитель на основе безводного хлорида магния, при этом у упомянутого твердого компонента катализатора площадь удельной поверхности (согласно измерению по способу Брунауэра-Эммета-Теллера) меньше, чем приблизительно 200 м2/г, а пористость (согласно измерению по способу Брунауэра-Эммета-Теллера) больше, чем приблизительно 0,2 мл/г.
Полиолефиновые композиции настоящего изобретения демонстрируют значительное улучшение прочности и ударной вязкости не за счет жесткости; в противоположность этому заявители к удивлению обнаружили, что жесткость композиций изобретения уменьшается в сравнении с характеристиками композиций предшествующего уровня техники, отличающихся меньшими уровнями содержания каучука. Говоря более конкретно, композициям изобретения придается высокое сопротивление разрыву, о чем свидетельствуют значения предела прочности при растяжении и относительного удлинения при разрыве, намного превышающие соответствующие параметры для термопластичных смол предшествующего уровня техники, используемых в гибких мембранных кровельных покрытиях, и одновременно с этим обнаруживается значительное уменьшение жесткости.
Композиции изобретения отличаются наличием очень хорошего баланса гибкости и механических свойств и, в частности, значений модуля упругости при изгибе и твердости D по Шору, и в то же самое время сохраняются хорошие свойства эластомера. Данные мягкие композиции, более легкие для укладки даже в области углов и дымовых труб, отличаются улучшенными прочностью и сопротивлением раздиру, а также улучшенными сопротивлением приподниманию под действием ветра и сопротивлением проколу; кроме этого им свойственна свариваемость в течение длительного времени.
Полиолефиновые композиции настоящего изобретения содержат от приблизительно 15 до приблизительно 40% (масс.), предпочтительно от приблизительно 15 до приблизительно 35%, а еще более предпочтительно от приблизительно 20 до приблизительно 30% кристаллического сополимера пропилена (А) и от приблизительно 60 до приблизительно 85% (масс.), предпочтительно от приблизительно 65 до приблизительно 85%, а еще более предпочтительно от приблизительно 70 до приблизительно 80% (масс.) эластомерной фракции (В).
В настоящем описании под «комнатной температурой» подразумевается приблизительно 25°С.
Кристаллический сополимер (А) композиций изобретения представляет собой сополимер пропилена с альфа-олефином Н2С=CHR1, где R1 представляет собой Н или линейный или разветвленный С2-8 алкил, содержащий, по меньшей мере, приблизительно 90% (масс.) пропилена, предпочтительно, по меньшей мере, приблизительно 95% пропилена. Данный сополимер характеризуется растворимостью в ксилоле при комнатной температуре, меньшей, чем приблизительно 15% (масс.), предпочтительно меньшей, чем приблизительно 10%, а еще более предпочтительно меньшей, чем приблизительно 8%. Упомянутым альфа-олефином предпочтительно являются этилен, 1-бутен, 1-пентен, 4-метилпентен, 1-гексен, 1-октен или их комбинации, а еще более предпочтительно им является этилен.
Эластомерная фракция (В) полиолефиновых композиций изобретения содержит сополимер пропилена с этиленом и сополимер этилена с альфа-олефином Н2С=CHR2, где R2 представляет собой линейный или разветвленный С2-8 алкил. Под «эластомерным» в настоящем документе понимается полимер, отличающийся низкой степенью кристалличности или аморфный и характеризующийся растворимостью в ксилоле при комнатной температуре, большей, чем приблизительно 40% (масс.).
Сополимер (1) пропилена с этиленом содержит от приблизительно 20 до приблизительно 35% (масс.) этилена, предпочтительно от приблизительно 25 до приблизительно 30%, и он характеризуется растворимостью в ксилоле при комнатной температуре, большей, чем приблизительно 70% (масс.), предпочтительно большей, чем приблизительно 75%; характеристическая вязкость фракции, растворимой в ксилоле, находится в диапазоне от приблизительно 3,0 до приблизительно 6,0 дл/г, более предпочтительно от приблизительно 3,5 до приблизительно 5,0 дл/г, а еще более предпочтительно от приблизительно 4,0 до приблизительно 4,5 дл/г.
Сополимер (2) представляет собой сополимер этилена, по меньшей мере, с одним альфа-олефином, описываемым формулой Н2С=CHR2, где R2 представляет собой линейный или разветвленный С2-8 алкил, не обязательно содержащий приблизительно от 0,5 до 5% (масс.) диена; при этом упомянутым альфа-олефином предпочтительно являются 1-бутен, 1-гексен или 1-октен, а еще более предпочтительно им является 1-бутен. Содержание альфа-олефина находится в диапазоне от приблизительно 15% до приблизительно 40% (масс.), а предпочтительно от приблизительно 20 до приблизительно 35%. Сополимер (2) характеризуется растворимостью в ксилоле при комнатной температуре, большей чем приблизительно 25% (масс.), предпочтительно большей чем приблизительно 30%, а значение характеристической вязкости у фракции, растворимой в ксилоле, находится в диапазоне от приблизительно 0,5 до приблизительно 5,0 дл/г, предпочтительно от приблизительно 1,0 до приблизительно 4,5 дл/г.
Как уже сообщалось ранее, сополимеры (1) и (2) эластомерной фракции (В) могут содержать диен, сопряженный или нет, такой как бутадиен, 1,4-гексадиен, 1,5-гексадиен и этилиденнорборнен-1. Диен, при его наличии, содержится в количестве от приблизительно 0,5 до приблизительно 5% (масс.) в расчете на массу фракции (В). Массовое отношение эластомерных сополимеров (1)/(2) находится в диапазоне от приблизительно 1:5 до приблизительно 5:1, а предпочтительно от приблизительно 1:2 до приблизительно 2:1.
Полиолефиновая композиция изобретения, предпочтительно полученная в результате последовательной полимеризации, по меньшей мере, в три стадии, обнаруживает высокое сопротивление разрыву, и, в частности, она характеризуется пределом прочности при растяжении ≥ 25 МПа (ASTM D 882, для мембраны толщиной 100 мкм), более предпочтительно ≥ 28 МПа; относительным удлинением при разрыве ≥ 700 МПа (ASTM D 882, для мембраны толщиной 100 мкм), более предпочтительно ≥ 750 МПа; и ударной вязкостью ≥ 150 МПа (ASTM D 638, для листа толщиной 1 мм), более предпочтительно ≥ 170 МПа.
Кроме этого, упомянутая композиция характеризуется модулем упругости при изгибе ≤ 130 МПа, предпочтительно ≤ 100 МПа, а более предпочтительно ≤ 80 МПа; твердостью D по Шору ≤ 40°S, а предпочтительно находящейся в диапазоне от приблизительно 25 до приблизительно 35; и величиной MFR < 1,0 г/10 мин, предпочтительно < 0,8 г/10 мин, а еще более предпочтительно < 0,6 г/10 мин.
В соответствии с предпочтительным вариантом реализации изобретения полиолефиновая композиция имеется в виде сферических частиц со средним диаметром в диапазоне от приблизительно 250 до приблизительно 7000 микрон, текучестью, меньшей, чем приблизительно 30 секунд, и объемной плотностью (после уплотнения), большей, чем приблизительно 0,4 г/мл.
Полиолефиновую композицию изобретения можно получить в результате последовательной полимеризации, по меньшей мере, в три стадии; в соответствии с предпочтительным вариантом реализации последовательную полимеризацию проводят в присутствии катализатора, включающего соединение триалкилалюминия, необязательно донор электрона, и твердый компонент катализатора, содержащий галогенид или галоген-алкоголят Ti и электронодонорное соединение, нанесенные на носитель на основе безводного хлорида магния.
Настоящее изобретение, кроме этого, относится к способу получения описанных ранее полиолефиновых композиций, при этом упомянутый способ включает, по меньшей мере, три стадии последовательной полимеризации, при этом каждую реакцию последовательной полимеризации проводят в присутствии полимерного материала, образованного в непосредственно предшествующей реакции полимеризации, где кристаллический сополимер (А) получают, по меньшей мере, на одной первой стадии, а эластомерную фракцию (В) получают, по меньшей мере, на двух последующих стадиях. Стадии полимеризации можно проводить в присутствии катализатора Циглера-Натта и/или металлоценового катализатора.
Подходящие катализаторы Циглера-Натта описываются в ЕР-А-45 977 и US 4399054, включаемых в настоящий документ для справки; другие примеры можно найти в US 4472524, включаемом в настоящий документ для справки. Твердые компоненты катализатора, используемые в упомянутых катализаторах, содержат в качестве доноров электронов (внутренних доноров) соединения, выбираемые из группы, состоящей из простых эфиров, кетонов, лактонов, соединений, содержащих атомы N, P и/или S, и сложных эфиров одно- и двухосновных карбоновых кислот. В особенности подходящими электронодонорными соединениями являются сложные эфиры фталевой кислоты, такие как диизобутил-, диоктил-, дифенил- и бензилбутилфталат. Другими подходящими донорами электронов являются простые 1,3-диэфиры, описываемые формулой:
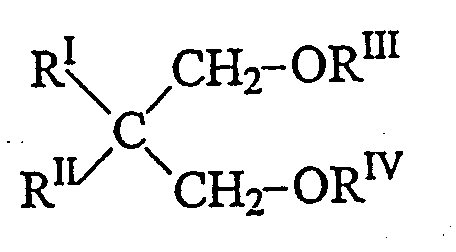
где RI и RII, одинаковые или отличающиеся друг от друга, представляют собой С1-С18 алкильные, С3-С18 циклоалкильные или С7-С18 арильные радикалы; RIII и RIV, одинаковые или отличающиеся друг от друга, представляют собой С1-С4 алкильные радикалы; или являются простые 1,3-диэфиры, у которых атом углерода в положении 2 входит в циклическую или полициклическую структуру, образованную 5, 6 или 7 атомами углерода и содержащую две или три ненасыщенности.
Простые эфиры данного типа описываются в ЕР-А-361493 и ЕР-А-728769.
Представительными примерами упомянутых простых диэфиров являются 2-метил-2-изопропил-1,3-диметоксипропан, 2,2-диизобутил-1,3-диметоксипропан, 2-изопропил-2-циклопентил-1,3-диметоксипропан, 2-изопропил-2-изоамил-1,3-диметоксипропан и 9,9-бис(метоксиметил)флуорен.
Получение описанных выше компонентов катализатора проводят в соответствии с различными способами.
В соответствии с предпочтительным способом получения аддукт MgCl2·nROH (в частности, в виде сфероидальных частиц), где n в общем случае находится в диапазоне от 1 до 3, а ROH представляет собой этанол, бутанол или изобутанол, вводят в реакцию с избытком TiCl4, содержащим электронодонорное соединение. Температура реакции в общем случае находится в диапазоне от 80 до 120°С. После этого твердую фазу выделяют и еще раз вводят в реакцию с TiCl4 в присутствии или в отсутствие электронодонорного соединения, после чего ее выделяют и промывают, используя аликвоты углеводорода, до тех пор, пока все ионы хлора не исчезнут.
В твердом компоненте катализатора соединение титана, выражаемое через Ti, в общем случае присутствует в количестве в диапазоне от приблизительно 0,5 до приблизительно 10% (масс.). Количество электронодонорного соединения, которое остается зафиксированным на твердом компоненте катализатора, в общем случае находится в диапазоне приблизительно от 5 до 20% (моль.) в расчете на дигалогенид магния.
Соединениями титана, которые можно использовать при получении твердого компонента катализатора, являются галогениды и галогеналкоголяты титана. Тетрахлорид титана является предпочтительным соединением.
Реакции, описанные выше, в результате приводят к получению галогенида магния в активной форме. В литературе известны и другие реакции, которые приводят к получению галогенида магния в активной форме из исходных магнийсодержащих соединений, отличных от галогенидов, таких как карбоксилаты магния.
Al-алкильные соединения, используемые в качестве сокатализаторов, включают Al-триалкилы, такие как Al-триэтил, Al-триизобутил, Al-три-н-бутил, и линейные или циклические Al-алкильные соединения, содержащие два или более атомов Al, связанных друг с другом при помощи атомов O или N, групп SO4 или SO3. Al-алкильное соединение в общем случае используют в таком количестве, чтобы соотношение Al/Ti находилось бы в диапазоне от приблизительно 1 до приблизительно 1000.
Электронодонорные соединения, которые можно использовать в качестве внешних доноров, включают сложные эфиры ароматических кислот, такие как алкилбензоаты, и, в частности, соединения кремния, содержащие, по меньшей мере, одну связь Si-OR, где R представляет собой углеводородный радикал. Примерами соединений кремния являются (трет-бутил)2Si(OCH3)2, (циклогексил)(метил)Si(OCH3)2, (фенил)2Si(OCH3)2 и (циклопентил)2Si(OCH3)2. С выгодой также можно использовать и простые 1,3-диэфиры, описываемые формулами, представленными выше. Если внутренним донором является один из данных простых диэфиров, то тогда внешние доноры можно будет не использовать.
У твердого компонента катализатора площадь удельной поверхности (согласно измерению по способу Брунауэра-Эммета-Теллера) предпочтительно меньше чем приблизительно 200 м2/г, а более предпочтительно она находится в диапазоне от приблизительно 80 до приблизительно 170 м2/г, и пористость (согласно измерению по способу Брунауэра-Эммета-Теллера) предпочтительно больше чем приблизительно 0,2 мл/г, а более предпочтительно она находится в диапазоне от приблизительно 0,25 до приблизительно 0,5 мл/г.
Катализаторы можно предварительно ввести в контакт с небольшими количествами олефина (форполимеризация), выдерживая катализатор в суспензии в углеводородном растворителе и проводя полимеризацию при температурах в диапазоне от комнатной до 60°С, получая, таким образом, количество полимера в диапазоне от приблизительно 0,5- до приблизительно 3-кратной массы катализатора. Данную операцию также можно провести и в жидком мономере с получением в данном случае количества полимера, соответствующего 1000-кратной массе катализатора.
В результате использования упомянутых выше катализаторов получают полиолефиновые композиции со сфероидальной формой частиц, при этом для частиц имеют место средний диаметр в диапазоне от приблизительно 250 до приблизительно 7000 микрон, текучесть, меньшая, чем приблизительно 30 секунд, и объемная плотность (после уплотнения), большая, чем приблизительно 0,4 г/мл.
Другими катализаторами, которые можно использовать в способе, соответствующем настоящему изобретению, являются катализаторы металлоценового типа, описанные в ЕР-А-0129368 и US 5324800, включаемых в настоящий документ для справки; в особенности выгодны мостиковые бис-инденильные металлоцены, например, описанные в ЕР-А-0485823 и US 5145819, включаемых в настоящий документ для справки. Еще одним классом подходящих катализаторов являются так называемые катализаторы с ограничением по геометрии, описанные в ЕР-А-0416815, ЕР-А-0420436, ЕР-А-0671404, ЕР-А-0643066 и WO 91/04257. Данные металлоценовые соединения можно использовать для получения сополимеров (1) и (2) эластомерной фракции (В).
В соответствии с предпочтительным вариантом реализации способ полимеризации изобретения включает три стадии, из которых все проводят в присутствии катализаторов Циглера-Натта, где: на первой стадии соответствующий мономер (мономеры) полимеризуют с получением кристаллического сополимера (А); на второй стадии смесь пропилена и этилена и необязательно диена полимеризуют с получением сополимера (В)(1); и на третьей стадии смесь этилена и альфа-олефина Н2С=CHR2 и необязательно диена полимеризуют с получением сополимера (В)(2).
Стадии полимеризации можно проводить в жидкой фазе, в газовой фазе или в газожидкостной фазе. Предпочтительно, когда полимеризацию с получением фракции кристаллического сополимера (А) проводят в жидком мономере (например, при использовании жидкого пропилена в качестве разбавителя), тогда как стадии сополимеризации с получением сополимеров (В)(1) и (В)(2) проводят в газовой фазе без промежуточных стадий за исключением частичной дегазации пропилена. В соответствии с наиболее предпочтительным вариантом реализации все три стадии последовательной полимеризации проводят в газовой фазе.
Температуры реакции на стадии полимеризации при получении кристаллического сополимера (А) и при получении сополимеров (В)(1) и (В)(2) могут быть одинаковыми или различающимися, и они предпочтительно находятся в диапазоне от приблизительно 40°С до приблизительно 90°С; более предпочтительно температура реакции находится в диапазоне от приблизительно 50 до приблизительно 80°С при получении фракции (А) и в диапазоне от приблизительно 40 до приблизительно 80°С при получении компонентов (В)(1) и (В)(2).
Давление на стадии полимеризации при получении фракции (А), при проведении реакции в жидком мономере, представляет собой давление, которое конкурирует с давлением паров жидкого пропилена при используемой рабочей температуре, и его возможно изменяют давлением паров небольшого количества инертного разбавителя, использованного для подачи каталитической смеси, и избыточным давлением мономеров и водорода, необязательно используемым в качестве регулятора степени полимеризации.
Давление для реакции полимеризации предпочтительно находится в диапазоне от приблизительно 33 до приблизительно 43 бар при проведении реакции в жидкой фазе и от приблизительно 5 до приблизительно 30 бар при проведении реакции в газовой фазе. Времена пребывания для трех стадий зависят от желательного соотношения между фракциями, и они могут находиться в диапазоне от приблизительно 15 минут до 8 часов. Можно использовать обычные регуляторы степени полимеризации, известные на современном уровне техники, такие как передатчики кинетической цепи (например, водород или ZnEt2).
Композиции изобретения можно использовать в комбинации с другими эластомерными полимерами, такими как этилен/пропиленовые сополимеры (EPR), этилен/пропилен/диеновые терполимеры (EPDM), сополимеры этилена с С4-С12 альфа-олефинами (например, сополимеры этилена/октена-1, такие как те, которые коммерчески доступны под наименованием Engage®) и их смеси. Такие эластомерные полимеры могут присутствовать в количестве в диапазоне от 5 до 80% (масс.) в расчете на полную композицию.
В дополнение к ее полимерным компонентам к композиции настоящего изобретения можно примешать и обычно используемые добавки, такие как армирующие и неармирующие наполнители, пигменты, зародышеобразователи, масла для наполнения, минеральные наполнители, антипирены (например, тригидрат алюминия), антиоксиданты, добавки, придающие стойкость к ультрафиолетовым лучам, (например, диоксид титана), стабилизаторы ультрафиолетового излучения, смазки (например, олеамид), вещества, препятствующие слипанию, антистатики, воски, связующие вещества для наполнителей и другие технологические добавки, известные на современном уровне техники в сфере составления полимерных смесей. Пигменты и другие добавки могут составлять вплоть до приблизительно 50 массовых процентов от полной композиции в расчете на полимерные компоненты плюс добавки; пигменты и наполнители предпочтительно присутствуют с содержанием, превышающим содержание в диапазоне от приблизительно 0 до приблизительно 30 массовых процентов от полной композиции.
С учетом свойств, описанных выше, полиолефиновые композиции настоящего изобретения представляют ценность при изготовлении мембранных кровельных покрытий для покрытия плоских кровель промышленного и гражданского назначения и в других применениях для кровельных покрытий. Собственно говоря, они отличаются высоким сопротивлением разрыву при сохранении мягкости и хороших механических свойств.
Кроме этого, мембранные кровельные покрытия, полученные с использованием полиолефиновых композиций изобретения, являются водонепроницаемыми, недорогими для изготовления и долговечными при средней продолжительности срока службы, намного превышающей соответствующую величину для современных коммерчески доступных кровельных гонтов, которые изготавливают с использованием стекловолокна или асфальта.
Мембранные кровельные покрытия, получаемые из полиолефиновых композиций настоящего изобретения, можно изготавливать по любому способу, обычно используемому при изготовлении мембранных кровельных покрытий из наполненных полимерных композиций. Например, мембраны можно изготавливать путем обычно используемых методик вальцевания, каландрования или экструдирования. Можно использовать и другие способы, в том числе нанесение покрытия методом распыления и формование с использованием роликовой головки. В соответствии с предпочтительным вариантом реализации полимерные композиции изобретения формуют в листы с толщиной в диапазоне от приблизительно 5 до приблизительно 200 мил. В это время листовое полотно можно разрезать на желательные размеры по длине и ширине.
Мембранные кровельные покрытия, изготовленные из композиций настоящего изобретения, необязательно можно армировать мешковиной. Помимо мембранных кровельных покрытий полиолефиновые композиции данного изобретения можно использовать для изготовления материалов для изолирующих слоев кровли, используемых в качестве замены для кровельного войлока.
Укладку мембранных кровельных покрытий можно проводить тем же самым способом и в тех же самых условиях, что и обычно используемые асфальтовые или стекловолоконные мембраны. Они имеют небольшую массу, что обеспечивает потенциальную экономию затрат на конструирование опорных структур для кровли, а также легкость укладки. Они обладают превосходной прочностью и долговечностью, вместе с превосходными температуростойкостью, атмосферостойкостью и эластичностью.
Способ покрытия кровли включает стадии укладки листов полимерного материала изобретения на покрываемую кровлю; перекрывание соседних кромок у слоев; нагревание областей перекрывания до температуры, несколько превышающей температуру размягчения листового материала, и соединение областей перекрывания при использовании тепла и под действием давления, достаточного для обеспечения приемлемой прочности шва. Полимерным композициям свойственна достаточная самоадгезия без использования клея.
Для определения свойств, описанных в подробном описании и в примерах, использовали следующие аналитические способы.
2) ASTM D 638, скорость в испытании 50 мм /мин для экструдированного листа толщиной 1 мм, при 23°С. Вырубленная заготовка Type IV, без экстензометра. Для испытаний при -40°С использовали камеру с искусственным климатом и образцы D1822 Type S
2) ASTM D 638, для экструдированного листа толщиной 1 мм. Вырубленная заготовка Type IV, без экстензометра. Для испытаний при -40°С использовали камеру с искусственным климатом и образцы D1822 Type S
Определение растворимости в ксилоле при комнатной температуре (% (масс.)):
В 250 мл ксилола при 135°С при перемешивании растворяли 2,5 г полимера. По истечении 20 минут раствор при перемешивании охлаждали до 25°С, а после этого ему давали возможность отстояться в течение 30 минут.
Осадок отфильтровывали при помощи фильтровальной бумаги; раствор упаривали в токе азота, а остаток высушивали в вакууме при 80°С до достижения постоянной массы. После этого рассчитывали массовый процент полимера, растворимого в ксилоле при комнатной температуре. Массовый процент полимера, нерастворимого в ксилоле при комнатной температуре, принимали за индекс изотактичности полимера. Данное значение по существу соответствовало индексу изотактичности, определенному в результате экстракции в кипящем н-гептане, который по определению представляет собой индекс изотактичности полипропилена.
Если только не будет указано другого, то образцы, используемые для различных физико-механических анализов, формовали при помощи пресса для литья под давлением Negri & Bossi 90 после стабилизации образца при помощи пространственно затрудненного фенольного стабилизатора IRGANOX R 1010 (0,05% (масс.)) и фосфитного стабилизатора IRGAFOS 168 (0,1% (масс.)) и гранулирования образца при использовании двухшнекового экструдера Berstorff (диаметр цилиндра 25 мм) при 210°С.
Условиями были следующие:
- температура расплава: 220°С;
- температура формы: 60°С;
- время впрыска: 9 с;
- время охлаждения: 15 с.
Размеры пластинок для испытаний были 127 х 127 х 2,5 мм. Из данных пластинок вырубали лопатки типа С и их подвергали испытаниям для определения предела прочности при растяжении при скорости головки, равной 500 мм/мин. Из данных пластинок также вырубали образцы для определения модуля упругости при изгибе и твердости D по Шору. Все образцы вырубали параллельно фронту подачи и, следовательно, перпендикулярно направлению течения.
Примеры 1 и 2
Получение каталитической системы
Компонент катализатора, содержащий аддукт MgCl2·3С2Н5OH, получали следующим образом: в колбу, погруженную в терморегулируемую баню, при 120°С при перемешивании в инертной атмосфере вводили 28,4 г безводного MgCl2, 49,5 г чистого безводного этанола, 100 мл вазелинового масла ROL OB/30 и 100 мл силиконового масла (вязкость 350 сСт) до тех пор, пока MgCl2 не растворялся полностью. После этого смесь в горячем состоянии, все время в инертной атмосфере, переводили в емкость объемом 150 мл, оснащенную обогревающей рубашкой и содержащую 150 мл вазелинового масла и 150 мл силиконового масла. Смесь выдерживали при 120°С при перемешивании, при этом перемешивание проводили при помощи мешалки Hanke & Kunkel K. G. Ika Werke Ultra Turrax T-45 N. Упомянутое перемешивание продолжали в течение 3 минут при 3000 об/мин. Смесь выгружали в емкость объемом 2 литра, содержащую 1000 мл безводного н-гептана, перемешанного и охлажденного таким образом, чтобы конечная температура не превышала бы 0°С. Таким образом полученные микросферы MgCl2·3EtOH отфильтровывали и высушивали в вакууме при комнатной температуре. Полученный таким образом высушенный аддукт после этого подвергали деалкоголированию в результате нагревания при температурах, постепенно увеличивающихся от 50°С до 100°С, в токе азота до тех пор, пока содержание спирта не становилось равным 1,1 моля на один моль MgCl2.
У полученного таким образом частично деалкоголированного аддукта площадь удельной поверхности была равна 11,5 м2/г, пористость - 0,13 мл/г, а объемная плотность - 0,564 г/мл.
25 г полученного аддукта при перемешивании при 0°С добавляли к 625 мл TiCl4. После этого смесь нагревали до 100°С в течение 1 часа. Когда температура достигала 40°С, добавляли диизобутилфталат в таком количестве, чтобы мольное отношение Mg/диизобутилфталат было бы равно 8. Полученную в результате смесь нагревали при 100°С еще в течение 2 часов, после этого ей давали возможность отстояться, а жидкость в горячем состоянии откачивали сифоном. Добавляли 550 мл TiCl4, и смесь нагревали при 120°C в течение 1 часа.
Полученной смеси давали возможность отстояться, а жидкость в горячем состоянии откачивали сифоном. Твердую фазу промывали 6 раз, используя 200 мл безводного гексана при 60°С, и еще три раза, используя 200 мл безводного гексана при комнатной температуре. После высушивания в вакууме твердая фаза демонстрировала пористость, равную 0,383 мл/г, и площадь удельной поверхности, равную 150 м2/г.
Общий способ полимеризации
Реакции полимеризации проводили в реакторах из нержавеющей стали с псевдоожиженным слоем.
Для того чтобы определить содержание этилена, пропилена и водорода, в ходе полимеризации газовую фазу в каждом реакторе непрерывно анализировали по методу газовой хроматографии. Используя приборы, которые измеряют и/или регулируют поток мономеров, этилен, пропилен, 1-бутен и водород подавали таким способом, чтобы в ходе полимеризации их концентрация в газовой фазе оставалась бы постоянной.
Процесс проводили непрерывно в три стадии, при этом каждая стадия включала полимеризацию мономеров в газовой фазе.
В петлевом реакторе из нержавеющей стали объемом 80 литров пропилен подвергали форполимеризации в жидком пропане при температуре внутри реактора 20°С в течение периода времени, равного 30 минутам, при подаче пропилена 30 кг/ч в присутствии каталитической системы, включающей твердый компонент (26 г/ч), полученный так, как описывалось выше, и смесь 75-80 г/ч Al-триэтил (TEAL) в 10%-ном растворе в гексане и подходящего количества донора дициклопентилдиметоксисилана (DCPMS), такую, чтобы массовое отношение TEAL/DCPMS было бы равно 5. Катализатор получали в соответствии со способом, описанным выше.
1-я стадия - Полученный таким образом форполимер выгружали в первый газофазный реактор, в котором температура была равна 60°С, а давление было равно 14 бар избыточного давления. После этого для получения состава газовой фазы, приведенного в таблице 1, проводили подачу водорода, пропилена, этилена и инертного газа с соотношением и в количествах, приведенных в таблице 1, и в течение времени, приведенного в таблице 1, проводили полимеризацию.
2-я стадия - После отбора образца для проведения различных анализов полимер, полученный на первой стадии, выгружали в реактор второй фазы, в котором температура была равна 60°С, а давление было равно 18 бар избыточного давления. После этого для получения состава газовой фазы, приведенного в таблице 1, проводили подачу водорода, пропилена, этилена и инертного газа с соотношением и в количествах, приведенных в таблице 1, и в течение времени, приведенного в таблице 1, проводили полимеризацию.
3-я стадия - После отбора образца для проведения различных анализов полимер, полученный на второй стадии, выгружали в реактор третьей фазы, в котором температура была равна 75°С, а давление было равно 14 бар избыточного давления. После этого для получения состава газовой фазы, приведенного в таблице 1, проводили подачу водорода, этилена, 1-бутена и инертного газа с соотношением и в количествах, приведенных в таблице 1, и в течение времени, приведенного в таблице 1, проводили полимеризацию.
По завершении полимеризации частицы полимера при атмосферном давлении выгружали в емкость, в которую противотоком подавали пар для того, чтобы отогнать остаточные мономеры. После этого полимер выгружали в емкость, в которую противотоком подавали азот при 80-90°С для того, чтобы высушить полимер.
Рабочие условия, использованные в описанном выше способе, и результаты анализов, проведенных для полимерных композиций, полученных при его проведении, приведены в таблицах 1 и 2 соответственно.
Примеры 3-5
Повторили методику полимеризации из примера 1 за исключением того, что на стадиях полимеризации использовали следующие условия:
- на стадии 1 полимеризацию проводили при температуре 60°С и при давлении 14 бар избыточного давления в течение периода времени, приведенного в таблице 1;
- на стадии 2 полимеризацию проводили при температуре 60°С и при давлении 18 бар избыточного давления в течение периода времени, приведенного в таблице 1; и
- на стадии 3 полимеризацию проводили при температуре 77°С и при давлении 14 бар избыточного давления в течение периода времени, приведенного в таблице 1.
Рабочие условия, использованные в описанном выше способе, и результаты анализов, проведенных для полимерных композиций, полученных при его проведении, приведены в таблицах 1 и 2 соответственно.
Примеры 6 и 7
Повторили методику полимеризации из примера 1 за исключением того, что на стадиях полимеризации использовали следующие условия:
- на стадии 1 полимеризацию проводили при температуре 60°С и при давлении 14 бар избыточного давления в течение периода времени, приведенного в таблице 1;
- на стадии 2 полимеризацию проводили при температуре 60°С и при давлении 18 бар избыточного давления в течение периода времени, приведенного в таблице 1; и
- на стадии 3 полимеризацию проводили при температуре 80°С и при давлении 14 бар избыточного давления в течение периода времени, приведенного в таблице 1.
Рабочие условия, использованные в описанном выше способе, и результаты анализов, проведенных для полимерных композиций, полученных при его проведении, приведены в таблицах 1 и 2 соответственно.
(% (масс.))
(% (моль.))
(% (масс.))
(% (масс.))
(% (моль.))
(% (моль.))
(% (масс.))
(% (масс.))
(г/10 мин)
(% (масс.))
(% (масс.))
(% (масс.))
(% (моль.))
(% (масс.))
* Рассчитано.
(г/10 мин)
(% (масс.))
1-бутена
(% (масс.))
Сравнительные примеры 1 и 2
В соответствии с методиками, описанными выше, полиолефиновые композиции, содержащие кристаллический пропилен/этиленовый сополимер (А) и пропилен/этиленовый эластомерный сополимер (В), подвергали испытаниям для целей сравнения. Характеристики данных композиций, полученных с марками продуктов, синтезированных в реакторе, продемонстрированы в таблице 3.
Приведенные выше результаты демонстрируют то, что композиции настоящего изобретения характеризуются очень хорошими значениями гибкости при одновременном увеличении сопротивления разрыву (предел прочности при растяжении и относительное удлинение при разрыве).
Испытания в случае применения для кровельных покрытий
Для композиций, полученных в приведенных выше примерах, были проведены следующие испытания для определения характеристик, специфических для применения для кровельных покрытий.
Получение образца и определение его характеристик
При помощи одношнекового экструдера Killion с одиночным шнеком 1,25" при температуре цилиндра, установленной на 230°С, тестируемую полимерную композицию стабилизировали для переработки в расплаве, используя фосфит (Irgafos 168), пространственно затрудненный фенол (Irganox 1010) и стеарат кальция в качестве акцептора кислоты. После этого перемешанную композицию экструдировали при помощи листового экструдера Killion с одиночным шнеком 1,5" с получением листа 1 мм х 200 мм шириной. Из листовых образцов изготавливали вырубленные заготовки в виде образцов для испытания на растяжение Type IV в соответствии с ASTM D 638. Механические свойства при растяжении измеряли при использовании динамометра Instron при скорости траверсы 500 мм/мин. Результаты описанных выше измерений приведены в следующей далее таблице 4.
Приведенные выше результаты демонстрируют улучшение гибкости композиций изобретения при соответствующем увеличении предела прочности при растяжении, ударной вязкости и сопротивления раздиру по сравнению с полиолефиновыми композициями, известными на современном уровне техники.
Испытание для определения свариваемости под действием горячего воздуха
Определение свариваемости под действием горячего воздуха представляет собой специальное испытание в случае приложений для однослойных кровельных покрытий. Сварные швы получали в результате перекрывания областей самого исходного листа шириной 25,4 мм. На межфазную поверхность двух листов подавали поток горячего воздуха при регулировании температуры. Регулировали скорость или время нагревания, а также давление смыкания на межфазной поверхности. Испытание проводили для экструдированных листов толщиной 1 мм при помощи прецизионного аппарата для сварки под действием горячего воздуха Leister X84. Для получения оценки окна сваривания протестировали температуры сварки 275°С, 345°С и 415°С при скорости для сварочного аппарата 1,3 м/мин.
Для сварных швов, полученных под действием горячего воздуха, при 23°С провели испытание на отслаивание с использованием вырубленных заготовок в виде полосок 25 мм при измерении усилия для отслаивания и относительного удлинения при разрыве. Характер разрушения оценивали в результате осмотра внешнего вида разрушившегося образца. Результаты описанных выше испытаний приведены в таблице 5.
удлинение (%)
отслаивания (Н)
удлинение (%)
отслаивания (Н)
удлинение (%)
Другие признаки, преимущества и варианты реализации изобретения, описанного в настоящем документе, станут легко понятными специалистам в соответствующей области после прочтения предшествующего описания. В этой связи необходимо сказать, что несмотря на то, что конкретные варианты реализации изобретения были описаны в значительной степени подробно, на практике можно будет осуществить и вариации, и модификации данных вариантов реализации без отклонения от объема и сущности описанного и заявленного изобретения.
| название | год | авторы | номер документа |
|---|---|---|---|
| СИЛЬНОНАПОЛНЕННЫЕ МЯГКИЕ ПОЛИОЛЕФИНОВЫЕ КОМПОЗИЦИИ | 2003 |
|
RU2318845C2 |
| КОМПОЗИЦИЯ МАТОЧНОЙ СМЕСИ ДЛЯ ПОЛУЧЕНИЯ ПОЛИОЛЕФИНОВЫХ ИЗДЕЛИЙ ЛИТЬЕВЫМ ФОРМОВАНИЕМ | 2003 |
|
RU2304598C2 |
| МЯГКИЕ ПОЛИОЛЕФИНОВЫЕ КОМПОЗИЦИИ | 2002 |
|
RU2300539C2 |
| МЕМБРАНЫ | 2011 |
|
RU2581869C2 |
| УДАРОПРОЧНАЯ ПОЛИОЛЕФИНОВАЯ КОМПОЗИЦИЯ, СПОСОБ ЕЕ ПОЛУЧЕНИЯ И ИЗДЕЛИЕ, СОДЕРЖАЩЕЕ УКАЗАННУЮ КОМПОЗИЦИЮ | 2003 |
|
RU2315069C2 |
| ПОЛИОЛЕФИНОВЫЕ КОМПОЗИЦИИ, ОБЛАДАЮЩИЕ ВЫСОКОЙ ТЕКУЧЕСТЬЮ | 2003 |
|
RU2305688C2 |
| ПОЛИОЛЕФИНОВЫЕ КОМПОЗИЦИИ | 2007 |
|
RU2451699C2 |
| УДАРОПРОЧНЫЕ ПОЛИОЛЕФИНОВЫЕ КОМПОЗИЦИИ | 2003 |
|
RU2308470C2 |
| ПОЛИМЕРНАЯ ОЛЕФИНОВАЯ КОМПОЗИЦИЯ ДЛЯ КРЫШЕК ДЛЯ БУТЫЛОК (ВАРИАНТЫ), КРЫШКА, ИЗГОТОВЛЕННАЯ ИЗ НЕЕ, СПОСОБ ПОЛУЧЕНИЯ ТАКОЙ КРЫШКИ, БУТЫЛКА, ЗАКРЫВАЕМАЯ ТАКОЙ КРЫШКОЙ | 1999 |
|
RU2230089C2 |
| ПОЛИМЕРНАЯ СМЕСЬ ДЛЯ ФОРМОВАНИЯ ПОЛЫХ ИЗДЕЛИЙ ЗАЛИВКОЙ И МЕДЛЕННЫМ ВРАЩЕНИЕМ ФОРМЫ | 1997 |
|
RU2186798C2 |
Полиолефиновая композиция, пригодная для получения однослойных мембранных кровельных покрытий, содержащая: (А) от приблизительно 15 до 40 мас.% кристаллического сополимера пропилена, по меньшей мере, с одним альфа-олефином, описываемым формулой H2C=CHR1, где R1 представляет собой Н или С2-8 линейный или разветвленный алкил, содержащего, по меньшей мере, приблизительно 90 мас.% пропилена и характеризующегося растворимостью в ксилоле при комнатной температуре, меньшей чем приблизительно 15 мас.%; (В) от приблизительно 60 до приблизительно 85 мас.% эластомерной фракции, содержащей: (1) сополимер пропилена с этиленом, содержащий от приблизительно 20 до приблизительно 35 мас.% этилена и характеризующийся растворимостью в ксилоле при комнатной температуре, большей чем приблизительно 70 мас.%, характеристической вязкостью фракции, растворимой в ксилоле, большей чем приблизительно 3,0 дл/г; и (2) сополимер этилена, по меньшей мере, с одним альфа-олефином, описываемым формулой Н2С=CHR2, где R2 представляет собой линейный или разветвленный C2-8 алкил, содержащий приблизительно от 15 до 40 мас.% альфа-олефина и характеризующийся растворимостью в ксилоле при комнатной температуре, большей чем приблизительно 25 мас.%, характеристической вязкостью фракции, растворимой в ксилоле, в диапазоне от приблизительно 0,5 до приблизительно 5,0 дл/г; при этом массовое отношение (1)/(2) находится в диапазоне от приблизительно 1:5 до приблизительно 5:1. Полиолефиновая композиция изобретения, предпочтительно получаемая в результате последовательной полимеризации, по меньшей мере, в три стадии, характеризуется пределом прочности при растяжении ≥25 МПа, относительным удлинением при разрыве ≥700 МПа и ударной вязкостью ≥150 МПа. 4 н. и 10 з.п. ф-лы, 5 табл.
(A) от приблизительно 15 до приблизительно 40 мас.% кристаллического сополимера пропилена, по меньшей мере, с одним альфа-олефином, описываемым формулой Н2С=CHR1, где R1 представляет собой Н или линейный или разветвленный C2-8 алкил, содержащего, по меньшей мере, приблизительно 90 мас.% пропилена и характеризующегося растворимостью в ксилоле при комнатной температуре, меньшей чем приблизительно 15 мас.%;
(B) от приблизительно 60 до приблизительно 85 мас.% эластомерной фракции, содержащей:
(1) сополимер пропилена с этиленом, необязательно содержащий приблизительно от 0,5 до 5 мас.% диена, содержащий от приблизительно 20 до приблизительно 35 мас.% этилена и характеризующийся растворимостью в ксилоле при комнатной температуре, большей, чем приблизительно 70 мас.%, характеристической вязкостью фракции, растворимой в ксилоле, большей, чем приблизительно 3,0 дл/г, и вплоть до приблизительно 6,0 дл/г; и
(2) сополимер этилена, по меньшей мере, с одним альфа-олефином, описываемым формулой Н2С=CHR2, где R2 представляет собой линейный или разветвленный С2-8 алкил, необязательно содержащий приблизительно от 0,5 до 5 мас.% диена, содержащий приблизительно от 15 до 40 мас.% альфа-олефина и характеризующийся растворимостью в ксилоле при комнатной температуре, большей, чем приблизительно 25 мас.%, характеристической вязкостью фракции, растворимой в ксилоле, в диапазоне от приблизительно 0,5 до приблизительно 5,0 дл/г;
при этом массовое отношение (1)/(2) находится в диапазоне от приблизительно 1:5 до приблизительно 5:1.
| US 5360868 А, 01.11.1994 | |||
| 1970 |
|
SU412534A1 | |
| Прибор для передвижения железнодорожных повозок | 1926 |
|
SU11057A1 |
| ПЛЕНОЧНЫЙ ИЛИ ЛИСТОВОЙ МАТЕРИАЛ, ПЛЕНОЧНОЕ ИЛИ ЛИСТОВОЕ ИЗДЕЛИЕ (ВАРИАНТЫ) | 1994 |
|
RU2124535C1 |

