Уровень техники
CGRP (Calcitonin Gene-Related Peptide; пептид, связанный с геном кальцитонина) представляет собой существующий в природе 37-аминокислотный пептид, который генерируется тканеспецифичной чередующейся обработкой РНК кальцитонинового мессенджера и широко распространен в центральном и периферическом отделах нервной системы. CGRP локализован преимущественно в сенсорных афферентных и центральных нейронах и опосредует некоторые биологические действия, включая вазодилатацию. CGRP экспрессирует в альфа- и бета-формах, которые отличаются одной или тремя аминокислотами, у крыс и человека соответственно. CGRP-альфа и CGRP-бета проявляют сходные биологические свойства. При высвобождении из клетки CGRP инициирует ее биологические реакции путем связывания с рецепторами определенных клеточных поверхностей, что преимущественно сопряжено с активацией аденилилциклазы. Рецепторы CGRP идентифицированы и фармакологически оценены в некоторых тканях и клетках, включая ткани и клетки головного мозга, ткани и клетки сердечно-сосудистого и эндотелиального происхождения и ткани и клетки гладких мышц.
Опосредуемая CGRP вазодилатация средней артерии мозговой оболочки крысы, как показано, сенсибилизирует нейроны тригеминальных ядерных caudalis (Williamson et al., The CGRP Family: Calcitonin Gene-Related Peptide (CGRP), Amylin and Adrenomedullin, Landes Bioscience, 2000, 245-247). Аналогично растяжение дуральных кровеносных сосудов при головной боли в случае мигрени может активировать тригеминальные нейроны. Некоторые связанные симптомы, в том числе экстракраниальная боль и лицевая allodynia, могут быть результатом сенсибилизированных тригеминальных нейронов (Burstein et al., Ann. Neurol. 2000, 47, 614-624). Антагонисты CGRP могут дать положительный результат при смягчении, предупреждении или реверсировании эффектов нейронной сенсибилизации.
Способность соединений настоящего изобретения действовать как антагонисты CGRP делает их полезными фармакологическими агентами в случае заболеваний, которые поражают CGRP у человека и животных, но особенно у человека. Такие заболевания включают мигрень и гистаминовую головную боль (Doods, Curr Opin Inves Drugs, 2001, 2(9), 1261-1268; Edvinsson et al., Cephalalgia, 1994, 14, 320-327); головную боль при хроническом давлении (Ashina et al., Neurology, 2000, 14, 1335-1340); боль (Yu et al., Eur. J. Pharm., 1998, 347, 275-282); хроническую боль (Hulsebosch et al., Pain, 2000, 86, 136-175); нейрогенное воспаление и воспалительную боль (Holzer, Neurosci., 1988, 24, 739-768; Delay-Goyet et al., Acta Physiol. Scanda., 1992, 146, 537-538; Salmon et al., Nature Neurosci., 2001, 4(4), 357-358); глазную боль (May et al., Cephalalgia, 2002, 22, 195-196), зубную боль (Awawdeh et al., Int. Endocrin. J., 2002, 35, 30-36), инсулиннезависимый сахарный диабет (Molina et al., Diabetes, 1990, 39, 260-265); сосудистые заболевания, воспаление (Zhang et al., Pain, 2001, 89, 265), артрит, астму (Foster et al., Ann. NY Acad. Sci., 1992, 657, 397-404; Schini et al., Am. J. Physiol., 1994, 267, H2483-H24290; Zheng et al., J. Virol., 1993, 67, 5786-5791); шок, сепсис (Beer et al., Crit. Care Med., 2002, 30(8), 1794-1798); синдром зависимости от опиума (Salmon et al., Nature Neurosci., 2001, 4(4), 357-358), толерантность к морфину (Menard et al., J. Neurosci., 1996, 16(7), 2342-2351); приливы крови у мужчин и женщин (Chen et al., Lancet, 1993, 342, 49; Spetz et al., J. Urology, 2001, 166, 1720-1723); аллергический дерматит (Wallengren, Contact Dermatitis, 2000, 43(3), 137-143); энцефалит, травму головного мозга, ишемию, шок, эпилепсию и нейродегенеративные заболевания (Rohrenbeck et al., Neurobiol. of Disease 1999, 6, 15-34); заболевания кожи (Geppetti and Holzer, Eds., Neurogenic Inflammation, 1996, CRC Press, Boca Raton, FL), нейрогенное кожное покраснение, розоватость кожи и эритему. Особую важность имеет острое или профилактическое лечение головной боли, включая мигрень и гистаминовую головную боль.
Настоящее изобретение относится к соединениям, которые могут быть полезны в качестве лигандов для рецепторов CGRP, в особенности в качестве антагонистов для рецепторов CGRP, к способам их получения, их применению при лечении, к содержащим их фармацевтическим композициям и к способам лечения с их применением.
Суть изобретения
Настоящее изобретение относится к соединениям формулы I:

(где переменные R1, R2 и R3 имеют определенные в описании значения), которые могут быть использованы в качестве антагонистов рецепторов CGRP и могут быть использованы для лечения или профилактики заболеваний, при которых поражаются CGRP, таких как головная боль, мигрень и гистаминовая головная боль. Изобретение также относится к фармацевтическим композициям, содержащим такие соединения, и к применению таких соединений и композиций для лечения или профилактики таких заболеваний, при которых поражаются CGRP.
Подробное описание изобретения
Настоящее изобретение относится к антагонистам CGRP, которые включают соединения формулы I:

где:
R1 выбран из:
Н, С1-С6-алкила, С3-С6-циклоалкила и гетероцикла, незамещенного или замещенного одним или несколькими заместителями, независимо друг от друга выбранными из:
С1-С6-алкила, С3-С6-циклоалкила, фенила, гетероарила, гетероцикла, (F)pС1-С3-алкила, галогена, OR4, O(CH2)sOR4, CO2R4, CN, NR10R11, O(CO)R4,
где указанный фенил, указанный гетероарил и указанный гетероцикл каждый независимо друг от друга является незамещенным или замещен 1-5 заместителями, где заместители независимо друг от друга выбраны из R4,
где указанный гетероарил выбран из: имидазола, изоксазола, оксазола, пиразина, пиразола, пиридазина, пиридина, пиримидина и тиазола;
где указанный гетероцикл выбран из: азетидина, диоксана, диоксолана, морфолина, оксетана, пиперазина, пиперидина, пирролидина, тетрагидрофурана и тетрагидропирана;
арил или гетероарил выбраны из: фенила, имидазола, изоксазола, оксазола, пиразина, пиразола, пиридазина, пиридина, пиримидина и тиазола,
где указанный арил и указанный гетероарил каждый независимо друг от друга является незамещенным или замещен одним или несколькими заместителями, независимо друг от друга выбранными из: С1-С6-алкила, С3-С6-циклоалкила, (F)pС1-С3-алкила, галогена, OR4, CO2R4, (CO)NR10R11, SO2NR10R11, N(R10)SO2R11, S(O)mR4, CN, NR10R11 и O(CO)R4;
R2 выбран из:
Н, С0-С6-алкила, С3-С6-циклоалкила и гетероцикла, незамещенного или замещенного одним или несколькими заместителями, независимо друг от друга выбранными из:
С1-С6-алкила, С3-С6-циклоалкила, фенила, гетероарила, гетероцикла, (F)pС1-С3-алкила, галогена, OR4, O(CH2)sOR4, CO2R4, CN, NR10R11 и O(CO)R4;
где указанный фенил, указанный гетероарил и указанный гетероцикл каждый независимо друг от друга является незамещенным или замещен 1-5 заместителями, независимо друг от друга выбранными из R4,
где указанный гетероарил выбран из: бензимидазола, бензотиофена, фурана, имидазола, индола, изоксазола, оксазола, пиразина, пиразола, пиридазина, пиридина, пиримидина, пиррола, тиазола, тиофена и триазола;
где указанный гетероцикл выбран из: азетидина, имидазолидина, имидазолина, изоксазолина, изоксазолидина, морфолина, оксазолина, оксазолидина, оксетана, пиразолидина, пиразолина, пирролина, тетрагидрофурана, тетрагидропирана, тиазолина и тиазолидина;
арил или гетероарил выбраны из: фенила, бензимидазола, бензотиофена, фурана, имидазола, индола, изоксазола, оксазола, пиразина, пиразола, пиридазина, пиридина, пиримидина, пиррола, тиазола, тиофена и триазола;
где указанный арил и указанный гетероарил каждый независимо друг от друга является незамещенным или замещен одним или несколькими заместителями, независимо друг от друга выбранными из: С1-С6-алкила, С3-С6-циклоалкила, (F)pС1-С3-алкила, галогена, OR4, CO2R4, (CO)NR10R11, SO2NR10R11, N(R10)SO2R11, S(O)mR4, CN, NR10R11 и O(CO)R4;
R10 и R11 независимо друг от друга выбраны из: Н, С1-С6-алкила, (F)pС1-С6-алкила, С3-С6-циклоалкила, арила, гетероарила и бензила, незамещенного или замещенного галогеном, гидрокси или С1-С6-алкокси, где R10 и R11 могут быть объединены вместе с образованием цикла, выбранного из: азетидинила, пирролидинила, пиперидинила, пиперазинила и морфолинила, который является незамещенным или замещен 1-5 заместителями, где заместители независимо друг от друга выбраны из R4;
R4 независимо друг от друга выбраны из: Н, С1-С6-алкила, (F)pС1-С6-алкила, С3-С6-циклоалкила, арила, гетероарила и фенила, незамещенного или замещенного гидрокси или С1-С6-алкокси;
R3 независимо друг от друга выбраны из Н, замещенного или незамещенного С1-С3-алкила, CN и CO2R4;
р принимает значения от 0 до 2q+1, для заместителей с q атомами углерода;
m принимает значения 0, 1 или 2;
s принимает значения 1, 2 или 3;
и их фармацевтически приемлемые соли и индивидуальные диастереомеры.
В одном из вариантов настоящее изобретение относится к соединениям формулы:

где:
R1 выбран из:
С1-С6-алкила, незамещенного или замещенного одним или несколькими заместителями, независимо друг от друга выбранными из: С1-С6-алкила, С3-С6-циклоалкила, фенила, гетероарила, гетероцикла, (F)pС1-С3-алкила, галогена, OR4, O(CH2)sOR4, CO2R4, CN, NR10R11 и O(CO)R4,
R2 выбран из:
арила, незамещенного или замещенного одним или несколькими заместителями, независимо друг от друга выбранными из: С1-С6-алкила, С3-С6-циклоалкила, (F)pС1-С3-алкила, галогена, OR4, CO2R4, (CO)NR10R11, SO2NR10R11, N(R10)SO2R11, S(O)mR4, CN, NR10R11 и O(CO)R4;
R10 и R11 независимо друг от друга выбраны из: Н, С1-С6-алкила, (F)pС1-С6-алкила, С3-С6-циклоалкила, арила, гетероарила и бензила, незамещенного или замещенного галогеном, гидрокси или С1-С6-алкокси, где R10 и R11 могут быть объединены вместе с образованием цикла, выбранного из: азетидинила, пирролидинила, пиперидинила, пиперазинила и морфолинила, который является незамещенным или замещен 1-5 заместителями, где заместители независимо друг от друга выбраны из R4;
R4 независимо друг от друга выбраны из: Н, С1-С6-алкила, (F)pС1-С6-алкила, С3-С6-циклоалкила, арила, гетероарила и фенила, незамещенного или замещенного гидрокси или С1-С6-алкокси;
р принимает значения от 0 до 2q+1, для заместителей с q атомами углерода;
m принимает значения 0, 1 или 2;
s принимает значения 1, 2 или 3;
и их фармацевтически приемлемые соли или индивидуальные диастереомеры.
Следует понимать, что, когда одна или несколько из приведенных выше структур или подструктур многократно повторяет заместители, имеющие одно и то же обозначение, то каждая их таких переменных может иметь одинаковое значение или отличаться от аналогичной указанной переменной. Например, заместитель R2 перечислен четыре раза в формуле I, и каждый из заместителей R2 в формуле I может независимо друг от друга представлять собой любую подструктуру, определенную под обозначением R2. Изобретение не ограничено структурами и подструктурами, в которых каждый из заместителей R2 должен быть одинаковым для данной структуры. То же самое справедливо относительно любой переменной, появляющейся множество раз в структуре или подструктуре.
Соединения настоящего изобретения могут содержать один или несколько асимметричных центров и, следовательно, могут существовать в виде рацемических смесей, единичных энантиомеров, диастереомерных смесей и индивидуальных диастереомеров. Могут присутствовать дополнительные асимметричные центры в зависимости от природы различных заместителей в молекуле. Каждый такой асимметричный центр будет независимо друг от друга образовывать два оптических изомера, и подразумевается, что все возможные оптические изомеры и диастереомеры в смесях и в виде чистых или частично очищенных соединений входят в объем настоящего изобретения. Настоящее изобретение, как подразумевается, охватывает все такие изомерные формы указанных соединений.
Некоторые соединения, описанные в данном изобретении, содержат олефиновые двойные связи и, если не оговорено особо, подразумевается, что они включают как Е, так и Z геометрические изомеры.
Независимые синтезы таких диастереомеров или их хроматографическое разделение могут быть осуществлены так, как известно в данной области техники, путем соответствующей модификации методологии, описанной в работе. Абсолютная стереохимия соединений может быть определена с помощью рентгеновской кристаллографии кристаллических продуктов или кристаллических промежуточных соединений, которые были дериватизированы, если это необходимо, с помощью реагента, содержащего асимметричный центр известной абсолютной конфигурации.
Если желательно, то рацемические смеси соединений могут быть разделены так, чтобы выделить индивидуальные энантиомеры. Разделение может быть проведено с использованием способов, хорошо известных в данной области, таких как реакция сочетания рацемической смеси соединений с энантиомерно чистым соединением с образованием диастереомерной смеси, после чего следует разделение индивидуальных диастереомеров стандартными способами, такими как дробная кристаллизация или хроматография. Реакция сочетания часто представляет собой образование солей с использованием энантиомерно чистой кислоты или энантиомерно чистого основания. Диастереомерные производные затем могут быть превращены в чистые энантиомеры путем расщепления введенного хирального остатка. Рацемическая смесь соединений также может быть разделена непосредственно хроматографическими способами с использованием хиральных стационарных фаз, и такие способы хорошо известны в данной области.
С другой стороны, любой энантиомер соединения может быть получен путем стереоселективного синтеза с использованием оптически чистых исходных материалов или реагентов известной конфигурации способами, которые хорошо известны в данной области.
Как очевидно специалистам в данной области, не все заместители R10 и R11 способны образовывать циклическую структуру. Более того, даже те заместители, которые способны к образованию цикла, могут образовывать или могут не образовывать циклическую структуру.
Также очевидно для специалиста в данной области, что термин «галоген» или «атом галогена», который используется в данном описании, включает хлор, фтор, бром и йод.
Используемый в данном описании термин «алкил», как подразумевается, означает линейную, разветвленную или циклическую структуры, не содержащие двойную или тройную связь. Следовательно, С1-6-алкил определяет группу, которая содержит 1, 2, 3, 4, 5 или 6 атомов углерода в линейном или разветвленном расположении, то есть С1-6-алкил конкретно включает метил, этил, н-пропил, изопропил, н-бутил, изобутил, трет-бутил, пентил и гексил. «Циклоалкил» представляет собой алкил, который частично или полностью образует кольцо из трех или более атомов. С0 или С0-алкил предназначен для определения наличия прямой ковалентной связи.
Термин «алкенил» означает линейную или разветвленную структуры и их комбинации из указанного числа атомов углерода, содержащую, по меньшей мере, одну двойную связь углерод-углерод, где водород может быть замещен дополнительной углерод-углеродной двойной связью. С2-6-алкенил, например, включает этенил, пропенил, 1-метилэтенил, бутенил и т.п.
Термин «алкинил» означает линейную или разветвленную структуры и их комбинации из указанного числа атомов углерода, содержащие, по меньшей мере, одну тройную связь углерод-углерод. Таким образом, С2-6-алкинил означает группу, содержащую 2, 3, 4, 5 или 6 атомов углерода в линейном или разветвленном расположении, то есть С2-6-алкинил конкретно включает 2-гексинил и 2-пентинил.
Используемый в данном описании термин «арил», как подразумевается, означает любое стабильное моноциклическое или бициклическое углеродное кольцо, содержащее до 7 элементов в каждом кольце, где, по меньшей мере, одно кольцо является ароматическим. Примеры таких арильных элементов включают фенил, нафтил, тетрагидронафтил, инданил или бифенил.
Термин «гетероцикл» или «гетероциклический», который используется в описании, за исключением указанных случаев, представляет собой стабильную 5-7-членную моноциклическую или стабильную 8-11-членную бициклическую гетероциклическую кольцевую систему, которая является или насыщенной или ненасыщенной и которая состоит из атомов углерода и одного-четырех гетероатомов, выбранных из группы, включающей N, O и S, и где гетероатомы азота и серы могут быть необязательно окислены, а азотный гетероатом может быть необязательно кватернизован, и включают любую бициклическую группу, в которой любое из определенных выше гетероциклических колец конденсировано с бензольным кольцом. Гетероциклическое кольцо может быть присоединено по любому гетероатому или атому углерода, который приводит к созданию стабильной структуры. Примеры таких гетероциклических групп включают, но без ограничения, азетидин, хроман, дигидрофуран, дигидропиран, диоксан, диоксолан, гексагидроазепин, имидазолидин, имидазолидинон, имидазолин, имидазолинон, индолин, изохроман, изоиндолин, изотиазолин, изотиазолидин, изоксазолин, изоксазолидин, морфолин, морфолинон, оксазолин, оксазолидин, оксазолидинон, оксетан, 2-оксогексагидроазепин, 2-оксопиперазин, 2-оксопиперидин, 2-оксопирролидин, пиперазин, пиперидин, пиран, пиразолидин, пиразолин, пирролидин, пирролин, хинуклидин, тетрагидрофуран, тетрагидропиран, тиаморфолин, тиазолин, тиазолидин, тиоморфолин и их N-оксиды.
Термин «гетероарил», используемый в описании, за исключением отмеченных случаев, определяет стабильную 5-7-членную моноциклическую или стабильную 9-10-членную конденсированную бициклическую гетероциклическую кольцевую систему, которая содержит ароматическое кольцо, любое кольцо которой может быть насыщенным, такую как пиперидинил, частично насыщенным или ненасыщенным, такую как пиридинил, и которое состоит из атомов углерода и одного-четырех гетероатомов, выбранных из группы, состоящей из N, O и S, и где гетероатомы азота и серы могут быть необязательно окислены, а азотный гетероатом может быть необязательно кватернизован, и включают любую бициклическую группу, в которой любое из определенных выше гетероциклических колец конденсировано с бензольным кольцом. Гетероциклическое кольцо может быть присоединено по любому гетероатому или атому углерода, который приводит к созданию стабильной структуры. Примеры таких гетероарильных групп включают, но без ограничения, бензимидазол, бензизотиазол, бензизоксазол, бензофуран, бензотиазол, бензотиофен, бензотриазол, бензоксазол, карболин, циннолин, фуран, фуразан, имидазол, индазол, индол, индолизин, изохинолин, изотиазол, изоксазол, нафтиридин, оксадиазол, оксазол, фталазин, птеридин, пурин, пиран, пиразин, пиразол, пиридазин, пиридин, пиримидин, пиррол, хиназолин, хинолин, хиноксалин, тетразол, тиадиазол, тиазол, тиофен, триазин, триазол и их N-оксиды.
Термин «алкокси», как в «С1-С6-алкокси», относится к алкоксигруппам, содержащим от 1 до 6 атомов углерода, в линейной, разветвленной и циклической конфигурации. Примеры включают метокси, этокси, пропокси, изопропокси, циклопропилокси, циклогексилокси и т.п.
Выражение «фармацевтически приемлемые» используется в данном случае для определения соединений, материалов, композиций и/или дозированных лекарственных форм, которые в рамках утвержденного медицинского законодательства подходят для применения в контакте с тканями тела человека или животного без избыточной токсичности, раздражения, аллергической реакции и других проблем или осложнений, соизмеримых с разумным соотношением положительный эффект/риск.
Используемое в данном описании определение «фармацевтически приемлемые соли» относится к производным, в которых исходное соединение модифицировано путем получения их кислотных или основных солей. Примеры фармацевтически приемлемых солей включают, но без ограничения, минеральные или органические кислотные соли основных остатков, таких как амины; щелочные или органические соли кислотных остатков, таких как карбоновые кислоты; и т.п. Фармацевтически приемлемые соли включают обычные нетоксичные соли или четвертичные аммонийные соли исходного соединения, полученные, например, из нетоксичных неорганических или органических кислот. Например, такие обычные нетоксичные соли представляют собой соли, полученные из неорганических кислот, таких как соляная, бромистоводородная, серная, сульфаминовая, фосфорная, азотная кислота и тому подобное; и соли, полученные из органических кислот, таких как уксусная, пропионовая, янтарная, гликолевая, стеариновая, молочная, малеиновая, винная, лимонная, аскорбиновая, памовая, малеиновая, гидроксималеиновая, фенилуксусная, глутаминовая, бензойная, салициловая, сульфаниловая, 2-ацетоксибензойная, фумаровая, толуолсульфоновая, метансульфоновая, этандисульфоновая, щавелевая, изэтионовая кислота и т.п.
Число определенных переменных, присутствующих в некоторых случаях, определяется по числу присутствующих атомов углерода. Например, переменную «р» иногда определяют следующим образом: «р принимает значения от 0 до 2q+1, для заместителей с q атомами углерода». Когда заместитель представляет собой (F)рС1-3-алкил, то это означает, что, когда присутствует один атом углерода, может присутствовать 2(1)+1=3 атома фтора. Когда присутствуют два атома углерода, может присутствовать 2(2)+1=5 атомов фтора, и, когда присутствуют три атома углерода, может присутствовать 2(3)+1=7 атомов фтора.
Когда соединение настоящего изобретения является основным, соли могут быть получены из фармацевтически приемлемых нетоксичных кислот, включая неорганические и органические кислоты. Такие кислоты включают уксусную, бензолсульфоновую, бензойную, камфорсульфоновую, лимонную, этансульфоновую, фумаровую, глюконовую, глутаминовую, бромистоводородную, соляную, изэтионовую, молочную, малеиновую, яблочную, миндальную, метансульфоновую, слизевую, азотную, памовую, пантотеновую, фосфорную, янтарную, серную, винную, п-толуолсульфоновую кислоту и т.п. В одном из аспектов настоящего изобретения соли представляют собой соли лимонной, бромистоводородной, соляной, малеиновой, фосфорной, серной, фумаровой и винной кислот. Следует понимать, что при использовании в данном случае подразумевается, что ссылки на соединения формулы I также включают и фармацевтически приемлемые соли.
Примером изобретения является применение соединений, описанных в примерах и в описании. Конкретными соединениями в рамках настоящего изобретения являются соединения, которые выбраны из группы, включающей соединения, описанные в приведенных ниже примерах, и их фармацевтически приемлемые соли и индивидуальные диастереомеры.
Соединения могут быть использованы в способе подавления рецепторов CGRP у пациента, такого как млекопитающее, нуждающегося в таком подавлении, который включает введение эффективного количества соединения. Настоящее изобретение относится к применению соединений, описанных в изобретении, в качестве антагонистов рецепторов CGRP. Помимо приматов, особенно людей, ряд других млекопитающих может подвергаться лечению в соответствии со способом настоящего изобретения.
Другой вариант осуществления настоящего изобретения относится к способу лечения, регуляции, ослабления или снижения риска заболевания или расстройства, при котором у больного поражаются рецепторы CGRP, и этот способ включает введение больному терапевтически эффективного количества соединения, которое представляет собой антагонист рецепторов CGRP.
Настоящее изобретение также относится к способу производства медикамента для подавления активности рецепторов CGRP у человека и животных, включающему смешение соединения настоящего изобретения с фармацевтически приемлемым наполнителем или разбавителем.
Субъект, который подвергается лечению способами настоящего изобретения, обычно является млекопитающим, например человеком, мужчиной или женщиной, которому необходимо подавление активности рецепторов CGRP. Термин «терапевтически эффективное количество» означает количество соединения, которое будет вызывать биологическую или медицинскую реакцию ткани, системы, животного или человека, которая предполагается исследователем, ветеринаром, лечащим врачом или другим клиницистом. Используемое в данном описании понятие «лечение» относится как к лечению, так и к предупреждению или профилактическому лечению упомянутых выше состояний, особенно у больного, который предрасположен к такому заболеванию или расстройству.
Термин «композиция», используемый в данном описании, как подразумевается, охватывает продукт, содержащий определенные ингредиенты в определенных количествах, а также продукт, который образуется, непосредственно или опосредованно, из сочетания определенных ингредиентов в определенных количествах. Такой термин относительно фармацевтической композиции, как подразумевается, включает продукт, содержащий активный(е) ингредиент(ы) и инертный(е) ингредиент(ы), который(е) представляе(ю)т собой носитель, а также продукт, который образуется, непосредственно или опосредованно, из сочетания, комплексообразования или агрегации любых двух или нескольких ингредиентов, или из диссоциации одного или нескольких ингредиентов, или из других типов реакций или взаимодействий одного или нескольких ингредиентов. Таким образом, фармацевтические композиции настоящего изобретения включают любую композицию, полученную смешением соединения настоящего изобретения и фармацевтически приемлемого носителя. Под «фармацевтически приемлемым» понимается, что носитель, разбавитель или эксципиент должны быть совместимы с другими ингредиентами рецептуры и не должны быть вредными для реципиента.
Термин «введение» и «или назначение» соединения следует понимать как доставку соединения настоящего изобретения или пролекарства соединения настоящего изобретения индивидууму, нуждающемуся в лечении.
Применимость соединений в соответствии с настоящим изобретением в качестве антагониста активности рецептора CGRP может быть показана с помощью методологии, известной в данной области. Ингибирование связывания 125I-CGRP с рецепторами и функциональный антагонизм рецепторов CGRP определяют следующим образом.
ОЦЕНКА СВЯЗЫВАНИЯ: Связывание 125I-CGRP с рецепторами в мембранах клеток SK-N-MC проводят по существу так, как описано в публикации Edvinsson et al. (2001), Eur.J.Pharmacol., 415, 39-44). Если говорить кратко, то мембраны (25 мкг) инкубируют в 1 мл связывающего буфера (10 мМ HEPES, рН 7,4, 5 мМ MgCl2 и 0,2% альбумина бычьей сыворотки (BSA), содержащего 10 пМ 125I-CGRP и ингибитор. После выдерживания при комнатной температуре в течение 3 часов опыт прерывают путем фильтрования через фильтрующие пластины из стекловолокна GFB (Millipore), которые были блокированы 0,5% полиэтиленимином в течение 3 часов. Фильтры промывают три раза буфером для анализа, охлажденным льдом, затем пластины сушат на воздухе. Добавляют сцинтилляционную жидкость (50 мкл) и подсчитывают радиоактивность на приборе Topcount (Packard Instrument). Данные анализа обрабатывают с использованием Prism, и Ki определяют с использованием уравнения Cheng-Prusoff (Cheng & Prusoff (1973), Biochem. Pharmacol. 22, 3099-3108).
ФУНКЦИОНАЛЬНАЯ ОЦЕНКА: Клетки SK-N-MC выращивают в минимальной поддерживающей среде (МЕМ), дополненной 10% фетальной бычьей сыворотки, 2 мМ L-глутамина, 0,1 мМ неосновных аминокислот, 5 мМ пирувата натрия, 100 ед./мл пенициллина и 100 мкг/мл стрептомицина, при 37°C, 95% влажности и 5% СО2. В случае анализа цАМФ клетки наносят из расчета 5×105 клетка/лунку на 96-луночный покрытый поли-D-лизином планшет (Becton-Dickinson) и культивируют в течение 18 час до проведения анализа. Клетки промывают физиологическим раствором с фосфатным буфером (PBS, Sigma), затем предварительно выдерживают с 300 мкМ изобутилметилксантина в бессывороточной МЕМ в течение 30 мин при 37°C. Добавляют CGRP (8-37 мкл) и клетки выдерживают в течение 10 мин до добавления CGRP. Инкубацию продолжают еще 15 мин, затем клетки промывают PBS и отрабатывают для определения цАМФ в соответствии с рекомендациями производителя. Максимальную стимуляцию в сравнении с базовым уровнем определяют с использованием 100 нМ CGRP. Кривые доза-реакция получают с помощью Prism. Рассчитывают зависимость доза-отношение (DR) и используют для создания завершенных Schild-графиков (Arunlakshana & Schild (1959), Br. J. Pharmacol. 14, 48-58).
В частности, соединения приведенных далее примеров имеют активность в качестве антагонистов рецепторов CGRP в упомянутых выше методах оценки обычно со значениями Ki или IC50 менее чем приблизительно 50 мкМ. Такие результаты указывают на значительную активность соединений при применении в качестве антагонистов рецепторов CGRP.
Способность соединений настоящего изобретения действовать в качестве антагонистов CGRP делает их полезными фармакологическими агентами в случае заболеваний, которые поражают CGRP у человека и животных, но в особенности у человека.
Соединения настоящего изобретения находят применение при лечении, профилактике, ослаблении, регулировании или снижении риска одного или нескольких из следующих состояний или заболеваний: головная боль; мигрень; гистаминовая головная боль; головная боль при хроническом давлении; боль; хроническая боль; нейрогенное воспаление и воспалительная боль; глазная боль; зубная боль; диабет; инсулиннезависимый сахарный диабет; сосудистые заболевания; воспаление; артрит; астма; шок, сепсис; синдром отказа от опиатов; толерантность к морфину; приливы крови у мужчин и женщин; аллергический дерматит; энцефалит; травма головного мозга; эпилепсия; нейродегенеративные заболевания; заболевания кожи; нейрогенное кожное покраснение; розоватость кожи и эритема; и другие состояния, которые можно лечить или предупреждать путем подавления рецепторов CGRP. Особенно важным является острое или профилактическое лечение головной боли, включая мигрень и гистаминовую головную боль.
Соединения также могут быть использованы в способе профилактики, лечения, регуляции, ослабления или уменьшения риска заболеваний, расстройств и состояний, указанных в описании.
Соединения также могут быть использованы в способе профилактики, лечения, регуляции, ослабления или уменьшения риска заболеваний, расстройств и состояний в комбинации с другими агентами.
Соединения настоящего изобретения могут быть использованы в комбинации с одним или несколькими другими лекарствами при лечении, профилактике, регуляции, ослаблении или уменьшении риска заболеваний или состояний, для которых соединения изобретения или другие лекарства могут находить применение, где комбинация лекарств вместе является безопаснее или более эффективной, чем любое лекарство отдельно. Такое другое лекарство (лекарства) может быть введено способом и в обычно используемом количестве одновременно или последовательно с заявляемыми соединениями. Когда заявляемое соединение используют одновременно с одним или несколькими другими лекарствами, может быть использована фармацевтическая композиция в стандартной лекарственной форме, содержащей такие другие лекарства и заявляемое соединение. Однако комбинированная терапия также может включать лечение, при котором заявляемые соединения и одно или несколько других лекарственных средств вводят по различным перекрывающимся схемам. Также подразумевается, что при использовании в комбинации с одним или несколькими другими активными ингредиентами соединения настоящего изобретения и другие активные ингредиенты могут быть использованы в более низких дозах, чем при использовании каждого лекарства по отдельности. Таким образом, фармацевтические композиции настоящего изобретения включают композиции, которые содержат один или несколько активных ингредиентов помимо заявляемого соединения или соединений.
Например, соединения настоящего изобретения могут быть использованы в комбинации с противовоспалительным или обезболивающим агентом или средством против мигрени, таким как, например, эрготамин или агонисты 5-НТ1, в особенности агонист 5-НТ1в/1D, например, суматриптан, наратриптан, золмитриптан, элетриптан, алмотриптан, фловатриптан, донитриптан и ризатриптан; ингибитором циклооксигеназы, таким как селективный ингибитор циклооксигеназы-2, например, рофекоксиб, эторикоксиб, целекоксиб, валдекоксиб или паракоксиб; нестероидным противовоспалительным агентом или цитокин-подавляющим противовоспалительным агентом, например, с таким соединением, как аспирин, ибупрофен, кетопрофен, фенопрофен, напроксен, индометацин, сулиндак, мелоксикам, пироксикам, теноксикам, лорноксикам, кеторолак, этодолак, мефенаминовая кислота, меклофенаминовая кислота, флуфенаминовая кислота, толфенаминовая кислота, диклофенак, оксапрозин, апазон, нимесулид, набуметон, тенидап, этанерцепт, толметин, фенилбутазон, оксифенбутазон, дифлунизал, салсалат, олсалазин или сулфазалазин и т.п.; или со стероидным анальгетиком. Аналогично соединения настоящего изобретения могут быть введены с болеутоляющим средством, таким как ацетаминофен, фенацетин, кодеин, фентанил, суфентанил, метадон, ацетилметадол, бупренорфин или морфин.
Кроме того, соединения настоящего изобретения могут быть использованы в сочетании с ингибитором интерлейкина, таким как ингибитор интерлейкина-1; антагонистом рецептора NK-1, например апрепитантом; антагонистом NMDA; антагонистом NR2B; антагонистом рецептора брадикинина-1; агонистом рецептора аденозина А1; блокатором натриевого канала, например ламотригином; опиатным агонистом, таким как левометадил-ацетат или метадил-ацетат; ингибитором липоксигеназы, таким как ингибитор 5-липоксигеназы; антагонистом альфа-рецептора, например индорамином; агонистом альфа-рецептора; антагонистом ваниллоидного рецептора; агонистом, антагонистом или потенцирующим средством mGluR5; модулятором рецептора GABA A, например акампросат-кальцием; никотиновыми антагонистами или агонистами, включая никотин; мускариновыми агонистами или антагонистами; селективными ингибиторами повторного поглощения серотонина, например флуоксетином, пароксетином, сертралином, дулоксетином, эсциталопрамом или циталопрамом; трициклическим антидепрессантом, например амитриптилином, доксепином, протриптилином, дезипрамином, тримипрамином или имипрамином; антагонистами лейкотриена, например монтелукастом или зафирлукастом; ингибиторами оксида азота или ингибиторами синтеза оксида азота.
Кроме того, соединения настоящего изобретения могут быть использованы в сочетании с алкалоидами спорыньи, такими как, например, эрготамин, эргоновин, метилэргоновин, метэрголин, эрголоидные мезилаты, дигидроэрготамин, дигидроэргокорнин, дигидроэргокристин, дигидроэргокриптин, дигидро-I-эргокриптин, дигидро-ϑ-эргокриптин, эрготоксин, эргокорнин, эргокристин, эргокриптин, I-эргокриптин, ϑ-эргокриптин, эргозин, эргостан, бромкриптин или метисергид.
Кроме того, соединения настоящего изобретения могут быть использованы в сочетании с бета-адренергическим антагонистом, таким как тимолол, пропанолол, атенолол или надолол и т.п.; ингибитором МАО, например, фенелзином; блокатором кальциевого канала, например, таким как флунаризин, нимодипин, ломеризин, верапамил, нифедипин, прохлорперазин или габапентин; с нейролептиками, такими как оланзапин и кветиапин; с антиконвульсантом, таким как топирамат, зонизамид, тонаберсат, караберсат или дивалроэкс-натрий; антагонистом ангиотензина II, например, таким как лозартан и кандесартан-цилексетил; ингибитором ангиотензин-преобразующего фермента, таким как лизиноприл; или токсином бутулизма типа А.
Соединения настоящего изобретения могут быть использованы в сочетании со стимулирующим средством, таким как кофеин, Н2-антагонист, симетикон, гидроксид алюминия или магния; с противоотечным средством, таким как фенилэфрин, фенилпропаноламин, псевдоэфедрин, оксиметазолин, эпинефрин, нафазолин, ксилометазолин, пропилгекседрин или лево-дезокси-эфедрин; с противокашлевым средством, таким как кодеин, гидрокодон, карамифен, карбетапентан или декстрометорфан; с диуретиком; прокинетическим средством, таким как метоклопрамид или домперидон, и с седативным или неседативным антигистамином.
В одном из вариантов соединения настоящего изобретения используются в сочетании со средствами против мигрени, такими как: эрготамин; агонист 5-НТ1, в особенности агонист 5-НТ1в/1D, в частности суматриптан, наратриптан, золмитриптан, элетриптан, алмотриптан, фловатриптан, донитриптан и ризатриптан; и ингибитором циклооксигеназы, таким как селективный ингибитор циклооксигеназы-2, в частности рофекоксиб, эторикоксиб, целекоксиб, мелоксикам, валдекоксиб или паракоксиб.
Приведенные выше комбинации включают комбинации соединения настоящего изобретения не только с одним другим активным соединением, но также с двумя или несколькими другими активными соединениями. Аналогично соединения настоящего изобретения могут быть использованы в комбинации с другими лекарственными средствами, которые используются для профилактики, лечения, регуляции, ослабления или снижения риска заболеваний или состояний, для которых могут быть использованы соединения настоящего изобретения. Такие другие лекарства могут быть введены с помощью способа и в обычно используемом количестве одновременно или последовательно с соединением настоящего изобретения. Когда соединение настоящего изобретения используется одновременно с одним или несколькими другими лекарствами, возможна фармацевтическая композиция, содержащая помимо соединения настоящего изобретения такие другие лекарства. Таким образом, фармацевтические композиции настоящего изобретения включают композиции, которые также содержат один или несколько других активных ингредиентов помимо соединения настоящего изобретения.
Массовое отношение соединения настоящего изобретения к другому(им) активному(ым) ингредиенту(ам) может меняться и будет зависеть от эффективной дозы каждого ингредиента. Обычно используют эффективную дозу каждого ингредиента. Следовательно, например, когда соединение настоящего изобретения смешано с другим агентом, массовое отношение соединения настоящего изобретения к другому агенту будет обычно находиться в интервале приблизительно от 1000:1 до 1:1000, или приблизительно от 200:1 до 1:200. Комбинации соединения настоящего изобретения и других активных ингредиентов обычно будут находиться в пределах упомянутого выше интервала, но в каждом случае должна быть использована эффективная доза каждого активного ингредиента.
В таких композициях соединение настоящего изобретения и другие активные агенты могут быть введены отдельно или в сочетании. Кроме того, введение одного элемента может быть проведено до, параллельно или последовательно с введением другого(их) агента(ов) и через такие же или другие пути введения.
Соединения настоящего изобретения могут быть введены перорально, парентерально (например, внутримышечной, внутрибрюшинной, внутривенной, интрацеребровентикулярной (ICV), интрацистернальной инъекцией или вливанием, подкожной инъекцией или с помощью имплантата), с помощью ингаляционного спрея, назальными, вагинальными, ректальными, подъязычными или местными способами введения, и могут быть приготовлены, отдельно или вместе, в виде подходящих дозированных стандартных лекарственных рецептур, содержащих обычные нетоксичные фармацевтически приемлемые носители, адъюванты и растворители, подходящие для каждого способа введения. Помимо лечения теплокровных животных соединения настоящего изобретения эффективны при использовании человеком.
Фармацевтические композиции для введения соединения настоящего изобретения обычно присутствуют в стандартной лекарственной форме и могут быть приготовлены любыми способами, которые хорошо известны в области фармации. Все способы включают стадию проведения объединения активного ингредиента с носителем, который включает один или несколько вспомогательных активных ингредиентов. В общем случае фармацевтические композиции готовят путем однородного и близкого объединения активного ингредиента с жидким носителем или тонкоизмельченным твердым носителем или с обоими, и затем, если необходимо, путем формования продукта в желаемый препарат. В фармацевтической композиции активное соединение находится в количестве, достаточном для получения желаемого воздействия на протекание или состояние заболевания. Используемый в данном описании термин «композиция», как подразумевается, охватывает продукт, содержащий определенные ингредиенты в определенных количествах, а также любой продукт, который образуется непосредственно или опосредованно из сочетания определенных ингредиентов в определенных количествах.
Фармацевтические композиции, содержащие активный ингредиент, могут находиться в форме, подходящей для перорального применения, например в виде таблеток, лепешек, водных или масляных суспензий, диспергируемых порошков или гранул, эмульсий, твердых или мягких капсул, или сиропов или эликсиров. Композиции, предназначенные для перорального применения, могут быть приготовлены способами, известными в данной области техники для производства фармацевтических композиций, и такие композиции могут содержать один или несколько агентов, выбранных из группы, включающей подслащивающие агенты, корригирующие вкус и запах агенты, красящие агенты и консерванты, чтобы получить фармацевтически отменные и приятные препараты. Таблетки содержат активный ингредиент в смеси с нетоксичными фармацевтически приемлемыми эксципиентами, которые подходят для производства таблеток. Такие эксципиенты могут представлять собой, например, инертные разбавители, такие как карбонат кальция, карбонат натрия, лактоза, фосфат кальция или фосфат натрия; гранулирующий и дезинтегрирующий агент, например кукурузный крахмал или альгиновая кислота; связующие агенты, например крахмал, желатин или аравийская камедь; и смазывающие агенты, например стеарат магния, стеариновая кислота или тальк. Таблетки могут быть без покрытия, или на них может быть нанесено покрытие с помощью известных технологий, чтобы задержать дезинтеграцию и абсорбцию в желудочно-кишечном тракте и, таким образом, обеспечить постоянное действие в течение более длительного периода. Например, может быть использован задерживающий время высвобождения материал, такой как моностеарат или дистеарат глицерина. Таблетки также могут быть покрыты с помощью методик, описанных в патентах США 4256108, 4166452 и 4265874, с получением осмотических терапевтических таблеток для контролируемого высвобождения. Пероральные таблетки могут быть приготовлены для немедленного высвобождения, например быстроплавящиеся таблетки или облатки, быстрорастворимые таблетки или быстрорастворимые пленки.
Препараты для перорального применения также могут быть получены в виде твердых желатиновых капсул, где активный ингредиент смешан с инертным твердым разбавителем, например с карбонатом кальция, фосфатом кальция или каолином, или в виде мягких желатиновых капсул, где активный ингредиент смешан с водой или масляной средой, например с кокосовым маслом, жидким вазелином или оливковым маслом.
Водные суспензии содержат активные материалы в смеси с эксципиентами, подходящими для производства водных суспензий. Такие эксципиенты представляют собой суспендирующие агенты, например натрий-карбоксиметилцеллюлозу, метилцеллюлозу, гидроксипропилметилцеллюлозу, альгинат натрия, поливинилпирролидон, трагакант и аравийскую камедь; диспергирующие или смачивающие агенты могут представлять собой существующий в природе фосфатид, например лецитин, или продукты конденсации алкиленоксида с жирными кислотами, например полиоксиэтиленстеарат, или продукты конденсации этиленоксида с длинноцепочечными алифатическими спиртами, например гептадекаэтиленоксицетанол, или продукты конденсации этиленоксида с неполными эфирами, полученными из жирных кислот и гексита, такие как полиоксиэтиленсорбитмоноолеат, или продукты конденсации этиленоксида с неполными эфирами, полученными из жирных кислот и ангидридов гексита, например полиэтиленсорбитанмоноолеат. Водные суспензии также могут содержать один или несколько консервантов, например этил- или н-пропил-п-гидроксибензот, один или несколько красящих агентов, один или несколько корригирующих вкус или запах агентов и один или несколько подслащивающих агентов, таких как сахароза или сахарин.
Фармацевтические композиции могут находиться в форме стерильной инъецируемой водной или масляной суспензии. Такая суспензия может быть приготовлена по известным в данной области методикам с использованием подходящих диспергирующих или смачивающих агентов и суспендирующих агентов, которые были упомянуты выше. Стерильные инъецируемые препараты также могут представлять собой стерильный инъецируемый раствор или стерильную инъецируемую суспензию в нетоксичном парентерально приемлемом разбавителе или растворителе, например в виде раствора в 1,3-бутандиоле. Среди приемлемых носителей и растворителей, которые могут быть использованы, находятся вода, раствор Рингера и изотонический раствор хлорида натрия. Кроме того, стерильные, нелетучие масла обычно используют в качестве растворителя или суспендирующей среды. Для этих целей может быть использовано любое нетоксичное нелетучее масло, в том числе синтетические моно- или диглицериды. Кроме того, жирные кислоты, такие как олеиновая кислота, находят применение при приготовлении инъецируемых средств.
Фармацевтическая композиция и способ настоящего изобретения могут дополнительно включать терапевтически активные соединения, упомянутые выше, которые обычно применяют для лечения описанных выше патологических состояний.
При лечении, профилактике, регуляции, ослаблении или снижении риска состояний, которые требуют подавления активности рецептора CGRP, подходящий уровень доз будет составлять обычно от 0,01 до 500 мг на кг массы тела пациента в день, и эти дозы могут быть введены в разовых или разделенных дозах. Подходящий уровень доз может составлять приблизительно от 0,01 до 250 мг/кг в день, приблизительно от 0,05 до 100 мг/кг в день или приблизительно от 0,1 до 50 мг/кг в день. В пределах указанного интервала доза может составлять от 0,05 до 0,5, от 0,5 до 5 или от 5 до 50 мг/кг в день. В случае перорального введения композиции могут быть приготовлены в форме таблеток, содержащих от 1,0 до 1000 мг активного ингредиента, в частности 1,0, 5,0, 10,0, 15,0, 20,0, 25,0, 50,0, 75,0, 100,0, 150,0, 200,0, 250,0, 300,0, 400,0, 500,0, 600,0, 750,0, 800,0, 900,0 и 1000,0 мг активного ингредиента, для регулирования дозы по симптомам для пациента, который подвергается лечению. Соединения могут быть введены по схеме от 1 до 4 раз в день или могут быть введены один или два раза в день.
При лечении, профилактике, регуляции, ослаблении или снижении риска головной боли, мигрени, гистаминовой головной боли или других заболеваний, для которых предназначены соединения настоящего изобретения, обычно удовлетворительные результаты получают, когда соединения настоящего изобретения вводят при ежедневной дозе приблизительно от 0,1 мг до 100 мг на кг массы тела животного, даваемой в виде разовой ежедневной дозы, или в виде разделенных доз от двух до шести раз в день, или в форме с постепенным высвобождением. Для большинства крупных млекопитающих суммарная дневная доза составляет приблизительно от 1,0 до 1000 мг или приблизительно от 1 до 50 мг. В случае взрослого человека массой 70 кг суммарная дневная доза обычно будет составлять приблизительно от 7 до 350 мг. Такой режим дозирования схема может быть адаптирован с целью достижения оптимальной терапевтической реакции.
Однако следует понимать, что определенный уровень доз и частота введения для любого конкретного пациента могут меняться и будут зависеть от различных факторов, в том числе от активности конкретного используемого соединения, метаболической стабильности и продолжительности действия данного соединения, возраста, массы тела, общего состояния здоровья, пола, питания, способа и времени введения, скорости выведения, лекарственной комбинации, серьезности конкретного состояния и реципиента, подвергающегося лечению.
Некоторые способы получения соединений настоящего изобретения представлены в следующих схемах и примерах. Исходные материалы приготовлены по методикам, известным в данной области или представленным в описании.
Соединения настоящего изобретения могут быть легко получены в соответствии с приведенными ниже схемами и конкретными примерами или их модификациями с использованием легко доступных исходных материалов, реагентов и обычных методик синтеза. В этих реакциях также возможны изменения, которые сами по себе известны специалистам в данной области, но не упоминаются при подробном описании. Общие методики получения соединений, заявленных в настоящем изобретении, могут быть легко поняты и восприняты специалистом в данной области при рассмотрении следующих схем.
Синтез капролактам-азабензимидазолоновых промежуточных соединений может быть осуществлен так, как описано на схемах 1-6.
Получение конечных соединений протекает через промежуточные соединения, которые представляют собой соединения формулы I и формулы II, и синтез каждого промежуточного соединения описан.
РЕАКЦИОННЫЕ СХЕМЫ
Получение конечных соединений протекает через промежуточные соединения, которые представляют собой соединения формулы III и формулы IV, и синтез каждого промежуточного соединения описан.

В общем случае промежуточные соединения формул III и IV могут быть соединены через мочевинный мостик, как показано на схеме I. Аминное промежуточное соединение I может быть превращено в реакционноспособный карбамат, например п-нитрофенилкарбамат 2, который затем взаимодействует с амином, подобным промежуточному соединению 3, с получением мочевины 4. Другие активные промежуточные соединения, известные специалистам в данной области, могут быть использованы для получения соединений типа соединения 4. Например, амин 1 может быть сразу же ацилирован подходящим карбамоилхлоридом.
СХЕМА 1
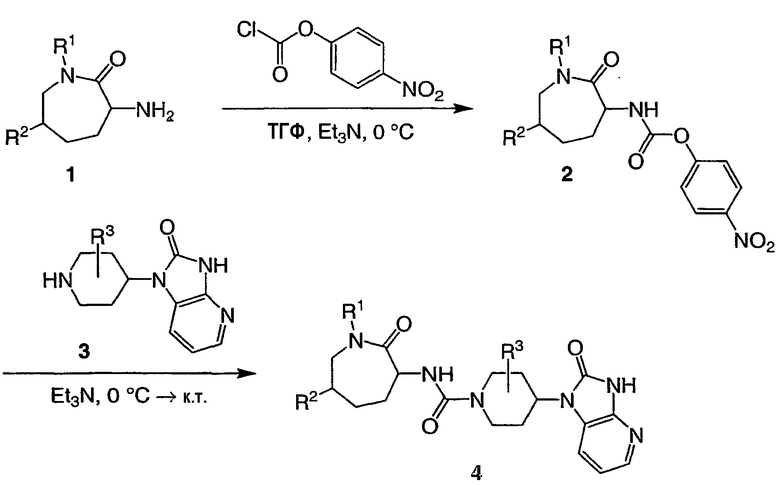
Синтез соединений, представленных промежуточным соединением II, может быть осуществлен с помощью методик, аналогичных методикам, описанным в публикациях Henning et al., J. Med. Chem., 1987, 30, 814-819; Carpino et al., WO 96/35713; Brown et al., J. Chem. Soc., 1957, 682-686; Barlin et al., Aust. J. Chem., 1982, 35(11), 2299-2306; и в приведенных в них ссылках.
Кроме того, синтез соединений, представленных промежуточным соединением II, может быть выполнен в соответствии со схемой 2. Например, диаминогетероцикл, такой как 2,3-диаминопиридин 5, может быть подвергнут восстановительному алкилированию кетонами, такими как соединение 6, с получением моноалкилированного продукта 7. Замыкание кольца карбонилдиимидазолом дает имидазолон 8. Окончательное снятие защиты в стандартных условиях дает конечный продукт 9.
СХЕМА 2

Капролактамы могут быть получены по стратегии олефиновой реакции обмена, которая показана на схеме 3. Гидрохлорид 2,4-диметоксибензиламина алкилируют 2,3-дибромпропеном в мягких основных условиях с получением амина 11. (2R)-2-{[(Бензилокси)карбонил]амино}пент-4-еновая кислота 12, полученная на одной стадии из коммерчески доступного D-аллилглицина в соответствии с известными методиками (J. Chem. Soc., 1962, 3963-3968), может быть сопряжена с амином 11 в различных условиях с получением амида 13. Ряд катализируемых переходным металлом перекрестных сопряжений может быть осуществлен на винилбромиде, например, опосредуемые палладием арилирования фенилбороновой кислотой и карбонатом натрия, с получением производного стирола 14. Реакция обмена с замыканием цикла, проводимая в присутствии рутениевого катализатора Груббса второго поколения в дихлорметане при мягком нагревании, дает лактам 15. Удаление диметоксибензильной группы и гидрирование c защитой in situ первичного амина дает соответствующий насыщенный лактам 17. После селективного алкилирования амидного азота различными электрофилами, такими как алкилбромиды, снятие защиты в кислых условиях дает соединения общей формулы 19.
СХЕМА 3

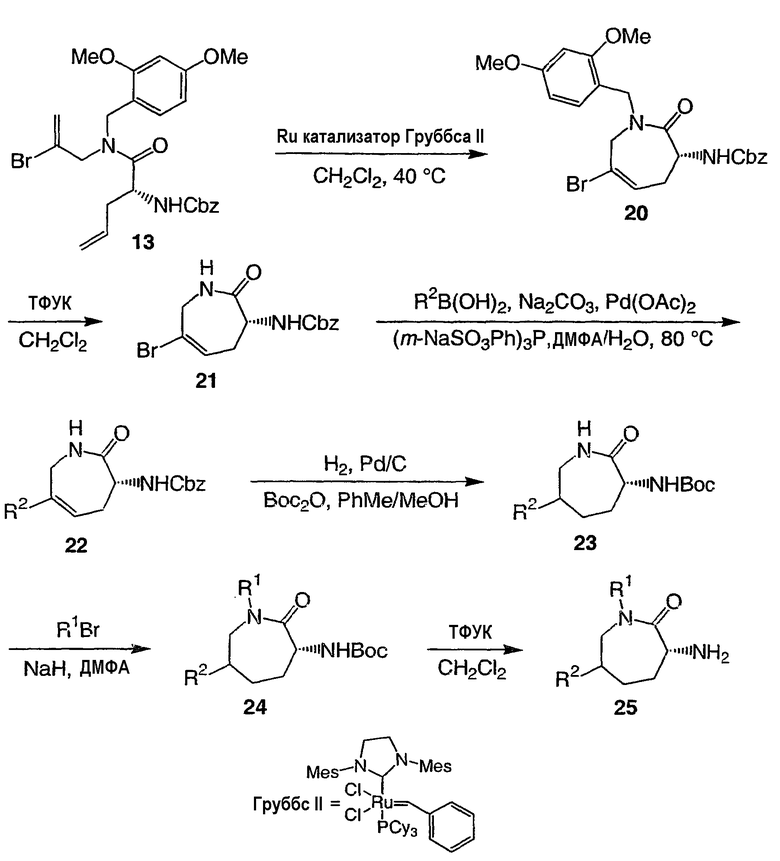
Переменные в положении 6 капролактамов могут быть введены с использованием аналогичной стратегии (схема 4). Реакция обмена с замыканием кольца может быть проведена непосредственно на винилбромиде 13 с использованием рутениевого катализатора Груббса второго поколения с получением циклического винилбромида 20. Удаление диметоксибензильной группы и опосредуемое палладием перекрестное сопряжение, в данном случае с бороновой кислотой, дает соединения общей формулы 22. Преобразование от соединения 21 до соединения 22 не ограничено производными бороновой кислоты. После стандартного гидрирования амидный азот может быть селективно алкилирован различными электрофилами, например алкилбромидами, с использованием гидрида натрия в качестве основания. Снятие защиты дает лактамы общей формулы 25.
СХЕМА 4
В некоторых случаях конечный продукт может быть дополнительно модифицирован, например, путем манипуляции с заместителями. Такие манипуляции могут включать, но не ограничиваются только ими, реакции восстановления, окисления, алкилирования, ацилирования и гидролиза, которые в целом известны специалистам в данной области.
ПРИМЕРЫ
В некоторых случаях порядок проведения приведенных выше реакционных схем может меняться для ускорения реакции или для исключения образования нежелательных продуктов реакции. Следующие примеры предложены для того, чтобы изобретение могло быть понято более полно. Эти примеры являются только иллюстративными, и их не следует рассматривать в качестве ограничивающих изобретение.
Промежуточное соединение 1

Дигидрохлорид 2-оксо-1-(4-пиперидинил)-2,3-дигидро-1Н-имидазо[4,5-b]пиридина
Стадия А. 2-Амино-3-[(1-трет-бутоксикарбонилпиперидин-4-ил)амино]пиридин
К раствору 2,3-диаминопиридина (5,00 г, 45,8 ммоль) и N-(трет-бутоксикарбонил)-4-пиперидона (9,58 г, 48,1 ммоль) в дихлорэтане (75 мл) при комнатной температуре добавляют триацетоксиборгидрид натрия (14,5 г, 68,7 ммоль). Через 5 час добавляют еще триацетоксиборгидрид натрия (1,8 г) и снова еще через 2,5 час. Реакционную смесь перемешивают в течение ночи и реакцию гасят 5% водным раствором гидроксида натрия. Реакционную массу экстрагируют метиленхлоридом и промывают 5% водным раствором гидроксида натрия, водой и насыщенным раствором хлорида натрия. После сушки над сульфатом натрия раствор фильтруют и упаривают, получая сырой продукт. Полученный продукт очищают хроматографией (силикагель, элюирование с градиентом от 3 до 5% метанола в метиленхлориде), получая указанное в заголовке соединение (4,44 г). МС 293 (М+1). 1H-ЯМР (500 МГц, CD3OD) δ 7,32 (дд, J=1, 5 Гц, 1H), 6,85 (дд, J=1, 8 Гц, 1H), 6,59 (дд, J=5, 8 Гц, 1H), 4,04 (д, J=13 Гц, 2H), 3,46 (м, 1H), 2,98 (шир.с, 2H), 2,01 (дд, J=2, 12 Гц, 2H), 1,46 (с, 9H), 1,37 (квд, J=4, 12 Гц, 2H).
Стадия В. 2-Оксо-1-(1-трет-бутоксикарбонилпиперидин-4-ил)-2,3-дигидро-1Н-имидазо[4,5-b]пиридин
Карбонилдиимидазол (0,70 г, 4,33 ммоль) добавляют к раствору 2-амино-3-[(1-трет-бутоксикарбонилпиперидин-4-ил)амино]пиридина (1,15 г, 3,93 ммоль) в ацетонитриле (150 мл) при комнатной температуре. Через несколько часов добавляют дополнительное количество карбонилдиимидазола (0,81 г) и реакционную смесь перемешивают в течение ночи. Ацетонитрил выпаривают в вакууме, остаток распределяют между водой и хлороформом, органическую фазу промывают насыщенным раствором соли и сушат над сульфатом магния. Сырой продукт очищают хроматографией (силикагель, элюирование с градиентом от 1,2 до 2,5% метанола в метиленхлориде), получая указанное в заголовке соединение (1,09 г). 1H-ЯМР (500 МГц, CDCl3) δ 9,39 (шир.с, 1H), 8,04 (дд, J=1, 5 Гц, 1H), 7,33 (дд, J=1, 8 Гц, 1H), 6,99 (дд, J=5, 8 Гц, 1H), 4,50 (м, 1H), 4,32 (шир.с, 2H), 2,86 (шир.с, 2H), 2,20 (м, 2H), 1,86 (д, J=12 Гц, 2H), 1,50 (с, 9H).
Стадия С. Дигидрохлорид 2-оксо-1-(4-пиперидинил)-2,3-дигидро-1Н-имидазол[4,5-b]пиридина
2-Оксо-1-(1-трет-бутоксикарбонилпиперидин-4-ил)-2,3-дигидро-1Н-имидазо[4,5-b]пиридин (1,03 г, 3,23 ммоль) растворяют в метаноле (25 мл) и при комнатной температуре добавляют 2 н. раствор соляной кислоты в эфире (8 мл). Через 2 часа летучие компоненты удаляют в вакууме, получая указанное в заголовке соединение (0,92 г). МС 219 (М+1). 1H-ЯМР (500 МГц, CD3OD) δ 8,01 (дд, J=1, 6 Гц, 1H), 7,83 (д, J=8 Гц, 1H), 7,28 (дд, J=6, 8 Гц, 1H), 4,60 (м, 1H), 3,59 (д, J=12 Гц, 2H), 3,21 (т, J=12 Гц, 2H), 2,70 (дкв, J=4, 13 Гц, 2H), 2,12 (д, J=13 Гц, 2H).
Промежуточное соединение 2
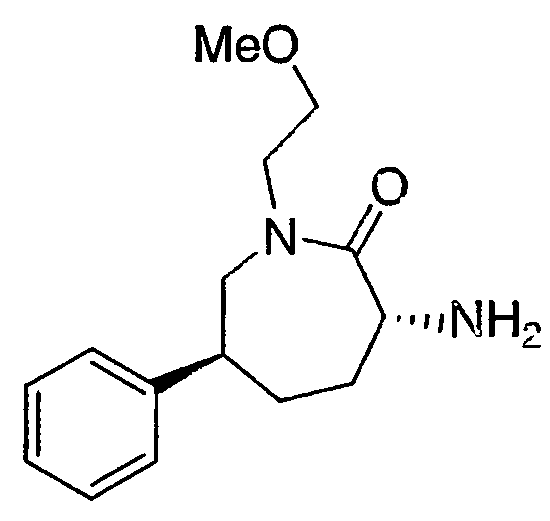
(3R,6S)-3-Амино-1-(2-метоксиэтил)-6-фенилазепан-2-он
Стадия А: 2-бром-N-(2,4-диметоксибензил)проп-2-ен-1-амин
Триэтиламин (16,0 мл, 114 ммоль) добавляют к раствору гидрохлорида 2,4-диметоксибензиламина (11,1 г, 54,5 ммоль) и 2,3-дибромпропена (10,9 г, 54,5 ммоль) в дихлорметане (200 мл). Через 18 час добавляют воду и смесь экстрагируют дихлорметаном (3×). Объединенные органические экстракты промывают насыщенным раствором соли, сушат над сульфатом магния, фильтруют и концентрируют. Очистка хроматографией на силикагеле [100% дихлорметан → 95% дихлорметан/5% (10% гидроксид аммония/метанол)] дает указанное в заголовке соединение (7,85 г).
Стадия В: бензил (1R)-1-{[(2-бромпроп-2-енил)(2,4-диметоксибензил)амино]карбонил}бут-3-енилкарбамат
Гидрохлорид 1-[3-(диметиламино)пропил]-3-этилкарбодиимида (55 мг, 0,285 ммоль) добавляют к раствору 2-бром-N-(2,4-диметоксибензил)проп-2-ен-1-амина (73 мг, 0,256 ммоль) и (2R)-2-{[(бензилокси)карбонил]амино}пент-4-еновой кислоты (71 мг, 0,285 ммоль) в дихлорметане (5 мл). Через 18 час смесь концентрируют. Очистка хроматографией на силикагеле [5% этилацетат/гексаны → 30% этилацетат/гексаны] дает указанное в заголовке соединение (77 мг). МС 517 (М+1).
Стадия С: бензил (1R)-1-{[(2,4-диметоксибензил)(2-фенилпроп-2-енил)амино]карбонил}бут-3-енилкарбамат
Тетракис(трифенилфосфин)палладий(0) (1,11 г, 0,962 ммоль) добавляют к раствору бензил (1R)-1-{[(2-бромпроп-2-енил)(2,4-диметоксибензил)амино]карбонил}бут-3-енилкарбамата (2,49 г, 4,81 ммоль), фенилбороновой кислоты (0,65 г, 5,29 ммоль) и карбоната натрия (2М в воде, 4,81 мл, 9,63 ммоль) в тетрагидрофуране (54 мл) и воде (20 мл) и смесь нагревают до 60°C. Через 1 час смеси дают охладиться до комнатной температуры и экстрагируют дихлорметаном (3×). Объединенные органические экстракты промывают насыщенным раствором соли, сушат над сульфатом магния, фильтруют и концентрируют. Очистка хроматографией на силикагеле [5% этилацетат/гексаны → 30% этилацетат/гексаны] дает указанное в заголовке соединение (2,02 г). МС 515 (М+1).
Стадия D: бензил (3R)-1-(2,4-диметоксибензил)-2-оксо-6-фенил-2,3,4,7-тетрагидро-1Н-азепин-3-илкарбамат
[1,3-бис-(2,4,6-Триметилфенил-2-имидазолидинилиден)дихлор(фенилметилен)-(трициклогексилфосфин)рутений] (катализатор Груббса второго поколения) (0,68 г, 0,79 ммоль) добавляют к раствору бензил (1R)-1-{[(2,4-диметоксибензил)(2-фенилпроп-2-енил)амино}карбонил}бут-3-енилкарбамата (2,02 г, 3,93 ммоль) в дихлорметане (395 мл) и нагревают до 40°C. Через 40 час смеси дают охладиться до комнатной температуры и концентрируют. Очистка хроматографией на силикагеле [5% этилацетат/гексаны → 30% этилацетат/гексаны] дает указанное в заголовке соединение (1,00 г). МС 487 (М+1).1H-ЯМР (500 МГц, CDCl3) δ 7,39-7,31 (м, 5H), 7,26-7,19 (м, 3H), 7,17 (д, J=8,3 Гц, 1H), 6,99 (д, J=7,1 Гц, 2H), 6,41 (дд, J=8,3, 2,0 Гц, 1H), 6,33 (с, 1H), 6,22 (д, J=6,4 Гц, 1H), 5,77-5,76 (м, 1H), 5,16-5,09 (м, 3H), 4,82 (д, J=14,7 Гц, 1H), 4,65 (дд, J=17,6, 2,7 Гц, 1H), 4,54 (д, J=14,4 Гц, 1H), 3,93 (д, J=17,6 Гц, 1H), 3,77 (с, 3H), 3,64 (с, 3H), 2,91-2,86 (м, 1H), 2,42-2,36 (м, 1H).
Стадия Е: бензил (3R)-2-оксо-6-фенил-2,3,4,7-тетрагидро-1Н-азепин-3-илкарбамат
Раствор L-метионина (2,56 г, 17,2 ммоль) в трифторуксусной кислоте (15 мл) добавляют к раствору бензил (3R)-1-(2,4-диметоксибензил)-2-оксо-6-фенил-2,3,4,7-тетрагидро-1Н-азепин-3-илкарбамата (0,84 г, 1,72 ммоль) в дихлорметане (20 мл). Через 18 час смесь концентрируют и добавляют воду. Смесь экстрагируют этилацетатом, промывают водой (2×), насыщенным водным раствором бикарбоната натрия (2×), насыщенным раствором соли, сушат над сульфатом магния, фильтруют и концентрируют. Очистка хроматографией на силикагеле [5% этилацетат/гексаны → 50% этилацетат/гексаны] дает указанное в заголовке соединение (0,44 г). МС 337 (М+1).
Стадия F: трет-бутил (3R,6S)-2-оксо-6-фенилазепан-3-илкарбамат
10% Палладий на угле (75 мг) добавляют к раствору бензил (3R)-2-оксо-6-фенил-2,3,4,7-тетрагидро-1Н-азепин-3-илкарбамата (596 мг, 1,77 ммоль) и ди-трет-бутилдикарбоната (773 мг, 3,54 ммоль) в этилацетате (30 мл). Реакционный сосуд вакуумируют и заполняют в обратном направлении азотом (3×), затем заполняют в обратном направлении водородом (1 атм). Через 2 часа смесь фильтруют и концентрируют. Очистка хроматографией на силикагеле [30% этилацетат/гексаны → 50% этилацетат/гексаны] дает указанное в заголовке соединение (289 мг).
Стадия G: трет-бутил (3R,6S)-1-(2-метоксиэтил)-2-оксо-6-фенилазепан-3-илкарбамат
Гидрид натрия (60% дисперсия в минеральном масле; 6,2 мг, 0,158 ммоль) добавляют к раствору трет-бутил (3R,6S)-2-оксо-6-фенилазепан-3-илкарбамата (40 мг, 0,131 ммоль) и 2-бромэтилметилового эфира (0,013 мл, 0,138 ммоль) в N,N-диметилформамиде (2 мл) при 0°C. После добавления смеси дают нагреться до комнатной температуры. Через 4 часа реакцию гасят водой и смесь экстрагируют этилацетатом. Органический слой промывают водой (3×), насыщенным раствором соли, сушат над сульфатом магния, фильтруют и концентрируют. Очистка хроматографией на силикагеле [5% этилацетат/гексаны → 30% этилацетат/гексаны] дает указанное в заголовке соединение (41 мг). МС 363 (М+1).
Стадия H: (3R,6S)-3-амино-1-(2-метоксиэтил)-6-фенилазепан-2-он
Трифторуксусную кислоту (2,5 мл) добавляют к раствору трет-бутил (3R,6S)-1-(2-метоксиэтил)-2-оксо-6-фенилазепан-3-илкарбамата (41 мг, 0,113 ммоль) в дихлорметане (5 мл). Через 1 час раствор концентрируют. Добавляют насыщенный водный раствор бикарбоната натрия и смесь экстрагируют дихлорметаном (3×). Объединенные органические экстракты промывают насыщенным раствором соли, сушат над сульфатом магния, фильтруют и концентрируют. МС 263 (М+1). 1H-ЯМР (500 МГц, CDCl3) δ 7,32 (т, J=7,3 Гц, 2H), 7,25-7,22 (м, 1H), 7,18 (д, J=8,3 Гц, 2H), 3,83-3,76 (м, 3H), 3,56-3,49 (м, 3H), 3,35 (с, 3H), 3,34-3,30 (м, 1H), 2,77-2,72 (м, 1H), 2,13-2,10 (м, 1H), 2,03-1,94 (м, 2H), 1,74-1,68 (м, 1H).
Следуя фактически методикам, описанным для получения промежуточного соединения 2, получают промежуточные соединения, представленные в таблице I-1.
ТАБЛИЦА I-1


Промежуточное соединение 12

(3R)-3-Амино-6-(4-гидроксифенил)азепан-2-он
Стадия А: бензил (3R)-6-бром-1-(2,4-диметоксибензил)-2-оксо-2,3,4,7-тетрагидро-1Н-азепин-3-илкарбамат
[1,3-бис-(2,4,6-Триметилфенил-2-имидазолидинилиден)дихлор(фенилметилен)-(трициклогексилфосфин)рутений] (катализатор Груббса второго поколения) (1,78 г, 2,05 ммоль) добавляют к раствору бензил (1R)-1-{[(2-бромпроп-2-енил)(2,4-диметоксибензил)амино]карбонил}бут-3-енилкарбамата (5,29 г, 10,2 ммоль) в дихлорметане (1000 мл) и нагревают до 40°C. Через 18 час смеси дают охладиться до комнатной температуры и концентрируют. Очистка хроматографией на силикагеле [5% этилацетат/гексаны → 30% этилацетат/гексаны] дает указанное в заголовке соединение (0,79 г). МС 489 (М+1). 1H-ЯМР (500 МГц, CDCl3) δ 7,36-7,35 (м, 4H), 7,33-7,30 (м, 1H), 7,17-7,15 (м, 1H), 6,46-6,43 (м, 2H), 6,13 (д, J=6,1 Гц, 1H), 6,04-6,03 (м, 1H), 5,13-5,07 (м, 2H), 4,93-4,88 (м, 1H), 4,75 (д, J=14,4 Гц, 1H), 4,64-4,60 (м, 1H), 4,47 (д, J=14,4 Гц, 1H), 3,86 (д, J=18,3 Гц, 1H), 3,81 (с, 3H), 3,80 (с, 3H), 2,68-2,63 (м, 1H), 2,24-2,05 (м, 1H).
Стадия В: бензил (3R)-6-бром-2-оксо-2,3,4,7-тетрагидро-1Н-азепин-3-илкарбамат
Раствор L-метионина (274 мг, 1,84 ммоль) в трифторуксусной кислоте (5 мл) добавляют к раствору бензил (3R)-6-бром-1-(2,4-диметоксибензил)-2-оксо-2,3,4,7-тетрагидро-1Н-азепин-3-илкарбамата (90 мг, 0,184 ммоль) в дихлорметане (5 мл). Через 18 час смесь концентрируют. Очистка ВЭЖХ с обращенной фазой [С-18, 95% вода/ацетонитрил → 5% вода/ацетонитрил с 0,1% трифторуксусной кислоты] дает указанное в заголовке соединение (17 мг). МС 339 (М+1).
Стадия С: бензил (3R)-6-(4-гидроксифенил)-2-оксо-2,3,4,7-тетрагидро-1Н-азепин-3-илкарбамат
Ацетат палладия (1 мг, 0,003 ммоль) добавляют к раствору бензил (3R)-6-бром-2-оксо-2,3,4,7-тетрагидро-1Н-азепин-3-илкарбамата (18 мг, 0,053 ммоль), 4-гидроксифенилбороновой кислоты (9 мг, 0,064 ммоль), карбоната натрия (2М в воде, 0,066 мл, 0,133 ммоль) и тринатрий-3-[бис(3-сульфонатофенил)фосфино]бензолсульфоната (5 мг, 0,088 ммоль) в N,N-диметилформамиде (0,45 мл) и воде (0,15 мл) и нагревают до 80°C. Через 1,5 часа смеси дают охладиться до комнатной температуры и фильтруют. Очистка ВЭЖХ с обращенной фазой [С-18, 95% вода/ацетонитрил → 5% вода/ацетонитрил с 0,1% трифторуксусной кислоты] дает указанное в заголовке соединение (15 мг). МС 353 (М+1).
Стадия D: (3R)-3-амино-6-(4-гидроксифенил)азепан-2-он
10% Палладий на угле (10 мг) добавляют к раствору бензил (3R)-6-(4-гидроксифенил)-2-оксо-2,3,4,7-тетрагидро-1Н-азепин-3-илкарбамата (15 мг, 0,043 ммоль) в толуоле (5 мл) и метаноле (1 мл). Реакционный сосуд вакуумируют и заполняют в обратном направлении азотом (3×), затем заполняют в обратном направлении водородом (1 атм). Через 18 час смесь фильтруют и концентрируют. МС 221 (М+1).
Следуя фактически методикам, описанным для получения промежуточного соединения 12, получают промежуточные соединения, представленные в таблице I-2.
ТАБЛИЦА I-2



Промежуточное соединение 22

(3R,6S)-3-Амино-6-(2,3-дифторфенил)азепан-2-он
Стадия А: 2-бром-N-(2,4-диметоксибензил)проп-2-ен-1-амин
Триэтиламин (16,0 мл, 114 ммоль) добавляют к раствору гидрохлорида 2,4-диметоксибензиламина (11,1 г, 54,5 ммоль) и 2,3-дибромпропена (10,9 г, 54,5 ммоль) в дихлорметане (200 мл). Через 18 час добавляют воду и смесь экстрагируют дихлорметаном (3×). Объединенные органические экстракты промывают насыщенным раствором соли, сушат над сульфатом магния, фильтруют и концентрируют. Очистка хроматографией на силикагеле [100% дихлорметан → 95% дихлорметан/5% (10% гидроксид аммония/метанол)] дает указанное в заголовке соединение (7,85 г).
Стадия В: бензил (1R)-1-{[(2-бромпроп-2-енил)(2,4-диметоксибензил)амино]карбонил}бут-3-енилкарбамат
Гидрохлорид 1-[3-(диметиламино)пропил]-3-этилкарбодиимида (55 мг, 0,285 ммоль) добавляют к раствору 2-бром-N-(2,4-диметоксибензил)проп-2-ен-1-амина (73 мг, 0,256 ммоль) и (2R)-2-{[(бензилокси)карбонил]амино}пент-4-еновой кислоты (71 мг, 0,285 ммоль) в дихлорметане (5 мл). Через 18 час смесь концентрируют. Очистка хроматографией на силикагеле [5% этилацетат/гексаны → 30% этилацетат/гексаны] дает указанное в заголовке соединение (77 мг). МС 517 (М+1).
Стадия C: бензил (1R)-1-{[[2-(2,3-дифторфенил)проп-2-енил](2,4-диметоксибензил)амино]карбонил}бут-3-енилкарбамат
Аддукт дихлор[1,1'-бис(дифенилфосфино)ферроцен]палладия и дихлорметана (0,726 г, 0,889 ммоль) добавляют к раствору бензил (1R)-1-{[(2-бромпроп-2-енил)(2,4-диметоксибензил)амино]карбонил}бут-3-енилкарбамата (9,2 г, 17,8 ммоль), 2,3-дифторфенилбороновой кислоты (2,95 г, 18,7 ммоль) и карбоната натрия (2М в воде; 19,6 мл, 39,1 ммоль) в N,N-диметилформамиде (60 мл) и смесь нагревают до 75°C. Через 2 часа смеси дают охладиться до комнатной температуры и экстрагируют дихлорметаном (3×). Объединенные органические экстракты промывают насыщенным раствором соли, сушат над сульфатом магния, фильтруют и концентрируют. Очистка хроматографией на силикагеле [5% этилацетат/гексаны → 55% этилацетат/гексаны] дает указанное в заголовке соединение (6,8 г). МС 551,2 (М+1).
Стадия D: бензил (3R)-6-(2,3-дифторфенил)-1-(2,4-диметоксибензил)-2-оксо-2,3,4,7-тетрагидро-1Н-азепин-3-илкарбамат
[1,3-бис-(2,4,6-Триметилфенил-2-имидазолидинилиден)дихлор(фенилметилен)-(трициклогексилфосфин)рутений] (катализатор Груббса второго поколения) (2,62 г, 3,09 ммоль) добавляют к раствору бензил (1R)-1-{[[2-(2,3-дифторфенил)проп-2-енил](2,4-диметоксибензил)амино]карбонил}бут-3-енилкарбамата (6,8 г, 12,35 ммоль) в дихлорметане (1800 мл) и раствор нагревают до 40°C. Через 48 час добавляют дополнительное количество катализатора (0,52 г, 0,61 ммоль) и реакционную смесь продолжают нагревать при 40°C еще 48 час. Смеси дают охладиться до комнатной температуры и концентрируют. Очистка хроматографией на силикагеле [5% этилацетат/гексаны → 55% этилацетат/гексаны] дает указанное в заголовке соединение (3,71 г). МС 523,1 (М+1).
Стадия Е: бензил (3R)-6-(2,3-дифторфенил)-2-оксо-2,3,4,7-тетрагидро-1Н-азепин-3-илкарбамат
Трифторуксусную кислоту (60 мл) добавляют к раствору бензил (3R)-6-(2,3-дифторфенил)-1-(2,4-диметоксибензил)-2-оксо-2,3,4,7-тетрагидро-1Н-азепин-3-илкарбамата (3,70 г, 7,08 ммоль) в дихлорметане (40 мл). Через 18 час смесь концентрируют при 25°C, добавляют метанол (150 мл) и осадок отфильтровывают. Фильтрат концентрируют, разбавляют дихлорметаном (100 мл), промывают водой (2×), насыщенным водным раствором бикарбоната натрия (2×), насыщенным раствором соли, сушат над сульфатом магния, фильтруют и концентрируют. Очистка хроматографией на силикагеле [5% этилацетат/гексаны → 65% этилацетат/гексаны] дает указанное в заголовке соединение (1,75 г). МС 373,1 (М+1).
Стадия F: трет-бутил (3R,6S)-6-(2,3-дифторфенил)-2-оксоазепан-3-илкарбамат
10% Палладий на угле (700 мг) добавляют к раствору бензил (3R)-6-(2,3-дифторфенил)-2-оксо-2,3,4,7-тетрагидро-1Н-азепин-3-илкарбамата (2,6 г, 6,98 ммоль) и ди-трет-бутилдикарбоната (5,03 г, 23,0 ммоль) в толуоле (200 мл). Реакционный сосуд вакуумируют и заполняют в обратном направлении азотом (3×), затем заполняют в обратном направлении водородом (1 атм). Через 24 часа смесь фильтруют и концентрируют. Очистка препаративной хроматографией с обращенной фазой (DeltaPak C18, 15 мкм, 47×300 мм, 70 мл/мин: 80% Н2О/NH4OAc:от 20% CH3CN до 100% CH3CN в течение 60 мин) дает чистый транс-изомер указанного в заголовке соединения (1,2 г). МС 341,2 (М+1).1H-ЯМР (500 МГц, CDCl3) δ 7,07-7,04 (м, 2H), 6,91-6,89 (м, 1H), 6,04 (шир.с, 1H), 5,93 (д, J=5,6 Гц, 1H), 4,46 (дд, J=10,5, 4,6 Гц, 1H), 3,65-3,59 (м, 1H), 3,21 (дд, J=15,1, 7,3 Гц, 1H), 3,05-3,00 (м, 1H), 2,25-2,20 (м, 1H), 2,17-2,10 (м, 2H), 1,79-1,71 (м, 1H), 1,46(с, 9H).
Стадия G: (3R,6S)-3-амино-6-(2,3-дифторфенил)азепан-2-он
Трифторуксусную кислоту (4 мл) добавляют к раствору трет-бутил (3R,6S)-6-(2,3-дифторфенил)-2-оксоазепан-3-илкарбамата (82 мг, 0,241 ммоль) в дихлорметане (4 мл). Через 1 час раствор концентрируют. Добавляют насыщенный водный раствор бикарбоната натрия и смесь экстрагируют дихлорметаном (3×). Объединенные органические экстракты промывают насыщенным раствором соли, сушат над сульфатом магния, фильтруют и концентрируют. МС 241,0 (М+1).
Промежуточное соединение 23

(3R,6S)-3-Амино-6-(2,3-дифторфенил)-1-[2-(метилтио)этил]азепан-2-он
Стадия А: трет-бутил (3R,6S)-6-(2,3-дифторфенил)-1-[2-(метилтио)этил]-2-оксоазепан-3-илкарбамат
Гидрид натрия (60% дисперсия в минеральном масле; 40 мг, 0,600 ммоль) добавляют к раствору трет-бутил-(3R,6S)-6-(2,3-дифторфенил)-2-оксоазепан-3-илкарбамата (170 мг, 0,500 ммоль) в N,N-диметилформамиде (4 мл) при 0°C. Через 5 мин смесь охлаждают до -30°C и добавляют 1-йод-2-(метилтио)этан [получен по известной методике: J. Org. Chem., 1987, 52, 2299-2301 (158 мг, 0,782 ммоль)]. Добавляют дополнительное количество гидрида натрия (33 мг, 0,50 ммоль) и через 4 часа добавляют избыток гидрида натрия (33 мг, 0,50 ммоль) и 1-йод-2-(метилтио)этана (75,6 мг, 0,374 ммоль). Через 3 часа добавляют конечные порции гидрида натрия (33 мг, 0,50 ммоль) и 1-йод-2-(метилтио)этана (75,6 мг, 0,374 ммоль) и смесь перемешивают при -20°C в течение ночи. Реакцию гасят водой и смесь концентрируют этилацетатом. Органический слой промывают водой (3×), насыщенным раствором соли, сушат над сульфатом магния, фильтруют и концентрируют. Очистка хроматографией на силикагеле [0% этилацетат/гексаны → 50% этилацетат/гексаны] дает указанное в заголовке соединение (77 мг). МС 415 (М+1).
Стадия В: (3R,6S)-3-амино-6-(2,3-дифторфенил)-1-[2-(метилтио)этил]азепан-2-он
Трифторуксусную кислоту (2 мл) добавляют к раствору трет-бутил (3R,6S)-6-(2,3-дифторфенил)-1-[2-(метилтио)этил]-2-оксоазепан-3-илкарбамата (77 мг, 0,186 ммоль) в дихлорметане (10 мл). Через 30 мин раствор концентрируют и сушат азеотропной отгонкой с толуолом (2×). Добавляют насыщенный водный раствор бикарбоната натрия и смесь экстрагируют дихлорметаном (3×). Объединенные органические экстракты промывают насыщенным раствором соли, сушат над сульфатом магния, фильтруют и концентрируют. МС 315,2 (М+1).
Промежуточное соединение 24

(3R,6S)-3-Амино-6-(2,3-дифторфенил)-1-[2-(метилсульфинил)этил]азепан-2-он
Стадия А: трет-бутил (3R,6S)-6-(2,3-дифторфенил)-1-[2-(метилсульфинил)этил]-2-оксоазепан-3-илкарбамат
Периодат натрия (11,3 мг, 0,053 ммоль) добавляют к раствору трет-бутил (3R,6S)-6-(2,3-дифторфенил)-1-[2-(метилтио)этил]-2-оксоазепан-3-илкарбамата (22 мг, 0,053 ммоль) в метаноле (2 мл) и воде (2 мл). Через 30 мин добавляют периодат натрия (22 мг, 0,11 ммоль). Через 18 час добавляют насыщенный водный раствор карбоната натрия и смесь экстрагируют этилацетатом. Органический слой промывают насыщенным водным раствором карбоната натрия (3×), насыщенным раствором соли, сушат над сульфатом магния, фильтруют и концентрируют, получая указанное в заголовке соединение. МС 431 (М+1).
Стадия В: (3R,6S)-3-амино-6-(2,3-дифторфенил)-1-[2-(метилсульфинил)этил]азепан-2-он
Трифторуксусную кислоту (1 мл) добавляют к раствору трет-бутил (3R,6S)-6-(2,3-дифторфенил)-1-[2-(метилсульфинил)этил]-2-оксоазепан-3-илкарбамата (23 мг, 0,053 ммоль) в дихлорметане (2 мл). Через 3 часа раствор концентрируют. Добавляют насыщенный водный раствор бикарбоната натрия и смесь экстрагируют дихлорметаном (3×). Объединенные органические экстракты промывают насыщенным раствором соли, сушат над сульфатом магния, фильтруют и концентрируют, получая указанное в заголовке соединение. МС 331 (М+1).
Промежуточное соединение 25

(3R,6S)-3-Амино-6-(2,3-дифторфенил)-1-[2-(метилсульфонил)этил]азепан-2-он
Стадия А: трет-бутил (3R,6S)-6-(2,3-дифторфенил)-1-[2-(метилсульфонил)этил]-2-оксоазепан-3-илкарбамат
Оксон (16,1 мг, 0,11 ммоль) добавляют к раствору трет-бутил (3R,6S)-6-(2,3-дифторфенил)-1-[2-(метилтио)этил]-2-оксоазепан-3-илкарбамата (22 мг, 0,053 ммоль) в метаноле (2 мл) и воде (2 мл). Через 6 час добавляют избыток оксона (32 мг, 0,22 ммоль). Через 18 час реакцию гасят водным раствором сульфита натрия и смесь экстрагируют этилацетатом. Органический слой промывают насыщенным водным раствором карбоната натрия (3×), насыщенным раствором соли, сушат над сульфатом магния, фильтруют и концентрируют, получая указанное в заголовке соединение. МС 447 (М+1).
Стадия В: (3R,6S)-3-амино-6-(2,3-дифторфенил)-1-[2-(метилсульфонил)этил]азепан-2-он
Трифторуксусную кислоту (1 мл) добавляют к раствору трет-бутил (3R,6S)-6-(2,3-дифторфенил)-1-[2-(метилсульфонил)этил]-2-оксоазепан-3-илкарбамата (23,7 мг, 0,053 ммоль) в дихлорметане (2 мл). Через 4 часа раствор концентрируют. Добавляют насыщенный водный раствор бикарбоната натрия и смесь экстрагируют дихлорметаном (3×). Объединенные органические экстракты промывают насыщенным раствором соли, сушат над сульфатом магния, фильтруют и концентрируют, получая указанное в заголовке соединение. МС 347 (М+1).
Промежуточное соединение 26

(3R,6S)-3-Амино-6-(2,3-дифторфенил)-1-(2-метоксиэтил)азепан-2-он
Стадия А: трет-бутил (3R,6S)-6-(2,3-дифторфенил)-1-(2-метоксиэтил)-2-оксоазепан-3-илкарбамат
Гидрид натрия (60% дисперсия в минеральном масле; 17,6 мг, 0,264 ммоль) добавляют к раствору трет-бутил (3R,6S)-6-(2,3-дифторфенил)-2-оксоазепан-3-илкарбамата (75 мг, 0,220 ммоль) в N,N-диметилформамиде (2 мл) при 0°C. Через 5 мин добавляют 2-бромэтилметиловый эфир (0,025 мл, 0,264 ммоль) и смеси дают нагреться до комнатной температуры. Через 3 часа реакцию гасят водой и смесь экстрагируют этилацетатом. Органический слой промывают водой (2×), насыщенным раствором соли, сушат над сульфатом магния, фильтруют и концентрируют, получая указанное в заголовке соединение. МС 421 (М+Na).
Стадия В: (3R,6S)-3-амино-6-(2,3-дифторфенил)-1-(2-метоксиэтил)азепан-2-он
Трифторуксусную кислоту (2,5 мл) добавляют к раствору трет-бутил (3R,6S)-6-(2,3-дифторфенил)-1-(2-метоксиэтил)-2-оксоазепан-3-илкарбамата (99 мг, 0,248 ммоль) в дихлорметане (5 мл). Через 1 час раствор концентрируют и сушат азеотропной отгонкой с толуолом (2×). Добавляют насыщенный водный раствор бикарбоната натрия и смесь экстрагируют дихлорметаном (3×). Объединенные органические экстракты промывают насыщенным раствором соли, сушат над сульфатом магния, фильтруют и концентрируют, получая указанное в заголовке соединение. МС 299,2 (М+1).
Промежуточное соединение 27

(3R,6S)-3-Амино-6-(2,3-дифторфенил)-1-(2,2,2-трифторэтил)азепан-2-он
Стадия А: трет-бутил (3R,6S)-6-(2,3-дифторфенил)-2-оксо-1-(2,2,2-трифторэтил)азепан-3-илкарбамат
Гидрид натрия (60% дисперсия в минеральном масле; 70,7 мг, 1,06 ммоль) добавляют к раствору трет-бутил (3R,6S)-6-(2,3-дифторфенил)-2-оксоазепан-3-илкарбамата (301 мг, 0,884 ммоль) в N,N-диметилформамиде (7 мл) при -35°C. Через 15 мин добавляют 2,2,2-трифторэтилтрихлорметансульфонат (0,314 мл, 1,91 ммоль) и реакционную смесь перемешивают при -35°C. Через 30 мин добавляют дополнительное количество гидрида натрия (27 мг, 0,40 ммоль) и 2,2,2-трифторэтилтрихлорметансульфоната (0,140 мл, 0,85 ммоль). Через 2 часа реакцию гасят водой и смесь экстрагируют этилацетатом. Органический слой промывают водой (3×), насыщенным раствором соли, сушат над сульфатом магния, фильтруют и концентрируют. Очистка хроматографией на силикагеле (0% этилацетат/гексаны → 30% этилацетат/гексаны) дает указанное в заголовке соединение (306 мг). МС 423 (М+1).
Стадия В: (3R,6S)-3-амино-6-(2,3-дифторфенил)-1-(2,2,2-трифторэтил)азепан-2-он
Трифторуксусную кислоту (2,5 мл) добавляют к раствору трет-бутил (3R,6S)-6-(2,3-дифторфенил)-2-оксо-1-(2,2,2-трифторэтил)азепан-3-илкарбамата (135 мг, 0,320 ммоль) в дихлорметане (5 мл). Через 30 мин раствор концентрируют и сушат азеотропной отгонкой с толуолом (2×). Добавляют насыщенный водный раствор бикарбоната натрия и смесь экстрагируют дихлорметаном (3×). Объединенные органические экстракты промывают насыщенным раствором соли, сушат над сульфатом магния, фильтруют и концентрируют. МС 323,1 (М+1). 1H-ЯМР (500 МГц, CDCl3) δ 7,11-7,03 (м, 2H), 6,93-6,89 (м, 1H), 4,21-4,13 (м, 1H), 4,10-3,98 (м, 2H), 3,85 (д, J=11,0 Гц, 1H), 3,35 (д, J=15,4 Гц, 1H), 3,04-2,99 (м, 1H), 2,13-2,09 (м, 2H), 2,08-2,02 (м, 1H), 1,78-1,70 (м, 3H).
Промежуточное соединение 28

(3R,6S)-3-Амино-6-(2,3-дифторфенил)-1-(пиридин-2-илметил)азепан-2-он
Стадия А: трет-бутил (3R,6S)-6-(2,3-дифторфенил)-2-оксо-1-(пиридин-2-илметил)азепан-3-илкарбамат
Гидрид натрия (60% дисперсия в минеральном масле; 30 мг, 1,175 ммоль) добавляют к раствору трет-бутил (3R,6S)-6-(2,3-дифторфенил)-2-оксоазепан-3-илкарбамата (160 мг, 0,470 ммоль) в N,N-диметилформамиде (6 мл) при 0°C. Через 30 мин добавляют 2-бромметилпиридин (0,125 мг, 0,494 ммоль). Через 1 час реакцию гасят водой и смесь экстрагируют этилацетатом. Органический слой промывают водой (2×), насыщенным раствором соли, сушат над сульфатом магния, фильтруют и концентрируют, получая указанное в заголовке соединение (202 мг). МС 432,2 (М+1).
Стадия В: (3R,6S)-3-амино-6-(2,3-дифторфенил)-1-(пиридин-2-илметил)азепан-2-он
Трифторуксусную кислоту (3 мл) добавляют к раствору трет-бутил (3R,6S)-6-(2,3-дифторфенил)-2-оксо-1-(пиридин-2-илметил)азепан-3-илкарбамата (202 мг, 0,468 ммоль) в дихлорметане (4 мл). Через 18 час раствор концентрируют. Добавляют насыщенный водный раствор бикарбоната натрия и смесь экстрагируют дихлорметаном (3×). Объединенные органические экстракты промывают насыщенным раствором соли, сушат над сульфатом магния, фильтруют и концентрируют. МС 332,2 (М+1).
Промежуточное соединение 29

(3R,6S)-3-Амино-6-(2,3-дифторфенил)-1-пиридин-4-илазепан-2-он
Стадия А: трет-бутил (3R,6S)-6-(2,3-дифторфенил)-2-оксо-1-пиридин-4-илазепан-3-илкарбамат
4-Бромпиридин (286 мг, 1,47 ммоль) добавляют к раствору трет-бутил (3R,6S)-6-(2,3-дифторфенил)-2-оксоазепан-3-илкарбамата (200 мг, 0,588 ммоль), 4,5-бис(дифенилфосфино)-9,9-диметилксантена (20 мг, 0,035 ммоль), трис(дибензилиденацетон)дипалладия(0) (22 мг, 0,024 ммоль) и карбоната цезия (268 мг, 0,823 ммоль) в диоксане (6 мл) и смесь нагревают при 150°C в микроволновом реакторе Personal Chemistry Smith Creator. Через 30 мин добавляют дополнительное количество 4,5-бис(дифенилфосфино)-9,9-диметилксантена (20 мг, 0,035 ммоль) и трис(дибензилиденацетон)дипалладия(0) (22 мг, 0,024 ммоль). Через 30 мин смесь распределяют между этилацетатом и водой. Органический слой промывают водой (2×), насыщенным раствором соли, сушат над сульфатом магния, фильтруют и концентрируют, получая указанное в заголовке соединение (55 мг). МС 418,2 (М+1).
Стадия В: (3R,6S)-3-амино-6-(2,3-дифторфенил)-1-пиридин-4-илазепан-2-он
Трифторуксусную кислоту (2 мл) добавляют к раствору трет-бутил (3R,6S)-6-(2,3-дифторфенил)-2-оксо-1-пиридин-2-илазепан-3-илкарбамата (55 мг, 0,132 ммоль) в дихлорметане (2 мл). Через 1 час раствор концентрируют. Добавляют насыщенный водный раствор бикарбоната натрия и смесь экстрагируют дихлорметаном (3×). Объединенные органические экстракты промывают насыщенным раствором соли, сушат над сульфатом магния, фильтруют и концентрируют. МС 318,2 (М+1).
Следуя по существу методикам, описанным для получения промежуточных соединений 23-28, получают промежуточные соединения, представленные в таблице I-3.
ТАБЛИЦА I-3
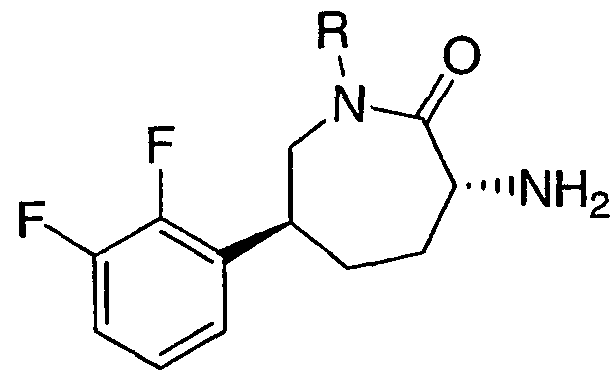


Пример 1

N-[(3R,6S)-1-(2-Метоксиэтил)-2-оксо-6-фенилазепан-3-ил]-4-(2-оксо-2,3-дигидро-1Н-имидазо[4,5-b]пиридин-1-ил)пиперидин-1-карбоксамид
Триэтиламин (0,015 мл, 0,107 ммоль) добавляют к раствору (3R,6S)-3-амино-1-(2-метоксиэтил)-6-фенилазепан-2-она (28 мг, 0,107 ммоль) и 4-нитрофенилхлорформиата (22 мг, 0,107 ммоль) в тетрагидрофуране (2 мл) при 0°C. Через 30 мин добавляют 2-оксо-1-пиперидиний-4-ил-2,3-дигидро-1Н-имидазо[4,5-b]пиридин-4-ийдихлорид (37 мг, 0,128 ммоль), диизопропилэтиламин (0,074 мл, 0,427 ммоль) и дихлорметан (2,5 мл) и смеси дают нагреться до комнатной температуры. Через 18 час добавляют насыщенный водный раствор карбоната натрия и смесь экстрагируют этилацетатом. Органический слой промывают насыщенным водным раствором карбоната натрия (3×), насыщенным раствором соли, сушат над сульфатом магния, фильтруют и концентрируют. Очистка хроматографией на силикагеле (100% дихлорметан → 93% дихлорметан/метанол) дает указанное в заголовке соединение (45 мг). МС 507,2737 (М+1).
Следуя по существу методикам, описанным для получения соединения примера 1, получают соединения, примеры которых представлены в таблице Е-1.
ТАБЛИЦА Е-1

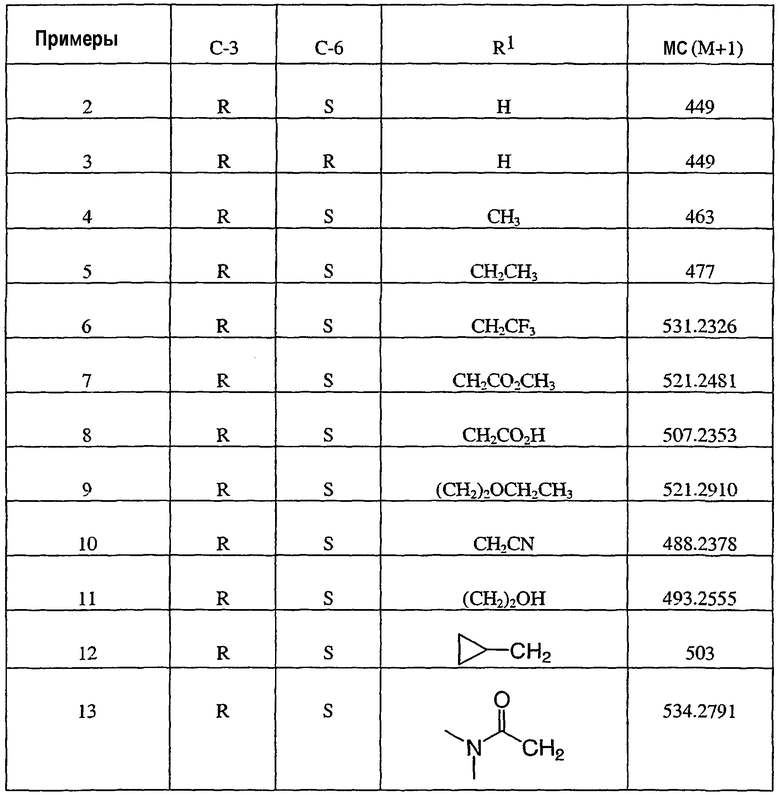
Пример 14

N-[(3R)-6-(4-Гидроксифенил)-2-оксоазепан-3-ил]-4-(2-оксо-2,3-дигидро-1Н-имидазо[4,5-b]пиридин-1-ил)пиперидин-1-карбоксамид
Триэтиламин (0,009 мл, 0,091 ммоль) добавляют к раствору (3R)-3-амино-6-(4-гидроксифенил)азепан-2-она (10 мг, 0,045 ммоль) и 4-нитрофенилхлорформиата (18 мг, 0,091 ммоль) в N,N-диметилформамиде (0,2 мл) и тетрагидрофуране (0,2 мл) при 0°C. Через 1 час добавляют 2-оксо-1-пиперидиний-4-ил-2,3-дигидро-1Н-имидазо[4,5-b]пиридин-4-ийдихлорид (26 мг, 0,091 ммоль) и диизопропилэтиламин (0,032 мл, 0,182 ммоль) и смеси дают нагреться до комнатной температуры. Через 18 час смесь очищают ВЭХЖ с обращенной фазой (С-18, 95% вода/ацетонитрил → 5% вода/ацетонитрил с 0,1% трифторуксусной кислоты), получая указанное в заголовке соединение (7 мг). МС 465,2243 (М+1).
Следуя по существу методикам, описанным для получения соединения примера 14, получают соединения, примеры которых представлены в таблице Е-2.
ТАБЛИЦА Е-2



Пример 34

N-{(3R,6S)-6-(2,3-Дифторфенил)-1-[2-(метилтио)этил]-2-оксоазепан-3-ил}-4-(2-оксо-2,3-дигидро-1Н-имидазо[4,5-b]пиридин-1-ил)пиперидин-1-карбоксамид
Триэтиламин (0,020 мл, 0,143 ммоль) добавляют к раствору (3R,6S)-3-амино-6-(2,3-дифторфенил)-1-[2-(метилтио)этил]азепан-2-она (45 мг, 0,143 ммоль) и 4-нитрофенилхлорформиата (29 мг, 0,143 ммоль) в тетрагидрофуране (3 мл) при 0°C. Через 15 мин добавляют 2-оксо-1-пиперидиний-4-ил-2,3-дигидро-1Н-имидазо-[4,5-b]пиридин-4-ийдихлорид (46 мг, 0,157 ммоль), триэтиламин (0,080 мл, 0,572 ммоль) и дихлорметан (5 мл) и смесь нагревают до 50°C. Через 30 мин смеси дают охладиться до комнатной температуры и концентрируют. Очистка хроматографией на силикагеле [(100% дихлорметан → 95% дихлорметан/метанол (10% гидроксид аммония/метанол)] дает указанное в заголовке соединение (60 мг). Указанное в заголовке соединение превращают в HCl-соль с помощью 2М раствора HCl в эфире. МС 559,2 (М+1). 1H-ЯМР (500 МГц, CD3OD) δ 8,11 (дд, J=7,8 Гц, 1,0 Гц, 1H), 7,99 (дд, J=6,1 Гц, 1,0 Гц, 1H), 7,38-7,35 (м, 1H), 7,18-7,14 (м, 3H), 4,76 (д, J=10,7 Гц, 1H), 4,61-4,54 (м, 1H), 4,31-4,26 (м, 2H), 4,15-4,09 (м, 1H), 3,85-3,80 (м, 1H), 3,67-3,58 (м, 1H), 3,37 (д, J=15,4 Гц, 1H), 3,20-3,16 (м, 1H), 3,08-2,96 (м, 2H), 2,75-2,70 (м, 2H), 2,49-2,41 (м, 1H), 2,34-2,26 (м, 1H), 2,21-2,16 (м, 1H), 2,14 (с, 3H), 2,11-2,07 (м, 2H), 1,90-1,85 (м, 3H).
Пример 35

N-{(3R,6S)-6-(2,3-Дифторфенил)-1-[2-(метилсульфинил)этил]-2-оксоазепан-3-ил}-4-(2-оксо-2,3-дигидро-1Н-имидазо[4,5-b]пиридин-1-ил)пиперидин-1-карбоксамид
Триэтиламин (0,010 мл, 0,027 ммоль) добавляют к раствору (3R,6S)-3-амино-6-(2,3-дифторфенил)-1-[2-(метилсульфинил)этил]азепан-2-она (8,9 мг, 0,027 ммоль) и 4-нитрофенилхлорформиата (5,4 мг, 0,027 ммоль) в тетрагидрофуране (0,700 мл) при 0°C. Через 45 мин добавляют 2-оксо-1-пиперидиний-4-ил-2,3-дигидро-1Н-имидазо[4,5-b]пиридин-4-ийдихлорид (7,9 мг, 0,027 ммоль) и триэтиламин (0,040 мл, 0,108 ммоль) и смеси дают нагреться до комнатной температуры. Через 16 час смесь концентрируют. Очистка ВЭХЖ с обращенной фазой (С-18, 95% вода/ацетонитрил → 5% вода/ацетонитрил с 0,1% трифторуксусной кислоты), дает указанное в заголовке соединение. МС 575 (М+1).
Пример 36

N-{(3R,6S)-6-(2,3-Дифторфенил)-1-[2-(метилсульфонил)этил]-2-оксоазепан-3-ил}-4-(2-оксо-2,3-дигидро-1Н-имидазо[4,5-b]пиридин-1-ил)пиперидин-1-карбоксамид
Триэтиламин (0,010 мл, 0,023 ммоль) добавляют к раствору (3R,6S)-3-амино-6-(2,3-дифторфенил)-1-[2-(метилсульфонил)этил]азепан-2-она (8 мг, 0,023 ммоль) и 4-нитрофенилхлорформиата (4,6 мг, 0,023 ммоль) в тетрагидрофуране (0,700 мл) при 0°C. Через 15 мин добавляют 2-оксо-1-пиперидиний-4-ил-2,3-дигидро-1Н-имидазо[4,5-b]пиридин-4-ийдихлорид (6,7 мг, 0,023 ммоль) и триэтиламин (0,040 мл, 0,092 ммоль) и смеси дают нагреться до комнатной температуры. Через 16 час смесь концентрируют. Очистка ВЭХЖ с обращенной фазой (С-18, 95% вода/ацетонитрил → 5% вода/ацетонитрил с 0,1% трифторуксусной кислоты), дает указанное в заголовке соединение. МС 591 (М+1).
Пример 37

N-[(3R,6S)-6-(2,3-Дифторфенил)-1-(2-метоксиэтил)-2-оксоазепан-3-ил]-4-(2-оксо-2,3-дигидро-1Н-имидазо[4,5-b]пиридин-1-ил)пиперидин-1-карбоксамид
Триэтиламин (0,030 мл, 0,218 ммоль) добавляют к раствору (3R,6S)-3-амино-6-(2,3-дифторфенил)-1-(2-метоксиэтил)азепан-2-она (65 мг, 0,218 ммоль) и 4-нитрофенилхлорформиата (44 мг, 0,218 ммоль) в тетрагидрофуране (3 мл) при 0°C. Через 30 мин добавляют 2-оксо-1-пиперидиний-4-ил-2,3-дигидро-1Н-имидазо[4,5-b]пиридин-4-ийдихлорид (70 мг, 0,240 ммоль), триэтиламин (0,120 мл, 0,872 ммоль) и дихлорметан (5 мл) и смеси дают нагреться до комнатной температуры. Через 4,5 часа смесь концентрируют. Очистка хроматографией на силикагеле [100% дихлорметан → 95% дихлорметан/метанол (10% гидроксид аммония/метанол)] дает указанное в заголовке соединение (88 мг). Указанное в заголовке соединение превращают в HCl-соль с помощью 2М раствора HCl в эфире. МС 543,3 (М+1). 1H-ЯМР (500 МГц, CD3OD) δ 8,09 (дд, J=7,8 Гц, 1,0 Гц, 1H), 7,99 (дд, J=6,1 Гц, 1,0 Гц, 1H), 7,36 (м, 1H), 7,17-7,10 (м, 3H), 4,78 (д, J=11,0 Гц, 1H), 4,61-4,54 (м, 1H), 4,31-4,24 (м, 2H), 4,18-4,13 (м, 1H), 3,94-3,89 (м, 1H), 3,57-3,53 (м, 2H), 3,48-3,43 (м, 1H), 3,39 (д, J=15,1 Гц, 1H), 3,34 (с, 3H), 3,19-3,15 (м, 1H), 3,07-2,96 (м, 2H), 2,49-2,41 (м, 1H), 2,35-2,26 (м, 1H), 2,19-2,06 (м, 3H), 1,90-1,76 (м, 3H).
Пример 38

N-[(3R,6S)-6-(2,3-Дифторфенил)-2-оксо-1-(2,2,2-трифторэтил)азепан-3-ил]-4-(2-оксо-2,3-дигидро-1Н-имидазо[4,5-b]пиридин-1-ил)пиперидин-1-карбоксамид
Триэтиламин (0,038 мл, 0,276 ммоль) добавляют к раствору (3R,6S)-3-амино-6-(2,3-дифторфенил)-1-(2,2,2-трифторэтил)азепан-2-она (89 мг, 0,276 ммоль) и 4-нитрофенилхлорформиата (56 мг, 0,276 ммоль) в тетрагидрофуране (3 мл) при 0°C. Через 30 мин добавляют 2-оксо-1-пиперидиний-4-ил-2,3-дигидро-1Н-имидазо[4,5-b]пиридин-4-ийдихлорид (88 мг, 0,304 ммоль), триэтиламин (0,152 мл, 1,104 ммоль) и дихлорметан (5 мл) и смеси дают нагреться до комнатной температуры. Через 48 час смесь концентрируют. Очистка хроматографией на силикагеле [100% дихлорметан → 95% дихлорметан/метанол (10% гидроксид аммония/метанол)] дает указанное в заголовке соединение (98 мг). Указанное в заголовке соединение превращают в HCl-соль с помощью 2М раствора HCl в эфире. МС 567,2 (М+1). 1H-ЯМР (500 МГц, CD3OD) δ 8,11 (дд, J=7,8 Гц, 1,0 Гц, 1H), 8,00 (дд, J=5,9 Гц, 1,0 Гц, 1H), 7,34-7,31 (м, 1H), 7,19-7,13 (м, 3H), 4,86-4,83 (м, 1H), 4,62-4,57 (м, 1H), 4,49-4,41 (м, 1H), 4,33-4,25 (м, 3H), 4,12-4,04 (м, 1H), 3,51-3,46 (м, 1H), 3,18-3,14 (м, 1H), 3,09-2,96 (м, 2H), 2,51-2,44 (м, 1H), 2,33-2,25 (м, 1H), 2,22-2,10 (м, 3H), 1,90-1,81 (м, 3H).
Пример 39

N-[(3R,6S)-6-(2,3-Дифторфенил)-2-оксо-1-(пиридин-2-илметил)азепан-3-ил]-4-(2-оксо-2,3-дигидро-1Н-имидазо[4,5-b]пиридин-1-ил)пиперидин-1-карбоксамид
Триэтиламин (0,065 мл, 0,468 ммоль) добавляют к раствору (3R,6S)-3-амино-6-(2,3-дифторфенил)-1-(пиридин-2-илметил)азепан-2-она (180 мг, 0,417 ммоль) и 4-нитрофенилхлорформиата (94 мг, 0,468 ммоль) в тетрагидрофуране (3 мл) при 0°C. Через 1 час добавляют 2-оксо-1-пиперидиний-4-ил-2,3-дигидро-1Н-имидазо[4,5-b]пиридин-4-ийдихлорид (136 мг, 0,468 ммоль) и триэтиламин (0,195 мл, 1,404 ммоль) и смеси дают нагреться до комнатной температуры. Через 18 час смесь распределяют между этилацетатом и водой. Органический слой промывают водой (2×), насыщенным раствором соли, сушат над сульфатом магния, фильтруют и концентрируют. Очистка хроматографией на силикагеле [100% дихлорметан → 95% дихлорметан/метанол (10% гидроксид аммония/метанол)] дает указанное в заголовке соединение (240 мг). Указанное в заголовке соединение превращают в HCl-соль с помощью 2М раствора HCl в эфире. МС 576,3 (М+1).1H-ЯМР (500 МГц, CD3OD) δ 8,78 (д, J=5,9 Гц, 1H), 8,62-8,59 (м, 1H), 8,07 (д, J=8,3 Гц, 1H), 8,03-7,98 (м, 3H), 7,32 (дд, J=8,1 Гц, 6,1, 1H), 7,18-7,15 (м, 3H), 5,43 (д, J=16,9 Гц, 1H), 4,75 (д, J=17,1 Гц, 1H), 4,56-4,51 (м, 1H), 4,36-4,28 (м, 3H), 3,51 (д, J=15,1 Гц, 1H), 3,48 (с, 1H), 3,22-3,17 (м, 1H), 3,05-2,98 (м, 2H), 2,44-2,40 (м, 1H), 2,34-2,23 (м, 2H), 2,18-2,14 (м, 2H), 1,97-1,92 (м, 1H), 1,89-1,88 (м, 2H).
Пример 40

N-[(3R,6S)-6-(2,3-Дифторфенил)-2-оксо-1-пиридин-4-илазепан-3-ил]-4-(2-оксо-2,3-дигидро-1Н-имидазо[4,5-b]пиридин-1-ил)пиперидин-1-карбоксамид
Триэтиламин (0,018 мл, 0,132 ммоль) добавляют к раствору (3R,6S)-3-амино-6-(2,3-дифторфенил)-1-пиридин-4-илазепан-2-она (42 мг, 0,132 ммоль) и 4-нитрофенилхлорформиата (27 мг, 0,132 ммоль) в тетрагидрофуране (2 мл) при 0°C. Через 1 час добавляют 2-оксо-1-пиперидиний-4-ил-2,3-дигидро-1Н-имидазо[4,5-b]пиридин-4-ийдихлорид (38 мг, 0,132 ммоль) и триэтиламин (0,054 мл, 0,396 ммоль) и смеси дают нагреться до комнатной температуры. Через 18 час смесь распределяют между этилацетатом и водой. Органический слой промывают водой (2×), насыщенным раствором соли, сушат над сульфатом магния, фильтруют и концентрируют. Очистка хроматографией на силикагеле [100% дихлорметан → 95% дихлорметан/метанол (10% гидроксид аммония/метанол)] дает указанное в заголовке соединение (53 мг). Указанное в заголовке соединение превращают в HCl-соль с помощью 2М раствора HCl в эфире. МС 562,2 (М+1). 1H-ЯМР (500 МГц, CD3OD) δ 8,76 (д, J=7,6 Гц, 2H), 8,11 (д, J=7,3 Гц, 2H), 8,00 (д, J=6,4 Гц, 2H), 7,30 (т, J=7,0 Гц, 1H), 7,27-7,21 (м, 3H), 5,11 (д, J=11,5 Гц, 1H), 4,72-4,65 (м, 1H), 4,60-4,55 (м, 1H), 4,35-4,33 (м, 2H), 4,14 (д, J=16,1 Гц, 1H), 3,42-3,39 (м, 1H), 3,09-3,02 (м, 2H), 2,47-2,44 (м, 1H), 2,37-2,30 (м, 2H), 2,24-2,18 (м, 2H), 2,06-2,03 (м, 1H), 1,91 (шир.с, 2H).
Следуя по существу методикам, описанным для получения соединений примеров 34-40, получают соединения, примеры которых представлены в таблице Е-4.
ТАБЛИЦА Е-4



Хотя настоящее изобретение описано и проиллюстрировано с помощью некоторых конкретных вариантов осуществления, специалисту в данной области будет очевидно, что могут быть проведены различные подгонки, изменения, модификации, замещения, детализации или добавления методик и протоколов без отступления от сути и объема изобретения. Например, эффективные дозы, отличные от конкретных установленных доз, могут быть применимы вследствие различной чувствительности млекопитающих, которые подвергаются лечению в случае любых симптомов с помощью описанных выше соединений настоящего изобретения.
| название | год | авторы | номер документа |
|---|---|---|---|
| ПРОИЗВОДНЫЕ [4-(1-АМИНОЭТИЛ)ЦИКЛОГЕКСИЛ]МЕТИЛАМИНА И [6-(1-АМИНОЭТИЛ)ТЕТРАГИДРОПИРАН-3-ИЛ]МЕТИЛАМИНА | 2009 |
|
RU2515906C2 |
| СОЕДИНЕНИЯ АЗАБЕНЗОФУРАНИЛА И СПОСОБ ИХ ПРИМЕНЕНИЯ | 2007 |
|
RU2448111C2 |
| ПРОИЗВОДНОЕ ПИПЕРИДИНА, СПОСОБ ЕГО ПОЛУЧЕНИЯ И ЕГО ФАРМАЦЕВТИЧЕСКОЕ ПРИМЕНЕНИЕ | 2016 |
|
RU2730508C2 |
| ЗАМЕЩЕННЫЕ ПРОИЗВОДНЫЕ КАРБОНУКЛЕОЗИДА, ПРИМЕНЯЕМЫЕ В КАЧЕСТВЕ ПРОТИВОРАКОВЫХ АГЕНТОВ | 2017 |
|
RU2712944C1 |
| ПИРАЗОЛ-4-ИЛ-ГЕТЕРОЦИКЛИЛ-КАРБОКСАМИДНЫЕ СОЕДИНЕНИЯ И СПОСОБЫ ПРИМЕНЕНИЯ | 2012 |
|
RU2638552C2 |
| НОВЫЕ СОЕДИНЕНИЯ В КАЧЕСТВЕ ИНГИБИТОРОВ КАТЕПСИНА, ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ НА ИХ ОСНОВЕ, ИХ ПРИМЕНЕНИЕ И СПОСОБ ИХ ПОЛУЧЕНИЯ | 2002 |
|
RU2316546C2 |
| ПРОИЗВОДНЫЕ ТИЕНОПИРРОЛА ДЛЯ ПРИМЕНЕНИЯ ДЛЯ НАЦЕЛИВАНИЯ НА БЕЛКИ, КОМПОЗИЦИИ С УКАЗАННЫМИ ПРОИЗВОДНЫМИ, СПОСОБЫ И ПРИМЕНЕНИЯ | 2017 |
|
RU2771166C2 |
| СПОСОБ ПОЛУЧЕНИЯ ПРОИЗВОДНЫХ 6-АЛКИЛИДЕНПЕНЕМА | 2003 |
|
RU2317297C2 |
| ПРОЛЕКАРСТВО ПРОИЗВОДНОГО АМИНОКИСЛОТЫ | 2017 |
|
RU2739318C2 |
| РЕАКЦИИ МАКРОЦИКЛИЗАЦИИ И ПРОМЕЖУТОЧНЫЕ СОЕДИНЕНИЯ И ДРУГИЕ ФРАГМЕНТЫ, ПРИГОДНЫЕ В ПОЛУЧЕНИИ АНАЛОГОВ ХАЛИХОНДРИНА B | 2014 |
|
RU2710545C2 |
Изобретение относится к соединению формулы I:

где: R1 выбран из: Н, C1-С6-алкила, С3-С6-циклоалкила и гетероцикла, незамещенного или замещенного одним или несколькими заместителями, независимо друг от друга выбранными из: C1-С6-алкила, С3-С6-циклоалкила, фенила, гетероарила, гетероцикла, (F)рС1-С3-алкила, галогена, OR4, O(CH2)sOR4, CN, NR10R11, где указанный фенил, указанный гетероарил и указанный гетероцикл каждый независимо друг от друга является незамещенным или замещен 1-5 заместителями, где заместители независимо друг от друга выбраны из R4, где указанный гетероарил выбран из: пиридина; где указанный гетероцикл выбран из: тетрагидрофурана; R2 выбран из: Н, С0-С6-алкила, С3-С6-циклоалкила и гетероцикла, незамещенного или замещенного одним или несколькими заместителями, независимо друг от друга выбранными из: фенила, гетероарила, галогена, OR4; где указанный фенил, указанный гетероарил и указанный гетероцикл каждый независимо друг от друга является незамещенным или замещен 1-5 заместителями, независимо друг от друга выбранными из R4, где указанный гетероарил выбран из: имидазола, пиридина; R10 и R11 независимо друг от друга выбраны из: Н, C1-С6-алкила, (F)рС1-С6-алкила, С3-С6-циклоалкила; R4 независимо друг от друга выбраны из: Н, C1-С6-алкила, (F)рС1-С6-алкила, С3-С6-циклоалкила; R3 независимо друг от друга выбраны из: Н; р принимает значения от 0 до 2q+1, для заместителей с q атомами углерода; s принимает значения 1,2 или 3; и его фармацевтически приемлемые соли и индивидуальные диастереомеры. Соединения формулы (I) могут быть использованы в качестве антагонистов рецепторов CGRP и могут быть использованы для лечения или профилактики заболеваний, при которых поражаются CGRP, таких как головная боль, мигрень и гистаминовая головная боль. Изобретение также относится к фармацевтическим композициям, содержащим такие соединения, и к применению таких соединений для профилактики или лечения таких заболеваний, при которых поражаются CGRP. 5 н.п. ф-лы, 6 табл.

где R1 выбран из Н, C1-С6-алкила, С3-С6-циклоалкила и гетероцикла, незамещенного или замещенного одним или несколькими заместителями, независимо друг от друга выбранными из
C1-С6-алкила, С3-С6-циклоалкила, фенила, гетероарила, гетероцикла, (F)pC1-С3-алкила, галогена, OR4, O(СН2)sOR4, CN, NR10R11, где указанный фенил, указанный гетероарил и указанный гетероцикл каждый независимо друг от друга является незамещенным или замещен 1-5 заместителями, где заместители независимо друг от друга выбраны из R4, где указанный гетероарил выбран из пиридина; где указанный гетероцикл выбран из тетрагидрофурана;
R2 выбран из Н, С0-С6-алкила, С3-С6-циклоалкила и гетероцикла, незамещенного или замещенного одним или несколькими заместителями, независимо друг от друга выбранными из фенила, гетероарила, галогена, OR4; где указанный фенил, указанный гетероарил и указанный гетероцикл каждый независимо друг от друга является незамещенным или замещен 1-5 заместителями, независимо друг от друга выбранными из R4, где указанный гетероарил выбран из имидазола, пиридина;
R10 и R11 независимо друг от друга выбраны из Н, C1-С6-алкила, (F)рС1-С6-алкила, С3-С6-циклоалкила;
R4 независимо друг от друга выбраны из Н, C1-С6-алкила, (F)рС1-С6-алкила, С3-С6-циклоалкила;
R3 независимо друг от друга выбраны из Н;
р принимает значения от 0 до 2q+1, для заместителей с q атомами углерода;
s принимает значения 1, 2 или 3;
и его фармацевтически приемлемые соли и индивидуальные диастереомеры.

где R1 выбран из C1-С6-алкила, незамещенного или замещенного одним или несколькими заместителями, независимо друг от друга выбранными из C1-С6-алкила, С3-С6-циклоалкила, фенила, гетероарила, гетероцикла, (F)рС1-С3-алкила, галогена, OR4, CN, NR10R11, где указанный гетероарил выбран из пиридина; где указанный гетероцикл выбран из тетрагидрофурана; R2 выбран из арила, незамещенного или замещенного одним или несколькими заместителями, независимо друг от друга выбранными из C1-С6-алкила, С3-С6-циклоалкила, (F)рС1-С3-алкила, галогена, OR4; где указанный арил выбран из фенила;
R10 и R11 независимо друг от друга выбраны из Н, C1-С6-алкила, (F)pC1-C6-алкила, С3-С6-циклоалкила;
R4 независимо друг от друга выбраны из Н, C1-С6-алкила, (F)рС1-С6-алкила, С3-С6-циклоалкила;
р принимает значения от 0 до 2q+1, для заместителей с q атомами углерода;
и его фармацевтически приемлемые соли и индивидуальные диастереомеры.
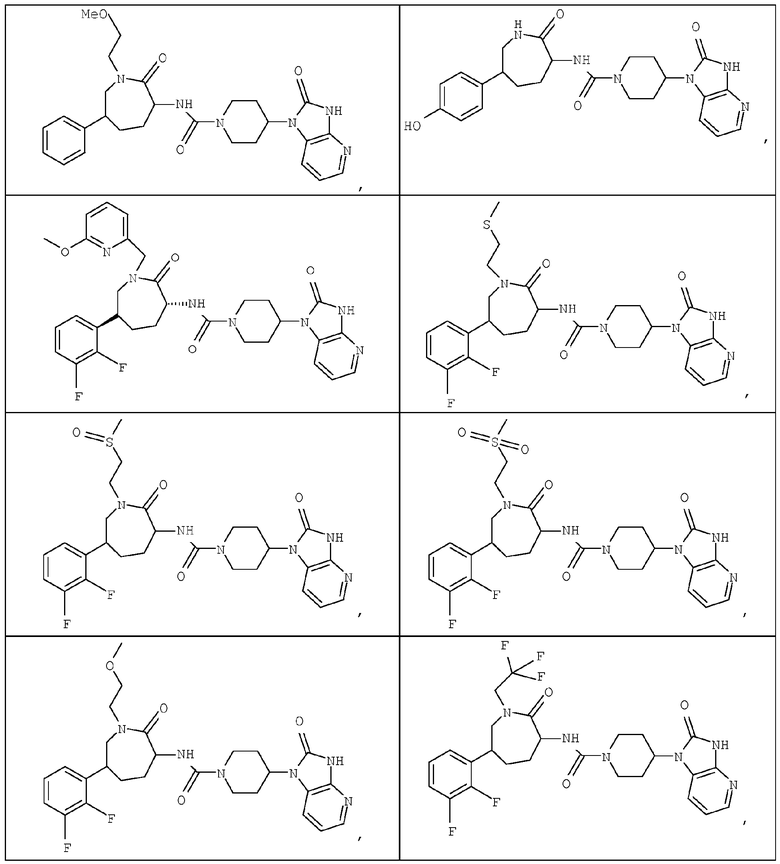
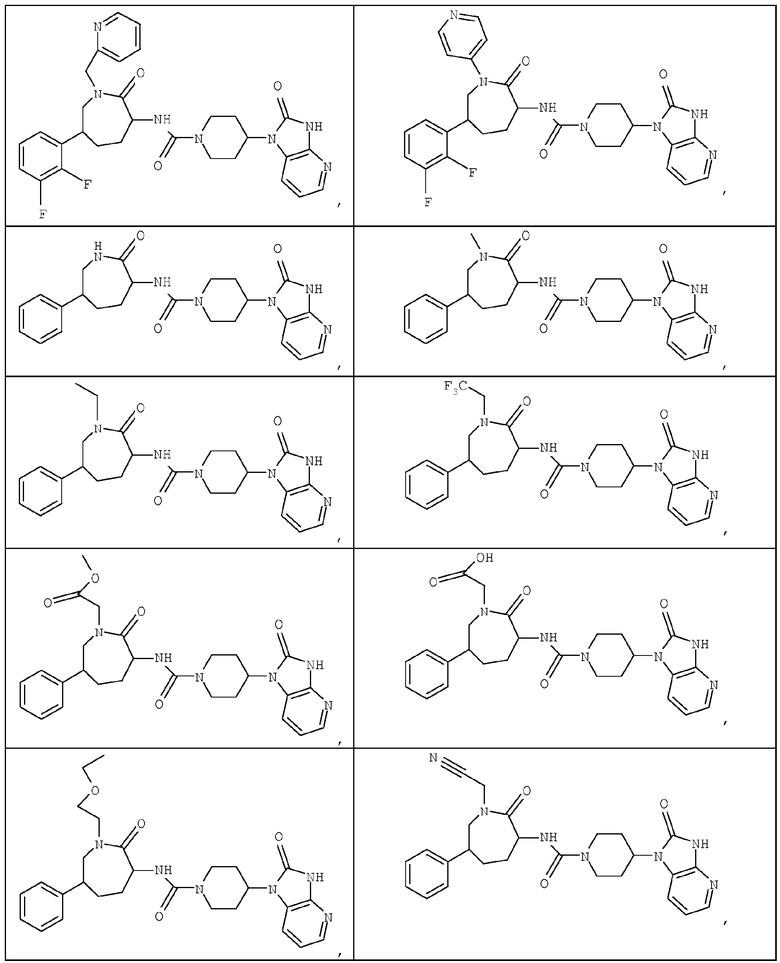


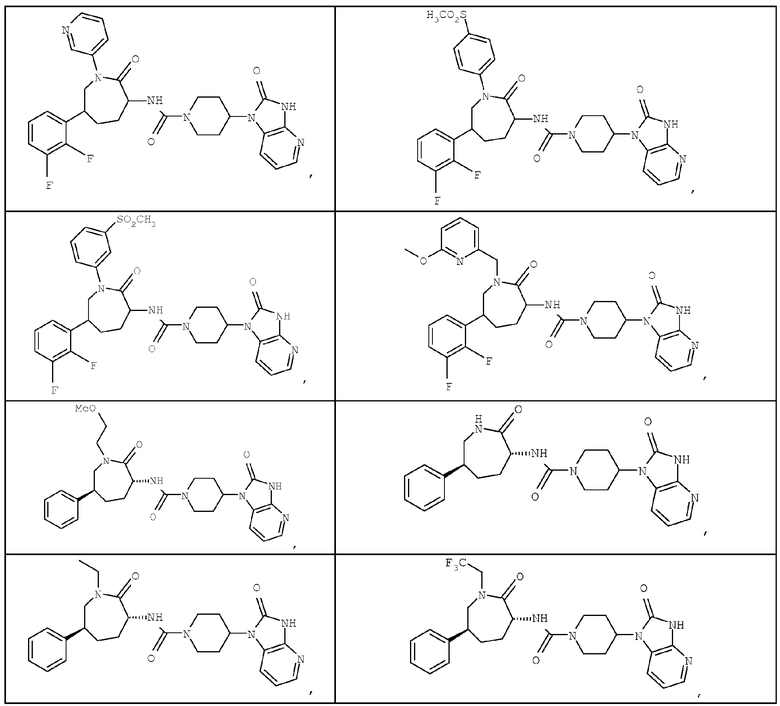


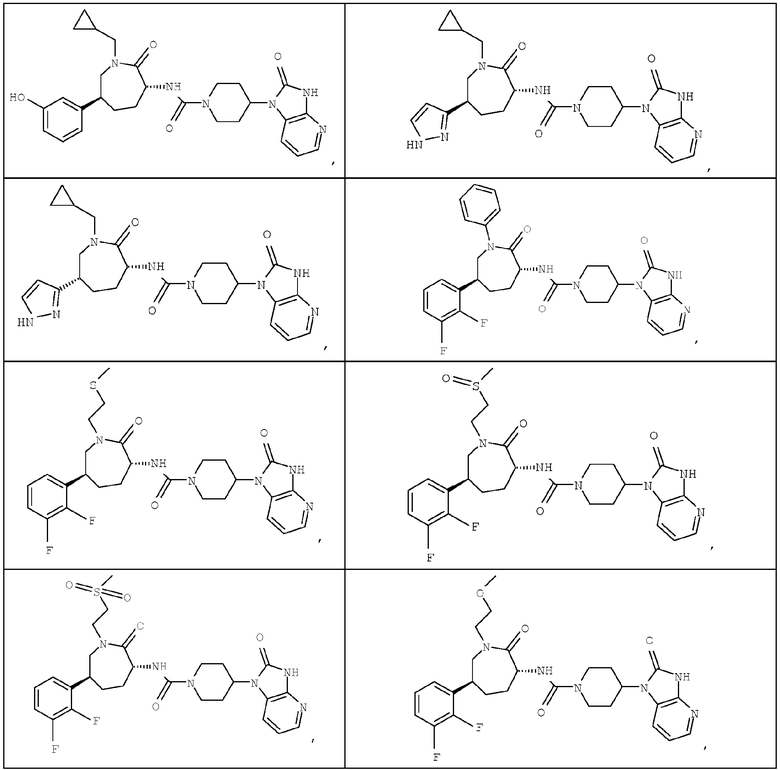


и его фармацевтических приемлемых солей и индивидуальных диастереомеров.
| WO 00/18764 A1, 06.04.2000 | |||
| ПРОИЗВОДНЫЕ АМИДА ФЕНИЛЦИКЛОГЕКСИЛКАРБОНОВОЙ КИСЛОТЫ, СМЕСЬ ИХ ИЗОМЕРОВ ИЛИ ОТДЕЛЬНЫЕ ИЗОМЕРЫ И ИХ СОЛИ, ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ С АНТИАРТЕРИОСКЛЕРОТИЧЕСКОЙ И АНТИРЕСТЕНОЗНОЙ АКТИВНОСТЬЮ | 1996 |
|
RU2158261C2 |

