Настоящее изобретение относится к полиолефиновым композициям, содержащим компонент в виде кристаллического пропиленового полимера, выбираемый из статистических сополимеров пропилена-этилена и/или другого α-олефина, и сополимер этилена с С4-С10 α-олефинами.
Композиции настоящего изобретения легко можно превратить в различные типы готовых изделий или полуфабрикатов, в частности, при использовании методик литьевого формования, поскольку композиции демонстрируют относительно высокие значения скорости течения расплава (MFR). Кроме того, поскольку им по существу не свойственно побеление под действием напряжения при сгибании пластинки толщиной 1 мм, упомянутые композиции можно широко использовать, в том числе для игрушек и посуды, в частности, для тех изделий, которым необходимо наличие у них ударопрочности при низких температурах, не позволяющее изделиям разрушиться. Упомянутые изделия в выгодном случае можно использовать в областях, где имеет место контакт с продуктами питания, примерами которых являются контейнеры для продуктов питания, подходящие для морозильных камер.
Композиции, содержащие полипропилен и каучукообразную фазу, образованную эластомерным сополимером этилена и α-олефинов, уже известны на современном уровне техники, и они описываются, в частности, в европейских патентах 170255 и 373660 и в WO 01/19915. Упомянутые композиции демонстрируют ударопрочность и в случае европейского патента 373660 и WO 01/19915 значения прозрачности, интересные для широкого применения, однако совокупный баланс свойств все еще не является полностью удовлетворительным в широком ассортименте возможного использования с учетом высоких стандартов, требуемых для рынка. Поэтому существует постоянная потребность в композициях данного типа, обладающих улучшенными свойствами.
В настоящее время был получен новый и ценный баланс свойств в результате создания полиолефиновых композиций настоящего изобретения, содержащих (в массовых процентах):
1) 55-80% кристаллического пропиленового гомополимера или сополимера, содержащего вплоть до 15% этилена и/или С4-С10 α-олефина (олефинов) и имеющего значение MFR, по меньшей мере, равное 15 г/10 мин; и
2) 20-45% сополимера этилена и одного или нескольких С4-С10 α-олефинов (олефина), содержащего от 10 до 40% упомянутого С4-С10 α-олефина (олефинов);
при этом упомянутые композиции имеют значения MFR (230°С, 2,16 кг), равные или больше 15 г/10 мин, полное содержание этилена, равное 20% или более, полное содержание С4-С10 α-олефина (олефинов), равное 4,5% или более, соотношение полного содержания этилена и полного содержания С4-С10 α-олефина (олефинов), равное 2,3 или более, и значение характеристической вязкости фракции, растворимой в ксилоле при комнатной температуре, равное 1,7 дл/г или менее, предпочтительно 1,5 дл/г или менее.
Из приведенных выше определений очевидно, что термин «сополимер» включает полимеры, содержащие более одного типа сомономеров.
Композиции настоящего изобретения предлагают, в частности, комбинацию очень высокой текучести и высокой ударопрочности (выраженных через температуру перехода от пластического разрушения к хрупкому и ударную вязкость в испытании по Изоду), и высокой прозрачности.
Предпочтительные полиолефиновые композиции представляют собой гибкие полиолефиновые композиции, содержащие (в массовых процентах):
1) 55-75%, предпочтительно 55-70%, кристаллического пропиленового гомополимера или сополимера, содержащего вплоть до 15% этилена и/или С4-С10 α-олефина (олефинов) и имеющего значение MFR в диапазоне от 15 до 80 г/10 мин; и
2) 25-45%, предпочтительно 30-45%, сополимера этилена и одного или нескольких С4-С10 α-олефинов (олефина), содержащего от 15 до 40% упомянутого С4-С10 α-олефина (олефинов);
при этом упомянутые композиции имеют значения MFR (230°С, 2,16 кг), равные или больше 15 г/10 мин, полное содержание этилена, равное 20% или более, полное содержание С4-С10 α-олефина (олефинов), равное 6% или более, соотношение полного содержания этилена и полного содержания С4-С10 α-олефина (олефинов), равное 2,3 или более, содержание совокупной фракции, растворимой в ксилоле при комнатной температуре, равное 18 мас.% или более, предпочтительно, по меньшей мере, равное 20 мас.%, и значение характеристической вязкости фракции, растворимой в ксилоле при комнатной температуре, равное 1,7 дл/г или менее, предпочтительно 1,5 дл/г или менее.
Композиции настоящего изобретения предпочтительно имеют значение MFR в диапазоне от 15 до 40 г/10 мин.
В особенности предпочтительными признаками композиций настоящего изобретения являются:
- содержание полимера, нерастворимого в ксилоле при комнатной температуре (23°С), (по существу эквивалентное индексу изотактичности) для компонента 1): не менее 90%, в особенности не менее 93%, при этом упомянутые процентные содержания являются массовыми и их получают в расчете на массу компонента 1);
- полное содержание этилена в диапазоне от 20 до 40 мас.%;
- полное содержание С4-С10 α-олефина (олефинов) в диапазоне от 6 до 15 мас.%;
- значение модуля упругости при изгибе меньше 770 МПа, но предпочтительно больше 600 МПа, более предпочтительно больше 650 МПа;
- содержание фракции, растворимой в ксилоле при комнатной температуре, менее 35%, более предпочтительно менее 30 мас.%;
- характеристическая вязкость фракции, растворимой в ксилоле при комнатной температуре, в диапазоне от 0,8 до 1,5 дл/г.
Температура перехода от пластического разрушения к хрупкому в общем случае равна или меньше -35°С, при этом нижний предел ориентировочно соответствует приблизительно -60°С.
Упомянутые С4-С10 α-олефины, которые присутствуют или могут присутствовать в качестве сомономеров в компонентах и фракциях композиций настоящего изобретения, описываются формулой CH2=CHR, где R представляет собой алкильный радикал, линейный или разветвленный, содержащий 2-8 атомов углерода, или же арильный (в частности, фенильный) радикал.
Примерами упомянутых С4-С10 α-олефинов являются 1-бутен, 1-пентен, 1-гексен, 4-метил-1-пентен и 1-октен. В особенности предпочтителен 1-бутен.
Композиции настоящего изобретения можно получить по способу ступенчатой полимеризации, включающему, по меньшей мере, две последовательные стадии, где компоненты 1) и 2) получают на отдельных последовательных стадиях, проводя реакцию на каждой стадии, за исключением первой стадии, в присутствии полимера, образовавшегося на предшествующей стадии, и катализатора, использованного на ней. Катализатор добавляют только на первой стадии, однако его активность такова, что он остается все еще активным и на всех последующих стадиях.
Компонент 1) предпочтительно получают до компонента 2).
Полимеризацию, которая может быть непрерывной или периодической, проводят, следуя известным методикам и проводя реакцию в жидкой фазе в присутствии или в отсутствие инертного разбавителя, или в газовой фазе, или по методикам с использованием смешанной газожидкостной фазы. Оба компонента 1) и 2) предпочтительно получают в газовой фазе.
Время, давление и температура реакции в том, что касается двух стадий, не являются критическими факторами, однако лучше всего, если температура будет находиться в диапазоне от 20 до 100°С. Давление может быть атмосферным или более высоким.
Регулирование молекулярной массы проводят при использовании известных регуляторов, в частности водорода.
Такую полимеризацию предпочтительно проводят в присутствии стереоспецифических катализаторов Циглера-Натта. Существенным компонентом упомянутых катализаторов является твердый компонент катализатора, содержащий соединение титана, имеющее, по меньшей мере, одну связь титан-галоген, и электронодонорное соединение, причем оба соединения наносят на носитель в виде галогенида магния в активной форме. Еще одним существенным компонентом (сокатализатором) является алюминийорганическое соединение, такое как алюминийалкильное соединение.
Необязательно добавляют внешний донор.
Катализаторы, в общем случае используемые в способе изобретения, способны приводить к получению полипропилена с индексом изотактичности, превышающим 90%, предпочтительно превышающим 95%. Катализаторы, обладающие упомянутыми выше характеристиками, хорошо известны в патентной литературе; в особенности выгодными являются катализаторы, описанные в патенте США 4399054 и европейском патенте 45977.
Твердые компоненты катализатора, используемые в упомянутых катализаторах, содержат в качестве доноров электронов (внутренних доноров) соединения, выбираемые из группы, состоящей из простых эфиров, кетонов, лактонов, соединений, содержащих атомы N, P и/или S, и сложных эфиров моно- и дикарбоновых кислот.
В особенности подходящими электронодонорными соединениями являются сложные эфиры фталевой кислоты, такие как диизобутил-, диоктил-, дифенил- и бензилбутилфталат.
Другими в особенности подходящими донорами электронов являются простые 1,3-диэфиры, описываемые формулой:
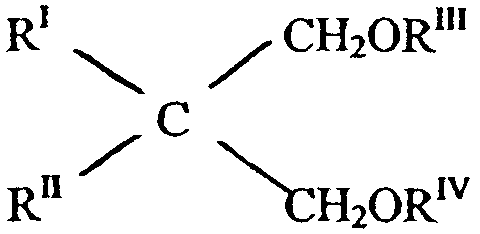
где RI и RII являются одинаковыми или различными, и они представляют собой С1-С18 алкильный, С3-С18 циклоалкильный или С7-С18 арильный радикалы; RIII и RIV являются одинаковыми или различными, и они представляют собой С1-С4 алкильные радикалы; или же простые 1,3-диэфиры, в которых атом углерода в положении 2 входит в циклическую или полициклическую структуру, образованную из 5, 6 или 7 атомов углерода и содержащую две или три ненасыщенности.
Простые эфиры данного типа описываются в опубликованных европейских патентных заявках 361493 и 728769.
Представительными примерами упомянутых простых диэфиров являются 2-метил-2-изопропил-1,3-диметоксипропан, 2,2-диизобутил-1,3-диметоксипропан, 2-изопропил-2-циклопентил-1,3-диметоксипропан, 2-изопропил-2-изоамил-1,3-диметоксипропан, 9,9-бис(метоксиметил)флуорен.
Получение упомянутых выше компонентов катализатора проводят в соответствии с различными способами.
Например, аддукт MgCl2·nROH (в частности, в виде сфероидальных частиц), где n в общем случае находится в диапазоне от 1 до 3, а ROH представляет собой этанол, бутанол или изобутанол, вводят в реакцию с избытком TiCl4, содержащего электронодонорное соединение. Температура реакции в общем случае находится в диапазоне от 80 до 120°С. После этого твердую фазу выделяют и еще раз вводят в реакцию с TiCl4 в присутствии или в отсутствие электронодонорного соединения, после чего ее выделяют и промывают аликвотами углеводорода до тех пор, пока не будут удалены все ионы хлора.
В твердом компоненте катализатора соединение титана, в расчете на Ti, в общем случае присутствует в количестве в диапазоне от 0,5 до 10 мас.%. Количество электронодонорного соединения, которое остается фиксированным на твердом компоненте катализатора, в общем случае находится в диапазоне от 5 до 20 мол.% в расчете на дигалогенид магния.
Соединениями титана, которые можно использовать для получения твердого компонента катализатора, являются галогениды и галогеналкоголяты титана. Тетрахлорид титана является предпочтительным соединением.
Реакции, описанные выше, в результате приводят к получению галогенида магния в активной форме. В литературе известны и другие реакции, которые приводят к получению галогенида магния в активной форме из исходных соединений магния, отличных от галогенидов, таких как карбоксилаты магния.
Al-алкильные соединения, используемые в качестве сокатализаторов, включают Al-триалкилы, такие как Al-триэтил, Al-триизобутил, Al-три-н-бутил, и линейные или циклические Al-алкильные соединения, содержащие два или более атомов Al, связанные друг с другом при помощи атомов O или N, или же групп SO4 или SO3. Al-алкильное соединение в общем случае используют в таком количестве, чтобы соотношение Al/Ti находилось в диапазоне от 1 до 1000.
Электронодонорные соединения, которые можно использовать в качестве внешних доноров, включают сложные эфиры ароматических кислот, такие как алкилбензоаты, и в особенности соединения кремния, содержащие, по меньшей мере, одну связь Si-OR, где R представляет собой углеводородный радикал. Примерами соединений кремния являются (трет-бутил)2Si(OCH3)2, (циклогексил)(метил)Si(OCH3)2, (фенил)2Si(OCH3)2 и (циклопентил)2Si(OCH3)2. В выгодном варианте также можно использовать и простые 1,3-диэфиры, описываемые формулами, приведенными выше. Если внутренним донором будет один из данных простых диэфиров, то внешние доноры можно и не использовать.
Катализаторы можно предварительно ввести в контакт с небольшими количествами олефинов (форполимеризация).
Другими катализаторами, которые можно использовать в способе, соответствующем настоящему изобретению, являются катализаторы металлоценового типа, описанные в работах USP 5324800 и ЕР-А-0129368; в особенности выгодными являются мостиковые бис-инденильные металлоцены, например, описанные в работах USP 514819 и ЕР-А-0485823. Еще одним классом подходящих катализаторов являются так называемые катализаторы с ограничением по геометрии, описанные в работах ЕР-А-0416815 (Dow), ЕР-А-0420436 (Exxon), EP-А-0671404, ЕР-А-0643066 и WO 91/04257. Данные металлоценовые соединения можно использовать, в частности, для получения сополимеров (а) и (b).
Композиции настоящего изобретения также можно получать в результате синтезирования упомянутых компонентов 1) и 2) по отдельности, проводя реакции с использованием тех же самых катализаторов и, по существу, в тех же самых условиях полимеризации, что и описанные прежде (за исключением того, что способ полностью ступенчатой полимеризации реализоваться не будет, а упомянутые компоненты будут получать на раздельных стадиях полимеризации), а после этого механически смешивая упомянутые компоненты в расплавленном или размягченном состоянии. Возможно использование обычных смесительных аппаратов, подобных шнековым экструдерам, в особенности двухшнековым экструдерам.
Композиции настоящего изобретения также могут содержать и добавки, обычно используемые на современном уровне техники, такие как антиоксиданты, светостабилизаторы, термостабилизаторы, зародышеобразователи, красители и наполнители.
В частности, добавление зародышеобразователей приводит к значительному улучшению важных физико-механических свойств, таких как модуль упругости при изгибе, теплостойкость (HDT), предел текучести при растяжении и прозрачность.
Типичными примерами зародышеобразователей являются п-трет-бутилбензоат и 1,3- и 2,4-дибензилиденсорбиты.
Зародышеобразователи предпочтительно добавляют в композиции настоящего изобретения в количествах в диапазоне от 0,05 до 2 мас.%, более предпочтительно от 0,1 до 1 мас.%, в расчете на полную массу.
Добавление неорганических наполнителей, таких как тальк, карбонат кальция и минеральные волокна, также приводит к улучшению некоторых механических свойств, таких как модуль упругости при изгибе и HDT. Тальк также может проявлять и действие зародышеобразователя.
Конкретные подробности приведены в следующих далее примерах, которые приводятся для иллюстрирования настоящего изобретения без его ограничения.
Примеры 1-3
В следующих далее примерах полиолефиновые композиции, соответствующие настоящему изобретению, получают по способу ступенчатой полимеризации.
Твердым компонентом катализатора, используемым в полимеризации, является компонент высокостереоспецифического катализатора Циглера-Натта, нанесенный на носитель в виде хлорида магния, содержащий приблизительно 2,5 мас.% титана и диизобутилфталат в качестве внутреннего донора, полученный по аналогии со способом, описанным в примере 1 опубликованной европейской патентной заявки 674991.
СИСТЕМА КАТАЛИЗАТОРА И ФОРПОЛИМЕРИЗАЦИОННАЯ ОБРАБОТКА
Твердый компонент катализатора, описанный выше, перед введением его в полимеризационные реакторы контактируют в течение 5 минут при -5°С с триэтилалюминием (TEAL) и дициклопентилдиметоксисиланом (DCPMS) при массовом соотношении TEAL/DCPMS, равном приблизительно 4, и в таком количестве, чтобы молярное соотношение TEAL/Ti было равно 65.
После этого систему катализатора подвергают форполимеризации, выдерживая ее в виде суспензии в жидком пропилене при 20°С в течение приблизительно 20 минут перед ее введением в первый полимеризационный реактор.
ПОЛИМЕРИЗАЦИЯ
Полимеризацию проводили в непрерывном режиме в каскаде из двух газофазных реакторов, оснащенных устройствами для переноса продукта, поступающего из реактора, непосредственно предшествующего, в реактор, непосредственно последующий.
В газовой фазе проводили непрерывный анализ и подачу водорода и мономера (мономеров) таким образом, чтобы желательная концентрация выдерживалась постоянной.
В первом газофазном полимеризационном реакторе получали сополимер пропилена/этилена в результате подачи непрерывного и постоянного потока системы подвергнутого форполимеризации катализатора, водорода (используемого в качестве регулятора степени полимеризации) и мономеров пропилена и этилена в газообразном состоянии, получая, таким образом, компонент 1).
Полимер, полученный в первом реакторе, выгружали во второй реактор, где в результате подачи мономера (мономеров) и водорода с надлежащими молярными соотношениями синтезировали сополимер этилена/бутена, получая, таким образом, компонент 2).
После этого частицы полимера вводили во вращающийся барабан, где их перемешивали с 0,05 мас.% вазелинового масла, 0,05 мас.% стеарата натрия, 0,15 мас.% Irganox® B215 (1 массовая часть пентаэритрил-тетракис[3-(3,5-ди-трет-бутил-4-гидроксифенила], смешанная с 1 массовой частью трис(2,4-ди-трет-бутилфенил)фосфита) и 0,2 мас.% 3,4-диметилбензилиденсорбита марки Millad® 3988.
После этого частицы полимера вводили в двухшнековый экструдер Berstorff™ ZE 25 (соотношение длина/диаметр у шнеков: 33) и экструдировали в атмосфере азота при следующих условиях:
Данные, относящиеся к конечным полимерным композициям, приведенные в таблицах 1 и 2, получены в ходе измерений, проведенных для таким образом экструдированных полимеров.
Данные, продемонстрированные в таблицах, получали при использовании следующих далее методов испытаний.
- Молярные соотношения подаваемых газов
Определяли по методу газовой хроматографии.
- Содержание этилена и 1-бутена в полимерах
Определяли по методу ИК-спектроскопии.
- Скорость течения расплава (MFR)
Определяли в соответствии с методом по ASTM D 1238, условие L (MFR "L").
- Содержание фракций, растворимых и нерастворимых в ксилоле
Определяли следующим образом.
В стеклянную колбу, оснащенную холодильником и магнитной мешалкой, вводили 2,5 г полимера и 250 мл ксилола. Температуру увеличивали в течение 30 минут вплоть до температуры кипения растворителя. Полученный таким образом прозрачный раствор после этого выдерживали при кипячении с обратным холодильником и перемешивании еще в течение 30 минут. Затем закрытую колбу выдерживали в течение 30 минут в бане со льдом и водой, а также в течение 30 минут в термостатированной водяной бане при 25°С. Полученную таким образом твердую фазу отфильтровывали на фильтровальной бумаге для быстрого фильтрования. 100 мл отфильтрованной жидкости выливали в предварительно взвешенный алюминиевый контейнер, который нагревали на нагревательной плитке в потоке азота для удаления растворителя в результате его выпаривания. После этого контейнер выдерживали в печи при 80°С в вакууме до тех пор, пока не достигали постоянной массы. После этого рассчитывали массовое процентное содержание полимера, растворимого в ксилоле при комнатной температуре. Массовое процентное содержание полимера, нерастворимого в ксилоле при комнатной температуре, принимали за индекс изотактичности полимера. Данное значение, по существу, соответствует индексу изотактичности, определенному при экстрагировании с использованием кипящего н-гептана, который по определению представляет собой индекс изотактичности полипропилена.
- Характеристическая вязкость (I.V.)
Определяли в тетрагидронафталине при 135°С.
- Модуль упругости при изгибе
Определяли в соответствии с методом по ISO 178.
- Температура перехода от пластического разрушения к хрупкому (D/B)
Определяли в соответствии с внутренним методом МА 17324, доступным по запросу.
В соответствии с данным методом ударопрочность по двум осям ориентации определяли, используя действие удара автоматического компьютеризированного ударного бойка.
Круглые образцы для испытаний получали, вырезая их при помощи круглого пробойника (диаметром 38 мм). Их кондиционировали в течение, по меньшей мере, 12 часов при 23°С и 50% относительной влажности, а после этого на 1 час помещали в термостатированную баню при температуре испытания.
В ходе развития удара ударным бойком (5,3 кг, полусферический пуансон с диаметром 1,27 см) по круглому образцу, располагающемуся на круглой опоре, фиксировали кривую усилие-время. Используемой была машина типа CEAST 6758/000, модель № 2.
Температура перехода от пластического разрушения к хрупкому обозначает температуру, при которой 50% образцов претерпевает хрупкое разрушение при проведении упомянутого испытания на ударопрочность.
- Получение образцов в виде пластинок
Пластинки для измерения D/B, имеющие размеры 127×127×1,5 мм, получали в соответствии с внутренним способом МА 17283; пластинки для измерения мутности с толщиной 1 мм получали, используя литьевое формование, в соответствии с внутренним способом МА 17335 при времени впрыска, равном 1 секунде, температуре 230°С, температуре формы 40°С, причем описание всех упомянутых способов доступно по запросу.
Способ МА 17283
Литьевая машина представляла собой машину типа Negri BossiТМ (NB 90) с усилием замыкания пресс-формы, равным 90 тоннам. Форма представляла собой прямоугольную пластинку (127×127×1,5 мм).
Основные технологические параметры приводятся ниже:
Метод МА 17335
Литьевая машина представляла собой машину типа BattenfeldТМ BA 500CD с усилием замыкания пресс-формы, равным 50 тоннам. Оформляющая вставка позволяла формовать две пластинки (55×60×1 или 1,5 мм каждая).
- Мутность пластинки
Определяли в соответствии со внутренним методом МА 17270, доступным по запросу.
Пластинки кондиционировали в течение 12-48 часов при относительной влажности 50±5% и температуре 23±1°С.
Использованным аппаратом был колориметр HunterТМ D25P-9. Принципы проведения измерений и вычислений приведены в стандарте ASTM-D1003.
Аппарат калибровали без образца, калибровку проверяли при использовании стандарта мутности. Измерение мутности проводили для пяти пластинок.
- Ударная вязкость в испытании по Изоду (с надрезом)
Определяли в соответствии с методом по ISO180/1А.
Сравнительный пример 1с
Повторяли пример 1, за исключением того, что полимеризацию проводили в каскаде из трех реакторов. В первом реакторе в результате подачи мономеров и водорода с надлежащими молярными соотношениями получали кристаллический сополимер пропилена-этилена (компонент (А')). Таким образом полученный сополимер выгружали во второй реактор, где в результате подачи мономеров и водорода с надлежащими молярными соотношениями получали сополимер пропилена-этилена (компонент (А")).
Сополимер, полученный во втором реакторе, выгружали в виде непрерывного потока и после обезгаживания с удалением не прореагировавших мономеров вводили в виде непрерывного потока в третий газофазный реактор вместе с количественно постоянными потоками водорода и мономеров этилена и 1-бутена в газообразном состоянии. Таким образом получали компонент (В).
Условия полимеризации, молярные соотношения, состав и свойства полученных сополимеров продемонстрированы в таблице 2. Сравнительная композиция продемонстрировала значение модуля упругости при изгибе в том же самом диапазоне, что и значение для композиций настоящего изобретения, - значение, которое получили только благодаря наличию в матрице, отличающейся низкой текучестью, области кристаллического полимера.
В сопоставлении со сравнительной композицией композиции, соответствующие настоящему изобретению, характеризуются сопоставимой или даже еще лучшей жесткостью и лучшей ударопрочностью, выраженной через температуру перехода от пластического разрушения к хрупкому, несмотря на значительно более высокие значения MFR, которые являются причиной улучшения перерабатываемости, поскольку в общем случае она оказывает негативное влияние на жесткость и ударопрочность.
Примечания к таблице.
Содержание компонента в композиции = массовая доля полимера, полученного в указанном реакторе; С2 - = этилен; С4 - = бутен; Н2/С2 - = молярное соотношение подаваемого водорода и подаваемого этилена; С2 -/(С2 -+С3 -) = молярное соотношение подаваемого этилена и подаваемого этилена плюс подаваемого пропилена; С4 -/(С4 -+С2 -) = молярное соотношение подаваемого бутена и подаваемого бутена плюс подаваемого этилена.
| название | год | авторы | номер документа |
|---|---|---|---|
| УДАРОПРОЧНЫЕ ПОЛИОЛЕФИНОВЫЕ КОМПОЗИЦИИ | 2003 |
|
RU2308470C2 |
| ПОЛИОЛЕФИНОВЫЕ КОМПОЗИЦИИ | 2010 |
|
RU2554352C2 |
| УДАРОПРОЧНЫЕ ПОЛИОЛЕФИНОВЫЕ КОМПОЗИЦИИ | 2000 |
|
RU2232783C2 |
| ПОЛИОЛЕФИНОВЫЕ КОМПОЗИЦИИ | 2007 |
|
RU2451699C2 |
| КОМПОЗИЦИЯ МАТОЧНОЙ СМЕСИ ДЛЯ ПОЛУЧЕНИЯ ПОЛИОЛЕФИНОВЫХ ИЗДЕЛИЙ ЛИТЬЕВЫМ ФОРМОВАНИЕМ | 2003 |
|
RU2304598C2 |
| ПОЛИОЛЕФИНОВАЯ КОМПОЗИЦИЯ ДЛЯ СИСТЕМ ТРУБОПРОВОДОВ И ЛИСТОВ | 2007 |
|
RU2458085C2 |
| УДАРОПРОЧНЫЕ ПОЛИОЛЕФИНОВЫЕ КОМПОЗИЦИИ | 2002 |
|
RU2309169C2 |
| ПРОЗРАЧНАЯ И ГИБКАЯ КОМПОЗИЦИЯ ПРОПИЛЕНОВЫХ ПОЛИМЕРОВ И ИЗДЕЛИЕ, ПОЛУЧЕННОЕ ИЗ НЕЕ | 2002 |
|
RU2296772C2 |
| УДАРОПРОЧНЫЕ ПОЛИОЛЕФИНОВЫЕ КОМПОЗИЦИИ | 2005 |
|
RU2371458C2 |
| ПОЛИОЛЕФИНОВАЯ МАТОЧНАЯ СМЕСЬ И КОМПОЗИЦИЯ, ПОДХОДЯЩАЯ ДЛЯ ЛИТЬЕВОГО ФОРМОВАНИЯ | 2010 |
|
RU2531352C2 |
Изобретение относится к полиолефиновой композиции, способу ее получения и к изделиям, содержащим полиолефиновую композицию, таким как игрушки, посуда, контейнеры для продуктов питания. Композиция содержит от 55 до менее чем 70 мас.% кристаллического пропиленового гомополимера или сополимера, содержащего до 15% этилена и/или С4-С10 α-олефина (олефинов), имеющего значение скорости течения расплава, определенное при 230°С и нагрузке 2,16 кг, в диапазоне от 15 до 80 г/10 мин, и от более чем 30 до 45 мас.% сополимера этилена и одного или нескольких С4-С10 α-олефина (олефинов), содержащего 15-40% С4-С10 α-олефина (олефинов). Композиция имеет значение скорости течения расплава, определенное при 230°С и нагрузке 2,16 кг, равное или больше 15 г/10 мин, полное содержание этилена, равное 20% или более, полное содержание С4-С10 α-олефина (олефинов), равное 6% или более, соотношение полного содержания этилена и полного содержания С4-С10 α-олефина (олефинов), равное 2,3 или более, содержание совокупной фракции, растворимой в ксилоле при комнатной температуре, равное 18 мас.% или более, и значение характеристической вязкости фракции, растворимой в ксилоле при комнатной температуре, равное 1,7 дл/г или менее. Способ получения композиции заключается в проведении двух последовательных стадий. При этом вышеуказанные компоненты получают на разных стадиях, а катализатор используют на первой стадии. Способом литьевого формования из вышеуказанной композиции получают изделия. Изобретение позволяет повысить текучесть, ударопрочность и прозрачность. 3 н. и 6 з.п. ф-лы, 2 табл.
1) от 55 до менее чем 70% кристаллического пропиленового гомополимера или сополимера, содержащего вплоть до 15% этилена и/или С4-С10 α-олефина(олефинов) и имеющего значение скорости течения расплава, определенное при 230°С и нагрузке 2,16 кг, в диапазоне от 15 до 80 г/10 мин; и
2) от более чем 30 до 45% сополимера этилена и одного или нескольких С4-С10 α-олефина(олефинов), содержащего от 15 до 40% упомянутого С4-С10 α-олефина (олефинов);
при этом упомянутая композиция имеет значение скорости течения расплава, определенное при 230°С и нагрузке 2,16 кг, равное или больше 15 г/10 мин, полное содержание этилена, равное 20% или более, полное содержание С4-С10 а-олефина(олефинов), равное 6% или более, соотношение полного содержания этилена и полного содержания С4-С10 α-олефина(олефинов), равное 2,3 или более, содержание совокупной фракции, растворимой в ксилоле при комнатной температуре, равное 18 мас.% или более, и значение характеристической вязкости фракции, растворимой в ксилоле при комнатной температуре, равное 1,7 дл/г или менее.
| US 5541260 A, 30.07.1996 | |||
| US 4734459 A, 29.03.1988 | |||
| ПЛЕНОЧНЫЙ ИЛИ ЛИСТОВОЙ МАТЕРИАЛ, ПЛЕНОЧНОЕ ИЛИ ЛИСТОВОЕ ИЗДЕЛИЕ (ВАРИАНТЫ) | 1994 |
|
RU2124535C1 |
| RU 94031477 A1, 10.07.1996. | |||

