Область техники
Данное изобретение относится к новым производным карбонуклеозида, применяемым в лечении аномального роста клеток, таких как раковые, у млекопитающих. Это изобретение также относится к способу применения таких соединений в лечении аномального роста клеток у млекопитающих, особенно человека, и к фармацевтическим композициям в качестве противораковых агентов.
Уровень техники
Пост-трансляционная модификация аргининовых остатков метилированием важная для многих критических клеточных процессов, включая ремоделирование хроматина, транскрипцию гена, трансляцию белка, трансдукцию сигнала, сплайсинг РНК и пролиферацию клеток. Метилирование аргинина катализируется ферментами белка аргининметилтрансферазы (PRMT). Существует всего девять членов PRMT, и восемь имеют описанную ферментную активность на целевых субстратах.
Семейство ферментов белка аргиниметилтрансферазы (PRMT) используют S-аденозилметионин (SAM) для переноса метильных групп на аргининовые остатки на целевых белках. PRMT I типа катализируют образование монометильного аргинина и асимметрических диметиларгининов, а PRMT II типа катализируют монометильный аргинин и симметрические диметиларгинины. PRMT5 является ферментов II типа, дважды переносящим метильную группу из SAM на два атома азота ω-гуанидино аргинина, что вызывает ω-NG, N'G дисимметрическое метилирование белковых субстратов.
PRMT5 белок найден в ядре и цитоплазме, и имеет множество белковых субстратов, таких как гистоны, факторы транскрипции и белки сплайсосомы. PRMT5 имеет партнера по связыванию, Mep50 (белок метилосомы 50) и действует во множестве белковых комплексов. PRMT5 ассоциируется с комплексами ремоделирования хроматина (SWI/SNF, NuRD) и эпигенетически контролирует гены, вовлеченные в развитие, пролиферацию и дифференциацию клеток, включая антионкогены, через метилирование гистонов (Karkhanis, V. et al., Versatility of PRMT5 Induced Methylation in Growth Control and Development, Trends Biochem Sci 36(12) 633-641 (2011)). PRMT5 также контролируют экспрессию генов через ассоциацию с белковыми комплексами, которые привлекают PRMT5 для метилирования нескольких факторов транскрипции p53 (Jansson, M. et al., Arginine Methylation Regulates the p53 Response, Nat. Cell Biol. 10, 1431-1439 (2008)); E2F1 (Zheng, S. et al., Arginine Methylation-Dependent Reader-Writer Interplay Governs Growth Control by E2F-1, Mol Cell 52(1), 37-51 (2013)); HOXA9 (Bandyopadhyay, S. et al., HOXA9 Methylation by PRMT5 is Essential for Endothelial Cell Expression of Leukocyte Adhesion Molecules, Mol. Cell. Biol. 32(7):1202-1213 (2012)); и NFκB (Wei, H. et al., PRMT5 dimethylates R30 of the p65 Subunit to Activate NFκB, PNAS 110(33), 13516-13521 (2013)). В цитоплазме, PRMT5 имеет другой набор субстратов, вовлеченных в другие клеточные функции, включая сплайсинг РНК (белки Sm), сборку Гольджи (gm130), биогенез рибосомы (RPS10), медиированное piRNA замалчивание гена (белки Piwi) и подачу сигналов РЭФР (Karkhanis, 2011).
Дополнительные источники, относящиеся к PRMT5, включают: Aggarwal, P. et al., (2010) Nuclear Cyclin D1/CDK4 Kinase Regulates CUL4B Expression and Triggers Neoplastic Growth via Activation of the PRMT5 Methyltransferase, Cancer Cell 18: 329-340; Bao, X.et al., Overexpression of PRMT5 Promotes Tumor Cell Growth and is Associated with Poor Disease Prognosis in Epithelial Ovarian Cancer, J Histochem Cytochem 61: 206-217 (2013); Cho E. et al., Arginine Methylation Controls Growth Regulation by E2F1, EMBO J. 31(7) 1785-1797 (2012); Gu, Z. et al., Protein Arginine Methyltransferase 5 Functions in Opposite Ways in the Cytoplasm and Nucleus of Prostate Cancer Cells, PLoS One 7(8) e44033 (2012); Gu, Z. et al., Protein Arginine Methyltransferase 5 is Essential for Growth of Lung Cancer Cells, Biochem J. 446: 235-241 (2012); Kim, J. et al., Identification of Gastric Cancer Related Genes Using a cDNA Microarray Containing Novel Expressed Sequence Tags Expressed in Gastric Cancer Cells, Clin. Cancer Res. 11(2) 473-482 (2005); Nicholas, C. et al., PRMT5 is Upregulated in Malignant and Metastatic Melanoma and Regulates Expression of MITF and p27(Kip1), PLoS One 8(9) e74710 (2012); Powers, M. et al., Protein Arginine Methyltransferase 5 Accelerates Tumor Growth by Arginine Methylation of the Tumor Suppressor Programmed Cell Death 4, Cancer Res. 71(16) 5579-5587 (2011); Wang, L. et al., Protein Arginine Methyltransferase 5 Suppresses the Transcription of the RB Family of Tumor Suppressors in Leukemia and Lymphoma Cells, Mol. Cell Biol. 28(20), 6262-6277 (2008).
PRMT5 сверхэкспрессируется при многих раках и был обнаружен в образцах пациента и колониях клеток, включающих лимфому В-клеток и лейкоз (Wang, 2008) и следующих солидных опухолях: желудка (Kim 2005), пищевода (Aggarwal, 2010), молочной железы (Powers, 2011), легкого (Gu, 2012), простаты (Gu, 2012), меланомы (Nicholas 2012), толстой кишки (Cho, 2012) и яичников (Bao, 2013). Во многих этих раках, сверхэкспрессия PRMT5 коррелирует с плохим прогнозом. Неправильное метилирование аргинина PRMT5 субстратов связано с другими показаниями в дополнение к раку, такими как метаболические расстройства, воспалительное и аутоиммунное заболевание и гемоглобинопатии.
Сущность изобретения
Учитывая его роль в регулировании биологических процессов, PRMT5 является привлекательной целью для модулирования ингибиторами малых молекул. В настоящее время, разработано несколько эффективных ингибиторов PRMT5, и ни один ингибитор PRMT5 не вышел в клинику.
Каждый вариант соединений в соответствии с данным изобретением, описанный ниже, может быть объединен с любым другим вариантом соединений в соответствии с данным изобретением, описанных здесь, не противоречащим варианту, с которым он объединен. Более того, каждый из приведенных ниже вариантов осуществления, описывающих изобретение, включает в свой объем фармацевтически приемлемые соли соединений в соответствии с данным изобретением. Следовательно, фраза ʺили его фармацевтически приемлемая сольʺ подразумевается в описании всех описанных здесь соединений.
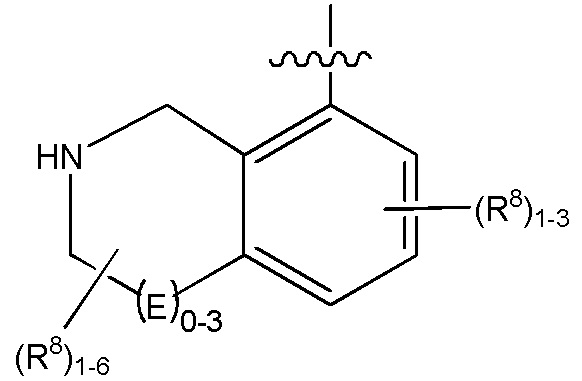
Изобретение включает варианты, в которых представлено соединение формулы (I):
Варианты данного изобретения включают соединения формулы (I):
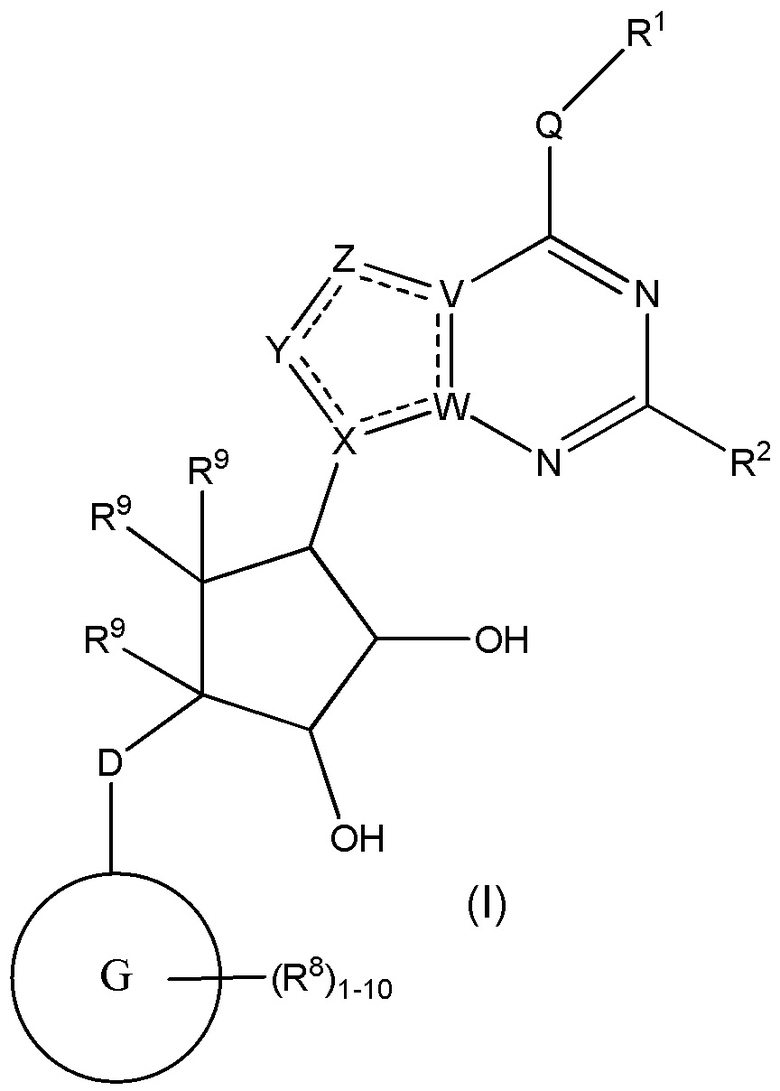
или их фармацевтически приемлемую соль, где:
R1 выбирают из группы, включающей (C1-C8)алкил, (C1-C8)галоалкил, гидрокси, (C1-C8)алкокси, (C5-C12)арил, 5-12-членный гетероарил, (C3-C10)циклоалкил, 3-12-членный гетероциклил, OR4, SR4 и N(R4)2, где каждый R4 независимо является A-R14, где A отсутствует, (C1-C3)алкил, -C(O)- или -SO2-, и R14 является водородом, (C1-C8)алкилом, (C5-C12)арилом, 5-12-членным гетероарилом, (C3-C10)циклоалкилом или 3-12-членным гетероциклилом, или два R4 объединены с получением 4-6-членного гетероциклического кольца, содержащего 1-3 гетероатома, выбранных из N, O и S;
R2 является водородом, галогеном, (C1-C8)алкилом, гидрокси, (C1-C8)алкокси или N(R5)2, где каждый R5 независимо является водородом или (C1-C8)алкилом, или два R5 объединены с получением 4-6-членного гетероциклического кольца, содержащего 1-3 гетероатома, выбранных из N, O и S;
каждый R3 независимо выбирают из водорода, гидрокси, NH2; (C1-C8)алкила или гетероалкила, имеющего 1-8 атомов, или если D является C(R3)2, R3 дополнительно выбирают из фтора, (C1-C8)алкилена или гетероалкилена, связанного с атомом на G с получением кольца, конденсированного с G, где R3 необязательно замещен 1-6 R8;
каждый R9 независимо является водородом или фтором;
D является C(R3)2, NR3, O, S или S(O)1-2;
G является (C5-C12)арилом или 5-12-членной гетероарильной кольцевой системой, конденсированной с (C3-C10)циклоалкильной или гетероциклильной кольцевой системой;
каждый R8 отсутствует или его независимо выбирают из группы, включающей (C1-C8)алкил, (C1-C8)галоалкил, гидрокси, (C1-C8)алкокси, (C5-C12)арил, 5-12-членный гетероарил, (C3-C10)циклоалкил, 3-12-членный гетероциклил, OR4, SR4, N(R4)2, CN, галоген и CON(R4)2, где два R8 необязательно объединены с получением 4-6-членного спиро-циклоалкильного кольца, циклоалкильного конденсированного кольца или алкиленового мостика, связующего G, и где два R8 необязательно объединены с образованием карбонила;
где каждый R4 независимо является A-R14, где A отсутствует, является (C1-C3)алкилом, -C(O)- или -SO2-, и R14 является водородом, (C1-C8)алкилом, (C5-C12)арилом, 5-12-членным гетероарилом, (C3-C10)циклоалкилом или 3-12-членным гетероциклилом, или два R4 объединены с получением 4-6-членного гетероциклического кольца, содержащего 1-3 гетероатома, выбранных из N, O и S;
Q отсутствует или является двухвалентной группой, выбранной из O, S, NH и (C1-C8)алкилена;
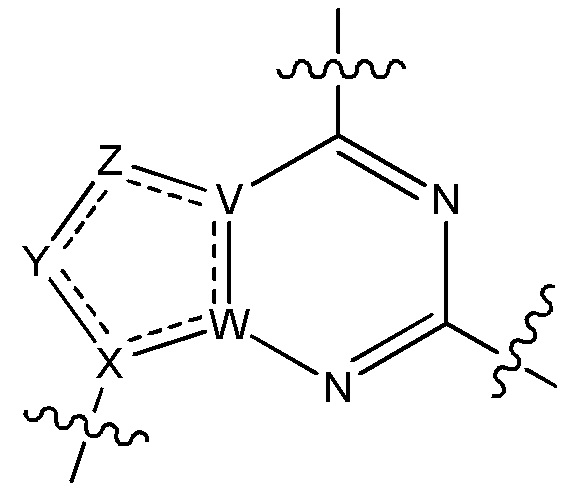
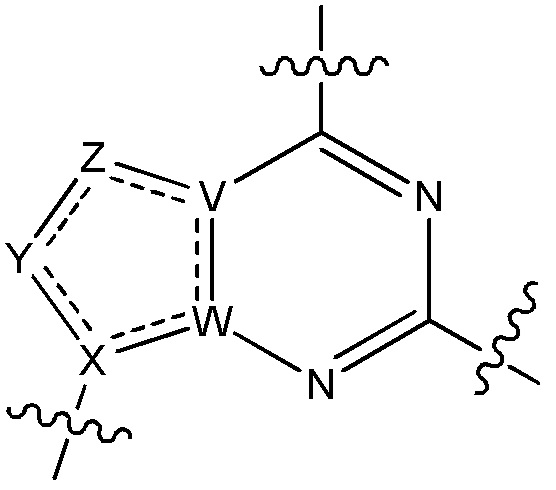
V является N или C, если Z присутствует, где если V образует двойную связь V является углеродом или V является N или CH и образует двойную связь с W, если Z отсутствует;
W является N или C, где если W образует двойную связь если W является углеродом;
X является N или C если Y присутствует, где если X образует двойную связь, X является углеродом или X является O, NR16 или C(R16)2 если Y отсутствует, где R16 является H или метилом;
Y отсутствует, является CR10, N, NR10, O или S или Y отсутствует, водородом или (C1-C8)алкилом, если Z отсутствует, где каждый R10 независимо выбирают из водорода, (C1-C8)алкила, гидрокси, (C1-C8)алкокси, галогена, SH, S-(C1-C8)алкила и N(R11)2, если Y является CR10, где Y образует двойную связь с соседним членом кольца, когда Y является CR10 или N, и где каждый R11 независимо является водородом, (C1-C8)алкилом, (C5-C12)арилом или 5-12-членным гетероарилом, или два R11 объединены с получением 4-6-членного гетероциклического кольца, содержащего 1-3 гетероатома, выбранных из N, O и S, или Y является C(R10)2 и два R10 и атом углерода, с которым они связаны, образуют карбонил или тиокарбонил;
Z отсутствует, CR12, N, NR12, O или S, или Z отсутствует, является водородом или (C1-C8)алкилом, если Y отсутствует, где каждый R12 независимо выбирают из водорода, (C1-C8)алкила, гидрокси, (C1-C8)алкокси, фтора, хлора, брома, SH, S-(C1-C8)алкила и N(R13)2, где Z образует двойную связь с соседним членом кольца, если он является CR12 или N, где каждый R13 независимо является водородом, (C1-C8)алкилом, (C5-C12)арилом или 5-12-членным гетероарилом, или два R13 объединены с получением 4-6-членного гетероциклического кольца, содержащего 1-3 гетероатома, выбранных из N, O и S, и где Z не является NR12, если X является N, V является C, W является C и Y является CR10, или Z является C(R12)2 и два R12 и атом углерода, с которым они связаны, образуют карбонил или тиокарбонил; и
каждый ----- отсутствует или является необязательной связью, где не более двух, не соседних ----- могут присутствовать.
Варианты данного изобретения также включают соединения формулы (II):
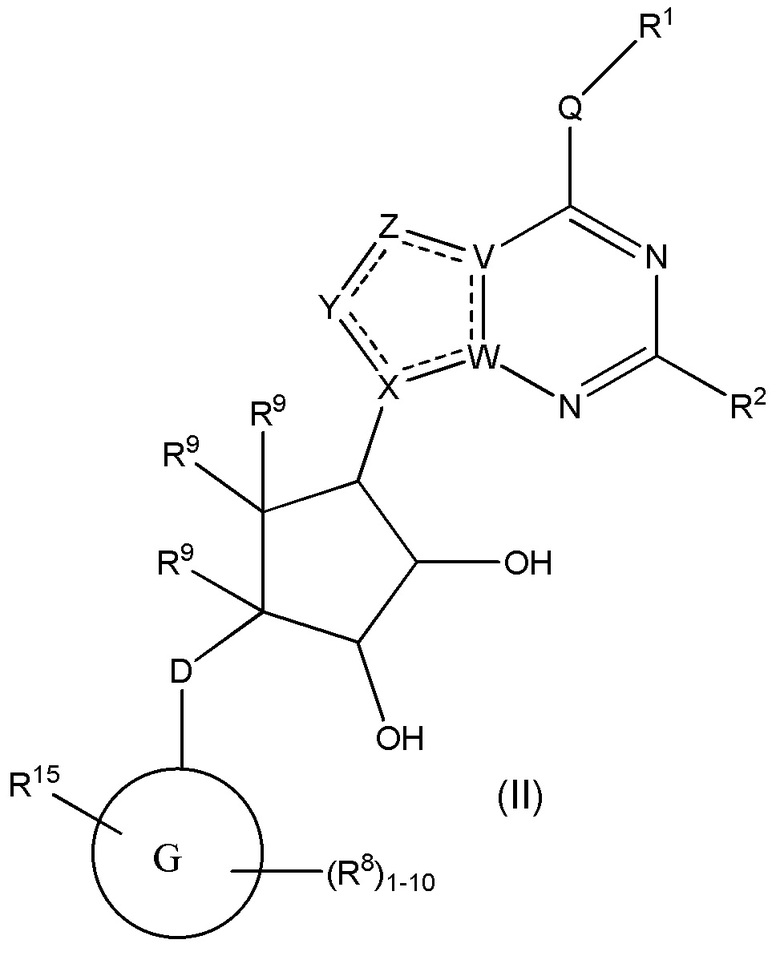
или его фармацевтически приемлемая соль, где:
R1 выбирают из группы, включающей (C1-C8)алкил, (C1-C8)галоалкил, гидрокси, (C1-C8)алкокси, (C5-C12)арил, 5-12-членный гетероарил, (C3-C10)циклоалкил, 3-12-членный гетероциклил, OR4, SR4 b N(R4)2, где каждый R4 независимо является A-R14, где A отсутствует, является (C1-C3)алкил, -C(O)- или -SO2-, и R14 является водородом, (C1-C8)алкилом, (C5-C12)арилом, 5-12-членным гетероарилом, (C3-C10)циклоалкилом или 3-12-членным гетероциклилом, или два R4 объединены с получением 4-6-членного гетероциклического кольца, содержащего 1-3 гетероатома, выбранных из N, O и S;
R2 является водородом, галогеном, (C1-C8)алкилом, гидрокси, (C1-C8)алкокси или N(R5)2, где каждый R5 независимо является водородом или (C1-C8)алкилом, или два R5 объединены с получением 4-6-членного гетероциклического кольца, содержащего 1-3 гетероатома, выбранных из N, O и S;
каждый R3 независимо выбирают из водорода, гидрокси, NH2; (C1-C8)алкила или гетероалкила, имеющего 1-8 атомов, или если D является C(R3)2, R3 дополнительно выбирают из фтора, (C1-C8)алкилена или гетероалкилена, связанного с атомом на G с получением кольца, конденсированного с G, где R3 необязательно замещен 1-6 R8;
каждый R9 независимо является водородом или фтором;
D является C(R3)2, O или S(O)1-2;
G является (C5-C12)арилом, 5-12-членной гетероарильной кольцевой системой;
R15 является гетероалкилом, имеющим 1-8 атомов, связанным с атомом на G и необязательно замещенным 1-6 R8, или R15 является гетероалкиленом, связанным с атомом на G, необязательно замещенным 1-6 R8, и связан с соседним атомом на G (т.е. два конца R15, если R15 является гетероалкиленом, связаны с соседними атомами углерода на G кольце);
каждый R8 отсутствует или его независимо выбирают из группы, включающей (C1-C8)алкил, (C1-C8)галоалкил, гидрокси, (C1-C8)алкокси, (C5-C12)арил, 5-12-членный гетероарил, (C3-C10)циклоалкил, 3-12-членный гетероциклил, OR4, SR4, N(R4)2, CN, галоген и CON(R4)2, где два R8 необязательно объединены с получением 4-6-членного спиро-циклоалкильного кольца, циклоалкильного конденсированного кольца или алкиленового мостика, связующего G, и где два R8 необязательно объединены с образованием карбонила;
где каждый R4 независимо является A-R14, где A отсутствует, является (C1-C3)алкилом, -C(O)- или -SO2-, и R14 является водородом, (C1-C8)алкилом, (C5-C12)арилом, 5-12-членным гетероарилом, (C3-C10)циклоалкилом или 3-12-членным гетероциклилом, или два R4 объединены с получением 4-6-членного гетероциклического кольца, содержащего 1-3 гетероатома, выбранных из N, O и S;
Q отсутствует или является двухвалентной группой, выбранной из O, S, NH и (C1-C8)алкилена;
V является N или C, где если V образует двойную связь, V является углеродом;
W является N или C, где если W образует двойную связь, W является углеродом;
X является N или C, где если X образует двойную связь, X является углеродом;
Y является CR10, N, NR10, O или S, где каждый R10 независимо выбирают из водорода, (C1-C8)алкила, гидрокси, (C1-C8)алкокси, или R10 необязательно выбирают из галогена, SH, S-(C1-C8)алкила и N(R11)2, если Y является CR10, где Y образует двойную связь с соседним членом кольца, когда Y является CR10 или N, и где каждый R11 независимо является водородом, (C1-C8)алкилом, (C5-C12)арилом или 5-12-членным гетероарилом или два R11 объединены с получением 4-6-членного гетероциклического кольца, содержащего 1-3 гетероатома, выбранных из N, O и S, или Y является C(R10)2 и два R10 и атом углерода, с которым они связаны, образуют карбонил или тиокарбонил;
Z является CR12, N, NR12, O или S, где каждый R12 независимо выбирают из водорода, (C1-C8)алкила, гидрокси, (C1-C8)алкокси, или R12 необязательно выбирают из фтора, хлора, брома, йода, SH, S-(C1-C8)алкила и N(R13)2, если Z является CR12, где Z образует двойную связь с соседним членом кольца, если он является CR12 или N, где каждый R13 независимо является водородом, (C1-C8)алкилом, (C5-C12)арилом или 5-12-членным гетероарилом, или два R13 объединены с получением 4-6-членного гетероциклического кольца, содержащего 1-3 гетероатома, выбранных из N, O и S, и где Z не является NR12, если X является N, V является C,
W является C и Y является CR10 или Z является C(R12)2 и два R12 и атом углерода, с которым они связаны, образуют карбонил или тиокарбонил; и
каждая ----- является необязательной связью, где не более двух, не соседних ----- могут присутствовать,
при условии, что D является S(O)1-2, если G является C10арилом или 10-членным гетероарилом.
Дополнительные варианты данного изобретения включают соединения формулы (III):
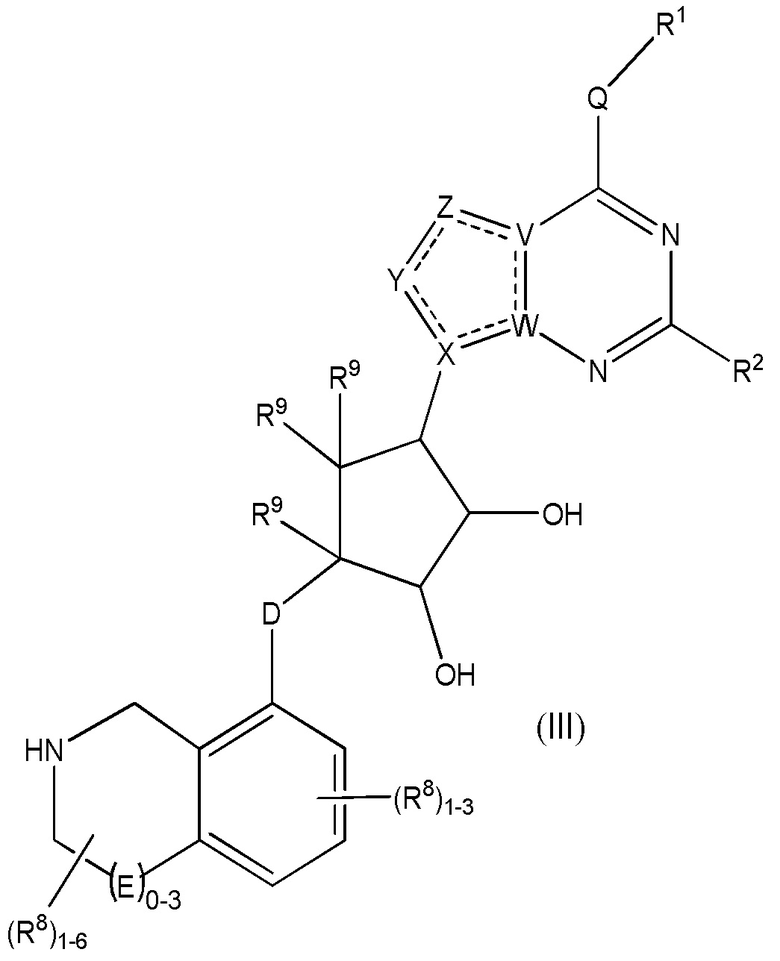
или их фармацевтически приемлемую соль, где:
каждый R1 независимо выбирают из группы, включающей (C1-C8)алкил, (C1-C8)галоалкил, гидрокси, (C1-C8)алкокси, (C5-C12)арил, 5-12-членный гетероарил, (C3-C10)циклоалкил, 3-12-членный гетероциклил, OR4, SR4 и N(R4)2, где каждый R4 независимо является A-R14, где A отсутствует, является (C1-C3)алкилом, -C(O)- или -SO2-, и R14 является водородом, (C1-C8)алкилом, (C5-C12)арилом, 5-12-членным гетероарилом, (C3-C10)циклоалкилом или 3-12-членным гетероциклилом, или два R4 объединены с получением 4-6-членного гетероциклического кольца, содержащего 1-3 гетероатома, выбранных из N, O и S;
R2 является водородом, галогеном, (C1-C8)алкилом, гидрокси, (C1-C8)алкокси или N(R5)2, где каждый R5 независимо является водородом или (C1-C8)алкилом, или два R5 объединены с получением 4-6-членного гетероциклического кольца, содержащего 1-3 гетероатома, выбранных из N, O и S;
каждый R3 независимо является водородом, гидрокси или NH2; или если D является C(R3)2, R3 дополнительно выбирают из фтора;
каждый R9 независимо является водородом или фтором;
D является C(R3)2, O или S(O)1-2;
E является NR1, CH2, C(R1)2, O или -S(O)2;
каждый R8 отсутствует или его независимо выбирают из группы, включающей (C1-C8)алкил, (C1-C8)галоалкил, гидрокси, (C1-C8)алкокси, (C5-C12)арил, 5-12-членный гетероарил, (C3-C10)циклоалкил, 3-12-членный гетероциклил, OR4, SR4, N(R4)2, CN, галоген и CON(R4)2, где два R8 необязательно объединены с получением 4-6-членного спиро-циклоалкильного кольца, циклоалкильного конденсированного кольца или алкиленового мостика, связующего G, и где два R8 необязательно объединены с образованием карбонила;
где каждый R4 независимо является A-R14, где A отсутствует, является (C1-C3)алкилом, -C(O)- или -SO2-, и R14 является водородом, (C1-C8)алкилом, (C5-C12)арилом, 5-12-членным гетероарилом, (C3-C10)циклоалкилом или 3-12-членным гетероциклилом, или два R4 объединены с получением 4-6-членного гетероциклического кольца, содержащего 1-3 гетероатома, выбранных из N, O и S;
Q отсутствует или является двухвалентной группой, выбранной из O, S, NH и (C1-C8)алкилена;
V является N или C, где если V образует двойную связь, V является углеродом;
W является N или C, где если W образует двойную связь, W является углеродом;
X является N или C, где если X образует двойную связь, X является углеродом;
Y является CR10, N, NR10, O или S, где каждый R10 независимо выбирают из водорода, (C1-C8)алкила, гидрокси, (C1-C8)алкокси, галогена, SH, S-(C1-C8)алкила и N(R11)2, если Y является CR10, где Y образует двойную связь с соседним членом кольца, когда Y является CR10 или N, и где каждый R11 независимо является водородом, (C1-C8)алкилом, (C5-C12)арилом или 5-12-членным гетероарилом, или два R11 объединены с получением 4-6-членного гетероциклического кольца, содержащего 1-3 гетероатома, выбранных из N, O и S, или Y является C(R10)2, и два R10 и атом углерода, с которым они связаны, образуют карбонил или тиокарбонил;
Z является CR12, N, NR12, O или S, где каждый R12 независимо выбирают из водорода, (C1-C8)алкила, гидрокси, (C1-C8)алкокси, фтора, хлора, брома, SH, S-(C1-C8)алкила и N(R13)2, где Z образует двойную связь с соседним членом кольца, если он является CR12 или N, где каждый R13 независимо является водородом, (C1-C8)алкилом, (C5-C12)арилом или 5-12-членным гетероарилом, или два R13 объединены с получением 4-6-членного гетероциклического кольца, содержащего 1-3 гетероатома, выбранных из N, O и S, и где Z не является NR12, если X является N, V является C, W является C и Y является CR10 или Z является C(R12)2 и два R12 и атом углерода, с которым они связаны, образуют карбонил или тиокарбонил; и
каждый ----- является необязательной связью, где не более двух, не соседних ----- могут присутствовать.
Дополнительные варианты данного изобретения включают соединения формулы (IV):
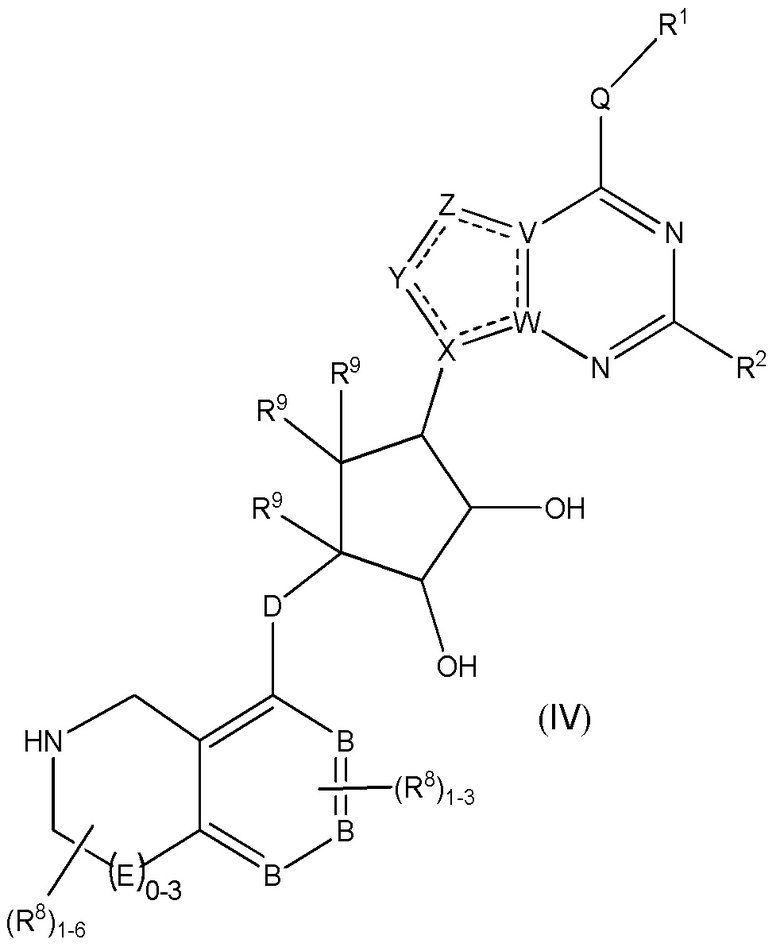
или их фармацевтически приемлемую соль, где:
каждый R1 независимо выбирают из группы, включающей (C1-C8)алкил, (C1-C8)галоалкил, гидрокси, (C1-C8)алкокси, (C5-C12)арил, 5-12-членный гетероарил, (C3-C10)циклоалкил, 3-12-членный гетероциклил, OR4, SR4 и N(R4)2, где каждый R4 независимо является A-R14, где A отсутствует, является (C1-C3)алкилом, -C(O)- или -SO2-, и R14 является водородом, (C1-C8)алкилом, (C5-C12)арилом, 5-12-членным гетероарилом, (C3-C10)циклоалкилом или 3-12-членным гетероциклилом, или два R4 объединены с получением 4-6-членного гетероциклического кольца, содержащего 1-3 гетероатома, выбранных из N, O и S;
R2 является водородом, галогеном, (C1-C8)алкилом, гидрокси, (C1-C8)алкокси или N(R5)2, где каждый R5 независимо является водородом или (C1-C8)алкилом, или два R5 объединены с получением 4-6-членного гетероциклического кольца, содержащего 1-3 гетероатома, выбранных из N, O и S;
каждый R3 независимо является водородом, гидрокси или NH2; или если D является C(R3)2, R3 дополнительно выбирают из фтора;
каждый R9 независимо является водородом или фтором;
B является N или C;
E является NR1, CH2, C(R1)2, O или -S(O)2;
каждый R8 отсутствует или его независимо выбирают из группы, включающей (C1-C8)алкил, (C1-C8)галоалкил, гидрокси, (C1-C8)алкокси, (C5-C12)арил, 5-12-членный гетероарил, (C3-C10)циклоалкил, 3-12-членный гетероциклил, OR4, SR4, N(R4)2, CN, галоген и CON(R4)2, где два R8 необязательно объединены с получением 4-6-членного спиро-циклоалкильного кольца, циклоалкильного конденсированного кольца или алкиленового мостика, связующего G, и где два R8 необязательно объединены с образованием карбонила;
где каждый R4 независимо является A-R14, где A отсутствует, является (C1-C3)алкилом, -C(O)- или -SO2-, и R14 является водородом, (C1-C8)алкилом, (C5-C12)арилом, 5-12-членным гетероарилом, (C3-C10)циклоалкилом или 3-12-членным гетероциклилом, или два R4 объединены с получением 4-6-членного гетероциклического кольца, содержащего 1-3 гетероатома, выбранных из N, O и S;
Q отсутствует или является двухвалентной группой, выбранной из O, S, NH и (C1-C8)алкилена;
V является N или C, где если V образует двойную связь, V является углеродом;
W является N или C, где если W образует двойную связь, W является углеродом;
X является N или C, где если X образует двойную связь, X является углеродом;
Y является CR10, N, NR10, O или S, где каждый R10 независимо выбирают из водорода, (C1-C8)алкила, гидрокси, (C1-C8)алкокси, галогена, SH, S-(C1-C8)алкила и N(R11)2, если Y является CR10, где Y образует двойную связь с соседним членом кольца, когда Y является CR10 или N, и где каждый R11 независимо является водородом, (C1-C8)алкилом, (C5-C12)арилом или 5-12-членного гетероарила, или два R11 объединены с получением 4-6-членного гетероциклического кольца, содержащего 1-3 гетероатома, выбранных из N, O и S, или Y является C(R10)2 и два R10 и атом углерода, с которым они связаны, образуют карбонил или тиокарбонил;
Z является CR12, N, NR12, O или S, где каждый R12 независимо выбирают из водорода, (C1-C8)алкила, гидрокси, (C1-C8)алкокси, фтора, хлора, брома, SH, S-(C1-C8)алкила и N(R13)2, где Z образует двойную связь с соседним членом кольца, если он является CR12 или N, где каждый R13 независимо является водородом, (C1-C8)алкилом, (C5-C12)арилом или 5-12-членным гетероарилом, или два R13 объединены с получением 4-6-членного гетероциклического кольца, содержащего 1-3 гетероатома, выбранных из N, O и S, и где Z не является NR12, если X является N, V является C, W является C и Y является CR10 или Z является C(R12)2 и два R12 и атом углерода, с которым они связаны, образуют карбонил или тиокарбонил; и
каждая ----- является необязательной связью, где не более двух, не соседних ----- могут присутствовать.
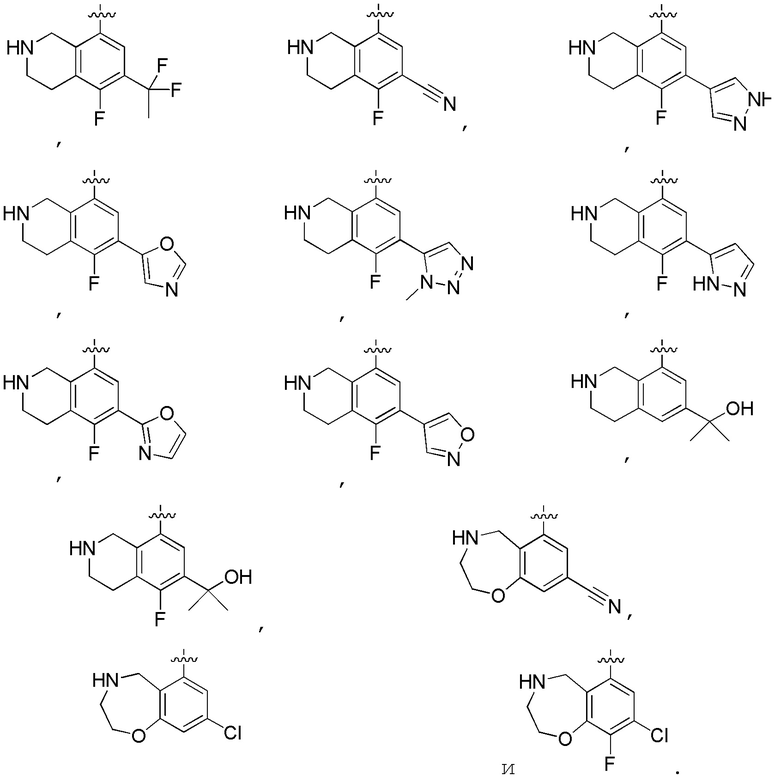
Другие варианты данного изобретения включают описанные соединения, где
выбирают из:
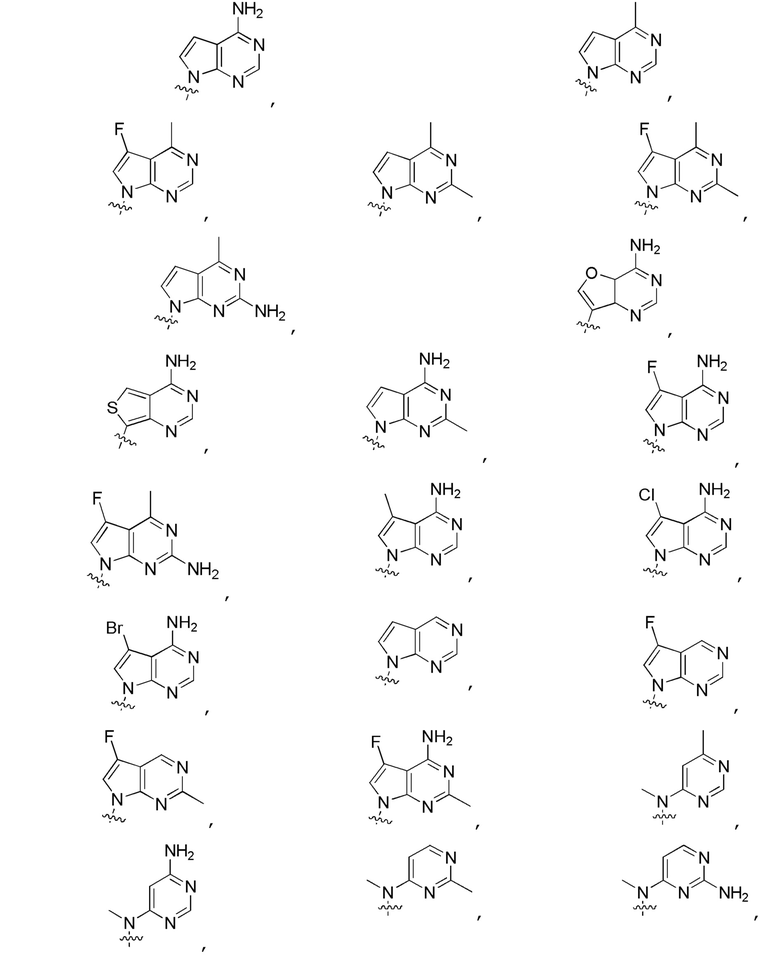
В определенных вариантах:
выбирают из:
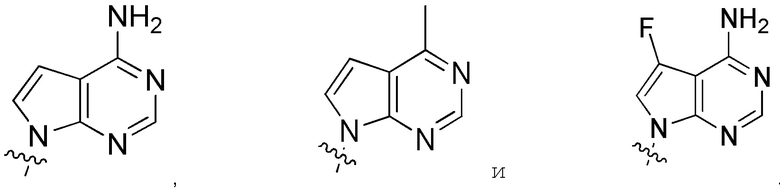
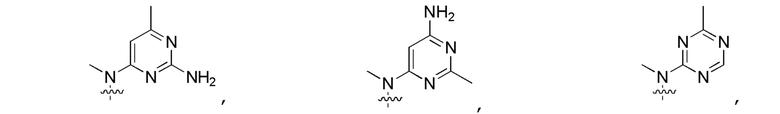
Другие варианты данного изобретения включают описанные соединения, где
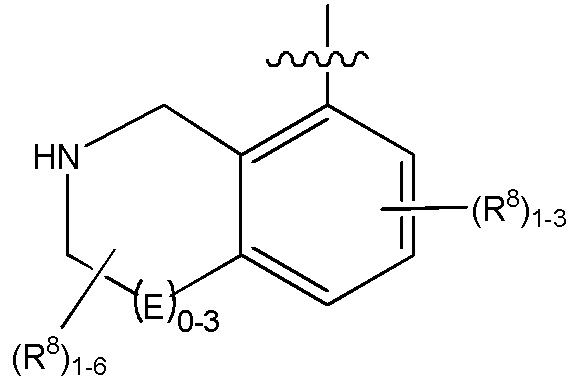
выбирают из:
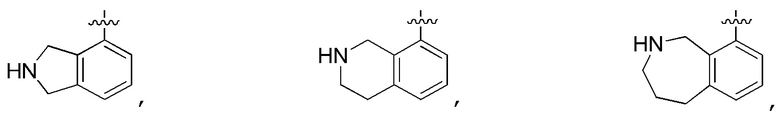
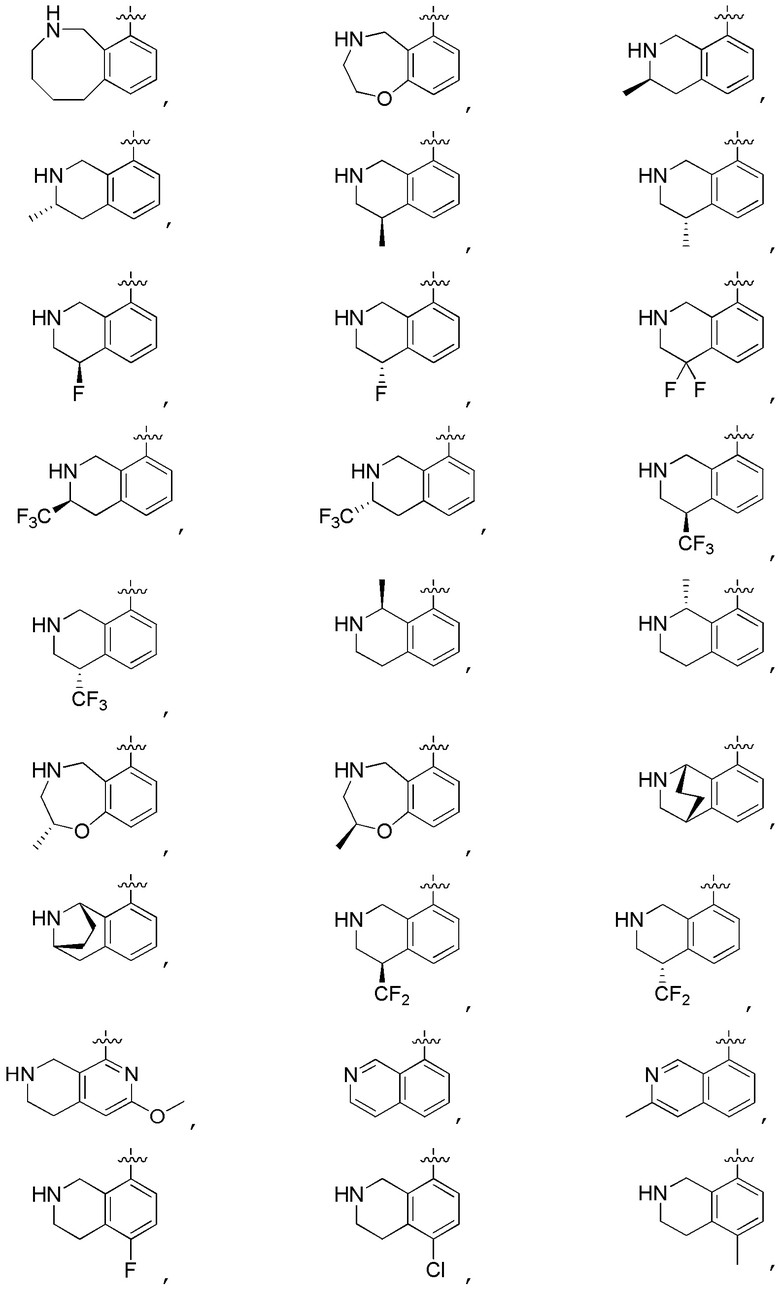
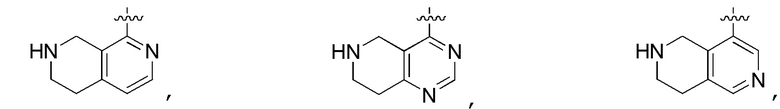
В определенных вариантах
является:
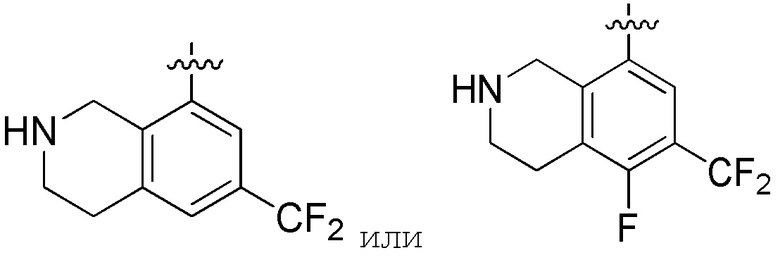 .
.
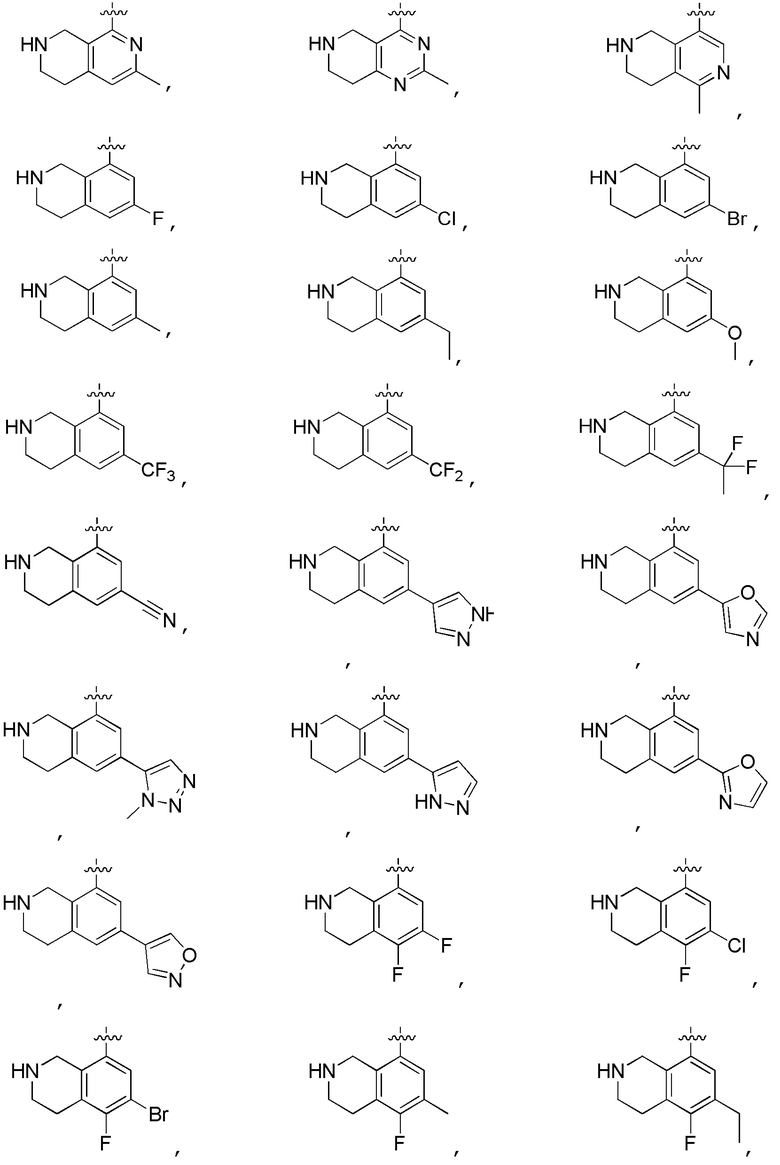
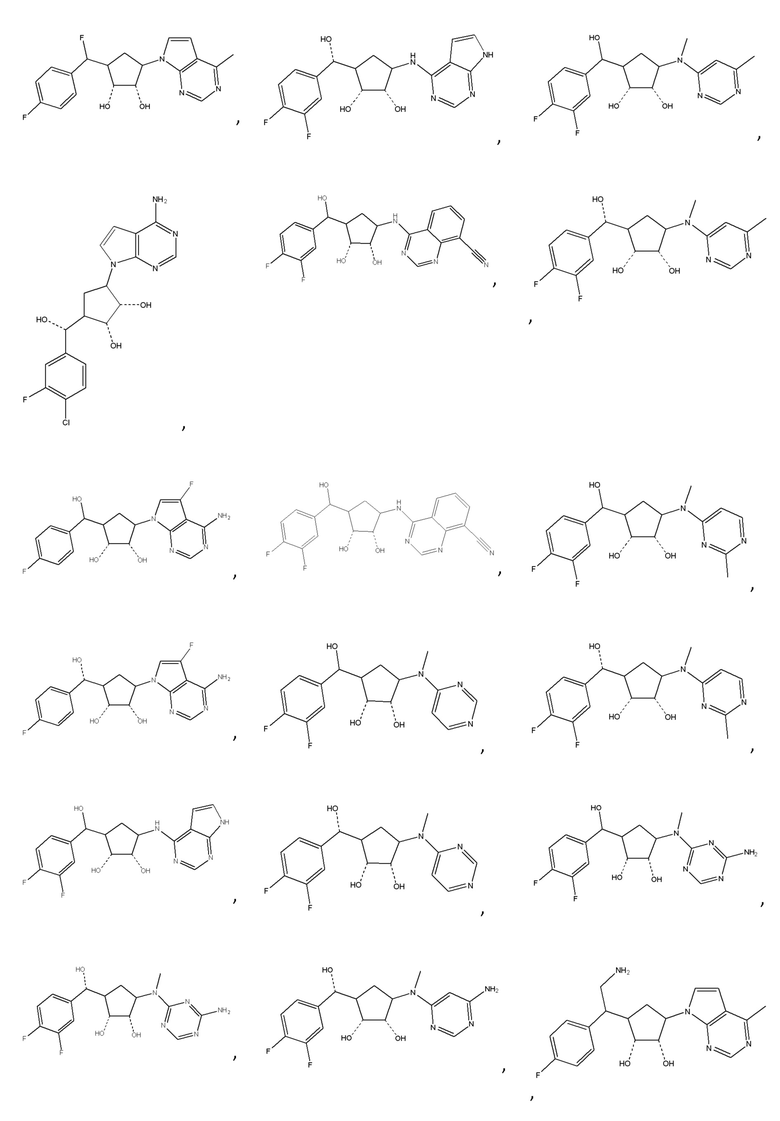
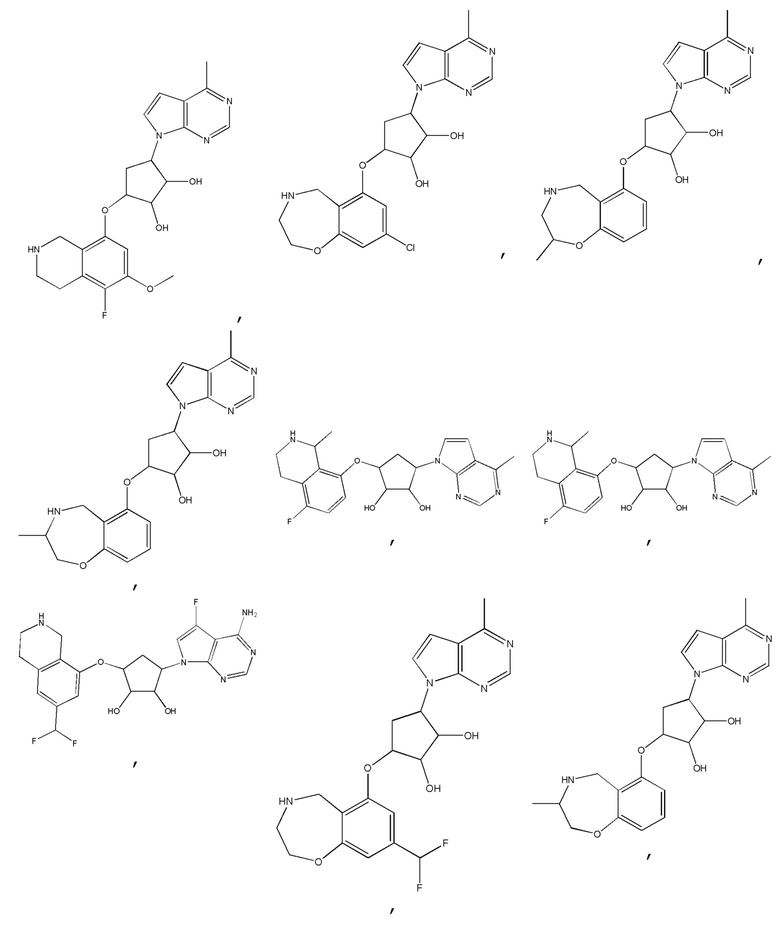
Другие варианты данного изобретения включают соединения, выбранные из:
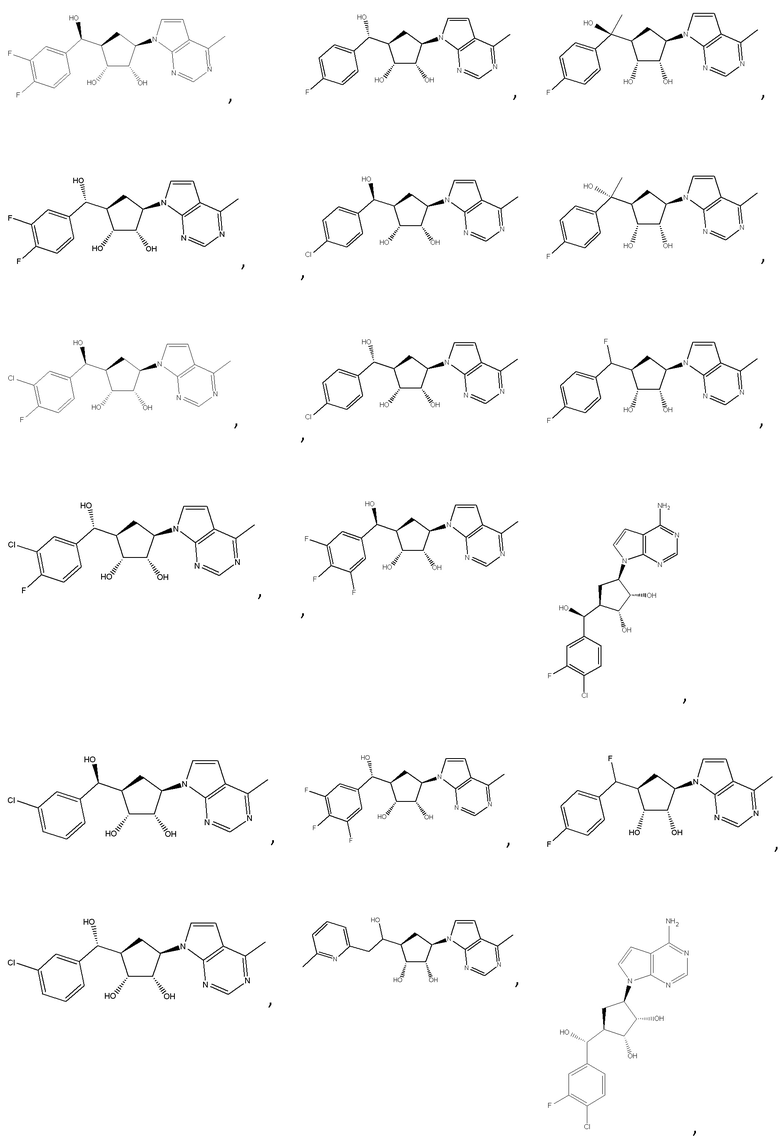
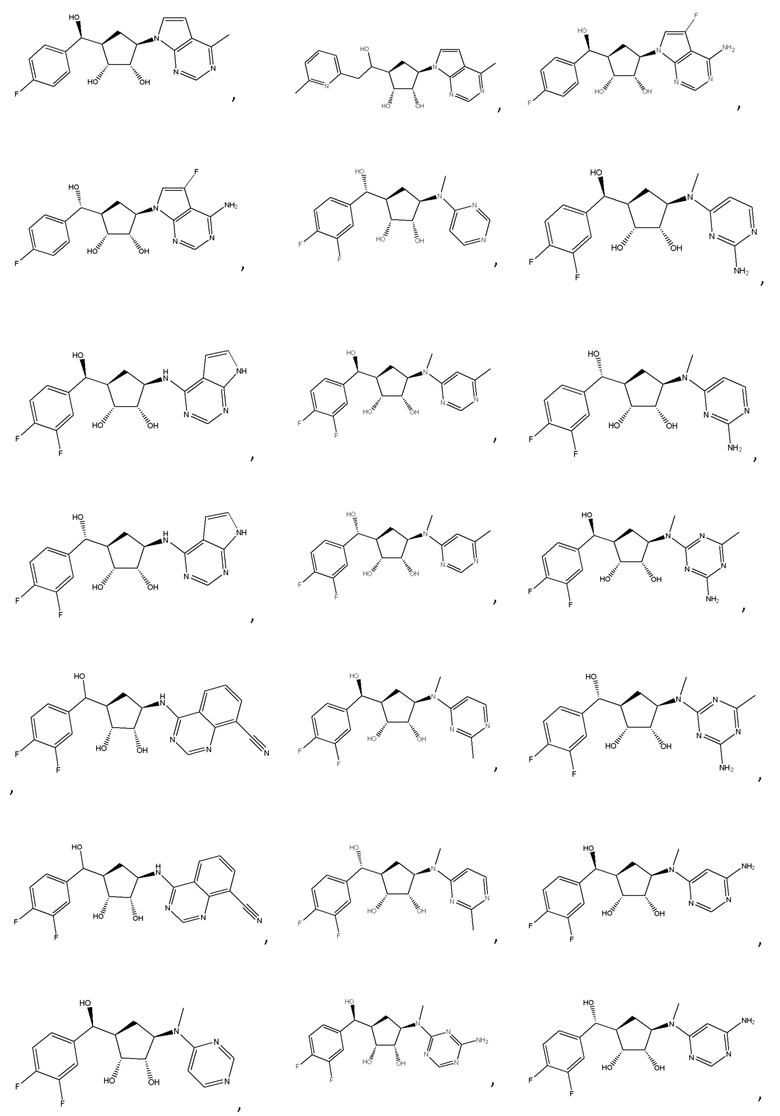
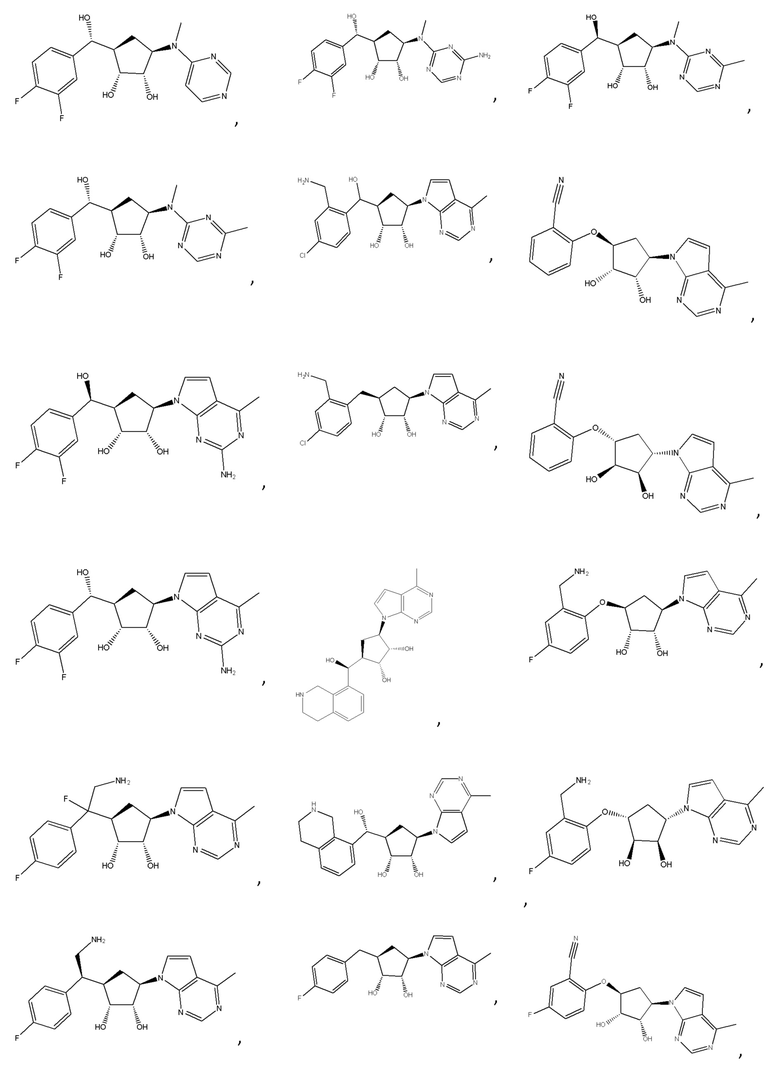
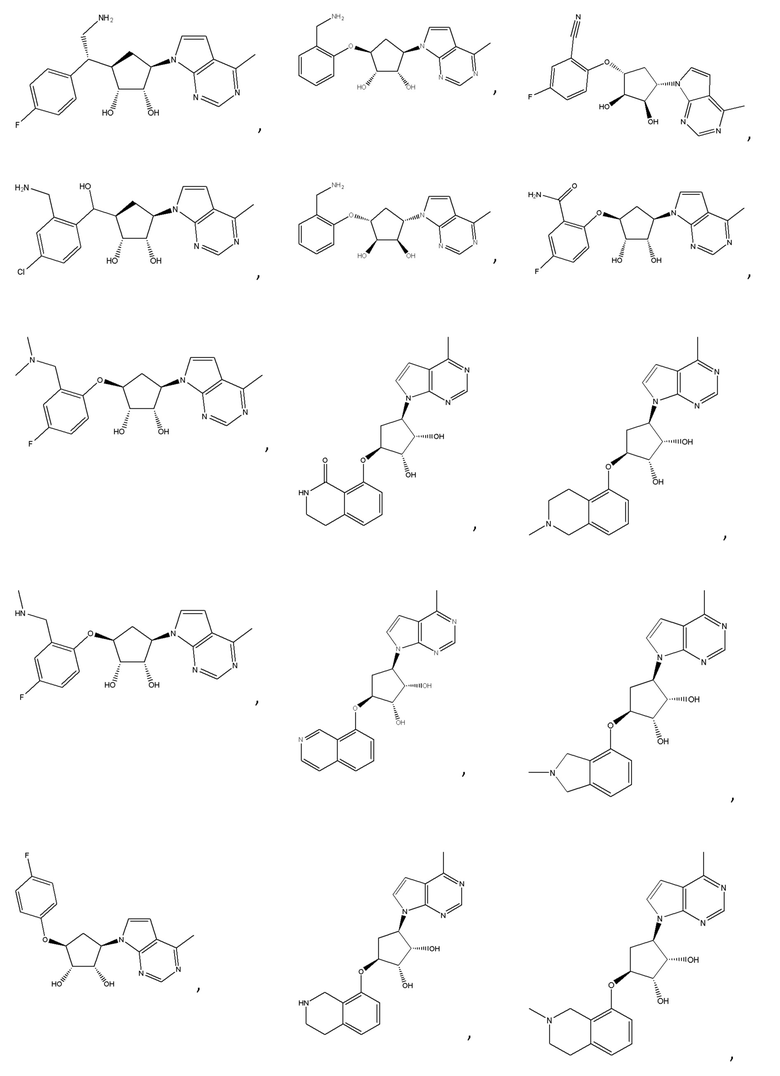
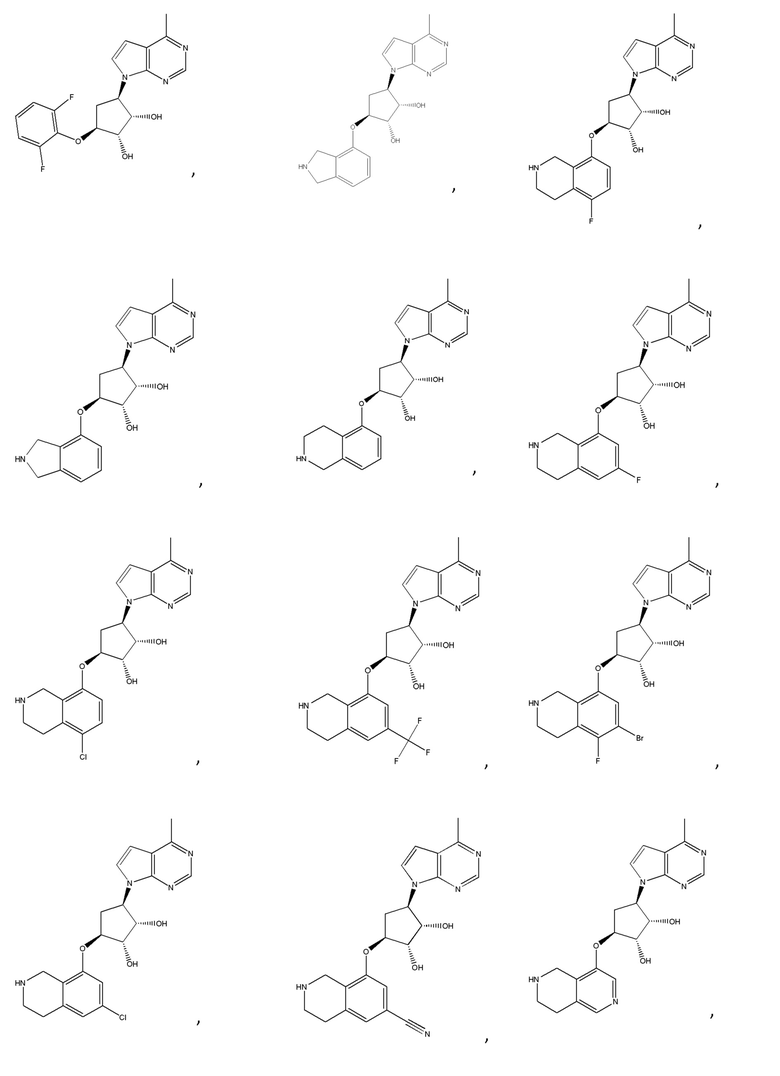
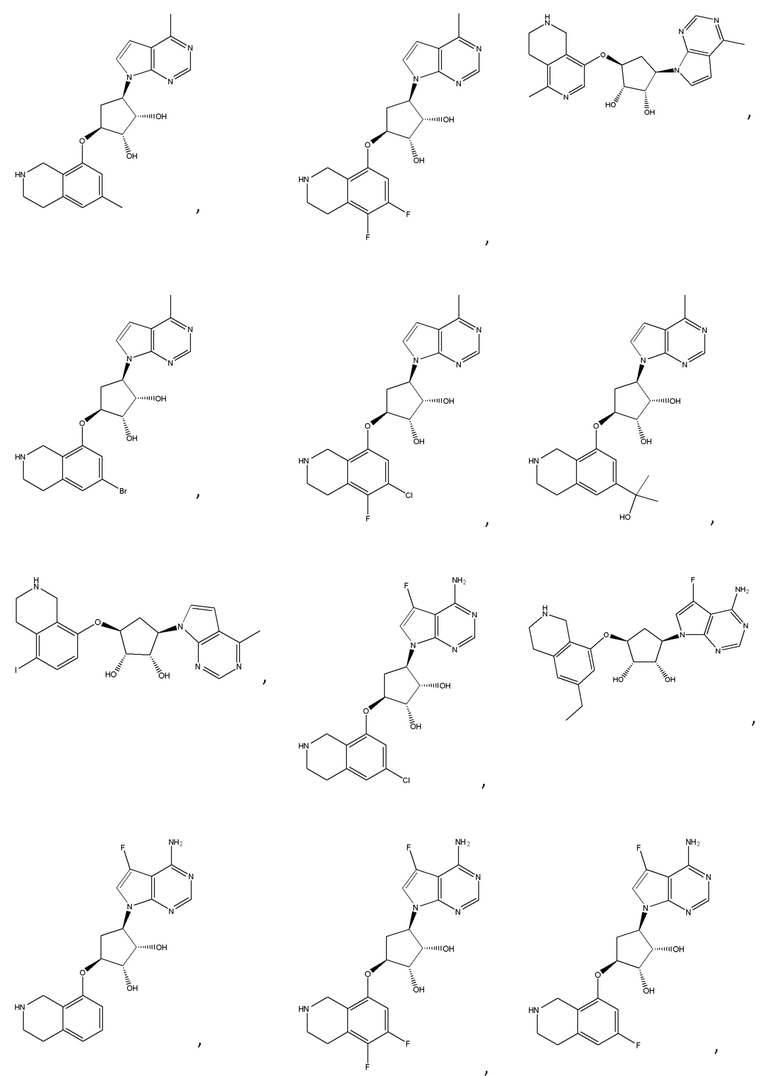
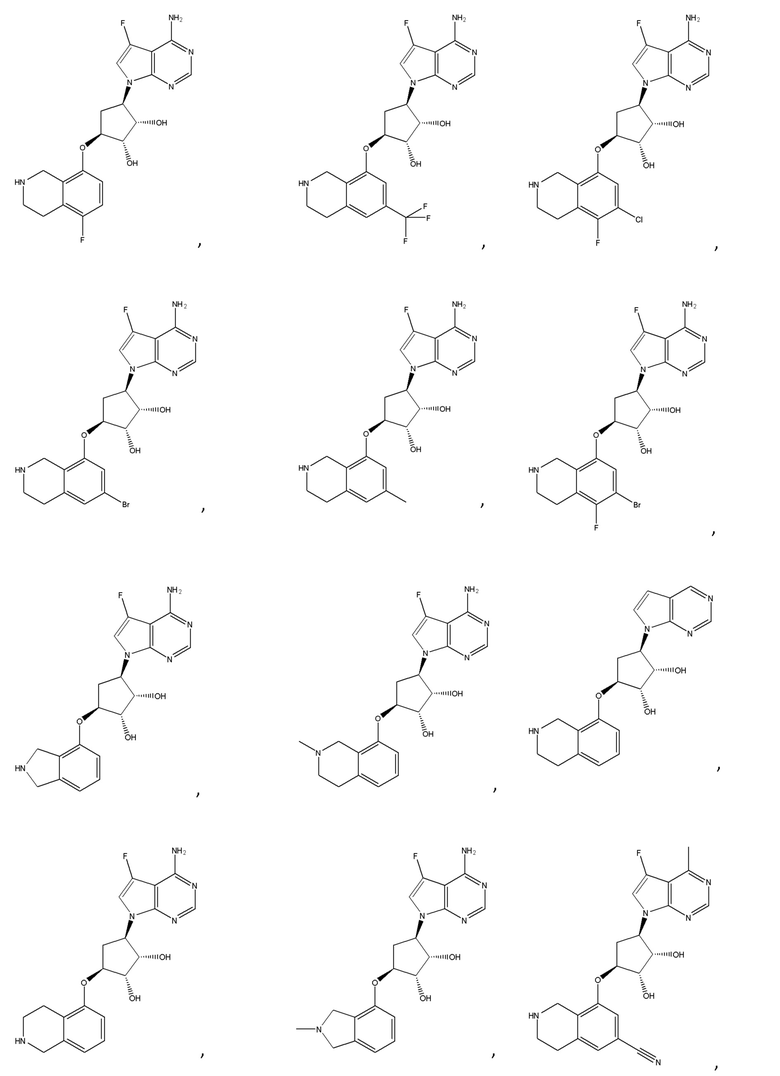
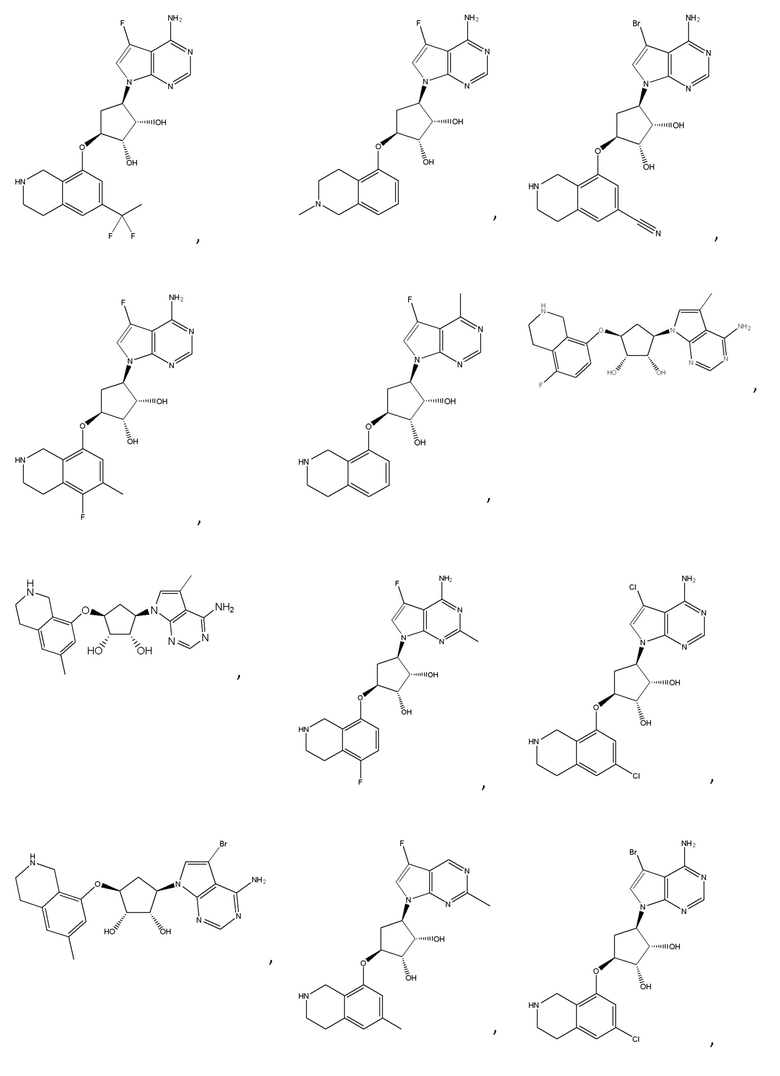
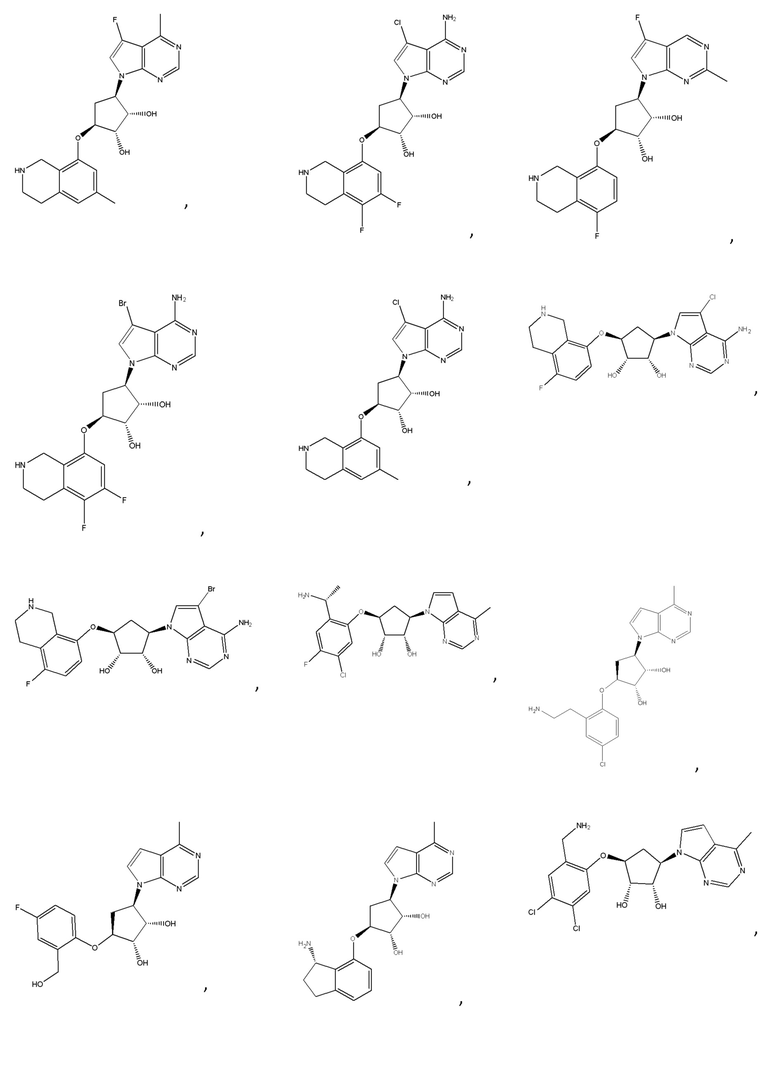
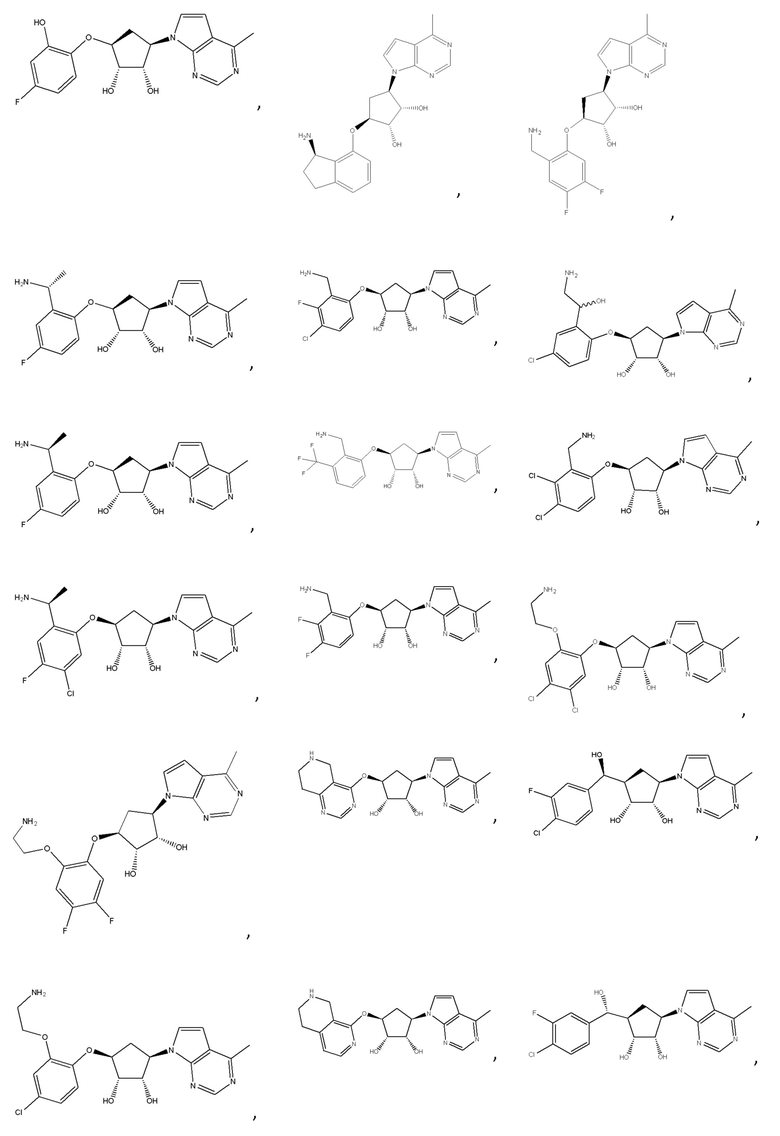
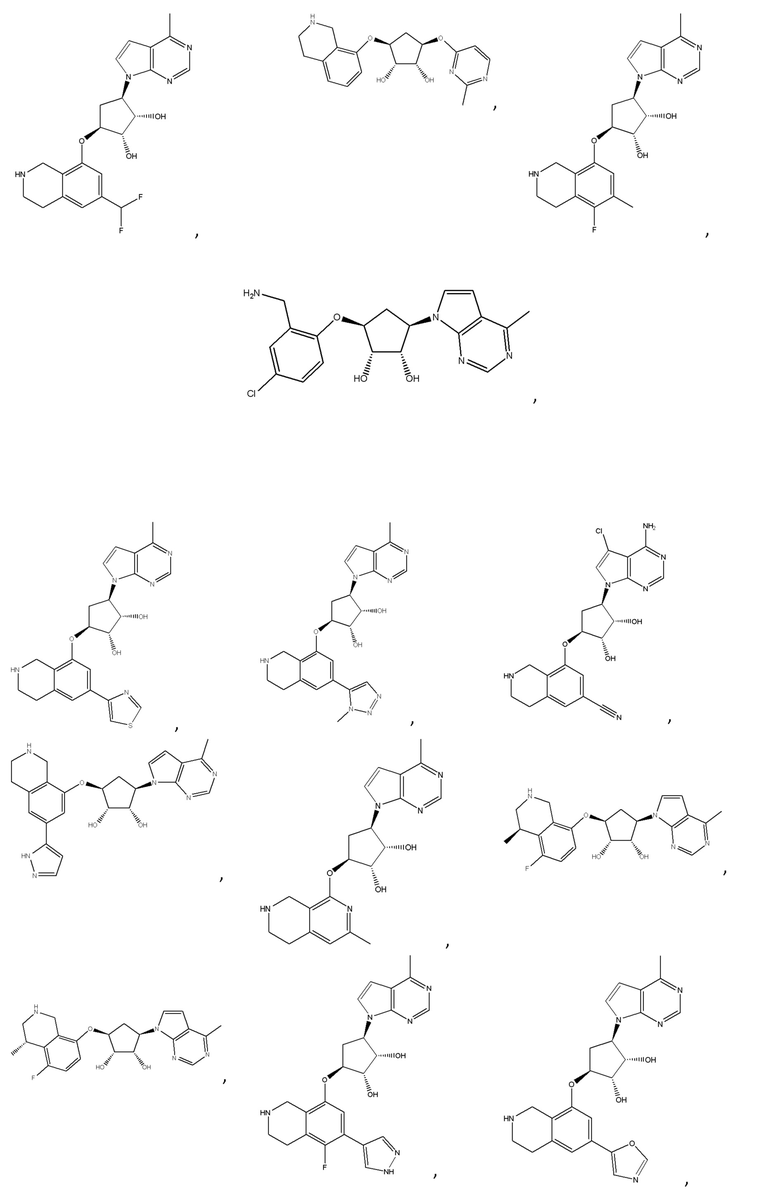
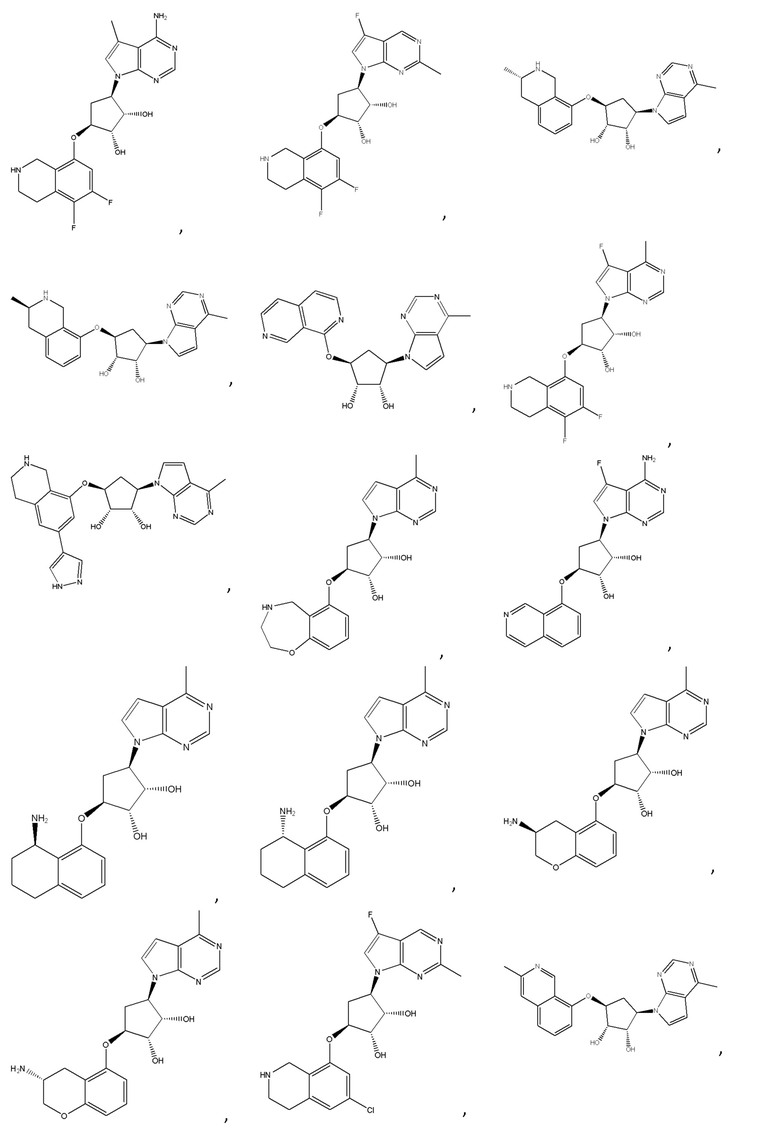
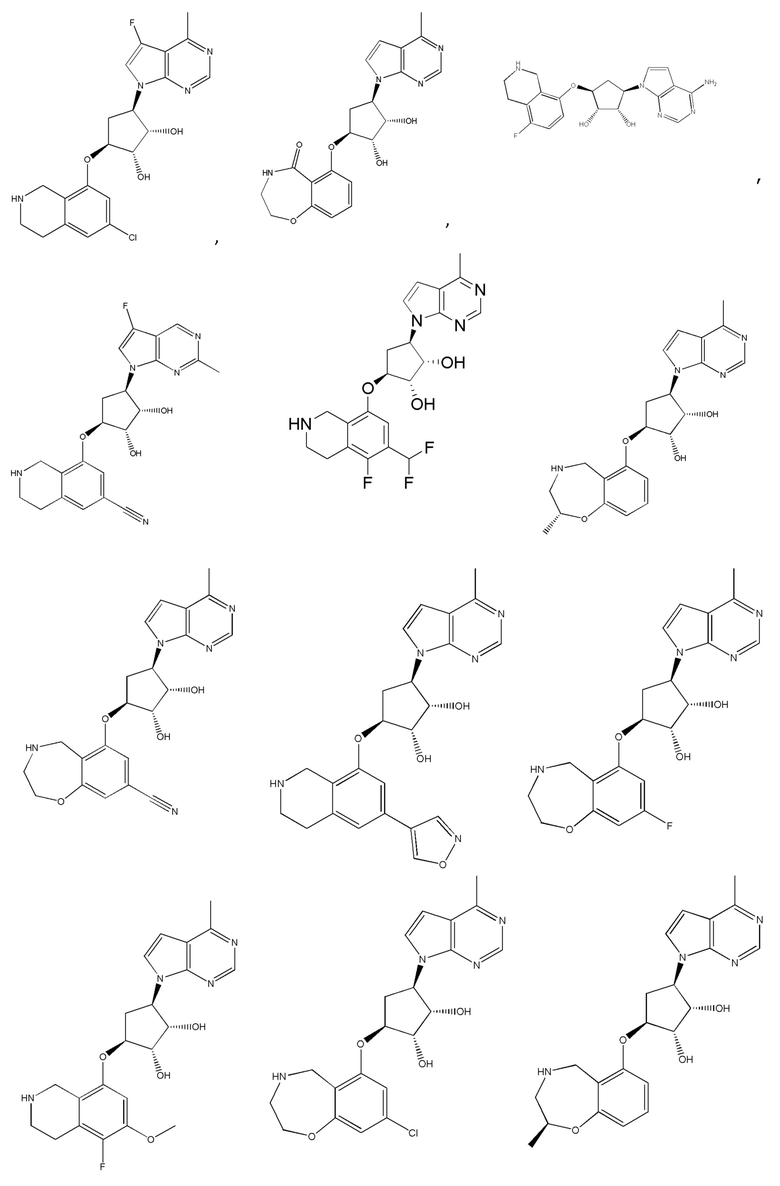
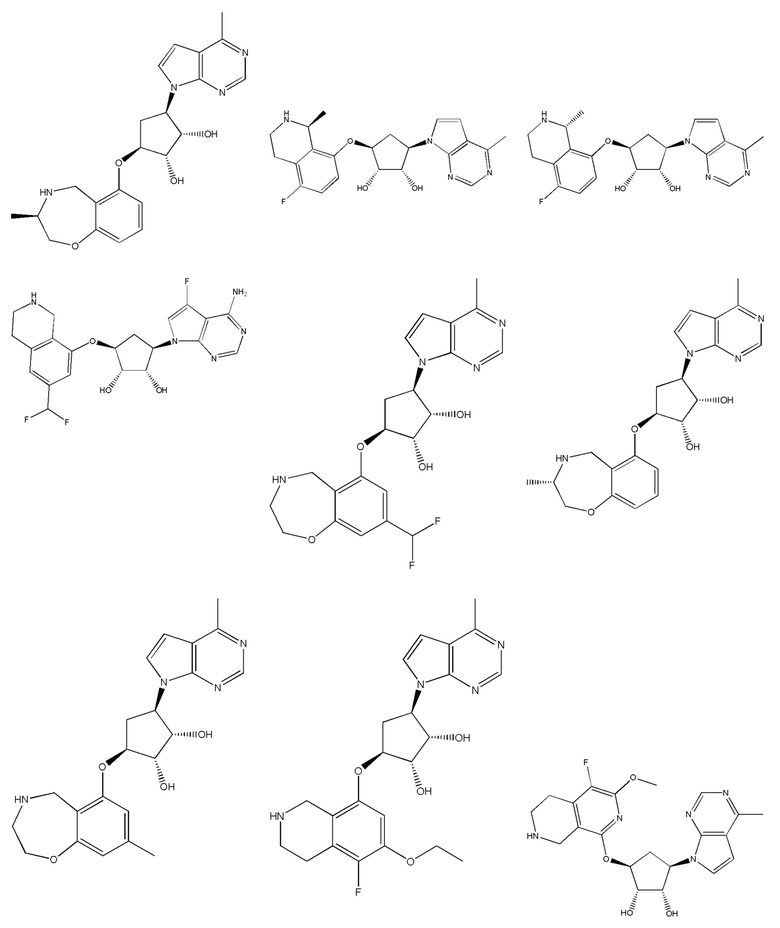
или их фармацевтически приемлемой соли.
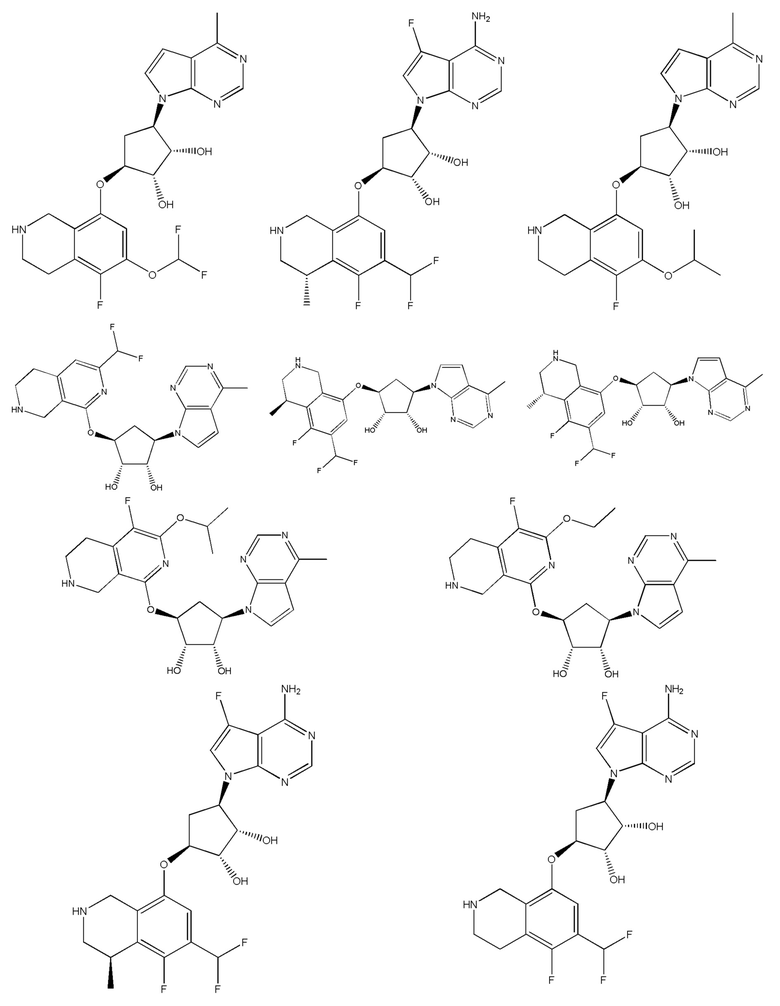
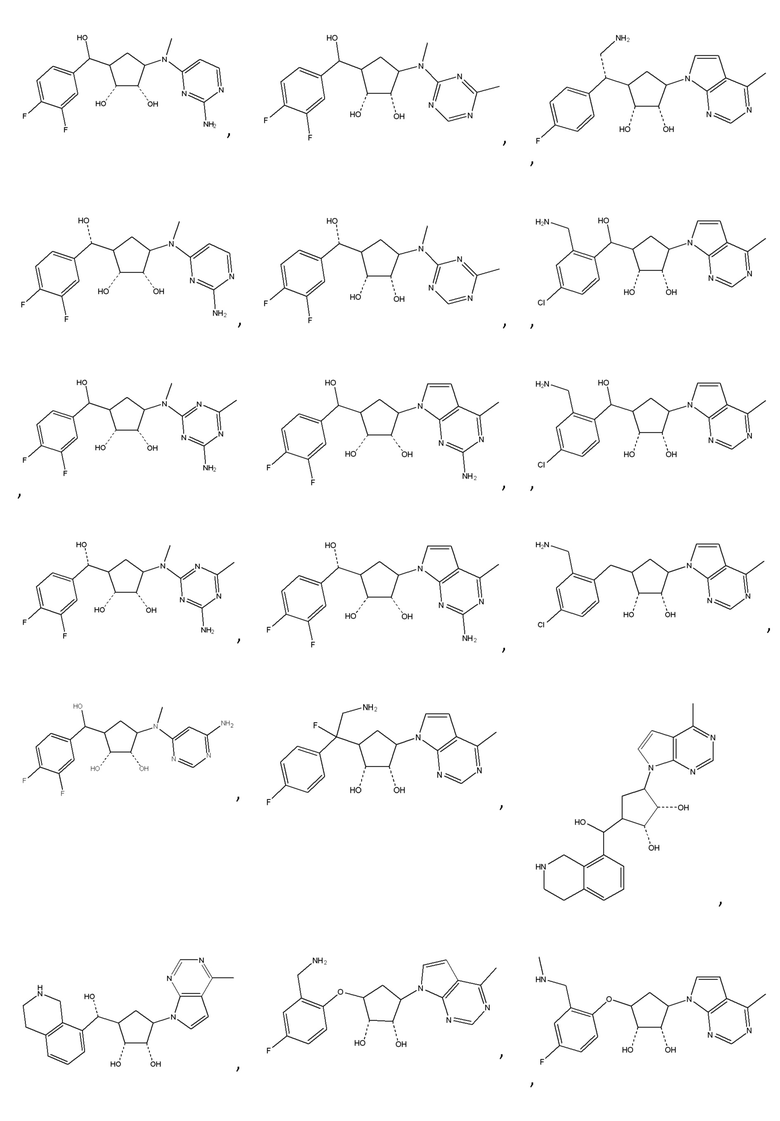
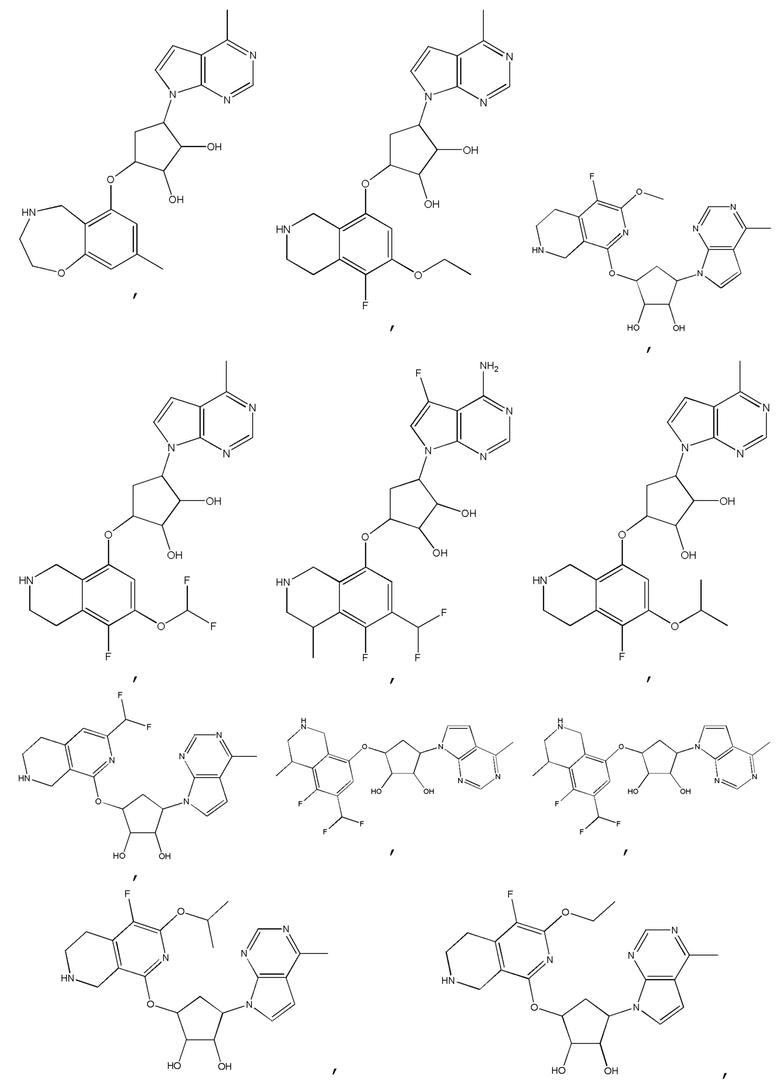
В определенных вариантах данного изобретения соединение выбирают из:
или их фармацевтически приемлемая соль.
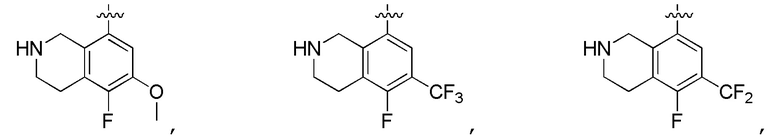
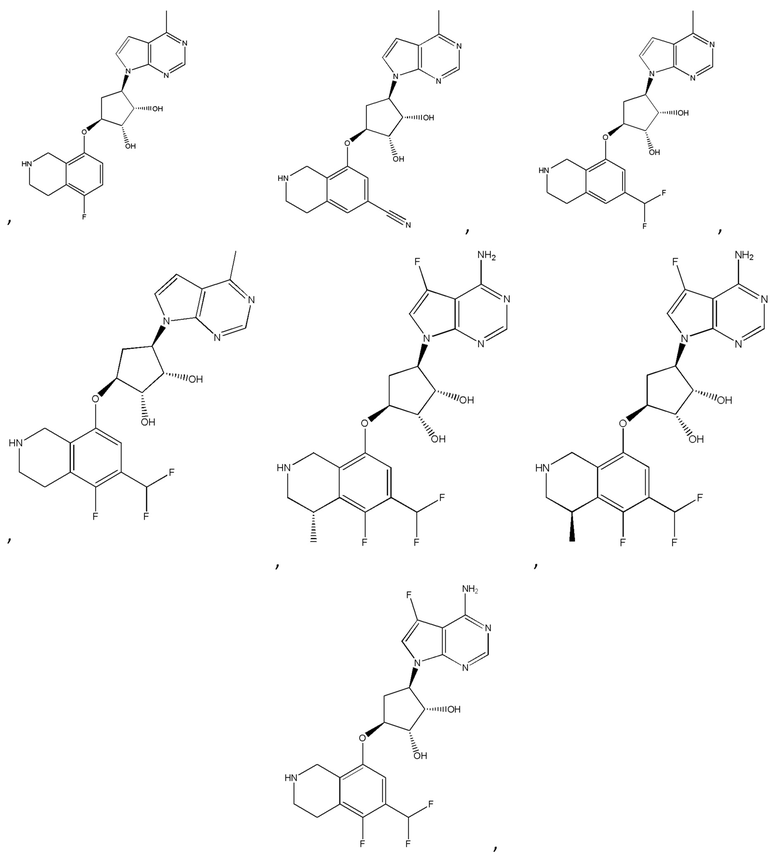
Другие варианты данного изобретения включают соединения, выбранные из:
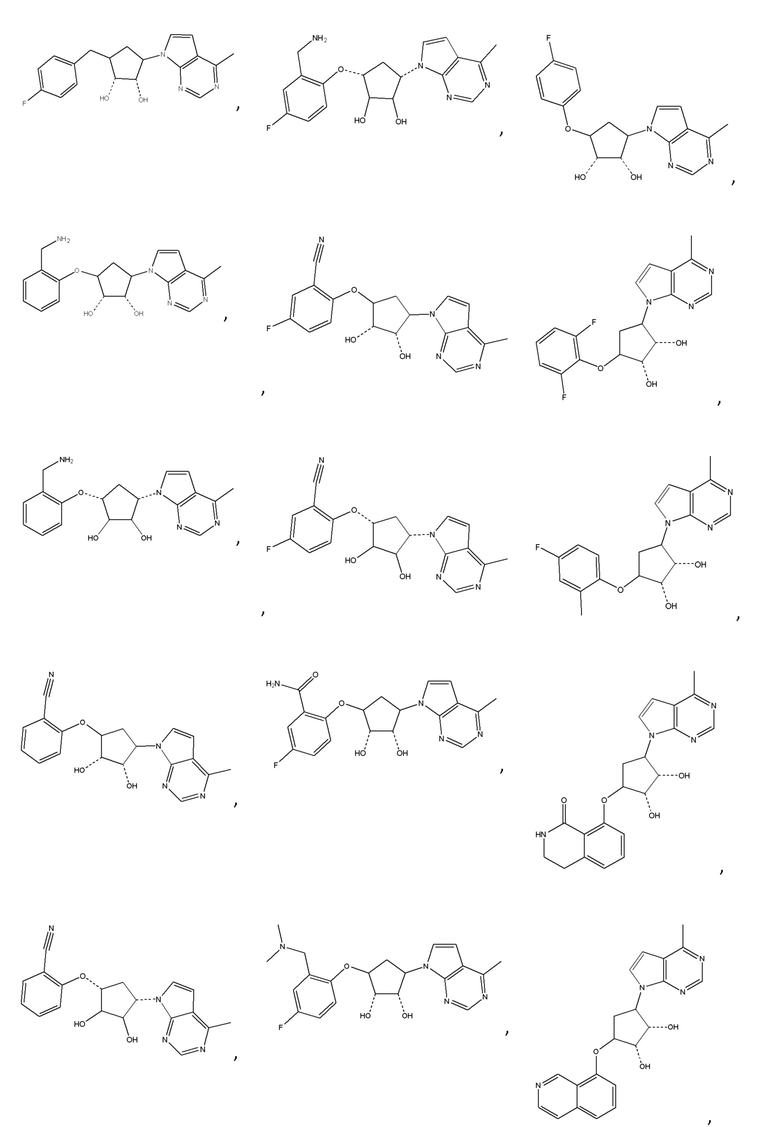
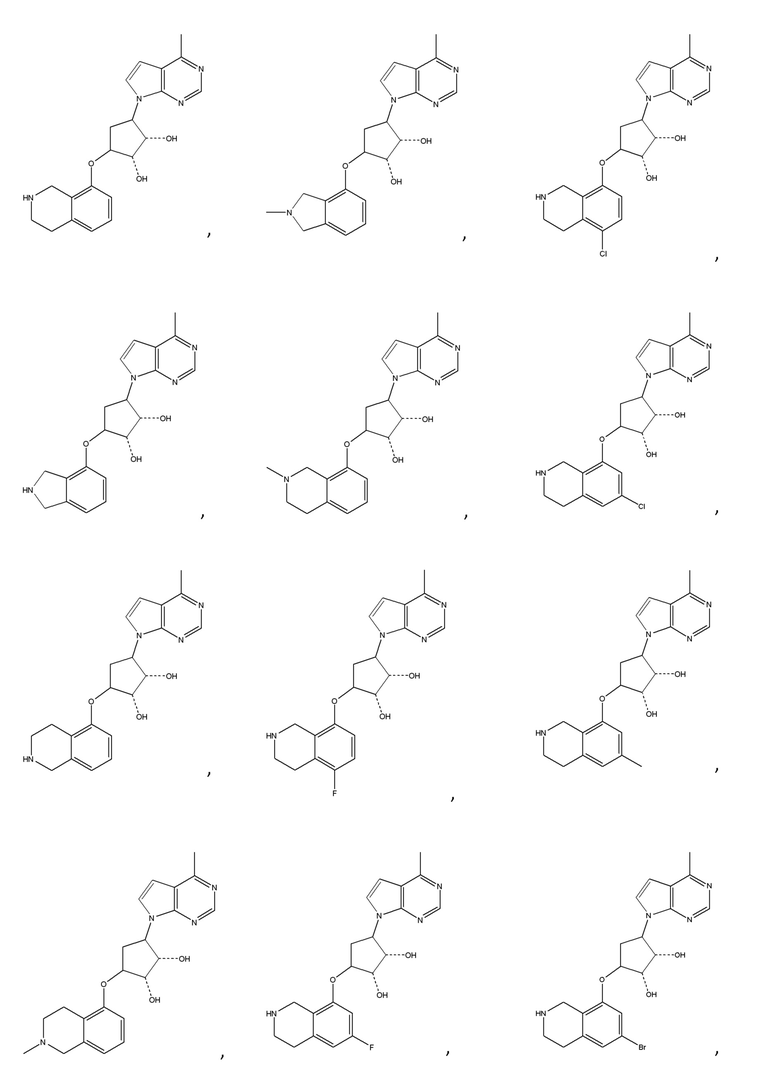
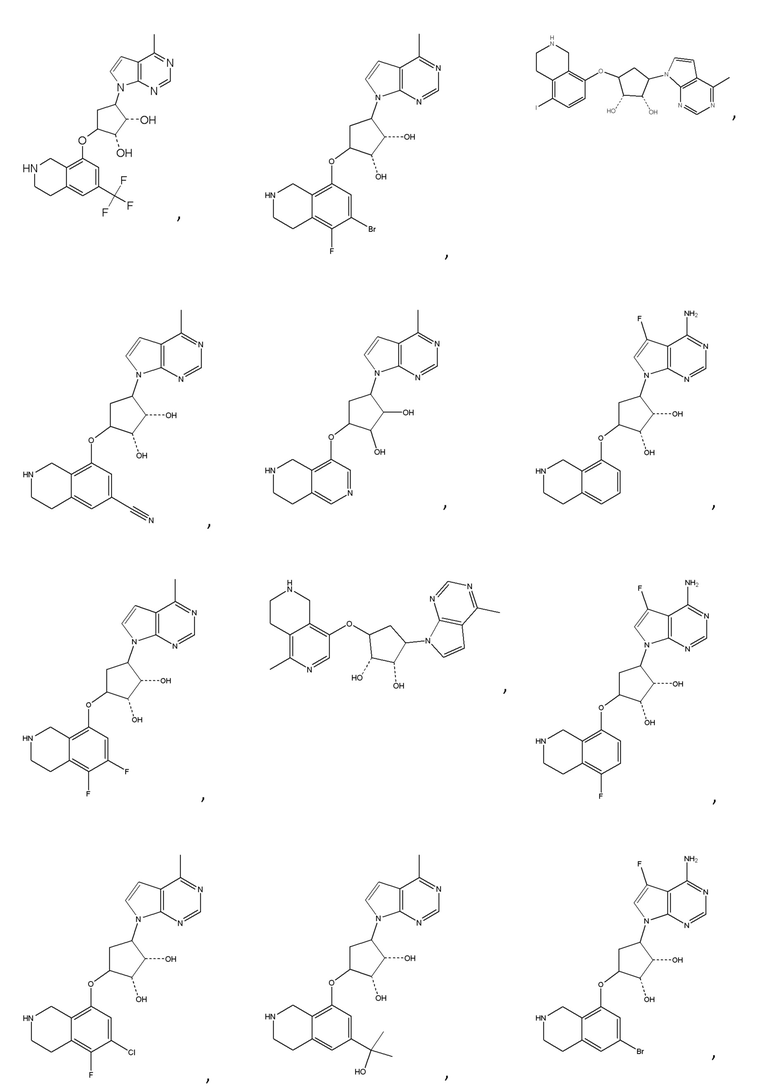
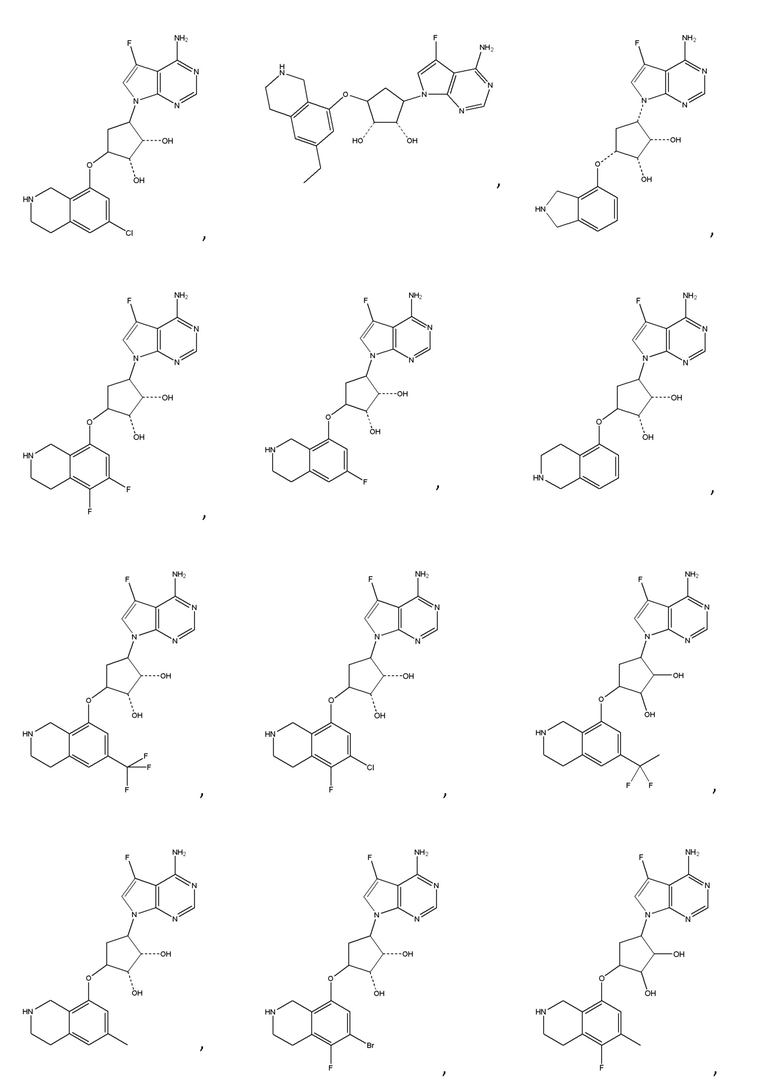
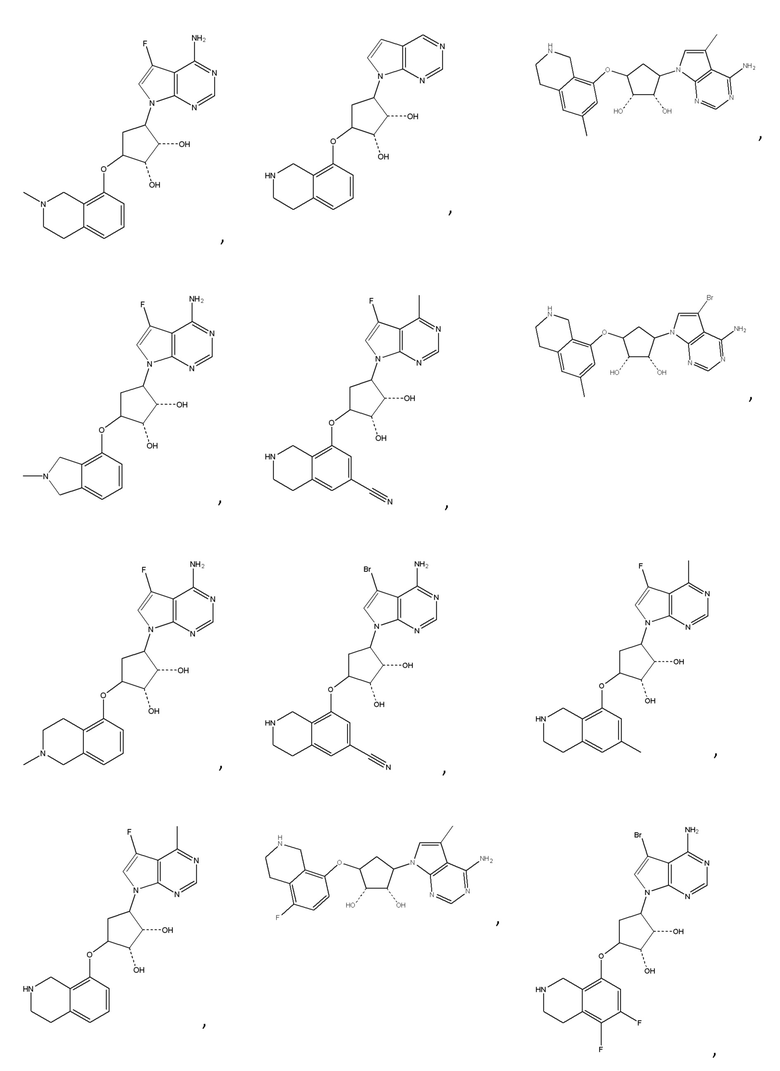
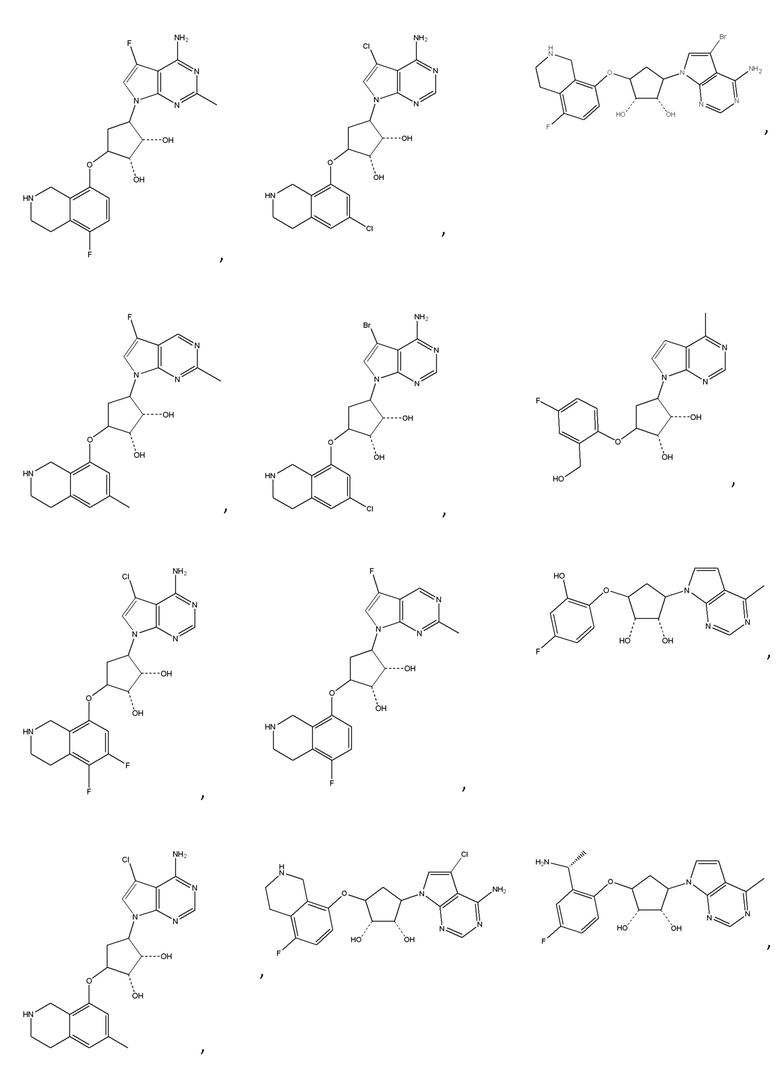
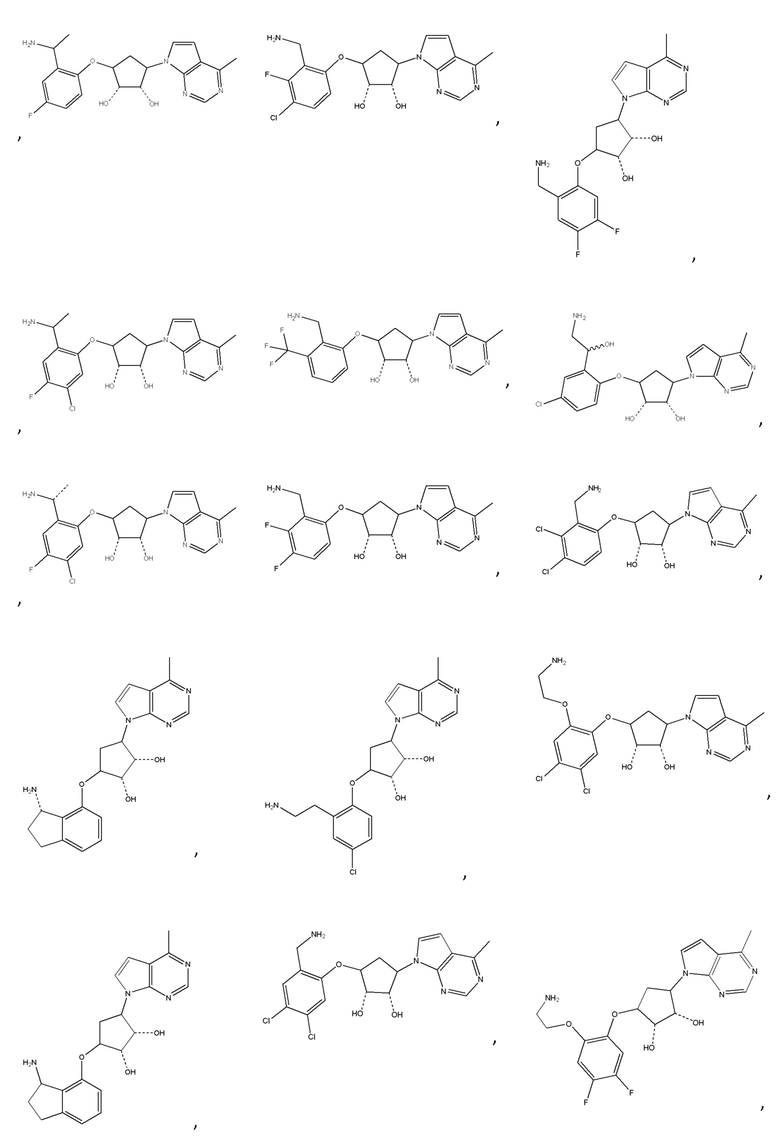
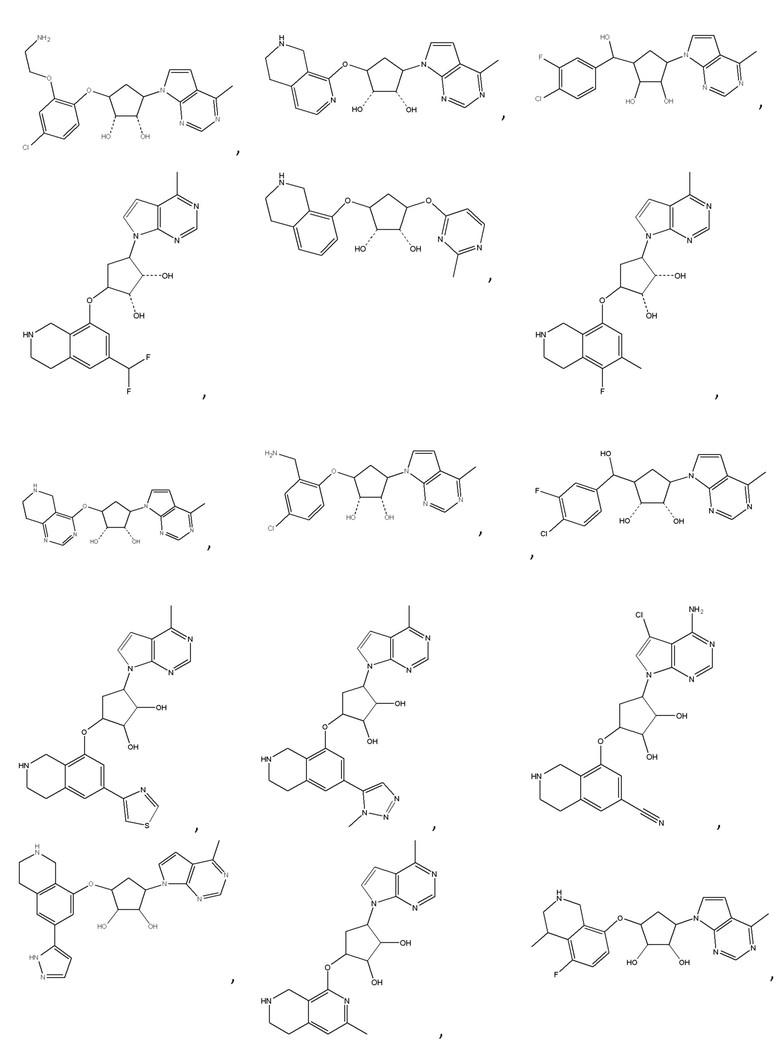
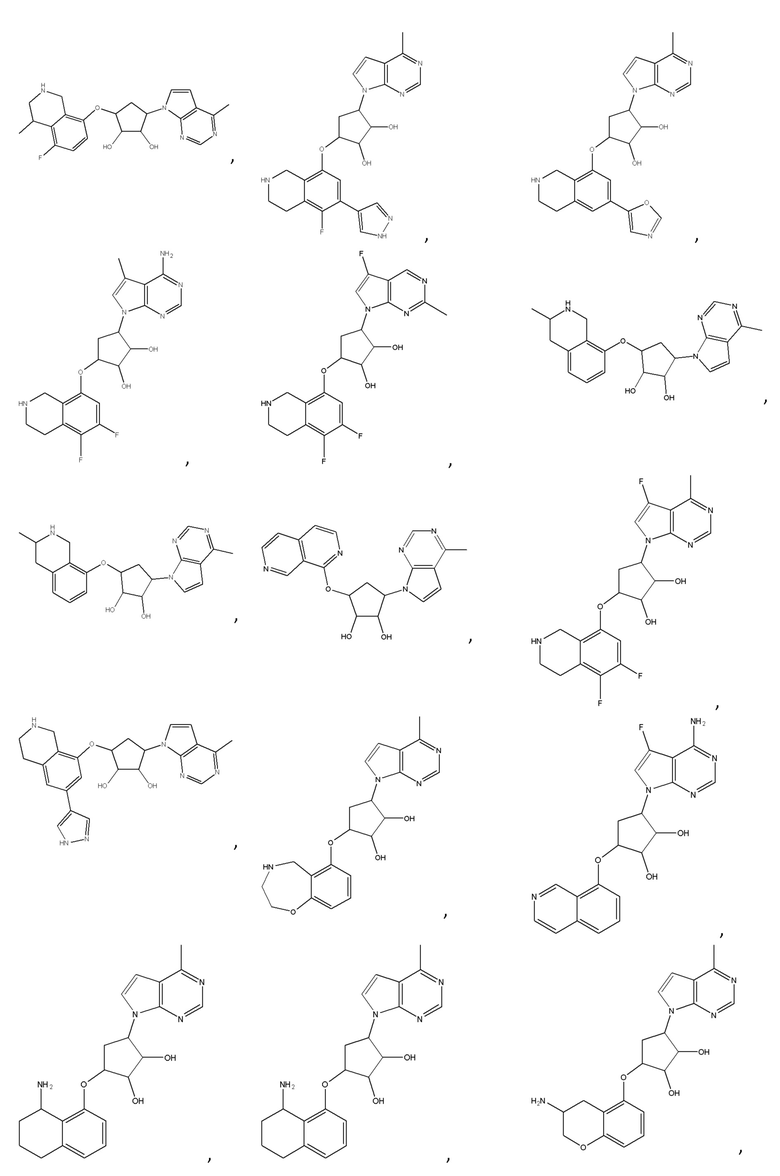
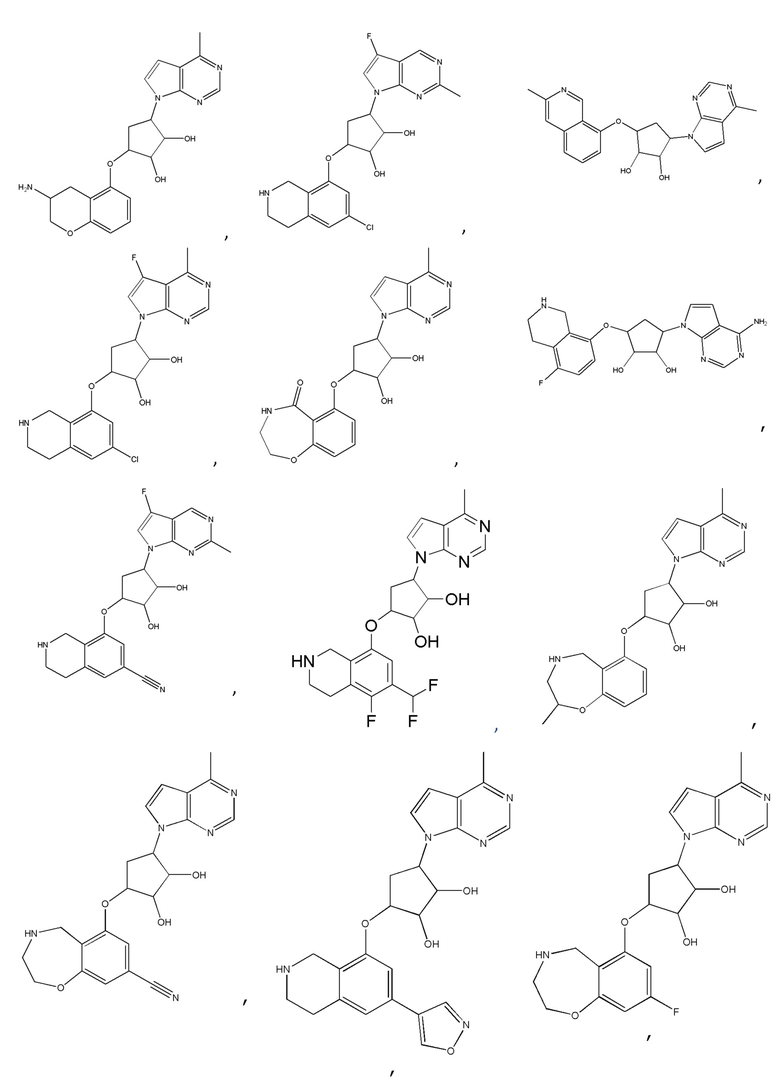
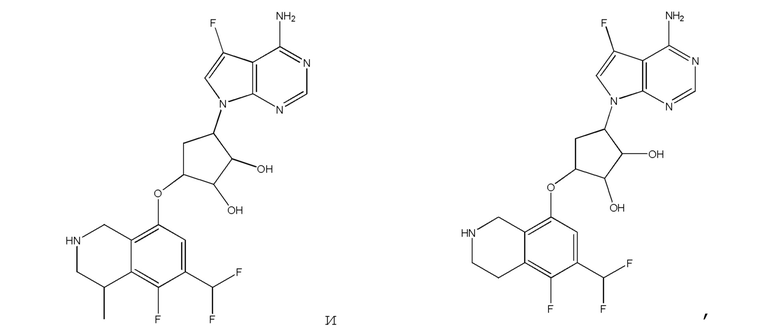
или их фармацевтически приемлемая соль.
Также в соответствии с данным изобретением представлены фармацевтические композиции, содержащие описанное здесь соединение и фармацевтически приемлемый носитель.
Дополнительно в данном изобретении представлены способы лечения аномального роста клеток у млекопитающих, включающие введение млекопитающему терапевтически эффективного количества описанного здесь соединения. Аномальным ростом клеток является рак. Описанным здесь раком может быть рак легких, рак костей, рак поджелудочной железы, рак кожи, рак головы или шеи, кожная или внутриглазная меланома, рак матки, рак яичников, рак прямой кишки, рак анальной области, рак желудка, рак толстой кишки, рак молочной железы, рак матки, рак маточных труб, рак эндометрия, рак шейки матки, рак влагалища, рак вульвы, болезнь Ходжкина, рак пищевода, рак тонкой кишки, рак эндокринной системы, рак эндокринной системы, рак щитовидной железы, рак околощитовидной железы, рак надпочечников, саркома мягких тканей, рак мочеиспускательного канала, рак полового члена, рак предстательной железы, хронический или острый лейкоз, лимфоцитарные лимфомы, рак мочевого пузыря, рак почки или мочеточника, почечно-клеточный рак, рак почечной лоханки, новообразования центральной нервной системы (ЦНС), первичная лимфома ЦНС, опухоли позвоночника, глиома ствола головного мозга или аденома гипофиза.
Кроме того, в данном изобретении представлено применение описанного здесь соединения для приготовления лекарственного средства для лечения аномального роста клеток у млекопитающего. Аномальным ростом клеток является рак. Описанным здесь раком может быть рак легких, рак костей, рак поджелудочной железы, рак кожи, рак головы или шеи, кожная или внутриглазная меланома, рак матки, рак яичников, рак прямой кишки, рак анальной области, рак желудка, рак толстой кишки, рак молочной железы, рак матки, рак маточных труб, рак эндометрия, рак шейки матки, рак влагалища, рак вульвы, болезнь Ходжкина, рак пищевода, рак тонкой кишки, рак эндокринной системы, рак эндокринной системы, рак щитовидной железы, рак околощитовидной железы, рак надпочечников, саркома мягких тканей, рак мочеиспускательного канала, рак полового члена, рак предстательной железы, хронический или острый лейкоз, лимфоцитарные лимфомы, рак мочевого пузыря, рак почки или мочеточника, почечно-клеточный рак, рак почечной лоханки, новообразования центральной нервной системы (ЦНС), первичная лимфома ЦНС, опухоли позвоночника, глиома ствола головного мозга или аденома гипофиза.
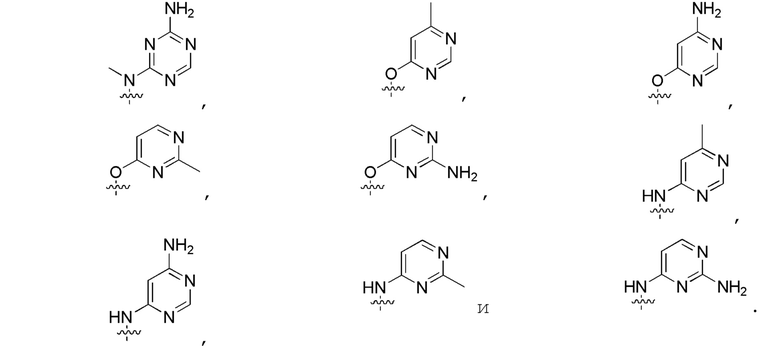
В изобретении также представлены варианты, в которых:
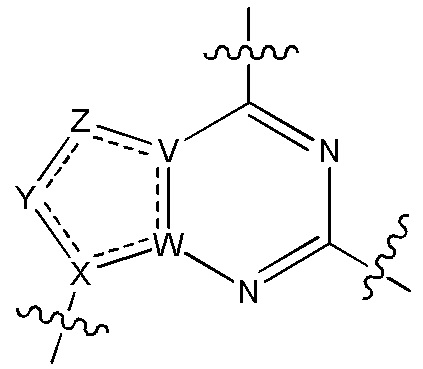
в формуле I или формуле II выбирают из:
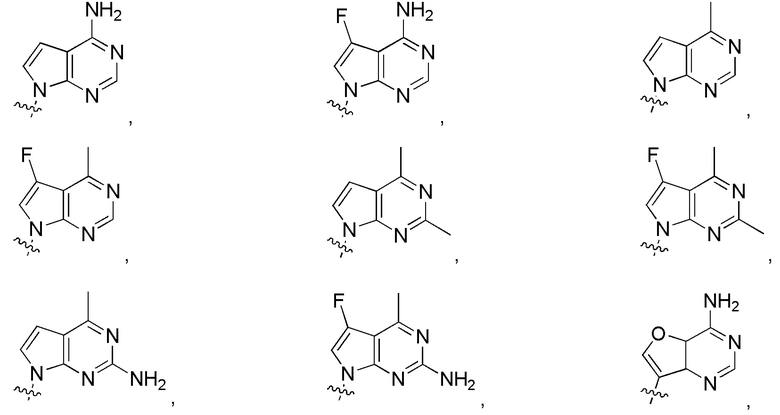
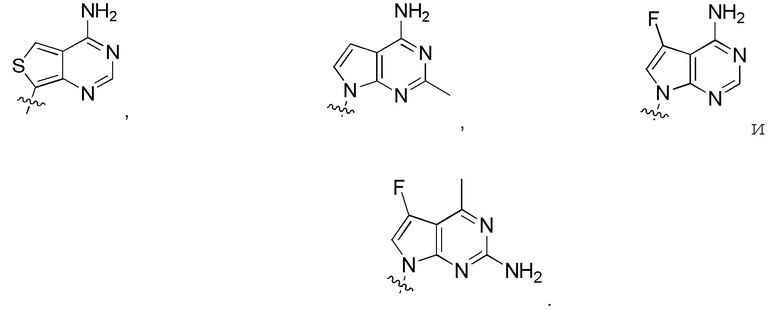
Дополнительные варианты изобретения включают фармацевтическую композицию, содержащую описанное здесь соединение или его фармацевтически приемлемую соль, и фармацевтически приемлемый носитель.
Дополнительные варианты изобретения включают способы лечения аномального роста клеток у млекопитающего, где способ включает введение млекопитающему терапевтически эффективного количества описанного здесь соединения или его фармацевтически приемлемой соли, и фармацевтически приемлемого носителя.
Дополнительные варианты изобретения включают описанные здесь способы лечения, где аномальным ростом клеток является рак. В частности, такие способы, где раком является рак легких, рак костей, рак поджелудочной железы, рак кожи, рак головы или шеи, кожная или внутриглазная меланома, рак матки, рак яичников, рак прямой кишки, рак анальной области, рак желудка, рак толстой кишки, рак молочной железы, рак матки, рак маточных труб, рак эндометрия, рак шейки матки, рак влагалища, рак вульвы, болезнь Ходжкина, рак пищевода, рак тонкой кишки, рак эндокринной системы, рак эндокринной системы, рак щитовидной железы, рак околощитовидной железы, рак надпочечников, саркома мягких тканей, рак мочеиспускательного канала, рак полового члена, рак предстательной железы, хронический или острый лейкоз, лимфоцитарные лимфомы, рак мочевого пузыря, рак почки или мочеточника, почечно-клеточный рак, рак почечной лоханки, новообразования центральной нервной системы (ЦНС), первичная лимфома ЦНС, опухоли позвоночника, глиома ствола головного мозга или аденома гипофиза.
Также представлен вариант данного изобретения, включающий применение описанного здесь соединения или его фармацевтически приемлемой соли для приготовления лекарственного средства, применяемого в лечении аномального роста клеток у млекопитающего, в частности, где аномальным ростом клеток является рак, и более конкретно, где раком является рак легких, рак костей, рак поджелудочной железы, рак кожи, рак головы или шеи, кожная или внутриглазная меланома, рак матки, рак яичников, рак прямой кишки, рак анальной области, рак желудка, рак толстой кишки, рак молочной железы, рак матки, рак маточных труб, рак эндометрия, рак шейки матки, рак влагалища, рак вульвы, болезнь Ходжкина, рак пищевода, рак тонкой кишки, рак эндокринной системы, рак эндокринной системы, рак щитовидной железы, рак околощитовидной железы, рак надпочечников, саркома мягких тканей, рак мочеиспускательного канала, рак полового члена, рак предстательной железы, хронический или острый лейкоз, лимфоцитарные лимфомы, рак мочевого пузыря, рак почки или мочеточника, почечно-клеточный рак, рак почечной лоханки, новообразования центральной нервной системы (ЦНС), первичная лимфома ЦНС, опухоли позвоночника, глиома ствола головного мозга или аденома гипофиза.
В другом варианте в изобретении представлена фармацевтическая композиция, содержащая соединение в соответствии с данным изобретением или его фармацевтически приемлемую соль, и фармацевтически приемлемый носитель.
В другом варианте, в изобретении представлен способ лечения аномального роста клеток у млекопитающего, включая человека, где способ включает введение млекопитающему терапевтически эффективного количества описанного здесь соединения или его фармацевтически приемлемой соли. В другом варианте, аномальным ростом клеток является рак. В другом варианте, раком является рак легких, рак костей, рак поджелудочной железы, рак кожи, рак головы или шеи, кожная или внутриглазная меланома, рак матки, рак яичников, рак прямой кишки, рак анальной области, рак желудка, рак толстой кишки, рак молочной железы, рак матки, рак маточных труб, рак эндометрия, рак шейки матки, рак влагалища, рак вульвы, болезнь Ходжкина, рак пищевода, рак тонкой кишки, рак эндокринной системы, рак эндокринной системы, рак щитовидной железы, рак околощитовидной железы, рак надпочечников, саркома мягких тканей, рак мочеиспускательного канала, рак полового члена, рак предстательной железы, хронический или острый лейкоз, лимфоцитарные лимфомы, рак мочевого пузыря, рак почки или мочеточника, почечно-клеточный рак, рак почечной лоханки, новообразования центральной нервной системы (ЦНС), первичная лимфома ЦНС, опухоли позвоночника, глиома ствола головного мозга или аденома гипофиза.
Определения
Если не указано иначе, в описании и формуле изобретения применяют следующие термины, имеющие описанные ниже значения. Переменные, определенные в этом разделе, такие как R, X, n и подобные, даны для ссылки только в этом разделе и не подразумевается, что они имеют те же значения при применении вне этого раздела описания. Также, многие определенные здесь группы могут быть необязательно замещены. Перечисление в этом разделе определений типовых заместителей является примерным и не ограничивает заместители, определенные где-либо еще в этом описании и формуле изобретения.
"Алкенил" относится к алкильное группе, определенной здесь, состоящей из, по крайней мере, одну двойную связь углерод-углерод. Типовые примеры включают, но не ограничены ими, этенил, 1-пропенил, 2-пропенил, 1-, 2- или 3-бутенил и подобные. ʺАлкениленʺ относится к двухвалентной форме алкенила.
ʺАлкоксиʺ относится к -O-алкилу, где алкилом предпочтительно является C1-C8, C1-C7, C1-C6, C1-C5, C1-C4, C1-C3, C1-C2 или C1алкил.
"Алкил" относится к насыщенному алифатическому углеводородному радикалу, включающему группы с прямой цепью и разветвленной цепью, содержащие от 1 до 20 атомов углерода. (ʺ(C1-C20)алкилʺ), предпочтительно, от 1 до 12 атомов углерода (ʺ(C1-C12)алкилʺ), более предпочтительно, от 1 до 8 атомов углерода (ʺ(C1-C8)алкилʺ) или от 1 до 6 атомов углерода (ʺ(C1-C6)алкилʺ) или от 1 до 4 атомов углерода (ʺ(C1-C4)алкилʺ). Примеры алкильных групп включают метил, этил, пропил, 2-пропил, н-бутил, изо-бутил, трет-бутил, пентил, неопентил и подобные. Алкил может быть замещенным или не замещенным. Типовые заместители включают циклоалкил, арил, гетероарил, гетероалицикл, гидрокси, алкокси, арилокси, меркапто, алкилтио, арилтио, циано, галоген, карбонил, тиокарбонил, O-карбамил, N-карбамил, O-тиокарбамил, N-тиокарбамил, C-амидо, N-амидо, C-карбокси, O-карбокси, нитро, силил, амино и -NRxRy, где Rx и Ry являются, например, водородом, алкилом, циклоалкилом, арилом, карбонилом, ацетилом, сульфонилом, трифторметансульфонилом и, объединенные, пяти- или шестичленным гетероалициклическим кольцом. ʺГалоалкилʺ, например, (C1-C8)галоалкил, относится к алкилу, имеющему один или более галогеновых заместителей. ʺАлкиленʺ относится к двухвалентной форме алкила.
"Алкинил" относится к алкильной группе, определенной здесь, включающей, по крайней мере, два атома углерода и, по крайней мере, одну тройную связь углерод-углерод. Типовые примеры включают, но не ограничены ими, этинил, 1-пропинил, 2-пропинил, 1-, 2- или 3-бутинил и подобные. ʺАлкиниленʺ относится к двухвалентной форме алкинила.
"Амино" относится к -NRxRy группе, где Rx и Ry оба являются водородом.
"(C6-C12)арил" относится к полностью углеродным моноциклическим или конденсированным кольцевым полициклическим группам, содержащим от 6 до 12 атомов углерода, имеющим полностью конъюгированную пи-электронную систему. Также, "(C5-C12)арил" относится к полностью углеродным моноциклическим или конденсированным кольцевым полициклическим группам, содержащим от 5 до 12 атомов углерода, имеющим полностью конъюгированную пи-электронную систему. Примеры, без ограничений, арильных групп включают фенил, нафталинил и антраценил. Арильная группа может быть замещена или не замещена. Типовые заместители включают галоген, тригалометил, алкил, гидрокси, алкокси, арилокси, меркапто, алкилтио, арилтио, циано, нитро, карбонил, тиокарбонил, C-карбокси, O-карбокси, O-карбамил, N-карбамил, O-тиокарбамил, N-тиокарбамил, C-амидо, N-амидо, сульфинил, сульфонил, амино и -NRxRy, где Rx и Ry такие, как определены выше.
"Циано" относится к -C≡N группе. Циано может быть обозначена как CN.
"(C3-C10)циклоалкил" относится к 3-10-членному полностью углеродному моноциклическому кольцу, -10-членному полностью углеродному бициклическому кольцу, полностью углеродному 5-членному/6-членному или 6-членному/6-членному конденсированному бициклическому кольцу, мультициклическому конденсированному кольцу (ʺконденсированнаяʺ кольцевая система означает, что каждое кольцо в системе имеет общую соседнюю пару атомов углерода и каждым другим кольцом в системе), где одно или более колец может содержать одну или более двойных связей, но ни одно из колец не имеет полностью конъюгированной пи-электронной системы, и мостиковую полностью углеродную систему. Примеры, без ограничений, циклоалкильных групп включают циклопропан, циклобутан, циклопентан, циклопентен, циклогексан, циклогексадиен, адамантан, циклогептан, циклогептатриен и подобные. Циклоалкильная группа может быть замещена или не замещена. Типовые заместители включают алкил, арил, гетероарил, гетероалицикл, гидрокси, алкокси, арилокси, меркапто, алкилтио, арилтио, циано, гало, карбонил, тиокарбонил, C-карбокси, O-карбокси, O-карбамил, N-карбамил, C-амидо, N-амидо, нитро, амино и -NRxRy, где Rx и Ry такие, как определены выше.
ʺГалогенʺ или префикс ʺгалоʺ относится к фтору, хлору, брому и йоду. Предпочтительно, галоген относится к фтору или хлору.
ʺГетероалкил" относится к алкильной группе с прямой цепью или разветвленной цепью, содержащей 1-20 атомов углерода, предпочтительно, 1-12 атомов углерода, более предпочтительно, 1-8 атомов углерода или 1-6 атомов углерода или 1-4 атома углерода, где один, два или три атома углерода замещены гетероатомом, выбранным из NRx, N, O, и S(O)n (где n равно 0, 1 или 2). Обычно гетероатомы, если имеется более одного гетероатома, не являются соседними друг с другом. Типовые гетероалкилы включают сложные эфиры алкила, вторичные и третичные алкиламины, амиды, алкил сульфиды и подобные. Группой может быть концевая группа или мостиковая группа. В данном описании ссылка на нормальную цепь при применении в контексте мостиковой группы, относится к прямой цепи из атомов, связывающей два концевых положения мостиковой группы. Для ʺалкилаʺ, типовые заместители на ʺгетероалкилеʺ включают циклоалкил, арил, гетероарил, гетероалицикл, гидрокси, алкокси, арилокси, меркапто, алкилтио, арилтио, циано, галоген, карбонил, тиокарбонил, O-карбамил, N-карбамил, O-тиокарбамил, N-тиокарбамил, C-амидо, N-амидо, C-карбокси, O-карбокси, нитро, силил, амино и -NRxRy, где Rx и Ry являются, например, водородом, алкилом, циклоалкилом, арилом, карбонилом, ацетилом, сульфонилом, трифторметансульфонилом и, объединенные, пяти- или шестичленным гетероалициклическим кольцом. ʺГетероалкенилʺ относится к гетероалкилу, имеющему одну или более двойных связей углерод-углерод. ʺГетероалкиленʺ относится к двухвалентной форме гетероалкила. ʺГетероалкениленʺ относится к двухвалентной форме гетероалкенила.
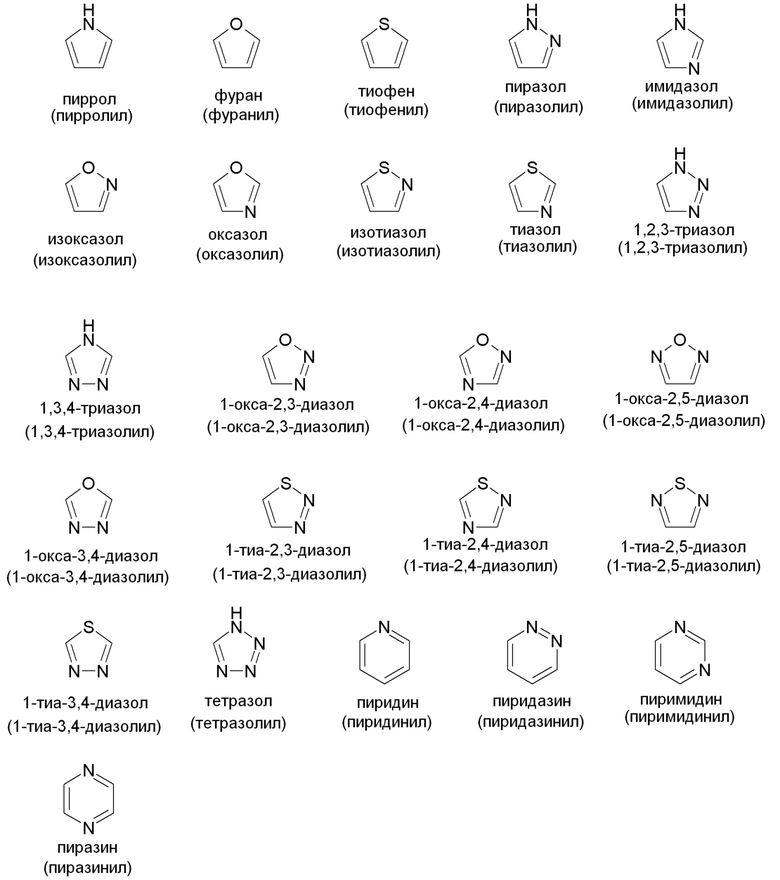
"Гетероарил" относится к моноциклической или конденсированной кольцевой группе, содержащей 5-12 атомов углерода в кольце, содержащей один, два, три или четыре гетероатома, выбранных из NRx, N, O и S(O)n (где n равно 0, 1 или 2) и, кроме того, имеющей полностью конъюгированную пи-электронную систему. Предпочтительные гетероарильные группы включают (C2-C7)гетероарил в соответствии с указанным выше определением. Примеры, без ограничений, незамещенных гетероарильных групп включают пиррол, фуран, тиофен, имидазол, оксазол, тиазол, пиразол, пиридин, пиримидин, хинолин, изохинолин, пурин, тетразол, триазин и карбазол. Гетероарильная группа может быть замещена или не замещена. Типовые заместители включают алкил, циклоалкил, гало, тригалометил, гидрокси, алкокси, арилокси, меркапто, алкилтио, арилтио, циано, нитро, карбонил, тиокарбонил, сульфонамидо, C-карбокси, O-карбокси, сульфинил, сульфонил, O-карбамил, N-карбамил, O-тиокарбамил, N-тиокарбамил, C-амидо, N-амидо, амино и -NRxRy, где Rx и Ry такие, как определены выше. Фармацевтически приемлемым гетероарилом является такой, который достаточно стабилен для присоединения к соединению в соответствии с данным изобретением, составления в фармацевтическую композицию и последующего введения пациенту, нуждающемуся в таковом. Примеры типовых моноциклических гетероарильных групп включают, но не ограничены ими:
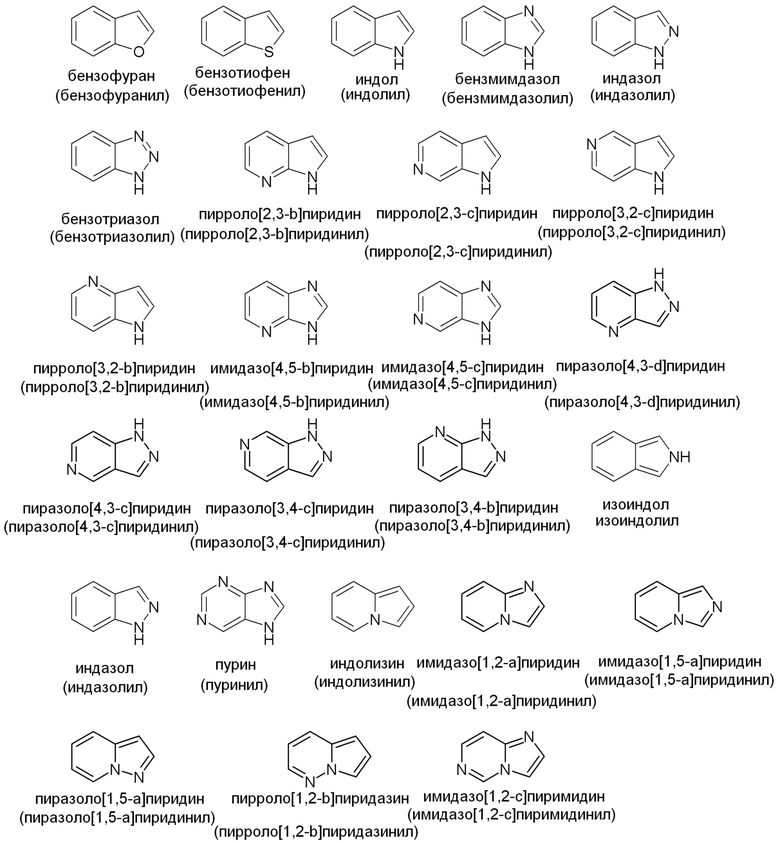
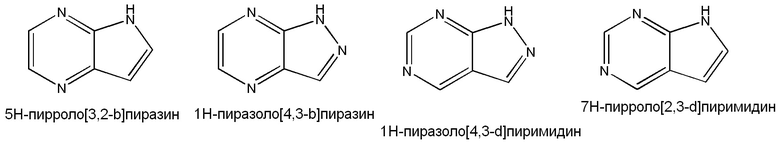
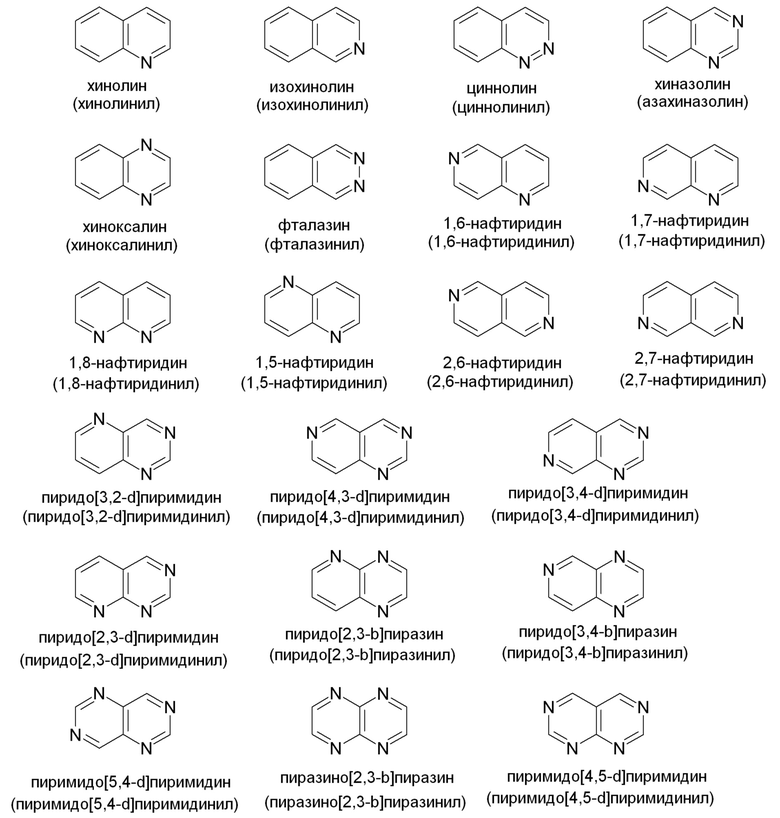
Примеры подходящих конденсированных кольцевых гетероарильных групп включают, но не ограничены ими:
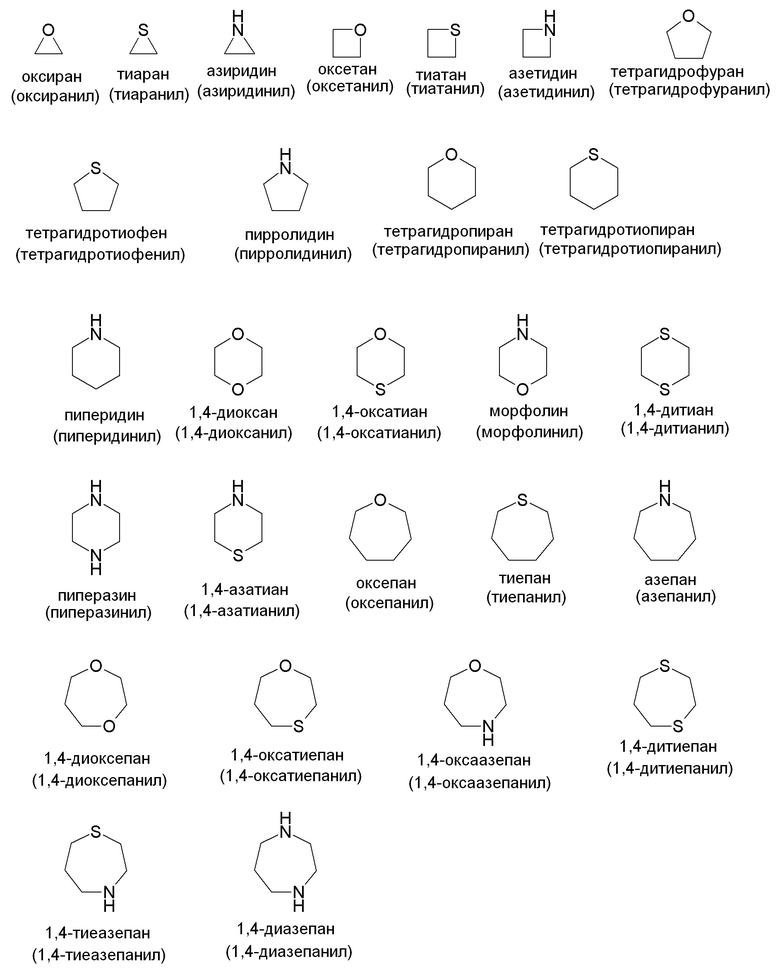
"Гетероциклилʺ относится к моноциклической или конденсированной кольцевой системе, содержащей 3-12 атомов в кольце, содержащей один, два, три или четыре гетероатома, выбранных из N, O и S(O)n (где n равно 0, 1 или 2), и 1-9 атомов углерода. Кольца также могут иметь одну или более двойных связей. Однако кольца не имеют полностью конъюгированную пи-электронную систему. Предпочтительные гетероциклы включают (C2-C6)гетероциклы в соответствии с представленным выше определением. Примеры подходящих насыщенных гетероалициклических групп включают, но не ограничены ими:
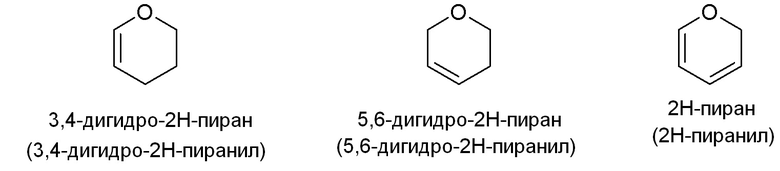
Примеры подходящих частично ненасыщенных гетероалициклических групп включают, но не ограничены ими:
Гетероциклильная группа необязательно замещена одним или двумя заместителями, независимо выбранными из гало, низшего алкила, низшего алкилзамещеного карбокси, сложного эфира гидрокси или моно- или диалкиламино. Более того, гетероцикл может содержать мостик, включая мостик между не соседними атомами углерода на гетероцикле, где мостик содержит 1-2 атома углерода и 0-1 гетероатомов, выбранных из NRx, O и S(O)n (где n равно 0, 1 или 2).
"Гидрокси" или ʺгидроксилʺ относится к -OH группе.
"In vitro" относится к методикам, проводимым в искусственной среде, такой как, например, без ограничений, в пробирке для тестирования или культуральной среде.
"In vivo" относится к методикам, проводимым в живом организме, таком как, без ограничений, мышь, крыса или кролик.
"Необязательный" или "необязательно" означает, что описываемое далее событие или обстоятельство может, но не обязательно случается, и что описание включает случаи, где событие или обстоятельство возникает, и случаи, где не возникает. Например, "гетероциклическая группа, необязательно замещенная алкильной группой" означает, что алкил может, но не обязательно присутствует, и описание включает ситуации, где гетероциклическая группа замещена алкильной группой и ситуации, где гетероциклическая группа не является замещенной алкильной группой.
"Организм" относится к любому живому существу, имеющему, по крайней мере, одну клетку. Живой организм может быть простым как, например, отдельная эукариотическая клетка или сложным, как млекопитающее, включая человека.
"Фармацевтически приемлемый наполнитель" относится к инертному веществу, добавляемому к фармацевтической композиции для облегчения введения соединения. Примеры, без ограничений, наполнителей включают карбонат кальция, фосфат кальция, различные сахара и типы крахмала, производные целлюлозы, желатин, растительные масла и полиэтиленгликоли.
В данном описании термин ʺфармацевтически приемлемая сольʺ относится к таким солям, которые сохраняют биологическую эффективность и свойства исходного соединения. Такие соли включают:
(i) кислотно-аддитивные соли, которые могут быть получены взаимодействием свободного основания исходного соединения с неорганическими кислотами, такими как хлористоводородная кислота, бромистоводородная кислота, азотная кислота, фосфорная кислота, серная кислота и хлорная кислота и подобные, или с органическими кислотами, такими как уксусная кислота, щавелевая кислота, (D) или (L) яблочная кислота, малеиновая кислота, метансульфоновая кислота, этансульфоновая кислота, п-толуолсульфоновая кислота, салициловая кислота, винная кислота, лимонная кислота, янтарная кислота или малоновая кислота и подобные; или
(ii) соли, образованные, когда кислый протон, присутствующий в исходном соединении, либо замещается ионом металла, например, ионом щелочного металла, ионом щелочноземельного металла или ионом алюминия; либо координируется с органическим основанием, таким как этаноламин, диэтаноламин, триэтаноламин, трометамин, N-метилглюкамин и подобные.
"Фармацевтическая композиция" относится к смеси одного или более описанных здесь соединений или их физиологически/фармацевтически приемлемых солей, сольватов, гидратов или пролекарств, с другими химическими компонентами, такими как физиологически/фармацевтически приемлемые носители и наполнители. Целью фармацевтической композиции является облегчение введения соединения в организм.
В данном описании "физиологически/фармацевтически приемлемый носитель" относится к носителю или разбавителю, который не вызывает значительное раздражение в организме и не отменяет биологическое действие и свойства вводимого соединения.
"Терапевтически эффективное количество" относится к такому количеству вводимого соединения, которое облегчает до некоторой степени один или более симптомов лечимого расстройства. При ссылке на лечение рака, терапевтически эффективное количество относится к такому количеству, которое оказывает, по крайней мере, одно из следующих действий:
снижение размера опухоли;
ингибирование (то есть замедление до некоторой степени, предпочтительно, остановку) метастазов опухоли;
ингибирование до некоторой степени (то есть замедление до некоторой степени, предпочтительно, остановку) роста опухоли, и
облегчение до некоторой степени (или, предпочтительно, устранение) одного или более симптомов, связанных с раком.
"Лечить", "лечение" и "обработка" относится к способу облегчения или отмены расстройства, медиированного метилтрансферазой и/или присущих ему симптомов. Что касается конкретно рака, эти термины просто означают, что продолжительность жизни индивидуума с раком увеличивается, или что один или более симптомов заболевания снижается.
Подробное описание
Общие схемы синтеза соединений в соответствии с данным изобретением могут быть найдены в разделе примеров.
Если не указано иначе, все ссылки на соединения в соответствии с данным изобретением включают ссылки на их соли, сольваты, гидраты и комплексы, и на сольваты, гидраты и комплексы солей, включая полиморфы, стереоизомеры и их изотопно меченные версии.
Фармацевтически приемлемые соли включают кислотно-аддитивные и основные соли (включая дисоли). Подходящие кислотно-аддитивные соли получают из кислот, которые образуют не токсичные соли. Примеры включают ацетат, аспартат, бензоат, бесилат, бикарбонат/карбонат, бисульфат/сульфат, борат, камсилат, цитрат, эдисилат, эсилат, формиат, фумарат, глюцептат, глюконат, глюкуронат, гексафторфосфат, гибензат, гидрохлорид/хлорид, гидробромид/бромид, гидройодид/йодид, изетионат, лактат, малат, малеат, малонат, мезилат, метилсульфат, нафтилат, 2-напсилат, никотинат, нитрат, оротат, оксалат, пальмитат, памоат, фосфат/гидрофосфат/дигидрофосфат, сахарат, стеарат, сукцинат, тартрат, тозилат и трифторацетат.
Подходящие основные соли получают из оснований, которые образуют не токсичные соли. Примеры включают соли алюминия, аргинина, бензатина, кальция, холина, диэтиламина, диоламина, глицина, лизина, магния, меглумина, оламина, калия, натрия, трометамина и цинка. Обзор подходящих солей представлен в ʺHandbook of Pharmaceutical Salts: Properties, Selection, and Useʺ by Stahl and Wermuth (Wiley-VCH, Weinheim, Germany, 2002), описание которой включено сюда в качестве ссылки полностью.
Тозилат, гидрохлорид и мезилат являются целевыми солями.
Фармацевтически приемлемая соль соединений в соответствии с данным изобретением может быть легко получена смешиванием растворов соединения и желаемой кислоты или основания, подходящим образом. Соль может выпадать в осадок из раствора и ее собирают фильтрацией или она может быть восстановлена выпариванием растворителя. Степень ионизации в соли может варьироваться от полностью ионизированной до практически не ионизированной.
Соединения в соответствии с данным изобретением могут существовать в несольватированной и сольватированной формах. Термин 'сольват' применяют здесь для описания молекулярного комплекса, содержащего соединение в соответствии с данным изобретением и одну или более молекул фармацевтически приемлемого растворителя, например, этанола. Термин 'гидрат' применяют, если растворителем является вода. Фармацевтически приемлемые сольваты в соответствии с данным изобретением включают гидраты и сольваты, где растворитель кристаллизации может быть изотопно замещен, например, D2O, d6-ацетон, d6-ДМСО.
Также в объем данного изобретения включены комплексы, такие как клатраты, комплексы включения лекарство-хозяин, где, в отличие от указанных выше сольватов, лекарство и хозяин присутствуют в стехиометрических или не стехиометрических количествах. Также включены комплексы лекарственного средства, содержащие два или более органических и/или неорганических компонента, которые могут быть в стехиометрических или не стехиометрических количествах. Полученные комплексы могут быть ионизированы, частично ионизированы или не ионизированы. Обзор таких комплексов представлен в J Pharm Sci, 64 (8), 1269-1288 by Haleblian (August 1975), описание которой включено сюда в качестве ссылки полностью.
Также в объем изобретения включены полиморфы, пролекарства и изомеры (включая оптические, геометрические и таутомерные изомеры) соединений в соответствии с данным изобретением.
Производные соединений в соответствии с данным изобретением, которые сами обладают незначительным или не обладают фармакологическим действием, могут, при введении пациенту, превращаться в заявленные соединения, например, гидролитическим расщеплением. Такие производные называют 'пролекарства'. Дальнейшая информация по применению пролекарств может быть найдена в 'Pro-drugs as Novel Delivery Systems, Vol. 14, ACS Symposium Series (T Higuchi and W Stella) и 'Bioreversible Carriers in Drug Design', Pergamon Press, 1987 (ed. E B Roche, American Pharmaceutical Association), описания которых включены сюда в качестве ссылки полностью.
Пролекарства в соответствии с данным изобретением могут, например, быть получены заменой подходящих функциональных групп, присутствующих в завяленных соединениях, определенными группами, известными специалистам в данной области техники как 'про-группы', описанными, например, в "Design of Prodrugs" by H Bundgaard (Elsevier, 1985), описание которой включено сюда в качестве ссылки полностью.
Некоторые примеры пролекарств в соответствии с данным изобретением включают:
(i) если соединение содержит функциональную группу карбоновой кислоты -(COOH), ее сложного эфира, например, заменой водорода (C1-C8)алкилом;
(ii) если соединение содержит функциональную группу спирта (-OH), его простого эфира, например, заменой водорода (C1-C6)алканоилоксиметила; и
(iii) если соединение содержит функциональную группу первичного или вторичного амино (-NH2 или -NHR, где R≠H), его амида, например, заменой обоих атомов водорода (C1-C10)алканоилом.
Другие примеры замещающих групп в соответствии с представленными выше примерами и примерами других типов пролекарств, могут быть найдены в указанных выше ссылках.
Наконец, определенные заявленные соединения сами могут действовать как пролекарства для других заявленных соединений.
Соединения в соответствии с данным изобретением, содержащие один или более асимметрических атомов углерода, могут существовать как два или более стереоизомеров. Если соединения в соответствии с данным изобретением имеют, по крайней мере, один хиральный центр, они могут, соответственно, существовать в виде энантиомеров. Если соединения имеют два или более хиральных центров, они могут дополнительно существовать в виде диастереомеров. Также, если соединение в соответствии с данным изобретением содержит циклопропильную группу или другую циклическую группу, где существует хиральность, и алкенильную и алкениленовую группу, возможны геометрические цис/транс (или Z/E) изомеры. Если соединение содержит, например, кето или оксимную группу или ароматическую группу, может существовать таутомерный изомеризм ('таутомеризм'). Одно соединение может демонстрировать более одного типа изомеризма.
В объем данного изобретения включены все стереоизомеры, геометрические изомеры и таутомерные формы заявленных соединений, включая соединения, демонстрирующие более одного типа изомеризма, и смеси одного или более из них. Также включены кислотно-аддитивные или основные соли, в которых противоион является оптически активным, например, D-лактат или L-лизин, или рацемическим, например, DL-тартрат или DL-аргинин.
Цис/транс изомеры могут быть разделены обычными методами, хорошо известными специалисту в данной области техники, например, хроматографией и фракционной кристаллизацией.
Обычные методы получения/выделения энантиомеров включают хиральный синтез из подходящего оптически чистого предшественника или разделение рацемата (или рацемата соли или производного) с применением, например, хиральной жидкостной хроматографии высокого давления (ЖХВД) или сверхкритической жидкостной хроматографии (СЖХ).
Альтернативно, рацемат (или рацемический предшественник) может взаимодействовать с подходящим оптически активным соединением, например, спиртом, или, если соединение содержит кислотную или основную группу, кислотой или основанием, таким как винная кислота или 1-фенилэтиламин. Полученная диастереомерная смесь может быть разделена хроматографией и/или фракционной кристаллизацией, и один или оба диастереоизомера превращают в соответствующий чистый энантиомер(ы) методами, хорошо известными специалисту в данной области техники.
Стереоизомерные конгломераты могут быть разделены обычными методами, известными специалисту в данной области техники; см., например, ʺStereochemistry of Organic Compoundsʺ by E L Eliel (Wiley, New York, 1994), описание которой включено сюда в качестве ссылки полностью.
Изобретение также включает изотопно-меченные соединения в соответствии с данным изобретением, в которых один или более атомов замещены атомом, имеющим то же атомное число, но отличающимся атомной массой или массовым числом от атомной массы или массового числа, обычно имеющихся в природе. Примеры изотопов, подходящих для включения в соединения в соответствии с данным изобретением, включают изотопы водорода, такие как 2H и 3H, углерода, такие как 11C, 13C и 14C, хлора, такие как 36Cl, фтора, такие как 18F, йода, такие как 123I и 125I, азота, такие как 13N и 15N, кислорода, такие как 15O, 17O и 18O, фосфора, такие как 32P, и серы, такие как 35S. Определенные изотопно-меченные соединения в соответствии с данным изобретением, например, содержащие радиоактивный изотоп, применяют в исследованиях распределения лекарственного средства и/или субстрата в тканях. Радиоактивные изотопы тритий, 3H, и углерод-14, 14C, особенно полезны для этой цели благодаря простоте их введения и готовым средствам определения. Замещение более тяжелыми изотопами, такими как дейтерий, 2H, может дать определенные терапевтические преимущества благодаря большей метаболической стабильности, например, увеличенный период полувыведения in vivo или пониженная доза, и, следовательно, могут быть предпочтительны в определенных обстоятельствах. Замещение позитронно-активными изотопами, такими как Substitution 11C, 18F, 15O и 13N, может применяться в позитронно-эмиссионной томографии (ПЭТ) для исследования степени занятости рецептора субстратом.
Изотопно-меченные соединения в соответствии с данным изобретением обычно получают обычными методами, известными специалисту в данной области техники, или способами, аналогичными тем, которые описаны здесь, с применением подходящего изотопно-меченного реагента вместо не меченного реагента, применяемого в других случаях.
Фармацевтически приемлемые сольваты в соответствии с данным изобретением включают такие, в которых растворитель кристаллизации может быть замещен изотопом, например, D2O, d6-ацетон, d6-ДМСО.
Соединения в соответствии с данным изобретением, предназначенные для фармацевтического применения, могут вводиться в виде кристаллических или аморфных или их смесей. Они могут быть получены, например, в виде твердых прессованных масс, порошков или пленок, способами, такими как осаждение, кристаллизация, сушка вымораживанием, сушка распылением или сушка выпариванием. Микроволновая или радиочастотная сушка может применяться для этой цели.
Соединения могут вводиться отдельно или в сочетании с одним или более соединениями в соответствии с данным изобретением. В общем, их вводят в виде композиции в сочетании с одним или более фармацевтически приемлемыми наполнителями. Термин ʺнаполнительʺ применяют здесь для описания любого ингредиента, отличного от соединения(ий) в соответствии с данным изобретением. Выбор наполнителя в значительной степени зависит от таких факторов, как конкретный способ введения, действие наполнителя на растворимость и стабильность, и природу лекарственной формы.
Фармацевтические композиции, подходящие для доставки соединений в соответствии с данным изобретением, и способы из получения очевидны специалистам в данной области техники. Такие композиции и способы их получения могут быть найдены, например, в 'Remington's Pharmaceutical Sciences', 19th Edition (Mack Publishing Company, 1995), описание которой включено сюда в качестве ссылки полностью.
Пероральное введение: Соединения в соответствии с данным изобретением могут вводиться перорально. Пероральное введение может включать проглатывание, при этом соединение попадает в желудочно-кишечный тракт, или может применяться буккальное или подъязычное введение, при котором соединение попадает в кровоток непосредственно изо рта.
Композиции, подходящие для перорального введения, включают твердые композиции, такие как таблетки, капсулы, содержащие порошки, жидкости или порошки, пастилки (включая заполненные жидкостью), жевательные формы, мульти- или нанопорошки, гели, твердые растворы, липосомы, пленки (включая мукоадгезивные), капсулы, спреи и жидкие композиции.
Жидкие композиции включают суспензии, растворы, сиропы и эликсиры. Такие композиции могут применяться в мягких или твердых капсулах и обычно включают носитель, например, воду, этанол, полиэтиленгликоль, пропиленгликоль, метилцеллюлозу или подходящее масло, и один или более эмульгирующих агентов и/или суспендирующих агентов. Жидкие композиции также могут быть получены восстановлением твердого вещества, например, из саше.
Соединения в соответствии с данным изобретением также могут применяться в быстрорастворимых, рассасываемых лекарственных формах, таких как описаны в Expert Opinion in Therapeutic Patents, 11 (6), 981-986 by Liang and Chen (2001), описание которой включено сюда в качестве ссылки полностью.
Для таблетированных лекарственных форм, в зависимости от дозы, лекарственное средство может составлять от 1% масс. до 80% масс. лекарственной формы, более предпочтительно, от 5% масс. до 60% масс. лекарственной формы. В дополнение к лекарственному средству, таблетки обычно содержат разрыхлитель. Примеры разрыхлителей включают гликолят крахмала натрия, карбоксиметилцеллюлозу натрия, карбоксиметилцеллюлозу кальция, кроскармеллозу натрия, кросповидон, поливинилпирролидон, метилцеллюлозу, микрокристаллическую целлюлозу, низшую или алкилзамещенную гидроксипропилцеллюлозу, крахмал, прежелатинизированный крахмал и альгинат натрия. Обычно разрыхлитель составляет от 1% масс. до 25% масс., предпочтительно, от 5% масс. до 20% масс. лекарственной формы.
Связующие вещества обычно применяют для улучшения связующих свойств таблетированной композиции. Подходящие связующие агенты включают микрокристаллическую целлюлозу, желатин, сахара, полиэтиленгликоль, природные и синтетические камеди, поливинилпирролидон, прежелатинизированный крахмал, гидроксипропилцеллюлозу и гидроксипропилметилцеллюлозу. Таблетки также могут содержать разбавители, такие как лактоза (моногидрат, высушенный распылением моногидрат, безводную и т.д.), маннит, ксилит, декстрозу, сахарозу, сорбит, микрокристаллическую целлюлозу, крахмал и двухосновный дигидрат фосфата кальция.
Таблетки могут необязательно включать поверхностно-активные агенты, такие как лаурилсульфат натрия и полисорбат 80, и глиданты, такие как двуокись кремния и тальк. Если присутствуют, поверхностно-активные агенты обычно составляют от 0,2% масс. до 5% масс. таблетки, и глиданты обычно составляют от 0,2% масс. до 1% масс. таблетки.
Таблетки также обычно содержат смазывающие агенты, такие как стеарат магния, стеарат кальция, стеарат цинка, стеарилфумарат натрия, и смеси стеарата магния с лаурилсульфатом натрия. Смазывающие агенты обычно присутствуют в количествах от 0,25% масс. до 10% масс., предпочтительно, от 0,5% масс. до 3% масс. таблетки.
Другие обычные ингредиенты включают антиоксиданты, красители, вкусовые добавки, консерванты и агенты, маскирующие вкус.
Типовые таблетки содержат вплоть до 80% масс. лекарственного средства, от около 10% масс. до около 90% масс. связующего агента, от около 0% масс. до около 85% масс. разбавителя, от около 2% масс. до около 10% масс. разрыхлителя и от около 0,25% масс. до около 10% масс. смазывающего агента.
Смеси для таблеток могут быть спрессованы непосредственно или валком для получения таблеток. Смеси для таблеток или части смесей альтернативно могут быть гранулированы влажным, сухим способом или в расплаве, отверждены в расплаве или экструдированы до таблетирования. Конечные композиции могут включать один или более слоев и могут быть в оболочке или без; или инкапсулированы.
Композиции таблеток более подробно обсуждаются в ʺPharmaceutical Dosage Forms: Tablets, Vol. 1ʺ, by H. Lieberman and L. Lachman, Marcel Dekker, N.Y., N.Y., 1980 (ISBN 0-8247-6918-X), описание которой включено сюда в качестве ссылки полностью.
Твердые композиции для перорального введения могут быть составлены для немедленного и/или модифицированного выделения. Композиция с модифицированным выделением включают отложенное, замедленное, периодическое, контролируемое, целевое и программированное выделение.
Подходящие композиции с модифицированным выделением описаны в патенте США № 6,106,864. Подробности других подходящих методик выделения, таких как высокоэнергетические дисперсии и осмотические и покрытые частицы, могут быть найдены в Verma et al., Pharmaceutical Technology On-line, 25(2), 1-14 (2001). Применение жевательной резинки для достижения контролируемого выделения описано в WO 00/35298. Содержания этих ссылок включено сюда в качестве ссылки полностью.
Парентеральное введение
Соединения в соответствии с данным изобретением также могут вводиться непосредственно в кровоток, в мышцы или во внутренний орган. Подходящие пути парентерального введения включают внутривенный, внутриартериальный, внутрибрюшинный, интратекальный, интравентрикулярный, интрауретральный, интрастернальный, внутричерепной, внутримышечный и подкожный. Подходящие устройства для парентерального введения включают иглу (включая микроиглу), шприцы, безигольные шприцы и методы вливания.
Парентеральные композиции обычно являются водными растворами, которые могут содержать наполнители, такие как соли, углеводороды и буферные агенты (предпочтительно, до pH от 3 до 9), но, для некоторых применений, они могут быть поле подходящим образом составлены в виде стерильного не водного раствора или в виде высушенной формы для применения в сочетании с подходящим носителем, таким как стерильная апирогенная вода.
Получение парентеральных композиций в стерильных условиях, например, лиофилизацией, может быть легко проведено с применением стандартных фармацевтических методов, хорошо известных специалисту в данной области техники.
Растворимость соединений в соответствии с данным изобретением, применяемых в получении парентеральных растворов, может быть повышена применением подходящих методик составления, таких как введение агентов, улучшающих растворимость. Композиции для парентерального введения могут быть составлены для немедленного и/или модифицированного выделения. Композиции с модифицированным выделением включают отложенное, замедленное, периодическое, контролируемое, целевое или программируемое выделение. Таким образом, соединения в соответствии с данным изобретением могут быть составлены в виде твердого вещества, полутвердого вещества или тиксотропной жидкости для введения в виде имплантируемого депо, обеспечивающего модифицированное выделение активного соединения. Примеры таких композиций включают покрытые лекарственным средством стенты и СМГК микросферы.
Местное введение
Соединения в соответствии с данным изобретением также могут вводиться местно на кожу или слизистую, то есть кожно или чрезкожно. Типовые композиции для этой цели включают гели, гидрогели, лосьоны, кремы, мази, тонкоизмельченные порошки, повязки, пены, пленки, кожные пластыри, вафли, импланты, губки, волокна, бандажи и микроэмульсии. Липосомы также могут применяться. Типовые носители включают спирт, воду, минеральное масло, жидкий вазелин, белый вазелин, глицерин, полиэтиленгликоль и пропиленгликоль. Могут быть введены улучшители проникновения; см., например, J Pharm Sci, 88 (10), 955-958 by Finnin and Morgan (October 1999). Другие средства местного введения включают введение электропорацией, ионтофорезом, фонофорезом, сонофорезом и инъекцией микроиглой или без иглы (например, Powderject™, Bioject™, и т.д.). Содержание этих ссылок включено сюда в качестве ссылки полностью.
Композиции для местного введения могут быть составлены для немедленного и/или модифицированного выделения. Композиции с модифицированным выделением включают отложенное, замедленное, периодическое, контролируемое, целевое или программируемое выделение.
Ингалируемое/интраназальное введение
Соединения в соответствии с данным изобретением также могут вводиться интраназально или ингаляцией, обычно в форме сухого порошка (отдельно, в виде смеси, например, в сухой смеси с лактозой или в виде частиц смешанного компонента, например, смешанного с фосфолипидами, такими как фосфатидилхолин) из сухого порошкового ингалятора или в виде аэрозольного спрея из контейнера под давлением, помпы, спрея, распылителя (предпочтительно, распылителя, в котором применяется электрогидродинамика для получения мелкодисперсного тумана) или небулайзера, с или без применения подходящего газа-вытеснителя, такого как 1,1,1,2-тетрафторэтан или 1,1,1,2,3,3,3-гептафторпропан. Для интраназального применения порошок может включать биоадгезивный агент, например, хитозан или циклодекстрин.
Контейнер под давлением, помпа, спрей, распылитель или небулайзер содержит раствор или суспензию соединения(ий) в соответствии с данным изобретением, включающую, например, этанол, водный этанол или подходящий альтернативный агент для диспергирования, солюбилизации или расширенного выделения активного агента, газ-вытеснитель в качестве растворителя и необязательное поверхностно-активное вещество, такое как сорбитантриолеат, олеиновая кислота или олигомолочная кислота.
До применения в композиции сухого порошка или суспензии, лекарственное средство микронизируют до размера, подходящего для доставки ингаляцией (обычно менее 5 микронов). Это может достигаться любым подходящим методом измельчения, таким как размол на спиральной струйной мельнице, размол на струйной мельнице с кипящим слоем, обработка сверхкритической жидкостью с получением наночастиц, гомогенизация под высоким давлением или сушка распылением.
Капсулы (сделанные, например, из желатина или ГПМЦ), блистеры и картриджи для применения в ингаляторе или инсуфляторе могут быть составлены так, чтобы содержать порошковую смесь соединения в соответствии с данным изобретением, подходящее порошковое основание, такое как лактоза или крахмал, и модификатор эффективности, такой как l-лейцин, маннит или стеарат магния. Лактоза может быть безводной или в форме моногидрата, предпочтительно, последнее. Другие подходящие наполнители включают декстран, глюкозу, мальтозу, сорбит, ксилит, фруктозу, сахарозу и трегалозу.
Подходящий раствор для применения в распылителе с электрогидролинамикой для получения мелкодисперсного тумана может содержать от 1 мкг до 20 мг соединения в соответствии с данным изобретением на введение, и объем введения может варьироваться от 1 мкл до 100 мкл. Типовая композиция включает соединение в соответствии с данным изобретением, пропиленгликоль, стерильную воду, этанол и хлорид натрия. Альтернативные растворители, которые могут применяться вместо пропиленгликоля, включают глицерин и полиэтиленгликоль.
Подходящие вкусовые добавки, такие как ментол и левоментол, или подсластители, такие как сахарин или сахарин натрий, могут быть добавлены в такие композиции в соответствии с данным изобретением, предназначенные для ингалируемого/интраназального введения.
Композиции для ингалируемого/интраназального введения могут быть составлены для немедленного и/или модифицированного выделения с применением, например, поли(DL-молочной-согликолевой кислоты (СМГА). Композиции с модифицированным выделением включают отложенное, замедленное, периодическое, контролируемое, целевое и программированное выделение.
Для сухих порошковых ингаляторов или аэрозолей, однократная доза определяется клапаном, который доставляет отмеренное количество. Аппараты в соответствии с данным изобретением обычно сделаны так, чтобы вводить отмеренную дозу или ʺпшикʺ, содержащую желаемое количество соединения в соответствии с данным изобретением. Общая суточная доза может вводиться в виде однократной дозы или, более часто, в виде нескольких доз в течение суток.
Ректальное/интравагинальное введение: соединения в соответствии с данным изобретением могут вводиться ректально или вагинально, например, в форме суппозиториев, пессариев или клизмы. Масло какао является традиционной основной для суппозиториев, но подходящим образом могут применяться различные альтернативы. Композиции для ректального/вагинального введения могут быть составлены для немедленного и/или модифицированного выделения. Композиции с модифицированным выделением включают отложенное, замедленное, периодическое, контролируемое, целевое и программированное выделение.
Глазное введение: соединения в соответствии с данным изобретением также могут вводиться непосредственно в глаз или ухо, обычно в форме капель или микронизированной суспензии или раствора в изотоническом, pH-скорректированном стерильном физиологическом растворе. Другие композиции для глазного и ушного введения включают мази, биоразлагаемые (например, абсорбируемые гелевые губки, коллаген) и не биоразлагаемые (например, силикон) импланты, вафли, линзы и порошковые и везикулярные системы, такие как ниосомы или липосомы. Полимер, такой как поперечно-сшитая полиакриловая кислота, поливиниловый спирт, гиалуроновая кислота, целлюлозный полимер, например, гидроксипропилметилцеллюлоза, гидроксиэтилцеллюлоза или метилцеллюлоза, или гетерополисахаридный полимер, например, гелановая камедь, может быть введен вместе с консервантом, таким как хлорид бензалкония. Такие композиции также могут вводиться ионтофорезом.
Композиции для глазного/ушного введения могут быть составлены для немедленного и/или модифицированного выделения. Композиции с модифицированным выделением включают отложенное, замедленное, периодическое, контролируемое, целевое и программированное выделение.
Другие методики
Соединения в соответствии с данным изобретением могут быть объединены с растворимыми макромолекулярными веществами, такими как циклодекстрин и его подходящие производные, или содержащие полиэтиленгликоль полимеры, для улучшения их растворимости, скорости растворения, маскировки вкуса, биодоступности и/или стабильности для применения в любых из указанных выше способах введения.
Комплексы лекарственное средство-циклодекстрин, например, обычно применяют для большинства лекарственных форм и способов введения. Могут применяться инклюзионные и не инклюзионные комплексы. В качестве альтернативы прямого получения комплекса с лекарственным средством, циклодекстрин может применяться в качестве вспомогательной добавки, т.е. в качестве носителя, разбавителя или солюбилизатора. Наиболее часто для этих целей применяют альфа-, бета- и гамма-циклодекстрины, примеры которых можно найти в публикациях PCT №№ WO 91/11172, WO 94/02518 и WO 98/55148, описания которых включены сюда в качестве ссылок полностью.
Доза: количество вводимого активного соединения зависит от лечимого субъекта, тяжести расстройства или состояния, скорости введения, характера соединения и мнения лечащего врача. Однако эффективная доза обычно составляет от около 0,001 до около 100 мг на кг массы тела в сутки, предпочтительно, от около 0,01 до около 35 мг/кг/сутки, одной или несколькими дозами. Для 70 кг человека доза будет составлять от около 0,07 до около 7000 мг/сутки, от около 0,7 до около 2500 мг/сутки. В некоторых случаях, дозы ниже нижнего предела указанного выше интервала могут быть более чем адекватны, в других случаях, даже большие дозы могут применяться без возникновения вредных побочных эффектов, где такие большие дозы обычно делят на несколько меньших доз для введения в течение суток.
Наборы частей: Если желательно вводить сочетание активных соединений, например, для целей лечения конкретного заболевания или состояния, в объем данного изобретения включен вариант, когда две или более фармацевтические композиции, по крайней мере, одна из которых содержит соединение в соответствии с данным изобретением, могут удобным образом быть объединены в форме набора, подходящего для совместного введения композиций. таким образом, набор в соответствии с данным изобретением включает две или более отдельные фармацевтические композиции, по крайней мере, одна из которых содержит соединение в соответствии с данным изобретением, и средства для раздельного хранения таких композиций, такие как контейнер, разделенная бутылка или разделенный пакет из фольги. Примером такого набора является обычная блистерная упаковка, применяемая для упаковки таблеток, капсул и подобных.
Набор в соответствии с данным изобретением особенно подходит для введения различных лекарственных форм, например, пероральной и парентеральной, для введения отдельных композиций с разными интервалами введения, или для капельного введения разных композиций друг против друга. Для соблюдения режима, набор обычно содержит инструкции по введению, а также может содержать памятку.
ПРИМЕРЫ
Для некоторых описанных стадий может быть необходимо защищать потенциально реакционноспособные функциональные группы, реакция которых нежелательна, и отщеплять такие защитные группы впоследствии. В таком случае, может применяться любой совместимый защитный радикал. В конкретных способах могут применяться способы защиты и снятия защиты, такие как описаны в T.W. Greene (Protective Groups in Organic Synthesis, A. Wiley-Interscience Publication, 1981) или by P. J. Kocienski (Protecting groups, Georg Thieme Verlag, 1994).
Все указанные здесь реакции и получение новых исходных соединений, применяемых здесь, являются обычными, и подходящие реагенты и условия реакции для их осуществления или получения, а также способы выделения желаемых продуктов хорошо известны специалистам в данной области техники и указаны в ссылках на литературные источники и примеры и примеры получения.
Могут применяться следующие аббревиатуры:
Ac (ацетил); AcCl (ацетилхлорид); AcOH или HOAc (уксусная кислота); Ac2O (уксусный ангидрид); водн. (водный); Boc или boc (трет-бутоксикарбонил); около (около или приблизительно); CDCl3 (дейтерированный хлороформ); CH2Cl2 и/или ДХМ (дихлорметан); ТДАС (трифторид диэтиламиносеры); ДХЭ (дихлорэтан); ДЭА (диэтиламин); DIBAL или DIBAL-H (гидрид диизобутилалюминия); ДИК (диизопропилкарбодиимид); ДИПЭА или основание Хюнига (N,N-диизопропилэтиламин); ДМА (диметилацетамид); ДМФ (диметилформамид); ДМЭ (этиленгликоль); ПДМ (периодинан Десса-Мартина); ДМАП (4-диметиламинопиридин); ДМСО (диметилсульфоксид); ДМСО-d6 (дейтерированный диметилсульфоксид); ЭДК или ЭДКИ (1-этил-3-(3-диметиламинопропил)карбодиимид); Et (этил); Et3N или ТЭА (триэтиламин); EtOH (этанол); EtOAc (этилацетат); Et2O (диэтиловый эфир); г или гм (грамм или граммы); ГАТУ (гексафторфосфат 2-(7-аза-1H-бензотриазол-1-ил)-1,1,3,3-тетраметилурония); ГБТУ (гексафторфосфат o-(бензотриазол-1-ил)-1,1,3,3-тетраметилурония); HMPT (трис(диметиламино)фосфин); ВЭЖХ (высокоэффективная жидкостная хроматография); ГОБТ (1-гидроксибензотриазол); ч или час (час или часы, соответственно); iBu (изобутил); ИПС (изо-пропиловый спирт); iPr (изопропил); iPrOAc (изопропилацетат); KHMDS (бис(триметилсилил)амид калия); KOAc (ацетат калия); ЖХМС (жидкостная хроматография-масс спектрометрия); LiHMDS (бис(триметилсилил)амид лития); Me (метил); MeOH (метанол); MeOD (дейтерированный метанол); MeCN (ацетонитрил); м или мин (минута или минуты, соответственно); мг (миллиграмм или миллиграммы); Ms (метилсульфонил); MsCl (метансульфонилхлорид); N (нормаль); NBS (N-Бромсукцинимид); NFSI (N-Фтордибензолсульфонамид); ЯМР (ядерный магнитный резонанс); nBu (н-бутил); nBuLi (н-бутиллитий); nPr (н-пропил); Pd/C (палладий на угле); Pd2(dba)3 (трис(дибензилиденацетон)дипалладий(0)); Pd(dppf)Cl2 ([1,1′-бис(дифенилфосфино)ферроцен]дихлорпалладий(II)); Ph (фенил); ПТСК или пТСК (п-толуолсульфоновая кислота); Ву (время удержания); кт (комнатная температура); RuCl(п-кумен)[(R,R)-Ts-DPEN] ([N-[(1R,2R)-2-(Амино-κN)-1,2-дифенилэтил]-4-метилбензолсульфонамидато-κN]хлор[(1,2,3,4,5,6-η)-1-метил-4-(1-метилэтил)бензол]рутений); с или сек (секунда или секунды, соответственно); Selectfluor (бис(тетрафторборат)N-Хлорметил-N′-фтортриэтилендиаммония); СЭМ (2-триметилсилилэтоксиметокси); СЖХ (сверхкритическая жидкостная хроматография); Si-Тиоl (1-пропантиол двуокиси кремния); T3P (пропилфосфоновый ангидрид); ФТБА (фторид тетрабутиламмония); TBDMSCl (т-бутилдиметилсилилхлорид); ТБМЭ или МТБЭ (трет-бутилметиловый эфир); т-BuOH (2-метил-2-пропанол, трет-бутанол или трет-бутиловый спирт); TDA-1 (трис[2-(2-метоксиэтокси)этил]амин или трис (3,6-диоксагептил)амин); ТЭА, NEt3 или Et3N (триэтиламин); ТФК (трифторуксусная кислота); ТГФ (тетрагидрофуран); ТГП (тетрагидропиран); ТСХ (тонкослойная хроматография); ТМС (триметилсилил); ТМСCl (триметилсилилхлорид); ТМСCF3 (триметил(трифторметил)силан); Tos или тозил (4-толуолсульфонил); TOSMIC (п-толуолсульфонилметил изоцианид); УФ (ультрафиолет).
Общая схема синтеза соединений карбонуклеозида
Схема 1:
Как представлено на схеме 1, соединение, такое как 1a, может быть куплено или синтезировано (Chem. Rev., 2012, 112 (8), pp 4642-4686). Обычно метил (1S,2R,3S,4R)-4-амино-2.3-дигидроксициклопентан-1-карбоксилат подвергают взаимодействию с подходящим образом функционализированным пиримидином или другим гетероциклом с получением соединений, таких как 1b. Эта реакция может осуществляться стандартным нуклеофильным ароматическим замещением с применением условий, таких как диизопропилэтиламин или триметиламин в ДМСО, ДМА или ДМФ, или условий с применением фторида цезия в ДМСО. Альтернативные условия реакции могут включать реакции сочетания металла, такие как палладиевые сочетания. Циклизация соединений, таких как 1b, до соединений, таких как 1c, может проходить во множестве условий, включая конденсации или металлические сочетания, а зависимости от типа атома A. Защита диола с получением 1d может быть выполнена с применением ацетона или 2,2-диметоксипропана в мягкой кислоте. Сложные эфиры, такие как 1d, превращают в алкил- и арилкетоны, такие как 1e, с применением алкильных и арильных металлических реагентов, таких как алкильные и арильные реагенты Гриньяра (M=Mg), алкильные и арильные литиевые реагенты, алкильные и арильные купраты, алкильные и арильные цинкаты, а также другие металлорганические реагенты. Обычно эти реакции проводят в эфирных растворителях, таких как ТГФ, MeТГФ, диоксан или подобный растворитель, при температурах от -78°C до 60°C. Алкил- и арилкетоны, такие как 1e, могут быть превращены во вторичные спирты, такие как 1f, с применением восстанавливающих агентов, таких как NaBH4, LiBH4, LiAlH4, DIBAL и другие. Обычно эти реакции проводят во множестве растворителей, таких как ДХМ, ТГФ, MeOH, EtOH или другие при различных температурах. Алкил- и арилкетоны, такие как 1e, могут быть преимущественно превращены в диастереомерно обогащенные вторичные спирты, такие как 1f, с применением хиральных восстанавливающих условий, таких как RuCl(п-кумен)[(R,R)-Ts-DPEN] и формиат натрия (J. Org. Chem, 2014, 79, 3238-3243). Обычно, эти реакции проводят в растворителе EtOAc и при комнатной температуре. Наконец, соединения, такие как 1f, могут быть лишены защиты для раскрытия триольных соединений, таких как 1g, обработкой кислотой, такой как ТФК или разбавленная HCl. Обычно, эти реакции проводят в присутствии воды при 0°C или кт. Соединения на каждой стадии могут быть очищены стандартными методами, такими как хроматография на колонке, кристаллизация, ВЭЖХ с обращенной фазой или СЖХ. Если необходимо, разделение диастеремеров 1f или 1g может быть проведено стандартными методами, известными в данной области техники, такими как хиральная СЖХ или ВЭЖХ с получением отдельных диастереомеров.
Схема 2:
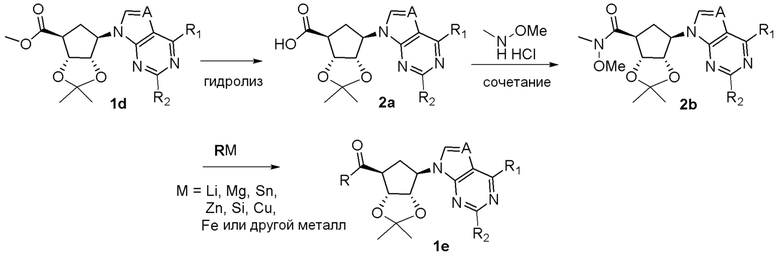
Как представлено на схеме 2, соединения, такие как 1d, могут быть гидролизованы с основанием с получением кислоты 2a. Обычно эти реакции проводят с применением источника гидроксида, такого как гидроксид лития, гидроксид натрия или другие. Соединения, такие как карбоновая кислота 2a, превращают в соединения, такие как амид Вейнреба 2b, обработкой N,O-диметилгидроксиламином HCl и стандартными реагентами амидного сочетания, такими как ГОБТ и ЭДКИ, T3P или ГАТУ с основанием, таким как ДИПЭА или ТЭА. Обычно, эти реакции проводят в растворителях, таких как ДМФ или ТГФ, и при температурах от 0°C до 60°C. Амиды Вейнреба, такие как 2b, превращают в алкил- и арилкетоны, такие как 1e, с применением алкильных и арильных металлических реагентов, таких как алкильные и арильные реагенты Гриньяра (M=Mg), алкильные и арильные реагенты лития, алкил- и арилкупраты, алкил- и арилцинкаты, а также другие металлорганические реагенты. Обычно эти реакции проводят в эфирном растворителе, таком как ТГФ, MeТГФ, диоксан или подобный растворитель, при температуре от -78°C до 60°C.
Схема 3:
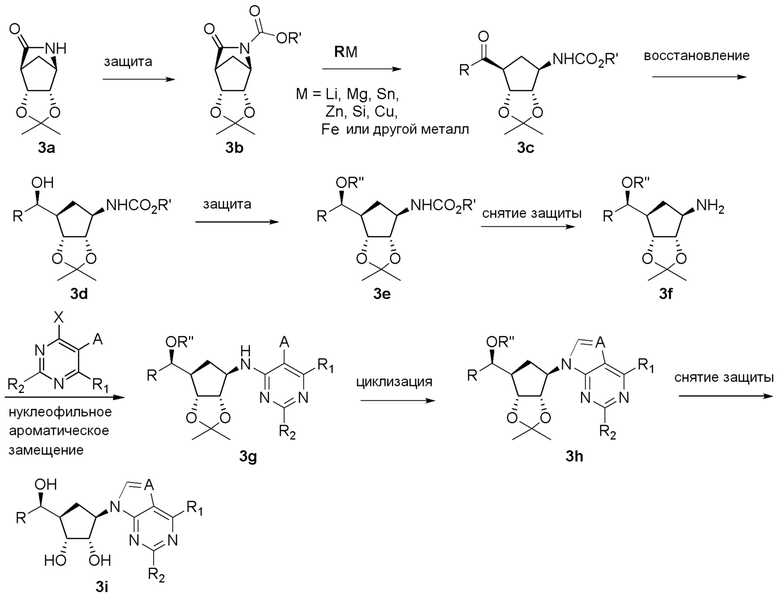
Как представлено на схеме 3, соединение, такое как 3a, [(3aS,4R,7S,7aR)-2,2-диметилтетрагидро-4,7-метано[1.3]диоксоло[4.5-c]пиридин-6(3aH)-он], может быть куплено или синтезировано (Chem. Rev., 2012, 112 (8), 4642-4686) и может быть защищено как карбамат с применением реагентов, таких как Boc ангидрид или бензоилхлорид с получением лактамов, таких как 3b. Эти активированные лактамы (3b) превращают в алкил- и арилкетоны, такие как 3c, с применением алкильных и арильных металлических реагентов, таких как алкильные и арильные реагенты Гриньяра (M=Mg), алкильные и арильные реагенты лития, алкил- и арилкупраты, алкил- и арилцинкаты, а также другие металлорганические реагенты. Обычно эти реакции проводят в эфирных растворителях, таких как ТГФ, MeТГФ, диоксан или подобный растворитель, при температурах от -78°C до 60°C. Алкил- и арилкетоны, такие как 3c, могут быть превращены во вторичные спирты, такие как 3d, с применением реагентов, таких как NaBH4, LiBH4, LiAlH4, DIBAL и другие. Обычно эти реакции проводят во множестве растворителей, таких как ДХМ, ТГФ, MeOH, EtOH или другие при различных температурах. Алкил- и арилкетоны, такие как 3c, преимущественно превращают в диастереомерно обогащенные вторичные спирты, такие как 3d, с применением хиральных восстанавливающих условий, таких как RuCl(p-кумен)[(S,S)-Ts-DPEN] и формиат натрия (J. Org. Chem, 2014, 79, 3238-3243). Обычно, эти реакции проводят в растворителе EtOAc и при комнатной температуре. Полученный вторичный спирт, такой как 3d, может быть защищен множеством реагентов, которые обеспечивают стратегии ортогонального снятия защиты карбамата, установленной ранее в способе. Такие реагенты включают ТМСCl, TESCl, TBDMSCl, TIPSCl и другие. Соединения, такие как 3e, могут быть лишены защиты с получением соединений, таких как 3f, множеством способов, в зависимости от того, какой карбамат установлен. Примеры включают применение разбавленного раствора трифторуксусной кислоты в случае Boc карбамата, или гидролиз с Pd катализатором и газообразным водородом в случае бензоилкарбамата. Соединения, такие как 3f, подвергают взаимодействию с подходящим образом замещенными гетероциклами в нуклеофильном ароматическом замещении с получением соединений, таких как 3g. Такие реакции обычно проводят с применением органических оснований, таких как диизопропилэтиламин или триметиламин, в ДМСО, ДМА или ДМФ, или в условиях с применением фторида цезия в ДМСО. Альтернативные условия реакции могут включать реакции металлического замещения, такие как палладиевые замещения. Циклизация соединений, таки как 3g, до соединений, таких как 3h, может проводиться во множестве состояний, включая конденсации или металлические сочетания в зависимости от типа атома A. Наконец, соединения, такие как 3h, могут быть лишены защиты с получением триольных соединений, таких как 3i, обработкой кислотой, такой как ТФК или разбавленная HCl. Обычно эти реакции проводят в присутствии воды при 0°C или кт. Соединения на каждой стадии могут быть очищены стандартными методами, такими как хроматография на колонке, кристаллизация, ВЭЖХ с обращенной фазой или СЖХ. При необходимости, разделение 3d или любого последующего соединения может проводиться стандартными методами, известными в данной области техники, такими как хиральная СЖХ или ВЭЖХ с получением отдельных диастереомеров.
Схема 4:
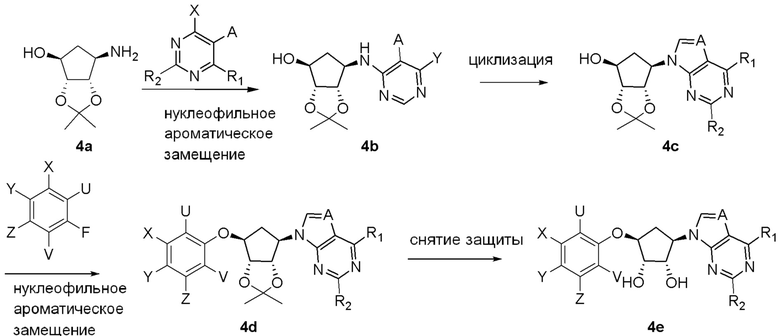
Как представлено на схеме 4, соединение, такое как 4a, [(3aR,4S,6R,6aS)-6-амино-2,2-диметилтетрагидро-4H-циклопента[d][1.3]диоксо-4-ол], может быть куплено или синтезировано [J. Perkin Транс. I, 1985, 1437; Tetrahedron Letters, 2000, (41) 9537-9530]. Соединения, такие как 4a, могут подвергаться нуклеофильному ароматическому замещению подходящим образом замещенным гетероциклом с получением соединений, таких как 4b. Циклизация соединений, таких как 4b, до соединений, таких как 4c, может проводиться во множестве состояний, включая конденсации или металлические сочетания, в зависимости от типа атома A. Соединения, такие как 4c, подвергают нуклеофильному ароматическому замещению подходящим образом замещенным фторбензолом с получением сложных эфиров фенола, таких как 4d. Наконец, соединения, такие как 4d, могут быть лишены защиты с получением диольных соединений, таких как 4e, обработкой кислотой, такой как ТФК или разбавленная HCl. Обычно эти реакции проводят в присутствии воды при 0°C или кт. Соединения на каждой стадии могут быть очищены стандартными методами, такими как хроматография на колонке, кристаллизация, ВЭЖХ с обращенной фазой или СЖХ. При необходимости, разделение диастереомеров 4a может проводиться стандартными методами, известными в данной области техники, такими как хиральная СЖХ или ВЭЖХ с получением отдельных диастереомеров.
Схема 5:
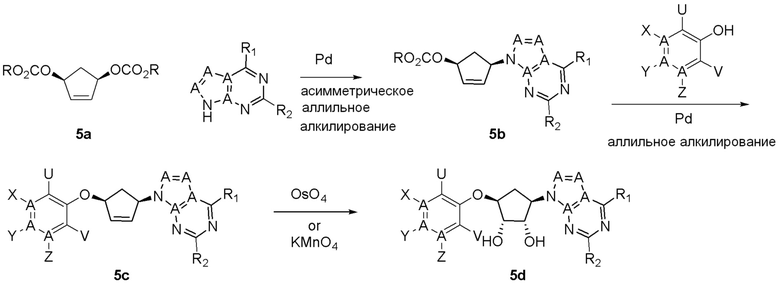
Как представлено на схеме 5, соединения, такие как 5a, могут быть куплены или синтезированы (Chemical Science, 2010, 1, p427; Journal of the American Chemical Society, 2000, 122, p5947; Organic Letters, 2012, 14(9), p2254). Соединения, такие как 5a, подвергают асимметрическому аллильному алкилированию с подходящим образом замещенными гетероциклами с применением множества палладиевых катализаторов и хиральных лигандов с получением соединений, таких как 5b. Соединения, такие как 5b, подвергают аллильному алкилированию с подходящим образом замещенными фенолами или гидроксигетероциклами с применением множества палладиевых катализаторов и лигандов с получением соединений, таких как 5c. Соединения, такие как 5c, могут быть дигидроксилированы с получением диолов, таких как 5d, с применением реагентов, таких как тетроксид осмия или перманганат калия. Соединения на каждой стадии могут быть очищены стандартными методами, такими как хроматография на колонке, кристаллизация, ВЭЖХ с обращенной фазой или СЖХ. При необходимости, разделение диастереомеров 5d может проводиться стандартными методами, известными в данной области техники, такими как хиральная СЖХ или ВЭЖХ с получением отдельных диастереомеров.
Схема 6:
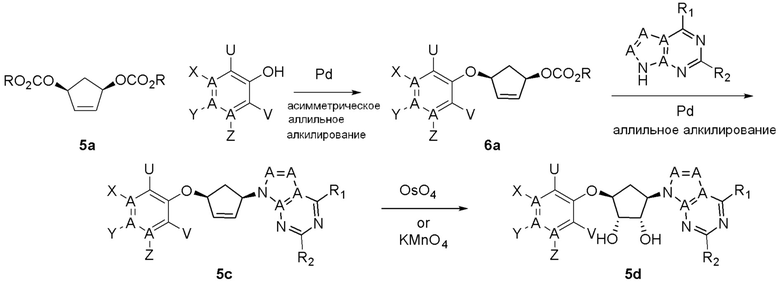
Как представлено на схеме 6, соединения, такие как 5a, подвергают асимметрическому аллильному алкилированию с подходящим образом замещенными фенолами или гидроксигетероциклами с применением множества палладиевых катализаторов и хиральных лигандов с получением соединений, таких как 6a. Соединения, такие как 6a, подвергают аллильному алкилированию с подходящим образом замещенными гетероциклами с применением множества палладиевых катализаторов и лигандов с получением соединений, таких как 5c. Соединения, такие как 5c, могут быть дигидроксилированы с получением диолов, таких как 5d, с применением реагентов, таких как тетроксид осмия или перманганат калия.
Схема 7:
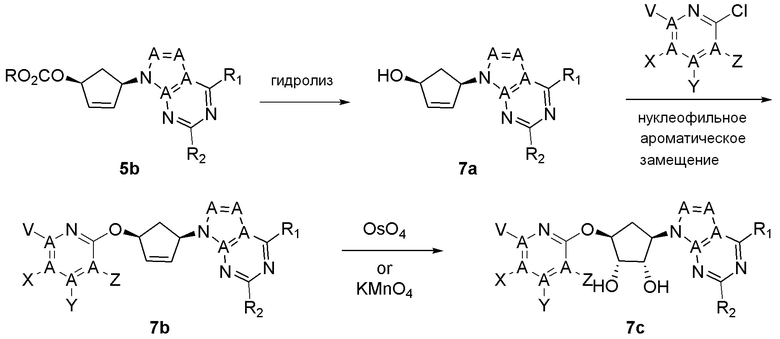
Как представлено на схеме 7, соединения, такие как 5b, могут быть гидролизованы до соединений, таких как 7a, с применением источника гидроксида, такого как гидроксид лития, гидроксид натрия или другие. Соединения, такие как 7a, подвергают нуклеофильному ароматическому замещению подходящим образом замещенным гетероциклом с получением соединений, таких как 7b. Соединения, такие как 7b, могут быть дигидроксилированы с получением диолов, таких как 7c, с применением реагентов, таких как тетроксид осмия или перманганата калия. Соединения на каждой стадии могут быть очищены стандартными методами, такими как хроматография на колонке, кристаллизация, ВЭЖХ с обращенной фазой или СЖХ. При необходимости, разделение диастереомеров 7c может проводиться стандартными методами, известными в данной области техники, такими как хиральная СЖХ или ВЭЖХ с получением отдельных диастереомеров.
Схема 8:
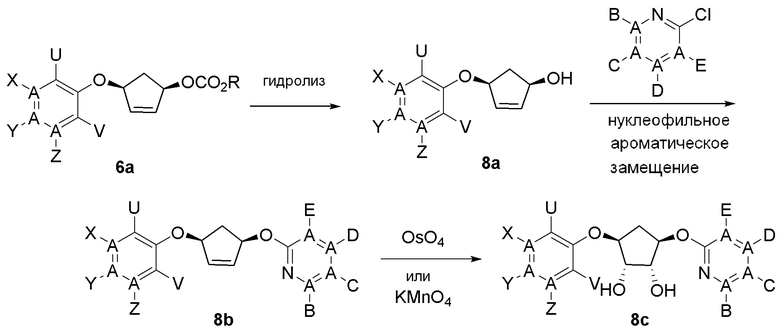
Как представлено на схеме 8; соединения, такие как 6a, могут быть гидролизованы до соединений, таких как 8a, с применением источника гидроксида, такого как гидроксид лития, гидроксид натрия или другие. Соединения, такие как 8a, подвергают нуклеофильному ароматическому замещению с подходящим образом замещенным гетероциклом с получением соединений, таких как 8b. Соединения, такие как 8b, могут быть дигидроксилированы с получением диолов, таких как 8c, с применением реагентов, таких как тетроксид осмия или перманганат калия. Соединения на каждой стадии могут быть очищены стандартными методами, такими как хроматография на колонке, кристаллизация, ВЭЖХ с обращенной фазой или СЖХ. При необходимости, разделение диастереомеров 8c может проводиться стандартными методами, известными в данной области техники, такими как хиральная СЖХ или ВЭЖХ с получением отдельных диастереомеров.
Пример 1 (Схема A): Синтез (1S,2R,3R,5R)-3-((R)-1-гидроксиэтил)-5-(4-метил-7H-пирроло[2.3-d]пиримидин-7-ил)циклопентан-1,2-диола (A-12)
Пример 2 (Схема A): Синтез (1S,2R,3R,5R)-3-((S)-1-гидроксиэтил)-5-(4-метил-7H-пирроло[2.3-d]пиримидин-7-ил)циклопентан-1,2-диола (A-13)
Схема A
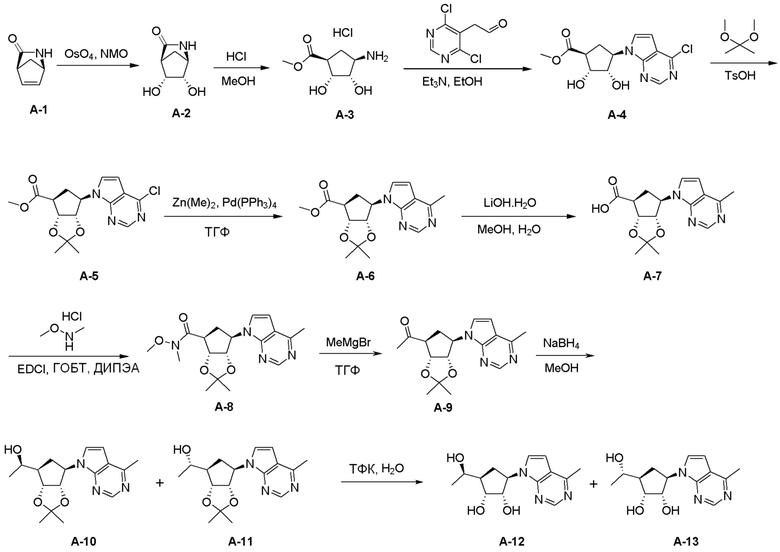
Стадия 1: Синтез (1R,4S,5R,6S)-5,6-дигидрокси-2-азабицикло[2.2.1]гептан-3-он (A-2)
К светло-желтой двухфазной смеси (1R,4S)-2-азабицикло[2.2.1]гепт-5-ен-3-она A-1 (Chemical Reviews, 2012, 112 (8), pp 4642-4686)(100 г, 916 ммоль) и NMO (118 г, 1,01 моль) в изоамиловом спирте (500 мл) и воде (500 мл) добавляют 2,5% OsO4 в т-BuOH (1,5 г, 5,9 ммоль, 76 мл) при кт (15°C). Смесь нагревают при 70°C в течение 2 ч. Смесь охлаждают до кт (15°C). NaHSO3 (12 г) добавляют и перемешивают при кт (15°C) в течение 45 мин. Смесь концентрируют в вакууме с получением неочищенного продукта (150 г) в виде темного твердого вещества, которое очищают хроматографией на силикагеле, элюируя MeOH в ДХМ=10% с получением A-2 (90 г, 69%) в виде светло-розового твердого вещества. 1H ЯМР (400 MГц, ДМСО-d6) δ ч./млн. 7,54 (шс, 1H), 5,00 (дд, J=21,3, 22,6 Гц, 2H), 3,81-3,64 (м, 2H), 3,43 (с, 1H), 2,25 (с, 1H), 1,93-1,67 (м, 2H)
Стадия 2: Синтез метил (1S,2R,3S,4R)-4-амино-2.3-дигидроксициклопентан-1-карбоксилата-HCl (A-3)
Соединение A-2 (33 г, 231 ммоль) добавляют к 4N HCl в MeOH (500 мл) при кт (15°C). Суспензию перемешивают при кт (15°C) в течение 20 ч. Смесь концентрируют в вакууме, и остаток суспендируют в ДХМ и фильтруют. Твердое вещество промывают ДХМ и сушат в вакууме с получением A-3 (45 г, 92%) в виде белого твердого вещества. ЖХМС [M+1] 176; 1H ЯМР (400 MГц, D2O) δ ч./млн. 4,27 (т, J=5,1 Гц, 1H), 4,04 (т, J=6,4 Гц, 1H), 3,72 (с, 3H), 3,55 (кв, J=8,4 Гц, 1H), 2,97 (дт, J=5,0, 8,8 Гц, 1H), 2,49 (тд, J=8,5, 13,8 Гц, 1H), 1,83 (тд, J=9,2, 13,7 Гц, 1H)
Стадия 3: Синтез метил (1S,2R,3S,4R)-4-(4-хлор-7H-пирроло[2.3-d]пиримидин-7-ил)-2.3-дигидроксициклопентан-1-карбоксилата (A-4)
К белой суспензии соединения A-3 (42 г, 200 ммоль) и 2-(4,6-дихлорпиримидин-5-ил)ацетальдегида (40 г, 209 ммоль) в EtOH (400 мл) добавляют Et3N (40,4 г, 400 ммоль). Полученный желтый раствор перемешивают при кипении с обратным холодильником в течение 2 ч. Смесь концентрируют в вакууме до около 100 мл. Остаток выливают в NH4Cl водн (500 мл) и экстрагируют EtOAc (200 мл×3). Экстракт промывают насыщенным раствором соли (200 мл), сушат над Na2SO4 и концентрируют в вакууме с получением неочищенного продукта A-4 (60 г, >99%) в виде красной камеди, которую применяют сразу на следующей стадии. ЖХМС [M+1] 312
Стадия 4: Синтез метил (3aR,4S,6R,6aS)-6-(4-хлор-7H-пирроло[2.3-d]пиримидин-7-ил)-2,2-диметилтетрагидро-4H-циклопента[d][1.3]диоксол-4-карбоксилата (A-5)
К раствору неочищенного продукта A-4 (60 г, 192 ммоль) в 2,2-диметоксипропане (300 мл) и ацетоне (300 мл) добавляют TsOH⋅H2O (40 г, 212 ммоль). Смесь перемешивают при кт (15°C) в течение 1 ч. Смесь выливают в NaHCO3 водн. (1000 мл) и экстрагируют EtOAc (500 мл). Экстракт промывают насыщенным раствором соли, концентрируют в вакууме с получением неочищенного продукта, который очищают хроматографией на силикагеле, элюируя EtOAc в петролейном эфире от 0-100% с получением A-5 (42 г, 62%) в виде желтой камеди, которая затвердевает при выстаивании. ЖХМС [M+1] 352; 1H ЯМР (400 MГц, CDCl3) δ ч./млн. 8,63 (с, 1H), 7,29 (д, J=3,5 Гц, 1H), 6,64 (д, J=3,8 Гц, 1H), 5,15-5,04 (м, 2H), 5,03-4,98 (м, 1H), 3,76 (с, 3H), 3,12 (ддд, J=5,3, 7,6, 10,7 Гц, 1H), 2,75-2,59 (м, 2H), 1,59 (с, 3H), 1,33 (с, 3H)
Стадия 5: Синтез метил (3aR,4S,6R,6aS)-2,2-диметил-6-(4-метил-7H-пирроло[2.3-d]пиримидин-7-ил)тетрагидро-4H-циклопента[d][1.3]диоксол-4-карбоксилата (A-6)
К раствору A-5 (15 г, 42,6 ммоль) в сухом ТГФ (125 мл) добавляют Pd(PPh3)4 (1,97 г, 1,71 ммоль). К полученному желтому раствору добавляют 1M раствор Zn(Me)2 (171 мл, 171 ммоль). Смесь дегазируют с N2 четыре раза. Желтый раствор нагревают при кипении с обратным холодильником в течение 3 ч, он меняется на темный раствор. Смесь выливают в охлажденный NH4Cl водн. (200 мл) осторожно и экстрагируют EtOAc (200 мл×3). Экстракт промывают насыщенным раствором соли (200 мл), сушат над Na2SO4 и концентрируют в вакууме с получением неочищенного продукта (18 г) в виде желтой камеди. Неочищенный продукт очищают хроматографией на силикагеле, элюируя EtOAc в петролейном эфире от 0 до 100% до A-6 (13,5 г, 96%) в виде желтой камеди. [α]20D -21,57° (c=8,4 мг/мл, метанол). ЖХМС [M+1] 332; 1H ЯМР (400 MГц, ДМСО-d6) δ ч./млн. 8,65 (с, 1H), 7,74 (д, J=3,5 Гц, 1H), 6,74 (д, J=3,8 Гц, 1H), 5,18-5,06 (м, 1H), 5,01-4,90 (м, 2H), 3,65 (с, 3H), 3,15-3,03 (м, 1H), 2,64 (с, 3H), 2,46-2,40 (м, 1H), 1,49 (с, 3H), 1,23 (с, 3H)
Стадия 6: Синтез (3aR,4S,6R,6aS)-2,2-диметил-6-(4-метил-7H-пирроло[2.3-d]пиримидин-7-ил)тетрагидро-4H-циклопента[d][1.3]диоксол-4-карбоновой кислоты (A-7)
Смесь A-6 (13 г, 33 ммоль) и LiOH (2,8 г, 66,7 ммоль) в ТГФ (100 мл)/H2O (100 мл) перемешивают при кт (15°C) в течение 4 ч. ТСХ (ДХМ/MeOH=10/1) показала расход большей части ИМ. Смесь разбавляют водой (50 мл) и промывают EtOAc (100 мл×2). К водному слою добавляют H3PO4 (3,6 г, 37 ммоль) и экстрагируют EtOAc/ТГФ (50 мл/50 мл×5). Экстракт сушат над Na2SO4 и концентрируют в вакууме с получением A-7 (8,9 г, 84%) в виде желтого твердого вещества. ЖХМС [M+1] 318; 1H ЯМР (400 MГц, CDCl3) δ ч./млн. 8,77 (с, 1H), 7,26 (шс, 1H), 6,59 (д, J=3,8 Гц, 1H), 5,25 (т, J=5,3 Гц, 1H), 5,14-4,98 (м, 2H), 3,18 (ддд, J=4,8, 7,8, 10,0 Гц, 1H), 2,77-2,60 (м, 5H), 1,60 (с, 3H), 1,35 (с, 3H)
Стадия 7: Синтез (3aR,4S,6R,6aS)-N-метокси-N,2,2-триметил-6-(4-метил-7H-пирроло[2.3-d]пиримидин-7-ил)тетрагидро-4H-циклопента[d][1.3]диоксол-4-карбоксамида (A-8)
К суспензии неочищенного продукта A-7 (7 г, 22,1 ммоль) и N,O-диметилгидроксиламина-HCl (4,30 г, 44,1 ммоль) в ТГФ (140 мл) добавляют ДИПЭА (11,4 г, 88,2 ммоль) и 50% T3P (28,1 г, 25,8 мл, 44,1 ммоль) при кт (15°C). Полученный красный раствор перемешивают при кт (15°C) в течение 20 ч. ЖХМС показала, что основной пик является желаемым соединением. Смесь разбавляют EtOAc и промывают NH4Cl водн, NaHCO3 водн, насыщенным раствором соли, сушат над Na2SO4 и концентрируют в вакууме до A-8 (7,3 г, 91,8%) в виде желтой камеди. [α]20D +27,30° (c=2,82 мг/мл, метанол); ЖХМС [M+1] 361; 1H ЯМР (400 MГц, CDCl3) δ ч./млн. 8,77 (с, 1H), 7,31 (д, J=3,5 Гц, 1H), 6,60 (д, J=3,5 Гц, 1H), 5,36-5,27 (м, 1H), 5,11-5,03 (м, 1H), 4,91 (дд, J=5,6, 7,2 Гц, 1H), 3,77 (с, 3H), 3,61-3,51 (м, 1H), 3,24 (с, 3H), 2,72 (с, 3H), 2,65-2,56 (м, 1H), 2,54-2,42 (м, 1H), 1,60 (с, 3H), 1,30 (с, 3H).
Стадия 8: Синтез 1-((3aR,4S,6R,6aS)-2,2-диметил-6-(4-метил-7H-пирроло[2.3-d]пиримидин-7-ил)тетрагидро-4H-циклопента[d][1.3]диоксол-4-ил)этан-1-она (A-9)
К светло-желтому раствору A-8 (130 мг, 0,361 ммоль) в ТГФ (5,39 мл) добавляют MeMgBr (3,0 M раствор в диэтиловом эфире, 0,144 мл, 0,433 ммоль) при 0°C. После добавления, смесь перемешивают при 0°C в течение 40 мин. Смесь гасят водн. NH4Cl (40 мл) на ледяной бане и экстрагируют EtOAc (30 мл×2). Экстракт промывают насыщенным раствором соли (20 мл), сушат над Na2SO4 и концентрируют в вакууме с получением неочищенного продукта A-9 (110 мг, 97%) в виде желтого масла и применяют, как есть на следующей стадии.
Стадия 9: Синтез (R)-1-((3aR,4R,6R,6aS)-2,2-диметил-6-(4-метил-7H-пирроло[2.3-d]пиримидин-7-ил)тетрагидро-4H-циклопента[d][1.3]диоксол-4-ил)этан-1-ола (A-10) и (S)-1-((3aR,4R,6R,6aS)-2,2-диметил-6-(4-метил-7H-пирроло[2.3-d]пиримидин-7-ил)тетрагидро-4H-циклопента[d][1.3]диоксол-4-ил)этан-1-ола (A-11)
К желтому раствору A-9 (110 мг, 0,349 ммоль) в сухом MeOH (6 мл) добавляют NaBH4 (26 мг, 0,698 ммоль) при 0°C. После добавления, смесь перемешивают при 0°C в течение 60 мин. Затем реакционную смесь концентрируют в вакууме с получением белого твердого вещества (150 мг), которое отделяют хиральной СЖХ с получением A-10 (62 мг, 56%) и A-11 (61 мг, 55%) и применяют, как есть.
Стадия 10: Синтез (1S,2R,3R,5R)-3-((R)-1-гидроксиэтил)-5-(4-метил-7H-пирроло[2.3-d]пиримидин-7-ил)циклопентан-1,2-диола (A-12) и (1S,2R,3R,5R)-3-((S)-1-гидроксиэтил)-5-(4-метил-7H-пирроло[2.3-d]пиримидин-7-ил)циклопентан-1,2-диола (A-13)
К желтой суспензии A-10 (74 мг, 0,02 ммоль) в H2O (1 мл) добавляют ТФК (1 мл) при 0°C. Смесь перемешивают при комнатной температуре (15°C) в течение 2 ч. Насыщенный водный K2CO3 (10 мл) добавляют в смесь (0°C) медленно, до pH 7-8. Очищают преп. ВЭЖХ с получением соединения A-12 (34 мг, 53%). ЖХМС [M+1] 278; 1H ЯМР (400 MГц, MeOD-d4) δ ч./млн. 8,60 (с, 1H), 7,60 (д, J=3,8 Гц, 1H), 6,73 (д, J=3,5 Гц, 1H), 5,12-5,00 (м, 1H), 4,37 (дд, J=5,8, 9,3 Гц, 1H), 4,17 (дд, J=2,9, 5,6 Гц, 1H), 3,81 (квин, J=6,2 Гц, 1H), 2,71 (с, 3H), 2,31 (тд, J=8,3, 12,7 Гц, 1H), 2,07 (ддт, J=2,8, 5,9, 9,0 Гц, 1H), 1,84 (ддд, J=9,3, 10,9, 12,7 Гц, 1H), 1,26 (д, J=6,3 Гц, 3H)
К желтой суспензии A-11 (74 мг, 0,23 ммоль) в H2O (1,5 мл) добавляют ТФК (1,5 мл) по каплям при 0°C. Затем реакционную смесь перемешивают при комнатной температуре (20°C) в течение 2ч, затем доводят до pH=7 20% K2CO3. Водную фазу was очищают преп. ВЭЖХ с получением A-13 (45 мг, 70%). ЖХМС [M+1] 278; 1H ЯМР (400 MГц, MeOD-d4) δ ч./млн. 8,59 (с, 1 H), 7,61 (д, J=3,53 Гц, 1 H), 6,72 (д, J=3,53 Гц, 1 H), 5,03-5,12 (м, 1 H), 3,92-4,30 (дд, J=8,60, 5,95 Гц, 1 H), 3,92-4,03 (м, 2 H), 2,70 (с, 3 H), 2,28 (дт, J=12,57, 8,27 Гц, 1 H), 2,06 (тт, J=8,71, 4,30 Гц, 1 H), 1,88-1,99 (м, 1 H), 1,22 (д, J=6,62 Гц, 3 H)
Примеры 3-16 получают по методике со схемы A с применением подходящего реагента Гриньяра на стадии 8.
бромид 3,4-дифторфенилмагния
[M+1]
1H ЯМР (700 MГц, ДМСО-d6) δ ч./млн. 8,61 (с, 1 H) 7,65 (д, J=3,74 Гц, 1 H) 7,39-7,45 (м, 1 H) 7,32-7,39 (м, 1 H) 7,19-7,28 (м, 1 H) 6,70 (д, J=3,52 Гц, 1 H) 5,69 (д, J=4,62 Гц, 1 H) 4,93-5,02 (м, 1 H) 4,80 (шс, 1 H) 4,51-4,64 (м, 2 H) 4,23-4,35 (м, 1 H) 3,94 (д, J=4,18 Гц, 1 H) 2,63 (с, 3 H) 2,20-2,30 (м, 1 H) 2,00 (дт, J=12,93, 8,72 Гц, 1 H) 1,60 (ддд, J=12,87, 10,78, 8,25 Гц, 1 H)
бромид 3,4-дифторфенилмагния
[M+1]
1H ЯМР (700 MГц, ДМСО-d6) δ ч./млн. 8,62 (с, 1 H) 7,68 (д, J=3,74 Гц, 1 H) 7,32-7,41 (м, 2 H) 7,14-7,23 (м, 1 H) 6,73 (д, J=3,52 Гц, 1 H) 5,68 (д, J=4,40 Гц, 1 H) 4,90-4,98 (м, 1 H) 4,86 (шс, 1 H) 4,80 (шс, 1 H) 4,67 (шс, 1 H) 4,23 (дд, J=8,14, 5,72 Гц, 1 H) 3,85-3,94 (м, 1 H) 2,65 (с, 3 H) 2,23 (тт, J=8,72, 4,37 Гц, 1 H) 1,85 (дт, J=12,98, 8,36 Гц, 1 H) 1,78 (дт, J=12,87, 9,74 Гц, 1 H)
бромид 3-хлор-4-фторфенилмагния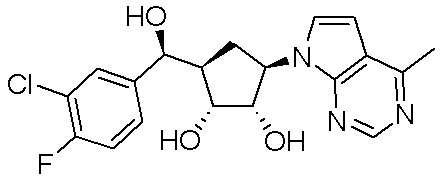
[M+1]
1H ЯМР (700 MГц, ДМСО-d6) δ ч./млн. 8,60 (с, 1 H) 7,64 (д, J=3,52 Гц, 1 H) 7,57 (дд, J=7,26, 1,76 Гц, 1 H) 7,37-7,42 (м, 1 H) 7,32-7,37 (м, 1 H) 6,69 (д, J=3,52 Гц, 1 H) 5,70 (д, J=4,62 Гц, 1 H) 4,93-5,02 (м, 1 H) 4,80 (д, J=6,82 Гц, 1 H) 4,58-4,62 (м, 1 H) 4,57 (д, J=3,52 Гц, 1 H) 4,28 (дт, J=9,57, 6,00 Гц, 1 H) 3,94 (шс, 1 H) 2,63 (с, 3 H) 2,22-2,28 (м, 1 H) 2,00 (дт, J=12,98, 8,80 Гц, 1 H) 1,60 (ддд, J=12,87, 10,78, 8,25 Гц, 1 H)
бромид 3-хлор-4-фторфенилмагния 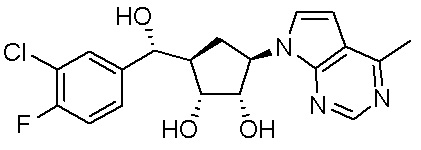
[M+1]
1H ЯМР (700 MГц, ДМСО-d6) δ ч./млн. 8,60 (с, 1 H) 7,66 (д, J=3,52 Гц, 1 H) 7,52 (д, J=6,82 Гц, 1 H) 7,30-7,38 (м, 2 H) 6,71 (д, J=3,52 Гц, 1 H) 5,68 (д, J=4,62 Гц, 1 H) 4,90-4,98 (м, 1 H) 4,86 (шс, 1 H) 4,80 (т, J=4,51 Гц, 1 H) 4,66 (шс, 1 H) 4,23 (шс, 1 H) 3,88 (шс, 1 H) 2,64 (с, 3 H) 2,19-2,27 (м, 1 H) 1,86 (дт, J=12,82, 8,45 Гц, 1 H) 1,78 (дт, J=12,87, 9,74 Гц, 1 H)
бромид 3-хлорфенилмагния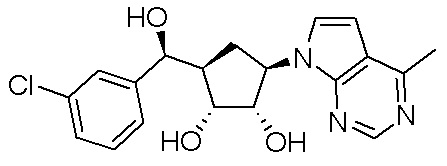
[M+1]
1H ЯМР (700 MГц, ДМСО-d6) δ ч./млн. 8,53-8,63 (м, 1 H) 7,59-7,68 (м, 1 H) 7,44 (с, 1 H) 7,31-7,39 (м, 2 H) 7,23-7,31 (м, 1 H) 6,66-6,73 (м, 1 H) 5,66 (д, J=4,62 Гц, 1 H) 4,93-5,03 (м, 1 H) 4,79 (д, J=6,60 Гц, 1 H) 4,57-4,62 (м, 1 H) 4,55 (шс, 1 H) 4,30 (д, J=3,30 Гц, 1 H) 3,95 (шс, 1 H) 2,58-2,67 (м, 3 H) 2,21-2,31 (м, 1 H) 1,96-2,05 (м, 1 H) 1,55-1,67 (м, 1 H)
бромид 3-хлорфенилмагния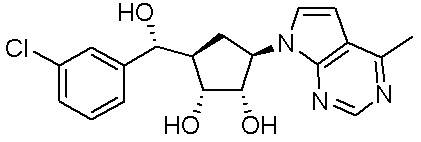
[M+1]
1H ЯМР (700 MГц, ДМСО-d6) δ ч./млн. 8,60 (с, 1 H) 7,67 (д, J=3,74 Гц, 1 H) 7,39 (с, 1 H) 7,32-7,37 (м, 1 H) 7,29-7,32 (м, 1 H) 7,27 (д, J=7,70 Гц, 1 H) 6,71 (д, J=3,52 Гц, 1 H) 5,65 (д, J=4,84 Гц, 1 H) 4,90-4,98 (м, 1 H) 4,85 (шс, 1 H) 4,81 (т, J=4,40 Гц, 1 H) 4,67 (шс, 1 H) 4,24 (шс, 1 H) 3,91 (шс, 1 H) 2,64 (с, 3 H) 2,24 (дт, J=8,47, 4,35 Гц, 1 H) 1,81-1,88 (м, 1 H) 1,75-1,81 (м, 1 H)
бромид 3-фторфенилмагния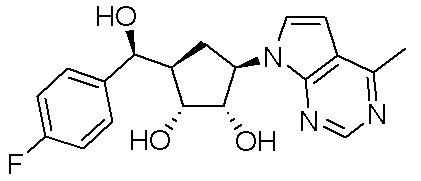
[M+1]
1H ЯМР (400 MГц, ДМСО-d6) δ ч./млн. 8,60 (с, 1 H) 7,62 (д, J=3,55 Гц, 1 H) 7,42 (дд, J=8,50, 5,81 Гц, 2 H) 7,13 (т, J=8,93 Гц, 2 H) 6,68 (д, J=3,55 Гц, 1 H) 5,51 (д, J=4,52 Гц, 1 H) 4,91-5,05 (м, 1 H) 4,76 (д, J=6,97 Гц, 1 H) 4,59 (дд, J=6,79, 4,71 Гц, 1 H) 4,53 (д, J=3,79 Гц, 1 H) 4,32 (дт, J=9,60, 6,08 Гц, 1 H) 3,99 (шс, 1 H) 2,63 (с, 3 H) 2,26 (кв, J=7,34 Гц, 1 H) 1,96 (дт, J=12,72, 8,80 Гц, 1 H) 1,49-1,64 (м, 1 H)
бромид 3-фторфенилмагния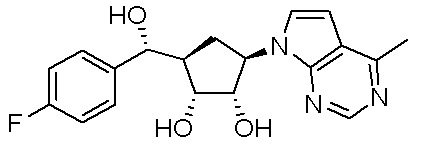
[M+1]
1H ЯМР (700 MГц, ДМСО-d6) δ ч./млн. 8,57 (с, 1 H) 7,65 (д, J=3,52 Гц, 1 H) 7,36 (т, J=6,60 Гц, 2 H) 7,11 (т, J=8,36 Гц, 2 H) 6,73 (д, J=3,74 Гц, 1 H) 5,70 (шс, 1 H) 4,93 (кв, J=9,02 Гц, 2 H) 4,78 (д, J=4,40 Гц, 2 H) 4,19-4,26 (м, 2 H) 2,63 (с, 3 H) 2,18-2,28 (м, 1 H) 1,86 (дт, J=13,04, 8,45 Гц, 1 H) 1,71-1,82 (м, 1 H)
бромид 4-хлорфенилмагния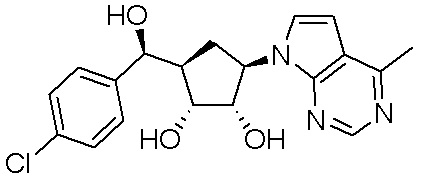
[M+1]
1H ЯМР (400 MГц, ДМСО-d6) δ ч./млн. 1,58 (ддд, J=12,90, 10,64, 8,13 Гц, 1 H) 1,98 (дт, J=12,93, 8,70 Гц, 1 H) 2,20-2,29 (м, 1 H) 2,58-2,66 (м, 3 H) 3,96 (д, J=3,79 Гц, 1 H) 4,25-4,35 (м, 1 H) 4,52 (шс, 1 H) 4,58 (д, J=4,28 Гц, 1 H) 4,76 (шс, 1 H) 4,91-5,04 (м, 1 H) 5,57 (шс, 1 H) 6,70 (д, J=3,55 Гц, 1 H) 7,32-7,45 (м, 4 H) 7,64 (д, J=3,67 Гц, 1 H) 8,61 (с, 1 H)
бромид 4-хлорфенилмагния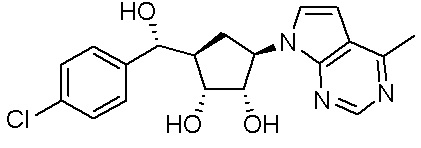
[M+1]
1H ЯМР (400 MГц, ДМСО-d6) δ ч./млн. 1,73-1,92 (м, 2 H) 2,22 (тт, J=8,67, 4,23 Гц, 1 H) 2,67 (с, 3 H) 3,84-3,96 (м, 1 H) 4,24 (дд, J=8,44, 5,50 Гц, 1 H) 4,64 (шс, 1 H) 4,73-4,89 (м, 2 H) 4,89-5,01 (м, 1 H) 5,56 (шс, 1 H) 6,76 (д, J=3,55 Гц, 1 H) 7,33-7,40 (м, 4 H) 7,71 (д, J=3,55 Гц, 1 H) 8,65 (с, 1 H)
бромид 3.4.5-трифторфенилмагния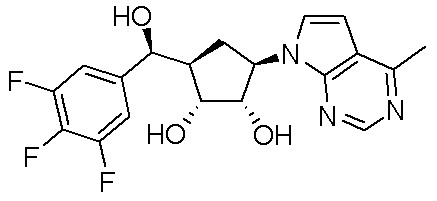
[M+1]
1H ЯМР (700 MГц, ДМСО-d6) δ ч./млн. 8,53 (с, 1 H) 7,59 (д, J=3,52 Гц, 1 H) 7,21-7,29 (м, 2 H) 6,63 (д, J=3,52 Гц, 1 H) 5,76 (д, J=4,40 Гц, 1 H) 4,88-4,95 (м, 1 H) 4,74 (д, J=6,82 Гц, 1 H) 4,48-4,56 (м, 2 H) 4,18 (дт, J=9,52, 6,02 Гц, 1 H) 3,84 (шс, 1 H) 2,56 (с, 2 H) 2,15-2,23 (м, 1 H) 1,97 (дт, J=12,82, 8,67 Гц, 1 H) 1,57 (ддд, J=12,60, 10,95, 8,58 Гц, 1 H)
бромид 3.4.5-трифторфенилмагния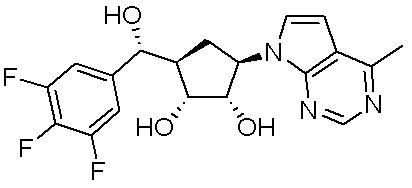
[M+1]
1H ЯМР (700 MГц, ДМСО-d6) δ ч./млн. 8,53 (с, 1 H) 7,58 (д, J=3,52 Гц, 1 H) 7,21 (дд, J=8,69, 6,93 Гц, 2 H) 6,64 (д, J=3,52 Гц, 1 H) 5,75 (д, J=4,84 Гц, 1 H) 4,86 (дт, J=10,07, 8,39 Гц, 1 H) 4,81 (д, J=6,60 Гц, 1 H) 4,75 (т, J=4,62 Гц, 1 H) 4,61 (д, J=4,62 Гц, 1 H) 4,15 (дт, J=8,14, 6,16 Гц, 1 H) 3,85 (кв, J=4,40 Гц, 1 H) 2,57 (с, 2 H) 2,17 (дкв, J=8,80, 4,40 Гц, 1 H) 1,77 (дт, J=12,98, 8,36 Гц, 1 H) 1,69 (дт, J=12,87, 9,85 Гц, 1 H)
Синтез бромида (3.4.5-трифторфенил)магния (для Примеров 13 & 14)
К смеси магния (264 мг, 11,0 ммоль) и ТГФ (5 мл) добавляют йод (5,0 мг) и раствор 5-бром-1.2.3-трифторбензола (211 мг, 1 ммоль) в ТГФ (0,5 мл). Смесь нагревают до 60°C, и раствор 5-бром-1.2.3-трифторбензола (1,9 г, 9 ммоль) в ТГФ (4,5 мл) добавляют по каплям. Реакционную смесь нагревают в течение двух часов, раствор охлаждают до комнатной температуры и применяют сразу на следующей стадии.
Примеры 15 & 16 (Схема B) получают по методике со схемы A с применением 2,6-диметилпиридина и nBuLi на стадии 8 вместо бромида метилмагния.
Схема B
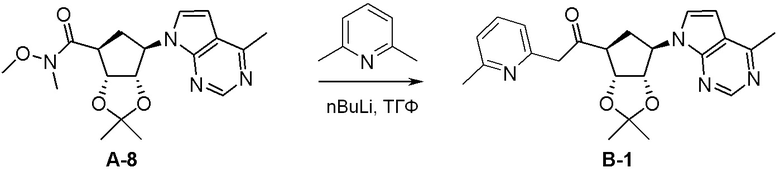
К раствору 2,6-диметилпиридина (168 мг, 1,56 ммоль) в сухом ТГФ (8 мл) при -75°C добавляют n-BuLi (125 мг, 1,96 ммоль, 1,22 мл, 1,6 M) по каплям, температуру поддерживают около -70°C. Полученную суспензию перемешивают при -75°C в течение 1 ч. Раствор A-8 (282 мг, 0,782 ммоль) в сухом ТГФ (4 мл) добавляют при -75°C по каплям. Полученную суспензию перемешивают при -75°C и нагревают до к.т. и перемешивают в течение ночи. ТГФ выпаривают, в неочищенный продукт добавляют H2O и экстрагируют EtOAc, концентрируют, очищают хроматографией на колонке с 5% MeOH/EtOAc с получением 169 мг B-1 (53% выход) в виде желтого масла. ЖХМС [M+1] 407,15.
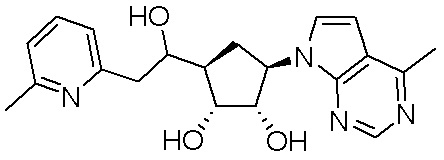
[M+1]
1H ЯМР (700 MГц, ДМСО-d6) δ ч./млн. 8,55 (с, 1 H) 7,60 (д, J=3,52 Гц, 1 H) 7,52 (т, J=7,70 Гц, 1 H) 7,00 (д, J=7,70 Гц, 1 H) 7,01 (д, J=7,48 Гц, 1 H) 6,65 (д, J=3,30 Гц, 1 H) 4,98-5,02 (м, 1 H) 4,92 (кв, J=8,80 Гц, 1 H) 4,73 (д, J=5,72 Гц, 1 H) 4,66 (д, J=3,08 Гц, 1 H) 4,06-4,12 (м, 1 H) 4,03 (дт, J=11,33, 5,56 Гц, 1 H) 3,83 (кв, J=4,25 Гц, 1 H) 2,66-2,75 (м, 2 H) 2,58 (с, 3 H) 2,38 (с, 3 H) 2,09 (дт, J=12,27, 8,06 Гц, 1 H) 1,84-1,89 (м, 1 H) 1,79-1,84 (м, 1 H)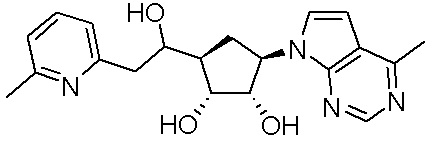
[M+1]
1H ЯМР (700 MГц, ДМСО-d6) δ ч./млн. 8,59 (с, 1 H) 7,66 (д, J=3,52 Гц, 1 H) 7,58 (т, J=7,70 Гц, 1 H) 7,10 (д, J=7,48 Гц, 1 H) 7,07 (д, J=7,48 Гц, 1 H) 6,69 (д, J=3,74 Гц, 1 H) 5,11 (д, J=5,06 Гц, 1 H) 4,93-5,00 (м, 1 H) 4,79 (д, J=6,82 Гц, 1 H) 4,72 (д, J=3,08 Гц, 1 H) 4,20 (дт, J=9,19, 6,30 Гц, 1 H) 4,07-4,12 (м, 1 H) 3,85-3,90 (м, 1 H) 2,87 (дд, J=13,64, 4,40 Гц, 1 H) 2,80 (дд, J=13,86, 8,36 Гц, 1 H) 2,63 (с, 3 H) 2,43 (с, 3 H) 2,13 (дт, J=12,54, 8,36 Гц, 1 H) 1,95-2,03 (м, 1 H) 1,68-1,76 (м, 1 H)
Пример 17 (Схема C): (1S,2R,3S,5R)-3-(1-гидроксициклопропил)-5-(4-метил-7H-пирроло[2.3-d]пиримидин-7-ил)циклопентан-1,2-диол (C-2)
Схема C
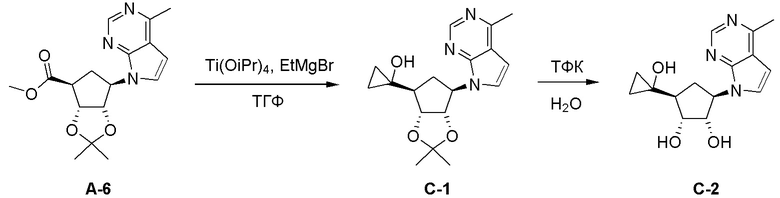
Стадия 1: Синтез 1-((3aR,4S,6R,6aS)-2,2-диметил-6-(4-метил-7H-пирроло[2.3-d]пиримидин-7-ил)тетрагидро-4H-циклопента[d][1.3]диоксол-4-ил)циклопропан-1-ола (C-1)
К светло-желтому раствору A-6 (140 мг, 0,422 ммоль) в сухом ТГФ (6,32 мл) добавляют Ti(OiPr)4 (168 мг, 0,591 ммоль) и EtMgBr (3,0 M раствор в диэтиловом эфире, 0,845 мл, 2,53 ммоль) при кт (13°C). После добавления, смесь перемешивают при 13°C в течение 10 мин. Смесь гасят H2O (50 мл) на ледяной бане и экстрагируют EtOAc (30 мл×2). Экстракт промывают насыщенным раствором соли (30 мл), сушат над Na2SO4 и концентрируют в вакууме с получением неочищенного остатка, который очищают флэш-хроматографией, элюируя EtOAc/ДХМ 0-100% с получением C-1 (90 мг, 65%) в виде желтого твердого вещества. ЖХМС [M+1] 330; 1H ЯМР (400 MГц, CDCl3) δ ч./млн. 8,72 (с, 1H), 7,22 (д, J=3,5 Гц, 1H), 6,55 (д, J=3,8 Гц, 1H), 5,06-4,99 (м, 1H), 4,94 (дд, J=3,3, 6,3 Гц, 1H), 4,86 (д, J=5,8 Гц, 1H), 2,74 (с, 3H), 2,68-2,57 (м, 2H), 2,11 (д, J=3,0 Гц, 1H), 1,59 (с, 3H), 1,34 (с, 3H), 1,25 (д, J=3,8 Гц, 1H), 0,93 (д, 1H), 0,88-0,78 (м, 1H), 0,77-0,67 (м, 1H), 0,60-0,51 (м, 1H)
Стадия 2: Синтез (1S,2R,3S,5R)-3-(1-гидроксициклопропил)-5-(4-метил-7H-пирроло[2.3-d]пиримидин-7-ил)циклопентан-1,2-диола (C-2)
К суспензии C-1 (65 мг, 0,20 ммоль) в H2O (3 мл) добавляют ТФК (3 мл) при 0°C. Смесь перемешивают при кт (13°C) в течение 1,5 ч. Смесь выливают в 20% K2CO3 водн. (50 мл) и экстрагируют EtOAc (30 мл×3). Экстракт промывают насыщенным раствором соли (50 мл×2), сушат над Na2SO4 и концентрируют в вакууме с получением C-2 (50 мг, 88%) в виде твердого вещества. ЖХМС [M+1] 290; 1H ЯМР (400 MГц, ДМСО-d6) δ ч./млн. 8,60 (с, 1H), 7,66 (д, J=3,8 Гц, 1H), 6,70 (д, J=3,5 Гц, 1H), 5,32 (с, 1H), 5,03-4,93 (м, 1H), 4,77 (д, J=6,8 Гц, 1H), 4,61 (д, J=4,3 Гц, 1H), 4,19 (тд, J=6,3, 8,9 Гц, 1H), 4,04-3,97 (м, 1H), 2,63 (с, 3H), 2,19 (тд, J=8,5, 12,8 Гц, 1H), 1,93-1,82 (м, 1H), 1,63 (дт, J=3,0, 8,8 Гц, 1H), 0,67-0,60 (м, 1H), 0,57-0,48 (м, 2H), 0,46-0,39 (м, 1H)
Пример 18 (Схема D): (1S,2R,3S,5R)-3-(2-гидроксипропан-2-ил)-5-(4-метил-7H-пирроло[2.3-d]пиримидин-7-ил)циклопентан-1,2-диол (D-2)
Схема D
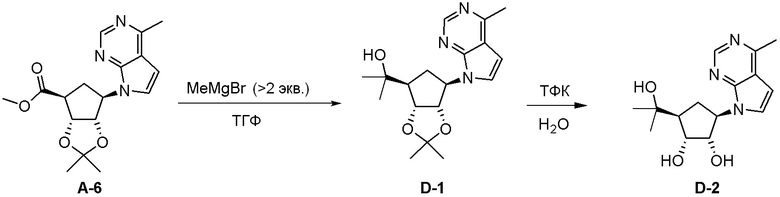
Стадия 1: Синтез 2-((3aR,4S,6R,6aS)-2,2-диметил-6-(4-метил-7H-пирроло[2.3-d]пиримидин-7-ил)тетрагидро-4H-циклопента[d][1.3]диоксол-4-ил)пропан-2-ола (D-1)
К светло-желтому раствору A-6 (150 мг, 0,453 ммоль) в сухом ТГФ (6,77 мл) добавляют MeMgBr (3,0 M раствор в диэтиловом эфире, 0,905 мл, 2,72 ммоль) при кт (13°C). После добавления смесь перемешивают при 13°C в течение 10 мин. Смесь гасят H2O (50 мл) на ледяной бане и экстрагируют EtOAc (30 мл×2). Экстракт промывают насыщенным раствором соли (30 мл), сушат над Na2SO4 и концентрируют в вакууме с получением неочищенного продукта (160 мг) в виде желтого масла, которое очищают флэш-хроматографией, элюируя EtOAc/ДХМ 0-100% с получением D-1 (120 мг, 80%) в виде белого твердого вещества. ЖХМС [M+1] 332; 1H ЯМР (400 MГц, CDCl3) δ ч./млн. 8,76 (с, 1H), 7,26 (д, J=3,8 Гц, 1H), 6,57 (д, J=3,5 Гц, 1H), 5,03 (ддд, J=6,0, 8,3, 10,8 Гц, 1H), 4,96-4,88 (м, 1H), 4,84 (дд, J=5,3, 7,3 Гц, 1H), 2,73 (с, 3H), 2,47-2,37 (м, 2H), 2,31-2,21 (м, 1H), 1,97 (с, 1H), 1,59 (с, 3H), 1,40 (с, 3H), 1,32 (с, 3H), 1,27 (с, 3H)
Стадия 2: Синтез (1S,2R,3S,5R)-3-(2-гидроксипропан-2-ил)-5-(4-метил-7H-пирроло[2.3-d]пиримидин-7-ил)циклопентан-1,2-диола (D-2)
К суспензии D-1 (100 мг, 0,302 ммоль) в H2O (5 мл) добавляют ТФК (5 мл) при 0°C. Смесь перемешивают при кт (13°C) в течение 1,5 ч. Смесь выливают в 20% K2CO3 водн. (50 мл) и экстрагируют EtOAc (30 мл×3). Экстракт промывают насыщенным раствором соли (50 мл×2), сушат над Na2SO4 и концентрируют в вакууме с получением D-2 (70 мг, 80%) в виде желтого твердого вещества. ЖХМС [M+1] 292; 1H ЯМР (400 MГц, ДМСО-d6) δ ч./млн. 8,60 (с, 1H), 7,65 (д, J=3,5 Гц, 1H), 6,69 (д, J=3,5 Гц, 1H), 4,92 (дт, J=7,3, 10,4 Гц, 1H), 4,73 (д, J=7,0 Гц, 1H), 4,53 (д, J=4,3 Гц, 1H), 4,32 (с, 1H), 4,08 (тд, J=6,7, 9,7 Гц, 1H), 3,92 (т, J=6,3 Гц, 1H), 2,63 (с, 3H), 2,03 (тд, J=7,8, 11,5 Гц, 1H), 1,88 (дт, J=2,3, 9,0 Гц, 1H), 1,84-1,73 (м, 1H), 1,19 (с, 3H), 1,08 (с, 3H)
Пример 19 (Схема E): (1S,2R,3S,5R)-3-[(1S)-1-(4-фторфенил)-1-гидроксиэтил]-5-(4-метил-7H-пирроло[2.3-d]пиримидин-7-ил)циклопентан-1,2-диол (E-3)
Пример 20 (Схема E): (1S,2R,3S,5R)-3-[(1R)-1-(4-фторфенил)-1-гидроксиэтил]-5-(4-метил-7H-пирроло[2.3-d]пиримидин-7-ил)циклопентан-1,2-диол (E-4)
Схема E
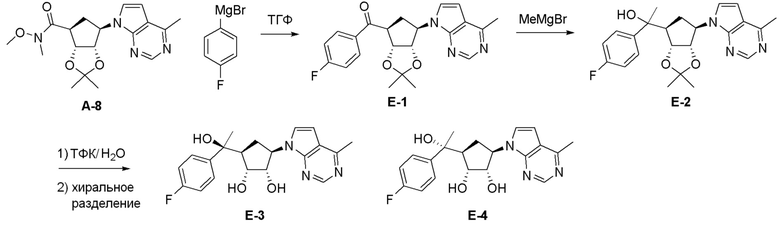
Стадия 1: Синтез ((3aR,4S,6R,6aS)-2,2-диметил-6-(4-метил-7H-пирроло[2.3-d]пиримидин-7-ил)тетрагидро-4H-циклопента[d][1.3]диоксол-4-ил)(4-фторфенил)метанона (E-1)
По методике стадии 8 со схемы A с применением бромида (4-фторфенил)магния получают E-1 (240 мг, 95%). ЖХМС [M+1] 395,80. 1H ЯМР (400 MГц, ХЛОРОФОРМ-d) δ ч./млн. 8,75 (с, 1 H) 8,12 (дд, J=8,86, 5,44 Гц, 2 H) 7,34 (дд, J=8,56, 5,50 Гц, 1 H) 7,15-7,22 (м, 2 H) 6,61 (д, J=3,67 Гц, 1 H) 5,28-5,37 (м, 1 H) 4,98-5,08 (м, 2 H) 3,71 (шс, 1 H) 2,70-2,76 (м, 4 H) 2,60-2,70 (м, 1 H) 1,68 (с, 3 H) 1,33 (с, 3 H)
Стадия 2: Синтез 1-((3aR,4S,6R,6aS)-2,2-диметил-6-(4-метил-7H-пирроло[2.3-d]пиримидин-7-ил)тетрагидро-4H-циклопента[d][1.3]диоксол-4-ил)-1-(4-фторфенил)этан-1-ола (E-2)
К раствору E-1 (66 мг, 0,17 ммоль) в сухом ТГФ (6,0 мл, c=0,106 M) при 50°C добавляют бромид метилмагния (99,5 мг, 0,835 ммоль, 0,278 мл, 3,0 M), полученный раствор перемешивают при 50°C в течение 0,5 ч. Смесь добавляют к станд. NH4Cl (20 мл) медленно, смесь экстрагируют EtOAc (25 мл×3). Экстракт промывают насыщенным раствором соли (25 мл), сушат над Na2SO4 и концентрируют в вакууме с получением 70 мг E-2 в виде бесцветного масла. ЖХМС [M+1] 411,80.
Стадия 3: Синтез (1S,2R,3S,5R)-3-[(1S)-1-(4-фторфенил)-1-гидроксиэтил]-5-(4-метил-7H-пирроло[2.3-d]пиримидин-7-ил)циклопентан-1,2-диол (E-3) и (1S,2R,3S,5R)-3-[(1R)-1-(4-фторфенил)-1-гидроксиэтил]-5-(4-метил-7H-пирроло[2.3-d]пиримидин-7-ил)циклопентан-1,2-диол (E-4)
По методике стадии 10 со схемы A и последующим разделением хиральной СЖХ получают E-3 (19 мг, 31%). ЖХМС [M+1] 371,90. 1H ЯМР (700 MГц, ДМСО-d6) δ ч./млн. 8,61 (с, 1 H) 7,61 (д, J=3,74 Гц, 1 H) 7,54 (дд, J=8,58, 5,72 Гц, 2 H) 7,13 (т, J=8,80 Гц, 2 H) 6,72 (д, J=3,52 Гц, 1 H) 5,26 (с, 1 H) 4,97-5,06 (м, 1 H) 4,70 (д, J=5,94 Гц, 1 H) 4,17 (д, J=3,30 Гц, 1 H) 4,04-4,12 (м, 1 H) 3,54 (шс, 1 H) 2,64 (с, 3 H) 2,39 (т, J=8,91 Гц, 1 H) 2,19 (дт, J=12,54, 8,58 Гц, 1 H) 1,95 (тд, J=11,83, 8,91 Гц, 1 H) 1,35 (с, 3 H)
и E-4 (12 мг, 19%). ЖХМС [M+1] 371,90. 1H ЯМР (700 MГц, ДМСО-d6) δ ч./млн. 8,51-8,59 (м, 1 H) 7,52-7,59 (м, 1 H) 7,40-7,47 (м, 2 H) 7,02-7,10 (м, 2 H) 6,65 (дд, J=3,52, 1,54 Гц, 1 H) 5,24 (с, 1 H) 4,78-4,88 (м, 2 H) 4,67-4,74 (м, 1 H) 4,10-4,19 (м, 2 H) 2,57-2,65 (м, 3 H) 2,35 (т, J=9,35 Гц, 1 H) 1,54 (с, 3 H) 1,39-1,51 (м, 2 H)
Пример 21 (Схема F): (1S,2R,3S,5R)-3-[фтор(4-фторфенил)метил]-5-(4-метил-7H-пирроло[2.3-d] пиримидин-7-ил)циклопентан-1,2-диол (F-3 Изомер A)
Пример 22 (Схема F): (1S,2R,3S,5R)-3-[фтор(4-фторфенил)метил]-5-(4-метил-7H-пирроло[2.3-d]пиримидин-7-ил)циклопентан-1,2-диол (F-4 Изомер B)
Схема F
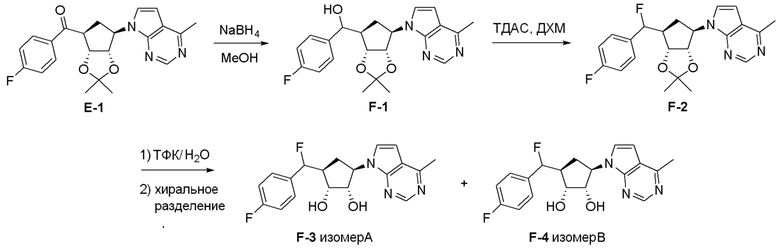
Стадия 1: Синтез ((3aR,4R,6R,6aS)-2,2-диметил-6-(4-метил-7H-пирроло[2.3-d]пиримидин-7-ил)тетрагидро-4H-циклопента[d][1.3]диоксол-4-ил)(4-фторфенил)метанола (F-1)
По методике стадии 8 со схемы A получают F-1 (110 мг, 99%). ЖХМС [M+1] 398,15.
Стадия 2: Синтез 7-((3aS,4R,6S,6aR)-6-(фтор(4-фторфенил)метил)-2,2-диметилтетрагидро-4H-циклопента[d][1.3]диоксол-4-ил)-4-метил-7H-пирроло[2.3-d]пиримидина (F-2)
К раствору F-1 (110 мг, 0,277 ммоль) в 5 мл ДХМ добавляют ТДАС (223 мг, 1,38 ммоль), перемешивают при кт. в течение 30 мин. Реакцию гасят станд. NaHCO3, фазы разделяют и водную фазу экстрагируют 3 порциями ДХМ. Органические фазы объединяют и промывают насыщенным раствором соли, концентрируют с получением неочищенного продукта F-2, который применяют сразу на следующей стадии. ЖХМС [M+1] 400,10.
Стадия 3: Синтез (1S,2R,3S,5R)-3-[фтор(4-фторфенил)метил]-5-(4-метил-7H-пирроло[2.3-d] пиримидин-7-ил)циклопентан-1,2-диола
По методике стадии 10 со схемы A и последующим разделением хиральной СЖХ получают
F-3 Изомер A (7,7 мг, 7,8% две стадии)
ЖХМС [M+1] 360,10. 1H ЯМР (700 MГц, ДМСО-d6) δ ч./млн. 1,40-1,49 (м, 1 H) 1,91 (дт, J=12,87, 8,64 Гц, 1 H) 2,55 (дд, J=17,39, 8,58 Гц, 1 H) 2,62 (с, 3 H) 4,07 (шс, 1 H) 4,36-4,48 (м, 1 H) 4,91-5,02 (м, 3 H) 5,59 (дд, J=1,00 Гц, 1 H) 6,67 (д, J=3,30 Гц, 1 H) 7,22 (т, J=8,58 Гц, 2 H) 7,45-7,56 (м, 2 H) 7,65 (д, J=3,30 Гц, 1 H) 8,60 (с, 1 H)
и F-4 Изомер B (1,3 мг, 1,3% две стадии)
ЖХМС [M+1] 360,15. 1H ЯМР (400 MГц, ДМСО-d6) δ ч./млн. 1,84-1,94 (м, 1 H) 2,09-2,16 (м, 1 H) 2,64 (с, 3 H) 3,83 (кв, J=4,40 Гц, 1 H) 4,24-4,35 (м, 1 H) 4,80 (д, J=4,52 Гц, 1 H) 4,89-5,04 (м, 2 H) 5,72-5,83 (м, 1 H) 6,71 (д, J=3,55 Гц, 1 H) 7,20-7,30 (м, 2 H) 7,47 (дд, J=8,13, 5,81 Гц, 2 H) 7,66 (д, J=3,55 Гц, 1 H) 8,61 (с, 1 H)
Пример 23 (Схема G): (1R,2S,3R,5R)-3-(4-амино-7H-пирроло[2.3-d]пиримидин-7-ил)-5-((S)-(4-хлор-3-фторфенил)(гидрокси)метил)циклопентан-1,2-диол (G-7)
Пример 24 (Схема G): (1R,2S,3R,5R)-3-(4-амино-7H-пирроло[2.3-d]пиримидин-7-ил)-5-((R)-(4-хлор-3-фторфенил)(гидрокси)метил)циклопентан-1,2-диол (G-9)
Схема G
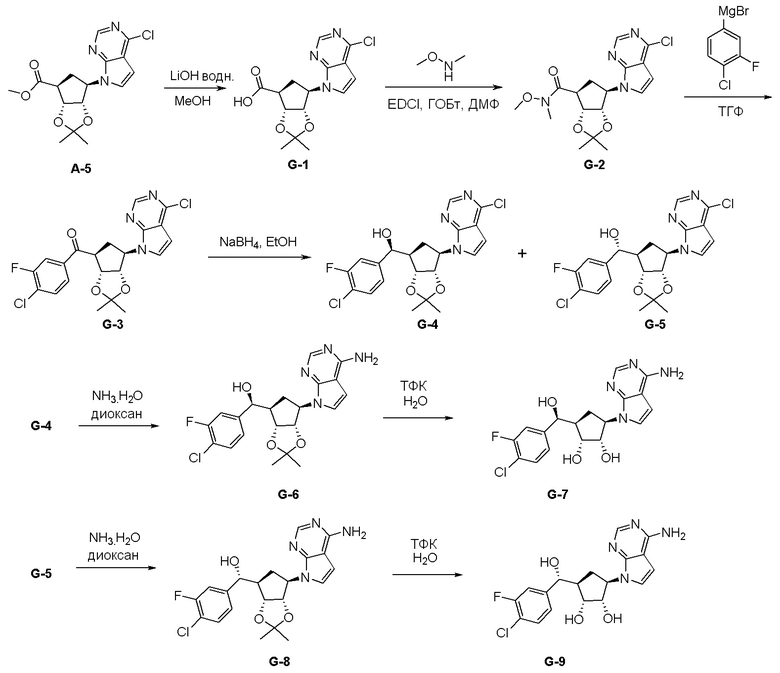
Стадия 1: Синтез (3aR,4S,6R,6aS)-6-(4-хлор-7H-пирроло[2.3-d]пиримидин-7-ил)-2,2-диметилтетрагидро-4H-циклопента[d][1.3]диоксол-4-карбоновой кислоты (G-1)
Смесь A-5 (200 мг, 0,48 ммоль) и LiOH (40,6 мг, 0,966 ммоль) в ТГФ (2 мл)/H2O (2 мл) перемешивают при кт (25°C) в течение 2 ч. Смесь разбавляют водой (5 мл) и доводят 1N HCl до pH=2 и экстрагируют EtOAc (10 мл×2). Экстракт сушат над Na2SO4 и концентрируют в вакууме с получением неочищенного продукта G-1 (200 мг) в виде желтой камеди, применяют сразу на следующей стадии. ЖХМС [M+1] 338; 1H ЯМР (400MГц, ДМСО-d6) δ ч./млн. 8,65 (с, 1H), 7,97 (д, J=3,8 Гц, 1H), 6,72 (д, J=3,5 Гц, 1H), 5,17-5,07 (м, 1H), 5,01-4,90 (м, 2H), 3,03-2,93 (м, 1H), 2,60-2,53 (м, 2H), 1,49 (с, 3H), 1,23 (с, 3H)
Стадия 2: Синтез (3aR,4S,6R,6aS)-6-(4-хлор-7H-пирроло[2.3-d]пиримидин-7-ил)-N-метокси-N,2,2-триметилтетрагидро-4H-циклопента[d][1.3]диоксол-4-карбоксамида (G-2)
К суспензии неочищенного продукта G-1 (200 мг, 461 ммоль) и N,O-диметилгидроксиламин-HCl (70 мг, 0,72 ммоль) в ТГФ (4 мл) добавляют ДИПЭА (186 мг, 1,44 ммоль) и 50% T3P (458 мг, 0,42 мл, 1,5 ммоль) при кт (15°C). Полученный красный раствор перемешивают при кт (15°C) в течение 20 ч. В реакционной смеси образуется некоторое количество твердого вещества. Смесь выливают в NaHCO3 водн. (15 мл) и экстрагируют EtOAc (10 мл×3). Экстракт промывают насыщенным раствором соли (10 мл×2), сушат над Na2SO4 и концентрируют в вакууме с получением G-2 (150 мг, 82% за две стадии) в виде желтой камеди. ЖХМС [M+1] 381; 1H ЯМР (400 MГц, CDCl3) δ ч./млн. 8,66 (с, 1H), 7,42 (д, J=3,5 Гц, 1H), 6,67 (д, J=3,8 Гц, 1H), 5,27 (дд, J=5,1, 7,2 Гц, 1H), 5,06 (дд, J=5,0, 7,0 Гц, 1H), 4,92 (дд, J=5,5, 7,0 Гц, 1H), 3,78 (с, 3H), 3,64-3,51 (м, 1H), 3,25 (с, 3H), 2,70-2,42 (м, 2H), 1,61 (с, 3H), 1,32 (с, 3H)
Стадия 3: Синтез (4-хлор-3-фторфенил)((3aR,4S,6R,6aS)-6-(4-хлор-7H-пирроло[2.3-d]пиримидин-7-ил)-2,2-диметилтетрагидро-4H-циклопента[d][1.3]диоксол-4-ил)метанона (G-3)
К раствору G-2 (150 мг, 0,394 ммоль) в сухом ТГФ (3 мл) добавляют бромид (4-хлор-3-фторфенил)магния (3,1 мл, 1,54 ммоль) при 0°C. Смесь перемешивают при 0°C в течение 3 ч. Смесь гасят NH4Cl водн. (20 мл) и разбавляют EtOAc (10 мл×2). Органический слой промывают насыщенным раствором соли (10 мл), сушат над Na2SO4 и концентрируют в вакууме с получением неочищенного продукта G-3 (250 мг, >99%) в виде желтого масла, которое применяют сразу на следующей стадии. ЖХМС [M+1] 450
Стадия 4: Синтез (S)-(4-хлор-3-фторфенил)((3aR,4R,6R,6aS)-6-(4-хлор-7H-пирроло[2.3-d]пиримидин-7-ил)-2,2-диметилтетрагидро-4H-циклопента[d][1.3]диоксол-4-ил)метанола (G-4) и (R)-(4-хлор-3-фторфенил)((3aR,4R,6R,6aS)-6-(4-хлор-7H-пирроло[2.3-d]пиримидин-7-ил)-2,2-диметилтетрагидро-4H-циклопента[d][1.3]диоксол-4-ил)метанола (G-5)
К раствору неочищенного продукта G-3 (150 мг, 0,4 ммоль) в MeOH (5 мл) добавляют NaBH4 (45 мг, 1,18 ммоль) при 0°C. Смесь перемешивают при 0°C в течение 1 ч. Смесь гасят NH4Cl водн. (10 мл) и разбавляют EtOAc (10 мл×3). Органический слой промывают насыщенным раствором соли (10 мл), сушат над Na2SO4 и концентрируют в вакууме, затем очищают хроматографией на силикагеле, элюируя EtOAc в петролейном эфире от 0 до 60% с получением G-4 (30 мг, 17%) и G-5 (50 мг, 28%) в виде светло-желтого твердого вещества.
G-4: ЖХМС [M+1] 452
G-5: ЖХМС [M+1] 452
Стадия 5: Синтез (S)-((3aR,4R,6R,6aS)-6-(4-амино-7H-пирроло[2.3-d]пиримидин-7-ил)-2,2-диметилтетрагидро-4H-циклопента[d][1.3]диоксол-4-ил)(4-хлор-3-фторфенил)метанола (G-6)
Смесь G-4 (30 мг, 0,0663 ммоль) в диоксане/NH3.H2O (1 мл/1 мл) нагревают при 120°C микроволнами в течение 2 ч. ЖХМС показала расход большей части ИМ, и получается хороший выход. Смесь концентрируют в вакууме с получением неочищенного продукта G-6 (50 мг, >100%) в виде желтого твердого вещества, применяют сразу на следующей стадии. ЖХМС [M+1] 433
Стадия 6: Синтез (1R,2S,3R,5R)-3-(4-амино-7H-пирроло[2.3-d]пиримидин-7-ил)-5-((S)-(4-хлор-3-фторфенил)(гидрокси)метил)циклопентан-1,2-диола (G-7)
Смесь G-6 (30 мг, 0,0663ммоль) в ТФК/H2O (1 мл/1 мл) перемешивают при кт (25°C) в течение 1 ч. ЖХМС показала расход большей части ИМ и основным пиком было желаемое соединение. Смесь выливают в 20% K2CO3 водн. (10 мл) и экстрагируют EtOAc (10 мл×2). Экстракт промывают насыщенным раствором соли (10 мл×2), сушат над Na2SO4 и концентрируют в вакууме с получением неочищенного продукта, который очищают преп.-ВЭЖХ с получением G-7 (10 мг, 37%) в виде белого твердого вещества. ЖХМС [M+1] 393; 1H ЯМР (400 MГц, ДМСО-d6) δ ч./млн. 8,01 (с, 1H), 7,52 (т, J=8,0 Гц, 1H), 7,35 (дд, J=1,6, 10,4 Гц, 1H), 7,27-7,18 (м, 2H), 6,92 (с, 2H), 6,55 (д, J=3,5 Гц, 1H), 5,74 (д, J=5,3 Гц, 1H), 4,85 (д, J=6,5 Гц, 1H), 4,84-4,73 (м, 2H), 4,62 (д, J=4,8 Гц, 1H), 4,24-4,10 (м, 1H), 3,91 (кв, J=4,9 Гц, 1H), 2,21 (тд, J=4,3, 8,5 Гц, 1H), 1,90-1,64 (м, 2H)
Соединение G-9 получают из G-5 (10 мг, 37%) в виде белого твердого вещества по методике получения G-7 из G-4.
G-9: ЖХМС [M+1] 393; 1H ЯМР (400 MГц, ДМСО-d6) δ ч./млн. 8,02 (с, 1H), 7,54 (т, J=8,0 Гц, 1H), 7,40 (дд, J=1,6, 10,7 Гц, 1H), 7,30-7,24 (м, 1H), 7,21 (д, J=3,5 Гц, 1H), 6,92 (с, 2H), 6,54 (д, J=3,5 Гц, 1H), 5,76 (д, J=4,5 Гц, 1H), 4,87-4,76 (м, 2H), 4,61 (т, J=5,5 Гц, 1H), 4,51 (д, J=3,8 Гц, 1H), 4,28-4,19 (м, 1H), 3,93-3,88 (м, 1H), 2,23 (д, J=8,5 Гц, 1H), 2,06-1,94 (м, 1H), 1,66-1,52 (м, 1H)
Пример 25 (Схема H): (1R,2S,3R,5R)-3-(4-амино-5-фтор-7H-пирроло[2.3-d]пиримидин-7-ил)-5-[(S)-(4-фторфенил)(гидрокси)метил]циклопентан-1,2-диол (H-5)
Пример 26 (Схема H): (1R,2S,3R,5R)-3-(4-амино-5-фтор-7H-пирроло[2.3-d]пиримидин-7-ил)-5-[(R)-(4-фторфенил)(гидрокси)метил]циклопентан-1,2-диол (H-6)
Схема H
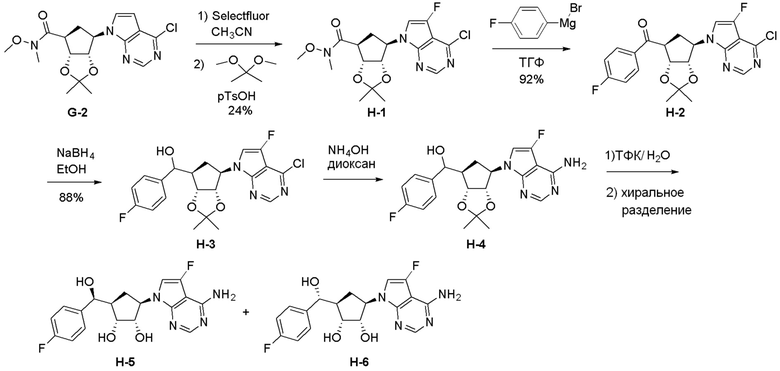
Стадия 1: Синтез (3aR,4S,6R,6aS)-6-(4-хлор-5-фтор-7H-пирроло[2.3-d]пиримидин-7-ил)-N-метокси-N,2,2-триметилтетрагидро-4H-циклопента[d][1.3]диоксол-4-карбоксамида (H-1)
К раствору G-2 (Схема г) (400 мг, 1,05 ммоль) в 20 мл безводного CH3CN добавляют Selectfluor (558 мг, 1,58 ммоль) и AcOH (10 мл). Смесь нагревают при 70°C в течение 6 часов в атмосфере N2. Реакционную смесь концентрируют, добавляют H2O и EtOAc, водный слой экстрагируют 3 раза EtOAc. Органический слой промывают насыщенным раствором соли, сушат над Na2SO4, концентрируют с получением коричневого масла.
В полученное выше коричневое масло добавляют 2,2-диметоксипропан (109 мг, 1,05 ммоль, 3,50 мл, 0,3 M) и моногидрат толуолсульфоновой кислоты (599 мг, 3,15 ммоль), желто-коричневую суспензию энергично перемешивают при кт в течение 15 мин. Реакционную смесь разбавляют 50 мл H2O, нейтрализуют твердым NaHCO3, летучие вещества осторожно удаляют в вакууме, и полученный коричневый водный раствор экстрагируют EtOAc 3×20 мл, органические слои объединяют и концентрируют, очищают хроматографией на колонке с 60% EtOAc/гептан с получением 100 мг H-1 (24% выход) в виде бесцветной камеди.
ЖХМС [M+1] 398,80. 1H ЯМР (400 MГц, ХЛОРОФОРМ-d) δ ч./млн. 8,63 (с, 1 H) 7,22 (д, J=2,57 Гц, 1 H) 5,25-5,36 (м, 1 H) 5,01 (дд, J=7,03, 4,95 Гц, 1 H) 4,83 (дд, J=6,97, 5,50 Гц, 1 H) 3,77 (с, 3 H) 3,58 (д, J=7,34 Гц, 1 H) 3,25 (с, 3 H) 2,58-2,68 (м, 1 H) 2,38-2,52 (м, 1 H) 1,61 (с, 3 H) 1,31 (с, 3 H)
Стадия 2: Синтез ((3aR,4S,6R,6aS)-6-(4-хлор-5-фтор-7H-пирроло[2.3-d]пиримидин-7-ил)-2,2-диметилтетрагидро-4H-циклопента[d][1.3]диоксол-4-ил)(4-фторфенил)метанона (H-2)
По методике стадии 8 со схемы A с применением бромида 4-фторфенилмагния и H-1 получают H-2 (100 мг, 92%).
ЖХМС [M+1] 433,70. 1H ЯМР (400 MГц, ХЛОРОФОРМ-d) δ ч./млн. 8,62 (с, 1 H) 8,07-8,16 (м, 2 H) 7,14-7,23 (м, 3 H) 5,25-5,37 (м, 1 H) 4,98 (дд, J=6,91, 4,58 Гц, 1 H) 4,86-4,93 (м, 1 H) 4,03 (ддд, J=10,30, 7,79, 4,52 Гц, 1 H) 2,58-2,76 (м, 2 H) 1,67 (с, 3 H) 1,32 (с, 3 H)
Стадия 3: Синтез ((3aR,4R,6R,6aS)-6-(4-хлор-5-фтор-7H-пирроло[2.3-d]пиримидин-7-ил)-2,2-диметилтетрагидро-4H-циклопента[d][1.3]диоксол-4-ил)(4-фторфенил)метанола (H-3)
По методике стадии 4 со схемы G, H-2 восстанавливают до H-3 (88 мг, 88%).
ЖХМС [M+1] 435,70.
Стадия 4: Синтез ((3aR,4R,6R,6aS)-6-(4-амино-5-фтор-7H-пирроло[2.3-d]пиримидин-7-ил)-2,2-диметилтетрагидро-4H-циклопента[d][1.3]диоксол-4-ил)(4-фторфенил)метанола (H-4)
По методике стадии 5 со схемы G, H-3 превращают в H-4 в виде неочищенного продукта для следующей стадии.
ЖХМС [M+1] 417,80.
Стадия 5: Синтез (1R,2S,3R,5R)-3-(4-амино-5-фтор-7H-пирроло[2.3-d]пиримидин-7-ил)-5-[(S)-(4-фторфенил)(гидрокси)метил]циклопентан-1,2-диола (H-5) и (1R,2S,3R,5R)-3-(4-амино-5-фтор-7H-пирроло[2.3-d]пиримидин-7-ил)-5-[(R)-(4-фторфенил)(гидрокси)метил]циклопентан-1,2-диола (H-6)
По методике стадии 6 со схемы G и последующей очисткой хиральной СЖХ, снимают защиту с H-4 и изомеры разделяют на H-5 и H-6 (26,5 мг, 35%).
H-5: ЖХМС [M+1] 376,90. 1H ЯМР (700 MГц, ДМСО-d6) δ ч./млн. 8,01 (с, 1 H) 7,40 (дд, J=8,36, 5,72 Гц, 1 H) 7,19 (с, 1 H) 7,13 (т, J=8,80 Гц, 1 H) 6,85 (шс, 1 H) 5,52 (д, J=4,62 Гц, 1 H) 4,87 (кв, J=9,68 Гц, 1 H) 4,77 (д, J=6,82 Гц, 1 H) 4,53 (дд, J=7,04, 4,62 Гц, 1 H) 4,51 (д, J=3,52 Гц, 1 H) 4,16 (дт, J=9,63, 5,97 Гц, 1 H) 3,93 (шс, 1 H) 2,16-2,22 (м, 1 H) 1,86 (дт, J=13,04, 8,78 Гц, 1 H) 1,42 (ддд, J=12,98, 10,56, 8,14 Гц, 1 H)
H-6: ЖХМС [M+1] 376,90. 1H ЯМР (700 MГц, ДМСО-d6) δ ч./млн. 8,01 (с, 1 H) 7,37 (дд, J=8,47, 5,61 Гц, 1 H) 7,23 (д, J=1,76 Гц, 1 H) 7,12 (т, J=8,80 Гц, 1 H) 6,87 (шс, 1 H) 5,51 (д, J=4,84 Гц, 1 H) 4,82-4,88 (м, 1 H) 4,79-4,82 (м, 1 H) 4,76 (т, J=4,84 Гц, 1 H) 4,58 (д, J=4,62 Гц, 1 H) 4,10 (дт, J=8,36, 6,05 Гц, 1 H) 3,80-3,86 (м, 1 H) 2,17 (тт, J=8,64, 4,46 Гц, 1 H) 1,79 (дт, J=13,09, 8,53 Гц, 1 H) 1,68 (дт, J=12,93, 9,60 Гц, 1 H)
Пример 27 (Схема I)-(1S,2R,3R,5R)-3-[(S)-(3,4-дифторфенил)(гидрокси)метил]-5-(7H-пирроло[2.3-d]пиримидин-4-иламино)циклопентан-1,2-диол (I-12)
Пример 28 (Схема I)-(1S,2R,3R,5R)-3-[(R)-(3,4-дифторфенил)(гидрокси)метил]-5-(7H-пирроло[2.3-d]пиримидин-4-иламино)циклопентан-1,2-диол (I-13)
Схема I
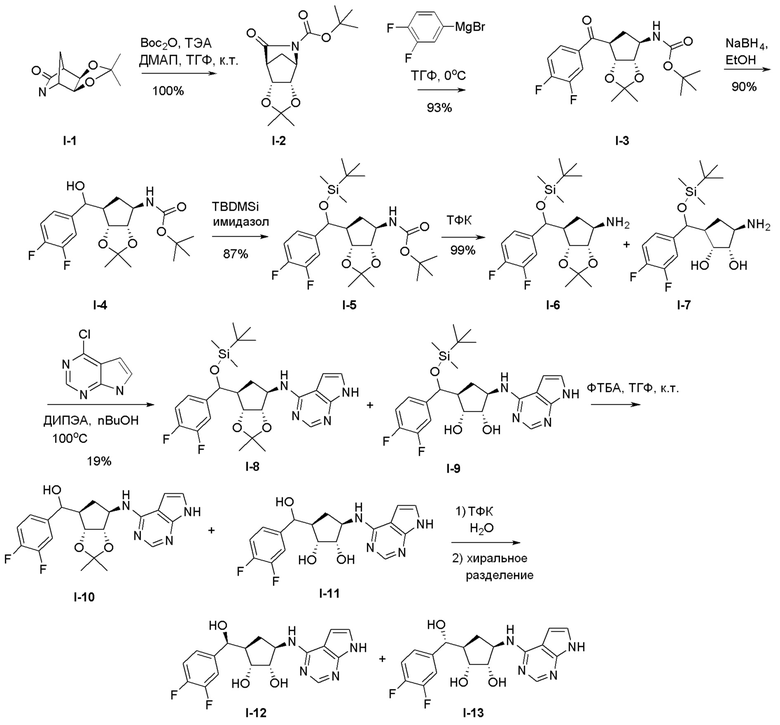
Стадия 1: Синтез трет-бутил (3aS,4R,7S,7aR)-2,2-диметил-6-оксотетрагидро-4,7-метано[1.3]диоксоло[4.5-c]пиридин-5(4H)-карбоксилата (I-2)
К раствору (3aS,4R,7S,7aR)-2,2-диметилтетрагидро-4,7-метано[1.3]диоксоло[4.5-c]пиридин-6(3aH)-она I-1 (Chemical Reviews, 2012, 112 (8), pp 4642-4686) (5000 мг, 27,29 ммоль) и ангидрида Boc (7,15 г, 32,7 ммоль) в ТГФ (54,6 мл, c=0,5 M) добавляют триэтиламин (3,31 г, 32,7 ммоль, 4,56 мл) и ДМАП (333 мг, 2,73 ммоль). Реакционную смесь перемешивают при кт в течение 3 ч. Смесь концентрируют до рыжевато-коричневого твердого вещества, которое очищают хроматографией на колонке с 30% EtOAc/гептаном с получением 7,9 г I-2 (100% выход) в виде белого твердого вещества.
ЖХМС [M+1-tBu] 228. 1H ЯМР (400 MГц, ХЛОРОФОРМ-d) δ ч./млн. 4,60 (д, J=5,4 Гц, 1H), 4,49 (д, J=5,4 Гц, 1H), 4,44 (с, 1H), 2,90 (с, 1H), 2,16-2,07 (м, 1H), 2,05-1,96 (м, 1H), 1,53 (с, 9H), 1,50 (с, 3H), 1,37 (с, 3H).
Стадия 2: Синтез трет-бутил ((3aS,4R,6S,6aR)-6-(3,4-дифторбензоил)-2,2-диметилтетрагидро-4H-циклопента[d][1.3]диоксол-4-ил)карбамат (I-3)
К раствору I-2 (1,42 г, 5,00 ммоль) в ТГФ (10,0 мл, c=0,5 M), охлажденному на бане с ледяной водой, добавляют бромид 4-фторфенилмагния (997 мг, 5 ммоль, 5,00 мл, 1 M). Реакционную смесь перемешивают на ледяной бане в течение 10 мин, гасят добавлением 50 мл MeOH и 50 мл станд. NH4Cl при охлаждении на ледяной бане. Водный слой экстрагируют EtOAc, органический слой концентрируют, очищают хроматографией на колонке с 30% EtOAc/гептан с получением 4,15 г I-3 (93% выход) в виде белого твердого вещества.
ЖХМС [M+1-Boc] 298. 1H ЯМР (400 MГц, ХЛОРОФОРМ-d) δ ч./млн. 7,84-7,95 (м, 2 H) 7,29-7,35 (м, 1 H) 5,43 (шс, 1 H) 4,77 (д, J=5,50 Гц, 1 H) 4,51 (д, J=5,62 Гц, 1 H) 4,18 (шс, 1 H) 3,87 (д, J=8,68 Гц, 1 H) 2,53 (д, J=6,85 Гц, 1 H) 2,04 (д, J=13,94 Гц, 1 H) 1,55 (с, 3 H) 1,45 (с, 9 H) 1,31 (с, 3 H)
Стадия 3: Синтез трет-бутил ((3aS,4R,6R,6aR)-6-((3,4-дифторфенил)(гидрокси)метил)-2,2-диметилтетрагидро-4H-циклопента[d][1.3]диоксол-4-ил)карбамата (I-4)
К раствору I-3 (4531 мг, 11,40 ммоль) в EtOH (57,0 мл, c=0,2 M) добавляют NaBH4 (2160 мг, 57,0 ммоль), перемешивают при кт в течение 30 мин. Реакционную смесь гасят добавлением станд. NH4Cl, экстрагируют EtOAc, органические слои концентрируют, очищают хроматографией на колонке, элюируя 50% EtOAc/гептаном с получением 4120 мг I-4 (90% выход) в виде белого твердого вещества. ЖХМС [M+1-Boc] 300.
Стадия 4: Синтез трет-бутил ((3aS,4R,6S,6aR)-6-(((трет-бутилдиметилсилил)окси)(3,4-дифторфенил)метил)-2,2-диметилтетрагидро-4H-циклопента[d][1.3]диоксол-4-ил)карбамат (I-5)
К раствору I-4 (4300 мг, 10,77 ммоль) и имидазола (2200 мг, 32,3 ммоль) в ДМФ (53,8 мл, c=0,2 M) добавляют хлорид т-бутилдиметилсилила (4870 мг, 32,3 ммоль). Смесь перемешивают при кт в течение ночи. Дополнительный TBSCI (1620 мг, 10,8 ммоль) добавляют и перемешивают при кт в течение ночи. Реакционную смесь гасят H2O и экстрагируют EtOAc (×3). Органический слой сушат над Na2SO4, концентрируют, очищают хроматографией на колонке с 15-20% EtOAc/гептаном с получением 5,27 г I-5 (87% выход) в виде бесцветного масла, которое затвердевает в вакууме. ЖХМС [M+1-Boc] 414.
Стадия 5: Синтез смеси (3aS,4R,6S,6aR)-6-(((трет-бутилдиметилсилил)окси)(3,4-дифторфенил)метил)-2,2-диметилтетрагидро-4H-циклопента[d][1.3]диоксол-4-амина (I-6) и (1R,2S,3R,5S)-3-амино-5-(((трет-бутилдиметилсилил)окси)(3,4-дифторфенил)метил)циклопентан-1,2-диола (I-7)
I-5 (1250 мг, 2,433 ммоль) в ТФК (5,0 мл, c=0,5 M) перемешивают при кт в течение 10-15 мин. В реакционную смесь добавляют EtOAc и нейтрализуют станд. NaHCO3 до pH около 7, водный слой экстрагируют EtOAc, органические слои объединяют и сушат над Na2SO4, концентрируют с получением 1 г (99% выход) в виде светло-желтого масла в виде смеси двух соединений I-6 и I-7.
Стадия 6: Синтез смеси N-((3aS,4R,6S,6aR)-6-(((трет-бутилдиметилсилил)окси)(3,4-дифторфенил)метил)-2,2-диметилтетрагидро-4H-циклопента[d][1.3]диоксол-4-ил)-7H-пирроло[2.3-d]пиримидин-4-амина (I-8) и (1R,2S,3R,5S)-3-((7H-пирроло[2.3-d]пиримидин-4-ил)амино)-5-(((трет-бутилдиметилсилил)окси)(3,4-дифторфенил)метил)циклопентан-1,2-диола (I-9)
К указанным выше соединениям I-6 и I-7 (154 мг, 0,372 ммоль) в 4 мл раствора nBuOH добавляют 4-хлор-7H-пирроло[2.3-d]пиримидин (85,8 мг, 0,559 ммоль) и ДИПЭА (96,3 мг, 0,745 ммоль, 0,123 мл), нагревают при 100°C в течение 2 дней. Реакционную смесь охлаждают до к.т., добавляют EtOAc. Органический слой промывают H2O три раза и затем насыщенным раствором соли, сушат над Na2SO4, концентрируют, очищают хроматографией на колонке, элюируя 80% EtOAc/гептаном до 10% EtOAc/MeOH с получением 35,3 мг (19% выход) смеси двух соединений I-8 и I-9 в виде коричневого масла.
Стадия 7: Синтез смеси ((3aR,4R,6R,6aS)-6-((7H-пирроло[2.3-d]пиримидин-4-ил)амино)-2,2-диметилтетрагидро-4H-циклопента[d][1.3]диоксол-4-ил)(3,4-дифторфенил)метанола (I-10) и (1R,2S,3R,5R)-3-((7H-пирроло[2.3-d]пиримидин-4-ил)амино)-5-((3,4-дифторфенил)(гидрокси)метил)циклопентан-1,2-диола (I-11)
К смеси полученных выше соединений I-8 и I-9 (49 мг, 0,10 ммоль) в 1 мл ТГФ добавляют ФТБА (39,2 мг, 0,150 ммоль, 0,15 мл, 1,0 M) по каплям, перемешивают при кт в течение 0,5 ч, концентрируют и применяют неочищенный продукт на следующей стадии.
Стадия 8: Синтез (1S,2R,3R,5R)-3-[(S)-(3,4-дифторфенил)(гидрокси)метил]-5-(7H-пирроло[2.3-d]пиримидин-4-иламино)циклопентан-1,2-диола (I-12) и (1S,2R,3R,5R)-3-[(R)-(3,4-дифторфенил)(гидрокси)метил]-5-(7H-пирроло[2.3-d]пиримидин-4-иламино)циклопентан-1,2-диола (I-13)
Смесь полученных выше соединений I-10 и I-11 (42 мг, 0,10 ммоль) растворяют в ТФК/H2O (1 мл/1 мл) и перемешивают при кт в течение 2 ч. После завершения, смесь концентрируют с получением неочищенного масла, которое растворяют в H2O, промывают EtOAc (1 мл×2). Водный слой лиофилизируют и смесь разделяют сверхкритической CO2 жидкостной хроматографией с получением (1S,2R,3R,5R)-3-[(S)-(3,4-дифторфенил)(гидрокси)метил]-5-(7H-пирроло[2.3-d]пиримидин-4-иламино)циклопентан-1,2-диола I-12 (5,88 мг, 15%) и (1S,2R,3R,5R)-3-[(R)-(3,4-дифторфенил)(гидрокси)метил]-5-(7H-пирроло[2.3-d]пиримидин-4-иламино)циклопентан-1,2-диола I-13 (5,72 мг, 15%).
I-12: ЖХМС [M+1] 376,90. 1H ЯМР (700 MГц, ДМСО-d6) δ ч./млн. 1,06-1,12 (м, 1 H) 1,88-1,96 (м, 1 H) 2,15-2,21 (м, 1 H) 2,88 (кв, J=6,83 Гц, 1 H) 3,77 (т, J=6,15 Гц, 1 H) 3,88 (шс, 1 H) 4,24-4,32 (м, 1 H) 4,47 (д, J=5,98 Гц, 1 H) 6,53 (шс, 1 H) 7,06 (шс, 1 H) 7,15 (шс, 1 H) 7,25-7,36 (м, 3 H) 7,99 (шс, 1 H)
I-13: ЖХМС [M+1] 376,90. 1H ЯМР (700 MГц, ДМСО-d6) δ ч./млн. 1,32-1,40 (м, 1 H) 1,81 (дт, J=12,55, 8,07 Гц, 1 H) 2,10-2,20 (м, 1 H) 2,88 (кв, J=6,83 Гц, 1 H) 3,75-3,81 (м, 1 H) 3,83 (шс, 1 H) 4,18-4,31 (м, 1 H) 4,73 (шс, 1 H) 6,55 (шс, 1 H) 7,07 (шс, 1 H) 7,14 (шс, 1 H) 7,24-7,41 (м, 3 H) 8,00 (шс, 1 H)
Примеры 29 и 30 получают по методике со схемы I с применением 4-хлорквинaзолин-8-карбонитрила на стадии 6 вместо 4-хлор-7H-пирроло[2.3-d]пиримидина.
Изомер A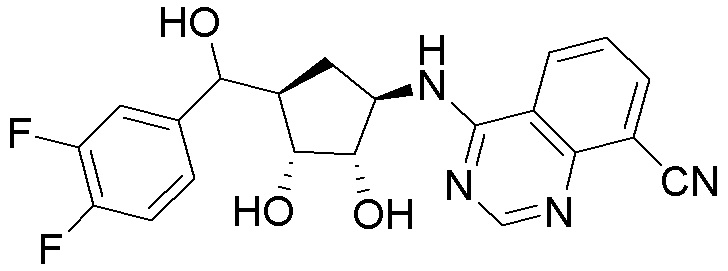
1H ЯМР (700 MГц, ДМСО-d6) δ ч./млн. 1,40 (дт, J=13,11, 8,82 Гц, 1 H) 1,85 (дт, J=13,15, 8,20 Гц, 1 H) 2,17 (дт, J=8,80, 4,31 Гц, 1 H) 3,86 (кв, J=4,61 Гц, 1 H) 3,88-3,95 (м, 1 H) 4,43 (т, J=7,69 Гц, 1 H) 4,75 (т, J=4,10 Гц, 1 H) 7,09-7,16 (м, 1 H) 7,22-7,32 (м, 2 H) 7,58 (т, J=7,86 Гц, 1 H) 8,21 (д, J=7,34 Гц, 1 H) 8,38 (д, J=7,34 Гц, 1 H) 8,40-8,46 (м, 2 H)
Изомер B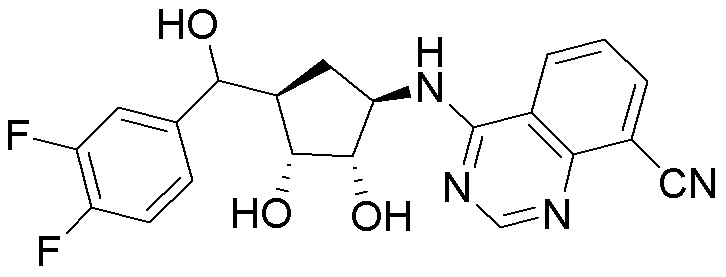
1H ЯМР (700 MГц, ДМСО-d6) δ ч./млн. 1,13-1,18 (м, 1 H) 1,92-1,99 (м, 1 H) 2,16-2,25 (м, 1 H) 3,92 (шс, 2 H) 4,42-4,52 (м, 2 H) 7,11-7,15 (м, 1 H) 7,22-7,26 (м, 1 H) 7,26-7,30 (м, 1 H) 7,55-7,60 (м, 1 H) 8,21 (дд, J=7,34, 1,20 Гц, 1 H) 8,40-8,44 (м, 2 H)
Пример 31 (Схема J)-(1S,2R,3R,5R)-3-[(S)-(3,4-дифторфенил)(гидрокси)метил]-5-[метил(пиримидин-4-ил)амино]циклопентан-1,2-диол (J-5)
Пример 32 (Схема J)-(1S,2R,3R,5R)-3-[(R)-(3,4-дифторфенил)(гидрокси)метил]-5-[метил(пиримидин-4-ил)амино]циклопентан-1,2-диол (J-5)
Схема J
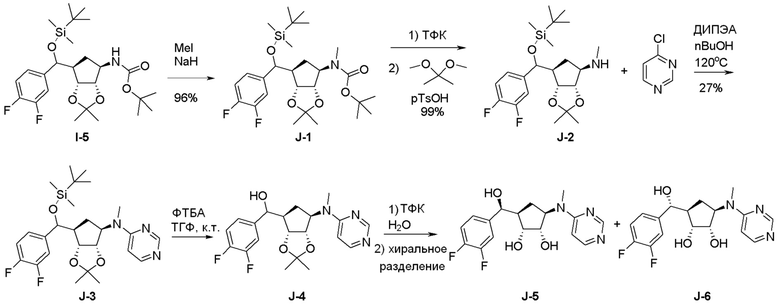
Стадия 1: Синтез трет-бутил ((3aS,4R,6S,6aR)-6-(((трет-бутилдиметилсилил)окси)(3,4-дифторфенил)метил)-2,2-диметилтетрагидро-4H-циклопента[d][1.3]диоксол-4-ил)(метил)карбамата (J-1)
Гидрид натрия (304 мг, 7,59 ммоль) добавляют порциями к раствору I-5 (1300 мг, 2,531 ммоль) в ДМФ (15 мл, 0,1 M) в атмосфере азота, через 5 мин добавляют йодметан (1080 мг, 7,59 ммоль), перемешивают при кт в течение 30 мин. Реакционную смесь добавляют H2O, экстрагируют EtOAc, концентрируют и очищают хроматографией на колонке с 20% EtOAc/гептаном с получением 1290 мг J-1 (96% выход) в виде бесцветного масла, ЖХМС [M+1-Boc] 428,1.
Стадия 2: Синтез (3aS,4R,6S,6aR)-6-(((трет-бутилдиметилсилил)окси)(3,4-дифторфенил)метил)-N,2,2-триметилтетрагидро-4H-циклопента[d][1.3]диоксол-4-амина (J-2)
J-1 (967 мг, 1,83 ммоль) в ТФК (5,0 мл, c=0,1 M) перемешивают при кт в течение 10-15 мин. В реакционную смесь добавляют EtOAc и станд. NaHCO3 для нейтрализации до pH около 7, экстрагируют EtOAc, органический слой сушат над Na2SO4, концентрируют с получением бесцветного масла.
Полученное выше масло растворяют в диметилацетале ацетона (10 мл, c=0,1 M), добавляют толуол-4-сульфоновую кислоту (349 мг, 1,83 ммоль), перемешивают при кт в течение 15 мин. В реакционную смесь добавляют стд. NaHCO3, экстрагируют EtOAc, органический слой промывают насыщенным раствором соли, сушат над Na2SO4, концентрируют с получением 0,78 г J-2 (99% выход) в виде светло-желтого масла. ЖХМС [M+1] 428,15.
Стадия 3: Синтез N-((3aS,4R,6S,6aR)-6-(((трет-бутилдиметилсилил)окси)(3,4-дифторфенил)метил)-2,2-диметилтетрагидро-4H-циклопента[d][1.3]диоксол-4-ил)-N-метилпиримидин-4-амина (J-3)
Смесь J-2 (200 мг, 0,468 ммоль), 4-хлорпиримидин (64,3 мг, 0,561 ммоль) и ДИПЭА (121 мг, 0,935 ммоль, 0,155 мл) в н-бутаноле (2,0 мл, c=0,2 M) нагревают при 120°C в течение ночи. Реакционную смесь охлаждают до к.т., растворитель удаляют. Остаток добавляют EtOAc, промывают H2O 3 раза и затем насыщенным раствором соли, сушат над Na2SO4 и концентрируют, очищают хроматографией на колонке с 70% EtOAc/гептаном с получением 64 мг J-3 (27% выход) в виде коричневого масла. ЖХМС [M+1] 506,20.
Стадия 4: Синтез (3,4-дифторфенил)((3aR,4R,6R,6aS)-2,2-диметил-6-(метил(пиримидин-4-ил)амино)тетрагидро-4H-циклопента[d][1.3]диоксол-4-ил)метанола (J-4)
По методике стадии 7 со схемы I, снимают защиту с J-3 с получением J-4 в виде неочищенного продукта для следующей стадии. ЖХМС [M+1] 392,15.
Стадия 5: Синтез (1S,2R,3R,5R)-3-[(S)-(3,4-дифторфенил)(гидрокси)метил]-5-[метил(пиримидин-4-ил)амино]циклопентан-1,2-диола (J-5) и (1S,2R,3R,5R)-3-[(R)-(3;4-дифторфенил)(гидрокси)метил]-5-[метил(пиримидин-4-ил)амино]циклопентан-1,2-диола (J-6)
По методике стадии 8 со схемы I, снимают защиту с J-4 (50 мг, 0,13 ммоль) с получением J-5 (19,73 мг, 44%) и J-6 (3,14 мг, 7%).
J-5: ЖХМС [M+1] 351,90. 1H ЯМР (700 MГц, ДМСО-d6) δ ч./млн. 1,18-1,31 (м, 1 H) 1,63 (д, J=9,68 Гц, 1 H) 2,08-2,19 (м, 1 H) 2,90 (шс, 3 H) 3,73-3,82 (м, 1 H) 3,90 (дд, J=9,35, 5,17 Гц, 1 H) 4,38 (шс, 1 H) 4,49 (д, J=5,94 Гц, 1 H) 4,63 (шс, 1 H) 5,60 (шс, 1 H) 6,57-6,69 (м, 1 H) 7,21 (шс, 1 H) 7,31-7,42 (м, 2 H) 8,02-8,14 (м, 1 H) 8,41 (шс, 1 H)
J-6: ЖХМС [M+1] 351,90. 1H ЯМР (700 MГц, ДМСО-d6) δ ч./млн. 1,42 (шс, 2 H) 2,10 (шс, 1 H) 2,90 (шс, 3 H) 3,76 (шс, 1 H) 3,86-3,97 (м, 1 H) 4,54 (шс, 1 H) 4,65 (шс, 1 H) 4,70-4,78 (м, 1 H) 5,58 (шс, 1 H) 6,63 (шс, 1 H) 7,17 (шс, 1 H) 7,28-7,40 (м, 2 H) 8,09 (шс, 1 H) 8,41 (шс, 1 H)
Примеры 33-36 получают по методике стадии со схемы J с применением хлорпиримидинов, перечисленных на стадии 3.
4-хлор-6-метилпиримидин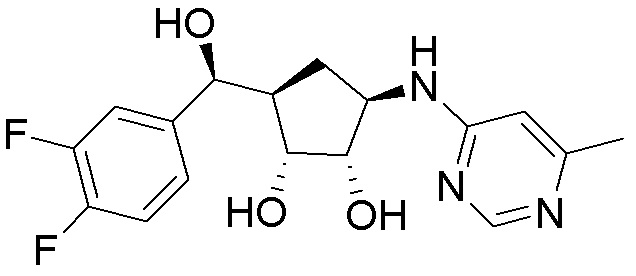
1H ЯМР (700 MГц, ДМСО-d6) δ ч./млн. 1,18-1,31 (м, 1 H) 1,61 (д, J=10,34 Гц, 1 H) 2,13 (д, J=7,04 Гц, 1 H) 2,22 (с, 3 H) 2,88 (шс, 3 H) 3,77 (шс, 1 H) 3,84-3,94 (м, 1 H) 4,32-4,41 (м, 1 H) 4,49 (т, J=5,06 Гц, 1 H) 4,61 (шс, 1 H) 5,61 (д, J=4,40 Гц, 1 H) 6,49 (шс, 1 H) 7,15-7,23 (м, 1 H) 7,32-7,43 (м, 2 H) 8,28 (с, 1 H)
4-хлор-6-метилпиримидин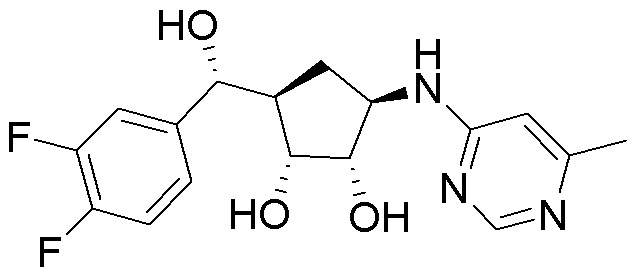
1H ЯМР (700 MГц, ДМСО-d6) δ ч./млн. 1,40 (т, J=9,24 Гц, 2 H) 2,09 (дт, J=8,14, 4,29 Гц, 1 H) 2,22 (с, 3 H) 2,89 (шс, 3 H) 3,71-3,78 (м, 1 H) 3,86-3,95 (м, 1 H) 4,46-4,54 (м, 1 H) 4,62 (шс, 1 H) 4,73 (т, J=4,40 Гц, 1 H) 5,58 (д, J=4,62 Гц, 1 H) 6,50 (шс, 1 H) 7,16 (шс, 1 H) 7,29-7,39 (м, 2 H) 8,28 (с, 1 H)
4-хлор-2-метилпиримидин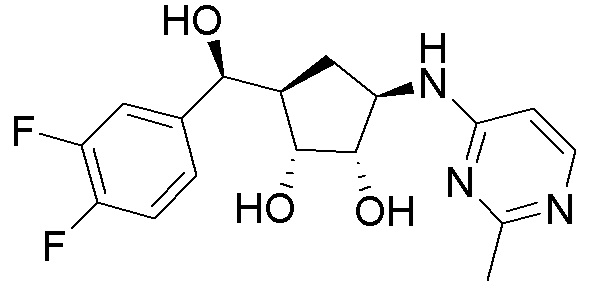
1H ЯМР (700 MГц, ДМСО-d6) δ ч./млн. 1,17-1,32 (м, 1 H) 1,57-1,68 (м, 1 H) 2,08-2,19 (м, 1 H) 2,31 (шс, 3 H) 2,88 (шс, 3 H) 3,77 (шс, 1 H) 3,84-3,94 (м, 1 H) 4,37 (шс, 1 H) 4,49 (шс, 1 H) 4,63 (шс, 1 H) 5,61 (шс, 1 H) 6,43 (шс, 1 H) 7,21 (шс, 1 H) 7,37 (д, J=8,58 Гц, 2 H) 7,99 (шс, 1 H)
4-хлор-2-метилпиримидин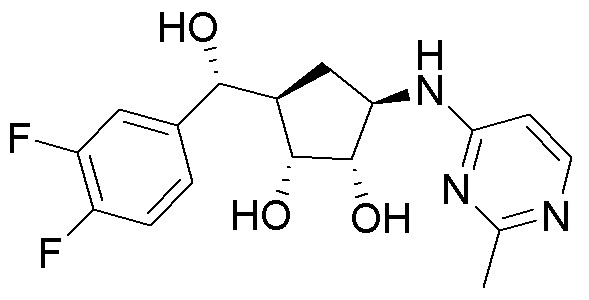
1H ЯМР (700 MГц, ДМСО-d6) δ ч./млн. 1,35-1,48 (м, 2 H) 2,10 (тт, J=8,53, 4,24 Гц, 1 H) 2,31 (с, 3 H) 2,89 (шс, 3 H) 3,71-3,79 (м, 1 H) 3,91 (дт, J=8,42, 6,02 Гц, 1 H) 4,52 (д, J=3,96 Гц, 1 H) 4,65 (шс, 1 H) 4,73 (т, J=4,51 Гц, 1 H) 5,59 (д, J=4,62 Гц, 1 H) 6,43 (д, J=5,06 Гц, 1 H) 7,12-7,19 (м, 1 H) 7,29-7,38 (м, 2 H) 7,99 (д, J=5,94 Гц, 1 H)
Пример 37 (Схема K)-(1R,2S,3R,5R)-3-[(4-амино-1,3,5-триазин-2-ил)(метил)амино]-5-[(S)-(3,4-дифторфенил)(гидрокси)метил]циклопентан-1,2-диол (K-4)
Пример 38 (Схема K)-(1R,2S,3R,5R)-3-[(4-амино-1,3,5-триазин-2-ил)(метил)амино]-5-[(R)-(3,4-дифторфенил)(гидрокси)метил]циклопентан-1,2-диол (K-5)
Схема K
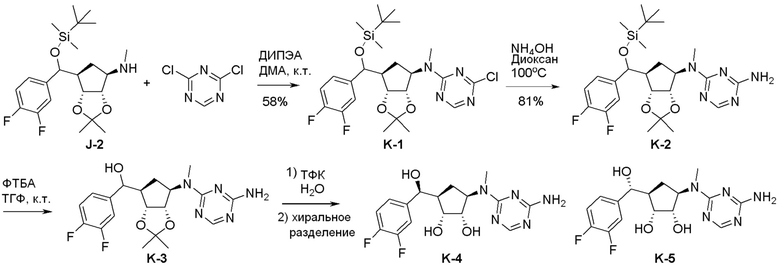
Стадия 1: Синтез N-((3aS,4R,6S,6aR)-6-(((трет-бутилдиметилсилил)окси)(3,4-дифторфенил)метил)-2,2-диметилтетрагидро-4H-циклопента[d][1.3]диоксол-4-ил)-4-хлор-N-метил-1,3,5-триазин-2-амина (K-1)
По методике стадии 3 со схемы J с применением 2,4-дихлор-[1,3,5]триазина с J-2 получают K-1 (267 мг, 58%).
Стадия 2: Синтез N2-((3aS,4R,6S,6aR)-6-(((трет-бутилдиметилсилил)окси)(3,4-дифторфенил)метил)-2,2-диметилтетрагидро-4H-циклопента[d][1.3]диоксол-4-ил)-N2-метил-1,3,5-триазинe-2,4-диамин (K-2)
По методике стадии 5 со схемы G, K-1 превращают в K-2 (86 мг, 81%). ЖХМС [M+1] 522.
Стадия 3: Синтез ((3aR,4R,6R,6aS)-6-((4-амино-1,3,5-триазин-2-ил)(метил)амино)-2,2-диметилтетрагидро-4H-циклопента[d][1.3]диоксол-4-ил)(3,4-дифторфенил)метанола (K-3)
По методике стадии 4 со схемы J, снимают защиту с K-2 с ФТБА с получением K-3 в виде неочищенного продукта для следующей стадии.
Стадия 4: Синтез (1R,2S,3R,5R)-3-[(4-амино-1,3,5-триазин-2-ил)(метил)амино]-5-[(S)-(3,4-дифторфенил)(гидрокси)метил]циклопентан-1,2-диола (K-4) и (1R,2S,3R,5R)-3-[(4-амино-1,3,5-триазин-2-ил)(метил)амино]-5-[(3R)-(3,4-дифторфенил)(гидрокси)метил]циклопентан-1,2-диола (K-5)
По методике стадии 5 со схемы J с последующим разделением хиральной СЖХ, K-3 превращают в K-4 (12 мг, 20%) и K-5 (48 мг, 80%).
K-4: ЖХМС [M+1] 367,90, 1H ЯМР (400 MГц, ДМСО-d6) δ ч./млн. 1,12-1,23 (м, 1 H) 1,52-1,64 (м, 1 H) 2,08 (шс, 1 H) 2,96 (д, J=9,78 Гц, 3 H) 3,78 (шс, 1 H) 3,87 (шс, 1 H) 4,48 (д, J=5,38 Гц, 1 H) 4,94 (шс, 1 H) 5,55 (шс, 1 H) 6,95 (с, 1 H) 7,08 (шс, 2 H) 7,21 (шс, 1 H) 7,30-7,45 (м, 2 H) 8,07 (шс, 1 H)
K-5: ЖХМС [M+1] 367,90. 1H ЯМР (700 MГц, ДМСО-d6) δ ч./млн. 1,31-1,45 (м, 2 H) 2,05 (шс, 1 H) 2,93 (м, 3 H) 3,72 (шс, 1 H) 3,90 (шс, 1 H) 4,65-4,72 (м, 1 H) 4,86 (д, J=15,03 Гц, 1 H) 7,14 (шс, 1 H) 7,25-7,35 (м, 2 H) 7,98 (шс, 1 H) 8,30 (шс, 2 H)
Примеры 39-42 получают по методике стадии со схемы K с применением 2,4-дихлорпиримидина (Примеры 39 & 40) или 2,4-дихлор-6-метил-1,3,5-триазина (Примеры 41 & 42), указанных на стадии 1.
2,4-дихлорпиримидин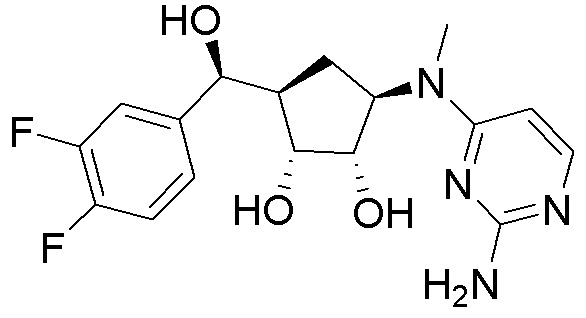
1H ЯМР (700 MГц, ДМСО-d6) δ ч./млн. 1,17-1,22 (м, 1 H) 1,53-1,60 (м, 1 H) 2,06-2,11 (м, 1 H) 2,81 (шс, 3 H) 3,75 (шс, 1 H) 3,84-3,89 (м, 1 H) 4,32 (д, J=3,08 Гц, 1 H) 4,48 (т, J=5,28 Гц, 1 H) 4,60 (д, J=5,94 Гц, 1 H) 5,59 (д, J=4,62 Гц, 1 H) 5,81 (с, 2 H) 5,87 (д, J=5,94 Гц, 1 H) 7,20 (шс, 1 H) 7,32-7,40 (м, 2 H) 7,67 (д, J=5,72 Гц, 1 H)
2,4-дихлорпиримидин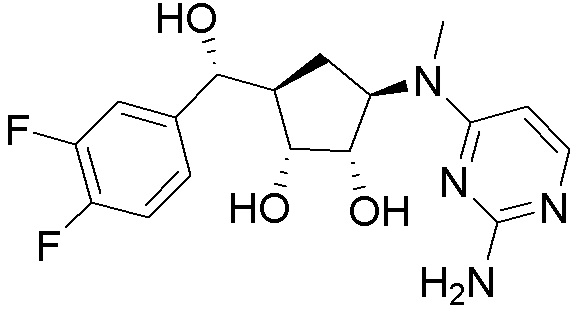
1H ЯМР (700 MГц, ДМСО-d6) δ ч./млн. 1,30-1,44 (м, 2 H) 2,05 (дт, J=8,25, 4,24 Гц, 1 H) 2,82 (шс, 3 H) 3,74 (шс, 1 H) 3,88 (шс, 1 H) 4,48 (шс, 1 H) 4,62 (шс, 1 H) 4,72 (шс, 1 H) 5,57 (д, J=4,40 Гц, 1 H) 5,81 (шс, 2 H) 5,87 (д, J=5,06 Гц, 1 H) 7,09-7,19 (м, 1 H) 7,28-7,38 (м, 2 H) 7,67 (шс, 1 H)
2,4-дихлор-6-метил-1,3,5-триазин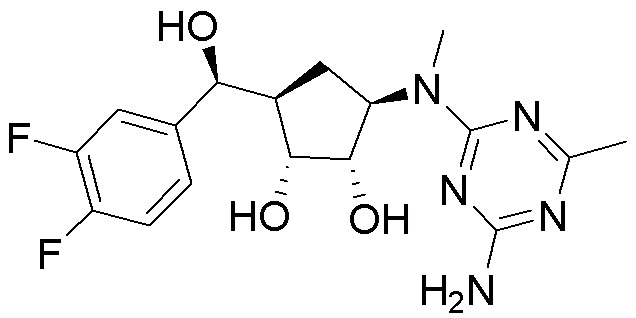
1H ЯМР (700 MГц, ДМСО-d6) δ ч./млн. 1,18 (д, J=13,66 Гц, 1 H) 1,49-1,59 (м, 1 H) 2,12 (д, J=14,69 Гц, 4 H) 2,91 (д, J=19,81 Гц, 3 H) 3,79 (шс, 1 H) 3,88 (шс, 1 H) 4,46 (шс, 1 H) 4,88 (шс, 1 H) 7,16 (шс, 1 H) 7,23-7,36 (м, 2 H)
2,4-дихлор-6-метил-1,3,5-триазин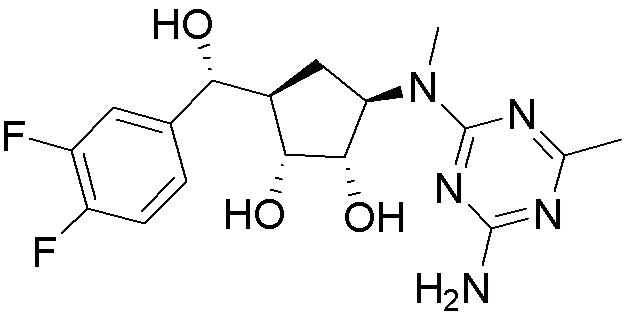
1H ЯМР (700 MГц, ДМСО-d6) δ ч./млн. 1,31-1,45 (м, 2 H) 2,07 (д, J=18,62 Гц, 4 H) 2,82-2,96 (м, 3 H) 3,72 (шс, 1 H) 3,87 (шс, 1 H) 4,68 (шс, 1 H) 4,94-4,78 (м, 1 H) 6,37 (шс, 2 H) 7,13 (шс, 1 H) 7,22-7,34 (м, 2 H)
Пример 43 (Схема L)-(1R,2S,3R,5R)-3-[(6-аминопиримидин-4-ил)(метил)амино]-5-[(S)-(3,4-дифторфенил)(гидрокси)метил]циклопентан-1,2-диол (L-4)
Пример 44 (Схема L)-(1R,2S,3R,5R)-3-[(6-аминопиримидин-4-ил)(метил)амино]-5-[(R)-(3,4-дифторфенил)(гидрокси)метил]циклопентан-1,2-диол (L-5)
Схема L
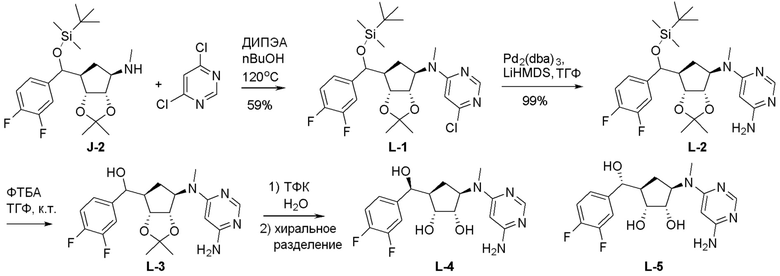
Стадия 1: Синтез N-((3aS,4R,6S,6aR)-6-(((трет-бутилдиметилсилил)окси)(3,4-дифторфенил)метил)-2,2-диметилтетрагидро-4H-циклопента[d][1.3]диоксол-4-ил)-6-хлор-N-метилпиримидин-4-амина (L-1)
По методике стадии 1 со схемы K, J-2 обрабатывают 4,6-дихлорпиримидином в н-бутанолом и ДИПЭА с получением L-1 (188 мг, 60%). ЖХМС [M+1] 540,20.
Стадия 2: Синтез N4-((3aS,4R,6S,6aR)-6-(((трет-бутилдиметилсилил)окси)(3,4-дифторфенил)метил)-2,2-диметилтетрагидро-4H-циклопента[d][1.3]диоксол-4-ил)-N4-метилпиримидин-4,6-диамина (L-2)
К раствору L-1 (188 мг, 0,348 ммоль) в ТГФ (10 мл, c=0,035 M) добавляют трис(дибензилиденацетон)дипалладий (41,4 мг, 0,045 ммоль) и дициклогексил(2-фенилфенил)фосфан (33,7 мг, 0,092 ммоль), закрывают крышкой и продувают азотом, затем добавляют LHMDS (153 мг, 0,905 ммоль, 0,90 мл, 1,0 M), нагревают при 75°C в течение 18 ч. Реакционную смесь охлаждают до к.т., разбавляют водой и EtOAc, слои разделяют. Органическую фазу сушат над Na2SO4, фильтруют и концентрируют. Продукт очищают хроматографией на колонке с 80%-100% EtOAc/гептаном с получением 180 мг L-2 (99% выход) в виде светло-желтого масла. ЖХМС [M+1] 521,30.
Стадия 3: Синтез ((3aR,4R,6R,6aS)-6-((6-аминопиримидин-4-ил)(метил)амино)-2,2-диметилтетрагидро-4H-циклопента[d][1.3]диоксол-4-ил)(3,4-дифторфенил)метанола (L-3)
По методике стадии 3 со схемы K, L-2 обрабатывают ФТБА с получением L-3 в виде неочищенного продукта для следующей стадии. ЖХМС [M+1] 407,20.
Стадия 4: Синтез (1R,2S,3R,5R)-3-[(6-аминопиримидин-4-ил)(метил)амино]-5-[(S)-(3,4-дифторфенил)(гидрокси)метил]циклопентан-1,2-диола (L-4) и (1R,2S,3R,5R)-3-[(6-аминопиримидин-4-ил)(метил)амино]-5-[(R)-(3,4-дифторфенил)(гидрокси)метил]циклопентан-1,2-диола (L-5)
По методике стадии 4 со схемы K, L-3 обрабатывают ТФК в воде. L-4 (31 мг, 25%) и L-5 (11 мг, 9%) выделяют после обработки и очистки хиральной СЖХ.
L-4: ЖХМС [M+1] 366,90. 1H ЯМР (700 MГц, ДМСО-d6) δ ч./млн. 1,13-1,26 (м, 1 H) 1,55 (дт, J=12,60, 8,56 Гц, 1 H) 2,04-2,13 (м, 1 H) 2,77 (с, 2 H) 3,71-3,79 (м, 1 H) 3,81-3,88 (м, 1 H) 4,27-4,35 (м, 1 H) 4,47 (т, J=5,28 Гц, 1 H) 4,55 (д, J=5,72 Гц, 1 H) 5,47 (с, 1 H) 5,58 (д, J=4,62 Гц, 1 H) 6,03 (шс, 2 H) 7,15-7,23 (м, 1 H) 7,30-7,40 (м, 2 H) 7,86 (с, 1 H)
L-5: ЖХМС [M+1] 366,90. 1H ЯМР (700 MГц, ДМСО-d6) δ ч./млн. 1,29-1,42 (м, 2 H) 1,99-2,09 (м, 1 H) 2,78 (шс, 3 H) 3,74 (шс, 1 H) 3,86 (д, J=7,04 Гц, 1 H) 4,41-4,49 (м, 1 H) 4,52-4,60 (м, 1 H) 4,72 (шс, 1 H) 5,47 (с, 1 H) 5,56 (д, J=3,96 Гц, 1 H) 6,03 (шс, 1 H) 7,15 (шс, 1 H) 7,28-7,38 (м, 2 H) 7,86 (с, 1 H)
Пример 46 (Схема M)-(1S,2R,3R,5R)-3-[(S)-(3,4-дифторфенил)(гидрокси)метил]-5-[метил(4-метил-1,3,5-триазин-2-ил)амино]циклопентан-1,2-диол (M-4)
Пример 47 (Схема M)-(1S,2R,3R,5R)-3-[(R)-(3,4-дифторфенил)(гидрокси)метил]-5-[метил(4-метил-1,3,5-триазин-2-ил)амино]циклопентан-1,2-диол (M-5)
Схема M
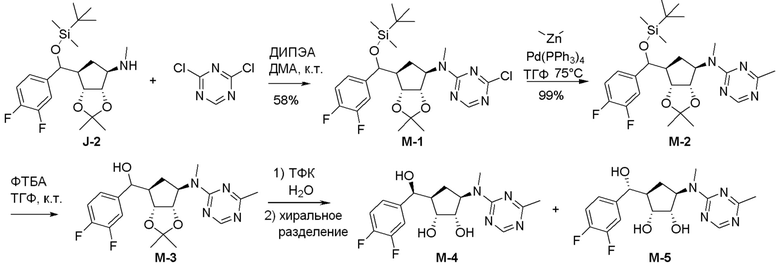
Стадия 1: Синтез N-((3aS,4R,6S,6aR)-6-(((трет-бутилдиметилсилил)окси)(3,4-дифторфенил)метил)-2,2-диметилтетрагидро-4H-циклопента[d][1.3]диоксол-4-ил)-4-хлор-N-метил-1,3,5-триазин-2-амина (M-1)
Реакционную смесь J-2 (360 мг, 0,842 ммоль), 2,4-дихлор[1,3,5]триазина (152 мг, 1,01 ммоль) и ДИПЭА (218 мг, 1,68 ммоль, 0,279 мл) в ДМА (5 мл, 0,16 M) перемешивают при кт в течение 5 ч. В реакционную смесь добавляют EtOAc, промывают H2O 3 раза и затем насыщенным раствором соли. Органический слой сушат над Na2SO4 и концентрируют, очищают хроматографией на колонке с 20% EtOAc/гептаном с получением 267 мг M-1 (58,6% выход) в виде бесцветного масла. ЖХМС [M+1] 541.
Стадия 2: Синтез N-((3aS,4R,6S,6aR)-6-(((трет-бутилдиметилсилил)окси)(3,4-дифторфенил)метил)-2,2-диметилтетрагидро-4H-циклопента[d][1.3]диоксол-4-ил)-N,4-диметил-1,3,5-триазин-2-амина (M-2)
M-2 (176 мг, 99%) получают из M-1 по методике стадии 5 со схемы A. ЖХМС [M+1] 521.
Стадия 3: Синтез (3,4-дифторфенил)((3aR,4R,6R,6aS)-2,2-диметил-6-(метил(4-метил-1,3,5-триазин-2-ил)амино)тетрагидро-4H-циклопента[d][1.3]диоксол-4-ил)метанола (M-3)
По методике стадии 3 со схемы K, M-2 обрабатывают ФТБА с получением M-3 в виде неочищенного продукта для следующей стадии. ЖХМС [M+1] 407.
Стадия 4: Синтез (1S,2R,3R,5R)-3-((S)-(3,4-дифторфенил)(гидрокси)метил)-5-(метил(4-метил-1,3,5-триазин-2-ил)амино)циклопентан-1,2-диола (M-4) и (1S,2R,3R,5R)-3-((R)-(3,4-дифторфенил)(гидрокси)метил)-5-(метил(4-метил-1,3,5-триазин-2-ил)амино)циклопентан-1,2-диола (M-5)
По методике стадии 4 со схемы K, M-3 обрабатывают ТФК в воде. M-4 (78,97 мг, 64%) и M-5 (24,48 мг, 20%) выделяют после обработки и очистки хиральной СЖХ.
M-4: ЖХМС [M+1] 366,90. 1H ЯМР (400 MГц, ДМСО-d6) δ ч./млн. 1,26-1,34 (м, 1 H) 1,56-1,69 (м, 1 H) 2,13 (шс, 1 H) 2,24-2,32 (м, 3 H) 3,00 (с, 3 H) 3,78 (шс, 1 H) 3,91 (шс, 1 H) 4,33 (д, J=15,89 Гц, 1 H) 4,48 (шс, 1 H) 4,60 (шс, 1 H) 4,92-5,04 (м, 1 H) 5,56 (д, J=4,40 Гц, 1 H) 7,22 (д, J=4,52 Гц, 1 H) 7,30-7,43 (м, 2 H) 8,39 (д, J=14,67 Гц, 1 H)
M-5: ЖХМС [M+1] 366,90. 1H ЯМР (700 MГц, ДМСО-d6) δ ч./млн. 1,36-1,48 (м, 2 H) 2,09 (шс, 1 H) 2,27 (д, J=23,74 Гц, 3 H) 2,98 (д, J=4,10 Гц, 3 H) 3,73 (шс, 1 H) 3,90 (м, 1 H) 4,70 (шс, 1 H) 4,93 (т, J=9,99 Гц, 1 H) 7,15 (шс, 1 H) 7,31 (д, J=9,39 Гц, 2 H) 8,34 (д, J=33,65 Гц, 1 H)
Пример 48 (Схема N)-(1R,2S,3R,5R)-3-(2-амино-4-метил-7H-пирроло[2.3-d]пиримидин-7-ил)-5-[(S)-(3,4-дифторфенил)(гидрокси)метил]циклопентан-1,2-диол (N-5)
Пример 49 (Схема N)-(1R,2S,3R,5R)-3-(2-амино-4-метил-7H-пирроло[2.3-d]пиримидин-7-ил)-5-[(R)-(3,4-дифторфенил)(гидрокси)метил]циклопентан-1,2-диол (N-6)
Схема N
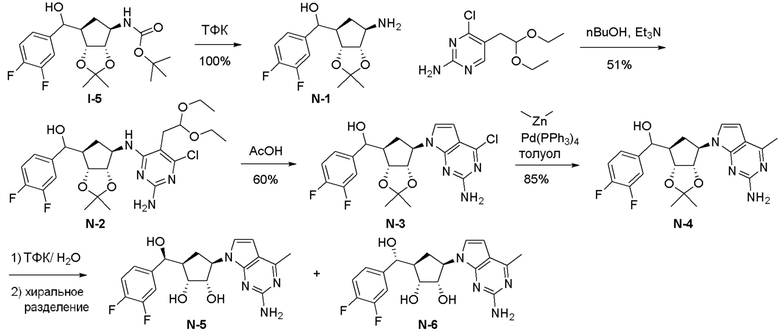
Стадия 1: Синтез ((3aR,4R,6R,6aS)-6-амино-2,2-диметилтетрагидро-4H-циклопента[d][1.3]диоксол-4-ил)(3,4-дифторфенил)метанола (N-1)
I-5 (616 мг, 1,54 ммоль) в ТФК (3 мл, c=0,5 M) перемешивают при кт в течение 10-15 мин. Реакционную смесь добавляют EtOAc и нейтрализуют станд. NaHCO3 до pH около 7, водный слой экстрагируют EtOAc 3 раза, органический слой сушат над Na2SO4, концентрируют с получением 460 мг N-1 (100% выход) в виде белого твердого вещества, применяют на следующей стадии. ЖХМС [M+1] 300,15.
Стадия 2: Синтез ((3aR,4R,6R,6aS)-6-((2-амино-6-хлор-5-(2,2-диэтоксиэтил)пиримидин-4-ил)амино)-2,2-диметилтетрагидро-4H-циклопента[d][1.3]диоксол-4-ил)(3,4-дифторфенил)метанола (N-2)
Смесь N-1 (214 мг, 0,714 ммоль), 4,6-дихлор-5-(2,2-диэтоксиэтил)пиримидин-2-амина (200 мг, 0,714 ммоль), и триэтиламина (289 мг, 2,86 ммоль) в 10 мл nBuOH нагревают при 135°C в герметично закрытой пробирке в течение 40 ч. Реакционную смесь охлаждают до к.т., концентрируют, добавляют H2O, экстрагируют EtOAc, продукт очищают хроматографией на колонке с 50-60% EtOAc/гептаном с получением 200 мг N-2 (51% выход) в виде желтого твердого вещества. ЖХМС [M+1] 543,20.
Стадия 3: Синтез ((3aR,4R,6R,6aS)-6-(2-амино-4-хлор-7H-пирроло[2.3-d]пиримидин-7-ил)-2,2-диметилтетрагидро-4H-циклопента[d][1.3]диоксол-4-ил)(3,4-дифторфенил)метанола (N-3)
N-2 (200 мг, 0,368 ммоль) добавляют к уксусной (15 мл, c=0,025 M), раствор нагревают при 100°C в течение 1 ч. Летучие вещества удаляют в вакууме, остаток добавляют EtOAc, нейтрализуют станд. NaHCO3, слои разделяют, и водный слой экстрагируют EtOAc 3 раза. Органический слой концентрируют, очищают хроматографией на колонке с 50% EtOAc/гептаном с получением 100 мг N-3 (60% выход) в виде беловатого твердого вещества. ЖХМС [M+1] 451,10.
Стадия 4: Синтез ((3aR,4R,6R,6aS)-6-(2-амино-4-метил-7H-пирроло[2.3-d]пиримидин-7-ил)-2,2-диметилтетрагидро-4H-циклопента[d][1.3]диоксол-4-ил)(3,4-дифторфенил)метанола (N-4)
По методике стадии 5 со схемы A, N-3 превращают в N-4 (69 мг, 85%). ЖХМС [M+1] 431,15.
Стадия 5: Синтез (1R,2S,3R,5R)-3-(2-амино-4-метил-7H-пирроло[2.3-d]пиримидин-7-ил)-5-[(S)-(3,4-дифторфенил)(гидрокси)метил]циклопентан-1,2-диола (N-5) и (1R,2S,3R,5R)-3-(2-амино-4-метил-7H-пирроло[2.3-d]пиримидин-7-ил)-5-[(R)-(3,4-дифторфенил)(гидрокси)метил]циклопентан-1,2-диола (N-6)
По методике стадии 4 со схемы K, N-4 обрабатывают ТФК в воде. N-5 (14,6 мг, 23%) и N-6 (6,6 мг, 11%) выделяют после последовательной обработки и очистки хиральной СЖХ.
N-5: ЖХМС [M+1] 390,85. 1H ЯМР (700 MГц, ДМСО-d6) δ ч./млн. 7,35-7,40 (м, 1 H) 7,29-7,35 (м, 1 H) 7,21 (шс, 1 H) 7,07 (д, J=3,74 Гц, 1 H) 6,38 (д, J=3,52 Гц, 1 H) 5,90 (с, 2 H) 5,74 (д, J=4,40 Гц, 1 H) 4,83 (д, J=6,60 Гц, 1 H) 4,71-4,79 (м, 1 H) 4,51-4,58 (м, 2 H) 4,15-4,21 (м, 1 H) 3,90 (шс, 1 H) 2,38 (с, 3 H) 2,18 (д, J=7,92 Гц, 1 H) 1,83-1,93 (м, 1 H) 1,44 (д, J=10,34 Гц, 1 H)
N-6: ЖХМС [M+1] 390,90. 1H ЯМР (700 MГц, ДМСО-d6) δ ч./млн. 7,29-7,36 (м, 2 H) 7,16 (шс, 1 H) 7,12 (д, J=3,52 Гц, 1 H) 6,40 (д, J=3,30 Гц, 1 H) 5,89 (с, 2 H) 5,76 (шс, 1 H) 4,86 (д, J=5,94 Гц, 1 H) 4,76 (т, J=4,62 Гц, 1 H) 4,73 (кв, J=8,88 Гц, 1 H) 4,68 (шс, 1 H) 4,08-4,12 (м, 1 H) 3,83-3,86 (м, 1 H) 2,39 (с, 3 H) 2,16 (дт, J=8,53, 4,21 Гц, 1 H) 1,76 (дт, J=12,98, 8,47 Гц, 1 H) 1,57-1,64 (м, 1 H)
Пример 50 (Схема O) - (1S,2R,3S,5R)-3-[2-амино-1-фтор-1-(4-фторфенил)этил]-5-(4-метил-7H-пирроло[2.3-d]пиримидин-7-ил)циклопентан-1,2-диол (O-5)
Пример 51 (Схема O) - (1S,2R,3R,5R)-3-[(1S)-2-амино-1-(4-фторфенил)этил]-5-(4-метил-7H-пирроло[2.3-d]пиримидин-7-ил)циклопентан-1,2-диол (O-6)
Пример 52 (Схема O) - (1S,2R,3R,5R)-3-[(1R)-2-амино-1-(4-фторфенил)этил]-5-(4-метил-7H-пирроло[2.3-d]пиримидин-7-ил)циклопентан-1,2-диол (O-7)
Схема O
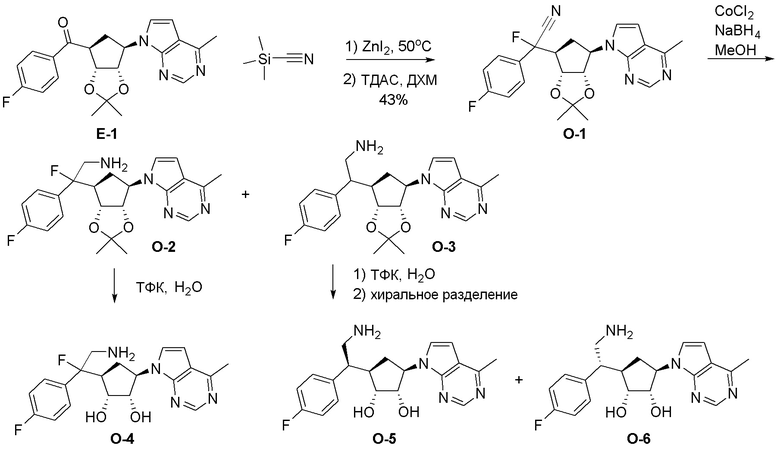
Стадия 1: Синтез 2-((3aR,4S,6R,6aS)-2,2-диметил-6-(4-метил-7H-пирроло[2.3-d]пиримидин-7-ил)тетрагидро-4H-циклопента[d][1.3]диоксол-4-ил)-2-фтор-2-(4-фторфенил)ацетонитрила (O-1)
К смеси E-1 (175 мг, 0,443 ммоль) и йодида цинка(II) (2,12 мг, 0,00664 ммоль) а герметично закрываемой пробирке добавляют 2 мл безводного CH2Cl2, затем цианид триметилсилила (0,25 мл) под азотом. Реакционную смесь герметично закрывают и нагревают при 50°C в течение 2 дней. Полученную смесь охлаждают на ледяной бане, ТДАС (78,5 мг, 0,487 ммоль) добавляют по каплям, перемешивают при кт в течение 15 мин. Реакционную смесь промывают H2O, 0,5 N HCl, станд. NaHCO3, насыщенным раствором соли и концентрируют. Продукт очищают хроматографией на колонке с 50% EtOAc/гептаном с получением 80 мг O-1 (43% выход) в виде желтого масла. ЖХМС [M+1] 425,10.
Стадия 2: Синтез 2-((3aR,4S,6R,6aS)-2,2-диметил-6-(4-метил-7H-пирроло[2.3-d]пиримидин-7-ил)тетрагидро-4H-циклопента[d][1.3]диоксол-4-ил)-2-фтор-2-(4-фторфенил)этан-1-амина (O-2) и 2-((3aR,4R,6R,6aS)-2,2-диметил-6-(4-метил-7H-пирроло[2.3-d]пиримидин-7-ил)тетрагидро-4H-циклопента[d][1.3]диоксол-4-ил)-2-(4-фторфенил)этан-1-амина (O-3)
O-1 (70 мг, 0,16 ммоль) растворяют в 0,5 мл MeOH. Добавляют гексагидрат хлорида кобальта (II) (118 мг, 0,495 ммоль) и смесь охлаждают на ледяной бане. Добавляют NaBH4 (32,8 мг, 0,825 ммоль) и реакционную смесь перемешивают при 0°C в течение 0,5 ч. Реакционную смесь гасят станд. NH4Cl, экстрагируют EtOAc, органический слой промывают насыщенным раствором соли, концентрируют, очищают преп. ТСХ на тарелке с 5% MeOH/CH2Cl2 с получением 20 мг O-2 (28% выход) в виде желтого масла, (ЖХМС [M+1] 429,10) и 22 мг O-3 (32% выход) в виде желтого масла. (ЖХМС [M+1] 411,20)
Стадия 3: Синтез (1S,2R,3S,5R)-3-[2-амино-1-фтор-1-(4-фторфенил)этил]-5-(4-метил-7H-пирроло[2.3-d]пиримидин-7-ил)циклопентан-1,2-диола (O-4)
По методике стадии 4 со схемы K, O-2 обрабатывают ТФК в воде. O-4 (1,2 мг, 6,6%) выделяют после последовательной обработки и очистки СЖХ.
ЖХМС [M+1] 388,90. 1H ЯМР (400 MГц, МЕТАНОЛ-d4) δ ч./млн. 1,62-1,73 (м, 1 H) 1,81 (с, 3 H) 2,54-2,73 (м, 4 H) 4,17 (шс, 1 H) 4,76-4,94 (м, 2 H) 6,59 (д, J=3,42 Гц, 1 H) 7,04 (т, J=8,68 Гц, 2 H) 7,29-7,42 (м, 3 H) 8,46 (с, 1 H)
Стадия 4: Синтез (1S,2R,3R,5R)-3-[(1S)-2-амино-1-(4-фторфенил)этил]-5-(4-метил-7H-пирроло[2.3-d]пиримидин-7-ил)циклопентан-1,2-диола (O-5) и (1S,2R,3R,5R)-3-[(1R)-2-амино-1-(4-фторфенил)этил]-5-(4-метил-7H-пирроло[2.3-d]пиримидин-7-ил)циклопентан-1,2-диоли (O-6)
По методике стадии 4 со схемы K, O-3 обрабатывают ТФК в воде. O-5 (5,4 мг, 27%) и O-6 (3,7 мг, 19%) выделяют после последовательной обработки и очистки хиральной СЖХ.
O-5: ЖХМС [M+1] 370,90. 1H ЯМР (400 MГц, МЕТАНОЛ-d4) δ ч./млн. 1,42-1,57 (м, 1 H) 1,65-1,79 (м, 1 H) 2,24 (шс, 1 H) 2,49-2,68 (м, 3 H) 2,83 (шс, 1 H) 2,96-3,10 (м, 1 H) 3,31 (д, J=13,20 Гц, 1 H) 4,02 (т, J=5,81 Гц, 1 H) 4,27 (т, J=6,79 Гц, 1 H) 4,75 (шс, 1 H) 6,56 (д, J=3,18 Гц, 1 H) 6,98 (т, J=8,38 Гц, 2 H) 7,13-7,28 (м, 2 H) 7,31 (д, J=2,93 Гц, 1 H) 8,45 (с, 1 H)
O-6: ЖХМС [M+1] 370,90. 1H ЯМР (400 MГц, МЕТАНОЛ-d4) δ ч./млн. 1,78 (д, J=9,29 Гц, 1 H) 2,26-2,40 (м, 2 H) 2,53-2,67 (м, 4 H) 2,91-3,00 (м, 1 H) 3,24-3,25 (м, 1 H) 3,67-3,78 (м, 1 H) 4,19-4,32 (м, 1 H) 4,82-4,89 (м, 1 H) 6,60 (д, J=3,67 Гц, 1 H) 7,08 (т, J=8,68 Гц, 2 H) 7,27-7,40 (м, 3 H) 8,44-8,52 (м, 1 H)
Пример 53 (Схема P)-(1S,2R,3R,5R)-3-((S)-(2-(аминометил)-4-хлорфенил)(гидрокси)метил)-5-(4-метил-7H-пирроло[2.3-d]пиримидин-7-ил)циклопентан-1,2-диол (P-8)
Пример 54 (Схема P)-(1S,2R,3R,5R)-3-((R)-(2-(аминометил)-4-хлорфенил)(гидрокси)метил)-5-(4-метил-7H-пирроло[2.3-d]пиримидин-7-ил)циклопентан-1,2-диол (P-9)
Схема P
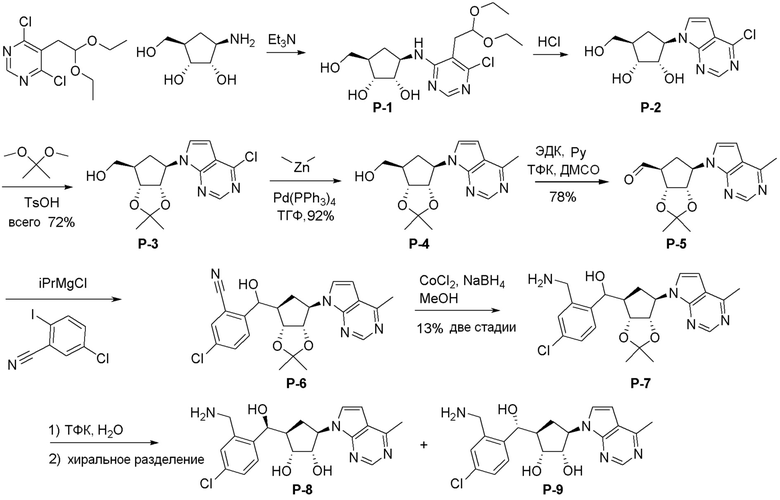
Стадия 1: Синтез (1R,2S,3R,5R)-3-((6-хлор-5-(2,2-диэтоксиэтил)пиримидин-4-ил)амино)-5-(гидроксиметил)циклопентан-1,2-диола (P-1)
(1R,2S,3R,5R)-3-амино-5-(гидроксиметил)циклопентан-1,2-диол (4160 мг, 22,6 ммоль) и 4,6-дихлор-5-(2,2-диэтоксиэтил)пиримидин (J. Med. Chem. 10, 665, 1967) (5000 мг, 18,86 ммоль) в 40 мл абсолютного этанола в герметично закрытой колбе обрабатывают Et3N (7630 мг, 75,4 ммоль), нагревают при 80°C в течение 18 ч. Реакционную смесь охлаждают на ледяной бане, твердое вещество фильтруют, и маточную жидкость концентрируют с получением P-1 в виде коричневой суспензии, которую применяют сразу на следующей стадии. ЖХМС [M+1] 375,80.
Стадия 2: Синтез (1R,2S,3R,5R)-3-(4-хлор-7H-пирроло[2.3-d]пиримидин-7-ил)-5-(гидроксиметил) циклопентан-1,2-диола (P-2)
Суспензию P-1 (7088 мг, 18,86 ммоль) в 30 мл диоксана, обрабатывают 10 мл 1 N HCl, нагревают при 80° в течение 30 мин. Реакционную смесь нейтрализуют NH4OH до pH 7, и летучие вещества удаляют в вакууме с получением P-2 в виде коричневой суспензии, которую применяют сразу на следующей стадии. ЖХМС [M+1] 283,85.
Стадия 3: Синтез ((3aR,4R,6R,6aS)-6-(4-хлор-7H-пирроло[2.3-d]пиримидин-7-ил)-2,2-диметилтетрагидро-4H-циклопента[d][1.3]диоксол-4-ил)метанола (P-3)
P-2 (5350 мг, 18,86 ммоль) и 2,2-диметоксипропан (50 мл, c=0,38 M) обрабатывают моногидратом п-толуолсульфоновой кислоты (7170 мг, 37,7 ммоль), и желто-коричневую суспензию доводят pH 4-5, энергично перемешивают при кт в течение 15 мин. Реакционную смесь разбавляют 100 мл H2O, нейтрализуют твердым NaHCO3. Летучие вещества осторожно удаляют в вакууме, и полученный коричневый водный раствор экстрагируют 20% изопропанолом/ДХМ, органические слои объединяют, концентрируют, очищают хроматографией на колонке с 50% EtOAc/ДХМ до 100% EtOAc с получением 4,4 г P-3 в виде желтой камеди (72% общий выход).
ЖХМС [M+1] 324,10. 1H ЯМР (400 MГц, ХЛОРОФОРМ-d) δ ч./млн. 8,63 (с, 1 H) 7,33 (д, J=3,55 Гц, 1 H) 6,63 (д, J=3,67 Гц, 1 H) 4,94-5,06 (м, 2 H) 4,72 (дд, J=6,66, 4,46 Гц, 1 H) 3,85-3,92 (м, 1 H) 3,78-3,85 (м, 1 H) 2,43-2,56 (м, 2 H) 2,28-2,42 (м, 1 H) 2,09 (шс, 1 H) 1,55-1,64 (м, 3 H) 1,33 (с, 3 H)
Стадия 4: Синтез ((3aR,4R,6R,6aS)-2,2-диметил-6-(4-метил-7H-пирроло[2.3-d]пиримидин-7-ил)тетрагидро-4H-циклопента[d][1.3]диоксол-4-ил)метанола (P-4)
По методике стадии 5 со схемы A P-3 превращают в P-4 (930 мг, 88%).
ЖХМС [M+1] 304,15. 1H ЯМР (400 MГц, ХЛОРОФОРМ-d) δ ч./млн. 1,25 (с, 3 H) 1,53 (с, 3 H) 2,21-2,34 (м, 2 H) 2,38-2,47 (м, 2 H) 2,75 (с, 3 H) 3,75 (шс, 1 H) 3,80 (шс, 1 H) 4,65 (дд, J=6,66, 4,10 Гц, 1 H) 4,86-5,00 (м, 2 H) 6,57 (шс, 1 H) 7,25 (шс, 1 H) 8,71 (с, 1 H)
Стадия 5: Синтез (3aR,4S,6R,6aS)-2,2-диметил-6-(4-метил-7H-пирроло[2.3-d]пиримидин-7-ил)тетрагидро-4H-циклопента[d][1.3]диоксол-4-карбальдегида (P-5)
ЭДК.HCl (3110 мг, 16,2 ммоль) добавляют к раствору P-4 (1230 мг, 4,055 ммоль) в безводном диметилсульфоксиде (22,5 мл, c=0,18 M). Пиридин (641 мг, 8,11 ммоль) добавляют, затем ТФК (462 мг, 4,05 ммоль), перемешивают при кт в течение 1,5 ч. Реакционную смесь разбавляют H2O, экстрагируют EtOAc дважды, объединенные органические фазы промывают H2O 3 раза, насыщенным раствором соли, сушат над Na2SO4, фильтруют и концентрируют с получением P-5 (950 мг) в виде коричневого масла (78% выход).
ЖХМС [M+1] 302,15. 1H ЯМР (400 MГц, ХЛОРОФОРМ-d) δ ч./млн. 1,28 (с, 3 H) 1,53 (с, 3 H) 2,47-2,57 (м, 1 H) 2,58-2,69 (м, 1 H) 2,85 (с, 3 H) 3,13 (тд, J=8,68, 4,28 Гц, 1 H) 4,94 (дд, J=6,60, 4,16 Гц, 1 H) 5,04 (тд, J=8,04, 4,34 Гц, 1 H) 5,14 (дд, J=6,42, 4,22 Гц, 1 H) 6,66 (д, J=3,42 Гц, 1 H) 7,26 (шс, 1 H) 8,73 (с, 1 H) 9,80 (с, 1 H)
Стадия 6: Синтез 5-хлор-2-(((3aR,4R,6R,6aS)-2,2-диметил-6-(4-метил-7H-пирроло[2.3-d]пиримидин-7-ил)тетрагидро-4H-циклопента[d][1.3]диоксол-4-ил)(гидрокси)метил)бензонитрила (P-6)
К раствору 5-хлор-2-йодбензонитрила (1570 мг, 5,97 ммоль) в сухом ТГФ (29,9 мл, c=0,2 M) при -78°C добавляют хлорид изопропилмагния (1080 мг, 7,47 ммоль, 5,74 мл, 1,3 M). Полученный раствор перемешивают при -78°C в течение 15 мин, P-5 (900 мг, 2,99 ммоль) в ТГФ (5,0 мл) добавляют по каплям. Реакционную смесь переносят на ледяную баню, нагревают до к.т. и перемешивают в течение ночи. Реакцию затем гасят станд. NH4Cl, экстрагируют EtOAc 3 раза; органические слои объединяют, промывают насыщенным раствором соли, сушат над Na2SO4, концентрируют и очищают хроматографией на колонке с 5% MeOH/ДХМ с получением P-6 (700 мг) в виде желтого масла. ЖХМС [M+1] 439,10.
Стадия 7: Синтез (2-(аминометил)-4-хлорфенил)((3aR,4R,6R,6aS)-2,2-диметил-6-(4-метил-7H-пирроло[2.3-d]пиримидин-7-ил)тетрагидро-4H-циклопента[d][1.3]диоксол-4-ил)метанола (P-7)
К раствору P-6 (200 мг, 0,456 ммоль) в MeOH (5 мл, c=0,09 M) добавляют гексагидрат хлорида кобальта(II) (545 мг, 2,28 ммоль) и NaBH4 (181 мг, 4,56 ммоль). Реакционную смесь перемешивают при кт в течение 0,5 ч, затем гасят станд. NH4Cl. Водный слой насыщают твердым NaCl и экстрагируют 20% изопропиловым спиртом/ДХМ множество раз. Органические слои концентрируют и очищают препаративной ВЭЖХ с получением 50 мг P-7 в виде бесцветного масла (13% две стадии). ЖХМС [M+1] 443,10.
Стадия 8: Синтез (1S,2R,3R,5R)-3-((S)-(2-(аминометил)-4-хлорфенил)(гидрокси)метил)-5-(4-метил-7H-пирроло[2.3-d]пиримидин-7-ил)циклопентан-1,2-диола (P-8) и (1S,2R,3R,5R)-3-((R)-(2-(аминометил)-4-хлорфенил)(гидрокси)метил)-5-(4-метил-7H-пирроло[2.3-d]пиримидин-7-ил)циклопентан-1,2-диола (P-9)
По методике стадии 4 со схемы K, P-7 обрабатывают ТФК в воде. P-8 (5,5 мг,12% выход) и P-9 (0,2 мг, 0,4% выход) выделяют после последовательной обработки и очистки хиральной СЖХ. ЖХМС [M+1] 403,00.
Пример 55 (Схема Q) - (1S,2R,3S,5R)-3-(2-(аминометил)-4-хлорбензил)-5-(4-метил-7H-пирроло[2.3-d]пиримидин-7-ил)циклопентан-1,2-диол (Q-4)
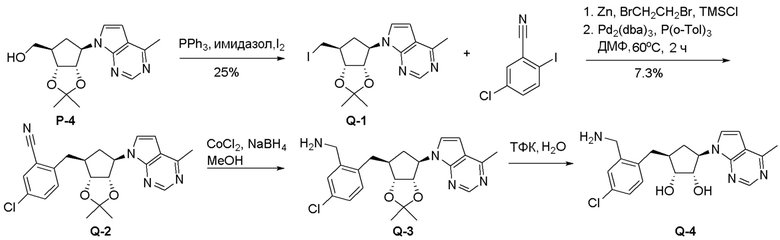
Стадия 1: Синтез 7-((3aS,4R,6S,6aR)-6-(йодметил)-2,2-диметилтетрагидро-4H-циклопента[d][1.3]диоксол-4-ил)-4-метил-7H-пирроло[2.3-d]пиримидина (Q-1)
Раствор трифенилфосфина (529 мг, 1,98 ммоль) и имидазола (135 мг, 1,98 ммоль) в метиленхлориде (8,24 мл, c=0,2 M) добавляют I2 (502 мг, 1,98 ммоль), затем по каплям добавляют P-4 (500 мг, 1,65 ммоль) в 2 мл метиленхлорида. Полученную реакционную смесь перемешивают при кт в течение ночи, и затем разбавляют водой (20 мл), экстрагируют метиленхлоридом (3×20 мл). Объединенные органические слои сушат над Na2SO4, концентрируют и очищают хроматографией на колонке с 60% EtOAc/гептаном с получением 280 мг Q-1 в виде желтого масла (25% выход).
ЖХМС [M+1] 414,00. 1H ЯМР (400 MГц, ХЛОРОФОРМ-d) δ ч./млн. 1,33 (с, 3 H) 1,60 (с, 3 H) 2,26-2,37 (м, 1 H) 2,40 (дд, J=11,55, 5,93 Гц, 1 H) 2,59 (дт, J=12,29, 6,08 Гц, 1 H) 2,80 (с, 3 H) 3,41 (дд, J=10,03, 6,72 Гц, 1 H) 3,50 (дд, J=10,09, 4,83 Гц, 1 H) 4,56 (т, J=6,11 Гц, 1 H) 5,02-5,13 (м, 2 H) 6,64 (д, J=3,55 Гц, 1 H) 7,28 (д, J=3,55 Гц, 1 H) 8,79 (с, 1 H)
Стадия 2: Синтез 5-хлор-2-(((3aR,4S,6R,6aS)-2,2-диметил-6-(4-метил-7H-пирроло[2.3-d]пиримидин-7-ил)тетрагидро-4H-циклопента[d][1.3]диоксол-4-ил)метил)бензонитрила (Q-2)
К суспензии цинка (318 мг, 4,87 ммоль) в сухом дегазированном ДМФ (8,11 мл, c=0,1 M) добавляют 1,2-дибромэтан (33 мг, 0,17 ммоль, 15 мкл) под азотом. Смесь нагревают тепловой пушкой около 30 сек до начала выделения газа из раствора, что показывает активацию цинка. Смесь охлаждают до к.т., добавляют ТМСCl (19 мг, 0,18 ммоль, 23 мкл) и реакционную смесь перемешивают при кт в течение 15 мин, затем добавляют раствор Q-1 (335 мг, 0,811 ммоль) в сухом дегазированном ДМФ (1 мл). Полученную смесь нагревают при 60°C в течение 5 мин, затем перемешивают при кт в течение 30 мин. После выпадения в осадок твердых частиц цинка, реакционную смесь фильтруют через шприцевой фильтр в смесь 5-хлор-2-йодбензонитрила (214 мг, 0,812 ммоль), Pd2(dba)3 (37,2 мг, 0,0406 ммоль) и три-o-толилфосфина (49,4 мг, 0,162 ммоль) в 1 мл сухого дегазированного ДМФ. Реакционную смесь промывают азотом и перемешивают при 80°C в течение 2 ч. После охлаждения до к.т. реакционную смесь разделяют между H2O (30 мл) и EtOAc (30 мл). Органическую фазу разделяют, промывают H2O (3×30мл) и насыщенным раствором соли (1×30мл), сушат над Na2SO4, концентрируют и очищают хроматографией на колонке с 70% EtOAc/гептаном с получением 25 мг Q-2 (7,3% выход).
ЖХМС [M+1] 423,10. 1H ЯМР (400 MГц, ХЛОРОФОРМ-d) δ ч./млн. 1,29 (с, 3 H) 1,56 (с, 3 H) 2,21-2,38 (м, 2 H) 2,48-2,60 (м, 1 H) 2,78 (с, 3 H) 2,95-3,04 (м, 1 H) 3,24 (дд, J=14,18, 6,72 Гц, 1 H) 4,62 (т, J=6,72 Гц, 1 H) 4,92-5,01 (м, 1 H) 5,04 (дд, J=7,09, 5,38 Гц, 1 H) 6,61 (д, J=3,67 Гц, 1 H) 7,25 (д, J=3,67 Гц, 1 H) 7,36 (д, J=8,31 Гц, 1 H) 7,52 (дд, J=8,44, 2,20 Гц, 1 H) 7,60 (д, J=2,20 Гц, 1 H) 8,76 (с, 1 H)
Стадия 3: Синтез (5-хлор-2-(((3aR,4S,6R,6aS)-2,2-диметил-6-(4-метил-7H-пирроло[2.3-d]пиримидин-7-ил)тетрагидро-4H-циклопента[d][1.3]диоксол-4-ил)метил)фенил)метанамина (Q-3)
По методике стадии 7 со схемы P, Q-2 восстанавливают до Q-3 в виде неочищенного продукта для следующей стадии. ЖХМС [M+1] 427,10.
Стадия 4: Синтез (1S,2R,3S,5R)-3-(2-(аминометил)-4-хлорбензил)-5-(4-метил-7H-пирроло[2.3-d]пиримидин-7-ил)циклопентан-1,2-диола (Q-4)
По методике стадии 4 со схемы K, Q-3 обрабатывают ТФК в воде. Q-4 (9,5 мг, 35%) выделяют после последовательной обработки и очистки хиральной СЖХ.
Q-4: ЖХМС [M+1] 386,85. 1H ЯМР (700 MГц, ДМСО-d6) δ ч./млн. 1,48-1,56 (м, 1 H) 1,97-2,04 (м, 1 H) 2,11 (шс, 1 H) 2,53-2,60 (м, 5 H) 2,91 (дд, J=14,09, 5,72 Гц, 1 H) 3,68-3,78 (м, 2 H) 4,29 (т, J=6,23 Гц, 1 H) 4,84 (кв, J=8,37 Гц, 1 H) 6,63 (д, J=3,42 Гц, 1 H) 7,09-7,17 (м, 2 H) 7,41 (шс, 1 H) 7,62 (д, J=2,22 Гц, 1 H) 8,53 (с, 1 H)
Пример 56 (Схема R) - (1S,2R,3R,5R)-3-((S)-гидрокси(1,2,3,4-тетрагидроизохинолин-8-ил)метил)-5-(4-метил-7H-пирроло[2.3-d]пиримидин-7-ил)циклопентан-1,2-диол (R-7)
Пример 57 (Схема R) - (1S,2R,3R,5R)-3-((R)-гидрокси(1,2,3,4-тетрагидроизохинолин-8-ил)метил)-5-(4-метил-7H-пирроло[2.3-d]пиримидин-7-ил)циклопентан-1,2-диол (R-8)
Схема R
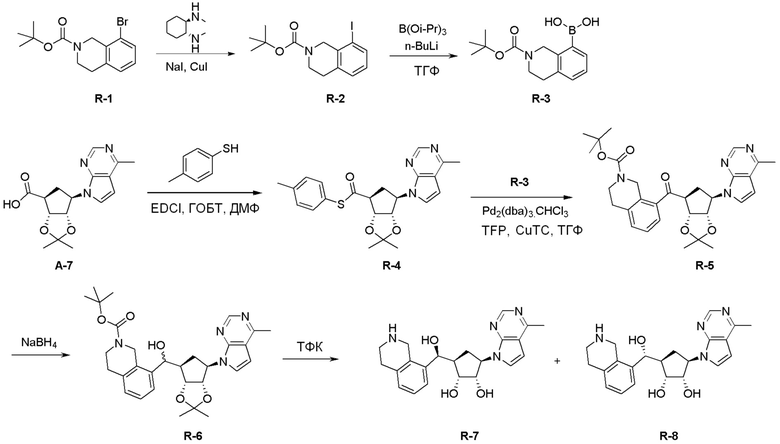
Стадия 1: Синтез трет-бутил 8-йод-3,4-дигидроизохинолин-2(1H)-карбоксилата (R-2)
Смесь трет-бутил 8-бром-3,4-дигидроизохинолин-2(1H)-карбоксилата (R-1) (1100 мг, 3,52 ммоль), NaI (4,76 г, 31,8 ммоль), CuI (402 мг, 2,12 ммоль) и транс-N,N-диметилциклогексана (602 мг, 4,22 ммоль) в диоксане (20 мл) продувают N2 в течение 10 мин. Полученную желтую суспензию перемешивают при 110°C в герметично закрытой пробирке в течение 48 ч. Реакционную смесь разбавляют петролейным эфиром (50 мл) и фильтруют. Фильтрат концентрируют в вакууме и остаток очищают хроматографией на силикагеле, элюируя EtOAC в петролейном эфире от 0 до 20% с получением R-2 (1200 мг, 94,8%) в виде светло-желтой камеди. ЖХМС [M+1-tBu] 304; 1H ЯМР (400 MГц, CDCl3) δ ч./млн. 7,70 (д, J=7,8 Гц, 1H), 7,45-7,39 (м, 1H), 7,12 (д, J=7,5 Гц, 1H), 6,88 (с, 1H), 4,44 (шс, 2H), 3,62 (т, J=5,5 Гц, 2H), 2,81 (д, J=4,8 Гц, 2H), 1,50 (с, 9H)
Стадия 2: Синтез (2-(трет-бутоксикарбонил)-1,2,3,4-тетрагидроизохинолин-8-ил)бороновой кислоты (R-3)
К раствору R-2 (250 мг, 0,696 ммоль) и триизопропилбората (262 мг, 0,321 ммоль) в сухом ТГФ (8 мл) добавляют 2,5 M н-BuLi (0,418 мл, 1,04 ммоль) при -60°C. Смесь перемешивают при -60°C в течение 30 мин. Смесь выливают в водн. NH4Cl (15 мл) и экстрагируют EtOAc (10 мл×3). Экстракт сушат над Na2SO4 и концентрируют в вакууме с получением неочищенного продукта. Неочищенный продукт очищают хроматографией на силикагеле, элюируя MeOH в ДХМ от 0 до 10% с получением R-3 (180 мг, 93%) в виде светло-желтой камеди, которую применяют сразу на следующей стадии.
Стадия 3: Синтез S-(п-толил)(3aR,4S,6R,6aS)-2,2-диметил-6-(4-метил-7H-пирроло[2.3-d]пиримидин-7-ил)тетрагидро-4H-циклопента[d][1.3]диоксол-4-карботиоата (R-4)
К соединению A-7 (270 мг, 0,851 ммоль) в ТГФ добавляют 4-метилбензолтиол (211 мг, 1,7 ммоль), ДИПЭА (440 мг, 3,4 ммоль) и T3P (1,08 г, 1,7 ммоль) при кт (15°C). Смесь перемешивают при кт 5-10°C в течение 2 дней. Смесь выливают в водн. NaHCO3 (20 мл) и экстрагируют EtOAc (10 мл×2). Экстракт промывают насыщенным раствором соли (15 мл) и концентрируют в вакууме, затем очищают хроматографией на силикагеле, элюируя EtOAc в петролейном эфире от 0 до 100% с получением R-4 (320 мг, 89%) в виде белого твердого вещества. ЖХМС [M+1] 424
Стадия 4: Синтез трет-бутил 8-((3aR,4S,6R,6aS)-2,2-диметил-6-(4-метил-7H-пирроло[2.3-d]пиримидин-7-ил)тетрагидро-4H-циклопента[d][1.3]диоксол-4-карбонил)-3,4-дигидроизохинолин-2(1H)-карбоксилата (R-5)
Смесь R-4 (125 мг, 0,3 ммоль) и R-3 (180 мг, 0,32 ммоль), CuTC (90 мг, 0,47 ммоль), Pd2(dba)3.CHCl3 (31 мг, 0,03 ммоль) и TFP (20,6 мг, 0,0886 ммоль) в сухом ТГФ (5 мл) дегазируют с N2 четыре раза. Смесь перемешивают при 50°C в герметично закрытой пробирке в течение 16 часов. Смесь разбавляют EtOAc (10 мл) и фильтруют. Фильтрат концентрируют в вакууме досуха. Остаток очищают хроматографией на силикагеле, элюируя EtOAc в петролейном эфире от 0 до 100% с получением R-5 (70 мг, 40%) в виде белого твердого вещества. ЖХМС [M+1] 533
Стадия 5: Синтез трет-бутил 8-(((3aR,4R,6R,6aS)-2,2-диметил-6-(4-метил-7H-пирроло[2.3-d]пиримидин-7-ил)тетрагидро-4H-циклопента[d][1.3]диоксол-4-ил)(гидрокси)метил)-3,4-дигидроизохинолин-2(1H)-карбоксилата (R-6)
К раствору R-5 (70 мг, 0,131 ммоль) в MeOH (2 мл) добавляют NaBH4 (86,9 мг, 2,30 ммоль) при кт 15°C. Смесь перемешивают при кт 15°C в течение 30 мин. Смесь разбавляют водой (10 мл) и экстрагируют EtOAc (10 мл×3). Экстракт промывают насыщенным раствором соли (10 мл), сушат над Na2SO4 и концентрируют в вакууме с получением неочищенного продукта R-6 (60 мг, 85%) в виде светло-желтого твердого вещества.
Стадия 6: Синтез (1S,2R,3R,5R)-3-((S)-гидрокси(1,2,3,4-тетрагидроизохинолин-8-ил)метил)-5-(4-метил-7H-пирроло[2.3-d]пиримидин-7-ил)циклопентан-1,2-диола (R-7) и (1S,2R,3R,5R)-3-((R)-гидрокси(1,2,3,4-тетрагидроизохинолин-8-ил)метил)-5-(4-метил-7H-пирроло[2.3-d]пиримидин-7-ил)циклопентан-1,2-диола (R-8)
К соединению R-6 (60 мг, 0,1 ммоль) добавляют ТФК/H2O (1 мл/1 мл, перед этим охладив до 0°C). Смесь перемешивают при кт (15°C) в течение 3 ч. Смесь концентрируют в вакууме с получением неочищенного продукта, который очищают СЖХ. После СЖХ, продукты повторно очищают преп. ТСХ с получением R-7 (4,5 мг, 13%) и R-8 (3,5 мг, 9%) в виде белых твердых веществ.
R-7: ЖХМС [M+1] 395; 1H ЯМР (400 MГц, MeOD-d4) δ ч./млн. 8,63 (с, 1H), 7,59 (д, J=3,8 Гц, 1H), 7,42 (д, J=7,5 Гц, 1H), 7,21 (т, J=7,5 Гц, 1H), 7,06 (д, J=7,3 Гц, 1H), 6,73 (д, J=3,8 Гц, 1H), 5,09-5,01 (м, 1H), 4,81 (д, J=7,0 Гц, 1H), 4,64 (дд, J=5,3, 9,5 Гц, 1H), 4,31 (дд, J=2,0, 5,5 Гц, 1H), 4,29-4,16 (м, 2H), 3,21-3,06 (м, 2H), 2,92 (д, J=4,0 Гц, 2H), 2,72 (с, 3H), 2,56-2,47 (м, 1H), 2,22 (тд, J=8,8, 13,1 Гц, 1H), 1,83 (ддд, J=8,3, 10,7, 13,1 Гц, 1H)
R-8: ЖХМС [M+1] 395; 1H ЯМР (400 MГц, MeOD-d4) δ ч./млн. 8,63 (с, 1H), 7,72 (д, J=3,8 Гц, 1H), 7,42 (д, J=8,0 Гц, 1H), 7,23-7,17 (м, 1H), 7,07 (д, J=7,5 Гц, 1H), 6,77 (д, J=3,8 Гц, 1H), 5,12-5,04 (м, 2H), 4,48-4,42 (м, 1H), 4,30-4,15 (м, 2H), 4,14-4,10 (м, 1H), 3,24-3,18 (м, 2H), 3,00-2,94 (м, 2H), 2,74 (с, 3H), 2,45-2,36 (м, 1H), 2,20-2,14 (м, 1H) 2,09-1,99 (м, 1H)
Пример 58 (Схема S) - (1S,2R,3S,5R)-3-(4-фторбензил)-5-(4-метил-7H-пирроло[2.3-d]пиримидин-7-ил)циклопентан-1,2-диол (S-1)
Схема S
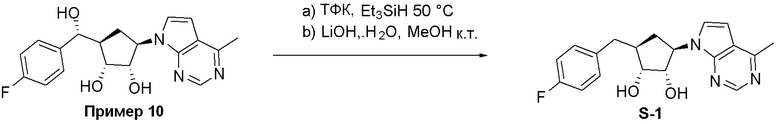
Смесь (1S,2R,3R,5R)-3-((R)-(4-фторфенил)(гидрокси)метил)-5-(4-метил-7H-пирроло[2.3-d]пиримидин-7-ил)циклопентан-1,2-диола (Пример 10) (30 мг, 0,084 ммоль), трифторуксусной кислоты (302 мкл) и триэтилсилана (268 мкл, 1,68 ммоль) нагревают до 50°C в течение 2 часов. Реакционную смесь концентрируют до масла, затем добавляют моногидрат гидроксида лития (20 мг, 0,48 ммоль) и метанол (1 мл) и перемешивают при кт в течение 45 мин. Реакционную смесь концентрируют для удаления метанола, затем разделяют между водой и EtOAc. Слой EtOAc промывают насыщенным раствором соли, сушат с Na2SO4, фильтруют и концентрируют с получением S-1 в виде масла с 52% выходом.
ЖХМС-ХИАД(+): MH+=342, 1H ЯМР (700 MГц, ДМСО-d6) δ ч./млн. 8,58 (с, 1H), 7,64 (д, J=3,5 Гц, 1H), 7,25 (дд, J=5,9, 8,1 Гц, 2H), 7,08 (т, J=8,8 Гц, 2H), 6,68 (д, J=3,5 Гц, 1H), 4,98-4,84 (м, 2H), 4,77 (д, J=4,4 Гц, 1H), 4,44-4,23 (м, 1H), 3,82-3,75 (м, 1H), 2,92 (дд, J=6,6, 13,6 Гц, 1H), 2,68-2,57 (м, 4H), 2,26-2,12 (м, 1H), 2,06 (тд, J=8,3, 13,0 Гц, 1H), 1,60-1,41 (м, 1H)
Пример 59 (Схема T) - (+/-)-(1S,2S,3S,5R)-3-метокси-5-(4-метил-7H-пирроло[2.3-d]пиримидин-7-ил)циклопентан-1,2-диол (T-5)
Пример 60 (Схема T) - (1S,2S,3S,5R)-3-метокси-5-(4-метил-7H-пирроло[2.3-d]пиримидин-7-ил)циклопентан-1,2-диол (T-6)
Пример 61 (Схема T) - (1R,2R,3R,5S)-3-метокси-5-(4-метил-7H-пирроло[2.3-d]пиримидин-7-ил)циклопентан-1,2-диол (T-7)
Схема T
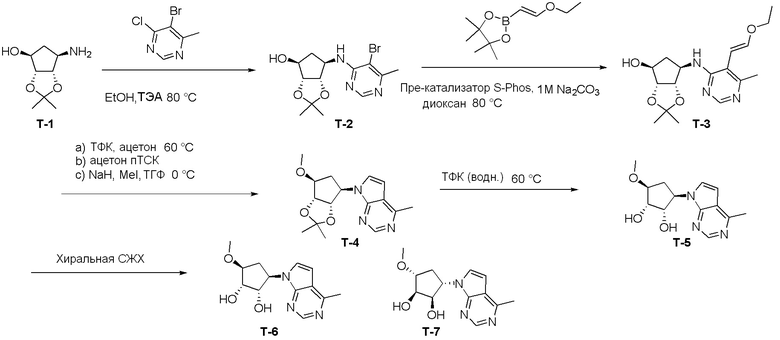
Стадия 1: (+/-)-(3aR,4S,6R,6aS)-6-((5-бром-6-метилпиримидин-4-ил)амино)-2,2-диметилтетрагидро-4H-циклопента[d][1.3]диоксол-4-ол (T-2)
Смесь (+/-)-(3aR,4S,6R,6aS)-6-амино-2,2-диметилтетрагидро-4H-циклопента[d][1.3]диоксол-4-ола (T-1) (786 мг, 4,54 ммоль), 5-бром-4-хлор-6-метилпиримидина (1,04 г, 4,99 ммоль) и триметиламина (0,822 мл, 5,9 ммоль) в этаноле (9,0 мл, 0,5 M) нагревают до 80°C в течение 20 часов. Реакционную смесь неочищенного продукта концентрируют до твердого вещества, затем очищают хроматографией на силикагеле с градиентом от 0% до 100% EtOAc в гептане с получением T-2 в виде белого твердого вещества, 1,35 г (87% выход).
ЖХМС-ИЭР(+): MH+=344/346, 1H ЯМР (400 MГц, МЕТАНОЛ-d4) δ ч./млн. 8,29 (с, 1H), 4,60 (д, J=6,4 Гц, 1H), 4,56-4,52 (м, 1H), 4,51-4,47 (м, 1H), 4,22 (д, J=3,9 Гц, 1H), 2,33-2,23 (м, 1H), 1,76 (д, J=14,2 Гц, 1H), 1,41 (с, 3H), 1,27 (с, 3H).
Стадия 2: Синтез (+/-)-(3aR,4S,6R,6aS)-6-((5-((E)-2-этоксивинил)-6-метилпиримидин-4-ил)амино)-2,2-диметилтетрагидро-4H-циклопента[d][1.3]диоксол-4-ола (T-3)
К раствору T-2 (2,06 г, 5,98 ммоль) и (E)-2-(2-этоксивинил)-4,4,5,5-тетраметил-1,3,2-диоксаборолана (1,19 г, 5,98 ммоль) в диоксане (19,9 мл, 0,3 M) промывают в вакууме азотом, затем добавляют 2N карбонат натрия (водн.) (8,98 мл), затем пре-катализатор S-Phos (136 мг, 0,180 ммоль). Полученную смесь нагревают до 80°C в течение 23 часов. Реакционную смесь охлаждают до к.т., затем разбавляют EtOAc и водой. Органический слой промывают насыщенным раствором соли, сушат с Na2SO4, фильтруют и концентрируют до масла, затем очищают хроматографией на силикагеле, элюируя градиентом 0%-100% EtOAc в гептане с получением T-3 в виде белого твердого вещества, 1,21 г (60% выход).
ЖХМС-ИЭР(+): MH+=336, 1H ЯМР (400MГц, ДМСО-d6) δ ч./млн. 8,23 (с, 1H), 6,66 (д, J=13,2 Гц, 1H), 6,38 (д, J=8,6 Гц, 1H), 5,63 (д, J=2,4 Гц, 1H), 5,32 (д, J=13,2 Гц, 1H), 4,50-4,33 (м, 3H), 4,07 (шс, 1H), 3,87 (кв, J=6,9 Гц, 2H), 2,24 (с, 3H), 2,12 (тд, J=5,2, 13,6 Гц, 1H), 1,62 (д, J=13,9 Гц, 1H), 1,34 (с, 3H), 1,25 (т, J=7,0 Гц, 3H), 1,18 (с, 3H)
Стадия 3: Синтез (+/-)-7-((3aS,4R,6S,6aR)-6-метокси-2,2-диметилтетрагидро-4H-циклопента[d][1.3]диоксол-4-ил)-4-метил-7H-пирроло[2.3-d]пиримидина (T-4)
Смесь (+/-)-(3aR,4S,6R,6aS)-6-((5-((E)-2-этоксивинил)-6-метилпиримидин-4-ил)амино)-2,2-диметилтетрагидро-4H-циклопента[d][1.3]диоксол-4-ола (111 мг, 0,331 ммоль) и трифторуксусной кислоты (127 мкл, 1,65 ммоль) в ацетоне (3,31 мл, 0,1 M) нагревают до 60°C в течение 43 часов. Реакционную смесь концентрируют до масла, затем подвергают азеотропной перегонке с толуолом. Неочищенное масло растворяют в ацетоне (3,31 мл), затем добавляют ПДМ (81 мкл, 0,662 ммоль) и ПТСК (3,1 мг, 0,016 ммоль). Смесь перемешивают при кт в течение 48 часов. Реакционную смесь концентрируют до масла, затем повторно растворяют в EtOAc. Слой EtOAc промывают насыщенным NaHCO3 (водн.), насыщенным раствором соли, сушат с Na2SO4, фильтруют и концентрируют до масла. Смесь неочищенного продукта очищают хроматографией на силикагеле, элюируя градиентом 0%-100% EtOAc в гептане с получением (3aR,4S,6R,6aS)-2,2-диметил-6-(4-метил-7H-пирроло[2.3-d]пиримидин-7-ил)тетрагидро-4H-циклопента[d][1.3]диоксол-4-ола в виде белого твердого вещества, 76 мг (79% выход, 80% чистота).
К суспензии (3aR,4S,6R,6aS)-2,2-диметил-6-(4-метил-7H-пирроло[2.3-d]пиримидин-7-ил)тетрагидро-4H-циклопента[d][1.3]диоксол-4-ола (76 мг, 80% чистый) в 2-метилтетрагидрофуране (1 мл) при кт добавляют 60% гидрид натрия (26 мг). Через 15 минут добавляют метилйодид (200 мкл). Через 1 час добавляют еще метилйодид (200 мкл) и смесь перемешивают при кт в течение еще 3 часов. Реакционную смесь гасят насыщенным хлоридом аммония (водн), затем экстрагируют EtOAc. Слой EtOAc промывают насыщенным раствором соли, сушат с Na2SO4, фильтруют и концентрируют до масла. Неочищенное масло очищают хроматографией на силикагеле, элюируя градиентом 0%-100% EtOAc в гептане с получением (+/-)-7-((3aS,4R,6S,6aR)-6-метокси-2,2-диметилтетрагидро-4H-циклопента[d][1.3]диоксол-4-ил)-4-метил-7H-пирроло[2.3-d]пиримидина в виде бесцветного масла, 39 мг (39% выход).
ЖХМС-ИЭР(+): MH+=304, 1H ЯМР (400 MГц, ХЛОРОФОРМ-d) δ ч./млн. 8,77 (с, 1H), 7,43 (д, J=3,7 Гц, 1H), 6,53 (д, J=3,7 Гц, 1H), 5,30 (ддд, J=2,6, 5,3, 7,8 Гц, 1H), 4,84 (дд, J=2,1, 6,2 Гц, 1H), 4,71 (д, J=6,4 Гц, 1H), 3,94 (т, J=4,2 Гц, 1H), 3,41 (с, 3H), 2,80-2,65 (м, 4H), 2,24 (тд, J=4,6, 14,5 Гц, 1H), 1,54 (с, 3H), 1,30 (с, 3H).
Стадия 4: Синтез (+/-)-(1S,2S,3S,5R)-3-метокси-5-(4-метил-7H-пирроло[2.3-d]пиримидин-7-ил)циклопентан-1,2-диола (T-5), (1S,2S,3S,5R)-3-метокси-5-(4-метил-7H-пирроло[2.3-d]пиримидин-7-ил)циклопентан-1,2-диола (T-6) и (1R,2R,3R,5S)-3-метокси-5-(4-метил-7H-пирроло[2.3-d]пиримидин-7-ил)циклопентан-1,2-диола (T-7).
Смесь T-4 (39 мг, 0,13 ммоль) и 50% водного ТФК (800 мкл) нагревают до 60°C в течение 2 часов. Реакционную смесь концентрируют до масла, затем очищают СЖХ (колонка 3HOP) с получением (+/-)-(1S,2S,3S,5R)-3-метокси-5-(4-метил-7H-пирроло[2.3-d]пиримидин-7-ил)циклопентан-1,2-диола (T-5) в виде белого твердого вещества, 25 мг (75% выход).
T-5: ЖХМС-ХИАД(+): MH+=264, 1H ЯМР (400 MГц, ХЛОРОФОРМ-d) δ ч./млн. 8,61 (с, 1H), 7,31 (д, J=3,7 Гц, 1H), 6,59 (д, J=3,4 Гц, 1H), 4,95 (кв, J=8,6 Гц, 1H), 4,54-4,39 (м, 1H), 4,23 (д, J=4,9 Гц, 1H), 3,87 (т, J=5,4 Гц, 1H), 3,47 (с, 3H), 2,98-2,81 (м, 1H), 2,71 (с, 3H), 2,09 (ддд, J=5,1, 9,0, 13,8 Гц, 1H).
T-5 разделяют хиральной СЖХ (колонка Lux Cellulose-4 4,6×100 мм 3мкм, 30% MeOH/ДЭА @ 120 бар, 4 мл/мин) с получением (1S,2S,3S,5R)-3-метокси-5-(4-метил-7H-пирроло[2.3-d]пиримидин-7-ил)циклопентан-1,2-диола (T-6) в виде белого твердого вещества, 6 мг (19% выход).
T-6: ЖХМС-ХИАД(+): MH+=264, 1H ЯМР (700 MГц, ДМСО-d6) δ ч./млн. 8,61 (с, 1H), 7,60 (д, J=3,5 Гц, 1H), 6,70 (д, J=3,5 Гц, 1H), 5,11-4,83 (м, 3H), 4,40-4,26 (м, 1H), 3,91 (шс, 1H), 3,61 (т, J=4,8 Гц, 1H), 3,31 (с, 3H), 2,63 (с, 3H), 2,56 (тд, J=8,0, 14,0 Гц, 1H), 1,77 (ддд, J=4,8, 9,1, 13,8 Гц, 1H)
и (1R,2R,3R,5S)-3-метокси-5-(4-метил-7H-пирроло[2.3-d]пиримидин-7-ил)циклопентан-1,2-диола (T-7) в виде белого твердого вещества, 6 мг (19% выход).
T-7: ЖХМС-ХИАД(+): MH+=264, 1H ЯМР (700 MГц, ДМСО-d6) δ ч./млн. 8,61 (с, 1H), 7,60 (д, J=3,5 Гц, 1H), 6,70 (д, J=3,5 Гц, 1H), 5,07-4,92 (м, 3H), 4,38-4,25 (м, 1H), 3,91 (шс, 1H), 3,66-3,52 (м, 1H), 3,31 (с, 3H), 2,63 (с, 3H), 2,56 (тд, J=8,1, 13,8 Гц, 1H), 1,76 (ддд, J=4,6, 8,9, 13,8 Гц, 1H)
Пример 62: (Схема U) - (1S,2S,3S,5R)-3-(2-(аминометил)фенокси)-5-(4-метил-7H-пирроло[2.3-d]пиримидин-7-ил)циклопентан-1,2-диол (U-6)
Пример 63: (Схема U) - (1R,2R,3R,5S)-3-(2-(аминометил)фенокси)-5-(4-метил-7H-пирроло[2.3-d]пиримидин-7-ил)циклопентан-1,2-диол (U-7)
Схема U
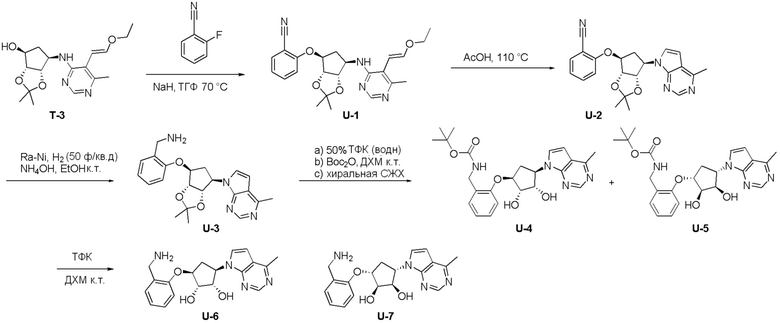
Стадия 1: Синтез 2-(((3aR,4S,6R,6aS)-6-((5-((E)-2-этоксивинил)-6-метилпиримидин-4-ил)амино)-2,2-диметилтетрагидро-4H-циклопента[d][1.3]диоксол-4-ил)окси)бензонитрила (U-1)
К смеси T-3 (181 мг, 0,54 ммоль) и 2-фторбензонитрила (57,4 мкл, 65,4 мг, 0,54 ммоль) в ТГФ (1,8 мл, 0,3 M) при кт добавляют 60% гидрид натрия (45,3 мг, 1,13 ммоль). После перемешивания в течение 5 минут реакционную смесь нагревают до 70°C в течение 6 часов. Реакционную смесь охлаждают до к.т., затем гасят насыщенным NH4Cl (водн), разбавляют водой и экстрагируют EtOAc. Слой EtOAc промывают насыщенным раствором соли, сушат с MgSO4, фильтруют, затем концентрируют до масла. Неочищенное масло очищают хроматографией на силикагеле, элюируя 0-100% EtOAc-Гептаном с получением U-1 в виде янтарного масла, 210 мг (89% выход).
ЖХМС-ИЭР(+): MH+=437, 1H ЯМР (400 MГц, ХЛОРОФОРМ-d) δ ч./млн. 8,48 (с, 1H), 7,68-7,49 (м, 2H), 7,19-6,98 (м, 2H), 6,47 (д, J=13,2 Гц, 1H), 5,45 (д, J=7,6 Гц, 1H), 5,41 (д, J=13,2 Гц, 1H), 4,82-4,69 (м, 4H), 3,86 (кв, J=6,8 Гц, 2H), 2,70 (тд, J=6,1, 15,0 Гц, 1H), 2,35 (с, 3H), 2,13 (д, J=15,2 Гц, 1H), 1,51 (с, 3H), 1,32 (с, 3H), 1,26 (т, J=7,0 Гц, 4H).
Стадия 2: 2-(((3aR,4S,6R,6aS)-2,2-диметил-6-(4-метил-7H-пирроло[2.3-d]пиримидин-7-ил)тетрагидро-4H-циклопента[d][1.3]диоксол-4-ил)окси)бензонитрил (U-2)
Раствор U-1 (210 мг, 0,481 ммоль в уксусной кислоте (4,81 мл, 0,1 M) нагревают до 110°C в течение 19 часов. Реакционную смесь концентрируют до масла, затем очищают хроматографией на силикагеле, элюируя 0-100% EtOAc-Гептаном с получением U-2 в виде темно-янтарного стекла, 166 мг (88% выход).
ЖХМС-ИЭР(+): MH+=391, 1H ЯМР (400 MГц, ХЛОРОФОРМ-d) δ ч./млн. 8,79 (с, 1H), 7,69 (д, J=3,4 Гц, 1H), 7,65-7,51 (м, 2H), 7,13 (д, J=8,6 Гц, 1H), 7,07 (т, J=7,6 Гц, 1H), 6,66 (д, J=3,7 Гц, 1H), 5,48 (дд, J=3,2, 6,8 Гц, 1H), 5,01 (д, J=4,6 Гц, 1H), 4,95 (дд, J=2,7, 6,1 Гц, 1H), 4,86 (д, J=5,9 Гц, 1H), 3,06 (тд, J=7,6, 14,7 Гц, 1H), 2,74 (с, 3H), 2,57 (д, J=14,9 Гц, 1H), 1,62 (с, 4H), 1,34 (с, 4H).
Стадия 3: Синтез (2-(((3aR,4S,6R,6aS)-2,2-диметил-6-(4-метил-7H-пирроло[2.3-d]пиримидин-7-ил)тетрагидро-4H-циклопента[d][1.3]диоксол-4-ил)окси)фенил)метанамина (U-3)
Смесь U-2 (139 мг, 0,356 ммоль) в этаноле (7,12 мл, 0,05 M) добавляют к 28% NH4OH (3 мл) никелю Ренея (70 мг, 1,2 ммоль). Смесь гидрируют при 50 ф/кв.д в автоклаве из нержавеющей стали в течение 24 часов. Реакционную смесь фильтруют через целит, затем фильтрат концентрируют до масла и очищают хроматографией на силикагеле, элюируя 0-100% MeOH в EtOAc с получением U-3 в виде бесцветного масла, 81 мг (58% выход).
ЖХМС-ХИАД(+): MH+=395, 1H ЯМР (400 MГц, МЕТАНОЛ-d4) δ ч./млн. 8,64 (с, 1H), 7,62 (д, J=3,7 Гц, 1H), 7,34-7,23 (м, 2H), 7,12 (д, J=8,6 Гц, 1H), 6,96 (т, J=7,5 Гц, 1H), 6,75 (д, J=3,7 Гц, 1H), 5,28 (дт, J=3,5, 7,0 Гц, 1H), 5,19 (дд, J=3,4, 6,4 Гц, 1H), 4,94 (д, J=6,4 Гц, 1H), 4,87 (т, J=5,9 Гц, 1H), 3,72 (шс, 2H), 2,95 (тд, J=7,0, 14,1 Гц, 1H), 2,73 (с, 3H), 2,65-2,54 (м, 1H), 1,60 (с, 3H), 1,35 (с, 3H).
Стадия 4: Синтез трет-бутил (2-(((1S,2S,3S,4R)-2.3-дигидрокси-4-(4-метил-7H-пирроло[2.3-d]пиримидин-7-ил)циклопентил)окси)бензил)карбамата (U-4) и трет-бутил (2-(((1R,2R,3R,4S)-2.3-дигидрокси-4-(4-метил-7H-пирроло[2.3-d]пиримидин-7-ил)циклопентил)окси)бензил)карбамата (U-5)
Раствор U-3 (81 мг, 0,21 ммоль) в 50% ТФК (водн) (600 мкл) перемешивают при кт в течение 16 часов. Реакционную смесь затем концентрируют до масла. Смесь неочищенного масла и Boc2O (30,8 мг, 1,41 ммоль) в ДХМ (30,8 мг, 0,141 ммоль) перемешивают при кт в течение 6 часов. Реакционную смесь концентрируют, затем очищают хроматографией на силикагеле, элюируя 40-100% EtOAc в гептане, затем 0-20% MeOH в EtOAc с получением рацемической смеси U-4 и U-5 в виде бесцветного масла, 73 мг. Энантиомеры разделяют хиральной СЖХ (колонка Chiralpak IC-3 4,6×100 мм 3 мкм, 30% MeOH @ 120 бар, 4 мл/мин) с получением U-4 (25,4 мг, 40% выход) и U-5 (24,4 мг, 38% выход) в виде белых твердых веществ.
U-4: ЖХМС-ИЭР(+): MH+=455, 1H ЯМР (400 MГц, МЕТАНОЛ-d4) δ ч./млн. 8,52 (с, 1H), 7,51 (шс, 1H), 7,23-7,09 (м, 2H), 6,92 (д, J=8,1 Гц, 1H), 6,84 (т, J=7,5 Гц, 1H), 6,64 (д, J=3,7 Гц, 1H), 5,15 (кв, J=8,6 Гц, 1H), 4,67-4,58 (м, 2H), 4,34-4,15 (м, 2H), 4,11 (д, J=4,4 Гц, 1H), 2,90 (ддд, J=7,3, 9,4, 14,5 Гц, 1H), 2,61 (с, 3H), 2,24-2,08 (м, 1H), 1,33 (с, 9H). [α]D22= +61,3° (c=0,1, MeOH).
U-5: ЖХМС-ИЭР(+): MH+=455, 1H ЯМР (400 MГц, МЕТАНОЛ-d4) δ ч./млн. 8,62 (с, 1H), 7,61 (шс, 1H), 7,33-7,17 (м, 2H), 7,02 (д, J=8,1 Гц, 1H), 6,94 (т, J=7,3 Гц, 1H), 6,74 (д, J=3,7 Гц, 1H), 5,25 (кв, J=9,0 Гц, 1H), 4,74 (д, J=7,1 Гц, 2H), 4,43-4,25 (м, 2H), 4,21 (д, J=4,4 Гц, 1H), 3,00 (ддд, J=7,3, 9,5, 14,7 Гц, 1H), 2,71 (с, 3H), 2,38-2,12 (м, 1H), 1,43 (с, 9H). [α]D22= -86,1° (c=0,1, MeOH)
Стадия 5: Синтез (1S,2S,3S,5R)-3-(2-(аминометил)фенокси)-5-(4-метил-7H-пирроло[2.3-d]пиримидин-7-ил)циклопентан-1,2-диола (U-6)
Смесь U-4 (24 мг, 0,053 ммоль) и трифторуксусной кислоты (120 мг, 1,06 ммоль) в ДХМ (0,176 мл, 0,3 M) перемешивают при кт в течение 3 часов, затем концентрируют до масла и очищают СЖХ (колонка ZymorSpher Pyridin Diol 150×21,2 мм с 20-40% MeOH @ 5%/мин, 100 бар, 60 мл/мин) с получением U-6 в виде белого твердого вещества, 6,54 мг (35% выход).
ЖХМС-ХИАД(+): MH+=355, 1H ЯМР (700 MГц, ДМСО-d6) δ ч./млн. 8,62 (с, 1H), 7,66 (д, J=2,2 Гц, 1H), 7,37 (д, J=7,0 Гц, 1H), 7,28 (т, J=7,5 Гц, 1H), 7,07 (д, J=7,9 Гц, 1H), 6,96 (т, J=7,0 Гц, 1H), 6,72 (д, J=2,6 Гц, 1H), 5,12 (кв, J=8,8 Гц, 1H), 4,61 (д, J=3,1 Гц, 1H), 4,06 (шс, 1H), 2,94-2,75 (м, 1H), 2,64 (с, 3H), 2,03 (т, J=9,7 Гц, 1H)
Стадия 6: Синтез (1R,2R,3R,5S)-3-(2-(аминометил)фенокси)-5-(4-метил-7H-пирроло[2.3-d]пиримидин-7-ил)циклопентан-1,2-диола (U-7)
U-7 получают по методике получения U-6, начиная с U-5, с получением белого твердого вещества, 9 мг (50% выход).
ЖХМС-ХИАД(+): MH+=355, 1H ЯМР (700 MГц, ДМСО-d6) δ ч./млн. 8,63 (с, 1H), 7,66 (д, J=3,5 Гц, 1H), 7,39 (д, J=7,5 Гц, 1H), 7,36 (т, J=7,7 Гц, 1H), 7,13 (д, J=8,4 Гц, 1H), 7,00 (т, J=7,5 Гц, 1H), 6,72 (д, J=3,5 Гц, 1H), 5,19-5,04 (м, 2H), 4,63 (д, J=4,4 Гц, 2H), 4,08 (д, J=4,4 Гц, 1H), 4,06 (с, 2H), 2,87 (тд, J=8,3, 14,2 Гц, 1H), 2,64 (с, 3H), 2,06 (ддд, J=4,0, 9,5, 13,9 Гц, 1H)
Пример 64 (Схема V) - 2-(((1S,2S,3S,4R)-2.3-дигидрокси-4-(4-метил-7H-пирроло[2.3-d]пиримидин-7-ил)циклопентил)окси)бензонитрил (V-1)
Пример 65 (Схема V) - 2-(((1R,2R,3R,4S)-2.3-дигидрокси-4-(4-метил-7H-пирроло[2.3-d]пиримидин-7-ил)циклопентил)окси)бензонитрил (V-2)
Схема V
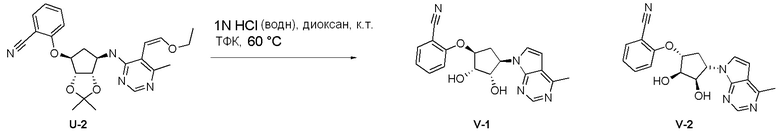
Стадия 1: Синтез 2-(((1S,2S,3S,4R)-2.3-дигидрокси-4-(4-метил-7H-пирроло[2.3-d]пиримидин-7-ил)циклопентил)окси)бензонитрила (V-1) и 2-(((1R,2R,3R,4S)-2.3-дигидрокси-4-(4-метил-7H-пирроло[2.3-d]пиримидин-7-ил)циклопентил)окси)бензонитрила (V-2)
V-1 (9,5 мг, 47% выход) и V-2 (9,4 мг, 47% выход) получают из U-2 по методике стадии 5 со схемы U.
V-1: ЖХМС-ИЭР(+): MH+=351, 1H ЯМР (400 MГц, МЕТАНОЛ-d4) δ ч./млн. 8,68 (шс, 1H), 7,75 (д, J=3,7 Гц, 1H), 7,71-7,60 (м, 2H), 7,30 (д, J=8,6 Гц, 1H), 7,12 (т, J=7,6 Гц, 1H), 6,82 (д, J=3,7 Гц, 1H), 5,36 (кв, J=8,5 Гц, 1H), 4,89 (д, J=6,1 Гц, 1H), 4,71 (дд, J=4,6, 8,3 Гц, 1H), 4,23 (д, J=4,2 Гц, 1H), 3,16-3,01 (м, 1H), 2,75 (с, 3H), 2,18 (ддд, J=2,4, 7,6, 14,7 Гц, 1H).
V-2: ЖХМС-ИЭР(+): MH+=351, 1H ЯМР (400 MГц, МЕТАНОЛ-d4) δ ч./млн. 8,73 (шс, 1H), 7,79 (д, J=3,7 Гц, 1H), 7,72-7,61 (м, 1H), 7,31 (д, J=8,6 Гц, 1H), 7,12 (т, J=7,6 Гц, 1H), 6,87 (д, J=3,7 Гц, 1H), 5,43-5,31 (м, 1H), 4,90 (д, J=5,9 Гц, 1H), 4,72 (дд, J=4,6, 8,6 Гц, 1H), 4,23 (д, J=4,4 Гц, 1H), 3,16-3,01 (м, 1H), 2,77 (с, 2H), 2,19 (ддд, J=2,3, 7,6, 14,8 Гц, 1H)
Пример 66 (Схема W) - (1S,2S,3S,5R)-3-(2-(аминометил)-4-фторфенокси)-5-(4-метил-7H-пирроло[2.3-d]пиримидин-7-ил)циклопентан-1,2-диол (W-5)
Пример 67 (Схема W) - (1R,2R,3R,5S)-3-(2-(аминометил)-4-фторфенокси)-5-(4-метил-7H-пирроло[2.3-d]пиримидин-7-ил)циклопентан-1,2-диол (W-6)
Схема W
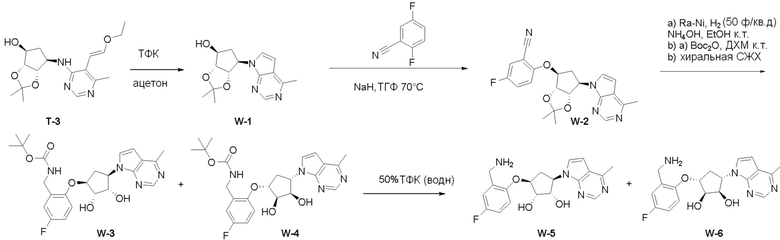
Стадия 1: Синтез (3aR,4S,6R,6aS)-2,2-диметил-6-(4-метил-7H-пирроло[2.3-d]пиримидин-7-ил)тетрагидро-4H-циклопента[d][1.3]диоксол-4-ола (W-1)
Смесь T-3 (111 мг, 0,331 ммоль) и трифторуксусной кислоты (127 мкл, 1,65 ммоль) в ацетоне (3,31 мл, 0,1 M) нагревают до 60°C в течение 43 часов. Реакционную смесь концентрируют до масла, затем растворяют в EtOAc. Слой EtOAc промывают насыщенным NaHCO3(водн), насыщенным раствором соли, сушат с Na2SO4, фильтруют и концентрируют до масла. Смесь неочищенного продукта очищают хроматографией на силикагеле, элюируя градиентом 0%-100% EtOAc в гептане с получением W-1 в виде белого твердого вещества, 76 мг (79% выход, 80% чистый).
Стадия 1: Синтез 2-(((3aR,4S,6R,6aS)-2,2-диметил-6-(4-метил-7H-пирроло[2.3-d]пиримидин-7-ил)тетрагидро-4H-циклопента[d][1.3]диоксол-4-ил)окси)-5-фторбензонитрила (W-2)
К смеси W-1 (67,1 мг, 0,2 ммоль) и 2,5-дифторбензонитрила (55,0 мг, 0,395 ммоль, 55,0 мкл) в ТГФ (1,01 мл, c=0,3 M) при кт добавляют 60% гидрид натрия (24,3 мг, 0,608 ммоль). После перемешивания в течение 3 минут реакционную смесь нагревают до 70°C в течение 6 часов, затем гасят насыщенным NH4Cl (водн), затем разбавляют EtOAc и водой. Слой EtOAc промывают насыщенным раствором соли, сушат с Na2SO4, фильтруют затем концентрируют до масла. Масло очищают хроматографией на силикагеле, элюируя 0-100% EtOAc в гептане с получением W-2 в виде белой пены, 101 мг (81% выход).
ЖХМС-ИЭР(+): MH+=409, 1H ЯМР (400 MГц, ХЛОРОФОРМ-d) δ ч./млн. 1,34 (с, 3 H) 1,61 (с, 3 H) 2,59 (д, J=15,41 Гц, 1 H) 2,78 (с, 3 H) 3,05 (дт, J=15,16, 7,34 Гц, 1 H) 4,84 (д, J=6,36 Гц, 1 H) 4,86-4,92 (м, 1 H) 5,04 (д, J=4,40 Гц, 1 H) 5,35-5,53 (м, 1 H) 6,68 (д, J=3,18 Гц, 1 H) 7,11 (дд, J=9,78, 3,67 Гц, 1 H) 7,32 (д, J=7,58 Гц, 2 H) 7,66 (шс, 1 H) 8,80 (с, 1 H).
Стадия 2: Синтез трет-бутил (2-(((1S,2S,3S,4R)-2.3-дигидрокси-4-(4-метил-7H-пирроло[2.3-d]пиримидин-7-ил)циклопентил)окси)-5-фторбензил)карбамата (W-3) и трет-бутил (2-(((1R,2R,3R,4S)-2.3-дигидрокси-4-(4-метил-7H-пирроло[2.3-d]пиримидин-7-ил)циклопентил)окси)-5-фторбензил)карбамата (W-4)
К смеси W-2 (101 мг, 0,247 ммоль) в этаноле (7,0 мл) добавляют 28% гидроксид аммония (3,0 мл) и никель Ренея (70 мг, 1,2 ммоль). Смесь гидрируют при 50 ф/кв.д в автоклаве из нержавеющей стали в течение 24 часов. Реакционную смесь фильтруют через целит, затем концентрируют до пены.
К этому неочищенному амину добавляют boc ангидрид (54 мг, 0,247 ммоль) в ДХМ (3 мл, 0,3 M) и перемешивают при кт в течение 2 часов. Смесь концентрируют до пены и очищают хроматографией на силикагеле с получением рацемической смеси W-3 и W-4 в виде бесцветного масла, 104 мг (82% выход). Энантиомеры разделяют хиральной СЖХ (колонка Chiralpak AS-3 4,6×100 мм 3 мкм, 8% MeOH+10 мМ NH3 @ 120 бар, 4 мл/мин) с получением W-3 (34,2 мг, 27% выход) и W-4 (35,9 мг, 28% выход) в виде белых твердых веществ.
W-3: ЖХМС-ИЭР(+): MH+=513, 1H ЯМР (400 MГц, ХЛОРОФОРМ-d) δ ч./млн. 8,86-8,76 (м, 1H), 7,45 (шс, 1H), 7,02 (д, J=8,6 Гц, 1H), 6,95 (д, J=5,1 Гц, 2H), 6,67 (д, J=3,2 Гц, 1H), 5,35 (шс, 1H), 5,07 (д, J=3,7 Гц, 1H), 4,89-4,71 (м, 3H), 4,21 (шс, 1H), 2,98 (тд, J=7,2, 14,7 Гц, 1H), 2,81 (с, 3H), 2,54 (д, J=15,2 Гц, 1H), 1,61 (с, 3H), 1,50-1,38 (м, 10H), 1,34 (с, 3H), [α]D22=+28,2° (c=0,1, MeOH)
W-4: ЖХМС-ИЭР(+): MH+=513 ~80% чистый.
Стадия 3: Синтез (1S,2S,3S,5R)-3-(2-(аминометил)-4-фторфенокси)-5-(4-метил-7H-пирроло[2.3-d]пиримидин-7-ил)циклопентан-1,2-диола (W-5)
Смесь W-3 (34 мг, 0,066 ммоль) в смеси 50% ТФК (водн) (0,4 мл) перемешивают при кт в течение 3 часов, затем концентрируют и очищают СЖХ (колонка ZymorSpher Pyridine Diol 150×21,2 мм с 20-40% MeOH @ 5%/мин, 100 бар, 60 мл/мин) с получением W-5 в виде белого твердого вещества, 16,5 мг (67% выход).
ЖХМС-ИЭР(+): MH+=373, 1H ЯМР (400 MГц, ДМСО-d6) δ ч./млн. 8,63 (с, 1H), 7,65 (д, J=3,7 Гц, 1H), 7,28 (дд, J=2,6, 9,2 Гц, 1H), 7,21-7,03 (м, 2H), 6,72 (д, J=3,4 Гц, 1H), 5,19-4,98 (м, 2H), 4,69-4,48 (м, 2H), 4,06 (д, J=4,6 Гц, 1H), 4,00 (с, 2H), 2,95-2,76 (м, 1H), 2,64 (с, 3H), 2,04 (ддд, J=4,0, 9,5, 13,8 Гц, 1H)
Стадия 4: Синтез (1R,2R,3R,5S)-3-(2-(аминометил)-4-фторфенокси)-5-(4-метил-7H-пирроло[2.3-d]пиримидин-7-ил)циклопентан-1,2-диола (W-6)
W-6 получают из W-4 по методике стадии 3 со схемы W с получением белого твердого вещества, 17,2 мг (70% выход).
ЖХМС-ИЭР(+): MH+=373, 1H ЯМР (400 MГц, ДМСО-d6) δ ч./млн. 8,63 (с, 1H), 7,65 (д, J=3,7 Гц, 1H), 7,28 (дд, J=2,6, 9,2 Гц, 1H), 7,21-7,07 (м, 2H), 6,72 (д, J=3,4 Гц, 1H), 5,18-5,02 (м, 2H), 4,69-4,54 (м, 2H), 4,06 (д, J=4,6 Гц, 1H), 4,01 (с, 2H), 2,94-2,74 (м, 1H), 2,64 (с, 3H), 2,04 (ддд, J=4,2, 9,5, 13,7 Гц, 1H).
Пример 68 (Схема X)-2-(((1S,2S,3S,4R)-2,3-дигидрокси-4-(4-метил-7H-пирроло[2.3-d]пиримидин-7-ил)циклопентил)окси)-5-фторбензонитрил (X-2)
Пример 69 (Схема X)-2-(((1R,2R,3R,4S)-2,3-дигидрокси-4-(4-метил-7H-пирроло[2.3-d]пиримидин-7-ил)циклопентил)окси)-5-фторбензонитрил (X-3)
Схема X
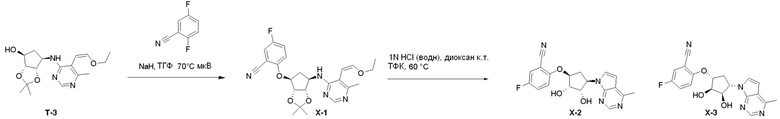
Стадия 1: Синтез 2-(((3aR,4S,6R,6aS)-6-((5-((Z)-2-этоксивинил)-6-метилпиримидин-4-ил)амино)-2,2-диметилтетрагидро-4H-циклопента[d][1.3]диоксол-4-ил)окси)-5-фторбензонитрила (X-1)
Смесь T-3 (67,1 мг, 0,2 ммоль) и O-фторбензонитрила (25,5 мкл, 0,240 ммоль) в ТГФ (0,67 мл, 0,3 M) при кт добавляют 60% гидрид натрия (16,8 мг, 0,42 ммоль). Смесь затем нагревают до 70°C в микроволновом реакторе в течение 30 минут. Реакционную смесь гасят насыщенным NH4Cl (водн), затем разбавляют водой и экстрагируют EtOAc. EtOAc промывают насыщенным раствором соли, сушат с Na2SO4, фильтруют и концентрируют до масла, затем очищают хроматографией на силикагеле, элюируя 0-100% EtOAc/Гептаном с получением X-1 в виде янтарного масла, 58 мг (64% выход).
ЖХМС-ИЭР(+): MH+=455, 1H ЯМР (400 MГц, ХЛОРОФОРМ-d) δ ч./млн. 8,47 (с, 1H), 7,35-7,28 (м, 2H), 7,16-7,06 (м, 1H), 6,47 (д, J=13,2 Гц, 1H), 5,43 (д, J=7,3 Гц, 1H), 5,39 (д, J=13,2 Гц, 1H), 4,81-4,65 (м, 4H), 3,87 (кв, J=7,0 Гц, 2H), 2,69 (тд, J=6,1, 14,9 Гц, 1H), 2,34 (с, 3H), 2,11 (д, J=14,9 Гц, 1H), 1,50 (с, 3H), 1,31 (с, 2H), 1,27 (т, J=7,0 Гц, 3H).
Стадия 2: Синтез 2-(((1S,2S,3S,4R)-2,3-дигидрокси-4-(4-метил-7H-пирроло[2.3-d]пиримидин-7-ил)циклопентил)окси)-5-фторбензонитрила (X-2) и 2-(((1R,2R,3R,4S)-2,3-дигидрокси-4-(4-метил-7H-пирроло[2.3-d]пиримидин-7-ил)циклопентил)окси)-5-фторбензонитрила (X-3)
Смесь X-1 (58 мг, 0,13 ммоль) и 1N HCl (водн) (255 мкл, 0,255 ммоль) в 1,4-диоксане (0,382 мл, 0,15 M) перемешивают при кт в течение 24 часов. Добавляют трифторуксусную кислоту (200 мкл), и смесь перемешивают при кт в течение 48 часов, затем нагревают до 60°C в течение 6 часов. Реакционную смесь концентрируют до масла, затем разделяют между EtOAc и водой. Добавляют насыщенный NaHCO3 (водн), и слой EtOAc промывают насыщенным раствором соли, сушат с Na2SO4, фильтруют, затем концентрируют до белого твердого вещества, 44 мг. Энантиомеры разделяют хиральной СЖХ (колонка Lux Cellulose-2 4,6×100 мм 3 мкм, 30% MeOH @ 120 бар, 4 мл/мин) с получением X-2 (17 мг, 36% выход) и X-3 (16,2 мг, 35% выход) в виде белых твердых веществ.
X-2: ЖХМС-ИЭР(+): MH+=369, 1H ЯМР (400 MГц, ДМСО-d6) δ ч./млн. 8,64 (с, 1H), 7,79 (дд, J=3,1, 8,2 Гц, 1H), 7,70 (д, J=3,4 Гц, 1H), 7,65-7,51 (м, 2H), 7,40 (дд, J=4,2, 9,3 Гц, 1H), 6,76 (д, J=3,4 Гц, 1H), 5,41 (шс, 1H), 5,15 (кв, J=8,9 Гц, 1H), 4,77 (шс, 1H), 4,53 (шс, 1H), 4,05 (шс, 1H), 2,97-2,75 (м, 1H), 2,65 (с, 3H), 2,11-1,93 (м, 1H), [α]D22= +47,3° (c=0,1, MeOH).
X-3: ЖХМС-ИЭР(+): MH+=369, 1H ЯМР (400 MГц, МЕТАНОЛ-d4) δ ч./млн. 7,72 (д, J=3,4 Гц, 1H), 7,51 (дд, J=2,9, 7,6 Гц, 1H), 7,47-7,38 (м, 1H), 7,32 (дд, J=4,0, 9,2 Гц, 1H), 6,80 (д, J=3,4 Гц, 1H), 5,33 (кв, J=8,6 Гц, 1H), 4,85 (шс, 1H), 4,71 (дд, J=4,8, 8,2 Гц, 1H), 4,22 (д, J=3,9 Гц, 1H), 3,12-2,97 (м, 1H), 2,74 (шс, 3H), 2,26-2,12 (м, 1H), [α]D22= -53,5° (c=0,1, MeOH).
Пример 70 (Схема Y) - 2-(((1S,2S,3S,4R)-2.3-дигидрокси-4-(4-метил-7H-пирроло[2.3-d]пиримидин-7-ил)циклопентил)окси)-5-фторбензамид (Y-3)
Схема Y
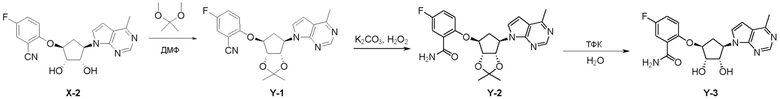
Стадия 1 - Синтез 2-(((3aR,4S,6R,6aS)-2,2-диметил-6-(4-метил-7H-пирроло[2.3-d]пиримидин-7-ил)тетрагидро-4H-циклопента[d][1.3]диоксол-4-ил)окси)-5-фторбензонитрила (Y-1)
К суспензии X-2 (4 г, 9,8 ммоль) в диметоксипропане (60 мл)/ДМФ (20 мл) добавляют TsOH.H2O (2990 мг, 15,75 ммоль) при кт (20°C). Полученный раствор перемешивают при кт (20°C) в течение 24 ч. Смесь очищают непосредственно хроматографией на силикагеле, элюируя EtOAc в петролейном эфире 0-100% с получением Y-1 (2,2 г, 55%) в виде белого твердого вещества. ЖХМС 409 [M+1]; 1H ЯМР (400 MГц, CDCl3) δ ч./млн. 8,77 (с, 1H), 7,60 (д, J=3,8 Гц, 1H), 7,33-7,25 (м, 2H), 7,15-7,06 (м, 1H), 6,63 (д, J=3,5 Гц, 1H), 5,44 (ддд, J=2,5, 5,0, 7,8 Гц, 1H), 5,02 (дд, J=2,3, 6,0 Гц, 1H), 4,90-4,85 (м, 1H), 4,82 (д, J=6,3 Гц, 1H), 3,04 (ддд, J=6,8, 8,0, 15,1 Гц, 1H), 2,72 (с, 3H), 2,63-2,53 (м, 1H), 1,60 (с, 3H), 1,33 (с, 3H)
Стадия 2 - Синтез 2-(((3aR,4S,6R,6aS)-2,2-диметил-6-(4-метил-7H-пирроло[2.3-d]пиримидин-7-ил)тетрагидро-4H-циклопента[d][1.3]диоксол-4-ил)окси)-5-фторбензамида (Y-2)
К раствору Y-1 (50 мг, 0,12 ммоль) в ДМСО (0,25 мл) добавляют K2CO3 (20 мг, 0,15 ммоль), затем H2O2 (0,25 мл) по каплям в льде-воде, затем реакционную смесь нагревают до комнатной температуры в течение 2 ч, наблюдается газообразование. К реакционной смеси добавляют воду и экстрагируют EtOAc два раза. Органический слой сушат над сульфатом натрия, фильтруют, и остаток очищают преп.-ТСХ с получением Y-2 (36 мг, 69%) и применяют как есть на следующей стадии.
Стадия 3-Синтез 2-(((1S,2S,3S,4R)-2.3-дигидрокси-4-(4-метил-7H-пирроло[2.3-d]пиримидин-7-ил)циклопентил)окси)-5-фторбензамида (Y-3)
К суспензии Y-2 (42 мг, 0,1 ммоль) в H2O (0,3 мл) добавляют ТФК (0,3 мл) по каплям при 0°C. Затем реакционную смесь перемешивают при комнатной температуре (20°C) в течение 16 ч. Реакционную смесь затем доводят до pH 7 добавлением 20% K2CO3, при этом образуется твердое вещество, затем фильтруют и промывают водой и МТБЭ. Твердое вещество сушат с получением Y-3 (32 мг, 84%). ЖХМС 387 [M+1]; 1H ЯМР (400 MГц, MeOD-d4) δ ч./млн. 8,63 (с, 1H), 7,62-7,65 (м, 1H), 7,60 (д, J=3,8 Гц, 1H), 7,24-7,30 (м, 2H), 6,75 (д, J=3,5 Гц, 1H), 5,16 (кв, J=8,8 Гц, 1H), 4,82-4,87 (м, 2H), 4,27 (д, J=4,8 Гц, 1H), 3,02 (ддд, J=14,6, 9,6, 7,4 Гц, 1H), 2,73 (с, 3H), 2,42 ч./млн. (ддд, J=13,7, 9,2, 4,3 Гц, 1H)
Пример 71 (Схема Z) - (1S,2S,3S,5R)-3-(2-((диметиламино)метил)-4-фторфенокси)-5-(4-метил-7H-пирроло[2.3-d]пиримидин-7-ил)циклопентан-1,2-диол (8)
Схема Z
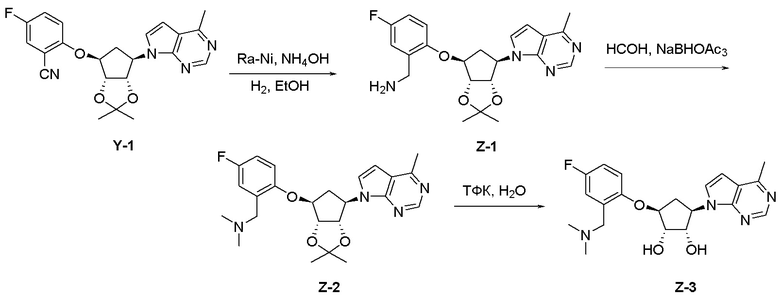
Стадия 1 - Синтез (2-(((3aR,4S,6R,6aS)-2,2-диметил-6-(4-метил-7H-пирроло[2.3-d]пиримидин-7-ил)тетрагидро-4H-циклопента[d][1.3]диоксол-4-ил)окси)-5-фторфенил)метанамина (Z-1)
Смесь Y-1 (500 мг, 1,22 ммоль) и Ra-Ni (100 мг) в EtOH (30 мл)/NH3.H2O (3 мл) дегазируют с H2 четыре раза. Смесь перемешивают при кт (20°C) под баллоном H2 в течение 20 ч, затем выдерживают при кт в течение 20 ч. Смесь фильтруют и концентрируют в вакууме с получением Z-1 (510 мг, >99%) в виде светло-желтой камеди. ЖХМС [M+1] 413; 1H ЯМР (400 MГц, CDCl3) δ ч./млн. 8,79 (с, 1H), 7,37 (д, J=3,5 Гц, 1H), 7,05 (дд, J=2,4, 8,7 Гц, 1H), 6,99-6,88 (м, 2H), 6,58 (д, J=3,8 Гц, 1H), 5,33 (ддд, J=2,6, 5,5, 8,0 Гц, 1H), 5,05 (дд, J=2,9, 6,1 Гц, 1H), 4,88-4,80 (м, 1H), 4,80-4,75 (м, 1H), 3,76 (шс, 2H), 3,06-2,89 (м, 1H), 2,73 (с, 3H), 2,62-2,47 (м, 1H), 1,61 (с, 3H), 1,34 (с, 3H)
Стадия 2-Синтез 1-(2-(((3aR,4S,6R,6aS)-2,2-диметил-6-(4-метил-7H-пирроло[2.3-d]пиримидин-7-ил)тетрагидро-4H-циклопента[d][1.3]диоксол-4-ил)окси)-5-фторфенил)-N,N-диметилметанамина (Z-2)
Смесь Z-1 (100 мг, 0,24 ммоль), 37% CH2O (59 мг, 0,73 ммоль) и NaBHOAc3 (206 мг, 0,970 ммоль) в ТГФ (2 мл) перемешивают при кт в течение 2 ч. Смесь выливают в NaHCO3 водн. (10 мл) и экстрагируют EtOAc (10 мл×3). Экстракт промывают насыщенным раствором соли (10 мл), сушат над Na2SO4 и концентрируют в вакууме с получением Z-2 (100 мг, 94%) в виде бесцветной камеди. ЖХМС 441 [M+1]; 1H ЯМР (400 MГц, CDCl3) δ ч./млн. 8,79 (с, 1H), 7,49 (д, J=3,8 Гц, 1H), 7,14-7,08 (м, 1H), 7,01-6,86 (м, 2H), 6,55 (д, J=3,5 Гц, 1H), 5,37 (т, J=2,8 Гц, 1H), 5,02 (д, J=4,0 Гц, 1H), 4,83 (д, J=6,3 Гц, 1H), 4,77 (дд, J=3,3, 5,8 Гц, 1H), 3,33 (с, 2H), 2,97 (д, J=8,0 Гц, 1H), 2,73 (с, 3H), 2,54 (д, J=13,8 Гц, 1H), 2,23 (с, 6H), 1,60 (с, 3H), 1,33 (с, 3H)
Стадия 3 - Синтез (1S,2S,3S,5R)-3-(2-((диметиламино)метил)-4-фторфенокси)-5-(4-метил-7H-пирроло[2.3-d]пиримидин-7-ил)циклопентан-1,2-диола (Z-3)
К ТФК/H2O (1 мл/2 мл) добавляют Z-2 (100 мг, 0,23 ммоль) при кт. Смесь перемешивают при кт в течение 1 ч. Смесь выливают в 20% K2CO3 (10 мл). Смесь промывают насыщенным NaCl и экстрагируют EtOAc/ТГФ (20 мл/20 мл) дважды. Экстракт промывают, концентрируют в вакууме. Остаток растворяют в CH3CN/H2O (10 мл/50 мл) и лиофилизируют с получением Z-3 (70 мг, 77%) в виде белого твердого вещества. ЖХМС 404 [M+1]; 1H ЯМР (400 MГц, ДМСО-d6) δ ч./млн. 8,61 (с, 1H), 7,69 (д, J=3,8 Гц, 1H), 7,13 (д, J=8,5 Гц, 1H), 7,05 (д, J=5,5 Гц, 2H), 6,72 (д, J=3,5 Гц, 1H), 5,36 (шс, 1H), 5,16-5,09 (м, 2H), 4,60-4,49 (м, 2H), 4,01 (д, J=3,3 Гц, 1H), 3,57-3,48 (м, 1H), 3,44 (шс, 1H), 2,90-2,78 (м, 1H), 2,63 (с, 3H), 2,23 (с, 6H), 1,96-1,86 (м, 1H)
Пример 72 (Схема AA) - (1S,2S,3S,5R)-3-(4-фтор-2-((метиламино)метил)фенокси)-5-(4-метил-7H-пирроло[2.3-d]пиримидин-7-ил)циклопентан-1,2-диол (11)
Схема AA
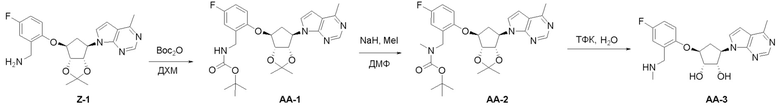
Стадия 1 - Синтез трет-бутил (2-(((3aR,4S,6R,6aS)-2,2-диметил-6-(4-метил-7H-пирроло[2.3-d]пиримидин-7-ил)тетрагидро-4H-циклопента[d][1.3]диоксол-4-ил)окси)-5-фторбензил)карбамата (AA-1)
К раствору Z-1 (140 мг,0,34 ммоль) и Et3N (34 мг, 0,34 ммоль) в ДХМ (5 мл) добавляют Boc2O (74 мг, 0,34 ммоль) при кт (20°C). Смесь перемешивают при кт (20°C) в течение 1 ч. Смесь очищают хроматографией на силикагеле, элюируя EtOAC в петролейном эфире 0-100% с получением AA-1 (140 мг, 81%) в виде бесцветного масла. ЖХМС 513 [M+1]; 1H ЯМР (400 MГц, CDCl3) δ ч./млн. 8,78 (с, 1H), 7,33 (с, 1H), 7,00 (д, J=8,8 Гц, 1H), 6,93 (д, J=5,2Гц, 2H), 6,58 (д, J=3,6Гц, 1H), 5,30-5,28 (м, 1H), 5,06-5,05 (м, 1H), 4,81 (д, J=6Гц, 2H), 4,76-4,75 (м, 1H), 4,18 (д, J=3,2Гц, 1H), 2,98-2,90 (м, 1H), 2,71 (с, 3H), 2,54-2,50 (м, 1H), 1,55 (с, 3H), 1,44 (с, 9H), 1,24 (с, 3H)
Стадия 2 - Синтез трет-бутил (2-(((3aR,4S,6R,6aS)-2,2-диметил-6-(4-метил-7H-пирроло[2.3-d]пиримидин-7-ил)тетрагидро-4H-циклопента[d][1.3]диоксол-4-ил)окси)-5-фторбензил)(метил)карбамата (AA-2)
К раствору AA-1 (140 мг, 0,27 ммоль) в сухом ДМФ (3 мл) добавляют 60% NaH (16,4 мг, 0,41 ммоль) при кт (0°C). Смесь перемешивают при кт (20°C) в течение 1 ч. CH3I (56 мг, 0,4 ммоль) добавляют к смеси и перемешивают при кт (20°C) в течение 20 ч. Смесь выливают в NH4Cl водн. (10 мл) и экстрагируют EtOAc (5 мл×3). Экстракт промывают насыщенным раствором соли (5 мл), сушат над Na2SO4 и концентрируют в вакууме с получением неочищенного продукта (200 мг) в виде коричневого масла, которое очищают хроматографией на силикагеле, элюируя EtOAc в петролейном эфире 0-100% с получением AA-2 (100 мг, 70%) в виде бесцветной камеди. ЖХМС 527 [M+1]
Стадия 3 - Синтез (1S,2S,3S,5R)-3-(4-фтор-2-((метиламино)метил)фенокси)-5-(4-метил-7H-пирроло[2.3-d]пиримидин-7-ил)циклопентан-1,2-диола (AA-3)
К ТФК/H2O (1 мл/2 мл) добавляют AA-2 (89 мг, 0,169 ммоль) при кт. Смесь перемешивают при кт в течение 2 ч, затем выливают в 20% K2CO3 водн. (5 мл). Смесь экстрагируют EtOAc (10 мл×3). Экстракт промывают насыщенным раствором соли (10 мл×2), сушат над Na2SO4, фильтруют и концентрируют в вакууме. Остаток растворяют в CH3CN/H2O (4 мл/10 мл) и лиофилизируют с получением AA-3 (55 мг, 84%) в виде белого твердого вещества. ЖХМС 387 [M+1]; 1H ЯМР (400 MГц, ДМСО-d6) δ ч./млн. 8,62 (с, 1H), 7,67 (д, J=3,8 Гц, 1H), 7,23-7,14 (м, 1H), 7,08-6,98 (м, 2H), 6,73 (д, J=3,8 Гц, 1H), 5,12 (кв, J=9,1 Гц, 2H), 4,57 (шс, 2H), 4,02 (д, J=4,8 Гц, 1H), 3,71 (с, 2H), 2,92-2,77 (м, 1H), 2,65 (с, 3H), 2,34 (с, 3H), 2,03-1,93 (м, 1H)
Пример 73 (Схема BB) - (1S,2S,3S,5R)-3-(4-фторфенокси)-5-(4-метил-7H-пирроло[2.3-d]пиримидин-7-ил)циклопентан-1,2-диол (BB-4)
Схема BB

Стадия 1: Синтез трет-бутил-((1S,4R)-4-(4-метил-7H-пирроло[2.3-d]пиримидин-7-ил)циклопент-2-ен-1-ил)карбоната (BB-2)
Пробирка A: В высушенную в печи реакционную пробирку, оборудованную магнитной мешалкой и охлаждаемым потоком аргона, добавляют продукт присоединения трис(бензилиденацетон)дипалладия(0) и хлороформа (78,7 мг, 0,0760 ммоль) и лиганд Троста (S,S)-DACH-нафтил (MFCD02684552) (180 мг, 0,228 ммоль). Пробирку продувают в вакууме аргоном в динамическом вакууме и ДХЭ (7,5 мл), который барботируют с аргоном в течение 30 минут. Раствор перемешивают в течение 30 минут при кт, получая в этот момент ярко оранжевый раствор лигированного катализатора. На этой стадии получают пробирку B.
Пробирка B: В высушенную в печи реакционную пробирку, оборудованную магнитной мешалкой и охлаждаемым потоком аргона, добавляют 4-метил-7H-пирроло[2.3-d]пиримидин (HG-1) (506 мг, 3,8 ммоль), ди-трет-бутил-((1R,3S)-циклопент-4-ен-1,3-диил)-бис(карбонат) (BB-1) (полученный как описано в J. Am. Chem. Soc, 2006, 128, 6054-6055) (1,37 г, 4,56 ммоль) и карбонат цезия (1,36 г, 4,18 ммоль). Пробирку продувают в вакууме аргоном в динамическом вакууме и добавляют ДХЭ (7,5 мл), который барботируют с аргоном в течение 30 минут, затем добавляют содержимое пробирки A через герметизированный шприц. Реакционную смесь перемешивают под аргоном при кт в течение 48 часов. Реакционную смесь переносят в делительную воронку с ДХМ. Раствор промывают 2 порциями воды и 1 порцией насыщенного раствора соли. Небольшое количество 1M HCl применяют рассеяния эмульсии в последней промывке. Органическую фазу сушат (MgSO4), фильтруют и концентрируют в вакууме. Неочищенный остаток очищают флэш-хроматографией на колонке (40 г SiO2, Isco, 100% гепт. до 100% EtOAc, 20 мл фракции) с получением соединения BB-2 (814 мг, 68%, >99% эи) в виде коричневой камеди, которая затвердевает при выстаивании. ЖХМС [M+H]=316 найдено; Хиральная ЖХМС (колонка Chiralcel OJ-3 4,6×100 мм 3 мк, 4% MeOH+10 нМ NH3 @ 120 бар, 4 мл/мин) пик 1 @ 0,63 мин, пик 2 @ 0,77 мин, наблюдаемый основной пик @ 0,65 мин, 99,8:0,2% площадь МС; 1H ЯМР (400 MГц, ХЛОРОФОРМ-d) δ ч./млн. 8,78 (с, 1H), 7,32-7,22 (м, 1H), 6,60 (д, J=3,7 Гц, 1H), 6,35-6,23 (м, 1H), 6,10 (д, J=5,4 Гц, 1H), 6,00 (шс, 1H), 5,66-5,57 (м, 1H), 3,21-3,07 (м, 1H), 2,75 (с, 3H), 1,89 (д, J=14,9 Гц, 1H), 1,57-1,46 (м, 9H).
Стадия 2: Синтез 7-((1R,4S)-4-(4-фторфенокси)циклопент-2-ен-1-ил)-4-метил-7H-пирроло[2.3-d]пиримидина (BB-3)
В сцинтилляционную пробирку, оборудованную магнитной мешалкой, добавляют BB-2 (56,8 мг, 0,180 ммоль), 4-фторфенол (22,2 мг, 0,198 ммоль), дифенилфосфинопропан (dppp) (4,5 мг, 0,0108 ммоль), продукт присоединения трис(дибензилиденацетон)дипалладия(0) с хлороформом (4,7 мг, 0,0045 ммоль), и карбонат цезия (64,6 мг, 0,198 ммоль). Пробирку продувают аргоном в динамическом вакууме, затем добавляют ДХЭ (0,9 мл), который барботируют с аргоном в течение 30 минут. Реакционную смесь перемешивают при кт под аргоном в течение 2,5 часов. Реакционную смесь берут с ДХМ и загружают непосредственно в упакованную колонку с силикагелем. Неочищенный остаток очищают флэш-хроматографией на колонке (12 г SiO2, Isco, 100% Гепт. до 100% EtOAc, 9 мл фракции) с получением соединения BB-3 (38,8 мг, 70% выход) в виде бесцветной камеди. ЖХМС [M+H]=310 наблюдаемый; 1H ЯМР (400 MГц, ХЛОРОФОРМ-d) δ ч./млн. 8,77 (с, 1H), 7,34 (д, J=3,55 Гц, 1H), 6,92-7,04 (м, 2H), 6,80-6,92 (м, 2H), 6,57 (д, J=3,55 Гц, 1H), 6,37 (д, J=5,38 Гц, 1H), 6,14 (д, J=4,03 Гц, 1H), 5,97-6,09 (м, 1H), 5,17-5,34 (м, 1H), 3,02-3,18 (м, 1H), 2,56-2,91 (м, 3H), 1,97 (тд, J=3,35, 14,70 Гц, 1H).
Стадия 3: Синтез (1S,2S,3S,5R)-3-(4-фторфенокси)-5-(4-метил-7H-пирроло[2.3-d]пиримидин-7-ил)циклопентан-1,2-диола (BB-4)
В сцинтилляционную пробирку, оборудованную магнитной мешалкой и содержащую BB-3 (38,8 мг, 0,125 ммоль), добавляют ДХМ (0,73 мл) и воду (0,03 мл). В раствор добавляют 4-Метилморфолин-N-оксид (NMO) (44,1 мг, 0,376 ммоль), затем по каплям добавляют тетраоксид осмия (32 мкл, 0,005 ммоль) в виде 4% масс. раствора в воде. Реакционную смесь перемешивают при кт в течение 8 часов. Реакцию гасят 1M NaHSO3 водн., перемешивают в течение 30 минут и переносят в делительную воронку с ДХМ. Раствор затем разбавляют водой, и продукт экстрагируют 4 порциями 3:1 смеси CHCl3/и-PrOH. Объединенные органические экстракты сушат (MgSO4), фильтруют и концентрируют в вакууме с получением неочищенного продукта в виде белого твердого вещества. Неочищенный остаток очищают препаративной ВЭЖХ (колонка Lux Cellulose-1 4,6×100 мм 3 мк, 15% MeOH @ 120 бар, 4 мл/мин) с получением соединения BB-4 (30,4 мг, 71%, >99% ди) в виде белого твердого вещества. ЖХМС [M+H]=344 наблюдаемый; [α]22D=-36,0° (c=0,1, MeOH); 1H ЯМР (400 MГц, МЕТАНОЛ-d4) δ ч./млн. 8,61 (с, 1H), 7,58 (д, J=3,67 Гц, 1H), 6,94-7,09 (м, 4H), 6,75 (д, J=3,67 Гц, 1H), 5,27 (кв, J=8,64 Гц, 1H), 4,58-4,68 (м, J=3,67 Гц, 2H), 4,17 (д, J=4,89 Гц, 1H), 2,96 (ддд, J=7,15, 9,32, 14,46 Гц, 1H), 2,71 (с, 3H), 2,09 (ддд, J=3,85, 8,53, 14,03 Гц, 1H). 19F ЯМР (377 MГц, МЕТАНОЛ-d4) δ ч./млн.=-125,65 (с, 1F).
Примеры 74-77 получают с применением методики, изображенной на схеме BB и с применением подходящего коммерческого фенольного реагента на стадии 2.
2,6-дифторфенол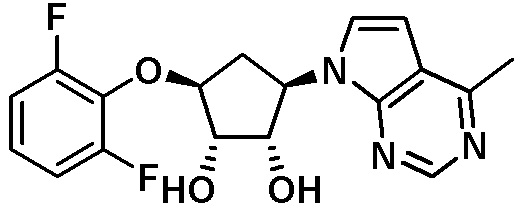
1H ЯМР (400 MГц, МЕТАНОЛ-d4) δ ч./млн. 8,64 (с, 1H), 7,70 (д, J=4,02 Гц, 1H), 7,18-7,02 (м, 3H), 6,82 (д, J=3,51 Гц, 1H), 5,39-5,31 (м, 1H), 4,75 (м, 1H), 4,68 (м, 1H), 4,23 (м, 1H), 3,03-2,94 (м, 1H), 2,75 (с, 3H), 2,19 (дд, J=14,8, 5,3 Гц, 1H).
4-фтор-2-метилфенол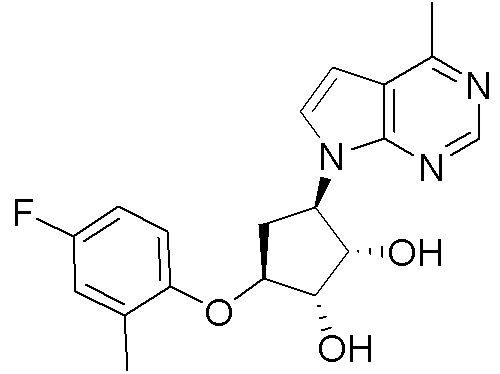
1H ЯМР (400 MГц, MeOD-d4) δ ч./млн. 8,63 (с, 1H), 7,59 (д, J=3,8 Гц, 1H), 6,96 (тд, J=9,5, 4,0 Гц, 2H), 6,85-6,91 (м, 1H), 6,78 (д, J=3,5 Гц, 1H), 5,29 (кв, J=8,8 Гц, 1H), 4,64-4,73 (м, 2H), 4,21 (д, J=4,5 Гц, 1H), 3,01 (ддд, J=14,6, 9,5, 7,0 Гц, 1H), 2,73 (с, 3H), 2,31 (с, 3H), 2,16 ч./млн. (ддд, J=14,5, 8,5, 3,4 Гц, 1H)
8-гидрокси-3,4-дигидроизохинолин-1(2H)-он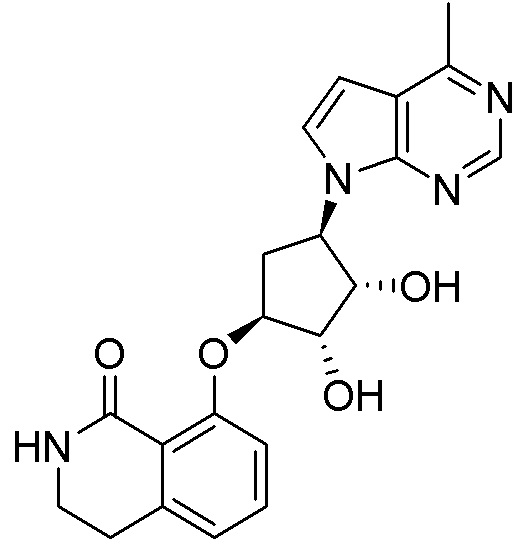
1H ЯМР (400 MГц, MeOD-d4) δ ч./млн. 8,60 (с, 1H), 7,97 (д, J=4,0 Гц, 1H), 7,42 (т, J=8,0 Гц, 1H), 7,08 (д, J=8,8 Гц, 1H), 6,91 (д, J=7,3 Гц, 1H), 6,75 (д, J=3,8 Гц, 1H), 5,47-5,33 (м, 1H), 4,83-4,71 (м, 2H), 4,24 (д, J=3,8 Гц, 1H), 3,42 (т, J=6,5 Гц, 2H), 3,06-2,98 (м, 1H), 2,97-2,90 (м, 2H), 2,71 (с, 3H), 2,11-2,02 (м, 1H)
изохинолин-8-ол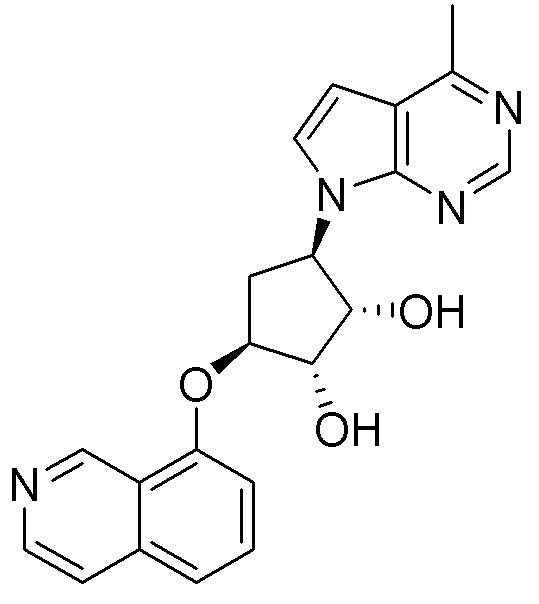
1H ЯМР (400 MГц, ДМСО-d6) δ ч./млн. 9,69 (шс, 1H), 8,62 (с, 1H), 8,56 (шд, J=4,5 Гц, 1H), 7,83 (шд, J=5,5 Гц, 1H), 7,79 (д, J=3,5 Гц, 1H), 7,72 (т, J=8,0 Гц, 1H), 7,53 (д, J=8,0 Гц, 1H), 7,24 (д, J=7,8 Гц, 1H), 6,76 (д, J=3,5 Гц, 1H), 5,50 (шс, 1H), 5,20 (кв, J=1,0 Гц, 2H), 4,89-4,83 (м, 1H), 4,72-4,66 (м, 1H), 4,18 (шд, J=4,3 Гц, 1H), 3,02-2,91 (м, 1H), 2,66 (с, 3H), 2,24-2,14 (м, 1H)
Пример 78 (Схема CC) - (1S,2S,3S,5R)-3-((6-хлор-5-фтор-1,2,3,4-тетрагидроизохинолин-8-ил)окси)-5-(4-метил-7H-пирроло[2.3-d]пиримидин-7-ил)циклопентан-1,2-диол (CC-3)
Схема CC
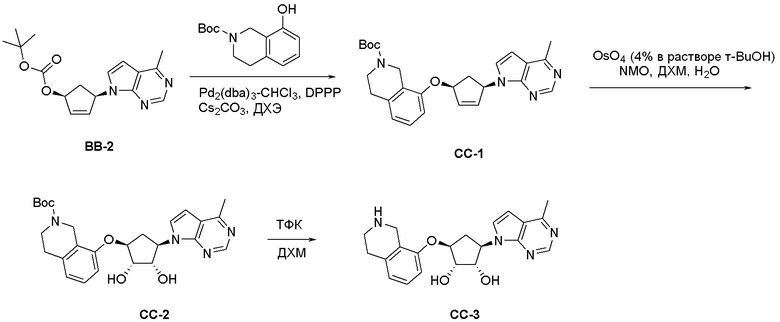
Стадия 1 - Синтез трет-бутил 8-(((1S,4R)-4-(4-метил-7H-пирроло[2.3-d]пиримидин-7-ил)циклопент-2-ен-1-ил)окси)-3,4-дигидроизохинолинe-2(1H)-карбоксилата (CC-1)
К смеси BB-2 (600 мг, 1,9 ммоль) и трет-бутил 8-гидрокси-3,4-дигидроизохинолинe-2(1H)-карбоксилата (474 мг, 1,9 ммоль), Cs2CO3 (682 мг, 2,09 ммоль) и DPPP (47,1 мг, 0,11 ммоль) в ДХЭ (15 мл) продувают N2 в течение 40 мин. К смеси добавляют Pd2(dba)3.CHCl3 (49 мг, 0,048 ммоль) под N2. Реакционную смесь продувают N2 в течение 5 мин, затем перемешивают при кт (20°C) под N2 в течение 40 мин. Смесь немедленно очищают (очищают непосредственно раствор) хроматографией на силикагеле, элюируя петролейным эфиром/EtOAc=8/1-1/1, затем ДХМ/MeOH=12/1 с получением CC-1 (735 мг, 87%) в виде желтой камеди. ЖХМС [M+1] 447; 1H ЯМР (400 MГц, CDCl3) δ ч./млн. 8,77 (с, 1H), 7,33 (д, J=3,5 Гц, 1H), 7,13 (т, J=7,9 Гц, 1H), 6,79-6,72 (м, 2H), 6,62-6,52 (м, 1H), 6,44-6,33 (м, 1H), 6,21-6,11 (м, 1H), 6,09-6,01 (м, 1H), 5,41-5,33 (м, 1H), 4,71-4,40 (м, 2H), 3,71-3,48 (м, 2H), 3,22-3,12 (м, 1H), 2,82 (шс, 2H), 2,73 (с, 3H), 1,98 (д, J=15,1 Гц, 1H), 1,51 (с, 9H)
Стадия 2 - Синтез трет-бутил 8-(((1S,2S,3S,4R)-2,3-дигидрокси-4-(4-метил-7H-пирроло[2.3-d]пиримидин-7-ил)циклопентил)окси)-3,4-дигидроизохинолин-2(1H)-карбоксилата (CC-2)
К смеси CC-1 (730 мг, 1,63 ммоль) в ДХМ (15 мл)/H2O (0,5 мл) добавляют NMO (575 мг, 4,9 ммоль) и OsO4 (4% в т-BuOH, 832 мг, 0,12 ммоль). Смесь перемешивают при кт (25°C) в течение 3 ч. Добавляют Na2SO3 (500 мг) и воду (20 мл). Смесь перемешивают при кт в течение 1 ч, затем смесь фильтруют. Фильтрат разбавляют водой (50 мл) и экстрагируют EtOAc (50 мл×2). Экстракт промывают насыщенным раствором соли (50 мл), сушат над Na2SO4 и концентрируют в вакууме и очищают хроматографией на силикагеле, элюируя MeOH в ДХМ 0-10% с получением CC-2 (560 мг, 71%) в виде белого твердого вещества. ЖХМС [M+1] 481; 1H ЯМР (400 MГц, CDCl3) δ ч./млн. 8,67 (с, 1H), 7,24 (д, J=3,8 Гц, 1H), 7,18 (т, J=7,9 Гц, 1H), 6,90 (д, J=8,0 Гц, 1H), 6,80 (д, J=7,8 Гц, 1H), 6,60 (д, J=3,3 Гц, 1H), 5,08-4,94 (м, 1H), 4,87-4,76 (м, 1H), 4,61-4,39 (м, 3H), 4,37-4,28 (м, 1H), 3,76-3,55 (м, 2H), 3,46-3,31 (м, 1H), 3,20-3,04 (м, 1H), 2,86-2,79 (м, 2H), 2,73 (с, 3H), 2,48-2,30 (м, 1H), 1,48 (с, 9H)
Стадия 3 - Синтез (1S,2S,3R,5S)-3-(4-метил-7H-пирроло[2.3-d]пиримидин-7-ил)-5-((1,2,3,4-тетрагидроизохинолин-8-ил)окси)циклопентан-1,2-диол (CC-3)
К раствору CC-2 (560 мг, 1,17 ммоль) в ДХМ (15 мл) добавляют ТФК (4,5 мл) при 0°C. Реакционную смесь перемешивают при 0°C в течение 1 часа. Реакционную смесь выпаривают и растворяют в воде (30 мл) и добавляют K2CO3 (1 г). Смесь разбавляют насыщенным раствором соли (30 мл) и экстрагируют EtOAc/ТГФ (1/1, 30 мл×3). Экстракт промывают насыщенным раствором соли (30 мл×2), сушат над Na2SO4, фильтруют и концентрируют в вакууме с получением CC-3 (400 мг, 90%) в виде белого твердого вещества. ЖХМС [M+1] 381; 1H ЯМР (400 MГц, ДМСО-d6) δ ч./млн. 8,64 (с, 1H), 7,62 (д, J=3,8 Гц, 1H), 7,11-7,02 (м, 1H), 6,81 (д, J=8,3 Гц, 1H), 6,73 (д, J=3,5 Гц, 1H), 6,68 (д, J=7,5 Гц, 1H), 5,33 (шс, 1H), 5,19-5,08 (м, 2H), 4,56 (шс, 2H), 3,99 (шс, 1H), 3,85 (с, 2H), 2,93 (т, J=5,8 Гц, 2H), 2,90-2,81 (м, 1H), 2,70-2,66 (м, 2H), 2,65 (с, 3H), 1,98-1,86 (м, 1H)
Примеры 79 & 80 получают с применением методики, изображенной на схеме CC и с применением подходящего коммерческого NBoc-защищенного фенольного реагента на стадии 1.
трет-бутил 4-гидроксиизоиндолин-2-карбоксилат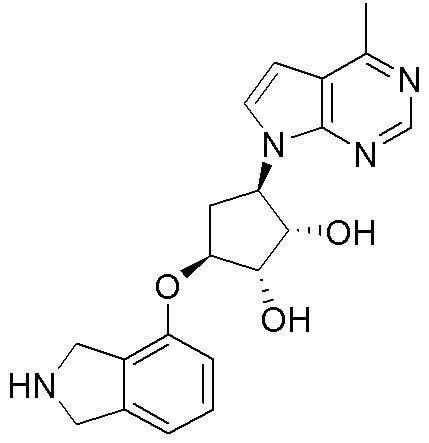
1H ЯМР (400 MГц, MeOD-d4) δ ч./млн. 8,63 (с, 1H), 8,43-8,33 (м, 1H), 7,59 (д, J=3,5 Гц, 1H), 7,45-7,36 (м, 1H), 7,13-7,08 (м, 1H), 7,07-7,01 (м, 1H), 6,77 (д, J=3,8 Гц, 1H), 5,28-5,19 (м, 1H), 4,82-4,78 (м, 1H), 4,76-4,71 (м, 1H), 4,67 (д, J=4,8 Гц, 4H), 4,24 (д, J=3,8 Гц, 1H), 3,08-2,94 (м, 1H), 2,73 (с, 3H), 2,28-2,15 (м, 1H)
трет-бутил 5-гидрокси-3,4-дигидроизохинолин-2(1H)-карбоксилат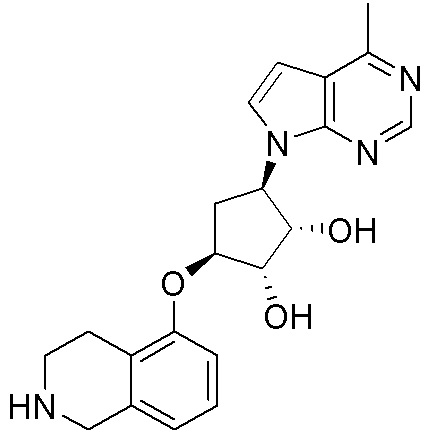
1H ЯМР (400 MГц, ДМСО-d6) δ ч./млн. 8,64 (с, 1H), 7,64 (д, J=3,5 Гц, 1H), 7,26-7,18 (м, 1H), 6,99 (д, J=8,0 Гц, 1H), 6,82 (д, J=7,8 Гц, 1H), 6,73 (д, J=3,5 Гц, 1H), 5,40 (шс, 1H), 5,19-5,08 (м, 2H), 4,66-4,54 (м, 2H), 4,22 (с, 2H), 4,02 (шс, 1H), 2,96-2,83 (м, 3H), 2,65 (с, 3H), 2,01-1,90 (м, 1H)
Пример 81 (Схема DD) - (1S,2S,3R,5S)-3-(4-метил-7H-пирроло[2.3-d]пиримидин-7-ил)-5-[(2-метил-1,2,3,4-тетрагидроизохинолин-5-ил)окси]циклопентан-1,2-диол (DD-1)
Схема DD
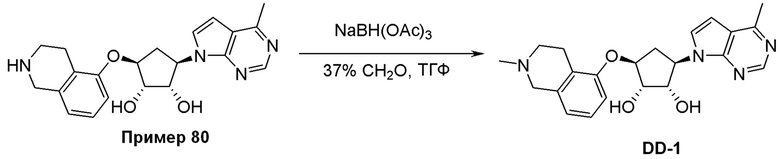
Стадия 1: Синтез(1S,2S,3R,5S)-3-(4-метил-7H-пирроло[2.3-d]пиримидин-7-ил)-5-[(2-метил-1,2,3,4-тетрагидроизохинолин-5-ил)окси]циклопентан-1,2-диола (DD-1)
Смесь соединения из Примера 80 (42 мг, 0,11 ммоль), 37% CH2O (26,9 мг, 0,331 ммоль) и NaBHOAc3 (93,6 мг, 0,442 ммоль) в ТГФ (1,2 мл) перемешивают при кт в течение 2 ч. Смесь выливают в NaHCO3 водн. (10 мл) и экстрагируют EtOAc (20 мл×4). Экстракт сушат над Na2SO4 и концентрируют в вакууме с получением остатка (50 мг) в виде белого твердого вещества, которое очищают преп.-ТСХ (ДХМ:MeOH=10:1 с NH3.H2O) с получением DD-1 (35 мг, 80%) в виде белого твердого вещества. ЖХМС 395 [M+1]; 1H ЯМР (400 MГц, ДМСО-d6) δ ч./млн. 8,63 (с, 1H), 7,63 (д, J=3,5 Гц, 1H), 7,16 (т, J=8,0 Гц, 1H), 6,92 (д, J=8,5 Гц, 1H), 6,75-6,69 (м, 2H), 5,36 (д, J=3,8 Гц, 1H), 5,18-5,09 (м, 2H), 4,64-4,54 (м, 2H), 4,02 (шс, 1H), 3,83 (шс, 2H), 2,99 (шс, 2H), 2,92-2,82 (м, 3H), 2,65 (с, 3H), 2,58 (шс, 3H), 1,95 (ддд, J=3,8, 9,5, 13,8 Гц, 1H)
Пример 82 & 83 получают по методике Примеров 79 & 78, соответственно
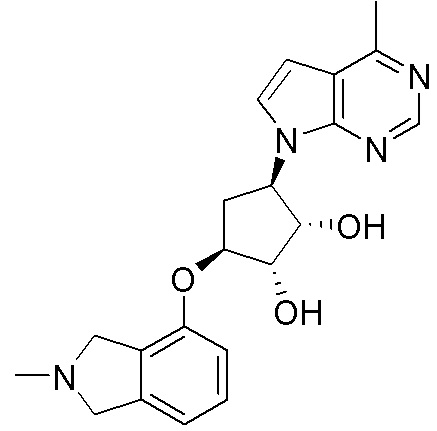
1H ЯМР (400 MГц, MeOD-d4) δ ч./млн. 8,62 (с, 1H), 7,56 (д, J=3,8 Гц, 1H), 7,21 (т, J=7,9 Гц, 1H), 6,89 (дд, J=7,8, 14,1 Гц, 2H), 6,77 (д, J=3,8 Гц, 1H), 5,29 (кв, J=8,7 Гц, 1H), 4,73 (ддд, J=1,8, 3,5, 7,0 Гц, 1H), 4,65 (дд, J=4,8, 8,8 Гц, 1H), 4,18 (д, J=4,5 Гц, 1H), 3,97 (д, J=12,5 Гц, 4H), 3,00 (ддд, J=7,0, 9,5, 14,6 Гц, 1H), 2,71 (с, 3H), 2,62 (с, 3H), 2,15-2,07 (м, 1H)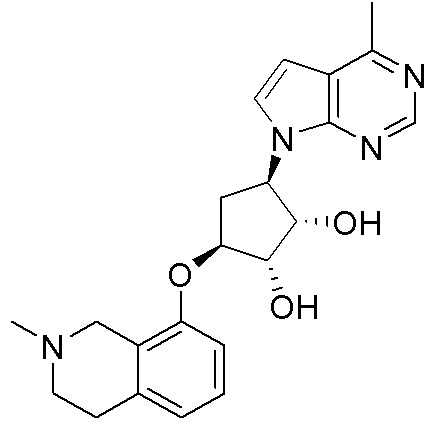
1H ЯМР (400 MГц, MeOD-d4) δ ч./млн. 8,62 (с, 1H), 7,57 (д, J=3,8 Гц, 1H), 7,22 (т, J=8,0 Гц, 1H), 6,93 (д, J=7,8 Гц, 1H), 6,84 (д, J=7,5 Гц, 1H), 6,76 (д, J=3,8 Гц, 1H), 5,23 (кв, J=8,9 Гц, 1H), 4,74-4,68 (м, 2H), 4,20 (д, J=5,5 Гц, 1H), 4,05 (с, 2H), 3,19-3,12 (м, 2H), 3,10-3,05 (м, 2H), 3,00 (ддд, J=7,0, 9,3, 14,3 Гц, 1H), 2,83 (с, 3H), 2,72 (с, 3H), 2,24-2,15 (м, 1H)
Синтез трет-бутил 6-фтор-8-гидрокси-3,4-дигидроизохинолин-2(1H)-карбоксилата (TP-1)
Схема EE
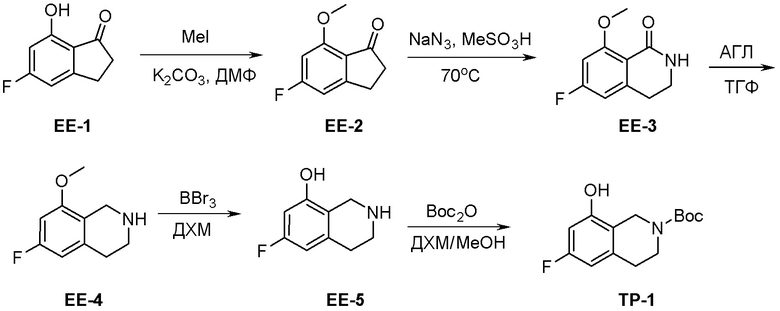
Стадия 1 - Синтез 5-фтор-7-метокси-2,3-дигидро-1H-инден-1-она (EE-2)
К раствору 5-фтор-7-гидрокси-2.3-дигидро-1H-инден-1-она (получен по методике, описанной в Bio. Med. Chem Letters, 20, 1004-1007, 2010) (1 г, 6,02 ммоль) и K2CO3 (2,5 г, 18 ммоль) в сухом ДМФ (10 мл) добавляют MeI (1710 мг, 12,0 ммоль) при 0°C. После добавления, реакционную смесь перемешивают при 25°C в течение 4 часов. Реакционную смесь разделяют между EtOAc и H2O. Водный слой экстрагируют EtOAc. Объединенные органические слои промывают насыщенным раствором соли, сушат над Na2SO4 и концентрируют с получением EE-2 (1 г, 92%) в виде желтого твердого вещества. ЖХМС 181 [M+1]; 1H ЯМР (400 MГц, CDCl3) δ ч./млн. 6,73-6,69 (м, 1H), 6,51 (дд, J=2,0, 11,3 Гц, 1H), 3,94 (с, 3H), 3,13-3,03 (м, 2H), 2,73-2,65 (м, 2H)
Стадия 2 - Синтез 6-фтор-8-метокси-3,4-дигидроизохинолин-1(2H)-она (EE-3)
К раствору EE-2 (950 мг, 5,27 ммоль) в 1,2-ДХЭ/MeSO3H (38 мл/29 мл) при 0°C порциями добавляют NaN3 (1,4 мг, 21,1 ммоль). После добавления, смесь перемешивают при 70°C в течение 16 ч. Смесь охлаждают до 0°C, доводят до pH 7-8 добавлением водного NaHCO3 (насыщ). Смесь экстрагируют ДХМ (15 мл×2), сушат над Na2SO4, фильтруют и концентрируют с получением EE-3 (1,15 г, 74%) в виде коричневого твердого вещества. ЖХМС 196 [M+1]; 1H ЯМР (400 MГц, ДМСО-d6) δ ч./млн. 7,70 (шс, 1H), 6,85 (дд, J=2,4, 11,9 Гц, 1H), 6,73 (дд, J=2,4, 8,9 Гц, 1H), 3,76 (с, 3H), 3,21 (дт, J=3,5, 6,3 Гц, 2H), 2,80 (т, J=6,3 Гц, 2H)
Стадия 3 - Синтез 6-фтор-8-метокси-1,2,3,4-тетрагидроизохинолина (EE-4)
Соединение EE-4 получают из EE-3 по методике получения стадии 5 со схемы FF с получением неочищенного продукта EE-4 (455 мг, >99%) в виде желтого масла и применяют сразу на следующей стадии. ЖХМС 182 [M+1]; 1H ЯМР (400 MГц, ДМСО-d6) δ ч./млн. 6,63 (дд, J=2,3, 11,3 Гц, 1H), 6,48 (дд, J=2,3, 9,5 Гц, 1H), 3,75 (с, 3H), 3,67-3,61 (м, 2H), 2,85 (т, J=5,8 Гц, 2H), 2,61 (т, J=5,6 Гц, 2H)
Стадия 4 - Синтез 6-фтор-1,2,3,4-тетрагидроизохинолин-8-ола (EE-5)
Соединение EE-5 получают из EE-4 по методике получения стадии 6 со схемы FF с получением неочищенного продукта EE-5 (228 мг, 54%) в виде желтого твердого вещества. ЖХМС 168 [M+1]; 1H ЯМР (400 MГц, ДМСО-d6) δ ч./млн. 6,39-6,28 (м, 2H), 3,63 (с, 2H), 2,85 (т, J=5,8 Гц, 2H), 2,63-2,57 (м, 2H)
Стадия 5 - Синтез трет-бутил 6-фтор-8-гидрокси-3,4-дигидроизохинолин-2(1H)-карбоксилата (TP-1)
Соединение TP-1 получают из EE-5 по методике получения стадии 7 со схемы FF с получением TP-1 (182 мг, 50%) в виде желтого твердого вещества. МС 212 [M-56+1]; 1H ЯМР (400 MГц, ДМСО-d6) δ ч./млн. 10,11 (с, 1H), 6,45 (д, J=10,5 Гц, 2H), 4,29 (с, 2H), 3,51 (м, J=5,8 Гц, 2H), 2,69 (м, J=5,6 Гц, 2H), 1,43 (с, 9H)
Синтез трет-бутил 5-фтор-8-гидрокси-3,4-дигидроизохинолин-2(1H)-карбоксилата (TP-2)
Схема FF
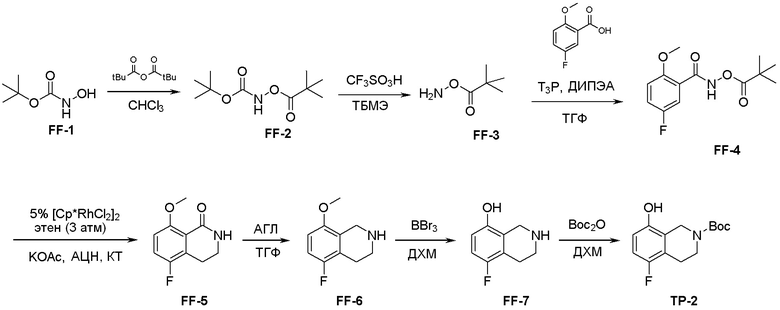
Стадия 1 - Синтез трет-бутил (пивалоилокси)карбамат (FF-2)
К раствору соединения FF-1 (20 г, 150 ммоль) в CHCl3 (200 мл) медленно добавляют ангидрид пивалиновой кислоты (34 г, 180 ммоль) на ледяной бане и затем перемешивают при 70°C в течение 16 ч. Реакционный раствор разбавляют ДХМ (200 мл) и промывают насыщенным NaHCO3 (200 мл×2) до pH ~7. Органический слой сушат над Na2SO4, фильтруют и концентрируют при пониженном давлении с получением светло-желтого масла. Неочищенный продукт кристаллизуют с петролейным эфиром (20 мл) с получением FF-2 (18 г, 55%) в виде белого твердого вещества. 1H ЯМР (400 MГц, CDCl3) δ ч./млн. 7,8 (шс, 1H), 1,5 (с, 9H), 1,3 (с, 9H)
Стадия 2 - Синтез O-пивалоилгидроксиламина (FF-3)
К раствору FF-2 (16 г, 70 ммоль) в ТБМЭ (32 мл) добавляют CF3SO3H (10,6 г, 70,7 ммоль) при 0°C. Реакционный раствор перемешивают при 15°C в течение 2 ч. Реакционную смесь выпаривают с получением неочищенного продукта FF-3 (20 г, >99%) в виде белого твердого вещества и применяют сразу в следующей реакции.
Стадия 3 - Синтез 5-фтор-2-метокси-N-(пивалоилокси)бензамида (FF-4)
К раствору 5-фтор-2-метоксибензойной кислоты (4,50 г, 26,4 ммоль) в ТГФ (90,0 мл) добавляют T3P (19 г, 29,1 ммоль) при 0°C. После добавления, реакционную смесь перемешивают в течение 30 мин при 25°C. В реакционную смесь добавляют ДИПЭА (13,8 мл, 8,82 ммоль), затем FF-3 (7,28 г, 29,1 ммоль). После добавления, реакционную смесь перемешивают при 25°C в течение 16 часов. Реакционную смесь фильтруют, и фильтрат разделяют между EtOAc и H2O. Органический слой разделяют, сушат над Na2SO4 и концентрируют с получением неочищенного продукта, который очищают флэш-хроматографией на колонке (EtOAc:петролейный эфир=1~100%, затем MeOH:ДХМ=1%~10%) с получением FF-4 (9 г, >99%) в виде белого твердого вещества и применяют сразу в следующей реакции.
Стадия 4 - Синтез 5-фтор-8-метокси-3,4-дигидроизохинолин-1(2H)-она (FF-5)
Суспензию FF-4 (8,00 г, 28,0 ммоль), KOAc (6,06 г, 61,7 ммоль) и [Cp*RhCl2]2 (867 мг, 1,40 ммоль) в MeCN (100 мл) в сосуде продувают N2 в течение 5 мин и затем охлаждают до -40°C сухим льдом/ацетоном. Этилен продувают в сосуд в течение 30 мин, затем герметично закрывают и перемешивают при 25°C в течение 16 ч. Темно-красную суспензию фильтруют, и фильтровальную лепешку промывают CH3CN. Объединенный фильтрат концентрируют с получением неочищенного продукта, который очищают флэш-хроматографией на колонке (EtOAc:петролейный эфир=1%~100%, затем MeOH:ДХМ=1~8%) с получением FF-5 (4,62 г, 84%) в виде желтого твердого вещества. 1H ЯМР (400 MГц, ДМСО-d6) δ ч./млн. 7,85 (шс, 1H), 7,32 (т, J=9,0 Гц, 1H), 6,99 (дд, J=4,3, 9,3 Гц, 1H), 3,76 (с, 3H), 3,31-3,22 (м, 2H), 2,79 (т, J=6,3 Гц, 2H)
Стадия 5 - Синтез 5-фтор-8-метокси-1,2,3,4-тетрагидроизохинолина (FF-6)
К раствору FF-5 (4,66 г, 23,9 ммоль) в сухом ТГФ (240 мл) добавляют LiAlH4 (3,62 г, 95,5 ммоль) порциями. После добавления, реакционную смесь нагревают при 65°C (кипение с обратным холодильником) в течение 2 часов. Реакционную смесь гасят 4 мл H2O. Смесь дистиллируют в EtOAc и фильтруют. Фильтровальную лепешку промывают EtOAc. Фильтрат сушат над Na2SO4 и концентрируют с получением неочищенного продукта, который очищают флэш-хроматографией на колонке (MeOH:ДХМ=1~8%) с получением FF-6 (2,9 г, 66%) в виде желтой камеди. 1H ЯМР (400 MГц, ДМСО-d6) δ ч./млн. 6,92 (т, J=9,2 Гц, 1H), 6,73 (дд, J=4,4, 8,9 Гц, 1H), 3,73 (с, 3H), 3,69 (с, 2H), 2,88 (т, J=5,8 Гц, 2H), 2,55 (т, J=5,8 Гц, 2H)
Стадия 6 - Синтез 5-фтор-1,2,3,4-тетрагидроизохинолин-8-ола (FF-7)
К раствору FF-6 (2,86 г, 15,8 ммоль) в ДХМ (28,6 мл) добавляют BBr3 (2,86 мл, 30,3 ммоль) при -10°C по каплям. После добавления, реакционную смесь перемешивают при 0°C в течение 2 часов. Реакционную смесь охлаждают до -10°C и гасят MeOH. Полученную смесь разбавляют в H2O, подщелачивают насыщ. K2CO3 до pH 9~10 и затем экстрагируют EtOAc (50 мл×3). Объединенные органические слои промывают насыщенным раствором соли, сушат над Na2SO4 и концентрируют с получением неочищенного продукта, который очищают флэш-хроматографией на колонке (MeOH:ДХМ=1:10) с получением FF-7 (1,0 г, 38%) в виде желтого твердого вещества. 1H ЯМР (400 MГц, ДМСО-d6) δ ч./млн. 9,24 (шс, 1H), 6,76 (т, J=9,0 Гц, 1H), 6,55 (дд, J=4,6, 8,7 Гц, 1H), 3,69 (с, 2H), 2,90 (т, J=5,9 Гц, 2H), 2,54 (т, J=6,4 Гц, 2H)
Стадия 7 - Синтез трет-бутил 5-фтор-8-гидрокси-3,4-дигидроизохинолин-2(1H)-карбоксилата (TP-2)
К раствору FF-7 (1,0 г, 6 ммоль) в ДХМ (25,00 мл) и MeOH (5 мл) добавляют Boc2O (1,4 г, 6,6 ммоль), затем Et3N (2,08 мл, 15,0 ммоль) при 0°C. После добавления, реакционную смесь перемешивают при 25°C в течение 16 часов. Реакционную смесь подкисляют водн. лимонной кислотой до pH 3~4, и полученную смесь разделяют между ДХМ и H2O. Органический слой разделяют, и водный слой экстрагируют ДХМ. Объединенные органические слои промывают насыщ. NaHCO3, насыщенным раствором соли, сушат над Na2SO4 и концентрируют с получением неочищенного продукта, который очищают флэш-хроматографией на колонке (MeOH:ДХМ=1:10) с получением TP-2 (744 мг, 47%) в виде белого твердого вещества. МС 212 [M-55+1]; 1H ЯМР (400 MГц, ДМСО-d6) δ ч./млн. 9,55 (с, 1H), 6,83 (т, J=9,0 Гц, 1H), 6,62 (дд, J=4,6, 8,9 Гц, 1H), 4,32 (с, 2H), 3,52 (т, J=5,9 Гц, 2H), 2,63 (т, J=5,6 Гц, 2H), 1,41 (с, 9H)
Методики со схемы FF применяют для синтеза промежуточных соединений гидрокситетрагидроизохинолина TP-3 - TP-7.
1H ЯМР (400 MГц, ДМСО-d6) δ ч./млн. 10,13 (с, 1H), 6,68 (с, 2H), 4,29 (с, 2H), 3,53-3,45 (т, J=5,6 Гц, 2H), 2,74-2,64 (т, J=5,5 Гц, 2H), 1,42 (с, 9H)
1H ЯМР (400 MГц, ДМСО-d6) δ ч./млн. 10,36 (шс, 1H), 7,00-6,96 (с, 1H), 6,95-6,91 (с, 1H), 4,40 (с, 2H), 3,55 (т, J=5,8 Гц, 2H), 2,79 (т, J=5,6 Гц, 2H), 1,44 (с, 9H)
1H ЯМР (400 MГц, CDCl3) δ ч./млн. 6,58-6,51 (м, 1H), 6,46 (с, 1H), 4,51 (шс, 2H), 3,64-3,60 (м, 2H), 2,80-2,76 (м, 2H), 2,25 (с, 3H), 1,51 (с, 9H)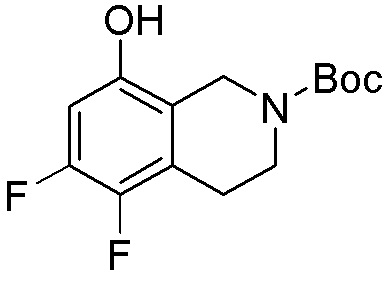
1H ЯМР (400 MГц, ДМСО-d6) δ ч./млн. 10,04 (с, 1H), 6,63 (дд, J=6,8, 12,3 Гц, 1H), 4,30 (с, 2H), 3,60-3,50 (т, J=5,8 Гц, 2H), 2,75-2,65 (т, J=5,9 Гц, 2H), 1,42 (с, 9H)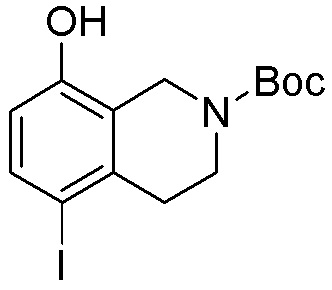
1H ЯМР (400 MГц, ДМСО-d6) δ ч./млн. 9,93 (с, 1H), 7,50 (д, J=8,3 Гц, 1H), 6,54 (д, J=8,5 Гц, 1H), 4,32 (с, 2H), 3,59-3,49 (м, 2H), 2,57 (т, J=5,8 Гц, 2H), 1,42 (с, 9H)
Синтез трет-бутил 8-гидрокси-6-(2-гидроксипропан-2-ил)-3,4-дигидроизохинолин-2(1H)-карбоксилата (TP-9)
Схема GG
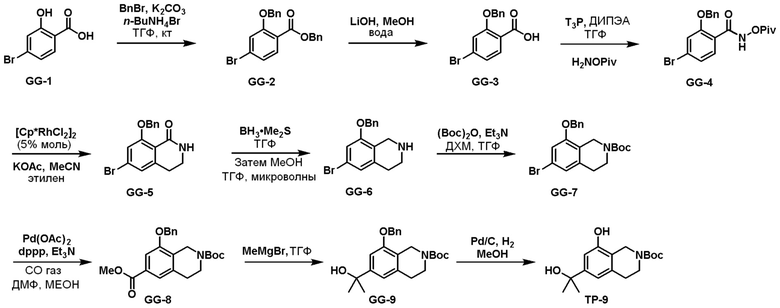
Стадия 1: Синтез бензил 2-(бензилокси)-4-бромбензоата (GG-2)
К раствору салициловой кислоты GG-1(50 г, 230 ммоль) в ТГФ (1000 мл) добавляют карбонат калия (95,5 г, 691 ммоль), бромид тетрабутиламмония (14,9 г, 46,1 ммоль) и бензилбромид (95,8 г, 576 ммоль). Реакционную смесь перемешивают при 20°C в течение 16 часов. Раствор фильтруют и концентрируют в вакууме с получением желаемого продукта GG-2 в виде неочищенного коричневого твердого вещества (150 г). Этот продукт применяют на следующей стадии без дальнейшей очистки. ТСХ (ПЭ/ЭА=10:1, Rf~0,5)
Стадия 2: Синтез 2-(бензилокси)-4-бромбензoйной кислоты (GG-3)
К раствору GG-2 (140 г, 352 ммоль, неочищенный продукт, ~53% чистота) в MeOH/воде/ДХМ (500 мл/500 мл/500 мл) добавляют гидроксид лития (44,4 г, 1060 ммоль) при 10°C. Реакционную смесь перемешивают при 70°C в течение 16 часов. Раствор концентрируют в вакууме, и из раствора выпадает в осадок желтое твердое вещество. Твердые вещества фильтруют, и фильтровальную лепешку промывают водой (50 мл×2). Желтое твердое вещество подкисляют 1N HCl до pH=2. Желтое твердое вещество фильтруют и сушат в инфракрасной печи с получением соединения GG-3 (43,3 г, 40%) в виде желтого твердого вещества. 1ЯМР (400 MГц, ДМСО-d6) δ ч./млн. 12,76 (шс, 1H), 7,61 (д, J=8,3 Гц, 1H), 7,52-7,47 (м, 2H), 7,44 (д, J=1,8 Гц, 1H), 7,43-7,38 (м, 2H), 7,37-7,31 (м, 1H), 7,23 (дд, J=1,8, 8,3 Гц, 1H), 5,25 (с, 2H).
Стадия 3: Синтез 2-(бензилокси)-4-бром-N-(пивалоилокси)-бензамида (GG-4)
К раствору GG-3 (31,2 г, 101,58 ммоль) в ТГФ (630 мл) добавляют T3P (71,1 г, 112 ммоль) при 0°C. После добавления, реакционную смесь перемешивают в течение 30 минут при 25°C. К полученному выше раствору добавляют ДИПЭА (78,8 г, 609 ммоль), затем O-пивалоилгидроксиламин (28 г, 112 ммоль). После добавления, реакционную смесь перемешивают при 25°C в течение 2 часов. Раствор переносят в делительную воронку с EtOAc (500 мл) и промывают 1 порцией воды (300 мл), 1 порцией водн. лимонной кислоты (300 мл), 1 порцией насыщ. NaHCO3 и 1 порцией насыщенного раствора соли (300 мл). Органический слой сушат над Na2SO4, фильтруют и концентрируют в вакууме. Неочищенный остаток очищают флэш-хроматографией на колонке (силикагель, петролейный эфир:EtOAc=7:1) с получением соединения GG-4 (30,4 г, 74%) в виде желтого твердого вещества. 1H ЯМР (400 MГц, ДМСО-d6) δ ч./млн. 10,74 (с, 1H), 8,06 (д, J=8,5 Гц, 1H), 7,52-7,35 (м, 5H), 7,28 (д, J=1,5 Гц, 1H), 7,26 (д, J=1,8 Гц, 1H), 7,23 (д, J=1,5 Гц, 1H), 5,27 (с, 2H), 1,34 (с, 9H)
Стадия 4: Синтез пивалата 8-(бензилокси)-6-бром-1-оксо-3,4-дигидроизохинолин-2(1H)-ила (GG-5)
Суспензию GG-4 (40,7 г, 100,18 ммоль), KOAc (10,8 г, 110 ммоль) и Cp2RhCl2 (3,1 г, 5,01 ммоль) в MeCN (1300 мл) охлаждают до 0°C и раствор продувают газообразным этиленом в течение 45 минут. Сосуд герметично закрывают и перемешивают при 10°C в течение 16 часов. Реакционную суспензию фильтруют и лепешку промывают 2 порциями 5:1 смесью ДХМ/MeOH (100 мл). Фильтрат концентрируют при пониженном давлении с получением неочищенного продукта (50 г) в виде желтого твердого вещества. Неочищенный остаток очищают флэш-хроматографией на колонке (силикагель, ДХМ:MeOH=40:1) с получением соединение GG-5 (28 г, 84%) в виде желтого твердого вещества. ЖХМС [M+H+Na] 356 наблюдаемый; 1H ЯМР (400 MГц, ДМСО-d6) δ ч./млн. 7,79 (т, J=3,1 Гц, 1H) 7,53-7,59 (м, 2H) 7,35-7,42 (м, 2H) 7,30-7,34 (м, 1H) 7,26 (д, J=2,0 Гц, 1H) 7,15 (д, J=2,0 Гц, 1H) 5,20 (с, 2H) 3,25 (тд, J=6,1, 3,6 Гц, 2H) 2,83 (т, J=6,3 Гц, 2H).
Стадия 5: Синтез 8-(бензилокси)-6-бром-3,4-дигидроизохинолин-1(2H)-она (GG-6)
К смеси GG-5 (36,5 г, 110 ммоль) в безводном ТГФ (700 мл) добавляют BH3·Me2S (10M, 33 мл, 330 ммоль) при 0°C по каплям под N2. После добавления, реакционную смесь перемешивают при 5°C в течение 12 часов. На этой стадии реакционную смесь нагревают при 78°C (кипение с обратным холодильником) в течение 3 часов до завершения. Реакционную смесь постепенно охлаждают до кт и осторожно гасят 70 мл MeOH при 0°C. Раствор концентрируют, затем помещают в ТГФ (2 мл) и MeOH (10 мл) и переносят в микроволновую пробирку. Раствор нагревают при 120°C в микроволновом реакторе в течение 1 часа. Раствор концентрируют с получением черного твердого вещества. Неочищенный остаток очищают флэш-хроматографией на колонке (120 г силикагель × 3, MeOH/ДХМ=10%~20%) с получением соединения GG-6 (8,28 г, 24%) в виде коричневого твердого вещества. ЖХМС [M+H] 319 наблюдаемый; 1H ЯМР (400 MГц, ДМСО-d6) δ ч./млн. 8,99 (шс, 1H) 7,37-7,50 (м, 5H) 7,23 (д, J=1,3 Гц, 1H) 7,11 (д, J=1,3 Гц, 1H) 5,21 (с, 2H) 4,11 (с, 2H) 2,97 (т, J=5,9 Гц, 2H).
Стадия 6: Синтез трет-бутил 8-(бензилокси)-6-бром-3,4-дигидроизохинолин-2(1H)-карбоксилата (GG-7)
К смеси GG-6 (10,99 г, 34,537 ммоль) в ДХМ (110 мл) и ТГФ (22 мл) добавляют Et3N (10,5 г, 104 ммоль) и (Boc)2O (11,3 г, 51,8 ммоль) при 10°C. Реакционную смесь перемешивают при 10°C в течение 16 часов. Смесь концентрируют в вакууме с получением неочищенного продукта в виде черной камеди. Неочищенный остаток очищают флэш-хроматографией на колонке (120 г силикагеля, EtOAc/петролейный эфир 4%-7%) с получением соединения GG-7 (9,9 г, 68,5%) в виде желтой камеди. ЖХМС [M+H-Boc] 319 наблюдаемый; 1H ЯМР (400 MГц, ХЛОРОФОРМ-d) δ ч./млн. 7,48-7,30 (м, 5H), 6,98-6,88 (м, 2H), 5,15-4,96 (м, 2H), 4,60-4,44 (м, 2H), 3,71-3,54 (м, 2H), 2,88-2,71 (м, 2H), 1,50 (с, 9H).
Стадия 7: Синтез 2-(трет-бутил) 6-метил 8-(бензилокси)-3,4-дигидроизохинолин-2,6(1H)-дикарбоксилата (GG-8)
Смесь GG-7 (600 мг, 1,43 ммоль), DPPP (296 мг, 0,717 ммоль), ТЭА (435 мг, 4,30 ммоль) и Pd(OAc)2 (161 мг, 0,717 ммоль) в MeOH (20 мл) и ДМФ (20 мл) нагревают при 120°C под 22 бар окиси углерода в течение 24 часов. Смесь концентрируют в вакууме и переносят в делительную воронку с EtOAc. Раствор промывают 5 порциями насыщенного раствора соли, сушат над сульфатом натрия, фильтруют и концентрируют в вакууме. Неочищенный остаток очищают флэш-хроматографией на колонке (12 г, силикагель, 20% EtOAc/петролейный эфир) с получением соединения GG-8 (440 мг, 77% выход) в виде белого твердого вещества. 1H ЯМР (400 MГц, ХЛОРОФОРМ-d) δ ч./млн. 7,54-7,28 (м, 7H), 5,22-5,08 (м, 2H), 4,67-4,54 (м, 2H), 3,91 (с, 3H), 3,70-3,57 (м, 2H), 2,93-2,77 (м, 2H), 1,50 (с, 9H).
Стадия 8: Синтез трет-бутил 8-(бензилокси)-6-(2-гидроксипропан-2-ил)-3,4-дигидроизохинолин-2(1H)-карбоксилата (GG-9)
К бесцветному раствору GG-8 (327 мг, 0,823 ммоль) в сухом ТГФ (15 мл) добавляют MeMgBr (3,0 M раствор в диэтиловом эфире, 1,65 мл, 4,94 ммоль) по каплям при 0°C. Смесь перемешивают при 17°C в течение 1,5 часов. Раствор охлаждают до 0°C и гасят водой (10 мл). Раствор переносят в делительную воронку с EtOAc и фазы разделяют. Водную фазу экстрагируют 2 порциями EtOAc (20 мл). Объединенные органические экстракты промывают 1 порцией насыщенного раствора соли (30 мл), сушат над сульфатом натрия, фильтруют и концентрируют в вакууме. Неочищенный остаток очищают флэш-хроматографией на колонке (силикагель, 12 г, петролейный эфир:ЭА=3:1) с получением соединения GG-9 (320 мг, 98%) в виде бесцветной камеди. 1H ЯМР (400 MГц, ХЛОРОФОРМ-d) δ ч./млн. 7,29-7,49 (м, 5H) 6,97 (с, 1H) 6,84 (с, 1H) 5,12 (шс, 2H) 4,58 (с, 2H) 3,65 (шс, 2H) 2,82 (шс, 2H) 1,57 (с, 6H) 1,49 (с, 9H).
Стадия 9: Синтез трет-бутил 8-гидрокси-6-(2-гидроксипропан-2-ил)-3,4-дигидроизохинолин-2(1H)-карбоксилата (TP-9)
Смесь GG-9 (290 мг, 0,73 ммоль) и Pd/C (105 mg) в MeOH (10 мл) перемешивают при 15°C в течение 5 часов под баллоном водорода. Смесь фильтруют через слой Целита, и фильтрат концентрируют в вакууме. Неочищенный остаток очищают флэш-хроматографией на колонке (4 г, силикагель, 60% EtOAc/петролейный эфир) с получением соединения TP-9 (144 мг, 64%) в виде белого твердого вещества. 1H ЯМР (400 MГц, ХЛОРОФОРМ-d) δ ч./млн. 6,91 (шс, 1H), 6,76 (с, 1H), 4,53 (шс, 2H), 3,63 (шс, 2H), 2,80 (шс, 2H), 1,55 (с, 6H), 1,50 (с, 9H).
Последовательность стадий 1-6 & 9 со схемы GG применяют для синтеза промежуточных соединений гидрокситетрагидроизохинолина TP-8 (Схема III), TP-10-12 из соответствующей салициловой кислоты.
(см. схему III)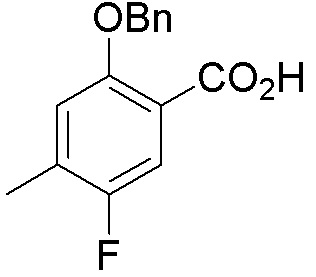
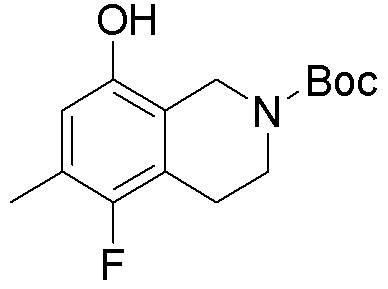
1H ЯМР (400 MГц, MeOD) δ ч./млн. 6,45 (д, J=6,3 Гц, 1H), 4,40 (с, 2H), 3,66-3,50 (м, 2H), 2,70 (т, J=5,9 Гц, 2H), 2,14 (д, J=1,5 Гц, 3H), 1,50-1,47 (м, 9H)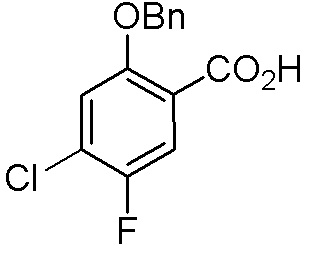
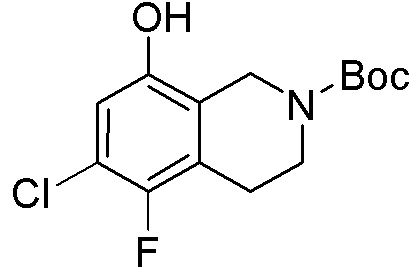
1H ЯМР (400 MГц, ДМСО-d6) δ ч./млн. 10,06 (с, 1H), 6,76 (д, J=6,3 Гц, 1H), 4,31 (с, 2H), 3,54 (т, J=5,8 Гц, 2H), 2,69 (т, J=5,8 Гц, 2H), 1,42 (с, 9H)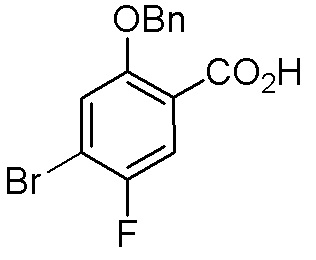
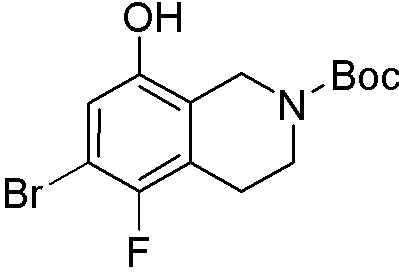
1H ЯМР (400 MГц, ДМСО-d6) δ ч./млн. 10,04 (с, 1H), 6,88 (д, J=5,8 Гц, 1H), 4,30 (с, 2H), 3,54 (т, J=5,8 Гц, 2H), 2,69 (т, J=5,9 Гц, 2H), 1,42 (с, 9H)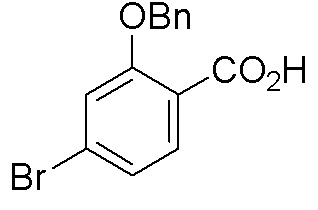
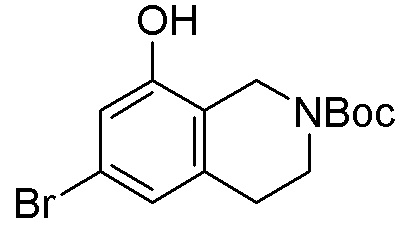
1H ЯМР (400 MГц, CDCl3) δ ч./млн. 6,94-6,66 (м, 2H), 4,46 (м, 2H), 3,61 (т, J=5,6 Гц, 2H), 2,76 (м, 2H), 1,51 (с, 9H)
Синтез трет-бутил 6-этил-8-гидрокси-3,4-дигидроизохинолин-2(1H)-карбоксилата (TP-13)
Схема HH
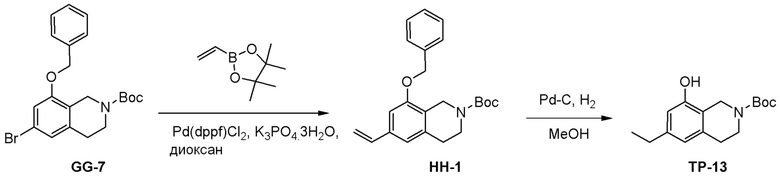
Стадия 1 - Синтез трет-бутил 8-(бензилокси)-6-винил-3,4-дигидроизохинолин-2(1H)-карбоксилата (HH-1)
Смесь GG-7 (530 мг, 1,27 ммоль) и 4,4,5,5-тетраметил-2-винил-1,3,2-диоксаборолана (410 мг, 2,67 ммоль), K3PO4,3H2O (675 мг, 2,53 ммоль), PdCl2(dppf) (93 мг, 0,013 ммоль) в диоксане (20 мл) и H2O (5 мл) дегазируют с N2, нагревают при 100°C в течение 2 часов. Смесь очищают пре-ТСХ (петролейный эфир/EtOAc=8/1) с получением HH-1 (370 мг, 80%) в виде бесцветной камеди. ЖХМС 266 [M-Boc+1]; 1H ЯМР (400 MГц, CDCl3) δ ч./млн. 7,51-7,28 (м, 5H), 6,83 (д, J=12,0 Гц, 2H), 6,65 (дд, J=10,9, 17,4 Гц, 1H), 5,69 (д, J=17,6 Гц, 1H), 5,22 (д, J=10,8 Гц, 1H), 5,12 (шс, 2H), 4,58 (шс, 2H), 3,73-3,56 (м, 2H), 2,90-2,73 (м, 2H), 1,50 (с, 9H)
Стадия 2 - Синтез трет-бутил 6-этил-8-гидрокси-3,4-дигидроизохинолин-2(1H)-карбоксилата (TP-13)
Смесь HH-1 (370 мг, 1,01 ммоль) и Pd/C (370 мг) в MeOH (20 мл) дегазируют с H2 и перемешивают при 50°C под H2 (50 ф/кв.д) в течение 24 ч. Смесь фильтруют, и фильтрат концентрируют в вакууме с получением неочищенного продукта, который очищают хроматографией на силикагеле, элюируя EtOAc в петролейном эфире 0-20% с получением TP-12 (210 мг, 75%) в виде белого твердого вещества. ЖХМС 222 [M-55+1]; 1H ЯМР (400 MГц, CDCl3) δ ч./млн. 6,57 (шс, 1H), 6,48 (с, 1H), 4,51 (шс, 2H), 3,63 (шс, 2H), 2,78 (шс, 2H), 2,62-2,45 (кв, J=7,5 Гц, 2H), 1,50 (с, 9H), 1,20 (т, J=7,5 Гц, 3H)
Синтез трет-бутил 6-(дифторметил)-8-гидрокси-3,4-дигидроизохинолин-2(1H)-карбоксилата (TP-14)
Схема II
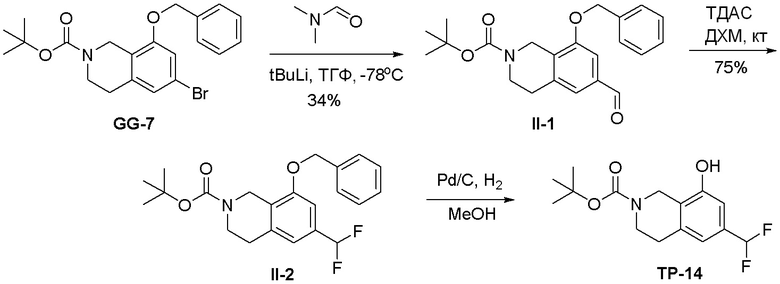
Стадия 1: Синтез трет-бутил 8-(бензилокси)-6-формил-3,4-дигидроизохинолин-2(1H)-карбоксилата (II-1)
GG-7 (1000 мг, 2,390 ммоль) в высушенной в печи двухгорлой колбе, оборудованной термометром, растворяют в тетрагидрофуране (22,0 мл, c=0,109 M), охлаждают до -78°C, медленное добавляют трет-бутиллитий (459 мг, 7,17 ммоль, 4,22 мл, 1,7 M) под N2, сохраняя внутреннюю температуру около -70°C, перемешивают при -78°C в течение 30 мин, N,N-диметилформамид (262 мг, 3,59 ммоль) добавляют по каплям и перемешивают при -78°C в течение 1,5 ч. Реакцию гасят станд. NH4Cl при -78°C, экстрагируют EtOAc 3 раза. Органические слои объединяют, промывают насыщенным раствором соли, сушат над Na2SO4, концентрируют и очищают хроматографией на колонке с 15% EtOAc/гептаном с получением II-1 (300 мг, 34% выход) в виде бесцветного масла.
ЖХМС [M+1-Boc] 268,10. 1H ЯМР (400 MГц, ХЛОРОФОРМ-d) δ ч./млн. 1,51 (с, 9 H) 2,91 (т, J=5,38 Гц, 2 H) 3,68 (т, J=5,69 Гц, 2 H) 4,65 (с, 2 H) 5,17 (шс, 2 H) 7,29 (д, J=9,05 Гц, 2 H) 7,31-7,37 (м, 1 H) 7,40 (т, J=7,27 Гц, 2 H) 7,43-7,48 (м, 2 H) 9,92 (с, 1 H)
Стадия 2: Синтез трет-бутил 8-(бензилокси)-6-(дифторметил)-3,4-дигидроизохинолин-2(1H)-карбоксилата (II-2)
К раствору II-1 (300 мг, 0,816 ммоль) в ДХМ (16,3 мл, c=0,05 M) при 0°C добавляют трифторид (диэтиламино)серы (1320 мг, 8,16 ммоль, 1070 мкл). Реакционную смесь вынимают с ледяной бани и перемешивают при кт в течение ночи. Реакционную смесь разбавляют ДХМ и гасят станд. NaHCO3, перемешивают при кт до превращения выделения CO2. Слои разделяют, и водный слой экстрагируют ДХМ. Органический слой сушат над Na2SO4 и концентрируют, очищают хроматографией на колонке с 15% EtOAc/гептаном с получением II-2 (240 мг, 75% выход) в виде прозрачного масла.
ЖХМС [M+1-Boc] 290,10. 1H ЯМР (400 MГц, ХЛОРОФОРМ-d) δ ч./млн. 1,51 (с, 9 H) 2,86 (т, J=5,26 Гц, 2 H) 3,66 (т, J=5,62 Гц, 2 H) 4,61 (с, 2 H) 5,13 (шс, 2 H) 6,58 (т, J=57,22 Гц, 1 H) 6,92 (д, J=8,31 Гц, 2 H) 7,31-7,48 (м, 5 H)
Стадия 3: Синтез трет-бутил 6-(дифторметил)-8-гидрокси-3,4-дигидроизохинолин-2(1H)-карбоксилата (TP-14)
К раствору II-2 (240 мг, 0616 ммоль) в метаноле (10 мл) добавляют палладий на угле (66 мг, 0,616 ммоль). Реакционный раствор дегазируют и повторно заполняют газообразным водородом. Смесь оборудуют водородным баллоном и перемешивают при кт в течение ночи. Реакционную смесь фильтруют, растворители удаляют в вакууме и продукт очищают хроматографией на колонке с 20% EtOAc/гептаном с получением TP-14 (175 мг, 95% выход) в виде белого твердого вещества.
ЖХМС [M+1-tBu] 242. 1H ЯМР (400 MГц, ХЛОРОФОРМ-d) δ ч./млн. 1,52 (с, 9 H) 2,84 (шс, 2 H) 3,66 (т, J=5,81 Гц, 2 H) 4,56 (шс, 2 H) 6,53 (т, J=57,22 Гц, 1 H) 6,76 (с, 1 H) 6,84 (шс, 1 H)
Синтез трет-бутил 6-(1,1-дифторэтил)-8-гидрокси-3,4-дигидроизохинолин-2(1H)-карбоксилата (TP-15)
Схема JJ
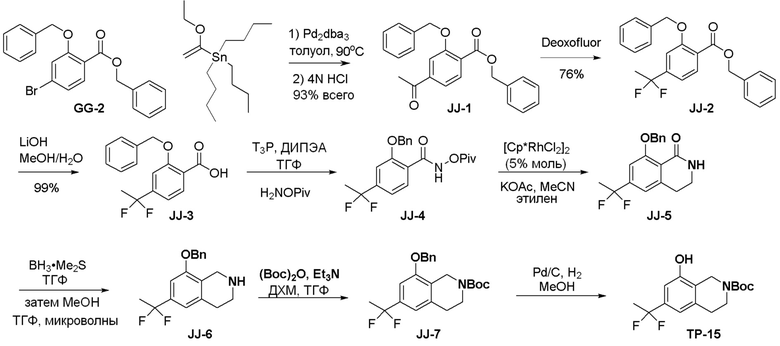
Стадия 1: Синтез бензил 4-ацетил-2-(бензилокси)бензоата (JJ-1)
Смесь GG-2 (8544 мг, 21,51 ммоль), трибутил(1-этоксивинил)олова (8160 мг, 22,6 ммоль), Pd2(dba)3 (394 мг, 0,430 ммоль) и 2,2'-бис(дифенилфосфино)-1,1'-бинафтила (536 мг, 0,860 ммоль) в толуоле (108 мл, c=0,2 M) дегазируют и нагревают при 90°C в течение 18 ч. Толуол удаляют, и неочищенный продукт переносят на следующую стадию.
Неочищенный продукт растворяют в ТГФ (108 мл, c=0,20 M), добавляют хлористоводородную кислоту (6670 мг, 183 ммоль, 45,7 мл, 4,0 M), перемешивают при кт в течение 2 ч. Органический растворитель удаляют; H2O добавляют, экстрагируют EtOAc 3 раза. Органические слои объединяют, промывают насыщенным раствором соли, сушат над Na2SO4, фильтруют и концентрируют, очищают хроматографией на силикагеле, элюируя 15% EtOAc/гептаном с получением 7180 мг желтого масла.
ЖХМС [M+1] 361,10. 1H ЯМР (400 MГц, ХЛОРОФОРМ-d) δ ч./млн. 2,60 (с, 3 H) 5,23 (с, 2 H) 5,37 (с, 2 H) 7,29-7,42 (м, 8 H) 7,45 (д, J=6,85 Гц, 2 H) 7,54 (дд, J=8,01, 1,28 Гц, 1 H) 7,62-7,65 (м, 1 H) 7,90 (д, J=7,95 Гц, 1 H)
Стадия 2: Синтез бензил 2-(бензилокси)-4-(1,1-дифторэтил)бензоата (JJ-2)
К JJ-1 (7,380 г, 20,48 ммоль) добавляют deoxofluor (22,7 г, 102 ммоль). Реакционную смесь нагревают до 80°C в течение 4,5 ч, охлаждают до к.т., выливают в станд. NaHCO3 и после завершения выделения CO2 водный слой экстрагируют EtOAc 3 раза. Органический слой концентрируют в вакууме, очищают хроматографией на колонке с 10% EtOAc/гептаном с получением JJ-2 (5,94 г, 76% выход) в виде бесцветного масла, которое затвердевает в вакууме.
1H ЯМР (400 MГц, ХЛОРОФОРМ-d) δ ч./млн. 1,89 (т, J=18,16 Гц, 3 H) 5,19 (с, 2 H) 5,36 (с, 2 H) 7,12 (д, J=8,07 Гц, 1 H) 7,17 (с, 1 H) 7,29-7,42 (м, 8 H) 7,45 (д, J=6,72 Гц, 2 H) 7,89 (д, J=8,07 Гц, 1 H)
Стадия 3: Синтез 2-(бензилокси)-4-(1,1-дифторэтил)бензойной кислоты (JJ-3)
По методике стадии 2 со схемы GG, JJ-2 гидролизуют JJ-3 (4,49 г, 99%) в виде белого твердого вещества.
1H ЯМР (400 MГц, ДМСО-d6) δ ч./млн. 1,97 (т, J=18,89 Гц, 3 H) 5,26 (с, 2 H) 7,19 (д, J=7,95 Гц, 1 H) 7,29-7,36 (м, 2 H) 7,40 (т, J=7,34 Гц, 2 H) 7,51 (д, J=7,21 Гц, 2 H) 7,71 (д, J=7,95 Гц, 1 H) 12,89 (шс, 1 H)
Стадия 4-8: Синтез трет-бутил 6-(1,1-дифторэтил)-8-гидрокси-3,4-дигидроизохинолин-2(1H)-карбоксилата (TP-15)
По методикам стадии 3 со схемы GG (1,30 г, 70%), стадии 4 со схемы GG (684 мг, 65%), стадии 5 со схемы FF и стадии 6 со схемы GG (575 мг, 77%), стадии 9 со схемы GG (400 мг, 90%), JJ-3 превращают в TP-15.
ЖХМС [M+1-Boc] 214,10. 1H ЯМР (400 MГц, ХЛОРОФОРМ-d) δ ч./млн. 1,52 (с, 9 H) 1,87 (т, J=18,10 Гц, 3 H) 2,83 (шс, 2 H) 3,65 (т, J=5,69 Гц, 2 H) 4,56 (шс, 2 H) 6,76 (с, 1 H) 6,83 (шс, 1 H)
Синтез трет-бутил 8-гидрокси-3,4-дигидро-2,6-нафтиридин-2(1H)-карбоксилата (TP-16)
Схема KK
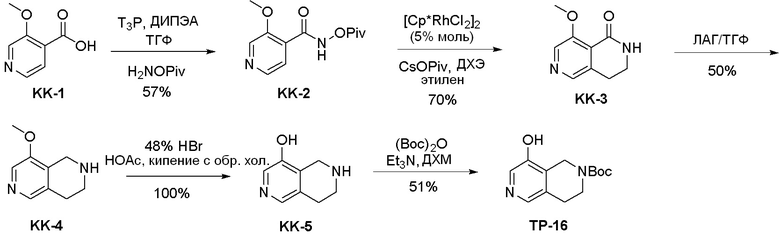
Стадия 1-3: Синтез 8-метокси-1,2,3,4-тетрагидро-2,6-нафтиридина (KK-4)
По методикам стадии 3 со схемы GG (939 мг, 57%), стадии 4 со схемы GG с применением пиволата цезия и дихлорэтана вместо ацетата калия и ацетонитрила (290 мг, 70%), и стадии 5 со схемы FF (136 мг, 50%), KK-1 превращают в KK-4.
ЖХМС [M+1] 165,10. 1H ЯМР (400 MГц, ХЛОРОФОРМ-d) δ ч./млн. 2,77 (т, J=5,81 Гц, 2 H) 3,11 (т, J=5,81 Гц, 2 H) 3,90 (с, 3 H) 3,94 (с, 2 H) 8,03 (д, J=2,93 Гц, 2 H)
Стадия 4: Синтез 5,6,7,8-тетрагидро-2,6-нафтиридин-4-ола (KK-5)
KK-4 (136,0 мг, 0,828 ммоль) в 5 мл 48% HBr и 3 мл ледяной уксусной кислоты герметично закрывают и кипятят с обратным холодильникомe при 120°C в течение 4 дней. Реакционную смесь охлаждают до кт, концентрируют уксусную кислоту азеотропной перегонкой из гептана 3 раза, нейтрализуют осторожным добавлением 5 N NaOH до pH около 9. Выпаривают на роторном испарителе для удаления большей части H2O, добавляют MeOH, сушат с силикагелем. Продукт очищают хроматографией на колонке с 10% MeOH/ДХМ с 0,5%NH4OH с получением KK-5 (124 мг, 100%). ЖХМС [M+1] 151,10.
Стадия 5: Синтез трет-бутил 8-гидрокси-3,4-дигидро-2,6-нафтиридин-2(1H)-карбоксилата (TP-16)
По методикам стадии 6 со схемы GG, KK-5 превращают в TP-16 (106 мг, 51%).
ЖХМС [M-55+1] 228. 1H ЯМР (400 MГц, ДМСО-d6) δ ч./млн. 10,13 (с, 1H), 6,68 (с, 2H), 4,29 (с, 2H), 3,53-3,45 (т, J=5,6 Гц, 2H), 2,74-2,64 (т, J=5,5 Гц, 2H), 1,42 (с, 9H)
Синтез трет-бутил 8-гидрокси-5-метил-3,4-дигидро-2,6-нафтиридин-2(1H)-карбоксилата (TP-17)
Схема LL
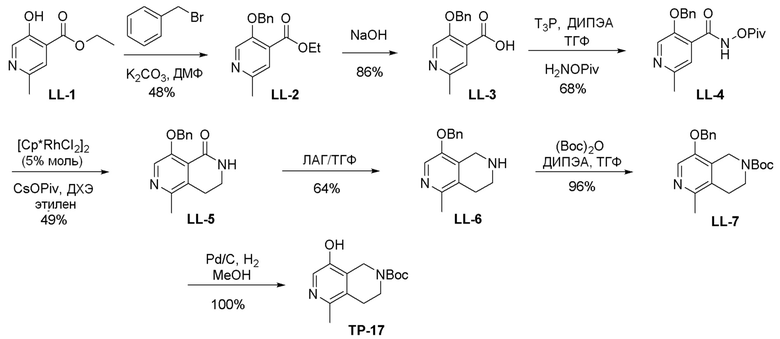
Стадия 1: Синтез (5-(бензилокси)-2-метилпиридин-4-ил)(этокси)метанола (LL-2)
Этил 5-гидрокси-2-метилизоникотинат (LL-1) (синтез описан в WO10100475) (2190 мг, 17,22 ммоль) и N-бензилбромид (3540 мг, 20,7 ммоль), K2CO3 (4810 мг, 34,4 ммоль) в 20 мл ДМФ нагревают при 80°C в течение ночи. Затем реакционную смесь охлаждают до к.т., добавляют EtOAc, промывают H2O 3 раза. Органический слой концентрируют, очищают хроматографией на колонке с 30% EtOAc/гептаном с получением LL-2 (16 г, 48% выход) в виде коричневого твердого вещества.
ЖХМС [M+1] 272,10. 1H ЯМР (400 MГц, ХЛОРОФОРМ-d) δ ч./млн. 1,37 (т, J=7,15 Гц, 3 H) 2,54 (с, 3 H) 4,39 (кв, J=7,13 Гц, 2 H) 5,23 (с, 2 H) 7,34 (д, J=7,09 Гц, 1 H) 7,36-7,43 (м, 2 H) 7,44-7,51 (м, 3 H) 8,35 (с, 1 H)
Стадия 2: Синтез (5-(бензилокси)-2-метилпиридин-4-ил)метандиола (LL-3)
К раствору LL-2 (2160 мг, 7,961 ммоль) в 20 мл MeOH добавляют гидроксид натрия (1590 мг, 39,8 ммоль, 7,96 мл, 5,0 M), и реакционную смесь нагревают при 50°C в течение 4 ч. Смесь охлаждают до к.т., MeOH выпаривают, H2O добавляют, нейтрализуют 1 N HCl до pH около 4, желтое твердое вещество измельчают, фильтруют и промывают H2O с получением LL-3 (1,66 г, 86% выход).
ЖХМС [M+1] 244,10. 1H ЯМР (400 MГц, ДМСО-d6) δ ч./млн. 2,42 (с, 3 H) 5,27 (с, 2 H) 7,33 (д, J=7,21 Гц, 1 H) 7,35-7,43 (м, 3 H) 7,44-7,52 (м, 2 H) 8,41 (с, 1 H)
Стадия 3-7:-Синтез трет-бутил 8-гидрокси-5-метил-3,4-дигидро-2,6-нафтиридин-2(1H)-карбоксилата (TP-17)
По методикам стадии 3 со схемы GG (1,60 г, 68%), стадии 4 со схемы GG с применением пиволата цезия и дихлорэтана вместо ацетата калия и ацетонитрила (620 мг, 49%), стадии 5 со схемы FF (366 мг, 64%), стадии 6 со схемы GG (483 мг, 96%) и стадии 9 со схемы GG (386 мг, 100%), LL-3 превращают в TP-17.
1H ЯМР (400 MГц, ХЛОРОФОРМ-d) δ ч./млн. 1,51 (с, 9 H) 2,41 (с, 3 H) 2,71 (т, J=5,38 Гц, 2 H) 3,69 (т, J=5,75 Гц, 2 H) 4,59 (с, 2 H) 7,95 (с, 1 H)
Синтез трет-бутил 6-циано-8-гидрокси-3,4-дигидроизохинолин-2(1H)-карбоксилата (TP-18)
Схема MM
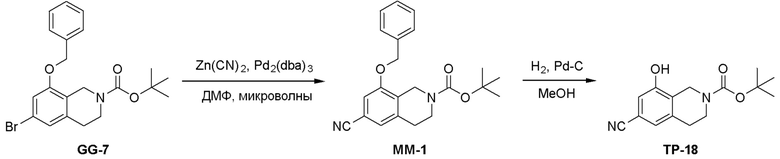
Стадия 1: Синтез трет-бутил 8-(бензилокси)-6-циано-3,4-дигидроизохинолин-2(1H)-карбоксилата (MM-1)
Соединение GG-7 (700 мг, 1,67 ммоль) растворяют в ДМФ (5,00 мл). Zn(CN)2 (236 мг, 2,01 ммоль) добавляют к раствору. Реакционный раствор дегазируют в течение 2 мин. Pd(PPh3)4 (580 мг, 0,502 ммоль) добавляют к смеси. Реакционную смесь дегазируют с N2 в течение 3 мин. Затем реакционную смесь нагревают микроволнами при 150°C в течение 30 мин. Реакционный раствор превращается из желтого в черный. Реакционный раствор охлаждают и разбавляют EtOAc/H2O (8 мл/8мл), затем смесь фильтруют и фильтрат экстрагируют. Органический слой разделяют, сушат и выпаривают с получением неочищенного продукта, который очищают флэш-хроматографией с петролейным эфиром/EtOAc 0-25% с получением MM-1 (350 мг, 57%) в виде белого твердого вещества. ЖХМС 265 [M-Boc]+; 1H ЯМР (400 MГц, ДМСО-d6) δ ч./млн. 7,52-7,32 (м, 6H), 7,29 (с, 1H), 5,22 (шс, 2H), 4,53-4,40 (м, 2H), 3,54 (т, J=5,6 Гц, 2H), 2,79 (т, J=5,5 Гц, 2H), 1,41 (с, 9H)
Стадия 2: Синтез трет-бутил 6-циано-8-гидрокси-3,4-дигидроизохинолин-2(1H)-карбоксилата (TP-18)
Соединение MM-1 (320 мг, 0,878 ммоль) растворяют в MeOH (3 мл). Pd/C (93 мг, 0,44 ммоль) добавляют к реакционному раствору и дегазируют H2 баллоном три раза, и перемешивают под H2 баллоном при 20°C в течение 1 часа. ДХМ (5 мл) добавляют к смеси, реакционную смесь фильтруют и концентрируют с получением неочищенного продукта, который очищают флэш-хроматографией с петролейным эфиром/EtOAc 0-50% с получением TP-18 в виде белого твердого вещества (125 мг, 51,9%). ЖХМС [219-tBu]+; 1H ЯМР (400 MГц, CDCl3) δ ч./млн. 7,45-7,28 (м, 1H), 7,10-6,95 (м, 1H), 6,87 (шс, 1H), 4,57 (шс, 2H), 3,66 (т, J=5,9 Гц, 2H), 2,82 (шс, 2H), 1,53 (с, 9H)
Примеры 84-98 получают по методике примера 78 со схемы CC с применением подходящего NBoc-защищенного тетрагидроизохинолина на стадии 1.
TP-2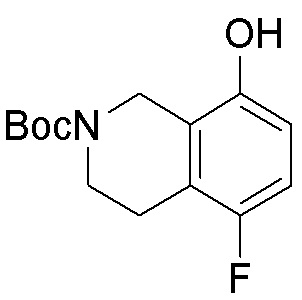
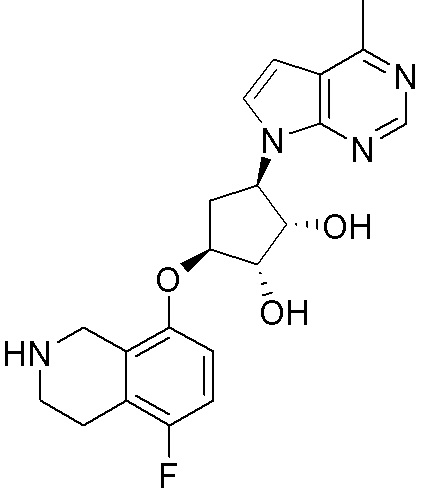
1H ЯМР (400 MГц, ДМСО-d6) δ ч./млн. 8,63 (с, 1H), 7,62 (д, J=3,5 Гц, 1H), 6,96-6,89 (м, 1H), 6,86-6,80 (м, 1H), 6,72 (д, J=3,5 Гц, 1H), 5,31 (д, J=3,8 Гц, 1H), 5,16-5,06 (м, 2H), 4,59-4,49 (м, 2H), 4,00-3,95 (м, 1H), 3,82 (с, 2H), 2,91 (т, J=5,8 Гц, 2H), 2,88-2,77 (м, 1H), 2,64 (с, 3H), 2,57 (т, J=5,8 Гц, 2H), 1,92 (ддд, J=3,8, 9,3, 13,6 Гц, 1H)
TP-1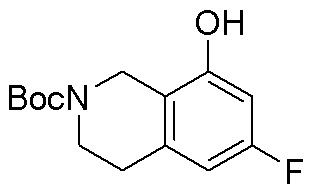
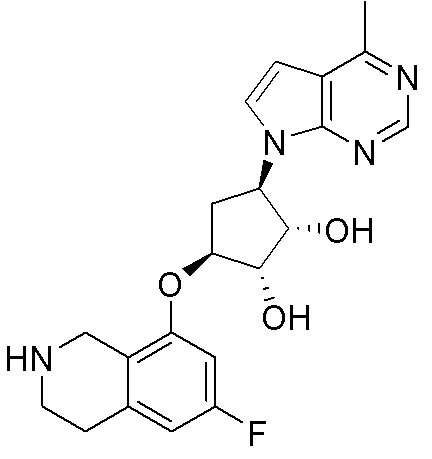
1H ЯМР (400 MГц, MeOD-d4) δ ч./млн. 8,61 (с, 1H), 7,55 (д, J=3,5 Гц, 1H), 6,75 (д, J=3,8 Гц, 1H), 6,68 (дд, J=2,3, 10,8 Гц, 1H), 6,49 (дд, J=2,1, 9,2 Гц, 1H), 5,24 (кв, J=8,8 Гц, 1H), 4,74-4,63 (м, 2H), 4,17 (д, J=4,8 Гц, 1H), 3,93 (с, 2H), 3,06 (т, J=5,9 Гц, 2H), 3,03-2,94 (м, 1H), 2,80 (т, J=5,8 Гц, 2H), 2,22-2,10 (м, 1H)
TP-19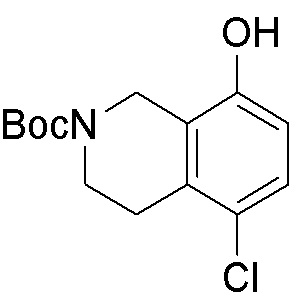
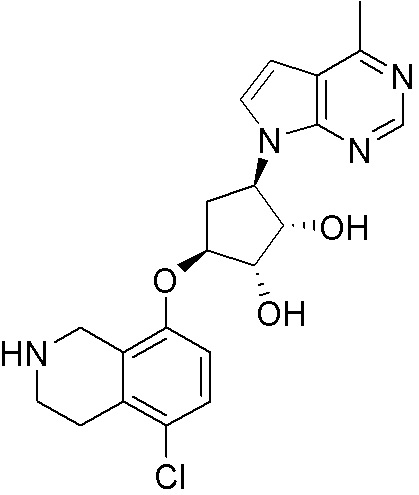
1H ЯМР (400 MГц, MeOD-d4) δ ч./млн. 8,64 (с, 1H), 7,57 (д, J=4,0 Гц, 1H), 7,25 (д, J=9,0 Гц, 1H), 6,92 (д, J=8,5 Гц, 1H), 6,77 (д, J=3,5 Гц, 1H), 5,25 (кв, J=9,0 Гц, 1H), 4,72 (дд, J=4,8, 8,3 Гц, 2H), 4,20 (д, J=4,5 Гц, 1H), 4,03 (с, 2H), 3,16 (т, J=6,0 Гц, 2H), 3,07-2,91 (м, 1H), 2,83 (т, J=5,8 Гц, 2H), 2,74 (с, 3H), 2,27-2,05 (м, 1H)
TP-3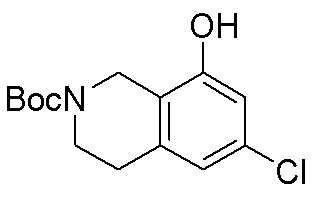
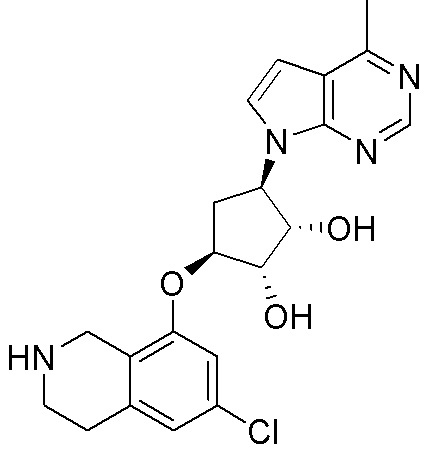
1H ЯМР (400 MГц, ДМСО-d6) δ ч./млн. 8,62 (с, 1H), 7,62 (д, J=3,8 Гц, 1H), 6,92 (с, 1H), 6,76 (с, 1H), 6,72 (д, J=3,5 Гц, 1H), 5,41-5,35 (м, 1H), 5,10 (д, J=9,3 Гц, 2H), 4,63-4,44 (м, 2H), 3,99-3,91 (м, 1H), 3,78 (с, 2H), 2,93-2,78 (м, 3H), 2,69-2,59 (м, 4H), 2,02-1,88 (м, 1H)
TP-5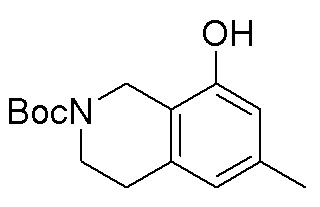
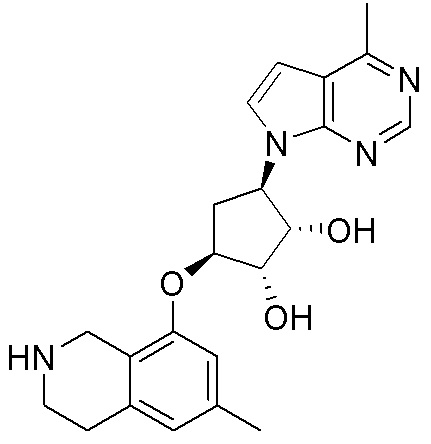
1H ЯМР (400 MГц, MeOD-d4) δ ч./млн. 8,64 (с, 1H), 7,56 (д, J=3,8 Гц, 1H), 6,77 (д, J=3,8 Гц, 1H), 6,70-6,66 (с, 1H), 6,61-6,55 (с, 1H), 5,34-5,23 (м, 1H), 4,73-4,66 (м, 2H), 4,22-4,16 (м, 1H), 3,96 (с, 2H), 3,07-3,06 (м, 3H), 2,84-2,76 (м, 2H), 2,73 (с, 3H), 2,30 (с, 3H), 2,18-2,07 (м, 1H)
TP-12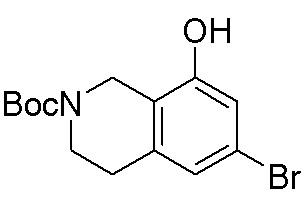
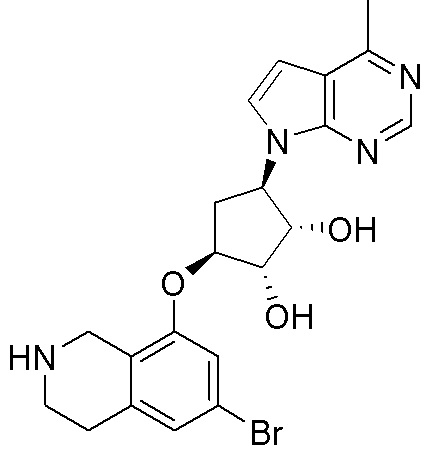
1H ЯМР (400 MГц, MeOD-d4) δ ч./млн. 8,63 (с, 1H), 7,57 (д, J=3,5 Гц, 1H), 7,13 (с, 1H), 7,05-6,94 (м, 1H), 6,76 (д, J=3,8 Гц, 1H), 5,22 (кв, J=9,0 Гц, 1H), 4,76-4,67 (м, 2H), 4,24-4,14 (м, 1H), 4,09 (с, 2H), 3,23 (т, J=6,0 Гц, 2H), 3,00 (ддд, J=7,3, 9,3, 14,3 Гц, 1H), 2,95-2,86 (м, 2H), 2,73 (с, 3H), 2,32-2,15 (м, 1H)
TP-4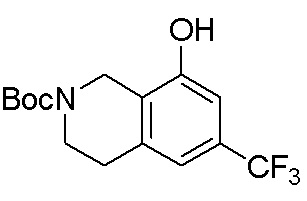
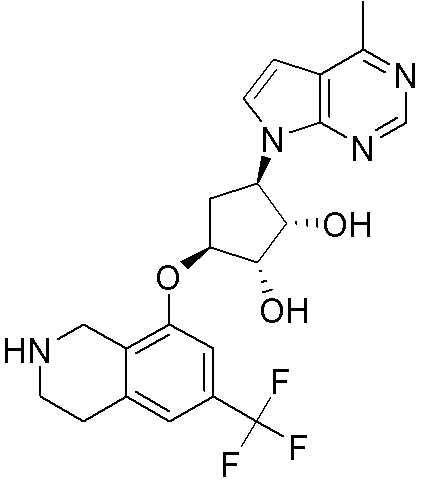
1H ЯМР (400 MГц, MeOD-d4) δ ч./млн. 8,62 (с, 1H), 7,57 (д, J=3,5 Гц, 1H), 7,13 (с, 1H), 7,07 (с, 1H), 6,75 (д, J=3,8 Гц, 1H), 5,23 (кв, J=9,0 Гц, 1H), 4,76 (дд, J=2,6, 4,6 Гц, 1H), 4,72 (дд, J=4,9, 8,9 Гц, 1H), 4,18 (д, J=5,0 Гц, 1H), 4,03 (с, 2H), 3,12-3,06 (м, 2H), 3,00 (ддд, J=7,3, 9,3, 14,3 Гц, 1H), 2,88 (т, J=5,6 Гц, 2H), 2,71 (с, 3H), 2,27-2,17 (м, 1H)
TP-18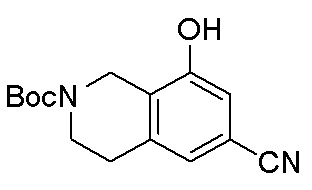
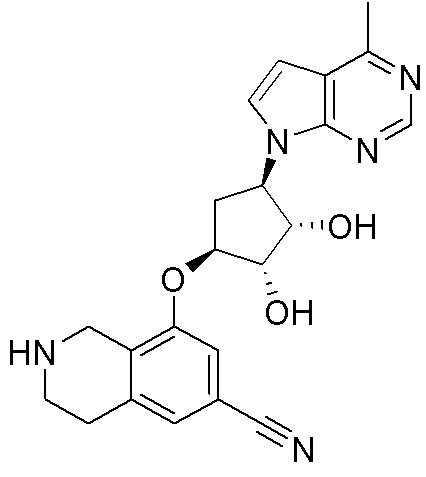
1H ЯМР (400 MГц, MeOD-d4) δ ч./млн. 8,64 (с, 1H), 7,58 (д, J=3,5 Гц, 1H), 7,24 (с, 1H), 7,17 (с, 1H), 6,77 (д, J=3,8 Гц, 1H), 5,29-5,19 (м, 1H), 4,80-4,69 (м, 2H), 4,19 (д, J=4,5 Гц, 1H), 4,03 (с, 2H), 3,10-3,07 (м, 2H), 3,05-2,97 (м, 1H), 2,90-2,84 (м, 2H), 2,74 (с, 3H), 2,27-2,23 (м, 1H)
TP-6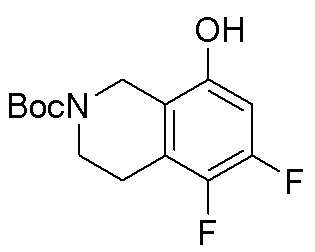
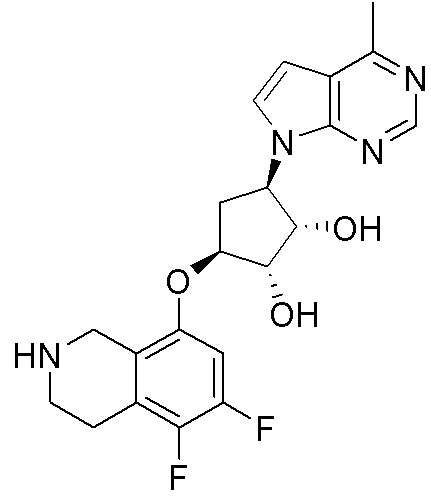
1H ЯМР (400 MГц, MeOD-d4) δ ч./млн. 8,62 (с, 1H), 7,55 (д, J=3,8 Гц, 1H), 7,02 (дд, J=6,9, 12,2 Гц, 1H), 6,75 (д, J=3,5 Гц, 1H), 5,21-5,11 (м, 1H), 4,76-4,70 (м, 1H), 4,70-4,63 (м, 1H), 4,22-4,15 (м, 3H), 3,37 (т, J=6,3 Гц, 2H), 3,03-2,90 (м, 3H), 2,72 (с, 3H), 2,31-2,21 (м, 1H)
TP-10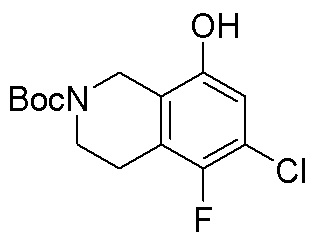
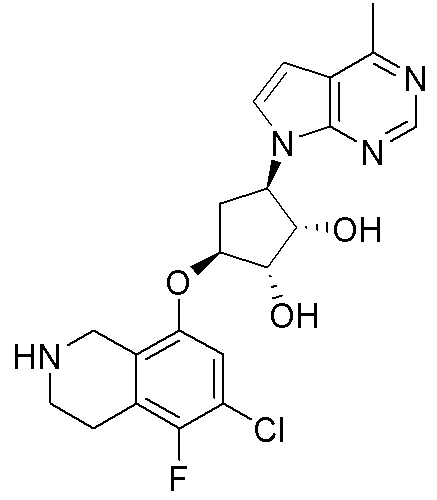
1H ЯМР (400 MГц, ДМСО-d6) δ ч./млн. 8,63 (с, 1H), 7,63 (д, J=3,5 Гц, 1H), 7,14 (д, J=6,3 Гц, 1H), 6,72 (д, J=3,8 Гц, 1H), 5,41 (д, J=3,0 Гц, 1H), 5,21-4,96 (м, 2H), 4,65-4,46 (м, 2H), 4,06-3,90 (м, 3H), 3,08 (т, J=5,8 Гц, 2H), 2,91-2,78 (м, 1H), 2,77-2,69 (м, 2H), 2,64 (с, 3H), 2,04-1,90 (м, 1H)
TP-11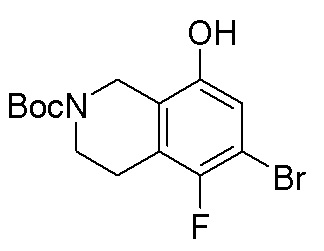
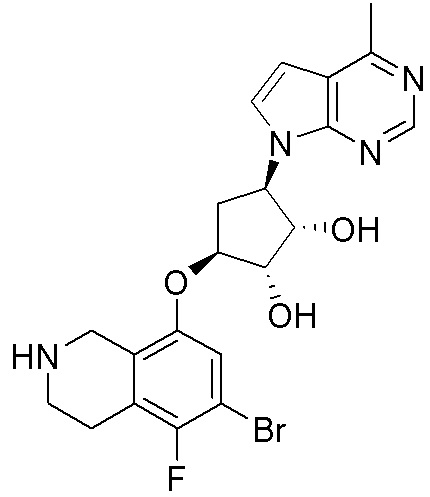
1H ЯМР (400 MГц, ДМСО-d6) δ ч./млн. 8,63 (с, 1H), 7,63 (д, J=3,76 Гц, 1H), 7,22 (д, J=5,52 Гц, 1H), 6,72 (с, 1H), 5,40 (д, J=3,76 Гц, 1H), 5,01-5,17 (м, 2H), 4,51-4,62 (м, 2H), 3,88-4,01 (м, 3H), 3,06 (т, J=5,90 Гц, 2H), 2,78-2,87 (м, 1H), 2,71 (т, J=5,27 Гц, 2H), 2,64 (с, 3H), 2,00-1,95 (м, 1H)
TP-16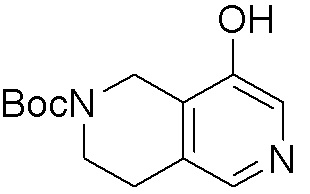
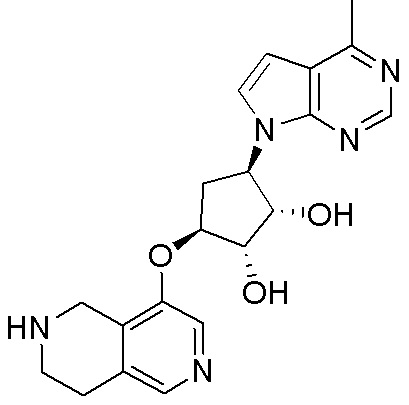
1H ЯМР (400 MГц, ДМСО-d6) δ ч./млн. 8,63 (с, 1H), 8,19 (с, 1H), 8,03 (с, 1H), 7,64 (д, J=3,8 Гц, 1H), 6,72 (д, J=3,5 Гц, 1H), 5,43 (д, J=4,3 Гц, 1H), 5,17-5,06 (м, 2H), 4,71 (шс, 1H), 4,62-4,53 (м, 1H), 4,04-3,97 (м, 3H), 3,10 (шс, 2H), 2,95-2,83 (м, 1H), 2,78 (шс, 2H), 2,64 (с, 3H), 2,08-1,94 (м, 1H)
TP-17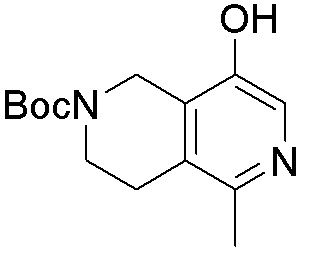
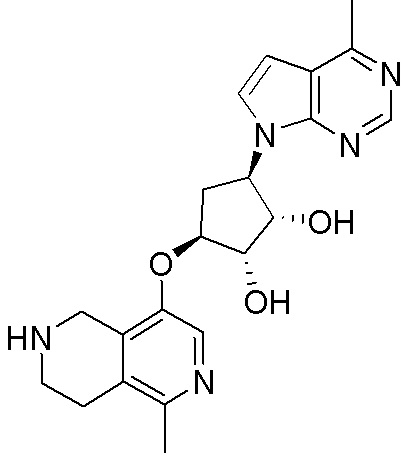
1H ЯМР (400 MГц, МЕТАНОЛ-d4) δ ч./млн. 8,62 (с, 1H), 8,03 (с, 1H), 7,56 (д, J=3,8 Гц, 1H), 6,75 (д, J=3,5 Гц, 1H), 5,29-5,12 (м, 1H), 4,75 (ддд, J=4,8, 9,0, 13,6 Гц, 2H), 4,20 (д, J=4,3 Гц, 1H), 4,09 (с, 2H), 3,22 (т, J=6,0 Гц, 2H), 3,05-2,94 (м, 1H), 2,79 (т, J=5,8 Гц, 2H), 2,72 (с, 3H), 2,40 (с, 3H), 2,30-2,19 (м, 1H)
TP-9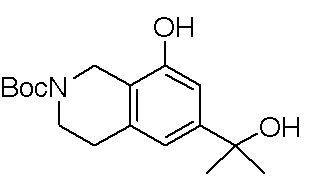
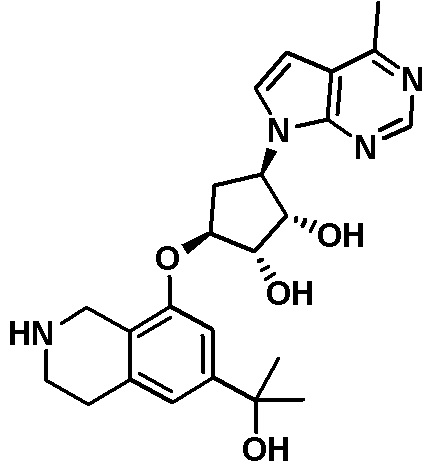
1H ЯМР (400 MГц, МЕТАНОЛ-d4) δ ч./млн. 8,64 (с, 1 H) 7,57 (д, J=3,5 Гц, 1 H) 7,00 (с, 1 H) 6,88 (с, 1 H) 6,77 (д, J=3,5 Гц, 1 H) 5,29 (кв, J=8,8 Гц, 1 H) 4,73 (с, 2 H) 4,22 (д, J=4,8 Гц, 1 H) 4,02 (с, 2 H) 3,12 (т, J=5,9 Гц, 2 H) 3,03 (ддд, J=14,6, 9,5, 7,0 Гц, 1 H) 2,87 (т, J=5,8 Гц, 2 H) 2,73 (с, 3 H) 2,17 (ддд, J=14,3, 8,4, 3,6 Гц, 1 H) 1,53 (с, 6 H)
TP-7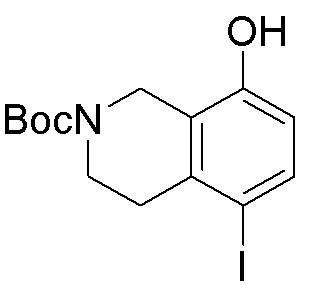
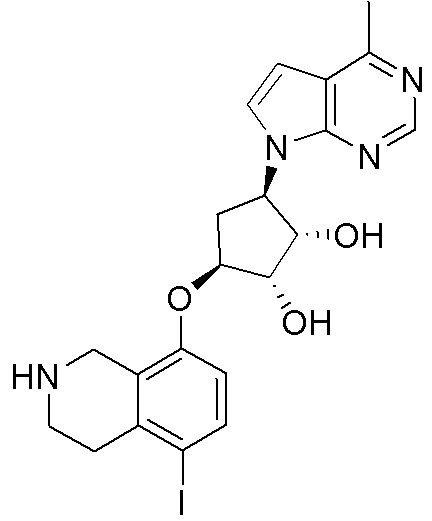
1H ЯМР (400 MГц, ДМСО-d6) δ ч./млн. 8,64 (с, 1H), 7,71 (д, J=8,4 Гц, 1H), 7,62 (с, 1H), 6,80 (д, J=8,8 Гц, 1H), 6,71 (с, 1H), 5,38 (с, 1H), 5,19 -5,02 (м, 2H), 4,60-4,55 (м, 2H), 4,08-3,93 (м, 3H), 3,18 (с, 2H), 2,91-2,78 (м, 1H), 2,64 (шс, 4H), 2,00-1,96 (м, 1H)
Пример 99 (Схема NN)-(1S,2S,3R,5S)-3-(4-амино-5-фтор-7H-пирроло[2.3-d]пиримидин-7-ил)-5-((1,2,3,4-тетрагидроизохинолин-8-ил)окси)циклопентан-1,2-диол (NN-5)
Схема NN
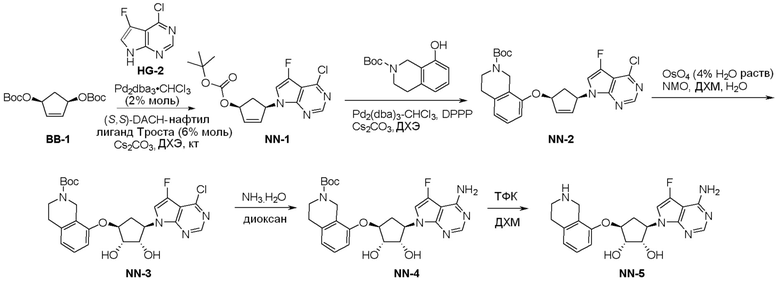
Стадия 1 - Синтез трет-бутил ((1S,4R)-4-(4-хлор-5-фтор-7H-пирроло[2.3-d]пиримидин-7-ил)циклопент-2-ен-1-ил)карбоната (NN-1)
Пробирка A: В сухую круглодонную колбу (продутую N2), добавляют лиганд Троста (S,S)-DACH-нафтил (1,13 г, 1,43 ммоль) и Pd2(dba)3.CHCl3 (493 мг, 0,48 ммоль). Пробирку продувают N2 четыре раза и добавляют ДХЭ (50 мл, орошают N2 в течение 30 мин). Черный раствор перемешивают в течение 30 мин при 12°C, в этот момент получают красно-коричневый раствор.
Пробирка B: В сухую круглодонную колбу (продутую N2) добавляют BB-1 (7155 мг, 23,82 ммоль), 4-хлор-5-фтор-7H-пирроло[2.3-d]пиримидин (4,5 г, 26,23 ммоль) и Cs2CO3 (8,54 г, 26,2 ммоль). Пробирку продувают N2 пять раз и добавляют ДХЭ (50 мл), затем добавляют содержимое пробирки A через шприц. Реакционную смесь перемешивают при 12°C под N2 в течение 24 часов.
Реакционную смесь фильтруют и концентрируют до коричневой камеди. Неочищенный остаток очищают флэш-биотаж (120 г, силикагель, EtOAc/петролейный эфир=15%) с получением NN-1 (6,4 г, 76%) в виде беловатого твердого вещества. ЖХМС [M+1] 354; 1H ЯМР (400 MГц, CDCl3) δ ч./млн. 8,62 (с, 1H), 7,08 (д, J=2,5 Гц, 1H), 6,35-6,29 (м, 1H), 6,10-6,00 (м, 2H), 5,63-5,55 (м, 1H), 3,11 (тд, J=7,8, 15,4 Гц, 1H), 1,87 (тд, J=3,8, 15,0 Гц, 1H), 1,52 (с, 9H)
Стадия 2 - Синтез трет-бутил 8-(((1S,4R)-4-(4-хлор-5-фтор-7H-пирроло[2.3-d]пиримидин-7-ил)циклопент-2-ен-1-ил)окси)-3,4-дигидроизохинолин-2(1H)-карбоксилата (NN-2)
В сухую микроволновую пробирку (продутую N2) добавляют NN-1 (200 мг, 0,57 ммоль), трет-бутил 8-гидрокси-3,4-дигидроизохинолин-2(1H)-карбоксилат (141 мг, 0,57 ммоль), Cs2CO3 (203 мг, 0,622 ммоль), Pd2(dba)3.CHCl3 (15 мг, 0,014 ммоль) и DPPP (14 мг, 0,03 ммоль). Затем пробирку продувают N2 три раза и добавляют ДХЭ (2,6 мл, продувают N2 в течение 30 мин). Черную смесь перемешивают при 20°C в течение 1 часа. Затем реакционную смесь сразу очищают преп.-ТСХ (петролейный эфир:EtOAc=4:1) с получением NN-2 (260 мг, 95%) в виде белой пены.
ЖХМС [M+23] 507; 1H ЯМР (400 MГц, CDCl3) δ ч./млн. 8,63 (с, 1H), 7,16 (д, J=2,0 Гц, 1H), 7,14-7,09 (м, 1H), 6,79 (д, J=7,3 Гц, 1H), 6,74 (д, J=7,5 Гц, 1H), 6,46-6,40 (м, 1H), 6,15-6,06 (м, 2H), 5,37-5,31 (м, 1H), 4,74-4,42 (м, 2H), 3,75-3,54 (м, 2H), 3,19-3,10 (м, 1H), 2,87-2,79 (м, 2H), 1,97 (д, J=14,6 Гц, 1H), 1,51 (с, 9H)
Стадия 2 - Синтез трет-бутил 8-(((1S,2S,3S,4R)-4-(4-хлор-5-фтор-7H-пирроло[2.3-d]пиримидин-7-ил)-2,3-дигидроксициклопентил)окси)-3,4-дигидроизохинолин-2(1H)-карбоксилата (NN-3)
К смеси NN-2 (260 мг, 0,536 ммоль) в ДХМ (9 мл)/H2O (0,3 мл) добавляют NMO (188 мг, 1,61 ммоль) и OsO4 (4% в т-BuOH, 204 мг, 0,0322 ммоль) при 20°C. Черную смесь перемешивают при 20°C в течение 2 часов. Смесь разбавляют ДХМ (10 мл) и гасят насыщ. Na2SO3 (5 мл) и разделяют. Водный слой экстрагируют ДХМ (10 мл). Объединенный органический слой промывают насыщенным раствором соли (10 мл), сушат над Na2SO4, фильтруют и концентрируют и очищают ISCO (12 г, силикагель, EtOAc:петролейный эфир=1:1) с получением NN-3 (230 мг, 83%) в виде бесцветной камеди.
ЖХМС [M+23] 541; 1H ЯМР (400 MГц, CDCl3) δ ч./млн. 8,62 (с, 1H), 7,17 (т, J=8,0 Гц, 1H), 7,13 (д, J=2,5 Гц, 1H), 6,86-6,79 (м, 2H), 5,21-5,05 (м, 1H), 5,02-4,75 (м, 2H), 4,67-4,56 (м, 1H), 4,53-4,42 (м, 2H), 4,37-4,29 (м, 1H), 3,72-3,57 (м, 2H), 3,32-2,99 (м, 2H), 2,87-2,78 (м, 2H), 2,33-2,22 (м, 1H), 1,50-1,44 (м, 9H)
Стадия 3 - Синтез трет-бутил 8-(((1S,2S,3S,4R)-4-(4-амино-5-фтор-7H-пирроло[2.3-d]пиримидин-7-ил)-2,3-дигидроксициклопентил)окси)-3,4-дигидроизохинолин-2(1H)-карбоксилата (NN-4)
Раствор NN-3 (105 мг, 0,202 ммоль) в NH3.H2O и диоксане (2,8 мл /2,8 мл) герметично закрывают в стеклянной пробирке при 90°C в течение 15 часов. Реакционную смесь концентрируют с получением NN-4 (101 мг, >99%) в виде желтой камеди и применяют сразу на следующей стадии. ЖХМС [M+23] 522
Стадия 4 - Синтез (1S,2S,3R,5S)-3-(4-амино-5-фтор-7H-пирроло[2.3-d]пиримидин-7-ил)-5-((1,2,3,4-тетрагидроизохинолин-8-ил)окси)циклопентан-1,2-диола (NN-5)
К светло-желтому раствору NN-4 (101 мг, 0,202 ммоль) в ДХМ (5 мл) добавляют ТФК (1 мл) по каплям при 0°C. Желтый раствор перемешивают при 20°C в течение 2 часов. Смесь концентрируют MeOH (4 мл), добавляют к остатку и подщелачивают твердым K2CO3 до pH 7-8. Смесь фильтруют и концентрируют, очищают преп.-ВЭЖХ с получением NN-5 (35 мг, 39%) в виде светло-желтого твердого вещества.
ЖХМС [M+1] 400; 1H ЯМР (400 MГц, MeOD) δ ч./млн. 8,09 (с, 1H), 7,35-7,23 (м, 1H), 7,15-7,06 (м, 1H), 7,03-6,96 (м, 1H), 6,94-6,83 (м, 1H), 5,16-5,07 (м, 1H), 4,75-4,68 (м, 1H), 4,63-4,55 (м, 1H), 4,37 (с, 2H), 4,22-4,16 (м, 1H), 3,54-3,44 (м, 2H), 3,17-3,08 (м, 2H), 3,04-2,92 (м, 1H), 2,16-2,02 (м, 1H)
Примеры 100-111 получают по методике примера 99 со схемы NN с применением подходящего NBoc-защищенного тетрагидроизохинолина на стадии 2.
TP-2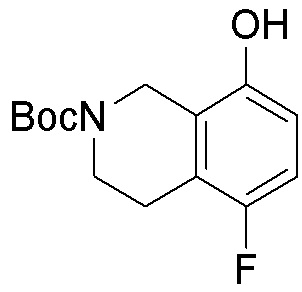
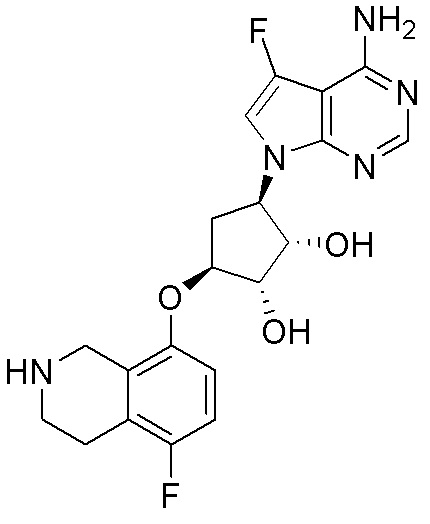
1H ЯМР (400 MГц, MeOD-d4) δ ч./млн. 8,09 (с, 1H), 7,08 (д, J=2,0 Гц, 1H), 7,02-6,83 (м, 2H), 5,13 (кв, J=8,8 Гц, 1H), 4,68-4,62 (м, 1H), 4,55 (дд, J=5,0, 8,8 Гц, 1H), 4,15 (д, J=5,5 Гц, 1H), 4,13 (с, 2H), 3,26 (т, J=6,0 Гц, 2H), 3,01-2,92 (м, 1H), 2,89 (т, J=6,1 Гц, 2H), 2,04 (ддд, J=4,3, 9,1, 14,0 Гц, 1H)
TP-12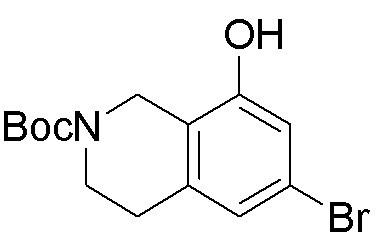
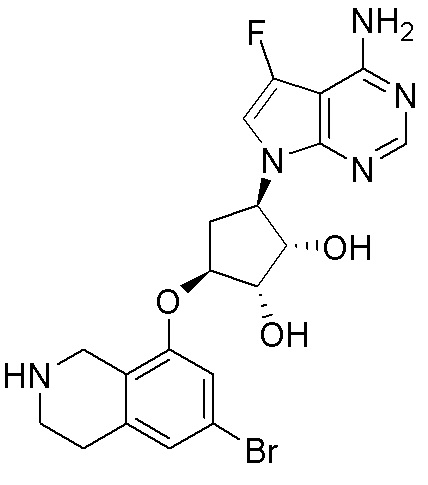
1H ЯМР (400 MГц, MeOD-d4) δ ч./млн. 8,08 (с, 1H), 7,17-7,10 (м, 1H), 7,09-7,05 (м, 1H), 7,04-7,00 (м, 1H), 5,10 (кв, J=8,8 Гц, 1H), 4,69-4,63 (м, 1H), 4,57-4,51 (м, 1H), 4,16-4,06 (м, 3H), 3,29-3,24 (м, 2H), 2,99-2,89 (м, 3H), 2,13-2,02 (м, 1H)
TP-3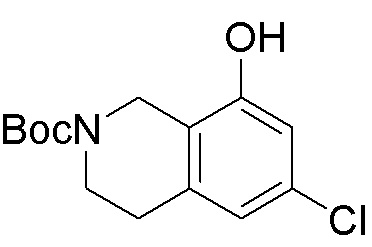
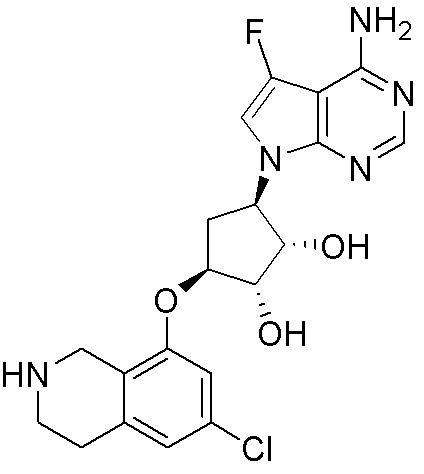
1H ЯМР (400 MГц, MeOD-d4) δ ч./млн. 8,08 (с, 1H), 7,07 (д, J=4,0 Гц, 2H), 6,93 (с, 1H), 5,07 (кв, J=8,8 Гц, 1H), 4,72-4,63 (м, 1H), 4,60-4,53 (м, 1H), 4,28 (с, 2H), 4,16-4,12 (м, 1H), 3,44 (т, J=6,1 Гц, 2H), 3,06 (т, J=6,3 Гц, 2H), 2,99-2,88 (м, 1H), 2,19-2,04 (м, 1H)
TP-6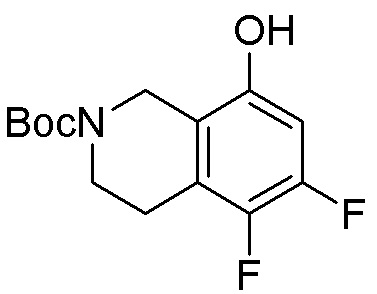
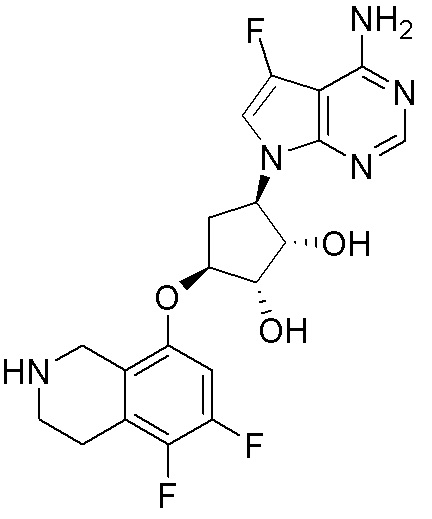
1H ЯМР (400 MГц, MeOD-d4) δ ч./млн. 8,07 (с, 1H), 7,07 (д, J=2,0 Гц, 1H), 7,03 (дд, J=6,7, 12,2 Гц, 1H), 5,06 (кв, J=9,0 Гц, 1H), 4,63 (тд, J=2,6, 4,8 Гц, 1H), 4,55 (дд, J=5,3, 8,8 Гц, 1H), 4,20 (с, 2H), 4,13 (д, J=4,0 Гц, 1H), 3,40 (т, J=6,3 Гц, 2H), 3,01 (т, J=6,0 Гц, 2H), 2,93 (тд, J=8,2, 14,3 Гц, 1H), 2,09 (ддд, J=4,8, 9,3, 14,1 Гц, 1H)
TP-4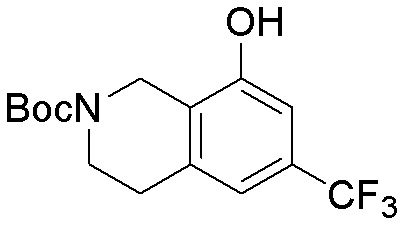
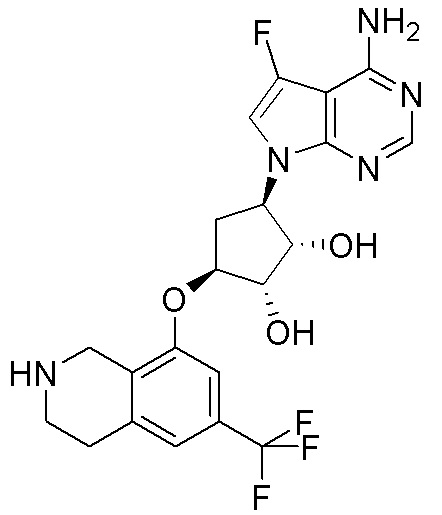
1H ЯМР (400 MГц, MeOD-d4) δ ч./млн. 8,08 (с, 1H), 7,12 (с, 1H), 7,08 (д, J=2,3 Гц, 1H), 7,06 (с, 1H), 5,14 (кв, J=9,3 Гц, 1H), 4,74-4,67 (м, 1H), 4,52 (дд, J=5,0, 8,8 Гц, 1H), 4,13 (д, J=5,5 Гц, 1H), 3,98 (с, 2H), 3,09-3,04 (м, 2H), 2,95 (ддд, J=7,3, 9,2, 14,4 Гц, 1H), 2,88-2,83 (м, 2H), 2,06 (ддд, J=4,1, 9,0, 13,7 Гц, 1H)
TP-5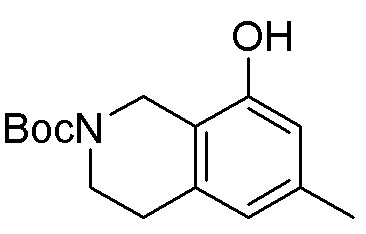
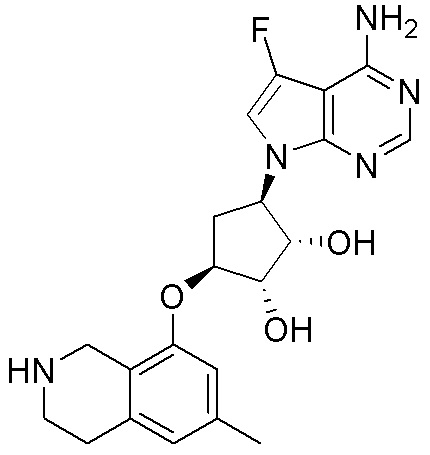
1H ЯМР (400 MГц, ДМСО-d6) δ ч./млн. 8,05 (с, 1H), 7,20 (с, 1H), 6,92 (шс, 2H), 6,66 (с, 1H), 6,53 (с, 1H), 5,06-4,98 (м, 1H), 4,50 (шс, 1H), 4,41-4,37 (м, 1H), 3,95-3,92 (м, 1H), 3,88 (шс, 2H), 3,05-2,95 (м, 3H), 2,84-2,67 (м, 2H), 2,23 (с, 3H), 1,84-1,73 (м, 1H)
TP-13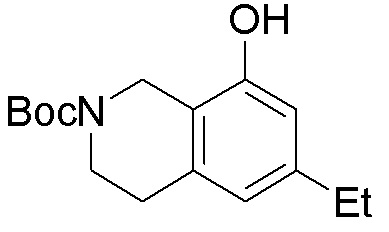
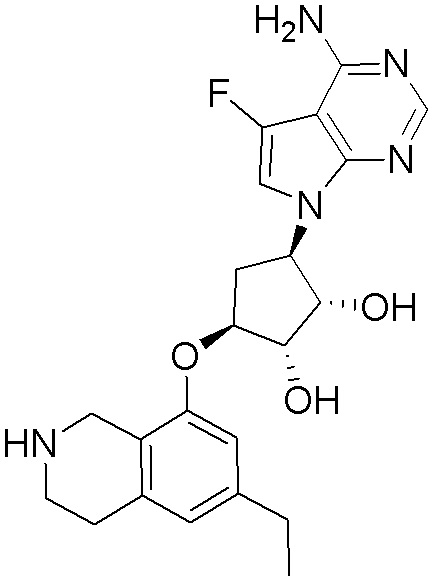
1H ЯМР (400 MГц, MeOD-d4) δ ч./млн. 8,14 (с, 1H), 7,22 (д, J=2,0 Гц, 1H), 6,85 (с, 1H), 6,74 (с, 1H), 5,17 (кв, J=9,0 Гц, 1H), 4,74-4,66 (м, 1H), 4,56 (дд, J=4,9, 8,9 Гц, 1H), 4,30 (с, 2H), 4,15 (д, J=5,0 Гц, 1H), 3,47 (т, J=6,4 Гц, 2H), 3,07 (т, J=6,1 Гц, 2H), 3,02-2,92 (м, 1H), 2,64 (кв, J=7,4 Гц, 2H), 2,12-2,02 (м, 1H), 1,23 (т, J=7,7 Гц, 3H)
TP-1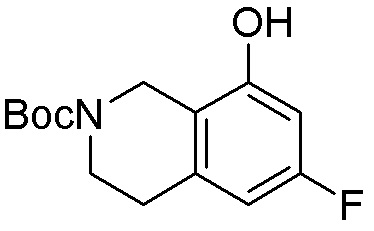
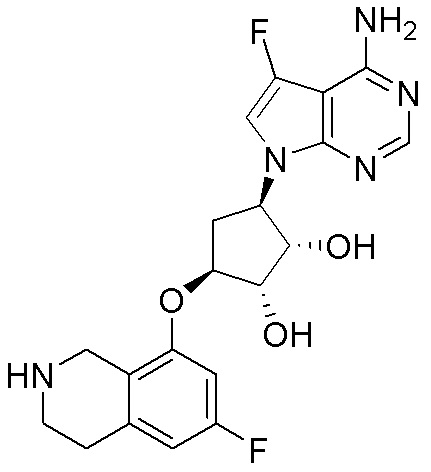
1H ЯМР (400 MГц, MeOD-d4) δ ч./млн. 8,16 (с, 1H), 7,27 (д, J=2,0 Гц, 1H), 6,86 (дд, J=2,3, 10,8 Гц, 1H), 6,67 (д, J=9,3 Гц, 1H), 5,21-5,12 (м, 1H), 4,68 (т, J=5,3 Гц, 1H), 4,56 (дд, J=5,0, 9,0 Гц, 1H), 4,30 (с, 2H), 4,15 (д, J=4,8 Гц, 1H), 3,48 (т, J=6,3 Гц, 2H), 3,10 (т, J=6,0 Гц, 2H), 3,02-2,91 (м, 1H), 2,12 (ддд, J=4,6, 9,5, 14,0 Гц, 1H)
TP-10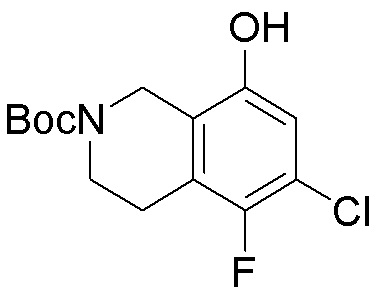
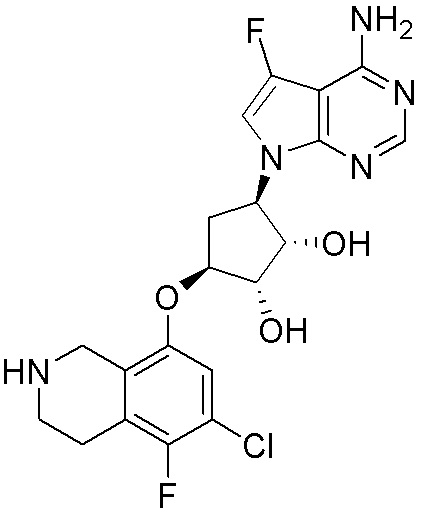
1H ЯМР (400 MГц, MeOD-d4) δ ч./млн. 8,08 (с, 1H), 7,08 (д, J=2,0 Гц, 1H), 7,01 (д, J=6,0 Гц, 1H), 5,16-5,09 (м, 1H), 4,63-4,58 (м, 1H), 4,53-4,47 (м, 1H), 4,14-4,08 (м, 1H), 3,92 (с, 2H), 3,07 (т, J=5,9 Гц, 2H), 2,98-2,88 (м, 1H), 2,82-2,73 (м, 2H), 2,07-1,97 (м, 1H)
TP-11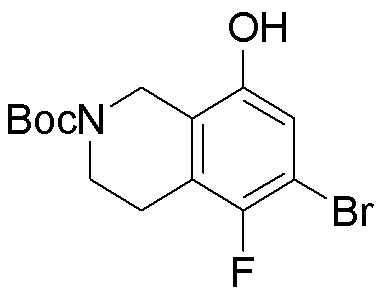
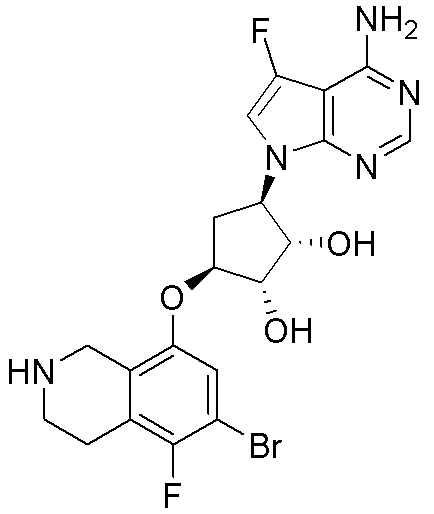
1H ЯМР (400 MГц, MeOD) δ ч./млн. 8,09 (с, 1 H), 7,04-7,20 (м, 2 H), 5,13 (м, J=9,00 Гц, 1 H), 4,59-4,68 (м, 1 H), 4,52 (м, J=8,80 Гц, 1 H), 4,13 (д, J=4,77 Гц, 1 H), 3,95 (с, 2 H), 3,11 (м, J=6,00 Гц, 2 H), 2,87-3,03 (м, 1 H), 2,81 (м, J=5,80 Гц, 2 H), 1,97-2,13 (м, 1 H)
трет-бутил 4-гидроксиизоиндолин-2-карбоксилат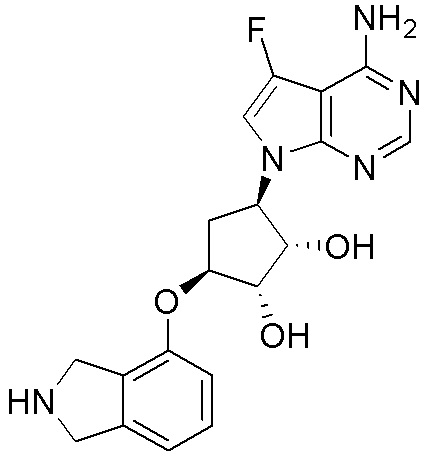
1H ЯМР (400 MГц, MeOD-d4) δ ч./млн. 8,09 (с, 1H), 7,45-7,38 (м, 1H), 7,14-7,07 (м, 2H), 7,07-7,01 (м, 1H), 5,18-5,08 (м, 1H), 4,78-4,72 (м, 1H), 4,66 (с, 4H), 4,58-4,52 (м, 1H), 4,20-4,15 (м, 1H), 3,03-2,92 (м, 1H), 2,13-2,02 (м, 1H)
трет-бутил 5-гидрокси-3,4-дигидроизохинолинe-2(1H)-карбоксилат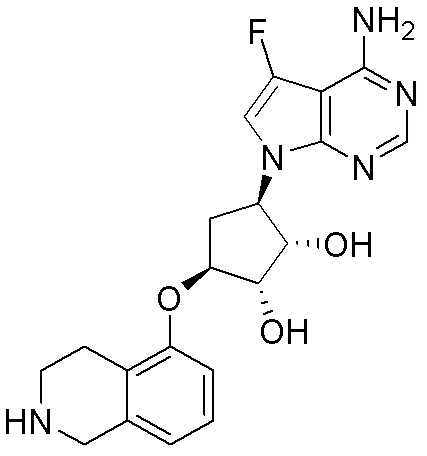
1H ЯМР (400 MГц, MeOD-d4) δ ч./млн. 8,09 (с, 1H), 7,29 (т, J=7,8 Гц, 1H), 7,08 (д, J=2,0 Гц, 1H), 7,04 (д, J=8,5 Гц, 1H), 6,86 (д, J=7,8 Гц, 1H), 5,22-5,09 (м, 2H), 4,74-4,68 (м, 1H), 4,57 (дд, J=5,0, 8,5 Гц, 1H), 4,37 (с, 2H), 4,18 (д, J=4,0 Гц, 1H), 3,56 (т, J=6,5 Гц, 2H), 3,10 (т, J=6,5 Гц, 2H), 3,04-2,93 (м, 1H), 2,11-1,98 (м, 1H)
TP-15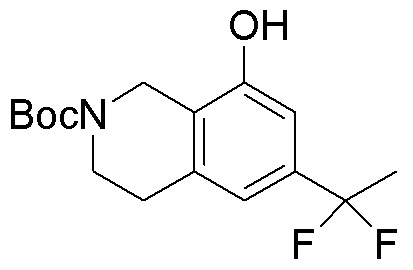
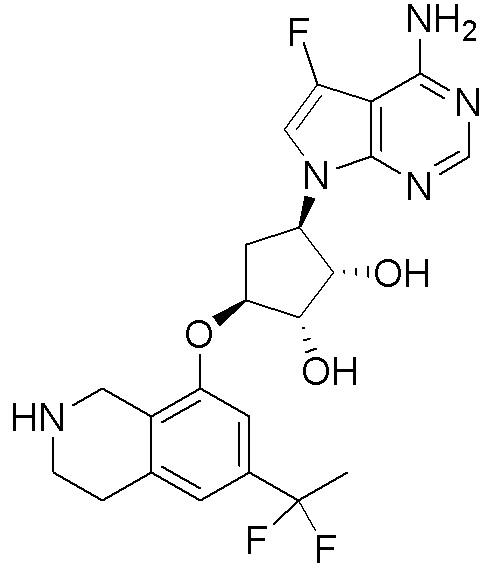
1H ЯМР (400 MГц, MeOD) δ ч./млн. 8,29 (с, 1H), 7,51 (д, J=2,3 Гц, 1H), 7,14 (с, 1H), 7,10 (с, 1H), 5,27 (кв, J=9,0 Гц, 1H), 4,79-4,75 (м, 1H), 4,60 (дд, J=5,0, 9,0 Гц, 1H), 4,39 (с, 2H), 4,17 (д, J=4,8 Гц, 1H), 3,52 (т, J=6,3 Гц, 2H), 3,17 (т, J=6,0 Гц, 2H), 3,00 (ддд, J=7,7, 9,0, 14,2 Гц, 1H), 2,17 (ддд, J=4,1, 9,7, 14,1 Гц, 1H), 1,93 (т, J=18,3 Гц, 3H)
TP-8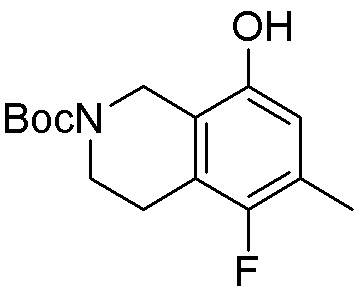
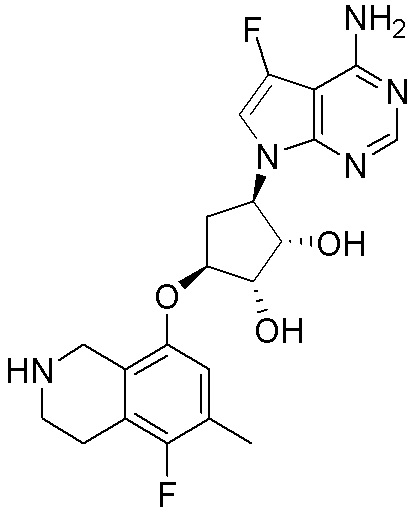
1H ЯМР с муравьиной кислотой (400 MГц, MeOD-d4) δ ч./млн. 8,50 (шс, 1H), 8,07 (с, 1H), 7,07 (д, J=2,3 Гц, 1H), 6,88 (д, J=6,3 Гц, 1H), 5,07 (кв, J=8,7 Гц, 1H), 4,67-4,61 (м, 1H), 4,55 (дд, J=5,0, 8,8 Гц, 1H), 4,28 (с, 2H), 4,16-4,11 (м, 1H), 3,49-3,41 (м, 2H), 3,01 (т, J=6,1 Гц, 2H), 2,96-2,87 (м, 1H), 2,27 (д, J=1,8 Гц, 3H), 2,10-2,01 (м, 1H)
Пример 114 (Схема OO) - (1S,2S,3R,5S)-3-(4-амино-5-фтор-7H-пирроло[2.3-d]пиримидин-7-ил)-5-((2-метил-1,2,3,4-тетрагидроизохинолин-8-ил)окси)циклопентан-1,2-диол (OO-1)
Схема OO
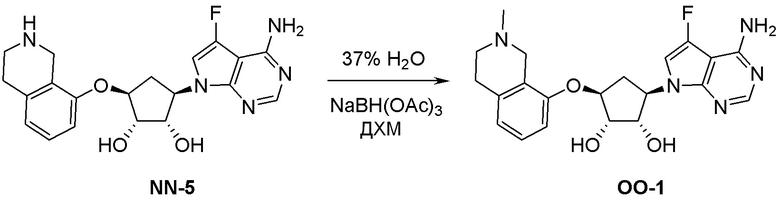
Стадия 1 - Синтез(1S,2S,3R,5S)-3-(4-амино-5-фтор-7H-пирроло[2.3-d]пиримидин-7-ил)-5-((2-метил-1,2,3,4-тетрагидроизохинолин-8-ил)окси)циклопентан-1,2-диол (OO-1)
Смесь NN-5 (100 мг, 0,25 ммоль), 37% CH2O (31 мг, 0,38 ммоль) и NaBH(OAc)3 (212 мг, 1,0 ммоль) в ТГФ (6 мл) перемешивают при 20°C в течение 2 ч. Смесь фильтруют и обрабатывают преп-ВЭЖХ с получением OO-1 (65 мг, 53%) в виде светло-желтого твердого вещества. ЖХМС 414 [M+1]; 1H ЯМР (400 MГц, MeOD-d4) δ ч./млн. 8,10 (с, 1H), 7,34-7,27 (м, 1H), 7,16-7,10 (м, 1H), 7,03-6,97 (м, 1H), 6,94-6,88 (м, 1H), 5,18-5,09 (м, 1H), 4,75-4,68 (м, 1H), 4,59-4,53 (м, 1H), 4,42-4,34 (м, 2H), 4,21-4,16 (м, 1H), 3,55-3,46 (м, 2H), 3,23-3,16 (м, 2H), 3,08 (с, 3H), 3,04-2,92 (м, 1H), 2,16-2,03 (м, 1H)
Примеры 115 & 116 получают по методикам схемы OO из Примеров 110 & 111 соответственно.
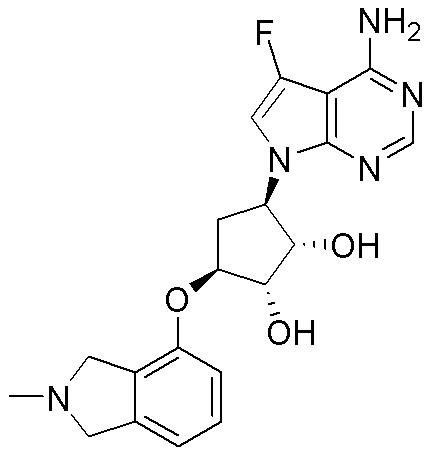
1H ЯМР (400 MГц, MeOD-d4) δ ч./млн. 8,09 (с, 1H), 7,41 (т, J=7,9 Гц, 1H), 7,15-7,06 (м, 2H), 7,02 (д, J=7,5 Гц, 1H), 5,21-5,10 (м, 1H), 4,77-4,70 (м, 1H), 4,66 (д, J=5,5 Гц, 4H), 4,53 (дд, J=5,1, 8,7 Гц, 1H), 4,18 (д, J=5,0 Гц, 1H), 3,11 (с, 3H), 2,98 (ддд, J=7,4, 9,0, 14,4 Гц, 1H), 2,05 (ддд, J=4,3, 9,3, 14,1 Гц, 1H)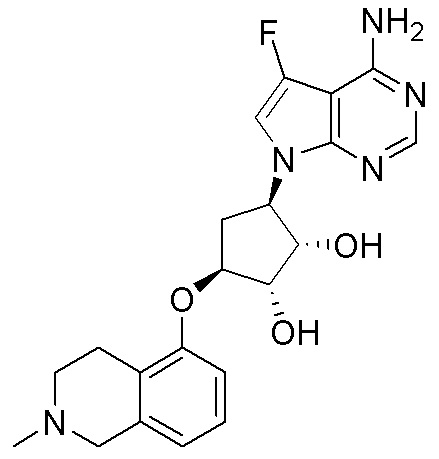
1H ЯМР (400 MГц, MeOD-d4) δ ч./млн. 8,09 (с, 1H), 7,27 (т, J=8,0 Гц, 1H), 7,08 (д, J=2,0 Гц, 1H), 7,01 (д, J=8,3 Гц, 1H), 6,82 (д, J=7,8 Гц, 1H), 5,22-5,10 (м, 1H), 4,71 (шс, 1H), 4,56 (дд, J=4,9, 8,7 Гц, 1H), 4,23 (шс, 2H), 4,18 (д, J=4,5 Гц, 1H), 3,46-3,37 (м, 2H), 3,10 (т, J=6,1 Гц, 2H), 2,99 (ддд, J=7,5, 9,2, 14,4 Гц, 1H), 2,93 (с, 3H), 2,05 (с, 1H)
Синтез 5-фтор-2-метил-7H-пирроло[2.3-d]пиримидина (HG-3)
Схема PP
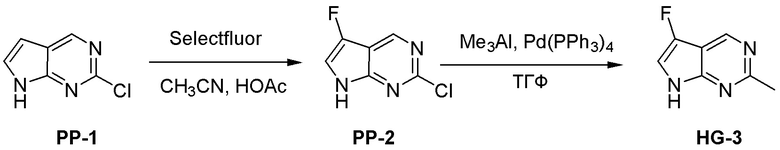
Стадия 1 - Синтез 2-хлор-5-фтор-7H-пирроло[2.3-d]пиримидина (PP-2)
К раствору 2-хлор-7H-пирроло[2.3-d]пиримидина (PP-1) (8,5 г, 55 ммоль) и selectfluor (29,4 г, 83,0 ммоль) в CH3CN (500 мл) добавляют AcOH (100 мл) под N2. Смесь перемешивают при 70°C в течение 16 часов. Цвет реакционной смеси превращается из желтого в оранжевый. Реакционную смесь концентрируют и подвергают азеотропной перегонке с толуолом (30 мл×3). Затем твердое вещество разбавляют CH2Cl2/EtOAc (1/1, 200 мл) и перемешивают при комнатной температуре (25°C) в течение 16 ч и фильтруют. Фильтрат концентрируют и промывают ДХМ с получением неочищенного продукта в виде коричневого твердого вещества. МТБЭ добавляют и перемешивают в течение ночи и фильтруют с получением PP-2 (1,5 г, 15%) в виде желтого твердого вещества. ЖХМС 172 [M+1]; 1H ЯМР (400 MГц, MeOD-d4) δ ч./млн. 8,89 (д, J=0,8 Гц, 1H), 7,31 (д, J=2,5 Гц, 1H)
Стадия 2 - Синтез 5-фтор-2-метил-7H-пирроло[2.3-d]пиримидина (HG-3)
Pd(PPh3)4 (835 мг, 0,723 ммоль) добавляют в раствор PP-2 (2,48 г, 14,46 ммоль) в сухом ТГФ (36 мл) при 10°C. Суспензию дегазируют с N2. Раствор Me3Al (2M, 14,5 мл, 28,9 ммоль) медленно добавляют в смесь при -10°C. После добавления, смесь нагревают при 70°C в течение 16 часов. Реакционную смесь осторожно добавляют в лед-воду. Смесь разбавляют EtOAc и фильтруют через целит. Фильтрат разделяют между EtOAc и H2O. Органический слой разделяют и промывают насыщенным раствором соли, сушат над Na2SO4 и концентрируют с получением неочищенного продукта и очищают на флэш-колонке (MeOH:ДХМ=1%~7,5%) с получением продукта HG-3 (1,2 г, 55%) в виде желтого твердого вещества. ЖХМС 152 [M+1]; 1H ЯМР (400 MГц, MeOD-d4) δ ч./млн. 11,76 (шс, 1H), 8,96 (с, 1H), 7,44 (т, J=2,5 Гц, 1H), 2,63 (с, 3H)
Синтез 5-фтор-4-метил-7H-пирроло[2.3-d]пиримидина (HG-4)
Схема QQ
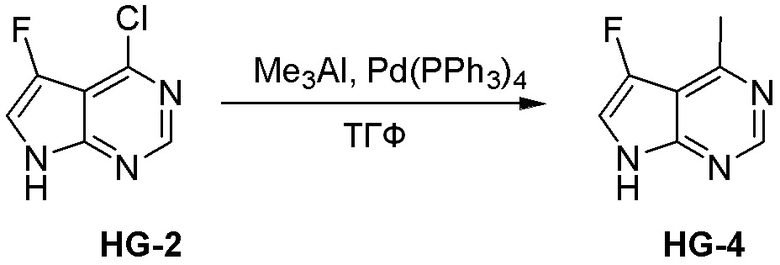
К Pd(PPh3)4 (2 г, 1,75 ммоль) добавляют раствор HG-2 (7,5 г, 43,7 ммоль) в сухом ТГФ (75 мл). Суспензию дегазируют с N2. Раствор AlMe3 (43,7 мл, 87,5 ммоль, 2M) добавляют в смесь в ледяной воде. После добавления желтый раствор нагревают при 80°C в течение 16 ч. Реакционную смесь гасят льдом и водным виннокислым калием-натрием. Смесь разделяют между EtOAc и H2O. Смесь фильтруют, и фильтровальную лепешку промывают EtOAc (100 мл×3). Органический слой разделяют и сушат над Na2SO4 и концентрируют, затем очищают на флэш-колонке (MeOH:ДХМ=1%~4%) с получением продукта. Продукт растирают в ДХМ и фильтруют. Фильтровальную лепешку промывают ТБМЭ и собирают с получением продукта HG-4 (2,00 г, 30%) в виде желтого твердого вещества. Фильтрат концентрируют, очищают на флэш-колонке (MeOH:ДХМ=1%~5%) с получением продукта. ЖХМС 152 [M+1]; 1H ЯМР (400 MГц, MeOD-d4) δ ч./млн. 11,89 (шс, 1H), 8,62 (с, 1H), 7,48 (т, J=2,5 Гц, 1H), 2,70 (с, 3H)
Синтез 4-хлор-5-фтор-2-метил-7H-пирроло[2.3-d]пиримидина (HG-5)
Схема RR
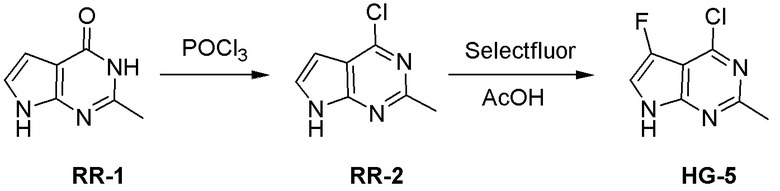
Стадия 1 - Синтез 4-хлор-2-метил-7H-пирроло[2.3-d]пиримидина (RR-2)
Смесь 2-метил-3,7-дигидро-4H-пирроло[2.3-d]пиримидин-4-она (RR-1) (2,7 г, 18,1 ммоль) в POCl3 (35 мл) нагревают при кипении с обратным холодильником в течение 4 часов. Смесь охлаждают до 20°C и концентрируют. К остатку добавляют ледяную воду (20 мл) и подщелачивают твердым Na2CO3 до pH~8. Смесь экстрагируют EtOAc (60 мл×2). Объединенные органические слои промывают насыщенным раствором соли (100 мл), сушат над Na2SO4, фильтруют и концентрируют с получением RR-2 (2,4 г, 78%) в виде беловатого твердого вещества. ЖХМС 168 [M+1]; 1H ЯМР (400 MГц, CDCl3) δ ч./млн. 10,70-10,41 (м, 1H), 7,36-7,29 (м, 1H), 6,67-6,56 (м, 1H), 2,81 (с, 3H)
Стадия 2 - Синтез 4-хлор-5-фтор-2-метил-7H-пирроло[2.3-d]пиримидина (HG-5)
Смесь RR-2 (2,35 г, 14 ммоль) и Selectfluor (7,45 г, 21 ммоль) в CH3CN (110 мл) и AcOH (22 мл) перемешивают при 70°C под N2 в течение 16 часов, реакционная смесь превращается из коричневой в розовую. Смесь концентрируют и подвергают азеотропной перегонке с толуолом (50 мл×2). Фильтрат концентрируют и очищают преп.-ВЭЖХ с получением HG-5 (600 мг, 23%) в виде желтого твердого вещества. ЖХМС 186 [M+1]; 1H ЯМР (400 MГц, MeOD-d4) δ ч./млн. 12,27-12,18 (м, 1H), 7,62-7,56 (м, 1H), 2,61 (с, 3H)
Примеры 117-119 получают по методике CC-3 (Пример 78) с применением подходящего пирролопиримидина на стадии 1 со Схемы BB и подходящего N-Boc защищенного тетрагидроизохинолина на стадии 1 со Схемы CC.
HG-4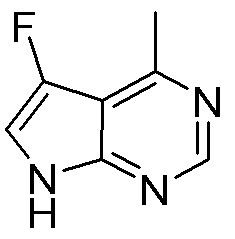
трет-бутил8-гидрокси-3,4-дигидроизохинолин-2(1H)-карбоксилат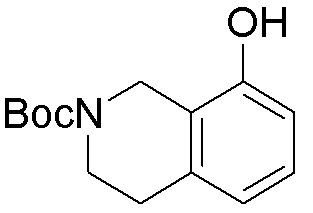
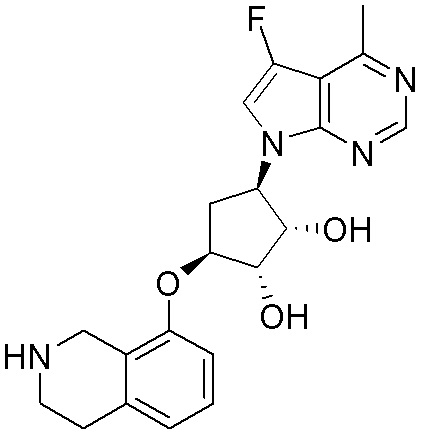
1H ЯМР (400 MГц, MeOD-d4) δ ч./млн. 8,62 (с, 1H), 8,50 (шс, 1H), 7,44 (д, J=1,5 Гц, 1H), 7,27 (т, J=7,8 Гц, 1H), 6,97 (д, J=8,5 Гц, 1H), 6,88 (д, J=7,5 Гц, 1H), 5,23 (кв, J=9,0 Гц, 1H), 4,74-4,69 (м, 1H), 4,69-4,62 (м, 1H), 4,36 (с, 2H), 4,18 (д, J=4,5 Гц, 1H), 3,47 (т, J=6,0 Гц, 2H), 3,10 (т, J=6,0 Гц, 2H), 3,04-2,90 (м, 1H), 2,78 (с, 3H), 2,21-2,07 (м, 1H)
7H-пирроло[2.3-d]пиримидин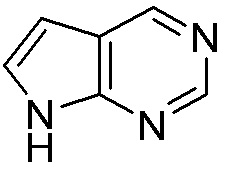
трет-бутил8-гидрокси-3,4-дигидроизохинолин-2(1H)-карбоксилат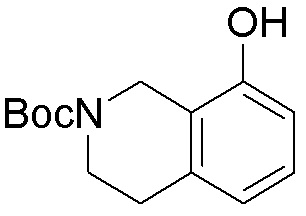
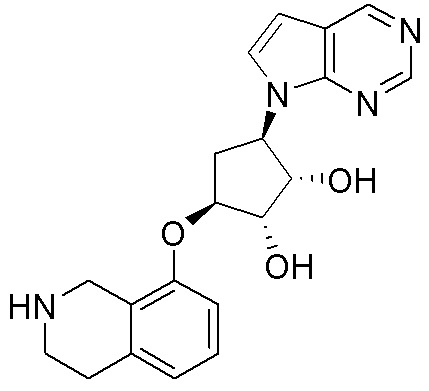
1H ЯМР (400 MГц, ДМСО-d6) δ ч./млн. 9,00 (с, 1H), 8,79 (с, 1H), 7,70 (д, J=3,6 Гц, 1H), 7,12-7,03 (м, 1H), 6,87-6,78 (д, J=8,0, 1H), 6,78-6,62 (м, 2H), 5,39-5,32 (м, 1H), 5,21-5,07 (м, 2H), 4,63-4,51 (м, 2H), 4,02-3,97 (м, 1H), 3,93-3,81 (м, 2H), 3,00-2,91 (м, 2H), 2,90-2,82 (м, 1H), 2,71-2,68 (м, 1H), 2,00-1,89 (м, 1H)
HG-4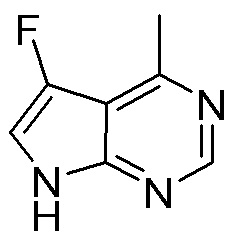
TP-18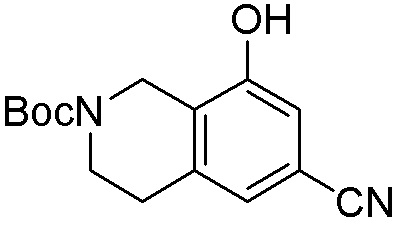
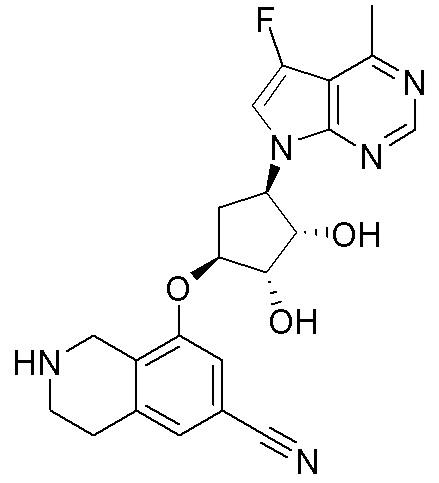
1H ЯМР с муравьиной кислотой (400 MГц, ДМСО-d6) δ ч./млн. 8,66 (с, 1H), 8,28 (с, 1H), 7,71 (с, 1H), 7,28 (с, 1H), 7,20 (с, 1H), 5,14 (кв, J=9,6 Гц, 1H), 4,63-4,60 (м, 1H), 4,45 (дд, J=4,8, 9,0 Гц, 1H), 3,96 (д, J=4,8 Гц, 1H), 3,89 (с, 2H), 2,95-2,90 (м, 2H), 2,88-2,80 (м, 1H), 2,70-2,67 (м, 5H), 1,95-1,90 (м, 1H)
Пример 120 получают по методике NN-5 (Пример 99) с применением подходящего пирролопиримидина на стадии 1 со Схемы BB и подходящего N-Boc защищенного тетрагидроизохинолина на стадии 1 со Схемы NN.
5-бром-4-хлор-7H-пирроло[2.3-d]пиримидин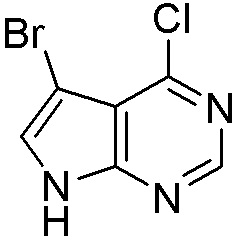
TP-18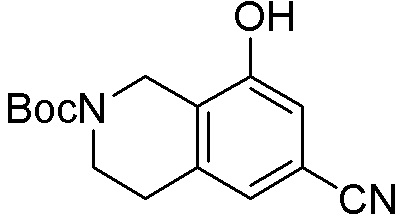
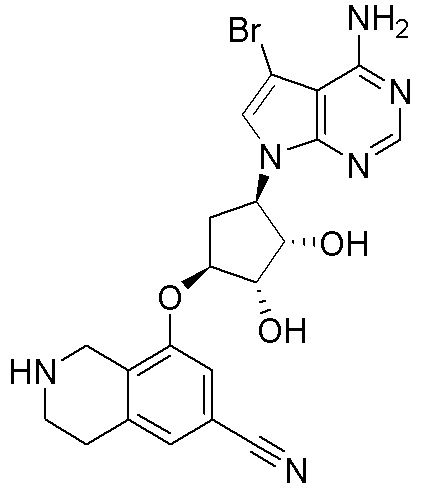
1H ЯМР (400 MГц, MeOD-d4) δ ч./млн. 8,12 (с, 1H), 7,39 (с, 1H), 7,38 (с, 1H), 7,31 (с, 1H), 5,05 (кв, J=9,2 Гц, 1H), 4,81-4,74 (м, 1H), 4,67 (дд, J=4,9, 8,9 Гц, 1H), 4,38 (с, 2H), 4,18 (шд, J=4,3 Гц, 1H), 3,48 (т, J=6,0 Гц, 2H), 3,13 (шт, J=5,9 Гц, 2H), 3,03-2,93 (м, 1H), 2,26-2,19 (м, 1H)
Пример 121 (Схема SS) - (1S,2S,3R,5S)-3-(4-амино-5-метил-7H-пирроло[2.3-d]пиримидин-7-ил)-5-((5-фтор-1,2,3,4-тетрагидроизохинолин-8-ил)окси)циклопентан-1,2-диол (SS-5)
Схема SS
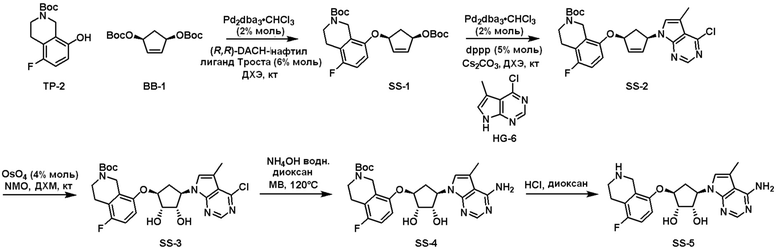
Стадия 1: Синтез трет-бутил 8-(((1S,4R)-4-((трет-бутоксикарбонил)окси)циклопент-2-ен-1-ил)окси)-5-фтор-3,4-дигидроизохинолин-2(1H)-карбоксилата (SS-1)
Пробирка A: В высушенную в печи реакционную пробирку, оборудованную магнитной мешалкой и охлаждаемую в потоке аргона, добавляют продукт присоединения Трис(бензилиденацетон)дипалладия(0) и хлороформа (62 мг, 0,060 ммоль) и MFCD02684551 лиганд Троста (R,R)-DACH-нафтил (142 мг, 0,180 ммоль). Пробирку продувают в вакууме аргоном в динамическом вакууме и добавляют ДХЭ (5,0 мл), который барботируют с аргоном в течение 30 минут. Раствор перемешивают в течение 30 минут при кт с получением ярко-оранжевого раствора лигированного катализатора. На этой стадии получают пробирку B.
Пробирка B: В высушенную в печи реакционную пробирку, оборудованную магнитной мешалкой и охлаждаемую в потоке аргона, добавляют TP-2 (800 мг, 2,99 ммоль), и ди-трет-бутил-((1R,3S)-циклопент-4-ен-1,3-диил)-бис(карбонат) (BB-1) (получен, как описано в J. Am. Chem. Soc, 2006, 128, 6054-6055) (1,08 г, 3,59 ммоль). Пробирку продувают в вакууме аргоном в динамическом вакууме и добавляют ДХЭ (5,0 мл), который барботируют с аргоном в течение 30 минут, затем добавляют содержимое пробирки A через герметичный шприц. Реакционную смесь перемешивают под аргоном при кт в течение 12 часов. Реакционную смесь концентрируют в вакууме и очищают флэш-хроматографией на колонке (24 г SiO2, Isco, 100% Гепт. до 100% EtOAc, 20 мл фракции) с получением SS-1 (1,33 г, >95%) в виде бледно-желтого твердого вещества. ЖХМС [M+H-Boc-изобутилен]=294 наблюдаемый; 1H ЯМР (400 MГц, ХЛОРОФОРМ-d) δ ч./млн. 6,84 (т, J=8,8 Гц, 1H), 6,65 (дд, J=4,3, 8,9 Гц, 1H), 6,20 (тд, J=1,5, 5,7 Гц, 1H), 6,17-6,11 (м, 1H), 5,46 (т, J=5,9 Гц, 1H), 5,09 (т, J=5,6 Гц, 1H), 4,57-4,40 (м, 2H), 3,72-3,54 (м, 2H), 3,02 (тд, J=7,4, 14,5 Гц, 1H), 2,77 (т, J=5,7 Гц, 2H), 1,94 (тд, J=4,5, 14,4 Гц, 1H), 1,50 (д, J=1,6 Гц, 18H).
Стадия 2: Синтез трет-бутил 8-(((1S,4R)-4-(4-хлор-5-метил-7H-пирроло[2.3-d]пиримидин-7-ил)циклопент-2-ен-1-ил)окси)-5-фтор-3,4-дигидроизохинолин-2(1H)-карбоксилата (SS-2)
В сцинтилляционную пробирку, оборудованную магнитной мешалкой, добавляют HG-6 (113 мг, 0,674 ммоль), SS-1 (303 мг, 0,674 ммоль), дифенилфосфинопропан (dppp) (13,9 мг, 0,034 ммоль), продукт присоединения трис(дибензилиденацетон)дипалладия(0) и хлороформа (14 мг, 0,014 ммоль) и карбонат цезия (242 мг, 0,741 ммоль). Пробирку продувают аргоном в динамическом вакууме, затем добавляют ДХЭ (2,25 мл), который барботируют с аргоном в течение 30 минут. Реакционную смесь перемешивают при кт под аргоном в течение 1,5 часов. Реакционную смесь переносят в делительную воронку с ДХМ и разбавляют водой. Фазы разделяют, и водную фазу экстрагируют 2 порциями ДХМ. Объединенные органические фазы сушат (MgSO4), фильтруют и концентрируют в вакууме. Неочищенный остаток очищают флэш-хроматографией на колонке (12 г SiO2, Isco, 100% Гепт. до 100% EtOAc, 9 мл фракции) с получением SS-2 (287,6 мг, 86%) в виде бесцветной камеди. ЖХМС [M+H]=499 наблюдаемый; 1H ЯМР (400 MГц, ХЛОРОФОРМ-d) δ ч./млн. 8,57 (с, 1H), 7,14 (с, 1H), 6,86 (т, J=8,8 Гц, 1H), 6,68 (дд, J=3,7, 8,4 Гц, 1H), 6,37 (д, J=5,4 Гц, 1H), 6,11 (д, J=4,4 Гц, 1H), 6,06-5,96 (м, 1H), 5,33-5,25 (м, 1H), 4,69-4,41 (м, 2H), 3,75-3,55 (м, 2H), 3,22-3,03 (м, 1H), 2,79 (т, J=5,4 Гц, 2H), 2,49 (с, 3H), 1,95 (тд, J=3,6, 14,8 Гц, 1H), 1,51 (с, 9H).
Стадия 3: Синтез трет-бутил 8-(((1S,2S,3S,4R)-4-(4-хлор-5-метил-7H-пирроло[2.3-d]пиримидин-7-ил)-2,3-дигидроксициклопентил)окси)-5-фтор-3,4-дигидроизохинолин-2(1H)-карбоксилата (SS-3)
В сцинтилляционную пробирку, оборудованную магнитной мешалкой и содержащую SS-2 (278 мг, 0,557 ммоль), добавляют ДХМ (2,79 мл). В раствор добавляют 4-Метилморфолин-N-оксид (NMO) (0,13 мл, 0,613 ммоль) в виде 50% масс. раствора в воде, затем по каплям добавляют тетраоксид осмия (100 мкл, 0,022 ммоль) в виде 4% масс. раствора в воде. Реакционную смесь перемешивают при кт в течение 4 часов. Реакционную смесь переносят в делительную воронку с ДХМ, разбавляют водой и затем разбавляют 1M NaHSO3. Фазы разделяют, и водную фазу экстрагируют 3 порциями ДХМ. Объединенные органические экстракты сушат (MgSO4), фильтруют и концентрируют в вакууме. Неочищенный остаток очищают флэш-хроматографией на колонке (12 г SiO2, Isco, 100% Гепт. до 100% EtOAc, 9 мл фракции) с получением SS-3 (169 мг, 57%) в виде белого твердого вещества. ЖХМС [M+H]=533 наблюдаемый; 1H ЯМР (400 MГц, ХЛОРОФОРМ-d) δ ч./млн. 8,49 (с, 1H), 7,09 (шс, 1H), 6,89 (т, J=8,8 Гц, 1H), 6,81 (дд, J=4,2, 8,8 Гц, 1H), 5,03 (кв, J=8,9 Гц, 1H), 4,73 (т, J=5,2 Гц, 1H), 4,55 (дд, J=5,2, 8,1 Гц, 1H), 4,53-4,40 (м, 2H), 4,30 (д, J=4,5 Гц, 1H), 3,73-3,54 (м, 2H), 3,13-2,97 (м, 1H), 2,79 (т, J=5,7 Гц, 2H), 2,49 (с, 3H), 2,31 (ддд, J=4,6, 9,4, 14,1 Гц, 1H), 1,48 (шс, 9H).
Стадия 4: Синтез трет-бутил 8-(((1S,2S,3S,4R)-4-(4-амино-5-метил-7H-пирроло[2.3-d]пиримидин-7-ил)-2,3-дигидроксициклопентил)окси)-5-фтор-3,4-дигидроизохинолин-2(1H)-карбоксилата (SS-4)
В реакционную пробирку, оборудованную магнитной мешалкой, добавляют SS-3 (159 мг, 0,298 ммоль) в виде раствора в диоксане (0,8 мл) и гидроксид аммония (0,8 мл, 5,00 ммоль). Реакционную смесь помещают в микроволновый реактор и нагревают до 120°C в течение 6 часов. Раствор переносят в делительную воронку с ДХМ и разбавляют водой. Фазы разделяют и водную фазу экстрагируют 3 порциями 3:1 смеси ДХМ:ИПС. Объединенные органические экстракты сушат (MgSO4), фильтруют и концентрируют в вакууме. Неочищенный остаток очищают флэш-хроматографией на колонке (12 г SiO2, Isco, 100% Гепт. до 10% MeOH/EtOAc, 9 мл фракции) с получением SS-4 (98,4 мг, 64%) в виде белого твердого вещества. ЖХМС [M+H]=514 наблюдаемый; [α]22D=-70,0° (C=0,1, MeOH); 1H ЯМР (400 MГц, ХЛОРОФОРМ-d) δ ч./млн. 8,13 (с, 1H), 6,98-6,81 (м, 2H), 6,76 (шс, 1H), 5,32 (шс, 2H), 4,93-4,80 (м, 1H), 4,73 (т, J=5,3 Гц, 1H), 4,54 (шс, 1H), 4,44 (д, J=16,8 Гц, 2H), 4,25 (шс, 1H), 3,75-3,51 (м, 2H), 3,11-2,92 (м, 1H), 2,79 (т, J=5,5 Гц, 2H), 2,43 (с, 3H), 2,38-2,25 (м, 1H), 1,48 (шс, 9H).
Стадия 5: Синтез (1S,2S,3R,5S)-3-(4-амино-5-метил-7H-пирроло[2.3-d]пиримидин-7-ил)-5-((5-фтор-1,2,3,4-тетрагидроизохинолин-8-ил)окси)циклопентан-1,2-диола (SS-5)
В сцинтилляционную пробирку, оборудованную магнитной мешалкой и содержащую SS-4 (79,4 мг, 0,155 ммоль), добавляют диоксан (0,4 мл). В раствор добавляют хлористоводородную кислоту (0,4 мл) в виде 4M раствора в диоксане, и реакционную смесь перемешивают при кт в течение 17 часов. Реакцию гасят наполовину насыщенным водным NaHCO3 и переносят в делительную воронку с ДХМ. Фазы разделяют, и водную фазу экстрагируют 3 порциями 3:1 смеси ДХМ/ИПС. Объединенные органические экстракты (MgSO4), фильтруют и концентрируют в вакууме. Выделенный продукт растворяют в минимальном количестве метанола и разбавляют водой. Образец замораживают и лиофилизируют в течение ночи с получением SS-5 (51,3 мг, 80%) в виде белого твердого вещества. ЖХМС [M+H] 414 наблюдаемый; 1H ЯМР (400 MГц, МЕТАНОЛ-d4) δ ч./млн. 8,03 (с, 1H), 6,98 (д, J=1,0 Гц, 1H), 6,93-6,81 (м, 2H), 5,07 (кв, J=8,7 Гц, 1H), 4,62 (ддд, J=1,8, 4,0, 7,0 Гц, 1H), 4,51 (дд, J=5,0, 8,5 Гц, 1H), 4,17-4,11 (м, 1H), 3,98 (с, 2H), 3,10 (т, J=6,0 Гц, 2H), 2,93 (ддд, J=7,3, 9,3, 14,5 Гц, 1H), 2,77 (т, J=5,9 Гц, 2H), 2,43 (д, J=1,1 Гц, 3H), 1,99 (ддд, J=4,0, 8,4, 13,8 Гц, 1H); 19F ЯМР (376 MГц, МЕТАНОЛ-d4) d=-131,36 (с, 1F).
Примеры 122-129 получают по методике SS-5 (Пример 121) с применением подходящего N-Boc защищенного тетрагидроизохинолина на стадии 1 S и подходящего пирролопиримидина на стадии 2 Схемы SS.
TP-5
4-хлор-5-метил-7H-пирроло[2.3-d]пиримидин
1H ЯМР (400 MГц, MeOD-d4) δ ч./млн. 8,05 (с, 1 H), 6,99 (с, 1 H), 6,70 (с, 1 H), 6,61 (с, 1 H), 5,17-5,06 (м, 1 H), 4,71-4,64 (м, 1 H), 4,53 (дд, J=8,53, 4,77 Гц, 1 H), 4,16 (д, J=4,27 Гц, 1 H), 3,99 (с, 2 H), 3,12 (д, J=5,90 Гц, 2 H), 2,97 (дд, J=14,50, 9,50, 7,20 Гц, 1 H), 2,83 (т, J=5,77 Гц, 2 H), 2,45 (с, 3 H), 2,30 (с, 3 H), 2,04-1,93 (м, 1 H)
TP-5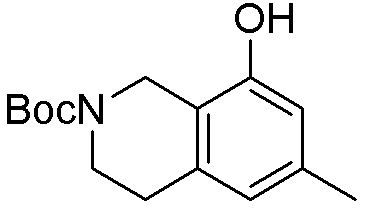
5-бром-4-хлор-7H-пирроло[2.3-d]пиримидин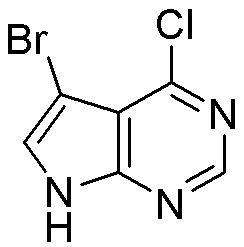
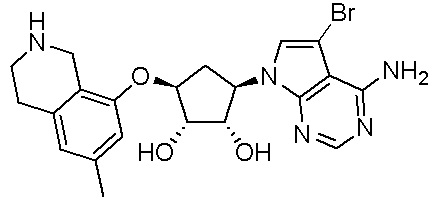
1H ЯМР (400 MГц, MeOD-d4) δ ч./млн. 8,12 (с, 1H), 7,35 (с, 1H), 6,77 (с, 1H), 6,67 (с, 1H), 5,11 (кв, J=9,0 Гц, 1H), 4,69 (ддд, J=1,6, 4,0, 7,2 Гц, 1H), 4,62 (дд, J=4,8, 8,8 Гц, 1H), 4,18-4,13 (м, 3H), 3,31-3,27 (м, 2H), 3,03-2,92 (м, 3H), 2,33 (с, 3H), 2,14-2,06 (м, 1H)
TP-5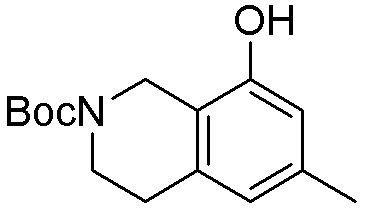
HG-4 (стадия 4 пропущена)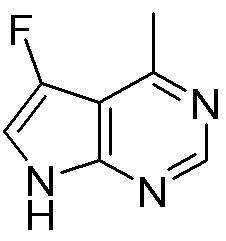
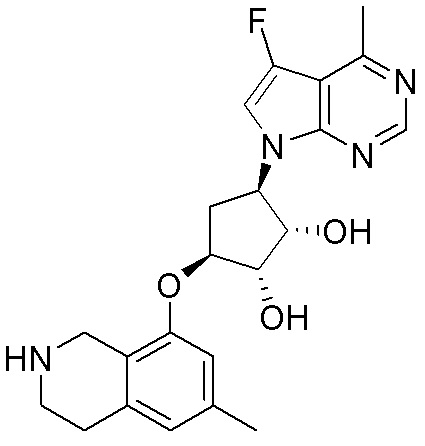
M+1
1H ЯМР (400 MГц, MeOD-d4) δ ч./млн. 8,65 (с, 1H), 7,43 (д, J=2,0 Гц, 1H), 6,81 (с, 1H), 6,70 (с, 1H), 5,25 (кв, J=9,0 Гц, 1H), 4,71 (дт, J=2,3, 3,6 Гц, 1H), 4,69-4,64 (м, 1H), 4,26 (с, 2H), 4,17 (д, J=4,8 Гц, 1H), 3,39 (т, J=6,1 Гц, 2H), 3,04-2,95 (м, 3H), 2,80 (с, 3H), 2,34 (с, 3H), 2,20-2,11 (м, 1H)
TP-6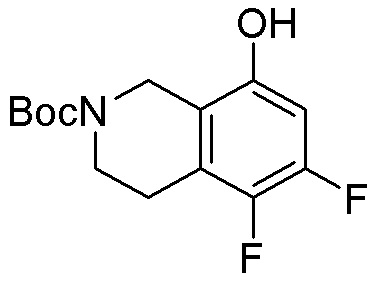
5-бром-4-хлор-7H-пирроло[2.3-d]пиримидин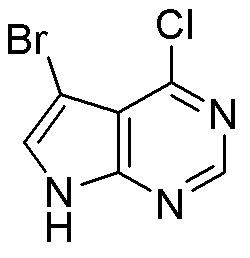
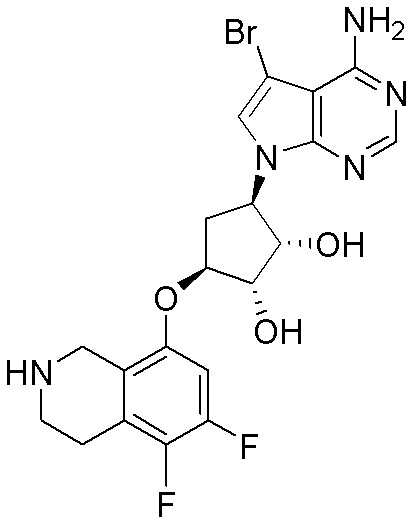
1H ЯМР (400 MГц, MeOD-d4) δ ч./млн. 8,10 (с, 1H), 7,35 (с, 1H), 6,87 (дд, J=6,8, 12,5 Гц, 1H), 5,09 (кв, J=8,8 Гц, 1H), 4,58-4,51 (м, 2H), 4,13 (д, J=5,0 Гц, 1H), 3,90 (с, 2H), 3,12-3,01 (м, 2H), 3,00-2,90 (м, 1H), 2,78 (т, J=5,8 Гц, 2H), 2,13-2,00 (м, 1H)
TP-2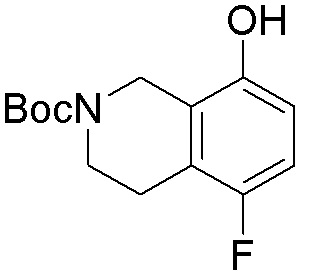
HG-5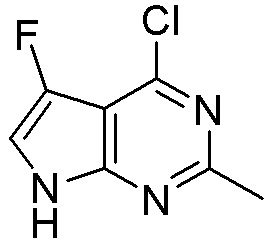
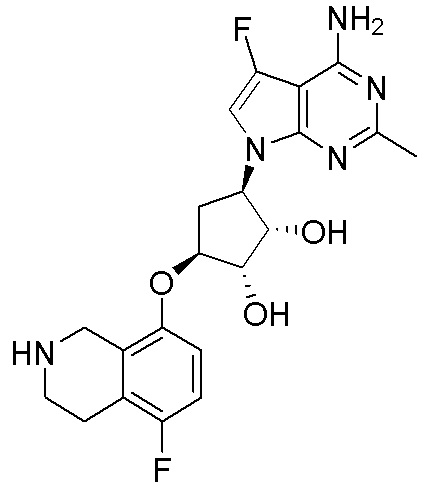
1H ЯМР (400 MГц, ДМСО-d6) δ ч./млн. 9,07 (шс, 2H), 7,27 (шс, 1H), 7,15 (т, J=9,2 Гц, 1H), 6,99 (дд, J=4,1, 9,2 Гц, 1H), 5,38 (шс, 1H), 5,18 (шс, 1H), 5,08 (кв, J=9,0 Гц, 1H), 4,53 (шс, 1H), 4,39-4,30 (м, 1H), 4,21 (шс, 2H), 3,95 (шс, 1H), 2,95-2,88 (м, 2H), 2,83-2,73 (м, 1H), 2,53-2,51 (м, 3H), 2,40 (с, 3H), 1,74 (ддд, J=4,4, 9,3, 13,7 Гц, 1H)
TP-5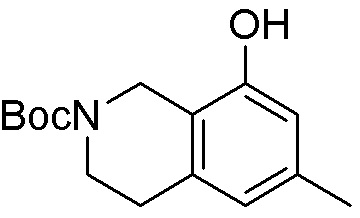
HG-3 (стадия 4 пропущена)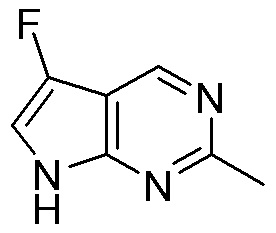
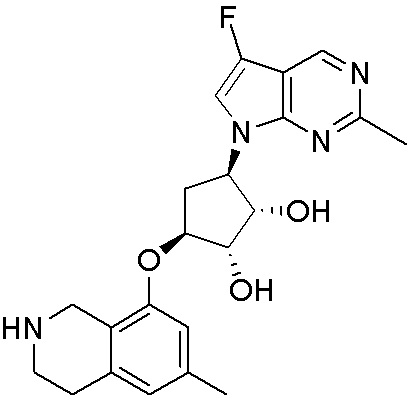
1H ЯМР (400 MГц, MeOD-d4) δ ч./млн. 8,90 (с, 1H), 7,39 (с, 1H), 6,76 (с, 1H), 6,66 (с, 1H), 5,35 (кв, J=9,2 Гц, 2H), 4,58 (дд, J=4,9, 8,7 Гц, 1H), 4,17 (д, J=4,3 Гц, 1H), 4,12 (с, 2H), 3,25 (т, J=6,0 Гц, 2H), 3,06-2,95 (м, 1H), 2,92 (т, J=5,9 Гц, 2H), 2,74 (с, 3H), 2,33 (с, 3H), 2,13-2,04 (м, 1H)
TP-6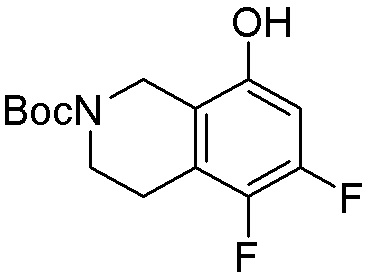
4,5-дихлор-7H-пирроло[2.3-d]пиримидин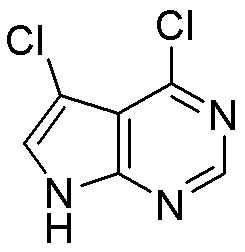
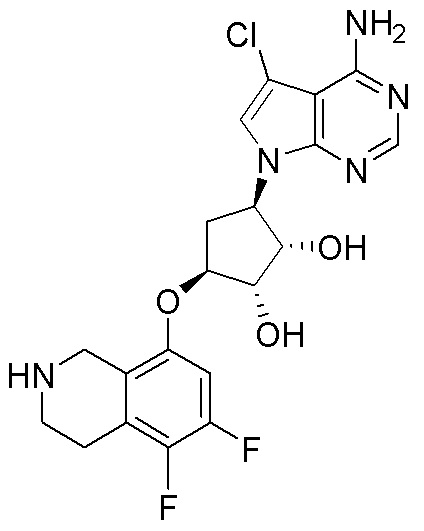
1H ЯМР (400 MГц, MeOD-d4) δ ч./млн. 8,09 (с, 1H), 7,29 (с, 1H), 6,89 (дд, J=6,7, 12,4 Гц, 1H), 5,15-5,01 (м, 1H), 4,60-4,50 (м, 2H), 4,16-4,07 (м, 1H), 3,94 (с, 2H), 3,11 (т, J=6,0 Гц, 2H), 2,99-2,88 (м, 1H), 2,81 (т, J=5,8 Гц, 2H), 2,12-2,02 (м, 1H)
TP-5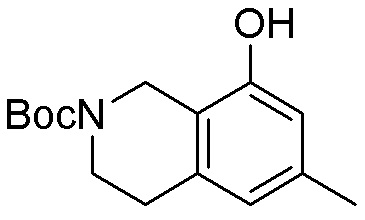
4.5-дихлор-7H-пирроло[2.3-d]пиримидин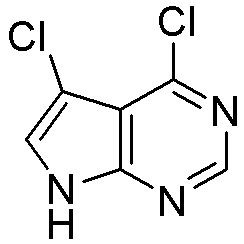
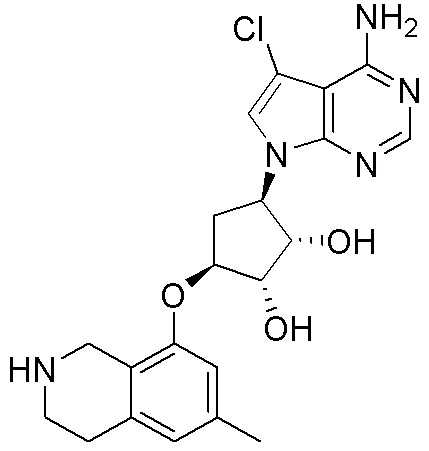
1H ЯМР (400 MГц, MeOD-d4) δ ч./млн. 8,12 (с, 1H), 7,30 (с, 1H), 6,83 (с, 1H), 6,72 (с, 1H), 5,08 (кв, J=9,0 Гц, 1H), 4,74-4,68 (м, 1H), 4,64 (дд, J=5,0, 8,8 Гц, 1H), 4,28 (с, 2H), 4,17 (д, J=4,8 Гц, 1H), 3,44 (т, J=6,1 Гц, 2H), 3,05 (т, J=6,1 Гц, 2H), 2,98 (ддд, J=7,4, 9,1, 14,4 Гц, 1H), 2,35 (с, 3H), 2,12 (ддд, J=4,0, 9,3, 14,0 Гц, 1H)
Примеры 130-132 получают с применением методики, изображенной на схеме SS с применением подходящего тетрагидроизохинолина на стадии 1 и пирролопиримидина на стадии 2. В модификации стадии 5, трифторуксусную кислоту (ТФК) применяют для снятия защиты.
Пример 130 (Схема TT) - (1S,2S,3R,5S)-3-(4-амино-5-хлор-7H-пирроло[2.3-d]пиримидин-7-ил)-5-((6-хлор-1,2,3,4-тетрагидроизохинолин-8-ил)окси)циклопентан-1,2-диол (TT-2)
Схема TT
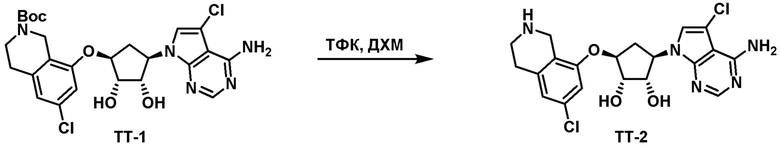
К охлаждаемому раствору TT-1 (50,0 мг, 0,091 ммоль) в ДХМ (2,0 мл) добавляют ТФК (0,50 мл). Желтый раствор перемешивают при 15°C в течение 1 часа. Реакционную смесь концентрируют в вакууме с получением неочищенного продукта, и pH раствора доводят до 7~8 с применением насыщенного водн. NaHCO3 (2 мл). Затем к реакционному раствору добавляют ФА (1%) водн. (2 мл). Смесь очищают преп.-ВЭЖХ, и желаемые фракции объединяют и лиофилизируют с получением TT-2 (35,0 мг, 86%) в виде белого твердого вещества.
TP-3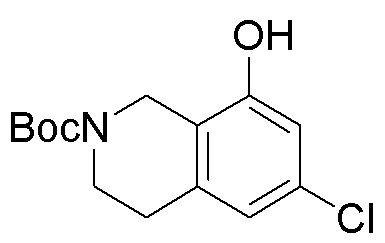
4,5-дихлор-7H-пирроло[2.3-d]пиримидин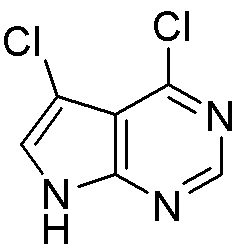
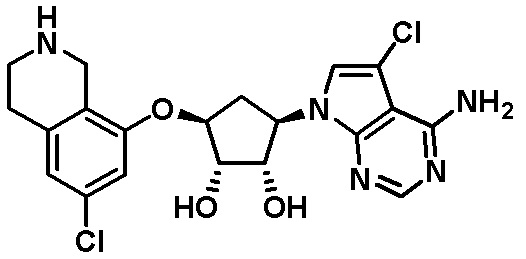
1H ЯМР (400 MГц, ДМСО-d6) δ ч./млн. 8,04 (с, 1H), 7,38 (с, 1H), 7,01-6,89 (м, 2H), 4,92 (кв, J=9,0 Гц, 1H), 4,56 (д, J=5,3 Гц, 1H), 4,47 (дд, J=4,9, 9,2 Гц, 1H), 4,13 (с, 2H), 3,95 (д, J=4,8 Гц, 1H), 3,29 (т, J=6,1 Гц, 2H), 2,94 (т, J=5,9 Гц, 2H), 2,86-2,76 (м, 1H), 1,89 (ддд, J=3,9, 9,5, 13,7 Гц, 1H)
TP-3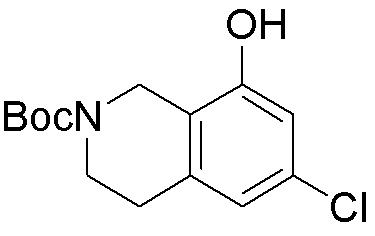
5-бром-4-хлор-7H-пирроло[2.3-d]пиримидин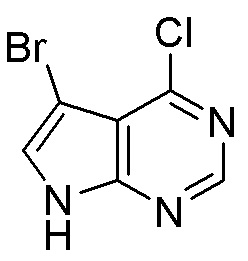
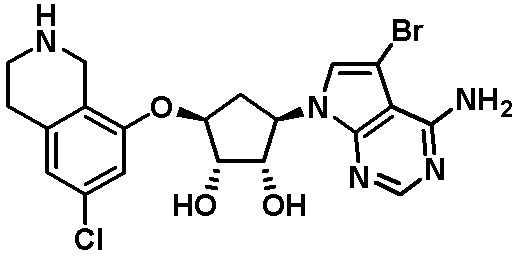
1H ЯМР (400 MГц, ДМСО-d6) δ ч./млн. 8,10 (с, 1H), 7,53 (с, 1H), 6,92 (д, J=1,5 Гц, 1H), 6,85-6,57 (м, 2H), 5,36 (шс, 1H), 5,16 (шс, 1H), 4,99 (кв, J=9,2 Гц, 1H), 4,58-4,51 (м, 1H), 4,50-4,42 (м, 1H), 3,93 (д, J=5,0 Гц, 1H), 3,79 (с, 2H), 2,91 (т, J=5,8 Гц, 2H), 2,84-2,73 (м, 1H), 2,70-2,62 (м, 2H), 1,96-1,84 (м, 1H)
TP-2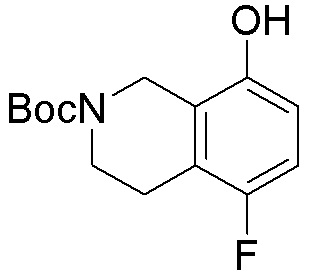
HG-3 (Стадия 4 пропущена)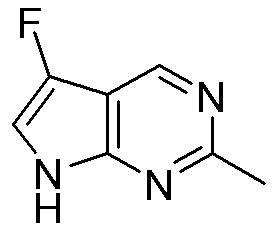
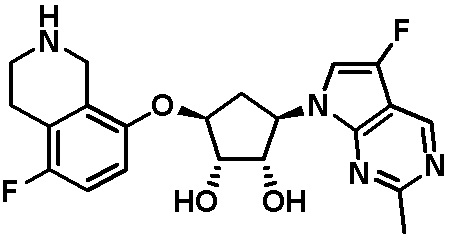
1H ЯМР (400 MГц, МЕТАНОЛ-d4) δ ч./млн. 8,87 (д, J=0,8 Гц, 1H), 7,38 (д, J=2,3 Гц, 1H), 6,92-6,81 (м, 2H), 5,37-5,27 (м, 1H), 4,65-4,60 (м, 1H), 4,56 (т, J=8,9 Гц, 1H), 4,14 (д, J=5,0 Гц, 1H), 3,96 (с, 2H), 3,10-3,04 (м, J=6,0, 6,0 Гц, 2H), 3,01-2,91 (м, J=7,2, 9,4, 14,6 Гц, 1H), 2,79-2,73 (м, J=6,0, 6,0 Гц, 2H), 2,72 (с, 3H), 2,11-2,03 (м, 1H)
Примеры 133 & 134 получают с применением методики, изображенной на схеме SS с применением подходящего тетрагидроизохинолина на стадии 1 и пирролопиримидина на стадии 2. В модификации общей методики стадии 4, ГОБТ применяют в качестве катализатора, как описано на схеме UU.
Синтез трет-бутил 8-(((1S,2S,3S,4R)-4-(4-амино-5-хлор-7H-пирроло[2.3-d]пиримидин-7-ил)-2,3-дигидроксициклопентил)окси)-5-фтор-3,4-дигидроизохинолин-2(1H)-карбоксилата (UU-2)
Схема UU
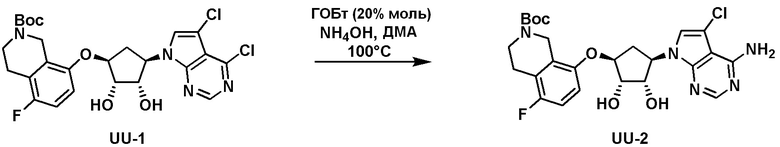
В реакционную пробирку, оборудованную магнитной мешалкой, добавляют UU-1 (147 мг, 0,266) в виде раствора в ДМА (2,5 мл), затем добавляют ГОБТ.гидрат (8,13 мг, 0,053 ммоль). В раствор добавляют гидроксид аммония (1,0 мл, 7,96 ммоль). Пробирку герметично закрывают тефлоновой крышкой и помещают в нагревательный блок. Реакционную смесь нагревают при 100°C в течение 19 часов. Раствор переносят в делительную воронку с ДХМ и разбавляют водой. Фазы разделяют, и водную фазу экстрагируют 3 порциями ДХМ. Объединенные органические экстракты сушат (MgSO4), фильтруют и концентрируют в вакууме. Неочищенный остаток очищают флэш-хроматографией на колонке (4 г SiO2, Isco, 100% Гепт. до 10% MeOH/EtOAc, 9 мл фракции). Фракции, содержащие продукт, разделяют и концентрируют в вакууме. Продукт лиофилизируют с получением продукта, содержащего минимальные примеси, в виде беловатого твердого вещества (140,7 мг, 99%). Продукт применяют на стадии 5, применяя ТФК/ДХМ для снятия защиты, как изображено на схеме TT, без дальнейшей очистки.
TP-2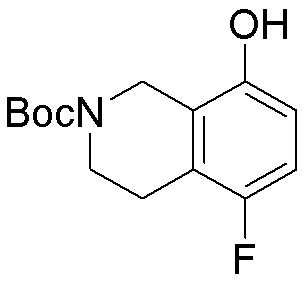
4,5-дихлор-7H-пирроло[2.3-d]пиримидин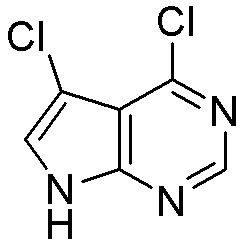
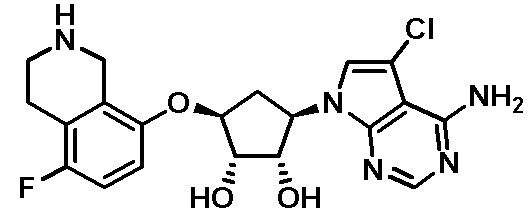
[M+H]
1H ЯМР (700 MГц, ДМСО-d6) δ ч./млн. 8,08 (шс, 1H), 7,44 (с, 1H), 6,97-6,89 (м, 1H), 6,82 (д, J=4,6 Гц, 1H), 5,26 (шс, 1H), 5,11 (шс, 1H), 4,99 (кв, J=8,6 Гц, 1H), 4,55-4,39 (м, 2H), 3,94 (шс, 1H), 3,83 (шс, 2H), 2,93 (шс, 2H), 2,84-2,70 (м, 2H), 2,58 (шс, 2H), 1,92-1,82 (м, 1H).
TP-2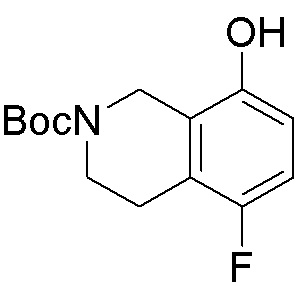
5-бром-4-хлор-7H-пирроло[2.3-d]пиримидин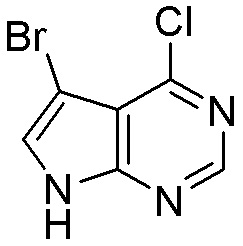
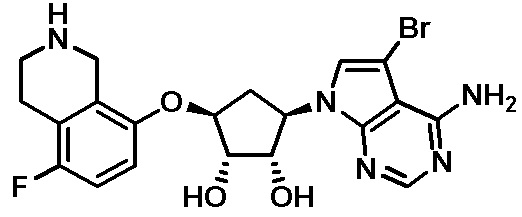
1H ЯМР (700MГц, ДМСО-d6) δ ч./млн. (с, 1H), 7,49 (с, 1H), 6,94-6,88 (м, 1H), 6,84-6,77 (м, 1H), 5,25 (шс, 1H), 5,15-5,05 (м, 1H), 4,99 (д, J=9,0 Гц, 1H), 4,47 (д, J=3,3 Гц, 2H), 3,94 (шс, 1H), 3,81 (шс, 2H), 2,92 (шс, 2H), 2,80-2,70 (м, 1H), 2,58 (шс, 2H), 1,86 (шс, 1H).
Пример 135 (Схема VV) - (1S,2S,3S,5R)-3-(4-фтор-2-(гидроксиметил)фенокси)-5-(4-метил-7H-пирроло[2.3-d]пиримидин-7-ил)циклопентан-1,2-диол (2)
Схема VV
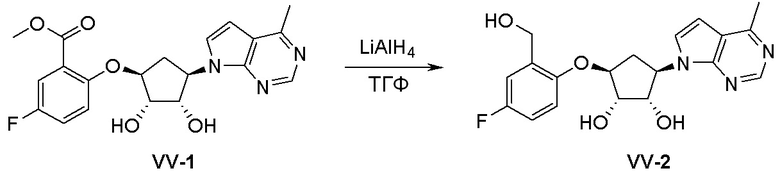
Соединение VV-1 получают с применением методик со стадий 2 и 3 со Схемы BB, начиная с BB-2, и с применением коммерчески доступного метил 5-фтор-2-гидроксибензоата. К раствору VV-1 (90 мг, 0,22 ммоль) в сухом ТГФ (5 мл) добавляют LiAlH4 (30 мг, 0,79 ммоль) при 0°C в течение 2 часов. H2O (30 мл) добавляют к реакционной смеси, и смесь экстрагируют EtOAc (30 мл×4). Органический слой промывают насыщенным раствором соли, сушат над Na2SO4 и концентрируют в вакууме до остатка, который очищают преп.-ТСХ с получением VV-2 (60 мг, 72%) в виде белого твердого вещества. ЖХМС 374 [M+1]; 1H ЯМР (400 MГц, ДМСО-d6) δ ч./млн. 8,62 (с, 1H), 7,65 (д, J=3,8 Гц, 1H), 7,18 (д, J=9,0 Гц, 1H), 7,04-6,99 (м, 2H), 6,72 (д, J=3,5 Гц, 1H), 5,32 (д, J=3,8 Гц, 1H), 5,23 (т, J=5,6 Гц, 1H), 5,16-5,07 (м, 2H), 4,58 (д, J=5,8 Гц, 2H), 4,56-4,50 (м, 2H), 3,99 (шс, 1H), 2,88-2,78 (м, 1H), 2,64 (с, 3H), 1,97-1,87 (м, 1H)
Пример 136 (Схема WW) - (1S,2S,3S,5R)-3-(4-фтор-2-гидроксифенокси)-5-(4-метил-7H-пирроло[2.3-d]пиримидин-7-ил)циклопентан-1,2-диол (WW-6)
Схема WW
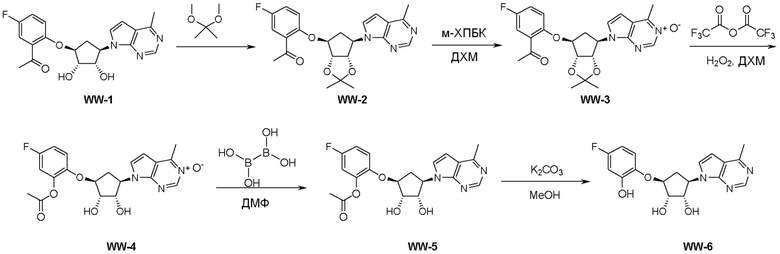
Стадия 1 - Синтез 1-(2-(((3aR,4S,6R,6aS)-2,2-диметил-6-(4-метил-7H-пирроло[2.3-d]пиримидин-7-ил)тетрагидро-4H-циклопента[d][1.3]диоксол-4-ил)окси)-5-фторфенил)этан-1-она (WW-2)
Соединение WW-1 получают с применением методик со стадий 2 и 3 со Схемы BB, начиная с BB-2 и с применением коммерчески доступного 1-(5-фтор-2-гидроксифенил)этан-1-она. К перемешиваемой белой суспензии WW-1 (1,06 г, 2,75 ммоль) в ацетоне (6 мл) добавляют 2,2-диметоксипропан (16 мл) и п-толуолсульфоновую кислоту (523 мг, 2,75 ммоль) при кт (25°C). Реакционную смесь перемешивают при 25°C в течение 15 ч. Водный NaHCO3 добавляют к реакционной смеси до pH 8,0. Затем смесь экстрагируют EtOAc (15 мл×5). Органические слои разделяют, сушат и выпаривают с получением неочищенного продукта, который очищают хроматографией, элюируя MeOH/ДХМ 0-5% с получением WW-2 (1,06 г, 91%) в виде белого твердого вещества. Продукт применяют сразу на следующей стадии.
Стадия 2 - Синтез 3-оксида 7-((3aS,4R,6S,6aR)-6-(2-ацетил-4-фторфенокси)-2,2-диметилтетрагидро-4H-циклопента[d][1.3]диоксол-4-ил)-4-метил-7H-пирроло[2.3-d]пиримидина (WW-3)
Соединение WW-2 (1,03 г, 2,42 ммоль) растворяют в ДХМ (15 мл). Затем м-ХПБК (1,97 г, 9,68 ммоль) добавляют в смесь. Реакционную смесь нагревают при 40°C в течение 16 часов. Смесь охлаждают до 25°C.Затем смесь разбавляют ДХМ (15 мл) и затем добавляют насыщенный тиосульфат натрия (15 мл), органический слой разделяют. Органический слой промывают насыщенным NaHCO3 (20 мл), сушат над Na2SO4, фильтруют и очищают флэш-хроматографией, элюируя 0-10% MeOH/ДХМ с получением WW-3 (600 мг, 56%) в виде желтого масла и применяют сразу на следующей стадии.
Стадия 3 - Синтез 7-((1R,2S,3S,4S)-4-(2-ацетокси-4-фторфенокси)-2,3-дигидроксициклопентил)-4-метил-7H-пирроло[2.3-d]пиримидин 3-оксида (WW-4)
Трифторуксусный ангидрид (3,56 г, 16,9 ммоль) охлаждают до -10°C в течение около 10-20 мин и 30% H2O2 (457 мг, 3,13 мл) добавляют по каплям и перемешивают в течение 10 мин (сохраняя температуру от 0 до -10°C). К этой смеси добавляют соединение WW-3 (570 мг, 1,29 ммоль) в ДХМ (5 мл) по каплям и перемешивают при 25°C в течение 10-30 мин. Насыщенный Na2S2O3/NaHCO3 водн. (15 мл) добавляют в смесь и перемешивают при 25°C в течение 10 мин. Реакционный раствор экстрагируют ДХМ (10 мл×2), сушат и выпаривают с получением неочищенного продукта WW-4 (540 мг, >99%) в виде желтого масла. ЖХМС 418 [M+1]
Стадия 4 - Синтез ацетата 2-(((1S,2S,3S,4R)-2,3-дигидрокси-4-(4-метил-7H-пирроло[2.3-d]пиримидин-7-ил)циклопентил)окси)-5-фторфенила (WW-5)
Соединение WW-4 (540 мг, 1,22 ммоль) растворяют в ДМФ (1 мл). Затем гидродиборную кислоту (658 мг, 7,34 ммоль) добавляют к смеси и перемешивают при 25°C в течение 30 мин. Реакционный раствор WW-5 применяют сразу на следующей стадии без дальнейшей очистки. ЖХМС 402 [M+1]
Стадия 5 - Синтез (1S,2S,3S,5R)-3-(4-фтор-2-гидроксифенокси)-5-(4-метил-7H-пирроло[2.3-d]пиримидин-7-ил)циклопентан-1,2-диола (WW-6)
Раствор соединения WW-5 (490 мг, 1,11 ммоль) в ДМФ добавляют MeOH (2 мл). K2CO3 (2,30 г, 16,6 ммоль) добавляют в смесь. Реакционную смесь перемешивают при 25°C в течение 2 часов. Растворитель выпаривают, и неочищенный продукт очищают преп.-ВЭЖХ с получением WW-6 (62 мг, 16%) в виде белого твердого вещества. ЖХМС 360 [M+1]; 1H ЯМР (400 MГц, ДМСО-d6) δ ч./млн. 9,53 (с, 1H), 8,63 (с, 1H), 7,75 (д, J=3,5 Гц, 1H), 7,01 (дд, J=5,9, 8,9 Гц, 1H), 6,75 (д, J=3,8 Гц, 1H), 6,68 (д, J=3,0 Гц, 1H), 6,66 (д, J=3,0 Гц, 1H), 6,56 (дт, J=3,0, 8,7 Гц, 1H), 5,26 (д, J=3,0 Гц, 1H), 5,16 (кв, J=8,9 Гц, 1H), 5,07 (д, J=6,8 Гц, 1H), 4,54 (шс, 2H), 4,00 (шс, 1H), 2,83-2,74 (м, 1H), 2,65 (с, 3H), 1,97-1,90 (м, 1H)
Пример 137 (Схема XX) - (1S,2S,3S,5R)-3-(2-((R)-1-аминоэтил)-4-фторфенокси)-5-(4-метил-7H-пирроло[2.3-d]пиримидин-7-ил)циклопентан-1,2-диол (XX-3)
Пример 138 (Схема XX) - (1S,2S,3S,5R)-3-(2-((S)-1-аминоэтил)-4-фторфенокси)-5-(4-метил-7H-пирроло[2.3-d]пиримидин-7-ил)циклопентан-1,2-диол (XX-4)
Схема XX
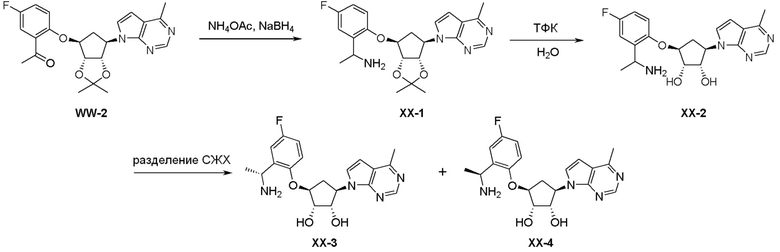
Стадия 1 - Синтез 1-(2-(((3aR,4S,6R,6aS)-2,2-диметил-6-(4-метил-7H-пирроло[2.3-d]пиримидин-7-ил)тетрагидро-4H-циклопента[d][1.3]диоксол-4-ил)окси)-5-фторфенил)этан-1-амина (XX-1)
Соединение WW-2 (100 мг, 0,235 ммоль) растворяют в MeOH (1 мл). Затем NH4OAc (181 мг, 2,35 ммоль) добавляют в смесь и перемешивают при 25°C в течение 1 часа. Затем добавляют NaBH3CN (89 мг, 1,41 ммоль) и нагревают до 80°C в течение 16 часов. Смесь охлаждают до 25°C, затем добавляют 1 мл насыщенного NaHCO3 и перемешивают при 25°C в течение 10 мин. Реакционную смесь экстрагируют ДХМ (5 мл×3), разделяют, сушат и выпаривают с получением неочищенного продукта, который очищают преп.-ТСХ (MeOH/ДХМ 0-10%) с получением XX-1 (35 мг, 35%) в виде бесцветного масла и применяют сразу на следующей стадии.
Стадия 2 - Синтез (1S,2S,3S,5R)-3-(2-(1-аминоэтил)-4-фторфенокси)-5-(4-метил-7H-пирроло[2.3-d]пиримидин-7-ил)циклопентан-1,2-диола (XX-2)
К соединению XX-1 (100 мг, 0,234 ммоль) в H2O (0,5 мл) добавляют ТФК (0,5 мл) при 0°C. Смесь перемешивают при 0°C в течение 60 мин. pH реакционного раствора доводят до pH=9 добавлением насыщенного водн. K2CO3. Конечный раствор разделяют преп-ВЭЖХ с получением 90 мг XX-2 (90 мг, 99%) в виде белого твердого вещества.
Стадия 3 - Разделение (1S,2S,3S,5R)-3-(2-((R)-1-аминоэтил)-4-фторфенокси)-5-(4-метил-7H-пирроло[2.3-d]пиримидин-7-ил)циклопентан-1,2-диола (XX-3) и (1S,2S,3S,5R)-3-(2-((S)-1-аминоэтил)-4-фторфенокси)-5-(4-метил-7H-пирроло[2.3-d]пиримидин-7-ил)циклопентан-1,2-диола (XX-4)
Разделяют соединение XX-2 хиральной СЖХ
XX-3: ЖХМС 370 [M-NH2]; 1H ЯМР (400 MГц, ДМСО-d6) δ ч./млн. 8,61 (с, 1H), 7,61 (д, J=3,7 Гц, 1H), 7,29 (дд, J=3,0, 10,0 Гц, 1H), 7,03-6,90 (м, 2H), 6,72 (д, J=3,7 Гц, 1H), 5,34 (шс, 1H), 5,18-5,01 (м, 2H), 4,61-4,49 (м, 2H), 4,33 (кв, J=6,4 Гц, 1H), 4,01 (д, J=4,5 Гц, 1H), 2,94-2,79 (м, 1H), 2,64 (с, 3H), 2,00 (ддд, J=4,0, 9,4, 13,8 Гц, 2H), 1,25 (д, J=6,5 Гц, 3H)
XX-4: ЖХМС 370 [M-NH2]; 1H ЯМР (400 MГц, ДМСО-d6) δ ч./млн. 8,62 (с, 1H), 7,60 (д, J=3,7 Гц, 1H), 7,29 (дд, J=3,0, 10,0 Гц, 1H), 7,04-6,87 (м, 2H), 6,71 (д, J=3,5 Гц, 1H), 5,35 (шс, 1H), 5,20-5,03 (м, 2H), 4,63-4,46 (м, 2H), 4,33 (кв, J=6,6 Гц, 1H), 4,02 (д, J=4,3 Гц, 1H), 2,91-2,78 (м, 1H), 2,64 (с, 3H), 2,04-1,91 (м, 2H), 1,26 (д, J=6,6 Гц, 3H)
Примеры 139-142 получают по методике получения XX-3 & XX-4 начиная с BB-2 и подходящего фенольного кетона. Начинают со стадий 2 (аллильное алкилирование) & 3 (дигидроксилирование) со Схемы BB, затем стадия 1 (получение ацетонида) со Схемы WW, и затем стадии 1 (восстановительное аминирование), 2 (снятие защиты) и 3 (хиральное разделение) со схемы XX.
1-(4-хлор-5-фтор-2-гидроксифенил)этан-1-он
[M+1]
1H ЯМР (400 MГц, MeOD-d4) δ ч./млн. 8,61 (с, 1H), 7,55 (д, J=3,5 Гц, 1H), 7,31 (д, J=10,0 Гц, 1H), 7,20 (д, J=6,3 Гц, 1H), 6,75 (д, J=3,5 Гц, 1H), 5,16 (кв, J=9,0 Гц, 1H), 4,80-4,73 (м, 1H), 4,72-4,64 (м, 1H), 4,49-4,39 (м, 1H), 4,22 (д, J=5,0 Гц, 1H), 3,04-2,92 (м, 1H), 2,72 (с, 3H), 2,36-2,25 (м, 1H), 1,44 (д, J=6,8 Гц, 3H)
1-(4-хлор-5-фтор-2-гидроксифенил)этан-1-он
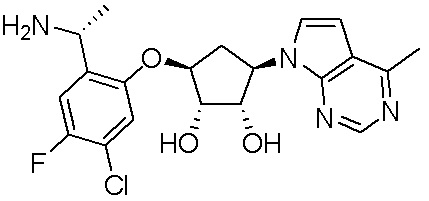
[M+1]
1H ЯМР (400 MГц, MeOD-d4) δ ч./млн. 8,62 (с, 1H), 7,56 (д, J=3,5 Гц, 1H), 7,31 (д, J=10,0 Гц, 1H), 7,19 (д, J=6,0 Гц, 1H), 6,75 (д, J=3,8 Гц, 1H), 5,17 (кв, J=9,0 Гц, 1H), 4,79 (дд, J=5,0, 8,8 Гц, 1H), 4,69 (шс, 1H), 4,42 (кв, J=6,8 Гц, 1H), 4,20 (д, J=4,8 Гц, 1H), 3,06-2,93 (м, 1H), 2,72 (с, 3H), 2,41-2,28 (м, 1H), 1,45 (д, J=6,8 Гц, 3H)
7-гидрокси-2,3-дигидро-1H-инден-1-он
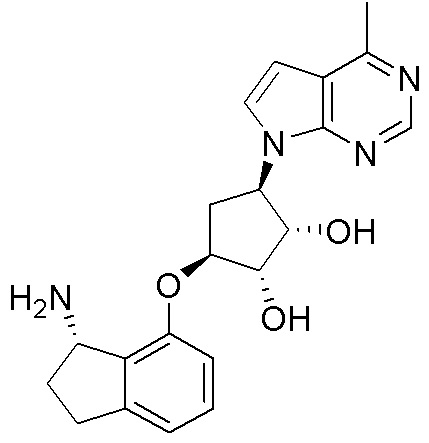
[M-NH2]
1H ЯМР (400 MГц, ДМСО-d6) δ ч./млн. 8,63 (с, 1H), 7,67 (д, J=3,5 Гц, 1H), 7,21-7,06 (м, 1H), 6,86-6,77 (м, 2H), 6,71 (д, J=3,5 Гц, 1H), 5,40 (шс, 1H), 5,15-5,04 (м, 2H), 4,65-4,57 (м, 2H), 4,51-4,45 (м, 1H), 4,04 (д, J=4,5 Гц, 1H), 3,03-2,93 (м, 1H), 2,90-2,79 (м, 1H), 2,70 (ддд, J=5,8, 9,1, 15,5 Гц, 1H), 2,64 (с, 3H), 2,34-2,22 (м, 1H), 2,15-1,81 (м, 2H), 1,76-1,65 (м, 1H)
7-гидрокси-2,3-дигидро-1H-инден-1-он
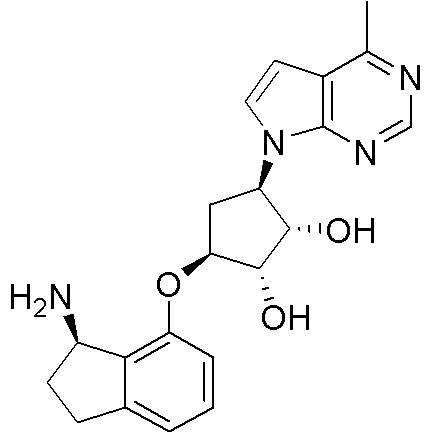
[M-NH2]
1H ЯМР (400 MГц, ДМСО-d6) δ ч./млн. 8,60 (с, 1H), 7,70 (д, J=3,5 Гц, 1H), 7,12 (т, J=7,8 Гц, 1H), 6,86-6,75 (м, 2H), 6,69 (с, 1H), 5,37 (шс, 1H), 5,23-5,03 (м, 2H), 4,66-4,55 (м, 2H), 4,48 (дд, J=4,5, 7,5 Гц, 1H), 4,02 (д, J=4,3 Гц, 1H), 3,02-2,93 (м, 1H), 2,90-2,80 (м, 1H), 2,76-2,66 (м, 1H), 2,66-2,59 (м, 3H), 2,28 (дт, J=8,2, 13,6 Гц, 1H), 2,06-1,97 (м, 1H), 1,77-1,64 (м, 1H)
Пример 143 (Схема YY) - (1S,2S,3S,5R)-3-(2-(аминометил)-4-хлор-3-фторфенокси)-5-(4-метил-7H-пирроло[2.3-d]пиримидин-7-ил)циклопентан-1,2-диол (YY-6)
Схема YY
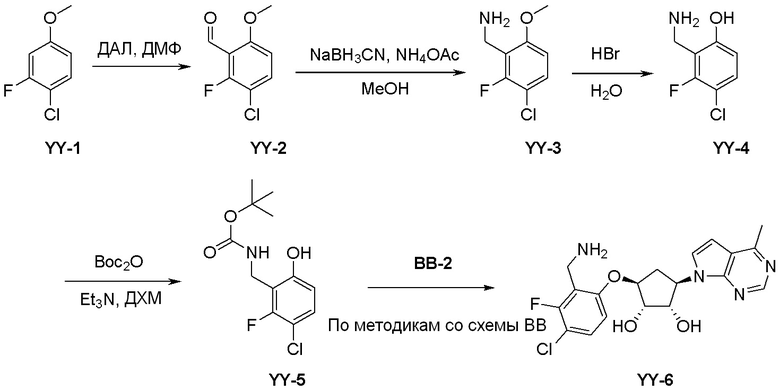
Стадия 1 - Синтез 3-хлор-2-фтор-6-метоксибензальдегида (YY-2)
К раствору 4-хлор-3-фторанизола (YY-1) (250 мг, 1,56 ммоль) в безводном ТГФ добавляют ДАЛ (0,86 мл, 2M, 1,71 ммоль) по каплям под N2 при -78°C и перемешивают при -78°C в течение 30 мин под N2. К раствору добавляют ДМФ (125 мг, 1,71 ммоль) при -78°C под N2, и полученную смесь перемешивают при -78°C в течение 20 мин. Реакцию гасят насыщ. NH4Cl при -78°C. Полученную смесь нагревают до 25°C. Смесь разделяют между EtOAc и H2O. Органический слой разделяют, и водный слой экстрагируют EtOAc. Объединенные органические слои промывают насыщенным раствором соли, сушат над Na2SO4 и концентрируют с получением неочищенного продукта. Неочищенный продукт очищают на флэш-колонке (EtOAc:петролейный эфир=1%-12%) с получением YY-2 (178 мг, 61%) в виде белого твердого вещества. 1H ЯМР (400 MГц, CDCl3) δ ч./млн. 10,41 (д, J=1,3 Гц, 1H), 7,54 (дд, J=8,0, 9,0 Гц, 1H), 6,76 (дд, J=1,5, 9,0 Гц, 1H), 3,95 (с, 3H)
Стадия 2 - Синтез (3-хлор-2-фтор-6-метоксифенил)метанамина (YY-3)
К раствору YY-2 (400 мг, 2,12 ммоль) в безводном MeOH (20,0 мл) добавляют NH4OAc (1,63 г, 21,2 ммоль). Смесь перемешивают при 25°C в течение 1 часа. К раствору добавляют NaCNBH3 (533 мг, 8,48 ммоль) при 25°C. Полученную смесь перемешивают в течение 16 часов при 25°C. Реакционную смесь разделяют между EtOAc и H2O. Органический слой разделяют, промывают насыщенным раствором соли, сушат над Na2SO4 и концентрируют с получением неочищенного продукта, который очищают на флэш-колонке (MeOH:ДХМ=1%-15%) с получением YY-3 (270 мг, 67%) в виде желтой камеди. 1H ЯМР (400 MГц, ДМСО-d6) δ ч./млн. 7,35 (т, J=8,8 Гц, 1H), 6,78 (д, J=9,0 Гц, 1H), 3,77 (с, 3H), 3,70 (с, 2H)
Стадия 3 - Синтез 2-(аминометил)-4-хлор-3-фторфенола (YY-4)
К раствору YY-3 (180 мг, 0,949 ммоль) в H2O (15,0 мл) добавляют водн. HBr (1,50 мл) по каплям при 0°C под N2. После добавления реакционную смесь нагревают при кипении с обратным холодильником (110°C) и перемешивают в течение 6 часов. Реакционную смесь охлаждают до 0°C, разбавляют в H2O и нейтрализуют насыщ. NaOH. Водный слой экстрагируют EtOAc. Органический слой промывают насыщенным раствором соли, сушат над Na2SO4 и концентрируют с получением неочищенного продукта, который очищают пре-ТСХ (MeOH:ДХМ=1:10) с получением YY-4 (50 мг, 30%) в виде белого твердого вещества. 1H ЯМР (400 MГц, ДМСО-d6) δ ч./млн. 7,18 (т, J=8,9 Гц, 1H), 6,52 (дд, J=1,5, 8,8 Гц, 1H), 3,92 (д, J=1,3 Гц, 2H)
Стадия 4 - Синтез трет-бутил (3-хлор-2-фтор-6-гидроксибензил)карбамата (YY-5)
К раствору YY-4 (50,0 мг, 0,285 ммоль) в ДХМ (5,00 мл) и MeOH (1,0 мл) добавляют Boc2O (68,4 мг, 0,313 ммоль) затем Et3N (72 мг, 0,71 ммоль) при 0°C. После добавления, реакционную смесь перемешивают при 25°C в течение 16 часов. Реакционную смесь подкисляют насыщ. лимонной кислотой до pH 5~6. Смесь разделяют между ДХМ и H2O. Органический слой разделяют, и водный слой экстрагируют ДХМ. Объединенные органические слои промывают насыщ.NaHCO3 и насыщенным раствором соли, сушат над Na2SO4 и концентрируют с получением неочищенного продукта, который очищают пре-ТСХ (EtOAc:Петролейный эфир=1:2) с получением YY-5 (45 мг, 57%) в виде белого твердого вещества. 1H ЯМР (400 MГц, ДМСО-d6) δ ч./млн. 10,22 (с, 1H), 7,27 (т, J=8,4Гц), 7,02 (шс, 1H), 6,67 (д, J=9,2 Гц), 4,14 (с, 2H), 1,37 (с, 9H)
Стадия 5 - Синтез (1S,2S,3S,5R)-3-(2-(аминометил)-4-хлор-3-фторфенокси)-5-(4-метил-7H-пирроло[2.3-d]пиримидин-7-ил)циклопентан-1,2-диола (YY-6)
Соединение YY-6 синтезируют с применением YY-5 и BB-2 по методикам стадий 2 & 3 со схемы BB.
ЖХМС 407 [M+1]; 1H ЯМР (400 MГц, ДМСО-d6+D2O) δ ч./млн. 8,57 (с, 1H), 7,61 (д, J=3,5 Гц, 1H), 7,56 (т, J=8,7 Гц, 1H), 7,00 (д, J=9,0 Гц, 1H), 6,72 (шс, 1H), 5,14-4,99 (м, 1H), 4,64 (шс, 1H), 4,59 (дд, J=4,6, 9,4 Гц, 1H), 4,08 (с, 3H), 2,90-2,78 (м, 1H), 2,62 (с, 3H), 2,06 (т, J=10,4 Гц, 1H)
Примеры 144-146 получают по методике получения YY-6, начиная с подходящего анизольного реагента и по методике стадий 3 & 4 со схемы YY.
2-метокси-6-(трифторметил)фенил)метанамин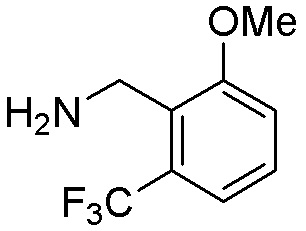
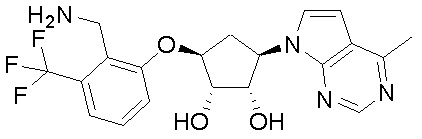
[M+1]
1H ЯМР (400 MГц, ДМСО-d6) δ ч./млн.=8,60 (с, 1H), 7,71 (д, J=3,5 Гц, 1H), 7,56-7,48 (м, 1H), 7,47-7,40 (м, 1H), 7,33 (д, J=7,8 Гц, 1H), 6,71 (д, J=3,5 Гц, 1H), 5,11 (кв, J=9,3 Гц, 1H), 4,71 (шс, 1H), 4,65 (дд, J=4,9, 9,2 Гц, 1H), 4,13 (д, J=4,3 Гц, 1H), 4,02 (шс, 2H), 2,97-2,82 (м, 1H), 2,64 (с, 3H), 2,11 (ддд, J=4,3, 9,7, 13,7 Гц, 1H)
(2,3-дифтор-6-метоксифенил)метанамин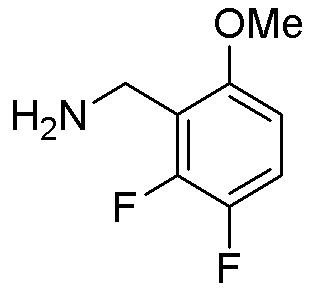
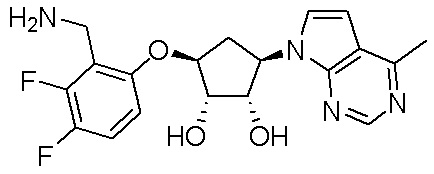
[M+1]
1H ЯМР (400 MГц, ДМСО-d6) δ ч./млн. 8,61 (с, 1H), 7,68 (с, 1H), 7,31-7,20 (м, 1H), 6,90-6,83 (м, 1H), 6,72 (д, J=3,5 Гц, 1H), 5,54-5,25 (м, 1H), 5,17-5,03 (м, 2H), 4,68-4,55 (м, 2H), 4,07-4,01 (м, 1H), 3,77 (д, J=1,3 Гц, 2H), 2,90-2,79 (м, 1H), 2,64 (с, 3H), 2,10-1,99 (м, 1H)
2-(5-хлор-2-метоксифенил)этан-1-амин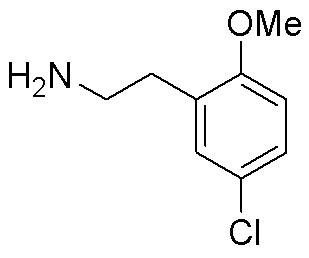
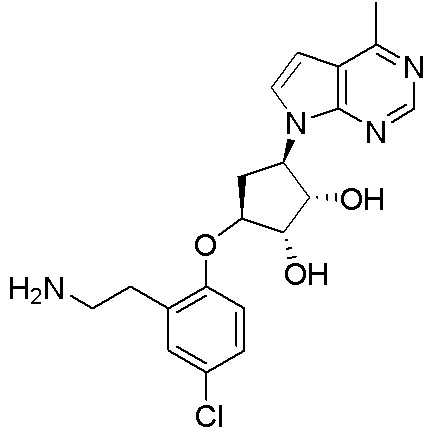
[M+1]
1H ЯМР (400 MГц, ДМСО-d6+D2O) δ ч./млн. 8,59 (с, 1H), 7,58 (д, J=3,8 Гц, 1H), 7,23-7,16 (м, 2H), 7,00 (д, J=8,5 Гц, 1H), 6,72 (д, J=3,8 Гц, 1H), 5,16-5,01 (м, 1H), 4,62-4,46 (м, 2H), 3,99 (д, J=5,3 Гц, 1H), 2,91-2,81 (м, 1H), 2,79-2,68 (м, 2H), 2,62 (с, 2H), 2,00-1,88 (м, 1H)
Пример 147 (Схема ZZ) - (1S,2S,3S,5R)-3-(2-(аминометил)-4,5-дихлорфенокси)-5-(4-метил-7H-пирроло[2.3-d]пиримидин-7-ил)циклопентан-1,2-диол (ZZ-7)
Схема ZZ
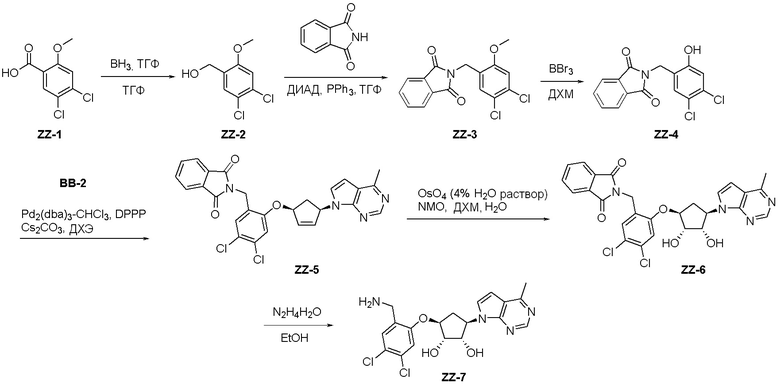
Стадия 1 - Синтез (4,5-дихлор-2-метоксифенил)метанола (ZZ-2)
К раствору метоксибензойной кислоты (ZZ-1) (500 мг, 2,26 ммоль) в безводном ТГФ (23 мл) добавляют BH3.ТГФ (6,79 мл, 6,79 ммоль) при 0°C под N2. После добавления, реакционную смесь перемешивают при 30°C под N2 в течение 3 часов. Реакционную смесь охлаждают до -20°C и гасят насыщ. NH4Cl. Реакционную смесь разделяют между EtOAc и H2O. Органический слой разделяют, и водный слой экстрагируют EtOAc. Объединенные органические слои промывают насыщенным раствором соли, сушат над Na2SO4 и концентрируют с получением неочищенного продукта, который очищают флэш-хроматографией на колонке (EtOAc:Петролейный эфир=1%-35%) с получением ZZ-2 (383 мг, 81,8%) в виде белого твердого вещества. 1H ЯМР (400 MГц, ДМСО-d6) δ ч./млн. 7,49 (с, 1H), 7,22 (с, 1H), 5,28 (т, J=5,6 Гц), 4,44 (д, J=6 Гц), 3,31 (с, 3H)
Стадия 2 - Синтез 2-(4,5-дихлор-2-метоксибензил)изоиндолин-1,3-диона (ZZ-3)
К раствору ZZ-2 (230 мг, 1,11 ммоль) и фталимида (163 мг, 1,11 ммоль) в безводном ТГФ (12 мл) добавляют PPh3 (291 мг, 2,30 ммоль). Реакционную смесь перемешивают при -20°C под N2 в течение 30 мин. К раствору добавляют ДЭАД (193 мг, 1,11 ммоль) при -10°C~-20°C. После добавления, реакционная смесь становится желтым раствором, который нагревают до 25°C и перемешивают в течение 2 часов. Реакционную смесь концентрируют, и остаток очищают на флэш-колонке (12,0 г гель, EtOAc:петролейный эфир=10%-35%) с получением неочищенного продукта и затем очищают преп-ТСХ (EtOAc:вазелин=1%~15%) с получением продукта ZZ-3 (30 мг, 8%) в виде белого твердого вещества, и применяют как есть на следующей стадии.
Стадия 3 - Синтез 2-(4,5-дихлор-2-гидроксибензил)изоиндолин-1,3-диона (ZZ-4)
К раствору ZZ-3 (50 мг, 0,15 ммоль) в ДХМ (2,00 мл) добавляют BBr3 (0,20 мл) по каплям при 0°C. После добавления, реакционную смесь перемешивают при 0°C в течение 2 часов. Реакционную смесь осторожно гасят MeOH. Полученную смесь разделяют между ДХМ и насыщ. NaHCO3. Органический слой промывают насыщенным раствором соли, сушат над Na2SO4 и концентрируют с получением неочищенного продукта, который очищают преп-ТСХ (EtOAc:петролейный эфир=1:1) с получением ZZ-4 (43 мг, 90%) в виде белого твердого вещества. 1H ЯМР (400 MГц, ДМСО-d6) δ ч./млн. 10,50 (с, 1H), 7,94-7,76 (м, 4H), 7,32 (с, 1H), 7,00 (с, 1H), 4,68 (с, 2H)
Стадия 4 - Синтез 2-(4,5-дихлор-2-(((1S,4R)-4-(4-метил-7H-пирроло[2.3-d]пиримидин-7-ил)циклопент-2-ен-1-ил)окси)бензил)изоиндолин-1,3-диона (ZZ-5)
К перемешиваемому раствору ZZ-4 (43 мг, 0,13 ммоль) и трет-бутил ((1S,4R)-4-(4-метил-7H-пирроло[2.3-d]пиримидин-7-ил)циклопент-2-ен-1-ил)карбоната (42 мг, 0,13 ммоль) в ДХЭ (1,50 мл) добавляют Cs2CO3 (52 мг, 0,16 ммоль), Pd2(dba)3.CHCl3, (3,5 мг, 0,003 ммоль) и dppp (3,3 мг, 0,008 ммоль). Реакционную смесь дегазируют и продувают N2 3 раза. Полученный раствор перемешивают в течение 1,5 ч при 25°C. Реакционную смесь концентрируют и очищают преп-ТСХ (EtOAc:петролейный эфир=1:1) с получением продукта ZZ-5 (62 мг, 89%) в виде желтой камеди и применяют сразу на следующей стадии.
Стадия 5 - Синтез 2-(4,5-дихлор-2-(((1S,2S,3S,4R)-2,3-дигидрокси-4-(4-метил-7H-пирроло[2.3-d]пиримидин-7-ил)циклопентил)окси)бензил)изоиндолин-1,3-диона (ZZ-6)
К раствору ZZ-5 (62 мг, 0,12 ммоль) в ТГФ (2,50 мл) и H2O (0,50 мл) добавляют NMO (29,4 мг, 0,251 ммоль), затем OsO4 (136 мг, 0,02 ммоль, 4% масс./масс. в т-BuOH). После добавления, реакционную смесь перемешивают при 28°C в течение 4 часов. Реакционную смесь разбавляют в H2O и гасят насыщ. NaHSO3. Темный раствор экстрагируют EtOAc, сушат над Na2SO4 и концентрируют с получением неочищенного продукта, который очищают преп-ТСХ (MeOH:ДХМ=1:10, УФ) с получением ZZ-6 (30 мг, 45%) в виде желтого твердого вещества. 1H ЯМР (400 MГц, ДМСО-d6) δ ч./млн. 8,61 (с, 1H), 7,95-7,77 (м, 4H), 7,60 (д, J=3,5 Гц, 1H), 7,56 (с, 1H), 7,36 (с, 1H), 6,73 (д, J=3,5 Гц, 1H), 5,43 (д, J=4,0 Гц, 1H), 5,18-5,05 (м, 2H), 4,81 (с, 2H), 4,70-4,61 (м, J=6,0 Гц, 1H), 4,52-4,41 (м, 1H), 3,99-3,94 (м, 1H), 2,91-2,78 (м, 1H), 2,65 (с, 3H), 1,98-1,86 (м, 1H)
Стадия 6 - Синтез (1S,2S,3S,5R)-3-(2-(аминометил)-4,5-дихлорфенокси)-5-(4-метил-7H-пирроло[2.3-d]пиримидин-7-ил)циклопентан-1,2-диола (ZZ-7)
К раствору ZZ-6 (30 мг, 0,05 ммоль) в ТГФ (2,5 мл) добавляют NH2NH2 (31 мг, 0,81 ммоль) по каплям. После добавления, реакционный раствор перемешивают при 28°C в течение 16 часов. Реакционную смесь фильтруют, и фильтрат концентрируют. Остаток очищают преп-ВЭЖХ с получением ZZ-7 (11 мг, 46%) в виде белого твердого вещества. ЖХМС 423 [M+1]; 1H ЯМР (400 MГц, ДМСО-d6) δ ч./млн. 8,62 (с, 1H), 7,66 (д, J=3,8 Гц, 1H), 7,60 (с, 1H), 7,31 (с, 1H), 6,72 (д, J=3,3 Гц, 1H), 5,14-5,05 (м, 2H), 4,63 (м, 1H), 4,00 (м, 1H), 3,74 (с, 2H), 2,89-2,81 (м, 1H), 2,64 (с, 3H), 2,02 (м, 1H)
Пример 148 (1S,2S,3S,5R)-3-(2-(аминометил)-4,5-дифторфенокси)-5-(4-метил-7H-пирроло[2.3-d]пиримидин-7-ил)циклопентан-1,2-диол получают по методике получения ZZ-7, начиная с (4,5-дифтор-2-метоксифенил)метанола и стадий 2-6 со схемы ZZ.
(4,5-дифтор-2-метоксифенил)метанол
[M+1]
1H ЯМР (400 MГц, ДМСО-d6) δ ч./млн. 8,62 (с, 1H), 7,75-7,57 (м, 1H), 7,51-7,32 (м, 1H), 7,25-7,05 (м, 1H), 6,72 (д, J=3,5 Гц, 1H), 5,39 (д, J=18,1 Гц, 1H), 5,20-4,98 (м, 2H), 4,57 (д, J=4,8 Гц, 2H), 4,01 (д, J=4,3 Гц, 1H), 3,72 (с, 2H), 2,95-2,72 (м, 1H), 2,62 (с, 3H), 2,00 (ддд, J=4,5, 9,1, 13,7 Гц, 1H)
Пример 149 (Схема AAA) - (1S,2S,3S,5R)-3-(2-(2-амино-1-гидроксиэтил)-4-хлорфенокси)-5-(4-метил-7H-пирроло[2.3-d]пиримидин-7-ил)циклопентан-1,2-диол (AAA-9)
Схема AAA
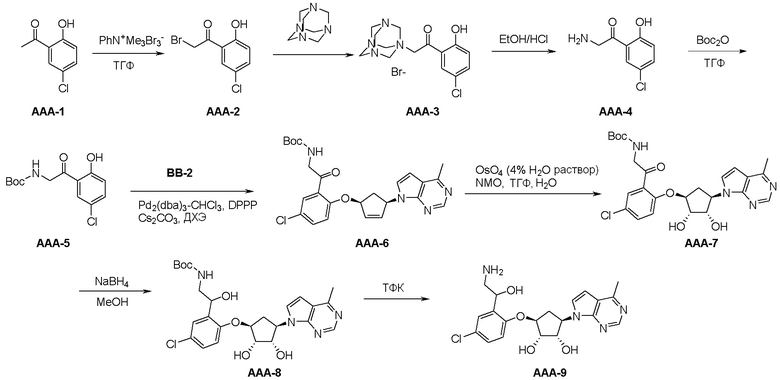
Стадия 1 - Синтез 2-бром-1-(5-хлор-2-гидроксифенил)этан-1-она (AAA-2)
К желтому раствору 1-(5-хлор-2-гидроксифенил)этан-1-она (AAA-1) (2 г, 11,72 ммоль) и в ТГФ (60 мл) добавляют PhMe3NBr3 (4,85 г, 12,9 ммоль) при кт (25°C). Полученный красный раствор перемешивают при кт в течение 12 ч с получением твердого вещества, затем смесь фильтруют. Фильтрат концентрируют в вакууме, и остаток очищают хроматографией на силикагеле, элюируя EtOAc в петролейном эфире 0-10% с получением AAA-2 (2,4 г) в виде желтого масла, которое применяют сразу на следующей стадии.
Стадия 2 - Синтез 2-((3r,5r,7r)-1l4,3,5,7-тетраазаадамантан-1-ил)-1-(5-хлор-2-гидроксифенил)этан-1-она, бромида (AAA-3)
К раствору 1,3,5,7-тетраазаадамантана (4,67 г, 33,3 ммоль) в CHCl3 (40 мл) добавляют AAA-2 (2,6 г, 10 ммоль) при кт (25°C). Смесь перемешивают при кт в течение 12 ч с получением твердого вещества. Твердое вещество собирают фильтрацией и промывают ДХМ и сушат в вакууме с получением неочищенного продукта AAA-3 (3 г, 74%) в виде белого твердого вещества, применяют сразу на следующей стадии.
Стадия 3 - Синтез 2-амино-1-(5-хлор-2-гидроксифенил)этан-1-она (AAA-4)
К суспензии SM1 (3 г, 8 ммоль) в EtOH (15 мл) добавляют конц. HCl (3 мл) при кт (25°C). Суспензию перемешивают при кт в течение 2 ч. Реакционную смесь выпаривают с получением AAA-4 (1,5 г, >100%) в виде желтого масла, которое применяют сразу без дальнейшей очистки. ЖХМС 186 [M+1]
Стадия 4 - Синтез трет-бутил (2-(5-хлор-2-гидроксифенил)-2-оксоэтил)карбамата (AAA-5)
К раствору (Boc)2O (2,29 г, 10,5 ммоль) в диоксане (15 мл) добавляют раствор AAA-4 (200 мг, раствор, нейтрализован водн. NaHCO3) при 0°C. Реакционный раствор перемешивают при 25°C. Реакционный раствор экстрагируют EtOAc (6 мл×3). Органические слои разделяют, сушат и выпаривают с получением неочищенного продукта, который очищают флэш-хроматографией с 0-25% EtOAc/петролейным эфиром с получением AAA-5 (600 мг, 22%) в виде белого твердого вещества, и применяют как есть на следующей стадии.
Стадия 5 - Синтез трет-бутил (2-(5-хлор-2-(((1S,4R)-4-(4-метил-7H-пирроло[2.3-d]пиримидин-7-ил)циклопент-2-ен-1-ил)окси)фенил)-2-оксоэтил)карбамата (AAA-6)
С применением BB-2 и по методикам стадии 2 со схемы BB получают AAA-6 (288 мг, 31%) в виде желтой камеди и применяют сразу на следующей стадии.
Стадия 6 - Синтез трет-бутил (2-(5-хлор-2-(((1S,2S,3S,4R)-2,3-дигидрокси-4-(4-метил-7H-пирроло[2.3-d]пиримидин-7-ил)циклопентил)окси)фенил)-2-оксоэтил)карбамата (AAA-7)
Начиная с AAA-6 и по методикам стадии 3 со схемы BB, получают AAA-7 (70 мг, 33%) в виде белого твердого вещества. ЖХМС 517 [M+1]
Стадия 7 - Синтез трет-бутил (2-(5-хлор-2-(((1S,2S,3S,4R)-2,3-дигидрокси-4-(4-метил-7H-пирроло[2.3-d]пиримидин-7-ил)циклопентил)окси)фенил)-2-гидроксиэтил)карбамата (AAA-8)
К смеси AAA-7 (70 мг, 0,14 ммоль) в MeOH (1 мл) добавляют NaBH4 (15,4 мг, 0,406 ммоль). Смесь перемешивают при 25°C в течение 2 ч. Смесь гасят 1N HCl водн. (5 мл). Смесь выпаривают с получением неочищенного продукта AAA-8 (70 мг, >99%) в виде белого твердого вещества.
Стадия 8 - Синтез (1S,2S,3S,5R)-3-(2-(2-амино-1-гидроксиэтил)-4-хлорфенокси)-5-(4-метил-7H-пирроло[2.3-d]пиримидин-7-ил)циклопентан-1,2-диола (AAA-9)
Соединение AAA-8 (62 мг, 0,12 ммоль) растворяют в ДХМ (0,6 мл). Смесь охлаждают до 0°C на ледяной бане. ТФК (0,2 мл) добавляют в смесь по каплям. Реакционную смесь перемешивают при 25°C в течение 2 часов. Реакционный раствор нейтрализуют насыщенным водн. NaHCO3 (1 мл), фильтруют и фильтрат сразу очищают преп.-ВЭЖХ с получением AAA-9 (21 мг, 42%) в виде белого твердого вещества. ЖХМС 441 [M+23]; 1H ЯМР (400 MГц, MeOD-d4) δ ч./млн. 8,62 (д, J=2,8 Гц, 1H), 7,58 (дд, J=3,3, 11,3 Гц, 1H), 7,48 (с, 1H), 7,26-7,22 (м, 1H), 7,03 (дд, J=2,1, 8,7 Гц, 1H), 6,75 (т, J=3,8 Гц, 1H), 5,20 (д, J=3,8 Гц, 1H), 5,10 (шс, 1H), 4,70 (шс, 2H), 4,22 (д, J=9,5 Гц, 1H), 3,04-2,94 (м, 2H), 2,77 (шс, 1H), 2,71 (с, 3H), 2,26 (шс, 1H)
Пример 150 (Схема BBB) - (1S,2S,3S,5R)-3-(2-(аминометил)-3,4-дихлорфенокси)-5-(4-метил-7H-пирроло[2.3-d]пиримидин-7-ил)циклопентан-1,2-диол (BBB-8)
Схема BBB
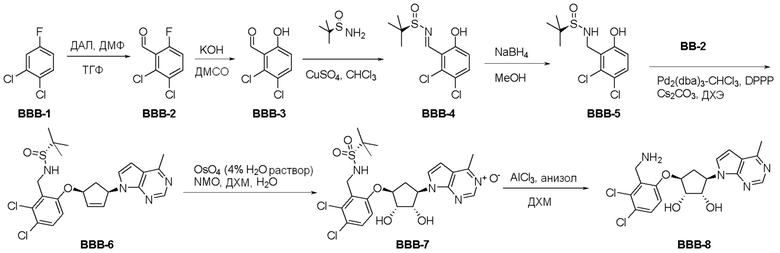
Стадия 1 - Синтез 2,3-дихлор-6-фторбензальдегида (BBB-2)
К раствору 1,2-дихлор-4-фторбензола (BBB-1) (2 г, 12,12 ммоль) в безводном ТГФ (30 мл) добавляют ДАЛ (6,67 мл, 2M, 13,3 ммоль) по каплям под N2, при -65°C. После добавления, реакционную смесь перемешивают при -65°C в течение 30 мин под N2. К полученному красному раствору добавляют ДМФ (1,77 г, 24,2 ммоль) при -65°C под N2 и полученную смесь перемешивают при -65°C в течение 20 мин. Реакцию гасят водой (50 мл) и экстрагируют EtOAc (30 мл×2). Экстракт промывают 1 N HCl (30 мл), насыщенным раствором соли (30 мл), сушат над Na2SO4 и концентрируют с получением неочищенного продукта, который экстрагируют петролейным эфиром (30 мл×3). Экстракт концентрируют в вакууме с получением BBB-2 (2,2 г, 94%) в виде желтого твердого вещества. 1H ЯМР (400 MГц, CDCl3) δ ч./млн. 10,44 (с, 1H), 7,66 (дд, J=5,3, 9,0 Гц, 1H), 7,10 (т, J=9,2 Гц, 1H)
Стадия 2 - Синтез 2,3-дихлор-6-гидроксибензальдегида (BBB-3)
К желтому раствору неочищенного продукта BBB-2 (2,2 г, 11,4 ммоль) в ДМСО (10 мл) медленно добавляют KOH (1280 мг, 22,8 ммоль) при 0°C. После добавления, смесь превращается в красную смеси и ее перемешивают при комнатной температуре (20°C) в течение 16 ч. Смесь разбавляют МТБЭ (100 мл). Жидкость декантируют, и остаток промывают МТБЭ (100 мл). Остаток затем разбавляют водой (30 мл) и доводят 1 N HCl до pH 2 и экстрагируют EtOAc (20 мл×2). Экстракт промывают насыщенным раствором соли (20 мл), сушат над Na2SO4 и концентрируют в вакууме с получением неочищенного продукта BBB-3 (390 мг, 18%) в виде желтого твердого вещества. 1H ЯМР (400 MГц, CDCl3) δ ч./млн. 11,98 (с, 1H), 10,44 (с, 1H), 7,56 (д, J=9,0 Гц, 1H), 6,90 (д, J=9,3 Гц, 1H)
Стадия 3 - Синтез (R,E)-N-(2,3-дихлор-6-гидроксибензилиден)-2-метилпропан-2-сульфинамида (4)
Смесь неочищенного продукта BBB-3 (150 мг, 0,79 ммоль), (R)-2-метилпропан-2-сульфинамида (143 мг, 1,2 ммоль) и CuSO4 (376 мг, 2,36 ммоль) в CHCl3 (3 мл) перемешивают при кт (30°C) в течение 5 дней. Смесь фильтруют и очищают хроматографией на силикагеле, элюируя EtOAc в петролейном эфире 0-30% с получением BBB-4 (70 мг, 30%) в виде светло-желтого твердого вещества. 1H ЯМР (400 MГц, CDCl3) δ ч./млн. 11,98 (с, 1H), 9,28 (с, 1H), 7,49 (д, J=8,8 Гц, 1H), 6,92 (д, J=9,3 Гц, 1H), 1,29 (с, 9H)
Стадия 4 - Синтез (R)-N-(2,3-дихлор-6-гидроксибензил)-2-метилпропан-2-сульфинамида (BBB-5)
К раствору BBB-4 (70 мг, 0,238 ммоль) в MeOH (3 мл) добавляют NaBH4 (27 мг, 0,714 моль) при кт (30°C). Смесь перемешивают при кт в течение 1 ч. Смесь концентрируют в вакууме и растворяют в воде (5 мл). К смеси добавляют NH4Cl водн. (2 мл) с получением некоторого количества твердого вещества. Смесь экстрагируют EtOAc (5 мл×3). Экстракт промывают насыщенным раствором соли (5 мл), сушат над Na2SO4 и концентрируют в вакууме с получением BBB-5 (55 мг, 78%) в виде желтого твердого вещества. 1H ЯМР (400 MГц, CDCl3) δ ч./млн. 9,31 (шс, 1H), 7,17 (д, J=8,8 Гц, 1H), 6,69 (д, J=9,0 Гц, 1H), 4,58-4,40 (м, 2H), 4,09-3,97 (м, 1H), 1,27 (с, 9H)
Стадия 5 - Синтез (R)-N-(2,3-дихлор-6-(((1S,4R)-4-(4-метил-7H-пирроло[2.3-d]пиримидин-7-ил)циклопент-2-ен-1-ил)окси)бензил)-2-метилпропан-2-сульфинамида (BBB-6)
Соединение BBB-6 (130 мг, 98%, содержит 1 экв. ДХМ) получают в виде желтой камеди из BBB-5 и BB-2 с применением методик со стадии 2 Схемы BB. ЖХМС 493 [M+1]; 1H ЯМР (400 MГц, CDCl3) δ ч./млн. 8,77 (с, 1H), 7,39-7,31 (м, 2H), 6,84 (д, J=9,0 Гц, 1H), 6,60 (д, J=3,5 Гц, 1H), 6,41 (д, J=5,5 Гц, 1H), 6,22 (д, J=4,8 Гц, 1H), 6,13-6,01 (м, 1H), 5,41-5,35 (м, 1H), 4,63-4,39 (м, 2H), 3,65 (т, J=6,7 Гц, 1H), 3,29-3,09 (м, 1H), 2,73 (с, 3H), 1,98 (тд, J=3,8, 14,7 Гц, 1H), 1,18 (с, 9H)
Стадия 6 - Синтез N-(2,3-дихлор-6-(((1S,2S,3S,4R)-2,3-дигидрокси-4-(4-метил-7H-пирроло[2.3-d]пиримидин-7-ил)циклопентил)окси)бензил)-2-метилпропан-2-сульфонамида (BBB-7)
Соединение BBB-6 обрабатывают по методике стадии 3 со схемы BB с получением BBB-7 (70 мг, 58%). ЖХМС 543 [M+16+1]; 1H ЯМР (400 MГц, CDCl3) δ ч./млн. 8,62 (с, 1H), 7,56 (шс, 1H), 7,19 (д, J=8,8 Гц, 1H), 6,77 (д, J=9,0 Гц, 1H), 6,54 (д, J=3,8 Гц, 1H), 5,64 (т, J=5,5 Гц, 1H), 5,14 (с, 1H), 5,13-5,04 (м, 1H), 4,56-4,50 (м, 1H), 4,47 (дд, J=4,8, 8,5 Гц, 1H), 4,44-4,34 (м, 2H), 4,04 (д, J=4,8 Гц, 1H), 2,83-2,75 (м, 1H), 2,62 (с, 3H), 2,14-2,04 (м, 1H), 1,20 (с, 9H)
Стадия 7 - Синтез (1S,2S,3S,5R)-3-(2-(аминометил)-3,4-дихлорфенокси)-5-(4-метил-7H-пирроло[2.3-d]пиримидин-7-ил)циклопентан-1,2-диола (BBB-8)
К раствору BBB-7 (35 мг, 0,064 ммоль) в сухом ДХМ (5 мл) добавляют анизол (170 мг, 1,56 ммоль) и AlCl3 (86 мг, 0,64 ммоль) при кт (25°C). Белую суспензию перемешивают при кт в течение 3 ч. Смесь гасят водн. NaHCO3 (3 мл), затем водн. тартратом калия-натрия (5 мл). Смесь экстрагируют EtOAc (5 мл×3). Экстракт концентрируют в вакууме и очищают ТСХ (ДХМ/MeOH=10/1) с получением BBB-8 (8 мг, 29%) в виде белого твердого вещества. ЖХМС 423 [M+1]; 1H ЯМР (400 MГц, ДМСО-d6) δ ч./млн. 8,61 (с, 1H), 7,69 (д, J=3,5 Гц, 1H), 7,49 (д, J=8,8 Гц, 1H), 7,11 (д, J=9,0 Гц, 1H), 6,72 (д, J=3,8 Гц, 1H), 5,44 (шс, 1H), 5,10 (кв, J=9,0 Гц, 2H), 4,64 (д, J=5,3 Гц, 2H), 4,06 (д, J=4,8 Гц, 1H), 3,92 (с, 2H), 2,92-2,81 (м, 1H), 2,64 (с, 3H), 2,05 (ддд, J=4,1, 9,2, 13,8 Гц, 1H)
Пример 151 (Схема CCC) - (1S,2S,3S,5R)-3-(2-(2-аминоэтокси)-4,5-дихлорфенокси)-5-(4-метил-7H-пирроло[2.3-d]пиримидин-7-ил)циклопентан-1,2-диол (CCC-4)
Схема CCC
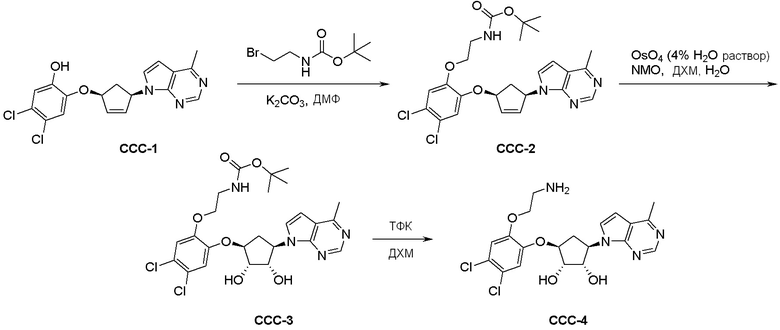
Стадия 1 - Синтез 4,5-дихлор-2-(((1S,4R)-4-(4-метил-7H-пирроло[2.3-d]пиримидин-7-ил)циклопент-2-ен-1-ил)окси)фенола (CCC-1)
Соединение CCC-1 получают с применением методик со стадии 2 со схемы BB с коммерчески доступным 4,5-дихлорбензол-1,2-диолом и BB-2.
Стадия 2 - Синтез трет-бутил (2-(4,5-дихлор-2-(((1S,4R)-4-(4-метил-7H-пирроло[2.3-d]пиримидин-7-ил)циклопент-2-ен-1-ил)окси)фенокси)этил)карбамата (CCC-2)
К перемешиваемому желтому раствору CCC-1 (78 мг, 0,21 ммоль) в сухом ДМФ (5 мл) добавляют K2CO3 (86 мг, 0,62 ммоль) и трет-бутил (2-бромэтил)карбамат (923 мг, 0,42 ммоль) при 25°C и перемешивают в течение 20 часов. Воду (10 мл) добавляют к реакционной смеси и экстрагируют EtOAc (2×20 мл). Объединенные органические слои промывают насыщенным раствором соли (5×20 мл), сушат над Na2SO4, фильтруют и концентрируют в вакууме с получением остатка, который очищают хроматографией на колонке (силикагель, элюируют EtOAc) с получением CCC-2 (108 мг, >99%) в виде бесцветной камеди. ЖХМС [M+1] 519; 1H ЯМР (400 MГц, CDCl3) δ ч./млн. 8,77 (с, 1H), 7,42 (д, J=3,5 Гц, 1H), 7,04 (с, 1H), 6,98 (с, 1H), 6,60 (д, J=3,5 Гц, 1H), 6,39 (тд, J=1,9, 5,5 Гц, 1H), 6,19 (дд, J=2,3, 5,3 Гц, 1H), 6,06 (дд, J=2,0, 4,8 Гц, 1H), 5,29 (д, J=7,0 Гц, 1H), 5,02 (шс, 1H), 4,03 (т, J=5,0 Гц, 2H), 3,59-3,47 (м, 2H), 3,10 (тд, J=7,7, 15,2 Гц, 1H), 2,73 (с, 3H), 2,07 (т, J=2,9 Гц, 1H), 1,42 (с, 9H)
Стадия 3 - Синтез трет-бутил (2-(4,5-дихлор-2-(((1S,2S,3S,4R)-2,3-дигидрокси-4-(4-метил-7H-пирроло[2.3-d]пиримидин-7-ил)циклопентил)окси)фенокси)этил)карбамата (CCC-3)
Соединение CCC-2 обрабатывают по методикам стадии 3 со схемы BB с получением CCC-3 (55 мг, 48%) в виде коричневой камеди. ЖХМС [M+23] 575
Стадия 4 - Синтез (1S,2S,3S,5R)-3-(2-(2-аминоэтокси)-4,5-дихлорфенокси)-5-(4-метил-7H-пирроло[2.3-d]пиримидин-7-ил)циклопентан-1,2-диола (CCC-4)
Соединение CCC-3 подвергают стандартному ТФК/водному снятию защиты по методике стадии 8 со схемы AAA, затем преп-ВЭЖХ с получением CCC-4 (35 мг, 75%). ЖХМС [M+23] 475; 1H ЯМР (400 MГц, MeOD-d4) δ ч./млн. 8,64 (с, 1H), 7,66 (д, J=3,8 Гц, 1H), 7,37 (с, 1H), 7,29 (с, 1H), 6,79 (д, J=3,5 Гц, 1H), 5,20 (кв, J=8,9 Гц, 1H), 4,76-4,68 (м, 1H), 4,66 (дд, J=5,3, 8,0 Гц, 1H), 4,33-4,23 (м, 3H), 3,40-3,36 (м, 2H), 3,01-2,90 (м, 1H), 2,74 (с, 3H), 2,29 (ддд, J=5,0, 8,8, 14,3 Гц, 1H)
Пример 152 (1S,2S,3S,5R)-3-(2-(2-аминоэтокси)-4,5-дифторфенокси)-5-(4-метил-7H-пирроло[2.3-d]пиримидин-7-ил)циклопентан-1,2-диол получают по методике получения CCC-4, начиная с 4,5-дифторбензол-1,2-диола и согласно стадиям 1-4 со схемы CCC.
4,5-дифторбензол-1,2-диол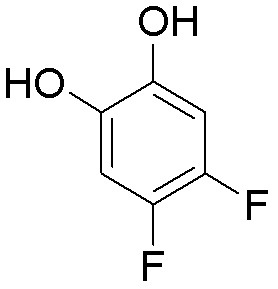
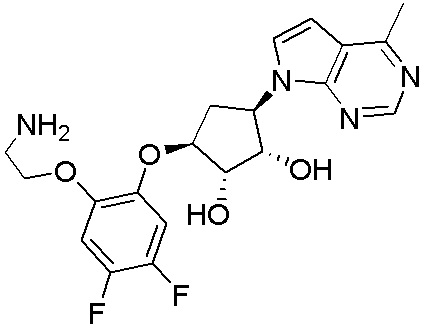
[M+1]
1H ЯМР (400 MГц, MeOD-d4) δ ч./млн. 8,64 (с, 1H), 7,67 (д, J=3,5 Гц, 1H), 7,26-7,07 (м, 2H), 6,80 (д, J=4,0 Гц, 1H), 5,19 (кв, J=8,9 Гц, 1H), 4,72-4,64 (м, 2H), 4,33-4,21 (м, 3H), 3,42-3,36 (м, 2H), 2,99-2,87 (м, 1H), 2,75 (с, 3H), 2,33-2,23 (м, 1H)
Пример 153 (Схема DDD) - (1S,2S,3S,5R)-3-(2-(2-аминоэтокси)-4-хлорфенокси)-5-(4-метил-7H-пирроло[2,3-d]пиримидин-7-ил)циклопентан-1,2-диол (DDD-6)
Схема DDD
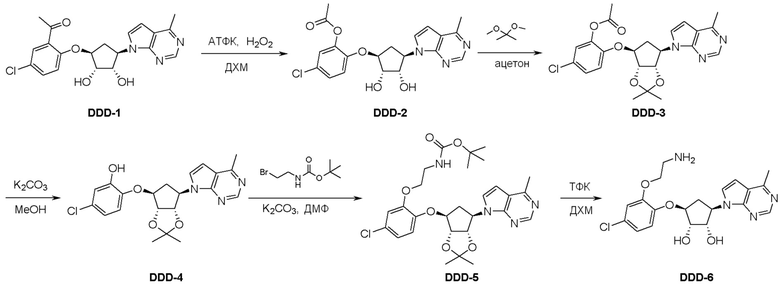
Стадия 1 - Синтез 1-(5-хлор-2-(((1S,2S,3S,4R)-2,3-дигидрокси-4-(4-метил-7H-пирроло[2.3-d]пиримидин-7-ил)циклопентил)окси)фенил)этан-1-она (DDD-1)
Соединение DDD-1 получают с применением методик со стадий 2 & 3 со схемы BB с коммерчески доступным 1-(5-хлор-2-гидроксифенил)этан-1-оном и BB-2.
Стадия 2 - Синтез ацетата 5-хлор-2-(((1S,2S,3S,4R)-2,3-дигидрокси-4-(4-метил-7H-пирроло[2.3-d]пиримидин-7-ил)циклопентил)окси)фенила (DDD-2)
Трифторуксусный ангидрид (1,46 г, 6,95 ммоль) охлаждают до -10°C добавляют по каплям и 30% H2O2 (185 мг, 1,63 ммоль) и раствор перемешивают в течение 10 мин. К этой смеси добавляют DDD-1 (210 мг, 0,523 ммоль) в ДХМ (3,5 мл) по каплям при 0°C и перемешивают при 25°C в течение 15 мин. Затем насыщенный тиосульфат натрия (2 мл) добавляют к реакционному раствору. Реакционный раствор подщелачивают насыщенным NaHCO3 до pH=7~8, экстрагируют ДХМ (30 мл×2). Объединенные органические слои промывают насыщенным раствором соли (40 мл), сушат над Na2SO4, фильтруют и выпаривают с получением неочищенного продукта, который очищают ISCO (силикагель, 12 г, MeOH/ДХМ=10%~14%) с получением желаемого продукта DDD-2 (160 мг, 73%) в виде белого твердого вещества. ЖХМС [M+1] 418; 1H ЯМР (400 MГц, CDCl3) δ ч./млн. 8,67 (с, 1H), 7,25 (м, 1H), 7,21 (дд, J=2,5, 8,8 Гц, 1H), 7,12-7,06 (м, 2H), 6,60 (д, J=3,8 Гц, 1H), 4,97 (кв, J=8,5 Гц, 1H), 4,78 (т, J=6,0 Гц, 1H), 4,50 (дд, J=5,3, 8,3 Гц, 1H), 4,29 (д, J=5,5 Гц, 1H), 3,12-3,01 (м, 1H), 2,72 (с, 3H), 2,36-2,27 (м, 1H), 2,24 (с, 3H)
Стадия 2 - Синтез ацетата 5-хлор-2-(((3aR,4S,6R,6aS)-2,2-диметил-6-(4-метил-7H-пирроло[2.3-d]пиримидин-7-ил)тетрагидро-4H-циклопента[d][1.3]диоксол-4-ил)окси)фенила (DDD-3)
К перемешиваемому бесцветному раствору DDD-2 (160 мг, 0,38 ммоль) в ацетоне (0,64 мл) добавляют 2,2-диметоксипропан (6,4 мл) и п-толуолсульфоновую кислоту (73 мг, 0,38 ммоль) при 25°C. Реакционную смесь перемешивают при 25°C в течение 1 часа. Затем водн. NaHCO3 (3 мл) добавляют к реакционной смеси до pH 8,0. Затем смесь экстрагируют EtOAc (15 мл×2). Органические слои разделяют, промывают насыщенным раствором соли (20 мл), сушат над Na2SO4 и фильтруют. Органический слой выпаривают с получением неочищенного продукта в виде бесцветной камеди, которую очищают хроматографией на колонке (эфир:EtOAc=1:1, Rf~0,55) с получением DDD-3 (126 мг, 72%) в виде белого твердого вещества. ЖХМС [M+1] 458; 1H ЯМР (400 MГц, CDCl3) δ ч./млн. 8,79 (с, 1H), 7,43 (д, J=3,8 Гц, 1H), 7,25-7,20 (м, 1H), 7,14-7,07 (м, 2H), 6,58 (д, J=3,5 Гц, 1H), 5,46-5,38 (м, 1H), 4,93 (д, J=5,8 Гц, 1H), 4,83-4,74 (м, 2H), 3,06-2,89 (м, 1H), 2,74 (с, 3H), 2,52-2,37 (м, 1H), 2,21 (с, 3H), 1,60 (шс, 3H), 1,32 (с, 3H)
Стадия 3 - Синтез 5-хлор-2-(((3aR,4S,6R,6aS)-2,2-диметил-6-(4-метил-7H-пирроло[2.3-d]пиримидин-7-ил)тетрагидро-4H-циклопента[d][1.3]диоксол-4-ил)окси)фенола (DDD-4)
Соединение DDD-3 (142 мг, 0,31 ммоль) растворяют в MeOH (3 мл) и H2O (1 мл). K2CO3 (85,7 г, 0,62 ммоль) добавляют в смесь. Реакционную смесь перемешивают при 25°C в течение 1 часа. Смесь нейтрализуют 10% лимонной кислотой до pH 6-7. Смесь разбавляют H2O (10 мл), экстрагируют EtOAc (10 мл×2). Объединенные органические слои промывают насыщенным раствором соли (10 мл), сушат над Na2SO4, фильтруют и выпаривают с получением DDD-4 (140 мг, >99%) в виде бесцветной камеди. ЖХМС [M+1] 416; 1H ЯМР (400 MГц, CDCl3) δ ч./млн. 8,79 (с, 1H), 7,22 (д, J=3,5 Гц, 1H), 6,96-6,90 (м, 2H), 6,87-6,81 (м, 1H), 6,61 (д, J=3,5 Гц, 1H), 5,95-5,74 (м, 1H), 5,27-5,19 (м, 2H), 4,87-4,78 (м, 2H), 2,99-2,87 (м, 1H), 2,74 (с, 3H), 2,61-2,50 (м, 1H), 1,59 (с, 3H), 1,34 (с, 3H)
Стадия 4 - Синтез трет-бутил (2-(5-хлор-2-(((3aR,4S,6R,6aS)-2,2-диметил-6-(4-метил-7H-пирроло[2.3-d]пиримидин-7-ил)тетрагидро-4H-циклопента[d][1.3]диоксол-4-ил)окси)фенокси)этил)карбамата (DDD-5)
К перемешиваемому раствору DDD-4 (140 мг, 0,34 ммоль) в сухом ДМФ (5 мл) добавляют K2CO3 (140 мг, 1,0 ммоль) и трет-бутил (2-бромэтил)карбамат (151 мг, 0,67 ммоль) при 25°C. Реакционную смесь перемешивают при 25°C в течение 15 часов. Воду (10 мл) добавляют к реакционной смеси и смесь экстрагируют EtOAc (20 мл×2). Объединенные органические слои промывают насыщенным раствором соли (20 мл×5), сушат над Na2SO4, фильтруют и концентрируют с получением неочищенного продукта, который очищают ISCO (EtOAc/петролейный эфир=60%) с получением DDD-5 (170 мг, 90%) в виде бесцветной камеди. ЖХМС [M+23] 581; 1H ЯМР (400 MГц, CDCl3) δ ч./млн. 8,80 (с, 1H), 7,77 (д, J=3,8 Гц, 1H), 6,96-6,84 (м, 3H), 6,59 (д, J=3,8 Гц, 1H), 5,50 (д, J=8,0 Гц, 1H), 5,18 (шс, 1H), 4,92-4,86 (м, 1H), 4,85-4,81 (м, 2H), 4,08-3,98 (м, 2H), 3,65-3,54 (м, 2H), 3,08-2,98 (м, 1H), 2,74 (с, 3H), 2,47 (д, J=14,6 Гц, 1H), 1,59 (с, 3H), 1,42 (с, 9H), 1,31 (с, 3H)
Стадия 5 - Синтез (1S,2S,3S,5R)-3-(2-(2-аминоэтокси)-4-хлорфенокси)-5-(4-метил-7H-пирроло[2.3-d]пиримидин-7-ил)циклопентан-1,2-диола (DDD-6)
Соединение DDD-5 подвергают стандартным ТФК/водным способам снятия защиты, затем преп-ВЭЖХ с получением DDD-6 (40 мг, 31%). ЖХМС [M+23] 441; 1H ЯМР (400 MГц, MeOD-d4) δ ч./млн. 8,64 (с, 1H), 7,78 (д, J=3,8 Гц, 1H), 7,14-7,05 (м, 2H), 6,96 (д, J=8,3 Гц, 1H), 6,82 (д, J=3,8 Гц, 1H), 5,41-5,27 (м, 1H), 4,74-4,70 (м, J=5,5 Гц, 1H), 4,67 (дд, J=4,6, 7,9 Гц, 1H), 4,21 (д, J=4,5 Гц, 1H), 4,15-4,07 (м, 2H), 3,18-3,08 (м, 2H), 3,06-2,94 (м, 1H), 2,74 (с, 3H), 2,14 (дд, J=6,8, 14,1 Гц, 1H)
(Схема EEE) - Синтез трет-бутил 5-хлор-8-гидрокси-3,4-дигидроизохинолин-2(1H)-карбоксилата (TP-19)
Схема EE
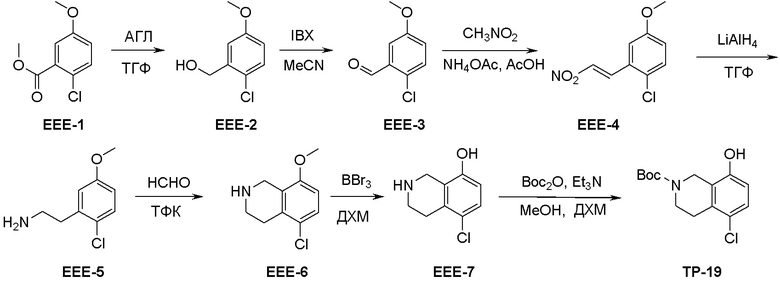
Стадия 1 - Синтез (2-хлор-5-метоксифенил)метанола (EEE-2)
К раствору метил 2-хлор-5-метоксибензоата EEE-1 (4,55 г, 22,7 ммоль) в безводном ТГФ (200 мл) добавляют LiAlH4 (1,72 г, 45,4 ммоль) при -10°C~-5°C порциями. Температуру поднимают до 0°C. После добавления, реакционную смесь перемешивают при 0°C в течение 2 часов. Реакционную смесь гасят 5% NaOH. Реакционную смесь фильтруют через слой Целита. К лепешке Целита добавляют ТГФ (100 мл) и EtOAc (100 мл) и перемешивают при 25°C в течение 0,5 часа. Смесь фильтруют, и объединенные органические слои сушат над Na2SO4 и концентрируют с получением неочищенного продукта EEE-2 (4,5 г) в виде бесцветного масла и применяют сразу на следующей стадии. 1H ЯМР (400 MГц, CDCl3) δ ч./млн. 7,25 (д, J=9,5 Гц, 1H), 7,05 (д, J=2,8 Гц, 1H), 6,78 (дд, J=2,9, 8,7 Гц, 1H), 4,75 (д, J=3,8 Гц, 2H), 3,81 (с, 3H), 2,04-1,94 (м, 1H)
Стадия 2 - Синтез 2-хлор-5-метоксибензальдегида (EEE-3)
К раствору EEE-2 (3,63 г, 16,16 ммоль) в CH3CN (120 мл) добавляют IBX (17,7 г, 63,1 ммоль) при 25°C. Полученную смесь нагревают при 80°C в течение 2 часов. Реакционную смесь охлаждают до 25°C, фильтруют, и промывают ДХМ (50 мл). Объединенный фильтрат концентрируют с получением неочищенного продукта, который очищают ISCO (силикагель, 80 г, EtOAc/петролейный эфир=17%) с получением EEE-3 (1,76 г, 49%) в виде белого твердого вещества. 1H ЯМР (400 MГц, CDCl3) δ ч./млн. 10,45 (с, 1H), 7,42 (д, J=3,0 Гц, 1H), 7,36 (д, J=8,8 Гц, 1H), 7,11 (дд, J=3,3, 8,8 Гц, 1H), 3,86 (с, 3H)
Стадия 3 - Синтез (E)-1-хлор-4-метокси-2-(2-нитровинил)бензола (EEE-4)
К раствору EEE-3 (1,76 г, 10,3 ммоль) в AcOH (17,0 мл) добавляют NH4OAc (0,795 г, 10,3 ммоль), затем MeNO2 (3,15 г, 51,6 ммоль). После добавления, реакционную смесь нагревают при 85°C в течение 10 часов, и затем охлаждают до 28°C. Реакционную смесь разбавляют в ДХМ и концентрируют с удалением AcOH с получением неочищенного продукта, который очищают на флэш-колонке (12 г гель, EtOAc:Петролейный эфир=1%~10%) с получением EEE-4 (1,78 г, 81%) в виде желтого твердого вещества. 1H ЯМР (400 MГц, CDCl3) δ ч./млн. 8,36 (д, J=13,8 Гц, 1H), 7,58 (д, J=13,8 Гц, 1H), 7,39 (д, J=9,0 Гц, 1H), 7,05 (д, J=3,0 Гц, 1H), 6,99 (дд, J=3,0, 9,0 Гц, 1H), 3,85 (с, 3H)
Стадия 4 - Синтез 2-(2-хлор-5-метоксифенил)этан-1-амина (EEE-5)
К раствору EEE-4 (862 мг, 4,04 ммоль) в безводном ТГФ (40 мл) добавляют LiAlH4 (613 мг, 16,1 ммоль) при -20°C под N2. После добавления, реакционную смесь перемешивают при 25°C в течение 1 часа. Реакционную смесь затем нагревают при 50°C и перемешивают в течение 2 часов. Реакционную смесь гасят каплями воды. Реакционную смесь разбавляют EtOAx, фильтруют и концентрируют с получением неочищенного продукта, который очищают флэш-хроматографией на колонке (40 г гель, MeOH:ДХМ=1%~8,0%) с получением EEE-5 (290 мг, 38,7%) в виде желтого масла. 1H ЯМР (400 MГц, CDCl3) δ ч./млн. 7,27-7,24 (м, 1H), 6,79 (д, J=2,8 Гц, 1H), 6,72 (дд, J=3,0, 8,8 Гц, 1H), 3,79 (с, 3H), 3,02-2,95 (м, 2H), 2,90-2,80 (м, 2H)
Стадия 5 - Синтез 5-хлор-8-метокси-1,2,3,4-тетрагидроизохинолина (EEE-6)
К раствору EEE-5 (195,0 мг, 0,679 ммоль) в ДХМ (7,00 мл) добавляют ТФК (0,70 мл), затем водный HCHO (37%, 110 мг, 1,36 ммоль). После добавления, реакционную смесь перемешивают при 25°C в течение 3 часов. Реакционную смесь разбавляют в H2O и нейтрализуют насыщ. Na2CO3. Смесь разделяют между EtOAc и H2O. Органический слой сушат над Na2SO4 и концентрируют с получением неочищенного продукта, который очищают преп.-ТСХ (EtOAc:петролейный эфир=1:1) с получением промежуточного соединения (170 мг), которое суспендируют в водн. HCl (24%, 2 мл) и нагревают при 110°C в течение 3 часов. Реакционную смесь нейтрализуют насыщ. Na2CO3, затем разделяют между EtOAc и H2O. Органический слой выпаривают с получением неочищенного продукта, который очищают преп.-ТСХ (EtOAc:петролейный эфир=1:0) с получением продукта EEE-6 (50,0 мг, 37%) в виде желтой камеди. 1H ЯМР (400 MГц, ДМСО-d6) δ ч./млн. 7,22 (д, J=8,8 Гц, 1H), 6,81 (д, J=8,8 Гц, 1H), 3,76 (с, 3H), 3,70 (с, 2H), 2,91 (т, J=6,0 Гц, 2H), 2,57 (т, J=5,8 Гц, 2H)
Стадия 6 - Синтез 5-хлор-1,2,3,4-тетрагидроизохинолин-8-ола (EEE-7)
К раствору EEE-6 (50,0 мг, 0,253 ммоль) в ДХМ (4,00 мл) добавляют BBr3 (0,40 мл, 4,20 ммоль) при 0°C. После добавления, реакционную смесь перемешивают при 25°C в течение 1 часа. Реакционную смесь гасят MeOH и подщелачивают насыщ. K2CO3 до pH 11-12. Смесь экстрагируют EtOAc. Органический слой сушат над Na2SO4 и концентрируют с получением неочищенного продукта, который очищают преп.-ТСХ (MeOH:ДХМ=1:10) с получением EEE-7 (50 мг, >99%) в виде желтой камеди и применяют сразу на следующей стадии. 1H ЯМР (400 MГц, CDCl3) δ ч./млн. 7,05 (д, J=8,5 Гц, 1H), 6,51 (д, J=9,3 Гц, 1H), 4,00 (с, 2H), 3,16 (т, J=6,0 Гц, 2H), 2,79 (т, J=6,1 Гц, 2H)
Стадия 7 - Синтез трет-бутил 5-хлор-8-гидрокси-3,4-дигидроизохинолин-2(1H)-карбоксилата (TP-19)
К раствору EEE-7 (50 мг, 0,272 ммоль) в ДХМ (5,00 мл) и MeOH (1,00 мл) добавляют Boc2O (65 мг, 0,300 ммоль), затем Et3N (68,9 мг, 0,681 ммоль). После добавления, реакционную смесь перемешивают при 25°C в течение 2,5 часов. Реакционную смесь нейтрализуют водн. HCl (0,1 M) до pH 4-5 при 0°C. Полученную смесь разделяют между ДХМ и H2O. Органический слой разделяют, промывают насыщенным раствором соли, сушат над Na2SO4 и концентрируют с получением неочищенного продукта, который очищают преп.-ТСХ (MeOH:ДХМ=1:10) с получением TP-19 (50 мг, 65%) в виде белого твердого вещества.
Примеры 154 & 155 получают по методике Примера 78 со схемы CC с применением подходящего NBoc-защищенного тетрагидроизохинолина на стадии 1.
TP-14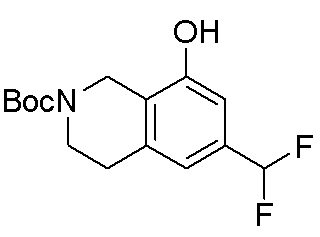
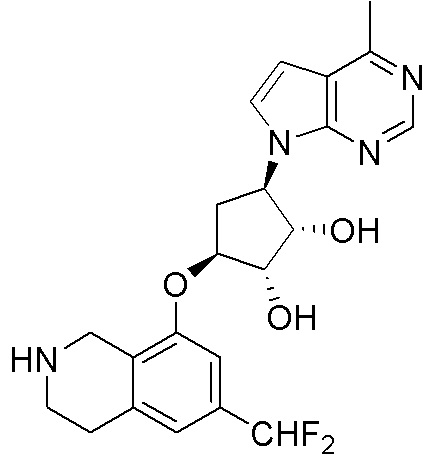
[M+1]
1H ЯМР (400 MГц, D2O) δ ч./млн. 8,85 (с, 1H), 7,81 (д, J=3,8 Гц, 1H), 7,14-7,05 (м, 3H), 6,89-6,59 (м, 1H), 5,35 (кв, J=9,0 Гц, 1H), 4,86-4,82 (м, 1H), 4,70-4,67 (м, 1H), 4,37 (с, 2H), 4,32 (д, J=4,8 Гц, 1H), 3,49 (т, J=6,1 Гц, 2H), 3,15-3,02 (м, 3H), 2,90 (с, 3H), 2,26-2,15 (м, 1H)
TP-8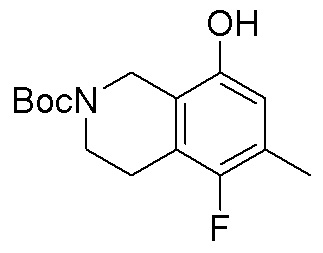
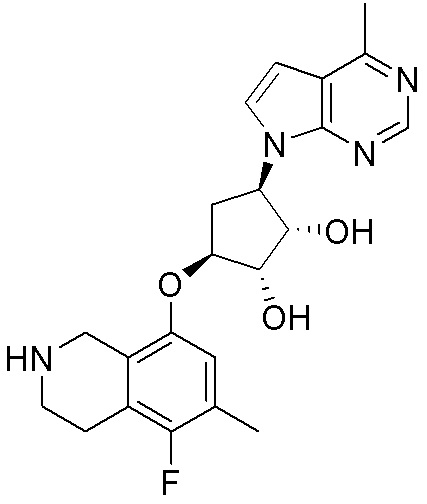
[M+1]
1H ЯМР (400 MГц, MeOD-d4) δ ч./млн. 8,62 (с, 1H), 7,54 (д, J=3,8 Гц, 1H), 6,86 (д, J=5,8 Гц, 1H), 6,74 (д, J=3,8 Гц, 1H), 5,18 (д, J=8,8 Гц, 1H), 4,73 (дд, J=5,0, 8,8 Гц, 1H), 4,66 (шс, 1H), 4,24 (с, 2H), 4,17 (д, J=4,5 Гц, 1H), 3,39 (т, J=6,3 Гц, 2H), 3,00-2,93 (м, 3H), 2,72 (с, 3H), 2,26 (д, J=1,8 Гц, 3H), 2,21 (ддд, J=4,1, 9,3, 13,9 Гц, 1H)
Пример 156 (Схема FFF) - (1S,2S,3R,5S)-3-(4-метил-7H-пирроло[2.3-d]пиримидин-7-ил)-5-((5,6,7,8-тетрагидропиридо[4,3-d]пиримидин-4-ил)окси)циклопентан-1,2-диол (FFF-4)
Схема FFF
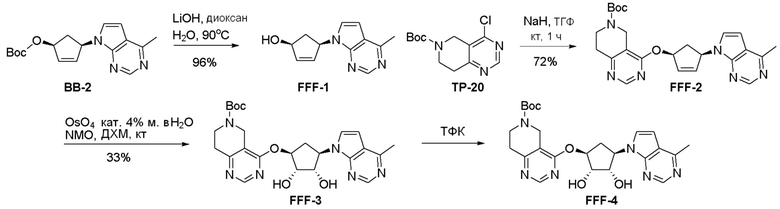
Стадия 1: Синтез (1S,4R)-4-(4-метил-7H-пирроло[2.3-d]пиримидин-7-ил)циклопент-2-ен-1-ола (FFF-1)
К раствору BB-2 (650 мг, 2,06 ммоль) в диоксане (0,3 мл) и H2O (0,3 мл) добавляют гидроксид лития (494 мг, 20,6 ммоль). Реакционную смесь нагревают при 90°C в течение ночи. Реакционную смесь охлаждают до к.т., разбавляют H2O, экстрагируют 20% изопропиловым спиртом/ДХМ, органические слои объединяют и очищают ISCO 4 г с 100% EtOAc - 10% MeOH/EtOAc с получением 424 мг FFF-1 (96% выход) в виде бесцветного масла, которое затвердевает при выстаивании.
ЖХМС [M+1] 216,15. 1H ЯМР (400 MГц, ХЛОРОФОРМ-d) δ ч./млн. 2,22 (дт, J=15,16, 2,14 Гц, 1 H) 2,73 (с, 3 H) 3,00 (ддд, J=15,25, 9,20, 7,70 Гц, 1 H) 4,89 (шс, 1 H) 5,37 (дкв, J=9,23, 2,22 Гц, 1 H) 5,58 (шс, 1 H) 5,85 (дд, J=5,50, 2,45 Гц, 1 H) 6,25-6,33 (м, 1 H) 6,54 (д, J=3,55 Гц, 1 H) 7,23 (д, J=3,55 Гц, 1 H) 8,69 (с, 1 H)
Стадия 2: Синтез трет-бутил 4-(((1S,4R)-4-(4-метил-7H-пирроло[2.3-d]пиримидин-7-ил)циклопент-2-ен-1-ил)окси)-7,8-дигидропиридо[4,3-d]пиримидин-6(5H)-карбоксилата (FFF-2)
К раствору FFF-1 (100 мг, 0,465 ммоль) в ТГФ (3 мл, c=0,2 M) добавляют гидрид натрия (27,9 мг, 0,697 ммоль) небольшими партиями. После перемешивания при кт в течение 10 мин добавляют трет-бутил 4-хлор-7,8-дигидропиридо[4,3-d]пиримидин-6(5H)-карбоксилат (TP-20) (125 мг, 0,465 ммоль). Полученную реакционную смесь перемешивают при кт в течение 3,5 ч, и затем гасят H2O, разделяют между EtOAc (20 мл) и H2O (20 мл). Органическую фазу разделяют, промывают насыщенным раствором соли, сушат над Na2SO4, концентрируют, очищают хроматографией на колонке с 5-10% MeOH/EtOAc с получением 150 мг FFF-2 (72% выход) в виде светло-желтого пенистого твердого вещества.
ЖХМС [M+1] 449,15. 1H ЯМР (400 MГц, ХЛОРОФОРМ-d) δ ч./млн. 1,44 (с, 9 H) 1,87 (д, J=15,04 Гц, 1 H) 2,66 (с, 3 H) 2,80 (т, J=4,77 Гц, 2 H) 3,15 (дт, J=15,31, 7,81 Гц, 1 H) 3,57-3,65 (м, 1 H) 3,65-3,75 (м, 1 H) 4,33 (д, J=17,61 Гц, 1 H) 4,44 (д, J=17,12 Гц, 1 H) 5,98 (дт, J=4,31, 1,94 Гц, 1 H) 6,01-6,07 (м, 1 H) 6,07-6,14 (м, 1 H) 6,33 (шс, 1 H) 6,52 (д, J=3,18 Гц, 1 H) 7,20 (д, J=3,67 Гц, 1 H) 8,51 (с, 1 H) 8,70 (с, 1 H)
Стадия 3: Синтез трет-бутил 4-(((1S,2S,3S,4R)-2,3-дигидрокси-4-(4-метил-7H-пирроло[2.3-d]пиримидин-7-ил)циклопентил)окси)-7,8-дигидропиридо[4,3-d]пиримидин-6(5H)-карбоксилата (FFF-3)
Соединение FFF-2 обрабатывают по методике со стадии 3 со схемы BB с получением FFF-3 (50 мг, 33%).
ЖХМС [M+1] 483,20. 1H ЯМР (400 MГц, ХЛОРОФОРМ-d) δ ч./млн. 1,49 (с, 9 H) 2,36-2,50 (м, 1 H) 2,76 (с, 3 H) 2,91 (т, J=5,81 Гц, 2 H) 3,06-3,22 (м, 1 H) 3,62-3,84 (м, 2 H) 4,13 (с, 1 H) 4,36-4,50 (м, 2 H) 4,53 (д, J=6,24 Гц, 1 H) 4,93-5,05 (м, 1 H) 5,45 (тд, J=7,12, 3,12 Гц, 1 H) 5,64 (шс, 1 H) 6,61 (д, J=3,67 Гц, 1 H) 7,23 (д, J=3,67 Гц, 1 H) 8,64 (с, 1 H) 8,76 (с, 1 H)
Стадия 4: Синтез (1S,2S,3R,5S)-3-(4-метил-7H-пирроло[2.3-d]пиримидин-7-ил)-5-((5,6,7,8-тетрагидропиридо[4,3-d]пиримидин-4-ил)окси)циклопентан-1,2-диола (FFF-4)
Соединение FFF-3 обрабатывают в стандартных условиях снятия защиты по методике стадии 3 со схемы CC с получением FFF-4 (24 мг, 100%).
ЖХМС [M+1] 383,10. 1H ЯМР (400 MГц, ДМСО-d6) δ ч./млн. 2,00-2,11 (м, 1 H) 2,92 (с, 3 H) 2,94-3,02 (м, 1 H) 3,06 (шс, 2 H) 3,48 (шс, 2 H) 4,11 (д, J=3,42 Гц, 1 H) 4,25 (шс, 2 H) 4,66 (дд, J=8,86, 4,58 Гц, 1 H) 5,22 (кв, J=8,93 Гц, 1 H) 5,28-5,37 (м, 1 H) 7,20 (шс, 1 H) 8,09 (шс, 1 H) 8,69 (с, 1 H) 9,18 (шс, 1 H) 9,71 (шс, 1 H) 9,87 (шс, 1 H)
Пример 157 (Схема GGG) - (1S,2S,3R,5S)-3-(4-метил-7H-пирроло[2.3-d]пиримидин-7-ил)-5-((5,6,7,8-тетрагидро-2,7-нафтиридин-1-ил)окси)циклопентан-1,2-диол (GGG-5)
Схема GGG
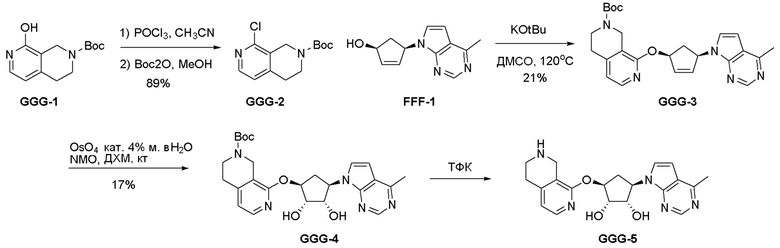
Стадия 1: Синтез трет-бутил 8-хлор-3,4-дигидро-2,7-нафтиридин-2(1H)-карбоксилата (GGG-2)
К раствору трет-бутил 8-гидрокси-3,4-дигидро-2,7-нафтиридин-2(1H)-карбоксилата (GGG-1) (250 мг, 0,999 ммоль) в CH3CN (2,5 мл) медленно добавляют POCl3 (2,5 мл) и нагревают при 90°C в течение ночи. Реакционную смесь нейтрализуют станд. NaHCO3, растворитель удаляют на роторном испарителе. Остаток добавляют MeOH 10 мл, в суспензию добавляют (BOC)2 (337 мг, 1,50 ммоль, 0,355 мл) и ДИПЭА (258 мг, 2,00 ммоль, 0,331 мл), перемешивают при кт в течение 30 мин. Органический растворитель выпаривают на роторном испарителе, добавляют EtOAc и H2O, экстрагируют EtOAc, органический слой концентрируют, очищают хроматографией на колонке с 30% EtOAc/гептаном с получением 240 мг GGG-2 (89% выход) в виде бесцветного масла.
ЖХМС [M+1] 269,10. 1H ЯМР (400 MГц, ХЛОРОФОРМ-d) δ ч./млн. 1,51 (с, 9 H) 2,84 (т, J=5,69 Гц, 2 H) 3,66 (т, J=5,81 Гц, 2 H) 4,56 (с, 2 H) 7,03 (д, J=5,01 Гц, 1 H) 8,17 (д, J=5,01 Гц, 1 H)
Стадия 2: Синтез трет-бутил 8-(((1S,4R)-4-(4-метил-7H-пирроло[2.3-d]пиримидин-7-ил)циклопент-2-ен-1-ил)окси)-3,4-дигидро-2,7-нафтиридин-2(1H)-карбоксилата (GGG-3)
Раствор FFF-1 (92 мг, 0,43 ммоль) и GGG-2 (115 мг, 0,427 ммоль) в ДМСО (4,27 мл, c=0,1 M) обрабатывают бутоксидом калия (61,8 мг, 0,534 ммоль, 0,534 мл, 1,0 M). Реакционную смесь нагревают до 120°C в течение 15 мин. Реакционную смесь охлаждают до к.т., разбавляют H2O и EtOAc (10 мл каждый). Водную фазу экстрагируют EtOAc (10 мл). Объединенные органические фазы промывают H2O (2×15 мл), насыщенным раствором соли (15 мл) и сушат над Na2SO4. Образец концентрируют и очищают препаративной ВЭЖХ с получением 40 мг GGG-3 в виде коричневого твердого вещества (21% выход).
ЖХМС [M+1] 448,20. 1H ЯМР (400 MГц, ХЛОРОФОРМ-d) δ ч./млн. 1,45-1,56 (м, 9 H) 1,94 (д, J=14,92 Гц, 1 H) 2,63 (с, 3 H) 2,68-2,81 (м, 2 H) 3,20 (дт, J=15,13, 7,78 Гц, 1 H) 3,55-3,69 (м, 2 H) 4,33-4,45 (м, 1 H) 4,49 (шс, 1 H) 6,06 (шс, 2 H) 6,11 (д, J=4,03 Гц, 1 H) 6,45 (шс, 1 H) 6,62 (шс, 1 H) 6,68 (д, J=5,01 Гц, 1 H) 7,39 (шс, 1 H) 7,92 (д, J=5,14 Гц, 1 H) 8,84 (шс, 1 H)
Стадия 3: Синтез трет-бутил 8-(((1S,2S,3S,4R)-2,3-дигидрокси-4-(4-метил-7H-пирроло[2.3-d]пиримидин-7-ил)циклопентил)окси)-3,4-дигидро-2,7-нафтиридин-2(1H)-карбоксилата (GGG-4)
Соединение GGG-3 обрабатывают по методике со стадии 3 со схемы BB с получением GGG-4 (7,3 мг, 17%).
ЖХМС [M+1] 481,90. 1H ЯМР (400 MГц, ХЛОРОФОРМ-d) δ ч./млн. 1,49 (с, 10 H) 2,43-2,54 (м, 1 H) 2,75 (с, 3 H) 2,81 (т, J=5,75 Гц, 2 H) 3,03 (шс, 1 H) 3,63-3,71 (м, 2 H) 4,38 (шс, 1 H) 4,47 (шс, 3 H) 4,66 (шс, 1 H) 5,03-5,12 (м, 1 H) 5,25-5,32 (м, 1 H) 5,48 (с, 1 H) 6,60 (д, J=3,55 Гц, 1 H) 6,77 (д, J=5,26 Гц, 1 H) 7,29 (д, J=3,79 Гц, 1 H) 7,93 (д, J=5,14 Гц, 1 H) 8,77 (с, 1 H)
Стадия 4: Синтез (1S,2S,3R,5S)-3-(4-метил-7H-пирроло[2.3-d]пиримидин-7-ил)-5-((5,6,7,8-тетрагидро-2,7-нафтиридин-1-ил)окси)циклопентан-1,2-диола (GGG-5)
Соединение GGG-4 обрабатывают в стандартных условиях снятия защиты по методике стадии 3 со схемы CC с получением GGG-5 (6 мг, 90%).
ЖХМС [M+1] 382,201H ЯМР (400 MГц, ДМСО-d6) δ ч./млн. 1,93-2,02 (м, 1 H) 2,86-2,92 (м, 3 H) 2,92-2,98 (м, 1 H) 3,01 (т, J=5,99 Гц, 2 H) 3,57 (с, 2 H) 4,08 (д, J=4,03 Гц, 1 H) 4,20 (шс, 2 H) 4,66 (дд, J=8,93, 4,52 Гц, 1 H) 5,15-5,23 (м, 1 H) 5,23-5,29 (м, 1 H) 6,91 (д, J=5,26 Гц, 1 H) 7,16 (д, J=3,30 Гц, 1 H) 7,99-8,06 (м, 2 H) 9,13 (с, 1 H) 9,52 (шс, 1 H) 9,64 (шс, 1 H)
Пример 158 (Схема HHH) - (1S,2R,3R,5S)-3-((2-метилпиримидин-4-ил)окси)-5-((1,2,3,4-тетрагидроизохинолин-8-ил)окси)циклопентан-1,2-диол (HHH-6)
Схема HHH
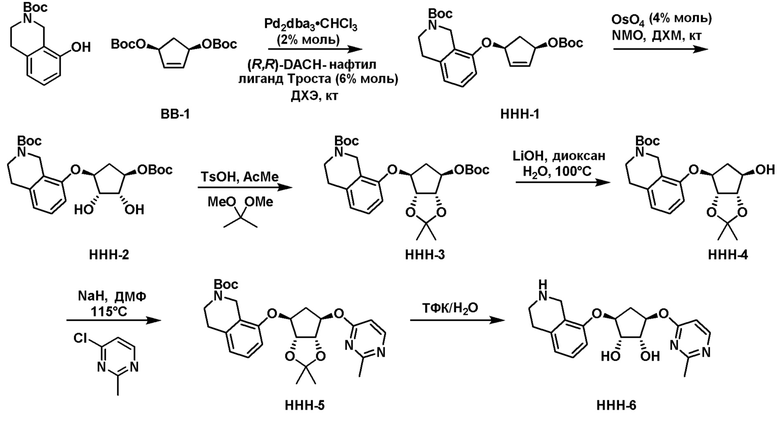
Стадия 1: Синтез трет-бутил 8-(((1S,4R)-4-((трет-бутоксикарбонил)окси)циклопент-2-ен-1-ил)окси)-3,4-дигидроизохинолин-2(1H)-карбоксилата (HHH-1)
Пробирка A: В высушенную в печи реакционную пробирку, оборудованную магнитной мешалкой и охлажденную в потоке аргона, добавляют продукт присоединения трис(бензилиденацетон)дипалладия(0) и хлороформа (44,7 мг, 0,043 ммоль) и MFCD02684551 (R,R)-DACH-нафтил лиганд Троста (102 мг, 0,129 ммоль). Пробирку продувают в вакууме аргоном в динамическом вакууме и добавляют ДХЭ (3,6 мл), который барботируют с аргоном в течение 30 минут. Раствор перемешивают в течение 30 минут при кт с получением ярко-оранжевого раствора лигированного катализатора. На этой стадии получают пробирку B.
Пробирка B: В высушенную в печи реакционную пробирку, оборудованную магнитной мешалкой и охлажденную в потоке аргона, добавляют трет-бутил 8-гидрокси-3,4-дигидроизохинолин-2(1H)-карбоксилат (538 мг, 2,16 ммоль) и ди-трет-бутил-((1R,3S)-циклопент-4-ен-1,3-диил)бис(карбонат) (BB-1) (полученный как описано в J. Am. Chem. Soc, 2006, 128, 6054-6055) (778 мг, 2,59 ммоль). Пробирку продувают в вакууме аргоном в динамическом вакууме и добавляют ДХЭ (3,6 мл), который барботируют с аргоном в течение 30 минут, затем добавляют содержимое пробирки A через герметичный шприц. Реакционную смесь перемешивают под аргоном при кт в течение 14 часов. Реакционную смесь концентрируют в вакууме и очищают флэш-хроматографией на колонке (24 г SiO2, Isco, 100% Гепт. до 100% EtOAc, 20 мл фракции) с получением HHH-1 (973 мг, >95%) в виде желтой пены. ЖХМС [M+H-Boc-изобутилен]=276 наблюдаемый; 1H ЯМР (400 MГц, ХЛОРОФОРМ-d) δ ч./млн. 7,12 (т, J=7,9 Гц, 1H), 6,75 (д, J=7,7 Гц, 1H), 6,72 (д, J=8,2 Гц, 1H), 6,22 (тд, J=1,4, 5,7 Гц, 1H), 6,18-6,09 (м, 1H), 5,47 (т, J=5,8 Гц, 1H), 5,15 (т, J=5,7 Гц, 1H), 4,59-4,40 (м, J=8,2 Гц, 2H), 3,74-3,54 (м, 2H), 3,05 (тд, J=7,4, 14,5 Гц, 1H), 2,81 (т, J=5,7 Гц, 2H), 1,96 (тд, J=4,5, 14,5 Гц, 1H), 1,52-1,47 (м, 18H).
Стадия 2: Синтез трет-бутил 8-(((1S,2S,3R,4R)-4-((трет-бутоксикарбонил)окси)-2,3-дигидроксициклопентил)окси)-3,4-дигидроизохинолин-2(1H)-карбоксилата (HHH-2)
В сцинтилляционную пробирку, оборудованную магнитной мешалкой и содержащую HHH-1 (225 мг, 0,521 ммоль), добавляют ДХМ (2,6 мл). В раствор добавляют 4-метилморфолин-N-оксид (NMO) (0,32 мл, 1,50 ммоль) в виде 50% масс. раствора в воде, затем по каплям добавляют тетраоксид осмия (130 мкл, 0,02 ммоль) в виде 4% масс. раствора в воде. Реакционную смесь перемешивают при кт в течение 23 часов. Реакционную смесь переносят в делительную воронку с ДХМ, разбавляют водой и затем разбавляют 1M NaHSO3. Фазы разделяют, и водную фазу экстрагируют 3 порциями ДХМ. Объединенные органические экстракты сушат (MgSO4), фильтруют, и концентрируют в вакууме. Неочищенный остаток очищают флэш-хроматографией на колонке (12 г SiO2, Isco, 100% Гепт. до 100% EtOAc, 9 мл фракции) с получением HHH-2 (211 мг, 87%) в виде белого твердого вещества. ЖХМС [M+H-Boc-изобутилен]=310 наблюдаемый; 1H ЯМР (400 MГц, ХЛОРОФОРМ-d) δ ч./млн. 7,12 (т, J=7,9 Гц, 1H), 6,76 (д, J=7,7 Гц, 1H), 6,71 (д, J=8,2 Гц, 1H), 4,90 (тд, J=5,8, 9,0 Гц, 1H), 4,69-4,59 (м, 1H), 4,45 (с, 2H), 4,34 (с, 1H), 4,26 (шс, 1H), 3,63 (д, J=5,7 Гц, 2H), 2,81 (т, J=5,7 Гц, 3H), 2,03-1,91 (м, J=5,7, 9,3 Гц, 1H), 1,53-1,48 (м, 18H).
Стадия 3: Синтез трет-бутил 8-(((3aR,4S,6R,6aS)-6-((трет-бутоксикарбонил)окси)-2,2-диметилтетрагидро-4H-циклопента[d][1.3]диоксол-4-ил)окси)-3,4-дигидроизохинолин-2(1H)-карбоксилата (HHH-3)
В реакционную пробирку, оборудованную магнитной мешалкой и содержащую HHH-2 (211 мг, 0,453 ммоль), добавляют ацетон (0,29 мл), 4-моногидрат толуолсульфоновой кислоты (172 мг, 0,906 ммоль) и 2,2-диметоксипропан (0,56 мл, 4,53 ммоль). Реакционную смесь перемешивают при кт в течение 1 часа. Реакционную смесь переносят в делительную воронку с EtOAc и водой. Двухфазную смесь разбавляют насыщ. NaHCO3 и фазы разделяют. Органическую фазу промывают 1 порцией наполовину насыщ. NaHCO3, и объединенные водные слои повторно экстрагируют 1 порцией EtOAc. Объединенные органические фазы сушат (MgSO4), фильтруют, и концентрируют в вакууме. Неочищенный остаток очищают флэш-хроматографией на колонке (12 г SiO2, Isco, 100% гепт. до 100% EtOAc, 9 мл фракции) с получением HHH-3 (140,5 мг, 61%) в виде белого твердого вещества. ЖХМС [M+H-Boc-изобутилен]=350 наблюдаемый; 1H ЯМР (400 MГц, ХЛОРОФОРМ-d) δ ч./млн. 7,12 (с, 1H), 6,73 (т, J=8,1 Гц, 2H), 4,94 (д, J=4,6 Гц, 1H), 4,80-4,66 (м, 3H), 4,61-4,43 (м, 2H), 3,63 (с, 2H), 2,80 (т, J=5,6 Гц, 2H), 2,47 (тд, J=5,4, 15,4 Гц, 1H), 2,25 (тд, J=1,5, 15,4 Гц, 1H), 1,49 (с, 12H), 1,45 (с, 9H), 1,31 (с, 3H).
Стадия 4: Синтез трет-бутил 8-(((3aR,4S,6R,6aS)-6-гидрокси-2,2-диметилтетрагидро-4H-циклопента[d][1.3]диоксол-4-ил)окси)-3,4-дигидроизохинолин-2(1H)-карбоксилата (HHH-4)
В микроволновую пробирку, оборудованную магнитной мешалкой и содержащую HHH-3 (134,5 мг, 0,266 ммоль), добавляют диоксан (1,3 мл) и воду (1,3 мл). В раствор добавляют гидроксид лития (63,7 мг, 2,66 ммоль) и пробирку герметично закрывают тефлоновой крышкой. Пробирку помещают в нагревательный блок и перемешивают при 100°C в течение 19 часов. Пробирку удаляют из нагревательного блока и охлаждают до кт. Раствор переносят в делительную воронку с EtOAc и разбавляют водой. Фазы разделяют, и водную фазу экстрагируют 3 порциями EtOAc. Объединенные органические экстракты сушат (MgSO4), фильтруют, и концентрируют в вакууме. Неочищенный остаток очищают флэш-хроматографией на колонке (4г SiO2, Isco, 100% Гепт. до 100% EtOAc, 9 мл фракции) с получением HHH-4 (97,3 мг, 90%) в виде белой пены. ЖХМС [M+H-Boc]=306 наблюдаемый; 1H ЯМР (400 MГц, ХЛОРОФОРМ-d) δ ч./млн. 7,17 (т, J=7,9 Гц, 1H), 6,84 (д, J=8,3 Гц, 1H), 6,81 (д, J=7,7 Гц, 1H), 4,75 (дд, J=5,4, 7,8 Гц, 2H), 4,67 (дд, J=1,3, 5,6 Гц, 1H), 4,55-4,33 (м, 2H), 4,27 (д, J=4,6 Гц, 1H), 3,78-3,49 (м, 2H), 2,82 (т, J=5,7 Гц, 2H), 2,42 (тд, J=5,0, 15,0 Гц, 1H), 2,10 (д, J=15,0 Гц, 1H), 1,49 (с, 9H), 1,46 (с, 3H), 1,31 (с, 3H).
Стадия 5: Синтез трет-бутил 8-(((3aR,4S,6R,6aS)-2,2-диметил-6-((2-метилпиримидин-4-ил)окси)тетрагидро-4H-циклопента[d][1.3]диоксол-4-ил)окси)-3,4-дигидроизохинолин-2(1H)-карбоксилата (HHH-5)
В высушенную в печи реакционную пробирку, оборудованную магнитной мешалкой и содержащую HHH-4 (43 мг, 0,110 ммоль), добавляют ДМФ (0,53 мл) и гидрид натрия (8,5 мг, 0,210 ммоль) в виде 60% масс. дисперсии в минеральном масле. Раствор перемешивают при кт в течение 1 часа с получением темно-коричневого раствора алкоксида натрия. В раствор добавляют 4-хлор-2-метилпиримидин (16,4 мг, 0,127 ммоль) и пробирку помещают в нагревательный блок и нагревают при 115°C в течение 16 часов. Реакционную смесь осторожно гасят добавлением по каплям воды. Раствор затем разбавляют водой и переносят в делительную воронку с EtOAc. Фазы разделяют, и водную фазу экстрагируют 4 порциями 3:1 смеси ДХМ/ИПС. Объединенные органические экстракты промывают 1 порцией насыщенного раствора соли, сушат (MgSO4), фильтруют и концентрируют в вакууме. Неочищенный продукт HHH-5 применяют на следующей стадии без дальнейшей очистки. ЖХМС [M+H-Boc]=398 наблюдаемый.
Стадия 6: Синтез (1S,2R,3R,5S)-3-((2-метилпиримидин-4-ил)окси)-5-((1,2,3,4-тетрагидроизохинолин-8-ил)окси)циклопентан-1,2-диола (HHH-6)
В круглодонную колбу, оборудованную магнитной мешалкой и содержащую HHH-5 (53 мг, 0,11 ммоль, неочищенный продукт со стадии 5) добавляют воду (1,0 мл) и ТФК (0,5 мл). Раствор перемешивают при кт в течение 1 часа. Реакционную смесь переносят в делительную воронку с ДХМ и доводят до основного pH насыщ. водн. NaHCO3. Фазы разделяют, и водную фазу экстрагируют 4 порциями 3:1 смеси ДХМ/ИПС. Объединенные органические экстракты промывают 1 порцией насыщ. водн. NaHCO3, сушат (MgSO4), фильтруют и концентрируют в вакууме. Неочищенный остаток очищают преп.-ВЭЖХ (колонка Lux Cellulose-1 4,6×100 мм 3мк, 20% MeOH/ДЭА @ 120 бар, 4 мл/мин) с получением HHH-6 (7,06 мг, 19% за 2 стадии) в виде белого твердого вещества. ЖХМС [M+H]=358 наблюдаемый; [α]22D=+3,7° (c=0,1, MeOH); 1H ЯМР (400 MГц, МЕТАНОЛ-d4) δ ч./млн. 8,05 (д, J=6,4 Гц, 1H), 7,15 (т, J=7,9 Гц, 1H), 6,84 (д, J=8,1 Гц, 1H), 6,79 (д, J=7,6 Гц, 1H), 6,67 (д, J=6,5 Гц, 1H), 4,68 (шс, 2H), 4,61 (тд, J=4,2, 8,1 Гц, 1H), 4,26 (т, J=4,3 Гц, 1H), 4,12 (тд, J=4,9, 7,4 Гц, 1H), 4,02-3,90 (м, 3H), 2,90 (т, J=5,7 Гц, 2H), 2,74 (тд, J=7,7, 15,0 Гц, 1H), 2,47 (с, 3H), 1,62 (тд, J=4,7, 14,6 Гц, 1H).
Синтез 2-(бензилокси)-5-фтор-4-метилбензойной кислоты (III-4)
Схема III
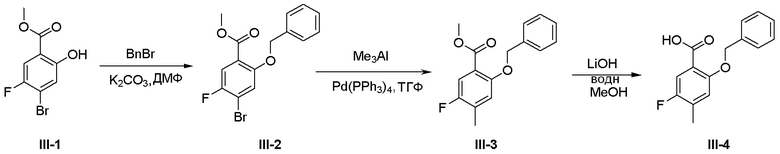
Стадия 1 - Синтез метил 2-(бензилокси)-4-бром-5-фторбензоата (III-2)
К раствору метил 4-бром-5-фтор-2-гидроксибензоата III-1 (1,62 г, 6,505 ммоль) в ДМФ (20 мл) добавляют K2CO3 (2,7 г, 19,5 ммоль) и BnBr (2,23 г, 13 ммоль). Смесь перемешивают при 16°C в течение 3 ч. Смесь разбавляют водой (100 мл). Затем смесь экстрагируют EtOAc (50 мл×2). Органические слои собирают, сушат и выпаривают с получением неочищенного продукта, который очищают флэш-хроматографией, элюируя петролейным эфиром/EtOAc 0-10% с получением III-2 (1,8 г, 82%) в виде белого твердого вещества, и применяют сразу на следующей стадии.
Стадия 2 - Синтез метил 2-(бензилокси)-5-фтор-4-метилбензоата (III-3)
Смесь III-2 (2 г, 5,87 ммоль), Pd(Ph3P)4 (339 мг, 0,293 ммоль) в ТГФ (2 0 мл) дегазируют с N2 четыре раза, затем добавляют AlMe3 (7,87 мл, 15,7 ммоль, 2M) при 0°, затем реакционную смесь перемешивают при 80°C в течение 24 часов. Реакционную смесь затем гасят водн. тетрагидратом тартрата калия-натрия, экстрагируют EtOAc три раза, объединенные органические слои сушат над Na2SO4, удаляют растворитель в вакууме, остаток очищают флэш-биотаж (петролейный эфир/EtOAc=0-5%) с получением III-3 (660 мг, 41%) в виде желтого масла. 1H ЯМР (400 MГц, CDCl3) δ ч./млн. 7,57-7,47 (м, 3H), 7,43-7,37 (м, 2H), 7,36-7,30 (м, 1H), 6,84 (д, J=6,0 Гц, 1H), 5,14 (с, 2H), 3,90 (с, 3H), 2,29 (д, J=2,0 Гц, 3H)
Стадия 3 - Синтез 2-(бензилокси)-5-фтор-4-метилбензойной кислоты (III-4)
К раствору III-3 (0,71 г, 2,59 ммоль) в MeOH (4 мл) добавляют раствор LiOH.H2O (326 г, 7,77 ммоль) в H2O (4 мл). Реакционную смесь перемешивают при 20°C в течение 2 часов. Реакционный раствор выпаривают для удаления большей части метанола, и затем остаток доводят до pH~2 1N HCl. Образовавшееся белое твердое вещество экстрагируют EtOAc (20 мл×2). Органические слои сушат и выпаривают с получением III-4 (670 мг, 99,5%) в виде белого твердого вещества. 1H ЯМР (400 MГц, CDCl3) δ ч./млн. 10,76 (шс, 1H), 7,83 (д, J=9,5 Гц, 1H), 7,52-7,37 (м, 5H), 6,96 (д, J=6,0 Гц, 1H), 5,26 (с, 2H), 2,35 (с, 3H)
Пример 159 (Схема JJJ) - (1S,2S,3S,5R)-3-(2-(аминометил)-4-хлорфенокси)-5-(4-метил-7H-пирроло[2.3-d]пиримидин-7-ил)циклопентан-1,2-диол (JJJ-3)
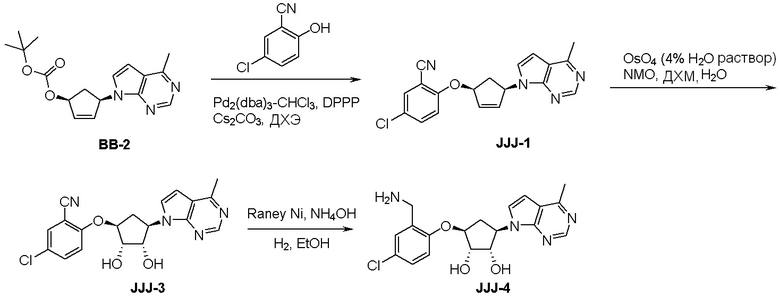
Стадия 1 - Синтез 5-хлор-2-(((1S,4R)-4-(4-метил-7H-пирроло[2.3-d]пиримидин-7-ил)циклопент-2-ен-1-ил)окси)бензонитрила (JJJ-1)
В сухую микроволновую пробирку (продувают N2) добавляют BB-2 (100 мг, 0,317 ммоль), 5-хлор-2-гидроксибензонитрил (56 мг, 0,37 ммоль), Cs2CO3 (114 мг, 0,35 ммоль), Pd2(dba)3 (8,2 мг, 0,008 ммоль) и DPPP (7,85 мг, 0,019 ммоль). Затем пробирку продувают N2 три раза и добавляют ДХЭ (1,5 мл, продувают N2 в течение 30 мин). Черную смесь перемешивают при 20°C в течение 1 часа. Затем реакционную смесь непосредственно очищают преп.-ТСХ (петролейный эфир:EtOAc=1/4) с получением JJJ-1 (83 мг, 75%, в виде желтой камеди. ЖХМС [M+1] 351; 1H ЯМР (400 MГц, CDCl3) δ ч./млн. 8,77 (с, 1H), 7,56 (д, J=2,8 Гц, 1H), 7,50 (дд, J=2,6, 8,9 Гц, 1H), 7,41 (д, J=3,8 Гц, 1H), 7,00 (д, J=9,0 Гц, 1H), 6,63 (д, J=3,5 Гц, 1H), 6,40 (тд, J=1,9, 5,5 Гц, 1H), 6,26 (дд, J=2,4, 5,6 Гц, 1H), 6,14-6,08 (м, 1H), 5,44 (д, J=7,0 Гц, 1H), 3,21-3,11 (м, 1H), 2,73 (с, 3H), 2,04 (тд, J=3,2, 14,9 Гц, 1H)
Стадия 2 - Синтез 5-хлор-2-(((1S,2S,3S,4R)-2,3-дигидрокси-4-(4-метил-7H-пирроло[2.3-d]пиримидин-7-ил)циклопентил)окси)бензонитрила (JJJ-2)
К смеси JJJ-1 (83 мг, 0,237 ммоль) в ДХМ (6 мл)/H2O (0,2 мл) добавляют NMO (96 мг, 0,71 ммоль) и OsO4 (4% в т-BuOH, 80 мг, 0,013 ммоль) при 20°C. Коричневую смесь перемешивают при 20°C в течение 6 часов. Смесь разбавляют ДХМ (5 мл) и гасят насыщ. Na2SO3 (2 мл) и разделяют. Органический слой промывают насыщенным раствором соли (5 мл). Объединенные водные слои экстрагируют ДХМ (5 мл×2). Объединенные органические слои сушат над Na2SO4, фильтруют и концентрируют с получением неочищенного продукта (100 мг) в виде светло-желтого твердого вещества, которое очищают преп.-ТСХ (EtOAc:MeOH=10:1) с получением JJJ-2 (40 мг, 44%) в виде белого твердого вещества. ЖХМС [M+1] 385; 1H ЯМР (400 MГц, MeOD) δ ч./млн. 8,60 (с, 1H), 7,73 (д, J=2,5 Гц, 1H), 7,66 (д, J=3,5 Гц, 1H), 7,63 (дд, J=2,6, 9,2 Гц, 1H), 7,30 (д, J=9,0 Гц, 1H), 6,76 (д, J=3,5 Гц, 1H), 5,35-5,26 (м, 1H), 4,69 (дд, J=4,9, 8,4 Гц, 1H), 4,21 (д, J=4,5 Гц, 1H), 3,04 (ддд, J=6,8, 9,8, 14,8 Гц, 1H), 2,71 (с, 3H), 2,17 (ддд, J=3,0, 7,7, 14,7 Гц, 1H)
Стадия 3 - Синтез (1S,2S,3S,5R)-3-(2-(аминометил)-4-хлорфенокси)-5-(4-метил-7H-пирроло[2.3-d]пиримидин-7-ил)циклопентан-1,2-диола (JJJ-3)
Смесь JJJ-2 (40 мг, 0,104 ммоль) и Raney-Ni (8 мг) в EtOH (7 мл)/NH3.H2O (0,5 мл) дегазируют с H2 четыре раза. Смесь перемешивают при 20°C под H2 баллоном в течение 20 ч. Смесь фильтруют, и фильтрат концентрируют в вакууме с получением белого твердого вещества, которое очищают преп.-ТСХ (ДХМ:MeOH:NH3.H2O=10:1:0,1) с получением продукта (~20 мг) в виде желтой камеди и лиофилизируют. Продукт очищают преп-ВЭЖХ с получением JJJ-3 (9 мг, 22%). ЖХМС [M+1] 389; 1H ЯМР (400 MГц, MeOD) δ ч./млн. 8,60 (с, 1H), 7,56 (д, J=3,8 Гц, 1H), 7,42 (д, J=2,5 Гц, 2H), 7,21 (д, J=8,8 Гц, 1H), 6,74 (д, J=3,5 Гц, 1H), 5,11 (кв, J=9,1 Гц, 1H), 4,82-4,76 (м, 2H), 4,29 (дд, J=2,1, 5,1 Гц, 1H), 4,26-4,15 (м, 2H), 3,06-2,92 (м, 1H), 2,72 (с, 3H), 2,37 (ддд, J=5,0, 9,7, 14,4 Гц, 1H)
Примеры 160 & 161 получают по методике со схемы A с применением бромида (4-хлор-3-фторфенил)магния на стадии 8.
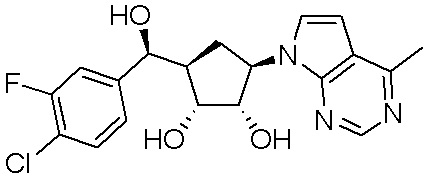
[M+1]
1H ЯМР (400 MГц, ДМСО-d6) δ ч./млн. 8,59 (с, 1H), 7,65 (д, J=3,8 Гц, 1H), 7,53 (т, J=8,0 Гц, 1H), 7,40 (дд, J=1,8, 10,5 Гц, 1H), 7,27 (дд, J=1,9, 8,2 Гц, 1H), 6,69 (д, J=3,5 Гц, 1H), 5,73 (д, J=4,8 Гц, 1H), 5,02-4,93 (м, 1H), 4,80 (д, J=7,0 Гц, 1H), 4,64-4,58 (м, 1H), 4,56 (д, J=3,8 Гц, 1H), 4,28 (тд, J=6,1, 9,9 Гц, 1H), 3,95-3,90 (м, 1H), 2,63 (с, 3H), 2,29-2,21 (м, 1H), 2,02 (тд, J=8,6, 12,9 Гц, 1H), 1,67-1,57 (м, 1H)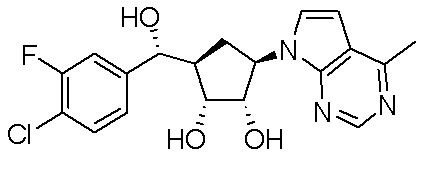
[M+1]
1H ЯМР (400 MГц, ДМСО-d6) δ ч./млн. 8,59 (с, 1H), 7,65 (д, J=3,5 Гц, 1H), 7,51 (т, J=8,0 Гц, 1H), 7,35 (дд, J=1,5, 10,5 Гц, 1H), 7,22 (дд, J=1,9, 8,2 Гц, 1H ), 6,71-6,67 (м, 1H), 5,71 (д, J=4,8 Гц, 1H), 4,97-4,88 (м, 1H), 4,86 (д, J=6,5 Гц, 1H), 4,82 (т, J=4,9 Гц, 1H), 4,67 (д, J=4,5 Гц, 1H), 4,26-4,18 (м, 1H), 3,90 (кв, J=4,9 Гц, 1H), 2,63 (с, 3H), 2,27-2,18 (м, 1H), 1,89-1,71 (м, 2H)
Пример 162 получают по методике Примера 99 со схемы NN с применением изохинолин-8-ола на стадии 2.
изохинолин-8-ол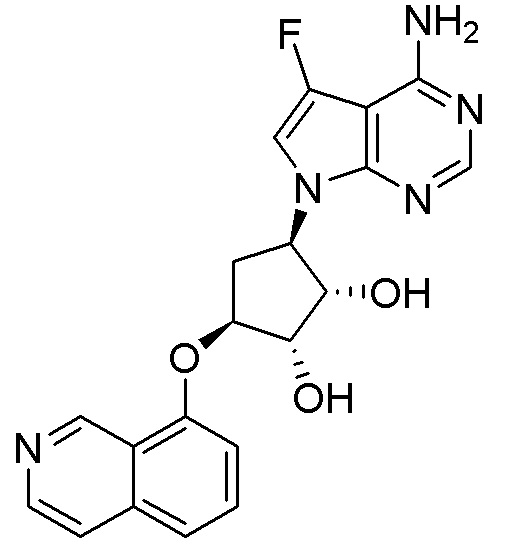
ЖХМС [M+1]
1H ЯМР (400 MГц, MeOD-d4) δ ч./млн. 9,62 (с, 1H), 8,46 (д, J=5,8 Гц, 1H), 8,04 (с, 1H), 7,79 (д, J=5,5 Гц, 1H), 7,72 (т, J=8,0 Гц, 1H), 7,52 (д, J=8,3 Гц, 1H), 7,21 (д, J=8,0 Гц, 1H), 7,13 (д, J=2,0 Гц, 1H), 5,19 (кв, J=9,0 Гц, 1H), 4,68 (шдд, J=4,9, 8,9 Гц, 2H), 4,33 (шд, J=4,8 Гц, 1H), 3,12-3,01 (м, 1H), 2,29-2,18 (м, 1H)
Синтез 6-гидрокси-3,4-дигидробензo[f][1,4]оксазепин-5(2H)-она (TP-21) и трет-бутил 6-гидрокси-2,3-дигидробензo[f][1,4]оксазепин-4(5H)-карбоксилата (TP-22) (Схема KKK)
Схема KKK
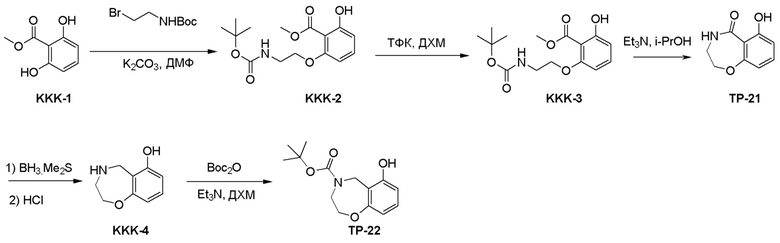
Стадия 1 - Синтез метил 2-(2-((трет-бутоксикарбонил)амино)этокси)-6-гидроксибензоата (KKK-2)
Раствор метил 2,6-дигидроксибензоата (1,2 г, 7,14 ммоль), трет-бутил (2-бромэтил)карбамата (2,33 г, 7,14 ммоль) и K2CO3 (2,5 г, 17,8 ммоль) в ДМФ(10 мл) перемешивают при 15°C в течение 32 ч. Воду добавляют к реакционной смеси и экстрагируют EtOAc три раза. Объединенные органические слои сушат над сульфатом натрия, концентрируют в вакууме, и остаток очищают флэш-хроматографией (петролейный эфир/EtOAc=10-20%) с получением соединения KKK-2 (900 мг, 41%) в виде белого твердого вещества и применяют сразу на следующей стадии.
Стадия 2 - Синтез метил 2-(2-((трет-бутоксикарбонил)амино)этокси)-6-гидроксибензоата (KKK-3)
К раствору соединения KKK-2 (900 мг, 2,89 ммоль) в ДХМ (10 мл) добавляют ТФК (2 мл) при 0-5°C, затем реакционную смесь перемешивают при 15°C в течение 2 ч. Растворитель удаляют и остаток (700 мг, >99%) сразу применяют на следующей стадии.
Стадия 3 - Синтез 6-гидрокси-3,4-дигидробензo[f][1,4]оксазепин-5(2H)-она (TP-21)
К раствору соединения KKK-3 (700 мг, 3,31 ммоль) в и-PrOH (6 мл) добавляют ТЭА (3,35 г, 33,1 ммоль) при 15°C, затем реакционную смесь перемешивают при 95°C в течение 6 ч. Растворитель удаляют, и остаток очищают флэш-хроматографией (20 г, петролейный эфир/EtOAc=10-50%) с получением соединения TP-21 (480 мг, 81%) в виде белого твердого вещества. 1H ЯМР (400 MГц, CDCl3) δ ч./млн. 12,61 (с, 1H), 7,33-7,29 (м, 1H), 6,70 (д, J=8,3 Гц, 1H), 6,57 (шс, 1H), 6,53 (д, J=8,0 Гц, 1H), 4,41-4,35 (м, 2H), 3,57 (кв, J=4,8 Гц, 2H)
Стадия 4 - Синтез 2,3,4,5-тетрагидробензo[f][1,4]оксазепин-6-ола (KKK-4)
К раствору соединения TP-21 (100 мг, 0,56 ммоль) в безводном ТГФ (2 мл) добавляют BH3-Me2S (127 мг, 1,67 ммоль) при 0°C по каплям под N2. После добавления, смесь нагревают до 70°C (кипение с обратным холодильником) в течение 3 часов. Реакционную смесь затем осторожно гасят 1 мл MeOH при -10~20°C, затем добавляют еще 6N HCl 10 мл, затем кипятят с обратным холодильникомeв течение 3 ч, удаляют основную часть растворителя в вакууме, затем остаток доводят до pH 8-9 K2CO3, экстрагируют EtOAc три раза. Объединенные органические слои сушат над Na2SO4, фильтруют и концентрируют с получением неочищенного соединения KKK-4 (0,5 г, >99%). ЖХМС [M+1] 166
Стадия 5 - Синтез трет-бутил 6-гидрокси-2,3-дигидробензo[f][1,4]оксазепин-4(5H)-карбоксилата (TP-22)
Неочищенное соединение KKK-4 (500 мг, 0,61 ммоль) растворяют в ДХМ (5 мл) и MeOH (5 мл). Добавляют (Boc)2O (132 мг, 0,605 ммоль) и Et3N (184 мг, 1,82 ммоль) и реакционную смесь перемешивают при 15°C в течение 16 часов. Добавляют ДХМ (10 мл), затем промывают уксусной кислотой (5 мл) и насыщенным NaCl (5 мл). Органический слой разделяют, сушат и выпаривают с получением остатка. Затем 10 мл MeOH добавляют для растворения остатка, затем K2CO3 (200 мг) добавляют к смеси. Реакционную смесь перемешивают при 15°C в течение 2 часов. ДХМ (25 мл×2) добавляют в раствор, раствор промывают уксусной кислотой (5 мл, pH <7) и насыщенным NaCl (5 мл). Органические слои объединяют, сушат и выпаривают с получением неочищенного продукта, который очищают преп.-ТСХ с получением соединения TP-22 (13 мг, 8%) в виде желтого твердого вещества. 1H ЯМР (400 MГц, CDCl3) δ ч./млн. 7,62 (шс, 1H), 7,07 (т, J=8,2 Гц, 1H), 6,73 (д, J=7,5 Гц, 1H), 6,64 (д, J=7,8 Гц, 1H), 4,37 (с, 2H), 4,01-3,92 (м, 2H), 3,79-3,70 (м, 2H), 1,40 (с, 9H) ЖХМС [M-Boc+1] 210.
Синтез трет-бутил 6-(1-(трет-бутоксикарбонил)-1H-пиразол-4-ил)-8-гидрокси-3,4-дигидроизохинолин-2(1H)-карбоксилата (TP-23) (Схема LLL)
Схема LLL
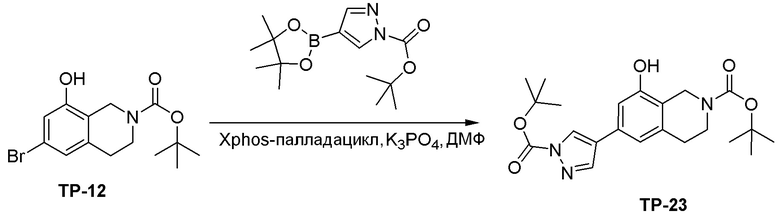
Под атмосферой N2, TP-12 (50 мг, 0,15 ммоль), трет-бутил 4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)-1H-пиразол-1-карбоксилат (67 мг, 0,23 ммоль), K3PO4 (97 мг, 0,46 ммоль) и XPhos-Палладацикл (13 мг, 0,015 ммоль) добавляют в пробирку. Добавляют ДМФ (1,60 мл), и реакционный раствор нагревают до 50°C в микроволновой печи в течение 16 часов. EtOAc и H2O добавляют в разбавленный реакционный раствор. Водный слой экстрагируют EtOAc (3 мл×2). Органические слои разделяют, сушат и выпаривают с получением неочищенного продукта, который очищают преп.-ТСХ (петролейный эфир/EtOAc=1/1) с получением желаемого соединения TP-23 (33 мг, 90%) в виде бесцветного масла. ЖХМС [M-Boc+1] 316.
Синтез трет-бутил 6-(1-(трет-бутоксикарбонил)-1H-пиразол-4-ил)-5-фтор-8-гидрокси-3,4-дигидроизохинолин-2(1H)-карбоксилата (TP-24) (Схема MMM)
Схема MMM
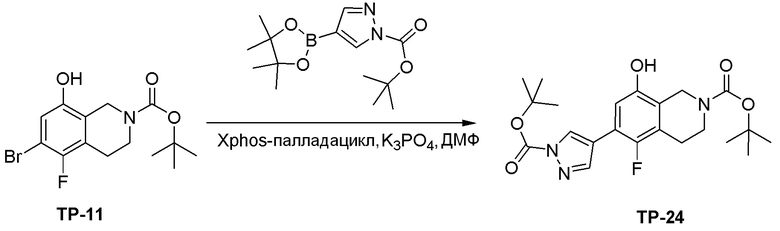
TP-24 синтезируют по методике получения TP-23 (Схема LLL) начиная с TP-11. ЖХМС [M-Boc+1] 334.
Синтез трет-бутил 8-гидрокси-6-(1-метил-1H-1,2,3-триазол-5-ил)-3,4-дигидроизохинолин-2(1H)-карбоксилата (TP-25) (Схема NNN)
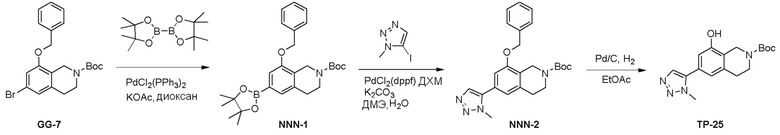
Стадия 1 - Синтез трет-бутил 8-(бензилокси)-6-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)-3,4-дигидроизохинолин-2(1H)-карбоксилата (NNN-1)
Смесь GG-7 (3,00 г, 7,17 ммоль), бис(пинаколато)диборона (2,73 г, 10,8 ммоль), KOAc (1,4 г, 14,3 ммоль) и PdCl2(dppf).CH2Cl2 (262 мг, 0,36 ммоль) в диоксане (30,0 мл) нагревают до 80°C в течение 16 часов. Воду (30 мл) добавляют к разбавленному реакционному раствору, затем экстрагируют EtOAc (30 мл×2). Органические слои объединяют, сушат и выпаривают с получением неочищенного продукта, который очищают флэш-хроматографией (120 г силикагель), элюируя петролейным эфиром/EtOAc 0-20%, с получением NNN-1 (3 г, 90%) в виде белого твердого вещества. ЖХМС [M-Boc+1] 366; 1H ЯМР (400 MГц, ДМСО-d6) δ ч./млн. 7,50-7,45 (м, 2H), 7,43-7,39 (м, 2H), 7,37-7,30 (м, 1H), 7,13 (д, J=2,5 Гц, 2H), 5,15 (шс, 2H), 4,54-4,38 (м, 2H), 3,59-3,50 (м, 2H), 2,77 (т, J=5,5 Гц, 2H), 1,42 (с, 9H), 1,29 (с, 12H)
Стадия 2 - Синтез трет-бутил 8-(бензилокси)-6-(1-метил-1H-1,2,3-триазол-5-ил)-3,4-дигидроизохинолин-2(1H)-карбоксилата (NNN-2)
Пробирку под аргоном, содержащую NNN-1 (800 мг, 1,72 ммоль), 5-йод-1-метил-1H-1,2,3-триазол (467 мг, 2,23 ммоль), PdCl2(dppf)-ДХМ (126 мг, 0,172 ммоль), K2CO3 (475 мг, 3,44 ммоль), ДМЭ (10,0 мл) и воду (1,00 мл) закрывают крышкой и нагревают при 80°C в течение 16 часов. Воду (10,0 мл) добавляют к реакционной смеси и экстрагируют EtOAc (10 мл×2). Органические слои разделяют, сушат и выпаривают с получением неочищенного продукта, который очищают флэш-хроматографией (80 г), элюируя петролейным эфиром/EtOAc (1:1) с получением NNN-2 (640 мг, 89%) в виде бесцветного масла. ЖХМС [M+1] 421.
Стадия 3 - Синтез трет-бутил 8-гидрокси-6-(1-метил-1H-1,2,3-триазол-5-ил)-3,4-дигидроизохинолин-2(1H)-карбоксилата (TP-25)
Соединение NNN-2 (640 мг, 1,52 ммоль) растворяют в EtOAc (3 мл). Pd/C (162 мг, 1,52 ммоль) добавляют в смесь, реакционную смесь перемешивают при 25°C в течение 48 часов. Раствор разбавляют EtOAc (10 мл) и фильтруют. Фильтрат выпаривают с получением неочищенного продукта, который очищают флэш-хроматографией, элюируя петролейным эфиром/EtOAc 0-30%, с получением желаемого продукта TP-25 (320 мг, 64%) в виде белого твердого вещества. ЖХМС [M+1] 331; 1H ЯМР (400 MГц, ДМСО-d6) δ ч./млн. 10,04 (с, 1H), 7,82 (с, 1H), 6,91-6,77 (м, 2H), 4,40 (с, 2H), 4,04 (с, 3H), 3,56 (м, 2H), 2,78 (м, 2H), 1,45 (с, 9H)
Синтез трет-бутил 8-гидрокси-6-(тиазол-4-ил)-3,4-дигидроизохинолин-2(1H)-карбоксилата (TP-26) (Схема OOO)
Схема OOO
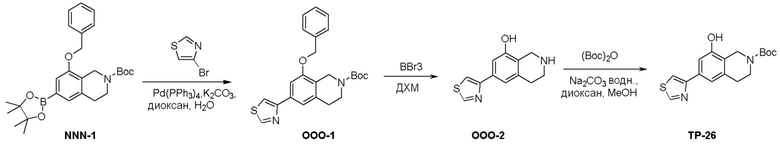
Стадия 1 - Синтез трет-бутил 8-(бензилокси)-6-(тиазол-4-ил)-3,4-дигидроизохинолин-2(1H)-карбоксилата (OOO-1)
Пробирку под аргоном, содержащую соединение NNN-1 (800 мг, 1,72 ммоль), 4-бромтиазол (367 мг, 2,23 ммоль), Pd(PPh3)4 (278 мг, 0,241 ммоль), K2CO3 (523 мг, 3,78 ммоль), диоксан (10 мл) и воду (1 мл) закрывают крышкой и нагревают при 80°C в течение 16 часов. Воду (10 мл) добавляют к реакционной смеси и экстрагируют EtOAc (10 мл×2). Органические слои разделяют, сушат и выпаривают с получением неочищенного продукта, который очищают флэш-хроматографией, элюируя петролейным эфиром/EtOAc 0-20%) с получением OOO-1 (370 мг, 51%) в виде бесцветного масла. ЖХМС [M+23] 445
Стадия 2 - Синтез 6-(тиазол-4-ил)-1,2,3,4-тетрагидроизохинолин-8-ола (OOO-2)
Соединение OOO-1 (320 мг, 0,76 ммоль) растворяют в ДХМ (10 мл) и охлаждают до 0°C на ледяной бане. BBr3 (1,14 г, 4,54 ммоль) добавляют к реакционному раствору и перемешивают при 25°C в течение 16 часов. Реакционный раствор охлаждают до 0°C. Метанол (3,00 мл) добавляют в реакционный раствор по каплям, затем воду (20 мл). Реакционный раствор промывают ДХМ (10 мл×2). Водный слой разделяют. Твердый Na2CO3 применяют для доведения pH до 9. Конечный раствор соединения OOO-2 применяют сразу для следующей стадии. ЖХМС [M+1] 233.
Стадия 3 - Синтез трет-бутил 8-гидрокси-6-(тиазол-4-ил)-3,4-дигидроизохинолин-2(1H)-карбоксилата (TP-26)
MeOH (5,00 мл) и диоксан (5,00 мл) добавляют в раствор соединения OOO-2 (180 мг, водный реакционный раствор, 40,0 мл), затем (Boc)2O (335 мг, 1,55 ммоль) добавляют к реакционной смеси и перемешивают при 25°C в течение 16 часов. ДХМ (20 мл) добавляют в разбавленный раствор. pH доводят до pH~3 добавлением 1N HCl водн. Раствор разделяют, и водный слой экстрагируют ДХМ (10 мл). Органические слои объединяют и промывают насыщенным NaCl (20,0 мл). Органические слои разделяют, сушат и выпаривают с получением неочищенного продукта, который очищают флэш-хроматографией, элюируя петролейным эфиром/EtOAc 0-50% с получением TP-26 (180 мг, 70%) в виде желтого твердого вещества. ЖХМС [M+1] 333; 1H ЯМР (400 MГц, ДМСО-d6) δ ч./млн. 9,78 (с, 1H), 9,16 (д, J=2,0 Гц, 1H), 7,99 (д, J=2,0 Гц, 1H), 7,33 (д, J=1,3 Гц, 1H), 7,24 (с, 1H), 4,38 (с, 2H), 3,56 (м, 2H), 2,77 (м, 2H), 1,44 (с, 9H)
Синтез трет-бутил 8-гидрокси-3-метил-3,4-дигидроизохинолин-2(1H)-карбоксилата (TP-28) (Схема PPP)
Схема PPP
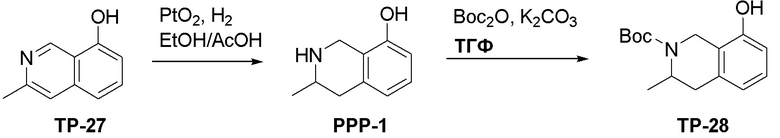
Стадия 1: Синтез 3-метил-1,2,3,4-тетрагидроизохинолин-8-ола (PPP-1)
В раствор 3-метилизохинолин-8-ола (TP-27) [получен как описано в Tetrahedron Letters 49 (2008) 3725-3728] (100 мг, 0,628 ммоль) в EtOH/AcOH (6 мл/0,2 мл) добавляют PtO2 (80 мг, 0,35 ммоль), гидрируют под 45 ф/кв.д H2 при кт в течение 16 ч. Реакционную смесь фильтруют и промывают EtOH, растворитель выпаривают с получением неочищенного продукта PPP-1 (103 мг, 100%) который применяют на следующей стадии без дальнейшей очистки. ЖХМС [M+1] 163,9.
Стадия 2: Синтез трет-бутил 8-гидрокси-3-метил-3,4-дигидроизохинолин-2(1H)-карбоксилата (TP-28)
К раствору PPP-1 (103 мг, 0,628 ммоль) в ТГФ (6 мл, 0,1 M) добавляют Boc2O (225 мг, 1,03 ммоль) и K2CO3 (356 мг, 2,57 ммоль) при 15°C, перемешивают при кт в течение 15 ч. Растворитель удаляют, остаток растворяют в MeOH, добавляют 0,1 г K2CO3, перемешивают в течение 2 ч. Твердое вещество фильтруют и промывают EtOAc, фильтрат концентрируют и очищают препаративной ТСХ с получением TP-28 (70 мг, 42%) в виде желтого твердого вещества.
ЖХМС [M+1-tBu] 207,9. 1H ЯМР (400 MГц, ХЛОРОФОРМ-d) δ ч./млн. 7,09-7,01 (м, 1H), 6,71 (д, J=7,8 Гц, 1H), 6,63 (д, J=8,0 Гц, 1H), 4,81 (шд, J=17,6 Гц, 1H), 4,63 (шс, 1H), 4,18 (д, J=17,6 Гц, 1H), 3,08 (шдд, J=5,5, 16,1 Гц, 1H), 2,55 (шддд, J=1,8, 14,6, 16,3 Гц, 1H), 1,54-1,43 (с, 9H), 1,09 (д, J=6,8 Гц, 3H)
Синтез трет-бутил 8-гидрокси-6-(оксазол-5-ил)-3,4-дигидроизохинолин-2(1H)-карбоксилата (TP-29) (Схема QQQ)
Схема QQQ
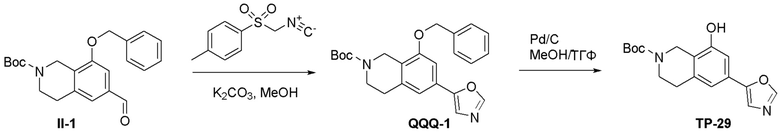
Стадия 1: Синтез трет-бутил 8-(бензилокси)-6-(оксазол-5-ил)-3,4-дигидроизохинолин-2(1H)-карбоксилата (QQQ-1)
В раствор II-1 (265 мг, 0,721 ммоль) и изоцианида п-толуолсульфонилметила (465 мг, 2,38 ммоль) в метаноле (14,4 мл, c=0,05 M) добавляют K2CO3 (199 мг, 1,44 ммоль), полученную суспензию кипятят с обратным холодильникомe в течение ночи. Реакционную смесь концентрируют, EtOAc и H2O добавляют. Слои разделяют; водный слой экстрагируют EtOAc. Органические слои концентрируют, очищают хроматографией на колонке с 35% EtOAc/гептаном с получением QQQ-1 (100 мг, 34%) в виде бесцветного масла.
ЖХМС [M+1-Boc] 307,15. 1H ЯМР (400 MГц, ХЛОРОФОРМ-d) δ ч./млн. 1,51 (с, 9 H) 2,86 (т, J=5,01 Гц, 2 H) 3,67 (т, J=5,62 Гц, 2 H) 4,62 (с, 2 H) 5,16 (шс, 2 H) 7,06 (с, 1 H) 7,08 (с, 1 H) 7,31 (с, 1 H) 7,35 (д, J=7,21 Гц, 1 H) 7,41 (т, J=7,34 Гц, 2 H) 7,44-7,50 (м, 2 H) 7,90 (с, 1 H)
Стадия 2: Синтез трет-бутил 8-гидрокси-6-(оксазол-5-ил)-3,4-дигидроизохинолин-2(1H)-карбоксилата (TP-29)
Соединение TP-29 получают из QQQ-1 по методике получения стадия 9 со схемы GG (79 мг, 100%).
ЖХМС [M+1-Boc] 217,10. 1H ЯМР (400 MГц, ХЛОРОФОРМ-d) δ ч./млн. 1,54 (с, 9 H) 2,83 (шс, 2 H) 3,67 (т, J=5,69 Гц, 2 H) 4,60 (шс, 2 H) 6,95 (шс, 2 H) 7,27 (с, 1 H) 7,90 (с, 1 H)
Синтез трет-бутил 8-гидрокси-6-(1H-пиразол-3-ил)-3,4-дигидроизохинолин-2(1H)-карбоксилата (TP-30) (Схема RRR)
Схема RRR
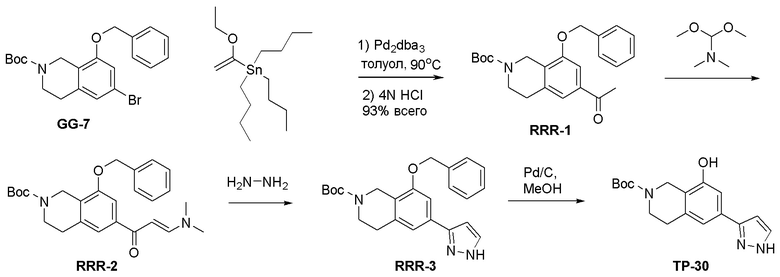
Стадия 1: Синтез трет-бутил 6-ацетил-8-(бензилокси)-3,4-дигидроизохинолин-2(1H)-карбоксилата (RRR-1)
Соединение RRR-1 получают из GG-7 по методике получения стадия 1 со схемы JJ (360,0 мг, 80,2%).
Стадия 2: Синтез трет-бутил (E)-8-(бензилокси)-6-(3-(диметиламино)акрилоил)-3,4-дигидроизохинолин-2(1H)-карбоксилата (RRR-2)
RRR-1 (65 мг, 0,17 ммоль) и диметилцеталь N,N-диметилформамид (0,5 мл) нагревают при 100°C в течение ночи. Смесь концентрируют, очищают хроматографией на колонке с 70% EtOAc/гептаном с получением RRR-2 70 мг (94% выход) в виде желтого масла.
ЖХМС [M+1] 437,20. 1H ЯМР (400 MГц, ХЛОРОФОРМ-d) δ ч./млн. 1,50 (с, 9 H) 2,77-2,92 (м, 2 H) 2,92-3,22 (м, 6 H) 3,66 (т, J=5,01 Гц, 2 H) 4,62 (с, 2 H) 5,17 (шс, 2 H) 5,66 (д, J=12,35 Гц, 1 H) 7,28 (шс, 1 H) 7,32 (д, J=7,21 Гц, 1 H) 7,38 (т, J=7,27 Гц, 3 H) 7,43-7,49 (м, 2 H) 7,80 (д, J=12,35 Гц, 1 H)
Стадия 3: Синтез трет-бутил 8-(бензилокси)-6-(1H-пиразол-3-ил)-3,4-дигидроизохинолин-2(1H)-карбоксилата (RRR-3)
RRR-2 (70 мг, 0,16 ммоль) и моногидрат гидразина (183 мг, 1,28 ммоль, 0,178 мл) в 2 мл EtOH перемешивают при кт в течение ночи. Реакционную смесь добавляют EtOAc и H2O, слои разделяют, водный слой экстрагируют EtOAc. Органические слои объединяют и промывают насыщенным раствором соли, сушат над Na2SO4, концентрируют с получением RRR-3 58,8 мг (90% выход) в виде твердого вещества.
ЖХМС [M+1-tBu] 350,10. 1H ЯМР (400 MГц, ХЛОРОФОРМ-d) δ ч./млн. 1,51 (с, 9 H) 2,76-2,93 (м, 2 H) 3,67 (т, J=5,50 Гц, 2 H) 4,62 (с, 2 H) 5,16 (шс, 2 H) 6,59 (д, J=2,32 Гц, 1 H) 7,15 (с, 1 H) 7,23 (с, 1 H) 7,30-7,36 (м, 1 H) 7,39 (т, J=7,27 Гц, 2 H) 7,44-7,49 (м, 2 H) 7,62 (д, J=2,20 Гц, 1 H)
Стадия 4: Синтез трет-бутил 8-гидрокси-6-(1H-пиразол-3-ил)-3,4-дигидроизохинолин-2(1H)-карбоксилата (TP-30)
Соединение TP-30 получают из RRR-3 по методике получения стадия 9 со схемы GG (32 мг, 70%).
ЖХМС [M+1-Boc] 216,10. 1H ЯМР (400 MГц, ХЛОРОФОРМ-d) δ ч./млн. 1,51 (с, 9 H) 2,77 (т, J=5,26 Гц, 2 H) 3,62 (т, J=5,50 Гц, 2 H) 4,58 (шс, 2 H) 6,46 (шс, 1 H) 6,97 (с, 1 H) 7,12 (шс, 1 H) 7,50-7,63 (м, 1 H)
Примеры 163-171 получают по методике Примера 78 со схемы CC с применением подходящего NBoc-защищенного тетрагидроизохинолина на стадии 1.
TP-21
Пропускают конечную стадию снятия защиты
ЖХМС
[M+1]
1H ЯМР (400 MГц, ДМСО-d6) δ ч./млн. 8,62 (с, 1H), 8,33 (т, J=6,1 Гц, 1H), 7,97 (д, J=3,8 Гц, 1H), 7,38 (т, J=8,3 Гц, 1H), 6,98 (д, J=8,3 Гц, 1H), 6,69 (дд, J=2,0, 5,8 Гц, 2H), 5,35 (д, J=3,5 Гц, 1H), 5,31-5,23 (м, 1H), 5,08 (д, J=6,8 Гц, 1H), 4,71 (шд, J=6,3 Гц, 1H), 4,63-4,56 (м, 1H), 4,11 (шд, J=4,8 Гц, 2H), 4,00 (шс, 1H), 3,25-3,16 (м, 2H), 2,90-2,80 (м, 1H), 2,64 (с, 3H), 1,72 (м, 1H)
TP-22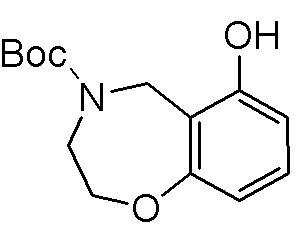
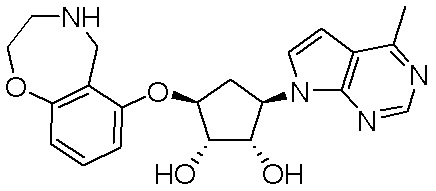
ЖХМС
[M+1]
соль HCl
1H ЯМР (400 MГц, MeOD-d4) δ ч./млн.=9,12-8,97 (м, 1H), 8,15-7,95 (м, 1H), 7,36-7,26 (м, 1H), 7,25-7,12 (м, 1H), 7,00-6,88 (м, 1H), 6,75 (д, J=6,5 Гц, 1H), 5,46-5,33 (м, 1H), 4,81-4,73 (м, 2H), 4,68-4,52 (м, 2H), 4,36-4,14 (м, 3H), 3,67-3,56 (м, 2H), 3,12-3,04 (м, 1H), 3,00 (шс, 3H), 2,40-2,25 (м, 1H)
TP-23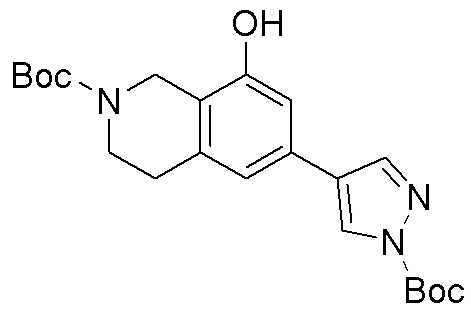
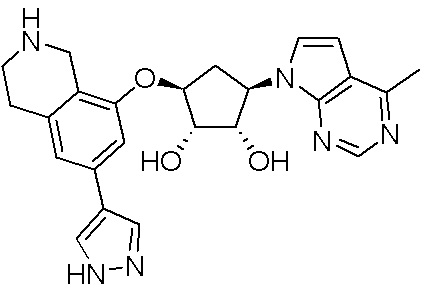
ЖХМС
[M+1]
соль HCl
1H ЯМР (400 MГц, D2O) δ ч./млн. 8,85-8,80 (м, 1H), 8,00-7,95 (м, 2H), 7,81 (шдд, J=1,4, 3,9 Гц, 1H), 7,14-7,07 (м, 2H), 7,05 (шд, J=3,3 Гц, 1H), 5,39-5,27 (м, 2H), 4,92-4,82 (м, 1H), 4,30 (шс, 3H), 3,50-3,39 (м, 2H), 3,11-2,99 (м, 3H), 2,88 (шс, 3H), 2,20 (шс, 1H)
TP-24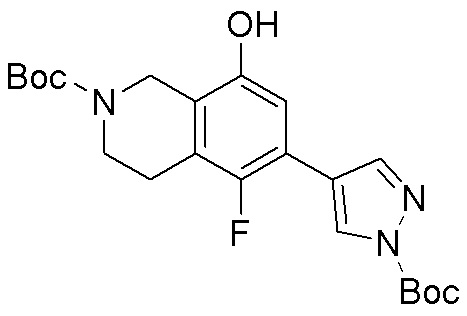
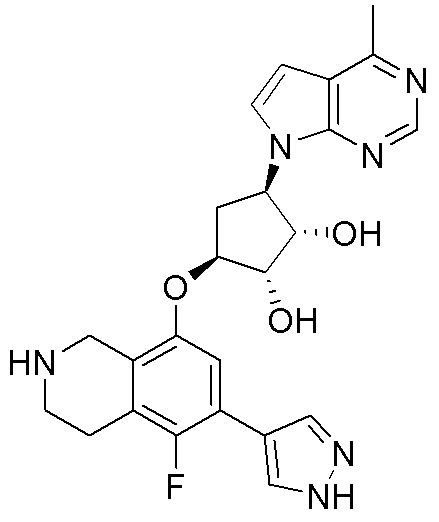
ЖХМС
[M+1]
соль HCl
1H ЯМР (400 MГц, D2O) δ ч./млн. 8,57 (с, 1H), 8,42 (с, 1H), 8,08 (с, 1H), 7,51 (д, J=3,8 Гц, 1H), 7,12 (д, J=5,8 Гц, 1H), 6,80 (с, 1H), 5,22 -5,13 (м, J=8,9, 8,9, 8,9 Гц, 2H), 4,89-4,84 (м, 1H), 4,66-4,61 (м, 1H), 4,39-4,27 (м, 3H), 3,54-3,49 (м, 2H), 3,11-3,04 (м, J=5,6, 5,6 Гц, 2H), 3,04-2,96 (м, 1H), 2,68 (с, 3H), 2,17-2,07 (м, 1H)
TP-25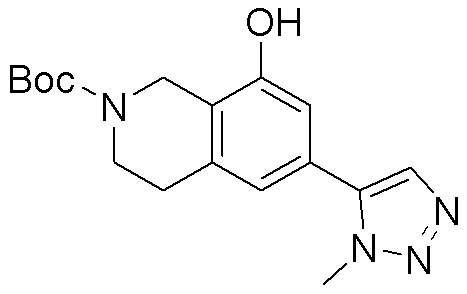
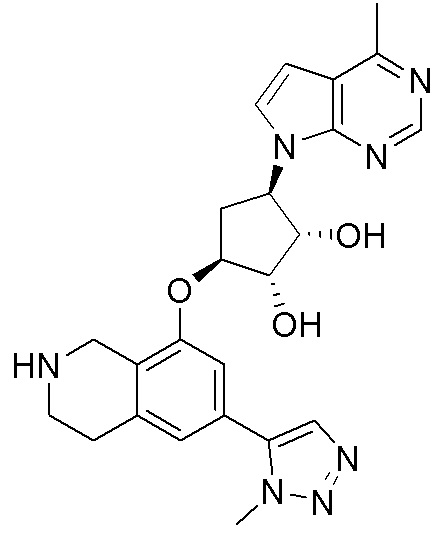
ЖХМС
[M+1]
соль HCl
1H ЯМР (400 MГц, D2O) δ ч./млн. 8,93-8,91 (с, 1H), 7,93-7,90 (д, J=4,0 Гц, 1H), 7,88 (с, 1H), 7,19 (д, J=4,0 Гц, 1H), 7,14-7,11 (с, 1H), 7,08 (с, 1H), 5,42 (м, 1H), 4,94-4,86 (м, 2H), 4,46 (с, 2H), 4,40 (м, 1H), 4,10 (с, 3H), 3,61-3,54 (м, 2H), 3,24-3,16 (м, 2H), 3,17-3,07 (м, 1H), 2,98 (с, 3H), 2,37-2,26 (м, 1H)
TP-26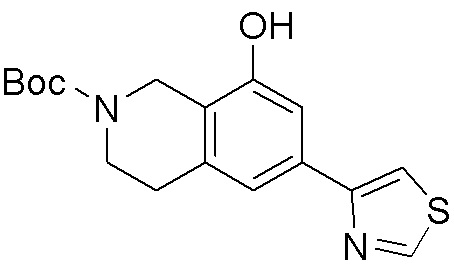
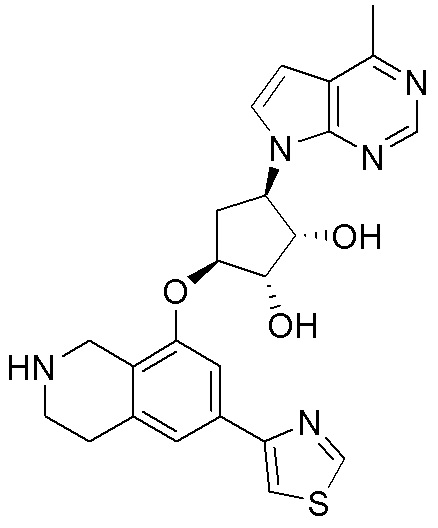
ЖХМС
[M+1]
соль HCl
1H ЯМР (400 MГц, D2O) δ ч./млн. 9,09 (д, J=1,8 Гц, 1H), 8,90 (с, 1H), 7,91 (д, J=2,0 Гц, 1H), 7,89 (д, J=4,0 Гц, 1H), 7,45 (с, 1H), 7,44 (с, 1H), 7,17-7,15 (м, 1H), 5,43 (м, 1H), 4,96 (м, 1H), 4,85-4,83 (м, 1H), 4,43 (с, 2H), 4,41 (м, 1H), 3,59-3,54 (м, 2H), 3,23-3,18 (м, 2H), 3,18-3,11 (м, 1H), 2,96 (с, 3H), 2,35-2,26 (м, 1H)
TP-27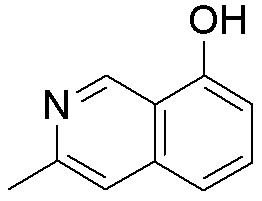
Пропускают конечную стадию снятия защиты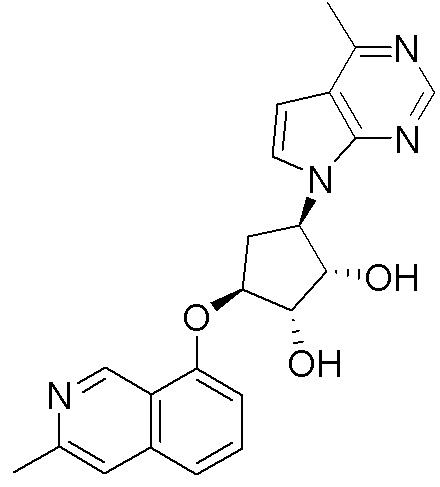
ЖХМС [M+1]
1H ЯМР (400 MГц, МЕТАНОЛ-d4) δ ч./млн. 9,56 (с, 1H), 8,57 (с, 1H), 7,71-7,60 (м, 3H), 7,43 (д, J=8,3 Гц, 1H), 7,13 (д, J=7,3 Гц, 1H), 6,75 (д, J=3,8 Гц, 1H), 5,30 (кв, J=8,9 Гц, 1H), 4,98 (м, 1H), 4,91 (м, 1H), 4,38 (д, J=5,0 Гц, 1H), 3,16-3,08 (м, 1H), 2,72 (с, 3H), 2,70 (с, 3H), 2,45-2,37 (м, 1H)
TP-29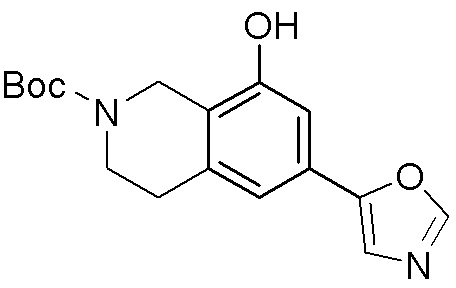
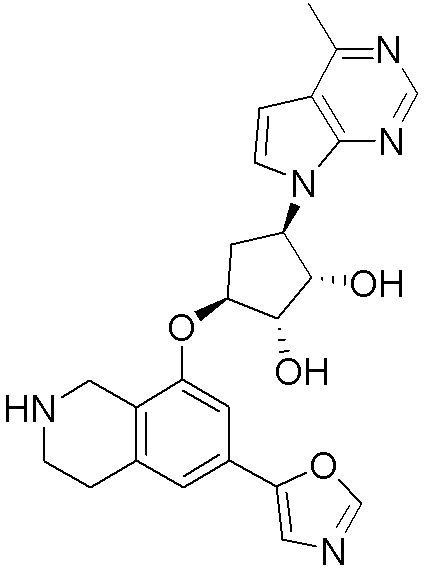
ЖХМС [M+1]
1H ЯМР (400 MГц, МЕТАНОЛ-d4) δ ч./млн. 8,99 (с, 1H), 8,30 (с, 1H), 8,01 (д, J=3,8 Гц, 1H), 7,63 (с, 1H), 7,35 (д, J=16,6 Гц, 2H), 7,17 (д, J=3,8 Гц, 1H), 5,40 (кв, J=9,3 Гц, 1H), 4,79 (дд, J=5,0, 9,0 Гц, 1H), 4,43 (с, 2H), 4,26 (д, J=4,5 Гц, 1H), 3,55 (т, J=6,3 Гц, 2H), 3,23-3,07 (м, 3H), 2,97 (с, 3H), 2,33 (ддд, J=4,1, 9,6, 13,9 Гц, 1H)
TP-30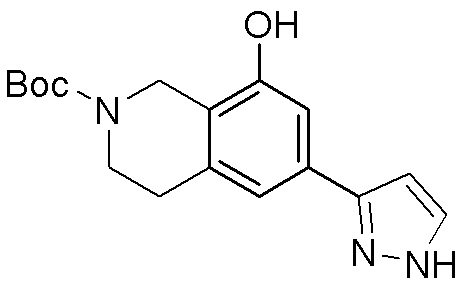
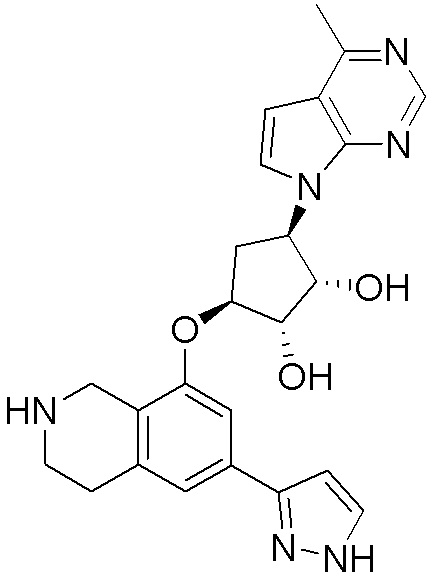
ЖХМС [M+1]
соль HCl
1H ЯМР (400 MГц, D2O) δ ч./млн.=8,88 (с, 1H), 7,86 (д, J=3,8 Гц, 1H), 7,79 (д, J=2,3 Гц, 1H), 7,34 (м, 2H), 7,14 (м, 1H), 6,77 (м, 1H), 5,41(м, 1H), 4,93 (м, 1H), 4,75-4,70 (м, 1H), 4,44-4,36 (м, 3H), 3,58-3,50 (м, 2H), 3,25-3,07 (м, 3H), 2,93 (с, 3H), 2,31 (м, 1H)
Пример 172 (Схема SSS) - Синтез (1S,2S,3S,5R)-3-(изохинолин-8-илокси)-5-(4-метил-7H-пирроло[2.3-d]пиримидин-7-ил)циклопентан-1,2-диола (SSS-6)
Схема SSS
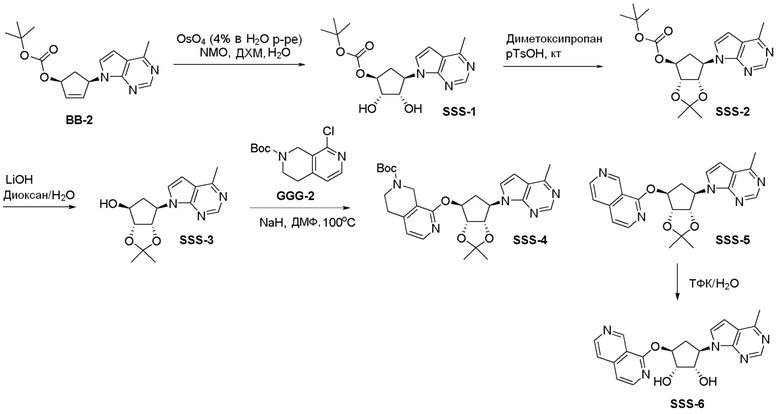
Стадия 1: Синтез трет-бутил ((1S,2S,3S,4R)-2,3-дигидрокси-4-(4-метил-7H-пирроло[2.3-d]пиримидин-7-ил)циклопентил)карбоната (SSS-1)
Соединение SSS-1 получают из BB-2 по методике получения стадии 2 со схемы CC с получением 274 мг (82% выход) в виде бесцветного масла.
ЖХМС [M+1] 350,10. 1H ЯМР (400 MГц, ХЛОРОФОРМ-d) δ ч./млн. 1,51 (с, 9 H) 2,18 (ддд, J=14,21, 9,14, 5,26 Гц, 1 H) 2,71 (с, 3 H) 3,02 (дт, J=14,18, 8,07 Гц, 1 H) 3,95-4,02 (м, 1 H) 4,30 (дд, J=5,38, 2,08 Гц, 1 H) 4,51 (дд, J=7,82, 5,50 Гц, 1 H) 4,96-5,08 (м, 2 H) 6,61 (д, J=3,67 Гц, 1 H) 7,29 (д, J=3,67 Гц, 1 H) 8,66 (с, 1 H)
Стадия 2: Синтез трет-бутил ((3aR,4S,6R,6aS)-2,2-диметил-6-(4-метил-7H-пирроло[2.3-d]пиримидин-7-ил)тетрагидро-4H-циклопента[d][1.3]диоксол-4-ил)карбоната (SSS-2)
Соединение SSS-2 получают из SSS-1 по методике получения стадии 3 со схемы P с получением 181 мг (59% выход) в виде бесцветного масла.
ЖХМС [M+1] 390,15. 1H ЯМР (400 MГц, ХЛОРОФОРМ-d) δ ч./млн. 1,32 (с, 3 H) 1,50 (с, 9 H) 1,58 (с, 3 H) 2,38-2,48 (м, 1 H) 2,73 (с, 3 H) 2,84-2,95 (м, 1 H) 4,82 (д, J=6,11 Гц, 1 H) 4,99 (дд, J=6,54, 3,12 Гц, 1 H) 5,06-5,13 (м, 1 H) 5,21-5,29 (м, 1 H) 6,57 (д, J=3,67 Гц, 1 H) 7,31 (д, J=3,67 Гц, 1 H) 8,78 (с, 1 H)
Стадия 3: Синтез (3aR,4S,6R,6aS)-2,2-диметил-6-(4-метил-7H-пирроло[2.3-d]пиримидин-7-ил)тетрагидро-4H-циклопента[d][1.3]диоксол-4-ола (SSS-3)
Соединение SSS-3 получают из SSS-2 по методике получения стадии 1 со схемы FFF с получением 127 мг (94% выход) в виде белого твердого вещества.
ЖХМС [M+1] 290,15. 1H ЯМР (400 MГц, ХЛОРОФОРМ-d) δ ч./млн. 1,33 (с, 3 H) 1,55 (с, 3 H) 2,20 (дд, J=15,65, 1,34 Гц, 1 H) 2,75 (с, 3 H) 2,95-3,08 (м, 1 H) 4,44-4,50 (м, 1 H) 4,80 (д, J=5,38 Гц, 1 H) 4,84 (дт, J=10,58, 2,48 Гц, 1 H) 4,98 (д, J=5,01 Гц, 1 H) 6,15 (д, J=9,41 Гц, 1 H) 6,55 (д, J=3,67 Гц, 1 H) 8,73 (с, 1 H)
Стадия 4: Синтез трет-бутил 8-(((3aR,4S,6R,6aS)-2,2-диметил-6-(4-метил-7H-пирроло[2.3-d]пиримидин-7-ил)тетрагидро-4H-циклопента[d][1.3]диоксол-4-ил)окси)-3,4-дигидро-2,7-нафтиридин-2(1H)-карбоксилата (SSS-4) и 1-(((3aR,4S,6R,6aS)-2,2-диметил-6-(4-метил-7H-пирроло[2.3-d]пиримидин-7-ил)тетрагидро-4H-циклопента[d][1.3]диоксол-4-ил)окси)-2,7-нафтиридина (SSS-5)
К раствору SSS-3 (80,7 мг, 0,279 ммоль) в ДМФ (5,58 мл, c=0,05 M) добавляют NaH (16,7 мг, 0,419 ммоль, 60%). После перемешивания в течение 10 мин добавляют GGG-2 (75,0 мг, 0,279 ммоль). Полученную реакционную смесь нагревают при 100°C в течение 1,5 ч. Реакцию гасят разбавленным NaHCO3 и затем разделяют между этилацетатом (20 мл) и водой (20 мл). Органическую фазу разделяют, промывают насыщенным раствором соли, сушат над сульфатом натрия, концентрируют и очищают хроматографией на колонке с 60% EtOAc/гептаном с получением SSS-4 (15 мг, 10%)
ЖХМС [M+1] 522,20. 1H ЯМР (400 MГц, ХЛОРОФОРМ-d) δ ч./млн. 1,34 (с, 3 H) 1,50 (с, 9 H) 1,60 (с, 3 H) 2,45 (д, J=14,18 Гц, 1 H) 2,72 (с, 3 H) 2,76 (т, J=5,38 Гц, 2 H) 3,05 (дт, J=14,61, 7,12 Гц, 1 H) 3,60 (шс, 1 H) 3,65 (шс, 1 H) 4,39 (шс, 2 H) 4,94 (д, J=6,24 Гц, 1 H) 5,05 (дд, J=6,17, 1,90 Гц, 1 H) 5,34 (ддд, J=7,70, 4,95, 2,38 Гц, 1 H) 5,60 (шс, 1 H) 6,61 (шс, 1 H) 6,69 (д, J=5,14 Гц, 1 H) 7,38 (д, J=3,67 Гц, 1 H) 7,95 (д, J=5,14 Гц, 1 H) 8,79 (м, 1 H)
и элюируя 5% MeOH/EtOAc с получением SSS-5 (35 мг, 30%)
ЖХМС [M+1] 418,15. 1H ЯМР (400 MГц, ХЛОРОФОРМ-d) δ ч./млн. 1,38 (с, 3 H) 1,63 (с, 3 H) 2,56 (дт, J=14,98, 3,82 Гц, 1 H) 2,72 (с, 3 H) 3,04-3,19 (м, 1 H) 5,11 (д, J=6,11 Гц, 1 H) 5,21 (д, J=6,11 Гц, 1 H) 5,35-5,46 (м, 1 H) 5,83 (дт, J=3,97, 2,05 Гц, 1 H) 6,61 (д, J=3,67 Гц, 1 H) 7,20 (д, J=5,87 Гц, 1 H) 7,39 (д, J=3,67 Гц, 1 H) 7,55 (д, J=5,62 Гц, 1 H) 8,22 (д, J=5,87 Гц, 1 H) 8,70 (шс, 1 H) 8,77 (с, 1 H) 9,38 (шс, 1 H)
Стадия 5: Синтез (1S,2S,3S,5R)-3-(изохинолин-8-илокси)-5-(4-метил-7H-пирроло[2.3-d]пиримидин-7-ил)циклопентан-1,2-диола (SSS-6)
Соединение SSS-6 получают из SSS-5 по методике получения стадия 10 со схемы A с получением 11,6 мг (36% выход) в виде белого твердого вещества.
ЖХМС [M+1] 378,10. 1H ЯМР (700 MГц, ДМСО-d6) δ ч./млн. 2,05-2,15 (м, 1 H) 2,64-2,72 (м, 3 H) 2,98 (ддд, J=14,75, 9,24, 7,48 Гц, 1 H) 4,25 (д, J=3,08 Гц, 1 H) 4,72 (дд, J=8,69, 4,73 Гц, 1 H) 5,21 (кв, J=8,88 Гц, 1 H) 5,48 (дд, J=5,94, 3,30 Гц, 1 H) 6,80 (д, J=3,52 Гц, 1 H) 7,43 (д, J=5,94 Гц, 1 H) 7,78-7,89 (м, 2 H) 8,24 (д, J=5,72 Гц, 1 H) 8,64 (с, 1 H) 8,80 (шс, 1 H) 9,72 (шс, 1 H)
Пример 173 получают по методике получения CC-3 (Пример 78) с применением подходящего пирролопиримидина на стадии 1 Схемы BB и подходящего N-Boc защищенного тетрагидроизохинолина на стадии 1 Схемы CC.
HG-3
TP-18
ЖХМС
[M+1]
1H ЯМР (400 MГц, MeOD-d4) δ ч./млн. 9,09 (шс, 1H), 7,66 (д, J=2,3 Гц, 1H), 7,40 (с, 1H), 7,33 (с, 1H), 5,38 (кв, J=9,1 Гц, 1H), 4,79 (т, J=5,1 Гц, 1H), 4,60 (дд, J=5,3, 9,3 Гц, 1H), 4,44 (с, 2H), 4,19 (д, J=4,8 Гц, 1H), 3,60-3,50 (м, 2H), 3,21-3,13 (м, 2H), 3,02 (тд, J=8,2, 14,5 Гц, 1H), 2,83-2,79 (м, 3H), 2,19 (ддд, J=4,6, 9,6, 14,1 Гц, 1H)
Пример 174 получают по методике NN-5 (Пример 99) с применением подходящего пирролопиримидина на стадии 1 Схемы BB и подходящего N-Boc защищенного тетрагидроизохинолина на стадии 1 Схемы NN.
4,5-дихлор-7H-пирроло[2.3-d]пиримидин
TP-18
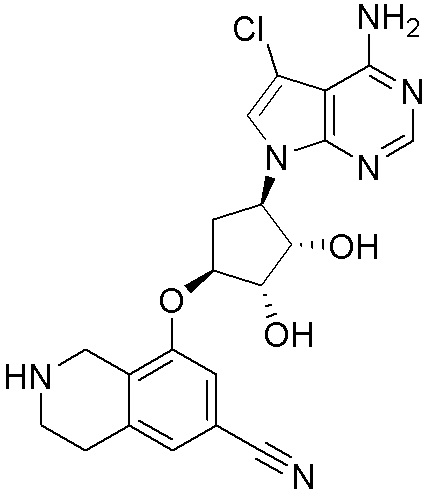
ЖХМС
[M+1]
соль HCl
1H ЯМР (400 MГц, MeOD-d4) δ ч./млн. 8,30 (с, 1H), 7,68 (с, 1H), 7,46 (с, 1H), 7,30 (с, 1H), 5,29-5,21 (м, 1H), 4,78 (т, J=5,2 Гц, 1H), 4,62 (дд, J=5,2, 9,2 Гц, 1H), 4,43 (с, 2H), 4,19 (д, J=5,1 Гц, 1H), 3,57-3,51 (м, 2H), 3,18 (т, J=6,1 Гц, 2H), 3,09-2,96 (м, 1H), 2,20 (ддд, J=4,5, 9,7, 14,0 Гц, 1H)
Примеры 175-179 получают по методике SS-5 (Пример 121) с применением подходящего N-Boc защищенного тетрагидроизохинолина на стадии 1 и подходящего пирролопиримидина на стадии 2 Схемы SS. Стадия 4 со схемы SS не требуется для этих примеров.
HG-3 (стадия 4 пропущена)
TP-3
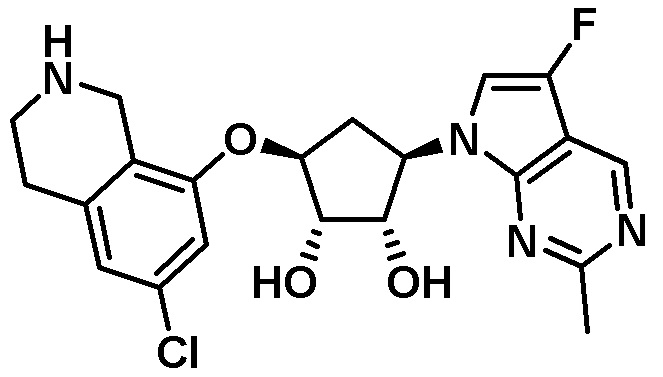
ЖХМС
[M+H]
1H ЯМР (400 MГц, ОКСИД ДЕЙТЕРИЯ) δ ч./млн. 9,13 (с, 1H), 7,61 (д, J=2,3 Гц, 1H), 6,97 (д, J=4,0 Гц, 2H), 5,40 (шд, J=8,5 Гц, 1H), 4,72 (шд, J=2,0 Гц, 1H), 4,58 (дд, J=4,8, 9,0 Гц, 1H), 4,32-4,27 (м, 3H), 3,46 (т, J=6,1 Гц, 2H), 3,09-3,05 (м, 2H), 3,01 (с, 1H), 2,83 (с, 3H), 2,17-2,07 (м, 1H).
HG-4 (стадия 4 пропущена)
TP-3
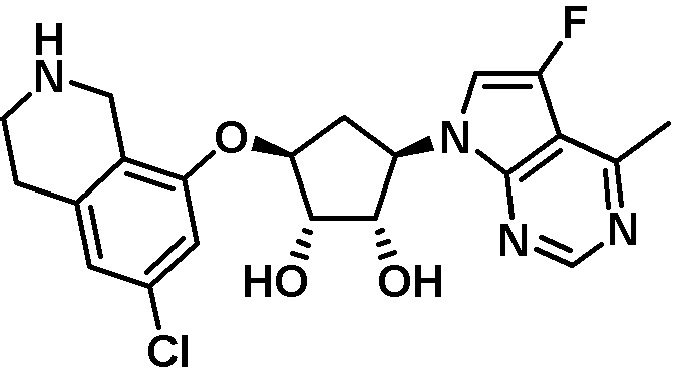
ЖХМС
[M+H]
1H ЯМР (400 MГц, ОКСИД ДЕЙТЕРИЯ) δ ч./млн. 8,88 (с, 1H), 7,69 (д, J=2,3 Гц, 1H), 7,01 (д, J=4,0 Гц, 2H), 5,43 (кв, J=9,0 Гц, 1H), 4,89-4,83 (м, 1H), 4,66 (дд, J=4,8, 8,8 Гц, 1H), 4,41-4,30 (м, 3H), 3,52 (т, J=6,3 Гц, 2H), 3,17-3,05 (м, 3H), 3,01 (с, 3H), 2,26-2,07 (м, 1H).
HG-4 (стадия 4 пропущена)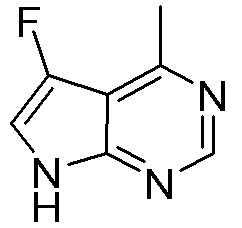
TP-6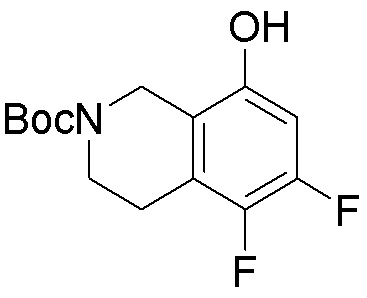
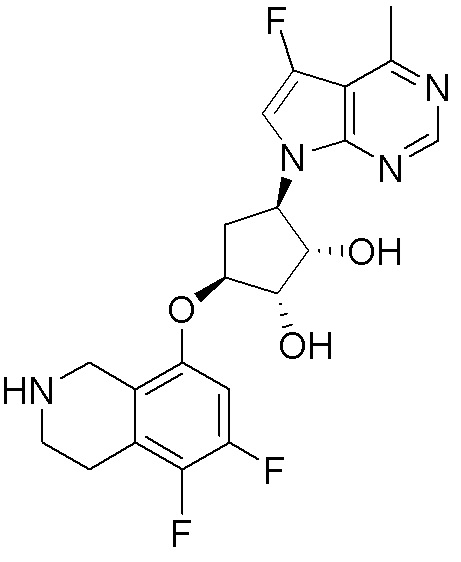
ЖХМС
[M+1]
соль HCl
1H ЯМР (400 MГц, D2O) δ ч./млн.=8,77 (с, 1H), 7,58 (шс, 1H), 6,92-6,85 (м, 1H), 5,32 (шд, J=8,3 Гц, 1H), 4,78-4,77 (м, 1H, под D2O), 4,61-4,47 (м, 1H), 4,26 (шс, 3H), 3,46 (шс, 2H), 3,04 (шс, 2H), 2,98 (шд, J=7,5 Гц, 1H), 2,90 (с, 3H), 2,09 (шс, 1H)
HG-4 (стадия 4 пропущена)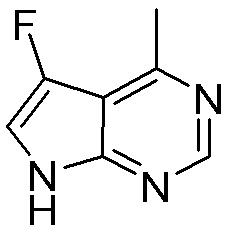
TP-6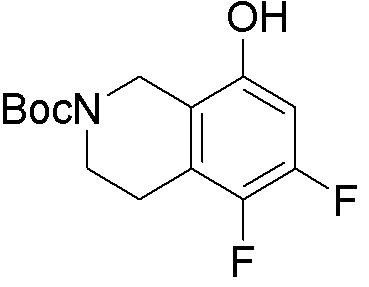
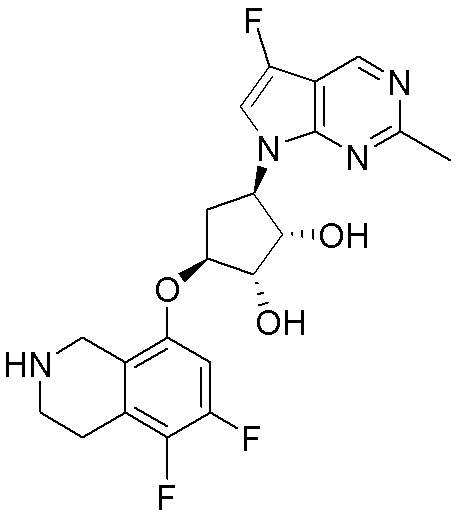
ЖХМС
[M+1]
соль HCl
1H ЯМР (400 MГц, D2O) δ ч./млн.=9,09 (с, 1H), 7,59 (с, 1H), 6,87 (шдд, J=6,5, 12,3 Гц, 1H), 5,39-5,30 (м, 1H), 4,58-4,57 (м, 1H), 4,24 (шс, 2H), 3,63 (шд, J=5,0 Гц, 1H), 3,56 (с, 2H), 3,45 (шт, J=5,8 Гц, 1H), 3,03 (шс, 2H), 2,99-2,92 (м, 1H), 2,78 (с, 3H), 2,08 (шд, J=10,5 Гц, 1H)
HG-6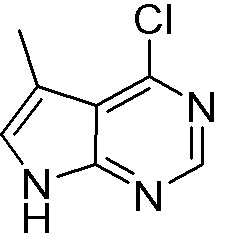
TP-6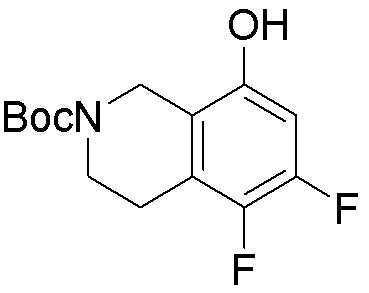
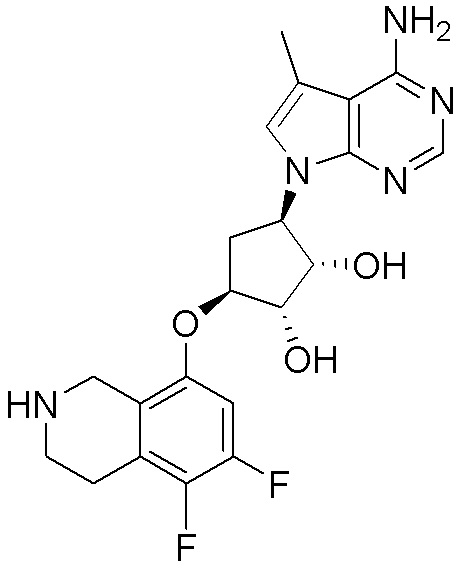
ЖХМС
[M+1]
соль HCl
1H ЯМР (400 MГц, MeOD-d4) δ ч./млн.=8,27 (с, 1H), 7,37 (с, 1H), 7,15-7,00 (м, 1H), 5,29-5,11 (м, 1H), 4,73-4,57 (м, 2H), 4,35 (шс, 2H), 4,17 (шд, J=3,5 Гц, 1H), 3,61-3,49 (м, 2H), 3,13 (шс, 2H), 3,02-2,91 (м, 1H), 2,55-2,44 (м, 3H), 2,23-2,09 (м, 1H)
Пример 180 (Схема TTT) - (1S,2S,3R,5S)-3-(4-амино-7H-пирроло[2.3-d]пиримидин-7-ил)-5-((5-фтор-1,2,3,4-тетрагидроизохинолин-8-ил)окси)циклопентан-1,2-диол (TTT-5)
Схема TTT
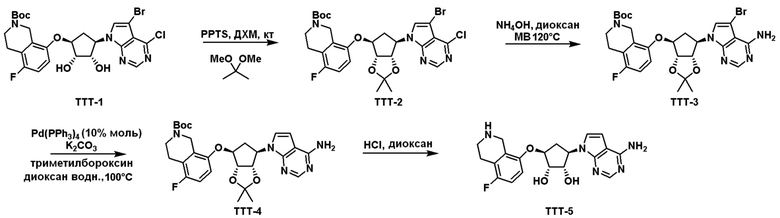
Стадия 1: Синтез трет-бутил 8-(((3aR,4S,6R,6aS)-6-(5-бром-4-хлор-7H-пирроло[2.3-d]пиримидин-7-ил)-2,2-диметилтетрагидро-4H-циклопента[d][1.3]диоксол-4-ил)окси)-5-фтор-3,4-дигидроизохинолин-2(1H)-карбоксилата (TTT-2)
Промежуточное соединение TTT-1 получают по общим методикам со стадий 1-3 со схемы SS с применением TP-2 и 5-бром-4-хлор-7H-пирроло[2.3-d]пиримидина в качестве подходящих исходных материалов.
В сцинтилляционную пробирку, оборудованную магнитной мешалкой и содержащую промежуточное соединение TTT-2 (281 мг, 0,470 ммоль), добавляют ДХМ (5,0 мл), 2,2-диметоксипропан (0,58 мл, 4,70 ммоль), и PPTS (11,8 мг, 0,047 ммоль). Реакционную смесь перемешивают при кт в течение 23 часов. Раствор переносят в делительную воронку с ДХМ и промывают 2 порциями полунасыщенного раствора соли. Органическую фазу сушат (MgSO4), фильтруют и концентрируют в вакууме с получением указанного в заголовке соединения TTT-2 (266 мг, 89%) в виде белого твердого вещества. ЖХМС [M+H]=637 наблюдаемый. 1H ЯМР (400 MГц, ХЛОРОФОРМ-d) δ ч./млн. 8,68 (с, 1H), 7,49 (шс, 1H), 6,97-6,86 (м, 1H), 6,84-6,71 (м, 1H), 5,35 (шс, 1H), 4,95 (дд, J=2,1, 6,1 Гц, 1H), 4,81 (д, J=6,1 Гц, 2H), 4,48 (шс, 2H), 3,65 (шс, 2H), 3,07-2,89 (м, 1H), 2,80 (шс, 2H), 2,50 (д, J=15,3 Гц, 1H), 1,60 (с, 3H), 1,47 (шс, 9H), 1,34 (с, 3H).
Стадия 2: Синтез трет-бутил 8-(((3aR,4S,6R,6aS)-6-(4-амино-5-бром-7H-пирроло[2.3-d]пиримидин-7-ил)-2,2-диметилтетрагидро-4H-циклопента[d][1.3]диоксол-4-ил)окси)-5-фтор-3,4-дигидроизохинолин-2(1H)-карбоксилата (TTT-3)
В микроволновую пробирку, оборудованную магнитной мешалкой, добавляют TTT-2 (152 мг, 0,238 ммоль) в виде раствора в диоксане (0,60 мл). В раствор добавляют гидроксид аммония (0,6 мл, 5,00 ммоль) и пробирку герметично закрывают тефлоновой крышкой. Пробирку помещают в микроволновый реактор и нагревают до 120°C в течение 6 часов, затем охлаждают до кт в течение ночи. Раствор переносят в делительную воронку с ДХМ и разбавляют водой. Фазы разделяют, и водную фазу экстрагируют 3 порциями ДХМ. Объединенные органические экстракты сушат (MgSO4), фильтруют и концентрируют в вакууме. Неочищенный остаток очищают флэш-хроматографией на колонке (12 г SiO2, Isco, 100% Гепт. до 100% EtOAc, 9 мл фракции) с получением указанного в заголовке соединения TTT-3 (81 мг, 55%) в виде белого твердого вещества. ЖХМС [M+H]=618 наблюдаемый. 1H ЯМР (400 MГц, ХЛОРОФОРМ-d) δ ч./млн. 8,31 (с, 1H), 7,16 (с, 1H), 6,98-6,85 (м, 1H), 6,84-6,73 (м, 1H), 5,61 (шс, 2H), 5,36-5,18 (м, 1H), 4,94 (дд, J=2,5, 6,1 Гц, 1H), 4,82-4,73 (м, 2H), 4,56-4,45 (м, 2H), 3,90-3,42 (м, 2H), 3,02-2,87 (м, 1H), 2,80 (т, J=4,9 Гц, 2H), 2,46 (тд, J=4,4, 14,8 Гц, 1H), 1,59 (с, 3H), 1,53-1,42 (м, 9H), 1,33 (с, 3H).
Стадия 3: Синтез трет-бутил 8-(((3aR,4S,6R,6aS)-6-(4-амино-7H-пирроло[2.3-d]пиримидин-7-ил)-2,2-диметилтетрагидро-4H-циклопента[d][1.3]диоксол-4-ил)окси)-5-фтор-3,4-дигидроизохинолин-2(1H)-карбоксилата (TTT-4)
В реакционную пробирку, оборудованную магнитной мешалкой, добавляют TTT-3 (81,0 мг, 0,131 ммоль), карбонат калия (54,3 мг, 0,393 ммоль) и Pd(PPh3)4 (15,1 мг, 0,013 ммоль). Пробирку герметично закрывают тефлоновой крышкой и продувают аргоном в динамическом вакууме. В пробирку добавляют диоксан (0,58 мл) и воду (0,07 мл). Пробирку переносят в нагревательный блок и нагревают при 100°C в течение 3,5 дней. Пробирку удаляют из нагревательного блока и охлаждают до кт. Реакционную смесь разбавляют водой и переносят в делительную воронку с ДХМ. Фазы разделяют, и водную фазу экстрагируют 3 порциями ДХМ. Объединенные органические экстракты сушат (MgSO4), фильтруют и концентрируют в вакууме. Неочищенный остаток очищают препаративной высокоэффективной жидкостной хроматографией (колонка Lux 5u Cellulose-2 30×250 мм, 33% MeOH масс./0,05% ДЭА в CO2, 100 бар, 80 мл/мин) с получением указанного в заголовке соединения TTT-4 (17,1 мг, 24%) в виде белого твердого вещества. ЖХМС [M+H]=540 наблюдаемый. [α]D22=+2,2° (C=0,1, MeOH). 1H ЯМР (400 MГц, ХЛОРОФОРМ-d) δ ч./млн. 8,35 (с, 1H), 7,19 (д, J=3,7 Гц, 1H), 6,94-6,85 (м, 1H), 6,84-6,75 (м, 1H), 6,44 (шс, 1H), 5,34-5,26 (м, 1H), 5,19 (шс, 2H), 5,01 (дд, J=2,6, 6,2 Гц, 1H), 4,79 (д, J=6,4 Гц, 1H), 4,75 (т, J=5,3 Гц, 1H), 4,44 (шс, 2H), 3,65 (шс, 2H), 3,07-2,86 (м, 1H), 2,78 (т, J=5,1 Гц, 2H), 2,59-2,42 (м, 1H), 1,60 (с, 3H), 1,49 (шс, 9H), 1,33 (с, 3H).
Стадия 4: Синтез (1S,2S,3R,5S)-3-(4-амино-7H-пирроло[2.3-d]пиримидин-7-ил)-5-((5-фтор-1,2,3,4-тетрагидроизохинолин-8-ил)окси)циклопентан-1,2-диола (TTT-5)
В сцинтилляционную пробирку, оборудованную магнитной мешалкой и содержащую TTT-4 (17,1 мг, 0,032 ммоль), добавляют воду (0,08 мл). В раствор добавляют хлористоводородную кислоту (0,4 мл, 4,0 M в диоксане, 2 ммоль), и реакционную смесь перемешивают при кт в течение 15 часов. Раствор переносят в делительную воронку с ДХМ, разбавляют водой и нейтрализуют насыщ. NaHCO3. Фазы разделяют и водную фазу экстрагируют 3 порциями 3:1 смеси ДХМ:ИПС. Объединенные органические экстракты сушат (MgSO4), фильтруют и концентрируют в вакууме. Полученный продукт сушат вымораживанием с получением указанного в заголовке соединения TTT-5 (12,5 мг, >95%) в виде белого твердого вещества. ЖХМС [M+H]=400 наблюдаемый. 1H ЯМР (400 MГц, МЕТАНОЛ-d4) δ ч./млн. 8,08 (с, 1H), 7,22 (д, J=3,7 Гц, 1H), 6,95-6,79 (м, 2H), 6,63 (д, J=3,7 Гц, 1H), 5,12 (кв, J=8,6 Гц, 1H), 4,69-4,61 (м, 1H), 4,57 (дд, J=4,8, 8,4 Гц, 1H), 4,16 (д, J=4,3 Гц, 1H), 3,96 (шс, 2H), 3,08 (т, J=5,1 Гц, 2H), 2,97 (ддд, J=7,3, 9,3, 14,6 Гц, 1H), 2,76 (т, J=5,9 Гц, 2H), 2,04 (ддд, J=3,9, 8,4, 14,1 Гц, 1H).
Пример 181 - (1S,2S,3S,5R)-3-((3-метил-5,6,7,8-тетрагидро-2,7-нафтиридин-1-ил)окси)-5-(4-метил-7H-пирроло[2.3-d]пиримидин-7-ил)циклопентан-1,2-диол
Схема UUU
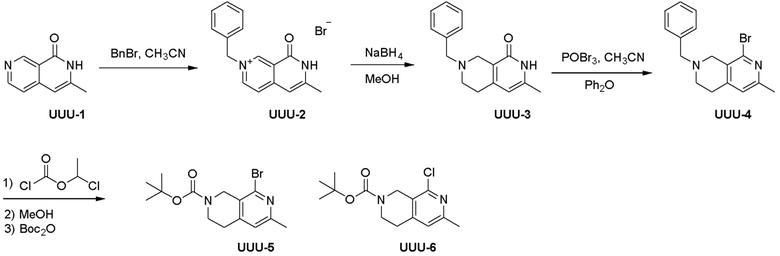
Стадия 1 - Синтез бромид 2-бензил-6-метил-8-оксо-7,8-дигидро-2,7-нафтиридин-2-ия (UUU-2)
К смеси 3-метил-2,7-нафтиридин-1(2H)-она UUU-1 (полученного по методике из патента WO2002068393) (3,9 г, 21,85 ммоль) в CH3CN (50 мл) добавляют BnBr (6,25 г, 36,5 ммоль) при кт (25°C). Затем смесь нагревают при кипении с обратным холодильникомe (85°C) в течение 16 часов. Осадок собирают фильтрацией, промывают этанолом (30 мл) и сушат в вакууме с получением UUU-2 (4,8 г, 60%) в виде желтого твердого вещества. ЖХМС [M-Br] 251; 1H ЯМР (400 MГц, ДМСО-d6) δ ч./млн.=12,57 (шс, 1H), 9,77 (с, 1H), 8,88 (д, J=6,8 Гц, 1H), 8,08 (д, J=6,8 Гц, 1H), 7,55 (шд, J=6,8 Гц, 2H), 7,44 (шд, J=7,3 Гц, 3H), 6,70 (с, 1H), 5,86 (с, 2H), 2,40 (с, 3H)
Стадия 2 - Синтез 7-бензил-3-метил-5,6,7,8-тетрагидро-2,7-нафтиридин-1(2H)-она (UUU-3)
К раствору соединения UUU-2 (4,8 г, 14,49 ммоль) в MeOH (50 мл) добавляют NaBH4 (7,68 г, 203 ммоль) порциями в течение 10 мин при 0°C затем перемешивают при кт (25°C) в течение 4 часов. Смесь концентрируют в вакууме для удаления растворителя. ДХМ (40 мл) добавляют и фильтруют. Фильтрат концентрируют в вакууме с получением неочищенного продукта, который очищают хроматографией на силикагеле (MeOH/ДХМ=0~10%) с получением UUU-3 (2,65 г,72%) в виде желтого твердого вещества. ЖХМС [M+1] 255; 1H ЯМР (400 MГц, ДМСО-d6) δ ч./млн.=11,33 (шс, 1H), 7,42-7,32 (м, 4H), 7,30-7,24 (м, 1H), 5,78 (с, 1H), 3,63 (с, 2H), 3,11 (с, 2H), 2,59 (шд, J=4,5 Гц, 2H), 2,56 (шд, J=3,5 Гц, 2H), 2,10 (с, 3H)
Стадия 3-2-бензил-8-бром-6-метил-1,2,3,4-тетрагидро-2,7-нафтиридин (UUU-4)
К раствору UUU-3 (2,15 г, 8,45 ммоль) в CH3CN (20 мл) и Ph2O (40 мл) при кт (25°C). Затем, POBr3 (12,1 г, 42,3 ммоль) добавляют порциями и нагревают при кипении с обратным холодильникомe (85°C) под N2 в течение 4 часов, через 20 мин начинает образовываться оранжевое твердое вещество. Осадок собирают фильтрацией, растворяют в воде (20 мл) и нейтрализуют раствором NaHCO3 до pH 8. Затем смесь экстрагируют ТБМЭ (50мл×2). Органический слой промывают насыщенным раствором соли (25 мл×2), сушат над Na2SO4, фильтруют и концентрируют в вакууме с получением неочищенного продукта, который очищают хроматографией (силикагель EtOAc/петролейный эфир=0~40%) с получением UUU-4 (1,55 г, 58%) в виде оранжевого твердого вещества. ЖХМС [M+1] 317; 1H ЯМР (400 MГц, CDCl3) δ ч./млн.=7,40-7,32 (м, 4H), 7,32-7,28 (м, 1H), 6,87 (с, 1H), 3,75 (с, 2H), 3,59 (с, 2H), 2,84-2,78 (м, 2H), 2,69-2,64 (м, 2H), 2,47 (с, 3H)
Стадия 4 - Синтез трет-бутил 8-бром-6-метил-3,4-дигидро-2,7-нафтиридин-2(1H)-карбоксилата (UUU-5) и трет-бутил 8-хлор-6-метил-3,4-дигидро-2,7-нафтиридин-2(1H)-карбоксилата (UUU-6)
К раствору соединения UUU-4 (1,30 г, 4,1 ммоль) в ДХМ (15 мл) добавляют по каплям 1-хлорэтилхлороформиат (0,62 мл, 5,74 ммоль) при кт (0°C) в течение 1 мин. Затем смесь нагревают до кт (20°C), перемешивают в течение 10 мин и нагревают при кипении с обратным холодильником (40°C) в течение 2 часов. Смесь концентрируют в вакууме для удаления растворителя. Затем твердое вещество растворяют в MeOH (15 мл) и нагревают при кипении с обратным холодильником (63°C) в течение 1,5 часов. Затем добавляют (Boc)2O (1,07 г 4,92 ммоль) и Et3N (1,71 мл, 12,3 ммоль). Смесь нагревают при (63°C) в течение 16 часов. Смесь концентрируют в вакууме с получением неочищенного продукта, который очищают хроматографией (силикагель MeOH/ДХМ=0~10%) с получением смеси соединений UUU-5 и UUU-6 в соотношении ~1:1 (872 мг) в виде белого твердого вещества, и применяют сразу на следующей стадии. ЖХМС [M+1] 327 и 283.
Стадии 5-7 проводят по методике стадий 2-4 со схемы GGG с применением FFF-1.
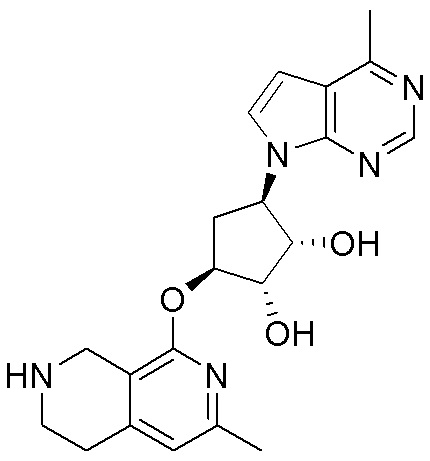
[M+1]
1H ЯМР (400 MГц, ДМСО-d6) δ ч./млн. 8,64 (с, 1H), 7,61 (д, J=3,8 Гц, 1H), 6,72 (д, J=3,8 Гц, 1H), 6,63 (с, 1H), 5,26-5,21 (м, 1H), 5,12 (кв, J=8,7 Гц, 1H), 4,56 (дд, J=4,9, 8,4 Гц, 1H), 4,05 (шд, J=2,3 Гц, 1H), 3,94-3,82 (м, 2H), 3,05 (шт, J=5,9 Гц, 2H), 2,86 (ддд, J=7,4, 9,6, 14,4 Гц, 1H), 2,72-2,66 (м, 2H), 2,64 (с, 3H), 2,31 (с, 3H), 1,92-1,78 (м, 1H)
Синтез трет-бутил-5-фтор-8-гидрокси-4-метил-3,4-дигидроизохинолин-2(1H)-карбоксилата (TP-31)
Схема VVV
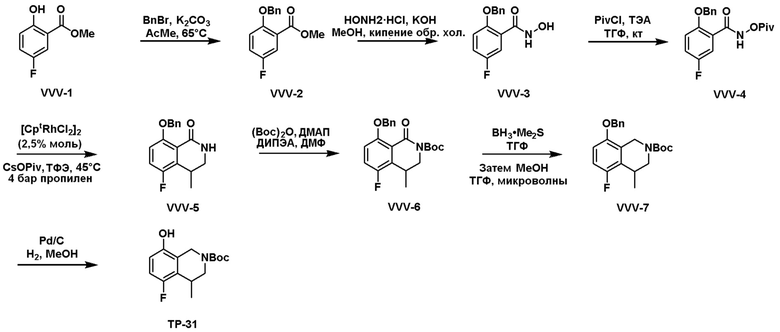
Стадия 1: Синтез метил 2-(бензилокси)-5-фторбензоата (VVV-2)
В круглодонную колбу, оборудованную магнитной мешалкой, добавляют метил 5-фтор-2-гидроксибензоат VVV-1 (3,68 г, 21,6 ммоль), карбонат калия (10,5 г, 75,7 ммоль) и ацетон (108 мл). В раствор добавляют бензилбромид (2,70 мл, 22,7 ммоль) и колбу оборудуют Findenser™. Колбу помещают в нагревательный кожух и нагревают при 65°C в течение 13 часов. Колбу удаляют из нагревательного блока и охлаждают до кт. Твердые вещества фильтруют через слой целлита и промывают несколькими порциями ацетона. Фильтрат концентрируют в вакууме и переносят в делительную воронку с EtOAc (~100 мл). Раствор промывают 2 порциями наполовину насыщенного раствора соли, сушат (MgSO4), фильтруют и концентрируют в вакууме с получением указанного в заголовке соединения VVV-2 (5,64 г, >95% выход) в виде белого кристаллического твердого вещества. Этот продукт применяют на следующей стадии без дальнейшей очистки. 1H ЯМР (400 MГц, ХЛОРОФОРМ-d) δ ч./млн. 7,54 (дд, J=3,3, 8,7 Гц, 1H), 7,51-7,45 (м, 2H), 7,44-7,36 (м, 2H), 7,36-7,29 (м, 1H), 7,14 (ддд, J=3,2, 7,5, 9,1 Гц, 1H), 6,97 (дд, J=4,3, 9,0 Гц, 1H), 5,16 (с, 2H), 3,92 (с, 3H). 19F ЯМР (376MГц, ХЛОРОФОРМ-d) δ ч./млн. -122,59 (с, 1F).
Стадия 2: Синтез 2-(бензилокси)-5-фтор-N-гидроксибензамида (VVV-3)
В круглодонную колбу, оборудованную магнитной мешалкой и содержащую VVV-2 (5,64 г, 21,7 ммоль), добавляют гидрохлорид гидроксиламина (4,52 г, 65,0 ммоль), гидроксид калия (7,29 г, 130 ммоль) и метанол (108 мл). Колбу оборудуют Findenser™ и помещают в нагревательный кожух. Реакционную смесь нагревают при 75°C в течение 3,5 часов. Реакционную смесь удаляют из нагревательного кожуха и постепенно охлаждают до кт. Раствор нейтрализуют уксусной кислотой и концентрируют в вакууме. Остаток переносят в делительную воронку с EtOAc и разбавляют водой. Фазы разделяют, и водную фазу экстрагируют 3 100 мл порциями EtOAc. Органические экстракты сушат (MgSO4), фильтруют и концентрируют в вакууме с получением указанного в заголовке соединения VVV-3 (5,62 г, >95% выход) в виде белого твердого вещества. Продукт применяют на следующей стадии без дальнейшей очистки. ЖХМС [M+H]=262 наблюдаемый. 1H ЯМР (400 MГц, ДМСО-d6) δ ч./млн. 10,71 (с, 1H), 9,18 (д, J=1,5 Гц, 1H), 7,47 (д, J=7,3 Гц, 2H), 7,39 (т, J=7,4 Гц, 2H), 7,35-7,27 (м, 2H), 7,25 (дд, J=3,2, 8,3 Гц, 1H), 7,19-7,13 (м, 1H), 5,20 (с, 2H). 19F ЯМР (376MГц, ДМСО-d6) δ ч./млн. -123,18 (с, 1F).
Стадия 3: Синтез 2-(бензилокси)-5-фтор-N-(пивалоилокси)бензамида (VVV-4)
В круглодонную колбу, оборудованную магнитной мешалкой и содержащую VVV-3 (3,0 г, 11,5 ммоль), добавляют ТГФ (35 мл) и триэтиламин (1,60 мл, 11,5 ммоль), затем по каплям добавляют пивалоилхлорид (1,55 мл, 12,6 ммоль). Реакционную смесь перемешивают при кт в течение 30 минут. Реакционную смесь переносят в делительную воронку с EtOAc. Раствор промывают 2 порциями 1 M HCl водн., 1 порцией наполовину насыщенного NaHCO3 и 2 порциями насыщенного раствора соли. Органический раствор затем сушат (MgSO4), фильтруют и концентрируют в вакууме. Неочищенный остаток очищают флэш-хроматографией на колонке (40 г SiO2, Isco, 100% Гепт. до 100% EtOAc, 20 мл фракции) с получением указанного в заголовке соединения (3,83 г, >95%) в виде бесцветного масла, сольватированного с EtOAc. Масло помещают в ДХМ и разбавляют гептаном, затем концентрируют в вакууме. Полученный продукт затем сушат под высоким вакуумом в течение ночи с получением указанного в заголовке соединения VVV-4 (3,59 г, 90%) в виде белого твердого вещества. ЖХМС [M+H]=346 наблюдаемый. 1H ЯМР (400 MГц, ХЛОРОФОРМ-d) δ ч./млн. 10,95 (с, 1H), 7,89 (дд, J=3,2, 9,1 Гц, 1H), 7,53-7,33 (м, 5H), 7,16 (ддд, J=3,3, 7,2, 9,0 Гц, 1H), 7,00 (дд, J=4,1, 9,1 Гц, 1H), 5,27 (с, 2H), 1,35 (с, 9H).
Стадия 4: Синтез 8-(бензилокси)-5-фтор-4-метил-3,4-дигидроизохинолин-1(2H)-она (VVV-5)
В сцинтилляционную пробирку, оборудованную магнитной мешалкой, добавляют VVV-4 (500 мг, 1,45 ммоль), пивалат цезия (678 мг, 2,90 ммоль) и [CptRhCl2]2 (25,4 мг, 0,036 ммоль). Содержимое пробирки переносят в реактор высокого давления и добавляют ТФЭ (7,0 мл). Реактор продувают азотом 3 раза, затем 3 циклами продувают газообразным пропиленом. Реакционную смесь нагревают до 45°C под давлением 4 бар пропилена в течение 20 часов. Раствор переносят в круглодонную колбу и концентрируют в вакууме. Неочищенный остаток очищают флэш-хроматографией на колонке (24 г SiO2, Isco, 100% Гепт. до 100% EtOAc, 9 мл фракции) с получением указанного в заголовке соединения VVV-5 (270 мг, 65%, 93:7 р.р.) в виде светло-розового твердого вещества. Анализы HSCQC и HOESY соответствуют обозначенному региоизомеру. ЖХМС [M+H]=286 наблюдаемый. 1H ЯМР (400 MГц, ХЛОРОФОРМ-d) δ ч./млн. 7,55 (д, J=7,5 Гц, 2H), 7,43-7,34 (м, 2H), 7,30 (д, J=7,3 Гц, 1H), 7,10 (т, J=8,8 Гц, 1H), 6,87 (дд, J=4,3, 9,0 Гц, 1H), 5,98 (д, J=3,7 Гц, 1H), 5,29-5,21 (м, 1H), 5,18-5,11 (м, 1H), 3,70 (ддд, J=0,7, 4,0, 12,6 Гц, 1H), 3,38-3,28 (м, 1H), 3,23 (ддд, J=1,5, 6,0, 12,6 Гц, 1H), 1,35 (д, J=7,0 Гц, 3H). 19F ЯМР (376MГц, ХЛОРОФОРМ-d) δ ч./млн. -129,24 (с, 1F).
Стадия 5: Синтез трет-бутил-8-(бензилокси)-5-фтор-4-метил-1-оксо-3,4-дигидроизохинолин-2(1H)-карбоксилата (VVV-6)
В круглодонную колбу, оборудованную магнитной мешалкой, добавляют VVV-5 (100 мг, 0,350 ммоль) в виде раствора в ДМФ (4,0 мл). Раствор охлаждают до 10°C, затем добавляют (Boc)2O (84,1 мг, 0,386 ммоль), ДИПЭА (92 мкл, 0,526 ммоль) и ДМАП (2,1 мг, 0,017 ммоль). Реакционную смесь перемешивают при кт в течение 16 часов, что дает низкое превращение в желаемый продукт. Реакционную смесь затем нагревают при 50°C в течение 24 часов и затем повышают до 75°C в течение еще 24 часов для достижения полного поглощения исходного материала. Реакционную смесь разбавляют водой и переносят в делительную воронку с EtOAc. Фазы разделяют, и органическую фазу промывают 1 порцией насыщенного раствора соли, сушат (Ns2SO4), фильтруют и концентрируют в вакууме. Неочищенный остаток очищают флэш-хроматографией на колонке (SiO2, 1% EtOAc/Pet. эфир до 40% EtOAc/пет. эфир) с получением указанного в заголовке соединения VVV-6 (100 мг, 74%) в виде бесцветной камеди. ЖХМС [M+H-Boc]=286 наблюдаемый.
Стадия 6: Синтез трет-бутил-8-(бензилокси)-5-фтор-4-метил-3,4-дигидроизохинолин-2(1H)-карбоксилата (VVV-7)
В круглодонную колбу, оборудованную магнитной мешалкой, добавляют VVV-6 (100 мг, 0,259 ммоль) в виде раствора в ТГФ. Раствор охлаждают до 0°C, затем the по каплям добавляют BH3·ТГФ. После завершения добавления реакционную смесь удаляют из ледяной бани и нагревают до 60°C под азотом в течение 6 часов. На этой стадии реакционную смесь охлаждают до -10°C и гасят осторожным добавлением по каплям MeOH. Реакционную смесь перемешивают при -10°C в течение 16 часов и затем концентрируют в вакууме. Неочищенный остаток очищают препаративной тонкослойной хроматографией (SiO2, 20% EtOAc/пет. эфир) с получением указанного в заголовке соединения VVV-7 (35,0 мг, 36%) в виде бесцветной камеди. ЖХМС [M+H-Изобутилен]=316 наблюдаемый.
Стадия 7: Синтез трет-бутил-5-фтор-8-гидрокси-4-метил-3,4-дигидроизохинолин-2(1H)-карбоксилата (TP-31)
В круглодонную колбу, оборудованную магнитной мешалкой, добавляют VVV-7 (70 мг, 0,190 ммоль) в виде раствора в MeOH (4,5 мл). В раствор добавляют Pd/C (10,0 мг, 0,094 ммоль) и верхнюю часть колбы продувают водородом 5 раз. Реакционную смесь перемешивают кт под 1 атм. водорода в течение 2,5 часов. Реакционную смесь фильтруют, и твердые вещества промывают ДХМ. Фильтрат концентрируют в вакууме, и неочищенный остаток очищают препаративной тонкослойной хроматографией (SiO2, 20% EtOAc/пет. эфир) с получением указанного в заголовке соединения TP-31 (45 мг, 85%) в виде бесцветной камеди. ЖХМС [M+H-Изобутилен]=226 наблюдаемый. 1H ЯМР (400 MГц, ХЛОРОФОРМ-d) δ ч./млн. 6,76 (шт, J=8,8 Гц, 1H), 6,55 (дд, J=4,4, 8,7 Гц, 1H), 5,14-4,78 (м, 2H), 4,22-3,95 (м, 2H), 3,23-2,94 (м, 2H), 1,54-1,46 (с, 9H), 1,23 (д, J=7,3 Гц, 3H).
Синтез трет-бутил (8-гидрокси-1,2,3,4-тетрагидронафталин-1-ил)карбамата (TP-32)
Схема WWW
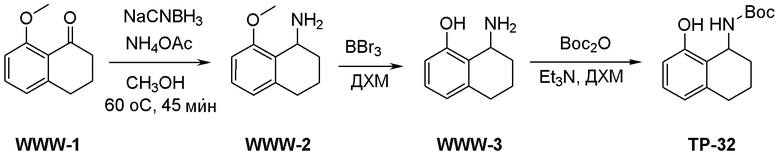
Стадия 1 - Синтез 8-метокси-1,2,3,4-тетрагидронафталин-1-амина (WWW-2)
К перемешиваемой желтой суспензии 8-метокси-3,4-дигидронафталин-1(2H)-она WWW-1 (600 мг, 3,4 ммоль) в MeOH (30 мл) добавляют CH3COONH4 (5,25 г, 68 ммоль) при 15°C. Смесь перемешивают при 15°C в течение 15 мин и затем добавляют NaBH3CN (1,5 г, 24 ммоль) при 15°C. Смесь облучают в микроволновом реакторе при 60°C в течение 45 мин. Смесь гасят насыщ. NaHCO3 (15 мл) и H2O (15 мл) и перемешивают при 15°C в течение 5 мин. Смесь концентрируют для удаления MeOH. Остаток экстрагируют ДХМ (20 мл×2). Объединенные органические слои промывают насыщенным раствором соли (10 мл), сушат над Na2SO4, фильтруют и концентрируют с получением неочищенного продукта WWW-2 (580 мг, 96%) в виде слегка розовой камеди и применяют как есть на следующей стадии.
Стадия 2 - Синтез 8-амино-5,6,7,8-тетрагидронафталин-1-ола (WWW-3)
Соединение WWW-2 (550 мг, 3,10 ммоль) растворяют в HCl/диоксане (5,00 мл), раствор перемешивают при 15°C в течение 10 мин. Затем раствор выпаривают с получением остатка. Остаток растворяют в ДХМ и охлаждают до 0°C на ледяной бане. Затем BBr3 (7,77 мг, 31,0 ммоль) добавляют к реакционному раствору по каплям. Реакционный раствор перемешивают при 20°C в течение 5 ч. Реакционный раствор гасят MeOH (8,00 мл), затем pH раствора доводят до pH=7 добавлением насыщенного водн. NaHCO3. Конечный раствор WWW-3 применяют сразу для следующей стадии (100 мл).
Стадия 3 - Синтез трет-бутил (8-гидрокси-1,2,3,4-тетрагидронафталин-1-ил)карбамата (TP-32)
К раствору (Boc)2O (730 мг, 3,34 ммоль) в диоксане (5,00 мл) добавляют раствор соединения WWW-3 (раствор в водн. насыщ. NaHCO3, 15 мл ~3,3 ммоль) при 0°C одной порцией. Реакционный раствор перемешивают при 20°C в течение 50 часов. Реакционную смесь экстрагируют ДХМ (2×) и органические слои объединяют и промывают насыщ. лимонной кислотой (2×15 мл), водн. насыщ. Na2CO3 (2×15 мл) и насыщенным раствором соли (2×15 мл). Органические слои сушат и выпаривают с получением остатка, который очищают преп.-ТСХ (петролейный эфир/EtOAc=4/1) с получением TP-32 (410 мг, 50%) в виде белого твердого вещества. ЖХМС [M-tBu+1] 207; 1H ЯМР (400 MГц, ДМСО-d6) δ ч./млн. 9,20 (с, 1H), 6,96 (т, J=7,8 Гц, 1H), 6,78 (шд, J=7,5 Гц, 1H), 6,57 (д, J=7,7 Гц, 1H), 6,51 (д, J=7,4 Гц, 1H), 4,80-4,71 (м, 1H), 2,72-2,63 (м, 1H), 2,61-2,53 (м, 1H), 1,89-1,74 (м, 2H), 1,67-1,46 (м, 2H), 1,39 (с, 9H)
Синтез трет-бутил (5-гидроксихроман-3-ил)карбамата (TP-33)
Схема XXX
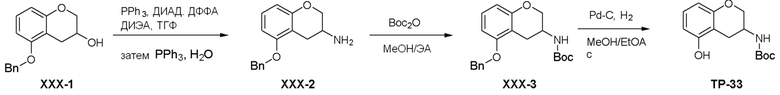
Стадия 1 - Синтез 5-(бензилокси)хроман-3-амина (XXX-2)
Соединение XXX-1 (700 мг, 2,73 ммоль, получено по точным методикам в литературе J. Org. Chem, 2013, 78, 7859-7884) вводят в подобные методики Мицунобу и Стаудингера в той же ссылке с получением продукта XXX-2 (520 мг, 75%) в виде бесцветной камеди, применяют сразу на следующей стадии. ЖХМС [M+1] 266; 1H ЯМР (400 MГц, CDCl3) δ ч./млн. 7,48-7,36 (м, 4H), 7,36-7,30 (м, 1H), 7,07 (т, J=8,3 Гц, 1H), 6,52 (дд, J=2,5, 8,3 Гц, 2H), 5,07 (с, 2H), 4,18-4,07 (м, 1H), 3,80 (ддд, J=1,0, 7,3, 10,5 Гц, 1H), 3,43-3,29 (м, 1H), 3,07 (дд, J=5,0, 16,6 Гц, 1H), 2,50 (дд, J=7,3, 16,8 Гц, 1H)
Стадия 2 - Синтез трет-бутил (5-(бензилокси)хроман-3-ил)карбамата (XXX-3)
К раствору XXX-2 (520 мг, 2,04 ммоль) в сухом MeOH (20 мл) добавляют Boc2O (889 мг, 4,07 ммоль) при кт и перемешивают в течение 30 мин (20 мл EtOAc добавляют для растворимости). Смесь концентрируют в вакууме с получением неочищенного продукта, который очищают хроматографией на силикагеле, элюируя EtOAc в петролейном эфире 0-50%, затем EtOAc в ДХМ 0-50% с получением XXX-3 (620 мг, 76%) в виде белого твердого вещества. ЖХМС [M-tBu+1] 300; 1H ЯМР (400 MГц, CDCl3) δ ч./млн. 7,45-7,36 (м, 4H), 7,34 (шд, J=6,8 Гц, 1H), 7,12-7,05 (м, 1H), 6,53 (д, J=8,3 Гц, 2H), 5,06 (с, 2H), 4,91-4,79 (м, 1H), 4,26-4,15 (м, 1H), 4,08 (шс, 2H), 3,01-2,92 (м, 1H), 2,81-2,69 (м, 1H), 1,44 (с, 9H)
Стадия 3 - Синтез трет-бутил (5-гидроксихроман-3-ил)карбамата (TP-33)
Смесь XXX-3 (620 мг, 2,43 ммоль) и Pd/C (300 мг) в MeOH/EtOAc (10 мл/10 мл) дегазируют с H2 четыре раза. Смесь перемешивают при кт под баллоном H2 в течение 16 ч. Смесь фильтруют и концентрируют (избыток Boc2O с предыдущей стадии также Boc-защищает фенол, поэтому добавляют MeOH (20 мл) и K2CO3 (2 г) и перемешивают в течение 2 ч, затем фильтруют). Смесь фильтруют и концентрируют. Неочищенный продукт разбавляют EtOAc и промывают насыщенным раствором соли (15 мл), сушат над Na2SO4 и концентрируют в вакууме, затем лиофилизируют с получением TP-33 (420 мг, 91%) в виде белого твердого вещества. ЖХМС [M-tBu+1] 210; 1H ЯМР (400 MГц, CDCl3) δ ч./млн. 6,99 (т, J=8,2 Гц, 1H), 6,48 (д, J=8,3 Гц, 1H), 6,40 (д, J=8,0 Гц, 1H), 5,19 (шс, 1H), 4,96-4,89 (м, 1H), 4,22 (шс, 1H), 4,15-4,03 (м, 2H), 2,91 (дд, J=5,5, 16,8 Гц, 1H), 2,71 (шд, J=17,3 Гц, 1H), 1,45 (с, 9H)
Пример 182 и Пример 183 (Схема YYY) -гидрохлорид (1S,2S,3S,5R)-3-((-5-фтор-4-метил-1,2,3,4-тетрагидроизохинолин-8-ил)окси)-5-(4-метил-7H-пирроло[2.3-d]пиримидин-7-ил)циклопентан-1,2-диола (YYY-3-изомер 1 и YYY-3-изомер 2)
Схема YYY
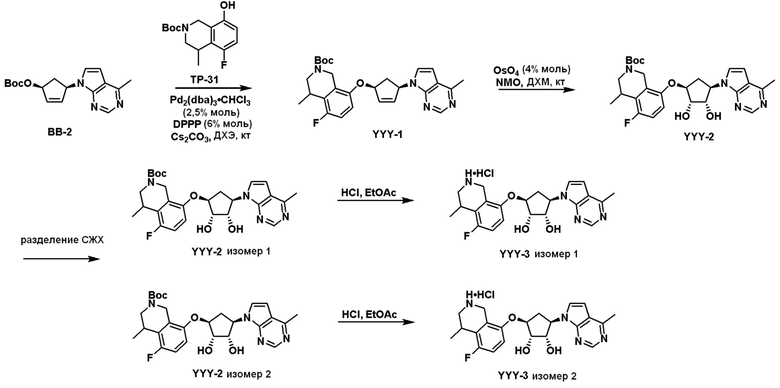
Стадия 1: Синтез трет-бутил-5-фтор-4-метил-8-(((1S,4R)-4-(4-метил-7H-пирроло[2.3-d]пиримидин-7-ил)циклопент-2-ен-1-ил)окси)-3,4-дигидроизохинолин-2(1H)-карбоксилата (YYY-1)
В микроволновую пробирку, оборудованную магнитной мешалкой, добавляют TP-31 (45 мг, 0,160 ммоль), BB-2 (50,4 мг, 0,160 ммоль), Pd2(dba)3·CHCl3 (4,14 мг, 0,004 ммоль), DPPP (3,96 мг, 0,010 ммоль) и карбонат цезия (57,3 мг, 0,176 ммоль). Пробирку продувают азотом под динамическим вакуумом и добавляют свежедегазированный ДХЭ (1,0 мл). Раствор перемешивают при кт под азотом в течение 1 часа. Раствор концентрируют в вакууме, и неочищенный остаток очищают препаративной тонкослойной хроматографией (SiO2, 33% EtOAc/пет. эфир) с получением указанного в заголовке соединения YYY-1 (60 мг, 86%) в виде камеди. ЖХМС [M+H]=579 наблюдаемый. 1H ЯМР (400 MГц, ХЛОРОФОРМ-d) δ ч./млн. 8,76 (с, 1H), 7,35-7,28 (м, 1H), 6,84 (шт, J=8,7 Гц, 1H), 6,73-6,51 (м, 2H), 6,37 (шс, 1H), 6,19-5,99 (м, 2H), 5,28 (шс, 1H), 4,24-3,95 (м, 2H), 3,22-3,05 (м, 3H), 2,73 (д, J=2,5 Гц, 3H), 1,96 (шд, J=14,6 Гц, 1H), 1,69-1,63 (м, 1H), 1,50 (с, 9H), 1,23 (т, J=7,0 Гц, 3H)
Стадия 2: Синтез трет-бутил-8-(((1S,2S,3S,4R)-2,3-дигидрокси-4-(4-метил-7H-пирроло[2.3-d]пиримидин-7-ил)циклопентил)окси)-5-фтор-4-метил-3,4-дигидроизохинолин-2(1H)-карбоксилата (YYY-2)
В реакционную пробирку, оборудованную магнитной мешалкой, добавляют YYY-1 (60,0 мг, 0,130 ммоль) в виде раствора в ДХМ (6,0 мл). В раствор добавляют воду (0,2 мл), NMO (44,1 мг, 0,376 ммоль) и OsO4 (112 мг, 0,0176 ммоль). Реакционную смесь перемешивают при кт в течение 5 часов. Реакцию гасят насыщ. Na2SO3 (5 мл) и переносят в делительную воронку с ДХМ. Фазы разделяют, и водную фазу экстрагируют 1 порцией ДХМ. Объединенные органические экстракты сушат (Na2SO4), фильтруют и концентрируют в вакууме. Неочищенный остаток очищают препаративной тонкослойной хроматографией с получением указанного в заголовке соединения YYY-2 (35 мг, 54%) в виде бесцветной камеди. ЖХМС [M+H]=513 наблюдаемый.
Стадия 3: Синтез трет-бутил-8-(((1S,2S,3S,4R)-2,3-дигидрокси-4-(4-метил-7H-пирроло[2.3-d]пиримидин-7-ил)циклопентил)окси)-5-фтор-4-метил-3,4-дигидроизохинолин-2(1H)-карбоксилата (YYY-2-изомер 1 & YYY-2-изомер 2)
YYY-2 (30 мг, 0,058 ммоль) далее очищают препаративной сверхкритической жидкостной хроматографией (OD 250мм × 30мм ×5 мкм, 0,1% NH4OH/ИПС до 30% NH4OH/ИПС, 60 мл/мин) с получением разделенных диастереомеров YYY-2-изомер 1 (15 мг, 50%) и YYY-2-изомер 2 (15 мг, 50%) в виде белых твердых веществ. ЖХМС [M+H]=513 наблюдаемый.
Стадия 4 с применением YYY-2-изомера 1: Синтез гидрохлорида (1S,2S,3S,5R)-3-((-5-фтор-4-метил-1,2,3,4-тетрагидроизохинолин-8-ил)окси)-5-(4-метил-7H-пирроло[2.3-d]пиримидин-7-ил)циклопентан-1,2-диола (YYY-3-изомер 1)
В реакционную пробирку, оборудованную магнитной мешалкой, добавляют YYY-2-изомер 1 (15 мг, 0,029 ммоль) в виде раствора в EtOAc (1,0 мл). Раствор охлаждают до 0°С, затем добавляют HCl (4M EtOAc, 59 мкл). Реакционную смесь удаляют из ледяной бани и перемешивают при кт в течение 16 часов. Раствор концентрируют в вакууме, затем сушат вымораживанием с получением указанного в заголовке соединения YYY-3-изомера 1 (8,26 мг, 63%) в виде белого твердого вещества. ЖХМС [M+H]=413 наблюдаемый. 1H ЯМР (400 MГц, МЕТАНОЛ-d4) δ ч./млн. 9,05 (д, J=3,3 Гц, 1H), 8,08 (д, J=2,4 Гц, 1H), 7,24 (д, J=3,7 Гц, 1H), 7,15-6,99 (м, 2H), 5,41 (кв, J=9,3 Гц, 1H), 4,76 (шд, J=4,8 Гц, 2H), 4,52-4,41 (м, 1H), 4,36-4,27 (м, 1H), 4,23 (д, J=4,8 Гц, 1H), 3,57-3,40 (м, 3H), 3,12-2,99 (м, 4H), 2,27 (дт, J=5,1, 9,5 Гц, 1H), 1,47 (д, J=6,7 Гц, 3H).
Стадия 4 с применением YYY-2-изомера 2: Синтез гидрохлорида (1S,2S,3S,5R)-3-((-5-фтор-4-метил-1,2,3,4-тетрагидроизохинолин-8-ил)окси)-5-(4-метил-7H-пирроло[2.3-d]пиримидин-7-ил)циклопентан-1,2-диола (YYY-3-изомер 2))
В реакционную пробирку, оборудованную магнитной мешалкой, добавляют YYY-2-изомер 2 (15 мг, 0,029 ммоль) в виде раствора в EtOAc (1,0 мл). Раствор охлаждают до 0°C, затем добавляют HCl (4M EtOAc, 59 мкл). Реакционную смесь удаляют из ледяной бани и перемешивают при кт в течение 16 часов. Раствор концентрируют в вакууме, затем сушат вымораживанием с получением указанного в заголовке соединения YYY-2-изомера 2 (8,48 мг, 70%) в виде белого твердого вещества. ЖХМС [M+H]=413 наблюдаемый. 1H ЯМР (400 MГц, МЕТАНОЛ-d4) δ ч./млн. 9,04 (д, J=3,0 Гц, 1H), 8,06 (д, J=3,2 Гц, 1H), 7,24 (д, J=3,7 Гц, 1H), 7,16-7,01 (м, 2H), 5,42 (кв, J=9,4 Гц, 1H), 4,77-4,70 (м, 2H), 4,48-4,39 (м, 1H), 4,36-4,27 (м, 1H), 4,21 (д, J=4,7 Гц, 1H), 3,55-3,40 (м, 3H), 3,17-2,96 (м, 4H), 2,33-2,18 (м, 1H), 1,46 (д, J=6,7 Гц, 3H).
Примеры 184-189 получают по методике Примеров 182 & 183 (Схема YYY), начиная с подходящего рацемического тетрагидроизохинолина на стадии 1 и разделяя диастереомеры до конечного снятия защиты.
TP-28
ЖХМС
[M+1]
Пример 184 (Изомер 1) - 1H ЯМР (400 MГц, МЕТАНОЛ-d4) δ ч./млн. 9,07 (с, 1H), 8,08 (д, J=3,8 Гц, 1H), 7,31 (т, J=8,0 Гц, 1H), 7,25 (д, J=3,8 Гц, 1H), 7,01 (д, J=8,3 Гц, 1H), 6,90 (д, J=7,8 Гц, 1H), 5,42 (кв, J=9,2 Гц, 1H), 4,83-4,73 (м, 2H), 4,54 (д, J=16,3 Гц, 1H), 4,34 (д, J=16,6 Гц, 1H), 4,24 (д, J=4,5 Гц, 1H), 3,73-3,62 (м, 1H), 3,23-3,04 (м, 2H), 3,03-2,91 (м, 4H), 2,32-2,23 (м, 1H), 1,54 (д, J=6,5 Гц, 3H).
Пример 185 (Изомер 2) - 1H ЯМР (400 MГц, МЕТАНОЛ-d4) 9,08 (с, 1H), 8,06 (д, J=3,8 Гц, 1H), 7,31 (т, J=8,0 Гц, 1H), 7,24 (д, J=3,8 Гц, 1H), 7,01 (д, J=8,3 Гц, 1H), 6,90 (д, J=7,5 Гц, 1H), 5,41 (кв, J=9,2 Гц, 1H), 4,84-4,73 (м, 2H), 4,59-4,50 (м, 1H), 4,34 (д, J=16,8 Гц, 1H), 4,22 (д, J=5,0 Гц, 1H), 3,70-3,60 (м, 1H), 3,21-3,05 (м, 2H), 3,04-2,92 (м, 4H), 2,34-2,25 (м, 1H), 1,55 (д, J=6,3 Гц, 3H)
TP-32
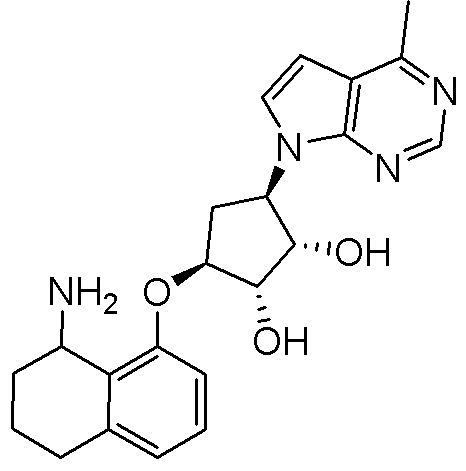
ЖХМС
[M-16+1]
Пример 186 (Изомер 1) - 1H ЯМР (400 MГц, MeOD-d4) δ ч./млн. 8,71 (с, 1H), 7,74 (д, J=3,8 Гц, 1H), 7,34 (т, J=8,0 Гц, 1H), 7,05 (д, J=8,3 Гц, 1H), 6,89 (д, J=7,8 Гц, 1H), 6,86 (д, J=3,5 Гц, 1H), 5,18 (кв, J=9,4 Гц, 1H), 4,84-4,79 (м, 3H), 4,22 (д, J=5,3 Гц, 1H), 3,06 (тд, J=8,4, 14,3 Гц, 1H), 2,98-2,88 (м, 1H), 2,88-2,81 (м, 1H), 2,80 (с, 3H), 2,50 (ддд, J=4,3, 10,0, 14,1 Гц, 1H), 2,25-2,10 (м, 2H), 1,98-1,89 (м, 2H)
Пример 187 (Изомер 2) - 1H ЯМР (400 MГц, MeOD-d4) δ ч./млн. 8,63 (с, 1H), 7,53 (д, J=3,5 Гц, 1H), 7,15 (т, J=8,0 Гц, 1H), 6,89 (д, J=8,3 Гц, 1H), 6,78-6,72 (м, 2H), 5,18 (кв, J=9,1 Гц, 1H), 4,82 (дд, J=5,0, 8,5 Гц, 1H), 4,78-4,72 (м, 1H), 4,40-4,29 (м, 2H), 3,04-2,93 (м, 1H), 2,89-2,74 (м, 2H), 2,73 (с, 3H), 2,36-2,23 (м, 1H), 2,02-1,86 (м, 3H), 1,80 (д, J=4,5 Гц, 1H)
TP-33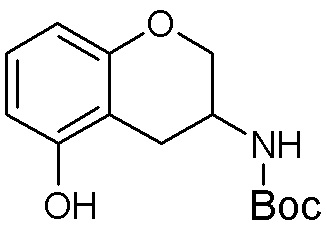
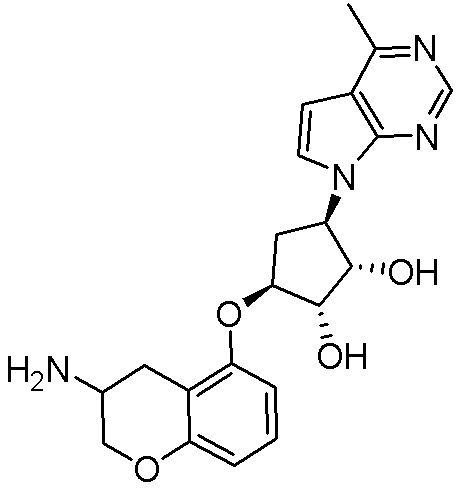
[M+1]
Пример 188 (Изомер 1) - 1H ЯМР (400 MГц, D2O) δ ч./млн. 8,82 (с, 1H), 7,81 (д, J=3,8 Гц, 1H), 7,15 (т, J=8,3 Гц, 1H), 7,08 (д, J=4,0 Гц, 1H), 6,65 (д, J=7,8 Гц, 1H), 6,56 (д, J=8,3 Гц, 1H), 5,33 (кв, J=9,1 Гц, 1H), 4,77-4,73 (м, 1H), 4,62 (дд, J=5,1, 8,7 Гц, 1H), 4,32-4,22 (м, 2H), 4,14 (д, J=11,3 Гц, 1H), 3,97 (шд, J=1,8 Гц, 1H), 3,13 (дд, J=6,3, 18,1 Гц, 1H), 3,02 (ддд, J=7,0, 9,8, 15,1 Гц, 1H), 2,87 (м, 4H), 2,21-2,12 (м, 1H)
Пример 189 (Изомер 2) - 1H ЯМР (400 MГц, D2O) δ ч./млн. 8,84 (с, 1H), 7,80 (д, J=4,0 Гц, 1H), 7,17 (т, J=8,3 Гц, 1H), 7,10 (д, J=3,8 Гц, 1H), 6,66 (д, J=8,0 Гц, 1H), 6,58 (д, J=8,5 Гц, 1H), 5,41-5,31 (м, 1H), 4,77 (шс, 2H), 4,36-4,28 (м, 2H), 4,17 (шд, J=12,0 Гц, 1H), 4,01 (шд, J=1,5 Гц, 1H), 3,20-3,10 (м, 1H), 3,09-2,99 (м, 1H), 2,94 (шс, 1H), 2,90 (с, 3H), 2,23-2,12 (м, 1H)
Пример 190 (Схема ZZZ) - (1S,2S,3S,5R)-3-((6-(дифторметил)-5-фтор-1,2,3,4-тетрагидроизохинолин-8-ил)окси)-5-(4-метил-7H-пирроло[2.3-d]пиримидин-7-ил)циклопентан-1,2-диол (ZZZ-16)
Схема ZZZ
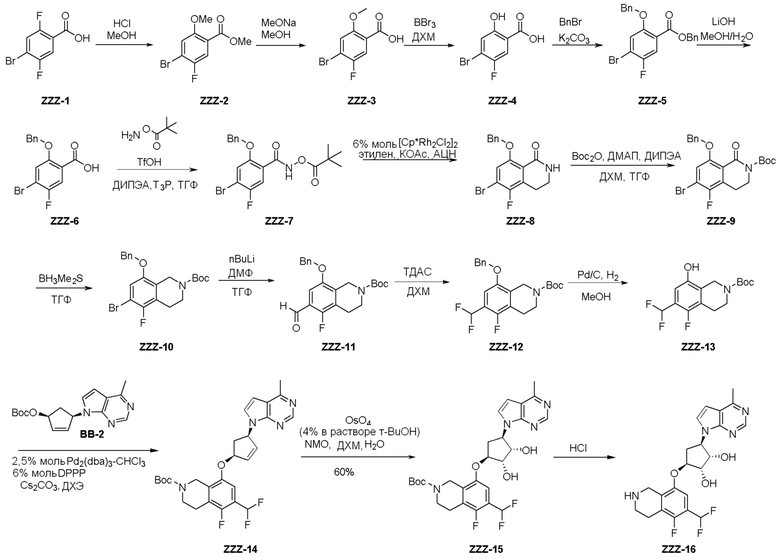
Стадия 1 - Синтез метил 4-бром-5-фтор-2-метоксибензоата (ZZZ-2)
Раствор 4-бром-2,5-дифторбензойной кислоты ZZZ-1 (95 г, 400,85 ммоль) в 4M HCl/MeOH (1500 мл) нагревают при 65°C в течение 3 часов. Смесь концентрируют в вакууме с получением ZZZ-2 (100 г, >99%) в виде светло-желтого твердого вещества. 1H ЯМР (400 MГц, ДМСО-d6) δ ч./млн. 7,93 (дд, J=5,5, 9,8 Гц, 1H), 7,81 (дд, J=6,0, 8,5 Гц, 1H), 3,92-3,85 (м, 3H)
Стадия 2 - Синтез 4-бром-5-фтор-2-метоксибензoйной кислоты (ZZZ-3)
К раствору ZZZ-2 (80 г, 7,97 ммоль) в сухом ДМФ (1200 мл) добавляют раствор MeONa (~17,2 г, 319 ммоль, 8 г Na растворяют в 80 мл MeOH полученного) при 0°C. Смесь перемешивают при 0°C в течение 10 мин, затем нагревают до кт (25°C) и перемешивают в течение 1 ч. К смеси добавляют ТБМЭ (1 л), затем выливают в ледяную воду (800 мл). Смесь экстрагируют ТБМЭ (500 мл×4). Органический слой промывают насыщенным раствором соли (300 мл×2) сушат над Na2SO4, фильтруют и концентрируют с получением продукта метилового эфира, метилового эфира (12,1 г) в виде светло-желтой камеди. Водный слой содержит карбоновую кислоту и его нейтрализуют HCl (1M) до pH=5, затем экстрагируют EtOAc (800 мл×3). Органический слой промывают насыщенным раствором соли (400 мл×2), сушат над Na2SO4, фильтруют и концентрируют с получением ZZZ-3 (100 г, >99% неочищенный продукт) в виде желтого масла и применяют сразу на следующей стадии. ЯМР содержит ДМФ. 1H ЯМР (400 MГц, ДМСО-d6) δ ч./млн. 7,59 (д, J=8,8 Гц, 1H), 7,46 (д, J=5,8 Гц, 1H), 3,83 (с, 3H)
Стадия 3 - Синтез 4-бром-5-фтор-2-гидроксибензoйной кислоты (ZZZ-4)
К раствору ZZZ-3 (~60 г неочищенного продукта, 240,93 ммоль) в сухом ДХМ (600 мл) добавляют BBr3 (68,3 мл растворяют в ДХМ 600 мл, 723 ммоль) при кт (20°C). Смесь перемешивают при кт (20°C) в течение 2 ч. Смесь выливают в воду (500 мл) и экстрагируют ДХМ (800 мл×3). Органический слой промывают насыщенным раствором соли (300 мл×2), сушат над Na2SO4, фильтруют и концентрируют с получением ZZZ-4 (35 г, 62%) в виде желтой камеди. 1H ЯМР (400 MГц, ДМСО-d6) δ ч./млн. 7,62 (д, J=9,0 Гц, 1H), 7,33 (д, J=5,8 Гц, 1H)
Стадия 4 - Синтез бензил 2-(бензилокси)-4-бром-5-фторбензоата (ZZZ-5)
К раствору ZZZ-4 (35 г, 148,93 ммоль) в сухом ДМФ (300 мл) добавляют K2CO3 (41,2 г, 298 ммоль) и BnBr (38,2 г, 223 ммоль). Смесь перемешивают при 25°C в течение 16 ч. Смесь разбавляют водой (200 мл) и экстрагируют EtOAc (300 мл×3). Органические слои собирают, сушат и концентрируют с получением неочищенного светло-желтого масла, которое очищают комби-флэш (силикагель EtOAc/петролейный эфир=0-8%) с получением ZZZ-5 (22 г, 36%) в виде светло-желтого твердого вещества. 1H ЯМР (400 MГц, CDCl3) δ ч./млн. 7,67 (д, J=8,6 Гц, 1H), 7,48-7,32 (м, 10H), 7,25 (д, J=5,5 Гц, 1H), 5,36 (с, 2H), 5,14 (с, 2H)
Стадия 5 - Синтез 2-(бензилокси)-4-бром-5-фторбензoйной кислоты (ZZZ-6)
К раствору ZZZ-5 (22 г, 52,98 ммоль) в MeOH (200 мл) добавляют раствор LiOH.H2O (6,67 г, 159 ммоль) в H2O (200 мл). Реакционную смесь перемешивают при 20°C в течение 4 часов. Реакционную смесь экстрагируют EtOAc (150 мл×2). Водный слой нейтрализуют 1 M водн. HCl при 0°C до pH 4-5, затем экстрагируют EtOAc (200 мл×2). Органический слой промывают насыщенным раствором соли (50 мл×2) сушат над Na2SO4, фильтруют и концентрируют с получением ZZZ-6 (12,5 г, 73%) в виде белого твердого вещества. ЖХМС [M+1] 324,96; 1H ЯМР (400 MГц, ДМСО-d6) δ ч./млн. 13,16 (шс, 1H), 7,66-7,56 (м, 2H), 7,53-7,46 (м, 2H), 7,45-7,37 (м, 1H), 7,45-7,37 (м, 1H), 7,37-7,30 (м, 1H), 5,22 (с, 2H)
Стадия 6 - Синтез 2-(бензилокси)-4-бром-5-фтор-N-(пивалоилокси)бензамида (ZZZ-7)
К раствору ZZZ-6 (23 г, 70,74 ммоль) в ТГФ (300 мл) добавляют O-пивалоилгидроксиламин (35,4 г, 141 ммоль), ДИПЭА (54,9 г, 424 ммоль) при кт (25°C). Затем добавляют T3P (113 г, 177 ммоль) при 0°C. После добавления смесь перемешивают при 0°C в течение 10 мин, затем нагревают до кт (20°C) и перемешивают в течение 16 часов. Смесь концентрируют для удаления большей части растворителя. Оставшуюся смесь разбавляют EtOAc (80 мл), промывают насыщ. водн. NaHCO3 (50 мл) и экстрагируют EtOAc (100 мл×2). Органический слой промывают насыщенным раствором соли (50 мл×2) сушат над Na2SO4, фильтруют и концентрируют с получением неочищенного продукта (42 г) в виде желтой камеди, которую очищают хроматографией (силикагель, EtOAc/петролейный эфир=0-25%) с получением ZZZ-7 (26 г, 87%) в виде светло-желтого твердого вещества. ЖХМС [M+1] 424; 1H ЯМР (400 MГц, CDCl3) δ ч./млн. 10,80 (шс, 1H), 7,93 (д, J=8,8 Гц, 1H), 7,51-7,34 (м, 5H), 7,27-7,23 (м, 1H), 5,24 (с, 2H), 1,33 (с, 9H)
Стадия 7 - Синтез 8-(бензилокси)-6-бром-5-фтор-3,4-дигидроизохинолин-1(2H)-она (ZZZ-8)
К суспензии ZZZ-7 (26 г, 64 ммоль) в MeCN (600 мл) добавляют KOAc (6,91 г, 70,4 ммоль) и [Cp*Rh2Cl2]2 (2,37 г, 3,84 ммоль), сосуд охлаждают до 0°C, в сосуд продувают этилен в течение 30 мин и герметично закрывают. Реакционную смесь перемешивают при кт (20°C) в течение 16 часов. Смесь концентрируют с получением неочищенного продукта (26 г) в виде желтого твердого вещества, которое очищают хроматографией (силикагель петролейный эфир:EtOAc=0-100%) с получением ZZZ-8 (12 г, 59%) в виде желтого твердого вещества. ЖХМС [M+1] 351; 1H ЯМР (400 MГц, ДМСО-d6) δ ч./млн. 7,92 (шс, 1H), 7,56 (д, J=7,0 Гц, 2H), 7,44-7,35 (м, 3H), 7,34-7,28 (м, 1H), 5,19 (с, 2H), 3,30 (дт, J=3,6, 6,2 Гц, 2H), 2,86 (т, J=6,1 Гц, 2H)
Стадия 8 - Синтез трет-бутил 8-(бензилокси)-6-бром-5-фтор-1-оксо-3,4-дигидроизохинолин-2(1H)-карбоксилата (ZZZ-9)
К раствору ZZZ-8 (11 г, 33,113 ммоль) в ТГФ (40 мл) и ДХМ (120 мл) добавляют Boc2O (11,6 г, 53 ммоль), ДИПЭА (15 г, 116 ммоль) и ДМАП (607 мг, 4,97 ммоль). После добавления, смесь перемешивают при кт (22°C) в течение 16 часов. Реакционную смесь концентрируют с получением неочищенного продукта (17,2 г,) в виде желтой камеди, которую затем очищают комби-флэш (EtOAc/петролейный эфир=0-20%) с получением ZZZ-9 (14,52 г, 97%) в виде светло-желтого твердого вещества. ЖХМС [M+23] 472; 1H ЯМР (400 MГц, ДМСО-d6) δ ч./млн. 7,63-7,22 (м, 6H), 5,32-5,14 (м, 2H), 3,86 (т, J=6,0 Гц, 2H), 2,97 (т, J=5,9 Гц, 2H), 1,50 (с, 9H)
Стадия 9 - Синтез трет-бутил 8-(бензилокси)-6-бром-5-фтор-3,4-дигидроизохинолин-2(1H)-карбоксилата (ZZZ-10)
К раствору ZZZ-9 (14,81 г, 32,9 ммоль) в ТГФ (150 мл) добавляют BH3Me2S (19,7 мл, 197 ммоль) при кт (25°C). Смесь нагревают при 70°C в течение 1 часа. Реакцию медленно гасят MeOH (40 мл). Смесь кипятят с обратным холодильникомe (65°C) в течение 16 ч. Смесь концентрируют с получением неочищенного продукта (26 г) в виде желтой камеди, в которую добавляют воду (60 мл), продукт экстрагируют EtOAc (100 мл×2). Органический слой промывают насыщенным раствором соли (50 мл×2), сушат над Na2SO4, фильтруют и концентрируют с получением неочищенного продукта (17 г), который очищают хроматографией (силикагель, EtOAc/петролейный эфир=0-25%) с получением ZZZ-10 (13,2 г) в виде светло-желтой камеди, который лиофилизируют с получением ZZZ-10 (13,06 г, 91%) в виде белого твердого вещества. ЖХМС [M-Boc+1] 337; 1H ЯМР (400 MГц, CDCl3) δ ч./млн. 7,44-7,31 (м, 5H), 6,92 (д, J=5,5 Гц, 1H), 5,04 (шс, 2H), 4,51 (с, 2H), 3,63 (шт, J=5,8 Гц, 2H), 2,88-2,72(м, 2H), 1,50 (с, 9H)
Стадия 10 - Синтез трет-бутил 8-(бензилокси)-5-фтор-6-формил-3,4-дигидроизохинолин-2(1H)-карбоксилата (ZZZ-11)
В высушенную в печи круглодонную колбу добавляют ZZZ-10 (285 мг, 0,653 ммоль) и сухой ТГФ (4,35 мл, 0,15 M). Добавляют раствор ДМФ (0,98 мл, 0,98 ммоль, 1M в ТГФ) и смесь охлаждают до -78°C сухим льдом/ацетоном. nBuLi (0,92 мл, 1,47 ммоль, 1,6M в гексане) добавляют по каплям и перемешивают при -78°C в течение 30 мин, реакционная смесь превращается из прозрачной в желтую. ЖХМС все еще показывает ~50% ZZZ-10 и поэтому добавляют еще 0,5 экв. ДМФ (0,49 мл, 0,49 ммоль, 1M в ТГФ) и nBuLi (0,46 мл, 0,735 ммоль, 1M в гексане) и перемешивают в течение 25 мин. Реакцию гасят водой и экстрагируют EtOAc. Водный слой экстрагируют EtOAc 3×. Объединенные органические фазы промывают водой и сушат над Na2SO4, фильтруют и концентрируют до неочищенного желтого масла (404 мг), которое очищают ISCO 25г 0-20% EtOAc/Гептан с получением ZZZ-11 в виде прозрачного масла (215 мг, 60%). ЖХМС [M+1] 286; 1H ЯМР (400 MГц, CDCl3) δ ч./млн. 10,35 (с, 1H), 7,49-7,32 (м, 5H), 7,23 (д, J=5,3 Гц, 1H), 5,13 (шс, 2H), 4,64 (с, 2H), 3,69 (т, J=5,7 Гц, 2H), 2,86 (т, J=5,1 Гц, 2H), 1,52 (с, 9H)
Стадия 11 - Синтез трет-бутил 8-(бензилокси)-6-(дифторметил)-5-фтор-3,4-дигидроизохинолин-2(1H)-карбоксилата (ZZZ-12)
К раствору ZZZ-11 (418 мг, 1,08 ммоль) добавляют ДХМ (10,8 мл, 0,1M). Раствор охлаждают до 0°C, затем ТДАС (0,36 мл, 2,71 ммоль) добавляют по каплям и перемешивают в течение 4 ч при кт. ЖХМС показывает ~25% продукта. Реакционную смесь охлаждают до 0°C и повторно добавляют ТДАС по каплям (0,36 мл, 2,71 ммоль) и перемешивают при кт в течение 2 часов. Реакция не завершена, и добавляют еще 2 экв. и перемешивают в течение 2 ч. Реакционную смесь затем нейтрализуют насыщ. водн. NaHCO3. Слои разделяют, и водный слой экстрагируют ДХМ 2×, сушат над Na2SO4, фильтруют и концентрируют, затем очищают ISCO 12г 0-15% EtOAc/Гептан с получением ZZZ-12 в виде белого твердого вещества (322 мг, 73%). ЖХМС [M-Boc+1] 308; 1H ЯМР (400 MГц, CDCl3) δ ч./млн. 7,47-7,32 (м, 5H), 7,03-6,75 (м, 2H), 5,10 (шс, 2H), 4,59 (шс, 2H), 3,66 (т, J=5,6 Гц, 2H), 2,84-2,75 (м, 2H), 1,50 (с, 9H)
Стадия 12 - Синтез трет-бутил 6-(дифторметил)-5-фтор-8-гидрокси-3,4-дигидроизохинолин-2(1H)-карбоксилата (ZZZ-13)
В круглодонную колбу добавляют ZZZ-12 (322 мг, 0,79 ммоль) и MeOH (15,8 мл, 0,05 M). Добавляют 10% Pd/C (30 мг) и добавляют водородный баллон. Реакционную смесь перемешивают при кт в течение ночи. Реакционную смесь фильтруют через шприц и фильтровальный наконечник. Неочищенный белый продукт (252 мг) растворяют в ДХМ:MeOH (~2:1) для полной растворимости для добавления в ISCO 12 г и очищают 0-25% EtOAc/гептаном с получением ZZZ-13 в виде белого твердого вещества (239 мг, 95%). ЖХМС [M-Boc+1] 218; 1H ЯМР (400 MГц, CDCl3) δ ч./млн. 6,99-6,62 (м, 2H), 4,55 (шс, 2H), 3,66 (т, J=5,9 Гц, 2H), 2,79 (шс, 2H), 1,52 (с, 9H)
Стадия 13 - Синтез трет-бутил 6-(дифторметил)-5-фтор-8-(((1S,4R)-4-(4-метил-7H-пирроло[2.3-d]пиримидин-7-ил)циклопент-2-ен-1-ил)окси)-3,4-дигидроизохинолин-2(1H)-карбоксилата (ZZZ-14)
В микроволновую пробирку (которую сушат тепловой пушкой, затем охлаждают в потоке N2) добавляют соединение BB-2 (238 мг, 0,753 ммоль), ZZZ-13 (239 мг, 0,753 ммоль), Cs2CO3 (270 мг, 0,829 ммоль), DPPP (18,7 мг, 0,0452 ммоль) и Pd2(dba)3-CHCl3 (19,5 мг, 0,02 ммоль), пробирку продувают N2 пять раз и добавляют ДХЭ (3 мл, продувают N2 в течение 30 мин). Пробирку продувают N2 еще три раза. Раствор перемешивают при 18°C под N2 в течение 55 мин. Реакционную смесь очищают преп.-ТСХ (EtOAc/петролейный эфир=1,5:1) с получением продукта ZZZ-14 (330 мг, 85%) в виде бесцветной камеди. ЖХМС [M+1] 515; 1H ЯМР (400 MГц, CDCl3) δ ч./млн. 8,78 (с, 1H), 7,29 (шд, J=3,4 Гц, 1H), 7,04-6,72 (м, 2H), 6,59 (шд, J=3,0 Гц, 1H), 6,39 (шд, J=5,3 Гц, 1H), 6,19 (шд, J=4,6 Гц, 1H), 6,08 (шдд, J=2,2, 4,3 Гц, 1H), 5,40-5,32 (м, 1H), 4,50 (шс, 2H), 3,73-3,57 (м, 2H), 3,26-3,16 (м, 1H), 2,80 (шт, J=5,4 Гц, 2H), 2,74 (с, 3H), 1,52 (с, 9H)
Стадия 14 - Синтез трет-бутил 6-(дифторметил)-8-(((1S,2S,3S,4R)-2,3-дигидрокси-4-(4-метил-7H-пирроло[2.3-d]пиримидин-7-ил)циклопентил)окси)-5-фтор-3,4-дигидроизохинолин-2(1H)-карбоксилата (ZZZ-15)
К смеси ZZZ-14 (330 мг, 0,641 ммоль) в ДХМ (10 мл)/H2O (0,4 мл) добавляют NMO (225 мг, 1,92 ммоль) и OsO4 (4% в т-BuOH, 285 мг, 0,045 ммоль) при 15°C. Черный раствор перемешивают при 18°C в течение 3 часов. Смесь гасят насыщ. водн. Na2SO3 (5 мл) и разбавляют ДХМ (10 мл) и разделяют. Водный слой экстрагируют ДХМ (5 мл). Объединенные органические слои промывают насыщенным раствором соли (10 мл), сушат над Na2SO4, фильтруют и концентрируют с получением неочищенного продукта (450 мг) в виде коричневого твердого вещества, которое очищают преп.-ТСХ (сначала, EtOAc/MeOH=20:1, затем, EtOAc) с получением ZZZ-15 (210 мг, 60%) в виде светло-желтого твердого вещества. ЖХМС [M+1] 549; 1H ЯМР (400 MГц, CDCl3) δ ч./млн. 8,69 (с, 1H), 7,22 (д, J=3,8 Гц, 1H), 7,10 (шд, J=5,3 Гц, 1H), 7,04-6,73 (м, 1H), 6,61 (д, J=3,8 Гц, 1H), 5,03- 4,88 (м, 1H), 4,80 (шс, 1H), 4,60-4,39 (м, 3H), 4,36-4,24 (м, 1H), 3,65 (шд, J=5,8 Гц, 2H), 3,46-3,29 (м, 1H), 3,20-3,02 (м, 1H), 2,80 (шт, J=5,4 Гц, 2H), 2,74 (с, 3H), 2,62 (с, 1H), 2,39 (с, 1H), 1,56-1,34 (м, 9H)
Стадия 15 - Синтез (1S,2S,3S,5R)-3-((6-(дифторметил)-5-фтор-1,2,3,4-тетрагидроизохинолин-8-ил)окси)-5-(4-метил-7H-пирроло[2.3-d]пиримидин-7-ил)циклопентан-1,2-диола (ZZZ-16)
К раствору ZZZ-15 (210 мг, 0,383 ммоль) в ДХМ (2 мл) добавляют HCl (г)/диоксан (4 N, 0,766 мл, 3,06 ммоль) при 0°C. Смесь перемешивают при 23°C в течение 2 часов. Твердое вещество выпадает в осадок. Растворитель декантируют, и твердое вещество сушат и лиофилизируют с получением ZZZ-16 в виде соли 2HCl+2 H2O (165 мг, 83%) в виде светло-желтого твердого вещества. ЖХМС [M+1] 449; 1H ЯМР (400 MГц, D2O) δ ч./млн. 8,93 (с, 1H), 7,91 (д, J=3,8 Гц, 1H), 7,24-6,88 (м, 3H), 5,41 (кв, J=9,0 Гц, 1H), 4,87 (шдд, J=2,4, 4,4 Гц, 1H), 4,75 (дд, J=5,0, 8,8 Гц, 1H), 4,45 (с, 2H), 4,39 (шд, J=5,0 Гц, 1H), 3,58 (т, J=6,3 Гц, 2H), 3,19-3,07 (м, 3H), 2,99 (с, 3H), 2,35-2,24 (м, 1H).
Пример 191 (Схема AAAA) - Синтез (1S,2S,3S,5R)-3-((4-фтор-3-метокси-5,6,7,8-тетрагидро-2,7-нафтиридин-1-ил)окси)-5-(4-метил-7H-пирроло[2.3-d]пиримидин-7-ил)циклопентан-1,2-диола (AAAA-6)
Схема AAAA
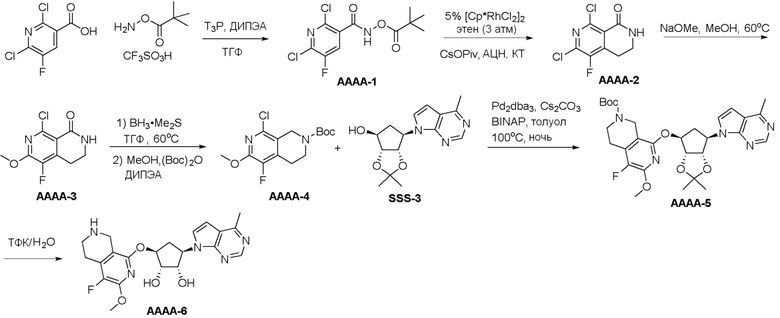
Стадия 1: Синтез 2,6-дихлор-5-фтор-N-(пивалоилокси)никотинамида (AAAA-1)
Соединение AAAA-1 получают по методике получения стадии 4 со схемы FF с применением 2,6-дихлор-5-фторникотиновой кислоты с получением 5,85 г (80% выход) в виде белого твердого вещества. 1H ЯМР (400 MГц, ХЛОРОФОРМ-d) δ ч./млн. 9,91 (шс, 1 H) 8,02 (д, J=5,14 Гц, 1 H) 1,37 (с, 9 H)
Стадия 2: Синтез 6,8-дихлор-5-фтор-3,4-дигидро-2,7-нафтиридин-1(2H)-она (AAAA-2)
Соединение AAAA-2 получают из AAAA-1 по методике получения стадии 4 со схемы GG с применением пивалата цезия с получением 1,47 г (33% выход) в виде желтого твердого вещества. ЖХМС [M+1-2Cl] 167,0. 1H ЯМР (400 MГц, ХЛОРОФОРМ-d) δ ч./млн. 6,13 (шс, 1 H) 3,59 (тд, J=6,45, 3,48 Гц, 2 H) 3,09 (т, J=6,42 Гц, 2 H)
Стадия 3: Синтез 8-хлор-5-фтор-6-метокси-3,4-дигидро-2,7-нафтиридин-1(2H)-она (AAAA-3)
К раствору AAAA-2 (200 мг, 0,85 ммоль) в 20 мл MeOH добавляют метоксид натрия (161 мг, 2,98 ммоль, 5,96 мл, 0,5 M), раствор нагревают при 60°C в течение 1 ч. Реакционную смесь концентрируют, повторно растворяют в ДХМ, промывают H2O дважды, ДХМ выпаривают, неочищенный продукт очищают хроматографией на колонке с 100% EtOAc с получением 100 мг AAAA-3 (51% выход) в виде беловатого твердого вещества. ЖХМС [M+1] 231,0. 1H ЯМР (400 MГц, ХЛОРОФОРМ-d) δ ч./млн. 5,96 (шс, 1 H) 4,09 (с, 3 H) 3,52 (тд, J=6,39, 3,48 Гц, 2 H) 3,03 (т, J=6,24 Гц, 2 H)
Стадия 4: Синтез трет-бутил 8-хлор-5-фтор-6-метокси-3,4-дигидро-2,7-нафтиридин-2(1H)-карбоксилата (AAAA-4)
Соединение AAAA-4 получают из AAAA-3 по методике получения стадии 5 со схемы GG с получением 105 мг (76% выход) в виде бесцветного масла. ЖХМС [M+1-Boc] 217,0. 1H ЯМР (400 MГц, ХЛОРОФОРМ-d) δ ч./млн. 4,48 (с, 2 H) 4,00 (с, 3 H) 3,63 (т, J=5,93 Гц, 2 H) 2,82 (т, J=5,69 Гц, 2 H) 1,50 (с, 9 H)
Стадия 5: Синтез трет-бутил 8-(((3aR,4S,6R,6aS)-2,2-диметил-6-(4-метил-7H-пирроло[2.3-d]пиримидин-7-ил)тетрагидро-4H-циклопента[d][1.3]диоксол-4-ил)окси)-5-фтор-6-метокси-3,4-дигидро-2,7-нафтиридин-2(1H)-карбоксилата (AAAA-5)
Смесь (3aR,4S,6R,6aS)-2,2-диметил-6-(4-метил-7H-пирроло[2.3-d]пиримидин-7-ил)тетрагидро-4H-циклопента[d][1.3]диоксол-4-ола (SSS-3) (98,0 мг, 0,34 ммоль), AAAA-4 (107 мг, 0,339 ммоль), карбоната цезия (221 мг, 0,677 ммоль), трис(дибензилиденацетон)дипалладия (31,0 мг, 0,0339 ммоль) и BINAP (42,2 мг, 0,0677 ммоль) в толуоле (6,77 мл, c=0,05 M) дегазируют и продувают N2 три раза. Реакционную смесь нагревают при 100°C в течение ночи, ЖХМС показывает, что реакция не завершена. Карбонат цезия (221 мг, 0,677 ммоль), трис(дибензилиденацетон)дипалладий (31,0 мг, 0,0339 ммоль) и BINAP (42,2 мг, 0,0677 ммоль) добавляют, дегазируют и продолжают нагревать при 100°C в течение 2 дней. Реакционную смесь охлаждают до кт, добавляют H2O, экстрагируют EtOAc, неочищенный продукт концентрируют и очищают хроматографией на колонке с 85% EtOAc/гептаном с получением 60 мг (31% выход) желтого масла. ЖХМС [M+1] 570,1. 1H ЯМР (400 MГц, ХЛОРОФОРМ-d) δ ч./млн. 8,79 (с, 1 H) 7,37 (д, J=3,67 Гц, 1 H) 6,61 (шс, 1 H) 5,47 (шс, 1 H) 5,35 (ддд, J=7,95, 5,26, 2,69 Гц, 1 H) 4,98-5,05 (м, 1 H) 4,89 (д, J=6,24 Гц, 1 H) 4,36 (шс, 2 H) 3,99 (с, 3 H) 3,56-3,64 (м, 2 H) 3,01 (дт, J=14,55, 7,27 Гц, 1 H) 2,78 (т, J=5,26 Гц, 2 H) 2,73 (с, 3 H) 2,41-2,52 (м, 1 H) 1,60 (с, 3 H) 1,50 (с, 9 H) 1,33 (с, 3 H)
Стадия 6: Синтез (1S,2S,3S,5R)-3-((4-фтор-3-метокси-5,6,7,8-тетрагидро-2,7-нафтиридин-1-ил)окси)-5-(4-метил-7H-пирроло[2.3-d]пиримидин-7-ил)циклопентан-1,2-диола (AAAA-6)
Соединение AAAA-6 получают из AAAA-5 по методике получения стадии 10 со схемы A с получением 19,7 мг (33% выход). ЖХМС [M+1] 430,0. 1H ЯМР (400 MГц, ДМСО-d6) δ ч./млн. 9,68 (шс, 1 H) 9,56 (шс, 1 H) 9,07 (с, 1 H) 7,99 (шс, 1 H) 7,10 (шс, 1 H) 5,11-5,26 (м, 2 H) 4,64 (дд, J=9,05, 4,65 Гц, 1 H) 4,15 (шс, 2 H) 4,10 (д, J=4,03 Гц, 1 H) 3,94 (с, 3 H) 3,38 (шс, 2 H) 2,91-3,03 (м, 3 H) 2,87 (с, 3 H) 1,95-2,05 (м, 1 H)
Примеры 192 & 193 получают по методике Примера 191 за исключением того, что этоксид натрия в этаноле применяют на стадии 3 Схемы AAAA для Примера 192, и изопропоксид натрия в изопропанол применяют на стадии 3 Схемы AAAA для Примера 193.
[M+1]
1H ЯМР (400 MГц, ДМСО-d6) δ ч./млн. 9,50 (шс, 1 H) 9,40 (шс, 1 H) 8,99 (шс, 1 H) 7,93 (шс, 1 H) 7,04 (шс, 1 H) 5,19 (кв, J=9,17 Гц, 1 H) 5,07-5,13 (м, 1 H) 4,63 (дд, J=9,05, 4,52 Гц, 1 H) 4,34-4,51 (м, 2 H) 4,15 (шс, 2 H) 4,09 (д, J=4,16 Гц, 1 H) 3,39 (шс, 2 H) 2,88-3,03 (м, 3 H) 2,83 (с, 3 H) 1,93-2,03 (м, 1 H) 1,33 (т, J=6,97 Гц, 3 H)
[M+1]
1H ЯМР (400 MГц, ДМСО-d6) δ ч./млн. 9,98 (шс, 1 H) 9,82 (шс, 1 H) 9,21 (с, 1 H) 8,10 (д, J=3,67 Гц, 1 H) 7,22 (д, J=3,67 Гц, 1 H) 5,18-5,30 (м, 2 H) 5,10 (д, J=5,14 Гц, 1 H) 4,67 (дд, J=9,11, 4,46 Гц, 1 H) 4,13 (д, J=4,52 Гц, 2 H) 4,09 (д, J=4,03 Гц, 1 H) 3,36 (шс, 2 H) 2,96-3,03 (м, 2 H) 2,94 (м, 4 H) 1,96-2,08 (м, 1 H) 1,35 (д, J=6,1 Гц, 3 H) 1,29 (д, J=6,1 Гц, 3 H)
Схема BBBB-Синтез 2-хлор-6-(дифторметил)никотиновая кислота (BBBB-4)
Схема BBBB
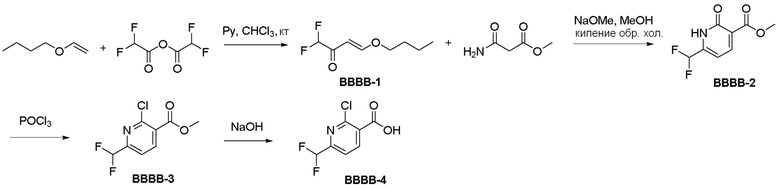
Стадия 1: Синтез (E)-4-бутокси-1,1-дифторбут-3-ен-2-она (BBBB-1)
Соединение BBBB-1 получают по WO20080269059. 1H ЯМР (400 MГц, ХЛОРОФОРМ-d) δ ч./млн. 7,86 (д, J=12,47 Гц, 1 H) 5,90 (д, 1 H) 5,77 (т, J=56 Гц, 1 H) 4,01 (т, J=6,48 Гц, 2 H) 1,68-1,80 (м, 2 H) 1,41-1,48 (м, 2 H) 0,97 (т, J=7,40 Гц, 3 H)
Стадия 2: Синтез метил 6-(дифторметил)-2-оксо-1,2-дигидропиридин-3-карбоксилата (BBBB-2)
Соединение BBBB-2 получают по WO20080269059. 1H ЯМР (400 MГц, ДМСО-d6) ä ч./млн. 12,18 (шс, 1 H) 8,20 (д, J=5,87 Гц, 1 H) 7,05 (шс, 1 H) 6,86 (т, J=52 Гц, 1 H) 3,81 (с, 3 H)
Стадия 3: Синтез метил 2-хлор-6-(дифторметил)никотината (BBBB-3)
Смесь BBBB-2 (3340 мг, 16,44 ммоль) и оксихлорида фосфора (10 мл) нагревают при 110°C в течение 24 ч, охлаждают до кт, добавляют ледяную воду, нейтрализуют твердым KOH. Реакционную смесь экстрагируют EtOAc три раза, органические слои объединяют и концентрируют, очищают хроматографией на колонке с 18% EtOAc/гептаном с получением 3,27 г (90% выход) BBBB-3 в виде белого твердого вещества. 1H ЯМР (400 MГц, ХЛОРОФОРМ-d) δ ч./млн. 8,31 (д, J=7,83 Гц, 1 H) 7,67 (д, J=7,83 Гц, 1 H) 6,61 (т, J=56 Гц, 1 H) 4,00 (с, 3 H); 19F ЯМР (376 MГц, ХЛОРОФОРМ-d) δ ч./млн. -116,82 (с, 1 F)
Стадия 4: Синтез 2-хлор-6-(дифторметил)никотиновой кислоты (BBBB-4)
К раствору BBBB-3 (1160 мг, 5,235 ммоль) в 20 мл MeOH добавляют гидроксид натрия (1050 мг, 26,2 ммоль, 5,23 мл, 5 M), нагревают при 65°C в течение 4 ч. Реакционную смесь нейтрализуют 1 N HCl до pH 4, растворитель удаляют, твердое вещество сушат над вакуумом и применяют на следующей стадии.
Пример 194 получают по методике Примера 191 (Схема AAAA), исключая стадию 3
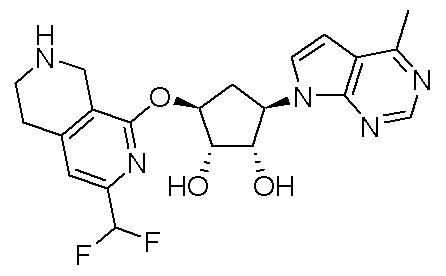
[M+1]
1H ЯМР (400 MГц, ДМСО-d6) δ ч./млн. 10,27 (шс, 1 H) 10,13 (шс, 1 H) 9,22 (с, 1 H) 8,12 (д, J=3,67 Гц, 1 H) 7,15-7,28 (м, 2 H) 6,84 (т, J=56 Гц, 1 H) 5,12-5,30 (м, 2 H) 4,69 (дд, J=8,93, 4,65 Гц, 1 H) 4,08-4,27 (м, 3 H) 3,38 (д, J=9,66 Гц, 2 H) 3,10 (шс, 2 H) 2,86-3,00 (м, 4 H) 1,92-2,10 (м, 1 H)
Примеры 195 & 196 получают по методике Примера 99 со схемы NN с применением подходящего NBoc-защищенного тетрагидроизохинолина на стадии 2.
TP-14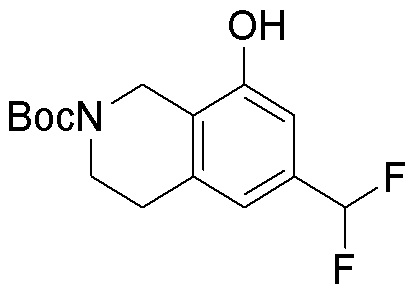
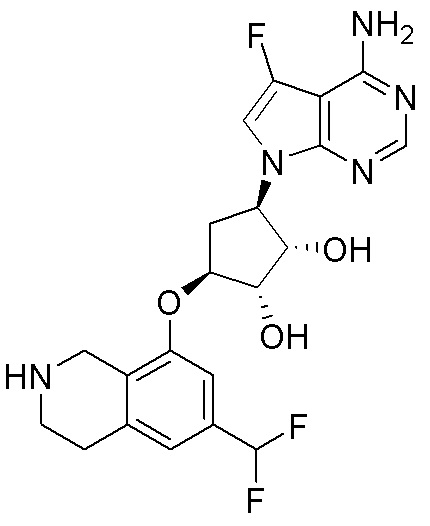
[M+1]
1H ЯМР (400 MГц, D2O, соль HCl) δ ч./млн. 8,20 (с, 1H), 7,25 (д, J=2,0 Гц, 1H), 7,12 (с, 1H), 7,09 (с, 1H), 6,77 (т, J=56,0 Гц, 1H), 5,29-5,17 (м, 1H), 4,84-4,81 (м, 1H), 4,56 (дд, J=5,0, 8,8 Гц, 1H), 4,37 (с, 2H), 4,29 (д, J=4,3 Гц, 1H), 3,50 (т, J=6,3 Гц, 2H), 3,13 (т, J=6,3 Гц, 2H), 3,03 (ддд, J=7,2, 9,3, 14,9 Гц, 1H), 2,15-2,01 (м, 1H)
ZZZ-13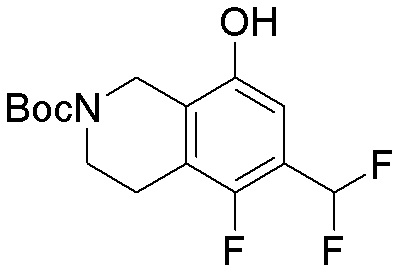
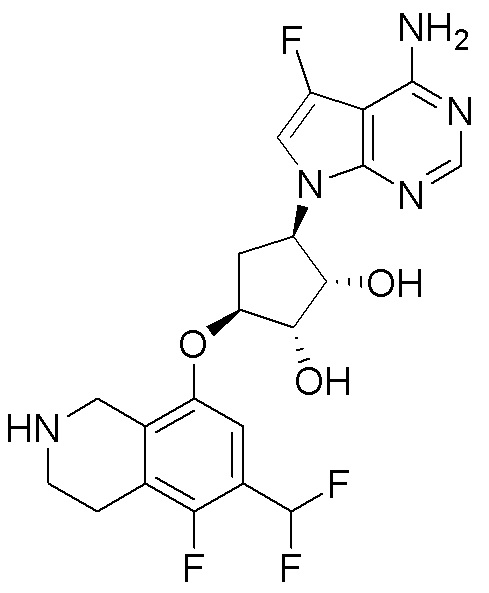
[M+1]
1H ЯМР (400 MГц, ДМСО-d6, соль HCl) δ ч./млн. 9,84 (шс, 2H), 8,76 (шс, 1H), 8,43 (с, 1H), 7,70 (д, J=1,8 Гц, 1H), 7,38-7,02 (м, 2H), 5,07 (кв, J=9,1 Гц, 1H), 4,71-4,56 (м, 1H), 4,47 (дд, J=4,9, 9,4 Гц, 1H), 4,23 (шс, 2H), 3,99 (шд, J=5,0 Гц, 1H), 3,37 (шс, 2H), 3,03-2,95 (м, 2H), 2,93-2,80 (м, 1H), 1,96-1,81 (м, 1H)
Схема CCCC - Синтез трет-бутил 8-гидрокси-6-(изоксазол-4-ил)-3,4-дигидроизохинолин-2(1H)-карбоксилата (TP-34)
Схема CCCC
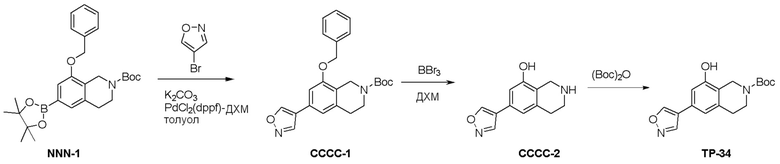
Стадия 1 - Синтез трет-бутил 8-(бензилокси)-6-(изоксазол-4-ил)-3,4-дигидроизохинолин-2(1H)-карбоксилата (CCCC-1)
Раствор NNN-1 (600 мг, 1,29 ммоль), 4-бромизоксазола (286 мг, 1,93 ммоль), K2CO3 (535 мг, 3,87 ммоль) и PdCI2(dppf)-ДХМ (94 мг, 0,129 ммоль) в толуоле (2,5 мл) и воде (1,5 мл) перемешивают под атмосферой N2 при 75°C в течение 16 ч. Реакционную смесь затем охлаждают до к.т. Воду добавляют, и смесь экстрагируют EtOAc (3×5 мл). Объединенные органические слои промывают насыщенным раствором соли (5 мл), сушат над Na2SO4, фильтруют и концентрируют. Неочищенный продукт очищают хроматографией на колонке с EtOAc/петролейным эфиром 15-85% с получением бесцветного масла CCCC-1 (160 мг, 31%). ЖХМС [M-tBu+1] 350,9
Стадия 2 - Синтез 6-(изоксазол-4-ил)-1,2,3,4-тетрагидроизохинолин-8-ола (CCCC-2)
Соединение CCCC-1 (140 мг, 0,344 ммоль) растворяют в ДХМ (10 мл). Реакционный раствор охлаждают до 0°C на ледяной бане. BBr3 (518 мг, 2,07 моль) добавляют. Реакционная смесь превращается из суспензии в прозрачный желтый раствор. Реакционную смесь перемешивают при 25°C в течение 16 часов. Реакционную смесь охлаждают до 0°C и MeOH (2 мл) добавляют в реакционную смесь по каплям, затем воду (20 мл). Реакционный раствор промывают ДХМ (10 мл×2). Водный слой разделяют и pH доводят до pH 9 применением твердого NaHCO3. Конечный раствор CCCC-2 применяют сразу для следующей стадии без дальнейшей очистки (23 мл, водн. раствор неочищенного продукта, >99%).
Синтез трет-бутил 8-гидрокси-6-(изоксазол-4-ил)-3,4-дигидроизохинолин-2(1H)-карбоксилата (TP-34)
MeOH (5 мл) и диоксан (5 мл) добавляют в раствор CCCC-2 (23 мл водн.), затем Boc2O (83 мг, 0,38 ммоль) добавляют в реакционный раствор, раствор перемешивают при 25°C в течение 16 часов. pH реакционного раствора доводят до pH~3 добавлением 1N HCl водн. Раствор разделяют, затем водный слой экстрагируют ДХМ (10 мл). Органические слои объединяют и промывают насыщенным NaCl (20,0 мл). Органический слой разделяют, сушат и выпаривают с получением неочищенного продукта, который очищают флэш-хроматографией, элюируя петролейным эфиром/EtOAc 0-50% с получением желаемого продукта в виде белого твердого вещества TP-34 (84 мг, 77% выход за 2 стадии). МС [M-Boc+1] 217,1; 1H ЯМР (400 MГц, ДМСО-d6) δ ч./млн. 9,83 (с, 1H), 9,32 (с, 1H), 9,02 (с, 1H), 6,97 (с, 1H), 6,89 (с, 1H), 4,36 (с, 2H), 3,55 (м, 2H), 2,75 (м, 2H), 1,44 (с, 9H)
Пример 197 получают с применением методики со схемы CC и с применением соединения TP-34.
TP-34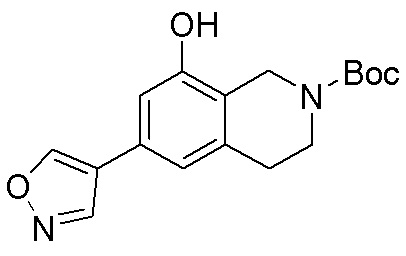
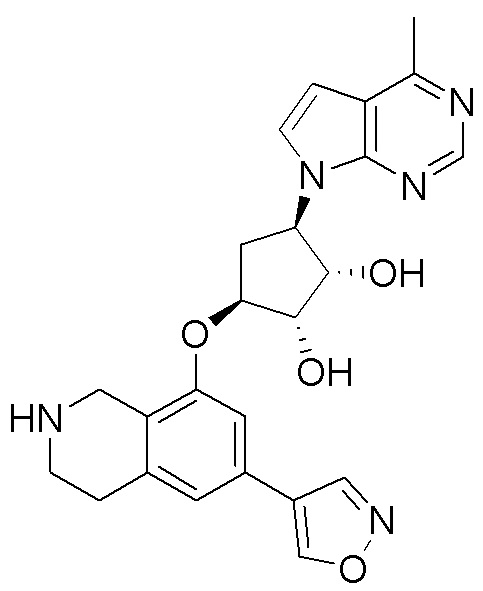
[M+1]
1H ЯМР (400 MГц, D2O) δ ч./млн. 8,98 (с, 1H), 8,83 (с, 1H), 8,80 (с, 1H), 7,80 (д, J=3,8 Гц, 1H), 7,15 (с, 1H), 7,12-7,07 (м, 2H), 5,35 (кв, J=9,0 Гц, 1H), 4,90-4,86 (м, 1H), 4,69-4,65 (м, 1H), 4,39-4,31 (м, 3H), 3,50 (т, J=6,3 Гц, 2H), 3,17-3,01 (м, 3H), 2,89 (с, 3H), 2,28-2,16 (м, 1H)
Схема DDDD - Синтез трет-бутил 5-фтор-8-гидрокси-6-метокси-3,4-дигидроизохинолин-2(1H)-карбоксилата (TP-35)
Схема DDDD
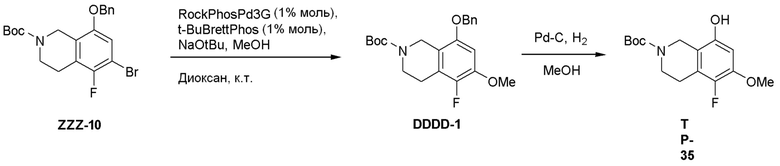
Стадия 1 - Синтез трет-бутил 8-(бензилокси)-5-фтор-6-метокси-3,4-дигидроизохинолин-2(1H)-карбоксилата (DDDD-1)
В пробирку A загружают t-BuBrettPhos (49 мг, 0,10 ммоль), NaOtBu (67,8 мг, 0,706 ммоль) и ZZZ-10 (220 мг, 0,504 ммоль) в диоксане (3 мл). Пробирку дегазируют N2 три раза. Затем в пробирку А добавляют MeOH (81 мг, 2,52 ммоль). В пробирку B загружают RockPhosPd3G (85 мг, 0,10 ммоль) и дегазируют N2 три раза, затем добавляют диоксан (2 мл) и перемешивают в течение 1 мин. Предкатализатор из пробирки B переносят в пробирку A и реакционный раствор перемешивают при 25°C в течение 16 часов. EtOAc (10 мл) добавляют для разбавления раствора и промывают водой (5 мл×2). Органические слои разделяют, сушат и выпаривают с получением неочищенного продукта, который очищают флэш-хроматографией, элюируя EtOAc/петролейным эфиром 0-15% с получением желаемого продукта в виде бесцветного масла DDDD-1 (110 мг, 56%). ЖХМС [M-Boc+1] 287,9; 1H ЯМР (400 MГц, ДМСО-d6) δ ч./млн. 7,54-7,27 (м, 5H), 6,83 (д, J=7,5 Гц, 1H), 5,15 (с, 2H), 4,43-4,26 (м, 2H), 3,82 (с, 3H), 3,53 (т, J=5,8 Гц, 2H), 2,71-2,66 (м, 2H), 1,42 (с, 9H)
Стадия 2 - Синтез трет-бутил 5-фтор-8-гидрокси-6-метокси-3,4-дигидроизохинолин-2(1H)-карбоксилата (TP-35)
Соединение DDDD-1 (110 мг, 0,28 ммоль) растворяют в MeOH (3 мл) и EtOAc (1 мл), затем добавляют Pd/C (15 мг, 0,142 ммоль). Раствор дегазируют H2 четыре раза и перемешивают при 25°C в течение 16 часов под H2 баллоном. Реакционную смесь разбавляют ДХМ (5 мл), фильтруют и концентрируют с получением TP-35 в виде белого твердого вещества (70 мг, 83%). ЖХМС [M-tBu+1] 242,1; 1H ЯМР (400 MГц, ДМСО-d6) δ ч./млн. 9,58 (с, 1H), 6,46 (д, J=7,3 Гц, 1H), 4,28 (с, 2H), 3,74 (с, 3H), 3,55-3,50 (м, 2H), 2,65 (м, 2H), 1,43 (с, 9H)
Соединение TP-36 получают с применением методики со Схемы DDDD с применением EtOH на Стадии 1
[M-tBu+1]
1H ЯМР (400 MГц, ДМСО-d6) δ ч./млн. 9,52 (с, 1H), 6,44 (д, J=7,0 Гц, 1H), 4,28 (с, 2H), 3,99 (кв, J=6,9 Гц, 2H), 3,55-3,49 (м, 2H), 2,64 (шт, J=6,0 Гц, 2H), 1,43 (с, 9H), 1,32 (т, J=6,9 Гц, 3H)
Примеры 198 & 199 получают с применением методики со схемы CC и с применением соединений TP-35 & TP-36, соответственно.
TP-35
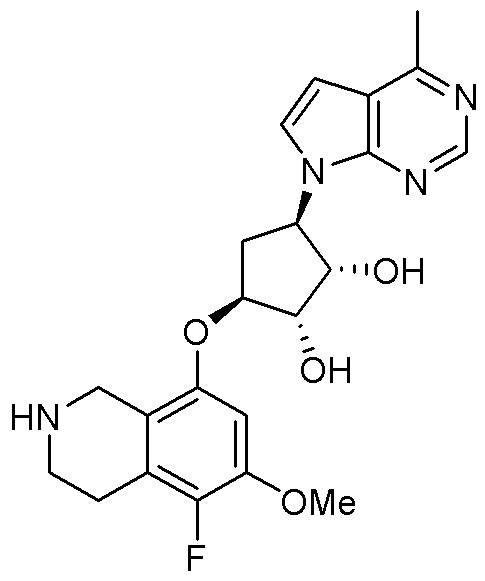
[M+1]
1H ЯМР (400 MГц, MeOD, соль HCl) δ ч./млн. 9,05 (с, 1H), 8,09 (д, J=3,7 Гц, 1H), 7,25 (д, J=3,7 Гц, 1H), 6,93-6,85 (м, 1H), 5,40 (кв, J=9,3 Гц, 1H), 4,94-4,90 (м, 1H), 4,81-4,66 (м, 1H), 4,33 (с, 2H), 4,21 (д, J=5,0 Гц, 1H), 3,93 (с, 3H), 3,57-3,47 (м, 2H), 3,14-3,05 (м, 3H), 3,02 (с, 3H), 2,37-2,24 (м, 1H)
TP-36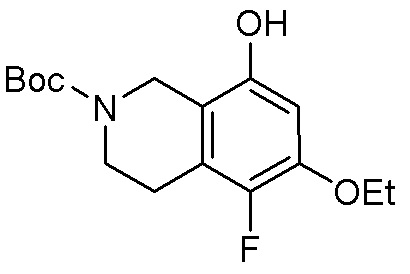
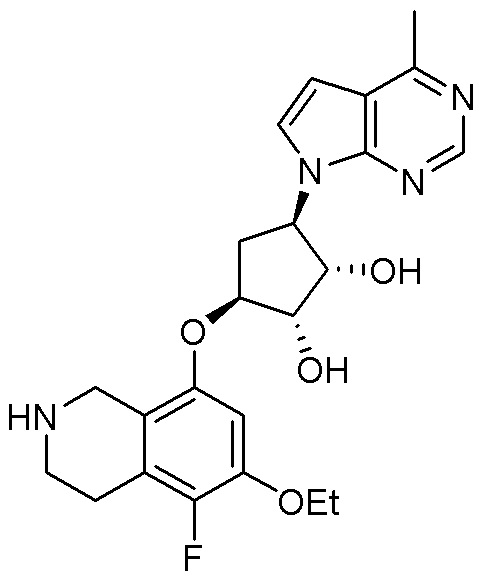
[M+1]
1H ЯМР (400 MГц, D2O, соль HCl) δ ч./млн. 8,82 (с, 1H), 7,84-7,74 (м, 1H), 7,08 (д, J=3,8 Гц, 1H), 6,70 (д, J=7,2 Гц, 1H), 5,28 (кв, J=9,1 Гц, 1H), 4,76-4,73 (м, 1H), 4,66-4,58 (м, 1H), 4,36-4,20 (м, 3H), 4,18-4,05 (м, 2H), 3,45 (т, J=6,4 Гц, 2H), 3,08-2,93 (м, 3H), 2,87 (с, 3H), 2,23-2,09 (м, 1H), 1,31 (т, J=7,0 Гц, 3H)
Схема EEEE - Синтез трет-бутил 6-(дифторметокси)-5-фтор-8-гидрокси-3,4-дигидроизохинолин-2(1H)-карбоксилат (TP-37)
Схема EEEE
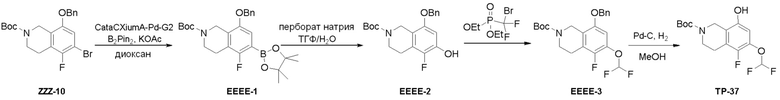
Стадия 1 - Синтез трет-бутил 8-(бензилокси)-5-фтор-6-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)-3,4-дигидроизохинолин-2(1H)-карбоксилата (EEEE-1)
К раствору ZZZ-10 (500 мг, 1,15 ммоль) в диоксане (29 мл) добавляют бис(пинаколато)диборон (1,46 г, 5,73 ммоль). Раствор дегазируют с N2 в течение 10 мин и cataCXium® A-Pd-G2 (76,6 мг, 0,12 ммоль) и KOAc (337 мг, 1,79 ммоль) добавляют в реакционный раствор под атмосферой N2. Реакционную смесь нагревают при 80°C в течение 16 часов. Добавляют воду (10 мл) и реакционную смесь экстрагируют EtOAc (10 мл×2). Органические слои разделяют, сушат и выпаривают с получением неочищенного продукта, который очищают флэш-хроматографией, элюируя петролейным эфиром/EtOAc 0-20% с получением EEEE-1(78%, 443 мг) в виде серого твердого вещества. ЖХМС [M-Boc+1] 384,0; 1H ЯМР (400 MГц, ДМСО-d6) δ ч./млн. 7,52-7,32 (м, 5H), 7,05 (д, J=4,3 Гц, 1H), 5,13 (с, 2H), 4,47 (шс, 2H), 3,57 (шт, J=5,8 Гц, 2H), 2,72-2,66 (м, 2H), 1,43 (с, 9H), 1,30 (с, 12H)
Стадия 2 - Синтез трет-бутил 8-(бензилокси)-5-фтор-6-гидрокси-3,4-дигидроизохинолин-2(1H)-карбоксилата (EEEE-2)
К раствору EEEE-1 (430 мг, 0,89 ммоль) в ТГФ (4 мл) и воде (4 мл) добавляют перборат натрия (306 мг, 3,74 ммоль) одной порцией при кт под N2. Реакционный раствор перемешивают при комнатной температуре в течение 8 часов. Смесь разбавляют EtOAc (10 мл) и водой (5 мл). Органический слой разделяют, сушат и выпаривают с получением неочищенного продукта, который очищают флэш-хроматографией, элюируя петролейным эфиром/EtOAc 0-20% с получением EEEE-2 (150 мг, 45%) в виде белого твердого вещества. ЖХМС [M+Na] 396.
Стадия 3 - Синтез трет-бутил 8-(бензилокси)-6-(дифторметокси)-5-фтор-3,4-дигидроизохинолин-2(1H)-карбоксилата (EEEE-3)
KOH (165 мг, 2,95 моль) суспендируют в смеси ацетонитрила (1 мл) и воды (1 мл) и охлаждают до -20°C. Соединение EEEE-2 (110 мг, 0,295 ммоль) добавляют порциями, затем диэтил (бром-дифторметил)фосфонат (157 мг, 0,59 ммоль) в течение более 15 мин. Смесь нагревают до 25°C в течение 2 дней. Воду добавляют в реакционный раствор и экстрагируют EtOAc (10 мл×3). Органические слои разделяют, сушат и концентрируют с получением неочищенного продукта, который очищают флэш-хроматографией, элюируя петролейным эфиром/EtOAc 0-10% с получением EEEE-3 (100 мг, 80%) в виде белого твердого вещества. ЖХМС [M-Boc+1] 324.
Стадия 4 - Синтез трет-бутил 6-(дифторметокси)-5-фтор-8-гидрокси-3,4-дигидроизохинолин-2(1H)-карбоксилата (TP-37)
К раствору EEEE-3 (100 мг, 0,23 ммоль) в MeOH (5 мл) добавляют Pd/C (25 мг, 0,024 ммоль). Смесь дегазируют и продувают H2 три раза. Полученную смесь перемешивают при кт (28°C) в течение 16 часов. Реакционную смесь разбавляют ДХМ (20 мл) и фильтруют и концентрируют с получением TP-37 в виде белого твердого вещества (75 мг, 95%). ЖХМС [M-tBu+1] 278,0; 1H ЯМР (400 MГц, ДМСО-d6) δ ч./млн. 9,99 (шс, 1H), 7,31 (т, J=74 Гц, 1H), 6,62 (д, J=8 Гц, 1H), 4,33 (с, 2H), 3,55 (т, J=8 Гц, 2H), 2,69 (т, J=8 Гц, 2H), 1,43 (с, 9H)
Пример 200 получают с применением методики со схемы CC и с применением соединения TP-37.
TP-37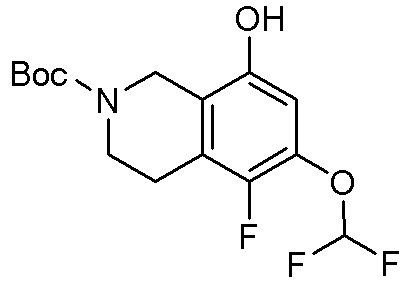
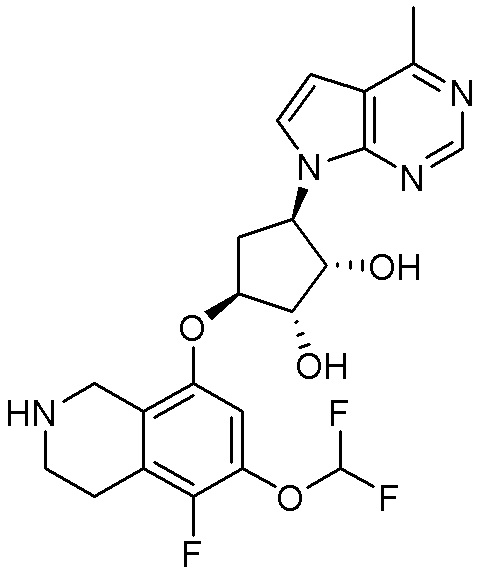
[M+1]
1H ЯМР (400 MГц, D2O, соль HCl) δ ч./млн. 8,85 (с, 1H), 7,82 (д, J=3,8 Гц, 1H), 7,11 (д, J=3,8 Гц, 1H), 6,98-6,58 (м, 2H), 5,32 (кв, J=9,0 Гц, 1H), 4,76-4,73 (м, 1H), 4,67-4,62 (м, 1H), 4,35-4,25 (м, 3H), 3,49 (т, J=6,3 Гц, 2H), 3,10-2,96 (м, 3H), 2,90 (с, 3H), 2,24-2,12 (м, 1H)
Схема FFFF - Синтез трет-бутил 5-фтор-8-гидрокси-6-изопропокси-3,4-дигидроизохинолин-2(1H)-карбоксилата (TP-38)
Схема FFFF
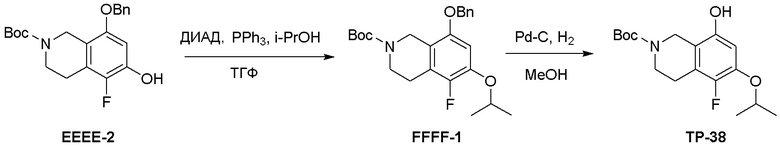
Стадия 1 - Синтез трет-бутил 8-(бензилокси)-5-фтор-6-изопропокси-3,4-дигидроизохинолин-2(1H)-карбоксилата (FFFF-1)
Раствор EEEE-2 (170 мг, 0,455 ммоль) в ТГФ (3 мл) охлаждают на ледяной бане и добавляют PPh3 (478 мг, 1,82 ммоль) под N2. ДИАД (368 мг, 1,82 ммоль) добавляют по каплям, и реакционную смесь перемешивают при 25°C в течение 30 мин с получением белой суспензии. Затем iPrOH (82,1 мг, 1,37 ммоль) добавляют в реакционный раствор одной порцией и перемешивают при 15°C в течение 16 часов. Смесь разбавляют EtOAc (50 мл) и H2O (100 мл). Смесь разделяют, и водный слой экстрагируют EtOAc (50 мл). Объединенные органические слои промывают насыщенным раствором соли (50 мл), сушат над Na2SO4, фильтруют и концентрируют. Неочищенный продукт очищают флэш-хроматографией, элюируя петролейным эфиром/EtOAc 0-20% с получением FFFF-1 (160 мг, 85%) в виде бесцветного масла. ЖХМС [M-tBu+1] 270,1; 1H ЯМР (400 MГц, ДМСО-d6) δ ч./млн. 7,50-7,29 (м, 5H), 6,78 (д, J=7,0 Гц, 1H), 5,15 (с, 2H), 4,65-4,53 (м, 1H), 4,38 (шд, J=2,8 Гц, 2H), 3,54 (т, J=5,9 Гц, 2H), 2,74-2,62 (м, 2H), 1,43 (с, 9H), 1,23 (д, J=6,0 Гц, 6H)
Стадия 2 - Синтез трет-бутил 5-фтор-8-гидрокси-6-изопропокси-3,4-дигидроизохинолин-2(1H)-карбоксилата (TP-38)
К раствору FFFF-1 (160 мг, 0,385 ммоль) в MeOH (5 мл) добавляют Pd/C (41 мг, 0,039 ммоль). Смесь дегазируют и продувают H2 три раза с применением H2 баллона, затем перемешивают при комнатной температуре (28°C) в течение 16 часов под H2 баллоном. Реакционную смесь разбавляют ДХМ (20,0 мл), фильтруют и концентрируют с получением TP-38 в виде желтого твердого вещества (118 мг, 94%). 1H ЯМР (400 MГц, ДМСО-d6) δ ч./млн. 9,50 (шд, J=2,5 Гц, 1H), 6,45 (д, J=7,3 Гц, 1H), 4,41 (тд, J=6,0, 12,0 Гц, 1H), 4,28 (с, 2H), 3,52 (т, J=5,6 Гц, 2H), 2,64 (шт, J=5,8 Гц, 2H), 1,42 (с, 9H), 1,26 (д, J=6,0 Гц, 6H)
Пример 201 получают с применением методики со схемы CC и с применением соединения TP-38.
TP-38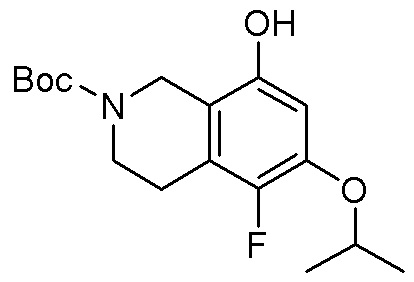
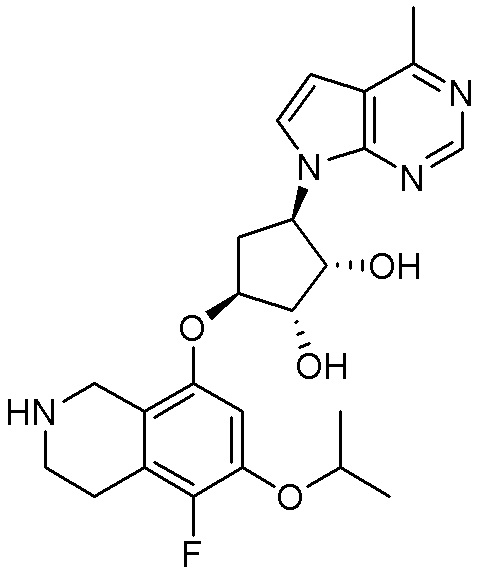
[M+1]
1H ЯМР (400 MГц, D2O, соль HCl) δ ч./млн. 8,90 (с, 1H), 7,88 (д, J=3,8 Гц, 1H), 7,16 (д, J=3,8 Гц, 1H), 6,80 (д, J=6,8 Гц, 1H), 5,37 (кв, J=9,0 Гц, 1H), 4,89-4,82 (м, 1H), 4,73-4,63 (м, 2H), 4,35-4,30 (м, 3H), 3,52 (т, J=6,3 Гц, 2H), 3,14-3,01 (м, 3H), 2,96 (с, 3H), 2,25 (ддд, J=4,0, 9,4, 13,9 Гц, 1H), 1,37-1,30 (м, 6H)
Схема GGGG - Синтез трет-бутил 8-циано-6-гидрокси-2,3-дигидробензo[f][1,4]оксазепин-4(5H)-карбоксилата (TP-39)
Схема GGGG
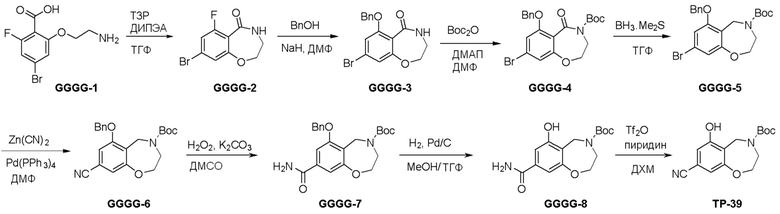
Стадия 1 - Синтез 8-бром-6-фтор-3,4-дигидробензo[f][1,4]оксазепин-5(2H)-она (GGGG-2)
К суспензии GGGG-1 (13,8 г, 43,87 ммоль, полученной с применением ссылочного патента US2015/64196) в сухом ТГФ (400 мл) добавляют ДИПЭА (28,3 г, 219 ммоль) и T3P (50% в EtOAc, 41,9 г, 65,8 ммоль) при 0°C. Смесь перемешивают при 0°C в течение 10 ч, затем при кт 25°C в течение 6 ч. Смесь выливают в воду (400 мл) и экстрагируют EtOAc (400 мл×2). Экстракт промывают насыщенным раствором соли (300 мл), сушат над Na2SO4 и концентрируют с получением GGGG-2 (9500 мг, 83%) в виде светло-желтого твердого вещества. ЖХМС [M+1] 261,6; 1H ЯМР (400 MГц, ДМСО-d6) δ ч./млн. 8,57 (шс, 1H), 7,44 (дд, J=1,8, 9,5 Гц, 1H), 7,22 (с, 1H), 4,22 (т, J=5,5 Гц, 2H), 3,27 (кв, J=5,6 Гц, 2H)
Стадия 2 - Синтез 6-(бензилокси)-8-бром-3,4-дигидробензo[f][1,4]оксазепин-5(2H)-она (GGGG-3)
К суспензии 60% NaH (6 г, 150 ммоль) в сухом ДМФ (140 мл) добавляют раствор GGGG-2 (6,5 г, 25 ммоль) и BnOH (5,41 г, 50 ммоль) в сухом ДМФ (70 мл) при кт 25°C под N2, затем перемешивают при кт 25°C под N2 в течение 16 с. Смесь выливают в ледяную воду (400 мл), в которой образуется твердое вещество. Смесь фильтруют. Твердое вещество промывают водой и сушат в вакууме с получением GGGG-3 (9500 мг, >99%) в виде белого твердого вещества. ЖХМС [M+1] 347,7; 1H ЯМР (400 MГц, ДМСО-d6) δ ч./млн. 8,27 (шт, J=5,7 Гц, 1H), 7,47 (д, J=6,6 Гц, 2H), 7,42-7,36 (м, 2H), 7,35-7,29 (м, 1H), 7,26-7,15 (м, 1H), 6,93 (д, J=1,4 Гц, 1H), 5,19 (с, 2H), 4,12 (т, J=5,5 Гц, 2H), 3,18 (кв, J=5,6 Гц, 2H).
Стадия 3 - Синтез трет-бутил 6-(бензилокси)-8-бром-5-оксо-2,3-дигидробензo[f][1,4]оксазепин-4(5H)-карбоксилата (GGGG-4)
К раствору GGGG-3 (7,3 г, 21 ммоль) в ДМФ (150 мл) добавляют Boc2O (6,86 г, 31,43 ммоль), ДИПЭА (8,13 г, 62,9 ммоль) и ДМАП (256 мг, 2,1 ммоль) при кт 25°C в течение 2 ч. Смесь выливают в воду. Твердое вещество собирают фильтрацией и промывают водой. Твердое вещество растворяют в EtOAc/ТГФ (50 мл/50 мл) и сушат над Na2SO4 и концентрируют в вакууме с получением неочищенного продукта GGGG-4 (10 г, >99%) в виде желтой камеди и применяют сразу на следующей стадии без дальнейшей очистки.
Стадия 4 - Синтез трет-бутил 6-(бензилокси)-8-бром-2,3-дигидробензo[f][1,4]оксазепин-4(5H)-карбоксилата (GGGG-5)
К раствору GGGG-4 (10 г, 21 ммоль) в ТГФ (150 мл) добавляют BH3.Me2S (8,39 мл, 83,9 ммоль) при кт 25°C. Смесь перемешивают при 60°C под N2 в течение 16 ч, затем выдерживают при кт в течение 2 дней. Реакцию медленно гасят MeOH (70 мл). Затем смесь кипятят с обратным холодильникомe в течение 16 ч и концентрируют. Неочищенный продукт очищают хроматографией на колонке (120 г колонка с силикагелем, EtOAc в петролейном эфире 0%-50%) с получением GGGG-5 (3130 мг, 34%) в виде белого твердого вещества. ЖХМС [M+23] 456; 1H ЯМР (400 MГц, CDCl3) δ ч./млн. 7,52-7,31 (м, 5H), 6,84 (шс, 1H), 6,82 (шс, 1H), 5,06 (шс, 2H), 4,64 (шс, 2H), 4,15-4,10 (м, 2H), 3,80 (шс, 2H), 1,29 (шс, 9H)
Стадия 5 - Синтез трет-бутил 6-(бензилокси)-8-циано-2,3-дигидробензo[f][1,4]оксазепин-4(5H)-карбоксилата (GGGG-6)
Соединение GGGG-5 (680 мг, 1,57 ммоль), Zn(CN)2 (368 мг, 3,13 ммоль) и Pd(PPh3)4 (181 мг, 0,157 ммоль) в ДМФ (10 мл) продувают N2 в течение 10 мин. Смесь перемешивают при 120°C в течение 3 ч. Смесь охлаждают, фильтруют и концентрируют. Остаток очищают хроматографией на силикагеле, элюируя EtOAc в петролейном эфире 0-50% с получением GGGG-6 (520 мг, 87%) в виде белого твердого вещества. ЖХМС [M-Boc+1] 280,9.
Стадия 6 - Синтез трет-бутил 6-(бензилокси)-8-карбамоил-2,3-дигидробензo[f][1,4]оксазепин-4(5H)-карбоксилат (GGGG-7)
К раствору GGGG-6 (520 мг, 1,37 ммоль) и K2CO3 (189 мг, 1,37 ммоль) в ДМСО (5 мл) медленно добавляют 30% H2O2 (465 мг, 4,1 ммоль) при кт 25°C. Примечание: испарение газа и экзотермическая реакция. Смесь перемешивают при кт 25°C в течение 1 ч. Смесь выливают в воду (10 мл) и фильтруют. Твердое вещество промывают водой. Твердое вещество суспендируют в MeOH (30 мл) и концентрируют в вакууме с получением GGGG-7 (520 мг, 96%) в виде белого твердого вещества. ЖХМС [M+Na] 421.
Стадия 7 - Синтез трет-бутил 8-карбамоил-6-гидрокси-2,3-дигидробензo[f][1,4]оксазепин-4(5H)-карбоксилата (GGGG-8)
Смесь GGGG-7 (520 мг, 1,31 ммоль) и Pd/C (125 мг) в MeOH/ТГФ (15 мл/15 мл) дегазируют с H2 четыре раза. Смесь перемешивают при 25°C под H2 баллоном в течение 16 ч. Смесь фильтруют и концентрируют в вакууме с получением GGGG-8 (400 мг, 99%) в виде белого твердого вещества. 1H ЯМР (400 MГц, ДМСО-d6, ротамеры) δ ч./млн. 9,85 (шс, 1H), 7,80 (шс, 1H), 7,22 (шс, 1H), 7,11-6,97 (м, 1H), 6,92 (шс, 1H), 4,57 (шс, 2H), 4,20-4,03 (м, 2H), 3,68 (шс, 2H), 1,42-1,13 (м, 9H)
Стадия 8 - Синтез трет-бутил 8-циано-6-гидрокси-2,3-дигидробензo[f][1,4]оксазепин-4(5H)-карбоксилата (TP-39)
К суспензии GGGG-8 (100 мг, 0,324 моль) и пиридина (128 мг, 1,62 моль) в безводном ДХМ (2 мл) добавляют Tf2O (275 мг, 0,973 моль) при 0°C и перемешивают в течение 0,5 ч. Реакционную смесь нагревают до 25°C и перемешивают в течение 16 ч. Смесь концентрируют в вакууме досуха. Остаток растворяют в MeOH (3 мл), затем K2CO3 (300 мг). Смесь перемешивают при кт 25°C в течение 16 ч. Реакционную смесь фильтруют и очищают преп.-ТСХ (петролейный эфир/EtOAc 1:1) с получением TP-39 (55 мг, 58%) в виде белого твердого вещества. ЖХМС [M-Boc+1] 190,8; 1H ЯМР (400 MГц, ДМСО-d6, ротамеры) δ ч./млн. 10,47 (шс, 1H), 6,83 (шс, 2H), 4,70-4,51 (м, 2H), 4,22-4,18 (м, 2H), 3,69 (шс, 2H), 1,46-1,21 (м, 9H)
Схема HHHH - Синтез трет-бутил 8-фтор-6-гидрокси-2,3-дигидробензo[f][1,4]оксазепин-4(5H)-карбоксилата (TP -40)
Схема HHHH
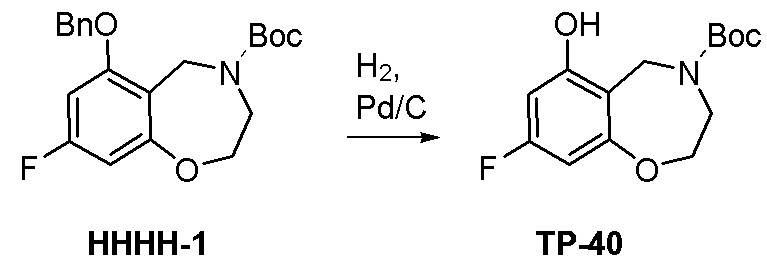
К раствору HHHH-1 (полученного по методике стадий 1-4 со схемы GGGG с применением 2-(2-аминоэтокси)-4,6-дифторбензoйной кислоты, 1980 мг, 5,302 ммоль) в MeOH (74 мл) добавляют 10% влажный Pd/C (590 мг) при 25°C. Смесь перемешивают при 25°C под баллоном H2 в течение 1,2 часов. Реакционную смесь фильтруют и концентрируют с получением TP-40 (1420 мг, 95%) в виде белого твердого вещества. ЖХМС [M-Boc+1] 184,8 1H ЯМР (400 MГц, ДМСО-d6, ротамеры) δ ч./млн. 10,60-9,69 (шс, 1H), 6,34-6,30 (м, 1H), 6,26-6,12 (м, 1H), 4,63-4,39 (м, 2H), 4,23-4,01 (м, 2H), 3,75-3,58 (м, 2H), 1,46-1,20 (м, 9H); 1H ЯМР (400 MГц, ДМСО-d6, переменная темп. 80°C) δ ч./млн. 6,33 (шд, J=10,3 Гц, 1H), 6,19 (дд, J=2,3, 10,3 Гц, 1H), 4,53 (с, 2H), 4,19-4,06 (м, 2H), 3,69 (шт, J=4,4 Гц, 2H), 1,36 (с, 9H)
Схема IIII - Синтез трет-бутил 8-хлор-6-гидрокси-2,3-дигидробензo[f][1,4]оксазепин-4(5H)-карбоксилата (TP-41)
Схема IIII
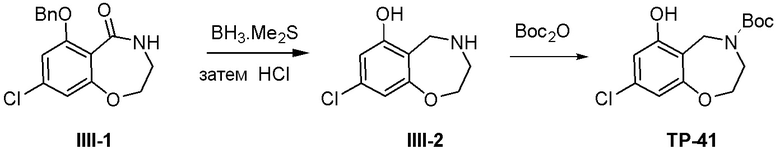
Стадия 1 - Синтез 8-хлор-2,3,4,5-тетрагидробензo[f][1,4]оксазепин-6-ола (IIII-2)
К раствору IIII-1 (полученного по методике стадий 1-3 со схемы GGGG с применением 2-(2-аминоэтокси)-4-хлор-6-фторбензoйной кислоты, 1000 мг, 3,292 ммоль) в сухом ТГФ (11 мл) добавляют BH3.Me2S (2,63 мл, 26,3 ммоль) при кт 25°C под N2. После добавления, смесь нагревают при 70°C в течение 16 часов. Смесь охлаждают до кт (25°C) и гасят MeOH (3 мл). Смесь концентрируют в виде белого твердого вещества. Неочищенный продукт растворяют в конц. HCl (13,7 мл) и нагревают при 110°C в течение 16 часов. Смесь концентрируют с получением IIII-2 (860 мг, >99%) в виде светло-желтого твердого вещества, которое применяют сразу на следующей стадии. ЖХМС [M+1] 199,7
Стадия 2 - Синтез трет-бутил 8-хлор-6-гидрокси-2,3-дигидробензo[f][1,4]оксазепин-4(5H)-карбоксилата (TP-41)
К раствору IIII-2 (1171 мг, 3,3 ммоль) в MeOH (21 мл) добавляют ТЭА (3,58 мл, 25,6 ммоль) и Boc2O (1,32 г, 6,02 ммоль) затем перемешивают при кт в течение 2 часов. Смесь концентрируют в вакууме с получением неочищенного продукта (1032 мг) в виде белого твердого вещества. Неочищенный продукт очищают комби-флэш (EtOAc/петролейный эфир=0~50%) с получением TP-41 (520 мг, 30%) в виде белого твердого вещества. ЖХМС [M-tBu+1] 243,6; 1H ЯМР (400 MГц, ДМСО-d6, ротамеры) δ ч./млн. 10,13 (с, 1H), 6,61-6,57 (м, 1H), 6,45 (шс, 1H), 4,61-4,46 (м, 2H), 4,25-4,05 (м, 2H), 3,66 (шс, 2H), 1,42-1,27 (м, 9H)
Примеры 202-204 получают по методике со схемы CC и с применением соединений TP-39, TP-40 & TP-41, соответственно
TP-39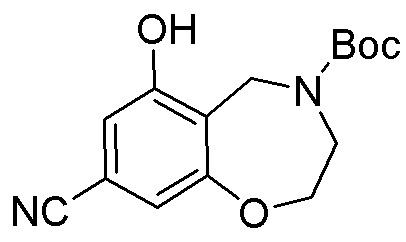
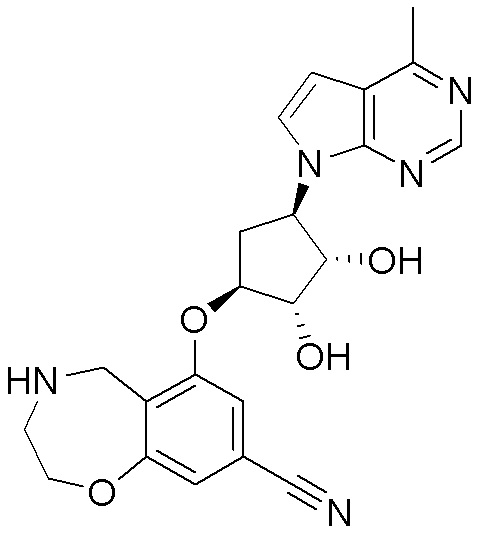
[M+1]
1H ЯМР (400 MГц, D2O, соль HCl) δ ч./млн. 8,86 (с, 1H), 7,85 (д, J=3,8 Гц, 1H), 7,25 (д, J=1,0 Гц, 1H), 7,20 (д, J=1,3 Гц, 1H), 7,11 (д, J=3,8 Гц, 1H), 5,35 (кв, J=9,2 Гц, 1H), 4,86-4,80 (м, 2H), 4,60 (кв, J=14,8 Гц, 2H), 4,43-4,26 (м, 3H), 3,66 (т, J=4,9 Гц, 2H), 3,15-3,04 (м, 1H), 2,91 (с, 3H), 2,28-2,18 (м, 1H)
TP-40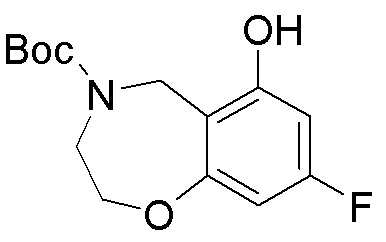
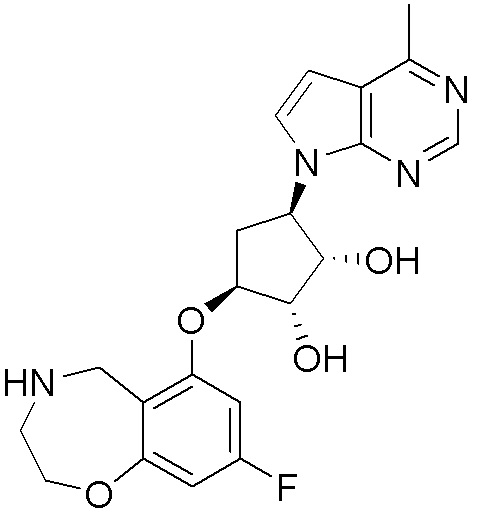
[M+1]
1H ЯМР (400 MГц, MeOD, соль HCl) δ ч./млн.=9,07 (с, 1H), 8,12 (д, J=4,0 Гц, 1H), 7,25 (д, J=3,8 Гц, 1H), 6,86 (дд, J=2,5, 10,8 Гц, 1H), 6,59 (дд, J=2,4, 9,4 Гц, 1H), 5,40 (кв, J=9,3 Гц, 1H), 4,77 (дд, J=5,0, 9,3 Гц, 2H), 4,62-4,50 (м, 2H), 4,34 (тдт, J=4,5, 9,3, 13,6 Гц, 2H), 4,25 (д, J=5,0 Гц, 1H), 3,65 (т, J=4,6 Гц, 2H), 3,13-3,04 (м, 1H), 3,02 (с, 3H), 2,36 (ддд, J=4,4, 9,7, 14,3 Гц, 1H)
TP-41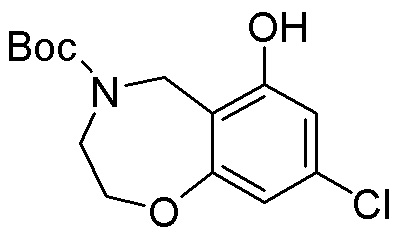
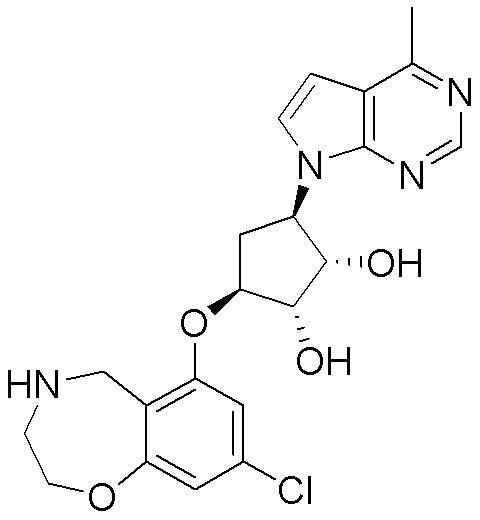
[M+1]
1H ЯМР (400 MГц, MeOD) δ ч./млн. 8,65 (с, 1H), 7,59 (д, J=3,7 Гц, 1H), 6,90 (д, J=1,9 Гц, 1H), 6,78 (д, J=3,6 Гц, 1H), 6,71 (д, J=1,9 Гц, 1H), 5,24 (кв, J=9,0 Гц, 1H), 4,73-4,66 (м, 2H), 4,21 (д, J=5,1 Гц, 1H), 4,16-4,04 (м, 4H), 3,22-3,17 (м, 2H), 3,00 (ддд, J=7,5, 9,2, 14,4 Гц, 1H), 2,74 (с, 3H), 2,29-2,15 (м, 1H)
Схема JJJJ - Синтез трет-бутил 8-(дифторметил)-6-гидрокси-2,3-дигидробензo[f][1,4]оксазепин-4(5H)-карбоксилата (TP-42)
Схема JJJJ
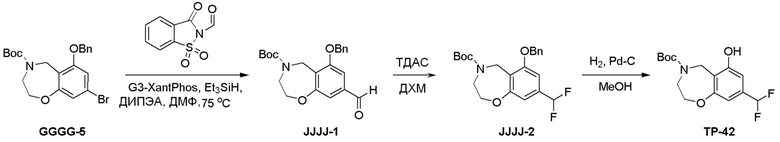
Стадия 1 - Синтез трет-бутил 6-(бензилокси)-8-формил-2,3-дигидробензo[f][1,4]оксазепин-4(5H)-карбоксилата (JJJJ-1)
Ref. Manabe, K. et al. Org. Lett., 2013, 5370-5373. В реакционную пробирку добавляют раствор GGGG-5 (1,64 г, 3,62 ммоль) в безводном ДМФ (18 мл) и N-формилсахарин (1200 мг, 5,66 ммоль), G3 Xantphos (107 мг, 0,113 ммоль) и ДИПЭА (634 мг, 4,91 ммоль). Смесь продувают N2 в течение 4 мин. К раствору добавляют Et3SiH (571 мг, 4,91 ммоль) при 0°C. После добавления, полученную смесь продувают N2 в течение 5 мин и затем нагревают при 75°C в течение 16 часов. Реакционную смесь разделяют между EtOAc и водой. Органический слой разделяют, и водный слой экстрагируют EtOAc (30 мл×3). Объединенные органические слои промывают насыщенным раствором соли, сушат над Na2SO4 и концентрируют с получением неочищенного продукта, который очищают на флэш-колонке (20 г, гель, EtOAc:петролейный эфир 1%-14%) с получением JJJJ-1 (730 мг, 53%) в виде белого твердого вещества. 1H ЯМР (400 MГц, CDCl3, ротамеры) δ ч./млн. 9,87 (шс, 1H), 7,59-7,30 (м, 5H), 7,21 (шс, 1H), 7,14 (шс, 1H), 5,16 (шс, 2H), 4,75 (шс, 2H), 4,24-4,17, (м, 2H), 3,85 (шс, 2H), 1,54-1,13 (м, 9H)
Стадия 2 - Синтез трет-бутил 6-(бензилокси)-8-(дифторметил)-2,3-дигидробензo[f][1,4]оксазепин-4(5H)-карбоксилата (JJJJ-2)
К раствору JJJJ-1 (930 мг, 2,43 ммоль) в безводном ДХМ (49 мл) добавляют ТДАС (3,91 г, 24,3 ммоль) при 0°C под N2. После добавления, реакционную смесь нагревают до комнатной температуры в течение 16 часов. Реакционный раствор разбавляют в ДХМ (50 мл) и насыщ. водн. NaHCO3 и перемешивают до превращения выделения CO2. Реакционную смесь разделяют между ДХМ и H2O. Органический слой разделяют, промывают насыщенным раствором соли, сушат над Na2SO4 и концентрируют с получением неочищенного продукта, который очищают на флэш-колонке (EtOAc:петролейный эфир 1%-15%) с получением JJJJ-2 (530 мг, 54%) в виде белого твердого вещества. 1H ЯМР (400 MГц, CDCl3, ротамеры) δ ч./млн. 7,58-7,29 (м, 5H), 6,82 (шс, 1H), 6,79 (шс, 1H), 6,54 (т, J=54 Гц, 1H), 5,12 (шс, 2H), 4,72 (шс, 2H), 4,21-4,07 (м, 2H), 3,83 (шс, 2H), 1,54-1,13 (м, 9H)
Стадия 3 - Синтез трет-бутил 8-(дифторметил)-6-гидрокси-2,3-дигидробензo[f][1,4]оксазепин-4(5H)-карбоксилата (TP-42)
К раствору JJJJ-2 (530 мг, 1,31 ммоль) в MeOH (13 мл) добавляют Pd/C (139 мг, 0,0163 ммоль). Смесь дегазируют и продувают H2 три раза, затем перемешивают при кт (35°C) в течение 16 часов. Реакционную смесь фильтруют через целит и концентрируют. Остаток лиофилизируют для удаления остаточного растворителя с получением TP-42 (360 мг, 87%) в виде белого твердого вещества. ЖХМС [M-tBu+1] 259,7; 1H ЯМР (400 MГц, ДМСО-d6, ротамеры) δ ч./млн. 10,21-9,89 (м, 1H), 7,02-6,47 (м, 3H), 4,65-4,49 (м, 2H), 4,22-4,04 (м, 2H), 3,74-3,62 (м, 2H), 1,46-1,11 (м, 9H)
Пример 205 получают с применением методики со схемы CC и с применением соединения TP-42.
TP-42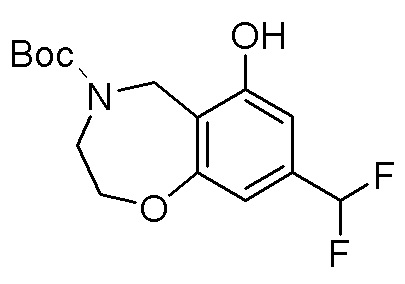
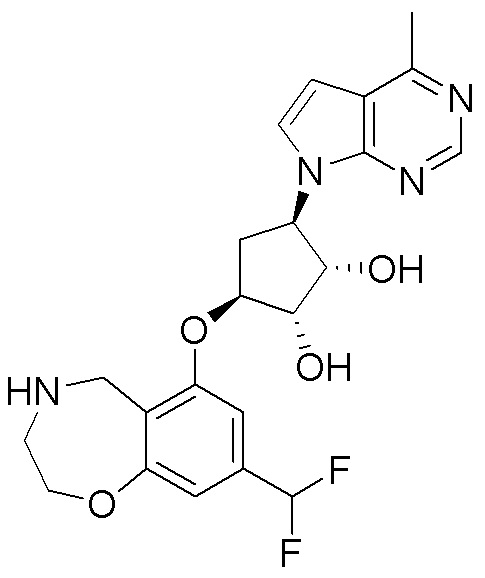
[M+1]
1H ЯМР (400 MГц, D2O, соль HCl) δ ч./млн. 8,90 (с, 1H), 7,89 (д, J=3,9 Гц, 1H), 7,15 (д, J=3,7 Гц, 1H), 7,08 (с, 1H), 7,02 (с, 1H), 6,77 (т, J=55,8 Гц, 1H), 5,37 (кв, J=9,0 Гц, 1H), 4,91-4,84 (м, 1H), 4,73-4,68 (м, 1H), 4,60 (дд, J=13,4, 30,7 Гц, 2H), 4,45-4,24 (м, 3H), 3,67 (т, J=4,8 Гц, 2H), 3,15-3,04 (м, 1H), 2,90 (с, 3H), 2,33-2,20 (м, 1H)
Схема KKKK - Синтез трет-бутил 6-гидрокси-8-метил-2,3-дигидробензo[f][1,4]оксазепин-4(5H)-карбоксилата (TP-43)
Схема KKKK
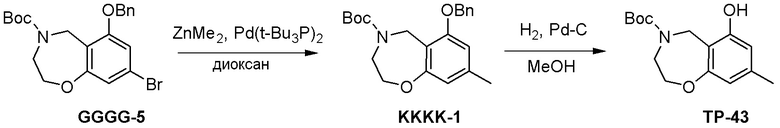
Стадия 1 - Синтез трет-бутил 6-(бензилокси)-8-метил-2,3-дигидробензo[f][1,4]оксазепин-4(5H)-карбоксилата (KKKK-1)
Раствор GGGG-5 (200 мг, 0,46 ммоль) в диоксане (9,21 мл) дегазируют с N2 в течение 5 мин. Затем бис(три-трет-бутилфосфин)Pd(0) (23,5 мг, 0,05 ммоль) и (Me)2Zn (0,921 мл, 0,92 ммоль) добавляют в реакционный раствор и нагревают при 90°C в течение 16 часов. Реакционный раствор выливают в EtOAc и 1N HCl (20 мл:20 мл). Смесь разделяют, и водный слой промывают EtOAc (20 мл×2). Органические слои разделяют, сушат и выпаривают с получением неочищенного продукта, который очищают флэш-хроматографией, элюируя петролейным эфиром/EtOAc 0-25% с получением KKKK-1 (160 мг, 94%) в виде желтого твердого вещества. ЖХМС [M+Na] 392,1; 1H ЯМР (400 MГц, ДМСО-d6, ротамеры) δ ч./млн. 7,57-7,30 (м, 5H), 6,62 (шс, 1H), 6,39 (с, 1H), 5,09 (шс, 2H), 4,65-4,60 (м, 2H), 4,17-4,04 (м, 2H), 3,68 (шс, 2H), 2,22 (с, 3H), 1,42-1,15 (м, 9H)
Стадия 2 - Синтез трет-бутил 6-гидрокси-8-метил-2,3-дигидробензo[f][1,4]оксазепин-4(5H)-карбоксилата (TP-43)
Соединение KKKK-1 (160 мг, 0,433 ммоль) растворяют в MeOH (8,00 мл). Затем Pd/C добавляют в реакционный раствор, затем дегазируют с H2 четыре раза и перемешивают при 30°C под H2 (баллон) в течение 16 часов. ДХМ (20 мл) добавляют для разбавления реакционного раствора, затем фильтруют и концентрируют с получением неочищенного продукта, который очищают флэш-хроматографией, элюируя петролейным эфиром/EtOAc 0-25%, с получением TP-43 (100 мг, 83%) в виде белого твердого вещества. ЖХМС [M-Boc+1] 179,9; 1H ЯМР (400 MГц, ДМСО-d6, ротамеры) δ ч./млн. 9,41 (с, 1H), 6,37-6,35 (м, 1H), 6,22-6,20 (м, 1H), 4,58-4,42 (м, 2H), 4,13-3,95 (м, 2H), 3,70-3,59 (м, 2H), 2,13 (с, 3H), 1,41-1,27 (м, 9H)
Пример 206 получают с применением методики со схемы CC и с применением соединения TP-43
TP-43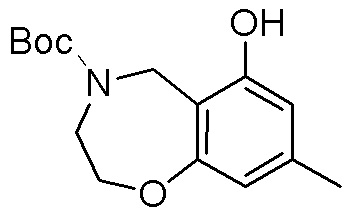
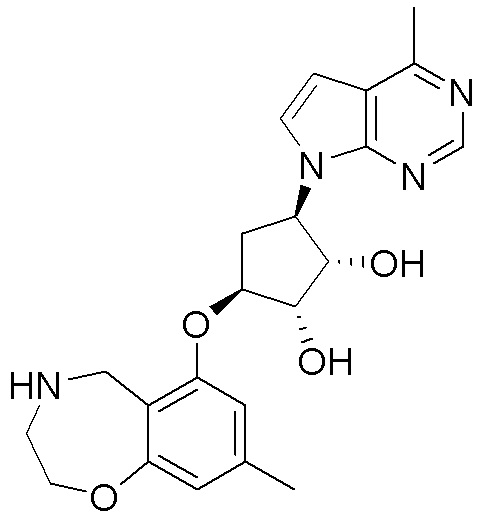
[M+1]
1H ЯМР (400 MГц, D2O, соль HCl) δ ч./млн. 8,89 (с, 1H), 7,87 (д, J=3,8 Гц, 1H), 7,14 (д, J=3,8 Гц, 1H), 6,78 (с, 1H), 6,71 (с, 1H), 5,38 (кв, J=9,0 Гц, 1H), 4,77-4,70 (м, 2H), 4,62-4,47 (м, 2H), 4,40-4,22 (м, 3H), 3,63 (т, J=4,8 Гц, 2H), 3,09 (ддд, J=7,3, 9,3, 14,7 Гц, 1H), 2,95 (с, 3H), 2,32 (с, 3H), 2,28-2,16 (м, 1H)
Схема LLLL - Синтез трет-бутил (S)-6-гидрокси-3-метил-2,3-дигидробензo[f][1,4]оксазепин-4(5H)-карбоксилата (TP-44)
Схема LLLL
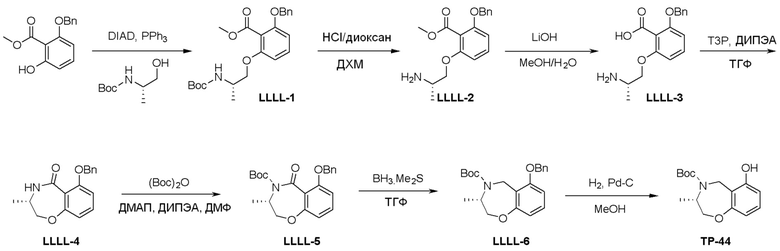
Стадия 1 - Синтез метил (S)-2-(бензилокси)-6-(2-((трет-бутоксикарбонил)амино)пропокси)бензоата (LLLL-1)
К раствору метил 2-(бензилокси)-6-гидроксибензоата (2,80 мг, 10,8 ммоль) в ТГФ (40,0 мл) добавляют PPh3 (7,11 г, 27,1 ммоль) на ледяной бане под N2. DIAD (5,48 г, 27,1 ммоль) добавляют в смесь по каплям, затем перемешивают при 25°C в течение 40 мин. Затем добавляют трет-бутил (S)-(1-гидроксипропан-2-ил)карбамат (5,70 г, 32,5 ммоль) в сухом ТГФ (20,0 мл), и реакционную смесь перемешивают при 28°C в течение 16 часов. Смесь разбавляют EtOAc (50 мл) и H2O (100 мл). Смесь разделяют, и водный слой экстрагируют EtOAc (50 мл). Объединенные органические слои промывают насыщенным раствором соли (50 мл), сушат над Na2SO4, фильтруют и концентрируют с получением неочищенного продукта, который очищают ISCO (120 г силикагель, петролейный эфир:EtOAc=4:1) с получением LLLL-1 (3,50 г, 78%) в виде бесцветной камеди. 1H ЯМР (400 MГц, ДМСО-d6) δ ч./млн. 7,42-7,35 (м, 4H), 7,34-7,29 (м, 2H), 6,83-6,75 (м, 2H), 6,71 (д, J=8,5 Гц, 1H), 5,16 (с, 2H), 3,95-3,92 (м, 1H), 3,80-3,70 (м, 5H), 1,43-1,34 (м, 9H), 1,08 (д, J=6,5 Гц, 3H)
Стадия 2 - Синтез метил (S)-2-(2-аминопропокси)-6-(бензилокси)бензоата (LLLL-2)
К раствору LLLL-1 (4,30 г, 10,3 ммоль) в ДХМ (10 мл) добавляют HCl(г)/диоксан (~4N, 20 мл) при 0°C. Смесь перемешивают при 25°C в течение 2 часов. Смесь концентрируют с получением LLLL-2 в виде соли HCl (4,00 г, >99%) в виде желтого твердого вещества, которое применяют сразу на следующей стадии.
Стадия 3 - Синтез (S)-2-(2-аминопропокси)-6-(бензилокси)бензoйной кислоты (LLLL-3)
К суспензии LLLL-2 (4,0 г, 3,17 ммоль) в MeOH (60 мл) и воде (12 мл) добавляют LiOH.H2O (3,46 г, 82,4 ммоль) при 25°C. Смесь перемешивают при 75°C в течение 15 часов. pH раствора доводят до pH~3 добавлением 1N. Раствор выпаривают с получением неочищенного продукта LLLL-3 (3,10 г, >99%) в виде белого твердого вещества, которое применяют сразу для следующей стадии без дальнейшей очистки.
Стадия 4 - Синтез (S)-6-(бензилокси)-3-метил-3,4-дигидробензo[f][1,4]оксазепин-5(2H)-она (LLLL-4)
К суспензии LLLL-3 (4,00 г, 5,77 ммоль) в сухом ТГФ (60 мл) добавляют ДИПЭА (3,73 г, 28,9 ммоль) и T3P (50% в EtOAc, 7,35 г, 11,5 ммоль) при 0°C. Смесь перемешивают при 25°C в течение 15 часов. Реакция не завершена, поэтому добавляют ДИПЭА (3,73 г, 28,9 ммоль) и T3P (50% в EtOAc, 7,35 г, 11,5 ммоль) и перемешивают в течение еще 16 часов. EtOAc (40,0 мл) добавляют для разбавления раствора. Раствор промывают 1N HCl (30 мл), насыщенным водн. NaHCO3 (30 мл) и насыщенным раствором соли (30 мл). Органические слои разделяют, сушат и выпаривают с получением неочищенного продукта, который очищают флэш-хроматографией, элюируя петролейным эфиром/EtOAc 0-50% с получением желаемого продукта LLLL-4 (900 мг, 55%) в виде белого твердого вещества. ЖХМС [M+1] 283,9; 1H ЯМР (400 MГц, ДМСО-d6) δ ч./млн. 8,09 (д, J=6,3 Гц, 1H), 7,51-7,44 (м, 2H), 7,41-7,25 (м, 4H), 6,99-6,91 (м, 1H), 6,68 (дд, J=0,9, 8,2 Гц, 1H), 5,15 (с, 2H), 3,97 (дд, J=4,3, 10,3 Гц, 1H), 3,86-3,77 (м, 1H), 3,46-3,44 (м, 1H), 1,05 (д, J=6,5 Гц, 3H)
Стадия 5 - Синтез трет-бутил (S)-6-(бензилокси)-3-метил-5-оксо-2,3-дигидробензo[f][1,4]оксазепин-4(5H)-карбоксилата (LLLL-5)
К раствору LLLL-4 (900 мг, 3,18 ммоль) в ДМФ (20,0 мл) добавляют ДИПЭА (1,23 г, 9,53 ммоль) и ДМАП (38,8 мг, 0,318 ммоль) при 25°C. Добавляют Boc2O (1,04 мг, 4,76 ммоль), и реакционную смесь перемешивают при 25°C в течение 16 часов. Смесь разбавляют ледяной водой (20 мл) и экстрагируют EtOAc (20 мл×2). Объединенные органические слои промывают насыщенным раствором соли (30 мл×5), сушат над Na2SO4 и фильтруют, концентрируют в вакууме с получением неочищенного продукта, который очищают ISCO (4 г силикагель, EtOAc:петролейный эфир=18%-20%) с получением LLLL-5 (720 мг, 59%) в виде белого твердого вещества, и применяют сразу на следующей стадии. ЖХМС [M-Boc+1] 238,9
Стадия 6 - Синтез трет-бутил (S)-6-(бензилокси)-3-метил-2,3-дигидробензo[f][1,4]оксазепин-4(5H)-карбоксилата (LLLL-6)
К раствору LLLL-5 (720 мг, 1,88 ммоль) в ТГФ (20 мл) добавляют BH3.Me2S (10 M, 0,751 мл, 7,51 ммоль) при 0°C. Смесь перемешивают при 74°C под N2 в течение 2 часов. Реакцию медленно гасят MeOH (5 мл) и нагревают при кипении с обратным холодильником в течение 16 часов. Смесь концентрируют с получением неочищенного продукта, который очищают комби-флэш (40 г колонка с двуокисью кремния, EtOAc в петролейном эфире 9-10%) с получением LLLL-6 (530 мг, 76,4%) в виде бесцветной камеди. ЖХМС [M-Boc+1] 270,0; 1H ЯМР (400 MГц, ДМСО-d6, ротамеры) δ ч./млн. 7,54-7,32 (м, 5H), 7,06 (т, J=8,2 Гц, 1H), 6,70 (д, J=8,3 Гц, 1H), 6,46 (дд, J=0,8, 8,3 Гц, 1H), 5,35-4,97 (м, 3H), 4,54-4,30 (м, 1H), 4,24-4,02 (м, 3H), 1,39-1,22 (м, 9H), 1,06 (м, 3H)
Стадия 7 - Синтез трет-бутил (S)-6-гидрокси-3-метил-2,3-дигидробензo[f][1,4]оксазепин-4(5H)-карбоксилата (TP-44)
К раствору LLLL-6 (530 мг, 1,43 ммоль) в MeOH (10,0 мл) добавляют влажный 10% Pd/C (100 мг). Реакционный раствор перемешивают при 30°C под H2 баллоном в течение 16 ч. Затем смесь фильтруют через целит, промывают MeOH, и концентрируют с получением TP-44 (360 мг, 90%) в виде белого твердого вещества. 1H ЯМР (400 MГц, ДМСО-d6, ротамеры) δ ч./млн. 9,61-9,49 (м, 1H), 6,87 (т, J=8,2 Гц, 1H), 6,44 (д, J=7,8 Гц, 1H), 6,26 (дд, J=1,0, 8,0 Гц, 1H), 5,23-4,94 (м, 1H), 4,54-4,28 (м, 1H), 4,15-4,00 (м, 3H), 1,39-1,25 (м, 9H), 1,06 (д, J=6,8 Гц, 3H)
Соединение TP-45 получают с применением методики со Схемы LLLL с применением трет-бутил (R)-(1-гидроксипропан-2-ил)карбамата на стадии 1
[M-Boc+1]
1H ЯМР (400 MГц, CDCl3, ротамеры) δ ч./млн. 8,15-7,57 (м, 1H), 6,81 (шт, J=7,9 Гц, 1H), 6,43 (шд, J=8,0 Гц, 1H), 6,39-6,20 (м, 1H), 4,96 (д, J=16,1 Гц, 1H), 4,71-4,31 (м, 1H), 4,17-4,03 (м, 2H), 3,87 (шдд, J=3,8, 12,5 Гц, 1H), 1,53-1,35 (м, 9H), 1,32 (шд, J=6,8 Гц, 3H)
Примеры 207 & 208 получают по методике со схемы CC и с применением соединений TP-44 & TP-45, соответственно
TP-44
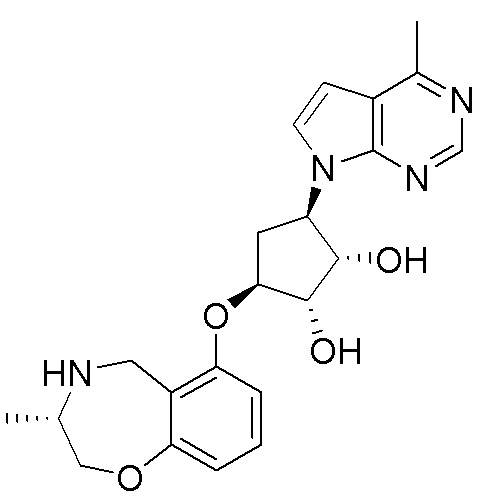
[M+1]
1H ЯМР (400 MГц, ДМСО-d6) δ ч./млн. 8,65 (с, 1H), 7,67 (д, J=3,6 Гц, 1H), 7,08 (т, J=8,1 Гц, 1H), 6,83-6,69 (м, 2H), 6,59 (д, J=8,0 Гц, 1H), 5,32 (д, J=4,0 Гц, 1H), 5,19-5,07 (м, 2H), 4,59-4,50 (м, 2H), 4,35 (д, J=14,9 Гц, 1H), 4,19 (дд, J=2,4, 11,8 Гц, 1H), 4,01 (шс, 1H), 3,59 (шд, J=14,7 Гц, 1H), 3,39 (шс, 1H), 3,15 (шд, J=6,5 Гц, 1H), 2,89-2,77 (м, 1H), 2,65 (с, 3H), 2,45-2,50 (м, 1H), 2,03-1,91 (м, 1H), 0,97 (д, J=6,5 Гц, 3H)
TP-45
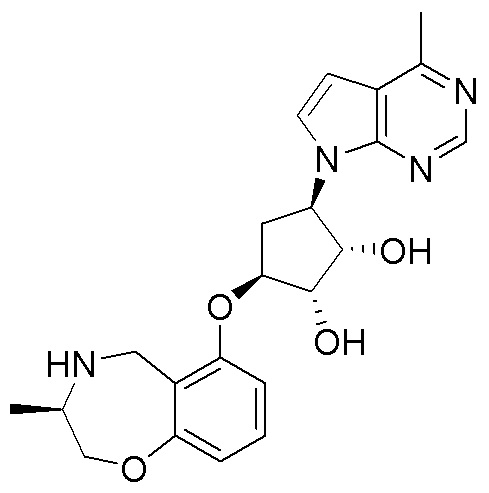
[M+1]
1H ЯМР (400 MГц, D2O, соль HCl) δ ч./млн. 8,81 (с, 1H), 7,77 (д, J=3,8 Гц, 1H), 7,32 (т, J=8,3 Гц, 1H), 7,07 (д, J=3,8 Гц, 1H), 6,86 (д, J=8,0 Гц, 1H), 6,77 (д, J=7,8 Гц, 1H), 5,31 (кв, J=8,9 Гц, 1H), 4,84-4,73 (м, 3H), 4,42 (дд, J=2,5, 13,3 Гц, 1H), 4,34 (дд, J=1,4, 4,9 Гц, 1H), 4,21 (д, J=14,6 Гц, 1H), 3,93-3,72 (м, 2H), 3,11-2,94 (м, 1H), 2,88 (с, 3H), 2,24-2,11 (м, 1H), 1,28 (д, J=6,5 Гц, 3H)
Схема MMMM - Синтез трет-бутил (R)-6-гидрокси-2-метил-2,3-дигидробензo[f][1,4]оксазепин-4(5H)-карбоксилата (TP-46)
Схема MMMM
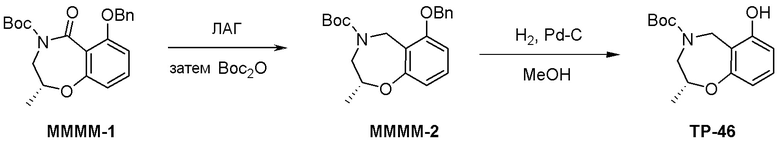
Стадия 1 - Синтез трет-бутил (R)-6-(бензилокси)-2-метил-2,3-дигидробензo[f][1,4]оксазепин-4(5H)-карбоксилата (MMMM-1)
К соединению MMMM-1 (полученного по методике получения стадий 1-5 со схемы LLLL с применением трет-бутил (R)-(2-гидроксипропил)карбамата), 500 мг, (1,5 ммоль) в сухом ТГФ (10,0 мл) добавляют ЛАГ (233 мг, 6,14 ммоль) при 25°C. Смесь перемешивают при 75°C в течение 2 ч. Реакционную смесь охлаждают до к.т., воду (2,0 мл) добавляют в реакционную смесь, затем Boc2O (670 мг, 3,07 ммоль). Смесь перемешивают в течение 2,5 ч при кт, затем добавляют воду и реакционную смесь экстрагируют EtOAc (10 мл×3), сушат с Na2SO4, концентрируют с получением неочищенного продукта, который очищают ISCO (20 г, силикагель, EtOAc/петролейный эфир=15%) с получением MMMM-2 (520 мг, 92%) в виде бесцветной камеди. ЖХМС [M-Boc+1] 269,9
Стадия 2 - Синтез трет-бутил (R)-6-гидрокси-2-метил-2,3-дигидробензo[f][1,4]оксазепин-4(5H)-карбоксилата (TP-46)
Соединение MMMM-2 (520 мг, 1,3 ммоль) растворяют в MeOH (10 мл) добавляют Pd/C (100 мг, 0,940 ммоль). Реакционный раствор перемешивают при 25°C под H2 в течение 2 ч. Затем смесь фильтруют через целит, промывают MeOH, и концентрируют. Остаток очищают ISCO (20 г, силикагель, EtOAc/петролейный эфир=25%) с получением TP-46 (234 мг, 67%) в виде белого твердого вещества. ЖХМС [M-Boc+1] 179,7; 1H ЯМР (400 MГц, CDCl3) δ ч./млн. 7,55 (с, 1H), 7,06 (т, J=8,1 Гц, 1H), 6,73 (д, J=8,1 Гц, 1H), 6,67-6,55 (м, 1H), 4,64 (д, J=14,7 Гц, 1H), 4,15-4,07 (м, 1H), 3,90 (шд, J=14,4 Гц, 1H), 3,84-3,76 (м, 1H), 3,29 (дд, J=8,9, 14,5 Гц, 1H), 1,41 (с, 9H), 1,29 (д, J=6,5 Гц, 3H)
Соединение TP-47 получают по методике получения со Схемы MMMM с применением трет-бутил (S)-(2-гидроксипропил)карбамата
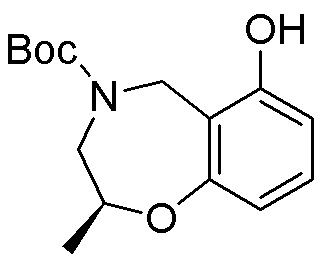
[M-Boc+1]
1H ЯМР (400 MГц, CDCl3) δ ч./млн. 7,56 (с, 1H), 7,05 (т, J=8,2 Гц, 1H), 6,72 (д, J=8,0 Гц, 1H), 6,66-6,59 (м, 1H), 4,63 (д, J=14,3 Гц, 1H), 4,14-4,07 (м, 1H), 3,89 (шд, J=14,6 Гц, 1H), 3,85-3,76 (м, 1H), 3,29 (дд, J=8,8, 14,3 Гц, 1H), 1,40 (с, 9H), 1,29 (д, J=6,5 Гц, 3H)
Примеры 209 & 210 получают по методике со схемы CC и с применением соединений TP-46 & TP-47, соответственно.
TP-46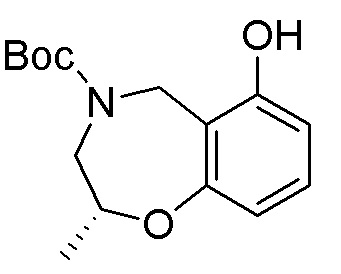
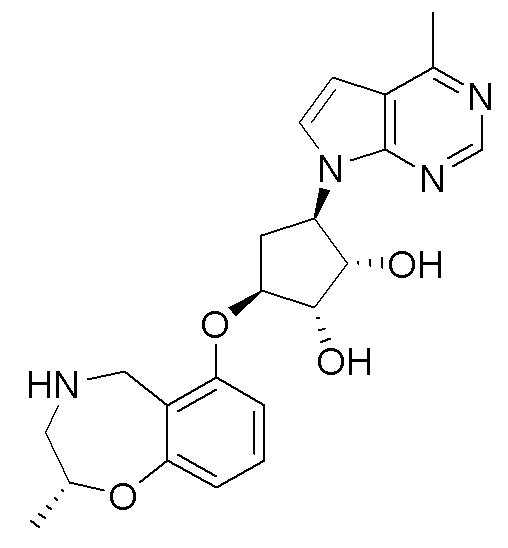
[M+1]
1H ЯМР (400 MГц, D2O, соль HCl) δ ч./млн. 8,80 (с, 1H), 7,77 (д, J=3,8 Гц, 1H), 7,30 (т, J=8,4 Гц, 1H), 7,06 (д, J=3,8 Гц, 1H), 6,84 (д, J=8,3 Гц, 1H), 6,76 (д, J=8,0 Гц, 1H), 5,28 (кв, J=9,0 Гц, 1H), 4,84-4,75 (м, 3H), 4,32 (дд, J=1,6, 4,9 Гц, 1H), 4,26-4,13 (м, 2H), 3,56 (шд, J=12,5 Гц, 1H), 3,34 (дд, J=10,5, 13,8 Гц, 1H), 3,06-2,94 (м, 1H), 2,87 (с, 3H), 2,23-2,09 (м, 1H), 1,38 (д, J=6,5 Гц, 3H)
TP-47
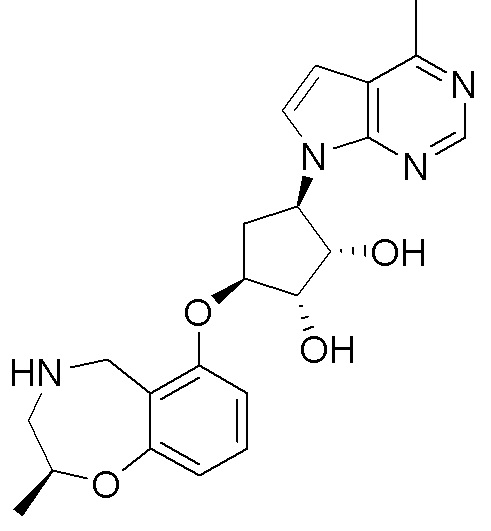
[M+1]
1H ЯМР (400 MГц, MeOD, соль HCl) δ ч./млн. 9,04 (с, 1H), 8,09 (д, J=3,8 Гц, 1H), 7,36 (т, J=8,2 Гц, 1H), 7,22 (д, J=3,8 Гц, 1H), 6,98 (д, J=8,3 Гц, 1H), 6,80 (д, J=8,3 Гц, 1H), 5,42 (кв, J=9,2 Гц, 1H), 4,81-4,72 (м, 3H), 4,33 (д, J=16 Гц, 1H), 4,29-4,22 (м, 2H), 3,63-3,60 (м, 1H), 3,42-3,39 (м, 1H), 3,14-3,05 (м, 1H), 3,01 (с, 3H), 2,33 (ддд, J=4,4, 9,8, 14,2 Гц, 1H), 1,48 (д, J=6,5 Гц, 3H)
Схема NNNN - Синтез трет-бутил-6-(дифторметил)-5-фтор-8-гидрокси-4-метил-3,4-дигидроизохинолин-2(1H)-карбоксилата (TP-48)
Схема NNNN
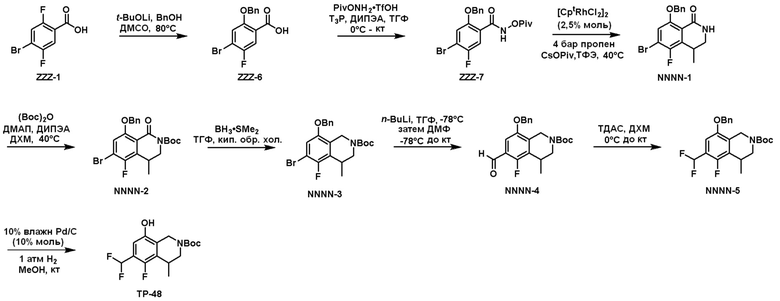
Стадия 1: Синтез 2-(бензилокси)-4-бром-5-фторбензoйной кислоты (ZZZ-6)
В круглодонную колбу, оборудованную магнитной мешалкой, добавляют трет-бутоксид лития (3,81 г, 47,6 ммоль) и ДМСО (119 мл, 0,2M). Колбу оборудуют findenser и добавляют бензиловый спирт (4,95 мл, 47,6 ммоль). Колбу помещают в нагревательный кожух и нагревают до 80°C в течение 5 минут. Колбу удаляют и добавляют ZZZ-1 (5,64 г, 23,8 ммоль). Колбу возвращают в нагревательный кожух и нагревают при 80°C в течение 17 часов. Реакционную смесь удаляют из нагревательного кожуха и охлаждают до кт. Раствор выливают в 1,2 л воды и подкисляют 48 мл 1M HCl водн. с получением рыжевато-коричневого осадка. Твердые вещества фильтруют и промывают водой. Твердые вещества собирают и сушат в вакуумной печи в течение 3,5 часов при 80°C с получением указанного в заголовке соединения ZZZ-6 (7,21 г, 93%) в виде светло-рыжевато-коричневого твердого вещества. 1H ЯМР (400 MГц, ДМСО-d6) δ ч./млн. 13,10 (шс, 1H), 7,61 (д, J=8,8 Гц, 1H), 7,57 (д, J=5,6 Гц, 1H), 7,48 (д, J=7,1 Гц, 2H), 7,44-7,36 (м, 2H), 7,35-7,28 (м, 1H), 5,22 (с, 2H).
Стадия 2: Синтез 2-(бензилокси)-4-бром-5-фтор-N-(пивалоилокси)бензамида (ZZZ-7)
В круглодонную колбу, оборудованную магнитной мешалкой, добавляют ZZZ-6 (5,73 г, 17,6 ммоль) и ТГФ (176 мл, 0,1M). Раствор охлаждают до 0°C и добавляют T3P (24,7 г, 38,8 ммоль) в виде 50% масс. раствора в EtOAc. После добавления, реакционную смесь перемешивают в течение 30 мин при 25°C. В раствор добавляют ДИПЭА (18,4 мл, 106 ммоль), затем FF-3 (5,18 г, 19,4 ммоль). После добавления, реакционную смесь перемешивают при 25°C в течение 1 часа. Реакцию гасят водой, разбавляют EtOAc и переносят в делительную воронку. Фазы разделяют, и органическую фазу промывают 1 порцией 10% лимонной кислоты, 1 порцией насыщ. NaHCO3 и 1 порцией насыщенного раствора соли. Органический экстракт затем сушат (MgSO4), фильтруют и концентрируют в вакууме. Неочищенный остаток очищают флэш-хроматографией на колонке (12г SiO2, Isco, 100% Гепт. до 100% EtOAc, 9 мл фракции) с получением указанного в заголовке соединения ZZZ-7 (6,24 г, 83%) в виде белого твердого вещества. 1H ЯМР (400 MГц, ХЛОРОФОРМ-d) δ ч./млн. 10,79 (с, 1H), 7,94 (д, J=8,9 Гц, 1H), 7,52-7,35 (м, 5H), 7,27-7,23 (м, 1H), 5,26 (с, 2H), 1,34 (с, 9H).
Стадия 3: Синтез 8-(бензилокси)-6-бром-5-фтор-4-метил-3,4-дигидроизохинолин-1(2H)-она (NNNN-1)
В 80 мл стальной реактор добавляют ZZZ-7 (4,00 г, 9,43 ммоль), пивалат цезия (4,41 г, 18,9 ммоль), [CptRhCl2]2 (166 мг, 0,236 ммоль) и трифторэтанол (47 мл, 0,2M). Реактор продувают азотом 3 раза, затем 3 циклами продувания газообразным пропиленом. Реакционную смесь нагревают до 40°C под 4 бар газообразного пропилена в течение 3 дней. Раствор гасят водн.NaHCO3 и переносят в делительную воронку с ДХМ. Фазы разделяют, и водную фазу экстрагируют 2 порциями ДХМ. Объединенные органические экстракты сушат (MgSO4), фильтруют и концентрируют в вакууме. Неочищенный остаток очищают флэш-хроматографией на колонке (40г SiO2, Isco, 100% Гепт. до 10% MeOH/EtOAc, 25 мл фракции) с получением указанного в заголовке соединения NNNN-1 (2,27 г, 66%, 9:1 р.р.) в виде коричневого твердого вещества. ЖХМС [M+H]=364 наблюдаемый. 1H ЯМР (400 MГц, ХЛОРОФОРМ-d) δ ч./млн. 7,55 (д, J=7,5 Гц, 2H), 7,47-7,36 (м, 2H), 7,35-7,28 (м, 1H), 7,13 (д, J=5,5 Гц, 1H), 5,92 (д, J=3,8 Гц, 1H), 5,27-5,19 (м, 1H), 5,16-5,10 (м, 1H), 3,69 (дд, J=4,0, 12,7 Гц, 1H), 3,40-3,30 (м, 1H), 3,23 (ддд, J=1,3, 6,0, 12,6 Гц, 1H), 1,36 (д, J=7,1 Гц, 3H).
Стадия 4: Синтез трет-бутил-8-(бензилокси)-6-бром-5-фтор-4-метил-1-оксо-3,4-дигидроизохинолин-2(1H)-карбоксилата (NNNN-2)
В круглодонную колбу, оборудованную магнитной мешалкой и содержащую NNNN-1 (1,20 г, 3,29 ммоль) добавляют ДХМ (11,0 мл, 0,3M), ДИПЭА (0,86 мл, 4,94 ммоль), (Boc)2O (1,08 г, 4,94 ммоль), и ДМАП (60,4 мг, 0,494 ммоль). Колбу оборудуют findenser и помещают в нагревательный блок. Реакционную смесь нагревают при 40°C в течение 16 часов. Колбу вынимают из нагревательного блока и охлаждают до кт. Реакцию гасят водой и переносят в делительную воронку с ДХМ. Фазы разделяют, и водную фазу экстрагируют 2 порциями ДХМ. Объединенные органические экстракты сушат (MgSO4), фильтруют и концентрируют в вакууме. Неочищенный остаток очищают флэш-хроматографией на колонке (40г SiO2, Isco, 100% гепт до 100% EtOAc, 9 мл фракции) с получением указанного в заголовке соединения NNNN-2 (1,41 г, 92%) в виде белой пены. ЖХМС [M+H-Boc]=364 наблюдаемый. 1H ЯМР (400 MГц, ХЛОРОФОРМ-d) δ ч./млн. 7,55 (д, J=7,5 Гц, 2H), 7,44-7,35 (м, 2H), 7,32 (д, J=7,3 Гц, 1H), 7,11 (д, J=5,6 Гц, 1H), 5,28-5,20 (м, 1H), 5,18-5,09 (м, 1H), 4,21 (дд, J=2,2, 13,2 Гц, 1H), 3,61 (дд, J=3,2, 13,1 Гц, 1H), 3,39-3,27 (м, 1H), 1,59 (с, 9H), 1,33 (д, J=7,1 Гц, 3H).
Стадия 5: Синтез трет-бутил-8-(бензилокси)-6-бром-5-фтор-4-метил-3,4-дигидроизохинолин-2(1H)-карбоксилата (NNNN-3)
В круглодонную колбу, оборудованную магнитной мешалкой и содержащую NNNN-2 (1,41 г, 3,04 ммоль), добавляют ТГФ (15 мл, 0,2M) и комплекс борана с диметилсульфидом (1,44 мл, 15,2 ммоль). Колбу оборудуют findenser и переносят в нагревательный блок. Реакционную смесь нагревают при 70°C в течение 30 минут. Колбу вынимают из нагревательного блока и постепенно охлаждают до кт. Реакционную смесь гасят добавлением по каплям метанола до завершения выделения газа, затем разбавляют гептаном. Раствор концентрируют в вакууме. Неочищенный остаток очищают флэш-хроматографией на колонке (40г SiO2, Isco, 100% гепт до 10% EtOAc/Гепт. до 100% EtOAc, 9 мл фракции) с получением указанного в заголовке соединения NNNN-3 (1,13 г, 82%) в виде белой пены. ЖХМС [M+H-Boc]=350 наблюдаемый. 1H ЯМР (400 MГц, ХЛОРОФОРМ-d) δ ч./млн. 7,47-7,30 (м, 5H), 6,93 (д, J=5,5 Гц, 1H), 5,18-4,79 (м, 3H), 4,26-3,96 (м, 2H), 3,28-2,95 (м, 2H), 1,51 (с, 9H), 1,25 (д, J=6,8 Гц, 3H).
Стадия 6: Синтез трет-бутил-8-(бензилокси)-5-фтор-6-формил-4-метил-3,4-дигидроизохинолин-2(1H)-карбоксилата (NNNN-4)
Примечание: nBuLi титруют, ТГФ сушат над активированными 4A молекулярными ситами, и иглы шприца сушат в печи при 85°C в вакууме до применения.
Круглодонную колбу, содержащую NNNN-3 (1,00 г, 2,22 ммоль) сушат под высоким вакуумом в течение ночи, оборудуют магнитной мешалкой и продувают аргоном в динамическом вакууме. В колбу добавляют ТГФ (11,0 мл, 0,2M) и раствор охлаждают до -78°C на бане AcMe/сухой лед. В охлажденный раствор добавляют н-бутиллитий (1,7 мл, 2,30 ммоль) по каплям для начала обмена металл-галоген. Реакционную смесь перемешивают при -78°C под аргоном в течение 30 минут.
Примечание: Реакционная смесь становится ярко-оранжевой при добавлении по каплям nBuLi.
В раствор добавляют ДМФ (0,26 мл, 3,4 ммоль) по каплям при -78°C. На этой стадии ледяную баню удаляют, и реакционную смесь постепенно нагревают до комнатной температуры.
Примечание: Реакционная смесь становится бледно-желтой при нагревании до комнатной температуры.
Реакционную смесь обратно гасят добавлением реакционного раствора в насыщ. водн. NH4Cl (10 мл). Водную фазу экстрагируют 4 порциями ДХМ. Объединенные органические экстракты сушат (MgSO4), фильтруют и концентрируют в вакууме. Неочищенный остаток очищают флэш-хроматографией на колонке (24 г SiO2, Isco, 100% Гепт. до 100% EtOAc, 20 мл фракции) с получением указанного в заголовке соединения NNNN-4 (885 мг, >95%) в виде белого парафинообразного твердого вещества. ЖХМС [M+H-Boc]=300 наблюдаемый. 1H ЯМР (400 MГц, ХЛОРОФОРМ-d) δ ч./млн. 10,34 (с, 1H), 7,48-7,31 (м, 5H), 7,21 (д, J=5,3 Гц, 1H), 5,29-4,87 (м, 3H), 4,32-4,03 (м, 2H), 3,35-3,18 (м, 1H), 3,17-3,00 (м, 1H), 1,52 (с, 9H), 1,29 (д, J=6,8 Гц, 3H).
Стадия 7: Синтез трет-бутил-8-(бензилокси)-6-(дифторметил)-5-фтор-4-метил-3,4-дигидроизохинолин-2(1H)-карбоксилата (NNNN-5)
Сцинтилляционную пробирку, оборудованную магнитной мешалкой и содержащую NNNN-4 (1,77 г, 4,43 ммоль), продувают аргоном в динамическом вакууме. Пробирку загружают ДХМ (44 мл, 0,1M) и раствор охлаждают до 0°C, затем по каплям добавляют ТДАС (1,46 мл, 11,1 ммоль). Ледяную баню удаляют, и раствор постепенно нагревают до кт. Реакционную смесь перемешивают под аргоном в течение 24 часов. Во время реакции добавляют 2 дополнительные аликвоты ТДАС (1,46 мл, 11,1 ммоль) в 12 и 17 часов, соответственно, для управления превращением (7,5 эквивалентов ТДАС всего). Реакционную смесь гасят добавлением по каплям насыщ. водн.NaHCO3. ОСТОРОЖНО: Во время гашения происходит быстрое выделение CO2. Содержимое пробирки переносят в делительную воронку с ДХМ и разбавляют водой. Фазы разделяют, и водную фазу экстрагируют 3 порциями ДХМ. Объединенные органические экстракты сушат (MgSO4), фильтруют и концентрируют в вакууме. Неочищенный остаток очищают флэш-хроматографией на колонке (40г SiO2, Isco, 100% Гепт. до 100% EtOAc, 20 мл фракции) с получением указанного в заголовке соединения NNNN-5 (1,59 г, 85%) в виде бледно-желтой камеди. ЖХМС [M+H-Boc]=322 наблюдаемый. 1H ЯМР (400 MГц, ХЛОРОФОРМ-d) δ ч./млн. 7,49-7,29 (м, 5H), 6,95 (д, J=5,3 Гц, 1H), 6,89 (т, J=55,1 Гц, 1H), 5,22-4,90 (м, 3H), 4,27-4,02 (м, 2H), 3,28-2,96 (м, 2H), 1,51 (с, 9H), 1,26 (д, J=7,0 Гц, 3H).
Стадия 8: Синтез трет-бутил-6-(дифторметил)-5-фтор-8-гидрокси-4-метил-3,4-дигидроизохинолин-2(1H)-карбоксилата (TP-48)
В 500 мл круглодонную колбу, оборудованную магнитной мешалкой и содержащую NNNN-5 (1,65 г, 3,91 ммоль), добавляют метанол (78 мл, 0,05M). В раствор добавляют Pd/C 10% масс. (417 мг, 0,391 ммоль) и раствор продувают водородом под динамическим вакуумом. Реакционную смесь энергично перемешивают под 1 атм. водорода в течение 1,5 часов. Реакционную смесь фильтруют через целит, твердое вещество промывают ДХМ, и фильтрат концентрируют в вакууме. Неочищенный остаток очищают флэш-хроматографией на колонке (24г SiO2, Isco, 100% Гепт. до 100% EtOAc, 20 мл фракции) с получением указанного в заголовке соединения TP-48 (1,15 г, 88%) в виде белой пены. ЖХМС [M+H-Boc]=232 наблюдаемый. 1H ЯМР (400 MГц, ХЛОРОФОРМ-d) δ ч./млн. 7,00-6,65 (м, 2H), 5,22-4,79 (м, 1H), 4,29-3,94 (м, 2H), 3,28-2,95 (м, 2H), 1,58-1,51 (м, 9H), 1,23 (д, J=7,0 Гц, 3H).
Схема OOOO - Синтез трет-бутил-5-фтор-8-гидрокси-1-метил-3,4-дигидроизохинолин-2(1H)-карбоксилата (TP-49)
Схема OOOO
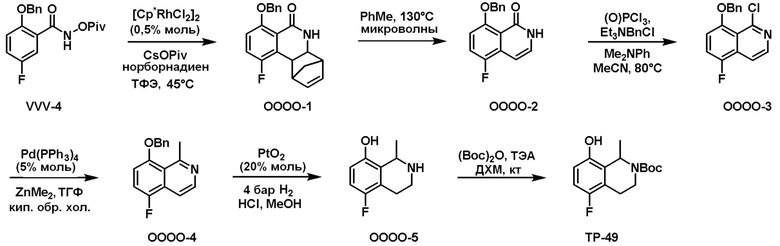
Стадия 1: Синтез 7-(бензилокси)-10-фтор-1,4a,5,10b-тетрагидро-1,4-метанофенантридин-6(4H)-она (OOOO-1)
В круглодонную колбу, оборудованную магнитной мешалкой, добавляют VVV-4 (2,29 г, 6,63 ммоль), пивалат цезия (3,10 г, 13,3 ммоль), [Cp*RhCl2]2 (20,5 мг, 0,0332 ммоль) и трифторэтанол (33 мл, 0,2M). Колбу закрывают крышкой с резиновой мембраной и добавляют норборнадиен (0,74 мл, 7,29 ммоль). Колбу помещают в нагревательный кожух, и реакционную смесь нагревают до 45°C в течение 40 минут. Колбу удаляют из нагревательного блока и охлаждают до кт. Раствор разбавляют водой, затем ДХМ и фазы разделяют. Органическую фазу промывают 1 порцией воды, сушат (MgSO4), фильтруют и концентрируют в вакууме. Неочищенный остаток затем сушат под высоким вакуумом с получением указанного в заголовке соединения OOOO-1 в виде оранжевого твердого вещества, которое применяют на следующей стадии без дальнейшей очистки. ЖХМС [M+H]=336 наблюдаемый.
Стадия 2: Синтез 8-(бензилокси)-5-фторизохинолин-1(2H)-она (OOOO-2)
В микроволновую пробирку, оборудованную магнитной мешалкой, добавляют OOOO-1 (~6,63 ммоль) в виде раствора в толуоле (15 мл, 0,44M). Пробирку герметично закрывают тефлоновой крышкой и помещают в микроволновый реактор. Реакционную смесь нагревают до 130°C в течение 1 часа. Раствор переносят в круглодонную колбу с ДХМ и концентрируют в вакууме. Неочищенный остаток затем сушат под высоким вакуумом с получением указанного в заголовке соединения OOOO-2 (1,79 г, >95%) в виде коричневого твердого вещества, которое применяют на следующей стадии без дальнейшей очистки. ЖХМС [M+H]=270 наблюдаемый. 1H ЯМР (400 MГц, ДМСО-d6) δ ч./млн. 11,07 (шс, 1H), 7,62 (д, J=7,2 Гц, 2H), 7,46 (т, J=9,3 Гц, 1H), 7,39 (т, J=7,5 Гц, 2H), 7,33-7,27 (м, 1H), 7,24-7,19 (м, 1H), 7,00 (дд, J=4,2, 9,0 Гц, 1H), 6,45 (д, J=7,1 Гц, 1H), 5,20 (с, 2H).
Стадия 3: Синтез 8-(бензилокси)-1-хлор-5-фторизохинолина (OOOO-3)
В реакционную пробирку, оборудованную магнитной мешалкой, добавляют хлорид бензилтриэтиламмония (846 мг, 3,71 ммоль) и OOOO-2 (500 мг, 1,86 ммоль) в виде раствора в ацетонитриле (19 мл, 0,1M). Пробирку герметично закрывают тефлоновой крышкой и добавляют диметиланилин (0,35 мл, 2,79 ммоль), затем по каплям добавляют оксихлорид фосфора (1,04 мл, 11,1 ммоль). Пробирку помещают в нагревательный блок и нагревают при 80°C в течение 10 минут. Раствор концентрируют, и неочищенный остаток переносят в делительную воронку с ДХМ и разбавляют водой. Фазы разделяют, и водную фазу экстрагируют 2 порциями ДХМ. Объединенные органические экстракты сушат (MgSO4), фильтруют и концентрируют в вакууме. Неочищенный остаток очищают флэш-хроматографией на колонке (24г SiO2, Isco, 100% Гепт. до 100% EtOAc, 9 мл фракции) с получением указанного в заголовке соединения OOOO-3 (465 мг, 87%) в виде светло-оранжевого твердого вещества. ЖХМС [M+H]=288 наблюдаемый. 1H ЯМР (400 MГц, ХЛОРОФОРМ-d) δ ч./млн. 8,32 (д, J=5,6 Гц, 1H), 7,78 (д, J=5,6 Гц, 1H), 7,56 (д, J=7,2 Гц, 2H), 7,48-7,28 (м, 4H), 6,97 (дд, J=4,3, 8,7 Гц, 1H), 5,27 (с, 2H).
Стадия 4: Синтез 8-(бензилокси)-5-фтор-1-метилизохинолина (OOOO-4)
В высушенную в печи реакционную пробирку, оборудованную магнитной мешалкой и охлажденную под потоком аргона, добавляют OOOO-3 (422 мг, 1,47 ммоль) и Pd(PPh3)4 (84,7 мг, 0,0733 ммоль). Пробирку закрывают крышкой с резиновой мембраной и продувают аргоном в динамическом вакууме. В пробирку добавляют ТГФ (7,33 мл, 0,2M), затем по каплям добавляют диметилцинк (2,20 мл, 4,40 ммоль). Пробирку герметично закрывают тефлоновой крышкой и помещают в нагревательный блок. Реакционную смесь кипятят с обратным холодильником при 75°C в течение 1 часа. Пробирку удаляют из нагревательного блока и охлаждают до кт. Реакционную смесь осторожно гасят добавлением по каплям насыщ. водн. NH4Cl. Раствор переносят в делительную воронку с ДХМ. Фазы разделяют, и водную фазу экстрагируют 3 порциями ДХМ. Объединенные органические экстракты промывают 1 порцией насыщенного раствора соли, сушат (MgSO4), фильтруют и концентрируют в вакууме. Неочищенный остаток очищают флэш-хроматографией на колонке (12г SiO2, Isco, 100% Гепт. до 100% EtOAc, 9 мл фракции) с получением (356 мг) светло-оранжевого твердого вещества. Выделенный продукт повторно подвергают флэш-хроматографии на колонке (12г SiO2, Isco, 100% Гепт. до 10% MeOH/EtOAc, 9 мл фракции) с получением указанного в заголовке соединения OOOO-4 (310 мг, 79%) в виде белого твердого вещества. Выделенное твердое вещество применяют на следующей стадии с ~80% чистотой на основе 1H ЯМР анализа. ЖХМС [M+H]=268 наблюдаемый. 1H ЯМР (400 MГц, ХЛОРОФОРМ-d) d=8,43 (д, J=5,7 Гц, 1H), 7,68 (д, J=5,9 Гц, 1H), 7,50 (д, J=7,1 Гц, 2H), 7,47-7,39 (м, 3H), 7,22 (т, J=8,9 Гц, 1H), 6,86 (дд, J=4,3, 8,6 Гц, 1H), 5,22 (с, 2H), 3,10 (с, 3H).
Стадия 5: Синтез 5-фтор-1-метил-1,2,3,4-тетрагидроизохинолин-8-ола (OOOO-5)
В стальной реактор добавляют OOOO-4 (250 мг, 0,935 ммоль), PtO2 (42,5 мг, 0,187 ммоль), этанол (15 мл) и уксусную кислоту (0,5 мл). Раствор гидрируют под 45 ф/кв.д водорода при кт в течение 20 часов. Раствор фильтруют через целит, и твердые вещества промывают этанолом. Фильтрат концентрируют в вакууме с получением указанного в заголовке соединения OOOO-5 (178 мг, >95%) в виде слегка желтого твердого вещества, которое применяют на следующей стадии без дальнейшей очистки. 1H ЯМР (400 MГц, МЕТАНОЛ-d4) δ ч./млн. 6,92-6,85 (м, 1H), 6,71-6,65 (м, 1H), 4,69 (кв, J=6,7 Гц, 1H), 3,51-3,37 (м, 2H), 3,06-2,84 (м, 2H), 1,62 (д, J=6,8 Гц, 3H)
Стадия 6: Синтез трет-бутил 5-фтор-8-гидрокси-1-метил-3,4-дигидроизохинолин-2(1H)-карбоксилата (TP-49)
В круглодонную колбу, оборудованную магнитной мешалкой и содержащую OOOO-5 (130 мг, 0,717 ммоль), добавляют метанол (10 мл, 0,07M), (Boc)2O (157 мг, 0,717 ммоль) и триметиламин (145 мг, 1,43 ммоль). Реакционную смесь перемешивают при 25°C в течение 2,5 часов. Раствор концентрируют в вакууме. Неочищенный остаток очищают флэш-хроматографией на колонке (20г SiO2, Isco, 25% EtOAc/пет. эфир) с получением указанного в заголовке соединения TP-49 (158 мг, 78%) в виде белого твердого вещества. 1H ЯМР (400 MГц, ХЛОРОФОРМ-d) δ ч./млн. 6,81-6,69 (м, 1H), 6,55 (дд, J=4,4, 8,7 Гц, 1H), 5,54-5,20 (м, 1H), 4,32-4,03 (м, 1H), 3,36-3,07 (м, 1H), 2,86-2,66 (м, 2H), 1,51 (с, 9H), 1,43 (д, J=6,6 Гц, 3H).
Примеры 211-214 получают по методике Примеров 182 & 183 (Схема YYY), начиная с подходящего рацемического тетрагидроизохинолина на стадии 1 и разделяя диастереомеры до конечного снятия защиты.
TP-48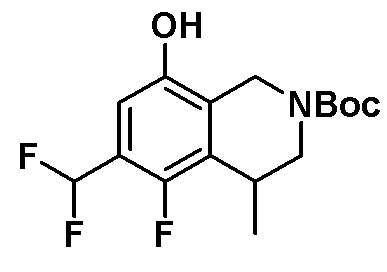
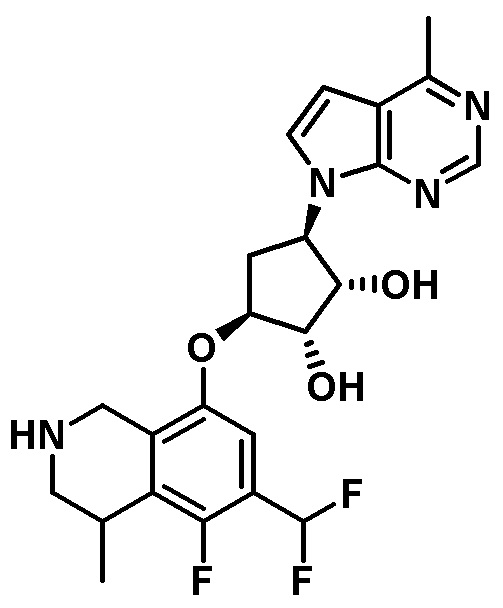
[M+H]
Пример 211 (Изомер 1) - 1H ЯМР (400 MГц, ОКСИД ДЕЙТЕРИЯ) δ ч./млн. 8,83 (с, 1H), 7,80 (шс, 1H), 7,14-6,80 (м, 3H), 5,32 (кв, J=9,0 Гц, 1H), 4,78 (шс, 2H), 4,48-4,37 (м, 1H), 4,32-4,18 (м, 2H), 3,50-3,32 (м, 3H), 3,10-2,98 (м, 1H), 2,89 (с, 3H), 2,24-2,13 (м, 1H), 1,33 (д, J=6,8 Гц, 3H).
Пример 212 (Изомер 2) - 1H ЯМР (400 MГц, ОКСИД ДЕЙТЕРИЯ) δ ч./млн. 8,83 (с, 1H), 7,80 (д, J=3,7 Гц, 1H), 7,14-6,77 (м, 3H), 5,32 (кв, J=9,1 Гц, 1H), 4,80-4,78 (м, 1H), 4,64 (шдд, J=5,0, 8,9 Гц, 1H), 4,47-4,35 (м, 1H), 4,30-4,20 (м, 2H), 3,51-3,30 (м, 3H), 3,09-2,95 (м, 1H), 2,88 (с, 3H), 2,26-2,10 (м, 1H), 1,32 (д, J=6,8 Гц, 3H).
TP-49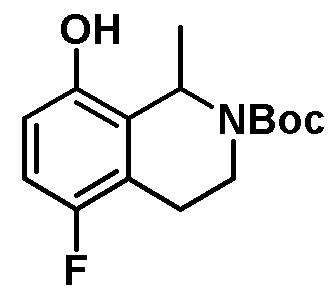
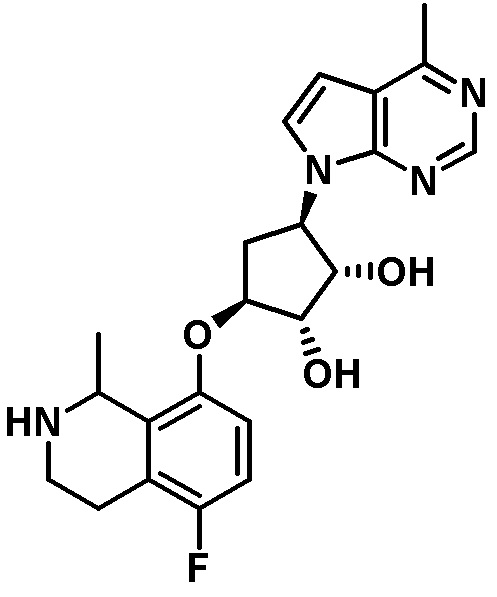
[M+H]
Пример 213 (Изомер 1) - 1H ЯМР (400 MГц, МЕТАНОЛ-d4) δ ч./млн. 9,04 (с, 1H), 8,03 (д, J=3,3 Гц, 1H), 7,23 (д, J=3,4 Гц, 1H), 7,14-6,96 (м, 2H), 5,39 (кв, J=9,1 Гц, 1H), 4,79 (шдд, J=4,9, 9,3 Гц, 3H), 4,27 (шд, J=4,2 Гц, 1H), 3,67-3,48 (м, 2H), 3,19-2,95 (м, 6H), 2,29 (шт, J=10,1 Гц, 1H), 1,75 (д, J=6,5 Гц, 3H).
Пример 214 (Изомер 2) - 1H ЯМР (400 MГц, МЕТАНОЛ-d4) d=9,03 (с, 1H), 8,08 (д, J=3,4 Гц, 1H), 7,25 (д, J=3,4 Гц, 1H), 7,14-7,01 (м, 2H), 5,37 (кв, J=9,2 Гц, 1H), 4,82-4,73 (м, 3H), 4,22 (шд, J=4,5 Гц, 1H), 3,63-3,54 (м, 2H), 3,20-2,99 (м, 6H), 2,38 (ддд, J=3,9, 9,7, 13,8 Гц, 1H), 1,75 (д, J=6,6 Гц, 3H)
Примеры 215&216 получают по методике Примеров 182 & 183 (Схема YYY), начиная с подходящего рацемического тетрагидроизохинолина на стадии 1, проводят аминолиз по методике стадии 4 (Схема NN), и разделяя диастереомеры до конечного снятия защиты.
TP-48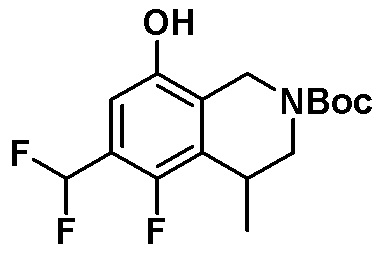
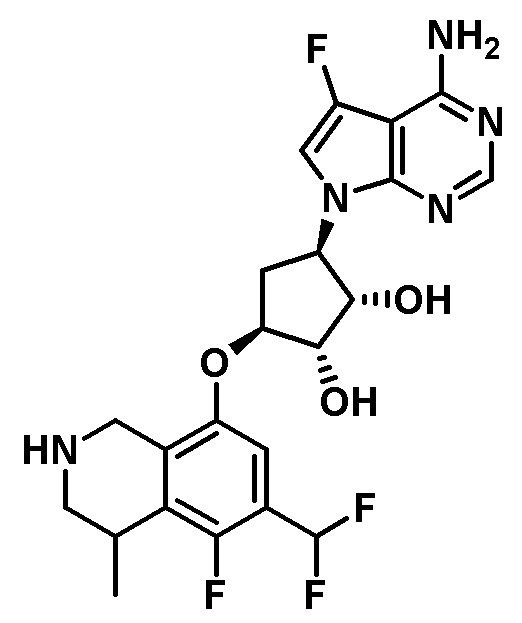
[M+H]
Пример 215 (Изомер 1) - 1H ЯМР (400 MГц, ОКСИД ДЕЙТЕРИЯ) δ ч./млн. 8,24 (с, 1H), 7,30 (с, 1H), 7,21-6,88 (м, 2H), 5,36-5,21 (м, 1H), 4,87 (шд, J=1,8 Гц, 1H), 4,57 (шдд, J=5,1, 8,7 Гц, 1H), 4,51-4,42 (м, 1H), 4,36-4,23 (м, 2H), 3,58-3,43 (м, 3H), 3,14-2,99 (м, 1H), 2,17-2,06 (м, 1H), 1,41 (д, J=6,5 Гц, 3H).
Пример 216 (Изомер 2) - 1H ЯМР (400 MГц, ОКСИД ДЕЙТЕРИЯ) δ ч./млн. 8,22 (с, 1H), 7,28 (д, J=2,0 Гц, 1H), 7,21-6,85 (м, 2H), 5,30-5,16 (м, 1H), 4,86 (шд, J=1,5 Гц, 1H), 4,56 (дд, J=5,0, 9,0 Гц, 1H), 4,46 (шд, J=16,8 Гц, 1H), 4,37-4,20 (м, 2H), 3,61-3,41 (м, 3H), 3,02 (ддд, J=7,5, 9,2, 14,9 Гц, 1H), 2,18-2,02 (м, 1H), 1,53-1,34 (м, 3H).
Биологические примеры
Биохимические способы исследования
Соединения солюбилизируют в ДМСО и серийно разводят, с применением 3-кратных разведений, в 100% ДМСО в концентрации в 50 раз больше, чем желаемая для анализа концентрация. После разведения 1 мкл добавляют в пустой 96-луночный титровальный микропланшет. Белковый комплекс PRMT5/MEP50 объединяют с H4(1-21) пептидом (SGRGKGGKGLGKGGAKRHRKV) в аналитическом буфере PRMT5 (50 мМ Трис pH 8,5, 50 мМ NaCl, 5 мМ MgCl2, 1 мМ ЭДТК, 1 мМ TCEP) и 44 мкл добавляют в титровальный микропланшет, содержащий соединение. S-Аденозил-L-метионин (SAM) получают объединением 3H меченного SAM с не меченным SAM в аналитическом буфере PRMT5 так, чтобы конечная концентрация SAM была 10 мкМ и специфическая активность была 0,2 мкКи/мкл. Реакционную смесь инициируют добавлением 5 мкл исходного раствора SAM в титровальный микропланшет. Конечные условия реакции включают 10 нМ комплекса PRMT5/MEP50, 200 нМ пептида и 1 мкМ SAM. Через 25 минут инкубирования при комнатной температуре, реакцию останавливают добавлением 100 мкл of 20% TCA. Продукт 3H-пептид поглощают с применением 96-луночного фильтровального планшета (MSIPN4B, Millipore) и промывают 5 раз ФРФБ буфером. Сцинтилляционную жидкость (100 мкл) добавляют в высушенный фильтровальный планшет и считают на жидкостном сцинтилляционном счетчике. Значения IC50 определяют сопоставлением данных со стандартной 4-параметрической кривой доза-реакция с применением собственного программного обеспечения Pfizer.
Результаты биохимического анализа суммированы в таблице 1, и показаны как значения IC50 в мкМ.
Таблица 1
Ингибирование фермента PRMT5
Анализ пролиферации A549
Клетки аденокарциномы легких A549 (American Type Culture Collection) сохраняют в среде для выращивания DMEM (Life Technologies) с добавлением 10% об./об. инактивированной теплом фетальный бычьей сыворотки (Sigma) и культивируют при 37°C, 5% CO2. Экспоненциально растущие клетки A549 высевают в 96-луночные черные обработанные культурой ткани планшеты (Corning) с плотностью 2500 клеток/мл в объеме 100 мкл культуральной среды и оставляют слипаться в течение ночи при 37°C, 5% CO2. На следующий день планшеты с соединением готовят, производят девятиточечные 3,3-кратные разведения в ДМСО с максимальной концентрацией 10 мМ. Соединения затем разводят в культуральной среде и 11 мкл добавляют к клеткам (конечная максимальная аналитическая концентрация 10 мкМ и ДМСО 0,2%). Клетки инкубируют с соединением при 37°C, 5%CO2 в течение 7 дней со средой, и соединение меняют на 4 день. Жизнеспособность клеток оценивают на 7 день добавлением 100 мкл реагента Cell Titer Glo (Promega) в планшет для измерения количества АТФ, присутствующей в клетках. Люминесценцию считывают с применением Envision 2104 Multilabel Reader (Perkin Elmer). 50% ингибирующую концентрацию (IC50) определяют с применением модели 4-параметрической подгонки, нормализованной до ДМСО контроля в дозе-реакции.
Результаты анализа пролиферации A549 из примеров суммированы в таблице 2, и показаны как значения IC50 в мкМ.
Таблица 2
IC50 пролиферации клеток A549
Молекулярная биология
Кодирующую ген полноразмерную PRMT5 открытую рамку считывания (ОРС) сливают непосредственно на Ala2 с последовательностью MDYKDDDDKGRAT, кодирующей маркер Flag (SEQ ID: 1) и полноцепной немаркированной MEP50 (SEQ ID: 2) с оптимизированным кодоном для экспрессии у млекопитающих и синтезированной в GenScript, Piscataway, NJ. Синтезированные гены клонируют в вектор экспрессии клеток насекомых pFASTBac Dual (Life Technologies) с применением стандартных методик клонирования на основе рестрикционного фермента. В конечной конструкции PRMT5 ОРС находится под контролем промотора полигедрина (polH), а MEP50 ОРС находится под контролем промотора p10. Дополнительно, MEP50 (SEQ ID: 2) субклонируют в вектор pFASTBac1 с применением стандартных методик клонирования на основе рестрикционного фермента.
Экспрессия белка
Вирусы создают с применением стандартных протоколов создания вирусов Bac-to-Bac (Life Technologies) и расширяют до неспецифических двухпроходных (P2) исходных растворов. Сверхэкспрессию белка проводят в экспоненциально растущих клетках Sf21, инфицированных 2×106 вирусным исходным раствором P2 при MOI=1 1:1 соотношении PRMT5-Mep50 двойной конструкции вируса и Mep50 конструкции вируса. Протокол совместной экспрессии применяют для дополнения дополнительного Mep50 для образования FlagPRMT5-Mep50 гетеродимера. Клетки собирают через 72 ч после заражения центрифугированием и замороженную лепешку хранят при -80°C.
Очистка белка
Комплекс FlagPRMT5-Mep50 очищают от лизата клеток с применением Flag сродственной хроматографии. Клетки лизируют в 50мМ Трис 7,5, 200 мМ NaCl, 10% глицерине, 0,25 мМ TCEP с добавлением не содержащего ЭДТК коктейля ингибитора протеазы (Roche). 1,5 мл лизисного буфера добавляют на 1 г замороженной лепешки. Очищенный лизат получают центрифугированием лизата клеток при 10000 g в течение 1 ч при 4°C. 5 мл Anti-FLAG M2 Agarose (Sigma) в течение 3 ч для выделения добавляют к очищенному лизату для выделения FlagPRMT5-Mep50. После связывания партий в течение 3 ч при 4°C, Flag смолу, связанную с FlagPRMT5-Mep50 промывают 20 объемами колонки (ОК) 50 мМ Трис 7,5, 200 мМ NaCl, 10% глицерина, 0,25 мМ TCEP, затем элюируют комплекс FlagPRMT5-Mep50 с применением 3 ОК 50 мМ Трис 7,5, 200 мМ NaCl, 10% глицерина, 0,25 мМ TCEP с добавлением 200 мкг/мл пептида FLAG (DYKDDDDK). FlagPRMT5-Mep50 затем очищают с применением колонки S300 26/600 (GE Healthcare) предварительно уравновешенной 2 ОК 25 мМ Трис pH7,5, 150 мМ NaCl, 5% глицерина, 0,5мМ TCEP буфера. Пиковую фракцию, содержащую комплекс FlagPRMT5-Mep50, концентрируют до 1,6 мг/мл, быстро замораживают в небольших аликвотах с применением жидкого азота, и хранят при -80°C.
Последовательности
SEQ ID: 1
MDYKDDDDKGRATAAMAVGGAGGSRVSSGRDLNCVPEIADTLGAVAKQGFDFLCMPVFHP RFKREFIQEPAKNRPGPQTRSDLLLSGRDWNTLIVGKLSPWIRPDSKVEKIRRNSEAAML QELNFGAYLGLPAFLLPLNQEDNTNLARVLTNHIHTGHHSSMFWMRVPLVAPEDLRDDII ENAPTTHTEEYSGEEKTWMWWHNFRТСХDYSKRIAVALEIGADLPSNHVIDRWLGEPIKA AILPTSЕСЛИLTNKKGFPVLSKMHQRLЕСЛИRLLKLEVQFIITGTNHHSEKEFCSYLQYLEYLS QNRPPPNAYELFAKGYEDYLQSPLQPLMDNLESQTYEVFEKDPIKYSQYQQAIYKCLLDR VPEEEKDTNVQVLMVLGAGRGPLVNASLRAAKQADRRIKLYAVEKNPNAVVTLENWQFEE WGSQVTVVSSDMREWVAPEKADIIVSELLGSFADNELSPECLDGAQHFLKDDGVSIPGEY TSFLAPISSSKLYNEVRACREKDRDPEAQFEMPYVVRLHNFHQLSAPQPCFTFSHPNRDP MIDNNRYCTLEFPVEVNTVLHGFAGYFETVLYQDITLSIRPETHSPGMFSWFPILFPIKQ PITVREGQTICVRFWRCSNSKKVWYEWAVTAPVCSAIHNPTGRSYTIGL*
SEQ ID: 2
MRKETPPPLVPPAAREWNLPPNAPACMERQLEAARYRSDGALLLGASSLSGRCWAGSLWL FKDPCAAPNEGFCSAGVQТЭАGVADLTWVGERGILVASDSGAVELWELDENETLIVSKFC KYEHDDIVSTVSVLSSGTQAVSGSKДИКIKVWDLAQQVVLSSYRAHAAQVTCVAASPHKD SVFLSCSEDNRILLWDTRCPKPASQIGCSAPGYLPTSLAWHPQQSEVFVFGDENGTVSLV DTKSTSCVLSSAVHSQCVTGLVFSPHSVPFLASLSЭДКSLAVLDSSLSELFRSQAHRDFV RDATWSPLNHSLLTTVGWDHQVVHHVVPTEPLPAPGPASVTE*
| название | год | авторы | номер документа |
|---|---|---|---|
| ИНГИБИТОРЫ МУТАЦИИ HER2 | 2022 |
|
RU2834124C2 |
| ИНГИБИТОРЫ RMT5 | 2019 |
|
RU2814198C2 |
| НИТРИЛСОДЕРЖАЩИЕ ПРОТИВОВИРУСНЫЕ СОЕДИНЕНИЯ | 2021 |
|
RU2786722C1 |
| НИТРИЛСОДЕРЖАЩИЕ ПРОТИВОВИРУСНЫЕ СОЕДИНЕНИЯ | 2021 |
|
RU2820150C2 |
| БИЦИКЛИЧЕСКИЕ СОЕДИНЕНИЯ В КАЧЕСТВЕ ИНГИБИТОРОВ ВЗАИМОДЕЙСТВИЯ/АКТИВАЦИИ PD1/PD-L1 | 2019 |
|
RU2777980C2 |
| ИНГИБИТОРЫ ТРАНСГЛУТАМИНАЗЫ 2 (TG2) | 2019 |
|
RU2781370C2 |
| ИНГИБИТОРНЫЕ СОЕДИНЕНИЯ | 2013 |
|
RU2673079C2 |
| ПИРРОЛОПИРИМИДИНЫ В КАЧЕСТВЕ ПОТЕНЦИАТОРОВ МВТР | 2017 |
|
RU2757457C2 |
| БИЦИКЛИЧЕСКИЕ ГЕТЕРОЦИКЛИЛЬНЫЕ СОЕДИНЕНИЯ И ИХ ПРИМЕНЕНИЯ | 2020 |
|
RU2811612C2 |
| ПРОИЗВОДНЫЕ 5-(7Н-ПИРРОЛО[2,3-d]ПИРИМИДИН-4-ИЛ)-5-АЗАСПИРО[2.5]ОКТАН-8-КАРБОНОВОЙ КИСЛОТЫ В КАЧЕСТВЕ НОВЫХ ИНГИБИТОРОВ JAK-КИНАЗЫ | 2018 |
|
RU2761626C2 |
Изобретение относится к соединениям общей формулы I-IV, обладающим свойством ингибитора фермента PRMT5, их применению, фармацевтической композиции на их основе, способу лечения аномального роста клеток, в том числе различных видов рака. В общей формуле I R1 выбирают из группы, включающей (С1-С8)алкил и N(R4)2, где каждый R4 независимо является A-R14, где А отсутствует и R14 является водородом; R2 является водородом, (С1-С8)алкилом или N(R5)2, где каждый R5 независимо является водородом; каждый R3 независимо выбирают из водорода, гидрокси, (С1-С8)алкила, или если D является C(R3)2, R3 дополнительно выбирают из фтора; каждый R9 независимо является водородом; D является C(R3)2, О; G является фенилом или 10-12-членной гетероарильной кольцевой системой с 1-2 атомами азота или 9-11-гетероциктильной кольцевой системой с 1-2 гетероатомами, выбранными из азота и кислорода; каждый R8 отсутствует или его независимо выбирают из группы, включающей (C1-С8) алкил, (С1-С8)галоалкил, гидрокси, (С1-С8)алкокси, 5-членный гетероарил с 2-3 гетероатомами, выбранными из N, О и S, CN, галоген и CON(R4)2, и где два R8 необязательно объединены с образованием карбонита; Q отсутствует; V является N или С, если Z присутствует, где если V образует двойную связь, V является углеродом или V является N или СН и образует двойную связь с W, если Z отсутствует; W является С, где если W образует двойную связь, если W является углеродом; X является N, если Y присутствует, где если X образует двойную связь, X является углеродом или X является NR16, если Y отсутствует, где R16 является Н или метилом; Y отсутствует, является CR10; Z отсутствует, является CR12, где каждый R12 независимо выбирают из водорода, фтора, хлора, брома. 15 н. и 24 з.п. ф-лы, 2 табл., 216 пр.
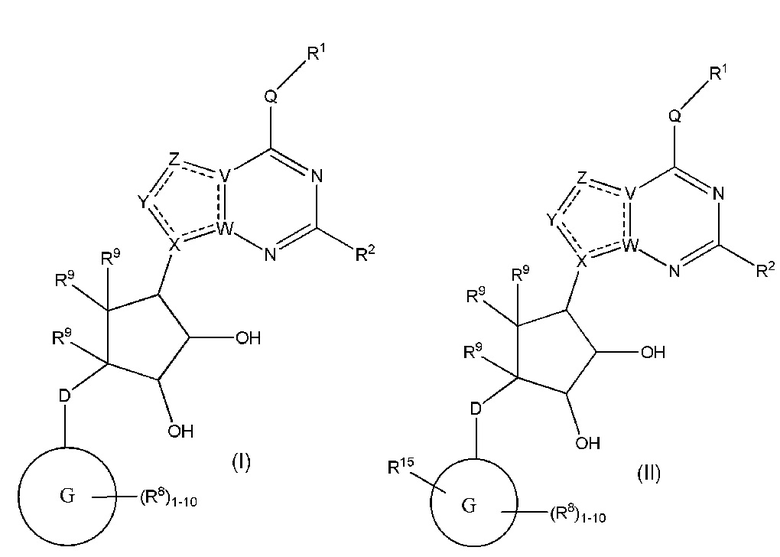
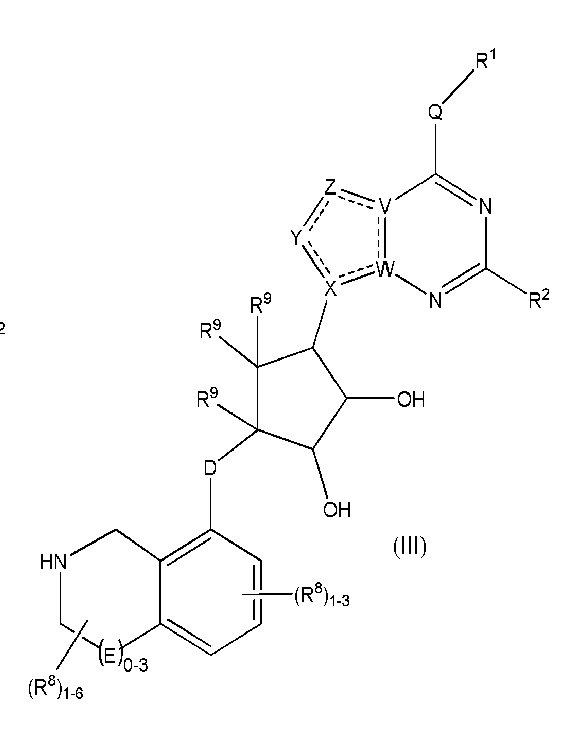
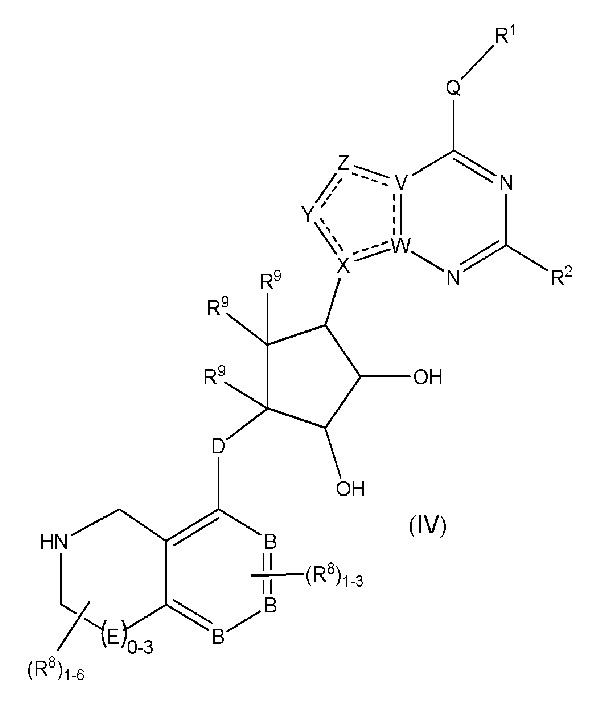
1. Соединение формулы (I):
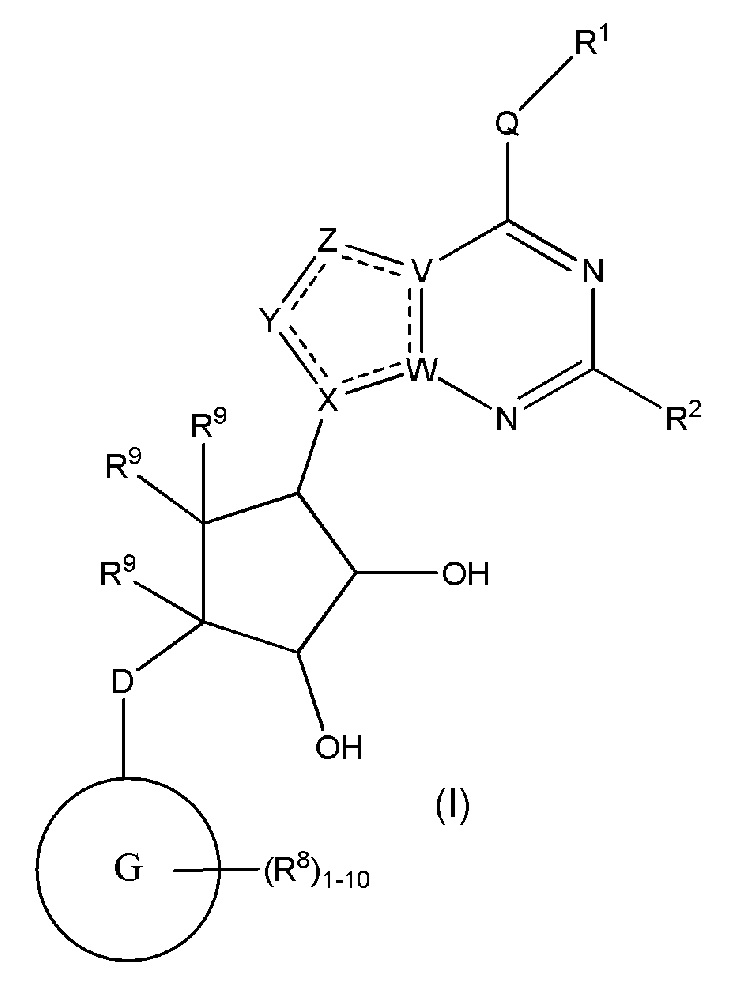
или его фармацевтически приемлемая соль, где:
R1 выбирают из группы, включающей (С1-С8)алкил и N(R4)2, где каждый R4 независимо является A-R14, где А отсутствует и R14 является водородом;
R2 является водородом, (С1-С8)алкилом или N(R5)2, где каждый R5 независимо является водородом;
каждый R3 независимо выбирают из водорода, гидрокси, (С1-С8)алкила, или если D является C(R3)2, R3 дополнительно выбирают из фтора;
каждый R9 независимо является водородом;
D является C(R3)2, О;
G является фенилом или 10-12-членной гетероарильной кольцевой системой с 1-2 атомами азота или 9-11-гетероциктильной кольцевой системой с 1-2 гетероатомами, выбранными из азота и кислорода;
каждый R8 отсутствует или его независимо выбирают из группы, включающей (C1-С8) алкил, (С1-С8)галоалкил, гидрокси, (С1-С8)алкокси, 5-членный гетероарил с 2-3 гетероатомами, выбранными из N, О и S, CN, галоген и CON(R4)2, и где два R8 необязательно объединены с образованием карбонита;
Q отсутствует;
V является N или С, если Z присутствует, где если V образует двойную связь, V является углеродом или V является N или СН и образует двойную связь с W, если Z отсутствует;
W является С, где если W образует двойную связь, если W является углеродом;
X является N, если Y присутствует, где если X образует двойную связь, X является углеродом или X является NR16, если Y отсутствует, где R16 является Н или метилом;
Y отсутствует, является CR10;
Z отсутствует, является CR12, где каждый R12 независимо выбирают из водорода, фтора, хлора, брома.
2. Соединение формулы (II):
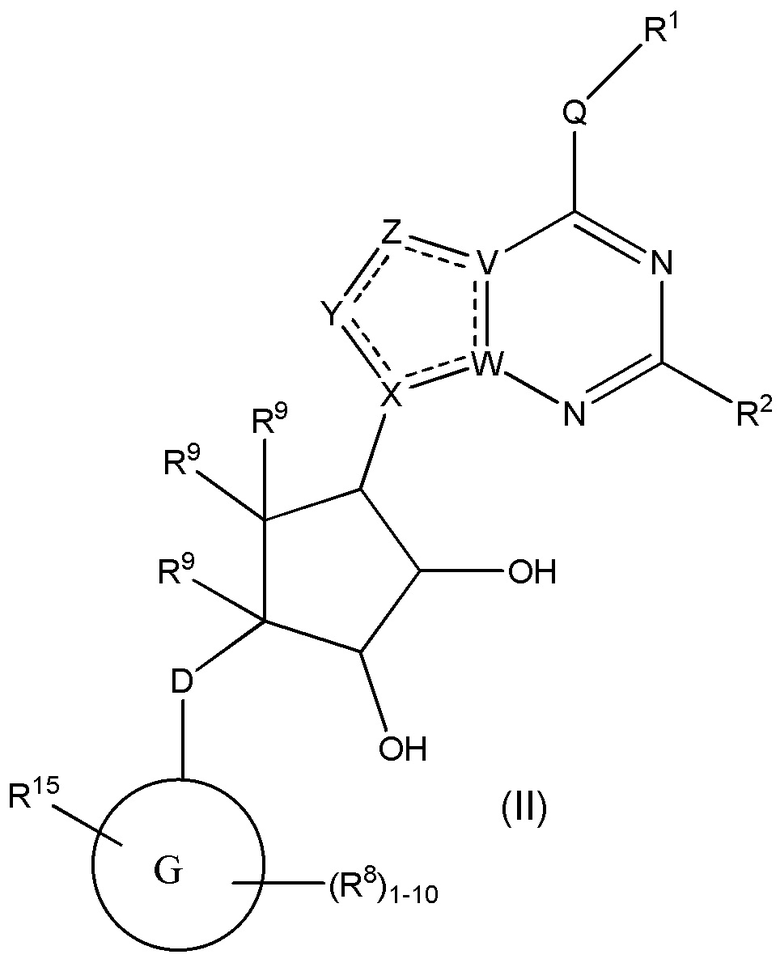
или его фармацевтически приемлемая соль, где:
R1 выбирают из группы, включающей (С1-С8)алкил и N(R4)2, где каждый R4 независимо является A-R14, где А отсутствует, и R14 является водородом;
R2 является водородом, (С1-С8)алкилом;
каждый R3 независимо выбирают из водорода, гидрокси;
D является C(R3)2, О;
G является фенилом;
R15 является 1-2 гетероатомами с 1-2 гетероатомами, выбранными из азота и кислорода, гетероалкилом, имеющим 2-4 атома, связанным с атомом на, или R15 является гетероалкиленом, связанным с атомом на G, необязательно замещенным 1-2 R8, и связан с соседним атомом на G (т.е. два конца R15, если R15 является гетероалкиленом, связаны с соседними атомами углерода на G кольце);
каждый R8 выбирают из группы, включающей (С1-С8)алкил, (С1-С8)галоалкил, гидрокси, (C1-С8)алкокси, 5-членный гетероарил 2-3 гетероатомами, выбранными из N, О и S, N(R4)2, CN, галоген, и где два R8 необязательно объединены с образованием карбонила;
где каждый R4 независимо является A-R14, где А отсутствует, и R14 является водородом;
Q отсутствует;
V является С, где если V образует двойную связь, V является углеродом;
W является С, где если W образует двойную связь, W является углеродом;
X является N;
Y является CR10, где каждый R10 независимо выбирают из водорода;
Z является CR12, где каждый R12 независимо выбирают из водорода, (С1-С8)алкила, или R12 необязательно выбирают из фтора, хлора, брома.
3. Соединение формулы (III):
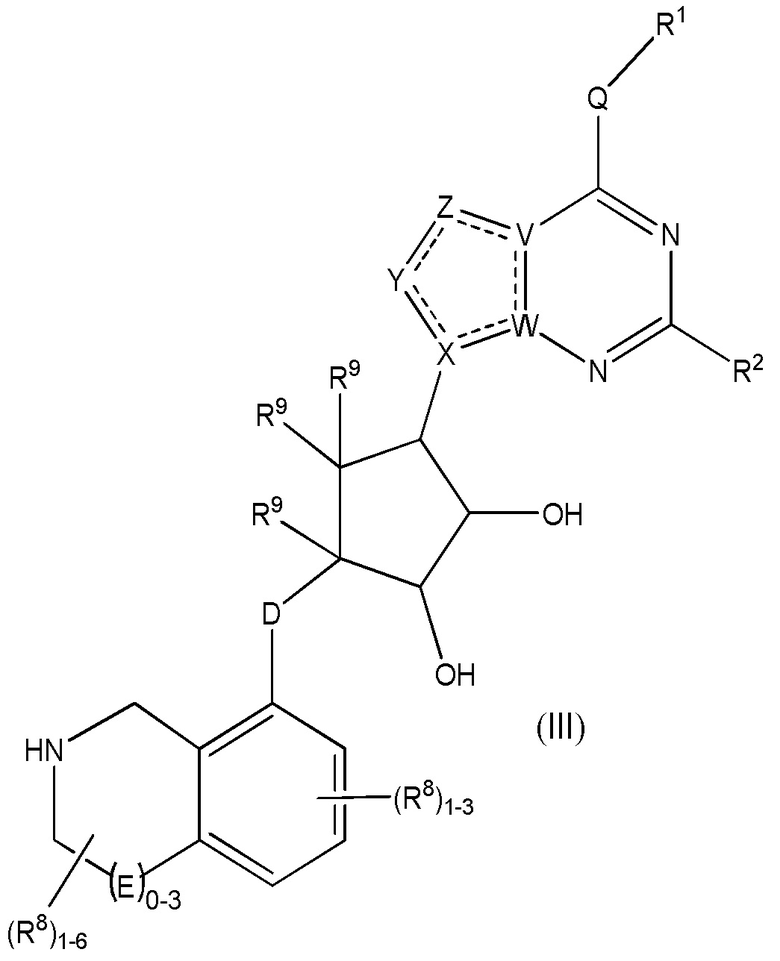
или его фармацевтически приемлемая соль, где:
каждый R1 независимо выбирают из группы, включающей (С1-С8)алкил, и N(R4)2, где каждый R4 независимо является A-R14, где А отсутствует, и R14 является водородом;
R2 является водородом, (С1-С8)алкилом;
каждый R3 независимо является водородом, гидрокси;
D является C(R3)2, О или S(О)1-2;
Е является СН2, C(R1)2, О;
каждый R8 отсутствует или его независимо выбирают из группы, включающей (С1-С8)алкил, (С1-С8)галоалкил, гидрокси, 5-членный гетероарил 2-3 гетероатомами, выбранными из N, О и S, N(R4)2, CN, и где два R8 необязательно объединены с образованием карбонила;
где каждый R4 независимо является A-R14, где А отсутствует, и R14 является водородом;
Q отсутствует;
V является С, где если V образует двойную связь, V является утлеродом;
W является С, где если W образует двойную связь, W является углеродом;
X является N;
Y является CR10, где каждый R10 независимо выбирают из водорода;
Z является CR12, где каждый R12 независимо выбирают из водорода, (С1-С8)алкила, фтора, хлора, брома.
4. Соединение формулы (IV):
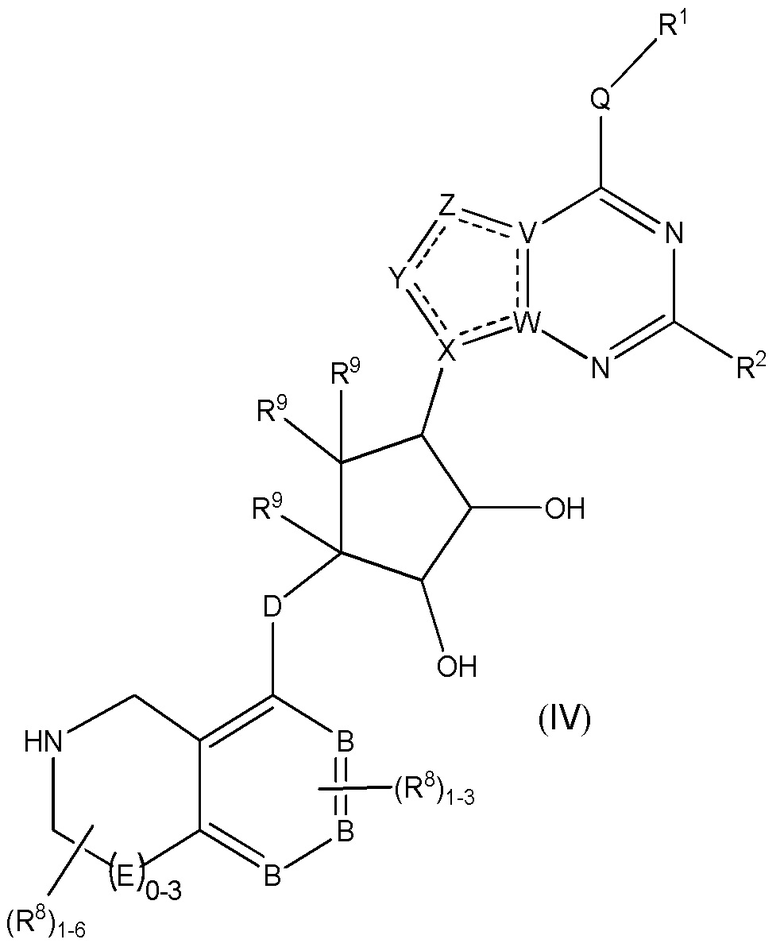
или его фармацевтически приемлемая соль, где:
каждый R1 независимо выбирают из группы, включающей (С1-С8)алкил, и N(R4)2, где каждый R4 независимо является A-R14, где А отсутствует, и R14 является водородом;
R2 является водородом, (С1-С8)алкилом;
D является C(R3)2, О или S(О)1-2;
каждый R3 независимо является водородом, гидрокси;
В является N или С;
Е является СН2, C(R1)2, О;
каждый R8 отсутствует или его независимо выбирают из группы, включающей (C1-С8)алкил, (С1-С8)галоалкил, гидрокси, (С1-С8)алкокси, 5-членный гетероарил 2-3 гетероатомами, выбранными из N, О и S, N(R4)2, CN и где два R8 необязательно объединены с образованием карбонила;
где каждый R4 независимо является A-R14, где А отсутствует, и R14 является водородом;
Q отсутствует;
V является С, где если V образует двойную связь, V является углеродом;
W является С, где если W образует двойную связь, W является углеродом;
X является N;
Y является CR10, где каждый R10 независимо выбирают из водорода;
Z является CR12, где каждый R12 независимо выбирают из водорода, (СС8)алкила, фтора, хлора, брома.
5. Соединение или фармацевтически приемлемая соль по любому из пп. 1-4, где
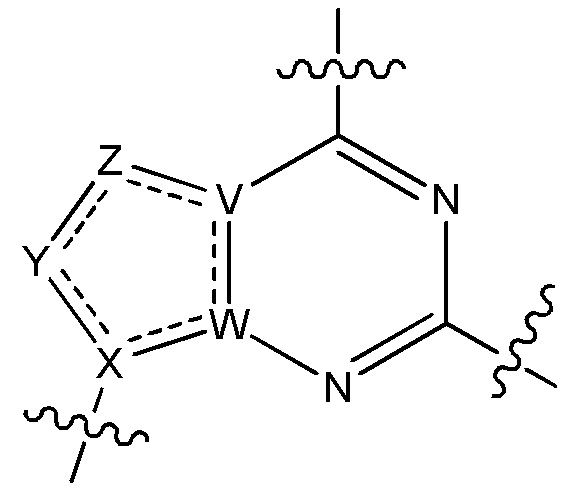
выбирают из:
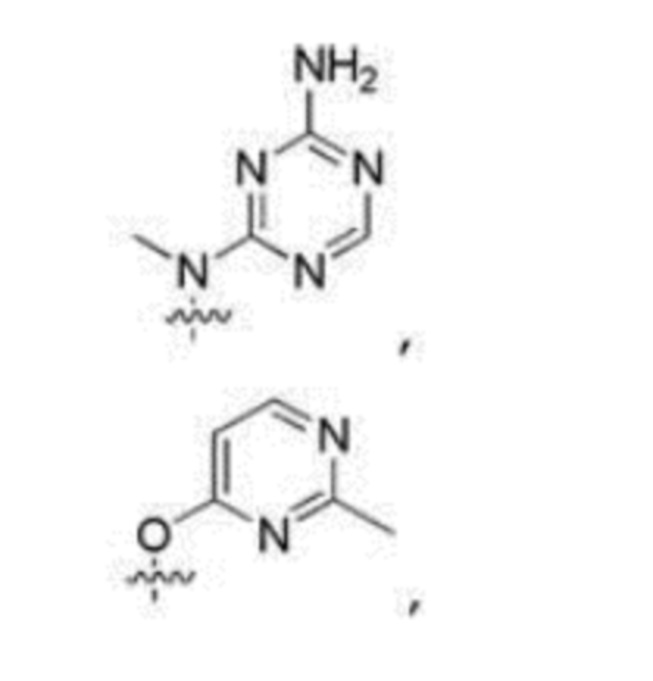
6. Соединение или фармацевтически приемлемая соль по пункту 5, где
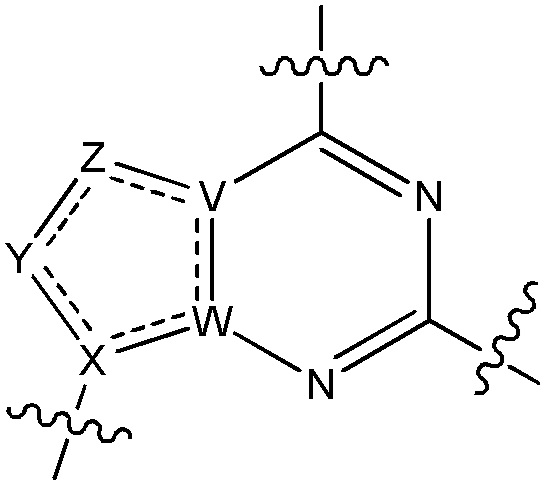
выбирают из:
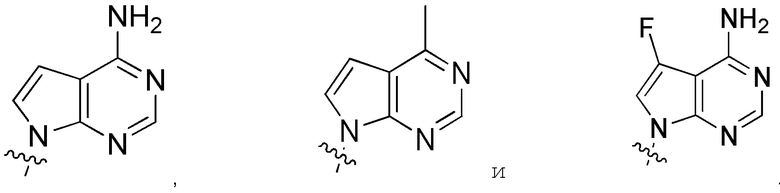
7. Соединение или фармацевтически приемлемая соль по пункту 3, где:
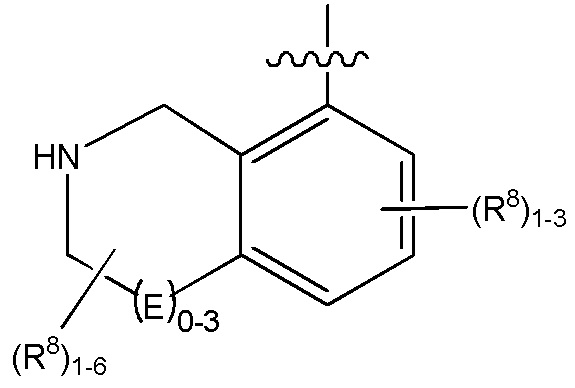
выбирают из:
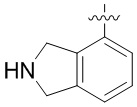 ,
,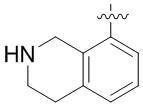 ,
,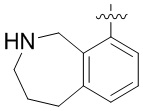 ,
,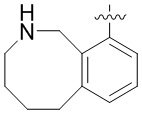 ,
,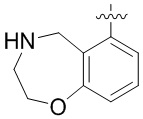 ,
,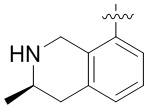 ,
,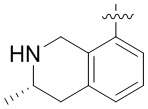 ,
,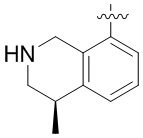 ,
,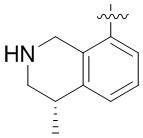 ,
,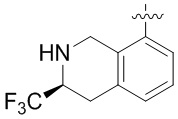 ,
,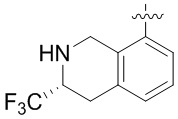 ,
,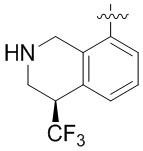 ,
,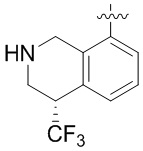 ,
,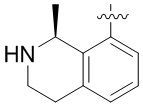 ,
,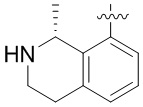 ,
,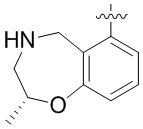 ,
,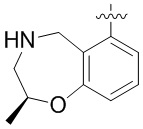 ,
,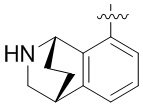 ,
,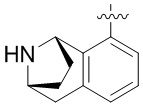 ,
,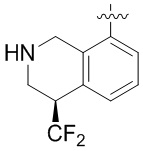 ,
,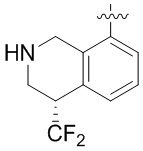 ,
,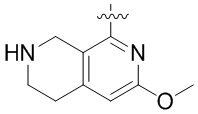 ,
,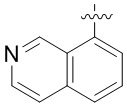 ,
,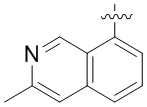 ,
,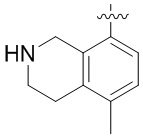 ,
,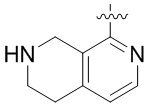 ,
,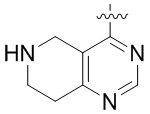 ,
,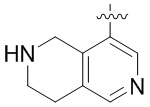 ,
,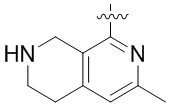 ,
,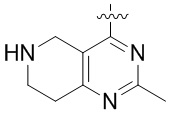 ,
,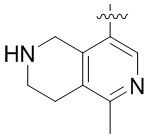 ,
,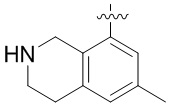 ,
,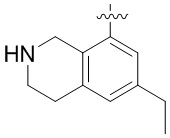 ,
,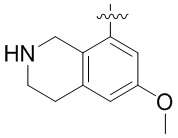 ,
,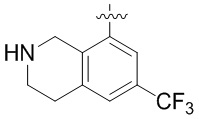 ,
,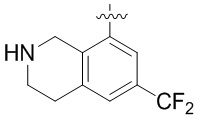
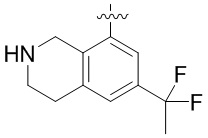 ,
,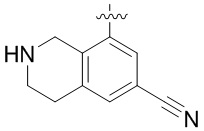 ,
,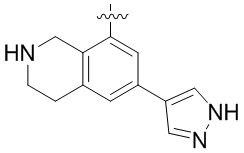 ,
,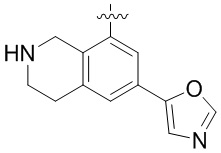 ,
,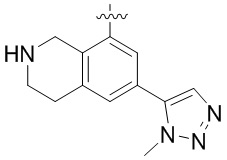 ,
,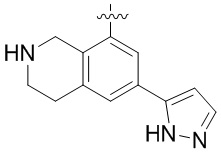 ,
,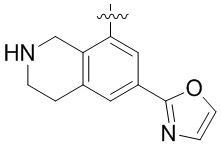 ,
,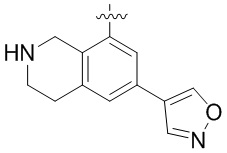 ,
,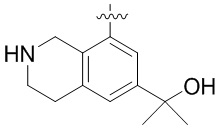 ,
,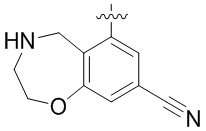 ,
,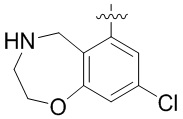
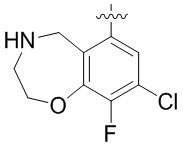 .
.
8. Соединение или фармацевтически приемлемая соль по пункту 7, где:
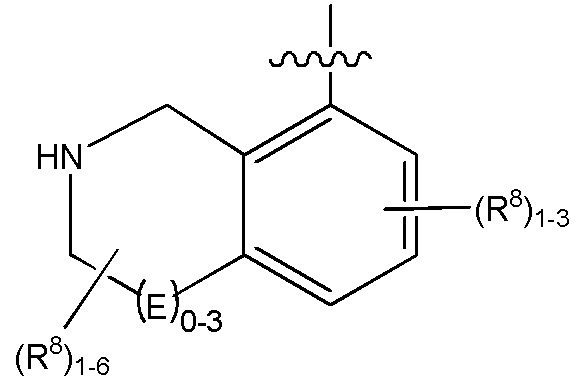
является:
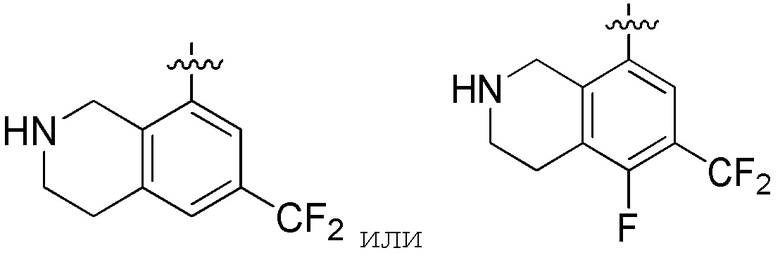 .
.
9. Соединение, выбранное из:
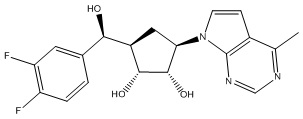 ,
,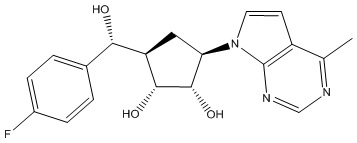 ,
,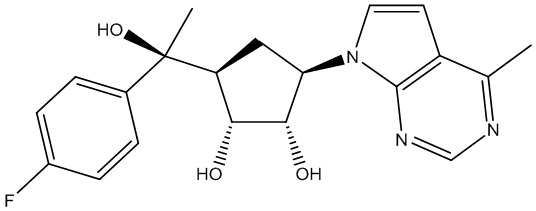 ,
,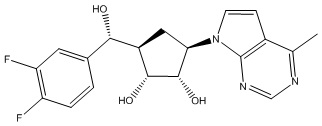 ,
,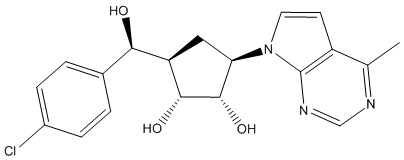 ,
,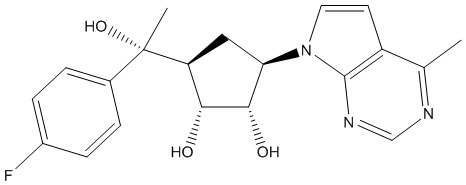 ,
,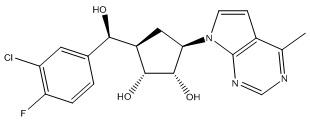 ,
,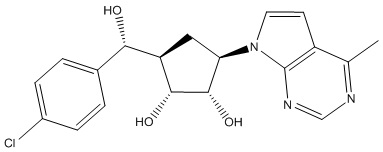 ,
,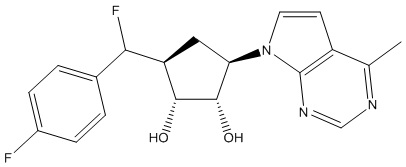 ,
,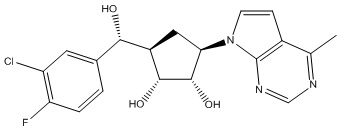 ,
,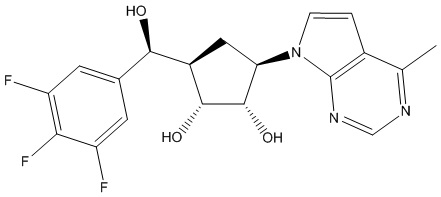 ,
,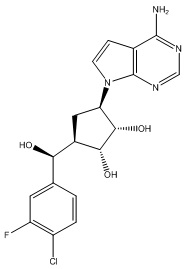 ,
,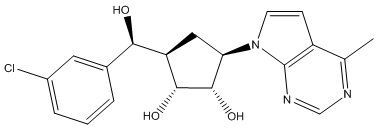 ,
,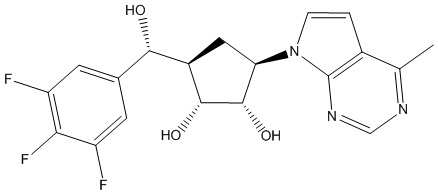 ,
,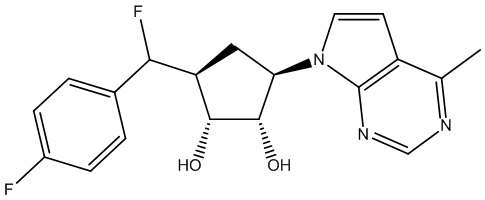 ,
,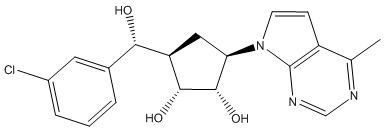 ,
,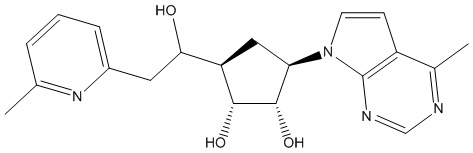 ,
,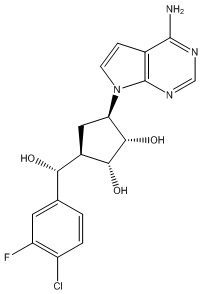 ,
,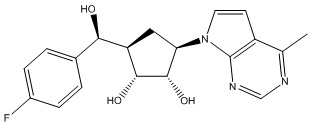 ,
,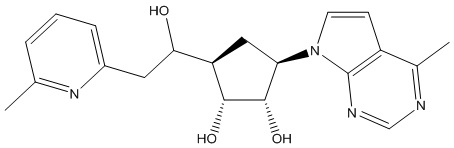 ,
,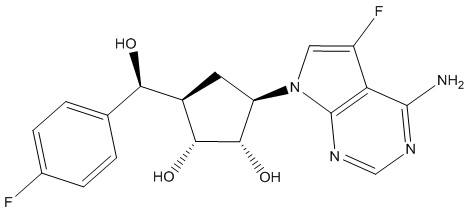 ,
,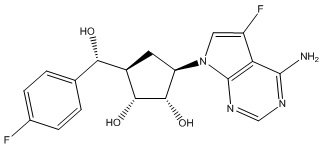 ,
,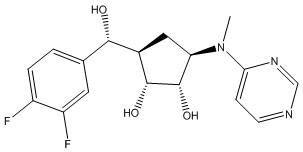 ,
,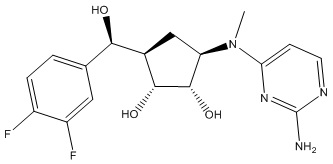 ,
,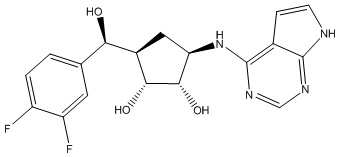 ,
,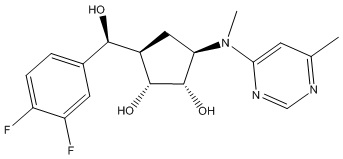 ,
,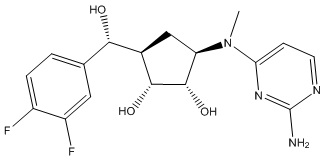 ,
,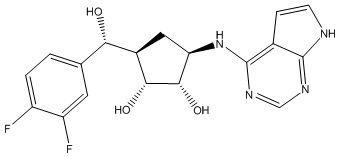 ,
,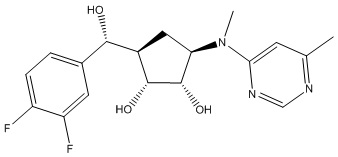 ,
,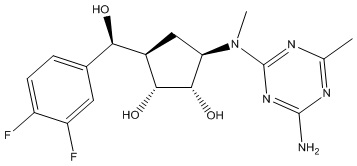 ,
,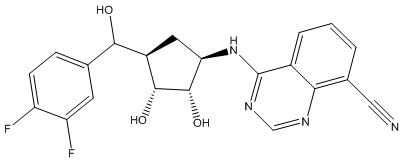 ,
,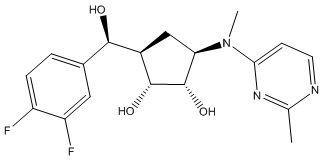 ,
,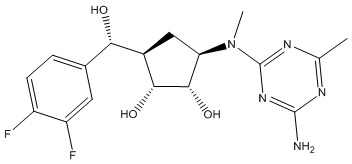 ,
,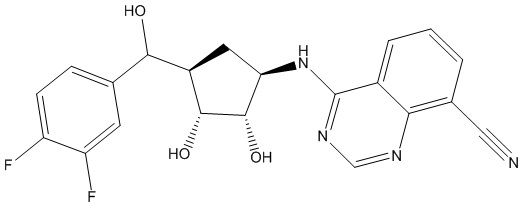 ,
,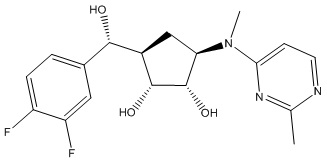 ,
,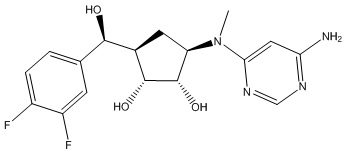 ,
,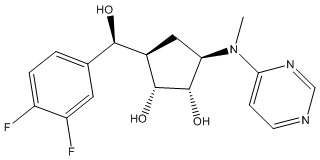 ,
,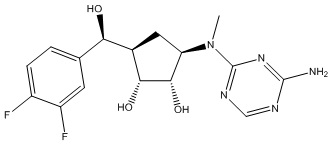 ,
,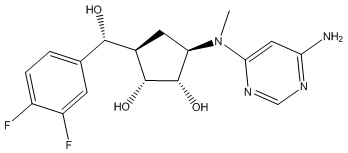 ,
,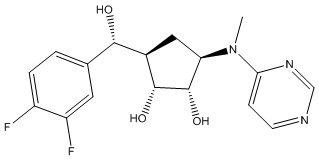 ,
,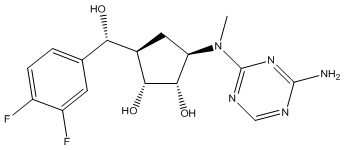 ,
,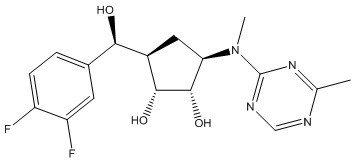 ,
,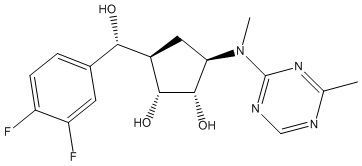 ,
,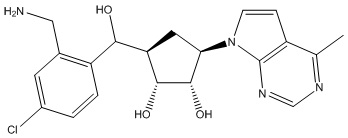 ,
,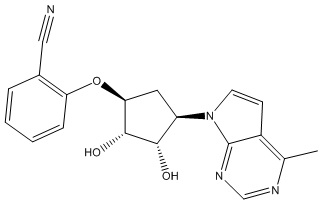 ,
,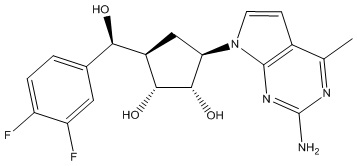 ,
,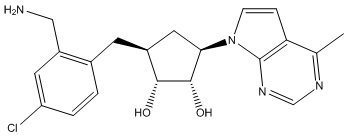 ,
,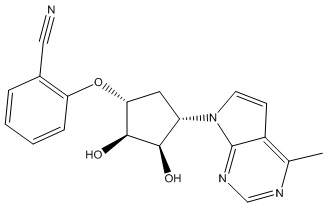 ,
,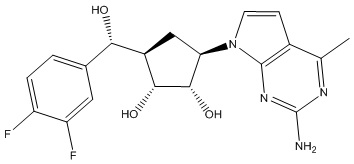 ,
,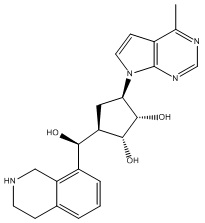 ,
,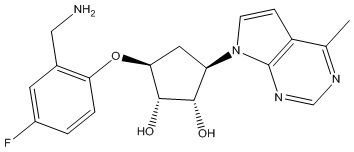 ,
,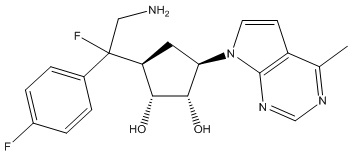 ,
,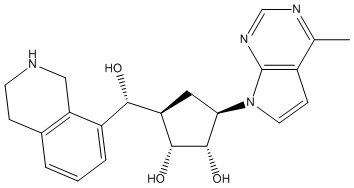 ,
,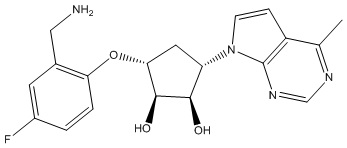
,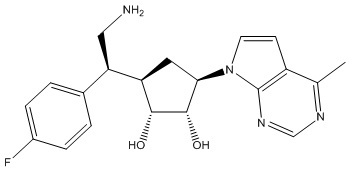 ,
,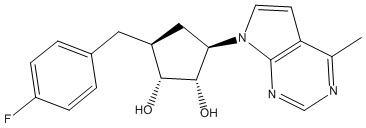 ,
,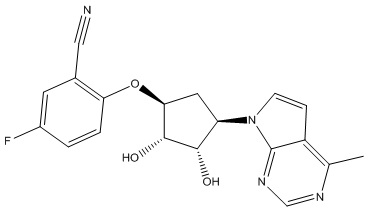 ,
,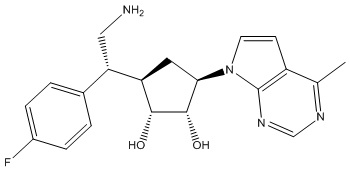 ,
,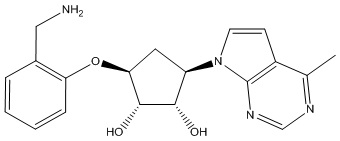 ,
,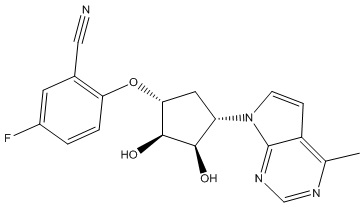 ,
,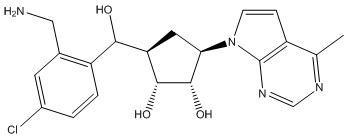 ,
,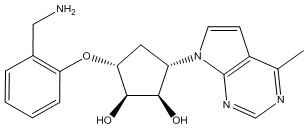 ,
,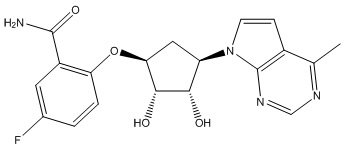 ,
,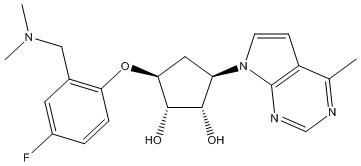 ,
,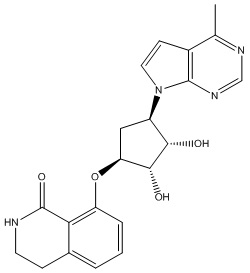 ,
,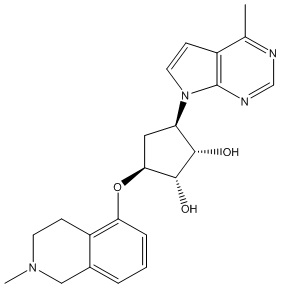 ,
,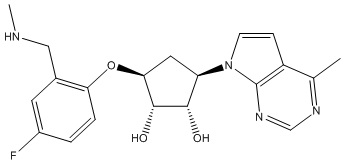
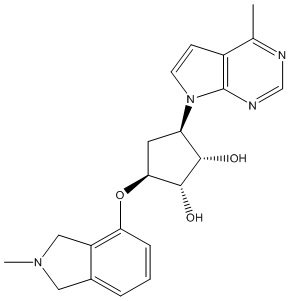
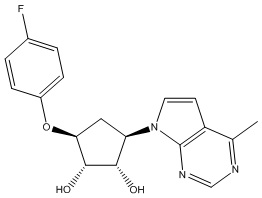 ,
,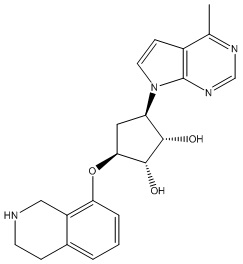 ,
,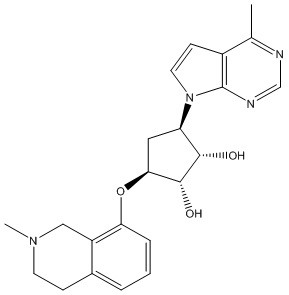 ,
,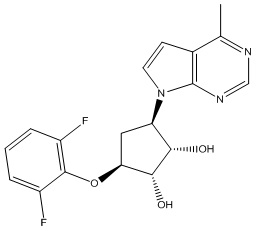 ,
,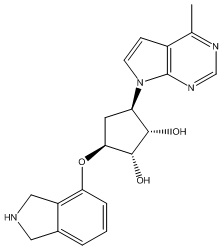 ,
,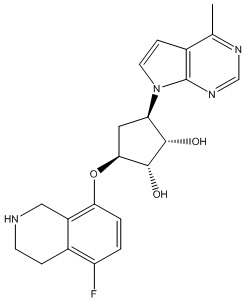 ,
,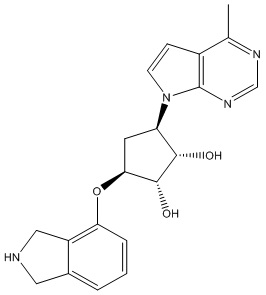 ,
,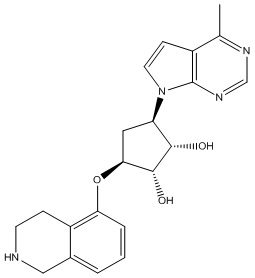 ,
,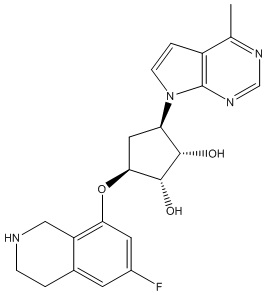 ,
,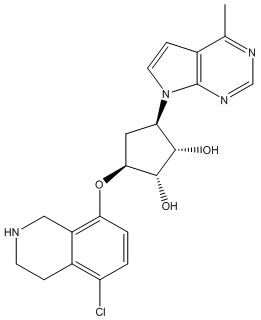 ,
,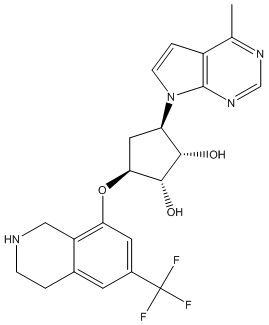 ,
,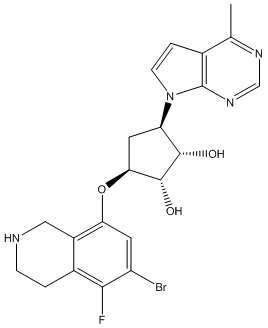 ,
,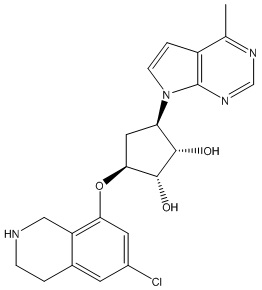 ,
,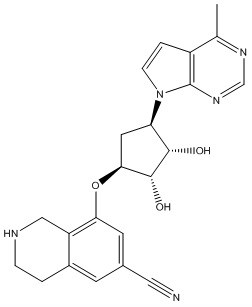 ,
,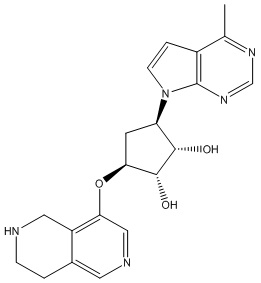 ,
,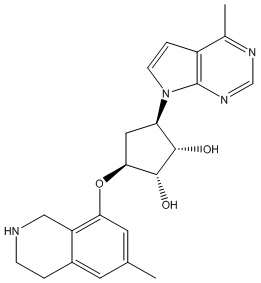 ,
,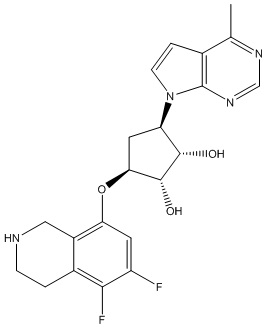 ,
,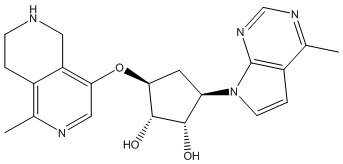 ,
,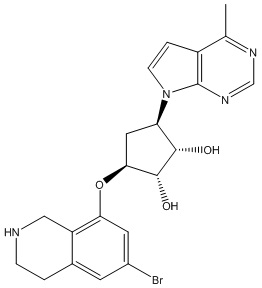 ,
,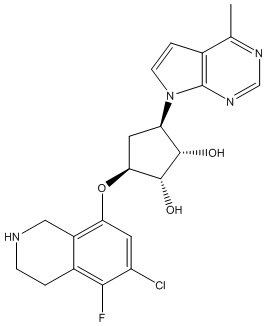 ,
,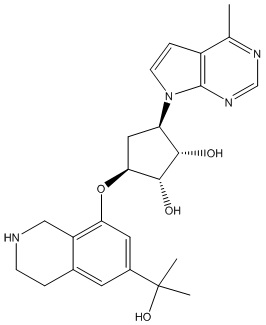 ,
,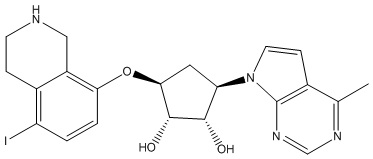 ,
,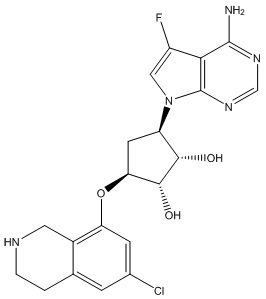 ,
,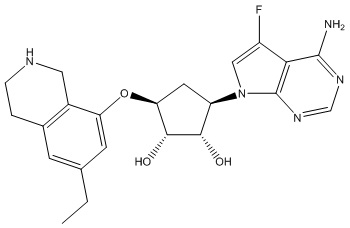 ,
,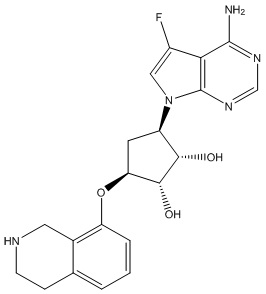 ,
,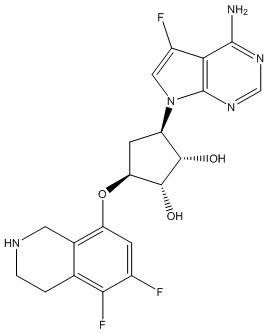 ,
,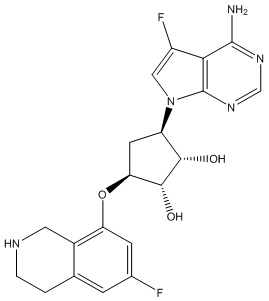 ,
,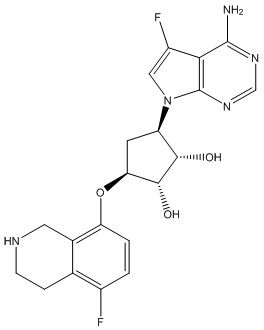 ,
,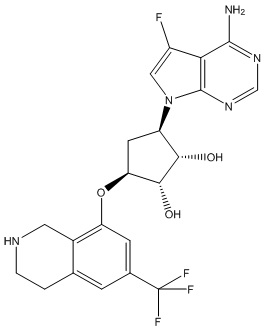 ,
,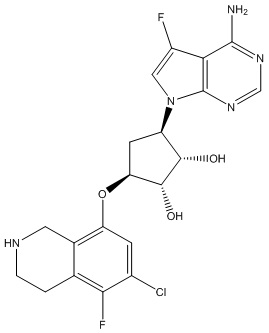 ,
,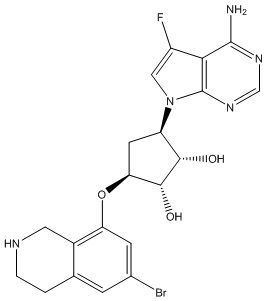 ,
,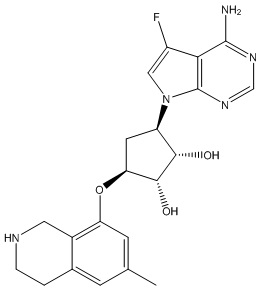 ,
,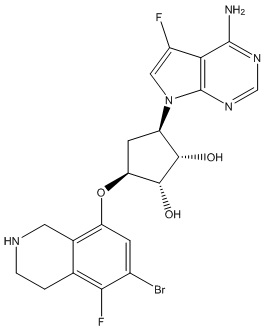 ,
,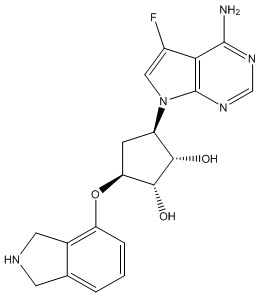 ,
,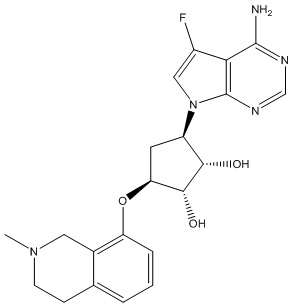 ,
,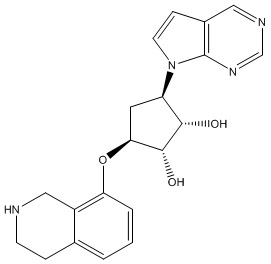 ,
,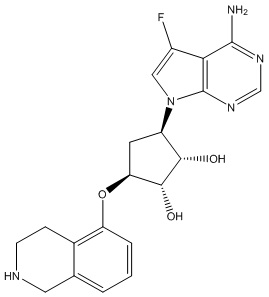 ,
,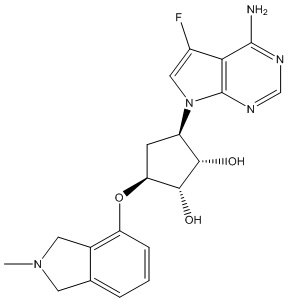 ,
,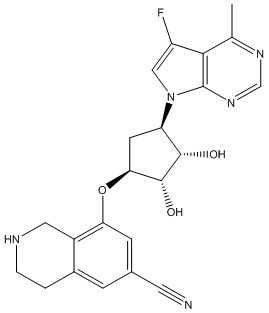 ,
,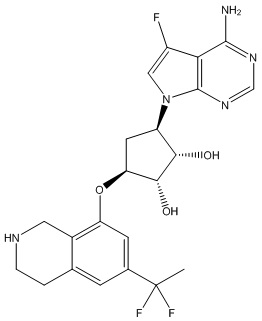 ,
,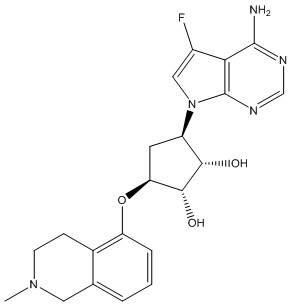 ,
,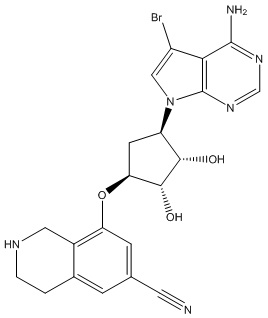 ,
,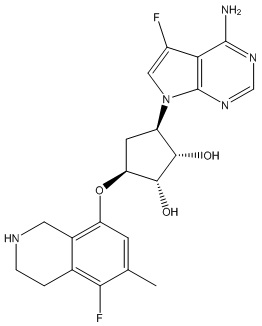 ,
,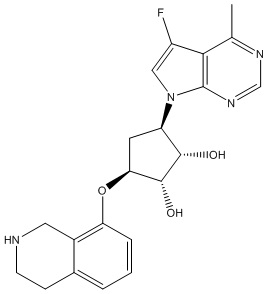 ,
,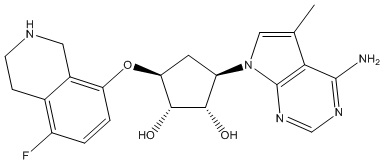 ,
,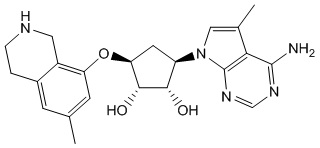 ,
,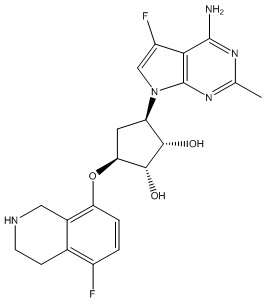 ,
,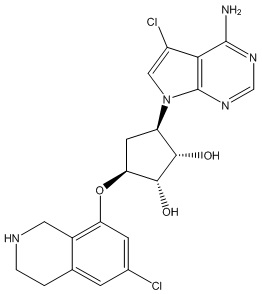 ,
,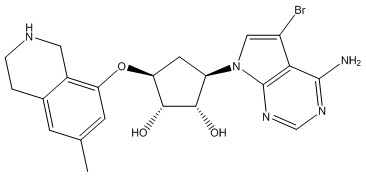 ,
,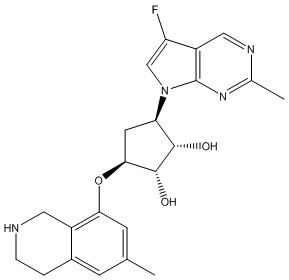 ,
,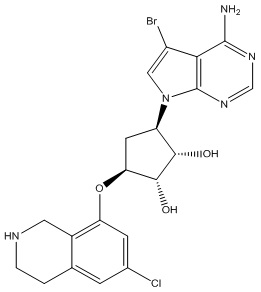 ,
,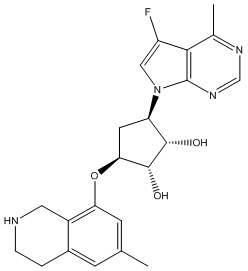 ,
,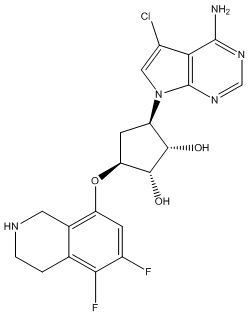 ,
,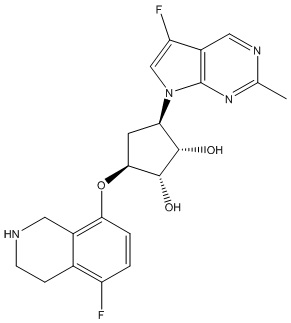 ,
,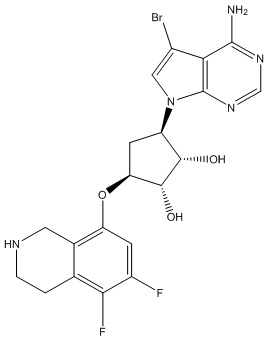 ,
,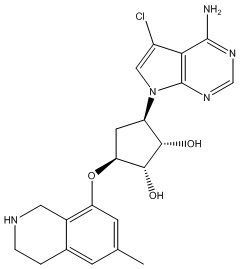 ,
,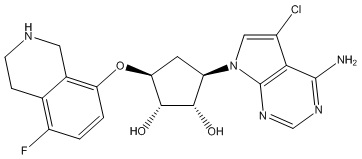 ,
,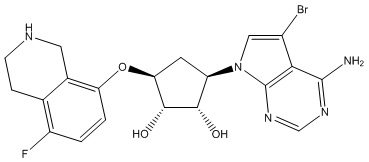 ,
,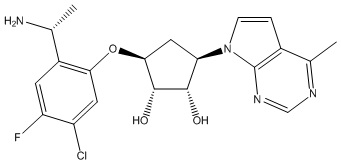 ,
,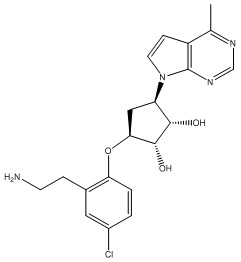 ,
,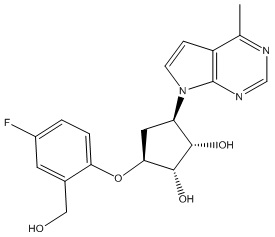 ,
,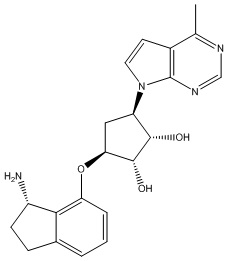 ,
,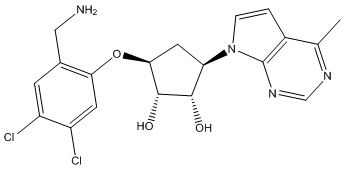 ,
,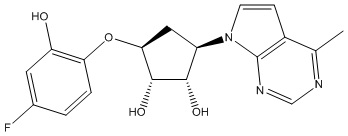 ,
,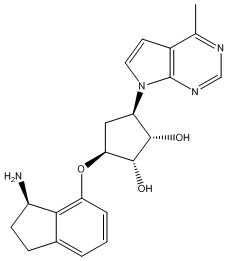 ,
,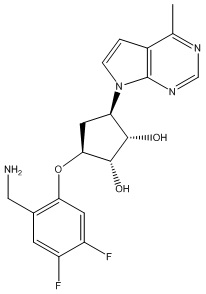 ,
,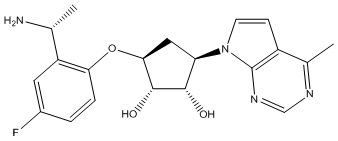 ,
,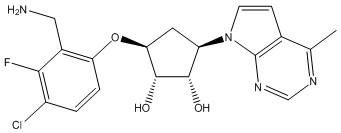 ,
,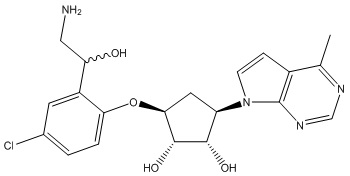 ,
,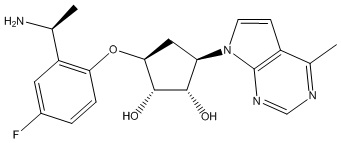 ,
,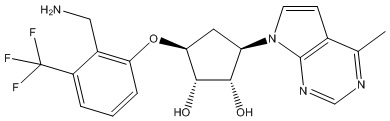 ,
,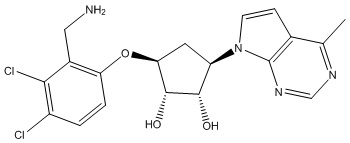 ,
,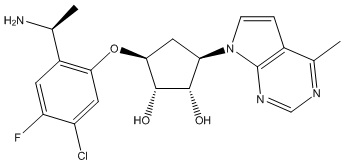 ,
,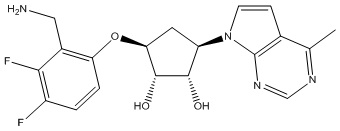 ,
,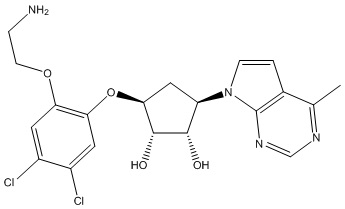 ,
,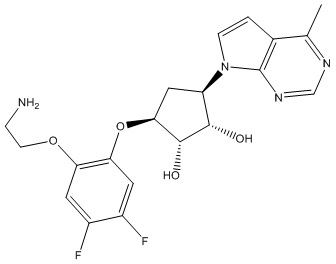 ,
,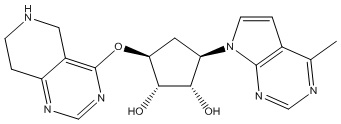 ,
,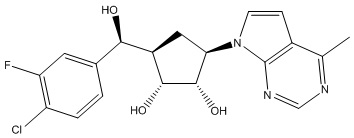 ,
,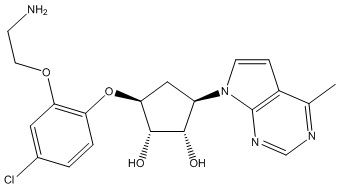 ,
,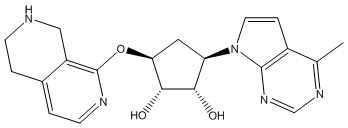 ,
,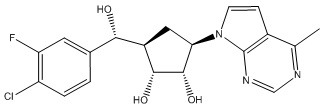 ,
,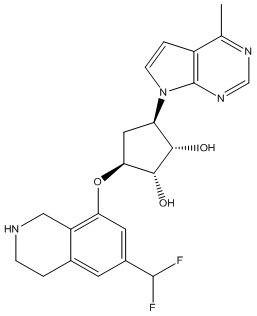 ,
,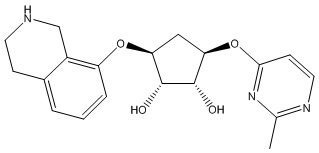 ,
,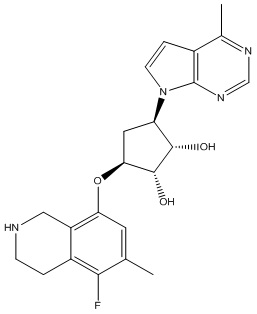 ,
,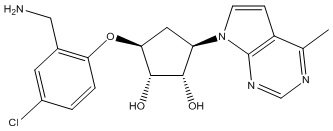 ,
,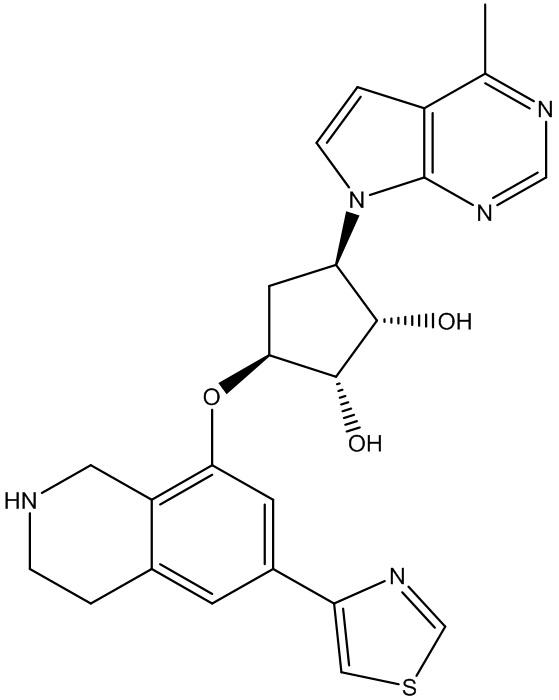 ,
,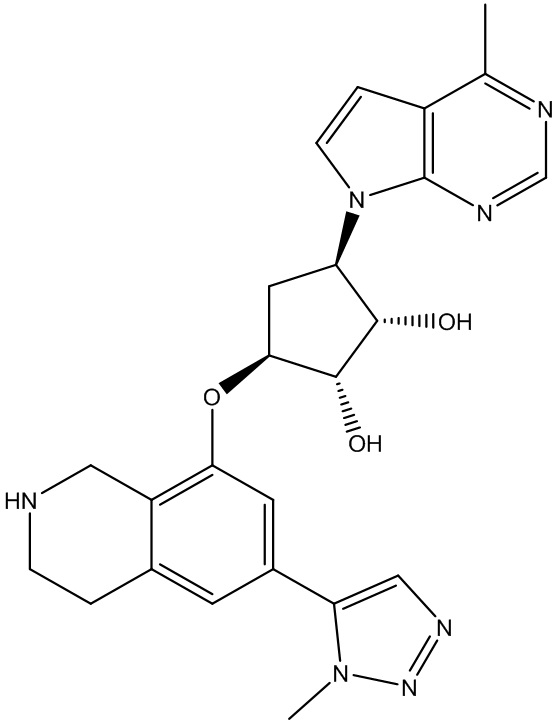 ,
,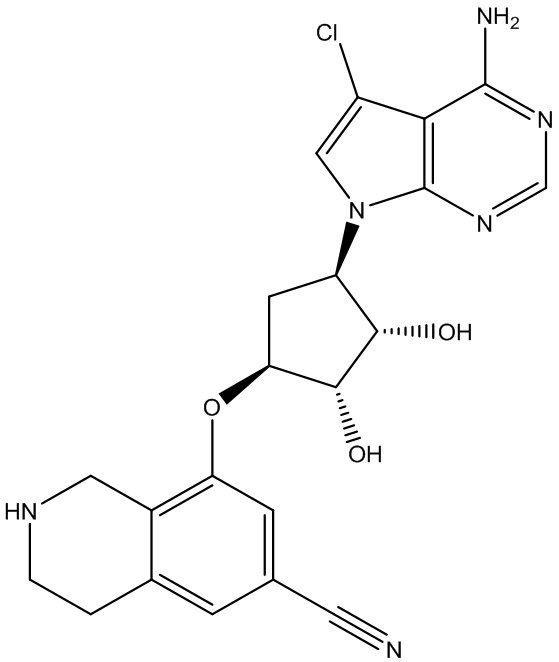 ,
,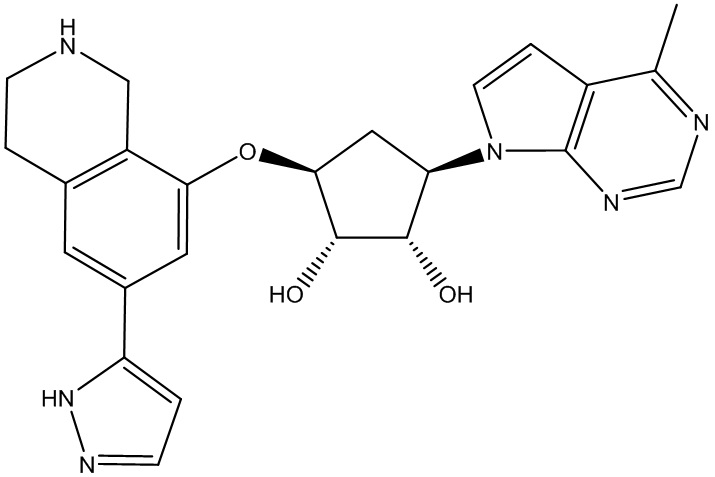 ,
,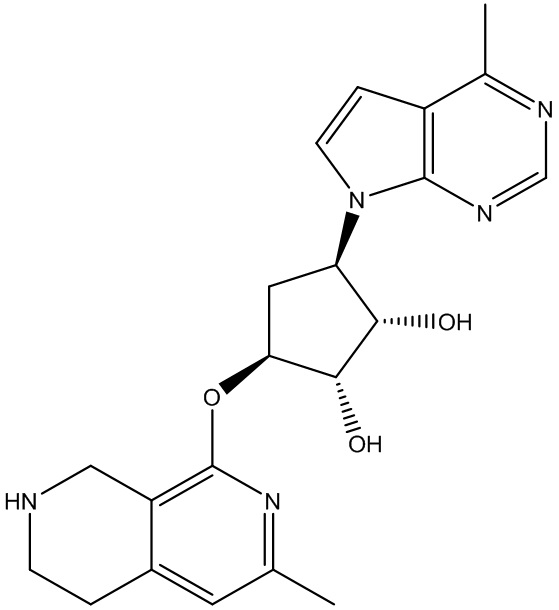 ,
,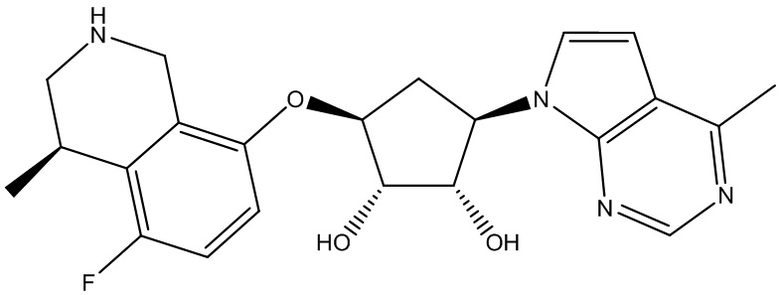 ,
,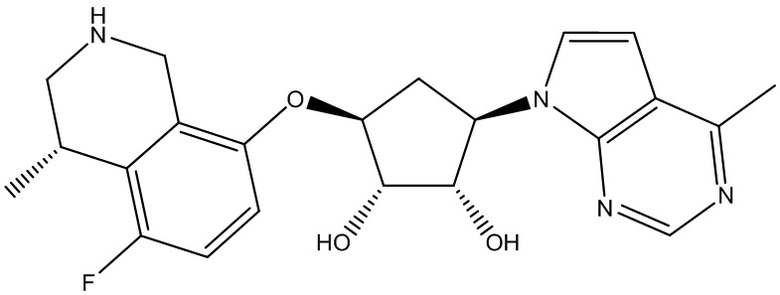 ,
,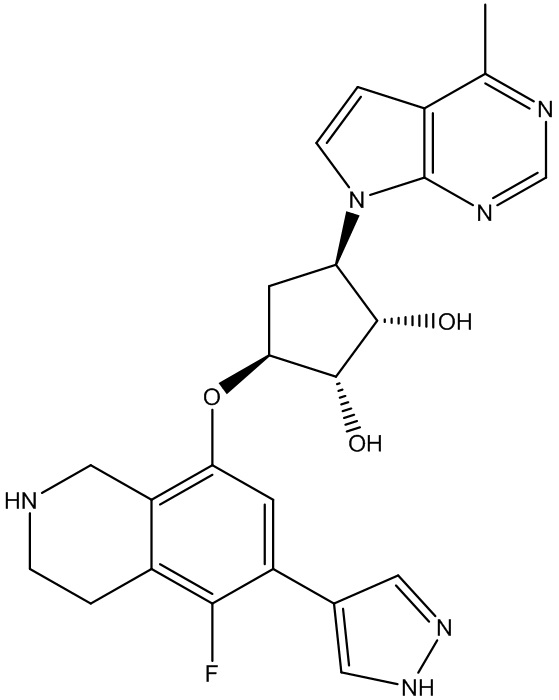 ,
,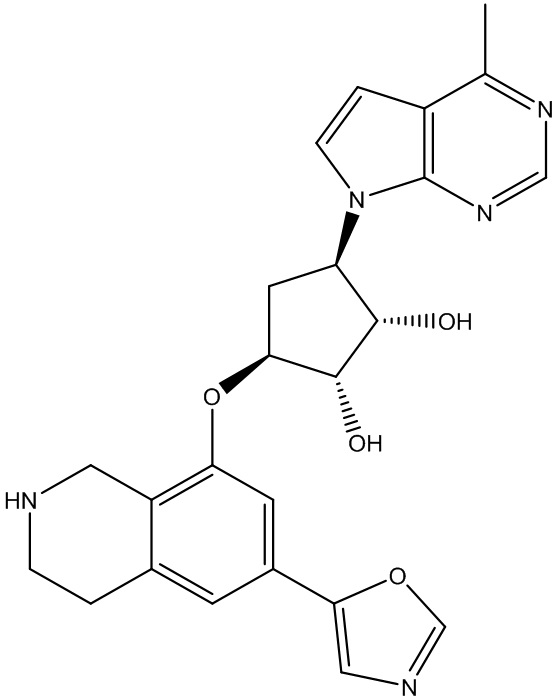 ,
,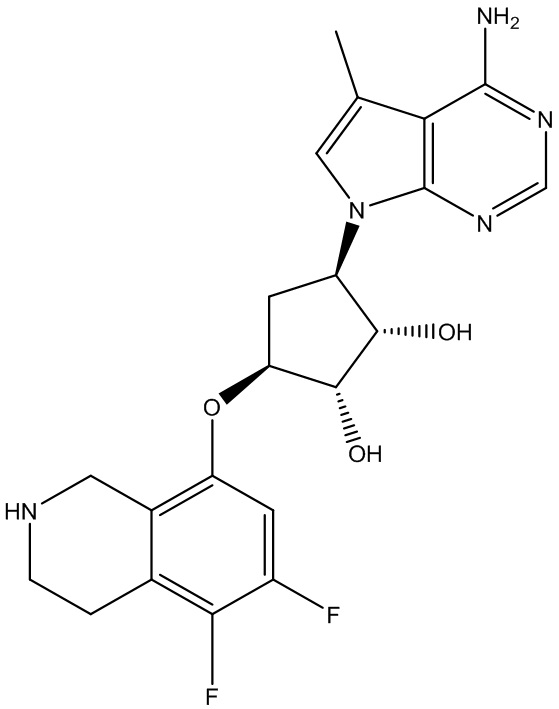 ,
,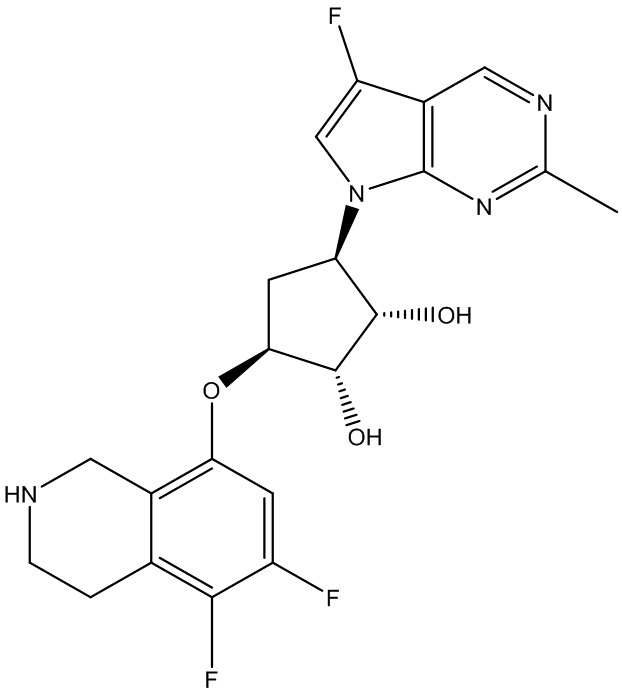 ,
,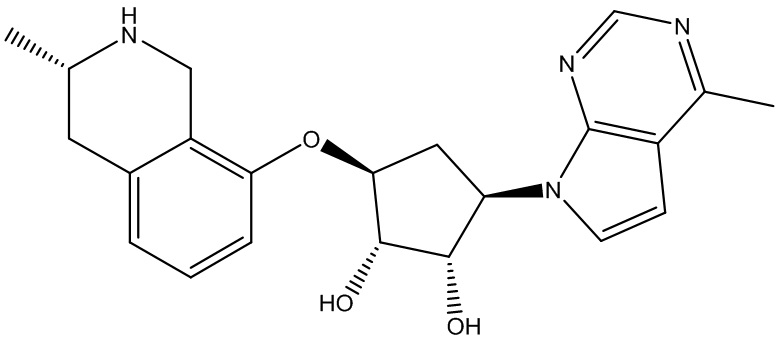 ,
,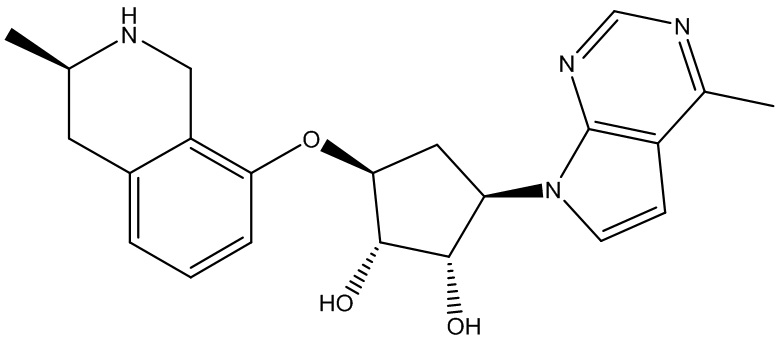 ,
,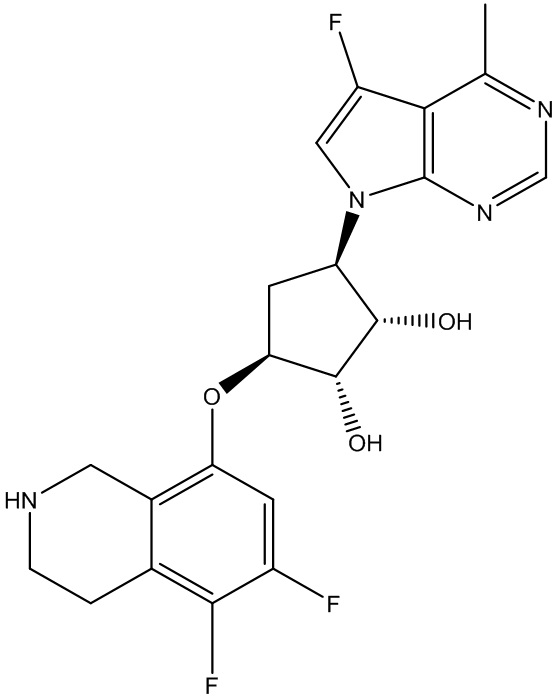 ,
,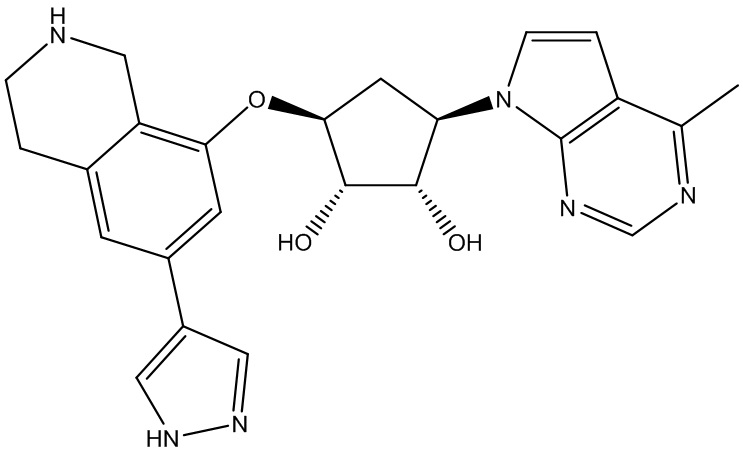 ,
,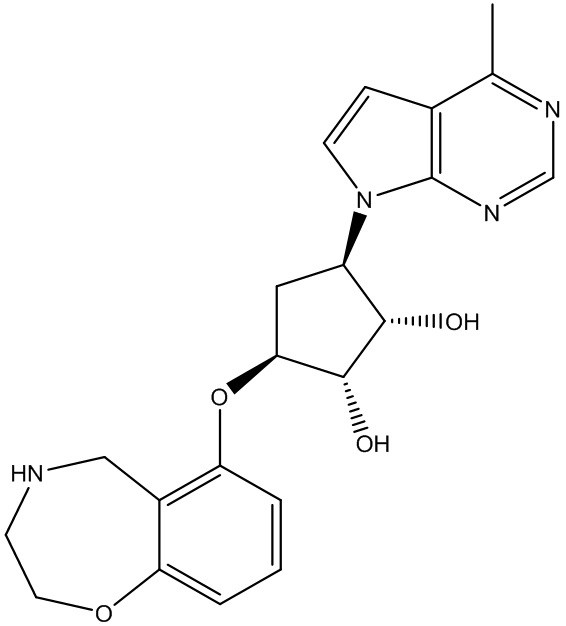 ,
,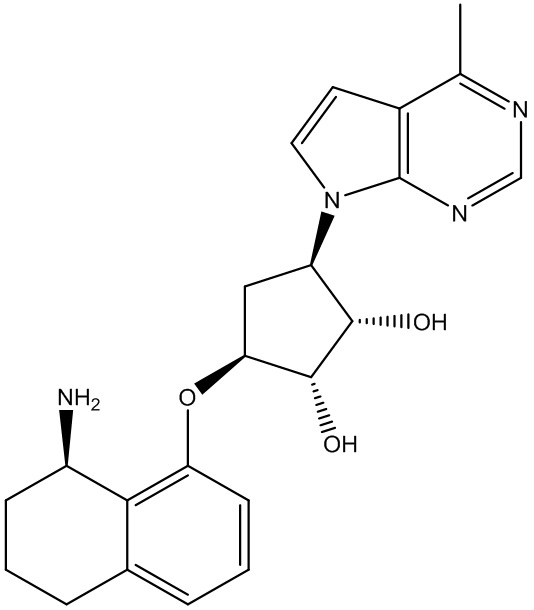 ,
,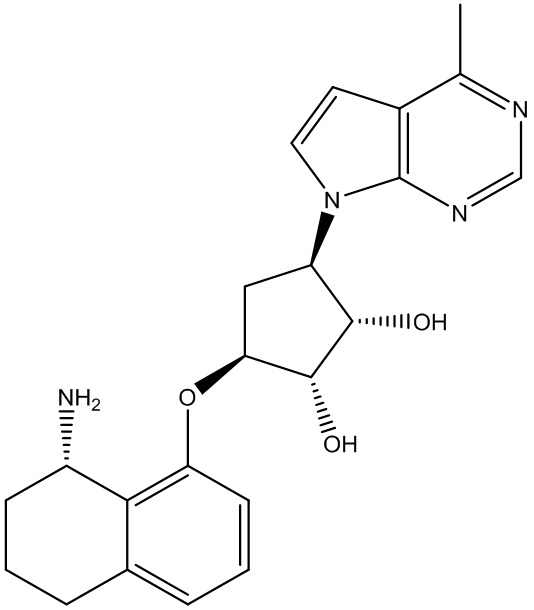 ,
,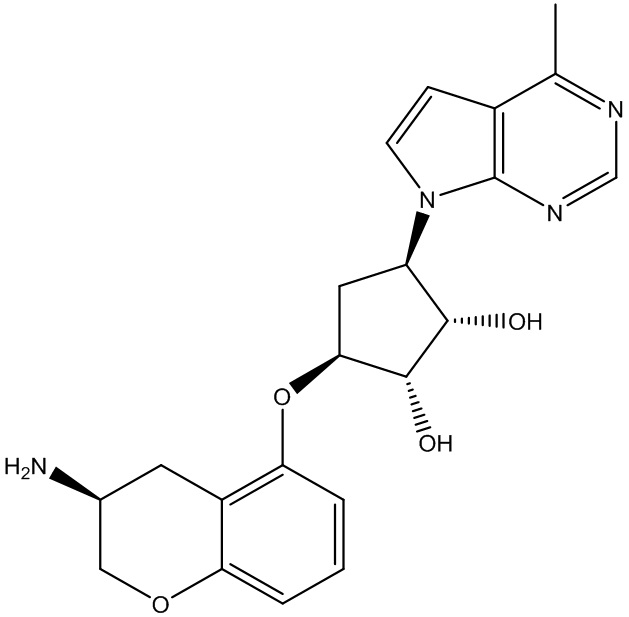 ,
,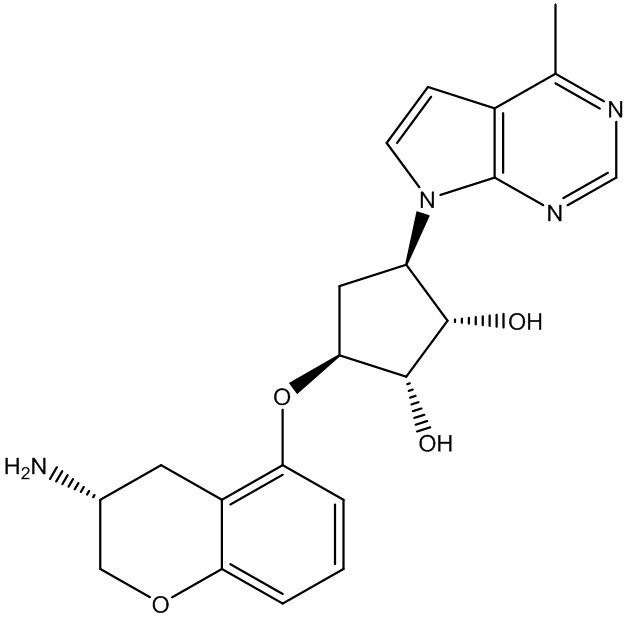 ,
,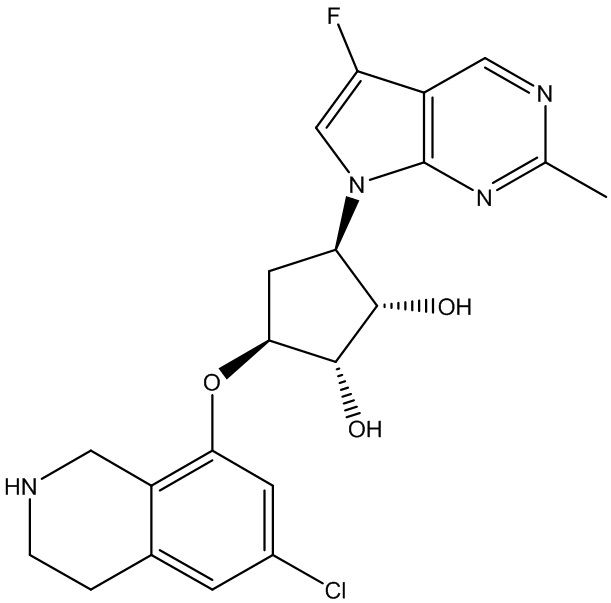 ,
,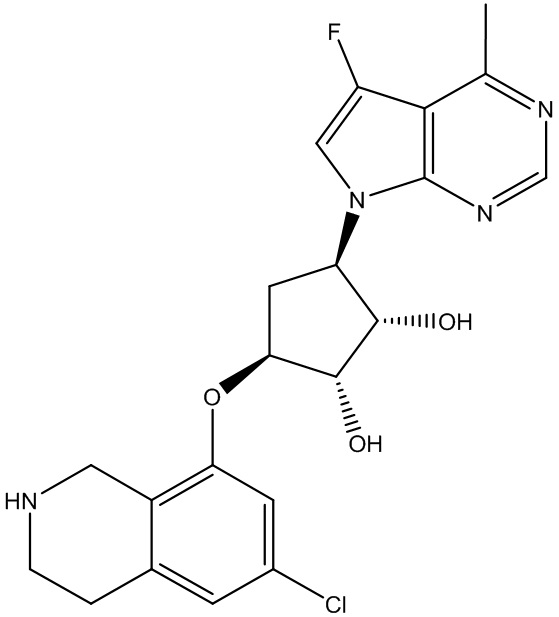 ,
,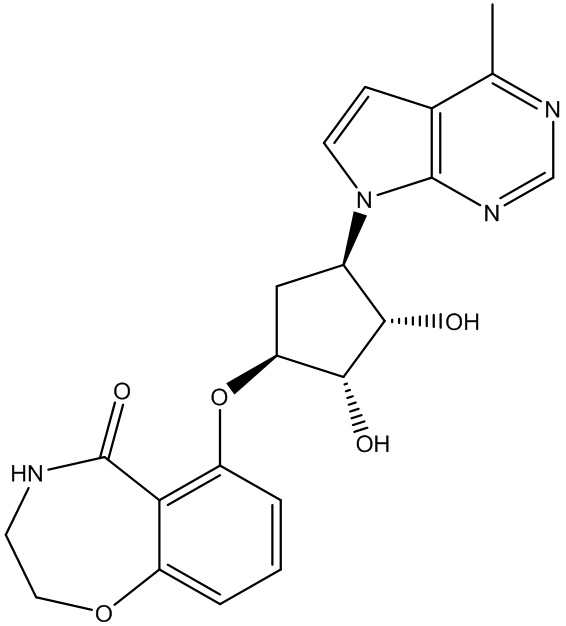 ,
,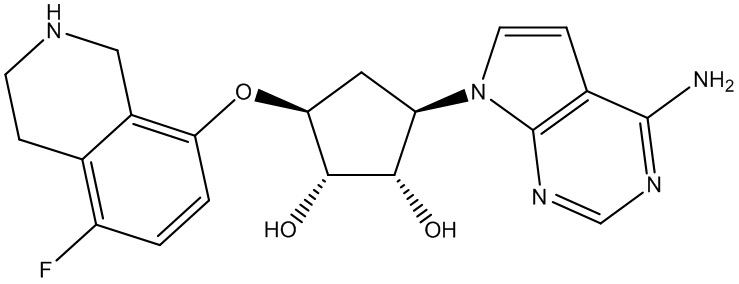
,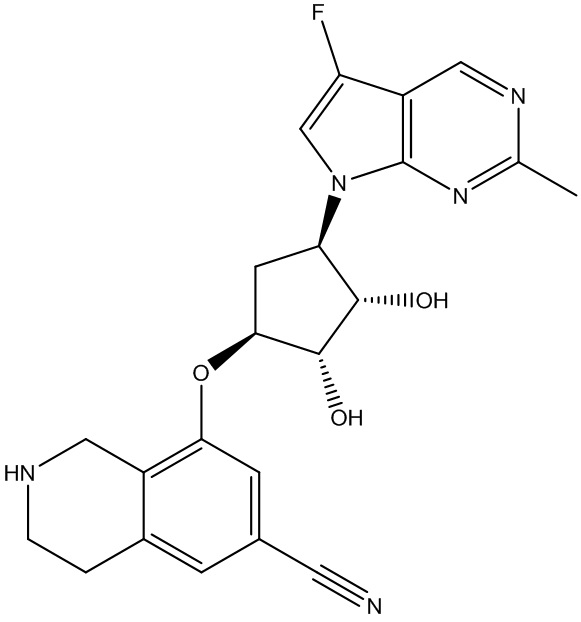 ,
,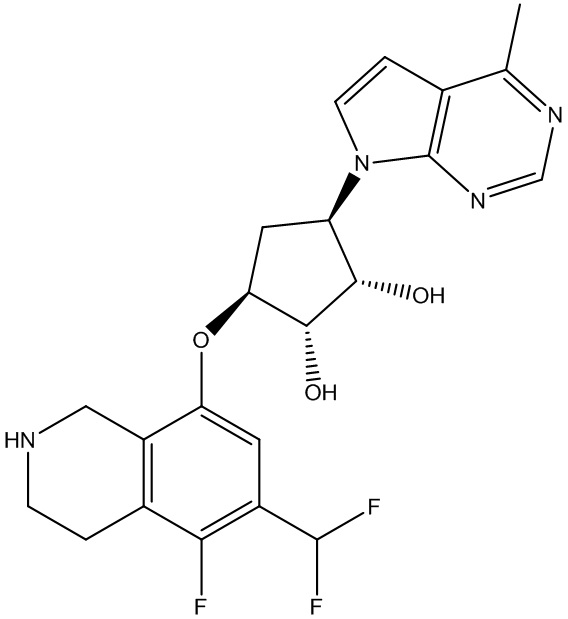 ,
,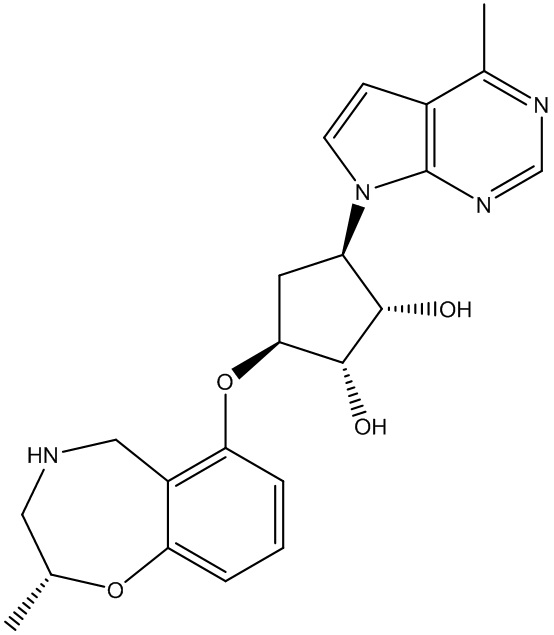 ,
,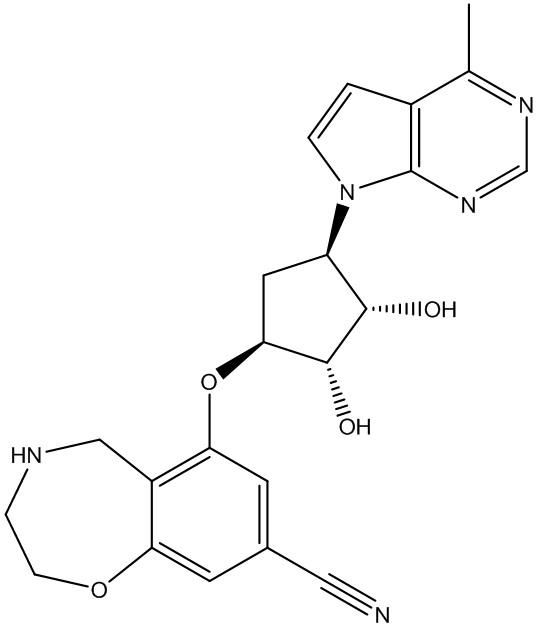 ,
,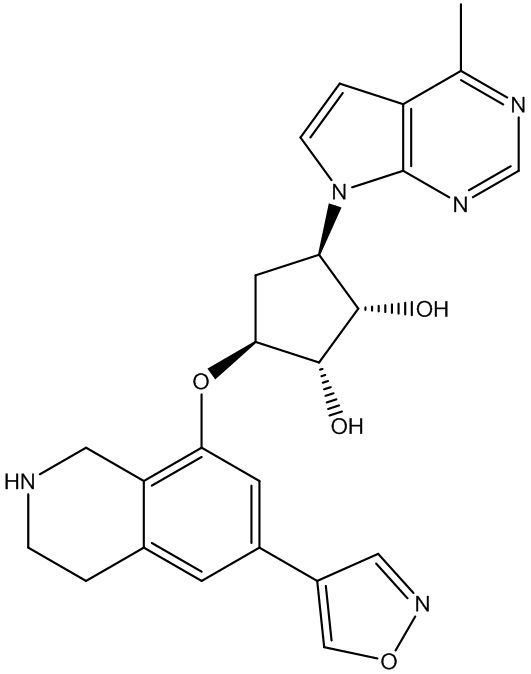 ,
,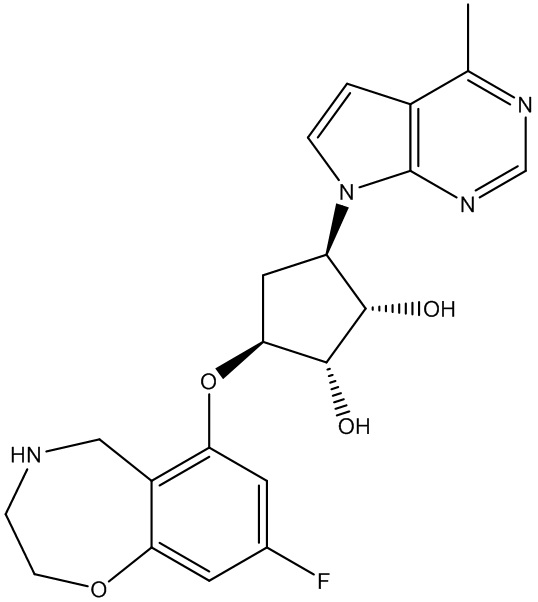 ,
,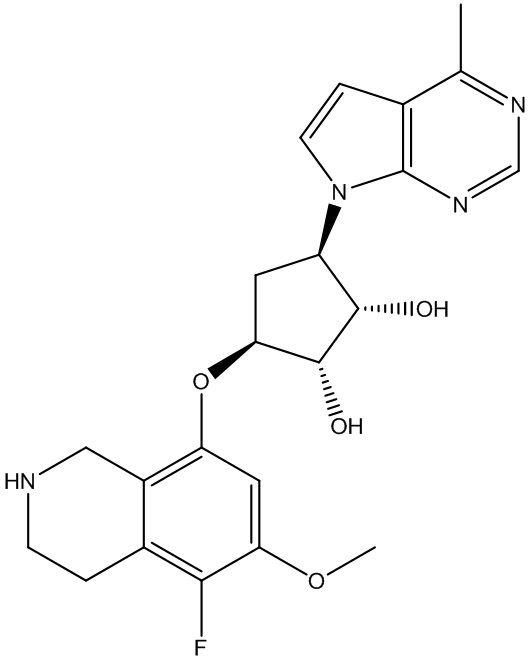 ,
,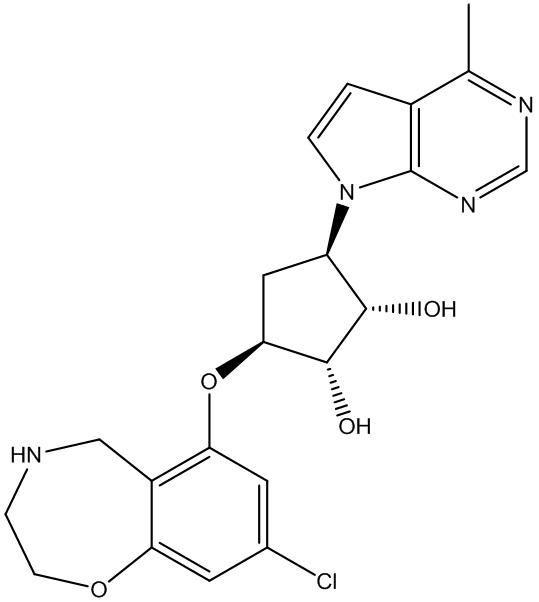 ,
,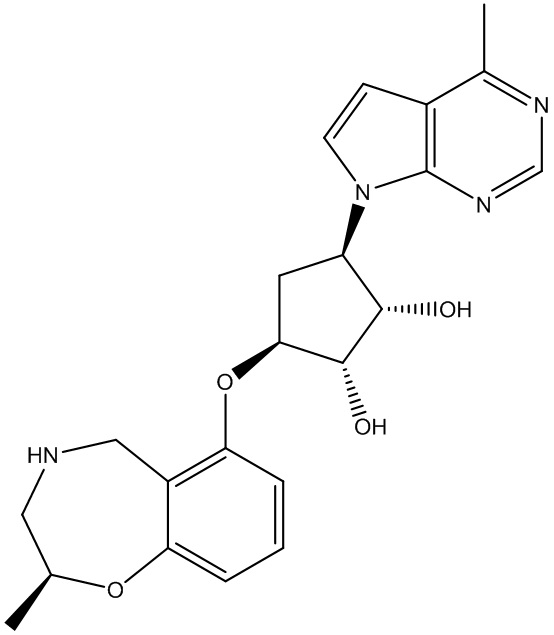 ,
,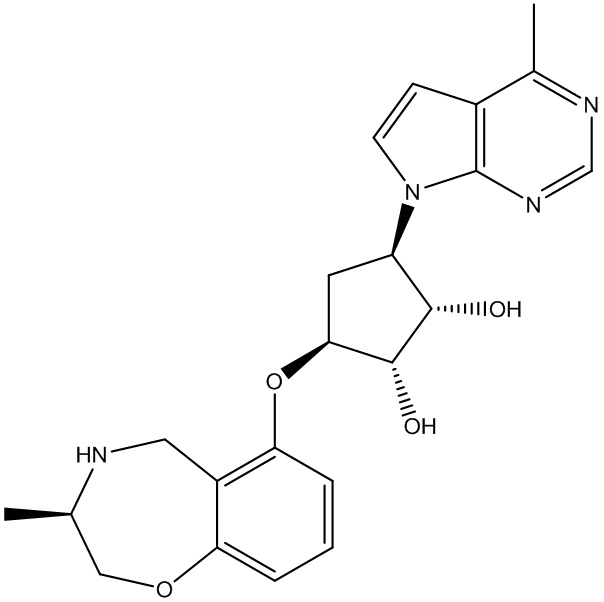 ,
,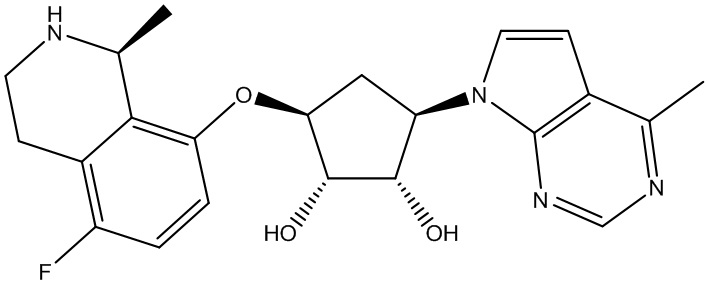 ,
,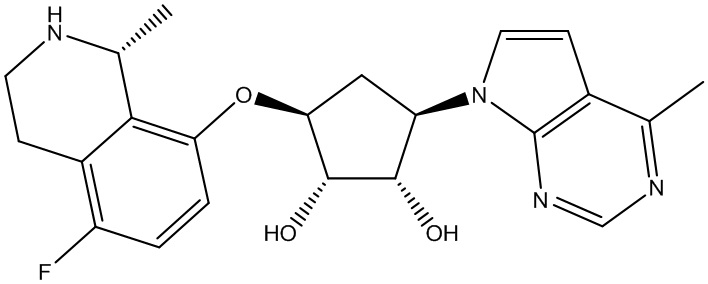 ,
,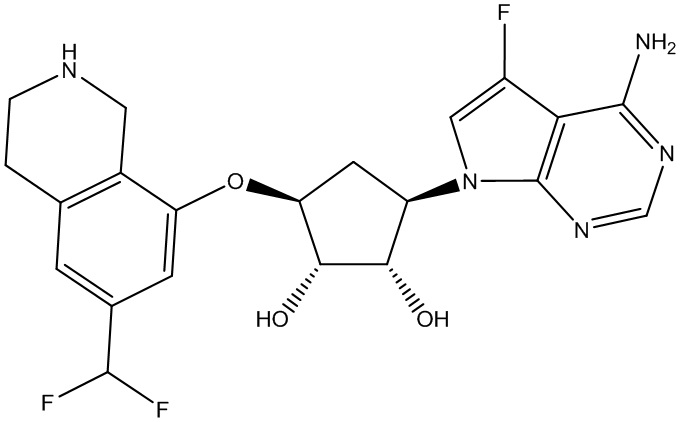 ,
,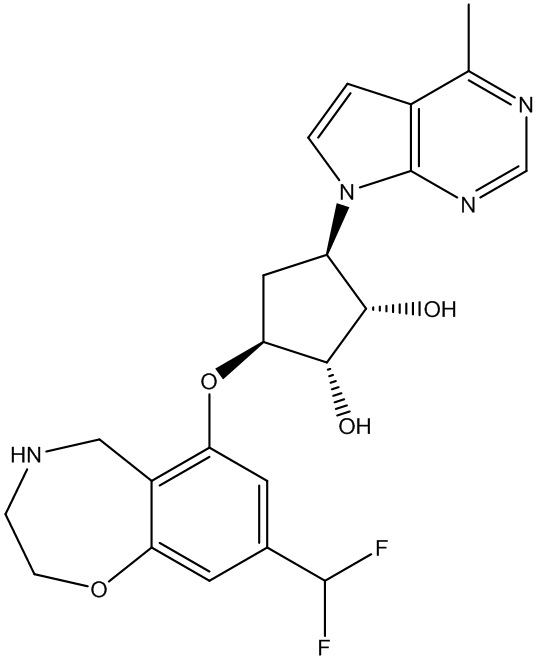 ,
,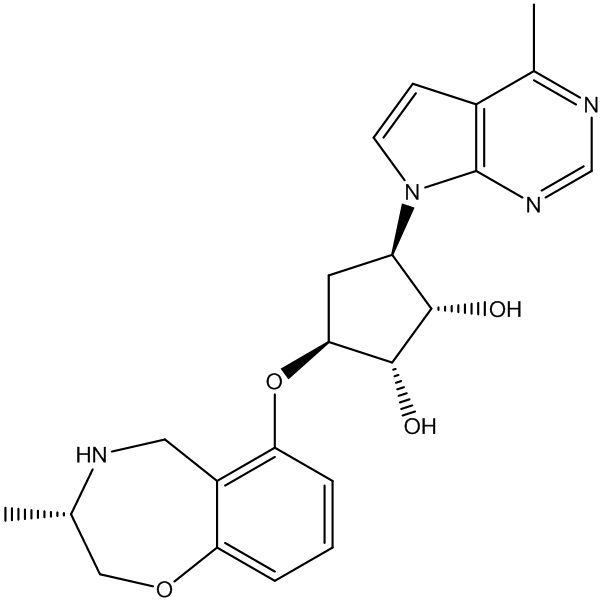 ,
,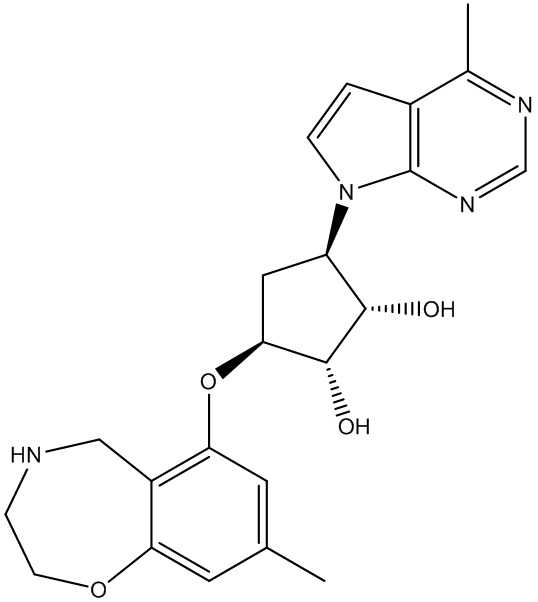 ,
,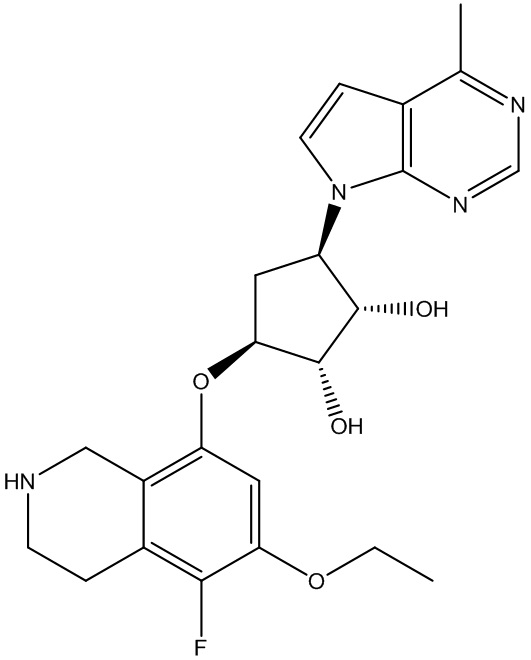 ,
,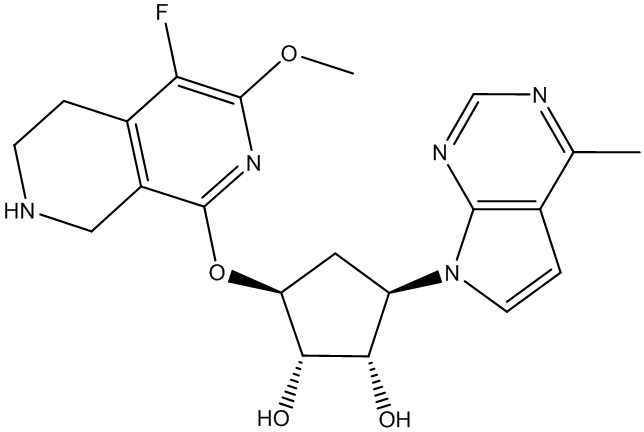 ,
,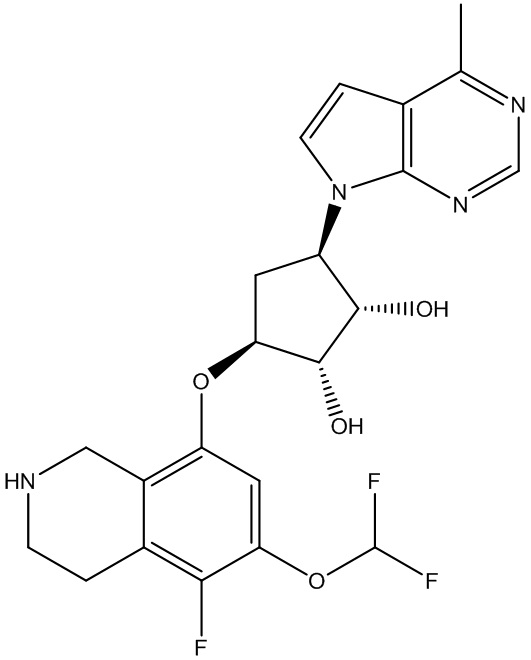 ,
,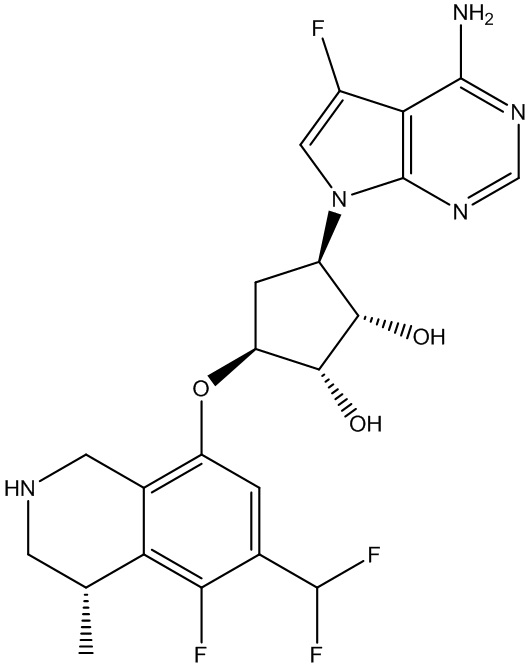 ,
,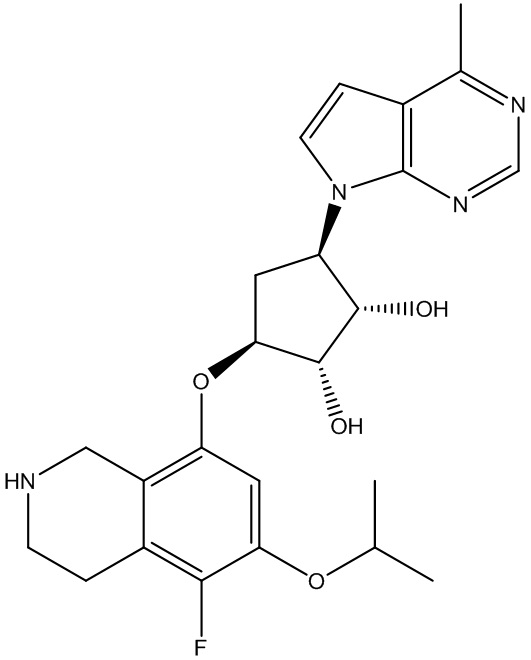 ,
,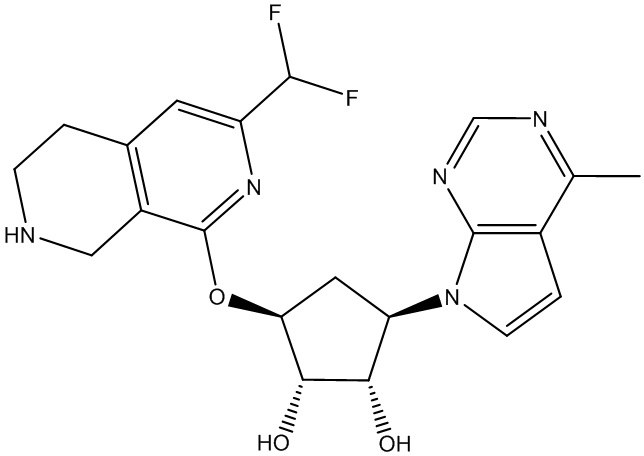 ,
,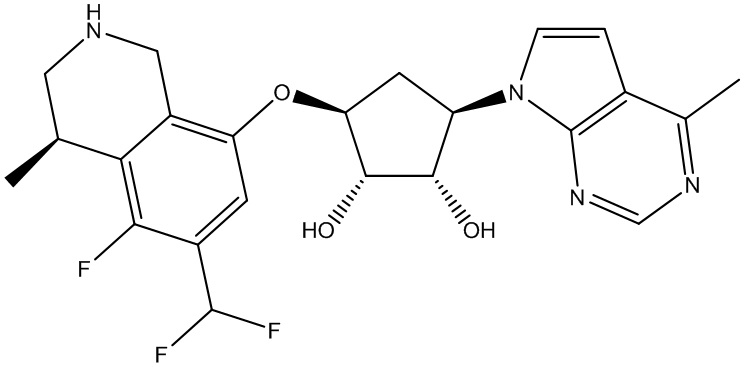 ,
,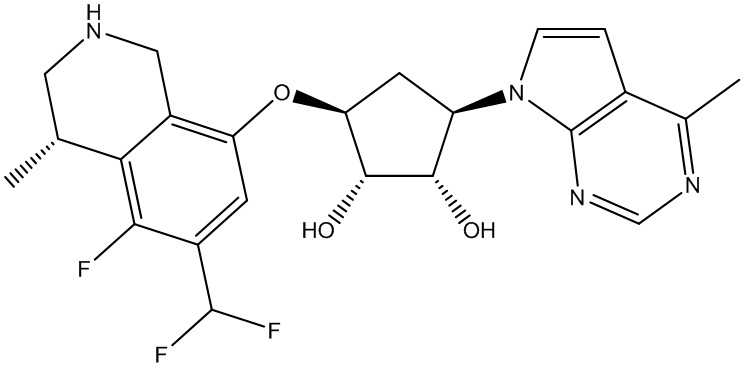 ,
,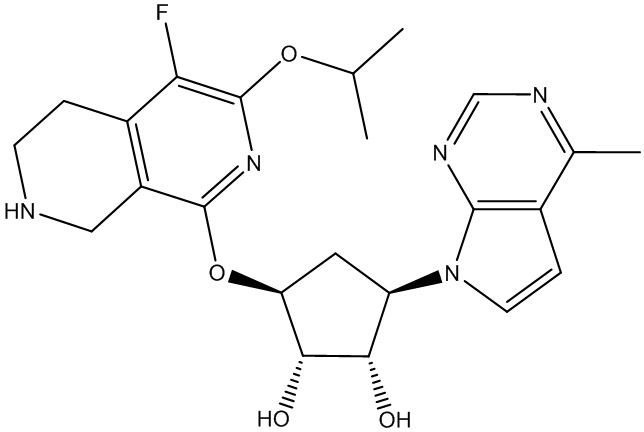 ,
,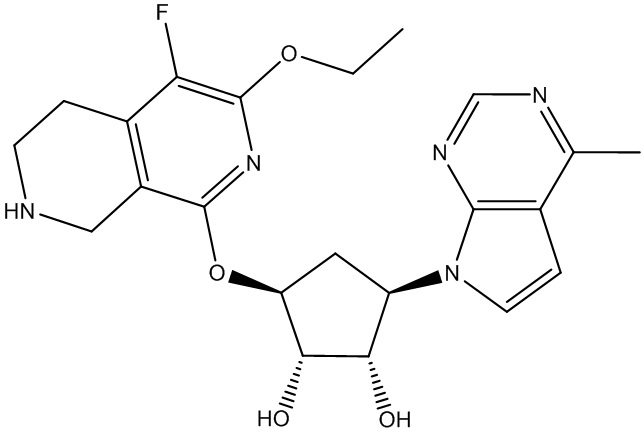 ,
,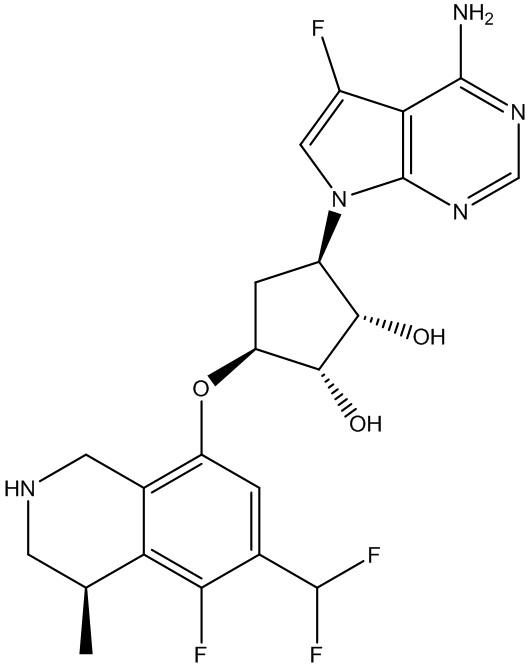 и
и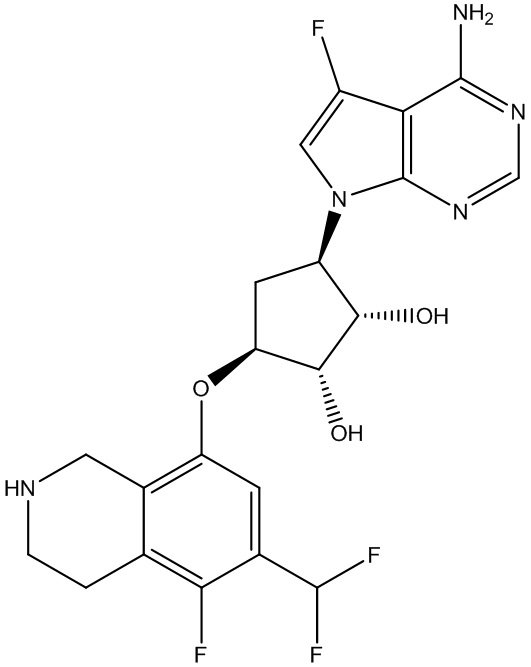 ,
,
или его фармацевтически приемлемой соли.
10. Соединение, выбранное из:
,,,,,,,
или его фармацевтически приемлемой соли.
11. Соединение, выбранное из:
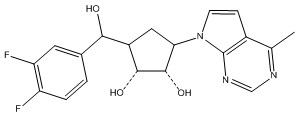 ,
,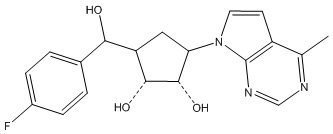 ,
,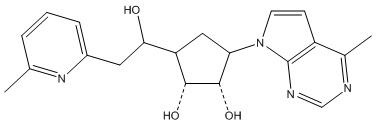 ,
,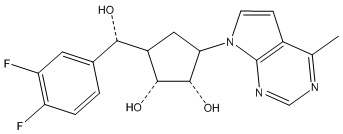 ,
,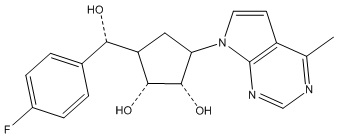 ,
,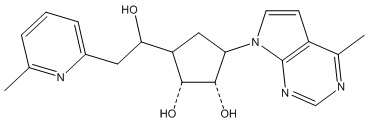 ,
,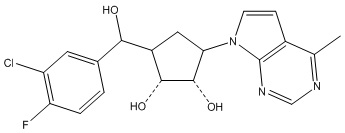 ,
,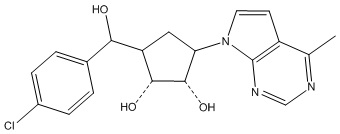 ,
,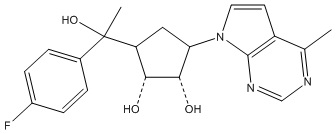 ,
,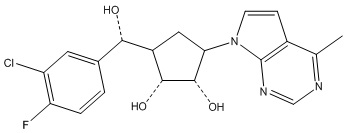 ,
,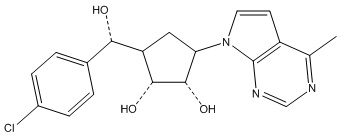 ,
,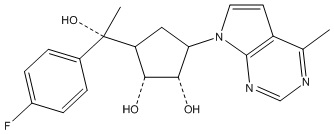 ,
,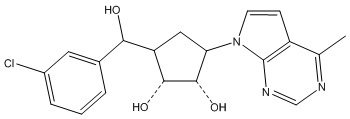 ,
,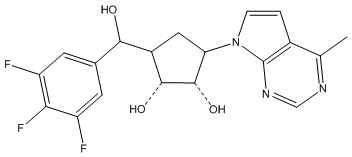 ,
,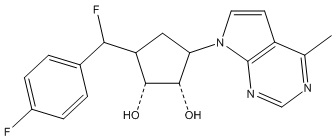 ,
,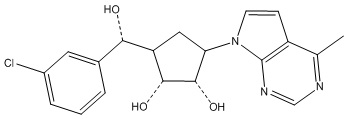 ,
,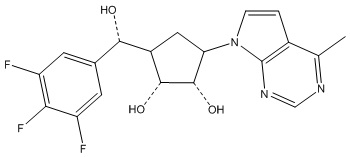 ,
,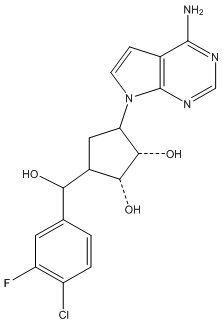 ,
,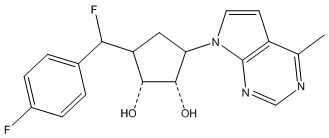 ,
,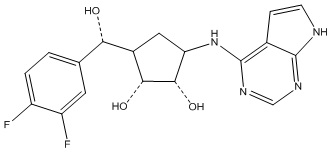 ,
,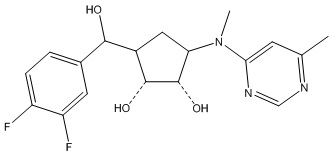 ,
,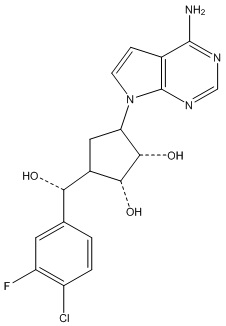 ,
,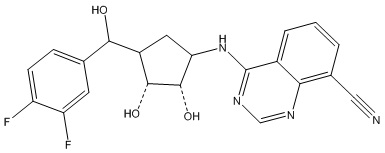 ,
,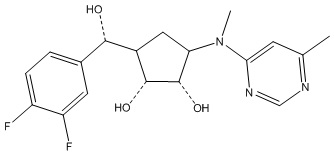 ,
,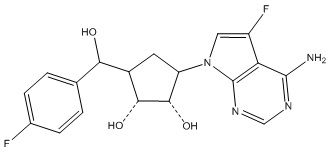 ,
,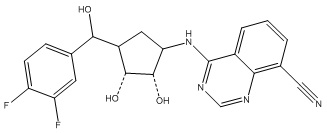 ,
,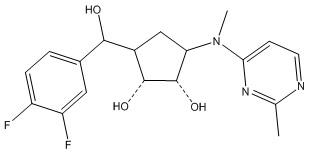 ,
,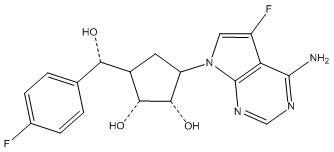 ,
,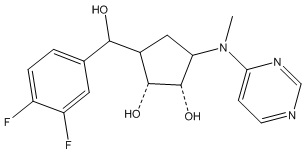 ,
,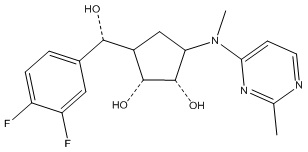 ,
,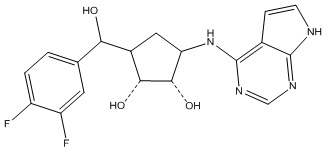 ,
,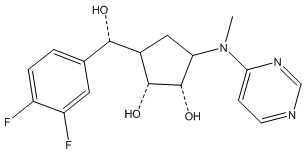 ,
,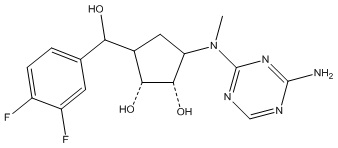 ,
,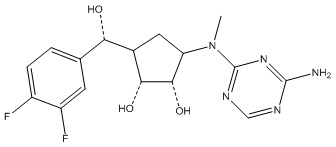 ,
,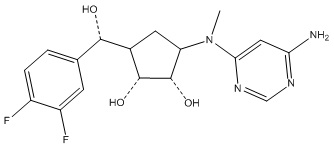 ,
,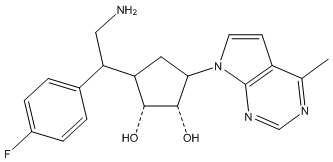 ,
,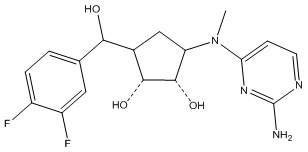 ,
,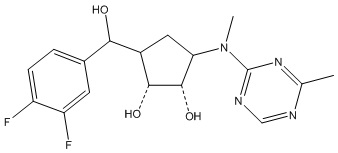 ,
,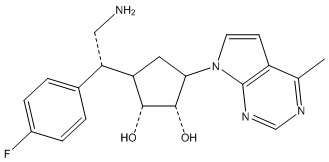 ,
,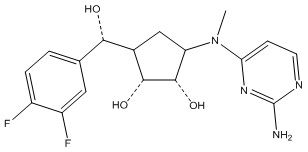 ,
,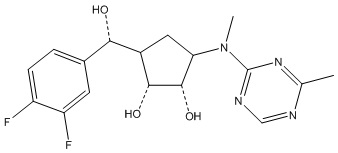 ,
,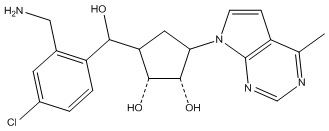 ,
,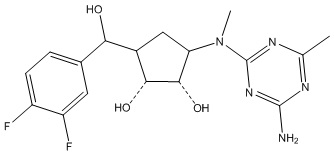 ,
,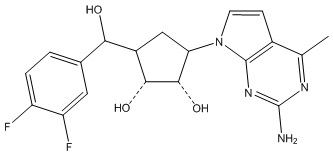 ,
,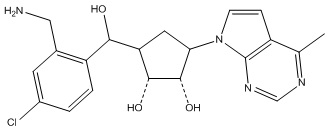 ,
,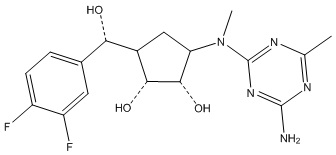 ,
,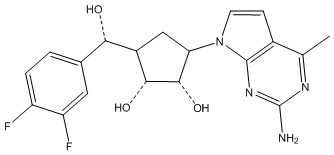 ,
,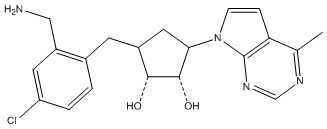 ,
,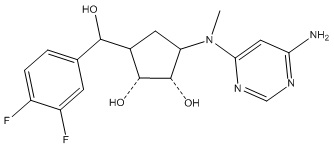 ,
,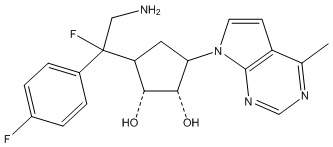 ,
,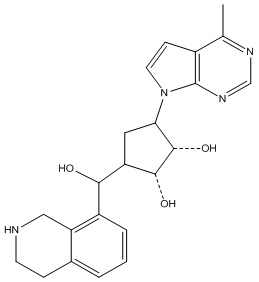 ,
,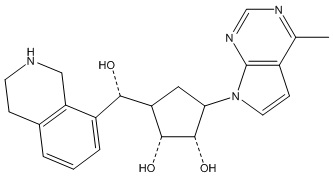 ,
,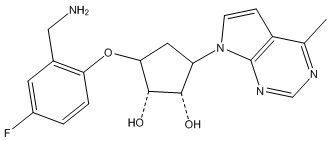 ,
,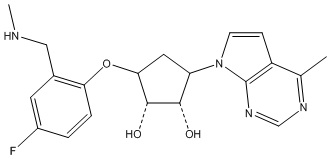 ,
,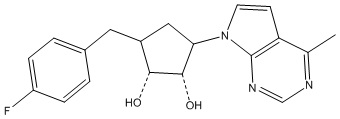 ,
,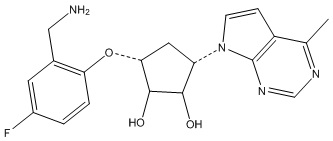 ,
,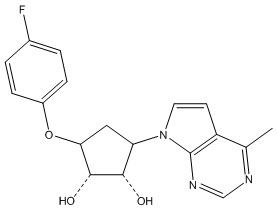 ,
,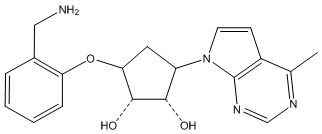 ,
,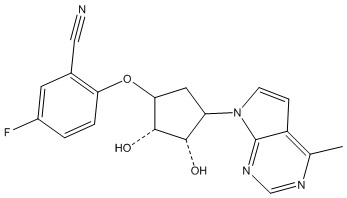 ,
,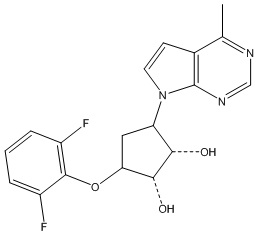 ,
,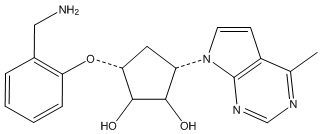 ,
,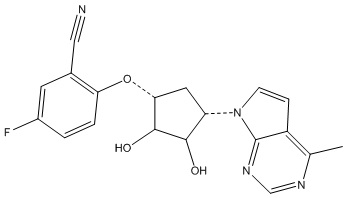 ,
,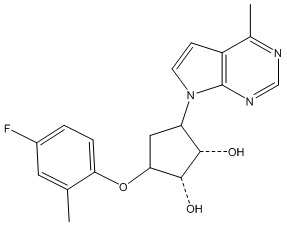 ,
,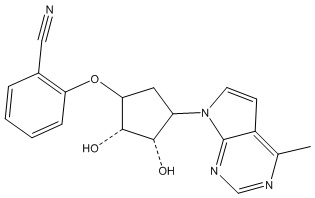 ,
,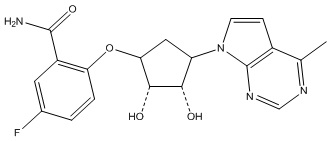 ,
,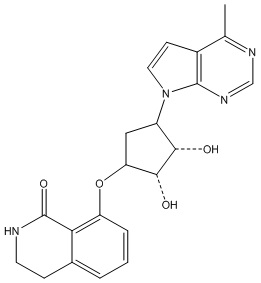 ,
,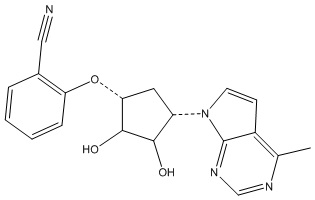 ,
,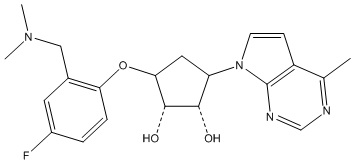 ,
,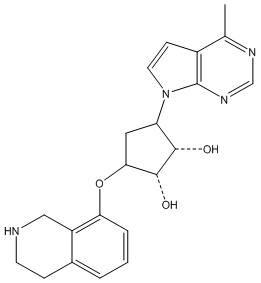 ,
,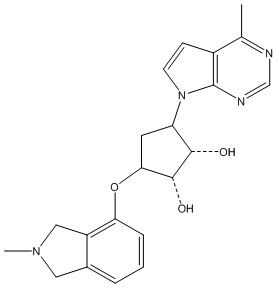 ,
,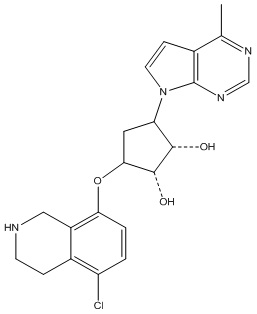 ,
,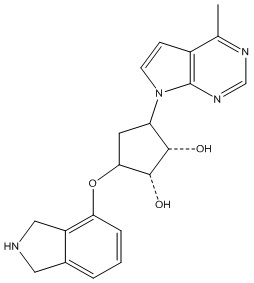 ,
,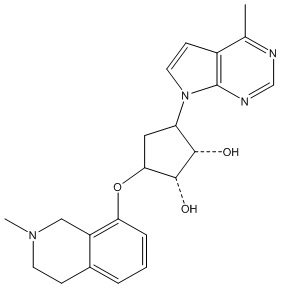 ,
,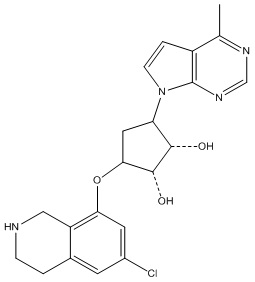 ,
,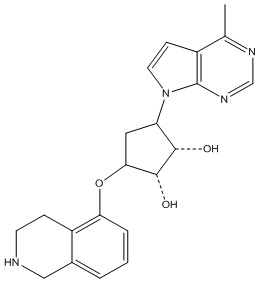 ,
,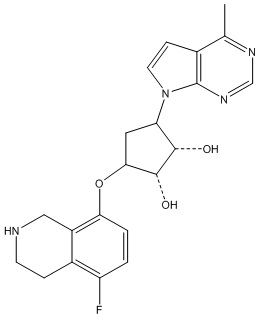 ,
,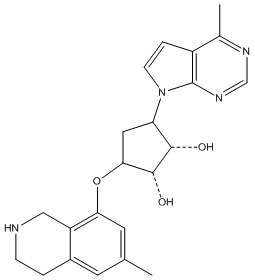 ,
,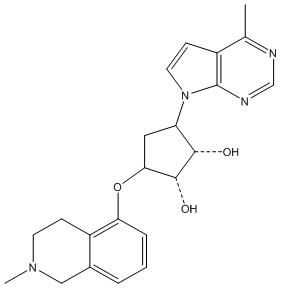 ,
,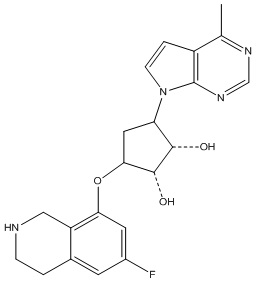 ,
,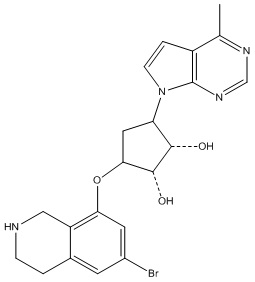 ,
,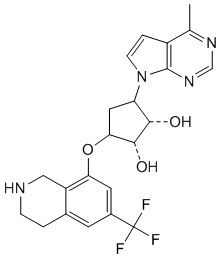 ,
,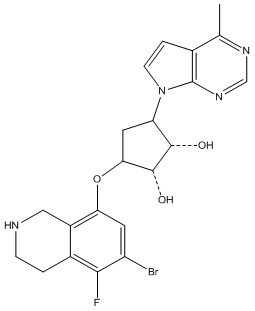 ,
,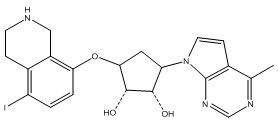 ,
,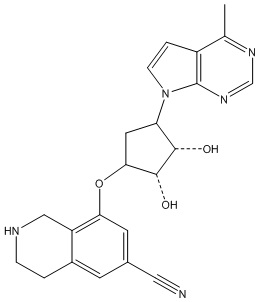 ,
,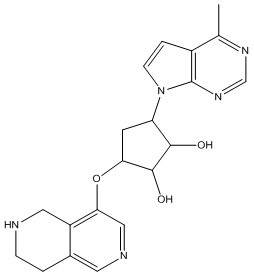 ,
,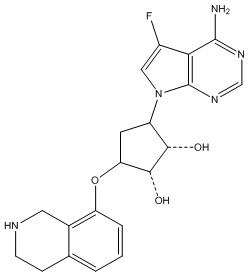 ,
,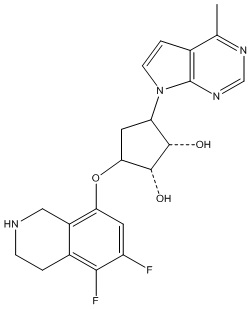 ,
,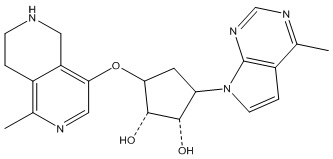 ,
,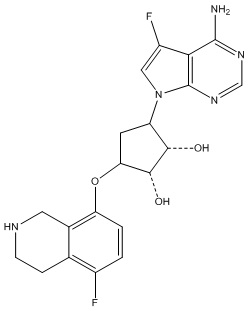 ,
,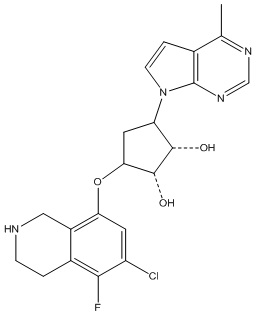 ,
,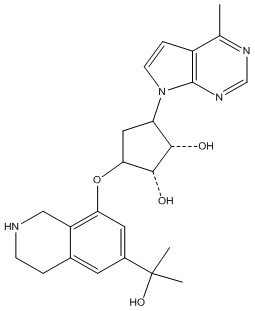 ,
,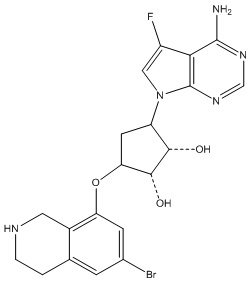 ,
,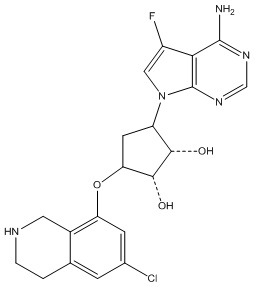 ,
,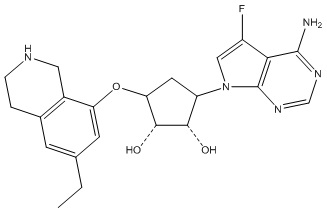 ,
,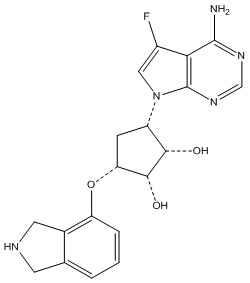 ,
,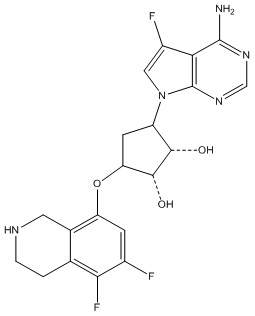 ,
,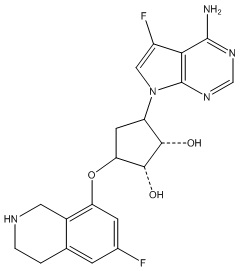 ,
,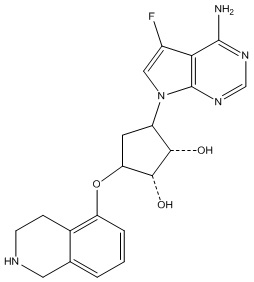 ,
,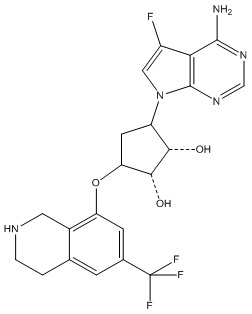 ,
,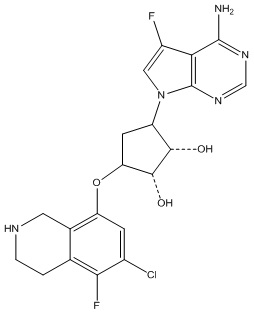 ,
,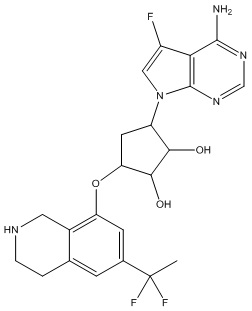 ,
,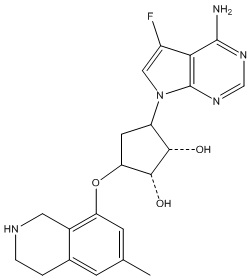 ,
,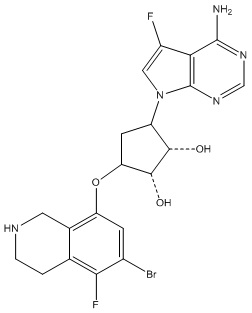 ,
,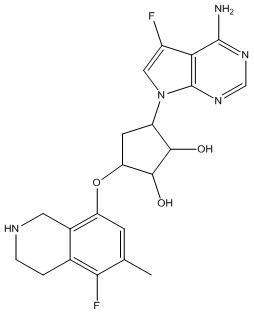 ,
,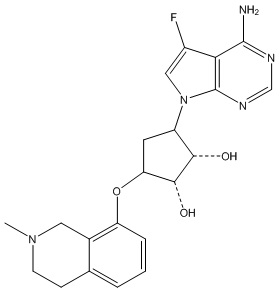 ,
,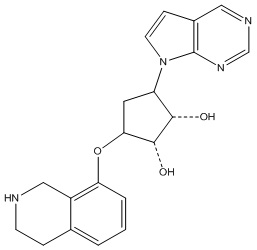 ,
,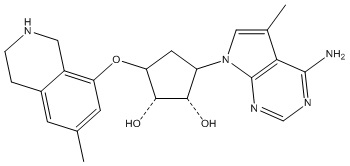 ,
,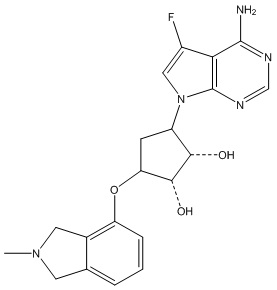 ,
,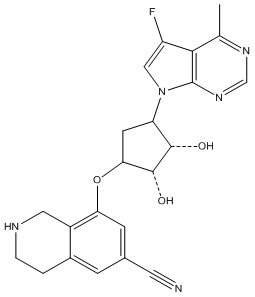 ,
,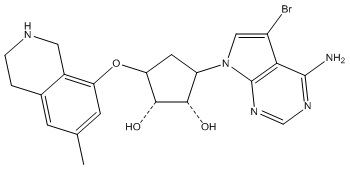 ,
,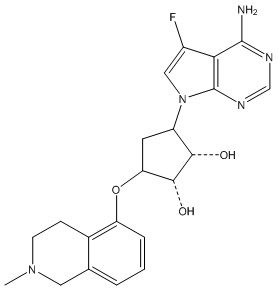 ,
,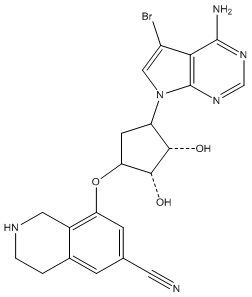 ,
,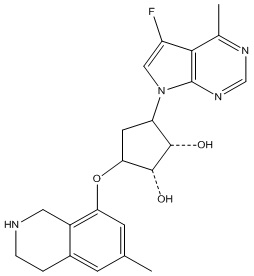 ,
,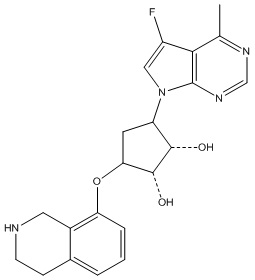 ,
,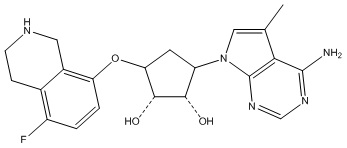 ,
,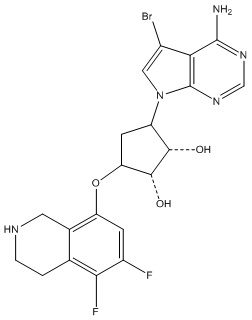 ,
,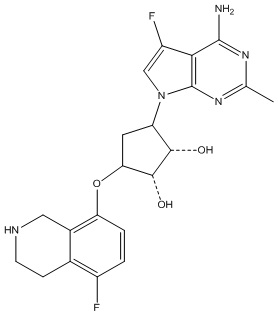 ,
,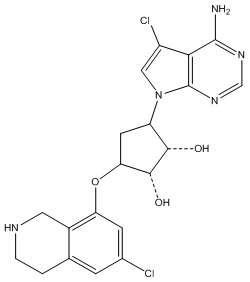 ,
,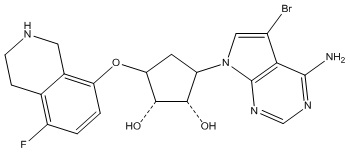 ,
,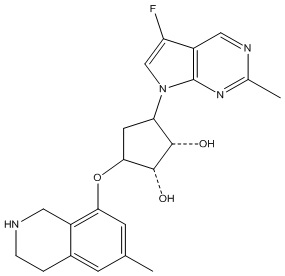 ,
,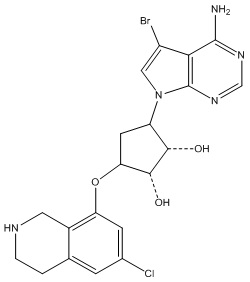 ,
,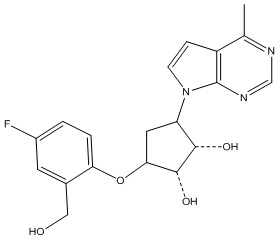 ,
,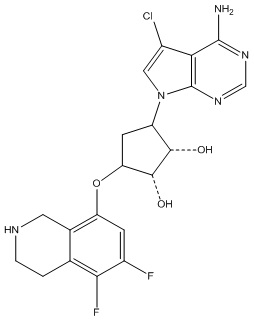 ,
,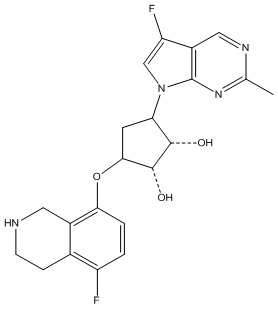 ,
,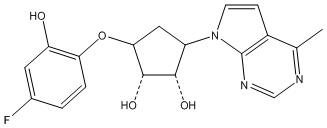 ,
,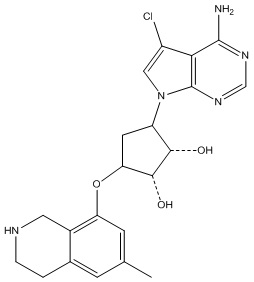 ,
,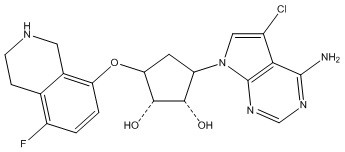 ,
,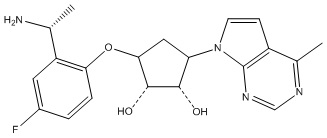 ,
,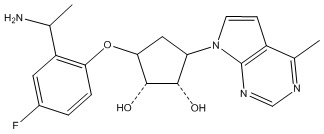 ,
,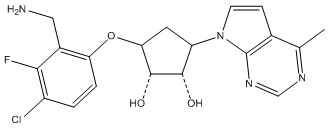 ,
,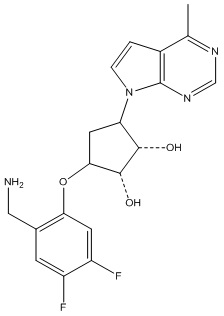 ,
,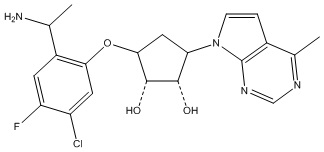 ,
,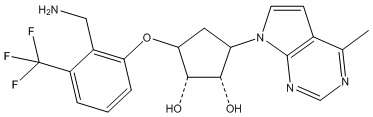 ,
,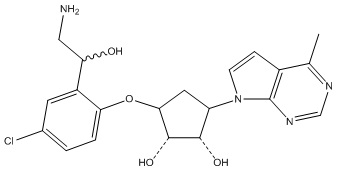 ,
,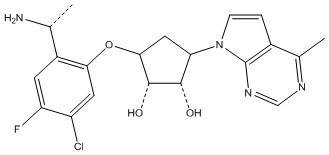 ,
,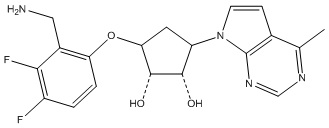 ,
,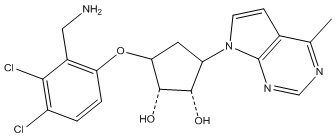 ,
,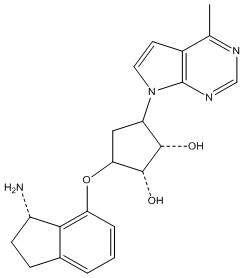 ,
,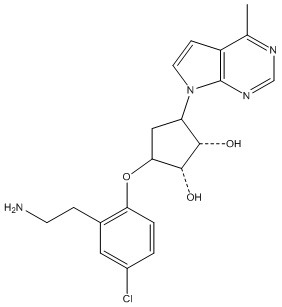 ,
,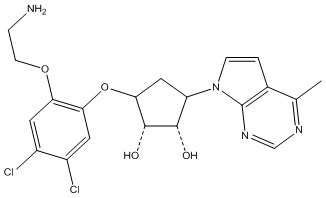 ,
,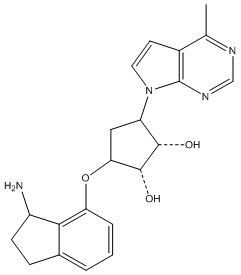 ,
,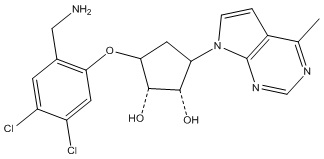 ,
,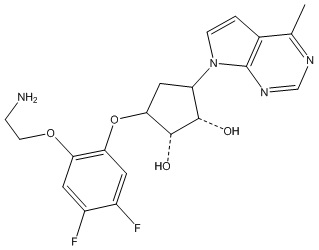 ,
,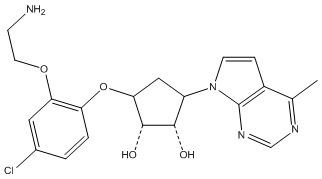 ,
,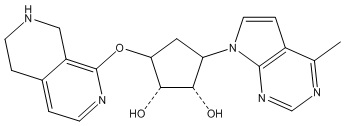 ,
,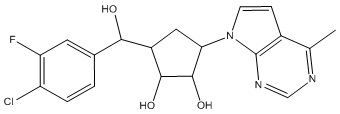 ,
,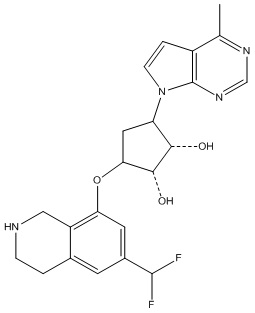 ,
,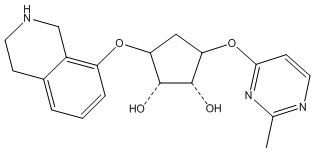 ,
,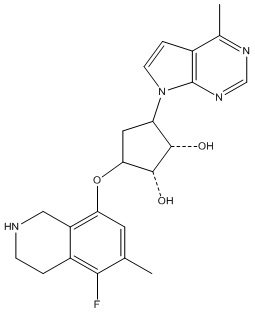 ,
,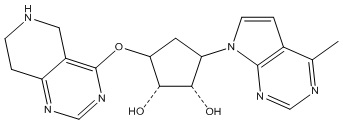 ,
,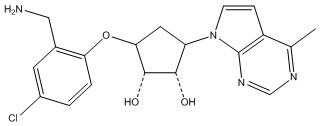 ,
,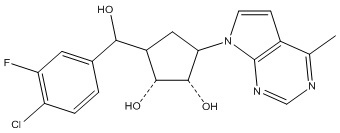 ,
,
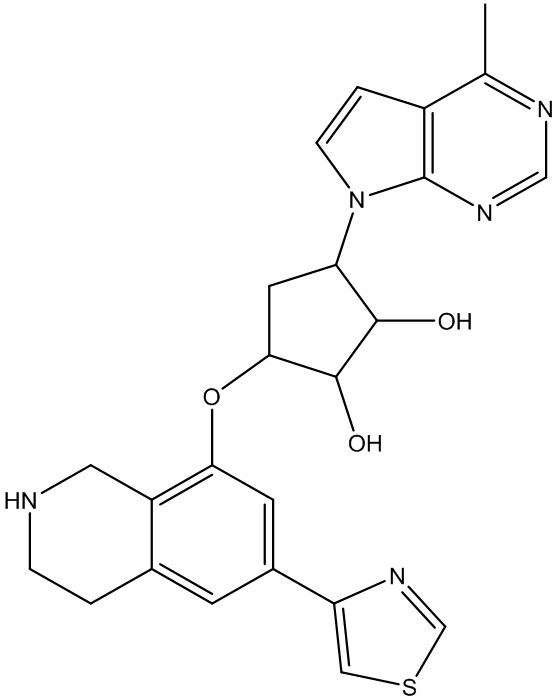 ,
,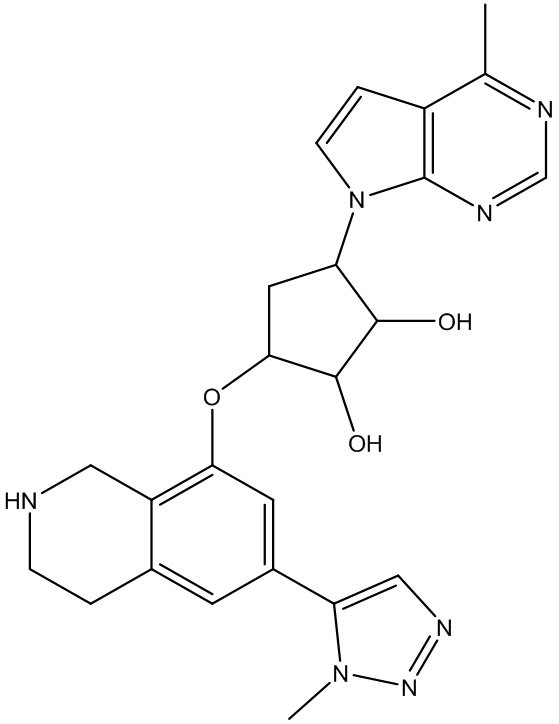 ,
,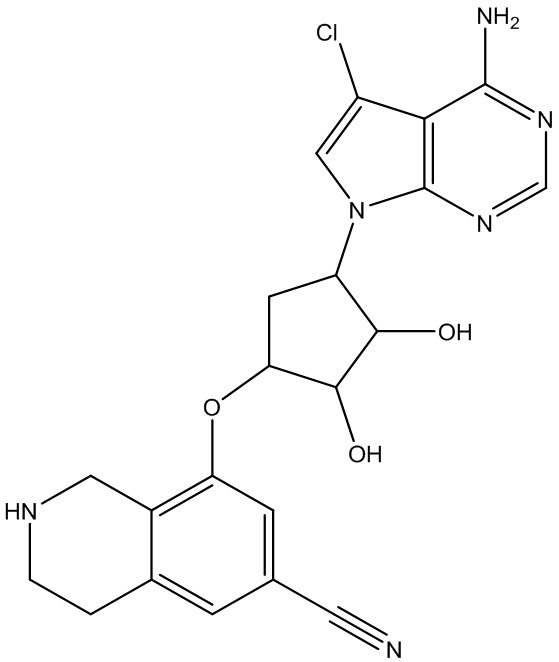 ,
,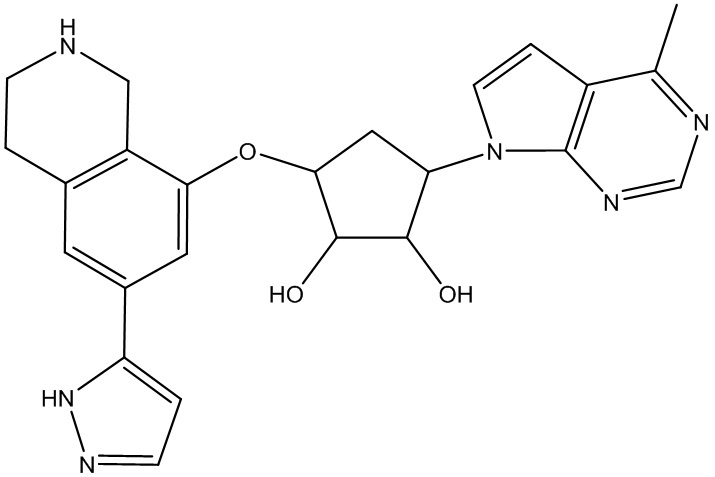 ,
,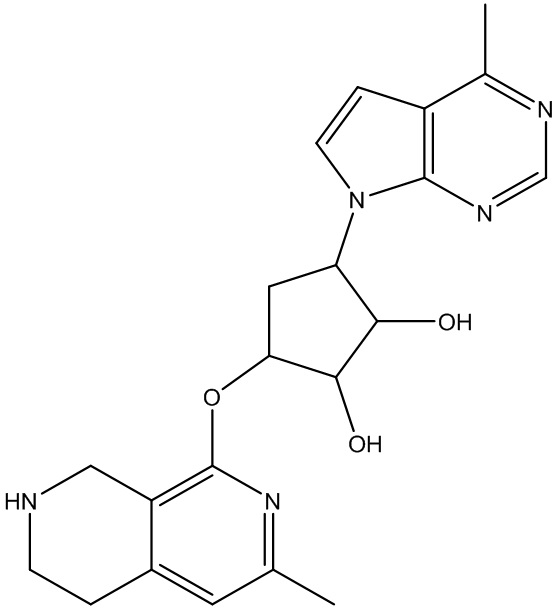 ,
,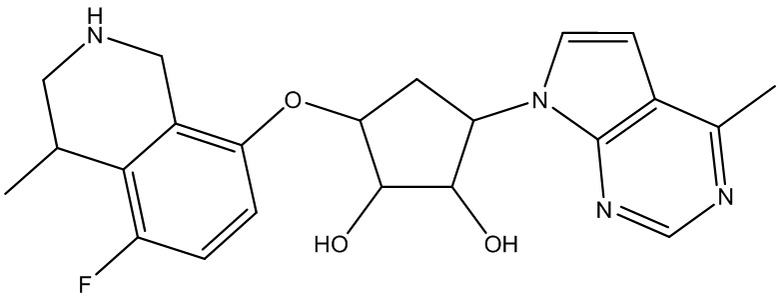 ,
,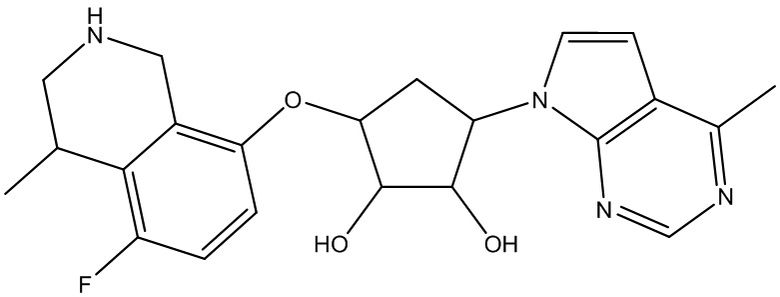 ,
,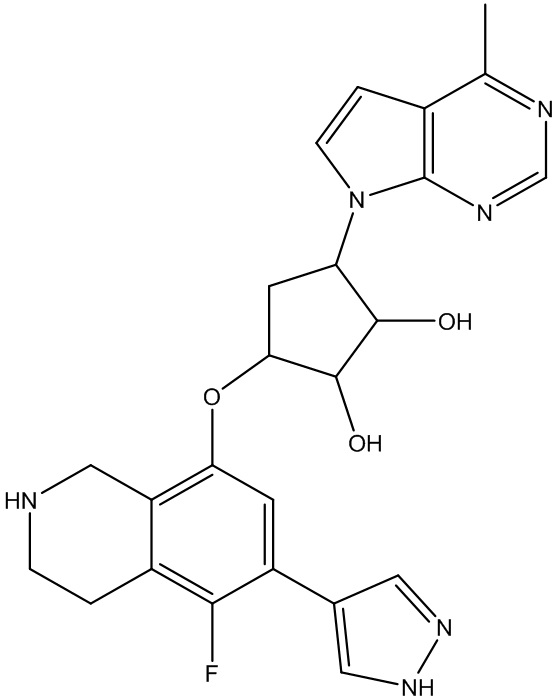 ,
,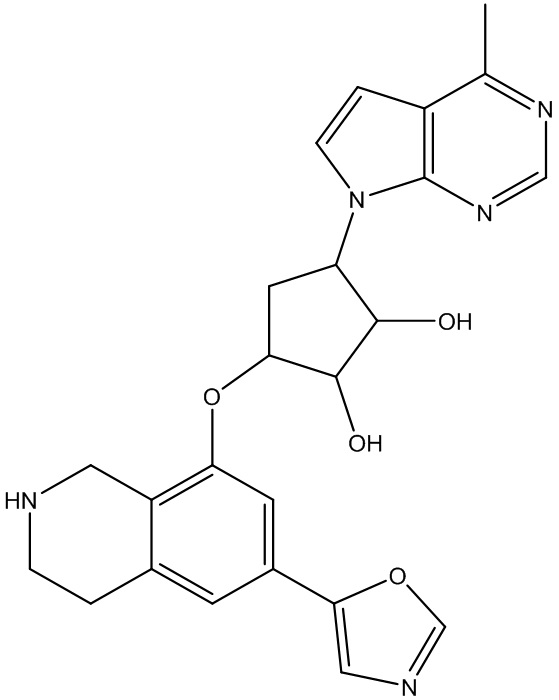 ,
,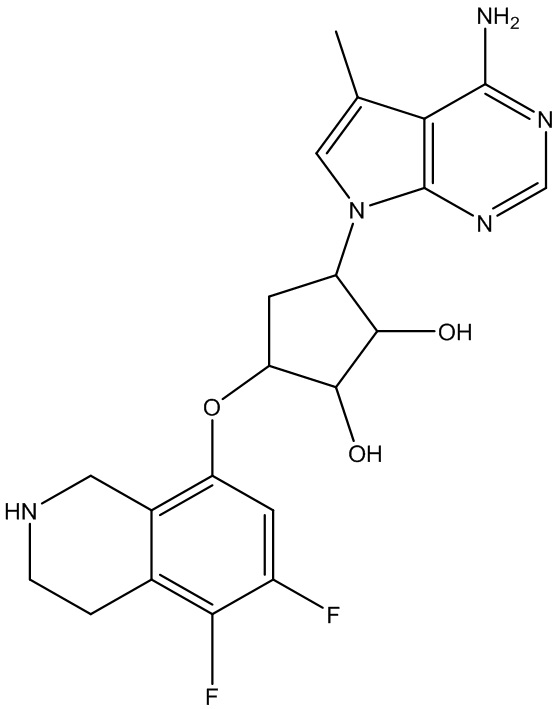 ,
,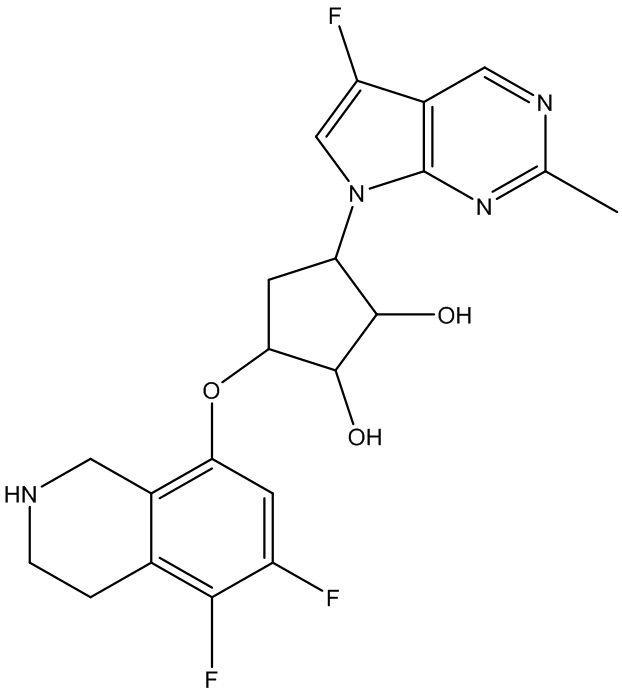 ,
,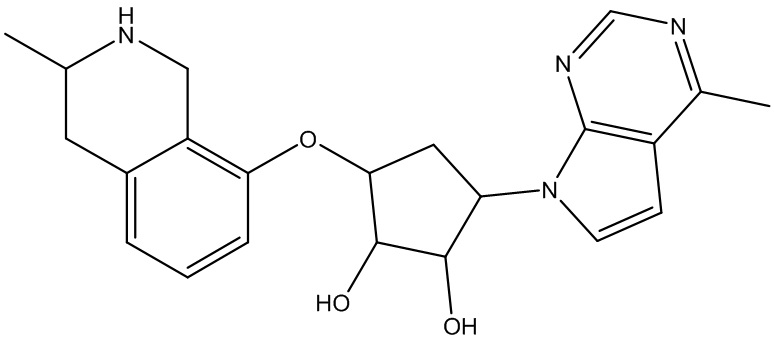 ,
, ,
,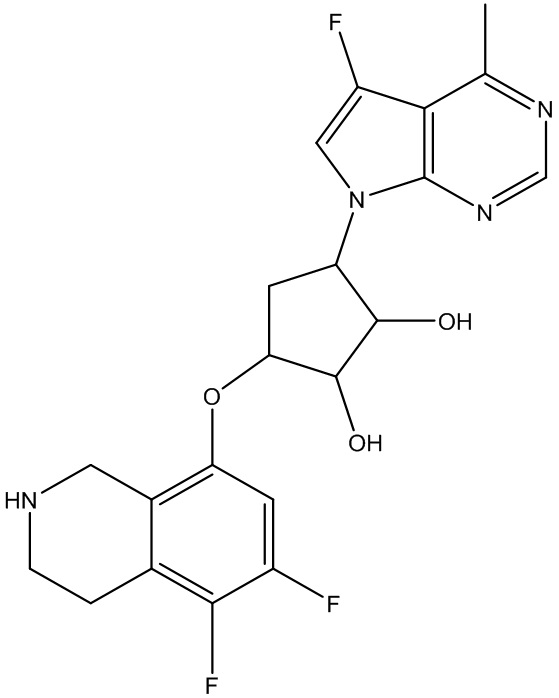 ,
,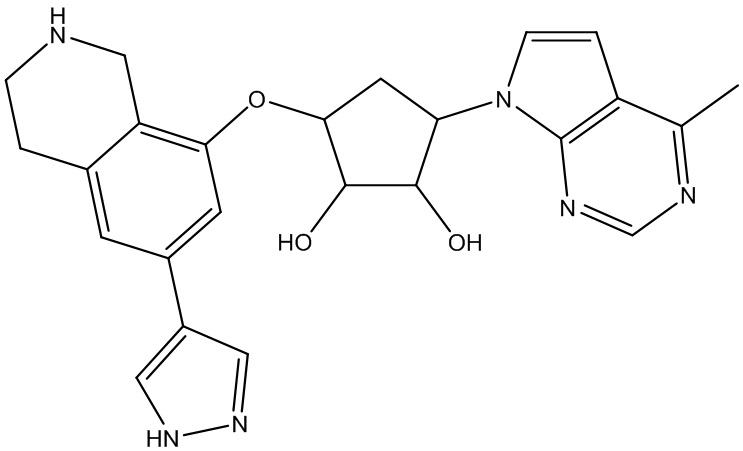 ,
,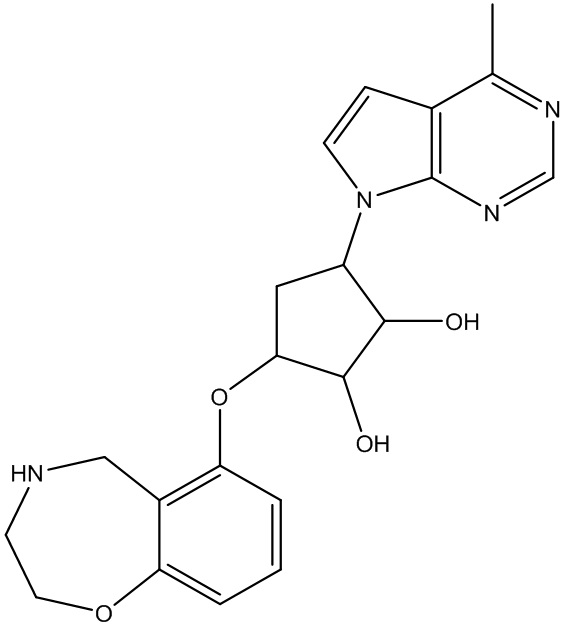 ,
,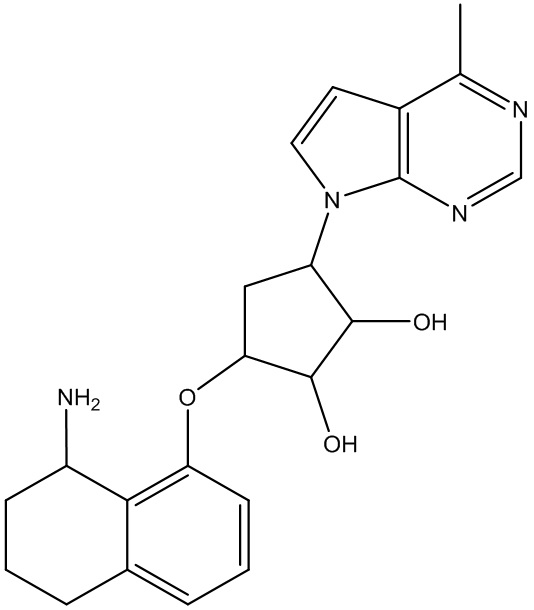 ,
,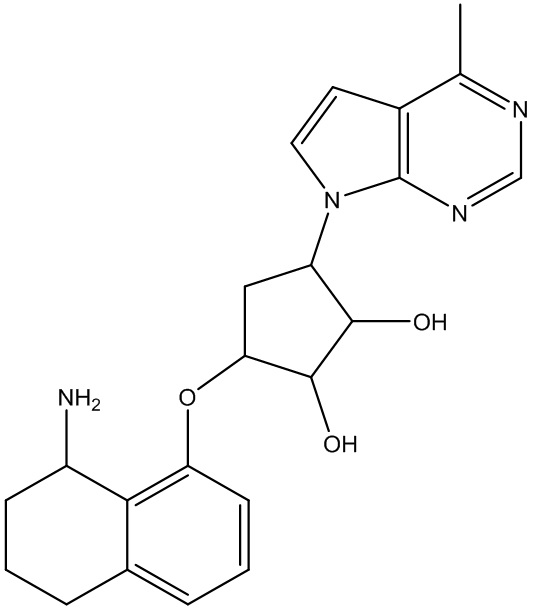 ,
,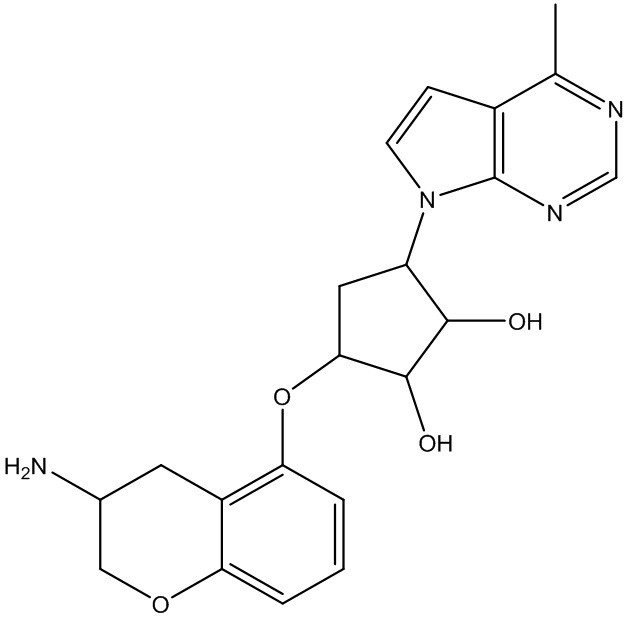 ,
, ,
,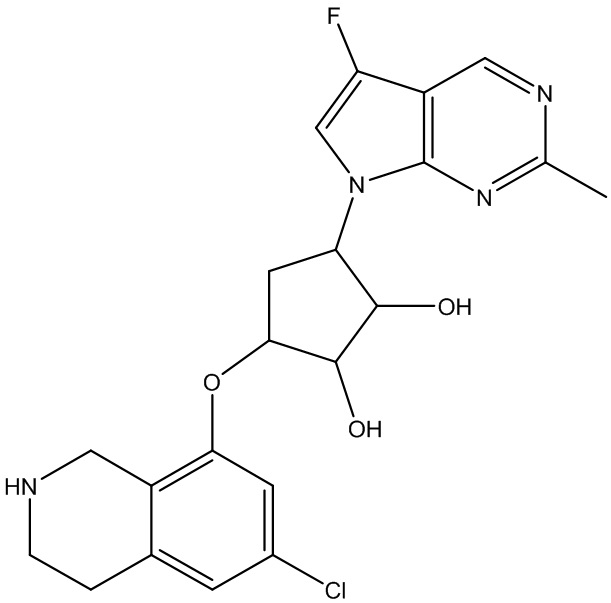 ,
,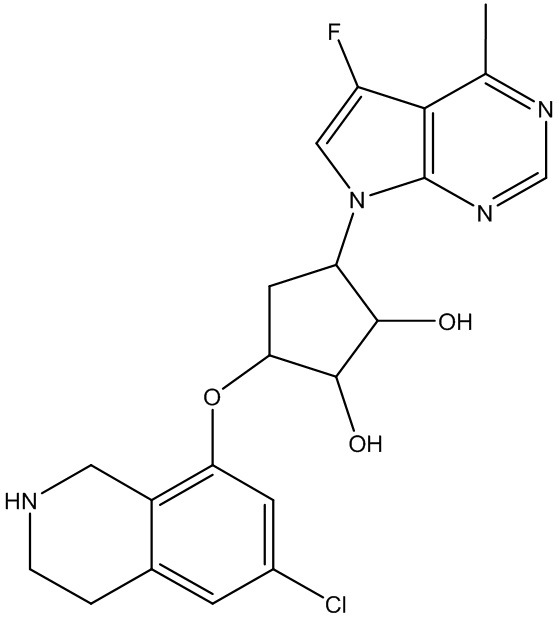 ,
,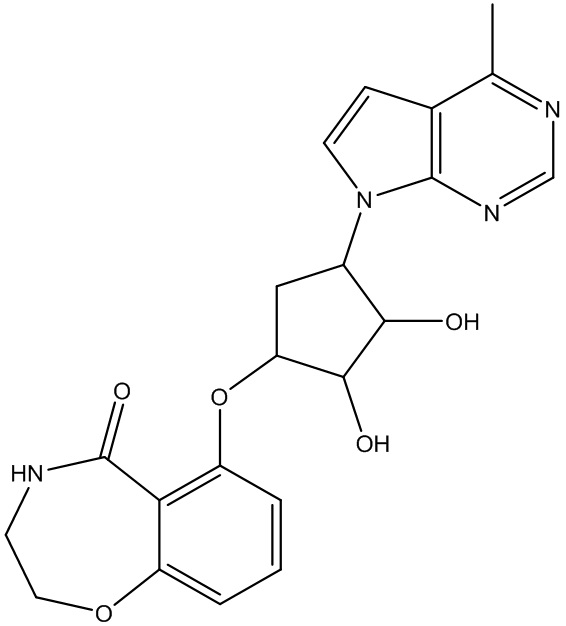 ,
,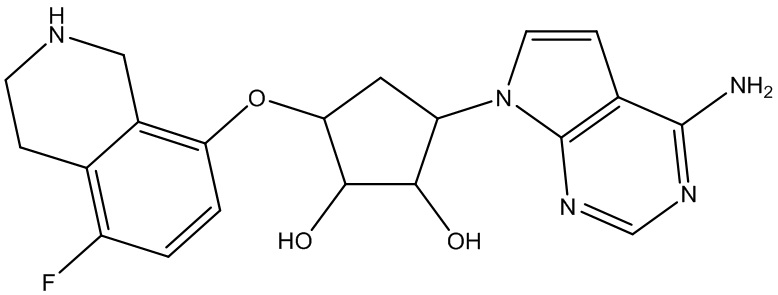
, 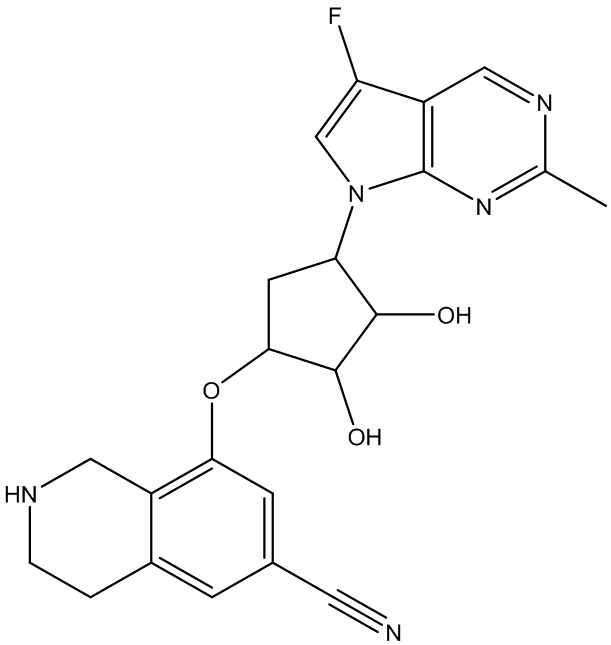 ,
,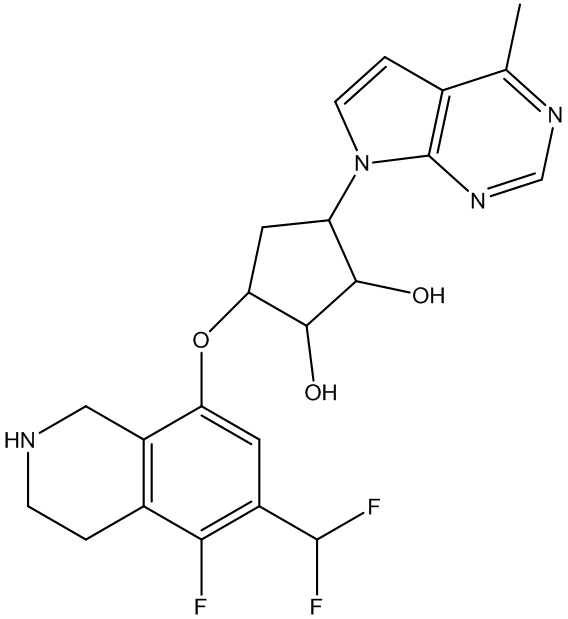 ,
,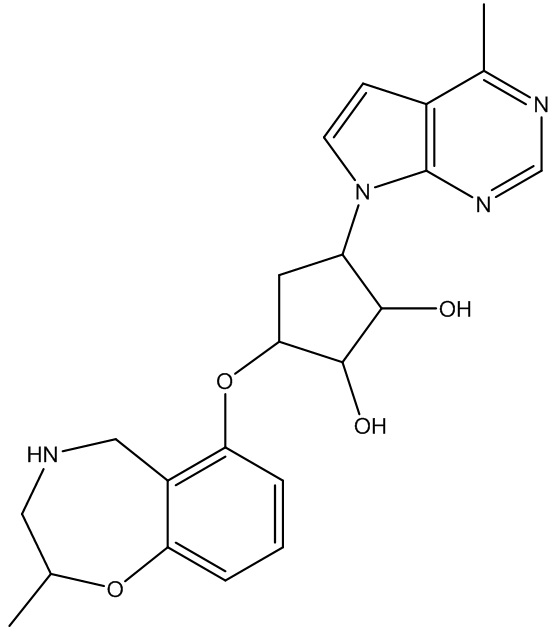 ,
,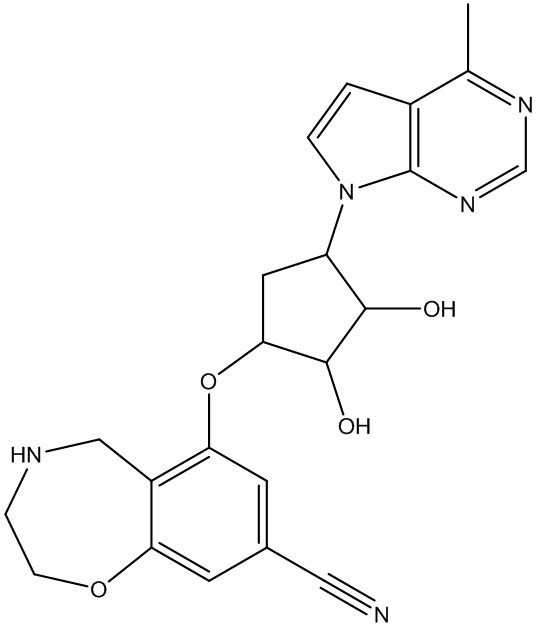 ,
,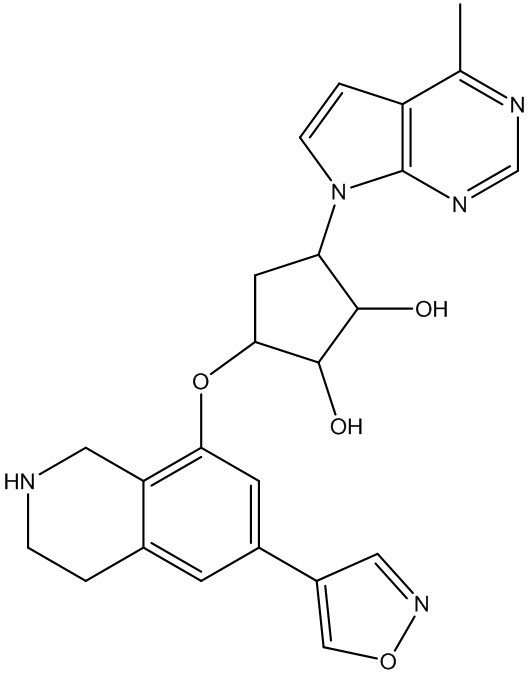 ,
,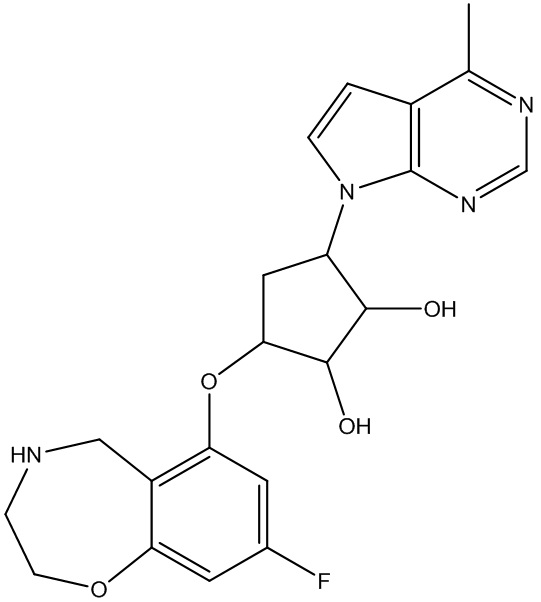 ,
,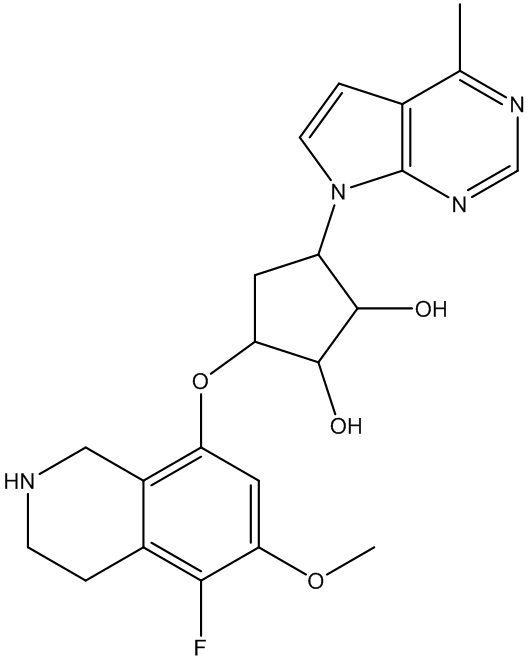 ,
,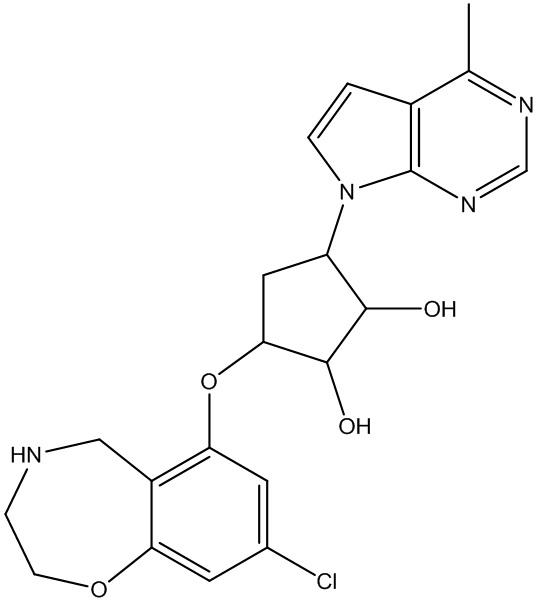 ,
,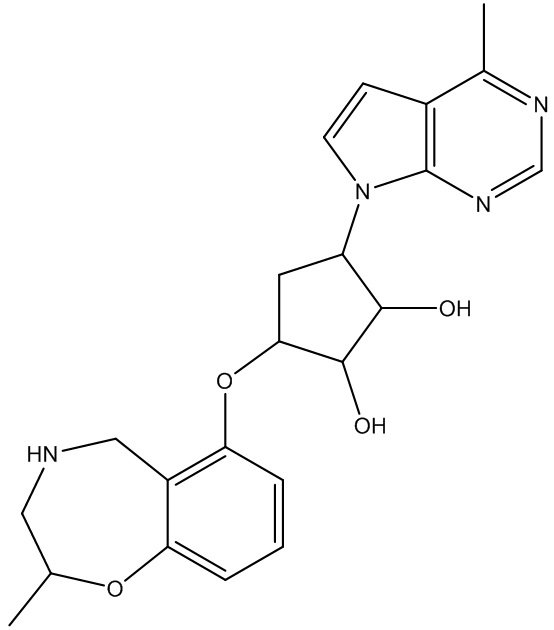 ,
,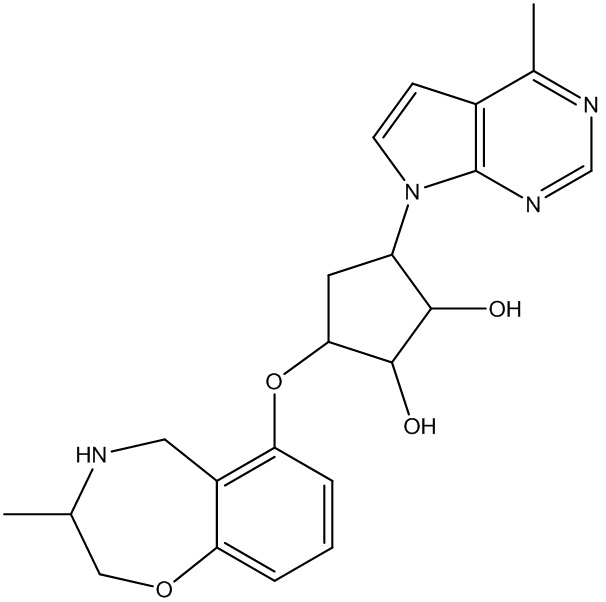 ,
,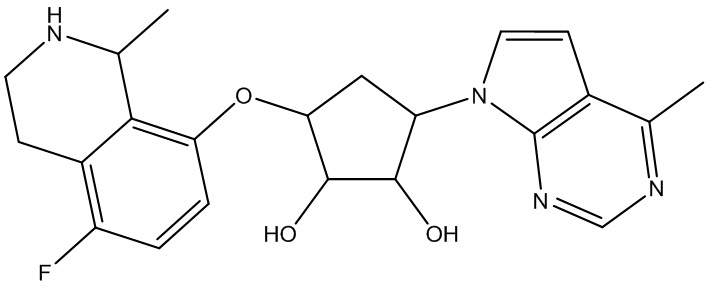 ,
,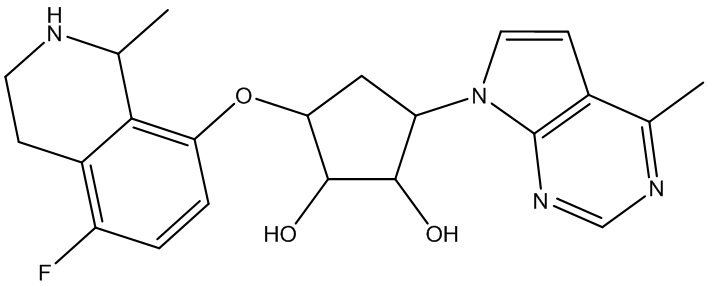 ,
,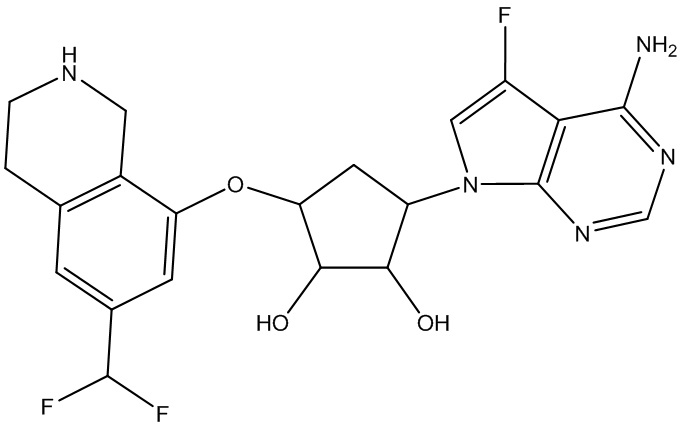 ,
,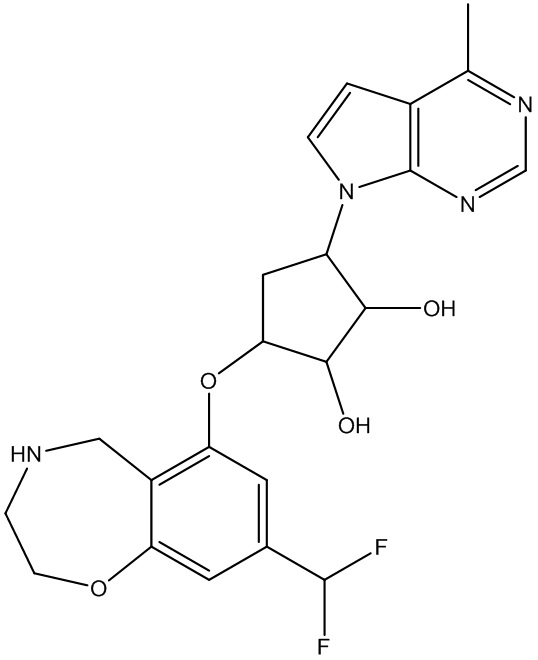 ,
,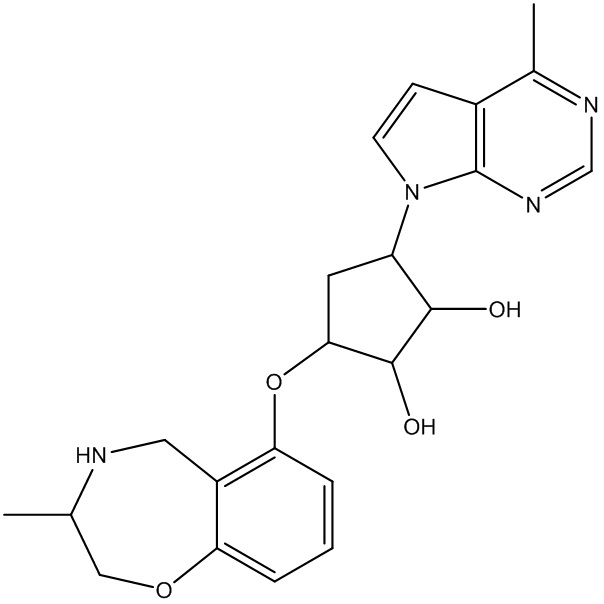 ,
,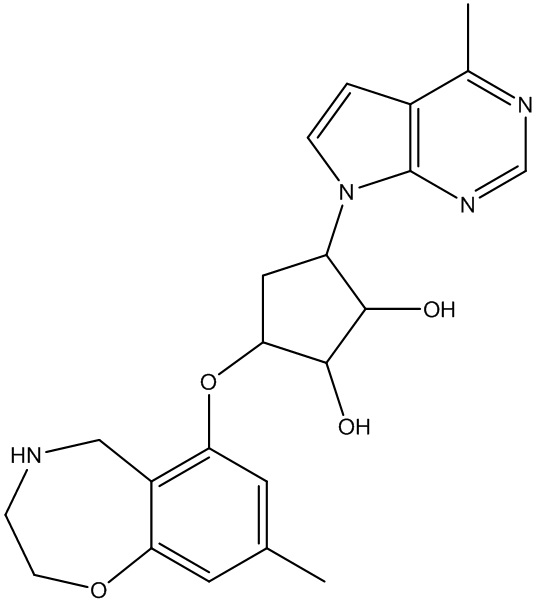 ,
,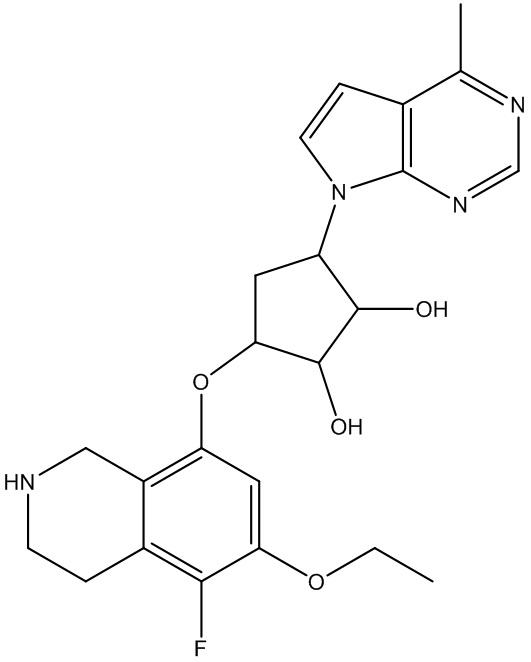 ,
,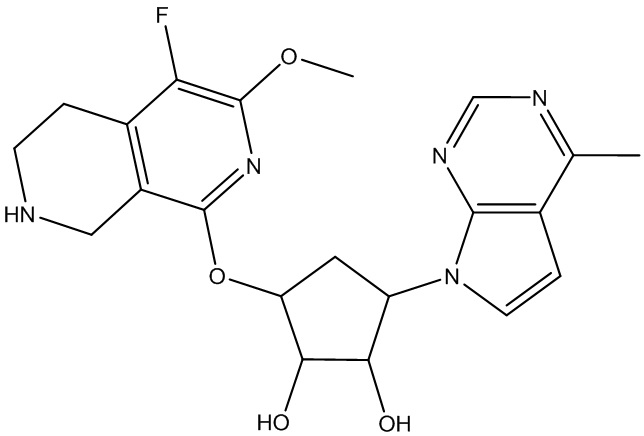 ,
,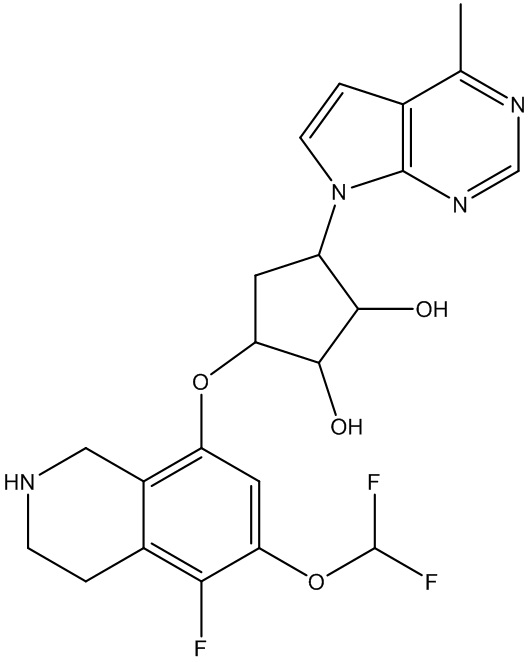 ,
,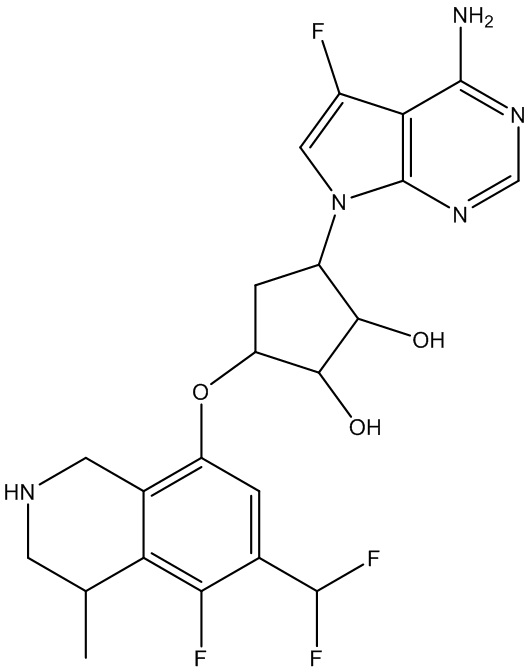 ,
,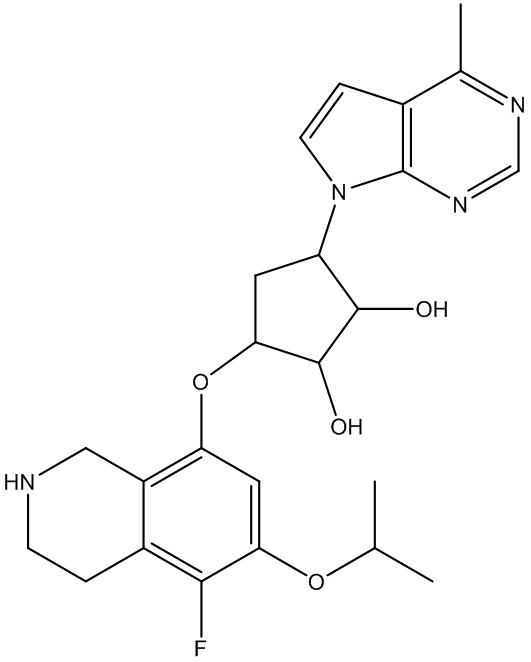 ,
,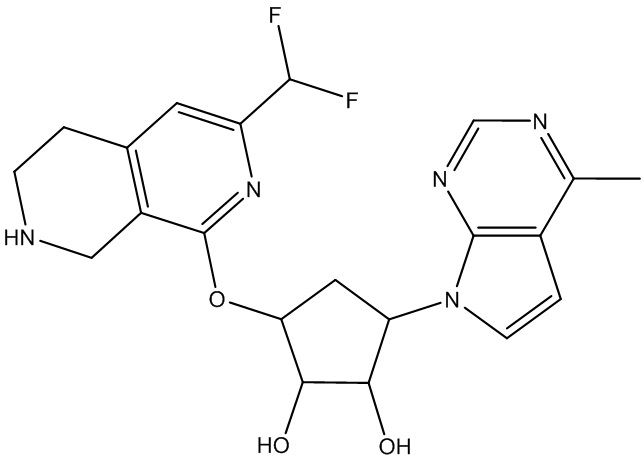 ,
,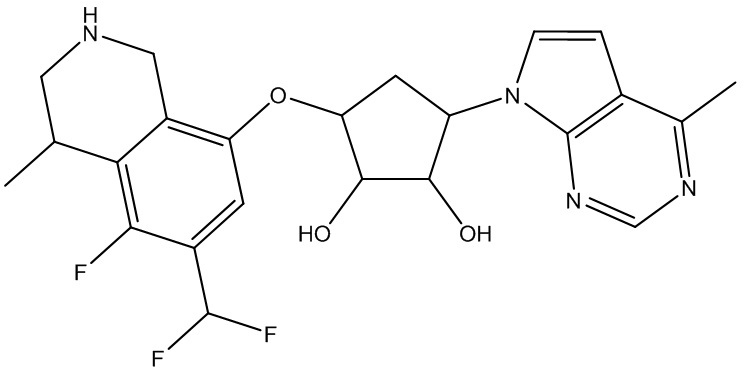 ,
,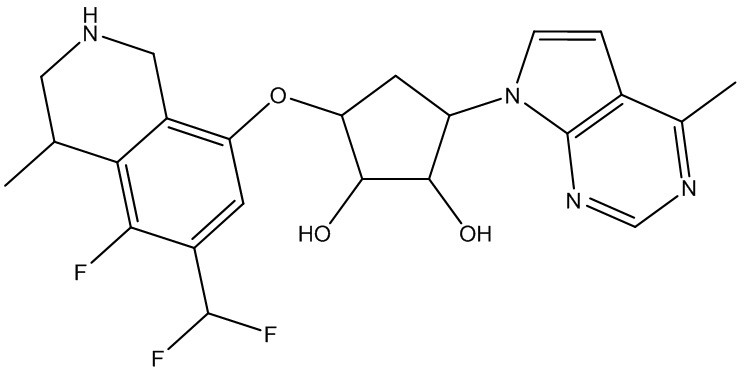 ,
,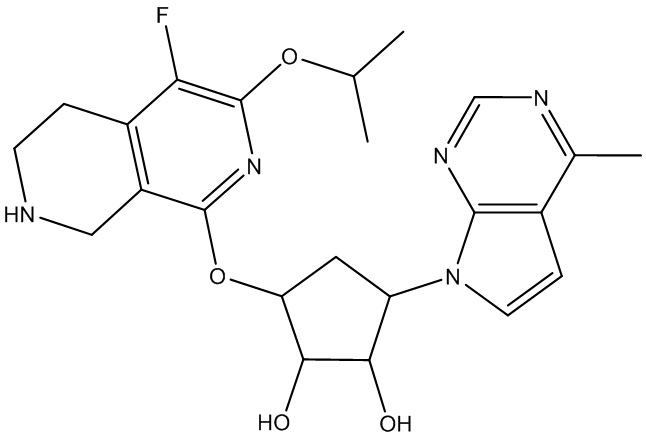 ,
,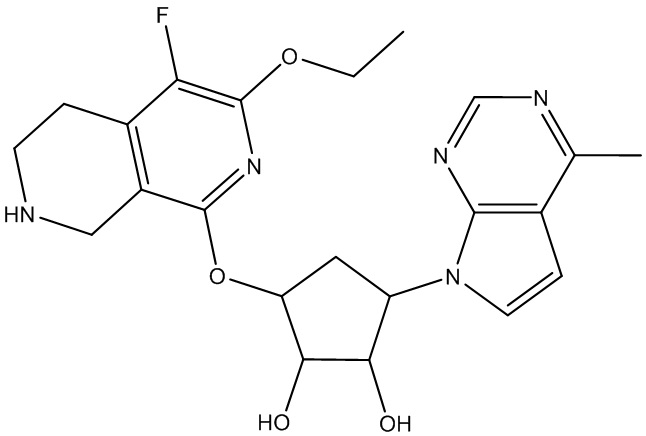 ,
,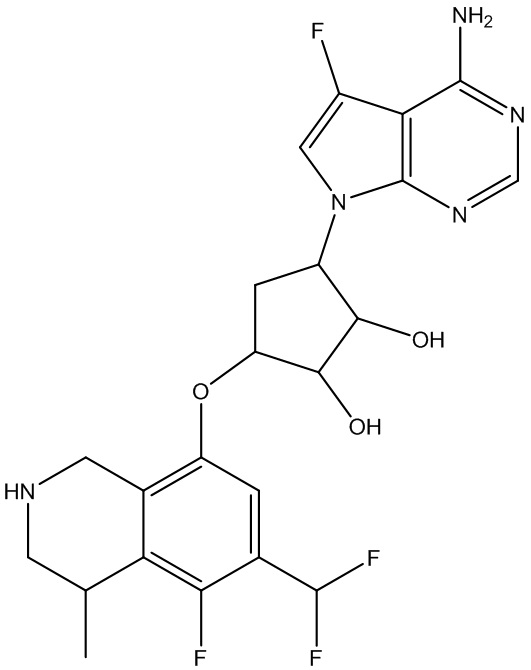 и
и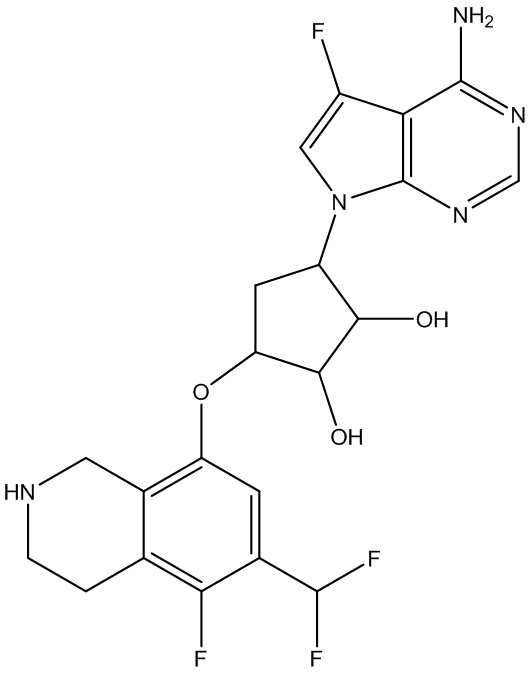 ,
,
или его фармацевтически приемлемой соли.
12. Соединение по любому из пп. 1-10, где указанную соль выбирают из ацетата, аспартата, бензоата, безилата, бикарбоната/карбоната, бисульфата/сульфата, бората, камзилата, цитрата, эдизилата, эзилата, формиата, фумарата, глюцептата, глюконата, глюкуроната, гексафторфосфата, гибензата, гидрохлорида/хлорида, гидробромида/бромида, гидройодида/йодида, изетионата, лактата, малата, малеата, малоната, мезилата, метилсульфата, нафтилата, 2-напсилата, никотината, нитрата, оротата, оксалата, пальмитата, памоата, фосфата/гидрофосфата/дигидрофосфата, сахарат, стеарата, сукцината, тартрата, тозилата, трифторацетата, солей алюминия, аргинина, бензатина, кальция, холина, диэтиламина, диоламина, глицина, лизина, магния, меглумина, оламина, калия, натрия, трометамина и цинка.
13. Соединение по любому из пп. 1-11, где указанную соль выбирают из гидрохлорида, тозилата и мезолата.
14. Фармацевтическая композиция, обладающая свойством ингибитора фермента РRMT5, содержащая эффективное количество соединения по любому из пп. 1-13 или его фармацевтически приемлемую соль, и фармацевтически приемлемый носитель.
15. Способ лечения аномального роста клеток у млекопитающих, где способ включает введение млекопитающему терапевтически эффективного количества соединения по любому из пп. 1-13 или его фармацевтически приемлемой соли.
16. Способ по п. 15, где аномальным ростом клеток является рак.
17. Способ по п. 16, где раком является рак легких, рак костей, рак поджелудочной железы, рак кожи, рак головы или шеи, кожная или внутриглазная меланома, рак матки, рак яичников, рак прямой кишки, рак анальной области, рак желудка, рак толстой кишки, рак молочной железы, рак матки, рак маточных труб, рак эндометрия, рак шейки матки, рак влагалища, рак вульвы, болезнь Ходжкина, рак пищевода, рак тонкой кишки, рак эндокринной системы, рак эндокринной системы, рак щитовидной железы, рак околощитовидной железы, рак надпочечников, саркома мягких тканей, рак мочеиспускательного канала, рак полового члена, рак предстательной железы, хронический или острый лейкоз, лимфоцитарные лимфомы, рак мочевого пузыря, рак почки или мочеточника, почечно-клеточный рак, рак почечной лоханки, новообразования центральной нервной системы (ЦНС), первичная лимфома ЦНС, опухоли позвоночника, глиома ствола головного мозга или аденома гипофиза.
18. Применение соединения по любому из пп. 1-13 или его фармацевтически приемлемой соли для получения лекарственного средства, применяемого в лечении аномального роста клеток у млекопитающих.
19. Применение по п. 18, где аномальным ростом клеток является рак.
20. Применение по п. 19, где раком является рак легких, рак костей, рак поджелудочной железы, рак кожи, рак головы или шеи, кожная или внутриглазная меланома, рак матки, рак яичников, рак прямой кишки, рак анальной области, рак желудка, рак толстой кишки, рак молочной железы, рак матки, рак маточных труб, рак эндометрия, рак шейки матки, рак влагалища, рак вульвы, болезнь Ходжкина, рак пищевода, рак тонкой кишки, рак эндокринной системы, рак эндокринной системы, рак щитовидной железы, рак околощитовидной железы, рак надпочечников, саркома мягких тканей, рак мочеиспускательного канала, рак полового члена, рак предстательной железы, хронический или острый лейкоз, лимфоцитарные лимфомы, рак мочевого пузыря, рак почки или мочеточника, почечно-клеточный рак, рак почечной лоханки, новообразования центральной нервной системы (ЦНС), первичная лимфома ЦНС, опухоли позвоночника, глиома ствола головного мозга или аденома гипофиза.
21. Соединение (1S,2S,3S,5R)-3-((6-(дифторметил)-5-фтор-1,2,3,4-тетрагидроизохинолин-8-ил)окси)-5-(4-метил-7Н-пирроло[2,3-d]пиримидин-7-ил)циклопентан-1,2-диол, имеющее структуру:
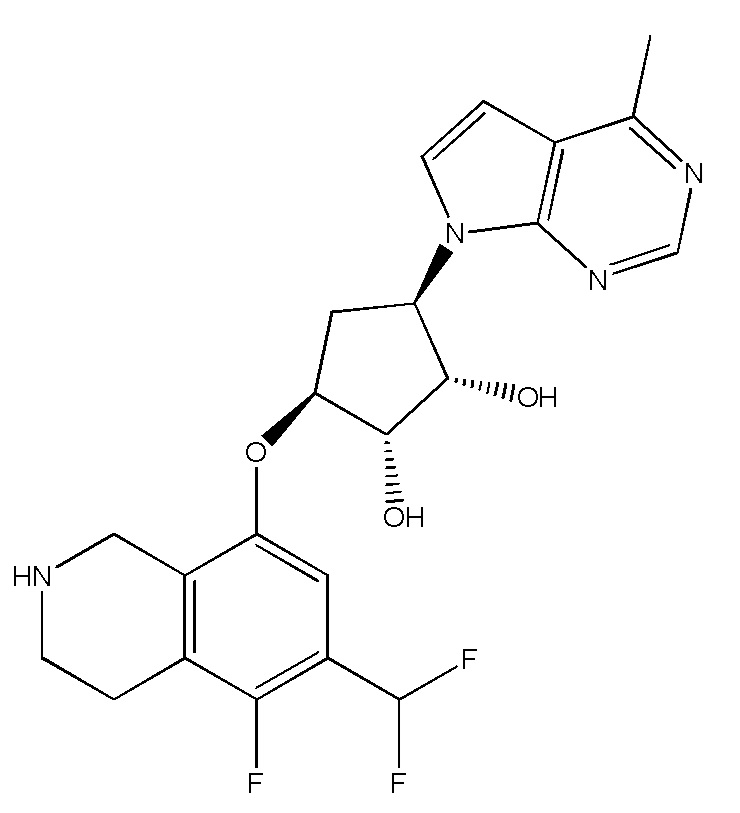
или его фармацевтически приемлемая соль.
22. Соединение по п.21, где указанную соль выбирают из ацетата, аспартата, бензоата, безилата, бикарбоната, карбоната, бисульфата, сульфата, бората, камзилата, цитрата, эдизилата, эзилата, формиата, фумарата, глюцептата, глюконата, глюкуроната, гексафторфосфата, гибензата, гидрохлорида, хлорида, гидробромида, бромида, гидройодида, йодида, изетионата, лактата, малата, малеата, малоната, мезилата, метилсульфата, нафтилата, 2-напсилата, никотината, нитрата, оротата, оксалата, пальмитата, памоата, фосфата, гидрофосфата, дигидрофосфата, сахарата, стеарата, сукцината, тартрата, тозилата, трифторацетата, солей алюминия, аргинина, бензатина, кальция, холина, диэтиламина, диоламина, глицина, лизина, магния, меглумина, оламина, калия, натрия, трометамина и цинка.
23. Фармацевтическая композиция, содержащая соединение (1S,2S,3S,5R)-3-((6-(дифторметил)-5-фтор-1,2,3,4-тетрагидроизохинолин-8-ил)окси)-5-(4-метил-7Н-пирроло[2,3-d]пиримидин-7-ил)циклопентан-1,2-диол, имеющего структуру:
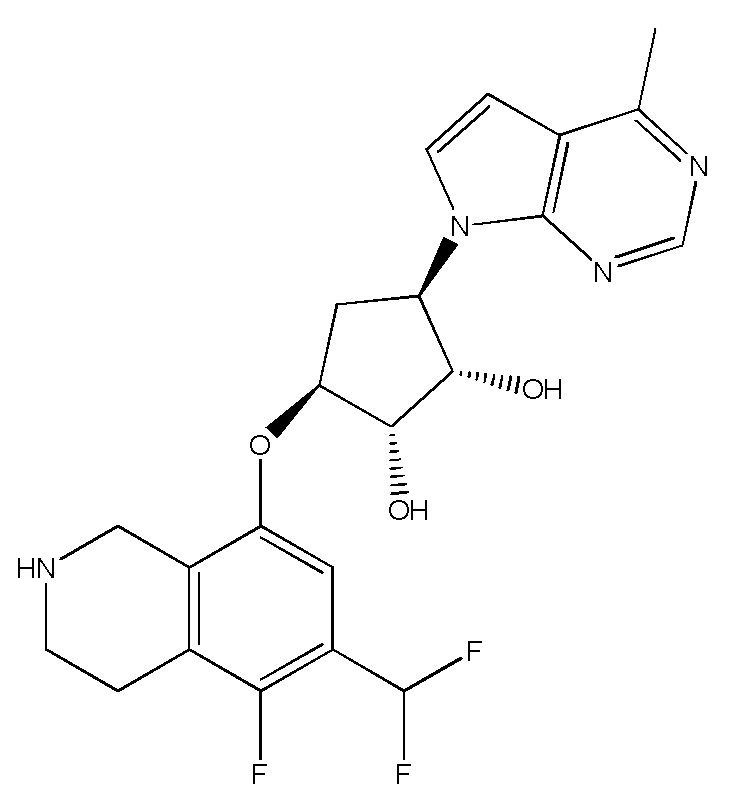
или его фармацевтически приемлемую соль и фармацевтически приемлемый носитель.
24. Соединение по любому из пп. 1-13 и 21, где указанную соль выбирают из фосфата, гидрофосфата и дигидрофосфата.
25. Соединение по любому из пп. 1-3 и 21, где указанную соль выбирают из фосфата, гидрофосфата и дигидрофосфата.
26. Способ лечения аномального роста клеток у млекопитающего, где способ включает введение млекопитающему терапевтически эффективного количества соединения (1S,2S,3S,5R)-3-((6-(дифторметил)-5-фтор-1,2,3,4-тетрагидроизохинолин-8-ил)окси)-5-(4-метил-7Н-пирроло[2,3-d]пиримидин-7-ил)циклопентан-1,2-диола, имеющего структуру:
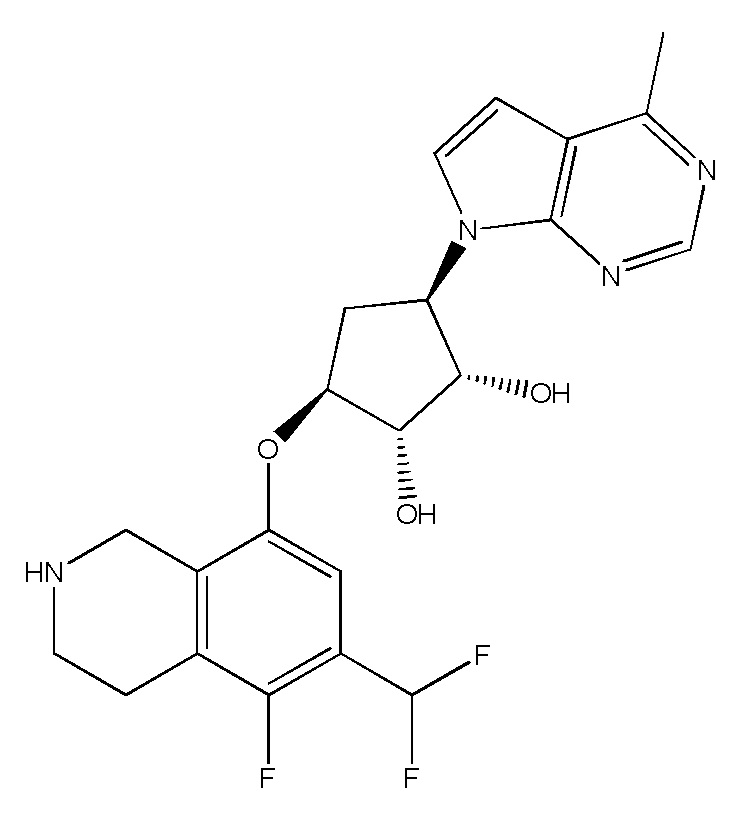
или его фармацевтически приемлемой соли.
27. Способ лечения рака у млекопитающего, где способ включает введение млекопитающему терапевтически эффективного количества соединения (1S,2S,3S,5R)-3-((6-(дифторметил)-5-фтор-1,2,3,4-тетрагидроизохинолин-8-ил)окси)-5-(4-метил-7Н-пирроло[2,3-d]пиримидин-7-ил)циклопентан-1,2-диола, имеющего структуру:
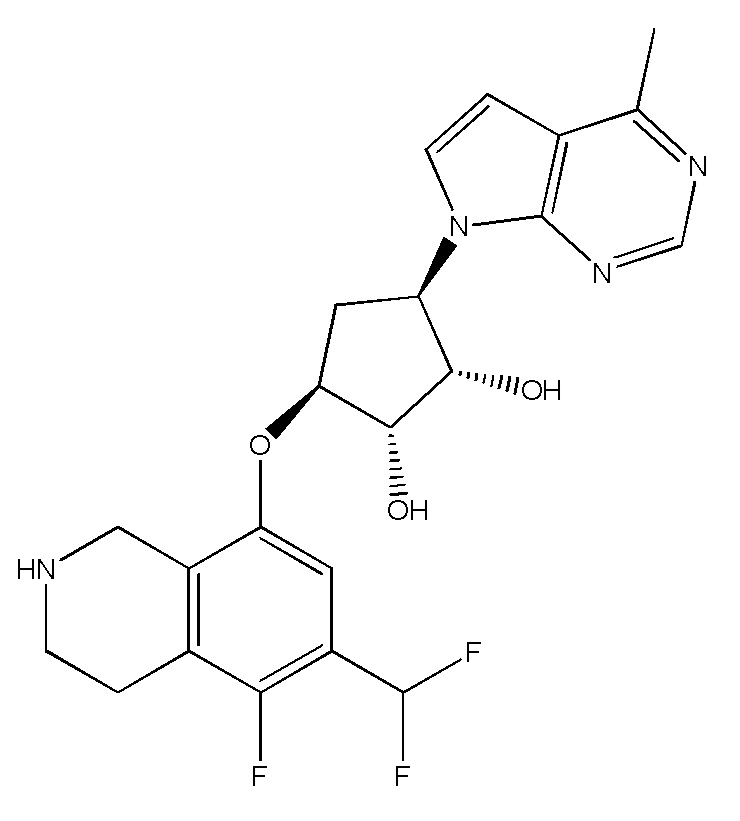
или его фармацевтически приемлемой соли.
28. Способ по п.27, где раком является рак легких, рак костей, рак поджелудочной железы, рак кожи, рак головы или шеи, кожная или внутриглазная меланома, рак матки, рак яичников, рак прямой кишки, рак анальной области, рак желудка, рак толстой кишки, рак молочной железы, рак матки, рак маточных труб, рак эндометрия, рак шейки матки, рак влагалища, рак вульвы, болезнь Ходжкина, рак пищевода, рак тонкой кишки, рак эндокринной системы, рак эндокринной системы, рак щитовидной железы, рак околощитовидной железы, рак надпочечников, саркома мягких тканей, рак мочеиспускательного канала, рак полового члена, рак предстательной железы, хронический или острый лейкоз, лимфоцитарные лимфомы, рак мочевого пузыря, рак почки или мочеточника, почечно-клеточный рак, рак почечной лоханки, новообразования центральной нервной системы (ЦНС), первичная лимфома ЦНС, опухоли позвоночника, глиома ствола головного мозга или аденома гипофиза.
29. Способ по п.27, где рак представляет собой лимфому.
30. Способ по п.27, где рак представляет собой рак толстой кишки.
31. Способ по п.27, где рак представляет собой рак пищевода.
32. Способ по п.27, где рак представляет собой рак желудка.
33. Способ по п.27, где рак представляет собой рак легких.
34. Способ по п.27, где рак представляет собой рак молочной железы.
35. Способ по п.27, где рак представляет собой рак яичников.
36. Способ по п.27, где рак представляет собой рак мочевого пузыря.
37. Способ по п.27, где рак представляет собой рак шейки матки или рак матки.
38. Способ по п.27, где рак представляет собой рак поджелудочной железы.
39. Способ по п.27, где рак представляет собой лейкоз.
| S.R | |||
| Das and A.W | |||
| Кипятильник для воды | 1921 |
|
SU5A1 |
| Приспособление для указания нагревания подшипников | 1924 |
|
SU668A1 |
| WO 2014100719 A2, 26.06.2014 | |||
| WO 2015013256 A1 A1 A1, 29.01.2015 | |||
| WO 2015200680 A2, 30.12.2015 | |||
| НОВОЕ ПРОИЗВОДНОЕ ПУРИНИЛПИРИДИНИЛАМИНО-2,4-ДИФТОРФЕНИЛСУЛЬФОНАМИДА, ЕГО ФАРМАЦЕВТИЧЕСКИ ПРИЕМЛЕМАЯ СОЛЬ, СПОСОБ ЕГО ПОЛУЧЕНИЯ И ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ, ОБЛАДАЮЩАЯ ИНГИБИРУЮЩЕЙ АКТИВНОСТЬЮ В ОТНОШЕНИИ RAF-КИНАЗЫ, СОДЕРЖАЩАЯ ДАННОЕ СОЕДИНЕНИЕ В КАЧЕСТВЕ АКТИВНОГО ИНГРЕДИЕНТА | 2011 |
|
RU2550038C2 |

