ОБЛАСТЬ ТЕХНИКИ
Данное изобретение относится к новому способу получения производных 6-алкилиденпенема, которые могут иметь значение в качестве ингибиторов β-лактамазы и антибактериальных агентов широкого спектра действия.
ПРЕДПОСЫЛКИ ДЛЯ СОЗДАНИЯ ИЗОБРЕТЕНИЯ
β-Лактамазы представляют собой ферменты, продуцируемые бактериями, которые гидролизуют β-лактамные антибиотики и поэтому являются основной причиной бактериальной резистентности. Пенициллины и цефалоспорины являются наиболее часто и широко используемыми в клинике β-лактамными антибиотиками. Однако развитие резистентности различных патогенов к β-лактамным антибиотикам действует разрушительным образом на эффективность лечения бактериальных инфекций (Coleman, K. Expert Opin. Invest. Drugs 1995, 4, 693; Sutherland, R. Infection 1995, 23 (4), 191; Bush, K. Cur. Pharm. Design 1999, 5, 839-845). Наиболее существенным известным механизмом развития устойчивости бактерий к β-лактамным антибиотикам является продуцирование сериновых β-лактамаз класса А, класса В и класса С. Эти ферменты разрушают β-лактамные антибиотики, приводя в результате к потере антибактериальной активности. Ферменты класса А предпочтительно гидролизуют пенициллины, тогда как лактамазы класса С имеют субстратный профиль, способствующий гидролизу цефалоспорина (Bush, K.; Jacoby, G.A.; Medeiros, A.A. Antimicrob. Agents Chemother. 1995, 39, 1211). К настоящему времени описано свыше 250 различных β-лактамаз (Payne, D.J,: Du, W and Bateson, J.H. Exp. Opin. Invest. Drugs 2000, 247) и существует потребность в новом поколении ингибиторов β-лактамаз с широким спектром действия. Бактериальную резистентность к рассматриваемым антибиотикам можно было бы значительно снизить введением β-лактамных антибиотиков в сочетании с соединением, которое ингибирует вышеуказанные ферменты.
Коммерчески доступные ингибиторы β-лактамаз, такие как клавулановая кислота, сулбактам и тазобактам, все эффективны в отношении патогенов, продуцирующих ферменты класса А. Выяснен механизм инактивации β-лактамаз класса А (таких как PCI и TEM-1) (Bush, K.; Antimicrob. Agents Chemother. 1993, 37, 851; Yang, Y.; Janota, K.; Tabei, K.; Huang, N.; Seigal, M.M.; Lin, Y.I.; Rasmussen, B.A. и Shalaes, D.M. J. Biol. Chem. 2000, 35, 26674-26682). Однако указанные соединения неэффективны в отношении организмов, продуцирующих ферменты класса С. Клавулановую кислоту используют в комбинации с амоксициллином и тикарциллином; аналогично сулбактам - с ампициллином, а тазобактам - с пиперациллином.
В патентах и заявках EP 0041768A, EP 0120613A, EP 0150781, EP 0210065, WO 87/00525, EP 0003960B1, GB 2042514A, GB 2042515A, EP 0087792A, EP 0115308A, GB 2124614B, EP 0150984A, EP 0087792, EP 0115308, EP 81301683.9, EP 84301255.0, EP 85100520.7, EP 85100521.5, EP 86305585.1, EP 86305584, EP 88311786.3, EP 88311787.1, EP 87300193.7, WO 93/03042, WO 94/10178, WO 95/28935 и WO 95/17184 в качестве ингибиторов β-лактамаз описываются пенемы с замещенной метиленовой группой в положении 6. В Европейском патенте EP 0232966B1 описан способ получения пенемов с замещенной метиленовой группой в положении 6 и их промежуточных соединений. Согласно этому патенту, соединения общей формулы I получают четырехстадийным способом из п-метоксибензил (5R,6S)-6-бромпенем-3-карбоксилата A и альдегида в присутствии пирофорных реагентов, таких как н-бутиллитий/дифениламин или бис(триметилсилил)амид лития при -78°C (Схема 1). Третья стадия указанного способа, стадия восстановительного элиминирования, дает Е и Z изомерные соединения F и D в соотношении 5:1 соответственно.
Схема 1

КРАТКОЕ ИЗЛОЖЕНИЕ ИЗОБРЕТЕНИЯ
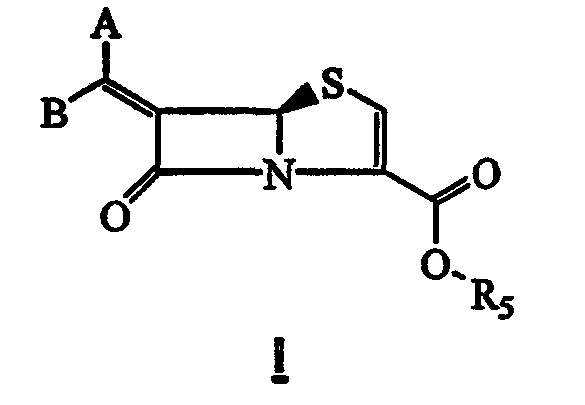
Данное изобретение относится к способу получения соединения формулы I

где:
один из заместителей A или В обозначает водород, а другой обозначает арил, необязательно замещенный одним или двумя R2, гетероарил, необязательно замещенный одним или двумя R2, конденсированный бициклический гетероарил, необязательно замещенный одним или двумя R2, конденсированный трициклический гетероарил, необязательно замещенный одним или двумя R2, циклоалкил, необязательно замещенный одним или двумя R2, алкил, необязательно замещенный одним или двумя R2, алкенил, необязательно замещенный одним или двумя R2, алкинил, необязательно замещенный одним или двумя R2, насыщенную или частично насыщенную гетероциклическую группу, необязательно замещенную одним или двумя R2; и где любой из указанных гетероарильных остатков, содержащий NH-группу в цикле, может быть необязательно замещен R1 при указанном атоме азота;
R5 представляет собой H, C1-C6-алкил, C5-C6-циклоалкил или CHR3OCO-C1-C6-алкил или соль;
R1 представляет собой H, необязательно замещенный алкил, необязательно замещенный арил, необязательно замещенные гетероарил или моно- или бициклические насыщенные гетероциклические группы, необязательно замещенный циклоалкил, необязательно замещенный алкенил, необязательно замещенный алкинил при условии, что атом углерода, который непосредственно связан с N, не должен входить в состав двойной или тройной связи; необязательно замещенный перфторалкил, необязательно -S(O)p-замещенный алкил или арил, где p равно 0-2, необязательно замещенный -C=O-гетероарил, необязательно замещенный -C=O-арил, необязательно замещенный -C=O-алкил, необязательно замещенный -C=O-циклоалкил, необязательно замещенную моно- или бициклическую насыщенную -C=O-гетероциклическую группу, необязательно замещенный C1-C6-алкиларил, необязательно замещенный C1-C6-алкилгетероарил, необязательно замещенный арил-С1-C6-алкил, необязательно замещенный гетероарил-C1-C6-алкил, необязательно замещенную моно- или бициклическую насыщенную C1-C6-алкилгетероциклическую группу, необязательно замещенный арилалкенил, содержащий 8-16 атомов углерода, -CONR6R7, -SO2NR6R7, необязательно замещенный арилалкоксиалкил, необязательно замещенный -алкил-O-алкиларил, необязательно замещенный -алкил-O-алкилгетероарил, необязательно замещенный арилоксиалкил, необязательно замещенный гетероарилоксиалкил, необязательно замещенный арилоксиарил, необязательно замещенный арилоксигетероарил, необязательно замещенный C1-C6-алкиларилоксиарил, необязательно замещенный C1-C6-алкиларилоксигетероарил, необязательно замещенные алкиларилоксиалкиламиносодержащие группы, необязательно замещенный алкоксикарбонил, необязательно замещенный арилоксикарбонил или необязательно замещенный гетероарилоксикарбонил;
R2 представляет собой водород, необязательно замещенный C1-C6-алкил, необязательно замещенный C2-C6-алкенил, необязательно замещенный C2-C6-алкинил, галоген, циано, N-R6R7, необязательно замещенный C1-C6-алкокси, гидрокси; необязательно замещенный арил, необязательно замещенный гетероарил, COOR6, необязательно замещенные алкиларилоксиалкиламиносодержащие группы, необязательно замещенный арилокси, необязательно замещенный гетероарилокси, необязательно замещенный C3-C6-алкенилокси, необязательно замещенный C3-C6-алкинилокси, C1-C6-алкиламино-C1-C6-алкокси, алкилендиокси, необязательно замещенную арилокси-C1-C6-алкиламиносодержащую группу, C1-C6-перфторалкил, необязательно S(О)q-замещенный С1-С6-алкил, необязательно S(O)q-замещенный арил, где q равно 0, 1 или 2, CONR6R7, гуанидино- или включенную в цикл гуанидиногруппу, необязательно замещенный алкиларил, необязательно замещенный арилалкил, необязательно замещенный C1-C6-алкилгетероарил, необязательно замещенный гетероарил-C1-C6-алкил, необязательно замещенную моно- или бициклическую насыщенную C1-C6-алкилгетероциклическую группу, необязательно замещенный арилалкенил, содержащий от 8 до 16 атомов углерода, SO2NR6R7, необязательно замещенный арилалкилоксиалкил, необязательно замещенный арилоксиалкил, необязательно замещенный гетероарилоксиалкил, необязательно замещенный арилоксиарил, необязательно замещенный арилоксигетероарил, необязательно замещенный гетероарилоксиарил, необязательно замещенный C1-C6-алкиларилоксиарил или необязательно замещенный C1-C6-алкиларилоксигетероарил;
R3 представляет собой водород, C1-C6-алкил, C5-C6-циклоалкил, необязательно замещенный арил, необязательно замещенный гетероарил;
R6 и R7 независимо представляют собой H, необязательно замещенный C1-C6-алкил, необязательно замещенный арил, необязательно замещенный гетероарил, необязательно замещенный C1-C6-алкиларил, необязательно замещенный арилалкил, необязательно замещенный гетероарилалкил, необязательно замещенный C1-C6-алкилгетероарил, R6 и R7 могут вместе образовывать 3-7-членную насыщенную циклическую систему, необязательно содержащую один или два гетероатома, таких как N-R1, O, S=(O)n, где n равно 0-2.
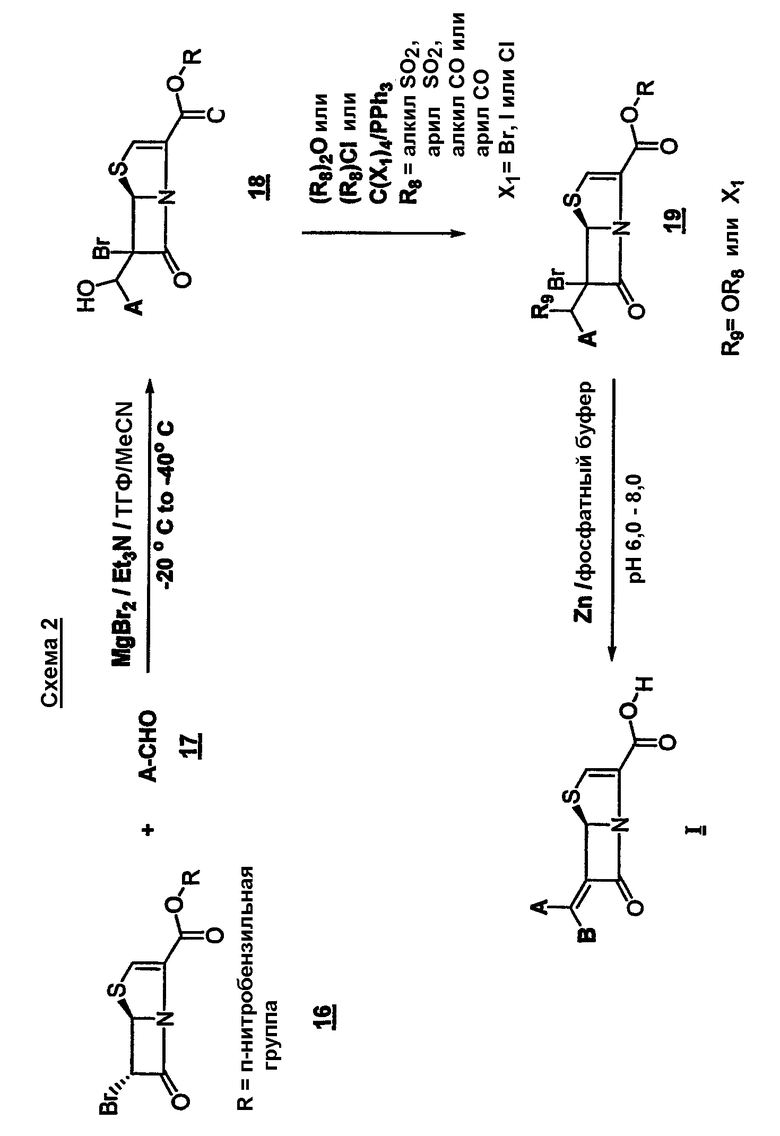
Указанный способ включает:
(а) конденсацию соответствующим образом замещенного альдегида 17

где A' представляет собой А, такой как определено выше, когда В представляет собой водород; или В, такой, как определено выше, когда А представляет собой водород,
с производным 6-бромпенема структуры 16
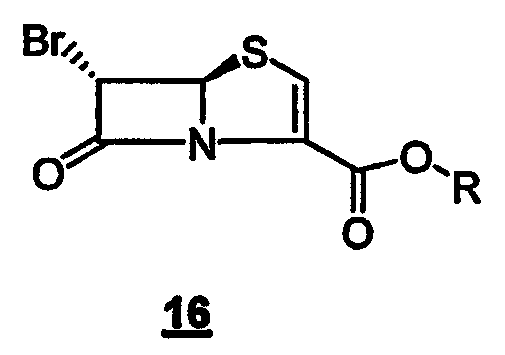
где R представляет собой пара-нитробензил, в присутствии кислоты Льюиса и мягкого основания при низкой температуре, которая дает промежуточный продукт альдольной конденсации 18
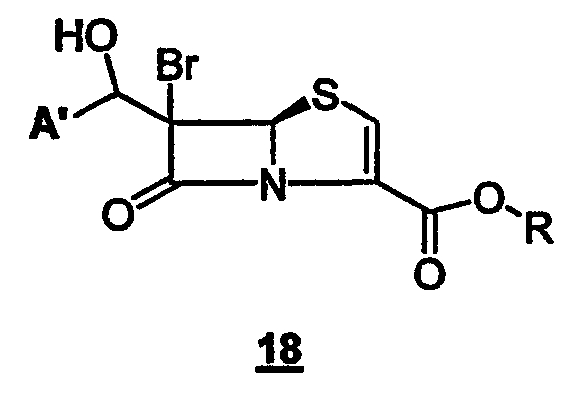
где A' и R такие, как определено выше;
(b) взаимодействие промежуточного соединения 18 с хлорангидридом или ангидридом кислоты формулы (R8)Cl или (R8)2O или с тетрагалогенметаном формулы C(X1)4 и трифенилфосфином, где R8 представляет собой алкил-SO2, арил-SO2, алкил-CO или арил-CO; X1 представляет собой Br, I или Cl; с образованием промежуточного соединения 19

где R9 представляет собой Х1или OR8, где R8, X1, A' и R такие, как определено выше; и
(c) превращение промежуточного соединения 19 в целевое соединение формулы I, где R5 представляет собой водород, посредством восстановительного элиминирования; и, если необходимо, дальнейшее превращение в фармацевтически приемлемую соль или в сложный эфир, где R5 представляет собой C1-C6-алкил, C5-C6-циклоалкил или CHR3OCO-C1-C6-алкил.
Данное изобретение также относится к производному 6-бром-пенема структуры 16,

где R представляет собой п-нитробензил, используемому на вышеуказанной стадии (а).
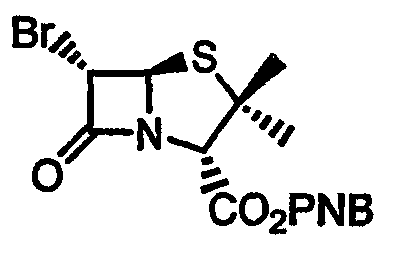
Данное изобретение, кроме того, относится к способу получения 4-нитробензил-(5R,6S)-6-бромпенем-3-карбоксилата формулы 16, который включает следующие стадии:
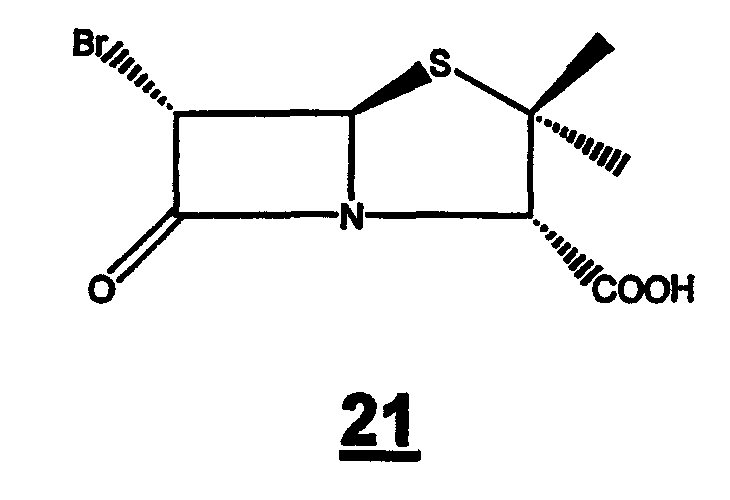
(А)(i) взаимодействие 6-аминопенициллановой кислоты с бромистоводородной кислотой в органическом растворителе и воде с образованием 6-бромпроизводного 21
и (ii) превращение 6-бромпенициллановой кислоты 21 в п-нитробензил 6-бромпеницилланат 22
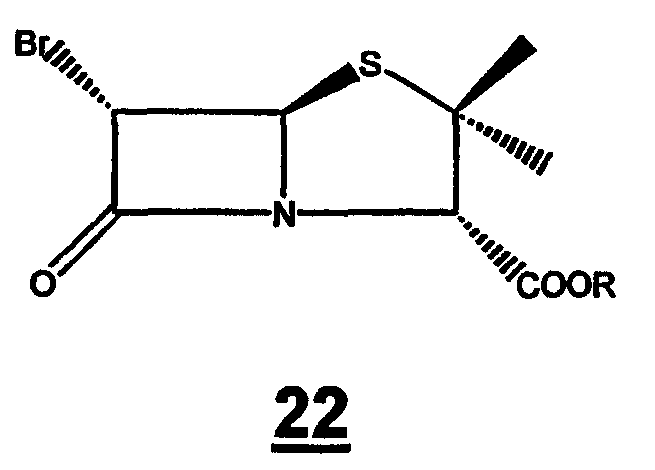
где R представляет собой п-нитробензил, действием 4-нитробензилбромида в присутствии основания в органическом растворителе;
(В) окисление п-нитробензил 6-бромпеницилланата 22 с образованием 4-нитробензил 6-бромпеницилланат-1-оксида 23

(С) кипячение с обратным холодильником 4-нитробензил 6-бромпеницилланат-1-оксида 23 с 2-меркаптобензотиазолом в ароматическом растворителе с образованием 4-нитробензил (2R)-2-[(3S,4R)-4-(бензотиазол-2-илдитио)-3-бром-2-оксоазетидин-1-ил]-3-метилбут-3-еноата 24;
(D) растворение 4-нитробензил (2R)-2-[(3S,4R)-4-(бензотиазол-2-илдитио)-3-бром-2-оксоазетидин-1-ил]-3-метилбут-3-еноата 24 в органическом растворителе и взаимодействие с органическим третичным основанием с образованием 4-нитробензил 2-[(3S,4R)-4-(бензотиазол-2-илдитио)-3-бром-2-оксоазетидин-1-ил]-2-метилбут-2-еноата 25;
(E) превращение 4-нитробензил 2-[(3S,4R)-4-(бензотиазол-2-илдитио)-3-бром-2-оксоазетидин-1-ил]-3-метилбут-2-еноата 25 в 4-нитробензил 2-[(3S,4R)-3-бром-4-формилтио-2-оксоазетидин-1-ил]-3-метилбут-2-еноат 26 в результате взаимодействия в ароматическом органическом растворителе с органической кислотой в присутствии смеси уксусный ангидрид/органическое третичное основание и триалкил- или триарилфосфина в интервале температур от около -10°С до около -30°С;
(F) пропускание озонированного кислорода через раствор 4-нитробензил 2-[(3S,4R)-3-бром-4-формилтио-2-оксоазетидин-1-ил]-3-метилбут-2-еноата 26 в органическом растворителе в течение 3-4 ч при температуре от -70°С до -90°С и последующую внутримолекулярную циклизацию с использованием фосфитного реагента, что приводит к образованию 4-нитробензил (5R,6S)-6-бромпенем-3-карбоксилата 16.
Способ удобно осуществлять, используя следующие стадии.
Стадия 1. Растворение 6-аминопенициллановой кислоты 20 в органическом растворителе (предпочтительно метаноле или ТГФ) и воде для образования 6-бромпроизводного 21 в присутствии 48 мас.% бромистоводородной кислоты и раствора нитрита натрия или калия при температуре от -10°С до -30°С. 6-Бромпроизводное пенициллановой кислоты 21 можно или выделить, или in situ превратить в п-нитробензил 6-бромпеницилланат 22 действием 4-нитробензилбромида в присутствии органических или неорганических оснований (предпочтительно карбоната натрия или калия) в органическом растворителе (предпочтительно ТГФ или ДМФА).
Стадия 2. 4-Нитробензил 6-бромпеницилланат 22, полученный способом, описанным для стадии 1, можно выделить или, не выделяя, превратить в 4-нитробензил 6-бромпеницилланат-1-оксид 23 в той же колбе ("in the same pot") (т.е. продолжая стадию 1) путем окисления 4-нитробензил 6-бромпеницилланата 22 любыми известными окислителями, такими как 3-хлорпероксибензойная кислота (mcpba) или пероксид водорода.
Стадия 3. Продукт стадии 2, а именно 4-нитробензил 6-бромпеницилланат-1-оксид 23, можно превратить в 4-нитробензил (2R)-2-[(3S,4R)-4-(бензотиазол-2-илдитио)-3-бром-2-оксоазетидин-1-ил]-3-метилбут-3-еноат 24 кипячением 4-нитробензил 6-бромпеницилланат-1-оксида 23 с 2-меркаптобензотиазолом в ароматическом растворителе (предпочтительно в толуоле или ксилоле).
Стадия 4. Продукт стадии 3, а именно 4-нитробензил (2R)-2-[(3S,4R)-4-(бензотиазол-2-илдитио)-3-бром-2-оксоазетидин-1-ил]-3-метилбут-3-еноат 24, можно растворить в органическом растворителе (предпочтительно в толуоле или ксилоле) и при взаимодействии с органическим третичным основанием (предпочтительно триэтиламином) при температуре окружающей среды получить 4-нитробензил 2-[(3S,4R)-4-(бензотиазол-2-илдитио)-3-бром-2-оксоазетидин-1-ил]-3-метилбут-2-еноат 25.
Стадия 5. Продукт стадии 4, а именно 4-нитробензил 2-[(3S,4R)-4-(бензотиазол-2-илдитио)-3-бром-2-оксоазетидин-1-ил]-3-метилбут-2-еноат 25, можно превратить в 4-нитробензил 2-[(3S,4R)-3-бром-4-формилтио-2-оксоазетидин-1-ил]-3-метилбут-2-еноат 26 взаимодействием с органической кислотой (предпочтительно муравьиной) в ароматическом органическом растворителе (предпочтительно в толуоле) в присутствии смеси уксусный ангидрид/органическое третичное основание (предпочтительно пиридин) и триалкил- или триарилфосфина (предпочтительно трифенилфосфина) при температуре примерно от -10°С до примерно -30°С (предпочтительно примерно от -15°С до -20°С).
Альтернативный вариант осуществления данного изобретения относится к последовательному превращению соединения 23 в соединение 26 без выделения промежуточных соединений.
Продукт стадии 2, а именно 4-нитробензил 6-бромпеницилланат-1-оксид 23, подвергают взаимодействию с меркаптобензотиазолом в ароматическом органическом растворителе (предпочтительно в толуоле) при кипячении с обратным холодильником в течение 1-3 ч и обрабатывают триэтиламином при температуре от 0 до -20°С в течение 3-4 ч. После указанной обработки в реакционную смесь загружают последовательно органическую кислоту (предпочтительно муравьиную кислоту) и ангидрид (уксусный ангидрид), органическое третичное основание (предпочтительно пиридин) и триалкил- или триарилфосфат при температуре от -10°С до -40°С.
Стадия 6. 4-Нитробензил 2-[(3S,4R)-3-бром-4-формилтио-2-оксоазетидин-1-ил]-3-метилбут-2-еноат 26 - продукт стадии 5 или вышеуказанной последовательности превращений - помещают в органический растворитель (предпочтительно этилацетат) при температуре от -70°C до -90°С и в течение 3-4 ч пропускают озонированный кислород, затем осуществляют внутримолекулярную циклизацию, используя фосфитный реагент (предпочтительно триметилфосфит). Продукт 4-нитробензил (5R,6S)-6-бромпенем-3-карбоксилат 16 кристаллизуют из смеси этилацетат:гексан.
Данное изобретение также относится к соединению, представленному следующей формулой (16):
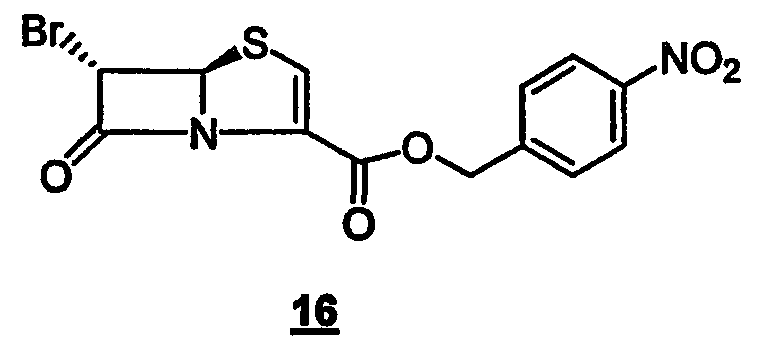
Вышеописанный способ (Стадия 1 - Стадия 6) получения соединения, представленного формулой 16, является промежуточным звеном в получении пенемов с замещенной метиленовой группой в положении 6 общей формулы I.
Соединение, представленное формулой 16, представляет собой кристаллическое производное (параметры дифракции рентгеновских лучей в порошкообразном образце приводятся в таблице 2).
Кристаллическая природа указанного промежуточного соединения придает ему стабильность и тем самым увеличивает срок хранения. Данные по стабильности соединения формулы 16 приведены в таблице 1.
Стадия 7. Взаимодействие 4-нитробензил (5R,6S)-6-бромпенем-3-карбоксилата 16 с соответствующим образом замещенными альдегидами (определенными выше) для осуществления стадии альдольной конденсации можно проводить в присутствии кислоты Льюиса (предпочтительно безводного MgBr2 или эфирата MgBr2)

и органического третичного основания (предпочтительно Et3N, пиридина, диметиламинопиридина (DMAP) или триалкиламина диизопропилэтиламина) в апротонном полярном органическом растворителе(ях) (предпочтительно ТГФ и/или ацетонитриле) при температуре от -10°С до -40°C. Промежуточные альдольные продукты общей формулы 18 функционализируют in situ ("in the same pot")при температуре от 0°C до -10°С и получают сложноэфирную уходящую группу: например, с хлорангидридом или ангидридом уксусной кислоты получают ацетат, с ангидридом трифторметансульфокислоты - трифлат, с тозилхлоридом - тозилат; или, если соединения формулы 18 выделяют, их можно превратить в галогенпроизводные взаимодействием с тетрагалогенметаном и трифенилфосфином при комнатной температуре в подходящем органическом растворителе, предпочтительно CH2Cl2.
Стадия 8. Последнюю стадию, а именно стадию восстановительного элиминирования, которая дает соединение общей формулы I, можно осуществить растворением смеси альдольных продуктов в органическом растворителе (предпочтительно ТГФ/ацетонитрил) при рН 6,0-8,5, предпочтительно при рН 6,5-7,0 фосфатного буфера, и действием активированного металла, такого как цинк, олово или алюминий, при температуре окружающей среды (предпочтительно при 20°С-35°C). Продукт в виде соли щелочного металла можно очистить колоночной хроматографией с обращенной фазой.
Альтернативно стадии 7 и 8 можно проводить последовательно в одной колбе ("in the same pot") без выделения промежуточного альдольного продукта.
Последнюю стадию, а именно стадию восстановительного элиминирования, можно осуществить растворением альдольного продукта в органическом растворителе (предпочтительно ТГФ или ацетонитриле) и фосфатном буфере с рН 6,5-7,0 и гидрированием над Pd/C при давлении 10-100 фунт/кв.дюйм (предпочтительно при 40 фунт/кв.дюйм).
Иллюстрацией данного изобретения служит типичный пример, приведенный на схеме А. На схеме А показаны подходящие условия реакции, однако можно использовать и другие реакционные условия, не выходя за границы объема данного изобретения. Например, можно использовать меньшую или большую длительность протекания реакции; обычно чем дольше длится реакция, тем более полно она проходит.

ОПИСАНИЕ ПРЕДПОЧТИТЕЛЬНЫХ ВАРИАНТОВ ОСУЩЕСТВЛЕНИЯ ИЗОБРЕТЕНИЯ
Данное изобретение относится к новому способу получения соединения формулы I
где
один из заместителей A или В обозначает водород, а другой обозначает арил, необязательно замещенный одним или двумя R2, гетероарил, необязательно замещенный одним или двумя R2, конденсированный трициклический гетероарил, необязательно замещенный одним или двумя R2, циклоалкил, необязательно замещенный одним или двумя R2, алкил, необязательно замещенный одним или двумя R2, алкенил, необязательно замещенный одним или двумя R2, алкинил, необязательно замещенный одним или двумя R2, насыщенную или частично насыщенную гетероциклическую группу, необязательно замещенную одним или двумя R2;
R5 представляет собой H, C1-C6-алкил, C5-C6-циклоалкил или CHR3OCO-C1-C6-алкил (образующие сложные эфиры, способные гидролизоваться in vivo) или металлы, такие как Na, K или Ca (образующие соли).
R1 представляет собой H, необязательно замещенный -C1-C6-алкил, необязательно замещенный арил, необязательно замещенные гетероарил или моно- или бициклические насыщенные гетероциклические группы, необязательно замещенный С3-С7-циклоалкил, необязательно замещенный С3-С6-алкенил, необязательно замещенный С3-С6-алкинил при условии, что атом углерода, который непосредственно связан с N, не должен входить в состав двойной или тройной связи; необязательно замещенный С1-С6-перфторалкил, необязательно -S(O)p-замещенный алкил или арил, где p равно 2, необязательно замещенный -C=O-гетероарил, необязательно замещенный -C=O-арил, необязательно замещенный -C=O-(С1-С6)-алкил, необязательно замещенный -C=O-(С3-С6)циклоалкил, необязательно замещенную моно- или бициклическую насыщенную -C=O-гетероциклическую группу, необязательно замещенный C1-C6-алкиларил, необязательно замещенный C1-C6-алкилгетероарил, необязательно замещенный арил-С1-C6-алкил, необязательно замещенный гетероарил-C1-C6-алкил, необязательно замещенную моно- или бициклическую насыщенную C1-C6-алкилгетероциклическую группу, необязательно замещенный арилалкенил, содержащий 8-16 атомов углерода, -CONR6R7, -SO2NR6R7, необязательно замещенный арилалкилоксиалкил, необязательно замещенный -алкил-O-алкиларил, необязательно замещенный -алкил-O-алкилгетероарил, необязательно замещенный арилоксиалкил, необязательно замещенный гетероарилоксиалкил, необязательно замещенный арилоксиарил, необязательно замещенный арилоксигетероарил, необязательно замещенный C1-C6-алкиларилоксиарил, необязательно замещенный C1-C6-алкиларилоксигетероарил, необязательно замещенные алкиларилоксиалкиламиносодержащие группы, необязательно замещенный алкоксикарбонил, необязательно замещенный арилоксикарбонил, необязательно замещенный гетероарилоксикарбонил. Предпочтительными R1 группами являются водород, необязательно замещенный алкил, необязательно замещенный арил, -С=O-(С1-С6)-алкил, С3-С6-алкенил, С3-С6-алкинил, необязательно замещенный циклоалкил, SO2-алкил, SO2-арил, необязательно замещенные гетероциклические группы, -CONR6R7 и необязательно замещенный гетероарил.
R2 представляет собой водород, необязательно замещенный C1-C6-алкил, необязательно замещенный C2-C6-алкенил, содержащий 1 или 2 двойных связи, необязательно замещенный C2-C6-алкинил, содержащий 1 или 2 тройных связи, галоген, циано, N-R6R7, необязательно замещенный C1-C6-алкокси, гидрокси; необязательно замещенный арил, необязательно замещенный гетероарил, COOR6, необязательно замещенные алкиларилоксиалкиламиносодержащие группы, необязательно замещенный арилокси, необязательно замещенный гетероарилокси, необязательно замещенный C3-C6-алкенилокси, необязательно замещенный C3-C6-алкинилокси, C1-C6-алкиламино-C1-C6-алкокси, алкилендиокси, необязательно замещенный арилокси-C1-C6-алкиламино, C1-C6-перфторалкил, необязательно S(О)q-замещенный С1-С6-алкил, необязательно S(O)q-замещенный арил, где q равно 0, 1 или 2, CONR6R7, гуанидино- или включенную в цикл гуанидиногруппу, необязательно замещенный C1-C6-алкиларил, необязательно замещенный арилалкил, необязательно замещенный C1-C6-алкилгетероарил, необязательно замещенный гетероарил-C1-C6-алкил, необязательно замещенную моно- или бициклическую насыщенную C1-C6-алкилгетероциклическую группу, необязательно замещенный арилалкенил, содержащий от 8 до 16 атомов углерода, SO2NR6R7, необязательно замещенный арилалкилоксиалкил, необязательно замещенный арилоксиалкил, необязательно замещенный гетероарилоксиалкил, необязательно замещенный арилоксиарил, необязательно замещенный арилоксигетероарил, необязательно замещенный гетероарилоксиарил, необязательно замещенный C1-C6-алкиларилоксиарил, необязательно замещенный C1-C6-алкиларилоксигетероарил, необязательно замещенный арилоксиалкил, необязательно замещенный гетероарилоксиалкил, необязательно замещенные алкиларилоксиалкиламиносодержащие группы. Предпочтительными R2 группами являются водород, необязательно замещенный алкил, необязательно замещенный алкокси, необязательно замещенный гетероарил, галоген, CN, гидрокси, необязательно замещенная гетероциклическая группа, -CONR6R7, COOR6, необязательно замещенный арил, S(O)q-алкил и S(O)q-арил;
R3 представляет собой водород, C1-C6-алкил, C5-C6-циклоалкил, необязательно замещенный арил, необязательно замещенный гетероарил;
R4 представляет собой водород, необязательно замещенный C1-C6-алкил; один из R4 представляет собой ОН, С1-С6-алкокси, -S-C1-C6-алкил, COOR6, -NR6R7, -CONR6R7; или R4R4 могут вместе представлять собой =O, или R4R4 вместе с атомом углерода, к которому они присоединены, могут образовывать 5-8-членную спиро-систему, содержащую или не содержащую гетероатомы, выбранные из N, O, S=(O)n (где n равно 0-2) и N-R1;
R6 и R7 независимо представляют собой Н, необязательно замещенный C1-C6-алкил, необязательно замещенный арил, необязательно замещенный гетероарил, необязательно замещенный C1-C6-алкиларил, необязательно замещенный арилалкил, необязательно замещенный гетероарилалкил, необязательно замещенный C1-C6-алкилгетероарил; R6 и R7 могут вместе образовать 3-7-членную насыщенную циклическую систему, необязательно содержащую один или два гетероатома, таких как N-R1, О, S=(O)n, где n равно 0-2.
Химические определения
Термин алкил означает алкильные группы как с линейной, так и с разветвленной цепью, состоящие из 1-12 атомов углерода, предпочтительно 1-6 атомов углерода.
Термин алкенил означает алкенильные группы как с линейной, так и с разветвленной цепью, состоящие из 2-8 атомов углерода, содержащие по меньшей мере одну двойную связь и не содержащие тройной связи, предпочтительно алкенильная группа содержит одну или две двойных связи. Такие алкенильные группы могут существовать в виде Е- и Z-изомеров; соединения по данному изобретению включают оба изомера. В случае алкенильной группы атом углерода, который входит в состав двойной связи, не должен быть соединен с гетероатомами, такими как O, S или N-R1.
Термин алкинил означает алкинильные группы как с линейной, так и с разветвленной цепью, состоящие из 2-6 атомов углерода, содержащие по меньшей мере одну тройную связь, предпочтительно алкинильная группа содержит одну или две тройных связи. В случае алкинильной группы атом углерода, который входит в состав двойной или тройной связи, не должен быть соединен с гетероатомами, такими как O, S или N-R1.
Термин циклоалкил относится к алициклической углеводородной группе, содержащей 3-7 атомов углерода. Термин перфторалкил в данной заявке используют для обозначения насыщенных алифатических углеводородных групп как с линейной, так и с разветвленной цепью, содержащих по меньшей мере один атом углерода и два или несколько атомов фтора. Примеры перфторалкильных групп включают CF3, CH2CF3, CF2CF3 и CH(CF3)2. Термин галоген обозначает Cl, Br, F и I.
Если алкил, алкенил, алкинил или циклоалкил определяют как «необязательно замещенный», возможными заместителями являются один или два из следующих: нитро, -арил, -гетероарил, алкоксикарбонил-, -алкокси, -алкоксиалкил, алкил-O-C2-C4-алкил-O-, -циано, -галоген, -гидрокси, -NR6R7, -трифторметил, -трифторметокси, арилалкил, алкиларил, R6R7N-алкил-, HO-C1-C6-алкил-, алкоксиалкил-, алкил-S-, -SO2NR6R7, -SO2NHR6, -CO2H, CONR6R7, арил-О-, гетероарил-О-, -S(O)s-арил (где s равно 0-2), -алкил-O-алкил-NR6R7, -алкиларил-O-алкил-N-R6R7, C1-C6-алкил, алкенил, алкинил, циклоалкил, алкоксиалкил-O-, R6R7N-алкил- и -S(O)s-гетероарил (где s равно 0-2). Предпочтительные заместители для алкила, алкенила, алкинила и циклоалкила включают галоген, нитро, арил, гетероарил, алкоксикарбонил-, алкокси, -алкоксиалкил, -циано, гидрокси и -N-R6R7.
Термин арил означает ароматический углеводородный остаток, выбранный из следующих групп: фенильной, α-нафтильной, β-нафтильной, бифенильной, антрильной, тетрагидронафтильной, флуоренильной, инданильной, бифениленильной и аценафтенильной.
Термином гетероарил определяется ароматическая гетероциклическая система (моноциклическая или бициклическая), где гетероарильные остатки выбирают из: (1) фурана, тиофена, индола, азаиндола, оксазола, тиазола, изоксазола, изотиазола, имидазола, N-метилимидазола, пиридина, пиримидина, пиразина, пиррола, N-метилпиррола, пиразола, N-метилпиразола, 1,3,4-оксадиазола, 1,2,4-триазола, 1-метил-1,2,4-триазола, 1H-тетразола, 1-метилтетразола, бензоксазола, бензотиазола, бензофурана, бензизоксазола, бензимидазола, N-метилбензимидазола, азабензимидазола, индазола, хиназолина, хинолина и изохинолина; (2) бициклических ароматических гетероциклов, где фенильный, пиридиновый, пиримидиновый или пиридазиновый цикл конденсирован с: (а) 5-членным ароматическим (ненасыщенным) гетероциклом, содержащим один атом азота; (b) 5- или 6-членным ароматическим (ненасыщенным) гетероциклом, содержащим два атома азота; (с) 5-членным ароматическим (ненасыщенным) гетероциклом, содержащим один атом азота вместе или с одним атомом кислорода, или с одним атомом серы; или (d) 5-членным ароматическим (ненасыщенным) гетероциклом, содержащим один гетероатом, выбранный из O, N или S.
Если арил или гетероарил определяют как «необязательно замещенный», один или два из нижеследующих являются возможными заместителями: нитро, -арил, -гетероарил, алкоксикарбонил-, -алкокси, -алкоксиалкил, алкил-O-C2-C4-алкил-O-, -циано, -галоген, -гидрокси, -N-R6R7, -трифторметил, -трифторметокси, арилалкил, алкиларил, R6R7N-алкил-, HO-C1-C6-алкил-, алкоксиалкил-, алкил-S-, -SO2N-R6R7, -SO2NH-R6, -CO2H, CONR6R7, арил-О-, гетероарил-О-, -S(O)s-арил (где s = 0-2), -алкил-O-алкил-NR6R7, -алкиларил-O-алкил-N-R6R7, C1-C6-алкил, алкенил, алкинил, циклоалкил, алкоксиалкил-O- и -S(O)s-гетероарил (где s равно 0-2). Предпочтительные заместители для арила и гетероарила включают алкил, галоген, -N-R6R7, трифторметил, -трифторметокси, арилалкил и алкиларил.
Арилалкил определяют как арил-C1-C6-алкил-. Арилалкильные группы включают бензил, 1-фенилэтил, 2-фенилэтил, 3-фенилпропил, 2-фенилпропил и им подобные. Термин «необязательно замещенный» относится к алкильной или арильной части, незамещенной или замещенной 1 или 2 заместителями, как определено выше.
Алкиларил определяют как C1-C6-алкиларил-. Термин «необязательно замещенный» относится к алкильной или арильной части, незамещенной или замещенной 1 или 2 заместителями, как определено выше.
Гетероарил-C1-C6-алкил обозначает гетероарилзамещенную алкильную группу, представляющую собой алкильную цепь из 1-6 атомов углерода (линейную или разветвленную). Алкилгетероарильные части включают гетероарил-(CH2)1-6- и им подобные группы. Термин «необязательно замещенный» относится к алкильной или гетероарильной части, незамещенной или замещенной 1 или 2 заместителями, как определено выше.
С1-С6-Алкилгетероарил обозначает алкильную цепь из 1-6 атомов углерода (линейную или разветвленную), присоединенную к гетероарильной группе, которая связана с остальной частью молекулы, т.е. C1-C6-алкилгетероарил-. Термин «необязательно замещенный» относится к алкильной или гетероарильной части, незамещенной или замещенной 1 или 2 заместителями, как определено выше.
Насыщенные или частично насыщенные гетероциклические группы обозначают такие гетероциклические остатки, как: азиридинил, азетидинил, 1,4-диоксанил, гексагидроазепинил, пиперазинил, пиперидинил, пирролидинил, морфолинил, тиоморфолинил, дигидробензимидазолил, дигидробензофуранил, дигидробензотиенил, дигидробензоксазолил, дигидрофуранил, дигидроимидазолил, дигидроиндолил, дигидроизооксазолил, дигидроизотиазолил, дигидрооксадиазолил, дигидрооксазолил, дигидропиразинил, дигидропиразолил, дигидропиридинил, дигидропиримидинил, дигидропирролил, дигидрохинолинил, дигидротетразолил, дигидротиадиазолил, дигидротиазолил, дигидротиенил, дигидротриазолил, дигидроазетидинил, дигидро-1,4-диоксанил, тетрагидрофуранил, тетрагидротиенил, тетрагидрохинолинил и тетрагидроизохинолинил.
Моно- или бициклическая насыщенная или частично насыщенная C1-C6-алкилгетероциклическая группа обозначает С1-С6-алкильную группу (линейную или разветвленную), присоединенную к гетероциклу (который определен выше) через атом углерода или атом азота, в которой другой конец алкильной цепи присоединен к остальной части молекулы. Термин «необязательно замещенный» относится к алкильной или гетероциклической части молекулы, незамещенной или замещенной 1 или 2 заместителями, определенными выше.
Арилалкилоксиалкил определяют как арил-C1-C6-алкил-O-C1-C6-алкил-. Термин «необязательно замещенный» относится к алкильной и/или арильной частям, незамещенным или замещенным 1 или 2 заместителями, определенными выше.
Алкилоксиалкил определяют как C1-C6-алкил-O-C1-C6-алкил-. Термин «необязательно замещенный» относится к алкильному остатку, незамещенному или замещенному 1 или 2 заместителями, определенными выше.
Арилоксиалкил определяют как арил-O-C1-C6-алкил-. Термин «необязательно замещенный» относится к алкильному или арильному остатку, незамещенному или замещенному 1 или 2 заместителями, определенными выше.
Гетероарилалкилоксиалкил определяют как гетероарил-C1-C6-алкил-О-C1-C6-алкил-. Термин «необязательно замещенный» относится к алкильной или гетероарильной части, незамещенной или замещенной 1 или 2 заместителями, определенными выше.
Арилоксиарил определяют как арил-О-арил-. Термин «необязательно замещенный» относится к арильному остатку, незамещенному или замещенному 1 или 2 заместителями, определенными выше.
Арилоксигетероарил определяют как арил-O-гетероарил- или -арил-O-гетероарил. В данном определении или арильный, или гетероарильный остаток может быть соединен с остальной частью молекулы. Термин «необязательно замещенный» относится к арильному остатку или гетероарильному остатку, незамещенному или замещенному 1 или 2 заместителями, как определено выше.
Алкиларилоксиарил определяют как арил-О-арил-С1-С6-алкил-. Термин «необязательно замещенный» относится к арильному остатку, незамещенному или замещенному 1 или 2 заместителями, определенными выше.
Алкиларилоксигетероарил определяют как гетероарил-О-арил-C1-C6-алкил-. Термин «необязательно замещенный» относится к арильному или гетероарильному остатку, незамещенному или замещенному 1 или 2 заместителями, определенными выше.
Алкиларилоксиалкиламиносодержащие группы определяют как R6R7N-C1-C6-алкил-O-арил-C1-C6-алкил-. Термин «необязательно замещенный» относится к алкильному или арильному остатку, незамещенному или замещенному 1 или 2 заместителями, определенными выше, R6 и R7 такие, как определено выше.
Алкоксикарбонил определяют как С1-С6-алкил-О-С=О-. Термин «необязательно замещенный» относится к алкильной группе алкоксильного остатка, незамещенной или замещенной 1 или 2 заместителями, определенными выше.
Арилоксикарбонил определяют как арил-О-С=О-. Термин «необязательно замещенный» относится к арильному остатку, незамещенному или замещенному 1 или 2 заместителями, определенными выше.
Гетероарилоксикарбонил определяют как гетероарил-О-С=О-. Термин «необязательно замещенный» относится к гетероарильному остатку, незамещенному или замещенному 1 или 2 заместителями, определенными выше.
Алкокси определяют как С1-С6-алкил-О-. Термин «необязательно замещенный» относится к алкильному остатку, незамещенному или замещенному 1 или 2 заместителями, определенными выше.
Арилокси определяют как арил-O-. Термин «необязательно замещенный» относится к арильному остатку, незамещенному или замещенному 1 или 2 заместителями, определенными выше.
Гетероарилокси определяют как гетероарил-О-. Термин «необязательно замещенный» относится к гетероарильному остатку, незамещенному или замещенному 1 или 2 заместителями, определенными выше.
Алкенилокси определяют как C3-C6-алкен-O-, например остатки, подобные аллил-O-, бутен-2-ен-O. Термин «необязательно замещенный» относится к алкеновому остатку, незамещенному или замещенному 1 или 2 заместителями, как определено выше, при условии, что атом углерода, входящий в состав двойной связи, не соединен с гетероатомом, таким как O, S или N-R1.
Алкинилокси определяют как C3-C6-алкин-O-, например группы, подобные HC≡C-CH2O-. Термин «необязательно замещенный» относится к алкиновому остатку, незамещенному или замещенному 1 или 2 заместителями, как определено выше, при условии, что атом углерода, входящий в состав двойной или тройной связи, не соединен с гетероатомом, таким как O, S или N-R1.
Алкиламиноалкокси определяют как R6R7N-C1-C6-алкил-O-C1-C6-алкил-, где терминальная алкильная группа, присоединенная к атому кислорода, соединена с остальной частью молекулы. R6 и R7 определены выше. Термин «необязательно замещенный» относится к алкильному остатку, незамещенному или замещенному 1 или 2 заместителями, определенными выше.
Алкилендиокси определяют как -O-(CH2)2-O-.
Арилоксиалкиламиносодержащие группы определяют как R6R7N-C1-C6-алкил-O-арил-, где арил присоединен к остальной части молекулы. Термин «необязательно замещенный» относится к алкильному или арильному остатку, незамещенному или замещенному 1 или 2 заместителями, определенными выше.
Арилалкенил определяют как арил-C2-C8-алкен-, при условии, что атом углерода, входящий в состав двойной связи, не соединен с гетероатомом, таким как O, S или N-R1. Термин «необязательно замещенный» относится к алкеновому или арильному остатку, незамещенному или замещенному 1 или 2 заместителями, определенными выше.
Гетероарилоксиалкил определяют как гетероарил-О-С1-С6-алкил-. Термин «необязательно замещенный» относится к гетероарильному остатку, незамещенному или замещенному 1 или 2 заместителями, определенными выше.
Гетероарилоксиарил определяют как гетероарил-О-арил-, где арильный остаток присоединен к остальной части молекулы. Термин «необязательно замещенный» относится к гетероарильному или арильному остатку, незамещенному или замещенному 1 или 2 заместителями, определенными выше.
Алкокси, алкоксиалкил, алкоксиалкилокси и алкилтиоалкилокси представляют собой группы, где алкильная цепь (линейная или разветвленная) состоит из 1-6 атомов углерода. Арилокси, гетероарилокси, арилтио и гетероарилтио содержат арильные и гетероарильные группы, определенные выше. Арилалкилокси, гетероарилалкилокси, арилалкилтио и гетероарилалкилтио содержат арильные и гетероарильные группы, определенные выше, и алкильные цепи (линейные или разветвленные) из 1-6 атомов углерода.
Арилоксиалкил, гетероарилоксиалкил, арилоксиалкилокси и гетероарилоксиалкилокси представляют собой заместители, где алкильный радикал содержит 1-6 атомов углерода. Термины моноалкиламино и диалкиламино относятся к остаткам с одной алкильной или двумя алкильными группами, где алкильная цепь содержит 1-6 атомов углерода, а группы могут быть одинаковыми или различными.
Термины моноалкиламиноалкил и диалкиламиноалкил относятся к моноалкиламино- и диалкиламиногруппам с одним или двумя алкильными заместителями (одинаковыми или различными) при атоме азота, который присоединен к алкильной группе, содержащей 1-3 атома углерода.
Выражение «конденсированная трициклическая гетероарильная группа» используют в описании и формуле изобретения для обозначения:
группы, содержащей три конденсированных цикла, из которых по меньшей мере один цикл ароматический (т.е. соответствует правилу Хюккеля (4n+2)). Конденсированная трициклическая гетероарильная группа содержит 1-6 гетероатомов, выбранных из группы, состоящей из O, S, N и N-R1. Конденсированная трициклическая гетероарильная группа должна быть связана с остальной частью молекулы формулы I через атом углерода, предпочтительно входящий в состав ароматического кольца системы. Конденсированная трициклическая гетероарильная группа может содержать 1-3 ароматических цикла и 0-2 неароматических цикла. Каждый ароматический цикл в конденсированной трициклической гетероарильной группе может содержать 5-7 атомов в своем составе (включая головные атомы мостиковой группы), которые выбраны из CR2, O, S, N и N-R1. Каждый из ароматических циклов конденсированной трициклической гетероарильной группы может содержать 0-3 гетероатома, выбранных из O, S, N или N-R1. Неароматические циклы, если они имеются в составе конденсированной трициклической гетероарильной группы, могут содержать 5-8 атомов в цикле (включая головные атомы мостиковой группы), из которых от 0 до 4 могут быть гетероатомами, выбранными из N, N-R1, О или S(O)n, где n равно 0-2. В каждом неароматическом цикле конденсированной трициклической гетероарильной группы один или два немостиковых атома углерода могут каждый иметь один или два заместителя R4, и R4 могут быть независимо одинаковыми или различными. Примеры конденсированных трициклических гетероарильных групп включают:
Размер циклов и тип соединения: (5-5-5)

В обеих формулах 1-А и 1-В Z1, Z2, Z3, Z4, Z5, Z6 и Z7 независимо выбраны из CR2, N, O, S или N-R1 и, как указано выше, один из Z1-Z7 является атомом углерода, к которому присоединена остальная часть молекулы пенема. Y1, Y2, Y3 и Y4 могут независимо представлять собой С или N.
Размер циклов и тип соединения: (5-5-6)

В обеих формулах 2-А и 2-В Z1, Z2, Z3, Z4, Z5, Z6, Z7 и Z8 независимо выбраны из CR2, N, O, S или N-R1 и, как указано выше, один из Z1-Z8 является атомом углерода, к которому присоединена остальная часть молекулы пенема. Y1, Y2, Y3 и Y4 могут независимо представлять собой С или N.
Размер циклов и тип соединения: (5-6-5)
В обеих формулах 3-А и 3-В Z1, Z2, Z3, Z4, Z5, Z6, Z7 и Z8 независимо выбраны из CR2, N, O, S или N-R1 и, как указано выше, один из Z1-Z8 является атомом углерода, к которому присоединена остальная часть молекулы пенема. Y1, Y2, Y3 и Y4 могут независимо представлять собой С или N.
Размер циклов и тип соединения (5-6-6)
В формулах 4-А, 4-В и 4-С Z1, Z2, Z3, Z4, Z5, Z6, Z7 и Z8 независимо выбраны из CR2, N, O, S или N-R1 и, как указано выше, один из Z1-Z8 является атомом углерода, к которому присоединена остальная часть молекулы пенема. Y1, Y2, Y3 и Y4 могут независимо представлять собой С или N.
Размер циклов и тип соединения: [5-5-неароматический)]
В формулах 5-А и 5-В Z1, Z2, Z3 и Z4 независимо выбраны из CR2, N, O, S или N-R1 и, как указано выше, один из Z1-Z4 является атомом углерода, к которому присоединена остальная часть молекулы пенема. Y1, Y2, Y3 и Y4 независимо представляют собой С или N. W1, W2 и W3 независимо выбраны из CR4R4, S(O)r (r равно 0-2), O или N-R1, при условии, что в состав насыщенной циклической системы не могут входить S-S, S-O или O-O связи;
и t равно 1-3.
Размер циклов и тип соединения: [5-6-неароматический)]

В формулах 6-А,6-В и 6-С Z1, Z2, Z3,Z4 и Z5 независимо выбраны из CR2, N, O, S или N-R1 и, как указано выше, один из Z1-Z5 является атомом углерода, к которому присоединена остальная часть молекулы пенема. Y1 и Y2 независимо представляют собой С или N. W1, W2 и W3 независимо выбраны из CR4R4, S(O)r (r равно 0-2), O или N-R1, при условии, что в состав насыщенной циклической системы не могут входить S-S, S-O или O-O связи;
и t равно 1-3.
Размер циклов и тип соединения: [5-(неароматический)-5]

В формулах 7-А и 7-В Z1, Z2, Z3, Z4, Z5 и Z6 независимо выбраны из CR2, N, O, S или N-R1 и, как указано выше, один из Z1-Z6 является атомом углерода, к которому присоединена остальная часть молекулы пенема. Y1, Y2, Y3 и Y4 независимо представляют собой С или N. W1 и W2 независимо выбраны из CR4R4, S(O)r (r равно 0-2), O или N-R1, при условии, что в состав насыщенной циклической системы не могут входить S-S, S-O или O-O связи; и t равно 1-3.
Размер циклов и тип соединения: [5-(неароматический)-6]

В формулах 8-А и 8-В Z1, Z2, Z3, Z4, Z5, Z6 и Z7 независимо выбраны из CR2, N, O, S или N-R1 и, как указано выше, один из Z1-Z7 является атомом углерода, к которому присоединена остальная часть молекулы пенема. Y1, Y2, Y3 и Y4 независимо представляют собой С или N. W1 и W2 независимо представляют собой CR4R4, S(O)r (r равно 0-2), O или N-R1, при условии, что в состав насыщенной циклической системы не могут входить S-S, S-O или O-O связи; и t равно 0-3.
Размер циклов и тип соединения: [5-(неароматический)-(неароматический)]

В формулах 9-А и 9-В Z1, Z2 и Z3 независимо выбраны из CR2, N, O, S или N-R1 и, как указано выше, один из Z1-Z3 является атомом углерода, к которому присоединена остальная часть молекулы пенема. Y1 и Y4 независимо представляют собой С или N. Y2 и Y3 независимо представляют собой СН или N. W1, W2, W3, W4 и W5 независимо представляют собой CR4R4, S(O)r (r равно 0-2), O или N-R1, при условии, что в состав насыщенной циклической системы не могут входить S-S, S-O или O-O связи; t равно 0-2 и u равно 1-3.
Размер циклов и тип соединения: (6-5-6)

В формулах 10-А и 10-В Z1, Z2, Z3,Z4, Z5, Z6, Z7, Z8 и Z9 независимо представляют собой CR2, N, O, S или N-R1 и, как указано выше, один из Z1-Z9 является атомом углерода, к которому присоединена остальная часть молекулы пенема. Y1, Y2, Y3 и Y4 независимо представляют собой С или N.
Размер циклов и тип соединения: (6-6-6)
В формулах 11-А, 11-В и 11-С Z1, Z2, Z3,Z4, Z5, Z6, Z7, Z8, Z9 и Z10 независимо представляют собой CR2, N, O, S или N-R1 и, как указано выше, один из Z1-Z10 является атомом углерода, к которому присоединена остальная часть молекулы пенема. Y1, Y2, Y3 и Y4 независимо представляют собой С или N.
Размер циклов и тип соединения: [6-5-(неароматический)]

В формулах 12-А и 12-В Z1, Z2, Z3,Z4 и Z5 независимо представляют собой CR2, N, O, S или N-R1 и, как указано выше, один из Z1-Z5 является атомом углерода, к которому присоединена остальная часть молекулы пенема. Y1, Y2, Y3 и Y4 независимо представляют собой С или N. W1, W2, W3 независимо представляют собой O, N-R1 или S=(O)r (r равно 0-2), при условии, что в состав насыщенной циклической системы не могут входить S-S, S-O или O-O связи; и t равно 1-4.
Размер циклов и тип соединения: [6-6-(неароматический)]
В формулах 13-А, 13-В и 13-С Z1, Z2, Z3,Z4, Z5 и Z6 независимо представляют собой CR2, N, O, S или N-R1 и, как указано выше, один из Z1-Z6 является атомом углерода, к которому присоединена остальная часть молекулы пенема. Y1, Y2, Y3 и Y4 независимо представляют собой С или N. W1, W2 и W3 независимо представляют собой CR4R4, S(O)r (r равно 0-2), O или N-R1 при условии, что в состав насыщенной циклической системы не могут входить S-S, O-O или S-O связи; и t равно 1-3.
Размер циклов и тип соединения: [6-(неароматический)-6]

В формулах 14-А, 14-В и 14-С Z1, Z2, Z3,Z4, Z5, Z6, Z7 и Z8 независимо представляют собой CR2, N, O, S или N-R1 и, как указано выше, один из Z1-Z8 является атомом углерода, к которому присоединена остальная часть молекулы пенема. Y1, Y2, Y3 и Y4 независимо представляют собой С или N. W1, W2 и W3 независимо представляют собой CR4R4, S(O)r (r равно 0-2), О или N-R1 при условии, что в состав насыщенной циклической системы не могут входить S-S, S-O или O-O связи; и t равно 1 или 2.
Размер циклов и тип соединения: [6-(неароматический)-(неароматический)]
В формулах 15-А, 15-В и 15-С Z1, Z2, Z3 иZ4 независимо представляют собой CR2, N, O, S или N-R1 и, как указано выше, один из Z1-Z4 является атомом углерода, к которому присоединена остальная часть молекулы пенема. Y1, Y2, Y3 и Y4 независимо представляют собой С или N. W1, W2, W3 и W4 независимо представляют собой CR4R4, S(O)r (r равно 0-2), O или N-R1 при условии, что в состав насыщенной циклической системы не могут входить S-S, S-O или O-O связи; t равно 1-3 и u равно 1-3.
Выражение «конденсированная бициклическая гетероарильная группа» используют в описании и формуле данного изобретения для обозначения:
группы, содержащей два конденсированных цикла, из которых один имеет ароматическую природу [т.е. подчиняется правилу Хюккеля (4n+2)], а другой - неароматическую.
Конденсированная бициклическая гетероарильная группа содержит от 1 до 6 гетероатомов, выбранных из O, S, N и N-R1.
Конденсированная бициклическая гетероарильная группа связана с остальной частью молекулы посредством атома углерода ароматического цикла, как показано в формуле I.
Ароматический цикл конденсированной бициклической гетероарильной группы содержит пять или шесть атомов в цикле (включая головные атомы мостиковой группы), выбранных из CR2, N, O, S или N-R1. Ароматический цикл конденсированной бициклической гетероарильной группы содержит от 0 до 3 гетероатомов, выбранных из О, S, N и NR1.
Неароматический цикл конденсированной бициклической гетероарильной группы содержит пять-восемь атомов в цикле (включая головные атомы мостиковой группы), выбранных из CR4R4, N, N-R1, O, S(O)n, где n равно 0-2. Неароматический цикл конденсированной бициклической гетероарильной группы содержит 0-4 гетероатома, выбранных из N, N-R1, О или S(O)n, где n равно 0-2.
Примеры конденсированных бициклических гетероарильных групп включают:
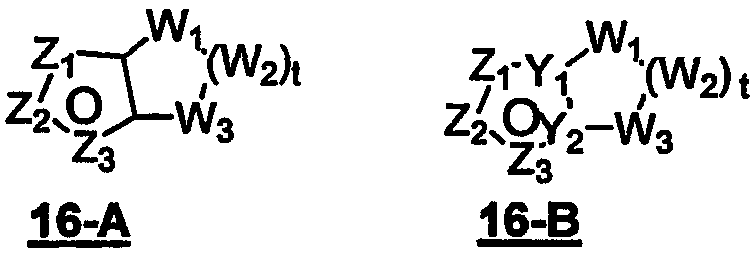

В формуле 16-А Z1, Z2 и Z3 независимо представляют собой CR2, N, O, S или N-R1 и один из Z1-Z3 является атомом углерода, к которому присоединена остальная часть молекулы, как показано в формуле I. Когда один из Z представляет собой CR2, два других Z могут представлять собой или два атома N, или один атом N и O, S, N-R1 в любых комбинациях без нарушения ароматичности; когда два Z представляют собой CR2, третий Z может быть произвольно выбран из N, O, S или N-R1 в любых комбинациях без нарушения ароматичности;
W1, W2 и W3 независимо представляют собой CR4R4, S, SO, SO2, O, N-R1, C=O при условии, что в состав насыщенной циклической системы не могут входить S-S, O-O или S-O связи; t равно 1-4.
В формуле 16-В Z1, Z2 и Z3 независимо представляют собой CR2, N, O, S или N-R1 и один из Z1-Z3 является атомом углерода, к которому присоединена остальная часть молекулы, как показано в формуле I.
Когда один из Z представляет собой CR2, два других Z могут независимо представлять собой CR2, N, O, S или N-R1 в любых комбинациях без нарушения ароматичности; когда два Z представляют собой N, тогда третий Z представляет атом углерода цикла, связанный с остальной частью молекулы пенема, как показано в формуле I.
W1, W2 и W3 независимо представляют собой CR4R4, S, SO, SO2, O, NR1;
t равно 1-4;
Y1 и Y2 представляют собой N или С; при этом, если ароматическим гетероциклом является имидазол, насыщенный цикл не может содержать атом S в положении, примыкающем к головному атому углерода мостиковой группы.
В формуле 16-С Z1, Z2, Z3 и Z4 независимо представляют собой CR2 или N и один из Z1-Z4 является атомом углерода, к которому присоединена остальная часть молекулы.
W1, W2 и W3 независимо представляют собой CR4R4, S, SO, SO2, O или N-R1 при условии, что в состав насыщенной циклической системы не могут входить S-S, O-O или S-O связи; t равно 1-4.
Y1 и Y2 независимо представляют собой С или N.
СПОСОБ ПО ДАННОМУ ИЗОБРЕТЕНИЮ
Соединения общей формулы I могут быть легко получены в мягких условиях конденсацией соответствующим образом замещенного альдегида 17 с производным 6-бромпенема структуры 16 (Схема 2) в присутствии кислоты Льюиса, предпочтительно безводного галогенида магния, более предпочтительно безводного MgBr2 или эфирата MgBr2, и мягкого основания, такого как триэтиламин, DMAP или диизопропилэтиламин, при низкой температуре, предпочтительно примерно от -20°C до -40°C. Промежуточный альдольный продукт 18 можно функционализировать действием хлорангидридов и ангидридов кислот, предпочтительно превратить их в ацетат, трифлат или тозилат 19; или, если соединение формулы 18 выделяют, его можно превратить в галогенпроизводное взаимодействием с тетрагалогенметаном и трифенилфосфином при комнатной температуре в подходящем органическом растворителе, предпочтительно CH2Cl2. Соединение 19 можно легко превратить в целевой продукт восстановительным элиминированием действием металла, такого как активированный цинк, и фосфатного буфера при умеренных температурах, предпочтительно от около 20°C до 35°C и при pH примерно 6,5-8,0 или гидрированием над катализатором, предпочтительно палладием на угле. Следует отметить, что стадию восстановительного элиминирования можно осуществить так, что произойдет снятие защиты карбоксильной группы. Если карбоксильную функцию защищает п-нитробензильная группа, тогда восстановительное элиминирование и снятие защиты выполняется в одну стадию. Однако если защитная группа не является п-нитробензильной, можно использовать методику из двух стадий, зависящую от природы защитной группы. Продукт может быть выделен в виде свободной кислоты или в виде соли щелочного металла. Вышеуказанную двухстадийную методику можно выполнять в одну стадию путем осуществления всего процесса без выделения промежуточного соединения 19. Данную относительно простую и крайне эффективную с точки зрения выхода и экономичности методику можно использовать для получения широкого круга соединений. Данная методика подходит также для крупномасштабного синтеза и применима к разнообразным альдегидам.
Один важный аспект данного изобретения относится к применению устойчивого промежуточного бромпенема 16. Стабильность промежуточного соединения имеет значение на стадии альдольной конденсации, где разложение сводят к минимуму, а также при хранении. В данном изобретении ключевое промежуточное соединение 16, где R представляет собой п-нитробензильную группу, является устойчивым и кристаллическим промежуточным соединением. В данном изобретении было найдено, что промежуточное соединение 16 является более стабильным, чем промежуточное соединение Q. Сравнительные данные по термической устойчивости промежуточных соединений 16 и Q приводятся ниже (таблица 1). Удивительная устойчивость промежуточного соединения 16 увеличивает время хранения соединения, а также возможность реализации способа в крупныхмасштабах. Как указано выше, защита карбоксильной группы п-нитробензильной группой уменьшает число стадий в данном способе получения конечного соединения структуры I. Кристаллическая структура промежуточного соединения установлена рентгеноструктурным анализом. В таблице 2 показаны параметры рентгеноструктурного анализа порошка соединения формулы 16.

1) Определено ВЭЖХ анализом.
Температура 40°С в твердом состоянии.
Условия ВЭЖХ:
Колонка: Inertsil SIL 100-5 4,6 × 150 мм (GL Science Inc.)
Подвижная фаза: этилацетат: н-гексан (10:35, об/об)
Длина волны: 312 нм; скорость потока: 1,0 мл/мин при комнатной температуре
Преимуществом является то, что на стадии восстановительного элиминирования в данном изобретении нужный "Z" изомер образуется крайне предпочтительно с селективностью 45-55:1 (около 50:1). В реакционной смеси не наблюдается образования "E" изомера. Стадию восстановительного элиминирования можно также осуществить гидрированием промежуточного соединения 19 над 10% Pd/C.
Промежуточное соединение 16, используемое в данном изобретении, может быть получено из коммерчески доступной 6-аминопенициллановой кислоты (6-APA) 20 (Схема 3). Она может быть превращена в бромпенем 16 по методике, приведенной на схеме 3. 6-Аминопенициллановую кислоту (6-APA) 20 превращают в бром-п-нитробензильное производное 22 по одностадийной (one pot)методике. Данное производное окисляют в сульфоксид 23, который подвергают реакции раскрытия цикла, и получают 24. Соединение 24 превращают в 16 по методике, кратко изложенной на Схеме 3. Преимуществом методики по данному изобретению является возможность превращения соединения 23 в 26 в одну стадию, без выделения промежуточных продуктов 24 и 25. Полученный таким образом бромпенем 16 подвергают взаимодействию с альдегидом 17 (см. Схему 2) в присутствии безводного MgBr2 или коммерчески доступного MgBr2:O(Et)2. Альдольный продукт выделяют в виде бромацетата 19, который можно легко превратить в конечный продукт I взаимодействием с активированным цинком (например, свежеактивированным 0,1 н. HCl) и фосфатным буфером (рН 6,5) при комнатной температуре. Продукт можно легко очистить известными методами, например колоночной хроматографией (с Diaion HP-21). Сначала элюирование проводят водой для удаления любых неорганических примесей, затем 10% раствором MeCN в воде. Полученный продукт имеет чистоту 98%, и его можно дополнительно очистить кристаллизацией. Альдольная конденсация с использование Et3N и MgBr2 крайне эффективна и имеет общий характер. Данное изобретение можно распространить на разнообразные альдегиды и получить конечный продукт общей структуры I.
Данное изобретение относится к способу получения соединения формулы I
где
один из заместителей A или В обозначает водород, а второй обозначает арил, необязательно замещенный одним или двумя R2, гетероарил, необязательно замещенный одним или двумя R2, конденсированный бициклический гетероарил, необязательно замещенный одним или двумя R2, конденсированный трициклический гетероарил, необязательно замещенный одним или двумя R2, циклоалкил, необязательно замещенный одним или двумя R2, алкил, необязательно замещенный одним или двумя R2, алкенил, необязательно замещенный одним или двумя R2, алкинил, необязательно замещенный одним или двумя R2, насыщенную или частично насыщенную гетероциклическую группу, необязательно замещенную одним или двумя R2;
R5 представляет собой H, C1-C6-алкил, C5-C6-циклоалкил, CHR3OCO-C1-C6-алкил, (образующие сложные эфиры, способные гидролизоваться in vivo) или металлы, такие как Na, K или Ca (образующие соли).
R2 представляет собой водород, необязательно замещенный C1-C6-алкил, необязательно замещенный C2-C6-алкенил, содержащий 1 или 2 двойных связи, необязательно замещенный C2-C6-алкинил, содержащий 1 или 2 тройных связи, галоген, циано, N-R6R7, необязательно замещенный C1-C6-алкокси, гидрокси; необязательно замещенный арил, необязательно замещенный гетероарил, COOR6, необязательно замещенные алкиларилоксиалкиламиносодержащие группы, необязательно замещенный арилокси, необязательно замещенный гетероарилокси, необязательно замещенный C3-C6-алкенилокси, необязательно замещенный C3-C6-алкинилокси, C1-C6-алкиламино-C1-C6-алкокси, алкилендиокси, необязательно замещенную арилокси-C1-C6-алкиламиносодержащую группу, C1-C6-перфторалкил, необязательно S(О)q-замещенный С1-С6-алкил, необязательно S(O)q-замещенный арил, где q равно 0, 1 или 2, CONR6R7, гуанидино- или включенную в цикл гуанидиногруппу, необязательно замещенный C1-C6-алкиларил, необязательно замещенный арилалкил, необязательно замещенный C1-C6-алкилгетероарил, необязательно замещенный гетероарил-C1-C6-алкил, необязательно замещенную моно- или бициклическую насыщенную C1-C6-алкилгетероциклическую группу, необязательно замещенный арилалкенил, содержащий от 8 до 16 атомов углерода, SO2NR6R7, необязательно замещенный арилалкилоксиалкил, необязательно замещенный арилоксиалкил, необязательно замещенный гетероарилоксиалкил, необязательно замещенный арилоксиарил, необязательно замещенный арилоксигетероарил, необязательно замещенный гетероарилоксиарил, необязательно замещенный C1-C6-алкиларилоксиарил, необязательно замещенный C1-C6-алкиларилоксигетероарил, необязательно замещенный арилоксиалкил, необязательно замещенный гетероарилоксиалкил, необязательно замещенные алкиларилоксиалкиламиносодержащие группы. Предпочтительными R2 группами являются водород, необязательно замещенный алкил, необязательно замещенный алкокси, необязательно замещенный гетероарил, галоген, CN, гидрокси, необязательно замещенная гетероциклическая группа, -CONR6R7, COOR6, необязательно замещенный арил, S(O)q-алкил и S(O)q-арил;
R3 представляет собой водород, C1-C6-алкил, C5-C6-циклоалкил, необязательно замещенный арил, необязательно замещенный гетероарил;
R6 и R7 независимо представляют собой Н, необязательно замещенный C1-C6-алкил, необязательно замещенный арил, необязательно замещенный гетероарил, необязательно замещенный C1-C6-алкиларил, необязательно замещенный арилалкил, необязательно замещенный гетероарилалкил, необязательно замещенный C1-C6-алкилгетероарил; R6 и R7 могут вместе образовать 3-7-членную насыщенную циклическую систему, необязательно содержащую один или два гетероатома, таких как N-R1, О, S=(O)n, где n равно 0-2.
Способ получения соединения формулы I включает следующие стадии.
Стадия 1. 6-Аминопенициллановую кислоту 20, растворенную в органическом растворителе (предпочтительно метаноле или ТГФ) и воде, превращают в 6-бромпроизводное в присутствии 48 мас.% бромистоводородной кислоты при температуре от -10°С до -30°C и раствора нитрита натрия или калия. 6-Бромпенициллановую кислоту 21 можно или выделить, или in situ превратить в п-нитробензил-6-бромпеницилланат 22, действием 4-нитробензилбромида в присутствии органических оснований или неорганических оснований (предпочтительно карбоната натрия или калия) в органическом растворителе (предпочтительно ТГФ или ДМФА).
Стадия 2. Продукт п-нитробензил 6-бромпеницилланат 22, полученный способом, кратко изложенным на стадии 1, можно выделить или, не выделяя, превратить в 4-нитробензил 6-бромпеницилланат-1-оксид 23 в той же колбе ("in the same pot") (т.е. последовательно продолжая стадию 1) окислением п-нитробензил 6-бромпеницилланата 22 в 1-оксид 4-нитробензил-6-бромпеницилланата 23, используя окислители, такие как 3-хлорпероксибензойную кислота (mcpba) или пероксид водорода.
Стадия 3. Продукт стадии 2, а именно 4-нитробензил 6-бромпеницилланат-1-оксид 23, можно превратить в 4-нитробензил (2R)-2-[(3S,4R)-4-(бензотиазол-2-илдитио)-3-бром-2-оксоазетидин-1-ил]-3-метилбут-3-еноат 24 кипячением 4-нитробензил 6-бромпеницилланат-1-оксида 23 с 2-меркаптобензотиазолом (HSBT) в ароматическом растворителе (предпочтительно в толуоле).
Стадия 4. Продукт стадии 3, а именно 4-нитробензил (2R)-2-[(3S,4R)-4-(бензотиазол-2-илдитио)-3-бром-2-оксоазетидин-1-ил]-3-метилбут-3-еноат 24, можно растворить в органическом растворителе (предпочтительно в толуоле) и при взаимодействии с органическим третичным основанием (предпочтительно триэтиламином) при температуре окружающей среды получить 4-нитробензил 2-[(3S,4R)-4-(бензотиазол-2-илдитио)-3-бром-2-оксоазетидин-1-ил]-3-метилбут-2-еноат 25.
Стадия 5. Продукт стадии 4, а именно 4-нитробензил 2-[(3S,4R)-4-(бензотиазол-2-илдитио)-3-бром-2-оксоазетидин-1-ил]-3-метилбут-2-еноат 25, можно превратить в 4-нитробензил 2-[(3S,4R)-3-бром-4-формилтио-2-оксоазетидин-1-ил]-3-метилбут-2-еноат 26 взаимодействием с органической кислотой (предпочтительно муравьиной) в ароматическом органическом растворителе (предпочтительно в толуоле) в присутствии смеси уксусный ангидрид/органическое третичное основание (предпочтительно пиридин) и триалкил- или триарилфосфина (предпочтительно трифенилфосфина) при температуре от -10°С до -30°С.
Последовательное превращение соединения 23 в соединение 26 без выделения промежуточных продуктов
Продукт стадии 2, а именно 1-оксид 4-нитробензил 6-бромпеницилланата 23, подвергают взаимодействию с меркаптобензотиазолом в кипящем ароматическом органическом растворителе (предпочтительно в толуоле) в течение 1-3 ч и обрабатывают триэтиламином при температуре от 0 до -20°С в течение 3-4 ч. После данной обработки в реакционную смесь последовательно загружают органическую кислоту (предпочтительно муравьиную кислоту) и ангидрид (уксусный ангидрид), органическое третичное основание (предпочтительно пиридин) и триалкил- или триарилфосфат при температуре от -10°C до -40°C.
Стадия 6. 4-Нитробензил 2-[(3S,4R)-3-бром-4-формилтио-2-оксоазетидин-1-ил]-3-метилбут-2-еноат 26 - продукт стадии 5 или последовательного одностадийного превращения - растворяют в органическом растворителе (предпочтительно этилацетате) при температуре от -70°C до -90°С и пропускают через раствор озонированный кислород в течение 3-4 ч, затем осуществляют внутримолекулярную циклизацию с помощью фосфитного реагента (предпочтительно триметилфосфита). Продукт, 4-нитробензил (5R,6S)-6-бромпенем-3-карбоксилат 16, кристаллизуют из смеси этилацетат:гексан.
Соединение, представленное следующей формулой (16):

Вышеописанный способ (Стадия 1 - Стадия 6) получения соединения, представленного формулой 16, является промежуточным звеном в получении пенемов с замещенной метиленовой группой в положении 6 общей формулы I.
Соединение, представленное формулой 16, представляет собой кристаллическое производное (параметры дифракции рентгеновских лучей в порошкообразном образце приводятся в таблице 2).
Кристаллическая природа указанного промежуточного соединения придает ему стабильность и тем самым увеличивает срок хранения. Данные по стабильности соединения формулы 16 приведены в таблице 1.
Стадия 7. Взаимодействие 4-нитробензил (5R,6S)-6-бромпенем-3-карбоксилата 16 с соответствующим образом замещенными альдегидами (определенными выше) для осуществления стадии альдольной конденсации можно проводить в присутствии кислоты Льюиса (предпочтительно безводного MgBr2 или эфирата MgBr2)

и органического третичного основания (предпочтительно Et3N, DMAP или диизопропилэтиламина) в апротонном полярном органическом растворителе (предпочтительно ТГФ и ацетонитриле) при температуре от -20°С до -40°C. Промежуточные альдольные продукты общей формулы 18 можно функционализировать in situ ("in the same pot")при температуре от 0°C до -10°С хлорангидридом или ангидридом уксусной кислоты в ацетат, ангидридом трифторметансульфокислоты - в трифлат или тозилхлоридом - в тозилат; или, если соединения формулы 18 выделяют, их можно превратить в галогенпроизводные взаимодействием 18 с тетрагалогенметаном и трифенилфосфином при комнатной температуре в подходящем органическом растворителе, предпочтительно CH2Cl2.
Стадия 8. Последнюю стадию, а именно стадию восстановительного элиминирования, которая дает соединение общей формулы I, можно осуществить растворением смеси альдольных продуктов в органическом растворителе (предпочтительно ТГФ/ацетонитрил) и фосфатном буфере (рН 6,5-7,0) и действием активированного металла, такого как цинк, олово или алюминий, при температуре окружающей среды (предпочтительно при 20°С-35°C). Продукт в виде соли щелочного металла можно очищать колоночной хроматографией с обращенной фазой.
Альтернативно стадии 7 и 8 можно проводить последовательно в одной колбе ("in the same pot") без выделения промежуточного альдольного продукта.
Последнюю стадию, а именно стадию восстановительного элиминирования, можно также осуществить растворением альдольного продукта в органическом растворителе и фосфатном буфере с рН 6,5-7,0 и гидрированием над Pd/C при давлении Н2 10-100 фунт/кв.дюйм (предпочтительно при 40 фунт/кв.дюйм).

Экспериментальная часть
Пример 1
Получение (5R)-(Z)-6-(2,3-дигидроимидазо[2,1-b]тиазол-6-илметилен)пенем-3-карбоксилата натрия
Стадия 1. п-Нитробензил-6-бромпеницилланат
6-Аминопенициллановую кислоту (5,0 г) добавляют к охлажденному раствору метанола (44 мл), воды (14 мл) и 48 мас.% бромистоводородной кислоты (14 мл) при -10°C. После завершения прибавления смесь охлаждают до -15°С и добавляют к ней в течение 15 мин раствор нитрита натрия (2,4 г в 6,6 мл воды) и полученный раствор перемешивают без охлаждения еще в течение 30 мин, затем растворяют в нем хлорид натрия (2,4 г). Реакционную смесь экстрагируют дихлорметаном (2 × 36 мл). Объединенные органические слои промывают насыщенным солевым раствором (36 мл), сушат (MgSO4) и концентрируют до объема 20 мл при пониженном давлении и 25°C. Оставшийся раствор содержит 6-бромпенициллановую кислоту и его непосредственно используют в следующей реакции.
Безводный карбонат калия (2,9 г), диметилформамид (40 мл) и 4-нитробензилбромид (5,0 г) последовательно добавляют к полученному выше раствору и смесь перемешивают при 35-40°C в течение 1 ч. Реакционный раствор выливают в смесь воды (33 мл) и дихлорметана (41 мл) и органический слой отделяют. Органический слой промывают водой (40 мл) и насыщенным солевым раствором (40 мл), сушат (MgSO4) и упаривают. Остаток наносят на хроматографическую колонку с силикагелем, элюируют смесью этилацетат-гексан (4/1) и получают указанное в заголовке соединение в виде бесцветного твердого вещества (7,2 г, 75%).
1H ЯМР (δ, CDCl3): 1,41 (с, 3H), 1,62 (с, 3H), 4,61 (с, 1H), 4,83 (д, 1H, J=1,5 Гц), 5,25 и 5,34 (AB, 2H, J=13,0 Гц), 5,41 (д, 1H, J=1,5 Гц), 7,56 (д, 2H, J=8,7 Гц), 8,26 (д, 2H, J=8,7 Гц).

Стадия 2. п-Нитробензил-6-бромпеницилланат-1-оксид
п-Нитробензил 6-бромпеницилланат (1,36 г) растворяют в дихлорметане (20 мл) и охлаждают до 0°C. К раствору добавляют 3-хлорпербензойную кислоту (0,56 г) и перемешивают в течение 10 мин. Реакционный раствор промывают насыщенным раствором гидрокарбоната натрия, водой и насыщенным солевым раствором, сушат (MgSO4) и упаривают при пониженном давлении. Остаток наносят на хроматографическую колонку с силикагелем, элюируют смесью этилацетат-гексан (1/1) и получают указанное в заголовке соединение в виде бесцветного твердого вещества (1,4 г, количественный выход).
1H ЯМР (δ, CDCl3): 1,17 (с, 3H), 1,69 (с, 3H), 4,60 (с, 1H), 5,04 (д, 1H, J=1,5 Гц), 5,10 (д, 1H, J=1,5 Гц), 5,30 и 5,37 (AB, 2H, J=12,9 Гц), 7,56 (д, 2H, J=8,7 Гц), 8,27 (д, 2H, J=8,7 Гц).
Последовательное одностадийное получение п-нитробензил 6-бромпеницилланат-1-оксида
6-Аминопенициллановую кислоту (500 г) добавляют к охлажденному раствору метанола (3,5 л), воды (1,36 л) и 48 мас.% бромистоводородной кислоты (1,36 л) при -10°C. После завершения прибавления смесь охлаждают до -15°C. Добавляют раствор нитрита натрия (235 г в 660 мл воды) в течение 30 мин и полученный раствор перемешивают без охлаждения еще в течение 30 мин. В реакционном растворе растворяют хлорид натрия (240 г) и реакционную смесь экстрагируют дихлорметаном (2 × 3 л). Объединенные органические слои промывают насыщенным солевым раствором (3 л), сушат (MgSO4) и упаривают до объема 1,6 л при пониженном давлении и температуре 25°C. Полученный раствор содержит 6-бромпенициллановую кислоту и его непосредственно используют в следующей реакции.
Безводный карбонат калия (288 г), диметилформамид (2,8 л) и 4-нитробензилбромид (500 г) последовательно добавляют к полученному раствору и смесь перемешивают при 35-40°C в течение 1 ч. Реакционный раствор выливают в смесь воды (2,8 л) и дихлорметана (3,4 л) и органический слой отделяют. Органический слой промывают водой (2,8 л) и насыщенным солевым раствором (3 л) и охлаждают до -2°С. Органический слой содержит п-нитробензил 6-бромпеницилланат и его непосредственно используют в следующей реакции.
К органическому слою в течение 15 мин добавляют 3-хлорпербензойную кислоту (355 г) и перемешивают в течение 10 мин. Реакционную смесь разбавляют этилацетатом (4,8 л) и промывают насыщенным раствором гидрокарбоната натрия (7,6 л). Органический слой отделяют и водный слой повторно экстрагируют этилацетатом (3,9 л). Объединенные органические слои промывают водой (3,9 л) и насыщенным солевым раствором (3,9 л), сушат (MgSO4) и упаривают при пониженном давлении при температуре 30°C. Полученное твердое вещество растирают в этилацетате (1,8 л) в течение 30 мин и к смеси по каплям добавляют гексан (5,4 л) в течение 1 ч. Твердое вещество отфильтровывают, промывают на фильтре смесью этилацетат-гексан (1/4, 0,8 л), сушат на воздухе в течение 1 ч и оставляют на ночь сушиться в вакууме при комнатной температуре. Получают указанное в заголовке соединение в виде бледно-желтого кристаллического вещества (768 г, 77%).
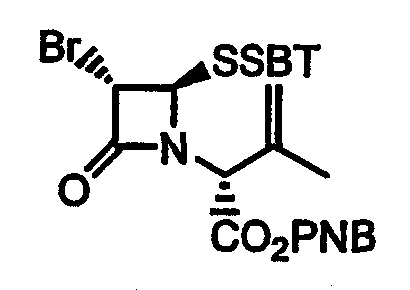
Стадия 3. п-Нитробензил (2R)-2-[(3S,4R)-4-(бензотиазол-2-илдитио)-3-бром-2-оксоазетидин-1-ил]-3-метилбут-3-еноат
п-Нитробензил 6-бромпеницилланат-1-оксид (4,31 г) и меркаптобензотиазол (1,67 г) нагревают в кипящем толуоле (14 мл) с азеотропным удалением воды (прибор Дина-Старка) в течение 1 ч. После охлаждения до комнатной температуры реакционный раствор упаривают при пониженном давлении. Остаток наносят на хроматографическую колонку с силикагелем, элюируют смесью этилацетат-гексан (2/5) и получают указанное в заголовке соединение в виде бледно-желтого твердого вещества (5,67 г, 98%).
1H ЯМР (δ, CDCl3): 1,92 (с, 3H), 4,87 (с, 1H), 5,05 (с, 1H), 5,10 (д, 1H, J=1,7 Гц), 5,21 (с, 4H), 7,39 (т, 1H, J=7,1 Гц), 7,41-7,50 (м, 3H), 7,81 (д, 1H, J=8,0 Гц), 7,90 (д, 1H, J=8,0 Гц), 8,20 (д, 2H, J=8,8 Гц).

Стадия 4. п-Нитробензил 2-[(3S,4R)-4-(бензотиазол-2-илдитио)-3-бром-2-оксоазетидин-1-ил]-3-метилбут-2-еноат
п-Нитробензил (2R)-2-[(3S,4R)-4-(бензотиазол-2-илдитио)-3-бром-2-оксоазетидин-1-ил]-3-метилбут-3-еноат (5,1 г) растворяют в толуоле (50 мл), охлаждают до 0°C и обрабатывают триэтиламином (0,12 мл). После перемешивания при 0°C в течение 2 ч реакционный раствор промывают 1M HCl, водой, насыщенным водным раствором гидрокарбоната натрия и насыщенным водным раствором хлорида натрия, сушат (MgSO4) и упаривают при пониженном давлении. Остаток наносят на хроматографическую колонку с силикагелем, элюируют смесью этилацетат-гексан (1/3) и получают указанное в заголовке соединение в виде бесцветного твердого вещества (4,16 г, 81%). Более полярная фракция содержит симметричный дисульфид в виде бесцветного твердого вещества (0,68 г, 19%).
1H ЯМР (δ, CDCl3): 2,01 (с, 3H), 2,24 (с, 3H), 5,01 (д, 1H, J=1,7 Гц), 5,24 и 5,30 (AB, 2H, J=13,1 Гц), 5,35 (д, 1H, J=1,7 Гц), 7,34-7,42 (м, 1H), 7,48-7,57 (м, 1H), 7,80 (д, 1H, J=8,0 Гц), 7,90 (д, 1H, J=8,0 Гц), 8,14 (д, 3H, J=8,7 Гц).
Симметричный дисульфид: 1H ЯМР (δ, CDCl3): 2,01 (с, 6H), 2,29 (с, 6H), 4,73 (д, 2H, J=1,7 Гц), 5,06 (д, 2H, J=1,7 Гц), 5,32 (с, 4H), 7,54 (д, 4H, J=8,6 Гц), 8,24 (д, 4H, J=8,6 Гц).

Стадия 5. п-Нитробензил 2-[(3S,4R)-3-бром-4-формилтио-2-оксоазетидин-1-ил]-3-метилбут-2-еноат
п-Нитробензил 2-[(3S,4R)-4-(бензотиазол-2-илдитио)-3-бром-2-оксоазетидин-1-ил]-3-метилбут-2-еноат (3,16 г) суспендируют в толуоле (20 мл) и охлаждают до -20°C. Добавляют последовательно муравьиную кислоту (1,10 г), уксусный ангидрид (2,45 г) и пиридин (0,44 г). Смесь охлаждают до -20°C. Трифенилфосфин (1,45 г) добавляют порциями в течение 5 мин и перемешивают при температуре от -15°С до -10°C в течение 1 ч, полученную суспензию охлаждают до -30°C и фильтруют. Твердое вещество на фильтре промывают холодным (-30°C) толуолом (15 мл). Объединенный фильтрат промывают последовательно смесью ледяной воды (8 мл) и насыщенного ледяного раствора хлорида натрия (1 мл), смесью ледяной воды (7 мл) и ледяного насыщенного раствора хлорида натрия (3 мл), холодным насыщенным раствором гидрокарбоната натрия (2 × 11 мл) и холодным насыщенным солевым раствором (11 мл). Органический слой сушат (MgSO4) и упаривают. Остаток наносят на хроматографическую колонку с силикагелем, элюируют смесью этилацетат-гексан (1/2) и указанное в заголовке соединение кристаллизуют из холодного изопропанола (10 мл). Твердое вещество отфильтровывают, промывают холодным изопропанолом (6 мл) и сушат в вакууме при комнатной температуре в течение ночи. Получают указанное в заголовке соединение в виде бесцветного кристаллического вещества (1,87 г, 77%), т.пл. 104-106°C;
1H ЯМР (δ, CDCl3): 2,00 (с, 3H), 2,30 (с, 3H), 4,82 (д, 1H, J=1,9 Гц), 5,32 и 5,38 (AB, 2H, J=13,3 Гц), 5,81 (д, 1H, J=1,9 Гц), 7,61 (д, 2H, J=8,6 Гц), 8,25 (д, 2H, J=8,6 Гц), 10,09 (с, 1H).
Последовательное одностадийное получение п-нитробензил 2-[(3S,4R)-3-бром-4-формилтио-2-оксоазетидин-1-ил]-3-метилбут-2-еноата из 1-оксида п-нитробензил-6-бромпеницилланата
п-Нитробензил 6-бромпеницилланат-1-оксид (766 г) и меркаптобензотиазол (294 г) нагревают в кипящем толуоле (2,4 л) с азеотропным удалением воды (прибор Дина-Старка) в течение 1 ч. Реакционную смесь охлаждают до 0°С и обрабатывают триэтиламином (25 мл). После перемешивания при 0°С в течение 2 ч полученную суспензию охлаждают до -20°С. Добавляют последовательно муравьиную кислоту (360 г), уксусный ангидрид (798 г) и пиридин (145 г), поддерживая температуру ниже -10°С. Смесь охлаждают до -20°С. Трифенилфосфин (473 г) добавляют порциями в течение 10 мин, поддерживая температуру между -15°C и -10°C. После перемешивания при -15˜-10°C еще в течение 1 ч полученную суспензию охлаждают до -30°C и затем фильтруют. Твердое вещество промывают холодным (-30°C) толуолом (500 мл). Объединенный фильтрат промывают последовательно смесью ледяной воды (2 л) и ледяного насыщенного раствора хлорида натрия (0,26 л), смесью ледяной воды (1,72 л) и ледяного насыщенного раствора хлорида натрия (0,8 л), холодным насыщенным раствором гидрокарбоната натрия (2 × 2,6 л) и холодным насыщенным солевым раствором (2,6 л). Органический слой сушат (MgSO4) и упаривают. Остаток наносят на хроматографическую колонку с силикагелем (6 кг), элюируют смесью этилацетат-гексан (1/2) и кристаллизуют указанное в заголовке соединение из холодного изопропанола (800 мл). Твердое вещество отфильтровывают, промывают холодным изопропанолом (400 мл), сушат на воздухе в течение 1 ч и в вакууме над P2O5 при комнатной температуре в течение ночи. Получают указанное в заголовке соединение в виде красновато-желтого кристаллического вещества (553 г, 70%)

Стадия 6. п-Нитробензил (5R,6S)-6-бромпенем-3-карбоксилат
п-Нитробензил 2-[(3S,4R)-3-бром-4-формилтио-2-оксоазетидин-1-ил]-3-метилбут-2-еноат (300 г) растворяют в этилацетате (7,5 л) и охлаждают до -70°C. Через энергично перемешиваемый раствор пропускают озонированный кислород в течение приблизительно 1,5 ч, до появления устойчивой синей окраски. Раствор продувают азотом в течение 2,5 ч и обрабатывают триметилфосфитом (384 мл) при -70°C. Смесь медленно доводят до температуры окружающей среды и продолжают перемешивание в течение 17 ч. Затем раствор осторожно нагревают до кипения и кипятят в течение 45 мин. После охлаждения до комнатной температуры смесь разбавляют этилацетатом (1,25 л) и промывают водой (2 × 2,5 л) и насыщенным солевым раствором (2,5 л). Органический слой сушат (MgSO4) и упаривают при пониженном давлении при 25°C. Твердый остаток растирают в этилацетате (0,6 л) и по каплям добавляют гексан (0,45 л) в течение 50 мин. Твердое вещество отфильтровывают и осадок на фильтре промывают смесью этилацетат-гексан (1/1, 2 × 200 мл) и смесью этилацетат-гексан (1/2, 300 мл). Затем твердое вещество сушат на воздухе в течение 1 ч и в вакууме при комнатной температуре в течение ночи. Получают указанное в заголовке соединение (109,6 г, 42%) в виде бесцветного кристаллического вещества. Т.пл. 148-151°С.
1H ЯМР (δ, CDCl3): 5,20 (д, 1H, J=0,9 Гц), 5,29 и 5,43 (АВ, 2Н, J=13,5 Гц), 5,81 (д, 1Н, J=1,5 Гц), 7,37 (д, 1H, J=0,9 Гц), 7,60 (д, 2H, J=8,7 Гц), 8,25 (д, 2H, J=8,7 Гц).

Стадия 7. Получение п-нитробензил (5R)-6-[ацетокси-(2,3-дигидроимидазо[2,1-b]тиазол-6-ил)метил]-6-бромпенем-3-карбоксилата с помощью Ph2NLi и эфирата MgBr2
Эфират MgBr2 (335 мг) добавляют к раствору п-нитробензил (5R,6S)-6-бромпенем-3-карбоксилата (385 мг) в сухом ТГФ (12 мл) в атмосфере аргона при комнатной температуре. Смесь охлаждают до -78°C. Реакционный сосуд закрывают фольгой, чтобы исключить воздействие света. Дифениламид лития, полученный прибавлением н-бутиллития (0,87 мл, 1,5 моль/л в н-гексане) к раствору дифениламина (220 мг) в сухом ТГФ (4 мл) при -20°C, добавляют в один прием к энергично перемешиваемой суспензии. Через 15-20 с энергично перемешиваемую смесь обрабатывают в один прием раствором 2,3-дигидроимидазо[2,1-b]тиазол-6-карбальдегида (170 мг) в сухом ацетонитриле (5 мл) одной порцией. Реакционную смесь перемешивают в течение 1 ч при -78°C и обрабатывают уксусным ангидридом (133 мг) одной порцией. Температуру реакционной смеси доводят до 0°C и перемешивают в течение 1 ч при 0°C. Смесь разбавляют этилацетатом и промывают водой и насыщенным солевым раствором. Органический слой сушат (MgSO4) и упаривают при пониженном давлении. Остаток наносят на хроматографическую колонку с силикагелем, элюируют смесью этилацетат-гексан (1/2˜1/1) и получают указанное в заголовке соединение в виде смеси двух диастереомеров (3/1, бесцветное аморфное твердое вещество, 447 мг, 77%).
Основной диастереоизомер: 1H ЯМР (δ, CDCl3): 2,00 (с, 3H), 3,76-3,86 (м, 2H), 4,10-4,21 (м, 2H), 5,27 и 5,45 (AB, 2H, J=13,6 Гц), 6,23 (с, 1H), 6,30 (с, 1H), 7,15 (с, 1H), 7,48 (с, 1H), 7,59-7,65 (м, 2H), 8,24 (д, 2H, J=8,6 Гц).
Минорный диастереоизомер: 1H ЯМР (δ, CDCl3): 2,24 (с, 3H), 3,76-3,86 (м, 2H), 4,10-4,21 (м, 2H), 5,27 и 5,45 (AB, 2H, J=13,6 Гц), 6,08 (с, 1H), 6,79 (с, 1H), 6,91 (с, 1H), 7,44 (с, 1H), 7,59-7,65 (м, 2H), 8,24 (д, 2H, J=8,6 Гц).
Получение п-нитробензил(5R)-6-[ацетокси(2,3-дигидроимидазо[2,1-b]тиазол-6-ил)метил]-6-бромпенем-3-карбоксилата с помощью безводного MgBr2 и Et3N
2,3-Дигидроимидазо[2,1-b]тиазол-6-карбальдегид (88 мг) добавляют к раствору безводного MgBr2 (115 мг) в сухом ацетонитриле (4 мл) в атмосфере аргона при комнатной температуре. В течение 30 мин осаждается бесцветный порошок. Добавляют раствор п-нитробензил (5R,6S)-6-бромпенем-3-карбоксилата (200 мг) в сухом ТГФ (4 мл), охлаждают до -20°C и добавляют триэтиламин (0,18 мл) в один прием. Реакционный сосуд покрывают фольгой, чтобы исключить воздействие света. Реакционную смесь перемешивают в течение 3 ч при -20°C и в один прием обрабатывают уксусным ангидридом (0,99 мл). Реакционной смеси дают согреться до 0°C и перемешивают в течение 16 ч при 0°C. Смесь разбавляют этилацетатом и промывают 5% водным раствором лимонной кислоты, насыщенным раствором гидрокарбоната натрия и насыщенным солевым раствором. Органический слой сушат над MgSO4 и фильтруют через рыхлый слой целита. Рыхлый слой промывают этилацетатом. Фильтрат концентрируют при пониженном давлении. Остаток наносят на хроматографическую колонку с силикагелем, элюируют смесью этилацетат-гексан (1/2˜1/1) и получают указанное в заголовке соединение в виде смеси двух диастереомеров (10/1, бесцветное аморфное твердое вещество, 286 мг, 90%).
Получение п-нитробензил (5R)-6-[ацетокси(2,3-дигидроимидазо[2,1-b]тиазол-6-ил)метил]-6-бромпенем-3-карбоксилата с помощью эфирата MgBr2 и Et3N
Эфират MgBr2 используют в реакции вместо безводного MgBr2 и выделяют продукт с 84% выходом.

Стадия 8. (5R)-(Z)-6-(2,3-Дигидроимидазо[2,1-b]тиазол-6-илметилен)пенем-3-карбоксилат натрия
п-Нитробензил (5R)-6-[ацетокси(2,3-дигидроимидазо[2,1-b]тиазол-6-ил)метил]-6-бромпенем-3-карбоксилат (500 мг) растворяют в ТГФ (7,0 мл) и ацетонитриле (3,2 мл). Быстро добавляют свежеактивированную цинковую пыль (2,0 г) с 0,5M фосфатным буфером (pH 6,5, 10,2 мл). Реакционный сосуд покрывают фольгой, чтобы исключить воздействие света. Реакционную смесь энергично перемешивают в течение 2 ч при комнатной температуре. Смесь разбавляют водой, охлаждают до 3°С и добавляют 1M NaOH, чтобы довести pH до 8. Реакционный раствор смешивают с этилацетатом и фильтруют через рыхлый слой целита (Celite), который промывают водой и водный слой отделяют. Водный слой содержит 224 мг (79%) указанного в заголовке соединения (определено ВЭЖХ), его концентрируют в высоком вакууме при 35°C. Концентрат наносят на хроматографическую колонку со смолой Daiaion HP-21 (20 мл, Mitsubishi Kasei Co. Ltd.). После абсорбции колонку элюируют водой, затем смесью 10% ацетонитрил-вода и получают очищенные активные фракции соединения, указанного в заголовке. Объединенные фракции концентрируют в высоком вакууме при 35°C, лиофилизуют и получают указанное в заголовке соединение в виде желтого аморфного твердого вещества (213 мг, 75%). Данное аморфное твердое вещество растворяют в воде (1,9 мл), к раствору добавляют ацетон (8 мл) при перемешивании при комнатной температуре, охлаждают до 3°C, затем перемешивают в течение 30 мин. Ацетон добавляют к смеси порциями по 2 мл каждые 30 мин в течение 4 ч при 3°C (всего добавляют 16 мл ацетона). После перемешивания в течение 1 ч при 3°C, твердое вещество отфильтровывают и осадок на фильтре промывают ацетоном (2 мл). Твердое вещество сушат на воздухе, в отсутствие света в течение 1 ч, затем в вакууме при комнатной температуре в течение ночи и получают указанное в заголовке соединение (200 мг, 71%) в виде желтого кристаллического вещества.
1H ЯМР (δ, D2O): 3,82 (д, 2H, J=7,4 Гц), 4,14-4,20 (м, 2H), 6,42 (с, 1H), 6,82 (с, 1H), 6,96 (с, 1H), 7,42 (с, 1H).
Последовательное получение (5R)-(Z)-6-(2,3-дигидроимидазо[2,1-b]тиазол-6-илметилен)пенем-3-карбоксилата натрия из п-нитробензил (5R)-6-[ацетокси(2,3-дигидроимидазо[2,1-b]тиазол-6-ил)метил]-6-бромпенем-3-карбоксилата
п-Нитробензил (5R)-6-[ацетокси(2,3-дигидроимидазо[2,1-b]тиазол-6-ил)метил]-6-бромпенем-3-карбоксилат (110,1 г) добавляют к раствору безводного MgBr2 (143,4 г) в сухом ацетонитриле (5 л) в атмосфере аргона при комнатной температуре. Бесцветный порошок осаждается в течение 30 мин. В полученную суспензию добавляют раствор п-нитробензил 6-бромпеницилланат-1-оксида (250 г) в сухом ТГФ (5 л) через ПТФЭ трубку при положительном давлении аргона в течение 12 мин. После охлаждения реакционной смеси до -20°C добавляют за один прием триэтиламин (217 мл). Реакционный сосуд покрывают фольгой, чтобы исключить воздействие света. Реакционную смесь перемешивают в течение 3 ч при -20°C и обрабатывают за один прием уксусным ангидридом (133 г). Реакционную смесь подогревают до 0°C и перемешивают в течение 16 ч при 0°C. Смесь разбавляют этилацетатом (20 л) и промывают 5% водным раствором лимонной кислоты (10 л), насыщенным раствором гидрокарбоната натрия (10 л) и насыщенным солевым раствором (10 л). Органический слой сушат (MgSO4) и фильтруют через рыхлый слой целита. Рыхлый слой промывают этилацетатом (1,4 л). Фильтрат концентрируют при пониженном давлении при 30°C. Остаток в виде коричневого масла содержит 327 г (86,7%) альдольного продукта (по данным ВЭЖХ).
Полученный продукт в виде коричневого масла растворяют в ТГФ (5,4 л) и ацетонитриле (2,5 л). Быстро добавляют свежеактивированную цинковую пыль (1,5 кг) с 0,5 Mфосфатным буфером (pH 6,5, 7,9 л). Реакционный сосуд покрывают фольгой, чтобы исключить воздействие света. Реакционную смесь энергично перемешивают в течение 2,5 ч, поддерживая температуру около 35°C, затем охлаждают до 3°C и добавляют ледяной 1 Mраствор NaOH (приблизительно 1750 мл) для доведения pH до 8 при температуре ниже 3°C. Смесь разбавляют этилацетатом (4 л) и фильтруют через рыхлый слой целита. Рыхлый слой промывают водой (2,5 л) и отделяют водный слой, который содержит 137 г (64%) указанного в заголовке соединения (по данным ВЭЖХ), водный слой концентрируют до 8,85 кг (около 8 л) в высоком вакууме при 35°C.
Концентрат наносят на хроматографическую колонку с Sepabeads SP-207 (7 л, Mitsubishi Kasei Co. Ltd.). После абсорбции осуществляют элюирование водой (14 л), затем смесью 20% ацетонитрил-вода и получают фракции очищенного (5R)-(Z)-6-(2,3-дигидроимидазо[2,1-b]тиазол-6-илметилен)пенем-3-карбоксилата натрия.
Объединенные фракции концентрируют в высоком вакууме при 35°C до 1,25 кг (примерно 1 л, содержит 130 г (61%) (5R)-(Z)-6-(2,3-дигидроимидазо[2,1-b]тиазол-6-илметилен)пенем-3-карбоксилата натрия (по данным ВЭЖХ). К концентрату добавляют ацетон (2,5 л) при перемешивании при комнатной температуре. Смесь охлаждают до 3°C, затем перемешивают в течение 30 мин. Ацетон добавляют к смеси порциями по 625 мл каждые 30 мин в течение 4 ч при 3°C (всего добавляют 5 л ацетона). После перемешивания в течение 1 ч при 3°C твердое вещество отфильтровывают и осадок на фильтре промывают ацетоном (800 мл). Твердое вещество сушат на воздухе в отсутствие света в течение 1 ч, затем в вакууме при комнатной температуре в течение ночи и получают указанное в заголовке соединение (112 г, 50%) из п-нитробензил (5R)-6-[ацетокси(2,3-дигидроимидазо[2,1-b]тиазол-6-ил)метил]-6-бромпенем-3-карбоксилата в виде желтого кристаллического вещества.
(5R),(Z)-6-(2,3-Дигидроимидазо[2,1-b]тиазол-6-илметилен)пенем-3-карбоксилат натрия из 4-нитробензил (5R)-6-[ацетокси(2,3-дигидроимидазо[2,1-b]тиазол-6-ил)метил]-6-бромпенем-3-карбоксилата по методике гидрогенолиза
4-Нитробензил (5R)-6-[ацетокси(2,3-дигидроимидазо[2,1-b]тиазол-6-ил)метил]-6-бромпенем-3-карбоксилат (116 мг) и 10% Pd/C (50% влаги, 58 мг) добавляют в смесь ТГФ (4 мл) и 0,5М фосфатного буфера с pH 6,5 (4 мл). Полученную смесь гидрируют при давлении водорода 40 фунтов/кв.дюйм при комнатной температуре в течение 1 ч. Реакционную смесь фильтруют, охлаждают до 3°С и добавляют 1M NaOH для доведения рН до 8. Смесь опять фильтруют и фильтрат промывают этилацетатом. Объединенный водный слой концентрируют при пониженном давлении и температуре 35°С и наносят на хроматографическую колонку со смолой Diaion HP-21 (7 мл, Mitsubishi Kasei Co. Ltd). После адсорбции осуществляют элюирование водой, затем 10% раствором ацетонитрила в воде. Объединенные фракции, содержащие чистый продукт, концентрируют при пониженном давлении при 35°C, лиофилизуют и получают указанное в заголовке соединение в виде желтого аморфного твердого вещества.
Пример 2
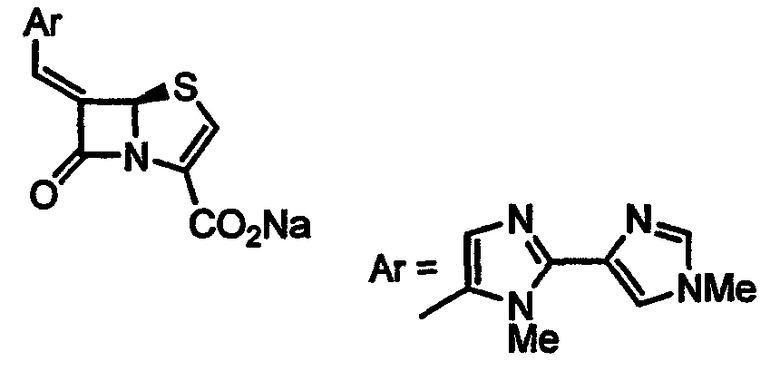
Стадия 1. 3,1′-Диметил-3H,1'H-[2,4']биимидазолил-4-карбальдегид
трет-Бутоксид калия (3,23 г) добавляют к смеси раствора 1Н,1'H-[2,4']биимидазолил-4-карбальдегида (1,95 г) в сухом ДМФА (140 мл) и 18-краун-6 (634 мг) при 0°C. Реакционную смесь перемешивают 10 мин и обрабатывают метилиодидом (1,85 мл). После перемешивания в течение 30 мин при 0°C и затем в течение 17 ч при комнатной температуре реакционную смесь концентрируют при пониженном давлении. Остаток растворяют в смеси H2O-CHCl3 и разделяют. Водный слой экстрагируют хлороформом. Объединенные органические слои сушат (MgSO4) и фильтруют. Фильтрат упаривают при пониженном давлении. Сырой продукт очищают колоночной хроматографией на силикагеле (соотношение хлороформ-метанол составляет 9/1). Указанное в заголовке соединение (887 мг, 39%) и его позиционный изомер 1,1'-диметил-1H,1'H-[2,4']биимидазолил-4-карбальдегид (296 мг, 13%) получают в виде бледно-розового твердого вещества.
3,1′-Диметил-3H,1′H-[2,4′]биимидазолил-4-карбальдегид: 1H ЯМР (δ, CDCl3) 3,78 (с, 3H), 4,37 (с, 3H), 7,51 (д, 1H, J=1,0 Гц), 7,62 (д, 1H, J=1,0 Гц), 7,76 (с, 1H), 9,71 (с, 1H).
1,1′-Диметил-1H,1′H-[2,4′]биимидазолил-4-карбальдегид: 1H ЯМР (δ, CDCl3) 3,76 (с, 3H), 4,10 (с, 3H), 7,47 (д, 1H, J=0,9 Гц), 7,57 (с, 1H), 7,59 (д, 1H J=0,9 Гц), 9,85 (с,1H).
Стадия 2. 4-нитробензиловый эфир (5R,6RS)-6-[(RS)-ацетокси(3,1′-диметил-3H,1′H-[2,4′]биимидазол-4-ил)метил]-6-бром-7-оксо-4-тиа-1-азабицикло[3.2.0]гепт-2-ен-2-карбоновой кислоты
3,1′-Диметил-3H,1′H-[2,4′]биимидазолил-4-карбальдегид (171 мг) добавляют к раствору безводного MgBr2 (800 мг) в сухом ацетонитриле (20 мл) в атмосфере азота при комнатной температуре. К полученной смеси добавляют раствор 4-нитробензилового эфира (5R,6S)-6-бром-7-оксо-4-тиа-1-азабицикло[3.2.0]гепт-2-ен-2-карбоновой кислоты (346 мг) в сухом ТГФ (20 мл), охлаждают до -20°C и добавляют в один прием триэтиламин (0,768 мл). Реакционный сосуд покрывают фольгой, чтобы исключить воздействие света. Реакционную смесь перемешивают 2 ч при -20°C и обрабатывают за один прием 4-диметиламинопиридином (22 мг) и уксусным ангидридом (0,17 мл). Реакционную смесь доводят до 0°C и перемешивают в течение 17 ч при 0°C. Смесь разбавляют этилацетатом и промывают 5% водным раствором лимонной кислоты, насыщенным раствором гидрокарбоната натрия и насыщенным солевым раствором. Органический слой сушат (MgSO4) и фильтруют. Фильтрат концентрируют при пониженном давлении. Остаток наносят на хроматографическую колонку с силикагелем, затем элюируют смесью хлороформ-метанол (9/1). Получают указанное в заголовке соединение (507,6 мг, 91,4%).
1H ЯМР (δ, CDCl3): 2,19 (с, 3H), 3,75 (с, 3H), 4,03 (с, 3H), 5,29 (д, 1H, J=13,5 Гц), 5,42 (д, 1H, J=13,5 Гц), 6,03 (с, 1H), 6,65 (с, 1H), 7,34 (с, 1H), 7,48 (д, 1H, J=1,2 Гц), 7,55 (д, 1H, J=1,2 Гц), 7,59-7,61 (м, 3H), 8,23-8,26 (м, 2H).
Стадия 3. Натриевая соль (5R),(6Z)-6-(3,1′-диметил-3H,1′H-[2,4′]биимидазол-4-илметилен)-7-оксо-4-тиа-1-азабицикло[3.2.0]гепт-2-ен-2-карбоновой кислоты
4-Нитробензиловый сложный эфир (5R,6RS)-6-[(RS)-ацетокси(3,1′-диметил-3H,1′H-[2,4′]биимидазол-4-ил)метил]-6-бром-7-оксо-4-тиа-1-азабицикло[3.2.0]гепт-2-ен-2-карбоновой кислоты (596 мг) растворяют в смеси ТГФ (8,3 мл) и ацетонитрила (3,9 мл). К полученной смеси добавляют свежеактивированную цинковую пыль (2,38 г) и 0,5Mфосфатный буфер (pH 6,5, 12,2 мл). Реакционный сосуд покрывают фольгой, чтобы исключить воздействие света. Реакционную смесь энергично перемешивают в течение 2 ч при комнатной температуре. Реакционный раствор смешивают с этилацетатом и фильтруют через рыхлый слой целита. Рыхлый слой промывают водой, водный слой отделяют и концентрируют в высоком вакууме при 35°C. Концентрат наносят на хроматографическую колонку со смолой Diaion HP-21 (60 мл, Mitsubishi Kasei Co. Ltd.). После адсорбции осуществляют элюирование водой, затем 5%-10% ацетонитрилом в воде. Объединенные фракции концентрируют в высоком вакууме при 35°C, лиофилизуют и получают указанное в заголовке соединение в виде желтого аморфного твердого вещества (66,3 мг, 19%). Т.пл. 113°C (с разложением).
1H ЯМР (δ, D2O): 3,31 (с, 3H), 3,52 (с, 3H), 5,74 (с, 1H), 6,43 (с, 2H), 6,70 (с, 1H), 7,27 (с, 1H), 7,46 (s, 1H).
Пример 3

Стадия 1. 2-Бензил-1Н-имидазол-4-карбальдегид
2-Фенилацетамидин (2 г) добавляют к раствору 2-бром-3-изопропоксипропеналя (2,88 г) в CHCl3 (30 мл). Реакционную смесь перемешивают в течение 1 ч при комнатной температуре. Затем к смеси добавляют триэтиламин (2,09 мл), нагревают и кипятят с обратным холодильником в течение 6 ч. Смесь охлаждают до комнатной температуры, разбавляют CHCl3 и промывают 10% раствором гидрокарбоната калия. Органический слой сушат (MgSO4) и концентрируют при пониженном давлении. Остаток наносят на хроматографическую колонку с силикагелем, затем элюируют смесью этилацетат-гексан (3/1). Неочищенное соединение кристаллизуют из этилацетата и н-гексана и получают указанное в заголовке соединение (1,10 г, 40%).
1H ЯМР (δ, CDCl3): 4,17 (с, 2H), 7,24-7,36 (м, 5H), 7,72 (уш.с, 1H), 9,60 (уш.с, 1H), 10,28 (уш.с, 1H).
Стадия 2. 4-Нитробензиловый эфир 2-бензил-4-формилимидазол-1-карбоновой кислоты
2-Бензил-1H-имидазол-4-карбальдегид (1 г) и гидрокарбонат натрия (1,13 г) растворяют в диоксане (30 мл) и воде (30 мл). К полученному раствору добавляют 48,7% раствор п-нитробензилхлорформиата (PNZCl) в диоксане (2,62 г) при 0°C и перемешивают в течение 2,5 ч. Реакционную смесь разбавляют этилацетатом, промывают насыщенным раствором гидрокарбоната натрия и насыщенным солевым раствором. Органический слой сушат (MgSO4) и концентрируют при пониженном давлении. Остаток наносят на хроматографическую колонку с силикагелем, затем элюируют смесью хлороформ-ацетон (9/1). Получают указанное в заголовке соединение в виде светло-коричневого масла (1,97 г, 100%).
1H ЯМР (δ, CDCl3): 4,46 (с, 2H), 5,42 (с, 2H), 7,19-7,30 (м, 5H), 7,44-7,47 (м, 2H), 8,06 (с, 1H), 8,21-8,25 (м, 2H), 9,91 (с, 1H).
Стадия 3. 4-Нитробензиловый эфир (5R,6RS)-6-{(RS)-ацетокси[2-бензил-1-(4-нитробензилоксикарбонил)-1Н-имидазол-4-ил]метил}-6-бром-7-оксо-4-тиа-1-азабицикло[3.2.0]гепт-2-ен-2-карбоновой кислоты
Раствор 4-нитробензилового эфира 2-бензил-4-формилимидазол-1-карбоновой кислоты (1,9 г) в сухом ацетонитриле (10 мл) добавляют к раствору безводного MgBr2 (2,41 г) в сухом ацетонитриле (50 мл) в атмосфере азота при комнатной температуре. К полученной смеси добавляют раствор 4-нитробензилового эфира (5R,6S)-6-бром-7-оксо-4-тиа-1-азабицикло[3.2.0]гепт-2-ен-2-карбоновой кислоты (2 г) в сухом ТГФ (60 мл), охлаждают до -20°C и добавляют триэтиламин (1,8 мл) в один прием. Реакционный сосуд покрывают фольгой, чтобы исключить воздействие света. Реакционную смесь перемешивают в течение 2 ч при -20°C и обрабатывают 4-диметиламинопиридином (127 мг) и уксусным ангидридом (0,98 мл) за один прием. Реакционную смесь отогревают до 0°C и перемешивают в течение 16 ч при 0°C. Смесь разбавляют этилацетатом и промывают H2O, насыщенным раствором гидрокарбоната натрия и насыщенным солевым раствором. Органический слой сушат (MgSO4) и фильтруют через рыхлый слой целита. Рыхлый слой промывают этилацетатом. Фильтрат концентрируют при пониженном давлении. Остаток наносят на хроматографическую колонку с силикагелем, осуществляют элюирование водой, затем смесью этилацетат-гексан (2/3˜1/1). Указанное в заголовке соединение получают в виде смеси двух диастереомеров (3/2), которая представляет собой бледно-желтое аморфное твердое вещество (2,8 г, 70%).
1H ЯМР (δ, CDCl3): 2,00 (с, 0,6 × 3H), 2,26 (с, 0,4 × 3H), 4,34-4,37 (м, 2H), 5,25-5,47 (м, 4H), 6,02 (с, 0,4 × 1Н), 6,22 (с, 0,6 × 1Н), 6,25 (с, 0,6 × 1Н), 6,85 (д, 0,4 × 1H, J=1,5 Гц), 7,14-7,62 (м, 11H), 8,21-8,25 (м, 4H).
Стадия 4. Натриевая соль (5R),(6Z)-6-(2-Бензил-1H-имидазол-4-илметилен)-7-оксо-4-тиа-1-азабицикло[3.2.0]гепт-2-ен-2-карбоновой кислоты
4-нитробензиловый эфир (5R,6RS)-6-{(RS)-ацетокси[2-бензил-1-(4-нитробензилоксикарбонил)-1Н-имидазол-4-ил]метил}-6-бром-7-оксо-4-тиа-1-азабицикло[3.2.0]гепт-2-ен-2-карбоновой кислоты (2,5 г) растворяют в ТГФ (35 мл) и ацетонитриле (16,3 мл). К полученной смеси добавляют свежеактивированную цинковую пыль (10 г) и 0,5Mфосфатный буфер с pH 6,4 (51,3 мл). Реакционный сосуд покрывают фольгой, чтобы исключить воздействие света. Реакционную смесь энергично перемешивают в течение 2 ч при комнатной температуре. Смесь охлаждают до 9°C и добавляют 1Mводный раствор NaOH, чтобы довести рН до 7,5. Реакционный раствор смешивают с этилацетатом и фильтруют через рыхлый слой целлита, слой промывают водой, отделяют водный слой и концентрируют его в высоком вакууме при 35°C. Концентрат наносят на хроматографическую колонку со смолой Diaion HP-21 (250 мл, Mitsubishi Kasei Co. Ltd.). После адсорбции осуществляют элюирование водой, затем смесью 5-10% ацетонитрил-вода. Объединенные фракции концентрируют в высоком вакууме при 35°C, лиофилизуют и получают указанное в заголовке соединение в виде желтого аморфного твердого вещества (780 мг, 67%). Т.пл.146°C (с разложением).
1H ЯМР (δ, D2O): 3,92 (с, 2H), 6,39 (д, 1H, J=0,8 Гц), 6,74 (с, 1H), 6,89 (с, 1H), 7,13-7,16 (м, 3H), 7,21-7,25 (м, 3H).
Пример 4
Стадия 1. 4-Нитробензиловый эфир 4-формил-2-тиазол-2-илимидазол-1-карбоновой кислоты
2-Тиазол-2-ил-1H-имидазол-4-карбальдегид (570 мг) растворяют в сухом CH2Cl2 (35 мл). К полученному раствору при 0°С последовательно добавляют N,N'-диизопропилэтиламин и 48,7% раствор п-нитробензилхлороформиата (PNZCl) в диоксане (1,69 мл) и перемешивают в течение 2 ч. Реакционную смесь разбавляют CHCl3 и промывают насыщенным раствором гидрокарбоната натрия и насыщенным солевым раствором. Органический слой сушат (MgSO4) и концентрируют при пониженном давлении. Остаток кристаллизуют из этилацетата и н-гексана и получают указанное в заголовке соединение (1,05 г, 92%).
1H ЯМР (δ, CDCl3): 5,49 (с, 2H), 7,50 (д, 2H, J=8,8 Гц), 7,55 (д, 1H, J=3,4 Гц), 7,89 (д, 1H, J=3,1 Гц), 8,19 (с, 1H), 8,24 (дкв, 2H, J=8,8 и 2,0 Гц), 9,97 (с, 1H).
Стадия 2. Натриевая соль (5R),(6Z)-6-(2-тиазол-2-ил-1H-имидазол-4-илметилен)-7-оксо-4-тиа-1-азабицикло[3.2.0]гепт-2-ен-2-карбоновой кислоты
4-Нитробензиловый эфир 4-формил-2-тиазол-2-илимидазол-1-карбоновой кислоты (940 мг) добавляют к раствору безводного MgBr2 (1,26 г) в сухом ацетонитриле (35 мл) в атмосфере азота при комнатной температуре. К полученной смеси добавляют раствор 4-нитробензилового эфира (5R,6S)-6-бром-7-оксо-4-тиа-1-азабицикло[3.2.0]гепт-2-ен-2-карбоновой кислоты (1,01 г) в сухом ТГФ (28 мл), охлаждают до -20°C и в один прием добавляют триэтиламин (0,942 мл). Реакционный сосуд покрывают фольгой, чтобы исключить воздействие света. Реакционную смесь перемешивают в течение 2,8 ч при -20°C и обрабатывают 4-диметиламинопиридином (64 мг) и уксусным ангидридом (0,495 мл) в один прием. Реакционную смесь оставляют согреваться до 0°C и перемешивают в течение 20 ч при 0°C. Смесь разбавляют этилацетатом и промывают 0,1M фосфатным буфером (pH 7), насыщенным раствором гидрокарбоната натрия и насыщенным солевым раствором. Органический слой сушат (MgSO4) и фильтруют через рыхлый слой целита. Рыхлый слой промывают этилацетатом. Фильтрат концентрируют при пониженном давлении. Остаток подвергают грубой очистке колоночной хроматографией на силикагеле (этилацетат-гексан 1/1˜чистый этилацетат) и получают сырой 4-нитробензиловый эфир (5R,6RS)-6-{(RS)-ацетокси[1-(4-нитробензилоксикарбонил)-2-тиазол-2-ил-1H-имидазол-4-ил]метил}-6-бром-7-оксо-4-тиа-1-азабицикло[3.2.0]гепт-2-ен-2-карбоновой кислоты (860 мг).
Сырой 4-нитробензиловый эфир (5R,6RS)-6-{(RS)-ацетокси[1-(4-нитробензилоксикарбонил)-2-тиазол-2-ил-1H-имидазол-4-ил]метил}-6-бром-7-оксо-4-тиа-1-азабицикло[3.2.0]гепт-2-ен-2-карбоновой кислоты (860 мг) растворяют в ТГФ (12 мл) и ацетонитриле (5,6 мл). К полученному раствору добавляют свежеактивированную Zn пыль (3,44 г) и 0,5Mфосфатный буфер (рН 6,4, 17,6 мл). Реакционный сосуд покрывают фольгой, чтобы исключить воздействие света. Реакционную смесь энергично перемешивают в течение 2 ч при комнатной температуре. Смесь смешивают с этилацетатом и фильтруют через рыхлый слой целита. Рыхлый слой промывают водой, отделяют водный слой и концентрируют его в высоком вакууме при 35°C. Концентрат наносят на хроматографическую колонку со смолой Diaion HP-21 (90 мл, Mitsubishi Kasei Co. Ltd.). После адсорбции осуществляют элюирование водой, затем 2,5-5% раствором ацетонитрила в воде. Объединенные фракции концентрируют в высоком вакууме при 35°C, лиофилизуют и получают указанное в заголовке соединение в виде желтого аморфного твердого вещества (126 мг, 11% из 7). Т.пл. 145°C (с разложением).
1H ЯМР (δ, D2O): 6,40 (д, 1H, J=0,7 Гц), 6,81 (с, 1H), 6,89 (с, 1H), 7,38 (с, 1H), 7,48 (д, 1H, J=3,1 Гц), 7,71 (д, 1H, J=3,3 Гц).
Пример 5

Стадия 1. 1-Метил-2-тиазол-2-ил-1Н-имидазол-4-карбальдегид
К раствору 2-тиазол-2-ил-1Н-имидазол-4-карбальдегида (717 мг) и 18-краун-6 (106 мг) в сухом ТГФ (30 мл) и сухом ДМФА (10 мл) добавляют трет-бутоксид калия (494 мг) при комнатной температуре. Реакционную смесь перемешивают в течение 10 мин и обрабатывают метилиодидом (0,274 мл). После перемешивания в течение 17,5 ч при комнатной температуре смесь концентрируют при пониженном давлении. Остаток растворяют в этилацетате и фильтруют. Фильтрат упаривают при пониженном давлении. Сырое вещество очищают колоночной хроматографией на силикагеле (этилацетат-гексан, 1/1). Указанное в заголовке соединение (410 мг, 53%) и его позиционный изомер 1-метил-2-тиазол-2-ил-1Н-имидазол-5-карбальдегид (240 мг, 31%) получают в виде белого твердого вещества.
1-Метил-2-тиазол-2-ил-1H-имидазол-4-карбальдегид: 1H ЯМР (δ, CDCl3): 4,21 (с, 3H), 7,44 (д, 1H, J=3,2 Гц), 7,68 (с, 1H), 7,89 (д, 1H, J=3,2 Гц), 9,91 (с, 1H).
1-Метил-2-тиазол-2-ил-1H-имидазол-5-карбальдегид: 1H ЯМР (δ, CDCl3) 4,48 (с, 3Н), 7,51 (д, 1Н, J=3,1 Гц), 7,82 (с, 1H), 7,97 (д, 1H, J=3,1 Гц), 9,82 (с, 1H).
Стадия 2. 4-нитробензиловый эфир (5R,6RS)-6-[(RS)-ацетокси(1-метил-2-тиазол-2-ил-1H-имидазол-4-ил)метил]-6-бром-7-оксо-4-тиа-1-азабицикло[3.2.0]гепт-2-ен-2-карбоновой кислоты
1-Метил-2-тиазол-2-ил-1Н-имидазол-4-карбальдегид (380 мг) добавляют к раствору безводного MgBr2 (1,03 г) в сухом ацетонитриле (28 мл) в атмосфере азота при комнатной температуре. В течение 10 мин выпадает в осадок бесцветный порошок. К полученной смеси добавляют раствор 4-нитробензилового эфира (5R,6S)-6-бром-7-оксо-4-тиа-1-азабицикло[3.2.0]гепт-2-ен-2-карбоновой кислоты (760 мг) в сухом ТГФ (28 мл), охлаждают до -20°C и в один прием добавляют триэтиламин (0,768 мл). Реакционный сосуд покрывают фольгой, чтобы исключить воздействие света. Реакционную смесь перемешивают в течение 2 ч при -20°C и обрабатывают в один прием 4-диметиламинопиридином (48 мг) и уксусным ангидридом (0,371 мл). Реакционной смеси дают согреться до 0°C и перемешивают в течение 22 ч при 0°C. Смесь разбавляют этилацетатом и промывают 5% водным раствором лимонной кислоты, насыщенным раствором гидрокарбоната натрия и насыщенным солевым раствором. Органический слой сушат (MgSO4) и фильтруют через рыхлый слой целита. Рыхлый слой промывают этилацетатом. Фильтрат концентрируют при пониженном давлении. Остаток наносят на хроматографическую колонку с силикагелем, затем элюируют смесью этилацетат-гексан (3/2). Указанное в заголовке соединение получают в виде смеси двух диастереомеров (1/1), которая представляет собой бледно-желтое аморфное твердое вещество (673,4 мг, 52%).
1H ЯМР (δ, CDCl3): 2,00 (с, 0,5 × 3H), 2,28 (с, 0,5 × 3H), 4,12 (с, 3H), 5,28 (д, 0,5 × 1H, J=13,5 Гц), 5,29 (д, 0,5 × 1H, J=13,5 Гц), 5,44 (д, 0,5 × 1H, J=13,5 Гц), 5,47 (д, 0,5 × 1H, J=13,5 Гц), 6,16 (с, 0,5 × 1H), 6,30 (с, 0,5 × 1H), 6,42 (с, 0,5 × 1H), 6,87 (д, 0,5 × 1H, J=0,8 Гц), 6,92 (д, 0,5 × 1H, J=0,8 Гц), 7,19 (с, 0,5 × 1H), 7,34-7,36 (м, 1H), 7,47 (с, 0,5 × 1H), 7,50 (с, 0,5 × 1H), 7,60-7,64 (м, 2H), 7,83-7,85 (м, 1H), 8,23-8,26 (м, 1H).
Стадия 3. Натриевая соль (5R),(6Z)-6-(1-метил-2-тиазол-2-ил-1H-имидазол-4-илметилен)-7-оксо-4-тиа-1-азабицикло[3.2.0]гепт-2-ен-2-карбоновой кислоты
4-Нитробензиловый эфир (5R,6RS)-6-[(RS)-ацетокси(1-метил-2-тиазол-2-ил-1H-имидазол-4-ил)метил]-6-бром-7-оксо-4-тиа-1-азабицикло[3.2.0]гепт-2-ен-2-карбоновой кислоты (604 мг) растворяют в ТГФ (8,5 мл) и ацетонитриле (3,9 мл). К полученной смеси добавляют свежеактивированную Zn пыль (2,41 г) и 0,5Mфосфатный буфер с pH 6,4 (23,6 мл). Реакционный сосуд покрывают фольгой, чтобы исключить воздействие света. Реакционную смесь энергично перемешивают в течение 2 ч при комнатной температуре. Реакционный раствор смешивают с этилацетатом и фильтруют через рыхлый слой целита. Рыхлый слой промывают водой, отделяют водный слой и концентрируют его в высоком вакууме при 35°C. Концентрат наносят на хроматографическую колонку со смолой Diaion HP-21 (60 мл, Mitsubishi Kasei Co. Ltd.). После адсорбции осуществляют элюирование водой, затем 5% и 10% раствором ацетонитрила в воде. Объединенные фракции концентрируют в высоком вакууме при 35°C, лиофилизуют и получают указанное в заголовке соединение в виде желтого аморфного твердого вещества (231 мг, 64%). Т.пл. 214°C (с разложением).
1H ЯМР (δ, D2O): 3,91 (с, 3H), 6,44 (с, 1H), 6,84 (с, 1H), 6,91 (с, 1H), 7,43 (с, 1H), 7,60 (д, 1H, J=3,3 Гц), 7,84 (д, 1H, J=3,3 Гц).
Пример 6
Стадия 1. 4-Нитробензиловый эфир 4-формил-2-фенилимидазол-1-карбоновой кислоты
4-Формил-2-фенилимидазол (624 мг) и гидрокарбонат натрия (791 мг) растворяют в диоксане (3,6 мл), ТГФ (3,6 мл) и воде (7,2 мл). К полученной смеси добавляют 48,7% раствор п-нитробензилхлорформиата (PNZCl) в диоксане (2,08 мл) при комнатной температуре и перемешивают в течение 2,5 ч. Смесь разбавляют этилацетатом и промывают насыщенным солевым раствором. Органический слой сушат (MgSO4) и концентрируют при пониженном давлении. Остаток кристаллизуют из этилацетата и н-гексана и получают указанное в заголовке соединение (956 мг, 75%).
1H ЯМР (δ, CDCl3): 5,41 (с, 2H), 7,32 (д, 2Н, J=8,6 Гц), 7,40-7,51 (м, 3H), 7,56-7,58 (м, 2H), 8,17-8,20 (м, 2H), 8,22 (с, 1H), 9,97 (с, 1H).
Стадия 2. 4-Нитробензиловый эфир (5R,6RS)-6-{(RS)-ацетокси[1-(4-нитробензилоксикарбонил)-2-фенил-1Н-имидазол-4-ил]метил}-6-бром-7-оксо-4-тиа-1-азабицикло[3.2.0]гепт-2-ен-2-карбоновой кислоты
4-Нитробензиловый эфир 4-формил-2-фенилимидазол-1-карбоновой кислоты (568 мг) и раствор 4-нитробензилового эфира (5R,6S)-6-бром-7-оксо-4-тиа-1-азабицикло[3.2.0]гепт-2-ен-2-карбоновой кислоты (587 мг) в сухом ТГФ (15 мл) последовательно добавляют к раствору безводного MgBr2 (622,4 мг) в сухом ацетонитриле (15 мл) в атмосфере азота при комнатной температуре. После охлаждения смеси до -20°C добавляют триэтиламин (0,494 мл) в один прием. Реакционный сосуд покрывают фольгой, чтобы исключить воздействие света. Реакционную смесь перемешивают в течение 1,5 ч при -20°C и обрабатывают в один прием уксусным ангидридом (0,277 мл). Температуру реакционной смеси доводят до 0°C и перемешивают в течение 27 ч при 0°C. Смесь разбавляют этилацетатом и промывают 5% водным раствором лимонной кислоты, насыщенным раствором гидрокарбоната натрия и насыщенным солевым раствором. Органический слой сушат (MgSO4) и фильтруют через рыхлый слой целита. Рыхлый слой промывают этилацетатом. Фильтрат концентрируют при пониженном давлении. Остаток наносят на хроматографическую колонку с силикагелем, затем элюируют смесью этилацетат-гексан (2/3˜1/1). Указанное в заголовке соединение получают в виде смеси трех диастереомеров (6/4/1,5), которая представляет собой бледно-желтое аморфное твердое вещество (986 мг, 86%).
1H ЯМР (δ, CDCl3): 2,04 (с, 0,35 × 3H), 2,10 (с, 0,13 × 3H), 2,30 (с, 0,52 × 3H), 5,25-5,43 (м, 4H), 6,09 (с, 0,52 × 1H), 6,22 (с, 0,13 × 1H), 6,31 (с, 0,35 × 1H), 6,33 (с, 0,35 × 1H), 6,87 (с, 0,13 × 1H), 6,92 (д, 0,52 × 1H, J=1,4 Гц), 7,31-7,76 (м, 11H), 8,17-8,25 (м, 4H).
Стадия 3. (5R),(6Z)-6-(2-Фенил-1Н-имидазол-4-илметилен)-7-оксо-4-тиа-1-азабицикло[3.2.0]гепт-2-ен-2-карбоновая кислота
4-Нитробензиловый эфир (5R,6RS)-6-{(RS)-ацетокси[1-(4-нитробензилоксикарбонил)-2-фенил-1Н-имидазол-4-ил]метил}-6-бром-7-оксо-4-тиа-1-азабицикло[3.2.0]гепт-2-ен-2-карбоновой кислоты (4) (1,15 г) растворяют в ТГФ (16,1 мл) и ацетонитриле (7,5 мл). К полученной смеси добавляют свежеактивированную Zn пыль (4,6 г) и 0,5Mфосфатный буфер (pH 6,4, 23,6 мл). Реакционный сосуд покрывают фольгой, чтобы исключить воздействие света. Реакционную смесь перемешивают в течение 2 ч при комнатной температуре. Реакционный раствор смешивают с этилацетатом и фильтруют через рыхлый слой целита. Рыхлый слой промывают водой и водный слой отделяют. Водный слой концентрируют в высоком вакууме при 35°C. Концентрат наносят на хроматографическую колонку со смолой Diaion HP-21 (100 мл, Mitsubishi Kasei Co. Ltd.). После адсорбции осуществляют элюирование водой и затем системой 5-10% ацетонитрил-вода. Объединенные фракции концентрируют в высоком вакууме при 35°C, лиофилизуют и получают указанное в заголовке соединение в виде желтого аморфного твердого вещества (322 мг, 63%). Т.пл. 281°C (с разложением);
1H ЯМР (δ, D2O): 6,32 (с, 1H), 6,76 (с, 1H), 6,79 (с, 1H), 7,22 (с, 1H), 7,24-7,33 (м, 3H), 7,60-7,63 (м, 2H).
Пример 7
Стадия 1. 4-Нитробензиловый эфир (5R,6S)-6-бром-6-[(S)-2,3-дигидроимидазо[2,1-b]тиазол-6-ил)гидроксиметил]-7-оксо-4-тиа-1-азабицикло[3.2.0]гепт-2-ен-2-карбоновой кислоты
2,3-Дигидроимидазо[2,1-b]тиазол-6-карбальдегид (2,63 г) добавляют к раствору безводного MgBr2 (3,8 г) в сухом ацетонитриле (124 мл) в атмосфере азота при комнатной температуре. Бесцветный порошок выпадает в осадок в течение 30 мин. К полученной смеси добавляют раствор 4-нитробензилового эфира (5R,6S)-6-бром-7-оксо-4-тиа-1-азабицикло[3.2.0]гепт-2-ен-2-карбоновой кислоты (6,2 г) в сухом ТГФ (124 мл). После охлаждения смеси до -20°C добавляют в один прием триэтиламин (5,21 мл). Реакционный сосуд покрывают фольгой, чтобы исключить воздействие света. Реакционную смесь перемешивают в течение 2,5 ч при -20°C, разбавляют этилацетатом и промывают 5% водным раствором лимонной кислоты, насыщенным раствором гидрокарбоната натрия и насыщенным солевым раствором. Органический слой сушат (MgSO4) и фильтруют через рыхлый слой целита. Рыхлый слой промывают этилацетатом. Фильтрат концентрируют при пониженном давлении. Остаток наносят на хроматографическую колонку с силикагелем, затем элюируют смесью этилацетат-гексан (3/1˜чистый этилацетат). Получают смесь указанного в заголовке соединения 3 и его диастереомера (10/1) в виде светло-коричневого аморфного твердого вещества (6,63 г, 79%) и указанное в заголовке соединение (3,20 г, 38%) выделяют из смеси колоночной хроматографией на силикагеле.
1H ЯМР (δ, CDCl3) 3,80-3,89 (м, 2H), 4,15-4,25 (м, 2H), 5,22 (с, 1H), 5,31 (д, 1H, J=13,5 Гц), 5,45 (д, 1H, J=13,5 Гц), 6,12 (с, 1H), 7,12 (с, 1H), 7,42 (с, 1H), 7,62 (д, 2H, J=8,6 Гц), 8,22-8,25 (м, 2H).
Стадия 2. (5R,6S)-6-[(S)-(2,3-Дигидроимидазо[2,1-b]тиазол-6-ил)гидроксиметил]-7-оксо-4-тиа-1-азабицикло[3.2.0]гепт-2-ен-2-карбоновая кислота
4-Нитробензиловый эфир (5R,6S)-6-бром-6-[(S)-(2,3-дигидроимидазо[2,1-b]тиазол-6-ил)гидроксиметил]-7-оксо-4-тиа-1-азабицикло[3.2.0]гепт-2-ен-2-карбоновой кислоты (2,0 г) растворяют в ТГФ (28 мл) и ацетонитриле (13 мл). К полученному раствору добавляют свежеактивированную Zn пыль (8,0 г) и 0,5Mфосфатный буфер (pH 6,4, 41 мл). Реакционный сосуд покрывают фольгой, чтобы исключить воздействие света. Реакционную смесь энергично перемешивают в течение 2 ч при комнатной температуре, затем охлаждают до 3°C и добавляют 1Mводный раствор NaOH, чтобы довести рН до 7,5. Реакционный раствор смешивают с этилацетатом и фильтруют через рыхлый слой целита. Рыхлый слой промывают водой, отделяют водный слой и концентрируют его в высоком вакууме при 35°C. Концентрат наносят на хроматографическую колонку со смолой Diaion HP-21 (200 мл, Mitsubishi Kasei Co. Ltd.). После адсорбции осуществляют элюирование водой и затем смесью 2,5% ацетонитрил-вода. Объединенные фракции концентрируют в высоком вакууме при 35°C, лиофилизуют и получают указанное в заголовке соединение в виде желтого аморфного твердого вещества (171 мг, 13%). Т.пл.152°C.
1H ЯМР (δ, D2O): 3,91 (т, 2H, J=7,4 Гц), 4,23 (т, 2H, J=7,4 Гц), 4,33-4,35 (м, 1H), 5,07 (д, 1H, J=5,5 Гц), 5,70 (д, 1H, J=1,6 Гц), 7,05 (д, 1H, J=1,0 Гц), 7,24 (с, 1H).
Пример 8
Получение (5R,6Z)-6-[(5-бензил-4,5,6,7-тетрагидротиено[3,2-c]пиридин-2-ил)метилен]-7-оксо-4-тиа-1-азабицикло[3.2.0]гепт-2-ен-2-карбоновой кислоты
Стадия 1. Этил 5-бензоил-4,5,6,7-тетрагидротиено[3,2-c]пиридин-2-карбоксилат
К перемешиваемому сухому ДМФА (7,3 г, 100 ммоль) медленно добавляют POCl3 (12,25 г, 80 ммоль) при температуре от 0°C до 5°С. По окончании прибавления загустевшую массу растворяют в CH2Cl2 (20 мл) и перемешивают при комнатной температуре в течение 2 ч. Вновь понижают температуру до 0°C и медленно добавляют 1-бензоил-4-пиперидон в CH2Cl2. После прибавления реакционную смесь перемешивают при комнатной температуре в течение 2 ч и выливают на измельченный лед и ацетат натрия. Перемешивают в течение 30 мин при комнатной температуре и экстрагируют CH2Cl2. Экстракт тщательно промывают водой, сушат над безводным MgSO4 и концентрируют. Сырой продукт растворяют в CH2Cl2 и медленно добавляют этилмеркаптоацетат (9,6 г, 80 ммоль)/Et3N (10,1 г, 100 ммоль) при комнатной температуре. Реакционную смесь кипятят с обратным холодильником в течение 2 ч и гасят водой. Дихлорметановый слой хорошо промывают водой, сушат над безводным MgSO4, фильтруют и концентрируют. Продукт очищают хроматографией на колонке с SiO2, элюируя смесью 50% этилацетата и гексана. Получают желтое масло. Выход 6,4 г (25%); M+H 316.
Стадия 2. (5-бензил-4,5,6,7-тетрагидротиено[3,2-с]пиридин-2-ил)метанол
К перемешиваемой суспензии LiAlH4 (2,0 г) в ТГФ медленно добавляют раствор этил (5-бензоил-4,5,6,7-тетрагидротиено[3,2-c]пиридин)-2-карбоксилата (6,0 г, 19 ммоль) в ТГФ при 0°С. После прибавления реакционную смесь перемешивают в течение 30 минут и гасят насыщенным NH4Cl. Реакционную смесь разбавляют CHCl3 и фильтруют. Фильтрат промывают насыщенным солевым раствором, сушат над безводным MgSO4, фильтруют, полученную желтую жидкость вводят в следующую стадию без очистки. Выход 4,5 г (91%).
Стадия 3. 2-Формил-(5-бензил-4,5,6,7-тетрагидротиено[3,2-c]пиридин
К перемешиваемому раствору (5-бензил-4,5,6,7-тетрагидротиено[3,2-с]пиридин-2-ил)метанола (4,0 г, 15,4 ммоль) в CH2Cl2 (300 мл) добавляют активный MnO2 (20 г, избыток) и перемешивают при комнатной температуре в течение 18 ч. В конце реакционную смесь фильтруют через целит, который затем промывают CHCl3. Реакционную смесь тщательно промывают водой, сушат и концентрируют. Продукт, как было показано, является чистым и его непосредственно используют на следующей стадии. Выход 3,0 г (76%). (M+H) 257.
Стадия 4. 4-Нитробензил-6-[(ацетокси)(5-бензил-4,5,6,7-тетрагидротиено[3,2-c]пиридин-2-ил)метил]-6-бром-7-оксо-4-тиа-1-азабицикло[3.2.0]гепт-2-ен-2-карбоксилат
2-Формил-(5-бензил-4,5,6,7-тетрагидротиено[3,2-c]пиридин (565 мг, 2,2 ммоль) и раствор 4-нитробензилового эфира (5R,6S)-6-бром-7-оксо-4-тиа-1-азабицикло[3.2.0]гепт-2-ен-2-карбоновой кислоты (772 мг, 2,0 ммоль) в сухом ТГФ (20 мл) последовательно добавляют в раствор безводного MgBr2: O(Et)2 (390 мг, 1,5 ммоль) в сухом ацетонитриле (15 мл) в атмосфере аргона при комнатной температуре. После охлаждения до -20°C добавляют за один прием Et3N (2,0 мл). Реакционный сосуд покрывают фольгой, чтобы исключить воздействие света. Реакционную смесь перемешивают в течение 2 ч при -20°C и обрабатывают уксусным ангидридом (1,04 мл) в один прием. Температуру реакционной смеси доводят до 0°C и перемешивают в течение 15 ч при 0°C. Смесь разбавляют этилацетатом и промывают 5% водным раствором лимонной кислоты, насыщенным раствором гидрокарбоната натрия и насыщенным солевым раствором. Органический слой сушат (MgSO4) и фильтруют через рыхлый слой целита. Рыхлый слой промывают этилацетатом. Фильтрат концентрируют при пониженном давлении. Остаток наносят на хроматографическую колонку с силикагелем, затем элюируют смесью этилацетат-гексан (1:1). Собранные фракции концентрируют при пониженном давлении и смесь диастереомеров используют на следующей стадии. Бледно-желтое аморфное твердое вещество. Выход 550 мг (40%), (M+H) 687.
Стадия-5. (5R,6Z)-6-[(5-Бензил-4,5,6,7-тетрагидротиено[3,2-c]пиридин-2-ил)метилен]-7-оксо-4-тиа-1-азабицикло[3.2.0]гепт-2-ен-2-карбоновая кислота
4-Нитробензил-6-[(ацетокси)(5-бензил-4,5,6,7-тетрагидротиено[3,2-c]пиридин-2-ил)метил]-6-бром-7-оксо-4-тиа-1-азабицикло[3.2.0]гепт-2-ен-2-карбоксилат (450 мг, 0,65 ммоль) растворяют в ТГФ (20 мл) и ацетонитриле (10 мл). Быстро добавляют свежеактивированную цинковую пыль (5,2 г) с 0,5 Mфосфатным буфером (pH 6,5, 28 мл). Реакционный сосуд покрывают фольгой, чтобы исключить воздействие света. Реакционную смесь энергично перемешивают в течение 2 ч при комнатной температуре. Реакционную смесь фильтруют, охлаждают до 3°C и добавляют 0,1M NaOH, чтобы довести рН до 8,5. Фильтрат промывают этилацетатом и отделяют водный слой. Водный слой концентрируют в высоком вакууме при 35°C и получают желтый осадок. Продукт очищают колоночной хроматографией с обращенной фазой (смола HP-21). Сначала осуществляют элюирование деионизированной водой (2 л), затем 10% CAN: вода. Фракции, содержащие продукт, объединяют и концентрируют при пониженном давлении при комнатной температуре. Твердое желтое вещество промывают ацетоном, отфильтровывают и сушат. Получают продукт в виде желтых кристаллов, выход 50 мг (18%), т.пл.198°C, (M+H) 411.
1H ЯМР (ДМСО-d6): δ 2,7 (м, 2H), 2,8 (уш.м, 2H), 3,4 (м, 2H), 3,8 (с, 2H), 6,3 (с, 1H), 6,5 (с, 1H), 7,1(с, 1H), 7,28(с, 1H), 7,4 (с, 5H).
Пример 9
Получение натриевой соли (5R),(6Z)-6-(7-метил-5,6,7,8-тетрагидроимидазо[1,2-a]пиразин-2-илметилен)-7-оксо-4-тиа-1-азабицикло[3.2.0]гепт-2-ен-2-карбоновой кислоты
Стадия 1. Этиловый эфир имидазо[1,2-a]пиразин-2-карбоновой кислоты
Этилбромпируват (62,9 г) добавляют к раствору 2-аминопиразина (24,8 г) в DME (258 мл) при комнатной температуре и перемешивают в течение 2,5 ч. Реакционную смесь охлаждают до 0°C, перемешивают 30 мин и получают светло-коричневый осадок. Осадок отфильтровывают, промывают Et2O и получают светло-коричневые кристаллы. Суспензию осадка (66,1 г) в EtOH (1,29 л) нагревают при температуре кипения для получения прозрачного раствора. После кипячения с обратным холодильником в течение 2 ч реакционную смесь концентрируют при пониженном давлении, затем смешивают с CHCl3 и насыщенным водным раствором NaHCO3. Смесь фильтруют через рыхлый слой целита, отделенный органический слой сушат (MgSO4) и фильтруют. Фильтрат концентрируют при пониженном давлении. Остаток наносят на хроматографическую колонку с силикагелем, затем осуществляют элюирование смесью CHCl3-MeOH (99/1˜97/3), полученные фракции концентрируют при пониженном давлении с последующей перекристаллизацией из смеси CHCl3-Et2O. Указанное в заголовке соединение получают в виде бледно-розовых кристаллов. Выход 10,9 г (22%).
1H ЯМР (CDCl3) δ: 1,46 (т, 3H, J=7,2 Гц), 4,49 (кв, 2H, J=7,2 Гц), 7,96 (д, 1H, J=4,7 Гц), 8,08 (дд, 1H, J=1,2 и 4,7 Гц), 8,26 (с, 1H), 9,21 (д, 1H, J=1,2 Гц).
Стадия 2. Гидрохлорид этилового эфира 5,6,7,8-тетрагидроимидазо[1,2-a]пиразин-2-карбоновой кислоты
К раствору этилового эфира имидазо[1,2-a]пиразин-2-карбоновой кислоты (13,7 г) в EtOH (546 мл) добавляют 0,46МHCl-EtOH (169 мл) и 10% Pd-C (50% влажность) (1,37 г). Смесь гидрируют при давлении водорода 40 фунт/кв.дюйм при комнатной температуре в течение 15 ч. Реакционную смесь фильтруют и Pd-C промывают этанолом. Фильтрат концентрируют при пониженном давлении. Остаток наносят на хроматографическую колонку с силикагелем, затем колонку элюируют смесью CHCl3-MeOH (9/1˜2/1). Указанное в заголовке соединение получают в виде коричневых кристаллов. Выход 10,4 г (63%).
1H ЯМР(CDCl3) δ: 1,38(т, 3H, J=7,1 Гц), 3,90(т, 2H, J=5,7 Гц), 4,40(кв, 2H, J=7,1 Гц), 4,59 (т, 2H, J=5,7 Гц), 4,80 (с, 2H), 8,20 (с, 1H).
Стадия 3. Этиловый эфир 7-метил-5,6,7,8-тетрагидроимидазо[1,2-a]пиразин-2-карбоновой кислоты
Триэтиламин (3,44 мл), 37% водный раствор HCHO (2,02 мл) и NaBH3CN (1,78 г) последовательно добавляют к раствору гидрохлорида этилового эфира 5,6,7,8-тетрагидроимидазо[1,2-a]пиразин-2-карбоновой кислоты (5,2 г) в MeOH (75 мл) при комнатной температуре и перемешивают в течение 3,5 ч в атмосфере азота. Смесь разбавляют CH2Cl2 и промывают 50% водным раствором K2CO3. Органический слой сушат (K2CO3) и фильтруют. Фильтрат концентрируют при пониженном давлении. Остаток наносят на хроматографическую колонку с силикагелем, затем осуществляют элюирование смесью CHCl3-ацетон(1/1-1/2). Указанное в заголовке соединение получают в виде оранжевого масла. Выход 2,68 г (57%).
1H ЯМР (CDCl3) δ: 1,37 (м, 3H, J=7,1 Гц), 2,50 (с, 3H), 2,85 (т, 2H, J=5,5 Гц), 3,69 (с, 2H), 4,06 (т, 2H, J=5,5 Гц), 4,36 (т, 2H, J=7,1 Гц), 7,52 (с, 1H).
Стадия 4: 7-Метил-5,6,7,8-тетрагидроимидазо[1,2-a]пиразин-2-карбальдегид
К раствору этилового эфира 7-метил-5,6,7,8-тетрагидроимидазо[1,2-a]пиразин-2-карбоновой кислоты (1,8 г) в CH2Cl2 (86 мл) добавляют 1,01 M раствор DIBAL в толуоле (13,6 мл) в атмосфере азота при -78°C и перемешивают в течение 2 ч. Реакционную смесь гасят 1М HCl и затем фильтруют через рыхлый слой целита. Фильтрат промывают 50% водным раствором K2CO3 и водный слой экстрагируют CH2Cl2 три раза. Объединенный органический слой сушат (K2CO3) и фильтруют. Фильтрат концентрируют при пониженном давлении. Остаток наносят на хроматографическую колонку с силикагелем, затем осуществляют элюирование смесью CHCl3-MeOH (19/1˜9/1). Указанное в заголовке соединение 5 получают в виде бесцветных кристаллов. Выход 591 мг (42%).
1H ЯМР (CDCl3) δ: 2,51 (с, 3H), 2,87 (т, 2H, J=5,5 Гц), 3,70 (с, 2H), 4,10 (т, 2H, J=5,5 Гц), 7,53 (с, 1H), 9,82 (д, 1H, J=1,4 Гц).
Стадия 5. 4-Нитробензиловый эфир (5R,6RS)-6-[(RS)-ацетокси(7-метил-5,6,7,8-тетрагидроимидазо[1,2-a]пиразин-2-ил)метил]-6-бром-7-оксо-4-тиа-1-азабицикло[3.2.0]гепт-2-ен-2-карбоновой кислоты (смесь диастереоизомеров)
7-Метил-5,6,7,8-тетрагидроимидазо[1,2-a]пиразин-2-карбальдегид (1,19 г) добавляют к раствору безводного MgBr2 (4,05 г) в сухом ацетонитриле (97 мл) в атмосфере азота при комнатной температуре. К полученной смеси добавляют раствор 4-нитробензилового эфира (5R,6S)-6-бром-7-оксо-4-тиа-1-азабицикло[3.2.0]гепт-2-ен-2-карбоновой кислоты (3,32 г) в сухом ТГФ (97 мл), охлаждают до -20°C и добавляют в один прием Et3N (3,0 мл). Реакционный сосуд покрывают фольгой, чтобы исключить воздействие света. Реакционную смесь перемешивают в течение 4,5 ч при -20°C и обрабатывают уксусным ангидридом (1,36 мл) в один прием. Температуру реакционной смеси доводят до 0°C и перемешивают при 0°С в течение 17 ч. Смесь разбавляют этилацетатом и промывают 5% водным раствором лимонной кислоты, насыщенным раствором гидрокарбоната натрия и насыщенным солевым раствором. Органический слой сушат (MgSO4) и фильтруют. Фильтрат концентрируют при пониженном давлении. Остаток наносят на хроматографическую колонку с силикагелем, затем элюируют смесью CHCl3-ацетон (9/1˜2/1). Указанное в заголовке соединение получают в виде смеси двух диастереомеров, которая представляет собой масло красного цвета. Выход 1,13 г.
1H ЯМР (CDCl3) δ: 1,20 (c, 0,81 × 3H), 2,24 (с, 0,19 × 3H), 2,48 (с, 3H), 2,80˜2,84 (м, 2H), 3,57˜3,67 (м, 2H), 3,97˜4,02 (м, 2H), 5,27 (д, 1H, J=13,6 Гц), 5,42 (д, 0,19 × 1H, J=13,6 Гц), 5,45 (д, 0,81 × 1H, J=13,6 Гц), 6,07 (с, 0,19 × 1H), 6,30 (c, 0,81 × 2H), 6,79 (с, 0,19 × 1H), 6,80 (с, 0,19 × 1H), 7,02 (с, 0,81 × 1H), 7,44 (с, 0,19 × 1H), 7,47 (с, 0,81 × 1H), 7,60 (д, 0,19 × 2H, J=8,2 Гц), 7,62 (д, 0,81 × 2H, J=8,6 Гц), 8,22˜8,26 (м, 2H).
Стадия 6. Натриевая соль (5R),(6Z)-6-(7-метил-5,6,7,8-тетрагидроимидазо[1,2-a]пиразин-2-илметилен)-7-оксо-4-тиа-1-азабицикло[3.2.0]гепт-2-ен-2-карбоновой кислоты
4-Нитробензиловый эфир (5R,6RS)-6-[(RS)-ацетокси(7-метил-5,6,7,8-тетрагидроимидазо[1,2-a]пиразин-2-ил)метил]-6-бром-7-оксо-4-тиа-1-азабицикло[3.2.0]гепт-2-ен-2-карбоновой кислоты (1,11 г) растворяют в ТГФ (32 мл) и ацетонитриле (32 мл). К полученной смеси добавляют свежеактивированную Zn пыль (4,46 г) и 0,5Mфосфатный буфер с pH 6,5 (48 мл). Реакционный сосуд покрывают фольгой, чтобы исключить воздействие света. Реакционную смесь энергично перемешивают в течение 2 ч при комнатной температуре. Реакционную смесь фильтруют через рыхлый слой целита, охлаждают до 3°C и добавляют 1MNaOH, чтобы довести pH до 7,5. Фильтрат промывают этилацетатом, отделяют водный слой и концентрируют его в высоком вакууме при 35°C. Концентрат наносят на хроматографическую колонку со смолой Diaion HP-21 (20 мл, Mitsubishi Kasei Co. Ltd.). После адсорбции осуществляют элюирование смесью H2O-MeCN (1/0˜95/5). Объединенные фракции концентрируют в высоком вакууме при 35°C, лиофилизуют и получают указанное в заголовке соединение в виде желтого аморфного твердого вещества. Выход 417 мг (65%). Т.пл. 200°C (с разложением).
1H ЯМР (D2O) δ: 2,32 (с, 3H), 2,79˜2,81 (м, 2H), 3,54 (с, 2H), 3,95 (т, 2H, J=5,6 Гц), 6,39 (с, 1H), 6,85 (с, 1H), 6,87 (с, 1H), 7,26 (с, 1H).
Пример 10
Получение натриевой соли (5R),(6Z)-7-оксо-6-(5,6,7,8-тетрагидроимидазо[1,2-a]пиразин-2-илметилен)-4-тиа-1-азабицикло[3.2.0]гепт-2-ен-2-карбоновая кислота
2-Кетопиперазин
2-Кетопиперазин получают по методу группы Merck (Патент США № 5629322).
Стадия 1. 4-п-Нитробензилоксикарбонил-2-кетопиперазин
К раствору 2-кетопиперазина (2,21 г) и диизопропилэтиламина (4,6 мл) в дихлорметане (110 мл) добавляют 48,7% раствор п-нитробензилоксикарбонилхлорида в 1,4-диоксане (10,7 мл) при 0°C и перемешивают в течение 0,5 ч при 0°C. В реакционную смесь добавляют воду (300 мл) и водный слой экстрагируют дихлорметаном (3 × 100 мл). Органический слой сушат (MgSO4) и фильтруют. Фильтрат концентрируют при пониженном давлении. Остаток наносят на хроматографическую колонку с силикагелем, затем элюируют смесью CHCl3-метанол (30:1) и указанное в заголовке соединение получают в виде белого твердого вещества (7,1 г, количественный выход).
1H ЯМР (δ, CDCl3): 3,42-3,45 (м, 2H), 3,74 (т, 2H, J=5,4 Гц), 4,19 (с, 2H), 5,26 (с, 2H), 6,39 (уш.с, 1H), 7,52 (д, 2H, J=8,6 Гц), 8,24 (д, 2H, J=8,6 Гц).
Стадия 2. 5-Метокси-4-п-нитробензилоксикарбонил-1,2,3,6-тетрагидропиразин
Тетрафторборат триметилоксония (97%, 3,7 г) добавляют к раствору 4-п-нитробензилоксикарбонил-2-кетопиперазина (6,7 г) в сухом дихлорметане (120 мл) при комнатной температуре и перемешивают в течение 17 ч. Реакционную смесь обрабатывают насыщенным водным раствором гидрокарбоната натрия и отделяют органический слой. Водный слой экстрагируют этилацетатом (3 × 100 мл), затем объединенный органический слой промывают насыщенным водным раствором гидрокарбоната натрия и насыщенным солевым раствором. Органический слой сушат (MgSO4) и фильтруют. Фильтрат концентрируют при пониженном давлении и указанное в заголовке соединение получают в виде светло-коричневого твердого вещества. Выход 5,7 г (80,6%).
1H ЯМР (δ, CDCl3): 3,48 (м, 2H), 3,57 (м, 2H), 3,70 (с, 3H), 3,97 (с, 2H), 5,26 (с, 2H), 7,52 (д, 2H, J=8,7 Гц), 8,23 (д, 2H, J=8,7 Гц).
Стадия 3. 2-Имино-4-п-нитробензилоксикарбонилпиперазин
Смесь 5-метокси-4-п-нитробензилоксикарбонил-1,2,3,6-тетрагидропиразина (5,7 г) и хлорида аммония (1,6 г) в сухом этаноле (100 мл) и кипятят с обратным холодильником в течение 4 ч. Реакционную смесь затем концентрируют при пониженном давлении. К остатку добавляют дихлорметан (100 мл) и экстрагируют водой (3 × 50 мл), затем объединенный водный слой промывают дихлорметаном. Водный слой нейтрализуют 10% водным раствором карбоната калия и затем экстрагируют дихлорметаном (8 × 50 мл). Объединенные органические экстракты сушат (MgSO4) и фильтруют. Фильтрат концентрируют при пониженном давлении и получают указанное в заголовке соединение в виде белого твердого вещества. Выход 4,9 г (91,2%).
1H ЯМР (δ, CDCl3): 3,49 (уш.с, 4H), 3,98 (уш.с, 2H), 5,26 (с, 2H), 7,52 (д, 2H, J=8,6 Гц), 8,23 (д, 2H, J=8,6 Гц).
Стадия 4. 7-п-Нитробензилоксикарбонил-5,6,7,8-тетрагидроимидазо[1,2-a]пиразин-2-карбальдегид (9) и 7-п-нитробензилоксикарбонил-5,6,7,8-тетрагидроимидазо[1,2-a]пиразин-3-карбальдегид
Из смеси 2-бром-3-гидроксипропеналя (2,8 г), моногидрата п-толуолсульфокислоты (33 мг) и 2-пропанола (3,5 мл) в циклогексане (28 мл) отгоняют азеотроп до тех пор, пока температура паров не поднимется до 80°C. Реакционную смесь концентрируют при пониженном давлении. Остаток растворяют в сухом ацетонитриле (30 мл). Добавляют раствор 2-имино-4-п-нитробензилоксикарбонилпиперазина (4,7 г) в сухом ацетонитриле (310 мл) при комнатной температуре. Реакционную смесь перемешивают при комнатной температуре в течение 3 ч и затем реакционный раствор упаривают в вакууме. Остаток растворяют в этилацетате (170 мл) и добавляют триэтиламин (2,4 мл), затем реакционную смесь кипятят с обратным холодильником в течение 1,5 ч. Реакционную смесь охлаждают до комнатной температуры, затем добавляют воду (170 мл) и разделяют слои. Водный слой экстрагируют дихлорметаном (2 × 100 мл). Объединенный органический слой сушат (MgSO4) и фильтруют. Фильтрат концентрируют при пониженном давлении. Остаток наносят на хроматографическую колонку с силикагелем, элюируют смесью CHCl3-метанол (50:1) и получают указанное в заголовке соединение в виде коричневого твердого вещества (выход 2,9 г, 51,6%) и его региоизомер (оранжевое аморфное твердое вещество, выход 0,8 г, 14,9%).
1H ЯМР (δ, CDCl3): 3,99 (т, 2H, J=5,4 Гц), 4,14 (т, 2H, J=5,4 Гц), 4,85 (с, 2H), 5,29 (с, 2H), 7,54 (д, 2H, J=8,6 Гц), 7,57 (с, 1H), 8,24 (д, 2H, J=8,6 Гц), 9,85 (с, 1H).
Региоизомер 1H ЯМР (δ, CDCl3): 3,95 (т, 2H, J=5,4 Гц), 4,44 (т, 2H, J=5,4 Гц), 4,87 (с, 2H), 5,29 (с, 2H), 7,54 (д, 2H, J=8,7 Гц), 7,78 (с, 1H), 8 24 (д, 2H, J=8,7 Гц), 9,71 (с, 1H).
Стадия 5. п-Нитробензиловый эфир (5R)-6-[ацетокси(7-п-нитробензилоксикарбонил-5,6,7,8-тетрагидроимидазо[1,2-a]пиразин-2-ил)метил]-6-бром-7-оксо-4-тиа-1-азабицикло[3.2.0]гепт-2-ен-2-карбоновой кислоты
Раствор 7-п-нитробензилоксикарбонил-5,6,7,8-тетрагидроимидазо[1,2-a]пиразин-2-карбальдегида (1,6 г) в сухом ацетонитриле (25 мл) добавляют к раствору MgBr2 (2,2 г) в сухом ацетонитриле (55 мл) в атмосфере азота при комнатной температуре, затем смесь перемешивают в течение 10 мин. Добавляют раствор 4-нитробензилового эфира (5R,6S)-6-бром-7-оксо-4-тиа-1-азабицикло[3.2.0]гепт-2-ен-2-карбоновой кислоты (1,8 г) в сухом ТГФ (80 мл), смесь охлаждают до -20°C, затем добавляют за один прием триэтиламин (1,6 мл). Реакционный сосуд покрывают фольгой, чтобы исключить воздействие света. Реакционную смесь перемешивают в течение 3 ч при -20°C и обрабатывают за один прием 4,4-диметиламинопиридином (58,3 г) и уксусным ангидридом (0,89 мл). Температуру реакционной смеси доводят до 0°C и перемешивают в течение 15 ч при 0°C. К реакционной смеси добавляют 10% водный раствор лимонной кислоты (320 мл) и водный слой экстрагируют этилацетатом (3 × 160 мл). Органический слой промывают водой, насыщенным раствором гидрокарбоната натрия и насыщенным солевым раствором, сушат (MgSO4) и фильтруют. Фильтрат концентрируют при пониженном давлении. Остаток наносят на хроматографическую колонку с силикагелем, затем элюируют смесью CH2Cl2-ацетон (20:1) и получают указанное в заголовке соединение в виде смеси двух диастереомеров (81:19, коричневое пенящееся аморфное твердое вещество). Выход 2,1 г (59,9%).
1H ЯМР (δ, CDCl3): 2,01 (с, 2,43H), 2,24 (с, 0,57H), 3,93-3,96 (м, 2H), 4,02-4,05 (м, 2H), 4,74-4,76 (м, 2H), 5,28 (д, 1H, J=13,5 Гц), 5,28 (с, 2H), 5,45 (д, 1H, J=13,5 Гц), 6,07 (с, 0,19H), 6,29 (с, 0,81H), 6,31 (с, 0,81H), 6,80 (с, 0,19H), 6,83 (с, 0,19H), 7,08 (с, 0,81H), 7,43 (с, 0,19H), 7,46 (с, 0,81H), 7,54 (д, 2H, J=8,6 Гц), 7,61 (д, 2H, J=8,8 Гц), 8,24 (д, 4H, J=8,3 Гц).
Стадия 6. Натриевая соль (5R),(6Z)-7-оксо-6-(5,6,7,8-тетрагидроимидазо[1,2-a]пиразин-2-илметилен)-4-тиа-1-азабицикло[3.2.0]гепт-2-ен-2-карбоновой кислоты
п-Нитробензиловый эфир (5R)-6-[ацетокси(7-п-нитробензилоксикарбонил-5,6,7,8-тетрагидроимидазо[1,2-a]пиразин-2-ил)метил]-6-бром-7-оксо-4-тиа-1-азабицикло[3.2.0]гепт-2-ен-2-карбоновой кислоты (2,0 г) растворяют в ТГФ (63 мл). К полученной смеси быстро добавляют свежеактивированную Zn пыль (7,9 г) и 0,5 моль/лфосфатный буфер с pH 6,5 (63 мл). Реакционный сосуд покрывают фольгой, чтобы исключить воздействие света. Реакционную смесь энергично перемешивают в течение 2 ч при комнатной температуре. Реакционный раствор фильтруют через рыхлый слой целлита, который затем промывают водой (150 мл) и н-бутанолом (150 мл). Водный слой отделяют, органический слой затем экстрагируют водой (2 × 50 мл). Объединенный водный слой концентрируют до массы 61 г и наносят на хроматографическую колонку со смолой Diaion HP-21 (80 мл, Mitsubishi Kasei Co. Ltd.). После адсорбции колонку элюируют водой и затем 5% водным раствором ацетонитрила. Объединенные фракции концентрируют в высоком вакууме при 35°C, лиофилизуют и получают указанное в заголовке соединение в виде желтого твердого аморфного вещества. Выход 172 мг (20,1%). Т.пл. 150°C (с разложением).
1H ЯМР (δ, D2O): 3,02 (т, 2H, J=5,6 Гц), 3,82 (с, 2H), 3,89 (д, 2H, J=5,6 Гц), 6,38 (с, 1H), 6,84 (с, 1H), 6,87 (с, 1H), 7,24 (с, 1H).
Пример 11
Получение (5R),(6Z)-6-{[5-(4-метоксибензил)-4,5,6,7-тетрагидротиено[3,2-с]пиридин-2-ил)]метилен}-7-оксо-4-тиа-1-азабицикло[3.2.0]гепт-2-ен-2-карбоновой кислоты
Стадия 1. 5-трет-Бутил 2-этил 6,7-дигидротиено[3,2-с]пиридин-2,5(4Н)-дикарбоксилат
5-трет-Бутил 2-этил 6,7-дигидротиено[3,2-с]пиридин-2,5(4Н)дикарбоксилат получают по методике, кратко изложенной в примере 8 (Стадия 1), исходя из трет-бутил-1-пиперидинкарбоксилата (9,9 г, 50 ммоль), POCl3 (6,3 г, 40 ммоль) и ДМФА (3,8 г, 50 ммоль). Хлорформильное промежуточное соединение подвергают взаимодействию с этилмеркаптоацетатом (6,0 г, 50 ммоль) и Et3N. Продукт очищают хроматографией на колонке с SiO2 при элюировании смесью гексан-этилацетат (3:1). Получают продукт в виде бесцветной жидкости с выходом 8,7 г (56%). (M+H) 312.
Стадия 2. трет-Бутил 2-(гидроксиметил)-6,7-дигидротиено[3,2-c]пиридин-5(4H)-карбоксилат
трет-Бутил 2-(гидроксиметил)-6,7-дигидротиено[3,2-c]пиридин-5(4H)-карбоксилат получают по методике, кратко изложенной в примере 8 (Стадия 2), исходя из 5-трет-бутил 2-этил 6,7-дигидротиено[3,2-c]пиридин-2,5(4H)-дикарбоксилата (1,0 г, 3,21 ммоль) и LiAlH4 (500 мг, избыток). Выделяют 807 мг (выход 92%) гидроксипроизводного в виде бесцветной жидкости (M+H) 270.
Стадия 3. трет-Бутил 2-(формил)-6,7-дигидротиено[3,2-c]пиридин-5(4H)карбоксилат
трет-Бутил 2-(формил)-6,7-дигидротиено[3,2-c]пиридин-5(4H)карбоксилат получают по методике, кратко изложенной в примере 8 (Стадия 3), исходя из трет-бутил 2-(гидроксиметил)-6,7-дигидротиено[3,2-c]пиридин-5(4H)-карбоксилата (1,0 г, 3,7 ммоль) в метиленхлориде (100 мл) и активного MnO2 (5 г, избыток). Выделяют 800 г (выход 81%) альдегида в виде коричневого твердого вещества.(M+H) 268.
Стадия 4. 2-(Формил)-6,7-дигидротиено[3,2-c]-5(4H)пиридин
2-(Формил)-6,7-дигидротиено[3,2-c]-5(4H)пиридин получают из трет-бутил 2-(формил)-6,7-дигидротиено[3,2-c]пиридин-5(4H)карбоксилата (1,0 г, 3,7 ммоль), который растворяют в CH2Cl2 (20 мл), MeOH (90%, 20 мл) и 1 н. HCl в диоксане (10 мл). Реакционную смесь перемешивают при комнатной температуре в течение 48 ч, затем упаривают досуха и используют на следующей стадии без дополнительной очистки. Выход 750 мг (гидрохлорид, количественный выход); (M+H) 168.
Стадия 5. 2-Формил[5-(4-метоксибензил)-4,5,6,7-тетрагидротиено][3,2-c]пиридин
К перемешиваемому раствору 2-(формил)-6,7-дигидротиено[3,2-c]-5(4H)пиридина (1,4 г, 5,2 ммоль) в ДМФА (20 мл) добавляют 4-метоксибензилхлорид (0,94 г, 6,2 ммоль) и N,N-диизопропилэтиламин (10 мл, избыток) при комнатной температуре. Реакционную смесь перемешивают в течение 24 ч и гасят водой. Реакционную смесь экстрагируют хлороформом, органический слой тщательно промывают водой, сушат над безводным MgSO4, фильтруют и концентрируют. Продукт очищают хроматографией на колонке с SiO2, элюируют этилацетатом. Получают светло-желтое масло, выход 470 мг (35%), (M+H) 288.
Стадия 6. 4-Нитробензил 6-[(ацетокси)[5(4-метоксибензил)-4,5,6,7-тетрагидротиено[3,2-c]пиридин-2-ил)метил]-6-бром-7-оксо-4-тиа-1-азабицикло[3.2.0]гепт-2-ен-2-карбоксилат
2-Формил[5-(4-метоксибензил)-4,5,6,7-тетрагидротиено][3,2-c]пиридин (574 мг, 2,0 ммоль) и раствор 4-нитробензилового эфира (5R,6S)-6-бром-7-оксо-4-тиа-1-азабицикло[3.2.0]гепт-2-ен-2-карбоновой кислоты (772 мг, 2,0 ммоль) в сухом ТГФ (20 мл) последовательно добавляют к раствору безводного MgBr2:O(Et)2 (390 мг, 1,5 ммоль) в сухом ацетонитриле (15 мл) в атмосфере аргона при комнатной температуре. После охлаждения до -20°C, добавляют за один прием Et3N (2,0 мл). Реакционный сосуд покрывают фольгой, чтобы исключить воздействие света. Реакционную смесь перемешивают в течение 2 ч при -20°C и обрабатывают уксусным ангидридом (1,04 мл) в один прием. Температуру реакционной смеси доводят до 0°C и перемешивают в течение 15 ч при 0°C. Смесь разбавляют этилацетатом и промывают 5% водным раствором лимонной кислоты, насыщенным раствором гидрокарбоната натрия и насыщенным солевым раствором. Органический слой сушат (MgSO4) и фильтруют через рыхлый слой целлита, слой целита промывают этилацетатом. Фильтрат концентрируют при пониженном давлении. Остаток наносят на хроматографическую колонку с силикагелем, элюируют смесью этилацетат:гексан (1:1). Собранные фракции концентрируют при пониженном давлении и полученную смесь диастереомеров в виде желтого аморфного твердого вещества используют на следующей стадии. Выход 550 мг (40%); (M+H) 714 и 716.
Стадия 7. (5R,6Z)-6-{[5-(4-Метоксибензил)-4,5,6,7-тетрагидротиено[3,2-c]пиридин-2-ил)метилен}-7-оксо-4-тиа-1-азабицикло[3.2.0]гепт-2-ен-2-карбоновая кислота
4-Нитробензил-6-[(ацетокси)[5(4-метоксибензил)-4,5,6,7-тетрагидротиено[3,2-c]пиридин-2-ил)метил]-6-бром-7-оксо-4-тиа-1-азабицикло[3.2.0]гепт-2-ен-2-карбоксилат (300 мг, 0,42 ммоль) растворяют в ТГФ (20 мл) и ацетонитриле (10 мл). Быстро добавляют свежеактивированную цинковую пыль (5,2 г) с 0,5M фосфатным буфером (pH 6,5, 28 мл). Реакционный сосуд покрывают фольгой, чтобы исключить воздействие света. Реакционную смесь энергично перемешивают в течение 2 ч при комнатной температуре. Реакционную смесь фильтруют, охлаждают до 3°C и 0,1М раствором NaOH доводят pH до 8,5. Фильтрат промывают этилацетатом, отделяют водный слой, концентрируют его в высоком вакууме при 35°C и получают желтый осадок. Продукт очищают хроматографией с обращенной фазой на колонке со смолой HP-21. Сначала колонку элюируют деионизированной водой (2 л) и затем смесью 10% CAN: вода. Собирают фракции, содержащие продукт, и упаривают при пониженном давлении при комнатной температуре. Полученное твердое желтое вещество промывают ацетоном, отфильтровывают, сушат и получают желтые кристаллы (50 мг, 18%). Т.пл. 127°C; (M+H) 441.
1H ЯМР (ДМСО-d6) δ: 2,7 (м, 2H), 2,8 (уш.м, 2H), 3,4 (м, 2H), 3,74 (с, 3H) 3,8 (с, 2H), 6,6 (с, 1H), 6,88 (дд, 2H), 7,14 (с, 1H), 7,24 (дд, 2H), 7,4 (с, 1H), 7,59 (с, 1H).
Пример 12
Получение натриевой соли (5R,6Z)-6-(5,6-дигидро-8Н-имидазо[2,1-c][1,4]тиазин-2-илметилен)-7-оксо-4-тиа-1-азабицикло[3.2.0]гепт-2-ен-2-карбоновой кислоты
Стадия 1. Гидроиодид 5-метилтио-3,6-дигидро-2Н-[1,4]тиазина
Гидроиодид 5-метилтио-3,6-дигидро-2H-[1,4]тиазина получают по методике, изложенной в патенте США № 5629322.
Стадия 2. Гидрохлорид 3-иминотиоморфолина
Гидроиодид 5-метилтио-3,6-дигидро-2H-[1,4]тиазина (7,1 г) растворяют в 10% водном растворе K2CO3 (150 мл) и водный слой экстрагируют CH2Cl2 (5 × 70 мл). Объединенный органический слой сушат (MgSO4), фильтруют и концентрируют при пониженном давлении. К полученному остатку в сухом этаноле (128 мл) добавляют хлорид аммония (1,7 г), кипятят с обратным холодильником в течение 1 ч. Реакционную смесь охлаждают до комнатной температуры. Растворитель удаляют в вакууме и получают гидрохлорид иминотиоморфолина в виде коричневого твердого вещества (4,3 г, количественный выход).
1H ЯМР (δ, ДМСО-d6): 3,15 (т, 2H, J=5,9 Гц), 3,74 (т, 2H, J=5,9 Гц), 3,83 (с, 2H), 8,97 (уш.с, 1H), 9,38 (уш.с, 1H), 9,99 (уш.с, 1H).
Стадия 3. 5,6-Дигидро-8Н-имидазо[2,1-c][1,4]тиазин-2-карбальдегид и 5,6-дигидро-8Н-имидазо[2,1-c][1,4]тиазин-3- карбальдегид
Из смеси 2-бром-3-гидроксипропеналя (7, 4,3 г), моногидрата п-толуолсульфокислоты (52 мг) и 2-пропанола (5,3 мл) в циклогексане (43 мл) отгоняют азеотроп до тех пор, пока температура паров не поднимется до 80°C. Реакционную смесь концентрируют при пониженном давлении. Остаток растворяют в сухом этаноле (28 мл). Добавляют смесь раствора гидрохлорида 3-иминотиоморфолина (4,3 г) в сухом этаноле (143 мл) и 28% метанольного раствора метилата натрия (5,0 мл) при комнатной температуре. Реакционную смесь перемешивают в течение 1 ч при комнатной температуре, затем реакционный раствор упаривают в вакууме. Остаток растворяют в хлороформе (128 мл), добавляют триэтиламин (3,6 мл), реакционную смесь кипятят с обратным холодильником в течение 2,5 ч, затем охлаждают до комнатной температуры и концентрируют при пониженном давлении. Остаток растворяют в дихлорметане (300 мл) и промывают 50% водным раствором K2CO3 (2 × 100 мл). Органический слой сушат (MgSO4) и фильтруют. Фильтрат концентрируют при пониженном давлении. Остаток наносят на хроматографическую колонку с силикагелем, элюируют смесью CHCl3-ацетон (10:1) и получают 5,6-дигидро-8H-имидазо[2,1-c][1,4]тиазин-2-карбальдегид (коричневое твердое вещество, 445 мг, 10,3%) и 5,6-дигидро-8Н-имидазо[2,1-c][1,4]тиазин-3-карбальдегид (коричневое твердое вещество, 872 мг, 20,2%).
5,6-Дигидро-8H-имидазо[2,1-c][1,4]тиазин-2-карбальдегид: 1H ЯМР (δ, CDCl3): 3,07 (т, 2H, J=5,7 Гц), 3,95 (с, 2H), 4,33 (т, 2H, J=5,7 Гц), 7,55 (с, 1H), 9,83 (с, 1H).
5,6-Дигидро-8Н-имидазо[2,1-c][1,4]тиазин-3-карбальдегид: 1H ЯМР (δ, CDCl3): 3,05 (т, 2H, J=5,7 Гц), 3,98 (с, 2H), 4,61 (т, 2H, J=5,7 Гц), 7,73 (с, 1H), 9,69 (с, 1H).
Стадия 4. Натриевая соль (5R),(6Z)-6-(5,6-дигидро-8Н-имидазо[2,1-c][1,4]тиазин-2-илметилен)-7-оксо-4-тиа-1-азабицикло[3.2.0]гепт-2-ен-2-карбоновой кислоты
Раствор 5,6-дигидро-8Н-имидазо[2,1-c][1,4]тиазин-2-карбальдегид (9, 392 мг) в сухом ацетонитриле (20 мл) добавляют к раствору MgBr2 (1,1 г) в сухом ацетонитриле (20 мл) в атмосфере азота при комнатной температуре, затем смесь перемешивают в течение 10 мин. Добавляют раствор 4-нитробензилового эфира (5R,6S)-6-бром-7-оксо-4-тиа-1-азабицикло[3.2.0]гепт-2-ен-2-карбоновой кислоты (1,0 г) в сухом ТГФ (40 мл) и смесь охлаждают до -20°C, затем в один прием добавляют триэтиламин (0,8 мл). Реакционный сосуд покрывают фольгой, чтобы исключить воздействие света. Реакционную смесь перемешивают в течение 3,5 ч при -20°C и обрабатывают в один прием 4-диметиламинопиридином (30 мг) и уксусным ангидридом (0,44 мл). Температуру реакционной смеси доводят до 0°C и перемешивают в течение 14 ч при 0°C. К реакционной смеси добавляют 10% водный раствор лимонной кислоты (240 мл) и водный слой экстрагируют этилацетатом (3 × 100 мл). Объединенный органический слой промывают водой, насыщенным раствором гидрокарбоната натрия и насыщенным солевым раствором, сушат (MgSO4) и фильтруют. Фильтрат концентрируют при пониженном давлении. Остаток предварительно очищают на колонке с силикагелем при элюировании смесью CH2Cl2-ацетон (50:1) и получают сырой п-нитробензиловый эфир (5R)-6-[ацетокси(5,6-дигидро-8H-имидазо[2,1-c][1,4]тиазин-2-ил)метил]-6-бром-7-оксо-4-тиа-1-азабицикло[3.2.0]гепт-2-ен-2-карбоновой кислоты в виде твердого вещества, которое дополнительно очищают хроматографией на колонке с SiO2, элюирование осуществляют смесью 50% этилацетат:гексан.
Полученное светло-желтое твердое вещество растворяют в ТГФ (17 мл), быстро добавляют свежеактивированную Zn пыль (2,2 г) с 0,5М фосфатным буфером (pH 6,5, 17 мл). Реакционный сосуд покрывают фольгой, чтобы исключить воздействие света. Реакционную смесь энергично перемешивают в течение 2 ч при комнатной температуре. Реакционный раствор фильтруют через рыхлый слой целита, слой целита промывают водой (40 мл) и н-бутанолом (30 мл). Водный слой отделяют, а органический слой затем экстрагируют 0,5М фосфатным буфером (pH 6,5; 2 × 10 мл). Объединенный водный слой концентрируют до 23 г, добавляют 1М раствор NaOH для доведения рН до 7,25 и наносят на хроматографическую колонку со смолой Diaion HP-21 (30 мл, Mitsubishi Kasei Co. Ltd.). После адсорбции осуществляют элюирование водой, затем 10% раствором ацетонитрила в воде. Объединенные фракции, содержащие продукт, концентрируют в высоком вакууме при 35°C, лиофилизуют и получают натриевую соль (5R),(6Z)-6-(5,6-дигидро-8H-имидазо[2,1-c][1,4]тиазин-2-илметилен)-7-оксо-4-тиа-1-азабицикло[3.2.0]гепт-2-ен-2-карбоновой кислоты в виде желтого аморфного твердого вещества (168 мг, 20,9%). Т.пл. 135°C (с разложением).
1H ЯМР (δ, D2O): 3,00 (т, 2H, J=5,7 Гц), 3,80 (AB, 2H, J=16,7 и 18,1 Гц), 4,19 (т, 2H, J=5,7 Гц), 6,44 (д, 1H, J=0,8 Гц), 6 89 (с, 1H), 6,93 (с, 1H), 7,29 (с, 1H). (M+H) 322.
Пример 13
Получение натриевой соли (5R),(6Z)-6-(6,7-дигидро-5Н-пирроло[1,2-а]имидазо-2-илметилен)-7-оксо-4-тиа-1-азабицикло[3.2.0]гепт-2-ен-2-карбоновой кислоты
Стадия 1. 6,7-Дигидро-5H-пирроло[1,2-a]имидазол-2-карбальдегид
К раствору гидрохлорида 4,5-дигидро-3H-пиррол-2-иламина (3,27 г) в EtOH (250 мл) добавляют 28% раствор метоксида натрия (5,26 г) при комнатной температуре. После перемешивания в течение 5 мин при комнатной температуре к полученной смеси добавляют 2-бром-3-пропоксипропеналь (5,79 г), затем реакционную смесь перемешивают в течение 1 ч при комнатной температуре и упаривают досуха в вакууме. Остаток растворяют в CHCl3 (300 мл) и добавляют триэтиламин (3,8 мл). Смесь нагревают и кипятят с обратным холодильником в течение 3 ч. Реакционную смесь охлаждают до комнатной температуры, промывают 50% K2CO3, сушат над безводным K2CO3, фильтруют и упаривают при пониженном давлении. Остаток наносят на хроматографическую колонку с силикагелем, элюируют смесью CHCl3-ацетон (2:1) и получают 6,7-дигидро-5H-пирроло[1,2-a]имидазол-2-карбальдегид (1,51 г, 41%) в виде светло-желтого твердого вещества.
1H ЯМР (δ, CDCl3): 2,62-2,7 (м, 2H), 2,90-2, 94 (м, 2H), 4,07 (т, 2H, J=7,2 Гц), 7,59 (с, 1H), 9,80 (с, 1H).
Стадия 2. Натриевая соль(5R),(6Z)-6-(6,7-дигидро-5H-пирроло[1,2-a]имидазол-2-илметилен)-7-оксо-4-тиа-1-азабицикло[3.2.0]гепт-2-ен-2-карбоновой кислоты
6,7-Дигидро-5H-пирроло[1,2-a]имидазол-2-карбальдегид (1,36 г) добавляют к раствору безводного MgBr2 (5,64 г) в сухом ацетонитриле (155 мл) в атмосфере аргона при комнатной температуре. К полученной смеси добавляют раствор 4-нитробензилового эфира (5R,6S)-6-бром-7-oxo-4-тиа-1-азабицикло[3.2.0]гепт-2-ен-2-карбоновой кислоты (3,86 г) в сухом THF (155 мл), охлаждают до -20°С и добавляют Et3N (4,18 мл) в один прием. Реакционный сосуд покрывают фольгой, чтобы исключить воздействие света. Реакционную смесь перемешивают в течение 3 ч при -20°С и обрабатывают уксусным ангидридом (1,89 мл) и DMAP (370 мг) в один прием. Температуру реакционной смеси доводят до 0°С и перемешивают в течение 14,5 ч при 0°C. Смесь разбавляют этилацетатом и промывают 1М водным раствором лимонной кислоты, насыщенным раствором гидрокарбоната натрия и насыщенным солевым раствором. Органический слой сушат (MgSO4) и фильтруют. Рыхлый слой промывают этилацетатом. Фильтрат концентрируют при пониженном давлении. Остаток растворяют в ТГФ (166 мл) и ацетонитриле (77 мл). Быстро добавляют свежеактивированную Zn пыль (23,2 г) с 0,5M фосфатным буфером (pH 6,5, 243 мл). Реакционный сосуд покрывают фольгой, чтобы исключить воздействие света. Реакционную смесь энергично перемешивают в течение 2 ч при комнатной температуре. Реакционную смесь фильтруют, охлаждают до 3°С и добавляют 1M NaOH, чтобы довести рН до 8. Фильтрат промывают этилацетатом и водный слой отделяют. К водному слою вновь добавляют 1M NaOH для доведения рН до 8. Полученную смесь концентрируют в высоком вакууме при 35°C. Концентрат наносят на хроматографическую колонку со смолой Diaion HP-21 (20 мл, Mitsubishi Kasei Co. Ltd.). После адсорбции осуществляют элюирование системой H2O-MeCN (1/0˜9/1) и получают фракции очищенной натриевой соли (5R),(6Z)-6-(6,7-дигидро-5H-пирроло[1,2-a]имидазол-2-илметилен)-7-оксо-4-тиа-1-азабицикло[3.2.0]гепт-2-ен-2-карбоновой кислоты. Объединенные фракции концентрируют в высоком вакууме при 35°C, лиофилизуют и получают указанное в заголовке соединение в виде желтого аморфного твердого вещества (681 мг, 24%, pH 7,8). Т.пл. 190°C (с разложением).
1H ЯМР (δ, D2O): 2,48-2,56 (м, 2H), 2,74-2,79 (м, 2H), 3,94-3,99 (м, 2H), 6,47 (д, 1H, J=0,7 Гц), 6,94 (с, 1H), 6,95 (с, 1H), 7,36 (с, 1H); (M+H) 291.
Пример 14
Получение (5R,6Z)-6-(имидазо[2,1-b][1,3]бензотиазол-2-илметилен)-7-оксо-4-тиа-1-азабицикло[3.2.0]гепт-2-ен-2-карбоновой кислоты
Стадия 1. Этил имидазо[2,1-b]бензотиазол-2-карбоксилат
Этил бромпируват (9,8 г, 50 ммоль) добавляют по каплям к перемешиваемому раствору 2-аминобензотиазола (7,5 г, 50 ммоль) в ДМФА (100 мл) при комнатной температуре. После добавления реакционную смесь кипятят с обратным холодильником в течение 6 ч. Реакционную смесь охлаждают до комнатной температуры и гасят ледяной водой. Водный слой нейтрализуют NH4OH, выделившееся твердое вещество отфильтровывают, тщательно промывают водой и сушат. Полученный сырой продукт представляет собой коричневое твердое вещество, выход 10 г (81%); M+H 248, т.пл. 97°C, используют на следующей стадии без очистки.
Стадия 2. Имидазо[2,1-b]бензотиазол-2-метанол
К перемешиваемой взвеси LiAlH4 (2,0 г, избыток) в сухом ТГФ медленно добавляют этил имидазо[2,1-b]бензотиазол-2-карбоксилат (4,9 г, 20 ммоль) в ТГФ (100 мл) при 0°C. После прибавления реакционную смесь перемешивают при комнатной температуре в течение 1 ч и гасят смесью насыщенных растворов NH4Cl/NH4OH. Отделенное твердое вещество смешивают со смесью хлороформ/MeOH (3:1) и фильтруют через рыхлый слой целита. Органический слой промывают один раз насыщенным раствором NaCl, сушат над безводным MgSO4, фильтруют и концентрируют. Полученное коричневое твердое вещество используют на следующей стадии без очистки. Выход 3,8 г (93%), (M+H) 205; т.пл. 131°С.
Стадия 3. 2-Формилимидазо[2,1-b]бензотиазол
К перемешиваемому раствору имидазо[2,1-b]бензотиазол-2-метанола (2,04 г, 10 ммоль) в метиленхлориде (200 мл) добавляют активированный MnO2 (15 г, избыток). Реакционную смесь перемешивают при комнатной температуре в течение 24 ч и фильтруют через рыхлый слой целита. Реакционную смесь концентрируют и продукт очищают хроматографией на колонке с силикагелем при элюировании системой 75% этилацетат:гексан. Получают коричневое твердое вещество, выход 800 мг (40%); (M+H) 203.
Стадия 4. 4-Нитробензил-6-[(ацетилокси)(имидазо[2,1-b][1,3]бензотиазол-2-ил)метил]-6-бром-7-оксо-4-тиа-1-азабицикло[3.2.0]гепт-2-ен-2-карбоксилат
2-Формилимидазо[2,1-b]бензотиазол (444 мг, 2,2 ммоль) и раствор 4-нитробензилового эфира (5R,6S)-6-бром-7-оксо-4-тиа-1-азабицикло[3.2.0]гепт-2-ен-2-карбоновой кислоты (772 мг, 2 ммоль) в сухом ТГФ (20 мл) последовательно добавляют к раствору безводный MgBr2: эфират (619 мг, 2,4 ммоль) в сухом ацетонитриле (15 мл) в атмосфере аргона при комнатной температуре. После охлаждения до -20°С добавляют за один прием Et3N (2,0 мл). Реакционный сосуд покрывают фольгой, чтобы исключить воздействие света. Реакционную смесь перемешивают в течение 2 ч при -20°C и обрабатывают уксусным ангидридом (1,04 мл) в один прием. Температуру реакционной смеси доводят до 0°C и перемешивают в течение 15 ч при 0°С. Смесь разбавляют этилацетатом и промывают 5% водным раствором лимонной кислоты, насыщенным раствором гидрокарбоната натрия и насыщенным солевым раствором. Органический слой сушат (MgSO4), фильтруют через рыхлый слой целита, который затем промывают этилацетатом. Фильтрат концентрируют при пониженном давлении. Остаток наносят на хроматографическую колонку с силикагелем, затем элюируют смесью этилацетат:гексан (1:1). Собранные фракции концентрируют при пониженном давлении и полученную смесь диастереомеров (в виде светло-желтого аморфного твердого вещества) используют на следующей стадии. Выход 850 мг (67%). Т.пл. 69°C; (M+H) 630.
Стадия 5. (5R),(6Z)-6-(Имидазо[1,2-b][1,3]бензотиазол-2-илметилен)-7-оксо-4-тиа-1-азабицикло[3.2.0]гепт-2-ен-2-карбоновая кислота
4-Нитробензил 6-[(ацетилокси)(имидазо[2,1-b][1,3]бензотиазол-2-ил)метил]-6-бром-7-оксо-4-тиа-1-азабицикло[3.2.0]гепт-2-ен-2-карбоксилат (500 мг, 0,79 ммоль) растворяют в ТГФ (17 мл) и ацетонитриле (36 мл). Быстро добавляют свежеактивированную Zn пыль (5,2 г) с 0,5M фосфатным буфером (pH 6,5, 28 мл). Реакционный сосуд покрывают фольгой, чтобы исключить воздействие света. Реакционную смесь энергично перемешивают в течение 2 ч при комнатной температуре. Реакционную смесь фильтруют, охлаждают до 3°C и добавляют 1 н. HCl для доведения pH до 7,5. Фильтрат промывают этилацетатом и водный слой отделяют. Водный слой концентрируют в высоком вакууме при 35°C и получают желтый осадок, который растворяют в ацетонитриле и загружают в колонку с обращенной фазой HP-21. Элюирование осуществляют сначала деионизированной водой (2 л), затем 10% ацетонитрилом в воде. Выход желтых кристаллов составляет 105 мг (35%). Т.пл. 233°C, (M+H) 356.
1H ЯМР (ДМСО-d6) δ: 6,51 (с, 1H), 6,53 (с, 1H), 7,09 (с, 1H), 7,47 (т, 1H, J=7,5 Гц), 7,54 (т, 1H, J=7,5 Гц), 8,06 (т, 1H), 8,62 (с, 1H).
Пример 15
Получение (5R,6Z)-6-[(7-метоксиимидазо[2,1-b][1,3]бензотиазол-2-илметилен)-7-оксо-4-тиа-1-азабицикло[3.2.0]гепт-2-ен-2-карбоновой кислоты
Стадия 1. Этил 7-метоксиимидазо[2,1-b]бензотиазол-2-карбоксилат
Этил 7-метоксиимидазо[2,1-b]бензотиазол-2-карбоксилат получают по методике, описанной в примере 1 (Стадия 1). Из исходных 6-метокси-2-аминобензотиазола (27 г, 0,15 моль) и этилбромпирувата (39,9 г, 0,2 моль) выделяют 24 г этил 7-метоксиимидазо[2,1-b]бензотиазол-2-карбоксилата (выход 43%) в виде коричневого твердого вещества, (M+H) 277.
Стадия 2. 7-Метоксиимидазо[2,1-b]бензотиазол-2-метанол
7-Метоксиимидазо[2,1-b]бензотиазол-2-метанол получают по методике, изложенной в примере 1 (Стадия 2). Из исходных этил 7-метоксиимидазо[2,1-b]бензотиазол-2-карбоксилата (12,5 г, 43,5 ммоль) и раствора LiAlH4 (43,5 мл, 0,5M раствора в ТГФ), выделяют 4,0 г (выход 40%) спиртового производного в виде коричневого твердого вещества. (M+H) 235.
Стадия 3. 2-Формил-7-метоксиимидазо[2,1-b]-бензотиазол
2-Формил-7-метоксиимидазо[2,1-b]-бензотиазол получают по методике, кратко изложенной в примере 1 (Стадия 3). Из исходных 7-метоксиимидазо[2,1-b]бензотиазол-2-метанола (4,0 г, 17 ммоль) в смеси метиленхлорид/ДМФА (300 мл:50 мл) и активного MnO2 (12 г, избыток), получают 822 мг (выход 21%) альдегидного производного в виде коричневого твердого вещества. (M+H) 233.
Стадия 4. 4-Нитробензил-6-[(ацетилокси)(7-метоксиимидазо[2,1-b][1,3]бензотиазол-2-ил)метил]-6-бром-7-оксо-4-тиа-1-азабицикло[3.2.0]гепт-2-ен-2-карбоксилат
2-Формил-7-метоксиимидазо[2,1-b]бензотиазол (822 мг, 3,5 ммоль) и раствор 4-нитробензилового эфира (5R,6S)-6-бром-7-оксо-4-тиа-1-азабицикло[3.2.0]гепт-2-ен-2-карбоновой кислоты (1,364 г, 3,54 ммоль) в сухом ТГФ (40 мл) последовательно добавляют к раствору безводный MgBr2: эфират (1,3 г, 5 ммоль) в сухом ацетонитриле (15 мл) в атмосфере аргона при комнатной температуре. После охлаждения до -20°C добавляют Et3N (2,0 мл) в один прием. Реакционный сосуд покрывают фольгой, чтобы исключить воздействие света. Реакционную смесь перемешивают в течение 2 ч при -20°C и обрабатывают уксусным ангидридом (1,04 мл) в один прием. Температуру реакционной смеси доводят до 0°C и перемешивают в течение 15 ч при 0°С. Смесь разбавляют этилацетатом и промывают 5% водным раствором лимонной кислоты, насыщенным раствором гидрокарбоната натрия и насыщенным солевым раствором. Органический слой сушат (MgSO4) и фильтруют через рыхлый слой целита. Рыхлый слой промывают этилацетатом. Фильтрат концентрируют при пониженном давлении. Остаток наносят на хроматографическую колонку с силикагелем, затем элюируют смесью этилацетат-гексан (1:1). Собранные фракции концентрируют при пониженном давлении и полученную смесь диастереомеров (в виде бледно-желтого аморфного твердого вещества) используют на следующей стадии. Выход 2,24 г (95%); (M+H) 660.
Стадия 5. (5R),(6Z)-6-[(7-Метоксиимидазо[1,2-b][1,3]бензотиазол-2-илметилен)]-7-оксо-4-тиа-1-азабицикло[3.2.0]гепт-2-ен-2-карбоновая кислота
4-Нитробензил 6-[(ацетокси)(7-метоксиимидазо[2,1-b][1,3]бензотиазол-2-ил)метил]-6-бром-7-оксо-4-тиа-1-азабицикло[3.2.0]гепт-2-ен-2-карбоксилат (659 мг, 1,0 ммоль) растворяют в ТГФ (17 мл) и ацетонитриле (36 мл). Быстро добавляют свежеактивированную Zn пыль (5,2 г) с 0,5M фосфатным буфером (pH 6,5, 28 мл). Реакционный сосуд покрывают фольгой, чтобы исключить воздействие света. Реакционную смесь энергично перемешивают в течение 2 ч при комнатной температуре. Реакционную смесь фильтруют, охлаждают до 3°C и добавляют 1 н. HCl для доведения рН до 7,5. Фильтрат промывают этилацетатом и отделяют водный слой. Водный слой концентрируют в высоком вакууме при 35°C и получают желтый осадок. Осадок отфильтровывают, промывают H2O, MeCN, ацетоном и получают указанное в заголовке соединение в виде желтых кристаллов, т.пл. 284°С. Выход 68 мг (23%); (M+H) 386.
1H ЯМР (ДМСО-d6) δ: 3,89 (с, 3H), 6,58 (с, 1H), 6,64 (с, 1H), 7,14 (с, 1H), 7,2 (дд, 1H, J=6,0 Гц), 7,75 (д, 1H, J=3,0 Гц), 8,03 (д, 1Н, J=6,0 Гц), 8,62 (с, 1H).
Пример 16
Получение (5R,6Z)-6-[(7-хлоримидазо[2,1-b][1,3]бензотиазол-2-илметилен)-7-оксо-4-тиа-1-азабицикло[3.2.0]гепт-2-ен-2-карбоновой кислоты
Стадия 1. Этил 7-хлоримидазо[2,1-b]бензотиазол-2-карбоксилат
Этил 7-хлоримидазо[2,1-b]бензотиазол-2-карбоксилат получают по методике, изложенной в примере 1 (Стадия 1). Из исходных 6-хлор-2-аминобензотиазола (9,2 г, 50 ммоль) и этилбромпирувата (11,6 г, 60 ммоль), выделяют 8,5 г (выход 60%) этил 7-хлоримидазо[2,1-b]-бензотиазол-2-карбоксилата в виде коричневого твердого вещества. (M+H) 281.
Стадия 2. 7-Хлоримидазо[2,1-b]-бензотиазол-2-метанол
7-хлоримидазо[2,1-b]-бензотиазол-2-метанол получают по методике, кратко изложенной в примере 1 (Стадия 2). Из исходных этил 7-хлоримидазо[2,1-b]бензотиазол-2-карбоксилата (9,0 г, 32,1 ммоль) и LiAlH4 (4,0 г, избыток), получают 5,5 г (выход 72%) спиртового производного в виде коричневого твердого вещества; т.пл. 166°C; (M+H) 239.
Стадия 3. 2-Формил-7-хлоримидазо[2,1-b]бензотиазол
2-Формил-7-хлоримидазо[2,1-b]бензотиазол получают по методике, изложенной в примере 1 (Стадия 3). Из исходных 7-хлоримидазо[2,1-b]бензотиазол-2-метанола (4,0 г, 16,8 ммоль) в смеси метиленхлорид/MeOH (300 мл/50 мл) и активного MnO2 (20 г, избыток) выделяют 2,2 г (выход 55%) альдегидного производного в виде коричневого твердого вещества.(M+H) 236.
Стадия 4. 4-Нитробензил 6-[(ацетилокси)(7-хлоримидазо[2,1-b][1,3]бензотиазол-2-ил)метил]-6-бром-7-оксо-4-тиа-1-азабицикло[3.2.0]гепт-2-ен-2-карбоксилат
2-Формил-7-хлоримидазо[2,1-b]бензотиазол (270 мг, 1,14 ммоль) и раствор 4-нитробензилового эфира (5R,6S)-6-бром-7-оксо-4-тиа-1-азабицикло[3.2.0]гепт-2-ен-2-карбоновой кислоты (500 мг, 1,14 ммоль) в сухом ТГФ (20 мл) последовательно добавляют к раствору безводного MgBr2:O(Et)2 (390 мг, 1,5 ммоль) в сухом ацетонитриле (15 мл) в атмосфере аргона при комнатной температуре. После охлаждения до -20°C добавляют в один прием Et3N (2,0 мл). Реакционный сосуд покрывают фольгой, чтобы исключить воздействие света. Реакционную смесь перемешивают в течение 2 ч при -20°C и обрабатывают уксусным ангидридом (1,04 мл) за один прием. Температуру реакционной смеси доводят до 0°C и перемешивают в течение 15 ч при 0°C. Смесь разбавляют этилацетатом и промывают 5% водным раствором лимонной кислоты, насыщенным раствором гидрокарбоната натрия и насыщенным солевым раствором. Органический слой сушат (MgSO4) и фильтруют через рыхлый слой целита. Рыхлый слой промывают этилацетатом. Фильтрат концентрируют при пониженном давлении. Остаток наносят на хроматографическую колонку с силикагелем, элюирование осуществляют смесью этилацетат:гексан (1:1). Собранные фракции концентрируют при пониженном давлении и получают смесь диастереомеров в виде бледно-желтого аморфного твердого вещества, которое используют на следующей стадии. Выход 495 мг (65%); (M+H) 665.
Стадия 5. (5R),(6Z)-6-[(7-Хлоримидазо[1,2-b][1,3]бензотиазол-2-ил)метилен]-7-оксо-4-тиа-1-азабицикло[3.2.0]гепт-2-ен-2-карбоновая кислота
4-Нитробензил-6-[(ацетилокси)(7-хлоримидазо[2,1-b][1,3]бензотиазол-2-ил)метил]-6-бром-7-оксо-4-тиа-1-азабицикло[3.2.0]гепт-2-ен-2-карбоксилат (450 мг, 0,67 ммоль) растворяют в ТГФ (20 мл) и ацетонитриле (10 мл). Быстро добавляют свежеактивированную Zn пыль (5,2 г) с 0,5M фосфатным буфером (pH 6,5, 28 мл). Реакционный сосуд покрывают фольгой, чтобы исключить воздействие света. Реакционную смесь энергично перемешивают в течение 2 ч при комнатной температуре. Реакционную смесь фильтруют, охлаждают до 3°C и добавляют 0,1 н. NaOH добавляют, доводя pH до 8,5. Фильтрат промывают этилацетатом, отделяют водный слой, концентрируют его в высоком вакууме при 35°C и получают желтый осадок. Продукт очищают хроматографией на колонке с обращенной фазой HP-21. Сначала элюирование проводят деионизированной водой (2 л) и потом 10% раствором ацетонитрила в воде. Фракции, содержащие продукт, собирают и концентрируют при пониженном давлении при комнатной температуре. Полученное желтое твердое вещество промывают ацетоном, отфильтровывают, сушат и получают желтые кристаллы с т.пл. 240°С (выход 80 мг, 18%); (M+H+Na) 412.
1H ЯМР (ДМСО-d6) δ: 6,6 (с, 2H), 7,1 (с, 1H), 7,62 (дд, 1H), 8,11 (д, 1H), 8,2 (с, 1H), 8,6 (с, 1H).
Пример 17
Получение (5R),(6Z)-6-имидазо[1,2-a]хинолин-2-илметилен-7-оксо-4-тиа-1-азабицикло[3.2.0]гепт-2-ен-2-карбоновой кислоты
Имидазо[1,2-a]хинолин-2-карбальдегид
Имидазо[1,2-a]хинолин-2-карбальдегид (4) получают по методу, разработанному Westwood и сотрудниками (J. Med. Chem. 1988, 31, 1098-1115).
Стадия 1. 4-Нитробензиловый эфир (5R,6RS)-6-[(RS)-ацетоксиимидазо[1,2-a]хинолин-2-илметил]-6-бром-7-оксо-4-тиа-1-азабицикло[3.2.0]гепт-2-ен-2-карбоновой кислоты
Имидазо[1,2-a]хинолин-2-карбальдегид (1,09 г) и раствор 4-нитробензилового эфира (5R,6S)-6-бром-7-оксо-4-тиа-1-азабицикло[3.2.0]гепт-2-ен-2-карбоновой кислоты (2,22 г) в сухом ТГФ (75,5 мл) последовательно добавляют к раствору безводного MgBr2 (2,5 г) в сухом ацетонитриле (75,5 мл) в атмосфере аргона при комнатной температуре. После охлаждения до -20°C добавляют Et3N (1,85 мл) в один прием. Реакционный сосуд покрывают фольгой, чтобы исключить воздействие света. Реакционную смесь перемешивают в течение 2 ч при -20°C и обрабатывают уксусным ангидридом (1,04 мл) за один прием. Температуру реакционной смеси доводят до 0°C и перемешивают в течение 15 ч при 0°С. Смесь разбавляют этилацетатом и промывают 5% водным раствором лимонной кислоты, насыщенным раствором гидрокарбоната натрия и насыщенным солевым раствором. Органический слой сушат (MgSO4), фильтруют через рыхлый слой целита, который затем промывают этилацетатом. Фильтрат концентрируют при пониженном давлении. Остаток наносят на хроматографическую колонку с силикагелем, затем элюируют смесью CHCl3-ацетон (1/0˜95/5). Собранные фракции концентрируют при пониженном давлении, затем перекристаллизовывают остаток из смеси CHCl3-Et2O и получают указанное в заголовке соединение в виде одного изомера, который представляет собой бледно-желтые кристаллы, выход 1,3 г (38%).
1H ЯМР (CDCl3) δ: 2,37 (с, 3H), 5,29 (д, 1H, J=13,5 Гц), 5,45 (д, 1H, J=13,5 Гц), 6,22 (с, 1H), 7,14 (с, 1H), 7,46-7,52 (м, 3H), 7,56 (д, 1H, J=9,6 Гц), 7,62 (д, 2H, J=8,6 Гц), 7,64-7,69 (м, 1H), 7,83 (дд, 1H, J=1,1 и 7,9 Гц), 7,93 (д, 1H, J=8,3 Гц), 7,99 (с, 1H), 8,25 (д, 2H, J=8,6 Гц).
Стадия 2. (5R),(6Z)-6-Имидазо[1,2-a]хинолин-2-ил)метилен-7-оксо-4-тиа-1-азабицикло[3.2.0]гепт-2-ен-2-карбоновая кислота
4-Нитробензиловый эфир (5R,6RS)-6-[(RS)-ацетоксиимидазо[1,2-a]хинолин-2-илметил]-6-бром-7-оксо-4-тиа-1-азабицикло[3.2.0]гепт-2-ен-2-карбоновой кислоты (1,3 г) растворяют в ТГФ (17 мл) и ацетонитриле (36 мл). Быстро добавляют свежеактивированную Zn пыль (5,2 г) с 0,5M фосфатным буфером (pH 6,5, 28 мл). Реакционный сосуд покрывают фольгой, чтобы исключить воздействие света. Реакционную смесь энергично перемешивают в течение 2 ч при комнатной температуре. Реакционную смесь фильтруют, охлаждают до 3°C и добавляют 1 н. HCl, доводя pH до 7,5. Фильтрат промывают этилацетатом, водный слой отделяют, концентрируют в высоком вакууме при 35°C и получают желтый осадок. Осадок отфильтровывают, промывают ацетоном и получают указанное в заголовке соединение в виде желтых кристаллов. Т.пл. 205°С, выход 297 мг (38%).
1H ЯМР (D2О) δ: 6,19 (с, 1H), 6,36 (с, 1H), 6,87 (с, 1H), 6,96 (д, 1H, J=9,5 Гц), 7,32 (д, 1H, J=9,5 Гц), 7,33 (с, 1H), 7,44-7,57 (м, 4H).
Пример 18
Получение (5R),(6Z)-6-(6,7-дигидро-5H-циклопента[d]имидазо[2,1-b][1,3]тиазол-2-илметилен)-7-оксо-4-тиа-1-азабицикло[3.2.0]гепт-2-ен-2-карбоновой кислоты
Стадия 1. Получение этил 6,7-дигидро-5H-циклопента[d]имидазо[2,1-b][1,3]тиазол-2-карбоксилата
Смесь 2-хлорциклопентанона (11,8 г, 100 ммоль) и тиомочевины (8,0 г, 101 ммоль) в смеси этанол: ТГФ (1:2) кипятят с обратным холодильником в течение 16 ч. Реакционную смесь охлаждают до комнатной температуры и выделившееся белое твердое вещество отфильтровывают (9,0 г отделено), затем растворяют в безводном этаноле (100 мл) и метоксиде натрия (1,9 г, 2,7 г, 51 ммоль). К полученному раствору добавляют этилбромпируват (10,0 г), перемешивают при комнатной температуре в течение 2 ч и кипятят с обратным холодильником в течение 48 ч. Затем реакционную смесь охлаждают до комнатной температуры и концентрируют. Остаток экстрагируют хлороформом и тщательно промывают водой. Продукт очищают хроматографией на колонке с силикагелем при элюировании смесью 50% этилацетат:гексан. Получают красное полутвердое вещество, выход 3,0 г, (M+H) 237.
Сложный эфир восстанавливают LiAlH4 и полученный спирт окисляют активным MnO2. Полученный альдегид используют на следующей стадии.
Стадия 3. Получение 4-нитробензил (5R)-6-[(ацетилокси)(6,7-дигидро-5H-циклопента[d]имидазо[2,1-b][1,3]тиазол-2-ил)метил-6-бром-7-оксо-4-тиа-1-азабицикло[3.2.0]гепт-2-ен-2-карбоксилата
2-Формил-6,7-дигидро-5H-циклопента[d]имидазо[2,1-b][1,3]тиазол (600 мг, 3,1 ммоль) и раствор 4-нитробензилового эфира (5R,6S)-6-бром-7-оксо-4-тиа-1-азабицикло[3.2.0]гепт-2-ен-2-карбоновой кислоты (1,2 г, 3 ммоль) в сухом ТГФ (20 мл) добавляют последовательно к раствору безводного MgBr2:O(Et)2 (1,2 г, 3,0 ммоль) в сухом ацетонитриле (15 мл) в атмосфере аргона при комнатной температуре. После охлаждения до -20°С добавляют в один прием Et3N (2,0 мл). Реакционный сосуд покрывают фольгой, чтобы исключить воздействие света. Реакционную смесь перемешивают в течение 2 ч при -20°С и обрабатывают уксусным ангидридом (1,04 мл) в один прием. Температуру реакционной смеси доводят до 0°С и перемешивают в течение 15 ч при 0°C. Смесь разбавляют этилацетатом и промывают 5% водным раствором лимонной кислоты, насыщенным раствором гидрокарбоната натрия и насыщенным солевым раствором. Органический слой сушат (MgSO4) и фильтруют через рыхлый слой целита, который затем промывают этилацетатом. Фильтрат концентрируют при пониженном давлении. Остаток наносят на хроматографическую колонку с силикагелем, элюируют смесью этилацетат:гексан (1:1). Собранные фракции концентрируют при пониженном давлении и полученную смесь диастереомеров в виде бледно-желтого аморфного твердого вещества используют на следующей стадии. Выход 850 мг (45%); (M+H) 620.
Стадия 4. Получение (5R),(6Z)-6-(6,7-дигидро-5H-циклопента[d] имидазо[2,1-b][1,3]тиазол-2-илметилен)-7-оксо-4-тиа-1-азабицикло[3.2.0]гепт-2-ен-2-карбоновой кислоты
4-Нитробензил (5R)-6-[(ацетилокси)(6,7-дигидро-5H-циклопента[d]имидазо[2,1-b][1,3]тиазол-2-ил)-6-бром-7-оксо-4-тиа-1-азабицикло[3.2.0]гепт-2-ен-2-карбоксилат (850 мг, 1,37 ммоль) растворяют в ТГФ (20 мл) и ацетонитриле (10 мл). Быстро добавляют свежеактивированную Zn пыль (5,2 г) с 0,5M фосфатным буфером (pH 6,5, 28 мл). Реакционный сосуд покрывают фольгой, чтобы исключить воздействие света. Реакционную смесь энергично перемешивают в течение 2 ч при комнатной температуре. Реакционную смесь фильтруют, охлаждают до 3°C и добавляют 0,1 н. NaOH, доводя рН до 8,5. Фильтрат промывают этилацетатом и отделяют водный слой. Водный слой концентрируют в высоком вакууме при 35°C и получают желтый осадок. Продукт очищают хроматографией на колонке с обращенной фазой со смолой HP-21. Сначала элюирование осуществляют деионизированной водой (2 л), затем смеью 10% ацетонитрил:вода. Собирают фракции, содержащие продукт, и концентрируют при пониженном давлении при комнатной температуре. Желтое твердое вещество промывают ацетоном, отфильтровывают, сушат и получают желтые кристаллы, т.пл. 192°С. Выход 138 мг (29%); (M+H+Na) 346.
1H ЯМР (ДМСО-d6) δ: 8,2 (с, 1H), 7,1 (с, 1H), 6,55 (с, 1H), 6,4 (с, 1H), 3,01 (м, 2H), 2,51 (м, 4H).
Пример 19
Получение натриевой соли (5R),(6Z)-6-(имидазо[1,2-a]хиноксалин-2-илметилен)-7-оксо-4-тиа-1-азабицикло[3.2.0]гепт-2-ен-2-карбоновой кислоты
Имидазо[1,2-a]хиноксалин-2-карбоксальдегид
Имидазо[1,2-a]хиноксалин-2-карбоксальдегид получают методом, разработанным Westwood и сотрудниками (J. Med. Chem. 1998, 31, 1098-1115).
Стадия 1. п-Нитробензиловый эфир (5R,6RS)-6-((RS)-ацетоксиимидазо[1,2-a]хиноксалин-2-илметил)-6-бром-7-оксо-4-тиа-1-азабицикло[3.2.0]гепт-2-ен-2-карбоновой кислоты
Раствор имидазо[1,2-a]хиноксалин-2-карбальдегида (505 мг) в сухом ацетонитриле (33 мл) добавляют к раствору MgBr2 (1,1 г) в сухом ацетонитриле (20 мл) в атмосфере азота при комнатной температуре и смесь перемешивают в течение 10 мин. Добавляют раствор 4-нитробензилового эфира (5R,6S)-6-бром-7-оксо-4-тиа-1-азабицикло[3.2.0]гепт-2-ен-2-карбоновой кислоты (931 мг) в сухом ТГФ (25 мл), затем смесь охлаждают до -20°C и добавляют триэтиламин (0,8 мл) в один прием. Реакционный сосуд покрывают фольгой, чтобы исключить воздействие света. Реакционную смесь перемешивают в течение 4 ч при -20°С и обрабатывают в один прием 4,4-диметиламинопиридином (58 мг) и уксусным ангидридом (0,44 мл). Температуру реакционной смеси доводят до 0°С и реакционную смесь перемешивают в течение 16 ч при 0°C. К реакционной смеси добавляют 10% водный раствор лимонной кислоты (200 мл) и водный слой экстрагируют этилацетатом (3 × 100 мл). Органический слой промывают водой, насыщенным раствором гидрокарбоната натрия и насыщенным солевым раствором, сушат (MgSO4) и фильтруют. Фильтрат концентрируют при пониженном давлении. Остаток очищают колоночной хроматографией на силикагеле при элюировании смесью CH2Cl2-ацетон (50:1) и получают указанное в заголовке соединение в виде смеси диастереомеров (78:22), которая представляет собой светло-коричневое пенящееся аморфное вещество (1,0 г, 68,9%).
1H ЯМР (CDCl3) δ: 2,07 (с, 0,66H), 2,38 (с, 2,34H), 5,30 (д, 1H, J=13,5 Гц), 5,45 (д, 0,78H, J=13,5 Гц), 5,48 (д, 0,22H, J=13,5 Гц), 6,24 (с, 0,78H), 6,46 (с, 0,22H), 6,63 (с, 0,22H), 7,18 (с, 0,78H), 7,50 (с, 0,78H), 7,52 (с, 0,22H), 7,61 (д, 1,56H, J=8,7 Гц), 7,63 (д, 0,44H, J=8,8 Гц), 7,64-7,67 (м, 1H), 7,68-7,73 (м, 1H), 7,92-7,95 (м, 1H), 8,08 (с, 0,78H), 8,13-8,16 (м, 1H), 8,24 (д, 1,56H, J=8,7 Гц), 8,25 (д, 0,44H, J=8,8 Гц), 8,33 (с, 0,22H), 9,05 (с, 0,78H), 9,09 (с, 0,22H).
Стадия 2. Натриевая соль (5R),(6Z)-6-(имидазо[1,2-a]хиноксалин-2-илметилен)-7-оксо-4-тиа-1-азабицикло[3.2.0]гепт-2-ен-2-карбоновой кислоты
п-Нитробензиловый эфир (5R,6RS)-6-((RS)-ацетоксиимидазо[1,2-a]хиноксалин-2-илметил)-6-бром-7-оксо-4-тиа-1-азабицикло[3.2.0]гепт-2-ен-2-карбоновой кислоты (951 мг) и 10% Pd-C (50% влажность, 477 мг) добавляют к смеси ТГФ (48 мл) и 0,5М фосфатного буфера (pH 6,5, 48 мл). Смесь гидрируют при давлении водорода 400 кПа при комнатной температуре в течение 4 ч. Реакционный раствор фильтруют и Pd-C промывают водой и н-бутанолом. Водный слой отделяют и органический слой затем экстрагируют водой. Объединенный водный слой концентрируют до массы 57 г и наносят на хроматографическую колонку со смолой Diaion HP-21 (60 мл, Mitsubishi Kasei Co. Ltd.) После адсорбции осуществляют элюирование водой, а затем 5, 10, 15 и 20% растворами ацетонитрила и воды (каждый раз порциями по 60 мл). Объединенные фракции концентрируют в высоком вакууме при 35°C, лиофилизуют и получают указанное в заголовке соединение в виде желтого аморфного твердого вещества, выход 148 мг (26,1%). Т.пл. 300°C (с разложением).
1H ЯМР(D2О) δ: 5,92 (с, 1H), 6,23 (с, 1H), 6,66 (с, 1H), 7,11-7,22 (м, 3H), 7,25 (д, 1H, J=7,9 Гц), 7,50 (с, 1H), 8,03 (с, 1H); ИК (KBr): 3413, 1748, 1592, 1553 см-1; λмакс.(H2O) 340, 293, 237, 218 нм.
Пример 20
Получение натриевой соли (5R),(6Z)-6-(5,6-дигидро-8H-имидазо[2,1-c][1,4]оксазин-2-илметилен)-7-оксо-4-тиа-1-азабицикло[3.2.0]гепт-2-ен-2-карбоновой кислоты
Стадия 1. Морфолин-3-он
Морфолин-3-он может быть получен по методу, описанному в патенте США № 5349045.
Стадия 2. Морфолин-3-тион
Смесь морфолин-3-она (4,7 г) и реагент Лоуссона (10,3 г) в сухом ТГФ (94 мл) кипятят с обратным холодильником в течение 1,5 ч. Реакционную смесь охлаждают до комнатной температуры и растворитель удаляют в вакууме. Остаток наносят на хроматографическую колонку с силикагелем, элюируют смесью CHCl3-метанол (50:1) и получают желтое твердое вещество. Перекристаллизация сырого продукта из смеси гексан-этилацетат дает указанное в заголовке соединение (4,0 г, 72,2%) в виде желтого порошка.
1H ЯМР (CDCl3) δ: 3,45 (т, 2H, J=5,1 Гц), 3,91 (т, 2H, J=5,1 Гц), 4,55 (с, 2H).
Стадия 3. 5-Метилтио-3,6-дигидро-2H-[1,4]оксазин
Смесь морфолин-3-тиона (4,7 г) и метилиодида (13 мл) в сухом CH2Cl2 (140 мл) перемешивают при комнатной температуре в течение 15 ч. Реакционную смесь фильтруют и твердое вещество промывают CH2Cl2. Полученное твердое вещество растворяют в 50% водном растворе K2CO3 (150 мл) и водный слой экстрагируют CH2Cl2 (8 × 100 мл). Объединенный CH2Cl2 слой сушат (MgSO4) и фильтруют. Фильтрат концентрируют при пониженном давлении и получают указанное в заголовке соединение в виде бледно-желтого масла (3,6 г, 67,8%).
1H ЯМР (CDCl3) δ: 2,32 (с, 3H), 3,71-3,74 (м, 4H), 4,14-4,15 (м, 2H).
Стадия 4. Гидрохлорид 3-иминоморфолина
Смесь 5-метилтио-3,6-дигидро-2H-[1,4]оксазина (3,6 г) и хлорида аммония (1,5 г) в сухом этаноле (136 мл) кипятят с обратным холодильником в течение 1 ч. Реакционную смесь охлаждают до комнатной температуры. Растворитель удаляют в вакууме и получают указанное в заголовке соединение в виде светло-коричневого твердого вещества (3,6 г, 97,7%).
1H ЯМР (ДМСО-d6) δ: 3,34 (м, 2H), 3,86 (т, 2H, J=5,2 Гц), 4,47 (с, 2H).
Стадия 5. 5,6-Дигидро-8H-имидазо[2,1-c][1,4]оксазин-2-карбальдегид (9) и 5,6-дигидро-8H-имидазо[2,1-c][1,4]оксазин-3-карбальдегид
Из смеси 2-бром-3-гидроксипропеналя (4,1 г), моногидрата п-толуолсульфокислоты (52 мг) и 2-пропанола (5,2 мл) в циклогексане (42 мл) отгоняют азеотроп до тех пор, пока температура паров не достигнет 80°C. Реакционную смесь концентрируют при пониженном давлении. Остаток растворяют в сухом этаноле (50 мл). Добавляют при комнатной температуре смесь раствора гидрохлорида 3-иминоморфолина (3,4 г) в сухом этаноле (200 мл) и 28% метанольного раствора метилата натрия (4,8 г). Реакционную смесь перемешивают при комнатной температуре в течение 2 ч и затем растворитель удаляют в вакууме. Остаток растворяют в хлороформе (125 мл) и добавляют триэтиламин (3,5 мл), затем реакционную смесь кипятят с обратным холодильником в течение 2 ч. Реакционную смесь охлаждают до комнатной температуры и концентрируют при пониженном давлении.
Остаток растворяют в дихлорметане (300 мл) и промывают 50% водным раствором K2CO3 (2 × 100 мл). Органический слой сушат (MgSO4) и фильтруют. Фильтрат концентрируют при пониженном давлении. Остаток наносят на хроматографическую колонку с силикагелем, элюируют смесью CHCl3-ацетон (4:1) и получают указанное в заголовке соединение (светло-оранжевое твердое вещество, 1,4 г, 36,3%) и другой региоизомер (светло-оранжевое твердое вещество, 609 мг, 16,1%).
Целевой продукт: 1H ЯМР (CDCl3) δ: 4,08-4,15 (м, 4H), 4,88 (с, 2H), 7,58 (с, 1H), 9,85 (с, 1H).
Нежелательный региоизомер: 1H ЯМР (CDCl3) δ: 4,06 (т, 2H, J=5,2 Гц), 4,40 (т, 2H, J=5,2 Гц), 4,90 (с, 2H), 7,75 (с, 1H), 9,72 (с, 1H).
Стадия 6. Натриевая соль (5R),(6Z)-6-(5,6-дигидро-8H-имидазо[2,1-c][1,4]оксазин-2-илметилен)-7-оксо-4-тиа-1-азабицикло[3.2.0]гепт-2-ен-2-карбоновой кислоты
Раствор 5,6-дигидро-8H-имидазо[2,1-c][1,4]оксазин-2-карбальдегида (1,2 г) в сухом ацетонитриле (66 мл) добавляют к раствору MgBr2 (3,6 г) в сухом ацетонитриле (66 мл) в атмосфере азота при комнатной температуре, затем смесь перемешивают в течение 10 мин. Добавляют раствор п-нитробензил (5R,6S)-6-бромпенем-3-карбоксилата (3,4 г) в сухом ТГФ (132 мл), смесь охлаждают до -20°C, затем в один прием добавляют триэтиламин (2,8 мл). Реакционный сосуд покрывают фольгой, чтобы исключить воздействие света. Реакционную смесь перемешивают 4 ч при -20°C и в один прием обрабатывают 4-диметиламинопиридином (100 мг) и уксусным ангидридом (1,5 мл). Температуру реакционной смеси доводят до 0°C и перемешивают в течение 18 ч при 0°C. К реакционной смеси добавляют 10% водный раствор лимонной кислоты (1 л) и водный слой экстрагируют этилацетатом (3 × 500 мл). Объединенный органический слой промывают водой, насыщенным раствором гидрокарбоната натрия и насыщенным солевым раствором, сушат (MgSO4) и фильтруют. Фильтрат концентрируют при пониженном давлении и получают сырой п-нитробензиловый эфир (5R)-6-[ацетокси(5,6-дигидро-8H-имидазо[2,1-c][1,4]оксазин-2-ил)метил]-6-бром-7-оксо-4-тиа-1-азабицикло[3.2.0]гепт-2-ен-2-карбоновой кислоты в виде коричневого аморфного твердого вещества.
К раствору полученного п-нитробензилового эфира (5R)-6-[ацетокси-(5,6-дигидро-8H-имидазо[2,1-c][1,4]оксазин-2-ил)метил]-6-бром-7-оксо-4-тиа-1-азабицикло[3.2.0]гепт-2-ен-2-карбоновой кислоты в ТГФ (72 мл) быстро добавляют свежеактивированную Zn пыль (14 г) с 0,5M фосфатным буфером (pH 6,5, 72 мл). Реакционный сосуд покрывают фольгой, чтобы исключить воздействие света. Реакционную смесь энергично перемешивают в течение 2,5 ч при комнатной температуре. Реакционный раствор фильтруют через рыхлый слой целита, который промывают водой (170 мл) и н-бутанолом (170 мл). Водный слой отделяют и затем органический слой экстрагируют 0,5М фосфатным буфером с pH 6,5 (2 × 50 мл). Объединенный водный слой концентрируют до 90 г, добавляют 1М NaOH, доводя pH до 7,5, и наносят на хроматографическую колонку со смолой Diaion HP-21 (120 мл, Mitsubishi Kasei Co. Ltd.). После адсорбции осуществляют элюирование водой, затем 5% водным раствором ацетонитрила. Объединенные активные фракции концентрируют в высоком вакууме при 35°C, лиофилизуют и получают указанное в заголовке соединение в виде желтого аморфного твердого вещества (756 мг, 29,1%). Т.пл. 130°C (с разложением).
1H ЯМР (ДМСО-d6) δ: 3,98-4,01 (м, 2H), 4,04-4,07 (м, 2H), 4,74 (AB, 2H, J=15,3 и 22,9 Гц), 6,40 (д, 1H, J=0,8 Гц), 6,55 (с, 1H), 6,95 (д, 1H, J=0,6 Гц), 7,54 (с, 1H); ИК (KBr): 3412, 1741, 1672, 1592, 1549 см-1; λмакс.(H2O) 304 нм.
Пример 21
Получение натриевой соли (5R),(6Z)-6-(5,6-дигидро-4H-пирроло[1,2-b]пиразол-2-илметилен)-7-оксо-4-тиа-1-азабицикло[3.2.0]гепт-2-ен-2-карбоновой кислоты
Стадия 1. Этиловый эфир 5,6-дигидро-4H-пирроло[1,2-b]пиразол-2-карбоновой кислоты
Указанное в заголовке соединение получают тем же путем, который использовали Ranganathan и сотрудники (Indian J. Chem. 1991, 30 B, 169-175).
Стадия 2. (5,6-Дигидро-4H-пирроло[1,2-b]пиразол-2-ил)метанол
MeOH (2,73 мл) добавляют к раствору LiBH4 (1,63 г) в ТГФ (180 мл) в атмосфере азота при комнатной температуре, затем к полученной суспензии добавляют этиловый эфир 5,6-дигидро-4H-пирроло[1,2-b]пиразол-2-карбоновой кислоты (8,11 г) и перемешивают в течение 2 ч при 40°C. Смесь гасят 1М HCl (до pH 1) и перемешивают в течение 1 ч при комнатной температуре. К раствору добавляют твердый K2CO3, доводя рН раствора до 8, и смесь экстрагируют AcOEt. Органический слой сушат (MgSO4) и фильтруют. Фильтрат концентрируют при пониженном давлении и получают указанное в заголовке соединение в виде коричневых кристаллов (4,87 г, 78%).
1H ЯМР (CDCl3) δ: 2,44 (т, 1H, J=5,8 Гц), 2,54-2,62 (м, 2H), 2,87 (т, 2H, J=7,4 Гц), 4,10 (т, 2H, J=7,2 Гц), 4,63 (д, 2H, J=5,8 Гц), 5,96 (с, 1H).
Стадия 3. 5,6-Дигидро-4H-пирроло[1,2-b]пиразол-2-карбальдегид
К раствору (5,6-дигидро-4H-пирроло[1,2-b]пиразол-2-ил)метанола (4,87 г) в CHCl3 (350 мл) добавляют MnO2 (активированный) (24,4 г) и кипятят с обратным холодильником в течение 1 ч в атмосфере азота. Реакционную смесь фильтруют через слой целита. Фильтрат упаривают при пониженном давлении. Остаток наносят на хроматографическую колонку с силикагелем, затем элюируют смесью н-гексан-AcOEt (1/1-1/2). Получают указанное в заголовке соединение в виде желтого масла (4,35 г, 91%).
1H ЯМР (CDCl3) δ: 2,63-2,71 (м, 2H), 2,95 (т, 2H, J=7,4 Гц), 4,22 (т, 2H, J=7,4 Гц), 6,52 (с, 1H), 9,89 (с, 1H).
Стадия 4. Натриевая соль (5R),(6Z)-6-(5,6-дигидро-4H-пирроло[1,2-b]пиразол-2-илметилен)-7-оксо-4-тиа-1-азабицикло[3.2.0]гепт-2-ен-2-карбоновой кислоты
5,6-Дигидро-4H-пирроло[1,2-b]пиразол-2-карбальдегид (1,36 г) добавляют к раствору безводного MgBr2 (5,52 г) в сухом ацетонитриле (148 мл) в атмосфере азота при комнатной температуре. К полученной смеси добавляют раствор 4-нитробензилового эфира (5R,6S)-6-бром-7-оксо-4-тиа-1-азабицикло[3.2.0]гепт-2-ен-2-карбоновой кислоты (97% чистоты) (3,97 г) в сухом ТГФ (148 мл), охлаждают до -20°C и в один прием добавляют Et3N (4,18 мл). Реакционный сосуд покрывают фольгой, чтобы исключить воздействие света. Реакционную смесь перемешивают в течение 4 ч при -20°C и в один прием обрабатывают уксусным ангидридом (1,89 мл) и DMAP (123 мг). Температуру реакционной смеси доводят до 0°C и перемешивают в течение 14 ч при 0°C. Смесь разбавляют этилацетатом и промывают 5% водным раствором лимонной кислоты, насыщенным раствором гидрокарбоната натрия, водой и насыщенным солевым раствором. Органический слой сушат (MgSO4) и фильтруют. Фильтрат концентрируют при пониженном давлении.
Остаток растворяют в ТГФ (106 мл) и ацетонитриле (49 мл). Быстро добавляют свежеактивированную Zn пыль (22,5 г) с 0,5M фосфатным буфером (pH 6,5, 155 мл). Реакционный сосуд покрывают фольгой, чтобы исключить воздействие света. Реакционную смесь энергично перемешивают в течение 1,5 ч при комнатной температуре. Реакционную смесь фильтруют через рыхлый слой целита. Фильтрат промывают этилацетатом и водный слой отделяют. Водный слой охлаждают до 3°C и добавляют 1M NaOH, доводя pH до 8,0, затем концентрируют в высоком вакууме при 35°С. Наносят на хроматографическую колонку со смолой Diaion HP-21 (79 мл, Mitsubishi Kasei Co. Ltd.). После адсорбции проводят элюирование смесью H2О-MeCN (1/0-9/1). Объединенные фракции концентрируют в высоком вакууме при 35°C, лиофилизуют и получают указанное в заголовке соединение в виде желтого аморфного твердого вещества (848 мг, 29%, pH 7,1). Т.пл. 190°C (с разложением).
1H ЯМР (D2О) δ: 2,49 (м, 2H), 2,78 (т, 2H, J=7,4 Гц), 4,02 (т, 2H, J=7,4 Гц), 6,01 (с,1H), 6,29 (с, 1H), 6,90 (с, 2H).
Пример 22
Получение натриевой соли (5R)(6Z)-7-оксо-6-(4,5,6,7-тетрагидропиразоло[1,5-a]пиридин-2-илметилен)-4-тиа-1-азабицикло[3.2.0]гепт-2-ен-2-карбоновой кислоты
Стадия 1. Тетрагидропиридино[1,2-с][1,2,3]оксадиазолон
Концентрированную HCl (1,96 мл) и NaNO2 (2,2 г) добавляют к раствору DL-пипеколиновой кислоты (3,04 г) в воде (21 мл) в атмосфере азота при 0°C и перемешивают в течение 1 ч. Раствор экстрагируют CH2Cl2 и органический слой промывают насыщенным солевым раствором. Смесь сушат над Na2SO4, концентрируют при пониженном давлении и получают сырую (2RS)-1-нитрозопиперидин-2-карбоновую кислоту в виде светло-желтых кристаллов.
Трифторуксусный ангидрид (1,93 г) добавляют к раствору сырой (2RS)-1-нитрозопиперидин-2-карбоновой кислоты в ТГФ (92 мл) в атмосфере азота при 0°C и перемешивают в течение 5 ч при 0°C и в течение 2 ч при комнатной температуре. Раствор концентрируют при пониженном давлении. Остаток наносят на хроматографическую колонку с силикагелем, затем элюируют смесью н-гексан-AcOEt (1/1-0/1). Получают указанное в заголовке соединение в виде бесцветных кристаллов (1,10 г, 33%).
1H ЯМР (CDCl3) δ: 1,93-1,99 (м, 2H), 2,08-2,15 (м, 2H), 2,65 (т, 2H, J=6,5 Гц), 4,26 (т, 2H, J=6,1 Гц).
Стадия 2. Этиловый эфир 4,5,6,7-тетрагидропиразоло[1,5-a]пиридин-2-карбоновой кислоты
Этилпропиолат (804 мг) добавляют к раствору тетрагидропиридино[1,2-с][1,2,3]оксадиазолона (1,04 г) в o-ксилоле (15 мл) в атмосфере азота и кипятят с обратным холодильником в течение 16 ч. Раствор концентрируют при пониженном давлении. Остаток наносят на хроматографическую колонку с силикагелем, затем элюируют смесью н-гексан-AcOEt (2/1-1/1). Получают указанное в заголовке соединение в виде желтого масла (871 мг, 65%) и этиловый эфир 4,5,6,7-тетрагидропиразоло[1,5-a]пиридин-3-карбоновой кислоты в виде желтого масла (345 мг, 26%).
1H ЯМР (CDCl3) δ: 1,39 (т, 3H, J=7,1 Гц), 1,84-1,91 (м, 2H), 2,02-2,09 (м, 2H), 2,82 (т, 2H, J=6,4 Гц), 4,22 (т, 2H, J=6,2 Гц), 4,39 (кв, 2H, J=7,1 Гц), 6,53 (с, 1H).
Стадия 3. (4,5,6,7-Тетрагидропиразоло[1,5-a]пиридин-2-ил)метанол
MeOH (0,29 мл) добавляют к раствору LiBH4 (чистота 90%) (174 мг) в ТГФ (19 мл) в атмосфере азота при комнатной температуре, затем к полученной суспензии добавляют этиловый эфир 4,5,6,7-тетрагидропиразоло[1,5-a]пиридин-2-карбоновой кислоты (862 мг) и перемешивают в течение 1 ч при комнатной температуре и 1,5 ч при 40°C. Смесь гасят 1М HCl (до pH 1) и перемешивают в течение 1 ч при комнатной температуре. К раствору добавляют твердый K2CO3, чтобы довести рН до 8 и смесь экстрагируют AcOEt. Органический слой сушат (MgSO4) и фильтруют. Фильтрат концентрируют при пониженном давлении и получают указанное в заголовке соединение в виде желтого масла (691 мг, 95%).
1H ЯМР (CDCl3) δ: 1,80-1,87 (м, 2H), 1,98-2,05 (м, 2H), 2,77 (т, 2H, J=6,4 Гц), 2,81-2,84 (уш., 1H), 4,09 (т, 2H, J=6,1 Гц), 4,62 (д, 2H, J=5,3 Гц), 5,96 (с, 1H).
Стадия 4. 4,5,6,7-Тетрагидропиразоло[1,5-a]пиридин-2-карбальдегид
К раствору (4,5,6,7-тетрагидропиразоло[1,5-a]пиридин-2-ил)метанола (673 мг) в CHCl3 (44 мл) добавляют MnO2 (активированный) (3,36 г) и кипятят с обратным холодильником в течение 1 ч в атмосфере азота. Реакционную смесь фильтруют через рыхлый слой целита. Фильтрат упаривают при пониженном давлении. Остаток наносят на хроматографическую колонку с силикагелем, затем элюируют смесью н-гексан-AcOEt (2/1-1/2). Получают указанное в заголовке соединение в виде бледно-желтого масла (510 мг, 77%).
1H ЯМР (CDCl3) δ: 1,90 (м, 2H), 2,10 (м, 2H), 2,84 (т, 2H, J=6,4 Гц), 4,23 (т, 2H, J=6,2 Гц), 6,52 (с, 1H), 9,92 (с, 1H).
Стадия 5. Натриевая соль (5R)(6Z)-7-оксо-6-(4,5,6,7-тетрагидропиразоло[1,5-a]пиридин-2-илметилен)-4-тиа-1-азабицикло[3.2.0]гепт-2-ен-2-карбоновой кислоты
4,5,6,7-Тетрагидропиразоло[1,5-a]пиридин-2-карбальдегид (483 мг) добавляют к раствору безводного MgBr2 (1,81 г) в сухом ацетонитриле (48 мл) в атмосфере азота при комнатной температуре. К полученной смеси добавляют раствор 4-нитробензилового эфира (5R,6S)-6-бром-7-оксо-4-тиа-1-азабицикло[3.2.0]гепт-2-ен-2-карбоновой кислоты (чистота 97%) (1,28 г) в сухом ТГФ (48 мл), охлаждают до -20°C и добавляют в один прием Et3N (1,35 мл). Реакционный сосуд покрывают фольгой, чтобы исключить воздействие света. Реакционную смесь перемешивают в течение 2 ч при -20°C и в один прием обрабатывают уксусным ангидридом (0,61 мл) и DMAP (40 мг). Температуру реакционной смеси доводят до 0°C и перемешивают в течение 16 ч при 0°C. Смесь разбавляют этилацетатом и промывают 5% водным раствором лимонной кислоты, насыщенным раствором гидрокарбоната натрия, водой и насыщенным солевым раствором. Органический слой сушат (MgSO4) и фильтруют. Фильтрат концентрируют при пониженном давлении.
Остаток растворяют в ТГФ (35 мл) и ацетонитриле (16 мл). Быстро добавляют свежеактивированную Zn пыль (7,43 г) с 0,5M фосфатным буфером (pH 6,5, 51 мл). Реакционный сосуд покрывают фольгой, чтобы исключить воздействие света. Реакционную смесь энергично перемешивают в течение 1,5 ч при комнатной температуре и фильтруют через рыхлый слой целита. Фильтрат промывают этилацетатом, водный слой отделяют, охлаждают до 3°С и добавляют 1M NaOH, чтобы довести pH до 8,0. Смесь концентрируют в высоком вакууме при 35°C. Концентрат наносят на колонку со смолой Diaion HP-21 (105 мл, Mitsubishi Kasei Co.Ltd.). После адсорбции проводят элюирование смесью H2О-MeCN (1/0-85/15). Объединенные фракции концентрируют в высоком вакууме при 35°C, лиофилизуют и получают указанное в заголовке соединение в виде желтого аморфного твердого вещества (427 мг, 41%, pH 7,7). Т.пл.190°C (с разложением).
1H ЯМР (D2O) δ: 1,67-1,71 (м, 2H), 1,85-1,89 (м, 2H), 2,64 (т, 2H, J=6,3 Гц), 3,97 (т, 2H, J=6,1 Гц), 5,97 (с, 1H), 6,25 (с, 1H), 6,85 (с, 1H), 6,88 (с, 1H).
Пример 23
Получение натриевой соли (5R),(6Z)-6-(7-метил-6-оксо-5,6,7,8-тетрагидроимидазо[1,2-a]пиразин-2-илметилен)-7-оксо-4-тиа-1-азабицикло[3.2.0]гепт-2-ен-2-карбоновой кислоты
Стадия 1. 5-Метокси-1-метил-3,6-дигидро-1H-пиразин-2-он
Указанное в заголовке соединение получают способом, который описан S. Rajappa and B. G. Advani (Tetrahedron. 1973, 29, 1299-1302).
Стадия 2. 5-Амино-1-метил-3,6-дигидро-1H-пиразин-2-он
Смесь 5-метокси-1-метил-3,6-дигидро-1H-пиразин-2-она (2,3 г) и хлорида аммония (936 мг) в сухом этаноле (32 мл) перемешивают при комнатной температуре в течение 1 ч и затем кипятят с обратным холодильником в течение 2 ч. Реакционную смесь охлаждают до комнатной температуры и упаривают при пониженном давлении. Остаток растирают в хлороформе при комнатной температуре в течение 30 мин. Осадок отфильтровывают и сушат в вакууме. Гидрохлорид 5-амино-1-метил-3,6-дигидро-1H-пиразин-2-она получают в виде светло-коричневого порошка (1,7 г, 66%).
Раствор гидрохлорида 5-амино-1-метил-3,6-дигидро-1H-пиразин-2-она (662 мг) в метаноле (10 мл) добавляют к 10% водному раствору карбоната калия при 0°С и затем перемешивают 40 мин при 0°С. Смесь концентрируют при пониженном давлении. Остаток растирают в хлороформе (18 мл) и метаноле (2 мл) при комнатной температуре в течение 30 мин. Осадок отфильтровывают и сушат в вакууме. Получают указанное в заголовке соединение в виде светло-коричневого порошка (515 мг, количественный выход).
1H ЯМР (ДМСО-d6) δ: 2,88 (с, 3H), 3,94 (с, 2H), 4,42 (с, 2H).
Стадия 3. 7-Метил-6-оксо-5,6,7,8-тетрагидроимидазо[1,2-a]пиразин-2-карбальдегид и 7-метил-6-оксо-5,6,7,8- тетрагидроимидазо[1,2-a]пиразин-3-карбальдегид
Раствор 2-бром-3-изопропоксипропеналя (1,3 г) в сухом ацетонитриле (60 мл) добавляют к раствору 5-амино-1-метил-3,6-дигидро-1H-пиразин-2-она (782 мг) в сухом ацетонитриле (60 мл) при комнатной температуре. Реакционную смесь перемешивают при комнатной температуре в течение 20 ч, добавляют триэтиламин (0,95 мл) и затем кипятят с обратным холодильником в течение 2 ч. Реакционную смесь охлаждают до комнатной температуры и затем упаривают при пониженном давлении. Остаток растворяют в хлороформе (10 мл) и промывают 50% водным раствором K2CO3 (10 мл). Водный слой экстрагируют хлороформом. Органический слой сушат (MgSO4) и фильтруют. Фильтрат упаривают при пониженном давлении. Остаток наносят на хроматографическую колонку с силикагелем, элюируют смесью CHCl3-MeOH (95:5) и получают указанное в заголовке соединение 7-метил-6-оксо-5,6,7,8-тетрагидроимидазо[1,2-a]пиразин-2-карбальдегид, в виде светло-желтого твердого вещества (541 мг, 49,1%) и его региоизомер, 7-метил-6-оксо-5,6,7,8-тетрагидроимидазо[1,2-a]пиразин-3-карбальдегид, в виде бледно-желтого твердого вещества (128 мг, 11,6%).
7-Метил-6-оксо-5,6,7,8-тетрагидроимидазо[1,2-a]пиразин-2-карбальдегид: 1H ЯМР (CDCl3) δ: 3,17 (с, 3H), 4,68 (с, 2H), 4,78 (с, 2H), 7,66 (с, 1H), 9,83 (с, 1H).
7-Метил-6-оксо-5,6,7,8-тетрагидроимидазо[1,2-a]пиразин-3-карбальдегид: 1H ЯМР (CDCl3) δ 3,16 (с, 3H), 4,70 (с, 2H), 5,03 (с, 2H), 7,82 (с, 1H), 9,73 (с, 1H).
Стадия 4. 4-Нитробензиловый эфир (5R,6RS)-6-[ацетокси(7-метил-6-оксо-5,6,7,8-тетрагидроимидазо[1,2-a]пиразин-2-ил)метил]-6-бром-7-оксо-4-тиа-1-азабицикло[3.2.0]гепт-2-ен-2-карбоновой кислоты
7-Метил-6-оксо-5,6,7,8-тетрагидроимидазо[1,2-a]пиразин-2-карбальдегид (319 мг) добавляют к раствору безводного MgBr2 (786 мг) в сухом ацетонитриле (32 мл) в атмосфере азота при комнатной температуре. К полученной смеси добавляют раствор 4-нитробензилового эфира (5R,6S)-6-бром-7-оксо-4-тиа-1-азабицикло[3.2.0]гепт-2-ен-2-карбоновой кислоты (687 мг) в ТГФ (32 мл), охлаждают до -20°C и в один прием добавляют триэтиламин (0,60 мл). Реакционный сосуд покрывают фольгой, чтобы исключить воздействие света. Реакционную смесь перемешивают в течение 3 ч при -20°С и в один прием обрабатывают 4-диметиламинопиридином (44 мг) и уксусным ангидридом (0,35 мл). Температуру реакционной смеси доводят до 0°C и перемешивают в течение 20 ч при 0°C. Смесь разбавляют этилацетатом и водой. После отделения органического слоя водный слой экстрагируют этилацетатом. Органические слои объединяют и промывают 5% водным раствором лимонной кислоты и насыщенным солевым раствором. Органический слой сушат (MgSO4) и фильтруют. Фильтрат концентрируют при пониженном давлении. Остаток наносят на хроматографическую колонку с силикагелем, затем проводят элюирование хлороформом. Получают указанное в заголовке соединение в виде смеси диастереомеров (желтое аморфное твердое вещество, 410 мг, 38%).
1H ЯМР (δ, CDCl3): 2,03 (с, 0,7 × 3 H), 2,09 (с, 0,3 × 3H), 3,15 (с, 3H), 4,59-4,62 (м, 2H), 4,66 (с, 0,3 × 2H), 4,67 (с, 0,7 × 2H), 5,28 (д, 1H, J=13,5 Гц), 5,43 (д, 0,3 × 1H, J=13,5 Гц), 5,45 (д, 0,7 × 1H, J=13,5 Гц), 6,07 (с, 0,3 × 1H), 6,28 (с, 0,7 × 1H), 6,32 (с, 0,7 × 1H), 6,83 (с, 0,3 × 1H), 6,86 (с, 0,3 × 1H), 7,10 (с, 0,7 × 1 H), 7,44 (с, 0,3 × 1H), 7,47 (с, 0,7 × 1H), 7,60 (д, 0,7 × 2H, J=8,6 Гц), 7,61 (д, 0,3 × 2H, J=8,6 Гц), 8,24 (д, 2H, J=8,6 Гц).
Стадия 5. Натриевая соль (5R),(6Z)-6-(7-метил-6-оксо-5,6,7,8-тетрагидроимидазо[1,2-a]пиразин-2-илметилен)-7-оксо-4-тиа-1-азабицикло[3.2.0]гепт-2-ен-2-карбоновой кислоты и натриевая соль (5R),(6Е)-6-(7-метил-6-оксо-5,6,7,8-тетрагидроимидазо[1,2-a]пиразин-2-илметилен)-7-оксо-4-тиа-1-азабицикло[3.2.0]гепт-2-ен-2-карбоновой кислоты
4-Нитробензиловый эфир (5R,6RS)-6-[ацетокси(7-метил-6-оксо-5,6,7,8-тетрагидроимидазо[1,2-a]пиразин-2-ил)метил]-6-бром-7-оксо-4-тиа-1-азабицикло[3.2.0]гепт-2-ен-2-карбоновой кислоты (481 мг) растворяют в ТГФ (6,7 мл) и ацетонитриле (3,1 мл). К полученной смеси добавляют свежеактивированную Zn пыль (1,92 г) и 0,5M фосфатный буфер (pH 6,5, 9,9 мл). Реакционный сосуд покрывают фольгой, чтобы исключить воздействие света. Реакционную смесь энергично перемешивают в течение 2 ч при комнатной температуре. Реакционный раствор смешивают с этилацетатом и фильтруют через рыхлый слой целита, который промывают водой. Водный слой отделяют, охлаждают до 3°C и добавляют 1M NaOH, доводя рН до 8,0. Смесь концентрируют в высоком вакууме при 35°C и лиофилизуют. Остаток разделяют препаративной ВЭЖХ (Inertsil ODS-2, GL Science Inc., 10 × 250 мм, 0,05М фосфатный буфер (pH 7,1): CH3CN = 93:7; 4,0 мл/мин). Разделенные фракции натриевой соли (5R),(6Z)-6-(7-метил-6-оксо-5,6,7,8-тетрагидроимидазо[1,2-a]пиразин-2-илметилен)-7-оксо-4-тиа-1-азабицикло[3.2.0]гепт-2-ен-2-карбоновой кислоты и натриевой соли (5R),(6Е)-6-(7-метил-6-оксо-5,6,7,8-тетрагидроимидазо[1,2-a]пиразин-2-илметилен)-7-оксо-4-тиа-1-азабицикло[3.2.0]гепт-2-ен-2-карбоновой кислоты охлаждают до 3°C и добавляют 1M раствор NaOH, чтобы довести рН до 8,0 соответственно. Каждый раствор концентрируют в высоком вакууме при 35°C. Каждый концентрат наносят на хроматографическую колонку со смолой Diaion HP-21 (60 мл, Mitsubishi Kasei Co. Ltd.). После адсорбции осуществляют элюирование водой, затем смесью 5% ацетонитрил-вода. Объединенные фракции концентрируют в высоком вакууме при 35°C, лиофилизуют и получают указанные в заголовке соединения: натриевую соль (5R),(6Z)-6-(7-метил-6-оксо-5,6,7,8-тетрагидроимидазо[1,2-a]пиразин-2-илметилен)-7-оксо-4-тиа-1-азабицикло[3.2.0]гепт-2-ен-2-карбоновой кислоты в виде желтого аморфного твердого вещества (125 мг, 44,4%, т.пл. 115-117°C (с разложением) и натриевую соль (5R),(6Е)-6-(7-метил-6-оксо-5,6,7,8-тетрагидроимидазо[1,2-a]пиразин-2-илметилен)-7-оксо-4-тиа-1-азабицикло[3.2.0]гепт-2-ен-2-карбоновой кислоты в виде желтого аморфного твердого вещества (19 мг, 6,7%).
Натриевая соль (5R),(6Z)-6-(7-метил-6-оксо-5,6,7,8-тетрагидроимидазо[1,2-a]пиразин-2-илметилен)-7-оксо-4-тиа-1-азабицикло[3.2.0]гепт-2-ен-2-карбоновой кислоты: 1H ЯМР (δ, D2О): 2,99 (с, 3H), 4,54 (с, 2H), 4,66 (с, 2H), 6,38 (с, 1H), 6,85 (с, 1H), 6,90 (с, 1H), 7,30 (с, 1H).
Натриевая соль (5R),(6Е)-6-(7-метил-6-оксо-5,6,7,8-тетрагидроимидазо[1,2-a]пиразин-2-илметилен)-7-оксо-4-тиа-1-азабицикло[3.2.0]гепт-2-ен-2-карбоновой кислоты: 1H ЯМР (δ, D2О): 2,94 (с, 3H), 4,45 (с, 2H), 4,56 (с, 2H), 6,22 (с, 1H), 6,48 (с, 1H), 6,94 (с, 1H), 7,69 (с, 1H).
Пример 24
Получение натриевой соли (5R),(6Z)-6-(6,7-дигидро-4Н-пиразоло[5,1-c][1,4]тиазин-2-илметилен)-7-оксо-4-тиа-1-азабицикло[3.2.0]гепт-2-ен-2-карбоновой кислоты
Стадия 1. (3R)-Тиоморфолин-3-карбоновая кислота
Указанное в заголовке соединение получают способом, который применяют Shiraiwa и сотрудники (Biosci. Biotechnol. Biochem. 1998, 62, 2382-2387).
Стадия 2. 3-Оксо-3a,4,6,7-тетрагидро-3H-2-окса-5-тиа-1-аза-7a-азониаинденид
К раствору (3R)-тиоморфолин-3-карбоновой кислоты (4,96 г) в 1М HCl (33,7 мл) добавляют NaNO2 (3,14 г) в атмосфере азота при 0°C и перемешивают в течение 0,5 ч. Раствор экстрагируют CHCl3 (5 раз) и органический слой промывают насыщенным солевым раствором. Смесь сушат над MgSO4, концентрируют при пониженном давлении и получают сырую (3R)-4-нитрозотиоморфолин-3-карбоновую кислоту в виде бледно-желтых кристаллов.
Трифторуксусный ангидрид (7,07 г) добавляют к раствору сырой (3R)-4-нитрозотиоморфолин-3-карбоновой кислоты в ТГФ (169 мл) в атмосфере азота при 0°C и перемешивают в течение 3 ч при 0°C и 17 ч при комнатной температуре. Раствор концентрируют при пониженном давлении. Остаток наносят на хроматографическую колонку с силикагелем, затем элюируют смесью н-гексан-AcOEt (1/1-0/1). Получают указанное в заголовке соединение в виде светло-коричневых кристаллов (3,41 г, 64%).
1H ЯМР (CDCl3) δ: 3,15 (т, 2H, J=5,5 Гц), 3,71 (с, 2H), 4,54 (т, 2H, J=5,5 Гц).
Стадия 3. Этиловый эфир 6,7-дигидро-4H-пиразоло[5,1-c][1,4]тиазин-2-карбоновой кислоты
Этилпропиолат (2,33 г) добавляют к раствору 3-оксо-3a,4,6,7-тетрагидро-3H-2-окса-5-тиа-1-аза-7a-азониаинденида (3,41 г) в o-ксилоле (72 мл) в атмосфере азота и кипятят с обратным холодильником в течение 15 ч. Раствор концентрируют при пониженном давлении. Остаток наносят на хроматографическую колонку с силикагелем, затем элюируют смесью н-гексан-AcOEt (2/1-1/1). Получают указанное в заголовке соединение в виде желтого масла (3,13 г, 68%) и второй нежелательный региоизомер, этиловый эфир 6,7-дигидро-4H-пиразоло[5,1-c][1,4]тиазин-3-карбоновой кислоты в виде желтого масла (556 мг, 12%).
1H ЯМР (CDCl3) δ: 1,31 (т, 3H, J=7,1 Гц), 3,04 (т, 2H, J=5,7 Гц), 3,81 (с, 2H), 4,32 (кв, 2H, J=7,1 Гц), 4,40 (т, 2H, J=5,7 Гц), 6,54 (с, 1H).
Стадия 4. (6,7-Дигидро-4H-пиразоло[5,1-c][1,4]тиазин-2-ил)метанол
Метанол (0,9 мл) и LiBH4 (чистота 90%) (536 мг) добавляют к раствору этилового эфира 6,7-дигидро-4H-пиразоло[5,1-c][1,4]тиазин-2-карбоновой кислоты (3,13 г) в ТГФ (59 мл) в атмосфере азота при комнатной температуре и перемешивают в течение 3 ч при 40°C. Смесь гасят 1М HCl до pH 1 и перемешивают в течение 1 ч при комнатной температуре. К раствору добавляют твердый K2CO3, доводя рН раствора до 8, и смесь экстрагируют AcOEt. Органический слой сушат (K2CO3) и фильтруют. Фильтрат концентрируют при пониженном давлении и получают указанное в заголовке соединение в виде бледно-желтого масла (2,51 г, количественный выход).
1H ЯМР (CDCl3) δ: 2,58 (уш, 1H), 3,07 (т, 2H, J=5,7 Гц), 3,84 (с, 2H), 4,33 (т, 2H, J=5,7 Гц), 4,63 (д, 2H, J=3,9 Гц), 6,05 (с, 1H).
Стадия 5. 6,7-Дигидро-4H-пиразоло[5,1-c][1,4]тиазин-2-карбальдегид
К раствору (6,7-дигидро-4H-пиразоло[5,1-c][1,4]тиазин-2-ил)метанола (2,31 г) в CHCl3 (135 мл) добавляют MnO2 (активированный) (11,46 г) и кипятят с обратным холодильником в течение 1 ч в атмосфере азота. Реакционную смесь фильтруют через рыхлый слой целита. Фильтрат концентрируют при пониженном давлении. Остаток наносят на хроматографическую колонку с силикагелем, затем элюируют смесью н-гексан-AcOEt (1/1) и получают указанное в заголовке соединение в виде бледно-желтых кристаллов (1,78 г, 78%).
1H ЯМР (CDCl3) δ: 3,15 (т, 2H, J=5,8 Гц), 3,90 (с, 2H), 4,48 (т, 2H, J=5,8 Гц), 6,58 (с, 1H), 9,92 (с,1H).
Стадия 6. Натриевая соль (5R)(6Z)-6-(6,7-дигидро-4H-пиразоло[5,1-c][1,4]тиазин-2-илметилен)-7-оксо-4-тиа-1-азабицикло[3.2.0]гепт-2-ен-2-карбоновой кислоты
6,7-Дигидро-4H-пиразоло[5,1-c][1,4]тиазин-2-карбальдегид (841 мг) добавляют к раствору безводного MgBr2 (1,88 г) в сухом ацетонитриле (39 мл) в атмосфере азота при комнатной температуре. К полученной смеси добавляют раствор 4-нитробензилового эфира (5R,6S)-6-бром-7-оксо-4-тиа-1-азабицикло[3.2.0]гепт-2-ен-2-карбоновой кислоты (чистота 99,7%) (1,93 г) в сухом ТГФ (39 мл), охлаждают до -20°C и добавляют в один прием Et3N (2,79 мл). Реакционный сосуд покрывают фольгой, чтобы исключить воздействие света. Реакционную смесь перемешивают в течение 2 ч при -20°C и в один прием обрабатывают уксусным ангидридом (0,94 мл) и DMAP (61 мг). Температуру реакционной смеси доводят до 0°C и перемешивают в течение 17 ч при 0°C. Смесь разбавляют этилацетатом и промывают 5% водным раствором лимонной кислоты, насыщенным раствором гидрокарбоната натрия, водой и насыщенным солевым раствором. Органический слой сушат (MgSO4) и фильтруют. Фильтрат концентрируют при пониженном давлении.
Остаток растворяют в ТГФ (83 мл) и ацетонитриле (39 мл). Быстро добавляют свежеактивированную Zn пыль (7,72 г) и 0,5M фосфатный буфер (pH 6,5, 122 мл). Реакционный сосуд покрывают фольгой, чтобы исключить воздействие света. Реакционную смесь энергично перемешивают в течение 1,5 ч при комнатной температуре и фильтруют через рыхлый слой целита. Фильтрат промывают этилацетатом, водный слой отделяют, охлаждают до 3°С и добавляют 1M раствор NaOH, чтобы довести pH до 8,0. Смесь концентрируют в высоком вакууме при 35°C. Концентрат наносят на колонку со смолой Diaion HP-21 (150 мл, Mitsubishi Kasei Co. Ltd.). После адсорбции проводят элюирование смесью H2О-MeCN (1/0-85/15). Объединенные фракции концентрируют в высоком вакууме при 35°C, лиофилизуют и получают указанное в заголовке соединение в виде желтого аморфного твердого вещества (371 мг, 22%, pH 8,0). Т.пл. 190°C (с разложением).
1H ЯМР (D2О) δ: 3,03 (т, 2H, J=5,7 Гц), 3,75 (с, 2H), 4,22 (т, 2H, J=5,7 Гц), 6,07 (с, 1 H), 6,27 (с, 1H), 6,86 (с, 1H), 6,89 (с, 1H).
Пример 25
Получение натриевой соли (5R),(6Z)-7-оксо-6-(4H-5-тиа-1,6a-диазапентален-2-илметилен)-4-тиа-1-азабицикло[3.2.1]гепт-2-ен-2-карбоновой кислоты
Стадия 1. 3-Оксо-3a,4-дигидро-3Н,6H-2-окса-5-тиа-1-аза-6a-азониапенталенид
К раствору L-тиопролина (24,3 г) в H2О (166 мл) добавляют концентрированную HCl (15 мл) и NaNO2 (16,6 г) в атмосфере азота при 0°C и перемешивают в течение 2 ч. Раствор экстрагируют CH2Cl2, органический слой сушат над MgSO4, концентрируют при пониженном давлении и получают неочищенное N-нитрозопроизводное в виде желтого твердого вещества.
Трифторуксусный ангидрид (5,0 мл) добавляют к раствору неочищенного N-нитрозотиопролина в ТГФ (350 мл) в атмосфере азота при 0°C и перемешивают в течение 5 ч при 0°C. Раствор концентрируют при пониженном давлении. Остаток наносят на хроматографическую колонку с силикагелем, затем элюируют смесью н-гексан-AcOEt (1:1). Получают указанное в заголовке соединение в виде светло-коричневого твердого вещества (4,0 г, 15,1%).
1H ЯМР (CDCl3) δ: 4,04 (т, 2H, J=1,7 Гц), 5,40 (т, 2H, J=1,7 Гц).
Стадия 2. Этиловый эфир 4H-5-тиа-1,6a-диазапентален-2-карбоновой кислоты
Этилпропиолат (3,1 мл) добавляют к раствору 3-оксо-3a,4-дигидро-3Н,6H-2-окса-5-тиа-1-аза-6a-азонио-3а-пенталенида (4,0 г) в o-ксилоле (130 мл) в атмосфере азота и кипятят с обратным холодильником в течение 19 ч. Раствор охлаждают до комнатной температуры и концентрируют при пониженном давлении. Остаток наносят на хроматографическую колонку с силикагелем, затем проводят элюирование смесью н-гексан-AcOEt (4:1). Получают указанное в заголовке соединение в виде желтого твердого вещества (2,7 г, 49,3%) и этиловый эфир 4H-5-тиа-1,6a-диазапентален-3-карбоновой кислоты в виде бледно-желтых кристаллов (1,2 г, 21,7%).
1H ЯМР (CDCl3) δ: 1,40 (т, 3H, J=7,1 Гц), 4,11 (д, 2H, J=2,1 Гц), 4,40 (кв, 2H, J=7,1 Гц), 5,24 (т, 2H, J=1,6 Гц), 6,61 (с, 1H).
Стадия 3. (4H-5-Тиа-1,6a-диазапентален-2-ил)метанол
К раствору этилового эфира 4H-5-тиа-1,6a-диазапентален-2-карбоновой кислоты (2,5 г) в эфире (126 мл) и MeOH (0,77 мл) добавляют LiBH4 (90% чистоты) (459 мг) в атмосфере азота при комнатной температуре, затем кипятят с обратным холодильником в течение 1,5 ч. Реакционную смесь гасят 1М HCl (25 мл) и перемешивают в течение 1 ч при комнатной температуре. Смесь нейтрализуют насыщенным раствором гидрокарбоната натрия и разделяют слои. Водный слой экстрагируют дихлорметаном (10 × 25 мл). Объединенный органический слой сушат (MgSO4) и фильтруют. Фильтрат концентрируют при пониженном давлении. Остаток наносят на хроматографическую колонку с силикагелем, затем элюируют AcOEt. Получают указанное в заголовке соединение в виде бледно-желтого твердого вещества (1,7 г, 87,9%).
1H ЯМР (CDCl3) δ: 2,95 (т, 1H, J=5,6 Гц), 4,07 (с, 2H), 4,62 (д, 2H, J=5,1 Гц), 5,13 (т, 1H, J=1,6 Гц), 6,04 (с, 1H).
Стадия 4. 4H-5-Тиа-1,6a-диазапентален-2-карбальдегид
Раствор диметилсульфоксида (2,2 мл) в сухом дихлорметане (8 мл) добавляют по каплям к раствору оксалилхлорида (2,0 мл) в сухом дихлорметане (110 мл) при -78°C и перемешивают при указанной температуре 15 мин. К полученной смеси добавляют по каплям раствор (4H-5-тиа-1,6a-диазапентален-2-ил)метанола (1,7 г) в сухом дихлорметане (40 мл) при -78°C и продолжают перемешивание еще в течение 15 мин. Реакционной смеси дают нагреться до -45°C и перемешивают в течение 1 ч. Добавляют по каплям триэтиламин (11,3 мл) и температуру реакционной смеси доводят до 0°C. Через 20 мин добавляют насыщенный раствор хлорида аммония (50 мл) и воду (100 мл) и слои разделяют. Водный слой экстрагируют AcOEt (3 × 150 мл). Объединенные органические слои промывают водой (200 мл) и насыщенным солевым раствором (200 мл), сушат (MgSO4) и фильтруют. Фильтрат концентрируют при пониженном давлении. Остаток наносят на хроматографическую колонку с силикагелем, затем элюируют смесью гексан-AcOEt (1:1). Получают указанное в заголовке соединение в виде желтого твердого вещества (1,7 г, количественный выход).
1H ЯМР (CDCl3) δ: 4,13 (с, 2H), 5,26 (д, 2H, J=1,4 Гц), 6,59 (с, 1H), 9,90 (с, 1H).
Стадия 5. Натриевая соль (5R),(6Z)-7-оксо-6-(4H-5-тиа-1,6a-диазапентален-2-илметилен)-4-тиа-1-азабицикло[3.2.0]гепт-2-ен-2-карбоновой кислоты
Раствор 4H-5-Тиа-1,6a-диазапентален-2-карбальдегида (1,7 г) в сухом ацетонитриле (92 мл) добавляют к раствору безводного MgBr2 (5,0 г) в сухом ацетонитриле (92 мл) в атмосфере азота при комнатной температуре, затем смесь перемешивают в течение 10 минут. Добавляют раствор п-нитробензил(5R,6S)-6-бромпенем-3-карбоксилата (4,3 г) в сухом ТГФ (184 мл), смесь охлаждают до -20°C, затем в один прием добавляют триэтиламин (7,4 мл). Реакционный сосуд покрывают фольгой, чтобы исключить воздействие света. Реакционную смесь перемешивают в течение 3 ч при -20°C и обрабатывают в один прием 4-диметиламинопиридином (138 мг) и уксусным ангидридом (2,1 мл). Температуру реакционной смеси доводят до 0°C и перемешивают в течение 15 ч при 0°C. К реакционной смеси добавляют 1М водный раствор лимонной кислоты (1000 мл) и водный слой экстрагируют этилацетатом (3 × 400 мл). Объединенные органические слои промывают водой, насыщенным раствором гидрокарбоната натрия и насыщенным солевым раствором, сушат (MgSO4) и фильтруют. Фильтрат концентрируют при пониженном давлении и получают неочищенный п-нитробензиловый эфир (5R)-6-[ацетокси(4H-5-тиа-1,6a-диазапентален-2-ил)метил]-6-бром-7-оксо-4-тиа-1-азабицикло[3.2.0]гепт-2-ен-2-карбоновой кислоты в виде коричневого аморфного твердого вещества.
К раствору неочищенного п-нитробензилового эфира (5R)-6-[ацетокси(4H-5-тиа-1,6a-диазапентален-2-ил)метил]-6-бром-7-оксо-4-тиа-1-азабицикло[3.2.0]гепт-2-ен-2-карбоновой кислоты в ТГФ (100 мл) быстро добавляют свежеактивированную Zn пыль (19,3 г) с 0,5М фосфатным буфером (pH 6,5, 100 мл). Реакционный сосуд покрывают фольгой, чтобы исключить воздействие света. Реакционную смесь энергично перемешивают в течение 2,5 ч при комнатной температуре. Реакционный раствор фильтруют через рыхлый слой целита, который промывают водой (200 мл) и н-бутанолом (200 мл). Водный слой отделяют и органический слой экстрагируют 0,5М фосфатным буфером (pH 6,5, 2 × 50 мл). Объединенные водные слои концентрируют до массы 90 г, добавляют 1М раствор NaOH и доводят рН до 8,0. Концентрат наносят на хроматографическую колонку с Diaion HP-21 смолой (180 мл, Mitsubishi Kasei Co. Ltd.). После адсорбции осуществляют элюирование водой, затем 15% водным раствором ацетонитрила. Объединенные активные фракции концентрируют в высоком вакууме при 35°C, лиофилизуют и получают указанное в заголовке соединение в виде желтого аморфного твердого вещества (634 мг, 17,4%, pH 7,25). Т.пл. 150°C (с разложением).
1H ЯМР (D2О) δ 4,00 (с, 2H), 5,09 (с, 2H), 6,14 (с, 1H), 6,36 (с, 1H), 6,91 (с, 1H), 6,92 (с, 1H). ИК (KBr): 3381, 1752, 1683, 1600, 1558 см-1. λмакс. (H2O): 292, 196 нм.
Пример 26
Получение натриевой соли (5R),(6Z)-6-(7H-имидазо[1,2-c]тиазол-2-илметилен)-7-оксо-4-тиа-1-азабицикло[3.2.0]гепт-2-ен-2-карбоновой кислоты
Стадия 1. Тиазолидин-4-он
Указанное в заголовке соединение получают способом, который применяют Marvin M. и Allen R. Harkness. (Tetrahedron Letters. 1994, 35, 6971-6974).
Стадия 2. Тиазолидин-4-тион
Реагент Лауссона (Lawesson) (33,5 г) добавляют к раствору тиазолидин-4-она (14,2 г) в сухом ТГФ (690 мл) и реакционную смесь кипятят с обратным холодильником в течение 2 ч. Смесь охлаждают до комнатной температуры и упаривают при пониженном давлении. Остаток растирают в растворе CHCl3: MeOH (7:3) (65 мл) при комнатной температуре в течение 30 мин. Осадок отфильтровывают, промывают раствором CHCl3:н-гексан (7:3) (15 мл) и сушат в вакууме. Получают тиазолидин-4-тион в виде бледно-желтого порошка (10,7 г, 65%).
1H ЯМР (CDCl3) δ: 4,08 (с, 2H), 4,70 (с, 2H).
Стадия 3. 4-Метилтио-2,5-дигидротиазол
Метилиодид (28,4 г) добавляют к кипящему раствору тиазолидин-4-тиона (9,5 г) в хлороформе (400 мл) и реакционную смесь кипятят с обратным холодильником в течение 1,5 ч. К реакционной смеси добавляют дополнительное количество метилиодида (56,8 г) пятью порциями с интервалами 30-60 мин. После кипячения в течение еще 1 часа реакционную смесь охлаждают до комнатной температуры. Затем добавляют 10% водный раствор карбоната калия (200 мл) и перемешивают в течение 15 мин при комнатной температуре. После отделения органического слоя водный слой экстрагируют CHCl3 (100 мл × 3). Органические слои объединяют, сушат (MgSO4) и фильтруют. Фильтрат концентрируют при пониженном давлении и сушат в вакууме. После сушки получают указанное в заголовке соединение в виде коричневого масла (11,0 г, количественный выход).
1H ЯМР (CDCl3) δ: 2,51 (с, 3H), 3,91 (т, 2H, J=3,5 Гц), 5,21 (т, 2H, J=3,5 Гц).
Стадия 4. Тиазолидин-4-илиденамин
Смесь 4-метилтио-2,5-дигидротиазола (10,7 г) и хлорида аммония (6,4 г) в сухом этаноле (400 мл) кипятят с обратным холодильником в течение 27,5 ч. Реакционную смесь охлаждают до комнатной температуры и упаривают при пониженном давлении. Остаток растворяют в хлороформе (300 мл) и 10% водном растворе карбоната калия (200 мл), затем перемешивают 20 мин при комнатной температуре. После отделения органического слоя водный слой экстрагируют хлороформом (100 мл × 5). Органические слои объединяют, сушат (MgSO4) и фильтруют. Фильтрат концентрируют при пониженном давлении, сушат в вакууме и получают сырой тиазолидин-4-илиденамин (5,5 г) в виде коричневого твердого вещества, который содержит побочный продукт, представляющий собой этоксипроизводное, и исходный 4-метилтио-2,5-дигидротиазол. Соотношение этих трех соединений, составляет 61:34:5 соответственно, как определено методом 1H ЯМР.
1H ЯМР (CDCl3) δ: 3,75 (т, 2H, J=2,8 Гц), 4,97 (т, 2H, J=2,9 Гц).
Стадия 5. 7H-Имидазо[1,2-c]тиазол-2-карбальдегид
Раствор 2-бром-3-изопропоксипропеналя (6,9 г) в сухом ацетонитриле (326 мл) добавляют к раствору неочищенного тиазолидин-4-илиденамина (3,3 г) в сухом ацетонитриле (326 мл) при комнатной температуре. Реакционную смесь перемешивают при комнатной температуре в течение 19,5 ч, добавляют триэтиламин (4,9 мл) и затем кипятят с обратным холодильником в течение 2 ч. Реакционную смесь охлаждают до комнатной температуры и затем упаривают при пониженном давлении. Остаток растворяют в дихлорметане (300 мл) и промывают 50% водным раствором карбоната калия (20 г). После фильтрования и отделения органического слоя водный слой экстрагируют дихлорметаном (50 мл × 4). Органические слои объединяют, сушат (MgSO4) и фильтруют. Фильтрат упаривают при пониженном давлении. Остаток наносят на хроматографическую колонку с силикагелем, затем элюируют смесью CHCl3-MeOH (100:3) и получают сырой 7H-имидазо[1,2-c]тиазол-2-карбальдегид в виде коричневого твердого вещества. Сырой продукт дважды перекристаллизовывают из смеси CHCl3-н-гексан (30:5 - 1-й раз и 30:60 - 2-й раз) при 0°C и получают целевой альдегид в виде светло-коричневых кристаллов (выход 1,84 г, 15%).
1H ЯМР (CDCl3) δ: 4,09 (т, 2H, J=1,3 Гц), 5,08 (т, 2H, J=1,2 Гц), 7,63 (с, 1H), 9,81 (с, 1H).
Стадия 6. Натриевая соль (5R),(6Z)-6-(7H-имидазо[1,2-c]тиазол-2-илметилен)-7-оксо-4-тиа-1-азабицикло[3.2.0]гепт-2-ен-2-карбоновой кислоты
7H-Имидазо[1,2-c]тиазол-2-карбальдегид (841 мг) добавляют к раствору безводного MgBr2 (2,93 г) в сухом ацетонитриле (116 мл) в атмосфере азота при комнатной температуре. К полученной смеси добавляют раствор 4-нитробензилового эфира (5R,6S)-6-бром-7-оксо-4-тиа-1-азабицикло[3.2.0]гепт-2-ен-2-карбоновой кислоты (чистота 99,7%) (2,51 г) в сухом ТГФ (116 мл), охлаждают до -20°C и в один прием добавляют Et3N (2,20 мл). Реакционный сосуд покрывают фольгой, чтобы исключить воздействие света. Реакционную смесь перемешивают в течение 4 ч при -20°C и обрабатывают в один прием уксусным ангидридом (1,26 мл) и DMAP (160 мг). Температуру реакционной смеси доводят до 0°С и перемешивают в течение 15 ч при 0°C. Смесь разбавляют этилацетатом и промывают 5% водным раствором лимонной кислоты, насыщенным раствором гидрокарбоната натрия, водой и насыщенным солевым раствором. Органический слой сушат (MgSO4) и фильтруют. Фильтрат концентрируют при пониженном давлении.
Остаток растворяют в ТГФ (53 мл) и ацетонитриле (25 мл). Добавляют свежеактивированную Zn пыль (15,1 г) и 0,5M фосфатный буфер (pH 6,5, 78 мл). Реакционный сосуд покрывают фольгой, чтобы исключить воздействие света. Реакционную смесь энергично перемешивают 1,5 ч при комнатной температуре и фильтруют через рыхлый слой целита. Фильтрат промывают этилацетатом и отделяют водный слой. Водный слой охлаждают до 3°C и добавляют 1M NaOH, доводя рН до 8,0. Смесь концентрируют в высоком вакууме при 35°C. Концентрат наносят на хроматографическую колонку с Diaion HP-21 (321 мл, Mitsubishi Kasei Co. Ltd.). После адсорбции осуществляют элюирование смесью H2O-MeCN (1/0-9/1). Объединенные фракции концентрируют в высоком вакууме при 35°C, лиофилизуют и получают указанное в заголовке соединение в виде желтого аморфного твердого вещества (1,1 г, 51%, pH 7,5). Т.пл. 145°C (с разложением).
1H ЯМР (D2О) δ 3,85 (с, 2H), 4,88 (с, 2H), 6,32 (с, 1H), 6,78 (с, 1H), 6,85 (с, 1H), 7,27 (с, 1H).
Пример 27
Получение (5R,6Z)-7-оксо-6-[(4-оксо-6,7-дигидро-4H-пиразоло[5,1-c][1,4]оксазин-2-ил)метилен]-4-тиа-1-азабицикло[3.2.0]гепт-2-ен-2-карбоновой кислоты
Стадия 1. Диэтил 1-(2-{[трет-бутил(диметил)силил]окси}этил)-1H-пиразол-3,5-дикарбоксилат
К раствору диэтил 3,5-пиразолдикарбоксилата (2,17 г, 10 ммоль) в ацетонитриле (10 мл) добавляют в атмосфере азота карбонат калия (2,07 г, 15 ммоль) и 2-бромэтокси-трет-бутилдиметилсилан (2,90 г, 12 ммоль). Смесь кипятят с обратным холодильником при перемешивании в течение 18 ч, затем охлаждают до комнатной температуры, разбавляют этилацетатом (20 мл) и фильтруют через Magnesol. Рыхлый слой на фильтре промывают этилацетатом (2 × 10 мл) и объединенный фильтрат упаривают. Остаток растворяют в смеси гексанов и пропускают через колонку с силикагелем (70 г). Элюируют сначала гексанами (100 мл), затем этилацетатом. Этилацетатные фракции упаривают и получают 3,71 г бесцветного масла; МС: m/e 371 (MH+).
Стадия 2. 1-(2-{[трет-Бутил(диметил)силил]окси}этил)-1H-пиразол-3,5-диметанол
К раствору диэтил 1-(2-{[трет-бутил(диметил)силил]окси}этил)-1H-пиразол-3,5-дикарбоксилата (0,74 г, 2 ммоль) в метиленхлориде (8 мл) в атмосфере азота добавляют при 0°C 12 мл 1,0M раствора диизобутилалюминийгидрида в метиленхлориде. После перемешивания при 0°C в течение 0,5 ч смесь доводят до комнатной температуры за 0,5 ч и затем гасят насыщенным раствором хлорида аммония (15 мл). Водный слой экстрагируют этилацетатом. Объединенный органический слой промывают насыщенным солевым раствором, сушат над безводным сульфатом натрия, упаривают и получают 0,44 г белого твердого вещества. Т.пл. 82-83°С. MC: m/e 287 (MH+).
Стадия 3. 1-(2-{[трет-Бутил(диметил)силил]окси}этил)-1H-пиразол-3,5-дикарбальдегид
К перемешиваемому раствору 1-(2-{[трет-бутил(диметил)силил]окси}этил)-1H-пиразол-3,5-диметанола (1,18 г, 4 ммоль) в метиленхлориде (20 мл) добавляют 4- метилморфолин-N-оксид (2,89 г, 24 ммоль) и молекулярные сита 4A (4 г). Реакционную смесь перемешивают при комнатной температуре в течение 10 мин и затем обрабатывают перрутенатом тетрапропиламмония (0,15 г, 0,4 ммоль). Продолжают перемешивание в течение 2 ч. Раствор в метиленхлориде концентрируют и разбавляют простым эфиром (40 мл). Смесь фильтруют через рыхлый слой силикагеля (40 г) и элюируют эфиром (2 × 20 мл). Объединенный элюат промывают 1 н. раствором HCl и насыщенным солевым раствором, сушат над безводным сульфатом натрия, упаривают и получают 0,79 г белого твердого вещества. Т.пл. 63-64°C. МС: m/e 283 (MH+).
Стадия 4. 4-Оксо-6,7-дигидро-4H-пиразоло[5,1-c][1,4]оксазин-2-карбальдегид
К раствору 1-(2-{[трет-бутил(диметил)силил]окси}этил)-1H-пиразол-3,5-дикарбальдегида (1,02 г, 6,07 ммоль) в ТГФ (30 мл) добавляют 6,68 мл 1,0M раствора фторида тетрабутиламмония в ТГФ при 0°C. После перемешивания в течение 1 ч смесь обрабатывают насыщенным раствором хлорида аммония (10 мл) и водный слой экстрагируют этилацетатом. Органический раствор промывают насыщенным солевым раствором, сушат над безводным сульфатом натрия, фильтруют через Magnesol и упаривают. Сырой смолообразный продукт промывают смесью гексанов, сушат в вакууме и затем растворяют в метиленхлориде (20 мл). К полученному раствору добавляют 4-метилморфолин-N-оксид (2,89 г, 24 ммоль) и молекулярные сита 4A (6 г). Смесь перемешивают при комнатной температуре 10 мин, затем обрабатывают перрутенатом тетрапропиламмония (0,11 г, 0,3 ммоль). Продолжают перемешивание в течение 2 ч. Раствор в метиленхлориде концентрируют и разбавляют этилацетатом (40 мл). Смесь фильтруют через рыхлый слой силикагеля (40 г) и элюируют этилацетатом (2 × 20 мл). Объединенный элюат промывают 1 н. раствором HCl и насыщенным солевым раствором, сушат над безводным сульфатом натрия, упаривают и получают 0,30 г белого твердого вещества. Т.пл. 135-136°C. МС: m/e 167 (MH+).
Стадия 5. 4-Нитробензил (5R)-6-[(ацетокси)(4-оксо-6,7-дигидро-4H-пиразоло[5,1-c][1,4]оксазин-2-ил)метил]-6-бром-7-оксо-4-тиа-1-азабицикло[3.2.0]гепт-2-ен-2-карбоксилат
К раствору MgBr2 (0,46 г, 2,52 ммоль) в ацетонитриле (13 мл) в атмосфере азота добавляют при перемешивании 4-оксо-6,7-дигидро-4H-пиразоло[5,1-c][1,4]оксазин-2-карбальдегид (0,14 г, 0,84 ммоль) при комнатной температуре. Затем добавляют раствор 4-нитробензилового эфира (5R,6S)-6-бром-7-оксо-4-тиа-1-азабицикло[3.2.0]гепт-2-ен-2-карбоновой кислоты (0,32 г, 0,84 ммоль) в ТГФ (13 мл) и смесь охлаждают до -20°С. Добавляют триэтиламин (0,35 мл, 2,52 ммоль) и смесь перемешивают при -20°C в темноте в течение 4 ч. Затем смесь обрабатывают уксусным ангидридом (0,2 мл, 2,0 ммоль) и 4-N,N-диметиламинопиридином (12 мг, 0,1 ммоль) и выдерживают при 0°C в течение 18 ч. Смесь концентрируют и остаток растворяют в этилацетате. Этилацетатный раствор промывают 5% раствором лимонной кислоты, насыщенным раствором бикарбоната натрия, насыщенным солевым раствором, сушат над безводным сульфатом натрия и упаривают. Полученное сырое вещество хроматографируют на силикагеле, элюируют смесью (EtOAc-CH2Cl2/1:5) и получают 0,27 г не совсем белого твердого вещества. Т.пл. 107-110°C. МС: m/e 595(MH+).
Стадия 6. (5R,6Z)-7-Оксо-6-[(4-оксо-6,7-дигидро-4H-пиразоло[5,1-c][1,4]оксазин-2-ил)метилен]-4-тиа-1-азабицикло[3.2.0]гепт-2-ен-2-карбоновая кислота
К раствору 4-нитробензил (5R)-6-[(ацетокси)(4-оксо-6,7-дигидро-4H-пиразоло[5,1-c][1,4]оксазин-2-ил)метил]-6-бром-7-оксо-4-тиа-1-азабицикло[3.2.0]гепт-2-ен-2-карбоксилата (0,22 г, 0,37 ммоль) в ТГФ (15 мл) в атмосфере азота добавляют 15 мл фосфатного буферного раствора (0,5M, pH 6,5), и 80 мг 10% Pd/C. Смесь гидрируют при давлении водорода 40-50 фунт/кв.дюйм в течение 3 ч и затем фильтруют через рыхлый слой целита, который промывают ТГФ, и фильтрат экстрагируют этилацетатом. Органический экстракт сушат над безводным сульфатом магния и упаривают. Остаток промывают эфиром и получают 0,07 г желтого твердого вещества. МС: m/e 320 (MH+).
1H ЯМР (ДМСО-d6) δ: 4,55-4,57 (м, 2H), 4,76-4,80 (м, 2H), 6,50 (с, 1H), 6,63 (с, 1H), 7,58 (с, 1H), 7,76 (с, 1H).
Пример 28
Получение 6-(6,7-дигидро-4H-тиено[3,2-c]пиран-2-илметилен)-7-оксо-4-тиа-1-азабицикло[3.2.0]гепт-2-ен-2-карбоновой кислоты
Стадия 1. Получение 6,7-дигидро-4H-тиено[3,2-c]пиран-2-карбальдегида
POCl3 (3,83 мл, 50 ммоль) добавляют по каплям к охлаждаемому льдом ДМФА (3,85 мл, 50 ммоль) в течение 3 мин. Добавляют DCM (20 мл) и, когда реакционная смесь становится вязкой, баню убирают. Реакционную смесь выдерживают при 23°С в течение 2 ч, затем опять охлаждают до 0°C и по каплям добавляют тетрагидропиран-4-он (5 г, 50 ммоль) в 10 мл DCM в течение 3 мин. Реакционную смесь выдерживают при 0°С в течение 2 ч и выливают в смесь льда и раствора ацетата натрия. Водный слой экстрагируют DCM (2 × 200 мл). Объединенные органические слои сушат над сульфатом магния. После удаления осушителя фильтрованием и концентрирования получают 5,0 г продукта, который растворяют в DCM (200 мл) и добавляют к 6,0 г этил 2-меркаптоацетата и 10 мл TEA. Смесь кипятят с обратным холодильником в течение 18 ч, промывают водой, сушат над сульфатом магния, затем фильтруют, концентрируют и подвергают флэш-хроматографии, используя 20% раствор этилацетата в гексане в качестве элюента. Полученное вещество растворяют в 100 мл ТГФ, вводят LAH (150 мл, 0,5M в ТГФ) и оставляют при 23°C на 10 мин. Затем реакционную смесь кипятят с обратным холодильником в течение 18 ч и гасят при 23°C водой и в конце 1 н. HCl до получения прозрачной смеси. Водный слой экстрагируют этилацетатом (2 × 200 мл) и объединенные органические слои сушат над сульфатом магния. После фильтрования и концентрирования получают 2,3 г сырого продукта, который растворяют в 300 мл DCM и к раствору добавляют диоксид марганца (15 г). Реакцию проводят при 23°C в течение 0,5 ч. Окислитель добавляют еще дважды порциями по 15 г с интервалом в 0,5 ч. Полученный раствор фильтруют через слой целита и фильтрат концентрируют. После колоночной флэш-хроматографии получают 1,206 г (выход 14%) продукта в виде масла.
1H ЯМР: δ 9,84 (с, 1H), 7,41 (с, 1H), 4,74 (с, 2H), 4,00 (т, 2H, J=5,6 Гц), 2,96 (т, 2H, J=5,6 Гц). MC: 169,1 (M+H).
Стадия 2. Получение 6-(6,7-дигидро-4H-тиено[3,2-c]пиран-2-илметилен)-7-оксо-4-тиа-1-азабицикло[3.2.0]гепт-2-ен-2-карбоновой кислоты
6,7-Дигидро-4H-тиено[3,2-c]пиран-2-карбальдегид (336 мг, 2 ммоль) растворяют в 20 мл ацетонитрила и добавляют бромид магния (516 мг, 2 ммоль) в атмосфере азота. Смесь перемешивают при 23°C в течение 0,5 ч. Затем вводят в один прием 4-нитробензиловый эфир 6-бром-7-оксо-4-тиа-1-азабицикло[3.2.0]гепт-2-ен-2-карбоновой кислоты (770 мг, 2 ммоль) в 20 мл ТГФ и смесь тотчас же охлаждают до -20°C. Вводят триэтиламин (1 мл) и смесь перемешивают при -20°C в течение 3 ч. Затем вводят уксусный ангидрид (0,4 мл) и смесь перемешивают при 0°С в течение 18 ч. Реакционную смесь разбавляют 400 мл этилацетата и промывают 100 мл 5% раствора лимонной кислоты, 100 мл насыщенного раствора бикарбоната натрия и 100 мл насыщенного раствора хлорида натрия. Органический слой сушат над сульфатом магния, фильтруют и концентрируют. После колоночной флэш-хроматографии с использованием 20% этилацетата в гексане получают 491 мг (41%) продукта. Продукт растворяют в 15 мл ТГФ и 15 мл водного фосфатного буфера (pH 6,5). Смесь гидрируют при давлении водорода 45 фунт/кв.дюйм в течение 1 ч с 0,5 г 10% палладия на угле, затем фильтруют через рыхлый слой целита и концентрируют в вакууме, удаляя большую часть ТГФ. Раствор охлаждают до 0°С и доводят рН до 8, добавляя 1 н. раствор гидроксида натрия и очищают методом ВЭЖХ с обращенной фазой, используя 2 л воды, затем 5% раствор ацетонитрила в воде в качестве элюента. Воду удаляют концентрированием в вакууме и выделяют 100 мг (38%) продукта. Т.пл. >250°C.
1H ЯМР δ: 7,36 (с, 1H), 7,15 (с, 1H), 6,55 (с, 1H), 6,44 (с, 1H), 4,61 (с, 2H), 3,88 (м, 2H), 2,86 (м, 2H), 2,27 (м, 2H), 1,43 (т, 3H). MC: 320,3 (M-H).
Пример 29
Получение 6-(6,7-дигидро-4H-тиено[3,2-c]тиопиран-2-илметилен)-7-оксо-4-тиа-1-азабицикло[3.2.0]гепт-2-ен-2-карбоновой кислоты
Стадия 1. Получение 6,7-дигидро-4H-тиено[3,2-c]тиопиран-2-карбальдегида
POCl3 (4,02 мл, 43 ммоль) добавляют по каплям к охлаждаемому льдом ДМФА (3,34 мл, 43 ммоль) в течение 3 мин, затем добавляют DCM (20 мл) и, когда реакционная смесь становится вязкой, баню удаляют. Реакционную смесь выдерживают при 23°С в течение 2 ч, затем опять охлаждают до 0°C и добавляют по каплям тетрагидротиопиран-4-он (5 г, 43 ммоль) в 10 мл DCM в течение 3 мин. Реакционную смесь выдерживают при 0°С в течение 2 ч. Разбавляют DCM (250 мл) и затем промывают ледяным насыщенным водным раствором ацетата натрия (200 мл). Органический слой сушат над сульфатом натрия. Отфильтровывают осушитель, фильтрат концентрируют, флэш-хроматографируют, используя 10% этилацетат в гексане, и получают 1,3 г (8 ммоль) продукта. Продукт растворяют в DCM (100 мл) и добавляют 1,2 мл (11 ммоль) этил (2-меркапто)ацетата и 1 мл TEA. Смесь кипятят с обратным холодильником в течение 18 ч, промывают водой и сушат над сульфатом магния. Фильтрованием, концентрированием и флэш-хроматографией, используя 20% этилацетат в гексане, получают 1,1 г (выход 11%) продукта.
1H ЯМР δ: 6,68 (с, 1H), 4,73 (с, 2H), 3,68 (с, 2H), 3,04 (т, 2H, J=7,6 Гц), 2,91 (т, 2H, J=7,6 Гц). MC (EI): 185,99 (M+).
Этиловый эфир 6,7-дигидро-4H-тиено[3,2-c]тиопиран-2-карбоновой кислоты (1,1 г, 4,8 ммоль) растворяют в 100 мл ТГФ и вводят LAH (40 мл, 0,5M раствор в DMG), выдерживают реакционную смесь при 23°C в течение 10 мин, затем кипятят с обратным холодильником в течение 18 ч и гасят при 23°C водой (10 мл). Органический слой декантируют и оставшуюся смесь промывают DCM (20 мл). Объединенные органические слои сушат над сульфатом натрия. Фильтруют, концентрируют фильтрат и после флэш-хроматографии с использованием 10-20% этилацетата в гексане в качестве элюента получают 940 мг сырого продукта, который растворяют в DCM (40 мл) и к раствору добавляют диоксид марганца (2 г). Реакцию проводят при 23°C в течение 0,5 ч. Реакционную смесь затем фильтруют через рыхлый слой целита и концентрируют. Флэш-хроматографией на колонке получают 320 мг (36%) продукта.
1H ЯМР: δ 9,82 (с, 1H), 7,46 (с, 1H), 3,56 (с, 2H), 3,15 (т, 2H, J=7,2 Гц), 2,95 (т, 2H, J=7,2 Гц). MC (EI): 228,02 (M+).
Стадия 2. Получение 6-(6,7-дигидро-4H-тиено[3,2-c]тиопиран-2-илметилен)-7-оксо-4-тиа-1-азабицикло[3.2.0]гепт-2-ен-2-карбоновой кислоты
6,7-Дигидро-4H-тиено[3,2-c]тиопиран-2-карбальдегид (320 мг, 1,72 ммоль) растворяют в 17 мл ацетонитрила и добавляют эфират бромида магния (450 мг, 1,74 ммоль) в атмосфере азота и смесь перемешивают при 23°С в течение 0,5 ч. Затем в один прием вводят 4-нитробензиловый эфир 6-бром-7-оксо-4-тиа-1-азабицикло[3.2.0]гепт-2-ен-2-карбоновой кислоты (660 мг, 1,72 ммоль) в 17 мл ТГФ и смесь немедленно охлаждают до -20°C. Добавляют триэтиламин (1 мл) и смесь перемешивают при -20°C в течение трех часов. Затем вводят уксусный ангидрид (0,4 мл) и смесь перемешивают при 0°C в течение 18 часов. Реакционную смесь затем разбавляют 400 мл этилацетата и промывают 5% раствором лимонной кислоты (100 мл), насыщенным раствором бикарбоната натрия (100 мл) и насыщенным солевым раствором (100 мл). Органический слой сушат над сульфатом магния, фильтруют и концентрируют. Флэш-хроматографией на колонке с использованием 20% этилацетата в гексане в качестве элюента получают 461 мг (44%) продукта. Полученный продукт растворяют в 20 мл ТГФ и 20 мл водного фосфатного буфера (pH 6,5). Полученную смесь гидрируют при давлении водорода 40 фунт/кв.дюйм в течение 1,5 ч в присутствии 0,5 г 10% палладия на угле, затем фильтруют через рыхлый слой целита и концентрируют в вакууме, удаляя большую часть ТГФ. Раствор охлаждают до 0°С и доводят рН до 8, добавляя 1 н. раствор гидроксида натрия. Продукт очищают методом ВЭЖХ с обращенной фазой, используя 2 л воды, и затем 5% раствор ацетонитрила в воде в качестве элюентов. Воду удаляют концентрированием в вакууме и выделяют 21 мг продукта (8,6%). Т.пл. >250°C.
1H ЯМР: δ 7,34(с, 1H), 7,18 (с, 1H), 6,59 (с, 1H), 6,44 (с, 1H), 3,71 (с, 2H), 2,93 (с, 2H), 2,50 (с, 2H). MC: 338,0 (M+H).
Пример 30
Получение 6-(5-метил-4,5,6,7-тетрагидротиено[3,2-c]пиридин-2-илметилен)-7-оксо-4-тиа-1-азабицикло[3.2.0]гепт-2-ен-2-карбоновой кислоты
Стадия 1. Получение (5-метил-4,5,6,7-тетрагидротиено[3,2-c]пиридин-2-ил)-метанола
Диэтиловый эфир 6,7-дигидро-4H-тиено[3,2-c]пиридин-2,5-дикарбоновой кислоты (46 г, 163 моль) растворяют в 200 мл ТГФ. Вводят 300 мл раствора LAH (1M, ТГФ) при 23°С и реакционную смесь перемешивают при 23°C в течение 18 ч, гасят 10 мл воды и сушат непосредственно над сульфатом натрия. После фильтрования и концентрирования получают 29,3 г (160 ммоль, 98%) сырого продукта.
1H ЯМР δ: 6,55 (с, 1H), 4,70 (с, 2H), 3,41 (с, 2H), 2,86 (т, 2H, J=5,6 Гц), 2,73 (т, 2H, J=5,6 Гц), 2,38 (с, 3H). MC: 184,0 (M+H).
Стадия 2. Получение 5-метил-4,5,6,7-тетрагидротиено[3,2-c]пиридин-2-карбальдегида
ДМСО (1,7 мл, 24 ммоль) в 5 мл CH2Cl2 охлаждают до -50 - -60°C и добавляют оксалилхлорид (1 мл, 11 ммоль) в 20 мл DCM в течение 5 мин при -50°С. Смесь выдерживают при -50°С в течение 5 мин, затем добавляют 1,67 г (9 ммоль) (5-метил-4,5,6,7-тетрагидротиено[3,2-c]пиридин-2-ил)метанола в 20 мл DCM при -50°С и смесь перемешивают еще 15 мин при -50°С. Затем добавляют триэтиламин (7 мл) при -50°С, через 5 мин баню убирают и смеси дают нагреться естественным образом до 23°С. Смесь промывают 100 мл воды и водный слой экстрагируют 100 мл этилацетата. Объединенные органические слои сушат над сульфатом магния, фильтруют, концентрируют и флэш-хроматографией на колонке с использованием 0-15% метанола в этилацетате получают целевой продукт (736 мг, выход 45%).
1H ЯМР δ: 9,81 (с, 1H), 7,42 (с, 1H), 3,56 (с, 2H), 3,00 (т, 2H, J=5,6 Гц), 2,91 (т, 2H, J=5,6 Гц), 2,51 (с, 3H). MC: 182,1 (M+H).
Стадия 3. Получение 6-(5-метил-4,5,6,7-тетрагидротиено[3,2-c]пиридин-2-илметилен)-7-оксо-4-тиа-1-азабицикло[3.2.0]гепт-2-ен-2-карбоновой кислоты
Этиловый эфир 2-формил-6,7-дигидро-4H-тиено[3,2-c]пиридин-5-карбоновой кислоты (724 мг, 4 ммоль) растворяют в 40 мл ацетонитрила и затем добавляют эфират бромида магния (1,2 г, 4,65 ммоль) в атмосфере азота. Смесь перемешивают при 23°C в течение 0,5 ч. Затем вводят за один прием 4-нитробензиловый эфир 6-бром-7-оксо-4-тиа-1-азабицикло[3.2.0]гепт-2-ен-2-карбоновой кислоты (1,54 г, 4 ммоль) в 40 мл ТГФ и смесь немедленно охлаждают до -20°C. Добавляют триэтиламин (2 мл) и смесь перемешивают при -20°C в течение 3 ч. Вводят уксусный ангидрид (0,66 мл) и смесь перемешивают при 0°C в течение 48 ч. Затем реакционную смесь разбавляют 500 мл этилацетата и промывают 50 мл 5% раствора лимонной кислоты, 50 мл насыщенного раствора гидрокарбоната натрия и 50 мл насыщенного раствора хлорида натрия. Для промывки каждого из водных растворов используют еще 300 мл этилацетата. Объединенные органические слои затем сушат над сульфатом натрия, фильтруют, упаривают и с помощью флэш-хроматографии на колонке, используя 20% этилацетат в гексане выделяют продукт (1,56 г, выход 64%). Указанный продукт затем растворяют в 20 мл ТГФ и 20 мл водного раствора фосфатного буфера (pH 6,5). Потом смесь гидрируют (при давлении водорода 40 фунт/кв.дюйм) в течение 2 ч в присутствии 0,5 г 10% палладия на угле. Затем реакционную смесь фильтруют через рыхлый слой целита и концентрируют в вакууме для удаления большей части ТГФ. Концентрированный раствор охлаждают до 0°С и подщелачивают до pH 8 1 н.раствором гидроксида натрия, затем очищают ВЭЖХ с обращенной фазой, используя сначала 2 л воды и затем 5% раствор ацетонитрила в воде в качестве элюентов. Воду затем удаляют в вакууме и получают 112 мг (13%) продукта. Т.пл. >250°С.
1H ЯМР δ: 7,48 (с, 1H), 7,37 (с, 1H), 7,21 (с, 1H), 7,10 (с, 1H), 3,41 (с, 2H), 2,88 (с, 2H), 2,68 (с, 2H), 2,37 (с, 3H). MC: 335,0 (M+H).
Пример 31
Получение этилового эфира 2-(2-карбокси-7-оксо-4-тиа-1-азабицикло[3.2.0]гепт-2-ен-6-илиденметил)-6,7-дигидро-4H-тиено[3,2-c]пиридин-5-карбоновой кислоты
Этиловый эфир 2-формил-6,7-дигидро-4H-тиено[3,2-c]пиридин-5-карбоновой кислоты (480 мг, 2 ммоль) растворяют в 20 мл ацетонитрила и добавляют эфират бромида магния (516 мг, 2 ммоль) в атмосфере N2. Смесь перемешивают при 23°C в течение 0,5 ч. Затем вводят за один прием 4-нитробензиловый эфир 6-бром-7-оксо-4-тиа-1-азабицикло[3.2.0]гепт-2-ен-2-карбоновой кислоты (770 мг, 2 ммоль) в 20 мл ТГФ и смесь немедленно охлаждают до -20°C. Добавляют триэтиламин (1 мл) и смесь перемешивают при -20°C в течение 3 ч. Затем вводят уксусный ангидрид (0,4 мл) и смесь перемешивают при 0°C в течение 48 ч. Реакционную смесь разбавляют 200 мл этилацетата и промывают последовательно 50 мл 5% раствора лимонной кислоты, 50 мл насыщенного раствора гидрокарбоната натрия и 50 мл насыщенного раствора хлорида натрия. Органический слой затем сушат над сульфатом натрия, фильтруют, концентрируют, флэш-хроматографируют на колонке, используя 20% этилацетат в гексане в качестве элюента и выделяют продукт (690 мг, выход 50%). Часть полученного продукта (456 мг, 0,69 ммоль) растворяют в 15 мл ТГФ и 15 мл водного фосфатного буфера (pH 6,5) и гидрируют при давлении водорода 40 фунт/кв.дюйм в течение 2 ч в присутствии 0,5 г 10% палладия на угле. Затем реакционную смесь фильтруют через рыхлый слой целита и концентрируют в вакууме для удаления большей части ТГФ. Концентрированный раствор охлаждают до 0°С, подщелачивают до pH 8 1 н.раствором гидроксида натрия и очищают ВЭЖХ с обращенной фазой, используя 2 л воды и затем 5% раствор ацетонитрила в воде в качестве элюента. Воду удаляют в вакууме и получают 18 мг (5%) продукта. Т.пл. >250°С.
1H ЯМР δ: 7,35 (с, 1H), 7,24 (с, 1H), 6,61 (с, 1H), 6,45 (с, 1H), 4,48 (с, 2H), 4,08 (квартет, 2H, J=7,2 Гц), 3,68 (м, 2H), 2,87 (м, 2H), 1,20 (т, 3H, J=7,2 Гц). MC: 393,0 (M+H).
Пример 32
Получение 7-оксо-6-(6,7,8,9-тетрагидро-5H-имидазо[1,2-a]азепин-2-илметилен)-4-тиа-l-азабицикло[3.2.0]гепт-2-ен-2-карбоновой кислоты
Стадия 1. Получение 6,7,8,9-тетрагидро-5H-имидазо[1,2-a]азепин-2-карбальдегида
Тиокапролактам (6,45 г, 50 ммоль) растворяют в 400 мл CH2Cl2 и добавляют метилиодид (16 мл, 5 экв.). Смесь перемешивают в атмосфере азота в течение 18 ч, затем обрабатывают 100 мл 50% водного раствора карбоната калия. Органический слой сушат над сульфатом магния. После фильтрования и концентрирования фильтрата получают 7,3 г вещества. Полученное вещество растворяют в 300 мл этанола и добавляют 2,83 г хлорида аммония. Смесь кипятят с обратным холодильником в течение 1 ч, затем растворитель удаляют в вакууме. К половине полученного вещества добавляют 200 мл этанола, затем 1,35 г (25 ммоль) метоксида натрия и 4,8 г (25 ммоль) 2-бром-3-изопропоксипропеналя и смесь перемешивают при 23°С в течение 2 ч. Затем растворитель удаляют и к остатку добавляют 200 мл хлороформа вместе с 10 мл триэтиламина. Смесь кипятят с обратным холодильником в течение 2 ч и затем охлаждают до 23°С. Реакционную смесь распределяют между 300 мл DCM и 50% карбонатом калия (2 × 150 мл). Органический слой сушат над сульфатом магния. После фильтрования и концентрирования получают 2,1 г продукта в виде масла.
1H ЯМР δ: 9,62 (с, 1H), 7,60 (с, 1H), 6,61 (с, 1H), 6,45 (с, 1H), 4,58 (с, 2H), 2,96 (2м, H), 1,90 (м, 2H), 1,72 (м, 2H). MC: 164,9 (M+H).
Стадия 2. Получение 7-оксо-6-(6,7,8,9-тетрагидро-5H-имидазо[1,2-a]азепин-2-илметилен)-4-тиа-1-азабицикло[3.2.0]гепт-2-ен-2-карбоновой кислоты
6,7,8,9-Тетрагидро-5H-имидазо[1,2-a]азепин-2-карбальдегид (1,312 г, 8 ммоль) растворяют в 80 мл ацетонитрила и добавляют эфират бромида магния (2,94 г, 8 ммоль) в атмосфере N2. Смесь перемешивают при 23°С в течение 0,5 ч. Затем в один прием вводят 4-нитробензиловый эфир 6-бром-7-оксо-4-тиа-1-азабицикло[3.2.0]гепт-2-ен-2-карбоновой кислоты (1,155 г, 3 ммоль) в 60 мл ТГФ и смесь немедленно охлаждают до -20°C. Добавляют триэтиламин (4 мл) и смесь перемешивают при -20°C в течение 4 ч. Затем вводят уксусный ангидрид (1 мл) и смесь перемешивают при 0°C в течение 20 ч. Реакционную смесь разбавляют 500 мл этилацетата и промывают 5% раствором лимонной кислоты (100 мл), насыщенным раствором гидрокарбоната натрия (100 мл) и насыщенным солевым раствором (100 мл). Органический слой сушат над сульфатом натрия, фильтруют, концентрируют и флэш-хроматографируют на колонке с использованием 20% этилацетата в гексане в качестве элюента. Получают 800 мг продукта, который растворяют в 20 мл ТГФ и 20 мл водного фосфатного буфера (pH 6,5). Смесь затем гидрируют при давлении водорода 40 фунт/кв.дюйм в течение 1 ч в присутствии 0,5 г 10% палладия на угле, затем фильтруют через рыхлый слой целита и концентрируют в вакууме для удаления большей части ТГФ. Раствор охлаждают до 0°С, доводят рН до 8, добавляя 1 н. раствор гидроксида натрия, и очищают ВЭЖХ с обращенной фазой, используя 2 л воды и затем 5% раствор ацетонитрила в воде в качестве элюента. Воду удаляют в вакууме и получают 131 мг (31%) продукта. Т.пл. >250°C.
1H ЯМР δ: 7,78 (с, 1H), 7,02 (с, 1H), 6,94 (с, 1H), 6,36 (с, 1H), 3,92 (м, 2H), 2,80 (м, 2H), 1,78 (м, 2H), 1,61 (м, 2H), 1,54 (м, 2H); MC: 318,2 (M+H).
Пример 33
Натриевая соль (5R),(6Z)-6-(7-бензил-5,6,7,8-тетрагидроимидазо[1,2-a]пиразин-2-илметилен)-7-оксо-4-тиа-1-азабицикло[3.2.0]гепт-2-ен-2-карбоновой кислоты
Стадия 1. Этиловый эфир 7-бензил-5,6,7,8-тетрагидроимидазо[1,2-a]пиразин-2-карбоновой кислоты
Et3N (6,27 мл), PhCHO (4,92 мл) последовательно добавляют к раствору гидрохлорида этилового эфира 5,6,7,8-тетрагидроимидазо[1,2-a]пиразин-2-карбоновой кислоты (9,47 г) в EtOH (81 мл) при комнатной температуре и перемешивают в течение 3 ч в атмосфере азота. Затем к реакционной смеси добавляют NaBH3CN (2,97 г) и перемешивают в течение 19 ч. Полученную смесь фильтруют через рыхлый слой целита, разбавляют CH2Cl2 и промывают 50% водным раствором K2CO3. Органический слой сушат (K2CO3) и фильтруют. Фильтрат концентрируют при пониженном давлении. Остаток наносят на хроматографическую колонку с силикагелем, затем элюируют смесью CHCl3-ацетон (1/0-9/1) и смесью CHCl3-MeOH (19/1-9/1). Получают указанное в заголовке соединение в виде бледно-желтых кристаллов (4,16 г, 36%).
1H ЯМР (CDCl3) δ: 1,36 (т, 3H, J=7,1 Гц), 2,87 (т, 2H, J=5,2 Гц), 3,71 (с, 2H), 3,75 (с, 2H), 4,01 (м, 2H), 4,34 (кв, 2H, J=7,1 Гц), 7,25-7,34 (м, 5H), 7,51 (с, 1H).
Стадия 2. 7-Бензил-5,6,7,8-тетрагидроимидазо[1,2-a]пиразин-2-карбальдегид
К раствору этилового эфира 7-бензил-5,6,7,8-тетрагидроимидазо[1,2-a]пиразин-2-карбоновой кислоты (283 мг) в сухом CH2Cl2 (5 мл) добавляют 1,01M раствор DIBAL в толуоле (1 мл + 0,2 мл + 0,3 мл) в атмосфере азота при -78°С и перемешивают в течение 1,5 ч. Смесь гасят 1M раствором HCl (5 мл). Реакционную смесь фильтруют через рыхлый слой целита. Фильтрат промывают 50% водным раствором K2CO3 и водный слой экстрагируют CH2Cl2. Объединенный органический слой сушат (K2CO3) и фильтруют. Фильтрат концентрируют при пониженном давлении. Остаток наносят на хроматографическую колонку с силикагелем, затем элюируют смесью CHCl3-ацетон (9/1˜4/1) и смесью CHCl3-MeOH (19/1). Получают указанное в заголовке соединение в виде бесцветных кристаллов (148 мг, 61%).
1H ЯМР (CDCl3) δ: 2,90 (т, 2H, J=5,5 Гц), 3,74 (с, 2H), 3,76 (с, 2H), 4,06 (т, 2H, J=5,5 Гц), 7,28-7,35 (м, 5H), 7,53 (с, 1H), 9,80 (с, 1H).
Стадия 3. 4-Нитробензиловый эфир (5R,6RS)-6-[(RS)-ацетокси(7-бензил-5,6,7,8-тетрагидроимидазо[1,2-a]пиразин-2-ил)метил]-6-бром-7-оксо-4-тиа-1-азабицикло[3.2.0]гепт-2-ен-2-карбоновой кислоты (смесь диастереомеров)
7-Бензил-5,6,7,8-тетрагидроимидазо[1,2-a]пиразин-2-карбальдегид (139 мг) добавляют к раствору безводного MgBr2 (325 мг) в сухом ацетонитриле (8,7 мл) в атмосфере азота при комнатной температуре. К полученной смеси добавляют раствор 4-нитробензилового эфира (5R,6S)-6-бром-7-оксо-4-тиа-1-азабицикло[3.2.0]гепт-2-ен-2-карбоновой кислоты (223 мг) в сухом ТГФ (8,7 мл), охлаждают до -20°C и добавляют Et3N (0,24 мл) в один прием. Реакционный сосуд покрывают фольгой, чтобы исключить воздействие света. Реакционную смесь перемешивают в течение 5 ч при -20°C и в один прием обрабатывают уксусным ангидридом (0,11 мл) и DMAP (7 мг). Температуру реакционной смеси доводят до 0°C и перемешивают в течение 15 ч при 0°C. Смесь разбавляют этилацетатом и промывают 5% водным раствором лимонной кислоты, насыщенным раствором гидрокарбоната натрия и насыщенным солевым раствором. Органический слой сушат (MgSO4) и фильтруют. Фильтрат концентрируют при пониженном давлении. Остаток наносят на хроматографическую колонку с силикагелем, затем элюируют смесью н-гексан-AcOEt(3/1˜1/1). Получают указанное в заголовке соединение в виде смеси двух диастереомеров (80/20, аморфное твердое вещество пурпурного цвета, 233 мг, 61%).
1H ЯМР (CDCl3), δ: 1,99 (с, 0,8 × 3H), 2,23 (с, 0,2 × 3H), 2,83-2,89 (м, 2H), 3,68 (д, 2H, J=4,9 Гц), 3,7l (с, 2H), 3,94-4,13 (м, 2H), 5,27 (д, 1H, J=13,6 Гц), 5,41 (д, 0,2 × 1H, J=13,6 Гц), 5,45 (д, 0,8 × 1H, J=13,6 Гц), 6,05 (с, 0,2 × 1H), 6,28 (с, 0,8 × 1H), 6,31 (с, 0,8 × 1H), 6,790 (с, 0,2 × 1H), 6,793 (с, 0,2 × 1H), 7,01 (с, 0,8 × 1H), 7,27-7,36 (м, 5H), 7,42 (с, 0,2 × 1H), 7,46 (с, 0,8 × 1H), 7,61 (д, 2H, J=8,6 Гц), 8,22 (д, 2H, J=8,6 Гц).
Стадия 4. Натриевая соль (5R),(6Z)-6-(7-бензил-5,6,7,8-тетрагидроимидазо[1,2-a]пиразин-2-илметилен)-7-оксо-4-тиа-1-азабицикло[3.2.0]гепт-2-ен-2-карбоновой кислоты
4-Нитробензиловый эфир (5R,6RS)-6-[(RS)-ацетокси(7-бензил-5,6,7,8-тетрагидроимидазо[1,2-a]пиразин-2-ил)метил]-6-бром-7-оксо-4-тиа-1-азабицикло[3.2.0]гепт-2-ен-2-карбоновой кислоты (1,27 г) растворяют в ТГФ (55 мл) и ацетонитриле (25 мл). Быстро добавляют свежеактивированную Zn пыль (5,08 г) вместе с 0,5M фосфатным буфером (pH 6,5, 80 мл). Реакционный сосуд покрывают фольгой, чтобы исключить воздействие света. Реакционную смесь энергично перемешивают в течение 2 ч при комнатной температуре. Реакционную смесь фильтруют через рыхлый слой целита. Фильтрат промывают этилацетатом и водный слой отделяют. Водный слой охлаждают до 3°C и добавляют 1M NaOH, доводя рН до 8,0. Смесь концентрируют в высоком вакууме при 35°C. Концентрат наносят на хроматографическую колонку со смолой Diaion HP-21 (79 мл, Mitsubishi Kasei Co. Ltd.). После адсорбции осуществляют элюирование смесью H2О-MeCN (1/0˜4/1). Объединенные фракции концентрируют в высоком вакууме при 35°C, лиофилизуют и получают указанное в заголовке соединение в виде желтого аморфного твердого вещества (390 мг, 49%, pH 7,7). Т.пл. 180°C (с разложением).
1H ЯМР (D2O) δ: 2,84-2,95 (м, 2H), 3,61 (д, 2H, J=7,2 Гц), 3,67 (с, 2H), 3,96 (т, 2H, J=5,7 Гц), 6,43 (с, 1H), 6,89 (с, 1H), 6, 93 (с, 1H), 7,28-7,37 (м, 6H).
Пример 34
Получение (5R),(6Z)-7-оксо-6-{[5-(пиридин-3-илметил)-4,5,6,7-тетрагидротиено[3,2-с]пиридин-2-ил)]метилен}-7-оксо-4-тиа-1-азабицикло[3.2.0]гепт-2-ен-2-карбоновой кислоты
Стадия 1. 2-Формил[5-(пиридин-3-илметил)-4,5,6,7-тетрагидротиено][3,2-c]пиридин
К перемешиваемому раствору 2-(формил)-6,7-дигидротиено[3,2-c]-5(4H)пиридина (1,05 г, 5,2 ммоль) в ДМФА (20 мл) гидрохлорид 3-пиколилхлорида (0,852 г, 5,2 ммоль) и N,N-диизопропилэтиламин (10 мл, избыток) добавляют при комнатной температуре. Реакционную смесь перемешивают в течение 24 ч и гасят водой. Реакционную смесь экстрагируют хлороформом, тщательно промывают водой, сушат над безводным MgSO4, фильтруют и концентрируют. Продукт очищают колоночной хроматографией на SiO2 при элюировании этилацетатом. Получают бледно-желтое полутвердое вещество. Выход 800 мг (59%); (M+H) 259.
Стадия 2. 4-Нитробензил-6-{(ацетокси)[5-(пиридин-3-илметил)-4,5,6,7-тетрагидротиено[3,2-c]пиридин-2-ил]метил}-6-бром-7-оксо-4-тиа-1-азабицикло[3.2.0]гепт-2-ен-2-карбоксилат
2-Формил [5-(пиридин-3-илметил)-4,5,6,7-тетрагидротиено[3,2-c]пиридин (516 мг, 2,0 ммоль) и раствор 4-нитробензилового эфира (5R,6S)-6-бром-7-оксо-4-тиа-1-азабицикло[3.2.0]гепт-2-ен-2-карбоновой кислоты (772 мг, 2,0 ммоль) в сухом ТГФ (20 мл) последовательно добавляют к раствору безводного MgBr2:O(Et)2 (390 мг, 1,5 ммоль) в сухом ацетонитриле (15 мл) в атмосфере аргона при комнатной температуре. После охлаждения до -20°C добавляют в один прием Et3N (2,0 мл). Реакционный сосуд покрывают фольгой, чтобы исключить воздействие света. Реакционную смесь перемешивают в течение 2 ч при -20°C и обрабатывают уксусным ангидридом (1,04 мл) в один прием. Реакционную смесь доводят до 0°C и перемешивают в течение 15 ч при 0°C. Смесь разбавляют этилацетатом и промывают 5% водным раствором лимонной кислоты, насыщенным раствором гидрокарбоната натрия и насыщенным солевым раствором. Органический слой сушат (MgSO4) и фильтруют через рыхлый слой целита. Рыхлый слой промывают этилацетатом. Фильтрат концентрируют при пониженном давлении. Остаток наносят на хроматографическую колонку с силикагелем, затем элюируют смесью этилацетат:гексан (1:1). Собранные фракции концентрируют при пониженном давлении и полученную смесь диастереомеров в виде желтого аморфного твердого вещества используют на следующей стадии. Выход 700 мг (51%). (M+H) 685 и 687.
Стадия 3. (5R,6Z)-6-{[5-(пиридин-3-илметил)-4,5,6,7-тетрагидротиено[3,2-c]пиридин-2-ил]метилен}-7-оксо-4-тиа-1-азабицикло[3.2.0]гепт-2-ен-2-карбоновая кислота
4-Нитробензил-6-[(ацетокси)[5(пиридин-3-илметил)-4,5,6,7-тетрагидротиено[3,2-c]пиридин-2-ил)метил]-6-бром-7-оксо-4-тиа-1-азабицикло[3.2.0]гепт-2-ен-2-карбоксилат (686 мг, 1,0 ммоль) растворяют в ТГФ (20 мл) и ацетонитриле (10 мл). Быстро добавляют свежеактивированную Zn пыль (5,2 г) вместе с 0,5M фосфатным буфером (pH 6,5, 28 мл). Реакционный сосуд покрывают фольгой, чтобы исключить воздействие света. Реакционную смесь энергично перемешивают в течение 2 ч при комнатной температуре. Реакционную смесь фильтруют, охлаждают до 3°C и добавляют 0,1M NaOH, чтобы довести рН до 8,0. Фильтрат промывают этилацетатом, водный слой отделяют, концентрируют в высоком вакууме при 35°С и получают желтый осадок. Продукт очищают хроматографией с обращенной фазой на колонке со смолой HP-21. Сначала проводят элюирование деионизированной водой (2 л) и затем 10% CAN: вода. Фракции, содержащие продукт, собирают и концентрируют при пониженном давлении при комнатной температуре. Полученное желтое твердое вещество промывают ацетоном, отфильтровывают и сушат. Выход продукта в виде желтых кристаллов 50 мг (12%), т.пл. 134-136°C. (M+H) 412.
1H ЯМР (ДМСО-d6) δ: 2,8 (м, 2H), 2,92 (уш.м, 2H), 3,6 (м, 2H), 3,86 (с, 2H), 6,3 (с, 1H), 6,41 (с, 1H), 7,17 (с, 1H), 7,29 (с, 1H), 7,35 (м, 1H), 7,7 (м, 1H), 8,48 (д, 1H), 8,54 (с, 1H).
Пример 35
Получение (5R,6Z)-7-оксо-6-{[5-(пиридин-3-илкарбонил)-4,5,6,7-тетрагидротиено[3,2-c]пиридин-2-ил)]метилен}-7-оксо-4-тиа-1-азабицикло[3.2.0]гепт-2-ен-2-карбоновой кислоты
Стадия 1. 2-Формил-[5-(пиридин-3-илкарбонил)-4,5 6,7-тетрагидротиено[3,2-c]пиридин
К перемешиваемому раствору 2-(формил)-6,7-дигидротиено[3,2-c]-5(4H)пиридина (606 мг, 3,0 ммоль) в ДМФА (20 мл) гидрохлорид никотиноилхлорида (531 мг, 3,0 ммоль) и N,N-диизопропилэтиламин (10 мл, избыток) добавляют при комнатной температуре. Реакционную смесь перемешивают в течение 24 ч и гасят водой. Реакционную смесь экстрагируют хлороформом, тщательно промывают водой, сушат над безводным MgSO4, фильтруют и концентрируют. Продукт очищают хроматографически на колонке с SiO2, элюирование проводят этилацетатом. Получают бледно-желтое полутвердое вещество. Выход 600 мг, 73%; (M+H) 273.
Стадия 2. 4-Нитробензил-6-[(ацетокси)[5-(пиридин-3-илкарбонил)-4,5,6,7-тетрагидротиено[3,2-c]пиридин-2-ил)метил]-6-бром-7-оксо-4-тиа-1-азабицикло[3.2.0]гепт-2-ен-2-карбоксилат
2-Формил-[5-(пиридин-3-илкарбонил)-4,5,6,7-тетрагидротиено[3,2-c]пиридин (400 мг, 1,4 ммоль) и раствор 4-нитробензилового эфира (5R,6S)-6-бром-7-оксо-4-тиа-1-азабицикло[3.2.0]гепт-2-ен-2-карбоновой кислоты (772 мг, 2,0 ммоль) в сухом ТГФ (20 мл) последовательно добавляют к раствору безводного MgBr2:O(Et)2 (619 мг, 2,4 ммоль) в сухом ацетонитриле (15 мл) в атмосфере аргона при комнатной температуре. После охлаждения до -20°C добавляют в один прием Et3N (2,0 мл). Реакционный сосуд покрывают фольгой, чтобы исключить воздействие света. Реакционную смесь перемешивают в течение 2 ч при -20°C и обрабатывают уксусным ангидридом (1,04 мл) в один прием. Температуру реакционной смеси доводят до 0°C и перемешивают в течение 15 ч при 0°C. Смесь разбавляют этилацетатом и промывают 5% водным раствором лимонной кислоты, насыщенным раствором гидрокарбоната натрия и насыщенным солевым раствором. Органический слой сушат (MgSO4) и фильтруют через рыхлый слой целита. Рыхлый слой промывают этилацетатом. Фильтрат концентрируют при пониженном давлении. Остаток наносят на хроматографическую колонку с силикагелем, затем элюируют смесью этилацетат:гексан (1:1). Собранные фракции концентрируют при пониженном давлении и полученную смесь диастереомеров в виде желтого аморфного твердого вещества используют на следующей стадии. Выход 300 мг (30%). Т.пл. 71°C; (M+H) 701.
Стадия 3. Натриевая соль (5R,6Z)-6-{[5-(пиридин-3-илкарбонил)-4,5,6,7-тетрагидротиено[3,2-c]пиридин-2-ил)метилен}-7-оксо-4-тиа-1-азабицикло[3.2.0]гепт-2-ен-2-карбоновой кислоты
4-Нитробензил-6-[(ацетокси)[5-(пиридин-3-илкарбонил)-4,5,6,7-тетрагидротиено[3,2-c]пиридин-2-ил)метил]-6-бром-7-оксо-4-тиа-1-азабицикло[3.2.0]гепт-2-ен-2-карбоксилат (800 мг, 1,14 ммоль) растворяют в ТГФ (20 мл) и ацетонитриле (10 мл). Быстро добавляют свежеактивированную Zn пыль (5,2 г) вместе с 0,5M фосфатным буфером (pH 6,5, 28 мл). Реакционный сосуд покрывают фольгой, чтобы исключить воздействие света. Реакционную смесь энергично перемешивают в течение 2 ч при комнатной температуре. Реакционную смесь фильтруют, охлаждают до 3°C и подщелачивают 0,1M NaOH до рН 8,5. Фильтрат промывают этилацетатом, водный слой отделяют, концентрируют в высоком вакууме при 35°С и получают желтый осадок, который очищают хроматографией с обращенной фазой на колонке с HP-21. Сначала проводят элюирование деионизированной водой (2 л) и затем 10% CAN в воде. Фракции, содержащие продукт, собирают и концентрируют при пониженном давлении при комнатной температуре. Полученное желтое твердое вещество промывают ацетоном, отфильтровывают и сушат. Выход желтых кристаллов 50 мг (12%), т.пл. 195°C. (M+H) 426.
Пример 36
Получение (5R,6Z)-7-оксо-6-{[5-(фенилацетил)-4,5,6,7-тетрагидротиено[3,2-c]пиридин-2-ил)]метилен}-7-оксо-4-тиа-1-азабицикло[3.2.0]гепт-2-ен-2-карбоновой кислоты
Стадия 1. 2-Формил[5-(фенилацетил)-4,5,6,7-тетрагидротиено[3,2-c]пиридин
К перемешиваемому раствору 2-(формил)-6,7-дигидротиено[3,2-c]-5(4H)пиридина (0,41 мг, 2,0 ммоль) в ДМФА (20 мл) добавляют фенилацетилхлорид (0,35 мг, 2,2 ммоль) и N,N-диизопропилэтиламин (10 мл, избыток) при комнатной температуре. Реакционную смесь перемешивают в течение 24 ч и гасят водой. Реакционную смесь экстрагируют хлороформом, тщательно промывают водой, сушат над безводным MgSO4, фильтруют и концентрируют. Продукт очищают хроматографически на колонке с SiO2, элюируя его этилацетатом. Получают белое твердое вещество. Выход 510 мг (89%); (M+H) 286.
Стадия 2. 4-Нитробензил-6-[(ацетокси)[5(фенилацетил)-4,5,6,7-тетрагидротиено[3,2-c]пиридин-2-ил)метил]-6-бром-7-оксо-4-тиа-1-азабицикло[3.2.0]гепт-2-ен-2-карбоксилат
2-Формил-[5-(фенилацетил)-4,5,6,7-тетрагидротиено[3,2-c]пиридин (340 мг, 1,2 ммоль) и раствор 4-нитробензилового эфира (5R,6S)-6-бром-7-оксо-4-тиа-1-азабицикло[3.2.0]гепт-2-ен-2-карбоновой кислоты (390 мг, 1,0 ммоль) в сухом ТГФ (20 мл) последовательно добавляют к раствору безводного MgBr2:O(Et)2 (310 мг, 1,2 ммоль) в сухом ацетонитриле (15 мл) в атмосфере аргона при комнатной температуре. После охлаждения до -20°C добавляют в один прием Et3N (2,0 мл). Реакционный сосуд покрывают фольгой, чтобы исключить воздействие света. Реакционную смесь перемешивают в течение 2 ч при -20°C и обрабатывают уксусным ангидридом (1,04 мл) в один прием. Температуру реакционной смеси доводят до 0°C и перемешивают в течение 15 ч при 0°C. Смесь разбавляют этилацетатом и промывают 5% водным раствором лимонной кислоты, насыщенным раствором гидрокарбоната натрия и насыщенным солевым раствором. Органический слой сушат (MgSO4) и фильтруют через рыхлый слой целита, который затем промывают этилацетатом. Фильтрат концентрируют при пониженном давлении. Остаток наносят на хроматографическую колонку с силикагелем, затем элюируют смесью этилацетат:гексан (1:1). Выделенные фракции концентрируют при пониженном давлении и полученную смесь диастереомеров в виде бледно-желтого аморфного твердого вещества используют на следующей стадии. Выход 360 мг (50%); (М+Н) 713.
Стадия 3. (5R,6Z)-6-{[5-(Фенилацетил)-4,5,6,7-тетрагидротиено[3,2-c]пиридин-2-ил)метилен}-7-оксо-4-тиа-1-азабицикло[3.2.0]гепт-2-ен-2-карбоновая кислота
4-Нитробензил-6-[(ацетокси)[5-(фенилацетил)-4,5,6,7-тетрагидротиено[3,2-c]пиридин-2-ил)метил]-6-бром-7-оксо-4-тиа-1-азабицикло[3.2.0]гепт-2-ен-2-карбоксилат (300 мг, 0,4 ммоль) растворяют в ТГФ (50 мл) и 0,5M фосфатном буфере (pH 6,5, 28 мл) и гидрируют при давлении водорода 40 фунт/кв.дюйм в присутствии 10% Pd/C (80 мг) в течение 2 ч, после чего реакционную смесь фильтруют через рыхлый слой целита и концентрируют. Полученное желтое твердое вещество растворяют в этилацетате и тщательно промывают водой. Органический слой сушат и концентрируют. Полученное желтое твердое вещество растирают в диэтиловом эфире, отфильтровывают и тщательно промывают диэтиловым эфиром и получают желтый твердый продукт 95% чистоты. Выход 160 мг (91%). Т.пл. 166-169°C, (M+H) 439.
Пример 37
Получение (5R,6Z)-6-[(7-метилимидазо[2,1-b][1,3]бензотиазол-2-илметилен)-7-оксо-4-тиа-1-азабицикло[3.2.0]гепт-2-ен-2-карбоновой кислоты
Стадия 1. Этил 7-метилимидазо[2,1-b]-бензотиазол-2-карбоксилат
Этил 7-метилимидазо[2,1-b]-бензотиазол-2-карбоксилат получают по методике, описанной в примере 1 (Стадия 1). Исходя из 6-метил-2-аминобензотиазола (3,2 г, 20 ммоль) и этилбромпирувата (4,0 г, 20,4 ммоль) получают этил 7-метилимидазо[2,1-b]бензотиазол-2-карбоксилат с выходом 3,0 г (57%) в виде коричневого твердого вещества. (M+H) 261.
Стадия 2. 2-Формил-7-метилимидазо[2,1-b]бензотиазол
К перемешиваемому раствору этил 7-метилимидазо[2,1-b]бензотиазол-2-карбоксилата (4,0 г, 15,38 ммоль) в сухом ТГФ при -78°C добавляют DIBAL (1M раствор в толуоле) (16,0 мл, 16 ммоль). Реакционную смесь перемешивают при -78°C и медленно доводят температуру до комнатной. Реакционную смесь перемешивают при комнатной температуре в течение 30 мин и гасят насыщенным раствором NH4Cl. Реакционную смесь экстрагируют хлороформом и тщательно промывают водой. Органический слой сушат над безводным MgSO4, фильтруют и концентрируют. Остаток очищают хроматографией на колонке с SiO2, элюирование проводят смесью хлороформ:метанол (20:1). Выделяют коричневое твердое вещество (800 мг, 24%). (M+H) 217.
Стадия 3. 4-Нитробензил-6-[(ацетилокси)(7-метилимидазо[2,1-b][1,3]бензотиазол-2-ил)метил]-6-бром-7-оксо-4-тиа-1-азабицикло[3.2.0]гепт-2-ен-2-карбоксилат
2-Формил-7-метилимидазо[2,1-b]бензотиазол (432 мг, 2,0 ммоль) и раствор 4-нитробензилового эфира (5R,6S)-6-бром-7-оксо-4-тиа-1-азабицикло[3.2.0]гепт-2-ен-2-карбоновой кислоты (772 мг, 2,0 ммоль) в сухом ТГФ (20 мл) последовательно добавляют к раствору безводного MgBr2:O(Et)2 (566 мг, 2,0 ммоль) в сухом ацетонитриле (15 мл) в атмосфере аргона при комнатной температуре. После охлаждения до -20°C добавляют в один прием Et3N (2,0 мл). Реакционный сосуд покрывают фольгой, чтобы исключить воздействие света. Реакционную смесь перемешивают в течение 2 ч при -20°C и обрабатывают уксусным ангидридом (1,04 мл) в один прием. Температуру реакционной смеси доводят до 0°C и перемешивают в течение 15 ч при 0°C. Смесь разбавляют этилацетатом и промывают 5% водным раствором лимонной кислоты, насыщенным раствором гидрокарбоната натрия и насыщенным солевым раствором. Органический слой сушат (MgSO4) и фильтруют через рыхлый слой целита. Рыхлый слой промывают этилацетатом. Фильтрат концентрируют при пониженном давлении. Остаток наносят на хроматографическую колонку с силикагелем, затем элюируют смесью этилацетат:гексан (1:1). Собранные фракции концентрируют при пониженном давлении и полученную смесь диастереомеров в виде бледно-желтого аморфного твердого вещества используют на следующей стадии. Выход 400 мг, 31%. (М+Н) 645.
Стадия 4. (5R),(6Z)-6-[(7-Метилимидазо[1,2-b][1,3]бензотиазол-2-илметилен)]-7-оксо-4-тиа-1-азабицикло[3.2.0]гепт-2-ен-2-карбоновая кислота
4-Нитробензил-6-[(ацетилокси)(7-метилимидазо[2,1-b][1,3]бензотиазол-2-ил)метил]-6-бром-7-оксо-4-тиа-1-азабицикло[3.2.0]гепт-2-ен-2-карбоксилат (350 мг, 0,54 ммоль) растворяют в ТГФ (20 мл) и ацетонитриле (10 мл). Быстро добавляют свежеактивированную Zn пыль (5,2 г) вместе с 0,5M фосфатным буфером (pH 6,5, 28 мл). Реакционный сосуд покрывают фольгой, чтобы исключить воздействие света. Реакционную смесь энергично перемешивают в течение 2 ч при комнатной температуре. Реакционную смесь фильтруют, охлаждают до 3°C и добавляют 0,1M NaOH, доводя рН до 8,5. Фильтрат промывают этилацетатом, водный слой отделяют, концентрируют в высоком вакууме при 35°С и получают желтый осадок. Продукт очищают хроматографией с обращенной фазой на колонке со смолой HP-21. Сначала проводят элюирование деионизированной водой (2 л) и затем 10% раствором ацетонитрила в воде. Фракции, содержащие продукт, собирают и концентрируют при пониженном давлении при комнатной температуре. Полученное желтое твердое вещество промывают ацетоном, отфильтровывают и сушат. Выход желтых кристаллов 110 мг (55%). Т.пл. 178°С (с разложением). (M+H+Na) 392.
1H ЯМР(ДМСО-d6) δ: 8,56 (с, 1H), 7,93 (д, 1H), 7,83 (с, 1H), 7,38 (д, 1H), 7,07 (с, 1H), 6,51 (с, 2H), 2,42 (с, 3H).
Стадия 4. (5R),(6Z)-6-[(7-Метилимидазо[1,2-b][1,3]бензотиазол-2-илметилен)]-7-оксо-4-тиа-1-азабицикло[3.2.0]гепт-2-ен-2-карбоновая кислота (Методика B)
4-Нитробензил-6-[(ацетокси)(7-метилимидазо[2,1-b][1,3]бензотиазол-2-ил)метил]-6-бром-7-оксо-4-тиа-1-азабицикло[3.2.0]гепт-2-ен-2-карбоксилат (350 мг, 0,54 ммоль) растворяют в ТГФ (40 мл) и фосфатном буфере с pH 6,5 (40 мл) и гидрируют над Pd/C (10%, 200 мг) при давлении водорода 40 фунт/кв.дюйм в течение 3 ч при комнатной температуре. По окончании гидрирования реакционную смесь фильтруют через рыхлый слой целита и промывают ацетонитрилом. Реакционную смесь концентрируют до 40 мл, охлаждают до 0°С и доводят рН до 8,5, добавляя 1 н. NaOH. Продукт очищают хроматографией с обращенной фазой на колонке со смолой HP-21. Сначала элюируют деионизированной водой (2 л) и затем 10% ацетонитрилом в воде. Фракции концентрируют и полученное желтое твердое вещество промывают ацетоном, отфильтровывают и сушат. Получают желтый твердый продукт (выход 110 мг, 55%).
Пример 38
Получение натриевой соли (5R),(6Z)-6-(4,5,6,7-тетрагидро-1,3a,3b,8-тетраазациклопента[a]инден-2-илметилен)-7-оксо-4-тиа-1-азабицикло[3.2.0]гепт-2-ен-2-карбоновой кислоты
Стадия 1. 5,6,7,8-Тетрагидро[1,2,4]триазоло[1,5-a]пиридин-2-иламин
К [1,2,4]триазоло[1,5-a]пиридин-2-иламину (2,5 г) в этаноле (72 мл) добавляют 12,7% раствор HCl в этаноле (5,35 мл) и 10% Pd-C (50% влажность) (2,5 г). Реакционную смесь гидрируют при давлении H2 400 кПа в течение 3 дней при комнатной температуре. Смесь фильтруют и концентрируют при пониженном давлении. Остаток обрабатывают насыщенным раствором карбоната калия и экстрагируют хлороформом. Органический слой сушат (Na2SO4) и концентрируют при пониженном давлении. Получают указанное в заголовке соединение в виде бледно-желтого твердого вещества (2,31 г, 90%).
1H ЯМР (400 МГц, CDCl3) δ: 1,88-1,94 (м, 2H), 1,98-2,05 (м, 2H), 2,77 (т, 2H, J=6,2 Гц), 3,95 (т, 2H, J=6,2 Гц), 4,09 (уш.с, 2H).
Стадия 2. Этиловый эфир 4,5,6,7-тетрагидро-1,3a,3b,8-тетраазациклопента[a]инден-2-карбоновой кислоты
Этилбромпируват (10,23 г) добавляют к 5,6,7,8-тетрагидро[1,2,4]триазоло[1,5-a]пиридин-2-иламину (5,8 г) в 1,2-диметоксиэтане (320 мл). Реакционную смесь перемешивают в течение 5 ч при комнатной температуре и концентрируют до 100 мл при пониженном давлении. Прибавлением диэтилового эфира (200 мл) получают осадок, который отфильтровывают, растворяют в этаноле (175 мл) и перемешивают в течение 20 ч при 110°С в экранированной ампуле. Реакционную смесь охлаждают до комнатной температуры и концентрируют при пониженном давлении. Остаток обрабатывают насыщенным раствором карбоната калия и экстрагируют хлороформом. Органический слой сушат (Na2SO4) и концентрируют при пониженном давлении. Остаток наносят на хроматографическую колонку с силикагелем, затем элюируют смесью этилацетат-метанол (1/1). Получают указанное в заголовке соединение в виде бледно-желтого твердого вещества (7,56 г, 77%).
1H ЯМР (400 МГц, CDCl3) δ: 1,42 (т, 3H, J=7,1 Гц), 2,14-2,25 (м, 4H), 3,11 (т, 2H, J=6,1 Гц), 4,37 (т, 2H, J=5,7 Гц), 4,41 (кв, 2H, J=7,1 Гц), 7,57 (с, 1H).
Стадия 3. 4,5,6,7-Тетрагидро-1,3a,3b,8-тетраазациклопента[a]инден-2-карбальдегид
Раствор (1,01M) диизобутилалюминийгидрида в толуоле (1,06 мл) добавляют по каплям к раствору этилового эфира 4,5,6,7-тетрагидро-1,3a,3b,8-тетраазациклопента[a]инден-2-карбоновой кислоты (100 мг) в сухом ТГФ (5 мл) при -78°C в атмосфере азота. Реакционную смесь перемешивают в течение 30 мин при -78°C и обрабатывают этанолом (примерно 1 мл). Температуру смеси доводят до 0°C и перемешивают в течение 1 ч при 0°C. Реакционный раствор разбавляют этилацетатом (20 мл), обрабатывают 0,5 мл насыщенного раствора хлорида аммония и подвергают действию ультразвука в течение примерно 5 мин (до тех пор, пока не образуется достаточное количество осадка). Смесь сушат (Na2SO4) и фильтруют через рыхлый слой целита. Фильтрат концентрируют при пониженном давлении. Остаток кристаллизуют из дихлорметана и диэтилового эфира и получают указанное в заголовке соединение (47,4 мг, 58%).
1H ЯМР (400 МГц, CDCl3) δ: 2,16-2,27 (м, 4H), 3,14 (т, 2H, J=6,1 Гц), 4,39 (т, 2H, J=5,7 Гц), 7,53 (с, 1H), 10,01 (с, 1H).
Стадия 4. 4-Нитробензиловый эфир (5R,6RS)-6-{(RS)-ацетокси[4,5,6,7-тетрагидро-1,3a,3b,8-тетраазациклопента[a]инден-2-ил]метил}-6-бром-7-оксо-4-тиа-1-азабицикло[3.2.0]гепт-2-ен-2-карбоновой кислоты
4,5,6,7-Тетрагидро-1,3a,3b,8-тетраазациклопента[a]инден-2-карбальдегид (2,97 г) добавляют к раствору безводного MgBr2 (4,45 г) в сухом ацетонитриле (110 мл) в атмосфере азота при комнатной температуре. К реакционной смеси добавляют раствор 4-нитробензилового эфира (5R,6S)-6-бром-7-оксо-4-тиа-1-азабицикло[3.2.0]гепт-2-ен-2-карбоновой кислоты (2,97 г) в сухом ТГФ (110 мл), охлаждают до -20°C и в один прием добавляют триэтиламин (6,45 мл). Реакционный сосуд покрывают фольгой, чтобы исключить воздействие света. Реакционную смесь перемешивают в течение 1,2 ч при -20°C и затем обрабатывают уксусным ангидридом (2,9 мл) в один прием. Реакционную смесь доводят до 0°C и перемешивают в течение 16,5 ч при 0°C. Смесь разбавляют этилацетатом и промывают Н2О и насыщенным солевым раствором. Органический слой сушат (MgSO4) и фильтруют через рыхлый слой целита. Рыхлый слой промывают этилацетатом. Фильтрат концентрируют при пониженном давлении. Остаток наносят на хроматографическую колонку с силикагелем и элюируют смесью этилацетат:н-гексан (3/1) и затем смесью этилацетат-метанол (5/1). Получают указанное в заголовке соединение в виде коричневого аморфного твердого вещества (651,6 мг, 13%).
1H ЯМР (400 МГц, CDCl3) δ: 2,10-2,24 (м, 4H), 2,29 (с, 3H), 3,04-3,07 (м, 2H), 4,28-4,32 (м, 2H), 5,27 (д, 1H, J=13,7 Гц), 5,43 (д, 1H, J=13,7 Гц), 6,19 (с, 1H), 6,91 (с, 1H), 7,01 (с, 1H), 7,49 (с, 1H), 7,59-7,62 (м, 2H), 8,23-8,25 (м, 2H).
Стадия 5. Натриевая соль (5R),(6Z)-6-(4,5,6,7-тетрагидро-1,3a,3b,8-тетраазациклопента[a]инден-2-илметилен)-7-оксо-4-тиа-1-азабицикло[3.2.0]гепт-2-ен-2-карбоновой кислоты
4-Нитробензиловый эфир (5R,6RS)-6-{(RS)-ацетокси[4,5,6,7-тетрагидро-1,3a,3b,8-тетраазациклопента[a]инден-2-ил]метил}-6-бром-7-оксо-4-тиа-1-азабицикло[3.2.0]гепт-2-ен-2-карбоновой кислоты (643,6 мг) растворяют в ТГФ (9 мл) и ацетонитриле (4,2 мл). Быстро добавляют свежеактивированную Zn пыль (2,57 г) и 0,5M фосфатный буфер (pH 6,4, 13,2 мл). Реакционный сосуд покрывают фольгой, чтобы исключить воздействие света. Реакционную смесь энергично перемешивают в течение 2 ч при комнатной температуре. Реакционную смесь охлаждают до 9°C и добавляют 0,1M NaOH, чтобы довести рН до 7,5. Реакционный раствор смешивают с этилацетатом и фильтруют через рыхлый слой целита. Рыхлый слой промывают водой. Водный слой концентрируют до объема 20 мл в высоком вакууме при 35°С. Концентрат наносят на хроматографическую колонку со смолой Diaion HP-21 (60 мл, Mitsubishi Kasei Co. Ltd.). После адсорбции осуществляют элюирование водой и затем 2,5-10% раствором ацетонитрила в воде. Объединенные фракции концентрируют в высоком вакууме при 35°С, лиофилизуют и получают указанное в заголовке соединение в виде желтого аморфного твердого вещества (68 мг, 18%, pH 7,4). Т.пл. 175°C (с разложением).
1H ЯМР (400 МГц, D2О) δ: 1,85-2,03 (м, 4H), 2,85-2,99 (м, 2H), 4,07-4,14 (м, 2H), 6,34 (с, 1H), 6,74 (с, 1H), 6,76 (с, 1H), 7,28 (с, 1H).
Пример 39
Получение (5R,6E)-6-[(10-бензил-11-оксо-10,11-дигидродибензо[b,f][1,4]оксазепин-8-ил)метилен]-7-оксо-4-тиа-1-азабицикло[3.2.0]гепт-2-ен-2-карбоновой кислоты
Стадия 1. Получение 8-(гидроксиметил)дибензо[b,f][1,4]оксазепин-11(10H)-она
Литийалюминийгидрид (11 мл, 11 ммоль) медленно добавляют к раствору метилового эфира 11-оксо-10,11-дигидродибензо[b,f][1,4]оксазепин-8-карбоновой кислоты (1,346 г, 5 ммоль) в ТГФ в атмосфере N2 при комнатной температуре. Реакционную смесь перемешивают в течение 1 ч 45 мин, затем гасят 2 н. раствором HCl до тех пор, пока рН раствора не станет 2-3. Удаляют весь ТГФ на роторном испарителе и пять раз экстрагируют реакционную смесь этилацетатом, органический слой сушат сульфатом натрия, фильтруют и концентрируют. Получают целевое соединение в виде твердого белого вещества с выходом 46%.
Стадия 2. Получение 11-оксо-10,11-дигидродибензо[b,f][1,4]оксазепин-8-карбальдегида
8-(Гидроксиметил)дибензо[b,f][1,4]оксазепин-11(10H)-он (0,241 г, 1 ммоль) в ацетонитриле добавляют к молекулярным ситам (1 г) в атмосфере азота при комнатной температуре, затем к смеси добавляют N-оксид 4-метилморфолина (0,175 г, 1,5 ммоль). После перемешивания полученной смеси в течение 10 мин добавляют к ней перрутенат тетрапропиламмония (0,0176 г, 0,05 ммоль) и доводят реакцию до завершения (ТСХ-контроль). Реакционную смесь разбавляют этилацетатом (10 мл) и быстро пропускают через небольшую колонку с силикагелем. Собирают все этилацетатные фракции, которые содержат целевой продукт, экстрагируют 1 н. раствором HCl и промывают насыщенным солевым раствором. Органический слой сушат над сульфатом натрия, фильтруют и концентрируют. Получают целевой продукт (белое твердое вещество) с выходом 83%.
Стадия 3. Получение 10-бензил-11-оксо-10,11-дигидродибензо[b,f][1,4]оксазепин-8-карбальдегида
Безводный карбонат калия (0,207 г, 1,5 ммоль) и бензилбромид (0,205 г, 1,2 ммоль) добавляют к раствору 11-оксо-10,11-дигидродибензо[b,f][1,4]оксазепин-8-карбальдегида (0,240 г, 1 ммоль) в ацетонитриле в атмосфере азота при комнатной температуре. Затем реакционную смесь кипятят с обратным холодильником в течение 4 ч, охлаждают до комнатной температуры, разбавляют этилацетатом, фильтруют через слой magnesol и концентрируют. Очищают на колонке с силикагелем при элюировании 50% этилацетатом в гексане. Получают целевое соединение (в виде светло-желтого масла) с выходом 63%.
Стадия 4. Получение 4-нитробензилового эфира 6-[ацетокси(10-бензил-11-оксо-10,11-дигидродибензо[b,f][1,4]оксазепин-8-ил)метил]-6-бром-7-оксо-4-тиа-1-азабицикло[3.2.0]гепт-2-ен-2-карбоновой кислоты
Раствор 10-бензил-11-оксо-10,11-дигидродибензо[b,f][1,4]оксазепин-8-карбальдегида (0,250 г, 0,759 ммоль) в ацетонитриле добавляют к бромиду магния (0,419 г, 2,28 ммоль) в атмосфере N2 при комнатной температуре. К полученной смеси затем добавляют раствор 4-нитробензилового эфира (5R,6S)-6-бром-7-оксо-4-тиа-1-азабицикло[3.2.0]гепт-2-ен-2-карбоновой кислоты (0,292 г, 0,758 ммоль) в сухом ТГФ. Через 15 мин реакционную смесь охлаждают до -20°C и добавляют триэтиламин (0,317 мл, 2,27 ммоль). Реакционную колбу покрывают фольгой, чтобы исключить воздействие света. После 4 ч при -20°C реакционную смесь обрабатывают уксусным ангидридом (0,358 мл, 3,795 ммоль) и DMAP (0,00927 г, 0,0759 ммоль), температуру реакционной смеси доводят до 0°С и на ночь помещают в холодильник. Реакционный раствор концентрируют, концентрат растворяют в этилацетате и промывают 5% водным раствором лимонной кислоты, насыщенным раствором NaHCO3, водой и насыщенным солевым раствором. Органический слой сушат сульфатом натрия, фильтруют и концентрируют. Очищают на колонке с силикагелем, элюируют смесью этилацетат/CH2Cl2 (1:15). Получают целевое соединение (светло-желтое масло) с выходом 41%.
Стадия 5. Получение натриевой соли 6-(10-бензил-11-оксо-10,11-дигидродибензо[b,f][1,4]оксазепин-8-илметилен)-7-оксо-4-тиа-1-азабицикло[3.2.0]гепт-2-ен-2-карбоновой кислоты
К раствору 4-нитробензилового эфира 6-[ацетокси(10-бензил-11-оксо-10,11-дигидродибензо[b,f][1,4]оксазепин-8-ил)метил]-6-бром-7-оксо-4-тиа-1-азабицикло[3.2.0]гепт-2-ен-2-карбоновой кислоты (0,210 г, 0,273 ммоль) в ТГФ добавляют 0,5М раствор фосфатного буфера (pH 6,5), затем добавляют 10% Pd-C (0,0546 г). Реакционную смесь гидрируют при давлении водорода 40 фунт/кв.дюйм в течение трех часов, фильтруют через рыхлый слой целита и удаляют ТГФ на роторном испарителе. Затем оставшуюся смесь экстрагируют этилацетатом и промывают водой и насыщенным солевым раствором. Органический слой сушат сульфатом натрия, фильтруют и концентрируют. Растворяют NaHCO3 в минимальном количестве дистиллированной воды и добавляют полученный раствор к реакционной смеси вместе с небольшим количеством этилацетата, до тех пор пока рН смеси не станет равным 7-8. Этилацетат отгоняют, остаток очищают на колонке с обращенной фазой (MCI Gel CHP20P), используя в качестве элюента различные растворы (0-20%) ацетонитрила в воде. Удаляют ацетонитрил и воду на роторном испарителе и полученное соединение сушат вымораживанием. Получают целевой продукт (желтое твердое вещество) с выходом 24%. Т.пл. 179°С.
1H ЯМР (ДМСО) δ: 1,755-1,825 (с, 1H), 2,497-2,506 (д, 2H), 5,243-5,434 (м, 2H), 6,516-6,770 (м, 1H), 7,039-7,792 (м, 11H).
Пример 40
Получение 6-(5-этокси-7,8-дигидро-6H-3,4,8b-триаза-асим-индацен-2-илметилен)-7-оксо-4-тиа-1-азабицикло[3.2.0]гепт-2-ен-2-карбоновой кислоты
Стадия 1. Получение 4-этокси-6,7-дигидро-5H-циклопентапиримидин-2-иламин
(SM: Ross, L.O.; Goodman, L.; Baker, B. R. J. Am. Chem. Soc. 1959, 81,3108)
4-Хлор-6,7-дигидро-5H-циклопентапиримидин-2-иламин (5,1 г) растворяют в 200 мл ксилола и 30 мл абсолютного этанола. Затем добавляют 6,8 г этоксида натрия и смесь кипятят с обратным холодильником в течение 3 ч. Затем растворитель удаляют в вакууме, к остатку добавляют 100 мл воды, фильтруют и промывают слой на фильтре водой (50 мл). Твердое вещество далее вакуумируют в течение нескольких часов для высушивания. Масса целевого продукта 5,3 г (выход 98%). Т.пл. 133,8-134,9°C.
1H ЯМР: (300 МГц, CDCl3) δ: 6,23 (с, NH2), 4,28 (квартет, 2H, J=6,9 Гц), 2,6 (м, 2H), 1,93 (м, 2H), 1,27 (т, CH3, J=6,9 Гц); MC: 180,0 (M+H).
Стадия 2. Получение этилового эфира 5-этокси-7,8-дигидро-6H-3,4,8b-триаза-асим-индацен-2-карбоновой кислоты
4-Этокси-6,7-дигидро-5H-циклопентапиримидин-2-иламин (5,2 г, 29 ммоль) растворяют в 100 мл сухого ТГФ. Затем по каплям добавляют этилбромпируват (5,4 мл) в течение 5 мин. Смесь перемешивают при 23°С в течение часа, затем фильтруют, промывают эфиром и получают 8,7 г твердого вещества. Данное твердое вещество затем растворяют в 50 мл этанола и кипятят с обратным холодильником в течение двух часов. Реакционную смесь охлаждают до комнатной температуры и распределяют между 350 мл хлороформа и 200 мл насыщенного раствора гидрокарбоната натрия. Органический слой отделяют и сушат над сульфатом магния. Отфильтровывают осушитель, фильтрат концентрируют и получают 6,5 г продукта. Т.пл. 168,6-168,7°C.
1H ЯМР (300 МГц, CDCl3) δ: 7,69 (с, 1H), 4,50 (квартет, 2H, J=7,2 Гц), 4,40 (квартет, 2H, J=7,2 Гц), 3,11 (т, 2H, J=9,6 Гц), 2,88 (т, 2H, J=9,6 Гц), 2,88 (м, 2H), 1,43 (т, 2H, J=7,2 Гц). MC: 276,2 (M+H).
Стадия 3. Получение 5-этокси-7,8-дигидро-6H-3,4,8b-триаза-асим-индацен-2-карбальдегида
Этиловый эфир 5-этокси-7,8-дигидро-6H-3,4,8b-триаза-асим-индацен-2-карбоновой кислоты (1,925 г) растворяют в 40 мл дихлорметана, охлаждают до -78°C. Затем добавляют DIBAL (1M, 21 мл, 3 экв.) в течение 5 мин. Реакционную смесь затем гасят 2 мл этанола и распределяют между 350 мл дихлорметана и 100 мл 1 н. раствора гидроксида натрия. Водный слой промывают еще 150 мл хлороформа и объединенный органический слой сушат над сульфатом магния, фильтруют, концентрируют и получают соответствующий спирт. Данный спирт растворяют в 150 мл дихлорметана и к раствору добавляют диоксид марганца (10 г). Смесь перемешивают при 23°C в течение 2 ч, затем фильтруют через рыхлый слой целита, концентрируют и получают целевой альдегид (1,1 г, 68%). Т.пл. 237,2-237,3°C.
1H ЯМР (300 МГц, CDCl3) δ: 9,94 (с, 1H, CHO), 8,39 (с, 1H), 4,46 (квартет, 2H, J=7,2 Гц), 3,2 (м, 2H, CH2), 2,85 (м, 2H, CH2), 2,24 (м, 2H, CH2), 1,38 (т, 3H, CH3, J=7,2 Гц). MC: 232,1 (M+H).
Стадия 4. Получение 4-нитробензилового эфира 6-[ацетокси(5-этокси-7,8-дигидро-6H-3,4,8b-триаза-асим-индацен-2-ил)метил]-6-бром-7-оксо-4-тиа-1-азабицикло[3.2.0]гепт-2-ен-2-карбоновой кислоты
Раствор 5-этокси-7,8-дигидро-6H-3,4,8b-триаза-асим-индацен-2-карбальдегида (693 мг, 3 ммоль) в ацетонитриле (30 мл) добавляют к эфирату бромида магния (1,03 г) и смесь перемешивают при 23°C в течение 0,5 ч. Затем за 1 мин вводят раствор 4-нитробензилового эфира 6-бром-7-оксо-4-тиа-1-азабицикло[3.2.0]гепт-2-ен-2-карбоновой кислоты (1,155 г, 1 экв.) в сухом ТГФ (30 мл) и затем реакционную смесь охлаждают до -20°C. Затем добавляют триэтиламин (0,7 мл) и реакционную смесь перемешивают в течение пяти часов при -20°C. Затем добавляют уксусный ангидрид (0,377 мл) и реакционную смесь выдерживают при 0°С в течение 18 ч. Затем реакционную смесь разбавляют 400 мл этилацетата и промывают 100 мл 5% лимонной кислоты, 100 мл насыщенного раствора гидрокарбоната натрия и 100 мл насыщенного раствора хлорида натрия. Органический слой сушат над сульфатом магния, фильтруют и концентрируют. Флэш-хроматографией на колонке с использованием 20% этилацетата в гексане в качестве элюента получают 1,1 г продукта. Т.пл. 118,7-119°С.
1H ЯМР (300 МГц, CDCl3) δ: 8,35 (д, 2H, J=11 Гц), 7,63 (м, 2H), 7,41 (д, 1H, J=6,9 Гц), 7,08 (д, 1H, J=11 Гц), 6,47 (с, 1H), 5,55 (4H, CH2), 4,54 (м, 2H), 3,09 (м, 2H), 2,93 (м, 2H), 2,32 (м, 2H), 1,41 (т, J=9,6 Гц). MC: 660,1 (M+H).
Стадия 5. Получение 6-(5-этокси-7,8-дигидро-6H-3,4,8b-триаза-асим-индацен-2-илметилен)-7-оксо-4-тиа-1-азабицикло[3.2.0]гепт-2-ен-2-карбоновой кислоты
4-Нитробензиловый эфир 6-[ацетокси(5-этокси-7,8-дигидро-6H-3,4,8b-триаза-асим-индацен-2-ил)метил]-6-бром-7-оксо-4-тиа-1-азабицикло[3.2.0]гепт-2-ен-2-карбоновой кислоты (1,03 г, 1,565 ммоль) суспендируют в 20 мл ТГФ и 20 мл водного фосфатного буфера (pH 6,5). Полученную смесь гидрируют в течение двух часов при давлении водорода 45 фунт/кв.дюйм, затем фильтруют через рыхлый слой целита и концентрируют в вакууме, удаляя большую часть ТГФ. Затем раствор охлаждают до 0°С, подщелачивают 1 н. раствором гидроксида натрия до рН 8 и очищают методом ВЭЖХ с обращенной фазой, используя сначала воду (1 л), а затем 5%-25% ацетонитрил в воде в качестве элюента. Воду затем удаляют в вакууме и получают 100 мг продукта. Т.пл. >250°C.
1H ЯМР (300 МГц, CDCl3) δ: 7,52 (с, 1H), 6,95 (с, 1H), 6,54 (с, 1H), 4,73 (м, 2H), 3,06 (м, 2H), 2,84 (м, 2H), 2,27 (м, 2H), 1,43 (т, 3H). MC: 383,2 (M+H).
Пример 41
Натриевая соль (5R,6E/Z)-7-оксо-6-(4H,10H-пиразоло[5,1-c][1,4]бензоксазепин-2-илметилен)-4-тиа-1-азабицикло[3.2.0]гепт-2-ен-2-карбоновой кислоты
Стадия 1. Получение диэтил 1-(2-фторбензил)-1H-пиразол-3,5-дикарбоксилата
2-Фторбензилбромид (2,0 мл, 16,58 ммоль) добавляют к смеси диэтил 3,5-пиразолдикарбоксилата (3,01 г, 14,18 ммоль), Cs2CO3 (5,57 г, 17,1 ммоль) и ацетонитрила (140 мл) в атмосфере азота, нагревают при 60°С в течение 2 ч и затем охлаждают до комнатной температуры. Реакционный раствор фильтруют и концентрируют. К остатку добавляют воду (примерно 200 мл) и экстрагируют EtOAc. Органический слой промывают водой и насыщенным солевым раствором, сушат над сульфатом натрия, фильтруют, концентрируют и получают диэтил 1-(2-фторбензил)-1H-пиразол-3,5-дикарбоксилат (светло-желтое масло) с количественным выходом.
Стадия 2. Получение 1-(2-фторбензил)-1H-пиразол-3,5-метандиола
К раствору диэтил 1-(2-фторбензил)-1H-пиразол-3,5-дикарбоксилата (4,80 г, 14,99 ммоль) в CH2Cl2 (90 мл) добавляют 1M раствор DIBAL-H в ТГФ (90 мл, 90 ммоль) при 0°C в атмосфере N2. Через 2 часа реакцию гасят водным раствором NH4Cl. Образовавшуюся суспензию фильтруют, фильтрат экстрагируют EtOAc и промывают насыщенным солевым раствором. Органический слой сушат над сульфатом натрия, фильтруют и концентрируют. Очищают на колонке с силикагелем, элюируя 5% MeOH в CH2Cl2, и получают 3,4 г указанного в заголовке диола (в виде прозрачного масла) с выходом 96%.
Стадия 3. Получение 4H,10H-пиразоло[5,1-c][1,4]бензоксазепин-2-карбальдегида
Вышеуказанный диол (3,83 г, 16,21 ммоль) в ГМФА (24 мл) добавляют к суспензии NaH (60%, 1,34 г, 33,5 ммоль) в толуоле (330 мл) в атмосфере N2, быстро нагревают до 95°С, выдерживают при этой температуре три часа и охлаждают до комнатной температуры, гасят водой и экстрагируют EtOAc. Органический слой промывают водой и насыщенным солевым раствором, сушат над сульфатом натрия, фильтруют и концентрируют. Остаток очищают на колонке с силикагелем, элюируя 2% MeOH в CH2Cl2, и получают 4H,10H-пиразоло[5,1-c][1,4]бензоксазепин-2-илметанол (белое твердое вещество). Выход 0,71 г (20%).
4H,10H-Пиразоло[5,1-c][1,4]бензоксазепин-2-илметанол (0,71 г, 3,28 ммоль), N-оксид 4-метилморфолина (1,198 г, 10,23 ммоль), молекулярные сита (порошок, 4А) (3,32 г) и ацетонитрил (0,07M) смешивают в атмосфере N2. Добавляют перрутенат тетрапропиламмония (0,113 г, 0,322 ммоль) и спустя три часа реакционный раствор фильтруют через целит и концентрируют. Остаток очищают на колонке с силикагелем, используя смесь EtOAc/гексан (1:1) в качестве элюента, и получают 4H,10H-пиразоло[5,1-c][1,4]бензоксазепин-2-карбальдегид (белое твердое вещество). Выход 0,31 г (44%).
Стадия 4. Получение 4-нитробензилового эфира 6-[ацетокси(4H,10H-пиразоло[5,1-c][1,4]бензоксазепин-8-ил)метил]-6-бром-7-оксо-4-тиа-1-азабицикло[3.2.0]гепт-2-ен-2-карбоновой кислоты
4H,10H-Пиразоло[5,1-c][1,4]бензоксазепин-2-карбальдегид (0,19 г, 0,887 ммоль) в ацетонитриле (14 мл) добавляют к MgBr2 (0,49 г, 2,66 ммоль) в атмосфере N2. Через 25 мин добавляют раствор 4-нитробензилового эфира 6-бром-7-оксо-4-тиа-1-азабицикло[3.2.0]гепт-2-ен-2-карбоновой кислоты (0,342 г, 0,888 ммоль) в ТГФ (14 мл). Через 15 мин реакционную смесь охлаждают до -20°С и 10 мин спустя добавляют Et3N (3 экв.) и помещают реакционную смесь в темноту. Через 6,5 ч добавляют уксусный ангидрид (0,42 мл, 4,45 ммоль) и DMAP (0,011 г, 0,09 ммоль). Реакционной смеси дают нагреться до 0°С и оставляют на ночь в холодильнике. Реакционный раствор концентрируют и получившийся остаток растворяют в EtOAc, промывают 5% водным раствором лимонной кислоты и насыщенным водным раствором NaHCO3; затем дополнительно промывают водой и насыщенным солевым раствором. Органический слой сушат над сульфатом натрия, фильтруют и концентрируют. Очищают на препаративных пластинках с силикагелем, используя смесь EtOAc/гексан (1:2) в качестве элюента. Получают продукт конденсации в виде желтого смолообразного твердого вещества. Выход 0,31 г (54%).
Стадия 5. Натриевая соль (5R,6E/Z)-7-оксо-6-(4H,10H-пиразоло[5,1-c][1,4]бензоксазепин-2-илметилен)-4-тиа-1-азабицикло[3.2.0]гепт-2-ен-2-карбоновой кислоты
К раствору продукта конденсации (5) (0,300 г, 0, 468 ммоль) в ТГФ (18 мл) добавляют 0,5M раствор фосфатного буфера (pH 6,5) (18 мл). Добавляют Pd на угле (0,102 г) и реакционную смесь гидрируют при давлении водорода 40 фунт/кв.дюйм в течение двух часов; фильтруют через целит и удаляют ТГФ на роторном испарителе. Остаток экстрагируют EtOAc. Органический слой сушат над сульфатом натрия, фильтруют и концентрируют. Растворяют NaHCO3 (0,08 г, 0,952 ммоль) в минимальном количестве воды и добавляют к концентрированному органическому слою вместе с небольшим количеством EtOAc; фильтруют и удаляют EtOAc на роторном испарителе. Очищают на колонке с обращенной фазой (MCI Gel CHP20P), варьируя количества ацетонитрила (от 0% до 15%) в воде. Из собранных фракций удаляют ацетонитрил и большую часть воды на роторном испарителе. Остаток сушат вымораживанием и получают 41 мг натриевой соли (5R,6E)-7-оксо-6-(4H,10H-пиразоло[5,1-c][1,4]бензоксазепин-2-илметилен)-4-тиа-1-азабицикло[3.2.0]гепт-2-ен-2-карбоновой кислоты (6) (в виде желтого твердого вещества) с выходом 22%. Методом ВЭЖХ найдено, что чистота продукта составляет 77%, а соотношение E/Z изомеров 3:2.
1H ЯМР (δ, ДМСО-d6): 5,366 (м, 4H), 5,649 (м, 4H), 6,326 (т, 2H), 6,444 (с, 2H), 6,551 (с, 2H), 6,640 (с, 2H), 6,810 (с, 2H), 6,974 (м, 2H), 7,249 (м, 2H), 7,355 (м, 2H). m/z: (M+H) 390,0.
Пример 42
Натриевая соль (5R),(6Z)-6-(5H-имидазо[2,1-a]изоиндол-2-илметилен)-7-оксо-4-тиа-1-азабицикло[3.2.0]гепт-2-ен-2-карбоновой кислоты
Стадия 1. Получение 5H-имидазо[2,1-a]изоиндол-2-карбальдегида
Раствор 2-бром-3-изопропоксипропеналя (4,97 г) в сухом ацетонитриле (3 мл) добавляют к смеси 3-амино-1H-изоиндола (3,4 г) в сухом ацетонитриле (100 мл). Реакционную смесь перемешивают в течение 3,25 ч при комнатной температуре. Затем к смеси добавляют триэтиламин (3,6 мл), нагревают и кипятят с обратным холодильником в течение 2 ч. Смесь охлаждают до комнатной температуры, разбавляют этилацетатом и промывают 20% раствором гидрокарбоната калия. После фильтрования через рыхлый слой целита органический слой сушат (MgSO4) и концентрируют при пониженном давлении. Остаток наносят на хроматографическую колонку с силикагелем, затем элюируют смесью этилацетат-гексан (3/1˜4/1). Неочищенное соединение кристаллизуют из этилацетата и н-гексана и получают указанное в заголовке соединение (1,04 г, 22%).
1H ЯМР (400 МГц, CDCl3) δ: 5,01 (с, 2H), 7,28-7,52 (м, 3H), 7,90 (с, 1H), 7,91-7,93 (м, 1H), 9,92 (с, 1H).
Стадия 2. Получение 4-нитробензилового эфира (5R,6RS)-6-[(RS)-ацетокси(5H-имидазо[2,1-a]изоиндол-2-ил)метил]-6-бром-7-оксо-4-тиа-1-азабицикло[3.2.0]гепт-2-ен-2-карбоновой кислоты
5H-Имидазо[2,1-a]изоиндол-2-карбальдегид (736,8 мг) добавляют к раствору безводного MgBr2 (1,8 г) в сухом ацетонитриле (50 мл) в атмосфере азота при комнатной температуре. К реакционной смеси добавляют раствор 4-нитробензилового эфира (5R,6S)-6-бром-7-оксо-4-тиа-1-азабицикло[3.2.0]гепт-2-ен-2-карбоновой кислоты (1,55 г) в сухом ТГФ (50 мл), охлаждают до -20°С и добавляют триэтиламин (1,34 мл) в один прием. Реакционный сосуд покрывают фольгой, чтобы исключить воздействие света. Реакционную смесь перемешивают в течение 2 ч при -20°С и обрабатывают уксусным ангидридом (0,76 мл) в один прием. Температуру реакционной смеси доводят до 0°С и перемешивают ее в течение 18 ч при 0°C. Смесь разбавляют этилацетатом и промывают H2O, насыщенным раствором гидрокарбоната натрия и насыщенным солевым раствором. Органический слой сушат (MgSO4) и фильтруют через рыхлый слой целита, который затем промывают этилацетатом. Фильтрат концентрируют при пониженном давлении. Остаток наносят на хроматографическую колонку с силикагелем, затем элюируют смесью этилацетат-гексан (2/3˜1/1). Указанное в заголовке соединение получают в виде смеси двух диастереомеров (5/1, бледно-желтое аморфное твердое вещество, 1,8 г, 73%).
1Н ЯМР (400 МГц, CDCl3) δ: 2,02 (с, 0,84 × 3H), 2,27 (с, 0,16 × 3H), 4,89-4,94 (м, 2H), 5,29 (д, 1H, J=13,6 Гц), 5,47 (д, 1H, J=13,6 Гц), 6,18 (с, 0,16 × 1H), 6,40 (с, 0,84 × 1H), 6,42 (с, 0,84 × 1H), 6,94 (д, 0,16 × 1 H, J=0, 9 Гц), 7,18 (д, 0,16 × 1H, J=0,7 Гц), 7,35-7,48 (м, 3H), 7,51 (с, 0,84 × 1H), 7,60-7,64 (м, 2H), 7,79-7,83 (м, 1H), 8,23-8,27 (м, 2H).
Стадия 3. Натриевая соль (5R),(6Z)-6-(5H-имидазо[2,1-a]изоиндол-2-илметилен)-7-оксо-4-тиа-1-азабицикло[3.2.0] гепт-2-ен-2-карбоновой кислоты
4-Нитробензиловый эфир (5R,6RS)-6-[(RS)-ацетокси(5H-имидазо[2,1-a]изоиндол-2-ил)метил]-6-бром-7-оксо-4-тиа-1-азабицикло[3.2.0]гепт-2-ен-2-карбоновой кислоты (1,5 г) растворяют в ТГФ (21 мл) и ацетонитриле (9,8 мл). К реакционной смеси добавляют свежеактивированную Zn пыль (6 г) и 0,5M фосфатный буфер (pH 6,4, 30,8 мл). Реакционный сосуд покрывают фольгой, чтобы исключить воздействие света. Смесь энергично перемешивают в течение 2 ч при комнатной температуре, затем охлаждают до 9°C и добавляют 1M водный раствор NaOH для доведения pH до 7,5. Реакционный раствор смешивают с этилацетатом и фильтруют через рыхлый слой целита, который затем промывают водой, водный слой отделяют и концентрируют до 25 мл в высоком вакууме при 35°C. Концентрат наносят на хроматографическую колонку со смолой Diaion HP-21 (100 мл, Mitsubishi Kasei Co. Ltd.). После адсорбции элюируют сначала водой, затем смесью 5-15% ацетонитрил-вода. Объединенные фракции концентрируют в высоком вакууме при 35°C, лиофилизуют и получают указанное в заголовке соединение в виде желтого аморфного твердого вещества (527 мг, 58%). Т.пл. 170°C (с разложением).
1H ЯМР (400 МГц, D2O) δ: 4,62 (с, 2H), 6,27 (с, 1H), 6,56 (с, 1H), 6,78 (с, 1H), 7,22-7,31 (м, 4H), 7,52 (д, 1H, J=6,7 Гц).
| название | год | авторы | номер документа |
|---|---|---|---|
| БИЦИКЛИЧЕСКИЕ 6-АЛКИЛИДЕНПЕНЕМЫ В КАЧЕСТВЕ ИНГИБИТОРОВ β-ЛАКТАМАЗ | 2003 |
|
RU2339640C2 |
| ПРОИЗВОДНЫЕ 3β-АЛКЕНИЛПЕНАМА И ИХ ФАРМАЦЕВТИЧЕСКИ СОВМЕСТИМЫЕ СОЛИ | 1994 |
|
RU2139290C1 |
| 2-ТИОЗАМЕЩЕННЫЕ КАРБАПЕНЕМЫ, ПРОМЕЖУТОЧНЫЕ СОЕДИНЕНИЯ ДЛЯ ПОЛУЧЕНИЯ, СПОСОБЫ ПОЛУЧЕНИЯ (ВАРИАНТЫ), ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ | 1994 |
|
RU2130457C1 |
| КОНДЕНСИРОВАННОЕ ЗАМЕЩЕННОЕ ПРОИЗВОДНОЕ АМИНОПИРРОЛИДИНА | 2007 |
|
RU2443698C2 |
| ЗАМЕЩЕННЫЕ 3-АМИНОХИНУКЛИДИНЫ | 1992 |
|
RU2092486C1 |
| ИНГИБИТОРЫ СЕТР | 2006 |
|
RU2513107C2 |
| КАРБАПЕНЕМОВЫЕ СОЕДИНЕНИЯ | 2017 |
|
RU2772909C2 |
| β-ЛАКТАМЫ | 1995 |
|
RU2143435C1 |
| БИЦИКЛИЧЕСКОЕ ПРОИЗВОДНОЕ ГАММА-АМИНОКИСЛОТЫ | 2008 |
|
RU2446148C2 |
| СПОСОБ ПОЛУЧЕНИЯ ХИНОЛИНИЛЛАКТАМОВ ИЛИ ИХ СОЛЕЙ | 1992 |
|
RU2130938C1 |
Изобретение относится к способу получения соединений формулы I, где один из заместителей А или В означает водород, а другой обозначает арил, необязательно замещенный одним или двумя R2, гетероарил, необязательно замещенный одним или двумя R2, и т.д., R5 представляет собой Н, С1-С6-алкил, С5-С6-циклоалкил или CHR3ОСО-С1-С6-алкил или соль; R2 представляет собой водород, необязательно замещенный C1-С6-алкил, необязательно замещенный С2-С6-алкенил, и т.д.; R3 представляет собой водород, C1-С6-алкил, С5-С6-циклоалкил, необязательно замещенный арил, необязательно замещенный гетероарил; включающему: (а) конденсацию замещенного альдегида (A'-CHO) (17), где А' представляет собой А, такой как определено выше, когда В представляет собой водород; или В, такой как определено выше, когда А представляет собой водород, с производным 6-бромпенема структуры 16, где R представляет собой п-нитробензил, в присутствии кислоты Льюиса и мягкого основания при низкой температуре, которая приводит к образованию промежуточного альдольного продукта 18; (b) взаимодействие промежуточного соединения 18 с хлорангидридом или ангидридом кислоты формулы (R8)Cl или (R8)2O или с тетрагалогенметаном формулы C(X1)4 и трифенилфосфином, где R8 представляет собой алкил-SO2, арил-SO2, алкил-СО или арил-СО; X1 представляет собой Br, I или Cl; с образованием промежуточного соединения 19, где R9 представляет собой X1 или OR8; и (с) превращение промежуточного соединения 19 в целевое соединение формулы I. Изобретение также относится к 4-нитробензил (5R,6S)-6-бромпенем-3-карбоксилата 16 и способу его получения, который включает следующие стадии: (A) (i) взаимодействие 6-аминопенициллановой кислоты с бромистоводородной кислотой в органическом растворителе и воде с образованием 6-бромпенициллановой кислоты 21 и ее превращение в п-нитробензил 6-бромпеницилланат 22, где R представляет собой п-нитробензил, действием 4-нитробензилбромида в присутствии основания в органическом растворителе; (В) окисление 22 с образованием п-нитробензил 6-бромпеницилланат-1-оксида 23; (С) кипячение 23 с 2-меркаптобензотиазолом в ароматическом растворителе с образованием 4-нитробензил (2R)-2-[(3S,4R)-4-(бензотиазол-2-илдитио)-3-бром-2-оксоазетидин-1-ил]-3-метилбут-3-еноата 24; (D) растворение 24 в органическом растворителе и взаимодействие с органическим третичным основанием с образованием 4-нитробензил-2-[(3S,4R)-4-(бензотиазол-2-илдитио)-3-бром-2-оксоазетидин-1-ил]-3-метилбут-2-еноата 25; (Е) превращение 25 в 4-нитробензил 2-[(3S,4R)-3-бром-4-формилтио-2-оксоазетидин-1-ил]-3-метилбут-2-еноата 26 в результате взаимодействия в ароматическом органическом растворителе с органической кислотой в присутствии смеси уксусный ангидрид/органическое третичное основание и триалкил- или триарилфосфина в интервале температур от около -10 до около -30°С; (F) пропускание озонированного кислорода через раствор 26 в органическом растворителе в течение 3-4 ч при температуре от -70°С до -90°С и последующую внутримолекулярную циклизацию с использованием фосфитного реагента. Технический результат - эффективный по выходу и экономичности способ получения производных 6-алкилиденпенема. 3 н. и 38 з.п. ф-лы, 2 табл.


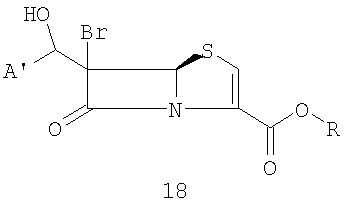

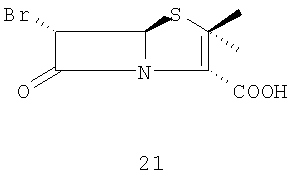
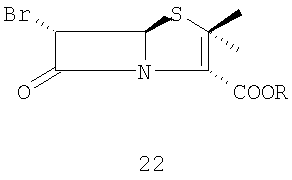

где один из заместителей А или В означает водород, а другой обозначает арил, необязательно замещенный одним или двумя R2, гетероарил, необязательно замещенный одним или двумя R2, конденсированный бициклический гетероарил, необязательно замещенный одним или двумя R2, конденсированный трициклический гетероарил, необязательно замещенный одним или двумя R2, циклоалкил, необязательно замещенный одним или двумя R2, алкил, необязательно замещенный одним или двумя R2, алкенил, необязательно замещенный одним или двумя R2, алкинил, необязательно замещенный одним или двумя R2, насыщенную или частично насыщенную гетероциклическую группу, необязательно замещенную одним или двумя R2; и где любой из указанных гетероарильных остатков, содержащий NH-группу в цикле, может необязательно быть замещен R1 при указанном атоме азота; R5 представляет собой Н, C1-С6-алкил, С5-С6-циклоалкил или CHR3OCO-C1-С6-алкил или соль;
R1 представляет собой Н, необязательно замещенный алкил, необязательно замещенный арил, необязательно замещенные гетероарил или моно- или бициклические насыщенные гетероциклические группы, необязательно замещенный циклоалкил, необязательно замещенный алкенил, необязательно замещенный алкинил при условии, что атом углерода, который непосредственно связан с N, не должен входить в состав двойной или тройной связи; необязательно замещенный перфторалкил, необязательно -S(O)p-замещенный алкил или арил, где р равно 0-2, необязательно замещенный -С=O-гетероарил, необязательно замещенный -С=O-арил, необязательно замещенный -С=O-алкил, необязательно замещенный -С=O-циклоалкил, необязательно замещенную моно- или бициклическую насыщенную -С=O-гетероциклическую группу, необязательно замещенный С1-С6-алкиларил, необязательно замещенный C1-С6-алкилгетероарил, необязательно замещенный арил-С1-С6-алкил, необязательно замещенный гетероарил-С1-С6-алкил, необязательно замещенную моно- или бициклическую насыщенную C1-С6-алкилгетероциклическую группу, необязательно замещенный арилалкенил, содержащий 8-16 атомов углерода, -COMR6R7, -SO2NR6R7, необязательно замещенный арилалкилоксиалкил, необязательно замещенный алкил-О-алкиларил, необязательно замещенный алкил-О-алкилгетероарил, необязательно замещенный арилоксиалкил, необязательно замещенный гетероарилоксиалкил, необязательно замещенный арилоксиарил, необязательно замещенный арилоксигетероарил, необязательно замещенный C1-С6-алкиларилоксиарил, необязательно замещенный C1-С6-алкиларилоксигетероарил, необязательно замещенные алкиларилоксиалкиламиносодержащие группы, необязательно замещенный алкоксикарбонил, необязательно замещенный арилоксикарбонил или необязательно замещенный гетероарилоксикарбонил;
R2 представляет собой водород, необязательно замещенный C1-С6-алкил, необязательно замещенный С2-С6-алкенил, необязательно замещенный С2-С6-алкинил, галоген, циано, N-R6R7, необязательно замещенный С1-С6-алкокси, гидрокси; необязательно замещенный арил, необязательно замещенный гетероарил, COOR6, необязательно замещенные алкиларилоксиалкиламиносодержащие группы, необязательно замещенный арилокси, необязательно замещенный гетероарилокси, необязательно замещенный С3-С6-алкенилокси, необязательно замещенный С3-С6-алкинилокси, С1-С6-алкиламино-С1-С6-алкокси, алкилендиокси, необязательно замещенную арилокси-С1-С6-алкиламиносодержашую группу, С1-С6-перфторалкил, необязательно S(O)q-замещенный C1-С6-алкил, необязательно S(O)q-замещенный арил, где q равно 0, 1 или 2, CONR6R7, гуанидино- или включенную в цикл гуанидиногруппу, необязательно замещенный алкиларил, необязательно замещенный арилалкил, необязательно замещенный C1-С6-алкилгетероарил, необязательно замещенный гетероарил-С1-С6-алкил, необязательно замещенную моно- или бициклическую насыщенную C1-С6-алкилгетероциклическую группу, необязательно замещенный арилалкенил, содержащий от 8 до 16 атомов углерода, SO2NP6R7, необязательно замещенный арилалкилоксиалкил, необязательно замещенный арилоксиалкил, необязательно замещенный гетероарилоксиалкил, необязательно замещенный арилоксиарил, необязательно замещенный арилоксигетероарил, необязательно замещенный гетероарилоксиарил, необязательно замещенный C1-С6-алкиларилоксиарил или необязательно замещенный C1-С6-алкиларилоксигетероарил;
R3 представляет собой водород, C1-С6-алкил, С5-С6-циклоалкил, необязательно замещенный арил, необязательно замещенный гетероарил;
R6 и R7 независимо представляют собой Н, необязательно замещенный C1-С6-алкил, необязательно замещенный арил, необязательно замещенный гетероарил, необязательно замещенный С1-С5-алкиларил, необязательно замещенный арилалкил, необязательно замещенный гетероарилалкил, необязательно замещенный C1-С6-алкилгетероарил, R6 и R7 вместе могут образовывать 3-7-членную насыщенную циклическую систему, необязательно содержащую один или два гетероатома, таких, как N-R1, О, S=(O)n, где n=0-2;
указанный способ включает
(а) конденсацию соответствующим образом замещенного альдегида 17
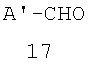
где А' представляет собой А такой, как определено выше, когда В представляет собой водород; или В такой, как определено выше, когда А представляет собой водород,
с производным 6-бромпенема структуры 16
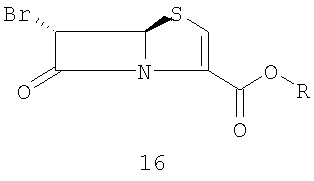
где R представляет собой п-нитробензил,
в присутствии кислоты Льюиса и мягкого основания при низкой температуре, которая приводит к образованию промежуточного альдольного продукта 18
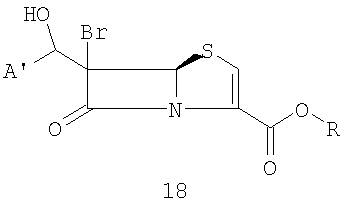
где А' и R такие, как определено выше,
(b) взаимодействие промежуточного соединения 18 с хлорангидридом или ангидридом кислоты формулы (R8)Cl или (R8)2O или с тетрагалогенметаном формулы C(X1)4 и трифенилфосфином, где R8 представляет собой алкил-SO2, арил-SO2, алкил-СО или арил-СО; Х1 представляет собой Br, I или Cl,
с образованием промежуточного соединения 19

где R9 представляет собой X1 или OR8, где R8, X1, А' и R такие, как определено выше; и
(с) превращение промежуточного соединения 19 в целевое соединение формулы I, где R5 представляет собой водород, посредством восстановительного элиминирования; и, если требуется, превращение в фармацевтически приемлемую соль или в сложный эфир, где R5 представляет собой C1-С6-алкил, С5-С6-циклоалкил или CHR3ОСО-С1-С6-алкил.

где Z1, Z2 и Z3 независимо представляют собой CR2, N, О, S или N-R1 при условии, что один из Z1, Z2 или Z3 является атомом углерода и связан с остальной молекулой так, как показано в формуле I;
W1, W2 и W3 независимо представляют собой CR4R4, S, SO, SO2, О, N-R1, С=O при условии, что не происходит образования ни S-S, ни O-O, ни S-O связи с образованием насыщенной кольцевой системы;
t равно 1-4;
R1, R2, R6 и R7 такие, как определено в п.1; и
R4 представляет собой Н, необязательно замещенный C1-С6-алкил, один из R4 представляет собой ОН, C1-С6-алкокси, -S-C1-С6-алкил, -COOR6, -NR6R7, -CONR6 R7; или R4R4 вместе могут представлять собой =O, или R4R4 вместе с атомом углерода, к которому они присоединены, могут образовывать 5-8-членную спиросистему, содержащую или не содержащую гетероатомы, выбранные из N, О, S=(O)n (где n равно 0-2), N-R1.

где Z1, Z2 и Z3 независимо представляют собой CR2, N, О, S или N-R1 при условии, что один из Z1-Z3 является атомом углерода и связан с остальной частью молекулы;
W1, W2 и W3 независимо представляют собой CR4R4, S, SO, SO2, О или N-R1;
t равно 1-4;
Y1 и Y2 независимо представляют собой N или С; при этом, если ароматическим кольцом бициклической гетероарильной группы является имидазол, неароматический цикл не может содержать атом серы в положении, примыкающем к атому углерода в голове мостика;
R1 и R2 такие, как определено в п.1;
R4 представляет собой Н, необязательно замещенный C1-С6-алкил, один из R4 представляет собой ОН, C1-С6-алкокси, -S-C1-С6-алкил, -COOR6, -NR6R7, -CONR6R7; или R4R4 вместе могут представлять собой =O, или R4R4 вместе с атомом углерода, к которому они присоединены, могут образовывать 5-8-членную спиросистему, содержащую или не содержащую гетероатомы, выбранные из N, О, S=(O)n (где n равно 0-2) и N-R1; и R6 и R7 независимо представляют собой Н, необязательно замещенный С1-С6-алкил, необязательно замещенный арил, необязательно замещенный гетероарил, необязательно замещенный C1-С6-алкиларил, необязательно замещенный арилалкил, необязательно замещенный гетероарилалкил, необязательно замещенный C1-С6-алкилгетероарил; или R6 и R7 могут вместе образовывать 3-7-членную насыщенную циклическую систему, необязательно содержащую один или два гетероатома, которые выбраны из N, О или S.

где Z1, Z2, Z3 и Z4 независимо представляют собой CR2 или N при условии, что один из Z1-Z4 является атомом углерода и связан с остальной частью молекулы;
W1, W2 и W3 независимо представляют собой CR4R4, S, SO, SO2, О или N-R1 при условии, что не происходит образования ни S-S, ни O-O, ни S-O связи с образованием насыщенной кольцевой системы;
t равно 1-4;
Y1 и Y2 независимо представляют собой С или N;
R1, R2, R4, R6 и R7 такие, как определено выше в п.13.
 или
или 
где Z1, Z2, Z3, Z4, Z5, Z6 и Z7 независимо представляют собой CR2, N, О, S или n-R1 при условии, что один из Z1-Z7 является атомом углерода, к которому присоединена остальная часть молекулы;
R1, R2, R6 и R7 такие, как определено в п.1;
Y1, Y2, Y3 и Y4 независимо могут быть С или N.
 или
или 
где Z1, Z2, Z3, Z4, Z5, Z6, Z7 и Z8 независимо представляют собой CR2, N, O, S или N-R1 при условии, что один из Z1-Z8 является атомом углерода, к которому присоединена остальная часть молекулы;
R1, R2, R6 и R7 такие, как определено в п.1;
и Y1, Y2, Y3 и Y4 независимо могут быть С или N.
 или
или 
где Z1, Z2, Z3, Z4, Z5, Z6, Z7 и Z8 независимо представляют собой CR2, N, O, S или N-R1 при условии, что один из Z1-Z8 является атомом углерода, к которому присоединена остальная часть молекулы;
R1, R2, R6 и R7 такие, как определено в п.1;
и Y1, Y2, Y3 и Y4 могут быть С или N.
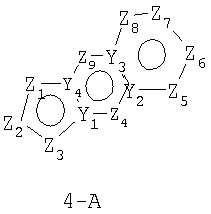


где Z1, Z2, Z3, Z4, Z5, Z6, Z7, Z8 и Z9 независимо представляют собой CR2, N, O, S или N-R1 при условии, что один из Z1-Z9 является атомом углерода, к которому присоединена остальная часть молекулы;
R1, R2, R6 и R7 такие, как определено в п.1; и
Y1, Y2, Y3 и Y4 независимо представляют собой С или N.
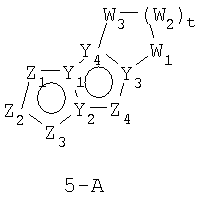 или
или 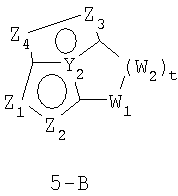
где Z1, Z2, Z3 и Z4 независимо представляют собой CR2, N, O, S или N-R1 при условии, что один из Z1-Z4 является атомом углерода, к которому присоединена остальная часть молекулы;
Y1, Y2, Y3 и Y4 независимо представляют собой С или N.
W1, W2 и W3 независимо представляют собой CR4R4, S(O)r (r равно 0-2), О или N-R1 при условии, что не происходит образования ни S-S, ни O-O, ни S-O связи с образованием насыщенной кольцевой системы;
R1, R2, R6 и R7 такие, как определено в п.1;
R4 представляет собой Н, необязательно замещенный C1-С6-алкил, ОН (при условии, что оба R4 не являются ОН), C1-С6-алкокси, -S-C1-С6-алкил, -COOR6, -NR6R7, -CONR6R7; или R4R4 могут вместе представлять собой =O, или R4R4 вместе с атомом углерода, к которому они присоединены, могут образовывать 5-8-членную спиросистему, содержащую или не содержащую гетероатомы, выбранные из N, О, S(O)n (где п равно 0-2), N-R1;
и t равно 1-3.
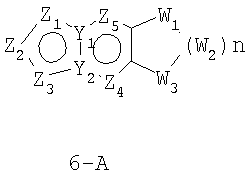
 или
или 
где Z1, Z2, Z3, Z4 и Z5 независимо представляют собой CR2, N, O, S или N-R1 при условии что один из Z1-Z5 является атомом углерода, к которому присоединена остальная часть молекулы;
Y1 и Y2 независимо представляют собой С или N;
W1, W2 и W3 независимо представляют собой CR4R4, S(O)r (r равно 0-2), О или N-R1 при условии, что не происходит образования ни S-S, ни O-O, ни S-O связи с образованием насыщенной кольцевой системы;
R1, R2, R6 и R7 такие, как определено в п.1;
R4 представляет собой Н, необязательно замещенный C1-С6-алкил, ОН (при условии, что оба R4 не являются ОН), C1-С6-алкокси, -S-C1-С6-алкил, -COOR6, -NR6R7, -CONR6R7; или R4R4 могут вместе представлять собой =O, или R4R4 вместе с атомом углерода, к которому они присоединены, могут образовывать 5-8-членную спиросистему, содержащую или не содержащую гетероатомы, выбранные из N, О, S(O)n (где n равно 0-2), N-R1;
и t равно 1-3.
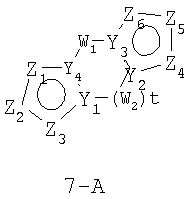 или
или 
где Z1, Z2, Z3, Z4, Z5 и Z6 независимо представляют собой CR2, N, O, S и N-R1 при условии, что один из Z1-Z6 является атомом углерода, к которому присоединена остальная часть молекулы;
Y1, Y2, Y3 и Y4 независимо представляют собой С или N;
W1 и W2 независимо представляют собой CR4R4, S(O)r (r равно 0-2), О, N-R1 при условии, что не происходит образования ни S-S, ни O-O, ни S-O связи с образованием насыщенной кольцевой системы;
R1, R2, R6 и R7 такие, как определено в п.1;
R4 представляет собой Н, необязательно замещенный C1-С6-алкил, ОН (при условии, что оба R4 не являются ОН), C1-С6-алкокси, -S-C1-С6-алкил, -COOR6, -NR6R7, -CONR6R7; или R4R4 могут вместе представлять собой =O, или R4R4 вместе с атомом углерода, к которому они присоединены, могут образовывать 5-8-членную спиросистему, содержащую или не содержащую гетероатомы, выбранные из N, О, S(O)n (где n равно 0-2), N-R1;
и t равно 1-3.
 или
или 
где Z1, Z2, Z3, Z4, Z5, Z6 и Z7 независимо представляют собой CR2, N, O, S или N-R1 при условии, что один из Z1-Z7 является атомом углерода, к которому присоединена остальная часть молекулы;
Y1, Y2, Y3 и Y4 независимо представляют собой С или N;
W1 и W2 независимо представляют собой CR4R4, S(O)r (r равно 0-2), О, N-R1 при условии, что не происходит образования ни S-S, ни O-O, ни S-O связи с образованием насыщенной кольцевой системы;
R1, R2, R6 и R7 такие, как определено в п.1;
R4 представляет собой Н, необязательно замещенный C1-С6-алкил, ОН (при условии, что оба R4 не являются ОН), C1-С6-алкокси, -S-C1-С6-алкил, -COOR6, -NR6R7, -CONR6R7; или R4R4 могут вместе представлять собой =O, или R4R4 вместе с атомом углерода, к которому они присоединены, могут образовывать 5-8-членную спиросистему, содержащую или не содержащую гетероатомы, выбранные из N, О, S(O)n (где n равно 0-2), N-R1;
и t равно 0-3.
 или
или 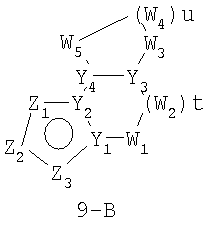
где Z1, Z2, и Z3 независимо представляют собой CR2, N, O, S или N-R1 при условии, что один из Z1-Z3 является атомом углерода, к которому присоединена остальная часть молекулы;
Y1 и Y4 независимо представляют собой С или N;
Y2 и Y3 независимо представляют собой CH или N;
W1, W2, W3, W4 и W5 независимо представляют собой CR4R4, S(O)r (r равно 0-2), О, N-R1 при условии, что не происходит образования ни S-S, ни O-O, ни S-O связи с образованием насыщенной кольцевой системы;
R1, R2, R6 и R7 такие, как определено в п.1;
R4 представляет собой Н, необязательно замещенный C1-С6-алкил, ОН (при условии, что оба R4 не являются ОН), C1-С6-алкокси, -S-C1-С6-алкил, -COOR6, -NR6R7, -CONR6R7; или R4R4 могут вместе представлять собой =O, или R4R4 вместе с атомом углерода, к которому они присоединены, могут образовывать 5-8-членную спиросистему, содержащую или не содержащую гетероатомы, выбранные из N, О, S(O)n (где n равно 0-2), N-R1;
t равно 0-2 и
u равно 1-3.
 или
или 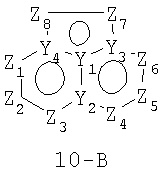
где Z1, Z2, Z3, Z4, Z5, Z6, Z7, Z8 и Z9 независимо представляют собой CR2, N, O, S или N-R1 при условии, что один из Z1-Z9 является атомом углерода, к которому присоединена остальная часть молекулы;
R1, R2, R6 и R7 такие, как определено в п.1;
Y1, Y2, Y3 и Y4 независимо представляют собой С или N.
 ,
,  или
или 
где Z1, Z2, Z3, Z4, Z5, Z6, Z7, Z8, Z9 и Z10 независимо представляют собой CR2, N, O, S или N-R1 при условии, что один из Z1-Z10 является атомом углерода, к которому присоединена остальная часть молекулы;
R1, R2, R6 и R7 такие, как определено в п.1;
Y1, Y2, Y3 и Y4 независимо представляют собой С или N.
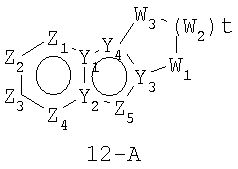 или
или 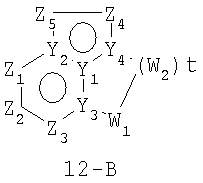
где Z1, Z2, Z3, Z4 и Z5 независимо представляют собой CR2, N, O, S или N-R1 при условии, что один из Z1-Z5 является атомом углерода, к которому присоединена остальная часть молекулы;
Y1, Y2, Y3 и Y4 независимо представляют собой С или N;
W1, W2 и W3 независимо представляют собой CR4R4, О, N-R1 или S(O)r (r равно 0-2), при условии, что не происходит образования ни S-S, ни O-O, ни S-O связи с образованием насыщенной кольцевой системы;
R1, R2, R6 и R7 такие, как определено в п.1;
R4 представляет собой Н, необязательно замещенный C1-С6-алкил, ОН (при условии, что оба R4 не являются ОН), C1-С6-алкокси, -S-C1-С6-алкил, -COOR6, -NR6R7, -CONR6R7; или R4R4 могут вместе представлять собой =O, или R4R4 вместе с атомом углерода, к которому они присоединены, могут образовывать 5-8-членную спиросистему, содержащую или не содержащую гетероатомы, выбранные из N, О, S(O)n (где n равно 0-2), N-R1;
и t равно 1-4.
 ,
, 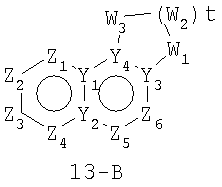 или
или 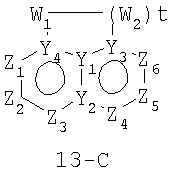
где Z1, Z2, Z3, Z4, Z5 и Z6 независимо представляют собой CR2, N, O, S или N-R1 при условии, что один из Z1-Z6 является атомом углерода, к которому присоединена остальная часть молекулы;
Y1, Y2, Y3 и Y4 независимо представляют собой С или N;
W1, W2 и W3 независимо представляют собой CR4R4, S(O)r (r равно 0-2), О, N-R1 при условии, что не происходит образования ни S-S, ни O-O, ни S-O связи с образованием насыщенной кольцевой системы;
R1, R2, R6 и R7 такие, как определено в п.1;
R4 представляет собой Н, необязательно замещенный C1-С6-алкил, ОН (при условии, что оба R4 не являются ОН), C1-С6-алкокси, -S-C1-С6-алкил, -COOR6, -NR6R7, -CONR6R7; или R4R4 могут вместе представлять собой =O, или R4R4 вместе с атомом углерода, к которому они присоединены, могут образовывать 5-8-членную спиросистему, содержащую или не содержащую гетероатомы, выбранные из N, О, S(O)n (где n равно 0-2), N-R1;
и t равно 1-3.
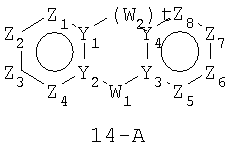 ,
, 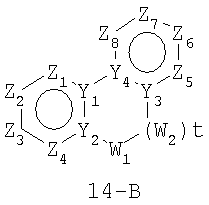 или
или 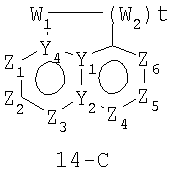
где Z1, Z2, Z3, Z4, Z5, Z6, Z7 и Z8 независимо представляют собой CR2, N, O, S или N-R1 при условии, что один из Z1-Z8 является атомом углерода, к которому присоединена остальная часть молекулы;
Y1, Y2, Y3 и Y4 независимо представляют собой С или N;
W1 и W2 независимо представляют собой CR4R4, S(O)r (r равно 0-2), О, N-R1 при условии, что не происходит образования ни S-S, ни O-O, ни S-O связи с образованием насыщенной кольцевой системы;
R1, R2, R6 и R7 такие, как определено в п.1;
R4 представляет собой Н, необязательно замещенный C1-С6-алкил, ОН (при условии, что оба R4 не являются ОН), C1-С6-алкокси, -S-C1-С6-алкил, -COOR6, -NR6R7, -CONR6R7; или R4R4 могут вместе представлять собой =O, или R4R4 вместе с атомом углерода, к которому они присоединены, могут образовывать 5-8-членную спиросистему, содержащую или не содержащую гетероатомы, выбранные из N, О, S(O)n (где n равно 0-2), N-R1;
и t равно 1 или 2.
 ,
,  или
или 
где Z1, Z2, Z3 и Z4 независимо представляют собой CR2, N, O, S или N-R1 при условии, что один из Z1-Z4 является атомом углерода, к которому присоединена остальная часть молекулы;
Y1, Y2, Y3 и Y4 независимо представляют собой С или N;
W1, W2, W3, W4 и W5 независимо представляют собой CR4R4, S(O)r (r равно 0-2), О, N-R1 при условии, что не происходит образования ни S-S, ни O-O, ни S-O связи с образованием насыщенной кольцевой системы;
R1, R2, R6 и R7 такие, как определено в п.1;
R4 представляет собой Н, необязательно замещенный C1-С6-алкил, ОН (при условии, что оба R4 не являются ОН), C1-С6-алкокси, -S-C1-С6-алкил, -COOR6, -NR6R7, -CONR6R7; или R4R4 могут вместе представлять собой =O, или R4R4 вместе с атомом углерода, к которому они присоединены, могут образовывать 5-8-членную спиросистему, содержащую или не содержащую гетероатомы, выбранные из N, О, S(O)n (где n равно 0-2), N-R1;
t равно 1-3;
u равно 1-3.
где R представляет собой п-нитробензил.
(A) (i) взаимодействие 6-аминопенициллановой кислоты с бромистоводородной кислотой в органическом растворителе и воде с образованием 6-бромпроизводного 21

и (ii) превращение 6-бромпенициллановой кислоты 21 в п-нитробензил 6-бромпеницилланат 22
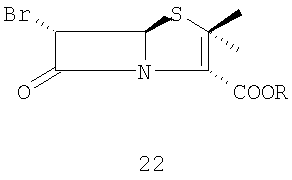
где R представляет собой п-нитробензил,
действием 4-нитробензилбромида в присутствии основания в органическом растворителе;
(B) окисление п-нитробензил 6-бромпеницилланата 22 с образованием п-нитробензил 6-бромпеницилланат-1-оксида 23

(C) кипячение п-нитробензил 6-бромпеницилланат-1-оксида 23 с 2-меркаптобензотиазолом в ароматическом растворителе с образованием 4-нитробензил (2R)-2-[(3S,4R)-4-(бензотиазол-2-илдитио)-3-бром-2-оксоазетидин-1-ил]-3-метилбут-3-еноата 24;
(D) растворение 4-нитробензил (2R)-2-[(3S,4R)-4-(бензотиазол-2-илдитио)-3-бром-2-оксоазетидин-1-ил]-3-метилбут-3-еноата 24 в органическом растворителе и взаимодействие с органическим третичным основанием с образованием 4-нитробензил-2-[(3S,4R)-4-(бензотиазол-2-илдитио)-3-бром-2-оксоазетидин-1 -ил]-3-метилбут-2-еноата 25;
(Е) превращение 4-нитробензил-2-[(3S,4R)-4-(бензотиазол-2-илдитио)-3-бром-2-оксоазетидин-1-ил]-3-метилбут-2-еноата 25 в 4-нитробензил 2-[(3S,4R.)-3-бром-4-формилтио-2-оксоазетидин-1-ил]-3-метилбут-2-еноата 26 в результате взаимодействия в ароматическом органическом растворителе с органической кислотой в присутствии смеси уксусный ангидрид/органическое третичное основание и триалкил- или триарилфосфина в интервале температур от около -10 до около -30°С;
(F) пропускание озонированного кислорода через раствор 4-нитробензил 2-[(3S,4R)-3-бром-4-формилтио-2-оксоазетидин-1-ил]-3-метилбут-2-еноата 26 в органическом растворителе в течение 3-4 ч при температуре от -70 до -90°С и последующую внутримолекулярную циклизацию с использованием фосфитного реагента, что приводит к образованию 4-нитробензил (5R,6S)-6-бромпенем-3-карбоксилата 16.
как определено в п.1, в котором 4-нитробензил (5R,6S)-6-бромпенем-3-карбоксилат 16, используемый на стадии (а), получают по п.33.
| СПОСОБ ПОЛУЧЕНИЯ НЕПРЕДЕЛЬНЫХ СПИРТОВ | 0 |
|
SU232966A1 |
| Экономайзер | 0 |
|
SU94A1 |
| СПОСОБ КЛАССИФИКАЦИИ МЕЛКОДИСПЕРСНЫХ МАТЕРИАЛОВ | 0 |
|
SU312186A1 |
| Торфодобывающая машина с вращающимся измельчающим орудием | 1922 |
|
SU87A1 |
| NEAL F.OSBORNE et al | |||
| Кипятильник для воды | 1921 |
|
SU5A1 |
| RU 94012854 A, 10.09.1995. | |||

